Embed Size (px)
Citation preview

I
République Algérienne Démocratique et Populaire
Ministère de l’enseignement supérieur et de la recherche scientifique
Université d’Oran Es-Sénia
Faculté des Sciences
Département de Biologie
Mémoire présenté pour l’obtention du diplôme de MAGISTER
en
AMÉLIORATION DES PLANTES
Mise au point de la technique d’électrophorèsebidimensionnelle appliquée à des espèces
sauvages du genre MedicagoPrésenté par :
FELLOUS Samir
Soutenu le: / / 2011
Jury:
Président KROUF Djamil Professeur Université d’Oran Es-Sénia
Examinateurs SAIDI Noureddine M.C.A Université d’Oran Es-Sénia
KACEM Mourad M.C.A Universitéd’OranEs-Sénia
Encadreur FYAD LAMECHE F.Z Professeur Université d’Oran Es-Sénia

II
DEDICACEÀ
À la mémoire de mes grands parents, et à celle de mon frère,
À mes amis, à mon neveu et mes cousins,
À tous mes enseignants,
À mes parents.

Remerciements

I
Table des matières
Introduction générale 01
Partie I : Revue bibliographique
1. Généralités sur le Medicago 03
1. 1. Taxonomie et génétique 03
1. 2. Origine géographique et aire de répartition 03
1. 3. Description botanique de la plante Medicago 04
1. 4. Germination et développement physiologique de la plante 05
1. 5. Intérêt de la plante 05
2. Préparation des échantillons 07
2. 1. Extraction non dénaturante 07
2. 1. 1. Dégradation protéolytique et inhibition des protéases 07
2. 1. 2. Composés phénoliques 08
2. 2. Extraction dénaturante 09
2. 2. 1. La méthode SDS 09
2. 2. 2. Précipitation directe 09
2. 2. 3. Extraction au phénol 10
2. 2. 4. Solubilisation différentielle 10
2. 2. 5. Cas des ponts disulfures 11
2. 2. 6. Rôle des Chaotropes et des surfactant dans l’extraction dénaturante 11
2. 3. Contaminants des préparations protéiques 12
2. 3. 1. Sels 12
2. 3. 2. Lipides 12
2. 3. 3. Acides nucléiques 13

II
2. 3. 4. Polysaccharides 13
3. Électrophorèse 14
3. 1. Historique 14
3. 2. Principe 15
3. 3. Migration électrophorètique 15
3. 4. Quelques méthodes classiques d’électrophorèse 16
3. 4. 1. Électrophorèses en zone 16
3. 4. 2. Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis (SDS-PAGE) 16
3. 4. 3. Électrophorèse discontinue 17
3. 4. 4. Électrophorèse en gel gradient 17
3. 5. Isoéléctrofocalisation en carriers ampholytes libres (CA-IEF) 18
3. 5. 1. Principe théorique 18
3. 5. 2. Facteurs influençant la formation du pH 19
A) Le phénomène de plateau « plateau phenomenon » 19
B) L’électrœndosmose 20
3. 5. 2. Autre types de gradient de pH 20
3. 6. Électrophorèse bidimensionnelle 20
3. 6. 1. Visualisation des protéines 21
3. 6. 1. 2. Méthodes de coloration par fluorescence 22
3. 6. 1. 3. Coloration négative par le zinc d’imidazole 22
3. 6. 1. 4. Coloration à l’argent 23
3. 6. 1. 5. Détection par radioactivité 23
3. 6. 2. Analyse de l’image 23
3. 6. 3. Évaluation des gels 2-D 24

III
3. 6. 4. Application de l’électrophorèse bidimensionnelle chez les plantes 24
Partie II: Matériels et méthodes
1. Échantillonnage et extraction des protéines totales 27
2. Électrophorèse bidimensionnelle: IEF/SDS-PAG 28
2. 1. Premier dimension: IEF-Carrier ampholytes 4% 28
2. 1. 1. Solubilisation des protéines en IEF 28
2. 1. 2. Préparation des tubes IEF 28
2. 1. 3. Préparation du gel IEF- Carriers ampholytes 28
2. 1. 4. Tampon d’électrodes de l’IEF-Carriers ampholytes 29
2. 1. 5. Focalisation 29
2. 1. 6. Démoulage des boudins 29
2. 2. Deuxième dimension: SDS-PAGE continu 10% 30
2. 2. 1. Préparation du gel et du tampon de la deuxième dimension 30
2. 2. 2. Séparation des protéines 30
2. 2. 3. Coloration, décoloration et fixation du gel 2-D IEF/SDS-PAGE 31
3. Électrophorèse bidimensionnelle: CN/SDS-PAGE 32
3.1 Premier dimension CN-PAGE 32
3. 1. 1 Solubilisation des protéines en CN-PAGE 32
3. 1. 2 Préparations des gels CN-PAGE 32
3. 1. 3. Tampon cuve de la CN-PAGE 33
2. 2. 4. Séparation des protéines en CN-PAGE 33
2. 2. 3. Coloration, décoloration et fixation du gel CN-PAGE 34
3. 2 Deuxièmes dimensions SDS-PAGE 34

IV
Partie III: Résultats et discussions
1. Préparation de l’échantillon
2. Préparation du gel IEF
2. 1. Cristallisation de l'urée
2. 2. Les Carriers ampholyte
2. 3. Coulage des gels d’IEF
2. 4. Temed et l’APS
3. Paramètre de focalisation et de séparation du gel 2-D
4. Électrophorèse bidimensionnelle Gel gradient 4-10%/SDS-PAGE
5. Électrophorèse bidimensionnelle Gel discontinu 4-10%/SDS-PAGE
6. Électrophorèse bidimensionnelle Gel discontinu 4-8%/SDS-PAGE
Partie IV: Conclusion générale et perspectives
Références bibliographiques

V
Liste des figures
Figure. 01 : Feuilles de différents écotypes du genre Medicago (Delalande, 2007) 04
Figure. 02 : Gousse de Medicago Truncatula (Ramakrishnan et al., 2006) 04
Figure. 03 : Réalisation du gradient du gel (Rothe et al., 1982) 18
Figure. 04 : Le principe d’électrophorèse à haute résolution (O’Farrell,1975) 21
Figure. 05 : Gel 2-D de graines matures de M. Truncatula coloré par le Coomassie BrilliantBlue G-250 (Zhentian et al, 2005) 22
Figure. 06 : Dispositif de séparation en éléctrophorèse bidimentionnelle.
Figure. 07: Partie du gel 2-d coloré au bleu de Coomassie représentant trois spots 36
Figure. 08: électrophorèse non dénaturante des protéines totales de graines sauvages deMedicago 40
Figure. 09-a: Gel d’electropphorese bidimensionnelle 1-D gradient
4-10%/SDS-PAGE continu 10% des proteines tatale de graines de Medicago. 42
Figure. 09-b : Profil protéique et Rfs des protéines totale des graines
sauvages de Medicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE 42
Figure 11-a: Gel d’electropphorese bidimensionnelle 1-D discontinu
4-10%/SDS-PAGE continu 10% des proteines tatale de graines de Medicago 47
Figure. 11-b: Profil des protéique et Rfs des protéines totale des graines sauvage deMedicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE 47
Figure. 12-a : Gel d’electropphorese bidimensionnelle 1-D discontinu
4-8%/SDS-PAGE continu 10% des proteines tatale de graines de Medicago 50
Figure. 12-b: Profil des protéique et Rfs des protéines totale des graines
sauvage de Medicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE 50
Figure. 13-a: Gel d’electropphorese bidimensionnelle 1-D discontinu 4-8%/SDS-PAGEcontinu 10% des proteines tatale de graines de Medicago 51
Figure. 13-b: Profil des protéique et Rfs des protéines totale des graines sauvage deMedicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE 51

VI
Liste des tableaux
Tableau. 01 : Rendement du blé, après association aux légumineuses (Hireche, 2006) 06
Tableau.02 : Solution d’extraction des protéines (Zivy,1986). 26
Tableau. 03: Tampon de solubilisation UKS (Granier,1988). 27
Tableau. 04: Solution du gel IEF-Carrier ampholytes (O’ Farrell, 2007). 27
Tableau. 05: Solutions des tampons d’électrodes (O’ Farrell, 2007) 28
Tableau. 06: Programme de voltage (Nagaraj. 2007). 28
Tableau. 07: Solutions utilisées pour la deuxième dimension Selon
(Laemmli, 1970. modifié). 29
Tableau. 08 : Solution de coloration et de décoloration au Coomassie G-250
(Blakesley et Boezi, 1977). 30
Tableau. 09: Tampon de solubilisation (Laemmli,1970. modifié). 31
Tableau. 10: Gel gradient 10%.(Laemmli, 1970. modifié). 32
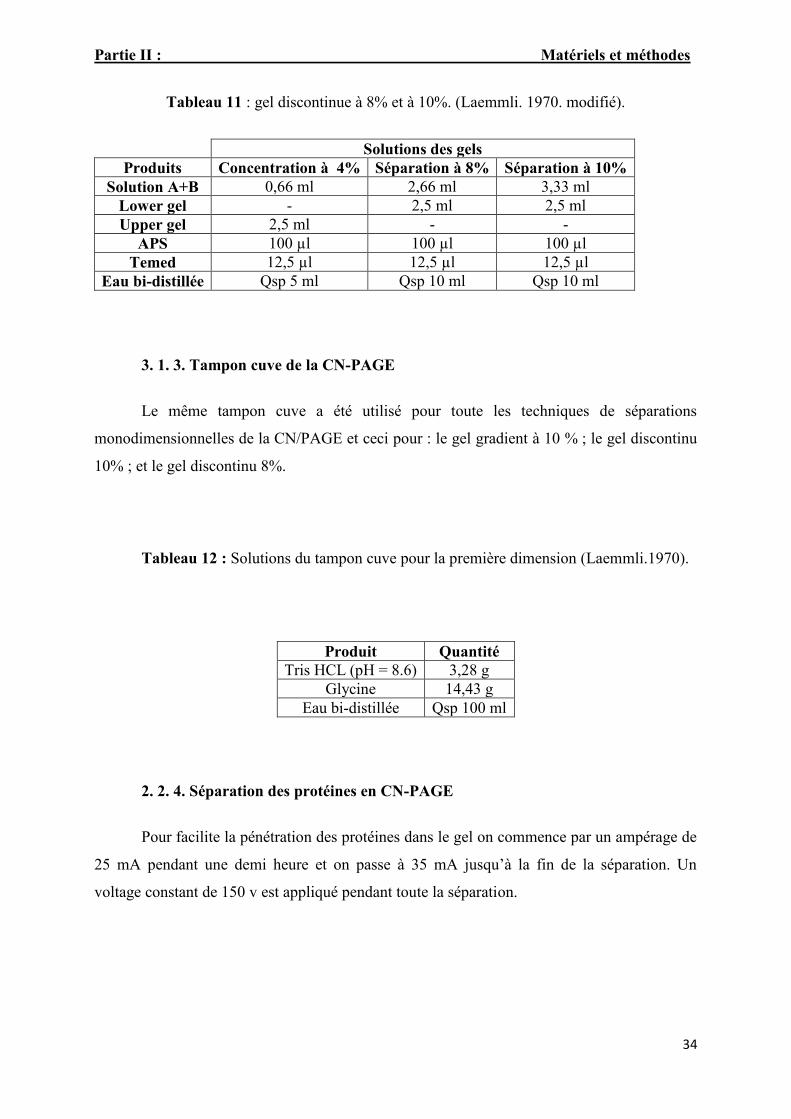
Tableau. 11: Gel discontinue à 8% et à 10%. (Laemmli,1970. modifié). 32
Tableau. 12: Solutions du tampon cuve pour la première dimension (Laemmli,1970). 33
Tableau. 13 : Solution de coloration au Coomassie R-250 (Schägger,2006). 34

VII
Abréviations
BN-PAGE: Bleu native polyacrylamide gel électrophorèsis
CBB: Coomassie Brillant Blue
CCD: Charged Coupled Device
CN-PAGE:Clear native polyacrylamide gel électrophorèsis
2-DE: L’électrophorèse bi-dimensionnelle
DTE: Dithioerythritol:
DTT: Dithiothreitol
E: Force du champ électrique
IEF : Isoelectrofocalisation
IPG: Immobilized PH Gradient
CHAPS: 3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
mep : Mobilité effective
Mr : Mass moléculaire
NEPHG-IEF : Non Equilibrum PH Gradient IsoElectroFocalisation).
Nonidet P-40 : Non ionique détergeant 40
pI : Point isoélectrique
SDS-PAGE : Sodium dodecyl sulfate
TBP: Tributyl phospahte
TCA : Tri-chloro-acetique acide
UKS : Urée potassium SDS
vep : Vitesse de migration
d SDS / PAGE : double SDS/PAGE

SUMMARY
Wild species of the genus Medicago, are of great economic interest in an intelligent
farming, in fact, they improve the yield of cereals such as wheat in rotation (Hireche. 2006).
This is relative to their varieties, it follows that their selection is of great importance, hence
the question of the development of two-dimensional electrophoresistechniquesuitedtothem.
Our work aims to present the different methods, largely modified this method for
proteomic analysis.
Keywords: Medicago, two-dimensional electrophoresis, proteomics

Résumé
Les espèces sauvages du genre Médicago, sont d’un grand intérêt économique dans un
cadre d’agriculture intelligente, en effet, elles permettent d’améliorer le rendement des
céréales tel que le blé, en système de rotation (Hireche. 2006). Ceci étant relatif à leurs
variétés, il en découle que leurs sélections est d’une grande importance, d’où la question de la
mise au point de la technique d’électrophorèse bidimensionnelle adaptée à ces dernières.
Nôtre travail consiste à présenter les différents procédés, en grande partie modifiés de cette
méthode d’analyse protéomique.
Mots clefs : Médicago, électrophorèse bidimensionnelle, protéomique.

Introduction générale

Introduction générale
1
Les espèces annuelles du genre Medicago L. peuvent jouer un rôle important dans
l’amélioration de la production fourragère en Algérie en produisant un fourrage en quantité et
de qualité supérieure. Elles assurent l’amélioration de la flore des jachères pâturées, entrent
facilement dans la rotation avec les céréales, se régénèrent par auto-semis, et constituent une
bonne réserve de semences dans le sol. Elles ont une capacité unique de fixer l'azote
atmosphérique à travers des relations symbiotique avec les bactéries du sol appelées
Rhizobium. Il en résulte de cette interaction, un apport important de nitrogène, donc une forte
quantité de protéines dans les légumes. C'est pourquoi elles sont considérer comme la source
majeur en alimentation humaine et animale, de plus elles assurent l'azote au sol et permettent
d'éviter l'utilisation de fertilisant chimique (Zhentian. 2005).
De la même façon que l’ensemble des gènes d’un organisme constituent son génome,
l’ensemble des protéines d’une cellule ou d’un tissu constituent son protéome. Alors que le
génome reste constant au cours de la vie d’un même organisme, le protéome de la cellule
change en permanence en réponse aux différentes conditions de vie de la cellule. L’analyse
protéomique, ou analyse du protéome d’une cellule, va donc étudier l’expression des
protéines cellulaires, ainsi que les variations quantitatives de cette expression au cours de
divers mécanismes cellulaires, tel que la différenciation, et l’adaptation aux stress biotique et
abiotiques (Rabilloud. 2003).
Afin d’étudier l’ensemble des protéines exprimées par un organisme ou un type
cellulaire donné, des methodes il convient de mettre en œuvre des méthodes analytiques et
reproductibles. L’électrophorèse bi-dimensionnelle (2-DE) introduite par O’Farrel en 1975
(O’Farrel. 1975) est l’une de ces méthodes. Elle permet, à l’issue de deux migrations
électrophorètiques successives, de visualiser un grand nombre de protéines de façon
simultanée. Cette technique permet l'exploration de la variabilité génétique intra et inter
spécifique qui est une étape préliminaire avant tout programme d'amélioration ou de sélection
variétale (Haynes et Yates. 2000).
La protéomique exige l'utilisation d'échantillons biologiques de qualité. L'extraction de
protéines à partir de tissus, de cellules isolées ou de liquides physiologiques est réalisée à
l'aide de tampons appropriés, mis au point en considérant la nature des protéines à étudier
(protéines cytosoliques, membranaires, nucléaires…), ces protéines doivent être maintenues à

Introduction générale
2
l'état soluble durant tout le processus analytique. En effet, les tampons d'extraction à pH bien
déterminé, sont constitués dans des proportions variables, de mélanges d'agents réducteurs, de
détergents, voire de solvants organiques. Ils sont généralement supplémentés d'inhibiteurs de
protéases. Les protocoles expérimentaux doivent éviter les contaminations par des acides
nucléiques, des lipides et les sels. Ces contaminants peuvent perturber la séparation des
protéines par électrophorèse bidimensionnelle (Sheoran. 2009).
L’objectif de ce travail est d'élaborer des protocoles efficaces et reproductibles
d'extraction de protéines totales et de préparation des gels de la premier dimension en
l'occurrence l’isoelectrofocalisation et de la deuxième dimension (SDS-PAGE), pour la mise
au point de la technique d'électrophorèse bidimensionnelle, sur des espèces annuelles
sauvages du genre Medicago.

1
Partie IRevue bibliographique

Partie I : Revue bibliographique
3
1. Généralités sur le Medicago
1. 1. Taxonomie et génétique
Le genre Medicago (luzerne) regroupe de nombreuses espèces de plantes proches des
trèfles, appartenant comme eux à la famille des Fabacées (ou Légumineuses), et à la sous
famille des Papilionideae. Selon Lesins et Lesins (1989), il comporte 20 espèces herbacées
pérennes, 34 herbacées annuelles et une arbustive (M. arborea). La luzerne cultivée comprend
deux espèces botaniques différentes, la luzerne faucille (Medicago falcata, L) et la luzerne
commune Medicago sativa, mais il existe une luzerne intermédiaire, Media pers ou Medicago
varia martyr, hybride de Medicago sativa x Medicago falcata (Camille. 1980. Cité par
Hireche. 2006).
Dans une étude taxonomique plus récente et complète du genre, Small et Jomphe
(Small et al., 1989) décrivent 83 espèces de Medicago. Environ les deux tiers de ces espèces
sont annuelles, tandis que les autres, dont le M. sativa, sont vivaces. Il existe au sein de ce
genre des espèces allogames et autogames, diploïdes avec 2 n= 16 chromosomes (le nombre
de base X = 8 et rarement X = 7) et d’autre sont tétraploïdes avec 2 n = 4 X = 32
chromosomes (Lesins et Lesins, 1979).
1. 2. Origine géographique et aire de répartition
Il est admis que le centre d'origine du genre Medicago est le Croissant fertile,
recouvrant les pays ou régions actuelles de Turquie, d’Iran, d’Irak, du Sud du Caucase et du
pourtour méditerranéen (Prosperi et al., 1993). Il existe plus spécialement sur l'ensemble du
pourtour méditerranéen 20 autres espèces de luzernes pérennes à allogamie non stricte,
diploïdes ou tétraploïdes (souvent les deux niveaux coexistent chez la même espèce) et 34
espèces annuelles, toutes autogames, et diploïdes à l'exception de Medicago sativa qui est
originaire des hauts plateaux iraniens, et de Medicago falcata qui est originaire de la Serbie
Occidentale, ce qui explique sa remarquable résistance au froid (Prosperi et al., 1995). Cette
double origine géographique et génétique fait que la luzerne est une des espèces les plus
répandues du globe (Hirech. 2005).

