• Le clonage de l’ADN – soit dans des cellules, soit in vitro – permet à des molécules d’ADN d’être sélectivement répliquées en un nombre de copies très élevé. Quand il est amplifié par ce moyen, l’ADN est efficacement purifié et le clonage de l’ADN permet d’étudier des séquences ADN isolées de l’ADN génomique.
• L’hybridation des acides nucléiques : approchecomplètement différente. Au lieu d’essayer de purifierindividuellement des séquences d’acides nucléiques,l’objectif est de les détecter spécifiquement et desuivre leur devenir au sein d’une population complexe quireprésente un échantillon biologique.

I, Hybridation moléculaire:
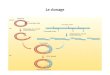
Association de 2 acides nucléiques simples brins de séquences complémentaires et qui conduit à la formation d'un double brin ou duplex par formation de liaisons H entre ces deux brins.Detection d'une molecule d'AND ou d'ARN dans un mélange contenant des milliers de similaires: "trouver une aiguille dans une meule de foin"
On distingues les hybrides:• ADN-ADN = Homoduplex• ADN-ARN = Hétéroduplex
La formation et la stabilité des duplex dépendent de nombreux facteurs :• la composition en bases • Longueur des duplex• complexité de la séquence.

ADN-ADN = Homoduplex
ADN-ARN = Hétéroduplex
Hybridation moléculaire

1) Principes de l’hybridation des acides nucléiques
Dans l’hybridation des acidesnucléiques, une population d’acidesnucléiques connus (SONDE) permetd’analyser une population d’acidesnucléiques mal connus
Une sonde est un ADN mono-brin ou ARN, ayant uneséquence complémentaire à laséquence à détecter.
En s’hybridant avec saséquence cible, elle permetde la détecter.

2) Types de sondes
Sonde génomique: fragment obtenu par coupure de l’ADN génomique.
Sonde ADNc : sonde ADN obtenue par transcription réverse d ’un ARNmmessager (= séquence codante).
Sonde oligonucléotides : ADN (ou ARN) simple brin de 18 à 50 nucléotidessynthétisé chimiquement.
3)Facteurs ayant une influence sur l’hybridation La température: la température optimale d’hybridation est en général
inférieure de 25% à la Tm de la sonde. Tm = 4 (C+G) + 2(A+T) ; Ta = Tm-5
La taille des fragments ou des sondes.
La force ionique: l’hybridation est accélérée en forte concentration saline.
La durée de l’hybridation : plus l’hybridation est longue, plus la rencontreentre séquences complémentaires est favorisée

C’est une température, appeléeégalement Température de demidénaturation (Tm) qui correspondà l’ouverture ou au déroulementde 50% de la chaîne de l’ADNchauffé.
C'est l'effet hyperchromiquequi correspond à la rupture desliaisons hydrogènes (liaisonsfaibles) et la séparation des 2chaînes entraînant uneaugmentation dans l'absorptiond'U.V de 40% à 260 nm.
La valeur de Tm des ADN estune fonction linéaire dupourcentage de (G + C) de l'ADN
3-a- La température de fusion de l’ADN Tm (melting Température)

II. Les agents de marquage des acides nucleiques (sondes)
Marquage : Méthode consistant à incorporer un nucléotide radioactif,fluorescent ou modifié chimiquement dans une molécule d’acidenucléique.
a) Isotopes radioactifs: qui permettent de constituer des sondes « chaudes »:ex: Le P32 est le plus utilisé pour le marquage des nucléotides. Il émet desrayonnements β qui vont impressionner des plaques photographiques lors del'autoradiographie
b) Marqueurs non-radioactifs: qui constitueront les sondes « froides » :La détection des sondes froides est basée sur une réaction enzymatiquequi fera apparaître un produit coloré ou fluorescent ou bien uneproduction de photons. Ex: biotine (interaction covalente avec l’avidine),digoxigénine (Ac-Ag) , fluorescéine (émet une lumière réfléchie defluorescence lorsqu'elle est excitée sous les ultraviolets)……

III. Techniques de marquage des sondes
1) Nick translation (déplacement de brèche)
But: couper des petits bous d’ADN pour les remplacer par de l’ADN marqué
a) Cassure aléatoire par une endonucléase , la Dnase
b) Agrandissement des cassures parl’activitéexonucléase 5’—3’ de l’ADN polyI
c) Synthèse d’ADN grace à l’activité polymerase de l’ADN poly I en utilisant des dNTP marquées
Résultat: obtention d’un ADN bicatenairecontenant des nucléotides marqués sur les deux brins

