Embed Size (px)
Citation preview

SPECTROSCOPIE IRThéorie et
ÉCOLE TECHNIQUE SUPÉRIEURE DU LABORATOIRE
95, rue du Dessous des Berges, 75013 PARIShttp://ligodin.free.fr
Licence des Industries Pharmaceutiques, Cosmétologiques, et de Santé : gestion, production et valorisation.Option APC - Année 2017/2018
1

SOMMAIRE
• 1) Théorie
• 1.1 Nature du rayonnement
• 1.2 Représentation d’une molécule : Niveaux d’énergie
• 1.3 Transitions roto-vibrationnelles des molécules vues comme des dipôles électriques
• 1.4 Modèle mécanique classique d’une vibration d’élongation d’une molécule diatomique
2

• 1.5 Traitement quantique des vibrations
• 1.6 L’oscillateur anharmonique
• 1.6.1 Formule générale : série de Mac Laurin
• 1.6.2 Potentiel de Morse
• 1.6.3 Oscillateur anharmonique quantifiée
• 1.7 Molécule polyatomique
• 1.7.1 Les modes de vibration
• 1.7.2 Notion de fréquence de groupe
• 1.7.3 Notion de fréquences fondamentales, harmoniques et de combinaison
• 1.7.4 Le couplage des vibrations
3

• 2) Instrumentation IR
• 2.1 Sources et détecteurs de rayonnement IR
• 2.1.1 Les sources
• 2.1.2 Les détecteurs
• 2.2 Appareils utilisés dans l’IR
• 2.2.1 Dispositifs d’analyse du rayonnement
• 2.2.2 Les appareils dispersifs
• 2.2.3 Les photomètres à filtres
4

INTRODUCTION
☛ L’objectif des spectroscopies optiques est d’obtenir des informations sur la matière à partir de son interaction avec le rayonnement.ˇ
☛ En pratique, l’analyse peut être qualitative : l’identification d’un composé est recherchée à partir de sa signature spectrale, ou quantitative : dans ce cas, c’est une méthode de dosage d’une substance, grâce à sa signature spectrale, qui est recherchée.
☛ S’intéresser à la spectroscopie IR, c’est s’intéresser à la spectroscopie de vibration des molécules.
5

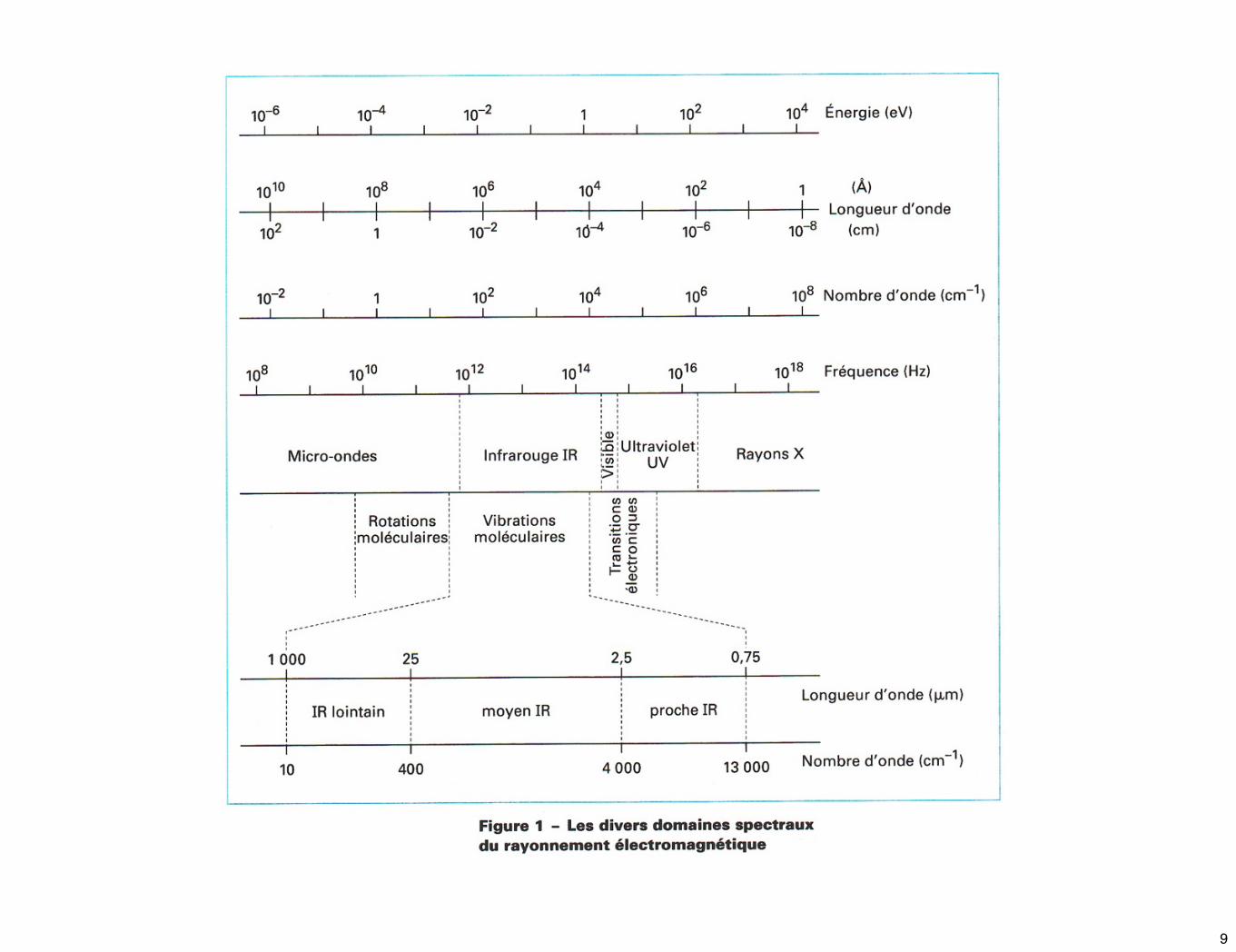
☛ Le rayonnement em est constitué par le couplage d’un champ électrique et magnétique qui lorsqu’il se propage, définit une onde em. Celle-ci est dite monochromatique si elle correspond à une radiation de fréquence ν donnée, parfaitement définie.
☛ Le formalisme décrivant la propagation d’ondes est donné par l’équation d’onde ou équation de d’Alembert :
1) Théorie1.1 Nature du rayonnement
6

Une solution de l’équation d’onde peut s’exprimer sous la forme d’ondes planes, c.a.d tel que les champs électrique et magnétique soient, à un instant donné, uniformes et perpendiculaires entre eux pour tous les points d’un plan donné (dit plan d’onde), et normaux à la direction de propagation.
Notes
7

☛ On définit aussi le nombre d’onde, exprimé en cm-1, par :
☛ Selon la mécanique quantique, le rayonnement em présente une double nature : ondulatoire et corpusculaire. Donc, le rayonnement peut être aussi considéré comme un flux de particules : les photons.
☛ L’énergie E d’un photon associé à une onde em de fréquence ν obéit à la relation de Planck :
E = h.ν
☛ Donc une onde peut être caractérisée par sa périodicité spatiale λ, sa périodicité temporelle ν ou par l’énergie des photons qui lui sont associées.
8
9

1.2 Représentation d’une molécule : Niveaux d’énergie
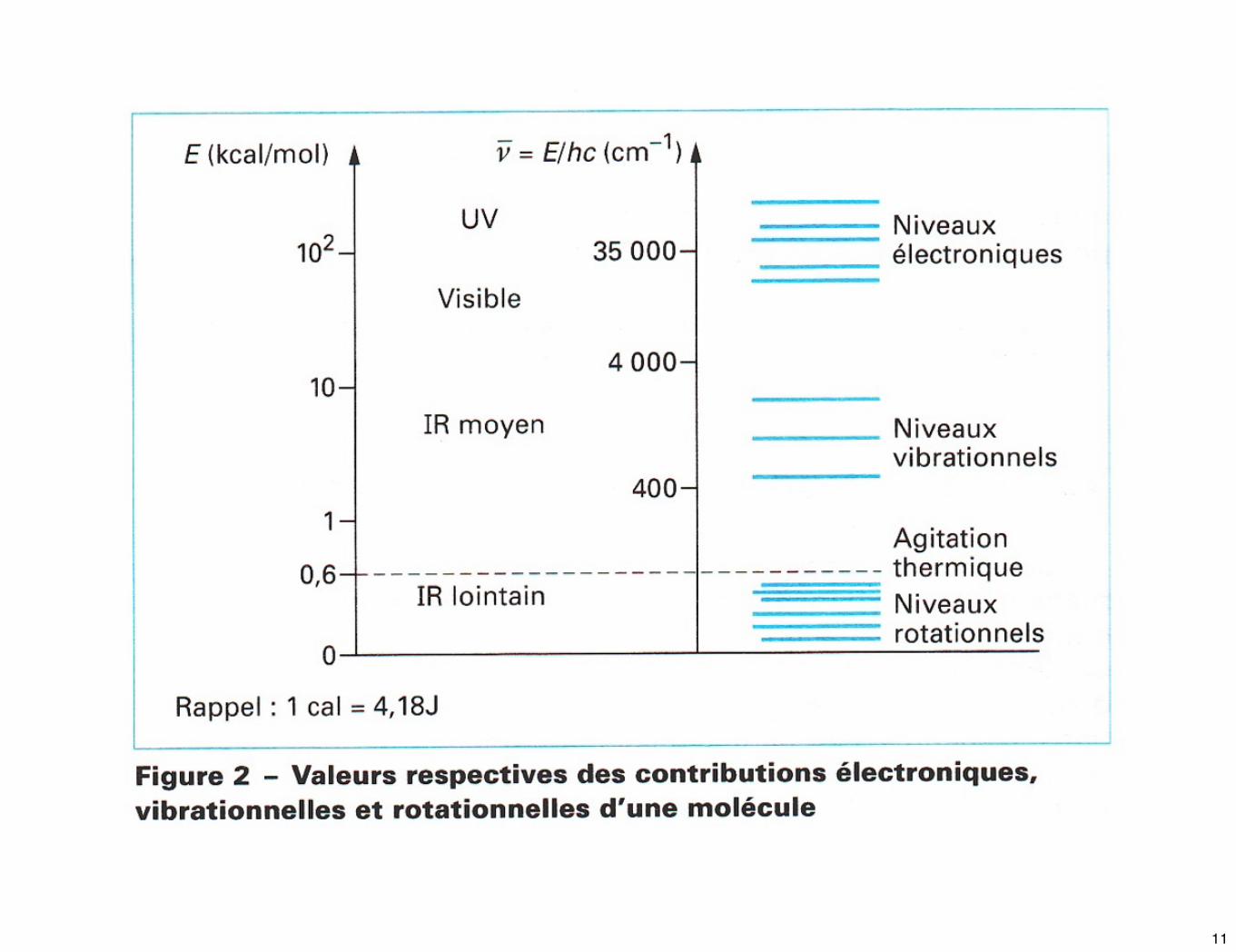
☛ L’énergie E d’une molécule est quantifiée et dépend de nombres entiers que l’on nomme nombres quantiques.
☛ Une molécule peut être considérée comme formée d’atomes dont les e- assurent la liaison chimique (liaison covalente).
☛ Pour simplifier la description moléculaire, on utilise l’approximation de Born-Oppenheimer qui revient à découpler le mouvement des noyaux de celui des e-, bien plus légers, et donc, à découpler leurs énergies respectives.
☛ En première approximation, E peut donc s’écrire sous la forme :
E = Ee + Ev + Er
Avec Ee >> Ev >> Er
10
11

