Embed Size (px)
Citation preview

Cinétique 2SESSION 2014-2015 (4 h)
Le sujet est composé de plusieurs parties largement indépendantes. Si vous estimez déjà avoir répondu à unequestion dans une autre partie, n’hésitez pas à ne pas y répondre de nouveau.
Toutes les réponses devront être justifiées et les résultats mis en valeur.
Table des matières
A. Éléments d’optimisation des réacteurs chimiques 2A.I. Étude de la marche isotherme d’un réacteur discontinu . . . . . . . . . . . . . . . . . . . . . . . . . . 2A.II. Étude du fonctionnement adiabatique d’un réacteur continu tubulaire . . . . . . . . . . . . . . . . . 5
B. Transfert intramoléculaire d’atome d’hydrogène 11B.I. Modèle cinétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11B.II. Résultats expérimentaux . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
C. Réacteur piston à recyclage 21C.I. Réacteur parfaitement agité continu . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21C.II. Réacteur piston en régime permanent . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21C.III.Réacteur piston à recyclage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23C.IV. Comparaison d’un R.P.A.C, d’un R.P. et d’un réacteur piston à recyclage . . . . . . . . . . . . . . . . 25C.V. Optimisation d’un réacteur piston à recyclage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
D. Théorie de l’état de transition 31D.I. Formulation de la constante de vitesse dans le cadre de la théorie de l’état de transition . . . . . . . 31D.II. Application à la détermination du mécanismes d’échange de molécules de solvant autour de cations
métalliques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
E. Constantes numériques 38
F. Formulaire 38

2
A. Éléments d’optimisation des réacteurs chimiques
Ce problème a pour but de présenter quelques aspects relatifs à la conception et à l’optimisation des réacteurschimiques. La réaction étudiée se déroule en phase gazeuse selon l’équilibre (1) :
A(g)
k1�k2
B(g) (1)
Les réactions directe et inverse sont supposées d’ordre 1 et k1 et k2 désignent les constantes de vitesse corres-pondantes. On donne :
k1 = k01 exp(− E1
RT
)avec k01 = 2,95 · 107 s−1 et E1 = 46,4 kJ ·mol−1 (2)
k2 = k02 exp(− E2
RT
)avec k02 = 1,57 · 1018 s−1 et E2 = 118,4 kJ ·mol−1 (3)
(4)
où R = 8,3145 J ·mol−1 ·K−1 est la constante des gaz parfaits et T est la température en kelvins.On peut démontrer et on admet donc que la relation est exothermique dans le sens A→ B .Toutes les courbes mentionnées dans ce problème doivent être tracées sur le même graphique, à partir de quelques points
calculés. L’échelle est imposée : en abscisse, 1 cm pour 10 K, T variant de 250 K à 420 K ; et en ordonnée, 10 cm pour uneunité.
A.I. Étude de la marche isotherme d’un réacteur discontinu
Un réacteur discontinu est l’appareillage le plus simple que l’on puisse envisager pour réaliser une transfor-mation chimique. Il consiste en un récipient de volume V dans lequel sont introduits les réactifs au début del’opération. Dans cette partie du problème, la température du réacteur est maintenue constante. À l’instant initial(t = 0) , la quantité du composé A est n0 et il n’y a pas de composé B . Pour l’instant t courant, on note α le taux detransformation de A, nA, la quantité de A dans le réacteur et nB la quantité de B.
A.1. La quantité de matière totale dépend-elle de α ?
Solution: Comme la réaction a une stœchiométrie 1 :1, la quantité de matière totale est conservée. Lapression est donc indépendante de l’avancement de la réaction.
A.2. Exprimer la vitesse volumique v =1V
d nB
d tet en déduire l’expression de r =
d α
d ten fonction de k1, k2 et α.
Solution:
v = k1 [A]− k2 [B] =1V
d αn0
d t=
1V
n0 (k1(1− α)− k2α) (5)
On en déduit :
r = k1(1− α)− k2α (6)
A.3. Donner l’expression de la constante d’équilibre K◦(T) en fonction de k1 et k2 à l’équilibre.
Solution:
K◦ =PB,éq
PA,éq=
[B]éq
[A]éq(7)

3
Remarque : Ici, l’égalité pour la constante d’équilibre dans l’échelle des pressions et celle dans l’échelle desconcentrations n’est valable que parce que les gaz sont considérés parfaits et qu’il y a une stœchiométrie1 :1.D’autre part, à l’équilibre, il n’y a plus d’évolution macroscopique :
k1 [A]éq − k2 [B]éq = 0⇐⇒[B]éq
[A]éq=
k1
k2(8)
Donc :
K◦ =k1
k2=
k01
k02exp
(E2 − E1
RT
)(9)
A.4. Quelle est l’influence sur l’équilibre (1) d’une élévation de température ?
Solution:
∂ ln K◦
∂ T= −E2 − E1
RT2 < 0 (10)
La constante d’équilibre décroit avec la température.
A.5. Calculer la valeur de K◦(T) pour T1 = 300 K et T2 = 400 K.
Solution: On trouve
K◦(T1) =2,95 · 107
1,57 · 1018 exp(
118,4 · 103 − 46,4 · 103
8,3145× 300
)= 64,6 (11)
et
K◦(T2) =2,95 · 107
1,57 · 1018 exp(
118,4 · 103 − 46,4 · 103
8,3145× 400
)= 4,74 · 10−2 (12)
A.6. Établir, en fonction de k1 , k2 et τ, l’expression de ατ , taux de transformation de A au bout d’un temps τ passédans le réacteur.
Solution: Il faut intégrer l’équation (6) :
d α
d t+ (k1 + k2)α = k1 (13)
Dont une solution particulière estk1
k1 + k2. Comme α(0) = 0, on en déduit :
α(t) =k1
k1 + k2
(1− e−(k1+k2)t
)(14)
A.7. Donner l’expression de αéq , taux de transformation de A lorsque l’équilibre thermodynamique est atteint.
Solution: L’équilibre correspond à t→ ∞ :
αéq =k1
k1 + k2=
KK + 1
(15)
4
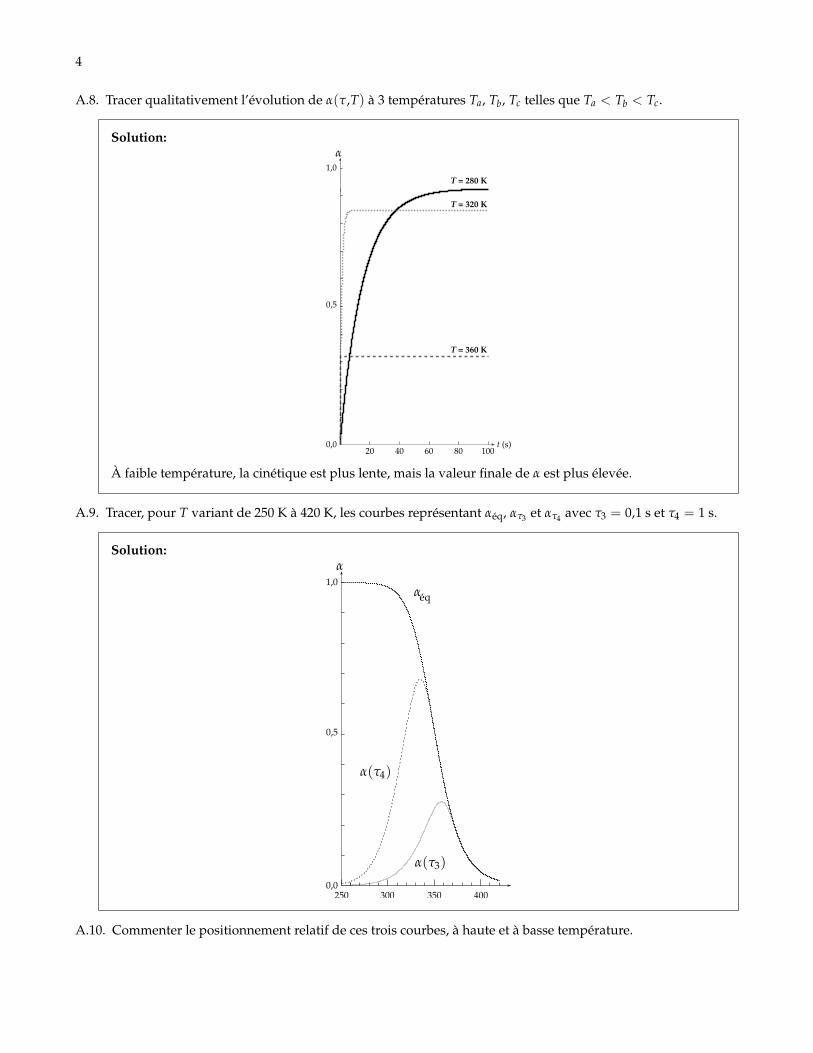
A.8. Tracer qualitativement l’évolution de α(τ,T) à 3 températures Ta, Tb, Tc telles que Ta < Tb < Tc.
Solution:
0,0
0,5
1,0T = 280 K
T = 320 K
T = 360 K
20 40 60 80 100t (s)
À faible température, la cinétique est plus lente, mais la valeur finale de α est plus élevée.
A.9. Tracer, pour T variant de 250 K à 420 K, les courbes représentant αéq, ατ3 et ατ4 avec τ3 = 0,1 s et τ4 = 1 s.
Solution:
250 300 350 4000,0
0,5
1,0
éq
A.10. Commenter le positionnement relatif de ces trois courbes, à haute et à basse température.

5
Solution: Pour αéq, plus la température augmente, plus la constante d’équilibre diminue. Pour les faiblestempératures (K◦ � 1), le taux de dissociation est proche de 1 alors qu’il tend vers 0 lorsque K◦ � 1 auxhautes-températures.Plus la valeur de τ est grande, plus la courbe se rapproche de la courbe correspondant à αéq. Commeα(τ,T = T0) est strictement croissante, on a donc toujours ατ3 < ατ3 < αéq.Pour toutes les courbes, au-delà d’une température, on rejoint la courbe αéq, en effet, lorsque τ est de
l’ordre de quelques1
k1(T) + k2(T), l’équilibre est atteint.
À l’inverse, aux faibles températures, la vitesse de réaction est très faible et l’équilibre n’a pas le tempsd’être atteint pour les valeurs de τ données. Par conséquent, le taux de conversion est lui aussi très faiblemême si la réaction est favorable thermodynamiquement.Sur cet exemple, il y a opposition entre cinétique et thermodynamique. Augmenter la températureaméliore la cinétique mais diminue le rendement maximal.
A.11. Déterminer graphiquement, pour τ3 et τ4, les températures optimales T3 et T4 de marche isotherme.
Solution: On optimise le processus pour le taux de conversion maximal, il faut donc regarder les maximades deux courbes.
T4 ≈ 335 K T3 ≈ 360 K (16)
Comme le maximum n’est pas sur la courbe αéq, ces optimums correspondent à des situations hors-équilibre.
A.II. Étude du fonctionnement adiabatique d’un réacteur continu tubulaire
Un réacteur tubulaire est constitué, dans sa forme élémentaire, d’un tube à l’intérieur duquel le milieu réactionnelcircule de façon isobare. Dans le cas d’un réacteur adiabatique (noté R.T.A.), les transferts thermiques avec l’ex-térieur sont nuls. De plus, la vitesse d’écoulement est suffisamment faible pour que l’on considère que le milieuréactionnel suit une évolution isenthalpique. La figure ci-après représente, pour un R.T.A., l’évolution entre lesinstants t et t + dt d’un système fermé au sein duquel se produit une réaction chimique.n0C◦P représente la capacité thermique à pression constante du système envisagé. Cette grandeur est indépendantede la constitution du système.
Sens de l'écoulement
parois adiabatiques
Système à l'instant t
T
Système à l'instant t + dt
Figure 1 – Évolution du système entre t et t + dt dans un R.T.A.
A.12. Faire un bilan d’enthalpie pour le système thermodynamique indiqué ci-dessus et en déduired Td α
en fonc-tion de ∆rH◦ et C◦P.

