Embed Size (px)
Citation preview

Ž .Chemical Geology 174 2001 291–305www.elsevier.comrlocaterchemgeo
Structural characterization of water-bearing silicate andaluminosilicate glasses by high-resolution solid-state NMR
Eric Robert a,1, Alan Whittington a,b,2, Franck Fayon a, Michel Pichavant c,Dominique Massiot a,)
a Centre de Recherche sur les Materiaux a Haute Temperature-CNRS, 1D aÕenue de la Recherche Scientifique,´ ` ´45071 Orleans cedex 2, France´
b Institut de Physique du Globe, 4 place Jussieu, 75252 Paris cedex 05, Francec Centre de Recherche sur la Chimie et la Synthese de Mineraux, CNRS, 45071 Orleans cedex 2, France` ´ ´
Accepted 9 February 2000
Abstract
Various one- and two-dimensional high-resolution solid-state NMR techniques have been applied to hydrous silicate andŽ . Ž .aluminosilicate glasses: simple acquisition, cross-polarization CP-MAS , heteronuclear correlation HETCOR , dipolar
Ž .dephasing, spin counting, double-quantum correlation, and rotational echo double resonance REDOR . The comparison ofthe results obtained for sodium tetrasilicate and phonolite glasses suggests that the water incorporation mechanisms arequalitatively similar for these two compositions. From proton NMR experiments, we observe no evidence of protonclustering and a wide range of chemical shifts ranging from 0 to 16 ppm, even for the aluminosilicate phonolite glass,
Ž .identifying at least three types of hydroxyl OH protons in addition to molecular H O. This variety of OH groups can be2
discussed in terms of hydrogen bonding strength. For both compositions, the results indicate some depolymerization of thetetrahedral network, but the picture cannot be so simple as to completely exclude any of the different previously proposedmodels for water incorporation in silicate glasses. q 2001 Elsevier Science B.V. All rights reserved.
Keywords: Silicate glass; Aluminosilicate glass; Hydrous glasses; Solid-state NMR; 1H; 23 Na; 27Al; 29Si; Dipolar interaction; Spin counting;Double-quantum correlation; HETCOR; CPMAS; REDOR
1. Introduction
The physical properties of silicate and aluminosil-Žicate melts and glasses e.g., liquidus and glass
) Corresponding author. Tel.: q33-238-25-55-18; fax: q33-238-63-81-03.
Ž .E-mail address: [email protected] D. Massiot .1 Present address: Union Minere, R&D Department, Kasteel-`
straat 7, B-2250 Olen, Belgium.2 Present address: Department of Geology, University of Illi-
nois, Urbana, IL 61801, USA.
transition temperatures, phase relations, viscosity,.density are strongly affected by small amounts of
water dissolved under pressure and at high tempera-Žture e.g., Burnham, 1979; Mysen et al., 1982; Ding-
well et al., 1996; Richet et al., 1996; Richet and.Polian, 1998 . However, despite many previous stud-
ies, the mechanism of water dissolution in silicateŽ .melts is still a matter of discussion McMillan, 1994 .
In situ measurements on hydrous melts are extremelydifficult to perform, and up to now limited to RamanŽ .Holtz et al., 1996 and near-infrared techniques
0009-2541r01r$ - see front matter q 2001 Elsevier Science B.V. All rights reserved.Ž .PII: S0009-2541 00 00321-1

( )E. Robert et al.rChemical Geology 174 2001 291–305292
Že.g., Nowak and Behrens, 1995; Shen and Keppler,.1995 . The latter techniques have confirmed the
Ž .proposal of Dingwell and Webb 1990 that waterspeciation changes upon heating through the glasstransition, moving towards higher concentrations of
Ž .hydroxyl OH groups and lower concentrations ofmolecular water. However, the glass structure frozenin on cooling through the glass transition is the sameas that of the liquid at the glass transition tempera-ture, hence, the study of hydrous glasses constitutesa valuable step towards understanding the dissolutionof water in silicate melts. Moreover, these glassesalso have important technological applications suchas nuclear waste glasses or fiber optic cables.
One of the most debated points is the question ofwhether or not the incorporation of water leads to therupture of Si–O–Si and Al–O–Si bonds to formSi–OH andror Al–OH. While it is widely acceptedthat water depolymerizes the network of alkali sili-
Žcate glasses e.g., Kohn et al., 1989a; Xu et al.,.1998; Zotov and Keppler, 1998 , it is not at all clear
that the same mechanism occurs in aluminosilicateŽglasses Burnham, 1975; Mysen and Virgo, 1986;
Kohn et al., 1989b, 1992, 1994, 1998; Sykes and.Kubicki, 1993, 1994 . Most of the models based on
vibrational spectroscopic results propose such de-Žpolymerization e.g., Stolper, 1982; Mysen and
.Virgo, 1986; Remmele et al., 1986 . However, astudy of albite, nepheline and anorthite–quartzglasses, performed using a variety of NMR tech-
Žniques, led to different conclusions Kohn et al.,.1989b, 1992 . These authors did not find conclusive
evidence for the presence of Al–OH or Si–OH in thehydrous samples and proposed a possible new inter-pretation of the vibrational data. Instead of depolym-erization of the T–O–T network, they suggested thatwater dissolution involves a simple exchange of Naq
for Hq as charge-balancing cation for AlOy tetrahe-4Ž qdra this charge-balancing H can also be seen as a
. Ž .bridging OH group , and formation of Na OH com-plexes.
A recent 17O NMR study observed a new peak inalbite glass with 10 wt.% water, which was tenta-tively identified as Si–OH, but there remains thepossibility that this new peak represents a protonated
Ž .bridging oxygen Xu et al., 1998 . Despite manystudies on this problem, it remains debated in thecase of aluminosilicate compositions, and new ad-
vances in spectroscopic techniques are probably re-quired to produce less equivocal results. Increasinglycomplex double-resonance experiments may be thekey to this problem. The recent 1Hr27Al and 1Hr23
Ž .Na TRAPDOR study of Zeng et al. 1999 investi-gated proton environments in several glasses on thejoint NaAlO –SiO and found three different proton2 2
OH resonances, which were all ascribed to the termi-nal T–OH groups, suggesting depolymerization ofthe network by water.
Our aim in this paper is to establish the feasibilityand use of a variety of possible NMR experimentsthat recently became available. We applied thesemethods to a hydrated sodium tetrasilicate and asynthetic iron-free phonolitic composition, with theaim of establishing the similarities and differences ofstructure and water incorporation mechanism in hy-drous silicate and aluminosilicate glasses. The sodiumtetrasilicate is a model compound, which has beenpreviously studied by several techniques, including
Ž .NMR Schaller and Sebald, 1995 , and the phonoliteis an iron-free synthetic analogue of an alkali-richvolcanic rock, which has recently been studied by
Ž .viscosity measurements Whittington et al., 2000 . Itis difficult or impossible to obtain well-resolvedspectra of iron-bearing compounds because the pres-ence of paramagnetic centers induces severe linebroadening. While several previous studies have con-
Ž .centrated on albite glass NaAlSi O , theoretically3 8
fully polymerized in the anhydrous state, the phono-lite is a more complex aluminosilicate composition,which is partially depolymerized even when anhy-drous. One aim of this study is to test whether resultsfrom albite are more generally applicable to othergeologically relevant compositions.
2. NMR background
Solid-state NMR is a quickly progressing tech-nique, sensitive to the environment of the selectively
Žobserved nucleus Engelhardt and Michel, 1987;Schmidt-Rohr and Spiess, 1994 and references
.therein . Observable nuclei have non-zero nuclearŽ 1 29 31 7spins Is1r2 for H, Si, or P; Is3r2 for Li,
11 23 69,71 17 27 . ŽB, Na, or Ga; Is5r2 for O or Al Table.1 , and their respective sensitivities depend on their

( )E. Robert et al.rChemical Geology 174 2001 291–305 293
Table 1NMR observable nuclei with their main characteristics
Nucleus Nuclear Natural LarmorŽ .spin I abundance % frequency
Ž .at 9.4 T MHz1H 1r2 99.99 400.229Si 1r2 4.7 79.523 Na 3r2 100 105.827Al 5r2 100 104.2
natural abundance, gyromagnetic ratios andŽ .quadrupolar momentum non-zero when I)1r2 .
