Embed Size (px)
Citation preview

MG 01
Facteurs influençant la composition d’un système en équilibre
chimique (équilibres ioniques exclus)
Biblio : BA, Bottin-Mallet tome 2
Introduction
Définitions système, équilibre chimique, variance (BA)
Il faut v>2 pour équilibre, sinon si on change un facteur, rupture d’équilibre.
Cet équilibre peut être influencé par de nombreux facteurs qu’il est important de maîtriser pour avoir le
meilleur rendement possible (industrie).
On envisage la modification d’un seul paramètre, tous les autres étant maintenus constants, cette
modification entraînant soit la variation de la constante d’équilibre thermodynamique (cas de T et du
solvant), soit la variation du quotient réactionnel.
Lors de la présentation, il faudra s’efforcer de bien mettre en évidence le fait que le système est à
l’équilibre chimique, et qu’un seul paramètre a été modifié.
I. Influence des paramètres physiques
a. Influence de la pression
BUP 879 2005
p. 1175
gaz Equilibre NO2 = N2O4
Montage : tricol avec septum. On
ajoute par au-dessus HNO3 et on
prélève par le septum dans une
seringue avec robinet pour pouvoir
fermer.
Couleur rapide + Hotte
Expérience :
On est à T fixée, composition fixée, on fait varier P.
On comprime, le gaz se colore -> NO2 et on détend, le gaz se décolore -> N2O4
Réaliser la compression et attendre que l’équilibre thermique soit atteint pour commenter le résultat
Interprétation :
Déplacement d’équilibre, loi de Le Chatelier
Transition : Ici on est restait à température atmosphérique, maintenant on change…

b. Influence de la température
BUP 879 2005
p. 1175
gaz Equilibre NO2 = N2O4
Montage : tricol avec septum. On
ajoute par au-dessus HNO3 et on
prélève par le septum dans une
seringue avec robinet pour pouvoir
fermer.
Couleur rapide + Hotte
Expérience :
On est à P fixée, composition fixée, on fait varier T.
On le met dans la glace, le gaz se décolore -> N2O4 ; On le met dans un bécher d’eau chaude, le gaz se
colore -> NO2
Interprétation :
Déplacement d’équilibre, loi de Le Chatelier
Fosset p. 106 La mesure de la solubilité de l’acide
benzoïque à différentes températures
permet de déterminer l’enthalpie et
l’entropie de dissolution de celui-ci.
Prélèvement de la solution : filtration
dans un bécher thermostaté à la même
température que le bain.
2h Dosage
colorimétrique
ou pH-mètrique
(pH mètre à
réétalonner à
chaque
température)
Expérience :
On est à P fixée, composition fixée, on fait varier T.
Interprétation :
Déplacement d’équilibre, loi de Le Chatelier. Détermination de l’enthalpie de dissolution.
Transition : Maintenant on fixe ces paramètres et on va faire varier la composition.
c. Influence du potentiel
Ici on a un équilibre d’oxydo-réduction faisant intervenir H+ et des électrons…. IONIQUE ??
Fosset p. 257 Le potentiel d’une électrode de platine
plongeant dans une solution contenant un
mélange équimolaire de quinone et
d’hydroquinone dépend de façon linéaire
du pH.
1h30 - Pas de
hotte
Expérience :
On est à P, T, composition fixées. On fait varier le potentiel.
Interprétation :

Loi de Nernst. Application -> électrode de pH.
Transition : Maintenant on fixe ces paramètres et on va faire varier la composition.
II. Influence de la composition
Attention, il faut supposer que l’équilibre se fait à chaque instant sinon jamais d’équilibre…
a. Estérification
TP 33 Bordas
p. 108
Liquide - Réaction
- Extraction
- Séchage
- Filtration
CCM
(condition ????)
Indice de réfraction
(droite
d’étalonnage), IR,
CPV ???
h ++ Hotte
* Description:
Manip à faire avec et sans Dean-Stark
Transition : Autre méthode, on distille…
b. Trans-etherification
JD 102 Liquide - Réaction et
distillation
simultanée
Indice de réfraction
IR
1 h ++ Hotte
* Description:
Distillation élimine le produit au fur et à mesure.
c. Ajout d’oxygène au Co-salen
JCE 1977, p.
443
solide On ajoute de O2 à un
complexe de Salen
h ++
* Description:
Ici on ajoute un des réactifs en excès
Bouteille d’O2 ?
Voir TP M1
Conclusion :

Maintenant qu’on a déplacé l’équilibre, on peut également accélérer l’atteinte de cet équilibre grâce à
un catalyseur. Compromis entre équilibre et cinétique
Ammoniac (BM)

MG 02
Exemples de détermination de grandeurs standard de réaction
(∆rG°, ∆rS°, ∆rH°)
Biblio : BA
Introduction
Définitions grandeurs standards de réaction (BA)
∆rG°= -T∆rS°+ ∆rH°
D’où on peut les déduire les uns des autres avec une étude de la température.
Reliés à la constante d’équilibre : ∆rG° = -RTlnK°
Important à connaître car détermine équilibre d’une réaction, et donc en partie son rendement !
(industrie) On va voir qu’il existe une large gamme d’expériences pour les déterminer.
Bien faire attention que l’on mesure ∆X, donc bien faire le lien avec ∆rX°
I. Etude calorimétrique
Dosage calorimétrique de l’acide phosphorique
Souil. TP, p73,
Fosset p.82
Wilson,
Newcombe
- on suit l’élévation de température du
mélange dans un calorimètre
- on utilise une base forte : volume
varie peu
- on prend des mesures très régulières
(fuites auront une contribution
constante)
Suivi de la
température
1h30 - Pas de
hotte
Expérience :
On a une courbe avec trois pentes. On en déduit à chaque fois la chaleur échangée et on remonte à
∆rH°
Transition : On a étudié l’enthalpie de neutralisation de l’acide, on peut également déterminer sa
constante d’acidité et remonter ainsi à son enthalpie libre.

II. Etude spectroscopique
Détermination du pKa duBBT
Daumarie p.
111
Déterminer une grandeur
thermodynamique par UV
Existence d’un point isobestique
1h30 UV
Expérience :
On détermine le pKa, donc le Ka et on remonte à ∆rG°
Transition : On a utilisé le caractère acide/base qui on a vu met en jeu des ions. Ces ions peuvent
modifier la conductivité de la solution…
III. Etude conductimétrique
Détermination du pKs de PbSO4
Voir si PbSO4 autorisé !
Fosset p. 104
BA p.104
La mesure de la conductivité d’une
solution saturée d’un sel très peu soluble
permet de déterminer son produit de
solubilité
Conductimétrie - Pas de
hotte
Expérience :
On détermine la fem, donc ∆rG° (à plusieurs températures différentes ce qui nous permet de remonter
à ∆rH° et ∆rS° ???????)
Transition : Ces composés étudiés présentent également des caractères oxred, qu’on peut étudier tout
de suite…
IV. Etude potentiométrique
Détermination des grandeurs thermodynamiques d’une pile
JCE, 1970, vol
47, p 365
Tables :
Bernard et
Busnod
- illustration de la relation entre
DrG°, DrS° et DrH° entre eux et
avec E°
- on suit l’évolution avec la
témpérature du potentiel entre une
électrode Ag/ AgCl et une
électrode à calomel saturé (car S
dépend de d(E°,T) )
Montage à 2
électrodes
3h - Pas de
hotte
Expérience :
On détermine la fem, donc ∆rG° à plusieurs températures différentes ce qui nous permet de remonter à
∆rH° et ∆rS°

Conclusion :
Importance des grandeurs de réaction mais il ne faut pas oublier cinétique

MG 03
Diagrammes binaires (solide-liquide ; liquide-vapeur) ; tracé ;
applications.
Biblio : BA, HP PC(pour applications), Bernard-Bunot ou Handbook (pour cryo)
Introduction
Souvent on a affaire à des mélange qui on des comportements différents. On peut montrer les deux
corps purs solides menthol-phénol on les mets ensemble : fusion. Et les propriétés du mélange
notamment les conditions de changement d’états vont dépendre de la composition du mélange. Dans le
cadre du montage deux composés dans deux phases. Deux cas différents miscibilité totale dans la phase
la plus condensée ou non miscibilité totale. La variance est alors v= +2-R. Tracé des diagrammes�
puisque donné expérimentale deux façon illustrées. 2. illustrer les applications de ces diagrammes.
Liquide/solide
JCE 1990
p.156
solide Phénol/menthol
On rapproche les
deux solides et hop
liquide
- hotte
I. Tracés de diagrammes binaires
a. Par courbe d’analyse thermique
Etude du binaire naphtalène-naphtol
BUP 573 p.
810
On ne fait
qu’un
mélange sur
deux, et on
divise les
quantités par
100
- on fait fondre dans des tubes des
mélanges de différentes
compositions
- on sort du bain d’huile et on suit
l’évolution de la température
toutes les 30 sec jusqu’à 50°c
- on prend des mesures de l’indice
de réfraction de la phase vapeur et
de la phase gaz
Suivit de la
température
pour courbe
d’analyse
thermique
3h00 - Hotte
b. Par mesure de composition
Etude du binaire acétone-chloroforme
BUP 879, vol
99, dec 2005,
p. 135
- on effectue une courbe
d’étalonnage pour relier indice de
réfraction et fraction massique de
chloroforme
- on place de l’acétone que l’on
porte à ébullition, on ajoute du
chloroforme
- on prend des mesures de l’indice
de réfraction de la phase vapeur et
de la phase gaz
Montage de
distillation avec
prise d’indic de
réfraction
3h00 - Hotte
Transition : Applications…
II. Applications
a. Hydrodistillation
Liquide/vapeur (non miscibles)
Chimie des
odeurs et des
couleurs p 207
liquide hydrodisillation
séparation
orga/aqueuse
séchage
IR, indice de
réfraction,
pouvoir
rotatoire
2h30 + Pas de
hotte
On distille l’hétéroazéotrope
b. Distillation fractionnée de l’éthanol
Liquide/vapeur (miscibles)
Maison : on mélange eau/éthanol et on distille (procédé industriel).
On fait une courbe d’étalonnage avec l’indice de réfraction et on observe qu’on distille (avec Vigreux)
l’azéotrope (d’où l’éthanol à 95%), si on le voulait pur, il faudrait un ternaire avec du toluène.
c. Abaissement cryoscoique
Solide/liquide (non miscibles)
Mettre de la glace, du sel et un thermomètre et montrer la chute de température.
Utilisée en synthèse organique (cf octanal JD 46)
Conclusion
Grand intérêt de l’étude de ces diagrammes binaires. Pour de nombreuses applications de séparations

de produits et purification de produits. Dans la vie courante le salage des routes, antigel dans les
radiateurs de voitures (eau/glycol), soudure (étain),…
Binaires d’énantiomère. Pour la séparation de racémique. Leçon binaire. Mais aussi ternaire expliquer
la différence de solubilité des énantiomère, dédoublement

MG 04
Interactions soluté-solvant et soluté-soluté.
Biblio : Gershell, Atkins, HP,…
Introduction
Une solution est constituée de plusieurs entités : solvant grande majorité et soluté minoritaire
Rôle du solvant : amener le soluté en contact entre eux
Interaction de différents types : solvant-soluté : permet solubilisation
soluté-soluté : pour des réaction futures.
Mais pouvant être de même nature. Il faut tenir compte de ces deux types d’interaction.
On va voir par énergie d’interaction croissante.
Il est important dans chacune des manipulations présentées de mettre en avant les caractéristiques du
solvant et celles du soluté afin de pouvoir discuter pour chacune des manipulation des interactions
soluté-solvant d’une part et soluté-soluté d’autre part.
I. Interactions faibles
a. Interactions de Van der Waals : solubilité et partage du diiode
Fosset p. 115 Le dosage du diiode dissous dans des
solutions saturées dans deux solvants
différents permet d’accéder aux
constantes des équilibres de
dissolution correspondants. On peut
alors également estimer la constante
de partage du diiode entre les deux
solvants
2h Extraction,
dosage
* Discussion
Diiode apolaire, très polarisable, préfère solvant organique…
* Application
Purification par extraction liquide/liquide en chimie organique.
Transition : Les solutés ne sont pas forcément des ions…

b Dimérisation par liaison hydrogène
Fosset p. 382 - Le partage de l’acide benzoïque
entre l’eau et le cyclohexane est
déterminé par dosage acido-basique
de chaque phase pour différentes
concentrations d’acide benzoïque. On
peut ainsi déterminer la constante de
dimérisation de l’acide benzoïque
dans le cyclohexane.
1h Dosage
acido-
basique
Hotte
* Discussion
Importance des liaisons hydrogènes.
* Application
Solubilisation de produit.
Transition : Des interactions plus fortes peuvent avoir lieu si les solutés sont chargés…
II. Interactions fortes électrostatiques
a. De type dipôle-ion : solvatation relative des cations alcalins
Fosset p. 354 - Les conductivités molaires à
dilution infinie des solutions de
chlorure de lithium, sodium et
potassium sont corrélés à la
solvatation relative des ions
alcalins
1h Conductimétrie
* Discussion
Interaction électrostatique entre les ions et les molécules d’eau. Eau diminue les interactions
électrostatiques entre les ions due à la forte constante diélectrique.
* Application
Permet aussi de renforcer la basicité d’une base -> capture de son contre-ion. O-alkylation, permet de
libérer la nucléophilie des alcoolates. Ou au contraire C-alkylation avec solvant très peu dissociant.
Transition : Dans des solvants peu dissociants, les ions vont rester ensemble !

b. De type ion-ion : mise en évidence de la formation de paires d’ions
* Discussion
Interaction électrostatique entre les ions, influence du solvant sur cette attraction.
* Application
Transfert de phase. On amène un ion d’intérêt dans une phase organique.
Conclusion
Grande importance en chimie orga : réactions, extraction,…
Chromato de partage, HPLC, CPV,…
Fosset p. 374 L’utilisation d’ions de couleurs
différente permet de visualiser et de
quantifier la formation de paires
d’ions et leur extraction par un
solvant organique.
2h UV Hotte

MG 05
Couples acide-base ; constantes d’acidité ; influence du milieu.
Biblio : Gershell, Atkins, HP,…
Introduction
Définition d’un couple acide-base de Brönstedt et de Lewis.
Définition constante d’acidité. Applications solution tampon et indicateur coloré.
Influence du milieu. Application au dosage et dénivellement des acides.
I. Couple acide-base
a. Détermination du Ka d’un acide
Daumarie p.
111
Déterminer une grandeur
thermodynamique par UV
Existence d’un point isobestique
1h30 UV
* Discussion
Très important, exemple indicateur coloré !
Transition : Nous avons utilisé la couleur des différents membres du couple pour déterminer le pKa
par spectrophotométrie, on peut également utiliser la mobilité des ions intervenant dans les couples
acide-base pour déterminé le pKa, notamment lorsque plusieurs acides sont présents.
b. Dosage d’un mélange d’acide
Fosset p. 53 L’étude de la conductivité de
solutions aqueuses d’un acide
faible en fonction de sa
concentration permet de
déterminer la constante de
dissociation de cet acide.
1h Conductimétrie
* Discussion
Certaines conditions sont à respecter pour doser un mélange d’acides
Transition : On utilise des indicateurs colorés pour doser des acides par exemples, mais que faire
quand ces acides sont faibles ? Nous allons voir qu’en changeant le milieu on peut changer la force
des acides. (ATTENTION : force et faiblesse des acides relatives….)


II. Influence du milieu
a. Influence de la complexation
Fosset p. 73 La présence d’un complexant d’un
acide modifie ses propriétés acido-
basiques. L’étude de la courbe de
dosage pH-mètrique permet d’évaluer
la constante de formation du
complexe
1h pH-
mètrie
* Discussion
En complexant, acide faible -> acide fort ! On peut doser !!
Transition : Nous avons vus comment rendre fort un acide. Cependant, la force d’un acide dépend
également également du solvant car l’eau nivelle tous les acides forts. Pour les différencier, on change
de milieu.
b. Influence du solvant
Fosset p. 68, BA p.
145
Des dosages
potentiométriques en solvant
acide acétique de HCl et HBr
permettent de mettre en
évidence leur différence de
comportement en solvant non
aqueux.
Dosage
potentiométrique
1h - hotte
* Discussion
Denivellation des acides forts !!
Intérêt : réaction en solvant organique,… il faut connaître la réactivité des acides !
Conclusion
Importance pour réactions acide-base : il faut connaître les pKa pour obtenir réaction quantitative en
chimie organique par exemple (Williamson, déprotonation d’alcool ; Wittig, déprotonation de l’ylure,
…)

MG 06
Techniques de titrage de mélanges d’acides et de bases.
Biblio : Gershell, Atkins, HP,…
Introduction
Définition d’un couple acide-base de Brönstedt et de Lewis. Définition titrage. Plein de façons de procéder.
I. Titrages de mélanges d’acides
a. Acides de Brönstedt
Wilson p. 100 Dosage de HCl et Acide acétique par de la soude
Dosage conductimètrique
1h - Hotte
(Daumarie p. 181, pH-mètrique si problème)
Application
Boisson gazeuse, additifs alimentaires acides (benzoique, ascorbique,…)
Transition : Il est également important de contrôler la teneur en acide de Lewis
b. Acides de Lewis
Dureté de l’eau
Olympiades de chimie, p36-38, Ecolochimie p 305-308Des
expériences
dans la famille
des A-B p.253
-Réaliser un dosage complexométrique- étalonner une solution d’EDTA- dosage en présence de NET- dosage en milieu ph 12
Dosage colorimétrique
1h - Hotte
Application
Importance de la dureté de l’eau pour les machines …
Transition : Les acides de Lewis peuvent servir à doser des bases de Lewis…

II. Titrages de mélanges de bases
a. Bases de Lewis
JFLM p. 85BA (pour calcul Ks)
On dose une solution contenant un mélange d’ions NaCl et d’ions KI par une solution d’AgNO3 par potentiométrie. On en déduit E° et Ks.
Potentiométrie
1h30 - Pas de hotte
Application
On en déduit le Ks et le potentiel Standard.
Transition : réactions acide-base exothermique…
b. Bases de Brönstedt
Souil p.73 ; JFLM p. 252
Dewar de 500mL : 100mL de soude à 1M, 100mL d'ammoniac à 1M, dosés par HCl à 5M (ou plus)] [3 segments,pentes voisines ; pentes des droites dépend des Cp ; pente augmente => plus de dégagement : il faut une grande concentration]
Dosage calorimétrique
1h -
Application
On en déduit l’enthalpie de neutralisation
Conclusion
Plein de moyens différents de titrer qu’on peut adapter.

MG 07
Techniques électrochimiques d'analyse : méthodes
potentiométriques. Exemples d'applications.
Introduction : Particulièrement intéressant de connaître la concentration d’un composé en solution. Question de pollution, contrôles des taux règlementés de certaines espèces etc. Il existe de nombreuses méthodes pour doser des solutés. Des techniques électrochimiques peuvent être utilisées pour cela. La détermination du potentiel de la solution va nous permettre de remonter la concentration d’espèce en solution, le suivi E=f(V) est plus précis que les dosages colorimétriques.
Dosage de la vitamine C dans le JFLM (Pt-ECS à courant nul)
Dosage de Cl- dans le serum phy BA
Fil conducteur comment doser les ions fluroures contenue dans une eau de badoit ?On commence par montrer une nouvelle technique : la potentiométrie qui a l’avantage que même sans couleur on peut connaître la concentration d’un ion.
I. Titrages potentiométriques
Fosset p. 298 Les ions Fe II sont dosés par oxydation par l’ion MnO4-. L’équivalence est mise en évidence par différentes méthodes potentiométriques : à courant nul ou à courant imposé entre différents types d’électrode.
Dosage potentiométrique
1h30 - Pas de hotte
Transition : Et pour F- ? Il faudrait trouver une réaction ou F- apparaît. On sait que les halogènes
forment un précipité avec Ag+. Peut-on doser un mélange d’halogène ? En effet, sur la bouteille de
Badoit, on voir qu’il y aussi des ions chlorures !

II. Détermination de grandeurs thermodynamiques
JFLM p. 85BA (pour calcul Ks)
On dose une solution contenant un mélange d’ions NaCl et d’ions KI par une solution d’AgNO3 par potentiométrie. On en déduit E° et Ks.
Potentiométrie
1h30 - Pas de hotte
Transition : mais AgF est soluble dans l’eau donc il n’y aura pas consommation de F- par
précipitation ! Il existe des électrodes particulières qui permettent de connaître la concentration d’une
seule espèce. Ce sont des électrodes spécifiques.
Comment fonctionne-t-elle ? Construction de l’électrode en verre son E dépend uniquement de la
concentration en H+
III. Electrodes spécifiques
a. Electrode spécifique aux protons
Fosset p. 257 Le potentiel d’une électrode de platine plongeant dans une solution contenant un mélange équimolaire de quinone et d’hydroquinone dépend de façon linéaire du pH.
1h30 - Pas de hotte
Transition : On peut faire de même pour doser les ions F- avec une électrode spécifique aux ions
fluorures.
b. Electrode spécifique aux fluorures
BUP 664 p1059 - Pas de hotte
TP DE M1 A RELIRE POUR LES AJOUTS DOSES
* Constitution
L’électrode à fluorures est une électrode à membrane, constituée par un monocristal de fluorure de lanthane LaF3, la solution interne d’activité constante étant une solution de fluorures, l’élément de référence interne du type Ag/AgCl
* Expérience
On maintient la force ionique constante grâce à une solution de NaCl molaire. Dans ce cas, la différence de potentiel est une fonction linéaire de pF.
Rq : aux faibles concentrations, la relation linéaire n’est plus applicable, la solubilité intrinsèque du cristal de LaF3 n’étant plus négligeable devant la molarité des ions F- introduits.On veillera à ne pas travailler en milieu basique, les ions HO- pouvant entrer en compétition avec les ions F-

Conclusion : Large utilisation de l’électrode de verre. Technique ampérométrique aussi exploitée pour doser une espèce.

