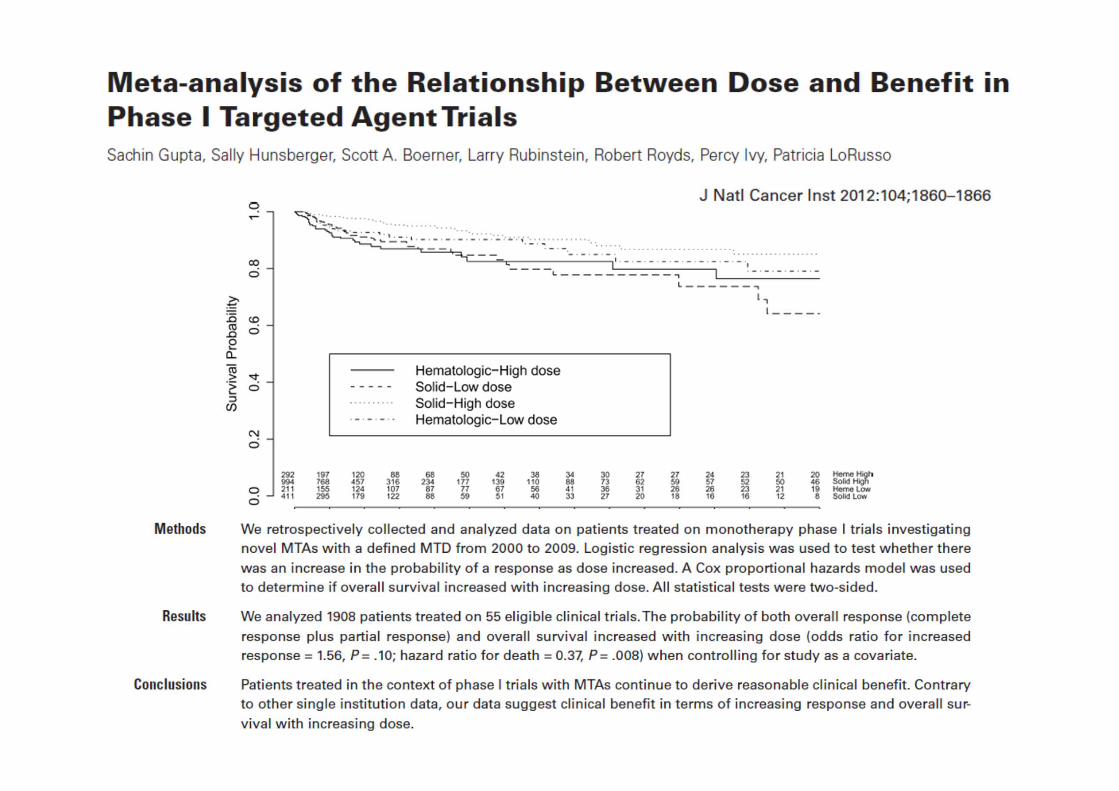
Embed Size (px)
Citation preview

Différentes phases d’étude clinique d’un nouveau
médicament
Norbert VeyDépartement d’Hématologie
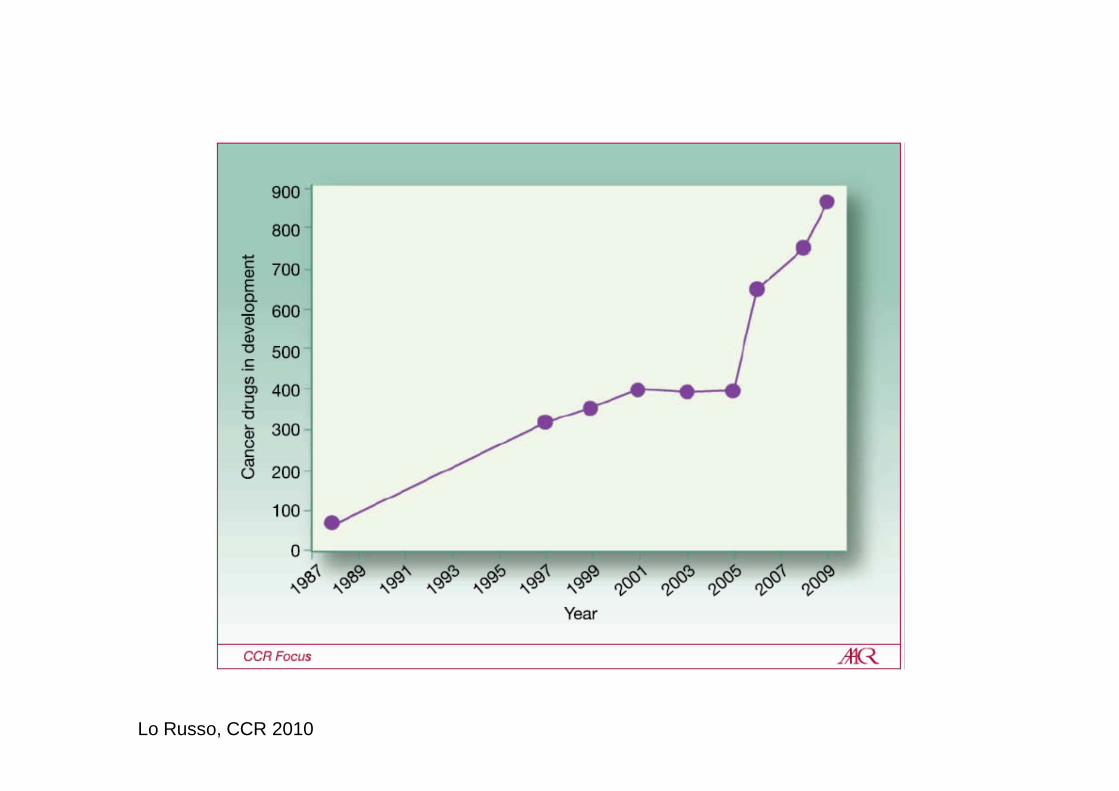
