Embed Size (px)
Citation preview

Hypertension artérielle pulmonaire
Pulmonary arterial hypertension
D. Montani a, O. Sitbon a, E. Fadel b, P. Dartevelle b, H. Nunes a,D. Lebrec c, G. Simonneau a, M. Humbert a,*
a Centre des maladies vasculaires pulmonaires, UPRES EA2705, service de pneumologie et réanimationrespiratoire, université Paris-Sud, hôpital Antoine-Béclère, Assistance publique-Hôpitaux de Paris, 157,rue de la Porte-de-Trivaux, 92140 Clamart, Franceb Centre chirurgical, hôpital Marie Lannelongue, service de chirurgie thoracique, 133, avenue de laRésistance,92350 Plessis-Robinson, Francec Laboratoire d’hémodynamique splanchnique et de biologie vasculaire, INSERM U-481, et serviced’hépatologie, hôpital Beaujon, 100 boulevard du Général-Leclerc, 92110 Clichy, France
MOTS CLÉSEmbolie pulmonaire ;Endartériectomie ;Hypertensionartérielle pulmonaire ;Hypertension portale ;Virus del’immunodéficiencehumaine
Résumé Sous le terme d’hypertension artérielle pulmonaire (HTAP) sont regroupéesdifférentes maladies touchant les artères pulmonaires de petit calibre, entraînant uneaugmentation progressive des résistances artérielles pulmonaires et une défaillanceventriculaire droite. L’HTAP peut survenir de façon sporadique ou dans un contextefamilial. Des mutations de BMPR2, un membre de la famille des récepteurs du transfor-ming growth factor (TGF)-b, sont responsables de plus de la moitié des cas familiaux et deplus de 10 % des formes apparemment sporadiques. D’autres facteurs génétiques ouenvironnementaux sont certainement impliqués dans le développement de cette mala-die. Ainsi, l’infection par le virus de l’immunodéficience humaine et l’hypertensionportale peuvent rarement se compliquer d’HTAP. La survenue de cette complication n’estpas liée à la sévérité de l’infection virale ou de la maladie hépatique, mais l’HTAPconstitue un facteur pronostique majeur. L’HTAP postembolique correspond à l’organi-sation fibreuse d’embolies pulmonaires aiguës chez de rares patients. L’endartériectomiepulmonaire sous circulation extracorporelle représente le traitement de référence desobstructions proximales des artères pulmonaires. Le traitement de l’HTAP repose sur desmesures simples (limitation des efforts, anticoagulation, diurétiques, oxygénothéra-pie...) associées à des thérapeutiques plus spécifiques (antagonistes calciques, prostacy-cline, antagonistes des récepteurs de l’endothéline). L’artériopathie plexiforme associedes lésions de fibrose et de prolifération endothéliale et musculaire lisse vasculairepulmonaire qui prédominent en général sur les phénomènes de vasoconstriction. Lesavancées médicales de ces dernières années, et en particulier l’apparition de traitementsaux propriétés mixtes vasodilatatrices et antiproliférantes (prostacycline et antagonistesdes récepteurs de l’endothéline), ont permis une amélioration significative du pronostic.© 2004 Publié par Elsevier SAS.
* Auteur correspondant.Adresse e-mail : [email protected] (M. Humbert).
EMC-Pneumologie 1 (2004) 46–68
www.elsevier.com/locate/emcpn
© 2004 Publié par Elsevier SAS.doi: 10.1016/S1762-4223(04)00017-0

KEYWORDSPulmonary embolism;Pulmonaryendarterectomy;Pulmonary arterialhypertension;Portal hypertension;Humanimmunodeficiencyvirus
Abstract Pulmonary arterial hypertension (PAH) is characterised by elevated pulmonaryarterial resistance leading to right heart failure. PAH has been described as eithersporadic or clustered in families. Defects within BMPR2, a receptor member of the TGF-bsuperfamily, underlie at least half of familial cases and more than 10 % of so-calledsporadic PAH. Additional genetic or environmental determinants presumably play a keyrole in the pathogenesis of this disorder. PAH rarely complicates the course of humanimmunodeficiency infection, as well as portal hypertension. In both situation PAH is notlinked to the severity of the underlying medical conditions, but it constitutes animportant prognosis factor. Chronic thromboembolic hypertension is the consequence ofacute pulmonary embolism in a rare subset of patients. Pulmonary endarterectomy is thetreatment of choice for cases with proximal obstruction of pulmonary arteries. PAHtreatment is based on simple measures (exercise limitation, anticoagulation, diuretics,oxygen...) and more specific drugs (calcium channel antagonists, prostacyclin, endothelinreceptor antagonists). Medical advances over the last decade, and particularly theavailability of novel drugs (prostaglandins and endothelin receptor antagonists) haveresulted in significant improvements in prognosis. Interestingly, these drugs have bothvasodilator and antiproliferative properties and the long-term effects of treatment maytherefore extend beyond simple acute vasodilator activity. The evolution of therapy fromvasodilators to antiproliferative agents reflects the advancement in our understanding ofthe mechanisms mediating PAH.© 2004 Publié par Elsevier SAS.
Introduction
Sous le terme d’hypertension artérielle pulmonaire(HTAP) sont regroupées différentes maladies tou-chant les artères pulmonaires de petit calibre en-traînant une augmentation progressive des résis-tances vasculaires pulmonaires et une défaillanceventriculaire droite. L’HTAP a une définition hémo-dynamique : pression artérielle pulmonairemoyenne supérieure à 25 mmHg au repos ou30 mmHg à l’effort associée à une pression arté-rielle pulmonaire occluse basse, inférieure à12 mmHg. En l’absence de traitement efficace,l’HTAP évolue inéluctablement vers le décès. Ainsi,la survie moyenne des patients souffrant d’HTAPidiopathique était de 2,8 ans avant l’avènementdes nouveaux traitements spécifiques. Des avan-cées significatives récentes dans la compréhensiondes mécanismes et des aspects génétiques del’HTAP ont permis une nouvelle approche physiopa-thologique et thérapeutique de la maladie. L’épo-prosténol intraveineux continu, associé au traite-ment conventionnel, a permis de transformer lepronostic de cette maladie mais au prix decontraintes importantes. Actuellement, le dévelop-pement de nouveaux traitements modifie de ma-nière importante la prise en charge des patientsatteints d’HTAP. Nous traiterons ici des différentespossibilités thérapeutiques et de l’algorithme déci-sionnel qui peut être proposé à partir des données
récentes de la littérature. Nous tenterons aussid’actualiser les informations concernant les HTAPassociées à l’infection par le virus de l’immunodé-ficience humaine (VIH) et les HTAP associées auxmaladies hépatiques. Enfin, nous aborderons lespossibilités chirurgicales dans la maladie throm-boembolique chronique (thromboendartériectomiepulmonaire).
Nouvelle classification (révision 2003)
La nouvelle classification définit l’HTAP idiopathi-que, anciennement HTAP « primitive », par la sur-venue de la maladie en l’absence de facteurs derisque classiques, tels que les connectivites, lescardiopathies congénitales, l’hypertension portale,l’infection par le VIH ou l’exposition à certainsmédicaments, notamment les anorexigènes de lafamille des dérivés de la fenfluramine1,2,3 (Ta-bleau 1). Par définition, les HTAP dites « secondai-res » à une cause bien identifiée, pouvant bénéfi-cier d’un traitement spécifique comme le cœurpulmonaire chronique postembolique (HTAP obs-tructive), l’insuffisance cardiaque gauche (HTAPpassive postcapillaire) ou l’insuffisance respiratoirechronique (HTAP hypoxique), n’appartiennent pasau groupe de maladies définies par le termed’HTAP.1
47Hypertension artérielle pulmonaire

Aspects génétiques
Hypertensions artérielles pulmonairesfamiliales et hypertensions artériellespulmonaires idiopathiques
L’HTAP idiopathique survient habituellement sousla forme de cas sporadiques inexpliqués.4,5,6 Sonincidence est difficile à établir du fait de l’absencede spécificité des symptômes et d’examen simplepour l’affirmer. Les HTAP idiopathiques sporadi-ques et les HTAP familiales ont une histoire natu-relle et une physiopathologie semblables. La pré-sentation clinique, la réponse aux thérapeutiquesvasodilatatrices et les lésions histologiques arté-rielles pulmonaires sont similaires.4,5,6,7 Le nombrede nouveaux cas annuels en France est estimé àenviron deux HTAP idiopathiques par million d’ha-bitants,4,5,6 dont au moins 5 % de formes familia-les.7 L’âge de survenue des HTAP idiopathiques esttrès variable avec un pic de fréquence entre 20 et40 ans, mais cette maladie peut se rencontrer àtous les âges.4,5,6 L’anticipation génétique, un phé-nomène caractérisé par l’apparition plus précocede la maladie au fil des générations successives, estobservé dans l’HTAP familiale. 7 La prédominance
féminine est une donnée habituelle des HTAP idio-pathiques sporadiques et des HTAP familiales avecenviron deux cas féminins pour un homme affecté,suggérant l’existence de cofacteurs de prédisposi-tion hormonaux ou génétiques liés au chromosomeX4,5,6 ou encore une perte des fœtus mâles inutero.7
« BMPR2 », un gène de l’hypertensionartérielle pulmonaire
L’analyse des formes familiales d’HTAP a permisd’identifier une transmission autosomique domi-nante avec une pénétrance incomplète de l’ordrede 10 à 20 %, expliquant la possibilité de sauts degénération compliquant l’identification des formesfamiliales de la maladie.7,8 L’étude des formesfamiliales a permis de mettre en évidence la res-ponsabilité du gène BMPR2 siégeant sur le bras longdu chromosome 2, en position 2q31-339 et codantpour le récepteur du facteur morphogène de l’os detype II, BMPR-II.10,11,12,13 La protéine BMPR-II cor-respond à un récepteur de la famille du transfor-ming growth factor b (TGF-b).14,15 Les membres dela famille du TGF-b agissent par le biais de l’inter-action avec des récepteurs de surface de type I et IIet l’activation de messagers intracellulaires, lesSmads.14,15 Des mutations des membres de la fa-mille du TGF-b ont été identifiées dans différentesaffections néoplasiques ou malformations vasculai-res au premier rang desquelles la maladie deRendu-Osler.15,16,17 Des mutations germinales deBMPR2 sont identifiées dans 60 % des formes fami-liales, et plusieurs dizaines de mutations germina-les hétérozygotes ont déjà été décrites dans cesfamilles.10,11,12 La diversité de ces mutations renddifficile le développement de tests génétiques sim-ples.8 L’absence de mutations détectables dans40 % des cas familiaux n’exclut pas l’existence demutations localisées au niveau de zones non sé-quencées en routine (introns ou séquences régula-trices). Enfin, il est possible que d’autres gènesencore méconnus puissent jouer un rôle dansl’HTAP.Dans les formes sporadiques d’HTAP idiopathi-
ques, des mutations germinales de BMPR2 sontidentifiées dans 10 à 30 % des cas12,13 soulignant laprobable sous-estimation des formes familiales enpratique quotidienne. Certaines mutations sont hé-ritées de parents phénotypiquement sains, d’autresmutations survenant de novo. Ces mutations sontsemblables à celles observées dans les formes fami-liales documentées.12,13
Les mutations germinales de BMPR2 induisentune susceptibilité à l’HTAP, mais elles ne sont ninécessaires ni suffisantes à la survenue de la mala-
Tableau 1 Nouvelle classification diagnostique de l’HTAPd’après les conclusions du Congrès mondial de Venise en2003.
1. HTAP « proliférantes »– HTAP idiopathique– HTAP familiale– HTAP associée à :- connectivite- infection par le VIH- hypertension portale- anorexigènes et toxiques- cardiopathie congénitale– HTAP persistante du nouveau-né– HTAP avec atteinte veineuse pulmonaire prédominante(maladie veino-occlusive)2. HTAP « passives » des cardiopathies gauches– Auriculaire ou ventriculaire– Valvulaire3. HTAP « hypoxiques »– BPCO– Pneumopathies infiltrantes– Apnée du sommeil– Altitude4. HTAP « obstructives » liées à une maladie tromboem-bolique– HTAP postembolique proximale– HTAP postembolique distale– Embolies non cruriques (tumorales...)
HTAP : hypertension artérielle pulmonaire ; BPCO : broncho-pneumopathie chronique obstructive.
48 D. Montani et al.

die. Des résultats récents suggèrent une réductiongénérale de l’expression de la protéine BMPR-II auniveau des artères pulmonaires de patients souf-frant d’HTAP idiopathique, cette réduction étantplus importante chez les patients présentant desmutations de BMPR2.18 Les mutations de BMPR2réduisent l’intensité de la liaison de BMPR-II avecses ligands comme BMP4 entraînant une réductionde l’activité BMP/Smad et un excès de proliférationdes cellules (endothéliales et musculaires lisses)portant les mutations de BMPR2 (Fig. 1).19 En fa-veur de cette hypothèse, il a été récemment dé-montré que les bone morphogenic proteins (BMP)diminuent la prolifération musculaire lisse vascu-laire pulmonaire de sujets sains mais pas des pa-tients souffrant d’HTAP idiopathique.20
Cofacteurs environnementaux et génétiques
Les anomalies de BMPR2 sont insuffisantes au déve-loppement de la maladie, comme l’atteste la péné-trance incomplète de cette mutation. Un deuxièmeévénement est donc probablement nécessaire àl’expression de la maladie chez des sujets généti-quement prédisposés.8 Diverses expositions envi-ronnementales ou facteurs génétiques surajoutéspourraient jouer un rôle fondamental. L’expositionaux réducteurs de l’appétit, en particulier les déri-vés de la fenfluramine, a été associée à de nom-breux cas d’HTAP en Europe et en Amérique duNord à la fin des années 1980 et au début desannées 1990.1 L’étude IPPHS (International PrimaryPulmonary Hypertension Study Group) a ainsi dé-montré que l’utilisation de dérivés anorexigènesétait associée à une augmentation significative durisque d’HTAP (odds-ratio à 6,3).21 Une utilisationprolongée pendant plus de 3 mois augmentait signi-ficativement l’odds-ratio à 23.21 Le risque de déve-lopper une HTAP est évalué à 1 sur 10 000 chez lessujets exposés aux dérivés de la fenfluramine, té-moignant de l’existence de facteurs de susceptibi-lité individuelle, possiblement d’origine généti-que.22 Nous avons récemment montré que 10 % despatients exposés développant une HTAP avaient desmutations germinales de BMPR2.22 La durée del’exposition aux anorexigènes était significative-ment plus courte chez les malades présentant desmutations de BMPR2.De même, une HTAP ne se développera que chez
0,1 % des sujets infectés par le VIH, chez 1 à 4 % despatients présentant une hypertension portale etchez 10 % des sclérodermies systémiques.1 Cepen-dant, les résultats les plus récents concernant lesprincipales conditions associées à l’HTAP (infectionpar le VIH, hypertension portale, syndrome d’Eisen-menger, sclérodermies, etc.) ne retrouvent que
très rarement des mutations BMPR2.8,22 Ledeuxième événement nécessaire à l’expression dela maladie pourrait être lui aussi d’origine généti-que. Deux grandes hypothèses ont été testées ré-cemment, l’influence du polymorphisme du trans-porteur de la sérotonine d’une part23 etl’acquisition de mutations somatiques au niveaudes cellules vasculaires pulmonaires d’autre part.24
Il a été démontré que les cellules musculaires lissesde patients souffrant d’HTAP idiopathique crois-
Figure 1 Voie de transduction du transforming growth factor(TGF)- b et localisation de mutations de BMPR-II identifiées chezdes patients souffrant d’hypertension artérielle pulmonaire(HTAP) idiopathique sporadique. Après liaison avec son ligand,BMPR-II forme un hétérodimère avec un récepteur de typeBMPR-I aboutissant à l’activation du domaine kinase de cedernier. Cela induit la phosphorylation de protéines intracellu-laires (les Smads) qui permettent la transduction du signal.Treize mutations germinales hétérozygotes de BMPR2 sont re-présentées sur ce schéma (en haut et à droite). Ces mutationspeuvent toucher toutes les portions de la protéine (intracellu-laire, transmembranaire et extracellulaire). Les mutations deBMPR2 réduisent l’intensité de la liaison de BMPR-II avec sesligands comme le BMP4. Cela aboutit à une réduction de l’acti-vité BMP/Smad et à un « gain de fonction » avec excès deprolifération des cellules portant les mutations de BMPR2. Ce« gain de fonction » pourrait contribuer aux anomalies histolo-giques décrites au niveau des artères de petit calibre au cours del’HTAP idiopathique, avec prolifération endothéliale et muscu-laire lisse. En faveur de cette hypothèse, il a été récemmentdémontré que les bone morphogenic proteins (BMP) diminuentla prolifération musculaire lisse vasculaire pulmonaire de sujetssains mais pas des patients souffrant d’HTAP idiopathique.
49Hypertension artérielle pulmonaire