Ž .In an intense principal field B typically ;10 T ,0
the selected nucleus is observed by applying aradio-frequency perturbation field oscillating at the
ŽLarmor frequency n sgB where g is the gyro-0 0.magnetic ratio, a constant for each nuclide and
registering the signal generated by the nuclear spinsŽcoming back to equilibrium. This signal free induc-
Ž .. Ž .tion decay FID is then Fourier-transformed FT toobtain the frequency spectrum of the observed nu-cleus. The selectivity of the experiment comes fromthe large differences in Larmor frequency for thedifferent nuclei, which allows selective excitationand recording of the spectra for each nuclei. NMRcharacterizes the perturbation of this resonance con-
Ž .dition due to a the shielding of the principal fieldŽby bonding electrons chemical shift anisotropy in-
. Ž .teraction , b the interaction between neighboringŽ . Ž .spins dipolar interaction or c the interaction of the
electric field gradient at the nucleus position with theŽnuclear quadrupolar momentum quadrupolar inter-
. Ž .action Table 2 . These perturbations are usually sosmall that they can be treated as first-order perturba-tions. The peak positions are expressed in ppm of theLarmor frequency compared to a reference com-pound. Nevertheless, quadrupolar interactions canbecome large enough to induce second-order effectsthat render the spectra of quadrupolar nuclei lessresolved, and more difficult to model and under-stand. In the solid state, all these interactions areanisotropic, depending on the orientation of the crys-tallite in the principal field, and, consequently, thestatic spectra of powdered samples are broad andunresolved. High-resolution spectra can be obtainedfor solids by averaging the anisotropic part of the
interaction by manipulating the sample with magic-Ž .angle spinning MAS , or manipulating the spin
system with radio-frequency pulses. This is the sourceof a large number of different experiments that aimat simplifying the spectra by removing or selectingthe different anisotropic interactions. Under fastMAS, which mimics the motional narrowing of aliquid sample, both dipolar interactions and thechemical shift anisotropy are averaged, and the MASNMR spectrum of a Is1r2 nucleus consists onlyof sharp peaks at the isotropic position. This positionis directly characteristic of the coordination number,bond angles, and bond lengths around the observednucleus. Quadrupolar interactions are reduced butnot eliminated under fast MAS, but higher resolutioncan be obtained using two-dimensional MQ-MAStechniques, which combine MAS spinning and multi-
Žple quantum evolution Frydman and Harwood, 1995;.Medek et al., 1995 . Because solid-state NMR is
mostly sensitive to the first-coordination spheres ofthe observed nuclei, it can still provide good resolu-tion when going from a perfectly ordered crystallinecompound to a more disordered amorphous or glassymaterial.
Using one-dimensional solid-state experimentsŽ .fast MAS or MQ-MAS , it is thus possible tocharacterize the first-coordination sphere of the ob-served nucleus in glasses, but this is obtained at thecost of averaging the dipolar interaction that contains
Table 2Physical background and information linked to the different inter-actions that can be observed by solid-state NMR in silicate oraluminosilicate glasses
Interaction Physical background Information
Dipolar Interaction between Distancesinteraction neighboring spinsChemical shift Shielding of principal Coordinationanisotropy field by electrons number
Bond angles anddistances
Quadrupolar Interaction between Geometryinteraction electric field
gradientand nucleusquadrupolarmomentum

( )E. Robert et al.rChemical Geology 174 2001 291–305294
information on longer range order. This dipolar inter-action is inversely proportional to the third power ofthe distance between interacting spins. Nevertheless,numerous solid-state one-dimensional experimentscan benefit from the dipolar interaction and enableits characterization, while retaining the high resolu-
Ž . Žtion of MAS spectra: a cross-polarization CP-.MAS transfers magnetization from abundant sensi-
Ž �1 4 29 .tive protons to a rare spin system e.g., H – Si ;Ž .b multiple-quantum experiments aim at acquiringindirectly the signature of groups of like spins cou-
Ž 1 . Ž .pled by dipolar interactions e.g., H pairs ; cŽdouble resonance experiments rotational echo dou-
Ž ..ble resonance REDOR aim at selecting from MASspectra the signature of nuclei, which are dipolar
Ž �1 4 29 �1 4 23 �1 4coupled to others e.g., H – Si, H – Na, H –27 .Al . Finally, from these experiments, it is alsopossible to build multiple-dimension experiments thatestablish correlations between the spectra of different
Ž Ž . 1nuclei heteronuclear correlation HETCOR H–
29 .Si or between double-quantum and single-quan-Žtum spectra spectrum of pairs of like nucleirspec-
.trum of individual nuclei .
3. Experimental techniques
3.1. Sample compositions and preparation
We selected two types of glasses: a model binaryŽsodium silicate Na O–4SiO further referred to as2 2
.NS4 and a synthetic iron-free analog of phonoliticŽ .composition further referred to as phonolite . The
NS4 dry and hydrated glasses are simple composi-tions which have already been studied by several
Žtechniques Schaller and Sebald, 1995; Zotov et al.,.1996; Zotov and Keppler, 1998 . The different envi-
Ž .ronments of silicon Q , Q , Q are known to be4 3 2
partly resolved in the 29Si MAS NMR spectra. In the
Table 3Compositions and synthesis conditions for the anhydrous and hydrous glass of NS4 and phonolite with the measured NMR parameters. For
2 <Ž . <C , AwidB indicates the half width of the quadrupolar coupling distribution. C se qQrhseqV rh; hs V yV rV ; C sQh Q z z x x y y z z Qh
2'C 1qh r3Q
Ž . Ž . Ž . Ž .Sample d ppm Width ppm C MHz C width MHzCS Qh Q
( )NS4 mol% SiO 80, Na O 20, NBOrT 0.52 2Ž .NS4 dry 0 wt.% H O2
29Si Q y105.6 14.0 – –429Si Q y92.0 11.3 – –323 Ž .Na 3.9 – 3.2 wid 0.7 0.7
Ž .NS4 1.28 2 kbar, 11008C, 1.28"0.07 wt.% H O229Si Q y104.0 13.6 – –429Si Q y93.0 11.4 – –329Si Q y83.4 10.7 – –223 Ž .Na 3.4 – 3.2 wid 0.7 0.7
( )Phonolite mol% SiO 65.4, Al O 12.7, MgO 3.1, CaO 2.8, Na O 10.0, K O 5.3, TiO 0.7, NBOrT 0.192 2 3 2 2 2Ž .Phonolite dry 0 wt.% H O2
29Si y94.6 15 – –23 Ž .Na y10 – 2.35 wid 0.6 0.527Al 61 – 7.1 1.6
Ž .Phonolite 0.9 3 kbar, 13008C, 0.90"0.1 wt.% H O229Si y92.7 19 – –23 Ž .Na y10 – 2.3 wid 0.5 0.4627Al 62 – 7.6 1.7
Ž .Phonolite 3.2 2 kbar, 12008C, 3.2"0.2 wt.% H O229Si y89.6 17 – –23 Ž .Na y7 – 2.3 wid 0.5 0.4827Al 61 – 6.0 1.2
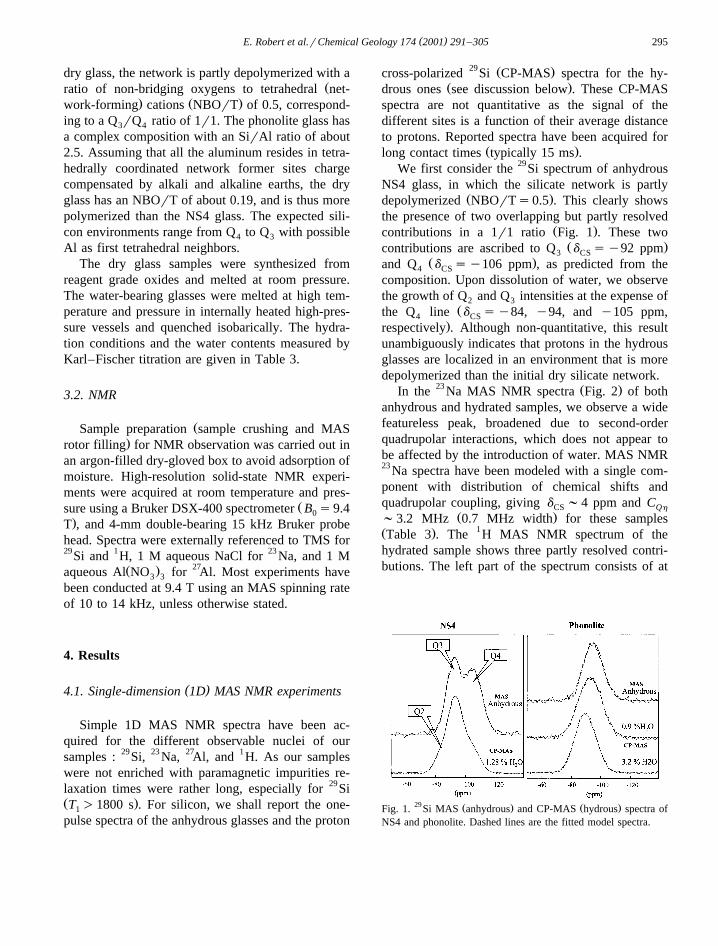
( )E. Robert et al.rChemical Geology 174 2001 291–305 295
dry glass, the network is partly depolymerized with aŽratio of non-bridging oxygens to tetrahedral net-
. Ž .work-forming cations NBOrT of 0.5, correspond-ing to a Q rQ ratio of 1r1. The phonolite glass has3 4
a complex composition with an SirAl ratio of about2.5. Assuming that all the aluminum resides in tetra-hedrally coordinated network former sites chargecompensated by alkali and alkaline earths, the dryglass has an NBOrT of about 0.19, and is thus morepolymerized than the NS4 glass. The expected sili-con environments range from Q to Q with possible4 3
Al as first tetrahedral neighbors.The dry glass samples were synthesized from
reagent grade oxides and melted at room pressure.The water-bearing glasses were melted at high tem-perature and pressure in internally heated high-pres-sure vessels and quenched isobarically. The hydra-tion conditions and the water contents measured byKarl–Fischer titration are given in Table 3.
