
Quand penser à une Maladie
Héréditaire du Métabolisme en
Néonatologie?
NECKER-ENFANTS MALADES

• Une centaine de maladies héréditaires du
métabolisme (MHM) actuellement connues
sont à révélation néonatale.
• Le néonatologiste est donc souvent en
première ligne pour évoquer la MHM,
organiser les prélèvements à visée
diagnostique et débuter un traitement.
• La survie et la prévention des séquelles
neurologiques dépendent de la précocité du
diagnostic et de la prise en charge.

Clinique NN à terme malade NN prématuré
infection anomalies
électrolytiques
(rein, endocrinologie)
traumatisme
hypoxie
néonatale
malformations association
fortuite MHM
• Symptômes non spécifiques
• Mais :
– Consanguinité
– Décès inexpliqués dans la famille
– Chronologie des évènements

• Les MHM peuvent être classées dans trois
grands cadres physiopathologiques :
– Intoxications endogènes
– Troubles du métabolisme énergétique
– Anomalies de synthèse ou du catabolisme des
molécules complexes

Groupe I : intoxications endogènes
• Accumulation d’un métabolite toxique par
interruption de sa dégradation suite à un
déficit enzymatique.
• Aminoacidopathies (leucinose)
• Aciduries organiques: acidurie
méthylmalonique, propionique, isovalérique
• Déficits du cycle de l’urée
• Intoxication par les sucres complexes
(galactosémie…)

Maladies d’intoxication: Mise en évidence d’un composé accumulé en amont du déficit
CAA
CAO
Intoxication exogène (lait) endogène (sevrage maternel-catabolisme)
Acides aminés
Acides organiques
NH3 pH iono
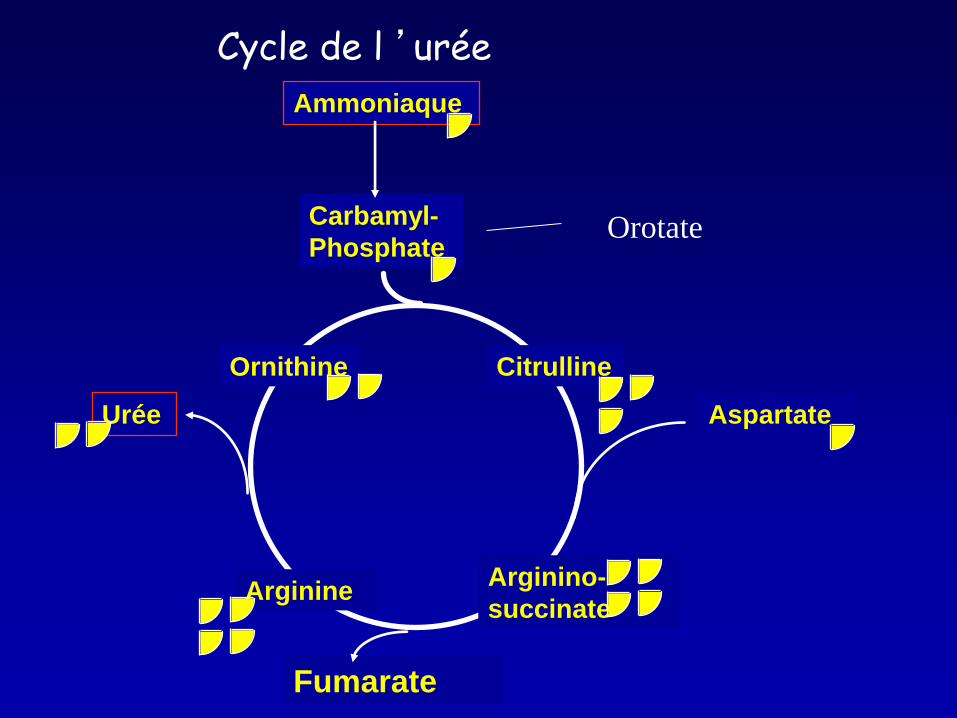
Ornithine Citrulline
Arginine Arginino-
succinate
Carbamyl-
Phosphate
Urée
Ammoniaque
Aspartate
Fumarate
Cycle de l ’urée
Orotate