sent plus rapidement en présence de sérotonineque celles de sujets contrôles.23 Cette caractéristi-que serait due au moins en partie à une augmenta-tion de l’expression du transporteur de la séroto-nine par ces cellules. L’analyse d’un poly-morphisme du promoteur du gène codant pour letransporteur de la sérotonine a mis en évidence unesur-représentation du génotype LL induisant uneaugmentation du transport de la sérotonine.23 Ilreste à démontrer si ce polymorphisme agit indé-pendamment ou en association avec les mutationsde BMPR2 ou d’autres facteurs.L’acquisition de mutations somatiques pourrait
aussi contribuer à la physiopathologie de l’HTAPidiopathique.24 L’analyse de lésions plexiformes desujets souffrant d’HTAP idiopathique a montrél’existence d’une prolifération monoclonale endo-théliale.25 Les cellules endothéliales au sein de ceslésions présentaient des mutations somatiques as-sociées à une diminution de l’expression protéiquedu récepteur au TGF-b de type II.24 Ces donnéessuggèrent que des anomalies génétiques somati-ques, similaires à celles observées dans certainesnéoplasies, pourraient participer à la constitutiondes lésions histologiques proliférantes caractéristi-ques de l’HTAP.24,25
Hypertension artérielle pulmonaireet maladie de Rendu-Osler
La maladie de Rendu-Osler ou télangiectasie hé-morragique héréditaire est une maladie génétiquede transmission dominante autosomique, à péné-trance quasi complète à l’âge de 40 ans, et dont lessignes sont essentiellement d’ordre vasculaire.16,17
Il s’agit d’épistaxis, de télangiectasies de la peau etdes muqueuses, de manifestations viscérales sousforme d’angiodysplasie, réalisant des fistules arté-rioveineuses pulmonaires, des shunts portocaves,des télangiectasies vésicales, des malformationsvasculaires cérébrales. La prévalence de cette pa-thologie est estimée à 1 sur 40 000.16,17 Deux gènescodant pour des protéines majeures de la voie detransduction du TGF-b ont été identifiés, l’endo-gline sur le chromosome 9q et ALK1 sur le chromo-some 12q.16,17 Le gène codant pour l’endogline estresponsable de la forme HHT1, et le gène ALK1 dela forme dite HHT2. Même si les tableaux cliniquessont très proches, les deux formes diffèrent par lafréquence des fistules artérioveineuses pulmonai-res de la maladie, plus fréquentes pour HHT1.16,17
L’existence de fistules artérioveineuses multiplesaboutit en général à un syndrome hyperkinétiquepouvant aboutir à des défaillances cardiaques gau-ches (insuffisance cardiaque gauche à haut débitavec HTAP postcapillaire).26 Dans une minorité de
cas, une HTAP précapillaire sévère peut paradoxa-lement s’observer.27,28,29 Six familles caractériséespar des cas avérés d’HTAP et de maladie de Rendu-Osler ont été récemment identifiées.29 Des muta-tions du gène ALK1 ont été retrouvées chez cinq deces familles. Des analyses immuno-histochimiquesont révélé une expression vasculaire pulmonaireendothéliale d’ALK1. Ces données permettentd’ajouter ALK1 au nombre des gènes associés àl’HTAP et soulignent le rôle critique de la voie detransduction du TGF-b au cours de l’HTAP.29
Vers le conseil clinique et génétique
Il est recommandé de proposer un dépistage et unesurveillance régulière chez les parents du premierdegré des sujets présentant une HTAP familiale.8
Outre l’examen clinique et l’interrogatoire, ce bi-lan inclut une radiographie thoracique, un électro-cardiogramme et une échocardiographie avec Dop-pler. Il est maintenant envisageable de rechercherdes mutations du gène BMPR2 chez un patient souf-frant d’HTAP et de réaliser, en cas de mutationidentifiée, un test génétique chez les parents dupremier degré.8 Cette attitude permettrait de ras-surer les sujets indemnes de mutation, de limiter ledépistage aux sujets génétiquement à risque, etpossiblement de proposer une intervention théra-peutique précoce chez les malades dépistés. Il estnéanmoins important de souligner les difficultéstechniques d’identification de ces mutations, cha-que cas pouvant être caractérisé par une mutationponctuelle située à n’importe quel niveau du gène.Dans les conditions actuelles, il est impossibled’exclure un risque de transmission familiale mêmeen cas de négativité du test génétique.8 La péné-trance incomplète constitue une difficulté supplé-mentaire ; l’identification d’une mutation ne tra-duisant qu’une probabilité relativement faible dedéveloppement de la maladie. De plus amples re-cherches sont nécessaires pour préciser la significa-tion de l’identification d’une mutation chez unsujet asymptomatique. Une étude allemande a ré-cemment démontré que dans les familles de mala-des, les porteurs de mutations ont des pressionsartérielles pulmonaires normales au repos, maismajorées à l’exercice, soulignant la possibilitéd’une restriction vasculaire pulmonaire encoreasymptomatique chez ces individus.30 Néanmoins,ce test est encore discutable, du fait de la fré-quence de faux positifs et de l’absence de reculsuffisant quant à la signification d’une élévation dela pression artérielle pulmonaire à l’effort.
Conclusion
Ces dernières années ont permis de transformernotre compréhension des mécanismes génétiques
50 D. Montani et al.

participant à la survenue de l’HTAP et d’apporterde nouvelles hypothèses physiopathologiques.L’implication des médiateurs de la famille du TGF-bet de la sérotonine permet d’entrevoir de nouvellespossibilités thérapeutiques pour cette maladie or-pheline. Néanmoins, de nombreuses incertitudespersistent et des travaux complémentaires sontencore nécessaires. Il semble maintenant essentielde proposer puis de valider des stratégies pour leconseil génétique des familles de sujets souffrantd’HTAP.
Nouveaux traitements de l’hypertensionartérielle pulmonaire
Le traitement conventionnel, seule possibilité thé-rapeutique, avant l’apparition de médicamentsspécifiques de l’HTAP reste d’actualité. Il associedes mesures générales (limiter les efforts, éviterl’altitude, contre-indiquer la grossesse) et des trai-tements associant diurétiques, anticoagulants etoxygénothérapie en cas d’hypoxie.Les inhibiteurs calciques ont représenté initiale-
ment un espoir d’améliorer le pronostic de l’HTAPmais il est vite apparu que ce traitement nes’adressait qu’à une minorité de patients « répon-deurs ». En effet, seules 7 % des HTAP idiopathiquesvues dans notre service bénéficient à long termedes effets des inhibiteurs calciques.31 La majoritéde ces patients présente, lors du test de vasodila-tation aiguë par le monoxyde d’azote effectué lorsdu cathétérisme initial, une quasi-normalisation deleur pression artérielle pulmonaire moyenne(PAPm < 40 mmHg) et un débit cardiaque conservéou augmenté.31,32 Pour les patients « non répon-deurs », les inhibiteurs calciques ne présententaucun intérêt et peuvent même représenter unecause d’aggravation de l’HTAP du fait de leurseffets systémiques en l’absence d’effets pulmonai-res. Bien que le traitement par inhibiteurs calci-ques soit réservé à un faible nombre de patients, ilgarde une place de choix dans l’arsenal thérapeu-tique de l’HTAP car le pronostic à long terme despatients « répondeurs » est transformé par le trai-tement. Par ailleurs, il s’y associe une améliorationtrès significative sur le plan fonctionnel.La prostacycline (époprosténol) administrée par
injection intraveineuse continue a représenté lepremier traitement spécifique de l’HTAP. Elle a étéutilisée pour la première fois au début des années198033 et a depuis prouvé son efficacité clinique,hémodynamique et en termes de survie chez lespatients atteints d’HTAP.34,35,36 Initialement,l’époprosténol avait été proposé comme traite-ment d’attente (« bridge-therapy ») de la trans-
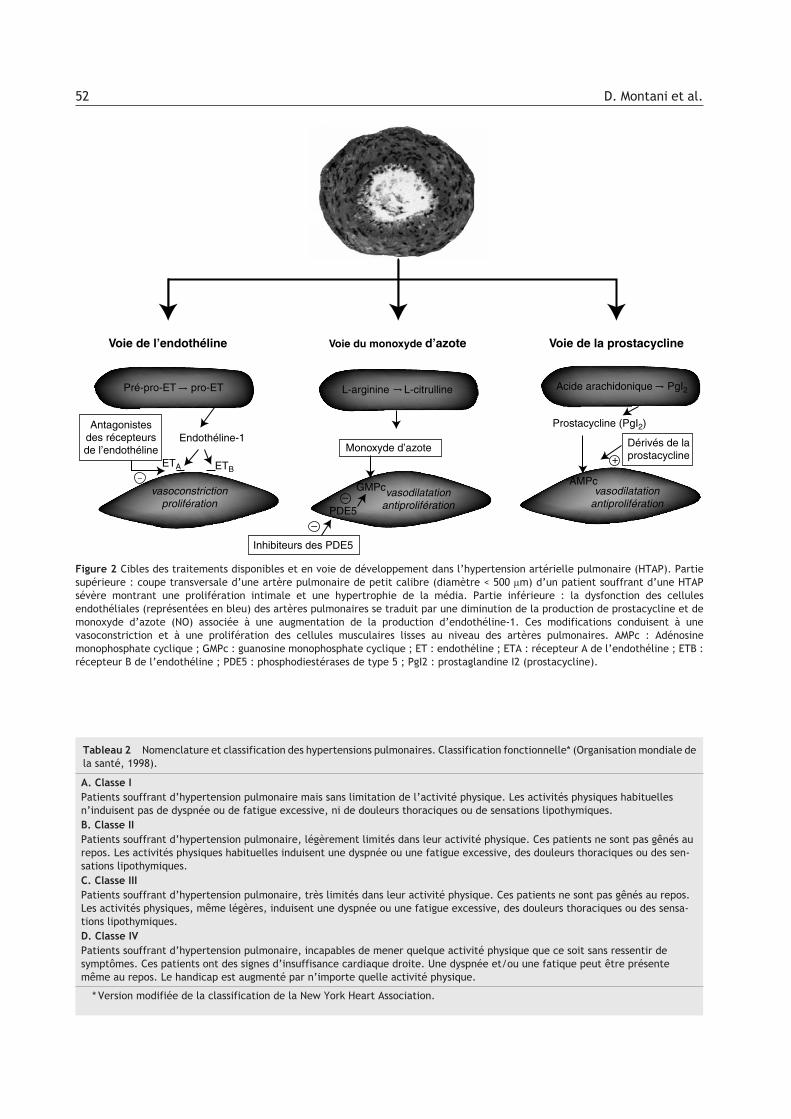
plantation pulmonaire, mais actuellement il estconsidéré comme le traitement de référence despatients souffrant d’HTAP sévère et s’inscritcomme une alternative à la transplantation. Endépit du bénéfice apporté, la perfusion continue deprostacycline est loin de constituer un traitementidéal, car il s’agit d’une thérapeutique complexe,inconfortable et dont le coût est très important.Ces inconvénients ont conduit au développementde nouveaux traitements utilisés sous des formesplus accessibles. La dysfonction endothéliale à la-quelle s’associent des anomalies musculaires lissesvasculaires pulmonaires constitue un élément fon-damental dans l’apparition de l’HTAP. Cette dys-fonction se caractérise entre autres par une vaso-constriction exagérée et un défaut devasodilatation. La diminution de production de mé-diateurs vasodilatateurs comme le monoxyded’azote (NO) ou la prostacycline, associée à unesurproduction de vasoconstricteurs commel’endothéline-1 affectent non seulement le tonusvasculaire mais entraînent aussi un remodelagevasculaire. Ces différents médiateurs constituentdes cibles privilégiées des traitements de l’HTAP.Les modalités d’action des différents traitementssont représentés sur la Figure 2.
Analogues de la prostacycline
Tréprostinil (voie sous-cutanée continue)Le tréprostinil (Remodulin ®)est un analogue de laprostacycline dont la demi-vie est de 27 minutes enadministration intraveineuse et de 58 à 83 minutesen administration sous-cutanée.37 Le tréprostinilest administré par voie sous-cutanée à l’aide d’unsystème de pompe semblable à celui utilisé pour ladélivrance de l’insuline chez le diabétique.38 L’effi-cacité du tréprostinil a été évaluée lors d’uneétude multicentrique, randomisée, versus placebosur 12 semaines, chez 470 patients souffrantd’HTAP « primitive », d’HTAP liée à un shunt congé-nital ou à une connectivite en classe fonctionnelleII, III ou IV de la New York Heart Association (NYHA)(Tableau 2).38 Une amélioration modeste mais si-gnificative de la distance parcourue en 6 minutes aété observée (amélioration moyenne de 16 m pourl’ensemble de la population et de 19 m pour lespatients ayant une HTAP « primitive »).38 Après12 semaines de suivi, le tréprostinil améliorait si-gnificativement les scores de dyspnée, les signes etsymptômes de l’HTAP ainsi que les paramètres hé-modynamiques. Pour le test de marche, lesmeilleures améliorations étaient observées chez lespatients qui avaient toléré les fortes doses de trai-tement (amélioration de 36 m lors du test de mar-che chez les patients traités par tréprostinil à une
51Hypertension artérielle pulmonaire
Acide arachidonique PgI2L-arginine L-citrullinePré-pro-ET pro-ET
Voie de l’endothéline Voie du monoxyde d’azote Voie de la prostacycline
Prostacycline (PgI2)
Dérivés de laprostacycline+
Monoxyde d’azote
vasodilatationantiprolifération
vasodilatationantiprolifération
GMPc
PDE5
vasoconstrictionprolifération
Endothéline-1
ETA ETB–
Antagonistesdes récepteursde l’endothéline
–
–
AMPc
Inhibiteurs des PDE5
Figure 2 Cibles des traitements disponibles et en voie de développement dans l’hypertension artérielle pulmonaire (HTAP). Partiesupérieure : coupe transversale d’une artère pulmonaire de petit calibre (diamètre < 500 lm) d’un patient souffrant d’une HTAPsévère montrant une prolifération intimale et une hypertrophie de la média. Partie inférieure : la dysfonction des cellulesendothéliales (représentées en bleu) des artères pulmonaires se traduit par une diminution de la production de prostacycline et demonoxyde d’azote (NO) associée à une augmentation de la production d’endothéline-1. Ces modifications conduisent à unevasoconstriction et à une prolifération des cellules musculaires lisses au niveau des artères pulmonaires. AMPc : Adénosinemonophosphate cyclique ; GMPc : guanosine monophosphate cyclique ; ET : endothéline ; ETA : récepteur A de l’endothéline ; ETB :récepteur B de l’endothéline ; PDE5 : phosphodiestérases de type 5 ; PgI2 : prostaglandine I2 (prostacycline).
Tableau 2 Nomenclature et classification des hypertensions pulmonaires. Classification fonctionnelle* (Organisation mondiale dela santé, 1998).
A. Classe IPatients souffrant d’hypertension pulmonaire mais sans limitation de l’activité physique. Les activités physiques habituellesn’induisent pas de dyspnée ou de fatigue excessive, ni de douleurs thoraciques ou de sensations lipothymiques.B. Classe IIPatients souffrant d’hypertension pulmonaire, légèrement limités dans leur activité physique. Ces patients ne sont pas gênés aurepos. Les activités physiques habituelles induisent une dyspnée ou une fatigue excessive, des douleurs thoraciques ou des sen-sations lipothymiques.C. Classe IIIPatients souffrant d’hypertension pulmonaire, très limités dans leur activité physique. Ces patients ne sont pas gênés au repos.Les activités physiques, même légères, induisent une dyspnée ou une fatigue excessive, des douleurs thoraciques ou des sensa-tions lipothymiques.D. Classe IVPatients souffrant d’hypertension pulmonaire, incapables de mener quelque activité physique que ce soit sans ressentir desymptômes. Ces patients ont des signes d’insuffisance cardiaque droite. Une dyspnée et/ou une fatique peut être présentemême au repos. Le handicap est augmenté par n’importe quelle activité physique.
* Version modifiée de la classification de la New York Heart Association.
52 D. Montani et al.

