Embed Size (px)
Citation preview

Wageningen Agricultural University
From the SelectedWorks of Pr. Mamoudou H. DICKO, PhD
Winter February 3, 2016
Cours Enzymologie Fondamentale et Appliquée(Niveau Master 1)Mamoudou H. DICKO, Prof.
Available at: https://works.bepress.com/dicko/54/

Notes de Cours Prof M. H. DICKO 1
ENZYMOLOGIE 1 Crédit 1 : Enzymologie Fondamentale et Appliquée
Code BCH4000
Notes de cours Pr. Mamoudou H. DICKO, PhD,
(Professeur Titulaire de Biochimie)
MASTERS DE SCIENCES BIOLOGIQUES Premier Semestre Parcours SVT :S7 (12 heures)
(Tronc commun)
Année académique 2015-2016
Deuxième Edition
UNIVERSITE DE OUAGA I Professeur Joseph Ki-Zerbo
------------------
Unité de Formation et de Recherche en Sciences de la Vie et de la Terre (UFR-SVT)
-----------------
Département de Biochimie-Microbiologie
-----------------
Laboratoire de Biochimie Alimentaire, Enzymologie,
Biotechnologie Industrielle et Bioinformatique (BAEBIB)
-------------------
09 BP. 848 Ouagadougou 09, Burkina Faso ; Fax : 226 50 307242,
Tel . +226 70272643/+22668542929, Email : [email protected]
URL: http://works.bepress.com/dicko
----------------

Notes de Cours Prof M. H. DICKO 2
Objectifs du cours
L'objectif principal de ce cours est de rappeler et donner aux étudiants les
connaissances indispensables en enzymologie appliquée, à savoir :
- connaître la structure des enzymes, les méthodes d’extraction, de purification,
de dosage et de caractérisation (électrophorèse, zymographie, etc.) des
enzymes ;
- comprendre les principes fondamentaux de la cinétique chimique et la
cinétique enzymatique y compris le rôle des inhibiteurs et autres effecteurs;
- comprendre les méthodes d’immobilisation des enzymes et leurs
conséquences positives ou négatives dans la biocatalyse enzymatique;
- comprendre les différends domaines d’utilisation des enzymes en biochimie
analytique, bio-industries, industrie agro-alimentaires, analyses bio-médicales,
en diagnostic, etc.
Les travaux scientifiques et Notes de cours sont gratuits, CEPENDANT, vous pouvez apporter votre contribution financière à leur élaboration. Vous pouvez contribuer financièrement ici en utilisant les références bancaires ci-dessous:
http://works.bepress.com/dicko/50
“Thank you for your contribution to higher education in Africa!”

Notes de Cours Prof M. H. DICKO 3
SOMMAIRE
CHAPITRE 1.
HISTORIQUE DE L’ENZYMOLOGIE (rappel):…………….…………Page 4
CHAPITRE 2.
BASES DE DONNEES INTERNET SUR LES ENZYMES……….. P a g e 1 1
CHAPITRE 3.
D E F I N I T I O N , C L A S S E S , S T R U C T U R E ( r a p p e l ) P a g e 2 4
CHAPITRE 4.
DOSAGE DES PROTEINES/ENZYMES (rappel)………..……..……Page 49
CHAPITRE 5.
EXTRACTION ET PURIFICATION DES ENZYMES………......……Page 68
C H A P I T R E 6 .
C I N E T I Q U E E N Z Y M A T I Q U E … … … … … … … … … . . P a g e 9 3
C H A P I T R E 7 .
I M M O B I L I S A T A I O N D E S E N Z Y M E S … … . … . . … . P a g e 1 3 9
C H A P I T R E 8 .
I N T R O - E N Z Y M O L O G I E I N D U S T R I E L L E . . … … . . P a g e 1 3 7
B I B L I O G R A P H I E … … … … … … … … … … … … … … . P a g e 1 8 4

Notes de Cours Prof M. H. DICKO 4
CHAPITRE 1.
HISTORIQUE DE L’ENZYMOLOGIE

Notes de Cours Prof M. H. DICKO 5
I. Historique de l’enzymologie Fondamentale
Les organismes vivants sont le siège d'un grand nombre de réactions biochimiques très
diverses.
Ces réactions s'effectuent dans des conditions où, normalement, elles ne pourraient se faire.
Si elles ont lieu, c'est parce qu'elles sont catalysées par des macromolécules biologiques : les
enzymes.
Le pouvoir de catalyse des enzymes est lié, entre autre, à la très haute spécificité de
reconnaissance des molécules sur lesquelles elles agissent.
L'enzymologie est la partie de la biochimie qui étudie les propriétés structurales et
fonctionnelles des enzymes (la relation structure - fonction).
En particulier, elle s'applique à décrire la vitesse des réactions catalysées par les enzymes.
Il est difficile de situer exactement la découverte de la notion d'enzyme et surtout d'enzyme
en tant que seul catalyseur des réactions chimiques qui se déroulent dans les organismes
vivants.
1783 : Lazzaro Spallanzani a rapporté que la viande est liquéfiée par un extrait gastrique. Il a
noté également que la température a un grand effet.
1814 : Kirchhoff a observé qu'un composant "glutineux" (comme il l'a appelé à l'époque) de
blé convertit l'amidon en sucre.
1833 : La première découverte d'une enzyme est d'habitude attribuée à Anselme Payen et
Jean-François Persozqui ont traité un extrait aqueux de malt à l'éthanol puis précipiter une
substance labile à la chaleur qui hydrolyse l'amidon.
Ils ont appelé cette fraction"diastase" (séparation en Grec) puisque cette fraction sépare le
sucre soluble de l'amidon insoluble. On sait maintenant que cette préparation était une
solution non purifiée d'amylase.
1834 : Theodor Schwann a obtenu le premier un agent actif d'origine animale (la pepsine)
qu'il a partiellement purifiée en traitant la paroi stomacale par l'acide.

Notes de Cours Prof M. H. DICKO 6
Au début du 20è siècle, de gros travaux furent entrepris pour purifier des enzymes et surtout
décrire leur activité catalytique en termes mathématiques.
Il est important de souligner qu'à l'époque les premières observations d'activité enzymatique
ont précédé une notion claire et précise de la catalyse. Le concept de catalyse provient de
l'observation de l'action de la diastase et de la pepsine parallèlement à celle de la levure
pendant la fermentation : dans tous les cas, une "substance était changée en une autre "sous
l'influence d'un agent actif : le catalyseur.
A l'époque, la levure n'était pas encore considérée comme une cellule vivante.
1838 : Charles Cagniard de Latour montra que le processus de fermentation est dû à des
organismes vivants.
1858 à 1871 : Les travaux de Louis Pasteur confirmèrent cette idée. Pasteur émit l'hypothèse
révolutionnaire que les changements chimiques lors de la fermentation résultaient des
processus de la vie des micro-organismes impliqués dans la fermentation.
A l'opposé, Liebig et Stahl privilégiaient une théorie purement chimique : un "ferment"était
une substance chimique produite par un organisme en décomposition et les atomes de ce
ferment étaient supposés en mouvement incessant. Cet état d'agitation élevé était transmis
aux atomes de la molécule de sucre (substrat du ferment) dont les éléments devaient être
maintenus par des forces faibles. Il en résultait une scission du sucre en CO2 et éthanol dont
les liaisons étaient plus fortes.
1860 : Berthelot fit macérer de la levure et obtint une fraction précipitable à l'alcool capable
de convertir le sucrose en glucose plus fructose. Il conclut que l'invertase (nom qu'il donna à
l'agent actif de cet extrait) était l'un des multiples ferments présents dans la levure.
1876 : Kühne proposa le nom d'enzyme (signifiant "dans la levure") pour qualifier ces
ferments. Le suffixe "ase" fût proposé par Duclaux en 1898.
1897 : Un chimiste allemand, Martin Hahn, tentait d'isoler des protéines de levure
(Saccharomyces cerevisiae) en broyant ces levure dans un mortier avec du sable fin et de
terre de diatomées. Cependant, cette préparation d'extrait de levure se conservait mal.
1897 : La même année, Bertrand observa que certaines enzymes requièrent des facteurs
dialysables pour leur activité : il les nomma coenzymes.

