
L’Evaluation Clinique du Dispositif Médical
Pour un domaine d’investigation très diversifié Pour une variété de DM (innovation, risque, etc.) Un contexte français/européen d’exigences Tout au long du cycle de vie du produit :
• du concept, • lors du pré-marquage CE, • pour le maintien du marquage CE, • pour le post-marquage CE • et pour le maintien de prise en charge
Post-inscription Faraj F. ABDELNOR www.acidim.asso.fr
des femmes, des hommes, vie et technologie

Pour un domaine d’investigation très diversifié
• x
Marché en France :
Entre 800.000 et 2.000.000 DM
LPPR : 80.000 produits &
prestations sous 3100 codes pour
6,8 M€ en 2010
(médicaments remboursés : 25,5M)
Marché hospitalier :
AP-HP : 90.000 références de DM en 2002
CHRU Tours : 14.000 références dont
4000 pour la liste en sus (3.000
médicaments dont 164 la liste en sus)
DMI liste en sus : 1,5 M€ en 2009
DM intra-GHS : 1,65 milliards en 2009

Pour une variété de DM de niveaux variables de : Risque / Nouveauté / Innovation
Un besoin de données cliniques qui doit être adapté à chaque situation
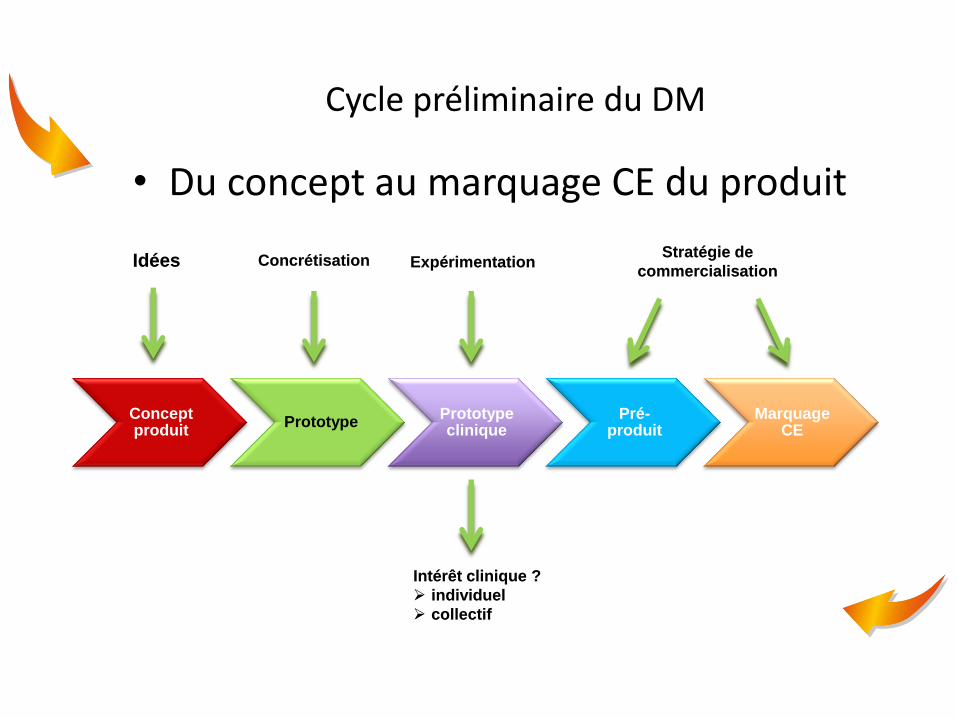
Cycle préliminaire du DM
• Du concept au marquage CE du produit
Concept produit
Prototype Prototype clinique
Pré-produit
Marquage CE
Idées Concrétisation Expérimentation Stratégie de
commercialisation
Intérêt clinique ?
individuel
collectif