MG 08
Piles électrochimiques et accumulateurs.
Biblio : BUP 633, Miomandre
Introduction : (BUP 633, Miomandre)Historiquement apparition des piles avec Galvani et Volta (empilement=>pile). Aujourd’hui600 millions de piles par an. Définition de pile, système électrolytique et accumulateur. Au cours de ce montage on va présenter quelques piles et accumulateurs et préciser les grandeurs caractéristiques qui vont déterminer leurs différentes utilisations.
I. Générateurs électrochimiques usuels
a. Pile Leclanché : non rechargeable
JFLM p. 198 ; BUP 633 p. 928 ; Sarrazin, BA 219
On prépare un gel d’agar-agar (40 % en masse) contenant du chlorure d’ammonium et du dioxyde de manganèse (IV). Dans un bécher on met une plaque de zinc que l’on tord et au centre un barreau de graphite. On mesure avec un voltmètre la ddp, on peut brancher une lampe sur une pile préalablement préparé.
Pas de hotte
Transition : Mais quand la pile a fini de débité morte, poubelle… Il existe d’autres générateurs
rechargeables : les accu !
b. Accumulateur au plomb : rechargeable
Sarrazin p. 280 ;
Besson et Guitton
p. 138
Charge et décharge de l’accumulateur avec Synchronie
Assez long
Synchronie Pas de hotte
SAVOIR LE SCHEMA DU MONTAGE
Transition : Cependant, tous ces générateurs sont polluants ! On cherche d’autres types de
générateurs…

II. Pile à combustible
Pile eau oxygénée - méthanol
Sarrazin p. 285 Pile - hotte
Demander s’il y a le matériel !
(Si problème, prendre la manip de la pile à concentration Brenon-Audat p. 125)
Transition : On peut également les utiliser pour déterminer des grandeurs caractéristiques de leur
fonctionnement.
III. Détermination de grandeurs caractéristiques
JCE, 1970, vol
47, p 365
Tables :
Bernard et
Busnod
- illustration de la relation entre DrG°, DrS° et DrH° entre eux et avec E°- on suit l’évolution avec la témpérature du potentiel entre une électrode Ag/ AgCl et une électrode à calomel saturé (car S dépend de d(E°,T) )
Montage à 2 électrodes
3h - Pas de hotte
Conclusion : Utilisation tout le temps, partout, indispensable. On essaye de nos jours de minimiser le rapport poids puissance des piles, donc on fait des piles au lithium !

MG 09
Electrolyse ; courbes intensité-potentiel ; réactions aux
électrodes.
Biblio : Miomandre, BUP 698 (chlore/soude)
Introduction : Précédemment on a vu les piles qui mettaient en jeu des réactions redox spontanées, aujourd’hui on va s’intéresser au processus inverse c'est-à-dire à l’électrolyse. Les électrolyses vont être très utiles dans de nombreux domaines industriels tels que la protection contre la corrosion, le dépôt décoratif, la purification de composés mais aussi l’électrosynthèse. Lors d’une électrolyse on va imposer une tension et l’objet de ce montage va être aussi de savoir comment choisir la valeur de cette tension.
Système électrochimique, importance des électrodes, phénomènes transport, convection, migration
I. Principe de l’électrolyse
JCE 1982 p. 586
Par coloration on montre les différents produits de l’oxydation de l’eau
2h
Discussion
Les couples de l’eau, utilité de l’électrolyte support, anode/cathode, circulation des électrons. D’après la thermodynamique on devrait avoir électrolyse pour 1,3V or il ne se passe rien. Cela va montrer l’importance de tracer des courbes I=f(E) pour connaître ce qui se passe dans le système.
Transition : L’électrolyse de l’eau peut être utilisée dans certains domaines mais le plus souvent on
cherche à électrolyser d’autres espèces chimiques donc on va chercher à l’éviter. Pour cela il faut
savoir à partir de quelle valeur de la tension elle va se produire et c’est dans ce domaine que l’on
pourra travailler.
II. Courbes intensité-potentiel
a. Domaine d’électroactivité de l’eau
BA p. 196-200 2h
Surtension sur différents métaux : électrodes de fer, de plomb de platine, graphite

On reprend le montage précédent avec la bonne surtension ! et ça marche.
Transition : Maintenant que on connaît le domaine dans lequel on peut travailler on va voir qu’il
existe différent types de couple redox sur lesquels on va agir.
b. Couples rapides/Couples lents
Sarrazin p.227 Fe III /Fe II 2h
Discussion :Couple lent/rapide ; limitation par transport de matière, diffusion ; mur du solvant …Remarque : on avait vu le couple O2/H2O qui est un couple lent.
Transition : On sait désormais grâce au courbe I=f(E) la tension ou l’intensité qu’il faut imposer on
va pouvoir alors les utiliser pour des applications industrielles comme l’électrodéposition
III. Applications
a. Dosage de l’acide ascorbique
JCE 1995, vol
12, p 445
- on dose un comprimé de vitamine C, on génère I2 qui réagit avec l’acide ascorbique jusqu’à ce que ce dernier soit entièrement consommé. On mesure le temps nécessaire à cette réaction, on remonte à la quantité de I2 généré et donc d’acide ascorbique présent
Montage à 2 électrodes avec garde et intentiostat
1h - Pas de hotte
Transition : Doser mais aussi produire
b. Obtention du manganèse par électrolyse
BA 222 ; Walton (en réf)
dans un poreux : un clou joue le rôle de la cathode, un morceau de magnésium métallique celui de l’anode. Une solution de carbonate de potassium joue le rôle d’électrolyte support. Fixer l’intensité et non la tension.+ Synchronie
Pas de hotte
Conclusion : Les courbes I=f(E) sont un très bon outil pour déterminer les conditions d’électrolyse. Les principales électrolyses réalisées actuellement sont le Zn, Al et le procédé Chlore/Soude mais de plus en plus on étudie les électrolyses à cause des applications dans les piles à combustibles par exemple.

MG 10
Méthodes non stationnaires en électrochimie :
chronoampérométrie et voltamétrie cyclique.
Biblio : Miomandre, Faulkner
Introduction : Dans les méthodes non stationnaires le courant dépend du temps et en particulier il va être régit par le transport de matière et notamment par le phénomène de diffusion qui varie avec le temps. 2 grandes méthodes se distinguent (chronoampérométrie et voltampérométrie cyclique) et on verra qu’elles sont utiles car, malgré la complexité des théories sous-jacentes, elles permettent d’obtenir une « signature » du système.
I. Principe des méthodes non stationnaires
a. Voltamétrie cyclique
BUP 899 dec 2007
Discussion
On essaye d’avoir une signature du couple pour cela il faut qu’il n’y ait pas d’impureté donc dégazage et on a vérifié que l’électrolyte était inerte. On fait un cycle et la voltamétrie cyclique a l’avantage de laisser l’espèce près de l’électrode (on peut représenter la couche de diffusion en fonction du temps). On peut donc déterminer un E1/2 et dire que le voltamogramme est caractéristique d’un système réversible. On peut observer l’influence de la vitesse de balayage.
Transition : Dans la loi de Randles-Sevcik il y a en général 2 inconnues le nombre d’électrons
échangés et le coefficient de diffusion. Pour tout déterminer correctement il faut donc une seconde
méthode.
b. Chronoampérométrie
BUP 899 dec 2007
Discussion
Grâce à la voltampérométrie on peut choisir les potentiels que l’on impose. Electrode tournante pour s’affranchir de la convection. On vérifie la loi de Cottrell en enlevant les premiers (pb de courants capacitifs) et les derniers points (pb de convection non négligeable). Cette fois on peut avoir n et D !!! Utilisation du DMSO pour avoir une grande fenêtre électroactive.

Transition : Les techniques non stationnaires permettent donc de caractériser une espèce chimique
mais elles ont surtout un immense avantage c’est que ce sont des méthodes dynamiques, on va pouvoir
observer l’évolution d’un système et en déduire notamment des informations sur les mécanismes
II. Applications
a. Etude d’un mécanisme
JCE 1983
BUP, 95, 830,
19 (info sup)
- étude d’un système irréversible : la thyronine- caractérisation d’espèces chimiques
Tracé de voltampérogramme
- Pas de hotte
L-thyronine est un antibiotique !
Transition : On peut étudier un mécanisme mais aussi produire…
b. Synthèse d’un polymère
BUP 830 p. 193
Cyclovoltamétrie Voltamétrie - Pas de hotte
Discussion sur le mécanisme, l’importance du tensioactif pour la mise en solution. Insister sur le côté chimie verte car on récupère juste le polymère sur l’électrode et on recommence. Exemple industriel avec l’adiponitrile pour le Nylon (Monsanto)
Conclusion : Les techniques non stationnaires sont particulièrement utilisées dans la détermination de mécanismes pour des systèmes biologiques. La recherche s’oriente aussi vers le dépôt de polymères conducteurs sur les électrodes ce qui permet d’obtenir de nouvelles électrodes avec des propriétés intéressantes.

MG 11
Méthodes stationnaires en électrochimie : polarographie et
voltamétrie sur électrode tournante.
Introduction : Définition mathématique : dX/dt=0. X grandeur intéressante ici, l’intensité ! Pourquoi ? parce que simplification et on peut en tirer des informations simplement comme coefficient de diffusion, concentration,…Nous allons voir au cours de ce montage comment arriver à ce I constant par la voltamétrie sur électrode tournante puis à l’aide de la polarographie.
I. Voltamétrie sur EDT
a. Obtention d’un régime stationnaire
BUP 2008 p. 7, JCE 2000 p. 1190
Fe(CN)63-/Fe(CN)64- EDT Pas de hotte
Comment l’obtenir ? Regardons ici un exemple avec l’oxydation de l’hexacyanoferrate II en III. #Faire une expérience de chronoampérométrie avec et sans agitation dans un bécher !. Pour ce faire on utilise un montage à trois électrodes :-une électrode de travail : EDT avec surface connue-une électrode de référence : ECSPotentiel imposé entre ces deux électrodes-une contre-électrode : platine, évite la polarisation de l’ECS (performances dégradées) en permettant le passage du courant dans l’ET en « fermant » le circuit. On ajoute un électrolyte support.
Plusieurs modes de transport : migration (affranchie avec électrolyte), convection et diffusion. Si on n’agite pas, couche de diffusion s’agrandit, transport de matière limitant. Si on agite, le milieu amène suffisamment vite de la matière pour que la couche de diffusion reste constante. Et donc la quantité de charge consommée à l’electrode est constante et I est constant.Mais convection par barreau magnétique pas top, on connaît pas l’épaisseur de la couche de diffusion ! On peut pas tirer grand-chose facilement !
EDT, loi de Levich.
#Faire une expérience de voltamétrie avec et sans agitation dans un bécher !. Gaine permet éviter effet de bord. Moteur pour faire tourner. Liquide expulsé vers l’extérieur. Couche de diffusion fixée. Loi de Levich dérive de Navier-Stokes. Montrer chrono toute belle. Qu’est-ce qu’on

peut en tirer ? Tracer une courbe i=f(E). La voltamétrie a pour but l’obtention d’un courant qui constitue une réponse du système, en fonction de la tension imposée, càd sous la contrainte imposée par l’opérateur. Normalement, il faudrait mettre un potentiel et attendre très longtemps pour que le régime stationnaire s’installe. Ici pour gagner du temps on va balayer en potentiel, donc on va injecter une dépendance temporelle !! Mais il suffit que le balayage soit suffisamment rapide pour ne pas trop appauvrir la couche de diffusion.On voit un palier de diffusion. Transport de matière limitant, i ne peut plus augmenter, la valeur est bloquée par la couche de diffusion.Grand avantage de l’EDT, on peut contrôler cette épaisseur !Montrer plusieurs graphes : si w trop bas, le transport par convection n’est pas assez rapide pour assurer une couche de diffusion constante (réaction trop rapide par rapport à l’apport).
Transition : Maintenant que l’on a un régime stationnaire, que peut-on en faire ?
b. Application
BUP 2008 p. 7, JCE 2000 p. 1190
Fe(CN)63-/Fe(CN)64- EDT Pas de hotte
Utilisation de la loi de Levich pour déterminer le coefficient de diffusion, soit en faisant varier C, soit en faisant varier w.
Dosage des ions Fe par Ce.
On se place à E fixé et on observe i en fonction de v. Interprétation avec les courbes i=f(E).
Transition : On s’est intéressé à l’oxydation, maintenant si on veut étudier la réduction, par exemple
comme on va le voir la réduction d’un mélange de cations métalliques, il faut changer d’électrode car
ici H+->H2 faible surtension (0,1 V sur platine, à part cations peu électropositifs Ag, Cu, Ni marche
pas) et bloque l’étude de la réduction. Donc électrode avec grande surtension pour H+, l’électrode à
goutte de mercure ! Qui d’ailleurs depuis sa découverte en 1922, a connu un grand essor !
II. Polarographie
a. Dosage d’un mélange d’ions
Prichard p.
170
Polaro
On va s’intéresser à un mélange d’ions Cd, Mn, Zn.#Présentation de l’appareil. Même principe que précédemment. On a toujours un montage à trois électrodes et on fait barboter N2 pour éliminer O2 et son pic de réduction en eau, et toujours un électrolyte support.On va utiliser une électrode liquide à goutte tombante ! Ici on peut voir le réservoir, le mercure descend

par un capillaire et le marteau fait tomber la goutte (avant capillaire très fin et on attendait que le poids l’emporte sur la capillarité). pas de souci d’irrégularité de surface, mais variation de l’aire de la goutte ! #Préparation de la solution. On va préparer ici la solution en pipettant 10 mL de chaque solution.Comme dit, avec la croissance de la goutte, on injecte une dépendance temporelle ! l’intensité augmente au cours du temps puis chute avec la chute de la goutte. En fait, non, car la couche de diffusion s’étend mais la goutte va « chercher » la matière et en fait globalement couche de diffusion constante. De plus, elle entraîne un mouvement de convection qui homogénéise la solution autour de la goutte. D’où le nom de pseudo-stationnaire.Ici on rajoute de la gélatine qui sert de surfactant pour avoir des gouttes avec une surface bien définie.#On lance la mesure. Il existe plusieurs méthodes de mesure. La plus courante à échelon, consiste à mesure le courant pendant un instant très court au même moment de la vie de chaque goutte. On s’affranchit de la dépendance temporelle issue de la croissance de la goutte. On obtient ces courbes. Palier de diffusion. On peut y lire la hauteur de la vague et le potentiel de demi-vague, caractéristique de l’espèce. On les a cherchés pour les trois espèces. Ensuite, on a effectué une régression linéaire de la hauteur de la vague en fonction de la concentration sur des étalons. Maintenant on effectue une mesure. On regarde la hauteur de la vague, on reporte et on a la concentration. On peut utiliser pour être plus précise le PID où on trace la dérivée. En effet, on mesure i(E) avant impulsion et i(E+dE) à la fin de l’impulsion et en divisant par dE, on a la dérivée.
Transition :
On peut déterminer la concentration d’espèce connue, que l’on rencontre en milieux aqueux, mais on
peut également mettre en évidence la présence d’espèces instables en milieu aqueux, comme le cuivre
b. Stabilisation du cuivre I par complexation
Fosset p. 174Utiliser KNO3
au lieu du
perchlorate
d’ammonium
Le degré d’oxydation I du cuivre peut être stabilisé en solution par complexation. L’étude polarographique permet de mettre en évidence ce phénomène par le changement de forme du polarogramme de réduction d’une solution de cuivre II en fonction de la quantité de ligand introduite.
Polaro
Deux vagues au lieu d’une ! En plus la hauteur de chaque vague est la moitié de celle sans ammoniac. Hauteur de vague proportionnelle au nombre d’électrons échangés ! On regarde les valeurs théoriques et effectivement, le Cu I ne se dismute pas avec l’ammoniac.
Conclusion : Sonde du système.Autres méthodes pour obtenir un régime stationnaire :- Micro-électrodes : 1970 miniaturisation d’électrodes en vue d’implantation in vivo pour la détection d’espèce d’intérêt biologique NO. Courant très faibles ! Plus de chutes ohmiques ! très utiles en biologie. Du fait de la taille micrométrique, courant de diffusion cylindrique. Diffusion pure. Couche de diffusion constante entièrement déterminée par la géométrie de l’électrode.

MG 12
Diagrammes potentiel-pH et potentiel pL ; applications.
Biblio : Miomandre, HP, Pourbaix
Introduction : Diagramme de prédominance des acides/bases et complexes montrent l’importance du pH et de pL mais ces espèces peuvent aussi intervenir dans des réactions redox. D’où dépendance en potentiel. Intéressant de tracer diagrammes pour voir influence de ces deux paramètres.
I. Diagramme potentiel-pH
a. Tracé du diagramme potentiel-pH du fer
Sarrazin p. 119
Potentiométrie, pH-mètrie
Pas de hotte
Discussion
Domaines d’immunité, de corrosion, de passivation.
Transition : Application…
b. Application : dosage de O 2 par la méthode de Winkler
JFLM p.77-81Autres : Ecolochimie p 297-299
- doser le diiode par le thiosulfate- précipitation de Mn(OH)2- oxydation par le O2 dissoud- acidification et dissolution en Mn3+- oxydation de I-- dosage de I3-
Dosage iodométrique
1h - Pas de hotte
Discussion :
Discussion sur la formation des oxydes de Manganèse, de l’Iode… qui explique les conditions d’acidité.
Transition : On a étudié l’échange de protons, maintenant on va étudié l’échange de ligands.
II. Applications
a. Diagramme potentiel-pNH3 de Ag
Du diagramme E-pH de l’argent en milieu ammoniacal, on déduit le diagramme E-pNH3
Sarrazin-
Verdaguer,
2.1/3 p121 (E-pH)
- obtention du diagramme EpH- expression de pNH3 en fonction des données- détermination de constantes thermodynamique
Montage à 2 * 2 électrodes (partage de l’ECS)
1h30 - Pas de hotte
Transition : Exploitons…
b. Exploitation du diagramme potentiel-pNH3 de Ag
On en déduit la stoechiométrie du complexe.
Transition : On peut également choisir les conditions expérimentales pour avoir l’espèce
prépondérante voulue…
c. Application : stabilisation du Cu(I) par complexation
Fosset p. 174Sarrazin 111
pour le
diagramme !!!
Le degré d’oxydation I du cuivre peut être stabilisé en solution par complexation. L’étude polarographique permet de mettre en évidence ce phénomène par le changement de forme du polarogramme de réduction d’une solution de cuivre II en fonction de la quantité de ligand introduite.
Polaro
Conclusion :
Grand intérêt, corrosion, hydrométallurgie du zinc, procédé Bayer,…
Possibilité de rajouter en application l’électrode de référence aux protons :
Fosset p. 257 Le potentiel d’une électrode de platine plongeant dans une solution contenant un mélange équimolaire de quinone et d’hydroquinone dépend de façon linéaire
1h30 - Pas de hotte

du pH.
pH dépend linéairement du potentiel, on a fabriquer une électrode spécifique aux protons.

MG 13
Conductivité des électrolytes ; mobilité des ions ; mesure et
applications.
Biblio : Atkins, Miomandre
Introduction
Les ions sont les porteurs de charges en solution. Plongé dans un champ électrique ils vont alors subir une attraction/répulsion qui va les mettre en mouvement. On va pouvoir alors définir une mobilité, grandeur caractéristique de chaque ion. Au cours de ce montage on montrera les utilisations de cette mobilité et on verra qu’elle est reliée à la conduction du courant en solution. On pourra alors définir la conductivité d’un ion et celle d’un électrolyte, c'est-à-dire une espèce capable de se dismuter sous forme ionique en solution. (Miomandre)
I. Mobilité des ions
a. Mise en évidence : frontière mobile
Atkins,
Defranchesci, p 35, Fosset p.363
- on impose un courant- on suit l’élévation de la coloration rose au cours du temps- on prend des mesures au cours du temps et on relie au nombre de transportNe pas mettre trop d’héliantine, vérifier que l’intensité reste constante au cours de la manip et appliquer unetension d’au moins 200V.
Montage artisanale de suivi de la coloration
Pas de hotte
(Atkins)*Définitions :
Mobilité : vitesse de migration d’un ion (v) = mobilité (u) * E ; u = ze/ffrottement
Nombre de transport : fraction du courant total transportée par les ions d’un type donné. Dans le cas simple où il n’y a que deux ions de charges opposées : t+ + t- = 1. et t+- = I+-/I = zu /( zu +zu )� � �
* Mesures :
L’élévation de la couleur rose indique le nombre d’ions qui se sont déplacés : n = c*V (volume lu). Donc la quantité de charge : z = z+*c*V*F. Ainsi t+ = z+*F*c*V/(I*∆t) (I mesurée sur ampèremètre).
* Discussion :
Mobilité augmente avec la charge, diminue avec la taille, dépend de la viscosité du solvant, rayon

hydrodynamique. Particularité de HO- et H3O+ avec le mécanisme de Grotus
Transition : on a vu que les ions se déplacent avec une valeur de mobilité différente, on peut donc les
séparer !
b. Application en biologie
Chimie petit dej p. 71
- 3 aa fournis- On les spot sur du papier Whatman préalablement imbibé d’une solution conductrice. (3 capillaires !)- Mettre des gants pour éviter de mettre des aa- On place le papier dans une cuve à électrophorèse
2h de migration
5 mA, 200 V
Pas de hotte sauf pour la révélation à la ninhydrine (il faut y aller !) et sécher au sèche-cheveux pendant 3 min très proche
* Principe :
Méthode de séparation des constituants d’un mélange en solution ; elle repose sur leur différence de vitesse de migration lorsqu’ils sont placés dans un champ électrique. Cette méthode s’applique aux espèces chargées. Selon le pH, les acides aminés sont porteurs de charge différente sous forme d’anions, de cations ou de zwittérions. Sous l’effet d’un champ électrique, ils migrent donc de façon différente puisque le sens et l’intensité de la force électrique de migration dépendent de leur charge. A pH égal au point isoélectrique, il n’y a pas de migration électrophorétique car la molécule est sous forme zwittérionique, globalement neutre.La vitesse de migration est liée à la mobilité u de chaque ion et aux interactions (adsorption) avec la phase stationnaire. Par ailleurs, le déplacement d est proportionnel à la différence de potentiel appliquée et au temps de migration : d = u*E*∆t
* Expérience :
Dépôt de spot sur papier humidifié d’une solution tampon de phosphate pour maintenir le pH et assurer le contact électrique. Il faut que le papier reste humide ! sinon plus de contact électrique.La solution électrolytique fixe le Ph. Il faut choisir un pH pour lequel les charges des différents constituants à séparer sont les plus différentes possible. La solution électrolytique fixe la force ionique. Plus elle est grande, plus faibles sont le champ électrique et la mobilité des ions, la migration électrophorétique est donc ralentie. En même temps les ions de la solution sont nécessaires pour véhiculer le courant. Un compromis doit donc être réalisé.
* Application :
Séquençage de l’ADN
Transition : A travers ces expériences on voit que les ions sont les porteurs de charge qui permettent
de boucler le circuit électrique, notamment dans l’électrophorèse, on a utilisé une solution d’ions
phosphates. Dans une solution, ils permettent donc le passage du courant électrique.

II. Conductivité des électrolytes
a. Electrolyte fort : loi de Kohlrausch
Daumarie p. 125
Vérifier la loi de KohlrauschMesure de conductivité après chaque ajout de KNO3 dans 100mL d’eau distillée.Tracer : conductivité=f(√[KNO3])Bien étalonner et thermostater
Conductimétrie,Chute de burette
Pas de hotte
(Atkins)* Principe
On va ici s’intéresser donc à la conduction électrique dans les solutions, assurée par les ions. Ces ions peuvent être issus de la dissolution d’un sel, appelé électrolyte. On s’intéressera d’abord aux électrolytes forts.
Ces ions, on l’a vu, permettent de conduire le courant de par leur mobilité ! On définit donc une grandeur pour caractériser cette propriété : Conductivité molaire ionique : = z*u*F en S.m²/mol et vaut en général 10 mS.m²/mol. �
Pour un électrolyte : = � � �i. Pour notre électrolyte, on a = ( (K� � +) + (NO� 3-)).
Cette valeur est donc caractéristique de la conductivité de l’électrolyte. Cependant elle varie avec la concentration. En effet, plus il y a d’ions, plus il y a d’interaction entre eux, freinant la mobilité. On va vérifier ici la loi de Kohlrausch pour les électrolytes forts : = ° - A√C� � . A ne dépend que de la stœchiométrie de l’électrolyte.
Pour avoir accès à cette valeur, on va donc mesurer la conductivité de la solution : = *C et ainsi on� � va pouvoir avec .�
* Principe du conductimètre
Voir FossetConductance : inverse de la résistance en SiemensConductivité : = G*l/S en Siemens/m. �
* Exploitation
On trace /C = f(√C) et on a la loi.�
Transition : Pour les électrolytes faibles, = *C* car la concentration d’ions est C* . Or � � � � �
dépend de C ! On a plus de loi linéaire mais la loi d’Ostwald.