Partie I : Revue bibliographique
4
1. 3. Description botanique de la plante Medicago
Chez le genre Medicago, la morphologie et l’architecture varient fortement entre les
génotypes de la même espèce annuelle, elles sont très dépendantes de l’environnement et
des conditions de culture (Delphine. 2006. Cités par Moulay).
Ce sont des plantes annuelles ou vivaces, le plus souvent herbacées, parfois aussi de
petits arbustes comme Medicago arborea, à feuilles trifoliolées, dont plusieurs espèces
sont cultivées comme plantes fourragères. Chaque axe sur une plante est composé de
phytomers. Un phytomer est l’unité élémentaire utilitaire d’un axe. Les feuilles (Figure 01)
sont trifoliées à folioles finement dentées au sommet et à inflorescence en grappe ou
racème de 10 à 20 fleurs, les fleurs sont violettes, pourpres ou bleuâtres chez la luzerne
commune, jaunes chez la luzerne faucille et violettes bigarrées de jaune chez la luzerne
intermédiaire (Camille. 1980).
M. laciniata M. ciliaris M. truncatula
Figure 01 : Feuilles de différents écotypes du genre Medicago (Delalande. 2007).
Le fruit est une gousse plus ou moins enroulée (Figure 02), soit spiralée (de 1 à 4
spires) pour Medicago sativa. La graine est plus ou moins réniforme est longue d’environ
2,5 mm (Camille. 1980).
Figure 02 : Gousse de Medicago Truncatula (Ramakrishnan et al. 2006).

Partie I : Revue bibliographique
5
1. 4. Germination et développement physiologique de la plante
La plante passe par différents stades végétatifs : le stade S1 est représenté par
l’apparition des deux cotylédons à la levée ; au cours du stade S2 l’émission des deux feuilles
cotylédonaires ou unifolié se fait ; au stade S3 (stade trifoliées), les feuilles composées de
trois folioles, rattachées à la tige par un pétiole apparaissent, et la tige grandit en produisant
des feuilles alternées ; puis au stade S4, les bourgeons émis forment des tiges secondaires ; Au
stade S5, les bourgeons donnent naissance à des tiges feuillées, alors que le bourgeon axillaire
de la première feuille aboutit à une tige secondaire, deux autres tiges secondaire pousse
depuis le niveau des cotylédons, les luzernes de type non dormant produisent plus de tiges
secondaires à partir du niveau du cotylédon que les types dormants, dont la croissance est
stoppé en hiver, c’est cet ensemble de tiges qui va former le collet, le développement des tiges
suit un ordre bien précis.
On distingue des tiges primaires, secondaires, et tertiaires ; Au stade S6, on observe
l’élongation des entrenœuds avec une croissance de plus en plus rapide, et l’apparition des
boutons floraux, au stade bourgeonnement, les fleurs apparaissent entre le 6ème et le 14ème
entrenœuds, selon les conditions du milieu de culture et le déterminisme génétique ; enfin, le
stade S7 est le stade de la floraison, de la fécondation, et de la maturité des graines,
l’accroissement en matière sèche se poursuit suivant une courbe en S, jusqu’à la pleine
floraison. Dès l’apparition des boutons floraux, l’élongation est très ralentie. Parallèlement la
proportion de matière sèche s’accroît dans la plante entière, mais celle des feuilles (riche en
protéines) diminue (Hireche. 2006).
1. 5. Intérêt de la plante
Les espèces annuelles du genre Medicago présentent un intérêt économique très
important. Cultivée pour la production de foin, la luzerne est utilisée depuis quelques
décennies par un nombre croissant d’usines de déshydratation, pour la fabrication d’aliments
d’excellente qualité. Elle est aussi utilisée sous forme de fourrages déshydratés, ou de farines
dans l’alimentation animale. Au-delà de cette fonction de base, les scientifiques s’intéressent
aux vertus d’un de ses constituants protéiques, la RuBisCO (Ribulose-1,5-bisphosphate
carboxylase oxygenase).

Partie I : Revue bibliographique
6
Celle-ci pourrait avantageusement remplacer les protéines de soja dans la nutrition
humaine, car elle est beaucoup plus riche en acides aminés essentiels, et se rapproche
davantage des protéines laitières (Hireche. 2006).
Ces plantes fourragères sont souvent utilisées dans les systèmes de rotation « céréales-
luzerne » (tableau 01). En Algérie, l'intégration céréaliculture pâturage temporaire d'espèces
annuelles de Medicago, nécessite la disponibilité d'écotypes adaptés, et ce au dépend des
jachères, peu productives en zones céréalières (Yahia et Fyad-Lameche. 2003).
L’interaction mutuelle entre les légumes et les microbes (Rhizobium, Sinorhizobium,
Brady rhizobium…etc.) représente un aspect important de la biologie des plantes, qui ne peut
être étudiée en utilisant le modèle Arabidopsis thaliana, car cette dernière ne peut établir une
symbiose avec les bactéries rhizobia, l’utilisation d’espèces tels que, le soja ou, l’alfa alfa
pour étudier la biologie des plantes est très compliquée, vu leurs génomes polyploïdes, et du
très grand nombre de séquences répétitives dans leur génomes. C’est pourquoi, Medicago
truncatula a émergé durant les dix dernières années comme modèle dans l’étude moléculaire
et génétique des légumineuses (Barker. 1990), cela est due en partie au petit nombre diploïde
de son génome et de sa pollinisation autogame, ainsi que sa capacité de se régénérer par
embryogenèse somatique (Bell. 2000 ; Cook. 1997).
Tableau 01 : Rendement du blé, après association aux légumineuses (Hireche. 2006).
Précédent cultural Rendement en blé en %Ecotype 1ère année 2ème année
M. littoralis 132 138M. scutellata 120 118M. truncatula 132 118
Trifolium Subterranéum 131 130M.sativa 118 116
Végétation spontanée 100 100
2. Préparation des échantillons
Les modalités de préparation de l’échantillon conditionnent la réussite d’une analyse
par électrophorèse bidimensionnelle (2-D). Les protéines cellulaires diffèrent par leur
solubilité en relation avec leurs caractéristiques physico-chimiques.

Partie I : Revue bibliographique
7
Les protéines cytoplasmiques sont généralement solubles dans l’eau, tandis que les
protéines nucléaires et membranaires sont souvent extrêmement hydrophobes et insolubles en
milieu aqueux (De Marqui. 2006 ; Stanley. 2003). L’absence de méthodes universelles
d’extraction des protéines implique une optimisation des conditions de préparation des
échantillons selon le type d’échantillon, et de la question biologique posée.
La solution de solubilisation est composée généralement d’un agent chaotrope (agent
biochimique désorganisant la structure protéique), d’un détergeant non ionique ou
zwitterionique, d’un agent réducteur (prévient l’oxydation des groupements SH (thiol) et
rompt les ponts disulfures), et d’un mélange d’ampholytes (composé chimique pouvant jouer
le rôle d’un acide ou d’une base) qui facilitent la séparation des protéines lors de
l’isoélectrofocalisation (Görg. 2000 ; Görg. 2004). Afin de minimiser les risques de
dégradation protéique lors de la préparation de l’échantillon, un mélange d’inhibiteurs de
protéases est couramment inclus dans la solution de solubilisation, afin de sauvegarder la
structure tridimensionnelle des protéines.
2. 1. Extraction non dénaturante
Ce type d'extraction est utilisé en particulier pour l’étude de la protéomique
fonctionnelle, et structurelle ainsi que pour les analyses ultérieures d’identification des
protéines (spectrométrie de masse). Dans cette méthode, la diminution du pH pendant la lyse
cellulaire effectuée pour la récupération des protéines solubles est compensée par des tampons
(Zivy. 1983).
2. 1. 1. Dégradation protéolytique et inhibition des protéases
La dégradation protéolytique est causée par les protéases, ces derniers sont des
enzymes qui catalysent l’hydrolyse des liaisons peptidiques (Huffaker. 1990). La présence des
protéases dans les graines dormantes a été confirmée par leur identification à ce stade de vie
(Duarte. 1996) où ils coexistent avec les protéines de réserve (St Angelo et al., 1969 ;
Mikkonen et al., 1986).
Généralement, des inhibiteurs de protéases sont utilisés pour éviter la dégradation des
protéines dans les tampons d’extraction non dénaturante.

Partie I : Revue bibliographique
8
Cashmore (Cashmore et al., 1976) ont utilisé un inhibiteur de protéases serine actif,
appelé PMSF (Phenylmethylsulfonyl Fluoride) pour extraire les protéines des graine du pois,
en effet, le PMSF est largement utilisé par de nombreux auteurs (Meza-Basso. 1986 ; Tingey.
1987). Cependant, l’utilisation d’un cocktail de protéases est plus efficace permettant 80%
d’inhibition, plutôt que 50% quand un seul inhibiteur est utilisé (Benchabane. 2008).
Pour éviter la protéolyse, il est possible de jouer sur la spécificité d’action des
protéases, par exemple, les protéases lysosomales sont actives sur des pH faibles et inhibés à
des pH élevé, il existe aussi des inhibiteurs de protéases qui s’attaquent aux substrats de
l’enzyme et empêchent la protéolyse (trypsine inhibiteur) (Rabilloud. 1996).
2. 1. 2. Composés phénoliques
Les polyphénols sont une famille de molécules organiques largement présente dans le
règne végétal, caractérisés par la présence de plusieurs groupements phénol associés en
structures plus ou moins complexes généralement de haut poids moléculaire, ces composés
sont les produits du métabolisme secondaire des plantes (Bate-Smith. 1962 ; Levin. 1971 ;
Meier et al., 2008 ).
Les plantes sont riches en composées phénoliques surtout dans les tissus verts, ou ils
s’accumulent principalement dans les vacuoles sous différentes formes, souvent solubles et
posent problème en extraction non dénaturante. En effet, l’oxydation des composés
phénoliques par la phénoloxydase ou par les peroxydases produit des traînais ou des stries
dans le gel (Hari. 1981).
Des inhibiteurs de la phenyloxydase tel que le PVP (Polyvinyl Pyrrolidone) sont
ajoutés au tampon d’extraction (Zivy et al., 1983 ; Mayer et al.,1987 ; Chory, 1987), pour
prévenir cette oxydation. Dans ce contexte, les résultats obtenus sur les bourgeons apicaux et
les jeunes plantules de la moutarde blanche (Sinapis alba) ont montré l’efficacité de ces
inhibiteurs (Cremer. 1985). Alternativement ou quelquefois en combinaison avec le PVP, des
agents réducteur tel que l’Ascorbate, β-mercaptoethanol (Cashmore. 1976 ; Meza-Basso.
1986 ; Baszczynski. 1982 ; Ginzburg. 1986 ; De Vries. 1982 ; Lin. 1984), le Potassium
Metabisulfite (Uemura. 1984), et le DTT (Zivy. 1983 ; Mayer. 1987) sont ajoutés pour éviter
l’activité phénoloxydase (Rabilloud. 1996).

Partie I : Revue bibliographique
9
2. 2. Extraction dénaturante
Cette méthode d'extraction est réalisée dans le cas des analyses en protéomique
analytique. Dans laquelle la structure tridimensionnelle des protéines est modifiée, et souvent
réduites en polypeptide pour faciliter leur solubilisation. Quelques méthodes et agents
chimiques sont décrits ci-dessous.
2. 2. 1. La méthode SDS
La méthode d’extraction par l’SDS à 60 C° suivis d’une précipitation à l’acétone a été
développée par Harrison et Black (Harrison et al., 1982) pour extraire les protéines foliaires et
des cellules du mésophile de digitaria sangunalis. En effet, la dénaturation thermique
fonctionne en synergie avec la dénaturation par l’SDS et offre une inactivation rapide des
protéases. Cependant cette procédure conduit à la précipitation des polypeptides de faible
poids moléculairen (Gallagher et Leonard. 1986). L’optimisation de cette technique (SDS 3
min à 100 C°) par Ames et Nikaido (Ames et al. 1976) a permis d'obtenir une meilleure
solubilisation des protéines membranaires de salmonella typhimurium, comparée au tampon
de lyse d’O Farrell, qui n’est pas efficace pour ce type de protéines. Une autre technique
combinant l’urée et le Nonidet P-40 (NP-40) avec le carbonate de potassium à un pH de 10.3
a permis une solubilisation efficace des protéines membranaires des graines végétale, sans
l’utilisation de l’SDS (Horst et al., 1980).
2. 2. 2. Précipitation directe
Cette procédure consiste en une précipitation des proteines par le TCA à 10% w/v,
suivis d’un rinçage à l’acétone des culots protéiques pour les solubiliser ensuite dans un
tampon UKS ( Urée, Potassium, SDS ) à fin de les analyser par électrophorèse 2-D (Granier.
1988). D'ailleurs, la précipitation à l’acétone a été utilisé par Vierling et Key (Vierling et al.
1985). Cependant, la combinaison de ces deux agents de précipitation et dénaturation (TCA,
Acétone) a été utilisé par Zivy (Zivy. 1986), et a donné des gels de bonnes qualités.
En outre, cette technique a été testé sur différent matériel végétal tels que la tomate,
pétunia, latex, noix de coco, pollen du blé, maïs, tournesol, levure, et même dans les tissus
animal (les nématodes), les résultats obtenus ainsi ont permis non seulement d'améliorer la

Partie I : Revue bibliographique
10
résolution des gels, mais aussi la disparition des stries. D'autre travaux ont montré que cette
technique s'est révélé plus rapide comparé à l’extraction au phénol permettant donc de
préparer plusieurs échantillons nécessaire dans les études de polymorphisme (Granier. 1988).
2. 2. 3. Extraction au phénol
Cette méthode était développée par Schuster et Davis (Schuster et Davis. 1983).
L'extraction au phénol consiste à extraire les protéines, aussi bien que les acides nucléiques
présents dans l’espace aqueux (Du Pont. 1988). Pour cela, on ajoute un tampon tris mélangé à
un volume égal de phénol hydro-saturé, puis on procède à une séparation des deux phases par
centrifugation (Hurkman et al., 1986).
Non seulement, cette technique inactive les enzymes sans recours à la chaleur, et
donne une résolution des gels et des spots comparable à la méthode SDS, mais aussi, elle
élimine les polysaccharides, et les détriments potentiels des extraits des plantes, ainsi que les
grosses molecules de glycoprotéines. (Rabilloud. 1996). De plus les culots préparés par cette
procédure semblent être les plus pure et les plus soluble (Cilia. 2009 ; Sheoran. 2009).
Cette méthode est utilisée par plusieurs auteurs, sur differents organes végetals tels
que les feuilles et les graines d’épinard (Guy. 1987), les racines et les bourgeons de l’orge
(Ramagopal. 1987) ou elle s'est avérée efficace pour la solubilisation des protéines
membranaires.
2. 2. 4. Solubilisation différentielle
Cette technique a été introduite par Moloy (Moloy et al., 1998) pour extraire les
protéines totales d’E. Coli. Elle est réalisée par la succession de trois étapes d’extraction dont
la dernière est la plus performante : la première étape est initiée par la lyse cellulaire en
utilisant le tris ; la deuxième étape, l’échantillon résultant est soumis à une extraction, cette
fois ci le tampon IEF conventionnel (Urée, CHAPS, DTT) ; la troisième étape, consiste à
traiter les culots (riches en protéines membranaires et représente 11% (w/w) du matériel de
départ ), par un tampon à base d’urée, thio-urée, CHAPS, TBP.
C'est ainsi qu'onze protéines de la surface membranaires d’ E. coli., ont été identifiées
dans un gel 2-D à partir de la dernière étape d’extraction (Herbert. 1999).

Partie I : Revue bibliographique
11
2. 2. 5. Cas des ponts disulfures
La rupture des ponts disulfures est réalisée par des agents thiols libres, de faible
volatilité et de petite concentration, tels que le β-mercaptoethanol, Dithiothreitol (DTT) ou
Dithioerythritol (DTE), Cysteamine (Rabilloud. 1996). Il faut signaler que le DDT ou DTE
présentent des inconvénients malgré leur utilisation à des concentrations assez faibles par
rapport au thiol protéiques, par exemple l’oxygène dissous est capable de ré-oxydé les thiols
en bisulfures de sorte que le thiol libre soit consommé, cela conduit à la ré-oxydation des
thiols protéiques en disulfures, et dans quelques cas a la suroxydation des protéines libres en
faux disulfures (Rabilloud. 1996).
2. 2. 6. Rôle des Chaotropes et des surfactant dans l’extraction dénaturante
Un agent chaotropique comme l’urée est une molécule qui détruit les faibles
interactions intramoléculaires (non-covalente), telles que les liaisons hydrogène, les forces de
van der Waals et les liaisons hydrophobe et dénature, ainsi les macromolécules biologiques
(protéine, l'ADN et l'ARN). L’introduction par Rabilloud (Rabilloud et al., 1997) de la thio-
urée et de l’urée s'est avéré très efficace dans la solubilisation des protéines membranaire et
des fractions cellulaires (chloroplaste, mitochondrie).
Un surfactant ou tensioactif est un composé qui modifie la tension superficielle entre
deux surfaces, Il permet ainsi de solubiliser deux phases non miscibles. Il existe plusieurs
substances ioniques non compatibles avec l’IEF, donc l’utilisation de l’SDS n’est pas
recommandée et on est limité aux surfactant non ionique ou zwitterionique, raison pour
laquelle le Triton X-100 et le Nonidet P-40 sont traditionnellement utilisés ainsi, que les
surfactants à base de sucre tel que l’octyl glucoside. Cependant dans les dernières années le
sulfobetaine CHAPS est devenus le surfactant de choix en association avec l’urée (Granier.
1988).
La combinaison de ces Chaotropes avec les surfactants tel que le monosulfobetaine
permet l’analyse d’une gamme d’échantillons variés, surtout en IEF où les surfactants sont
indispensable pour solubiliser les résidus hydrophobes en exposition après leur traitement par
les chaotropes (Granier. 1988).