dose supérieure à 13,8 ng kg–1 min–1.38 La douleurau point d’injection était la complication la plusfréquente (plus de 85 % des patients), représentantun facteur limitant à l’augmentation des doses etconduisant à l’arrêt du traitement dans 8 % descas.38 À noter que le traitement par tréprostinil apu être instauré sans problème chez des patientsayant présenté des complications infectieuses gra-ves de l’époprosténol intraveineux (transition de laforme intraveineuse à la forme sous-cutanée).39
Depuis 2002, le tréprostinil est enregistré pour letraitement de l’HTAP aux États-Unis.
Béraprost (voie orale)Le béraprost sodique (Beradrak ®) est le premieranalogue de la prostacycline stable sous formeorale.40 Le pic de concentration survient rapide-ment, environ 30 minutes après l’absorption et lademi-vie du béraprost est de l’ordre de 35 à 40 mi-nutes.37 Une étude randomisée en double aveugleversus placebo a évalué son efficacité sur 12 semai-nes chez 130 patients présentant une HTAP « primi-tive » ou évoluant dans un contexte de cardiopathiecongénitale, de connectivite, d’hypertension por-tale ou d’infection par le VIH41 en classe fonction-nelle II et III de la NYHA. Après 12 semaines, lebéraprost à la dose moyenne de 80 lg répartie enquatre prises, a permis d’améliorer la distanceparcourue lors du test de marche de 25 m enmoyenne pour l’ensemble de la population. En fait,l’analyse des sous-groupes retrouve un bénéficesignificatif du béraprost dans l’HTAP « primitive »(+ 45 m) mais pas de différence dans les HTAPassociées.41 Dans les deux cas, il n’est observéaucune modification hémodynamique au repos.41
Les effets secondaires liés à la vasodilatation péri-phérique ont été fréquemment observés, principa-lement à la phase initiale de titration (augmenta-tion rapide des doses jusqu’à la dose maximaletolérée), la tolérance pouvant devenir un facteurlimitant à long terme.41 Une étude randomisée, endouble aveugle, versus placebo a analysé l’effet àlong terme (12 mois) du béraprost mettant en évi-dence une diminution du bénéfice de ce traitementavec le temps.42 En effet, le test de marche despatients en classe fonctionnelle II ou III de la NYHAétait significativement amélioré à 3 et 6 mois, maisla différence n’était plus significative à 9 et12 mois.42 Le béraprost a reçu une autorisationdans le traitement de l’HTAP au Japon.
Iloprost (voie inhalée)L’iloprost (Ventavis ®) est un analogue stable de laprostacycline qui peut être administré sans risquepar inhalation chez les patients présentant uneHTAP.43 Il est essentiel que le système d’inhalation
produise des particules en suspension de taille ap-propriée (diamètre de 0,5 à 3 lm) afin de s’assurerd’un dépôt alvéolaire satisfaisant permettantd’améliorer la déposition pulmonaire.44 La courtedurée d’action de l’iloprost constitue le principaldésavantage de ce mode d’administration puisqu’ilnécessite la réalisation de 6 à 12 inhalations parjour.43,45 Récemment, une étude multicentriquerandomisée versus placebo incluant 207 patients declasse fonctionnelle III ou IV de la NYHA a analysé lebénéfice après 12 semaines d’un traitement pariloprost inhalé.45 Dans cette étude, les sujets inclusprésentaient une HTAP « primitive », liée à uneconnectivite ou un cœur pulmonaire chronique pos-tembolique non opérable car trop distal. Le critèreprincipal de jugement était un score combiné asso-ciant une amélioration de 10 % de la distanceparcourue en 6 minutes et une amélioration de laclasse fonctionnelle NYHA en l’absence d’aggrava-tion clinique. Cet objectif a été réalisé chez 17 %des sujets traités contre 4 % dans le groupe pla-cebo, cette différence étant significative.45 L’amé-lioration du test de marche était en moyenne de36 m chez l’ensemble des sujets traités et de 59 mau cours de l’HTAP « primitive ».45 Par rapport auxvaleurs de base, après 12 semaines de traitement,les paramètres hémodynamiques dans le groupetraité étaient améliorés après inhalation, maisidentiques avant celle-ci, alors qu’ils étaient dimi-nués dans le groupe placebo.45 La toux et les symp-tômes liés à la vasodilatation représentaient lesprincipaux effets secondaires observés lors de lanébulisation d’iloprost.45 De plus, le nombre desyncopes était significativement plus importantdans le groupe traité par iloprost.45 L’efficacité àlong terme du traitement nécessite de plus amplesétudes. L’iloprost inhalé est actuellement en coursd’enregistrement par l’agence européenne du mé-dicament.
Antagonistes des récepteursde l’endothéline : bosentan (voie orale)
L’endothéline-1 agit par liaison à deux types derécepteurs spécifiques ETA et ETB.46,47,48 Le bosen-tan (Tracleer®) est un antagoniste de ces deuxrécepteurs, actif par voie orale.49,50 Deux essais endouble aveugle, randomisés, versus placebo, ontévalué l’efficacité du bosentan chez des patientsprésentant une HTAP (« primitive » ou liée à lasclérodermie).49,50 Dans la première étude, 33 su-jets de classe fonctionnelle III de la NYHA ont étérandomisés pour recevoir un placebo ou du bosen-tan initié à la dose de 62,5 mg deux fois par jourpendant 4 semaines puis augmenté à 125 mg deuxfois par jour pendant au moins 12 semaines.50 Dans
53Hypertension artérielle pulmonaire

le groupe traité par bosentan, une amélioration de76 m de la distance parcourue lors du test demarche a été observée. Ce résultat était associé àune amélioration significative des paramètres hé-modynamiques avec une diminution de la pressionartérielle pulmonaire moyenne et une augmenta-tion du débit cardiaque.50 Dans la seconde étude,213 patients en classe fonctionnelle III ou IV de laNYHA ont été randomisés entre un placebo et dubosentan débuté à la dose de 62,5 mg deux fois parjour pendant 4 semaines puis augmenté à la dose de125 mg ou 250 mg deux fois par jour.49 Dans legroupe traité par bosentan, il a été observé uneamélioration du test de marche de 44 m pour l’en-semble de la population traitée et de 52 m chez lespatients présentant une HTAP primitive. L’étude aaussi permis de mettre en évidence dans le groupebosentan un allongement de la durée avant dégra-dation clinique (définie comme la survenue d’undécès, d’une transplantation pulmonaire, d’unehospitalisation pour décompensation de la maladie,d’une aggravation nécessitant l’arrêt du traite-ment, de la réalisation d’une atrioseptostomie oude la nécessité de débuter un traitement par injec-tion continue d’époprosténol).49 Ce travail n’a pasmontré de relation dose-effet, mais les perturba-tions du bilan hépatique ont, quant à elles, étédose-dépendantes.49 L’élévation des transaminasesà plus de huit fois la normale est survenue chez 3 %des patients traités à la dose de 125 mg deux foispar jour et chez 7 % des patients recevant une dosede 250 mg deux fois par jour. Une analyse échocar-diographique exhaustive a démontré une améliora-tion de l’index cardiaque et de la fonction ventri-culaire droite sous bosentan.51 Malgré des donnéesintéressantes en faveur d’une efficacité persistanteà 12 mois, l’évaluation à long terme du bosentannécessite plus d’informations. Le bosentan à ladose de 125 mg deux fois par jour est un traitementde l’HTAP approuvé aux États-Unis et en Europedepuis 2001 et 2002 respectivement. En septembre2003, plus de 10 000 patients avaient été traités parle bosentan dans le monde.Les inhibiteurs sélectifs du récepteur ETA (sitax-
sentan, ambrisentan) sont actuellement en coursd’évaluation dans ces indications.52 Plusieurs casd’élévation des transaminases (dont un fatal) ontété rapportés avec ces inhibiteurs sélectifs del’ETA, soulignant l’importance de la surveillancemensuelle de la fonction hépatique avec les médi-caments de cette classe thérapeutique.52
Inhibiteurs des phosphodiestérases de type5 (voie orale)
Les cellules contractiles des artères pulmonairescontiennent principalement des phosphodiestéra-
ses de type 3, 4 et 5. Les phosphodiestérases detype 5 sont spécifiques de la guanosine monophos-phate cyclique (GMPc) et peuvent être inhibées.53
Les inhibiteurs des phosphodiestérases de type5 tels que le sildénafil (Viagra®) ont un effet vaso-dilatateur pulmonaire démontré lors de tests devasodilatation effectués chez des patients présen-tant une HTAP « primitive » ou secondaire à unefibrose pulmonaire.53 Des équipes ont rapporté unpossible effet au long cours d’un traitement pro-longé par le sildénafil.54 Il n’existe pour l’instantque des résultats préliminaires et des études sonten cours afin d’évaluer l’efficacité de ce traite-ment dans l’HTAP. Ce traitement prometteur doitêtre évalué rigoureusement afin de tenter d’établirsa place dans la pharmacopée de l’HTAP. Il a l’inté-rêt de son administration par voie orale et de sabonne tolérance en particulier hépatique. Le sildé-nafil est administré en trois prises quotidiennes. Ladose optimale reste à définir.
Autres traitements
Inhibiteur de la thromboxane synthétaseet antagoniste du récepteur du thromboxane A2Le thromboxane A2 est un vasoconstricteur, unagrégant plaquettaire et un agent mitogène descellules musculaires lisses qui pourrait participer àla physiopathologie de l’HTAP.55 Une étude rando-misée, en double aveugle, versus placebo, a évaluéles effets dans l’HTAP « primitive » du terbogrel, uninhibiteur de la thromboxane synthétase et antago-niste du thromboxane A2, actif par voie orale.
56
L’étude a été interrompue prématurément devantl’apparition de violentes douleurs invalidantes desmembres inférieurs, représentant un facteurconfondant pour l’objectif primaire que constituaitle test de marche de 6 minutes.56
L-arginineCertaines observations laissent supposer que laL-arginine, le substrat de la NO synthétase, pour-rait diminuer la pression artérielle pulmonaire etaméliorer la tolérance à l’exercice au cours del’HTAP.57 Une étude contre placebo est actuelle-ment en cours pour évaluer l’efficacité potentiellede la L-arginine dans cette indication.
Conclusion
Malgré les progrès récents, les traitements actuelsde l’HTAP sont purement symptomatiques et lavéritable cure de l’HTAP n’est pas encore envisa-geable à partir de ces molécules. Néanmoins, lesprogrès réalisés dans la prise en charge des patientssouffrant d’HTAP ont été considérables au cours de
54 D. Montani et al.

