Embed Size (px)
Citation preview

SYNTHESIS AND CHARACTERIZATION OF NANOPOROUS MATERIALS:
NANOZEOLITES AND METAL-ORGANIC FRAMEWORKS
Thèse
Thanh Vuong Gia
Doctorat en Génie Chimique
Philosophiae Doctor (Ph.D)
Québec, Canada
© Thanh Vuong Gia, 2013


iii
Résumé
Dans ce travail, deux types de nanomatériaux poreux ont été obtenues: des
nanozéolithes et des matériaux à réseau organométallique (MOF). Pour les nanozéolithes,
deux nouvelles méthodes de synthèses ont été développé: une méthode à phase unique et
une méthode biphasique. Dans la méthode à phase unique, une quantité du gel-zéolithique
est ajoutée à une solution de toluène/n-butanol contenant l‟agent silylant organosilane.
Après 12 heures à 60oC, une phase homogène est obtenue. Ce mélange est traité
hydrothermalement pour produire une nanozéolithe fonctionalisée. En revanche, la
méthode de synthèse à deux phases, implique l‟introduction de l‟organosilane mélangé à un
solvant organique dans le gel de zéolithe aqueux conduisant ainsi à un mélange biphasique.
Après mélange et traitement hydrothermal, des nanozéolithes fonctionalisées par silylation
sont obtenu dans la phase organique et de larges cristaux de zéolithes sont obtenus dans la
phase aqueuse. En principe, les deux méthodes utilisent l‟organosilane pour empêcher la
croissance des cristaux. Le solvant organique joue le rôle de dispersant des nanozéolithes
fonctionalisées avec l‟organosilane à partir de la phase aqueuse, et contrôle le processus de
croissance des nanozéolithes. Ces deux méthodes de synthèse sont applicables autant aux
zéolithes MFI que FAU, telles que silicatite-1, ZSM-5 et NaY. Elles peuvent être étendues
à la synthèse d‟autres types de zéolithes. L‟activité catalytique de ces nanozéolithes a été
évaluée pour le craquage de FCC. Les résultats indiquent que la nanozéolithe de type FAU
montre une bonne activité dans cette réaction.
Pour l‟étude des matériaux à réseau organométallique (MOF), une nouvelle
approche a été développé pour la synthèse de MIL-88B en utilisant un cluster neutre de
métaux mixtes bimétalliques Fe2Ni(µ3-O). Les clusters occupent les nœuds du réseau MIL-
88B à la place du mono-métal Fe3 (µ3-O) avec un anion compensateur. Ce dernier est le
cluster formant le réseau du Fe3MIL-88B non-poreux qui est obtenu par la méthode
conventionnelle. De ce fait, en absence des anions compensateurs dans la structure, Fe2Ni
MIL-88B devient un matériau poreux. De plus, avec la combinaison de la flexibilité de
MIL-88B et des métaux mixtes comme nœuds dans le réseau, la porosité peut être contrôlée
par échange avec des ligands terminaux du réseau. Ceci nous a permis de moduler d‟une

iv
manière réversible la porosité de MIL-88B à différents niveaux, ainsi que la surface
spécifique et le volume de pores dépendant de taille de ligands échangés. Le mécanisme de
synthèse a été aussi étudié pour les matétiaux Fe3-MIL88B et Fe2Ni-MIL88B. Les résultats
montrent que pour la synthèse de Fe3-MIL88B, le mono-métal Fe3-MOF-235 est comme le
précurseur pour la formation de MIL-88B. Dans le cas d‟utilisation de métaux mixtes
Fe2Ni(µ3-O), les mono-métal Fe3-MOF-235 est formés en premier lors de la synthèse du
métal mixte Fe2Ni-MIL88B. Il est montré que la présence de l‟anion FeCl4- est déteminante
dans la formation de la phase initiale MOF-235 et dans le succès de la synthèse du MIL-
88B mono- ou bimétallique
L‟anion FeCl4- est très important pour le succès de la formation de MOF-235. Un
mécanisme d‟anion médiateur dans la formation de MOF-235 a été suggéré.

v
Abstract
In this thesis, two types of nanoporous materials: nanozeolites and metal-organic
frameworks were studies. For nanozeolites, two novel methods e.g. single-phase and two-
phases were reported for the synthesis of nanozeolites. In the single-phase synthesis
method, a proper amount of zeolite gel solution was added to a toluene/n-butanol solution
containing an organosilane. After 12 hours at 60oC, a single phase mixture was obtained.
This mixture was then subjected to hydrothermal crystallization to produce uniform
functionalized nanozeolites. In contrast, the two-phase synthesis method involved the
introduction of an organic solvent containing organosilane to the aqueous zeolite gel
solution, resulting in a two-phase mixture. Upon mixing and hydrothermal treatment of this
mixture, organosilane-functionalized nanozeolites were obtained in the organic phase
whereas, large zeolite crystals were found in the aqueous phase. In principle, both methods
employed the use of organosilane to inhibit the crystal growth. The organic solvent acted as
the medium for the dispersion of nanozeolites functionalized with organosilane from the
aqueous phase, which led to the complete halt of the growth process. These two methods
were demonstrated to be applicable to the synthesis of MFI and FAU nanozeolites such as
silicalite-1 and NaY, and could be applied to the synthesis of other types of zeolites.
Catalytic activity of the synthesized nanozeolites was evaluated by the cracking reaction of
FCC feed. The result showed that FAU nanozeolites can be good catalysts for the cracking
reaction.
For the study of the metal-organic frameworks (MOF), a new rational approach was
developed for the synthesis of mixed metal MIL-88B metal organic framework based on
the use of neutral bimetallic cluster, such as Fe2Ni(µ3-O) cluster. Unlike the conventional
negative charged single metal cluster, the use of neutral bimetallic cluster as a framework
node avoids the need of compensating anion inside porous MIL-88B system; thus such a
bimetallic MIL-88B becomes porous. The flexibility of the mixed metal MIL-88B can be
controlled by terminal ligands with different steric hindrance. This allows us to reversibly
customize the porosity of MIL-88B structure at three levels of specific surface area as well
as the pore volume. Synthesis mechanism was also studied. It was found that the

vi
monometallic Fe3-MOF-235 is the precursors to the formation of MIL-88B. MOF-235
comes first then later transforms to Fe3-MIL-88B or acts as seeds for the formation of
mixed Fe2Ni-MIL88B. FeCl4- anion is very important to the successful formation of MOF-
235. An anion mediated mechanism of the formation of MOF-235 is suggested

vii
List of Contents
Résumé .................................................................................................................................. iii Abstract ................................................................................................................................... v List of Contents .................................................................................................................... vii List of Tables .......................................................................................................................... x List of Figures ........................................................................................................................ xi
Acknowledgement ................................................................................................................ xv Preface ................................................................................................................................. xvi Chapter 1. Introduction ........................................................................................................... 1
1.1. Zeolite .......................................................................................................................... 1 1.1.1. Background ........................................................................................................... 1
1.1.2. Structure ................................................................................................................ 2 1.1.3. General Properties of Zeolites .............................................................................. 5
1.1.4. Applications .......................................................................................................... 6 1.2. Nanozeolites ................................................................................................................. 7
1.2.1. Background ........................................................................................................... 7 1.2.2. Synthesis ............................................................................................................... 8
1.2.3. Recent advances in application of nanozeolites ................................................. 26 1.3. Metal-organic frameworks ......................................................................................... 29
1.3.1. Background ......................................................................................................... 29 1.3.2. Design principles of MOFs ................................................................................. 33 1.3.3. Synthesis ............................................................................................................. 36
1.4. Some applications of MOFs ...................................................................................... 38
1.4.1. Adsorption .......................................................................................................... 38 1.4.2. MOF as catalysts ................................................................................................. 45
References ......................................................................................................................... 53
Chapter 2. Experimental ....................................................................................................... 61 2.1. Synthesis .................................................................................................................... 61
2.1.1. Preparation of clear zeolite gel solution ............................................................. 61 2.1.2. Synthesis of nanozeolites using clear gel solution in aqueous medium
(conventional method) .................................................................................................. 62 2.1.3. Synthesis of nanozeolites in organic medium..................................................... 63 2.1.4. Preparation of silica containing nanozeolites ..................................................... 65 2.1.5. Synthesis of MIL-88B metal-organic framework ............................................... 65
2.2. Characterization ......................................................................................................... 66
2.2.1. FTIR Spectroscopy ............................................................................................. 66 2.2.2. Raman spectroscopy ........................................................................................... 68
2.2.3. UV-Vis spectroscopy .......................................................................................... 68 2.2.4. Energy-dispersive X-ray spectroscopy ............................................................... 71 2.2.5. X-ray Diffraction (XRD) .................................................................................... 71 2.2.6.
29Si Magic Angle Spinning Nuclear Magnetic Resonance Spectroscopy (MAS
NMR) ............................................................................................................................ 73
2.2.7. Scanning electron microscope (SEM) ................................................................ 74 2.2.8. Transmission Electron Microscope (TEM) ........................................................ 75 2.2.9. Nitrogen Adsorption/Desorption Isotherms........................................................ 75

viii
2.2.10. Cracking reaction ............................................................................................. 78
References ........................................................................................................................ 79
Chapter 3. A New Route for the Synthesis of Uniform Nanozeolites with Hydrophobic
External Surface in Organic Solvent Medium ..................................................................... 81 Résumé ............................................................................................................................. 81 Abstract ............................................................................................................................ 81 References ........................................................................................................................ 88
Supporting information .................................................................................................... 89 Chapter 4. Synthesis of Silylated Nanozeolites in the Presence of Organic Phase: Two-
Phase and Single Phase Methods ......................................................................................... 95 Résumé ............................................................................................................................. 95 Abstract ............................................................................................................................ 95
4.1. Introduction ............................................................................................................... 96 4.2. Experimental ............................................................................................................. 99
4.2.1. Synthesis of silylated silicalite-1 using the two phase and single-phase methods
...................................................................................................................................... 99
4.2.2. Two-phase method ............................................................................................. 99 4.2.3. Single-phase method ........................................................................................ 100
4.2.4. Conventional method (synthesis of nanozeolites in aqueous medium) ........... 101 4.2.5. Characterization ............................................................................................... 101
4.3. Results and discussion ............................................................................................. 102
4.4. Conclusion ............................................................................................................... 113 Acknowledgments .......................................................................................................... 114
References ...................................................................................................................... 115
Chapter 5. Synthesis of Nanozeolite-Based FCC Catalysts and their Catalytic Activity in
Gasoil Cracking Reaction ................................................................................................... 117 Résumé ........................................................................................................................... 117
Abstract .......................................................................................................................... 118 5.1. Introduction ............................................................................................................. 119 5.2. Materials and methods ............................................................................................ 121
5.2.1. Synthesis of nanofaujasite ................................................................................ 121 5.2.2. Synthesis of nanofaujasite-based FCC catalysts .............................................. 121
5.2.3. Characterization ............................................................................................... 121 5.2.4. MAT cracking evaluation ................................................................................. 122
5.3. Results and discussion ............................................................................................. 123 5.3.1. Synthesis of nanozeolites ................................................................................. 123
5.3.2. Synthesis of FCC .............................................................................................. 133 5.3.3. Catalytic test ..................................................................................................... 136
5.4. Conclusion ............................................................................................................... 144
References ...................................................................................................................... 146 Chapter 6. Synthesis and Engineering Porosity of Mixed Metal Fe2Ni-MIL-88B Metal-
Organic Framework ............................................................................................................ 149 Résumé ........................................................................................................................... 149 Abstract .......................................................................................................................... 149
6.1. Introduction ............................................................................................................. 150 6.2. Experiments ............................................................................................................. 154 6.3. Results ..................................................................................................................... 155

ix
6.3.1. Synthesis of Mixed Metal Fe2Ni-MIL-88B with Different Terminal Ligands . 155
6.3.2. Reversible Breathing Control Using Terminal Ligand ..................................... 159
6.3.3. Adsorption Analysis ......................................................................................... 160 6.4. Discussion ................................................................................................................ 163 6.5. Conclusion ............................................................................................................... 167 Refrences ........................................................................................................................ 168 Supporting information ................................................................................................... 170
Chapter 7. Direct Synthesis and Mechanism for the Formation of Mixed Metal Fe2Ni-MIL-
88B ...................................................................................................................................... 185 Résumé ............................................................................................................................ 185 Abstract ........................................................................................................................... 185 7.1. Introduction .............................................................................................................. 186
7.2. Experimental Section ............................................................................................... 187 7.3. Results ...................................................................................................................... 188 7.4. Discussion ................................................................................................................ 194
7.5. Conclusion ............................................................................................................... 198
References ....................................................................................................................... 199 Supporting Information ................................................................................................... 219
Chapter 8. Conclusion ......................................................................................................... 233 List of Pulications ............................................................................................................... 239

x
List of Tables
Table 1.1. Molecular sieve types synthesized in nanosized form, synthesis conditions, and
crystal size[9] ............................................................................................................... 16 Table 2.1. Structure insensitive and sensitive framework vibrations of zeolites [4] ........... 67
Table 2.2. FTIR band assignment in the wavenumber 400 – 800 cm-1
............................... 67 Table 4.1. Physico-chemical properties of the calcined silylated nanozeolite and zeolite
samples prepared from the same clear zeolite gel, using different methods: the two-
phase, single-phase and conventional methods. ......................................................... 102 Table 5.1. Physicochemical properties of nanofaujasite samples. .................................... 133
Table 5.2. BET analysis of nanozeolite-based FFC catalyst samples. .............................. 134 Table 6.1. IR analysis of the MIL-88B samples ................................................................ 156
Table 6.2. Crystal parameters of the Fe2Ni-MIL-88B samples ......................................... 158 Table 6.3. Porosity of Fe2Ni-MIL-88B ............................................................................. 163 Table 7.1. FTIR band assignment in the wavenumber 400 – 800 cm
-1 ............................. 217
Table 7.2. Raman band assignments ................................................................................. 217
Table 7.3. Fe and Ni atomic percentages calculated from EDS spectra ............................ 217 Table 7.4. Comparison of the crystal parameters of MIL-88B and MOF-235 .................. 218

xi
List of Figures
Figure 1.1. Secondary building units and their symbols. Number in parenthesis indicates
frequency of occurrence .................................................................................................. 3 Figure 1.2. (a) MFI and (b) FAU structures ........................................................................... 4
Figure 1.3. Inter-conversion of Brönsted and Lewis acid sites .............................................. 5 Figure 1.4. Calculated surface to bulk atom ratios for spherical nanocrystals[13] ................ 8 Figure 1.5. Synthesis of nanozeolites from clear solutions [18] ............................................ 9 Figure 1.6. Two pathways of zeolite formation ................................................................... 10 Figure 1.7. A scheme for the crystallization mechanism of silicalite-1[28] ........................ 12
Figure 1.8. The "nanoslab" hypothesis: (a) the precursor unit containing one TPA cation
and (b) schematic representation of nanoslab formation by aggregation of precursor
units, as determined by XRS and GPC [32] ................................................................. 12
Figure 1.9. Nucleation and growth model of zeolite A and zeolite Y as represented by
TEM [34, 35] ................................................................................................................ 13 Figure 1.10. Schematic illustration of the mechanism proposed for nanoparticle evolution
and crystal growth by aggregation. ............................................................................... 14 Figure 1.11. Schematic illustration of confined space synthesis.[101]................................ 21 Figure 1.12. Schematic representation of synthesis of template-free zeolite nanocrystals by
using in situ thermoreversible polymer hydrogels.[104] .............................................. 23 Figure 1.13. Schematic representation of revered microemulsion ...................................... 24
Figure 1.14. Schematic representation of microemulsion-microwave synthetic method[115]
...................................................................................................................................... 26
Figure 1.15. Rational design of zeotype and MOF from the basis of zeolite ...................... 29 Figure 1.16. Crystal structure of HKUST-1 (left) and MOF-5 (right) ................................. 31
Figure 1.17. Number of metal–organic framework (MOF) structures reported in the
Cambridge Structural Database (CSD) from 1978 through 2006. The bar graph
illustrates the recent dramatic increase in the number of reports, while the inset shows
the natural log of the number of structures as a function of time, indicating the
extraordinarily short doubling time for MOF structures compared to the total number
of structures archived in the database.[129] ................................................................. 32
Figure 1.18. Two geometrically different but topologically identical nets. ......................... 34 Figure 1.19. MOF-5 structure from linking octahedral SBUs ............................................. 35
Figure 1.20. Design of MOF-5 from simple fcc structure ................................................... 36 Figure 1.21. Structure of NU-100 and its linker .................................................................. 40
Figure 1.22. The two different linkers used in MOF-210 (left) and the crystal structure of
MOF-210 displaying its unique topology with two different types of pores (right). ... 40 Figure 1.23. Excess hydrogen uptake at 77 K versus BET specific surface area (BET ssa)
for various high-porosity MOFs. The symbols denote measurements conducted by
different research groups (circles: at 20 bar by Hirscher et al.[193] triangles: at 60 bar
by Kaskel et al.[194]; squares: saturation values by Yaghi et al.).[195] ...................... 41 Figure 1.24. MOF-117 structure and comparison of the volumetric CO2 capacity of
crystalline MOF-177 relative to zeolite 13X pellets, MAXSORB carbon powder, and
pressurized CO2.[200] ................................................................................................... 42 Figure 1.25. ZIF-69 structure ............................................................................................... 43

xii
Figure 1.26. A portion of the structure of the sodalite-type framework of Cu-BTTri (1)
showing surface functionalization of a coordinatively unsaturated Cu(II) site with
ethylenediamine, followed by attack of an amino group on CO2 . .............................. 44 Figure 1.27. Detail of Pd-MOF, showing the 4-membered and the two 6-membered rings.
3D arrangement of the sodalite cages in sodalite-type frameworks. ............................ 46 Figure 1.28. (a) Basic building block of Cu(2-pymo)2 and (b) Diagram of the asymmetric
unit of the Co(PhIM)2 framework ................................................................................ 47
Figure 1.29. Transformation of ZIF-90 (A) by Reduction with NaBH4, and reaction with
ethanolamine to give ZIF-91 (B) and ZIF-92 (C) [213] ............................................... 49 Figure 1.30. (a) Eight-coordinate molecular building block that could be represented as a
tetrahedral building unit, (b) [H2TMPyP]4+
porphyrin, (c) crystal structure of rho-
ZMOF (left), hydrogen atoms omitted for clarity, and schematic presentation of
[H2TMPyP]4+
porphyrin ring enclosed in rho-ZMOF R-cage (right, drawn to scale)
[217] ............................................................................................................................. 51 Figure 2.1. Methods studied for the synthesis of nanozeolites ........................................... 61
Figure 2.2. Scheme of the autoclave: (1) a cylindrical stainless steel vessel, (2) a Teflon
cylindrical beaker, (3) a flat Teflon cover for closing the Teflon beaker, (4) a flat
stainless steel cover which was tightened up to part (1) by six screws.[3] .................. 63
Figure 2.3. The excitation process ....................................................................................... 69 Figure 2.4. Electronic energy levels and transitions. .......................................................... 69 Figure 2.5. Principle of EDX spectroscopy ......................................................................... 71
Figure 2.6. Diffraction of X-ray beams on a crystal lattice ................................................. 72 Figure 2.7. Types of sorption isotherms.[18] ...................................................................... 75
Figure 2.8. t-plot method. .................................................................................................... 78
Figure 3.1. XRD patterns of the as-made silylated zeolite and zeolite samples prepared
from the same zeolite gel in solvent medium in the presence of organosilane and in
aqueous medium in the absence of organosilane, respectively: (A) silicalite-1; (B)
faujasite. ....................................................................................................................... 85 Figure 3.2. TEM images of the as-made samples: (A) silylated nanosilicalite-1, (B)
silylated nanofaujasite. ................................................................................................. 86
Figure 4.1. Schematic representation of the two-phase synthesis method. ....................... 100 Figure 4.2. Schematic representation of the single-phase synthesis method. ................... 103
Figure 4.3. XRD patterns of the as-made silicalite-1 samples, (a) sample prepared using the
conventional method in aqueous medium, (b) AP silicalite-1, (c) OP silicalite-1 using
the two-phase method and (d) SOP silicalite-1 using single-phase method. ............. 104
Figure 4.4. SEM micrographs of the as-made samples, (A) silylated OP silicalite-1 and (B)
silylated AP silicalite-1. ............................................................................................. 106 Figure 4.5. TEM micrograph of the as-made SOP nanosilicalite-1 sample prepared using
the single-phase method. ............................................................................................ 107
Figure 4.6. FTIR spectra of the silicalite-1 samples prepared using the single-phase method
(A) and the conventional method (B). ........................................................................ 108
Figure 4.7 29
Si MAS NMR spectra of the silicalite-1 samples prepared from the same
zeolite gel solution using (a) the conventional method in aqueous medium without
organosilane, (b) as-made SOP nanosilicalite-1 using single-phase method in organic
solvent and (c) calcined SOP nanosilicalite-1. ........................................................... 109

xiii
Figure 4.8 29
Si MAS NMR spectra of the as-made silicalite-1 samples prepared from the
same zeolite gel solution using the two-phase method: (a) as-made AP nanosilicalite-1
and (b) as-made OP nanosilicalite-1. .......................................................................... 110 Figure 4.9. Nitrogen adsorption/desorption isotherms of the calcined samples: (A) SOP
silicalite-1, (B) OP silicalite-1 and (C) AP silicalite-1 (inset: t-plot curve). .............. 112 Figure 5.1. FT-IR spectra of the prepared nanofaujasite samples: (A) FAU–TOL2D
prepared using toluene and pre-heated zeolite gel for 2 days at 90 °C, (B) FAU–
FOR2D prepared using formamide and pre-heated zeolite gel for 2 days at 90 °C, (C)
FAU–FOR4D prepared using formamide and pre-heated zeolite gel for 4 days at
90 °C, and (D) zeolite Y reference. ............................................................................ 126 Figure 5.2. XRD patterns of nanofaujasite samples prepared: (A) FAU–TOL2D in toluene,
(B) FAU–FORM2D in formamide from the zeolite gel pre-heated at 90 °C for 2 days,
(C) FAU–FORM4D in formamide from the zeolite gel pre-heated at 90 °C for 4 days,
and (D) zeolite Y standard. ......................................................................................... 127 Figure 5.3. TEM images of (A) the sample FAU–TOL2D prepared in toluene from the
zeolite gel pre-heated at 90 °C for 2 days, (B) sample FAU–FOR2D prepared in
formamide from the zeolite gel pre-heated at 90 °C for 2 days, and (C) the sample
FAU–FOR4D prepared in formamide from the zeolite gel pre-heated at 90 °C for 4
days. ............................................................................................................................ 128 Figure 5.4.
29Si MAS NMR spectra of the as-made faujasite prepared in aqueous medium
in the absence of organosilane (conventional method) and silylated faujasite samples:
(A) FAU–TOL4D using toluene pre-heated for 4 days, (B) FAU–FOR2D using
formamide pre-heated for 2 days, (C) FAU–TOL2D using toluene pre-heated for 2
days, and (D) FAU-Standard using conventional method. ......................................... 130
Figure 5.5. N2 adsorption desorption isotherms of (A) FAU–TOL2D prepared in toluene
from the zeolite gel pre-heated at 90 °C for 2 days, (B) FAU–FOR2D prepared in
formamide from the zeolite gel pre-heated at 90 °C for 2 days, and (C) FAU-FOR4D
prepared in formamide from the zeolite gel pre-heated at 90 °C for 4 days. .............. 132 Figure 5.6. XRD patterns of the nanozeolite-based FCC catalyst samples prepared from the
corresponding 40, 24 and 100 nm nanozeolites: (A) FCC–TOL2D, (B) FCC–FOR2D
and (C) FCC–FOR4D. ................................................................................................ 133 Figure 5.7. SEM image of (A) FCC–FAU–TOL2D, (B) FCC–FAU–FOR2D and (C) FCC–
FAU-FOR4D. ............................................................................................................. 134 Figure 5.8. N2 adsorption desorption isotherms of (A) FCC–FAU–TOL2D, (B) FCC–
FAU–FOR2D and (C) FCC–FAU–FOR4D. .............................................................. 135 Figure 5.9. Relationship between conversion and catalyst-to-oil ratio of different prepared
FCC-samples. .............................................................................................................. 137 Figure 5.10. Correlation of dry gas yield with conversion of different prepared FCC-
samples. ....................................................................................................................... 138
Figure 5.11. Correlation of LPG yield with conversion of different prepared FCC-samples.
.................................................................................................................................... 139 Figure 5.12. Correlation between gasoline yield and conversion of different prepared FCC-
samples. ....................................................................................................................... 140 Figure 5.13. Relationship between gasoline selectivity and conversion of different prepared
FCC-samples. .............................................................................................................. 141 Figure 5.14. Relationship between LCO yield and conversion. ........................................ 142

xiv
Figure 5.15. Relation between HCO yield and conversion of different prepared FCC-
samples. ...................................................................................................................... 143
Figure 5.16. Relationship between coke yield and conversion of different prepared FCC-
samples. ...................................................................................................................... 144
Figure 6.1. XRD patterns of Fe2Ni-MIL-88B.H2O (a) and XRD simulation of the Fe3-MIL-
88B (b). ....................................................................................................................... 155 Figure 6.2. UV-Vis spectra of Fe2Ni-MIL-88B.Bp (a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-
MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) Fe2Ni-MIL-88B.H2O (e) and Fe3-MIL-
88B (f) ........................................................................................................................ 157
Figure 6.3. XRD patterns of Fe2Ni-MIL-88B samples, the planes of open phase are in
black, the planes of dense phase are in red and placed in boxes. Fe2Ni-MIL-88B.Bp
(a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) and
Fe2Ni-MIL-88B.H2O (e) ............................................................................................ 159 Figure 6.4. N2 adsorption isotherms at 77 K (A) and pore size distributions (B) of Fe2Ni-
MIL-88B.Bp (a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-
88B.DMF (d) and Fe2Ni-MIL-88B.H2O (e). .............................................................. 162 Figure 6.5. CO2 adsorption isotherms at 273 K of Fe2Ni-MIL-88B.Bp (a), Fe2Ni-MIL-
88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) and Fe2Ni-MIL-
88B.H2O (e) ................................................................................................................ 163 Figure 7.1. UV-Vis spectra of the samples Fe3-NO3-x (A) and Fe3-Cl-x (B) prepared using
Fe(NO3)3.9H2O and FeCl3.6H2O, respectively, at different synthesis times. ............ 202 Figure 7.2. UV-Vis spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) samples prepared
using Fe(NO3)3.9H2O and FeCl3.6H2O, respectively at different synthesis times ..... 204 Figure 7.3. Transmittance FTIR spectra of the samples of Fe3-NO3-x (A) and Fe3-Cl-x (B)
at different synthesis times ......................................................................................... 205 Figure 7.4. Transmittance FTIR spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) at
different synthesis times ............................................................................................. 207 Figure 7.5. Raman spectra of the samples Fe3-NO3-x (A) and Fe3-Cl-x (B) at different
synthesis times ............................................................................................................ 209
Figure 7.6. Raman spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) at different synthesis
times. .......................................................................................................................... 211
Figure 7.7. XRD patterns of Fe3-NO3-x (A) and Fe3-Cl-x (B) at different synthesis times.
(*) MOF-235 phase, (#): MIL-88B phase. ................................................................. 212
Figure 7.8. XRD patterns of Fe2Ni-NO3 (A) and Fe2Ni-Cl (B) at different synthesis time.
(*) MOF-235 phase, (#): MIL-88B phase .................................................................. 213 Figure 7.9. Representative HRTEM and EDS acquiring positions of Fe2Ni-Cl-12h crystal
.................................................................................................................................... 214

xv
Acknowledgement
I would like to thank the Laval Foundation for granting me the scholarship for this
study.
I would like to my deep and sincere gratitude to my supervisor, Professor Trong-On
Do. His wide knowledge and creative thoughts have been of great value for me. His
understanding, encouraging and patience guidance have provided a good basis for the
present thesis. It was a great pleasure to me to conduct this thesis under his supervision.
But it would have never come to such a happy ending if I hadn‟t met the love of my
life Phuong Trinh Nguyen midway of this study. It was only with her that I found the
courgage to go on with the rest of the study. She even gave up on her own academic carrier
to support me. I am indebted to her love forever.
I would like to thank Professor Serge Kaliaguine for giving me the access to his
laboratory where some of my experiments were carried out.
I wish to express my warm and sincere thanks to Dr. Hoang-Vinh Thang, Dr.
Bousselham Echchahed and my colleagues Minh-Hao Pham and Dinh-Cao Thang who
gave me invaluable thoughtful insights, advice, support, discussions and encouragements.
Special gratitude is also given to all the professors, staff and graduate students of the
Department of Chemical Engineering from Laval University for their great assistance and
cooperation.

xvi
Preface
This thesis documents our study in two most interesting nanoporous materials:
zeolites and metal-organic framework (MOF).
As one of the most important nanoporous materials, zeolites have been widely used
in catalysis, adsorption and ion exchange. Since their industrial introduction in 1954, the
annual market for zeolites has grown to 1.7 million tons. The reason zeolites can “enjoy”
such a great success is largely due to their interesting features: (i) high surface area, (ii)
uniform pore size structure, (iii) controllable acidity/basicity, and (iv) high
hydrothermostability. Inspired by zeolites, there are continuous attempts to search for other
materials which can duplicate zeolites‟ features, hence their name zeotypes. These attempts
have found some interesting zeotypes, however even AlPO4, which is regarded as one of
the best zeotypes, yet falls short on catalytic activity due to its neutral nature. That is why
when it comes to nanoporous materials, zeolites are still the first choice for researchers.
Nevertheless it is not to say that zeolites have no drawbacks. In fact due to the pore size
constraints, the use of zeolites are less effective when large molecules are involved.
Mesoporous materials can cope with large molecules but their amorphous nature leads to
their low hydrothermostablity and low catalytic activity. So the challenge for zeolite
science nowadays is to expand the application of zeolites to include large and bulky
molecules.
Most of new materials can be found with inspiration from nature as in case of
zeolite: with the knowledge of natural zeolites new successful methods to synthesize
zeolites were proposed. Rarely a new material comes out of solely intellectual vigor
without any precedent clues in nature. And the discovery of MOF was done in that unusual
fashion. While zeolites are solely inorganic materials, MOFs are “ambitious” ones
combining the whole two main categories of chemistry into crystalline structures: organic
entities connecting to each other via inorganic metal clusters. The resulting products are
huge and growing collections of MOFs with uniform nanoporous structure, extremely high
surface area. With MOFs, the nanoporous material researchers can have both organic part
and inorganic part to tinker with. Versatility and flexibility are the key and attractive
features of MOFs, as the choice of metal cluster and organic entities is almost infinite.

xvii
Researches on MOF are explosively growing, but there are still many problems in the
synthesis of MOF needed to be addressed: how the MOF forms, how to control porosity
etc.
In this thesis, we look at both zeolites and MOFs and choose to solve some of the
most pressing issues in these great materials. The thesis is built largely on our published or
submitted papers in recent years. The first author of these papers is also the author of this
thesis.
The thesis starts with an introduction in chapter 1 and then chapter 2 is an overview
of the techniques used in our study. In chapter 3, pulished in Journal of the American
Chemical Society 2007, 129 (13), 3810-3811, we suggest an approach to overcome the
pore size limit of zeolites: reducing zeolite crystal size from microns down to tens of
nanometers thus gaining more exposure of active sites to large molecules. Synthesis of
nanozeolites is not an easy task, without protection nanozeolites tend to aggregate or
dissolve to form large and stable crystals. We devise a new method to obtain highly
uniform nanozeolites with the hydrophobic surface and controlled crystal size. The main
distinction in our approaches is that an organic solvent is used as a medium for the
crystallization instead of water. The zeolite precursors (nanoslabs) are functionalized with
organic silane groups thus become hydrophobic and able to be well dispersed in the organic
solvent. Because the crystallization occurs in the organic phase and the zeolite precursors
are protected by functional groups. The aggregation can be avoided, hence, resulting in
small and uniform nanozeolites with the hydrophobic external surface. Chapter 4, published
in Microporous and Mesoporous Materials 2009, 120 (3), 310-316, pushes forward our new
method of synthesis of nanozeolites. Attention was put onto the organic solvent. We found
that depending on the amount of solvent one can have a single phase or two phase system.
The results revealed that the single-phase method allows producing uniform/small
nanosilicalite-1, whereas the two-phase one can bring two separate products: nanosized and
microsized zeolite crystals in organic phase and in aqueous phase, respectively. Chapter 5,
published in Applied Catalysis A: General 2010, 382 (2), 231-239, is our ultimate catalytic
test of our nanozeolite. A series of FCC catalyst containing our nanozeolites with different
sizes were prepared and tested on the gasoil cracking. The catalytic test results confirmed

xviii
our anticipation: the nanozeolites exhibit higher activity, this is due to higher external
surface area and higher number of active sites available.
Chapter 6 and 7 are dedicated to MOF. In chapter 6, Published in Dalton
Transactions 2013, 42 (2), 550-557, we take on one of the well-known MOFs: MIL-88B.
MIL-88B is best known for its structural breathing effect upon adsorption. That is, upon
interaction with various solvent molecules, the framework of MIL-88B can “swell” up and
down greatly and reversibly without breaking structure. However, MIL-88B is not actually
porous since solvent molecules are needed to pack its pores to sustain its swelling, when
solvent is removed, the structure shrinks and its pores are blocked by the charge balancing
anions. To overcome these drawbacks, the original MIL-88B was modified rationally.
Neutral mixed metal cluster was used instead of the single metal cluster which requires
compensating anion. Thus the anion blockage issue is avoided, the new MIL-88B is
genuinely porous upon removal of solvent. Next, taking advantage of the breathing effect,
we use rigid terminal ligands as “pillars” to sustain the structure. Now there is no need of
solvent molecules and the swelling degree of the structure will be determined by the size of
the terminal ligand. This rational approach has allowed us to control the porosity of MIL-
88B at three distinct levels in terms of surface area and pore size. Chapter 7, which was
submmited to CrystEngComm, continues our adventure with MIL-88B structure, we will
delve into very essential problems in the synthesis of MOFs: why just a seemingly trivial
substitution of iron chloride with iron nitrate results in a complete failure in the synthesis;
and how the phase competition wears on to finalize the desired products. It was amazing to
us that we could answer these problems with the knowledge coming from zeolite science. I
propose the established mechanisms of the synthesis of zeolites are very helpful to
understand the surprising mechanism by which chroride ion promotes the formation of
MIL-88B structure. Alike zeolites, the synthesis of MIL-88B starts with the formation of a
kinetically favored phase, and then if the synthesis time suffices, the thermokinetically
stable phase will come out.
Finally chapter 8 will concluded our thesis with some additional recommendations.
Bellow is the list of publications of which the contents are used in this thesis.

xix
1. G.T. Vuong and T.O. Do, A new route for the synthesis of uniform nanozeolites
with hydrophobic external surface in organic solvent medium. Journal of the
American Chemical Society, 2007. 129(13): p. 3810-3811.
2. G.T. Vuong and T.O. Do, Synthesis of silylated nanozeolites in the presence of
organic phase: Two-phase and single-phase methods. Microporous and
Mesoporous Materials, 2009. 120(3): p. 310-316.
3. G.T. Vuong., V.T. Hoang, D.T. Nguyen, and T.O. Do, Synthesis of nanozeolites
and nanozeolite-based FCC catalysts, and their catalytic activity in gas oil
cracking reaction. Applied Catalysis A: General, 2010. 382(2): p. 231-239.
4. G.T. Vuong, M.H. Pham, and T.O. Do, Synthesis and Engineering Porosity of
mixed metal Fe2Ni- MIL-88B Metal-Organic Framework. Dalton Transactions,
2013, 42, 550-557.
5. G.T. Vuong, M.H. Pham, and T.O. Do, Direct Synthesis and Mechanism for the
Formation of Mixed Metal Fe2Ni-MIL-88B. CrystatEngComm, submitted, 2013.


1
Chapter 1. Introduction
1.1. Zeolite
1.1.1. Background
Zeolites (Greek, zein,"to boil", lithos, "a stone") are aluminosilicates that have well-
defined porous structures. The term was originally coined in the 18th
century by a Swedish
mineralogist named Axel Fredrik Cronstedt who observed, upon rapidly heating a natural
mineral that the stones began to dance about as the water evaporated. Using the Greek
words which mean "stone that boils", he called this material as zeolite.[1]
Strictly speaking, zeolites are defined as crystalline microporous aluminosilicates
with pore structures consisting of sharing TO4 tetrahedra, where T is Si or Al. Zeolites can
be described with the following empirical formula:[2]
Mn+
1/n . AlO2- . x SiO2 . yH2O
Where M – counter ion
n – counter ion valence
x – silicon/aluminum ratio
y – content of hydrate water
Owing to the well-defined pore structure, zeolites are also known as "molecular
sieves". The term molecular sieve refers to a particular property of these materials, i.e., the
ability to selectively adsorb molecules based primarily on a size exclusion process. This is
due to a very regular pore structure of molecular dimensions. The maximum size of the
molecular or ionic species that can enter the pores of a zeolite is controlled by the diameter
of the pore channels.

2
1.1.2. Structure
The flexibility of the zeolite Si-O-Si bond explains the fact that about 200 structures
have been determined. Indeed, there is little energetic difference (10-12 kJ/mol) between
these remarkable porous silicates and higher density phases such as quartz. More than 150
zeolite types have been synthesized and 48 naturally occurring zeolites are known.[3]
The structure commission of the International Zeolite Association (IZA) provides
up to date classification by framework type. Each framework is assigned a three-letter code,
recognized by the IUPAC Commission on Zeolite Nomenclature.[3] According to the IZA
structure commission, zeolite frameworks can be thought to consist of finite or infinite (i.e.,
chain- or layer-like) component units. The primary building units are single TO4 tetrahedra.
The finite units which have been found to occur in tetrahedral frameworks are shown in
Figure 1.1. These secondary building units (SBU), which contain up to 16 T-atoms, are
derived assumption that the entire framework is made up of one type of SBU only. A unit
cell always contains an integral number of SBUs. In some instances, combinations of SBUs
have been encountered. However, it should be noted that the SBUs are only theoretical
topological building units and should not be considered to be or equated with species that
may be in the solution/gel during the crystallization of a zeolitic material.[3]
Zeolites can be also classified on grounds of their pore openings and the
dimensionality of their channels. Thus, one distinguishes small pore zeolites (eight-
membered-ring pores), medium pore zeolites formed by ten-membered rings, large pore
zeolites with twelve-membered-ring pores and extra-large pore zeolite category. This
classification simplifies comparisons in terms of adsorptive, molecular sieving and catalytic
properties.

3
Figure 1.1. Secondary building units and their symbols. Number in parenthesis indicates
frequency of occurrence
Two important and industrially relevant structures, MFI and FAU are depicted in
Figure 1.2. The channels in the MFI structure are formed by 5-1 building units linked

4
together. These building units render a framework of zigzag 10-membered ring channel
(5.1 x 5.5 Å) and intersecting straight 10-membered ring channels (5.3 x 5.6 Å).
(a)
(b)
Figure 1.2. (a) MFI and (b) FAU structures
The FAU structure (structure of zeolite Y) is made up of 6-6 SBUs. In addition, it is
possible to consider the sodalite cage, a truncated octahedron that has eight hexagonal and
six square faces, as basic structure of zeolite Y. The FAU structure is formed when half of
the octahedral faces are joined together to form hexagonal prisms. The spherical internal

5
cavity generated when eight sodalite cages are joined is called the -cage (or supercage)
and is about 13 Å in diameter. Entry into the spherical -cage can occur through four
identical openings that are 7.4 Å wide. The dimensions of zeolite Y allow reasonably large
molecules to penetrate the internal pores, since compounds may extend through a prism
into two connecting -cages.
1.1.3. General Properties of Zeolites
Figure 1.3. Inter-conversion of Brönsted and Lewis acid sites
As mentioned above, the presence of Al in the structure of zeolites results in the
formation of anion sites within the framework. Charge neutralization may occur by either
protonation or by interaction with a metal cation or a hydronium ion. Thus, both Brönsted
and Lewis acidities may be present within the zeolite framework. The protonation of the
Al-O-Si oxygen center can result in Brönsted acidity in the zeolites structure. Lewis acidity
is typically related to the compensating metal ions and defects in the aluminosilicate
framework. Brönsted acid sites in zeolites can change into Lewis acid sites through
dehydroxylation on heating.[4]
Although zeolites are usually considered acid catalysts, cation substitution with Rb
and Cs, as well as metal doping, creates a basic zeolite.[5] The presence of heavy metal
cations is believed to increase the negative charge on the aluminum center, which is
transferred to the adjacent oxygen atom, creating a basic site.[6]

6
The hydrophobicity is an important characteristic of zeolites since it can have a
profound influence on their chemical reactivity. Zeolites containing charges are normally
hydrophilic materials that, depending on the framework Si/Al ratio, can be more or less
selective adsorbents for polar or nonpolar molecules. However, silicalite-1 which is a pure
silica zeolite is a highly hydrophobic material. In contrast, FAU zeolite with the Si/A1 ratio
between 2 and 5 is a highly hydrophilic absorbent. It is then clear that the polarity of a
given zeolite could be controlled by controlling the Si/Al ratio by direct synthesis or by
postsynthesis treatments, and this, together with appropriate control of the number of
silanol groups by synthesis or postsynthesis treatments, should make it possible to prepare
zeolite catalysts within a wide range of surface polarities.[7]
1.1.4. Applications
Since their successful introduction as commercial molecular sieves in 1954,
synthetic zeolites have grown to an estimate $1.6-1.7 billion industry of which detergents
represent the largest volume.[8] LTA-type zeolites have been used to substitute phosphate
compounds in the water softening process in laundry. However, the largest market value for
zeolites is in refinery catalysis. FCC (Fluid Catalytic Cracking) catalysts account for more
than 95% of zeolite catalyst consumption and consist of various forms of zeolite Y. MFI-
type zeolites are the second most used catalyst, primarily because they are added to FCC
catalysts for octane number enhancement. Zeolites are also employed in the drying and
purification of natural gas, separation of paraffins and desulfurization processes. Despite
being in a relatively early stage of development, zeolites are also used in fine chemicals
production such as oxidation and acylation.[8]
Zeolite science appears to be a mature science and is still a very dynamic field.
Discoveries of new zeolites continuously open new areas of development. New trends at
the beginning of this century include environmental applications such as De-NOx catalysis
and hydrocarbon storage in vehicles powered with diesel or gasoline engines, and
biopharmaceutical applications. Zeolites can also be used in the nuclear industry for
radioactive waste storage. Applications of zeolite material science still play an important
role in many areas of technology.