Notes de Cours Prof M. H. DICKO 7
1902 : V. Henri et Adrian Brown suggérèrent indépendamment que la formation d'un
complexe enzyme - substrat est un intermédiaire obligatoire de la réaction enzymatique.
Cette suggestion s'appuyait sur la forme de la courbe obtenue quand on reporte la vitesse
initiale de la réaction en fonction de la concentration en substrat. A. Brown avait étudié la
vitesse d'hydrolyse du sucrose par la β-fructofuranosidase, l'invertase de la levure.
De plus cette suggestion était en accord avec le concept de reconnaissance enzyme - substrat
du type "clé - serrure" proposé par Emil Fisher en 1894.
Henri fût donc le premier à décrire l'équation mathématique reliant l'effet de la concentration
du substrat à la vitesse de catalyse.
1913 : Leonor Michaelis et Maud Menten redécouvrirent l'équation de V. Henri et établirent la
relation connue sous le nom d'équation de Henri - Michaelis - Menten.
Le point important est que l'obtention de cette équation repose sur l'hypothèse qu'il s'établit un
équilibre rapide entre les concentrations de l'enzyme, du substrat et du complexe enzyme -
substrat (E + S <==> ES).
1925 : George Briggs et James Haldane généralisèrent l'équation précédente en introduisant le
concept d'état stationnaire pour la concentration du complexe enzyme - substrat.
Le fait que les enzymes sont des protéines ne fût accepté qu'à partir de la fin des années 20.
De nouvelles techniques chimiques et physiques furent employées pour analyser la structure
des protéines.
1926 : James Summer (Prix Nobel en 1946) cristallisât l'uréase.
Années 30 : John Northrop (Prix Nobel en 1946) et ses collaborateurs cristallisèrent le
pepsinogène, la pepsine et plusieurs isoformes de la trypsine et de la chymotrypsine et
démontrèrent la pureté des cristaux obtenus.
Années 40 et 50 : Des centaines d'enzymes furent purifiées et cristallisées permettant ainsi
l'élucidation de dizaines de voies métaboliques.
1955 : Frédéric Sanger (1er Prix Nobel en 1958) publia la séquence complète en acides aminés
d'une petite protéine : l'insuline (masse molaire 6000 Da).
1957 : La première structure cristallographique d'une protéine (la myoglobine), déduite de la
diffraction des rayons X, fût obtenue par John Kendrew (Prix Nobel en 1962 avec Max
Perutz).

Notes de Cours Prof M. H. DICKO 8
Années 60: La première séquence d'une enzyme (la ribonucléase, masse molaire 13700 Da)
fût publiée en 1960 et la première synthèse chimique (également de la ribonucléase) fût
obtenue en 1969.
Les biochimistes focalisèrent alors sur le mécanisme de l'activité enzymatique et son mode de
régulation.
1958 : Daniel Koshland a proposé le modèle de l'ajustement induit. Le substrat induit un
changement conformationnel du site actif de l'enzyme.
1961 : Christian Anfinsen (Prix Nobel en 1972) et ses collaborateurs ont montré que, pour un
grand nombre de protéines, c'est la séquence en acides aminés qui détermine le repliement de
la chaîne polypeptidique dans son unique conformation native donc active. Cette conformation
est la plus stable parce qu'elle a une énergie libre minimale.
1963 : Cleland proposa une procédure claire et uniforme pour écrire les équations des
cinétiques des systèmes enzymatiques à plusieurs substrats.
1965 : Jacques Monod (Prix Nobel en 1965), Jeffries Wyman et Jean-Pierre Changeux
proposèrent un modèle cinétique (modèle MWC) pour les enzymes allostériques (enzymes
dont la courbe de vitesse en fonction de la concentration en substrat est une sigmoïdale et non
une hyperbole).
1966 : Daniel Koshland, Georges Nemethy et Filmer généralisèrent le modèle précédent
(modèle KNF) en incluant la notion d'ajustement induit proposé par Koshland.
1968 , Robert Holley, Har Gobind Khorana, Marshall Nirenberg , Prix Nobel de Médecine ou
physiologie pour leur interprétation du code génétique et de ses fonctions dans la
synthèse protéique.
1978 Werner Arber, Daniel Nathans, Hamilton O. Smith, Prix Nobel de Médecine ou
physiologie pour « la découverte des enzymes de restriction et leur application aux
problèmes de génétique moléculaire ».
Années 80 à nos jours :
1980, Paul Berg¹, Walter Gilbert² et Frederick Sanger, Prix Nobel de Chimie, Pour ses études
fondamentales de la biochimie des acides nucléiques, et en particulier de l'ADN recombinant,
²Pour leurs contributions à la détermination des séquences de bases dans les acides nucléiques.

Notes de Cours Prof M. H. DICKO 9
1987, Découverte des protéines chaperonnes (Ron Laskey - 1978; John Ellis - 1987) aident au
repliement de certaines protéines ou les maintiennent dans la conformation native quand la
cellule est soumise à certains stress. Ces protéines nativement non-repliées : elles n'ont pas de
structure tridimensionnelle à l'état libre et n'acquière leur structure repliée donc fonctionnelle
que quand elles interagissent avec leurs partenaires cellulaires.
Parmi les chaperonnes, on peut citer :
• les protéines de choc thermique ("Heat Shock Protein - HSP")
• les chaperonines
• les protéines LEA ("Late Abundant Embryogenesis")
1988 , Johann Deisenhofer, Robert Huber et Hartmut Michel, Prix Nobel de Chimie Pour la
détermination de la structure tridimensionnelle d'un site de la réaction photosynthétique
1989 , Sidney Altman et Thomas Robert Cech , Prix Nobel de Chimie Pour la découverte des
propriétés catalytiques de l'acide ribonucléique (Ribozyme)
1992 , Edmond Fischer et Edwin G. Krebs , Prix Nobel de Médecine ou de physiologie
pour « leurs découvertes concernant les phosphorylations réversibles de protéines comme un
mécanisme de régulation biologique ».
1993 , Kary B. Mullis¹, Michael Smith², Prix Nobel de Chimie 1Pour son invention de la
réaction en chaîne impliquant la polymérase et ² Pour ses contributions fondamentales à la
connaissance de la mutagenèse et à l'étude des protéines (enzymes).
1997 Paul D. Boyer¹, John Ernest Walker¹ et Jens Christian Skou², Prix Nobel de Chimie,
¹Pour leur élucidation du mécanisme enzymatique de la synthèse de l'adénosine triphosphate
(ATP), ²Pour la première découverte d'une enzyme transporteuse d'ions, la Na+, K+-ATPase
2001, Leland H. Hartwell et R. Timothy Hunt, Paul M. Nurse, Prix Nobel de Médecine ou de
physiologie pour « leur découverte de la cycline et des enzymes kinases dépendantes de la
cycline, des molécules fondamentales de la régulation du cycle cellulaire ».