.b Electrolyte faible : loi d’Ostwald
Fosset p. 53 L’étude de la conductivité de solutions aqueuses d’un acide faible en fonction de sa concentration permet de déterminer la constante de dissociation de cet acide.
Conductimétrie Pas de hotte
(Atkins)* Exploitation
Loi d’Ostwald : = °.� � �
Avec l’écriture du Ka, on a 1/ = 1/� ° + C/(Ka* ° ²)� � �
* Applications : On peut en déduire le pKa. Dans les deux cas, on voit que la conductivité de la solution d’électrolyte dépend de sa concentration. On peut donc effectuer des dosages (parfois plus précis que le pH-mètre).
Transition : On voit que la mesure de conductimétrie nous apporte des infos sur le système comme ici
le pKa ou encore une concentration et on peut en tirer d’autres grandeurs comme la CMC
c. Application : détermination d’une CMC
Fosset p. 390 - on prépare des échantillons de concentration en SDS différents- on mesure la conductance- on trace G=f(c) et la rupture de pente indique la CMCTHERMOSTATE
conductimétrie Pas de hotte
* Explication
SDS possède une longue chaîne hydrophobe. Il a donc tendance à former des micelles, mais pour cela il doit y avoir assez de molécules, d’où une CMCTaille de la micelle pas très bien définie mais grande ce qui implique une mobilité faible donc une faible conductivité. On repère donc CMC par brusque variation de conductivité.Rq : ici l’étalonnage ne sert pas, car on étudie une variation de conductivité. Conductivité varie avec T et CMC varie avec T, donc thermostaté
* Application
Utilisation en tant que surfactant, il est inutile de mettre plus de surfactant une fois que l’on a atteint la CMC.

Conclusion : Lors d’expérience en électrochimie ou on veut s’affranchir du courant de migration on utilise un électrolyte support fort.

MG 14
Exemples de dosages des ions métalliques en solution.
Introduction
Cation de différents métaux. En solution. Métaux lourds toxique, ou métaux essentiels à la vie on veut connaître leur concentration en solution.Dosage : détermination de la concentration d’une espèce en solution. Courbe d’étalonnageTitrage : détermination de la concentration d’une espèce en solution en faisant réagir l’espèce à doser. Réaction quantitative ! Totale ! et Rapide !
I. Titrage d’ion métalliques
a. Dosage colorimétrique
Olympiades de chimie, p36-38, Ecolochimie p 305-308Des
expériences
dans la famille
des A-B p.253
-Réaliser un dosage complexométrique- étalonner une solution d’EDTA- dosage en présence de NET- dosage en milieu ph 12
Dosage colorimétrique
1h - Hotte
Transition : les ions peuvent également transporter du courant…
b. Dosage conductimètrique
JFLM p.109 Al3+ Dosage conductimètre(à adapter)
Transition : On remarque qu’on a utilisé leur couleur, en effet souvent coloré !

II. Dosage d’ion métalliques
a. Dosage spectrophotométrique
JFLM p.135 Dosage du fer dans le vin blanc grâce à une échelle de teinte.
UV Pas de hotte
Transition : Pour un mélange d’ions, une méthode très utile est la polaro…
b. Méthode des ajouts dosés polarographie
Prichard p. 170
Méthodes des ajouts dosés Polaro Pas de hotte
Ou si un problème avec polaro, dosage potentiométrique de Fe par Ce (Sarrazin p. 87)
Conclusion : Diverses méthodes de dosage pour connaître la concentration en un ion métalliques. Il en existe beaucoup d’autres mises au point pour chaque situation.

MG 15
Complexation : applications aux dosages et aux extractions.
Introduction : Définition d’un complexe : entité moléculaire formée par l’association de deux ou de plusieurs entités moléculaires, ioniques ou neutres. Les énergies de liaisons sont en général de l’ordre de la centaine de kilojoule par mole à comparer avec les énergies de liaisons covalentes qui sont de l’ordre de 300 à 400 kJ par mole.Grâce à leurs nombreuses propriétés, les réactions de complexation vont être très intéressantes pour réaliser des dosages et des extractions.Définition d’un dosage : on détermine la quantité de matière de la substance d’intérêt. On verra que les complexes peuvent alors agir dans la réaction de dosage, comme c’est les cas dans la détermination de la dureté d’une eau car il est très important de contrôler la concentration en ion métalliques dans l’eau commerciale ou l’eau des canalisations, ou encore en tant qu’indicateurs colorés, ce qui sera mis en évidence dans le dosage de l’acide ascorbique dans un comprimé de vitamine C afin de vérifier la teneur en médicament des espèces actives.On pourra ensuite s’intéresser à l’utilité des complexes lors d’extractions, c’est-à-dire lorsque l’on souhaite faire passer un composé d’une phase à une autre. On réalisera une extraction liquide liquide des ions cuivre II d’une phase aqueuse à une phase organique en présence d’un ligand, suivie d’une extraction d’intérêt industriel, l’extraction solide liquide de l’aluminium contenu dans la bauxite
I. La complexation appliquée aux dosages
Les dosages ont de nombreuses applications dans la vie quotidienne, et permettent de vérifier par exemple qu’une eau soit conforme à la législation.
a. Dosage colorimétrique : détermination de la dureté de l’eau
Olympiades de chimie, p36-38, Ecolochimie p 305-308Des
expériences
dans la famille
des A-B p.253
-Réaliser un dosage complexométrique- étalonner une solution d’EDTA- dosage en présence de NET- dosage en milieu ph 12
Dosage colorimétrique
1h - Hotte
On se propose ici de doser la dureté d’une eau Contrex, et de comparer le résultat aux valeurs indiquées sur l’étiquette. La dureté de l’eau (titre hydrotimétrique de l’eau) est due dans la plupart des cas aux ions Ca2+ et Mg2+ .Sans être nocive pour l’homme une eau dure présente des inconvénients, surtout reliés au dépôt de calcaire dans les conduits de certains appareils ménagés. Rq : CaSO4 : solubilité diminue quand température augmente….Titre hydrométrique français : 1°f = 0,1 mmol/L de cation métalliques

Le titre prend en compte tous les ions métalliques.Les ions Mg et Ca sont déjà sous forme de complexes dans l’eau entouré de 6 H2O, aquacomplexe.
On réalise le dosage du calcium et du magnésium par le EDTA(avoir prévu 2 tubes à essai pour avoir la coloration de référence). Le NET complexe Mg et Ca !!! constantes d’équilibre proches. Vérifier avec papier pHEn préparation on a réalisé le dosage des ions calciums par le Patton en faisant précipiter le magnésium (II), essayer pH=11 (10 mL de soude à 5 M)On en déduit la concentration des ions magnésiums. Comparaison avec la valeur indiquée sur l’étiquette.Contrex : 148 TH !! eau très dure !!! Normale, les eaux minérales sont très riches en cations métalliques (d’où il est conseillé de boire de l’eau minérale……)
Problème : On n’a pas Y4-, l’indicateur coloré est pas efficace car pas assez de différence de stabilité entre les complexes entre l’EDTA et le NET. D’ailleurs la couleur revient au rose après un certain temps : réaction réversible. D’où également l’intérêt de mettre très peu d’indicateur coloré !!
Dans le calcul d’incertitude, prendre en compte la zone de virage !!!!!!!! C’est la plus grosse incertitude !!!!
Transition : On a dosé une espèce cationique au cours de cette première expérience grâce à la
formation de complexes métalliques, on va à présent s’intéresser au dosage d’une molécule organique
grâce à un autre type de complexe, un complexe d’intercalation.
b. Dosage pH-mètrique : dosage de l’acide borique
Fosset p. 73 La présence d’un complexant d’un acide modifie ses propriétés acido-basiques. L’étude de la courbe de dosage pH-mètrique permet d’évaluer la constante de formation du complexe
1h pH-mètrie
Transition : On a utilisé les complexes pour réaliser des réactions de dosage, en mettant à profit les
couleurs des complexes considérés ainsi que leur stabilité en phase aqueuse. Nous allons voir que
certains complexes métalliques sont plus solubles dans des phases organiques permettant l’extraction
de cation dans la phase organique.

II. Les complexes appliqués aux extractions
a. Extraction liquide liquide : extraction du cuivre II
Fosset p. 226 Une solution aqueuse contenant un ion métallique, un ligand ayant des propriétés acido-basiques et le complexe formé est en équilibre avec une phase organique. Le dosage par spectrophotométrie du complexe dans la phase organique en fonction du pH permet de déterminer la stoechiométrie et la stabilité de celui-ci.
2h UV Hotte
La première technique d’extraction que nous allons voir est basée sur la différence de stabilité d’un complexe de cuivre (II) en phase organique, le dichlorométhane, et en phase aqueuse. Le dosage par spectrophotométrie du complexe dans la phase aqueuse en fonction du pH donnera accès à la constante de partage entre les phases.
Le cuivre en phase aqueuse est présent sous forme d’aquacomplexe (6 H20). On met en présence une solution aqueuse contenant de ions Cu(II) sous forme d’aqua complexe et 2 mL de soude à 1 M et une phase organique contenant de ligand 8-hydroxyquinoléine dans du dichlorométhane. On agite. Le complexe formé entre la 8 hydroxyquinoléine et le cuivre est plus stable en phase organique. On extrait donc le cuivre de la phase aqueuse vers la phase organique. Prélever la phase organique et la phase aqueuse dans un erlenmeyer et un bécher.
L’efficacité de l’extraction est fonction du pH (cf la feuille). On prend le pH d’un pHmètre précédemment étalonné pour des pH acides. On mesure l’absorbance de la phase organique. On a fait des mesures à d’autres pH. On ajoutera le point sur la courbe. Visuellement, on peut comparer les couleurs des phases organiques et aqueuses à un pH de 1 puis à un pH de 2. On considère que l’extraction est totale, c’est-à-dire que tout le cuivre (II) est en phase organique, lorsque deux mesures d’absorbance à des pH sucessives sont invariantes. On reste dans un intervalle de pH acide, dans lequel le ligand reste sous une seule forme. De plus sa concentration est bien plus élevée que celles du cuivre. On trace log P en fonction du pH et on vérifie que l’on a bien une fonction croissante. Application en chimie organique lors d’une activation par transfert de phase.
En effet pour maintenir l’élecroneutralité, si un cation, grâce à une complexation, passe en phase organique, son anion suivra. On peut utiliser ceci lors de l’oxydation du styrène par le permangante en présence d’éther couronne (JFLM 2 : chimie organique p.39)
Transition : Nous avons vu que les complexes de cations métalliques permettaient de faire passer des
cations en solution aqueuse en phase organique. Nous avons également vu tout au long de ce montage
que les ions métalliques étaient présents dans l’eau sous forme d’aquacomplexe, nous allons voir
comment on peut utiliser cette propriété à des fins industrielles.

b. Extraction solide liquide : procédé Bayer
BUP 791 p. 38 Procédé industriel. On fabrique la bauxite de départ puis on extraie l’alumine.
1h+1h Filtration
On se propose d’extraire l’aluminium de la bauxite qui est composée essentiellement d’alumine et d’hématite. Ici, nous l’avons préparé dans le laboratoire, elle contient uniquement ces deux espèces. On broie finement la bauxite obtenue après passage à l’étuve, afin d’obtenir une poudre que l’on dilue dans de la soude. On se basant sur le diagramme de stabilité et d’existence d’espèces hydratées, on remarque que le fer précipitera sous forme de trihydroxyde de fer, ce qui donne la coloration rouge que l’on observe. L’aluminium, à un pH bien élevé pourra former un complexe et sera donc soluble dans la soude. On a réalisé une extraction de l’aluminium. On filtre pour séparer physiquement les deux phases. Vérifier avec papier pH Ce procédé est utilisé industriellement pour traiter la bauxite, minerais naturelle dont la couleur varie selon le pourcentage d’oxyde de fer. Il s’agit du procédé Bayer mis au point en 1894. On broie la bauxite, qui est ensuite mise en présence de soude. Les boues rouges sont éliminées et l’oxyde d’aluminium (alumine) subit un traitement électrochimique pour donner l’aluminiumOn met en évidence la présence d’oxyde d’aluminium est abaissant le pH de la solution afin d’obtenir le précipité blanc de trihydroxyde d’aluminium.
Conclusion ; En utilisant des propriétés fondamentales des complexes, telles que leur couleur et leur stabilité, on a pu réaliser des dosages et des extractions. Néanmoins le champ d’application des complexes est bien plus vaste. Ils peuvent être utilisé dans un but thérapeutique afin d’éliminer le plomb dans l’organisme en cas de saturnisme, sous forme de complexes dans les urines. Les propriétés des complexes sont également mises à profit dans l’imagerie médicale. Des complexes de gadolinium sont utilisés comme agents de contrastes, afin d’améliorer l’intensité du signal et sa qualité. La stabilité du complexe est alors fondamentale pour minimiser la toxicité in vivo.

Infos
Limitations du NET : ses solutions se décomposent lentement au cours du temps. Utilisation de la calamgite cf Skoog Historiquement, la dureté de l’eau était définie pour tenir compte de la capacité des cations présents dans l’eau à remplacer les ion sodium ou potassium des savons et à former des produits peu solubles. La plupart des cations de plusieurs charges possèdent cette propriété indésirable.Pour déterminer la concentration en ions calcium et en ions magnésium dans une eau minérale on utilise une réaction de complexation avec l’ion éthylènediaminetétraacétique (EDTA) que l’on note Y4-
(voir annexe)
Ca2+ (aq) + Y4-
(aq) = [CaY]2- (aq) constante de stabilité K1 = 4,0 . 1010 pKd = 10.6
Mg2+(aq ) + Y4-
(aq) = [MgY]2-(aq) constante de stabilité K2 = 5,0 .108 pKd = 8
- L’EDTA est un tétraacide noté H4Y . Les pKa successifs sont les suivants :H4Y / H3Y- pK1 = 2,1 H2Y2-/ HY3- pK3 = 6,2H3Y-/ H2Y2- pK2 = 2,8 HY3-/ Y4- pK4 = 10,3La base Y4- est capable de donner un complexe hexacoordiné avec les cations métalliques (chélate).Dans la pratique, on utilise le sel disodique de l’EDTA Na2H2Y H2Y2-
(aq) + Ca2+(aq) = [ CaY]2- (aq) + 2 H+
(aq) H2Y2-
(aq) + Mg2+(aq ) = [ MgY]2- (aq) + 2 H+
(aq)Le milieu doit être basique pour détruire les ions H+ formés et par suite déplacer l’équilibre vers la droite.On se placera à pH 10
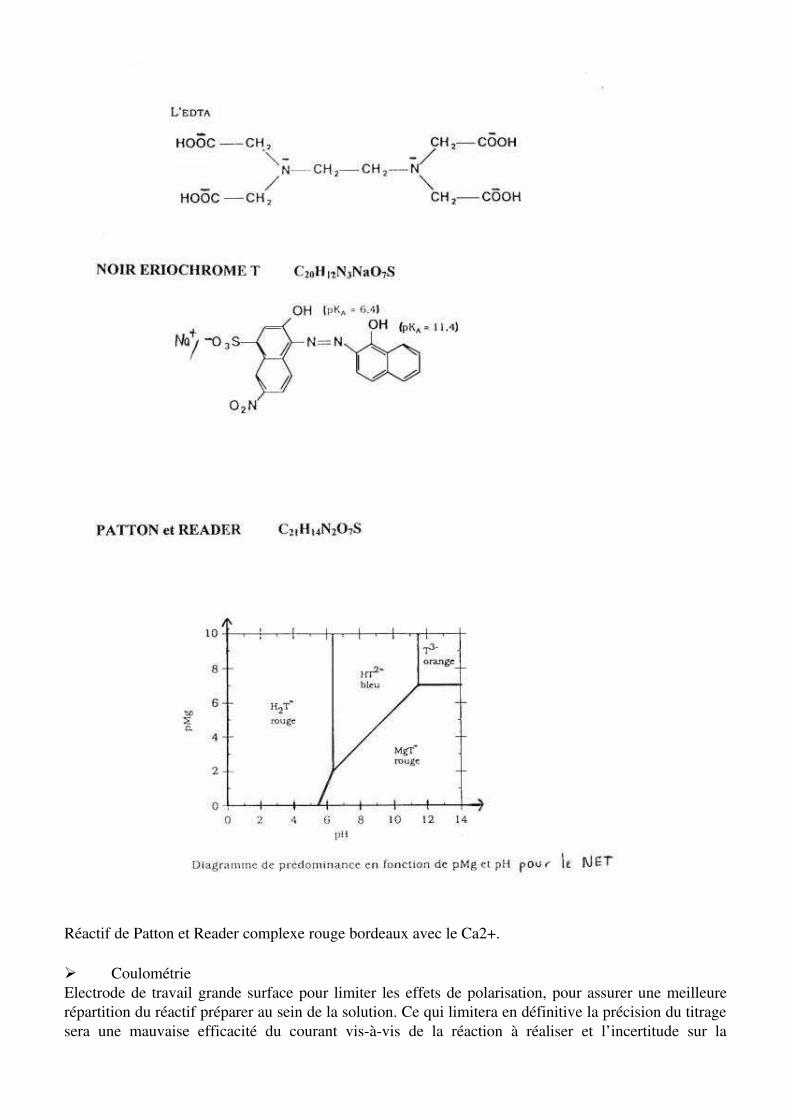
Réactif de Patton et Reader complexe rouge bordeaux avec le Ca2+.
� CoulométrieElectrode de travail grande surface pour limiter les effets de polarisation, pour assurer une meilleure répartition du réactif préparer au sein de la solution. Ce qui limitera en définitive la précision du titrage sera une mauvaise efficacité du courant vis-à-vis de la réaction à réaliser et l’incertitude sur la
localisation du point équivalent.

On va doser l’acide ascorbique de 20 mg du comprimé en effectuant une réaction d’oxydoréduction. L’acide ascorbique est oxydé en acide déshydroascorbique par du diiode produit électrochimiquement. On utilise comme technique la coulométrie pour avoir accès à la quantité d’acide ascorbique en fonction du temps et du courant. On déterminera l’équivalence grâce au complexe empois d’amidon I2.
Réaction aux électrodes : grande de platine pour générer du I2, on le voit avec l’indicateur coloré. Limite effets de polarisation, assure une meilleure répartition du réactifi au sein d la solution. De l’autre côté, on a une garde pour ne pas avoir la réduction de I2. On a un dégagement gazeux de H2. Le I2 qui est généré est utilisé pour oxyder l’acide ascorbique. Lorsque tout l’acide ascorbique est oxydé, on aura formation de complexe coloré. On a par ailleurs réalisé un blanc, afin de connaître le temps nécessaire à la coloration de la solution en l’absence de l’acide ascorbique.On prend le temps, on effectue le calcul + le calcul d’incertitude.
Il y a complexation des ions iodure I- avec I2 ce qui permet la solubilisation du diiode sous forme d'ions triiodures I3-. Dans cette représentation, le diiode est vu comme un acide de Lewis et l'ion iodure comme une base de Lewis.Diiode-empois d’amidon : complexe d’intercalation. L’amidon est formé d’une hélice constituée de molécule de glucose qui s’enchaîne. Le polyodure s’y insère, forme un complexe, et un transfert de charge permet de donner à ce complexe une couleur très intense.
//En dissolvant du diiode et de l'iodure de potassium dans l'eau on obtient de l'eau iodée. Cette solution s'appelle une solution de lugol.En dissolvant du diiode dans de l'éthanol, on obtient de la teinture d'iode qui est un antiseptique. Il permet en outre de mettre en évidence l'amidon. En effet, la teinture d'iode forme un complexe bleu foncé avec l'amidon. De façon générale, les solutions de diiode dans un solvant oxygéné sont brunes et celles dans un solvant non oxygéné sont de couleur violette.//
Empois d’amidon en grande quantité car I2 produit sur l’électrode met du temps à passer en solution (diffusion, agitation).
// La vitamine C intervient dans de nombreuses réactions d'oxydo-réduction dans l'organisme, dans le métabolisme du fer et des acides aminés, le maintien sous forme active de nombreuses enzymes. Elle est impliquée dans la lutte contre les radicaux libres oxygénés et favorise les défenses anti-bactériennes des globules blancs. La vitamine C est présente dans de nombreux aliments (fruits, légumes).Antioxydant dans les conservateurs alimentaires (E=0,36 V)Une vitamine est une substance organique nécessaire (en dose allant du microgramme à plusieurs milligrammes par jour) au métabolisme des organismes animaux et donc de l'homme. //
Problème : i varie, donc il faudrait intégrer (synchronie….). Si i augmente, t diminue et on a beaucoup d’imprécision sur le temps. Si i trop grand, oxydation du solvant !! Penser à plonger toute l’électrode et le diiode ne traverse pas le fritté pendant la durée de l’expérience.

