Embed Size (px)
Citation preview

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 1/14
Chapitre 3 : Méthodes électroanalytique.
I. Introduction.
Les méthodes électroanalytiques sont très répandues tant elles peuvent être utilisées pour des
analyses différentes (mesure de pH, détection de métaux…) que pour une échelle de temps très large (
de 10-6 à 106 secondes). En effet, le simple fait de pouvoir mesurer le signal électrique en réponse à
une perturbation imposée permet la grande simplicité instrumentale et le coût faible de ces méthode.De plus, celles-ci sont très sensibles (1µ A .1s = 1µ C lui même, selon F=NAV.e donne 10-11 mol ou
10-9g) et spécifiques pour une forme de l’élément (Fe, Fe2+, Fe3+…)
Classification des méthodes :
• Potentiométriques : Méthode à ‘courant nul’ où l’on mesurera le potentiel par un voltmètre
(grande impédance). On remarquera que le fait de ne pas avoir de circulation d’électrons, induit la
représentation non-flèchée.
• Ampérométriques : On mesurera le courant par un ampèremètre (impédance très faible)
II. Notions fondamentales d’électrochimie.
1. Rappels
Réaction redox, réducteur, oxydant, cathode, anode, cellule galvanique (pile), cellule
d’électrolyse, électrolyte, chaîne électrochimique, interface, potentiel de jonction liquide,
potentiel d’oxydoréduction (potentiel d’électrolyse ou potentiel redox), potentiel standard
d’oxydoréduction...
Electrode Standard Hydrogène
Equation de Nernst
2. Notion de courant
- I = déplacement d’électron dans tout le circuit extérieur
- I = mouvement d’anions et cations dans la jonction et l’électrolyte des 2 compartiments
- A la surface même des électrodes s’opèrent des processus de transferts électroniques
(réduction-oxydation)
1) Courant Faradique
Le courant Faradique est le courant qui circule à travers l’interface électrode-solution lorsd’un processus d’oxydoréduction.
Le courant d’électrolyse est une mesure directe de la vitesse à laquelle procède la réaction
d’électrode.
Loi de Faraday : puisque I = dQ/dt et Q = ne- . F . nmol
⇒ I = ne- .F.A.dN/dt A = Aire de l’électrode
N = nombre de moles par unité de surface (= nmol/A)
⇒dN/dt = FLUX de matière
Remarques :1
Le courant peut être contrôlé par le transfert de matière, c’est à dire par la vitesse d’arrivéedes substances à l’électrode.2 Il n’y a aucune relation entre charge de l’ion électroactif et nature anodique ou cathodiquede la réaction d’électrode (anode ou cathode peuvent être + ou -).
1

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 2/14
3 Le courant d’oxydation (+→ -) est positif, le courant de réduction (-→ +) est négatif.4 Le courant mesuré à l’électrode est la somme algébrique des courants des différentes
réactions procédant parallèlement à cette électrode.
2) Courant Capacitif
On peut observer un courant dans des circonstances où il ne se produit pas de réaction
électrochimique. L’origine de ce courant est en fait la formation d’une couche d’hydratation à
la surface de l’électrode qui constitue alors un diélectrique et produit une accumulation de
charge de part et d’autre de l’interface électrode-solution. Celle ci se comporte alors comme
un condensateur plan, qui induira un courant capacitif lorsqu’une variation de charge
interfaciale se produit au cours du temps.A= aire des armatures du condensateur
⇒I = dQ/dt avec Q = C.V et C = ε .A/d ε = constante diélectrique (≈épaisseur
des molécules d’eau ≈ 3Å)d = épaisseur du diélectriqueC= capacité du condensateur
⇒ I = C.dV/dt et pour un circuit R(la solution n’est pas infinimentconductrice)C série : I = V/R.exp-t/RC
ou bien
⇒I = V.dC/dt où la variation de capacité est induite par la variation de A.
⇒Le courant mesuré n’est pas une grandeur spécifique, car il est constitué de la somme de
tous les courants capacitifs et faradiques.
3. Tension des cellules électrochimiques
1) Dispositif à 2 électrodes.
La tension mesurée entre 2 électrodes correspond bien sur à un équilibre (pas de
courant) et se mesure par Eel. indic – Eel ref . Les interfaces des solutions sont considérées comme
des condensateurs placés en série ( fig.3) et ont une équation qui peut se ramener à 1/C = 1/CT
si 1/CSC E est rendu négligeable. CSC E doit donc être rendu très grande et pour ce faire, on
pourra jouer sur sa taille.
Effet du courant sur la tension des cellules électrochimiques.
Si un courant passe à travers une cellule, la tension de celle-ci n’est plus simplement égale à la
différence de potentiels rédox des électrodes. 2 facteurs nouveaux, la chutte ohmique et la polarisation nécessitent l’application d’une tension supérieure à la valeur thermodynamique
∆ V appliquée El 1 Solution El 2 El 1 El 2
1 2
∆ V Utile R sol . I (cas idéal !)
OX RED OX RED
1 : Il se produit ici une chute de potentiel des condensateurs. En effet, si le courant
passe, la tension sera diminuée (chute Ohmique : ∆ V = R.I .Cette chutte est normale, puisqu’à chaque interface, il y a toujours une ∆ V). Or, la ∆ V utile à la réaction est celle
après la première chute puisque le transfert d’électrons se fait là. On va donc diminuer les
2

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 3/14
pertes ailleurs pour ressentir la chute de l’électrode 1. Il s’agira donc de minimiser R en
ajoutant de l’électrolyte concentré
2 : Si la C est grande, la chute de potentiel sera petite, et donc, l’électrode 2 sera
grande. C’est un cas idéal : la première chute, qui correspond à l’arrachage d’électrons est
maximale. Le ∆ V utile sera donc maximal et il n’y aura plus de pertes après... ( ?)
