Embed Size (px)
Citation preview

Communications cellulaires et signalisation
Michel Vignes

INTRODUCTION GENERALE LES RECEPTEURS COUPLES AUX PROTEINES G 1-Introduction 2-Voies de transduction des RCPG de1986 à aujourd’hui et classes de RCPGs 3-Rappels de Pharmacologie 4-Rappels sur le cycle des protéines G 5-La désensibilisation des RCPG 5.1-Mécanisme général 5.2-La désensibilisation hétérologue 5.3-Le mécanisme d’endocytose 5.4-Les voies de signalisation activées par l’arrestine 5.5-Arrestine et cardiomyocytes 5.6-Notion de ligand ‘biaisé’ 5.7-Phénomène de tolérance: le cas des récepteurs opiacés 6-Régulation de l’activité des RCPGs par des protéines d’interaction (GIPs) 6.1-Protéines RGS 6.2-Protéines à domaines PDZ 6.3-Les protéines interagissant avec i3 et C-term 6.4-Les protéines HOMER 7-Oligomérisation des RCPG 7.1-Couplages fonctionnels entre deux récepteurs 7.2-Mise en évidence de l’intéraction entre 2 RCPGs: aspects techniques 7.3-Exemple de l’oligomérisation des récepteurs D2 / CB1 / A2A 7.4-Exemple de la dimérisation 5HT2 et mGlu2 7.5-Diméres des récepteurs opiacés: cibles de futurs analgésiques? 8-RCPGs et pathologies 8.1-Exemples de pathologies 8.2-Exemple des récepteurs GnRH

• La communication entre les cellules est requise tout au long de leur existence pour leur permettre de s’adapter en permanence à leur environnement et de produire une activité coordonnée dans les réseaux cellulaires complexes comme les réseaux de neurones, les épithélia, les organes en général
• La communication entre cellules peut être directe par la formation de jonctions communicantes (‘gap
junctions’) qui permet d’échanger rapidement une information comme par exemple des ions. Cette forme de communication se rencontre par exemple dans les épithélia à l’interface entre deux milieux (tube digestif par exemple), dans les réseaux d’astrocytes du SNC, dans les réseaux d’hépatocytes. Elle présente l’avantage d’être rapide pour permettre une réponse ‘en masse’ de plusieurs cellules à la fois mais assez peu spécifique.
• Une communication assez spécifique est obtenue par l’intermédiaire d’un médiateur extracellulaire physique (ions, pression, température, lumière, etc…) ou chimique (neurotransmetteur, hormone, ions, molécules odorantes, phéromones, lipides, etc…). Ces médiateurs activent des récepteurs présents à la surface des cellules. L’interaction entre ces médiateurs et récepteurs activent des processus intracellulaires conduisant à des réponses:
1. ‘très rapides’ comme une réponse ‘électrique’ telle l’émission d’un potentiel d’action ou la synthèse d’un second messager (milliseconde/seconde);
2. ‘rapides’ comme l’activation de phosphorylations ou déphosphorylations par des kinases ou phosphatases;
3. ‘lentes’ qui nécessiteront l’expression génique et la synthèse de nouvelles protéines (actions génomiques).
• Les cellules sont équipées d’un grand nombre de récepteurs lui permettant de répondre de manière
plus ou moins rapide à des stimuli externes. Ces récepteurs comportent des récepteurs–canaux, des récepteurs métabotropes, des récepteurs à activité tyrosine kinase, des récepteurs d’adhésion,…
INTRODUCTION GENERALE

LES RECEPTEURS COUPLES AUX PROTEINES G (ou RCPG) 1-Introduction
•Les RCPGs associent des signaux extracellulaires comme des neurotransmetteurs, hormones, Ca2+, photons, ... à des voies de signalisations intracellulaires jusqu’à des effets génomiques.
•Les RCPGs sont les récepteurs les plus abondants parmi les récepteurs membranaires. Ils constituent une famille d’au moins 800 membres distincts.
•Entre 3 et 5% du génome code pour des RCPGs.
•On sait que 30% des médicaments actuellement utilisés sont des ligands pour les RCPGs (agonistes ou antagonistes), quelques exemples: les beta-bloquants, des anti-hypertenseurs, les anti-psychotiques, les analgésiques morphiniques, des anti nauséeux, ...
•Au plan structural, ces récepteurs sont tous des protéines à 7 domaines transmembranaires. Certains d’entre eux ont été cristallisés et leur structure révélée par rayons X.
•Leur activation entraîne l’activation d’une protéine hydrolysant le GTP (d’où leur nom!). Toutefois, ils peuvent s’associer à d’autres protéines ce qui leur confère d’autres propriétés de signalisation.
•Au plan fonctionnel, les RCPGs sont fortement régulés par d’autres protéines (ou GIP ‘GPCRs Interacting Protein’), dont les RCPGs eux-mêmes (formation d’oligomères). Ces régulations sont cruciales, notamment, pour la désensibilisation des RCPGs.
•Une vision historique … voir les diapos suivantes…
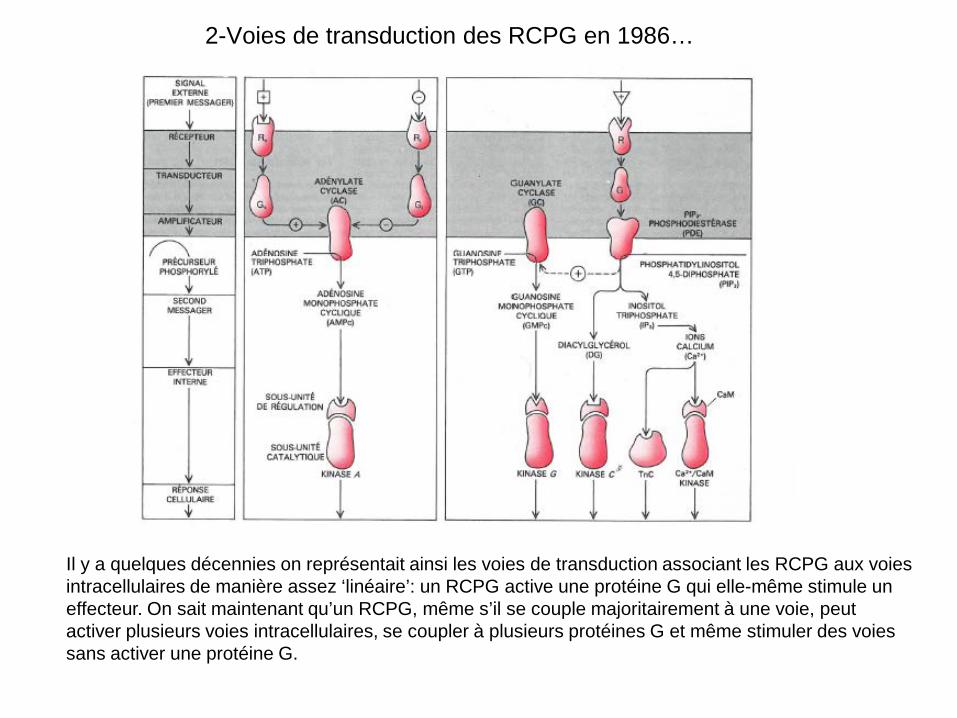
2-Voies de transduction des RCPG en 1986…
Il y a quelques décennies on représentait ainsi les voies de transduction associant les RCPG aux voies intracellulaires de manière assez ‘linéaire’: un RCPG active une protéine G qui elle-même stimule un effecteur. On sait maintenant qu’un RCPG, même s’il se couple majoritairement à une voie, peut activer plusieurs voies intracellulaires, se coupler à plusieurs protéines G et même stimuler des voies sans activer une protéine G.
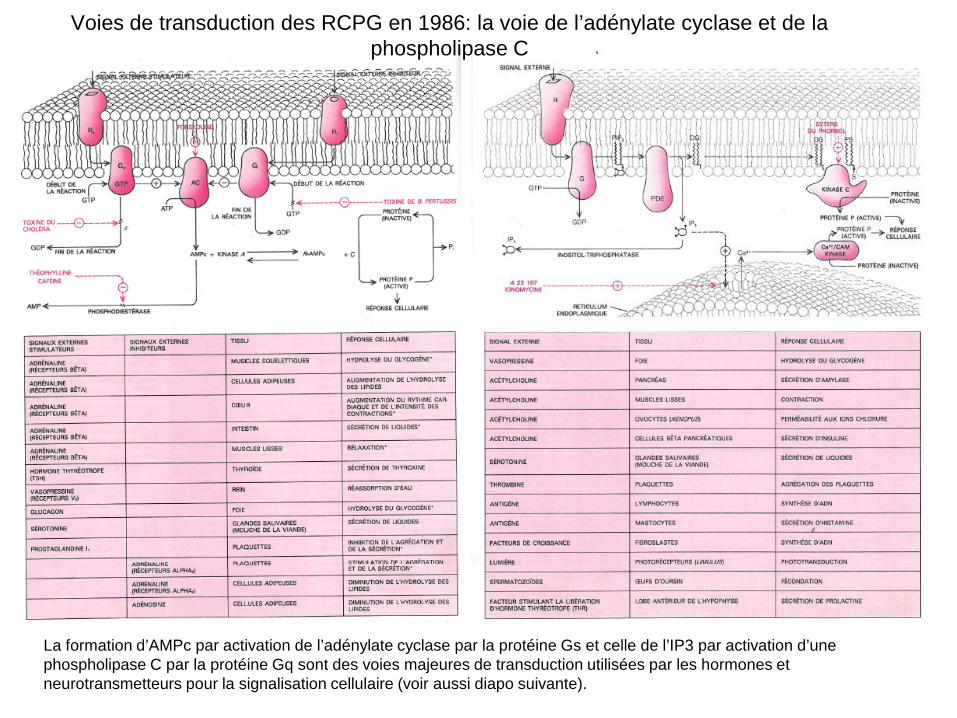
La formation d’AMPc par activation de l’adénylate cyclase par la protéine Gs et celle de l’IP3 par activation d’une phospholipase C par la protéine Gq sont des voies majeures de transduction utilisées par les hormones et neurotransmetteurs pour la signalisation cellulaire (voir aussi diapo suivante).
Voies de transduction des RCPG en 1986: la voie de l’adénylate cyclase et de la phospholipase C
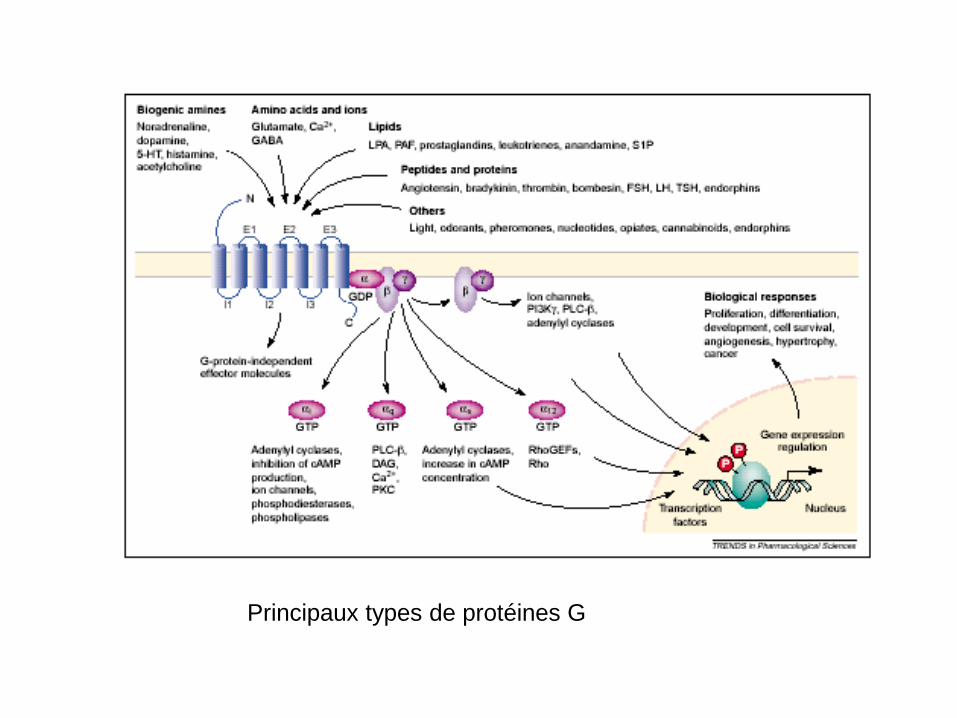
Principaux types de protéines G
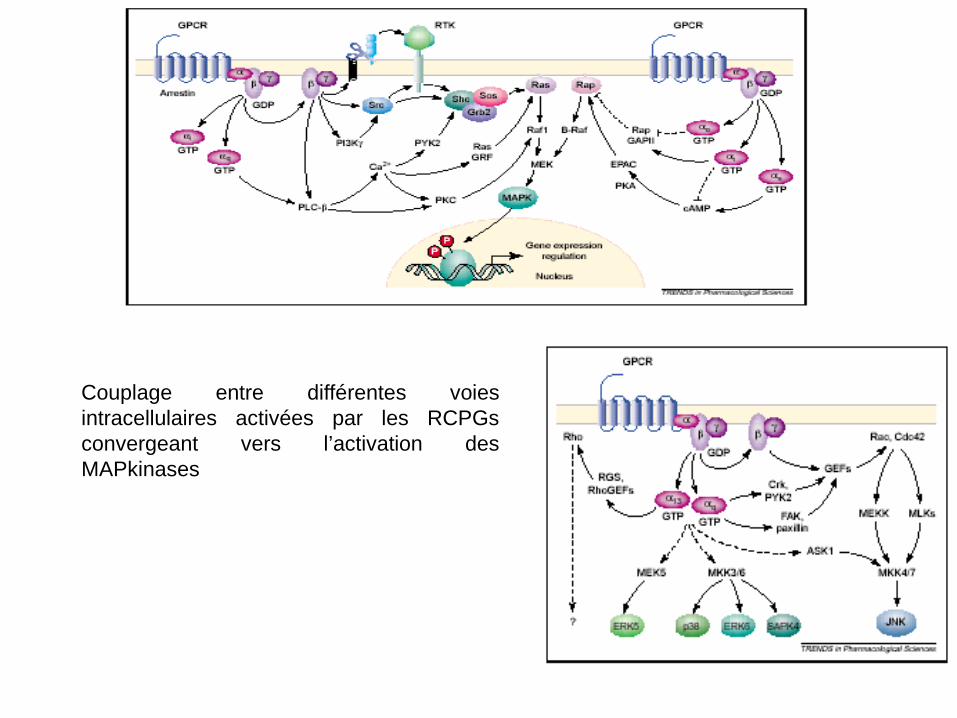
Couplage entre différentes voies intracellulaires activées par les RCPGs convergeant vers l’activation des MAPkinases
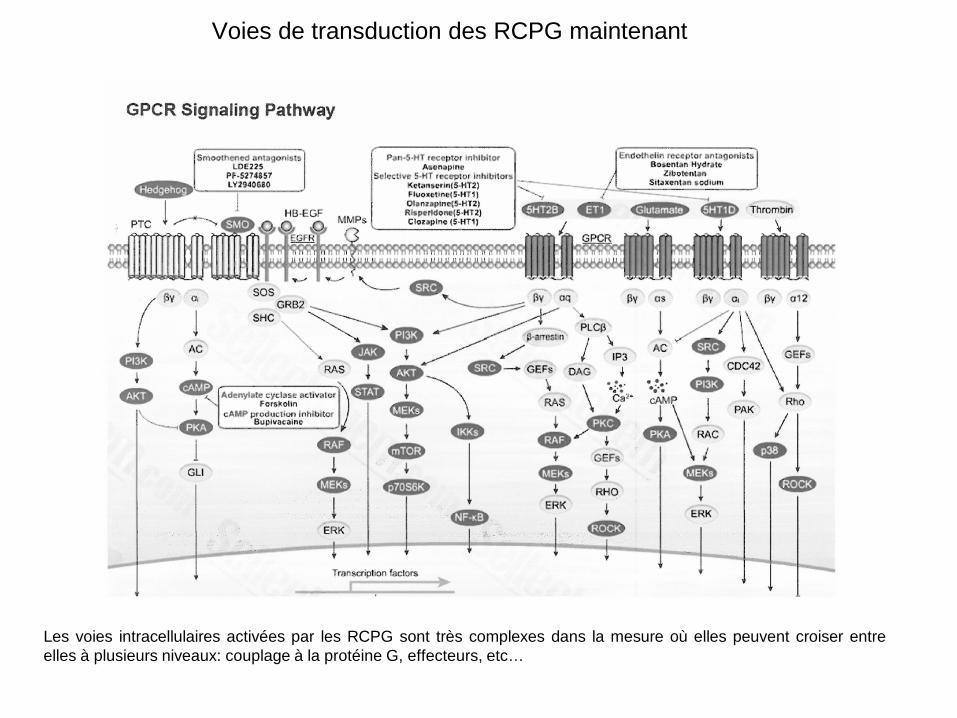
Voies de transduction des RCPG maintenant
Les voies intracellulaires activées par les RCPG sont très complexes dans la mesure où elles peuvent croiser entre elles à plusieurs niveaux: couplage à la protéine G, effecteurs, etc…