Notes de Cours Prof M. H. DICKO 10
2002, Sydney Brenner, H. Robert Horvitz et John E. Sulston, Prix Nobel de Médecine ou de
physiologie pour « leurs découvertes concernant la régulation génétique du développement des
organes et la mort cellulaire programmée ».
2004 Aaron Ciechanover, Avram Hershko et Irwin Rose, Prix Nobel de Chimie, Pour leurs
travaux sur la dégradation enzymatique des protéines contrôlée par l'ubiquitine.
2006 , Roger Kornberg, Prix Nobel de Chimie, Pour ses travaux sur les bases moléculaires de
la transcription chez les eucaryotes.
2009 , Venkatraman Ramakrishnan, Thomas Steitz et Ada Yonath, Prix Nobel de Chimie,
Pour leurs études de la structure et de la fonction du ribosome
2015 Tomas Lindahl, Paul L. Modrich et Aziz Sancar , Prix Nobel de Chimie, Pour leurs
études mécanistiques de la réparation de l'ADN impliquant des DNA polymérases à capacité
d’édition (synthèse et réparation).
Des approches plus récentes et plus vastes sont venues compléter les connaissances acquises
pendant plus d'un siècle :
• le développement de divers modèles du repliement de la chaine polypeptidique
• en parallèle, les moyens informatiques de plus en plus puissants ont permis de
développer des simulations utilisant la modélisation moléculaire, la mécanique moléculaire ou
la dynamique moléculaire (GROMACS, méthode de Monte-Carlo, méthode ab-initio, ...).
• des disciplines naissantes viennent compléter les connaissances acquises en
enzymologie: la génomique structurale, la génomique fonctionnelle, la protéomique, la
métabolomique et la BIOLOGIE SYNTHETIQUE.
• enfin, le développement d'Internet, la création de bases de données de plus en plus
complètes et spécialisées et la bioinformatique constituent autant d'outils modernes pour
développer les moyens d'étude en enzymologie.

Notes de Cours Prof M. H. DICKO 11
CHAPITRE 2.
BASES DE DONNEES INTERNET SUR LES ENZYMES

Notes de Cours Prof M. H. DICKO 12
I. Introduction
Les travaux menés sur les protéines en général et sur les enzymes en particulier sont d’une
importance cruciale pour la compréhension du vivant et leur étude pratique. La somme de
connaissances accumulées dans ce domaine est impressionnante. Ainsi, la série de
publications scientifiques sur Internet comporte aune quantité inestimable de données. Les
différentes bases de données ont été créées par les structures scientifiques afin de stocker ces
milliers d’informations concernant les protéines en générale et les enzymes en particulier. Il
serait passer en revue quelques sites Internet les plus importants dans le domaine de
l’enzymologie fondamentale.
II. BRENDA : http://www.brenda-enzymes.org/
BRENDA (Braunschweig Enzyme Database) est une banque de données d’informations générales
sur les enzymes. Elle est développée et entretenue par l'Institut de biochimie de l'Université de
Cologne (Technische Universität Braunschweig, Department of Biochemistry and
Bioinformatics). Le site donne des informations relatives aux données, sur les fonctions
enzymatiques, les structures, la nomenclature, les réactions et spécificités, les paramètres
fonctionnels, la préparation et l’isolement, les sources d’enzymes. Le site comporte également
des informations sur les différentes applications et utilisation pratique des enzymes.
Les logiciels et algorithmes et autres liens utilisés pas le site sont :
- SwissProt
- TREMBL
- UniProt Accessions
Contenu et caractéristique
La base de données contient plus de 40 champs, avec des informations spécifiques aux
enzymes sur plus de 4800 numéros EC (Enzyme Commission) qui sont classés en fonction
de l’IUBMB (International Union of Biochemistry and Molecular Biology). Les différents
champs de données couvrent les informations sur la nomenclature de l'enzyme, la réaction et
la spécificité, la structure de l'enzyme , l'isolement et la préparation, la stabilité de l'enzyme,
les paramètres cinétiques tels que la valeur de Km pour certains substrats, les enzymes
modifiées, l'application d'enzymes et les données relatives aux ligands.
Actuellement, BRENDA contient des données annotées manuellement de plus de 130 000
enzymes provenant de différentes publications scientifiques. Chaque entrée de l'enzyme est

Notes de Cours Prof M. H. DICKO 13
clairement liée à au moins une référence de la littérature, à sa source d’origine , et, le cas
échéant, à la séquence protéique de l'enzyme.
Une partie importante de BRENDA représente plus de 107 000 ligands d'enzymes, qui sont
disponibles et on les obtient à partir de leurs noms, synonymes ou via la structure
chimique. Le terme «ligand» est utilisé dans ce contexte à tous les composés qui
interagissent avec des enzymes.
L'origine de ces molécules varie d’antibiotiques naturels aux composés synthétiques qui ont
été synthétisés pour l'élaboration de médicaments ou de pesticides. En outre, des renvois à des
sources d'informations externes, telles que la séquence et la structure tridimensionnel (3D) des
bases de données, ainsi que des ontologies biomédicales, sont fournis.
Depuis 2006, les données de BRENDA sont complétées par des informations extraites de la
littérature scientifique. A cet effet, d’autres sources telles que text-mining FRENDA (Full
Reference Enzyme Data), AMENDA (Automatic Mining of Enzyme Data), DRENDA
(Disease-Related Enzyme information Database) et KENDA (Kinetic Enzyme Data) ont été
introduites. Ces résultats d'exploration de texte ont été tirés des titres et des résumés de tous
les articles de la base de données de PubMed .
Figure 1 : Page d’accueil de BRENDA
1.2. DISPONIBILITE DU SITE
L'utilisation de BRENDA est gratuite. En outre, FRENDA et AMENDA sont gratuits pour les
utilisations à but non-lucratif. Les utilisateurs commerciaux ont besoin d'une licence pour ces
bases de données par le biais de BIOBASE.
Les autres sites d’information générale sur les enzymes :

Notes de Cours Prof M. H. DICKO 14
http://home.nas.net/~dbc/cic_hamilton/enzyme.html
http://www.chem.qmul.ac.uk/iubmb/enzyme/
http://mbs.cbrc.jp/EzCatDB/
III. EXPASY. http://enzyme.expasy.org/
Expert ProteinAnalysis System (ExPASy) ou Bioinformatic Ressource Portal est une base de
données de l'Institut Suisse de Bioinformatique (en anglais Swiss Institute of Bioinformatics,
SIB) permettant l'analyse de la séquence et de la structure des protéines ainsi que leur
électrophorèse bidimensionnelle. Cette base de données fonctionne en collaboration avec
l'Institut Européen de Bio-informatique (EBI, http://www.ebi.ac.uk), Membre
du laboratoire Européen de Biologie Moléculaire (EMBL). ExPASy produit également la base
d’informations des logiciels UniProtKB/Swiss-Prot sur les séquences des protéines ainsi que
les suppléments informatiques annotés par les logiciels UniProtKB/TrEMBL. A partir du site
on peut avoir accès à plusieurs autres bases de données telles que la Nomenclature des
Enzymes, et des logiciels d’analyses de protéines (Protein digest) et des acides nucléiques.