2) Dispositif à 3 électrodes (fig.4)
On aura ici 3 électrodes, une électrode de travail ET, une électrode de référence ER, et
une contre-électrode CE, et 2 parties, une pour mesure électrique (I entre ET et CE) et une
pour mesure de potentiel (V entre ET et ER). En effet, avec ET et ER (le couple redox, qui
doit être à l’équilibre électrochimique) on ne peut pas mesurer le courant mais seulement le
potentiel. Pour mesurer ce courant, on aura donc besoin d’une troisième électrode (CE) placée
entre ET et ER (ampèremètre).
Ce dispositif permet un contrôle du potentiel de l’électrode lors de l’électrolyse.
III. Potentiomètre.
Méthode basée sur la relation liant le potentiel d’une cellule électrochimique et les
concentrations ou activités des espèces en solution en l’absence de courant. C’est donc une
méthode de mesure à courant nul (sauf un petit pour faire fonctionner le voltmètre) qui permet
d’effectuer facilement des titrages.
Remarque sur le schéma d’une cellule utilisée pour un dosage potentiométrique ( U = E ind –E ref +E j ):Pour améliorer le fonctionnement, il faudra minimiser E j, c’est à dire le potentiel de jonction.
Donc, soit Eind est lié à la concentration de l’analyte et il faudra essayer de minimiser E j, soit
Eref est lié à la concentration de l’analyte et E j est constant.
1. Electrodes de référence (rappel).
Une électrode de référence est une demi-cellule dont le potentiel est connu et reste constant,
indépendamment de la composition de la solution d’analyte.
Dans les tables, les conditions standards ne fixent ni la température, ni le solvant.
A contrario, les E° sont souvent donnés à 25° en solution aqueuse, donc si les conditions de
travail ne sont pas identiques, on utilisera l’équation de Nernst.
Bien que EH+/H2 = 0 on utilise une électrode au calomel car celle-ci est beaucoup plus facile à
réaliser. Dans cette électrode, on fixera [Cl-
] grâce au KCl saturé. Cependant, la température peut faire varier cette saturation de manière assez forte. Dés lors, dans des conditions de
changement de température, on utilisera une autre électrode de référence que celle au calomel.
De la même manière, certaines électrodes de référence sont sensibles à la variation de pH. Le
choix de l’électrode de référence est donc déterminant!
2. Electrodes indicatrices (de travail).
Une électrode indicatrice est une demi-cellule dont le potentiel varie de manière connue en
fonction de l’activité de l’analyte.
1) Electrode métallique.
Ces électrodes permettent le transport d’électrons et le dosage de l’activité ou de la
concentration des ions.
3

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 4/14
• Electrodes de la première espèce : Elles permettent un dosage DIRECT des cations
métalliques, l’électrode étant constituée du cation lui-même (Pb2+/Pb(s)).
Remarque : l’équation de Nernst prend cette forme puisque a i d’un solide ou liquide pur = 1!
• Electrodes de la deuxième espèce : Cette fois, le dosage se fera de manière INDIRECTE
par des espèces différentes.
Exemple : dosage de Cl- avec une électrode d’Ag. Cette réaction est couplée à une autre
réaction, un équilibre de précipitation (même principe que piles de C).
2) Electrode à membrane
Ces électrodes sont, cette fois, composées de matériaux conducteurs non pas
d’électrons, mais d’ions. Il s’agit donc ici d’une fine membrane séparant deux solutions et de
deux électrodes de référence (une en contact direct avec la solution d’analyte et l’autre séparée
par une membrane) dont la différence de potentiel dépendra de la nature et de la
concentration/activité des ions présents en solution. La mesure est donc plus facile, mais peut
subir des interférences.
• Electrodes de verre.
Ce type d’électrode est basé sur la dissociation du SiO2 en SiO44- qui va induire un échange
de cations monovalents mobiles (Na+, H+...) dans la membrane de verre (pénétration dans
les interstices). Un équilibre va alors s’établire (du type Na+ + H+verre ↔ Na+
verre +H+) et, les
deux interfaces ne subissant pas la même compensation de charge, une nouvelle différence
de potentiel liée aux échange d’ions va apparaître.
L’équation de NICOLSKY a ensuite été introduite pour palier à l’erreur alcaline. Celle-ci
est liée au fait qu’en solution basique, les électrodes de verre répondent à la fois aux ions
H+ et aux ions alcalins. Ces électrodes sont donc sélectives mais non-spécifiques (réponseà plusieurs espèces, mais uniquement aux cations monovalents. Donc, si l’on veut calculer
le pH en milieu basique, il va falloir jouer sur la composition du verre pour modifier sa
sélectivité...).
Eind = L + R.T/(ziF) . ln ( ai + ∑ j k ij a(zi/zj) )
k ij = coefficient de sélectivité. Il mesure en fait la réponse de la membrane sélective à
d’autres ions.
Tout ce qui est dans le ln = lié à la présence d’agents interférents (rarement le cas).
L = est une constante liée aux caractéristiques de la membrane. Puisque celle-ci est
impossible à calculer dans la manipulation, un étalonnage a été établit, qui fixe sa valeur.
• Electrodes à membrane cristalline
Il s’agit toujours de membranes conductrices d’ion dont la composition variera selon
l’analyte. Les électrodes les plus répandues sont celles aux halogénures et aux sulfures.
L’électrode la plus fiable est celle au fluorure de lanthane dopée au EuF 2 (dosage des F-),
mais il existe aussi des électrodes au sulfure d’Ag, Cu permettant de doser Ag+, Cu2+ et S2.
Ces électrodes permettent donc de doser les ions en solution dont elles sont composées (≈métalliques de première espèce)
• Electrodes à membrane liquide
La membrane poreuse permet ici un échange d’ions liquides. Celle-ci s’imbibe de
l’échangeur d’ions liquides et permet son contact avec la solution.