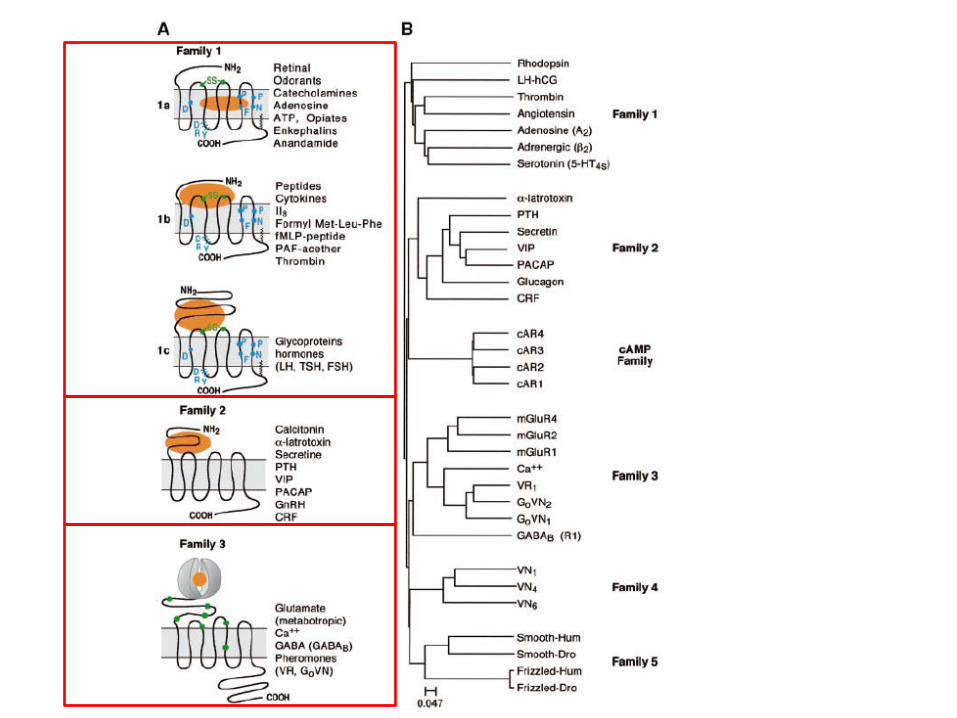
Les différentes familles des RCPGs en fonction de leur structure
Famille 1 / Classe A: la famille de récepteurs regroupant le plus grand nombre de RCPGs. La Rhodopsine est le premier à avoir été caractérisé, puis β-adrénergique, 5HT, chimiokines, morphiniques. Ces récepteurs présentent des ponts dissulfure entre TMIII et TMIV. On peut distinguer 3 sous-familles: -1ale site de l’agoniste est entre les segments TM; -1ble site de l’agoniste est entre le domaine N-terminal et les boucles ECL1 et ECL2; -1cle site de l’agoniste est entre le domaine N-terminal et ECL1. Famille 2 / Classe B: cette famille regroupe beaucoup de récepteurs de peptides. Ces RCPGs ont en général un domain N-terminal plus long que ceux de la famille 1 avec des cystéines conservées dans ce domaine ce qui facilite la liaison de certains peptides comme PTH, VIP, GnRH, sécrétine,… Famille 3 / Classe C: les récepteurs appartenant à cette classe sont caractérisés par un très grand domaine extracellulaire N-terminal. Les principaux membres de cette classe sont les récepteurs métabotropes du glutamate, les récepteurs du GABA GABAB, les récepteurs du Ca2+… Le site de liaison de l’agoniste se trouve dans le domaine extracellulairedomaine Venus Fly trap car il ressemble à celui du piège de la dionée carnivore…
Les RCPG ont tous une structure à 7 domaines transmembranaires (7TM) avec 3 boucles extracellulaires (ECL) et 3 boucles intracellulaires (ICL). Toutefois, on peut distinguer plusieurs familles en fonction de leur séquence en acides aminés et leur structure 3D. Un des critères de séparation en différentes familles est le site de fixation de l’agoniste sur le RCPG. De nombreux RCPGs ont été cristallisés et la structure 3D ainsi établie permet de confirmer l’appartenance à une des trois familles.
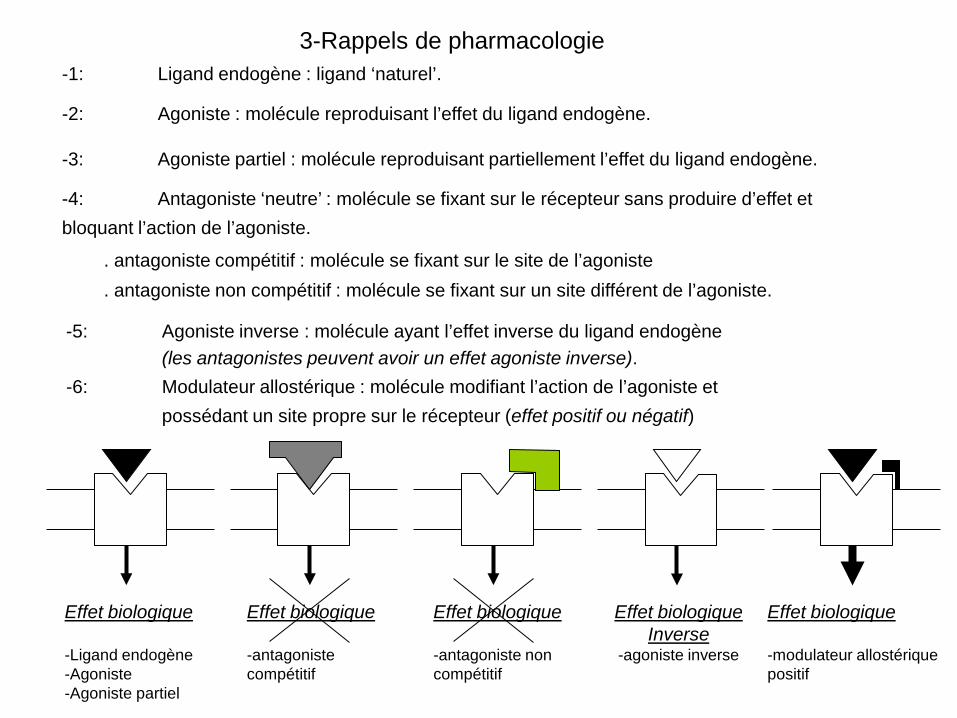
3-Rappels de pharmacologie -1: Ligand endogène : ligand ‘naturel’.
Effet biologique -Ligand endogène -Agoniste -Agoniste partiel
Effet biologique -antagoniste compétitif
Effet biologique Inverse
-agoniste inverse
Effet biologique -modulateur allostérique positif
-2: Agoniste : molécule reproduisant l’effet du ligand endogène.
-6: Modulateur allostérique : molécule modifiant l’action de l’agoniste et possédant un site propre sur le récepteur (effet positif ou négatif)
-5: Agoniste inverse : molécule ayant l’effet inverse du ligand endogène (les antagonistes peuvent avoir un effet agoniste inverse).
. antagoniste non compétitif : molécule se fixant sur un site différent de l’agoniste.
. antagoniste compétitif : molécule se fixant sur le site de l’agoniste
-4: Antagoniste ‘neutre’ : molécule se fixant sur le récepteur sans produire d’effet et bloquant l’action de l’agoniste.
-3: Agoniste partiel : molécule reproduisant partiellement l’effet du ligand endogène.
Effet biologique -antagoniste non compétitif
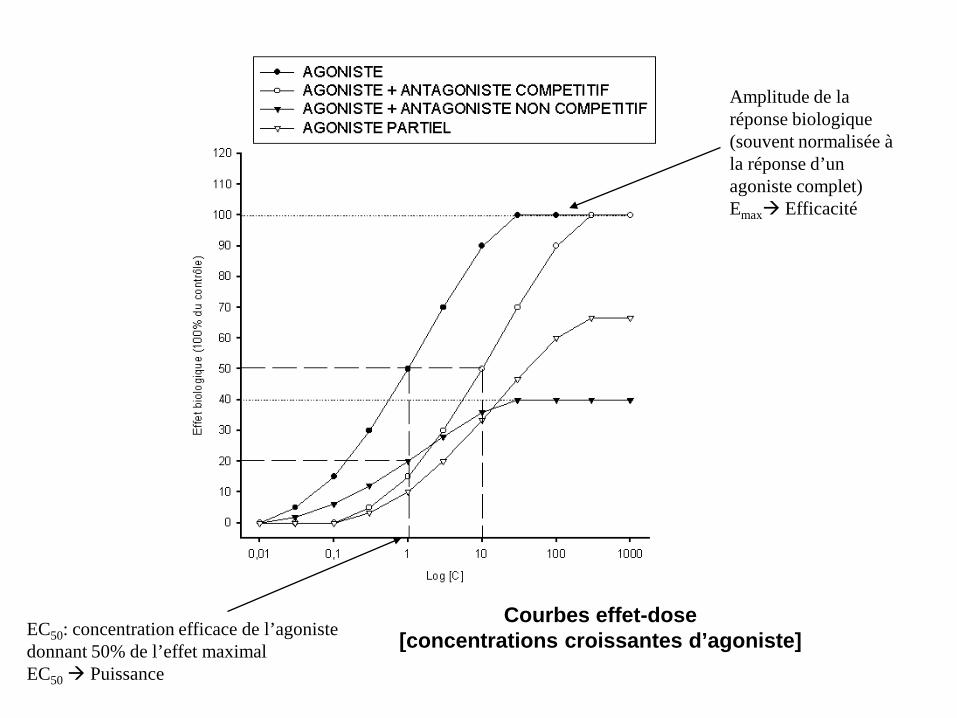
EC50: concentration efficace de l’agoniste donnant 50% de l’effet maximal EC50 Puissance
Courbes effet-dose [concentrations croissantes d’agoniste]
Amplitude de la réponse biologique (souvent normalisée à la réponse d’un agoniste complet) Emax Efficacité
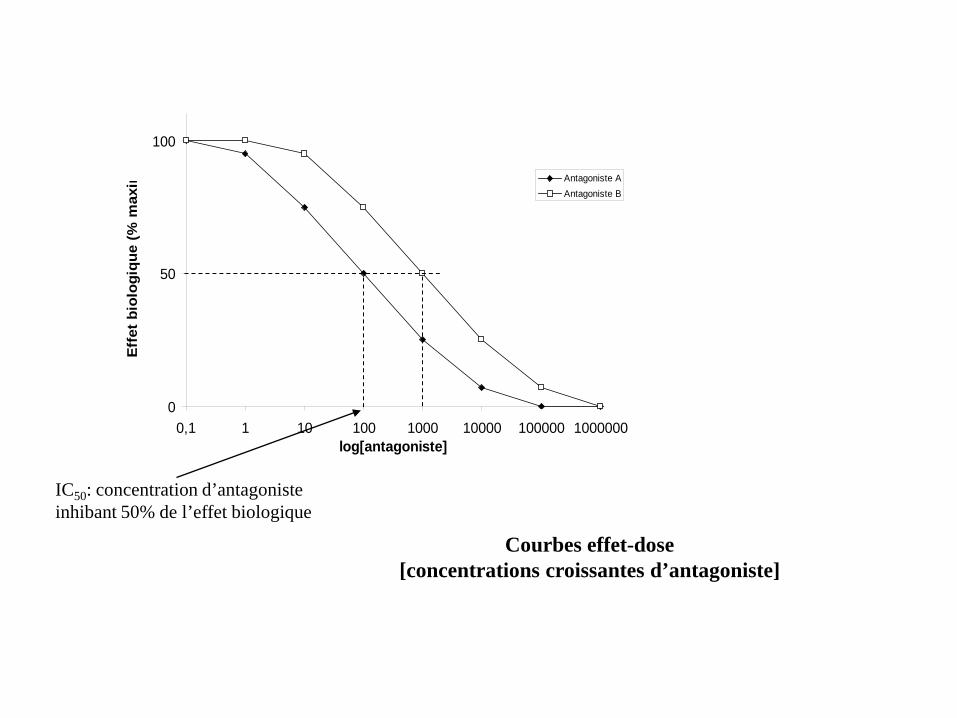
0
50
100
0,1 1 10 100 1000 10000 100000 1000000log[antagoniste]
Effe
t bio
logi
que
(% m
axim Antagoniste A
Antagoniste B
IC50: concentration d’antagoniste inhibant 50% de l’effet biologique
Courbes effet-dose [concentrations croissantes d’antagoniste]
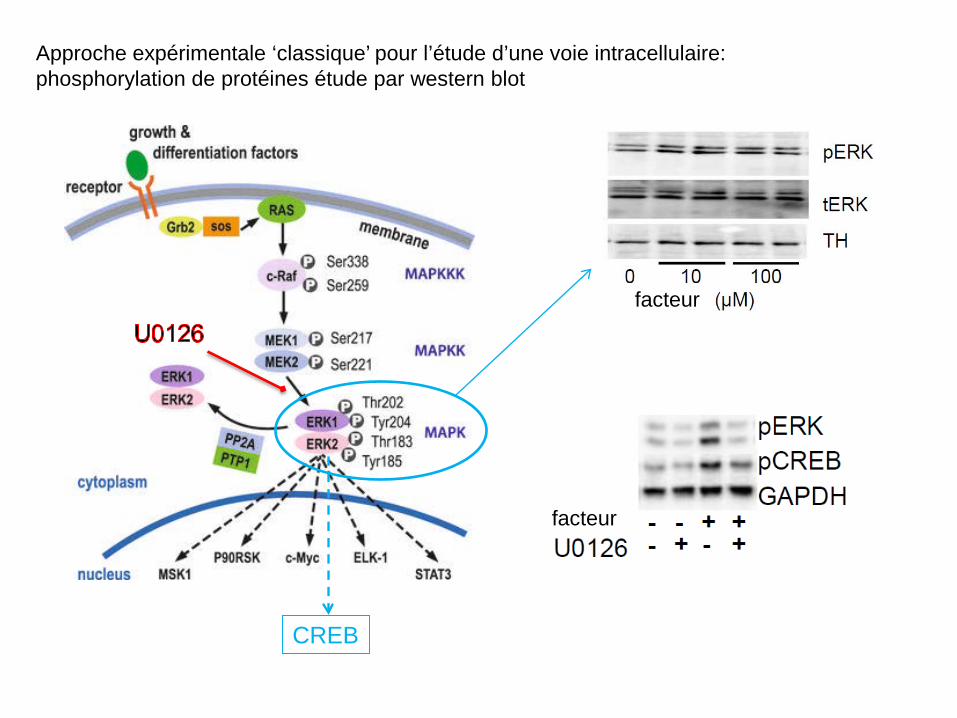
Approche expérimentale ‘classique’ pour l’étude d’une voie intracellulaire: phosphorylation de protéines étude par western blot
facteur
facteur
CREB
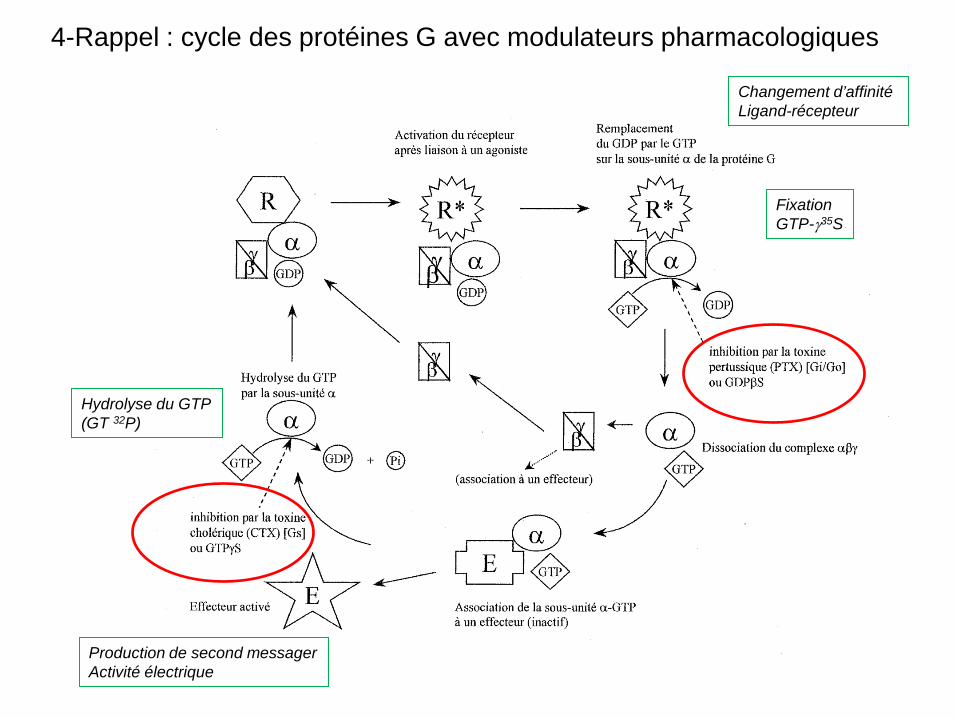
4-Rappel : cycle des protéines G avec modulateurs pharmacologiques
Fixation GTP-γ35S
Changement d’affinité Ligand-récepteur
Production de second messager Activité électrique
Hydrolyse du GTP (GT 32P)
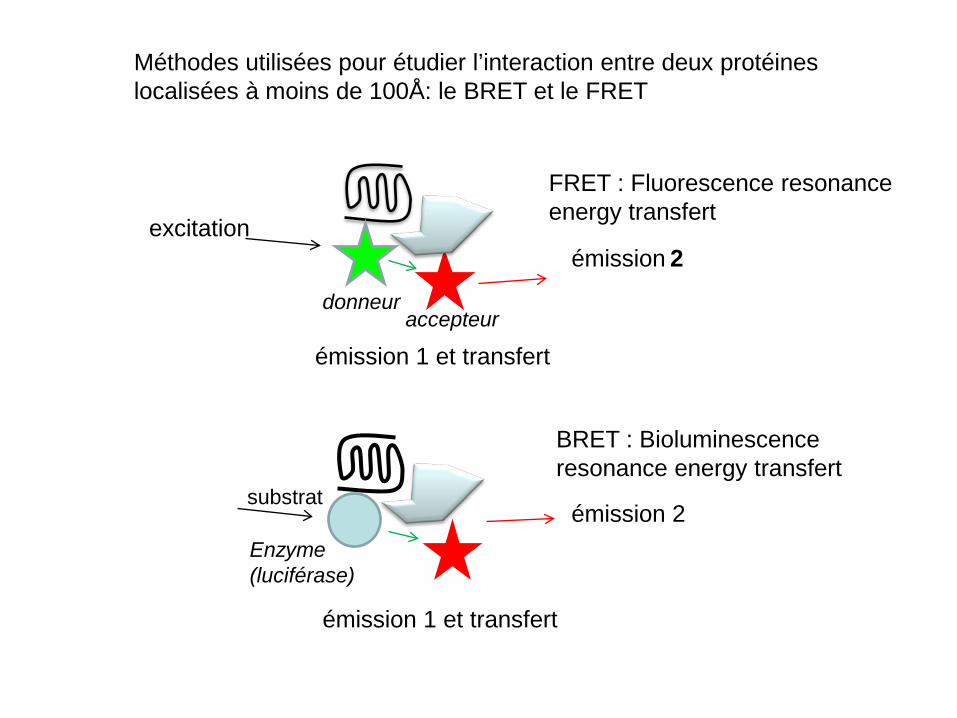
excitation
émission 1 et transfert
émission 2
Enzyme (luciférase)
émission 1 et transfert
émission 2 substrat
donneur accepteur
Méthodes utilisées pour étudier l’interaction entre deux protéines localisées à moins de 100Å: le BRET et le FRET
FRET : Fluorescence resonance energy transfert
BRET : Bioluminescence resonance energy transfert
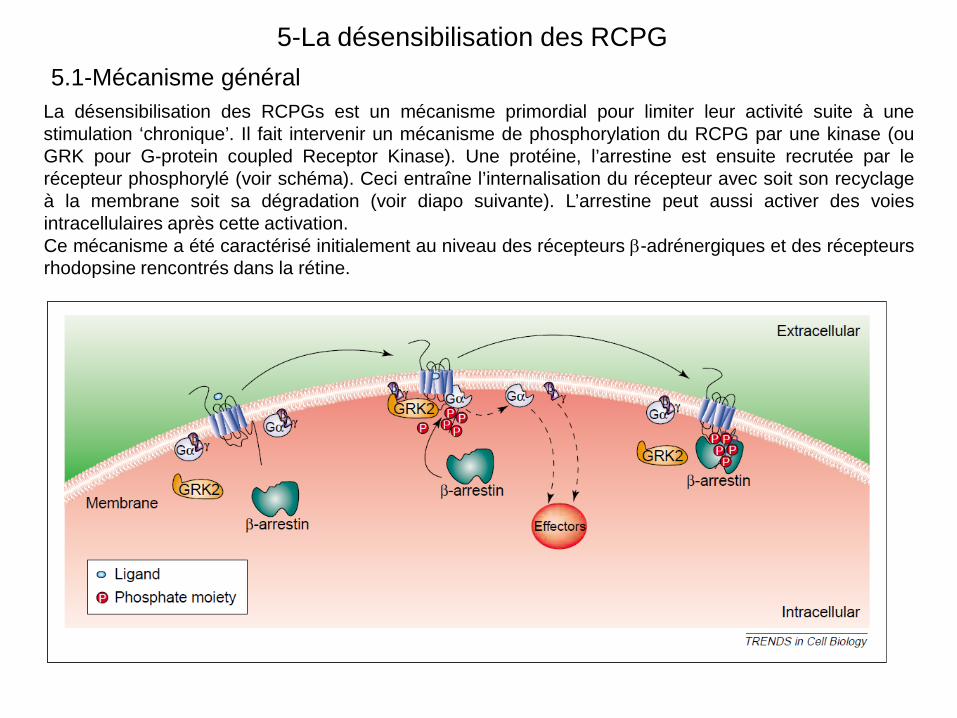
5-La désensibilisation des RCPG
La désensibilisation des RCPGs est un mécanisme primordial pour limiter leur activité suite à une stimulation ‘chronique’. Il fait intervenir un mécanisme de phosphorylation du RCPG par une kinase (ou GRK pour G-protein coupled Receptor Kinase). Une protéine, l’arrestine est ensuite recrutée par le récepteur phosphorylé (voir schéma). Ceci entraîne l’internalisation du récepteur avec soit son recyclage à la membrane soit sa dégradation (voir diapo suivante). L’arrestine peut aussi activer des voies intracellulaires après cette activation. Ce mécanisme a été caractérisé initialement au niveau des récepteurs β-adrénergiques et des récepteurs rhodopsine rencontrés dans la rétine.
5.1-Mécanisme général
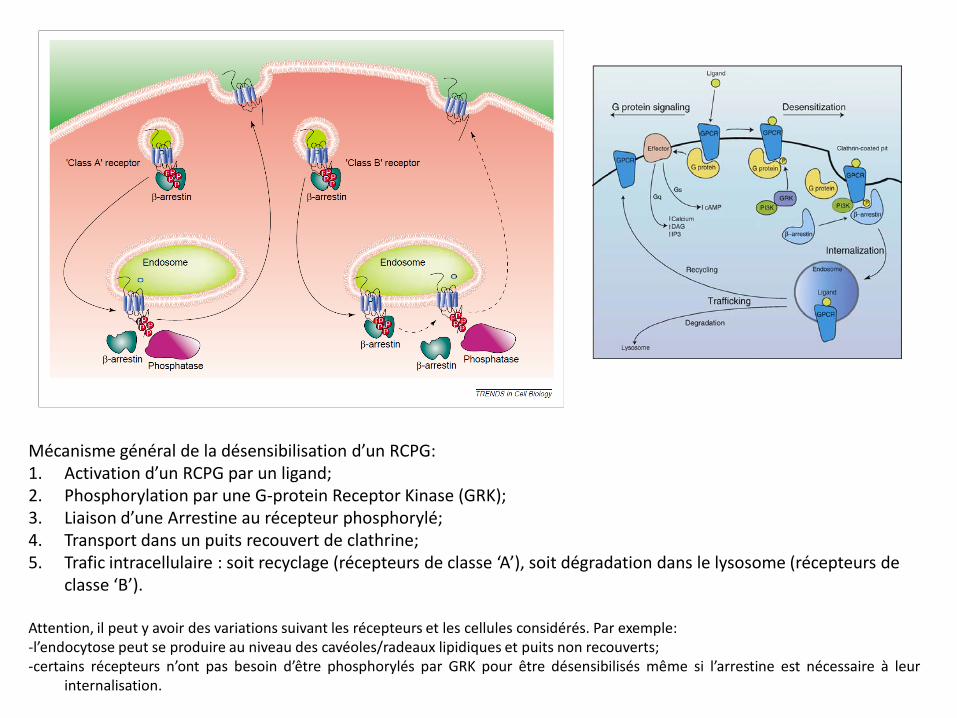
Mécanisme général de la désensibilisation d’un RCPG: 1. Activation d’un RCPG par un ligand; 2. Phosphorylation par une G-protein Receptor Kinase (GRK); 3. Liaison d’une Arrestine au récepteur phosphorylé; 4. Transport dans un puits recouvert de clathrine; 5. Trafic intracellulaire : soit recyclage (récepteurs de classe ‘A’), soit dégradation dans le lysosome (récepteurs de
classe ‘B’). Attention, il peut y avoir des variations suivant les récepteurs et les cellules considérés. Par exemple: -l’endocytose peut se produire au niveau des cavéoles/radeaux lipidiques et puits non recouverts; -certains récepteurs n’ont pas besoin d’être phosphorylés par GRK pour être désensibilisés même si l’arrestine est nécessaire à leur
internalisation.
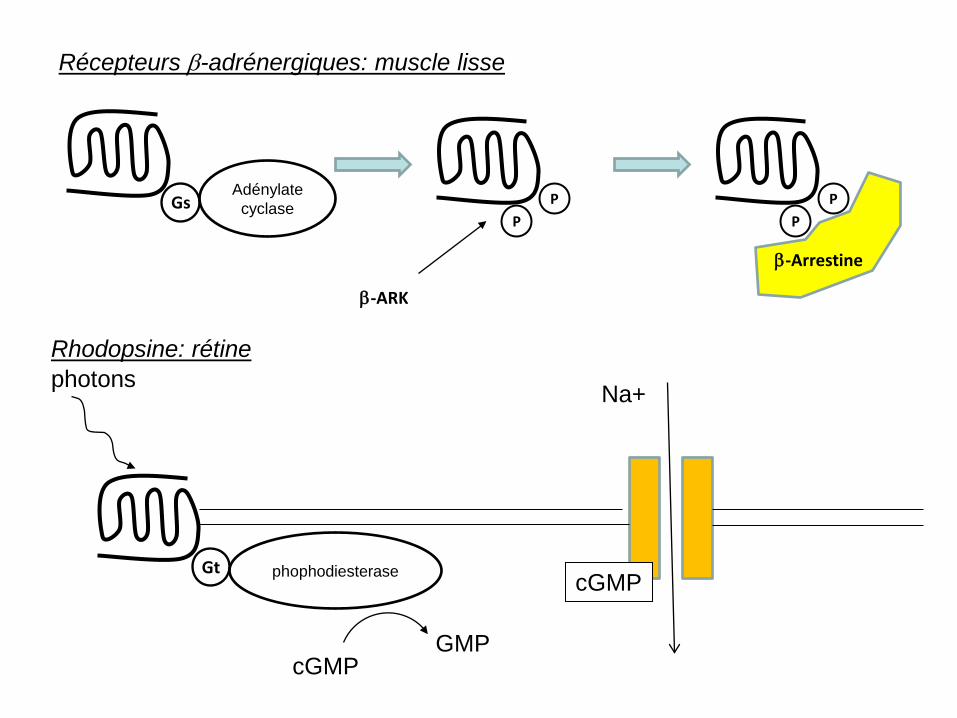
Gs Adénylate
cyclase P
β-ARK
P P
P
β-Arrestine
Gt phophodiesterase
cGMP GMP
cGMP
Na+ photons Rhodopsine: rétine
Récepteurs β-adrénergiques: muscle lisse