6
Solution: Comme le système suit une évolution isenthalpique :
H(t) = H(t + dt) (17)n0 (αHB(T) + (1− α)HA(T)) = n0 ((α + dα)HB(T + dT) + (1− α− dα)HA(T + dT)) (18)α (HB(T)− HA(T)) + HA(T) = (α + dα) (HB(T + dT)− HA(T + dT)) + HA(T + dT) (19)
De plus :
Hi(T + dT) = Hi(T) + C◦P,idT (20)
et la capacité calorifique du système est égale à :
n0(αCP,B + (1− α)CP,A) = n0C◦P (21)
Comme cette grandeur est indépendante de la constitution du système, cela indique que :
C◦P = C◦P,A = C◦P,B (22)
Ces deux relations permettent de ré-écrire l’équation (19) avec ∆rH◦ = HB(T)− HA(T) qui est indépen-dant de la température :
α∆rH◦ + HA(T) = (α + dα)∆rH◦ + HA(T) + C◦PdT (23)
⇐⇒ d Td α
= − ∆rH◦
C◦P(24)
A.13. Exprimer alors T en fonction de α, en notant T0 la température à l’entrée du réacteur pour laquelle α = 0.
Solution: L’intégration de la relation précédente donne :
T − T0 = α×−∆rH◦
C◦P(25)
Remarque : Ici, l’intégration est directe car la relation (22) indique que ∆rC◦p = 0 et donc que ∆rH◦ estindépendant de la température.
A.14. Tracer, sur le graphique précédent, la courbe correspondant à une marche adiabatique à partir d’une tem-
pérature d’entrée T0 = 320 K et pour
∣∣∣∣∣∆rH◦
C◦p
∣∣∣∣∣ = 50 K. Indiquer, par une flèche, le sens d’évolution du système
sur cette courbe.
Solution: Ici, la réaction est exothermique, la pente est donc positive et affine.
7
250 300 350 4000,0
0,5
1,0
éq
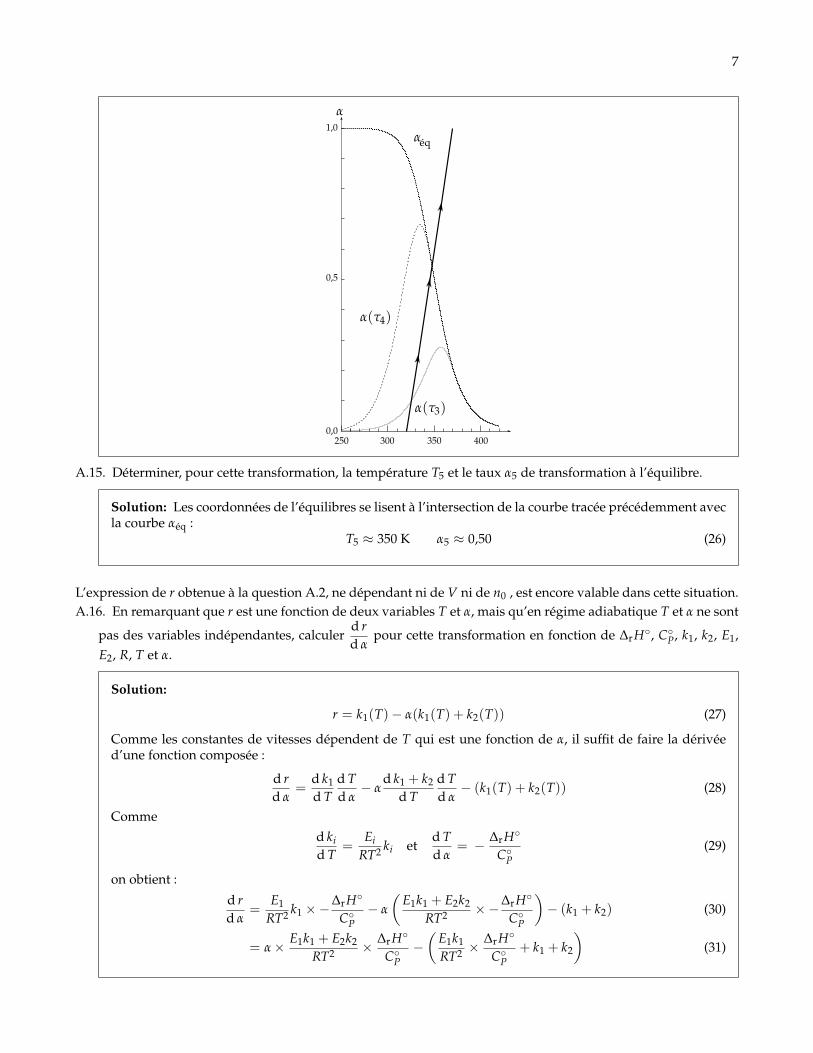
A.15. Déterminer, pour cette transformation, la température T5 et le taux α5 de transformation à l’équilibre.
Solution: Les coordonnées de l’équilibres se lisent à l’intersection de la courbe tracée précédemment avecla courbe αéq :
T5 ≈ 350 K α5 ≈ 0,50 (26)
L’expression de r obtenue à la question A.2, ne dépendant ni de V ni de n0 , est encore valable dans cette situation.A.16. En remarquant que r est une fonction de deux variables T et α, mais qu’en régime adiabatique T et α ne sont
pas des variables indépendantes, calculerd rd α
pour cette transformation en fonction de ∆rH◦, C◦P, k1, k2, E1,E2, R, T et α.
Solution:
r = k1(T)− α(k1(T) + k2(T)) (27)
Comme les constantes de vitesses dépendent de T qui est une fonction de α, il suffit de faire la dérivéed’une fonction composée :
d rd α
=d k1
d Td Td α− α
d k1 + k2
d Td Td α− (k1(T) + k2(T)) (28)
Commed kid T
=Ei
RT2 ki etd Td α
= − ∆rH◦
C◦P(29)
on obtient :
d rd α
=E1
RT2 k1 ×−∆rH◦
C◦P− α
(E1k1 + E2k2
RT2 ×−∆rH◦
C◦P
)− (k1 + k2) (30)
= α× E1k1 + E2k2
RT2 × ∆rH◦
C◦P−(
E1k1
RT2 ×∆rH◦
C◦P+ k1 + k2
)(31)
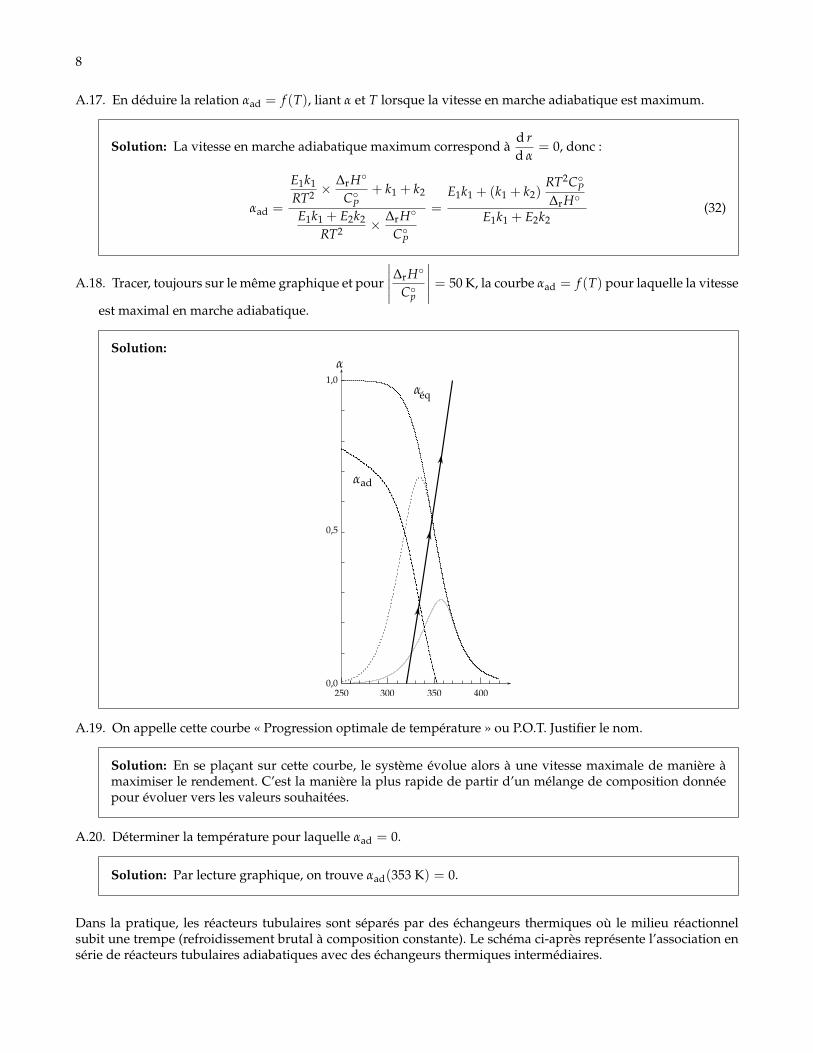
8
A.17. En déduire la relation αad = f (T), liant α et T lorsque la vitesse en marche adiabatique est maximum.
Solution: La vitesse en marche adiabatique maximum correspond àd rd α
= 0, donc :
αad =
E1k1
RT2 ×∆rH◦
C◦P+ k1 + k2
E1k1 + E2k2
RT2 × ∆rH◦
C◦P
=E1k1 + (k1 + k2)
RT2C◦P∆rH◦
E1k1 + E2k2(32)
A.18. Tracer, toujours sur le même graphique et pour
∣∣∣∣∣∆rH◦
C◦p
∣∣∣∣∣ = 50 K, la courbe αad = f (T) pour laquelle la vitesse
est maximal en marche adiabatique.
Solution:
250 300 350 4000,0
0,5
1,0
éq
A.19. On appelle cette courbe « Progression optimale de température » ou P.O.T. Justifier le nom.
Solution: En se plaçant sur cette courbe, le système évolue alors à une vitesse maximale de manière àmaximiser le rendement. C’est la manière la plus rapide de partir d’un mélange de composition donnéepour évoluer vers les valeurs souhaitées.
A.20. Déterminer la température pour laquelle αad = 0.
Solution: Par lecture graphique, on trouve αad(353 K) = 0.
Dans la pratique, les réacteurs tubulaires sont séparés par des échangeurs thermiques où le milieu réactionnelsubit une trempe (refroidissement brutal à composition constante). Le schéma ci-après représente l’association ensérie de réacteurs tubulaires adiabatiques avec des échangeurs thermiques intermédiaires.
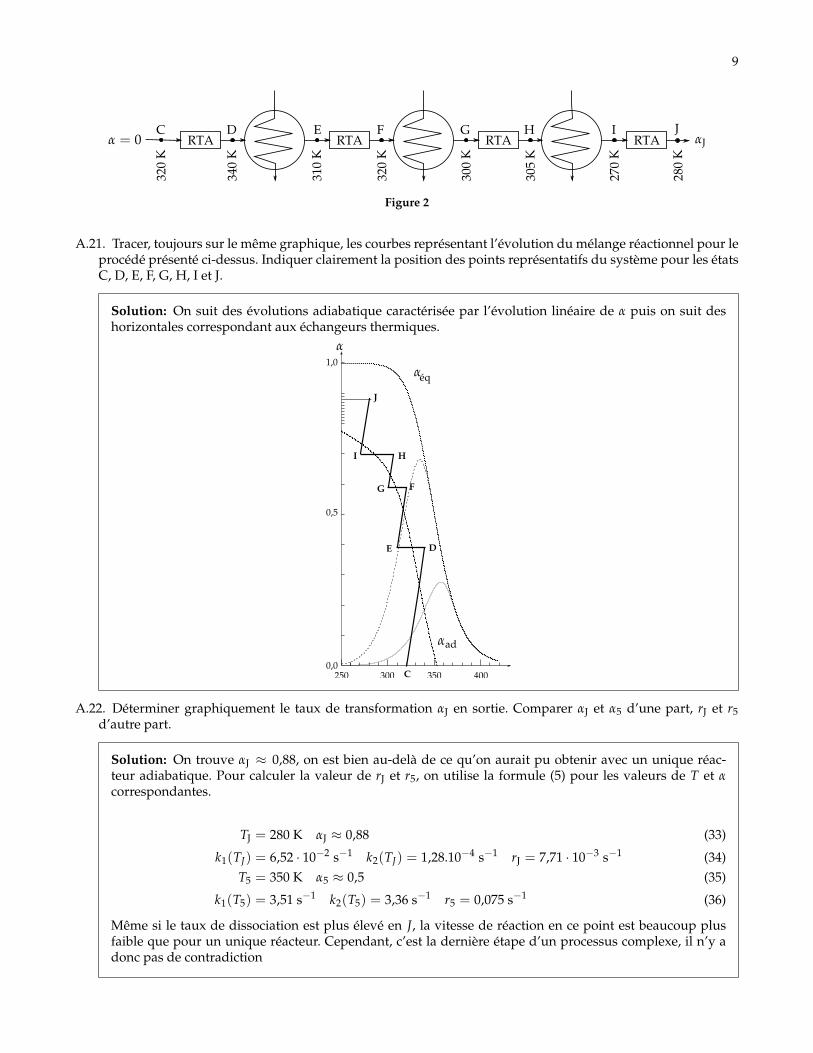
9
RTA RTA RTA RTAC D E F G H I J
320
K
340
K
310
K
320
K
300
K
305
K
270
K
280
K
Figure 2
A.21. Tracer, toujours sur le même graphique, les courbes représentant l’évolution du mélange réactionnel pour leprocédé présenté ci-dessus. Indiquer clairement la position des points représentatifs du système pour les étatsC, D, E, F, G, H, I et J.
Solution: On suit des évolutions adiabatique caractérisée par l’évolution linéaire de α puis on suit deshorizontales correspondant aux échangeurs thermiques.
250 300 350 4000,0
0,5
1,0
éq
C
DE
FG
HI
J
A.22. Déterminer graphiquement le taux de transformation αJ en sortie. Comparer αJ et α5 d’une part, rJ et r5d’autre part.
Solution: On trouve αJ ≈ 0,88, on est bien au-delà de ce qu’on aurait pu obtenir avec un unique réac-teur adiabatique. Pour calculer la valeur de rJ et r5, on utilise la formule (5) pour les valeurs de T et αcorrespondantes.
TJ = 280 K αJ ≈ 0,88 (33)
k1(TJ) = 6,52 · 10−2 s−1 k2(TJ) = 1,28.10−4 s−1 rJ = 7,71 · 10−3 s−1 (34)T5 = 350 K α5 ≈ 0,5 (35)
k1(T5) = 3,51 s−1 k2(T5) = 3,36 s−1 r5 = 0,075 s−1 (36)
Même si le taux de dissociation est plus élevé en J, la vitesse de réaction en ce point est beaucoup plusfaible que pour un unique réacteur. Cependant, c’est la dernière étape d’un processus complexe, il n’y adonc pas de contradiction

