Embed Size (px)
Citation preview

TECHNIQUE INSTRUMENTALERT
ICLE
S
46 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
A
RMN
2DRÉSUMÉLa RMN à deux dimensions (2D) présente un fort potentiel pour l’analyse quantitative de mélanges complexes, en raison de sa capacité à réduire le recouvrement spectral. Deux aspects majeurs doivent toutefois être pris en compte pour l’obtention de résultats justes et précis par RMN 2D quantitative. Le volume des pics dépend d’un grand nombre de facteurs qui rendent indispensable le développement d’approches correctives ou de calibration. D’autre part, la durée des expériences de RMN 2D limite leur application et affecte leur performance quantitative. Heureusement, la combinaison d’approches rigoureuses de chimie analytique à des développements méthodologiques innovants permet d’obtenir aujourd’hui des résultats répétables et reproductibles dans un temps expérimental compatible avec l’analyse de routine. Cet article présente quelques-uns des développements les plus récents dans ce domaine, illustrés par leur application à l’étude d’échantillons complexes pour lesquels la RMN 1D se révèle insuffi sante.
MOTS-CLÉSRMN 2D - Analyse quantitative - RMN ultrarapide – Calibrage - Ajouts dosés - M3S - Entrelacement
Fast and Ultrafast quantitative 2D NMR
SUMMARYTwo-dimensional (2D) NMR shows a high potential for quantitative analysis of complex mixtures, as it considerably
reduces spectral overlap. However, two major aspects should be considered to obtain accurate and precise results
by quantitative 2D NMR. First, the peak volumes depend on a number of factors which require the use of calibration
or correction strategies. Second, the quantitative performances –and therefore the application– of 2D NMR are
limited by long experiment durations. However, the recent development of meticulous analytical procedures,
associated with novel methodological developments, lead to repeatable and reproducible results in a reasonable
time, compatible with routine analysis. This article highlights some of the most recent developments in this area,
illustrated by their application to complex samples where 1D NMR fails due to spectral overlap.
KEYWORDS 2D NMR - quantitative analysis - ultrafast NMR – Calibration - Standard additions - M3S - Interleaving
La RMN 2D quantitative rapide et ultrarapideSerge AKOKA1 et Patrick GIRAUDEAU1,2
1 EBSI Team, Chimie et Interdisciplinarité : Synthèse, Analyse, Modélisation (CEISAM), Université de Nantes, CNRS, UMR 6230, LUNAM Université, Nantes, France2 Institut Universitaire de France, Paris, France – Tél : 0251125709 – Fax : 02 51 12 57 12 – Email : [email protected]
I - Introduction
La Résonance Magnétique Nucléaire (RMN) est sans doute l’une des méthodes d’analyse les plus polyvalentes car elle off re de nombreuses modalités (état liquide, état solide, in vivo, une ou plusieurs dimensions spectrales, relaxométrie, imagerie) mais aussi parce que ses applications touchent une large gamme de disciplines qui vont de la physique à la médecine en passant par la chimie et la biologie. La RMN est aujourd’hui la méthode de référence pour l’élucidation structurale de molécules de plus en plus complexes. Ses performances en termes de détermination de concentrations sont nettement moins connues alors que cette techni-que spectroscopique présente, de ce point de vue, des avantages indéniables ; c’est en eff et une méthode non destructive et non spécifi que (toutes les espèces présentes peuvent être détectées
simultanément).Dès les premières heures de la RMN, son aspect quantitatif a été mis à profi t pour déterminer le nombre relatif de noyaux sur les diff érents sites d’une molécule (1). La première application de chimie analytique fut proposée, quant à elle, par Hollis et col. dès 1963 (2). Depuis lors la RMN du proton a été régulièrement utilisée à des fi ns quantitatives dans des domaines aussi variés que l’analyse pharmaceutique (3), celle des produits naturels (4), la spectroscopie in vivo (5) ou la métabolomique (6) ; et cette liste est loin d’être exhaustive. Aujourd’hui, le terme de RMN quantitative ne recouvre pas uniquement la détermination de concentrations mais concerne également la mesure de nombreuses grandeurs telles que les teneurs isotopiques (en abondance naturelle ou après enrichissement) ou des paramètres physiques ou chimiques comme le
TECHNIQUE INSTRUMENTALE
47SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
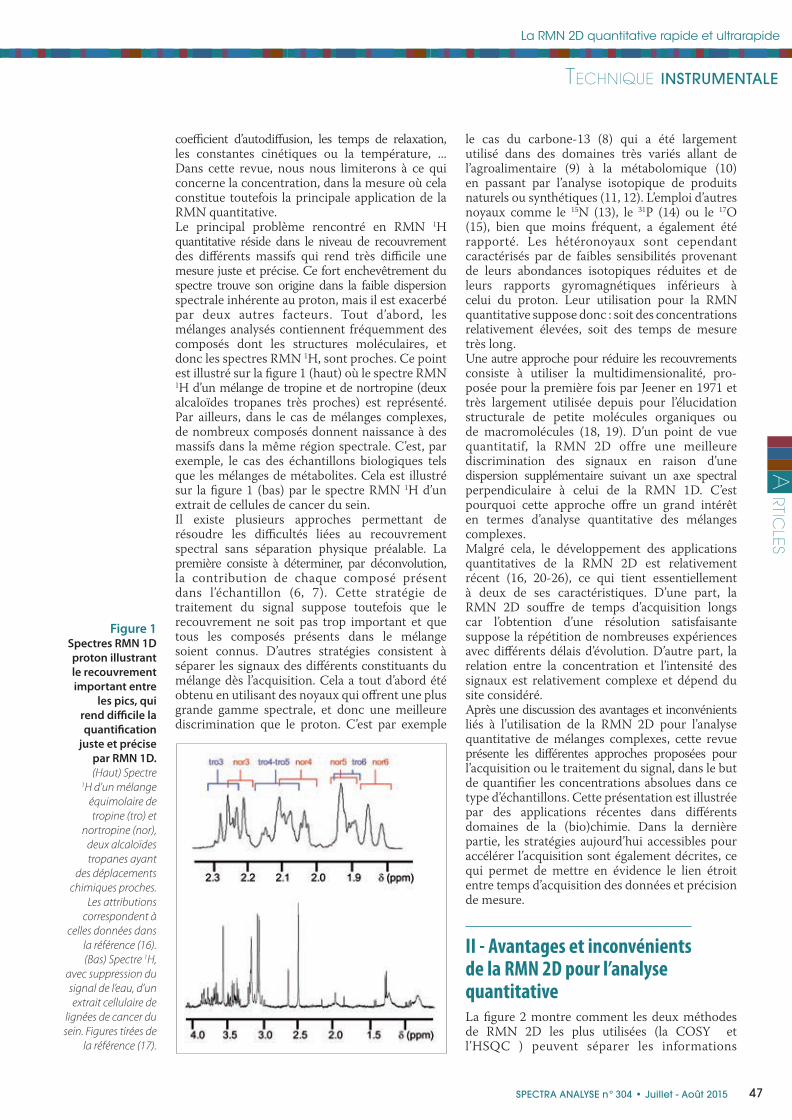
coeffi cient d’autodiff usion, les temps de relaxation, les constantes cinétiques ou la température, ... Dans cette revue, nous nous limiterons à ce qui concerne la concentration, dans la mesure où cela constitue toutefois la principale application de la RMN quantitative.Le principal problème rencontré en RMN 1H quantitative réside dans le niveau de recouvrement des diff érents massifs qui rend très diffi cile une mesure juste et précise. Ce fort enchevêtrement du spectre trouve son origine dans la faible dispersion spectrale inhérente au proton, mais il est exacerbé par deux autres facteurs. Tout d’abord, les mélanges analysés contiennent fréquemment des composés dont les structures moléculaires, et donc les spectres RMN 1H, sont proches. Ce point est illustré sur la fi gure 1 (haut) où le spectre RMN 1H d’un mélange de tropine et de nortropine (deux alcaloïdes tropanes très proches) est représenté. Par ailleurs, dans le cas de mélanges complexes, de nombreux composés donnent naissance à des massifs dans la même région spectrale. C’est, par exemple, le cas des échantillons biologiques tels que les mélanges de métabolites. Cela est illustré sur la fi gure 1 (bas) par le spectre RMN 1H d’un extrait de cellules de cancer du sein.Il existe plusieurs approches permettant de résoudre les diffi cultés liées au recouvrement spectral sans séparation physique préalable. La première consiste à déterminer, par déconvolution, la contribution de chaque composé présent dans l’échantillon (6, 7). Cette stratégie de traitement du signal suppose toutefois que le recouvrement ne soit pas trop important et que tous les composés présents dans le mélange soient connus. D’autres stratégies consistent à séparer les signaux des diff érents constituants du mélange dès l’acquisition. Cela a tout d’abord été obtenu en utilisant des noyaux qui off rent une plus grande gamme spectrale, et donc une meilleure discrimination que le proton. C’est par exemple
le cas du carbone-13 (8) qui a été largement utilisé dans des domaines très variés allant de l’agroalimentaire (9) à la métabolomique (10) en passant par l’analyse isotopique de produits naturels ou synthétiques (11, 12). L’emploi d’autres noyaux comme le 15N (13), le 31P (14) ou le 17O (15), bien que moins fréquent, a également été rapporté. Les hétéronoyaux sont cependant caractérisés par de faibles sensibilités provenant de leurs abondances isotopiques réduites et de leurs rapports gyromagnétiques inférieurs à celui du proton. Leur utilisation pour la RMN quantitative suppose donc : soit des concentrations relativement élevées, soit des temps de mesure très long.Une autre approche pour réduire les recouvrements consiste à utiliser la multidimensionalité, pro-posée pour la première fois par Jeener en 1971 et très largement utilisée depuis pour l’élucidation structurale de petite molécules organiques ou de macromolécules (18, 19). D’un point de vue quantitatif, la RMN 2D offre une meilleure discrimination des signaux en raison d’une dispersion supplémentaire suivant un axe spectral perpendiculaire à celui de la RMN 1D. C’est pourquoi cette approche off re un grand intérêt en termes d’analyse quantitative des mélanges complexes.Malgré cela, le développement des applications quantitatives de la RMN 2D est relativement récent (16, 20-26), ce qui tient essentiellement à deux de ses caractéristiques. D’une part, la RMN 2D souff re de temps d’acquisition longs car l’obtention d’une résolution satisfaisante suppose la répétition de nombreuses expériences avec diff érents délais d’évolution. D’autre part, la relation entre la concentration et l’intensité des signaux est relativement complexe et dépend du site considéré.Après une discussion des avantages et inconvénients liés à l’utilisation de la RMN 2D pour l’analyse quantitative de mélanges complexes, cette revue présente les diff érentes approches proposées pour l’acquisition ou le traitement du signal, dans le but de quantifi er les concentrations absolues dans ce type d’échantillons. Cette présentation est illustrée par des applications récentes dans diff érents domaines de la (bio)chimie. Dans la dernière partie, les stratégies aujourd’hui accessibles pour accélérer l’acquisition sont également décrites, ce qui permet de mettre en évidence le lien étroit entre temps d’acquisition des données et précision de mesure.
