Embed Size (px)
Citation preview

FLAMMARION MÉDECINE-SCIENCES — ACTUALITÉS NÉPHROLOGIQUES 2006(www.medecine.flammarion.com)
MICROANGIOPATHIE ET CONTRÔLE DE LA VOIE ALTERNE DU COMPLÉMENT
par
V. FRÉMEAUX-BACCHI*, F. BIENAIMÉ*, C. LOIRAT** et M.-A. DRAGON-DUREY*
Le terme de microangiopathie thrombotique (MAT) désigne une lésion del’endothélium des petites artérioles et des capillaires artériolaires entaînant la for-mation d’agrégats plaquettaires et de thromboses. La thrombopénie et l’anémiehémolytique résultent de la consommation des plaquettes et de la fragmentationdes hématies dans les vaisseaux lésés. Le plus souvent, ces signes biologiques sontassociés à des signes d’ischémie viscérale qui peuvent toucher tous les organes,mais prédominent au niveau du rein et du cerveau [1]. Les deux formes de MATles plus classiques sont le purpura thrombotique thrombocytopénique (PTT) et lesyndrome hémolytique et urémique (SHU). Le PTT, dans sa forme la plus fré-quente est une MAT de l’adulte dominée par la gravité de l’atteinte neurologiqueinitiale et est associé à un déficit acquis en ADAMTS 13, métalloprotéase spéci-fique du facteur Willebrand [2]. La présence d’anticorps anti-ADAMTS 13 sug-gère fortement que le PTT de l’adulte représente une maladie auto-immune [3]. Àl’inverse le SHU est plus fréquemment observé chez l’enfant et est caractérisé parl’existence d’une insuffisance rénale aiguë le plus souvent sévère, et volontiersoligo-anurique. L’atteinte rénale est constante. Les SHU dits typiques font suite àune infection intestinale à Escherichia coli producteur de toxines Shiga [4]. Cesformes représentent plus de 90 p. 100 des cas de SHU de l’enfant de moins de3 ans. Les adultes sont rarement atteints. Les SHU dits atypiques, c’est-à-dire quine sont pas secondaires à une infection bactérienne, une maladie auto-immune, unetransplantation rénale, une prise médicamenteuse ou un cancer sont rares et demauvais pronostic. Ils surviennent à tous les âges, de la période néonatale à l’âgeadulte, évoluent souvent par rechutes successives, et aboutissent fréquemment àl’insuffisance rénale terminale. Toutefois, une évolution favorable, avec de longues
* Service d’Immunologie, Hôpital Européen Georges Pompidou, Paris ; **Service de Néphrologie,Hôpital Robert Debré, Paris.

68 V. FREMEAUX-BACCHI ET COLL.
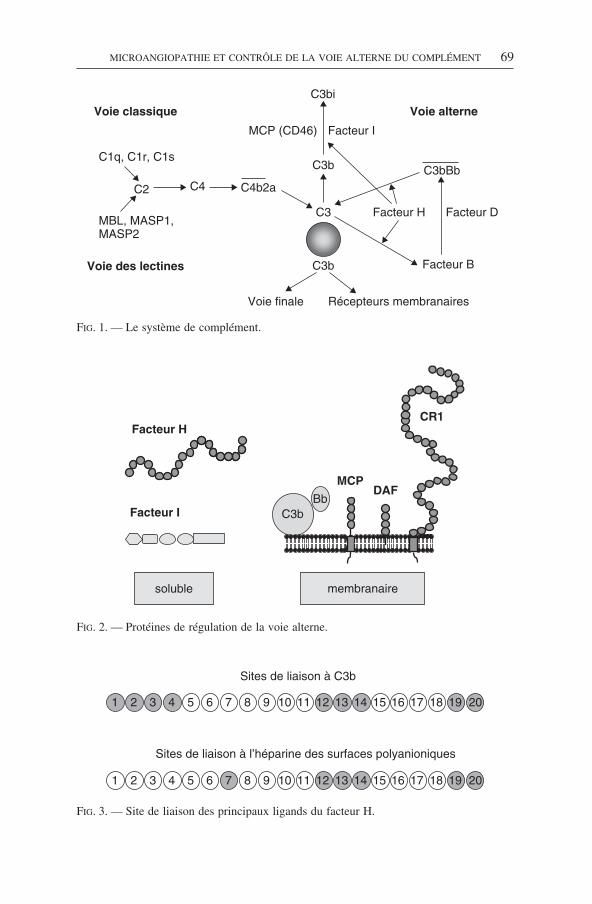
périodes de rémission, est parfois observée. Les SHU atypiques sont le plus sou-vent sporadiques, mais peuvent être familiaux, avec une transmission dominanteou le plus souvent récessive. Le risque de récidive après transplantation rénale,conduisant à la perte du greffon, est très élevé, mais actuellement imprévisible aucas par cas. Des études récentes ont montré que les SHU atypiques familiaux, maiségalement sporadiques, peuvent être associés à des anomalies génétiques de diver-ses protéines impliquées dans la régulation de l’activation de la voie alterne ducomplément : le facteur H (FH), la membrane cofactor protein (MCP) ou CD46et le facteur I (FI) (fig. 1) [5-7].