4

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 5/14
Graphique(p.12)U = f(ai)
Graphique
(p.13)-i = f(-E)
-i = courant de
L’électrode la plus utilisée est celle pour doser les Ca2+. Malgré tout, beaucoup
d’interférences sont possibles et, au fur et à mesure, on perd de l’échangeur d’ions qui se
dissout dans la solution. La durée de vie de cette électrode est donc assez faible.
• Electrodes modifiées et...
Ce type d’électrode utilisera un greffage de substance organique sur la surface de
l’électrode ou bien l’immobilisation d’une enzyme dans la membrane.
Exemple dosage de l’urée : urée + uréase ↔ NH4+ (détecté par l’él. en verre) + ...
• ...Senseurs à gaz
Ce type d’électrode est constitué d’une électrode à membrane de verre plus d’une
membrane perméable uniquement au gaz.
Exemple dosage de SO2 : SO2 + H2O ↔ HSO3- + H+ (dosé par l’électrode de verre)
L’étalonnage tient donc compte de la fraction de SO2 traversant la membrane et du dosage
à l’électrode.
Remarque : Tous les senseurs ne sont pas potentiométriques, il en existe des
ampérométriques mesurant un courant lié à la concentration.
• Evaluation des caractéristiques des électrodes indicatrices d’ions
- Facilité d’utilisation (matériel simple)
- Dosage de nombreux ions et composés biochimiques
- Méthode sélective et non-spécifique
- L’erreur peut être importante, et comporte toujours une erreur inhérente à la procédure
d’étalonnage.
Equation de base : U = K + RT/Z1 ln a1L’erreur relative sur a1 (∆ a1/a1) est liée à ∆ K, l’incertitude absolue sur K. Par
exemple, si ∆ K vaut ± 0.001 V, on peut s’attendre à une erreur relative
d’environ ± 4% sur l’activité. Cette erreur ne peut pas être éliminée : en effet,
on a ( fig.12) une déviation de la relation linéaire, même sans agents interférant
car à de petites concentrations, la membrane n’est plus assez sensible.
- Réponse souvent lente
- Durée de vie des électrodes limitée
IV. Courbes de Polarisation→Cas idéal sans transfert de matière : 1 étape = transfert d’électron
On a vu préalablement que Nernst n’était valable que pour les utilisation de potentiel (V/C,
lorsque I est nul). Pour les méthodes d’analyse ampérométriques, on devra donc établir la
relation I/C.
Dans un processus électrochimique, la réaction aux électrodes implique 2 étapes :
- Transport de matière vers la surface.
- Transfert d’électrons (oxydation)
-Production d’une espèce à l’électrode qui est éloignée (réduction)
5

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 6/14
Oxelectrode Oxsolution
R electrode R solution
L’une ou l’autre de ces étapes peut être déterminante, et dans un cas idéal, la solution reste
homogène de C∞ (cœur de phase) jusqu’à la surface de l’électrode, et n’est donc pas limitante
ni pour le transport ni pour la réaction (pas vraiment consommée)! La seule étape
déterminante est donc le transfert d’électrons.
⇒le transport de Oxsol se fait spontanément, donc [Oxelectrode] ≈ [Oxsolution], et malgré la
transformation Oxel→R el , la concentration de Ox reste plus ou moins constante.
C∞
El.
x
⇒la courbe de polarisation qui liera I et E sera du type I(E)
On définira alors le domaine idéalement polarisable comme celui dans lequel pas ou peu de
courant est présent (sauf peut-être un petit courant capacitif) et qui dépend de la nature du
métal, de l’électrolyte et du solvant.
-I I capacitif I de reduction (<0)
I d’oxydation (>0) 0 -E
E
↔
I Faradique (partout où ce n’est pas IC)
+I
Remarque : Le courant capacitif étant connu, on peut le soustraire
- Dans le domaine du courant capacitif, on peut appliquer à l’électrode n’importe quel courant ⇒E↔
est le
potentiel d’équilibre (comme on a pu voir pour les méthodes potentiométriques).
- Du coté -E, le domaine est limité par la réduction des cations/protons métalliques.
- Du coté +E, le domaine est limité par l’oxydation du métal de l’électrode.
Fig.13 : A peut être réduit en B et l’on voit que I est directement proportionnel a la
concentration de A. En effet, I est supérieur quand C1 que quand C2 (⇒C1>C2) : dN/dt = K.C
Expression des courbes I-E :
A un même potentiel, le relation entre I et C est définie par la loi de FARADAY.
I = n.F.A.dN/dt
Et I = I→
+ I←
= Ired + Iox
Et puisque l’on ne regarde qu’un transfert d’électron : v = k.C = ± dN/dt
On introduit ici k, constante de vitesse du transfert d’électron (en cm.s -1)
Avec dN/dt qui correspond toujours à une variation de la concentration surfacique.
* k →= k 0 exp - α →.F.(E-E0)/R.T α = coefficient du transfert d’électrons
* k ←
= k 0 exp α ←.F.(E-E0)/R.T
⇒I→
= -n.F.A.k →.C
→(- car sens de la consommation)
6

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 7/14
Graphiquesc/c° = f(xdistance
El.)
I←
= n.F.A.k ←.C
←
⇒ L’allure exponentielle de la courbe est lié au fait que k dépend exponentiellement de E.
C’est pourquoi pour I→, on doit appliquer une différence de potentiel supérieure aux plus
grandes concentrations (pour I←
c’est l’inverse...)
Quand à la mesure des courants on utilisera la résultante des courants de réduction et
d’oxydation. En effet, dans le cas d’une différence de potentiel, celle-ci s’appliquant aussi bien à la réduction qu’à l’oxydation, on aura α →+ α ←
= 1.