Groupe II: troubles du métabolisme
énergétique
• Défaut de production ou d’utilisation de l’énergie
atteintes pluri-viscérales mais surtout foie, muscle, cœur,
cerveau.
• Dans ce groupe :
– L’hypoglycémie peut être le symptôme majeur :
• Déficits de l’ oxydation des AG (OAG), glycogénoses...
– L’ hyperlactatémie peut être le symptôme majeur :
• Acidoses lactiques congénitales:
– Déficit en pyruvate carboxylase (PC)
– Déficit en pyruvate déshydrogénase (PDH)
– Cytopathies mitochondriales

Groupe III: anomalies de synthèse ou
du catabolisme des molécules
complexes
• Atteinte permanente, progressive, indépendante
de facteurs intercurrents
• Maladies lysosomales, peroxysomales, troubles
de glycosylation des protéines (CDG), anomalies
de la synthèse du cholestérol...
• Pas de traitement en urgence
• Nécessité d’établir le diagnostic SANS urgence

Clinique : intoxications endogènes
• Altération de l’état général d’un nouveau-né jusque là
bien portant
• Intervalle libre de 48 à 72 h = accumulation d’un
métabolite toxique épuré auparavant par le placenta
• Troubles de la prise alimentaire, léthargie, coma
• Tableau neurologique prédominant :
– Coma hypertonique, opisthotonos
– Mouvements anormaux de type boxe, pédalage,
élévation des membres, myoclonies, trémulations de
grande amplitude
• Tout coma avec une hypertonie ou un tonus normal
doit faire évoquer et rechercher une MHM

Tableaux neurologiques :
détérioration neurologique
• Tableau moins marqué sans intervalle libre net,
hypotonie généralisée :
– déficit énergétique
– anomalies des molécules complexes
• Évolution possible vers un coma
• Signes associés : dysmorphie, cœur, foie, rein
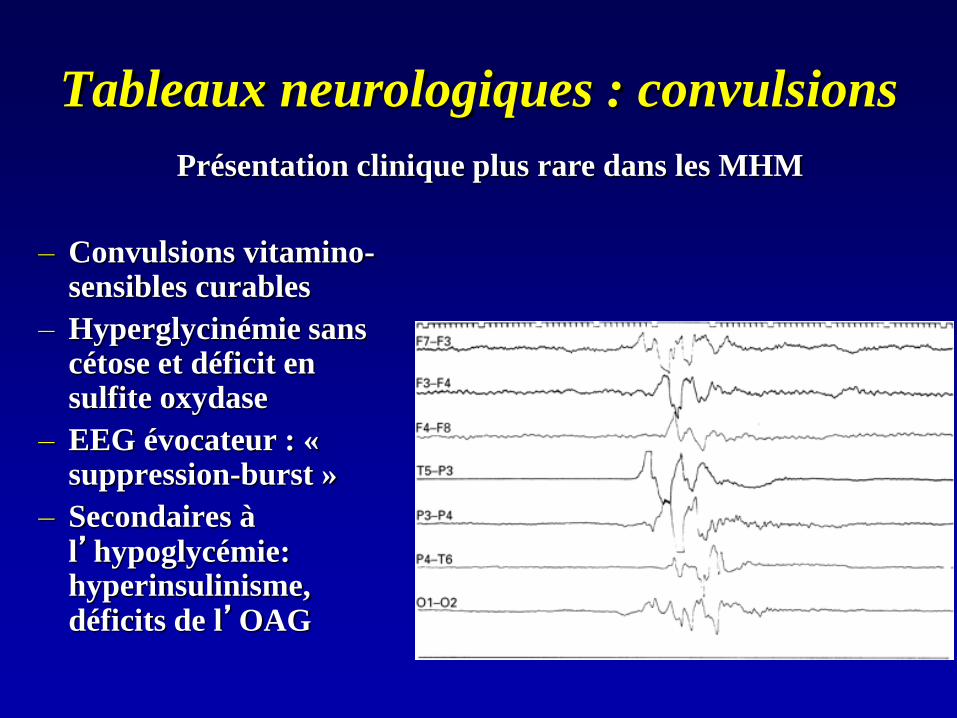
Tableaux neurologiques : convulsions
– Convulsions vitamino-sensibles curables
– Hyperglycinémie sans cétose et déficit en sulfite oxydase
– EEG évocateur : « suppression-burst »
– Secondaires à l’hypoglycémie: hyperinsulinisme, déficits de l’OAG
Présentation clinique plus rare dans les MHM