SHU ATYPIQUE ET COMPLÉMENT
Des taux sériques bas de la fraction 3 du complément (C3) liés à une activationde la voie alterne, ont été rapportés dès 1974 dans des cas de SHU atypiques fami-liaux ou sporadiques [8, 9]. L’effondrement permanent du C3, même en dehors despoussées de SHU ou chez des apparentés non atteints, suggérait un déficit hérédi-taire aboutissant à une dérégulation de la voie alterne du complément.
Le système du complément consiste en plusieurs protéines plasmatiques etmembranaires, organisées en trois voies d’activation : la voie classique, la voielectine, et la voie alterne. L’activation de ces voies aboutit à la formation des C3convertases, qui clivent le C3 et génèrent du C3b. Au niveau de la voie alterne, leC3b participe à la formation de la C3 convertase (C3bBb) qui active la boucled’amplification et permet la formation du complexe d’attaque membranaire, etla libération d’anaphylatoxines (voir fig. 1) [10]. Le système du complémenthumain est très régulé afin d’éviter des lésions non spécifiques des cellules del’hôte. Plusieurs protéines plasmatiques et membranaires régulent l’activité de laC3 convertase alterne. Cette régulation repose d’une part sur plusieurs protéinesancrées sur les membranes : CR1 (complement receptor 1 ou CD35), MCP (mem-brane co-factor protein, ou CD46), le DAF (decay accelereting factor ou CD55),d’autre part sur des régulateurs présents dans la circulation : le FH et le FI (fig. 2).Le FH est un élément clé de la régulation de la C3 convertase alterne. Il inhibe laformation de la C3 convertase alterne et accélère sa dissociation en agissantcomme cofacteur du FI pour dégrader le C3b. Le FH est synthétisé par le foie, etcodé par un gène unique situé sur le bras long du chromosome 1, en 1q32, dansun locus appelé RCA (regulators of complement activation) : ce locus regroupeune famille de gènes codant pour différentes protéines régulatrices de l’activationdu complément. Ces protéines ont la particularité d’être composées de séquencesrépétitives d’environ 60 acides aminés en tandem, appelés SCR (short concensusrepeat). La structure du FH est indiquée sur la fig. 3, avec les sites de liaison auC3b, et à l’héparine des surfaces polyanioniques des membranes cellulaires. Lesdomaines du FH qui participent à la régulation de la voie alterne du complémenten phase fluide sont localisés dans les SCR 1-4 N-terminaux. L’inactivation duC3b fixé aux surfaces cellulaires fait intervenir deux sites de liaison au C3b sur leFH, localisés dans les SCR 12-14 et les SCR 19-20. Le FH comprend trois sitesde liaisons pour des surfaces riches en polyanions (comme l’héparine), localisésdans les SCR 7, 12 à 14, et 19-20 [11, 12]. Le FH est une molécule qui a donc lacapacité d’interagir avec des membranes cellulaires.
MICROANGIOPATHIE ET CONTRÔLE DE LA VOIE ALTERNE DU COMPLÉMENT 69
MCPDAF
CR1
C3bBb
Facteur H
Facteur I
membranairesoluble
FIG. 2. — Protéines de régulation de la voie alterne.
Sites de liaison à C3b
1 2 98763 4 5 151413121110 2019181716
1 2 98763 4 5 151413121110 2019181716
Sites de liaison à l’héparine des surfaces polyanioniques
FIG. 3. — Site de liaison des principaux ligands du facteur H.
