Embed Size (px)
Citation preview

A R T I C L E
11isuma
ILLU
STR
ATI
ON
S:
KA
TY L
EM
AY
RÉSUMÉ ¨ Il existe désormais une séquence préliminaire de
l’ensemble du génome humain et de la plupart des gènes encodés.
Dans le présent article, nous expliquons ce qu’est le génome, ce
que représente son séquençage et comment il a été réalisé, avant
d’aborder une réflexion sur l’incidence de cette découverte sur
notre façon de faire de la recherche scientifique et, sur un plan plus
fondamental, sur notre connaissance de nous-mêmes. La descrip-
tion complète du génome humain constitue le fondement de la
biologie humaine et la condition essentielle à la connaissance
approfondie des mécanismes de la maladie. Comme telle, l’infor-
mation découlant du séquençage du génome humain équivaudra à
un guide de référence pour la science biomédicale XXIe siècle. Elle
aidera les chercheurs et les cliniciens à comprendre, diagnostiquer
et finalement traiter nombre des 5 000 maladies génétiques qui
affligent l’humanité, notamment les maladies plurifactorielles dans
lesquelles la prédisposition génétique et les éléments environne-
mentaux jouent un rôle important. (Traduction : www.isuma.net)
ABSTRACT ¨ We now have a draft sequence of the entire human
genome and most of the genes it encodes. Here we explain what
the genome is, what it means to sequence it, outline how the
sequencing was done, and reflect on what this means for the way
we do science and, more fundamentally, how we understand
ourselves. A comprehensive description of the human genome is
the foundation of human biology and the essential prerequisite for
an in-depth understanding of disease mechanisms. As such,
information generated by the sequenced human genome will
represent a source book for biomedical science in the 21st century.
It will help scientists and clinicians to understand, diagnose and
eventually treat many of the 5000 genetic diseases that afflict
humankind, including the multifactorial diseases in which genetic
predisposition and environmental cues play an important role.
B Y S T E P H E N S C H E R E R
Th e H u m a n
G e n o m e
P r o j e c t

12 Autumn • Automne 2001
the human genome project
Genes are instructions thatgive organisms their charac-teristics. The instructions are
stored in each cell of every livingorganism in a long string-like mole-cule called Deoxyribonucleic Acid(dna). dna molecules are subdividedinto finite structures called chromo-somes (Figure 1). Each organism has acharacteristic number of chromo-somes. For humans the number is 46(23 pairs) and this complete set ofgenetic information is called thegenome (Figure 2).
DNA, genes and genomesHuman dna looks like a twisted lad-der with three billion rungs. Ifunwound, your dna would stretchover five feet, but it is only 50 tril-lionths of an inch wide. The totalamount of dna in the 100 trillion orso cells in the human body laid end toend would run to the sun and backsome 20 times. The three billion rungsare made up of chemical units, called“base pairs,” of nucleotides —adenines, thymines, cytosines and gua-nines, represented by the letters A, T,C and G. Particular combinations ofthese dna base pairs (or genes) consti-tute coded instructions for the forma-tion and functioning of proteins, whichmake up the body and govern its bio-logical functioning (examples of pro-teins include insulin, collagen, diges-tive enzymes, etc.). Ribonucleic acid,(rna) is a single stranded copy of dnathat acts as an intermediate messengermolecule that allows dna sequence tobe translated to protein. This centralprocess, whereby dna transcribes torna, which in turn transcribes to pro-tein, underlies all of life.
Some genes are made up of only afew hundred base pairs, others run toa couple of million base pairs. It is nowestimated that we have around 30,000to 40,000 genes (much more work willbe required to determine the precisenumber), but those genes account forless than five percent of our dna, withthe rest being filler. For lack of a betterterm, the non-genic dna is oftendubbed “junk” since its role is not ful-ly known (in fact this might be a goodterm since just like the cell we oftenkeep our “junk” just in case we need it
contrary to “garbage” which we, andthe cell, throw away).