S’agit il d’une exception/initiative française ….
Une forte préoccupation des autorités depuis des années
Homologation (CNEH, MS/5D, MS/TI1) fin des années 80 Création de l’AdM (1992) et la Division EM 1993 Transposition des MDD et création de l’Unique ON Français Création de l’AFSSaPS (loi du 1er juillet 1998) Transformation de l’AFSSaPS en ANSM (29 décembre 2011) À venir !?.... (règlements européens et ‘toilettage du CSP!)
Domaine des DM en mutation continue……..
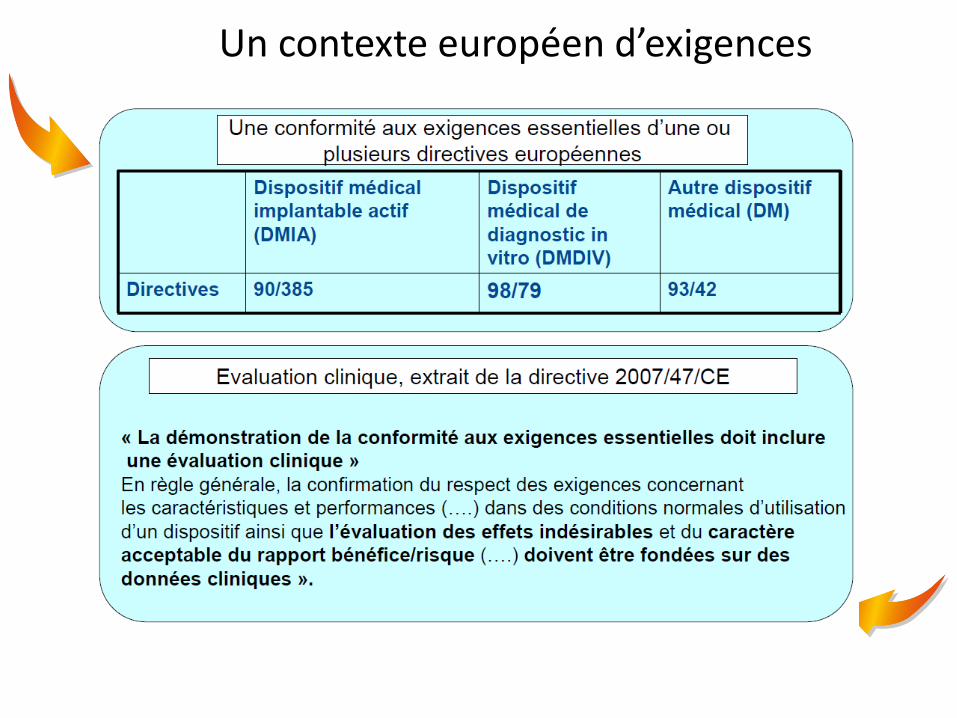
Un contexte européen d’exigences
• x

• Chapitre VI : Articles 49 à 60
– Evaluation clinique & investigation clinique • + détaillé que dans les directives actuelles
• Enregistrement électronique des investigations cliniques
• Délai soumission : max 35 jours + 6 jours
• Si plusieurs soumissions en UE : possibilité d’une soumission unique à
l’échelle Européenne
• Clarifications pour soumissions de modifications substantielles
• Communication entre les Etats Membres
• Communication de l’arrêt de la recherche et des évènements survenant
pendant les investigations cliniques
• Investigation clinique de suivi après-commercialisation
• Annexe XIII: – Evaluation clinique et suivi clinique post-commercialisation
– Exigences mieux détaillées que dans les directives
• Annexe XIV: – Investigations cliniques
Description des méthodes
Documentation
Contenu IB / Protocole
REGLEMENT 2012/0266 DMs et DMIAs En matière d’Investigations cliniques