MCP (CD46) Facteur I
C3
C1q, C1r, C1s
C2 C4 C4b2a
MBL, MASP1, MASP2
Facteur B
Voie alterne
C3bBb
Voie finale
Voie classiqueC3bi
C3b
C3b
Récepteurs membranaires
Facteur H Facteur D
Voie des lectines
FIG. 1. — Le système de complément.

70 V. FREMEAUX-BACCHI ET COLL.
DÉCOUVERTE DE MUTATIONS SUR TROIS GÈNES IMPLIQUÉS DANS LA RÉGULATION
DE LA VOIE ALTERNE DU COMPLÉMENT
L’association entre SHU atypique éventuellement familial et taux bas de facteur Hayant été rapportée dans les années 1980 [13-15], le premier gène candidat étu-dié a été le FH. En 1998, Warwicker et al. ont pu, à partir de trois familles, établirle lien génétique entre SHU atypique et le locus RCA situé sur le chromosome1q32 [5].
Il a été rapporté ensuite que des patients, enfants ou adultes, présentant un SHUatypique sporadique ou familial, avaient, en dépit d’un taux circulant de FH nor-mal, des mutations sur le gène du FH, localisées essentiellement dans les SCR 18,19 et 20 [16, 17]. Une fréquence particulièrement importante de mutations dansle SCR 20 a été rapportée [18]. Les cellules endothéliales des glomérules humainset les membranes basales glomérulaires sont riches en molécules polyanioniques,et la fixation du FH à leur surface est donc essentielle pour leur protection contrel’attaque par le complément. Il a été démontré que le FH muté a une capacitéréduite à inter-agir avec les polyanions et avec le C3b lié aux surfaces cellulaires,entraînant une perte de l’activité régulatrice du complément au niveau des mem-branes cellulaires [19]. En revanche, ces mutants gardent leur capacité de contrôlede l’activation du complément dans le plasma [20]. Cette propriété explique enpartie la fréquence des mutations retrouvées au niveau de la région C-terminaledu FH chez les patients.
Depuis le premier rapport de Warwicker [5], plusieurs études réalisées par4 groupes indépendants, dont le nôtre, ont identifié au moins une cinquantaine demutations du facteur H chez environ 200 patients atteints de SHU atypique, fami-lial et sporadique [21]. Ces études ont été effectuées principalement chez l’adulteet les descriptions cliniques sont hétérogènes et difficilement exploitables.
Certains patients atteints de SHU atypique n’ont pas de mutation du FH, bienqu’ils aient des stigmates d’activation de la voie alterne du complément. Desmutations localisées sur les exons des gènes de deux autres protéines régulatricesde la voie alterne ont été observées. Treize patients porteurs de mutations hété-rozygotes ou homozygotes du gène de MCP ont été rapportées [6, 22, 23]. Lesanomalies génétiques mises en évidence sont hétérogènes et comprennent desdélétions de petite taille et des mutations ponctuelles. Cette protéine est présenteà la surface de toutes les membranes cellulaires sauf celles des globules rouges,et est fortement exprimée dans le rein humain. À l’exception d’un cas, l’expres-sion de la protéine MCP est diminuée à la surface des cellules mononucléées despatients avec des mutations. Les patients qui ont la substitution S206P exprimentune quantité normale de protéines. Des expériences de transfection ont montréque la mutation affecte sélectivement la liaison de MCP à C3b [6]. La maladieest familiale avec une transmission autosomique récessive dans quatre cas(9 patients). Le SHU semble survenir à un âge plus tardif et avoir un pronosticmoins sévère que dans les cas avec mutation du FH. De plus, le rein greffé appor-tant la protéine MCP normale, il ne devrait pas y avoir de récidive après greffe.Le nombre limité de patients rapportés (seulement 3 patients greffés, sans réci-dive) doit rendre prudentes ces conclusions et invite évidemment à étudier le gène

MICROANGIOPATHIE ET CONTRÔLE DE LA VOIE ALTERNE DU COMPLÉMENT 71