1.3 Transitions roto-vibrationnelles dans les molécules vues comme des dipôles électriques
➫ Modifications des dipôles au cours des vibrations et des rotations
☛ Le rayonnement IR n’est pas assez énergétique pour induire les transitions électroniques que l’on peut rencontrer en spectroscopie X, UV, et visible.
☛ L’absorption IR n’est possible qu’entre les états de rotation et de vibration moléculaires.
☛ L’absorption IR par une molécule requiert que ses mvts de vibration et de rotation modifient son moment dipolaire. C’est uniquement dans ces conditions que le champ électrique alternatif peut interagir avec la molécule et entraîner une modification de l’amplitude de l’un de ces mvts.
12

☛ La molécule HCl est animée de mvts de vibration, son moment dipolaire varie et ce phénomène est à l’origine d’un champ variable qui peut interagir avec le champ électrique de l’onde incidente.
☛ Si la fréquence de celle-ci est exactement égale à celle de la vibration naturelle de la molécule, un transfert d’énergie se produit ce qui entraîne une modification de l’amplitude de la vibration moléculaire et, par conséquent, l’absorption de l’onde.
➫ Transitions de rotation
☛ L’énergie nécessaire pour observer une transition de rotation est faible : elle correspond à un rayonnement de 400 cm-1 à 10 cm-1. Les niveaux rotationnels étant quantifiés, l’absorption par les gaz dans l’IR lointain est caractérisée par la présence de raies discrètes bien définies.
☛ Dans les liquides ou les solides, les collisions intermoléculaires et les interactions entraînent un élargissement des raies jusqu’à former un continuum.
13

➫ Transitions de vibration
☛ Les niveaux d’énergie de vibration sont eux aussi quantifiés; pour la plupart des molécules, les différences d’énergie entre les différents états quantiques correspondent au domaine du MIR.
➫ Différents types de vibration moléculaire
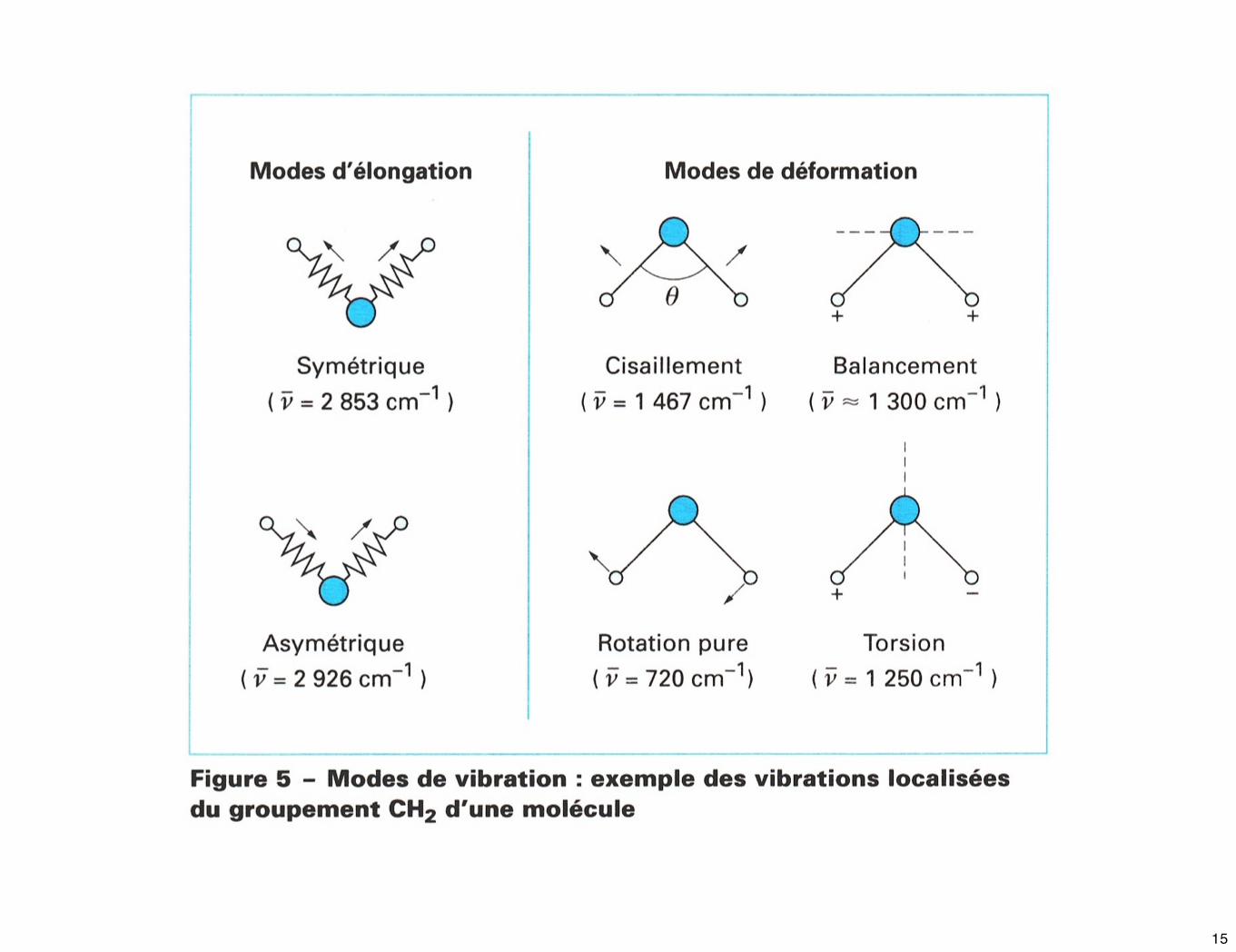
☛ Les vibrations peuvent être classées en 2 catégories : les vibrations d’élongation et les vibrations de déformation angulaire. Une vibration d’élongation correspond à une variation continue de la distance interatomique dans la direction de l’axe de la liaison entre les atomes.
☛ Les vibrations de déformation angulaire sont caractérisées par une modification de l’angle entre deux liaisons, il en existe de 4 types : le cisaillement, la rotation, le balancement et la torsion.
14
15

1.4 Modèle mécanique classique d’une vibration d’élongation dans une molécule diatomique
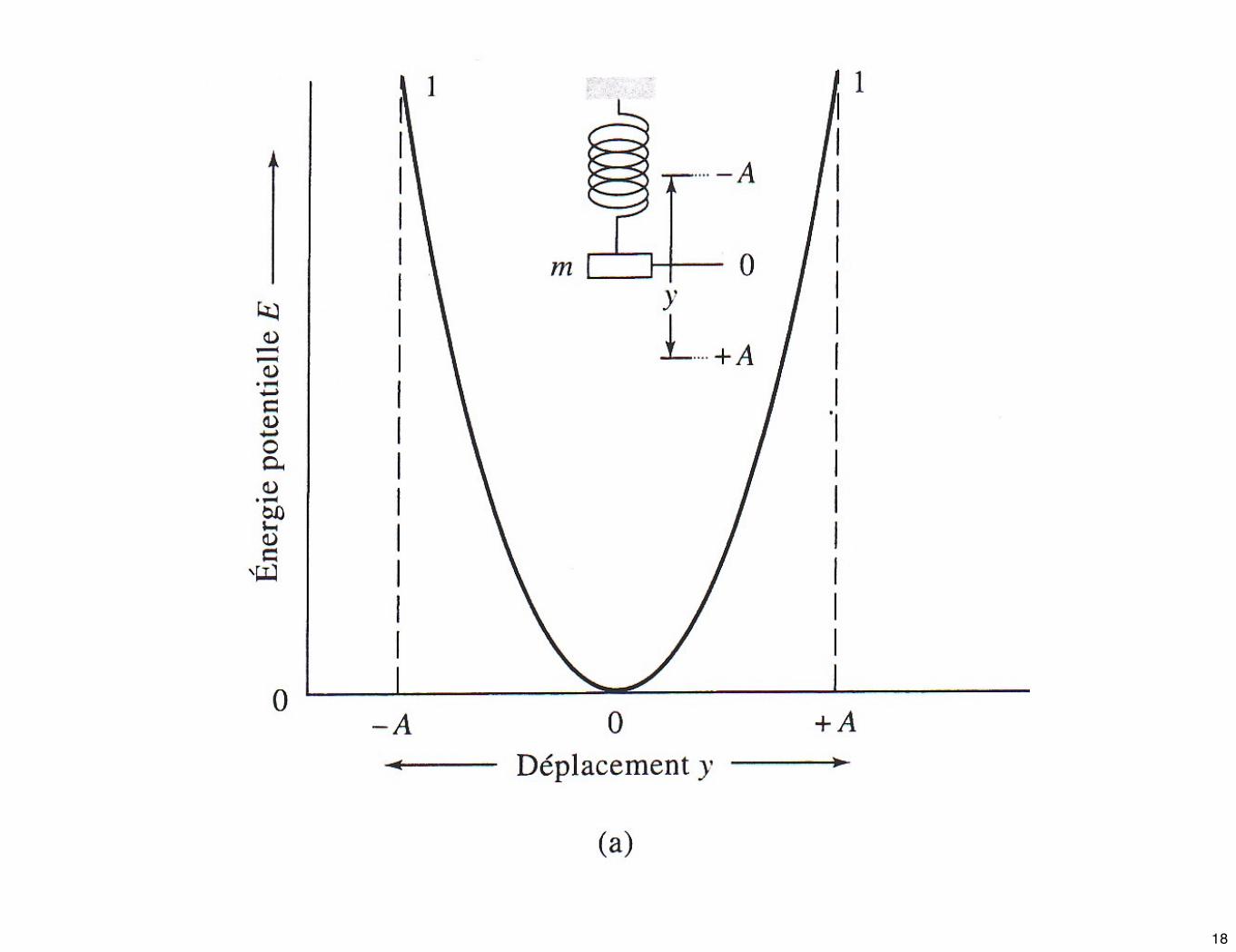
☛ Les propriétés d’une vibration d’élongation peuvent être décrites par un modèle mécanique constitué de deux masses reliées par un ressort. Une perturbation de l’une des deux masses le long de l’axe du ressort entraîne une vibration appelée mvt harmonique simple.
☛ Examinons la vibration d’une masse simple attachée à un ressort accroché à un point fixe. Si l’on écarte la masse d’une distance y = + A de son point d’équilibre par application d’une force dirigée selon l’axe du ressort, la force de rappel est proportionnelle à l’élongation (loi de Hooke), on a :
16

➫ Énergie potentielle d’un oscillateur harmonique
☛ On attribue arbitrairement la valeur zéro à l’énergie potentielle E du système lorsque la masse est au repos dans sa position d’équilibre. Lorsque l’on comprime ou l’on étire le ressort, l’énergie potentielle du système est égale à l’opposée du travail fourni pour effectuer l’une ou l’autre de ces actions.
☛ Si elle est déplacée, par exemple, de y à y + dy, la variation d’énergie potentielle dE s’écrira :
dE = - dW = - F.dy = k.y.dy
☛ L’intégration entre la position d’équilibre y = 0 et y donne : Notes
17
18