MG 16
Indicateurs de fin de réaction : caractéristiques ; utilisations.
Biblio : Skoog, Tec& Doc
Introduction :
Nécessaire de savoir quand une réaction est finie de manière quantitative. Particulièrement pour dosage
ou on veut savoir précisément quand la réaction est finie. Proportions stoechiométriques ainsi
déterminées -> quantité de matière.
Nécessité d’un indicateur de fin de réaction. On va voir les différents types d’indicateurs dans ce
montage : indicateurs colorés (historiquement, premiers indicateurs utilisés) et d’autres techniques
comme saut de pH, potentiel. On va comparer ces techniques : avantage, inconvénients, précisions,…
I. Indicateurs de fin de réaction acido/basique
Réaction de titrage acido-basique implique un saut de pH, un changement important de pH. Donc un
indicateur qui conviendrait serait par exemple un indicateur dont l’acide et la base ont des couleurs
différentes. Le pKa doit se situer autour du saut de pH.
On veut utiliser BBP comme indicateur de fin de réaction : nécessité de connaître son pKa car c’est lui
va définir dans quelles réactions on va pouvoir l’utiliser.
a. Détermination du pKa de l’indicateur
Daumarie p.
111
Déterminer une grandeur
thermodynamique par UV
Existence d’un point isobestique
1h30 UV
Autres : JFLM p 143 (info théoriques, [c] pas bonnes)
Description
Mesure pKa du BBP par spectro UV en faisant une échelle de teinte par mélanges de la forme acide
(jaune) et basique (violet) du BBP. Chaque solution ayant une concentration totale en indicateur coloré
constante. On mesure pH de chaque teinte et on fait spectre UV.
Commentaire
Zone de virage à pKa +- 1 (Skoog p. 191, T&D). En dehors de cette zone, l’œil n’est pas sensible au
changement de couleur du mélange.
Transition : On aimerait utiliser BBP pour doser une solution d’acide phosporique. On va voir si cet
indicateur convient.

b. Application au dosage de l’acide phosphorique
Souil capes p.
124
Dosage de la première acidité de
l’acide orthophosphorique utilisant
comme indicateur le BBP
1h pH-mètrie et
colorimétrie
Acide phosphorique a trois acidités qu’on dose séparément car la différence de pKa > 4 (2,12 ; 7,21 ;
12,67). Première acidité moyenne car absence de point d’inflexion. Troisième acidité pas mesurable
(trop faible).
Comment choisir l’indicateur ? (Tec & Doc)
Zone de virage doit être autour de la zone du saut de pH.
Première idée de la zone de virage : pH(équivalence) = (pKa1+pKa2)/2 = 4,6 ;
Indicateur coloré :
Peu précis car si zone de virage un peu décalée, on a début de virage avant équivalence et pareil si le
saut de pH est pas très vertical. Calcul d’incertitude.
Mais très pratique, rapide
Saut de pH :
Demande plus d’appareillage
Mais plus précis (méthode des tangentes, dérivée, cercle osculateur,…) Calcul d’incertitude.
Transition : Un indicateur peut changer de couleur en fonction du potentiel !
II. Indicateur de fin de réaction rédox
a. Détermination du potentiel standard de l’o-phen ferreuse
Sarrazin p. 87 Titrage potentiométrique
des ions Fe par Ce avec
indicateur coloré, l’o-
phénantroline ;
Pas de pipette à double
trait ! solutions trop
colorées !
Potentiomé
trie
-
On détermine le potentiel standard et la stoechiométrie du complexe.
Zone de virage : E +- = E° +- 0,06/n. (Skoog)
Transition : On applique…

b. Application au dosage de Fe par Ce
Sarrazin p. 87 Titrage potentiométrique
des ions Fe par Ce avec
indicateur coloré, l’o-
phénantroline ;
Pas de pipette à double
trait ! solutions trop
colorées !
Potentiométrie
-
Comment choisir l’indicateur ?
Zone de virage doit être autour de la zone du saut de potentiel.
Indicateur coloré :
Peu précis car si zone de virage un peu décalée, on a début de virage avant équivalence et pareil si le
saut de potentiel est pas très vertical. Calcul d’incertitude.
Mais très pratique, rapide
Saut de pH :
Demande plus d’appareillage
Mais plus précis (méthode des tangentes, dérivée, cercle osculateur,…) Calcul d’incertitude.
Transition : Un indicateur peut changer de couleur en se complexant !!
III. Indicateur de fin de réaction de complexation
Ecolochimie p
305-308
Cachau
acide/base
p.253
JFLM
Skoog 299
- Réaliser un dosage
complexométrique
- étalonner une solution d’EDTA
- dosage en présence de NET
- dosage en milieu pH 12 ; faire le
calcul de précipitation.
Dosage
colorimétrique
1h - Hotte
Compétition de complexation entre le réactif titrant et l’indicateur coloré.
La plus grosse incertitude est sur la lecture du volume équivalent ! Indicateur coloré pas bien adapté.
Conclusion
Autres méthodes : Suivi photométrique, CCM, …

Dureté de l’eau
Historiquement, la dureté de l’eau était définie pour tenir compte de la capacité des cations présents
dans l’eau à remplacer les ions sodium ou potassium des savons et à former ainsi des produits peu
solubles. La plupart des cations porteurs de plusieurs charges possèdent cette propriété indésirable.
Cependant, dans les eaux naturelles, les concentrations en ions calcium et magnésium dépassent
généralement de loin celle de tout autre ion métallique. C’est pourquoion exprime à présent la dureté en
termes de la concentration hypothétique en carbonate de calcium qui serait équivalente à la
concentration totale de tous les caitons polyvalents présents dans l’échantillon.
La détermination de la dureté est un test analytique fréquemment utilisé pour évaluer la qualité de l’eau
affectée à des usages ménagers ou industriels. Ce test est d’importance, car le chauffage des eaux dures
cause la précipitation du carbonate de calcium, ce qui entraîne l’obstruction des chaudières et des
tuyauteries.
On détermine habituellement la dureté de l’eau en titrant par l’EDTA un échantillon tamponné à pH 10.
Le magnésium forme avec l’EDTA le complexe le moins stable de tous les cations polyvalents présents
habituellement dans les échantillons d’eau : il n’est donc pas titré aussi longtemps qu’on n’a pas ajouté
assez de réactifs pour complexer tous les autres cations de l’échantillon. Dès lors, un indicateur d’ion
magnésium, tel que la calmagite ou le noir d’érichrome T, peut servir d’indicateur pour évaluer la
dureté de l’eau. On ajoute souvent une petite quantité du complexe EDTA-magnésium dans le tampn
ou dans le titrant pour qu’il y ait assez d’ions magnésium pour que l’indicateur fonctionne
correctement.
Trousses pour le dosage de la dureté de l’eau
Des trousses permettant la détermination de la dureté de l’eau de distribution sont disponibles chez les
distributeurs d’adoucisseurs d’eau et d’accessoires de plomberie. Elles contiennent habituellement un
récipient calibré pour contenir un volume connu d’eau, une mesurette permettant de prélever la quantité
appropriée d’un mélange tampon en poudre, une solution d’indicateur et un flacon de solution étalon
d’EDTA muni d’un compte-gouttes. Il suffit de compter le nombre de gouttes de réactif étalon
nécessaires pour entraîner le virage de l’indicateur. La concentration de la solution d’EDTA est
généralement telle qu’une goutte correspond à un degré hydrotimétrique français, càd 10-4 mol d’ion
calcium par litre d’eau.

MG 17
Solubilité et produit de solubilité : étude et applications.
Biblio : HP PCSI II
Introduction :
Définition de Ks et de s. On dit qu’on va se limité à la solubilité des solides mais que les gaz aussi on
une solubilité. Important à connaître car paramètre à maîtriser pour solubiliser un produit pour le faire
réagir ou au contraire pour le faire précipiter pour l’éliminer.
D’après la définition, on fait intervenir le solvant. Voyons tout de suite son influence.
I. Influence de divers facteurs sur s et Ks
a. Influence du solvant
Daumarie p.
131
Etudier la solubilité de NaCl dans
l’eau et dans l’éthanol à l’aide du
dosage des ions chlorure par les
ions argent (I). Appliquer la
conclusion de cette expérience à
la purification du sel gris de
Guérande.
2h Dosage
potentiométrique
On dose les ions en solutions par AgNO3 avec électrode d’argent.
Eau étant plus dissociante, elle solubilise mieux que l’éthanol.
Transition : Le Ks est une constante d’équilibre, elle dépend donc de la température…
b. Influence de la température
Fosset p. 106 La mesure de la solubilité d’un
composé à différentes températures
permet de déterminer l’enthalpie et
l’entropie de dissolution de celui-
ci.
Prélèvement de la solution :
filtration dans un bécher
thermostaté à la même température
que le bain.
2h Dosage
colorimétrique
On a Ks = [PhCOOH]. On dose l’acide benzoïque restant dans l’eau. On trace ln Ks = f(T). On en
déduit l’enthalpie et l’entropie de dissolution.

Transition : On voit ici que le Ks dépend de la forme protonée de l’acide et donc du pH.
c. Influence du pH
BUP 791 p. 38 Procédé industriel. On fabrique la
bauxite de départ puis on extraie
l’alumine.
Diagramme HP PCSI II
1h+1h Filtration
On fait solubilise sélectivement les ions Al dans une solution basique, puis on diminue le pH pour faire
précipiter ces ions.
Transition : Ce procédé est un procédé industriel…
II. Applications
a. Séparation de Al/Fe : procédé Bayer
BUP 791 p. 38 Procédé industriel. On fabrique la
bauxite de départ puis on extraie
l’alumine.
1h+1h Filtration
Grâce à ce procédé Bayer, on sépare les ions Fe et Al présents dans la bauxite.
Transition : On utilise également le phénomène de solubilité en chimie organique pour purifier des
produits.
b. Recristallisation d’une chalcone
JD n°90 solide - mélange intime des
deux solides
- lavage
Point de fusion,
IR, CCM
1h - Hotte
Principe d’une recristallisation.
Conclusion
En industrie c’est très important de connaître tout les pKs car on va s’en servir pour faire précipiter
sélectivement les impuretés comme par exemple pour la purif du Zn

Hydrométallurgie du zinc
BUP 770
p.111
Lixiviation : élimination des impuretés
sous formes de précipité
filtration 3h - Pas de
hotte
On se place en milieu acide et hop, plein de précipité sauf Zn.
Influence de la force ionique
Daumarie p.
136
On dose par conductimétrie une solution
de PbSO4 saturée avec concentration
différente en KNO3
conductimétr
ie
3h - Pas de
hotte
Wikipedia
Les sucres des bonbons, des boissons sucrées et même des jus de fruits jouent un rôle important dans la
carie dentaire et par conséquent dans la destruction de l'émail. La bouche contient un grand nombre et
une grande variété de bactéries, et quand le saccharose, le plus commun des sucres, couvre la surface
de la dent, certaines bactéries buccales interagissent avec lui pour former de l'acide lactique, qui
diminue le pH dans la bouche. Les cristaux d'hydroxyapatite de l'émail sont alors déminéralisés,
permettant une invasion bactérienne plus importante et plus en profondeur dans la dent. La bactérie la
plus impliquée dans la carie dentaire est Streptococcus mutans, mais le nombre et les espèces de
bactéries varient en fonction de la progression de la destruction dentaire.

MG 18
Facteurs influençant les équilibres hétérogènes ; dissolution et
partage ; applications
Introduction :
Définition équilibres hétérogènes. Les équilibres hétérogènes correspondent aux équilibres existant
entre plusieurs phases. On va s’intéresser à liquide/solide (dissolution) et liquide/liquide (partage).
Au cours de ce montage, on étudiera l’influence de différent facteur sur un exemple (mais qui
s’appliquera à d’autres équilibres) et comment on peut les appliquer.
L’équilibre donné par : K(T) = a� i avec ai = 1 pour les solides, ai = �ici pour les solutés.
Donc on peut jouer sur la température, sur les forces ioniques, les concentrations via d’autres
équilibres.
I. Facteur influençant la valeur de la constante d’équilibre
Influence de la température : application à la recristallisation
JD n°90
Dissolution
solide - mélange intime des
deux solides
- lavage
Point de fusion,
IR, CCM
1h - Hotte
Principe d’une recristallisation, prouvée par les CCM et points de fusion.
II. Facteurs influençant les paramètres de la constante d’équilibre
a. Influence de la force ionique
Daumarie p.
136
Dissolution
On dose par conductimètre une solution de
CaSO4 saturée avec concentration
différente en KNO3.
Prendre PbSO4
conductimètr
e
3h - Pas de
hotte
On détermine le Ks pour différentes forces ioniques.
b. Influence des équilibres acido-basique : application à l’extraction
Daumarie p.
142
Partage
Indicateur coloré, partage entre l’eau
et le choroforme dépend du pH
Ampoule
à
décanter
Principe d’une extraction.

Application : chimie organique. Par exemple pour enlever un acide, on fait un lavage en milieu
basique pour le déprotonner et le faire passer en phase aqueuse.

c. Influence des équilibres de complexation
PrincipeFosset p. 226
Partage
Une solution aqueuse contenant un
ion métallique, un ligand ayant des
propriétés acido-basiques et le
complexe formé est en équilibre avec
une phase organique. Le dosage par
spectrophotométrie du complexe dans
la phase organique en fonction du pH
permet de déterminer la
stoechiométrie et la stabilité de celui-
ci.
2h UV Hotte
La présence d’un ligand permet de faire passer un ion en phase organique.
ApplicationBUP 791 p. 38 Procédé industriel. On fabrique la
bauxite de départ puis on extraie
l’alumine.
1h+1h Filtration
On se propose d’extraire l’aluminium de la bauxite qui est composée essentiellement d’alumine et
d’hématite. Ici, nous l’avons préparé dans le laboratoire, elle contient uniquement ces deux espèces. On
broie finement la bauxite obtenue après passage à l’étuve, afin d’obtenir une poudre que l’on dilue dans
de la soude. On se basant sur le diagramme de stabilité et d’existence d’espèces hydratées, on remarque
que le fer précipitera sous forme de trihydroxyde de fer, ce qui donne la coloration rouge que l’on
observe. L’aluminium, à un pH bien élevé pourra former un complexe et sera donc soluble dans la
soude. On a réalisé une extraction de l’aluminium. On filtre pour séparer physiquement les deux
phases.
Vérifier avec papier pH
Ce procédé est utilisé industriellement pour traiter la bauxite, minerais naturelle dont la couleur varie
selon le pourcentage d’oxyde de fer. Il s’agit du procédé Bayer mis au point en 1894. On broie la
bauxite, qui est ensuite mise en présence de soude. Les boues rouges sont éliminées et l’oxyde
d’aluminium (alumine) subit un traitement électrochimique pour donner l’aluminium
On met en évidence la présence d’oxyde d’aluminium est abaissant le pH de la solution afin d’obtenir
le précipité blanc de trihydroxyde d’aluminium.
Conclusion
Deux équilibres très utilisés. Par exemple, hydrométallurgie du zinc ou HPLC.
On peut remplacer Bayer par Hydrométallurgie du zinc
BUP 770 p.111
+ livres
Terminale S
Lixiviation : élimination des impuretés
sous formes de précipité
filtration 3h - Pas de
hotte
On se place en milieu acide et hop, plein de précipité sauf Zn.

MG 19
Méthodes de séparation de constituants d’un mélange homogène
ou d’une solution.
Introduction :
Définitions mélange homogène, solution.
A l’issue des réactions de synthèse d’une molécule, on obtient généralement un mélange de
constituants, comprenant par exemple le produit désirés et des produits secondaires des réactifs n’ayant
pas réagi. Le but est d’isoler le produit. Pour cela différentes méthodes de séparations s’offrent aux
chimistes.
I. Séparation par changement de phase
a. Extraction liquide/liquide
Florilège p.
149
Création d’un mélange de 4
composés. Extraction puis
distilations. Etapes de purification si
il y a le temps
long
Transition : Il reste encore d’autres impuretés qui ont des affinités trop similaires pour les différentes
phases liquides pour les séparer ! On continue avec distillation.
b. Distillation
Florilège p.
149
Création d’un mélange de 4
composés. Extraction puis
distillations. Etapes de purification si
il y a le temps
long
Transition : Cependant, des composés peuvent avoir des températures d’ébullition trop proches ou
trop élevées. Il faut jouer sur autre chose, la chromato, différence d’affinité pour phases stationnaires.
II. Séparation par chromotagraphie
Chimie du petit déjeuner p
271
Florilège de chimie
pratique p. 159
solide - piler les épinards
- extraire au DCM,
évaporer
- colonne d’alumine
- évaporer
UV,
CCM
2h - Hotte

Solvants : 90/10 ether de
pétrole AcOEt, 90/10
DCM, MeOH
Illustration pédagogique du principe de chromatographie.
Transition : On a joué sur la gravité pour séparer les produits, s’ils sont chargés, on peut jouer sur
leur charge.
III. Séparation par électrophorèse
Chimie petit
def p. 71
BUP 664
- 3 aa fournis
- On les spotte sur du papier
Whatman préalablement imbibé
d’une solution conductrice. (3
capillaires !)
- Mettre des gants pour éviter de
mettre des aa
- On place le papier dans une cuve à
électrophorèse
2h de
migration
5 mA
Pas de hotte sauf
pour la révélation à
la ninhydrine (il
faut y aller !) et
sécher au sèche-
cheveux pendant 3
min très proche
Séquençage de protéine
Conclusion :
En industrie, distillation pour séparer coupes de pétrôle, extraction par procédé Bayer,…

MG 20
Chromatographies. Principes physicochimiques de la
chromatographie. Applications.
Biblio : Skoog, Chavanne
Introduction :
Définitions chromatographie. A l’issue des réactions de synthèse d’une molécule, on obtient
généralement un mélange de constituants, comprenant par exemple le produit désirés et des produits
secondaires des réactifs n’ayant pas réagi. Le but est de pourvoir évaluer la composition du mélange
qualitativement et quantitativement, et de procéder à la séparation des produits. La technique de
chromatographie intéressante. Définition de la chromatographie. 4 types que nous allons détaillé.
Distinguer les chromatographie préparatives et analytiques.
I. Chromatographie d’adsorption
a. Chromatographie sur couche mince
Chimie du petit déjeuner p
271
Florilège de chimie
pratique p. 159
Solvants : 90/10 ether de
pétrole AcOEt, 90/10
DCM, MeOH
solide - piler les épinards
- extraire au DCM,
évaporer
- colonne d’alumine
- évaporer
UV,
CCM
2h - Hotte
Principe de la CCM.
Transition : Maintenant, on va pouvoir l’utiliser pour faire la colonne.
b. Chromatographie sur colonne
Chimie du petit déjeuner p
271
Florilège de chimie
pratique p. 159
Solvants : 90/10 ether de
pétrole AcOEt, 90/10
DCM, MeOH
solide - piler les épinards
- extraire au DCM,
évaporer
- colonne d’alumine
- évaporer
UV,
CCM
2h - Hotte

Principe de la colonne
Transition : Autres techniques d’analyses.
II. Chromatographie de partage
Chromatographie en phase vapeur
Blanchard p.135
Vernin p.125
Brut réactionnel
Ajout de 1ml au lieu de
10 gouttes de toluène
Utiliser un étalon : le
nonane, on peut injecter o
et p ensemble
- extraction
- lavage
CPV 2h30 - Hotte
Chromatographie sur papier (si problème)
Chimie du petit
déj p. 66
Séparation d’acide aminé
sur papier
Chromato
sur papier
2h -
Transition : On s’est intéressé à des composés neutres, mais on peut également séparer les ions avec
une chromato toute adaptée.
III. Chromatographie échangeuse d’ions
Chimie tout p.
97
On sépare Co et Ni 2h
Principe. Domaines d’utilisation : purification de l’eau. Par ex les filtre Britta, retiennent les métaux.
Transition : On a joué sur les charges, on peut aussi jouer sur la taille.
IV. Chromatographie d’exclusion stérique
Feneuil p. 113
Interdit à
l’agreg,
apprendre par
cœur !
Séparation des constituants du lait
(caséïne, lactase) Tamponner à pH =
8,9. Elution longue.
2h

Principe
Conclusion :
Reste la chromatographie d’exclusion stérique. Méthode de séparation très utile. Basées sur les
différences de propriétés chimiques et physiques des constituants. HPLC chromatographie très
performantes sépare des dia. Apparition de phase chirale pour séparer des énantiomères.

MG 21
Systèmes dispersés et systèmes micellaires : mise en évidence et
propriétés physicochimiques.
Biblio : Juste argile, Cabane
Introduction : Cabane
Nous connaissons les corps purs qui peuvent être sous plusieurs phases liquide, solide, gaz (phase =
état de la matière qui est uniforme dans sa composition chimique et dans sont état physique). Pour les
mélanges, on peut considérer des mélanges homogènes mais également hétérogènes qui sont très
importants ! On va s’intéresser à ces derniers, et plus particulièrement aux systèmes dispersés :
Systèmes dispersés = système dans lequel des particules sont dispersées dans une phase continue de
composition (ou d’état) différente. Phase dispersée et dispersante
Tableau de la Juste argile 88 + exemples
Dispersant\Dispersé g l s
g Aerosol liquide Aerosol solide
l Mousse Emulsion (colloïdale) Sol ou suspension
s Mousse solide Gel ou émulsion solide Sol solide ou
dispersions solides
Nous nous intéressons ici aux systèmes dispersés colloïdaux, c’est-à-dire des systèmes dispersés dans
lesquels les particules sont de tailles inférieures à 1µm. En effet, nous verrons que le contrôle de leurs
propriétés passe par la physico-chimie alors que pour les particules plus grosses, cela révèle plutôt du
génie des procédés.
Il ressort de la définition des dispersions que celles-ci ne peuvent pas être des états stables au sens
thermodynamique. En effet, les particules dispersées ont la composition et la structure d’une phase à
l’équilibre. Idem pour dispersant. Ces phases sont séparées par des interfaces qui ont un coût en énergie
libre : en effet, les atomes, ions ou molécules disposés aux interfaces ont une coordination moins
favorable que ceux qui sont en volume. S’il n’en était pas ainsi, l’état d’équilibre du système serait une
solution, et les particules solides se dissoudraient spontanément. L’excès d’énergie libre des interfaces
est mesuré par leur tension interfaciale. Ces systèmes sont métastables.
Elle peut être diminuée par l’ajout d’agents tensioactifs (molécules amphiphiles) qui ont tendance à
former des agrégats de dimensions colloïdales et qui existent en équilibre avec les molécules ou les
ions à partir desquels ils sont formés. De tels agrégats se nomment micelles.
On a alors des systèmes micellaires.
Nous allons mettre en évidence ces systèmes, s’intéresser à leur propriétés et voir comment on peut
s’en servir.