MCP dans une grande cohorte de patients [6]. Par ailleurs, notre groupe et Kava-nagh et al. ont rapporté des mutations sur le gène du FI, chez 5 patients atteintsde SHU atypique [7]. Le facteur I est une sérine-protéase circulant sous formeactive dans la plasma, qui possède une étroite spécificité pour le C3b. Elle cliveà deux endroits la chaîne α du C3b pour permettre la formation de C3bi, fragmenthémolytiquement inactif de C3b (c’est-à-dire qui a perdu la capacité de se lier aufacteur B). Le facteur I coupe ensuite la protéine C3bi pour générer un fragmentC3c libéré en phase fluide et un fragment C3dg qui reste liée à la surface activa-trice. La présence d’un cofacteur (FH, MCP ou CR1) est indispensable à l’actiondu facteur I. Le gène codant pour le facteur I est localisé sur le chromosome 4en position 4q25 et comprends 13 exons. L’activité sérine-protéase est localiséesur la chaîne légère de la protéine, alors que la chaîne lourde est composée dequatre domaines dont les fonctions sont inconnues à ce jour. Les dosages antigé-niques du facteur I ont permis de porter le diagnostic de déficit partiel chez quatrepatients ayant présenté un SHU atypique à l’âge adulte. Dans trois cas, la mutationmise en évidence a pour conséquence un codon stop (W127X, R456X, W528X)[7, 24]. Dans un cas, il s’agit d’une délétion d’un nucléotide (del922C) responsabledu changement du cadre de lecture entraînant l’apparition d’un codon stop pré-maturé [24]. Une mutation localisée au cœur du site actif de la protéine (D506V),non associée à un déficit a été mise en évidence chez un enfant qui présente depuisl’âge de 18 mois un SHU sous la forme récidivante [7]. Il s’agit dans les cinq casde formes sporadiques.
IMPACT DU POLYMORPHISME GÉNÉTIQUE DU FH ET DE MCP DANS LA MALADIE
Un grand nombre d’apparentés des patients qui présentent une mutation auniveau du FH ont la même mutation que le patient, mais sont asymptomatiques.Ceci a conduit à s’interroger sur la responsabilité de variants polymorphiques dugène du FH, entraînant une susceptibilité au SHU [25]. Les variations de séquencedans le génome humain sont pour l’essentiel des changements d’une seule base,nommés SNP (pour single nucleotide polymorphism). Une étude détaillée portantsur 10 SNP localisés au niveau du locus RCA a été effectuée par Esparza-Gordilloet al. [23]. La contribution des variants polymorphiques du FH et de MCP dans lagénétique du SHU atypique a été recherchée chez des patients caucasiens français(n = 77), anglais (n = 75) et espagnols (n = 41) [23, 26]. L’allèle T du polymor-phisme situé au niveau du promoteur du gène du FH en position – 257, a montréune liaison forte avec le SHU (p < 0,001, dans les trois groupes). Ceci est en accordà la première observation faite par l’équipe italienne [25] et suggère que des poly-morphismes dans cette région promotrice du gène du FH peuvent contribuer aurisque génétique du SHU. Un haplotype défini par la combinaison de trois SNP,dont celui localisé au niveau du promoteur, a permis de déterminer un allèle duFH qui influence la prédisposition génétique au SHU. L’analyse par notre groupede cinq SNP liés au gène de MCP a permis d’individualiser un haplotype, définipar la présence d’un G au niveau du promoteur du gène de MCP en position –261, plus fréquemment présent chez les patients atteints de SHU que dans la popu-lation contrôle (p < 0,001) [26].

72 V. FREMEAUX-BACCHI ET COLL.
À la différence des maladies monogéniques dont la caractérisation génétique aconnu un succès considérable dans les dernières années, les avancées dans ledomaine des maladies multifactorielles ont été beaucoup plus ténues et les straté-gies pour identifier les gènes de susceptibilité à ces maladies sont diverses.L’approche traditionnelle qui consiste à étudier les gènes un par un et à mettre enrelation quelques polymorphismes avec la maladie a de nombreuses limites. Ilapparaît de plus en plus évident que l’étude des maladies complexes requiert nonseulement d’analyser la variabilité complète des gènes candidats, mais aussid’aborder la variabilité génétique des systèmes biologiques dans leur ensemble. Ilest probable que c’est l’accumulation et/ou l’interaction de plusieurs anomaliesqui conduit au dysfonctionnement du système. Le challenge actuel afin de modé-liser au mieux la relation génotype/phénotype réside dans l’analyse multivariéed’un grand nombre de polymorphismes.
