Embed Size (px)
Citation preview

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 1 sur 11
Item 220 : Adénome hypophysaire
Objectifs pédagogiques terminaux : « diagnostiquer un adénome hypophysaire »
- Bénins, les adénomes hypophysaires sont des tumeurs bien différenciées, de croissance habituellement
lente sur plusieurs années, développées de manière monoclonale à partir des cellules endocrines anté-hypophysaires. Ils représentent 10 % des tumeurs intracrâniennes.
- On distingue les microadénomes dont le plus grand diamètre est inférieur à 10 mm, et les macroadénomes qui peuvent représenter de volumineuses tumeurs envahissantes.
- Les adénomes peuvent être non-sécrétants, révélés alors par le syndrome tumoral associé éventuellement à des signes d’hypopituitarisme, ou sécrétants : les prolactinomes, les plus fréquents, entraînent le classique syndrome aménorrhée-galactorrhée; les adénomes somatotropes sont responsables de l’acromégalie; les adénomes corticotropes entraînent une maladie de Cushing et les adénomes thyréotropes, plus rares, une hyperthyroïdie.
Etiopathogénie Elle a fait l’objet de nombreux travaux récents. La démonstration du caractère monoclonal de ces tumeurs a montré qu’elles prenaient leur origine dans une prolifération des cellules hypophysaires, même si des facteurs extérieurs à ces cellules (facteurs de croissance, hormones hypothalamiques...) peuvent jouer un rôle promoteur. L’immense majorité des adénomes hypophysaires survient de manière sporadique, mais certaines pathologies familiales peuvent s’accompagner d’une fréquence accrue d’adénomes hypophysaires. C’est le cas de la Néoplasie Endocrinienne Multiple de type 1, dont le gène est désormais connu, qui associe une hyperparathyroïdie, quasi-constante, à d’autres atteintes glandulaires, concernant essentiellement le pancréas endocrine (gastrinomes, insulinomes) et l’hypophyse. Diagnostic Selon leurs caractéristiques morphologiques (taille, extension tumorale) et fonctionnelles, les adénomes hypophysaires peuvent se manifester par un ou plusieurs des éléments de la triade symptomatique : syndrome tumoral, avec ses manifestations cliniques et radiologiques, hypersécrétion d'une ou plusieurs hormones anté-hypophysaires, déficit hormonal touchant une ou plusieurs des lignées hormonales hypophysaires, avec leurs manifestations cliniques et biologiques. En dehors de ces circonstances classiques un nombre croissant d’adénomes est découvert lors de l’exploration d’une masse hypophysaire de découverte fortuite. La démarche diagnostique face a cette situation est schématisée sur la figure 1.

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 2 sur 11
Ces différentes manifestations résultent directement des bases anatomo-physiologiques de la région hypothalamo-hypophysaires (Tableau 1). L'hypophyse est en effet une glande formée d’un lobe antérieur (antéhypophyse) sécrétant plusieurs hormones dans la circulation générale, et d’un lobe postérieur (post-hypophyse) dans lequel aboutissent les axones de neurones hypothalamiques sécrétant ocytocine et vasopressine (hormone anti-diurétique ou ADH ou AVP). L’antéhypophyse est sous le contrôle d’hormones hypothalamiques sécrétées dans le système porte hypothalamo-hypophysaire et reliée à la région hypothalamique par la tige pituitaire au niveau du troisième ventricule. Elle est située dans une loge inextensible, constituée par la selle turcique de l'os sphénoïde en bas, en avant et en arrière, le diaphragme sellaire tapissé par les méninges en haut, et les parois du sinus caverneux latéralement. Celui-ci est traversé par la carotide interne intracrânienne, les portions ophtalmique et maxillaire du nerf trigéminé (V1 et V2), et par les nerfs oculomoteurs (III, IV et VI). La proximité du chiasma optique, croisement des voies optiques, au-dessus de la loge hypophysaire explique le risque visuel en cas de pathologie expansive suprasellaire Tableau 1 Bases fonctionnelles de l’antéhypophyse, montrant les différents types cellulaires (lignées), leur sécrétion hormonale, et leurs régulateurs hypothalamiques principaux, stimulateurs (+) ou inhibiteurs (-). lignée hormone hypophysaire hormone hypothalamique lactotrope prolactine (PRL) dopamine (-) somatotrope hormone de croissance somatostatine (-) (GH =Growth hormone) GHRH (+) (GH Releasing Hormone) GH sécrétagogue (+) (Ghréline) thyréotrope thyréotropine (TSH = TRH (+) (Thyrotropin Releasing Hormone) thyreo Stimulating Hormone) somatostatine (-) corticotrope corticotropine (ACTH= CRH (+) (Corticotropin Releasing Hormone) Adrenocorticotropic Hormone) AVP (+) (Arginine Vasopressin) gonadotrope gonadotropines (FSH= LHRH ou GnRH (+) (LH ou Gonadotropin Releasing Hormone) Folliculo Stimulating Hormone, LH= Luteotropic Hormone Syndrome tumoral 1 - Clinique Les céphalées sont typiquement frontales ou orbitaires, avec irradiations fréquentes au vertex. Peu spécifiques, non pulsatiles, elles sont généralement calmées par les antalgiques habituels. Elles sont présentes même en cas de microadénomes, par mise en tension du diaphragme sellaire. Les réductions du champ visuel sont observées seulement en cas de macroadénome ayant une extension suprasellaire atteignant les voies optiques. Du fait de la répartition des fibres nerveuses au niveau du chiasma, le champ temporal de chaque côté est le premier touché. L'intensité de l'atteinte est croissante avec le degré de la compression : exclusion de la tâche aveugle (Fig 2), aplatissement des isoptères, quadranopsie temporale supérieure, puis la typique hémianopsie bitemporale, jusqu'à la cécité. L'expansion suprasellaire étant souvent médiane et le retentissement est le plus souvent bilatéral. L’examen du champ visuel (campimétrie de Goldman, ou encore champ visuel automatisé) sera complété par une mesure de l’acuité visuelle et un examen du fond d’œil. La diplopie est observée en cas de compression d'un nerf oculomoteur du fait d'une extension tumorale dans le sinus caverneux. Elle peut être explorée par un test de Lancaster.