Lo Russo, CCR 2010
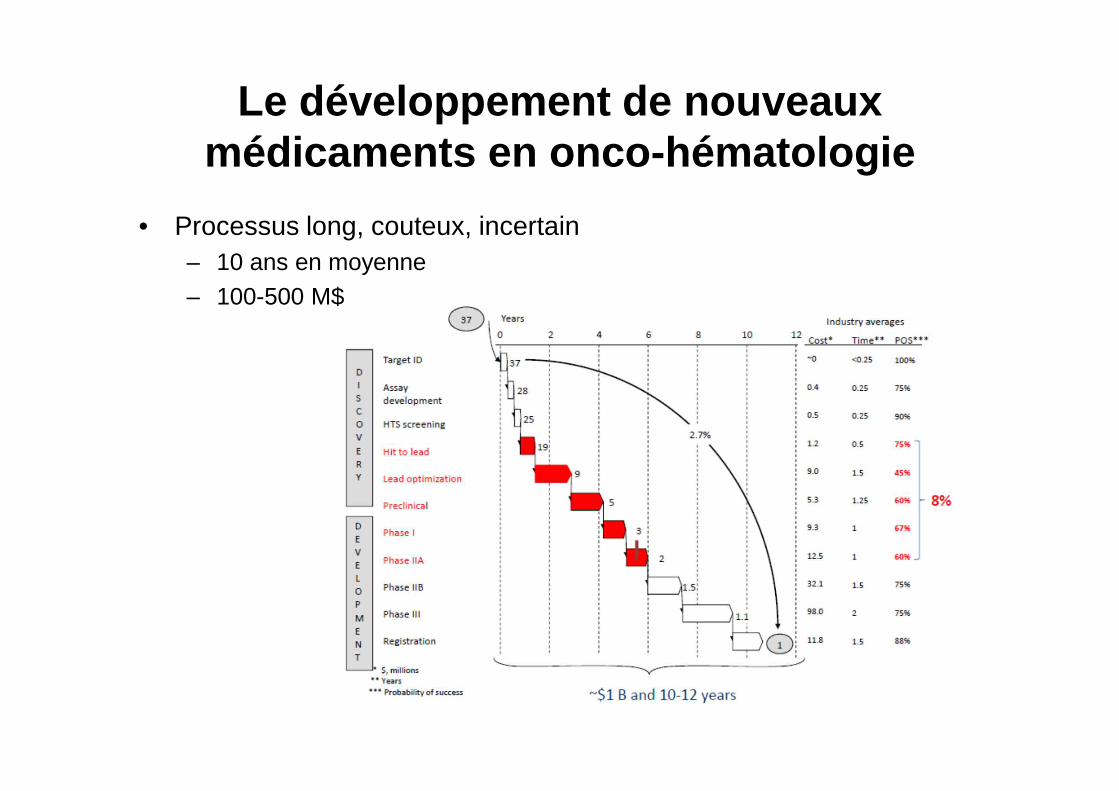
Le développement de nouveaux médicaments en onco -hématologie
• Processus long, couteux, incertain– 10 ans en moyenne– 100-500 M$