3.2. NMR
ŽSample preparation sample crushing and MAS.rotor filling for NMR observation was carried out in
an argon-filled dry-gloved box to avoid adsorption ofmoisture. High-resolution solid-state NMR experi-ments were acquired at room temperature and pres-
Žsure using a Bruker DSX-400 spectrometer B s9.40.T , and 4-mm double-bearing 15 kHz Bruker probe
head. Spectra were externally referenced to TMS for29Si and 1H, 1 M aqueous NaCl for 23 Na, and 1 M
Ž . 27aqueous Al NO for Al. Most experiments have3 3
been conducted at 9.4 T using an MAS spinning rateof 10 to 14 kHz, unless otherwise stated.
4. Results
( )4.1. Single-dimension 1D MAS NMR experiments
Simple 1D MAS NMR spectra have been ac-quired for the different observable nuclei of oursamples : 29Si, 23 Na, 27Al, and 1H. As our sampleswere not enriched with paramagnetic impurities re-laxation times were rather long, especially for 29SiŽ .T )1800 s . For silicon, we shall report the one-1
pulse spectra of the anhydrous glasses and the proton
29 Ž .cross-polarized Si CP-MAS spectra for the hy-Ž .drous ones see discussion below . These CP-MAS
spectra are not quantitative as the signal of thedifferent sites is a function of their average distanceto protons. Reported spectra have been acquired for
Ž .long contact times typically 15 ms .We first consider the 29Si spectrum of anhydrous
NS4 glass, in which the silicate network is partlyŽ .depolymerized NBOrTs0.5 . This clearly shows
the presence of two overlapping but partly resolvedŽ .contributions in a 1r1 ratio Fig. 1 . These two
Ž .contributions are ascribed to Q d sy92 ppm3 CSŽ .and Q d sy106 ppm , as predicted from the4 CS
composition. Upon dissolution of water, we observethe growth of Q and Q intensities at the expense of2 3
Žthe Q line d sy84, y94, and y105 ppm,4 CS.respectively . Although non-quantitative, this result
unambiguously indicates that protons in the hydrousglasses are localized in an environment that is moredepolymerized than the initial dry silicate network.
23 Ž .In the Na MAS NMR spectra Fig. 2 of bothanhydrous and hydrated samples, we observe a widefeatureless peak, broadened due to second-orderquadrupolar interactions, which does not appear tobe affected by the introduction of water. MAS NMR23 Na spectra have been modeled with a single com-ponent with distribution of chemical shifts andquadrupolar coupling, giving d ;4 ppm and CCS Qh
Ž .;3.2 MHz 0.7 MHz width for these samplesŽ . 1Table 3 . The H MAS NMR spectrum of thehydrated sample shows three partly resolved contri-butions. The left part of the spectrum consists of at
29 Ž . Ž .Fig. 1. Si MAS anhydrous and CP-MAS hydrous spectra ofNS4 and phonolite. Dashed lines are the fitted model spectra.
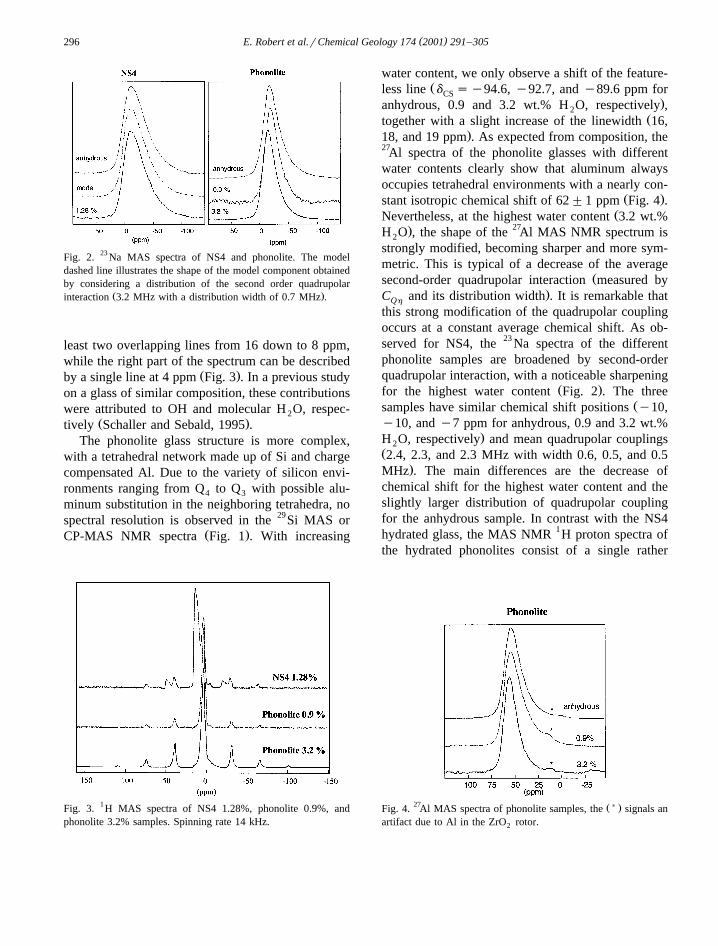
( )E. Robert et al.rChemical Geology 174 2001 291–305296
Fig. 2. 23 Na MAS spectra of NS4 and phonolite. The modeldashed line illustrates the shape of the model component obtainedby considering a distribution of the second order quadrupolar
Ž .interaction 3.2 MHz with a distribution width of 0.7 MHz .
least two overlapping lines from 16 down to 8 ppm,while the right part of the spectrum can be described
Ž .by a single line at 4 ppm Fig. 3 . In a previous studyon a glass of similar composition, these contributionswere attributed to OH and molecular H O, respec-2
Ž .tively Schaller and Sebald, 1995 .The phonolite glass structure is more complex,
with a tetrahedral network made up of Si and chargecompensated Al. Due to the variety of silicon envi-ronments ranging from Q to Q with possible alu-4 3
minum substitution in the neighboring tetrahedra, nospectral resolution is observed in the 29Si MAS or
Ž .CP-MAS NMR spectra Fig. 1 . With increasing
Fig. 3. 1H MAS spectra of NS4 1.28%, phonolite 0.9%, andphonolite 3.2% samples. Spinning rate 14 kHz.