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 3 sur 11
Une apoplexie hypophysaire, correspondant à une brusque hémorragie intra-adénomateuse, peut entraîner un tableau évoquant une hémorragie méningée, avec céphalées intenses d'apparition brutale, fébricule, diplopie, syndrome confusionnel. 2 - Neuroradiologie En cas de suspicion d'adénome hypophysaire fondée sur des éléments cliniques, éventuellement confirmés par des éléments biologiques l'exploration morphologique à pratiquer est une imagerie en résonance magnétique (IRM) hypophysaire. L'IRM a prouvé sa supériorité sur la tomodensitométrie dans les microadénomes comme dans les macroadénomes. Le scanner peut parfois être utilisé pour des raisons d'accessibilité ou pour mieux explorer le cadre osseux. L'IRM permet actuellement de détecter des microadénomes de 2 ou 3 mm, sous la forme d'une anomalie de signal arrondie intra-parenchymateuse. Toutefois, ce type d'image peut être observé chez près de 10 % de sujets témoins (" incidentalomes hypophysaires ") et ne doit donc être interprété qu'en fonction du contexte clinique et biologique. La radiographie simple du crâne, même centrée sur la selle turcique ne possède pas une sensibilité suffisante et ne doit donc pas être demandée à titre diagnostique. Syndromes d'hypersécrétion 1 - Hyperprolactinémie Le retentissement endocrinien de l'hyperprolactinémie se manifeste assez précocement chez la femme non ménopausée sous la forme de troubles du cycle (oligospanioménorrhée, aménorrhée), d'une galactorrhée, de troubles sexuels (baisse de la libido, sécheresse vaginale, dyspareunie), et parfois seulement sous forme d'une infertilité par anovulation avec conservation des cycles. Le mécanisme de l'atteinte de la fonction gonadique est une inhibition de la libération de LHRH (luteinizing hormone releasing hormone) hypothalamique induite par l'excès de prolactine. Chez la femme ménopausée, la galactorrhée est rare et c'est le syndrome tumoral qui est révélateur. Chez l'homme, les manifestations, conduisant plus tardivement au diagnostic que chez la femme jeune, sont représentées par des troubles sexuels (baisse de libido, dysérection, impuissance érectile), raréfaction de la pilosité faciale ou somatique et rarement gynécomastie voire galactorrhée. Les complications de l'hyperprolactinémie sont liées à l’atteinte de la fonction gonadique: essentiellement l’infertilité et l’ostéoporose en cas d’hyperprolactinémie prolongée. Sur le plan biologique, la prolactinémie basale est trouvée élevée, supérieure à 20 µg/l. Le taux basal de prolactine est généralement bien corrélé avec le volume tumoral, un taux supérieur à 200 µg/L étant quasi-spécifique d'un macroprolactinome. Au contraire, un taux inférieur à 100 µg/L en présence d'un macroadénome volumineux est en faveur d'une hyperprolactinémie accompagnant un adénome non-sécrétant par un mécanisme de compression de la tige pituitaire. À la différence des hypogonadismes d'origine ovarienne, les taux de gonadotrophines (LH et FSH) ne sont pas augmentés. En cas d'insuffisance gonadotrope lésionnelle associée, les gonadotrophines seront même abaissées en base ou après stimulation par LHRH exogène (test au LHRH). 