➫ Fréquence de vibration
Notes
19
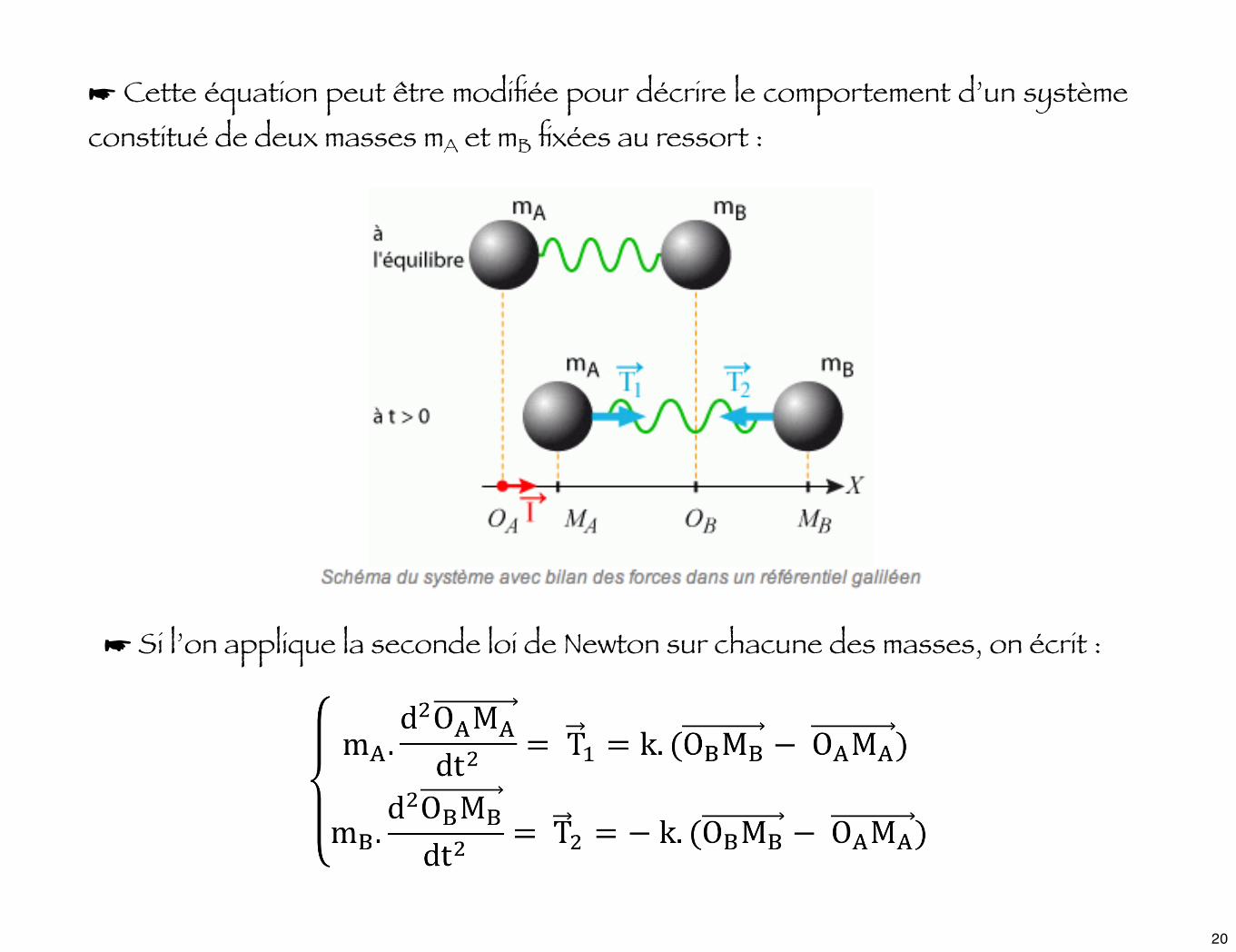
☛ Si l’on applique la seconde loi de Newton sur chacune des masses, on écrit :
☛ Cette équation peut être modifiée pour décrire le comportement d’un système constitué de deux masses mA et mB fixées au ressort :
20

Notes
21

☛ On obtient finalement,
☛ Equation similaire à celle que l’on a vu dans le cas d’une masse, la fréquence naturelle de vibration de ce système est donnée par :
1.5 Traitement quantique des vibrations
☛ Les équations de la mécanique classique ne décrivent pas complètement le mouvement des particules de dimensions atomiques.
☛ La quantification des énergies de vibration moléculaire n’apparait pas dans ces équations.ˇ
22

☛ L’équation de Schrödinger vient remplacer l’équation du mouvement et s’écrit :
HΨ(q) = EvΨ(q)
☛ Cette équation joue le même rôle en mécanique quantique que les équations du mouvement de Newton en mécanique classique.
où H représente l’opérateur Hamiltonien définie par :
23

☛ La résolution de l’équation permet de trouver les niveaux d’énergie Ev de la molécule diatomique.
☛ Ces niveaux quantifiés sont repérés par un nombre quantique ν qui est appelé nombre quantique de vibration.
☛ On obtient,
Avec ν ∈ N.ˇ
24
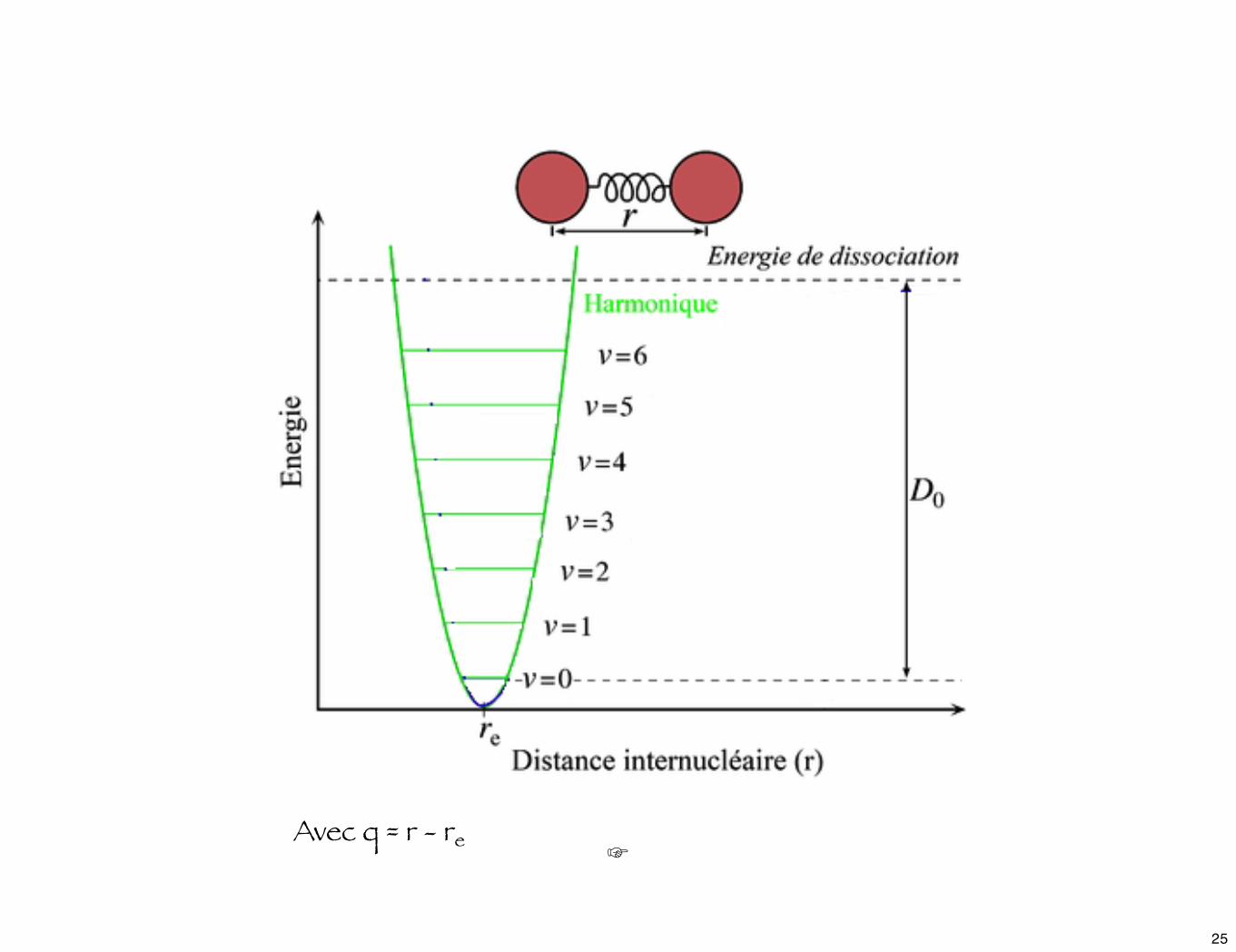
☞Avec q = r - re
25
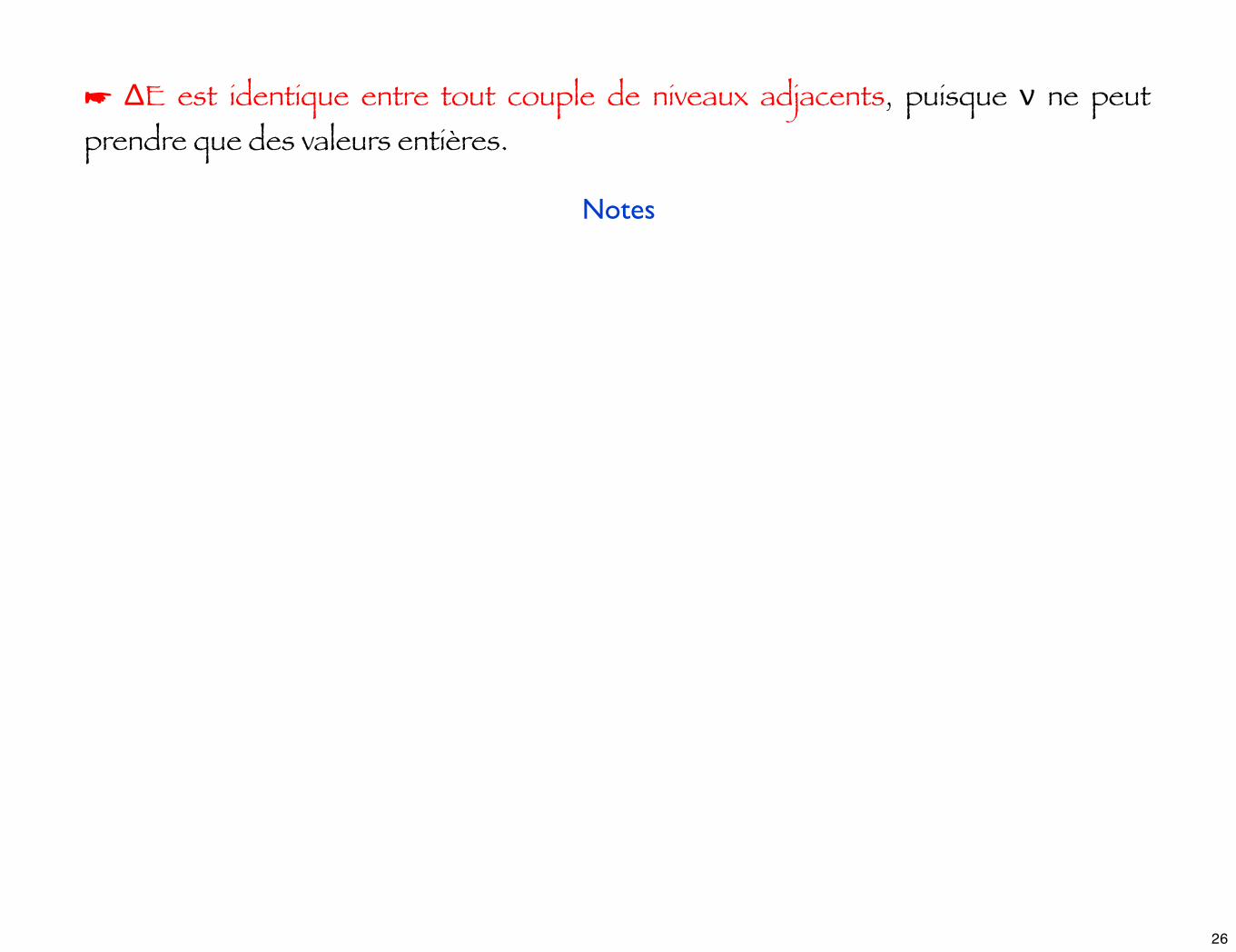
☛ ΔE est identique entre tout couple de niveaux adjacents, puisque ν ne peut prendre que des valeurs entières.
Notes
26