DÉFICIT ACQUIS EN FH ET SHU ATYPIQUE
Nous avons récemment démontré que le SHUa pouvait survenir dans le contexted’une maladie auto-immune avec le développement d’auto-anticorps anti-facteurH induisant un déficit acquis en facteur H [27]. Nous avons détecté la présenced’IgG anti-FH chez trois enfants âgés de 3 à 11 ans au début de la maladie (deuxgarçons et une fille). Cliniquement, cette forme de SHU auto-immun, ne présentepas de particularité notable à la période aiguë. Tous les trois ont présenté la triadeclassique du SHU. Des signes prodromiques étaient notés chez deux d’entre euxcomprenant des signes digestifs. Les trois enfants ont présenté une forme récur-rente de la maladie (1 à 3 rechutes) avec le développement d’une insuffisancerénale terminale (IRT) pour l’un et une évolution favorable avec des séquellesrénales modérées chez les deux autres.
Biologiquement, des stigmates d’activation par la voie alterne, attestés par unediminution du dosage antigénique plasmatique du C3 étaient présents chez deuxtiers des enfants. L’activité fonctionnelle du FH était diminuée (10 à 70 p. 100 desvaleurs normales) chez les trois patients et nous avons pu montré chez deux d’entreeux qu’elle était corrélée avec les titres d’IgG anti-FH. Chez les trois enfants,l’étude génétique du FH, du FI et de MCP était normale. Les expérimentationsréalisées pour étudier in vitro l’influence des anticorps anti-facteur H sur les fonc-tions de celui-ci ont montré que ces auto-anticorps perturbaient spécifiquementl’activité de dissociation de la C3 convertase alterne, probablement en touchant lacapacité de H à se lier à C3b fixé sur une surface activatrice et non pas sa capacitéà fixer C3b en phase fluide.
Deux enfants ont été traités par échanges plasmatiques avec contrairement à cequi est généralement décrit dans les SHU typiques ou atypiques [1], une bonneefficacité. Les échanges plasmatiques ont permis de réduire efficacement le titredes IgG anti-FH. Dans les deux cas une rechute est survenue approximativement15 jours après l’arrêt des échanges motivant l’introduction de traitements immu-nosuppresseurs.
La grande fréquence de déficits acquis en ADAMTS 13 chez les patients atteintsde PTT est apparemment en contraste avec la faible fréquence d’anticorps anti-FHrapportée chez les patients atteints de SHUa pour lequel les facteurs génétiques

MICROANGIOPATHIE ET CONTRÔLE DE LA VOIE ALTERNE DU COMPLÉMENT 73
semblent prédominants. Néanmoins ces anticorps, qui ne sont pas encore recher-chés de façon systématique, pourraient être responsables d’un nombre importantde cas de SHUa, particulièrement dans le groupe de patients ne présentant pas defacteur génétique de susceptibilité identifié. De plus, il est possible que, commedans le cas du PTT, ces anticorps puissent être présents uniquement de façon tran-sitoire et détectables uniquement à la période aiguë de la maladie.
En conclusion, cette nouvelle étiologie de SHUa devrait entraîner une modifi-cation de la conduite diagnostique avec inclusion de la recherche précoce des auto-anticorps anti-FH et, quand elle est positive, la mise en place rapide de traitementsspécifiques incluant les échanges plasmatiques et/ou des traitements immunosup-presseurs. Leur recherche systématique à la période aiguë de la maladie, devraitpermettre d’établir la fréquence réelle du SHU auto-immun.
RÔLE DU COMPLÉMENT DANS LA PHYSIOPATHOLOGIE DE LA MALADIE
Ces nouvelles données nous permettent d’affirmer qu’une dérégulation de lavoie alterne du complément participe aux mécanismes de la lésion de microangio-pathie thrombotique du SHU atypique. Il est admis que dans le SHU, les lésionstissulaires sont la conséquence d’une activation des cellules endothéliales et desplaquettes qui, sous leur phénotype procoagulant, sont à l’origine de la formationdes micro-thrombi. Le point commun des trois protéines impliquées, le facteur H,le facteur I et MCP est la fonction de clivage de C3b en C3bi. Le facteur I estl’enzyme de la réaction, CD46 est un cofacteur qui exerce cette fonction sur duC3b fixé sur une surface cellulaire tandis que le facteur H peut également êtrecofacteur en phase fluide et effectuer cette fonction sur toute surface riche en poly-anions. Ainsi, une défaillance d’une de ces trois protéines aboutit à un défaut dedégradation de C3b en C3bi. Les principales conséquences de cette dérégulationde la voie alterne du complément vont être la libération excessive des anaphyla-toxines C3a et C5a et la formation aux sites d’activation de complexes d’attaquemembranaires C5b9. Les anaphylatoxines C3a et C5a sont capables d’activerdirectement les plaquettes et les cellules endothéliales. Elles possèdent d’impor-tantes capacités chimiotactiques sur les polynucléaires neutrophiles et sur lesmonocytes/macrophages avec induction de cytokines pro-inflammatoires [28].