2 - Acromégalie Le tableau clinique lié à l’hypersécrétion chronique de GH est caractérisé par l'installation progressive et insidieuse de modifications morphologiques : prognathisme (Fig 3), élargissement des mains et des pieds nécessitant des changements de pointure de chaussures, épaississement des traits, en particulier le nez et les lèvres. Ces signes passent souvent inaperçus du patient et de son entourage, et seront mis en évidence par la comparaison de clichés successifs (documents d'identité par exemple). On note également une hypersudation, une hyperséborrhée, parfois une hypertrichose ; des troubles de l'articulé dentaire et une macroglossie, avec fréquents ronflements nocturnes, une raucité de la voix ; des arthralgies, un syndrome
©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 4 sur 11
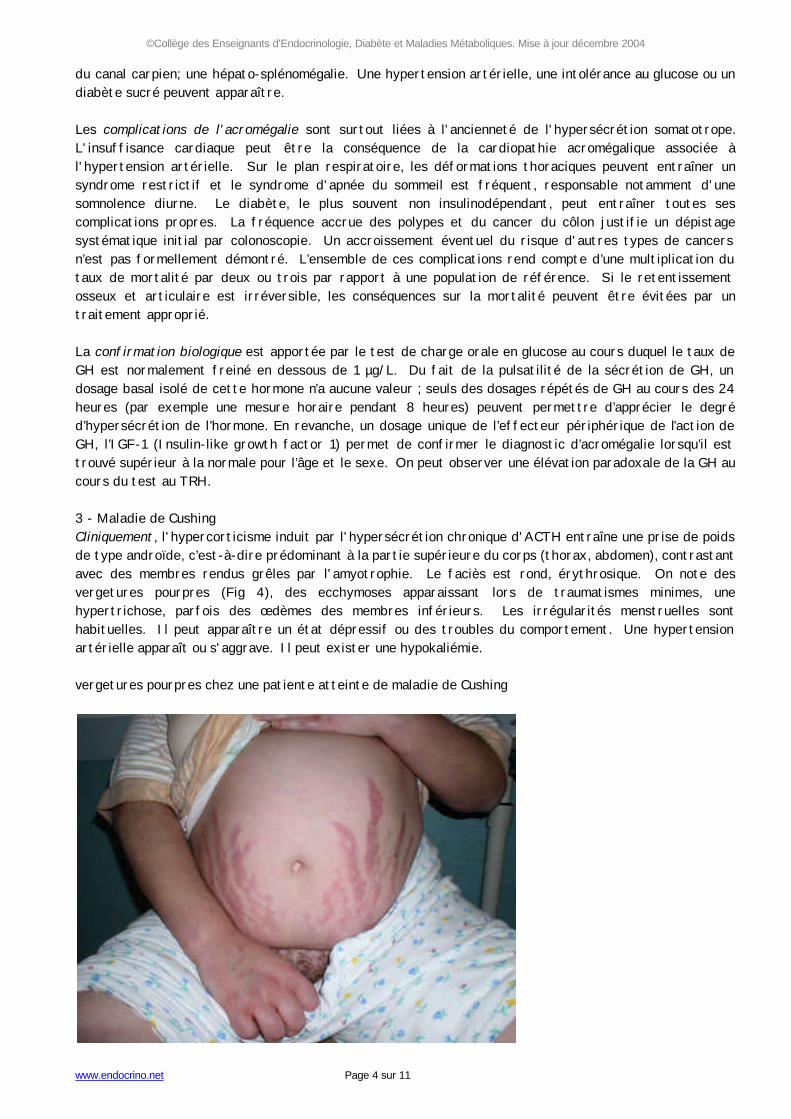
du canal carpien; une hépato-splénomégalie. Une hypertension artérielle, une intolérance au glucose ou un diabète sucré peuvent apparaître. Les complications de l'acromégalie sont surtout liées à l'ancienneté de l'hypersécrétion somatotrope. L'insuffisance cardiaque peut être la conséquence de la cardiopathie acromégalique associée à l'hypertension artérielle. Sur le plan respiratoire, les déformations thoraciques peuvent entraîner un syndrome restrictif et le syndrome d'apnée du sommeil est fréquent, responsable notamment d'une somnolence diurne. Le diabète, le plus souvent non insulinodépendant, peut entraîner toutes ses complications propres. La fréquence accrue des polypes et du cancer du côlon justifie un dépistage systématique initial par colonoscopie. Un accroissement éventuel du risque d'autres types de cancers n’est pas formellement démontré. L’ensemble de ces complications rend compte d’une multiplication du taux de mortalité par deux ou trois par rapport à une population de référence. Si le retentissement osseux et articulaire est irréversible, les conséquences sur la mortalité peuvent être évitées par un traitement approprié. La confirmation biologique est apportée par le test de charge orale en glucose au cours duquel le taux de GH est normalement freiné en dessous de 1 µg/L. Du fait de la pulsatilité de la sécrétion de GH, un dosage basal isolé de cette hormone n’a aucune valeur ; seuls des dosages répétés de GH au cours des 24 heures (par exemple une mesure horaire pendant 8 heures) peuvent permettre d’apprécier le degré d’hypersécrétion de l’hormone. En revanche, un dosage unique de l’effecteur périphérique de l’action de GH, l’IGF-1 (Insulin-like growth factor 1) permet de confirmer le diagnostic d’acromégalie lorsqu’il est trouvé supérieur à la normale pour l’âge et le sexe. On