Embed Size (px)
Citation preview

In Vitro Model Assemblies To Study the Impact ofLignin-Carbohydrate Interactions on the Enzymatic Conversion
of Xylan
Imen Boukari,†,‡ Jean-Luc Putaux,§ Bernard Cathala,| Abdellatif Barakat,†,‡ Bodo Saake,⊥
Caroline Remond,†,‡ Michael O’Donohue,#,∇,O and Brigitte Chabbert*,†,‡
INRA, UMR 614, Fractionnement des AgroRessources et Environnement, F-51686 Reims, France,University of Reims Champagne Ardenne, UMR 614, Fractionnement des AgroRessources et
Environnement, F-51686 Reims, France, Centre de Recherches sur les Macromolecules Vegetales(CERMAV-CNRS), affiliated with Universite Joseph Fourier and member of the Institut de Chimie
Moleculaire de Grenoble, BP 53, F-38041, Grenoble Cedex 9, France, INRA, UR1268 Biopolymeres,Interactions et Assemblages, F-44300, Nantes, France, VTI, Institute for Wood Technology and Wood
Biology, 21031 Hamburg, Germany, Universite de Toulouse, INSA, UPS, INP, LISBP, F-31077 Toulouse,France, INRA, UMR792 Ingenierie des Systemes Biologiques et des Procedes, F-31400 Toulouse, France,
and CNRS, UMR5504, F-31400 Toulouse, France
Received April 21, 2009; Revised Manuscript Received June 30, 2009
Endo-�-1,4-xylanases (EC 3.2.1.8) are the main enzymes involved in the hydrolysis of xylans, the most abundanthemicelluloses in plant biomass. However, the development of efficient endoxylanases for use in biorefineryprocesses is currently hampered by insufficient knowledge regarding the impact of the cell wall network organizationon the action of the enzyme at the supramolecular level. The action pattern of a GH11 endoxylanase fromThermobacillus xylanilyticus (Tx-xyl) was investigated by means of in vitro reconstituted model systems whichcan mimic certain cell wall structures. The action of Tx-xyl was evaluated on polymer assemblies displayingincreasing complexity using delignified glucuronoarabinoxylan (GAX), then GAX-DHP model complexes obtainedby oxidative polymerization of coniferyl alcohol into dehydrogenation polymers (DHP: lignin model compounds)in the presence of GAX. At a high concentration of GAX, interchain associations are formed leading to highmolecular weight aggregates. These structures did not appear to affect the action of endoxylanase, which inducesdisaggregation of the self-aggregates along with polymer depolymerization. To mimic lignin-carbohydrateinteractions, two different GAX-DHP nanocomposites were prepared and incubated with endoxylanase. In bothcases, free GAX was hydrolyzed, while the GAX-DHP complexes appeared to be resistant. In the case of thenoncovalently linked GAX-DHPZL complexes, enzyme action favored a decrease in particle size, owing to theremoval of their relatively exposed carbohydrate chains, whereas the complex supramolecular organization ofthe covalently linked GAX-DHPZT complexes severely hampers the enzyme’s access to carbohydrate. Overall,these results establish the negative impact of DHP on the endoxylanase action and provide new knowledge regardingthe limitations of the enzyme action in the lignocellulose bioconversion processes.
Introduction
Lignocellulosic biomass is an abundant, renewable resourcethat can be used for the production of fuel ethanol andindustrially relevant chemicals. However, its use as a rawmaterial for biorefining constitutes a considerable technologicalchallenge, particularly because of its chemical complexity andits recalcitrance. Thermochemical technologies have beendeveloped for the fractionation and upgrading of lignocellulosicbiomass, but enzymatic bioconversion offers an alternative,environmentally friendly strategy. However, the presence oflignin and the cell wall network provide limitations to efficientbioconversion of lignocelluloses.
The lignified plant cell walls are indeed composite materialsresulting from the assembly of different biopolymers (cellulose,hemicelluloses, lignin, etc.). These components are intercon-nected through a variety of covalent and noncovalent interac-tions, giving rise to a highly organized network. Lignin is acomplex aromatic polymer composed of phenylpropane units1
that impregnate the preexisting hemicellulose-cellulose net-work,2 thereby imparting both rigidity and biological resistanceto the lignocellulosic structure. Over the last few decades, theassociations between lignin and hemicelluloses have beenextensively studied to improve biomass delignification, a keytechnology for the pulp and paper industry. Lignin associateswith hemicelluloses through noncovalent and covalent linkagesto form a dense and highly organized network. Covalently linkedstructures form the so-called lignin-carbohydrate complexes(LCCs).3 Previously, it has been shown that these LCC structuresare particularly problematic for lignocellulose bioconversionprocesses because lignin impedes enzyme-mediated hydrolysisof carbohydrates. Evidence in the literature suggests that ligninacts as a physical barrier, restricting enzyme access to carbo-hydrates,4 and may also interact with enzymes through possibly
* To whom correspondence should be addressed. E-mail: [email protected].
† INRA, UMR 614.‡ University of Reims Champagne Ardenne.§ CERMAV-CNRS.| INRA, UR1268.⊥ Institute for Wood Technology and Wood Biology.# Universite de Toulouse.∇ INRA, UMR792 Ingenierie des Systemes Biologiques et des Procedes.O CNRS, UMR5504.
Biomacromolecules 2009, 10, 2489–2498 2489
10.1021/bm9004518 CCC: $40.75 2009 American Chemical SocietyPublished on Web 08/05/2009

hydrophobic interactions resulting in nonproductive binding.5
Enzyme inactivation by lignin has also been suggested becausephenolic compounds can form soluble inactive enzyme-inhibitorcomplexes at very low concentrations and insoluble protein-phenolic complexes at high concentrations.6-9
All these limitations have prompted the emergence anddevelopment of a variety of chemical and physical pretreatmentsaimed at reducing the lignin inhibitory effects, as recentlyreviewed by Chandra et al.10 However, development of sustain-able biorefinery technologies requires reduction in chemicals,toxic effluents, and energy-costly processes and would takeadvantage of the use of highly efficient glycoside hydrolases.In this context, a more comprehensive view of the mechanismsthat hamper enzymatic conversion would be required to designnew efficient enzymes. Although the lignin content and thepresence of phenolic cross-linkages have often been negativelycorrelated with the susceptibility of lignocellulosic biomass tobiodegradation,11-13 only a few studies have addressed the wayin which lignin interactions with polysaccharides and thesupramolecular organization of the cell wall network may impactenzyme action.14-17 This is because the high complexity andvariability of lignified cell walls is an obstacle for the clearinterpretation of data. In this respect, the in vitro design of modelsystems constitutes an attractive experimental tool that may beuseful to understand the effect of the cell wall polymersassembly on the enzymatic breakdown of lignocelluloses. Thesemimetic assemblies have proven to be useful tools to investigatethe effect of pre-existing polysaccharides on lignification, givingfurther understanding on the cell wall assembly and biosynthesis.One of the simplest approaches involves in vitro oxidativepolymerization of lignin precursors (monolignols) into “lignin-like polymers” or DHPs (dehydrogenation polymers) in thepresence of natural or artificially synthesized cell wallcomponents.18-24 This strategy was shown to be advantageousbecause the characteristics of the lignin or the lignin-carbohydrateinteractions can be controlled through the use of differentmonolignols, carbohydrates, and polymerization conditions.Notably, Barakat et al.25 have successfully modeled lignin-heteroxylan interactions by performing in vitro oxidative poly-merization of lignin precursors in the presence of glucuronoara-binoxylan (GAX) using the well-known “bulk” (Zulaufverfahren,ZL) and “end-wise” (Zutropfverfahren, ZT) DHP polymerizationmodes. Two complex systems were obtained, GAX-DHPZL andGAX-DHPZT, which are characterized by the predominance ofnoncovalent and covalent interactions, respectively.
To study the cell-wall restrictions to hemicellulose biocon-version, we have addressed the action of a thermostable GH11endo-�-1,4-xylanase (Tx-Xyl) from Thermobacillus xylanilyti-cus.26 This enzyme, which does not possess any carbohydratebinding module,27 is quite active on both isolated polymers andcomplex natural lignocelluloses, but its efficiency does appearto be correlated with lignin content.28-31 Endoxylanases are usedin food and feed industries as well as pulp and paper technolo-gies32 and are receiving increasing interest in bioethanolproduction,33 either as added enzymes (with cellulases) or aspart of an integrated bioprocess (pentose fermentation, etc.).However, in contrast, with the abundant literature on the actionpattern of cellulases,17 fewer data are available on the organi-zational factors that attenuate endoxylanase action in planta.To acquire a more comprehensive view on these limitations,the impact of the supramolecular organization of the cell wallpolymers on Tx-Xyl was investigated. To this end, we proposea new approach that is based on the enzymatic breakdown ofin vitro reconstructed model systems. The action pattern of Tx-
Xyl was evaluated on polymer assemblies displaying increasedcomplexity using self-aggregates of glucuronoarabinoxylan(GAX) and bioinspired GAX-DHP nanocomposites. In additionto experiments conducted with high levels of enzyme, the useof a low concentration of enzyme was chosen to highlightpossible distinct restriction patterns.