10
A.23. Conclure quant à l’intérêt de l’utilisation d’un tel dispositif.
Solution: Avec ce dispositif, on suit du mieux possible la courbe αad pour laquelle la vitesse de dissocia-tion est maximale. De plus, les étapes de refroidissement permettent d’aller vers une situation où le tauxde dissociation est élevé.Avec ce processus, on optimise donc simultanément la contrainte cinétique en suivant le maximum devitesse pour le réacteur adiabatique et la contrainte thermodynamique qui indique qu’il faut se placer àfaible température pour maximiser le rendement.Plus on avance dans l’enchaînement de réacteurs, plus la vitesse de réaction est lente mais plus α aug-mente.

11
B. Transfert intramoléculaire d’atome d’hydrogène
Ce transfert que l’on peut schématiser par : Oa −H · · ·Ob → Oa · · ·H−Ob a été l’objet de nombreuses étudespour déterminer notamment s’il s’agit d’un mouvement localisé (seul H se déplace) ou couplé à d’autres mouve-ments intramoléculaires.
Les expériences ayant des temps de résolution de l’ordre de quelques femtosecondes permettent de créer despaquets d’ondes localisés et d’en préciser l’évolution : un « mouvement direct » de H selon la coordonnée deréaction Oa −H · · ·Ob ou bien un mouvement faisant intervenir plusieurs modes de vibration de la molécule.
Le système étudié est le 2-hydroxybenzoate de méthyle (ou salicylate de méthyle) gazeux, à une pressionsuffisamment basse pour que la dynamique étudiée ne soit pas affectée par les collisions. Oa représente l’atomed’oxygène de la fonction phénol et Ob celui du carbonyle de la fonction ester.
Le salicylate de méthyle possède deux rotamères A et B, dont les propriétés spectroscopiques schématisées surla Figure 3 sont les suivantes :
— la forme excitée de B, notée B∗ émet dans l’U.V. (pic de fluorescence pour λmax = 330 nm) ;— la fluorescence associée à A∗, état excité de A, est décalée vers le bleu (λmax = 440 nm) à cause du passage
d’une forme excitée ester (A∗) vers une forme excitée de type énol, notée T∗, qui émet dans le bleu ;— la possibilité de passage ester→ énol n’existe pas pour le rotamère B.
Figure 3 – Schéma simplifié des propriétés spectroscopiques des deux rotamères A et B du salicylate de méthyle.λ0−0 correspond à la transition entre les deux niveaux d’énergie de point zéro.
L’expérience comporte une première impulsion laser (pompe) de longueur d’onde λpu ajustable (compriseentre 280 et 330 nm) qui a pour effet de former les états excités A∗ et B∗. Après un délai variable td, une deuxièmeimpulsion (sonde) de longueur d’onde λpr = 610 nm agit sur les populations des états excités A∗, B∗ et T∗. Laconséquence est une diminution de l’intensité de fluorescence mesurée à 440 ou à 330 nm, dont l’importanceest reliée aux populations des états excités avant l’impulsion sonde à td. On peut ainsi remonter à l’évolutiontemporelle des populations des divers états excités.
B.I. Modèle cinétique
Pour analyser les résultats des expériences, on envisage le modèle cinétique suivant :— Rotamère A :
L’état fondamental de la molécule correspond à un état noté |a〉, A∗ (resp. T∗.) correspond à |b〉 (resp. |d〉)

12
— kt est la constante de vitesse de transfert de l’état |b〉 vers l’état |d〉.— fb, (resp. fd) représente la constante de vitesse pour la fluorescence de |b〉 vers |a〉 à 330 nm (resp. de |d〉
vers |e〉 à 440 nm).— knr est la constante de vitesse de transfert non radiatif de l’état |d〉 vers un état noté | f 〉.— βσ0 est la section efficace d’absorption moléculaire pour le faisceau sonde de l’état |b〉 vers un état noté|c〉 et (1− β)σ0 celle pour l’absorption associée au faisceau sonde de l’état |d〉 vers un état noté |g〉, avec0 6 β 6 1 et σ0 représentant une section efficace constante.
On indique que la fluorescence est un processus beaucoup plus lent que le transfert d’atome d’hydrogèneétudié ici : f−1
b et f−1d sont de l’ordre de grandeur de la nanoseconde alors que k−1
t et k−1nr appartiennent au
domaine de la picoseconde ou de la femtoseconde.— Rotamère B :
Pour le rotamère B, le même modèle est applicable en prenant kt = knr = 0 et en ne considérant que lesétats |a〉, |b〉 et |c〉 pour décrire respectivement B, B∗ et l’état résultant de l’excitation par l’impulsion sondede B∗ (la section efficace est alors σ0).
B.1. Proposer une structure moléculaire pour A, B et T∗.
Solution:
OH
O
O
O
O
H
OOH
O
O
A T*B
La forme A doit pouvoir conduire à une structure de type énol, c’est ce qui permet de déterminer le lienentre conformère et dénomination. En effet, la structure B ne conduit pas à une forme énol après transfertde l’hydrogène.
B.2. Représenter, à l’aide d’un schéma énergétique, les états |a〉 à |g〉 et les transitions entre ces états pour lerotamère A. Même question pour le rotamère B.
Solution: À l’aide du texte, on aboutit au deux schémas suivants :— Schéma pour le rotamère A :
A
A*
T
T*
kt
fbfd
knr
— Schéma pour le rotamère B :

13
B
B*
fb
L’excitation a lieu pour passer de A à A∗ (B à B∗) avec le pulse à λpu
On suppose que chaque impulsion a une durée infiniment courte, l’impulsion pompe étant appliquée à t = 0,l’impulsion sonde à td. On distingue différents régimes de temps :
1. t < 0 :— na(t) = N nombre de molécules dans la région d’interaction avec le faisceau laser.— nb(t) = nc(t) = · · · = ng(t) = 0 où ni(t) représente la population de l’état |i〉 à l’instant t.
2. t = 0— na(0) = N − n0
— nb(0) = n0 =NσabλpuEpu
hcπR2 où σab représente la section efficace d’adsorption moléculaire correspondant aufaisceau pompe, d’énergie Epu localisé dans une sphère de rayon R.
— nc(0) = · · · = ng(0) = 0.
3. 0 6 t 6 td c’est à dire entre les impulsions pompe et sonde.
B.3. Comment s’exprimentd na
d t,
d nbd t
,d ndd t
,d ne
d ten fonction des populations nb, nd et des différentes constantes
cinétiques. On étudiera le cas du rotamère A pour 0 6 t 6 td.
Solution:— L’état |a〉 est peuplé uniquement par fluorescence depuis |b〉
d na
d t= fbnb (37)
— Il y a désexcitation depuis |b〉 vers |a〉 par fluorescence et vers |d〉 :
d nbd t
= − fbnb − ktnb = − (kt + fb) nb (38)
— l’état |d〉 est peuplé par transfert depuis |b〉 et dépeuplé par fluorescence vers |e〉 ou vers | f 〉 par unphénomène non radiatif :
d ndd t
= ktnb − knrnd − fdnd = ktnb − (knr + fd)nd (39)

14
— l’état |e〉 est peuplé par fluorescence depuis |d〉 :
d ne
d t= fdnd (40)
B.4. Même question pour le rotamère B.
Solution: De manière analogue, on obtient :
d na
d t= fbnb (41)
d nbd t
= − fbnb (42)
B.5. Pour le rotamère A, simplifier les équations obtenues dans le cas (auquel on se limitera par la suite) où fb � ktet fd � knr.
Solution: On peut négliger les termes liés à la fluorescence devant les autres termes :
d na
d t= fbnb (43)
d nbd t≈ − ktnb (44)
d ndd t≈ ktnb − knrnd (45)
d ne
d t= fdnd (46)
(47)
B.6. Quelles sont, pour 0 6 t 6 td, les expressions de nb(t), na(t), nd(t) et ne(t) pour le rotamère A ?
Solution: On commence par la population nb(t) pour laquelle on intègre la relation précédente avecnb(0) = n0 :
nb(t) = n0 exp(−ktt)) (48)
On en déduit l’expression de na(t) pour laquelle on doit avoir na(0) = N − n0 :
na(t) = A︸︷︷︸solution sans second membre
− fbn0
ktexp (−ktt)︸ ︷︷ ︸
solution particulière
(49)
na(t) = N − n0 +fbn0
kt(1− exp(−ktt)) (50)
On peut ensuite en déduire l’expression de nd(t) pour lequel nd(0) = 0 :
nd(t) = B exp (−knrt) Solution générale (51)
Pour trouver une solution particulière, on peut utiliser la méthode de variation de la constante en cher-chant une solution particulière de la forme nd(t) = B(t) exp (−knrt) :
d Bd t
= ktn0 exp ((knr − kt) t) (52)
B(t) =ktn0
knr − ktexp ((knr − kt) t) + C (53)

15
On en déduit l’expression finale de nd(t) :
nd(t) =ktn0
knr − kt(exp (−ktt)− exp (−knrt)) (54)
Pour ne(t) avec ne(0) = 0 :
ne(t) = D +fdktn0
knr − kt
(exp (−ktt)−kt
− exp (−knrt)−knr
)(55)
=fdktn0
knr − kt
(1− exp (−ktt)
−kt− exp (−knrt)− 1
−knr
)(56)
Pour résumer :
nb(t) = n0 exp(−ktt)) (57)
na(t) = N − n0 +fbn0
kt(1− exp(−ktt)) (58)
nd(t) =ktn0
knr − kt(exp (−ktt)− exp (−knrt)) (59)
ne(t) =fdktn0
knr − kt
(1− exp (−ktt)
−kt− exp (−knrt)− 1
−knr
)(60)
B.7. Même question pour nb(t) et na(t) dans le cas du rotamère B.
Solution: Ici, on ne peut pas négliger la fluorescence et nb(t) + na(t) = N donc :
nb(t) = n0 exp(− fbt) (61)na(t) = N − n0 exp(− fbt)) (62)
Pour t = td on a :
nb(t+d ) = nb(t−d ) (1− αβσ0) (63)
nc(t+d ) = nc(t−d )αβσ0 (64)
nd(t+d ) = nd(t−d ) (1− α (1− β) σ0) (65)
ng(t+d ) = nd(t−d )α (1− β) σ0 (66)
(67)
avec α =λprEpr
hcπR2 où Epr représente l’énergie totale de l’impulsion sonde et R le rayon de la sphère où est focaliséle faisceau.
B.8. Quelle est l’expression, pour le rotamère A, de nb(t) pour t > td ? Même question pour le rotamère B.