Essais thérapeutiques (études cliniques)
• Expérimentation qui a pour but de tester des traitements sur des humains– Sous contrôle des facteurs pouvant contribuer à la variabilité,
biais, application des traitements, mesure du devenir et analyse
• Il s’agit d’une démarche expérimentale– Hypothèses– Objectifs – Critères de jugement– Conditions expérimentales…

Spécificité des ET
• Éthique– Encadrement règlementaire et législatif– CPP– Rôle et responsabilités de l’IP+++
• Bénéfice pour le participant?– Intérêt collectif > individuel– Accès traitements innovants encadré– Soins de support optimisés
• Bénéfices pour les intervenants– Rigueur de la démarche– École de la PV
• Impact des résultats sur la pratique médicale
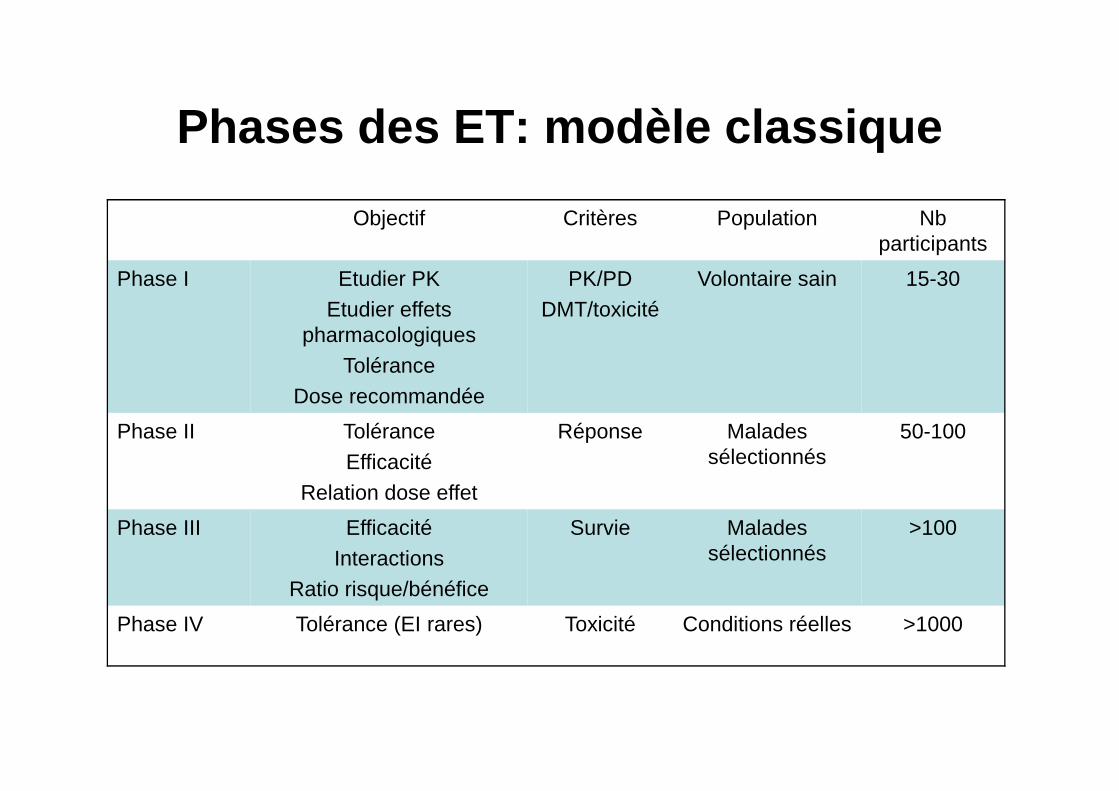
Phases des ET: modèle classique
Objectif Critères Population Nb participants
Phase I Etudier PKEtudier effets
pharmacologiquesTolérance
Dose recommandée
PK/PDDMT/toxicité
Volontaire sain 15-30
Phase II ToléranceEfficacité
Relation dose effet
Réponse Malades sélectionnés
50-100
Phase III EfficacitéInteractions
Ratio risque/bénéfice
Survie Malades sélectionnés
>100
Phase IV Tolérance (EI rares) Toxicité Conditions réelles >1000