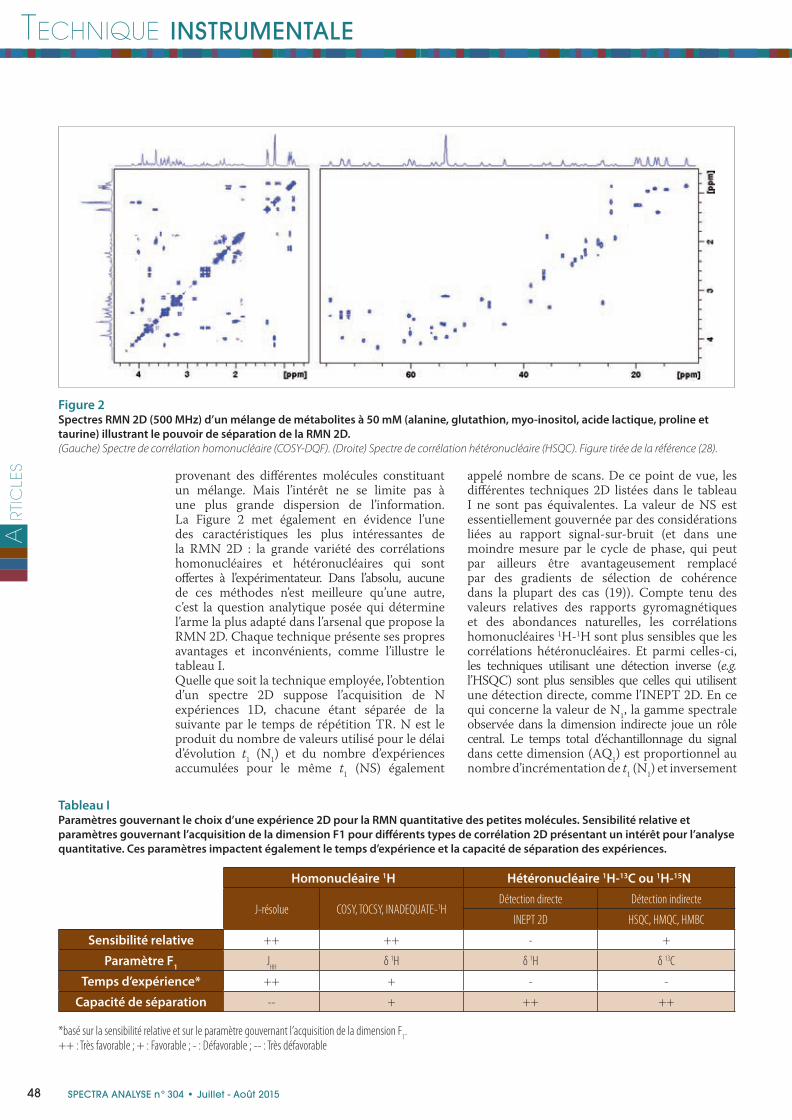
II - Avantages et inconvénients de la RMN 2D pour l’analyse quantitativeLa fi gure 2 montre comment les deux méthodes de RMN 2D les plus utilisées (la COSY et l’HSQC ) peuvent séparer les informations
Figure 1Spectres RMN 1D
proton illustrant
le recouvrement
important entre
les pics, qui
rend diffi cile la
quantifi cation
juste et précise
par RMN 1D.
(Haut) Spectre 1H d’un mélange
équimolaire de
tropine (tro) et
nortropine (nor),
deux alcaloïdes
tropanes ayant
des déplacements
chimiques proches.
Les attributions
correspondent à
celles données dans
la référence (16).
(Bas) Spectre 1H,
avec suppression du
signal de l’eau, d’un
extrait cellulaire de
lignées de cancer du
sein. Figures tirées de
la référence (17).
TECHNIQUE INSTRUMENTALE
48 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
provenant des diff érentes molécules constituant un mélange. Mais l’intérêt ne se limite pas à une plus grande dispersion de l’information. La Figure 2 met également en évidence l’une des caractéristiques les plus intéressantes de la RMN 2D : la grande variété des corrélations homonucléaires et hétéronucléaires qui sont off ertes à l’expérimentateur. Dans l’absolu, aucune de ces méthodes n’est meilleure qu’une autre, c’est la question analytique posée qui détermine l’arme la plus adapté dans l’arsenal que propose la RMN 2D. Chaque technique présente ses propres avantages et inconvénients, comme l’illustre le tableau I.Quelle que soit la technique employée, l’obtention d’un spectre 2D suppose l’acquisition de N expériences 1D, chacune étant séparée de la suivante par le temps de répétition TR. N est le produit du nombre de valeurs utilisé pour le délai d’évolution t
1 (N
1) et du nombre d’expériences
accumulées pour le même t1 (NS) également
appelé nombre de scans. De ce point de vue, les diff érentes techniques 2D listées dans le tableau I ne sont pas équivalentes. La valeur de NS est essentiellement gouvernée par des considérations liées au rapport signal-sur-bruit (et dans une moindre mesure par le cycle de phase, qui peut par ailleurs être avantageusement remplacé par des gradients de sélection de cohérence dans la plupart des cas (19)). Compte tenu des valeurs relatives des rapports gyromagnétiques et des abondances naturelles, les corrélations homonucléaires 1H-1H sont plus sensibles que les corrélations hétéronucléaires. Et parmi celles-ci, les techniques utilisant une détection inverse (e.g. l’HSQC) sont plus sensibles que celles qui utilisent une détection directe, comme l’INEPT 2D. En ce qui concerne la valeur de N
1, la gamme spectrale
observée dans la dimension indirecte joue un rôle central. Le temps total d’échantillonnage du signal dans cette dimension (AQ
1) est proportionnel au
nombre d’incrémentation de t1 (N
1) et inversement
Figure 2Spectres RMN 2D (500 MHz) d’un mélange de métabolites à 50 mM (alanine, glutathion, myo-inositol, acide lactique, proline et
taurine) illustrant le pouvoir de séparation de la RMN 2D.
(Gauche) Spectre de corrélation homonucléaire (COSY-DQF). (Droite) Spectre de corrélation hétéronucléaire (HSQC). Figure tirée de la référence (28).
Homonucléaire 1H Hétéronucléaire 1H-13C ou 1H-15N
J-résolue COSY, TOCSY, INADEQUATE-1HDétection directe Détection indirecte
INEPT 2D HSQC, HMQC, HMBC
Sensibilité relative ++ ++ - +
Paramètre F1
JHH
δ 1H δ 1H δ 13C
Temps d’expérience* ++ + - -
Capacité de séparation -- + ++ ++
*basé sur la sensibilité relative et sur le paramètre gouvernant l’acquisition de la dimension F1.
++ : Très favorable ; + : Favorable ; - : Défavorable ; -- : Très défavorable
Tableau IParamètres gouvernant le choix d’une expérience 2D pour la RMN quantitative des petites molécules. Sensibilité relative et
paramètres gouvernant l’acquisition de la dimension F1 pour diff érents types de corrélation 2D présentant un intérêt pour l’analyse
quantitative. Ces paramètres impactent également le temps d’expérience et la capacité de séparation des expériences.

TECHNIQUE INSTRUMENTALE
49SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
proportionnel à la gamme spectrale observée (SW
1). Ainsi, pour une valeur donnée de AQ
1,
N1 est d’autant plus grand que SW
1 est grande.
Par ailleurs, une valeur trop petite de AQ1 induit
des artéfacts de troncature dans la dimension indirecte (27). La durée d’une expérience de RMN 2D dépend donc du paramètre spectral qui doit être échantillonné dans la dimension indirecte. L’observation des couplages homonucléaires 1H-1H nécessite un nombre plus faible de points (N
1)
que l’observation des déplacements chimiques 1H, qui à son tour impose un N
1 plus petit que
l’échantillonnage des déplacements chimiques 13C. Une expérience J-résolue 1H sera donc plus courte qu’une COSY 1H-1H qui prendra elle-même moins de temps qu’une HSQC 1H-13C. In fi ne, la durée d’expérience est donc gouvernée par la sensibilité intrinsèque de la séquence utilisée et par le paramètre spectral observé dans la direction indirecte. Il est également essentiel de prendre en compte la capacité de l’expérience à séparer des résonances proches. Cette caractéristique augmente avec un accroissement de la gamme spectrale et est donc inversement proportionnelle au temps nécessaire pour l’acquisition des données.L’une des principales limitations de la RMN 2D réside donc dans le temps de mesure relativement long. L’impact de cette succession de nombreuses
expériences est particulièrement sévère dans le cas d’acquisitions quantitatives car le temps de répétition (qui sépare deux expériences consécutives) doit être grand devant les T
1
afi n de permettre une relaxation complète. Un temps d’acquisition des données aussi long a plusieurs conséquences. La plus immédiate est un engorgement des spectromètres et une explosion du coût de l’analyse. D’un point de vue plus fondamental, cela rend impossible l’observation d’échantillons dont la composition évolue en raison d’une cinétique chimique ou biochimique par exemple. La dernière conséquence est peut-être moins évidente mais n’en a pas moins un impact signifi catif d’un point de vue analytique. Plus le temps d’acquisition d’une expérience RMN 2D est long et plus celle-ci est exposée aux instabilités de l’appareillage (29, 30). Cela inclut notamment des variations électroniques (angle de nutation, phases, gain du récepteur, etc) mais aussi les instabilités de champ magnétique à l’intérieur ou à l’extérieur de l’aimant. Ces petites variations ont un impact négligeable dans la dimension directe, comme en RMN 1D, car l’ensemble des points d’un FID (décroissance libre de l’induction) est acquis dans un intervalle de temps très court ; mais il en va tout autrement dans la dimension indirecte où ces variations sont responsables d’un bruit additionnel en raison du temps relativement
Figure 3Stratégies permettant d’accéder à des concentrations absolues à partir de spectres RMN 2D.
Le volume des pics 2D dépend d’un certain nombre de facteurs (temps de relaxation, constantes de couplage, délais, angles
d’impulsions, eff ets d’off -résonance, etc.) et le facteur k refl ète cette dépendance. p est le nombre de spins équivalents par
site, [c] est la concentration à déterminer, et VS est le volume sensible sur lequel la détection est opérée. La première stratégie
(fl èches de gauche) permet de rendre k indépendant des facteurs susmentionnés en modifi ant la séquence d’impulsions. Les
deux autres stratégies reposent sur des calculs théoriques (fl èches de droite) ou des approches de calibrage (fl èches du centre)
afi n de déterminer la valeur de k pour chaque pic d’intérêt. Figure tirée de la référence (17).