L’activation de C5 a également pour conséquence l’association de C5b avec lesprotéines de la voie finale commune C6 à C9 et la formation du complexe d’attaquemembranaire C5b9. Il a été montré que C5b9 est capable d’activer les cellulesendothéliales avec induction de l’expression membranaire de la P sélectine, de lasécrétion de facteur von Willebrand de haut poids moléculaire [29] et de la sécré-tion de chimiokines comme l’IL-8 et MCP-1 (monocyte chemoattractant protein)[30]. C5b9 est également capable d’activer les plaquettes avec induction del’expression de la P sélectine à la surface des plaquettes, sécrétion de granules α,riches en ATP et mise en place d’un statut procoagulant avec activation de laprothrombinase [31].
Au total, la capacité de C3a, C5a et C5b9 à activer directement les cellules endo-théliales, les plaquettes mais aussi les leucocytes, en font de très bons agents induc-teurs d’un phénotype procoagulant à l’origine des micro-thrombi du SHU atypique.

74 V. FREMEAUX-BACCHI ET COLL.
Les cellules endothéliales glomérulaires, du fait de leur implication dans denombreuses néphropathies ont fait l’objet de plusieurs études « in vitro » et « invivo ». Ainsi, un modèle de microangiopathie thrombotique a été développé chezle rat à l’aide d’anticorps dirigés contre des cellules endothéliales glomérulaires.Ces auteurs ont démontré que ces lésions étaient induites par le complexe C5b9[32]. Enfin, en utilisant le même modèle animal, Hugues et al. ont montré que lescomplexes C5b9 étaient capables d’induire l’apoptose des cellules endothélialesglomérulaires ayant pour conséquence leur détachement de la membrane basaleglomérulaire [33]. Cet unique modèle animal de MAT a permis de démontrer queles lésions induites étaient complément-dépendantes.
VERS LA RECHERCHE DE MUTATIONS DANS D’AUTRES GÈNES
À ce jour aucun facteur de susceptibilité n’a été identifié dans plus de la moitiédes SHU atypiques avec ou en absence d’antécédents familiaux. Ces formes,comme celles associées aux mutations au niveau du gène du facteur H ou du fac-teur I sont de mauvais pronostic. L’objectif actuel est d’identifier de nouveauxgènes de susceptibilité associés à cette maladie. L’ensemble des intervenants dela voie alterne du complément constitue une source de candidats potentiels. Plu-sieurs gènes comme le facteur B ou CR1 ont été étudiés et n’ont pas permis d’iden-tifier de nouveaux facteurs de susceptibilité [22, 34]. Les mécanismes de régulationfont intervenir des couples dont le point commun est le C3. Marder et al. ont émisen 1983 l’hypothèse qu’un déficit acquis en C3 pourrait être secondaire à uneincapacité du C3 à interagir correctement avec ses protéines de régulation [35].Plusieurs candidats sont actuellement à l’étude comme la Properdine, la C4 bin-ding-protein ou le C3. L’activation incontrôlée de la voie alterne, systémique ouuniquement localement, pourrait être responsable de l’atteinte rénale. Il est impor-tant de définir si cette maladie est restreinte uniquement aux conséquences del’activation incontrôlée du complément ou pourrait être secondaire à d’autresmécanismes dont les conséquences sur la cellule endothéliale seraient identiques.