Materials and Methods
Glucuronoarabinoxylan Sample (GAX). Delignified water-solubleglucuronoarabinoxylans (GAXs) were obtained from oat spelt, aspreviously described.34 Briefly, GAXs were extracted from oat speltswith 5% (w/v) NaOH at 90 °C and further purified by washing withmethanol/water (60/40, v/v), methanol, and ether. ClO2 bleaching wasthen carried out at 70 °C for 3 h; water-soluble GAXs were thenrecovered by centrifugation.
Basically, the analytical and structural characterization of the GAXsample indicated an average molecular weight of 22650 g/mol, anarabinose/xylose ratio of 0.23, and a 4-methyl glucuronic acid contentof 8.2%. The lignin content was estimated to 4.7% using the Klasonprocedure.34
Endo-�-1,4-xylanase (Tx-Xyl). The thermostable GH11 endoxy-lanase (Tx-Xyl) was produced from Thermobacillus xylanilyticus andpurified to homogeneity using a two-step chromatographic procedure(ion-exchange (Q Sepharose fast flow) followed by hydrophobicinteraction (phenyl Sepharose CL4B) chromatography) according tothe previously established protocol.26 The specific activity of the pureprotein was 2000 IU/mg protein, where one IU is defined as the amountof endoxylanase required to release 1 µmol of reducing xyloseequivalent from birchwood xylan per min at 60 °C.
Fluorescence Probe Study of GAX Aggregation. Fluorescencespectroscopy of pyrene was used to probe GAX self-aggregation. Pyreneis a hydrophobic molecule whose fluorescence properties depend onthe polarity of the environment. Following excitation at 335 nmwavelength, the emission spectra of pyrene showed vibronic peaks at372 nm (intensity I1) and 382 nm (intensity I3). The change in the I1/I3
ratio is sensitive to the hydrophobicity of pyrene’s environment (forexample, I1/I3 is equal to 0.6 in hexane, to 1.3 in methanol and to 1.7in water) and was used to examine the aggregation behavior of GAXand to estimate the critical concentration of aggregation (CAC). Forthis purpose, solutions of increased concentration of GAX (0.1-10 g/L)dissolved in water were mixed with pyrene (5 × 10-7 M finalconcentration) and absorption spectra were recorded on a Perkin-ElmerLS50B spectrofluorimeter.
Synthesis of Carbohydrate-Lignin Complexes (GAX-DHP). Co-niferyl alcohol (4-hydroxy-3-methoxy cinnamyl alcohol) used in thesynthesis of GAX-DHP complexes was prepared according to theprocedure described by Ludley and Ralph.35 Dehydrogenation polymers(DHPs, lignin model compounds) were synthesized by oxidativepolymerization of coniferyl alcohol (1 g/L) using horseradish peroxidase(Sigma) and hydrogen peroxide in the presence of glucuronoarabi-noxylan (GAX; 1 g/L), as described previously.25 Two polymerizationmodes were used according to the speed of reagent addition: bulk(Zulaufverfahren or ZL) mode consists of the simultaneous adding ofall reactants, whereas in the end-wise (Zutropfverfahren or ZT)polymerization, reactants were added gradually (dropwise). Bothpolymerization methods yielded stable colloidal suspensions of GAX-DHP complexes.
Endoxylanase Assays. Enzyme-mediated hydrolysis of free delig-nified GAX and GAX-DHP complexes was carried out at 60 °C undercontinuous stirring. To determine maximal hydrolysis rates, sampleswere incubated with a large excess of Tx-Xyl (2 IU/mg GAX), whereaskinetic studies were performed using limiting amounts of Tx-Xyl (0.05IU/mg GAX) in order to monitor the enzyme action pattern on eachsubstrate. For all tests, substrates were preincubated at 60 °C for 15min before Tx-Xyl was added. After, aliquots were removed from thereaction mixture at regular intervals, boiled for 15 min to inactivateTx-Xyl, and submitted to both HP-SEC and reducing sugar analyses.
2490 Biomacromolecules, Vol. 10, No. 9, 2009 Boukari et al.
Determination of Hydrolysis Rates. The rates of hydrolysis of GAXand GAX-DHP complexes were determined by measuring the amountof reducing sugars (xylose and xylo-oligosaccharides) released by theendoxylanase. Reducing sugars were quantified as alditol acetates usinggas-liquid chromatography according to the procedure developed byCourtin et al.36 except that samples were hydrolyzed using sulphuricacid (1 M final concentration) at 100 °C for 2 h and dichoromethanewas used to extract alditol acetates. Chromatographic analysis wasperformed using a gas chromatograph (GC Hewlett-Packard 6890A)equipped with an autosampler and a flame ionizing detector. Separationof alditol acetates was achieved at 220 °C on a Supelco Sp-2380 polarcolumn (0.25 mm × 30 m) using He (at 1 bar) as the carrier gas.Detection and injection were performed at 250 °C. Appropriate mixturesof monomeric reducing sugars were used for calibration and inositolwas included as the internal standard.
Multidetector Size Exclusion Chromatography (HP-SEC/MALLS). High performance size exclusion chromatographic (HP-SEC)system was connected online to a UV detector (Waters 2996), arefractive index detector (RI) (Waters 410) and a multiangle laser lightscattering (MALLS) detector working simultaneously at 18 angles(Dawn MALLS, 632.8 nm, Wyatt corporation).37 Chromatographicseparation of 100-200 µL injected solutions (filtered on 0.45 µm PTFEfilter) was performed with two elution systems: NaNO3 solution (50mM) containing 0.02% NaN3 and a mixture of dimethyl sulfoxide(DMSO)/water (90:10) containing 50 mM of LiBr (abbreviated asDMSO). When using the aqueous eluent, thermostatically controlled(50 °C) SHODEX OH pack (802, 803, and 805; each 4.6 × 300 mm)columns set were used (flow rate of 1 mL/min). In the DMSO eluentsystem SHODEX KD (802, 804, 806 M; each 8 × 300 mm) columnsset were used at 50 °C (flow rate of 0.5 mL/min). For the analysis ofGAX-self-aggregates, the aqueous nitrate solution was employed andsamples displayed concentrations from 0.1 to 5.0 g/L.
The software used (Astra for Windows 4.73, Wyatt technology, SantaBarbara, CA) allowed online data collection during HP-SEC runs, aswell as calculation of the molecular weights (Mw) and the root-mean-square radii of gyration (r.m.s) distributions and averages.
The sample recovery rates after HP-SEC analysis were determinedas the ratio of the eluted mass (determined according to RI signal andknown dn/dc values37) and the injected mass. Chromatographic yieldsrange about or higher than 50%.
Transmission Electron Microscopy (TEM). Droplets of samplesuspensions were deposited on glow-discharged carbon-coated grids.The liquid in excess was blotted away with filter paper and a drop of2% (w/v) uranyl acetate negative stain was added prior to drying.Samples were observed using a Philips CM200 microscope operatingat 80 kV. Images were recorded on Kodak SO163 films. The negativeswere digitized offline with a Kodak Megaplus CCD camera and theparticle diameter was measured using the ImageJ software.