TECHNIQUE INSTRUMENTALE
50 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
long qui sépare l’acquisition de deux points consécutifs du FID (temps de recyclage : TR). En raison de ce « bruit t
1 », le rapport signal-sur-bruit
est toujours inférieur dans la dimension indirecte (F
1) par rapport à la dimension directe (F
2). Le
« bruit t1 » est très dépendant de la confi guration
et de la génération du spectromètre, il peut signifi cativement aff ecter la précision d’expérience 2D quantitative (28, 31), et donc la concentration limite qui peut être mesurée avec la précision cible. La dernière partie de cet article sera consacrée à la manière dont le spectroscopiste peut composer avec cette contrainte temporelle pour améliorer à la fois le temps de mesure et la précision des résultats obtenus.Par ailleurs, quelle que soit la séquence utilisée pour une acquisition de RMN 2D, il s’agit inévitablement d’une séquence multi-impulsionnelle et, de ce fait, le volume sous les raies dépend de nombreux paramètres spécifi ques de la résonance considérée : temps de relaxation longitudinale et transversale, valeurs relatives des constantes de couplage (homo ou hétéronucléaire) par rapport aux délais de la séquence, angle de nutation des impulsions RF, off -résonance, etc (32). La situation est donc plus complexe que dans le cas d’une acquisition 1D après une simple impulsion, où la surface des raies est directement proportionnelle au nombre de noyaux équivalents et à la concentration, pourvu que la saturation partielle soit évitée. C’est la raison pour laquelle la RMN 2D est souvent cataloguée, un peu rapidement, comme « non-quantitative » ; de nombreux spectroscopistes considérant qu’elle ne peut pas être utilisée pour déterminer une concentration. Fort heureusement, diff érentes stratégies ont été proposées pour contourner cet inconvénient, et elles seront décrites dans la suite de cet article.L’aspect multiparamétrique de la RMN 2D rend impossible la formulation d’une recette universelle pour le choix de la meilleure technique à des fi ns d’analyse quantitative. Ce choix est en particulier éminemment dépendant de l’échantillon ; alors qu’une acquisition J-résolue homonucléaire sera suffi sante pour quantifi er toutes les résonances d’un mélange avec peu de composés (16), l’analyse d’un mélange complexe nécessitera une HSQC 1H-13C (22) et impliquera donc un temps de mesure plus important. Les dix dernières années ont été l’occasion d’eff orts considérables pour contourner les deux contraintes mentionnées plus haut : temps de mesure et dépendance du signal 2D à de nombreux paramètres spécifi ques de la raie considérée. La plupart des solutions proposées sont en fait applicables quelle que soit la technique RMN utilisée, c’est pourquoi nous avons choisi de les présenter du point de vue analytique plutôt qu’en partant de la séquence d’impulsion utilisée pour proposer telle ou telle amélioration.
III - Approches analytiques de détermination de concentration par RMN 2D
1. Approches de calibrage et d’ajouts dosésComme décrit plus haut, les volumes des pics en RMN 2D dépendent de nombreux facteurs (32), liés à la séquence d’impulsions (délais, angles, eff ets d’off -résonance) ou au système de spins étudié (constantes de couplage et temps de relaxation). Les temps de relaxation sont eux-mêmes dépendants de facteurs externes tels que la température, le pH (milieux aqueux) ou le champ B
0. La Figure 3 présente les principales approches
permettant de s’aff ranchir de ces dépendances afi n de déterminer la concentration absolue des analytes. De manière générale, le volume des pics peut s’exprimer en fonction de la concentration [c] de l’analyte ciblé, de la manière suivante :
V = k(T1, T
2, J
CH, J
HH, délais…)·p·[c]·V
S
Dans cette expression, p est le nombre de spins équivalents (connu a priori) et VS est le volume sensible, en principe identique pour tous les spins et pour une confi guration matérielle donnée. Le coeffi cient de proportionnalité k dépend, de manière complexe, des paramètres cités plus haut. Le coeffi cient k est fortement site-spécifi que en RMN 2D, c’est-à-dire qu’il est diff érent pour chaque pic du spectre. Afi n de réaliser des mesures quantitatives, il est donc indispensable de déterminer ce coeffi cient pour chaque pic d’intérêt, ou de s’aff ranchir de son caractère site-spécifi que. Trois familles d’approches ont été décrites dans ce but. La première stratégie consiste à faire en sorte que le coeffi cient de proportionnalité soit identique pour tous les pics du spectre. Cette approche, développée notamment par Koskela et col. (20, 32), permet en principe une quantification absolue directe à partir des spectres 2D. Cependant, elle est peu appliquée en pratique car ses performances sont limitées. Les séquences d’impulsions (basées sur l’expérience HSQC) qui ont été proposées pour la mettre en œuvre, ne parviennent que partiellement à atteindre leur objectif. Mentionnons également l’approche HSQC
0, récemment développée par Markley et
col., qui pourrait améliorer signifi cativement les performances de cette stratégie (33).Une seconde approche consiste à calculer le coeffi cient k de chaque pic d’intérêt, à partir de leur expression théorique dépendant des constantes de couplage et des temps de relaxation (34, 35). L’applicabilité de ces méthodes semble toutefois relativement limitée par la nécessité d’une connaissance préalable approfondie des paramètres de relaxation et de couplage de l’échantillon étudié, pouvant s’avérer très fastidieuse, voire impossible, dans le cas d’un
TECHNIQUE INSTRUMENTALE
51SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
mélange complexe.La troisième famille d’approches repose sur l’utilisation d’une procédure de calibrage, de manière similaire aux approches employées pour de nombreuses autres techniques analytiques comme la chromatographie ou les spectroscopies optiques. Seule cette approche est détaillée ici, car elle est de loin la plus utilisée en pratique, ses performances surpassant largement celles des autres méthodes. La méthode la plus simple consiste à enregistrer au préalable une série de courbes de calibrage pour tous les pics d’intérêt, en enregistrant les spectres 2D d’une série d’échantillons modèles contenant les analytes cibles en concentration connue, comme cela se fait dans les dosages par spectroscopie UV ou IR. De récentes études ont démontré l’excellente performance de cette approche en termes de justesse, précision et linéarité. Elle présente l’avantage de ne nécessiter qu’une série d’expériences de calibration, quel que soit le nombre d’échantillons à analyser. Cette approche a été appliquée avec succès à la quantifi cation absolue des métabolites les plus abondants dans des échantillons biologiques, en utilisant notamment l’expérience HSQC 1H-13C (22, 36, 37). Une approche similaire a permis la quantifi cation des composés organiques dans le lait (38). Une justesse et une précision de quelques pourcents ont été reportées pour cette méthode parfaitement adaptée à la quantifi cation simultanée de composés multiples dans un grand nombre d’échantillons.L’approche de calibrage externe souff re toutefois d’inévitables diff érences entre les échantillons utilisés pour le calibrage et les échantillons d’intérêt. C’est notamment le cas pour les échantillons biologiques, dont le milieu est
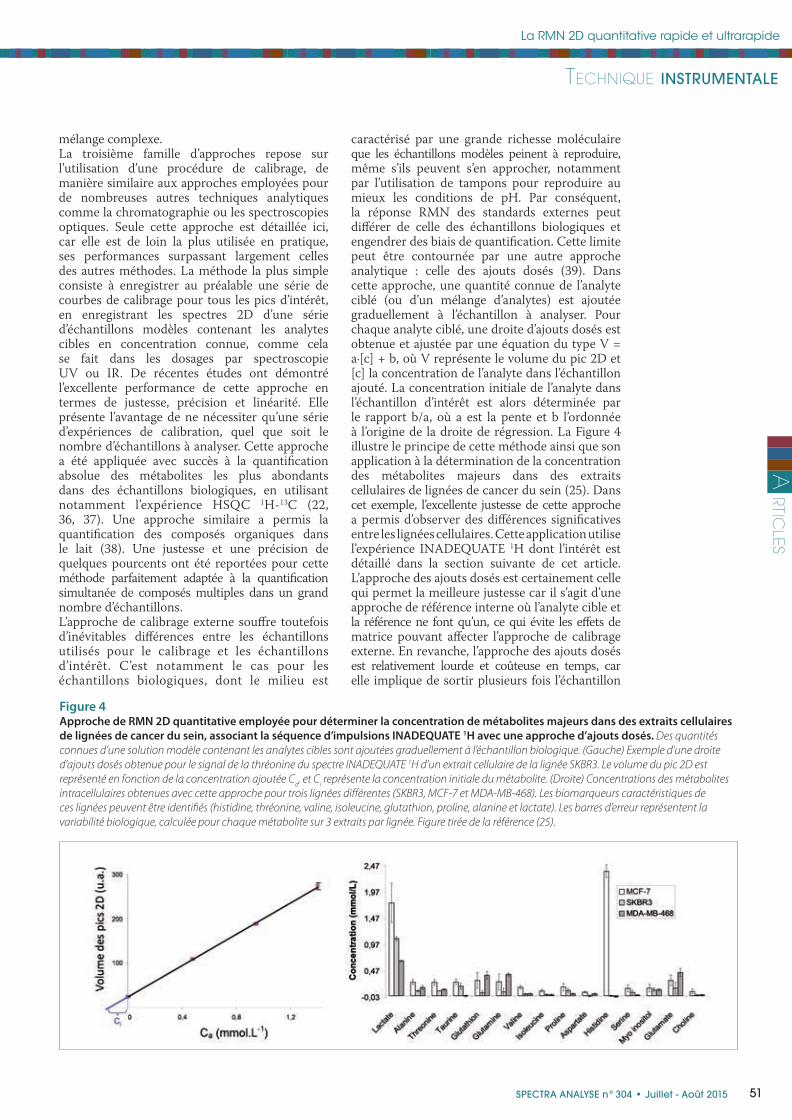
caractérisé par une grande richesse moléculaire que les échantillons modèles peinent à reproduire, même s’ils peuvent s’en approcher, notamment par l’utilisation de tampons pour reproduire au mieux les conditions de pH. Par conséquent, la réponse RMN des standards externes peut diff érer de celle des échantillons biologiques et engendrer des biais de quantifi cation. Cette limite peut être contournée par une autre approche analytique : celle des ajouts dosés (39). Dans cette approche, une quantité connue de l’analyte ciblé (ou d’un mélange d’analytes) est ajoutée graduellement à l’échantillon à analyser. Pour chaque analyte ciblé, une droite d’ajouts dosés est obtenue et ajustée par une équation du type V = a·[c] + b, où V représente le volume du pic 2D et [c] la concentration de l’analyte dans l’échantillon ajouté. La concentration initiale de l’analyte dans l’échantillon d’intérêt est alors déterminée par le rapport b/a, où a est la pente et b l’ordonnée à l’origine de la droite de régression. La Figure 4 illustre le principe de cette méthode ainsi que son application à la détermination de la concentration des métabolites majeurs dans des extraits cellulaires de lignées de cancer du sein (25). Dans cet exemple, l’excellente justesse de cette approche a permis d’observer des diff érences signifi catives entre les lignées cellulaires. Cette application utilise l’expérience INADEQUATE 1H dont l’intérêt est détaillé dans la section suivante de cet article. L’approche des ajouts dosés est certainement celle qui permet la meilleure justesse car il s’agit d’une approche de référence interne où l’analyte cible et la référence ne font qu’un, ce qui évite les eff ets de matrice pouvant aff ecter l’approche de calibrage externe. En revanche, l’approche des ajouts dosés est relativement lourde et coûteuse en temps, car elle implique de sortir plusieurs fois l’échantillon
Figure 4Approche de RMN 2D quantitative employée pour déterminer la concentration de métabolites majeurs dans des extraits cellulaires
de lignées de cancer du sein, associant la séquence d’impulsions INADEQUATE 1H avec une approche d’ajouts dosés. Des quantités
connues d’une solution modèle contenant les analytes cibles sont ajoutées graduellement à l’échantillon biologique. (Gauche) Exemple d’une droite
d’ajouts dosés obtenue pour le signal de la thréonine du spectre INADEQUATE 1H d’un extrait cellulaire de la lignée SKBR3. Le volume du pic 2D est
représenté en fonction de la concentration ajoutée Ca, et C
i représente la concentration initiale du métabolite. (Droite) Concentrations des métabolites
intracellulaires obtenues avec cette approche pour trois lignées diff érentes (SKBR3, MCF-7 et MDA-MB-468). Les biomarqueurs caractéristiques de
ces lignées peuvent être identifi és (histidine, thréonine, valine, isoleucine, glutathion, proline, alanine et lactate). Les barres d’erreur représentent la
variabilité biologique, calculée pour chaque métabolite sur 3 extraits par lignée. Figure tirée de la référence (25).