Phases des essais en cancérologie
• Sous-types:– Phase 0 (endpoint= biomarqueur)– IB: étude association de molécules– IIA: phase II avec recherche de dose– IIB: phase II randomisée (comparative)

Essais de phase I
• Centre de recherche habilité, unité de lieu pour permettre l’intégration optimale des informations– Agrément (règlementaire)– Labellisation (label CLIPP, INCa)
• Petits effectifs, volontaires sains (ou malades en OH)
• Contraintes (en OH)– Ne pas exposer trop de sujets à des doses toxiques– Ne pas exposer trop de sujets à des doses inefficaces

Pré-requis pour un essai FIH
• Le nombre de volontaire recevant en même temps la substance• l’intervalle entre un volontaire et le suivant• les critères d’administration ou de non administration au suivant• Choix des doses
– choix de la première dose– progression de doses (géométrique, arythmétique)– dose maximale– le délai entre une dose et la dose suivante (second bloc)
• Les modalités de décision (qualification, personnes responsables..)
http://ansm.sante.fr/var/ansm_site

Objectifs d’une phase I
• Obtenir des informations sur– La pharmacocinétique– La pharmacodynamique
• Déterminer une dose maximale tolérée (DMT)– Dose maximale à laquelle ≤33% des patients présentent une DLT (US)– Première dose pour laquelle ≥33% des patients présentent une DLT (EU, Japon)– Définition variable de la DLT (en fonction de la pathologie, le médicament, le
contexte…)
• (Déterminer une dose biologiquement active)
• Pour proposer une dose recommandée en phase II (RP2D)– MTD (US)– 1 palier au-dessous MTD (EU, Japon)