I. Systèmes dispersés
a. Mise en évidence
* Formation d’un gel
summerlin p165 De l’éthanol dans l’acétate de calcium saturé. Formation d’un gel.
On rajoute HCl, dissolution du gel.
Interprétation
Gel = réseau qui piège le liquide. Ici, l’acétate de calcium qui forme un réseau qui piège l’éthanol.
Avec l’acide, on forme l’acide acétique, le réseau est brisé.
Application
On brûle l’éthanol emprisonné -> sert à allumer les fondues, (les barbecues) (Nom commercial :
Sterno)
Autre exemple de gel : jelly, gélatine, agar-agar.
Transition : ici dispersion d’un liquide dans un solide on peut disperser un gaz dans un liquide pour
former une mousse comme les œufs en neige.
* Formation d’une mousse
summerlin p167 Dans de l’eau, Al2(SO4)3 et NaHCO3. Formation d’une mousse
Interprétation
Al acide, libère H+ et donc HCO3 -> CO2, ca mousse !
Production d’une surface CO2-liquide grande. On verra plus tard comment stabiliser une mousse.
Application
Problème de tension superficielle et métastable.
Autre ex : bière, champagne et si plus dense : œuf en neige (on piège CO2). Or on sait que les œufs en
neige ont tendance à retomber ! en effet, on retrouve la métastabilité.
Transition : Ces systèmes ont des propriétés physiques particulières.

b. Propriétés optiques
Le coucher de soleil
Summerlin p.
163 ; JFLM
Montage : QI + cuve avec Na2S2O3 + écran (lentille ?)
Puis, on ajoute HCl, on agite vite et hop !
Discussion sur les propriétés optiques des systèmes dispersés.
Particules dispersées ont en général un indice de réfraction différent de celui du milieu de dispersion.
Pour des particules plus petites que la longueur d’onde de la lumière, cette diffusion est d’autant plus
importante que les particules sont grosses.
Diffusion de la lumière pour certaines longueurs d’onde selon la taille des particules.
2H+ + S2O32- -> H2S2O3
H2S2O3 -> H2SO3 + soufre colloïdal
Le soufre colloïdal diffuse la lumière et produit différente couleur suivant sa taille.
Plus le colloïde croît, plus la lumière est bloquée et différentes couleurs sont produites.
La concentration C du soufre en solution croît d’abord très rapidement dans le temps. Elle dépasse la
valeur C0 limite de solubilité dans le milieu des particules de soufre de taille macroscopique sans que le
solide précipite. Les premiers nuclei n’apparaissent qu’à la concentration Cs.
En effet, pour des systèmes très divisés, il ne devient plus possible de négliger la contribution de leur
surface dans l’énergie interne totale. En conséquence, quand le système est suffisamment divisé, toutes
les grandeurs thermodynamiques (solubilité, tension de vapeur,…) deviennent dépendantes de l’aire
des particules !! Donc Cs ≠ C0
Du moment que les nuclei sont formés, il y a alors croissance de ces nuclei tandis que l’apparition de
nouveaux germes continue. La concentration du soufre en solution continue d’augmenter mais pour peu
de temps car si la vitesse de production du soufre est sensiblement constante sa vitesse de condensation
n’arrête pas de croître par suite de l’augmentation du nombre de nuclei ainsi que de leur surface
(puisque leur taille augmente). En conséquence, la concentration de soufre en solution passe par un
maximum puis redevient inférieure à Cs et en conséquence la formation de nouveau nuclei est stoppée.
Ensuite, on observe seulement un grossissement des nuclei existants qui sont alimentés en soufre par
diffusion moléculaire du soufre en solution. Le système colloïdal est d’autant plus homogène en taille
que l’étape de nucléation est courte devant l’étape de grossissement.
Dans la nature, ce sont les particules de l’atmosphère qui diffusent la lumière.
Dans les peintures et dans tous les revêtements blancs ou colorés (papier), certaines particules servent
ainsi à renvoyer la lumière vers l’œil (dioxyde de titane, carbonate de calcium), d’autres à sélectionner
les longueurs d’onde renvoyées (pigments), et éventuellement à absorber les UV (dioxyde de titane,
oxyde de zinc). En mélangeant dans la même formule des particules de dioxyde de titane, de carbonate
de calcium et de pigments colorés, on met à profit une autre propriété des dispersions, qui est la
possibilité de mélanger à l’échelle colloïdale des composants qui ne sont pas miscibles à l’échelle
moléculaire.
Transition : les émulsions pas stables on peut cependant stabiliser certaines émulsions de différentes
façons.

II. Systèmes micellaires
a. Mise en évidence
Fosset p. 390 - on prépare des échantillons de
concentration en SDS différents
- on mesure la conductance
- on trace G=f(c) et la rupture de
pente indique la CMC
Détermination
de CMC par
conductimétrie
3h00 - Hotte
Il faut la cuve à ultra son, pour homogénéiser les solutions il ne faut pas que ça mousse de trop.
En effet, état de surface du bécher peut fausser le résultat.
Principe de la conductimètrie. Structures des tensioactifs. CMC = concentration en tensioactifs à partir
de laquelle se forment des micelles. Tensioactifs, d’abord à la surface d’abord puis forme des micelles
dans la solution.
Principe du savon. Peut aider à faire tenir des émulsions instables (moutarde dans la vinaigrette, œuf
dans la mayonnaise).
Transition : On s’en sert pour de nombreuses application notamment pour former de la silice
mésoporeuse ou bien pour la catalyse comme on peut le voir avec une catalyse micellaire
électrochimique en permettant la dissolution d’un composé.

b. Propriété physique : stabilité des microémulsions
Fosset p. 394 Pour certaines compositions de mélange quaternaire dodécylsulfate de
sodium/eau/toluène/butanol, on peut obtenir des systèmes monophasiques appelés
microémulsions. Un diagramme pseudo-ternaire mettant en évidence les limites du
domaine de stabilité des microémulsions peut être tracé.
Attention pour la solution D qui doit être une microémulsion :
(quantité divisée par 2) 57 g d’eau, 33 g butanol, 16,5 g SDS
* Expérience
On fait des mélanges eau/toluène et toluène/butanol/SDS
Après agitation, chacun de ses mélanges prend un aspect blanc laiteux (émulsion pas stable). On ajoute
lentement le mélange des quatre espèces jusqu’à disparition du blanc laiteux (microémulsion). On note
V le volume de ce changement. Remarque, dans tous les mélanges, la proportion de butanol par rapport
au SDS est constante. Donc on a un pseudo-ternaire : eau/toluène/(butanol+SDS).
On trace un diagramme pseudo-ternaire. On obtient le domaine de stabilité = domaine monophasique
(isotrope) appelé micro-émulsion
Sans SDS, métastable. Comment peut-on stabiliser ? Par ajout d’un tensioactif.
Le SDS permet de cacher l’eau et le toluène l’un à l’autre pour faire qqc de stable.
* Micro-émulsion
Dans le cas des microémulsions, on dissout dans un liquide des molécules amphiphiles et d’autres
molécules qui seront transportées dans les micelles. Cette solution est l’état d’équilibre du système, càd
que pour cette composition, le système n’abaisserait pas son énergie libre en se séparant en deux phases
macroscopiques. Le terme « microémulsion » employé dans ce cas prête à confusion, comme nous
l’avons déjà noté, puisque ces solutions micellaires sont formées d’objets beaucoup plus petits (moins
de 30 nm de diamètre) que les émulsions (y compris les nanoémulsions), et que ce sont des systèmes à
l’équilibre. Conditions très sévères d’obtention car il faut que l’amphiphile forme des structures
correspondant à la courbure spontanée des monocouches dans les conditions présentes de solvatation.
Différent d’une dispersion qui n’est pas à l’équilibre, car les deux phases ont une tension interfaciale, et
l’état d’équilibre correspond à une aire d’interface minimale, donc à la séparation des deux phases.
Remarque : Microémulsion : phase dispersée enrobée par tensioactif (de l’ordre du nanomètre,
n’absorbe pas dans le visible, donc paraît limpide). Stable thermodynamiquement (parfum que l’on
veut limpide,…)
Lait de beauté, pas microémulsion car on veut pas limpide !
Transition On rencontre une application de la stabilisation des émulsions dans la vie courante avec la
vinaigrette car l’huile et le vinaigre forment une émulsion avec une durée de vie très courte donc il
faut chercher à la stabiliser

c. Propriété chimique : catalyse micellaire
Fosset p. 343 La réaction du cristal violet avec l’ion
hydroxyde est suivie par spectrophotométrie.
L’introduction de bromure de
cétyltributylammonium (CTAB) se traduit par
une accélération de la réaction si la
concentration en CTAB est suffisante pour
produire des micelles en solution.
UV - Hotte
* Principe
Le cristal violet sous sa forme acide est violet et sous sa forme basique incolore. Donc en ajoutant de la
soude, on étude sa décoloration. En se plaçant à la longueur d’onde maximum d’absorption, on en
déduit l’évolution de la concentration (puisque le cristal violet est la seule espèce à absorber).
Si la réaction est une étape élémentaire, on doit avoir une vitesse du second ordre. Dans les conditions
expérimentales choisies, on constate qu’il y a dégénéresence de l’ordre par rapport à HO-. On a donc
une réaction du pseudo-premier ordre. -d[CVH+]/dt = k’[CVH+]
On trace –lnA = -lnA° + k’t
* Observation
Il a été démontré que le rôle du tensioactif (SDS ou CTAB) est limité à une modification de la vitesse
de réaction : le bilan de la réaction est inchangé.
On remarque que le CTAB catalyse la réaction et le SDS l’inhibe.
* Interprétation
La courbe donnant les variations de la constante de vitesse avec la concentration de CTAB nous permet
d’observer que l’activité catalytique du CTAB n’est sensible qu’à partir d’une certaine concentration.
Cette concentration limite correspond à la CMC et l’on peut donc admettre que l’activité catalytique du
CTAB est essentiellement liée à l’existence de micelles.
Le CTAB forme des micelles cationiques et le SDS des micelles anioniques. Le mécanisme suivant
peut alors expliquer les phénomènes observés. Le cristal violet est un ion possédant des noyaux
aromatiques qui le rendent soluble dans les milieux organiques. Il peut donc s’insérer facilement dans
la partie hydrophobe des micelles.
L’activité catalytique (ou inhibitrice) s’explique alors par des interactions électrostatiques : pour une
micelle cationique, comme pour le CTAB, il y a attraction électrostatique des ions hydroxyde, chargés
négativement, vers la micelle et donc vers le cristal violet dans la micelle. Au contraire pour une
micelle anionique, il y a répulsion entre la micelle et l’ion hydroxyde. Ce mécanisme de catalyse
permet également d’expliquer le rôle atténuateur du NaCl : celui-ci exerce un effet d’écran qui diminue
aussi bien les répulsions que les attractions. Il y a donc une diminution de l’activité catalytique du
CTAB et de l’activité inhibitrice du SDS.
Lorsque la concentration en tensioactifs augmente, l’activité catalytique ne continue pas à croître aussi
vite que précédemment. On peut attribuer cette diminution relative notamment aux changements des
tailles de micelles (le nombre de micelles augmente moins vite).

c. Application à la polymérisation (si cristal violet interdit)
BUP 830 p. 193 Le PEDOT, synthèse en milieux aqueux : à partir d’un monomère piégé dans
micelles de SDS (au-dessus de la cmc)
PEDOT s’oxyde puis se dimérise
Hauteur du pic augmente, polymère récupérable
Mettre en solution, puis volta-cyclique
Chimie verte dans l’eau : mais problème des TA
Le monomère est peu soluble dans l’eau, alors on utilise un tensioactif pour le stabiliser.
Conclusion
Cette brève revue suffit à faire comprendre pourquoi les propriétés des dispersions sont originales par
rapport à celles des liquides homogènes : elles ne sont pas déterminées principalement par la
composition globale, mais plutôt par l’état de division de la matière, càd par l’état d’agrégation des
particules, et par leurs interactions. Cela permet un contrôle efficace des propriétés par des actions
portant sur l’état de dispersion.
Les systèmes dispersés présentent donc une grande diversité d’applications. Dans l’industrie ils
répondent à un demande précise par exemple il faut avoir une microémulsion dans le lave-vaisselle
mais une mousse pour le shampoing !
Recherche : silices mésoporeuses

MG 22
Structures et propriétés physico-chimiques des complexes des
métaux de transition.
Biblio : Shriver, Huheey
Introduction :
Définitions complexes, métaux de transition.
Historique : découverte en 1913 par Werner.
Propriétés et structures intimement liés. On va donc au cours de ce montage discuter des différentes
propriétés que présentent les complexes et voir leur lien avec leur structure.
I. Propriétés physiques et structure
a. Propriétés optiques
Fosset p. 186 Détermination de delta et Td ou Oh.
Préparation de solution
Cobalt
2h UV
Utilisation des propriétés optiques. Tanabe-Sugano, Série spectrochimique.
Ligand à champ fort ou faible déterminent la structure du composé (géométrie et stoechiométrie).
Grâce aux propriétés optiques, on peut déterminer la structure d’un complexe.
Transition : Pas le seul moyen de prévoir structure du complexe. On a vu sur les diagrammes
énergétiques qui suivant la structure, électron célibataire ou pas…
b. Propriétés magnétiques
JCE 1980 p.
826 ; JCE
1980 p. 900 ;
BUP 2008 p.
41
Synthèses de plusieurs complexes,
puis études IR et UV. Interprétation
avec Tanabe. Balance de Gouy.
Montrer une filtration pour faire
qqc ?
Nickel
long UV, IR hotte
Champ fort, champ faible, Haut spin, Bas spin ; Principe de la balance de Gouy.
Transition : Les complexes de par le métal ainsi porteurs d’électrons peut participer à des réactions
d’oxydo-réduction.


II. Propriétés chimiques et structure
a. Propriétés oxydo-réductrices
Sarrazin p. 87 Titrage potentiométrique
des ions Fe par Ce avec
indicateur coloré, l’o-
phénantroline ;
Pas de pipette à double
trait ! solutions trop
colorées !
Potentiométrie
-
Propriétés oxydo-réductrices modifiées par la complexation.
Permet la détermination de la stoechiométrie du complexe.
Transition : Les complexes peuvent participer à d’autres réactions chimiques et peuvent même les
accélérer.
b. Propriétés catalytiques
JD 102 ;
BUP 2004
p. 163
solide - Réaction
- Extraction
- Filtration
Palladium
Point de fusion,
IR
h + Hotte
Principe de la catalyse. Utilité des métaux de transition à valence très varié, degré d’oxydation très
varié, structure facilement modifiable…
Conclusion :
Grand intérêt des complexes en catalyse, notamment en catalyse supportée pour gagner la facilité de
séparation de la catalyse hétérogène. Leur chimie est très riche : le corps humain les utilise. Grand
interêt de comprendre leur structure, leur propriétés complexantes et leur réactivité pour en faire des
médicaments ou des sondes RMN…
On peut rajouter dans Propriétés optiques mais risque de faire répétition et c’est encore du Nickel.
Fosset p. 192 Le solvant peut jouer le rôle de
ligand. Les propriétés
spectroscopiques et la couleur de
certains complexes dépendent alors
2h UV ;
Gouy

du solvant utilisé. Nickel

MG 23
Corrosion, protection contre la corrosion ;
passivation des métaux.
Biblio : Miomandre, HP PC, Lamoureux
Introduction Miomandre et HP
Parmi les réactions spontanées les réactions de corrosion et leur contrôle thermodynamique et cinétique
sont d’une énorme importance pratique et économique : chaque année la corrosion humide provoque la
destruction d’environ 150 millions de tonnes de fer ou d’acier (1/5 de la production). Conséquences
économiques importantes (dizaines de milliards d’euros par an) : la protection du fer et de l’acier et
plus généralement de tous les métaux corrodables, est donc un objectif primordial (HP intro courbe i-E)
De façon générale, la corrosion est une réaction d’oxydoréduction qui se produit entre un métal et son
environnement lorsque celui-ci contient des agents oxydants.
Définition : la corrosion désigne l’ensemble des phénomènes par lesquels un métal ou un alliage
métallique tend à s’oxyder sous l’influence des réactifs gazeux ou en solution. Elle est dite sèche
lorsque les agents oxydants ne sont pas en solution (ex par le dioxygène de l’air); elle est dite humide
dans le cas contraire (corrosion en milieu marin).
On va s’intéresser à deux aspects de la corrosion humide : la thermodynamique qui grâce à l’utilisation
de potentiels rédox et de diagrammes E-pH va pouvoir offrir des prévisions. C’est ensuite l’aspect
cinétique qui doit venir valider ces prévisions et permettre d’explorer les facteurs qui vont déterminer
la vitesse d’échange d’électrons. On utilisera notre connaissance de la corrosion pour essayer de
protéger les métaux de ce phénomène.
I. Etude de la corrosion
a. Mise en évidence
Sarrazin p293 et
Fosset p253
on décape des plaques de fer ou des
clous, on prépare un gel d’agar-agar
contenant de la phénolphtaléïne et du
ferricyanure. Quand le gel commence à
prendre, on le verse dans un tube à essai,
on maintient un clou jusqu’à la prise du
gel. On verra une zone rose vers le haut
et une zone bleue vers le bas du clou. On
fait la manip pour un gel sans NaCl
Pas de
hotte

* Expérience : L’utilisation d’indicateurs colorés permet de mettre en évidence des phénomènes de
corrosion du fer.
* Interprétation : le ferricyanure est un indicateur de la présence de fer(II) issu de l’oxydation du fer
car Fe(II) donne une coloration dite bleu de Prusse en présence d’ion hexacyanoferrate(III). La
phénolphtaléine permet de visualiser les zones où se produit la réduction de l’eau ou celle du dioxygène
car le milieu devient basique.
* Bilan :
- on a montré qu’il y a formation de Fe(II), bien une corrosion.
- l’oxydation du métal libère des électrons, et donc on a également une autre réaction, cette fois-ci de
réduction du dioxygène, visible grâce à la phénolphtaléine (en corrosion humide les principaux agents
oxydants sont le dioxygène dissous et les protons)
� On identifie le site anodique et le site cathodique. Il y a nécessairement un lien électrique entre
l’anode et la cathode et un lien ionique par l’électrolyte. En effet les électrons produits dans la zone
d’oxydation du fer sont consommés dans la zone de réduction du dioxygène, car ils se déplacent à
l’intérieur de la pièce métallique. De plus l’intensité de la corrosion augmente si les milieux
contiennent une grande quantité d’ions (ex du milieu marin) qui permettent de fermer le circuit
électrique.
Ici, l’oxydation du fer et la réduction du dioxygène ont lieu simultanément mais dans des zones
différentes. Ce fait expérimental montre que la corrosion différentielle est une réaction électrochimique
dans laquelle les zones d’échange électronique sont distinctes, et non pas une réaction chimique
d’oxydoréduction. Il existe un autre type de corrosion, dit uniforme, au cours de laquelle l’échange
d’électrons est directement réalisé entre le réducteur et l’oxydant.
* Est-ce que c’était prévisible thermodynamiquement? oui
-> La prévision de la corrosion d’un métal est liée, en première approximation, à la position du métal
sur l’échelle des potentiels standards. Plus l’oxydation du métal est aisée (E° tout petit) plus la tendance
à la corrosion va être élevée. Ex du fer et du zinc
-> Les réactions observées en corrosion obéissent aux lois de la thermodynamique : un métal ne peut se
corroder que s’il est instable par rapport à son produit de corrosion, les diagrammes de Pourbaix
indiquent les limites thermodynamique de la stabilité du métal considéré par rapport à ses ions dans le
milieu et aux produits de réactions, en fonction du pH et du potentiel. Examen du diagramme du fer
montre deux zones de protection : soit la passivation et l’immunité et une zone de corrosion. Il
n’indique pas la cinétique de ces équations.
* Bilan sur les facteurs influençant la vitesse de réaction :
Lorsque le fer est dans un milieu où existe un gradient de concentration en dioxygène, la corrosion se
produit dans les zones les moins oxygénées. La corrosion va donc tendre à faire disparaître cette
différence de concentration de dioxygène. En plus de ce facteur, il faut aussi tenir compte de
l’électrolyte (ex beaucoup d’ions ou non), des gradients de température, les surfaces des anodes et des
cathodes…
Transition : comment quantifier cet aspect cinétique ? on veut avoir accès à la valeur du potentiel de
corrosion et de l’intensité de corrosion

b. Cinétique de la corrosion
Sarrazin p 296 dans une solution acidifiée à pH 2, on
trempe lame de fer et de platine. On
mesure les tensions et le courant. on
trace E en fonction de i.
Pas de
hotte
* Expérience :
Il s’agit d’un phénomène électrochimique, d’où l’importance de la cinétique. On va avoir recours aux
droites d’Evans avec Fer.
* Interprétation :
On trace en fonction du ln, pourquoi ?
On a deux réactions électrochimiques qui se déroulent simultanément sur l’électrode. Au potentiel
d’abondon, E=Ecorr et i=0, on s’appuie sur les courbes intensité-potentiel. Butler Volmer avec une des
exponentielles à négliger devant l’autre dès que la surtension est suffisamment positive ou négative. On
obtient une variation sensiblement linéaire de E avec lnI, qui s’appelle le diagramme d’Evans.
Attention, les pentes des droites sont indépendantes, elles correspondent à deux couples différents. De
plus la droite a une pente d’autant plus faible que le système est rapide. La cinétique globale est
contrôlée par la réaction la plus lente. On est ici, et c’est souvent le cas, sous contrôle cathodique car
c’est la réduction de l’agent oxydant qui est la plus lente. On obtient avec le diagramme d’Evans
l’intensité de corrosion de la plaque de fer.
Diagramme d’Evans car on a un phénomène électrochimique. Du fait de sa simplicité il est
particulièrement commode pour illustrer les phénomènes fondamentaux de la corrosion (contrôle
anodique ou cathodique selon la pente la plus grande) (Lamoureux p 28).
Transition : on a vu que le fer pouvait être le siège d’un phénomène de corrosion. Mais lorsque l’on a
mis en présence d’une lame de zinc, c’est le zinc qui s’est oxydé. Peut-on utiliser ce fait pour protéger
les édifices de la corrosion.

II. Protection de la corrosion
a. Anode sacrificielle
Sarrazin p 287 dans une solution HCl, on place une
lame de fer et de zinc. D’abord on les
sépare puis on les connecte entre elles
avec un fil et ampèremètre pour montrer
le sens du courant.
Pas de
hotte
* Interprétation :
Sur le zinc, il y a des bulles O2, on ajoute le fer, également un dégagement gazeux, on les relie, plus de
formation de gaz à la surface du zinc. On les interprète avec les courbes intensité-potentiel, et la
position relative des courbes anodiques d’oxydation des métaux et des courbes cathodique de la
réduction de H+ sur les différents métaux.
* Bilan :
Le fer est corrodé là où il joue le rôle d’anode, pour le protéger, on constitue un circuit électrique où il
joue le rôle de cathode. Il reçoit alors un courant d’électrons et réduit le dioxygène sans être lui-même
attaqué. On le relie à un métal plus réducteur que lui (ici Zn, mais ça peut être le magnésium) qui subit
la corrosion à sa place. Les coques en acier des navires ou les conduites enterrées sont protégés par des
électrodes de zinc convenablement disposées. Environ tous les 100 mètres pour les tubes. Quand ces
morceaux sont presque dissous, on les remplace (HP et miomandre)
On peut aussi faire jouer au fer le rôle de cathode en imposant un courant (méthode dite active), le
métal à protéger est alors relié au métal protecteur au moyen d’un générateur. Dans ce cas, la protection
est totale, mais la consommation de l’anode est plus rapide. Lorsque le milieu présente des
caractéristiques mal définies, ou si la surface à protéger est importante, on privilégie cette deuxième
méthode.
* Bilan : action réductrice, courant électrique ou anode réactive « sacrificielle ». On s’est placé dans le
domaine théorique d’immunité.
* Transition : On peut les relier électriquement ou même les mettre en contact par électrodéposition.

b. Nickelage du cuivre
Sarrazin p 254 Cu, Pt, ECS dans acide borique à
différents pH et en présence de Ni (II).
Rincer la plaque à l’eau et la sécher au
sèche-cheveux pour déterminer la masse
déposée.
Synchronie pour avoir i précisément et
comparer avec la masse déposée.
Pas de
hotte
* Principe :
Intérêt industriel : casserole, tube, éléments thermochauffant en cuivre nickeléOn a également une méthode passive, où l’on applique sur le métal un revêtement protecteur, tels que
des peintures, des vernis, des revêtements par d’autres métaux. Par électrolyse d’une solution contenant
le cation à déposer : électrozingage, chromage, le nickelage…
Bien que le zinc soit plus réducteur que le fer, il résiste mieux à la corrosion atmosphérique car il se
recouvre d’une couche d’hydrocarbonate de zinc, adhérente et imperméable.
Deux cas de figures :
- si le métal protecteur est plus oxydable (pas de gros soucis s’il y a des défauts, mais grosse
couche est nécessaire) Le nickelage du cuivre et l’électrozingage du fer.
- si le métal protecteur est moins oxydable (pas de défauts, sinon corrosion rapide, mais une fine
couche suffit).
* Expérience :
On effectue les droites de polarisation dans plusieurs conditions : à différents pH sans Ni, aux mêmes
pH avec Ni. On voit que le plus pH augmente, plus il y a de différence entre les droites de réduction du
Ni et de H+. On préfère donc travailler à pH élevé pour que H+ ne se réduise pas à la place du Ni. Mais
pas trop élevé pour éviter que le Ni II ne précipite sous forme d’hydroxyde.
* Remarque :
On peut former une couche superficielle protectrice grâce à une réaction chimique, on plonge une pièce
d’acier dans une solution chaude avec des ions phosphates, on provoque ainsi l’apparition d’une
couche de phosphate de fer imperméable. Il s’agit de la parkérisation, utilisée dans l’industrie
automobile pour protéger les carrosseries (HP)
Transition : Des métaux sont naturellement protégés de la corrosion par leur couche d’oxyde ex
aluminium et alumine.