ces dernières années. L’époprosténol a été le pre-mier traitement spécifique de l’HTAP utilisé, per-mettant d’améliorer la survie et la qualité de vie deces malades. Le développement de nouveaux trai-tements laisse entrevoir de nouvelles options thé-rapeutiques, mais leur efficacité à long terme né-cessite cependant encore d’être évaluée. Enl’absence de données comparant les différents pro-duits, le choix du traitement initial dépend doncautant de l’expérience des équipes et des régle-mentations locales, que de l’état clinique du pa-tient et de ses préférences. La plupart des expertsrecommandent pour les HTAP sévères instables untraitement par époprosténol en perfusion continue.En dehors de cette situation dramatique, les possi-bilités de traitement en Europe en première inten-tion comprennent le bosentan administré par voieorale ou l’iloprost nébulisé, débutés sous sur-veillance dans un centre spécialisé dans la prise encharge des maladies vasculaires pulmonaires. Dansles stades précoces d’HTAP, peu de données sontactuellement disponibles. Il n’existe pas de traite-ment enregistré chez les patients en classe fonc-tionnelle I ou II de la NYHA en Europe. Dans un futurproche, de nouveaux traitements comme le béra-prost, les inhibiteurs sélectifs de l’ETA ou le sildé-nafil pourront peut-être trouver leur place dansl’arsenal thérapeutique de l’HTAP, cela après avoirété évalués de manière rigoureuse et avoir étéreconnus par les agences du médicament. À partirdes données actuelles, un algorithme résumant laprise en charge de l’HTAP est proposé (Fig. 3). Nousespérons qu’une meilleure compréhension des mé-canismes conduisant à cette maladie permettra ledéveloppement de nouveaux traitements ciblantdirectement la prolifération anormale des cellulesendothéliales et musculaires lisses vasculaires pul-monaires. De telles avancées pourraient faire envi-sager l’émergence d’un traitement curatif de cettemaladie orpheline.
Hypertension artérielle pulmonaireassociée à l’infection par le virusde l’immunodéficience humaine
Le premier cas d’HTAP survenant au cours de l’in-fection par le VIH a été rapporté en 1987.58 Avec unrecul de 15 ans, plus de 200 patients ayant uneHTAP associée au VIH ont été recensés dans lalittérature, essentiellement sous la forme de casisolés ou de petites séries. L’infection par le VIH estdonc actuellement une cause clairement reconnued’HTAP (Tableau 1). Depuis le début de l’épidémiepar le VIH, de nombreux progrès ont été réalisés
dans la prise en charge de ces malades, grâce àl’utilisation de combinaisons d’antirétroviraux ouhighly active antiretroviral therapy (HAART) et letraitement prophylactique des infections opportu-nistes. Ces modifications ont permis d’améliorer lasurvie des sujets infectés par le VIH,59,60 dévoilantde nouvelles complications à long terme. Parmi cescomplications, l’HTAP est une manifestation raremais probablement sous-estimée, les causes dedyspnée étant multiples chez ces patients. L’appa-rition d’une HTAP au cours de l’évolution d’uneinfection par le VIH représente un tournant dans lamaladie car la survie de ces patients en est consi-dérablement diminuée.61
Épidémiologie
La fréquence de l’HTAP au cours de la maladie VIHest difficile à établir avec précision. Speich et al.62
ont examiné par échodoppler cardiaque 74 patientsayant des signes cardiaques ou respiratoires parmi1 200 sujets infectés par le VIH étudiés pendant9 mois. Six patients présentaient une HTAP, soitune incidence de 0,5 %. De même, dans une impor-tante cohorte de 3 349 patients séropositifs suivispendant 5 ans, 19 cas d’HTAP étaient dépistés,correspondant à une incidence identique de l’ordrede 0,5 %.61 La prévalence de l’HTAP associé au VIHest actuellement sous-estimée, mais elle devraitprobablement augmenter du fait d’une meilleurereconnaissance de cette association morbide et del’amélioration de la survie des patients séroposi-tifs. L’âge de survenue de l’HTAP associée au VIHest de 33 ans en moyenne, survenant à tout âge (2 à56 ans).60,63,64 La prédominance féminine, bienconnue dans l’HTAP « primitive » 2, n’est pas re-trouvée dans cette population, le sex-ratio étant de1/1,264 Il existe cependant un biais important se-condaire à la distribution même de l’infection parle VIH dans la population générale (homo- et bi-sexualité masculine, toxicomanie, hémophilie).L’HTAP peut toucher tous les groupes à risqued’infection par le VIH, mais les toxicomanes sontles plus fréquemment concernés puisqu’ils repré-sentent de 42 à 58 % de l’ensemble des mala-des.60,63,64 L’importance de l’immunodépression,la charge virale ou les antécédents d’infectionsopportunistes ne semblent pas corrélés au risque dedévelopper une HTAP. 59,60,63,64,65,66,67 L’HTAP estdiagnostiquée en moyenne 33 mois après la décou-verte de la séropositivité64, mais elle peut égale-ment la révéler (25 % des cas de la série de Petit-pretz et al.),68 ce qui justifie de réaliser unesérologie VIH dans le bilan initial de toute HTAPapparemment idiopathique.
55Hypertension artérielle pulmonaire

Physiopathologie
Les mécanismes à l’origine du développementd’une HTAP associée à l’infection VIH sont actuel-lement inconnus.66 La première hypothèse qui a étéévoquée est celle de la responsabilité non pas du
VIH lui-même mais d’un cofacteur, éventuellementlié à un comportement à risque particulier. Environ18 % de l’ensemble des patients publiés ont, outrel’infection par le VIH, une ou plusieurs autres cau-ses possibles d’HTAP.60,64 Parmi ces causes, on noteles cirrhoses hépatiques le plus souvent virales (hé-
Inhibiteurs des PDE5 ?5
Hypertension artérielle pulmonaire (classe fonctionnelle III ou IV de la NYHA)1
Traitement conventionnel(anticoagulants oraux ± diurétiques ± oxygène)
Réponse au test de vasodilatation aiguë ?2
oui non
Inhibiteurs calciques (voie orale)
Réponse persistante ?3
oui non
Poursuivre le traitement
Classe III4 Classe IV4
Antagonistes des récepteursde l’endothéline
ouAnalogues de la prostacycline
ouépoprosténol intraveineux
Absence d’améliorationou dégradation
(traitement combiné ?)6
Époprosténol intraveineuxou antagonistes des récepteurs
de l’endothélineou analogues de la prostacycline
Atrioseptostomieet/ou
transplantation pulmonaire7
Figure 3 Algorithme décisionnel dans le traitement de l’hypertension artérielle pulmonaire (HTAP).1. Cet algorithme est assezlargement accepté pour les patients de classe fonctionnelle III ou IV de la New York Heart Association (NYHA), alors qu’il existe peude données pour les patients en classe fonctionnelle I ou II. Les différents traitements ont été essentiellement évalués dans l’HTAPidiopathique (HTAP primitive), et dans l’HTAP associée à la sclérodermie ou à la prise d’anorexigènes. L’extrapolation de cetalgorithme aux autres types d’HTAP doit donc être réalisée avec prudence. Cet algorithme inclut les médicaments enregistrés dans aumoins un pays ou approuvés dans d’autres indications. 2. Les substances utilisées pour tester la vasoréactivité sont des agents decourte demi-vie comme la prostacycline intraveineuse, l’adénosine intraveineuse ou le monoxyde d’azote (NO) inhalé. Lestraitements au long cours par les inhibiteurs calciques doivent être proposés seulement aux patients considérés comme « répondeurs »au test de vasodilatation. Cette « réponse » se définit par une importante réduction de la pression artérielle pulmonaire (PAP)moyenne au cours du test (baisse de plus de 10 mmHg aboutissant à une PAP moyenne inférieure à 40 mmHg) associée à un débitcardiaque conservé ou augmenté. 3. Certains patients traités par les inhibiteurs calciques ont un bénéfice clinique persistant. Cespatients sont en classe fonctionnelle I ou II de la NYHA avec des paramètres hémodynamiques proches de la normale après 1 an detraitement. 4. La plupart des experts recommandent de traiter les patients en classe fonctionnelle IV de la NYHA par l’époprosténolen intraveineux. En dehors de cette situation particulière, le traitement de première intention peut inclure le bosentan par voie oraleou un analogue de la prostacycline. La Food and Drug Administration a enregistré l’époprosténol intraveineux, le bosentan oral et letréprostinil sous-cutané dans le traitement des patients en classe fonctionnelle III ou IV de la NYHA. En Europe, l’époprosténolintraveineux est indiqué pour le traitement des patients en classe fonctionnelle III ou IV de la NYHA, tandis que le bosentan oral etl’iloprost nébulisé sont reconnus pour le traitement des patients en classe fonctionnelle III. 5. Il existe peu de données sur letraitement par sildénafil à long terme dans l’HTAP à l’exception de quelques cas et de courtes séries. Des études sont actuellementen cours afin d’évaluer l’efficacité et la place de ce médicament dans la prise en charge de l’HTAP. 6. Les traitements « combinés »représentent de nouvelles possibilités thérapeutiques par association d’agents dont les mécanismes d’action sont différents.L’adjonction de sildénafil ou de bosentan chez des patients recevant des analogues de la prostacycline a déjà permis d’obtenir desrésultats favorables. Néanmoins, de nouvelles études sont nécessaires pour confirmer l’intérêt des traitements « combinés » dans laprise en charge de l’HTAP. 7. La transplantation pulmonaire représente une alternative pour les patients potentiellement opérablesqui restent en classe fonctionnelle III ou IV de la NYHA après 3 mois de traitement par époprosténol. Il est essentiel de prendre encompte les délais d’attente relativement longs avant transplantation dans la décision d’inscription sur liste de transplantation.L’atrioseptostomie représente une alternative pour des patients ayant une HTAP sévère qui s’aggrave malgré un traitement médicalmaximal.
56 D. Montani et al.

patite B ou C),60 la prise d’amphétamines, de co-caïne ou d’anorexigènes,69,70,71 les embolies pul-monaires de talc liées à l’injection intraveineuse decomprimés écrasés,72,73,74, voire une maladiethromboembolique chronique d’origine cruori-que.75 L’existence de ces mécanismes lésionnelsdivers pourrait expliquer la fréquence élevée del’HTAP chez les patients toxicomanes. Le rôled’une co-infection virale, notamment par l’herpèsvirus humain 8 (HHV8), pourrait également êtreenvisagé dans l’HTAP associé à l’infection VIHcomme dans le sarcome de Kaposi.76 Une étuderécente77 a retrouvé des marqueurs d’infection àHHV8 dans les cellules endothéliales de patientsprésentant des HTAP. La prévalence des marqueursde l’infection HHV8 n’était pas différente entre lespatients souffrant d’HTAP idiopathique ou associéeau VIH, cette observation ne permettant pas d’ex-pliquer le lien entre VIH et HTAP.La seconde hypothèse, communément admise,
est celle de l’existence d’une relation de cause àeffet, directe ou indirecte, entre l’HTAP et l’infec-tion par le VIH. Une étude autopsique a révélé chez19 macaques sur 85 infectés par le « simian immu-nodeficiency virus », une artériopathie pulmonairecaractérisée par des lésions de fibrose, d’épaissis-sement de l’intima et de la média, et par desinfiltrats inflammatoires périvasculaires alorsqu’aucune particule virale ni matériel génétiquen’a été mis en évidence.78 Contrairement àd’autres complications cardiovasculaires du VIHcomme la cardiomyopathie dilatée,79 le virus n’ajamais été directement retrouvé au niveau desvaisseaux pulmonaires.80,81 Le virus du VIH pourraitagir indirectement en favorisant la libération, pardes lymphocytes ou les macrophages activés, demédiateurs ayant un effet prolifératif sur les cellu-les endothéliales ou les cellules musculaires lisses.L’analyse de l’artériopathie de trois patients ayantune HTAP associée à l’infection VIH a permis deconstater que les lésions plexiformes étaient pré-dominantes.82 Ces lésions se constituaient essen-tiellement de cellules endothéliales et s’accompa-gnaient d’un important infiltrat périvasculaire delymphocytes T et de macrophages, ce qui suggéraitle rôle de ces cellules inflammatoires dans le remo-delage vasculaire.82 De nombreux facteurs de pro-lifération sont libérés de manière accrue dans l’in-fection VIH (platelet derived growth factor,83
vascular endothelial growth factor, tissue necrosisfactor a).84 De plus, la gp-120, glycoprotéine d’en-veloppe du VIH-1, provoque une production abon-dante de TNF-a et d’endothéline-1 par les monocy-tes.85 La protéine tat, produit d’un gène régulateurdu VIH-1, peut aussi stimuler la croissance cellu-laire (cellules de sarcome de Kaposi86 et cellulesendothéliales humaines).87
Seule une faible proportion de patients séroposi-tifs développent une HTAP, laissant supposer quecette affection requiert une ou plusieurs conditionsassociées pour se développer. L’existence de muta-tions du gène BMPR2 dans l’HTAP associée au VIHreprésentait une hypothèse séduisante qui n’a pasété confirmée puisque dans notre série de 19 pa-tients testés, aucun n’avait de mutations retrou-vées.67 Enfin, le système human leucocyte antigen(HLA) pourrait également être un élément prédis-posant puisqu’une augmentation significative de lafréquence de l’allèle HLA-DR6 et DR52 a été obser-vée chez les patients développant une HTAP dans lecadre d’une infection VIH.88
Histologie
Les lésions histologiques de l’HTAP associée au VIHsont similaires à celles de l’HTAP « primitive ». Uneartériopathie plexiforme est l’aspect le plus cou-rant, retrouvé dans plus de 85 % des cas publiés.58,59,63,64,65,66 Une artériopathie thrombotique estdécrite dans un cas89 et une maladie veino-occlusive dans trois cas.70,90,91
Diagnostic
La présentation clinique de l’HTAP associée à l’in-fection VIH est semblable à celle de l’HTAP « pri-mitive ».68,92 Le délai entre les premiers signescliniques et la découverte de l’HTAP est plus courtchez les patients séropositifs (9,9 ± 13,6 moiscontre 21,4 ± 26,5 mois), témoignant probablementd’une surveillance médicale accrue.68 Le diagnosticest le plus souvent évoqué lors d’une échographiecardiaque et confirmé par la réalisation d’un cathé-térisme cardiaque droit. Il est important d’éliminercertaines situations fréquentes chez le séropositifpouvant être à l’origine de l’HTAP, notammentl’hypertension portale (association fréquente d’hé-patite B ou C chronique). En revanche, dans notreexpérience, la réponse au test de vasodilatationaiguë par le monoxyde d’azote est exceptionnelledans l’HTAP associée au VIH67,92 et significative-ment moins fréquente que dans l’HTAP idiopathi-que,92 ce qui suggère que les lésions de remodelagevasculaire fixées apparaissent probablement plusrapidement dans le cadre de l’infection par le VIH.
Évolution
Le pronostic de l’HTAP associée à l’infection VIHest conditionné par l’aggravation de l’HTAP plutôtque par les complications liées à l’immunodépres-sion, l’HTAP étant responsable d’environ 75 % desdécès de ces patients.59,60,63,64,65,66 La première
57Hypertension artérielle pulmonaire
étude analysant la survie de ces patients retrouvaitun taux de survie de 46 % à 2 ans similaire à celledes patients atteints d’HTAP idiopathique.68 Ce-pendant, notre équipe a démontré, sur une sérieplus importante, que la survie des patients séropo-sitifs est moins bonne que celle des séronégatifs.92
Opravil et al.61 retrouvent, quant à eux, une surviede 58 % à 1 an, 32 % à 2 ans et 21 % à 3 ans,significativement moins bonne que celle des pa-tients séropositifs sans HTAP (médiane de survie de1,3 contre 2,6 ans), l’HTAP représentant un facteurprédictif de mortalité indépendant. Il semble néan-moins que l’on assiste depuis quelques années à unediminution de la mortalité de l’HTAP associée àl’infection VIH grâce à l’effet combiné des nouvel-les thérapies antirétrovirales et des nouvelles pos-sibilités thérapeutiques dans le traitement del’HTAP.
Traitement
Le traitement conventionnel (limitation des ef-forts, anticoagulation orale, diurétiques et oxygènesi nécessaire) est similaire à celui de l’HTAP idiopa-thique.2 La prescription des antivitamines K peutêtre délicate, des troubles de l’hémostase étantassez fréquents dans cette population (thrombopé-nie du VIH, hépatopathie concomitante). Les inhi-biteurs calciques à fortes doses n’ont démontréleur bénéfice dans l’HTAP idiopathique que chez lespatients répondeurs en aigu aux vasodilatateurs.2
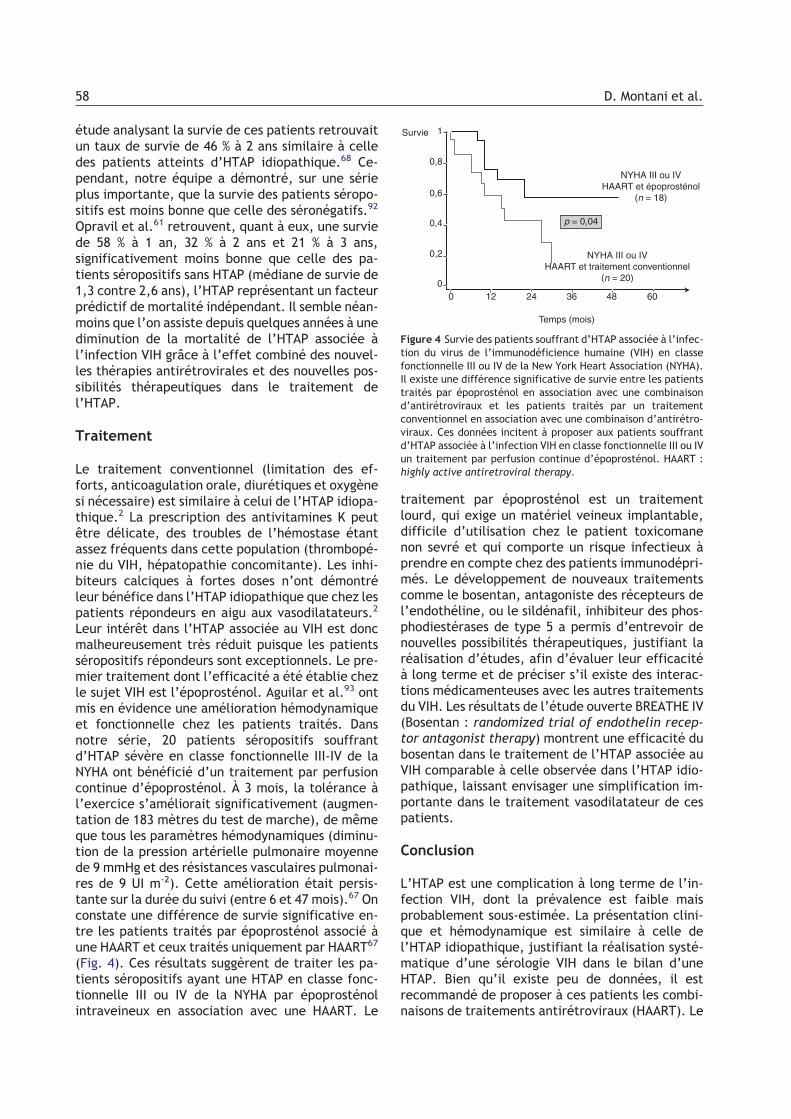
Leur intérêt dans l’HTAP associée au VIH est doncmalheureusement très réduit puisque les patientsséropositifs répondeurs sont exceptionnels. Le pre-mier traitement dont l’efficacité a été établie chezle sujet VIH est l’époprosténol. Aguilar et al.93 ontmis en évidence une amélioration hémodynamiqueet fonctionnelle chez les patients traités. Dansnotre série, 20 patients séropositifs souffrantd’HTAP sévère en classe fonctionnelle III-IV de laNYHA ont bénéficié d’un traitement par perfusioncontinue d’époprosténol. À 3 mois, la tolérance àl’exercice s’améliorait significativement (augmen-tation de 183 mètres du test de marche), de mêmeque tous les paramètres hémodynamiques (diminu-tion de la pression artérielle pulmonaire moyennede 9 mmHg et des résistances vasculaires pulmonai-res de 9 UI m-2). Cette amélioration était persis-tante sur la durée du suivi (entre 6 et 47 mois).67 Onconstate une différence de survie significative en-tre les patients traités par époprosténol associé àune HAART et ceux traités uniquement par HAART67
(Fig. 4). Ces résultats suggèrent de traiter les pa-tients séropositifs ayant une HTAP en classe fonc-tionnelle III ou IV de la NYHA par époprosténolintraveineux en association avec une HAART. Le
traitement par époprosténol est un traitementlourd, qui exige un matériel veineux implantable,difficile d’utilisation chez le patient toxicomanenon sevré et qui comporte un risque infectieux àprendre en compte chez des patients immunodépri-més. Le développement de nouveaux traitementscomme le bosentan, antagoniste des récepteurs del’endothéline, ou le sildénafil, inhibiteur des phos-phodiestérases de type 5 a permis d’entrevoir denouvelles possibilités thérapeutiques, justifiant laréalisation d’études, afin d’évaluer leur efficacitéà long terme et de préciser s’il existe des interac-tions médicamenteuses avec les autres traitementsdu VIH. Les résultats de l’étude ouverte BREATHE IV(Bosentan : randomized trial of endothelin recep-tor antagonist therapy) montrent une efficacité dubosentan dans le traitement de l’HTAP associée auVIH comparable à celle observée dans l’HTAP idio-pathique, laissant envisager une simplification im-portante dans le traitement vasodilatateur de cespatients.
Conclusion
L’HTAP est une complication à long terme de l’in-fection VIH, dont la prévalence est faible maisprobablement sous-estimée. La présentation clini-que et hémodynamique est similaire à celle del’HTAP idiopathique, justifiant la réalisation systé-matique d’une sérologie VIH dans le bilan d’uneHTAP. Bien qu’il existe peu de données, il estrecommandé de proposer à ces patients les combi-naisons de traitements antirétroviraux (HAART). Le
Survie
Temps (mois)
1
0,8
0,6
0,4
0,2
00 12 24 36 48 60
NYHA III ou IVHAART et traitement conventionnel
(n = 20)
NYHA III ou IVHAART et époprosténol
(n = 18)
p = 0,04
Figure 4 Survie des patients souffrant d’HTAP associée à l’infec-tion du virus de l’immunodéficience humaine (VIH) en classefonctionnelle III ou IV de la New York Heart Association (NYHA).Il existe une différence significative de survie entre les patientstraités par époprosténol en association avec une combinaisond’antirétroviraux et les patients traités par un traitementconventionnel en association avec une combinaison d’antirétro-viraux. Ces données incitent à proposer aux patients souffrantd’HTAP associée à l’infection VIH en classe fonctionnelle III ou IVun traitement par perfusion continue d’époprosténol. HAART :highly active antiretroviral therapy.
58 D. Montani et al.