Exemple de définitions de DLT
• Extra-hématologiques– EI grade 2 irréversible – Ou EI grade 3-4 réversible– Sont généralement exclus: SLT, alopécie, fièvre/infections
• Hématologiques– Cytotoxiques (hors leucémie)
• Neutropénie grade 4 >2 semaines• Thrombopénie grade 4 > 1 semaine
– Leucémie/SMD• Grade 4 > 6 semaines en l’absence d’infiltration blastique
• Limites– Risque de faux négatifs: méconnaissance des risques– Risque de faux positif: élimination d’un médicament
potentiellement efficace
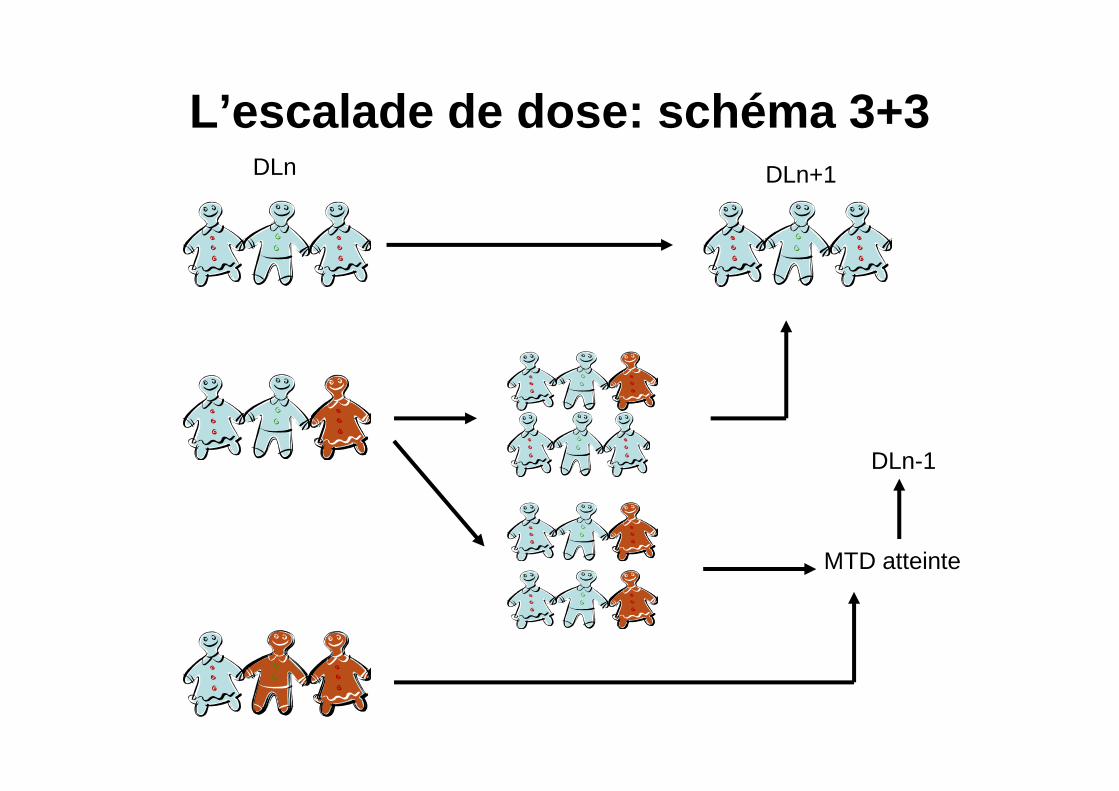
L’escalade de dose: schéma 3+3DLn DLn+1
DLn-1
MTD atteinte