water content, we only observe a shift of the feature-Žless line d sy94.6, y92.7, and y89.6 ppm forCS
.anhydrous, 0.9 and 3.2 wt.% H O, respectively ,2Žtogether with a slight increase of the linewidth 16,
.18, and 19 ppm . As expected from composition, the27Al spectra of the phonolite glasses with differentwater contents clearly show that aluminum alwaysoccupies tetrahedral environments with a nearly con-
Ž .stant isotropic chemical shift of 62"1 ppm Fig. 4 .ŽNevertheless, at the highest water content 3.2 wt.%
. 27H O , the shape of the Al MAS NMR spectrum is2
strongly modified, becoming sharper and more sym-metric. This is typical of a decrease of the average
Žsecond-order quadrupolar interaction measured by.C and its distribution width . It is remarkable thatQh
this strong modification of the quadrupolar couplingoccurs at a constant average chemical shift. As ob-served for NS4, the 23 Na spectra of the differentphonolite samples are broadened by second-orderquadrupolar interaction, with a noticeable sharpening
Ž .for the highest water content Fig. 2 . The threeŽsamples have similar chemical shift positions y10,
y10, and y7 ppm for anhydrous, 0.9 and 3.2 wt.%.H O, respectively and mean quadrupolar couplings2
Ž2.4, 2.3, and 2.3 MHz with width 0.6, 0.5, and 0.5.MHz . The main differences are the decrease of
chemical shift for the highest water content and theslightly larger distribution of quadrupolar couplingfor the anhydrous sample. In contrast with the NS4hydrated glass, the MAS NMR 1H proton spectra ofthe hydrated phonolites consist of a single rather
27 Ž) .Fig. 4. Al MAS spectra of phonolite samples, the signals anartifact due to Al in the ZrO rotor.2
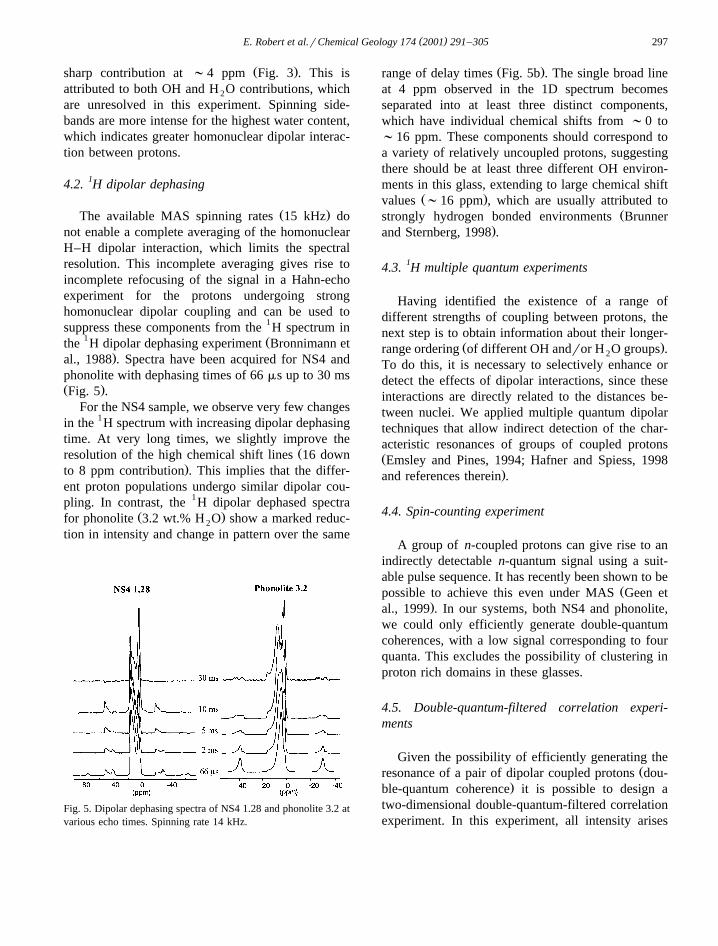
( )E. Robert et al.rChemical Geology 174 2001 291–305 297
Ž .sharp contribution at ;4 ppm Fig. 3 . This isattributed to both OH and H O contributions, which2
are unresolved in this experiment. Spinning side-bands are more intense for the highest water content,which indicates greater homonuclear dipolar interac-tion between protons.
4.2. 1H dipolar dephasing
Ž .The available MAS spinning rates 15 kHz donot enable a complete averaging of the homonuclearH–H dipolar interaction, which limits the spectralresolution. This incomplete averaging gives rise toincomplete refocusing of the signal in a Hahn-echoexperiment for the protons undergoing stronghomonuclear dipolar coupling and can be used tosuppress these components from the 1H spectrum in
1 Žthe H dipolar dephasing experiment Bronnimann et.al., 1988 . Spectra have been acquired for NS4 and
phonolite with dephasing times of 66 ms up to 30 msŽ .Fig. 5 .
For the NS4 sample, we observe very few changesin the 1H spectrum with increasing dipolar dephasingtime. At very long times, we slightly improve the
Žresolution of the high chemical shift lines 16 down.to 8 ppm contribution . This implies that the differ-
ent proton populations undergo similar dipolar cou-pling. In contrast, the 1H dipolar dephased spectra
Ž .for phonolite 3.2 wt.% H O show a marked reduc-2
tion in intensity and change in pattern over the same
Fig. 5. Dipolar dephasing spectra of NS4 1.28 and phonolite 3.2 atvarious echo times. Spinning rate 14 kHz.
Ž .range of delay times Fig. 5b . The single broad lineat 4 ppm observed in the 1D spectrum becomesseparated into at least three distinct components,which have individual chemical shifts from ;0 to;16 ppm. These components should correspond toa variety of relatively uncoupled protons, suggestingthere should be at least three different OH environ-ments in this glass, extending to large chemical shift
Ž .values ;16 ppm , which are usually attributed toŽstrongly hydrogen bonded environments Brunner
.and Sternberg, 1998 .
4.3. 1H multiple quantum experiments
Having identified the existence of a range ofdifferent strengths of coupling between protons, thenext step is to obtain information about their longer-
Ž .range ordering of different OH andror H O groups .2
To do this, it is necessary to selectively enhance ordetect the effects of dipolar interactions, since theseinteractions are directly related to the distances be-tween nuclei. We applied multiple quantum dipolartechniques that allow indirect detection of the char-acteristic resonances of groups of coupled protonsŽEmsley and Pines, 1994; Hafner and Spiess, 1998
.and references therein .
4.4. Spin-counting experiment
A group of n-coupled protons can give rise to anindirectly detectable n-quantum signal using a suit-able pulse sequence. It has recently been shown to be
Žpossible to achieve this even under MAS Geen et.al., 1999 . In our systems, both NS4 and phonolite,
we could only efficiently generate double-quantumcoherences, with a low signal corresponding to fourquanta. This excludes the possibility of clustering inproton rich domains in these glasses.
4.5. Double-quantum-filtered correlation experi-ments
Given the possibility of efficiently generating theŽresonance of a pair of dipolar coupled protons dou-
.ble-quantum coherence it is possible to design atwo-dimensional double-quantum-filtered correlationexperiment. In this experiment, all intensity arises
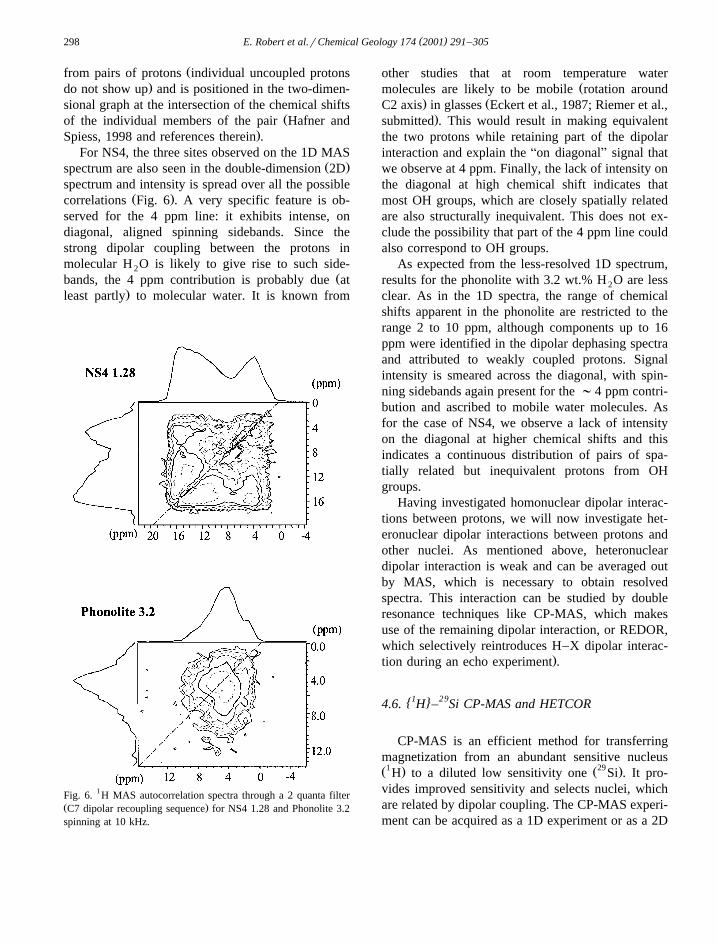
( )E. Robert et al.rChemical Geology 174 2001 291–305298
Žfrom pairs of protons individual uncoupled protons.do not show up and is positioned in the two-dimen-
sional graph at the intersection of the chemical shiftsŽof the individual members of the pair Hafner and
.Spiess, 1998 and references therein .For NS4, the three sites observed on the 1D MAS
Ž .spectrum are also seen in the double-dimension 2Dspectrum and intensity is spread over all the possible
Ž .correlations Fig. 6 . A very specific feature is ob-served for the 4 ppm line: it exhibits intense, ondiagonal, aligned spinning sidebands. Since thestrong dipolar coupling between the protons inmolecular H O is likely to give rise to such side-2
Žbands, the 4 ppm contribution is probably due at.least partly to molecular water. It is known from
Fig. 6. 1H MAS autocorrelation spectra through a 2 quanta filterŽ .C7 dipolar recoupling sequence for NS4 1.28 and Phonolite 3.2spinning at 10 kHz.
other studies that at room temperature waterŽmolecules are likely to be mobile rotation around
. ŽC2 axis in glasses Eckert et al., 1987; Riemer et al.,.submitted . This would result in making equivalent
the two protons while retaining part of the dipolarinteraction and explain the Aon diagonalB signal thatwe observe at 4 ppm. Finally, the lack of intensity onthe diagonal at high chemical shift indicates thatmost OH groups, which are closely spatially relatedare also structurally inequivalent. This does not ex-clude the possibility that part of the 4 ppm line couldalso correspond to OH groups.