16
Solution: Il suffit de reprendre les deux questions précédente en changeant la population initiale :
nb,rotamère A(t > td) = n0 exp(−kttd)︸ ︷︷ ︸nb(t
−d )
(1− αβσ0) exp(−kt(t− td)) (68)
nb,rotamère B(t > td) = n0 exp(− fbtd)︸ ︷︷ ︸nb(t
−d )
(1− αβσ0) exp(− fb(t− td)) (69)
(70)
Pour un délai td donné, le signal détecté dans l’U.V., dû à un rotamère est proportionnel au nombre Ma(td) demolécules qui fluorescent de l’état |b〉 vers l’état |a〉 entre t = 0 et t→ ∞.
B.9. Montrer que pour le rotamère A on a :
Ma(td) =n0 fb
kt[1− αβσ0 exp(−kttd)] (71)
Solution:
Ma,A(td) =∫ td
0fbnb(t 6 td)dt︸ ︷︷ ︸
molécules fluorescant avant td
+∫ ∞
td
fbnb(t > td)dt︸ ︷︷ ︸molécules fluorescant après td
(72)
=∫ td
0fbn0 exp(−ktt))dt +
∫ ∞
td
fbn0 exp(−kttd) (1− αβσ0) exp(−kt(t− td))dt (73)
=fbn0
−kt(exp(−kttd)− 1)− fbn0 (1− αβσ0)
−ktexp(−kttd) (74)
=n0 fb
kt[1− αβσ0 exp(−kttd)] (75)
B.10. Calculer Ma(td) pour le rotamère B. Quel rotamère a la contribution la plus importante à la fluorescencedans l’U.V. ?
Solution:
Ma,B(td) =∫ td
0fbn0 exp(− fbt))dt +
∫ ∞
td
fbn0 exp(− fbtd) (1− αβσ0) exp(− fb(t− td))dt (76)
= n0 [1− αβσ0 exp(− fbtd)] (77)
Comme fb � kt, le facteur pré-exponentiel ainsi que le terme exponentiel sont beaucoup plus grands pourle rotamère B que pour le rotamère A. C’est donc le rotamère B qui aura une plus grosse contribution à lafluorescence dans l’U.V.

17
Le signal détecté dans le bleu est proportionnel au nombre de molécules Me(td) qui fluorescent de |d〉 vers |e〉, onadmet l’expression suivante :
Me(td) =n0 fdknr
[1− ασ0
(β exp (−kttd) +
kt
kt − knr(1− β) [exp (−knrtd)− exp (−kttd)]
)](78)
B.II. Résultats expérimentaux
Les résultats d’expérience de dépeuplement sont représentés sur la Figure 4. Aux temps courts (0 < td < 300 fs),la courbe expérimentale (a), correspondant à l’émission à 440 nm, a pu être ajustée avec une seule fonction expo-nentielle contenant un terme exp(−t/τ1), avec une constante de temps τ1 = 60 fs.B.11. Le modèle cinétique permet-il de rendre compte des résultats expérimentaux de la Figure 4. Peut-on attribuer
les courbes (a) ou (b) à l’un ou l’autre des rotamères ?
Solution: Pour le rotamère B, seule la fluorescence est responsable de la population du fondamental avecune échelle de temps relativement longue, à l’inverse, pour le rotamère A, l’échelle de temps est contrôléepar kt qui est beaucoup plus courte. On en déduit que la courbe (a) correspond à la courbe de fluores-cence du rotamère A et que la courbe (b) correspond à la courbe de fluorescence du rotamère B. De plus,cela correspond au modèle cinétique pour lequel on avait montré que la contribution dans l’U.V. vientmajoritairement du rotamère B alors que la contribution dans le bleu vient du rotamère A.
B.12. Montrer que l’évolution aux temps longs de la courbe (a) peut également être ajustée par une seule expo-nentielle contenant un terme de la forme exp(−t/τ2). Déterminer numériquement τ2.
Solution: Au temps longs, on trouve également une allure exponentielle pour la courbe expérimentale.Avec la méthode de la tangente initiale, on trouve τ2 de l’ordre de 28 ps.
B.13. En déduire les valeurs de kt et knr pour cette expérience.
Solution: Au temps court, c’est la formation de T∗ qui est l’étape cinétiquement déterminante alors qu’autemps longs, c’est la dépopulation de |d〉 vers | f 〉 qui va contrôler la fluorescence. On en déduit :
kt =1τ1
=1
60 · 10−15 = 1,7 · 1013 Hz (79)
knr =1τ2
=1
28 · 10−12 = 3,6 · 1010 Hz (80)
(81)
La précision est moindre sur kt vu l’échelle de temps extrêmement courte.
B.14. Les courbes de fluorescence enregistrées dans l’U.V. ont pu être ajustées avec une seule exponentielle ayantune constante de temps longue, de l’ordre de 1 ns. En utilisant les courbes de la Figure 4, peut-on attribuer àT∗ l’émission dans l’U.V. ?
Solution: Ici, la courbe peut être paramétrisée avec l’exponentielle en 1/ fb. Et comme fb � kt,knr, lesignal UV ne peut pas être attribué à T∗ dont l’évolution suit des échelles de temps beaucoup plus courtes.
B.15. Que peut-on en conclure pour l’existence et la vitesse de transfert d’hydrogène dans la forme A∗ ?
Solution: On en déduit que pour le rotamère dans la forme A∗, il y a un transfert rapide (avec uneconstante de temps de l’ordre de kt � 1) vers la forme T∗ qui n’a pas lieu pour la forme B∗ (désexci-tation uniquement par fluorescence).
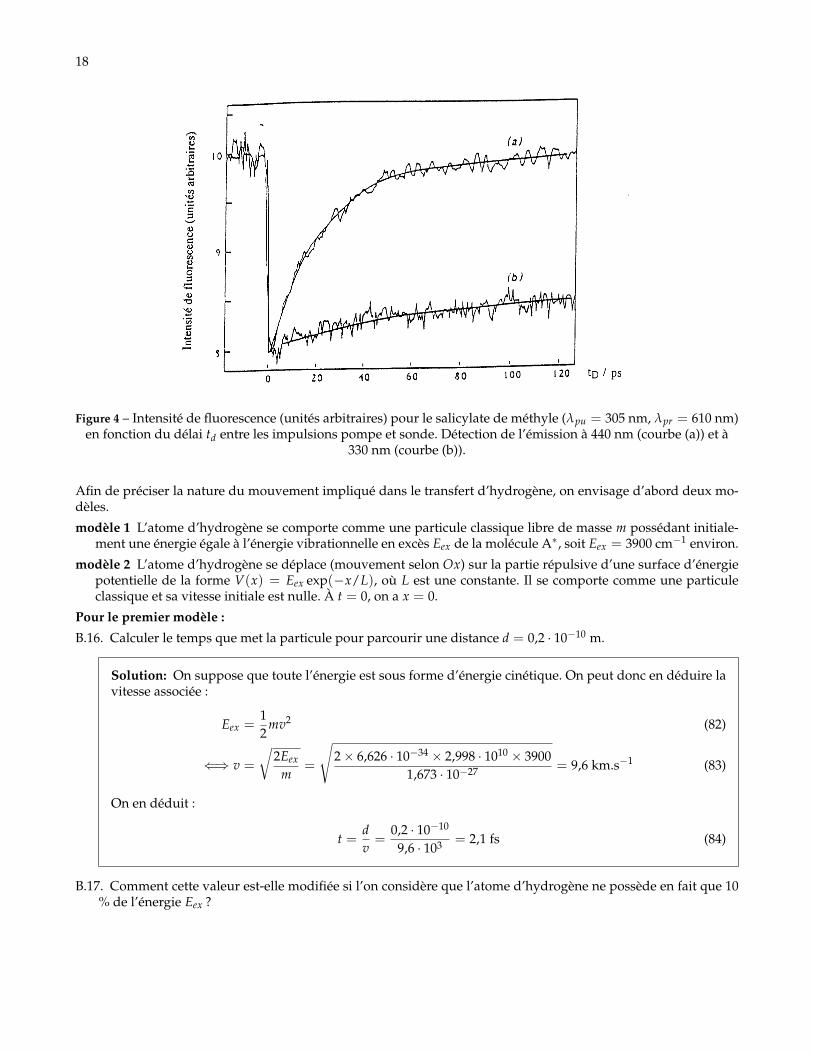
18
Figure 4 – Intensité de fluorescence (unités arbitraires) pour le salicylate de méthyle (λpu = 305 nm, λpr = 610 nm)en fonction du délai td entre les impulsions pompe et sonde. Détection de l’émission à 440 nm (courbe (a)) et à
330 nm (courbe (b)).
Afin de préciser la nature du mouvement impliqué dans le transfert d’hydrogène, on envisage d’abord deux mo-dèles.
modèle 1 L’atome d’hydrogène se comporte comme une particule classique libre de masse m possédant initiale-ment une énergie égale à l’énergie vibrationnelle en excès Eex de la molécule A∗, soit Eex = 3900 cm−1 environ.
modèle 2 L’atome d’hydrogène se déplace (mouvement selon Ox) sur la partie répulsive d’une surface d’énergiepotentielle de la forme V(x) = Eex exp(−x/L), où L est une constante. Il se comporte comme une particuleclassique et sa vitesse initiale est nulle. À t = 0, on a x = 0.
Pour le premier modèle :
B.16. Calculer le temps que met la particule pour parcourir une distance d = 0,2 · 10−10 m.
Solution: On suppose que toute l’énergie est sous forme d’énergie cinétique. On peut donc en déduire lavitesse associée :
Eex =12
mv2 (82)
⇐⇒ v =
√2Eex
m=
√2× 6,626 · 10−34 × 2,998 · 1010 × 3900
1,673 · 10−27 = 9,6 km.s−1 (83)
On en déduit :
t =dv=
0,2 · 10−10
9,6 · 103 = 2,1 fs (84)
B.17. Comment cette valeur est-elle modifiée si l’on considère que l’atome d’hydrogène ne possède en fait que 10% de l’énergie Eex ?

19
Solution:
t′ =t√0,1
=2,1√0,1
= 6,6 fs (85)
Pour le second modèle :
B.18. Montrer que le temps t nécessaire pour parcourir une distance d a pour expression :
t =
∫ d
0
d x√2Eex
m[1− exp (−x/L)]
(86)
Solution: On part de la conservation de l’énergie mécanique :
E =12
mv2 + V(x) (87)
v =d xd t
=
√2(Eex −V(x))
m(88)
On en déduit l’expression demandée :
t =
∫ d
0
d x√2Eex
m[1− exp (−x/L)]
(89)
On donne : ∫d x√
1− exp (−x/L)= 2L ln
(exp
( x2L
)+
√exp
( xL
)− 1)
(90)
et on prend L = 0,1 · 10−10 m.
B.19. Calculer t pour d = 0,2 · 10−10 m et Eex = 3900 cm−1.
Solution:
t =√
m2Eex
2L ln(
exp( x
2L
)+
√exp
( xL
)− 1)
(91)
=
√1,673 · 10−27
2× 6,626 · 10−34 × 2,998 · 1010 × 3900× 2× 0,1 · 10−10 ln
(e +
√exp(2)− 1
)(92)
= 3,4 fs (93)
B.20. Peut-on rendre compte des données expérimentales présentées ci-dessus à l’aide du premier modèle ? Mêmequestion pour le second modèle ?