III. Protection par passivation des métaux
a. Passivation du fer
Sarrazin et
Fosset p283
mise en évidence de la passivation d’un acier
grâce à la courbe i-potentiel. Donc montage à
trois électrodes, avec une électrode de fer
plongée dans une solution d’acide nitrique.
Par volampérométrie cyclique, ça peut
marche 120mV/s. Rouille apparait
Voltampérométrie
cyclique
Pas de
hotte
* Expérience :
Certains métaux peuvent en présence d’un oxydant former à leur surface un film d’oxyde fin, adhérant
et continu. Ils sont pratiquement inertes en milieu agressif ! chrome, aluminium, titane. On trace la
courbe de passivation anodique du fer
* Interprétation :
On distingue plusieurs domaines (acitivté, passivité..) D’abord oxydation du fer en Fe(II) et dépôt en
Fe(OH)2, potentiel de Flade, l’électrode est entièrement recouverte de cette couche, Fe(II) donne Fe(III)
hydroxyde donne oxyde. L’oxydation cesse (la couche ne conduit pas les ions !) et on a une chute de
l’intensité.
On peut regarder sur le diagramme potentiel pH domaine de passivation là où l’on a les oxyde
(Miomandre p.67 ou p.247)
Couche d’oxyde pas solide, pas le cas pour d’autres oxydes
On peut ajouter du chrome, si les aciers contiennent un pourcentage massique dépasse 12% environ, il
s’agira d’aciers inoxydables.
Transition : ça ne marche pas super bien pour le fer, mais pour d’autre oui

b. Passivation de l’aluminium
Sarrazin p.
298 et JFLM
p. 184
Nettoyage de l’aluminium avec de l’acétone.
On met la plaque à l’anode (1cm) pour
l’électrolyse d’une solution d’acide
sulfurique. On impose un courant de 50 à 100
mA par cm² pendant 10 à 15 minutes, la
plaque est rincée à l’eau, puis à l’ammoniac
puis à l’eau. On trempe la plaque dans une
solution chaude d’alizarine (ligand coloré)
plus profondément que la marque
d’anodisation.
Synchronie pour avoir i précis et pouvoir
comparer à la masse obtenue.
Pas de
hotte
* Expérience :
Anodiser une pièce métallique c’est mettre une pièce à l’anode d’une électrolyse pour l’oxyder en
surface. Epaisseur de la couche protectrice estimée (JFLM p187)
* Remarque :
Intérêt car la couche d’oxyde peut subir un traitement chimique et se faire colorer. Le film de Al(OH)3
est mis en contact avec des ligands colorés d’Al3+. Une réaction de complexation a lieu entre Al3+ et les
ligands. Le film est durci en déshydratant l’hydroxyde en oxyde (alumine) les molécules de colorants
se trouvent emprisonnées. Là où il n’y a pas eu anodisation, il n’y a pas fixation de la couleur.
Conclusion :
Selon le métal considéré, différentes stratégies pour avoir une protection contre la corrosion. Méthode
de prévention : attention aux raccords, aux agencements, des revêtements protecteurs, des protections
cathodiques (immunisation) et anodiques (passivation). Une dernière méthode, les inhibiteurs
d’adsorption, molécules qui s’adsorbent sur le métal.

MG 24
Spectrophotométrie IR, UV et visible : principes, applications.
Biblio : Skoog
Introduction :
Important de pouvoir caractériser produits. Il faut des méthodes que l’on va voir aujourd’hui.
Définition de la spectroscopie en générale. Manipulation d’introduction la matière absorbe le
rayonnement avec un réseau et rétroprojecteur. KMnO4 absorbe certaines longueurs d’onde. Utilisation
en chimie. UV transition électronique. IR transition entre état vibrationnels. Ou Rotationnels.
I. Principes des spectrophotométrie IR et UV-visible
a. La loi de Beer-Lambert
JFLM p. 135 Droite d’étalonnage,
détermination d’une
concentration de Fe dans le
vin.
Préparation d’une solution
UV -
On effectue une droite d’étalonnage et on vérifie la loi de Beer-Lambert
Application : On prend une mesure, on report sur la droite et hop concentration.
Transition : Même principe dans l’IR sauf qu’on étudiera pas l’intensité des pics mais plus leur
position et leur éventuel décalage.
b. La loi de Hooke
Spectre de l’acétone deutéré (effet isotopique)
Spectre de l’acétone et du propanol (effet de
liaison)
Spectre de différents dérivés CH3CO-X (X=OH,
Cl, NH2, OCH3) (effet inductif et mésomère)
IR -
Transition : Appliquons ceci toutd e suite !

II. Applications
a. En chimie organique : structure d’un composé organique
JD n°90 solide - mélange intime des
deux solides
- lavage
Point de fusion,
IR, CCM
1h - Hotte
OU
JFLM orga
p. 149,
Daumarie
solide - Réaction
- Filtration
- Recristallisation
Point de fusion,
IR, CCM, test
aux phénols
3 h + Hotte
Point de fusion, première caractérisation. Ensuite, il en faut une autre l’IR. On voit que la double
liaison C=O s’est déplacé par conjugaison. On a donc bien eu réaction.
Transition : Ces spectrophotométries peuvent aussi servir à élucider des structures en chimie
inorganique
b. En chimie inorganique : structure d’un complexe
JCE 1980 p.
826 ; JCE
1980 p. 900 ;
BUP 2008 p.
41
Synthèses de plusieurs complexes,
puis études IR et UV. Interprétation
avec Tanabe.
long UV, IR hotte
UV : Série spectrochimique. Détermine structure.
IR : permet de connaître l’atome de fixation du ligand.
Conclusion :
Grand intérêt, riches en information. Autres : RMN.

MG 25
La réaction chimique : mise en évidence des caractéristiques
cinétiques à partir de quelques exemples.
Biblio : Skoog, Miomandre, Atkins
Introduction :
Définition de la cinétique. Enjeu loi de la cinétique à déterminer. Pour optimiser les réactions et
comprendre le mécanisme des réactions. Nombreuses techniques que l’on peut exploiter pour suivre la
cinétique d’une réaction. Apparition ou disparition d’un composé coloré, d’un composé ionique, d’un
changement du pourvoir rotatoire.
On s’intéressera tout d’abord au facteur régissant la cinétique d’une réaction : ordre partiel, constante
de vitesse, puis comment on peut l’augmenter.
I. Paramètres cinétiques
a. Ordre partiel
BUP 762 p.
457
Détermination d’une loi de vitesse
par UV-visible (ordre partiel et
constante de vitesse)
2h UV
OU si possible (plus simple au niveau de la loi de vitesse)
BA p. 235 Détermination d’une loi de vitesse
par UV-visible (ordre partiel et
constante de vitesse). Utilisation de
cristal violet !
2h UV
Définition loi de vitesse, ordre partiel et constante de vitesse ; Détermination d’un ordre partiel.
Transition : Il faut également tenir compte de la constante de vitesse.
b. Constante de vitesse
BUP 762 p.
457
Détermination d’une loi de vitesse
par UV-visible (ordre partiel et
constante de vitesse)
2h UV
Détermination d’une constante de vitesse à une température donnée…

Transition : Voyons voir les facteurs influençant ces caractéristiques cinétiques.
II. Facteurs influençant la cinétique
a. Influence de la température
Solvolyse du tertiobutyle
Blanchard
p.167
Daumarie p.71
brut - préparation
- suivi
conductimétrique
2h30 - Hotte
Loi d’Arrhenius, détermination d’une énergie d’activation. Autre caractéristique cinétique mise en
évidence.
Transition : La cinétique est aussi sensible aux ions présents dans le milieu
b. Influence de la force ionique
Fosset p. 324 Détermination
colorimétrique avec
chronomètre
-
Loi de Debye-Hückel…
Transition : On peut accélérer une réaction, voyons les caractéristiques d’une réaction catalysée.
III. Catalyse
Artero p. 135 Montrer un intermédiaire réactionnel 2h
On montre l’augmentation de la vitesse : tube à essai et après, avec catalyseurs.
On montre l’existence d’un intermédiaire réactionnel, typique lorsqu’on a catalyse.
Conclusion :
Enjeu industriel d’avoir de connaître la loi de vitesse des réactions pour optimiser un réacteur. Et
produire plus vite.
Enjeu dans les laboratoires : Permet l’étude des mécanismes réactionnels puisque un mécanisme sera
rejeté si la loi de vitesse théorique ne correspond pas à la loi de vitesse mesurée expérimentalement.

MG 26
Méthodes de détermination de l'ordre d'une réaction chimique.
Biblio : Skoog, Miomandre, Atkins
Introduction :
Définition de la cinétique. Enjeu loi de la cinétique à déterminer pour obtenir des informations quand
au mécanisme. La loi de vitesse. Celle pour un acte élémentaire simple mais pour une réaction faisant
intervenir de nombreuses étapes plus complexe.
Nombreuses technique que l’on peut exploiter pour suivre la cinétique d’une réaction. Apparition ou
disparition d’un composé coloré, d’un composé ionique, d’un changement du pourvoir rotatoire.
Hypothèses sur une loi de vitesse. Intégration. Puis vérification expérimentale. Et on doit aussi vérifier
que la réaction n’a pas un autre ordre.
I. Méthode intégrale
a. Détermination d’un ordre global
Blanchard
p.167
Daumarie p.71
Solvolyse du
tertiobutyle
- préparation
- suivi conductimétrique
2h30 - Hotte
Définition loi de vitesse, ordre partiel et constante de vitesse ; Détermination d’un ordre global
Transition : On peut également affiner l’étude et déterminer un ordre partiel.
b. Détermination d’un ordre partiel
BUP 762 p.
457
Iodation de la propanone.
Détermination d’une loi de vitesse
par UV-visible (ordre partiel et
constante de vitesse)
2h UV
OU si possible (plus simple au niveau de la loi de vitesse)
BA p. 235 Décoloration du cristal violet.
Détermination d’une loi de vitesse
par UV-visible (ordre partiel et
constante de vitesse). Utilisation de
cristal violet !
2h UV
Détermination d’un ordre partiel par dégénérescence de l’ordre.

Transition : On suit en fonction du temps, fastidieux…

II. Méthode du demi-temps de réaction
Brenon et Audat
p. 163
Mutarotation du glucose Long - Polarimètre
On peut remonter à k grâce au temps de demi-réaction et également à l’ordre en acide, catalyseur, en
effectuant la manipulation à différente concentration en acide. On peut montrer ainsi la catalyse acide.
Transition : On avait supposé que toutes les réactions avait un ordre constant, ce n’est pas toujours le
cas.
III. Méthode des vitesses initiales
Fosset p. 324 ; Bernard
et Maire p. 101
Réaction diiode et peroxodisulfate.
Détermination colorimétrique avec
chronomètre
Il faut excès de peroxodisulfate
-
Ici, la réaction garde son ordre, mais on veut illustrer la méthode des vitesses initiales. On peut
remonter à k.
Conclusion :
Enjeu dans les laboratoires : Permet l’étude des mécanismes réactionnels puisque un mécanisme sera
rejeté si la loi de vitesse théorique ne correspond pas à la loi de vitesse mesurée expérimentalement.
Facteur influençant la cinétique comme la température, les conditions de solvants ou bien la présence
d’un catalyseur pourront être étudié.

MG 27
Catalyse par les métaux de transition et leurs composés.
Introduction
Définition d’un catalyseur. Courbe d’énergie potentielle=f(CR). Un catalyseur : plus vite mieux et
moins cher ! Deux types de catalyse : homogène et hétérogène
Définition du métal de transition. DO nombreux, catalyse d’oxydoréduction. Soit sous forme de solide,
soit sous forme de complexe soluble (coordination des ligands change ses propriétés). Notamment,
grâce à leurs nombreux degrés d’oxydation et leur nombre de coordination élevé, ils peuvent
rapprocher les réactifs et catalyser la réaction.
Pour chaque manip, écrire la quantité de catalyseur utilisé et le nombre d’équivalent !
I. Etude de la catalyse
a. Mise en évidence : les sels de Seignette
Artero p. 135 Réaction entre tartrate et H2O2
Montrer un intermédiaire réactionnel et la régénération
du catalyseur par changement de couleur
Tube à essai : montre que sans catalyseur rien ne se
passe et que si pas les deux réactifs et catalyseur, rien
ne se passe non plus.
Ajout de H2O2 : passe du rose au vert puis du vert au
rose.
Trempe : pour isoler IR et prendre UV
2h UV Pas de hotte
Observation :
On montre grâce à des tubes à essais que si on ne met pas de catalyseur, rien ne se passe.
Dans le ballon, il y a un complexe de Co (4e ligne, II, d7) auquel on a ajouté le tartrate : rose (complexe
hexaaqua) -> rose fushia. Un complexe tartrate-Co s’est formé dont le potentiel redox est plus faible
que l’aqua, notamment il devient inférieur à celui de H2O2 et ce dernier peut oxyder le cobalt. Quand on
ajoute H2O2, La solution passe du rose au vert puis au rose et on a un dégagement de CO2 vérifié par
eau de chaux. Grâce au degré d’oxydation III, la rupture des liaisons C-C est activée.
Interprétation :
Le dégagement de CO2 montre qu’il y a eu la réaction attendue. Pendant, cette réaction le mélange
réactionnel a changé de couleur. Il y a donc eu formation d’un nouveau composé caractérisé par son
spectre UV (trempe effectué pour l’isoler). Il montre les transitions d-d. Il a ensuite disparu pour
revenir à la couleur initiale a priori et vérifions tout de suite si c’est bien la même couleur et donc le
même composé.
Ainsi le complexe tartrate-Co a formé un IR avec le peroxyde d’hydrogène. Il a donc permis de passer

par un autre chemin réactionnel avec Ea plus faible. Ensuite il a été régénéré. Donc on a bien eu
catalyse.
Transition : Comment peut-on quantifier cette accélération ?
b. Influence sur la cinétique : dismutation de H 2O2
JFLM p. 280 MnO2 et H2O2. On suit le volume
d’oxygène libéré.
Loi de vitesse : ordre partiel par
rapport au catalyseur : 2, 3, 5 mL de
cat. Bien déclencher le chrono au
début. Puis vitesse à t1 fixé.
2h Pas de hotte
* Observations :
On place du peroxyde d’hydrogène dans le montage. On voit qu’il ne se passe rien. On ajoute du MnO2
et là on constate un dégagement gazeux : du dioxygène. Il y a dismutation du peroxyde en présence de
MnO2 (4e ligne, IV, d3)
* Etude cinétique :
On peut mesurer la quantité de dioxygène libérée en fonction du temps et ceci pour différentes masses
de catalyseur. On voit que plus il y a de catalyseur, plus la réaction est rapide. Il intervient donc bien
dans la cinétique. On peut également vérifier que la masse de catalyseur est la même qu’initialement.
On peut également calculer le rapport des constantes de vitesse.
* Application :
Introduction d’une vitesse importante : turnover number pour les chercheurs (v par site de cat), vitesse
spécifique massique pour les industriels (v par m de cat)
Transition : En industrie, le mieux c’est d’avoir une vitesse grandement accélérée par peu de
catalyseur car en général cher comme on va le voir. En effet, on utilise souvent des métaux nobles
comme le Pd, Ru, Rh, Ni,…
II. Applications
a. Catalyse homogène : couplage de Suzuki
JD 102 ;
BUP 2004 p. 163
(numéro du BUP
dans le JD)
(mécanisme)
solide - Réaction
- Extraction
- Filtration
Point de fusion,
IR, CCM
2h + Hotte
* Description:
On va effectuer une catalyse homogène en utilisant un complexe de Pd (5e ligne, II, d8). Quantité
catalytique. Description du cycle catalytique. Intervient dans le mécanisme, régénéré.
* Intérêt de la catalyse homogène :
Chimie fine, grande sélectivité, facilité de changement de structure.

* Intérêt synthétique :
Autres : Heck, Stille,…. Grand intérêt de coupler pour synthèse convergente avec grande
stéréosélectivité.
Transition : On peut également utiliser le Pd sous sa forme solide.
b. Catalyse hétérogène : hydrogénation
BUP à paraître liquide - filtration sur célite
- extraction
- lavage
IR, CCM, indice
de réfraction,
CPV
2h + Hotte
OU
JD 20
Mêmes réactifs, pas de
NaBH4, dissoudre les
réactifs avant et les
mélanger dans l’insert.
liquide - filtration sur célite
- extraction
- lavage
IR, CCM,
indice de
réfraction, CPV
2h + Hotte
* Description :
On va effectuer une catalyse hétérogène en utilisant un complexe de Pd (5e ligne, II, d8) ou de Ni (4e
ligne, II, d8). Quantité catalytique. Dihydrogène se chimisorbe sur le Pd ou sur le Ni. Permet d’affaiblir
la liaison H-H (400 kJ/mol). Puis chimisorption de l’alcyne et création de C-H. Intervention du
catalyseur dans le mécanisme et régénération.
* Intérêt de la catalyse hétérogène :
Filtration facile !
* Application industrielle: hydrogénation des acides gras pour faire la margarine. Utilisation de
catalyseur dans l’industrie (TI J1255 Pd ou Pt supporté sur Al2O3 et avec In ou Sn pour améliorer
activité). Gros tonnage ! On ne peut pas faire ça avec homogène
Conclusion :
Catalyse organosupportée avantage de la catalyse homogène sélectivité et hétérogène filtration. Lier
efficacité et productivité. Catalyse enzymatique.

MG 28
Catalyse hétérogène : principes et applications.
Biblio : Skoog, Miomandre, Atkins
Introduction :
Définition d’un catalyseur. Courbe d’énergie potentielle=f(CR). Un catalyseur : plus vite mieux et
moins cher !
I. Principe
a. Mise en évidence
JFLM p. 280 MnO2 et H2O2. On suit le volume
d’oxygène libéré.
Loi de vitesse : ordre partiel par
rapport au catalyseur : 2, 3, 5 mL de
cat. Bien déclencher le chrono au
début. Puis vitesse à t1 fixé.
2h Pas de hotte
Transition : On peut également affiner l’étude et déterminer un ordre partiel.
b. Sélectivité
Blanchard p.
206
Mesplède p.
213
Sélectivité de la réaction sur l’éthanol
suivant le catalyseur (Cu ou Al2O3)
Il faut deux fours, prend du temps.
(Test à la 2,4 DNPH ou réactif de
Tollens)
Avec Cu, on a oxydation et avec l’alumine réduction.
Transition : On vient de montrer une déshydrogénation mais aussi hydrogénation.

II. Application
a. Hydrogénation
BUP à
paraître
liquide - filtration sur célite
- extraction
- lavage
IR, CCM,
indice de
réfraction, CPV
2h + Hotte
Transition : Le métal en lui-même catalyse, mais des fois ça peut être du à son état de surface, à ce
qu’il a en surface.
b. Acétalisation
JD 63 Catalyse par la
montmorillonite
IR, indice de
réfraction, CCM
1 h Hotte
A faire avec ou sans catalyseur.
Montmorillonite = argile, acide de Lewis ou de Brönstedt
Conclusion :
Catalyse hétérogène très intéressant dans l’industrie, nombreux procédés utilisent ces catalyseurs.
Problème de sélectivité bien souvent. Les catalyseurs homogènes permettent une meilleure sélectivité
car on peut jouer sur les ligands autour d’un métal et faire des réactions stéréo sélectives. Une autre
catalyse homogène non illustrée ici est la catalyse supportée. On fixe le catalyseur sur une résine.
Faciliter de purification !

MG 29
Le magnésium et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Introduction :
(Pascal) Le magnésium, métal de la colonne des alcalino-terreux, n’existe pas à l’état libre dans la
nature, mais l’abondance de ses composés est telle qu’il occupe le huitième rang dans la lithosphère
(chlorophylle, eau de mer, roches). Il est considéré comme un élément biogénétique indispensable.
Le magnésium a pour configuration électronique 1s22s22p63s2 et il est donc caractérisé par une seule
valence habituelle de 2, qui lui donne un nombre très restreint de composés.
Au cours du montage, on s’intéressera aux propriétés du magnésium au degré 0, qui est un puissant
réducteur, puis à celle de magnésium au degré +II en mettant en avant l’importance biologique de cet
élément ainsi que son rôle en chimie organique.
I. Caractère réducteur du métal
1. Détermination du potentiel standard
Sarrazin p76-
77, JFLM p252
On mesure l’élévation de T induite par
l’oxydation de Mg dans HCl à 6M, et
on en déduit l’enthalpie de la réaction
Introduction de Mg
dans le Dewar
Visualisation de
l’élévation de
température
Calorimétrie
1h
NB on peut travailler avec des morceaux de tournure, et prendre plutôt [HCl]=1M (la dissolution est
alors moins violente)
En première approximation on assimile HCl 1M à de l’eau
On détermine la masse en eau du calorimètre (dans le JFLM : on pèse le Dewar vide, on ajoute de l’eau
chaude, on pèse à nouveau et on prend la température. On ajoute un quantité d’eau froide de
température et masse connue. On mesure la température finale du mélange)
On mène l’expérience et on a accès à l’élévation de la température.
Mg + 2H+ � Mg2+ + H2
On relie l’élévation de la température et les échanges thermiques
Q=(Meau+MDewar)*C*(Tf-Ti) on néglige le Mg
On détermine l’enthalpie de la réaction on l’assimile à l’enthalpie standard, on connaît S°, on a accès à
enthalpie libre standard de la réaction et donc au potentiel d’oxydoréduction
Transition : on a un métal très réducteur, une autre façon de le voir, c’est avec la formation de MgO
(En présence d’air il brûle avec production de lumière intense et trouve son application dans les
flash.)