Results and Discussion
Characterization of GAX Self-Aggregation. Isolated xylansare rarely perfectly “soluble” in a thermodynamic sense.38
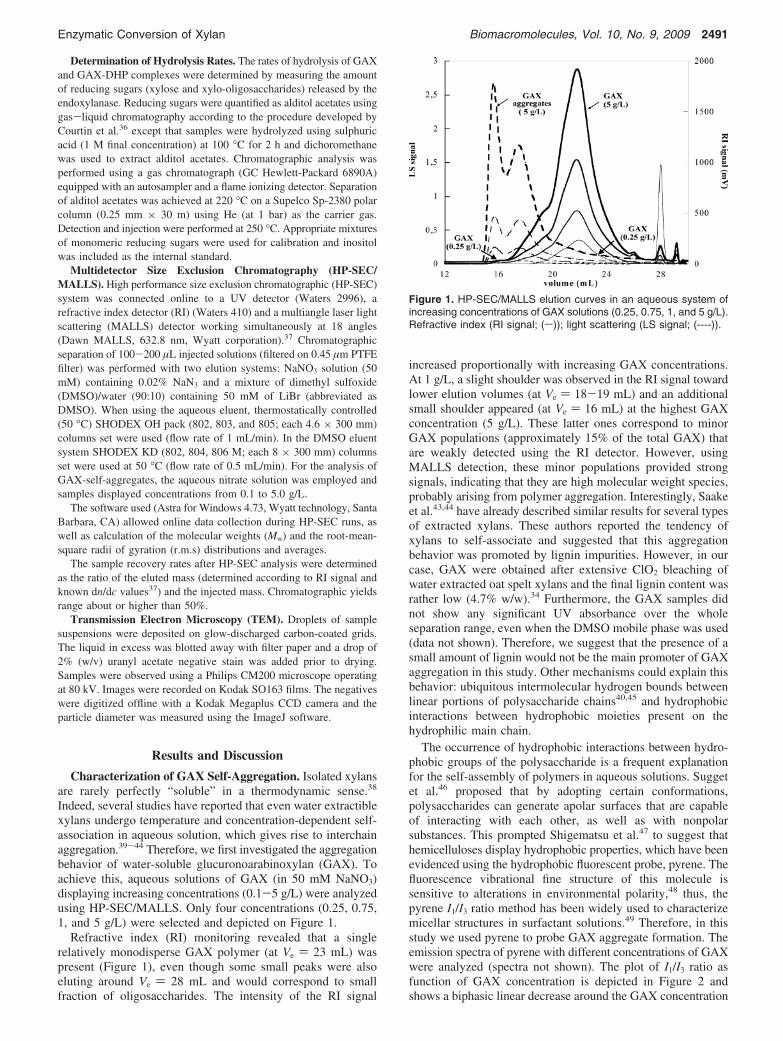
Indeed, several studies have reported that even water extractiblexylans undergo temperature and concentration-dependent self-association in aqueous solution, which gives rise to interchainaggregation.39-44 Therefore, we first investigated the aggregationbehavior of water-soluble glucuronoarabinoxylan (GAX). Toachieve this, aqueous solutions of GAX (in 50 mM NaNO3)displaying increasing concentrations (0.1-5 g/L) were analyzedusing HP-SEC/MALLS. Only four concentrations (0.25, 0.75,1, and 5 g/L) were selected and depicted on Figure 1.
Refractive index (RI) monitoring revealed that a singlerelatively monodisperse GAX polymer (at Ve ) 23 mL) waspresent (Figure 1), even though some small peaks were alsoeluting around Ve ) 28 mL and would correspond to smallfraction of oligosaccharides. The intensity of the RI signal
increased proportionally with increasing GAX concentrations.At 1 g/L, a slight shoulder was observed in the RI signal towardlower elution volumes (at Ve ) 18-19 mL) and an additionalsmall shoulder appeared (at Ve ) 16 mL) at the highest GAXconcentration (5 g/L). These latter ones correspond to minorGAX populations (approximately 15% of the total GAX) thatare weakly detected using the RI detector. However, usingMALLS detection, these minor populations provided strongsignals, indicating that they are high molecular weight species,probably arising from polymer aggregation. Interestingly, Saakeet al.43,44 have already described similar results for several typesof extracted xylans. These authors reported the tendency ofxylans to self-associate and suggested that this aggregationbehavior was promoted by lignin impurities. However, in ourcase, GAX were obtained after extensive ClO2 bleaching ofwater extracted oat spelt xylans and the final lignin content wasrather low (4.7% w/w).34 Furthermore, the GAX samples didnot show any significant UV absorbance over the wholeseparation range, even when the DMSO mobile phase was used(data not shown). Therefore, we suggest that the presence of asmall amount of lignin would not be the main promoter of GAXaggregation in this study. Other mechanisms could explain thisbehavior: ubiquitous intermolecular hydrogen bounds betweenlinear portions of polysaccharide chains40,45 and hydrophobicinteractions between hydrophobic moieties present on thehydrophilic main chain.
The occurrence of hydrophobic interactions between hydro-phobic groups of the polysaccharide is a frequent explanationfor the self-assembly of polymers in aqueous solutions. Suggetet al.46 proposed that by adopting certain conformations,polysaccharides can generate apolar surfaces that are capableof interacting with each other, as well as with nonpolarsubstances. This prompted Shigematsu et al.47 to suggest thathemicelluloses display hydrophobic properties, which have beenevidenced using the hydrophobic fluorescent probe, pyrene. Thefluorescence vibrational fine structure of this molecule issensitive to alterations in environmental polarity,48 thus, thepyrene I1/I3 ratio method has been widely used to characterizemicellar structures in surfactant solutions.49 Therefore, in thisstudy we used pyrene to probe GAX aggregate formation. Theemission spectra of pyrene with different concentrations of GAXwere analyzed (spectra not shown). The plot of I1/I3 ratio asfunction of GAX concentration is depicted in Figure 2 andshows a biphasic linear decrease around the GAX concentration
Figure 1. HP-SEC/MALLS elution curves in an aqueous system ofincreasing concentrations of GAX solutions (0.25, 0.75, 1, and 5 g/L).Refractive index (RI signal; (s)); light scattering (LS signal; (----)).
Enzymatic Conversion of Xylan Biomacromolecules, Vol. 10, No. 9, 2009 2491
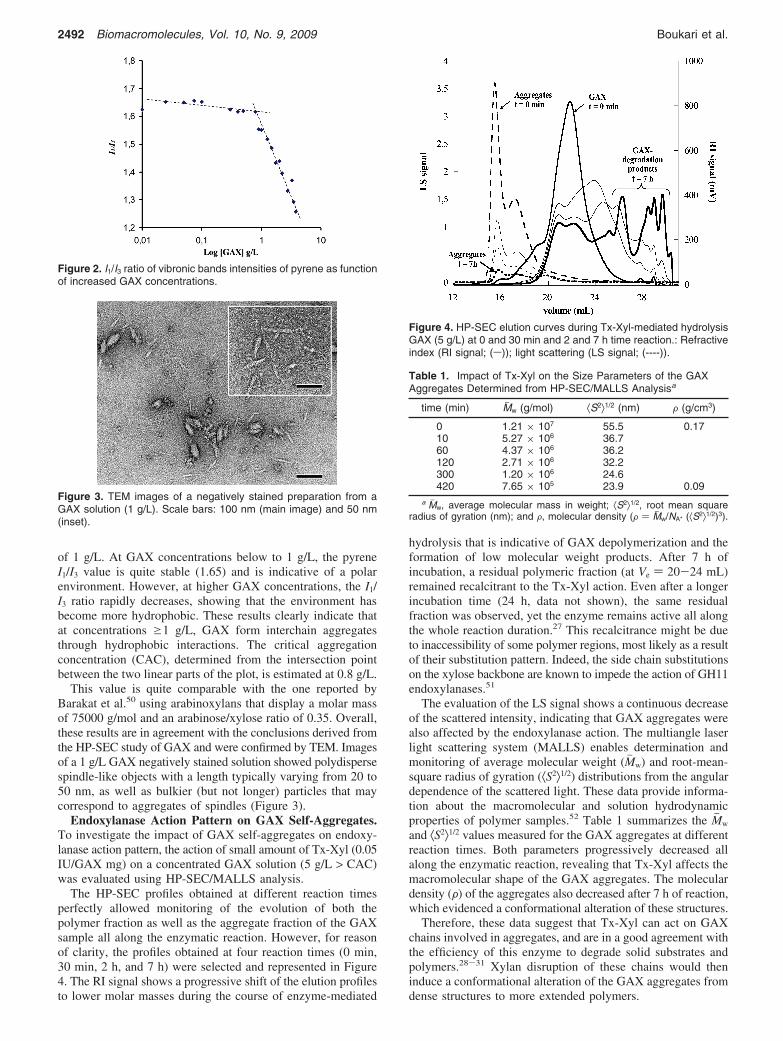
of 1 g/L. At GAX concentrations below to 1 g/L, the pyreneI1/I3 value is quite stable (1.65) and is indicative of a polarenvironment. However, at higher GAX concentrations, the I1/I3 ratio rapidly decreases, showing that the environment hasbecome more hydrophobic. These results clearly indicate thatat concentrations g1 g/L, GAX form interchain aggregatesthrough hydrophobic interactions. The critical aggregationconcentration (CAC), determined from the intersection pointbetween the two linear parts of the plot, is estimated at 0.8 g/L.