20
Solution: Dans les deux cas, l’ordre de grandeur du temps nécessaire est de l’ordre de la femtosecondealors qu’expérimentalement, le temps du transfert est de l’ordre de τ1 = 60 fs. Donc aucun des deuxmodèles ne permet de rationaliser le transfert d’hydrogène.
B.21. On dispose d’une estimation des nombres d’onde des modes normaux de vibration de la forme excitée A∗ :2582 cm−1 pour l’élongation O-H et 176 cm−1 pour le mode normal de plus basse fréquence. Pour chacun deces deux modes, calculer la durée correspondant à une demi période.
Solution:
t1/2,2582 =1
2cσ=
12× 2,998 · 1010 × 2582
= 6,5 fs (94)
t1/2,176 =1
2cσ=
12× 2,998 · 1010 × 176
= 95 fs (95)
(96)
B.22. Comparer les différentes échelles de temps obtenues ci-dessus avec celles de l’expérience.
Solution: L’échelle de temps pour le transfert d’hydrogène donné par l’expérience correspond plutôt àcelle d’une demi vibration du mode normal le moins énergétique.
B.23. Quelles conclusions peut-on tirer pour décrire le mouvement de transfert d’atome d’hydrogène : localisé,couplé à d’autres mouvements ou « direct » notamment ?
Solution: À priori, on a plutôt un couplage avec d’autres mouvements car le mode localisé et le modedirect donnent des échelles de temps trop courtes.

21
C. Réacteur piston à recyclage
Pour les notations, on choisit :— rA : vitesse de disparition du réactif A ;— Fi : le débit molaire de l’espèce i ;
— X =C0 − C
C0: le taux de conversion de A ;
— Q : le débit volumique.
C.I. Réacteur parfaitement agité continu
Pour un réacteur parfaitement agité continu (R.P.A.C.), la constitution du mélange en sortie correspond à celledu réacteur. On considère un système pour lequel le débit volumique en entrée est égal au débit volumique desortie.
C.1. Donner la définition du temps de passage, quelle est sa signification physique ?
Solution:
τ =VQ
(97)
C’est le temps moyen que passe une particule dans le réacteur.
C.2. À l’aide d’un bilan de matière sur le R.P.A.C. pendant un intervalle de temps dt, donner une relation entre Fe,Fs, rA et V.
Solution: En régime stationnaire, la constitution du réacteur ne dépend pas du temps donc ce qui entrepar le réacteur est donc soit consommé par la réaction, soit est récupéré en sortie :
Fedt = Fsdt + VrAdt (98)
⇐⇒ rA =Fe − Fs
V(99)
C.3. En déduire l’expression du temps de séjour en fonction de C0, Xe, Xs, et rA.
Solution: On a Fi = QCi = QC0(1− Xi)
rA =Fe − Fs
V= Q
C0 (Xs − Xe)
V(100)
⇐⇒ τ =VQ
=C0 (Xs − Xe)
rA(101)
C.II. Réacteur piston en régime permanent
On s’intéresse ensuite à un réacteur piston idéal en régime stationnaire pour lequel on considère que la constitutionet les grandeurs physiques sont homogènes pour une section de volume infinitésimal. La section du réacteur estconstante et notée S et la longueur est notée L. La réaction étudiée est :
A→ P (102)
Le schéma du réacteur est donné figure 5.

22
Fe
Qe
Fs
Qs
dV
Xe Xs
F(x)
Q(x)
X(x)
Figure 5 – Schéma d’un réacteur piston.
C.4. Pour respecter les hypothèses d’un réacteur piston idéal, le nombre de Reynolds associé à l’écoulement doit-ilêtre faible ou élevé ?
Solution: L’écoulement doit être turbulent et non laminaire. Car pour un écoulement à bas nombre deReynolds, la vitesse adopte un profil parabolique qui est incompatible avec le fait d’avoir une constitutionhomogène au sein des tranches successives.
C.5. À l’aide d’un bilan de matière sur un volume dV fixe, établir le lien entre dFA, dV et rA la vitesse de disparitionde A.
Solution: Le système est en régime permanent, il n’y a donc pas d’évolution temporelle. Ce qui rentre enx correspond à ce qui sort en x + dx plus ce qui a réagi.
F(x) = F(x + dX) + rA(x)dV (103)
On en déduit :
rA(x) = −d F(x)d V
(104)
C.6. Ré-exprimer la vitesse de réaction en fonction de Q(x), C0, XA(x) et dV.
Solution:
F(x) = Q(x)CA(x) (105)
et
CA(x) = C0(1− X(x)) (106)
On en déduit :
rA(x) = C0d (Q(x)XA(x))
d V(107)
C.7. Quel est le temps de séjour dans l’élément de volume dV ? En déduire le temps de séjour dans le réacteurpiston.
Solution:
dts =dV
Q(x)(108)

23
Le temps de séjour total est donc égal à :
ts =∫ L
x=0
dVQ(x)
(109)
C.8. Le temps de séjour dépend-il du réactif considéré ? Justifier.
Solution: Le temps de séjour est indépendant du réactif considéré, en effet, pour un réacteur piston idéal,chaque particule de fluide passe le même temps dans le réacteur.
C.9. Dans le cas le plus général, y-a-t-il un lien entre le temps de séjour et le temps de passage pour un réacteurpiston ?
Solution: Non. Il n’y a égalité que si le débit volumique est constant.
C.10. Montrer que la composition d’un réacteur fermé parfaitement agité, de même volume que le réacteur pis-ton, après un temps ts est la même que la composition du réacteur piston en sortie si le débit volumique estconstant.
Solution: Pour un réacteur fermé :
rA = C0d XA
d t(110)
⇐⇒ ts = C0
∫ Xs
X=Xe
dXA
rA(111)
Pour un réacteur piston dont le débit volumique ne varie pas, alors les équations (107) et (113) donnent :
ts =∫ L
x=0
dVQ(x)
= C0
∫ L
x=0
dXA
rA(112)
L’équation d’évolution est la même. Dans un cas, on a un temps de réaction, dans l’autre le temps detraversée du réacteur.
C.III. Réacteur piston à recyclage
On considère dans la suite que le débit volumique en entrée et en sortie est constant : Qe = Qs = Q.Dans certains cas, il est intéressant de réinjecter en entrée du réacteur une partie du mélange de sortie. On parledonc de réacteur piston à recyclage.
Le rapport des débits volumiques de fluide retourné en entré du R.P. et du fluide quittant le système est appelé
coefficient de recyclage R =Q′′′
Qs. Sa valeur peut varier de zéro à l’infini.
C.11. À quelles situations physiques correspondent les situations R = 0, R = ∞ ?
Solution: La situation R = 0 correspond à un réacteur piston idéal classique. La situation R = ∞ corres-pond à un réacteur parfaitement agité continu (RPAC). En effet, dans ce cas, la concentration du réactifchute directement à sa valeur en sortie.

24
Xe = 0
Fe
Qe
Fs
Qs = Qe
Xs
Q' = (R + 1)Qe
X'
F'Q" = (R + 1)Qe
X" = Xs
F" = (R + 1)Fs
Q"' = R Qe
X"' = Xs
F"' = R Fs
E F G
I
S
Figure 6 – Schéma d’un réacteur piston à recyclage. On suppose qu’en entrée, Xe = 0.
C.12. Donner l’expression du temps de séjour dans le réacteur piston à recyclage ts en fonction de V, Q et R puisen fonction de C0, X′, Xs, et rA.
Solution:
ts =VQ”
=V
Q (R + 1)= C0
∫ Xs
X=X′
dXA
rA(113)
C.13. Donner une relation entre Fs, Fe et Xs.
Solution:Fs = QC0 (1− Xs) = Fe (1− Xs) (114)
C.14. Exprimer la concentration en A au point F en fonction de Q, R, Fe et Fs puis en déduire une expression enfonction de C0, R et Xs.
Solution: La concentration en F correspond au barycentre des concentrations provenant de l’entrée et dela sortie du réacteur.
CA(F) =F′
Q′=
Fe + RFs
Q (1 + R)=
Fe (1 + R(1− Xs))
Q (1 + R)= C0
1 + R(1− Xs)
(1 + R)(115)
C.15. Donner une autre relation entre CA (F), C0 et X′.
Solution:
FF = Q′CA (F) = Q′C0(1− X′
)(116)
CA (F) = C0(1− X′
)(117)
C.16. En déduire l’expression de X′ en fonction de R et Xs
Solution: L’égalité entre les deux expressions pour CA (F) donne :
C0(1− X′
)= C0
1 + R(1− Xs)
(1 + R)(118)
⇐⇒ X′ =RXs
1 + R(119)