7
1.2. Nanozeolites
1.2.1. Background
Nanozeolites are a type of zeolites which have the particle distribution and sizes of
less than 200 nm.[9] Compared to “ordinary” zeolites of which the particle diameters are of
micrometer order, nanozeolites represent very small particle size, the narrowness of their
particle size distributions (often monodisperse) and especially, the fact that they are
composed of discrete particles (single crystal) rather than aggregates.
One of the advantages of nanozeolites is their higher external surface area. The
external surface is of vital important in numerous processes, including adsorption and
catalysis. For example, in the fluidized catalytic cracking (FCC) process, the commercial
catalysts are manufactured by dispersion of 1 micron FAU and MFI zeolites in an
amorphous alumina-silica matrix. For cracking to occur, gasoil molecules must pass
through the matrix and reach the surface of the zeolite crystals. The molecules then diffuse
through the micropores of zeolites until they reach an active site. Due to the zeolite
structure, molecules larger than 7.4 Å cannot reach active sites located inside the zeolites.
This problem can be solved by replacing the micrometer-sized zeolites with the
corresponding nanozeolites. The substitution could lead to the decrease in the diffusional
resistance and the increase in the external surface area, hence raising the number of active
sites available for large molecules.[10-12] Zeolite particles in the 10-100 nm range can
bring in new applications of zeolites. The huge surface areas of the nanosized materials
dictate that many of the atoms are on the surface, thus allowing good “atom economy” in
surface-gas and surface-liquid reactions. Figure 1.4 illustrates the calculated numbers of
atoms on spherical solid nanoparticles (iron) that are surface or bulk (interior) atoms. The
ratio of atoms available on the surface increases as the crystal size decreases. A 20 nm
particle has about 10% atoms present on the surface. This feature demonstrates that it is
necessary to be very small in order to benefit from the atom economy desired.[13]

8
Figure 1.4. Calculated surface to bulk atom ratios for spherical nanocrystals[13]
Besides the improvement on the external surface areas, nanozeolites have been
found to be excellent “building blocks” for constructing structured materials.[14]
Hierarchical porous materials with controlled porosity microstructure are of great interest
for catalysis and separation applications.[15-17] These porous structures can be fabricated
by templated self-assembly of silicalite nanocrystals (nanosilicalite-1) and the structures so
obtained include long zeolite fibers, micro-patterned zeolite films and micro-macroporous
zeolite structures. The use of preformed zeolite nanocrystals for preparation of supported
zeolite films and membranes is one of major applications of nanozeolites. The small size of
nanozeolites offers high homogeneity and intactness of the zeolite layer and reduces the
number of defects in the film, such as crack and pinholes.
1.2.2. Synthesis
1.2.2.1. Principles
A typical synthesis of nanozeolites using this method can be described as follows
(Figure 1.5):

9
- Amorphous reactants containing silica and alumina are mixed together with a
structure directing agent (SDA) source, usually in a basic (high pH) medium, resulting in a
clear solution.
- The aqueous reaction mixture is heated, often (for reaction temperatures around
100 °C) in a sealed autoclave.
- For some time after raising to synthesis temperature, the reactants remain
amorphous.
- After the above “induction period”, crystalline zeolite product can be detected.
- Gradually, essentially all amorphous material is replaced by an approximately
equal mass of zeolite crystals (which are recovered by filtration, washing and drying).
Figure 1.5. Synthesis of nanozeolites from clear solutions [18]
It is clear that a substantial understanding of the mechanism of the formation of
zeolite is necessary for the synthesis of nanozeolite. However, the study on the formation of
zeolites, although dates back to its earliest days,[2] has not reached its conclusion yet.[18-
20] The formation of zeolite is still a developing subject which receives a lot of interest and

10
its study would not only benefit the zeolite science but also contribute to the understanding
of the crystallization and growth of materials. The difficulties in the study of zeolite
synthesis originate from its inherent synthesis conditions which involve high temperature
and closed system. Hence in situ studies often require sophisticated equipment and setup.
However, thanks to the application of new characterization techniques and the rapid
development in computation chemistry in the last two decades, some significant discoveries
have been found, giving us a better picture of zeolite formation mechanism.[21, 22]
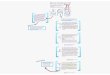
Figure 1.6. Two pathways of zeolite formation
Unlike the crystallization of common solid materials which often involves only two
distinct phases, the liquid phase and the solid phase,[23] the crystallization of zeolite
implicates three phases: liquid phase, amorphous phase (or gel) and crystalline phase of
Aluminosilica gel
Zeolite
Solution pathway Solid pathway

11
zeolite.[2] And it is the determination of the role of the amorphous phase that is the main
issue in the study of the crystallization of zeolite.[18, 19] Based on the role of the
amorphous phase there are two possible pathways in zeolite formation (Figure 1.6): (i) the
solid pathway in which the amorphous phase is a precursor to zeolite, the transformation to
zeolite taking place inside the amorphous phase via structure rearrangement; and (ii) the
solution pathway in which the amorphous phase is merely a nutrient source for crystal
growth by its dissolution to release active aluminosilicate monomer to the solution.
The solid pathway was suggested by Flanigen and Breck as early as 1960[2, 24, 25].
However subsequent studies on synthesis of zeolite A by Kerr and Zhdanov [19, 26, 27] in
the next two decades favored the solution pathway. By the end of 1980s the solution
pathway had been accepted, the nature of gel had very little impact on the final zeolite
structure. The new clear solution synthesis of zeolite and the application of advanced
characterization methods such as NMR and TEM in 1990s saw a numerous reports
emphasizing the role of the intermediate amorphous phase. The clear solution synthesis
method utilizes a clear solution of starting materials, hence facilitating the feasibility of in
situ techniques, allowing better imaging of the nucleation.
For example, in the crystallization of silicalite-1, an all-silica and hydrophobic
zeolite, it was revealed by de Moor et al[28] that hydrophobic silicates and structure
directing agents (SDA) are assembled by hydrophobic interaction, which results in the
formation of primary units (ca. 2.8 nm) in the solution prior to nucleation. Subsequently,
the nucleation occurs via aggregation of primary units. The primary units were also
incorporated directly into the crystalline phase during crystal growth. As the result,
nucleation and subsequent crystal growth mechanisms are described by a cluster
aggregation scheme (Figure 1.7). Further study was carried out by Jacobs et al.[29-33]. The
silica species in an aged clear sol (which crystallizes silicalite upon heating) were extracted.
The resulting powder was characterized by a wide variety of methods leading to the
identification of constituent "nanoslabs" having dimensions 1.3 × 4.0 × 4.0 nm and having
the MFI structure with nine intersections per particle, each constituent unit containing a
TPA cation (Figure 1.8a). Aggregation of precursor units leads to larger particles
measuring up to 15.6 × 8 × 8 nm and eventually to the crystalline colloidal MFI-type

12
material which forms the final product of the synthesis (Figure 1.8b). Hence, the authors
proposed that the formation of the final silicalite-1 crystals was resulted from stacking the
nanoslabs.
Figure 1.7. A scheme for the crystallization mechanism of silicalite-1[28]
Figure 1.8. The "nanoslab" hypothesis: (a) the precursor unit containing one TPA cation
and (b) schematic representation of nanoslab formation by aggregation of precursor units,
as determined by XRS and GPC [32]

13
Figure 1.9. Nucleation and growth model of zeolite A and zeolite Y as represented by
TEM [34, 35]

14
In the crystallization of hydrophilic zeolites (low Si/Al zeolites) using clear solution
method, the presence of nanosized amorphous gel particles in the pre-crystallized solution
was also found.[34, 35] From the TEM observations, Mintova et al[34, 35] revealed that,
these amorphous gel particles have different sizes, depending on the starting materials and
the zeolite structure. The particles in the synthesis of nanozeolite A was about 5 nm
whereas, those in the synthesis of nanozeolite Y was 25-35 nm. The authors proposed that
the mechanism involved the aggregation of these particles (Figure 1.9).
The nucleation and growth of zeolite can be slowed down by lowering synthesis
temperature thus allowing detailed observation. In a synthesis of silicalite-1 at room
temperature that lasted over a year by Tsapatsis et al and other groups, the evolution of the
intermediate gel from the starting clear solution was well documented.[36] The authors
found that 5 – 10 nm nanoparticle gels, although appearing at early stages[37], are subject
to slow structure rearrangement steps before reaching a critical precursor state that allows
their aggregation to form zeolite nanoparticle (Figure 1.10). Model calculations based on
the proposed mechanism are in good agreement with observation data.[38, 39]
Figure 1.10. Schematic illustration of the mechanism proposed for nanoparticle evolution
and crystal growth by aggregation.
These studies using clear solution synthesis hence emphasize the important role of
the intermediate amorphous phase, implying the preference of the solid pathway. However,
even in the clear solution synthesis, evidence for the solution pathway was also found.[40-
42]. In the synthesis of zeolite A from clear solution, Bronić et al[42] found that the zeolite

15
formation is similar to the synthesis involving heterogeneous aluminosilicate gel, namely
by (i) precipitation of an amorphous aluminosilicate gel precursor, (ii) formation of the
particles of quasi-crystalline phase (nuclei) inside the gel matrix, (iii) „„releasing‟‟ of the
nuclei from the gel matrix during its dissolution (autocatalytic nucleation) and (iv) growth
of the nuclei (crystals) from the liquid phase. The remark for the clear solution synthesis is
the need of high temperature (60 oC) to induce the formation of the gel while in the
conventional synthesis, the gel comes readily at ambient temperature.
In summary, the two pathways: solution and solid ones are likely the two extremes
of the formation of zeolite. Indeed, the formation of zeolite would always involve the
cooperation of these both pathways, however, in certain conditions that one pathway will
dominate over the other one. The conventional synthesis of micron-sized zeolite in which
the amorphous phase forms fast at large quantity would favor the solution pathway while
the clear solution synthesis would prefer the solid pathway.
Generally in the synthesis of nanoparticle materials, the crucial step is to intercept
the crystal growth of the particle right after the end of the nucleation stage, thus allowing
the control of the desired particle size and shape. For nanozeolite synthesis, the dominate
formation pathways are very important because it is the factor to determine whether it is
possible to effectively intercept the growth process. By its nature due to the presence of
large gel phase in the conventional synthesis, there is no clear separation between the
nucleation and the growth, in fact both processes wear on in parallel. It is possible to
control the growth process in this case but the undesired impact is that the nucleation is
affected also. Keep in mind that the formation of zeolite is very sensitive to the nucleation
step, the interception of zeolite growth would even prevent the formation of zeolite,
yielding amorphous products. In contrast the clear solution synthesis allows separated
nucleation and growth, hence the control of the growth process would be possible without
disturbing the nucleation process. A number of nanozeolites with different structures have
been synthesized such as FAU, MFI, LTA, MOR… Most of them were prepared using
clear solutions or gels, however, other methods such as confined space synthesis and
synthesis using growth inhibitor have been found to be useful to synthesize these materials.

16
1.2.2.2. Synthesis from clear solutions
The synthesis of nanozeolites from clear solutions was first discovered by Shoeman
et al[43] and Verduijn.[44] Since then this method has been widely used in the synthesis of
nanozeolites (Table 1.1).
Table 1.1. Molecular sieve types synthesized in nanosized form, synthesis conditions, and
crystal size[9]
Type Molar composition of the clear synthesis solution Temp, oC
Crystal
size range,
nm
Ref
AEI 1Al2O3:3.16P2O5:3.16(TEA)2O:186H2O 100,150,1
70 120-240 [45]
AEL 1.6i-Pr2NH:1.3-1.73P2O5:1.1Al2O3:35/70H2O:0/0.8-1.2HF 160,200 100-800 [46]
AFI 0.7-1.1(TEA)2O:0.6-1.0Al2O3:1.1P2O5:50H2O 160, 150-
160(mw) 50-300 [47, 48]
AFI 1(TEA)2O:1Al2O3:1.32P2O5:110H2O 90,110,16
0(mw) 80-600 [49]
BEA Al2O3:16-400SiO2:5.16-105(TEA)2O:240-6400H2O 140 10-200 [50]
BEA 0.48Na2O:9TEAOH:0.25Al2O3:25SiO2:295H2O 100 60 [51, 52]
BEA SiO2:0.2(TEA)2O:11.8H2O 100 100 [53]
FAU 5.5Na2O:1.0Al2O3:4.0SiO2:190H2O 60 20-100 [54]
FAU
2.46(TMA)2O:0.04Na2O:Al2O3:3.4SiO2:370H2O
1.576(TMA)2O:0.044Na2O:Al2O3:3.62SiO2:246H2O
100
100
[55]
FAU 3.4SiO2:0.83-1.7Al2O3:2.3(TMA)2O:0.1NaCl:300H2O 100 80 [56]
FAU 4Na2O:0.2Al2O3:1.0SiO2:200H2O 25 100-300 [56]
FAU 1.00Al2O3:4.35SiO2:1.40-3.13(TMA)2O(2OH
-):0-
2.40(TMA)2O(2Br-): 0.048Na2O:249.00H2O
100 32-120 [57]
FAU 2.46(TMA)2O:0.032-
0.43Na2O:1.0Al2O3:3.40SiO2:370H2O:13.6EtOH
100,130,
100+130 75-137 [58, 59]
FAU 5.5 Na2O:Al2O3:10 SiO2:180 H2O:x NaCl 100 80 - 200 [60]
GIS 1Al2O3:4.17SiO2:2.39(TMA)2O:253H2O 100 30-50 [61]
LTA
2.0-2.3(TMA)2O:0.2-0.5Na2O:Al2O3:3.4SiO2:370H2O
1.2(TMA)2O:0.42Na2O:Al2O3:3.62SiO2:246H2O
100
230-240
[43, 55]
LTA 1.12-3.6SiO2:1.0Al2O3:1.5-7(TMA)2O:0.007-0.28NaCl:276-
500H2O 100
50+130-
900 [34, 62]
LTA 6.1-15.8SiO2:Al2O3:17Na2O:0.9-6.5(TMA)2O:389H2O:3iPrO2 80 50-100 [63]
LTA 0.3Na2O:11.25SiO2:1.8Al2O3:13.4(TMA)2O:700H2O 22 40-80 [35]
LTA 0.22Na2O:5.0SiO2:Al2O3:8.0(TMA)2O:400H2O 63 130 [64]
LTL 10-12.5K2O:1.0Al2O3:16-40SiO2:250-450H2O 140-190 30-70 [48]
LTL 10K2O:1Al2O3:20SiO2:400H2O 175 50-60 [65]
LTL 8.0K2O:Al2O3:20SiO2:200H2O 72.5,82.5,
92.5 30-75 [66]
LTL 0.005BaO:0.25K2O:0.08Al2O3:1.0SiO2:15H2O 170 140 [67]
MEL 0.55Na2O:1.26(TBA)2O:10SiO2:150H2O 67.5,90 50-200 [68]
MEL SiO2:0.3TBAOH:4.0EtOH:18H2O 22+100 90 [69]
MEL 0.35TBAOH:1.0TEOS:12H2O 60-90 114 [69]

17
MFI 5-9TPAOH:0-0.3Na2O:25SiO2:0-0.25Al2O3:480-1500H2O 98 130-230 [70]
MFI 1Al2O3:60SiO2:21.4TPAOH:650H2O 170 10-100 [71]
MFI 9TPAOH:0.16Na2O:1Al2O3:50Si:300-495H2O:0/100EtOH 165 15-60 [72]
MFI Al2O3:60SiO2:11TPAOH:900H2O 70,90 10-20 [73]
MFI 0/0.53Na2O:0.62-1.52(TPA)2O:10SiO2:60/143H2O 50,60, 80 25-80 [44]
MFI 9TPAOH:0/0.1Na2O:25SiO2:480/1500H2O:100EtOH 98 95-180 [74]
MFI 0.01-0.443TPAOH:20-80H2O:TEOS 115 >90 [75, 76]
MFI 9TPAOH:25SiO2:480H2O:100EtOH
60,80,60+
100,
80+100
60-80 [77]
MFI 3-13TPAOH:25SiO2:480H2O:100EtOH 95 100 [78]
MFI 3.0/4.5/9.0TPAOH:16NaOH:50SiO2:495/2000H2O:100EtOH 60-98,165 20-1000 [79]
MFI 0.27Na2O:5TPAOH:25SiO2:420H2O 22+230 130-260 [80]
MFI 9TPAOH:25SiO2:0.13Na2O:595H2O:100EtOH 60,100
60-
170,100-
300
[81]
MFI 9TPAOH:25SiO2:0.13Na2O:595H2O:100EtOH 22+60,22
+100
55-
160,70-
230
[82]
MFI 9TPAOH:0.25TiO2:25SiO2:404H2O:100EtOH 100 85 [83]
MFI 0.36TPAOH:0.06TiO2:1.00SiO2:16.2H2O:4EtOH:0.24BuOH 22+175(m
w) <100 [84]
MFI 25 SiO2:3-9TPAOH:480-1500H2O:100EtOH. 100 100 – 200 [85-87]
MFI 20SiO2 : 9TPAOH : 9500H2O 40 100 [88]
MFI 20SiO2 : 7.2TPAOH : 360H2O 92 20 - 50 [89]
MFI 25SiO2 : 9TPAOH : 400H2O 95 100 [90]
MOR 6Na2O:2Al2O3:30SiO2:780H2O+seeds 150 63 [91]
OFF 2.78(TMA)2O:0.47-0.98K2O:0-
0.5Na2O:Al2O3:9.90H2O:91H2O 85 45-60 [44, 92]
OFF 10SiO2:1.0Al2O3:110-220H2O:0.12Na2O:0.5K2O:3-
4.5(TMA)2O 100 30-250 [92, 93]
SOD 14(TMA)2O:0.85Na2O:1.0Al2O3:40SiO2:805H2O 100 37 [43]
ZSM-
22 1.52(TMA)2O:0.53Li2O:0-0.08Na2O:3.4SiO2:315H2O 100 49-108 [61]
Parameters affecting the crystal size
From the perspective of crystallization theory, the crystal size is a function of the
ratio between rate of nucleation and rate of growth.[23] Thus, to obtain nanozeolites, one
should optimize these following conditions: (i) attaining very high nucleation rates and (ii)
providing stabilization of nuclearsized entities. The first condition is controlled by many
parameters such as temperature, alkalinity, aging… whereas the second one depends
chiefly on the role of SDAs. Here is a brief review of these parameters.
- Temperature: a low crystallization temperature (80 – 100oC) is often applied. This
is because temperature raises growth rate more than nucleation rates. However, it should be

18
noted that a too low temperature usually results in poor crystallinity, low efficiency, and
longer crystallization time.
- Aging: the aging of the synthesis mixture at room temperature has significant
influence on the nucleation rates. This is due to the fact that nucleation rate is favored at
room temperature, but the growth rate is negligible, and thus the nuclei prevail until the
temperature is raised.
- Alkalinity: the concentration of OH- ions strongly increases the solubility of
silicate species. In general, smaller zeolite crystals tend to formed at higher alkalinity.[60]
- The concentration of the clear solution strongly affects the degree of saturation of
the system. At lower supersaturation, growth is favored at the expense of nucleation.
Dilution of the solution can cause large crystal to form. Hence a high concentration is a
desired parameter.
- The solubility of the silica source plays an important role in the synthesis of
nanozeolites. Smaller crystals are formed from monomeric silicate solutions than by
dissolution of colloidal silica.
- Metal cations usually facilitate the crystal growth. Hence, they should be present
in the synthesis solution at low concentration. For example, it has been found that sodium is
the growth-limiting nutrient in the formation of Y-type zeolite.[43, 59] The crystallizations
of Y-type and A-type zeolites are very sensitive to sodium content. In some cases, a small
variation in this factor results in the formation of a different crystal phase, for the synthesis
of zeolites with the clear synthesis gel having molar composition of 2.46 (TMA)2O: x
Na2O: 1.0 Al2O3 : 3.40 SiO2 : 370 H2O:13.6 EtOH (0.03 < x < 0.43), the sodium
concentration of the batch is crucial for controlling which zeolite phase crystallizes. Greater
Na2O/Al2O3 ratio (0.43) in the batch favors the formation of zeolite A with a higher yield of
56.5% after a shorter crystallization time. Lower Na2O/Al2O3 ratio (0.03) in the batch
produces smaller zeolite Y crystals with a lower yield of 8.1% after a longer crystallization
time.

19
- Structure directing agent (SDA): SDAs are often quaternary of the type [R4N]+OH
-
(where R is an alkyl group, typically CH3, C2H5, C3H7 or C4H9). The presence of SDA in
the synthesis solution helps assist the formation of desired zeolite structure. Furthermore,
SDA is responsible to the stabilization of silicate subcolloidal particles[94] as well as the
nanozeolites.[95] In the synthesis of nanosilicalite-1 using tetrapropylammonium hydroxide
(TPAOH) as SDA, it was revealed[95] that, there were two different environments for the
TPA+ cation: (i) TPA
+ occluded in the channel intersections and (ii) TPA
+ adsorbed on to
the external surface of the particles. Bulky quaternary ammonium cations adsorbed on to
particle surfaces provide steric stabilization, preventing aggregation upon collision. In an
aqueous medium, zeolite particles will acquire a negative surface charge due to dissociation
of the surface silanol groups. Such a surface charge will cause organic cations in the
surrounding solution to align along the particles‟ surface, creating an electric double layer.
This stabilizing barrier of bulky organic cations restricts the close approach of similar
particles, such that the attractive potential between them is insufficient to cause aggregation
or flocculation.
1.2.2.3. Synthesis using growth inhibitor
The synthesis of nanozeolites using growth inhibitor is in fact derived from the
synthesis from clear solution. In this method, an organic additive other than SDA is
introduced to inhibit the growth process, thus resulting in small zeolite crystals. The
reactivity of the additive and its content in the synthesis mixture are two significant factors.
The addictive should be able to adsorb on to or react with the surface of the silicate
particles, thus, protects them from further aggregation. If the concentration of the additive
is too high, zeolite might not be obtained since there would be not sufficiently free
aluminosilicate species for the formation of zeolite structure. In contrast, if the
concentration is too low, the inhibition effect might be inadequate.
Hosokawa et al[96] revealed that, a nonionic surfactant (polyoxyethylene lauryl
ether, C12E6) and polyethylene glycol (PEG 600) were able to act as the growth inhibitors
in the synthesis of nanosized A-type zeolites. The inhibitor was added with the synthesis
mixture prior to crystallization. TEM investigation showed that the resulting zeolite A was

20
in the form of aggregated 30 – 40 nm particles which was nearly similar to the size of the
aluminosilicate precursor species available in the synthesis gel prior to crystallization. This
observation indicated that the growth of the zeolite crystals should be inhibited.
The introduction of a growth inhibitor can be postponed until viable zeolite
precursors become prevalent. Thus the undesired interference of the inhibitor in the
formation of precursors is avoided. This approach was developed by Naik et al[97] for
preparation of nanosilicalite-1. The procedure involves: (i) prepare a clear solution that is
known to produce colloidal TPA-silicalite upon extended hydrothermal reaction; (ii)
subject the solution to hydrothermal condition but stop before the appearance of colloidal
silicalite; (iii) protect the TPA-silicalite precursor nanoparticles with cationic surfactant
(CTABr) and collect them as flocculated mass; (iv) convert the precursor/surfactant hybrid
into nanocrystals via high temperature steaming. A steaming temperature of 150 oC was
found enough to convert the collected precursor into nanocrystals. The obtained
nanocrystals were smaller than 30 nm. However these particles are hard aggregated and can
not redispersed in water.
Serrano et al[98] reported the use of organosilane as the growth inhibitor.
Organosilane is a good silylating agent which has been widely used for the
functionalization of zeolites. According to the author, MFI and beta zeolites were
successfully synthesized, using phenylaminopropyl-trimethoxysilane (PHAPTMS). The
synthesis is based on reducing the growth of zeolite crystals by silanization of the zeolitic
seeds to hinder their further aggregation. Typically, the synthesis of MFI zeolite is as
follows: a clear solution of TPA-aluminosilicate was produced. The precursor solution was
precrystallized under reflux with stirring (100 rpm) at 90 ºC for 20 h. Then, the zeolite
seeds obtained were functionalized by reaction with PHAPTMS at 90 ºC for 6 h. Finally,
the resulting solution was subjected to hydrothermal treatment at 170 oC for 5 days.
However, as investigated by TEM analysis, the MFI sample obtained consisted of particles
of about 300 – 400 nm which were formed by aggregation of ultra small primary units of
10 nm. Having that large size, the sample was hardly considered as true nanozeolite.

21
1.2.2.4. Confined space synthesis
In the field of zeolite science, the term “confined space” was first used by Jabobsen
et al. [99-102] in 1999 to describe a novel method for zeolites synthesis which allows
preparation of nanosized zeolites crystals with a controlled crystal size distribution. The
principle of confined-space synthesis is to synthesize the zeolite inside the mesoporous of
an inert matrix. The maximum crystal size is limited by the diameter of the mesopores as
shown in Figure 1.11
The zeolite gel is introduced into the mesopores of the matrix by sequential
incipient wetness impregnations the matrix with the gel precursor solution. For the
synthesis of ZSM-5, the carbon black was impregnated to incipient wetness with a clear
solution of TPAOH, H2O, NaOH and ethanol. After aging for 3 h at room temperature, the
carbon black was subjected to hydrothermal treatment at 180 oC for 48 h in an autoclave.
The product was then recovered by calcinations to remove the matrix.[99]
Figure 1.11. Schematic illustration of confined space synthesis.[101]
This method has also been applied to the synthesis of beta, X and A zeolites.[101,
102] Two carbon black matrixes were used with a pore diameter of 31.6 and 45.6 nm,
respectively. Generally, the crystal size distributions of the zeolites obtained were governed

22
by the pore size of the carbon black matrix and were typically in the range 30-45 nm.
Crucial factors in the synthesis are (i) restriction of the crystallization of the zeolite gel
within the pore system of the matrix, which was achieved by the incipient wetness
impregnation method employed to load the mesopores with a synthesis gel, and (ii)
prevention of diffusion of the zeolite gel species from the mesopores, which was ensured
by avoiding direct contact between the impregnated carbon black matrix and the water at
the bottom of the autoclave.[9] Jacobsen et al. later reported that the failure in controlling
these two factors could lead to the formation of mesoporous zeolites rather than
nanozeolites.[103] Although the confined space synthesis by Jacobsen et al. eliminates the
problem of recovering nanocrystals form solution, it has its own drawback. For example the
carbon matrix must have a uniform distribution of mesopores to ensure the size distribution
of the product. Careful procedures were needed to impregnate the synthesis solution just
inside not outside. And finally, the zeolite particles are aggregated and not discrete.
Another type of confined space synthesis has been developed by Wang et al.[104]
The technique works on the thermoreversibility of gelling polymers. These polymer gels
are hydrogels with three-dimensional networks of polymer chains that are cross-linked via
either physical or chemical bonds, and they can entrap a large volume of water. The
interesting gelation behavior of thermoreversible polymer hydrogels is that it is reversibly
responsive to temperature. In particular, the polymers gel at elevated temperatures and turn
back to solution at room temperature. This feature is attractive because the temperature
profile of their solution-gel transition can very nicely fit that of hydrothermal synthesis of
zeolite. The three-dimensional pores of polymer hydrogels can potentially serve as
microreactors or nanoreactors for controlling zeolite growth. The general synthesis
procedure is illustrated in Figure 1.12.[104] A (20 – 180 nm) and X (10 – 100 nm) zeolites
were prepared using this technique, however, the size distributions were broad. In addition,
this method encounters the following difficulties: (i) Gelation of methyl cellulose is
complicated. The thermoreversibility depends strongly on the heating and cooling rates.
Any variation in these rates can lead to different gel properties.[105] (ii) It is difficult for
the gelation of methyl cellulose to occur in the presence of zeolite synthesis gel since the
high basic content of the gel facilitates the solubility of methyl cellulose.[106] (iii) The
authors applied slight hydrothermal conditions to prepare A and X zeolites, i.e. at 80oC for

23
2 – 3 h. However, for the synthesis of other zeolite structure, the conditions generally
involve the temperature higher or equal to 100oC and the crystallization time of several
days. It is likely that the gel structure of methyl cellulose would not be able to maintain
under these hard conditions.
Figure 1.12. Schematic representation of synthesis of template-free zeolite nanocrystals by
using in situ thermoreversible polymer hydrogels.[104]

24
Figure 1.13. Schematic representation of revered microemulsion
Finally, it would be worthy of mentioning the synthesis of nanozeolites using
microemulsion. Microemulsions are colloidal „nano-dispersions‟ of water in oil (or oil in
water) stabilized by surfactants.[107] These thermodynamically stable dispersions can be
considered as truly nanoreactors which can be used to carry out chemical reactions and, in
particular, to synthesize nanomaterials. The main idea behind this technique is that by
appropriate control of the synthesis parameters one can use these nanoreactors to produce
tailor-made products down to a nanoscale level with new and special properties (Figure
1.13). Reversed microemulsions have been wildly used to prepare inorganic
nanoparticles.[107, 108] Thus, the application of this technique in the synthesis of
nanozeolites is of interest. The first attempt of using microemulsion to prepare zeolites can
be credited to Dutta et al.[109, 110] The authors reported the synthesis of 1 – 2 m A-type
zeolite and 0.6 m zeolite-type zicophosphate. Nevertheless, since the preparation of
Surfactant
Water droplets
Increase
[water]/[surfactant]

25
zeolites is somewhat different from that of inorganic particles. The technique has the
following difficulties:
(i) The crystallization of zeolites often involves heating at high temperature. Under
these conditions the microemulsion becomes unstable, the nanoreactor effect is not
attained. Manna et al.[111] reported the synthesis of silicalite-1 in a microemulsion of
tetraethylenepentamine–sodium bis(2-ethylhexly) sulfosuccinate (AOT)–water system
containing fluoride ions. At the crystallization temperature of 170 oC, the microemulsion
system was destroyed and turned into bicontinuous emulsion. Hence the resulting silicalite-
1 crystals were twinned and had large diameter of 4 m.
(ii) The interaction between surfactant and aluminosilicate species is complicated
and can affect the stability of microemulsion and the morphology of zeolites. Shantz et
al.[112-114] found that, initially, the microemulsion acts as a confined space, effectively
inhibiting zeolite growth in the early stages of synthesis as compared to bulk syntheses.
However, once the particles reach a critical size, approximately 100 nm, the effect of
surfactant adsorption at the aluminosilicate surface becomes so important that the small
particles formed in the microemulsions aggregate to form large particles.
Chen et al.[115] reported the successful synthesis of 40-80 nm A-type nanozeolite.
To overcome these above problems, the microemulsion system containing the syntheis gel
was crystallized at a low temperature of 75 oC in very short duration less than 60 min. To
facilitate the crystallization, microwave heating was applied instead of conventional
heating. In spite of that, the size distribution is still broad (Figure 1.14).

26
Figure 1.14. Schematic representation of microemulsion-microwave synthetic method[115]
1.2.3. Recent advances in application of nanozeolites
The expected application of nanozeolite is its catalytic activity due to three main
factors. First is the accessibility. The external surface of nanozeolite is higher, hence
exposing catalytically active to large molecule. Second is higher activity. Also increasing
with the external surface is the number of active sites.[116] As the low-coordinated corner
and edge sites are more active, the catalytic activity should increase with decrease in size of
the nanozeolite. And third is the improvement in the diffusion into the internal active sites
of nanozeolites thanks to their smaller size.
Beside direct applications in catalysts, in the last five years there has been a great
interest in the use of nanozeolite to build advanced catalysts. The search for mesoporous
materials with zeolitic walls has received a lot of attention and effort from the microporous
and mesoporous materials community.[117, 118] These hierarchically nanoporous zeolites

27
would combine the hydrothermal stability, catalytic activity of zeolite with the accessibility
of mesopores, thus it is possible to design efficient catalyst for reactions involving large
and bulky molecules. However, the synthesis of these materials is not easy. The straight
forward methods which introduce both zeolite template and mesoporous template do not
work as the mesoprous template and zeolite cancel each other out. In 2006, a report by
Ryoo et al suggested a novel method to synthesize hierarchically nanoporous zeolites.[119,
120] Nanozeolite ZSM-5 was synthesized in such a long duration that affords sufficient
time for the freshly formed nanoparticles to gather into large aggregates. The aggregates
feature the intraparticle mesopores as well as the zeolitic micropore. NH3-TPD and
spectroscopy analyses of the hierarchically nanoporous zeolite showed its acidity as high as
conventional zeolites.[121] Thus as the catalyst for reactions of small reactants and
products such as methanol to olefin/gasoline conversion, it exhibits a catalytic activity on
par with bulk zeolite. However the superior activity of this hierarchically nanoporous
zeolite is revealed when larger molecules that cannot diffuse easily inside bulk zeolites are
introduced. The mesoporous MFI zeolite exhibits much higher catalytic activity and
selectivity in the jasminaldehyde (α-n-amylcinnamaldehyde) synthesis reaction than bulk
zeolite. It also displayed an outstanding catalytic activity in the synthesis of vesidryl
(2‟,4,4‟-trimethoxychalcone).[119] Not only activity but the stability of nanozeolite-based
materials is also improved. Hierarchical structure exhibited remarkably high resistance to
deactivation in catalytic activity of various reactions such as isomerization of 1,2,4-
trimethylbenzene, cumene cracking, and esterification of benzylalcohol with hexanoic acid,
as compared with conventional MFI and mesoporous aluminosilicate MCM-41.[122] Ryoo
et al also showed that nanozeolites are also better support than bulk zeolite or mesoporous
silica.[123] Palladium acetate was immobilized on the mesopore wall of hierarchical
MFI nanozeolite, and tested as a catalyst for Suzuki coupling reaction in water. The
catalyst exhibited very high activity in the coupling of various aryl bromides with
arylboronic acids. Moreover, the catalyst could be recycled without a significant loss
of catalytic activity.[124] There have been reports on the synthesis using MFI, SOD, BEA,
LTA zeolites.[123]
The year 2009 saw another breakthrough in nanozeolite applications.[125, 126] A
special type of nanozeolite, the zeolite nanosheet of ZSM-5 which is composed of only two

28
layers of a microporous channel in the perpendicular direction was successfully
prepared.[125, 126] The author found that, in the case of the methanol-to-hydrocarbon
conversion, the MFI zeolite nanosheet exhibited much longer catalytic lifetime than the
bulk zeolite. This result illustrates the facile diffusion of coke precursors in the internal
acidic catalytic sites out of nanozeolite.

29
1.3. Metal-organic frameworks
1.3.1. Background
The advent of zeolite has inspired researches for zeolite analogs or zeotype. From
the basis of zeolite structure which is built on the oxygen connected tetrahedral TO4 (T =
Si, Al) it is a rational step to propose possible structure which is built on other polyhedra of
other atom other than Si and Al such as octahedral TO6 pentacoordinated TO5, or pyramidal
TO4, TO3 units. Several zeotype structures have been found. These include a large number
of main block phosphates, such as those of gallium, indium, and tin, as well as several
transition metal phosphates, including systems based upon vanadium, molybdenum, cobalt,
and iron.[127, 128] Attempts to preplace the oxygen atom with sulfur, chloride, and
nitroten were also reported. [127]
Figure 1.15. Rational design of zeotype and MOF from the basis of zeolite
But in retrospective, the most radical design of porous materials which can be traced
back to zeolite is metal-organic framework (MOF). By definition, MOF is
a crystalline compound consisting of metal ions or clusters coordinated to often
Replace SiO4 with PO4
Zeotype AlPO4
Replace Al, Si with M
Replace O with L
MOF
Zeolite

30
rigid organic molecules to form one-, two-, or three-dimensional structures.[129, 130] In
the MOF framework, the metal or metal cluster is called the node and the organic molecule
linking these nodes is called linker.[131] Hence, MOF is a completely new redesign of
zeotype structure: replacing both the TO4 and the oxygen atom with the node and linker,
respectively. The design principle of MOF is illustrated in Figure 1.15.
From the point of coordination chemistry, MOF is often called coordination
polymer. In most cases, both of these terms, MOF and coordination polymer can be
interchangeably used. However, their use and scope are still subject to debate.[132] MOF is
often used to imply a 3D crystalline structure while coordination polymer refers to 1D and
2D infinitive array. An IUPAC project was initiated in 2009 to address the terminology
issues in this area. A progress report has been published.[133]
There are some similarities in history of MOF discovery and that of zeolite. Both
were found by accident without deliberate purpose to obtain porous structures. While
zeolite was discovered about 200 years before its full potentials were realized, the time it
took chemists to appreciate the amazing world of MOF is even astonishingly longer. The
first successful synthesis of MOF, the Prussian Blue dates back to 1705 by Diesbach but it
would be more than 270 years later, the structure of Prussian Blue, Fe4[Fe(CN)6]3.xH2O
was resolved.[134] In the intervene and sometime after, some preparations of MOF such as
M(imidazole)2 (M = Ni, Cu, Zn and Ag)[135] and iron(III) dicarboxylate[136] were
reported with suggestion of their polymeric nature. However, the general consensus was
that they are just a kind of coordination compounds with expected and typical properties
such as magnetism.[137] The framework aspect of these compounds which would be later
considered as MOFs received little attention. It was only after 1989 with the landmark
paper of Robson and Hoskin and their subsequent paper in 1990[138, 139] that the real
exciting framework of MOFs was realized.
The breakthrough in the discovery of Robson and Hoskin is that it is possible to
design coordination polymers or MOFs.[140, 141] Applying the idea of Wells[142] which
describes the crystal in terms of network with nodes and linkers, they showed that it is
possible to produce MOF of predetermined topology and pore size and functionality by
rational selection of node and linker and appropriate experiment conditions. Thus the

31
porosity and the catalytic activity of MOF can be engineered. The idea was soon picked up
and developed by several prominent researchers[143-149] making important contributions.
However, it would take another decade since the first report of Robson and Hoskin on
MOFs before the interest in MOFs took off and soon exploded thanks to the two separate
discoveries of HKUST-1 (Cu3(btc)2.3H2O) (btc: benzenetricarboxylic acid) and MOF-5
(ZnO4(bdc)3) (bdc: benzenedicarboxylic acid) reported in Science[150] and Nature[151],
respectively, in 1999.
Figure 1.16. Crystal structure of HKUST-1 (left) and MOF-5 (right)
Before the discoveries of HKUST-1 and MOF-5, despite early promising reports of
new MOF structures[149, 152], the stability and the porosity of MOFs were met with
skepticism because of the weak coordination bonds in MOFs.[132] But the coming of
HKUST-1 and MOF-5 changed everything. MOF-5 exhibits spectacular and unprecedented
specific area of 4400 m2/g[153] which would be easily five times the average specific area
of zeolite and put MOF-5 in the class of ultraporous materials. HKUST-1 displays a much
lower specific area of 690 m2/g which is on par with zeolites but exhibits thermostability up
to 240 oC.[150] In addition catalytic potentials and its flexibility in ligand exchange were
clearly demonstrated. But HKUST-1 and MOF-5 are not only striking in their properties,
what is also startling is their rational design. Both the nodes of HKUST-1 and MOF-5, the
copper cluster and the zinc cluster, have been known in copper acetate and basic zinc

32
acetate, respectively for decades.[154] The beauty of the design of HKUST-1 and MOF-5
is that it was simple and logical: replacing acetic acid with ditopic linker terephtalic (bdc)
or tritopic linker trimesic acid (btc) to obtaine MOF-5 or HKUST-1. In a retrospective view
10 years later, even one of the co-inventors of MOF-5, Michael O‟Keeffe still wondered
why no one had come up with that idea before.[132]
Figure 1.17. Number of metal–organic framework (MOF) structures reported in the
Cambridge Structural Database (CSD) from 1978 through 2006. The bar graph illustrates
the recent dramatic increase in the number of reports, while the inset shows the natural log
of the number of structures as a function of time, indicating the extraordinarily short
doubling time for MOF structures compared to the total number of structures archived in
the database.[129]
Fast forwards to the present, the research of MOF is now at an explosive mode with
an exponential growth in the number of research papers and reviews appearing in literature
(Figure 1.17).[129] Almost every issue of any chemistry related journal features at least one
research paper or review on MOF. Exciting properties of MOF in every aspect seem to be
reported daily and non-stop: catalysis,[155-158] adsorption,[159-165] separations,[166-

33
168] optics,[169] drug delivery,[170] magnetism[171] and luminescence[172]… MOFs
also begin to see their industrial application.[173] Still the chemistry of MOF is not
matured yet, there are lot of wonders left to be found.[174] And even new discoveries of
MOF sometimes could catch their veteran researchers by surprise, raising questions
whether there is a limit to MOF.[175]
1.3.2. Design principles of MOFs
The crystal structure is a complicated result of the connectivity, the geometry, and
the shape and position of its elements. Among these parameters, the connectivity between
the linkers and the nodes is the most important one. This parameter can be described by
using the network concept.
A network (net) is a topology classification of MOFs that can aid the description
and the understanding of MOFs structure. A network is a polymeric collection of
interlinked nodes; each link connects two nodes and each node is linked to three or more
other nodes. A node cannot be connected to only two nodes; in this case it then becomes a
link. Similarly, a link can only connect two nodes; if it connects more than two it is a node.
The network must also have a repeating pattern and thus a finite number of unique nodes
and links.
A MOF structure can be described by a corresponding net which provides with
information on the number of different nodes and there connectivities. Since the net does
not involve the geometry parameters of the structure, it is often much simpler than the
crystal structure it represents while still keeping vital information of how the structure can
be constructed. For example two hypothetical structures are showed in Figure 1.18, which
are clearly different, but in fact they share the same net, a 3-connecting net.[131]

34
Figure 1.18. Two geometrically different but topologically identical nets.
The naming of a net name as a three letter code often refers to the most well-known
structure that it represents. For example the dia net represent the four-connecting
framework of diamond. The sod is that of sodalite zeolite.[176]
In addition to the net concept, to facilitate the design of MOF, Yaghi et al suggested
the use of the concept secondary building unit (SBU).[176, 177] The SBU of a MOF
structure is the abstraction of the repeating metal cluster in the MOF structure, the metal
cluster is regarded merely as a node in the net with its number of connectivities.
For example, the MOF-5 structure is formed by the Zn4O clusters are linked by bdc
linkers. The Zn4O as an SBU can be represented as an octahedron with its 6 vertices being
the 6 connectors. Hence the structure of MOF-5 is the linking of these octohedra (Figure
1.19)

35
Figure 1.19. MOF-5 structure from linking octahedral SBUs
To obtain a desired structure, it is necessary to apply two transformations to its net:
decoration and expansion. Decoration is to replace a vertex by a group of vertices. A
special case of decoration is augmentation; this is a replacement of the vertices of an N-
connected net by a group of N-vertices. Expansion is to increase the spacing between
vertices in a net by using longer links, which in principle means that a bond is replaced by a
sequence of bonds. Both decoration and expansion are the main principles of the reticular
synthesis which have produced series of MOF having the same topology.[178] To
illustrate, the design of MOF-5 from the simple structure of NaCl is explained. NaCl
structure is a 6 connecting framework, by augmentation, the node of NaCl is replaced with
the Zn4O cluster octahedra with the same 6 connectors. Then in the expansion step the
links between these octahedra are increased by replacing the link with bdc, thus the MOF-5
structure is obtained.