Clinique : troubles du métabolisme
énergétique
• Signes cliniques présents parfois dès la naissance
– Hypotonie généralisée => absence d’autonomie respiratoire
– +/- dysmorphie, malformations
• Hypoglycémie prédominante :
– Déf. de l’OAG : myocardiopathie, troubles du rythme,
syndrome de Reye (coma, hyper NH3, IHC)
• Hyperlactatémie prédominante:
– déficit en PDH, déficit en PC, déficit de la chaîne respiratoire
• Hypoglycémie+ acidose lactique
– Déficits glycogénolyse /néoglucogenèse, déficit en PC

Tableaux hépatiques
• Hépatomégalie + hypoglycémie :
– Glycogénoses type I et III
– Déficit de l’ OAG
– Déficits de la néoglucogénèse
– CDG Ib (hyperinsulinisme)

Tableaux hépatiques
• Hépatomégalie + splénomégalie + ascite
± anasarque foetoplacentaire:
– Maladies lysosomales
– Déficit en transaldolase (polyols urinaires)
– Hémochromatose néonatale
– CDG

Tableaux hépatiques
• Insuffisance hépato-cellulaire :
– 1ère semaine : hémochromatose, cytopathies mitochondriales, déficit en transaldolase
– 2ème semaine : galactosémie, tyrosinémie, fructosémie
– 2ème mois : déficit de synthèse des acides biliaires
– Les cytopathies mitochondriales, les déficits de l’ OAG (LCHAD) et les maladies du cycle de l’urée sont possibles durant toute la période néonatale.
– Les causes infectieuses (herpès +++) doivent toujours être éliminées.

Tableaux cardiaques
• Cardiomyopathie hypertrophique,
– + Troubles du rythme et de la conduction (mort
subite+++):
• Déficit de l’ OAG
– + Hypotonie :
• Chaîne respiratoire
– + Hypotonie+hépatomégalie+ macroglossie
• Maladie de Pompe (plutôt nourrisson)

MHM
Penser d’abord aux maladies CURABLES
Débuter un traitement d’urgence parallèlement aux investigations
Détérioration
neurologique
type intoxication
Convulsions
prédominantes
Ictère
Insuffisance
hépatique
Défaillance
cardiaque
Troubles du rythme
Hypoglycémies
persistantes
•Leucinose
•Acid. Organique •AMM, AP
•AIV, DCM
•cycle de l’urée
•convulsions
vitamino-
sensibles
•vit B6, PLP
•biotine
•acide folinique
•galactosémie
•fructosémie
•CDG type I b
•synthèse des
acides biliaires
•OAG
•Déf.OAG •Glycogénoses
•NGG
•Hyperinsulinisme
•Déf.OAG

Orienter le diagnostic
• Les urines : recueil immédiat +++
– Odeur « sirop d’érable » ou « curry »= leucinose,
ou « pieds en sueurs »= AIV ou acidurie
glutarique II
– DNPH + = leucinose (acides a cétoniques)
– ACETEST®(KETODIABUR®)
• corps cétoniques (acides b cétoniques)
• ! toujours anormal chez le nouveau-né