Notes de Cours Prof M. H. DICKO 15
Figure 2 : Page d’accueille de ExPASy
Le serveur ExPASy est mis en service depuis le 1er
août 1993. Le site contient des
informations sur
Protéomiques
Génomique
Bioinformatique Structurale
Systèmes Biologiques
Phylogénie/Evolotion
Génétique des populations
Transcriptomique

Notes de Cours Prof M. H. DICKO 16
Biophysique
Imagérie
Infrastructure de l’Information et de la Technologie
La conception des médicaments
Glyco-biologie
CONTENTU ET CARACTERISQUES DE LA BASE DE DONNEES
Le SIB développe et maintien des bases de données de réputation internationale,
incluant UniProtKB / Swiss-Prot (base de données de séquences de protéines qui ont été
organisées et classées manuellement, offrant ainsi un haut niveau d'annotation), neXtProt (base
de données des protéines humaines), le répertoire SWISS-MODEL (modèles de structures
protéiques tridimensionnelles), STRING (réseaux d'interactions protéiques), SwissRegulon
(réseaux de régulation de transcription à l'échelle du génome), EPD (une base de données des
promoteurs eucaryotes), SWISS-2DPAGE (base de données de gels 2D), le répertoire
WORLD-2DPAGE (répertoire de gels 2D), mirz (atlas d'expression des petits ARNs), CLIPZ
(bases de données des sites de liaison des protéines liant l'ARN), PROSITE (familles de
protéines et domaines), MyHits (séquences de protéines et motifs), CleanEx (données
d'expression génique), Bgee (base de données pour extraire et comparer les profils
d'expression des gènes entre espèces animales), OpenFlu (base de données pour le virus de la
grippe humaine et animale), ViralZone (portail vers les protéines virales UniProtKB),
GlycoSuiteDB (base de données de glycanes), SugarBindDB (liste des séquences de glucides
connus auxquels les organismes pathogènes adhèrent spécifiquement), OrthoDB (catalogue
hiérarchique des eucaryotes orthologues), miROrtho (catalogue des gènes des micro-ARNs
animaux), ImmunoDB (base de données de gènes et familles de gènes liés au système
immunitaire des insectes), DNA and RNA transcription inverse, etc.
LOGICIELS DEVELOPPES PAR SIB
Le SIB développe et fournit des logiciels pour la communauté des chercheurs en sciences de la
vie, tels que SWISS-MODEL (modélisation par homologie des structures de protéines), Swiss
Dock/EADock (service web pour l'ancrage d'une petite molécule à une protéine cible), ISA
(outil de bi-clustering intégratif), PPA (modularisation couplée de plusieurs ensembles de
données) , Mélanie (plateforme d'analyse de gels 2D), MSight (logiciel d'imagerie et d'analyse

Notes de Cours Prof M. H. DICKO 17
pour la spectrométrie de masse), OMA (base de données des orthologues extraits de génomes
complets disponibles), DeepView/Swiss-PdbViewer (visualisation de protéines, modélisation
et analyse), et Newick (traitement d'arbre phylogénétique à haut débit), etc...
IV. ExplorEnz The Enzyme Database, http://www.enzyme-
database.org/
ExplorEnz a été développé par School of Biochemistry & Immunology, Trinity College,
Dublin 2, (Ireland) en 2005 pour simplifier l'accès aux bases de données d'enzyme de UIBBM.
Les données, qui sont stockées dans une base de données MySQL, de préserver la mise en
forme des noms chimiques selon les normes IUPAC. C’est une interface de requête internet
simple, facile à utiliser, et est disponible, avec un moteur de recherche avancé pour les
requêtes plus complexes.
Figure 3 : Page accueille EplorEnz
http://www.ebi.ac.uk/thornton-srv/databases/enzymes/
1.1.Contenu et caractéristique
L'Institut européen de bioinformatique (en anglais European Bioinformatics Institute, EBI)
est une organisation qui constitue une sous-division du Laboratoire européen de biologie
moléculaire (European Molecular Biology Laboratory, EMBL). Il propose un certain nombre
de bases de données d'acides nucléiques, de séquences protéiques et de structures
macromoléculaires.

Notes de Cours Prof M. H. DICKO 18
Le site a une ébauche sur plusieurs bases de données déjà existantes sur les protéines et les
enzymes. EBI donne des informations sur la structure des enzymes.
L’algorithme utilisé est le BLAST, les logiciels de séquençage, d’alignement et de phylogénie
moléculaire.
Cette base de données contient les structures enzymatiques connus qui ont été déposées dans
la Protein Data Bank (PDB).
En la date du 11 Janvier 2014Il y avait 45,960 PDB-enzymes entrées dans la base de données
du PDB impliquant 38 549 fichiers PDB distincts.
Figure 4 : Page d’accueille EBI
V. La base de données CAZY, http://www.cazy.org/
La base de données Carbohydrate-Active enZYmes Database (CAZY) décrit les familles de
motifs catalytiques structurelle et les domaines fonctionnels des enzymes qui
dégradent, modifient ou créent des liaisons glycosidiques. Mis en ligne par le
laboratoire de l’Architecture et Fonction des Macromolécules Biologiques (AFMB) de
l’Université de la Méditerranée (Luminy), Prof Bernard Henrissat à Marseille, depuis
1998, CAZy est spécialement dédiée à la présentation et l'analyse de l'information
génomique, structurelle et biochimique sur les carbohydrases.

Notes de Cours Prof M. H. DICKO 19
Figure 5 : Page d’accueil EzCatDB
VI. REBASE, http://rebase.neb.com/rebase/rebase.html
Une enzyme de restriction est une protéine enzymatique qui peut couper un fragment
d'ADN au niveau d'une séquence généralement un palindrome de nucléotides, caractéristique
appelée site de restriction. Chaque enzyme de restriction reconnaît ainsi un site spécifique.
Plusieurs centaines d'enzymes de restriction sont actuellement connues, on en retrouve
naturellement dans un grand nombre d'espèces de bactéries. Ces enzymes, naturellement
présentes chez les bactéries, sont devenues des outils important en génie génétique.
Figure 6 : Page d’accueille REBASE

Notes de Cours Prof M. H. DICKO 20
VII. Le National Center for Biotechnology Information (NCBI),
http://www.ncbi.nlm.nih.gov/pubmed/14681451
Le NCBI, est un centre américain pour les informations biotechnologiques. C’est un institut
national américain pour l'information biologique moléculaire. Cet organisme, fondé en 1988 et
situé à Bethesda dans le Maryland, fait partie de la Bibliothèque américaine de médecine, un
des Instituts américains de la santé.
Le NCBI propose et contient de nombreuses bases de données qui ne sont souvent
qu'indicatives.
Figure 5 : Page d’accueille NCBI
VIII. Biomedical Enzyme Database, http://www.science.co.il/Biomedical/Enzyme-
Databases.asp
Ce site israélien a des liens vers la quasi-totalité des bases de données en enzymologie et
certaines bases de données génomiques. Cette base de données nationale est aussi un
répertoire et lien de site de la science et de la technologie en Israël mais aussi dans le monde.
Le site comprend également les documents scientifiques juifs de la diaspora. Il a été créé en
1996 et a pour but de fournir un accès facile, rapide et épuré à l'information en sciences
chimiques et sciences de la vie et un lien vers les entreprises œuvrant dans ce domaine.
Les Bases de données dédiées à la médecine et à biologie moléculaire
http://www.meddb.info/index.php.en?cat=2&subcat=85
o http://www.genome.jp/dbget-bin/www_bfind?enzyme
o http://mbcf.dfci.harvard.edu/cmsmbr/biotools/biotools3.html
o http://www.drugbank.ca/databases
o http://www.iuphar-db.org/DATABASE/ReceptorFamiliesForward?type=ENZYME

