-
8/9/2019 Thal Donna
1/21
THALASSEMIAS
-
8/9/2019 Thal Donna
2/21
Basic Features Thallasemia syndromes are characterized by
varying degrees of ineffective hematopoiesis and
increased hemolysis Clinical syndromes are devided into - and
-
thallasemias
Most -thallasemias are due to point mutations inone or both of
the two -globin genes(chromosome 11)
Most -thallasemias syndromes are due todeletion of one or more
of the -globin genesrather than to point mutations
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
3/21
Epidemiology
Although -thallasemia has >200
mutations, most are rare Approximately 20 common alleles
constitute 80 of the known thallasemias
worldwide; 3% of the worlds population
carries gene for -thallasemia, and in
Southeast Asia 5-10% of the population
carries genes for -thallasemia
DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
4/21
-Thalassemia
0-Thallasemia
+
-Thallasemia -Thallasemia
-Thallasemia
Hb Lepore
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
5/21
-Thalassemia
Silent carrier -thallasemia
-thallasemia trait Hb Constant Spring
HbH disease
Hydrops fetalis
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
6/21
-Thalassemia: Homozygous or
Doubly Heterozygous Forms
Pathogenesis
Variable reduction of -chain synthesis
Relative -globin chain excess resulting inintracellular
precipitation of insoluble -chains
Increased but ineffective erythropoiesis withmany red cell
precurcors prematurely destroyed;related to -chain excess
Shortened red cell life span; variable splenicsequestration
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
7/21
Sequelae
Hyperplastic marrow
Increased iron absorption and iron overload Hypersplenism
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
8/21
Hematology Anemia: Hypochromic, microcytic
Reticulocytosis
Leukopenia and thrombocytopenia Blood smear: target cells and
nucleated red cells, extreme
anisocytosis, contracted red cells, polychromasia,punctate
basophilia, circulating normoblast
Hemoglobin Fraised; hemoglobin A2 increased
Bone marrow: May be megaloblastic (due to folatedepletion);
eryhtoid hyperplasia
Osmotic fragility: decreased
Serum ferritin: raised
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
9/21
Clinical Features Failure to thrive in early childhood
Anemia
Jaundice Hepatosplenomegaly
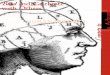
Abnormal facies, prominence of malar eminences, frontalbossing,
depression of bridge of the nose, and exposure ofupper central
teeth
Growth retardation, delayed puberty, primary amenorrheain
females
Leg ulcers
Skin bronzing
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
10/21
Management Hypertransfusion Protocol, is used to maintain a
pretransfusionHb between 10.5 and 11.0 g/dL
Hypertransfusion results in: Maximizing growth and
development
Minimazing extramedullary hematopoiesis anddecreasing facial and
skeletal abnormalities
Reducing excessive iron absorption from gut Retarding the
development of slenomegaly and
hypersplenism
Reducing and/or delaying the onset of complications
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
11/21
management
Chelation Therapy
The objectives:
To bind free extracellular iron
To remove excess intracellular iron
To attain a negative iron balance
Iron overload results from: Ongoing transfusion therapy
Increased gut absorption of iron
Chronic hemolysis
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
12/21
management
Desferrioxamine (Desferal):
Ch
elation sh
ould be instituted when t
heferritin level is >1000 ng/mL and adequate
iron is excreted into the urine with the
desferrioxamine challenge
Dose: 40-60 mg/kg/day, is infusedsubcutaneously over 8-10
hours
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
13/21
management Splenectomy
Splenectomy reduces the transfusion requirements in
patients withhypersplenism Two weeks prior to splenectomy, a
polyvalent
pneumococcal and meningococcal vaccine should begiven
Indications:
Persistent increase in blood requirements by 50% or moreover
initial needs for more than 6 months
Annual packed cell transfusion >250 mL/kg/year
Evidence of severe leukopenia and/or thrombocytopenia
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
14/21
management
Supportive Care
Folic acid Hepatitis B vaccination
Endocrine intervention
Genetic counceling and antenatal diagnosis
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
15/21
management
Deferiprone (L1)
Dose: 75 mg/kg/day ICL-670
Hematopoietic Stem Cell Transplantation
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005DeBaun M, Vichinsky E. Nelson Textbook of Pediatrics. 2007
-
8/9/2019 Thal Donna
16/21
-Thalassemia IntermediaClinical Features
Patients generally do not require transfusions and
maintain a Hb between 7 and 10 g/dL Marked medullary
expansion,
hepatosplenomegaly, growth retardation, facialanomalies, and
hyperbilirubinemia occur if
patients are not adequately transfused Patients are most healthy
if managements is as
vigorous as that for thallasemia major
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
17/21
-Thalassemia Minor orTrait
(Heterozygous 0 or +)
Clinical Features
Asymptomatic (physical examination isnomal)
Thalassemia trait or unusual severity
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
18/21
-Thalassemia
Hemoglobin H disease is clinically milder
than homozygous -thalassemia and does
not require a hypertransfusion protocol
Hydrops fetalis is not compatible with life
and presents with intrauterine or neonatal
death
Lanzkowsky P. Manual of Pediatric Hematology and Oncology.
2005
-
8/9/2019 Thal Donna
19/21
Thank You
-
8/9/2019 Thal Donna
20/21
Indikasi Rawat pada Penderita ITP
Akut:
Jumlah trombosit
-
8/9/2019 Thal Donna
21/21
Indikasi Pemberian Trombosit
Trombosit

















![[Donna Dewberry] Roses](https://img.pdfslide.fr/doc/110x75/577cd8df1a28ab9e78a22e11/donna-dewberry-roses.jpg)