36
Figure 1.20. Design of MOF-5 from simple fcc structure
1.3.3. Synthesis
The synthesis of MOF bears some similarities to that of zeolites. Both employ
solvothermal technique. While water is almost the exclusive solvent in the synthesis of
zeolites, the use of other solvents are often encountered in the synthesis of MOF. The
equipment for the synthesis of zeolites thus can be used for the synthesis of MOF without
any modification. In a typical synthesis, a solution containing metal salts and the linkers are
prepared at room temperature, the solution is then transferred into a capped autoclave
which is afterwards heated at determined temperature for certain duration. Solid product is
recovered by filtration or centrifugation. Just like the synthesis of zeolites, the parameters:
solvent, temperature, pH, concentration, synthesis time and even anion types are important
factors affecting the formation of MOF. Eventually, the right synthesis parameters should
favor the formation of the SBUs while maintaining the integrity of the organic linkers.
Solvent: water would be the first to be considered for the synthesis due to its
convenience: high availability, ease of handling and non-toxicity. However, as the organic
linkers often exhibit low solubility in water, other organic solvents such as
dimethylformamide (DMF), diethylformamide (DEF) and alcohol are often used.
Sometimes a cosolvent is necessary to tune the solubility. Solvents have a great impact on
the structure of MOFs. For example, in the synthesis of MOF-5, the SBU Zn4O is very
sensitive to solvents, and it was found that alkyl formamide such as DMF and DEF favor its
formation.[151, 178] That‟s why DMF is often used in the synthesis involving Zn4O.[178,

37
179] In contrast, water is detrimental to Zn4O, even with a trace amount of water in DMF
could render the synthesis of MOF-5 fruitless.[153]
Temperature of the synthesis should be high enough to increase the solubility of the
reactants and accelerate the reaction, however, the temperature should not be too high to
avoid the thermal decomposition of the organic linker. The synthesis temperature can be
elevated to 210 oC as in the synthesis of Cr-MIL-101 of which the formula is
Cr3O(bdc)3F.3H2O.[180] One of the reasons for such a high temperature is the low
solubility of bdc in water which is used as solvent at low temperature. Sometimes from the
same synthesis composition change in temperature can result in different MOF structures.
This is the case when there is a competition between the kinetically favored structure and
the thermodynamically favored one. The two MOFs: MIL-88B and MIL-101 have the same
formula Fe3O(bdc)3Cl.xDMF but different structures.[180, 181] In addition, MIL-88B is
the kinetically favored structure while MIL-101 is thermodynamically favored one. From
the same mixture of FeCl3 and bdc in DMF, one can obtain MIL-88B at temperature less
than 100 oC and MIL-101 at temperature higher than 170
oC.[182]
pH is another important factor to consider. When acidic organic linkers such as
polycarboxylic are used, a basic environment is needed to facilitate the deprotonation of the
linkers, so that they can be assembled into the MOF frameworks. The syntheses of MIL-
88B, MIL-88A structure would demand a basic strength of NaOH to initiate.[183, 184] It
should be noted that the agent used to change the pH does not interfere with the SBU by
directly bonding to it, that‟s why trimethyl amine is a common choice to increase pH due to
its high basicity and low ability in forming a coordination bond to metal.[149, 177] In most
of the syntheses not only the protonation of linkers but the desired SBU only coexist in a
very short pH range, hence control of pH is a vital factor. A classic example of the careful
control of pH is the synthesis of MOF-5 in DMF using Zn(NO3)2.6H2O and bdc without
any additional pH agent.[151] The linker bdc needs to be deprotonated, hence a basic pH is
required, but a strong basic environment does not favor the SBU Zn4O. The use of DMF as
solvent perfectly overcomes this problem. At high temperature, DMF slowly and partly
decomposes to release dimethyl amine, which in return deprotonate bdc.[185, 186] Due to
its slow release, the amine is almost used up by the deprotonation and thus the overall

38
increase in pH is very small and does not have any detrimental impact on the SBU. In this
case the combined factors of both solvent and temperature allow a precise control over pH.
Sometimes the need of deprotonation is outweighed by the need to maintain the integrity of
the synthesis mixture. In the synthesis of MIL-101 as the temperature is as high as 210 oC,
thus the chance is that Cr(III) ion would undergo hydrolysis yielding oxide, to prevent this,
HF is added, reducing the pH, hence preventing the hydrolysis.[180, 184]
Concentration in general affects the speed of the synthesis. Traditionally, the
ultimate goal of the synthesis of MOF is to obtain high quality single crystals for
subsequent structural analysis and characterization. Thus the synthesis is carried at high
dilution so that the crystal growth be controlled. But when high yield is desired over the
crystal quality, one can increase concentration.[130] In some instance increase in
concentration would promote the formation of kinetically favored structure over the
thermodynamically favored one. Fe3O-MIL-101 can be prepared in a diluted solution,
increase in the concentration results in the exclusive formation of MIL-88B.[182, 187]
For thermodynamically stable products, longer crystallization would increase the
crystal quality, however for kinetically favored products, care should be taken to choose the
right crystallization time, to obtain the maximum yield in product with high quality in
crystallinity. Jhung et al observed that the synthesis of MIL-101 should be in one-day
course, upon prolonging to two days, MIL-101 dissolve, giving room for another more
stable MOF: MIL-53 to come.[188]
1.4. Some applications of MOFs
1.4.1. Adsorption
Specific surface areas of MOFs often come at thousands m2/g. In fact, the material
having the highest specific surface area per a mass unit is MOF, up to 6000 - 7000
m2/g.[175, 179, 189] So adsorption is the obvious application of MOFs. Earlier attempts
involved adsorption of hydrogen[190] due to interest in the hydrogen-powered technology
for mobile vehicles.[191] Now adsorption analyses of MOFs have embraced almost every
common gases: N2, CO, CO2, alkanes, alkenes, aromatics, H2O and H2S. [159-165, 192]

39
Adsorption of hydrogen: as an alternative to mobile transportation based on
combustion engine, hydrogen technology is very promising. The engine built on the
hydrogen fuel cell would be completely “green” since it only takes in hydrogen and oxygen
and exhausts water. Oxygen is freely available in air, so the issue for the fuel cell is the
storage of hydrogen. The storage of hydrogen is in fact the biggest problem. Other factors
in hydrogen technology such as the price of the fuel cell and cost and safety requirement of
hydrogen production plants are also important but they are merely cost driven factors and
would be relieved when the fuel cell automobiles are produced at mass scale. But the
storage of hydrogen is a scientific and technological problem. An on-board storage of
hydrogen should allow a running course of 500 km or greater for one charge of hydrogen
while meeting all performance metrics. Unfortunately, this is impossible for current storage
technology, regardless of cost.[191] The U.S. Department of Energy (DOE) target of
hydrogen storage by the year 2017 is 5.5 wt % in gravimetric capacity, 40 g/L of
volumetric capacity at an operating temperature of 40 - 60 oC under a maximum delivery
pressure of 100 atm. The targets are for a complete system, including tank, material, valves,
regulators, piping, mounting brackets, insulation, added cooling capacity, and/or other
balance-of-plant components. So the actual adsorption capacity of the materials should be
greater.[163]
Due to its high porosity, studies of MOFs as hydrogen storage materials have been
extensively carried out. The highest excess hydrogen storage capacity reported so far for
MOFs is 99.5 mg/g at 56 bar and 77 K in NU-100 (NU = North-western University, Figure
1.21), which has a total capacity of 164 mg/g at 77 K and 70 bar.[193] The highest total
hydrogen storage capacity reported is 176 mg/g (excess 86 mg/g) in MOF-210 at 77 K and
80 bar (Figure 1.22).[189]

40
Figure 1.21. Structure of NU-100 and its linker
Figure 1.22. The two different linkers used in MOF-210 (left) and the crystal structure of
MOF-210 displaying its unique topology with two different types of pores (right).

41
Figure 1.23. Excess hydrogen uptake at 77 K versus BET specific surface area (BET ssa)
for various high-porosity MOFs. The symbols denote measurements conducted by different
research groups (circles: at 20 bar by Hirscher et al.[193] triangles: at 60 bar by Kaskel et
al.[194]; squares: saturation values by Yaghi et al.).[195]
A general trend is that there is a linear correlation between the hydrogen uptake at
77 K and the specific area (Figure 1.23).[194, 195] Hence the strategy to improve hydrogen
storage of MOFs is to increase its surface area and pore volume. This can be done by
elongation of the linker as in the case of NU-100 or use of mixed linker as in the case of
MOF-210. In fact at 77 K NU-100, MOF-210 and other MOFs readily exhibits hydrogen
storage capacity greater than the DOE target, however, at room temperature the storage
capacity drops drastically and does not meet the DOE target. This is due to the nature of
physical adsorption of hydrogen on MOFs. The heat of adsorption is a useful parameter
representing the stability of the adsorption of hydrogen on MOFs. The heat of hydrogen
adsorption on MOFs is small and in the range of 5 – 9 kJ/mol.[196] Optimal heat of
adsorption should be about 15 kJ/mol.[197] One could overcome this problem by
introducing chemically active site into MOFs to strengthen the bond between MOF and
hydrogen. The active sites can be the metal site or the function group in the organic linkers.

42
Recent theoretical studies showed that impregnation MOFs with light metal that can form
hydride with H2 can improve greatly the heat of adsorption.[198, 199]
Figure 1.24. MOF-117 structure and comparison of the volumetric CO2 capacity of
crystalline MOF-177 relative to zeolite 13X pellets, MAXSORB carbon powder, and
pressurized CO2.[200]

43
Figure 1.25. ZIF-69 structure
Another application in adsorption is CO2 capture. There is growing concern that
anthropogenic CO2 emissions are contributing to global climate change. Therefore, it is
critical to develop technologies to mitigate this problem. One very promising approach to
reducing CO2 emissions is CO2 capture at a power plant, transport to an injection site, and
sequestration for long-term storage in any of a variety of suitable geologic formations.[201]
Compared with common solid porous materials MOFs exhibit superior CO2 adsorption
capacity. Zeolite 13X and activated carbon MAXSORB are those conventional materials
that exhibit the highest reported gravimetric CO2 capacity of 7.4 mmol/g and 25 mmol/g,
respectively and at 32 – 35 bar.[202, 203] But the gravimetric CO2 capacity of MOF-177 of
33.5 mmol/g readily exceeds these standard materials, having 150% of their capacity.[200]
In terms of volume, one liter of MOF-177 could hold 9 liters of CO2 at ambient
temperature. This was a record for MOF back in 2005, however it was soon surpassed by
an exceptional MOF: the ZIF-69 structure (Figure 1.25) which is composed

44
of tetrahedrally-coordinated Zn ion connected by organic imidazole derivative linkers. One
liter of ZIF-69 can hold 83 liters of CO2.[204]
The selective CO2 adsorption of MOFs can be improved by using metal ion with
high affinity to CO2.[205] Among the family of MOF-74 built on different metal ion: Mg,
Zn, Ni, Co, the Mg-MOF-74 exhibits the highest selectivity in adsorption of CO2 at room
temperature and very low pressure of 0.1 atm. One unit cell of Mg-MOF-74 can hold 12
CO2 molecules, twice as much as the other MOF-74.[206]
Figure 1.26. A portion of the structure of the sodalite-type framework of Cu-BTTri (1)
showing surface functionalization of a coordinatively unsaturated Cu(II) site with
ethylenediamine, followed by attack of an amino group on CO2 .
Functionalization of MOFs with amine group can lead to strong binding of CO2. As
illustrated in Figure 1.26, alkylamine incorporation onto the open metal sites of Cu-BTTri
(BTTri: 1,3,5-benzenetristriazolate) was found to be an effective method for
postsynthetically modifying this MOF to enhance the CO2 binding.[207] A 3.5-fold
increase in gravimetric capacity at 0.15 bar (to ∼ 9.5 wt %) compared with the

45
nonfunctionalized Cu-BTTri framework was achieved. Significantly, the functionalized
framework exhibits a higher uptake of CO2 at very low pressures compared with the
nongrafted material and displays a record isosteric heat of adsorption of 90 kJ/mol.
1.4.2. MOF as catalysts
Catalysis is potentially one of the most important applications of MOF. As is the
case of zeolites, the catalytic applications of MOFs come from the following factors:
catalytic activity of the metal ions, catalytic activity of the organic linkers, and catalyst
support. Moreover some distinctive catalytic properties of zeolites such as shape selectivity,
confinement effect are expected to be also available for MOFs [208].
1.4.2.1 Metal ions as catalytic sites
It is well known that transition metal complexes can be used as active homogeneous
catalysts in selective synthetic routs under mild operating conditions for valuable chemicals
from basic organic precursors. Some of major reactions catalyzed by transition metal
complexes are hydrogenation, oxidation, hydroformylation, carbonylation, carbon-carbon
bond formation reactions. Some of the important commercial applications of homogeneous
catalysis are: hydroformylation of olefins to aldehydes/alcohols, carbonylation of methanol
to acetic acid, synthesis of L-dopa by asymmetric hydrogenation, oxidation of p-xylene to
terephthalic acid, hydrocyanation of butadiene to adiponitrile, ethylene oligomerization etc.
Though these homogeneous catalysts play an extremely important role in highly efficient
processes, yet there are some serious drawbacks, mainly in terms of catalyst-product
separation from the reaction mixture and the reusability of the catalyst.
The use of MOF as alternative catalysts can overcome these shortcoming. In this
approach, the active transitional metals are introduced into the MOF structure, playing the
role of connectors, being coordinated by imidazolate linkers. Thus a heterogeneous catalyst
which emulates the catalytic properties of homogeneous complex is obtained. Compared
with the counterpart homogeneous catalysts, these MOF heterogeneous catalysts provide
obvious advantages: the ease of recovery and the ability of being used repeatedly.
It should be kept in mind that, the MOF structure must be activated in an
appropriate way to make sure that the coordination spheres of the metal ion are not

46
completely blocked by the organic linkers or solvent molecules, so that they are accessible
for the reactant molecules [209]. The use metal complexes in MOF structures as active sites
for catalysis has been reported recently by the group of A. Corma [209, 210].
Figure 1.27. Detail of Pd-MOF, showing the 4-membered and the two 6-membered rings.
3D arrangement of the sodalite cages in sodalite-type frameworks.
They found that, [210] Pd-MOF is a very active catalyst for alcohol oxidation,
Suzuki C-C coupling, and olefin hydrogenation for which several homogeneous Pd
complexes are known to perform well. The Pd-MOF is composed of palladium and 2-
hydroxypyrimidinolate as organic linker, [Pd(2-pymo)2]·3H2O(2-pymo = 2-hydroxy-
pyrimidinolate). This material is topologically related to a 3D cubic sodalite-type
framework, with accessible Pd(II) ions in a square planar coordination [211] as illustrated
in Figure 1.27. Moreover, the regular pore system of the material (accessible through two
different hexagonal windows with free openings of 4.8 and 8.8 Å) conferred potential
shape-selective properties to the material that was demonstrated for the hydrogenation of
olefins with different steric hindrance. The structure of Pd-MOF was found to remain intact
under the reaction conditions tested, allowing recovery and reuse for successive catalytic
cycles.

47
The Corma‟s group also probed the catalytic activity of Co-MOF and Cu-MOF
[209]. Two metal–organic frameworks, [Cu(2-pymo)2] and [Co(PhIM)2] (2-pymo = 2-
hydroxypyrimidinolate; PhIM = phenylimidazolate) as shown in (Figure 1.28), containing
respectively Cu2+
and Co2+
ions and anionic diazaheterocyclic ligands (pyrimidinolate and
phenylimidazolate) as organic linkers, were successfully used for the aerobic oxidation of
tetralin, yielding α-tetralone (T=O) as the main product. Both materials are stable and
recyclable under the reaction conditions. Kinetic studies revealed significant differences
between the two MOFs, as a consequence of the different catalytic behavior of their central
metal ions. [Cu(2-pymo)2] is highly active for the activation of tetralin to produce
tetralinhydroperoxide (T–OOH), and less efficient in reacting the peroxide. Meanwhile, the
use of the cobalt catalyst involves a long induction period for the reaction. However, once
T–OOH is formed, Co2+
rapidly and efficiently transforms this into T=O, with high
tetralone-to-tetralol ratio (T=O/T–OH of ca. 7). As a result of this, a compromise between
the two effects has been reached with a mixture containing 10 wt% Cu-MOF and 90 wt%
Co-MOF, that yields the best performance in terms of activity, selectivity and low level of
hydroperoxides [209].
Figure 1.28. (a) Basic building block of Cu(2-pymo)2 and (b) Diagram of the asymmetric
unit of the Co(PhIM)2 framework
1.4.2.2 Ligands as catalytic sites
Just like their counterpart metal connectors, ligand linkers can play the role of
catalytic sites. The functionalization of the zeolitic imidazolate frameworks (ZIF) a special
(a) (b)

48
type of MOFs illustrates the importance of the organic linkers in catalysis. Basically, ZIFs
are composed of tetrahedrally-coordinated Zn ion connected by organic imidazole
derivative linkers.[204, 212] The interesting in the structure of ZIFs is that it is similar to
zeolitic structures: (i) Im bridges make the M–Im–M angle, close to 145°, which is
coincident with the Si–O–Si angle which is preferred and commonly found in many
zeolites; (ii) the metal ion should have a tetrahedral configuration so that the bond angle
Im-M-Im is of 109.5, equal to that of O-Si-O. Hence there is a perfect analogy between
zeolites and ZIFs, metal ion plays the role of T ion whereas Im is the O bridges. For each
imidazole used as a linker, beside two N atoms forming the bonds, three C-H bonds are left
available for tailoring with functional groups, hence providing the linkers with desired
catalytic properties.
Interestingly, the introduction of active sites into linkers can be done easily by
choosing the imidazole derivatives that already possess the functional groups. For example,
to increase the hydrophilicity of ZIFs, one may want to substitute the H atom at the C-H
bond with an aldehyde group. To achieve that, the use of imidazolecarboxylicaldehyde in
the synthesis of ZIFs is an obvious choice. Furthermore, there are many options for this
imidazolecarboxylicaldehyde: (i) the aldehyde can be attached at one of three positions in
the heterocylic of imidazole, each of these positions has different contribution to the
hydrophilicity of the resulting ZIF (ii) the number of attached aldehyde group can be as
many as three, as the number of aldehyde group attached increases, it is expected that the
hydrophilicity increases. Hence, in this case, the properties of the ZIF material have been
predetermined by picking up the right imidazole derivative, or its properties have been
designed.
Other small functional groups such as -Cl, -Br, -NO2 can be attached with the
linkers using this design method, provided that their attachments with imidazole ring do not
affect the formation of ZIF structures.
However, this method may become difficult to carry out when large functional
groups are desired. This is due to the fact that large functional groups may cause steric
hindrance and change the polarity of the bond N-H in the imidazole ring. Hence, the ZIF

49
structure could not be obtained. In addition, the functional groups may become vulnerable
to chemical conversion under the synthesis conditions.
This drawback can be overcome by using post synthesis treatment methods. The
principal of this method is to introduce functional groups bearing active components onto
the organic linkers in the already-formed ZIF structure. The organic linkers now become
anchoring sites to immobilize the desired functional group. The anchoring can occur via
covalent or noncovalent bonding. When the covalent bond obtained, it can be robust
enough to withstand the harsh conditions of catalytic reactions.
Figure 1.29. Transformation of ZIF-90 (A) by Reduction with NaBH4, and reaction with
ethanolamine to give ZIF-91 (B) and ZIF-92 (C) [213]
An interesting example which illustrates the capacity of immobilization of
functional groups onto ZIF structure has been recently reported [213]. The ZIF-90 structure
which is consisted of tetrahedral Zn(II) joined by imidazolate-2-carboxilyaldehye linkers
was employed to modify with different functional group (Figure 1.29). The anchoring site
of interest is the aldehyde group which is already available in the imidazole linkers. Two
kinds of reactions were applied for the functionalization, which in return, give two different
modified ZIFs from the same starting ZIF-90. When the modification involves a strong

50
reducing agent e.g. NaBH4 the aldehyde group is reduced to the corresponding alcohol
derivative, resulting in a new ZIF-91. If other reactant e.g. ethanolamine is used, the
aldehyde group converts to an imine, giving to a ZIF-92. Remarkably, XRD and BET
analysis of these two functionalized ZIF showed that, the zeolitic structure and the high
specific surface area of ZIF-90 are maintained during the transformation.
1.4.3.3. Support and encapsulator for catalysts
Thanks to their well-defined pore structures and high thermal stability, it would be
logical to use MOFs as catalyst supports. Given that zeolite supports have been widely
employed as an ideal medium for dispersion and stabilization of active metal and metal
oxide nanoparticles [214-216], one may want to try these nanoparticles on MOF supports.
Although having a uniform pore system structure similar to zeolites, the difference of
components in MOF structures (metal ions and organic linkers) would have different effect
on the supported nanoparticles. Because of that, new catalytic properties can be obtained.
An advantage of MOF structures is that their uniform pore systems can selectively
immobilize an individual molecule inside their cages, in this case the molecule is said to be
encapsulated. The encapsulation is possible thanks to the shape selectivity nature and the
confinement effect of zeolitic structure [208]. Hence, if a homogeneous catalyst molecule
can be encapsulated in a MOF structure, the potential applications would be overwhelming.
The encapsulation of a molecule can be carried out in a post synthesis treatment.
The molecule is inserted via diffusion provided that it is small enough to travel through the
pore system of the MOF.
Another approach is to introduce the desired guest molecule during the synthesis of
MOF. Under proper conditions, the obtained MOF can have the guest molecules
encapsulated inside its pore structure.

51
Figure 1.30. (a) Eight-coordinate molecular building block that could be represented as a
tetrahedral building unit, (b) [H2TMPyP]4+
porphyrin, (c) crystal structure of rho-ZMOF
(left), hydrogen atoms omitted for clarity, and schematic presentation of [H2TMPyP]4+
porphyrin ring enclosed in rho-ZMOF R-cage (right, drawn to scale) [217]
Using this approach, Alkordi et al[217] have successfully encapsulated
metalloporphyrins inside a MOF material (Figure 1.30). Metalloporphyrins are very active
homogeneous catalysts which have been used in many reactions such as hydroxylation
epoxidation of hydrocarbons. Many attempts to immobilize metalloporphyrins on
conventional host materials such as metal oxide, mesoporous silica and zeolites have been
made [218-225]. However the catalytic activity of the obtained systems is limited by low
loading, leaching problem as well as the dispersion of metalloporphyrins. To overcome
these limitations the authors propose the use of recently synthesized (indium-imidazoledi-

52
carboxylate)-based rho-MOF (topologically analogous to zeolite RHO) as the host matrix.
Indeed, the large voids inside its periodic R-cages and their anionic nature can afford the
ability for encapsulation of cationic porphyrins. In addition, the synthesis of this porphyrin-
encapsulated MOF is facile, porphyrin, 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin
tetra(p-toluenesulfonate) ([H2TMPyP] [p-tosyl]4), was mixed together with starting
materials In(NO3)3.xH2O and 4,5-imidazoledicarboxylic acid in organic solvent to produce
the desired product. The obtained product showed high catalytic activity in the oxidation of
cyclohexane without any trace of leaching and outperformed the catalytic activity of rho-
MOF impregnated with Mn-metallated porphyrin. These facts clearly suggest the
outstanding property of the porphyrin-encapsulated MOF.

53
References
1 M. E. Davis, R. F. Lobo, Chem. Mater. 1992, 4, 756.
2 D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, Wiley,
London 1973.
3 C. Baerlocher, W. M. Meier, D. H. Olson, Atlas of Zeolite Framework Types,
Elsevier, New York 2001.
4 P. A. Jacobs, Carboniogenic Activity of Zeolites, Elsevier New York 1977.
5 Y. Ono, T. Baba, Catal. Today 1997, 38, 321.
6 M. Huang, A. Adnot, S. Kaliaguine, J. Catal. 1992, 137, 322.
7 A. Corma, J. Catal. 2003, 216, 298.
8 H. International, Zeolites: industry trends and worldwide markets in 2010, Wiley,
New York 2000.
9 L. Tosheva, V. P. Valtchev, Chem. Mater. 2005, 17, 2494.
10 K. Rajagopalan, A. W. Peters, G. C. Edwards, Applied Catalysis 1986, 23, 69.
11 T. Sano, H. Ikeya, T. Kasuno, Z. B. Wang, Y. Kawakami, K. Soga, Zeolites 1997,
19, 80.
12 M. A. Camblor, A. Corma, A. Martínez, F. A. Mocholí, J. P. Pariente, Applied
Catalysis 1989, 55, 65.
13 K. J. Klabunde, J. Stark, O. Koper, C. Mohs, D. G. Park, S. Decker, Y. Jiang, I.
Lagadic, D. Zhang, The Journal of Physical Chemistry 1996, 100, 12142.
14 M. E. Davis, Nature 2002, 417, 813.
15 P. Yang, T. Deng, D. Zhao, P. Feng, D. Pine, B. F. Chmelka, G. M. Whitesides, G.
D. Stucky, Science 1998, 282, 2244.
16 B. T. Holland, C. F. Blanford, A. Stein, Science 1998, 281, 538.
17 O. D. Velev, P. M. Tessier, A. M. Lenhoff, E. W. Kaler, Nature 1999, 401, 548.
18 C. S. Cundy, P. A. Cox, Microporous Mesoporous Mater. 2005, 82, 1.
19 C. S. Cundy, P. A. Cox, Chem. Rev. 2003, 103, 663.
20 R. A. van Santen, Nature 2006, 444, 46.
21 A. Aerts, C. E. A. Kirschhock, J. A. Martens, Chem. Soc. Rev. 2010, 39, 4626.
22 S. Bordiga, F. Bonino, K. P. Lillerud, C. Lamberti, Chem. Soc. Rev. 2010, 39, 4885.
23 J. W. Mullin, Crystallization, Butterworth-Heinemann, Oxford 1993.
24 E. M. Flanigen, D. W. Breck, presented at 137th Meeting of the ACS, Cleveland,
OH, 1960.
25 D. W. Breck, J. Chem. Ed. 1964, 41.
26 G. T. Kerr, J. Phys. Chem. 1966, 70.
27 G. T. Kerr, Zeolites 1989, 9.
28 P.-P. E. A. de Moor, T. P. M. Beelen, B. U. Komanschek, L. W. Beck, P. Wagner,
M. E. Davis, R. A. van Santen, Chemistry – A European Journal 1999, 5, 2083.
29 R. Ravishankar, C. Kirschhock, B. J. Schoeman, P. Vanoppen, P. J. Grobet, S.
Storck, W. F. Maier, J. A. Martens, F. C. De Schryver, P. A. Jacobs, The Journal of
Physical Chemistry B 1998, 102, 2633.
30 R. Ravishankar, C. E. A. Kirschhock, P.-P. Knops-Gerrits, E. J. P. Feijen, P. J.
Grobet, P. Vanoppen, F. C. De Schryver, G. Miehe, H. Fuess, B. J. Schoeman, P. A.
Jacobs, J. A. Martens, The Journal of Physical Chemistry B 1999, 103, 4960.
31 C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs, J. A.
Martens, The Journal of Physical Chemistry B 1999, 103, 4965.

54
32 C. E. A. Kirschhock, R. Ravishankar, L. V. Looveren, P. A. Jacobs, J. A. Martens,
The Journal of Physical Chemistry B 1999, 103, 4972.
33 C. E. A. Kirschhock, R. Ravishankar, P. A. Jacobs, J. A. Martens, The Journal of
Physical Chemistry B 1999, 103, 11021.
34 S. Mintova, N. H. Olson, T. Bein, Angew. Chem., Int. Ed. 1999, 38, 3201.
35 S. Mintova, N. H. Olson, V. Valtchev, T. Bein, Science 1999, 283, 958.
36 T. M. Davis, T. O. Drews, H. Ramanan, C. He, J. Dong, H. Schnablegger, M. A.
Katsoulakis, E. Kokkoli, A. V. McCormick, R. L. Penn, M. Tsapatsis, Nat Mater
2006, 5, 400.
37 A. Rivas-Cardona, M. Chovanetz, D. F. Shantz, Microporous Mesoporous Mater.
2012, 155, 56.
38 G. Xomeritakis, S. Nair, M. Tsapatsis, Microporous Mesoporous Mater. 2000, 38,
61.
39 T. O. Drews, M. Tsapatsis, Microporous Mesoporous Mater. 2007, 101, 97.
40 S. Bosnar, J. Bronic, T. Antonic Jelic, B. Subotic, CrystEngComm 2012, 14, 3069.
41 A. Palcic, J. Bronic, D. Brlek, B. Subotic, CrystEngComm 2011, 13, 1215.
42 J. Bronić, A. Mužic, T. Antonić Jelić, J. Kontrec, B. Subotić, J. Cryst. Growth 2008,
310, 4656.
43 B. J. Schoeman, J. Sterte, J. E. Otterstedt, Zeolites 1994, 14, 110.
44 J. P. Verduijn, WO9308125,, 1993.
45 M. Vilaseca, S. Mintova, K. Karaghiosoff, T. H. Metzger, T. Bein, Appl. Surf. Sci.
2004, 226, 1.
46 G. Zhu, S. Qiu, F. Gao, G. Wu, R. Wang, B. Li, Q. Fang, Y. Li, B. Gao, X. Xu, O.
Terasaki, Microporous Mesoporous Mater. 2001, 50, 129.
47 C. S. Cundy, J. O. Forrest, J. Microporous Mesoporous Mater. 2004, 72, 67.
48 H. Du, M. Fang, W. Xu, X. Meng, W. Pang, J. Mater. Chem. 1997, 7, 551.
49 S. Mintova, S. Mo, T. Bein, Chem. Mater. 1998, 10, 4030.
50 M. A. Camblor, A. Corma, A. Mifsud, J. Pérez-Pariente, S. Valencia, Stud. Surf.
Sci. Catal. 1997, 105, 341.
51 M. A. Camblor, A. Corma, S. Valencia, Microporous Mesoporous Mater. 1998, 25,
59.
52 B. J. Schoeman, E. Babouchkina, S. Mintova, V. P. Valtchev, J. Sterte, J. Porous
Mater. 2001, 8, 13.
53 S. Mintova, M. Reinelt, T. H. Metzger, J. Senker, T. Bein, Chem. Commun. 2003,
326.
54 B. Z. Zhan, M. A. White, M. Lumsden, J. Mueller-Neuhaus, K. N. Robertson, T. S.
Cameron, M. Gharghouri, Chem. Mater. 2002, 14, 3636.
55 S. Mintova, V. Valtchev, Stud. Surf. Sci. Catal. 1999, 125, 141.
56 V. P. Valtchev, K. N. Bozhilov, J. Phys. Chem. B 2004, 108, 15587.
57 B. A. Holmberg, H. Wang, J. M. Norbeck, Y. Yan, Microporous Mesoporous
Mater. 2003, 59, 13.
58 B. A. Holmberg, H. Wang, Y. Yan, Microporous Mesoporous Mater. 2004, 74, 189.
59 Q. Li, D. Creaser, J. Sterte, Chem. Mater. 2002, 14, 1319.
60 T. F. Chaves, H. O. Pastore, D. Cardoso, Microporous Mesoporous Mater. 2012,
161, 67.
61 J. Kecht, B. Mihailova, K. Karaghiosoff, S. Mintova, T. Bein, Langmuir 2004, 20,
5271.

55
62 G. Zhu, S. Qiu, J. Yu, Y. Sakamoto, F. Xiao, R. Xu, O. Terasaki, Chem. Mater.
1998, 10, 1483.
63 Q. Li, D. Creaser, J. Sterte, Stud. Surf. Sci. Catal. 2001, 135, 02.
64 J. Hedlund, B. Schoeman, J. Sterte, Chem. Commun. 1997, 1193.
65 M. Tsapatsis, M. Lovallo, T. Okubo, M. E. Davis, M. Sadakata, Chem. Mater. 1995,
7, 1734.
66 M. H. Anthonis, M. Mertens, J. P. Verduijn, WO9703021, 1997.
67 O. Larlus, V. P. Valtchev, Chem. Mater. 2004, 16, 3381.
68 S. Mintova, N. Petkov, K. Karaghiosoff, T. Bein, Microporous Mesoporous Mater.
2001, 50, 121.
69 J. P. Dong, J. Zou, Y. C. Long, Microporous Mesoporous Mater. 2003, 57, 9.
70 A. E. Persson, B. J. Schoeman, J. Sterte, J. E. Otterstedt, Zeolites 1995, 15, 611.
71 R. Van Grieken, J. L. Sotelo, J. M. Menéndez, J. A. Melero, Microporous
Mesoporous Mater. 2000, 39, 135.
72 W. Song, R. E. Justice, C. A. Jones, V. H. Grassian, S. C. Larsen, Langmuir 2004,
20, 8301.
73 J. Aguado, D. P. Serrano, J. M. Escola, J. M. Rodríguez, Microporous Mesoporous
Mater. 2004, 75, 41.
74 A. E. Persson, B. J. Schoeman, J. Sterte, J. E. Otterstedt, Zeolites 1994, 14, 557.
75 B. J. Schoeman, J. Sterte, KONA 1997, 15, 150.
76 C. S. Tsay, A. S. T. Chiang, Microporous Mesoporous Mater. 1998, 26, 89.
77 Q. Li, D. Creaser, J. Sterte, Microporous Mesoporous Mater. 1999, 31, 141.
78 S. Yang, A. Navrotsky, D. J. Wesolowski, J. A. Pople, Chem. Mater. 2004, 16, 210.
79 W. Song, R. E. Justice, C. A. Jones, V. H. Grassian, S. C. Larsen, Langmuir 2004,
20, 4696.
80 V. P. Valtchev, A. C. Faust, J. Lézervant, Microporous Mesoporous Mater. 2004,
68, 91.
81 Q. Li, B. Mihailova, D. Creaser, J. Sterte, Microporous Mesoporous Mater. 2000,
40, 53.
82 Q. Li, B. Mihailova, D. Creaser, J. Sterte, Microporous Mesoporous Mater. 2001,
43, 51.
83 G. Zhang, J. Sterte, B. Schoeman, J. Chem. Mater. 1997, 9, 210.
84 C. S. Cundy, J. O. Forrest, R. Plaisted, J. Microporous Mesoporous Mater. 2003,
66, 143.
85 B. Tokay, M. Somer, A. Erdem-Şenatalar, F. Schüth, R. W. Thompson,
Microporous Mesoporous Mater. 2009, 118, 143.
86 B. Tokay, O. Karvan, A. Erdem-Şenatalar, Microporous Mesoporous Mater. 2010,
131, 230.
87 B. Tokay, A. Erdem-Şenatalar, Microporous Mesoporous Mater. 2012, 148, 43.
88 S. Kumar, R. L. Penn, M. Tsapatsis, Microporous Mesoporous Mater. 2011, 144,
74.
89 S. A. Pelster, R. Kalamajka, W. Schrader, F. Schüth, Angew. Chem. 2007, 119,
2349.
90 A. Aerts, M. Haouas, T. P. Caremans, L. R. A. Follens, T. S. van Erp, F. Taulelle, J.
Vermant, J. A. Martens, C. E. A. Kirschhock, Chemistry – A European Journal
2010, 16, 2764.

56
91 B. O. Hincapie, L. J. Garces, Q. Zhang, A. Sacco, S. L. Suib, Microporous
Mesoporous Mater. 2004, 67, 19.
92 J. P. Verduijn, WO9703020, 1997.
93 J. Hedlund, E. Kurpan, Stud. Surf. Sci. Catal. 2001, 135, 224.
94 R. K. Iler, The Chemistry of Silica, Wiley, New York 1979.
95 R. Ravishankar, C. Kirschhock, B. J. Schoeman, P. Vanoppen, P. J. Grober, S.
Storck, W. F. Maier, J. A. Martens, F. C. De Schryver, P. A. A. Jacobs, J. Phys.
Chem. B 1998, 102, 2633.
96 H. Hosokawa, K. Oki, Chem. Lett. 2003, 32, 586.
97 S. P. Naik, J. C. Chen, A. S. T. Chiang, Microporous Mesoporous Mater. 2002, 54,
293.
98 D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez, Á. Peral, Chem. Mater.
2006, 18, 2462.
99 I. Schmidt, A. Krogh, K. Wienberg, A. Carlsson, M. Brorson, C. J. H. Jacobsen,
Chem. Commun. 2000, 2157.
100 C. Madsen, C. J. H. Jacobsen, Chem. Commun. 1999, 673.
101 I. Schmidt, A. Boisen, E. Gustavsson, K. Ståhl, S. Pehrson, S. Dahl, A. Carlsson, C.
J. H. Jacobsen, Chem. Mater. 2001, 13, 4416.
102 I. Schmidt, C. Madsen, C. J. H. Jacobsen, Inorg. Chem. 2000, 39, 2279.
103 C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt, A. Carlsson, J. Am. Chem.
Soc. 2000, 122, 7116.
104 H. Wang, B. A. Holmberg, Y. Yan, J. Am. Chem. Soc. 2003, 125, 9928.
105 L. Li, P. M. Thangamathesvaran, C. Y. Yue, K. C. Tam, X. Hu, Y. C. Lam,
Langmuir 2001, 17, 8062.
106 D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht, Comprehensive
Cellulose Chemistry, Vol. 1, Wiley-VCH, New York 2004.
107 M. A. López-Quintela, Current Opinion in Colloid & Interface Science 2003,
8, 137.
108 B. L. Cushing, V. L. Kolesnichenko, C. J. O'Connor, Chem. Rev. 2004, 104, 3893.
109 P. K. Dutta, D. Robins, Langmuir 1991, 7, 1048.
110 P. K. Dutta, M. Jakupca, K. S. N. Reddy, L. Salvati, Nature 1995, 374, 44.
111 A. Manna, B. D. Kulkarni, R. K. Ahedi, A. Bhaumik, A. N. Kotasthane, J. Colloid
Interface Sci. 1999, 213, 405.
112 C. S. Carr, S. Kaskel, D. F. Shantz, Chem. Mater. 2004, 16, 3139.
113 S. Lee, D. F. Shantz, Chem. Commun. 2004, 680.
114 C. S. Carr, D. F. Shantz, Microporous Mesoporous Mater. 2005, 85, 284.
115 Z. Chen, S. Li, Yan, Chem. Mater. 2005, 17, 2262.
116 J. M. Thomas, W. J. Thomas, J. Anderson, M. Boudart, Principles and practice of
heterogeneous catalysis, Vol. 638, VCH New York, 1997.
117 D. T. On, S. Kaliaguine, Angew. Chem., Int. Ed. 2001, 40, 3248.
118 D. T. On, S. Kaliaguine, Angew. Chem., Int. Ed. 2002, 41, 1036.
119 M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi, R. Ryoo, Nat Mater
2006, 5, 718.
120 M. Choi, R. Srivastava, R. Ryoo, Chem. Commun. 2006, 4380.
121 K. Suzuki, Y. Aoyagi, N. Katada, M. Choi, R. Ryoo, M. Niwa, Catal. Today 2008,
132, 38.
122 R. Srivastava, M. Choi, R. Ryoo, Chem. Commun. 2006, 4489.

57
123 D.-H. Lee, M. Choi, B.-W. Yu, R. Ryoo, Chem. Commun. 2009, 74.
124 M.-J. Jin, A. Taher, H.-J. Kang, M. Choi, R. Ryoo, Green Chemistry 2009, 11, 309.
125 M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki, R. Ryoo, Nature 2009, 461,
246.
126 A. Corma, Nature 2009, 461, 182.
127 A. K. Cheetham, G. Férey, T. Loiseau, Angew. Chem. Int. Ed. 1999, 38, 3268.
128 Y. Ma, W. Tong, H. Zhou, S. L. Suib, Microporous Mesoporous Mater. 2000, 37,
243.
129 J. R. Long, O. M. Yaghi, Chem. Soc. Rev. 2009, 38, 1213.
130 J. L. C. Rowsell, O. M. Yaghi, Microporous Mesoporous Mater. 2004, 73, 3.
131 S. R. Batten, S. M. Neville, D. R. Turner, Coordination polymers : design, analysis
and application, Royal Society of Chemistry, 2009.
132 M. O'Keeffe, Chem. Soc. Rev. 2009, 38, 1215.
133 S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L.
Ohrstrom, M. O'Keeffe, M. P. Suh, J. Reedijk, CrystEngComm 2012, 14, 3001.
134 H. J. Buser, D. Schwarzenbach, W. Petter, A. Ludi, Inorg. Chem. 1977, 16, 2704.
135 J. E. Bauman, J. C. Wang, Inorg. Chem. 1964, 3, 368.
136 C. T. Dziobkowski, J. T. Wrobleski, D. B. Brown, Inorg. Chem. 1981, 20, 671.
137 D. M. L. Goodgame, D. J. Williams, R. E. P. Winpenny, Angewandte Chemie
International Edition in English 1987, 26, 1044.
138 B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1989, 111, 5962.
139 B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1990, 112, 1546.
140 S. R. Batten, R. Robson, Angew. Chem. Int. Ed. 1998, 37, 1460.
141 R. Robson, J. Chem. Soc., Dalton Trans. 2000, 3735.
142 A. F. Wells, A. Wells, Three-dimensional nets and polyhedra, Wiley New York,
1977.
143 S. Kitagawa, S. Matsuyama, M. Munakata, T. Emori, J. Chem. Soc., Dalton Trans.
1991, 2869.
144 M. Fujita, Y. J. Kwon, S. Washizu, K. Ogura, J. Am. Chem. Soc. 1994, 116, 1151.
145 L. R. MacGillivray, S. Subramanian, M. J. Zaworotko, J. Chem. Soc., Chem.
Commun. 1994, 1325.
146 L. Carlucci, G. Ciani, D. M. Proserpio, A. Sironi, J. Chem. Soc., Chem. Commun.
1994, 2755.
147 O. M. Yaghi, G. Li, Angewandte Chemie International Edition in English 1995, 34,
207.
148 O. M. Yaghi, G. Li, H. Li, Nature 1995, 378, 703.
149 O. M. Yaghi, H. Li, C. Davis, D. Richardson, T. L. Groy, Acc. Chem. Res. 1998, 31,
474.
150 S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen, I. D. Williams, Science
1999, 283, 1148.
151 H. Li, M. Eddaoudi, M. O'Keeffe, O. M. Yaghi, Nature 1999, 402, 276.
152 H. Li, M. Eddaoudi, T. L. Groy, O. Yaghi, J. Am. Chem. Soc. 1998, 120, 8571.
153 S. S. Kaye, A. Dailly, O. M. Yaghi, J. R. Long, J. Am. Chem. Soc. 2007, 129,
14176.
154 F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced inorganic
chemistry, Vol. 5, Wiley New York, 1988.
155 L. Ma, C. Abney, W. Lin, Chem. Soc. Rev. 2009, 38, 1248.

58
156 A. Corma, H. Garc a, F. X. Llabrés i Xamena, Chem. Rev. 2010, 110, 4606.
157 M. Yoon, R. Srirambalaji, K. Kim, Chem. Rev. 2012, 112, 1196.
158 J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen, J. T. Hupp, Chem. Soc.
Rev. 2009, 38, 1450.
159 J. Graetz, Chem. Soc. Rev. 2009, 38, 73.
160 S. S. Han, J. L. Mendoza-Cortes, W. A. G. Iii, Chem. Soc. Rev. 2009, 38, 1460.
161 J.-R. Li, R. J. Kuppler, H.-C. Zhou, Chem. Soc. Rev. 2009, 38, 1477.
162 L. J. Murray, M. Dinca, J. R. Long, Chem. Soc. Rev. 2009, 38, 1294.
163 M. P. Suh, H. J. Park, T. K. Prasad, D.-W. Lim, Chem. Rev. 2012, 112, 782.
164 K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm,
T.-H. Bae, J. R. Long, Chem. Rev. 2012, 112, 724.
165 H. Wu, Q. Gong, D. H. Olson, J. Li, Chem. Rev. 2012, 112, 836.
166 J.-R. Li, J. Sculley, H.-C. Zhou, Chem. Rev. 2012, 112, 869.
167 D. Zacher, O. Shekhah, C. Woll, R. A. Fischer, Chem. Soc. Rev. 2009, 38, 1418.
168 O. Shekhah, J. Liu, R. A. Fischer, C. Woll, Chem. Soc. Rev. 2011, 40, 1081.
169 C. Wang, T. Zhang, W. Lin, Chem. Rev. 2012, 112, 1084.
170 P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R.
E. Morris, C. Serre, Chem. Rev. 2012, 112, 1232.
171 W. Zhang, R.-G. Xiong, Chem. Rev. 2012, 112, 1163.
172 Y. Cui, Y. Yue, G. Qian, B. Chen, Chem. Rev. 2012, 112, 1126.
173 A. U. Czaja, N. Trukhan, U. Muller, Chem. Soc. Rev. 2009, 38, 1284.
174 New J. Chem. 2010, 34, 2355.
175 O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R.
Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın, J. T. Hupp, J. Am. Chem. Soc. 2012, 134,
15016.
176 M. O‟Keeffe, M. A. Peskov, S. J. Ramsden, O. M. Yaghi, Acc. Chem. Res. 2008,
41, 1782.
177 M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe, O. M.
Yaghi, Acc. Chem. Res. 2001, 34, 319.
178 O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi, J. Kim,
Nature 2003, 423, 705.
179 K. Koh, A. G. Wong-Foy, A. J. Matzger, J. Am. Chem. Soc. 2009, 131, 4184.
180 G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé, I.
Margiolaki, Science 2005, 309, 2040.
181 S. Surble, C. Serre, C. Mellot-Draznieks, F. Millange, G. Ferey, Chem. Commun.
2006, 284.
182 S. Bauer, C. Serre, T. Devic, P. Horcajada, J. r. m. Marrot, G. r. Férey, N. Stock,
Inorg. Chem. 2008, 47, 7568.
183 T. Chalati, P. Horcajada, R. Gref, P. Couvreur, C. Serre, J. Mater. Chem. 2011, 21,
2220.
184 P. F. G. S. C. G. R. C. P. Horcajada-cortes, US Patent 20100226991, 2010.
185 S. Hausdorf, F. Baitalow, J. Seidel, F. O. R. L. Mertens, The Journal of Physical
Chemistry A 2007, 111, 4259.
186 S. Hausdorf, J. r. Wagler, R. Moβig, F. O. R. L. Mertens, The Journal of Physical
Chemistry A 2008, 112, 7567.