Orienter le diagnostic
• Le sang : bilan simple
– pH, base déficit
– Lactate
– Ionogramme (trou anionique)
– Ammoniémie
– Glycémie
– Bilan hépatique, hémostase
– CPK
– NFS

NH3 cétonurie glucose lactate pH autre
Cycle de l‘urée
Leucinose
Acid.Organiques
Déf. Oxydation
AG
Glycogénose I
Déf en PDH
Chaîne Respi
Déf en PC
N ou N
N
, N,
++à+++
N ou N N N + ou N
Neutropénie, thrombopénie, Ca
CPK AG/CC <2
+ ou N
Ou triglycerides
Acide urique
DNPH++
, N,
N
+ ou N
+ ou ++
N
Lactate/ pyruvate:N
BOB/AcAc:
BOB/AcAc :
Quelques profils biologiques typiques

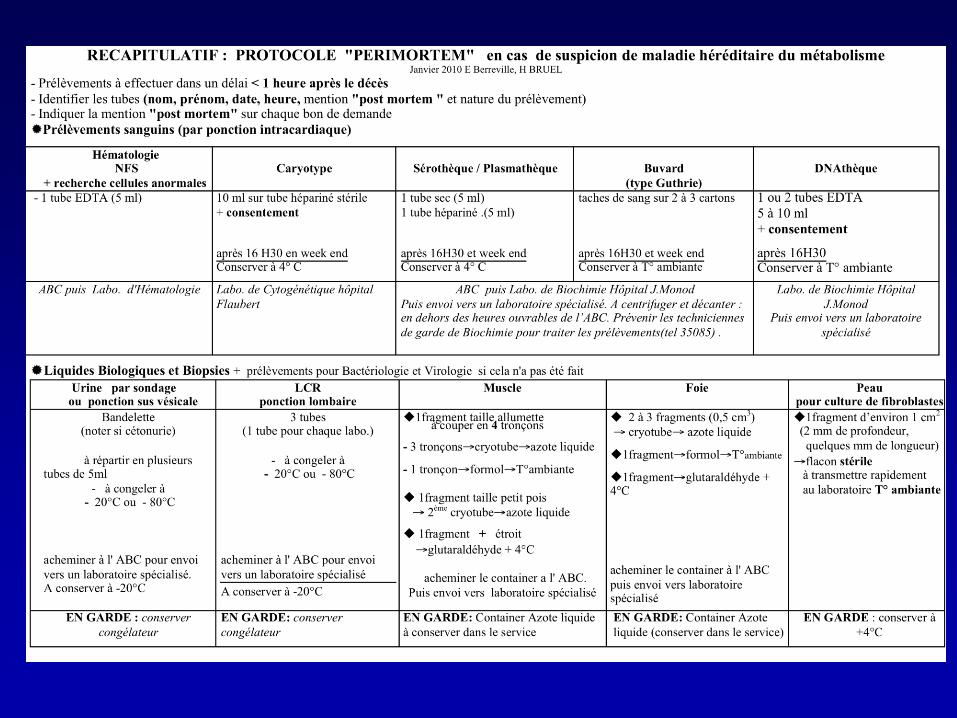
… et stocker
• Plasma, urines, LCR congelés :
– Chromatographies des acides aminés plasma, urines et
LCR des acides organiques urinaires, acylcarnitines
plasma
• Sang total sur EDTA et papier buvard
– Etude de l’ADN, profil des acylcarnitines
• Biopsie de peau dans le sérum physiologique à
T° ambiante :
– Culture de fibroblastes, étude enzymatique
• ± tissus congelés : foie, muscle, cœur, rein...

Conclusion • Penser aux MHM au même titre que
les autres urgences néonatales
• Un bilan biologique simple permet
d’orienter vers ce type de maladie
• Penser à prélever et à stocker y
compris en cas de décès
• Débuter sans tarder un traitement
d’urgence
• Organiser la prise en charge avec une
unité spécialisée
Recommended

