peut observer une élévation paradoxale de la GH au cours du test au TRH. 3 - Maladie de Cushing Cliniquement, l'hypercorticisme induit par l'hypersécrétion chronique d'ACTH entraîne une prise de poids de type androïde, c’est-à-dire prédominant à la partie supérieure du corps (thorax, abdomen), contrastant avec des membres rendus grêles par l'amyotrophie. Le faciès est rond, érythrosique. On note des vergetures pourpres (Fig 4), des ecchymoses apparaissant lors de traumatismes minimes, une hypertrichose, parfois des œdèmes des membres inférieurs. Les irrégularités menstruelles sont habituelles. Il peut apparaître un état dépressif ou des troubles du comportement. Une hypertension artérielle apparaît ou s'aggrave. Il peut exister une hypokaliémie. vergetures pourpres chez une patiente atteinte de maladie de Cushing

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 5 sur 11
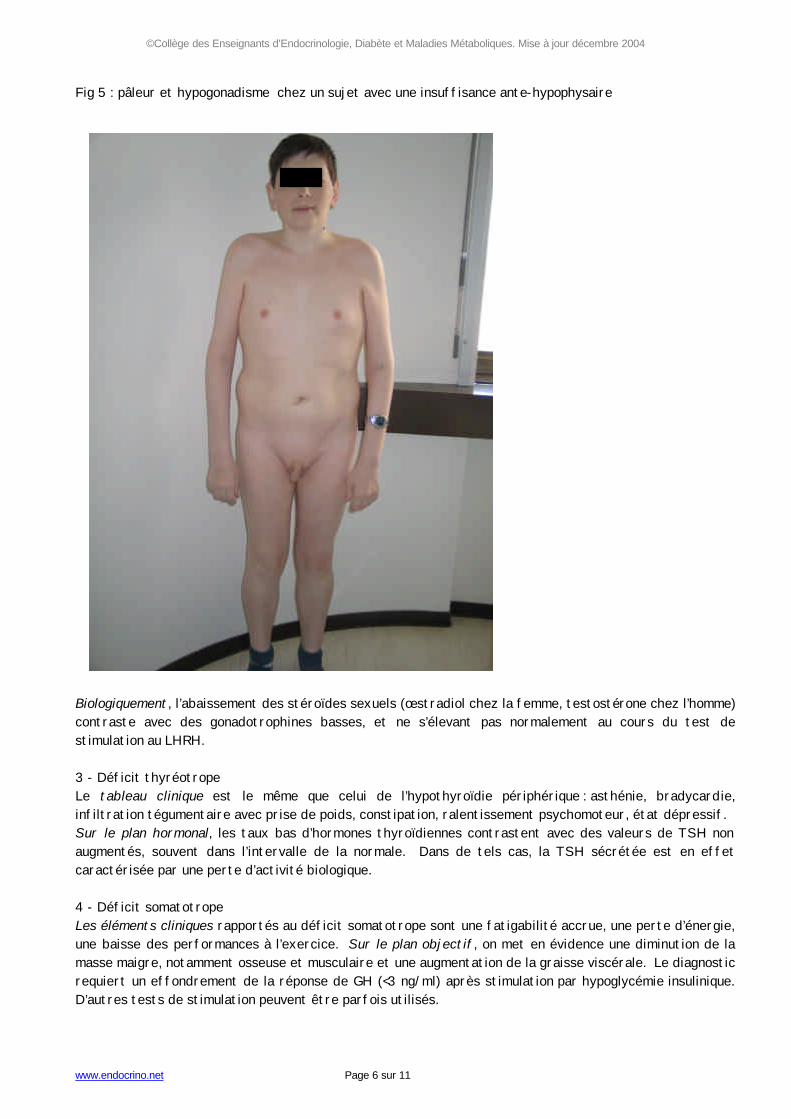
Les complications de la maladie de Cushing sont les mêmes que celles d'une corticothérapie au long cours, avec un risque de mortalité : risque accru d'infection notamment à germes opportunistes; déminéralisation osseuse avec risque de fractures vertébrales, cervico-fémorales ou des os longs ; décompensation psychiatrique ; hypokaliémie sévère, hypertension artérielle ou diabète compliqués ; phlébites ou embolies pulmonaires. Le diagnostic biologique d’hypercortisolisme est souvent difficile et comporte deux aspects. Le diagnostic positif de l’hypercorticisme repose sur l’augmentation de la cortisolémie basale, de préférence mesurée le soir, ou à plusieurs reprises au cours des 24 heures, montrant une perte du rythme nycthéméral ; une élévation du cortisol libre urinaire des 24 heures ; et sur l’absence de freinage de l’hypercorticisme au cours d’un test à la dexaméthasone “ minute ” (1 mg au coucher, et mesure du cortisol le lendemain à 8 heures), ou “ faible ”, (“ test de Liddle ” faible comportant la prise de 2 mg/jour de dexaméthasone à raison de 0,5 mg toutes les 6 heures pendant 48 heures). Le diagnostic étiologique repose sur un faisceau d’arguments cliniques, biologiques et radiologiques qui permettent de distinguer l’hypercorticisme lié à un adénome corticotrope (dénommé “ maladie ” de Cushing), qui représente environ 2/3 des causes de syndromes de Cushing endogènes, d’une autre cause, essentiellement adénome surrénalien ou sécrétion ectopique d’ACTH. Les moyens de ce diagnostic sont donc développés dans la partie “ diagnostic différentiel ”. 