The Human Genome Project and DNA sequencing“Sequencing” is the process of deter-mining the specific order and identityof the three billion base pairs in thegenome with the ultimate goal of iden-tifying all of the genes. “Mapping” isthe process of identifying discrete dnasegments of known position on achromosome which are then used forsequencing (mapping is a crucial stepfor proper reconstruction of thegenome; it usually precedes sequenc-ing but is also necessary in post-sequencing). To give one a sense of theenormity of the task to decode thehuman genome, a simple listing of allof the atcg combinations would fillhundreds of phone books. And hav-ing the sequence is only a first step infinding the genes, understanding howthey operate, and how particulargenetic discrepancies are associatedwith diseases. Using this dna sequenceinformation to diagnose, prevent ortreat those genetic predispositions orcauses of disease is an even more com-plex task.
When the idea of sequencing thehuman genome was first proposedabout 15 years ago, it was highly con-troversial. For one thing, given thetechnology at the time, sequencing
dna was painstakingly slow andexpensive. Sequencing the entirehuman genome seemed impractical.Others felt it made more sense tofocus research effort and money onlyon discovering particular genesthought to be associated with dis-eases, rather than trying to spell outthe entire genome. However, even fordisease projects aimed at finding sin-gle gene disorders (such as that forcystic fibrosis) the worldwide costcould often run in the tens of millionsdollars before success was achieved.The idea for the human genome pro-ject eventually won support, justifiedas an investment in fundamental bio-logical infrastructure upon which thescientific community around theworld would build exciting newresearch that would lead to the diag-nosis and treatment of disease.
The enormity of the challengeinspired the creation of an interna-tional, publicly funded research ini-tiative called the Human GenomeProject (hgp), officially launched inthe early 1990s. The Human GenomeOrganization (hugo) brings togetherover 1000 scientists from 50 countriesworldwide, and has a mandate thatincludes promoting international col-laboration within the hgp. The earli-est genome meetings were attended bya rag-tag group of usually fewer than100 biologists of diverse background
FIGURE 1
Protein
Gene
CellNucleus
Chromosomes
DNA
Figure 1. The human genome consists of 23 pairs of chromosomes that are comprised of DNA. Approximately 5% of the DNA along the chromosome encodes for genes. Genes make proteins. Proteins provide cellular structure and functional activity within the body.

13isuma
the human genome project
and research interests, only a few ofwhom had some type of vision of howto accomplish this distant goal (Fig-ure 3). Since 1990, the strategies andmeasures of successes of the hgp havemost often been debated at an annualmeeting held in Cold Spring HarborLaboratories, New York (directed byJames Watson, co-discover of dna).The most accurate documentation ofthe science and progress behind thegenome project can be found withinthe abstracts of this meeting. The ear-ly consensus was that before large-scale and cost-effective sequencing ofthe entire genome could be complet-ed, numerous technological develop-ments, associated with mapping andsequencing processes as well as withthe computational capacity to makesense of the resulting information,were essential.
With developments in technologyover the last few years, the seqencingis now done through a process calledfluorescence-based dideoxy sequenc-ing (Figure 4a). Fragments of dna arefirst cloned in bacteria. They are thenput into a chemical reaction with freenucleotides, some of which are taggedwith fluorescent dyes. Nucleotidesattach themselves to the dna frag-ments in a particular order. Similarly,
dyed nucleotides can attach them-selves to the dna fragments, but othernucleotides will not adhere to the dyednucleotides. Thus the chemical reac-tion generates dna fragments of vari-ous length that each terminate at flu-ourescently dyed A, T, C or Gnucleotides. The underlying sequencefor the range of the dna fragmentscreated in the chemical reaction is thendetermined by an automated sequenc-ing machine. The synthesized dnafragments are negatively charged. Thesequencing machine sets up an electri-cal field. The dna fragments movethrough a pourous gel toward the pos-itive charge, with shorter fragmentsmoving more quickly through the gel.The fluorescent, tagged bases at theend of the fragments can be detectedwith the help of a laser, and the result-ing information stored on a comput-er. As a result, the order in which theparticular tagged nucleotides are readreflects their order on the stretch ofdna that has been replicated in thechemical reaction.
Each reaction reveals the sequenceof about 500 letters (A,T,C,G) of dnabefore the process runs its course. Oncethese relatively tiny sequences areobtained, their place in the overallgenome dna sequence must be deter-
FIGURE 2
Figure 2. The 23 pairs of human chromosomes one from each pair being inherited from one parent. Note the interchange of material between chromosomes 7 and 13 which disrupts a gene leading to disease.
1
6 7 8 9 10 11 12
181716151413
19 20 21 22 X Y
2 3 4 5
It will likely
take two to three
years for the draft
form of the dna
sequence to reach
what is considered
to be in a finished
state.