Partie I : Revue bibliographique
12
2. 3. Contaminants des préparations protéiques
2. 3. 1. Sels
En général, une haute quantité de sel est présente dans les organismes halophiles ou
dans certains fluides biologiques (urine, sueur, plasma…etc.), des méthodes de concentration
des protéines et de dialyses (dialyseurs sous vide, centrifugation, etc.) sont souvent utilisées
(Manabe. 1982). Cependant, une perte de protéines par adsorption et par diffusion à travers
les membranes de dialyse des molécules de faible poids moléculaire est observée (Rabilloud.
1996). Des techniques prometteuses basées sur la précipitation des protéines avec des
colorants ont été mises au point, ces méthodes sont des substituts efficaces de la précipitation
par la TCA pour l’élimination des sels et sur d’autre techniques modernes (Kole. 2010 ;
Marshall. 1995 ; Bollag. 1991).
2. 3. 2. Lipides
Les lipides se lient aux protéines par des interactions hydrophobes affectant leur
charge et leur Mr (masse moléculaire), ce complexe protéine-lipide est insoluble dans les
solutions aqueuses, conduisant généralement à l‘échec de la pénétration des protéines dans le
gel IEF (Shaw et al., 2003), cette interaction peut être concurrencé par l’addition de quantité
importante de détergents et par la précipitation à base de TCA/acétone (Nagaraj. 2007 ;
Hopkinson. 2005).
L’élimination finale du solvant est très importante pour éviter les problèmes
d’émulsion ou de précipitation. Aussi, si les culots obtenus sont séchés excessivement pour
l’élimination complète du solvant, il serait impossible de ré-solubilisé les protéines même
dans les tampons fortement dénaturant. Ainsi l’achèvement d’un cycle propre et reproductible
de délipidation/ré-solubilisation est difficile du moment qu’une délipidation par des solvants
organique est requise (Rabilloud. 1996).
2. 3. 3. Acides nucléiques
Les acides nucléiques sont de grosses molécules, qui augmentent considérablement la
viscosité de la solution et obstruent les pores du gel (Shaw et al., 2003). Ils se comportent
comme des poly-anions, et se lient aux protéines par des interactions électrostatiques. Le
complexe formé se lie aux carriers ampholytes du tampon IEF (Galante. 1976). Cet ensemble

Partie I : Revue bibliographique
13
se lie à son tour aux protéines et donne une fausse focalisation, de plus, il augmente le nombre
de stries et de traînées dans le gel 2-D (Heizmann. 1980).
La digestion enzymatique RNase, DNase est utilisée pour éliminer ces acides
nucléiques (O’Farrell. 1975). Elle est accompagnée de l’addition d’ampholytes au tampon
d’extraction, et suivie d’une précipitation/centrifugation des produits de la digestion par la
TCA (Shirey. 1969 ; Chaudhury. 1973 ; Rabilloud. 1986 ; Shaw et al., 2003). Cependant,
certains acides nucléiques se lient fortement aux protéines même en présence d’une grande
quantité d’urée (qui dénature les protéines) (Sanders. 1980). Ces protéines peuvent être
solubilisées par des compétiteurs de cations tel que la prolamine (Sanders. 1980) ou la
lécithine (Willard. 1979) à pH acide, ou bien le pH d’extraction peut être augmenté de sorte
que les protéines se comportant comme des anions et sont répulsées par les acides nucléiques
(Rabilloud. 1996).
2. 3. 4. Polysaccharides
Tout comme les acides nucléiques mais à moindre titre, les polysaccharides (amidon,
glycogène, etc.) augmentent la viscosité de la solution et obstruent les pores du gel vu leur
charge et leur Mr. Les méthodes d’ultracentrifugation utilisées pour éliminer les acides
nucléiques, le sont pour les polysaccharides. Aussi, la précipitation sélective des protéines par
la TCA, sulfate d’ammonium, phénol/ammonium, acétate (Hurkman. 1986) ou par des
colorant (Marshall. 1992 ; Marshall. 1995) suivie d’une ré-solubilisation, peut être envisagé
comme solution pour leur l’élimination (Rabilloud. 1996).

Partie I : Revue bibliographique
14
3. Électrophorèse
3.1. Historique
Le début du XVIIIème siècle, marqué par les travaux de Charles de Coulomb (1736-
1806), Alessandro Volta (1745-1827) et André-Marie Ampère (1775-1836) ont permit
l’émergence des premières lois de l’électro-statistique et de l’électricité. En 1859, l’Allemand
Georg Hermann Quincke (1834-1924) découvre qu’il est possible de déplacer des particules
colloïdales (sous forme de colle gélatineuse) sous l’action d’un champ électrique : c’est la
cataphorèse. Par la suite, Hermann Von Helmholtz (1821-1894) développe l’électro-osmose :
sous un champ électrique, il observe que des particules chargées se déplacent vers le pôle de
signe opposé à leur charge. En 1892, S.E. Linder et H. Picton, imaginent d’exploiter cette
observation pour la séparation de particules chargées. En 1937, c’est le Suédois Arne
Wilhelm Kaurin Tiselius (1902-1971) qui met en œuvre cette technique de séparation pour les
protéines du sérum sanguin et du lait, ce qui lui vaudra le prix Nobel en 1948. Sa méthode de
fractionnement en phase liquide est alors trop onéreuse en matériel et en personnel pour la
pratique courante, mais il la perfectionne et en 1950 il met au point l’électrophorèse sur
papier, plus simple et permettant une utilisation plus large (Groulade. 1978 b). Depuis 1957,
l’électrophorèse sur acétate de cellulose autorise une meilleure individualisation des fractions
et ainsi l’établissement de tracés plus expressifs.
Les années suivantes, l’utilisation de gel de polyacrylamide permet l’obtention d’un
fractionnement plus important, de plus l'introduction de l'électrophorèse bidimensionnelle par
O' Farrell (O'Farrell. 1975) et le développement d’autre variante de cette technique (Schägger
et von Jagow, 1991; Schägger et al., 1994) comme la BN-PAGE et CN-PAGE a amélioré le
pouvoir résolutif de l'électrophorèse et a permit la détection des modifications post
traductionnelle jusque là impossible à mettre en évidence par les méthodes
monodimensionnelle.
Aujourd’hui cette technique, qui ne cesse de s’améliorer par la diversité des supports
et des techniques de réalisation, est devenue un outil indispensable dans de nombreux
laboratoires, au niveau de la recherche en génomique et en protéomique au sens large. En
amélioration des plantes, cette technique est utilisée dans l'identification variétale et la mise
en évidence des protéines de stress biotique et abiotique.

Partie I : Revue bibliographique
15
3. 2. Principe
Les méthodes connues sous le nom d’électrophorèse, sont basées sur le déplacement
de molécules chargées sous l’effet d’un champ électrique. Quand les molécules sont de charge
négative (anions) se dirige vers l’électrode positive ou anode. Quand les molécules sont de
charge positive (cation), ils se dirigent vers l’électrode négative (cathode). Le champ
électrique est obtenu par un générateur de courant continu, dont les bornes sont reliées à la
cathode et à l’anode. Le support du champ électrique est constitué par un tampon d’un pH et
d’une concentration convenables, dont les ions conduisent le courant. Les phénomènes de
séparation se produisent dans un support poreux approprié, il peut s’agir de papier filtre
(électrophorèse sur papier), d’un dérivé de la cellulose (acétate de cellulose), d’un gel extrait
d’algue (agarose) ou d’un gel de polymère organique (polyacrylamide) où le calibre des
micro-canalicules internes des mailles du réseau de polymère, gouverne la vitesse de
migration des molécules à travers ce réseau.
3. 3. Migration électrophorètique
En électrophorèse, la séparation est permise par les différences de vitesses de
migrations existant entre les différents analytes. La vitesse de migration électrophorétique
d’un analyte (vep) mesurée en mètre par seconde (m.s-1). Elle s’exprime en fonction de sa
mobilité effective (mep) mesurée en mètre carré par seconde par volte (m2.s-1.V-1) multipliée
de son champ électrique appliqué (E) mesurée volte en par mètre (v.m-1) : vep = mep E
En réalité, la mobilité électrophorètique d’un composé dépend de plusieurs paramètres
: la mobilité absolue du composé ; le pH et la force ionique de l’électrolyte ; et de l’interaction
avec des molécules présentes dans le milieu de séparation. Si l’analyte présente des propriétés
acido-basiques, le pH du tampon est important, car dans ce cas, son degré d’ionisation et par
conséquent sa mobilité électrophorètique sera influencée par le pH. En outre, quelles que
soient les propriétés acido-basiques de l’analyte considéré, si ce dernier est chargé, sa
mobilité électrophorétique dépendra de la force ionique de l’électrolyte support. Enfin, la
mobilité relative des molécules peut être spécifiée, en la calculant relativement à la distance
de migration de substances standard, généralement des colorants tel que le bleu de
bromophénol (Chrambach. 1985).

Partie I : Revue bibliographique
16
3. 4. Quelques méthodes classiques d’électrophorèse
3. 4. 1. Électrophorèses en zone
L’électrophorèse en zone regroupe toutes les techniques dont le principe de séparation
est basé sur la différence de mobilité électrophorétique. Deux particules de vitesses de
migration données parcourent des distances distinctes en un intervalle de temps similaire,
elles vont ainsi être localisées dans des zones différentes. L'échantillon (un mélange d'espèces
anioniques et cationiques) est introduit dans le système qui contient une solution d’électrolyte
support. En général, la concentration de l'électrolyte support est élevée par rapport à celle des
espèces ioniques de l'échantillon et permet de maintenir un pH relativement constant, et un
gradient de potentiel uniforme dans le système entier. Les espèces ioniques de l'électrolyte et
de l'échantillon ont une certaine mobilité effective, et quand un courant électrique passe à
travers le système, ces espèces ioniques migrent avec des vitesses spécifiques : les cations
migrent vers la cathode et les anions vers l'anode. Il en résulte un flux d'ions de l'électrolyte
support suivi par un flux d'ions de l'échantillon sous forme de bandes (Viera-Nunes. 2006).
3. 4. 2. Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis (SDS-PAGE)
Cette technique a été introduite par Shapiro (Shapiro et al., 1967). Dans cette méthode
les protéines sont exclusivement séparées selon leur poids moléculaire, car l’ajout du SDS
(détergent anionique) masque la charge des protéines, pour donner une charge nette constante
par unité de mass qui est de 1.4 gramme SDS par gramme de protéine, et rompt les liaisons
hydrogène, donc, déplie la structure tertiaire et quaternaire des protéines. Il existe une relation
linéaire entre le logarithme du poids moléculaire et la distance de migration relative de la
micelle SDS-polypeptide. Par rapport aux gels homogènes, les gels gradués permettent la
séparation d'une large gamme de protéines, et une meilleure linéarité, et les bandes sont mieux
définit grâce à la diminution des phénomènes de diffusion (Westermeier. 2004).
3. 4. 3. Electrophorese native (CN-) et (BN-) PAGE
L’électrophorèse native en gel de polyacrylamide est un outil performant pour
l’analyse des complexes de protéines membranaire, la plus grande révolution dans cette
technique a été l’introduction de la BN-PAGE. Quoique ces techniques soient très
performantes, ils ne peuvent être appliqués à tous les systèmes d’analyse avec la même
résolution. Dans la CN-PAGE les protéines migrent selon leur poids moléculaire et leur

Partie I : Revue bibliographique
17
charge native puisque les protéines ne sont pas dénaturées, dans ce cas seul les protéines
acides pI <5.7 sont séparées, pour la BN, le colorant bleu de Coomassie présent dans le
tampon cuve (cathode) se lie aux protéines leur conférant une charge négative comme pour le
cas de l’SDS, cependant le bleu de Coomassie n’altère pas la structure tridimensionnelle,
c’est une methode à decalage de charge qui permet la séparation des protéines basiques.
(Schägger et von Jagow. 1991; Schägger et al., 1994).
3. 4. 4. Électrophorèse discontinue
Elle a été introduite par Ornstein et Davis (Ornstein et Davis. 1964). La discontinuité
est basée sur quatre paramètres : la structure du gel (diamètre des pores) ; le pH du tampon ;
la force ionique du tampon ; la nature des ions dans le gel et dans les tampons électrodes.
Dans cette technique, deux gels sont superposés, le gel de concentration (stacking gel), aux
larges pores, et le gel de résolution aux mailles plus fines. L’SDS peut être ajouté lors de la
préparation des solutions du gel et aux tampons d’électrodes pour donner une charge négative
globale aux protéines (Ornstein. 1964).
3. 4. 5. Électrophorèse en gel gradient
Il ne s’agit pas à proprement parler d’une nouvelle méthode d’électrophorèse mais
plutôt d’un autre mode de fonctionnement des différentes méthodes décrites précédemment
(électrophorèse de zone, électrophorèse en disque…etc.). Pour obtenir une concentration
graduelle (figure 03), on mélange continuellement à l’aide d’un agitateur, deux solutions
monomérique d’acrylamide de différentes concentrations. La densité de la solution la plus
concentrée peut être augmentée par le glycérol ou le sucrose de sorte que les deux couches ne
se mélangent pas dans les plaques. Ainsi, on obtient un gradient linéaire (Westermeier. 2004).
Le gel gradient a un effet d’affûtage sur les bandes « sharpening effect », il peut être utilisé
pour déterminer le diamètre moléculaire des protéines dans leurs états natifs (Rothe et al.,
1982).

Partie I : Revue bibliographique
18
Figure 03 : Réalisation du gradient du gel (Rothe et al., 1982).
3. 5. Isoéléctrofocalisation en carriers ampholytes libres (CA-IEF)
3. 5. 1. Principe théorique
L’IEF est basée sur la séparation des molécules amphotères, tel que les peptides et les
protéines dans un gel à gradient de pH et sous l’action d’un champ électrique. En effet, dans
ce champ, les molécules migrent vers l’anode ou la cathode jusqu’à ce qu’elles atteignent une
position dans le gradient du pH, où leurs charges nettes sont nulles. La vitesse de migration
est proportionnelle à cette charge qui est elle-même proportionnelle à la différence entre le pH
du milieu et le pI (point isoélectrique) de la protéine.
Quoique, la base théorique de la réalisation d’un gradient de pH naturel (formé
naturellement par un champ électrique) a été décrite par Svensson en 1961 (Svensson. 1961),
la réalisation pratique, est l’œuvre de Vesterberg (Vesterberg, 1969) qui avait synthétisé un
mélange d’isomères aliphatiques d’acides oligoamino-oligocarboxylique. Ces tampons
forment un spectre d’ampholytes de faible poids moléculaire avec des points isoélectriques
qui se rapproche d’une façon continue. Leur formule chimique générale est la suivante :

Partie I : Revue bibliographique
19
Le nombre de racines (x) de la formule (CH2) x–COOH, peut prendre la valeur de 2
ou 3.
Ces carriers ampholytes présentent un avantage majeur au niveau de leur point
isoélectrique : une haute capacité tampon ; une parfaite solubilité ; une conductivité régulière
du courant électrique ; et l’absence d’effets biologique. Presque tous les carriers ampholytes
sont chargés soit positivement (pI élevé) soit négativement (pI faible). La plupart des
solutions commerciales disponibles contiennent 40% (w/v) d'ampholytes. Donc, la
concentration peut ne pas être spécifiée. Usuellement, une concentration de 2 à 2.5% (w/v) de
carriers ampholytes est utilisée pour un gradient moyennement uniforme de pH 3 à 10
(Westermeier. 2004).
Pour la réalisation de l’IEF carriers ampholytes, les gels sont coulés dans des tubes de
1 mm de diamètre et d’une langueur de 24 cm en moyenne, après polymérisation et séparation
des protéines, les gels ont une forme cylindrique après leur démoulage (Westermeier. 2004).
3. 5. 2. Facteurs influençant la formation du pH
La formation du pH peut être influencée par plusieurs facteurs tel que la température et
le voltage. Cependant, il existe deux phénomènes importants qui rendent très difficile
l’établissement de ce pH et limitent l’utilisation de l’IEF en carrier ampholyte, ces deux
facteurs sont le phénomène de plateau et l’éléctrœndosmose.
A) Le phénomène de plateau « plateau phenomenon » : expliqué en détail en 1986 par
Mosher (Mosher et al., 1986), il consiste en une déformation progressive du gradient de pH,
qui tend à être aplati au niveau des pH neutres, et donne des pentes élevées aux pH extrêmes
du gradient. Ce phénomène est dû à la compression des ampholytes les plus acides vers
l’anode et les plus basiques vers la cathode, ce qui conduit à un aplatissement des zones
d’ampholytes dans la région neutre du gradient, et donc à la formation d’un plateau (Poitevin.
2008).
B) L’électrœndosmose : Les supports ou milieux de séparation (gel ou surface), et les
équipements (les plaques de verres, les tubes ou capillaires), peuvent comporter des groupes
chargés (oxyde de silicium dans les surfaces en verres) (Westermeier. 2004).