4 - Hyperthyroïdie haute L'adénome thyréotrope entraîne les mêmes signes que les autres causes de thyrotoxicose : tachycardie, amaigrissement, hypersudation et thermophobie, diarrhée motrice, nervosité, fatigabilité. Il s'y associe un goitre le plus souvent de volume modéré. Les complications des hyperthyroïdies hautes sont les mêmes que celles des autres causes de thyrotoxicose, essentiellement le risque de cardiothyréose. Le profil biologique typique est celui d’une élévation des fractions libres des hormones thyroïdiennes T3 et T4, associée à un taux de TSH dans les limites de la normale (mais dans ce cas inapproprié au taux de T3 et T4) ou élevé, en général de façon modeste. La sous-unité alpha libre de la TSH est élevée, avec un rapport molaire par rapport à la TSH supérieur à 1. Syndromes d'hyposécrétion 1 - Déficit corticotrope Cliniquement, asthénie croissante au cours de la journée, hypotension orthostatique, pâleur, anorexie ou nausées sont les principaux symptômes. Une perte de pilosité sexuelle peut être observée chez la femme. À la différence des insuffisances surrénales périphériques, il n'y a pas de mélanodermie, et du fait de la préservation de la fonction minéralocorticoïde, il n'y a pas d'anomalie ionique en dehors d'une éventuelle décompensation. L’exploration hormonale montre un cortisol libre urinaire bas, une cortisolémie abaissée le matin, et des mesures répétées pendant les 24 heures caractérisées par des taux bas de cortisol en regard de taux d’ACTH bas ou “ normaux ” mais inappropriés. L’atténuation de la réponse d’ACTH et cortisol au cours d’une hypoglycémie insulinique, ou encore lors d’un test à la métopirone (dosage du composé S), peut aider à confirmer le diagnostic en cas de doute. 2 - Déficit gonadotrope Au plan clinique, les troubles du cycle chez la femme, une dépilation chez l’homme (Fig 5), des troubles de la fonction sexuelle et de la fertilité dans les deux sexes sont les conséquences de l’hypogonadisme par atteinte lésionnelle des cellules gonadotropes.
©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 6 sur 11
Fig 5 : pâleur et hypogonadisme chez un sujet avec une insuffisance ante-hypophysaire Biologiquement, l’abaissement des stéroïdes sexuels (œstradiol chez la femme, testostérone chez l’homme) contraste avec des gonadotrophines basses, et ne s’élevant pas normalement au cours du test de stimulation au LHRH. 3 - Déficit thyréotrope Le tableau clinique est le même que celui de l’hypothyroïdie périphérique : asthénie, bradycardie, infiltration tégumentaire avec prise de poids, constipation, ralentissement psychomoteur, état dépressif. Sur le plan hormonal, les taux bas d’hormones thyroïdiennes contrastent avec des valeurs de TSH non augmentés, souvent dans l’intervalle de la normale. Dans de tels cas, la TSH sécrétée est en effet caractérisée par une perte d’activité biologique. 4 - Déficit somatotrope Les éléments cliniques rapportés au déficit somatotrope sont une fatigabilité accrue, une perte d’énergie, une baisse des performances à l’exercice. Sur le plan objectif, on met en évidence une diminution de la masse maigre, notamment osseuse et musculaire et une augmentation de la graisse viscérale. Le diagnostic requiert un effondrement de la réponse de GH (<3 ng/ml) après stimulation par hypoglycémie insulinique. D’autres tests de stimulation peuvent être parfois utilisés.