As expected from the less-resolved 1D spectrum,results for the phonolite with 3.2 wt.% H O are less2
clear. As in the 1D spectra, the range of chemicalshifts apparent in the phonolite are restricted to therange 2 to 10 ppm, although components up to 16ppm were identified in the dipolar dephasing spectraand attributed to weakly coupled protons. Signalintensity is smeared across the diagonal, with spin-ning sidebands again present for the ;4 ppm contri-bution and ascribed to mobile water molecules. Asfor the case of NS4, we observe a lack of intensityon the diagonal at higher chemical shifts and thisindicates a continuous distribution of pairs of spa-tially related but inequivalent protons from OHgroups.
Having investigated homonuclear dipolar interac-tions between protons, we will now investigate het-eronuclear dipolar interactions between protons andother nuclei. As mentioned above, heteronucleardipolar interaction is weak and can be averaged outby MAS, which is necessary to obtain resolvedspectra. This interaction can be studied by doubleresonance techniques like CP-MAS, which makesuse of the remaining dipolar interaction, or REDOR,which selectively reintroduces H–X dipolar interac-
.tion during an echo experiment .
{1 } 294.6. H – Si CP-MAS and HETCOR
CP-MAS is an efficient method for transferringmagnetization from an abundant sensitive nucleusŽ1 . Ž29 .H to a diluted low sensitivity one Si . It pro-vides improved sensitivity and selects nuclei, whichare related by dipolar coupling. The CP-MAS experi-ment can be acquired as a 1D experiment or as a 2D
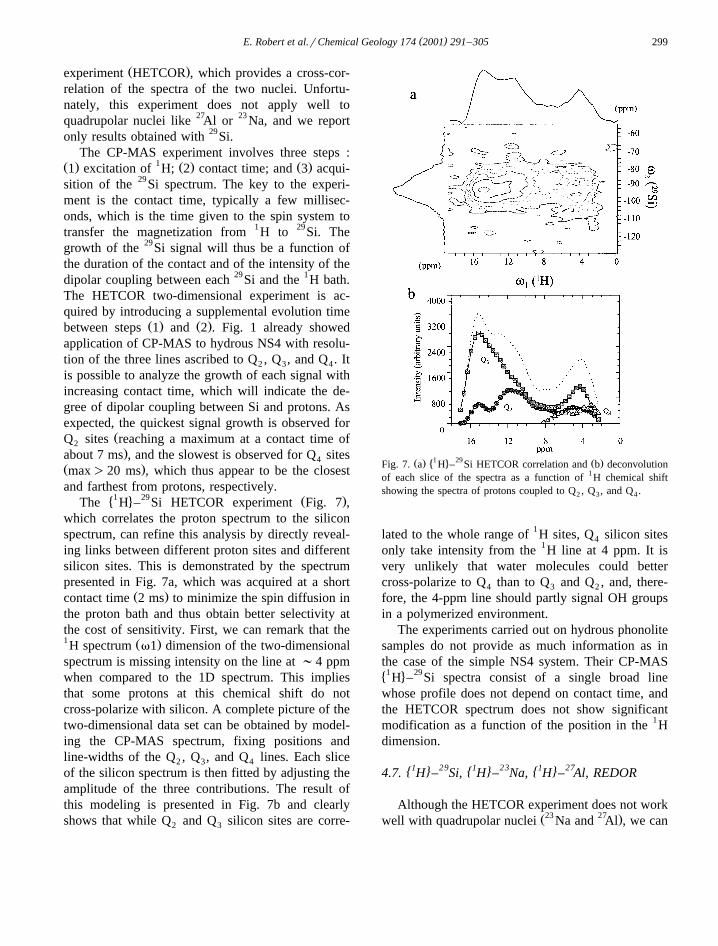
( )E. Robert et al.rChemical Geology 174 2001 291–305 299
Ž .experiment HETCOR , which provides a cross-cor-relation of the spectra of the two nuclei. Unfortu-nately, this experiment does not apply well toquadrupolar nuclei like 27Al or 23 Na, and we reportonly results obtained with 29Si.
The CP-MAS experiment involves three steps :Ž . 1 Ž . Ž .1 excitation of H; 2 contact time; and 3 acqui-sition of the 29Si spectrum. The key to the experi-ment is the contact time, typically a few millisec-onds, which is the time given to the spin system totransfer the magnetization from 1H to 29Si. Thegrowth of the 29Si signal will thus be a function ofthe duration of the contact and of the intensity of thedipolar coupling between each 29Si and the 1H bath.The HETCOR two-dimensional experiment is ac-quired by introducing a supplemental evolution time
Ž . Ž .between steps 1 and 2 . Fig. 1 already showedapplication of CP-MAS to hydrous NS4 with resolu-tion of the three lines ascribed to Q , Q , and Q . It2 3 4
is possible to analyze the growth of each signal withincreasing contact time, which will indicate the de-gree of dipolar coupling between Si and protons. Asexpected, the quickest signal growth is observed for
ŽQ sites reaching a maximum at a contact time of2.about 7 ms , and the slowest is observed for Q sites4
Ž .max)20 ms , which thus appear to be the closestand farthest from protons, respectively.
�1 4 29 Ž .The H – Si HETCOR experiment Fig. 7 ,which correlates the proton spectrum to the siliconspectrum, can refine this analysis by directly reveal-ing links between different proton sites and differentsilicon sites. This is demonstrated by the spectrumpresented in Fig. 7a, which was acquired at a short
Ž .contact time 2 ms to minimize the spin diffusion inthe proton bath and thus obtain better selectivity atthe cost of sensitivity. First, we can remark that the1 Ž .H spectrum v1 dimension of the two-dimensionalspectrum is missing intensity on the line at ;4 ppmwhen compared to the 1D spectrum. This impliesthat some protons at this chemical shift do notcross-polarize with silicon. A complete picture of thetwo-dimensional data set can be obtained by model-ing the CP-MAS spectrum, fixing positions andline-widths of the Q , Q , and Q lines. Each slice2 3 4
of the silicon spectrum is then fitted by adjusting theamplitude of the three contributions. The result ofthis modeling is presented in Fig. 7b and clearlyshows that while Q and Q silicon sites are corre-2 3
Ž . �1 4 29 Ž .Fig. 7. a H – Si HETCOR correlation and b deconvolutionof each slice of the spectra as a function of 1H chemical shiftshowing the spectra of protons coupled to Q , Q , and Q .2 3 4
lated to the whole range of 1H sites, Q silicon sites4
only take intensity from the 1H line at 4 ppm. It isvery unlikely that water molecules could bettercross-polarize to Q than to Q and Q , and, there-4 3 2
fore, the 4-ppm line should partly signal OH groupsin a polymerized environment.
The experiments carried out on hydrous phonolitesamples do not provide as much information as inthe case of the simple NS4 system. Their CP-MAS�1 4 29H – Si spectra consist of a single broad linewhose profile does not depend on contact time, andthe HETCOR spectrum does not show significantmodification as a function of the position in the 1Hdimension.
{1 } 29 {1 } 23 {1 } 274.7. H – Si, H – Na, H – Al, REDOR
Although the HETCOR experiment does not workŽ23 27 .well with quadrupolar nuclei Na and Al , we can
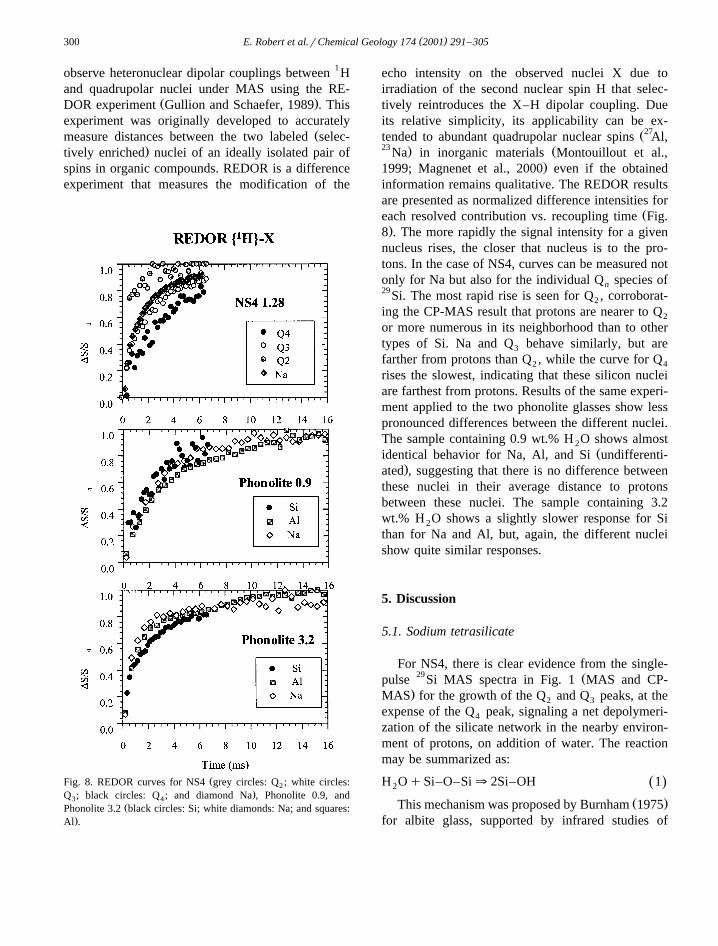
( )E. Robert et al.rChemical Geology 174 2001 291–305300
observe heteronuclear dipolar couplings between 1Hand quadrupolar nuclei under MAS using the RE-
Ž .DOR experiment Gullion and Schaefer, 1989 . Thisexperiment was originally developed to accurately
Žmeasure distances between the two labeled selec-.tively enriched nuclei of an ideally isolated pair of
spins in organic compounds. REDOR is a differenceexperiment that measures the modification of the
ŽFig. 8. REDOR curves for NS4 grey circles: Q ; white circles:2.Q ; black circles: Q ; and diamond Na , Phonolite 0.9, and3 4
ŽPhonolite 3.2 black circles: Si; white diamonds: Na; and squares:.Al .