This value is quite comparable with the one reported byBarakat et al.50 using arabinoxylans that display a molar massof 75000 g/mol and an arabinose/xylose ratio of 0.35. Overall,these results are in agreement with the conclusions derived fromthe HP-SEC study of GAX and were confirmed by TEM. Imagesof a 1 g/L GAX negatively stained solution showed polydispersespindle-like objects with a length typically varying from 20 to50 nm, as well as bulkier (but not longer) particles that maycorrespond to aggregates of spindles (Figure 3).
Endoxylanase Action Pattern on GAX Self-Aggregates.To investigate the impact of GAX self-aggregates on endoxy-lanase action pattern, the action of small amount of Tx-Xyl (0.05IU/GAX mg) on a concentrated GAX solution (5 g/L > CAC)was evaluated using HP-SEC/MALLS analysis.
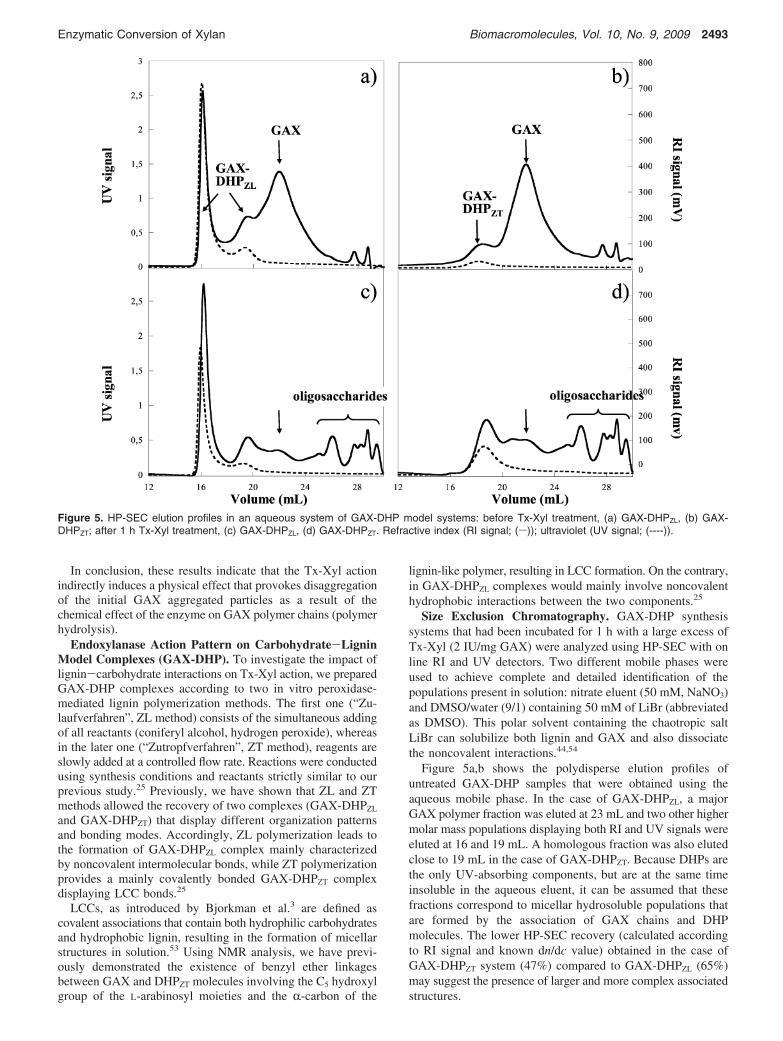
The HP-SEC profiles obtained at different reaction timesperfectly allowed monitoring of the evolution of both thepolymer fraction as well as the aggregate fraction of the GAXsample all along the enzymatic reaction. However, for reasonof clarity, the profiles obtained at four reaction times (0 min,30 min, 2 h, and 7 h) were selected and represented in Figure4. The RI signal shows a progressive shift of the elution profilesto lower molar masses during the course of enzyme-mediated
hydrolysis that is indicative of GAX depolymerization and theformation of low molecular weight products. After 7 h ofincubation, a residual polymeric fraction (at Ve ) 20-24 mL)remained recalcitrant to the Tx-Xyl action. Even after a longerincubation time (24 h, data not shown), the same residualfraction was observed, yet the enzyme remains active all alongthe whole reaction duration.27 This recalcitrance might be dueto inaccessibility of some polymer regions, most likely as a resultof their substitution pattern. Indeed, the side chain substitutionson the xylose backbone are known to impede the action of GH11endoxylanases.51
The evaluation of the LS signal shows a continuous decreaseof the scattered intensity, indicating that GAX aggregates werealso affected by the endoxylanase action. The multiangle laserlight scattering system (MALLS) enables determination andmonitoring of average molecular weight (Mj w) and root-mean-square radius of gyration (⟨S2⟩1/2) distributions from the angulardependence of the scattered light. These data provide informa-tion about the macromolecular and solution hydrodynamicproperties of polymer samples.52 Table 1 summarizes the Mj w
and ⟨S2⟩1/2 values measured for the GAX aggregates at differentreaction times. Both parameters progressively decreased allalong the enzymatic reaction, revealing that Tx-Xyl affects themacromolecular shape of the GAX aggregates. The moleculardensity (F) of the aggregates also decreased after 7 h of reaction,which evidenced a conformational alteration of these structures.
Therefore, these data suggest that Tx-Xyl can act on GAXchains involved in aggregates, and are in a good agreement withthe efficiency of this enzyme to degrade solid substrates andpolymers.28-31 Xylan disruption of these chains would theninduce a conformational alteration of the GAX aggregates fromdense structures to more extended polymers.
Figure 2. I1/I3 ratio of vibronic bands intensities of pyrene as functionof increased GAX concentrations.
Figure 3. TEM images of a negatively stained preparation from aGAX solution (1 g/L). Scale bars: 100 nm (main image) and 50 nm(inset).
Figure 4. HP-SEC elution curves during Tx-Xyl-mediated hydrolysisGAX (5 g/L) at 0 and 30 min and 2 and 7 h time reaction.: Refractiveindex (RI signal; (s)); light scattering (LS signal; (----)).
Table 1. Impact of Tx-Xyl on the Size Parameters of the GAXAggregates Determined from HP-SEC/MALLS Analysisa
time (min) Mj w (g/mol) ⟨S2⟩1/2 (nm) F (g/cm3)
0 1.21 × 107 55.5 0.1710 5.27 × 106 36.760 4.37 × 106 36.2120 2.71 × 106 32.2300 1.20 × 106 24.6420 7.65 × 105 23.9 0.09
a Mj w, average molecular mass in weight; ⟨S2⟩1/2, root mean squareradius of gyration (nm); and F, molecular density (F ) Mj w/NA* (⟨S2⟩1/2)3).
2492 Biomacromolecules, Vol. 10, No. 9, 2009 Boukari et al.
In conclusion, these results indicate that the Tx-Xyl actionindirectly induces a physical effect that provokes disaggregationof the initial GAX aggregated particles as a result of thechemical effect of the enzyme on GAX polymer chains (polymerhydrolysis).
Endoxylanase Action Pattern on Carbohydrate-LigninModel Complexes (GAX-DHP). To investigate the impact oflignin-carbohydrate interactions on Tx-Xyl action, we preparedGAX-DHP complexes according to two in vitro peroxidase-mediated lignin polymerization methods. The first one (“Zu-laufverfahren”, ZL method) consists of the simultaneous addingof all reactants (coniferyl alcohol, hydrogen peroxide), whereasin the later one (“Zutropfverfahren”, ZT method), reagents areslowly added at a controlled flow rate. Reactions were conductedusing synthesis conditions and reactants strictly similar to ourprevious study.25 Previously, we have shown that ZL and ZTmethods allowed the recovery of two complexes (GAX-DHPZL
and GAX-DHPZT) that display different organization patternsand bonding modes. Accordingly, ZL polymerization leads tothe formation of GAX-DHPZL complex mainly characterizedby noncovalent intermolecular bonds, while ZT polymerizationprovides a mainly covalently bonded GAX-DHPZT complexdisplaying LCC bonds.25
LCCs, as introduced by Bjorkman et al.3 are defined ascovalent associations that contain both hydrophilic carbohydratesand hydrophobic lignin, resulting in the formation of micellarstructures in solution.53 Using NMR analysis, we have previ-ously demonstrated the existence of benzyl ether linkagesbetween GAX and DHPZT molecules involving the C5 hydroxylgroup of the L-arabinosyl moieties and the R-carbon of the
lignin-like polymer, resulting in LCC formation. On the contrary,in GAX-DHPZL complexes would mainly involve noncovalenthydrophobic interactions between the two components.25
Size Exclusion Chromatography. GAX-DHP synthesissystems that had been incubated for 1 h with a large excess ofTx-Xyl (2 IU/mg GAX) were analyzed using HP-SEC with online RI and UV detectors. Two different mobile phases wereused to achieve complete and detailed identification of thepopulations present in solution: nitrate eluent (50 mM, NaNO3)and DMSO/water (9/1) containing 50 mM of LiBr (abbreviatedas DMSO). This polar solvent containing the chaotropic saltLiBr can solubilize both lignin and GAX and also dissociatethe noncovalent interactions.44,54
Figure 5a,b shows the polydisperse elution profiles ofuntreated GAX-DHP samples that were obtained using theaqueous mobile phase. In the case of GAX-DHPZL, a majorGAX polymer fraction was eluted at 23 mL and two other highermolar mass populations displaying both RI and UV signals wereeluted at 16 and 19 mL. A homologous fraction was also elutedclose to 19 mL in the case of GAX-DHPZT. Because DHPs arethe only UV-absorbing components, but are at the same timeinsoluble in the aqueous eluent, it can be assumed that thesefractions correspond to micellar hydrosoluble populations thatare formed by the association of GAX chains and DHPmolecules. The lower HP-SEC recovery (calculated accordingto RI signal and known dn/dc value) obtained in the case ofGAX-DHPZT system (47%) compared to GAX-DHPZL (65%)may suggest the presence of larger and more complex associatedstructures.