Notes de Cours Prof M. H. DICKO 21
o http://www.dd-database.org/other-diseases/enzymes.html
o http://www.enzyme-database.org/
IX. La base de données MEROPS, http://merops.sanger.ac.uk/
C’est une ressource d'information pour les peptidases (protéases ou protéinases et les enzymes
protéolytiques) et les protéines qui les inhibent. La page peut résumer et décrire une peptidase
donné et peut être atteint par l'utilisation d'un index sous son nom, MEROPS identifie ou
organisme les sources. Le résumé décrit la classification et la nomenclature de la peptidase et
propose des liens vers des pages supplémentaires montrant identificateurs de séquence, la
structure si elle est connue, la littérature et plus.
Liste d’autres Bases de données
- Peroxidase database : http://peroxibase.toulouse.inra.fr/index.php
ASD Allosteric Database
PROCOGNATE Database of cognate ligands for the domains of enzyme structures
MACiE Database of enzyme reaction mechanisms
ORENZA database of ORphan ENZyme Activities
ExplorEnz Enzyme Database
BRENDA Enzyme Information System
Enzyme Nomenclature
Enzyme Nomenclature database
EC2PDB Enzyme Structures Database
Kinomer Eukaryotic Protein Kinase Groups

Notes de Cours Prof M. H. DICKO 22
FunTree evolution of enzyme function within domain super families
HCS Hierarchical Classification of Hydrolases Catalytic Sites
IntEnz Integrated relational Enzyme database
Nomenclature of Multiple Forms of Enzymes (isozymes)
MEROPS Peptidase database
PeroxiBase Plant Peroxidase Database
PSDB Plant Subtilase Database of plant subtilisin-like serine proteases
Proteases of E. coli
PTP Protein Tyrosine Phosphatases
REBASE Restriction Enzyme Database
Restriction Enzymes Glossary
Symbolism and Terminology in Enzyme Kinetics
eQuilibrator Thermodynamics calculator for biochemical reactions
Thermodynamics of Enzyme-Catalyzed Reactions
http://www.biochem.ucl.ac.uk/bsm/dbbrowser/protocol/ecenzfrm.html
http://metacyc.org/
http://xpdb.nist.gov/enzyme_thermodynamics/
http://csbl.bmb.uga.edu/dbCAN/
http://biochemweb.org/enzymes.shtml
http://www.kegg.jp/kegg/document/help_bget_enzyme.html
http://sourceforge.net/projects/intenz/
http://priam.prabi.fr/
http://rle.cbi.pku.edu.cn/home.cgi
http://www.orphanenzymes.org/database/
http://www.geneinfinity.org/sp/sp_resenzym.html
http://www.orenza.u-psud.fr/
http://www.ebi.ac.uk/intenz/
http://www.worthington-biochem.com/index/manual.html
http://www.mrc-lmb.cam.ac.uk/genomes/madanm/pres/rebase1.htm
http://sfld.rbvi.ucsf.edu/django/
http://www.dude-db.org/
http://bioinformatics.ca/links_directory/tool/9734/intenz-integrated-relational-enzyme-database
http://www.enzymedirectory.com/dbandsoft.html
http://databib.org/repository/250
http://2013.igem.org/Team:Wageningen_UR/Backbone_enzyme_database
http://www.amano-enzyme.co.jp/eng/enzyme/
http://www.matrixscience.com/help/enzyme_help.html
http://www.enzymedirectory.com/dbandsoft.html
http://sites.univ-provence.fr/~wabim/biologie/cata.html
http://www.wisdom.weizmann.ac.il/~biospi/metDB_links.htm
http://www.bioinfo.de/isb/gcb01/poster/fleischmann.html
http://www.biodbs.info/De.html
http://flandersbio.be/life-sciences-database/category/enzyme/

Notes de Cours Prof M. H. DICKO 23
http://neurolex.org/wiki/Category:Resource:IntEnz-_Integrated_relational_Enzyme_database
http://www.hsls.pitt.edu/obrc/index.php?page=URL1097006648
http://supfam.org/SUPERFAMILY/EC.html
http://biocyc.org/
http://doqcs.ncbs.res.in/template.php?&y=pathwaydetails&pn=85&page=enzyme
http://eprints.bbk.ac.uk/1106/
http://www.enzyme.chem.msu.ru/hcs/
http://cagt.bu.edu/page/PRECISE_faq
http://www.arabidopsis.org/biocyc/
http://www.lightsources.org/publication-databases
http://cssb2.biology.gatech.edu/EFICAz/
http://www.bioon.com/experiment/data/58766.shtml
http://88proof.com/synthetic_biology/blog/archives/142
http://www.bioinformatics.fr/resources.php?tag=enzymes&P=15
http://home.nas.net/~dbc/cic_hamilton/enzyme.html
http://secure.megazyme.com/carbohydrate-active-enzymes
http://www.teed.uni-stuttgart.de/cgi-bin/new_THDP/index.pl
http://www.rcsb.org/pdb/home/home.do
http://ecocyc.org/enzymes.shtml
http://www.colorado.edu/chemistry/bioinfo/RestrictionEnzymeMapping.htm
http://www.cazypedia.org/index.php/Glycoside_hydrolases
http://www.bio.net/mm/bio-software/1991-December/001110.html
http://www.naturalstandard.com/news/news201012058.asp
http://telomerase.asu.edu/
https://www.cas.org/news/product-news/updated-enzyme-nomenclature
http://jcggdb.jp/GlycoPOD/protocolShow.action?nodeId=t124
http://pec.biodbs.info/PectinaseClassification.html
http://www.physics.iisc.ernet.in/~dichome/seqdb.htm
http://uucd.biocuckoo.org/
http://pathway.yeastgenome.org/
http://www.academia.edu/1602374/MACiE_a_database_of_enzyme_reaction_mechanisms
http://www.genenames.org/genefamilies/UBE2
http://www.bcmsu.ac.in/BDBASE.HTM
http://www.labtimes.org/labtimes/product/j2011/2011_07.lasso
http://biopython.org/DIST/docs/cookbook/Restriction.html
http://www.enzyme.cbirc.iastate.edu/
L’avènement de la technologie a eu un impact favorable sur les sciences biologiques. Avec
l’outil informatique les activités, la recherches et le traitement des données qui prenaient un
long temps et qui étaient pénibles ont été simplifié. Avec un terminal on peut accéder aux

Notes de Cours Prof M. H. DICKO 24
informations, les traiter, les comparer, faire des annotations et même faire une prédiction grâce
aux différentes bases de données
CHAPITRE 3 .
Déf in i t ions , Class i f icat ion e t St ructure