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 7 sur 11
5 – Panhypopituitarisme L’association des différents déficits hormonaux réalise le panhypopituitarisme. Il combine les signes déjà évoqués, avec en particulier une asthénie marquée, une peau pâle (Fig 5), sèche et fine, des troubles sexuels. Le diabète insipide ne complique un adénome hypophysaire, en règle, qu’à la suite d’une lésion post-hypophysaire ou de la tige au cours d’une exérèse chirurgicale. Les Complications des hypopituitarismes sont dominées par l'insuffisance surrénalienne aiguë est rarement révélatrice. Elle doit être prévenue par une bonne information du patient et de son entourage sur les risques de décompensation de l'insuffisance corticotrope, même traitée, que peuvent entraîner un stress important, par exemple chirurgical ou accidentel, un état de déshydratation, une pathologie grave intercurrente. Elle se manifeste par une asthénie majeure, une hypotension artérielle entraînant un collapsus cardiovasculaire, des troubles digestifs à type de nausées, douleurs abdominales, vomissements. Une hyponatrémie avec natriurèse conservée est alors présente. Un accroissement de la morbi-mortalité globale et cardiovasculaire a été observé chez des patients hypopituitaires recevant un traitement substitutif des fonctions thyroïdienne, surrénalienne et gonadique. Le déficit somatotrope associé, non traité, pourrait représenter une des raisons de cette situation. Diagnostic du type d'adénome Le diagnostic précis repose sur l'étude histologique et immunohistochimique de l'adénome lorsque celui-ci est retiré chirurgicalement. Dans le cas contraire, le diagnostic est fondé sur des arguments de présomption représentés par les données cliniques, biologiques et radiologiques. 1 - prolactinome : Il représente la plus fréquente des tumeurs hypophysaires (40 %) et certaines études autopsiques ont trouvé des prolactinomes méconnus chez 10 à 20 % des sujets. Le diagnostic repose sur l'existence d'une hyperprolactinémie, typiquement non stimulable par le TRH, le métoclopramide ou la dompéridone (ces agents entraînant une élévation du taux de PRL inférieure à 100 % de la valeur basale), associée à une lésion tumorale hypophysaire sur les clichés neuroradiologiques (scanner ou imagerie par résonance magnétique). Le volume de l’adénome est en règle assez bien corrélé avec les taux de PRL. La forme la plus fréquente est le microprolactinome (Fig 6) de la femme jeune. Chez l’homme ou la femme ménopausée, il s’agit le plus souvent d’un macroprolactinome (Fig 7), qui peut être parfois très volumineux, atteignant les lobes temporaux ou le tronc cérébral. La grande majorité des microprolactinomes (environ 95 %) restant stables au cours du temps, l’objectif principal de traitement est dans ces cas la restauration d’une fonction gonadique normale. En cas d'exérèse chirurgicale, le diagnostic est définitivement confirmé par l'analyse anatomo-pathologique avec marquage immunohistochimique. Certains adénomes mixtes à PRL sécrètent d'autres hormones provenant ou non du même contingent cellulaire, avant tout la GH (adénomes somato-lactotropes). 2 – adénome somatotrope Les adénomes somatotropes représentent environ 15 % des adénomes hypophysaires. Il s’agit dans la majorité des cas de macroadénomes avec des extensions supra- ou para-sellaires. Pourtant, du fait du caractère insidieux des déformations progressives, le retard diagnostique est en moyenne de 5 à 10 ans, et il existe souvent un hypopituitarisme et une atteinte campimétrique au moment du diagnostic. 3 – adénome corticotrope Ils représentent environ 10 % des adénomes hypophysaires. La plupart des adénomes corticotropes (environ 90 %) sont des microadénomes. Il n’est pas rare qu’ils ne soient pas visualisés même par des examens IRM de qualité optimale en coupes millimétriques.
©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 8 sur 11
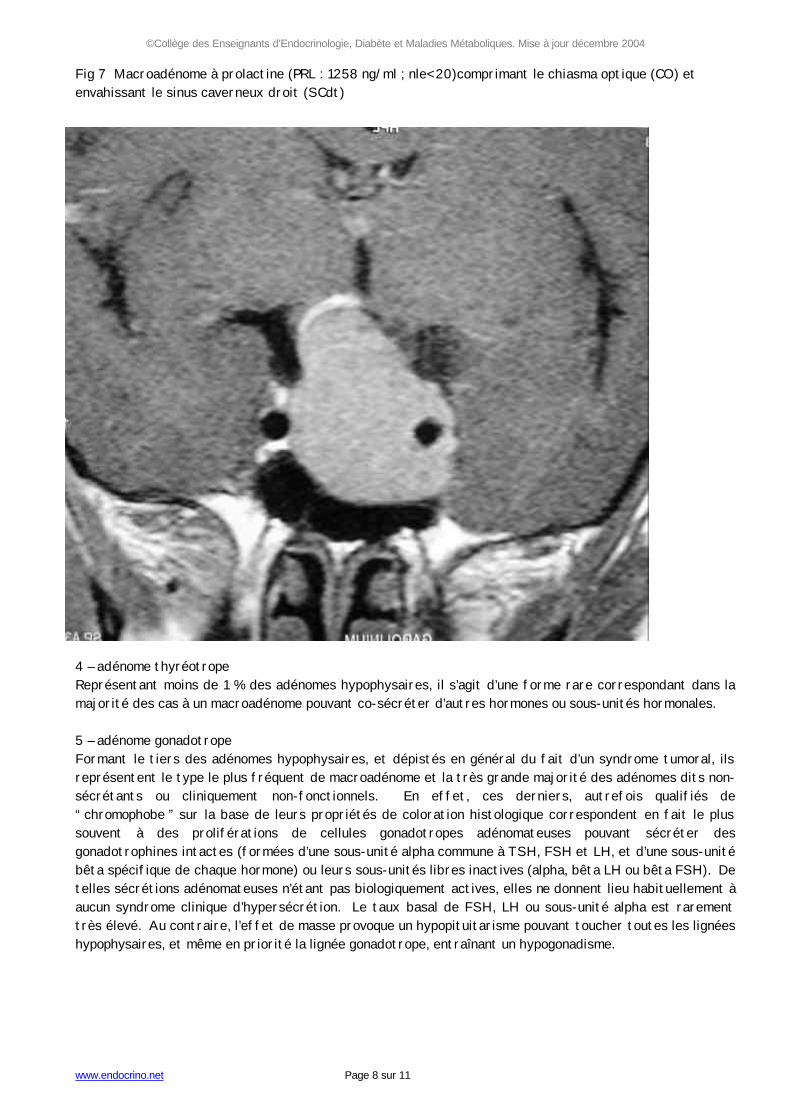