59
187 P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont,
M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G.
Férey, C. Serre, J. Am. Chem. Soc. 2011, 133, 17839.
188 N. A. Khan, S. H. Jhung, Crystal Growth & Design 2010, 10, 1860.
189 H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R.
Q. Snurr, M. O'Keeffe, J. Kim, O. M. Yaghi, Science 2010, 329, 424.
190 Dinca, Mircea, J. R. Long, Angew. Chem. Int. Ed. 2008, 47, 6766.
191 S. Satyapal, J. Petrovic, C. Read, G. Thomas, G. Ordaz, Catal. Today 2007, 120,
246.
192 J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard, O. M. Yaghi, Science
2005, 309, 1350.
193 O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G.
Kanatzidis, S. T. Nguyen, R. Q. Snurr, J. T. Hupp, Nat Chem 2010, 2, 944.
194 A. G. Wong-Foy, A. J. Matzger, O. M. Yaghi, J. Am. Chem. Soc. 2006, 128, 3494.
195 B. Panella, M. Hirscher, H. Pütter, U. Müller, Adv. Funct. Mater. 2006, 16, 520.
196 Y. E. Cheon, M. P. Suh, Chem. Commun. 2009, 2296.
197 S. K. Bhatia, A. L. Myers, Langmuir 2006, 22, 1688.
198 S. S. Han, W. A. Goddard, J. Am. Chem. Soc. 2007, 129, 8422.
199 S. S. Han, W. A. Goddard, The Journal of Physical Chemistry C 2008, 112, 13431.
200 A. R. Millward, O. M. Yaghi, J. Am. Chem. Soc. 2005, 127, 17998.
201 J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried, R. D. Srivastava, Int. J.
Greenhouse Gas Ctr. 2008, 2, 9.
202 S. Cavenati, C. A. Grande, A. E. Rodrigues, J. Chem. Eng. Data 2004, 49, 1095.
203 S. Himeno, T. Komatsu, S. Fujita, J. Chem. Eng. Data 2005, 50, 369.
204 R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe, O. M.
Yaghi, Science 2008, 319, 939.
205 J. An, N. L. Rosi, J. Am. Chem. Soc. 2010, 132, 5578.
206 S. R. Caskey, A. G. Wong-Foy, A. J. Matzger, J. Am. Chem. Soc. 2008, 130, 10870.
207 A. Demessence, D. M. D‟Alessandro, M. L. Foo, J. R. Long, J. Am. Chem. Soc.
2009, 131, 8784.
208 A. Corma, Chem. Rev. 1995, 95, 559.
209 F. X. Llabres i Xamena, O. Casanova, R. Galiasso Tailleur, H. Garcia, A. Corma, J.
Catal. 2008, 255, 220.
210 F. X. Llabres i Xamena, A. Abad, A. Corma, H. Garcia, J. Catal. 2007, 250, 294.
211 J. A. R. Navarro, E. Barea, J. M. Salas, N. Masciocchi, S. Galli, A. Sironi, C. O.
Ania, J. B. Parra, Inorg. Chem. 2006, 45, 2397.
212 A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O‟Keeffe, O. M.
Yaghi, Acc. Chem. Res. 2010, 43, 58.
213 W. Morris, C. J. Doonan, H. Furukawa, R. Banerjee, O. M. Yaghi, J. Am. Chem.
Soc. 2008, 130, 12626.
214 Y. Xu, L. Lin, Applied Catalysis A: General 1999, 188, 53.
215 E. P. Reddy, L. Davydov, P. Smirniotis, Applied Catalysis B: Environmental 2003,
42, 1.
216 V. Meille, Applied Catalysis A: General 2006, 315, 1.
217 M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank, M. Eddaoudi, J. Am. Chem. Soc.
2008, 130, 12639.

60
218 W. S. Cardoso, M. S. P. Francisco, R. Landers, Y. Gushikem, Electrochimica Acta
2005, 50, 4378.
219 T. A. Khan, J. A. Hriljac, Inorganica Chimica Acta 1999, 294, 179.
220 Z. Li, C.-G. Xia, X.-M. Zhang, Journal of Molecular Catalysis A: Chemical 2002,
185, 47.
221 H. Nur, H. Hamid, S. Endud, H. Hamdan, Z. Ramli, Materials Chemistry and
Physics 2006, 96, 337.
222 I. L. V. Rosa, C. M. C. P. Manso, O. A. Serra, Y. Iamamoto, Journal of Molecular
Catalysis A: Chemical 2000, 160, 199.
223 F. C. Skrobot, I. L. V. Rosa, A. P. A. Marques, P. R. Martins, J. Rocha, A. A.
Valente, Y. Iamamoto, Journal of Molecular Catalysis A: Chemical 2005, 237, 86.
224 F. S. Vinhado, C. M. C. Prado-Manso, H. C. Sacco, Y. Iamamoto, Journal of
Molecular Catalysis A: Chemical 2001, 174, 279.
225 W. Xu, H. Guo, D. L. Akins, J. Phys. Chem. B 2001, 105, 1543.

61
Chapter 2. Experimental
2.1. Synthesis
A number of methods were studied in this thesis. The preparation of clear gel
solution was the first step of all these methods. The obtained solutions were then processed
in different procedures depending on the method applied. The use of the same starting
solution among these methods can provide the ease of comparison between them (Figure
2.1).
Figure 2.1. Methods studied for the synthesis of nanozeolites
2.1.1. Preparation of clear zeolite gel solution
Silicalite-1
In a typical recipe according to Trong-On Do et al,[1] 14 g of tetrapropylammonium
hydroxide (TPAOH) 20% in water was added to 7.8 g of tetraorthosilicate Si(OC2H5)4. The
mixture was stirred vigorously for 24 h at room temperature. The molar gel composition
was: 2.68 SiO2 : 1 TPAOH : 168 H2O.
FAU zeolites
Clear gel solution
Synthesis in
aqueous media Single phase
synthesis
Two phase
synthesis

62
The preparation of clear synthesis solution for FAU nanozeolites followed Mintova
et al.[2] First a 38.4 g solution of NaOH 0.05 N (Fischer) was diluted with 121.6 g H2O.
Then 52.3 g tetramethylammonium hydroxide solution (Aldrich, 25% in water) and
aluminium isopropoxide, Al(iPr)3, (Aldrich, 98%) were added in that order, and stirred
vigorously until the solution became clear. To this solution 21.66 g of tetraorthosilicate was
added. This clear solution was aged for 3 days under vigorous stirring at room temperature.
The final molar composition was: 2.46 (TMA)2O : 0.032 Na2O : 1 Al2O3 : 3.4 SiO2 : 370
H2O.
2.1.2. Synthesis of nanozeolites using clear gel solution in aqueous
medium (conventional method)
In this method, a Teflon-lined stainless steel autoclave (Figure 2.2) was employed
to perform the crystallization under hydrothermal conditions. Before use, the Teflon beaker
was cleaned by immersing in hydrofluoric acid (HF, 20%) for 24 h, and then washed with
distilled water. This is to remove the crystalline residue on the surface of the Teflon beaker
from previous synthesis. After being filled with the prepared clear solution, the autoclave
was completely sealed and heated in a convection oven at desired temperature.
For the synthesis of silicalite-1, the temperature was 100oC and the crystallization
time was 24 h, whereas, for that of FAU zeolites, the temperature and crystallization time
were 100oC and 6 days, respectively.
The solid product was recovered by centrifugation at the speed of 20.000 rpm for 1
h, washed several times with distilled water, dried over night at 80 oC, and calcined in air at
550 oC for 8 h.
In this thesis the zeolites produced using this method were referred to as reference
samples or nanozeolites synthesized using conventional method.

63
Figure 2.2. Scheme of the autoclave: (1) a cylindrical stainless steel vessel, (2) a Teflon
cylindrical beaker, (3) a flat Teflon cover for closing the Teflon beaker, (4) a flat stainless
steel cover which was tightened up to part (1) by six screws.[3]
2.1.3. Synthesis of nanozeolites in organic medium
2.1.3.1. Synthesis of nanozeolites using single-phase method
The procedure is consisted of the following steps:
(i) Preparing a clear zeolite gel solution. This clear gel was heated at 80ºC for 12 hours
to produce zeolite seeds.
(ii) To this clear zeolite gel solution, an organic solvent containing organosilanes was
added and stirred for several hours at 40-100ºC.

64
(iii) The organic phase containing organosilane-functionalized zeolite seeds was
extracted. This organic phase was transferred into an autoclave and then heated at desired
temperature for crystallization.
(iv) After the crystallization, the resulting nanozeolite product was recovered by
centrifugation and then washed with ethanol and water for several times. The product was
then dried at 80ºC for 24 hours and calcined at 550ºC for 5 hours.
Typically, 10 g of zeolite gel solution was added to 500 ml of a solution of toluene
containing n-butanol (30% wt) and a certain amount of hexadecyltrimethoxysilane. The
organosilane was in a proportion of less than 10% mol in regards to the silica content in the
gel. After vigorous stirring at 60 oC for 48 h a mixture clear to naked eyes was obtained.
This mixture was then transferred to an autoclave for further hydrothermal treatment at
150oC for 3 days and 170
oC for 1 day for faujasite and silicalite-1, respectively.
2.1.3.2. Synthesis of nanozeolites using two-phase method
The procedure is consisted of the following steps
i) Preparing a clear zeolite gel solution. This clear solution was heated at 80 ºC for
12 hours to produce zeolite seeds.
ii) To this clear solution, an organic solvent (such as n-octane, toluene, etc.)
containing oganosilanes (for example, hexadecyltrimethoxysilane) in an organic solvent
was added. The organosilane was in proportion of less than 10% mol with respect to the
silica content in the gel. Since the solvent is insoluble in water, a two-phase system was
obtained and was stirred for 12 hours at 80 ºC.
iii) The two-phase mixture was transferred into an autoclave and then heated at
elevated temperature (100-200 ºC) for crystallization.
iv) After the crystallization, the organic phase containing organosilane-
functionalized nanozeolite was extracted. In the organic phase, the resulting nanozeolite
product was recovered by centrifugation and then washed with ethanol and water for
several times. The product was then dried at 80 ºC for 24 hours and calcined at 550 ºC for 5
hours.

65
For the synthesis of nanosilicalite-1, typically, 21.8 g of clear zeolite gel solution
was heated at 80oC for 12 h, the obtained solution was added with 93 g of a solution of
toluene containing 1.22% wt hexadecyltrimethoxysilane, resulting in a two-phase mixture.
After stirring for 12 hours at 60oC, the organic phase mixture was extracted and transferred
into an autoclave; and heated at 170oC for 24 hours.
2.1.4. Preparation of silica containing nanozeolites
Two types of silica containing zeolites which had the zeolite content of 20 %wt and
50% wt respectively were prepared. These two samples were referred to as nano-faujasite-
20 and nano-faujasite-50, respectively. Typically, for the synthesis of silica containing
20%wt zeolite, 30 g of distilled water was added with 100 g of ethanol (Aldrich 98%)
under stirring. Next, 14.16 g of TEOS (Aldrich, 98%) was added and stirred until the
solution became clear. Subsequently, 1 g of FAU nanozeolite, which was prepared using
conventional method, was added. The obtained mixture was then heated at 80 oC with
reflux under vigorous stirring overnight. The product was separated by filtration and
washed with distilled water for several times, dried overnight at 120 oC, then calcined at
550 oC for 8 h.
2.1.5. Synthesis of MIL-88B metal-organic framework
In a typical synthesis, 0.67 mmol of FeCl3.6H2O 99%, 0.33 mmol of corresponding
Ni(NO3)2.6H2O 97% and 1 mmol of bdc 98% were dissolved in 10 ml of DMF. To this
clear solution, 0.4 mmol of NaOH was added under stirring for 15 min. The mixture was
then transferred into a Teflon-lined autoclave and heated at 100 oC for 15 h. Solid product
was then recovered by filtration and washed several times with DMF. The sample was
treated with water, pyridine (Py), pyrazine (Pz) and 4-,4‟-bipyridine (Bp) to obtain Fe2Ni-
MIL-88B.H2O, Fe2Ni-MIL-88B.Py, Fe2Ni-MIL-88B.Pz and Fe2Ni-MIL-88B.Bp,
respectively (see Supporting Information). For comparison, the single metal Fe3-MIL-
88B.DMF was also prepared using the procedure of Férey et al
For the mechanism investigation of MIL-88B the syntheses have the same molar
ratios with respect to the metal cluster (Fe3O or Fe2NiO). In addition, the molar ratio of bdc
(benezendicarboxylic) over metal cluster is kept stoichiometric (and is of 3). Single metal

66
synthesis: vials of 10 ml DMF solution of 10 mmol of FeCl3.6H2O 99% or Fe(NO3)3.9H2O
98% were added with 10 mmol bdc under stirring. Next 4 ml of NaOH 2M was rapidly
injected with continuous stirring. The vials were then capped and heated at 100 oC for
different times: 0 h (5 min after the addition of NaOH at room temperature), 1h, 2h, 3h, and
12 h. Mixed metal synthesis: vials of 10 ml DMF solution of 3,33 mmol of Ni(NO3)2.6H2O
and 6.67 mmol of FeCl3.6H2O 99% or Fe(NO3)3.9H2O 98% were added with 10 mmol bdc
under stirring. Next 4 ml of NaOH 2M was rapidly injected with continuous stirring. The
vials were then capped and heated at 100 oC for different times: 0 h (5 min after the
addition of NaOH at room temperature), 1h, 2h, 3h, and 12 h. Solids products were
recovered by centrifugation at 5000 rpm for 5 min. The solids were then dried in vacuum
for 24 h at 50 oC. In general, the samples prepared with FeCl3.6H2O yield firm solids.
However, those prepared with Fe(NO3)3.9H2O become thick gel during the heat treatment
and thus, their corresponding solid products are not as firm as the solids from the Cl- based
samples.
2.2. Characterization
2.2.1. FTIR Spectroscopy
Fourier transform infrared spectroscopy (FTIR) is the subset of spectroscopy that
deals with the infrared part of the electromagnetic spectrum; it can be used to identify a
compound and to investigate the composition of a sample. Typically, when a molecule is
exposed to infra-red (IR) radiation, it absorbs specific frequencies of radiation. The
frequencies which are absorbed are dependent upon the functional groups within the
molecule and the symmetry of the molecule. IR radiation can only be absorbed by bonds
within a molecule, if the radiation has exactly the right energy to induce a vibration of the
bond. This is the reason why only specific frequencies are absorbed.
Infrared spectroscopy focuses on the frequency range 400 - 4000 cm-1
, where cm-1
is known as wavenumber (1/wavelength), which is a unit of measure for the frequency. To
generate the infrared spectrum, radiation containing all frequencies in the IR region is
passed through the sample. Those frequencies which are absorbed appear as a decrease in
the detected signal. This information is displayed as a spectrum of percentage transmitted

67
radiation plotted against wavenumber. When determining structure types of zeolites, the
region of 400-1400 cm-1
is interesting. This region contains the framework vibrations of
zeolite structure, and stretching and bending modes of the silica-alumina TO4 tetrahedra.
The framework vibrations of zeolite-type materials can be divided into structure insensitive
and structure sensitive vibrations, as shown in Table 2.1. For the MIL-88B, the region of
400 – 800 cm-1
is important since it includes the vibrations of the metal clusters (Table 2.2).
Table 2.1. Structure insensitive and sensitive framework vibrations of zeolites [4]
Structure insensitive vibrations Wavenumber (cm-1
)
Asymmetric stretching vibrations 1200 – 1000
Symmetric stretching vibrations 850 – 700
Bending vibrations 600 – 400
Structure sensitive vibrations Wavenumber (cm-1
)
Asymmetric stretching vibrations 1050 – 1150
Symmetric stretching vibrations 750 – 820
Double ring vibrations 500 – 650
Pore opening vibrations 300 – 420
Table 2.2. FTIR band assignment in the wavenumber 400 – 800 cm-1
Band (cm-1
) Assignment
750 C-H[5, 6]
720 Fe2NiO[7, 8]
690 C-C[5, 6]
660 OCO[5, 6]
624 Fe3O[9, 10]
550 Fe-O, Ni-O[11]
To measure a sample, 1 mg of the sample was well-mixed with 99 mg of KBr
powder (1 % sample in KBr) by grinding using an agate mortar and pestle. The obtained

68
powder was then crushed in a mechanical die press to form a translucent wafer. Finally, the
wafer was placed in a FTIR spectrometer for measurement. A pure KBr wafer was also
made for the background corrections.
2.2.2. Raman spectroscopy
The Raman scattering technique is a vibrational molecular spectroscopy which
is derived from an inelastic light scattering process. With Raman spectroscopy, a laser
photon is scattered by a sample molecule and loses (or gains) energy during the process.
The amount of energy lost is seen as a change in energy (wavelength) of the
irradiating photon. This energy loss is characteristic for a particular bond in the molecule.
Raman can provide a precise spectral fingerprint, unique to a molecule or an individual
molecular structure. In this respect it is similar to the more commonly found FT-IR
spectroscopy. Raman analysis was carried out with a Horiba U100 Raman spectrometer
using excitation wavelength of 514 nm.
2.2.3. UV-Vis spectroscopy
While IR and Raman spectroscopy techniques allow studying the vibrations of the
atoms and the molecules, the UV-Vis (ultraviolet and visible, with the wavelength ranging
from 200 to 800 nm) spectroscopy provides information about the electron transition inside
them. When continuous radiation strike a material, a portion of the radiation may be
absorbed. If that occurs, the residual or reluctant radiation, when it is passed through a
prism, yields a spectrum with gaps in it, called an absorption spectrum. As a result of
energy absorption, atoms or molecules pass from a state of low energy (the initial, or
ground state) to a state of higher energy (the excited state). Figure 2.3 depicts this
excitation process, which is quantized.

69
Figure 2.3. The excitation process
The electromagnetic radiation that is absorbed has energy exactly equal to the
energy difference between the excited and ground states. In the case of ultraviolet and
visible spectroscopy, the transitions that result in the absorption of electromagnetic
radiation in this region of the spectrum are transitions between electronic energy levels. As
a molecule absorbs energy, an electron is promoted from an occupied orbital to an
unoccupied orbital of greater potential energy. Generally, the most probable transition is
from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular
orbital (LUMO). The energy differences between electronic levels in most molecules vary
from 125 to 650 kJ/mole (kilojoules per mole).
Figure 2.4. Electronic energy levels and transitions.

70
For most molecules, the lowest-energy occupied molecular orbitals are the
orbitals, which correspond to s bonds. The orbitals lie at somewhat higher energy levels,
and orbitals that holdunshared pairs, the nonbonding (n) orbitals, lie at even higher
energies. The unoccupied, or antibonding orbitals ( * and *), are the orbitals of highest
energy. Figure 2.4a shows a typical progression of electronic energy levels. In all
compounds other than alkanes, the electrons may undergo several possible transitions of
different energies. Some of the most important transitions are
Figure 2.4b illustrates these transitions. Clearly, the energy required to bring about
transitions from the highest occupied energy level (HOMO) in the ground state to the
lowest unoccupied energy level (LUMO) is less than the energy required to bring about a
transition from a lower occupied energy level. Thus, in Figure 2.4b an n * transition
would have a lower energy than a * transition. For many purposes, the transition of
lowest energy is the most important. For transition metal complex the UV-Vis spectra can
yield information about the d – d transfer and the charge transfer band. In our study, UV-
Vis analysis was carried out in a Cary 300 instrument, using MgO wafer.

71
2.2.4. Energy-dispersive X-ray spectroscopy
Figure 2.5. Principle of EDX spectroscopy
Energy-dispersive X-ray spectroscopy (EDX) is an analytical technique used for the
elemental analysis or chemical characterization of a sample. When a high-energy beam of
charged particles such as electrons or protons, or a beam of X-rays, is focused into the
sample being studied, the incident beam may excite an electron in an inner shell of the
atoms, ejecting it from the shell while creating an electron hole where the electron was. An
electron from an outer, higher-energy shell then fills the hole, and the energy released from
this may be released in the form of an X-ray. The number and energy of the X-rays emitted
from a specimen can be measured by an energy-dispersive spectrometer. As the energy of
the X-rays is characteristic of the difference in energy between the two shells, and of the
atomic structure of the element from which they were emitted, this allows the elemental
composition of the specimen to be measured. The EDX analysis was carried out using the
Tecnai G2 F20 200 kV scanning transmission electron microscope. The probe diameter is
about 3 nm and the acquiring time was set to 200 s.
2.2.5. X-ray Diffraction (XRD)
X-ray Diffraction (XRD) is a technique in crystallography in which the pattern
produced by the diffraction of X-rays through the closely spaced lattice of atoms in a

72
crystal is recorded and then analyzed to reveal the nature of that lattice. XRD is a powerful
technique for determining zeolite structure. Moreover, sample preparation is relatively
easy, and the test itself is often rapid and non-destructive.
Figure 2.6. Diffraction of X-ray beams on a crystal lattice
The mechanism of XRD was well explained by Bragg in 1913.[12] A crystal can be
considered to be composed of discrete parallel planes or layers of atoms. As the wave
enters the crystal, it will be partially reflected by the first layer of atoms, while the rest of it
will continue through to the second layer, where the process continues. The separately
reflected waves will remain in phase if the difference in the path length of each wave
(2dsin) is equal to an integer multiple of the wavelength (n) (Figure 2.6).
nd sin2 (2.1)
This equation is known as Bragg‟s law, where:
- n is an integer,
- λ is the wavelength of x-rays, and moving electrons, protons and neutrons,
- d is the spacing between the planes in the atomic lattice, and
- θ is the angle between the incident ray and the scattering planes.
Waves that satisfy this condition interfere constructively and result in a reflected
wave of significant intensity, causing a peak in the pattern diffraction.

73
The pattern of powder diffraction peaks can be used to quickly identify materials
(thanks to the zeolite pattern database [13]), and changes in peak width or position can be
used to determine crystal size, crystallinity of zeolites.[12]
The crystallinity of zeolite can be derived from XRD as follow:
(hkl)
(hkl)
Intensity of peak sampleCrystallinity (%) 100
Intensity of peak reference
(2.2)
The crystal size, a is calculated according to Scherrer‟s equation:
cos
91.0a
(2.3)
Where, is the full width at half maximum of the corresponding peaks. However,
as noticed by Jacobsen et al,[14] the large crystals contribute more to the average size
determined by XRD than the small crystals. Hence, crystal sizes determined from XRD of
very small zeolite crystals are not very accurate and should be taken as first approximations
to the true crystal size.
In a typical test, zeolite sample was characterized by powder wide-angle XRD,
recorded on a Siemens D5000 X-ray diffractometer using CuKα radiation (λ = 1.54184 Å).
The samples were scanned over a range of 2 values from 5 to 50o with a scan step size of
0.02o and a scan step time of 1 s.
2.2.6. 29
Si Magic Angle Spinning Nuclear Magnetic Resonance
Spectroscopy (MAS NMR)
The 29
Si MAS NMR spectroscopy now belongs among the most powerful
techniques for the characterization of molecular sieves such as zeolites and related
materials. The basis of this success was the invention of effective line narrowing techniques
and two-dimensional experiments that make the detection of highly resolved solid-state
NMR spectra and the separation of different spectral parameters possible. The technique

74
allows the direct investigation of the framework of zeolites and related materials, of extra-
framework cations and of the different types of hydroxyl groups.[15]
Lippmaa et al were among the first to show that the chemical shifts of the 29
Si in
solid silicates were approximately equal to the chemical shifts of species in solution.
Therefore it is easy to differentiate between Si(OSi)4 (Q4), (HO)Si(OSi)3 (Q
3),
(HO)2Si(OSi)2 (Q2) silicate species because the chemical shifts lie approximately 10 ppm
apart (Q4 at -110 ppm, Q
3 at -100 ppm, Q
2 at -90 ppm etc).[16] The
29Si MAS NMR
technique is also useful when investigating the incorporation of organosilane species.
Exchange of one Si-O bond against a Si-C bond (the transformation of a Q-silicon into a T-
silicon) causes a shift of about 45 ppm and again there is a separation of approximately 10
ppm between the silicon T3 RCSi(OSi)3, T
2 (RC)2(SiOSi)2 and T
1 (RC)3SiOSi. Thus T
3 can
be found close to -110 + 45 = -65 ppm T2 = -100 + 45 = -55 ppm and T
1 at -90 + 45 = 45
ppm.[17]
For measurement, 29
Si MAS NMR spectra were recorded at a frequency of 59.60
MHz using 30 o pulses of 3 μs duration, 2600 scans and 30 s recycle delays at room
temperature on a Bruker ASX 300 spectrometer.
2.2.7. Scanning electron microscope (SEM)
Scanning electron microscope (SEM) is a microscope that uses electrons rather than
light to form an image. As the primary electron beam “scans” across the sample, the
electrons on the surface of the sample are excited. This excitation leads to the emission of
the secondary electron beam from the surface which produces the image. The SEM can
produces images of high resolution, which means that closely spaced features can be
examined at a high magnification. Images obtained from this technique can provide
information about the surface and particle size of the samples. Preparation of the samples is
relatively easy since most SEMs only require the sample to be conductive. In this study, a
JEOL JSM-840 scanning electron microscope operated at 15 kV was used.

75
2.2.8. Transmission Electron Microscope (TEM)
Unlike the SEM technique, where electrons are detected by beam emission,
transmission electron microscopy (TEM) make use of the electron beam that has been
partially transmitted through the very thin (and so semitransparent for electrons) specimen.
This beam is detected to provide the image of the sample. TEM allows investigating the
information about the inner structure of the specimen as well as their particle size.
Generally, the TEM resolution is about an order of magnitude better than the SEM
resolution. All the TEM images reported in this thesis were obtained on a JEOL 2011
transmission electron microscope.
2.2.9. Nitrogen Adsorption/Desorption Isotherms
The isotherms of nitrogen adsorption and desorption at 77K is the technique which
has been often used for surface characterization of zeolites. It can provide information
about: specific surface, external surface, internal surface as well as the diameter of the
mesopores (if available). According to IUPAC, the isotherms are classified into six groups
as shown in Figure 2.7. The isotherms of zeolites fall into the type I group. This group are
distinguished by a plateau which is nearly or quite horizontal, and which may cut the P/Po =
1 axis sharply or may show a “tail” as saturated pressure is approached.[18]
Figure 2.7. Types of sorption isotherms.[18]

76
The specific surface area of the specimen is derived from the isotherm using BET
equation:[19]
00
0 *11
1 p
p
CV
C
CVppV
pp
mm
(2.4)
Where,
- V is the volume adsorbed,
- Vm is the volume adsorbed on a monolayer
- P is the pressure of the gas
- Po is the saturation pressure of adsorbates at the temperature of adsorption
- c is the BET constant, which is expressed by:
RT
EEc L1exp . E1 is the heat of
adsorption for the first layer, and EL is that for the second and higher layers and is equal to
the heat of liquefaction.
Equation (2.4) can be plotted as a straight line with 1/v[(Po/P) − 1] on the y-axis and
P/Po on the x-axis according to experimental results. This plot is called a BET plot. The
linear relationship of this equation is maintained only in the range of 0.05 < P/Po < 0.30.
The value of the slope and the y-intercept of the line are used to calculate the monolayer
adsorbed gas quantity Vm (reduced in STP) and the BET constant c. Thus the specific
surface area S is given by:
201022414
LaV
S mm [m
2/g]
(2.5)
where, am is the average area occupied by a molecule of adsorbate in the completed
monolayer for nitrogen am = 16.2 Ǻ2 and L is the Avogadro constant.

77
The external surface area which is strongly dependent on the particle size of zeolites
is calculated from the isotherm using t-plot method. This method was developed by de Boer
et al.[20, 21] It assumes that during the adsorption when the micropores are already filled-
up, the adsorption proceeds to occur on the external area. This stage of adsorption can be
regarded as an adsorption on a flat surface. Then the adsorption within this pressure region
may be described by a simple linear dependence:
o
extmicro
o P
PtSka
P
Pa .max,
(2.6)
where:
-
oP
Pa : volume adsorbed, reduced to STP,
- amicro,max - adsorption in saturated micropores, corresponding to total volume of
micropores,
- Sext - "external" surface area; here it is the surface area of pores larger than
micropores,
-
oP
Pt - estimated statistical thickness of adsorbed layer, according to Harkins et
al.[22] :
2/1
log034.0
99.13
o
o
P
PP
Pt
- k - coefficient which depends on units used for the values of adsorption a, layer
thickness t and surface area Sext, e.g. for t [nm], S [m2/g] and a [cm
3/g STP] we obtain: k =
0.6489.
The t-plot is illustrated in Figure 2.8.

78
In this study, the nitrogen adsorption/desorption measurements were carried out
using an Omnisorp-100 automatic analyser at -196 oC after degassing about 30 mg of
calcined sample at 200 oC for at least 4 h under vacuum (10
-4 -10
-5 torr).
Figure 2.8. t-plot method.
2.2.10. Cracking reaction
Cracking experiments were performed in an automated fixed-bed microactivity test
(MAT) unit (Zeton Automat IV), which was a modified version of ASTM D 5154. The unit
was equipped with collection systems for gas and liquid products. The distribution of
gaseous products was analyzed by gas chromatographies. The boiling point (bp) range of
the liquid products was determined by simulated distillation gas chromatography.
The catalysts were tested in the MAT unit at 510 °C with a weight hourly space
velocity (WHSV) of 8 h-1
. MAT results reported include conversion, yields of dry gas (H2,
H2S, C1 and C2), liquefied petroleum gas (LPG, i.e., C3-C4), gasoline (> C5, bp up to 215
°C), LCO (bp 215-345 °C), heavy cycle oil (HCO, bp above 345 °C) and coke. Conversion
was determined from the difference between the amount of feed and the amount of
amico,max
a
t
a = k.S.t
a = amicro,max + k.Sext.t

79
unconverted material defined as liquid product boiling above 215 °C (i.e., LCO + HCO).
The same vacuum gaz oil (VGO) was used to all MAT runs
References
1 D. Trong On, S. M. J. Zaidi, S. Kaliaguine, Microporous Mesoporous Mater. 1998,
22, 211.
2 S. Mintova, N. H. Olson, T. Bein, Angew. Chem., Int. Ed. 1999, 38, 3201.
3 V.-T. Hoang, Vol. PhD, Laval University, Quebec 2005.
4 A. Jentys, J. A. Lercher, Elsevier, Amsterdam 2001.
5 J. F. Arenas, J. I. Marcos, Spectrochim. Acta, Part A 1980, 36, 1075.
6 J. F. Arenas, J. I. Marcos, Spectrochim. Acta, Part A 1979, 35, 355.
7 L. Meesuk, U. A. Jayasooriya, R. D. Cannon, Spectrochim. Acta, Part A 1987, 43,
687.
8 R. Wu, U. A. Jayasooriya, R. D. Cannon, Spectrochim. Acta, Part A 2000, 56, 575.
9 M. K. Johnson, D. B. Powell, R. D. Cannon, Spectrochim. Acta, Part A 1981, 37,
995.
10 L. Montri, R. D. Cannon, Spectrochim. Acta, Part A 1985, 41, 643.
11 K. Nakamoto, Infrared spectra of inorganic and coordination compounds, Wiley-
Interscience New York 1986.
12 H. Koningsveld, J. M. Bennett, in Structures and Structure Determination, Vol. 2
(Eds: C. Baerlocher, J. M. Bennett, W. Depmeier, A. N. Fitch, H. Jobic, H.
Koningsveld, W. M. Meier, A. Pfenninger, O. Terasaki), Springer Berlin
Heidelberg, 1999, 1.
13 C. Baerlocher, W. M. Meier, D. H. Olson, Atlas of Zeolite Framework Types,
Elsevier, New York 2001.
14 I. Schmidt, C. Madsen, C. J. H. Jacobsen, Inorg. Chem. 2000, 39, 2279.
15 M. Hunger, E. Brunner, in Characterization I, Vol. 4 (Eds: H. Karge, J. Weitkamp),
Springer Berlin Heidelberg, 2004, 201.
16 D. T. On, S. Kaliaguine, Angew. Chem., Int. Ed. 2002, 41, 1036.
17 A. van Blaaderen, A. Vrij, J. Colloid Interface Sci. 1993, 156, 1.
18 S. Gregg, K. S. W. Sing, 1983.
19 S. Brunauer, P. H. Emmett, E. Teller, J. Am. Chem. Soc. 1938, 60, 309.
20 B. C. Lippens, J. H. de Boer, J. Catal. 1965, 4, 319.
21 B. C. Lippens, B. G. Linsen, J. H. d. Boer, J. Catal. 1964, 3, 32.
22 W. D. Harkins, G. Jura, J. Am. Chem. Soc. 1944, 66, 1366.


81
Chapter 3. A New Route for the Synthesis of Uniform
Nanozeolites with Hydrophobic External Surface in
Organic Solvent Medium
Gia-Thanh Vuong and Trong-On Do *
Department of Chemical Engineering, Laval University, Quebec G1K 7P4, Canada
Published in Journal of the American Chemical Society 2007, 129 (13), 3810-3811
DOI: 10.1021/ja069058p
Résumé
Des nanozéolithes silylées de tailles uniformes ont été synthétisés dans un milieu
organique en présence d‟hexadecytrimethylsilane. Les nanozéolithes silylées présentent en
plus des petits cristaux uniformes, avec une surface externe hydrophobe. Ces nanozéolithes
présentent des applications potentielles en catalyse dans les réactions impliquant de grosses
molécules. Ces applications sont directement liées à leur grande surface externe, à la
réduction de diffusion et à l‟exposition des sites actifs.
Abstract
Uniform silylated nanozeolites were synthesized in organic solvent medium in the
presence of hexadecytrimethylsilane. The resulting zeolites have not only small/uniform
crystal sizes, but also a hydrophobic external surface. These zeolites are considered of
potential for application as catalysts in reactions involving bulky molecules, owing to their
high external surface area, reduced diffusion pathways, and exposed active sites.

82
Nanosized zeolite crystals (nanozeolites) with narrow particle-size distributions and
sizes less than 100 nm have received much attention because of their great potential
applications in catalysis and adsorption. The decrease in the crystal sizes results in high
external surface areas, reduced diffusion path lengths, and more exposed active sites. For
example, small Y-type zeolite crystals have been reported to increase catalytic activity and
improve the selectivity of intermediate cracked products such as gasoline and light gas oil
in catalytic cracking of heavy gas oil. This catalyst exhibited lower deactivation rates since
the coke formation was suppressed.[1] Moreover, nanozeolites can be used as “building
units” for constructing hierarchical materials. Zeolite nanoclusters have been employed for
assembling mesoporous alumosilicates.[2] Materials with semicrystalline zeolitic mesopore
walls[3] and nanozeolite coated mesoporous aluminosilicates[4] were reported by our
group. Recently, zeolites with intracrystal mesopores and strong acidity were also
synthesized.[5] The resulting materials are considered of potential for application in
catalysis and separation, owing to easier transport of guest molecules through the
mesopores and shorter diffusion pathways in the zeolitic walls.
Syntheses of nanozeolites are often carried out in the aqueous phase. During the
crystallization, once the nanozeolite precursors are formed, the aqueous phase acts as an
effective environment for the incorporation of soluble aluminosilicate species and the
aggregation of zeolite crystals. This could lead to the formation of large crystals and
aggregates.[6] Direct synthesis using a clear gel solution of aluminosilicates can also
produce nanozeolites by careful control of the gel composition and crystallizing
conditions.[7] Another method, which is called confined space synthesis, has been
developed for the preparation of nanosized zeolite crystals. The synthesis is conducted
within an inert matrix such as porous carbon matrices,[8] thermoreversible polymer
hydrogels, or microemulsions[9] which provides a steric hindered space for zeolite crystal
growth.
Several synthetic routes have been reported for the preparation of nanocrystalline
zeolites. However, none of these attempts has produced an easy means of controlling the
small size. Furthermore, the external surface of nanocrystalline zeolites is hydrophilic and

83
thereby has mostly silanol groups that limit catalytic reactivity to the internal pore
surface.[6]
Serrano et al.[10] have recently reported the use of organosilane as growth inhibitor.
In this study, MFI and β zeolites were synthesized in the aqueous medium, using
phenylaminopropyl−trimethoxysilane (PHAPTMS). The synthesis is based on reducing the
growth of zeolite crystals by silanization of the zeolitic seeds to hinder their further
aggregation. However, as investigated by TEM analysis, the obtained MFI sample
consisted of particles of about 300−400 nm which were formed by the aggregation of
ultrasmall units of 10 nm. Having that large size, the sample was hardly considered as true
nanozeolite.
Herein, we demonstrate a new route for the synthesis of controlled uniform size
nanozeolites with the hydrophobic external surface. An organic solvent is used as the
medium for crystallization instead of water. The zeolite precursors are functionalized with
organic silane groups. They thus become hydrophobic and highly dispersed in the organic
solvent. Because the crystallization occurs in the organic phase and the zeolite precursors
are protected by functional groups, catastrophic aggregation can be prevented hence,
resulting in small and uniform nanozeolites with hydrophobic external surface.
The MFI and faujasite zeolites were selected to illustrate our approach, because they
are widely recognized for their unique properties as catalysts. As seen in Scheme 3.1, this
approach is simple and was found to be reproducible when using
hexadecyltrimethoxysilane as organosilane agent and the mixture of toluene and n-butanol
as organic medium.

84
Scheme 3.1. Schematic Representation of the Single-Phase Synthesis Method
The XRD patterns of the as-made nanozeolite samples prepared from silylated seeds
are shown in Figure 3.1 Samples prepared by the conventional method in aqueous medium
from the same clear zeolite gel solution without organosilane were used as references. The
XRD pattern of the nanosilicalite-1 sample is identical to that of the reference, indicating
the MFI structure of this nanosilicalite-1 sample. However, there is a clear broadening of
the reflections, which is attributed to small crystals. In addition, no significant peak at 2θ =
20−30° which is characteristic of amorphous phase was observed, indicating a relatively
high crystallinity of this sample. A similar trend was also observed for the nanofaujasite
sample (Figure 3.1B). Furthermore, the FTIR spectra of both as-made nanosilicalite-1 and
nanofaujasite match well with the typical FTIR peaks assigned to silicalite-1 and zeolite Y,
respectively (not shown).[11]

85
Figure 3.1. XRD patterns of the as-made silylated zeolite and zeolite samples prepared
from the same zeolite gel in solvent medium in the presence of organosilane and in aqueous
medium in the absence of organosilane, respectively: (A) silicalite-1; (B) faujasite.
Representative TEM micrographs of the as-made silylated zeolite samples are
shown in Figure 3.2 and exhibit very uniform nanocrystal sizes mostly with spherical and
cubic shaped particles for silicalite-1 and zeolite Y, respectively. The particle size is about
21 nm for silicalite-1 and 27 nm for faujasite. The standard deviation established from the
analysis of more than 1500 particles in representative TEM pictures of each sample showed
a standard deviation of less than 10% (Supporting Information (SI) Table 1). Interestingly,
the samples are composed of discrete particles rather than aggregates. Owing to the
organosilane being grafted on the zeolite precursor, the nanoparticles remain highly
dispersed in the organic medium and protected against drastic aggregation during
crystallization.

86
Figure 3.2. TEM images of the as-made samples: (A) silylated nanosilicalite-1, (B)
silylated nanofaujasite.
The 29
Si MAS NMR spectra of the as-made samples, silicalite-1 and silylated
silicalite-1, prepared from the same zeolite gel solution were investigated (SI Figure 1A).
The NMR spectrum of the as-made silicalite-1 sample shows a main resonance at ca. −110
ppm and a weak resonance peak at ca. −100 ppm which are attributed to Si(OSi)4, Q4 and
Si(OSi)3OH, Q3 species, respectively. For the as-made silylated silicalite sample, only one
resonance peak at ca. −110 ppm attributable to Q4 species was observed; however, an
additional peak at −65 ppm assigned to R-C-Si-(OSi)3 species is present.[11] This
additional peak is the result of the reaction between the silicon in the organosilane and the
silanol groups of zeolite nuclei during the synthesis. This also suggests the silanization on
the external surface of nanosilicalite-1, which acts to heal defect sites (e.g., silanol groups)
in the zeolite surface. Furthermore this calcined sample also shows essentially a single
resonance peak Q4 at ca. −110 ppm (SI Figure 1A). Thus, it can be concluded that the
presence of only one resonance Q4 even after calcination of the silylated silicalite-1 sample
suggests its hydrophobic surface character. Similar results were also obtained for the
silylated faujasite sample (SI Figure 1B). For the silylated faujasite sample, besides the
resonance peaks at −88, −95, −100, and −103 ppm corresponding to Si(3Al), Si(2Al),
Si(1Al), and Si(0Al), respectively,[11] the peak attributed to R-C-Si-(OSi)3 species at −65
ppm was also observed. This peak at −65 ppm is absent in the faujasite sample prepared in
aqueous medium in the absence of organosilane. For the silylated faujasite sample,
Q4 signals became much broader with higher intensity as compared to those of the faujasite

87
one. This means that the silanization led to the transformation of Q3 to Q
4 silicon species
during the synthesis.
The physicochemical properties and crystal size of the nanozeolite samples are
presented in SI Table 1 and SI Figure 2. The crystal sizes determined using the Scherrer
equation correspond reasonably well to those estimated from the TEM pictures. The BET
surface area is 570 and 545 m2/g for the nanosicalite-1 and nanofaujasite samples,
respectively. The external surface area based on t-plot calculation is 150 and 96 m2/g, in
that order. This high external surface value also indicates the small crystal size of the
sample. Detailed studies of the sorption behavior and catalytic properties are underway.
In conclusion, a new approach has been developed for the synthesis of uniform
zeolite nanocrystals with hydrophobic external surface. We anticipate that with this
approach the syntheses of other types of nanozeolites should also be possible. Furthermore,
a large variety of silylating agents allows also the tailoring of nanozeolite properties such as
crystal size and chemical nature of the surface.
Acknowledgment
We thank Prof. S. Kaliaguine for stimulating discussions and comments and Dr. B.
Nohair for assistance in connection with MAS NMR spectra. We thank the Natural
Sciences and Engineering Research Council of Canada (NSERC) for financial support of
this research and the Vietnam ministry of education and training for the scholarship
(G.T.V.)
Supporting Information Available
Experimental procedure, SI Figures 1−3, SI Table 1.

88
References
1. (a) Sano, T.; Ikeya, H.; Kasuno, T. Zeolites 1997, 19, 80. (b) Camblor, M. A.; Corma,
A.; Martinez, A.; Mocholi, F. A.; Pariente, J. P. Appl. Catal. 1989, 55, 65. (c) Tonetto,
G.; Atias, J.; de Lasa, H. Appl. Catal. 2004, 270, 9. (d) Do, T. O.; Kaliaguine, S.
In Nanoporous Materials Science and Engineering; Imperial College Press: London,
2004; Vol. 4, p 47.
2. (a) Liu, Y.; Zhang, W.; Pinnavaia, T. J. Angew. Chem., Int. Ed. 2001, 40, 1255. (b)
Zhang, Z.; Han, Y.; Zhu, L.; Wang, R.; Yu, Y.; Qui, S.; Zhao, D.; Xiao, F.-S. Angew.
Chem., Int. Ed. 2001, 40, 1258.
3. (a) Do, T. O.; Kaliaguine, S. Angew. Chem. Int. Ed. 2001, 40. (b) Do, T. O.; Kaliaguine,
S. Angew. Chem., Int. Ed. 2002, 41, 1036.
4. (a) Do, T. O.; Kaliaguine, S. J. Am. Chem. Soc. 2003, 125, 618. (b) Do, T. O.; Nossov,
A.; Springuel, M. A.; Schneider, C.; Bretherton, J. L.; Fyfe, C. A.; Kaliaguine, S. J. Am.
Chem. Soc. 2004, 126, 14324.
5. (a) Choi, M.; Cho, H. S.; Srivastava, R.; Venkatesan, C.; Choi, D. H.; Ryoo, R. Nature
Mater. 2006, 5, 718. (b) Wang, H.; Pinnavaia, T. J. Angew.Chem., Int. Ed. 2006, 45,
7603.
6. Tosheva, L.; Valchev, V. P. Chem. Mater. 2005, 17, 2494.(b) Cundy, C. S.; Cox, P.
A. Microporous Mesoporous Mater. 2005, 82, 1.
7. (a) Schoeman, B. J.; Sterte, J.; Otterstedt, J. Zeolites 1994, 14, 110. (b) Mintova, S.;
Olson, N. H.; Valtchev, V.; Bein, T. Science, 1999, 283, 958. (c) Davis, T. M.; Drews,
T. O.; Ramanan, H.; He, C.; Dong, J.; Schnablegger, H.; Katsoulakis, M.; Kokkoli, E.;
Mccormick, A. V.; Penn, R. L.; Tsapatsis, M. Nat. Mater. 2006, 5, 400.
8. (a) Schmidt, I.; Krogh, A.; Wienberg, K.; Carlsson, A.; Brorson, M.; Jacobsen, C. J. H.
Chem. Commun. 2000, 2157. (b) Jacobsen, C. J. H.; Madsen, C.; Houzvicka, J.;
Schmidt, I.; Carlson, A. J. Am. Chem. Soc. 2000, 122, 7116.
9. Wang, H.; Holmberg, A.; Yan, Y. J. Am. Chem. Soc. 2003, 125, 9928. (b) Chen, Z.; Li,
S.; Yan, Y. Chem. Mater. 2005, 17, 2262.
10. Serrano, D. P.; Aguado, J.; Escola, J. M.; Rodriguez, J. M.; Peral, A. Chem.
Mater. 2006,18, 2462.
11. (a) Holmberg, B. A.; Wang, H.; Norbeck, J. M.; Yan. Y. Microporous Mesoporous
Mater. 2003, 59, 13. (b) Ravishankar, R.; Kirschhock, C.; Schoeman, B. J.; Vanoppen,
P.; Grobet, P. J.; Stork, S.; Maier, W. F.; Martens, J. A.; Schryver, F. C.; Jacobs, P.
A. J. Phys. Chem. B 1998, 102, 2633.