Notes de Cours Prof M. H. DICKO 25
I. DEFINITIONS
A. Les Enzymes
Les enzymes sont des catalyseurs biologiques ou biocatalyseurs de nature protéique. Ce sont
des acteurs omniprésents dans la vie de la cellule. Sans ces protéines, aucune réaction du
métabolisme (anabolisme et catabolisme) des cellules ne serait possible. Toutes les réactions
chimiques se déroulent dans la cellule ou le milieu cellulaire, en présence d’une enzyme.
L’enzyme présente des propriétés de catalyse spécifiques d’une réaction chimique du
métabolisme de l’être vivant qui la produit.
Comme tous les catalyseurs, les enzymes agissent à des concentrations très petites. Elles
augmentent la vitesse des réactions chimiques, sans en modifier le résultat ou l’équilibre
thermodynamique. A la fin de la réaction, la structure de l’enzyme se retrouve inchangée.
Une enzyme donnée est spécifique d’une réaction, c’est-à-dire qu’elle catalyse toujours
la même transformation, se produisant sur les mêmes corps chimiques initiaux.
Les protéines enzymatiques sont synthétisées par des êtres vivants. Cette synthèse est
déterminée génétiquement: sa conservation dans le génome est favorisée par le besoin
· L’enzymologie est l’étude des enzymes.
· Le substantif « enzyme » est du genre féminin.
· Toutes les enzymes sont des protéines.
· Les protéines enzymatiques sont des catalyseurs, c’est-à-dire qu’en agissant à des
concentrations très petites, elles augmentent la vitesse des réactions chimiques, sans en
modifier l’équilibre thermodynamique. A la fin de la réaction la structure de l’enzyme se
retrouve inchangée.
Une enzyme donnée est spécifique d’une réaction, c’est-à-dire qu’elle catalyse toujours la
même transformation, se produisant sur les mêmes corps chimiques initiaux.
Les protéines enzymatiques sont synthétisées par des êtres vivants. Cette synthèse est
déterminée génétiquement : sa conservation dans le génome est favorisée par le besoin
qu’éprouve cet être vivant de faire cette réaction.
LES PRO-ENZYMES ou zymogènes sont enzymes synthétisés sous forme inactive ; l’activité
ne s’exerce que lorsqu’elle perd une partie de sa structure. Prenons l’exemple de quelques
enzymes digestives :
Pepsinogène (proenzme)+ HCL------- Pepsine (enzyme)
Trypsinogène (proenzyme) en présence de la pepsine-------------Trypsine (enzyme)

Notes de Cours Prof M. H. DICKO 26
Il existe dans la nature des biocatalystes non protéiques tels que les Ribozymes (catalyseurs
constitués de RNA) et des DNAzymes (catalyseurs formés de DNA). On les appelle aussi
acides nucléiques catalyseurs.
A. Substrat
Molécule qui entre dans une réaction pour y être transformée grâce à l’action
catalytique d’une enzyme. Toutes les molécules qui entrent dans une réaction enzymatique et
sont définitivement modifiées sont appelées substrats.
B. Produit
Molécule qui apparaît au cours d’une réaction catalysée par une enzyme.
· La nouvelle molécule qui résulte de cette transformation est appelée produit.
C. Ligand
Corps chimique ayant une liaison spécifique avec une enzyme. Toutes les molécules ayant
une liaison spécifique avec une protéine sont appelées ligands. Pour chaque ligand, il existe au
moins un site de fixation sur la protéine qui le reçoit.
D. Cofacteur
Corps chimique intervenant obligatoirement dans une réaction enzymatique :
- pour transporter ou compléter un substrat ;
- pour accepter un produit ;
- comme participant à la structure de l’enzyme.
· Les cofacteurs peuvent être des ions comme l’atome de Zinc de l’anhydrase carbonique ou
de petites molécules minérales habituellement présentes dans les milieux biologiques, à
commencer bien sûr par la molécule d’eau.
· Certains cofacteurs sont des molécules plus complexes synthétisées par les cellules: nous les
appellerons coenzymes.
E. Coenzymes

Notes de Cours Prof M. H. DICKO 27
C’est une molécule biologique intervenant comme cofacteur indispensable dans la catalyse
enzymatique d’une réaction. Les coenzymes libres interviennent stœchiométriquement dans la
réaction de manière catalytique. Les coenzymes sont des molécules biologiques c’est à dire
que leur synthèse naturelle ne peut être faite que par des cellules vivantes. Lorsque cette
synthèse n’est pas inscrite dans le patrimoine génétique d’une espèce, alors tout ou partie de la
molécule du coenzyme doit être apportée à cette espèce par son alimentation : cet aliment
indispensable s’appelle une vitamine. Les coenzymes sont des cofacteurs donc des molécules
indispensables à la catalyse enzymatique.
· Lorsque les coenzymes sont liés à l’enzyme par des liaisons de type électrostatique ou plus
faiblement encore, cette liaison est renouvelée à chaque réaction effectuée : en effet, l’énergie
mise en jeu par la liaison enzyme - coenzyme est du même ordre de grandeur que l’énergie
mise en jeu dans la liaison enzyme substrat; dans ce cas, la concentration des coenzymes doit
être du même ordre de grandeur que celle du substrat (on dit stœchiométrique). Ces
coenzymes sont appelés coenzymes libres parce qu’ils se dissocient de l’enzyme à chaque
réaction catalysée.
· Lorsque au contraire les coenzymes sont liés aux enzymes par des liaisons fortes de type
covalent, leur concentration est nécessairement la même que celle de l’enzyme, c’est à dire
très petite (on dit catalytique). Ces coenzymes sont appelé coenzymes liés parce qu’ils ne se
dissocient pas de l’enzyme.
F. Vitesse de réaction
Pour mesurer l’activité d’une réaction enzymatique n’ayant qu’un substrat et un produit, dans
un milieu défini, on observe les concentrations du substrat ou du produit, qui sont des
fonctions du temps écoulé : la concentration du substrat décroit au cours du temps, celle du
produit croit au cours du temps.
• Lorsqu’on fait ces mesures à des temps t1 puis t2 séparés par un délai dt, on appelle S1 et P1
les concentrations du substrat et du produit au temps t1, et S2 et P2 les concentrations du
substrat et du produit au temps t2. La différence entre les concentrations du substrat dS est
l’opposé de la différence entre les concentrations du produit dP. On appelle vitesse de la
réaction le rapport moins dS sur dt, qui est égal au rapport dP sur dt. La vitesse de réaction
représente le nombre de moles de substrat transformées en moles de produit, dans un volume
donné et dans un temps donné.

Notes de Cours Prof M. H. DICKO 28
G. Spécificité
L’enzyme est spécifique pour le substrat et pour le type de réaction. Théoriquement, une
seule réaction possible, sur un seul substrat. Mais en réalité, il existe une relation structure
activité : il faut que la bonne liaison soit placée au bon endroit :
• accès possible en fonction de la conformation
• réaction possible chimiquement (grandeur et nature des forces en jeu). Il est fréquent qu’une
enzyme intervienne non sur une molécule unique, mais sur une classe de substrats.
Exemple 1 : Galactose épimère en C4 du glucose, non pris en charge par les enzymes de la
glycolyse mais les amylases hydrolyses différents maltosaccharides.
• Exemple 2 : ß - oxydation des acides gras - Les enzymes prennent en charge des acyl - CoA.
A chaque dégradation, perte d’un motif à 2 carbones, mais les mêmes enzymes fonctionnent
sur le nouvel acyl - CoA Þ le même motif est reconnu.
12
12
12
12
tt
PP
tt
SS
dt
dP
dt
dSV

Notes de Cours Prof M. H. DICKO 29
En fonction de l’environnement atomique du site catalytique, un seul type de réaction sera
possible :
• acide - base
• redox, par échange d’électrons avec des ions métalliques (cytochromes)
• transferts de groupements, etc.
Les enzymes sont énantiospécifiques (distinction entre les configurations R ou S, ou les
anomères alpha ou béta) et régio-sélectives (distinction entre les positions ortho, para ou méta
au niveau des dérivés du benzène).
- L’Enantiosélectivité
Les acides aminés les animaux et végétaux sont de la série L (lafonction aminée sur le carbone
a asymétrique est orientée à gauche)
• Les protéases de ces organismes hydrolysent spécifiquement les
protéines contenant les acides aminés de la série L
• Si le polypeptide contient des acides aminés de la série D (parois des
bactéries, peptides antibiotiques) : pas de coupure
-La stéréospécificité
Beaucoup des réactions catalysées enzymatiquement démontrent de la stéréospécificité, par
ex., les déshydrogénases NAD+ et NADP+ transfèrent un hydrogène du substrat d'un seul côté
de la fonction nicotinamide. La stéréospécificité est dictée par leur liaison au site actif qui
contrôle l’accessibilité à l’hydrogène transféré.