echo intensity on the observed nuclei X due toirradiation of the second nuclear spin H that selec-tively reintroduces the X–H dipolar coupling. Dueits relative simplicity, its applicability can be ex-
Ž27tended to abundant quadrupolar nuclear spins Al,23 . ŽNa in inorganic materials Montouillout et al.,
.1999; Magnenet et al., 2000 even if the obtainedinformation remains qualitative. The REDOR resultsare presented as normalized difference intensities for
Žeach resolved contribution vs. recoupling time Fig..8 . The more rapidly the signal intensity for a given
nucleus rises, the closer that nucleus is to the pro-tons. In the case of NS4, curves can be measured notonly for Na but also for the individual Q species ofn29Si. The most rapid rise is seen for Q , corroborat-2
ing the CP-MAS result that protons are nearer to Q2
or more numerous in its neighborhood than to othertypes of Si. Na and Q behave similarly, but are3
farther from protons than Q , while the curve for Q2 4
rises the slowest, indicating that these silicon nucleiare farthest from protons. Results of the same experi-ment applied to the two phonolite glasses show lesspronounced differences between the different nuclei.The sample containing 0.9 wt.% H O shows almost2
Židentical behavior for Na, Al, and Si undifferenti-.ated , suggesting that there is no difference between
these nuclei in their average distance to protonsbetween these nuclei. The sample containing 3.2wt.% H O shows a slightly slower response for Si2
than for Na and Al, but, again, the different nucleishow quite similar responses.
5. Discussion
5.1. Sodium tetrasilicate
For NS4, there is clear evidence from the single-29 Žpulse Si MAS spectra in Fig. 1 MAS and CP-.MAS for the growth of the Q and Q peaks, at the2 3
expense of the Q peak, signaling a net depolymeri-4
zation of the silicate network in the nearby environ-ment of protons, on addition of water. The reactionmay be summarized as:
H OqSi–O–Si´2Si–OH 1Ž .2
Ž .This mechanism was proposed by Burnham 1975for albite glass, supported by infrared studies of

( )E. Robert et al.rChemical Geology 174 2001 291–305 301
Žwater speciation in aluminosilicate glasses Stolper,.1982 , and confirmed by NMR studies of hydratedŽNS4 Kohn et al., 1989a,b; Kummerlen et al., 1992;¨
.Schaller and Sebald, 1995; Zotov and Keppler, 1998Ž .and neutron diffraction Zotov et al., 1996 . In fur-
ther agreement with these studies, we have alsofound evidence for some water dissolved as molecu-lar H O. The 1H MAS spectra show two main2
contributions, the first at ;4 ppm and the second atŽ .higher chemical shift 16 down to 8 ppm , the latter
consisting of at least two different lines. Part of the4-ppm contribution shows spinning sidebands andstrong autocorrelation in the double-quantum-filtered1H dipolar correlation experiment, indicating strongdipolar coupling between two equivalent protons, aswould be anticipated in a rotating H O molecule.2
ŽThe other protons pertaining to the whole spectral.width show neither autocorrelation nor spinning
sidebands, and signal a distribution of pairs of unlikeprotons that should be ascribed to weaker OH–OHpairs. This assignments partly concur with those of
Ž . 1Schaller and Sebald 1995 , based on H MAS and1H MAS CRAMPS spectroscopy, but our interpreta-tion is that only part of the 4-ppm line can beunambiguously ascribed to molecular H O. This is2
supported by our observation that Q silicon species4Ž .cross-polarizes from this line HETCOR experiment .
While the relative distances from individual protonsto 29Si can be probed consistently by varying contacttime in the CP-MAS experiment or varying therecoupling time in the REDOR experiment, it canonly be assessed from REDOR experiments in thecase of a quadrupolar nucleus. 23 Na appears to havea rising behavior similar to that of Q species, which3
could possibly be linked to one OH group. Thisbehavior differs from that of the Q species, which2
could be linked to two OH. As Q is the dominant Si3
contribution, this means that sodium does not seemore protons in its close neighborhood than silicondoes, and, therefore, protons play a typical networkmodifying role and depolymerize the silicon net-work.
5.2. Phonolite
Interpretation of results from the phonolite ismuch less clear-cut, in part because it is a much
more complex composition. The most significantstructural difference is the participation of Al, aswell as Si in the formation of an aluminosilicatenetwork, and the concomitant increase in the numberof chemical environments seen by each of these
Žnuclei Q and Q species with possible substitution3 4.of Al as first neighbor . Resolution of separate Q
species in 29Si NMR spectra of albite glasses hasŽproved impossible Kohn et al., 1989b; Zavel’sky et
.al., 1998 , and the spectra in Fig. 1 show this to becase for the phonolite also. The chemical shift distri-bution increases slightly upon hydration, and averagechemical shifts move to less shielded values withincreasing water content, as was the case for NS4.These data suggest depolymerization of the alu-minum–silicon network upon hydration. This is no-ticeably different to what was observed in albite,which showed no change in average chemical shift
Ž .with increasing water content Kohn et al., 1989b .From proton NMR, we showed that there is a muchwider distribution of chemical shift than observed inthe simple single-pulse experiment, as the dipolardephasing experiment reveals a wide range of differ-ent lines up to 16 ppm. The OH and H O contribu-2
tions appear to strongly overlap mostly around 4ppm. No contrast was obtained from the HETCORexperiment but REDOR shows very similar rising
Ž23 27behavior for the three observed nuclei Na, Al,29 .and Si , at least for low water contents. Hence,
consistently with what we observed for NS4, weconsider that all nuclei see the same average protonsurrounding and that the dissolution of water leads toa depolymerization of the network. At higher watercontent, there seems to be an indication that the 29SiREDOR signal rises slightly slower. In the model
Ž .previously proposed by Kohn et al. 1989b , protonswould be preferentially associated with Al and Naeven at low water content, and this should appear inthe REDOR experiments.
5.3. Quadrupolar nuclei
Some additional information could be expectedfrom the interpretation of the spectra of the twoobservable quadrupolar nuclei 23 Na and 27Al. Twosources of broadening could be invoked: distributionof isotropic chemical shift and distribution of

( )E. Robert et al.rChemical Geology 174 2001 291–305302
quadrupolar interaction. In simple cases, they couldpossibly be analyzed with a systematic use of the
ŽMQ-MAS experiment Frydman and Harwood, 1995;.Medek et al., 1995 . However, multiple fieldrspin-
ning rate experiments show that the distribution ofquadrupolar interaction is likely to be dominant un-der our observation conditions. The sharpening ofboth 23 Na and 27Al lines upon hydration is wellestablished, even if the evaluation of the quadrupolarparameters is highly model dependent. It seems to bemostly linked to a reduction of the mean quadrupolar
Žcoupling Kohn et al., 1989b, 1992, 1998; Schmidt et.al., 2000 . This is very similar to what is observed
for zeolite structures where hydration diminishes theAl quadrupolar coupling. If we consider that most ofthe EFG viewed by Al or Na is due to point chargesin the network, this could be understood as a relax-ation of long-range order constraints. This is instrong contrast with what is observed for 17O incovalent, low coordination number environments,where most of the quadrupolar interaction comes
Žfrom the geometry of the binding orbitals Grandinetti.et al., 1995 .
5.4. Hydrogen bonding
It is known that strength and geometry of hydro-gen bonds influence proton chemical shift, deuteriumquadrupolar coupling or infrared absorption frequen-
Ž .cies Brunner and Sternberg, 1998 . The high valuesof proton chemical shifts that we observe directly inNS4 or in dipolar dephasing experiments in phono-lite can be interpreted in these terms.