Figure 5. HP-SEC elution profiles in an aqueous system of GAX-DHP model systems: before Tx-Xyl treatment, (a) GAX-DHPZL, (b) GAX-DHPZT; after 1 h Tx-Xyl treatment, (c) GAX-DHPZL, (d) GAX-DHPZT. Refractive index (RI signal; (s)); ultraviolet (UV signal; (----)).
Enzymatic Conversion of Xylan Biomacromolecules, Vol. 10, No. 9, 2009 2493
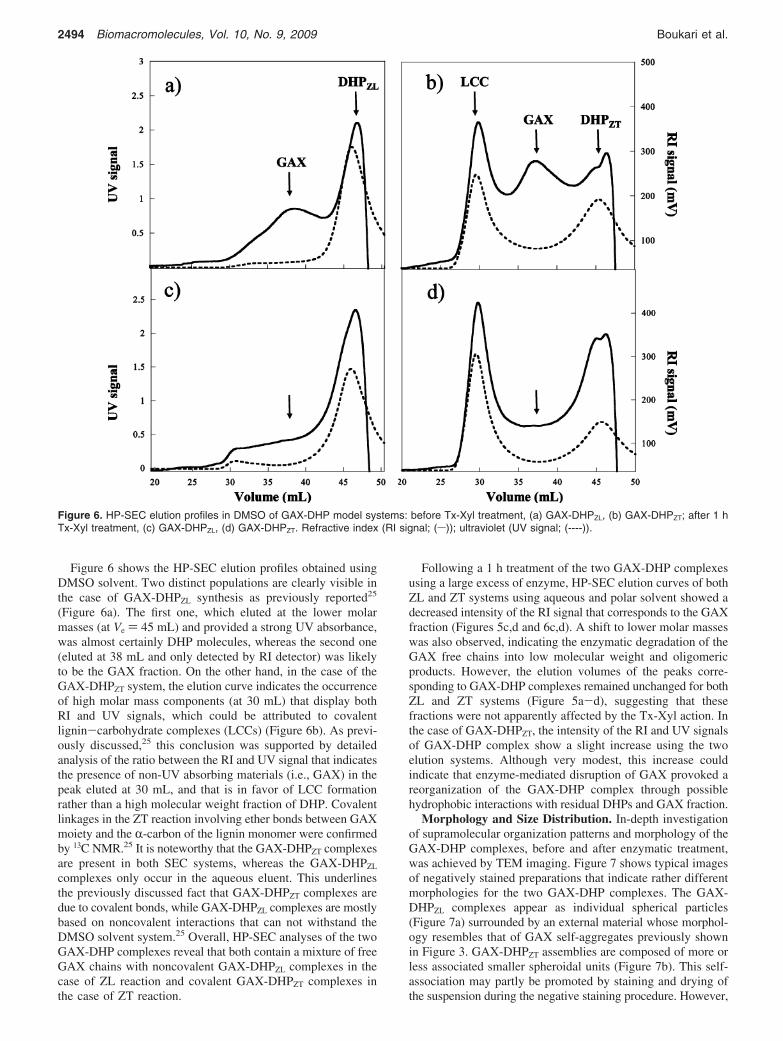
Figure 6 shows the HP-SEC elution profiles obtained usingDMSO solvent. Two distinct populations are clearly visible inthe case of GAX-DHPZL synthesis as previously reported25
(Figure 6a). The first one, which eluted at the lower molarmasses (at Ve ) 45 mL) and provided a strong UV absorbance,was almost certainly DHP molecules, whereas the second one(eluted at 38 mL and only detected by RI detector) was likelyto be the GAX fraction. On the other hand, in the case of theGAX-DHPZT system, the elution curve indicates the occurrenceof high molar mass components (at 30 mL) that display bothRI and UV signals, which could be attributed to covalentlignin-carbohydrate complexes (LCCs) (Figure 6b). As previ-ously discussed,25 this conclusion was supported by detailedanalysis of the ratio between the RI and UV signal that indicatesthe presence of non-UV absorbing materials (i.e., GAX) in thepeak eluted at 30 mL, and that is in favor of LCC formationrather than a high molecular weight fraction of DHP. Covalentlinkages in the ZT reaction involving ether bonds between GAXmoiety and the R-carbon of the lignin monomer were confirmedby 13C NMR.25 It is noteworthy that the GAX-DHPZT complexesare present in both SEC systems, whereas the GAX-DHPZL
complexes only occur in the aqueous eluent. This underlinesthe previously discussed fact that GAX-DHPZT complexes aredue to covalent bonds, while GAX-DHPZL complexes are mostlybased on noncovalent interactions that can not withstand theDMSO solvent system.25 Overall, HP-SEC analyses of the twoGAX-DHP complexes reveal that both contain a mixture of freeGAX chains with noncovalent GAX-DHPZL complexes in thecase of ZL reaction and covalent GAX-DHPZT complexes inthe case of ZT reaction.
Following a 1 h treatment of the two GAX-DHP complexesusing a large excess of enzyme, HP-SEC elution curves of bothZL and ZT systems using aqueous and polar solvent showed adecreased intensity of the RI signal that corresponds to the GAXfraction (Figures 5c,d and 6c,d). A shift to lower molar masseswas also observed, indicating the enzymatic degradation of theGAX free chains into low molecular weight and oligomericproducts. However, the elution volumes of the peaks corre-sponding to GAX-DHP complexes remained unchanged for bothZL and ZT systems (Figure 5a-d), suggesting that thesefractions were not apparently affected by the Tx-Xyl action. Inthe case of GAX-DHPZT, the intensity of the RI and UV signalsof GAX-DHP complex show a slight increase using the twoelution systems. Although very modest, this increase couldindicate that enzyme-mediated disruption of GAX provoked areorganization of the GAX-DHP complex through possiblehydrophobic interactions with residual DHPs and GAX fraction.
Morphology and Size Distribution. In-depth investigationof supramolecular organization patterns and morphology of theGAX-DHP complexes, before and after enzymatic treatment,was achieved by TEM imaging. Figure 7 shows typical imagesof negatively stained preparations that indicate rather differentmorphologies for the two GAX-DHP complexes. The GAX-DHPZL complexes appear as individual spherical particles(Figure 7a) surrounded by an external material whose morphol-ogy resembles that of GAX self-aggregates previously shownin Figure 3. GAX-DHPZT assemblies are composed of more orless associated smaller spheroidal units (Figure 7b). This self-association may partly be promoted by staining and drying ofthe suspension during the negative staining procedure. However,
Figure 6. HP-SEC elution profiles in DMSO of GAX-DHP model systems: before Tx-Xyl treatment, (a) GAX-DHPZL, (b) GAX-DHPZT; after 1 hTx-Xyl treatment, (c) GAX-DHPZL, (d) GAX-DHPZT. Refractive index (RI signal; (s)); ultraviolet (UV signal; (----)).