☛ Cette dernière relation permet de déterminer les constantes de force des différentes liaisons chimiques à partir de mesures dans l’IR.
☛ La valeur moyenne de k des liaisons simples est de l’ordre de 5.102 N.m-1. Les constantes de force des doubles et triples liaisons sont environ deux à trois fois plus élevées (respectivement 103 et 1,5.103 N.m-1).
☛ À partir de ces valeurs moyennes, on peut estimer les nombres d’onde des pics d’absorption fondamentaux, correspondant à la transition au premier état excité à partir de l’état fondamental des différentes liaisons.
Calculez approximativement le nombre d’onde et la l.o du pic fondamental d’absorption dû à la vibration d’élongation du groupement C=O :
27

➫ Règles de sélection
☛ La mécanique quantique montre qu’une transition vibrationnelle est observée seulement si : Δν = ± 1
Remarque : Dans la pratique, d’une part, on sait qu’une liaison peut se rompre, et d’autre part, on observe des bandes qui ne sont pas interprétables !!!
Ce modèle ne permet donc pas de décrire la dissociation et l’observation des bandes dites harmoniques. En effet, elles ne sont pas permises car les règles de sélection autorisent seulement Δν = ± 1
28

1.6 L’oscillateur anharmonique
☛ Le modèle mécanique harmonique est très simple et peut donner une idée sur la position des bandes fondamentales ou sur la force des liaisons, mais il n’est pas très représentatif des molécules réelles.
☛ On a admis que les liaisons étaient parfaitement élastiques. On sait cependant que ces liaisons peuvent se briser quand l’amplitude des vibrations devient importante.
☛ La forme de la courbe d’Ep de la molécule en fonction du déplacement est donc plus compliquée qu'une simple parabole. De nombreuses expressions mathématiques ont été proposées pour l'Ep des oscillateurs anharmoniques.
29

1.6.1 Formule générale : série de Mac Laurin
Notes
30

1.6.2 Potentiel de Morse
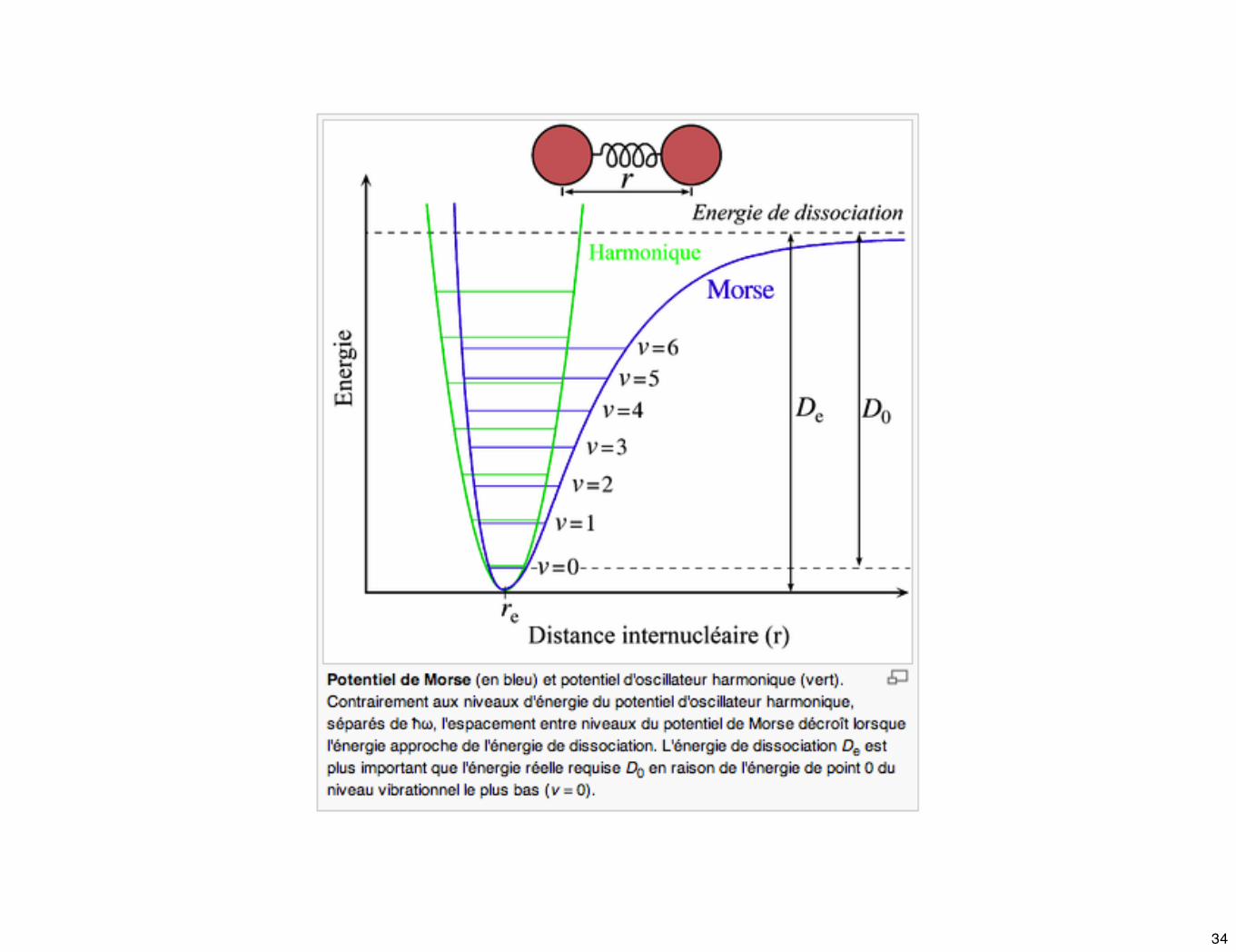
☛ Une expression purement empirique, qui suit avec une bonne approximation la courbe d'énergie potentielle, a été obtenue en 1929 par Morse et est connu sous le nom de potentiel de Morse :
où α est une cte caractéristique de la liaison entre les atomes.De l'énergie de dissociation.
31
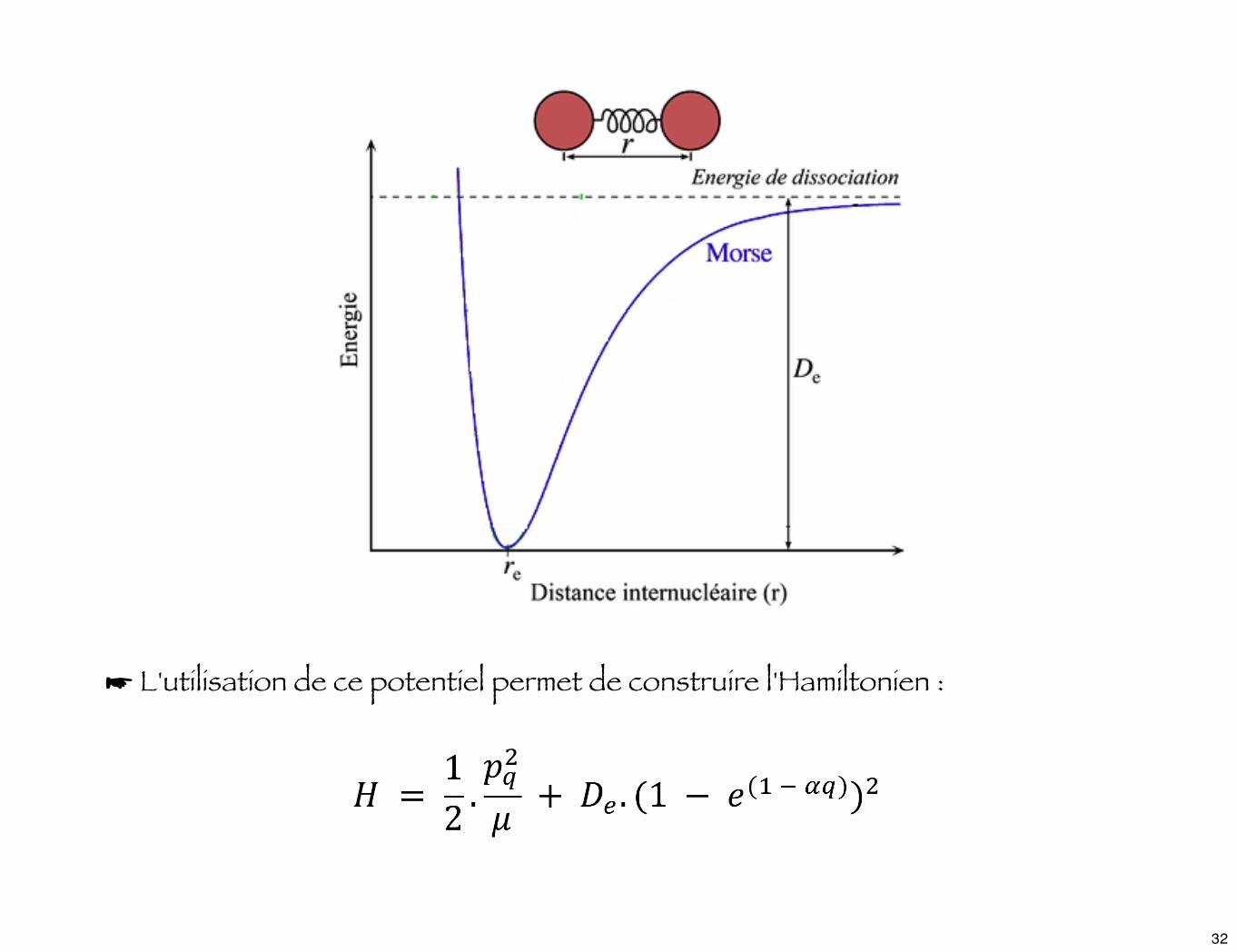
☛ L'utilisation de ce potentiel permet de construire l'Hamiltonien :
32

1.6.3 Oscillateur anharmonique quantifié
☛ Il faut de nouveau utiliser la mécanique quantique pour déterminer les niveaux d'énergie. La résolution de l'équation de Schrödinger est délicate. L'avantage important du potentiel de Morse est qu'il permet d'obtenir une solution presque exacte.
☛ On obtient les niveaux d'énergie autorisée suivant :
xe représente la constante d'anharmonicité > 0 : à cause de cette cte, l'espace entre les niveaux d'énergie ➘ quand ν ➚ et ces niveaux convergent vers une valeur limite qui correspond à la dissociation. ˇ
33
34