Au potentiel d’équilibre (E↔
), I=0. Or, comme I = I→
+ I←
, I→
= -I←
= I0 = I
↔( Fig.14)
⇒La résultante est la courbe mesurée et le point ou I=0 définit le potentiel d’équilibre
du système (et le domaine idéalement polarisable ? ?)
On peut encore y déterminer le courant d’échange I↔
, valeur qui va permettre de définir le
système comme réversible ou irréversible.
• Fig.14 : Dans la même gamme de potentiels (assez étroite), on pourra avoir oxydation
ou réduction. Le système sera donc d’autant plus réversible que le courant d’échange est
grand (la courbe de la somme de I est plus redressée)• Au contraire, dans un système où les courbes de réduction/oxydation sont éloignées,
on définira clairement un domaine du courant d’oxydation et un domaine du courant de
réduction. Le système possède un très petit courant d’échange et est donc pratiquement
irréversible
⇒Lier cela au domaine idéalement polarisable…
Un bon couple d’oxydoréduction doit avoir un potentiel plus ou moins constant. Le choix se
portera donc sur ceux dont le courant d’échange est important.
Malgré tout, il faut encore tenir compte de la nature de l’électrode. Par exemple, H+/H2 sur Pt
est très efficace contrairement au même couple sur Hg.⇒on doit définir couple et électrode
V. Transport de matière et courant limite de diffusion.→Cas réél avec transfert de matière (limitant)
On admettra ici les hypothèses suivantes : le transfert électronique est très rapide et
le transport de matière est le facteur limitant. C’est donc opposé aux courbes de polarisation.
En effet, on établit les courbes de polarisation, théoriquement, dans le cas idéal, et on y définit
un domaine idéalement polarisable. Mais dans les cas réels, on aura un courant limite de
diffusion, limitant, dû au transport de matière.
1. Modes de transport.
1) Convection.
La convection est le transport mécanique d’ions ou de molécules effectué par la
solution qui se déplace sous l’effet d’une agitation forcée (interférences mécaniques), ou
simplement induite par des gradients de densité. C’est un paramètre contrôlable qui peut être
évité si on empêche l’agitation et que la température est homogénéisée.
En chimie analytique, la convection ne sera pas induite par température (∆ T) puisque l’on
veut une température constante.
2) Migration.
7

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 8/14
La migration est le mouvement des ions à travers une solution sous l’action d’un
champ électrique. Paramètre contrôlable, évité si on ajoute un électrolyte support (sel dissocié
de très haute concentration), solution conductrice qui va homogénéiser le potentiel. En
d’autres termes, on va ajouter volontairement beaucoup d’ions spectateurs pour uniformiser le
champ électrique et éviter la migration.
Si la migration était le seul mode de transport des réactions chimiques, on ne pourrait
qu’oxyder des anions et réduire des cations.
3) Diffusion.
La diffusion et le mode de transport par lequel des ions ou des molécules se déplacent
à travers une solution d’une zone concentrée vers une zone plus diluée. La force motrice est
donc le gradient de concentration et permet une uniformisation du système.
La diffusion est le mode de transport des réactions électrochimiques car il permet la
consommation d’analyte à l’électrode.
Lois de diffusion :
Le courant mesuré : I = n.F.A (dN/dt)x=0 x=0 (à l’électrode)
On considère le cas de la diffusion semi-infinie linéaire (c’est à dire la diffusion vers
une surface plane uniquement en fonction de la distance x)
On considère que l’électrode est le plan et que d’un coté, la solution est infinie.
⇒Lois de FICK (applicables dans tous les domaines où il y a transport de matière):
D est le coefficient de diffusion et dépend de la nature de l’espèce qui diffuse (taille),
de la température et du milieu (surtout de la viscosité).
dN/dt : nombre de moles par unité de surface = FLUX de matière!
• dN/dt = -D dC/dx : le flux de matière est proportionnel et inverse au gradientde concentration.
(Le - signifie que le flux se fait dans le sens des concentrations décroissantes.)
• δ C/δ t = D δ ²C/δ x² (utilisée en quand le régime est non-stationnaire (puisque C/t),
elle exprime les variations du système) : La variation des concentration est proportionnelle à
la courbure du profil c = f(x). La diffusion tend à raboter les courbures et à
réhomogénéiser ainsi la solution.
C
C∞
Electrode
x x+dx x
Remarque : On doit aller chercher de la matière de plus en plus dans le cœur de la
solution. En tout point, on peut connaître le flux (cfr première équation de Fick = pente
de la courbe). La concentration entre x et x+dx diminue car ce qui sort de l’intervalle
est inférieur à ce qui rentre, venant de la solution externe (flux en x > flux en x+dx :
donc, plus on se rapproche de l’électrode, plus le flux sera important).
⇒la concentration varie au cours du temps (dans cet intervalle).