TECHNIQUE INSTRUMENTALE
52 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
du spectromètre afi n de procéder aux ajouts, et de répéter cette procédure pour chaque échantillon à analyser. Elle est donc incompatible avec les études à haut-débit impliquant un grand nombre d’échantillons. Dans ce dernier cas, l’approche de calibrage externe est sans doute préférable, même si elle implique une réduction de la justesse et de la précision.Les approches de calibrage externe et d’ajouts dosés reposent sur l’hypothèse que le facteur de réponse du pic 2D, et notamment les temps de relaxation, ne varient pas avec la concentration. Cette hypothèse semble raisonnable pour les petites molécules (23), et rend possible l’acquisition des spectres 2D en conditions de saturation partielle. Toutefois, une étude récente a montré que pour des molécules de haut poids moléculaire, la réponse du signal en RMN 2D ne varie pas toujours linéairement avec la concentration (40). Cela implique donc l’utilisation de courbes de calibrage non-linéaires, qui permettent d’atteindre une très bonne justesse mais rendent impossible l’utilisation d’une procédure d’ajouts dosés. Une attention toute particulière doit donc être portée à la linéarité de la réponse RMN sur la gamme de concentrations étudiées, et une étude préliminaire sur un cas modèle peut permettre de s’en assurer.Les applications récentes mentionnées ci-dessus démontrent l’effi cacité des approches analytiques pour obtenir des résultats quantitatifs à partir de spectres de RMN 2D. Ces approches présentent l’avantage d’être indépendantes de la séquence d’impulsions, ce qui permet de choisir la séquence la mieux adaptée au problème étudié indépendamment de la stratégie de quantifi cation choisie. Toutefois, les approches de calibrage et d’ajouts dosés requièrent de multiples expériences et peuvent donc être relativement coûteuses en temps. L’optimisation du temps d’expérience, décrite plus loin, est donc un facteur clé dans le développement d’approches quantitatives par RMN 2D.
2. Traitement du signal
Indépendamment de la séquence d’impulsions et de l’approche analytique employées, les conditions de traitement du signal et d’intégration jouent un rôle majeur dans la précision et la justesse des mesures quantitatives en RMN, et la RMN 2D ne fait pas exception. Les paramètres de traitement optimaux dépendent de la séquence d’impulsions et des paramètres d’acquisition, et il est impossible de lister ici des conditions de traitement génériques (17, 28). Toutefois, il est utile de garder à l’esprit un certain nombre de considérations générales permettant de minimiser l’impact des paramètres de traitement sur la quantitativité des résultats. Un premier point important porte sur le choix des fonctions d’apodisation : celles-ci doivent préserver au mieux le volume des pics 2D d’intérêt, eux-mêmes déterminés par l’amplitude maximale des signaux temporels correspondants .
La fonction d’apodisation doit donc être optimisée en fonction du nombre de points du FID, notamment dans la dimension F1 où celui-ci est généralement tronqué. Pour cela, la fonction d’apodisation doit respecter les critères suivants : son maximum doit coïncider avec le maximum du signal temporel donnant naissance au(x) pic(s) d’intérêt, et son amplitude doit décroître à zéro à la fi n du FID pour éviter la troncature de ce dernier.La détermination du volume des pics en RMN 2D est également très sensible aux distorsions de ligne de base dans les deux dimensions . La plupart des corrections de ligne de base sont appliquées dans le domaine fréquentiel en modélisant la ligne de base par une fonction polynômiale ou spline (41, 42). Des travaux récents ont mis en évidence l’impact majeur de la correction de ligne de base sur la précision des mesures quantitatives. Les résultats obtenus, qui ne sont pas forcément spécifi ques à la RMN 2D, montrent que la correction est généralement plus effi cace lorsqu’elle est restreinte à la zone contenant les pics d’intérêt, et lorsque le degré du polynôme employé pour la correction est inférieur ou égal à trois (16, 23). Lorsque le degré de correction de ligne de base est plus élevé, l’algorithme de correction risque de considérer certains pics larges comme faisant partie de la ligne de base, entraînant une distorsion des pics correspondants.Il est également utile de mentionner l’impact des procédures de traitement automatisées telles que la symétrisation (pour les spectres homonucléaires) ou le « tilt » (dans le cas de spectres J-résolus). Bien que ces procédures permettent d’améliorer la qualité des spectres homonucléaires et de faciliter l’élucidation structurale, elles peuvent être génératrices d’artéfacts (27); de plus, elles modifi ent le volume des pics 2D et peuvent considérablement affecter la précision des mesures (28). Ces approches doivent donc être évitées en RMN 2D quantitative.Enfi n, les méthodes utilisées pour déterminer le volume des pics 2D jouent également un rôle très important dans l’implémentation d’une stratégie de quantifi cation répétable et reproductible. Plusieurs méthodes d’intégration des pics 2D ont été proposées et sont actuellement utilisées. La plus simple est l’addition pure et simple de l’intensité des points situés à l’intérieur d’une zone d’intégration défi nie par l’utilisateur. Cette approche ne nécessite aucune connaissance préalable de la forme des pics, et des outils d’intégration 2D sont inclus dans la plupart des logiciels commerciaux. Toutefois, cette méthode est sensible aux distorsions de ligne de base, au bruit t
1 et aux signaux résiduels des
pics voisins. Elle est particulièrement délicate en cas de recouvrement entre des pics voisins, bien que Romano et col. aient proposé une méthode permettant de contourner ce problème (43). Une variante de cette approche consiste à réaliser une projection 1D en sommant les lignes contenant le pic d’intérêt. Les aires des pics sont ensuite

TECHNIQUE INSTRUMENTALE
53SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
déterminées comme en RMN 1D, par intégration ou déconvolution (24).Les approches de détermination des volumes 2D les plus prometteuses sont probablement celles reposant sur la déconvolution des pics par une approche d’ajustement à un modèle paramétrique (44). En eff et, ces stratégies tiennent compte du recouvrement spectral et utilisent de manière optimale l’information disponible a priori sur les déplacements chimiques et les largeurs de raies. Plusieurs publications récentes ont démontré l’intérêt de ces approches en RMN 2D quantitative. Dans un contexte biomoléculaire, Chylla et col. ont démontré l’intérêt d’utiliser une approche hybride temps/fréquence, nom-mée « maximum likelihood fi tting » (45). Plus récemment, la même équipe a développé un algorithme nommé FMLR (fast maximum likelihood reconstruction) qui réalise une déconvolution des spectres RMN 2D dans un but quantitatif. Cette approche permet d’obtenir une meilleure justesse (inférieure à 5%) que l’approche d’intégration, et de réduire l’impact de l’opérateur (46). Ces outils de déconvolution apparaissent donc très prometteurs, malheureusement ils ne sont pas encore implémentés au sein d’un logiciel commercial, et présentent également l’inconvénient d’être sensibles à la qualité de la forme des pics.
IV - Réduire le temps d’expérience pour améliorer la précision
Les stratégies analytiques que nous venons de décrire permettent de s’aff ranchir du premier inconvénient de la RMN 2D vis-à-vis de l’analyse quantitative : le caractère site-spécifi que du volume des pics 2D. Cette dernière section est dédiée au second inconvénient de la RMN 2D, la durée d’expérience, et à son impact sur le caractère quantitatif des données. La durée d’expérience élevée qui caractérise la RMN 2D entraîne bien sûr des conséquences matérielles (i.e. une occupation plus importante des spectromètres), alors que du point de vue quantitatif, la conséquence majeure de cette durée est que les expériences 2D sont plus sensibles aux instabilités temporelles de l’appareillage (29, 30). Même si le « bruit t
1 » qui
en résulte n’est pas toujours visible, le rapport S/B est toujours plus faible dans la dimension F
1, ce qui aff ecte la précision des expériences
2D. Le développement d’approches permettant de réduire la durée des expériences 2D est donc indispensable.