☛ L'anharmonicité conduit à deux types d'écart. Aux valeurs élevées du nombre quantique, ΔE ➘ et la règle de sélection n'est pas rigoureusement vérifiée.
Donc des transitions Δν = ± 2 ou ± 3 ne sont pas improbables. elles sont responsables de l'apparition de bandes harmoniques supérieures à des fréquences approximativement égales à 2 ou 3 fois la fréquence fondamentale. L'intensité de ces bandes est en général faible; elles peuvent même échapper à l'observation.
35
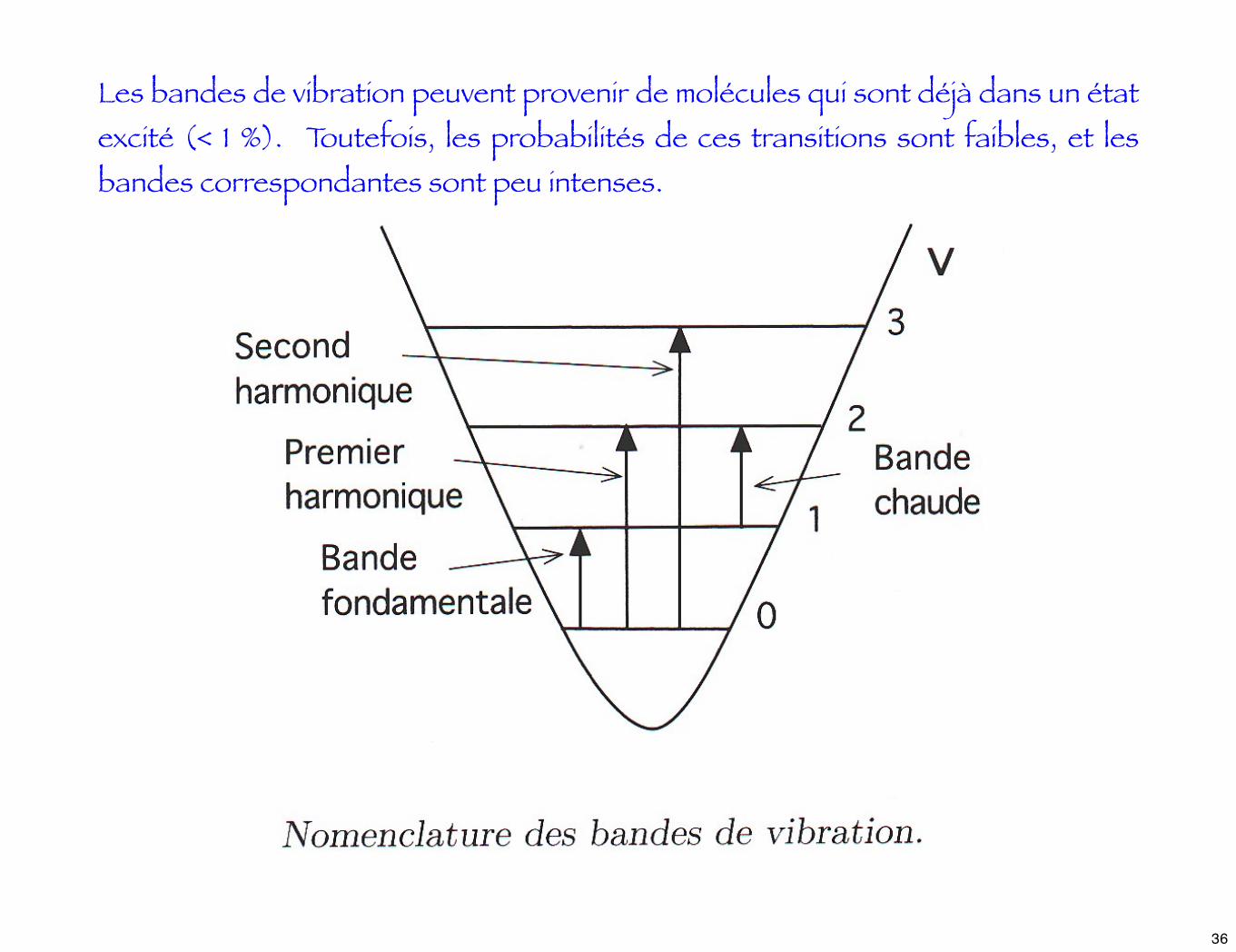
Les bandes de vibration peuvent provenir de molécules qui sont déjà dans un état excité (< 1 %). Toutefois, les probabilités de ces transitions sont faibles, et les bandes correspondantes sont peu intenses.
36

1.7 Molécule polyatomique
1.7.1 Les modes de vibration
☛ Une molécule formée de N atomes possède 3N degrès de liberté. Parmi eux, 3 représentent la translation de la molécule dans son ensemble (le long des 3 axes du repère x, y, z) et 3 autres définissent la rotation de la molécule autour de chacun de ces axes. Finalement, les mvts internes de vibration de la molécule seront déterminés par les 3N - 6 coordonnées restantes (mvts de vibration interatomiques).
☛ La molécule linéaire est un cas à part puisque, par définition, tous les atomes sont disposés en ligne droite. La rotation autour de l'axe de la liaison n'est pas possible, 2 degrès de liberté suffisent alors pour décrire la rotation de la molécule. Le nombre de modes de vibration est ainsi égal à 3N - 5.
37

☞ Ces modes de vibration sont appelés modes normaux.
☞ On a vu que l’on distingue, d'une part, les modes de vibrations de valence ou
d'élongation (symétriques ou antisymétriques) qui font intervenir une (des) variation(s) de(s) longueur(s) de liaison(s), les angles que forment ces liaisons restant constants, et, d'autre part, les modes de déformation, pour lesquels, les liaisons gardent leur longueur, mais les angles qu'elles forment varient.
☞ À chaque mode normal correspond une courbe d'Ep. Dans la mesure où l'on
peut considérer une vibration comme étant harmonique, les différences entre les niveaux d'énergie d'un mode donné sont égales, ce qui signifie qu'il n'apparaîtra qu'une seule bande d'absorption par mode pour lequel il y a modification du dipôle.
38

☞ 4 facteurs tendent à rendre le nombre de pics expérimentaux inférieur au
nombre théorique de modes normaux :
1) la symétrie des molécules est telle qu'un mode de vibration particulier ne modifie pas le dipôle,
2) les énergies de 2 modes de vibration ou plus sont égales ou quasi-égales,
3) l'intensité du pic est tellement faible qu'on ne peut le détecter,
4) l'énergie du mode de vibration est située en dehors du domaine spectral.
39

1.7.2 Notion de fréquence de groupe
☛ On s'est rendu compte expérimentalement que certaines molécules contenant, par exemple, le groupe R3C-H, présentent une bande d'absorption à 2960 cm-1, alors que d'autres molécules possédant une liaison C-H dans un groupe de type R2C-H ont une bande à 3020 cm-1, et, enfin d'autres ayant une liaison C-H dans un motif de type RC-H ont un mode de vibration à 3300 cm-1.
☛ Donc la présence dans un spectre de certaines fréquences caractéristiques sont considérées comme significatives de la présence d'un groupe chimique donné.
☛ L'existence de fréquences caractéristiques de groupements chimiques repose sur le fait que la cte de force d'une liaison chimique particulière semble transférable d'une molécule à une autre.
☛ Cela revient à dire que le mvt de vibration de la molécule est essentiellement localisé dans le groupe chimique considéré, qui constitue, en quelque sorte, un oscillateur indépendant.
40

41

☛ Occasionnellement, on peut rencontrer plus de bandes que prévu. Il peut exister des bandes harmoniques supérieures.
☛ De plus, on peut parfois rencontrer des bandes de combinaison quand un photon excite simultanément 2 modes de vibration. La fréquence de ces bandes est approximativement égale à la somme ou à la différence de 2 fréquences fondamentales.
☛ Ce phénomène se produit lorsqu'un quantum d'énergie est absorbée par 2 liaisons plutôt que par une seule.
42

☛ Une autre conséquence de cette anharmonicité est le couplage de deux vibrations de fréquences ν1 et ν2 en une vibration de fréquence somme ν1 + ν2 ou de fréquence différence ∣ν1 - ν2∣. On parle alors de combinaisons respectivement additive ou soustractive des bandes de fréquences ν1 ou ν2.
1.7.3 Notion de fréquences fondamentales, harmoniques et de combinaison
☛ On nomme harmoniques les fréquences nν0 (n entier) multiples de la fréquence fondamentale (la plus petite) ν0 d'un oscillateur.
Nous avons vu précédemment que l'anharmonicité des vibrations modifie l'expression de la fréquence d'une transition (puisque les niveaux se resserrent quand l'énergie ➚). Elle a aussi pour conséquence de provoquer l'apparition de bandes dites harmoniques correspondant à des transitions Δν > 1, de fréquence 2ν, 3ν, ... d'intensité faible par rapport à la bande de fréquence ν.
43

1.7.4 Le couplage des vibrations
☛ L'énergie d'une vibration peut être influencée (donc couplée) par d'autres oscillateurs dans la molécule. Un certain nombre de facteurs influencent ce couplage :
1) Le couplage est important entre deux vibrations d'élongation uniquement lorsqu'un atome participe aux 2 modes.
2) L'interaction entre des vibrations de déformation angulaire nécessite une liaison commune entre les 2 atomes oscillants.
3) Le couplage entre les vibrations d'élongation et de déformation angulaire ne peut se produire que si la liaison subissant l'élongation forme le côté d'un angle qui varie avec la vibration de déformation angulaire.
44