Exemple: cloretazine
Vey, N Exp Rev Anticancer Ther, 2006
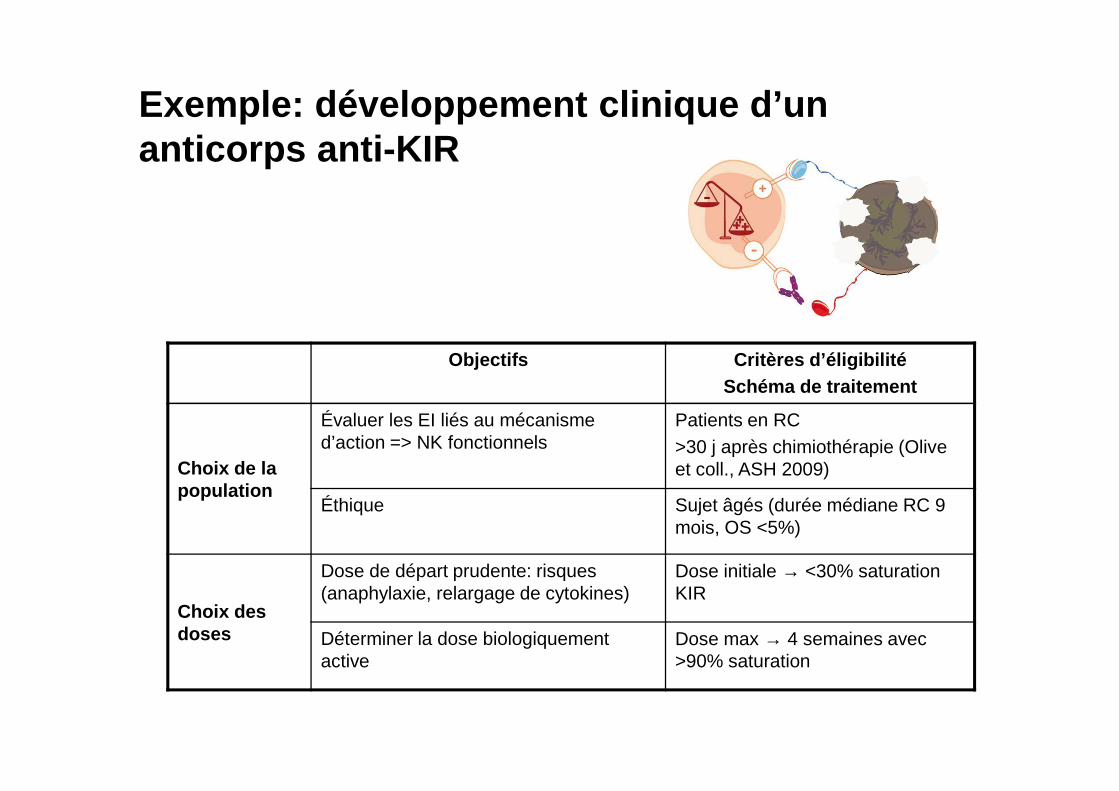
Exemple: développement clinique d’un anticorps anti-KIR
Objectifs Critères d’éligibilitéSchéma de traitement
Choix de la population
Évaluer les EI liés au mécanisme d’action => NK fonctionnels
Patients en RC>30 j après chimiothérapie (Olive et coll., ASH 2009)
Éthique Sujet âgés (durée médiane RC 9 mois, OS <5%)
Choix des doses
Dose de départ prudente: risques (anaphylaxie, relargage de cytokines)
Dose initiale → <30% saturation KIR
Déterminer la dose biologiquement active
Dose max → 4 semaines avec >90% saturation
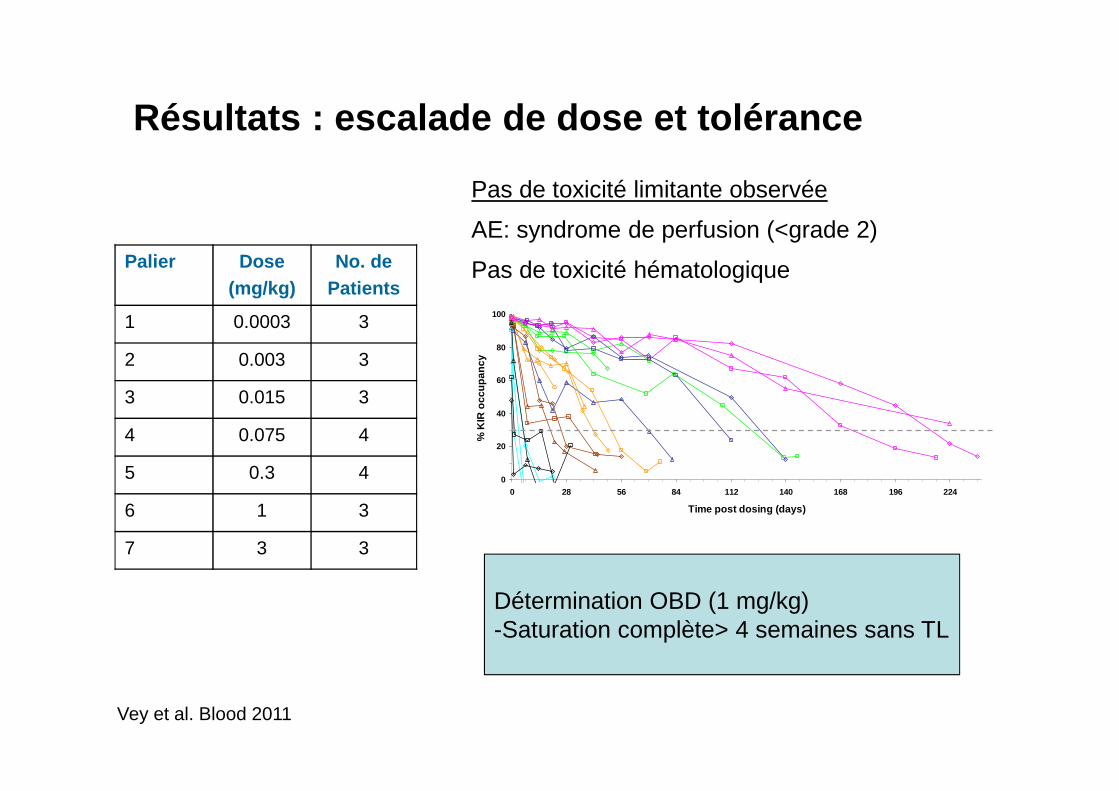
Résultats : escalade de dose et tolérance
Pas de toxicité limitante observée
AE: syndrome de perfusion (<grade 2)
Pas de toxicité hématologiquePalier Dose (mg/kg)
No. dePatients
1 0.0003 3
2 0.003 3
3 0.015 3
4 0.075 4
5 0.3 4
6 1 3
7 3 3
Vey et al. Blood 2011
0
20
40
60
80
100
0 28 56 84 112 140 168 196 224
Time post dosing (days)
% K
IR o
ccup
ancy
Détermination OBD (1 mg/kg)-Saturation complète> 4 semaines sans TL