2. Application à la protection du fer
Sarrazin p290 et
296
Gel avec clou et phénophtaléine et bleu
de prusse, puis clou entouré de
magnésium
Préparation du gel 30min
On montre un test avec le fer seul, la phénolphtaléine pour montrer la réduction de l’eau ou celle du
dioxygène et le ferricyanure de potassium comme indicateur « bleu de prusse » de la présence de Fe(II)
résultant de l’oxydation
On peut aussi prendre des lames de magnésium et de fer parallèle et proches. On les plonge dans un
bécher d’eau distillée, et on regarde le sens du courant : du fer vers le magnésium à l’extérieur de la
pile.
Discussion sur le E° et sur l’importance de la protection du fer (ex dans le Miomandre)
Transition : deuxième degré d’oxydation a un grand nombre d’applications. On va voir dans un
premier temps ses propriétés complexantes, qui peuvent être utilisées pour réaliser un dosage.
II. Le degré d’oxydation +II
1. En chimie analytique
Les eaux minérales contiennent du sulfate, du chlorure et du carbonate de magnésium
Ecolochimie
p305, Cachau p
253 Flashka
p101, Charlot
Dosage de la dureté de l’eau
Prendre un milieu tamponné
Colorimétrie, chute de
burette
2h
La dureté de l’eau (titre hydrotimétrique de l’eau) est due dans la plupart des cas aux ions Ca2+ et Mg2+
.Sans être nocive pour l’homme une eau dure présente des inconvénients, surtout reliés au dépôt de
calcaire dans les conduits de certains appareils ménagés.
En préparation on a réalisé le dosage des ions calciums par le Patton en faisant précipiter le magnésium
(II). On en déduit la concentration des ions magnésiums. Comparaison avec la valeur indiquée sur
l’étiquette.
Transition : on retrouve les propriétés complexantes dans la chimie du vivant

2. En biochimie
(Pascal ) Les animaux sont moins riches que les plantes : le magnésium existe dans la chlorophylle
sous forme d’une combinaison organo-magnésienne. Tous les tissus animaux renferment du
magnésium : l’os est le plus riche, puis viennent le rein, le cœur les muscles, le foie et le pancréas.
Daumarie p153,
JCE nov 75
p742
On extrait la chlorophylle des épinards
et on démétalle avec HCl 0,1M
(chauffer pour accélérer), on peut
ajouter de NET pour doser
Colonne et UV 2h
On extrait la chlorophylle des épinards sur colonne de silice, puis on ajoute HCl concentré, on teste
ensuite la présence de Mg avec le Net. On parle de l’importance pour la photosynthèse, , on peut aussi
se lancer dans une comparaison mollesse/dureté pour expliquer la facilité de la démetallation.
3. En chimie organique
Le magnésium a acquis une importance prodigieuse en chimie organique depuis la découverte faite par
Grignard des composés organométalliques.
JCE 86 p92 Champignon matsutake Lancement de la
synthèse
2h-3h
Parler des conditions, de sa réactivité
Conclusion :
Le magnésium présent une grande réactivité chimique et ses ions sont assez largement répandus dans la
nature. Le principal emploi du magnésium est la fabrication d’alliages légers utilisés en particulier dans
l’industrie automobile, aéronautique et spatiale. Le magnésium fait partie des oligo éléments
nécessaires à la vie.
Année 2008-2009
I) Propriétés redox du magnésium
a. Détermination du E° de Mg2+/Mg
b. Application à la protection du fer
II) Propriétés complexantes
Dosage de la dureté de l’eau
III) Utilisation en chimie organique
Synthèse et utilisation d’un organomagnésien
Sarrazin p76-77,
JFLM p252
On mesure l’élévation de T induite par
l’oxydation de Mg dans HCl à 6M
colorimétrie 1h
Sarrazin Manip très visuelle tout courte
Ecolochimie
p305, Flashka
p101, Charlot
Dosage de la dureté de l’eau
Prendre un milieu tamponnéColorimétrie, chute de
burette
2h
JCE 86 p92 Lancement de la
synthèse
2h
Année 2007-2008 :
I- Propriétés réductrices
a. Courbe intensité potentiel
b. Application à la protection par anode sacrificielle
II- Propriétés complexantes
a. La chlorophylle
b. Détermination de la dureté de l’eau
III- Utilisation en chimie orga
Matsutake
Sarrazin, p294 On trace l’oxydation de Mg et du fer pour
montrer que le Mg est plus réducteur que le
fer
Voltalab, montage 30min
Sarrazin p290 et
296
Gel avec clou et phénophtaléine et bleu de
prusse, puis clou entouré de magnésium
Préparation du gel 30min
Daumarie p153,
JCE nov 75 p742,
Souil p20
On extrait la chlorophylle des épinards et
on complexe avec Mg2+
Colorimétrie, chute de
burette
2h
Ecolochimie
p305
Dureté de l’eau Dosage de Ca2+ et Mg2+ 30 min
JCE 86 p92 Lancement de la
synthèse
2h-3h
Dans les cahiers verts :
I- caractère réducteur du métal
a. Oxophilie
b. Determination du E° par calorimétrie
c. Protection anodique
II- Le degré d’oxydation II. Propriétés complexantes
a. Dosage : détermination de la dureté de l’eau
Flashka EDTA titration
b. Extraction des pigments de la chlorophylle et mise en évidence de Mg2+ chelatant
JCE p1975 p742
I- le magnésium en tant que réducteur
II- Les complexes de magnésium
III- Utilisation du magnésium en chimie orga
a. Formation d’un magnésien

b. Dosage en retour
c. réactivité

MG 30
L’aluminium et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Introduction :
(Pascal) L’aluminium, qui possède une configuration électronique 1s22s22p63s23p1, est extrêmement
répandu dans la nature, il entre pour 8,3% dans la composition de l’écorce terrestre. Son nom
« aluminium » vient de « alumine », on verra à quels points la transformation de l’un en l’autre est
importante au cours du montage. L’alumine Al203, est au do III, c’est le do stable, on va étudier
d’autres composés à ce do et leur applications
I. Elaboration industrielle
Le principal minerai d’aluminium est la bauxite, composé d’oxyde hydraté d’aluminium, de silice et
d’oxyde de fer qui lui donne sa couleur rouge. On regarde une première étape d’extraction de l’alumine
de la bauxite
BUP 790 On fabrique la bauxite, puis on sépare le
fer de l’aluminium par précipitation
sélective.
Filtration et
précipitation
3h
HP PCSI Chimie 2 montre la dépendance de la solubilité par rapport au pH. On utilise les propriétés
amphotères de l’aluminium.
Transition : deuxième étape, faire subir une électrolyse en sel fondu à l’alumine pour donner
l’aluminium. Il s’agit de l’aluminium. Pourquoi vouloir l’obtenir ? Application en industrie grâce à sa
légèreté et ductibilité. Précaution
II. Propriétés réductrices
1. Mise en évidence
Sarrazin, p288 Manip qualitative où l’on trempe une
lame d’aluminium dans de la soude
tout 2min
Exploitation dans le Sarrazin avec les couples, on observe un dégagement de H2, on écrit les couples,
on justifie l’utilité d’un protection. L’aluminium qui a été protégé par une couche d’alumine, qui n’est
pas très rapidement soluble dans HCl.

2. Protection contre la corrosion
* anodisation : Un métal conducteur peut être protéger de la corrosion par la formation par exemple par
électrolyse d’une couche compacte et adhérente d’oxyde.
Fosset p292
JFLM
Anodisation : on nettoie une partie de la
plaque d’aluminium avec de l’acide
sulfurique, on la relie au pôle + du
générateur et deux autres plaques
d’aluminium au pôle -. J=5mA/cm2. On
mesure le temps d’électrolyse
électrolyse 30
min
On peut calculer en pesant avant et après la plaque le rendement faradique, ou le supposer de 100% et
regarder l’épaisseur de la couche.
Si l’aluminium se recouvre naturellement d’alumine, elle est relativement fragile. Ici on forme un
couche d’amine poreuse, on fixe le colorant. Plongée dans l’eau bouillante l’alumine cristallise en
böhmite. Utilisée dans l’industrie
Transition : on a vu apparaître à différents moments le do III, stable (cf configuration électronique).
III. Aluminium au do III
1. Application à la chimie organique
JD92 On fait l’acylation avec AlCl3 et sans Lancement,
observation du
dégagement
3h
La Friedel et Crafts est la plus importante et la plus générale des méthodes de préparation des cétones
aromatiques. La régiosélectivité de la réaction vient de la taille du réactif acylant : Ac2O/AlCl3.
NB ici on s’intéresse à la chimie orga, on peut aussi réaliser des synthèses inorganiques de complexes
Al(III) (ex dans le Feneuil)
Transition : en synthèse, il est souvent impératif de séparer des produit, une technique la
chromatographie, très répandu sur silice, mais parfois pas adapter, autre support possible
2. Application physico-chimique de l’alumine
chromatographie :
Florilège p159 Faire la colonne après avoir fait la CCM colonne 2h
On explique l’intérêt de l’alumine si on a un produit sensible.
Conclusion : L’aluminium a de nombreuses propriétés chimiques (pouvoir réducteur, acidité) que l’on
peut mettre à profit pour exploiter ses intéressantes propriétés physiques. Il est présent dans la plupart
des objets du quotidien, car par constitution d’alliage, il peut s’adapter à des nombreuses contraintes.

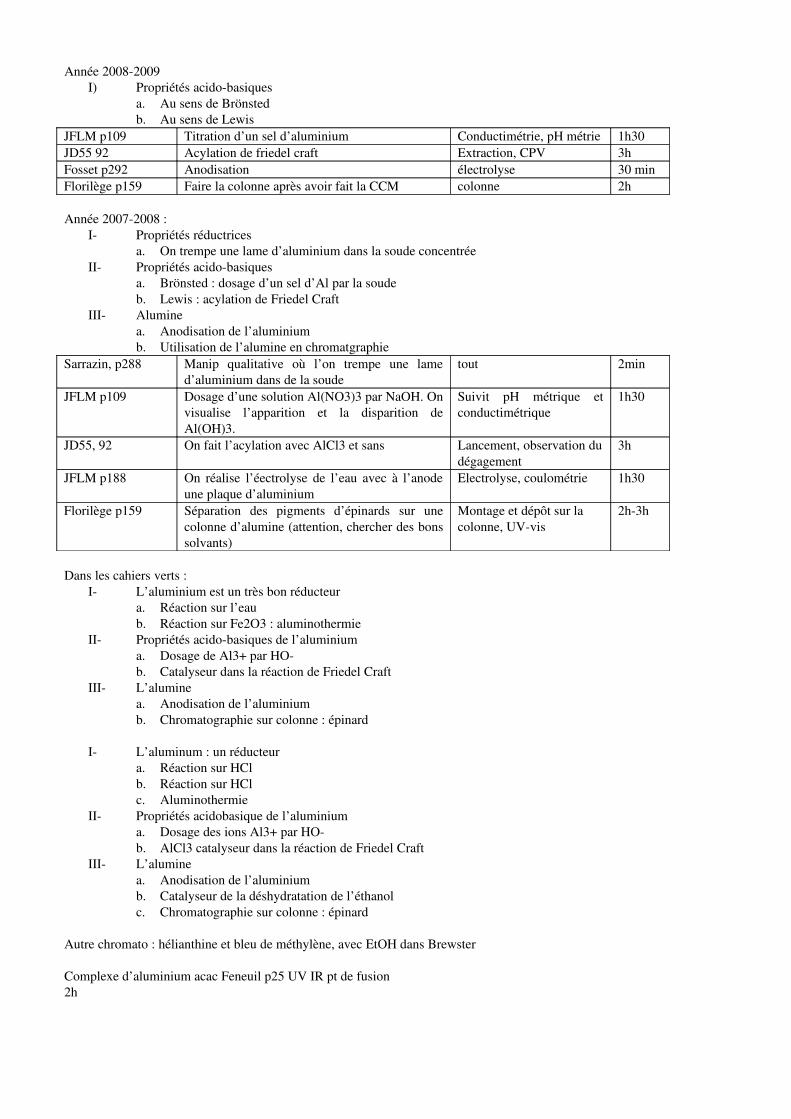
Année 2008-2009
I) Propriétés acido-basiques
a. Au sens de Brönsted
b. Au sens de Lewis
JFLM p109 Titration d’un sel d’aluminium Conductimétrie, pH métrie 1h30
JD55 92 Acylation de friedel craft Extraction, CPV 3h
Fosset p292 Anodisation électrolyse 30 min
Florilège p159 Faire la colonne après avoir fait la CCM colonne 2h
Année 2007-2008 :
I- Propriétés réductrices
a. On trempe une lame d’aluminium dans la soude concentrée
II- Propriétés acido-basiques
a. Brönsted : dosage d’un sel d’Al par la soude
b. Lewis : acylation de Friedel Craft
III- Alumine
a. Anodisation de l’aluminium
b. Utilisation de l’alumine en chromatgraphie
Sarrazin, p288 Manip qualitative où l’on trempe une lame
d’aluminium dans de la soude
tout 2min
JFLM p109 Dosage d’une solution Al(NO3)3 par NaOH. On
visualise l’apparition et la disparition de
Al(OH)3.
Suivit pH métrique et
conductimétrique
1h30
JD55, 92 On fait l’acylation avec AlCl3 et sans Lancement, observation du
dégagement
3h
JFLM p188 On réalise l’éectrolyse de l’eau avec à l’anode
une plaque d’aluminium
Electrolyse, coulométrie 1h30
Florilège p159 Séparation des pigments d’épinards sur une
colonne d’alumine (attention, chercher des bons
solvants)
Montage et dépôt sur la
colonne, UV-vis
2h-3h
Dans les cahiers verts :
I- L’aluminium est un très bon réducteur
a. Réaction sur l’eau
b. Réaction sur Fe2O3 : aluminothermie
II- Propriétés acido-basiques de l’aluminium
a. Dosage de Al3+ par HO-
b. Catalyseur dans la réaction de Friedel Craft
III- L’alumine
a. Anodisation de l’aluminium
b. Chromatographie sur colonne : épinard
I- L’aluminum : un réducteur
a. Réaction sur HCl
b. Réaction sur HCl
c. Aluminothermie
II- Propriétés acidobasique de l’aluminium
a. Dosage des ions Al3+ par HO-
b. AlCl3 catalyseur dans la réaction de Friedel Craft
III- L’alumine
a. Anodisation de l’aluminium
b. Catalyseur de la déshydratation de l’éthanol
c. Chromatographie sur colonne : épinard
Autre chromato : hélianthine et bleu de méthylène, avec EtOH dans Brewster
Complexe d’aluminium acac Feneuil p25 UV IR pt de fusion
2h

MG 31
Propriétés comparées des halogènes.
Biblio : Greenwood
Introduction :
Définir: élément halogène (élément=concept, pas de propriétés!) corps simple (dihalogènes) ions
(halogénures) corps composés (ex: chlorates) . On peut choisir de ne pas parler de ces derniers
Evoquer les isotopes, mais n influencent pas la réactivité
Présentation de la colonne des halogènes configuration électronique. Pourquoi on ne considère que Cl,
I et Br. F toxique, corrosif (verrerie spéciale) et As radioactif, rare (44mg dans tout le premier
kilomètre de la croûte terrestre). On ne considère que les degrés 0 et –I.
Place dans la classification périodique. Famille: propriétés similaires, mais évolution au sein d'une
colonne! Connaître les évolutions de l'électronégativité, et des rayons atomiques dans une colonne (les
mettre sur papier !)
Manipuler avec précaution sous hotte des X2 , avec gants nitriles et cristallisoir de thiosulfate de
sodium (réducteur des X2) ou NaOH ( formation des corps composés non toxiques, voir diagramme
potentiel pH)
Plan : même colonne, donc même type de réactivité chimique et de propriétés physicochimiques, donc
des caractères généraux en commun qualitatifs….mais des nuances, que nous montrerons dans la
deuxième partie quantitative ou comparative
Dans un rapport de jury : ce montage ne doit pas devenir « propriétés comparées des
halogénures ! »
I. Propriété comparée des dihalogènes (0)
a. Etats physiques
Toute première observation : Cl2 gazeux, Br2 liquide, I2 solide. Pas même état physique !
Transition : Et si on les met en solution ??
b. Solubilité
Bib: Sarrazin p 148+ BUP684 + la chimie expérimentale ( JFLM/Barbe) (un chapitre entier sur X2) :
A montrer: I2 aqueux (solution peu concentré car I2 peu soluble dans eau) passe presque totalement
en phase organique, ex : cyclohexane
Br2 aqueux ou eau de brome (plus concentrée), est plus soluble dans un solvant apolaire..
Discussion: X2 soluble préférentiellement dans un solvant apolaire -->dans le bilan des interactions

soluté-soluté, soluté-solvant et solvant-solvant, les interactions de London entre X2 explique ce
comportement. Ensuite, I2 plus soluble car plus polarisable.
Transition : Ces composés peuvent se trouver sous plusieurs états d’oxydation, dont –I. Ce sont des
oxydants !
c. Propriétés oxydo-réductrices
BUP 684 p.
889
Le dichlore oxyde I- et Br-
Montage spécial avec fioles de gardes
et barboteur pour éliminer dichlore.
2h ++ Hotte
Montage : ballon avec KMnO4 ; ampoule à brome avec HCl ; Le tout relié à des barboteurs :
Solution aqueuse de bleu d’indigo + KI, empois d’amidon + KBr, fluoréceisne + NaOH concentré +
fiole de garde
On voit que Cl2 oxyde les autres, plus fort oxydant.
Transition : En général, on trouve l’élément halogène sous forme d’halogénure (-I)
II. Propriétés comparées des halogénures (-I)
a. Bases de Brönstedt
Fosset p. 68, BA p.
145
Des dosages
potentiométriques en solvant
acide acétique de HCl et HBr
permettent de mettre en
évidence leur différence de
comportement en solvant non
aqueux.
Dosage
potentiométrique
1h - hotte
HBr acide plus fort que HCl car liaison plus polarisable.
Transition : C’est aussi une base de Lewis qui peut complexer !
b. Bases de Lewis
Fosset p. 213
JCE1971, p482
Synthèse de Co(py)2X2 avec
analyse UV et Gouy. Pas
même géométrie !
UV, Gouy Long si
on met
dans
four
- hotte
Transition : Les halogènes peuvent également se retrouver dans des composés organiques.


c . Nucléofuges
Blanchard
p.167
Daumarie p.71
Solvolyse
de
tertiobutyl
- préparation
- suivi
conductimétrique
2h30 - Hotte
Conclusion :
Utilisation industrielle production Cl2 et eau de javel, blanchissement du papier, toxicité des X2
(pollution), intérêt théorique en chimie orga pour les composés halogénés…
Si problème, solubilité comparée comparée des halogénures d’argent
JFLM p. 85 Dosage de I- et Cl- par Ag+. Dosage
potentiométrique

MG 32
Le chrome et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Biblio : Greenwood, Pascal, Bernard-Bunot, Atkins (pour Tanabe-Sugano)
Introduction :
Cr Z=24 structure électronique isolé au début du 19e sous forme d’une poudre grise à partir de la
crocoïte, minerai de couleur orangé, PbCrO4. Vauquelin prépare l’acide chromique et l’oxyde de
chrome (jaune-vert). Devant toutes ces couleurs propose chrome comme nom à ce nouvel élément.
Degrés d’oxydation : 0 III et VI.
Très utilisé industrie de colorant et depuis que la réduction aluminothermique a été découverte. Il a été
possible de préparer du Cr en gde quantité utilisation comme métal très intéressant. Présentation du
plan
I. Le chrome à l’état métallique : Cr(0)
Sarrazin p. 237 Passivation du chrome. Potentiel de
Flade
2h ++ Hotte
Potentiel de Flade plus faible que dans le cas du fer, d’où son utilisation dans l’acier inoxydable
(alliage chrome/fer)
Transition : Le chrome apparaît dans les aciers sous forme de DO 0. Mais il peut aussi être oxydé.
II. Le chrome dans les complexes
a. Complexe de Cr (III)
Fosset p189 Artero
p41 Gref p63
Complexes de Cr III.
La spectro UV permet de mettre en
évidence des différences structurales entre
deux complexes possédant des ligands
diffrents. Le nombre de ligands liés au
cation central peut varier suivant la nature
du ligand.
Attention on peut faire que 3 sur 4, un
nécessitant du Cr (VI) !
UV 3h - hotte

Série spectrochimique, déduire octa ou tétra, utiliser Tanabe-Sugano.
Transition : comme dans le complexe avec En3 on génère insitu Cr II plus réactif, On peut maintenant
voir comment stabilisé le Cr II
b. Cr (II) stabilisé par complexation
Artero p130 Souil
Agreg p45
Synthèse de Co(py)2X2 avec
analyse UV et Gouy. Pas
même géométrie !
UV, Gouy Long si
on met
dans
four
- hotte
Formation in situ de H2 pour réduire Cr III. Formation d’un dimère.
III. Le chrome comme agent oxydant : Cr (VI)
Blanchard p.
208
Remplacer le tube par un vrai
éthylotest !
2h ++ Hotte
Conclusion :
Chrome dans l’organisme, cofacteur de l’insuline.
Couleur des rubis et des émeraudes, et utilisation des transition du Cr pour faire les laser à rubis

MG 33
Le manganèse et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Biblio : Greenwood, Pascal
Introduction : (Greenwood p1211)
*Abondance
- manganèse : le 12ème élément le plus abondant, le 3e élément de transition le plus abondant.
- son minerai le plus répandu (pyrolusite MnO2) utilisé depuis l’Egypte antique dans la fabrication de
verres.
- des millions de tonnes annuelles
* Propriétés
- configuration électronique [Ar] 3d5 4s2
- structure métallique à l’état solide
* Principaux degrés d’oxydation :
Degré d’oxydation : le plus stable +II (couche à demie-remplie), jusqu’au degré +VII
Montrer le diagramme de Forst (Sarrazin p. 53)
Oxydation
VII Peu de composés, oxohalide (agent oxydant très fort)
VI Fluoro et oxo
V
IV Plus haut degré ou le Mn est stable en tant que complexe (bande audio)
III Peut être stabilisé (Mn(OH)2 blanc donne Mn2O3), préparation Mn(acac)3,
octaédrique et HS, distorsion Jahn Teller (BS avec CN)
II Très bien étudié, solution aqueuse, utilisé comme fertilisant (dans les endroits
où il n’y en a pas assez), résiste à l’oxydation et la réduction grâce à sa
couche ½ remplie, intervient dans la photosynthèse (plan). BS pour CN ce
qui permet une oxydation
0 95% dans l’industrie de l’acier, essentiellement sous forme de
ferromanganèse :
-> réagit avec le sulfure (et évite la formation de FeS : matériau friable)
-> réagit avec l’oxygène (évite la formation de bulles dans l’acier)
-> augmente la dureté de l’acier.
Principale caractéristique : grand nombre de degré d’oxydation que nous allons illustrer tout au long
de ce montage. On commence par son degré d’oxydation le plus stable.