Partie I : Revue bibliographique
20
Quand les groupes fixés sont négativement chargés en l’occurrence dans les tampons
basiques et neutres. Dans un champ électrique, ils sont attirés par l’anode, mais comme ils
sont fixés ils ne peuvent pas migrés, cela mène à une compensation par un contre flux d’ions
H3O+ en direction de la cathode, c’est le phénomène de l’électrœndoosmose. Dans le gel, cet
effet est observé comme un flux d’eau en direction de la cathode durant la migration
électrophorétique qui emporte les substances solubilisées. L’électrophorèse et
l’électrœndosmose sont ainsi additives, cela donne une mauvaise résolution et provoque la
déshydratation du gel du coté de l’anode (Westermeier. 2004).
Quand les groupes fixés sont positivement chargés, le flux électroendosmotique est en
direction de l’anode (Westermeier. 2004).
Dans ce cas, le gradient de pH ne peut être supérieur à 7,5 – 8, ce qui représente une
limite pour la séparation des protéines basiques. Une méthode spéciale a été développé pour
la séparation de ces protéines (O’Farrell. 1977), il s’agit de la NEPHG-IEF (Non Equilibrum
PH Gradient IsoElectroFocalisation). Cependant, cette technique est faiblement reproductible
(Kuhn. 1980).
3. 5. 3. Autre types de gradient de pH
L’utilisation de gradients de pH immobilisés (IPG pour Immobilized PH Gradient)
permet de s’affranchir des déformations observées avec l’utilisation d’ampholytes porteurs en
solution (Righetti. 2007). Cette technique est principalement utilisée en gel de
polyacrylamide, et les ampholytes porteurs de différents pI, sont liés de manière covalente à
des monomères d’acrylamide (Görg et al., 2000).
3. 6. Électrophorèse bidimensionnelle
Cette technique a été introduite pour la premier fois par O’ Farrell en 1975 (O’ Farrell.
1975), afin de séparer les protéines bactériennes d’e coli, il existe plusieurs variante pour cette
technique selon les paramètres physiquo-chimiques utilisés pour la séparation des protéines
c’est ainsi que pour L’IEF/PAGE ou SDS-PAGE, les polypeptides sont séparés selon leurs
charges native en premier dimension et selon leur poids moléculaire, le même principe est
utilisé dans l’électrophorèse bidimensionnelle CN/PAGE ou SDS-PAGE. Pour la BN/ PAGE
ou SDS-PAGE les protéines sont séparées selon leur poids moléculaire dans les deux
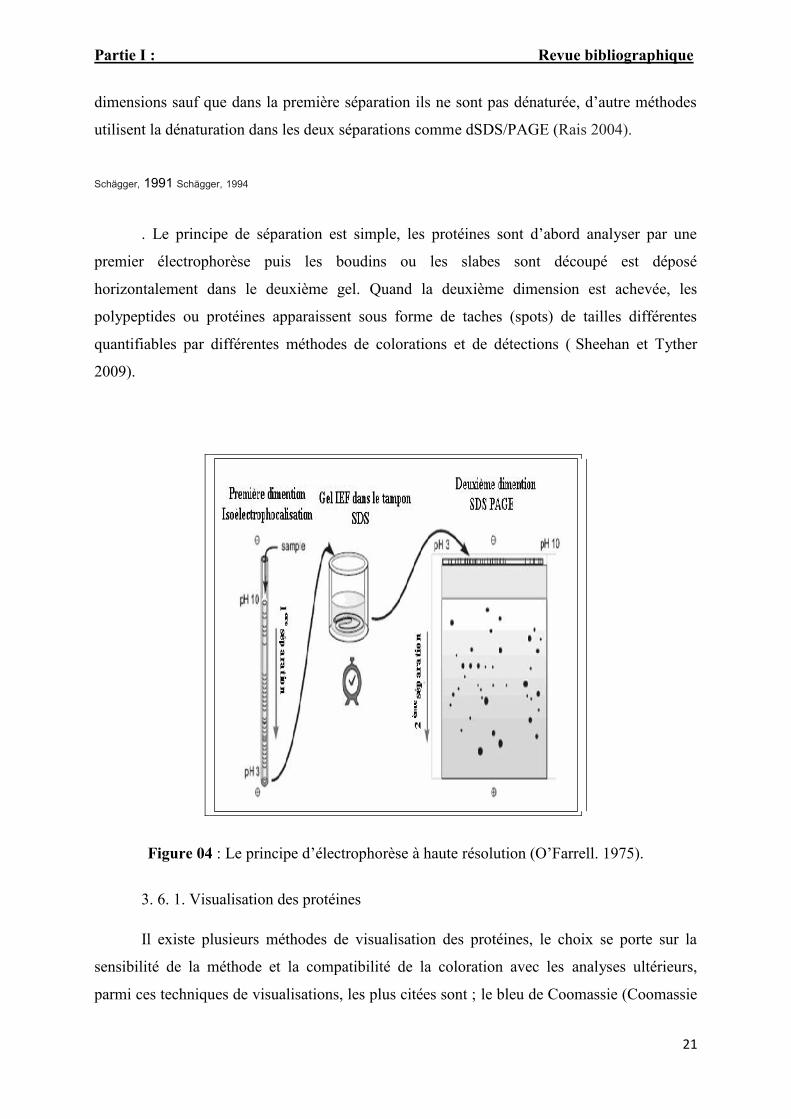
Partie I : Revue bibliographique
21
dimensions sauf que dans la première séparation ils ne sont pas dénaturée, d’autre méthodes
utilisent la dénaturation dans les deux séparations comme dSDS/PAGE (Rais 2004).
Schägger, 1991 Schägger, 1994
. Le principe de séparation est simple, les protéines sont d’abord analyser par une
premier électrophorèse puis les boudins ou les slabes sont découpé est déposé
horizontalement dans le deuxième gel. Quand la deuxième dimension est achevée, les
polypeptides ou protéines apparaissent sous forme de taches (spots) de tailles différentes
quantifiables par différentes méthodes de colorations et de détections ( Sheehan et Tyther
2009).
Figure 04 : Le principe d’électrophorèse à haute résolution (O’Farrell. 1975).
3. 6. 1. Visualisation des protéines
Il existe plusieurs méthodes de visualisation des protéines, le choix se porte sur la
sensibilité de la méthode et la compatibilité de la coloration avec les analyses ultérieurs,
parmi ces techniques de visualisations, les plus citées sont ; le bleu de Coomassie (Coomassie

Partie I : Revue bibliographique
22
Brillant Blue G-250 et R-250) qui est un triphenylméthane anionique, attiré
électrostatiquement par les acides aminés basiques. Cette technique colore la plupart des
protéines et peptides, avec une bonne linéarité quantitative, elle est compatible avec la
spectrométrie de masse, mais elle n’est pas très sensible, car elle requière au moins 1 µg de
protéines par spot. Elle est basée sur les propriétés colloïdales des colorants CBB (Coomassie
Brillant Blue) G-250 et R-250, la sensibilité de cette technique est de l’ordre du ng (Neuhoff.
1988) (Figure 05).
Figure 05 : Gel 2-D de graines matures de M. Truncatula coloré par le Coomassie BrilliantBlue G-250 (Zhentian, et al. 2005).
3. 6. 1. 2. Méthodes de coloration par fluorescence
La méthode la plus récente de coloration est l’utilisation de la fluorescence par
couplage covalent de molécules fluorescentes (Ünlü et al., 1997) ou l’utilisation des
complexes organométalliques fluorescents du type Sypro-Ruby (Berggren et al., 2000 ;
Rabilloud et al., 2001). Ces colorations fluorescentes sont moins sensibles que les techniques
utilisant les ions d’argent. Cependant, elles présentent une très grande gamme de linéarité et
sont parfaitement compatibles avec la spectrométrie de masse (Westermeier. 2004). Deep
PurpleTM est le colorant le plus sensible (Mackintosh et al., 2003), Suivi du Sypro-Ruby
(Berggren et al., 2002). D’autre sont de sensibilité similaire au Bleu de Coomassie, comme le
Nile-Red (Alba et al., 1996), le Sypro-Red et le Sypro-Orange (Steinberg et al., 1996). Ces
colorant sont relativement coûteuses et des scanners à fluorescence et camera CCD sont
nécessaire pour la réalisation de ces techniques (Sheehan et Tyther 2009).

Partie I : Revue bibliographique
23
3. 6. 1. 3. Coloration négative par le zinc d’imidazole
La sensibilité de cette méthode atteint plus de 15 ng (Hardy et al., 1996), elle est
appelée coloration négative, car elle colore uniquement le fond du gel sans les protéines, la
visualisation des spots est plus adaptée pour l’analyse par spectrométrie (Matsui et al., 1997).
Cependant elle ne peut être utilisée pour la quantification.
3. 6. 1. 4. Coloration à l’argent
Introduite pour les gels polyaccrylamide 2-D par Merril (Merril et al., 1979), sa
sensibilité atteint 0.2 ng. Il existe deux principale variantes de cette technique, la coloration au
nitrate d’argent, ou la coloration diamine d’argent. Cette dernière montre une très bonne
sensibilité pour les protéines basiques, mais elle contient quelques solutions caustiques, et
n’est pas compatible avec le tampon tricine (Westermeier. 2004). La coloration à l’argent,
présente une bonne sensibilité, une faible linéarité, et est moyennement compatible avec
l’analyse par spectrométrie de mass (Gharahdaghi et al., 1999 ; Shevchenko et al., 1996).
3. 6. 1. 5. Détection par radioactivité
Comme la coloration les méthodes de détection des protéines par radioactivité (radio-
labelling) varient aussi en termes de sensibilité, l’autoradiographie est presque la méthode la
plus commune de détection, car elle permet l’utilisation d’un nombre variés d’isotopes (P32,
I125, C14, et S35) (Dunn. 1993). Cependant, la détection peut prendre des jours, voir des
semaines, si un grand degré de sensibilité est requis (Dunn. 1993 ; Link. 1999).
3. 6. 2. Analyse de l’image
Avant l’analyse, l’électrophoregram doit être connecté à un ordinateur, lié à une
camera, ou un scanneur ou bien à un densitomètre. Parmi les méthodes d’imagerie disponible
la cameras CCD (Charged Coupled Device) est la plus polyvalente (Stochaj et al., 2003 ).
Cependant, des scanners sont préférés à ces derniers pour l’acquisition d’images de haute
résolution et pour obtenir un fond homogène allant du centre du gel jusqu’aux deux bords.
L’image est enregistrée par un autre scanneur munie de fonctions de modifications du mode
de transmission de lumière, la fluorescence ou la radioactivité sont détectées par des lasers
spéciaux. Pour l’automatisation, des spots protéiques de référence (marqueurs interne) sont
scanner ensemble avec les protéines à analyser, les coordonnée x et y sont envoyés à une liste
« picking liste » pour les stocker (Westermeier. 2004). Du moment que les techniques de

Partie I : Revue bibliographique
24
coloration sont compatibles avec la spectrométrie de mass, n’importe qu’elle changement
dans l’expression des protéines peut être identifié par la technique de Western Blot (Celis.
1999), et/ou, par l’analyse des peptides des spots excisées (Pandey. 2000), qui sont ensuite
comparés avec des bases de données de séquences protéiques de référence (Sanchez. 2001).
3. 6. 3. Évaluation des gels 2-D
L’évaluation des gels complexes requiert des logiciels qui corrigent l’arrière plan du
gel et quantifient les spots. Cependant, l’objectif principal des logiciels 2-D est la détection
automatique des spots, leur comparaison avec d’autres gels 2-D du même échantillon, et
l’identification des différences dans l’expression des protéines d’échantillons variés.
Les outils d’analyses statistiques sont aussi introduits dans les logiciels pour évaluer
par exemple la signification de la variabilité de l’expression protéique, les données générées
par les gels 2-D peuvent être analysés par les méthodes d’analyse uni/multi variées. La
résolution dépend de la finesse et de la clarté de l’image, elle est communément quantifiée par
le dpi «dots per inch» qui représentent le nombre de pixel par surface de gel. Cependant, les
scanners actuelle définissent les taille de pixels en μg 50, 100, ou 200, le pixel est un point
unique dans l’image (pix : picture, el : élément). En théorie, on peut avoir plus de 15 000
spots dans un seul gel, mais en pratique, il existe 5 000 spots seulement moyennant une bonne
séparation (Sheehan. 2009).
Il est impossible de détecter l’apparition ou la disparition de nouveaux spots, par
simple comparaison de deux gels contenant plusieurs milliers de spots, c’est pour cela que
l’utilisation de logiciel d’évaluation d’image est très utile (Berkelman et Stenstedt. 1998).
3. 6. 4. Application de l’électrophorèse bidimensionnelle chez les plantes
Le premier point d’étude de la protéomique est l’identification exhaustive des
protéines exprimées par un organisme (Malmstrom et al., 2004 ; McGregor et Dunn 2006).
Ceci a été possible grâce au développement simultané des techniques séparatives, ainsi que
des différents appareillages de spectrométrie de masse. De plus, cette approche a
considérablement été dopée par tous les programmes de séquençage de génomes, ainsi que
des outils et moteurs de recherche bioinformatique (Wheelock et Wheelock. 2008).
L’application d’une telle technologie de séparation des protéines, couplée à la spectrométrie
de masse a permis à un grand nombre de groupes de se lancer dans l’analyse systématique,

Partie I : Revue bibliographique
25
afin de réaliser des cartes protéiques de gel 2-D, et donc la mise en place de bases de donnés
construites autour des gels 2-D (Toda et al., 1998 ; Mollenkopf et al., 1999).
Le deuxième aspect important de l’analyse protéomique est l’analyse différentielle qui
consiste par exemple à comparer les protéomes de deux états distincts (stressé/témoin ;
traitement/ pas de traitement ; ...etc.) en observant l’apparition, la disparition ou les variations
des quantités des protéines (Komatsu. 2007).
L’approche de quantification la plus classique consiste à séparer les mélanges
protéiques sur des gels d’électrophorèse bidimensionnelle, et après coloration à comparer les
différences entre les gels. Les spots contenants les protéines exprimées de façon différentielle
seront analysés par spectrométrie de masse (Larrainza. 2007).
Un troisième point très important pour l’étude protéomique par spectrométrie de
masse est la recherche des modifications post-traductionnelles portées par les protéines
d’intérêts (Mateos. 2008). En effet, le faible nombre de gènes eucaryotes laisse à penser que
la complexité des organismes va résulter entre autre de la maturation et des modifications
post-traductionnelles des protéines. Ces modifications post-traductionnelles regroupent les
modifications chimiques covalentes, ainsi que la perte d’acides aminés sous l’action
d’exopeptidases (Biemann. 1990). La spectrométrie de masse est aujourd’hui l’outil de choix
pour répondre aux questions des modifications post- traductionnelles portées par les protéines.
Elle peut en effet identifier le type de modification (phosphorylation, glycosylation,
oxydation...), déterminer son emplacement sur la séquence des protéines, voir sa structure
(Mann et Jensen 2003 ; Heintz.2004).

3
Partie IIMatériels et méthodes

Partie II : Matériels et méthodes
26
1. Échantillonnage et extraction des protéines totales
Pour mettre au point la technique d’électrophorèse bidimensionnelle, 0.5 g de matériel végétal
(graines d’écotypes annuels sauvages, du genre Medicago) sont récupérées sur le site du
campus Mourad Taleb Salim ex l’Institut du Génie Maritime d’Oran (IGMO) en début
Septembre, pour intervenir avant la germination des graines. Les étapes (Zivy. 1986) de
l’extraction des protéines totales (tableau 02) sont valables pour la première dimension
electrophoretique IEF-Carrier ampholytes 4%, et CN-PAGE et se font comme suivant :
1- Des tubes eppendorfs de 1.5 ml vides sont pesés ;
2- Les graines sont finement broyer à l’aide d’un mortier dans de l’azote liquide,
jusqu'à obtention d’une poudre ;
3- On ajoute 10 ml (8+2 ml) de solution de précipitation par 0.5 g de poudre dans les
tubes eppendorfs, on place l’extrait dans un congélateur à -20 °C.
4- Les extraits sont centrifugés à 14 000 t/min pendant 15 min, pour séparer le culot
protéique du surnageant ;
5- Le surnageant est éliminé, le culot est couvert part 200 µl de solution de rinçage
éliminant les résidus de TCA, contenue dans la solution de précipitation. Les tubes
sont placés 1 heure à -20 °C.
6- Le rinçage est refait jusqu’à obtention d’un culot blanc.
7- Les culots sont séchés sous cloche à vide (1 heure).
8- Les tubes sont pesés pour déterminer le poids de chacun des culots secs.
Tableau 02 : Solution d’extraction des protéines (Zivy. 1986).
Solution Produit Quantité
Précipitation des protéinesTCA 10 g
β-Mercaptoéthanol 70 µlAcétone Qsp 100 ml

Partie II : Matériels et méthodes
27
Rinçage de TCA β-Mercaptoéthanol 70 µlAcétone Qsp 100 ml

Partie II : Matériels et méthodes
28
2. Électrophorèse bidimensionnelle : IEF/SDS-PAGE
2. 1. Premier dimension : IEF-Carrier ampholytes 4%
2. 1. 1. Solubilisation des protéines en IEF
La solubilisation des protéines extraites se réalise juste avant la séparation. On a utilisé
le tampon UKS conventionnel, qui est préparer dans l’ordre définit dans le tableau 03.
Tableau 03 : Tampon de solubilisation UKS (Granier. 1988).
Produit QuantitéUrée 2.85 g
K2CO3 0.125 µlSDS 0.625 µl
Triton X-100 1.5 mlDTT 125 µl
Ampholyte 3-10 250 µlEau bi-distillée Qsp 5 ml
2. 1. 2. Préparation des tubes IEF
Une journée avant utilisation, les tubes sont lavés par une solution de HCL 1 fois
normale, rincés en abondance par de l’alcool et par l’eau bi-distillée, puis séchés dans l’étuve
à 26 °C. Juste avant le coulage des gels, l’une des extrémités des tubes IEF est obstruée par la
paraffine.
2. 1. 3. Préparation du gel IEF- Carriers ampholytes
Le gel IEF à base d’urée est préparé dans un bécher de 25 ml. La solution (tableau 04)
est doucement agitée pendant cinq minutes, et mise dans un sonicateur (3 fois une minute, à
moins de 28°C), afin de solubiliser l’urée, puis le persulfate est ajouté. On introduite jusqu’au
fond du tube IEF une seringue munie d’un tube capillaire, pour couler le gel, en laissant un
espace afin de déposer les protéines.

Partie II : Matériels et méthodes
29
Tableau 04 : Solution du gel IEF-Carrier ampholytes (O’ Farrell. 2007).
Produit QuantitéUrée 3.75 g
Solution A+B (30%) 0.93 mlNP-40 1.38 ml
Ampholytes (2%) :
pH 3-10
- pH 3-10
pH 5-7 400 µlpH 3-10 100 µl
Persulfate (10%) 20 µlEau bi-distillée 1,37 ml
2. 1. 4. Tampon d’électrodes de l’IEF-Carriers ampholytes
Deux tampons d’électrode (tableau 05) sont utilisés pour la séparation en IEF : le
tampon cathodique (0.1 M NaOH), est versé dans la chambre supérieure où est plongée la
cathode ; et le tampon anodique (0.08 M H3PO4) remplit la chambre inferieur (anode).
Tableau 05 : Solutions des tampons d’électrodes (O’ Farrell. 2007).
Tampon cathodique Tampon anodiqueNaOH 4 g Acide phosphorique 85% 5.5 ml
Eau bi-distillée 1 l Eau bi-distillée 1 l
2. 1. 5. Focalisation
L’isoélectrofocalisation utilise un dispositif de 24 tubes Hoefer.USA-SE600-151.5
connecté à un générateur Consort E700 programmable (tableau 06) pour le voltage, l’intensité
du courant et la durée de migration. L’expérience qui porte sur huit gels nécessite huit tubes
IEF.
Les protéines quantifiés par la méthode de Bradford (1976) indiquent une
concentration de 0.56 µg/ µl, les volumes de protéines déposées sont de l’ordre de 20, 30 et
40 μl, par gel, dont la quantité est respectivement de 11,2 μg, 16,8 μg et 22,4 μg. Ceci afin de
mettre en évidence les protéines présentes en quantités différentes chez le même écotype.