CONCLUSION
Actuellement environ 50 p. 100 des patients qui présentent un SHU atypiqueont une mutation sur un des trois gènes de régulation de la voie alterne du com-plément. Les études de cohorte suggèrent qu’environ 30 p. 100 des patients pré-sentent une mutation sur le gène du facteur H. Il est donc clair que des progrèsrestent à faire dans le domaine de la génétique des facteurs de susceptibilité pourcette maladie. Dans la plupart des cas, les facteurs déclenchants de la maladie sontà ce jour inconnus. Il semble que le post-partum puisse être un exemple. Aucunedonnée n’est actuellement disponible sur l’étiologie des SHU secondaires à la prisemédicamenteuse, au cancer ou à la transplantation. La connaissance des facteursde susceptibilité et un début de compréhension de leurs implications dans la phy-siopathologie de la maladie devraient permettre d’orienter les options thérapeuti-ques dans de nouvelles directions.

MICROANGIOPATHIE ET CONTRÔLE DE LA VOIE ALTERNE DU COMPLÉMENT 75
BIBLIOGRAPHIE
1. MOAKE JL. Thrombotic microangiopathies. N Engl J Med, 2002 ; 347 : 589-600.
2. FURLAN M, ROBLES R, GALBUSERA M, et al. von Willebrand factor-cleaving protease in thromboticthrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med, 1998 ; 339 : 1578-1584.
3. COPPO P, BENGOUFA D, VEYRADIER A et al. Severe ADAMTS13 deficiency in adult idiopathicthrombotic microangiopathies defines a subset of patients characterized by various autoimmunemanifestations, lower platelet count, and mild renal involvement. Medicine, 2004 ; 83 : 233-244.
4. TARR PI, GORDON CA, CHANDLER WL. Shiga-toxin-producing Escherichia coli and haemolyticuraemic syndrome. Lancet, 2005 ; 365 : 1073-1086.
5. WARWICKER P, GOODSHIP TH, DONNE RL et al. Genetic studies into inherited and sporadic hemo-lytic uremic syndrome. Kidney Int, 1998 ; 53 : 836-844.
6. RICHARDS A, KEMP EJ, LISZEWSKI MK et al. Mutations in human complement regulator, membranecofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. ProcNatl Acad Sci U S A, 2003 ; 100 : 12966-12971.
7. FREMEAUX-BACCHI V, DRAGON-DUREY MA, BLOUIN J et al. Complement factor I : a susceptibilitygene for atypical haemolytic uraemic syndrome. J Med Genet, 2004 ; 41 : e84.
8. STUHLINGER W, KOURILSKY O, KANFER A et al. Letter : Haemolytic-uraemic syndrome : evidencefor intravascular C3 activation. Lancet, 1974 ; 2 : 788-789.
9. CARRERAS L, ROMERO R, REQUESENS C et al. Familial hypocomplementemic hemolytic uremicsyndrome with HLA-A3,B7 haplotype. JAMA, 1981 ; 245 : 602-604.
10. WALPORT MJ. Complement. First of two parts. N Engl J Med, 2001 ; 344 : 1058-1066.
11. PANGBURN MK. Cutting edge : localization of the host recognition functions of complement factorH at the carboxyl-terminal : implications for hemolytic uremic syndrome. J Immunol, 2002 ; 169 :4702-4706.
12. HELLWAGE J, JOKIRANTA TS, FRIESE MA et al. Complement C3b/C3d and cell surface polyanionsare recognized by overlapping binding sites on the most carboxyl-terminal domain of complementfactor H. J Immunol, 2002 ; 169 : 6935-6944.
13. THOMPSON RA, WINTERBORN MH. Hypocomplementaemia due to a genetic deficiency of beta 1Hglobulin. Clin Exp Immunol, 1981 ; 46 : 110-119.
14. PICHETTE V, QUERIN S, SCHURCH W et al. Familial hemolytic-uremic syndrome and homozygousfactor H deficiency. Am J Kidney Dis, 1994 ; 24 : 936-941.
15. ROUGIER N, KAZATCHKINE MD, ROUGIER JP et al. Human complement factor H deficiency asso-ciated with hemolytic uremic syndrome. J Am Soc Nephrol, 1998 ; 9 : 2318-2326.
16. RICHARDS A, BUDDLES MR, DONNE RL et al. Factor H mutations in hemolytic uremic syndromecluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet, 2001 ; 68 :485-490.
17. CAPRIOLI J, BETTINAGLIO P, ZIPFEL PF et al. The molecular basis of familial hemolytic uremicsyndrome : mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20.J Am Soc Nephrol, 2001 ; 12 : 297-307.