14 Autumn • Automne 2001
the human genome project
mined. To achieve a working draft dnasequence of the genome, two approach-es were followed. The hgp began bycreating a detailed map that would pro-vide a framework for the subsequentdna sequencing. Numerous dna“markers,” that is segments with anidentifiable location on the chromo-some (Figure 4b), were generatedthrough many worldwide initiatives.
This enabled the hgp to breakdown the genome into about 30,000sections, each containing an average of100,000 to 200,000 base pairs (Figure5). For the actual sequencing, each ofthese sections was broken down intostill smaller fragments, of about 2000base pairs. Initially, the plan was toput the fragments to be sequenced inorder, followed by complete sequencedetermination of each fragment in asystematic manner so that the entirehuman dna sequence would berevealed. This ‘clone-by-clone’ methodproduces highly accurate sequencewith few gaps. However, the upfrontprocess of building the sequence mapsis costly, time consuming and there-fore can be progress-limiting.
In 1998, to accelerate progress, amajor deviation from this plan was
the decision to collect only partialdata from each dna fragment, hence,a working or rough draft (the changeof strategy was partly due to thelaunching of a privately funded com-pany, Celera, which decided to deter-mine the dna sequence of small piecesof human dna totally at randomusing the “whole genome shotgun”(wgs) approach described below).Working draft dna sequence usuallycovers 95 percent of the genome(maintaining 99% accuracy) but it isdivided into many unordered seg-ments with gaps between. Additionalsequencing is required to generate thefinished dna sequence such that thereare no gaps or ambiguities, and thefinal product is greater than 99.99percent accurate. At the time of writ-ing, close to 90 percent of the humangenome sequence has been determined(approximately 70% of this in work-ing draft and 30% in finished form).
A second approach to generating adraft sequence of the human genomeis the whole genome shotgun (wgs)associated with Celera Corporation.In wgs, sufficient dna sequencing isperformed so that each nucleotide ofdna in the genome is covered numer-
ous times in fragments of about 500base pairs. Identifying where thoseindividual fragments fit in the overalldna is accomplished through the useof powerful computers that analyzethe raw data to find overlaps.
The wgs approach was used by sci-entists at The Institute for GenomicResearch to generate the first completesequence of a self-replicating organismcalled Haemophilus influenzae as wellas many other single-cell organisms.The advantage of wgs is that theupfront steps of constructing maps arenot needed. For organisms with muchlarger and more complex genomes,such as the fruitfly Drosophilamelanogaster and humans, assembliesof sequences are expected to be com-plicated by the presence of a vast num-ber of repetitive elements (approxi-mately 50% of human dna is repeats!).Notwithstanding, Celera initiated awgs project of the Drosophila genomeand in doing so, 3.2 million sequencereads were completed (giving a 12.8times coverage of the 120 Mb (millionbases) genome). Based on these data,115 Mb of dna sequence were assem-bled and, although it was quite com-prehensive, the genome was still divid-
FIGURE 3
Figure 3. Timeline of the Human Genome Project (HGP). A historical summary of some of the enabling technologies (on the left) and the achievements (on the right) leading up to and including the HGP are shown. The formal international HGP began around 1990. Other aspects of the HGP are the study of model organisms, analysis of genome variation, and the development of bioinformatics. Establishing training and public education programs, as well as the study of the ethical, legal, and social issues of genetics research in society are also important priorities.
DNA cloning and sequencing
Polymerase chain reaction
Automated DNA sequencing
Cloning large DNA molecules
Posit ional c loning
New genetic markers
Gene predict ion algorithms
Whole genome shotgun
SNPs and microarrays
Comparative DNA sequencing
1970-80s
1985
1986
1987
1989-1990
1990-1991
1992-1996
1993-1998
1998-2000
2000
2000-2005
Concept for human l inkage maps
Discussions of a human genome effort
Human Gene Mapping Workshops
First human l inkage map
Human Genome Organizat ion forms
Human Genome Project begins
Genetic maps completed
Physical maps completed
DNA sequencing begins
“Draft” DNA sequence
DNA sequence annotat ion
Having the
sequence is only a
first step in finding
the genes, under-
standing how they
operate, and how
particular genetic
discrepancies are
associated with
diseases.