Fig 7 Macroadénome à prolactine (PRL : 1258 ng/ml ; nle< 20)comprimant le chiasma optique (CO) et envahissant le sinus caverneux droit (SCdt) 4 – adénome thyréotrope Représentant moins de 1 % des adénomes hypophysaires, il s’agit d’une forme rare correspondant dans la majorité des cas à un macroadénome pouvant co-sécréter d’autres hormones ou sous-unités hormonales. 5 – adénome gonadotrope Formant le tiers des adénomes hypophysaires, et dépistés en général du fait d’un syndrome tumoral, ils représentent le type le plus fréquent de macroadénome et la très grande majorité des adénomes dits non-sécrétants ou cliniquement non-fonctionnels. En effet, ces derniers, autrefois qualifiés de “ chromophobe ” sur la base de leurs propriétés de coloration histologique correspondent en fait le plus souvent à des proliférations de cellules gonadotropes adénomateuses pouvant sécréter des gonadotrophines intactes (formées d’une sous-unité alpha commune à TSH, FSH et LH, et d’une sous-unité bêta spécifique de chaque hormone) ou leurs sous-unités libres inactives (alpha, bêta LH ou bêta FSH). De telles sécrétions adénomateuses n’étant pas biologiquement actives, elles ne donnent lieu habituellement à aucun syndrome clinique d’hypersécrétion. Le taux basal de FSH, LH ou sous-unité alpha est rarement très élevé. Au contraire, l’effet de masse provoque un hypopituitarisme pouvant toucher toutes les lignées hypophysaires, et même en priorité la lignée gonadotrope, entraînant un hypogonadisme.

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 9 sur 11
Diagnostic différentiel 1 - Devant un syndrome de masse hypophysaire : Les adénomes hypophysaires en sont la principale cause chez l'adulte. Les principales autres étiologies sont rassemblées dans le Tableau 2. Parmi les plus importantes, les craniopharyngiomes sont des tumeurs bénignes, de nature solide, kystique ou mixte, issues de résidus embryonnaires de la poche de Rathke, étagées du nasopharynx à la région diencéphalique. Plus fréquents dans l'enfance et l'adolescence, près de 50 % sont néanmoins diagnostiqués chez l'adulte devant des troubles visuels associés parfois à un diabète insipide et à des signes d'hypopituitarisme. Le taux de récidive après exérèse chirurgicale est élevé. Souvent révélés par une hypertension intracrânienne, un diabète insipide ou un hypopituitarisme chez un adulte jeune, les germinomes peuvent sécréter un marqueur biologique : bêta HCG (human chorionic gonadotropin); ils métastasent en particulier dans la moelle épinière, mais sont très radiosensibles. Les métastases hypophysaires sont souvent révélées par un diabète insipide d'apparition brutale ; elles concernent de nombreux cancers primitifs, principalement les cancers du sein et du poumon, et sont de mauvais pronostic (survie moyenne de 6 mois). Tableau 2 Lésions responsables de syndrome de masse hypophysaire, en dehors des adénomes hypophysaires. Cause physiologique : Hyperplasie lactotrope gravidique Hypothyroïdie périphérique Autres tumeurs bénignes crâniopharyngiomes méningiomes Tumeurs malignes germinomes (pinéalomes ectopiques) sarcomes chordomes adénocarcinomes hypophysaires métastases hypophysaires lymphomes Kystes Kyste de la poche de Rathke Kyste dermoïde Kyste arachnoïdien Lésions inflammatoires et infiltratives Hypophysite lymphocytaire Histiocytose X abcès hypophysaire tuberculome hypophysaire sarcoïdose 2 - Devant une hyperprolactinémie: La première cause à toujours évoquer est physiologique : la grossesse. L'hyperprolactinémie pathologique peut être due à un dysfonctionnement du tissu lactotrope normal, notamment par levée du frein tonique inhibiteur dopaminergique. Environ 25 % des cas d'aménorrhée secondaire sont reliés à une hyperprolactinémie. La moitié des hyperprolactinémies étant en rapport avec une lésion hypothalamo-hypophysaire, le dosage de la PRL basale doit être un examen de dépistage systématique dans les troubles de la fonction gonadique.