1. Optimisation des paramètres
La stratégie la plus simple pour réduire la durée des expériences de RMN 2D quantitative consiste en une optimisation minutieuse des paramètres d’acquisition, afi n de réduire leur durée tout en
préservant (ou en améliorant) leurs performances quantitatives. Une telle optimisation est loin d’être triviale car les paramètres généralement recommandés pour l’analyse de routine ne sont pas toujours ceux qui donnent une performance quantitative optimale. Selon l’expérience considérée (Tableau I), la durée d’expérience peut être réduite en ajustant le nombre N
1
d’incréments dans la dimension indirecte et/ou le nombre de scans (28). La réduction de N
1 impacte
la résolution dans la dimension indirecte, mais des valeurs relativement faibles peuvent être atteintes tout en préservant une séparation suffi sante entre les pics d’intérêt. D’autre part, lorsque la sensibilité n’est pas critique, le nombre de scans peut être réduit à une valeur minimale (souvent 1 ou 2) imposée par le cycle de phase de base. Dans ce cas, le cycle de phase peut être avantageusement remplacé par l’utilisation de gradients permettant de sélectionner le signal d’intérêt , ou encore en ayant recours à des impulsions composites ou adiabatiques (47) afi n de réduire les eff ets des imperfections d’impulsions.De récentes études ont démontré qu’une stratégie d’optimisation soignée permettait d’améliorer signifi cativement la précision des expériences de RMN 2D. Ainsi, nous avons montré que la précision des expériences 2D J-résolue et COSY-DQF peut être améliorée lorsque la durée d’expérience est réduite, grâce à une plus grande immunité des expériences optimisées vis-à-vis des instabilités de l’appareillage (16). Des expériences plus courtes off rent ainsi une meilleure répétabilité, tant que le rapport S/B et la résolution restent suffi sants pour mesurer avec une bonne précision le volume des pics d’intérêt. Dans cette étude, des spectres J-résolu et COSY-DQF d’un mélange équimolaire de tropine et nortropine ont été obtenus en une durée de 2,7 et 12 minutes, respectivement.Plus récemment, nous avons appliqué une approche similaire à l’expérience INADEQUATE 1H (23). Cette séquence d’impulsions, géné-ralement utilisée dans sa version 13C, présente un intérêt en RMN du proton car elle permet d’obtenir des spectres de corrélation homonucléaires dépourvus de pics diagonaux, un avantage majeur lorsque les pics à quantifi er sont proches de la diagonale. Dans cette étude, des spectres INADEQUATE 1H d’un mélange de métabolites ont pu être obtenu en seulement 7 minutes avec une répétabilité inférieure à 2% et une excellente linéarité, pour des concentrations de l’ordre de 100 μmol.L-1 (23). Notons que la présence d’un délai de refocalisation (1/4J
HH) nécessaire à la
création des cohérences double-quanta renforce le caractère site-spécifi que du signal et la nécessité d’une approche de calibrage. Ce protocole rapide a été appliqué avec succès à la détermination et à la comparaison du contenu métabolique de trois lignées cellulaires de cancer du sein exprimant diff érents récepteurs (Figure 4) (25).Cette stratégie d’optimisation des paramètres d’acquisition a également été appliquée avec succès
TECHNIQUE INSTRUMENTALE
54 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
en RMN hétéronucléaire. Lewis et col. ont ainsi proposé un protocole rapide de détermination de concentrations dans des mélanges complexes, basé sur une version rapide (12 min) de la séquence HSQC 1H-13C (21). Ce protocole, baptisé FMQ (Fast Metabolite Quantifi cation) et reposant sur une approche de calibrage, a été validé sur un mélange de 26 métabolites, avec une erreur moyenne de 2.7% et une erreur maximale de 10.3% sur les concentrations, contre 16.2% et 44.5% par RMN 1D, respectivement. Le protocole FMQ a ensuite été appliqué à des extraits métaboliques végétaux, permettant de mesurer simultanément la concentration d’environ 40 métabolites, dans une gamme de concentrations allant de 40 μmol.L-1 à 230 μmol.L-1.En conclusion, ces approches d’optimisation des paramètres permettent d’obtenir une bonne précision (généralement inférieure à 10%) en un temps d’expérience raisonnable. Jusqu’ici, de telles approches ont été systématiquement couplées aux stratégies analytiques de calibrage ou d’ajouts dosés décrites plus haut. Dans ce contexte, optimiser le temps d’expérience présente l’avantage de réduire la durée totale d’analyse, impactée par la nécessité d’enregistrer l’ensemble des points de la gamme d’étalonnage. Toutefois, la réduction du temps d’expérience reste limitée par le schéma d’échantillonnage inhérent à la RMN 2D ; mais il est possible de repousser cette limite en utilisant de nouvelles méthodes alternatives pour l’acquisition des spectres de RMN 2D, comme nous allons le voir.
2. La RMN 2D ultrarapide
Au cours des vingt dernières années, de nombreuses stratégies ont été développées pour réduire la durée d’acquisition des spectres multidimensionnels. On peut notamment citer les approches de type SOFAST (band-Selective Optimized-Flip-Angle Short-Transient) (48), le repliement spectral (49), la spectroscopie Hadamard (50) ou l’échantillonage non-uniforme (NUS) (51), cette dernière approche étant souvent associé à des approches novatrices de reconstruction des données. Toutefois, les applications de ces approches dans le domaine quantitatif sont restées limitées à quelques exemples. En particulier, le repliement spectral permet de raccourcir la durée des expériences de RMN 2D quantitative tout en préservant leurs performances (52). Toutefois, une approche rapide récente suscite un intérêt croissant pour l’analyse quantitative : la RMN 2D ultrarapide (RMN 2D UF), proposée en 2002 par L. Frydman et ses collaborateurs (53).La RMN 2D UF est une approche générique permettant d’obtenir tout type de spectre 2D en un seul scan, et donc en une fraction de seconde. Inspirée de l’imagerie, la RMN 2D UF utilise une logique très diff érente de la RMN 2D conventionnelle. Elle consiste à diviser l’échantillon
en N1 sous-ensembles, où les spins subissent une
période d’évolution t1 diff érente, le tout au cours
d’un seul et même scan. Le principe détaillé de la RMN 2D ultrarapide a été abondamment décrit (28, 54, 55) et ne sera pas expliqué ici. Le point clé de cette méthodologie est l’utilisation d’un bloc de codage spatial (combinant des impulsions à fréquence variable à des gradients de champ magnétique), suivi d’une détection analogue au motif EPSI (Echo-Planar Spectroscopic Imaging) utilisé en imagerie rapide. Alors que l’approche initiale souff rait de performances relativement limitées, elle a été considérablement améliorée au cours des dix dernières années, notamment en termes de sensibilité, résolution, largeur spectrale et forme des raies (56-64). De plus, le développement d’outils et de protocoles facilitant l’implémentation de cette technique (54, 65, 66), a permis son application dans de nombreux domaines où le temps d’expérience est un facteur clé, tel que le suivi de processus chimiques (67-69) ou biochimique rapides (70, 71), le couplage avec d’autres techniques (72-74), ou l’acquisition de spectres en champ inhomogène (75). Mais la RMN 2D UF est également un outil prometteur pour l’analyse quantitative rapide de haute précision (76).La première évaluation quantitative de la RMN 2D ultrarapide a été réalisée en 2009 sur une série d’échantillons modèles (76). La répétabilité et la reproductibilité de deux techniques homonucléaires, la spectroscopie J-résolue et la TOCSY, ont été évaluées. Une très bonne précision (1% pour la J-résolue et 7% pour la TOCSY) et une excellente linéarité ont été obtenues pour ces deux méthodes, pour des spectres acquis en
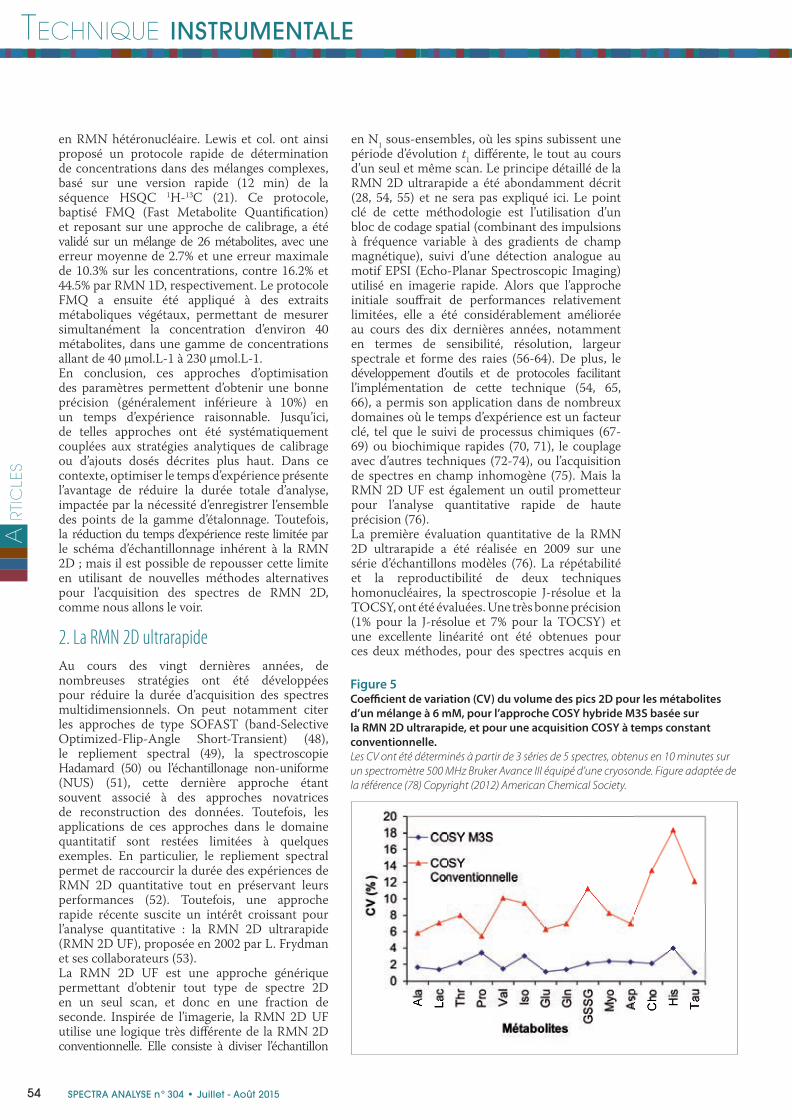
Figure 5Coeffi cient de variation (CV) du volume des pics 2D pour les métabolites
d’un mélange à 6 mM, pour l’approche COSY hybride M3S basée sur
la RMN 2D ultrarapide, et pour une acquisition COSY à temps constant
conventionnelle.
Les CV ont été déterminés à partir de 3 séries de 5 spectres, obtenus en 10 minutes sur
un spectromètre 500 MHz Bruker Avance III équipé d’une cryosonde. Figure adaptée de
la référence (78) Copyright (2012) American Chemical Society.