4) L'interaction est maximale quand les énergies individuelles des groupes couplés sont à peu près égales.
5) Il n'y a pratiquement pas de couplage entre des groupes séparés par deux liaisons ou plus.
6) Le couplage nécessite que les vibrations appartiennent au même groupe de symétrie.
45
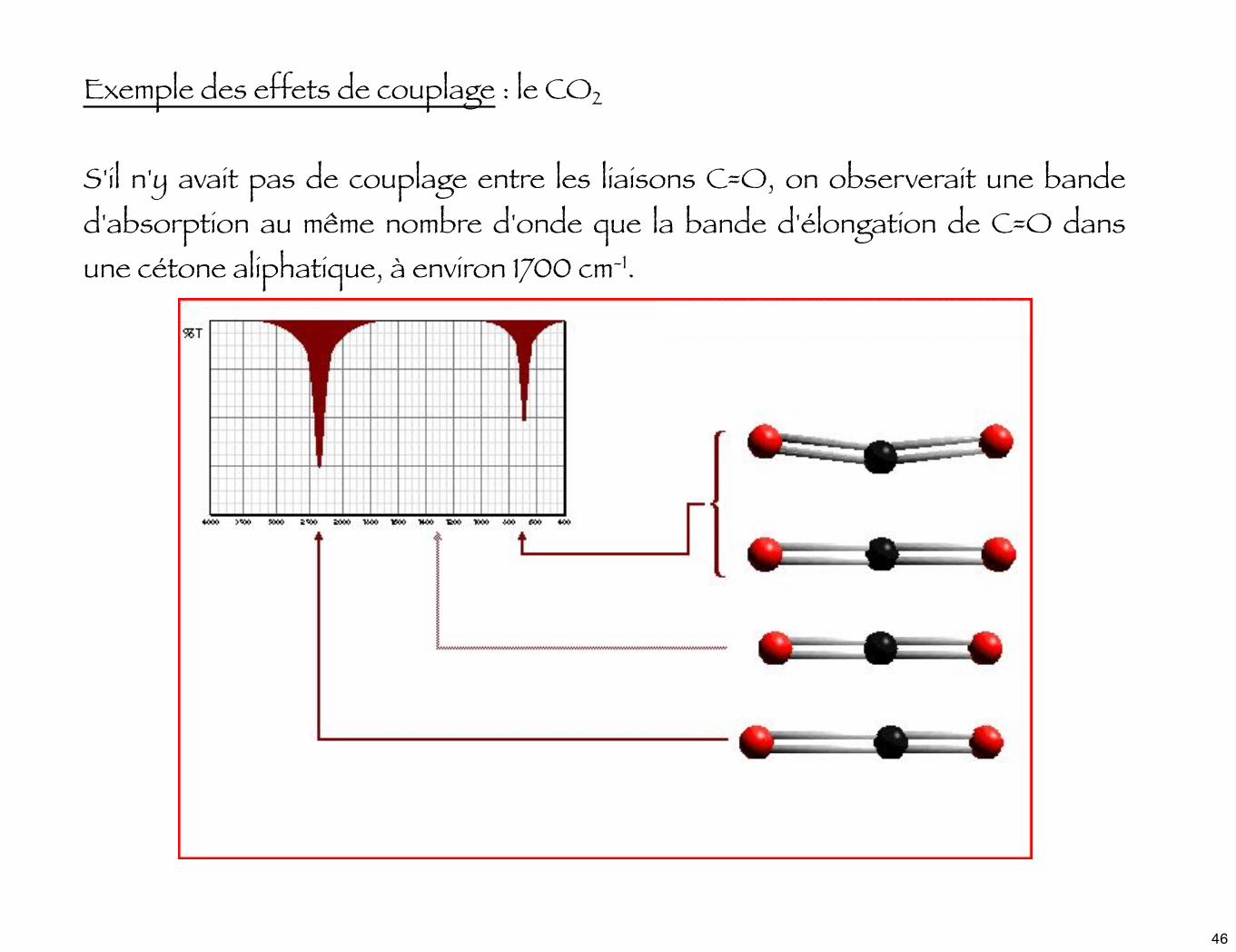
Exemple des effets de couplage : le CO2
S'il n'y avait pas de couplage entre les liaisons C=O, on observerait une bande d'absorption au même nombre d'onde que la bande d'élongation de C=O dans une cétone aliphatique, à environ 1700 cm-1.
46
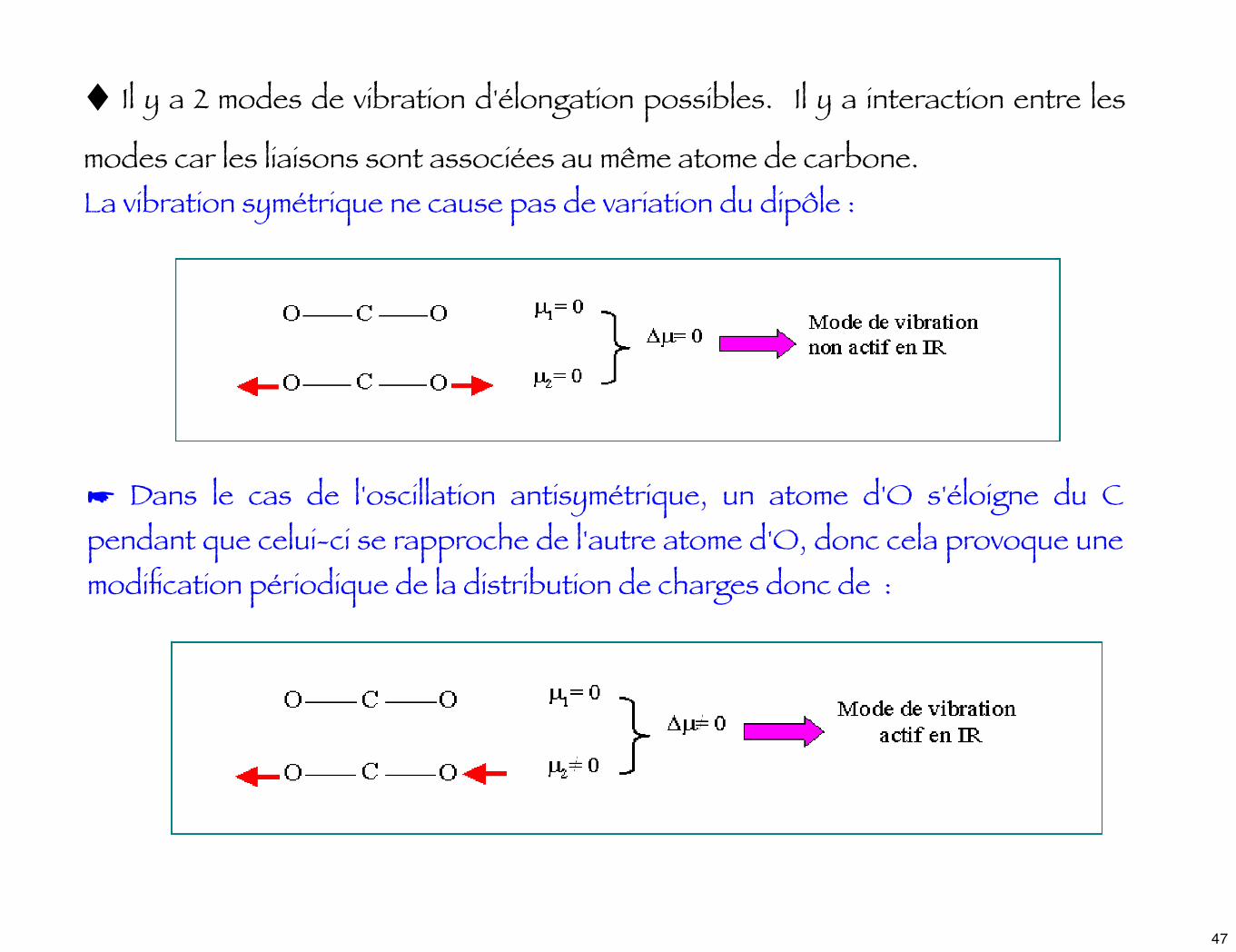
♦ Il y a 2 modes de vibration d'élongation possibles. Il y a interaction entre les
modes car les liaisons sont associées au même atome de carbone.La vibration symétrique ne cause pas de variation du dipôle :
☛ Dans le cas de l'oscillation antisymétrique, un atome d'O s'éloigne du C pendant que celui-ci se rapproche de l'autre atome d'O, donc cela provoque une modification périodique de la distribution de charges donc de :
47
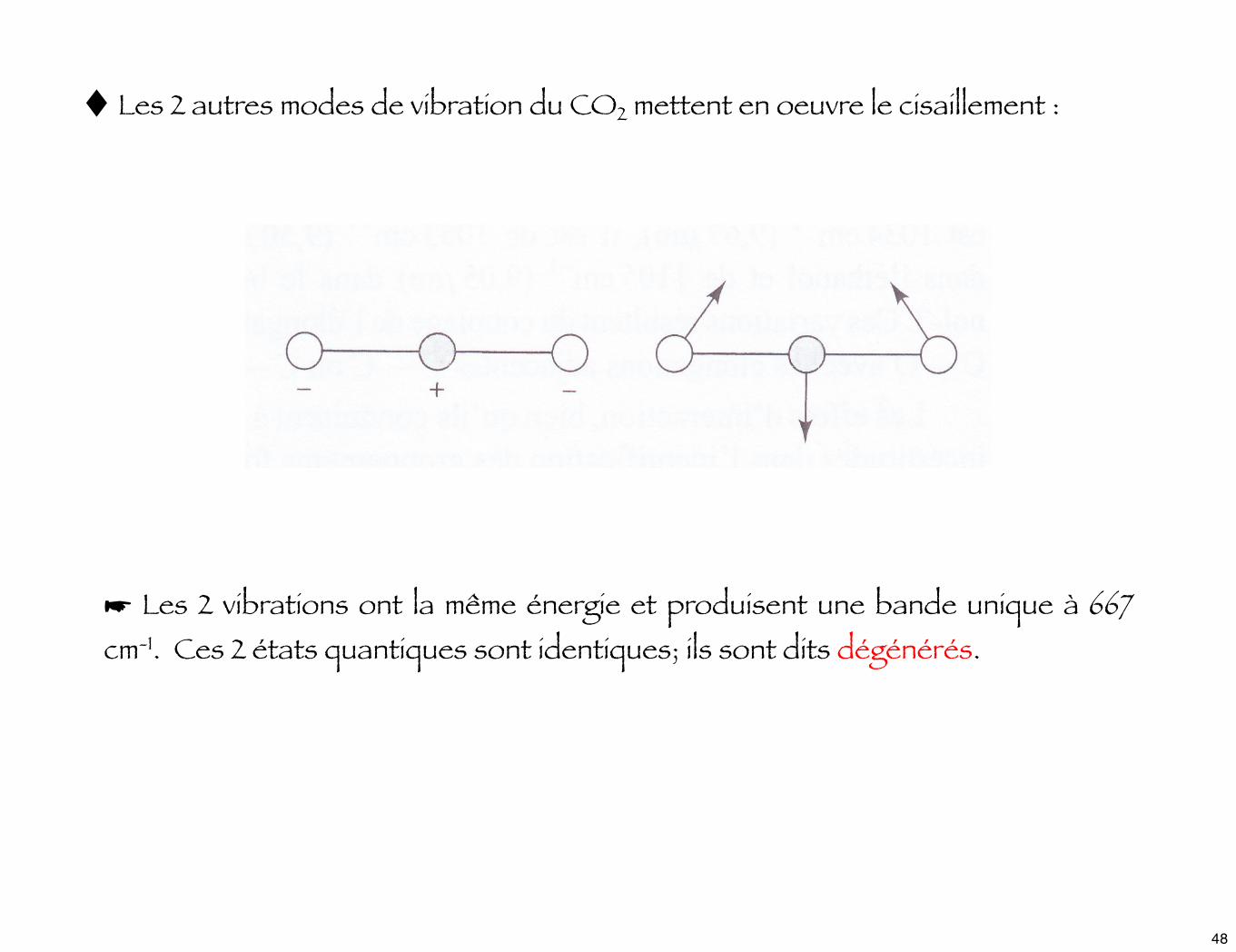
♦ Les 2 autres modes de vibration du CO2 mettent en oeuvre le cisaillement :
☛ Les 2 vibrations ont la même énergie et produisent une bande unique à 667 cm-1. Ces 2 états quantiques sont identiques; ils sont dits dégénérés.
48

☛ Le couplage des oscillations est un phénomène courant ; par conséquent, la position de la bande d'absorption correspondant à un groupement fonctionnel organique ne peut pas toujours être spécifiée avec exactitude.
Exemple : le nombre d'onde d'élongation de C-O dans le méthanol est de 1034 cm-1, il est de 1053 cm-1 dans l'éthanol, et de 1105 cm-1 dans le butan-2-ol. Ces variations résultent du couplage de l'élongation C-O avec les élongations adjacentes C-C ou C-H.
☛ Les effets d'interaction, bien qu'ils conduisent à des incertitudes dans l'identification des groupements fonctionnels, sont à l'origine de l'unicité de chaque spectre d'absorption IR ; cette propriété est d'une importance capitale et permet l'identification d'un composé donné.
49