18. PERKINS SJ, GOODSHIP TH. Molecular modelling of the C-terminal domains of factor H of humancomplement : a correlation between haemolytic uraemic syndrome and a predicted heparin bindingsite. J Mol Biol, 2002 ; 316 : 217-224.
19. MANUELIAN T, HELLWAGE J, MERI S et al. Mutations in factor H reduce binding affinity to C3band heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J ClinInvest, 2003 ; 111 : 1181-1190.
20. SANCHEZ-CORRAL P, PEREZ-CABALLERO D, HUARTE O et al. Structural and functional characteriza-tion of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet,2002 ; 71 : 1285-1295.
21. DRAGON-DUREY MA, FREMEAUX-BACCHI V. Atypical haemolytic uraemic syndrome and mutationsin complement regulator genes. Springer Semin Immunopathol, 2005 ; 1-16.
22. NORIS M, BRIOSCHI S, CAPRIOLI J et al. Familial haemolytic uraemic syndrome and an MCP muta-tion. Lancet, 2003 ; 362 : 1542-1547.

76 V. FREMEAUX-BACCHI ET COLL.
23. ESPARZA-GORDILLO J, GOICOECHEA de JORGE E, BUIL A et al. Predisposition to atypical hemolyticuremic syndrome involves the concurrence of different susceptibility alleles in the regulators ofcomplement activation gene cluster in 1q32. Hum Mol Genet, 2005 ; 14 : 1107.
24. KAVANAGH D, KEMP EJ, MAYLAND E et al. Mutations in complement factor I predispose to deve-lopment of atypical hemolytic uremic syndrome. J Am Soc Nephrol, 2005 ; 16 : 2150-2155.
25. CAPRIOLI J, CASTELLETTI F, BUCCHIONI S et al. Complement factor H mutations and gene polymor-phisms in haemolytic uraemic syndrome : the C-257T, the A2089G and the G2881T polymor-phisms are strongly associated with the disease. Hum Mol Genet, 2003 ; 12 : 3385-3395.
26. FREMEAUX-BACCHI V, KEMP EJ, GOODSHIP JA et al. The development of atypical HUS is influencedby susceptibility factors in factor H and membrane cofactor protein- evidence from two indepen-dent cohorts. J Med Genet, 2005 ; 22 : 22
27. DRAGON-DUREY MA, LOIRAT C, CLOAREC S et al. Anti-Factor H autoantibodies associated withatypical hemolytic uremic syndrome. J Am Soc Nephrol, 2005 ; 16 : 555-563.
28. SCHRAUFSTATTER IU, TRIEU K, SIKORA L et al. Complement c3a and c5a induce different signaltransduction cascades in endothelial cells. J Immunol, 2002 ; 169 : 2102-2110.
29. HATTORI R, HAMILTON KK, MCEVER RP et al. Complement proteins C5b-9 induce secretion ofhigh molecular weight multimers of endothelial von Willebrand factor and translocation of granulemembrane protein GMP-140 to the cell surface. J Biol Chem, 1989 ; 264 : 9053-9060.
30. KILGORE KS, SCHMID E, SHANLEY TP et al. Sublytic concentrations of the membrane attack com-plex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1through nuclear factor-kappa B activation. Am J Pathol, 1997 ; 150 : 2019-2031.
31. WIEDMER T, SIMS PJ. Participation of protein kinases in complement C5b-9-induced shedding ofplatelet plasma membrane vesicles. Blood, 1991 ; 78 : 2880-2886.
32. NANGAKU M, ALPERS CE, PIPPIN J et al. Renal microvascular injury induced by antibody to glo-merular endothelial cells is mediated by C5b-9. Kidney Int, 1997 ; 52 : 1570-1578.
33. HUGHES J, NANGAKU M, ALPERS CE et al. C5b-9 membrane attack complex mediates endothelialcell apoptosis in experimental glomerulonephritis. Am J Physiol Renal Physiol, 2000 ; 278 : F747-757.
34. KAVANAGH D, KEMP EJ, RICHARDS A et al. Does complement factor B have a role in the pathoge-nesis of atypical HUS ? Mol Immunol, 2005 ; 29 : 29.
35. MARDER HK, COLEMAN TH, FORRISTAL J et al. An inherited defect in the C3 convertase, C3b,Bb,associated with glomerulonephritis. Kidney Int, 1983 ; 23 : 749-758.