8
5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 9/14
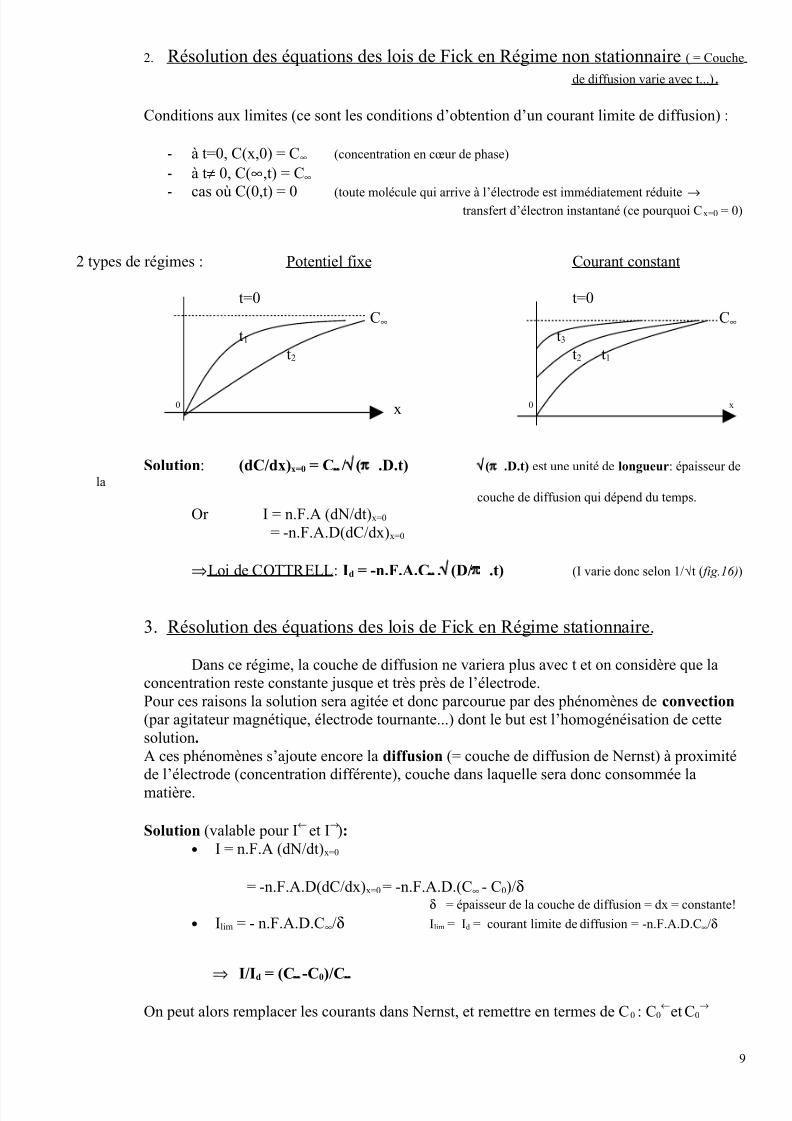
2. Résolution des équations des lois de Fick en Régime non stationnaire ( = Couche
de diffusion varie avec t...) .
Conditions aux limites (ce sont les conditions d’obtention d’un courant limite de diffusion) :
- à t=0, C(x,0) = C∞ (concentration en cœur de phase)
- à t≠ 0, C(∞,t) = C∞
-cas où C(0,t) = 0 (toute molécule qui arrive à l’électrode est immédiatement réduite →
transfert d’électron instantané (ce pourquoi Cx=0 = 0)
2 types de régimes : Potentiel fixe Courant constant
t=0 t=0
C∞ C∞
t1 t3
t2 t2 t1
0 x 0 x
Solution: (dC/dx)x=0 = C∞/√(π .D.t) √(π .D.t) est une unité de longueur: épaisseur de
la
couche de diffusion qui dépend du temps.
Or I = n.F.A (dN/dt)x=0
= -n.F.A.D(dC/dx)x=0
⇒Loi de COTTRELL : Id = -n.F.A.C∞.√(D/π .t) (I varie donc selon 1/√t ( fig.16))
3. Résolution des équations des lois de Fick en Régime stationnaire.
Dans ce régime, la couche de diffusion ne variera plus avec t et on considère que la
concentration reste constante jusque et très près de l’électrode.
Pour ces raisons la solution sera agitée et donc parcourue par des phénomènes de convection
(par agitateur magnétique, électrode tournante...) dont le but est l’homogénéisation de cette
solution.
A ces phénomènes s’ajoute encore la diffusion (= couche de diffusion de Nernst) à proximité
de l’électrode (concentration différente), couche dans laquelle sera donc consommée la
matière.
Solution (valable pour I←
et I→):
• I = n.F.A (dN/dt)x=0
= -n.F.A.D(dC/dx)x=0 = -n.F.A.D.(C∞ - C0)/δδ = épaisseur de la couche de diffusion = dx = constante!
• Ilim = - n.F.A.D.C∞/δ Ilim = Id = courant limite de diffusion = -n.F.A.D.C∞/δ
⇒ I/Id = (C∞-C0)/C∞
On peut alors remplacer les courants dans Nernst, et remettre en termes de C 0 : C0←
et C0→
9
5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 10/14
Graphique(p.17)
-i = f(-E)
-i = courant deréduction
-E = E appliqué vsECS
+
Ired = f(V)= Titrage !