But : synthèse de l’ADN marquéa) Chauffage de l’ADNdbpuis refroidissement
brutal entrainant une séparation définitive des 2 brins
b) Synthèse de façon aléatoire d’une multitude d’amorces
c) Parmi ces amorces, certaines vont forcement rencontrer leurs séquences complémentaires sur l’un des brins d’ADN
d) Synthèse d’ADN par l’ADN poly en utilisant des dNTP marquées
Résultat: obtention de 2 molécules d’ADN bicentenaires (dont un seul brin est marqué par molécule)
2) Multi-Random priming= multi amorcage au hasard

3) Marquage aux extrémités
• Marquage 5’P: grâce à une polynucléotide kinase Échange de phosphates par la polynucléotide kinase , en présence de γp32 d‘ATP, la polynucléotide kinase échange le phosphate en 5’ de la sonde, ce qui permet de la marquer.
• Marquage 3’OH: grâce à une polynucléotide kinaseLa transférase terminale catalyse l’addition de désoxynucléotides sur unefonction alcool 3’OH terminale de l’ADN

Annexe: Extraction et Purification de l’ADN


• Séparer des fragments d’ADN de différentes tailles en les faisant migrer dans un gel en les soumettant à un courant électrique.
L'électrophorèse sur gel d'agarose sépare les fragments d'ADN par taille. Les molécules d'ADN chargées négativement sont attirées par l'électrode positive. Lorsque l'ADN migre, les fragments d'ADN sont empêchés par le maillage du gel réticulé. Plus le fragment d’ADN est petit, moins il risque d’être ralenti. Par conséquent, les plus petits fragments d'ADN migrent plus rapidement.
ELECTROPHORESE DE L’ADN:
La charge relative étant constante, le système de discrimination entre les molécules est :
l'effet de filtration du gel utilisé « Tamisage moléculaire »
leur masse moléculaire ou nombre de paires de bases pb

L'électrophorèse sur
gel utilise un champ
électrique et des
électrodes positives et
négatives pour séparer
les molécules d'ADN en
fonction de leur taille.

• Le gel utilisé en électrophorèse est constitué d'une matrice de
polymère baignant dans un tampon conducteur.
• Deux principaux polymères sont utilisés : l'agarose (ADN:0,1-60kb)
et le polyacrylamide (ADN <0,1kb).
• La taille des mailles varie selon la concentration du gel d’agarose
Correspondance tailles des brins d’ADN/ concentration d’agarose

Séparation de fragments d’ADN de tailles différentes dans un gel d’éléctrophorèse

Séparation d'ADN sur gel d'agarose: coloration et standards
• Pour visualiser l'ADN, le gel d'agarose contenant les fragments d'ADN séparés est
plongé dans une solution de bromure d'éthidium, qui s'intercalera entre les paires
de bases de l'ADN (EtBr à la fois un agent intercalaire et fluochrome).
• Le gel est placé sous une source de lumière UV.
• La lumière ultraviolette excite le bromure d’éthidium et lui confère une
fluorescence orange.
Le bromure d'éthidium
se lie à l'ADN
bicaténaire par
intercalation.
Les bandes d'ADN sur un gel depolyacrylamide ou d'agarose ne sont pasvisibles si l'ADN n'est pas marqué ou coloré.

Exemples de marqueurs de taille
- Phage λ coupé par BstEII (0.12 à 8.45)
- Phage λ coupé par HindIII (0.13à23.13)
- pBR322 coupé par BstNI (0.12 à 1.86)
- 100 pb Ladder (100 1000)
La détermination de la taille d'un fragment se fait par rapport à la
migration d'un marqueur approprié contenant des fragments de tailles
connues (dans le puits de référence A).

Marqueursde taille(en pb)
Echantillond’AND de tailleinconnue
par rapport au puits d’inclusion
Pour déterminer la taille d’un
fragment d’ADN à l’aide d’un gel
d’agarose, il suffit de tracer une
courbe de la taille des molécules
sur une échelle logarithmique en
ordonnée (l’axe des y) en fonction
de la distance de migration en
abscisse (l’axe des x). Le
marqueur de poids moléculaire
permet de tracer une telle
courbe, qui sera utilisée pour
déterminer la taille de fragments
inconnus.
Distance de migration en fonction du logarithme de la taille des fragments d’ADN
Détermination de la taille des fragments d’ADN
Recommended