☛ Il existe plusieurs méthodes pour enregistrer le spectre IR d'une substance. Les spectromètres optiques ont en commun les éléments suivants :
1) Une source de radiations, qui doit émettre dans le domaine IR;
2) Le dispositif de sélection des l.o ;
3) Le dispositif de positionnement de l'échantillon dans le faisceau ;
4) Le détecteur et l'enregistreur.ˇ
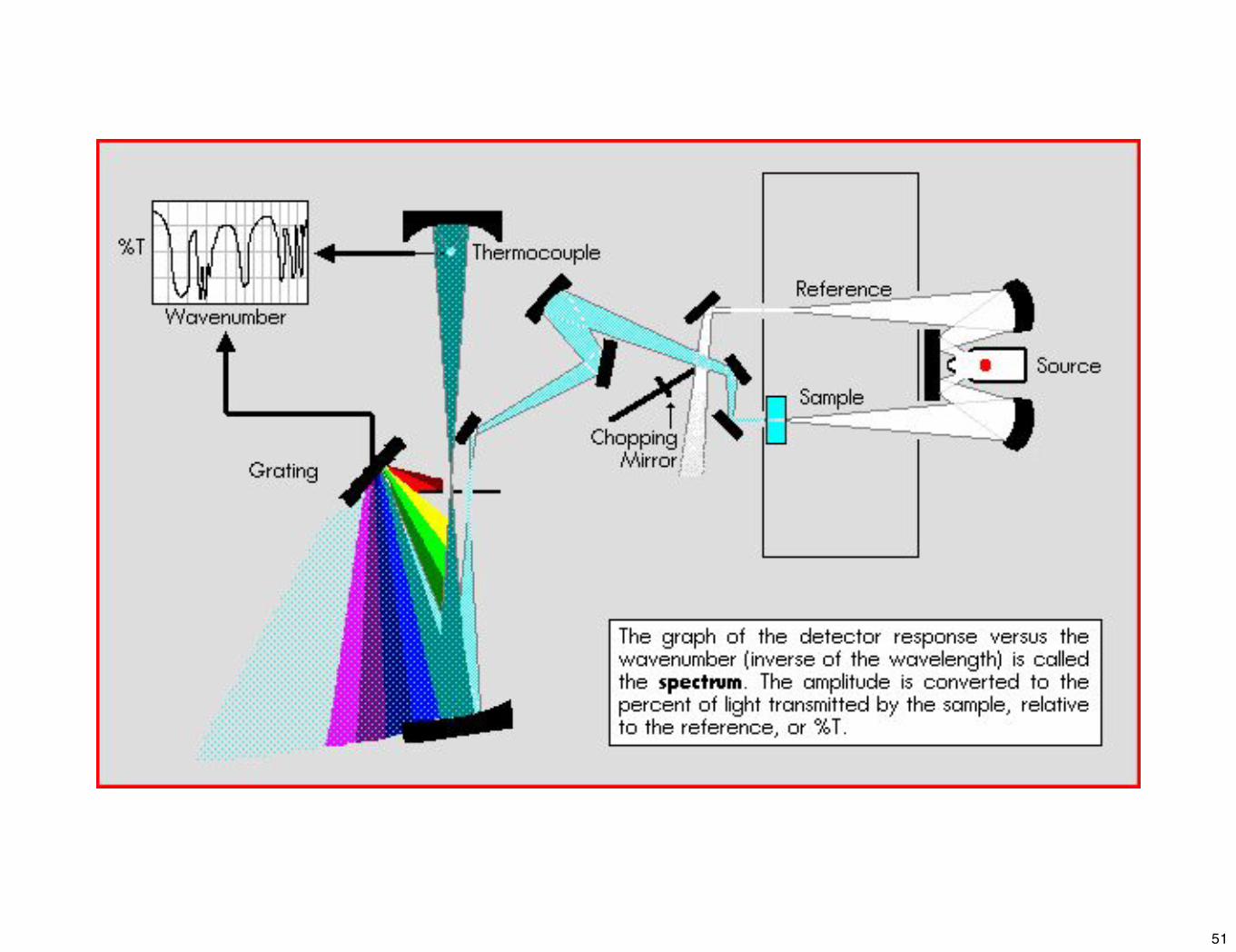
➤ La manière la plus naturelle d'enregistrer un spectre consiste à irradier l'échantillon séquentiellement dans le domaine spectral considéré et à enregistrer le rapport entre l'intensité du faisceau ayant traversé l'échantillon et l'intensité d'un signal de référence.
2) Instrumentation IR
50
51
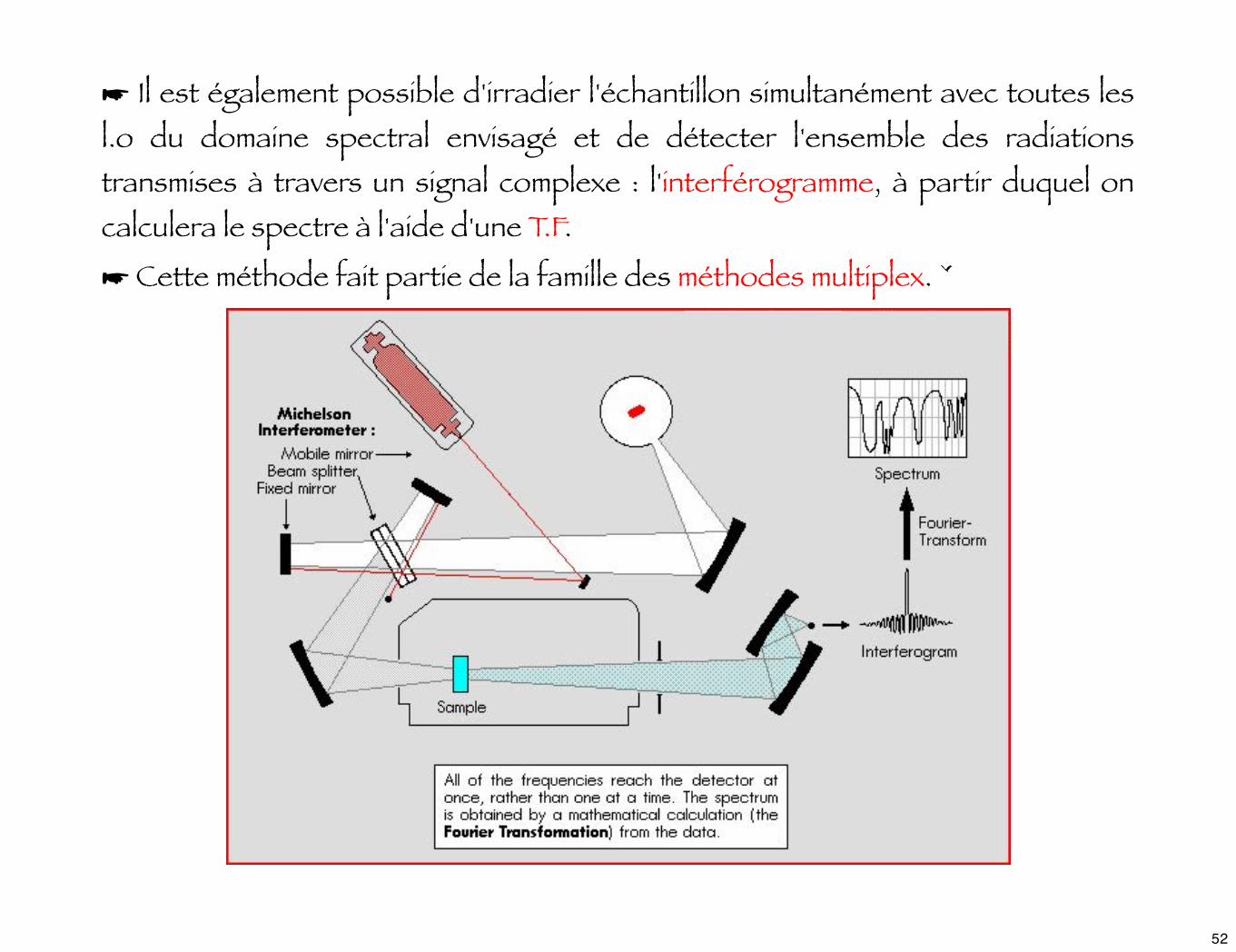
☛ Il est également possible d'irradier l'échantillon simultanément avec toutes les l.o du domaine spectral envisagé et de détecter l'ensemble des radiations transmises à travers un signal complexe : l'interférogramme, à partir duquel on calculera le spectre à l'aide d'une T.F.
☛ Cette méthode fait partie de la famille des méthodes multiplex. ˇ
52

☛ Les sources de rayonnement IR usuelles sont constituées par des solides portés à haute température qui rayonnent par incandescence. (température entre 800 et 2200 K).
2.1 Sources et détecteurs de rayonnement IR
2.1.1 Les sources
53

➤ Le bruit est commun à tous les détecteurs. On distingue :
● le bruit de détection (variations aléatoires du flux autour du signal) ;● le signal d'obscurité, détecté même lorsque le détecteur ne reçoit aucun photon.
Un détecteur performant minimise toutes ces sources de bruit et est caractérisé par un rapport S/N élevé.
2.1.2 Les détecteurs
Ils sont caractérisés par leur sensibilité, leur domaine spectral et leur efficacité quantique. La sensibilité représente le plus petit signal mesurable par le détecteur. Elle est exprimée comme la quantité de lumière correspondant au bruit intrinsèque. ˇ
54

➫ Le photoconducteur PbS est plutôt employé dans le PIR et on peut l'utiliser à température ambiante.
➫ Le photoconducteur MCT (Mercury Cadmium Telluride) est utilisé dans le MIR et LIR et nécessite un refroidissement à l'azote liquide (77 K) pour minimiser le bruit thermique. ˇˇ
➤ Les transducteurs photoconducteur :
Ils sont constitués d'un film mince de matériau semi-conducteur tel que le PbS, le tellure mixte de Hg et Cd. Le film est déposé sur une surface de verre et scellé sous vide dans une enceinte pour le protéger de l'atmosphère. ˇ
55

2.2 Appareils utilisés dans l'IR
☛ 3 types d'appareils sont disponibles :
1) Les spectrophotomètres dispersifs à réseaux ;
2) Les appareils multiplex basés sur la TF ;
3)Les photomètres mis au point pour l'analyse quantitative de nombreuses molécules organiques dans l'atmosphère.
☛ De nos jours, ce sont les IRTF qui sont les plus utilisés car plus rapides, fiables, et faciles d'utilisation. C'est uniquement lorsque le prix pose problème que les appareils dispersifs continuent à être utilisés (quasi-exclusivement dans le PIR).
56

2.2.1 Dispositifs d'analyse du rayonnement
☛ Ils permettent de discriminer les diverses fréquences d'un rayonnement polychromatique.
☛ Le 1er dispositif utilisé pour séparer les l.o d'un rayonnement IR a été le prisme, bloc transparent en NaCl ou KBr, matériaux hygroscopiques. Ce dispositif est actuellement abandonné.
➤ Les filtres interférentiels :
☛ Il est basé sur le principe de l'interféromètre de Fabry-Pérot, c.a.d qu'il combine de multiples réflexions entre 2 faces semi-réfléchissantes : selon les conditions d'interférences constructives ou destructives, seules certaines l.o sont transmises.
☛ Ce dispositif ne permettant des mesures qu'à une seule l.o, son utilisation est réservé aux photomètres.
57