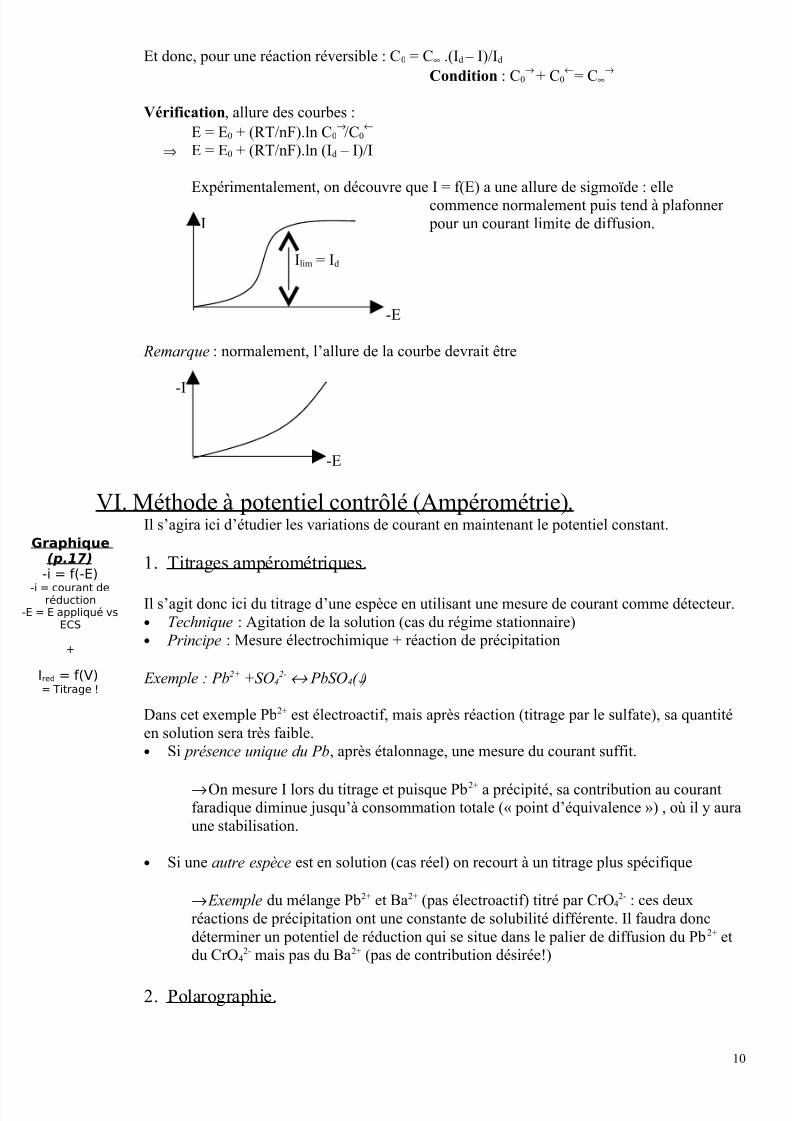
Et donc, pour une réaction réversible : C0 = C∞ .(Id – I)/Id
Condition : C0→
+ C0←
= C∞→
Vérification, allure des courbes :
E = E0 + (RT/nF).ln C0→/C0
←
⇒ E = E0 + (RT/nF).ln (Id – I)/I
Expérimentalement, on découvre que I = f(E) a une allure de sigmoïde : elle
commence normalement puis tend à plafonner
-I pour un courant limite de diffusion.
Ilim = Id
-E
Remarque : normalement, l’allure de la courbe devrait être
-I
-E
VI. Méthode à potentiel contrôlé (Ampérométrie).Il s’agira ici d’étudier les variations de courant en maintenant le potentiel constant.
1. Titrages ampérométriques.
Il s’agit donc ici du titrage d’une espèce en utilisant une mesure de courant comme détecteur.
• Technique : Agitation de la solution (cas du régime stationnaire)
• Principe : Mesure électrochimique + réaction de précipitation
Exemple : Pb2+ +SO42- ↔ PbSO4( ↓ )
Dans cet exemple Pb2+ est électroactif, mais après réaction (titrage par le sulfate), sa quantité
en solution sera très faible.
• Si présence unique du Pb, après étalonnage, une mesure du courant suffit.
→On mesure I lors du titrage et puisque Pb2+ a précipité, sa contribution au courant
faradique diminue jusqu’à consommation totale (« point d’équivalence ») , où il y aura
une stabilisation.
• Si une autre espèce est en solution (cas réel) on recourt à un titrage plus spécifique
→ Exemple du mélange Pb2+ et Ba2+ (pas électroactif) titré par CrO42- : ces deux
réactions de précipitation ont une constante de solubilité différente. Il faudra donc
déterminer un potentiel de réduction qui se situe dans le palier de diffusion du Pb2+ et
du CrO42- mais pas du Ba2+ (pas de contribution désirée!)
2. Polarographie.
10

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 11/14
La polarographie est une voltampérométrie (méthode électrochimique basée sur la
mesure du courant en fonction du potentiel appliqué à une microélectrode. Elle nécessite donc
une variation contrôlée du potentiel.) effectuée à l’aide d’une électrode à goutte de mercure.
Cette méthode est une des plus ancienne et été à la base de beaucoup d’autres. Elle n’est
pourtant plus autant utilisée aujourd’hui à cause du mercure, métal qui n’est malgré tout pas si
toxique que ça…
En résumé, quand une goutte tombe, une nouvelle se forme. La variation de capacité de
l’électrode étant liée à la variation de l’air de l’élément, on peut détecter un courant capacitif.On remarquera encore que la mesure est prise quand la goutte est encore attachée (une goutte
tombée ne sert à rien…puisque alors, plus d’électrode!) et que la mesure est celle d’un courant
total (capacitif + faradique) à volume fixe.
1) Courant capacitif
On obtient un courant capacitif si C ou ∆ V varient.
Ici, le courant est directement lié à la croissance de la goutte (A varie ⇒C varie). Donc, V est
constant et A varie au cours du temps.
On peut considérer la goutte comme une sphère dont le débit massique (m = masse écoulée
par unité de temps) est constant.
V = 4/3 π .r² = m/d avec d (densité volumique) = 13.6 kg/l
= m .t/d
A = 4π r² = 4π .(3/4π )2/3 . (mt/d)2/3
= 0.852 m2/3.t2/3
L’intensité du courant capacitif est donc donnée par
IC = dQ/dt = C(E-Epcn) dA/dtpcn : point de charge nulle (=potentiel d’équilibre) et C en µ F/cm²
IC = 0.598 C(∆ Epcn).m2/3.t-1/3
On remarque donc que IC décroît en t-1/3 et augmente avec E-E pcn ( fig.21 : au pcn, I C =0, a droite du pcn, I C < 0, et à gauche, I C > 0). Or, il faut minimiser IC pour ne garder
que le courant faradique IF (ou bien augmenter IF, qui est lié à la concentration, mais
pas d‘action possible sur la concentration!).
Fig.21: Lorsque IC est positif, E > E pcn et le sommet d‘une courbe est le moment où la goutte
est la plus grande. Inversement, lorsque IC est négatif, E < E pcn et le minimum de la courbe est
le moment où la goutte est la plus grande.
!!Remarque : En l’abscence de substances électroactives, le courant résiduel
polarographique se limite au seul courant capacitif qui charge les gouttes de mercure à
mesure qu’elles se forment.