15isuma
the human genome project
ed by 1630 gaps. Closure of the gaps isbeing completed with the help of themaps generated by the hgp.
Following the experience gainedfrom the Drosophila melanogasterproject, Celera planned a wholegenome assembly of human dna. Theaim of the project was to producehighly accurate, ordered sequencespanning more than 99.9 percent ofthe human genome. Based on the sizeof the human genome and the resultsfrom the Drosophila experiment, itwas predicted that over 70 millionsequencing reactions would need to becompleted. The alignment of theresulting sequence assemblies alongthe genome would be accomplishedusing the large number of dna mark-ers (including genes, see figures 4b and5) and physical maps generated by theongoing hgp. Since sequencing effortswere escalated by the publicly fundedhgp (and released each night on theWeb) using its approach, Celera couldalso easily integrate this data into theirassemblies thereby greatly reducing theamount of sequencing required. In theend, Celera completed approximately28 million reads and merged thesedata with dna sequence in publicdatabases. The final sequence assem-blies were ordered along chromosomes
using the dna markers and maps fromthe hgp.
It will likely take another two tothree years for the draft form of thedna sequence to reach what is con-sidered to be in a finished state. Thiswork is ongoing around the world inbig and small laboratories alike. Thecompleteness and accuracy of theseversions of the human genomesequence will be tested by many typesof experimentation over the nextdecade. In future dna sequencing pro-jects, as is currently ongoing for themouse genome, a combination of theclone-by-clone and wgs strategies willlikely become the standard becausethey are largely complementary.
Annotation of the human genome DNA sequenceAscribing functional meaning to allthe genes and other important biolog-ical information encoded by the dnain our genome will be the ultimategoal of the hgp. This process is oftencalled “annotation.” With this infor-mation, it will be possible to investi-gate the roles of all of the gene prod-ucts, how they are controlled, howthey interact, and how they might beinvolved in disease. The process ofannotating the human dna sequence
FIGURE 4
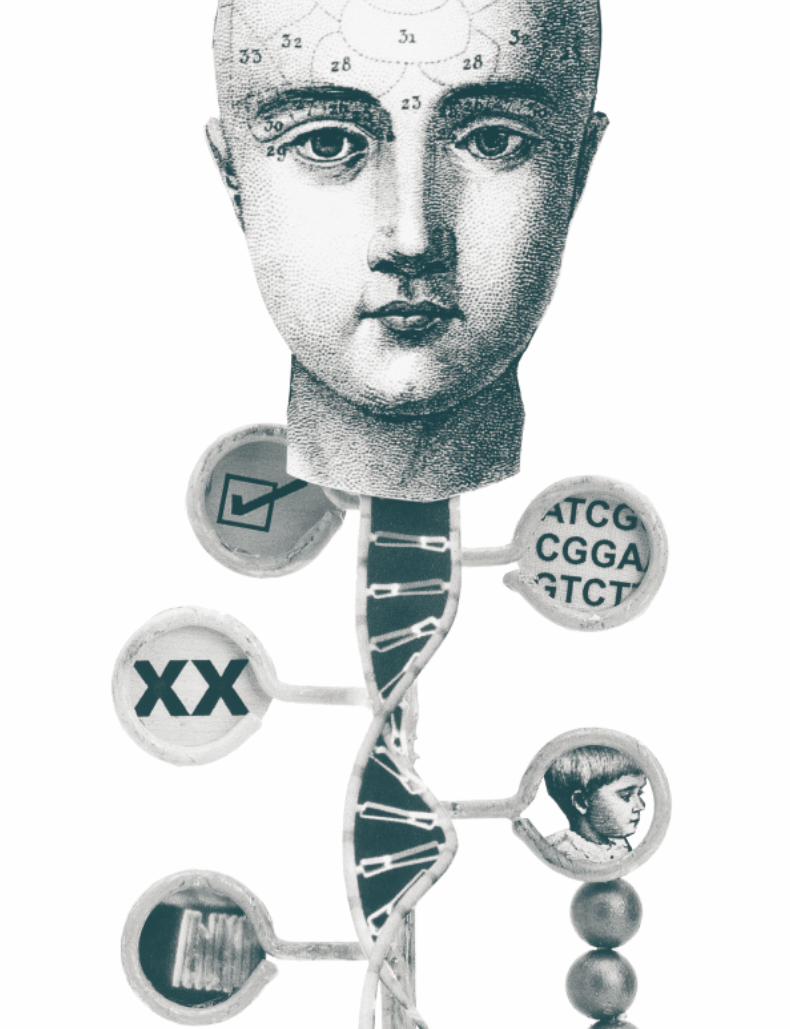
Figure 4. Techniques of the HGP. (A) DNA sequencing and (B) positioning a gene on a chromosome, in this case the CASPR2 gene on human chromosome 7.