2494 Biomacromolecules, Vol. 10, No. 9, 2009 Boukari et al.
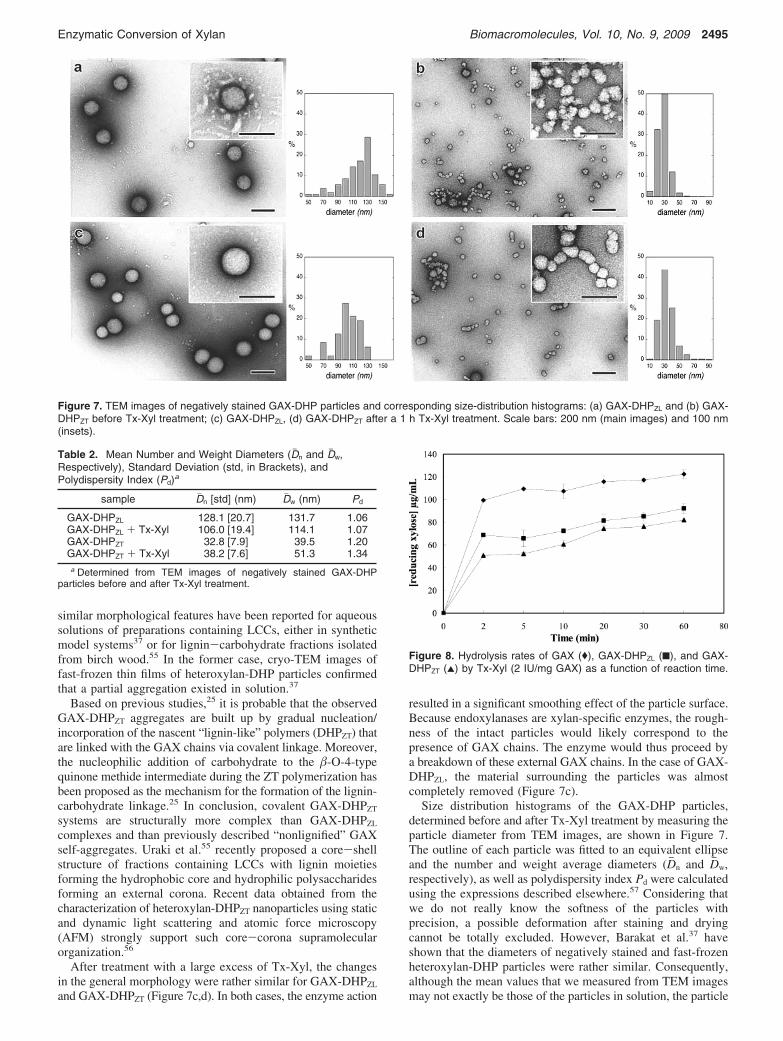
similar morphological features have been reported for aqueoussolutions of preparations containing LCCs, either in syntheticmodel systems37 or for lignin-carbohydrate fractions isolatedfrom birch wood.55 In the former case, cryo-TEM images offast-frozen thin films of heteroxylan-DHP particles confirmedthat a partial aggregation existed in solution.37
Based on previous studies,25 it is probable that the observedGAX-DHPZT aggregates are built up by gradual nucleation/incorporation of the nascent “lignin-like” polymers (DHPZT) thatare linked with the GAX chains via covalent linkage. Moreover,the nucleophilic addition of carbohydrate to the �-O-4-typequinone methide intermediate during the ZT polymerization hasbeen proposed as the mechanism for the formation of the lignin-carbohydrate linkage.25 In conclusion, covalent GAX-DHPZT
systems are structurally more complex than GAX-DHPZL
complexes and than previously described “nonlignified” GAXself-aggregates. Uraki et al.55 recently proposed a core-shellstructure of fractions containing LCCs with lignin moietiesforming the hydrophobic core and hydrophilic polysaccharidesforming an external corona. Recent data obtained from thecharacterization of heteroxylan-DHPZT nanoparticles using staticand dynamic light scattering and atomic force microscopy(AFM) strongly support such core-corona supramolecularorganization.56
After treatment with a large excess of Tx-Xyl, the changesin the general morphology were rather similar for GAX-DHPZL
and GAX-DHPZT (Figure 7c,d). In both cases, the enzyme action
resulted in a significant smoothing effect of the particle surface.Because endoxylanases are xylan-specific enzymes, the rough-ness of the intact particles would likely correspond to thepresence of GAX chains. The enzyme would thus proceed bya breakdown of these external GAX chains. In the case of GAX-DHPZL, the material surrounding the particles was almostcompletely removed (Figure 7c).
Size distribution histograms of the GAX-DHP particles,determined before and after Tx-Xyl treatment by measuring theparticle diameter from TEM images, are shown in Figure 7.The outline of each particle was fitted to an equivalent ellipseand the number and weight average diameters (Dj n and Djw,respectively), as well as polydispersity index Pd were calculatedusing the expressions described elsewhere.57 Considering thatwe do not really know the softness of the particles withprecision, a possible deformation after staining and dryingcannot be totally excluded. However, Barakat et al.37 haveshown that the diameters of negatively stained and fast-frozenheteroxylan-DHP particles were rather similar. Consequently,although the mean values that we measured from TEM imagesmay not exactly be those of the particles in solution, the particle
Figure 7. TEM images of negatively stained GAX-DHP particles and corresponding size-distribution histograms: (a) GAX-DHPZL and (b) GAX-DHPZT before Tx-Xyl treatment; (c) GAX-DHPZL, (d) GAX-DHPZT after a 1 h Tx-Xyl treatment. Scale bars: 200 nm (main images) and 100 nm(insets).
Table 2. Mean Number and Weight Diameters (Dj n and Djw,Respectively), Standard Deviation (std, in Brackets), andPolydispersity Index (Pd)a
sample Dj n [std] (nm) Djw (nm) Pd
GAX-DHPZL 128.1 [20.7] 131.7 1.06GAX-DHPZL + Tx-Xyl 106.0 [19.4] 114.1 1.07GAX-DHPZT 32.8 [7.9] 39.5 1.20GAX-DHPZT + Tx-Xyl 38.2 [7.6] 51.3 1.34a Determined from TEM images of negatively stained GAX-DHP
particles before and after Tx-Xyl treatment.
Figure 8. Hydrolysis rates of GAX ((), GAX-DHPZL (9), and GAX-DHPZT (2) by Tx-Xyl (2 IU/mg GAX) as a function of reaction time.
Enzymatic Conversion of Xylan Biomacromolecules, Vol. 10, No. 9, 2009 2495
size before and after the enzyme treatment can be relativelycompared. The values summarized in Table 2 clearly show thesize differences between the two systems. The GAX-DHPZL
particles are almost four times larger than GAX-DHPZT particles(Dj n ) 128.1 and 32.8 nm, respectively). The polydispersityindexes reveal that both initial GAX-DHP particles are relativelyhomogeneous in diameter. In the case of GAX-DHPZL particles,the apparent particle dimensions (Dj n and Djw) decreased afterthe Tx-Xyl treatment, most likely as a result of the hydrolysisof external GAX chains. Conversely, following Tx-Xyl actionthe apparent diameters of GAX-DHPZT particles slightly in-creased (Dj n increases from 32.8 to 38.2 nm), probably as a resultof a self-aggregation of the LCC rich complexes. At thedifference of GAX-DHPZL particles, this could be explained bya limited accessibility of the enzyme to the GAX fraction.
Enzymatic Hydrolysis Rates. The hydrolysis extent of GAX-DHPZL and GAX-DHPZT samples was monitored as functionof reaction time using a reducing sugar assay (Figure 8). Thefinal yield, estimated after 1 h of reaction with a large excessof enzyme (2 IU/mg GAX), revealed that, when compared toTx-Xyl treatment of free GAX, 24.5 ((3.9%) and 33% ((0.6%)less reducing xylose equivalents were released from GAX-DHPZL and GAX-DHPZT, respectively. These results highlightand confirm the general negative impact of lignin (DHPs) onxylanase-mediated GAX degradation but reveal that this phe-nomenon occurs irrespective of the exact nature of the interac-tions between GAX and DHPs. Therefore, a covalent lignin-carbohydrate bond is not a prerequisite for the restrictiveimpact of lignin on enzyme action as previously suggested.58
However, xylanase was less efficient on GAX-DHPZT whoseassembly involves covalent interactions.
It is important to note that the two polymerization modesthat were used to prepare the GAX-DHP complexes not onlygive rise to different interactions between the GAX and theDHPs, but they also generate different DHP polymer structures.Indeed, rapid bulk (ZL) polymerization, which is likely to berepresentative of the earliest stages of lignification, favors C-Ccoupling of monolignols into highly branched polymers, whereasgradual end-wise (ZT) polymerization, that probably reflectsprocesses that occur at latter stages favors �-O-4 coupling ofmonolignols into relatively linear polymers.1 Therefore, the factthat Tx-Xyl is inhibited in both cases indicates that the patternof intermonomer linkage is not a critical determining factoreither. In their studies on enzymatic degradability of cell wallmodel systems (DHP-CW) obtained by in situ polymerizationof coniferyl alcohol into primary maize cell walls, Grabber et
al.59 have also concluded that DHP-CW degradability usingenzyme cocktails was not influenced by the mode of coniferylalcohol polymerization, because they have observed similareffects with lignin (DHPs) formed by “bulk” or “end-wise”polymerization.