• Rester à l’écoute des modifications à venir
….. afin de s’assurer d’être en conformité
avec la réglementation parce que …..
• Un nouveau règlement se prépare pour la
suite de la directive 2001/20/CE (investigation
clinique pour les médicaments)
REGLEMENT 2012/0266 DMs et DMIAs Conclusions en matières d’investigations cliniques
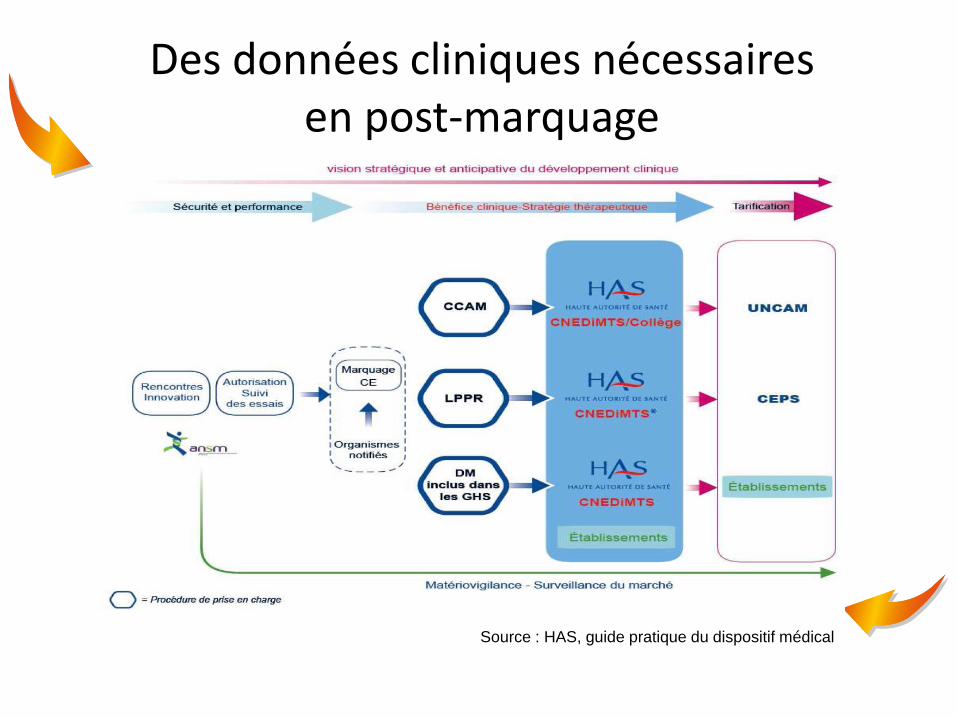
Des données cliniques nécessaires en post-marquage
Source : HAS, guide pratique du dispositif médical
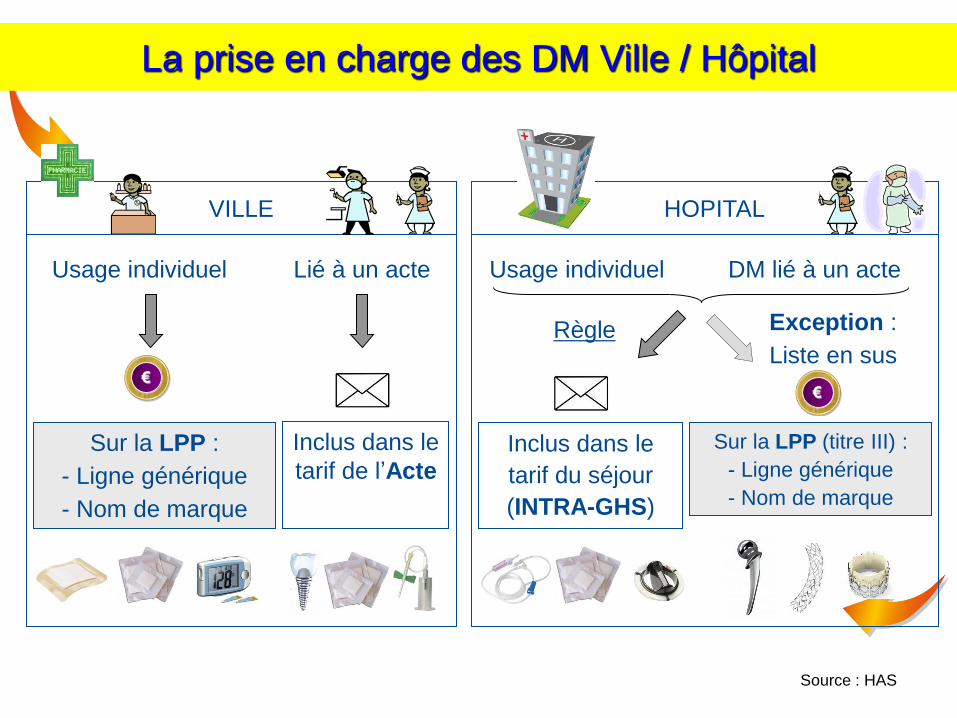
Source : HAS
HOPITAL VILLE
Usage individuel DM lié à un acte Lié à un acte
Inclus dans le
tarif de l’Acte
Règle
Inclus dans le
tarif du séjour
(INTRA-GHS)
Sur la LPP (titre III) :
- Ligne générique
- Nom de marque