➤ Les réseaux :
Un réseau est constitué d'une série de traits // gravés sur une surface réfléchissante. Soit un faisceau incident à angle α sur le réseau, on trouve un faisceau correspondant à la "réflexion spéculaire" selon - α, mais aussi d'autres faisceaux qui émergent selon les angles βk : ce phénomène porte le nom de diffraction lié à la répétition d'une structure optique.
☞ Le réseau est intégré dans un ensemble optique appelé monochromateur
comportant une fente d'entrée, un collimateur qui éclaire le réseau avec un faisceau // et dirige les rayons diffractés vers la fente de sortie.
58
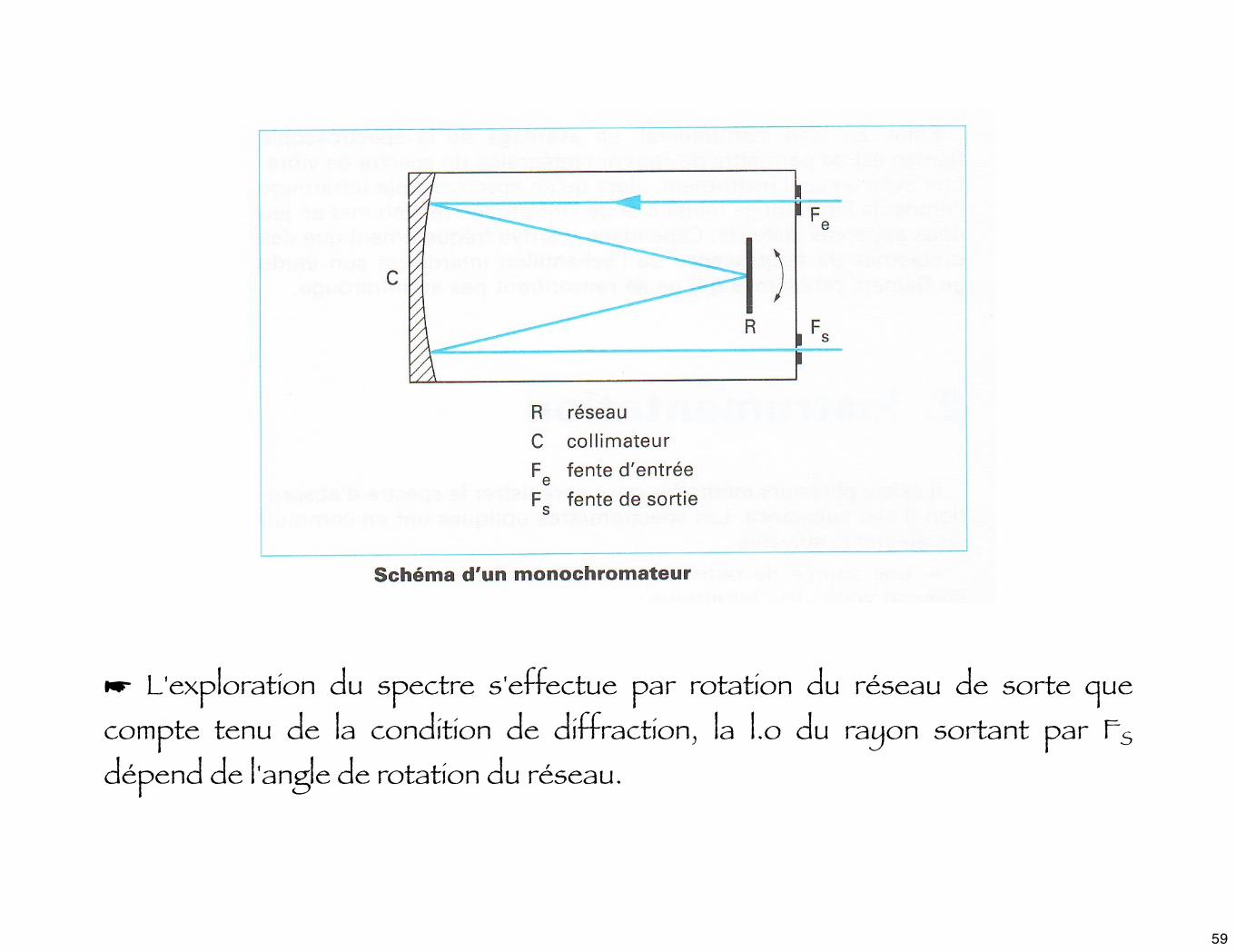
☛ L'exploration du spectre s'effectue par rotation du réseau de sorte que compte tenu de la condition de diffraction, la l.o du rayon sortant par FS dépend de l'angle de rotation du réseau.
59
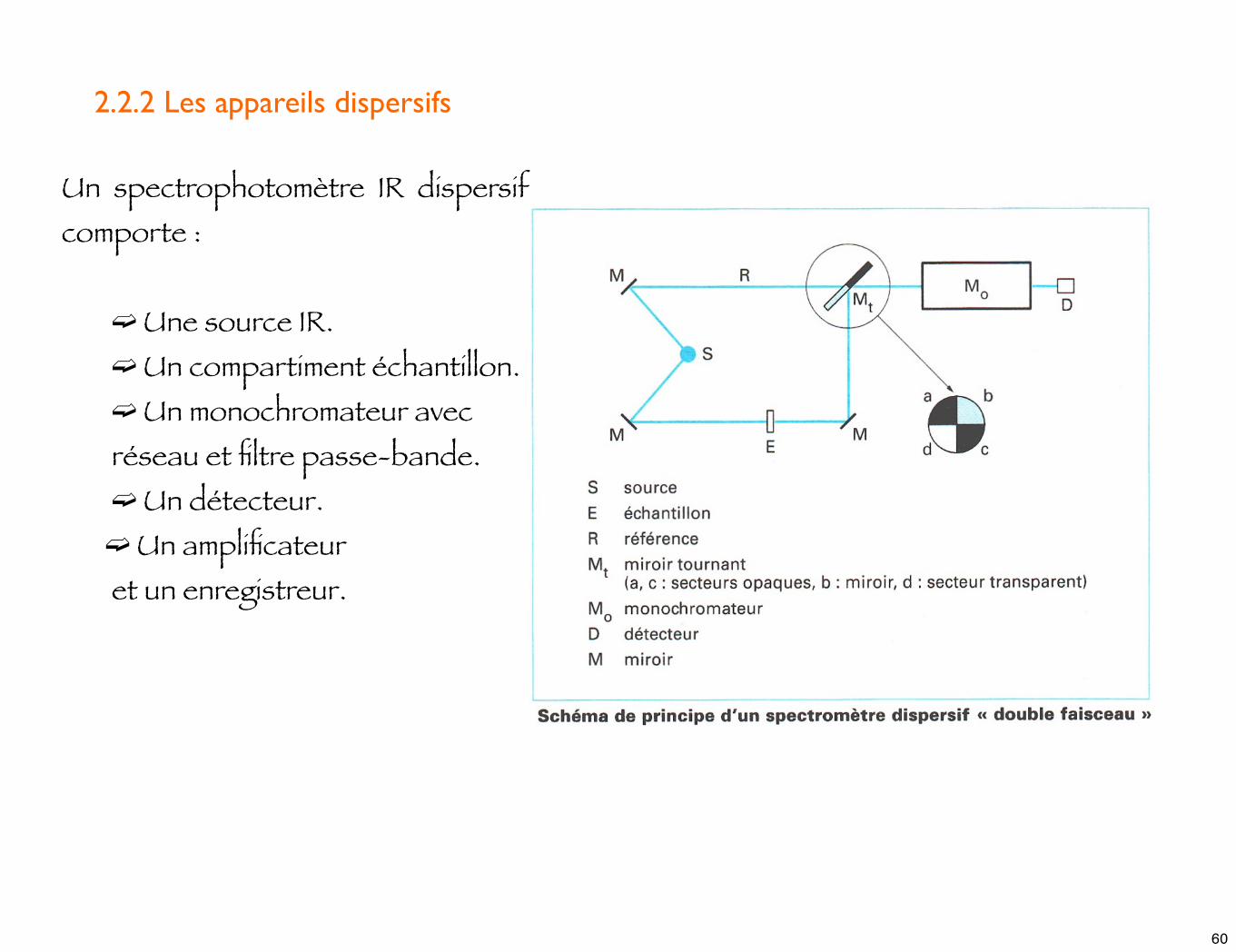
Un spectrophotomètre IR dispersif comporte :
➫ Une source IR. ➫ Un compartiment échantillon. ➫ Un monochromateur avec
réseau et filtre passe-bande. ➫ Un détecteur.
➫ Un amplificateur et un enregistreur.
2.2.2 Les appareils dispersifs
60

☛ la construction des systèmes optiques des appareils dispersifs n'est pas très différente de celle des spectro. UV/Visible.
☛ Une différence existe cependant : les compartiments échantillon et référence sont toujours situées entre la source et le monochromateur. Cette disposition est possible car le rayonnement IR, à l'opposé de l'UV/Visible, n'est pas suffisamment énergétique pour entraîner une photodécomposition de l'échantillon. Cela présente l'avantage d'éliminer efficacement le rayonnement diffusé par les parois de la cellule, qui n'atteint pas le détecteur.
☛ Avec un tel instrument, l'obtention du spectre nécessite 3 étapes :
➫ L'enregistrement du spectre "référence" obtenu sans échantillon. ➫ L'enregistrement du spectre "échantillon" obtenu avec l'échantillon. ➫ Le calcul, point par point, du rapport de ces 2 courbes précédentes.
61
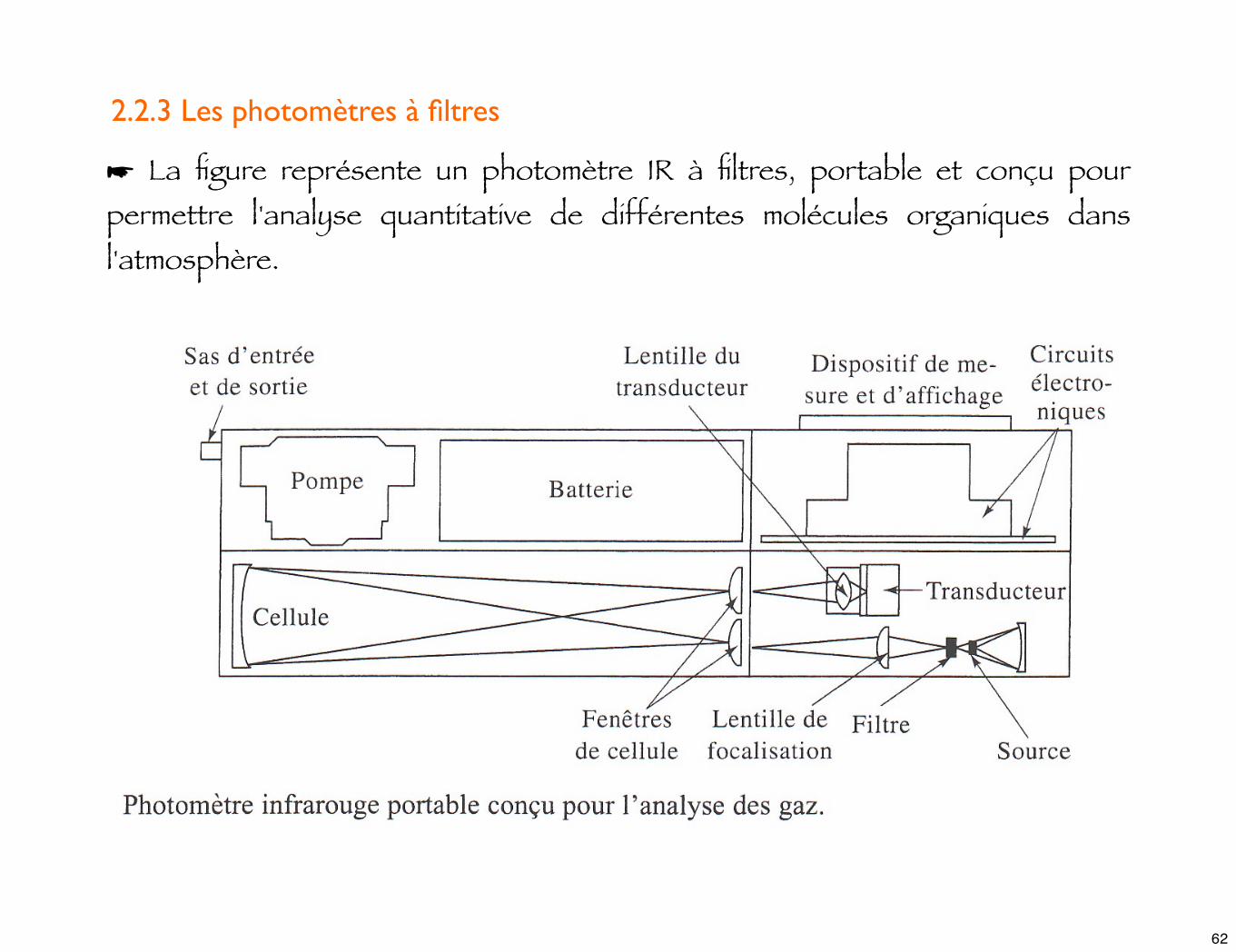
2.2.3 Les photomètres à filtres
☛ La figure représente un photomètre IR à filtres, portable et conçu pour permettre l'analyse quantitative de différentes molécules organiques dans l'atmosphère.
62

☛ La source est un filament de nichrome enroulé sur un barreau en céramique. Le transducteur est un système pyroélectrique.
☛ Une série de filtres interférentiels interchangeables couvre la région du spectre comprise entre 3000 et 750 cm-1. Chaque filtre est adapté à l'analyse d'une molécule spécifique.
☛ L'échantillon gazeux est introduit dans la cellule par une pompe alimentée par une batterie. La longueur du trajet optique est de 0,5 m ; une série de miroirs, qui ne sont pas représentés ici, permet d'accroître le trajet optique jusqu'à 20 m, par pas de 1,5 m. Cette caractéristique élargit de façon importante le domaine des concentrations accessibles.
63


























![Futurbig4 Ir Reg[1]](https://img.pdfslide.fr/doc/110x75/544f54f9af7959c4068b8e99/futurbig4-ir-reg1.jpg)