GRK et Arrestines : Appartiennent à la famille des G-protein coupled receptors Interacting Proteins ou GIP.
GRK: -7 isoformes -GRK1, GRK4 et GRK7: expression limitée à certains tissus -GRK1, GRK7: rétine -GRK4:testicules, cervelet, reins -GRK2, GRK3, GRK5 et GRK6: ubiquitaires -GRK4, GRK5 et GRK6: activité constitutive
Arrestines: -4 isoformes -Arrestines 1 et 4: rétine -Arrestines 2 et 3 : ubiquitaires (=β-arrestine 1 et β-arrestine 2)
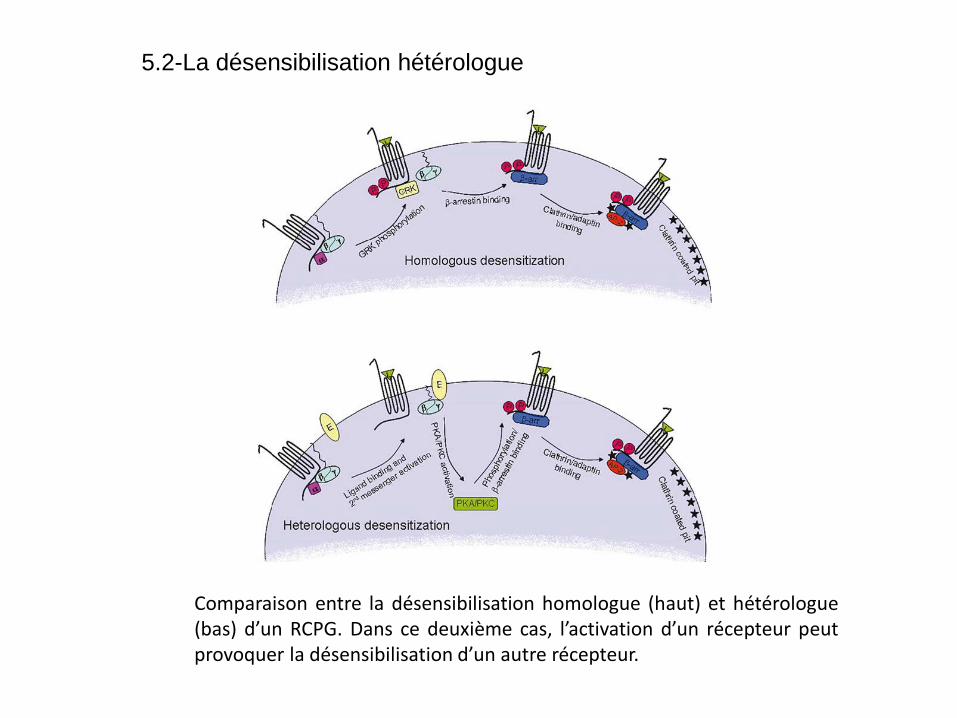
Comparaison entre la désensibilisation homologue (haut) et hétérologue (bas) d’un RCPG. Dans ce deuxième cas, l’activation d’un récepteur peut provoquer la désensibilisation d’un autre récepteur.
5.2-La désensibilisation hétérologue
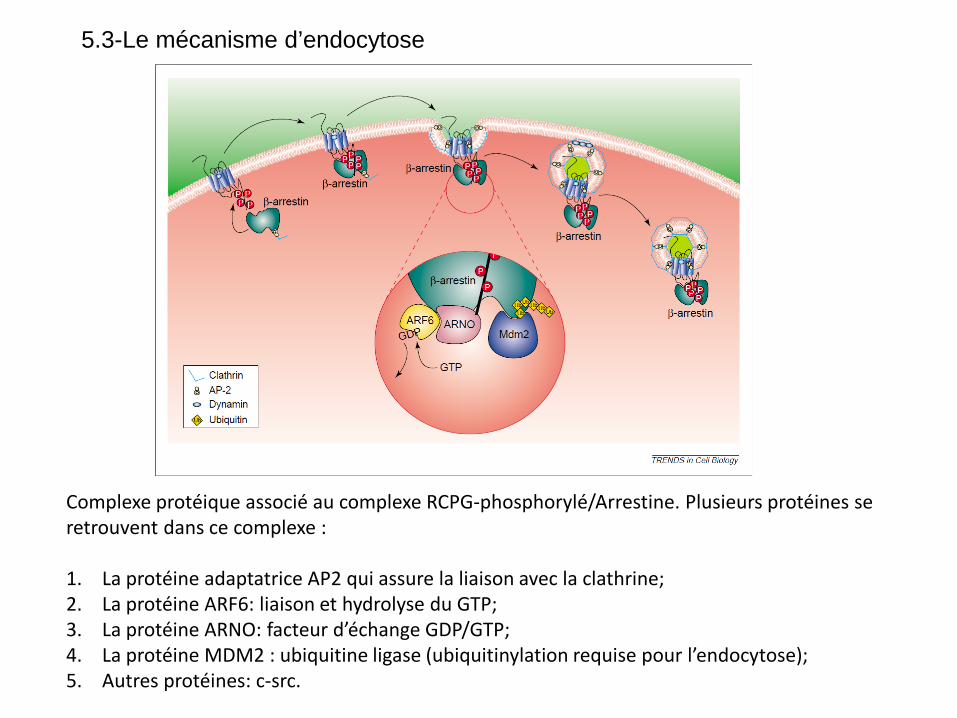
Complexe protéique associé au complexe RCPG-phosphorylé/Arrestine. Plusieurs protéines se retrouvent dans ce complexe : 1. La protéine adaptatrice AP2 qui assure la liaison avec la clathrine; 2. La protéine ARF6: liaison et hydrolyse du GTP; 3. La protéine ARNO: facteur d’échange GDP/GTP; 4. La protéine MDM2 : ubiquitine ligase (ubiquitinylation requise pour l’endocytose); 5. Autres protéines: c-src.
5.3-Le mécanisme d’endocytose
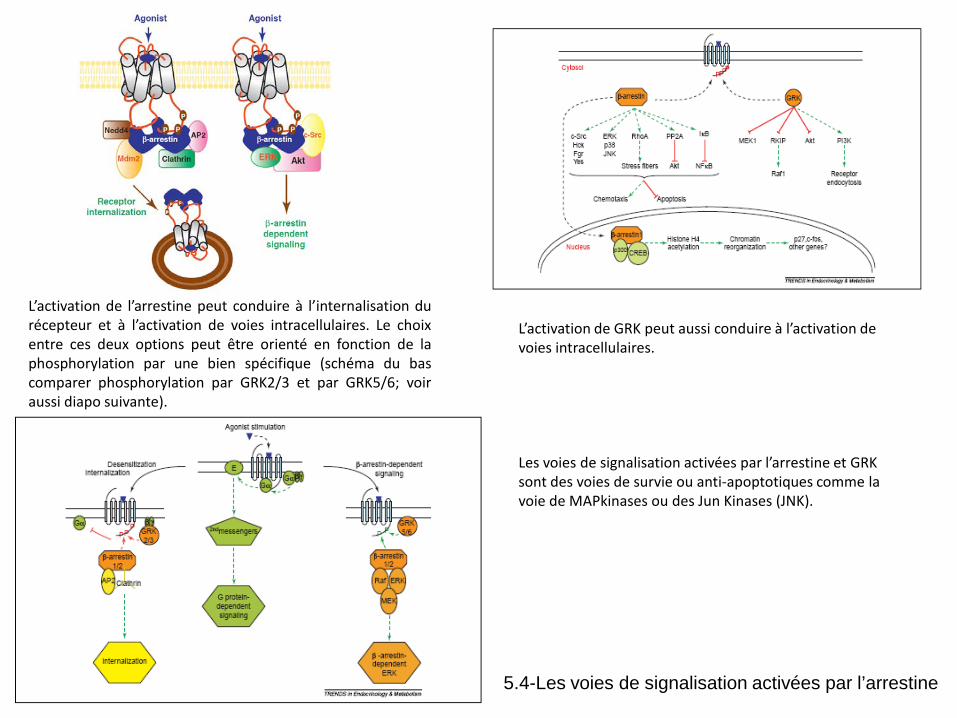
L’activation de GRK peut aussi conduire à l’activation de voies intracellulaires. Les voies de signalisation activées par l’arrestine et GRK sont des voies de survie ou anti-apoptotiques comme la voie de MAPkinases ou des Jun Kinases (JNK).
L’activation de l’arrestine peut conduire à l’internalisation du récepteur et à l’activation de voies intracellulaires. Le choix entre ces deux options peut être orienté en fonction de la phosphorylation par une bien spécifique (schéma du bas comparer phosphorylation par GRK2/3 et par GRK5/6; voir aussi diapo suivante).
5.4-Les voies de signalisation activées par l’arrestine
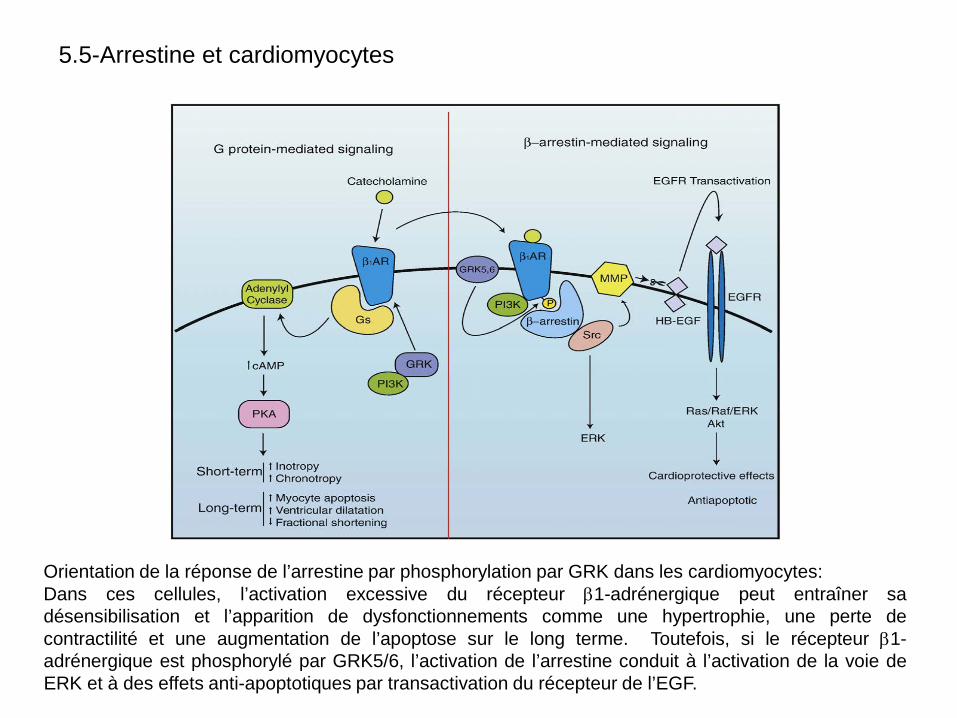
Orientation de la réponse de l’arrestine par phosphorylation par GRK dans les cardiomyocytes: Dans ces cellules, l’activation excessive du récepteur β1-adrénergique peut entraîner sa désensibilisation et l’apparition de dysfonctionnements comme une hypertrophie, une perte de contractilité et une augmentation de l’apoptose sur le long terme. Toutefois, si le récepteur β1-adrénergique est phosphorylé par GRK5/6, l’activation de l’arrestine conduit à l’activation de la voie de ERK et à des effets anti-apoptotiques par transactivation du récepteur de l’EGF.
5.5-Arrestine et cardiomyocytes
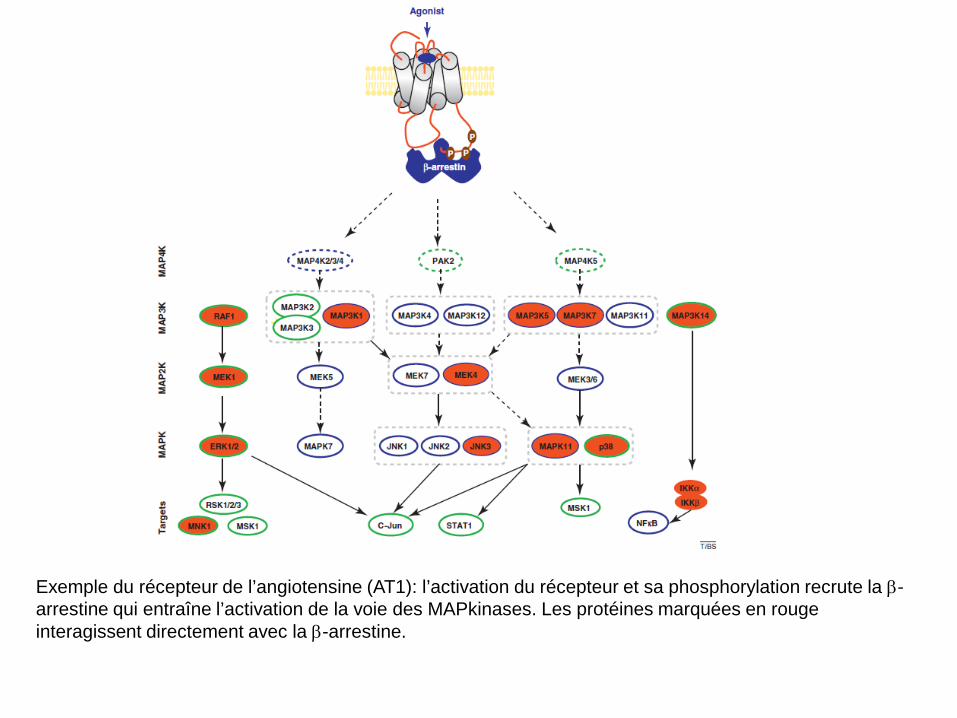
Exemple du récepteur de l’angiotensine (AT1): l’activation du récepteur et sa phosphorylation recrute la β-arrestine qui entraîne l’activation de la voie des MAPkinases. Les protéines marquées en rouge interagissent directement avec la β-arrestine.
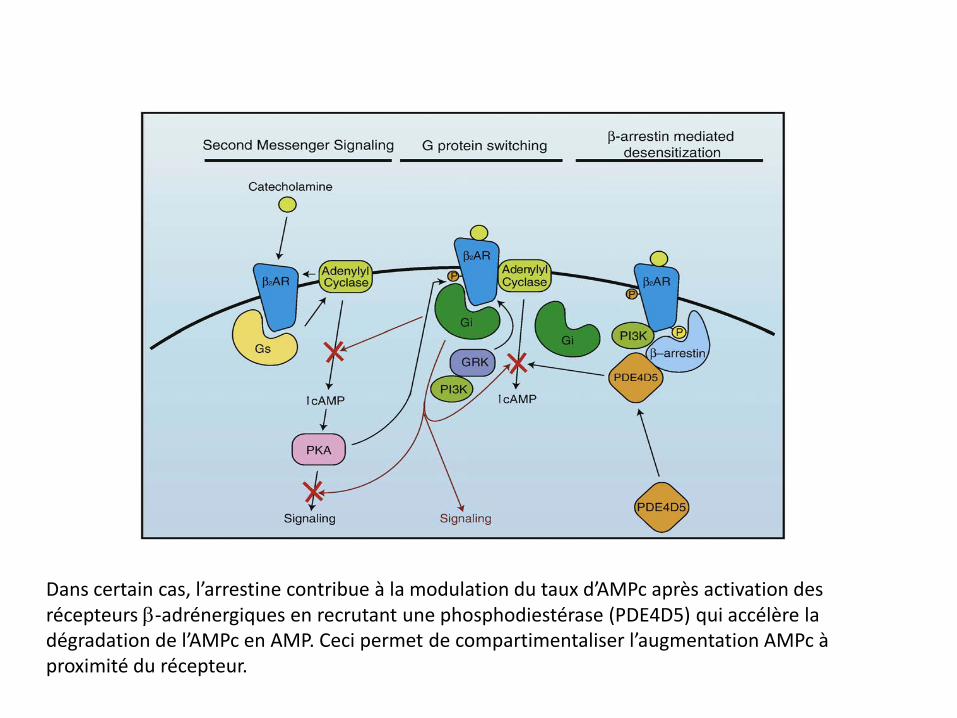
Dans certain cas, l’arrestine contribue à la modulation du taux d’AMPc après activation des récepteurs β-adrénergiques en recrutant une phosphodiestérase (PDE4D5) qui accélère la dégradation de l’AMPc en AMP. Ceci permet de compartimentaliser l’augmentation AMPc à proximité du récepteur.
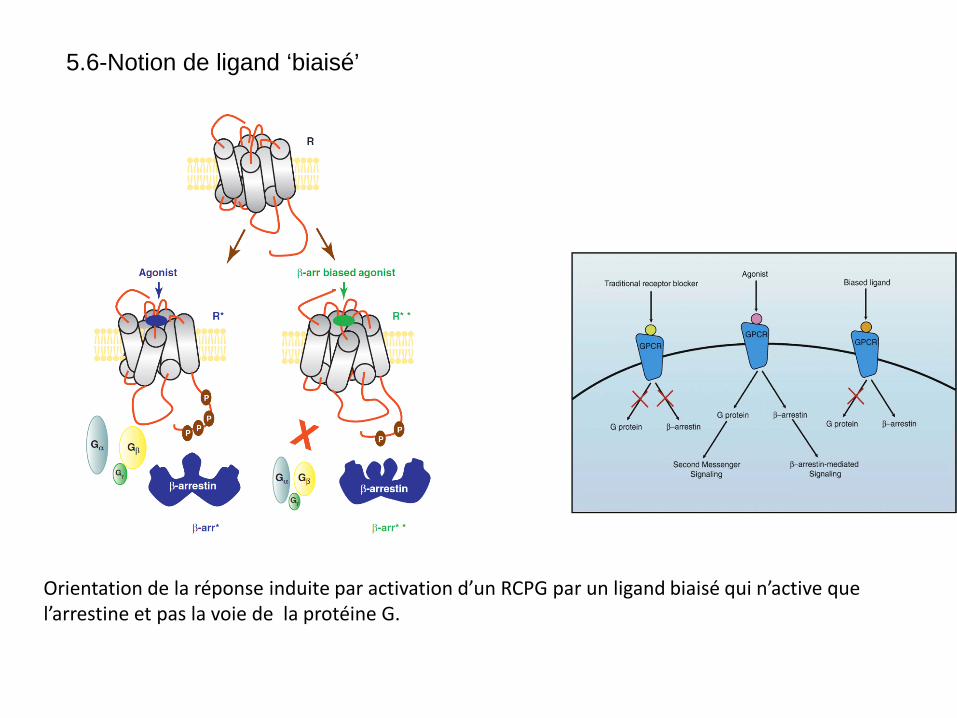
Orientation de la réponse induite par activation d’un RCPG par un ligand biaisé qui n’active que l’arrestine et pas la voie de la protéine G.
5.6-Notion de ligand ‘biaisé’

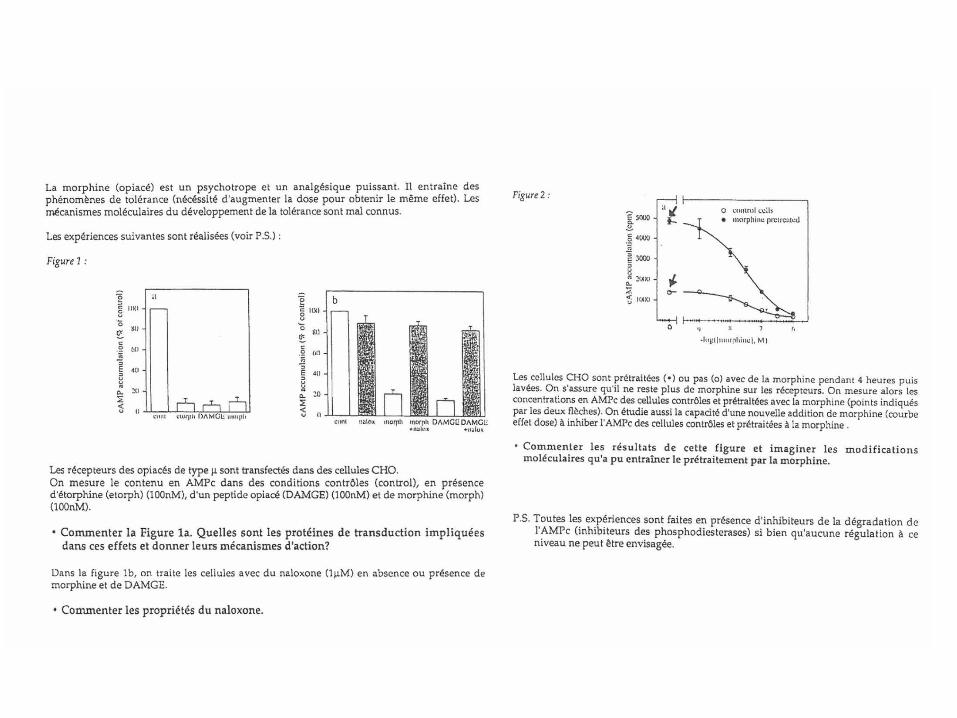
5.7-Phénomène de tolérance: le cas des récepteurs opiacés
1-Les opiacés sont les analgésiques les plus utilisés et les plus efficaces à ce jour. 2-Toutefois, une utilisation chronique conduit aux phénomènes de tolérance et de dépendance physique et psychique. 3-Aux Etats-Unis la crise des opiacés est devenue un vrai problème de santé publique: environ 250000 décès par jour par overdose en 10 ans. En cause, la prescription massive d’analgésiques puissants, tels que l’oxycodone, l’hydrocodone, la codéine, la morphine, le fentanyl qui provoquent la dépendance chez les patients et l’usage d’héroïne. 4-Au plan neurophysiologique, les peptides opiacés (enképhalines, dynorphine) agissent au niveau de la moelle épinière pour bloquer la nociception. Ces peptides sont libérés par les interneurones ‘descendants’. 5-Ils peuvent aussi agir sur le ‘circuit de la récompense’. 6-Les mécanismes de la tolérance sont encore mal connus.