TECHNIQUE INSTRUMENTALE
55SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
une fraction de seconde. Si cette première étude ouvre des perspectives prometteuses, elle permet également de souligner la sensibilité réduite de la RMN 2D UF par rapport à une acquisition conventionnelle. Cette réduction du rapport signal-sur-bruit est essentiellement liée à la durée d’analyse très courte, et également à des considérations d’échantillonnage du signal (77). D’autre part, la largeur spectrale accessible en un seul scan avec une bonne résolution est également limitée par la performance du système de gradients. Il est donc rapidement apparu que des expériences 2D en un seul scan éteint peu adaptées à l’analyse quantitative de mélanges complexes, où les composés d’intérêt sont souvent présents en faible concentration, et où les pics d’intérêt sont souvent étalés sur une grande gamme spectrale. Des approches hybrides ont alors été développées, consistant à combiner plusieurs expériences ultrarapides pour améliorer leurs performances.Le premier type d’approche hybride proposé est la méthode M3S (Multi-Scan Single Shot), consistant à accumuler plusieurs scans ultrarapides, en répétant plusieurs expériences séparées d’un délai de retour à l’équilibre (31). Bien que cette approche ne soit plus ultrarapide, elle off re une meilleure sensibilité par unité de temps que son équivalent conventionnel, au moins dans le cas de la RMN 2D homonucléaire. Cela est dû à l’absence totale de bruit t
1 en RMN 2D ultrarapide, l’ensemble
de la matrice 2D étant enregistrée en un laps de temps très court. L’approche M3S est donc bien mieux immunisée contre les instabilités de l’appareillage que son équivalent conventionnel. Une conséquence importante est que l’approche M3S off re une performance analytique supérieure à la RMN 2D conventionnelle pour un même temps d’expérience, notamment en termes de répétabilité (78). Comme le montre la fi gure 5,
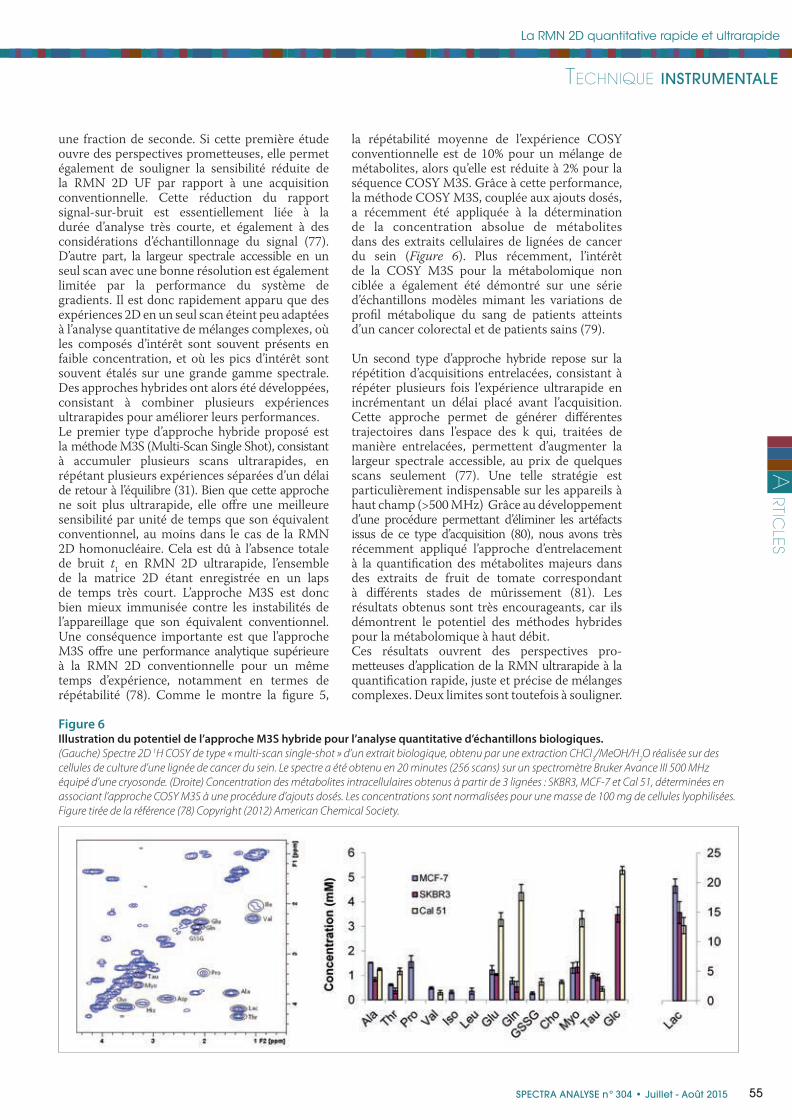
la répétabilité moyenne de l’expérience COSY conventionnelle est de 10% pour un mélange de métabolites, alors qu’elle est réduite à 2% pour la séquence COSY M3S. Grâce à cette performance, la méthode COSY M3S, couplée aux ajouts dosés, a récemment été appliquée à la détermination de la concentration absolue de métabolites dans des extraits cellulaires de lignées de cancer du sein (Figure 6). Plus récemment, l’intérêt de la COSY M3S pour la métabolomique non ciblée a également été démontré sur une série d’échantillons modèles mimant les variations de profi l métabolique du sang de patients atteints d’un cancer colorectal et de patients sains (79).
Un second type d’approche hybride repose sur la répétition d’acquisitions entrelacées, consistant à répéter plusieurs fois l’expérience ultrarapide en incrémentant un délai placé avant l’acquisition. Cette approche permet de générer diff érentes trajectoires dans l’espace des k qui, traitées de manière entrelacées, permettent d’augmenter la largeur spectrale accessible, au prix de quelques scans seulement (77). Une telle stratégie est particulièrement indispensable sur les appareils à haut champ (>500 MHz) Grâce au développement d’une procédure permettant d’éliminer les artéfacts issus de ce type d’acquisition (80), nous avons très récemment appliqué l’approche d’entrelacement à la quantifi cation des métabolites majeurs dans des extraits de fruit de tomate correspondant à diff érents stades de mûrissement (81). Les résultats obtenus sont très encourageants, car ils démontrent le potentiel des méthodes hybrides pour la métabolomique à haut débit.Ces résultats ouvrent des perspectives pro-metteuses d’application de la RMN ultrarapide à la quantifi cation rapide, juste et précise de mélanges complexes. Deux limites sont toutefois à souligner.
Figure 6Illustration du potentiel de l’approche M3S hybride pour l’analyse quantitative d’échantillons biologiques.
(Gauche) Spectre 2D 1H COSY de type « multi-scan single-shot » d’un extrait biologique, obtenu par une extraction CHCl3/MeOH/H
2O réalisée sur des
cellules de culture d’une lignée de cancer du sein. Le spectre a été obtenu en 20 minutes (256 scans) sur un spectromètre Bruker Avance III 500 MHz
équipé d’une cryosonde. (Droite) Concentration des métabolites intracellulaires obtenus à partir de 3 lignées : SKBR3, MCF-7 et Cal 51, déterminées en
associant l’approche COSY M3S à une procédure d’ajouts dosés. Les concentrations sont normalisées pour une masse de 100 mg de cellules lyophilisées.
Figure tirée de la référence (78) Copyright (2012) American Chemical Society.

TECHNIQUE INSTRUMENTALE
56 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
D’une part, les conclusions décrites ci-dessus sont principalement valables pour les techniques ultrarapides homonucléaires, car les approches ultrarapides hétéronucléaires (e.g. HSQC) n’off rent pas, pour le moment, de performances suffi santes pour l’étude d’échantillons dilués tels que des mélanges de métabolites. Les travaux en cours devraient toutefois permettre de rendre ces expériences compétitives. La seconde limite est technologique : même si les approches hybrides décrites ci-dessus sont plus répétables que la RMN 2D conventionnelle, elles restent limitées par le nombre de scans successifs que peut supporter la bobine de gradients. Les applications actuelles sont limitées à quelques dizaines de minutes et sont diffi cilement compatibles avec de très faibles concentrations qui nécessiteraient plusieurs heures d’acquisition. Les limites de concentration de ces approches sont évidemment très dépendantes de la sensibilité de l’appareillage (champ, sonde), mais des valeurs typiques peuvent être trouvées dans la littérature récente (78). Dans le futur, l’amélioration de la performance de ces approches passera certainement par le développement de systèmes de gradients plus robustes, mais également par la combinaison de ces approches hybrides avec d’autres stratégies de réduction du temps d’expérience (82, 83). Notons également que la RMN ultrarapide forme une base prometteuse pour l’acquisition de spectres de dimensionnalité supérieure à 2 en un temps raisonnable, comme l’ont démontré quelques travaux récents (84).
VII - Conclusions
Les travaux récents mentionnés dans cet article soulignent les progrès réalisés dans le développement d’approches quantitatives par RMN 2D. Les approches rapides, couplées à des stratégies analytiques rigoureuses, permettent maintenant d’atteindre une précision et une justesse de quelques pourcents en un temps raisonnable. Le lien entre précision et temps d’expérience est particulièrement intéressant, et souligne l’intérêt de réduire la durée d’expérience, pas seulement pour des raisons matérielles, mais également pour améliorer la précision des mesures.
Dans le domaine quantitatif, la sensibilité, et particulièrement la limite de quantifi cation (LOQ), sont des facteurs clés pour évaluer la performance d’une méthode. Toutefois, ces aspects n’ont volontairement pas été détaillés dans cet article, pour deux raisons. Tout d’abord, le manque de sensibilité n’est pas spécifi que à la RMN 2D : c’est un inconvénient connu de la RMN vis-à-vis d’autres techniques comme la spectrométrie de masse. D’autre part, la LOQ est extrêmement dépendante de l’appareillage et les exemples décrits plus haut ont été obtenus sur des spectromètres, avec des sondes très diff érentes. Comparer la sensibilité d’une méthode appliquée sur un spectromètre 400 MHz équipé d’une sonde à température ambiante, à celle d’une autre méthode appliquée sur un spectromètre 800 MHz équipé d’une cryosonde, n’aurait pas de sens. L’intérêt de travailler à plus haut champ et avec une cryosonde pour être plus sensible tombe sous le sens, et n’est pas spécifi que aux applications quantitatives. Toutefois, les très hauts champs peuvent poser un certain nombre de problèmes, notamment en termes de gamme de découplage, pouvant aff ecter spécifi quement les performances quantitatives. En conclusion, des tests circulaires réalisés sur des échantillons et avec des méthodes identiques, mais sur des spectromètres diff érents, seraient intéressants pour évaluer la robustesse et la reproductibilité des méthodes de RMN 2D quantitative.Enfi n, soulignons que la RMN quantitative bénéfi ciera certainement à terme des avancées récentes dans le domaine de l’hyperpolarisation. En particulier, les approches basées sur le parahydrogène (85) ou la polarisation dynamique nucléaire (DNP) (86) sont particulièrement prometteuses et ont été couplées avec succès aux approches single-scan (72, 87, 88). Ce domaine reste toutefois largement inexploré en termes quantitatifs, probablement car ces approches d’hyperpolari-sation augmentent considérablement la variabilité du signal. Une première évaluation de la répétabilité des expériences de DNP-RMN fait état de coeffi cients de variation de l’ordre de 10% (89). L’obtention de résultats quantitatifs juste et précis en RMN hyperpolarisée nécessitera certainement une optimisation minutieuse des paramètres contrôlant l’hyperpolarisation, et de nombreux développements peuvent être attendus dans ce domaine.
REMERCIEMENTSLes travaux
présentés dans
cet article
mettent en
jeu diverses
collaborations
scientifi ques,
notamment
avec les
membres de
l’équipe EBSI
du laboratoire
CEISAM à
l’Université de
Nantes, que
les auteurs
tiennent à
remercier cha-
leureusement.
Les travaux
présentés ont
notamment
été fi nancés
par l’ANR
(Projet 2010-
JCJC-0804-
01), la Région
Pays de la
Loire (Projet
Résonantes),
et Biogenouest
(plateforme
métabolomique
CORSAIRE).