G A C A C C C G GG A C A C C C G G
G A C A T C C G GG A C A T C C G G
A. B. 7
1413
11.2
11.21
22
21
15.315.215.1
1211.1 11.1
11.2211.23
21.121.221.322
31.131.231.3
3233343536
CASPR2
If a segment of dna
has been conserved
throughout
evolution, it almost
always encodes a
gene or a regulatory
element controlling
the expression of
that gene.

16 Autumn • Automne 2001
the human genome project
will take several forms including thecatalogue of the genes; the identifica-tion of the genes and dna sequencevariations that either directly cause orare associated with disease; and thestudy of variation between individu-als in the population.
Cataloguing the genes: Just as diffi-cult as constructing maps or determin-ing the dna sequence of the humangenome will be identifying all of thegenes it encodes. As mentioned earli-er, the genes comprise less than fivepercent of the dna scattered through-out the three billion nucleotides ofgenetic information. To complicatematters, human genes almost alwaysappear as discontinuous segments ofdna along a chromosome divided intogene-coding regions (exons) and non-coding regions (introns) (Figure 6). Tocomplicate matters further, there aredna stretches that do not encode forproteins but, instead, the rna moleculeperforms a biological function withinthe cell. Notwithstanding, most geneshave characteristic features that can beidentified using computer programstrained to search for these identifiers.For example, genes usually start with
the sequence atg, end with tag, tgaor taa, and the boundaries betweenintrons and exons are often defined bythe dinucleotides at and cg, respec-tively (Figure 6).
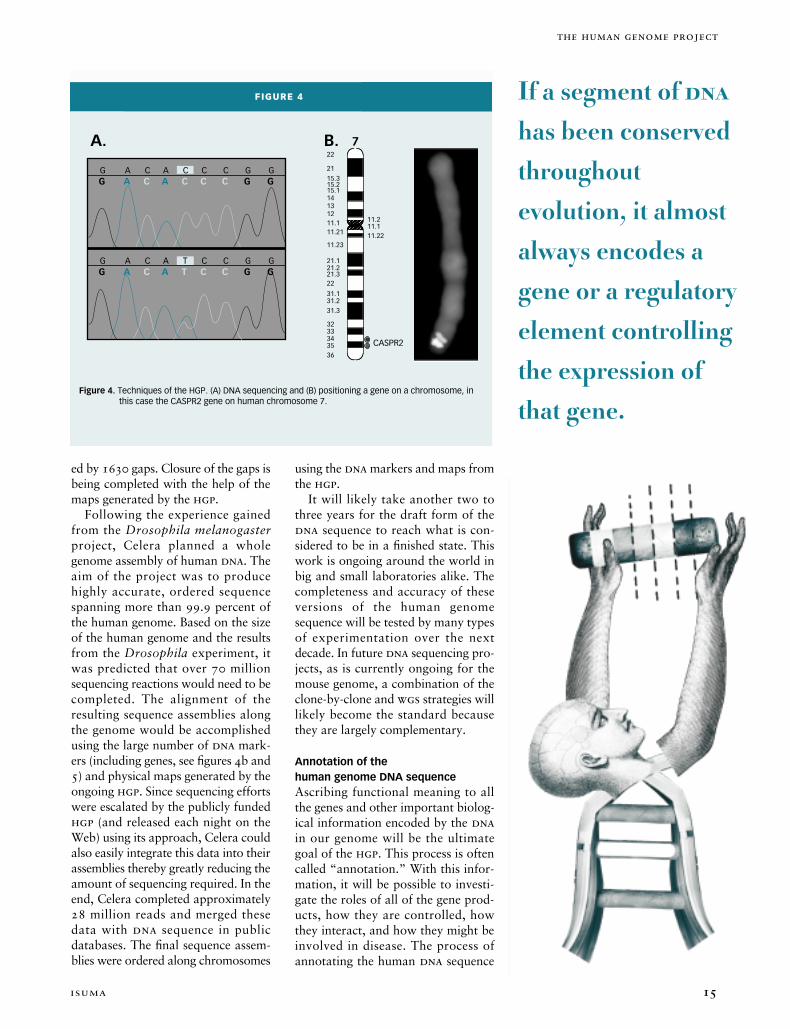
Another powerful approach to find-ing genes is to compare the dnasequence between two organisms tosearch for shared elements (Figure 7).If a segment of dna has been con-served throughout evolution it almostalways encodes a gene or a regulatoryelement controlling the expression ofthat gene. Almost every human geneknown can also be found in thegenomes of other mammalian species(e.g., mice and dogs) with the similar-ity of the two sequences often exceed-ing 85 percent. The genomes ofhumans and chimpanzees are 98.5percent identical. Some genes, includ-ing those that encode proteins involvedin replicating dna (and are thusabsolutely crucial to survival of everyorganism), are highly conserved inevery species from human all the waydown the evolutionary tree to yeastand bacteria. Other genes that origi-nated from a common progenitor haveevolved between species to have
unique functions such as specific fer-tilization (Figure 7).