Partie II : Matériels et méthodes
30
Tableau 06 : Programme de voltage (Nagaraj. 2007).
Programme de voltage de la pré-focalisation Programme de voltage de la focalisationVoltage Temps Voltage Temps
500 v 30 min 500 v 30 min1000 v 60 min 1000 v 60 min
16 000 v 21 h
La pré-focalisation permet d’établir un gradient de pH dans le gel. Les huit tubes sont
placés dans la cuve remplie par les tampons IEF, les protéines sont déposées et le dispositif
est fermer, avant sa connexion au générateur pour initier la séparation (tableau 06).
2. 1. 6. Démoulage des boudins
Les gels sont démoulés par jet d’une seringue remplie d’eau. Ils sont conservé à
- 20 °C dans de la paraffine couverte d’aluminium.
2. 2. Deuxième dimension : SDS-PAGE continu 10%
2. 2. 1. Préparation du gel et du tampon de la deuxième dimension
Le gel de la deuxième dimension est continu avec 10 % d’acrylamide avec une
épaisseur de 1,5 mm, le tampon utilisé est un tampon tris glycine homogène (pH = 8,6) pour
les deux cuves, quand au gel on a utilisé un tampon tris-Hcl (pH = 8,8) (tableau 07). Le gel est
préparé une heure avant la fin de l’IEF, pour lancer la deuxième dimension.
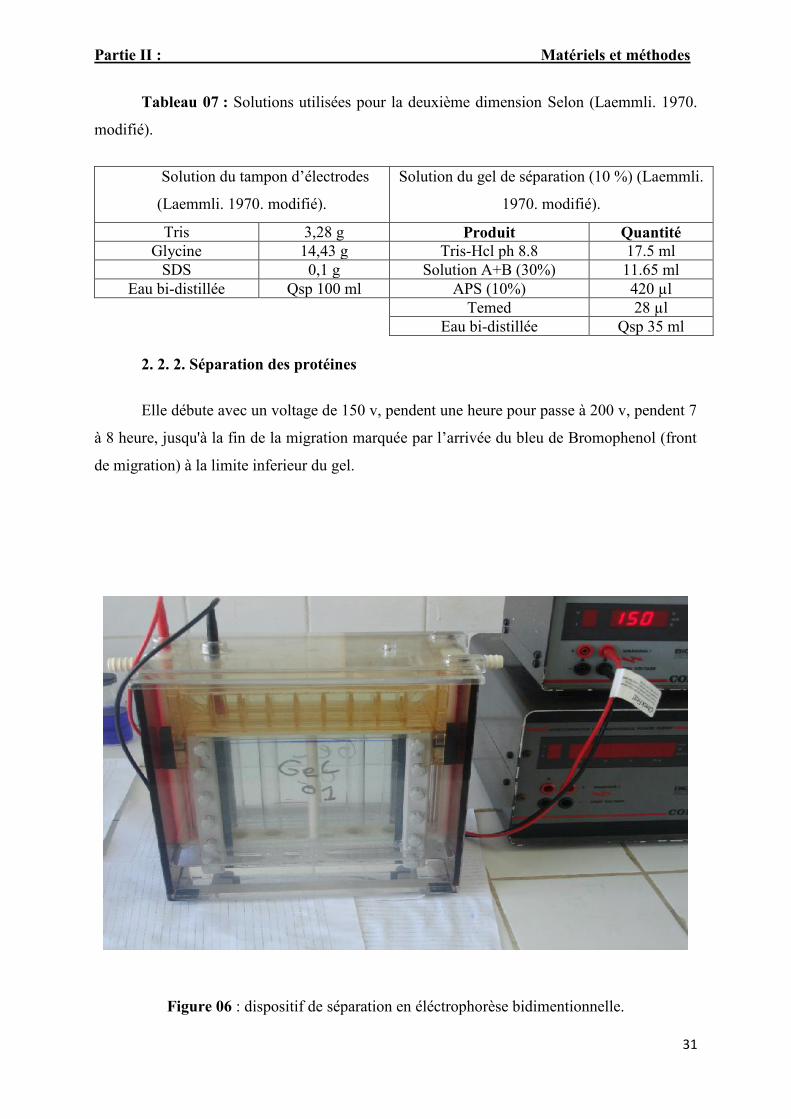
Partie II : Matériels et méthodes
31
Tableau 07 : Solutions utilisées pour la deuxième dimension Selon (Laemmli. 1970.
modifié).
Solution du tampon d’électrodes
(Laemmli. 1970. modifié).
Solution du gel de séparation (10 %) (Laemmli.
1970. modifié).
Tris 3,28 g Produit QuantitéGlycine 14,43 g Tris-Hcl ph 8.8 17.5 ml
SDS 0,1 g Solution A+B (30%) 11.65 mlEau bi-distillée Qsp 100 ml APS (10%) 420 µl
Temed 28 µlEau bi-distillée Qsp 35 ml
2. 2. 2. Séparation des protéines
Elle débute avec un voltage de 150 v, pendent une heure pour passe à 200 v, pendent 7
à 8 heure, jusqu'à la fin de la migration marquée par l’arrivée du bleu de Bromophenol (front
de migration) à la limite inferieur du gel.
Figure 06 : dispositif de séparation en éléctrophorèse bidimentionnelle.
Partie II : Matériels et méthodes
32
2. 2. 3. Coloration, décoloration et fixation du gel 2-D IEF/SDS-PAGE
La solution à base de Coomassie G 250 et de sulfate d’ammonium (tableau 08) colore
les gels, qu’on laisse reposer (48 heures) sous une douce agitation et à l’abri de la lumière. Le
G-250 est saupoudré de façon homogène sur les gels préalablement trempés dans la solution
de coloration. La solution de décoloration baigne les gels pendant une heure.
Tableau 08 : Solution de coloration et de décoloration au Coomassie G-250 (Blakesley et
Boezi. 1977).
Solution de coloration Solution de décolorationProduit Quantité QuantitéG-250 0.1 g -
Méthanol 20 ml 20 mlSulfate d’ammonium 10 g 10 g
Acide Octophosphorique (85%) 2 ml 2 mlEau Qsp 100 ml Qsp 100 ml
Les protéines sont fixées par une solution d’acide acétique à 7%. Pour visualiser les
spots, les gels sont mis dans du papier cellophane et scannés.
3. Électrophorèse bidimensionnelle : CN/SDS-PAGE
Pour optimiser l’electrophorèse 2-D, nous avons utilisé en premier dimension un gel
gradient non-dénaturant (4-10 %), et deux gels discontinus 8 % et 10 % ont été préparé, le
premier est utiliser pour la visualisation des protéines dans la premier dimension et le
deuxième est coupé en slabes est mit dans un tampon tris pour être conservé.
Partie II : Matériels et méthodes
33
3.1 Premier dimension CN-PAGE
3. 1. 1 Solubilisation des protéines en CN-PAGE
La solubilisation des protéines extraites se réalise juste avant la séparation. On a utilisé
le tampon (Laemmli. 1970. modifié) qui est préparé dans l’ordre définit dans le tableau 09.
Apres l’ajout du tampon (15µl/ µg) les proteines sont passée au bain marie pendant 3 min à
60°C.
Tableau 09 : Tampon de solubilisation (Laemmli. 1970. modifié).
3. 1. 2 Préparations
des gels CN-PAGE
Pour la réalisation de
la première dimension
electrophoretique (CN-
PAGE), nous avons réalisé
trois types de gels, un gel gradient de 10% (tableau 10), un gel discontinue de 10% et un gel
discontinue de 8% (tableau 11).
Tableau 10: gel gradient 10%.(Laemmli. 1970. modifié).
Produit Solution du gel4% 10%
Solution A+B 1,32 ml 3,33 mlLower gel 5 ml 2,5 ml
APS 100 µl 100 µlTemed 12,5 µl 12,5 µl
Eau bi-distillée Qsp 10 ml Qsp 10 ml
Produit QuantitéTris-HCl pH 7.2 500 µl
MgCl2 30 µlGlycerol 2 ml
Triton X-100 1 mlPMSF 200 µlNaCl 600 µlSDS 1%
β-Mercaptoethanol 5%H2O-bidistillée 10 ml
Partie II : Matériels et méthodes
34
Tableau 11 : gel discontinue à 8% et à 10%. (Laemmli. 1970. modifié).
Solutions des gelsProduits Concentration à 4% Séparation à 8% Séparation à 10%
Solution A+B 0,66 ml 2,66 ml 3,33 mlLower gel - 2,5 ml 2,5 mlUpper gel 2,5 ml - -
APS 100 µl 100 µl 100 µlTemed 12,5 µl 12,5 µl 12,5 µl
Eau bi-distillée Qsp 5 ml Qsp 10 ml Qsp 10 ml
3. 1. 3. Tampon cuve de la CN-PAGE
Le même tampon cuve a été utilisé pour toute les techniques de séparations
monodimensionnelles de la CN/PAGE et ceci pour : le gel gradient à 10 % ; le gel discontinu
10% ; et le gel discontinu 8%.
Tableau 12 : Solutions du tampon cuve pour la première dimension (Laemmli.1970).
Produit QuantitéTris HCL (pH = 8.6) 3,28 g
Glycine 14,43 gEau bi-distillée Qsp 100 ml
2. 2. 4. Séparation des protéines en CN-PAGE
Pour facilite la pénétration des protéines dans le gel on commence par un ampérage de
25 mA pendant une demi heure et on passe à 35 mA jusqu’à la fin de la séparation. Un
voltage constant de 150 v est appliqué pendant toute la séparation.

Partie II : Matériels et méthodes
35
2. 2. 3. Coloration, décoloration et fixation du gel CN-PAGE
La solution à base de Coomassie R 250 (tableau 13) colore les gels, qu’on laisse
reposer (48 heures) sous une douce agitation et à l’abri de la lumière. Le R-250 est saupoudré
de façon homogène sur les gels préalablement trempés dans la solution de coloration. La
solution de décoloration baigne les gels pendant une heure.
Tableau 13 : Solution de coloration au Coomassie R-250 (Schägger. 2006).
Solution de coloration Solution de décolorationProduit Quantité QuantitéR-250 0.1 g -
Méthanol 50 ml 50 mlAcide acétique 40 ml 40 mlEau bi-distillée Qsp 100 ml Qsp 100 ml
3. 2 Deuxièmes dimensions SDS-PAGE
Les gels conservés dans du tampon tris-SDS (voir. Composition du lower gel) sont
découpés par une lame très fine et d’une façon franche pour obtenir une surface linéaire afin
d’assurer un contact optimal entre les interfaces des gels de la première et de la deuxième
dimension. La préparation du gel est de la même composition que celle utilisée en deuxième
dimension de l’IEF (cf. supra). Le voltage est lui aussi semblable à celui de la CN-PAGE. La
coloration décoloration et fixation des protéines se fait de la même façon que pour la première
dimension CN-PAGE (cf. supra).

Partie IIIRésultats et discussion

Partie III Résultats et discussions
36
Électrophorèse bidimensionnelle IEF-CA/SDS-PAGE
Le gel de résolution à 10% d’acrylamide sépare les protéines dans une gamme de poids allant
de 14-205 kDa.
L’ammonium sulfate permet d’avoir un fond plus claire et homogène, car il aide à la
précipitation des fragments de protéines membranaires, lors de la centrifugation.
Dans cette étude nous avons réalisés plusieurs essais pour la mise au point de la
technique d'électrophorèse bidimensionnelle (IEF/SDS-PAGE et CN/SDS-PAGE) des
protéines totales, afin de mettre en évidence la variabilité génétique existante entre les
espèces annuelles de genre Medicago. Mais pour maitriser la technique nous avons utilisé des
espèces sauvages non répertoriées.
La préparation des gels IEF selon le protocole Monribot et Boucherie (Monribot et
Boucherie. 1999 modifié), c'est avérée difficile, car ce dernier utilise un volume important de
solution (25 ml) pour réaliser 24 gels IEF.
Dans notre étude on a utilisé 8 tubes pour la mise au point de la technique
d'isoelectrophocalisation de la première dimension. 7 ml de solution de gel IEF sont suffisant
pour couler les gels des 8 tubes (il faut 800μl de solution du gel pour remplir un tube IEF de
24 cm).
La concentration de l'urée (9.5 M) ne peut être réduite si on veut obtenir une
solubilisation optimale des protéines surtout les molécules hydrophobes dans une solution
aqueuse.
Dans notre cas et pour améliorer la solubilisation de l'urée on a mit d'avantage d'eau,
en déduisant la proportion de l’eau bi-distillée nécessaire pour une quantité suffisante pour 7
ml. Les résultats obtenus, montrent que l'urée peut se solubilisée en présence de quantité
suffisante d'eau, avec disparition des deux phases rencontré précédemment de plus on a utilisé
du NP-10 au lieu de NP-40 pour diluer la solution du gel.
Malgré les modifications introduite, cette solution n'est pas pure et on remarque
l'existence de petits granules insolubles suspendus dans la solution, ce qui peut obstruée les

Partie III Résultats et discussions
37
mailles des gels et empêchent les protéines de se séparées correctement, de plus on a observé
que l'urée cristallise rapidement à une température inférieur à 20°C.
Pour résoudre ce problème on a préparé les gels IEF dans une chambre à température
contrôlée (26°C), ensuite, la solution est passée au sonicateur 3 fois 1 minute, pour le
dégazage et une bonne solubilisation de l'urée, mais vu que la température du sonicateur
augmente avec les ultrasons et dépasse le seuil critique de 28°C, on a remarqué un début de
polymérisation.
La réduction du temps de sonication (3×10 seconde) et l'ajout du persulfate à la
solution après l'étape de sonication nous a permis d'obtenir une solution IEF homogène, et
évité l'initiation de la polymérisation dans le récipient.
Les gels sont coulés horizontalement et mit dans l'étuve à 26°C, pour la
polymérisation qui dure 24 heures, les résultats obtenus indique une polymérisation partielle
et dans la plupart des cas les gels sont complètement à l'état liquide.
La modification de la proportion des ampholytes de la gamme de Ph 3-10 dans la
solution par rapport aux autres gammes (5-7; 5-8), nous a permit d'obtenir une polymérisation
efficace des gels.
Après plusieurs essais effectuer pour l'amélioration de la polymérisation et
l'élimination des bulle d'airs on obtenait seulement 2 gels correct, sur 8, et qui sont de
consistance assez rigide et flexible, permettant leurs manipulation avec aisance surtout pour
leur transfert en deuxième dimension.
On changeant le mode de coulage des gels en horizontale par le coulage en verticale,
on a observé que les bulles d'aire remontées rapidement en surface, et les gels obtenus étaient
complètement dépourvues de ces derniers.
Les résultants indiquent que l'étape de pré-focalisation est très utile car, malgré le
nombre insuffisant et très peu représentatif de spots obtenus, ces dernier ne sont révélés que
par une IEF avec une pré-focalisation.

Partie III Résultats et discussions
38
Figure 07 : Partie du gel 2-d coloré au bleu de Coomassie représentant trois spots
Après transfère du gel IEF en deuxième dimension on remarque qu'une partie du gel
flotte dans la solution cathodique en dessus du gel SDS-PAGE et qu'il n’adhère pas à celui-ci,
il se détache progressivement au fur et à mesure de la deuxième séparation. Pour cette raison
on a utilisé une solution de 0.5 d'agar pour sceller les deux gels ensemble et permettre leur
contact pour assurer le transfère des protéines.
Les gels de la deuxième dimension obtenus sont plus rigides, avec une épaisseur de
1.5 mm, les résultats obtenus par la coloration au bleu de Coomassie G-250 montrent des gels
de couleur homogène, cette coloration dure 48 heures et elle est simple à réaliser par rapport à
la coloration au nitrate d'argent qui est plus compliqué.
Pour extraire les protéines totales, nous avons utilisé des solutions compatible
avec l'IEF c'est à dire qu'ils ne confèrent pas de charges supplémentaires aux molécules
analysées car cela pourraient faussées leurs points isoelectriques.
La solution à base de TCA/Acétone est fréquemment utilisé dans l'extraction
dénaturante, car elle offre un double avantage celui de la précipitation des protéines et de
l'inhibition de la protéolyse.
Au début de notre expérience, l'absence de polymérisation nous a laissé supposée
qu'elle est due à la présence du β- mercapthoetanol qui est un agent réducteur puissant (enlève
les ponts disulfure).
D’autres agents réducteurs tels que le DTT et le DTE qui sont des agents thiols de
faible puissance par rapport au β- mercapthoetanol ont été utilisés dans certains protocoles en
association avec des détergents zwitterionique plus performant comme le CHAPS et les
sulfobetaines SB-13.

Partie III Résultats et discussions
39
Cependant la non disponibilité de ces détergents pour notre étude ne nous permet pas
d’utiliser le DTT au risque de ne pas avoir une meilleurs solubilisation des protéines totales,
surtout pour les grosses molécules et les protéines hydrophobes.
Pour ne pas remplacer le β- mercapthoetanol très utile pour notre étude nous avons
réalisé des essais pour savoir s’il est le seul responsable de l’inhibition de la polymérisation
des gels IEF. Nous avons cherché dans la littérature, s’il n’existe pas d’autres agents
responsables de cette inhibition (Brewer. 1967).
Les essais effectuer par la suite pour la préparation des gels IEF, nous ont permit de
confirmer que deux facteur peuvent jouer un rôle crucial dans la polymérisation de
l'acrymlamide avec le bis-acrylamide qui sont respectivement; les ampholytes de la gamme de
Ph 3-10 et l’oxygène (Westermeier. 2004).
Les ampholytes de la gamme de pH 3-10, une concentration élevée entraine
l’augmentation de la concentration des Ph très acide et des pH très basiques constitutif (très
basique 8.5-10 et très acide 3- 4.5) de ce spectre, ces pH extrêmes inhibent la polymérisation
car le phénomène de pontage entre l’acrylamide et le bis-acrylamide initier par l’ammonium
persulfate (APS) nécessite un environnement neutre (Westermeier. 2004). De ce fait,
l’ajustement de la concentration de pH 3-10 en diminuant sa proportion a permis d'améliorer
la polymérisation.
En ce qui concerne l’oxygène, le mode horizontale de polymérisation conduit à
l'emprisonnement de l’air dans la solution, l’oxygène étant dissous dans le gel, il se
transforme en bulles plus au moins abondantes qui une fois piégées dans le gel polymérisé,
créent des lacunes entravant le passage du courant électrique (electric gap) empêchant ainsi la
séparation des protéines, ce phénomène est constaté lors du passage du bleu de méthylène
(étalon de migration).
Si ces bulles sont de grosses tailles, elles déstabilisent la structure des gels provoquant
des cassures à plusieurs endroits. Le mode de polymérisation en verticale semble plus efficace
car l’absorption d’oxygène est moins importante.
D'autre part, la sonication, procédé permettant à la fois le dégazage de la solution et la
dissolution de l'urée, se traduit par une meilleure homogénéisation du mélange et l’obtention
de meilleurs gels.