However, in addition to the divergence in the lignin structure,the mode of monolignols polymerization determines lignin-carbohydrate interactions of different nature and consequentlyresults in distinct supramolecular assemblies, as it has beenevidenced for GAX-DHPZL and GAX-DHPZT complexes. In-terestingly, both systems have the same DHP content, but alower hydrolysis yield is obtained in the case of GAX-DHPZT
complexes. This could be manifestly explained by the complex-ity of the supramolecular organization of these systems. Theoccurrence of intimate intra- and inter- connections betweenlignin and GAX within the LCCs may enhance entrapment ofGAX and induce a steric hindrance, thereby limiting theaccessibility of the polysaccharide chains to the endoxylanase.
Kinetic Assay. To ascertain the extent to which the supramo-lecular organization and thus substrate (GAX) accessibilityaffects Tx-Xyl action pattern, kinetic analyses were performedon GAX, GAX-DHPZL, and GAX-DHPZT as substrates andlimiting amounts of enzyme (0.05 IU/mg of GAX). The extentof enzymatic hydrolysis of GAX fraction in each sample wasmonitored as a function of incubation time, using HP-SECanalysis operating in aqueous conditions.
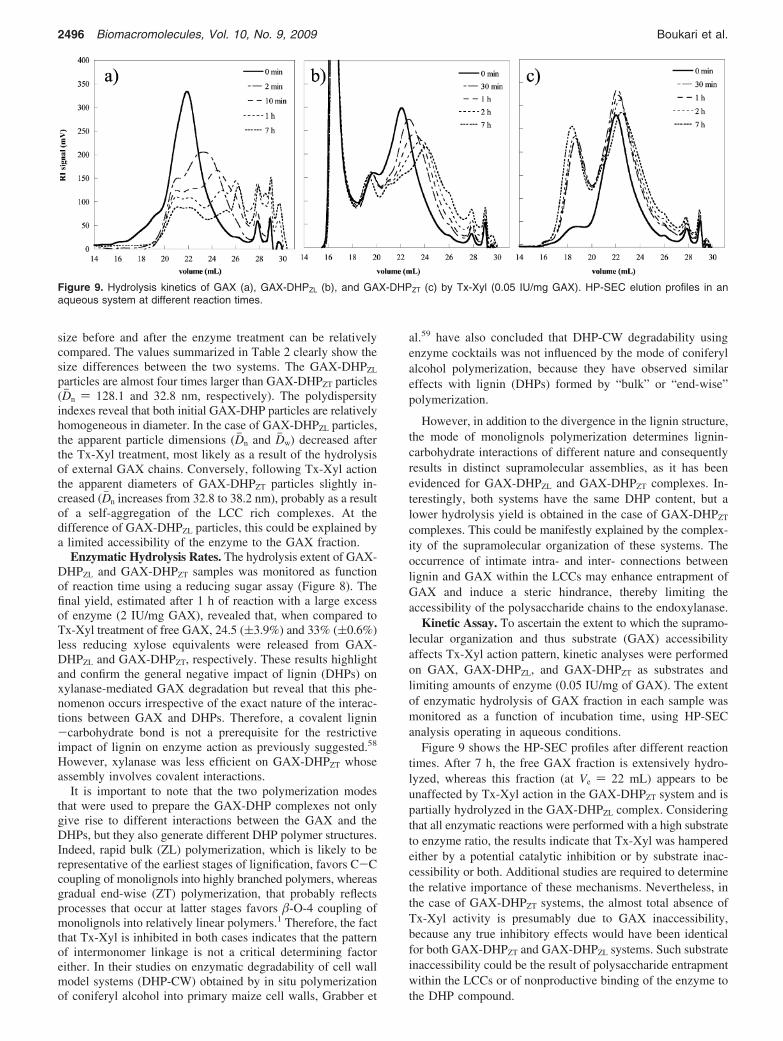
Figure 9 shows the HP-SEC profiles after different reactiontimes. After 7 h, the free GAX fraction is extensively hydro-lyzed, whereas this fraction (at Ve ) 22 mL) appears to beunaffected by Tx-Xyl action in the GAX-DHPZT system and ispartially hydrolyzed in the GAX-DHPZL complex. Consideringthat all enzymatic reactions were performed with a high substrateto enzyme ratio, the results indicate that Tx-Xyl was hamperedeither by a potential catalytic inhibition or by substrate inac-cessibility or both. Additional studies are required to determinethe relative importance of these mechanisms. Nevertheless, inthe case of GAX-DHPZT systems, the almost total absence ofTx-Xyl activity is presumably due to GAX inaccessibility,because any true inhibitory effects would have been identicalfor both GAX-DHPZT and GAX-DHPZL systems. Such substrateinaccessibility could be the result of polysaccharide entrapmentwithin the LCCs or of nonproductive binding of the enzyme tothe DHP compound.
Figure 9. Hydrolysis kinetics of GAX (a), GAX-DHPZL (b), and GAX-DHPZT (c) by Tx-Xyl (0.05 IU/mg GAX). HP-SEC elution profiles in anaqueous system at different reaction times.
2496 Biomacromolecules, Vol. 10, No. 9, 2009 Boukari et al.

Conclusion
Using in vitro reconstituted systems displaying increasedcomplexity, we have been able to provide new insights on themechanism of endoxylanase action at a supramolecular level.
First, the Tx-Xyl action pattern on glucuronoarabinoxylan(GAX) self-aggregates revealed an interesting “disaggregating”effect as a result to the well-known depolymerization mechanismof this endoglycoside hydrolase. Then the use of in vitroreconstituted lignin-glucuronoarabinoxylan (GAX-DHP) nano-composites provided evidence that the way in which cell wallpolymers are interconnected can impact on the enzymaticprocess, in addition to the restriction due to DHP. We showthat the supramolecular organization of the lignin-carbohydratecomplexes, which is a direct consequence of interactions thatoccur between the two components, influences the enzymeaction pattern. Principally, Tx-Xyl action is restricted to theperipheral carbohydrate chains of the GAX-DHP micellarstructures. Even though a covalent lignin-carbohydrate bound(LCC) within the GAX-DHPZT assemblies is not a prerequisitefor the restrictive action of lignin, these associations severelyhamper the access of the enzyme to GAX. With regard to theextrapolation of these observations for the analysis of biomassconversion scenarios, a certain amount of prudence is necessary.This is because, when compared to naturally occurring lignin,the “lignin-like” DHP polymers are structurally different andrather simple.60 In addition, the complexes would only mimicpart of the structures encountered in lignified cell walls.However, the fundamental observations of enzyme inhibition,substrate inaccessibility and enzyme-induced supramolecularstructural reorganization are all phenomena that should bepinpointed as obstacles for the bioconversion of lignocellulosicbiomass. Further investigations of enzyme inhibition andnonproductive binding by phenolic components are underprogress to get a more comprehensive view on the way ligninaffects the enzymatic conversion of lignocelluloses. All theseconsiderations should be helpful in designing and engineeringof more “adapted” and effective new enzymatic tools.
References and Notes
(1) Sarkanen, K. V. In Lignins, occurrence, formation, structure andreactions; Sarkanen, K. V., Ludwig, C. H., Eds.; Wiley-Interscience:New York, 1971.
(2) Atalla, R. H. Abstracts of Papers, 211th National Meeting of theAmerican Chemical Society, New Orleans, LA, March 24-28, 1996;American Chemical Society: Washington, DC, 1996; 24.
(3) Bjorkman, A. SVensk Papperstidning 1957, 60, 243–251.(4) Mooney, C. A.; Mansfield, S. D.; Touhy, M. G.; Saddler, J. N. Biores.
Technol. 1998, 64, 113–119.(5) Sutcliffe, R.; Saddler, J. N. Biotechnol. Bioeng. Symp. 1986, 17, 749–
762.(6) Sharma, A.; Milstein, O.; Vered, Y.; Gressel, J.; Flowers, H. M.
Biotechnol. Bioeng. 1985, 27, 1095–1101.(7) Senior, D. J.; Mayers, P. R.; Breuil, C.; Saddler, J. N. In Biotechnology
in Pulp and Paper Manufacture; Kirk, T. K., Chang, H.-M., Eds.;Butterworth-Heinemann: Boston, MA, 1990; Vol. 16, pp 9-182.
(8) Berlin, A.; Balakshin, M.; Gilkes, N.; Kadla, J.; Maximenko, V.; Kubo,S.; Saddler, J. J. Biotechnol. 2006, 125, 198–209.