25
L’expression de X′ permet de déduire les bornes d’intégration pour le réacteur à recyclage.
C.17. Montrer que le temps de passage pour un réacteur piston obéit à la relation suivante :
τrec =VQ
= (R + 1)C0
∫ Xs
RR+1 Xs
dXA
rA(120)
Solution: On réutilise l’expression de ts donnée équation (113) pour en déduire l’expression de τrec.
C.IV. Comparaison d’un R.P.A.C, d’un R.P. et d’un réacteur piston à recyclage
C.18. Montrer que le temps de passage d’un R.P.A.C. et d’un réacteur piston peut être lu graphiquement si on
traceC0
rAen fonction de X. L’illustrer sur le cas d’une réaction d’ordre 1 par rapport à A.
Solution: Dans le cas d’un R.P.A.C. :
τ =C0 (Xs − Xe)
rA(121)
Ce qui correspond à l’aire du rectangle de hauteurC0
rAet de largeur Xs − Xe.
Pour le R.P. :
τ = C0
∫ Xs
X=Xe
dXA
rA(122)
qui correspond à l’aire sous la courbeC0
rAentre Xe et Xs.
C.19. Montrer que τrec correspond à l’aire du rectangle OABC sur la figure 7, on pourra s’aider de l’aire du rec-tangle BCDE.
Solution: L’aire du rectangle BCDE correspond à l’intégrale∫ Xs
RR+1 Xs
C0dXA
rA. En effet, par construction on
s’est ramené de l’aire sous la courbe entre X′ et Xs à l’aire d’un rectangle de hauteur BC.
Ensuite, l’écart entre X′ et Xs vautXs
R + 1alors que celle entre X = 0 et X′ vaut
RXs
R + 1. L’aire du rectangle
OAED est donc R fois plus grande que l’aire du rectangle BCDE.L’aire de OABC est R+1 fois l’aire de BCDE donc égale à τrec.
C.20. En déduire que le réacteur piston peut être vu comme la combinaison d’un R.P.A.C. et d’un R.P. dont onprécisera les conditions opératoires (combinaison en série ou en parallèle, valeur de X en entrée et en sortiepour les deux réacteurs et valeur de rA pour le R.P.A.C.).
Solution: L’aire du rectangle OAED correspond au temps de passage d’un R.P.A.C. pour passer de X = 0à X = X′ avec une vitesse rA égale à celle du point de coordonnée F.De plus, l’aire du rectangle BCDE correspond au temps de passage pour un réacteur piston de compositionX = X′ en entrée et X = Xs en sortie.Le réacteur piston à recyclage correspond donc aux R.P.A.C et R.P. décris ci-dessus mis en série.
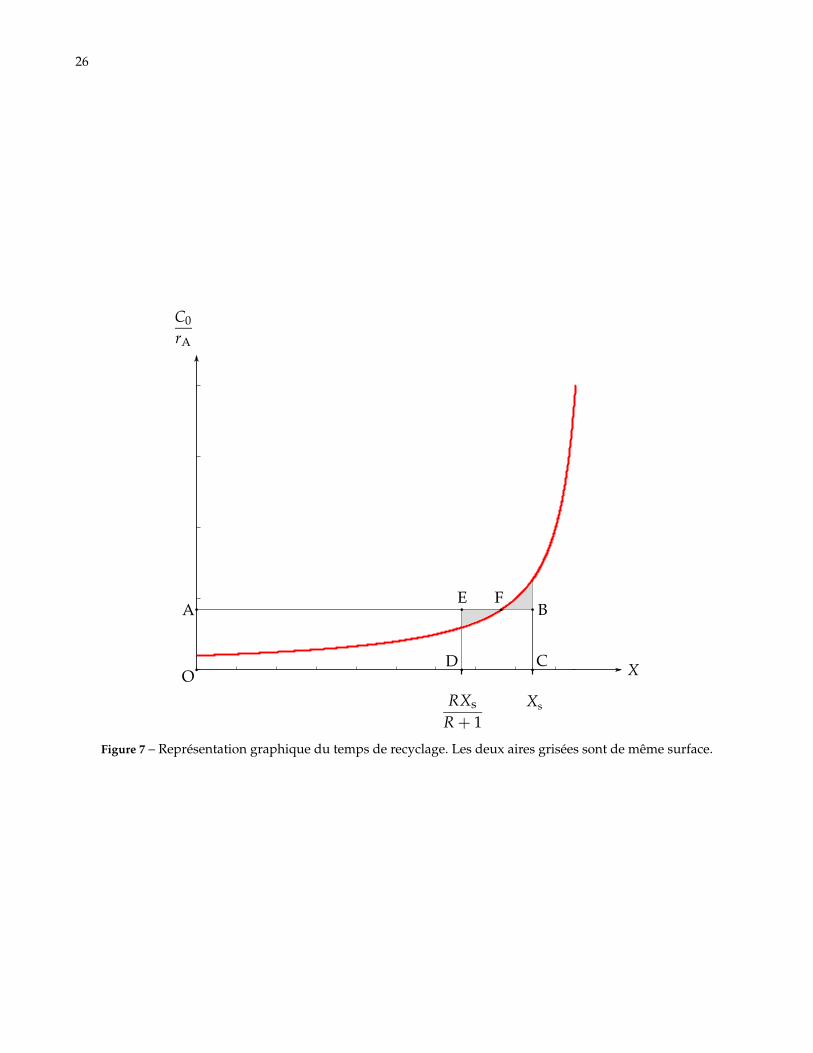
26
X
Xs
O
AE F
CD
B
Figure 7 – Représentation graphique du temps de recyclage. Les deux aires grisées sont de même surface.
27
Le réacteur piston peut donc être vu comme un modèle plus réaliste de réacteur dont les cas extrêmes corres-pondent à un R.P.A.C. ou un R.P.
C.V. Optimisation d’un réacteur piston à recyclage
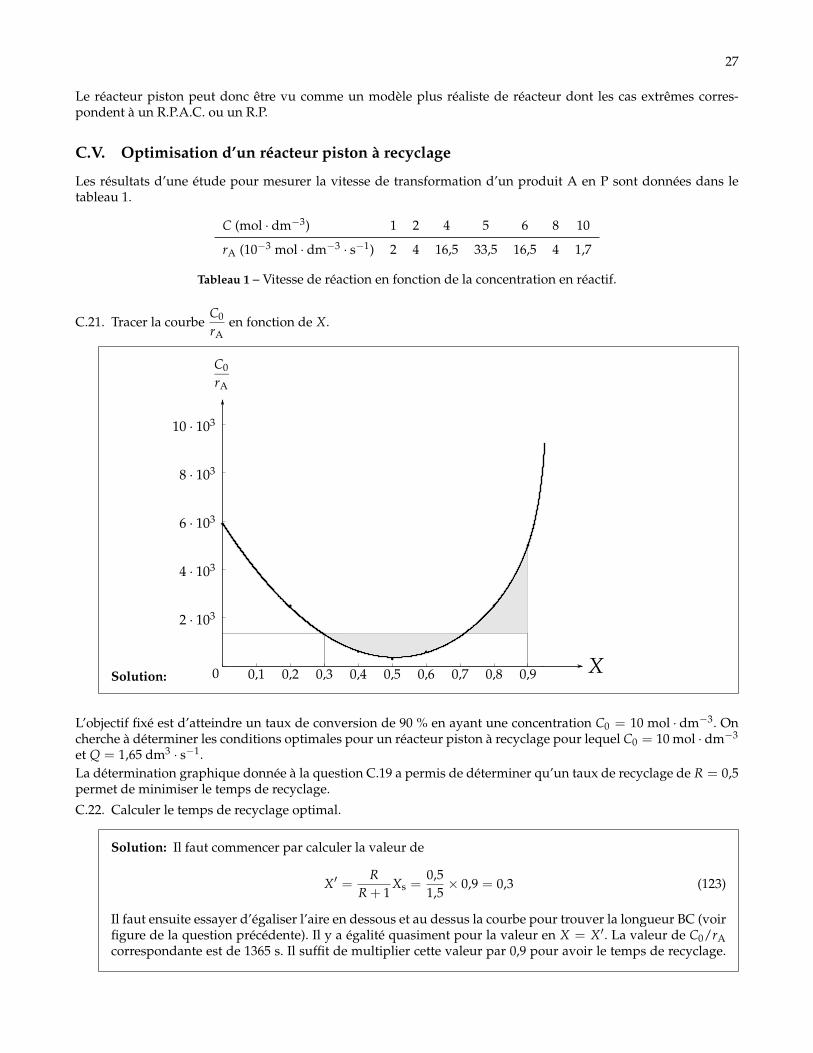
Les résultats d’une étude pour mesurer la vitesse de transformation d’un produit A en P sont données dans letableau 1.
C (mol · dm−3) 1 2 4 5 6 8 10
rA (10−3 mol · dm−3 · s−1) 2 4 16,5 33,5 16,5 4 1,7
Tableau 1 – Vitesse de réaction en fonction de la concentration en réactif.
C.21. Tracer la courbeC0
rAen fonction de X.
Solution: 0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 X
L’objectif fixé est d’atteindre un taux de conversion de 90 % en ayant une concentration C0 = 10 mol · dm−3. Oncherche à déterminer les conditions optimales pour un réacteur piston à recyclage pour lequel C0 = 10 mol · dm−3
et Q = 1,65 dm3 · s−1.La détermination graphique donnée à la question C.19 a permis de déterminer qu’un taux de recyclage de R = 0,5permet de minimiser le temps de recyclage.
C.22. Calculer le temps de recyclage optimal.
Solution: Il faut commencer par calculer la valeur de
X′ =R
R + 1Xs =
0,51,5× 0,9 = 0,3 (123)
Il faut ensuite essayer d’égaliser l’aire en dessous et au dessus la courbe pour trouver la longueur BC (voirfigure de la question précédente). Il y a égalité quasiment pour la valeur en X = X′. La valeur de C0/rAcorrespondante est de 1365 s. Il suffit de multiplier cette valeur par 0,9 pour avoir le temps de recyclage.
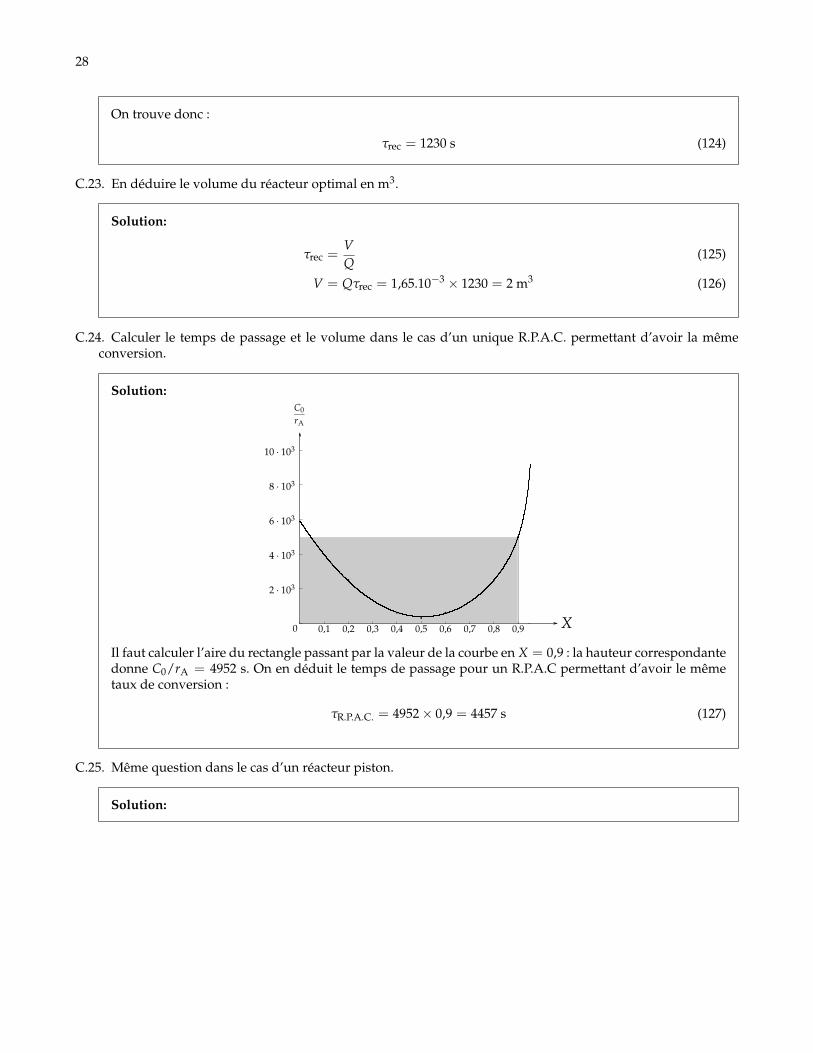
28
On trouve donc :
τrec = 1230 s (124)
C.23. En déduire le volume du réacteur optimal en m3.
Solution:
τrec =VQ
(125)
V = Qτrec = 1,65.10−3 × 1230 = 2 m3 (126)
C.24. Calculer le temps de passage et le volume dans le cas d’un unique R.P.A.C. permettant d’avoir la mêmeconversion.
Solution:
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 X
Il faut calculer l’aire du rectangle passant par la valeur de la courbe en X = 0,9 : la hauteur correspondantedonne C0/rA = 4952 s. On en déduit le temps de passage pour un R.P.A.C permettant d’avoir le mêmetaux de conversion :
τR.P.A.C. = 4952× 0,9 = 4457 s (127)
C.25. Même question dans le cas d’un réacteur piston.
Solution:
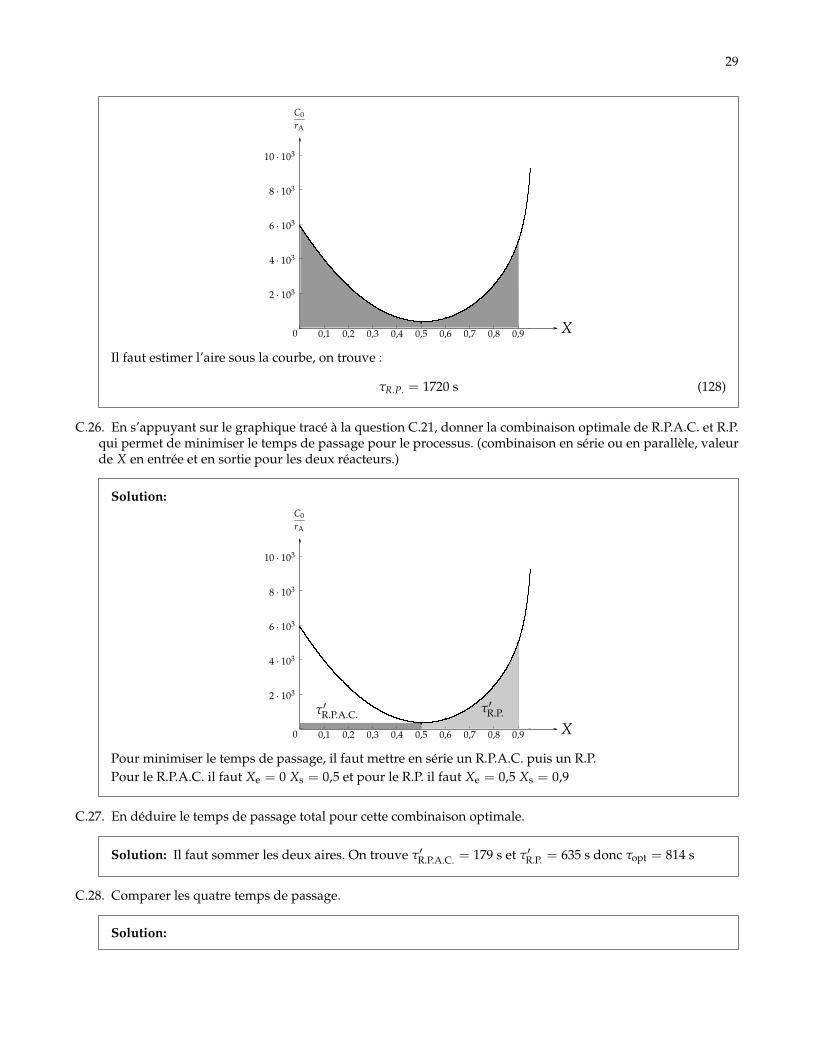
29
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 X
Il faut estimer l’aire sous la courbe, on trouve :
τR.P. = 1720 s (128)
C.26. En s’appuyant sur le graphique tracé à la question C.21, donner la combinaison optimale de R.P.A.C. et R.P.qui permet de minimiser le temps de passage pour le processus. (combinaison en série ou en parallèle, valeurde X en entrée et en sortie pour les deux réacteurs.)
Solution:
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 X
Pour minimiser le temps de passage, il faut mettre en série un R.P.A.C. puis un R.P.Pour le R.P.A.C. il faut Xe = 0 Xs = 0,5 et pour le R.P. il faut Xe = 0,5 Xs = 0,9
C.27. En déduire le temps de passage total pour cette combinaison optimale.
Solution: Il faut sommer les deux aires. On trouve τ′R.P.A.C. = 179 s et τ′R.P. = 635 s donc τopt = 814 s
C.28. Comparer les quatre temps de passage.
Solution:

30
τR.P.A.C. = 4457 s (129)τR.P. = 1720 s (130)τrec = 1230 s (131)τopt = 814 s (132)
Il faut donc privilégier un réacteur piston ou un réacteur piston à recylage pour s’approcher au mieux dutemps optimal. La gain associé à l’utilisation d’un réacteur piston à recyclgage par rapport à un réacteurpiston est tout de même de l’ordre de 30 %.
C.29. Expliquez la différence entre le temps de passage pour « la combinaison R.P.A.C. et R.P. optimale » et celledu réacteur piston à recyclage optimal.
Solution: Le réacteur piston à recyclage ne peut pas faire aussi bien que la combinaison optimale car lahauteur du rectangle est forcément supérieure au minimum de la courbe. C’est ce qui explique la diffé-rence significative entre les deux temps de passage.

31
D. Théorie de l’état de transition
D.I. Formulation de la constante de vitesse dans le cadre de la théorie de l’état de transition
On se place dans l’ensemble micro-canonique. Le but est de retrouver la formulation de la constante de vitessedans le cadre de la théorie de l’état de transition.
La fonction de partition d’un système est définie selon :
Z = ∑i
exp(−EikBT
)(133)
où :— i est un état vibrationnel de la molécule ;— Ei est l’énergie de l’état par rapport au fondamental ;— T est la température.Le calcul de cette dernière permet d’avoir accès à l’énergie libre :
F = −kBT ln Z (134)
D.1. Calculer la fonction de partition zvib pour un oscillateur harmonique. On rappelle que les niveaux énergé-tiques pour un oscillateur en mécanique quantique sont donnés par :
E = hν
(n +
12
)(135)
où n est un entier positif ou nul et ν la fréquence de vibration de l’oscillateur.
Solution: On utilise la formule donnée pour le calcul de Z, comme le fondamental a une énergie nonnulle, il faut corriger les énergies pour la sommation :
Z =∞
∑n=0
exp(− hνn
kBT
)=
1
1− exp(− hν
kBT
) (136)
C’est la somme du suite géométrique de raison exp(− hν
kBT
).
Pour un système de N particules indiscernables, la fonction de partition de l’ensemble du système peut s’écrire
Z =zN
N!où z est la fonction de partition pour une molécule.
D.2. Calculer l’expression du potentiel chimique en fonction de kB, T, z et N.
Solution:
F = − kBT ln Z = −kBT ln(
zN
N!
)= −NkBT ln z + kBT ln N! (137)
= − NkBT ln z + kBTN ln N − NkBT (138)
Ce qui permet d’en déduire l’expression du potentiel chimique :
µ =∂ F∂ N
∣∣∣∣T,V
= −kBT ln z + kBT ln N = −kBT ln( z
N
)(139)

32
Cette expression du potentiel chimique permet de montrer que la constante d’équilibre dans l’échelle de concen-tration peut s’exprimer sous la forme :
Kc =
(1V
)∑i νi
∏i(zi)
νi exp(−∆rU◦(0 K)
kBT
)= exp
(−∆rF
RT
)(140)
où— νi est le coefficient stœchiométrique algébrique ;— c◦ est la concentration standard ;— zi est la fonction de partition moléculaire de l’espèce i ;— V est le volume ;— ∆rU◦(0 K) est l’énergie interne de réaction à 0 K.On considère l’équilibre entre le complexe activé et les réactifs :
A + B� C‡ (141)
où C‡ est le complexe activé.
D.3. Donner l’expression de la constante d’équilibre dans l’échelle des concentrations.
Solution:
Kc =
[C‡] c◦
[A] [B]γC‡
γAγB= V
zC‡
zAzBexp
(−∆rU◦(0 K)
kBT
)(142)
Dans le cadre de la théorie du complexe activé, on peut factoriser la fonction de partition du complexe activé sousla forme zC‡ = z′C‡ × zvib où zvib est la fonction de partition associée à la vibration responsable de la réaction.
D.4. Ré-écrire la constante d’équilibre en faisant apparaître zvib.
Solution:
Kc = zvibVz′C‡
zAzBexp
(−∆rU◦(0 K)
kBT
)(143)
=1
1− exp(− hν
kBT
)Vz′C‡
zAzBexp
(−∆rU◦(0 K)
kBT
)(144)
D.5. Sachant que dans l’état de transition, la fréquence de vibration est petite devant kBT, donner une expressionsimplifié de zvib.
Solution:
zvib =1
1− exp(− hν
kBT
) ≈ kBThν
(145)
Kc =kBThν
Vz′C‡
zAzBexp
(−∆rU◦(0 K)
kBT
)(146)
La vitesse de réaction s’écrit v = ν[C‡].