traitement vasodilatateur le mieux évalué est ac-tuellement l’époprosténol mais l’utilisation desnouveaux traitements, notamment les antagonistesdes récepteurs de l’endothéline, va probablementmodifier la prise en charge de ces patients dans lesannées à venir.
Hypertension artérielle pulmonaireassociée aux maladies hépatiques
En cas d’hypertension portale, deux types d’at-teinte pulmonaire peuvent survenir en dehors desépanchements pleuraux. Il s’agit du syndrome hé-patopulmonaire entraînant une hypoxémie rare-ment symptomatique survenant chez 15 % des ma-lades atteints de cirrhose, et de l’hypertensionportopulmonaire plus rare mais présente et symp-tomatique chez environ 1 % des malades atteints decirrhose.94 Il n’existe pas de relation entre ces deuxcomplications pulmonaires de mécanisme diffé-rent.
Prévalence de l’hypertensionportopulmonaire
L’association entre hypertension portale et HTAPest bien établie et est reconnue comme une asso-ciation « très probable ». L’HTAP modérée etasymptomatique dite « forme passive » (pressionartérielle pulmonaire PAPm < 35 mmHg), est plusfréquente que l’HTAP sévère et symptomatiquedite « forme active » (PAPm > 35 mmHg). La préva-lence de l’HTAP de forme active chez les maladesatteints de cirrhose a été estimée à 0,25 %.95 Dansune série autopsique, des lésions plexiformes desartères pulmonaires ont été observées dans 0,26 %des cas.96 Plus récemment, il a été estimé que laprévalence de l’HTAP chez les malades atteints decirrhose était plus élevée, de l’ordre de 0,60 %.97
Cependant, la recherche systématique d’HTAPchez les malades candidats à la transplantationhépatique a trouvé une prévalence de 3 à 4 %.98 Lesrésultats de ces études suggèrent que chez lesmalades atteints d’hypertension portale, une HTAPpassive est présente chez 2 à 4 % des malades et uneHTAP sévère ou forme active, symptomatique estplus rare, probablement inférieure à 0,5 %. Il a étémontré par ailleurs que l’hypertension portale étaitresponsable de 10 % environ d’HTAP d’allure « pri-mitive ».94
Histologie
Les lésions histologiques pulmonaires observéeschez les malades atteints d’hypertension portopul-
monaire ne sont pas différentes de celles observéeschez les malades atteints d’HTAP idiopathique.99
Des lésions plexiformes des artères pulmonairesassociées à une hypertrophie de la média des artè-res, avec ou sans fibrose de l’intima, sont classique-ment retrouvées chez ces patients. Des lésionsthromboemboliques artérielles ont rarement étéobservées chez ces malades.
Mécanismes physiopathologiques
Les mécanismes responsables du développementd’une hypertension portopulmonaire sont proba-blement multifactoriels. La présence d’une circula-tion portosystémique secondaire à l’hypertensionportale semble nécessaire au développement d’uneHTAP. En effet, une circulation collatérale porto-systémique existe quelle que soit la cause d’hyper-tension portale et la survenue d’une HTAP est plusfréquente chez les malades ayant une anastomoseportocave chirurgicale.95 Il n’existe pas toutefoisde relation entre le niveau de pression artériellepulmonaire et l’intensité de la circulation collaté-rale évaluée par le débit sanguin azygos.100 Il a étésuggéré que des microemboles provenant du terri-toire porte pouvaient être responsables de l’HTAPpuisque ceux-ci pouvaient atteindre la circulationpulmonaire à travers les shunts portosystémiques,mais ce mécanisme n’a pas été confirmé par lesétudes autopsiques.101 Il n’existe pas de relationentre la survenue d’une HTAP et la sévérité de lamaladie du foie ni avec la cause de l’hypertensionportale (présence ou non d’une maladie du foie,bloc intra- ou extrahépatique).101 Le degré de l’hy-pertension portale mesurée par le gradient de pres-sion hépatique n’est pas corrélé avec la pressionartérielle pulmonaire.100 En cas d’hypertensionportale, l’augmentation importante du débit san-guin pulmonaire par une augmentation du débitcardiaque pourrait participer à la survenue d’uneHTAP, les forces de cisaillement pouvant être res-ponsables de remaniements vasculaires pulmonai-res.102 Il semble que dans les formes passives,l’augmentation du débit cardiaque soit à l’origined’une élévation modérée de la PAPm. Il existetoutefois une corrélation inverse entre le débitcardiaque et la pression artérielle pulmonaire chezles malades atteints d’hypertension portopulmo-naire symptomatique.100 Après transplantation hé-patique, une diminution du débit cardiaque sansmodification de la pression artérielle pulmonaire aété observée.103 De même, l’augmentation impor-tante du débit cardiaque après réalisation d’unshunt chirurgical n’augmente pas significativementla pression artérielle pulmonaire mais est associéeà une diminution des résistances vasculaires pulmo-
59Hypertension artérielle pulmonaire

naires.104 La présence de circulations collatéralesportosystémiques entraîne la circulation, au niveaupulmonaire, de substances produites dans le terri-toire splanchnique, normalement épurées par lefoie. En cas de cirrhose, les concentrations plasma-tiques de certains médiateurs vasoactifs comme lescytokines pro-inflammatoires ou les facteurs decroissance sont augmentées.105 La sérotonine pour-rait notamment être à l’origine d’une vasoconstric-tion pulmonaire,106 d’autant plus que le stockageplaquettaire est altéré dans la cirrhose.107 L’ab-sence de ces substances endogènes, observée chezles malades atteints d’hypertension portale parthrombose porte, suggère que ces mécanismes nepeuvent pas expliquer seuls la survenue d’uneHTAP. Une réduction de l’expression de la NO syn-thétase endothéliale pulmonaire ou une hyperex-pression de l’endothéline dans ces vaisseaux pour-rait modifier les parois des artères pulmonaires etaugmenter leur résistance.108,109 Il est probablequ’il existe des facteurs individuels en particuliergénétiques qui pourraient favoriser la survenued’une hypertension portopulmonaire. Dans notreexpérience, les mutations du gène BMPR2 ne sontpas identifiées chez les patients souffrant d’hyper-tension portopulmonaire.
Diagnostic
Chez les malades avec une forme passive ou asymp-tomatique d’HTAP, le diagnostic peut être fait aucours d’un cathétérisme cardiaque droit effectué àl’occasion d’une biopsie hépatique par voie trans-jugulaire100 ou au cours d’un bilan de prétransplan-tation hépatique. Chez les malades avec une formeactive d’HTAP, les manifestations cliniques sontidentiques à celles de l’HTAP idiopathique.94 Dansune étude récente, l’échodoppler cardiaque, effec-tué chez 165 patients souffrant d’hypertension por-tale, a retrouvé des signes d’HTAP chez 17 maladessoit 10 % et le cathétérisme droit a confirmé laprésence d’HTAP chez 10 de ces malades, soit 6 %des patients avec une hypertension portale.110 Cesrésultats confirment la sensibilité élevée et la va-leur prédictive positive faible sur ce terrain del’échographie cardiaque et souligne l’importancedu cathétérisme pour confirmer le diagnostic.
Évolution
L’histoire naturelle de l’HTAP chez les maladesatteints d’hypertension portale n’est pas connue.Elle dépend de la sévérité de l’HTAP, mais aussi descomplications de l’hypertension portale et de lasévérité de la maladie du foie. Chez les maladesavec une HTAP asymptomatique, il n’existe pas
d’information dans la littérature sur l’évolution deces malades, en particulier sur le passage de laforme passive à la forme active. Chez les maladessymptomatiques, le délai entre le diagnostic del’hypertension portale et le diagnostic d’HTAP estde 28 ± 38 mois et le délai entre la premièremanifestation et le diagnostic d’HTAP de 7 à20 mois.100 Les études autopsiques ont confirméque les manifestations de l’HTAP pouvaient précé-der celles de l’hypertension portale. La présenced’une forme active d’HTAP est de pronostic défa-vorable chez les malades atteints d’hypertensionportale, la survie de ces malades étant relative-ment courte, le plus souvent inférieure à24 mois.100
Traitement
Le traitement de l’hypertension portopulmonaireest identique à celui de l’HTAP idiopathique. Letraitement médical comprend des anticoagulants,des diurétiques, l’administration d’oxygène si né-cessaire ainsi qu’un traitement spécifique del’HTAP, le plus souvent dérivé de la prostacycline.Chez les malades atteints de cirrhose, les anticoa-gulants ne sont habituellement pas administréslorsqu’il existe une insuffisance hépatocellulairesévère. L’efficacité du bosentan, antagoniste nonspécifique des récepteurs de l’endothéline,49 chezles malades atteints d’hypertension portopulmo-naire est inconnue. Toutefois, étant donné le risquede cytolyse hépatique, il n’est pas utilisé actuelle-ment dans cette indication. Chez les patients symp-tomatiques, seuls les dérivés de la prostacyclinesont utilisés en pratique courante.2 Le traitementest administré par voie orale (Beraprost ®), inhalée(Iloprost®) ou le plus souvent en perfusion continueintraveineuse (Flolan®). Le traitement permetd’améliorer les signes fonctionnels, le périmètre demarche et la qualité de vie de ces malades.111 Dansune étude effectuée chez des malades atteintsd’hypertension portopulmonaire, l’époprosténol apermis d’améliorer l’hémodynamique de la plupartdes malades.112 La transplantation hépatique n’estpas indiquée en cas d’hypertension portopulmo-naire bien que quelques cas aient été rapportésmontrant une amélioration de l’HTAP après trans-plantation.113,114 Il existe peu d’arguments physio-pathologiques pour penser que la transplantationhépatique permette de guérir l’HTAP. Seule unediminution du débit cardiaque observée aprèstransplantation hépatique pourrait expliquer unediminution progressive et modérée de la pressionartérielle pulmonaire. En revanche, lorsqu’il existeune indication à une transplantation hépatique liéeà une insuffisance hépatocellulaire, il est établi
60 D. Montani et al.