Partie III Résultats et discussions
40
Dans notre étude nous avons utilisé l’urée, qui est nécessaire à l'amélioration de la
solubilisation des protéines hydrophobes et les protéines de grand poids moléculaire, en
association avec des tensioactifs non ioniques classiques tels que NP-40 et le Triton X-100,
cependant, ces tensioactifs même s'ils sont compatible avec l’IEF (car ils ne confèrent
aucunes charges aux protéines) ils ne sont pas très performant (Granier. 1988) et on a pas pu
séparer les protéines par ces tensioactifs sont largement remplacés dans les protocoles récent
par des surfactants zwitterioniques tels que le CHAPS, et les sulfobetaines SB-14.
D’un autre coté, les concentrations élevées d’urée (9M) permettant une meilleur
solubilisation des extraits protéiques tout en augmentant la viscosité du gel va paradoxalement
diminuer la mobilité des protéines ceci a été constaté lors de nos essais ou l'on a constaté que
les séparations prennent beaucoup plus de temps comparées aux gels SDS-PAGE de la
deuxième dimension, et dépassent même les 12 heures, période au delà de laquelle le Ph
commence à se dégrader. Ces phénomènes conjugués à l'établissement hasardeux du pH lors
de la préfocalisation peuvent expliquer les résultats négatifs obtenus.
La perte des protéines extraite par précipitation peut se produire à différents niveaux
du processus electrophoretique:
lors de l'extraction et de la solubilisation des protéines.
Lors du dépôt de protéines, ou la pénétration dans le gel.
À l'intérieur du gel lui même au point isoélectrique
Pendant le transfert du gel de la première a la deuxième dimension (Rabilloud. 1996).
Une perte très importante est en effet signalée due aux détergents et tensioactif
utilisés pour éliminer les contaminants des échantillons comme les lipides et les acides
nucléiques. Mais cette perte n'explique pas à elle seule les résultats insatisfaisants obtenu car
même s'il y'a une perte de protéines une partie devrait être présente dans le gel (Rabilloud.
1996).
Il est a indiqué que plusieurs phénomènes expliquent la dégradation et l'instabilité du
ph tel que l’eletroendosmose ou le phénomène de plateau, mais aussi la qualité de séparation
qui varie beaucoup d’une solution d’ampholyte à une autre confirmant la différence de qualité
qui existe entre les différentes marques commercialisées, cela a été signalé par le groupe

Partie III Résultats et discussions
41
Righetti (Righetti. 2007) qui ont fractionné des ampholytes en étudiant leurs propriétés de
focalisation par la CZE-MS( capilarity zone electrophoresis- mass spectrometry). Ils ont
conclus qu’une part non négligeable de ces ampholytes, s'étendant sur tout l'intervalle que
couvrent les différentes gammes, est de mauvaise qualité.
Dans notre études les ampholines 3-10, nous ont permis d’avoir les seuls et uniques
spots présent dans le gel et de toutes les expériences réalisées. Les pharmalytes et les
servalytes n’étaient pas efficace car aucune séparation n’a pu être réussie par ces solutions, ce
qui est en accord avec les travaux de Poitevin (2008). Par contre le groupe Righetti signale
que les meilleurs résultats sont obtenus par les pharmalytes, cela peut s'explique par le fait
qu'il a utilisé des Ph étroit pour son étude alors que nous avons utilise des pH étendu.
Les différents points discutés précédemment nous fons prendre conscience que cette
technique est très difficile à mettre au point et le fait que les résultant ne soient pas
reproductibles entre laboratoires et même au sein du même laboratoire (Zilberstein. 2010) fait
qu'elle est largement abandonner en faveur d'une nouvelle technique qui utilise un gradient
de Ph immobiliser c'est à dire qui est fixe car il se polymérise avec les molécules
d’acrylamide, ce principe a permis de révolutionner la technique d'électrophorèse
bidimensionnelle, de plus les progrès réaliser en matière de solubilisation des protéines
hydrophobe et de gros poids moléculaire ces réalisation en protéomique ont fortement
contribuer aux remplacement de l’IEF en carrier ampholytes par l'IPG.
A-Électrophorèse 2-D CN- gradient (4-10%) / SDS-PAGE continu (10%)
La technique d’électrophores native permet la séparation des protéines sans l’altération
de la structure tridimensionnelle. L’utilisation du protocole de précipitation à base de TCA et
la solubilisation des protéines par le tampon Laemmli (1970) a permis d’obtenir les résultats
présentes par les gels : gel 1-g gradient 4-10% ; gel bidimensionnel gradient 4-10%/SDS-
PAGE ; gel discontinu 4-10%/SDS-PAGE, gel discontinu 4-8 %/SDS-PAGE.
Pour le gel 1-D (figure 08), nous pouvons distinguer cinq zones de migration
Zone de migration lente : 0.56
Zone de migration moyenne : 0.71
Zone de migration rapide : 0.81 et 0.84
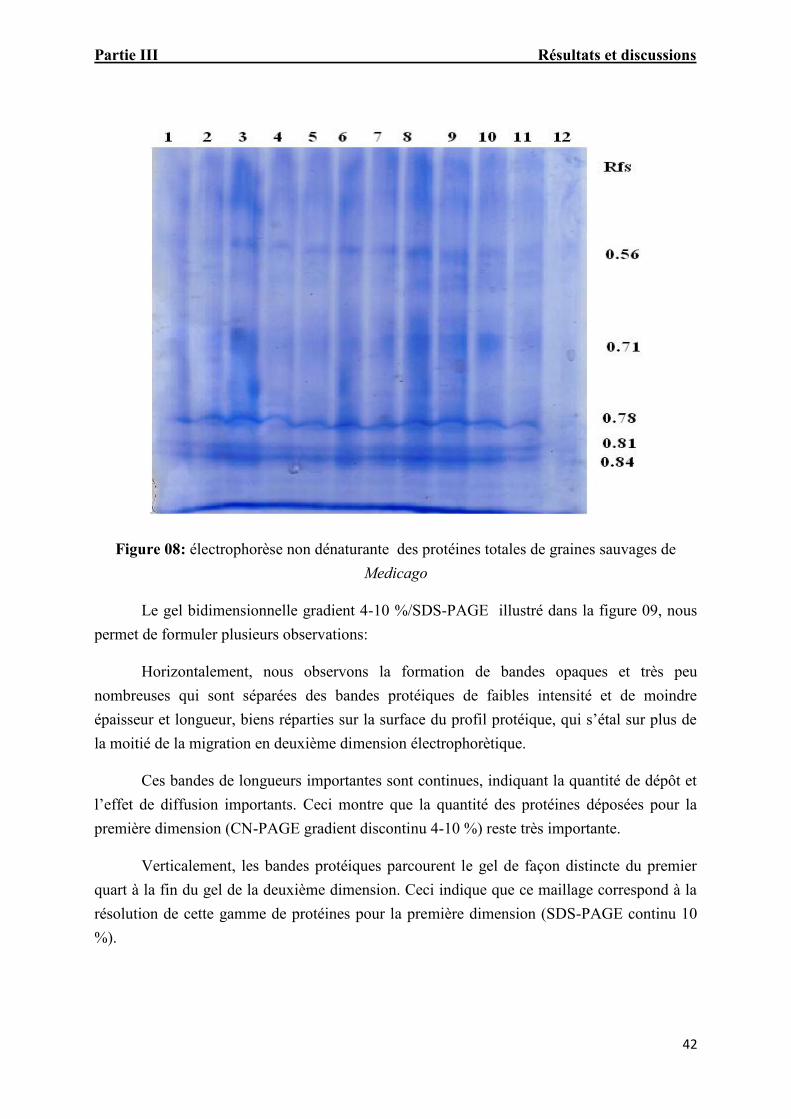
Partie III Résultats et discussions
42
Figure 08: électrophorèse non dénaturante des protéines totales de graines sauvages deMedicago
Le gel bidimensionnelle gradient 4-10 %/SDS-PAGE illustré dans la figure 09, nouspermet de formuler plusieurs observations:
Horizontalement, nous observons la formation de bandes opaques et très peunombreuses qui sont séparées des bandes protéiques de faibles intensité et de moindreépaisseur et longueur, biens réparties sur la surface du profil protéique, qui s’étal sur plus dela moitié de la migration en deuxième dimension électrophorètique.
Ces bandes de longueurs importantes sont continues, indiquant la quantité de dépôt etl’effet de diffusion importants. Ceci montre que la quantité des protéines déposées pour lapremière dimension (CN-PAGE gradient discontinu 4-10 %) reste très importante.
Verticalement, les bandes protéiques parcourent le gel de façon distincte du premierquart à la fin du gel de la deuxième dimension. Ceci indique que ce maillage correspond à larésolution de cette gamme de protéines pour la première dimension (SDS-PAGE continu 10%).

Partie III Résultats et discussions
43
En absence de marqueur de poids moléculaire, nous avons calculé les fronts demigrations de chaque bande, pour obtenir une représentation de la taille des molécules. Lesrésultats sont présentés dans la figure 10.
Ce profil schematise les bandes les plus représentatifs de notre gel dont le nombre estde neufs classé de a à h, dont les fronts de migration sont : a, b : 0.39, c : 0.54 ; d : 0.62 e :0.78, f, h 0.8, i : 0.81 g : 0.84.
Il montre l’existence de bandes protéiques dont le front de migration est similaire (a,b) et (f, h) et des bandes qui n’ont pas le même front de migration mais qui sont dans la mêmerangée verticale (e, f, g) et (h, i).
Figure 09-a: Gel d’electropphorese bidimensionnelle 1-D gradient 4-10%/SDS-PAGEcontinu 10% des proteines tatale de graines de Medicago.
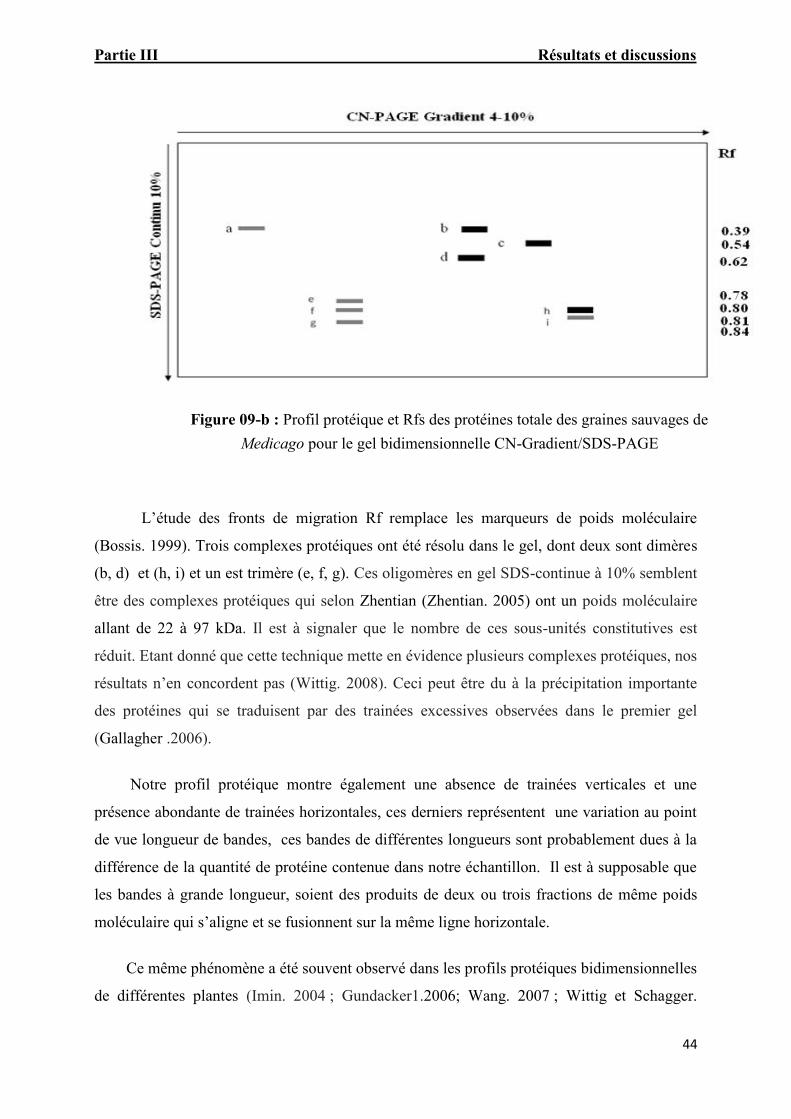
Partie III Résultats et discussions
44
L’étude des fronts de migration Rf remplace les marqueurs de poids moléculaire
(Bossis. 1999). Trois complexes protéiques ont été résolu dans le gel, dont deux sont dimères
(b, d) et (h, i) et un est trimère (e, f, g). Ces oligomères en gel SDS-continue à 10% semblent
être des complexes protéiques qui selon Zhentian (Zhentian. 2005) ont un poids moléculaire
allant de 22 à 97 kDa. Il est à signaler que le nombre de ces sous-unités constitutives est
réduit. Etant donné que cette technique mette en évidence plusieurs complexes protéiques, nos
résultats n’en concordent pas (Wittig. 2008). Ceci peut être du à la précipitation importante
des protéines qui se traduisent par des trainées excessives observées dans le premier gel
(Gallagher .2006).
Notre profil protéique montre également une absence de trainées verticales et une
présence abondante de trainées horizontales, ces derniers représentent une variation au point
de vue longueur de bandes, ces bandes de différentes longueurs sont probablement dues à la
différence de la quantité de protéine contenue dans notre échantillon. Il est à supposable que
les bandes à grande longueur, soient des produits de deux ou trois fractions de même poids
moléculaire qui s’aligne et se fusionnent sur la même ligne horizontale.
Ce même phénomène a été souvent observé dans les profils protéiques bidimensionnelles
de différentes plantes (Imin. 2004 ; Gundacker1.2006; Wang. 2007 ; Wittig et Schagger.
Figure 09-b : Profil protéique et Rfs des protéines totale des graines sauvages deMedicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE

Partie III Résultats et discussions
45
2009; Castillejo. 2010). Cette variabilité quantitative (longueur des protéines) que possède
les graines de Medicago reflète sa richesse protéique notamment en celles de réserve qui sont
stockés pendant le développement de la graine d’où l’intérêt de leur utilisation dans cette
étude. Ces résultats se conforment avec ceux trouvés par Mahnane (Mahnane. 2010) : Les
graines de Medicago renferment un très grand nombre de protéines différentes (protéines de
structure, protéines biologiquement actives et protéines de réserve. D’ailleurs, ce même auteur
rapporte en travaillant sur les graine de Medicago plante modèle des légumineuse qu’elle est
constituée des protéines essentiellement d’albumines et de globulines.
Donc, les bandes révélées dans le gel ayant une intensité plus importante pourrait bien
s’agir de globulines qui représentent 60 à 90 % de l’azote totale des légumineuses, elles ont
une structure spatiale complexe et peuvent se lier ou se dissocier selon les conditions du
milieu. Par ailleurs les bandes ayant une intensité moins importante pourrait probablement
être des albumines car, ces derniers représentent 10 à 20% des fractions protéiques des
légumineuse, ce sont des molécules ayant un rôle fonctionnel dans la cellule.
Il est fort probable de détecter des bandes non visibles dans le gel correspondant à des
protéines minoritaires. Cette non apparition des bandes semble être du à la concentration des
protéines majoritaires qui en atteignant certains seuil de concentration vont masquer les
protéines minoritaires présentes à ces endroits. Ce phénomène est d’autant plus important
dans les gels gradient ou les bandes sont très proches, car ce dernier sépare une large gamme
de poids moléculaire des protéines de poids moléculaire très proches ainsi que celles ayant
des masses très proches qui se dilatent empêchant ainsi leurs distinctions (Westermeir. 2004).
Notre recours à utiliser la TCA réside non seulement dans le fait qu’il constitue la
meilleure procédure de précipitation, mais aussi il donne le meilleur rendement en matière de
précipitation des protéines récalcitrantes. De plus, il élimine les sels, les lipides, les poly
phénols, les polysaccharides et les protéases, ce qui fait de lui un produit à large utilisation
dans la préparation des échantillons végétaux destinés aux analyses protéomiques. Des
recherches (Zhentian. 2005 ; Jorrín. 2007 ; Méchin, 2007) à ces fins protéomiques ont été
menés en utilisant la TCA et non pas l’éthanol, le méthanol/ammonium acétate, phénol–
chloroform– isoamyl alcool qui sont des produit alternative de la précipitation.