REFERENCES
(1) JUNGNICKEL J.L., FORBES J.W., Quantitative Measurement of Hydrogen Types by Integrated Nuclear Magnetic Reso-nance Intensities. Anal. Chem., 1963, 35, 938-942.
(2) HOLLIS D.P., Quantitative Analysis of Aspirin, Phenacetin, and Caff eine Mixtures by Nuclear Magnetic Resonance Spec-trometry. Anal. Chem., 1963, 35, 1682-1684.
(3) KWAKYE J.K., Use of NMR for quantitative analysis of phar-maceuticals. Talanta, 1985, 32, 1069-1071.
(4) PAULI G.F., JAKI B.U., LANKIN D.C., Quantitative 1H NMR: De-veloppement and potential of a method for natural products analysis. J. Nat. Prod., 2005, 68, 133-149.
(5) PODO F., HENRIKSEN O., BOVÉE W.M.M.J., LEACJ M.O., LEIBFRITZ D., DE CERTAINES J.D., Absolute metabolite quantifi -cation by in vivo NMR spectroscopy: I. Introduction, objectives and activities of a concerted action in biomedical research. Magn. Reson. Imaging, 1998, 16, 1085-1092.

TECHNIQUE INSTRUMENTALE
57SPECTRA ANALYSE n° 304 • Juillet - Août 2015
La RMN 2D quantitative rapide et ultrarapide
RTICLES
A
(6) WISHART D.S., Quantitative metabolomics using NMR. Trac-Trend. Anal. Chem., 2008, 27, 228-237.
(7) WELJIE A.M., NEWTON J., MERCIER P., CARLSON E., SLUPSKY C.M., Targeted Profi ling: Quantitative Analysis of 1H NMR Metabolomics Data. Anal. Chem., 2006, 78, 4430-4442.
(8) MARECI T.H., SCOTT K.N., Quantitative analysis of mixtures by carbon-13 nuclear magnetic resonance spectroscopy. Anal. Chem., 1977, 49, 2130-2136.
(9) VLAHOV G., Application of NMR to the study of olive oils. Prog. NMR Spec-trosc., 1999, 35, 341-357.
(10) AURSAND M., JORGENSEN L., GRASDALEN H., Quantitative high-reso-lution 13C Nuclear Magnetic Resonance of anserine and lactate in white muscle of Atlantic salmon (salmo salar). Comp. Biochem. Physiol., 1995, 112B, 315-321.
(11) BUSSY U., THIBAUDEAU C., THOMAS F., DESMURS J.-R., JAMIN E., RE-MAUD G., SILVESTRE V., AKOKA S., Isotopic fi nger-printing of active pharma-ceutical ingredients by 13C NMR and polarization transfer techniques as a tool to fi ght against counterfeiting. Talanta, 2011, 85, 1909-1914.
(12) TENAILLEAU E., LANCELIN P., ROBINS R.J., AKOKA S., Authentication of the origin of vanillin using quantitative natural abundance 13C NMR. J. Agric. Food Chem., 2004, 52, 7782-7787.
(13) LEVY G.C., PEHK T., SRINIVASAN P.R., Quantitative 15N NMR spectrosco-py. Org. Magn. Reson., 1980, 14, 129-132.
(14) DAIS P., SPYROS A., 31P NMR spectroscopy in the quality control and authentication of extra-virgin olive oil: A review of recent progress. Magn. Reson. Chem., 2007, 45, 367-377.
(15) LONNON D.G., HOOK J.M., 17O Quantitative Nuclear Magnetic Reso-nance Spectroscopy of Gasoline and Oxygenated Additives. Anal. Chem., 2003, 75, 4659-4666.
(16) GIRAUDEAU P., GUIGNARD N., HILLION H., BAGUET E., AKOKA S., Optimi-zation of homonuclear 2D NMR for fast quantitative analysis: Application to tropine-nortropine mixtures. J. Pharm. Biomed. Anal., 2007, 43, 1243-1248.
(17) GIRAUDEAU P., Quantitative 2D liquid-state NMR. Magn. Reson. Chem., 2014, 52, 259-272.
(18) JEENER J., Lecture presented at Ampere International Summer School II, Basko Polje, Yugoslavia, 1971.
(19) AUE W.P., BARTHOLDI E., ERNST R.R., Two-dimensional spectroscopy. Ap-plication to nuclear magnetic resonance. J. Chem. Phys., 1976, 64, 2229-2246.
(20) KOSKELA H., KILPELÄINEN I., HEIKKINEN S., Some aspects of quantitative 2D NMR. J. Magn. Reson., 2005, 174, 237-244.
(21) LEWIS I.A., KARSTEN R.H., NORTON M.E., TONELLI M., WESTLER W.M., MARKLEY J.L., NMR Method for Measuring Carbon-13 Isotopic Enrichment of Metabolites in Complex Solutions. Anal. Chem., 2010, 82, 4558-4563.
(22) LEWIS I.A., SCHOMMER S.C., HODIS B., ROBB K.A., TONELLI M., WESTLER W., SUSSMAN M., MARKLEY J.L., Method for determining molar concentra-tions of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem., 2007, 79, 9385-9390.
(23) MARTINEAU E., GIRAUDEAU P., TEA I., AKOKA S., Fast and precise quanti-tative analysis of metabolic mixtures by 2D 1H INADEQUATE NMR. J. Pharm. Biomed. Anal., 2011, 54, 252-257.
(24) MASSOU S., NICOLAS C., LETISSE F., PORTAIS J.-C., NMR-based fl uoxo-mics: Quantitative 2D NMR methods for isotopomers analysis. Phytoche-mistry, 2007, 68, 2330-2340.
(25) MARTINEAU E., TEA I., AKOKA S., GIRAUDEAU P., Absolute quantifi cation of metabolites in breast cancer cell extracts by quantitative 2D 1H INADE-QUATE NMR. NMR Biomed., 2012, 25, 985-992.
(26) ZHANG L., GELLERSTEDT G., Quantitative 2D HSQC NMR determination of polymer structures by selecting suitable internal standard references. Magn. Reson. Chem., 2007, 45, 37-45.
(27) ERNST R.R., BODENHAUSEN G., WOKAUN A., Principles of nuclear magnetic resonance in one and two dimensions, Oxford Science Publications ed., Oxford, 1987.
(28) GIRAUDEAU P., AKOKA S., Fast and ultrafast quantitative 2D NMR: vital tools for effi cient metabolomic approaches. Adv. Bot. Res., 2013, 67, 99-158.
(29) MEHLKOPF A.F., KORBEE D., TIGGELMAN T.A., FREEMAN R., Sources of t1 noise in two-dimensional NMR. J. Magn. Reson., 1984, 58, 315-323.
(30) MORRIS G.A., Systematic sources of signal irreproducibility and t1 noise in high fi eld NMR spectrometers. J. Magn. Reson., 1992, 100, 316-328.
(31) PATHAN M., AKOKA S., TEA I., CHARRIER B., GIRAUDEAU P., Multi-scan single shot» quantitative 2D NMR: a valuable alternative to fast conventional quantitative 2D NMR. Analyst, 2011, 136, 3157-3163.
(32) KOSKELA H., Quantitative 2D NMR studies. Annu. Rep. NMR Spectrosc., 2009, 66, 1-31.
(33) HU K., WYCHE T.P., BUGNI T.S., MARKLEY J.L., Selective Quantifi cation by 2D HSQC0 Spectroscopy of Thiocoraline in an Extract from a Sponge-De-rived Verrucosispora sp. J. Nat. Prod., 2011, 74, 2295-2298.
(34) RAI R.K., TRIPATHI P., SINHA N., Quantifi cation of metabolites from two-dimensional Nuclear MAgnetic Resonance spectroscopy: Application to human urine samples. Anal. Chem., 2009, 81, 10232-10238.
(35) BINGOL K., ZHANG F., BRÜSCHWEILER-LI L., BRÜSCHWEILER R., Quantita-tive Analysis of Metabolic Mixtures by Two-Dimensional 13C Constant-Time TOCSY NMR Spectroscopy. Anal. Chem., 2013, 85, 6414-6420.
(36) GOWDA G.A.N., TAYYARI F., YE T., SURYANI Y., WEI S., SHANAIAH N., RAF-TERY D., Quantitative Analysis of Blood Plasma Metabolites Using Isotope Enhanced NMR Methods. Anal. Chem., 2010, 82, 8983-8990.
(37) GRONWALD W., KLEIN M.S., KASPAR H., FAGERER S.R., NURNBERGER N., DETTMER K., BERTSCH T., OEFNER P.J., Urinary Metabolite Quantifi cation Em-ploying 2D NMR Spectroscopy. Anal. Chem., 2008, 80, 9288-9297.
(38) HU F., FURIHATA K., KATO Y., TANOKURA M., Nondestructive quantifi ca-tion of organic compounds in whole milk without pretreatment by two-di-mensional NMR spectroscopy. J. Agric. Food Chem., 2007, 55, 4307-4311.
(39) RAJABZADEH M., Determination of Unknown Concentrations of Sodium Acetate Using the Method of Standard Addition and Proton NMR: An Experiment for the Undergraduate Analytical Chemistry Laboratory. J. Chem. Educ., 2012, 89, 1454-1457.
(40) MARTINEAU E., EL KHANTACHE K., PUPIER M., SEPULCRI P., AKOKA S., GI-RAUDEAU P., Non-linear eff ects in quantitative 2D NMR of polysaccharides: pitfalls and how to avoid them. J. Pharm. Biomed. Anal., 2015, in press, doi: 10.1016/j.jpba.2015.01.056
(41) COBAS J.C., BERNSTEIN M.A., MARTÍN-PASTOR M., TAHOCES P.G., A new general-purpose fully automatic baseline-correction procedure for 1D and 2D NMR data. J. Magn. Reson., 2006, 183, 145-151.
(42) DIETRICH W., RÜDEL C.H., NEUMANN M., Fast and precise automatic ba-seline correction of one and two-dimensional spectra. J. Magn. Reson., 1991, 91, 1-11.
(43) ROMANO R., PARIS D., ACERNESE F., BARONE F., MOTTA A., Fractional volume integration in two-dimensional NMR spectra: CAKE, a Monte Carlo approach. J. Magn. Reson., 2008, 192, 294-301.
(44) MILLER M.I., GREENE A.S., Maximum-likelihood estimation for nuclear magnetic resonance spectroscopy. Journal of Magnetic Resonance, 1989, 83, 525-548.
(45) CHYLLA R.A., VOLKMAN B.F., MARKLEY J.L., Practical Model Fitting Ap-proaches to the Direct Extraction of NMR Parameters Simultaneously from All Dimensions of Multidimensional NMR Spectra. Journal of Biomolecular NMR, 1998, 12, 277-297.
(46) CHYLLA R.A., HU K., ELLINGER J.J., MARKLEY J.L., Deconvolution of Two-Di-mensional NMR Spectra by Fast Maximum Likelihood Reconstruction: Appli-cation to Quantitative Metabolomics. Anal. Chem., 2011, 83, 4871-4880.