que l’HTAP sévère (PAPm > 35 mmHg) est unecontre-indication. Il a été possible toutefois, danscertains cas, de diminuer la pression artérielle pul-monaire par les dérivés de la prostacycline permet-tant d’effectuer dans un second temps une trans-plantation hépatique. Actuellement, la tripletransplantation foie-cœur-poumons ne peut êtrerecommandée, car il existe une importante morta-lité opératoire de 50 % et une survie faible de 10 %à 5 ans.
Conclusion
L’HTAP symptomatique est une complication rarede l’hypertension portale, survenant dans 0,5 % descas. Il faut distinguer l’HTAP passive, reflet del’augmentation du débit cardiaque sans conséquen-ces thérapeutiques, de l’HTAP active, proche del’HTAP idiopathique. La survenue d’une HTAP n’estpas liée à la sévérité de la maladie hépatique etconstitue un élément pronostique majeur. Le trai-tement de référence est actuellement constituépar les analogues de la prostacycline, l’efficacitédes nouvelles thérapeutiques restant à évaluerdans cette indication.
Traitement chirurgical de la maladiethromboembolique
L’HTAP postembolique ou cœur pulmonaire chroni-que postembolique (CPCPE) est liée à la persistanceet l’organisation fibreuse des caillots après une ouplusieurs embolies pulmonaires aiguës.115 Cettemaladie, autrefois considérée comme exception-nelle, est actuellement de plus en plus fréquem-ment diagnostiquée probablement du fait de laréalisation plus fréquente d’échographie cardia-que. La prise en charge est chirurgicale si la situa-tion le permet, la thombo-endartériectomie pulmo-naire ayant considérablement transformé lepronostic de ces malades.
Physiopathologie
L’évolution naturelle de l’embolie pulmonaire sefait vers la résorption des caillots par une fibrino-lyse locale spontanée ou pharmacologique avecrestauration ad integrum du lit artériel pulmonaire.Dans 0,1 à 0,4 % des cas, pour des raisons encoremal connues (anomalies de l’hémostase ou du sys-tème fibrinolytique, embolies de caillots organisésou embolies répétées), cette résorption ne se pro-duit pas et l’évolution se fait vers une organisationfibreuse du caillot.116,117 La conséquence en est
une obstruction définitive et irréversible du lit ar-tériel pulmonaire pouvant entraîner une HTAP.L’apparition de la dyspnée après un intervalle librede plusieurs années (« lune de miel ») est habituelledans le CPCPE.115,118,119 Les symptômes ne sont engénéral pas liés à des récidives emboliques, il s’agitle plus souvent des conséquences du développe-ment d’une artériopathie dans les territoires nonobstrués et de la formation de thromboses in situsecondaires au bas débit.120 Des thrombus d’allu-vionnement, situés en amont de l’obstruction arté-rielle, sont présents dans 20 % des cas et sontfacilement visibles en angiographie et au scanner.Ils sont une conséquence de la maladie et non sacause initiale. Les lésions observées dans les terri-toires non obstrués sont identiques à celles obser-vées dans l’HTAP idiopathique. L’examen anatomo-pathologique des artères obstruées retrouve unvéritable moule de l’arbre artériel pulmonaire, re-couvert de fibres élastiques, soulignant la nécessitéde réaliser une endartériectomie et non une throm-bectomie.121,122
Diagnostic
Classiquement, il existe deux contextes de diagnos-tic de CPCPE, soit lors de l’exploration d’une dysp-née persistante après une embolie pulmonaire, soitlors du bilan d’une dyspnée faisant découvrir unemaladie thromboembolique passée inaperçue. Ilexiste souvent un retard diagnostique, les décom-pensations de cœur pulmonaire chronique étantparfois considérées comme des épisodes aigusthromboemboliques. Comme dans l’HTAP idiopa-thique, l’examen de référence pour le dépistage deCPCPE est l’échocardiographie cardiaque associéeau doppler pulsé. L’angioscanner peut montrer uneobstruction ou une diminution du diamètre de lalumière artérielle par rapport au diamètre externede l’artère pulmonaire, mais sa sensibilité pour leslésions situées en aval des premières branches estfaible.123 Un angioscanner normal ne suffit doncpas à éliminer le diagnostic d’HTAP postembolique.La scintigraphie pulmonaire de ventilation et deperfusion plus sensible est indispensable pourorienter le diagnostic. L’angiographie pulmonaire,associée à une étude hémodynamique complèteafin d’estimer la sévérité, est systématique. Elleconfirme le diagnostic et détermine, en fonction ducaractère proximal ou distal des lésions, les possi-bilités chirurgicales.124 Les images angiographiquesévoquant une maladie thromboembolique chroni-que comprennent l’arrêt sacciforme par obstruc-tion de l’artère pulmonaire, les irrégularités de laparoi artérielle, les changements brutaux de cali-bre de l’artère ou le manque de branches artériel-
61Hypertension artérielle pulmonaire

les segmentaires ou lobaires avec absence de pa-renchymographie dans ces territoires (Fig. 5).
Endartériectomie pulmonaire
Il existe deux possibilités chirurgicales dans leCPCPE : l’endartériectomie et la transplantationpulmonaire.125,126,127,128,129,130,131 L’endartériec-tomie est le traitement de choix chaque fois qu’elleest possible car elle restaure une fonction cardio-respiratoire proche de la normale. Cette interven-tion est limitée aux formes accessibles car proxima-les. Il s’agit d’une intervention chirurgicale lourdemais dont les bénéfices sont indiscutables en ter-mes cliniques, hémodynamiques et de survie. Lesformes distales sont inaccessibles au traitementchirurgical, leur évolution et leur prise en charge serapprochent alors de celles de l’HTAP idiopathique.La transplantation peut constituer une alternativechez ces patients. L’endartériectomie consiste àdésobstruer sous circulation extracorporelle cha-que artère pulmonaire et ses branches lobaires etsegmentaires soit environ 20 à 30 branches, afind’obtenir une diminution d’au moins 50 % des résis-tances vasculaires pulmonaires. Le matériel intra-luminal est, à ce stade, un tissu fibreux faisant
corps avec l’intima, inaccessible à toute tentativede thrombectomie ou de dilatation (Fig. 6). La« désobstruction » est donc une véritable endarté-riectomie, amorcée au niveau des troncs des artè-res pulmonaires droite et gauche en intrapéricardi-que et étendue progressivement vers la distalitédans chacune des nombreuses branches de l’arbreartériel pulmonaire (Fig. 7, 8). Les patients atteintsd’HTAP postembolique développent rapidement,après les épisodes emboliques, une hypervasculari-sation systémique à partir des artères bronchiqueset des artères de la paroi thoracique à travers lesadhérences pleurales séquellaires des embolies. Le
Figure 5 Angiographie pulmonaire sélective droite chez un pa-tient souffrant d’une hypertension artérielle pulmonaire post-embolique proximale accessible à la thromboendartériectomiepulmonaire. Cette angiographie montre un aspect irrégulier del’artère pulmonaire droite et un arrêt cupuliforme de l’artère dusegment apical du lobe inférieur droit, ainsi que des irrégularitésde nombreuses artères pulmonaires segmentaires et sous-segmentaires.
Figure 6 Matériel endoluminal retiré lors d’une endartériecto-mie. Dans le cœur pulmonaire chronique, le matériel intralumi-nal est, à ce stade, un tissu fibreux faisant corps avec l’intima.La « désobstruction » est donc une véritable endartériectomie,amorcée au niveau des troncs des artères pulmonaires droite etgauche en intrapéricardique et étendue progressivement vers ladistalité dans chacune des nombreuses branches de l’arbreartériel pulmonaire (soit environ 20 à 30 branches).
Figure 7 Dans le cœur pulmonaire chronique postembolique, onobserve un rétrécissement progressif de la lumière des artèrespulmonaires conduisant à leur obstruction complète. L’hyper-tension artérielle pulmonaire consécutive de l’obstruction en-traîne une hypertrophie et une dilatation du ventricule droit(VD), entraînant un septum paradoxal et une compression duventricule gauche (VG).
62 D. Montani et al.
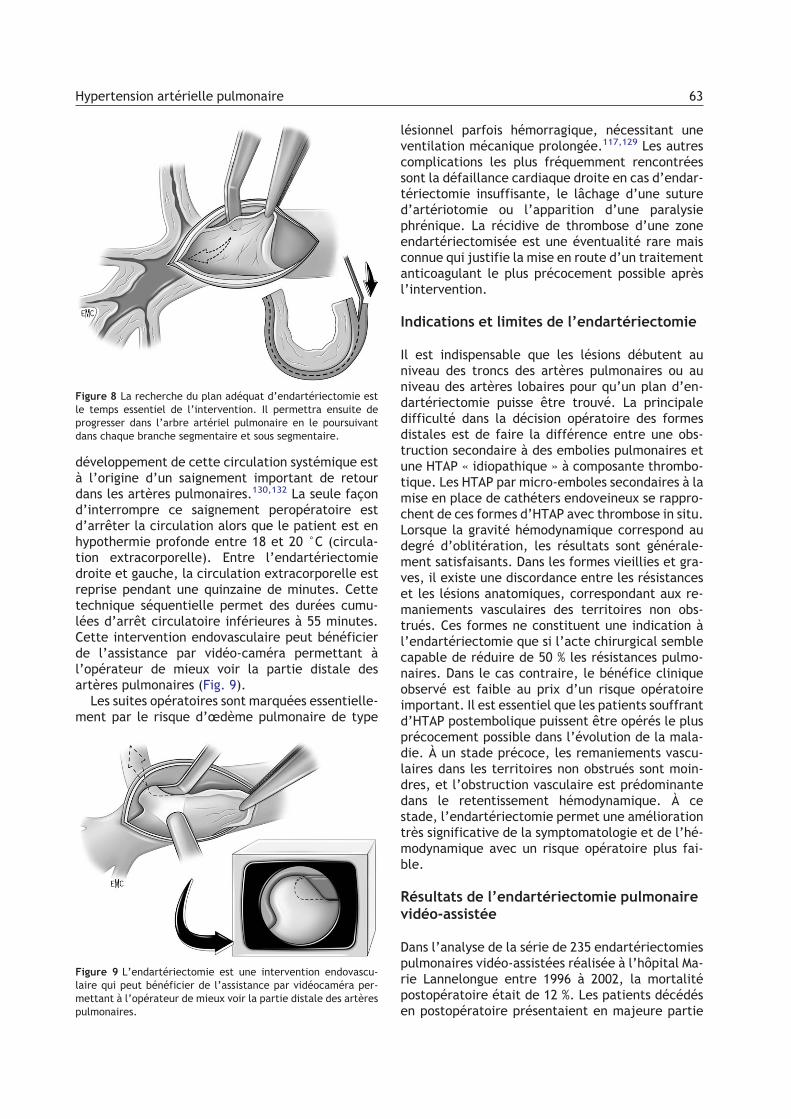
développement de cette circulation systémique està l’origine d’un saignement important de retourdans les artères pulmonaires.130,132 La seule façond’interrompre ce saignement peropératoire estd’arrêter la circulation alors que le patient est enhypothermie profonde entre 18 et 20 °C (circula-tion extracorporelle). Entre l’endartériectomiedroite et gauche, la circulation extracorporelle estreprise pendant une quinzaine de minutes. Cettetechnique séquentielle permet des durées cumu-lées d’arrêt circulatoire inférieures à 55 minutes.Cette intervention endovasculaire peut bénéficierde l’assistance par vidéo-caméra permettant àl’opérateur de mieux voir la partie distale desartères pulmonaires (Fig. 9).Les suites opératoires sont marquées essentielle-
ment par le risque d’œdème pulmonaire de type
lésionnel parfois hémorragique, nécessitant uneventilation mécanique prolongée.117,129 Les autrescomplications les plus fréquemment rencontréessont la défaillance cardiaque droite en cas d’endar-tériectomie insuffisante, le lâchage d’une sutured’artériotomie ou l’apparition d’une paralysiephrénique. La récidive de thrombose d’une zoneendartériectomisée est une éventualité rare maisconnue qui justifie la mise en route d’un traitementanticoagulant le plus précocement possible aprèsl’intervention.
Indications et limites de l’endartériectomie
Il est indispensable que les lésions débutent auniveau des troncs des artères pulmonaires ou auniveau des artères lobaires pour qu’un plan d’en-dartériectomie puisse être trouvé. La principaledifficulté dans la décision opératoire des formesdistales est de faire la différence entre une obs-truction secondaire à des embolies pulmonaires etune HTAP « idiopathique » à composante thrombo-tique. Les HTAP par micro-emboles secondaires à lamise en place de cathéters endoveineux se rappro-chent de ces formes d’HTAP avec thrombose in situ.Lorsque la gravité hémodynamique correspond audegré d’oblitération, les résultats sont générale-ment satisfaisants. Dans les formes vieillies et gra-ves, il existe une discordance entre les résistanceset les lésions anatomiques, correspondant aux re-maniements vasculaires des territoires non obs-trués. Ces formes ne constituent une indication àl’endartériectomie que si l’acte chirurgical semblecapable de réduire de 50 % les résistances pulmo-naires. Dans le cas contraire, le bénéfice cliniqueobservé est faible au prix d’un risque opératoireimportant. Il est essentiel que les patients souffrantd’HTAP postembolique puissent être opérés le plusprécocement possible dans l’évolution de la mala-die. À un stade précoce, les remaniements vascu-laires dans les territoires non obstrués sont moin-dres, et l’obstruction vasculaire est prédominantedans le retentissement hémodynamique. À cestade, l’endartériectomie permet une améliorationtrès significative de la symptomatologie et de l’hé-modynamique avec un risque opératoire plus fai-ble.
Résultats de l’endartériectomie pulmonairevidéo-assistée
Dans l’analyse de la série de 235 endartériectomiespulmonaires vidéo-assistées réalisée à l’hôpital Ma-rie Lannelongue entre 1996 à 2002, la mortalitépostopératoire était de 12 %. Les patients décédésen postopératoire présentaient en majeure partie
Figure 8 La recherche du plan adéquat d’endartériectomie estle temps essentiel de l’intervention. Il permettra ensuite deprogresser dans l’arbre artériel pulmonaire en le poursuivantdans chaque branche segmentaire et sous segmentaire.
Figure 9 L’endartériectomie est une intervention endovascu-laire qui peut bénéficier de l’assistance par vidéocaméra per-mettant à l’opérateur de mieux voir la partie distale des artèrespulmonaires.
63Hypertension artérielle pulmonaire