Exception :
Liste en sus
€
Usage individuel
Sur la LPP :
- Ligne générique
- Nom de marque
€
La prise en charge des DM Ville / Hôpital
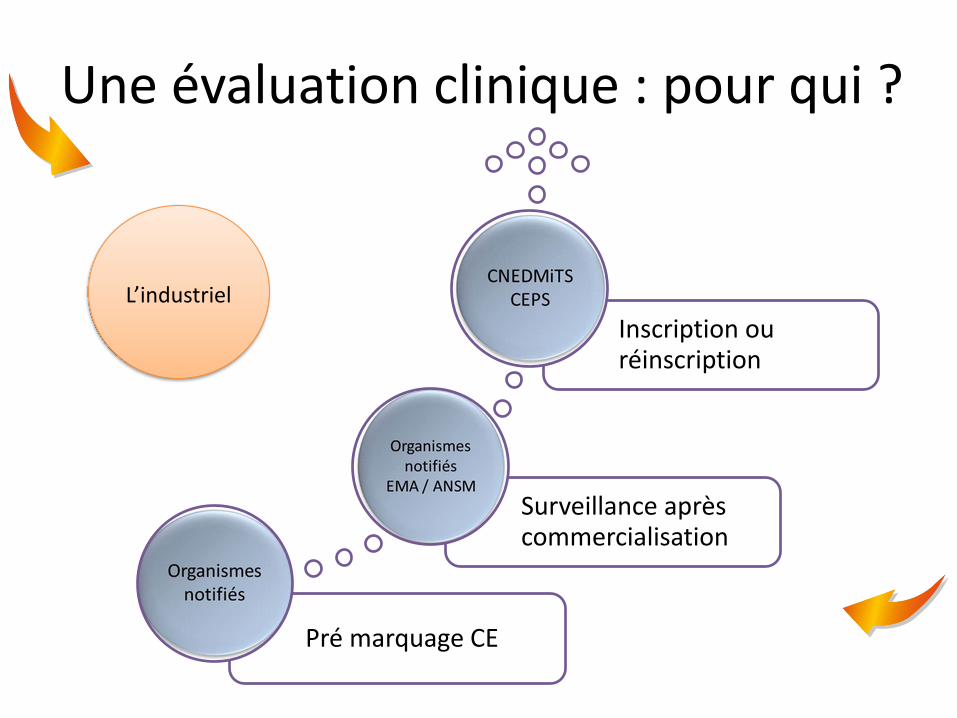
Une évaluation clinique : pour qui ?
Pré marquage CE
Surveillance après commercialisation
Inscription ou réinscription
L’industriel
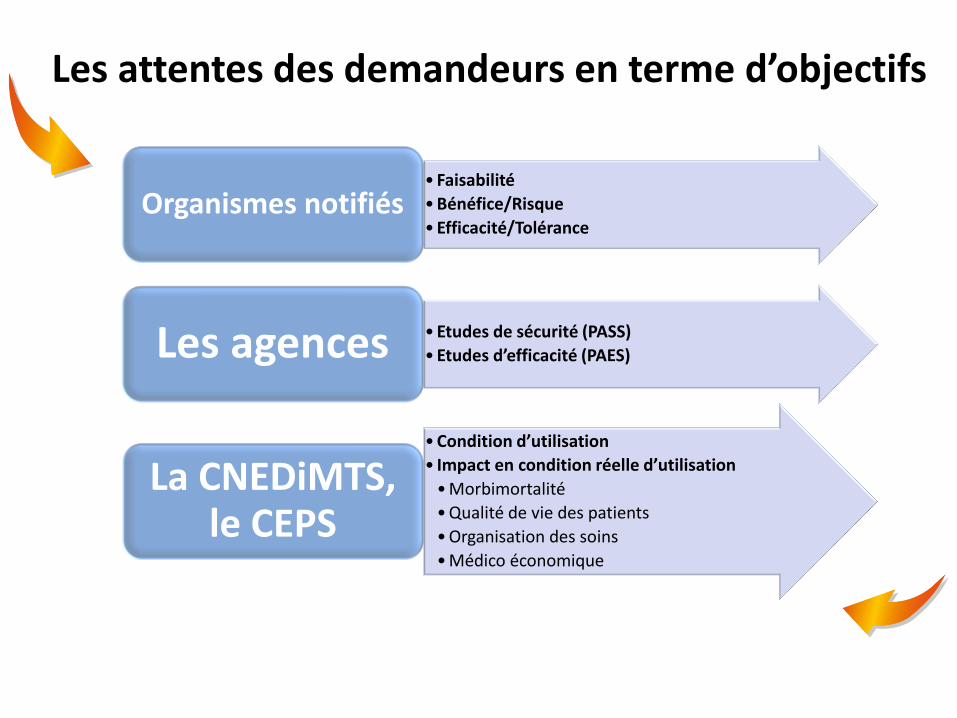
Les attentes des demandeurs en terme d’objectifs
• Faisabilité
• Bénéfice/Risque
• Efficacité/Tolérance Organismes notifiés
• Etudes de sécurité (PASS)
• Etudes d’efficacité (PAES) Les agences
• Condition d’utilisation
• Impact en condition réelle d’utilisation
• Morbimortalité
• Qualité de vie des patients
• Organisation des soins
• Médico économique
La CNEDiMTS, le CEPS