Partie III Résultats et discussions
46
Le profil protéique révèle des sous-unités ou fractions protéiques en disposition
verticale sous forme de piles. Ceci indique la présence d’états oligomeriques qui ne se sont
pas dénaturés dans le tampon de préparation des protéines à base d’SDS ou réduit par le β-
mercapthoethanol. Ces oligomères se sont séparées sous forme de complexes protéiques dans
la deuxième dimension. Cette dernière a permis d’avantage la dénaturation de ces complexes
par l’SDS présent à la fois dans les tampons cuve et dans les tampons du gel, ce qui a conduis
à leurs séparation sous forme de sous-unité en deuxième dimension, d’où leurs apparences
sous forme de piles verticales de fractions protéiques.
Pour ce qui est des fractions sur la même rangée horizontale, il pourrait bien s’agir des
sous-unités protéiques de même poids moléculaire qui se trouvent dans des complexes
protéiques différents (a, b) et (f, h), cela a été rapporté par (Reisinger et Eichacker. 2007). En
effet ces bandes peuvent être soit des protéines iso-formes ou des protéines similaires. On
suppose qu’ils sont des allèles produits du même locus (Bossis. 1996).
Dans notre études nous avons l’atout d’utiliser le SDS comme détergeant car il a non
seulement l’avantage de sa simplicité (Bianchi-Bosisio. 1991 ; Le Bricon. 1998 ; Pesce.
1972) mais aussi, il permet la séparation des protéines basiques qui est moins évidente par
d’autres détergents (). De plus, ce détergeant fortement anionique s’associe avec les protéines
dans un rapport de 1,4 g de SDS/g de protéines, quantité suffisante pour masquer leur charge
intrinsèque (Trivin.2002).
Ce détergent SDS a fait l’objet de pas mal d’étude d’électrophorèse bidimensionnelle
(Eubel et al. 2005 ; Williams et al. 2006 ; Swamy et al. 2006 ; Reisinger et al. 2007. Wittig
et al. 2008) Sur différents matériels végétales comme le blé (Zhao .1996) le riz (Agrawal et
al. 2005) et le Medicago (Daher et al. 2010) l’orge (Hynek.2006).
Des études pareils ont été menés par des chercheurs (Ossenbühl et al., 2004; Schwenkert
et al., 2006; Danielsson et al., 2006) travaillant sur des complexes protéiques de chloroplaste
et thylacoïde des algues et des plantes. Dans le même contexte d’autres auteurs ont signalé
pour la Medicago sativa et la Pisum sativum, une différence significative entre la résolution
des protéines selon les détergeant utilisés dans la solubilisation des protéines (Ladig. 2010).
En effet, des détergents tels que le DDM et le digitonin (mild detergent) n’altérant pas la
structure native des protéines permettent à la fois leur migration selon leurs charges charge

Partie III Résultats et discussions
47
nette et leurs poids moléculaire. L’utilisation de ces derniers a permis une meilleur
performance en matière de résolution (Eubel. 2005 ; Ladig.2010).
A titre de comparaison le nombre de fractions résolu dans une 2-D BN/SDS PAGE est
normalement moins important que dans les gels 2-D IEF/SDS-PAGE, Ceci est principalement
due au fait que les protéines ne sont pas séparées sur la surface total des gels mais plutôt
arrangées sous forme de piles verticales, ce qui est en accord avec les résultats observés dans
notre gel. Cela indique la complexité de la résolution dans la séparation native.
B-Électrophorèse 2-D CN- Discontinu (4-10%)/SDS-PAGE continu (10%)
Le gel présente des bande encore plus épaisse que le gel gradient et au lieu d’avoir des
spots on a obtenus des bandes très diffus, cependant ils sont bien distinct par rapport au gel
gradient. Par ailleurs on observe que les bande n’occupent que la moitié du gel et ne sont pas
séparé dans la totalité du gel.
Le profil des Rfs présente les bandes sous une formes schématique dans le gel, les
bandes avec une coloration noire représente les fractions qui apparaissent très dense et les
bande grise celles qui sont mois claire, il y’a sept bande de a à g dont le Rfs sont
respectivement : 0.28, 0.31 0.43 0.5 0.53 0.64 0. 91. Six de ces bande se disposent en ligne
verticale (b à g), la bande a, c et d sont très épaisses et laissent pensé que c’est une fusion de
deux bandes sous-jacente par diffusion, les bondes c présente un centre bien définit qui
correspond à un spot mais la diffusion est très importante autours.
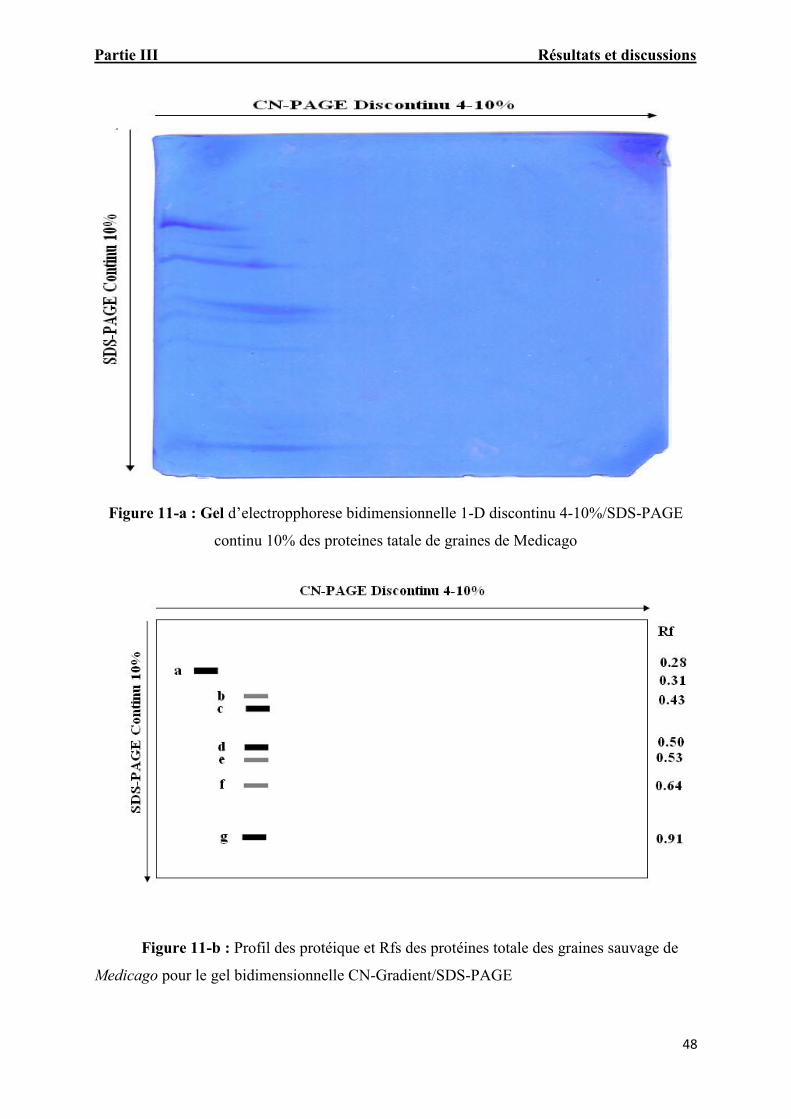
Partie III Résultats et discussions
48
Figure 11-a : Gel d’electropphorese bidimensionnelle 1-D discontinu 4-10%/SDS-PAGE
continu 10% des proteines tatale de graines de Medicago
Figure 11-b : Profil des protéique et Rfs des protéines totale des graines sauvage de
Medicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE

Partie III Résultats et discussions
49
B-Électrophorèse 2-D Cn- Discontinu (4-8%)/SDS-PAGE Continu (10%)
Pour améliorer la résolution et d’obtenir des spots avec un centre bien définis nous
avons réalisé un gel discontinue de 4-10%. Les résultats obtenus révèlent des bandes de plus
large épaisseur par rapport au gel gradient. De plus, ces bandes ne sont pas reparties sur toute
la surface du gel bidimensionnelle. Ceci pourrait se traduire par le fait que ces protéines n’ont
pas migré jusqu’au bout de la première dimension et sont restées bloquer dans la partie
supérieure du gel de résolution qui semble exercer un effet de restriction sur les protéines à
analyser.
Malgré que, les protéines sont disposées en ligne verticale, nous ne pouvons pas
confirmer ni infirmer le fait qu’ils appartiennent ou non au même complexe, car il est supposé
que l’obstruction des gels par les protéines de plus haut poids moléculaire (à cause du
maillage resserré de 10% de la première dimension) conduit au piégeage des molécules dans
la zone supérieur du gel discontinue, ce qui indique la répartition des bandes sur une partie du
gel 2-D et non pas sur toute la surface du gel.
Il est important de noter que la concentration dans le stacking gel peut causer
l’agrégation des protéines (Walker. 2000). D’un autre coté, quand la quantité des protéines
sont importante, des trainées verticales et horizontales sont observées dans le gel 2-D (), ce
qui explique la taille importantes des bandes et leur apparition sous formes de fractions
étalées horizontalement.
Des études comparatives entre le maillage des gels de résolution à concentration de
7.5% et de 9% sur deux complexes mitochondriales I et V montrent qu’ils sont bien séparés
l’un de l’autre. Une concentration de gel supérieure ou égal à 10% conduit à une faible
résolution du complexe I ou du complexe V qu’ils soient séparés ou réunis. Cet effet est aussi
observé dans un gel de 12% de concentration d’acrylamide (Yan .2009). Cependant, ces
complexes ont été très bien résolus par une concentration entre 7.5% et 9%. En générale dans
un gel de 7,5% les protéines ayant un poids moléculaire moins de 140 kDa devraient être
séparées, alors qu’un gel de 12% sépare les protéines ayant poids moléculaire inferieure à 60
kDa.

Partie III Résultats et discussions
50
L’absence du SDS dans la première dimension n’a pas facilité la solubilisation des
protéines de haut poids moléculaire, ce qui pourrait expliquer l’obstruction des mailles
d’acrylamide.
Ces résultats montrent que le maillage de 10% du gel de résolution de la première
dimension ne convient pas à la séparation de toute la gamme de taille des protéines de notre
échantillon, à ce moment là, il pourrait être mieux adapté pour analyser les protéines de faible
poids moléculaire. Par ailleurs la quantité des protéines déposées semble être en excès par
rapport au seuil de résolution.
C-Electrophorèse 2-D CN- Discontinu (4-8%)/SDS-PAGE
Pour améliorer la séparation et obtenir plus de bandes dans le gel et un profil protéique
étendu sur toute la surface du gel, nous avons réalisé une électrophorèse bidimensionnelle CN
à gel discontinu 4-8%, dont le gel de concentration est de 4% et le gel de séparation est de
8%. On a obtenu deux gels bidimensionnels.
Figure 12-a : Gel d’electropphorese bidimensionnelle 1-D discontinu 4-8%/SDS-PAGE continu 10% des proteines tatale de graines de Medicago
continu à 10% de graines savage de Medicago.
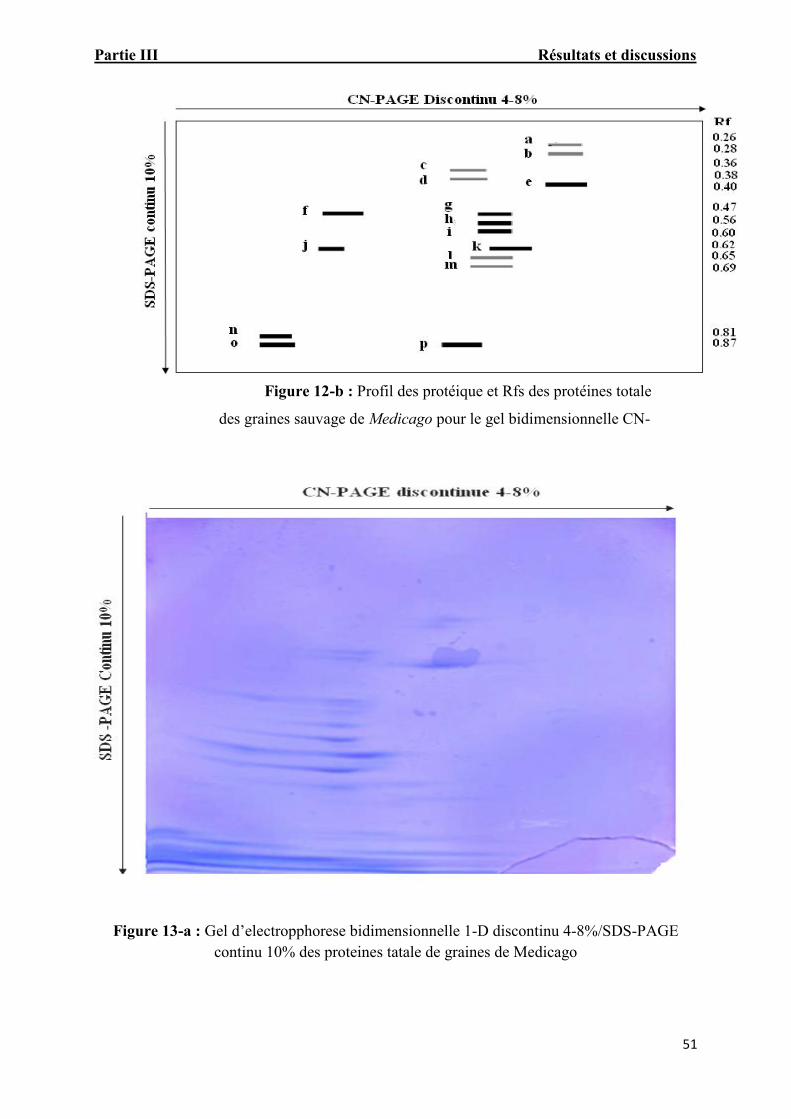
Partie III Résultats et discussions
51
Figure 13-a : Gel d’electropphorese bidimensionnelle 1-D discontinu 4-8%/SDS-PAGEcontinu 10% des proteines tatale de graines de Medicago
Figure 12-b : Profil des protéique et Rfs des protéines totale
des graines sauvage de Medicago pour le gel bidimensionnelle CN-
Gradient/SDS-PAGE

Partie III Résultats et discussions
52
Figure 13-b : Profil des protéique et Rfs des protéines totale des graines sauvage de
Medicago pour le gel bidimensionnelle CN-Gradient/SDS-PAGE
Les résultats obtenus des deux gels (figure 13-a et 13-b) ont permis d’avoir la
meilleure résolution que tous les gels réalisés précédemment (gel graduant et gel discontinu à
10%). cette résolution se manifeste par 16 fractions dans le gel 12-a (figure 12-b) et 15
fractions dans le gel 13-a (figure 13- b).
Dans le gel a, le phénomène de fusion des bandes (g et i) paru dans le gel
bidimensionnelle à gradient de concentration (b, c, d) persiste avec des spots à un centre plus
dance disposée cote à cote qui forme une seule ligne horizontale. Au point de vue résolution,
ce gel se révèle plus performant par rapport aux gels précédents. Dans le gel 13-a, le
phénomène de fusion des bandes est absent, les spots se révèlent sous forme ronde avec un
centre bien défini. Au point de vue résolution, ce gel se révèle plus performant aussi bien par
rapport aux gels précédents que par rapport à celui du gel 12-a.
Ce contraste en matière de résolution entre les gels précédents (gradient, discontinue à
10%) et ceux discontinus à 8% semble être est du au maillage moins restrictif que celui de
10%, ce qui permet de conclure que ce maillage est plus adapté pour la séparation de nos
échantillons. En effet la littérature rapporte qu’un maillage de 7.5 % pourrait servir de
concentration optimale de départ pour la séparation des protéines dont la taille est inconnue
(Walker. 1994). Dans notre étude les gels réalisés à 8% se sont traduits par des résultats
satisfaisants.

Partie III Résultats et discussions
53
Des résultats similaires ont été rapporté (Krause. 2006 ; Aivaliotis et al., 2007) qui
montre l’efficacité de cette technique en gel discontinue pour la séparation de type particulier
de protéines (protéine membranaire, protéines des mitochondriale et chloroplastique).
Pour le contraste en matière de résolution entre le gel 12-a et le gel 13-a, nous
soupçonnons un manque de reproductibilité qui a fait que le gel 13-a soit plus résolu que le
12-a. D’autres expériences restent à faire pour améliorer cette résolution.

Bibliographie

Partie V Bibliographie
54
Alba, F. J., Bermudez, A., Daban, J-R. (1996). Fluorescent Labeling of Proteins and its
Application to SDS-PAGE and Western Blotting. BioTechniques 21: 625–626.
Ames, G. F et Nikaido K. (1976). Two-dimensional gel electrophoresis of membrane
proteins. Biochemistry 15: 616-623.
Barker, D. G et al,. (1990). Medicago truncatula, a model plant for studying the molecular
geneticsof the Rhizobium-legume symbiosis. Plant Mol Biol. 8: 40–49.
Baszczynski, C. L et Walden, D. B. (1982). Regulation of gene expression in corn (Zea
Mays L.) by heat shock. Can J Biochem 60 : 569–579.
Bate-Smith, E. C. (1962). The phenolic constituents of plants and their taxonomic
significance: I Dicotyledons. Journal of the Linnean Society of London 58:95–173.
Bell, C. A et al., (2000). The Medicago genome initiative: a model legume database. Nucleic
Acid Res 29: 1–4.
Benchabane, M et al., (2008). Preventing unintended proteolysis in plant protein
biofactories. Plant Biotechnology Journal 6: 633–648.6: 633–648
Berggren, K et al., (2000). Background-free, high sensitivity staining of proteins in one- and
twodimensional sodium dodecyl sulfate-polyacrylamide gels using a luminescent
ruthenium complex. Electrophoresis 21: 2509-2521.
Berggren, K. N et al., (2002). An improved formulation of SYPRO Ruby protein gel stain:
Comparison with the original formulation and with a ruthenium II tris
(bathophenanthroline disulfonate) formulation. Proteomics 2: 486–498.
Berkman, T et Stenstedt, T. (1998). Handbook: 2D electrophoresis using immobilized pH
gradients. Principles & methods. Ed. Amersham Biosciences, New York, pp : 6429-
60.
Biemann, K. (1990). Peptides and proteins: overview and strategy. Methods Enzymol 193:
351-60.
Bollag, D. M., Edelstein, S. J., (1991). Protein Methods, Ed. Wiley Liss, New York, pp : 71-
93.
Camille, M., (1980). Fourrages, Ed. La maison rustique, Paris, pp : 302.
Campo, S et al., (2004). The defense response of germinating maize embryos against fungal
infection: A proteomics approach. Proteomics 4 : 383–396.
Cashmore, A. R., (1976). Protein synthesis in plant leaf tissue. The sites of synthesis of the
major proteins. J Biol Chem 251: 2848-2853.
Catusse, J. (2008). Transcriptome- and proteome-wide analyses of seed germination. C. R.
Biologies 331: 815–822.

Partie V Bibliographie
55
Chaudhury, S. (1973). Fractionation of chromatin nonhistone proteins. Biochim Biophys.
Acta 322: 155-165.
Chory, J et al., (1987). Gibberellin induced changes in the populations of translatable
mRNAs and accumulated polypeptides-in dwarfs of maize and pea. PlantPhysiol 83:
15-23.
Celis, J. E., et Gromov, P. (1999). 2D protein electrophoresis: can it be perfected?. Curr
Opin Biotechnol. 10: 16 – 21.
Cilia, M et al., (2009). A Comparison of Protein Extraction Methods Suitable for Gel-Based
Proteomic Studies of Aphid Proteins. Journal of Biomolecular Techniques 20: 201–
215.
Chrambach, A. (1985).The Practice of Quantitative Gel Electrophoresis. In. Westermeier,
R., (2004). Electrophoresis in the practice A Guide to Methods and Applications of
DNA and Protein Separations 4ème éd. Wiley-VCH, Germany, pp: 3.
Cook, D. R et al., (1997). Model legumes get the nod. Plant Cell 9: 275–28.
Cremer, F et Vande Walla, C. (1985). Method for extraction of proteins from green plant
tissues for two-dimensional polyacrylamide gel electrophoresis. Anal. Biochem 147:
22-26.
Davis, B. J. (1964). Disc Electrophoresis Method and Application to human serum proteins.
Ann NY Acad Sci 121: 404–427.
Delalande, M. (2007).Wild accessions / populations. Medicago truncatula handbook, pp: 1-
27.
Delphine, M. (2006). Morphology, development and plant architecture of M. Truncatula.
Medicago truncatula handbook, pp : 1-6.
De Marqui et al., (2006). Solubilization of proteins from human lymph node tissue and
twodimensional gel storage. J Biochem Mol Biol 39 : 216-22.
De Vries, S. C., Springer, J., Wessels, J. G. H. (1982). Diversity ofabundant mRNA
sequences and patterns of protein synthesis inetiolated and greened pea seedlings.
Planta 156: 129-135.
Duarte, I. (1996). Proteolysis in the quiescent seed. Physiologia plantarum 96: 519-
525.
Du Pont, F. M., Tanaka, C. K et Hurkman, W. J. (1988). Séparation and Immunological
Characterization of Membrane Fractions from Barley Roots. Plant Physiol 86:717-724.