89
Supporting information
Synthesis: The synthesis of nanozeolites involves two steps: (i) In the fist step,
clear zeolite gel solutions were prepared with molar composition of 2.5 TPAOH: 10 SiO2 :
250 H2O and 0.07 Na2O : 2.4 (TMA)2O : Al2O3 : 4 SiO2 : 264 H2O for silicalite-1 and
faujasite, respectively. In a typical silicalite-1 gel synthesis, 35g of 20% aqueous solution
of TPAOH was added to 19.5 g of TEOS in 25 g of water. The resulting clear solution was
stirred for 24 h at room temperature. These gel solutions were then heated at 80oC for 24 h
to speed up the formation of protozeolitic species (known as zeolite seeds). (ii) In the
second step, 10 g of gel solution was added to 500 ml of a solution of toluene containing n-
butanol (30% wt) and a proper amount of hexadecyltrimethylsilane. The organosilane was
in a proportion of less than 10 mol% in regards to the silica content in the gel. Tolulene is a
suitable medium for the modification of zeolite with organosilane and n-butanol acts as a
surfactant. The functionalization reaction was carried out batchwise in a glass reactor under
stirring at 60oC for 12 h and reflux. After 12 h of stirring, a mixture of only one clear liquid
phase was observed. This mixture was then transferred to an autoclave for further
hydrothermal treatment at 150oC for 3 days, and 180
oC for 5 days for faujasite and
silicalite-1 respectively. After the crystallization, the crude solution of nanozeolite product
was precipitated with ethanol and further isolated by centrifugation and then washed with
ethanol for several times. The product was then dried at 100ºC for 24 hours and calcined at
550ºC for 5 hours.
The conventional synthesis of the zeolites in aqueous medium was carried out
according to the procedure described in the literature.7 For the synthesis of silicalite-1, the
temperature was 150oC and the crystallization time was 3 days, whereas, for that of FAU
zeolites, the temperature and crystallization time were 100oC and 5 days.
Characterization: The FTIR spectra were recorded using a Biorad FTS-60
spectrometer on sample wafers. Powder XRD patterns of the materials were recorded on a
Philips X-ray diffractometer (PW 1010 generator and PW 1050 computer assisted
goniometer) using nickel-filtered CuKa ( = 1.5406 Ǻ) radiation, 0.0258 step size and a 1 s
step time. The crystal size of the zeolites was estimated from the broadening of the XRD

90
peaks using the Scherrer equation: d = 0.9λ /(w-w1) cos θ ; where d is the crystal diameter,
w and w1 are the half-intensity width of the relevant diffraction peak and the instrumental
broadening, respectively, λ is the X-ray wavelength and θ is the angle of diffraction. The
following reflections were used for the crystal size determination: for silicalite-1, [501] and
[151] plans corresponding to the 2θ peaks at 23.10 and 23.75º; and for faujasite, [440] and
[733] planes corresponding to those at 20.30 and 29.55º.
Nitrogen adsorption/desorption isotherms at -196oC were established using an
Omnisorp-100 apparatus. The specific surface area (SBET) was determined from the linear
part of the BET equation (P/Po = 0.05 - 0.15). High-resolution TEM images were obtained
on a JEOL 200 CX transmission electron microscope operated at 120 kV. The samples for
TEM were prepared by dispersing the fine powders of the products in slurry in ethanol onto
honeycomb carbon copper grids. Solid-state 29
Si MAS NMR spectra were recorded at room
temperature on a Bruker ASX 300 spectrometer.

91
S-Table 1. Physicochemical properties of the calcined silylated nanozeolite and zeolite
samples prepared from the same zeolite gel, in solvent medium in the presence of
organosilane, and in aqueous medium in absence of organosilane, respectively.
Sample
SBET
(m2/g)
SEXT
(m2/g)
SMIC
(m2/g)
VMIC
(cm3/g)
VMESO
(cm3/g)
RPORE
(nm)
Crystal size
(nm)-TEM
Crystal size*
(nm)-XRD
Nanosilicalite-1
570
150
420
0.154
0.230
3.7
~20
~23
Silicalite-1
512
15
479
0.162
-
-
~300
-
Nanofaujasite
545
96
449
0.149
0.195
6.5
~30
~24
Faujasite
479
19
460
0.173
-
-
~400
-
*The crystal sizes were calculated by applying the Scherrer’s equation to the XRD
reflections of [501] [151] planes for nanosilicalite-1 and those of [440], [733] planes for
nanofaujasite.

92
S-Figure 1. 29
Si MAS NMR spectra of the as-made silylated samples prepared from the
same zeolite gel, in solvent medium in the presence of organosilane, and in aqueous
medium in absence of organosilane, respectively.
(A) silicalite-1: a) as-made silicalite-1, b) as-made nanosilicalite-1, c) calcined
nanosilicalite-1.
(B) faujasite: a) as-made faujasite, b) as-made nanofaujasite.

93
S-Figure 2. Nitrogen adsorption/desorption isotherms and BJH pore radius distribution
(inset) of the calcined nanosilicalite-1 and nanofaujasite samples

94
S-Figure 3. TEM image of the calcined silylated nano faujasite sample

95
Chapter 4. Synthesis of Silylated Nanozeolites in the
Presence of Organic Phase: Two-Phase and Single Phase
Methods
Gia-Thanh Vuong and Trong-On Do *
Department of Chemical Engineering, Laval University, Quebec G1K 7P4, Canada
Published in Microporous and Mesoporous Materials 2009, 120 (3), 310-316
DOI: 10.1016/j.micromeso.2008.11.029
Résumé
Deux méthodes de synthèse, l‟une mono- l‟autre biphasique ont été développées
pour la préparation des nanozéolithes silylées. Dans ces méthodes, un solvant organique a
été utilisé comme milieu de cristallisation de ces nanozéolithes. Pour illustrer ces deux
méthodes, la zéolithe avec la structure MFI telle que silicalite-1 a été utilisée dans cette
étude. Différentes techniques de caractérisation incluant XRD, MET, MEB, BET et RMN
ont été utilisés pour caractériser les propriétés physico-chimiques de ces matériaux. Les
résultats montrent que la méthode à phase unique produit (des nanocristaux uniformes) de
silicalite-1, tandis que la méthode bi-phasique donne deux produits : nanozéolithe et
microzéolithe dans la phase organique et la phase aqueuse respectivement. Cependant, la
RMN indique que le caractère hydrophobe de ces silicalites-1 est conservé après
calcination.
Abstract
Two synthesis methods: two-phase method and single-phase one, for the
preparation of silylated nanozeolites were reported. In these methods an organic solvent
was used as an effective medium for the crystallization of zeolites. To illustrate these two
methods, silicalite-1 was selected in this study. Various techniques including XRD, TEM,
SEM, BET and NMR techniques were used to monitor the physico-chemical properties of
these synthesized materials. The results revealed that the single-phase method allows
producing uniform/small nanosilicalite-1, whereas the two-phase one can bring two
separate products: nanosized and microsized zeolite crystals in organic phase and in
aqueous phase, respectively. Furthermore, the NMR results indicate that the hydrophobic
surface character of these silicalite-1 samples can be obtained even after calcination.
Keywords: nanozelites, nanosilicalite-1, synthesis, organic medium, surface modification.

96
4.1. Introduction
Zeolites are crystalline aluminosilicate molecular sieves with uniform pores of
molecular dimensions. They have been widely used as heterogeneous catalysts and
adsorbents in the field of oil refining and in the petrochemical industry. Conventional
zeolites with crystal sizes of several micrometers are synthesized under hydrothermal
conditions. However, due to the pore size constraints (<15 Å), the unique catalytic
properties of zeolites are limited to reactant molecules having kinetic diameters below
10 Å [1-3]. To overcome this limitation, there is an intensive research to either minimize
the size of the zeolite crystals or to generate hierarchical zeolites containing a bimodal
porosity, both micro-/mesopores [4-11] or micro-/macropores [12, 13]. The former strategy
has led to the discovery of various types of zeolitic porous materials such as composite
MFI/MCM-41 materials, UL-zeolites and MSU-S [4-11].
Nanozeolite with crystal sizes of less than 100 nm, has been receiving much of
interest due to the fact that the reduction of zeolite crystal size from the micrometer to the
nanometer scale leads to increase the external surface area and reduce diffusion path
lengths [14, 15]. Furthermore, the reduction of crystal size to the nanometer scale results in
substantial changes in the properties of materials, which have an impact on the performance
of zeolites in traditional application areas such as catalysis and separation[16, 17].
Therefore, the synthesis of zeolite nanocrystals with small uniform diameter is highly
desirable.
A number of synthesis methods has been reported, which allows the syntheses of
nanozeolites with different structures, such as FAU, MFI, LTA and MOR [15, 18-20].
Synthesis methods are often carried out in the aqueous phase, and the key to the success
was the use of so-called “clear gel solution” [15]. Another method, which is called confined
space synthesis, has been developed for the preparation of nanosized zeolite crystals. The
synthesis is conducted within an inert matrix such as porous carbon matrices [21-23],
thermo-reversible polymer hydrogels, or microemulsions [24-29] which provides a steric
hindered space for zeolite crystal growth. However, the external surface of the nanozeolites
is generally hydrophilic and has high concentration of silanol groups that limit the catalytic

97
activity to the internal pore surface. Therefore, the synthesis of nanozeolites with
hydrophobic external surface is highly interesting [30-32]. To achieve that, the nanozeolites
have to undergo the post-synthesis treatment which often involves the silanization of the
nanozeolite surfaces with organosilane agents [33-35].
Serrano et al. [36] have reported the use of organosilane as growth inhibitor. MFI
and beta zeolites were synthesized in the aqueous medium, using phenylaminopropyl-
trimethoxysilane (PHAPTMS). The synthesis is based on reducing the growth of zeolite
crystals by silanization of the zeolitic seeds to hinder their further aggregation. However, as
investigated by TEM analysis, the silylated MFI samples were prepared with a broad
crystal size distribution in the range of 200–400 nm, which were formed by aggregation of
ultra small units of 10 nm. Recently, we reported a new method, namely a single organic
phase for the synthesis of controlled uniform size nanozeolites with the hydrophobic
external surface. An organic solvent is used as the medium for crystallization instead of
water. The zeolite precursors are functionalized with organic silane groups. They thus
become hydrophobic and well dispersed in the organic solvent. Because the crystallization
occurs in the organic phase and the zeolite precursors are protected by functional groups,
catastrophic aggregation can be prevented hence resulting in small and uniform silylated
nanozeolites with hydrophobic external surface [37]. Clear zeolite gel precursor was
prepared using the method described in the literature [38, 39]. The as-made silylated
nanozeolites are readily hydrophobic, hence, there is no need for the post-synthesis
treatment. Therefore it can be suggested that organic solvent is an effective medium for the
synthesis of nanozeolites.
The two-phase organic solvent/water synthesis is an appealing method which has
been used in the synthesis of transition metal nanoparticles such as Pt, TiO2, ZrO2,
CdS [40-47]. For the synthesis of TiO2 nanocrystals, for example, a toluene solution
containing titanium n-propoxide and oleic acid (OA) as a capping agent and an aqueous
solution of tert-butylamine are mixed and heated. The formation of nanoparticles takes
place at the toluene-water interface. The freshly formed nanoparticles capped with OA
become hydrophobic and could be dispersed in toluene. They therefore stop growing and
finally, monodisperse nanoparticles can be achieved [41-43]. The two phase synthesis can

98
be carried out with an appropriate choice of capping agents. These agents contain both
hydrophilic groups at one end that can be bound to the nanoparticle surface and
hydrophobic alkyl-chains at the other end [48].
The prospect of applying this strategy for the synthesis of nanozeolites is attractive,
it would be an effective approach for spatial isolation of zeolitic seeds from further growth
and the size could be tuned via changing the capping agent content. However, the
crystallization of zeolites is somewhat different and difficult as compared to the synthesis
of transition metal nanoparticles. Care should be taken in choosing the appropriate capping
agent and solvent for the synthesis of nanozeolites. One of the problems is that common
capping agents such as oxalic acid, stearic acid are incapable of “capping” zeolitic seeds. In
the synthesis of transition metal nanoparticles, these acids that contain donor group can
attach to metal atoms by coordinate bonding. Thus, a transition metal of which the atom has
an incomplete d shell is more likely to form the complex with the acids, compared to the
main group Si and Al ions (e.g., no transition metal ions) in the structure of zeolites.
To overcome this problem, the synthesis strategy is based on hindering the growth
of zeolite crystals by silanization of the zeolitic seeds using an organosilane. An aqueous
clear solution containing zeolitic seeds was added in an organic solvent containing
organosilane agent. As the organic solvent is insoluble in water, the organic phase stays on
top of the aqueous phase. Since the zeolitic seeds are rich in silanol groups, they can react
with the organosilane agents at the interface between the two phases. Because of the
protecting organic groups, the functionalized zeolitic precursors become more hydrophobic
and can diffuse into the organic phase.
In this paper, we report a two-phase method approach in which silylated
nanosilicalite-1 was prepared in the presence of both organic and aqueous phases.
Hexadecyltrimethoxysilane and toluene were used as silylating agent and organic solvent,
respectively. These materials were characterized using XRD, TEM, BET and 29
Si MAS
NMR techniques, and compared to those obtained by the single organic solvent phase
method.

99
4.2. Experimental
4.2.1. Synthesis of silylated silicalite-1 using the two phase and single-
phase methods
The synthesis of nanozeolites involves two steps: (i) In the fist step, clear zeolite gel
solution was prepared according to the method described in the literature [38, 39]. (ii) The
obtained clear gel solutions were then processed in different procedures depending on the
method applied. The use of the same starting zeolite gel solution can provide the ease of
comparison between these synthesis methods.
4.2.2. Two-phase method
The procedure is consisting of the following two steps (see Figure 4.1): (i) A clear
zeolite gel solution was prepared with a molar gel composition of 2.68 SiO2:1 TPAOH:168
H2O. In a typical recipe, 14 g of tetrapropylammonium hydroxide (TPAOH) 20% in water
was added to 7.8 g of tetraethoxysilane, Si(OC2H5)4. The mixture was stirred vigorously for
24 h at room temperature. This clear solution was then heated at 80 °C for 12 h to speed up
the formation of protozeolitic species (known as zeolite seeds). (ii) Typically, 21.8 g of
clear zeolite gel solution was added with 93 g of a solution of toluene containing 1.22 wt%
hexadecyltrimethoxysilane (note that the organosilane was in proportion of less than 10%
mol with respect to the silica content in the gel). Since the solvent is insoluble in water, a
two-phase system was obtained. The functionalization reaction was carried out batchwise
in a glass reactor under stirring at 60 °C for 12 h and reflux. This two-phase mixture was
transferred into a Teflon-lined stainless steel autoclave and then heated at 180 °C for 5
days. After crystallization, the two silicalite-1 products in organic phase (toluene) and
aqueous phase were recovered separately by centrifugation and then washed with ethanol
and water for several times. The products were then dried at 80 °C for 24 h and were
denoted as OP (organic phase) and AP (aqueous phase) samples.

100
Figure 4.1. Schematic representation of the two-phase synthesis method.
4.2.3. Single-phase method
The synthesis of silylated nanosilicalite-1 was carried out according to the
procedure described in reference [37]. 10 g of clear gel solution containing zeolitic seeds
was added to 500 ml of a solution of toluene containing n-butanol (30 wt%) and a proper
amount of hexadecyltrimethoxysilane. An amount of n-butanol is introduced in the organic
phase, which is expected to increase the miscibility of the organic phase toward the
aqueous one, since n-butanol is miscible in hydrocarbon solvent, however it is moderately
soluble in water. The mixture was heated in a glass reactor under stirring at 60 °C for 12 h
and reflux. After 12 h of stirring, a mixture of only one clear liquid phase was observed.

101
This mixture was then transferred to a Teflon-lined stainless steel autoclave for further
hydrothermal treatment at 180 °C for 5 days. After the crystallization, the crude solution of
nanosilicalite-1 product was precipitated with ethanol and further isolated by centrifugation
and then washed with ethanol for several times. The product was then dried at 80 °C for
24 h and is denoted as SOP sample.
4.2.4. Conventional method (synthesis of nanozeolites in aqueous
medium)
The conventional synthesis of the zeolites in aqueous medium was carried out
according to the procedure described in the literature [19, 38, 39]. After being filled with
the same starting silicalite-1 gel solution for the single-phase and two-phase methods, the
Teflon-lined stainless steel autoclave was completely sealed and heated in a convection
oven at 150 °C for 3 days. The solid product was recovered by centrifugation, washed
several times with distilled water, dried over night at 80 °C. The products synthesized using
this conventional method, were denoted as reference or conventional silicalite-1 samples.
4.2.5. Characterization
The FTIR spectra were recorded using a Biorad FTS-60 spectrometer on sample
wafers. Powder XRD patterns of the materials were recorded on a Philips X-ray
diffractometer (PW 1010 generator and PW 1050 computer assisted goniometer) using
nickel-filtered Cu Kα (λ = 1.5406 Ǻ) radiation, 0.0258° step size and a 1 s step time.
The nitrogen adsorption/desorption measurements were carried out using an
Omnisorp-100 automatic analyzer at −196 °C after degassing about 30 mg of calcined
sample at 200 °C for at least 4 h under vacuum (10−4
to 10−5
torr). The specific surface area
(SBET) was determined from the linear part of the BET equation (P/Po = 0.05 − 0.15).
High-resolution TEM images were obtained on a JEOL 200 CX transmission electron
microscope operated at 120 kV. The samples for TEM were prepared by dispersing the fine
powders of the products in slurry in ethanol onto honeycomb carbon copper grids.
However, for scanning electron microscope (SEM), JEOL JSM-840 scanning electron
microscope operated at 15 kV was used. Solid-state29Si MAS NMR spectra were recorded
at room temperature on a Bruker ASX 300 spectrometer.

102
4.3. Results and discussion
The synthesis of nanosilicalite-1 was selected to demonstrate the two phase method
approach (Figure 4.1). Hexadecyltrimethoxysilane and toluene were used as silylating
agent and organic solvent, respectively[29, 36]. After crystallization, the products were
recovered and denoted as OP in the organic phase and AP in the aqueous phase. The mass
ratio of the OP and AP products is about 6/4 in this work. For the single-phase method [37],
the water/oil ratio was reduced significantly (see Figure 4.2). To obtain a mixture of only
one clear liquid phase after stirring at 60 °C for 12 h, an appropriate amount of clear
aqueous zeolite gel solution is added to a solution of toluene containing both n-butanol and
hexadecyltrimethoxysilane (see Section 5.2 for details). The product is denoted as the SOP
sample.
The physico-chemical properties of the calcined silylated nanozeolite and zeolite
samples prepared from the same clear zeolite gel, using different methods: the two-phase,
single-phase and conventional methods are summarized in Table 4.1.
Table 4.1. Physico-chemical properties of the calcined silylated nanozeolite and zeolite
samples prepared from the same clear zeolite gel, using different methods: the two-phase,
single-phase and conventional methods.
*Product recovered in aqueous phase
The specific surface area, SBET determined from the linear part of the BET equation (P/P0 = 0.05–0.15). The mesopore size distribution
calculated using the desorption branch of the N2 adsorption/desorption isotherms and the Barrett–Joyner–Halenda (BJH) formula. The mesopore
surface area (SEXT) and mesopore volume (VBJH) obtained from the pore size distribution curves. The micropore surface area (SMIC) obtained
as SBET–SEXT and the average mesopore radius, (RPORE) calculated as 4VBJH/SBJH[49].

103
Figure 4.2. Schematic representation of the single-phase synthesis method.

104
2
5 10 15 20 25 30
Intensity
(d) Conventional method
(c) AP
(b) OP
(a) SOP
Figure 4.3. XRD patterns of the as-made silicalite-1 samples, (a) sample prepared using the
conventional method in aqueous medium, (b) AP silicalite-1, (c) OP silicalite-1 using the
two-phase method and (d) SOP silicalite-1 using single-phase method.
Figure 4.3 shows the X-ray powder pattern of the as-made silylated SOP sample
using the single-phase method, and as-made silylated OP and AP samples using the two

105
phase method. Sample prepared by the conventional method in aqueous medium from the
same clear zeolite gel solution without organosilane was used as a reference (d). The XRD
patterns of these samples are identical to that of the reference sample, indicating the MFI
structure of the samples. This also suggests that the presence of organosilane in the
synthesis mixture did not affect the formation of the desired structure. However, for the
SOP sample prepared using the single-phase method and the OP sample, there is a clear
broadening of the reflections, which is attributed to small crystals. In addition, no
significant peak at 2θ = 20–30° which is characteristic of amorphous phase was observed
suggesting a relatively high crystallinity of the silylated samples.
The crystal size of these samples was also investigated by scanning and
transmission electron microscope (SEM and TEM) techniques. The SEM micrographs of
the as-made silylated OP and AP silicalite-1 samples (e.g., the products in organic phase
and aqueous phase, respectively) are shown in Figure 4.4. The crystal size of the OP
silicalite-1 sample was uniform, ranging from 30 to 50 nm. In contrast, large crystals of
5μm were observed for the AP silicalite-1 sample. A difference in crystal size between the
two samples suggests the effect of media for crystallization on crystal size of the product.
Further, for the as-made silylated SOP silicalite-1 sample, a representative TEM
micrograph is also shown in Figure 4.5 and exhibits very uniform nanocrystal sizes with
mostly spherical and cubic shaped particles. The particle size is about 21 nm. Owing to the
organosilane being grafted on the zeolite precursors, the nanoparticles remain well
dispersed in the organic medium and are protected from drastic aggregation during
crystallization.

106
Figure 4.4. SEM micrographs of the as-made samples, (A) silylated OP silicalite-1 and (B)
silylated AP silicalite-1.

107
Figure 4.5. TEM micrograph of the as-made SOP nanosilicalite-1 sample prepared using
the single-phase method.
It is well known that water phase which is used as a crystallization medium, plays a
role of transporting monomers to the surface of zeolite crystals and large crystal sizes were
obtained. However, using an organic solvent instead of water, the zeolite precursors are
functionalized with organic silane groups. They thus become hydrophobic and highly
dispersed in the organic phase. Because of the crystallization occurs in the organic phase
with a limited water amount, catastrophic aggregation can be prevented. As a result, small
and uniform nanozeolites were observed for the OP silicalite-1 and SOP silicalite-1
samples. Figure 4.6 also shows the FTIR spectra of the samples prepared using the
conventional and single-phase methods (the calcined SOP sample). The FTIR peak
positions are identical to those of the MFI structure. In particular, the band at 550 cm−1
is
assigned to the asymmetric stretching mode of the five-membered ring present in ZSM-5
which is an indication of the MFI structure. Splitting of this lattice-sensitive band into a
doublet has been observed in nanophase silicalite-1 [39]. The FTIR spectrum of the SOP
silicalite-1 sample prepared using the single-phase method in Figure 4.6 shows the doublet
band at 561/547 cm−1
which indicates the formation of nanocrystals.

108
Wavenumber (cm-1
)
400600800100012001400
Tra
nsm
itta
nce
544
555 450
795
1080
1220
(A)
(B)
Figure 4.6. FTIR spectra of the silicalite-1 samples prepared using the single-phase method
(A) and the conventional method (B).

109
Figure 4.7 29
Si MAS NMR spectra of the silicalite-1 samples prepared from the same
zeolite gel solution using (a) the conventional method in aqueous medium without
organosilane, (b) as-made SOP nanosilicalite-1 using single-phase method in organic
solvent and (c) calcined SOP nanosilicalite-1.

110
Figure 4.8 29
Si MAS NMR spectra of the as-made silicalite-1 samples prepared from the
same zeolite gel solution using the two-phase method: (a) as-made AP nanosilicalite-1 and
(b) as-made OP nanosilicalite-1.
The 29
Si MAS NMR spectra of the as-made silicalite-1 samples prepared using
different methods from the same zeolite gel solution were investigated (Figure 4.7
and Figure 4.8). The NMR spectrum of the as-made silicalite-1 sample prepared using the
conventional method, in the absent of organosilane, shows a main resonance at
approximately −110 ppm and a weak resonance peak at approximately −100 ppm which are
attributed to Si(OSi)4, Q4 and Si(OSi)3OH, Q
3 species, respectively (Figure 4.7a). However,
for the as-made SOP silicalite-1 sample, only one resonance peak at approximately
−110 ppm attributable to Q4 species was observed, an additional intense peak at −65 ppm
assigned to R-C-Si-(OSi)3 species is present [33]. This additional peak is the result from the
reaction between the silicon in the organosilane and the silanol groups of zeolite nuclei

111
during the crystallization. This NMR broad peak at 50–70 ppm could be contributed
to T2 and T
3 which correspond to two and three OH groups consumed by one organosilane
molecule. The calcined sample also shows essentially a single resonance peak Q4 at
approximately −110 ppm (Figure 4.7b and c). It clearly indicates that the silanol groups
which disappear in the chemical interaction with the organosilane do not reappear upon
calcination. This could be due to two or three OH groups on the nanoparticle surface
consumed by one organosilane molecule. As a result, only one resonance Q4 of this sample,
even after calcination, suggests its hydrophobic surface character.
Furthermore, the as-made OP and AP silicalite-1 samples exhibit also only one
NMR peak at approximately −110 ppm characteristic of Q4 along with a NMR broad peak
at 50–70 ppm attributed to T2 and T
3 for the OP sample and a very weak peak for the AP
sample (Figure 4.8). This also suggests the silanization on the external surface of silicalite-
1, which acts to heal defect sites (e.g., silanol groups) on the zeolite surface. Thus, it can be
concluded that the presence of only one resonance Q4 even after calcination of the silylated
silicalite-1 samples suggests their hydrophobic surface character.

112
Figure 4.9. Nitrogen adsorption/desorption isotherms of the calcined samples: (A) SOP
silicalite-1, (B) OP silicalite-1 and (C) AP silicalite-1 (inset: t-plot curve).
Figure 4.9 shows the N2 adsorption/desorption isotherms and the BJH pore radius
distribution of the calcined SOP nanosilicalite-1 and OP nanosilicalite-1 samples. At low
relative P/Po pressure, a steep rise in uptake, followed by a flat curve, corresponds to filling
Micropore volume
Micropore volume
Micropore volume

113
of micropores with nitrogen. An inflection at higher pressures (e.g., in P/Po range from 0.7
to 0.9) and a hysteresis loop for the OP nanosilicalite-1 sample are characteristic of
capillary condensation and are related to the range of mesopores due to the interparticles.
For the AP silicalite-1 sample, mesopores were not observed (see Figure 4.9B) owing to its
large particle size. However, for the SOP nanosilicalite-1 sample no hysteresis loop is
present; even its particle size is smaller than that of the OP sample. This can be explained
by uncompacted nanoparticles of this sample [49]. The specific surface areas are 570 and
520 m2/g, and the external surface areas based on t-plot calculation are 150 and 106 m
2/g
for the SOP nanosilicalite-1 and OP nanosilicalite-1 samples, respectively. However, for
the AP silicalite-1 sample with large crystal size, the external surface area is very low, only
10 m2/g (Table 4.1). The high external surface values indicate the small crystal size of the
sample.
Indeed, silanol groups on silicalite-1 are mainly located on the external surface and
therefore the quantity of silanol sites is related to the zeolite crystal size. Due to the bulky
size of hexadecyltrimethoxysilane, only the silanol groups on the external surface of zeolite
crystals are accessible for the silanization. Hence the high external surface of nanozeolites
provides silanol sites which are available for the chemical functionalization. The order of
the external surface areas and of the amount of silanol sites reacted with organic silane
groups based on the NMR peak intensity at −65 ppm was found to be: SOP silicalite-
1 > OP silicalite-1 > AP silicalite-1. In contrast, the order of crystal size is: AP silicalite-
1 > OP silicalite-1 > SOP silicalite-1. Furthermore, as seen the NMR results, the silylated
silicalite-1 sample synthesized by these methods exhibit a hydrophobic character, even after
calcination.
4.4. Conclusion
In conclusion, we have demonstrated two methods in the presence of organic
medium for the synthesis of nanozeolites with hydrophobic external surface. Depending on
the method of synthesis, different crystal sizes can be synthesized: Uniform and small
nanozeolites using the single-phase method, however, using the two phase method, large
nanosized and microsized zeolite crystals in organic phase and in aqueous phase,
respectively. We believe that these methodologies can be applied to the synthesis of other

114
types of nanozeolites. Detailed studies of the sorption behavior and catalytic properties of
this class of nanozeolites are underway.
Acknowledgments
We thank Prof. S. Kaliaguine and Prof. F. Kleitz and Dr. V.T. Hoang for
stimulating discussions and comments. This study was supported by the Natural Sciences
and Engineering Research Council of Canada (NSERC).

115
References
1. D. Breck Zeolite Molecular Sieves: Structure, Chemistry and Use Wiley
Interscience, New York (1974)
2. C. Baerlocher, W.M. Meier, D. Olson Atlas of Zeolite Framework Types Elsevier
(2001)
3. L.B. McCusker, C. Baerlocher H.V. Bekkum, P.A. Jacobs, E.M. Flanigen, J.C.
Jansen (Eds.), Introduction to Zeolite Science and Practice (second ed.), Elsevier,
Amsterdam (2001), p. 37
4. Y. Liu, W. Zhang, T.J. Pinnavaia Angew. Chem. Int. Ed., 40 (2001), p. 1255
5. Z. Zhang, Y. Han, L. Zhu, R. Wang, Y. Yu, S. Qiu, D. Zhao, F.-S. Xiao Angew.
Chem. Int. Ed., 40 (2001), p. 1258
6. A. Karlsson, M. Stöcker, R. Schmidt Micropor. Mesopor. Mater., 27 (1999), p. 181
7. S. Kaliaguine, T.O. Do, Mesoporous Zeolitic Materials with Microporous Crystalline
Mesopore Walls: UL-zeolites, US 6,669,924 B1.
8. S. Kaliaguine, T.O. Do, Mesoporous Zeolitic Materials with Microporous Crystalline
Mesopore Walls: UL-zeolites, EP 1237818 B1.
9. T.O. Do, S. Kaliaguine Angew. Chem. Int. Ed., 40 (2001), p. 3248
10. T.O. Do, S. Kaliaguine Angew. Chem. Int. Ed., 41 (2002), p. 1036
11. T.O. Do, A. Nossov, M.A. Springuel-Huet, C. Schneider, J.L. Bretherton, C.A. Fyfe,
S. Kaliaguine J. Am. Chem. Soc., 126 (2004), p. 14324
12. S. Vaudreuil, B. Echchahed, P. Reinert, M. Bousmina, S. Kaliaguine, L. Bonneviot J.
Porous Mater., 14 (2007), p. 173
13. B.T. Holland, L. Abrams, A. Stein J. Am. Chem. Soc., 121 (1999), p. 4308
14. T.O. Do, S. Kaliaguine G.Q. Lu, X.S. Zhao (Eds.), Nanoporous Materials Science
and Engineering, Imperial College Press Publisher (2004), p. 47
15. L. Tosheva, V.P. Valtchev, Chem. Mater., 17 (2005), p. 2494
16. S.S. Kim, J. Shah, T.J. Pinnavaia, Chem. Mater., 15 (2003), p. 1664
17. E.F.S. Aguiar, M.L.M. Valle, M.P. Silva, D.F. Silva, Zeolites, 15 (1995), p. 620
18. C.S. Cundy, P.A. Cox, Chem. Rev., 103 (2003), p. 663
19. C.S. Cundy, J.O. Forrest, R.J. Plaisted, Micropor. Mesopor. Mater., 66 (2003), p. 143
20. T.M. Davis, T.O. Drews, H. Ramanan, C. He, J. Dong, H. Schnablegger, M.A.
Katsoulakis, E. Kokkoli, A.V. McCormick, R.L. Penn, M. Tsapatsis, Nat. Mater., 5
(2006), p. 400
21. C. Madsen, C. Madsen, C.J.H. Jacobsen, Chem. Commun. (1999), p. 673
22. C.J.H. Jacobsen, C. Madsen, T.V.W. Janssens, H.J. Jakobsen, J. Skibsted, Micropor.
Mesopor. Mater., 39 (2000), p. 393
23. I. Schmidt, C. Madsen, C.J.H. Jacobsen, Inorg. Chem., 39 (2000), p. 2279
24. H. Wang, B.A. Holmberg, Y. Yan, J. Am. Chem. Soc., 125 (2003), p. 9928
25. C.S. Carr, D.F. Shantz, Micropor. Mesopor. Mater., 85 (2005), p. 284
26. S.P. Naik, J.C. Chen, A.S.T. Chiang, Micropor. Mesopor. Mater., 54 (2002), p. 293
27. S. Lee, D.F. Shantz, Chem. Commun. (2004), p. 680
28. S. Lee, D.F. Shantz, Chem. Mater., 17 (2005), p. 409
29. Z. Chen, S. Li, Y.S. Yan, Chem. Mater., 17 (2005), p. 2262
30. R.S. Bowman, Micropor. Mesopor. Mater., 61 (2003), p. 43
31. P. Huttenloch, K.E. Roehl, K. Czurda, Environ. Sci. Technol., 35 (2001), p. 4260

116
32. W. Song, G. Li, V.H. Grassian, S.C. Larsen, Environ. Sci. Technol., 39 (2005), p.
1214
33. B.Z. Zhan, M.A. White, M. Lumsden, Langmuir, 19 (2003), p. 4205
34. W. Song, J.F. Woodworth, V.H. Grassian, S.C. Larsen, Langmuir, 21 (2005), p. 7009
35. B.Z. Zhan, M.A. White, P. Fancy, C.A. Kennedy, M. Lumsden, Macromolecules, 37
(2004), p. 2748
36. D.P. Serrano, J. Aguado, J.M. Escola, J.M. Rodriguez, A. Peral, Chem. Mater., 18
(2006), p. 2462
37. G.T. Vuong, T.O. Do, J. Am. Chem. Soc., 129 (2007), p. 3810
38. B.J. Schoeman, J. Sterte, J.E. Otterstedt, Zeolites, 14 (1994), p. 110
39. R. Ravishankar, C. Kirschhock, B.J. Schoeman, P. Vanoppen, P.J. Grobet, S. Storck,
W.F. Maier, J.A. Martens, F.C. De Schryver, P.A. Jacobs, J. Phys. Chem. B, 102
(1998), p. 2633
40. M. Niederberger, G. Garnweitner, F. Krumeich, R. Nesper, H. Colfen, M. Antonietti,
Chem. Mater., 16 (2004), p. 1202
41. D. Pan, S. Jiang, L. An, B. Jiang, Adv. Mater., 16 (2004), p. 982
42. D. Pan, Q. Wang, S. Jiang, X.J.L. An, Adv. Mater., 17 (2005), p. 176
43. D. Pan, N. Zhao, Q. Wang, S. Jiang, X. Ji, L. An, Adv. Mater., 17 (2005), p. 1991
44. Q. Wang, D. Pan, S. Jiang, X. Ji, L. An, B. Jiang, Chem. Eur. J., 11 (2005), pp. 3843–
3848
45. S.L. Horswell, C.J. Kiely, I.A. O‟Neil, D.J. Schiffrin, J. Am. Chem. Soc., 121 (1999),
p. 5573
46. M. Niederberger, M.H. Bartl, G.D. Stucky, J. Am. Chem. Soc., 124 (2002), p. 13642
47. M. Niederberger, M.H. Bartl, G.D. Stucky, Chem. Mater., 14 (2002), p. 4364
48. B.L. Cushing, V.L. Kolesnichenko, C.J. O‟Connor, Chem. Rev., 104 (2004), p. 3893
49. S.J. Gregg, K.S.W. Sing, Adsorption, Surface Area and PorosityAcademic Press,
London (1982)

117
Chapter 5. Synthesis of Nanozeolite-Based FCC
Catalysts and their Catalytic Activity in Gasoil Cracking
Reaction
Gia-Thanh Vuong, Vinh-Thang Hoang, Dinh-Tuyen Nguyen and Trong-On Do *
Department of Chemical Engineering, Laval University, Quebec G1K 7P4, Canada
Institute of Chemistry, Vietnamese Academy of Science and Technology, Viet Nam
Published in Applied Catalysis A: General 2010, 382 (2), 231-239
DOI: 10.1016/j.apcata.2010.04.049
Résumé
Une nouvelle méthode de synthèse de nanozéolithe en milieu organique utilisant le
formamide et le toluène comme milieu de cristallisation a été développé. Ces deux solvants
ont été utilisé à la place de l‟eau, en présence d‟agent de silylation. L‟effet des solvants
organiques est majeur dans la synthèse des nanozéolithes. Le formamide qui a des
propriétés similaires à l‟eau est un bon candidat de solvant pour la synthèse de
nanozéolithes. Cette méthode de synthèse nous facilite le contrôle de la taille des
nanoparticules des zéolithes. Dans cette étude, nanoparticules de zéolithe avec différentes
tailles; 25, 40 et 100 nm ont été préparées dans le toluène et le formamide agissant comme
solvant. À fin d‟étudier l‟effet de la taille des nanoparticules zéolithiques sur l‟activité
catalytique, une série de nanozéolithes a été utilisée pour le craquage catalytique. Les
résultats montrent une très bonne corrélation entre les tailles des nanozéolithes et l‟activité
catalytique. Les nanozéolithe de taille petite ont montré une activité catalytique supérieure
à celle des nanozéolithe de taille supérieure.

118
Abstract
A new method for the synthesis of nanosized zeolites in organic solvents, such as
formamide and toluene as crystallization medium instead of water, in the presence of
organosilane has been developed. Organic solvents have a great impact on the synthesis of
nanozeolites. Formamide, which has similar properties to water, is a good candidate as the
solvent for the synthesis of nanosized zeolites. This synthetic method allows easy
manipulation with the control of crystal sizes. In this study, different crystal sizes such as
25, 40 and 100 nm were prepared in toluene and formamide solvents. To study the effect of
crystal nanosizes on the catalytic performance of nanosized zeolites, nanozeolite-based
FCC catalysts were also prepared using different nanozeolite sizes as active component and
silica as inactive matrix. The activity of these catalysts was evaluated with FCC feedstock.
The results revealed a good correlation between the crystal size of zeolites and the activity:
smaller nanozeolite-based FCC catalyst exhibits higher catalytic activity.
Keywords: Nanozeolites; Formamide; Non-aqueous synthesis; FCC catalysts; FCC
cracking

119
5.1. Introduction
Nanozeolites with the size of less than 200 nm have received much of interest
recently, because of their great potential applications not only in catalysis and adsorption,
but also in a variety of new applications including chemical sensing, medicine,
optoelectronics etc. [1, 2]. The decrease in the crystal sizes results in higher external
surface areas, reduced diffusion path lengths, and more exposed active sites, which have an
impact on the performance of the nanosized zeolites as compared to that of conventional
zeolites of which the size is often of microns [1, 3]. Besides the well known applications of
such zeolites in catalysis and adsorption, nanozeolites can also find their applications as
seeds and as building blocks for the preparation of mesoporous zeolitic materials [1, 4-9].
Crystalline structure of zeolites with tridimensional network of well-defined micropores
(pore diameter less than 15 Å) brings both (i) advantage and (ii) disadvantage. (i) This
feature provides zeolite with a consistent adsorption behavior toward guest molecules. Only
molecules of size less than or equal to pore size aperture can have access to the vast internal
surface area of zeolites. Thus, when the catalytic reaction occurs inside the zeolite pores,
zeolites can exhibit high selectivity toward small guest molecules [2, 10, 11]. (ii) However,
the unique catalytic properties of zeolites are limited to reactant molecules having kinetic
diameters below 15 Å, due to the pore size constraints. Reactions involving large molecules
on zeolites hence must resort to only the external surface of zeolite [12].
The use of nanosized zeolites could overcome this limitation, the ratio of external to
internal number of atoms increases rapidly as the particle size decreases, and zeolite
nanoparticles have large external surface areas and high surface activity. The external
surface acidity is of importance, when the zeolite is used as catalyst in reactions involving
bulky molecule. The nanosized zeolites could bring better performance due to a high
accessibility of active phase and high external surface area. For example, in catalytic
cracking of gas oil, most of the hydrocarbon molecules are barred from zeolite pores and
thus only the external surface of zeolite contributes to the gas oil conversion. Most of
cracking of these molecules is realized on the interface of zeolite–matrix component of the
FCC catalysts [13, 14]. Rajagopalan et al. have shown that in cracking gas oil, when the
crystallite size of zeolite decreases, both conversion and selectivity clearly increase [15].

120
On this aspect, the use of nanozeolites is a workaround and an improvement for FCC
catalysts. Since the external surface of nanozeolites is expectedly higher and this type of
surface is accessible, cracking of large hydrocarbon molecules on nanozeolites with high
efficiency is possible. Hence a study of a nanozeolite-based FCC catalyst is of great
interest.
Synthesis of nanozeolites has been studied extensively [1]. A common approach is
to modify the general method of synthesis of zeolites, which is carried out in an aqueous
phase [18-20]. Careful adjustment of the parameters such as gel composition, temperature,
crystallization time, aging time etc. can allow nanozeolites to form. The principle of the
synthesis is derived from the classic nucleation and crystallization theory: facilitating the
nucleation, which produces nuclei as much as possible; and controlling a subsequent slow
growth of crystal particles. Ideally, the nucleation and growth processes should be
completely separate from each other.
There are two possible mechanisms of nucleation in the synthesis of zeolites [21]:
homogeneous nucleation and heterogeneous nucleation. Homogeneous nucleation occurs
from the mother liquid while heterogeneous nucleation happens within the gel.
Heterogeneous nucleation and growth are hardly separate process. Hence, regarding the
synthesis of nanozeolites, it is very important to obtain the starting synthesis gel in the state
of a “clear solution” or a clear gel solution in the hope that the homogeneous nucleation
would take place instead of the heterogeneous one. Other factors such as aging, pH,
crystallization time, gel composition are also subject to change to control the nucleation
and growth process.
In this paper, we report a new route for the synthesis of nanozeolites of FAU by the
soft controlling method using different types of solvents as crystallization medium instead
of water. The crystal size of the nanozeolites can be manipulated to some extent by
changing the solvent type. To evaluate the potential application, a series of FCC catalysts
based on these nanozeolites with various particle sizes are also prepared. The obtained
catalysts were tested against commercial catalysts in a standard test of gas oil cracking.

121
5.2. Materials and methods
5.2.1. Synthesis of nanofaujasite
Three kinds of samples were prepared. The synthesis followed what we have
reported [16]. In a typical procedure, Al(iPr)3 (19.5 g) was added into 78.36 g of TMAOH
25% under stirring for 3 h. Then 40.68 g of TEOS 98% was added. The stirring was
continued overnight to make sure TEOS was completely hydrolyzed. Then, 64 mL of
NaOH 0.1 M was added and stirred for another 3 h. The resulting clear solution was then
aged at 90 °C for 2 (or 4) days to speed up the formation of protozeolitic species known as
zeolite seeds. Subsequently, 10 g of the aged gel was added into 100 mL of
hexadecyltrimethoxysilane (HDMT, 10%) containing toluene (or formamide). The clear
homogeneous mixture was then transferred into an autoclave and heated for 5 days at
160 °C temperature. The silylated nanozeolite product was then recovered by centrifuge
and washed with ethanol three times before drying at 100 °C for 24 h. The samples,
prepared using toluene, were designated as FAU–TOLxD, while the ones using formamide
were designated as FAU–FORxD, where x is the aging time in day; the yield of synthesis
was 41% and 47%, respectively. Zeolite Y reference was used from Strem Chemical.
5.2.2. Synthesis of nanofaujasite-based FCC catalysts
35 g of TEOS was dissolved in 100 mL of ethanol. To this mixture 10 g of as-made
nanofaujasite was added. The mixture was stirred overnight and then evacuated under
reduced pressure. The collected solid was dried at 100 °C for 24 h then calcinated at 600 °C
for 6 h. The FCC catalyst samples were designated as FCC–FAU–TOLxD and FCC–FAU–
FORxD, where x is the aging time of zeolite gel in day.
5.2.3. Characterization
The FT-IR spectra were recorded using a Biorad FTS-60 spectrometer on sample
wafers. Powder XRD patterns of the materials were recorded on a Philips X-ray
diffractometer using nickel-filtered CuKα(λ = 1.5406 Ǻ) radiation.
The nitrogen adsorption/desorption measurements were carried out using an
Omnisorp-100 automatic analyzer at −196 °C after degassing about 30 mg of calcined
sample at 200 °C for at least 4 h under vacuum (10−4
–10−5
Torr). The specific surface area

122
(SBET) was determined from the linear part of the BET equation (P/Po = 0.05–0.15). TEM
images were obtained on a JEOL 200 CX transmission electron microscope operated at
120 kV. The samples for TEM were prepared by dispersing the fine powders of the
products in slurry in ethanol onto honeycomb carbon copper grids. For scanning electron
microscope (SEM), JEOL JSM-840 scanning electron microscope operated at 15 kV was
used. Solid-state 29Si MAS NMR spectra were recorded at room temperature on a Bruker
ASX 300 spectrometer.
5.2.4. MAT cracking evaluation
Cracking experiments were performed in an automated fixed-bed microactivity test
(MAT) unit (Zeton Automat IV), which was a modified version of ASTM D 5154. A
simplified drawing of the MAT unit is shown in Scheme 5.1. The unit was equipped with
collection systems for gas and liquid products. The distribution of gaseous products was
analyzed by gas chromatographies. The boiling point (bp) range of the liquid products was
determined by simulated distillation gas chromatography.
The catalysts were tested in the MAT unit at 510 °C with a weight hourly space
velocity (WHSV) of 8 h−1
. All samples were steamed with 20% water vapor in N2 at
550 °C for 24 h before the catalytic tests. MAT results reported include conversion, yields
of dry gas (H2, H2S, C1 and C2), liquefied petroleum gas (LPG, i.e., C3–C4), gasoline (>C5,
bp up to 215 °C), LCO (bp 215–345 °C), heavy cycle oil (HCO, bp above 345 °C) and
coke. Conversion was determined from the difference between the amount of feed and the
amount of unconverted material defined as liquid product boiling above 215 °C (i.e.,
LCO + HCO). The same vacuum gas oil (VGO) was used to all MAT runs [17].