Courant faradique
On cherche ici à obtenir le courant limite de diffusion (cfr. Cottrell, c’est à dire pour le régime
non stationnaire). Cependant, la goutte (sphère) grandit et change les conditions puisque le
‘plan de l’électrode’ s’avance. Dés lors, des corrections devront donc être apportées à la loi de
Fick (et donc à la loi de Cotrell):
A résoudre : dC/dt = D.d2C/dx2 + 2/3 . x/t . dC/dx nouveau facteur
11

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 12/14
Solution : (dC/dx)x=0 = C∞/√(3.π .D.t /7)
L’équation d’ILKOVIC apportera ensuite la valeur du courant de diffusion :
Id = 706.n.m2/3.t1/6.C∞.√D
On remarquera que la dépendance en t1/6 provient de la combinaison des deux
dépendances en temps (équation de Fick – Variation de A avec t)
Fig.22: La grande différence ici entre IF et IC provient de la concentration importante en
analyte. On pourra également dire qu’ici, le minimum de courant n’a pas de sens puisque
directement lié à la vitesse de réponse.
Fig.23: Le débit volumique doit être le même pour les deux mesures (étalon – analyte). On
utilisera donc la même hauteur de colonne de Hg et des réservoirs assez longs pour que la
consommation ne diminue pas trop la hauteur.
• Analyse quantitative basée sur I d (p.21).
Facteurs affectant Id :
- Effet de température (2% par °C pour D et puisque on prend la racine carrée : 1,44% d’erreur )
- Effet de la hauteur du réservoir (m identique)
- Maxima polarographiques
Analyse qualitative.
E1/2 = potentiel de demi-onde du système (à Id/2)
• Analyse de mélange d’espèces électroactives (I = I C + ∑ i I F i ).
Fig.25 : On voit uniquement des petites valeurs de µ A pour des gouttes de surface de 20mm²
et de durée de vie (temps de mesure) de 4s. Cette méthode est non-destructive puisque aucune
matière n’est consommée pendant la mesure.
Si les vagues sont assez séparées, on peut isoler les comportements, déterminer E1/2 et calculer
la concentration de l’espèce en connaissant son k (Id = k x.[X]).
• Evaluation de la méthode
- Dosage d’un grand nombre de cations, anions et molécules neutres (on peut par
exemple doser les O2 dissous en solution. Cependant si on ne commence pas par désoxygéner l’eau au N2, l’O2 interagira avec les métaux en solution et provoquera des
interférences).
- Méthode précise à 99%
- La limite de détection se situe à ± 10-5 M. Cette limite est due a IC, qui peut atteindre
l’ordre de IF. Il faudra donc augmenter le IF par rapport au IC (on peut combiner les 2).
Fig.29 : Plus la concentration en analyte est petite, plus IC sera grand. La limite de
détection est donc atteinte lorsque IC devient trop important.
3. Polarographie impulsionelle.
• But : améliorer les performance de la polarographie en augmentant le rapport I F/IC .
• Principe.
12
5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 13/14
Graphiques(p.23)E = f(t)I = f(t)
Et I = f(-E)toujours…
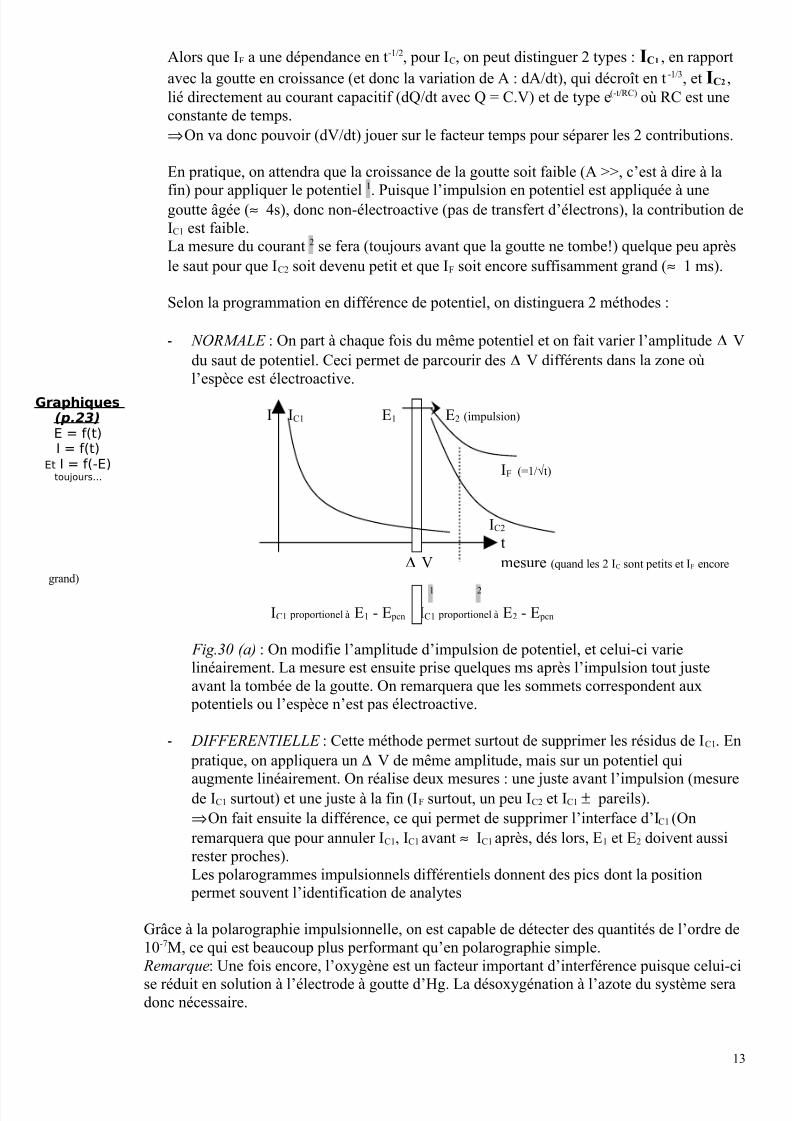
Alors que IF a une dépendance en t-1/2, pour IC, on peut distinguer 2 types : IC1 , en rapport
avec la goutte en croissance (et donc la variation de A : dA/dt), qui décroît en t -1/3, et IC2 ,
lié directement au courant capacitif (dQ/dt avec Q = C.V) et de type e(-t/RC) où RC est une
constante de temps.