Récepteurs opiacés 1-Quatre types de récepteurs opiacés (RCPG) : δ (DOR), κ (KOR), µ (MOR), Nociceptine (NOR) 2-Les récepteurs µ sont les plus représentés. Ce sont les principaux médiateurs de l’effet analgésique des opiacés. 3-Couplage principal: Gi. 4-Possibilité d’hétérodimérisation. 5-Localisation anatomique: -MOR: .SNCforte densité noyaux gris centraux (striatum), moelle épinière (corne dorsale). .système nerveux entérique. -DOR: .SNCBulbe olfactif, cortex, striatum, noyau accumbens .système nerveux entérique. -KOR: .SNCmoelle épinière, noyau accumbens -NOP: .SNC et système nerveux entérique
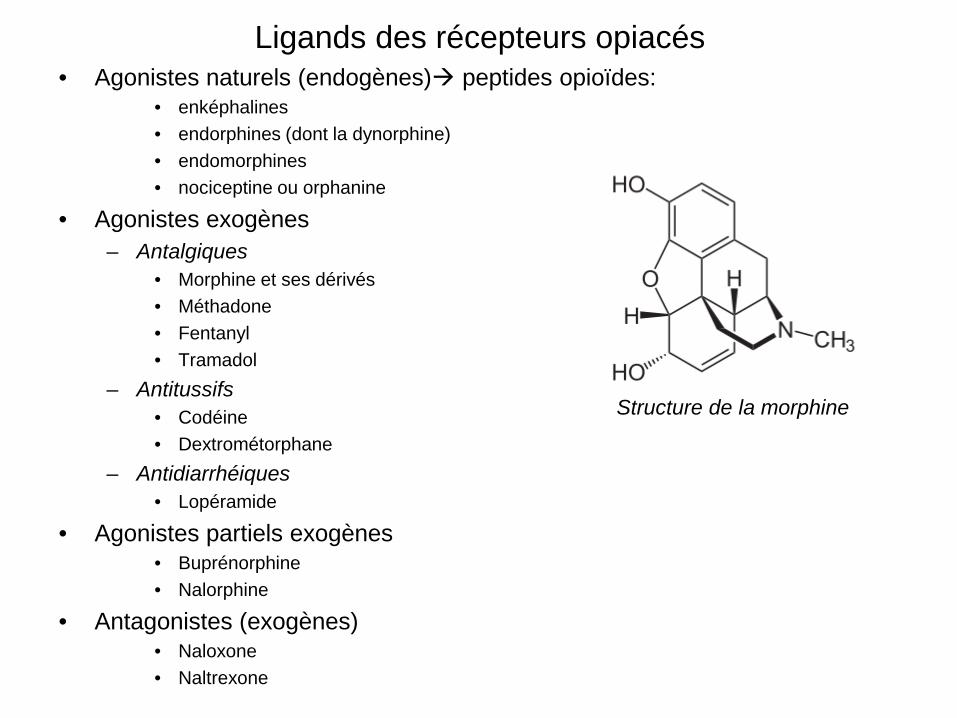
Ligands des récepteurs opiacés • Agonistes naturels (endogènes) peptides opioïdes:
• enképhalines • endorphines (dont la dynorphine) • endomorphines • nociceptine ou orphanine
• Agonistes exogènes – Antalgiques
• Morphine et ses dérivés • Méthadone • Fentanyl • Tramadol
– Antitussifs • Codéine • Dextrométorphane
– Antidiarrhéiques • Lopéramide
• Agonistes partiels exogènes • Buprénorphine • Nalorphine
• Antagonistes (exogènes) • Naloxone • Naltrexone
Structure de la morphine
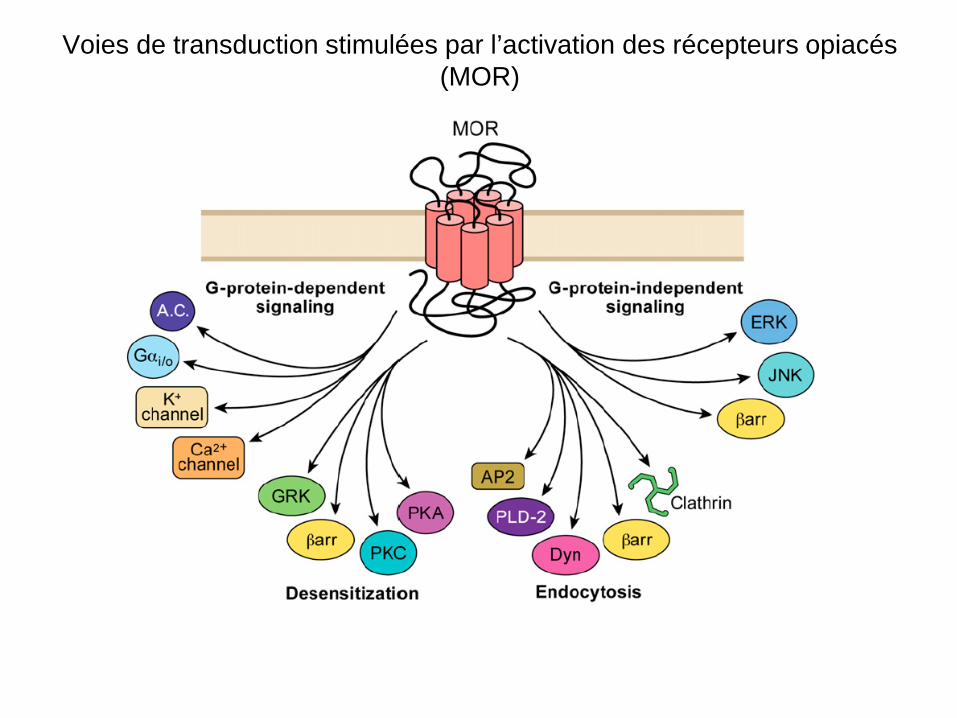
Voies de transduction stimulées par l’activation des récepteurs opiacés (MOR)
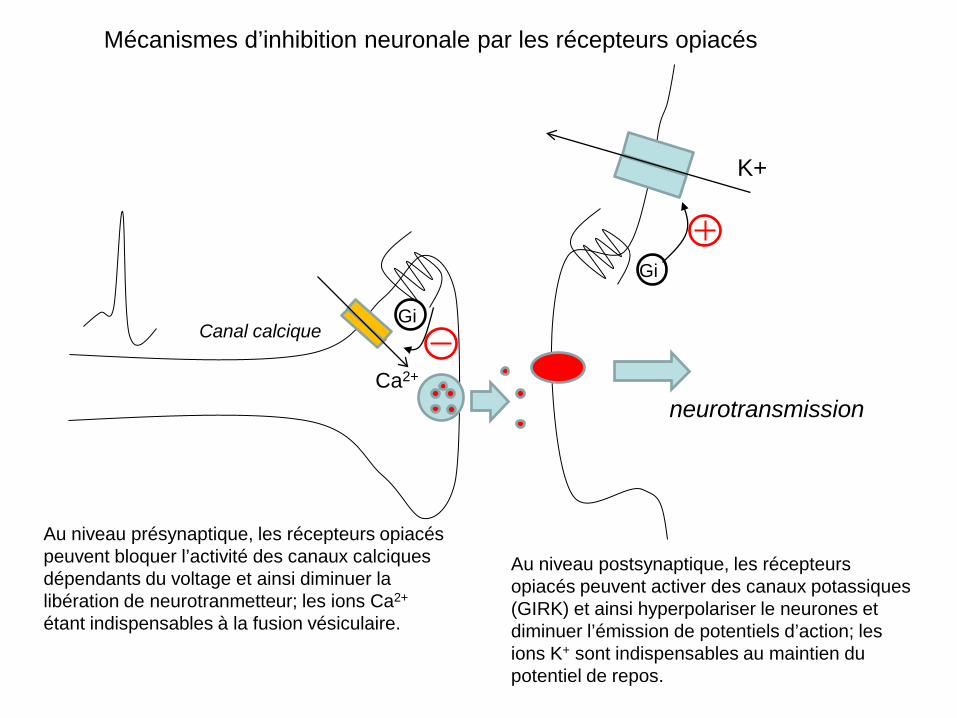
Mécanismes d’inhibition neuronale par les récepteurs opiacés
Gi Canal calcique
Ca2+
Gi
K+
neurotransmission
Au niveau présynaptique, les récepteurs opiacés peuvent bloquer l’activité des canaux calciques dépendants du voltage et ainsi diminuer la libération de neurotranmetteur; les ions Ca2+ étant indispensables à la fusion vésiculaire.
Au niveau postsynaptique, les récepteurs opiacés peuvent activer des canaux potassiques (GIRK) et ainsi hyperpolariser le neurones et diminuer l’émission de potentiels d’action; les ions K+ sont indispensables au maintien du potentiel de repos.
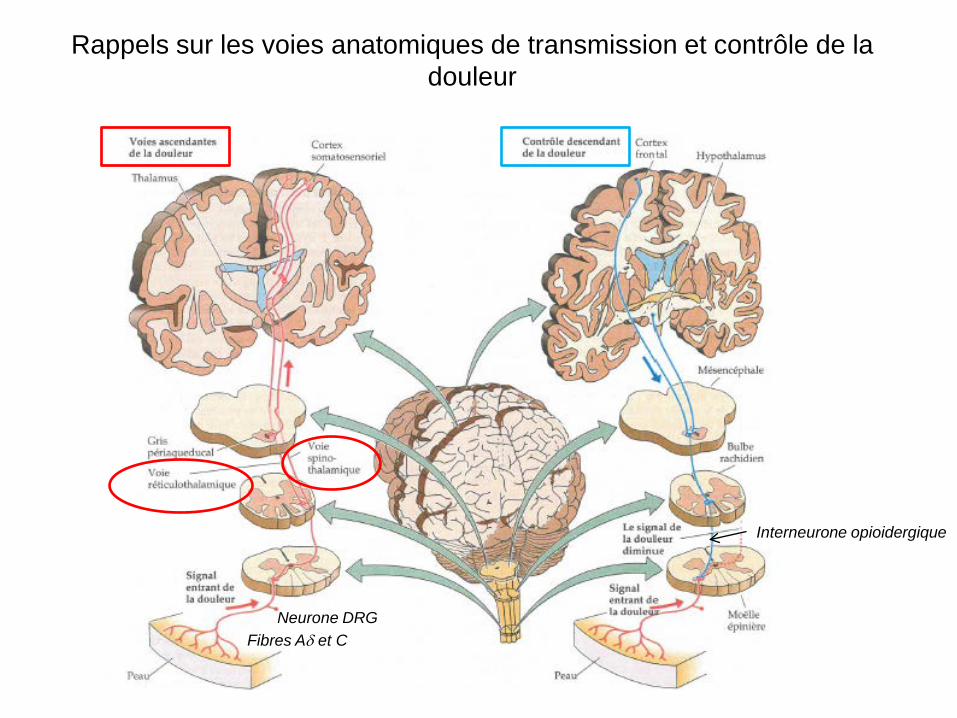
Rappels sur les voies anatomiques de transmission et contrôle de la douleur
Fibres Aδ et C Neurone DRG
Interneurone opioidergique
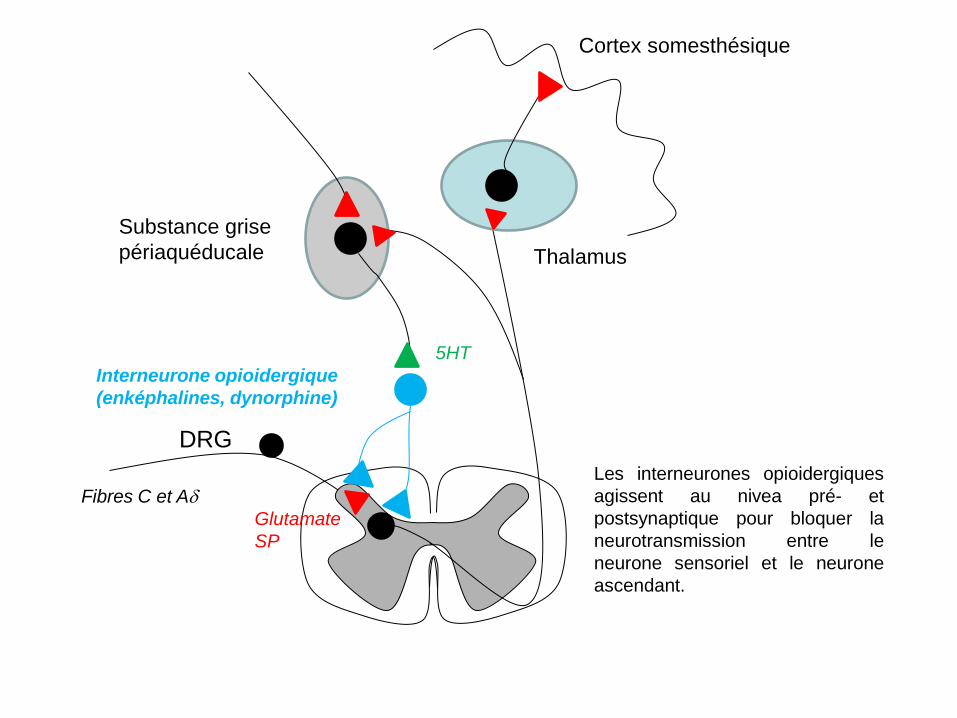
DRG
Fibres C et Aδ
Interneurone opioidergique (enképhalines, dynorphine)
Thalamus
Cortex somesthésique
Substance grise périaquéducale
5HT
Glutamate SP
Les interneurones opioidergiques agissent au nivea pré- et postsynaptique pour bloquer la neurotransmission entre le neurone sensoriel et le neurone ascendant.
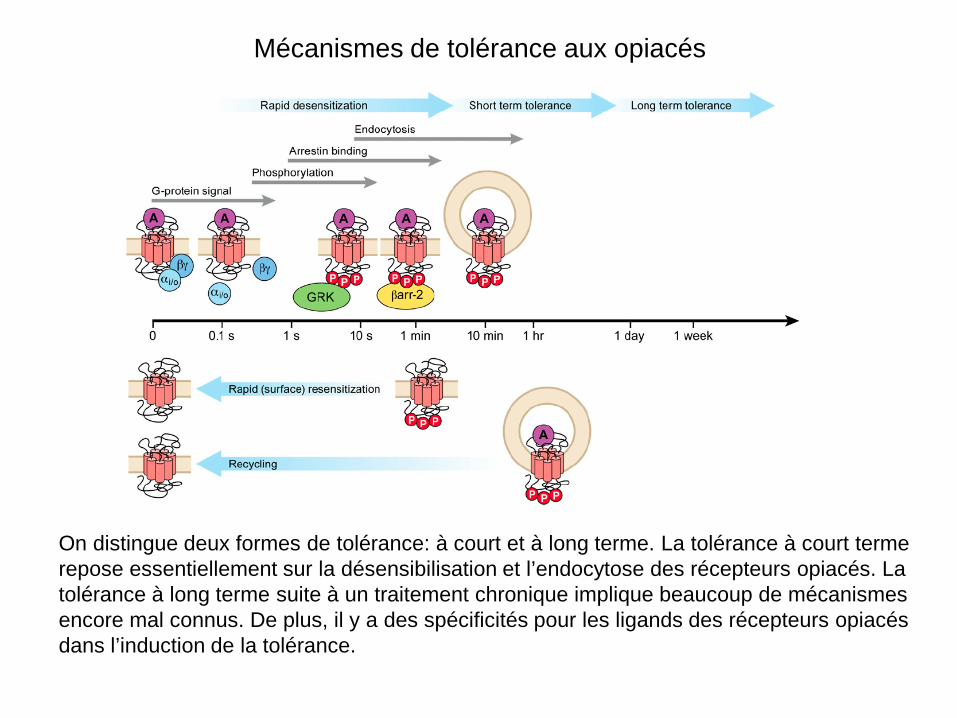
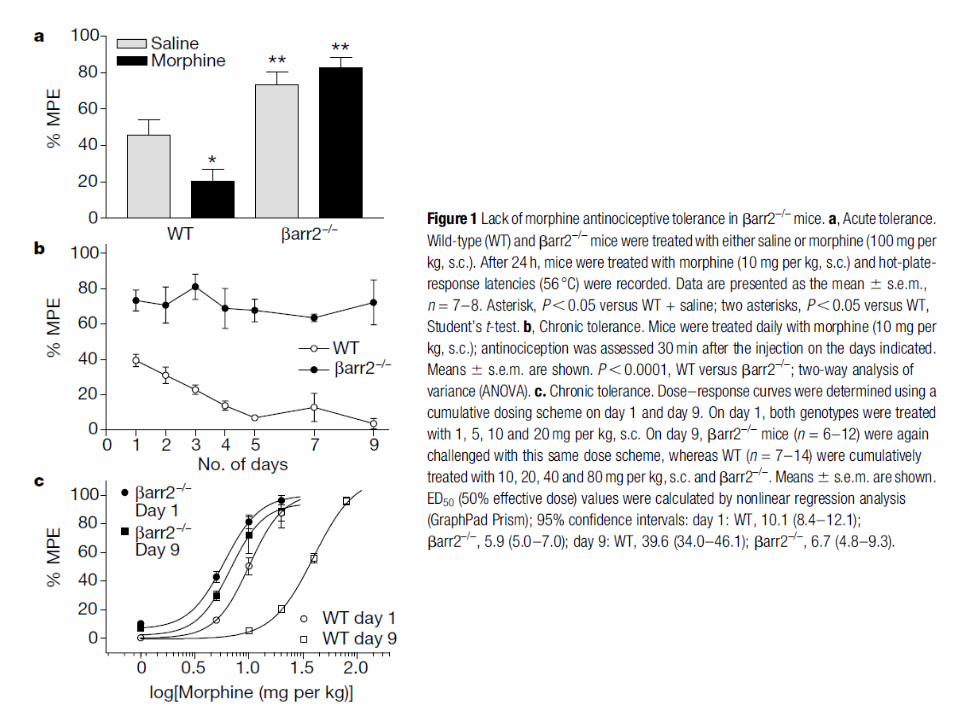
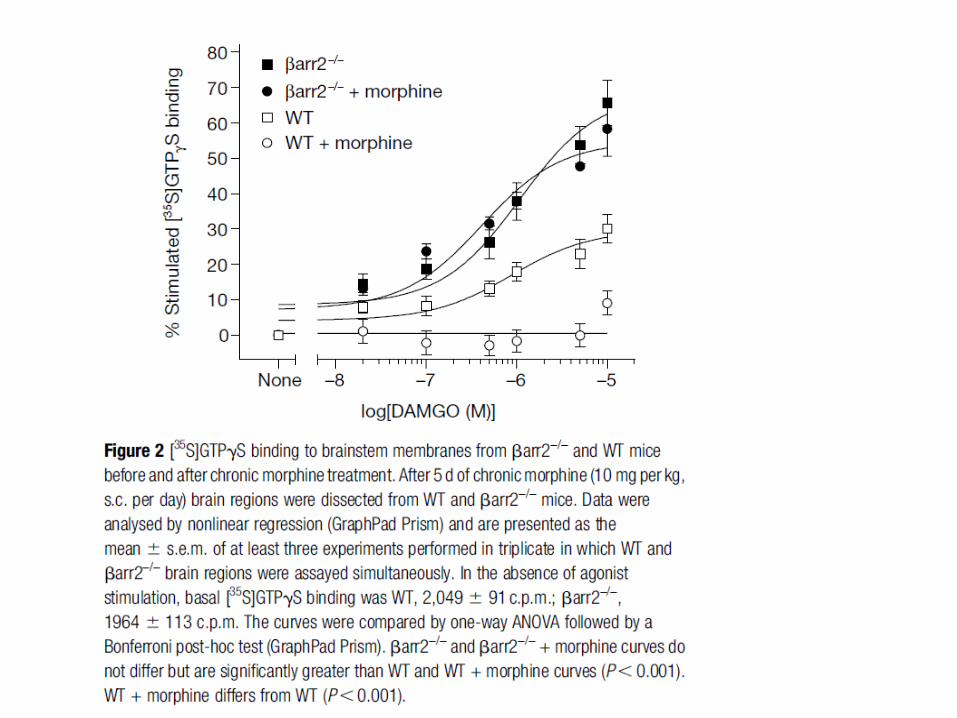
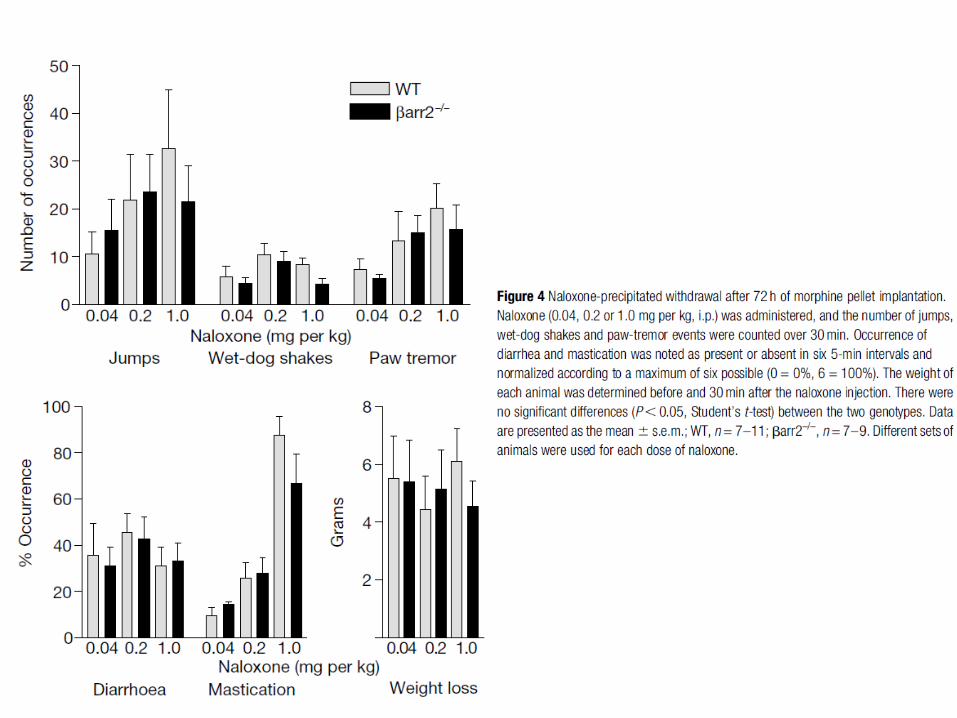
Mécanismes de tolérance aux opiacés
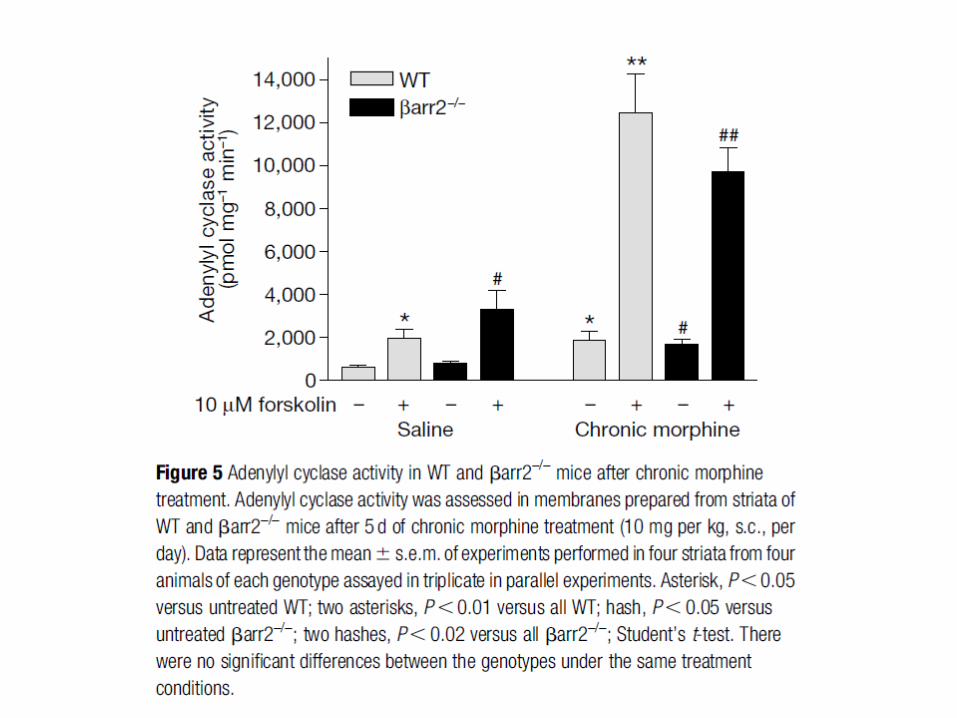
On distingue deux formes de tolérance: à court et à long terme. La tolérance à court terme repose essentiellement sur la désensibilisation et l’endocytose des récepteurs opiacés. La tolérance à long terme suite à un traitement chronique implique beaucoup de mécanismes encore mal connus. De plus, il y a des spécificités pour les ligands des récepteurs opiacés dans l’induction de la tolérance.
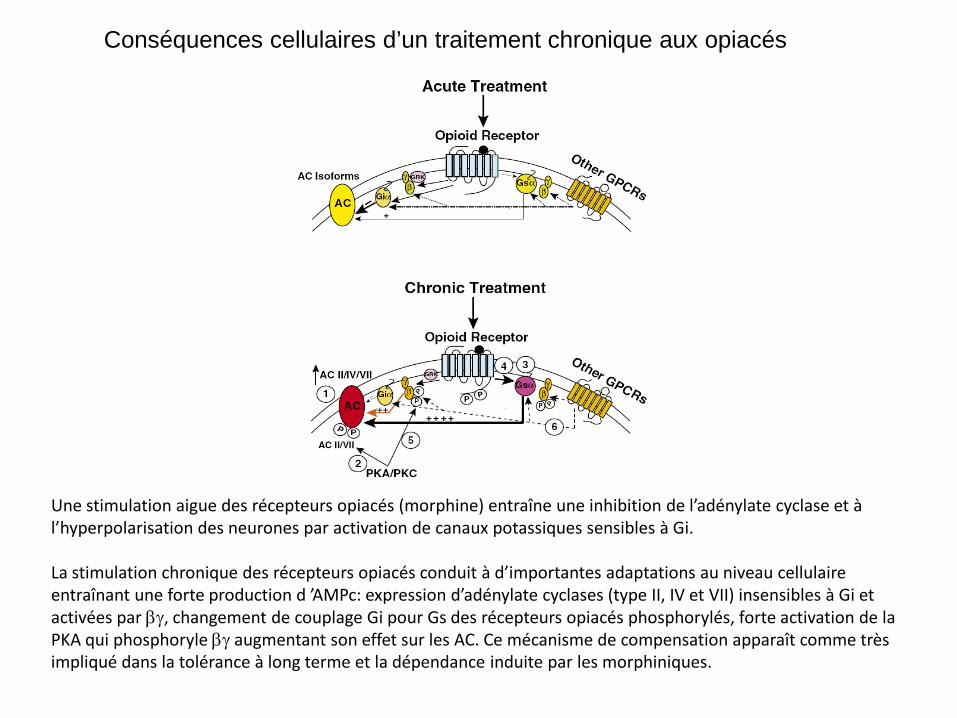
Une stimulation aigue des récepteurs opiacés (morphine) entraîne une inhibition de l’adénylate cyclase et à l’hyperpolarisation des neurones par activation de canaux potassiques sensibles à Gi. La stimulation chronique des récepteurs opiacés conduit à d’importantes adaptations au niveau cellulaire entraînant une forte production d ’AMPc: expression d’adénylate cyclases (type II, IV et VII) insensibles à Gi et activées par βγ, changement de couplage Gi pour Gs des récepteurs opiacés phosphorylés, forte activation de la PKA qui phosphoryle βγ augmentant son effet sur les AC. Ce mécanisme de compensation apparaît comme très impliqué dans la tolérance à long terme et la dépendance induite par les morphiniques.
Conséquences cellulaires d’un traitement chronique aux opiacés
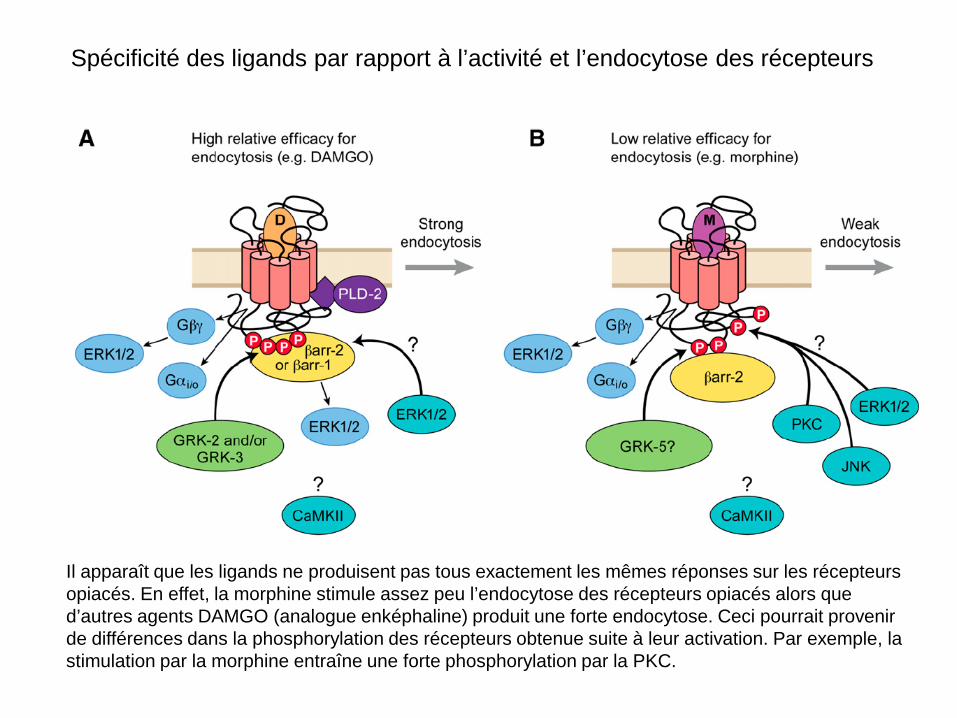
Spécificité des ligands par rapport à l’activité et l’endocytose des récepteurs
Il apparaît que les ligands ne produisent pas tous exactement les mêmes réponses sur les récepteurs opiacés. En effet, la morphine stimule assez peu l’endocytose des récepteurs opiacés alors que d’autres agents DAMGO (analogue enképhaline) produit une forte endocytose. Ceci pourrait provenir de différences dans la phosphorylation des récepteurs obtenue suite à leur activation. Par exemple, la stimulation par la morphine entraîne une forte phosphorylation par la PKC.
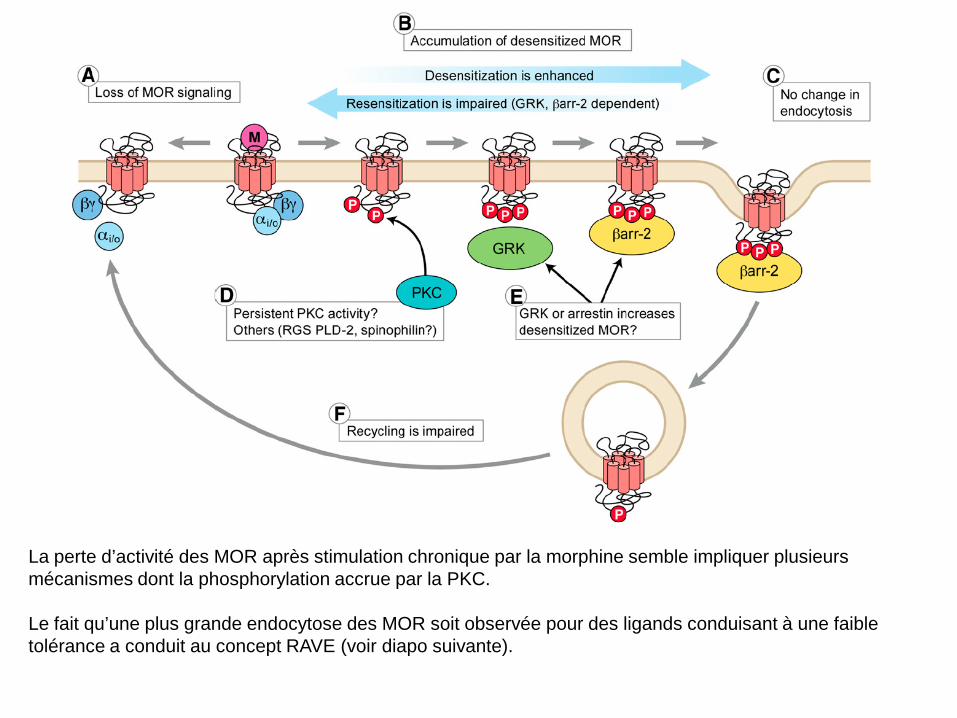
La perte d’activité des MOR après stimulation chronique par la morphine semble impliquer plusieurs mécanismes dont la phosphorylation accrue par la PKC. Le fait qu’une plus grande endocytose des MOR soit observée pour des ligands conduisant à une faible tolérance a conduit au concept RAVE (voir diapo suivante).
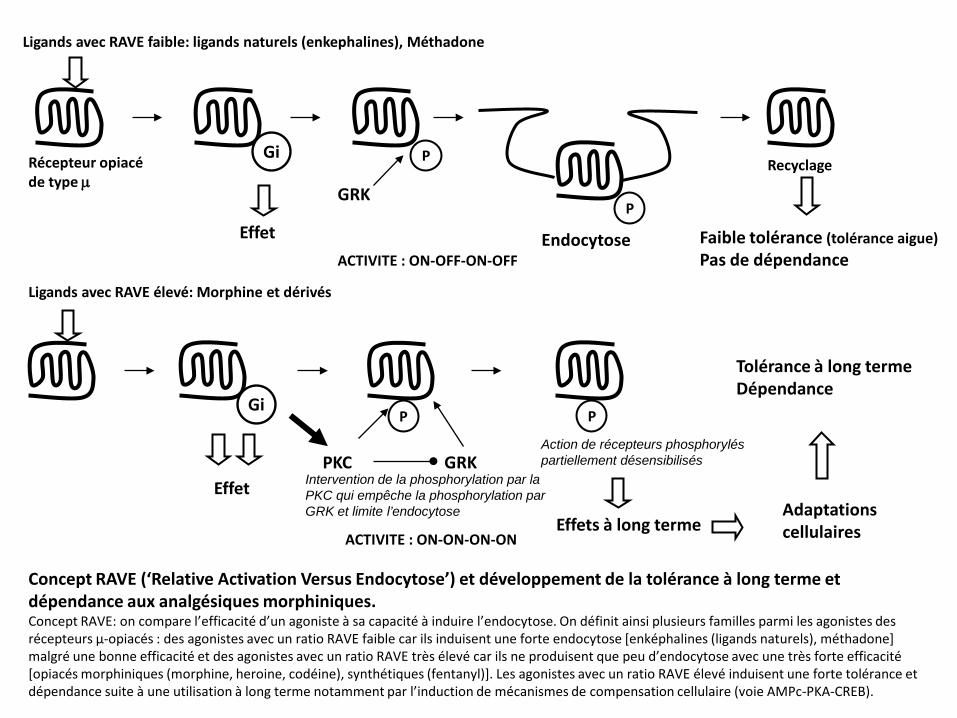
Gi P
Concept RAVE (‘Relative Activation Versus Endocytose’) et développement de la tolérance à long terme et dépendance aux analgésiques morphiniques. Concept RAVE: on compare l’efficacité d’un agoniste à sa capacité à induire l’endocytose. On définit ainsi plusieurs familles parmi les agonistes des récepteurs µ-opiacés : des agonistes avec un ratio RAVE faible car ils induisent une forte endocytose [enképhalines (ligands naturels), méthadone] malgré une bonne efficacité et des agonistes avec un ratio RAVE très élevé car ils ne produisent que peu d’endocytose avec une très forte efficacité [opiacés morphiniques (morphine, heroine, codéine), synthétiques (fentanyl)]. Les agonistes avec un ratio RAVE élevé induisent une forte tolérance et dépendance suite à une utilisation à long terme notamment par l’induction de mécanismes de compensation cellulaire (voie AMPc-PKA-CREB).
GRK P
Effet Endocytose
Recyclage
ACTIVITE : ON-OFF-ON-OFF
Gi
Effet PKC
P
GRK Intervention de la phosphorylation par la PKC qui empêche la phosphorylation par GRK et limite l’endocytose
P
Action de récepteurs phosphorylés partiellement désensibilisés
Effets à long terme ACTIVITE : ON-ON-ON-ON
Adaptations cellulaires
Tolérance à long terme Dépendance
Ligands avec RAVE faible: ligands naturels (enkephalines), Méthadone
Ligands avec RAVE élevé: Morphine et dérivés
Récepteur opiacé de type µ
Faible tolérance (tolérance aigue) Pas de dépendance