Problématique de l’évaluation clinique • Que faire au stade pré-marquage ? aux stades ultérieurs ?
• Quels critères utiliser pour démontrer un bénéfice clinique pour les patients ?
• Quelle méthodologie choisir à chaque étape ? Selon la classe et le type de DM ?
• Quel cadre réglementaire pour la collecte des données ?
• Comment faire pour les DM combinés (médicament ou DM prépondérant ?)
• Comment mettre en œuvre l’évaluation clinique ?
• En France, dans d’autres pays, en Europe ou ailleurs ?
• Délais ? Coûts ? Faire preuve de beaucoup d’anticipation est INDISPENSABLE

Etapes de l’évaluation clinique
• Analyse bibliographique • Conception du protocole de l’évaluation • Comité scientifique • Financement de l’étude • Autorisations réglementaires • Conduite de l’étude • Assurance qualité • Choix des investigateurs • Gestion des données • Analyse des données • Rapport d’étude • Exploitation réglementaire • Publication scientifique

L’évaluation clinique en pratique
• Typologie des études
• Méthodologie de l’évaluation clinique
• Les exigences essentielles en évaluation clinique
• La dimension économique dans les études sur le DM

Typologie des Essais cliniques
Dépend de la phase du développement du DM
• Etude Pré-Marquage CE
• Post-Marquage CE
– PMCF demandé par Organisme Notifié
– Remboursement
– Post-Inscription (HAS / CEPS)
– Autres situations:
• Extension d’indication
• FDA (étude IDE)

Etude en Inscription et Post-inscription
• Innovation / absence de recul dans la vraie vie
• Demandée par la HAS
• Protocole et design d’étude peuvent être validés par la HAS
• Données pragmatiques:
– aspects cliniques (bénéfices / risques patients)
– Aspects collectifs (paramètres économiques, sociétaux…)

Les études post-inscriptions
Quelques chiffres (source HAS): • Des résultats d’études pour 19% demandes (< 2011)
• Toutes les spécialités sont concernées
• Objectifs : Prédominance de la confirmation d’efficacité
• >25% de demande d’étude pour les ASA V
Ce qui a changé depuis Décembre 2011
• L’accord cadre intégre les études post-inscriptions dans la convention entre le CEPS et l’industriel rendant obligatoire la réalisation de l’étude demandée
Source : HAS

Les études post-inscriptions: La procédure
La CNEDIMTS demande une étude et l’indique dans l’avis
• Elle pointe les données manquantes
• Elle fixe les objectifs
La réalisation de l’étude est à la charge de l’industriel
Une rencontre Industriel-HAS-CEPS est possible, fixant: – Le calendrier – Un « avis » est donné par la HAS/CNEDIMTS sur la:
• méthodologie du protocole • Adéquation protocole/objectifs, • Comité scientifique: médecins et méthodologiste • Envoi d’un courrier précisant les points à améliorer • Les modifications sont de la responsabilité de l’industriel
Cela ne présage en rien des conclusions de l’évaluation Source : HAS

Les études post-inscriptions: points clés
Utilisation du DM en conditions réelles sur les patients de la population cible
EN CONSEQUENCE++
• Centres participants représentatifs • La description des patients
– représentativité ou exhaustivité – Patients non-inclus – Patients perdus de vue
• Les données manquantes • La pertinence clinique des critères de
jugement : mortalité, morbidité
• Le plan d’analyse
• Le rapport d’étude
Source : HAS

Les études post-inscriptions: conclusions
De point de vue HAS :
1. Ce n’est pas la démonstration de l’efficacité
2. Au cours de la réinscription, les résultats de l’étude post-inscription sont indispensables pour l’attribution du service rendu et de l’ASR.
Les éléments favorables:
– Les réunions CEPS/CNEDIMTS/INDUSTRIELS
– Accord cadre
Les points à améliorer :
– L’élaboration du protocole
– L’implication des professionnels de santé
Source : HAS

EN CONSULTATION
ACTUELLEMENT
GUIDE METHODOLOGIQUE
Choix méthodologiques pour le
développement clinique des
dispositifs médicaux
Projet de rapport d’évaluation
18 juillet 2013

L’Evaluation Clinique du Dispositif Médical les études post-inscriptions
Je vous remercie de votre attention
Questions ?
Faraj F. ABDELNOR www.acidim.asso.fr
des femmes, des hommes, vie et technologie
Recommended

