une HTAP persistante après endartériectomie. Dansce groupe de patients sont retrouvées principale-ment les maladies thromboemboliques sur cathé-ters, sondes de pacemaker ou dérivations ventricu-loatriales et des patients présentant probablementune HTAP idiopathique thrombotique mimant laprésentation clinique d’un CPCPE. Il est intéressantde remarquer que le taux de mortalité de cetteintervention est étroitement lié à la sévérité hémo-dynamique et à la valeur des résistances vasculairespulmonaires totales. La mortalité observée était de4, 10 et 20 % lorsque les résistances vasculairespulmonaires totales étaient respectivement infé-rieures à 900, entre 900 et 1 200 et supérieures à1200 dynes s m-5 . Il faut cependant interpréter cestaux de mortalité en fonction du degré d’obstruc-tion anatomique. Ainsi, un patient avec des résis-tances pulmonaires très élevées et une obstructionanatomique faible est à très haut risque alors quepour les mêmes résistances, celui dont l’obstruc-tion anatomique est très importante présente unrisque plus faible. En cas de sélection adéquate despatients, l’endartériectomie est habituellementsuivie d’une diminution considérable des résistan-ces pulmonaires et d’une amélioration importantede l’état fonctionnel du patient. L’améliorationfonctionnelle et hémodynamique se poursuit dansles mois qui suivent l’intervention, témoignant del’importance des remaniements vasculaires dansles territoires non obstrués.
Traitement médical
Les anticoagulants sont indispensables dans le trai-tement de l’HTAP postembolique, ils doivent êtredébutés dès la suspicion du diagnostic et poursuivisà vie (objectif international normalized ratio [INR]entre 2 et 3) que le patient ait bénéficié ou nond’une thromboendartériectomie. Dans les formesdistales non opérables ou en l’absence d’améliora-tion suffisante après endartériectomie, la prise encharge est similaire à celle de l’HTAP idiopathique.L’époprosténol est parfois proposé en attente de latransplantation pulmonaire (« bridge-therapy »).Par ailleurs, certaines équipes préparent leurs pa-tients les plus sévères par une injection continue deprostacycline afin de réduire les résistances pulmo-naires avant thromboendartériectomie. Un despoints essentiels à prendre en compte lors de l’ins-cription sur liste consiste à intégrer dans la décisionles délais d’attente relativement longs avant trans-plantation, conséquence du faible nombre de don-neurs en France (durée médiane d’attente de l’or-dre de 18 mois). Les nouveaux traitements, commeles analogues de la prostacycline, les antagonistesdes récepteurs de l’endothéline ou le sildénafil,nécessitent d’être évalués dans cette indication.
Conclusion
L’endartériectomie pulmonaire constitue le traite-ment de référence de l’HTAP postembolique asso-ciée à des obstructions proximales. Il s’agit d’uneintervention lourde nécessitant une circulation ex-tracorporelle mais qui permet d’améliorer signifi-cativement les symptômes, l’hémodynamique et lasurvie des patients souffrant de CPCPE. L’impor-tance des remaniements vasculaires dans les terri-toires non obstrués constitue un élément pronosti-que essentiel, justifiant la réalisation le plus tôtpossible de l’endartériectomie dans l’évolution dela maladie.
Références
1. Fishman AP. Clinical classification of pulmonary hyperten-sion. Clin Chest Med 2001;22:385–391.
2. Rubin LJ. Primary pulmonary hypertension. N Engl J Med1997;336:111–117.
3. Humbert M, Nunes H, Sitbon O, Parent F, Hervé P, Simon-neau G. Risk factors for pulmonary arterial hypertension.Clin Chest Med 2001;22:459–475.
4. Humbert M, Sitbon O, Simonneau G. Les hypertensionspulmonaires. In: Cohen A, Belmatoug N, editors. Cœur etmédecine interne. Paris: ESTEM; 2002. p. 1275–1309.
5. Rubin LJ. Primary pulmonary hypertension. Chest 1993;104:236–250.
6. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH,Detre M, et al. Primary pulmonary hypertension: a nationalprospective study. Ann Intern Med 1987;107:216–223.
7. Loyd JE, Butler MG, Foround TM, Conneally PM, Philips JA,Newman JH. Genetic anticipation and abnormal genderratio at birth in familial primary pulmonary hypertension.Am J Respir Crit Care Med 1995;152:93–97.
8. Humbert M, Trembath RC. Genetics of pulmonaryhypertension: from bench to bedside. Eur Respir J 2002;20:741–749.
9. Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH,Arnold ND, et al. Localization of the gene for familialprimary pulmonary hypertension to chromosome 2q31-32.Nat Genet 1997;15:277–280.
10. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phil-lips 3rd JA, Loyd JE, et al. Heterozygous germline muta-tions in a TGF--beta receptor, BMPR2, are the cause offamilial primary pulmonary hypertension. Nat Genet 2000;26:81–84.
11. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Vene-tos G, et al. Familial primary pulmonary hypertension(gene PPH1) is caused by mutations in the bone morphoge-netic protein receptor-II gene. Am J Hum Genet 2000;67:737–744.
12. Machado RD, Pauciulo MW, Thomson JR, Lane KB, Mor-gan NV, Wheeler L, et al. BMPR2 haploinsufficiency as theinherited molecular mechanism for primary pulmonaryhypertension. Am J Hum Genet 2001;68:92–102.
13. Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Hum-bert M, Elliott GC, et al. Sporadic primary pulmonaryhypertension is associated with germline mutations of thegene encoding BMPR-II, a receptor member of the TGF-beta-family. J Med Genet 2000;37:741–745.
64 D. Montani et al.

14. Massague J, Chen YG. Controlling TGF-beta signaling.Genes Dev 2000;14:627–644.
15. Humbert M, Soubrier F, Simonneau G. Un gène pourl’hypertension artérielle pulmonaire primitive. Rev MalRespir 2001;18:473–478.
16. Guttmacher AE, Marchuk DA, White Jr RI. Hereditary hem-orrhagic telangiectasia. N Engl J Med 1995;333:918–924.
17. Shovlin CL, Letarte M. Hereditary hemorrhagic telangi-ectasia and pulmonary arteriovenous malformations:issues in clinical management and review of pathogenicmechanisms. Thorax 1999;54:714–729.
18. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR,Trembath RC, et al. Primary pulmonary hypertension isassociated with reduced pulmonary vascular expression oftype II bone morphogenetic protein receptor. Circulation2002;105:1672–1678.
19. Rudarakanchana N, Flanagan JA, Chen H, Upton PD,Machado R, Patel D, et al. Functional analysis of bonemorphogenetic protein type II receptor mutations underly-ing primary pulmonary hypertension. HumMol Genet 2002;11:1517–1525.
20. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N,Sheares KK, et al. Altered growth responses of pulmonaryartery smooth muscle cells from patients with primarypulmonary hypertension to transforming growth factor-beta (1) and bone morphogenetic proteins. Circulation2001;104:790–795.
21. Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J,Kurz X, et al. Appetite-suppressant drugs and the risk ofprimary pulmonary hypertension. N Engl J Med 1996;335:609–616.
22. Humbert M, Deng Z, Simonneau G, Barst R, Sitbon O,Wolf M, et al. Bone morphogenetic protein receptor2 mutations in fenfluramine-induced pulmonary arterialhypertension. Eur Respir J 2002;20:518–523.
23. Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M,Capron F, et al. Serotonin transporter overexpression isresponsible for pulmonary artery smooth muscle hyperpla-sia in primary pulmonary hypertension. J Clin Invest 2001;108:1141–1150.
24. Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM.Microsattelite instability of endothelial cell growth andapoptosis genes within plexiform lesions in primary pulmo-nary hypertension. Circ Res 2001;88:e2–e11.
25. Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF,Tuder RM. Monoclonal endothelial cell proliferation ispresent in primary but not secondary pulmonary hyperten-sion. J Clin Invest 1998;101:927–934.
26. Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ,Jain D, Byrd B, et al. Liver disease in patients with heredi-tary hemorrhagic telangiectasia. N Engl J Med 2000;343:931–936.
27. Sapru RP, Hutchison DC, Hall JI. Pulmonary hypertension inpatients with pulmonary arteriovenous fistulae. Br Heart J1969;31:559–569.
28. Trell E, Johansson BW, Linell F, Ripa J. Familial pulmonaryhypertension and multiple abnormalities of large systemicarteries in Osler’s disease. Am J Med 1972;53:50–63.
29. Trembath RC, Thomson J, Machado RD, Morgan NV, Atkin-son C, Winship I, et al. Clinical and molecular geneticfeatures of pulmonary hypertension in hereditary hemor-rhagic telangiectasia. N Engl J Med 2001;345:325–334.
30. Grüning E, Jansen B, Mereles D, Barth U, Borst MM,Vogt IR, et al. Abnormal pulmonary artery pressureresponse in asymptomatic carriers of primary pulmonaryhypertension gene. Circulation 2000;102:1145–1150.
31. Sitbon O, Humbert M, Ioos V, Jaïs X, Parent F, Gar-cia G, et al. Who benefits from long-term calcium-channelblocker therapy in primary pulmonary hypertension?[abstract]. Am J Respir Crit Care Med 2003;167:A440.
32. Rich S, Kaufmann E, Levy PS. The effect of high doses ofcalcium-channel blockers on survival in primary pulmonaryhypertension. N Engl J Med 1992;327:76–81.
33. Higenbottam T, Wheeldon D, Wells F, Wallwork J. Long-term treatment of primary pulmonary hypertension withcontinuous intravenous epoprostenol (prostacyclin). Lan-cet 1984;1:1046–1047.
34. McLaughlin VV, Shillington A, Rich S. Survival in primarypulmonary hypertension: the impact of epoprostenoltherapy. Circulation 2002;106:1477–1482.
35. Shapiro SM, Oudiz RJ, Cao T, Romano MA, Beckmann XJ,Georgiou D, et al. Primary pulmonary hypertension:improved long-term effects and survival with continuousintravenous epoprostenol infusion. J Am Coll Cardiol 1997;30:343–349.
36. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G,Herve P, et al. Long-term intravenous epoprostenol infu-sion in primary pulmonary hypertension: prognostic factorsand survival. J Am Coll Cardiol 2002;40:780–788.
37. Galiè N, Manes A, Branzi A. The new clinical trials onpharmacological treatment in pulmonary arterial hyper-tension. Eur Respir J 2002;20:1037–1049.
38. Simonneau G, Barst RJ, Galiè N, Naeije R, Rich S,Bourge RC, et al. Continuous subcutaneous infusion oftreprostinil, a prostacyclin analogue, in patients with pul-monary arterial hypertension: a double-blind randomizedcontrolled trial. Am J Respir Crit Care Med 2002;165:800–804.
39. Vachiéry JL, Hill N, Zwicke D, Barst R, Blackburn S,Naeije R. Transitioning from IV epoprostenol to subcutane-aous treprostinil in pulmonary arterial hypertension. Chest2002;121:1561–1565.
40. Okano Y, Yoshioka T, Shimouchi A, Satoh T, Kunieda T.Orally active prostacyclin analogue in primary pulmonaryhyertension. Lancet 1997;349:1365.
41. Galiè N, Humbert M, Vachiéry JL, Vizza CD, Kneuss M,Manes A, et al. Effects of beraprost sodium, an oral pros-tacyclin analogue, in patients with pulmonary arterialhypertension: a randomized, double-blind placebo-controlled trial. J Am Coll Cardiol 2002;39:1496–1502.
42. Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S,Rubin L, et al. Beraprost therapy for pulmonary arterialhypertension. J Am Coll Cardiol 2003;41:2119–2125.
43. Hoeper MM, Schwarze M, Ehlerding S, Adler-Schuermeyer A, Spickerkoetter E, et al. Long-term treat-ment of primary pulmonary hypertension with aerosolizediloprost, a prostacyclin analogue. N Engl J Med 2000;342:1866–1870.
44. Gessler T, Schmehl T, Hoeper MM, Rose F, Ghofrani HA,Olschewski H, et al. Ultrasonic versus jet nebulization ofiloprost in severe pulmonary hypertension. Eur Respir J2001;17:14–19.
45. Olschewski H, Simonneau G, Galiè N, Higenbottam T,Naeije R, Rubin LJ, et al. Inhaled iloprost in severe pulmo-nary hypertension. N Engl J Med 2002;347:322–327.
46. Yanagisagawa M, Kurihara H, Kimura S, Tomobe Y, Koba-yashi M, Mitsui Y, et al. A novel potent vasoconstrictorpeptide produced by vascular endothelial cells. Nature1988;332:411–415.
47. Hirata Y, Katagi Y, Fukuda Y, Marumo F. Endothelin is apotent mitogen for rat vascular smooth muscle cells. Athe-rosclerosis 1989;78:225–228.
65Hypertension artérielle pulmonaire