33
D.6. En déduire que la constante de vitesse peut s’écrire :
k =kBThc◦
γAγB
γC‡exp
(−∆rF‡
RT
)(147)
où γi est le coefficient d’activité de l’espèce i, c◦ la concentration standard et ∆rF‡ l’énergie libre d’activationdont on donnera le sens.
Solution:
v = ν[C‡]= k [A] [B] (148)
= νKc
c◦[A] [B]
γAγB
γC‡(149)
= νkBThν
1c◦
γAγB
γC‡V
z′C‡
zAzBexp
(−∆rU◦(0 K)
kBT
)[A] [B] (150)
On en déduit l’expression de k demandée avec :
exp(−∆rF‡
RT
)= V
z′C‡
zAzBexp
(−∆rU◦(0 K)
kBT
)(151)
Qui correspond à l’expression de Kc pour laquelle on a enlevé la contribution de la vibration.
D.7. Cette expression dépend-t-elle de la vibration donnée ?
Solution: La fréquence ν n’apparaît plus explicitement, en effet, la contribution liée à la fonction de parti-tion se simplifie avec celle liée à la vitesse. On n’a donc pas besoin de connaître explicitement la vibrationresponsable pour obtenir l’expression de la constante de vitesse.
D.II. Application à la détermination du mécanismes d’échange de molécules de solvantautour de cations métalliques
Ici, on travaille dans l’ensemble grand-canonique, on admettra que l’expression de k trouvée précédemment reste valable si onremplace l’énergie libre d’activation par l’enthalpie libre d’activation.On s’intéresse ici à la détermination du volume d’activation par suivi RMN.La mesure de la constante de vitesse de l’échange d’une molécule de solvant est difficile à mesurer car l’équationbilan ne fait intervenir aucun changement :
M(S)z+n + S∗
kex� M(S∗)(S)z+
n−1 + S (152)
où S∗ est une molécule de solvant arbitrairement différenciée et M est un cation métallique. Une méthode de choixpour l’analyse cinétique de l’échange est la spectroscopie RMN et la mesure d’une grandeur caractéristique derelaxation : le T2.
La valeur de 1/T2 correspond à la somme de deux termes :
1T2
= kex +1
T2,Q(153)
où kex est la constante de vitesse de l’échange de molécule de solvant et 1/T2,Q la relaxation quadrupolaire. Enfonction du domaine de température, c’est l’un ou l’autre des mécanismes qui est prédominant.
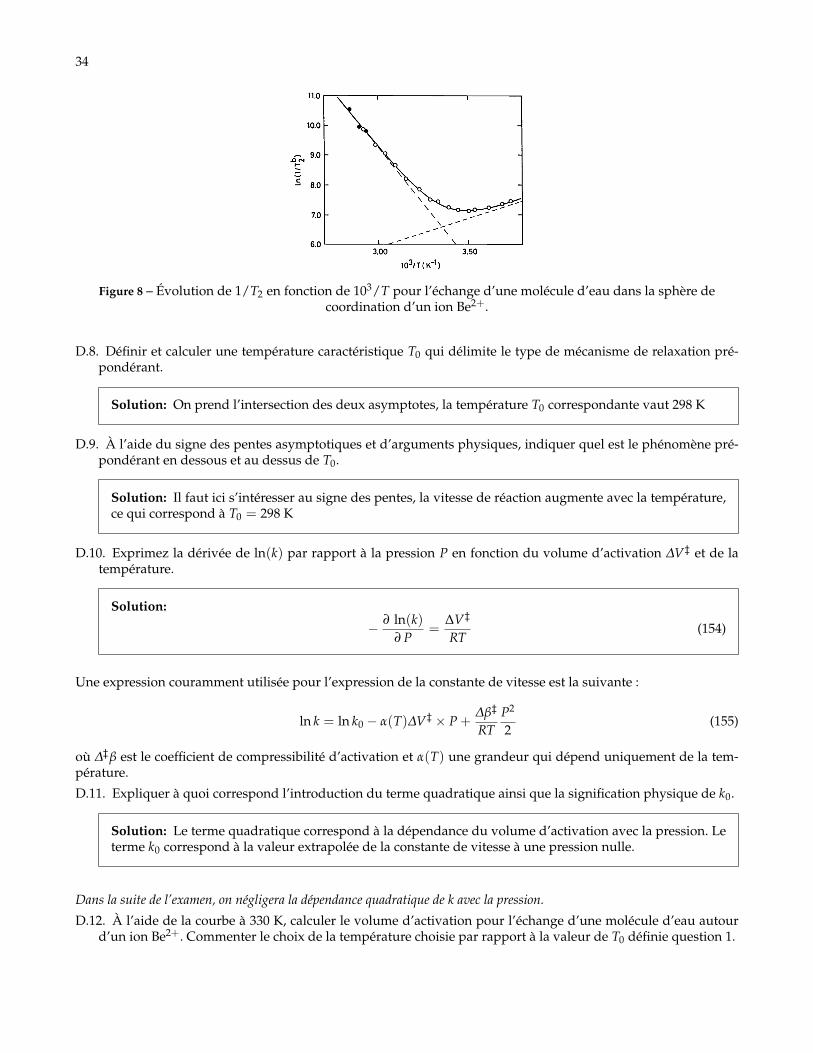
34
Figure 8 – Évolution de 1/T2 en fonction de 103/T pour l’échange d’une molécule d’eau dans la sphère decoordination d’un ion Be2+.
D.8. Définir et calculer une température caractéristique T0 qui délimite le type de mécanisme de relaxation pré-pondérant.
Solution: On prend l’intersection des deux asymptotes, la température T0 correspondante vaut 298 K
D.9. À l’aide du signe des pentes asymptotiques et d’arguments physiques, indiquer quel est le phénomène pré-pondérant en dessous et au dessus de T0.
Solution: Il faut ici s’intéresser au signe des pentes, la vitesse de réaction augmente avec la température,ce qui correspond à T0 = 298 K
D.10. Exprimez la dérivée de ln(k) par rapport à la pression P en fonction du volume d’activation ∆V‡ et de latempérature.
Solution:
− ∂ ln(k)∂ P
=∆V‡
RT(154)
Une expression couramment utilisée pour l’expression de la constante de vitesse est la suivante :
ln k = ln k0 − α(T)∆V‡ × P +∆β‡
RTP2
2(155)
où ∆‡β est le coefficient de compressibilité d’activation et α(T) une grandeur qui dépend uniquement de la tem-pérature.
D.11. Expliquer à quoi correspond l’introduction du terme quadratique ainsi que la signification physique de k0.
Solution: Le terme quadratique correspond à la dépendance du volume d’activation avec la pression. Leterme k0 correspond à la valeur extrapolée de la constante de vitesse à une pression nulle.
Dans la suite de l’examen, on négligera la dépendance quadratique de k avec la pression.
D.12. À l’aide de la courbe à 330 K, calculer le volume d’activation pour l’échange d’une molécule d’eau autourd’un ion Be2+. Commenter le choix de la température choisie par rapport à la valeur de T0 définie question 1.
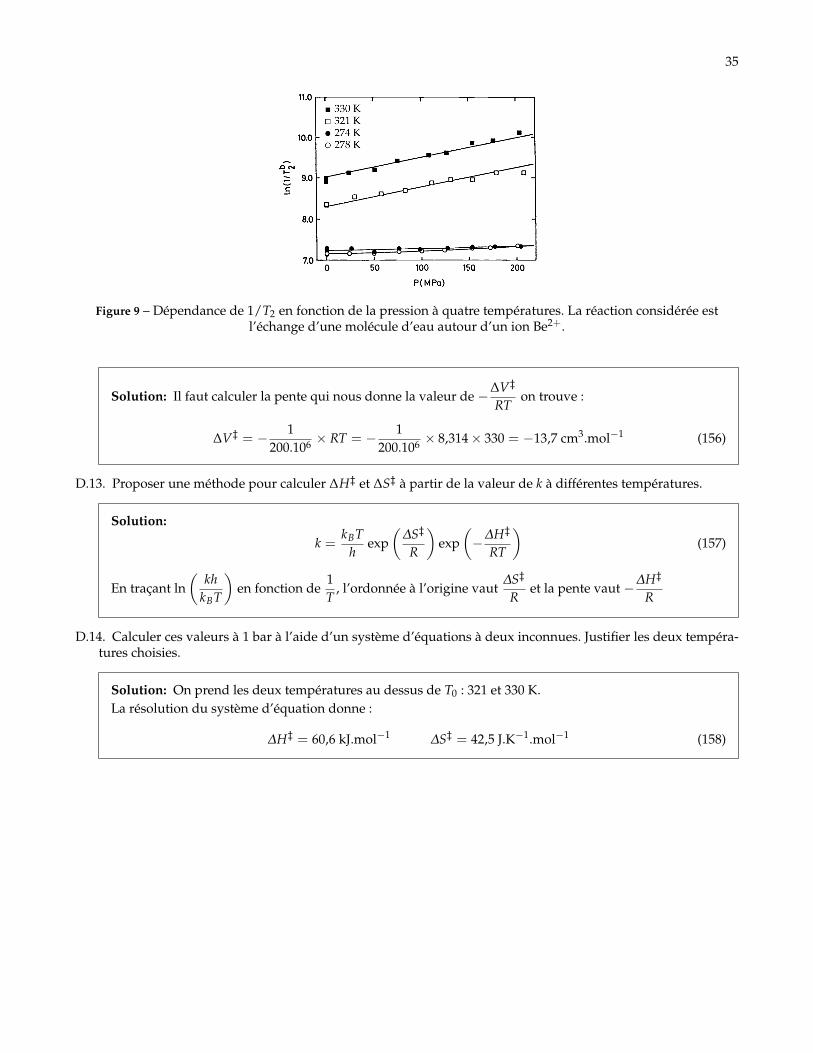
35
Figure 9 – Dépendance de 1/T2 en fonction de la pression à quatre températures. La réaction considérée estl’échange d’une molécule d’eau autour d’un ion Be2+.
Solution: Il faut calculer la pente qui nous donne la valeur de −∆V‡
RTon trouve :
∆V‡ = − 1200.106 × RT = − 1
200.106 × 8,314× 330 = −13,7 cm3.mol−1 (156)
D.13. Proposer une méthode pour calculer ∆H‡ et ∆S‡ à partir de la valeur de k à différentes températures.
Solution:
k =kBT
hexp
(∆S‡
R
)exp
(−∆H‡
RT
)(157)
En traçant ln(
khkBT
)en fonction de
1T
, l’ordonnée à l’origine vaut∆S‡
Ret la pente vaut −∆H‡
R
D.14. Calculer ces valeurs à 1 bar à l’aide d’un système d’équations à deux inconnues. Justifier les deux tempéra-tures choisies.
Solution: On prend les deux températures au dessus de T0 : 321 et 330 K.La résolution du système d’équation donne :
∆H‡ = 60,6 kJ.mol−1 ∆S‡ = 42,5 J.K−1.mol−1 (158)

36
Il existe trois grands types de mécanismes limites pour l’échange d’une molécule de solvant :— le mécanisme associatif (A) :
M(S)nz+ + S∗ → M(S)n(S∗)
z+ (159)
M(S)n(S∗)z+ → M(S∗)(S)n−1
z+ + S (160)
— Le mécanisme dissociatif (D) :
M(S)nz+ → M(S)n−1
z+ + S (161)
M(S)n−1z+ + S∗ → M(S∗)(S)n−1
z+ (162)
— Le mécanisme d’inter-échange concerté (I) lorsqu’aucun intermédiaire n’est identifiable :
M(S)nz+ + S∗ →
[S--M(S)n−1--(S∗)z+
]‡→ M(S∗)(S)n−1
z+ + S (163)
D.15. Indiquer qualitativement le signe de ∆V‡ pour chacun de ces mécanismes.
Solution:— Pour le mécanisme associatif ∆V‡ < 0— Pour le mécanisme dissociatif ∆V‡ > 0— Pour le mécanisme d’inter-échange ∆V‡ ≈ 0
D.16. En déduire le type de mécanisme d’échange de solvant pour l’ion Be2+ dans l’eau et les différents solvantspour lesquels ∆V‡ a été mesuré.
Solution: Dans l’ordre, de haut en bas :— A (eau) ;— A, Ia ;— A, Ia ;— A, Ia ;— D ;— D (DMPU) ;
D.17. Faire de même pour les différents ions métalliques et expliquer le lien entre le rayon des différents cationset le mécanisme d’échange des molécules de solvant.
Solution: Plus le rayon de l’ion est gros, plus il va être possible d’ajouter une molécule de solvant àla sphère de coordination. Les gros ions privilégient le mécanisme associatif et donc les petit volumesd’activation.À l’inverse, pour les ions de petit rayon, il n’est pas possible d’ajouter une molécule de solvant. on va doncavoir un mécanisme qui est plutôt de type dissociatif avec de grands volumes d’activation.
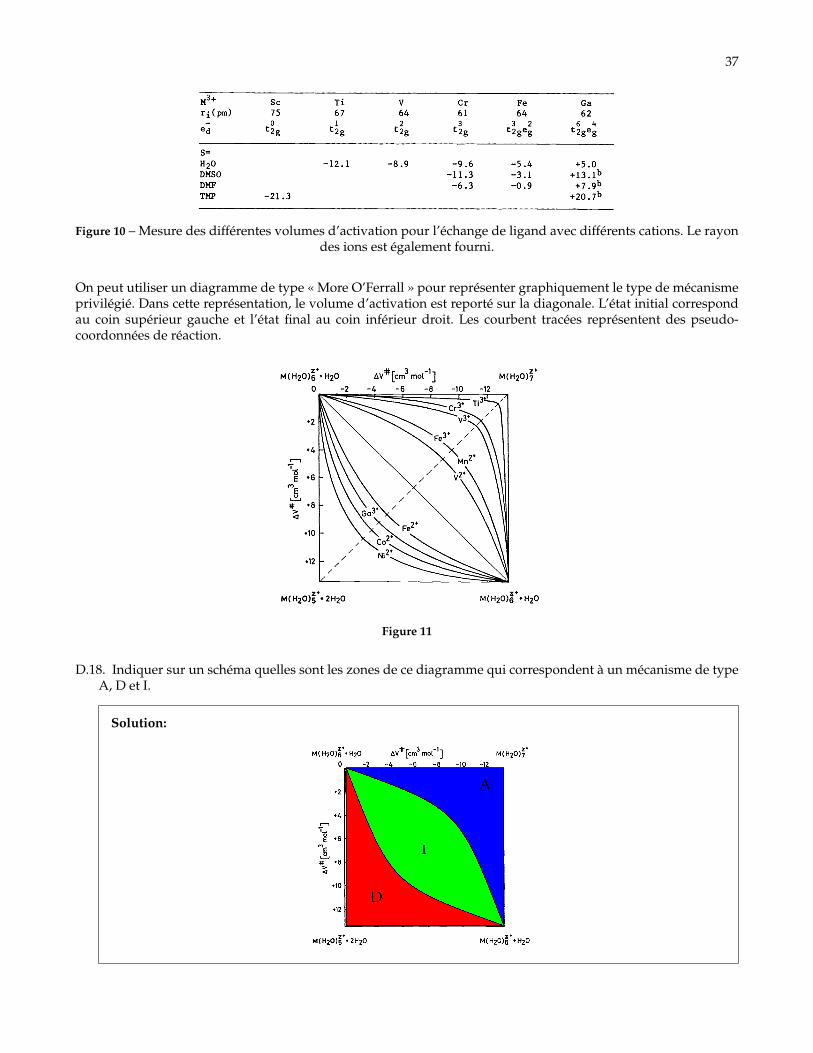
37
Figure 10 – Mesure des différentes volumes d’activation pour l’échange de ligand avec différents cations. Le rayondes ions est également fourni.
On peut utiliser un diagramme de type « More O’Ferrall » pour représenter graphiquement le type de mécanismeprivilégié. Dans cette représentation, le volume d’activation est reporté sur la diagonale. L’état initial correspondau coin supérieur gauche et l’état final au coin inférieur droit. Les courbent tracées représentent des pseudo-coordonnées de réaction.
Figure 11
D.18. Indiquer sur un schéma quelles sont les zones de ce diagramme qui correspondent à un mécanisme de typeA, D et I.
Solution:

38
D.19. Quelle serait à priori la valeur absolue maximale du volume d’activation pour un mécanisme de type A ouD.
Solution: Les valeurs limites possibles sont de l’ordre de 13 cm3.mol−1. En effet, pour un mécanisme dis-sociatif avec le solvant, le volume à l’état de transition ne peut pas augmenter indéfiniment sans qu’uneautre molécule vienne se coordonner, il est donc de l’ordre du volume initial moins le volume d’une mo-lécule d’eau. De même, pour un mécanisme associatif le volume de l’état de transition ne peut approxi-mativement pas être supérieur à celui du cation haxahydraté plus le volume d’une molécule d’eau.
E. Constantes numériques
constante de Planck h = 6,626 · 10−34 J.sconstante de Boltzmann kB = 1,381 · 10−23 J.K−1
vitesse de la lumière dans le vide c = 2,998.108 m.s−1
masse de l’électron me = 9,11.10−31 kgmasse du proton mp = 1,673 · 10−27 kgmasse du neutron mn = 1,675 · 10−27 kgcharge de l’électron q = −1,602 · 10−19 C
F. Formulaire
— h̄ =h
2π— Formule de Stirling : ln N! ' N ln N − N
— µ =d Fd N
∣∣∣∣T,V






