⇒On va donc pouvoir (dV/dt) jouer sur le facteur temps pour séparer les 2 contributions.
En pratique, on attendra que la croissance de la goutte soit faible (A >>, c’est à dire à la
fin) pour appliquer le potentiel1
. Puisque l’impulsion en potentiel est appliquée à unegoutte âgée (≈ 4s), donc non-électroactive (pas de transfert d’électrons), la contribution de
IC1 est faible.
La mesure du courant 2 se fera (toujours avant que la goutte ne tombe!) quelque peu après
le saut pour que IC2 soit devenu petit et que IF soit encore suffisamment grand (≈ 1 ms).
Selon la programmation en différence de potentiel, on distinguera 2 méthodes :
- NORMALE : On part à chaque fois du même potentiel et on fait varier l’amplitude ∆ V
du saut de potentiel. Ceci permet de parcourir des ∆ V différents dans la zone où
l’espèce est électroactive.
I IC1 E1 E2 (impulsion)
IF (=1/√t)
IC2
t
∆ V mesure (quand les 2 IC sont petits et IF encore
grand)1 2
IC1 proportionel à E1 - E pcn IC1 proportionel à E2 - E pcn
Fig.30 (a) : On modifie l’amplitude d’impulsion de potentiel, et celui-ci varie
linéairement. La mesure est ensuite prise quelques ms après l’impulsion tout juste
avant la tombée de la goutte. On remarquera que les sommets correspondent aux
potentiels ou l’espèce n’est pas électroactive.
- DIFFERENTIELLE : Cette méthode permet surtout de supprimer les résidus de IC1. En
pratique, on appliquera un ∆ V de même amplitude, mais sur un potentiel qui
augmente linéairement. On réalise deux mesures : une juste avant l’impulsion (mesurede IC1 surtout) et une juste à la fin (IF surtout, un peu IC2 et IC1 ± pareils).
⇒On fait ensuite la différence, ce qui permet de supprimer l’interface d’IC1 (On
remarquera que pour annuler IC1, IC1 avant ≈ IC1 après, dés lors, E1 et E2 doivent aussi
rester proches).
Les polarogrammes impulsionnels différentiels donnent des pics dont la position
permet souvent l’identification de analytes
Grâce à la polarographie impulsionnelle, on est capable de détecter des quantités de l’ordre de
10-7M, ce qui est beaucoup plus performant qu’en polarographie simple.
Remarque: Une fois encore, l’oxygène est un facteur important d’interférence puisque celui-ci
se réduit en solution à l’électrode à goutte d’Hg. La désoxygénation à l’azote du système seradonc nécessaire.
13

5/11/2018 Chimie Analytique - (Chap 3) - slidepdf.com
http://slidepdf.com/reader/full/chimie-analytique-chap-3 14/14
4. Redissolution anodique.
1) Principe.
Cette méthode est utilisée lorsque l’on désire mesurer des traces de cations métalliques
(dans des concentrations de l’ordre du ppb). Elle n’est en fait pas vraiment polarographie mais
dérivée car les premières mesures se font toujours avec une électrode d’Hg, toujours dans un
système à trois électrodes.
De l’autre coté, un capillaire, connecté à une seringue, rempli d’une quantité connue de Hg
(fonction de la hauteur et des caractéristiques du capillaire), permet d’avoir une goutte d’Hg
statique (goutte pendante ou film déposé, qui se forme vite et dont l’aire ne varie plus après).
Les performances de ce système sont améliorées car on introduira une étape supplémentaire,
l’étape de préconcentration. Il s’agira ici d’amener le plus d’espèces métalliques possibles
dans la goutte d’Hg, donc dans un volume plus petit d’un liquide. Seul le métal étant soluble
dans Hg, les ions métalliques subiront une réaction de réduction afin d’être solubilisés.
On donne le nom d’amalgame à ce mélange (métal + Hg) dans lequel la concentration
métallique est de fait plus élevée.
L’analyse en elle-même se fera par réoxydation (donc redissolution) des espèces métalliques
et mesure de ce courant d’oxydation (on arrêtera quelques secondes l’agitation pour que le
courant mesuré soit seulement lié à la diffusion...).
Fig.32: La réponse à la mesure est donnée sous forme de pics dont la position (∆ V) donne la
nature du métal et l’amplitude donne la quantité (concentration), après étalonnage.
2) Evaluation.
Il existe d’autres méthodes de redissolution (cathodique, par exemple...) mais dont
l’étape de préconcentration est différente. Elles sont également moins puissantes et nécessitent beaucoup plus d’attention de l’expérimentateur.
Fig.34: Dans ce graphe, on a pu détecter la présence de Cu et de Pb. La redissolution anodique
permet donc de détecter plusieurs espèces métalliques, en une seule expérience. Cette méthode
est donc très performante quantitativement et qualitativement.
Fig.35: Dans ce graphe, la position des pics est différente car le potentiel redox est différent.
Mais l’amplitude aussi est différente alors que les concentrations sont identiques. Cette
variabilité de l’amplitude est en fait due aux variations des coefficients de diffusion et aux
différences de solubilité des espèces dans le milieu.
14





