123
Scheme 5.1. Simplified diagram of the microactivity test MAT unit for cracking
experiments.
5.3. Results and discussion
5.3.1. Synthesis of nanozeolites
Crystallization of zeolites is complicated and sensitive to synthesis conditions. Its
mechanism is still under debate. And a small change in the synthesis parameters could
result in fruitless products. Hence it is very often that the products of the syntheses of

124
nanozeolites using clear gel method are poorly crystalline and sometimes desired structures
cannot be obtained [22, 23].
An alternative approach is to apply a physical restriction into the synthesis
environment [24-27]. The physical restriction provides a nanospace for the crystallization
of zeolites inside it but prevents them from growing larger than the size of the nanospace.
Porous carbon matrices, micro emulsion and methyl cellulose have been found being a
good physical restrictor. Nevertheless, there are some difficulties that needed to be
overcome: (i) the uniformity in the nanospace size of the restrictor of carbon matrix and
methyl cellulose is not perfect, (ii) full introduction of synthesis gel into the restricting
environment is almost impossible and (iii) the stability of the restrictor under the synthesis
conditions are not acceptable.
Recently, we and other authors [16, 28-30] have proposed a novel approach for the
synthesis of nanozeolites. The idea is to apply a “soft” restriction on the crystal growth
process. This is done using an organosilane to silanize the freshly formed nanozeolites
during the crystallization, the resulting functionalized nanozeolites thus become stable
toward the subsequent growth process. In our method, an organic solvent is introduced
which can disperse these functionalized nanozeolites and completely protect them from the
growth process. Hence, fine nanoparticles can be obtained. The introduction of organic
solvent is an attractive option; the dispersion of the synthesis gel into the organic solvent
depends largely on the affinity of the solvent toward water. A study of the influence of the
solvent on the preparation of nanozeolites would be necessary and worthwhile. When a
hydrophobic solvent is used, a large amount of the solvent is needed to obtain a complete
dispersion of the synthesis gel. But for a hydrophilic solvent, the expectation is that gel
dispersion would be easier. And thanks to the higher affinity toward the gel, higher impact
on the crystal size of the final product is anticipated.
In our previous study, we used toluene as the solvent, which is hydrophobic [16, 29,
30]; hence it was difficult to obtain a homogeneous mixture of the aqueous synthesis gel in
toluene. Thus, to adjust the affinity of this solvent to water, an addition of butanol as an
additive was necessary. However, as the content of butanol increases the crystal size
becomes larger; this is due to the fact that alcoholic systems tend to favor formation of

125
large crystals [31]. So there is a compromise of butanol content; it should be sufficient for a
complete dispersion of the synthesis gel but not too high so as the effect on crystal size is
not significant. According to Qiu et al. [31], alcohol with dielectric constant lower than that
of water would slow down the polymerization and thus the crystallization rate; hence large
crystals are favored. So a good alternative solvent for the synthesis of functionalized
nanozeolites should meet the following requirements: (i) high polarity and (ii) high
solvating capacity. In short, the solvent must resemble water in terms of physicochemical
properties as much as possible while maintaining dissolution capacity of organosilane
agent.
Bearing that in mind it is clear that formamide would be a perfect solvent. The
ability of formamide as a water replacement has been well established [32-34]. It should be
noted that as formamide is an aprotic solvent, it contributes no protons to the synthesis gel.
Hence, it is expected that the role of formamide would be neutral during the synthesis
process.
To demonstrate the advantage of using formamide, we show here three
representative samples of FAU nanozeolite, the first sample FAU–TOL prepared using
toluene as the main solvent and the last two samples FAU–FOR prepared using formamide.
The obtained FT-IR spectra in the region of framework vibrations are shown in Figure 5.1.
The band at 460 cm−1
is assigned to the internal vibration of TO4 (T = Si or Al) tetrahedra.
This vibration is always observable on aluminosilicate species [10]. The band at
565 cm−1
is attributed to the vibration of the double-ring D6R units [35]. This band can be
regarded as a confirmation of the presence of a zeolitic structure. The bands at 685 and
775 cm−1
are assigned to external linkage symmetrical stretching and internal tetrahedral
symmetrical stretching, respectively. Furthermore, the bands at 1010 and 1080 cm−1
are
assigned to internal tetrahedral asymmetrical stretching and external linkage asymmetrical
stretching, respectively [20]. Overall, the FT-IR spectra of these samples match well with
the typical FT-IR absorption peaks of zeolite Y (Figure 5.1).

126
Wavenumber (cm-1)
400600800100012001400
Tra
nsm
itta
nce
(A)
(B)
(C)
Figure 5.1. FT-IR spectra of the prepared nanofaujasite samples: (A) FAU–TOL2D
prepared using toluene and pre-heated zeolite gel for 2 days at 90 °C, (B) FAU–FOR2D
prepared using formamide and pre-heated zeolite gel for 2 days at 90 °C, (C) FAU–FOR4D
prepared using formamide and pre-heated zeolite gel for 4 days at 90 °C, and (D) zeolite Y
reference.
The XRD patterns of the samples (Figure 5.2) are identical to that of the FAU
structure. There is a clear broadening of the reflections from the sample, which is attributed
to small crystals. Furthermore, no evident peak at around 2θ = 20–30° which is

127
characteristic of amorphous phase, was observed indicating that the samples are highly
crystalline.
2 theta
5 10 15 20 25 30 35
(A)
(B)
(C)
Figure 5.2. XRD patterns of nanofaujasite samples prepared: (A) FAU–TOL2D in toluene,
(B) FAU–FORM2D in formamide from the zeolite gel pre-heated at 90 °C for 2 days, (C)
FAU–FORM4D in formamide from the zeolite gel pre-heated at 90 °C for 4 days, and (D)
zeolite Y standard.

128
Figure 5.3. TEM images of (A) the sample FAU–TOL2D prepared in toluene from the
zeolite gel pre-heated at 90 °C for 2 days, (B) sample FAU–FOR2D prepared in formamide
from the zeolite gel pre-heated at 90 °C for 2 days, and (C) the sample FAU–FOR4D
prepared in formamide from the zeolite gel pre-heated at 90 °C for 4 days.
Representative micrographs of the as-made nanofaujasite samples are shown
in Figure 5.3. The crystals appear very uniform. This is expected since the nanozeolite
particles were protected from aggregation during the crystallization. The crystal size values

129
of these samples FAU–TOL2D, FAU–FOR2D and FAU–FOR4D are 40, 25 and 100 nm,
respectively. For the samples prepared in the presence of formamide, for example, the
sample FAU–FOR4D which was prepared from the clear gel that was pre-heated for 4 days
at 90 °C has larger crystal size than that of the sample FAU–FOR2D which was prepared
from the gel pre-heated for 2 days at 90 °C. It is interesting to note that, while the FAU–
FOR2D sample exhibits typical cubic single nanocrystals, the FAU–FOR4D sample shows
spherical particles. The formation of these spherical particles is attributed to the Ostwald
ripening effect, which aggregates the nanocrystals into larger one.
Figure 5.4 shows the 29
Si MAS NMR spectra of the as-made faujasite prepared in
aqueous medium in the absence of organosilane (conventional method) and silylated
nanofaujasite samples prepared in solvent medium in the presence of organosilane. For the
as-made silylated nanozeolite samples, besides the resonance peaks at −88, −95, −100 and
−103 ppm corresponding to Si(3Al), Si(2Al), Si(1Al) and Si(0Al), respectively, the peak at
−65 ppm attributed to R–C–Si–(OSi)3 species. This peak results in the reaction between the
silicon in the organosilane and the silanol groups of zeolite nuclei during the crystallization.
The NMR broad peak at 50–70 ppm could be contributed to T2 and T
3 which correspond to
two and three OH groups consumed by one organosilane molecule. This peak at −65 ppm is
absent in the faujasite sample prepared in aqueous medium in the absence of
organosilane [36,37]. As seen in Figure 5.4 for the silylated nanofaujasite samples,
Q4 signals became much broader with higher intensity as compared to those of the faujasite
one. This means that the silanization led to the transformation of Q3 to Q
4 silicon species
during the crystallization. Thus, it can be concluded that the 3 samples of functionalized
nanozeolites were obtained.

130
Figure 5.4. 29
Si MAS NMR spectra of the as-made faujasite prepared in aqueous medium
in the absence of organosilane (conventional method) and silylated faujasite samples: (A)
FAU–TOL4D using toluene pre-heated for 4 days, (B) FAU–FOR2D using formamide pre-
heated for 2 days, (C) FAU–TOL2D using toluene pre-heated for 2 days, and (D) FAU-
Standard using conventional method.
The pre-heating treatment of gel at 90 °C was an attempt to populate the
protozeolitic species which were functionalized with organosilane agent for the next
process of crystallization. The duration of the pre-heating process of zeolite gel is a
significant parameter. It should be long to make sure that the population of protozeolitic
species becomes sufficient. As the pre-heating treatment of zeolite gel was done, for the
process of crystallization in the organic solvent, larger nanoparticles obviously grow at the
expense of smaller ones. As a result, these large species even functionalized with
organosilane agent would be precipated. In this case, they settle down on the bottom of the
teflon-line, and these species aggregate into larger ones.

131
However, the preparation using formamide allows production of nanozeolites with
controlled crystal sizes. This fact is of important interest since it opens up a new method to
synthesize nanozeolite crystals with predetermined crystal size. As discussed above, it is
expected that protozeolitic species in synthesis gel pre-heated at 90 °C for 4 days would be
larger in size than those in synthesis gel pre-heated for 2 days. Hence the dispersion of the
gel pre-heated for 4 days in an organic solvent such as toluene would be more difficult
since large protozeolitic species tend to aggregate at higher extent. Nevertheless, using
formamide allows a tolerance toward these zeolite gels; hence it is well dispersed into the
solvent. This is due to the fact that formamide has physicochemical properties similar to
water, while still retaining great dissolution power toward the organosilane agents.
However, the drawback could be the increase in crystal size. Figure 5.5 shows the
N2 adsorption/desorption isotherms of different silylated nanofaujasite samples after
calcination: FAU–TOL2D, FAU–FOR2D and FAU–FOR4D. The isotherms represent a
steep rise in uptake at low relative P/Po pressure and a flat curve following, which is typical
for microporous materials. However, for FAU–TOL2D and FAU–FOR4D (Figure 5.5A
and C), an inflection at P/Po of 0.7–0.9 and a hysteresis loop are characteristic of capillary
condensation and are related to the range of mesopores owing to the interparticles, while
for FAU–FOR2D, a hysteresis loop was essentially not observed (Figure 5.5B). This could
be due to its smaller particle size (25 nm), as compared to the 40 and 100 nm size of the
other ones. The specific surface areas are 505, 515 and 570 m2/g, and the external surface
areas based on t-plot calculation are 80, 115 and 65 m2/g for FAU–TOL2D, FAU–FOR2D
and FAU–FOR4D, respectively. In addition, the external surface areas of the samples are in
agreement with the TEM analysis. The sample with a smaller size as indicated by TEM
images shows higher external surface area. Some physicochemical properties of the
faujasite samples are tabulated in Table 5.1.

132
Figure 5.5. N2 adsorption desorption isotherms of (A) FAU–TOL2D prepared in toluene
from the zeolite gel pre-heated at 90 °C for 2 days, (B) FAU–FOR2D prepared in
formamide from the zeolite gel pre-heated at 90 °C for 2 days, and (C) FAU-FOR4D
prepared in formamide from the zeolite gel pre-heated at 90 °C for 4 days.

133
Table 5.1. Physicochemical properties of nanofaujasite samples.
Sample Particle size (nm) SBET [m2/g] Sexternal[m
2/g] Pore volume [cm
3/g]
FAU–TOL2D 40 505 80 0.43
FAU–FOR2D 25 520 130 0.60
FAU–FOR4D 100 570 65 0.45
5.3.2. Synthesis of FCC
Figure 5.6. XRD patterns of the nanozeolite-based FCC catalyst samples prepared from the
corresponding 40, 24 and 100 nm nanozeolites: (A) FCC–TOL2D, (B) FCC–FOR2D and
(C) FCC–FOR4D.
XRD patterns of the nanozeolite-based FFC catalyst samples with different
nanozeolite sizes are shown in Figure 5.6. The presence of the FAU structure is observed;
however, a broad peak at 2θ = 20–30° is available, implying the presence of amorphous
matrix. The SEM images of these samples show that the FCC catalyst samples are
aggregated into micro-size particles, which are composed of uniform spheres of ~200 nm.
These spheres are merely silica, and nanozeolites are well dispersed and incorporated along

134
the silica spheres (Figure 5.7). For these resulting FCC catalysts, the silica matrix could
stabilize nanozeolites and increase the resistance of zeolite to steam deactivation and
therefore increase the FCC catalyst real-life. The N2 adsorption/desorption isotherms of
these samples are shown in Figure 5.8. The specific surface area values are 360, 355 and
315 m2/g and the pore volumes are 0.90, 0.60 and 1.10 cm
3/g for FCC–FAU–TOL2D,
FCC–FAU–FOR2D and FCC–FAU–FOR4D, respectively (Table 5.2) which are also
namely FCC-40, FCC-25 and FCC-100.
Table 5.2. BET analysis of nanozeolite-based FFC catalyst samples.
Sample SBET [m2/g] Sexternal[m
2/g] Pore volume [cm
3/g]
FCC–FAU–TOL2D 360 312 0.90
FCC–FAU–FOR2D 355 254 0.60
FCC–FAU–FOR4D 315 312 1.10
Figure 5.7. SEM image of (A) FCC–FAU–TOL2D, (B) FCC–FAU–FOR2D and (C) FCC–
FAU-FOR4D.

135
Figure 5.8. N2 adsorption desorption isotherms of (A) FCC–FAU–TOL2D, (B) FCC–
FAU–FOR2D and (C) FCC–FAU–FOR4D.

136
5.3.3. Catalytic test
Before discussing the catalytic test results we should mention here two points: (i)
nanozeolite particles are the main active components of these FCC catalysts and their
activity in the cracking reaction is of our interest. The cracking of single hydrocarbon over
nanozeolite has been reported by several authors [12, 38, 39]. This kind of reaction can
provide a general suggestion on the potential of nanozeolite. However, it is necessary to
evaluate the activity of nanozeolites in real-life application; hence the cracking of a typical
feed for FCC cracking over nanozeolite-based catalysts was carried out. (ii) the matrix
component of our nanozeolite-based catalysts was deliberately made almost neutral
(amorphous silica) to the cracking reaction so that the impact of silica matrix as inactive
matrix on the overall activity of the catalyst is negligent. Consequently, the activity of the
catalysts can be supposed to stem from only the zeolite component. In a typical FCC
catalyst, matrix component also plays an active role in the cracking of large hydrocarbon,
contributing to the conversion as a whole [17]. Thus the activity of our nanozeolite-based
catalysts regarding conversion is expected to be lower than that of the commercial ones.
The relation between conversion and catalyst-to-oil ratio is shown in Figure 5.9. A
general trend can be observed. The conversion increased with the catalyst-to-oil ratio and
eventually it reached a plateau. This trend is explainable: as the catalyst-to-oil ratio rises the
number of active sites available for the cracking reaction becomes higher resulting in
higher conversion. As the ratio reaches a critical value, this effect is less pronounced; the
conversion approaches a steady state. In agreement with our anticipation, the conversion
over these catalysts is not very high. The highest value was observed on the sample FCC-
25 (nanozeolite size ~25 nm), which is about 50%.

137
Figure 5.9. Relationship between conversion and catalyst-to-oil ratio of different prepared
FCC-samples.
The most appealing conclusion drawn from the changed course of conversion is that
it clearly demonstrates the ability of the impact of nanoparticles on catalytic activity.
Reaction activity over the catalysts rises with the decrease in zeolite particle size. At the
same catalyst-to-oil ratio the catalyst that bears the smallest nanoparticle size has the
highest value among the three samples. Since the matrix components are identical and
neutral among the catalysts, the change in activity is attributed to the larger external surface
area, hence giving higher accessibility for large hydrocarbon molecules.
The correlation of dry gas with conversion is plotted in Figure 5.10. The dry gas is
the lightest fraction of the cracking reaction. It contains C1–C2 hydrocarbons and other light
gaseous molecules H2, H2S, CO and CO2 etc. Dry gas is undesired since it has low value
and hence its amount should be kept as low as possible. In most cases, dry gas is the
product of thermal cracking and the overcracking of gasoline. Thus, it is reasonable that an
increase in dry gas is observed at higher conversion. All the three catalyst samples
exhibited a very low production of dry gas. As the conversion reaches the maximum value,
10
20
30
40
50
60
2 4 6 8 10 12 14 16 18
C/O RATIO, g/g
MA
T C
ON
VE
RS
ION
, w
t %
FCC-100
FCC-40
FCC-25

138
the highest value of dry gas obtained over these catalysts is about only 2.5% wt. However,
at the same given conversion, the yield of dry gas is in the following order: FCC-
100 > FCC-40 > FCC-25. This order suggests that on nanozeolite, the secondary cracking
and thermal cracking reactions are subdued.
Figure 5.10. Correlation of dry gas yield with conversion of different prepared FCC-
samples.
LPG fraction is one of the products of cracking reaction that is valuable. The as-
produced LPG contains C3 and C4 hydrocarbons which can be used in the commercial LPG
and as a feedstock for further chemical upgrade to other chemicals of great value such as
the octane boosters: MTBE and ETBE etc. As shown in Figure 5.11, the LPG content
increased with the rise of the conversion. Although the profiles of the yield curve of LPG
over FCC-100 and FCC-40 are similar, in general, at a given conversion, the FCC-100 gave
the highest LPG yield, followed by FCC-40 and FCC-25.
1.0
1.2
1.4
1.6
1.8
2.0
2.2
2.4
2.6
2.8
20 25 30 35 40 45 50 55
MAT CONVERSION, wt %
DR
Y G
AS
, w
t %
FCC-100
FCC-40
FCC-25

139
Figure 5.11. Correlation of LPG yield with conversion of different prepared FCC-samples.
Gasoline is the objective of the FCC process. The relation between conversion and
gasoline yield is shown in Figure 5.12. The relation profiles clearly demonstrate the
advantage of nanozeolites. The catalyst containing smaller zeolite particles give higher
gasoline yield, and the yield gap among these catalysts rises with the increase of
conversion. Furthermore, an important parameter to evaluate the efficiency of an FCC
catalyst is the gasoline selectivity, which is defined as the ratio of gasoline to conversion.
The relationship between gasoline selectivity and conversion is shown in Figure 5.13.
Generally, the selectivity of gasoline decreased as the conversion increased. However, the
catalyst with smaller zeolite particles retained its higher selectivity.
2
4
6
8
10
12
14
16
20 25 30 35 40 45 50 55
LP
G, w
t %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

140
Figure 5.12. Correlation between gasoline yield and conversion of different prepared FCC-
samples.
LCO (Light Cycle Oil) is the product of which the value changes seasonally. LCO
is used as the feedstock to be upgraded to diesel and/or fuel oils. In an ideal cracking
process, LCO is the intermediate product of a chain cracking reaction:
HCO => LCO => gasoline. LCO is both the product of the cracking of HCO and the
reactant for the cracking to gasoline. The content of LCO produced can be taken as a
parameter reflecting the competition between these two reactions. Cracking of large
molecule cannot be done inside zeolite pores due to the small opening of its pores. In
addition, the matrix component of the catalyst is essentially inactive. Hence, cracking of
LCO and HCO must be realized on the external surface of nanozeolites. Thus, the catalyst
offering more of external surface area would give higher efficiency in cracking of large
molecules. Taking into account the chain cracking scheme above, it is expected that, at the
low conversion, the content of large molecules that are likely to be cracked is high; hence,
the LCO produced rises as the conversion rises. However, with an increase of the
conversion, the source of these molecules depleted; hence, after a maximum value of
0
10
20
30
40
20 25 30 35 40 45 50 55
GA
SO
LIN
E,
wt %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

141
conversion, the rate of the cracking of large molecule is exceeded by the rate of cracking
LCO; thus the content of LCO is decreased.
Figure 5.13. Relationship between gasoline selectivity and conversion of different prepared
FCC-samples.
Figure 5.14 shows the relationship between the LCO yield and the conversion. The
convex curves of the LCO profiles over the FCC-100 and FCC-25 are noticed. For the
FCC-40, the trend is different; the LCO yield exhibits a continuous decrease with the
increase of conversion. But there is a consistent order among these three catalysts: at a
given conversion, the yield of LCO is as follows: FCC-25 > FCC-40 > FCC-100.
40
45
50
55
60
65
70
20 25 30 35 40 45 50 55
GA
SO
LIN
E S
ELE
CT
IVIT
Y, w
t %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

142
Figure 5.14. Relationship between LCO yield and conversion.
HCO fraction is the undesired product of the FCC process. It contains the aromatic
hydrocarbons that are difficult to crack and sulfur. Hence, HCO yield should be diminished
to minimum. The relationships between HCO and conversion are in agreement with our
expectation (Figure 5.15): HCO yield is reduced using nanosized zeolites; the smaller the
zeolite particles the lower the HCO yield.
Coke is an inevitable product and the only product that cannot be recovered. Being
the catalyst poison and apparently giving no value in commercial applications, the coke
formation is undesired and its amount should be as low as possible. The relationship
between coke yield and conversion is shown in Figure 5.16. The FCC-25 showed the least
coke selectivity among the three catalysts.
15
17
19
21
23
25
20 25 30 35 40 45 50 55
LC
O, w
t %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

143
Figure 5.15. Relation between HCO yield and conversion of different prepared FCC-
samples.
In conclusion of the evaluation of FCC cracking, a clear trend has been noticed: the
activity increases with the decrease in crystal size of the nanozeolites. This is due to the fact
that cracking of FCC feed is heavily realized on external surface, which is higher on
nanozeolite. The activity of the catalyst as a whole (the conversion) was not very high; it is
deliberate since the matrix component was made neutral, and it is likely that the acidity of
nanozeolites is not sufficient. However, the addition of nanozeolite in FCC catalyst as main
component or additive is an interesting option.
20
30
40
50
60
20 25 30 35 40 45 50 55
HC
O, w
t %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

144
Figure 5.16. Relationship between coke yield and conversion of different prepared FCC-
samples.
5.4. Conclusion
In this study, we have reported new methods of preparing nanozeolites using
toluene and formamide solvents as crystallization medium instead of water. Different
crystal sizes, e.g., 25, 40 and 100 nm, were prepared in toluene and formamide solvents. It
was demonstrated that the solvents play an important role in giving the zeolite crystal with
desired size. The key parameter that is important to choose the suitable solvent is its
solvating power and its hydrophobicity. Nanozeolite-based FCC catalysts were prepared
using silica as inactive matrix in order to study the effect of crystal size on the performance
of nanozeolites. These FCC catalysts were evaluated with FCC feedstock. The relationship
between gasoline selectivity and conversion is a function of nanozeolite size. In general, the
performance of these catalyst is in the following order FCC-25 > FCC-40 > FCC-100.
Acknowledgments
1
2
3
4
5
6
7
8
20 25 30 35 40 45 50 55
CO
KE
, w
t %
MAT CONVERSION, wt %
FCC-100
FCC-40
FCC-25

145
This work was supported by the Natural Sciences and Engineering Research
Council of Canada (NSERC) and the Research Application Development Society
(SOVAR-Quebec). G.T.V. thanks the foundation of Laval University (FUL) for the
scholarship. The authors thank Prof. S. Kaliaguine for stimulating discussions and
comments.

146
References
1. L. Tosheva, V.P. Valtchev, Chem. Mater., 17 (2005), pp. 2494–2513
2. A. Corma, Chem. Rev., 97 (1997), pp. 2373–2420
3. A. Corma, E. Sastre, J. Catal., 129 (1991), pp. 177–185
4. T.-O. Do, A. Nossov, M.-A. Springuel-Huet, C. Schneider, J.L. Bretherton, C.A. Fyfe,
S. Kaliaguine, J. Am. Chem. Soc., 126 (2004), pp. 14324–14325
5. T.-O. Do, S. Kaliaguine, Angew. Chem. Int. Ed., 40 (2001), pp. 3248–3251
6. T.-O. Do, S. Kaliaguine, Angew. Chem. Int. Ed., 41 (2002), pp. 1036–1040
7. A. Karlsson, M. Stöcker, R. Schmidt, Micropor. Mesopor. Mater., 27 (1999), pp. 181–
192
8. L. Yu, Z. Wenzhong, J.P. Thomas, Angew. Chem. Int. Ed., 40 (2001), pp. 1255–1258
9. Z. Zongtao, H. Yu, Z. Lei, W. Runwei, Y. Yi, Q. Shilun, Z. Dongyuan, X. Feng-Shou,
Angew. Chem. Int. Ed., 40 (2001), pp. 1258–1262
10. D. Breck, Zeolite Molecular Sieves: Structure, Chemistry, and Use, Wiley Interscience,
New York (1974)
11. W.M.M.Ch. Baerlocher, D.H. Olson, Atlas of Zeolite Framework Types, Elsevier
(2001)
12. B.W. Wojciechowski, A. Corma, Catalytic Cracking: Catalysts, Chemistry, and
Kinetics (1986)
13. E.F.S. Aguiar, M.L.M. Valle, M.P. Silva, D.F. Silva, Zeolites, 15 (1995), pp. 620–623
14. M.A. Camblor, A. Corma, A. Martínez, F.A. Mocholí, J.P. Pariente, Appl. Catal., 55
(1989), pp. 65–74
15. K. Rajagopalan, A.W. Peters, G.C. Edwards, Appl. Catal., 23 (1986), pp. 69–80
16. G.-T. Vuong, T.-O. Do, Micropor. Mesopor. Mater., 120 (2009), pp. 310–316
17. S. Al-Khattaf, Energy Fuels, 17 (2003), p. 62
18. R. Ravishankar, C. Kirschhock, B.J. Schoeman, P. Vanoppen, P.J. Grobet, S. Storck,
W.F. Maier, J.A. Martens, F.C. De Schryver, P.A. Jacobs, J. Phys. Chem. B, 102
(1998), pp. 2633–2639
19. P. Morales-Pacheco, F. Alvarez-Ramirez, P. Del Angel, L. Bucio, J.M. Dominguez, J.
Phys. Chem. C, 111 (2007), pp. 2368–2378
20. P. Morales-Pacheco, F. Alvarez, L. Bucio, J.M. DominguezJ. Phys. Chem. C, 113
(2009), pp. 2247–2255
21. T.M. Davis, T.O. Drews, H. Ramanan, C. He, J. Dong, H. Schnablegger, M.A.
Katsoulakis, E. Kokkoli, A.V. McCormick, R.L. Penn, M. Tsapatsis, Nat. Mater., 5
(2006), pp. 400–408
22. C.S. Cundy, P.A. Cox, Chem. Rev., 103 (2003), pp. 663–702
23. C.S. Cundy, J.O. Forrest, R.J. Plaisted, Micropor. Mesopor. Mater., 66 (2003), pp.
143–156
24. C.J.H. Jacobsen, C. Madsen, T.V.W. Janssens, H.J. Jakobsen, J. Skibsted, Micropor.
Mesopor. Mater., 39 (2000), pp. 393–401
25. I. Schmidt, C. Madsen, C.J.H. Jacobsen, Inorg. Chem., 39 (2000), pp. 2279–2283
26. H. Wang, B.A. Holmberg, Y. Yan, J. Am. Chem. Soc., 125 (2003), pp. 9928–9929
27. Z. Chen, S. Li, Y.S. Yan, Chem. Mater., 17 (2005), pp. 2262–2266
28. D.P. Serrano, J. Aguado, J.M. Escola, J.M. Rodriguez, A. Peral, Chem. Mater., 18
(2006), pp. 2462–2464

147
29. G. T. Vuong, T.-O. Do, in: US (Ed.), US, 2008.
30. G.T. Vuong, T.-O. Do, J. Am. Chem. Soc., 129 (2007), pp. 3810–3811
31. S. Qiu, J. Yu, G. Zhu, O. Terasaki, Y. Nozue, W. Pang, R. Xu, Micropor. Mesopor.
Mater., 21 (1998), pp. 245–251
32. A. Lattes, E. Perez, I. Rico-Lattes, C. R. Chim., 12 (2009), pp. 45–53
33. M. Almgren, S. Swarup, J.E. Loefroth, J. Phys. Chem., 89 (1985), pp. 4621–4626
34. I. Rico, A. Lattes, J. Phys. Chem., 90 (1986), pp. 5870–5872
35. G. Coudurier, C. Naccache, J.C. Vedrine, J. Chem. Soc., Chem. Commun. (1982), pp.
1413–1415
36. B.Z. Zhan, M.A. White, M. Lumsden, Langmuir, 19 (2003), pp. 4205–4210
37. B.Z. Zhan, M.A. White, P. Fancy, C.A. Kennedy, M. Lumsden, Macromolecules, 37
(2004), pp. 2748–2753
38. P.B. Venuto, E.T. Habib Jr. Fluid Catalytic Cracking with Zeolite Catalysts, CRC
Press (1979)
39. B.C. Gates, J.R. Katzer, G.C.A. Schuit Chemistry of Catalytic Processes, McGraw-Hill
College (1979)


149
Chapter 6. Synthesis and Engineering Porosity of Mixed
Metal Fe2Ni-MIL-88B Metal-Organic Framework
Gia-Thanh Vuong, Minh-Hao Pham and Trong-On Do *
Department of Chemical Engineering, Laval University, Quebec G1K 7P4, Canada
Published in Dalton Transactions 2013, 42 (2), 550-557
DOI: 10.1039/C2DT32073H
Résumé
Une nouvelle approche a été développé pour la synthèse de Fe2Ni MIL-88B en
utilisant des clusters neutres de métaux mixtes, Fe2Ni(µ3-O). Ces clusters occupent les
nœuds du réseau MIL-88B, au lieu du mono-métal, Fe3 (µ3-O) avec un anion compensateur
présent dans le matériau Fe3MIL-88B non-poreux qui est obtenu par la méthode
conventionnelle. De ce fait, en absence des anions compensateurs dans la structure, Fe2Ni
MIL-88B devient un matériau poreux. De plus, la combinaison de la flexibilité de MIL-
88B et des métaux mixtes comme nœuds dans le réseau, la porosité peut être contrôlée par
échange des ligands terminaux du réseau. Ceci nous a permis de moduler d‟une manière
réversible la porosité ainsi que la surface spécifique du Fe2Ni MIL-88B à différents
niveaux, dépendamment de la taille des ligands échangés.
Abstract
A new rational approach has been developed for the synthesis of mixed metal MIL-
88B metal organic framework based on neutral mixed metal cluster, such as Fe2Ni(µ3-O)
cluster. Unlike the conventional negative charged single metal cluster, the use of neutral
mixed metal cluster as nodes in the framework avoids the need of compensating anion
inside porous MIL-88B system; thus mixed metal MIL-88B becomes porous. The
flexibility of the mixed metal MIL-88B can be controlled by terminal ligands with different
steric hindrance. This allows us to reversibly customize the porosity of MIL-88B structure
at three levels of specific surface area as well as the pore volume

150
6.1. Introduction
MOFs (Metal-organic frameworks) are ordered structures of metal clusters
connected by organic linkers.1 This combination of an inorganic entity and an organic one
can result in highly porous MOF crystals which show promising applications in adsorption,
catalysis and drug delivery.2-6
A classic example is the combination of trimeric M3(µ3-O)
clusters (M = Fe, Cr) with benzenedicarboxylate which gives rise to MIL-101, MIL-88B
and MOF-235 structures of which only MIL-101 shows porosity.7-13
As the metal is
trivalent, a guest anion is needed to balance the charge of the cluster14
. With regard to
porosity, this anion is not desired. It blocks the pores, rendering the MOF structure non-
porous as in the case of MIL-88B and MOF-235.9,11
Only MIL-101 which has sufficiently
large pore size can afford the high porosity.10
In return, the dense MIL-88B structure is not
rigid but flexible, the network can expanse upon adsorption of particular solvents. Such a
great increase of 125% of unit cell volume can be obtained.10,15
This distinctive feature
coined breathing effect by Férey et al. is of great interest since it provides a facile control
over the porosity by changing solvents.10,15
Unfortunately, the flexibility of MIL-88B
structure is not permanent without solvent molecules. Solvent molecules must remain in its
pores to sustain the expansion hence despite there is a gain in pore size and pore volume,
the pores are filled with solvent molecules and thus become inaccessible for adsorbates.
Upon removal of solvents, the structure shrinks, and MIL-88B returns to its dense state. A
workaround to sustain the porosity of MIL-88B structure has been recently reported.16
Instead of the straight and simple linker benzenedicarboxylate, functional groups were
introduced to the phenyl ring of the linker thus giving a steric hindrance effect to the linker.
The obtained series of functionalized MIL-88B can resist the shrinkage, and one of them,
MIL-88B(4CH3), can yield specific area up to 1200 m2/g.
16 However the reversibility of the
breathing effect was not reported by the authors. And it is likely that the breathing
magnitude in functionalized MIL-88B could become less due to the hindrance effect of the
functional group, the structure is more likely “fixed”.
On the other hand, most of MOFs are based on single metals.17,18
Many properties
of MOFs are dependent on the metal component such as the stability, magnetic behavior,
density etc. Therefore, a successful preparation of mixed metal MOFs would provide an

151
effective way to fine tune the properties of MOFs. Direct syntheses of mixed metal MOFs
are difficult, due to the high complexity upon addition of a second metal. Each metal can
form separate clusters irrelevant and unconnected to the other metal clusters, resulting in a
mixture of discrete MOFs. Rare-earth metals with high and flexible coordination capacity
are able to join together with transition metals in some MOF structures.19-21
However, it is
not the case for main group and transition metals. The syntheses of mixed transition metal
MOFs often employ linkers bearing free reactive functional groups that can coordinate with
other metal in a pre- or post-synthesis treatment.22-28
In this fashion, the second metal
contributes nothing to the construction of the framework structure, but resides within it as
an attachment. Few attempts which involve the selective introduction of the second metal
are reported to include both metals in the nodes of the framework. In these mixed metal
MOFs, the transition metal and the other one are distributed periodically among the nodes
of the framework.18
Although mixed metal clusters such as trimeric metal carboxylate have been
reported since 1911,14,29
however, to the best of our knowledge, there has been no report on
MOF structures built on them. In fact, synthesis and characterization of trimeric mixed
metal acetates of Fe(III) and first row divalent transition metal such as Co, Mn, Ni and Zn
have been well established.14
Thanks to the presence of the divalent metal, the mixed metal
cluster is balanced in charge without a compensating anion. Judging from the similarity
between the mixed metal cluster and the single metal cluster, we believe that it is possible
to obtain analogs of those MOF structures such as MIL-101, MIL-88 from these mixed
metal trimeric clusters. Because no compensating anion is needed in the framework, the
pore blockage by anions is avoided, and the mixed metal MOF could become porous
(Scheme 6.1).

152
Scheme 6.1. Mixed metal building unit forming porous MOF. Black: C, yellow: divalent
metal (Ni), green: Fe, olive: balancing anion (Cl, Br etc.), Red: O

153
Scheme 6.2. Reversible ligand exchange and porosity control of Fe2Ni-MIL-88B, for
clarity, only terminal ligands bonding to Ni are showed.
In this paper, we report a new approach for the synthesis of mixed metal MIL-88B
using mixed metal cluster (Scheme 6.1) and the control of the breathing of the obtained
mixed metal MOF via the steric hindrance of the terminal ligands (Scheme 6.2). To
illustrate our approach, we have selected the synthesis of the MIL-88B structure based on
Fe2Ni(µ3-O) cluster (denoted as Fe2Ni-MIL-88B). Our Fe2Ni-MIL-88B product shows
drastic change over the original single metal Fe3-MIL-88B in regard to porosity, and

154
considerately high N2 and CO2 adsorption. But the most interesting feature of Fe2Ni-MIL-
88B is its switchable porosity and specific surface area (30 m2/g to 1120 m
2/g) and pore
volume (10 x 10-3
cm3/g to 448 x 10
-3 cm
3/g). Unlike the conventional single metal Fe3-
MIL-88B, Fe2Ni-MIL-88B can switch reversibly and permanently from dense state to
porous one upon ligand exchange with certain terminal ligands.
6.2. Experiments
Sample preparation: In a typical synthesis, 0.67 mmol of FeCl3.6H2O 99%, 0.33
mmol of corresponding Ni(NO3)2.6H2O 97% and 1 mmol of bdc 98% were dissolved in 10
ml of DMF. To this clear solution, 0.4 mmol of NaOH was added under stirring for 15 min.
The mixture was then transferred into a Teflon-lined autoclave and heated at 100 oC for 15
h. Solid product was then recovered by filtration and washed several times with DMF.
Elemental analysis of the synthesized sample showed Fe 12.5 wt%, Ni 6.2 wt%, N 5.3
wt%. Thus, the suggested formula Fe2NiO(OOC-C6H4-COO)3.3DMF is designated as
Fe2Ni-MIL-88B.DMF. The sample was treated with water, pyridine (Py), pyrazine (Pz) and
4-,4‟-bipyridine (Bp) to obtain Fe2Ni-MIL-88B.H2O, Fe2Ni-MIL-88B.Py, Fe2Ni-MIL-
88B.Pz and Fe2Ni-MIL-88B.Bp, respectively (see Supporting Information). For
comparison, the single metal Fe3-MIL-88B.DMF was also prepared using the procedure of
Férey et al.30
Characterization: N2 and CO2 adsorption tests were carried out in an Autosorb 1
instrument, before analysis the samples were outgassed in vacuum for 3 hours at 150 oC.
Specific surface area was calculated with the BET model in the linear range of P/Po =
0 – 0.15. FTIR was carried in a FT-BIORAD 450s system using KBr disc. UV-VIS was
carried in a Cary 300 instrument using MgO disc. Powder X-ray diffraction (XRD) patterns
were collected on a Bruker SMART APEX II X-ray diffractometer with Cu Kα radiation (λ
= 1.5406 Å) in the 2θ range of 1 – 50° at a scan rate of 1.0° min–1
. For XRD measurement
of samples in Figure 1 and for crystal lattice calculation, the samples were dried in vacuum
overnight at 100 oC, then the analysis was taken immediately. Scanning electron
microscopy (SEM) images were taken on a JEOL 6360 instrument at an accelerating
voltage of 3 kV.

155
6.3. Results
6.3.1. Synthesis of Mixed Metal Fe2Ni-MIL-88B with Different Terminal
Ligands
Details of the synthesis and ligand exchanges are described in the experimental
section and in Supporting Information. Elemental analysis of the synthesized sample
showed Fe 12.5 wt%, Ni 6.2 wt%, N 5.3 wt%. Thus, the suggested formula Fe2NiO(OOC-
C6H4-COO)3.3DMF is designated as Fe2Ni-MIL-88B.DMF. The sample was treated with
water, pyridine (Py), pyrazine (Pz) and 4-,4‟-bipyridine (Bp) to obtain Fe2Ni-MIL-
88B.H2O, Fe2Ni-MIL-88B.Py, Fe2Ni-MIL-88B.Pz and Fe2Ni-MIL-88B.Bp, respectively.
For comparison, the pristine Fe3-MIL-88B.DMF was also prepared using the procedure of
Férey et al.30
2
5 10 15 20 25 30 35 40 45 50
(a)
(b)
Figure 6.1. XRD patterns of Fe2Ni-MIL-88B.H2O (a) and XRD simulation of the Fe3-MIL-
88B (b).