Current estimates put the number ofgenes in the human genome at between30,000 and 40,000. The precise num-ber continues to be hotly debated, andit could very well be that there areupwards of 50,000 to 60,000 or moregenes in the genome. Discrepancy innumbers can be attributed to differentinterpretations in the definitions, andthe use of different datasets which, inevery case, are still largely incomplete.For example, at present we only knowthe complete sequence from start toend of about 25 percent of the 30,000to 40,000 genes that have been pre-dicted using computer algorithms.
One of the most interesting obser-vations to come out of genomesequencing is that the number of genesdoes not necessarily correlate withorganism complexity or its place in theevolutionary hierarchy. For example,the fruitfly Drosophila melanogasterhas 13,000 genes and comprises 10times more cells than the worm C. ele-gans, which has 19,000 genes.
Disease gene identification: Theimmediate medical application of
FIGURE 5
Figure 5. Systematic mapping and sequencing of human chromosomes using a clone-by-clone approach. DNA markers from a chromosome region are positioned and simultaneously used to order large cloned DNA molecules (called YACs and BACs). BACs are broken down in ‘sub-clones’ for DNA sequencing. Obtaining partial sequence of the BACs represents ‘draft’ sequence. In the whole genome shotgun approach (WGS) the intermediate steps of cloning and mapping BACs and YACs are not completed. It is estimated there are 3 billion A,T,C,and G’s in the human genome.
YAC clones
(1,000,000 bp)
BAC clones
(100,000 bp)
Sub-clones forDNA sequencing(1000 bp)
ATCGGGTCTTTCGGAACGT
Chromosome
Chromosome regionGenetic marker
Expressed sequence tag
Gene
About 5,000
human diseases are
known to have a
genetic component,
and 1,000 disease-
associated markers
or genes have
already been
isolated.
17isuma
the human genome project
human genome information is in iden-tification of genes associated with dis-ease, and the pursuit of new diagnos-tics and treatments for these diseases(Figure 8). About 5,000 human dis-eases are known to have a geneticcomponent, and 1,000 disease-associ-ated markers or genes have alreadybeen isolated over the years. Until the1980s, the primary strategy for iden-tifying human genetic disease geneswas to focus on biochemical and phys-iological differences between normaland affected individuals. However, forthe vast majority of inherited single-gene disorders in humans, biochemi-cal information was either insufficientor too complicated to give insight intothe basic biological defect.
In the early 1980s an alternativestrategy was introduced that suggest-ed disease genes could be isolated sole-ly on their location along the chromo-some. This approach, now called“positional cloning,” consists of firstdetermining which of the 23 chromo-somes the disease gene resides on.Then genes on the chromosome aresystematically tested for dna sequencechanges (or mutations) that occur onlyin the family members having the dis-ease but which are not found in unaf-fected individuals.
Prior to the initiation of the hgp,the disease gene for Duchenne/Beckermuscular dystrophy, cystic fibrosis anda few others were identified. With thedevelopment of new mapping resourcesand technologies generated by the hgp,the positional cloning process wasgreatly simplified. This has acceleratedthe pace of discovery of new diseasegenes including those for fragile x syn-drome, Huntington disease, inheritedbreast cancer, early onset Alzheimerdisease and many others.