(9) Pan, X. J. Biobased Mater. Bioenergy 2008, 2, 25–32.(10) Chandra, R. P.; Bura, R.; Mabee, W. E.; Berlin, A.; Pan, X.; Saddler,
J. N. AdV Biochem. Eng./Biotechnol. 2007, 108, 67–93.(11) Grabber, J. H.; Ralph, J.; Lapierre, C.; Barriere, Y. C. R. Biol. 2004,
327, 455–465.(12) Akin, D. E. Biofuels, Bioprod. Biorefin. 2008, 2, 288–303.(13) Zhu, L.; O’Dwyer, J. P.; Chang, V. S.; Granda, C. B.; Holtzapple,
M. T. Bioresour. Technol. 2008, 99, 3817–3828.(14) Eriksson, J.; Malmsten, M.; Tiberg, F.; Callisen, T. H.; Damhus, T.;
Johansen, K. S. Colloid Interface Sci. 2005, 284, 99–106.(15) Ciolacu, D.; Ciolacu, F.; Dumitriu, R.; Vasile, C.; Popa, V. I. Cellul.
Chem. Technol. 2007, 41, 37–42.
(16) Ciolacu, D.; Ciolacu, F.; Popa, V. I. Macromol. Symp. 2008, 272,136–142.
(17) Ahola, S.; Turon, X.; Osterberg, M.; Laine, J.; Rojas, O. J. Langmuir2008, 24, 11592–11599.
(18) Higuchi, T.; Ogino, K.; Tanahashi, M. Wood Res. 1971, 51, 1–11.(19) Ohnishi, J.; Watanabe, N.; Koshijima, T. Phytochemistry 1992, 31,
1185–1190.(20) Ralph, J.; Helm, R. F.; Quideau, S.; Hatfield, R. D. J. Chem. Soc.,
Perkin Trans. 1992, 1, 2961–2969.(21) Terashima, N.; Atalla, R. H.; Ralph, S. A.; Landucci, L. L.; Lapierre,
C.; Monties, B. Holzforschung 1995, 49, 521–527.(22) Grabber, J. H.; Ralph, J.; Hatfield, R. D.; Quideau, S.; Kuster, T.;
Pell, A. N. J. Agric. Food Chem. 1996, 44, 1453–1459.(23) Touzel, J. P.; Chabbert, B.; Monties, B.; Debeire, P.; Cathala, B. J.
Agric. Food Chem. 2003, 51, 981–986.(24) Cathala, B.; Rondeau-Mouro, C.; Lairez, D.; Belval, F. B.; Durand,
H.; Gorrichon, L.; Touzel, J. P.; Chabbert, B.; Monties, B. PlantBiosyst. 2005, 139, 93–97.
(25) Barakat, A.; Winter, H.; Rondeau-Mouro, C.; Saake, B.; Chabbert,B.; Cathala, B. Planta 2007, 226, 267–281.
(26) Debeire-Gosselin, M.; Loonis, M.; Samain, E.; Debeire, P. In Xylansand Xylanases; Visser, J., Beldman, G., Kusters-van Someren, M. A.,Voragen, A. G. J., Eds.; Elsevier Science Publishers: New York, 1992;Vol. 46, pp 3-466.
(27) Harris, G. W.; Pickersgill, R. W.; Connerton, I.; Debeire, P.; Touzel,J.-P.; Breton, C.; Perez, S. Proteins 1997, 29, 77–86.
(28) Beaugrand, J.; Chambat, G.; Wong, V.; Goubet, F.; Remond, C.; Paes,G.; Paes, G.; Benamrouche, S.; Debeire, P.; O’Donohue, M.; Chabbert,B. Carbohydr. Res. 2004, 339, 2529–2540.
(29) Benamrouche, S.; Cronier, D.; Debeire, P.; Chabbert, B. J. CerealSci. 2002, 36, 253–260.
(30) Lequart, C.; Nuzillard, J. M.; Kurek, B.; Debeire, P. Carbohydr. Res.1999, 319, 102–111.
(31) Zilliox, C.; Debeire, P. Enzyme Microb. Technol. 1998, 22, 58–63.(32) Beg, Q. K.; Kapoor, M.; Mahajan, L.; Hoondal, G. S. Appl. Microbiol.
Biotechnol. 2001, 56, 326–338.(33) Saha, B. C. J. Ind. Microbiol. Biotechnol. 2003, 30, 279–291.(34) Winter, H.; Barakat, A.; Cathala, B.; Saake, B. Macromol. Symp. 2006,
232, 74–84.(35) Ludley, F. H.; Ralph, J. J. Agric. Food Chem. 1996, 44, 2942–2943.(36) Courtin, C. M.; Van den Broeck, H.; Delcour, J. A. J. Chromatogr.,
A 2000, 866, 97–104.(37) Barakat, A.; Putaux, J. L.; Saulnier, L.; Chabbert, B.; Cathala, B.
Biomacromolecules 2007, 8, 1236–1245.(38) Westbye, P.; Kohnke, T.; Glasser, W.; Gatenholm, P. Cellulose 2007,
14, 603–613.(39) Lebel, R. G.; Goring, D. A. I.; Timell, T. E. J. Polym. Sci., Part C:
Polym. Symp. 1963, 2, 9–28.(40) Blake, J. D.; Richards, G. N. Carbohydr. Res. 1971, 18, 11–21.(41) Ebringerova, A.; Hromadkova, Z.; Alfoldi, J.; Berth, G. Carbohydr.
Polym. 1992, 19, 99–105.(42) Esker, A.; Becker, U.; Jamin, S.; Beppu, S.; Renneckar, S.; Glasser,
W. Hemicelluloses: science and technology. ACS Symp. Ser. 2004(Gatenholm, P., Tenkanen, M. e., Eds.).
(43) Roubroeks, J.; Saake, B.; Glasser, W.; Gatenholm, P. Hemicelluloses:science and technology. ACS Symp. Ser. 2004 (Gatenholm, P.,Tenkanen, M. e., Eds.).
(44) Saake, B.; Kruse, T.; Puls, J. Bioresour. Technol. 2001, 80, 195–204.(45) Linder, A.; Bergman, R.; Bodin, A.; Gatenholm, P. Langmuir 2003,
19, 5072–5077.(46) Suggett, A. In Polysaccharides; Franks, F., Ed.; Plenum: New York,
N.Y., 1975; Vol. 4.(47) Shigematsu, M.; Goto, A.; Yoshida, S.; Tanahashi, M.; Shinoda, Y.
Mokuzai Gakkaishi 1994, 40, 1214–18.(48) Kalyanasundaram, K.; Thomas, J. K. J. Am. Chem. Soc. 1977, 99,
2039–2044.(49) Winnik, F. M.; Regismond, S. T. A. Colloids Surf., A 1996, 118, 1–
39.(50) Barakat, A.; Chabbert, B.; Cathala, B. Phytochemistry 2007, 68, 2118–
2125.(51) Biely, P.; Vrsanska, M.; Tenkanen, M.; Kluepfel, D. J. Biotechnol.
1997, 57, 151–166.(52) Wyatt, P. J. Anal. Chim. Acta 1993, 272, 1–40.(53) Yaku, F.; Tsuji, S.; Koshijima, T. Holzforschung 1979, 33, 54–9.(54) Ringena, O.; Lebioda, S.; Lehnen, R.; Saake, B. J. Chromatogr., A
2006, 1102, 154–163.(55) Uraki, Y.; Usukura, Y.; Kishimoto, T.; Ubukata, M. Holzforschung
2006, 60, 659–664.
Enzymatic Conversion of Xylan Biomacromolecules, Vol. 10, No. 9, 2009 2497

(56) Barakat, A.; Gaillard, C.; Lairez, D.; Saulnier, L.; Chabbert, B.;Cathala, B. Biomacromolecules 2008, 9, 487–493.
(57) Putaux, J.-L.; Buleon, A.; Borsali, R.; Chanzy, H. Int. J. Biol.Macromol. 1999, 26, 145–150.
(58) Sewalt, V. J. H.; Glasser, W. G.; Beauchemin, K. A. J. Agric. FoodChem. 1997, 45, 1823–1828.
(59) Grabber, J. H.; Hatfield, R. D.; Ralph, J. J. Agric. Food Chem. 2003,51, 4984–498.
(60) Saake, B.; Argyropoulos, D. S.; Beinhoff, O.; Faix, O. Phytochemistry1996, 43, 499–507.
BM9004518
2498 Biomacromolecules, Vol. 10, No. 9, 2009 Boukari et al.


