Notes de Cours Prof M. H. DICKO 30
H. L’Allostérie
L’allostérie est une propriété qu’ont certaines protéines actives dont les enzymes allostériques
qui peuvent changer de structure spatiale lorsqu’elles se lient à un effecteur en un site différent
du site actif. Cette liaison se traduisant par une modification de l’activité. L’allostérie
concerne des protéines douées d'activité : enzymes, transporteurs, canaux, pompes, récepteurs,
protéines contractiles, etc...
• Les effecteurs allostériques sont des ligands dont le site de fixation est différent du site de
fixation du substrat (site actif, site catalytique). Cela peut être une autre molécule de substrat
ou de produit, différente de celle qui participe à la réaction enzymatique ou au transport : on
parle alors d'effet allostérique homotrope. Si au contraire l'effecteur est une molécule
différente, on parle alors d'effet allostérique hétérotrope.
Lorsqu'une protéine allostérique comporte plusieurs sous-unités, la fixation du substrat ou de
l'effecteur sur une sous-unité se traduit par un effet sur les autres sous-unités de la protéine. La
modification de l'activité est toujours quantitative : augmentation, on parle alors d'activation
allostérique ou ralentissement, ou inhibition allostérique. La cinétique allostérique est plus
lente que la cinétique michaélienne pour les petites concentrations du substrat et devient plus
rapide au-delà. Aux environs du point d’inflexion de cette sigmoïde la pente de la courbe est
plus accusée, ce qui signifie que pour une même différence entre deux concentrations du
substrat, l’accélération de la réaction sera plus grande dans le cas de l’enzyme allostérique.
Cette propriété de coopérativité des protomères donne un avantage aux systèmes allostériques
par rapport aux enzymes à cinétique michaélienne pour la régulation de la vitesse des
réactions enzymatiques.
I. Unités Enzymatique
�Il est difficile de mesurer la quantité d’enzyme en unités de masse ou de concentration molaire;
l’activité enzymatique est définie en terme de vitesse de réaction.

Notes de Cours Prof M. H. DICKO 31
�L’Unité International (I.U.) est la quantité d’enzyme requise pour convertir 1μmol de substrat en
produit en 1 min à une température et pH spécifiés (ex. pH 7.0 et 25°C) et sousdes conditions de
saturation du substrat:
I.U. = 1-μmol substrat consommé/min
� le katal (kat) est l’unité d’activité catalytique. C’est la quantité d’enzyme qui transforme 1 mole de
substrat par seconde. C’est l’unité la plus cohérente avec le système International mais pas trop utilisée.
Il existe une relation entre l’IU et le katal :
1 IU= 1 µmol/min = 16,69 nmol/s = 16,67 nkat
� L’activité spécifique d’une enzyme est l’activité catalytique par unité de masse de protéine (I.U./mg
d’enzyme solide mesurée expérimentalement)
� Le taux de roulement (« turnover number » en anglais) exprime
l’activité catalytique en terme d’unités par mol d’enzyme pure (au lieu de mg de protéine).

Notes de Cours Prof M. H. DICKO 32
II. Nomenclature et Classification des enzymes
Depuis 1961, l'Union Internationale de Biochimie et biologie moléculaire a codifié la
nomenclature et la classification des enzymes sous une nomenclature dite officielle
(http://www.chem.qmul.ac.uk/iubmb/enzyme/index.html). Un certain nombre d’appellations
traditionnelles subsistent, mais l’emploi des numéros de code est aujourd’hui obligatoire dans
toutes les publications scientifiques et les documentations
techniques et commerciales.
Toutes les enzymes actuellement connues sont répertoriées sous un numéro portant 4 nombres séparés
par des points et précédés de EC soit (EC x1.x2.x3.x4). La signification des nombres est la suivante
X1- indique la réaction catalysée (1-6)
X2- indique le substrat général (ou groupe de substrats) impliqué
X3- indique le substrat spécifique ou la coenzyme
X4- le numéro de série de l’enzyme
X1 : Le premier nombre pouvant varier de 1 à 6 indique les six grandes classes d’enzymes
correspondant aux types de réactions :
1 : Classe des Oxydoréductases (transfert d'électrons, d’atomes d’hydrogène ou fixation d’oxygène)
2 : Classe des Transférases (transfert d'atomes ou de groupes d'atomes autres que ceux 1)
3 : Classe des Hydrolases (Coupure des liaisons avec fixation de radicaux H et OH issus de l’eau)
4 : Classe des Lyases (Coupure des liaisons par d'autres modes autres que l'hydrolyse).
5 : Classe des Isomérases (réaction conservant la formule brute du composé)
6 : Classe des Ligases (formation des liaisons entre C et un autre métalloïde en utilisant l'énergie de
l'ATP)
X2 : Le deuxième désigne la sous-classe de l'enzyme qui est définie suivant son mécanisme
d'action. Dans le cas des oxydoréductases on distingue les déshydrogénases, les monooxygénases et les
dioxygénases.

Notes de Cours Prof M. H. DICKO 33
X3 : Le 3e nombre désigne la nature de la molécule qui sert d'accepteur, lorsqu'il s'agit d'un transfert
d'électrons. X4 : Le 4e nombre est un numéro d'ordre dans le groupe et dans le sous-groupe.
Lorsqu'une enzyme se termine par 99, cela signifie qu'elle est incomplètement caractérisée.
Cette classification officielle précise et complète la nomenclature fonctionnelle. Dans un
rapport ou une publication le nom fonctionnel continue à être utilisé mais ce dernier est toujours suivi
entre parenthèses par son numéro dans la nomenclature officielle.
En métabolisme, lorsqu'on rencontre, dans un texte, un nom d'enzyme qu'on ne connaît pas il
faut essayer d'imaginer son rôle en procédant de la façon suivante :
déterminer le substrat et sa structure.
déterminer le type de réaction et essayer d'adapter le type de réaction à la structure trouvée, en
se demandant quelle est la partie de la molécule qui réagit.
Enfin reconstituer la réaction et les produits formés.
La plupart des réactions que nous allons rencontrer au cours du métabolisme sont simples. Les
noms des enzymes dans la nomenclature fonctionnelle vont nous permettre d'écrire facilement les
réactions catalysées.
III. LES DIFFERENTS TYPES D'ENZYMES
. 1. LES ENZYMES D'OXYDOREDUCTION ET DE FIXATION D'OXYGENE : Classe I.
EC.1.X2.X3.X4
Les réactions, qui échangent des électrons ou des hydrogènes, sont les plus fréquentes en
biochimie, celles de fixation d’oxygène sont rares. Les enzymes qui catalysent ces réactions sont
appelées des déshydrogénases. Lorsque les enzymes fixent préférentiellement des électrons ou des
hydrogènes sur des substrats elles sont appelées des déshydrogénases ou des réductases. Voyons les
principales enzymes d'Oxydoréduction.
3.1.1 - Déshydrogénases des fonctions alcool, carbonyles ou carboxyliques
Dans les réactions de dégradation le coenzyme ou cofacteur des déshydrogénases est le
NAD+ chargé de récupérer les électrons. Dans les réactions de biosynthèses les réactions de réductions
sont les plus fréquentes. Les électrons sont alors apportés par le coenzyme NADPH,H+ . La séquence
est la suivante :
R-CH2-OH R-CHO R-COOH