Encore beaucoup de questions…
• La désensibilisation des récepteurs et tolérance aux opiacés sont bien associées à une diminution du nombre de récepteurs fonctionnels.
• Les agonistes opiacés n’ont pas tous les mêmes effets sur les récepteurs et
voies de transduction (agonisme biaisé?) mais ceci n’est pas valable dans toutes les régions du SNC.
• Le concept RAVE apparaît limité: il ne prend en compte que l’activité et
l’endocytose pour expliquer le risuqe d’addiction. En fait la désensibilisation des récepteurs peut être aussi obtenue sans endocytose, après phosphorylation.
• La relation entre endocytose, tolérance et addiction reste encore à démontrer…

6-Régulation de l’activité des RCPGs par des protéines d’interaction (GIPs)
• Un grand nombre de protéines peuvent interagir avec des RCPGs de manière plus ou moins directe pour réguler non seulement leur activité, leur trafic intracellulaire et à la membrane, leur couplage, l’activité des protéines G auxquelles ils sont couplés ainsi que la spécificité de leur couplage;
• De nombreuses GIPs sont des protéines d’échafaudage (‘scaffold proteins’) qui couplent les RCPGs aux éléments du cytosquelette comme le réseau d’actine;
• Les protéines impliquées dans la désensibilisation, la GRK et l’arrestine, sont des GIPs;
• On distingue plusieurs familles de GIPs en fonction de leur structure et leur mode d’interaction avec les RCPG;
• Un RCPG peut aussi être une GIP vis-à-vis d’un autre RCPG lors d’une
oligomérisation.

6.1-Les protéines RGS (‘regulator of G-protein signaling’)
• La première activité des RGS découverte est l’activation de l’hydrolyse du GTP et GDP (activité GTPase Accelerating Protein ou ‘GAP’);
• Ces protéines possèdent toutes un domaine RGS qui leur permet
d’interagir avec α-GTP (voir tableau suivant); • Certaines protéines ont d’autres domaines fonctionnels (PDZ,
Kinase, etc…) qui leur confèrent d’autres fonctions; • Ces protéines peuvent aussi interagir directement avec les RCPG
par liaison avec la 3e boucle intracellulaire (i3) et le segment C-terminal: ceci leur confère un rôle modulateur sur l’activité des RCPGs.
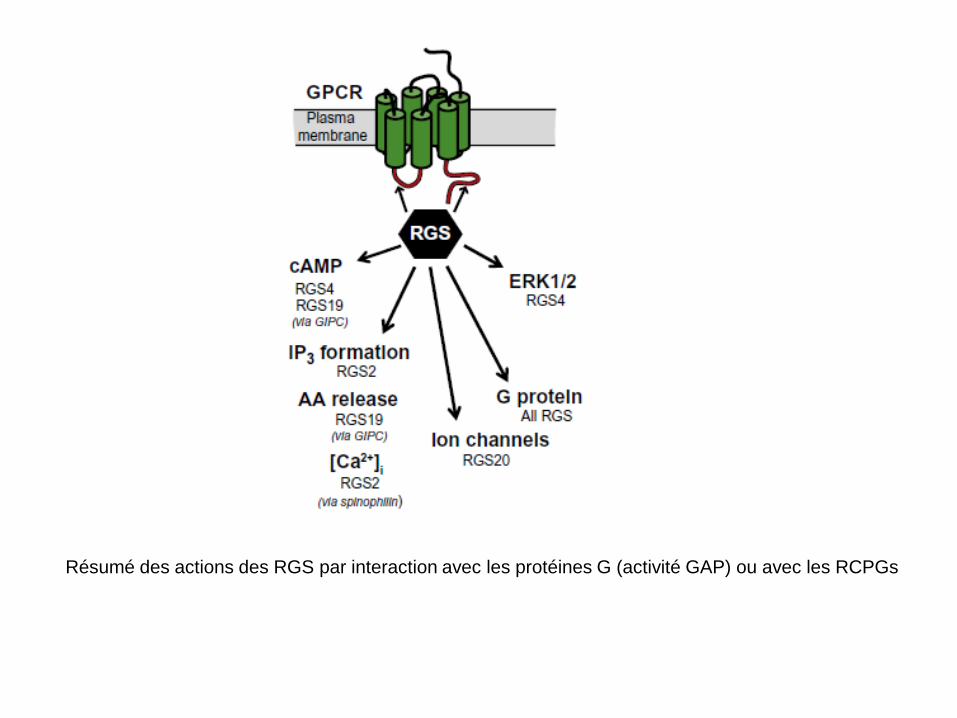
Résumé des actions des RGS par interaction avec les protéines G (activité GAP) ou avec les RCPGs
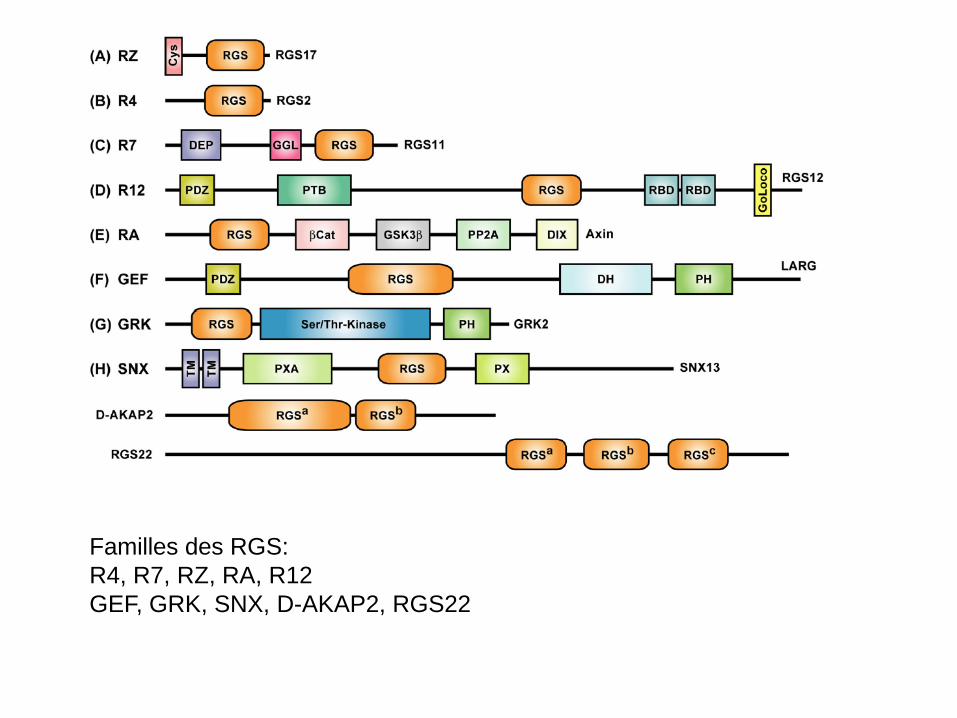
Familles des RGS: R4, R7, RZ, RA, R12 GEF, GRK, SNX, D-AKAP2, RGS22
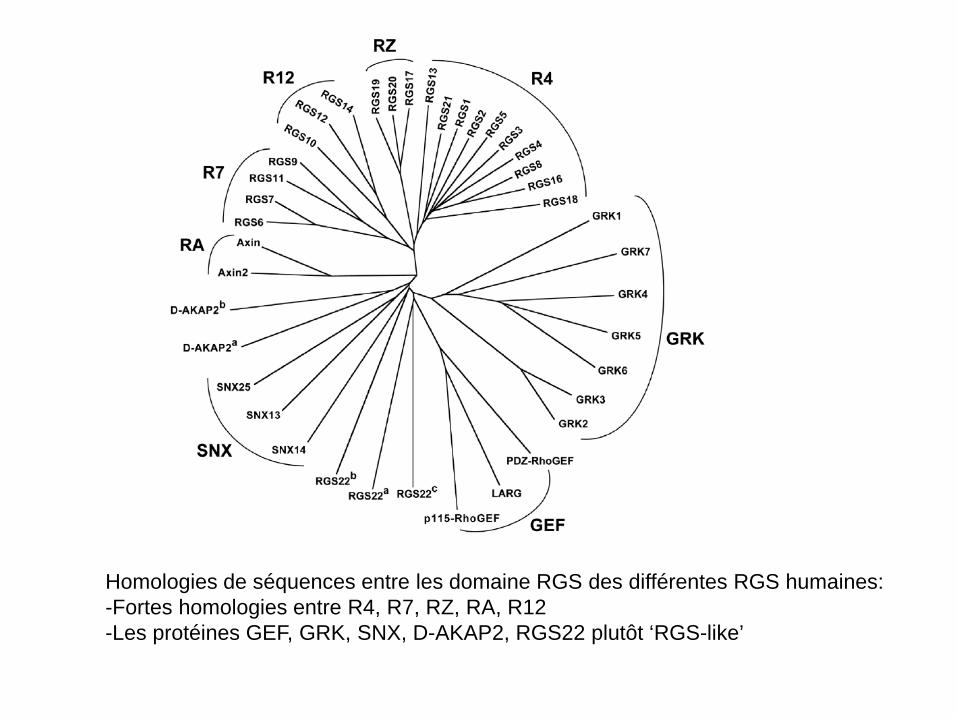
Homologies de séquences entre les domaine RGS des différentes RGS humaines: -Fortes homologies entre R4, R7, RZ, RA, R12 -Les protéines GEF, GRK, SNX, D-AKAP2, RGS22 plutôt ‘RGS-like’
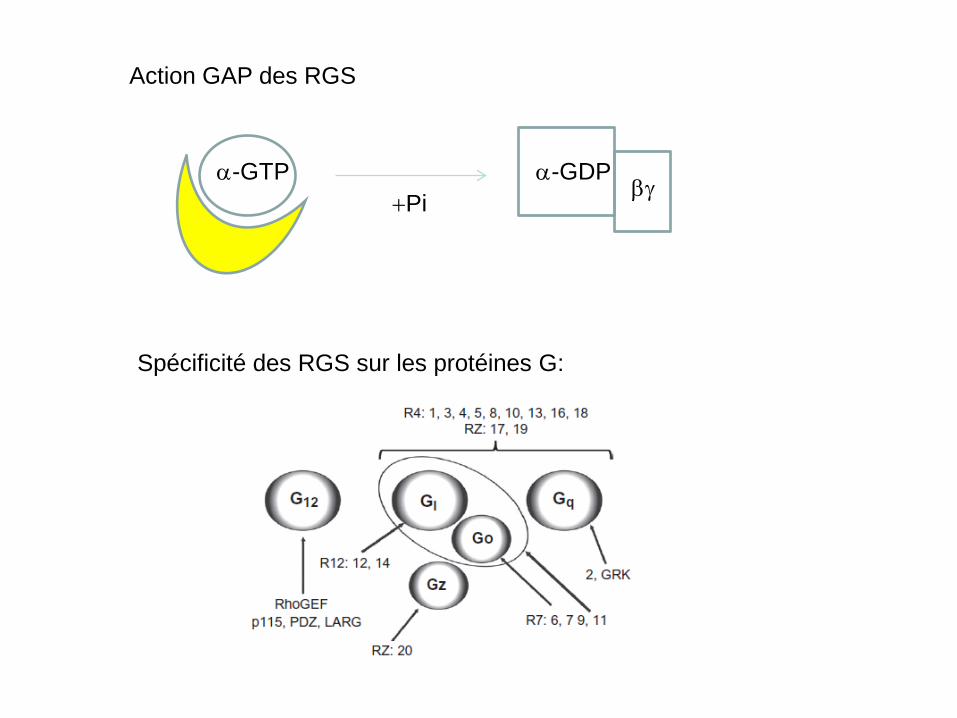
Action GAP des RGS
Spécificité des RGS sur les protéines G:
α-GTP α-GDP βγ
+Pi