156
The XRD patterns of all of Fe2Ni-MIL-88B samples were collected, however, due
to the flexibility of the MIL-88B structures, which will be explained in the next session, the
best way to determine the structure of the samples is to compare the completely dense
phase Fe2Ni-MIL-88B.H2O with the XRD simulation of the standard Fe3-MIL-88B. As
showed in Figure 6.1, the XRD of the Fe2Ni-MIL-88B.H2O is identical to the simulation
one, in addition no guest phase is found, implying the MIL-88B structure with the high
purity of the sample.
Table 6.1. IR analysis of the MIL-88B samples
Sample
Wavenumber (cm-1)
asymC sym(OCO) 31 DMF 32,33 Py 34-36 Pz 34 Bp 35 Fe2NiO/Fe3O 37,38
Fe2Ni-MIL-88B.Py 1606 1382 224 1486,
1447,
703 633
718
Fe2Ni-MIL-88B. H2O 1590 1381 209 717
Fe2Ni-MIL-88B.DMF 1609 1385 224 1660 718
Fe2Ni-MIL-88B.Pz 1603 1383 220 1416 717
Fe2Ni -MIL-88B.Bp 1609 1385 224 1431,
1408, 635
718
Fe3-MIL-88B 1601 1393 208 1666 624
The FTIR analysis of the samples is in agreement with the suggested MIL-88B
formula (for details, see Table 6.1 and Figure S1-S6, Supporting Information). There are
four remarks: (i) no free acid (no band at 1700 cm-1
) is found, (ii) the value = asym(OCO)
–sym(OCO) corresponds well to the bridge coordination mode of metal carboxylate,31
(iii)
the bands characteristic of H2O, DMF, Py, Pz and Bp are present on the samples Fe2Ni-
MIL-88B.H2O, Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py, Fe2Ni-MIL-88B.Pz and Fe2Ni-
MIL-88B.Bp, respectively; and (iv) the vibration of cluster Fe2Ni(µ3-O) (~718 cm-1
) is
observed, while the vibration of Fe3(µ3-O) (620cm-1
) is only noticed in the single metal
Fe3-MIL-88B sample.37,38

157
Figure 6.2. UV-Vis spectra of Fe2Ni-MIL-88B.Bp (a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-
MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) Fe2Ni-MIL-88B.H2O (e) and Fe3-MIL-88B (f)

158
Table 6.2. Crystal parameters of the Fe2Ni-MIL-88B samples
Sample a [Å] c [Å] Unit cell volume
[Å3] V/V (%)[a]
Fe2Ni-MIL-88B.H2O 11.2 19.1 2075 ~
Fe2Ni-MIL-88B.DMF
- Open phase 13.7 17.8 2893 39.4
- Dense phase 11.3 19.1 2112 1.8
Fe2Ni-MIL-88B.Py
- Open phase 14.2 17.4 3039 46.4
- Dense phase 11.3 18.5 2046 -1.4
Fe2Ni-MIL-88B.Pz
- Open phase 14.4 17.8 3197 54.1
- Dense phase 11.0 19.0 1991 -4.0
Fe2Ni-MIL-88B.Bp 14.1 17.4 2996 44.4
[a] The increase in unit cell volume compared with the Fe2Ni-MIL-88B.H2O sample
The UV-Vis spectra of the Fe2Ni-MIL-88B samples with different terminal ligands
are shown in Figure 6.2. The UV-Vis analysis also confirms the presence of Ni as well as
its octahedral coordination mode in the MIL-88B structures. The band at 244 nm is
assigned to the ligand-to-metal charge transfer, implying the bonding of carboxylate
oxygen to metal. Most of the transition bands of Ni2+
are obscured by those of Fe3+
.
However, the presence of the band at 760 nm, which is characteristic of the transition [3A2g
=> 1Eg(D)] of Ni in the tri-nuclear complex is observed. The transition [
6A1g =>
4A1g +
4Eg(G)] in Fe
3+ is also found at 350 - 500 nm.
39,40 Especially, the [
6A1g =>
4T2g] transitions
at 550 - 650 nm of Fe3+
, which are sensitive to the ligand field energy, clearly reveal the
bonding of the terminal ligand to the metal and the effect of the divalent metal Ni in the
complex. These transitions of the samples Fe2Ni-MIL-88B.H2O and Fe2Ni-MIL-88B.DMF
which involve the weak field terminal ligand H2O and DMF, are observed at 575 nm.
Accordingly, for the samples involving the strong field ligands Py, Pz and Bp, these bands
are shifted to lower energy (higher wavelength at 625 nm). This behavior is in agreement
with the higher ligand field energy of Py, Pz, and Bp than that of H2O and DMF.40
In the
Fe2Ni complex under the effect of Ni, the ligand field in Fe reduces,39
thus the [6A1g =>
4T2g] transition in Fe3-MIL-88B.DMF is at 525 nm while it is observed at 575 nm in the
Fe2Ni-MIL-88B.DMF. As seen in Figure S7, Supporting Information, the change in color

159
of the samples depends on the nature of terminal ligand (a-d) as well as on the presence of
Ni in the structure (d, f).
Figure 6.3. XRD patterns of Fe2Ni-MIL-88B samples, the planes of open phase are in
black, the planes of dense phase are in red and placed in boxes. Fe2Ni-MIL-88B.Bp (a),
Fe2Ni-MIL-88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) and Fe2Ni-MIL-
88B.H2O (e)
6.3.2. Reversible Breathing Control Using Terminal Ligand
The breathing behavior of the MIL-88B has been well documented by Férey et al.8
and can be studied in details by investigation of the XRD patterns in the 2 range from 7 to
12o. Figure 6.3 shows the XRD patterns of the mixed metal samples with different terminal
ligands, which also confirms the MIL-88B structure. For comparison the XRD pattern of

160
Fe3-MIL-88B is also displayed. As Férey et al. have reported,8 the swelling up of MIL-88B
structure causes the splitting and shifting to low 2 of the plane (100) and (101), while the
plane (002) is shifted to higher 2. Thus these planes can be used as the indicators of the
swollen (open) phase as well as the dense phase in the samples. In Figure 6.3, the planes
assigned to the dense phase are in red and in box, while the ones assigned to the open phase
are in black and without box. For the Fe2Ni-MIL-88B.H2O sample, only the dense phase is
observed. The Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py and Fe2Ni-MIL-88B.Pz samples
feature both open phase and dense phase. The Fe2Ni-MIL-88B.Bp sample exhibits an open
structure without any trace of the dense phase. Assuming the Fe2Ni-MIL-88B samples
taking the same hexagonal lattice structure as the original Fe3-MIL-88B, calculations of the
unit cell parameters using the assigned planes in Figure 6.3 were carried out. The results
listed in Table 6.2 are in agreement with the plane assignment. Upon swelling, the lattice
constant a increases from 11.0 Å to 14.4 Å while the c constant decreases from 19.1 Å to
17.5 Å. Consequently, these changes are reflected in the unit cell volume. The unit cell
volume of the open phase is about 40 - 50 % higher than that of the dense phase.
Obviously, the XRD patterns have demonstrated the effect of the terminal ligand on the
swelling degree of the samples. In terms of steric hindrance, the terminal ligands can be
classified into three groups: low hindrance of H2O, intermediate of DMF, Py and Pz and
high hindrance of Bp. The small ligand water with low steric hindrance hence cannot swell
up the structure. In fact, the Fe2Ni-MIL-88B.H2O sample exhibits a dense phase. The
introduction of larger ligands DMF, Py and Pz with higher steric hindrance brings about the
openness of the structure of the obtained Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py and
Fe2Ni-MIL-88B.Pz samples, but the openness is not full yet as some of the dense phase still
remains. The samples show both open and dense phases. Eventually, the ligand Bp, which
has the largest size and the steric hindrance among the terminal ligands used in this study,
yields the fully opened Fe2Ni-MIL-88B.Bp.
6.3.3. Adsorption Analysis
Figure 6.4A shows the nitrogen adsorption isotherms of the Fe2Ni-MIL-88B
samples with different terminal ligands, which are also in agreement with the XRD results.
All the isotherms of the Fe2Ni-MIL-88B samples show the characteristic of microporous

161
materials with the isotherm reaching a plateau at very low partial pressure. As the steric
hindrance of the terminal ligand increases from the small size of water to the middle size of
DMF, Py, Pz and finally to the large size of Bp, the BET specific surface area of the
samples also exhibits three levels: (i) dense (non-porous to N2), 30 m2/g for Fe2Ni-MIL-
88B.H2O; (ii) porous, 355 - 550 m2/g for Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py,
Fe2Ni-MIL-88B.Pz; and (iii) highly porous, 1120 m2/g for Fe2Ni-MIL-88B.Bp. The same
trend for their micropore volumes is also observed, showing three levels of pore volume. If
the micropore volume is taken as a factor measuring the breathing, then the pore volume of
the Fe2Ni-MIL-88B.Bp sample is about 44 times higher than that of the Fe2Ni-MIL-
88B.H2O sample. Interestingly, the micropore diameters calculated by the HK model41
are
also consistent with the steric hindrance of the corresponding terminal ligands (Figure
6.4B). The Fe2Ni-MIL-88B.Bp sample shows an average pore size of 6.3 Å, the highest
value among the samples. The Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py and Fe2Ni-MIL-
88B.Pz samples exhibit the value of 5.2 Å, while the pore volume of the dense structure
Fe2Ni-MIL-88B.H2O is negligible. Details of the porosity and the specific surface area of
the samples are shown in Table 6.3.
In addition to N2 adsorption, the CO2 adsorption measurements at low pressure (up
to 1 atm) at 273 K were also carried out (Figure 6.5). Again, the CO2 adsorption capacity of
the samples increases with the increase in porosity of the samples. The Fe2Ni-MIL-88B.Bp
sample shows the highest capacity, 101 cc/g STP of CO2, corresponding to a capacity of 20
wt %. This is one of the highest values (at 273 K at 1 atm) among MOF materials to date.42
The high CO2 adsorption capacity of the Fe2Ni-MIL-88B.Bp could be also due to the
presence of free pyridyl group of Bp in Fe2Ni-MIL-88B.Bp, which has a high affinity with
CO2.

162
P/Po
0.0 0.2 0.4 0.6 0.8 1.0
Vo
lum
e a
ds
orb
ed
[cc/g
]
0
100
200
300
400
(a)
(b)
(c)
(d)
(e)
Pore diameter [Å]
4 5 6 7 8
dD
(w)
[a.u
.]
(a)
(b)
(c)
(d)
(e)
(A) (B)
Figure 6.4. N2 adsorption isotherms at 77 K (A) and pore size distributions (B) of Fe2Ni-
MIL-88B.Bp (a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d)
and Fe2Ni-MIL-88B.H2O (e).

163
P [torr]
0 76 152 228 304 380 456 532 608 684 760
Volu
me a
dsorb
ed [
cc/g
]
0
20
40
60
80
100(a)
(b)
(c)
(d)
(e)
Figure 6.5. CO2 adsorption isotherms at 273 K of Fe2Ni-MIL-88B.Bp (a), Fe2Ni-MIL-
88B.Pz (b), Fe2Ni-MIL-88B.Py (c), Fe2Ni-MIL-88B.DMF (d) and Fe2Ni-MIL-88B.H2O (e)
Table 6.3. Porosity of Fe2Ni-MIL-88B
Samples Specific surface area
[m2/g]
Micropore volume
[10-3
cm3/g]
Pore width [Å]
Fe2Ni-MIL-88B.H2O 30 10 ~
Fe2Ni-MIL-88B.DMF 355 140 5.2
Fe2Ni-MIL-88B.Py 549 216 5.2
Fe2Ni-MIL-88B.Pz 465 186 5.2
Fe2Ni-MIL-88B.Bp 1120 448 6.3
6.4. Discussion
It is clear from the XRD analysis that our synthesized samples have the MIL-88B
structure with high purity. Both FTIR and UV-Vis analyses and elemental analysis indicate
the presence of Ni in the mixed metal MIL-88B samples. Interestingly, the spectroscopy

164
techniques provide us with unambiguous evidence of the presence of Fe2NiO cluster which
is the building unit of the mixed metal MIL-88B. The presence of Fe3O cluster which
implies Fe3-MIL-88B was not detected by FTIR and UV-Vis analyses of the mixed metal
samples. Hence this suggests the mixed metal Fe2Ni-MIL-88B structure.
Mixed metal Fe2Ni-MIL-88B exhibits stable, controllable and permanent porosity,
much better than single metal Fe3-MIL-88B. The spectroscopy results revealed that the
substituting ligands Py, Pz, and Bp are bound to the framework via a chemical bond to the
metal atom, not physically packing. In fact, DMF, H2O, Py, Pz and Bp are the terminal
ligands in the Fe2NiO cluster. The XRD analysis and adsorption isotherm measurements of
the samples Fe2Ni-MIL-88B.H2O, Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.Py, Fe2Ni-MIL-
88B.Pz and Fe2Ni-MIL-88B.Bp demonstrate that the porosity of the mixed metal Fe2Ni-
MIL-88B can be controlled by the size of the terminal ligands. Since the terminal ligand is
chemically bonded to the metal rather than a weak physical packing, the impact is
permanent. The samples retained their porosity after various types of treatment: drying in
vacuum at 150 °C for 24h, and/or exposure to air at room temperature for days. In contrast,
this behavior was not observed on single metal Fe3-MIL-88B treated with Py or Bp under
the same conditions; no improvement in porosity was found on Fe3-MIL-88B after the Py
exchange. This drastic difference in the breathing behavior between the Fe2Ni-MIL-88B
and original Fe3-MIL-88B stems from their different breathing mechanisms. While
breathing effect in Fe3-MIL-88B originates from the packing of solvent molecules, this
effect in Fe2Ni-MIL-88B comes from the bonding of neutral ligands to the trinuclear
clusters. The “swelling up” in Fe2Ni-88B would be dependent on the size of the ligand and
its orientation in bonding with the ligand. The larger the ligand is the higher is the swelling
up. And since bonding to each metal atom in MIL-88B structure is a terminal ligand (H2O
or DMF), which is ready to be replaced, it is possible to introduce larger terminal ligand
into the MIL-88B structure which helps retain the breathing effect. Common terminal
ligands for trimeric clusters are pyridine and its derivatives such as pyrazine and 4,4‟-
bipyridine,14,43
which have different steric hindrances and thus can be used as the breathing
agent of the MIL-88B structure, as illustrated in Scheme 7.2.

165
It should be noted that for the original single metal Fe3-MIL-88B, the activation
conditions affect strongly the specific surface area. For example badly activated Fe3-MIL-
88B(CH3) solid that still contains some DMF, exhibit a significant BET surface area while
the fully water exchange sample is non porous when dried.16
This behaviour is attributed to
the dependency of Fe3-MIL-88B on the solvent to retain its porosity, as long as the solvent
is present, the pores are opened. Thus solvent molecule is the pore opening agent for Fe3-
MIL-88B. In case of Fe2Ni-MIL-88B it is the terminal ligand that keeps the pores open and
since the terminal ligand is chemically bonded to the framework, it is expected that the
porosity of Fe2Ni-MIL-88B would be more stable. However, a more quantitative work such
as PXRD simulation will be necessary to confirm the mechanism of the pore opening in
mixed metal MIL-88B.
For rigid structure, addition of guest molecule into the pores always results in
decrease in surface area; this is due to the fact that the guest molecule will partially block
the pores, thus reducing the accessible surface. For example the attachment of chiral
organic ligand to Cr3-MIL-101 reduces the surface area by 70%.44
However this trend is
reversed in mixed metal Fe2Ni-MIL-88B, the larger the ligand is, the higher the surface
area is obtained. It is the flexibility of the mixed metal Fe2Ni-MIL-88B that gives this
interesting behavior. As the XRD patterns shows (Figure 7.3), the large ligand can trigger
the mixed metal Fe2Ni-MIL-88B framework to expand further, opening the pores, thus
gaining surface area.
The secondary divalent metal (Ni) in the mixed metal Fe2Ni-MIL-88B is also very
important to the obtained structure. As discussed earlier, the introduction of the divalent
metal helps avoid the need of the blocking anions, in addition the bond strength of the
cluster to the terminal ligand can be improved by selection of the second divalent metal
which has higher affinity to the ligand. For the trimeric mixed metal Fe2III
MII(µ3-O)(µ2-
O2CCH3)6L3 with L being pyridine derivative terminal ligand and M divalent metal,
Novitchi et al.43
found that the stability of the bond Ni-L is 44 times higher than Fe-L; and
Co-L is 6 times higher than Fe-L. Moreover, in a classic paper by Irving and Williams the
stability constants of the pyridine-based [MII(Py)(H2O)5]
2+ follow the order: Mn
2+(0.14) <
Fe2+
(0.6) < Co2+
(1.14) < Ni2+
(1.78). 45
In fact, the improvement in stability of Ni-based

166
complexes has been well established, thanks to the maximal crystal field stabilization
energy of Ni2+
.45
Hence, it is suggested that the mixed metal MOF based on Fe2Ni(µ3-O)
cluster would exhibit stronger binding affinity to pyridine-like terminal ligands. Moreover,
taking into account the anion-free state of the mixed metal MOF, the ligand can orient with
less restriction inside the pores to attain a proper bond to the metal.
Beside the exceptional advantage of permanent porosity, the use of terminal ligand
as a swelling agent has another fascinating feature: reversibility. The terminal ligand is
exchangeable without affecting the linkers and the nodes of the framework. As illustrated
in Scheme 2, the samples of Fe2Ni-MIL-88B.DMF, Fe2Ni-MIL-88B.H2O, Fe2Ni-MIL-
88B.Py, and Fe2Ni-MIL-88B.Pz can be mutually converted with the corresponding ligand
exchange. The XRD and N2 adsorption isotherm results show that the samples retain their
crystallinity as well as porosity after the various conversion cycles. The Fe2Ni-MIL-88B.Bp
sample can be converted to Fe2Ni-MIL-88B.DMF and then from this state it can be
converted to bear any other ligand of Py, Pz, DMF and H2O. It means that the porosity of
Fe2Ni-MIL-88B can be switched in-situ to yield the necessary porosity, one of the desired
features of smart porous materials.
Unlike zeolites and mesoporous inorganic materials regarded as rigid and fixed-
pore structures, the reversible change in pore size of Fe2Ni-MIL-88B is achieved by the
breathing of the whole structure with the use of the terminal ligands to sustain it.
Consequently, Fe2Ni-MIL-88B can also provide at least three different states of porosity
(Scheme 6.2). In fact, breathing can bring about 300 % change in volume in some MOFs.
In addition, the terminal ligands are available in many sizes as well as the bond strength.
Hence we believe the pore size control in MOFs is much easier and higher in magnitude
and importantly not exclusive to Fe2Ni-MIL-88B, but available to other MOFs capable of
breathing.
Another general advantage of trimeric mixed metal is that the ability to tether
functional groups to the MOF structure. In this study, with the use of Pz and Bp, the free
pyridyl group becomes available in MIL-88B structure. It is very likely that other
functional groups such as carboxyl, aldehyde can be introduced in the same manner using
the corresponding ligands such as nicotinic acid and pyridinecarboxaldehyde. The key

167
point about trimeric mixed metal MOFs is that the functional group can be prepared
separately, and then attaches to the MOF structure as a removable module, the MOF hence
becomes a flexible docking station for various types of functional modules.
6.5. Conclusion
In the field of adsorption and membrane technologies, a smart material which can
switch itself from high to low porosity or vice versa could be extremely beneficial. Imagine
an adsorbent that can take in desired gas molecules with high capacity, but it can also
become inaccessible for them upon terminal ligand exchange; or a membrane which can let
certain gas molecules pass through can be shut off entirely to them if necessary. Many
applications could benefit from this kind of “on and off” materials.
We regard the Fe2Ni-MIL-88B material as one step toward the creation of a truly
smart porous material. Its versatility lies in its switchable and reversible three-level
porosity. Different levels could be attained depending on the nature of stimulant terminal
ligand. Their obvious applications are of course adsorption and separation in which the
pore size of the material can be reversibly controlled to be wide opened, half opened or be
completely closed at will.
In conclusion, we have succeeded synthesizing MIL-88B structure based on mixed
metal cluster of Fe(III) and Ni(II). This mixed metal cluster helps bring in the porosity to
the MOF product and an exact control over the porosity and surface area by using simple
stimulant terminal ligands. We believe that this rationale approach is not restricted to the
MIL-88B structure but it can cover all other flexible MOF structures which are based on
trinuclear metal carboxylates MIL-101, MIL-100, MIL-88 and MOF-235 etc. With the rich
choices of the mixed metal clusters from a large collection of trimeric mixed metal cluster
M2III
MII (M
III: Fe, Cr, Mn, Rh and M
II: Ca, Ba, Mg, Ni, Mn, Co) and the great selections of
terminal ligands among N-based, S-based O-based ligands new fascinating properties of
trimeric mixed metal MOFs are more to come
† Electronic Supplementary Information (ESI) available: Details of synthesis, XRD
patterns, FTIR spectra SEM images and reversible ligand exchange. See
DOI: 10.1039/b000000x/

168
Refrences
1. O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim,
Nature, 2003, 423, 705-714.
2. A. Corma, H. Garc a and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606-4655.
3. J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc.
Rev., 2009, 38, 1450-1459.
4. J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477-1504.
5. L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294-1314.
6. P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D.
Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories,
L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat Mater, 2010, 9, 172-178.
7. M.-H. Pham, G.-T. Vuong, A.-T. Vu and T.-O. Do, Langmuir, 2011, 27, 15261-15267.
8. C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk and G. Ferey,
Science, 2007, 315, 1828-1831.
9. S. Surble, C. Serre, C. Mellot-Draznieks, F. Millange and G. Ferey, Chem. Commun.,
2006, 284-286.
10. G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I.
Margiolaki, Science, 2005, 309, 2040-2042.
11. A. C. Sudik, A. P. Côté and O. M. Yaghi, Inorg. Chem., 2005, 44, 2998-3000.
12. J. H. Yoon, S. B. Choi, Y. J. Oh, M. J. Seo, Y. H. Jhon, T.-B. Lee, D. Kim, S. H. Choi
and J. Kim, Catal. Today, 2007, 120, 324-329.
13. M.-H. Pham, G.-T. Vuong, F.-G. Fontaine and T.-O. Do, Crystal Growth & Design,
2011, 12, 1008-1013.
14. R. D. Cannon and R. P. White, in Prog. Inorg. Chem., John Wiley & Sons, Inc., 1988,
vol. 36, pp. 195-298.
15. G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380-1399.
16. P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M.
Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey
and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839-17847.
17. D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem.
Soc. Rev., 2009, 38, 1257-1283.
18. S. R. Caskey and A. J. Matzger, Inorg. Chem., 2008, 47, 7942-7944.
19. B. Zhao, X.-Y. Chen, P. Cheng, D.-Z. Liao, S.-P. Yan and Z.-H. Jiang, J. Am. Chem.
Soc., 2004, 126, 15394-15395.
20. B. Zhao, P. Cheng, Y. Dai, C. Cheng, D.-Z. Liao, S.-P. Yan, Z.-H. Jiang and G.-L.
Wang, Angew. Chem. Int. Ed., 2003, 42, 934-936.
21. J.-W. Cheng, J. Zhang, S.-T. Zheng, M.-B. Zhang and G.-Y. Yang, Angew. Chem. Int.
Ed., 2006, 45, 73-77.
22. E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long
and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382-14384.
23. S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem.
Commun., 2006, 2563-2565.

169
24. C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131,
9492-9493.
25. S. J. Garibay, Z. Wang and S. M. Cohen, Inorg. Chem., 2010, 49, 8086-8091.
26. F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132,
15390-15398.
27. K. C. Szeto, K. P. Lillerud, M. Tilset, M. Bjørgen, C. Prestipino, A. Zecchina, C.
Lamberti and S. Bordiga, The Journal of Physical Chemistry B, 2006, 110, 21509-
21520.
28. K. C. Szeto, C. Prestipino, C. Lamberti, A. Zecchina, S. Bordiga, M. Bjørgen, M. Tilset
and K. P. Lillerud, Chem. Mater., 2007, 19, 211-220.
29. R. Weinland and H. Holtmeier, Z. Anorg. Allg. Chem., 1928, 173, 49-62.
30. S. Bauer, C. Serre, T. Devic, P. Horcajada, J. r. m. Marrot, G. r. Férey and N. Stock,
Inorg. Chem., 2008, 47, 7568-7576.
31. U. Kumar, J. Thomas and N. Thirupathi, Inorg. Chem., 2009, 49, 62-72.
32. A. Fateeva, P. Horcajada, T. Devic, C. Serre, J. Marrot, J.-M. Grenèche, M. Morcrette,
J.-M. Tarascon, G. Maurin and G. Férey, Eur. J. Inorg. Chem., 2010, 2010, 3789-3794.
33. Y. J. Kim and C. R. Park, Inorg. Chem., 2002, 41, 6211-6216.
34. M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001,
190, 261-274.
35. K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination
Compounds, John Wiley & Sons, Inc., 2008, pp. 1-273.
36. K. N. Wong and S. D. Colson, J. Mol. Spectrosc., 1984, 104, 129-151.
37. M. Yazdanbakhsh, H. Tavakkoli, M. Taherzadeh and R. Boese, J. Mol. Struct., 2010,
982, 176-180.
38. L. Meesuk, U. A. Jayasooriya and R. D. Cannon, Spectrochimica Acta Part A:
Molecular Spectroscopy, 1987, 43, 687-692.
39. A. B. Blake and A. Yavari, J. Chem. Soc., Chem. Commun., 1982, 1247-1249.
40. A. B. Blake, A. Yavari, W. E. Hatfield and C. N. Sethulekshmi, J. Chem. Soc., Dalton
Trans., 1985, 2509-2520.
41. G. Horváth and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470-475.
42. K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-
H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724-781.
43. G. Novitchi, F. Riblet, R. Scopelliti, L. Helm, A. Gulea and A. E. Merbach, Inorg.
Chem., 2008, 47, 10587-10599.
44. M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim,
J. Am. Chem. Soc., 2009, 131, 7524-7525.
45. H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192-3210.

170
Supporting information
Preparation
In a typical synthesis, 0.67 mmol of FeCl3.6H2O 99%, 0.33 mmol of corresponding
Ni(NO3)2.6H2O 97% and 1 mmol of bdc 98% were dissolved in 10 ml of DMF. To this
clear solution, 0.4 mmol of NaOH was added under stirring for 15 min. The mixture was
then transferred into an Teflon-lined autoclave and heated at 100 oC for 15 h. Solid product
was then recovered by filtration and washed several times with DMF.
Characterization
N2 and CO2 adsorption tests were carried out in an Autosorb 1 instrument, before analysis
the samples were outgassed in vacuum for 3 hours at 150 oC. Specific surface area was
calculated with the BET model in the linear range of P/Po = 0 – 0.15. KBr solid state
FTIR was carried in a FT-BIORAD 450s system. MgO solid state UV-VIS was carried in a
Cary 300 instrument. Powder X-ray diffraction (XRD) patterns were collected on a Bruker
SMART APEX II X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) in the 2θ
range of 4 – 20° at a scan rate of 1.0° min–1
. For XRD measurement of samples in Figure 3
and for crystal lattice calculation, the samples were dried in vacuum overnight at 100 oC,
then the analysis was taken immediately. Peak fitting was carried out using Jade software
package (http://www.materialsdata.com/).Simulation of Fe3-MIL-88B XRD pattern was
done on the crystalography data reported by Férey et al 1 using Mercury software package
(https://www.ccdc.cam.ac.uk/products/mercury/) Scanning electron microscopy (SEM)
images were taken on a JEOL 6360 instrument at accelerating voltage of 3 kV
FTIR

171
Figure S1. FTIR spectra of Fe2Ni-MIL-88B.DMF
Figure S2. FTIR spectra of Fe2Ni-MIL-88B.Py
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
asym
C
Py
sym
(OCO)
asym
C
Fe2NiO
sym
(OCO)
DMF
Fe2NiO Py

172
Figure S3. FTIR spectra of Fe2Ni-MIL-88B.Pz
Figure S4. FTIR spectra of Fe2Ni-MIL-88B.Bp
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
asym
C
asym
C
sym
(OCO)
sym
(OCO)
Fe2NiO
Fe2NiO
Pz
Bp
Bp

173
Figure S5. FTIR spectra of Fe2Ni-MIL-88B.H2O
Figure S6. FTIR spectra of Fe3-MIL-88B.DMF
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
400600800100012001400160018002000
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Wavenumbers [1/cm]
Tra
nsm
itta
nce
asym
C
asym
C
sym
(OCO)
sym
(OCO)
Fe2NiO
Fe2NiO
Bp

174
Figure S7. Images of Fe2Ni-MIL-88B.Bp (a), Fe2Ni-MIL-88B.Pz (b), Fe2Ni-MIL-88B.Py
(c), Fe2Ni-MIL-88B.DMF (d) Fe2Ni-MIL-88B.H2O (e) and Fe-MIL-88B.DMF (f).
d
b
d c
f e
a

175
Figure S8. SEM image of as-synthesized Fe2Ni-MIL-88B.DMF
Ligand exchange reactions:
+ DMF => H2O: 0.25 g of Fe2Ni-MIL-88B.DMF was added with 10 ml of water. The
obtained mixture was stirred at room temperature for 3 hours, and then the solid was
recovered by filtration and dried at 100 ⁰C overnight. XRD pattern showed in Figure 3e
+ H2O=>DMF: 0.25 g of Fe2Ni-MIL-88B.H2O was added with 10 ml of DMF. After
stirring for 30 min, the mixture was transferred into an autoclave and it was placed in oven

176
at 110 ⁰C for 3 days. The product was recovered by filtration and washed several times
with DMF (Figure S9), BET specific surface area: 320 m2/g.
Figure S9: XRD pattern of Fe2Ni-MIL-88B.DMF obtained from Fe2Ni-MIL-
88B.H2O
+ DMF=>Py: 0.25 g of Fe2Ni-MIL-88B.DMF was added with 10 g of pyridine. Very
quickly the solid changed its color from brown to olive green. The mixture was stirred for 3
hours. The product was filtrated and dried in vacuum at 100 oC overnight. XRD pattern
showed in Figure 3c.

177
+ Py = > DMF. 0.25 g of Fe2Ni-MIL-88B.Py was added with 10 ml of DMF. The mixture
was stirred at 100 oC for 3 days while the color gradually changed from olive green to
yellow. The product was filtrated, washed with DMF and dried in vacuum at 100 oC
overnight (Figure S10). BET specific surface area: 340 m2/g.
Figure S10: XRD pattern of Fe2Ni-MIL-88B.DMF obtained from Fe2Ni-MIL-88B. Py
+ DMF = > Pz: 0.25 g of Fe2Ni-MIL-88B.DMF was added with 10 g of pyrazine. The
mixture was heated to 70 oC as pyrazine melted, stirring was applied for 3 hours. The olive
green product was recovered by hot filtration and dried in vacuum at 100 oC overnight.
XRD pattern showed in Figure 3b.

178
+ Pz => DMF: 0.25 g Fe2Ni-MIL-88B.Pz was added with 10 ml of DMF, the mixture was
transferred into an autoclave and heated at 100 oC for 3 days. Brown solid product was
filtrated and washed with DMF before drying in vacuum at 100 o
C overnight (Figure S11).
BET specific surface area: 325 m2/g.
Figure S11: XRD pattern of Fe2Ni-MIL-88B.DMF obtained from Fe2Ni-MIL-88B.
Pz

179
+ Pz = > H2O: 0.25 of Fe2Ni-MIL-88B.Pz in a vial was added with 15 ml of water. The vial
was then sealed and stirred at 95 oC. After 3 hours the brown product was filtrated and
dried in vacuum at 100 oC overnight (Figure S12). BET specific surface area: 15 m
2/g.
Figure S12: XRD pattern of Fe2Ni-MIL-88B.H2O obtained from Fe2Ni-MIL-88B. Pz
+ H2O => Pz: 0.25 g of Fe2Ni-MIL-88B.H2O was added with 10 g of pyrazine. The mixture
was heated at 100 oC for 3 days. The olive green product was recovered by filtration and
dried in vacuum at 100 oC overnight (Figure S13). BET specific surface area: 420 m
2/g.

180
Figure S13: XRD pattern of Fe2Ni-MIL-88B.Pz obtained from Fe2Ni-MIL-88B. H2O
+ Py => H2O: 0.25 of Fe2Ni-MIL-88B.Py in a vial was added with 15 ml of water. The vial
was then sealed and stirred at 95 oC. After 3 hours the brown product was filtrated and
dried in vacuum at 100 oC overnight (Figure S14), BET specific surface area: 10 m
2/g.

181
Figure S14: XRD pattern of Fe2Ni-MIL-88B.H2O obtained from Fe2Ni-MIL-88B. Py
+ H2O => Py: 0.25 g of Fe2Ni-MIL-88B.H2O was added with 10 ml of pyridine. The
mixture was sealed in a vial and stirred at 100 oC for 4 days. Olive green product was
filtered and dried in vacuum at 100 oC overnight (Figure S14). BET specific surface area:
530 m2/g

182
Figure S15: XRD pattern of Fe2Ni-MIL-88B.Py obtained from Fe2Ni-MIL-88B. H2O
+ DMF => Bp: 0.16 g of bipyridine was introduced to 2 ml of DMF, to this solution 0.12 g
of Fe2Ni-MIL-88B.DMF was added. The mixture was then stirred at 100 oC for 4 days.
Olive green product was filtered and dried in vacuum at 100 oC. XRD pattern showed in
Figure 3a

183
Figure S16: XRD pattern of Fe2Ni-MIL-88B.DMF obtained from Fe2Ni-MIL-88B.Bp
+ Bp => DMF: 0.12 g of Fe2Ni-MIL-88B.Bp was added with 15 ml of DMF, the mixture as
transferred into an autoclave and it was placed in an oven at 100 oC for 6 days. Brown
product was filtered and washed with DMF before drying in vacuum overnight at 100 oC
(Figure S16). BET specific surface area: 330 m2/g.
References
(1) S. Bauer, C. Serre, T. Devic, P. Horcajada, J. r. m. Marrot, G. r. Férey, N. Stock,
Inorg. Chem. 2008, 47, 7568


185
Chapter 7. Direct Synthesis and Mechanism for the
Formation of Mixed Metal Fe2Ni-MIL-88B
Gia-Thanh Vuong, Minh-Hao Pham and Trong-On Do *
Department of Chemical Engineering, Laval University, Quebec G1K 7P4, Canada
Submitted to CrystEngComm 2013.
Résumé
Le mécanisme de synthèse a été étudié pour la synthèse Fe3-MIL88B et Fe2Ni-
MIL88B. Ces matériaux ont été caractérisés par différentes techniques, telles que la
spectroscopie UV-Vis, IR et Raman et la diffraction des RX. Les résultats montrent que
pour la synthèse de Fe3-MIL88B, le mono-métal Fe3-MOF-235 se forme en première étape
de synthèse et joue le rôle de précurseur (germe) pour la formation de Fe3-MIL-88B: les
germes MOF-235 sont formés, ensuit se transforment en Fe3-MIL-88B. Dans le cas
d‟utilisation du cluster de métaux mixtes Fe2Ni(µ3-O), les mono-métaux Fe3-MOF-235
formés en premier étape jouent le rôle comme germes pour la croissance de matériau
Fe2Ni-MIL88B. L‟anion FeCl4- est très important pour le succès de la formation de MOF-
235. Un mécanisme d‟anion médiateur dans la formation de MOF-235 a été suggéré.
Abstract
The direct synthesis of Fe3-MIL-88B and Fe2Ni-MIL-88B was analyzed using
different characterization techniques including UV-Vis, IR, Raman spectroscopies and
XRD. It was found that single metal Fe3-MOF-235 seeds which were formed from the first
stage of synthesis are as precursors for the formation of MIL-88B. Fe3-MOF-235 seeds
formed in the first stage of synthesis were then transformed to Fe3-MIL-88B in the case of
single metal, and to mixed Fe2Ni-MIL88B in the case of mixed metal synthesis. In the both
cases of Fe3-MIL-88B and Fe2Ni-MIL-88B, FeCl4- anion is a key feature to the formation
of MOF-235. An anion mediated mechanism for the formation of MOF-235 structure is
also suggested.

186
7.1. Introduction
Metal-organic frameworks (MOFs) are one of the fastest growing fields of
chemistry.[1] The structure of MOFs is formed by a polymeric connection of a metal
cluster in coordination bond with an organic linker, which results in a vast collection of
MOFs.[2] As a typical MOF structure, MIL-88B is of special interest due to its potentials in
adsorption, catalysis, biomedicine.[3-7] Its structure is built on the connection of 1,4-
bezenedicarboxylate (bdc) with trinuclear oxo-centered metal cluster (Me3O, Me = Fe, Cr,
Sc). Several trinuclear metal clusters have been reported in successful synthesis of MIL-
88B yielding Fe3-MIL-88B, Cr3-MIL-88B and Sc3-MIL88B.[6, 8, 9] In our recent
publication,[10] a novel route to prepare a new type of mixed metal MIL-88B structure was
reported. Unlike the conventional negative charged single metal cluster, the use of neutral
mixed metal cluster as nodes in the framework avoids the need of compensating anion
inside porous MIL-88B system. As a result, this mixed metal MIL-88B becomes porous.
Furthermore, the flexibility of the mixed metal MIL-88B can be controlled by terminal
ligands with different steric hindrance. This allows us to reversibly customize the porosity
of MIL-88B structure at three levels of specific surface area as well as the pore volume.[10]
In the synthesis of trinuclear-based MOFs such as MIL-88B and MIL-101, when
iron and 1,4-bezenedicarboxylic acid (bdc) are used in the presence of DMF, the kinetically
and thermodynamically stable phases are MIL-88B and MIL-101 which form at low
temperatures (≤100 °C), and thermodynamically stable MIL-53, which forms at higher.[11,
12] When a second metal is introduced in the reaction medium to obtain the mixed metal
MOF, the situation becomes more complicated. There are competing reactions to yield
single metal and mixed metal MOFs. It is also necessary to determine which factors
promote the single metal MOF and which factors promote the mixed metal MOFs and why.
A detailed study of the synthesis of both the single metal and mixed metal of trinuclear
based MOF could contribute to the understanding of the kinetics and mechanism of the
MOF formation
In this study, as the continuing part of the previous publication,[10] we report the
effect of several factors on the formation of both single metal Fe3-MIL-88B and mixed
metal Fe2Ni-MIL-88B. It occurred to us that even though the synthesis of mixed metal

187
MOFs is more complicated than that of the single metal one, the presence of the second
metal was found to be helpful in determination of the important initial solid Fe3-MOF-235,
revealing a vital template effect from a surprising source: halogen anion. The impacts of
pH, concentration, second metal were considered and illustrated. We also found similarities
in the principles of synthesis between zeolites and MOFs, which can be used as a guideline
for the synthesis of porous materials.
7.2. Experimental Section
Chemicals: FeCl3.6H2O (99%) and Fe(NO3)3.9H2O (98%), 1,4-bezenedicarboxylic
acid (bdc, 98%), NaOH (99%), N,N-dimethyl-formamide (DMF) were used as purchased.
Synthesis: Syntheses of Fe3-MIL-88B and Fe2Ni-MIL-88B were carried out
following our previous work.[10] Two series of samples with different times of
crystallization were prepared using two different iron sources, FeCl3.6H2O and
Fe(NO3)3.9H2O: single metal and mixed metal based MIL-88B. (i) For single metal based
MIL-88B synthesis: 5 vials of 10 ml DMF solution containing 10 mmol of FeCl3.6H2O
99% (or Fe(NO3)3.9H2O 98%) was added with 10 mmol 1,4-bezenedicarboxylic acid (bdc)
under stirring at room temperature. Subsequently, 4 ml of NaOH 2M was rapidly injected
under continuous stirring. The vials were then capped and heated at 100 oC for different
times: 0 h (e.g., 5 min after the addition of NaOH at room temperature), 1h, 2h, 3h, and 12
h. (ii) For mixed metal based MIL-88B synthesis: the same procedure was also applied for
the synthesis of mixed metal MIL-88B, except that 10 ml DMF solution containing 3.33
mmol of Ni(NO3)2.6H2O and 6.67 mmol of FeCl3.6H2O 99% (or Fe(NO3)3.9H2O 98%)
were used. Solids products were recovered by centrifugation at 5000 rpm for 5 min. The
solids were then dried in vacuum for 24 h at 50 oC. In general, the samples prepared with
FeCl3.6H2O yield firm solids. However, those prepared from Fe(NO3)3.9H2O become thick
gel during the heat treatment and thus, their corresponding solid products are not as firm as
the solids prepared from FeCl3.6H2O. The samples are designated as [Metal cluster]-[Anion
type]-[Synthesis time]. For example, Fe2Ni-Cl-5h is the mixed metal MIL-88B sample
prepared at 100 oC for 5 h using FeCl3
.6H2O.

188
Characterization Methods: FTIR was carried in a FT-BIORAD 450s instrument
using KBr disc. FTIR spectra were normalized by setting the transmittance value of the
band at 750 cm-1
which represents the strong vibration of the C-H bond to 0.05. UV-Vis
analysis was carried out in a Cary 300 instrument using MgO disc as the reference sample.
Normalization of the spectra was done by setting the value of the strongest absorbance
band at 350 nm to 1. Raman analysis was carried out with a Horiba U100 Raman
spectrometer using excitation wavelength of 514 nm. The spectra were normalized by
setting the value of the strong absorbance band of the benzene ring at 1615 cm-1
to 1.
Powder X-ray diffraction (XRD) patterns were collected on a Bruker SMART APEX II X-
ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) in the 2θ range of 1 - 20° at a scan
rate of 1.0° min-1
. HRTEM analysis was carried out with a Hitachi HF-2000 Field Emission
TEM. EDS analysis was done with a fine electron probe of 3 nm, the acquiring time was set
to 200 sec. Samples were dispersed on a copper grid.
7.3. Results
UV-Vis spectra: UV-Vis spectra of the two series of single metal and mixed metal
based samples are shown in Figure 7.1 and Figure 7.2. For the single metal synthesis, the
UV-Vis spectra of the samples do not change much over the synthesis times; it is likely that
the Fe3+
species (octahedral configuration) remains the same throughout the synthesis. For
the mixed metal samples, most of the transition bands of Ni2+
are obscured or overlapped
by those of Fe3+
, however, the presence of octahedral Fe3+
is verified. The transition [6A1g
=> 4A1g +
4Eg(G)] in Fe
3+ is found at 350 - 500 nm and the [
6A1g =>
4T2g] transition at 550 -
650 nm is also attributed to Fe3+
.[13, 14] In contrast to the single metal, the UV-Vis
spectra of the mixed metal samples change considerably with the synthesis time. In the
Fe2Ni complex under the effect of Ni,[13] the ligand field in Fe reduces, thus the [6A1g =>
4T2g] transition in single metal samples (Fe3O) is at 525 nm while it is observed at 575 nm
in the Fe2Ni. In the spectra of mixed metal Fe2Ni-MIL-88B, the band at 760 nm is
observed. This band is characteristic of the transition [3A2g =>
1Eg(D)] of Ni in the tri-
nuclear cluster [13] and thus it can be used as an indicator of the formation of the mixed
metal cluster Fe2NiO. The evolution of the UV-Vis spectra over synthesis time of both
syntheses using Fe(NO3)3.9H2O and FeCl3.6H2O exhibits a similar trend. At 0 h, the spectra

189
are similar to the single metal samples; no band at 760 nm is observed. However after 3 h,
the band 760 nm is found in two series of mixed metal samples prepared using
Fe(NO3)3.9H2O and FeCl3.6H2O, the intensity of this band increases with synthesis time,
up to 12 h (Fig. 2). This suggests that at the early stage of synthesis, only Fe3+
is present in
the solid, and after that the mixed metal cluster Fe2Ni is formed. In other words, the
formation of Fe3O cluster takes place first, followed by that of the mixed metal cluster
Fe2NiO. The difference between the mixed metal syntheses using Fe(NO3)3.9H2O and
FeCl3.6H2O is the rate of the formation of Fe2NiO cluster. For the sample prepared using
Fe(NO3)3.9H2O, the band at 760 nm is very weak even after 12 h of synthesis; however,
for the sample prepared using FeCl3.6H2O, this band is readily observed after only 2 h of
synthesis and it still remains its strong intensity after 12 h (Figure 7.2).
FT-IR spectra: The FTIR band at 1660 cm-1
which is characteristic of DMF was
observed on all of the samples (Supporting information), as also reported in ref [15]. The
presence of free bdc linker (FTIR band at 1700 cm-1
) [16] was observed on the samples
prepared from Fe(NO3)3.9H2O but not on those prepared from FeCl3.6H2O. This absence of
free acid bdc in the Cl- based samples is interesting given the fact that no effort was made
to wash the obtained solid off free bdc acid. This observation implies that under these
investigated conditions; H2BDC is mostly deprotonated when Cl- is used. The free bdc
observed in the NO3- based samples also relates to its gel-like behavior of the products
while the Cl- based synthesis produces firmly solid products.
Detailed analysis was focused on the wavelength range from 400 – 800 cm-1
which
includes the well-documented framework vibration of the trinuclear cluster.[17, 18] The
FTIR band assignments in this range are showed in Table 7.1. Beside the presence of the
vibrations of the organic ligand bdc, the vibration of the central oxygen in single Fe3O and
mixed Fe2NiO clusters at 720 and 620 cm-1
was observed, respectively. Since the central
oxygen is available only in the trinuclear clusters, these FTIR bands are considered as their
indicators. And the presence of Fe3O and Fe2NiO can be distinguished.
For the single metal samples at different synthesis times, only the Fe3O vibration
(600 – 625 cm-1
) which is of interest is shown in Figure 7.3. The vibration of Fe3O is
observed in all the samples. The band is well defined at synthesis time 0 h and continues to

190
remain strong at the synthesis time 12 h. This implies that the cluster Fe3O is formed fast
and remains stable under the synthesis conditions. For the samples that employ Cl-, the
FTIR band characterisric of Fe3O is more intense and well-defined than those using NO3-.
The FT-IR spectra of the mixed metal samples are shown in Figure 7.4. The spectra
exhibit a gradual development of Fe2NiO clusters, and concomitantly with a decrease of
Fe3O ones. For the samples using Cl-, at 0 h, (Figure 7.4B) after the addition of NaOH at
room temperature, the FTIR band of Fe3O was visible, while the band of Fe2NiO was not
found. However, after 1h, the FTIR band of Fe2NiO was observed and the one of Fe3O
decreased. At 2 h and 3 h, the FTIR band of Fe2NiO is prominent while that of Fe3O
becomes very weak. Finally after 12 h, only the well-defined band Fe2NiO was observed
while the band of Fe3O almost disappeared. For the samples using NO3- (Figure 7.4A), the
same trend was also observed. The FTIR band of Fe3O appears first right at 0 h and
decreases with the synthesis time. In contrast, the band of Fe2NiO was observed only after
3 h. Thus, using Fe(NO3)3.9H2O, the formation of Fe2NiO clusters is much slower than that
using FeCl3.6H2O. After 12 h, both FTIR bands of Fe3O and Fe2NiO were still observed.
In short, FTIR analysis of the mixed metal synthesis using NO3- (e.g.,
Fe(NO3)3.9H2O) or Cl- (e.g., FeCl3.6H2O) shows that both Fe3O and Fe2NiO clusters were
produced, first Fe3O then followed by Fe2NiO. For the mixed metal synthesis using Cl-,
with increasing synthesis time, the Fe2NiO cluster becomes the main one, while the Fe3O
cluster diminishes. In contrast, for the mixed metal synthesis using NO3-, both types of the
clusters are observed at the end of the synthesis.
Raman spectra: The similar results were also observed for the Raman spectra. The
bdc linker was found on all of the samples. The band at 631 cm-1
is assigned to the in-plane
bending of the carboxylate group OCO.[19-21] The weak band at 1125 cm-1
is attributed to
the vibration of C-COO.[22] The benzene ring in bdc gives rise to the vibrations at 860,
1146 and 11616 cm-1
. [19-21] The Raman spectra also reveal some key inorganic
components in the samples. The vibrations of the trinuclear cluster are found at two weak
bands 175 and 267 cm-1
.[23] The medium band at 430 cm-1
is assigned to the metal oxygen
bond.[17] The bands at around 567cm-1
found only on the mixed metal samples are
tentatively assigned to the asymmetric stretching mode of Fe2NiO cluster.[19-21]