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 10 sur 11
- lésions hypophysaires non lactotropes ou lésions suprahypophysaires: l'hyperprolactinémie résulte de l'interruption de la voie tubéro-infundibulaire par une lésion tumorale (craniopharyngiome, méningiome, adénome hypophysaire non-sécrétant,...), infiltrative (sarcoïdose, histiocytose X,...) ou mécanique (arachnoïdocèle, séquelles de traumatisme ou de radiothérapie,...) - hyperprolactinémies iatrogènes : très banales, elles sont le fait d'un grand nombre de médicaments antidopaminergiques (neuroleptiques, antidépresseurs, antiémétiques,...) ou oestrogéniques (contraceptifs oraux,...). - hyperprolactinémies d'accompagnement: une hyperprolactinémie peut être associée à une hypothyroïdie périphérique, à une dystrophie ovarienne polykystique, à une insuffisance rénale chronique, à certains traumatismes thoraciques. - macroprolactinémies, correspondant à un excès de formes lourdes de PRL, liées à des autoanticorps antiprolactine sans retentissement pathologique. 3 - Devant une thyrotoxicose: Les causes périphériques seront en pratique éliminées par les dosages biologiques de base qui montrent une TSH non freinée en regard de valeurs élevées d'hormones thyroïdiennes (T3 et/ou T4). Devant un tel profil biologique, on peut discuter l'éventualité d'autoanticorps anti-hormones thyroïdiennes ou, surtout en cas de contexte familial, un rare syndrome de résistance aux hormones thyroïdiennes 4 - Devant un hypercorticisme La détermination du caractère ACTH-dépendant ou non de l’hypercorticisme repose sur le dosage immunoradiométrique de l’ACTH. En regard d’une cortisolémie supérieure à 15 µg/dl (415 nmol/l), un taux d’ACTH inférieur à 5 pg/ml (1,1 pmol/l) signe l’origine surrénalienne de l’hypersécrétion de cortisol, qui freine l’ACTH. Il faut alors rechercher une masse surrénalienne par un scanner ou une IRM des surrénales. Dans les syndromes de Cushing ACTH-dépendants, cet examen montre un aspect typique d’hyperplasie surrénalienne bilatérale dans seulement 70 % des cas. Si le dosage d’ACTH est en faveur d’une tumeur à ACTH, il faut déterminer si celle-ci est hypophysaire ou ectopique. Classiquement, la résistance à l’inhibition par les glucocorticoïdes étant partielle dans les adénomes corticotropes et totale dans les tumeurs ectopiques, on utilise pour les différencier le test à la dexaméthasone fort, ou “ test de Liddle ” fort (8 mg par jour à raison de 2 mg toutes les 6 heures pendant 48 heures). De même, il existe typiquement une non réponse de l’ACTH au CRH en cas de sécrétion ectopique, alors que le test est positif dans la maladie de Cushing d’origine hypophysaire. En l’absence de visualisation d’une image hypophysaire par l’IRM, on peut réaliser un cathétérisme des sinus pétreux pour s’assurer de l’origine hypophysaire de l’hypersécrétion d’ACTH. Traitement Le traitement fait appel à une prise en charge multidisciplinaire spécialisée La thérapeutique dépend du type et de la taille de l’adénome. Les microprolactinomes relèvent soit d’un traitement chirurgical qui peut seul être curateur, soit d’un traitement médical dopaminergique au long cours. Du fait de leur très faible évolutivité, sous réserve d’une surveillance régulière, ils ne représentent plus une contre-indication absolue à une contraception oestroprogestative. Les macroprolactinomes doivent être traités en premier par dopaminergiques, la chirurgie étant réservée, sauf urgence compressive, aux cas de résistance ou d’intolérance au traitement médical. Les adénomes somatotropes ou thyréotropes ou corticotropes relèvent toujours d’un abord transsphénoïdal lorsqu’il est possible, éventuellement précédé d’un traitement médical de quelques mois. Dans l’acromégalie, en cas d’hypersécrétion résiduelle de GH, le traitement médical fait appel aux formes retard des somatostatinergiques, et beaucoup plus rarement aux dopaminergiques. Un antagoniste de la GH (pegvisomant) sera prochainement disponible en cas d’échec des somatostatinergiques. Dans la maladie de Cushing non guérie par une intervention hypophysaire initiale, un traitement anticortisolique au long cours est parfois proposé. Une hypophysectomie totale, une radiothérapie hypophysaire ou une surrénalectomie endoscopique bilatérale peuvent cependant constituer

©Collège des Enseignants d’Endocrinologie, Diabète et Maladies Métaboliques. Mise à jour décembre 2004
www.endocrino.net Page 11 sur 11
une alternative, sachant que la correction de l’hypercorticisme représente une priorité thérapeutique. Les adénomes gonadotropes et non sécrétants relèvent d’une exérèse chirurgicale lorsqu’ils sont volumineux, et plus rarement d’une simple surveillance. La radiothérapie est surtout utilisée comme traitement complémentaire d’un volumineux résidu post-chirurgical dans les macroprolactinomes résistants, les adénomes somatotropes, thyréotropes ou cliniquement non fonctionnels. Certains plus petits résidus sécrétants seront traités par approche stéréotaxique ou Gamma-Unit.