Autres actions des RGS (1) par interaction directe avec un RCPG Exemples: -RGS7: même si elle interagit préférentiellement avec Gi, elle produit une diminution de l’activité des récepteurs 5HT2c par interférence avec Gq ou en se fixant sur le récepteur 5HT2c grâce à son domaine DEP; -RGS12: la présence du domaine ‘GoLoco’ lui permet de se fixer sur αi-GDP et ainsi de prolonger l’effet de βγ. Dans ce cas on dit qu’elle a un rôle de GDI ou ‘guanine nucleotide dissociation inhibitor. Cet effet a été mis en évidence sur les récepteurs dopaminergiques de type D2 qui sont couplés à Gi et à l’ouverture d’un canal potassique de type Kir. Cette protéine peut aussi interférer avec les canaux calciques de type N qui sont bloqués par Gi, grâce à leur domaine PTB (phospho-tyrosine binding domain). -RGS 2: liaison aux récepteurs muscariniques M1 et M5 couplés à Gq et inhibition du couplage à la PLC. -RGS4: la liaison aux récepteurs µ et δ opiacés inhibe leur couplage à Gi et accélère l’endocytose des récepteurs δ opiacés.

Autres actions des RGS (2): par couplage indirect avec une protéine d’échafaudage Exemples: RGS19 (ou GAIP pour G-alpha interacting protein): cette protéine peut être recrutée par GIPC (pour GAIP interacting protein C terminus) qui se lie au récepteur. On peut ainsi avoir une interaction de RGS19 et un découplage de Gi. RGS2: elle peut être recrutée par la spinophiline liée au RCPG (adrénergique par exemple) et moduler la réponse de Gq.

6.2-Les protéines PDZ
Les protéines PDZ ou à domaines PDZ sont des protéines cytoplasmiques adaptatrices qui permettent de construire des échafaudages transitoires pour assembler des protéines en complexes de signalisation. La structure PDZ vient de l’homologie entre les protéines PSD95, Disc large de la Drosophile et ZO1 la protéine des jonctions adhérentes. Les domaines PDZ contiennent 80 à 90 AA et forment 6 feuillets β et 2 hélices α. Ces domaines interagissent avec des motifs de 3 à 5 AA sur les cibles (donc ici les RCPGs).

Familles de protéines PDZ
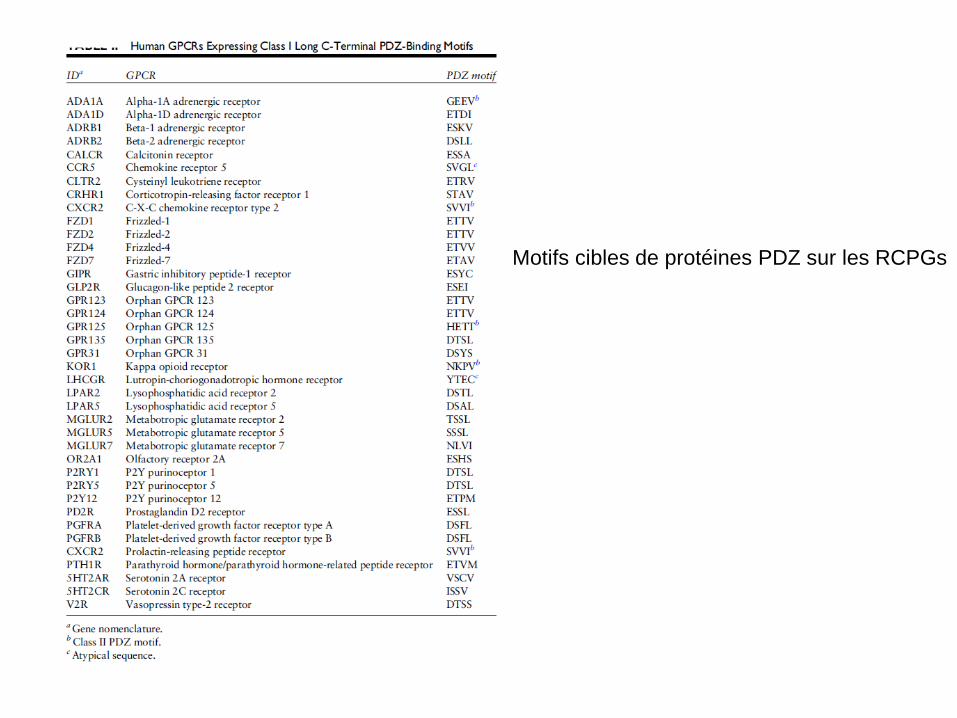
Motifs cibles de protéines PDZ sur les RCPGs
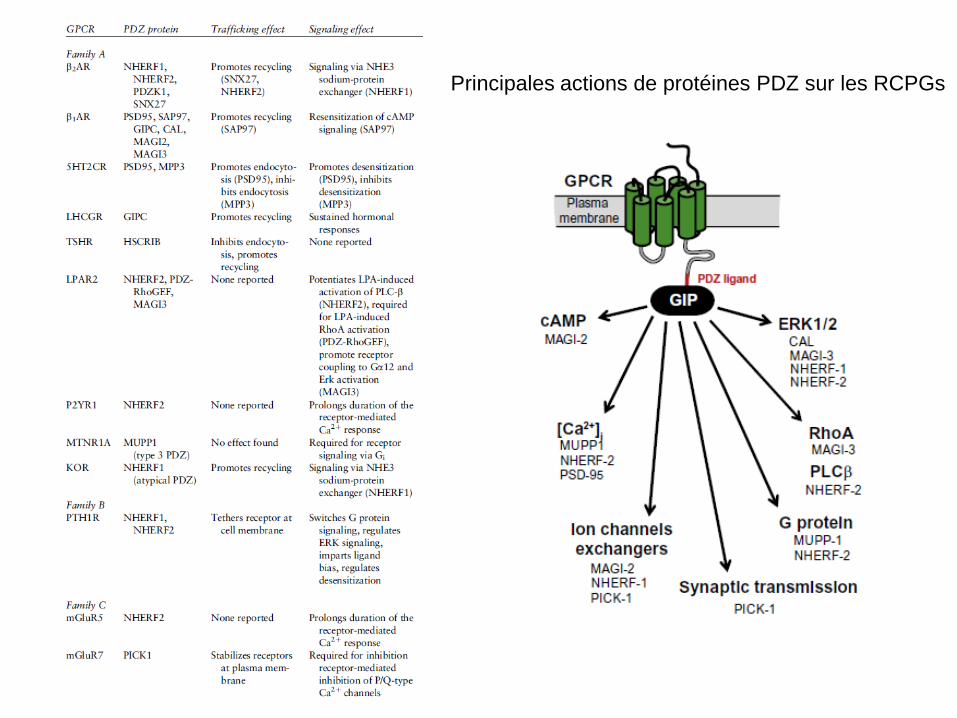
Principales actions de protéines PDZ sur les RCPGs

Actions des protéines PDZ sur les RCPGs Exemples: MUPP1: cette protéine peut se fixer sur les récepteurs 5HT2 et augmenter leur signal calcique NHERF1 et 2: les protéines NHERF pour ‘Na+/H+ exchange regulator factor’ peuvent se fixer sur les RCPG et moduler profondément leur activité: -par liaison sur les récepteurs β2 ADR, NHERF1 réprime inhibition de l’échangeur Na+/H+ induite par le récepteur β2 ADR; -par liaison sur les récepteurs opiacés κ, NHERF1 augmente l’effet sur cet échangeur; -par liaison sur les récepteurs purinergiques et mGlu5, NERHF2 augmente le signal calcique qu’ils stimulent; -ces protéines peuvent orienter le couplage d’un récepteur: ainsi le récepteur de l’hormone parathyroïde PTH1 active l’AC et pas la PLC dans les cellules musculaires lisses. Par contre, NHERF1 active la PLC dans les keratinocytes, myocytes et lymphocytes et pas l’AC. Dans les osteoblastes et cellules rénales il active les deux voies. En fait, NHERF1 facilite le couplage à Gq et NHERF2 inhibe le couplage à Gs, facilite Gq et Gi. PICK1: son couplage au récepteur métabotrope du glutamate de type mGlu7 est absolument requis pour sa fonction (épilepsie absence si cette interaction n’existe pas).
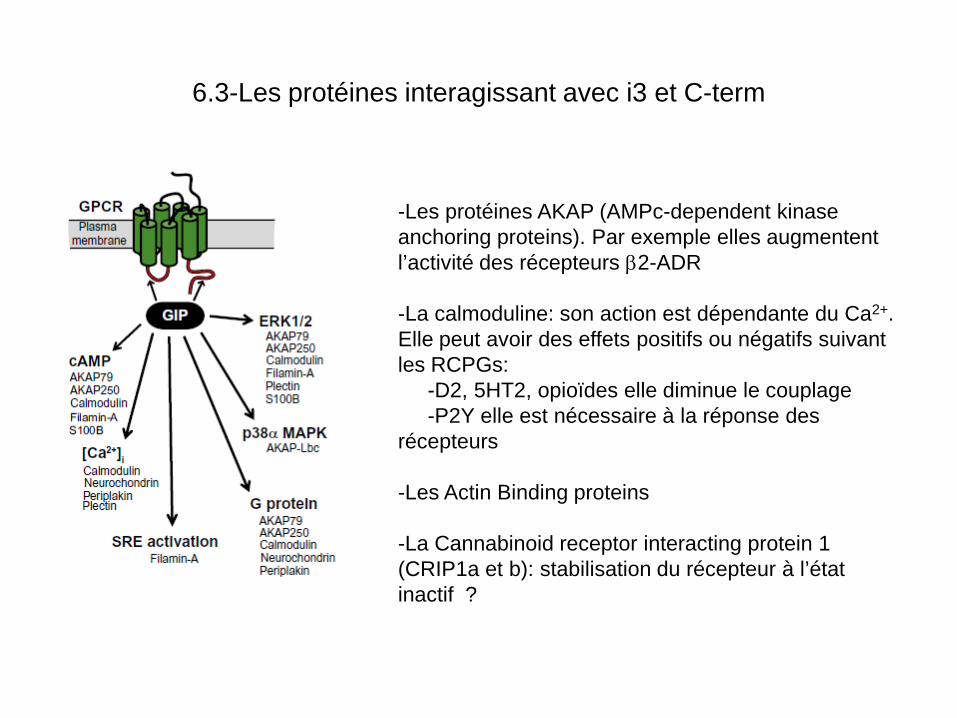
6.3-Les protéines interagissant avec i3 et C-term
-Les protéines AKAP (AMPc-dependent kinase anchoring proteins). Par exemple elles augmentent l’activité des récepteurs β2-ADR -La calmoduline: son action est dépendante du Ca2+. Elle peut avoir des effets positifs ou négatifs suivant les RCPGs: -D2, 5HT2, opioïdes elle diminue le couplage -P2Y elle est nécessaire à la réponse des récepteurs -Les Actin Binding proteins -La Cannabinoid receptor interacting protein 1 (CRIP1a et b): stabilisation du récepteur à l’état inactif ?
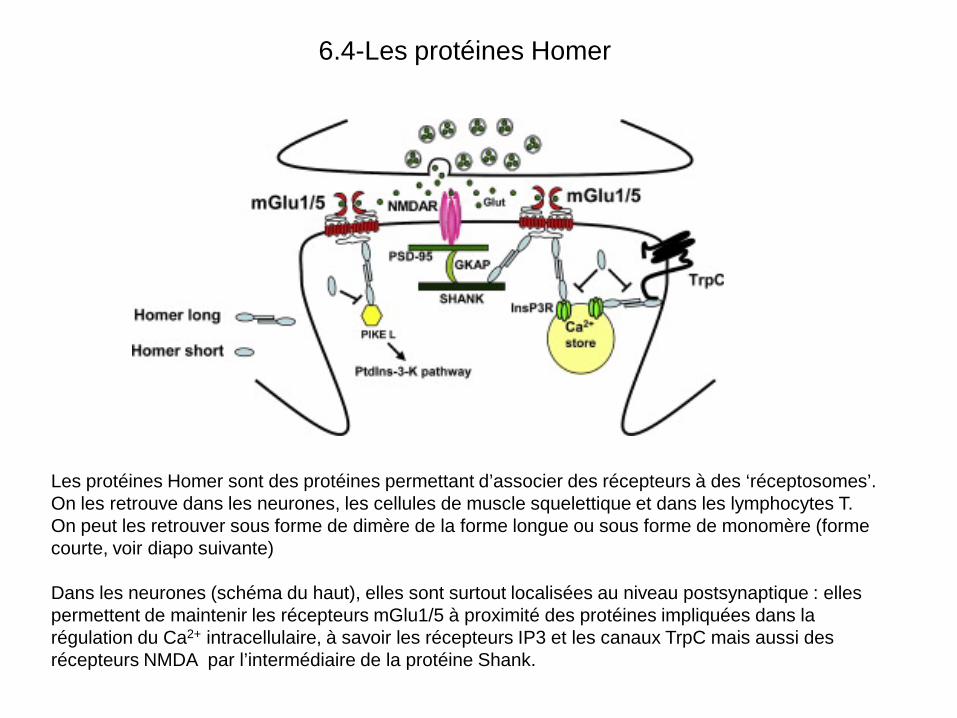
6.4-Les protéines Homer
Les protéines Homer sont des protéines permettant d’associer des récepteurs à des ‘réceptosomes’. On les retrouve dans les neurones, les cellules de muscle squelettique et dans les lymphocytes T. On peut les retrouver sous forme de dimère de la forme longue ou sous forme de monomère (forme courte, voir diapo suivante) Dans les neurones (schéma du haut), elles sont surtout localisées au niveau postsynaptique : elles permettent de maintenir les récepteurs mGlu1/5 à proximité des protéines impliquées dans la régulation du Ca2+ intracellulaire, à savoir les récepteurs IP3 et les canaux TrpC mais aussi des récepteurs NMDA par l’intermédiaire de la protéine Shank.
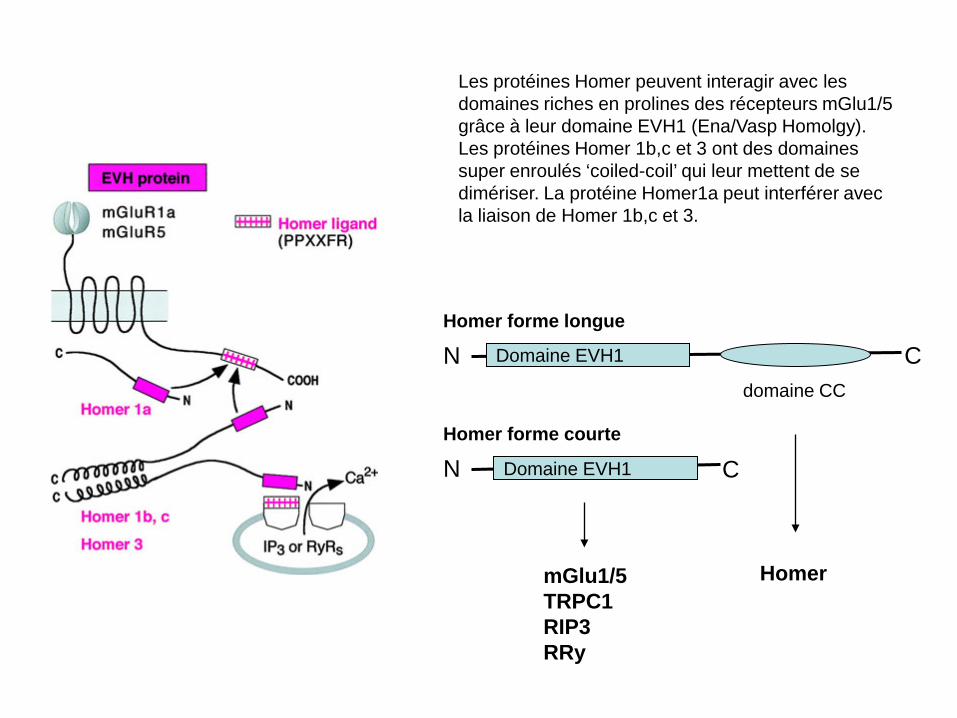
Les protéines Homer peuvent interagir avec les domaines riches en prolines des récepteurs mGlu1/5 grâce à leur domaine EVH1 (Ena/Vasp Homolgy). Les protéines Homer 1b,c et 3 ont des domaines super enroulés ‘coiled-coil’ qui leur mettent de se dimériser. La protéine Homer1a peut interférer avec la liaison de Homer 1b,c et 3.
Domaine EVH1
Homer forme longue
N C
Homer forme courte
N C
domaine CC
mGlu1/5 TRPC1 RIP3 RRy
Homer
Domaine EVH1
7-Oligomérisation des RCPGs
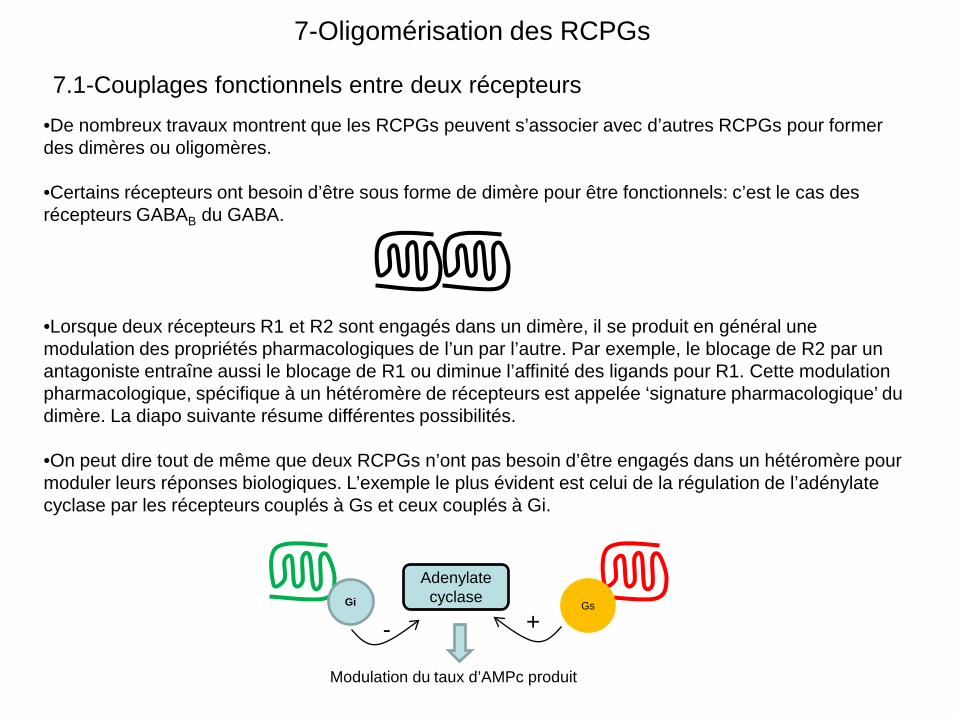
•De nombreux travaux montrent que les RCPGs peuvent s’associer avec d’autres RCPGs pour former des dimères ou oligomères.
•Certains récepteurs ont besoin d’être sous forme de dimère pour être fonctionnels: c’est le cas des récepteurs GABAB du GABA.
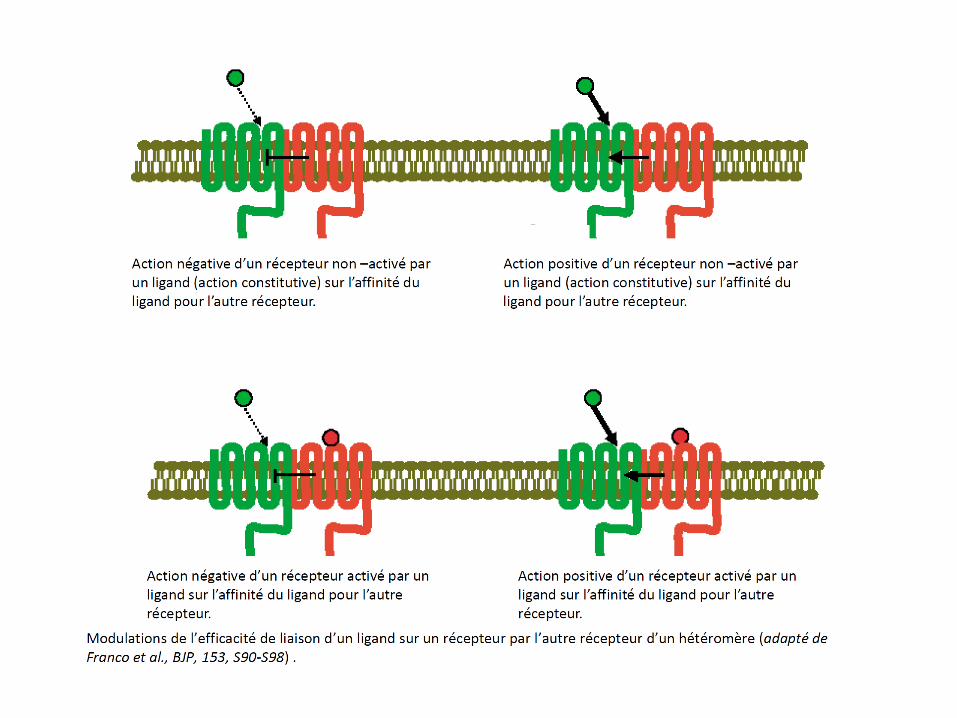
•Lorsque deux récepteurs R1 et R2 sont engagés dans un dimère, il se produit en général une modulation des propriétés pharmacologiques de l’un par l’autre. Par exemple, le blocage de R2 par un antagoniste entraîne aussi le blocage de R1 ou diminue l’affinité des ligands pour R1. Cette modulation pharmacologique, spécifique à un hétéromère de récepteurs est appelée ‘signature pharmacologique’ du dimère. La diapo suivante résume différentes possibilités.
•On peut dire tout de même que deux RCPGs n’ont pas besoin d’être engagés dans un hétéromère pour moduler leurs réponses biologiques. L’exemple le plus évident est celui de la régulation de l’adénylate cyclase par les récepteurs couplés à Gs et ceux couplés à Gi.
Gi Gs
Adenylate cyclase
- +
Modulation du taux d’AMPc produit
7.1-Couplages fonctionnels entre deux récepteurs