With the hgp sequence now welladvanced, it becomes possible to dis-sect the molecular genetics of multi-factorial diseases such as cancer andcardiovascular disease which involvemultiple combinations of genes andstrong environmental components.The study of these common diseaseswill require careful patient and familydata collection, thorough clinicalexamination, systematic dna analysisand complex statistical modeling.
FIGURE 6
Figure 6. Anatomy of a gene. While genes occupy less than 5% of the DNA on a chromosome they often have characteristic features that allow them to be identified amongst the so-called ‘junk’ DNA. Promoters are the regions of DNA that turn genes on and off. Most genes start with ATG and end with TAA, TGA, or TAG. The GT and AG di-nucleotides usually flank the parts of genes (exons) that code for proteins. RNA and messenger RNA (mRNA) are intermediates to the production of proteins.
DNA
AAAAAAAAA
RNA
mRNA
promoter exons intron
Transcription
Processing
protein
ATG TAAGT GTAG AG
FIGURE 7
Figure 7. Genes conserved between species. Thirteen human genes on human chromosome 7 are compared to the equivalent genes in the mouse genome. A 45 degree line between indicates the DNA sequence of the human and equivalent mouse gene is >85% identical. The deviation at the intersection of the zonadhesin (ZAN) genes occurs because each encodes a protein that is involved in the process of species-specific fertilization of sperm to egg. Therefore, during evolution the DNA sequences of these genes diverged from each other to fulfill the role of making a protein unique to the survival of that organism.
ACHE
BAF53BTFR2
PERQ1
GNB2
EPORPP20
EPHB4
ZAN
TRIP6CIP1
Hs.157779ASR2
Ach
e
Baf
53b
Tfr2
PerQ
1
Gnb
2
Epo
Rpp2
0
Eph4
b
ZAN
Trip
6C
ip1
Hs.
1577
79A
SR2
1
1 250,000
250,000
Mouse gene
Hu
man
gen
e

18 Autumn • Automne 2001
the human genome project
Human genome sequence variation:There are many types of variation inthe human genome with singlenucleotide changes (snps) being themost frequent occurring in one of1000 nucleotides. Therefore, on aver-age, each genome contains approxi-mately three million snps. Other dnasequence variations include copy num-ber changes, insertions, deletions,duplications, translocations, inver-sions and more complex rearrange-ments, but these are not as frequent.There are, thus, significant differencesbetween the genome of the two par-ents whose chromosomes cometogether to form new life. This genet-ic variation is the basis of evolution.It is this genetic variation that con-tributes, in part, to health and to eachindividual’s unique traits. However,when dna sequence variants occurwithin genes or affect their expression,disease can sometimes result.
Technological advances, availabili-ty of genetic markers and higher reso-lution snp maps generated by the hgphave revolutionized the study ofhuman genetic variation. The bio-medical applications include identify-ing variations within specific genesthat cause or predispose to disease,finding gene-environment interactionsthat might have toxologic implicationsand identifying variations in immuneresponse genes that will have implica-tions for transplantation and vaccinedevelopment.
The study of human genome vari-ability has also greatly improved our
grasp of human history, evolution ofthe human gene pool and our basicunderstanding of genome evolution.For example, studies of dna variationhave revealed that the vast majority ofgenetic diversity (>80%) occursbetween individuals in the same popu-lation even in small or geographicallyisolated groups. Moreover, most vari-ation seems to predate the time whenhumans migrated out of Africa around200,000 years ago. Therefore, the con-cept of homogeneous groups (or races)having major biological differences isnot consistent with genetic evidence.
The Human Genome Project and society With the success of the hgp, we haveovercome the psychological barrier ofcracking nature’s code and now facethe more daunting responsibility ofhaving power over the genetic destinyof our own species. As such, the hgpjoins the ranks of the other massivescientific undertakings of the 20thcentury — splitting of the atom andthe conquest of space—in transform-ing civilization. And, just as Galileo’swork was foundational to proving theCopernican theory which debunkedthe notion that the earth was the cen-tre of the universe, the hgp provesthere are human-to-animal dnasequence links, thus substantiatingDarwin’s theory that we are not aunique life form. With this informa-tion, the hgp promises to give us pro-found knowledge as the basis tounderstanding how our minds work,
FIGURE 8
Figure 8. Applications of the Human Genome Project. The immediate application of the human genome information are identification of genes associated with disease, and development of diagnostic and predictive tests. Based on this information the pursuit of new treatments for these diseases can be pursued.