(47) KUPCE E., FREEMAN R., Adiabatic pulses for wideband inversion and broadband decoupling. J. Magn. Reson. A, 1995, 115, 273-276.
(48) SCHANDA P., BRUTSCHER B., Very fast two-dimensional NMR spectros-copy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc., 2005, 127, 8014-8015.

TECHNIQUE INSTRUMENTALE
58 SPECTRA ANALYSE n° 304 • Juillet - Août 2015
RTIC
LES
A
(49) JEANNERAT D., High resolution in heteronuclear 1H-13C NMR experi-ments by optimizing spectral aliasing with one-dimensional carbon data. Magn. Reson. Chem., 2003, 41, 3-17.
(50) KUPCE E., FREEMAN R., Two-dimensional Hadamard spectroscopy. J. Magn. Reson, 2003, 162, 300-310.
(51) KAZIMIERCZUK K., STANEK J., ZAWADZKA-KAZIMIERCZUK A., KOŹ-MIŃSKI W., Random sampling in multidimensional NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 420-434.
(52) MARTINEAU E., AKOKA S., BOISSEAU R., DELANOUE B., GIRAUDEAU P., Fast quantitative 1H-13C 2D NMR with very high precision. Anal. Chem., 2013, 85, 4777-4783.
(53) FRYDMAN L., SCHERF T., LUPULESCU A., The acquisition of multidimen-sional NMR spectra within a single scan. Prod. Natl. Acad. Sci. USA, 2002, 99, 15858-15862.
(54) GAL M., FRYDMAN L., Ultrafast Multidimensional NMR: Principles and Practice of Single-scan Methods, in: G.A. Morris, J.W. Emsley (Eds.) Ency-clopedia of Magnetic Resonance, Wiley, Chichester, 2010, pp. 43-60.
(55) TAL A., FRYDMAN L., Single-scan multidimensional magnetic resonance Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 241-292.
(56) GIRAUDEAU P., AKOKA S., Resolution and sensitivity aspects of ultrafast J-resolved 2D NMR spectra. J. Magn. Reson., 2008, 190, 339-345.
(57) GIRAUDEAU P., AKOKA S., A new gradient-controlled method for im-proving the spectral width of ultrafast 2D NMR experiments. J. Magn. Reson., 2010, 205, 171-176.
(58) GIRAUDEAU P., AKOKA S., Sensitivity and lineshape improvement in ul-trafast 2D NMR by optimized apodization in the spatially encoded dimen-sion. Magn. Reson. Chem., 2011, 49, 307-313.
(59) PELUPESSY P., Adiabatic single scan two-dimensional NMR spectrocopy. J Am Chem Soc, 2003, 125, 12345-12350.
(60) PELUPESSY P., DUMA L., BODENHAUSEN G., Improving resolution in single-scan 2D spectroscopy. J. Magn. Reson., 2008, 194, 169-174.
(61) SHROT Y., FRYDMAN L., Spatial encoding strategies for ultrafast multidi-mensional nuclear magnetic resonance. J. Chem. Phys., 2008, 128, 052209.
(62) SHROT Y., FRYDMAN L., Spatial/spectral encoding of the spin interac-tions in ultrafast multidimensional NMR. J. Chem. Phys., 2009, 131, 224516.
(63) TAL A., SHAPIRA B., FRYDMAN L., Single-scan 2D Hadamard NMR spec-troscopy. Angew. Chem. Int. Ed., 2009, 48, 2732-2736.
(64) ROUGER L., LOQUET D., MASSOU S., AKOKA S., GIRAUDEAU P., Limitation of diff usion eff ects in ultrafast 2D NMR by encapsulation of analytes in phos-pholipidic vesicles. ChemPhysChem, 2012, 13, 4124-4127.
(65) PATHAN M., CHARRIER B., TEA I., AKOKA S., GIRAUDEAU P., New practi-cal tools for the implementation and use of ultrafast 2D NMR experiments. Magn Reson Chem, 2013, 51, 168-175.
(66) QUEIROZ JUNIOR L.H.K., FERREIRA A.G., GIRAUDEAU P., Optimization and practical implementation of ultrafast 2D NMR experiments. Quim. Nova., 2013, 26, 577-581.
(67) QUEIROZ JUNIOR L.H.K., GIRAUDEAU P., DOS SANTOS F.A.B., OLIVEIRA K.T., FERREIRA A.G., Real-time mechanistic monitoring of an acetal hydrolysis using ultrafast 2D NMR. Magn. Reson. Chem., 2012, 50, 496-501.
(68) HERRERA A., FERNÁNDEZ-VALLE E., MARTÍNEZ-ÁLVAREZ R., MOLERO D., PARDO Z.D., SÁEZ E., GAL M., Real-Time Monitoring of Organic Reactions with Two-Dimensional Ultrafast TOCSY NMR Spectroscopy13. Angew. Chem. Int. Edit., 2009, 48, 6274-6277.
(69) PARDO Z.D., OLSEN G.L., FERNÁNDEZ-VALLE M.E., FRYDMAN L., MARTÍNEZ-ÁLVAREZ R., HERRERA A., Monitoring Mechanistic Details in the Synthesis of Pyrimidines via Real-Time, Ultrafast Multidimensional NMR Spectroscopy. J. Am. Chem. Soc., 2012, 134, 2706-2715.
(70) CORAZZA A., RENNELLA E., SCHANDA P., MIMMI M.C., CUTUIL T., RAI-MONDI S., GIORGETTI S., FOGOLARI F., VIGLINO P., FRYDMAN L., GAL M., BELLOTTI V., BRUTSCHER B., ESPOSITO G., Native-unlike Long-lived Interme-
diates along the Folding Pathway of the Amyloidogenic Protein b2-Micro-globulin Revealed by Real-time Two-dimensional NMR. J. Biol. Chem., 2010, 285, 5827-5835.
(71) LEE M.-K., GAL M., FRYDMAN L., VARANI G., Real-time multidimensional NMR follows RNA folding with second resolution. Proc.Natl. Acad. Sci. USA, 2010, 107, 9192-9197.
(72) GIRAUDEAU P., SHROT Y., FRYDMAN L., Multiple Ultrafast, Broadband 2D NMR Spectra of Hyperpolarized Natural Products. J. Am. Chem. Soc., 2009, 131, 13902-13903.
(73) QUEIROZ JUNIOR L.H.K., QUEIROZ D.P.K., DHOOGHE L., FERREIRA A.G., GIRAUDEAU P., Real-time separation of natural products by ultrafast 2D NMR coupled to on-line HPLC. Analyst, 2012, 137, 2357-2361.
(74) BOISSEAU R., BUSSY U., GIRAUDEAU P., BOUJTITA M., In Situ Ultrafast 2D NMR Spectroelectrochemistry for Real-Time Monitoring of Redox Reactions. Anal. Chem., 2015, 87, 372-375.
(75) PELUPESSY P., RENELLA E., BODENHAUSEN G., High-Resolution NMR in Magnetic Fields with Unknown Spatiotemporal Variations. Science, 2009, 324, 1693-1697.
(76) GIRAUDEAU P., REMAUD G.S., AKOKA S., Evaluation of Ultrafast 2D NMR for Quantitative Analysis. Anal. Chem., 2009, 81, 479-484.
(77) FRYDMAN L., LUPULESCU A., SCHERF T., Principles and features of single-scan two-dimensional NMR spectroscopy. J. Am. Chem. Soc., 2003, 125, 9204-9217.
(78) LE GUENNEC A., TEA I., ANTHEAUME I., MARTINEAU E., CHARRIER B., PA-THAN M., AKOKA S., GIRAUDEAU P., Fast determination of absolute meta-bolite concentrations by spatially-encoded 2D NMR: application to breast cancer cell extracts. Anal. Chem., 2012, 84, 10831-10837.
(79) LE GUENNEC A., GIRAUDEAU P., CALDARELLI S., Evaluation of fast 2D NMR for metabolomics. Anal. Chem., 2014, 86, 5946-5954.
(80) ROUGER L., CHARRIER B., PATHAN M., AKOKA S., GIRAUDEAU P., Proces-sing strategies to obtain clean interleaved ultrafast 2D NMR spectra. J. Magn. Reson., 2014, 238, 87-93.
(81) JÉZÉQUEL T., DEBORDE C., MAUCOURT M., ZHENDRE V., MOING A., GIRAU-DEAU P., Absolute quantifi cation of metabolites in tomato fruit extracts by fast 2D NMR. Metabolomics, 2015, In press, doi: 10.1007/s11306-015-0780-0.
(82) GAL M., SCHANDA P., BRUTSCHER B., FRYDMAN L., UltraSOFAST HMQC NMR and the Repetitive Acquisition of 2D Protein Spectra at Hz Rates. J. Am. Chem. Soc., 2007, 129, 1372-1377.
(83) TAL A., SHAPIRA B., FRYDMAN L., Single-Scan 2D Hadamard NMR Spec-troscopy. Angew. Chem. Int. Ed., 2009, 48, 2732-2736.
(84) GIRAUDEAU P., CAHOREAU E., MASSOU S., PATHAN M., PORTAIS J.-C., AKOKA S., UFJCOSY: a Fast 3D NMR Method for Measuring Isotopic Enrich-ments in Complex Samples. ChemPhysChem, 2012, 13, 3098-3101.
(85) EISENSCHMID T.C., KIRSS R.U., DEUTSCH P.P., HOMMELTOFT S.I., EISEN-BERG R., BARGON J., LAWLER R.G., BALCH A.L., Para hydrogen induced pola-rization in hydrogenation reactions. J. Am. Chem. Soc., 2002, 109, 8089-8091.
(86) ARDENKJAER-LARSEN J.H., FRIDLUND B., GRAM A., HANSSON G., HANS-SON L., LERCHE M.H., SERVIN R., THANING M., GOLMAN K., Increase in signal-to-noise ratio of >10,000 times in liquid state NMR. Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158-10163.
(87) FRYDMAN L., BLAZINA D., Ultrafast two-dimensional nuclear magnetic resonance spectroscopy of hyperpolarized solutions. Nature Physics, 2007, 3, 415-419.
(88) LLOYD L.S., ADAMS R.W., BERNSTEIN M., COOMBES S., DUCKETT S.B., GREEN G.G.R., LEWIS R.J., MEWIS R.E., SLEIGH C.J., Utilization of SABRE-De-rived Hyperpolarization To Detect Low-Concentration Analytes via 1D and 2D NMR Methods. J. Am. Chem. Soc., 2012, 134, 12904–12907.
(89) YON M., LALANDE-MARTIN J., HARRIS T., TEA I., GIRAUDEAU P., FRYDMAN L., 13C NMR detection of metabolic mixtures enhanced by dynamic nuclear polarization. ScienceJet, 2015, 5, 1.





