Several different correlations have been reported,based on studies of crystalline hydrates where bondlengths can be investigated by other means. Eckert et
Ž .al. 1988 fitted available experimental data with therelation:
d ppm s79.05y255d nm 2Ž . Ž . Ž .H ,iso OH . . . O
Strictly speaking, the chemical shift should beinversely proportional the bond polarization energy,and hence to bond lengths, but empirical equationsassuming direct proportionality provide similarlygood fits over relatively wide ranges of chemical
Ž .shift. Brunner and Sternberg 1998 fitted a largerdatabase with the following relation:
d ppm s4.65rr nm y17.4 3Ž . Ž . Ž .H ,iso HB
where r is the H . . . O distance, rather than theHBŽ .OH . . . O distance of Eq. 2 . To allow direct com-
Ž .parison with previous studies, we will use Eq. 2 inthe following discussion.
In NS4, there are distinct signals for molecular`water at 4 ppm, corresponding to an O H . . . O bond
length of 0.294 nm. It has been argued above thatintensity at this chemical shift does not result en-tirely from molecular water, so that some OH is alsohydrogen bonded with a similar bond length, and,hence, strength. These two signals may result fromhydrogen bonding between water and Si–OH speciesas described for hydrous silica glass by Kohn et al.Ž .1989a .
Ž .Much higher chemical shifts 8 to 16 ppm areobserved directly in NS4 or through dipolar dephas-ing experiments in phonolite. These would corre-spond to hydrogen bond lengths of about 0.28 and0.25 nm and could be related to the weak vibration at2350 cmy1 observed in the Raman spectrum of NS4glass containing 1 wt.% H O by McMillan and2
Ž .Remmele 1986 . They attributed this signal, corre-sponding to a hydrogen bond length of about 0.26nm, to intratetrahedral hydrogen bonding betweenSiOH groups and NBO. These authors also identifieda band extending from a maximum at about 2700down to 2500 cmy1, corresponding to OH . . . O bondlengths of about 0.265 nm. Neither band was seen inthe Raman spectra of silica glass, and intensity atthese chemical shifts was not observed in a 1H MAS
Ž .study of silica glass by Kohn et al. 1989a , indicat-ing that NBO must be involved.
To try to better understand the high proton chemi-cal shifts that we measured for both compositions,we can refer to the literature. From a multinuclear
Ž .MAS NMR study, Kohn et al. 1989b ruled outŽ .more than a very small fraction -1% of silicon
atoms being in Q –OH groups in hydrous albite3Ž .glasses. Xu et al. 1998 , making use of the newly
developed MQ-MAS experiment applied to 17O,identified a line in hydrous NS4 glass ascribed toSiOH. These authors also observed an apparent in-crease in intensity at the same position for hydrous

( )E. Robert et al.rChemical Geology 174 2001 291–305 303
albite glasses. This signal was small and obscured bythe nearby Al–O–Si signal, which is much moreintense, and while no firm conclusions were drawn,it seems possible that at least a small amount ofSi–OH may be present in hydrous albite. In contrastwith albite, some NBO are already present in theanhydrous NS4 and phonolite glasses prior to hydra-tion. The question then arises of the attribution of the8 to 16 ppm H lines that we observe in NS4 andphonolite glasses and that preferentially cross-polarize to the depolymerized part of the networkŽ�1 4 29 .H – Si HETCOR . Such chemical shifts werereported neither in hydrous silica nor in hydrous
Ž .albite glasses using simple experiments . Our resultsare in agreement with a SiOH group hydrogen bondedto a NBO as depicted by McMillan and RemmeleŽ .1986 . This confirms that at least some of theprotons act as network modifiers.
Nevertheless, this discussion is based on a largelysimplified picture of the aluminosilicate network. Foranhydrous glasses, it is known that bridging oxygenscan have modifier cations in their neighborhood, andthat the environment of a NBO is much more com-plex than a single link to one specific modifierŽ .Florian et al., 1996; Stebbins et al., 1997 . Ourresults do not exclude any of the previously pro-posed mechanisms and are consistent with recentviscosity measurements, which suggest that several
Žmechanisms may operate simultaneously Whitting-.ton et al., 2000 . This preliminary investigation
indicates directions for future advances in the under-standing of hydrous glass structures using high-reso-lution solid-state NMR.
Most previous studies have dealt with largeŽ .amounts ;5 to 10 wt.% of dissolved water added
to a given composition. This introduces two types ofŽ .difficulties: i a large proportion of protons remain
in water molecules and dominate in spectroscopicŽ .studies and ii if protons act as network modifiers, it
would be of interest to work at constant NBOrTratio.
6. Conclusions
We have studied two anhydrous and hydrousŽ .silicate sodium tetrasilicate and aluminosilicate
Ž .synthetic iron free phonolite analog glasses by avariety of high-resolution solid-state NMR tech-niques. While the usual one-dimensional NMR ex-periments provide information on the structure of thefirst-coordination shell of the observed nuclei, somemore sophisticated experiments, which take advan-
Ž .tage of the reintroduction of the homonuclear H–HŽ .or heteronuclear H–X dipolar interaction, can now
characterize longer-range order: CP-MAS, HET-COR, H dipolar dephasing, H spin counting, Hdouble-quantum correlation, and H–X REDOR. Itclearly appears that these proton-based NMR experi-ments provide a new insight to the mechanism ofdissolution of water.
The comparison of results obtained for sodiumtetrasilicate and phonolite glasses suggests that thewater incorporation mechanisms are qualitativelysimilar for these two compositions. Due to the quali-tative nature of many of the more complex experi-ments, quantitative comparisons are beyond the scopeof the present study. From proton NMR experiments,we observe no evidence of proton clustering and awide range of chemical shifts ranging from 0 to 16ppm, even for the aluminosilicate phonolite glass,identifying at least three types of OH protons inaddition to molecular H O. This variety of OH2
groups can be interpreted in terms of hydrogen bond-ing strength. For both compositions, we see evidenceof depolymerization of the tetrahedral network, andthe picture is more complicated than described bypreviously proposed models.
This preliminary work opens the way for moredetailed studies of the network modifier role of theproton, especially at low water content. High-resolu-tion proton-based solid-state NMR experimentsshould provide efficient tools for studying local and
Žmedium range order in these glasses e.g., Zeng et.al., 1999 .
Acknowledgements
We thank Thomas Riemer and Pascal Richet forhelpful discussions and Simon Kohn, Don Dingwell,and an anonymous reviewer for their comments. Weacknowledge financial support from the CNRSChemistry Department, Region Centre and European´

( )E. Robert et al.rChemical Geology 174 2001 291–305304
Community Training and Mobility Research pro-gram AHydrous silicate melts in situ at high tempera-ture and pressureB TMR ERBFMRX 960063.
References
Burnham, C.W., 1975. Water and magmas, a mixing model.Geochim. Cosmochim. Acta 52, 2659–2669.
Burnham, C.W., 1979. The importance of volatile constituents. In:Ž .Yoder, H.S. Ed. , The Evolution of the Igneous Rocks:
Fiftieth Anniversary Perspectives. Princeton Univ. Press, pp.439–482.
Bronnimann, C.E., Zeigler, R.C., Maciel, G.E., 1988. ProtonNMR study of dehydration of the silica gel surface. J. Am.Chem. Soc. 110, 2023–2026.
Brunner, E., Sternberg, U., 1998. Solid-state NMR investigationsof the nature of hydrogen bonds. J. Progr. Nucl. Magn. Res.Spectrosc. 32, 21–57.
Dingwell, D.B., Webb, S.L., 1990. Relaxation in silicate melts.Eur. J. Mineral. 2, 427–449.
Dingwell, D.B., Romano, C., Hess, K.-U., 1996. The effect ofwater on the viscosity of a haplogranitic melt under P–T–Xconditions relevant to silicic volcanism. Contrib. Mineral.Petrol. 124, 19–28.
Eckert, H., Yesinowski, J.P., Stolper, E.M., Stanton, T.R., Hol-loway, J., 1987. The state of water in rhyolitic glasses. Adeuterium NMR study. J. Non-Cryst. Solids 93, 93–114.
Eckert, H., Yesinowski, J.P., Silver, L.A., Stolper, E.M., 1988.Water in silicate glasses: quantitation and structural studies by1H solid echo and MAS-NMR methods. J. Phys. Chem. 92,2055–2064.
Emsley, L., Pines, A., 1994. Lectures on Pulsed NMR, Proceedingof the International School of Physics AEnrico FermiB, SocietaItaliana di Fisica CXXIII.
Engelhardt, G., Michel, D., 1987. High Resolution Solid StateNMR of Silicates and Zeolites. Wiley, New York.