7.2-Mise en évidence de l’interaction entre 2 RCPGs: aspects techniques
• Plusieurs méthodes existent pour mettre en évidence la formation d’un hétéromère de RCPG. D’abord des méthodes mettant en évidence la proximité de deux RCPGs:
– La co-immunoprécipitation qui revient à purifier les protéines d’une préparation avec un anticorps dirigé contre une protéine (ici un RCPG) et puis à révéler la présence d’une protéine partenaire (ici un autre RCPG) avec un anticorps dirigé contre cette protéine après électrophorèse (Western blot);
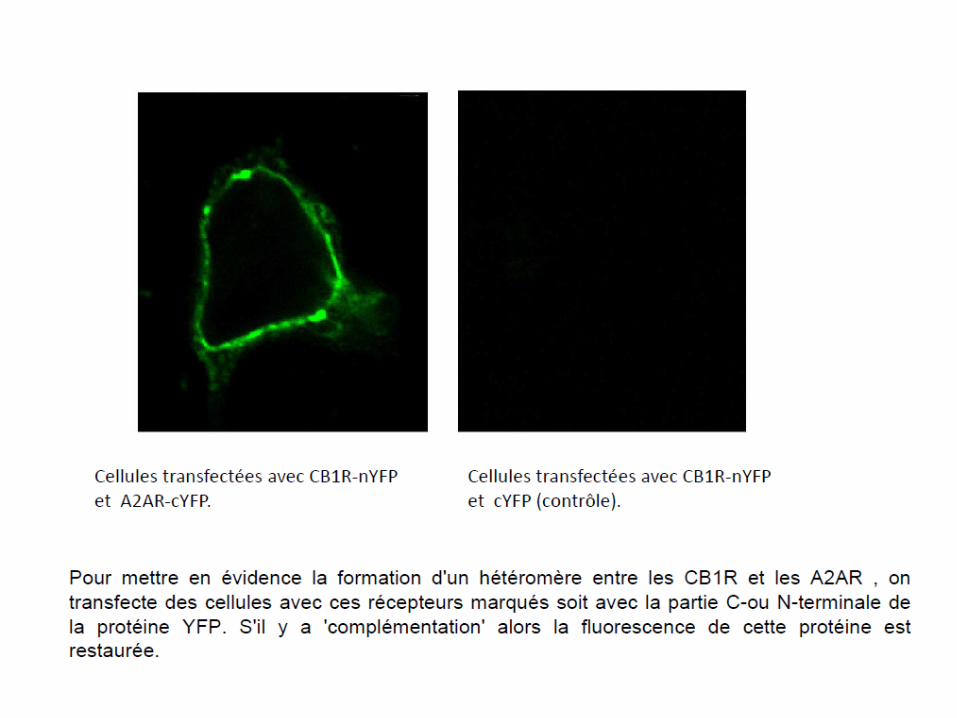
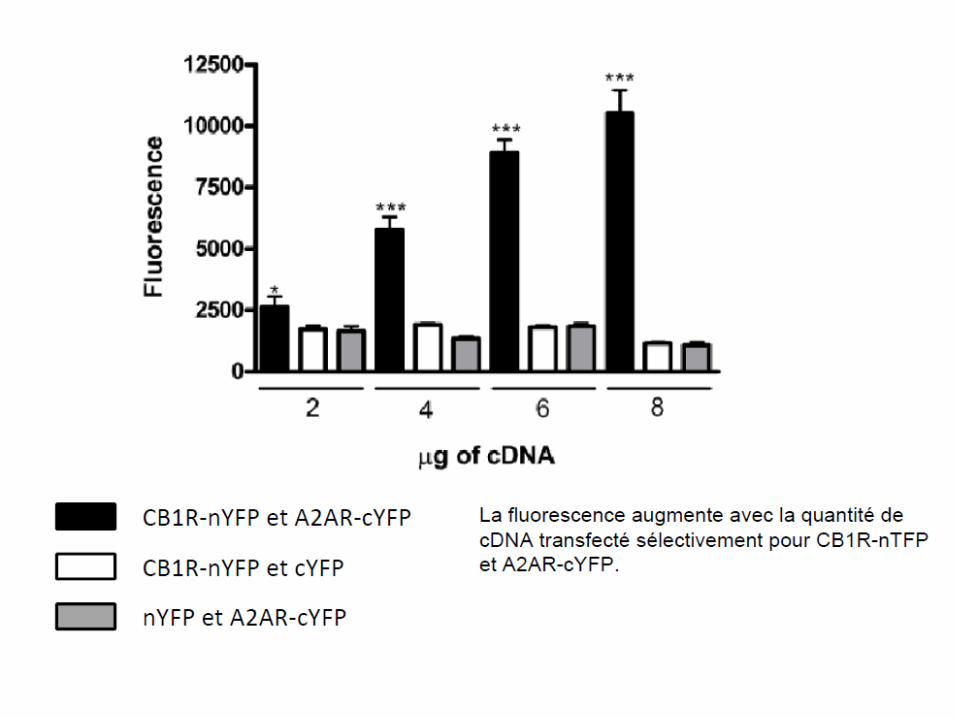
– Le marquage des deux récepteurs avec des marqués spécifiques marqués avec un fluorophore: on peut apprécier la co-localisation par épifluorescence.
– La méthode FRET (et de BRET) qui consiste à marquer un récepteur avec un fluorophore donneur de photons et l’autre récepteur avec un fluorophore accepteur de photons.Donc si les deux récepteurs sont à proximité, il y a transfert de fluorescence (FRET) entre les deux. On peut aussi générer la lumière par bioluminescence dans ce cas on parlera de BRET. On peut aussi faire une complémentation en marquant un récepteur avec la N-terminale et l’autre avec la partie C-terminale d’une protéine fluorescente (GFP par exemple); Quand les récepteurs sont à proximité, alors il y a reconstitution de cette protéine et fluorescence (voir diapo suivante).
• Pour voir si l’hétéromère est fonctionnel il faut étudier sa signature pharmacologique en mesurant ses réponses biologiques en présence de ligands de l’autre récepteur.
excitation
émission 1 et transfert
émission 2 Enzyme (luciférase)
émission 1 et transfert
émission 2
substrat
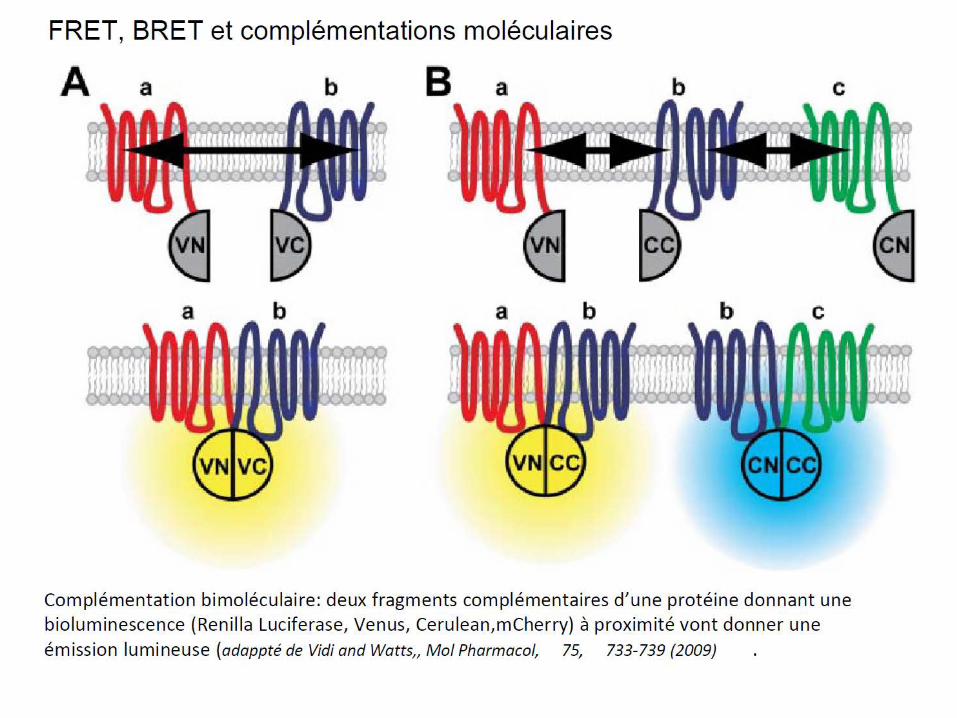
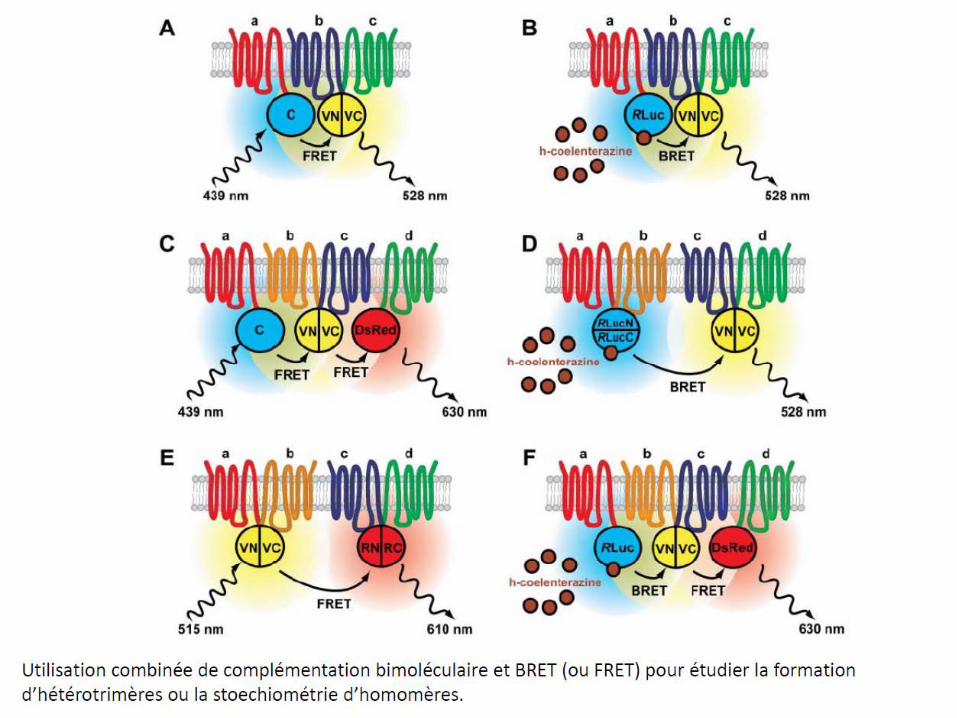

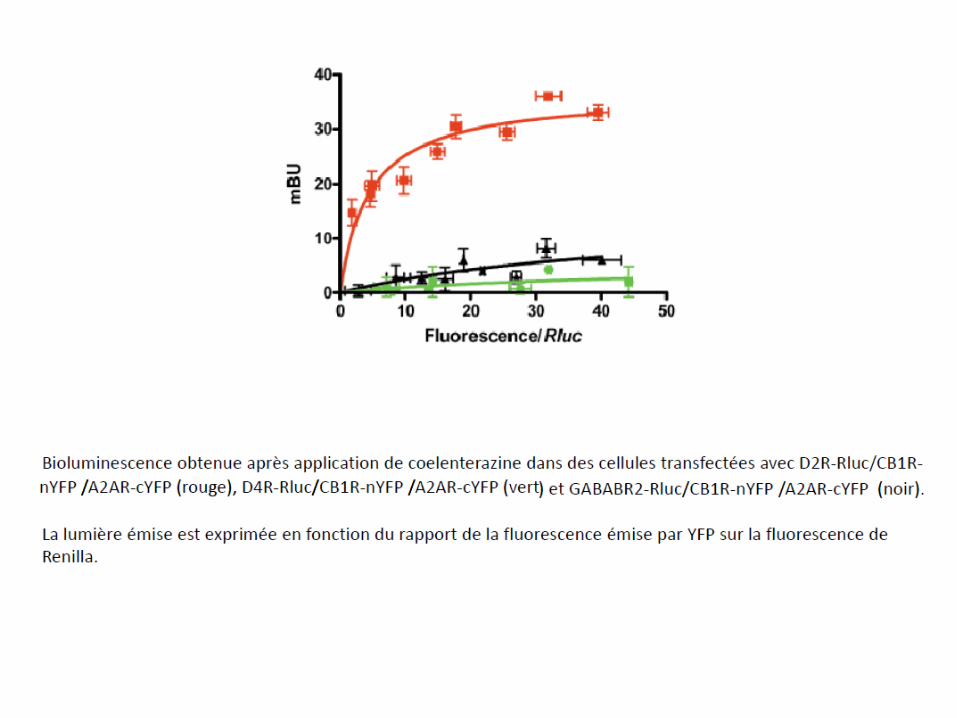
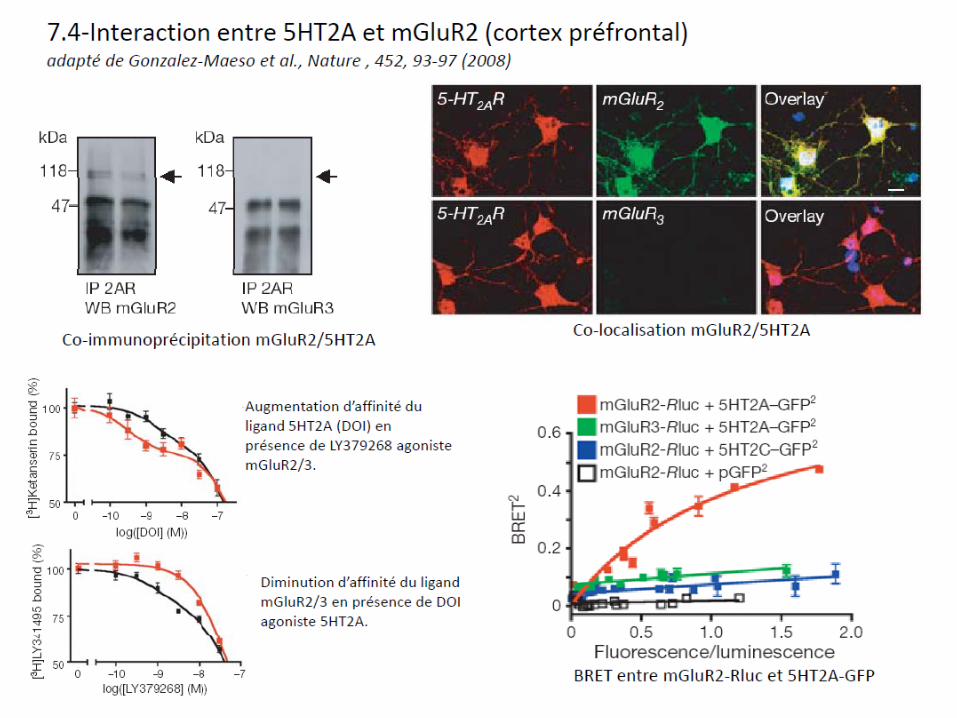
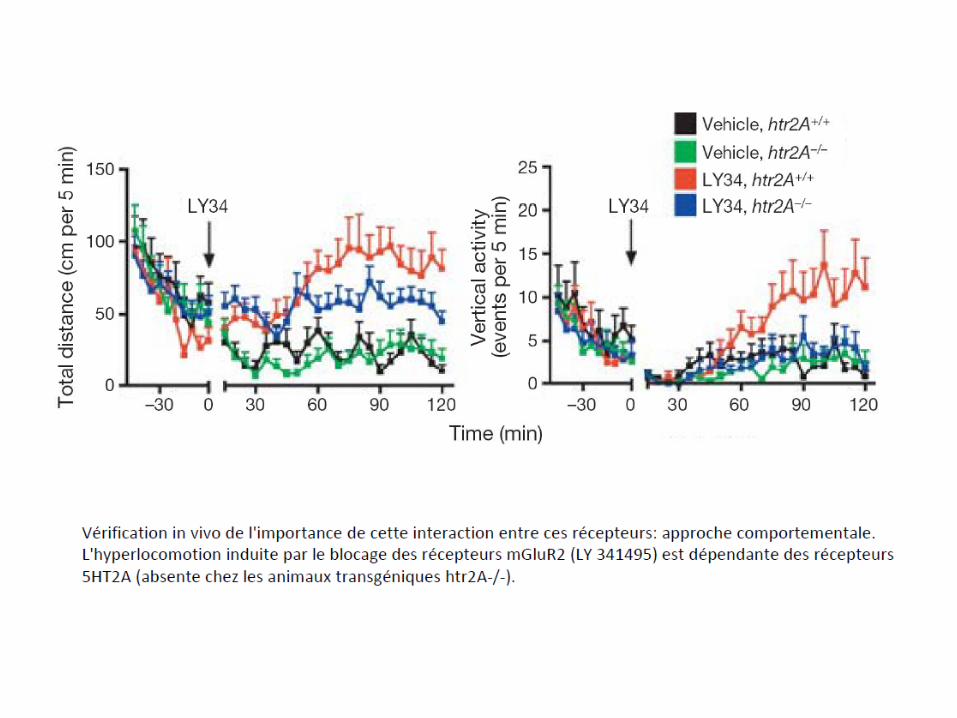