Diagnostic tests
Carrier tests
Population screening Basic defect
Disease pathology
Gene/Protein therapy
cure
We now face the
more daunting
responsibility of
having power
over the genetic
destiny of our
own species.

19isuma
the human genome project
to be able to quantitate nature andnurture, and increasingly, to be ableto alter our genetic constitution.
Our preoccupation with dna hasalready shed light on historical puzzlessuch as the roots and migrations ofancient peoples, historical demographyof cultures and human genetic diversi-ty. We now know that the age-old prej-udices of caste, race or royalty, whichgranted undue importance to biologi-cal inheritance, have no substantialgenetic basis. The hgp gives the scien-tific basis for promoting the concept ofone race and of equal opportunity forall at the beginning of life. Sadly, thisknowledge, on its own, is unlikely tobe enough to overcome deeply embed-ded racial and other prejudices.
Each success of the hgp, from con-ceptualization through to mappingand dna sequencing, followed closebehind advances in technology andimplementation of new strategies, onebuilding on the other. This large-scale,technology-driven approach will epit-omize how science is to be conductedin the 21st century. To identify genesinvolved in disease we will determinethe differences in dna specific to thoseafflicted. To identify traits unique toHomo sapiens we will study differ-ences in dna between species. To sort
through all of this information we willdesign even more massive computersand newer technologies. And the timebetween acceptance of a new technol-ogy and its application will onlybecome shorter. To keep up with thisrevolution we may be forced toredesign ourselves using the sametools that brought on the change.
It took less than 50 years from thediscovery of the structure of the dnamolecule in 1953 to the cracking ofthe human genetic code. In theupcoming years, scientists will workvigorously on the hgp to find newgenes, to determine the function of thegene products and to apply all of thisinformation to the study of commondiseases. While the hgp will not be acure-all, for some diseases there willbe cures in our lifetime. And becauseof the hgp, preventive medicinethrough early diagnosis and directivegenetic counselling will, over time,move to the forefront of health care,replacing our current reactive system.
Perhaps more difficult than thegenetic science will be the social andmoral implications of this knowledge.It raises issues of genetic privacy andownership of our genome, not tomention human cloning, social Dar-winism and perhaps even eugenics. In
the past, our biological evolution(based on genes) was slow and ran-dom caused by the changes in theenvironment, whereas, our culturalevolution (based on ideas) was fastand purposeful due to frequent for-mation of new ideas and technologiesto disseminate them. In the future, wemay be able to catalyze evolution inthe same way as cultural change hasoccurred. It will be the responsibilityof all in society to ensure that there isproper education and adequate dis-cussion of these topics to ensure thebest decisions are made for ourspecies as a whole as we move for-ward. Therefore, programs studyingethical, legal and societal implicationsof genetic information will be asimportant to success in applying thehgp knowledge as continued supportof the science.
Stephen W. Scherer is a Senior Scientist inGenetics and Genomic Biology and AssociateDirector of The Centre for Applied Genomics at TheHospital for Sick Children in Toronto. He is alsoAssociate Professor in Molecular and MedicalGenetics at the University of Toronto. His labora-tory has contributed to mapping, sequencing andgene identification studies on human chromosome7 as part of the HGP. He is currently Chair of theHuman Genome Organization (HUGO) AnnotationCommittee.
Unique en son genre, cet ouvrage constitue une initiation auvaste et complexe domaine de la bioéthique. S’adressant auxprofessionnels de la santé comme aux généralistes, GuyDurand étudie les concepts de base, les principes et les grillesd’analyse des principaux auteurs et courants contemporains.
INTRODUCTION GÉNÉRALE À LA BIOÉTHIQUEHistoire, concepts et outilsguy durand
«»
Il s’agit d’une entreprise d’envergure, d’un travail titanesque, d’une œuvre majeure qui aura un impact considérable. Ce livre
sera probablement la référence en bioéthique pendant de nombreuses années, la bible de la bioéthique. Il n’est pas
demain le jour où un autre reprendra cette entreprise…
Michelle Dallaire, médecin
576 pages • 39,95 $
Fidesf i d e s
• cerf