Partie V Bibliographie
56
Galante, E., Caravaggio, T., Righetti, P.G. (1976). Binding of ampholine to
transfer RNA. Biochim Biophys Acta 442: 309–315.
Gallagher, S. R et al., (1986). A Sensitive Diffusion Plate Assay for Screening Inhibitors of
Protease Activity in Plant Cell Fractions. Plant Physiol 81: 869-874.
Gharahdaghi, F et al., (1999). Mass spectrometric identification of proteins from silver
stainedpolyacrylamide gel: A method for a removal of silver ions to enhance
sensitivity. Electrophoresis 20: 601-5.
Giavalisco et al., (2005). Proteome analysis of Arabidopsis thaliana by two dimensional gel
electrophoresis and matrix-assisted laser desorption/ionisation-time of flight mass
spectrometry. Proteomics 5 : 1902–1913.
Ginzburg, C. et Salomon, R. (1986). The Effect of Dormancy on the Heat Shock Response
in Gladiolus Cormels. Plant Physiol 8I: 259-267.
Görg, A et al., (2000). The current state of two-dimensional electrophoresis with
immobilized pH gradients. Electrophoresis 21 : 1037-53.
Görg, A., et Wiess, W. (2004). Protein profile comparisons of microorganisms, cells and
tissues using 2D gels, in Proteome Analysis Interpreting the Genome ( Speicher , D.
W. (eds), Elsevier , Amsterdam, pp: 19 – 73.
Görg, A et al., (2004). Current two-dimensional electrophoresis technology for proteomics.
Proteomics 4 : 3665-85.
Granier, F. (1988). Extraction of plant proteins for two-dimensional electrophoresis.
Electrophoresis 9: 712-718.
Groulade, P. (1978b). L’électrophorèse des protéines sériques chez le chien à l’état normal et
pathologique. Rec Med Vét 154 : 833-846.
Guy, C. L et Haskell, D. (1987). Induction of Freezing Tolerance in Spinach Is Associated
with the Synthesis of Cold Acclimation Induced Proteins. Plant Physiol 84: 872-878.
Hardy, E et al., (1996). Recovery of Biologically Active Proteins Detected with Imidazole–
Sodium Dodecyl Sulfate–Zinc (Reverse Stain) on Sodium Dodecyl Sulfate Gels. Anal
Biochem 240: 150–152.
Hari, V. (1981). A method for the two-dimensional electrophoresis of leaf proteins. Anal
Biochem 113: 332-335.

Partie V Bibliographie
57
Harrison, P. A et Black, C. C. (1982). Two-Dimensional Electrophoretic Mapping of
Proteinsof Bundle Sheath and Mesophyll Cells of the C4 Grass Digitaria sanguinalis
(L.)Scop. Plant Physiol 70: 1359- 1366.
Herbert, B. (1999). Advances in protein solubilisation for two-dimensional electrophoresis.
Electrophoresis 20: 660-663.
Hireche, Y. (2006). Réponse de la luzerne cultivée au stress hydrique et à la profondeur de
semis. Mémoire de magister en sciences agronomiques. Université de Batna, pp : 19.
Horst, M. N et al., (1980). Alkaline urea solubilization, two-dimensional electrophoresis and
lectin staining of mammalian cell plasma membrane and plant seed proteins. Anal
Biochem 102:399-408.
Huffaker, R. C. (1990). Proteolytic activity during senescence of plants. New Phytol 116 :
199-231.
Hurkman, W. J et Tanaka, C. K. (1986). Solubilization of Plant Membrane Proteins for
Analysis by Two-Dimensional Gel Electrophoresis. Plant Physiol 81: 802-806.
Kim S.T et al ., (2004a). Proteomic analysis of pathogen-responsive proteins from rice leaves
induced by rice blast fungus, Magnaporthe grisea. Proteomics 4: 3569–3578.
Kim S. T et al., (2004b). Proteome analysis of rice blast fungus (Magnaporthe grisea) proteome
during appressorium formation. Proteomics 4 : 3579–3587.
Kole, L. P et al., (2010). Recent advances in sample preparation techniques for effective
bioanalytical methods. Biomed Chromatogr 25: 199–217.
Komatsu, S. (2007). Proteomic analysis of rice seedlings during cold stress. Proteomics 7: 1293–
1302.
Kuhn, 0 et Wilt, F. H. (1980). Double labeling of chromatin proteins, in vivo and in vitro,
and their two-dimensional electrophoretic resolution. Anal Biochem 105: 274-280.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature 227: 680 – 685.
Larrainzar, E. (2007). Medicago truncatula Root Nodule Proteome Analysis Reveals
Differential Plant and Bacteroid Responses to Drought Stress. Plant Physiology,
144 :1495–1507.
Lesins, K.A et Lisens I. (1979). Genus Medicago (Leguminosae): A taxogenetic study. W.
Junk, the Hauge. London.

Partie V Bibliographie
58
Lesins, K. A et Lisens, I. (1989). Medicago (Leguminosae), A Taxogenetic study Dr
W.Junk,The Hague. London.
Levin, D. (1971). Plant phenolics – An ecological perspective. American Naturalist 105:157–
181.
Lin, C. Y., Roberts, J. K., et Key, J. L. (1984). Acquisition of Thermotolerance in Soybean
Seedling. Plant Physiol 7: 152-160.
Link, A. J. (1999). Autoradiography of 2D gels. Methods Mol Biol. 112: 285 – 290.
Mackintosh, J. A et al., (2003). A fluorescent natural product for ultra sensitive detection of
proteins in one-dimensional and two-dimensional gel electrophoresis. Proteomics 3:
2273–2288.
Malmstrom, J et al., (2004). Proteome annotations and identifications of the human
pulmonary fibroblast. J Proteome Res 3: 525-37.
Manabe, T., Hayama, E., Okuyama, T. (1982). Microscale multisample two-dimensional
electrophoresis of proteins in human serum, cerebrospinal fluid, and urine. Clin Chem
28: 824-827.
Marshall, T., Williams, K. M. (1992). Recovery of protein by Coomassie Brilliant Blue
precipitation prior to electrophoresis. Electrophoresis 13: 887-888.
Marshall, T et al., (1995). Protein concentration by precipitation with pyrogallol red prior to
electrophoresis. Electrophoresis 16: 28-31.
Mateos, A. (2008). Etudes proteomique de la micoheterogeneite des caseines αs1 te β
equines : identification dees variants post traductionnels et de phosphorylation :
identification des cites phosphorylé et des β caseines. Thèse de Doctorat. Institut
national de loraine, pp : 31.
Matsui, N. M et al., (1997). Immobilized pH gradient two-dimensional gel electrophoresis
and mass spectrometric identification of cytokine-regulated proteins in ME-180
cervical carcinoma cells. Electrophoresis 18: 409–417.
Mayer, J. E et al., (1987). A simple and general plant tissue extraction procedure for
twodimensional gel electrophoresis. Plant Cell Reports 6: 77-81.
McGregor, E et Dunn, M. J. (2006). Proteomics of the heart : unraveling disease.Circ Res
98 : 309-21.
Meier, C. L., Suding, K. N., Bowman, W. D. (2008). Carbon flux from plants to soil: roots
are a below-groundsource of phenolic secondary compounds in an alpineecosystem.
Journal of Ecology 96: 421–430.

Partie V Bibliographie
59
Merril, C. R., Switzer, R. C., Van Keuren, M. L. (1979). Trace polypeptides in cellular
extracts and human body fluids detected by two-dimensional electrophoresis and a
highly sensitive silver stain. Proc Nat Acad Sci 76: 4335-4339.
Meza-Basso, L et al., (1986). Changes in Protein Synthesis in Rapeseed (Brassica napus)
Seedlings during a Low Temperature Treatment. Plant Physiol 82: 733-738.
Mikkonen, A et al. (1986). Intracellular localization of some peptidases and a-matmosidase
in cotyledons of resting kidney bean, Phaseolus vulgaris. Physiol Plant. 68 : 75-80.
Mollenkopf, H. J et al., (1999). A dynamic two-dimensional polyacrylamide gel
electrophoresis database: the mycobacterial proteome via Internet. Electrophoresis 20:
2172-80.
Moloy, M. P et al., (1998). Extraction of membrane proteins by differential solubilization for
separation using two-dimensional gel electrophoresis. Electrophoresis 19: 837-844.
Monribot, C et Boucherie H. (1999). Two-dimensional electrophoresis with carrier
ampholytes in Proteome research: two-dimensional electrophoresis and identification
methods T. Rabilloud (eds.) Springer, pp : 31-55.
Mosher, R. A., Thormann, W. & M. (1986). Bier: An Explanation for the Plateau
Phenomenon in Isoelectric Focusing. J Chromatogr 351: 31-38.
Moulay, D. (2008). Recherche de marqueurs génétiques liés à la tolérance à la salinité chez
des écotypes d’espèces annuelles de Medicago. Mémoire de magister en Amélioration
des plantes. Université d'Oran-Es-Sénia, pp : 6.
Nagaraj, S et al. (2007). Proteomics of Medicago truncatula. Medicago truncatula handbook,
pp : 6-16.
Nalivaeva, N.N., Turner, A.J. (2001). Post-translational modifications of proteins : Acetyl
choline estérase as a model system. Proteomics 1 : 735-747.
Neuhoff, V. (1988). Improved staining of proteins in polyacrylamide gels including
isoelectricfocusing gels with clear background at nanogram sensitivity using
Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9: 255-262.
O’Farrell, P. H. (1975). High resolution two-dimensional electrophoresis of proteins. J. Biol.
Chem 250: 4007-4021.
O'Farrell P. Z., Goodman, H. M., O'Farrel, P. H. (1977). High resolution two-dimensional
electrophoresis of basic as well as acidic proteins. Cell 12: 1133–1142.
Ornstein, L et Davis, B.J. (1964). "Disc Electrophoresis, 1, Background and Theory". Ann
New York Acad Sci 121:321-349.

Partie V Bibliographie
60
Pandey, A et Mann, M. (2000). Proteomics to study genes and genomes. Nature 405: 837 –
846.
Pionneau et al., (1998). Electrophorese bidimensionnelle des proteines. protocol utilisé à
l’INRA Pierroton, pp: 1-52.
Poitevin, M. (2008). Contribution au développement d’un microsystème pour la séparation
bidimensionnelle de protéines par électrophorèse. Thèse de doctorat de Chimie
Analytique. Université Paris VI – Pierre et Marie Curie, pp: 31.
Prosperi, J. M et al. (1993). Diversité génétique, conservation et utilisation des ressources
génétiques des luzernes méditerranéennes. Les Sauve qui peut, S4, 17-24.
Prosperi, J.M et al. (1995). Ressources génétiques des plantes fourragères et à gazon, Ed.
INRA. Paris.
Rabilloud, T. (1996). Solubilization of proteins for electrophoretic analyses. Electrophoresis
17: 813-829.
Rabilloud, T et al. (1997). Improvement of the solubilization of proteins in two-dimensional
electrophoresis with immobilized pH gradients. Electrophoresis, 18: 307-316.
Rabilloud, T et al. (2001). A comparison between Sypro Ruby and rutheniumII tris
(bathophenanthroline disulfonate) asfluorescent stains for protein detection in gels.
Proteomics 1: 699 – 704.
Rabilloud, T. (2003). • 10-12 septembre 2003 / September 10-12, 2003 Analyse protéomique
: nouvelles approches d’études des interactions protéine-protéine Proteomic Analysis:
new approaches to studying protein-protein interactions. Atelier 146 : pp: 7.
Radin, N. S. (1981). Extraction of tissue lipids with a solvent of low toxicity. Methods
Enzymol 72:5-7.
Ramagopal, S. (1987). Salinity Stress Induced Tissue-Specific Proteins in Barley Seedlings.
Plant Physiol 84:324-331.
Ramakrishnan M. N et al. (2006). Medicago truncatula Stock centres. Medicago truncatula
handbook. pp : 1-18.
Rampitsch C et Srinivasan, M. (2006). The application of proteomics to the plant biology :
a review. Can J Bot 84: 883-892.
Righetti, P. G et al. (2007). Carrier ampholytes for IEF, on their fortieth anniversary (1967–2007),
brought to trial in court: The verdict (pages 3799–3810) Electrophoresis 28: 3799-3810.
Rockwell, K. R. (2009). Biologically distinct conformations of Bcl-x can be resolved using
2D isoelectric focusing. Molecular Immunology 46 : 1605–1612.

Partie V Bibliographie
61
Rothe, G. M., Purkhanbaba, M. (1982). Determination of molecular weights and Stokes'
radii of non-denatured proteins by polyacrylamide gradient gel electrophoresis. 1. An
equation relating total polymer concentration, the molecular weight of proteins in the
range of 10-4-10-6, and duration of electrophoresis. Electrophoresis 3: 33–42.
Sanchez, J-C et al.,(2001). The mouse SWISS-2D PAGE database: a tool for proteomics of
diabetes and obesity. Proteomics 1: 136 – 163.
Sanders, M. M., Groppi, V. E., Browning, E. T. (1980). Resolution of basic cellular
proteins including histone variants by two-dimensional gel electrophoresis: Evaluation
of lysine to arginineratios and phosphorylation. Anal Biochem 103: 157-165.
Schuster, A. M. et Davies, E. (1983). Ribonucleic Acid and Protein Metabolism in Pea
Epicotyls : I. The Aging Process. Plant Physiol 73: 809-816.
Shapiro, A. L., Vi Tuela, E., Maizel, J. V. (1967). Molecular weight estimation of
polypeptide chains by electrophoresis in SDS polyacrylamide gels. Biochem Biophys
Res Commun 28: 815–822.
Shaw M. M. Riederer1, B. M. (2003). Sample preparation for two-dimensional gel
electrophoresis. Proteomics, 3 :1408–1417.
Sheehan, D et Tyther, R. (2009). Two-Dimensional Electrophoresis Protocols. Ed. Methods
in Molecular Biology, Two-Dimensional Electrophoresis Protocols, vol. 519 ©
Humana Press, a part of Springer Science + Business Media, Irland, pp: 9.
Sheoran, I. S et al., (2009). Compatibility of plant protein extraction methods with mass
spectrometry for proteome analysis. Plant Science 176: 99–104.
Shevchenko, A et al., (1996). Mass spectrometric sequencing of proteins from silver stained
polyacrylamide gels. Anal Chem 68: 850–858.
Shirey, T., Huang, R. C. C. (1969). Use of sodium dodecyl sulfate, alone to separate
chromatin proteins from deoxyribonucleoprotein of Arbacia punctulata
spermchromatin. Biochemistry 8: 4138-4148.
Small, E et Jomphe, M. (1979). A synopsis of the genus Medicago (Leguminosae). Can. J.
Bot 67: 3260-3294.
St Angelo, A. J., Ory, R. L., Hansen, H. J. (1969). Localization of an acid protease In
hempseed. Phytochemistry 8: 1135.
Stanley, B. A et al., (2003). Optimizing protein solubility for two-dimensional gel
electrophoresis analysis of human myocardium. Proteomics 3 : 815-820.

Partie V Bibliographie
62
Steinberg, T. H., Hangland, R. P., Singer, V. I. (1996). Applications of SYPRO orange and
SYPRO red protein gel stains. Anal Biochem 239: 238–245.
Stochaj, W. R., Berkelman, T et Laird, N. (2003). Chapitre 4. Preparative two-dimensional
gel electrophoresis with immobilised pH gradients, in Simpson, R. J.Proteins and
Proteomics, A Laboratory Manual (eds). Cold Spring Harbor , New York, pp : 143 –
218.
Svensson, H. (1961). Isoelectric focalisation analysis and caracterisation of ampholytes in
naturalph gradientI the differential equation of solute concentration at a steady state
and its solution for thesimple cases. Acta Chem Scand 15: 325–341.
Tingey, S.V. et Coruzzi, G.M. (1987). Glutamine Synthetase of Nicotiana
plumbaginifolia:Cloning and in Vivo Expression. Plant Physiol 84: 366-373.
Toda, T., Kaji, K et Kimura, N. (1998). TMIG-2DPAGE: a new concept of twodimensional
gel protein database for research on aging. Electrophoresis 19: 344-8.
Uemura, M. et Yoshida, S. (1984). Involvement of Plasma Membrane Alterations in
ColdAcclimation of Winter Rye Seedlings (Secale cereale L. cv Puma). Plant Physiol
75:818-826.
Ünlü, M., Morgan, M. E., et Minden, J. S. (1997). Difference gel electrophoresis: a single
gel method for detecting changes in protein extracts. Electrophoresis 18: 2071 – 2077.
Vesterberg, O. (1969). Synthesis and Isoelectric Fractionation of Carrier Ampholytes. Acta
Chem Scan 23: 2653–2666.
Vieira-Nunes, A.I. (2006). Transport d’ions sous l’effet d’un champ electrique en milieu
poreux : application à la separation de terres rares par électrophorèse à focalisation.
Thèse de doctorat à Institut National Polytechnique de Lorraine - Nancy-Université, pp: 18.
Vierling, E et Key, J. L. (1985). Ribulose 1,5-Bisphosphate Carboxylase Synthesis during
Heat Shock. Plant Physiol 78: 155-162.
Wasinger, V.C et al., (1995). Progress with gene-product mapping of the Mollicutes:
Mycoplasma genitalium. Electrophoresis 16: 1090–1094.
Wheelock, A .M et Wheelock, C.E. (2008). Bioinformatics in gel- based proteomics book of
Plant Proteomics: Technologies, Strategies, and Applications. Ed. John Wiley &
Sons, Inc. New jersey, pp:108.
Westermeier, R., Naven, T. (2004). Electrophoresis in the practice A Guide to Methods
andApplications of DNA and Protein Separations 4ème éd. Wiley-VCH, Germany.

Partie V Bibliographie
63
Willard, K.E et al., (1979). Analytical techniques for cell fractions. XXVI. A two-
dimentionalelectrophoretic analysis of basic proteins using phosphatidyl choline/urea
solubilization. Anal Biochem 100: 289-98.
Yahia, N et Fyad-Lameche, F.Z. (2003). Evaluation de la variabilité de jeunes plants de
Medicago soumis à un régime de basse température. Acta botanica gallica 150: 3-17.
Yan, S. (2005). Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics
5 : 235–244.
Zhentian, L et al., (2005). A Two-dimensional Electrophoresis Proteomic Reference Map
and Systematic Identification of 1367 Proteins from a Cell Suspension Culture of the
Model Legume Medicago truncatula. Molecular & Cellular Proteomics 4: 1812-1825.
Zivy, M., Thiellement, H., Vienne, D., et Hofmann, J. P. (1983 ). Study on nuclear and
cytoplasmic genome expression in wheat by two-dimensional gel electrophoresis.
Theor Appl Genet 66: 1-7.
Zivy, M. (1986). Electrophoraise¨ bidimensionnelle, Presses Universitaires In: Galteau, M. M
et Siest, C., Eds., Nancy, pp : 69-72.





