7.5-Dimères des récepteurs opiacés: cibles de futurs analgésiques?
• Les récepteurs opiacés µ, δ, κ et nociceptine peuvent former des homomères et des hétéromères. • Les hétéromères mis en évidence sont µ-δ, δ-κ, µ-κ, et µ-nociceptine. On observe aussi la
formation d’homomères µ-µ par exemple. Toutefois la présence de ces hétéromères reste à confirmer in vivo.
• Les problèmes associés à la prise chronique d’opiacés comme le développement de la tolérance
semble associé à une mauvaise endocytose des récepteurs stimulés par certains opiacés comme la morphine (voir concept ‘RAVE’).
• Donc d’un point de vue pharmacologique, il pourrait être intéressant de cibler les hétéromères,
notamment les hétéromères µ-δ pour produire un effet analgésique et ce pour plusieurs raisons: d’abord ces deux récepteurs sont souvent co-localisés, ensuite le blocage des récepteurs δ avec un antagoniste diminue la tolérance de la morphine sans diminuer son effet analgésique. Enfin, les récepteurs δ sont dégradés après désensibilisation alors que les µ recyclent. Donc l’activation des hétéromères favoriserait l’internalisation. Des ligands bivalents ont ainsi été synthétisés: ce sont des agonistes-antagonistes des récepteurs opiacés comme le MDAN (voir diapo suivante).
• Les homomères de récepteurs µ stimulés à la fois par un ligand à faible RAVE comme le
DAMGO, la méthadone ou les endorphines par un ligand à fort RAVE comme la morphine ont une meilleure endocytose que les récepteurs µ sous forme monomérique.
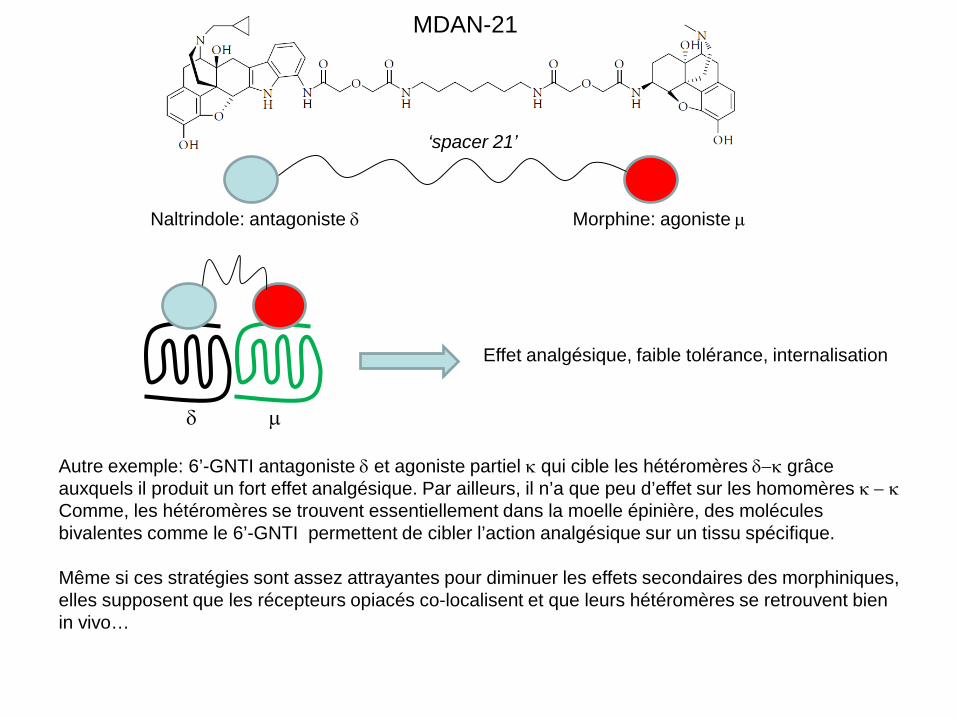
Naltrindole: antagoniste δ Morphine: agoniste µ
MDAN-21
Effet analgésique, faible tolérance, internalisation
δ µ
Autre exemple: 6’-GNTI antagoniste δ et agoniste partiel κ qui cible les hétéromères δ−κ grâce auxquels il produit un fort effet analgésique. Par ailleurs, il n’a que peu d’effet sur les homomères κ − κ Comme, les hétéromères se trouvent essentiellement dans la moelle épinière, des molécules bivalentes comme le 6’-GNTI permettent de cibler l’action analgésique sur un tissu spécifique. Même si ces stratégies sont assez attrayantes pour diminuer les effets secondaires des morphiniques, elles supposent que les récepteurs opiacés co-localisent et que leurs hétéromères se retrouvent bien in vivo…
‘spacer 21’

8-RCPGs et pathologies
• On recense un grand nombre de pathologies liées à des mutations des RCPGs ou des protéines appartenant aux voies qui leurs sont associées.
• Les mutations des RCPGs affectent leur activité de différentes manières:
elles interfèrent directement avec leur fonction (gain ou perte), avec leur trafic à la membrane ou encore leur trafic intracellulaire (voir diapo suivante).
• On recense plus de 600 mutations différentes pouvant conduire à l’inactivation des RCPG et 100 conduisant à une activation des RCPG.
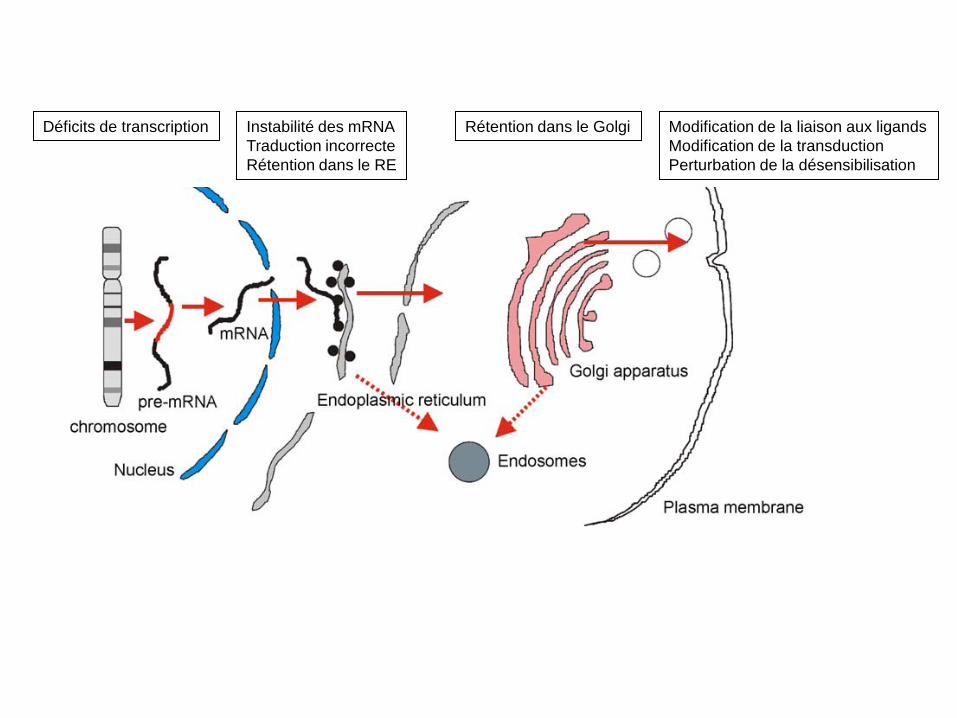
Déficits de transcription Instabilité des mRNA Traduction incorrecte Rétention dans le RE
Rétention dans le Golgi Modification de la liaison aux ligands Modification de la transduction Perturbation de la désensibilisation
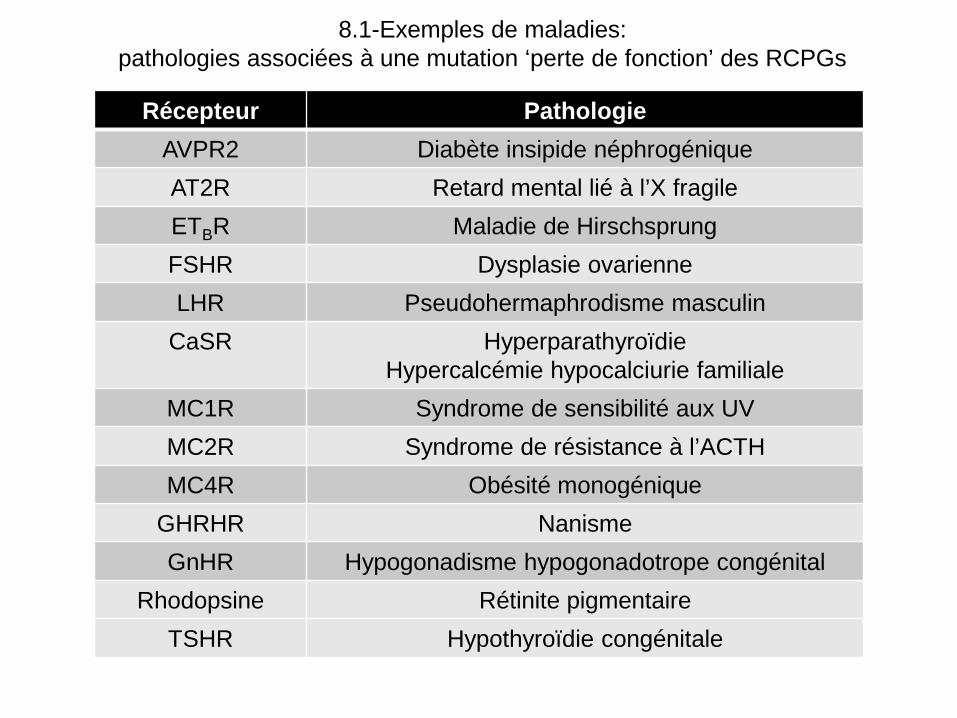
8.1-Exemples de maladies: pathologies associées à une mutation ‘perte de fonction’ des RCPGs
Récepteur Pathologie
AVPR2 Diabète insipide néphrogénique AT2R Retard mental lié à l’X fragile ETBR Maladie de Hirschsprung FSHR Dysplasie ovarienne LHR Pseudohermaphrodisme masculin
CaSR Hyperparathyroïdie Hypercalcémie hypocalciurie familiale
MC1R Syndrome de sensibilité aux UV MC2R Syndrome de résistance à l’ACTH MC4R Obésité monogénique
GHRHR Nanisme GnHR Hypogonadisme hypogonadotrope congénital
Rhodopsine Rétinite pigmentaire TSHR Hypothyroïdie congénitale
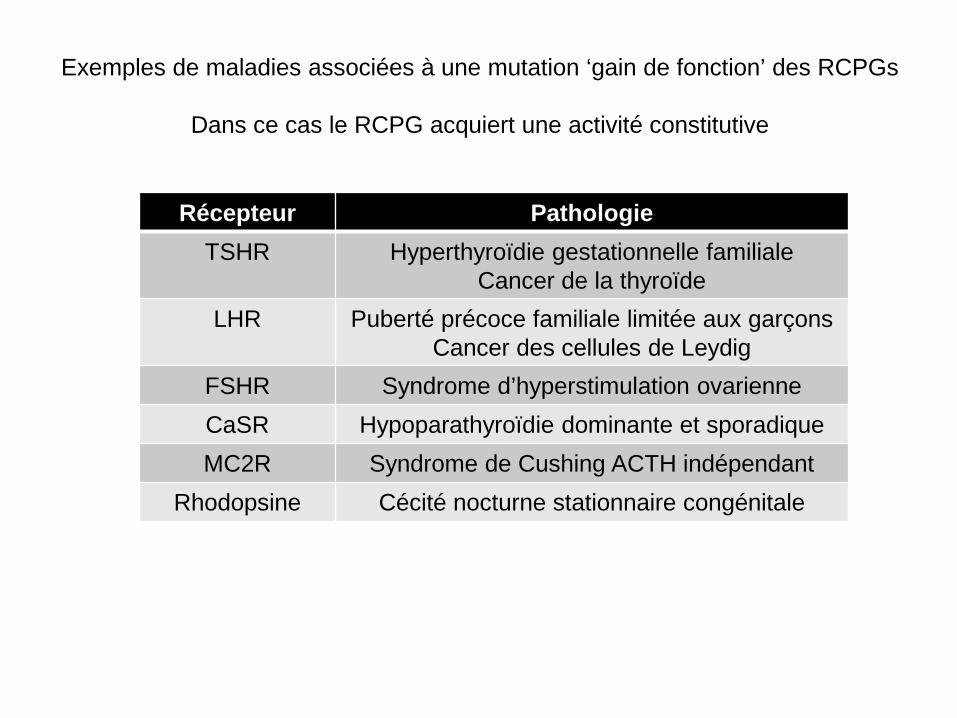
Exemples de maladies associées à une mutation ‘gain de fonction’ des RCPGs
Dans ce cas le RCPG acquiert une activité constitutive
Récepteur Pathologie TSHR Hyperthyroïdie gestationnelle familiale
Cancer de la thyroïde LHR Puberté précoce familiale limitée aux garçons
Cancer des cellules de Leydig FSHR Syndrome d’hyperstimulation ovarienne CaSR Hypoparathyroïdie dominante et sporadique MC2R Syndrome de Cushing ACTH indépendant
Rhodopsine Cécité nocturne stationnaire congénitale
Mutation entraînant une perte partielle de fonction
Mutation entraînant une perte totale de fonction
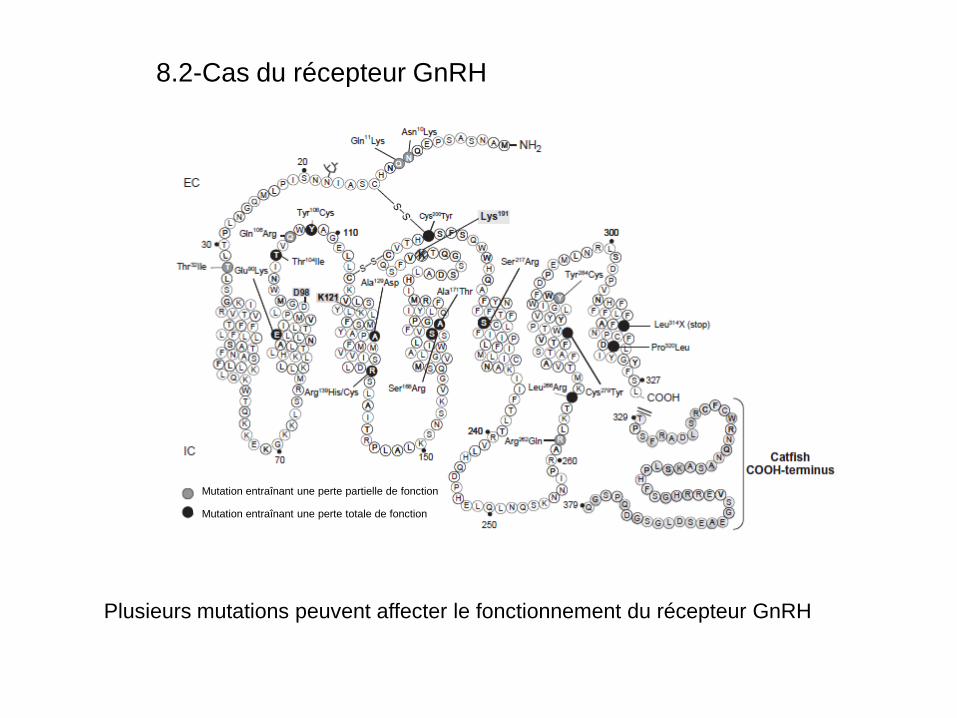
8.2-Cas du récepteur GnRH
Plusieurs mutations peuvent affecter le fonctionnement du récepteur GnRH
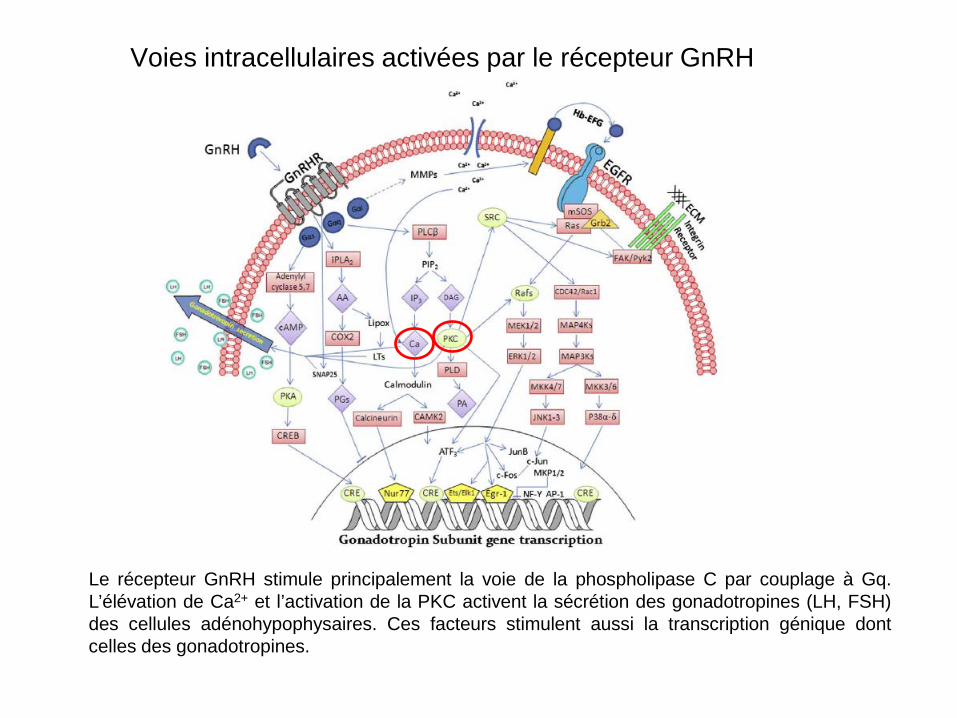
Voies intracellulaires activées par le récepteur GnRH
Le récepteur GnRH stimule principalement la voie de la phospholipase C par couplage à Gq. L’élévation de Ca2+ et l’activation de la PKC activent la sécrétion des gonadotropines (LH, FSH) des cellules adénohypophysaires. Ces facteurs stimulent aussi la transcription génique dont celles des gonadotropines.
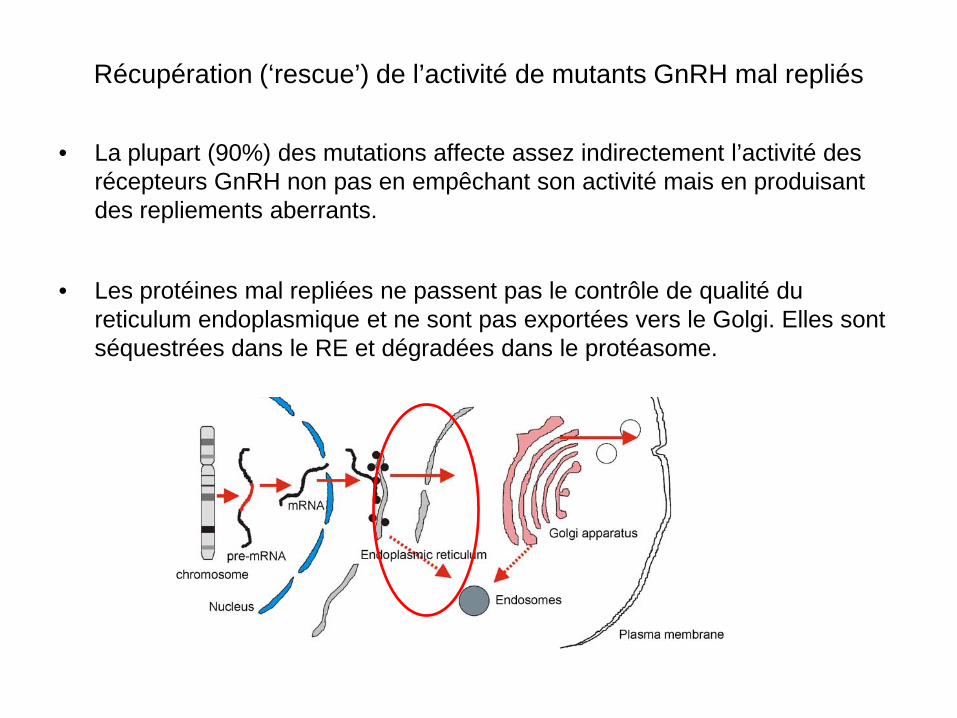
• La plupart (90%) des mutations affecte assez indirectement l’activité des récepteurs GnRH non pas en empêchant son activité mais en produisant des repliements aberrants.
• Les protéines mal repliées ne passent pas le contrôle de qualité du
reticulum endoplasmique et ne sont pas exportées vers le Golgi. Elles sont séquestrées dans le RE et dégradées dans le protéasome.
Récupération (‘rescue’) de l’activité de mutants GnRH mal repliés
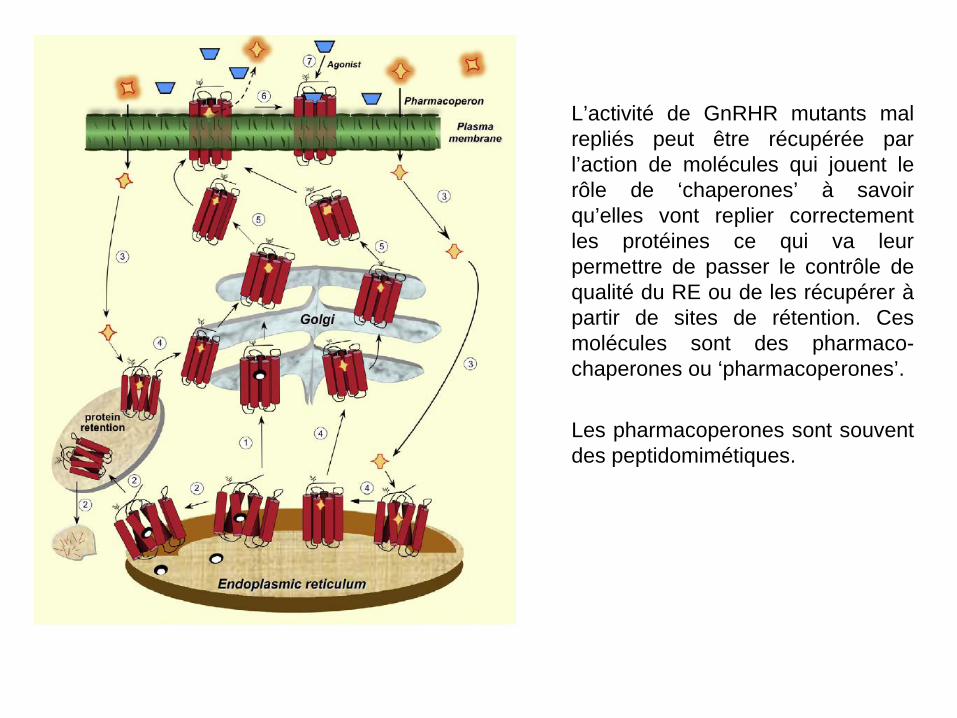
L’activité de GnRHR mutants mal repliés peut être récupérée par l’action de molécules qui jouent le rôle de ‘chaperones’ à savoir qu’elles vont replier correctement les protéines ce qui va leur permettre de passer le contrôle de qualité du RE ou de les récupérer à partir de sites de rétention. Ces molécules sont des pharmaco-chaperones ou ‘pharmacoperones’. Les pharmacoperones sont souvent des peptidomimétiques.