I. Utilisation du Mn (II) en chimie analytique : méthode de Winkler
JFLM p.77-81 - Dans l’eau à analyser, on ajoute MnCl2
et NaOH. On bouche rapidement. On
laisse 30 min.
- Un solide brun précipite (Mn III).
- On ajoute H2SO4, redissolution des sels.
- On ajoute KI
- On dose un volume précis de la solution.
Dosage
iodométrique
1h - Pas de
hotte
* Expérience
Le dioxygène dissout oxyde le manganèse : 4 Mn (II) + O2 + 4 H+ -> 4 Mn (III) + 2 H2O
On ajoute I- pour reformer Mn II: 4 Mn (III) + 4 I- -> 4 Mn (II) + 2 I2
On dose le diiode formé : 2 I2 + 4 S2O32- -> 4 I- + 2 S4O6
2-
* Remarque :
Pour l’oxydation, on se place en milieu basique pour que le dioxygène soit plus oxydant que le
manganèse : Mn (III)/Mn(II) : 1,51 V à pH = 0 et 0,13 V à pH = 14
O2/H2O : 1,23 à pH = 0 et 0, 39 V à pH = 14
On revient à pH acide pour dissoudre les sels et parce que I2 n’existe pas à pH basique (dismutation en
iodate et iodure). De plus, à partir de là, on peut laisser l’erlenmeyer ouvert car le dioxygène dissout ne
réagit plus avec le manganèse à pH trop acide.
* Intérêt
Permet de rendre compte de la qualité d’une eau : O2 > 5 mg/L
Transition : On vient de voir le degré III, peut-on le stabiliser ?
II. Stabilisation du Mn(III) par complexation
BA p. 215,
Pascal,
BUP 908
(pour Gouy)
- MnCl2 + acac. (Isolation du complexe Mn II possible ?)
- Puis ajout de MnO4-.
- on chauffe 10 min à 60°C, on a précipitation d’un solide
noir que l’on lave à l’eau glacée (enlever l’acétate de
sodium ?).
IR affaiblissement de la CO
UV dans CHCl3, Et2O, AcOEt
Balance de Gouy (4e- célib : spin fort, 4,98)
IR, UV,
balance
de
Gouy,
pF
Hotte
La précipitation que nous étudions a été historiquement prouvé en 1899 l’existence de ce degré
d’oxydation.
* Interprétation :
Les degrés d’oxydation II et VII réagissent pour donner III. La structure est donnée (complexe II avec 2
acac est oxydé par VII et stabilisé par un énolate d’acac)
* Application (Pascal) : La stabilité de ce complexe a été utilisée à des fins analytiques pour doser les
ions Mn3+ en présence de Mn4+. (principe du dosage en plus)

Transition : Ce degré III apparaît aussi au côté du IV dans une énorme utilisation du manganèse : la
pile Leclanché.
III. Utilisation industrielle du Mn (IV) : pile Leclanché
JFLM p. 198 ;
BUP 633 p. 928 ;
Sarrazin, BA 219
On prépare un gel d’agar-agar (40 % en masse)
contenant du chlorure d’ammonium et du dioxyde de
manganèse (IV). Dans un bécher on met une plaque de
zinc que l’on tord et au centre un barreau de graphite. On
mesure avec un voltmètre la ddp, on peut brancher une
lampe sur une pile préalablement préparé.
Pas de
hotte
* Principe :
Il s’agit d’une pile habituelle (sauf les piles boutons ou l’accumulateurs au plomb ou Cd-Ni).
(-) Zn / Zn2+ : électrolyte : MnO(OH) (III) /MnO2 (IV) / C (+)
- à l’anode : Zn donne Zn2+, l’enveloppe de zinc d’une pile usagée a quasiment disparu
- à la cathode : obtention de MnO(OH) (on a une baguette de carbone pour faire le contact électrique)
* Remarque :
Le terme de pile sèche vient de la comparaison avec les piles liquides (Daniel), mais le gel doit rester
humide pour conduire.
* Difficultés techniques :
-> avoir un grand courant : surtout à cause du pont salin, on utilise une mince pellicule de carton
imbibée d’électrolyte
-> consommer totalement les réactifs
Transition : Ce degré IV n’est pas le plus grand, on peut encore oxyder le manganèse pour en faire un
composé très oxydant.
IV. Préparation du Mn (VI) à partir du Mn (0)
BA 222 ; Walton
(en réf)
dans un poreux : un clou joue le rôle de la cathode, un
morceau de magnésium métallique celui de l’anode.
Une solution de carbonate de potassium joue le rôle
d’électrolyte support. Fixer l’intensité et non la tension.
+ Synchronie
Pas de
hotte
* Interprétation :
Il apparaît à l’anode une coloration violette : Mn + 8HO- = MnO4- + 4 H2O + 7e-
Il apparaît un dégagement gazeux : 2 H2O +2e- = 2 H2 + 2 OH- (l’agitation peut être obtenu avec un
bullage) Mesurer le pH, montrer les courbes i-E
On peut prélever des échantillons toutes les 20 minutes, on peut les titrer avec UV 425 à 700 nm avec
une courbe étalonnage (10-5 à 10-4 M). On trace le graphe du nombre d’équivalents de manganèse
obtenu par rapport au courant. Calcul du rendement faradique = Cexp/Cth et Cth = I*t/(7*F*V)
(attention : après 2h, on peut voir se cristalliser du permanganate solide Walton)
* Interprétation : On a un rendement très faible, car on a besoin de 7 e- pour faire cette réaction !

Industriellement on utilise plutôt l’oxydation du manganate de sodium ou potassium (obtenue par
oxydation basique de MnO2) sur une anode de fer.
Conclusion : aspect biologique (Pascal intro): Le manganèse est extrêmement répandu dans la nature.
On le trouve parmi les constituants minéraux des plantes et en petite quantité chez les animaux (sang
des mammifères) Métallation de la porphyrine avec Mn (III) ?

MG 34
Le fer et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Biblio : Greenwood, Pascal
Introduction : (Greenwood p1211)
*Abondance
6,2 % dans la croûte terrestre, noyau de la Terre en fer. Lié à l’histoire de l’homme : âge de fer.
Révolution industrielle, lié à la réduction des oxydes de fer par la coke.
* Propriétés
- configuration électronique
- structure métallique à l’état solide
* Principaux degrés d’oxydation :
Degré d’oxydation : 0, II, III
I. Principaux degrés d’oxydation
a. Diagramme de potentiel-pH
Sarrazin p.
119
Potentiométrie,
pH-mètrie
Pas de
hotte
Transition : On voit plusieurs degré d’oxydation non nul, mais quand est-il du degré 0 dans l’eau
b. Corrosion : mise en évidence et protection
Fosset p. 279 +
283
Mise en évidence avec une boîte
de Pétri + potentiel de Flade
Voltametrie
cyclique
Pas de
hotte
Potentiel de Flade, protection par couche d’oxyde, doit être forte.
Transition : On a vu que les DO élevés peuvent former des oxydes mais aussi des complexes.

II. Synthèses et propriétés des complexes
a. Synthèse de Fe(acac) 3 et ses propriétés magnétiques
Microchimie p.
158
Synthèse d’un complexe de Fer et
analyse par UV et Gouy
UV,
Gouy
Hotte
Transition : D’autres complexes avec des proprétés redox intéressantes.
b. Synthèse de Fe(o-phen) 3+ x et ses propriétés rédox
Sarrazin p. 87 Titrage potentiométrique
des ions Fe par Ce avec
indicateur coloré, l’o-
phénantroline ;
Pas de pipette à double
trait ! solutions trop
colorées !
Potentiomé
trie
-
Détermination de la stoechiométrie, utilisation comme indicateur coloré.
Transition : Autre complexe intéressant…
c. Formation de Fe(phenolate)3 : test des phénols
Blanchard p. 212 Dans tube à essai, qualitatif -
Conclusion :
Autre complexe Sel de Mohr pour stabiliser Fe II dans l’eau.
Rouille = 1% de l’économie mondiale
Ferrocène = PN 1973 pour Wilkinson
Synthèse de NH3 sur catalyseur de Fe

MG 35
Le cobalt et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Biblio : Greenwood
Introduction :
Connu depuis 2600 ans pour coloration des verres en Egypte en Iran
Naturellement : arseniures, oxydes, sulfures (smaltite, cobaltite, …). On l’en extrait. Production
mondiale 33 000 tonnes par an en 1995. 30 % servent toujours à colorer les céramiques et les peintures.
Co (0) : [Ar] 4s² 3d7
Co (II) : [Ar] 4s² 3d5
Co (III) : [Ar] 4s1 3d5
On voit une première explication à la stabilité de ses degrés d’oxydation (couche plein ou demi-
pleine), mais on va en voir d’autre grâce à ses complexes.
I. Principaux degrés d’oxydation
JCE 1987
fevrier, p. 165
Diagramme E-pH du cobalt
Faire les manipulations décrites
et suivre par E-pH.
Potentiométrie,
pH-mètrie
Pas de
hotte
* Expérience
Ne pas mettre de peroxyde dans la solution initiale de chlorure de Cobalt.
Utiliser KOH à 36% pour la deuxième partie, suffisant.
Faire un suivi potentiométrique et pH-mètrique
Se placer dans un bécher avec électrode et la solution initiale. Ajouter de la soude, formation d’un
précipité bleu (Td) puis rose (octa) d’un complexe neutre non soluble de Co(HO)2.
Dans un ballon de 100mL contenant 50 mL de KOH chauffé à 90°C, on ajoute la solution de CoCl2
pour obtenir une solution bleue limpide : Co(OH)42-
Puis, on ajoute du peroxyde (attention ça mousse, dégagement de O2, produit de réduction) pour avoir
le complexe neutre non soluble Co(HO)3.
On peut faire le chemin inverse en chauffant et avec HCl puis en laissant refroidir assez longtemps.
La cinétique est très lente pour revenir car complexe de Co III très stable, surtout en champ fort car d6
et tous les électrons sont « stabilisés » même si thermodynamique très favorable.
Co est un métal ferromagnétique peu courant car il est assez fortement réducteur donc on trouvera Co
principalement à l’état d’oxydation II et III qui sont stables en milieu aqueux. Il faut dire à ce moment
que le degré II aura des propriétés de réducteurs puisqu’il va par exemple facilement s’oxyder à l’air,

alors que le degré III sera plutôt oxydant, surtout en milieu acide.
Transition : On a vu plusieurs degrés d’oxydation. Dans l’eau, complexes aqua qui peuvent être très
utiles…
II. Synthèses et propriétés des complexes
a. Complexes de Co (II) : testeurs d’humidité
JFLM p. 99 ; Fosset p. 186 Test d’humidité des poupées
bretonnes et analyse avec le fosset
en UV sur la géométrie.
Pas de Tanabe-Sugano ici car
transition dans l’IR Allure du
spectre de CoCl4- soumis à doute.
UV -
* Expérience
Solution de Cobalt en milieu aqueux ou HCl. Spectres. Un pic à chaque fois sur les trois possibles. Les
autres en IR, donc pas de Tanabe-Sugano. Discuter de la position des pics sur les différents spectres
UV pour dire que correspond bien à la couleur observer. On peut seulement discuter sur les
coefficients d’extinction molaires. En déduire Td et Oh.
Bien montrer les deux fioles, les deux couleurs (bleu plus intense !)
* Discussion
Caractéristique du Cobalt de pouvoir facilement passer de la géométrie Td à la Oh. En effet, d’après la
théorie du champ cristallin, la stabilisation dans les deux géométries est très importante.
Td : -3/5 ∆t = - 12/45 ∆o
Oh : - 2/5 ∆o
Ni fait ça aussi.
Labilité des ligands de Co2+. De ce fait, complexe de Cl, très dur à isoler ! Ligands s’échangent très
rapidement ! C’est pour ça qu’on ne regarde que des spectres !
* Application
Testeurs d’humidité : complexe de butanol, quand il y a de l’eau, l’eau remplace le butanol,
changement de couleur !
Transition : Sur E-pH, Co (III) peu stable, pour le stabiliser, la complexation.

b. Synthèse de Co(acac) 3 (III)
Artero p. 99 Synthèse d’un complexe de
Cobalt, puis SEAr.
Bien prendre CoCO3 sinon pas de
base dans le milieu. Pas faire la
recri trop sale.
UV, IR,
Gouy
Hotte
* Expérience
On ajoute pentadione au Co II avec peroxyde d’hydrogène et hop dégagement de O2 (effervescence).
Filtration, on obtient un solide vert très fonce, des cristaux brillants. IR montre déplacement de la CO,
UV on voit la bande à transfert de charge vers 400nm.
* Interprétation
Stabilité du Co III !! Inerte, difficile d’échanger des ligands. Donc on passe par Co II, pour échanger
ligands Cl, eau avec acetoacetate. Puis on oxyde le cobalt en Co III.
La brillance des cristaux vient du fait qu’ils sont de l’ordre du millimètre, gros cristaux avec face bien
défini qui renvoie la lumière que dans une seule direction au contraire des microcristaux qui la renvoie
dans toutes les directions.
La bande à transfert de charge alors qu’elle ne l’est pas pour Co II.
Gouy confirme que diamagnétique, donc acetoacetate champ fort !! Complexe particulièrement stable.
* Application
Nitration de l’acetoacetate : le Co active le ligand pour qu’il se fasse nitré plus rapidement et dans des
conditions plus douces ! Rôle important des complexes !
IR confirme C-N, UV déplacé (augmentation de la force du champ par effet électroattracteur ?)
Substitution de NO2 très difficile sans cette activation.
Transition : En activant ligand, il peut ainsi catalyser réaction…
III. Propriétés catalytiques des complexes de Cobalt
Artero p. 135
������������� ����������� ������ �����
Montrer un intermédiaire réactionnel 2h UV,
trempe
Trouver E du tartrate pour montrer la thermodynamique possible ou non.
E(Co III/Co II) = 1,98 V > E(H2O2/O2) = 1,78 V > E(Co III tartrate/Co II tartrate)
Ainsi le Co peut être oxydé en III et il active le ligand tartrate pour qu’il soit décomposé en CO2
(affaiblissement des liaisons C-C)
L’intermédiaire réactionnel est particulièrement stable car le Co III en champ fort est très stabilisé.

Conclusion :
Utilisé en procédé oxo pour hydroformylation.
Complexe de la vitamine B12 : liaison C-Co organométallique naturel

MG 36
Le nickel et ses composés. Principaux degrés d'oxydation ;
structure électronique ; synthèse et propriétés des complexes.
Biblio : Greenwood, Pascal
Introduction : Greenwood
*Abondance
Résistant à la corrosion, dans les alliages
Etait dans pièces de monnaie mais très allergisant.
* Propriétés
- configuration électronique
* Principaux degrés d’oxydation :
Degré d’oxydation : 0, II
I. Principaux degrés d’oxydation
Feneuil
Pas trouvé…
Diagramme E-pH du nickel Potentiométrie,
pH-mètrie
Pas de
hotte
.
Transition : On a vu plusieurs degrés d’oxydation. Par electrolyse on peut déposer du 0 à partir du II.
II. Electrodéposition du Ni (0)
Sarrazin p 254
Besson ?
Cu, Pt, ECS dans acide borique à
différents pH et en présence de Ni (II).
Rincer la plaque à l’eau et la sécher au
sèche-cheveux pour déterminer la masse
déposée.
Synchronie pour avoir i précisément et
comparer avec la masse déposée.
Pas de
hotte
* Principe :
Intérêt industriel : casserole, tube, éléments thermochauffant en cuivre nickelé
On a également une méthode passive, où l’on applique sur le métal un revêtement protecteur, tels que
des peintures, des vernis, des revêtements par d’autres métaux. Par électrolyse d’une solution contenant

le cation à déposer : électrozingage, chromage, le nickelage…
Bien que le zinc soit plus réducteur que le fer, il résiste mieux à la corrosion atmosphérique car il se
recouvre d’une couche d’hydrocarbonate de zinc, adhérente et imperméable.
Deux cas de figures :
- si le métal protecteur est plus oxydable (pas de gros soucis s’il y a des défauts, mais grosse
couche est nécessaire) Le nickelage du cuivre et l’électrozingage du fer.
- si le métal protecteur est moins oxydable (pas de défauts, sinon corrosion rapide, mais une fine
couche suffit).
* Expérience :
On effectue les droites de polarisation dans plusieurs conditions : à différents pH sans Ni, aux mêmes
pH avec Ni. On voit que le plus pH augmente, plus il y a de différence entre les droites de réduction du
Ni et de H+. On préfère donc travailler à pH élevé pour que H+ ne se réduise pas à la place du Ni. Mais
pas trop élevé pour éviter que le Ni II ne précipite sous forme d’hydroxyde.
* Remarque :
On peut former une couche superficielle protectrice grâce à une réaction chimique, on plonge une pièce
d’acier dans une solution chaude avec des ions phosphates, on provoque ainsi l’apparition d’une
couche de phosphate de fer imperméable. Il s’agit de la parkérisation, utilisée dans l’industrie
automobile pour protéger les carrosseries (HP)
Transition : Ni allergisant, important de pouvoir le doser.
III. Utilisation du Ni (II) en chimie analytique
Brenon-Audat
p. 90-91
2h Dosage
colorimétrique
Transition : On l’a complexé pour le doser, mais on peut également faire d’autres complexes
IV.Synthèses et propriétés des complexes
a. Synthèse des complexes de Ni (II)
JCE 1980 p.
826 ; JCE
1980 p. 900 ;
BUP 2008 p.
41
Synthèses de plusieurs complexes,
puis études IR et UV. Interprétation
avec Tanabe. Balance de Gouy.
long UV, IR hotte

On décrit la synthèse de plusieurs complexes.
Transition : Propriétés, la première que l’on voit : leurs couleurs !
b. Propriétés optiques
JCE 1980 p.
826 ; JCE
1980 p. 900 ;
BUP 2008 p.
41 ; Fosset
Synthèses de plusieurs complexes,
puis études IR et UV. Interprétation
avec Tanabe. Balance de Gouy.
long UV, IR hotte
Série spectrochimique, recherche de leur structure.
Transition : Vérifié par propriétés magnétiques.
c. Propriétés magnétiques
JCE 1980 p.
826 ; JCE
1980 p. 900 ;
BUP 2008 p.
41
Synthèses de plusieurs complexes,
puis études IR et UV. Interprétation
avec Tanabe. Balance de Gouy.
long UV, IR hotte
Balance de Gouy, recherche d’électrons célibataires. Colle avec les résultats précédents.
Conclusion :
Le nickel est le plus allergisant de tous les métaux. Pour cette raison, le nickel a été exclu de l'alliage
utilisé pour les nouvelles pièces de monnaie européennes. Il y a des polémiques sur l'utilisation du
Nickel dans les amalgames dentaires sous la forme de Ni/Cr.
Rôle en catalyse : Ni de Raney

MG 37
Le cuivre et ses composés. Principaux degrés d’oxydation ;
structure électronique ; synthèse et propriétés de complexes.
Biblio : Pascal, Greenwood, …
Introduction :
Connu depuis très longtemps (référence dans la Bible et l’Iliade) car la métallurgie du cuivre natif est
simple et le cuivre est l’un des éléments les plus abondants dans la nature. Alliage avec l’étain pour donner
le bronze. Pascal
Produit à des millions de tonnes par an et c’est un des métaux de transition bon marché. Il est
principalement utilisé pour ses propriétés de bon conducteur. (cf Greenwood et BUP 790 p12)
Z=28 Configuration électronique.
Le cuivre possède une sous-couche d remplie et un électron s. Ses propriétés sont très différentes des
alcalins, car les électrons de la couche d écrantent de façon bien moins efficaces la charge nucléaire
ressentie par l’électron s.
Il existe des degrés 0, I, II III ou IV, les deux derniers étant beaucoup moins fréquents. Cotton
I. Degré 0 : préparation du cuivre par électrolyse
L’affinage industriel du cuivre par voie sèche était insuffisant à cause des impuretés (fer, nickel, cobalt,
manganèse plomb parmi d’autres). Aujourd’hui la purification est réalisée par affinage électrolytique.
Le cuivre est le meilleur conducteur électrique après l’argent, ainsi qu’un très bon conducteur de chaleur.
De nombreuses applications :
- fils et câbles électriques
- canalisation
- toitures…
BUP 790
Miomandre p318
La lixiviation du minerai de cuivre avec agitation des
cuves
On réalise un minerai en mélangeant du sable, de
l’oxyde de cuivre II et du sulfate de fer III puis on
dissout les cations métalliques dans de l’acide
sulfurique. On fait précipiter le fer par ajout de la
soude. On filtre et on plonge une lame de fer qui
permet la réduction du cuivre
suivi pH-métrique possible
1h30 Pas de
hotte

peser les électrodes à la fin
Tension 2 volt
Transition : On a vu que le cuivre II servait à produire du cuivre 0 et donne une couleur bleue,
pourquoi ? complexe !
II. Synthèse et étude des complexes de Cu (II)
a. Synthèse
Synthèse du [Cu(en)2(H2O)2]2+ Artéro p31
����������������������������� ������ ������������ �������� ���������������������������������� ����������������������������������
Transition : Puis étude spectroscopique de ce complexe ainsi que Cu(H20)6 et Cu(EDTA)
b. Etude specstroscopique
On retrouve la série spectrochimique (les azotes sont à champ plus fort que les oxygènes)
En ce qui concerne l’absorbance, le complexe aqua a un très faible coefficient (ce qui est en accord avec
Oh). Pour les deux autres complexes, le coefficient est bien plus grand mais relativement faible (<100). Ils
sont Oh également mais présentent des déformations de Jahn-Teller qui abaisse la symétrie (effet de pince
des ligands bidentates et hexadentates qui différencient les ligands).

III. Degré I : stabilisation
Configuration électronique : Cuivre I d10
� Ces composés sont diamagnétiques et bien souvent incolores. La stabilité relative du cuivre II et I en
solution aqueuse dépend des anions ou autres ligands présents. Cu I instable dans l’eau (sauf CuCl et
CuCN très insolubles dans l’eau). On peut déplacer l’équilibre : agent complexant pour déplacer en faveur
de Cu II, ligands CN-, I-, Me2S en faveur de Cu I. La géométrie peut également intervenir, on peut
regarder le rapport de concentration à l’équilibre pour l’éthylènediamine et pour l’ammoniaque : 10^5
versus 10^-2.
Fosset p. 174
Utiliser KNO3
au lieu du
perchlorate
d’ammonium
Le degré d’oxydation I du cuivre peut
être stabilisé en solution par
complexation. L’étude
polarographique permet de mettre en
évidence ce phénomène par le
changement de forme du
polarogramme de réduction d’une
solution de cuivre II en fonction de la
quantité de ligand introduite.
Polaro
Deux vagues au lieu d’une ! En plus la hauteur de chaque vague est la moitié de celle sans ammoniac.
Hauteur de vague proportionnelle au nombre d’électrons échangés ! On regarde les valeurs théoriques et
effectivement, le Cu I ne se dismute pas avec l’ammoniac.
Conclusion
�� ������� ����������������������� ��!�������������� ����������"�����������������������##� ���� �� ����������������� ������������ ��$����� ����� ������� ��"���%%���� (JD33, ou réaction de Sonogashira, test à la liqueur de Fehling). &���������#������'�� ������ ����� ��������������� ��� ����� ����'�(������#� ������������ ������)� ������������' ������%(�������� ��� ������ ������� ���# ���������� ������) �)��%* �)�% )
Degré non présentés : III et les supraconducteurs à haute température YBaCu3Oè-x
������� ����� ������ ��+��������&�����������,-�.��/01���� �'� �#� ��� �+������23��� ��4* ���������������������5��4*5��





