Florian, P., Vermillion, K.E., Grandinetti, P.J., Farnan, I., Steb-bins, J.F., 1996. Cation distribution in mixed alkali disilicateglasses. J. Am. Chem. Soc. 118, 3493.
Frydman, L., Harwood, J.S., 1995. Isotropic spectra of half-in-teger quadrupolar spins from bidimensional magic-angle spin-ning NMR. J. Am. Chem. Soc. 117, 5367–5368.
Geen, H., Graf, R., Heindrichs, A.S.D., Hickman, B.S., Schnell,I., Spiess, H.W., Titman, J.J., 1999. Spin counting with fastMAS. J. Magn. Reson. 138, 167–172.
Grandinetti, P.J., Baltisberger, J.H., Farnan, I., Stebbins, J.F.,Werner, U., Pines, A., 1995. Solid-state 17O magic-angle anddynamic-angle spinning NMR study of the SiO polymorph2
coesite. J. Phys. Chem. 99, 12341.Ž .Gullion, T., Schaefer, J., 1989. In: Warren, S. Ed. , Advances in
Magnetic Resonance vol. 13. Academic Press, San Diego.Hafner, S., Spiess, H.W., 1998. Advanced solid-state NMR spec-
troscopy of strongly dipolar coupled spins under fast magicangle spinning. Concepts Magn. Reson. 10, 99–128.
Holtz, F., Beny, J.M., Mysen, B.O., Pichavant, M., 1996. High-´temperature Raman spectroscopy of silicate and aluminosili-cate hydrous glasses: implications for water speciation. Chem.Geol. 128, 25–39.
Kohn, S.C., Dupree, R., Smith, M.E., 1989a. Proton environmentsand hydrogen bonding in hydrous silicate glasses from protonNMR. Nature 337, 539–541.
Kohn, S.C., Dupree, R., Smith, M.E., 1989b. A multinuclearresonance study of the structure of hydrous albite glasses.Geochim. Cosmochim. Acta 53, 2925–2935.
Kohn, S.C., Dupree, R., Mortuza, M.G., 1992. The interactionbetween water and aluminosilicate magmas. Chem. Geol. 96,399–409.
Kohn, S.C., Smith, M.E., Dupree, R., 1994. Comment on Aamodel for H O solubility mechanisms in albite melts from2
infrared spectroscopy and molecular orbital calculationsB byD. Sykes and J.D. Kubicki. Geochim. Cosmochim. Acta 58,1377–1380.
Kohn, S.C., Smith, M.E., Dirken, P.J., van Eck, E.R.H., Kentgens,A.P.M., Dupree, R., 1998. Sodium environments in dry andhydrous albitic glasses: improved 23 Na solid state NMR dataand their implications for water dissolution mechanisms.Geochim. Cosmochim. Acta 62, 79–87.
Kummerlen, J., Merwin, L.H., Sebald, A., Keppler, H., 1992.¨Structural role of H O in sodium silicate glasses: results from229Si and 1H NMR spectroscopy. J. Phys. Chem. 96, 6405–6410.
Magnenet, C., Massiot, D., Klur, I., Coutures, J.P, 2000. Charac-terization of alumino silicate on rutile surface by high-resolu-tion solid state NMR. J. Mater. Sci. 35, 115–121.
McMillan, P., Remmele Jr., R.L., 1986. Hydroxyl sites in SiO2glass: a note on infrared and Raman spectra. Am. Mineral. 71,772–778.
McMillan, P., 1994. Water solubility and speciation models. Min.Soc. Am. Rev. Min. 30, 131–156.
Medek, A., Harwood, J.S., Frydman, L., 1995. Multiple-quantummagic-angle spinning NMR: a new method for the study ofquadrupolar nuclei in solids. J. Am. Chem. Soc. 117, 12779–12787.
Montouillout, V., Massiot, D., Douy, A., Coutures, J.P., 1999.Characterization of MgAl O precursor powders prepared by2 4
27 27 �1 4aqueous routes: Al MAS, MQ-MAS and Al– H REDORinvestigations. J. Am. Ceram. Soc., in press.
Mysen, B.O., Virgo, D., Seifert, F.A., 1982. The structure ofsilicate melts: implications for chemical and physical proper-ties of natural magmas. Rev. Geophys. Space Phys. 20, 353–383.
Mysen, B.O., Virgo, D., 1986. Volatiles in silicate melts at highpressure and temperature: 2. Water in melts along the joinNaAlO –SiO and a comparison of solubility mechanisms of2 2
water and fluorine. Chem. Geol. 57, 333–358.Nowak, M., Behrens, H., 1995. The speciation of water in haplo-
granitic glasses and melts determined by in situ near-infraredspectroscopy. Geochim. Cosmochim. Acta 59, 3445–3450.
Remmele, R., Stanton, T., McMillan, P., Holloway, J., 1986.Raman spectra of hydrous glasses along the quartz–albite join.
Ž . Ž .EOS Trans. Am. Geophys. Union 67, 1274 abstract .

( )E. Robert et al.rChemical Geology 174 2001 291–305 305
Richet, P., Lejeune, A.M., Holtz, F., Roux, J., 1996. Water andthe viscosity of andesite melts. Chem. Geol. 128, 185–197.
Richet, P., Polian, A., 1998. Water as a dense ice-like componentin silicate glasses. Science 281, 396–398.
Riemer, T., Schmidt, B., Behrens, H., Dupree, R., submitted.H OrOH ratio determination in hydrous silicate glasses by2
static proton NMR and the effect of chemical shift anisotropy.Schaller, T., Sebald, A., 1995. One- and two-dimensional 1H
magic-angle spinning experiments on hydrous silicate glasses.Solid State Nucl. Magn. Reson. 5, 89–102.
Schmidt, B.C., Riemer, T., Kohn, S.C., Behrens, H., Dupree, R.,2000. The effects of water on the structure of quartz–albiteglasses quenched from hydrous melts at high P and T: anNMR spectroscopic study. Geochim. Cosmochim. Acta, inpress.
Schmidt-Rohr, K., Spiess, H.W, 1994. Multi-Dimensional SolidState NMR and Polymers. Academic Press, New York.
Shen, A., Keppler, H., 1995. Infrared spectroscopy of hydroussilicate melts to 10008C and 10 kbar: direct observation ofH O speciation in a diamond–anvil cell. Am. Mineral. 80,2
1335–1338.Stebbins, J.F., Oglesby, J.V., Xu, Z., 1997. Disorder among
network-modifier cations in silicate glasses: new constraintsfrom triple-quantum 17O NMR. Am. Mineral. 82, 1116–1124.
Stolper, E., 1982. Water in silicate glasses: an infrared spectro-scopic study. Contrib. Mineral. Petrol. 81, 1–17.
Sykes, D., Kubicki, J.D., 1993. A model for H O solubility2
mechanisms in albite melts from infrared spectroscopy andmolecular orbital calculations. Geochim. Cosmochim. Acta 57,1039–1052.
Sykes, D., Kubicki, J.D., 1994. Reply to the comment by Kohn,S.C., Smith, M.E. and Dupree, R. on Aa model for H O2
solubility mechanisms in albite melts from infrared spec-troscopy and molecular orbital calculationsB. Geochim. Cos-mochim. Acta 58, 1381–1384.
Whittington, A., Richet, P., Linard, Y., Holtz, F., 2000. Theviscosity of hydrous phonolites and trachytes. Chem. Geol.,this volume.
Xu, Z., Maekawa, Oglesby, J.V., Stebbins, J.F., 1998. Oxygenspeciation in hydrous silicate glasses: an oxygen-17 NMRstudy. J. Am. Chem. Soc. 20, 9894–9901.
Zavel’sky, V.O., Bezmen, N.I., Zharikov, V.A., 1998. Water inalbite glasses: OH-groups, isolated molecules, and clusters. J.Non-Cryst. Solids 224, 225–231.
Zeng, Q., Nekvasil, H., Grey, C.P., 1999. Proton environments inhydrous aluminosilicate glasses: a 1H MAS, 1Hr27Al, and1Hr23 Na TRAPDOR NMR study. J. Phys. Chem. B 103,7406–7415.
Zotov, N., Keppler, H., Hannon, A.C., Soper, A.K., 1996. Theeffect of water on the structure of silicate glasses — a neutrondiffraction study. J. Non-Cryst. Solids 202, 153–163.
Zotov, N., Keppler, H., 1998. The influence of water on thestructure of hydrous sodium tetrasilicate glasses. Am. Mineral.83, 823–834.


















![Comportement sous irradiation des géopolymères · [2] Phair et al (2002) Effect of the silicate activator pH on the microstructural characteristics of waste-based geopolymer. International](https://img.pdfslide.fr/doc/110x75/601776993a62b811e91e2dd0/comportement-sous-irradiation-des-gopolym-2-phair-et-al-2002-effect-of-the.jpg)