191
However, the most interesting results in the Raman analysis are the determination of
the compensating anions for the Fe3O cluster, e.g. NO3- and FeCl4
-. As discussed earlier, for
the single metal Fe3O-based material, each Fe3O carboxylate cluster in the framework
needs an anion to balance the charge. A presence of Fe3O clusters in the framework implies
the necessary accompany of these anions. For synthesis using Fe(NO3)3.9H2O, nitrate is
likely the only anion available.
For the mixed metal syntheses using FeCl3.6H2O, beside the available nitrate which
comes from Ni(NO3)2, the source of anions can also include Cl- and FeCl4
-. Although it is
impossible to determine Cl- in Raman spectra, the detection of FeCl4
- and NO3
- is
feasible.[17] The stretching vibration of FeCl4- leads to a well-defined band at 330 cm
-
1.[17, 24, 25] For NO3
-, the symmetric N-O stretching vibrations give rise to the strong
band at 1044 cm-1
. [17, 26, 27] Details of the band assignments are listed in Table 2. For
the single and mixed metal syntheses using Fe(NO3)3.9H2O thus having only NO3- as the
anion source, the presence of nitrate is found on all the samples, as observed by the Raman
band at 1044 cm-1
. Nitrate anion content correlates well with Fe3O clusters in the
framework by the intensity of this Raman band at1044 cm-1
(Figure 7.6A). In the single
metal synthesis, the band 1044 cm-1
of nitrate remains stable, in correlation with the readily
formed Fe3O clusters (Figure 7.5A). In the mixed metal synthesis using Fe(NO3)3.9H2O,
the band 1044 cm-1
grows in strength and shape over the synthesis time, implying an
increase of the single metal cluster Fe3O (Figure 7.6A). Taking into account of the FTIR
spectra results, it suggests that although there is a competition from the formation of
Fe2NiO, the formation of the Fe3O cluster is still favored when Fe(NO3)3.9H2O is used. In
the mixed metal syntheses using FeCl3.6H2O, Raman spectra (Figure 7.6B) shows the
presence of anion FeCl4- by the Raman band at 330 cm
-1, which diminished during the
synthesis; however, there is no nitrate found in the sample, this fact implies the high
selectivity of FeCl4- and Cl
- anions over NO3
- of the clusters. In the single metal synthesis,
FeCl4- is found right at 0 h, however the Raman band at 330 cm
-1 of FeCl4
- decreases
sharply during the synthesis, after 3 h, this band of FeCl4- is vanished (Figure 7.5B). It is
likely that FeCl4- decomposes, providing additional iron source for the growing Fe3O
clusters and leaving Cl- as the balancing anion in the cluster. In the mixed metal synthesis
using FeCl3.6H2O, this Raman band of FeCl4- at 330 cm
-1 is much smaller. Also the

192
decomposition of FeCl4- is much faster, after 2 h, no significant band of FeCl4
- was
observed, implying the formation of mixed metal cluster readily dominates and no
compensating anion is needed as compared to that of the single metal one.
Thus, the spectroscopy data have allowed us to determine and distinguish Fe3O and
Fe2NiO clusters as well as the balancing anions that accompany the formation of Fe3O
clusters. The results from the spectroscopy analysis suggest that: (i) in the single metal
synthesis, Fe3O clusters as nodes in the MOF structure were formed, regardless of anion
used; (ii) in the mixed metal syntheses, when Cl- was used in the synthesis mixture, single
Fe3O clusters were formed at the first stage, followed by the formation of mixed metal
Fe2NiO in the framework, which subsequently becomes the main clusters with increasing
the synthesis time. At the initial stage, FeCl4- is the main balancing anion for the Fe3O
clusters in the framework. It is then decomposed. Finally, the Cl- becomes the balancing
anion for the Fe3O cluster. When NO3- was used, both Fe2NiO and Fe3O clusters were
produced, however, free bdc was found. Trinuclear Fe3O cluster exhibits high preference of
Cl- based anion (FeCl4
- or Cl
-) as balancing anion over NO3
-. It is however noted that the
spectroscopy data cannot reveal how the nodes and linkers arrange in space, in other words,
they cannot confirm the crystalline or amorphous structure of the solids. To determine the
structure and phase, the XRD analysis is needed.
XRD analysis: XRD patterns of the Fe3- NO3-x (A) and Fe3-Cl-x (B) samples as a
function of synthesis time are shown in Figure 7.7. The results revealed that, for single
metal syntheses at 100 o
C, the use of NO3- as anion in the synthesis mixture yields no
definitive structure even after 12 h of synthesis, while using Cl-, the Fe3-Cl-x sample
exhibits a gradual structure change from MOF-235 to MIL-88B as a function of synthesis
time. As seen in Figure 7.7B, the solid obtained right after the addition of NaOH (at 0 h)
exhibits readily the XRD pattern of the MOF-325 structure. And then in the next three
hours at 100 oC, its XRD pattern is much more intense implying the increase of its
crystallinity; and only MOF-235 phase was observed. However after 12 hours at 100 oC,
the XRD pattern shows a mixture of both MIL-88B and MOF-235.
The similar trend is also observed for the synthesis of mixed metal MIL-88B
(Figure 8). Again, Fe2Ni-NO3-x samples show essentially noncrystalline phase regardless

193
of the synthesis time (Figure 8A). In contrast, the sample Fe2Ni-Cl-x clearly exhibits faster
phase transition than the single metal synthesis. At 0 h after the addition of NaOH, although
the MOF-235 phase was dominant, the MIL-88B was observed with the presence of weak
peaks of the plane (101) and (002) (Figure 8B). After 1 h, the MIL-88B structure was
established as the prominent and characteristic planes (100) (101) (002) appeared.
However, the MOF-235 phase is still pronounced as its (101) plane is still intense. After 3
h, the MIL-88B phase became the major phase over the MOF-235 phase. The peaks
characteristic of MOF-235 phase were very weak or disappeared. Eventually after 12 h,
only MIL-88B in open form is present. Hence, the transformation from MOF-235 to MIL-
88B in the mixed metal synthesis is faster than that of the single metal synthesis.
High-resolution transmission electron microscopy (HRTEM) and energy-
dispersive X-ray spectroscopy (EDS) analysis: Other important information is whether
the Fe/Ni ratio varies along its crystal. For this purpose, HRTEM and EDS techniques were
employed; different Fe2Ni-Cl-12h crystals were observed. A representative HRTEM image
of a crystal of Fe2Ni-Cl-12h sample is shown in Figure 7.9. The crystal is an elongated
hexagonal bipyramid, which is 500 nm long and 80 nm wide. This crystal shape is
frequently encountered for MIL-88B.[28, 29] EDS spectra were acquired on a large number
of positions in the crystal. For example, as shown in Figure 7.9, a selection of 5 different
positions are shown, they are two positions (1 and 4) near the external part of the crystal
and three positions (2, 3 and 5) approaching the crystal center. The atomic ratios of Fe and
Ni from the EDS spectra are also displayed in
Table 7.3. The results exhibit that the Fe/Ni ratio is not identical but does vary
throughout the crystal. In terms of Fe/Ni ratio, the crystal is rich in Fe in the center but Ni
content increases as one move outward. At the outer part of the crystal, the Fe/Ni reaches
the value of 2, in agreement with the stoichiometric ratio of Fe2NiO cluster. This behavior
implies that the Fe2Ni-MIL-88B crystal indeed includes both Fe3O and Fe2NiO clusters in
the framework.

194
7.4. Discussion
General remarks: The phase selection and transition in the synthesis of MIL
materials have been investigated by several groups.[30-33] The first report on this matter
by Férey et al.[32] dealt with the synthesis of Fe based MIL-53 using dimeric metal cluster
as its framework nodes. The authors found that, MOF-235 is formed at the first stage of
synthesis, followed by the formation of MIL-53 product. As the SBUs of MIL-53 and
MOF-235 are different, it would be impossible for a solid phase transition from MOF-235
to MIL-53. It suggests that during the MIL-53 synthesis, MOF-235 dissolves releasing free
monomers to yield subsequent MIL-53. For the synthesis Cr-based MIL-53, Jhung et
al.[30, 31] reported that MIL-101, but not MOF-235 is the transient phase and is
subsequently converted into MIL-53 under the studied synthetic conditions. Recently,
Stavitski et al. [33] studied on the synthesis of MOFs based on tri-nuclear cluster of Al, the
authors‟ findings are in agreement with the previous reports by Férey and Jhung groups. In
fact, they also suggest that MOF-235 is the first to appear and then forms the MIL-101, and
finally they dissolve to yield MIL-53. Thus regardless of the metal cluster, a general rule is
observed: MOF-235, which is kinetically favored, will be appeared first and play as the
precursor to form MIL-101. Finally the most thermodynamically stable MIL-53 is formed
at the expense of both MIL-101 and MOF-235. And as MIL-101 and MOF-235 are built on
the same trinuclear cluster while MIL-53 employs the dimeric oxo-cluster, it is likely that
trinuclear cluster is kinetically favored over dimeric one, however, tri-nuclear cluster is
thermodynamically less stable than the dimeric one. To the best of our knowledge, no
information about the phase transformation and the formation MIL-88B during the
synthesis has been reported. Table 7.4 summarizes the crystal parameters of MIL-88B and
MOF-235. For single metal Fe, both MOF-235 and MIL-88B structures are cationic
framework of the same formula Fe3O(bdc)3. The difference in terms of composition is the
compensating anions which are Cl- in the MIL-88B and FeCl4
- in MOF-235. In regard to
topology, MOF-235 and MIL-88B are identical; they are both built on the acs net,[34]
having the same crystal and space group. In addition, thanks to the breathing capacity,[35]
the a lattice constant can increase from 11 Å to 14 Å, while the c constant decreases from
19 Å to 14 Å, accordingly. This flexible range of lattice constant of MIL-88B structure
comprehensively encompasses the lattice parameters of MOF-235. Hence topologically

195
speaking, structures of MIL-88B and MOF-235 are likely to be inter-convertible; one could
be converted to other by distortion without breaking the linker bdc and the tri-nuclear
cluster. And with this high similarity in structure of MIL-88B and MOF-235, it is likely
that MIL-88B would form fast at initial stages, but does it come before or after MOF-235?
In our study of the MIL-88B synthesis, a secondary metal (Ni) which can form stable
mixed metal complex with Fe, is introduced, the situation would become more
complicated, there would be the additional competition between mixed metal MIL and
single metal MIL. However, as it will be explained later, the secondary metal Ni turns out
to be useful, playing a role of spectroscopically labeled atom in the detection and
distinction of single and mixed trinuclear clusters and thus it is possible to distinguish
MOF-235 and MIL-88B structures.
Determination of phase transformation during the synthesis: As seen above in
the FTIR, Raman and UV-Vis spectroscopy analyses, all the syntheses at first produce
single metal solid (Fe) even in the case with the presence of secondary metal Ni. FTIR
spectra of the solids also revealed the tri-nuclear Fe3O cluster which is the building unit of
MOF-235 and MIL-88B. However, based on the XRD results, these initial solids exhibit
that only those prepared using the FeCl3.6H2O source are structural and were identified as
MOF-235, while no structural products were observed using Fe(NO3)3.9H2O as the Fe
source during the synthesis. Furthermore, for the Cl- based samples, the solid structure
gradually changes from MOF-235 to MIL-88B with increasing the synthesis time. Along
with this phase transformation, for the mixed metal synthesis, the mixed metal tri-nuclear
cluster Fe2NiO begins to appear. As seen in the FTIR spectra (Figure 7.4), the vibration of
mixed metal cluster becomes pronounced over the synthesis time, while the vibration of
single metal cluster diminishes. This accompanies with the decrease of FeCl4- anions as
shown in Raman spectra (Figure 7.6). Hence, the trend observed for the samples using
FeCl3.6H2O is as follows: MOF-235 comes first, and then MIL-88B which is more stable
comes later and gradually takes over the MOF-235. The MIL-88B develops thanks to the
transformation from the MOF-235 structure. The phase transformation from MOF-235 to
MIL-88B is also observed and is found to be fast. In addition, there are competing reactions
forming Fe3-MIL-88B and Fe2Ni-MIL-88B. The formation of single metal cluster Fe3O
dominates at the initial stages with the formation of MIL-235, and then Fe3-MIL-88B.

196
Subsequently, it is surpassed by the formation of mixed metal cluster Fe2NiO for the Fe2Ni-
MIL-88B synthesis. However, this behavior is not observed for the mixed metal synthesis
using Fe(NO3)3.9H2O. No MOF-235 was produced during the synthesis; especially,
amorphous solid product was yielded. Strictly speaking, Cl- is required in composition of
MOF-235. This also implies that MOF-235 would play an important role for the phase
transformation from MOF-235 to MIL-88B.
The HRTEM and EDS analyses show that in the mixed metal synthesis using Cl-,
the Fe3 and Fe2NiO clusters are not distributed separately in two kinds of crystals Fe3-MIL-
88B and Fe2Ni-MIL-88B, respectively. In fact these clusters reside in the same crystal in
which the Fe3O cluster prefers the center while the Fe2Ni ones take the outward place.
Taking into account the findings of the spectroscopy data and XRD analysis, it is suggested
that, the formation of Fe2Ni-MIL-88B at first starts with single Fe3O cluster in form of
MOF-235 and then Fe2NiO cluster comes in as the crystal grows. The kinetically favored
MOF-235 could be seeds or in other word precursors to subsequent growing of
thermodynamically stable MIL-88B structure. Without MOF-235, the formation of the
coordination framework is much more difficult as in the case of nitrate based synthesis.
Although there are readily Fe3 and/or Fe2NiO cluster in the synthesis mixture, the lack of
MOF-235 seeds as precursor results in an amorphous gel in the final product of the NO3-
based synthesis. The possible whole transformation is illustrated in Scheme 8.1.
Anion effect: When only nitrate is used, the solid products are amorphous on all
runs. However, when chlorate is introduced in the form of FeCl3.6H2O, MOF structures
were obtained under certain conditions, and high selectivity of Cl- is also observed, even in
the case of mixed metal synthesis, there are both NO3- and Cl
-, only Cl
- is present in the
final product. In our syntheses involving FeCl3.6H2O, FeCl4- as balancing anion is found at
the beginning of the syntheses then it gradually disappears. In fact, the necessity of halogen
for the synthesis of trinuclear MOF has been reported in the synthesis of the trinuclear
based MIL by several authors. [30-33],46-48
The first report on the synthesis of Cr3-MIL-101
mentioned the use of Cr(NO3)3.9H2O without chlorate, however, other halogen anion in the
form of HF is required.[36] Later, Jhung et al. found out that when CrCl3 is used, there is
no need of HF to obtain MIL-101(Cr).[30, 31] Reports on the synthesis of Al3-MIL-101

197
also emphasized the successful use of AlCl3 and the infertile use of Al(NO3)3.[33, 37]
Successful preparations of Fe3-MIL-101 also involve the use of FeCl3.6H2O.[12, 38, 39]
But from the experimental point of view, the difference in products obtained from different
anions deserves a rational explanation. The role of ion type in the synthesis of MOF has
been emphasized,[11] but so far, to the best of our knowledge, there is few comprehensive
investigation published. Férey et al. suggested a vague mineral role of F- and a possible
template effect of Cl-, but the detail is unknown.[11, 12, 40] The original formula of MOF-
235 is Fe3O(bdc)3.FeCl4 which requires Cl-, thus strictly speaking, nitrate cannot give
original MOF-235 structure. But the question is why a similar structure based one nitrate
Fe3O(bdc)3.NO3 was not observed, instead a gel is form. We believe that the difference lies
in the template effect of the anion. Let‟s consider the case when FeCl4- and NO3
- residing in
the pore structure. FeCl4- is a tetrahedron of which four vertexes are Cl anion and the center
is the Fe cation. The -1 charge of FeCl4- is distributed evenly in the four Cl anions. The
NO3- features a triangle structure with nitrogen in the center and three oxygens at the
corners. The -1 charge of NO3- is distributed evenly in the three O anions, each carrying -
2/3 charge. For every 3 Fe atoms in the cluster, one anion is needed to balance the charge.
It would be safe to assume that the charge is distributed evenly among the Fe atoms in the
framework. Hence a tetrahedral structure of FeCl4- would be better at balancing the charge
of its MOF surrounding than a flat structure of nitrate. However, to confirm this suggestion,
comprehensive theoretical calculation and simulation would be necessary.
A similar role of compensating anion can be found in zeolite science, the role of
counter ions is not only to balance the framework but also to initiate the ordering structure
in the nucleation.[41-43] As zeolite framework is positive charged, the cations will assume
the template role, organizing around themselves negative charged oligomers in an
energetically favored fashion, thus forming certain favored geometry. This idea has been
suggested since the early days of the zeolite science and has been consolidated and
developed ever since, and become widely accepted.[41-44] In a similar fashion, the
formation of MOF-235 could be started with the assembly of the positive charged metal
carboxylate clusters around the negative charged FeCl4- in a geometry that favors the
formation of the acs net of MIL-235. The bulkier and more spacious of FeCl4- is a template
guiding the formation of ordered structure, while the nitrate is less effective resulting in

198
disordered structure. The possible mechanism is illustrated in Scheme 8.2. Beside the
anions, the template effect can be drawn from the use of bulky and appropriate terminal
ligand such as CH3CN as reported by Choi et al.[28] Other possible ligands could be
pyridine, THF. Bulkier linkers could also provide a template effect.[45] The use of
functional bdc (NH2-bdc) could improve the selection of MOF-235 as well as MIL-88B
without the need of chlorate.[29] The steric hindrance of the function group could stabilize
and increasing the preference of MOF-235 structure over the amorphous solid.
7.5. Conclusion
The synthesis of Fe2Ni-MIL-88B provides us an opportunity to have a detailed
investigation of the synthesis of MOF. We found in it many aspects of crystal nucleation
and growth: phase transformation, phase selectivity, precursor and template. The key to the
explanation of all these phenomena is to understand the kinetic and thermodynamic
difference between Fe3O and Fe2NiO. Fe3O is kinetically favored while Fe2NiO is
thermodynamically favored. FeCl4- anion is suggested to be template for the formation of
MOF-235 as well as the MIL-88B structure. Although our suggestions on the mechanisms
and the anion effect are in agreement with the experiment results, a theoretical calculation
is actually needed.
We also notice resemblances between zeolite science and MOF science. In fact,
concepts and ideas that have been well developed in the synthesis of zeolite such as:
template, SBU (secondary unit building), seeding, aging etc. can be used and applied to
MOF synthesis. In return, it is hoped that advances in MOF science could also inspire new
discovery in zeolites.
† Electronic Supplementary Information (ESI) available: Details of FTIR spectra HRTEM
and EDS spectra. See DOI: 10.1039/b000000x/

199
References
1 H.-C. Zhou, J. R. Long, O. M. Yaghi, Chem. Rev. 2012, 112, 673.
2 H. Li, M. Eddaoudi, M. O'Keeffe, O. M. Yaghi, Nature 1999, 402, 276.
3 orma ar a X la r s i Xamena Chem. Rev. 2010, 110, 4606.
4 P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E.
Morris, C. Serre, Chem. Rev. 2011, 112, 1232.
5 P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D.
Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories,
L. Cynober, S. Gil, G. Ferey, P. Couvreur, R. Gref, Nat Mater 2010, 9, 172.
6 L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N.
Acerbi, M. L. Clarke, P. A. Wright, Catal. Sci. Tech. 2013, 3, 606.
7 A. Dhakshinamoorthy, M. Alvaro, H. Chevreau, P. Horcajada, T. Devic, C. Serre, H.
Garcia, Catal. Sci. Technol. 2012, 2, 324.
8 P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M.
Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey,
C. Serre, J. Am. Chem. Soc. 2011, 133, 17839.
9 S. Surble, C. Serre, C. Mellot-Draznieks, F. Millange, G. Ferey, Chem. Commun. 2006,
284.
10 G.-T. Vuong, M.-H. Pham, T.-O. Do, Dalton Trans. 2013, 42, 550.
11 G. Ferey, Chem. Soc. Rev. 2008, 37, 191.
12 auer erre Devi or aja a r m arrot r re to k Inorg.
Chem. 2008, 47, 7568.
13 A. B. Blake, A. Yavari, J. Chem. Soc., Chem. Commun. 1982, 1247.
14 A. B. Blake, A. Yavari, W. E. Hatfield, C. N. Sethulekshmi, J. Chem. Soc., Dalton
Trans. 1985, 2509.
15 Y. J. Kim, C. R. Park, Inorg. Chem. 2002, 41, 6211.
16 A. Fateeva, P. Horcajada, T. Devic, C. Serre, J. Marrot, J.-M. Grenèche, M. Morcrette,
J.-M. Tarascon, G. Maurin, G. Férey, Eur. J. Inorg. Chem. 2010, 2010, 3789.
17 K. Nakamoto, Infrared spectra of inorganic and coordination compounds, Wiley-
Interscience New York 1986.
18 R. D. Cannon, R. P. White, in Prog. Inorg. Chem., Vol. 36, John Wiley & Sons, Inc.,
1988, 195.
19 G. Xue, Prog. Polym. Sci. 1994, 19, 317.
20 J. F. Arenas, J. I. Marcos, Spectrochim. Acta, Part A 1979, 35, 355.
21 J. F. Arenas, J. I. Marcos, Spectrochim. Acta, Part A 1980, 36, 1075.
22 P. Larkin, Infrared and Raman Spectroscopy; Principles and Spectral Interpretation,
Elsevier, Amsterdam 2011.
23 M. K. Johnson, D. B. Powell, R. D. Cannon, Spectrochim. Acta, Part A 1981, 37, 995.
24 H. Hellman, R. S. Laitinen, L. Kaila, J. Jalonen, V. Hietapelto, J. Jokela, A. Sarpola, J.
Rämö, J. Mass Spectrom. 2006, 41, 1421.
25 M. S. Sitze, E. R. Schreiter, E. V. Patterson, R. G. Freeman, Inorg. Chem. 2001, 40,
2298.
26 J. Sanders-Loehr, W. D. Wheeler, A. K. Shiemke, B. A. Averill, T. M. Loehr, J. Am.
Chem. Soc. 1989, 111, 8084.
27 A. Ianoul, T. Coleman, S. A. Asher, Anal. Chem. 2002, 74, 1458.
28 W. Cho, S. Park, M. Oh, Chem. Commun. 2011, 47, 4138.

200
29 M.-H. Pham, G.-T. Vuong, A.-T. Vu, T.-O. Do, Langmuir 2011, 27, 15261.
30 N. A. Khan, S. H. Jhung, Cryst. Growth Des 2010, 10, 1860.
31 N. A. Khan, J. W. Jun, S. H. Jhung, Eur. J. Inorg. Chem. 2010, 2010, 1043.
32 F. Millange, M. I. Medina, N. Guillou, G. Férey, K. M. Golden, R. I. Walton, Angew.
Chem. Int. Ed. 2010, 49, 763.
33 E. Stavitski, M. Goesten, J. Juan-Alcañiz, A. Martinez-Joaristi, P. Serra-Crespo, A. V.
Petukhov, J. Gascon, F. Kapteijn, Angew. Chem. Int. Ed. 2011, 50, 9624.
34 N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe, O. M. Yaghi, Acc. Chem. Res.
2005, 38, 176.
35 C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk, G. Ferey,
Science 2007, 315, 1828.
36 G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé, I.
Margiolaki, Science 2005, 309, 2040.
37 P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon, F. Kapteijn, Chem. Mater. 2011,
23, 2565.
38 P. F. G. S. C. G. R. C. P. Horcajada-cortes, US Patent 20100226991, 2010.
39 K. M. L. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran, W. Lin, J. Am. Chem. Soc.
2009, 131, 14261.
40 D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey, J.-S. Chang, Adv. Funct. Mater. 2009,
19, 1537.
41 D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, Wiley, London
1973.
42 C. S. Cundy, P. A. Cox, Chem. Rev. 2003, 103, 663.
43 C. S. Cundy, P. A. Cox, Microporous Mesoporous Mater. 2005, 82, 1.
44 G.-T. Vuong, T.-O. Do, J. Am. Chem. Soc. 2007, 129, 3810.
45 P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M.
Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey,
C. Serre, J. Am. Chem. Soc. 2011, 133, 17839.
46 L. Meesuk, U. A. Jayasooriya, R. D. Cannon, Spectrochim. Acta, Part A 1987, 43, 687.
47 R. Wu, U. A. Jayasooriya, R. D. Cannon, Spectrochim. Acta, Part A 2000, 56, 575.
48 L. Montri, R. D. Cannon, Spectrochim. Acta, Part A 1985, 41, 643.
49 A. C. Sudik, A. P. Côté, O. M. Yaghi, Inorg. Chem. 2005, 44, 2998.

201
FIGURE CAPTIONS, SCHEMES AND TABLES
0 h
Wavelength (nm)
200 300 400 500 600 700 800
1h
2 h
3 h
Adsorp
tion (
a.u
.)
12 h
(A)

202
0 h
Wavelength (nm)
200 300 400 500 600 700 800
1h
2 h
3 h
12 h
(B)
Figure 7.1. UV-Vis spectra of the samples Fe3-NO3-x (A) and Fe3-Cl-x (B) prepared using
Fe(NO3)3.9H2O and FeCl3.6H2O, respectively, at different synthesis times.

203
0 h
Wavelength (nm)
200 300 400 500 600 700 800
3 h
2 h
1 h
Adsorp
tion (
a.u
.)
12 h
(A)

204
0 h
Wavelength (nm)
200 300 400 500 600 700 800
1h
2 h
3 h
Adsorp
tion (
a.u
.)
12 h
(B)
Figure 7.2. UV-Vis spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) samples prepared
using Fe(NO3)3.9H2O and FeCl3.6H2O, respectively at different synthesis times

205
Wavenumber cm-1
600610620630640650
Fe3O Fe3O
12 h12 h
3 h
3 h
2 h
2 h
1h 1 h
0 h 0h
(A)(B)
Wavenumber cm-1
600610620630640650
Figure 7.3. Transmittance FTIR spectra of the samples of Fe3-NO3-x (A) and Fe3-Cl-x (B)
at different synthesis times

206
Fe2NiO Fe3O
3 h
2 h
1 h
0 h
12 h (A)
Wavenumber [1/cm]
600625650675700725750

207
Fe2NiO Fe3O
3 h
2 h
1 h
0 h
12 h
Wavenumber [1/cm]
600625650675700725750
(B)
Figure 7.4. Transmittance FTIR spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) at
different synthesis times

208
0 h
Wavelength (1/cm)
10030050070090011001300150017001900
1 h
2 h
3 h
12 h
(A)

209
0 h
Wavelength (1/cm)
10030050070090011001300150017001900
1 h
2 h
3 h
12 h
(B)
Figure 7.5. Raman spectra of the samples Fe3-NO3-x (A) and Fe3-Cl-x (B) at different
synthesis times

210
0 h
Wavelength (1/cm)
10030050070090011001300150017001900
1 h
2 h
3 h
12 h
(A)

211
0 h
Wavelength (1/cm)
10030050070090011001300150017001900
1 h
2 h
3 h
12 h
(B)
Figure 7.6. Raman spectra of Fe2Ni-NO3-x (A) and Fe2Ni-Cl-x (B) at different synthesis
times.

212
0 h
3 h
12 h
(A)
2
5 8 11 14 17 20
2
5 8 11 14 17 20
12 h
3 h
2 h
1 h
0 h
#
*
#
#
*
#*
#
#
*
(B)
Figure 7.7. XRD patterns of Fe3-NO3-x (A) and Fe3-Cl-x (B) at different synthesis times.
(*) MOF-235 phase, (#): MIL-88B phase.

213
2
5 8 11 14 17 20
#
#
#
##
#
#
#
#
#
*
#
#
##
*
*#
# #
*
*
*
#
#
#
**
*
*
*
*
#
#
#
#
#
#
#
#
*
*
0 h
1 h
2 h
3 h
12 h
(B)
2
5 8 11 14 17 20
0 h
3 h
12 h
(A)
Figure 7.8. XRD patterns of Fe2Ni-NO3 (A) and Fe2Ni-Cl (B) at different synthesis time.
(*) MOF-235 phase, (#): MIL-88B phase

214
Figure 7.9. Representative HRTEM and EDS acquiring positions of Fe2Ni-Cl-12h crystal
1
2
3
5 4

215
SCHEMES
Scheme 8.1. Possible mechanism of the formation of MIL-88B samples using FeCl3.6H2O

216
Schem 8.2. Possible proposition of anion mediated formation of MIL-88B

217
Table 7.1. FTIR band assignment in the wavenumber 400 – 800 cm-1
Band (cm-1
) Assignment
750 C-H [20, 21]
720 Fe2NiO [46, 47]
690 C-C [20, 21]
660 OCO [20, 21]
624 Fe3O [23, 48]
550 Fe-O, Ni-O [17]
Table 7.2. Raman band assignments
Band (cm-1
) Assignment
175 nsym(M3O)
267 δasym(M3O)
330 FeCl4-
440 M-O
567 Fe2NiO
631 OCO
860 CC
1050 NO3-
1125 CX
1146 CH
1431 CH3 (bending mode in DMF)
1454 CH3 (bending mode in DMF)
1616 CC
Table 7.3. Fe and Ni atomic percentages calculated from EDS spectra

218
Position Fe (atomic %) Ni (atomic %) Fe/Ni
1 75 36 2.1
2 67 35 2.2
3 81 29 2.8
4 78 37 2.1
5 89 19 4.7
Table 7.4. Comparison of the crystal parameters of MIL-88B and MOF-235
Parameter MIL-88B [9, 12] MOF-235 [49]
Chemical formula Fe3O(bdc)3Cl.3DMF Fe3O(bdc)3FeCl4.3DMF
Molar weight (g.mol-1
) 905.8 1092.8
Net acs acs
Crystal system Hexagonal Hexagonal
Space group P -6 2 c P -6 2 c
a (Å) 11.1075 12.531
b (Å) 11.1075 12.531
c (Å) 19.0925 18.476
90 90
90 90
120 120
Volume (Å3) 2040.8 2512.6

219
Supporting Information
1. FTIR spectra of samples with wavelength 400 – 4000 cm-1
1.1.Single metal synthesis using Fe(NO3)3.9H2O
0 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
1 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

220
2 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
3 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

221
12 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
1.2.Single metal synthesis using FeCl3.6H2O
0 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

222
1 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
2 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

223
3 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
12 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
1.3.Mixed metal synthesis using Fe(NO3)3.9H2O

224
0 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
1 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

225
2 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
3 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

226
12 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
1.4.Mixed metal synthesis using FeCl3.6H2O
0 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

227
1 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
2 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

228
3 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce
12 h
Wavenumber [cm-1
]
5001000150020002500300035004000
Tra
nsm
itta
nce

229
2. HRTEM and EDS analysis
EDS spectra of 5 positions taken on the same Fe2Ni-MIL-88B crystal (see Figure 9 in the
article).
Since the samples were dispersed on a copper grid for analysis. Signal of copper was
observed in the EDS spectra.
EDS spectroscopy of position 1
EDS spectroscopy of position 2
EDS spectroscopy of position 3

230
EDS spectroscopy of position 4
EDS spectroscopy of position 5
3. XPS analysis of Fe2Ni-MIL-88B-12h, dried
The X-ray photoelectron spectra (XPS) were taken on a photoelectron spectrometer
(KRATOS AXIS-ULTRA) with a monochromatic X-ray source of Al K . The operating
conditions for recording high-resolution spectra were as follows: 1486.6 eV and 225 W;
pass energy of 160 eV with anoperating pressure of 10-9
Torr.
CasaXPS software was use to analys the collected XPS spectra. 1 All spectra were
calibrated using the adventitious C 1s peak with a fixed value of 284.8 eV. Shirley
background was then applied and subtracted.
As the Fe cation in trinuclear cluster is at high spin state,2,3
envelope of Fe 2p3/2 spectrum
was fit with peaks corresponding to the GS multiplets, surface structures and shake-up-
related satellites.4-6
All the four GS multiplets have the same full width at half-maximum
(FWHM) of 1.6 eV and their peak areas are in similar to those of multiplets. The rest of the
envelop was filled with one surface structure peak and one satellites peak.
Fitting result is in agreement with Fe3+
GS multiplets , suggesting the presence of only Fe3+
not Fe2+
in the sample.
Gupta and Sen (GS) multiplet peak parameters used to fit the high-spin Fe3+

231
Sample
Peak 1 Peak 2 Peak 3 Peak 4
eV %
area E eV
%
area E
(eV) eV
%
area E
(eV) eV
%
area E
(eV)
Fe2Ni-MIL-88B-12h 713.2 7 1.3 711.9 21 1.1 710.8 35 1.5 709.3 37
GS Fe3+ multiplets 10 0.6 20 1.3 30 1.6 40
References
1. Fairley N. CasaXPS Version 2.2.19, copyright 1999–2003
2. Boudalis A. K. et al Polyhedron 2005, 24, 1540
3. Psycharis, V et al Eur. J. Inorg. Chem 2006, 2006, 3710-
4. Gupta RP, Sen SK. Phys. Rev. B. 1975; 12: 15
5. Grosvenor A.P et al. Surf. Interface Anal. 2004, 36, 1564
6. Mullet M. et al. Surf. Interface Anal. 2008, 40, 323


233
Chapter 8. Conclusion
For the study of nanozeolites, two novel methods, single-phase and two-phase
synthesis, have been developed for the synthesis of nanozeolites. Both of them employed
the use of organosilane agent to restrain the growth of zeolite during the crystallization
process thus, resulting in uniform nanozeolites. This growth control works on the principle
that organosilane can react with silanol groups which are abundant on the external surface
of nanozeolites. Upon reacting with organosilane, zeolite nanocrystals are functionalized
with the organic group of the organosilane. These functionalized crystals therfore cannot
incorporate more aluminosilicate species which would lead to the larger crystal.
In the two-phase synthesis, in addition to the aqueous medium, an organic solvent
was introduced. As the nanocrystals are functionalized with organic group and thus become
hydrophobic, it can disperse into the organic phase, and thus the growth process is
completely stopped. It is clear that the diffusion is strongly dependent on the hydophobicity
of the functionalized crystals. This parameter is governed by two opposite factors, the
crystal size (the hydrophilic factor) and the degree of functionalization (the hydrophobic
factor) of the functionalized crystals. Generally, increase in crystal size results in higher
value of the hydrophibicity factor, given the same degree of functionalization. In contrast,
the hydrophobic factor is favored by the increase in the external surface. As the results in
the two-phase synthesis, functionalized nanozeolites are more likely to be found in the
organic solvent than in the aqueous phase.
The two-phase synthesis provides a useful way to produce nanozeolites dispersed in
the organic phase. In contrast to the two-phase synthesis, the aim of the single-phase
method is to well disperse the zeolites synthesis solution in an organic solvent. This is
carried out with the aid of organosilane and n-butanol. Organosilane is used with the same
purpose as in the two-phase synthesis, i.e. to functionalize and “hydophobilize” the
produced nanozeolites. The introduction of n-butanol is intended to increase the dispersion
of the aqueous medium in the organic phase. In our study, careful selecting the organic
phase/inorganic phase ratio and mixing at proper temperature could lead to a single-phase

234
solution for further crystallization. Due to the absence of the separate aqueous phase as in
the case of the two-phase synthesis, only nanozeolites are produced, there is no trace of
large crystal in the obtained product. Using this method we have successfully synthesized
silicalite-1 and FAU nanozeolites.
The application potentials of our synthesized zeolites were also evaluated. The
catalyst was prepared by hydrolysis of TEOS in the presence of FAU nanozeolites to
incorporate the zeolites in the amorphous silica. The cracking reaction of FCC feed stock
was chosen to determine the catalytic activity of nanozeolites. The results showed that our
catalysts gave high activity and it is recommended that it can be used to improve the
activity of FCC catalysts. Direct application of nanozeolites as catalyst is not the only way
to utilize it. Another approach to maximize the potential of nanozeolite is to use them as
nano-unit to build superstructure materials. Some pioneering studies by prominent
researchers in the field have emphasized the significance of this approach: nanozoelite to
form zeolitic mesoporous-nanoporous materials, nanozeolite to form zeolitic nanosheets.
For our study of MOFs, we have showed that there are many opportunities to
customize these materials. As MOFs are composed of two components: the metal cluster
and the organic linker, the general strategy would be focused on these two. The common
one is to play with the organic linker while keeping the metal cluster. Due to its organic
nature, the linker can be modified in many aspects without much change in its reactivity in
regards to the connectivity to the metal clusters. Hence connectivities between metal cluster
and linker remain intact, thus the synthesis is likely to be successful. Almost all of
approaches to modify a linker have been used: functionalization, prolonging, isomerization
etc. yielding explosive number of new MOFs. The second strategy to modify the metal
clusters, are much less popular and less successful. That is not because the modification of
clusters is any less interesting but in fact it is much more difficult than modification of
linkers. Having second metal into MOF structure, hence mixed metal MOF is great but, the
complexity of the synthesis would rise abruptly. Different reactivities of metals do not
allow the incorporation of multi metals into one single MOF structure.
We believe that to increase the chance of success in the synthesis of mixed metal
MOF, it would be better to start with an original single metal MOF structure and apply the

235
modification of the metal cluster with the introduction of the second metal such that the
mixed metal cluster will retain the original shape and connectivity, hence, the obtained
mixed metal MOF will not suffer any disruption in connection due to the second metal. We
focused on MIL-88B which had been regarded as a dense structure to illustrate this
approach. The single metal cluster Fe3O in the original MIL-88B was replaced with mixed
metal Fe2NiO successfully to obtaine mixed metal Fe2Ni-MIL-88B. The real benefit of this
modification is clear as mixed MIL-88B becomes porous because the need of
accompanying and blocking anion is avoided by using mixed metal cluster. But another
significant change in the mixed metal MIL-88B is the improvement in bond strength
between the mixed metal clusters and the terminal ligands. This boost in bond strength has
allowed us to take advantage of the breathing effect of MIL-88B to control its porosity.
Strongly attached terminal ligands in mixed metal MIL-88B now become pillars to sustain
its structure. The larger the ligands are the greater the porosity is. We can control the
porosity of MIL-88B at three different levels. One may find this discovery contradicts
common sense that large molecules would block the pores instead of enlarging them. But
this fact just perfectly embodies how powerful and exciting the MOF materials are:
interesting and surprising properties are often found in MOFs.
Of course, this approach of using mixed metal can be applied to other MOFs. MIL-
88A, MIL-88C and MIL-88D are the obvious candidates. In fact it is possible to use mixed
metal cluster Fe2NiO to replace any single metal trimeric cluster of the type M3O. Other
mixed metal clusters of Fe(III), Cr(III) with divalent metal such as: Ca, Mg, Ba, Sr, Mn, Co
and Zn should be able for the synthesis of mixed metal MOFs. The breathing effect
combined with the controllable porosity of mixed metal MIL-88B would be of great
interests for membrane technology. It could allow creation of smart membrane that can
change its pore size “on the fly” increasing the selectivity. The terminal ligands attached to
mixed metal clusters are also a useful agent to functionalize MOFs. Function groups could
be introduced via terminal ligands and in return could be removable by ligand exchange.
Thus we can envisage mixed metal MIL-88B like a chemical “main board” which carries
various function group modules.

236
The study on the synthesis of mixed metal MOFs also led us to the discovery of the
underlying synthesis mechanism. The importance of anion sources has been recognized, but
never been well explained. In the synthesis of MOFs based trimeric cluster such as Fe3O as
in the case of MIL-88, MIL-101 families, the necessary of halogen anion was remarked but
how it contributes to the synthesis was not mentioned in literature. With the synthesis of
MIL-88B we found that a template should be available for the structure to form. The
template for MIL-88B is FeX4- (X is halogen) and that is why halogen ion is required.
FeX4- should be available in the synthesis so that the cationic frameworks of MIL-88B, the
precursor MOF-235 can be built around it. It would be better if we could do some
theoretical calculations to prove the preference in energy of our MIL-88B structure bearing
FeX4 over other anions such as nitrate, sulphate… But we would like to emphasize the
similarity between the well known cation template mechanism suggested by Breck and our
proposal of anion template mechanism. It turns out that knowledge of zeolites could be
very helpful in dealing with MOFs.
Another similar feature to zeolites in the synthesis of MOFs is that, the synthesis
involves phase transformations in which the more stable phase replace the less stable one,
the less stable phase is thus the precursor to the stable phase. For the synthesis of MIL-88B,
MOF-235 is the precursor phase, one should make sure the synthesis conditions favor the
formation of MOF-235 otherwise MIL-88B will not come out. In the synthesis of zeolite a
reactive gel phase may come and go in the middle of the process, acting as intermediate
phase before the formation of zeolite phase. This is possibly due to the gain in energy from
gel phase to zeolite phase is great enough. For MOFs, the weaker coordination bond does
not afford enough energy necessary for a transformation from a gel phase to a MOF phase.
This is why the intermediate phase, the precursor phase of MIL-88B should be another
MOF phase, as we can obtaine MOF-235 in a matter of several minutes of synthesis. Any
formation of gel phase at the initial stage of the synthesis would signal its failure since the
chance the gel phase can transform to MOF phase is unlikely.
In conclusion, we have showed in this study that both nanozeolites and MOFs are
potential nanoporous materials. There are plenty of ways to exploit them. Together they
form the foundation of the science of nanoporous materials. But is it possible to obtain

237
materials composed of both zeolites and MOFs? We think yes. As our nanozeolites are
readily functionalized with organic functions, it would possible to attach them to a MOF
“main board”. The result would be an ultimate super nanoporous materials and that is what
we are dreaming about now.

238

239
List of Pulications
1. G.T. Vuong and T.O. Do, A new route for the synthesis of uniform nanozeolites with
hydrophobic external surface in organic solvent medium. Journal of the American
Chemical Society (JACS), 2007. 129(13): p. 3810-3811.
2. G.T. Vuong and T.O. Do, Nanozeolites and process for preparation thereof, 2008,
WO Patent WO/2008/058,398.
3. G.T. Vuong., S. Kaliaguine, and T.O. Do, A strategy towards macroporous sponge-like
networks of metal oxide-surfactant mesophases and bulk metal oxides. Journal of
Porous Materials, 2008. 15(6): p. 679-683.
4. G.T. Vuong and T.O. Do, Synthesis of silylated nanozeolites in the presence of organic
phase: Two-phase and single-phase methods. Microporous and Mesoporous Materials,
2009. 120(3): p. 310-316.
5. G.T. Vuong., V.T. Hoang, D.T. Nguyen, and T.O. Do, Synthesis of nanozeolites and
nanozeolite-based FCC catalysts, and their catalytic activity in gas oil cracking
reaction. Applied Catalysis A: General, 2010. 382(2): p. 231-239.
6. Pham, M.H., G.T. Vuong, F.G. Fontaine, and T.O. Do, A Route to Bimodal Micro-
Mesoporous Metal–Organic Frameworks Nanocrystals. Crystal Growth & Design,
2011. 12(2): p. 1008-1013.
7. Pham, M.H., G.T. Vuong, A.T. Vu, and T.O. Do, Novel Route to Size-Controlled Fe-
MIL-88B–NH2 Metal–Organic Framework Nanocrystals. Langmuir, 2011. 27(24): p.
15261-15267.
8. Pham, M.H., G.T. Vuong, F.G. Fontaine, and T.O. Do, Rational Synthesis of Metal-
Organic Framework Nanocubes and Nanosheets Using Selective Modulators and Their
Morphology-Dependent Gas-Sorption Properties. Crystal Growth & Design, 2012.
12(6): p. 3091-3095.
9. G.T. Vuong, M.H. Pham, and T.O. Do, Synthesis and Engineering Porosity of mixed
metal Fe2Ni- MIL-88B Metal-Organic Framework. Dalton Transactions, 2013, 42, 550-
557.
10. G.T. Vuong, M.H. Pham, and T.O. Do, Direct Synthesis and Mechanism for the
Formation of Mixed Metal Fe2Ni-MIL-88B. CrystatEngComm, submitted, 2013.