Essais de phase II
• Objectif = estimation du taux de réponse d’une dose fixe (efficacité)– Vise à établir la dose optimale (efficacité la meilleure pour le minimum
d’EI)
• Faibles effectifs
• Non comparatifs (phase IIA)
• Comparatifs (phase IIB)– Diffèrent des phase III par
• La quantité d’information disponible avant essai• Le nombre de sujets• Le critère de jugement (intermédiaire)

Limites et interprétation
• Design classique– Montrer un taux de réponse >20%– Plan de Gehan à 2 étapes
• 0/14 réponse: arrêt pour inefficacité• ≥1/14 réponses: poursuivre avec 15-20 patients supplémentaires pour
affiner l’estimation du taux de réponses(Gehan E, J Chron Dis. 1991)
• Comparaison avec données historiques, mais– Biais de sélection– Biais de suivi– Biais d’évaluation– Effet placebo– Régression vers la moyenne…

Essais de phase III
• Apportent la preuve de l’intérêt d’un nouveau médicament/stratégie– Enregistrement (EMEA, FDA…)
• Sont comparatifs– placebo – ou traitement de référence
• Permettent:– D’évaluer la pharmacodynamie en situation thérapeutique– D’ajuster les doses– D’évaluer la sécurité et la tolérance dans ces conditions

Randomisation
• Résultat imprévisible (pas de sélection pour l’allocation dans un groupe de traitement)
• Acceptable sous le principe d’incertitude (ou clause d’équivalence)
• La randomisation ne suffit pas– Non exclusion des randomisés (ITT)– Contrôle de l’effet placebo (simple ou double insu)– Procédures (suivi) identique des groupes– Mesures des effets identiques (évaluation en aveugle)– critères de jugement robustes (ie survie)