48. DiCarlo VS, Chen SJ, Meng QC, Durand J, Yano M,Chen YF, et al. ETA-receptor antagonist prevents andreverses chronic hypoxia-induced pulmonary hypertensionin rat. Am J Physiol 1995;269:L690–697.
49. Channick RN, Simonneau G, Sitbon O, Robbins JM, Frost A,Tapson VF, et al. Effects of the dual endothelin-receptorantagonist bosentan in patients with pulmonaryhypertension: a randomised placebo-controlled study.Lancet 2001;358:1119–1123.
50. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM,Keogh A, et al. Bosentan therapy for pulmonary arterialhypertension. N Engl J Med 2002;346:896–903.
51. Galiè N, Hinderliter AL, Torbicki A, Fourme T, Simon-neau G, Pulido T, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic anddoppler measures in patients with pulmonary arterialhypertension. J Am Coll Cardiol 2003;41:1380–1386.
52. Barst RJ, Rich S A, Horn EM, McLaughlin VV, McFarlin J.Clinical efficacy of sitaxsentan, an endothelin-A receptorantagonist, in patients with pulmonary arterial hyperten-sion. Chest 2002;121:1860–1868.
53. Ghofrani HA, Wiedemann R, Rose F, Schermuly RT,Olschewski H, Weissmann N, et al. Sildenafil for teatmentof lung fibrosis and pulmonary hypertension: a randomisedcontrolled trial. Lancet 2002;360:895–900.
54. Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primarypulmonary hypertension. N Engl J Med 2000;343:1342.
55. Christman BW, McPherson CD, Newman JH, King GA, Ber-nard GR, Groves BM, et al. An imbalance between theexcretion of thromboxane and prostacyclin metabolites inpulmonary hypertension. N Engl J Med 1992;327:70–75.
56. Langleben D, Christman BW, Barst RJ, Dias VC, Galiè N,Higenbottam TW, et al. Effects of the thromboxane syn-thetase inhibitor and receptor antagonist terbogrel inpatients with primary pulmonary hypertension. Am Heart J2002;143:E4.
57. Nagaya N, Uematusu M, Oya H, Sato N, Sakamaki F, Kyot-ani S, et al. Short-term oral administration of L-arginineimproves hemodynamics and exercise capacity in patientswith precapillary pulmonary hypertension. Am J RespirCrit Care Med 2001;163:887–891.
58. Kim KK, Factor SM. Membranoproliferative glomerulone-phritis and plexogenic pulmonary arteriopathy in a homo-sexual man with acquired immunodeficiency syndrome.Hum Pathol 1987;18:1293–1296.
59. Palella Jr FJ, Delaney KM, Moorman AC, Loveless MO,Fuhrer J, Satten GA, et al., the HIV outpatient studyinvestigators. Declining morbidity and mortality amongpatients with advanced human immunodeficiency virusinfection. N Engl J Med 1998;338:853–860.
60. Mocroft A, Vella S, Benfield TL, Chiesi A, Miller V, Garga-lianos P, et al., for the EuroSida study group. Changingpatterns of mortality across Europe in patients infectedwith HIV-1. Lancet 1998;352:1725–1730.
61. Opravil M, Pechere M, Speich R, Joller-Jemelka HI, Jenni R,Russi EW, et al., the Swiss HIV cohort study. HIV-associatedprimary pulmonary hypertension. A case control study. AmJ Respir Crit Care Med 1997;155:990–995.
62. Speich R, Jenni R, Opravil M, Pfab M, Russi EW. Primarypulmonary hypertension in HIV infection. Chest 1991;100:1268–1271.
63. Mesa RA, Edell ES, Dunn WF, Edwards WD. Human immu-nodeficiency virus infection and pulmonary hypertension:two new cases and a review of 86 reported cases. MayoClin Proc 1998;73:37–45.
64. Mehta NJ, Khan IA, Metha RN, Sepkowitz DA. HIV-relatedpulmonary hypertension. Analytic review of 131 cases.Chest 2000;118:1133–1141.
65. Pellicelli AM, Barbaro G, Palmieri F, Girardi E,D’Ambrosio C, Rianda A, et al. Primary pulmonary hyper-tension in HIV patients: a systematic review. Angiology2001;52:31–41.
66. Seoane L, Shellito J, Welsh D, de Boisblanc BP. Pulmonaryhypertension associated with HIV infection. South Med J2001;94:635–639.
67. Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z,Knowles JA, et al. Prognostic factors for survival in humanimmunodeficiency virus-associated pulmonary arterialhypertension. Am J Respir Crit Care Med 2003;167:1433–1439.
68. Petitpretz P, Brenot F, Azarian R, Parent F, Rain B,Hervé P, et al. Pulmonary hypertension in patients withhuman immunodeficiency virus infection. Comparison withprimary pulmonary hypertension. Circulation 1994;89:2722–2727.
69. Rouveix E, Job C, Delorme G, Jouin H, Franc B, Saveuse H.Lethal pulmonary arterial hypertension in a heroin andamphetamine addict. Ann Med Interne 1989;140:153.
70. Pétureau F, Escamilla R, Hermant C, Mourlanette P, Ber-jaud J, Krempf M. Pulmonary artery hypertension in HIVseropositive drug addicts. Rev Mal Respir 1998;15:97–102.
71. Speich R, Jenni R, Opravil M, Jaccard R. Regression ofHIV-associated pulmonary arterial hypertension and long-term survival during antiretroviral therapy. Swiss MedWkly 2001;131:663–665.
72. Magnan A, Ottomani A, Garbe L, Arnaud A, Manelli JC.Respiratory failure in a HIV seropositive heroin addictfemale. Ann Fr Anesth Reanim 1991;10:74–76.
73. Saadjian A, Gueunoun M, Philip-Joet F. Pulmonary hyper-tension secondary to talc microemboli in a HIV seropositiveheroin-addict woman. Arch Mal Cœur 1991;84:1369–1373.
74. Ferrari E, Drai E, Taillan B, Talbodec A, Baudouy M,Morand P. Pulmonary hypertension caused by talcoma in acouple of drug addicts. Ann Cardiol Angeiol 1995;44:14–15.
75. Maliakkal R, Friedman SA, Sridhar S. Progressive pulmonarythromboembolism in association with HIV disease. N YState J Med 1992;92:403–404.
76. Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C,Thomas A, et al. Kaposi’s sarcoma-associated herpesvirusinfects endothelial and spindle cells. Nat Med 1995;1:1274–1278.
77. Cool CD, Rai PR, Yeager ME, Hernandez-Saavedra D,Serls AE, Bull TM, et al. Expression of human herpes virus8 in primary pulmonary hypertension. N Engl J Med 2003;349:1113–1122.
78. Chalifoux LV, Simon MA, Pauley DR, Mackey JJ, Wyand MS,Ringler DJ. Arteriopathy in macaques infected with simianimmunodeficiency virus. Lab Invest 1992;67:338–349.
79. Barbaro G, Di Lorenzo G, Grisorio B, Barbarini G. Incidenceof cardiomyopathy and detection of HIV in myocardial cellsof HIV positive patients. N Engl J Med 1998;339:1093–1099.
80. Mette SA, Palevsky HI, Pietra GG, Williams TM, Bruder E,Prestipino AJ, et al. Primary pulmonary hypertension inassociation with pulmonary hypertension infection. A pos-sible viral etiology for some forms of hypertensive pulmo-nary arteriopathy. Am Rev Respir Dis 1992;145:1196–1200.
81. Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F,Rain B, et al. Platelet-derived growth factor expression inprimary pulmonary hypertension: comparison of HIV serop-ositive and HIV seronegative patients. Eur Respir J 1998;11:554–559.
82. Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesisand evolution plexiform lesions in pulmonary hypertensionassociated with scleroderma and human immunodeficiencyvirus infection. Hum Pathol 1997;28:434–442.
66 D. Montani et al.

83. Ascherl G, Hohenadl C, Schatz O, Shumay E, Bogner J,Eckhart L, et al. Infection with human immunodeficiencyvirus-1 increases expression of vascular endothelial cellgrowth factor in T cells: implications for acquired immu-nodeficiency syndrome-associated vasculopathy. Blood1999;93:4232–4241.
84. Rosemberg ZF, Fauci AS. Immunopathogenic mechanismsof HIV infection: cytokine induction of HIV expression.Immunol Today 1990;11:176–180.
85. Ehrenreich H, Rieckmann P, Sinowatz F, Weih KA,Arthur LO, Goebel FD, et al. Potent stimulation of mono-cytic endothelin-1 production by HIV-1 glycoprotein 120. JImmunol 1993;150:4601–4609.
86. Ensoli B, Barillari G, Salahuddin SZ, Gallo RC, Wong-Staal F. Tat protein of HIV-1 stimulates growth of cellsderived from Kaposi’s sarcoma lesions of AIDS patients.Nature 1990;345:84–86.
87. Hofman FM, Wright AD, Dohadwala MM, Wong-Staal F,Walker SM. Exogenous tat protein activates human endot-hélial cells. Blood 1993;82:2774–2780.
88. Morse JH, Barst RJ, Itescu S, Flaster ER, Sinha G,Zhang Y, et al. Primary pulmonary hypertension in HIVinfection. An outcome determined by particular HLA classII alleles. Am J Respir Crit Care Med 1996;153:1299–1301.
89. Heron E, Laaban JP, Capron F, Quieffin J, Brechot JM,Rochemaure J. Thrombotic primary pulmonary hyperten-sion in an HIV+ patient. Eur Heart J 1994;15:394–396.
90. Ruchelli ED, Nojadera G, Rutstein RM, Rudy B. Pulmonaryveno-occlusive disease. Another vascular disorder associ-ated with human immunodeficiency virus infection? ArchPathol Lab Med 1994;118:664–666.
91. Escamilla R, Hermant C, Berjaud J, Mazerolles C, Daussy X.Pulmonary veno-occlusive disease in a HIV-infected intra-venous drug abuser. Eur Respir J 1995;8:1982–1984.
92. Nunes H, Humbert M, Jagot JL, Sitbon O, Hervé P, Simon-neau G. Hypertension artérielle primitive et infection parle VIH. STV 1998;10:555–562.
93. Aguilar RV, Farber HW. Epoprostenol (prostacyclin)therapy in HIV-associated pulmonary hypertension. Am JRespir Crit Care Med 2000;162:1846–1850.
94. Hervé P, Lebrec D, Brenot F, Simonneau G, Humbert M,Sitbon O, et al. Pulmonary vascular disorders in portalhypertension. Eur Respir J 1998;11:1153–1166.
95. Lebrec D, Capron JP, Dhumeaux D, Benhamou JP. Pulmo-nary hypertension complicating portal hypertension. AmRev Respir Dis 1979;120:849–856.
96. Rüttner JR, Bärtschi JP, Niedermann R, Schneider J. Plexo-genic pulmonary arteriopathy and liver cirrhosis. Thorax1980;35:133–136.
97. McDonnel PJ, Toye PA, Hutchins GM. Primary pulmonaryhypertension and cirrhosis: are they related? Am RevRespir Dis 1983;127:437–441.
98. Castro M, Krowka MJ, Schroeder DR, Beck KC, Plevack DJ,Rettke SR, et al. Frequency and clinical implications ofincreased pulmonary artery pressures in liver transplanta-tion. Mayo Clin Proc 1996;71:543–551.
99. Mandell SM, Groves BM. Pulmonary hypertension in liverdisease. Clin Chest Med 1996;17:17–33.
100. Hadengue A, Benhayoun M, Lebrec D, Benhamou JP. Pul-monary hypertension complicating portal hypertension:prevalence and relation to splanchnic hemodynamics. Gas-troenterology 1991;100:520–528.
101. Sallam M, Watson WC. Pulmonary hypertension due tomicro-thromboembolism from splenic and portal veinsafter portacaval anastomosis. Br Heart J 1970;32:269–271.
102. Botney MD. Role of hemodynamics in pulmonary vascularremodeling. Implications for primary pulmonary hyperten-sion. Am J Respir Crit Care Med 1999;159:361–364.
103. Rassiat E, Barriere E, Durand F, Bernuau J, Belghiti J,Valla D, et al. Pulmonary hemodynamics and gas exchangeafter liver transplantation in patients with cirrhosis. DigDis Sci 2002;47:746–749.
104. Luca A, Garcia-Pagan JC, de Lacy AM, Escorsell A, Feu F,Visa J, et al. Effects of end-to-side portacaval shunt anddistal splenorenal shunt on systemic and pulmonary hae-modynamics in patients with cirrhosis. J GastroenterolHepatol 1999;14:1112–1118.
105. Panos RJ, Backer SK. Mediators, cytokines, and growthfactors in liver-lung interactions. Clin Chest Med 1996;17:151–169.
106. Herve P, Launay JM, Scrobohaci ML, Brenot F, Simon-neau G, Petitpretz P, Hervé P, et al. Increased plasmaserotonin in patients with primary pulmonary hyperten-sion. Am J Med 1995;99:249–254.
107. Beaudry P, Hadengue A, Callebert J, Gaudin C, Soliman H,Moreau R, et al. Blood and plasma 5-hydroxytryptaminelevels in patients with cirrhosis. Hepatology 1994;20(4Pt1):800–1203.
108. Giaid A, Saleh D. Reduced expression of endothelial nitricoxide synthase in the lungs of patients with pulmonaryhypertension. N Engl J Med 1995;333:214–221.
109. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R,Shennib H, et al. Expression of endothelin-1 in the lungs ofpatients with pulmonary hypertension. N Engl J Med 1993;328:1732–1739.
110. Colle IO, Moreau R, Godinho E, Belghiti J, Ettori F, Cohen-Solal A, et al. Diagnosis of portopulmonary hypertension incandidates for liver transplantation: a prospective study.Hepatology 2003;37:401–409.
111. Castelain V, Chemla D, Humbert M, Sitbon O, Simonneau G,Lecarpentier Y, et al. Pulmonary artery pressure-flow rela-tions after prostacyclin in primary pulmonary hyperten-sion. Am J Respir Crit Care Med 2002;165:334–440.
112. Krowka MJ, Frantz RP, McGoon MD, Severson C, Plevak DJ,Wiesner RH. Improvement in pulmonary hemodynamicsduring intravenous epoprostenol (prostacyclin): a study of15 patients with moderate to severe portopulmonaryhypertension. Hepatology 1999;30:641–648.
113. Kuo PC, Plotkin JS, Gaine S, Schroeder RA, Rustgi VK,Rubin LJ, et al. Portopulmonary hypertension and the livertransplant candidate. Transplantation 1999;67:1087–1093.
114. Krowka MJ, Plevak DJ, Findlay JY, Rosen CB, Wiesner RH,Krom RA. Pulmonary hemodynamics and perioperativecardiopulmonary-related mortality in patients with por-topulmonary hypertension undergoing liver transplanta-tion. Liver Transpl 2000;6:443–450.
115. Moser KM, Auger WR, Fedullo PF, Jamieron SW. Chronicthromboembolic pulmonary hypertension: clinical pictureand surgical treatment. Eur Respir J 1992;5:334–342.
116. DeSoyza ND, Murphy ML. Persistent post-embolic pulmo-nary hypertension. Chest 1972;62:665–668.
117. Fedullo F, Auger WR, Channick RN, Moser KM, Jamie-son SW. Chonic thromboembolic pulmonary hypertension.Clin Chest Med 1995;16:353–374.
118. Moser KM, Auger WR, Fedullo PF. Chronic major-vesselthromboembolic pulmonary hypertension. Circulation1990;81:1735–1743.
119. Azarian R, Brenot F, Sitbon O, Musset D, Grimon G, Boyer-Neumann C, et al. Pulmonary arterial hypertension ofchronic thrombo-embolic origin. Presse Med 1994;23:1017–1022.
120. Moser KM, Bloor CM. Pulmonary vascular lesions occurringin patients with chronic major vessel thromboembolic pul-monary hypertension. Chest 1993;103:685–692.
67Hypertension artérielle pulmonaire

121. Cabrol C, Cabrol A, Acar J, Gandibackhch I, Guiraudon G,Laughlin L, et al. Surgical correction of chronic post-embolic obstructions of the pulmonary arteries. J ThoracCardiovasc Surg 1978;76:620–628.
122. Dor V, Jourdan J, Schmitt R, Sabatier M, Arnulf JJ, Kreit-mann P, et al. Delayed pulmonary thrombectomy via aperipheral approach in the treatment of pulmonary embo-lism and sequelae. Thorac Cardiovasc Surg 1981;29:227.
123. Bergin CJ, Sirlin CB, Hauschildt JP, Huynh TV, Auger WR,Fedullo PF, et al. Chronic thromboembolism: diagnosiswith helical CT and MR imaging with angiographic andsurgical correlation. Radiology 1997;204:695–702.
124. Nicod P, Peterson KM, Moser KM, Levine MS, Dittrich H,Buchbinder M, et al. Pulmonary angiography in severechronic pulmonary hypertension. Ann Intern Med 1987;107:565–568.
125. Palevsky HI. Therapeutic options for severe pulmonaryhypertension. Clin Chest Med 1997;18:595–609.
126. Azarian R, Parent F, Musset D, Dartevelle P, Duroux P,Simonneau G. Chronic pulmonary heart disease: therapeu-tic indications. Arch Mal Cœur Vaiss 1995;88(suppl2):1799–1805.
127. Chapelier A, Vouhé P, Macchiarini P, Lenot B, Cerrina J,Leroy-Ladurie F, et al. Comparative outcome of heart-lungand lung transplantation for pulmonary hypertension. JThorac Cardiovasc Surg 1993;106:399–407.
128. Chitwood WR, Sabiston DC, Wechsler AS. Surgical treat-ment of chronic unresolved pulmonary embolism. ClinChest Med 1984;5:507–536.
129. Hartz RS, Byrne JG, Levitsky S, Park J, Rich S. Predictors ofmortality in pulmonary thromboendarterectomy. Ann Tho-rac Surg 1996;62:1255–1259.
130. Jamieson SW, Auger WR, Fedallo PF, Channick RN, Kri-ett JM, Tazari RY, et al. Experience and results with150 pulmonary thromboendarteriectomy operations over a29-month period. J Thorac Cardiovasc Surg 1993;106:116–127.
131. Mayer E, Dahm M, Hake U, Schmid FX, Pitton M, Kupfer-wasser I, et al. Mid-term results of pulmonary thromboen-darterectomy for chronic thromboembolic pulmonaryhypertension. Ann Thorac Surg 1996;61:1788–1792.
132. Moser KM, Daily PO, Peterson K, Dembitsky W, Vapnek JM,Shure D, et al. Thromboendarteriectomy for chronic,major-vessel thromboembolic pulmonary hypertension.Ann Intern Med 1987;107:560–565.
68 D. Montani et al.





