Supériorité versus équivalence versus non -infériorité

Essais de phase 0
• Répondent aux problématiques nouvelles posées par les thérapies ciblées– Toxicité faible– Effets biologiques connus– Nombreuses molécules candidates
• Principe– Précèdent phase I– Très faibles doses (guidées par données précliniques),
exposition de courte durée– Analyses PK/PD en temps réel

Essais de phase 0
• Avantages– Rapidité– Screening de plusieurs molécules– Choix du meilleur composé– Informations pour l’optimisation de la dose– Anticipation des effets in vivo de la molécule

Conclusions
• Importance des essais• Importance de la qualité des données• Importance des investigateurs
– Qualité du suivi– Interprétation des résultats– Évaluation de sécurité
• Du patient++• Mais aussi de la molécule…
• Décalage entre modèles proposés par les statisticiens et leur utilisation

L’interne dans une unité de phase I
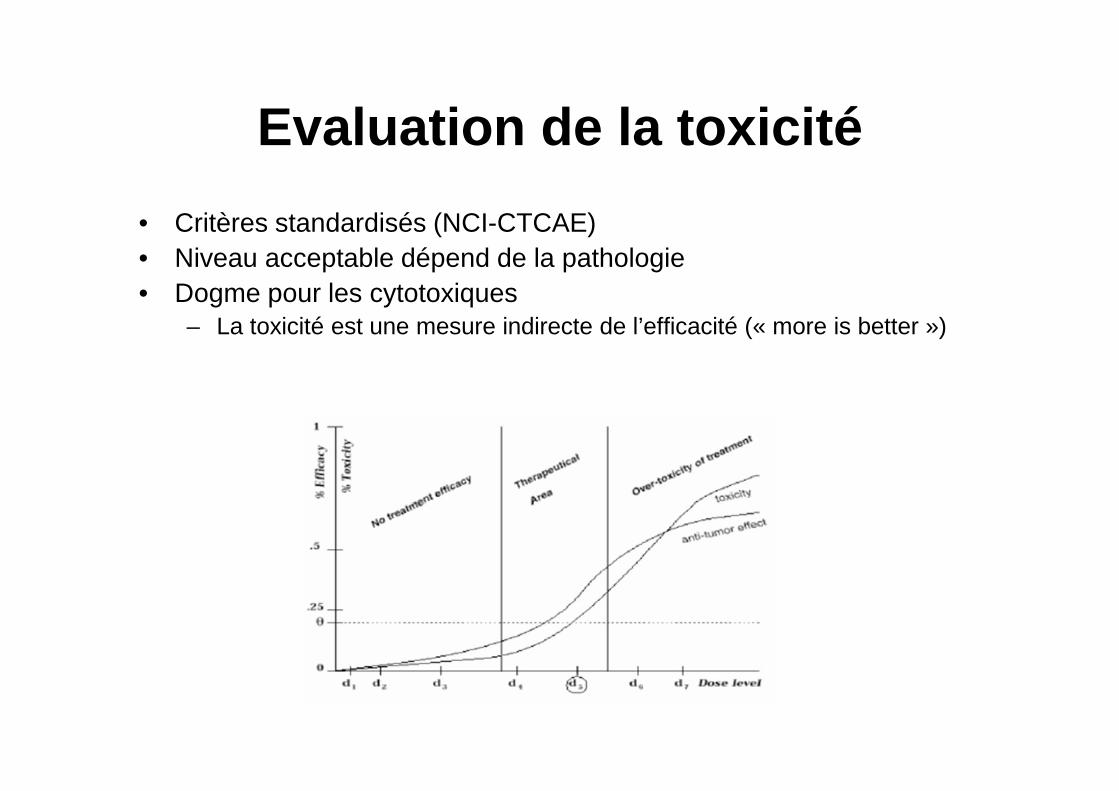
Evaluation de la toxicité
• Critères standardisés (NCI-CTCAE)• Niveau acceptable dépend de la pathologie• Dogme pour les cytotoxiques
– La toxicité est une mesure indirecte de l’efficacité (« more is better »)