Notes de Cours Prof M. H. DICKO 34
Quelques exemples
- alcool déshydrogénase (EC 1.1.1.1)
CH3-CH2OH + NAD+ CH3-CHO + NADH,H+
- Malate déshydrogénase (EC .1.1.37)
HOOC-CH2-CHOH-COOH + NAD+ HOOC-CH2-CO-COOH + NADH,H+
- Aldéhyde déshydrogénase (EC 1.2.1.3.)
R-CHO + NAD+ + H2O R-COOH + NADH,H+
- Glycéraldéhyde 3- déshydrogénase (EC : 1.2.1.12)
-O-CH2-CHOH-CHO + NAD+ + Pi -O-CH2-CHOH-COO +
NADH,H+
- Pyruvate déshydrogénase (EC 1.2.4.1)
CH3-CO-COOH + HSCoA + NAD+ CH3-COSCoA + CO2 + NADH,H+
2. LES TRANSFERASES : Classe II. EC.2.X2.X3.X4
Ces enzymes transfèrent des radicaux ou des groupes d'atomes d'une molécule (substrat
donneur) à une molécule (substrat accepteur). Leur nom complet comporte le donneur, l'accepteur, le
radical transféré suivi de transférase. Les groupes transportés sont :
- le méthyle (-CH3), l'hydroxyméthyle (-CH2OH), Carboxyle (-COOH), groupement carboné
comportant des fonction aldéhydes ou cétones (-CHO) ou (-CO-R), groupe acyle, groupe osidyle,
amine, phosphoryle, etc...
-
3.2.1 - Enzymes transférant un groupe méthyle
Ce sont les méthyltransférases ou méthylases. Le donneur est souvent la S-
adénosylméthionine. On peut citer comme exemple :
- Protéines -N-méthyl transférase (EC 2.1.1.23) : elles méthylent les résidus basiques comme
arginine et la lysine dans des protéines spécifiques.
- ARNt-méthyl transférases (EC 2.1.1.29) : elles méthylent les résidus cytosine du ARNt.
- ADN-méthyltransférases (EC 2.1.1.37). La méthylation porte sur la cytosine du DNA.
3 - LES HYDROLASES. Classe III. EC :3.X2.X3.X4
Ce sont des enzymes de dégradation. Elles provoquent la coupure d'une molécule et fixent les
radicaux H et OH de l’eau sur les valences libérées. Ce sont des enzymes sans coenzymes. Elles
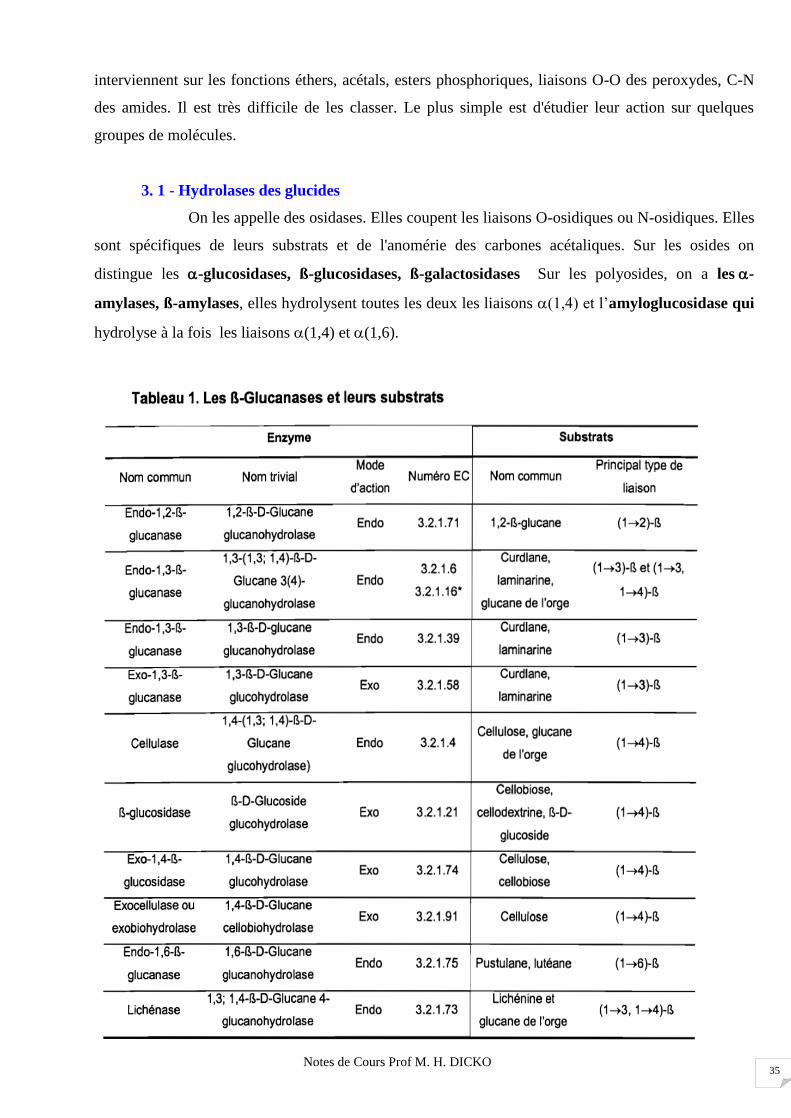
Notes de Cours Prof M. H. DICKO 35
interviennent sur les fonctions éthers, acétals, esters phosphoriques, liaisons O-O des peroxydes, C-N
des amides. Il est très difficile de les classer. Le plus simple est d'étudier leur action sur quelques
groupes de molécules.
3. 1 - Hydrolases des glucides
On les appelle des osidases. Elles coupent les liaisons O-osidiques ou N-osidiques. Elles
sont spécifiques de leurs substrats et de l'anomérie des carbones acétaliques. Sur les osides on
distingue les -glucosidases, ß-glucosidases, ß-galactosidases Sur les polyosides, on a les -
amylases, ß-amylases, elles hydrolysent toutes les deux les liaisons (1,4) et l’amyloglucosidase qui
hydrolyse à la fois les liaisons (1,4) et (1,6).

Notes de Cours Prof M. H. DICKO 36
3.2 - Hydrolases des esters phosphoriques d'oses
On y rencontre les phosphatases qui enlèvent le groupement phosphorique. Leur rôle est
inverse de celui des kinases :
- Glucose 6-phosphatase
Glucose 6- + H2O Glucose + Pi
- Glucose 1-phosphatase
Glucose 1- + H2O Glucose + Pi
- Fructose 1,6 bisphosphatase
Fructose-1,6-bis + H2O Fructose 6- + Pi
3.3 Hydrolases des lipides
* Hydrolases des triglycérides
- Triglycéride lipase
Triglycéride + H2O 2 Acides gras + 2-monoacylglycérol
- Diglycéride lipase)
Diglycéride + H2O 2 Acides gras + glycérol
* Phospholipases : ces enzymes hydrolysent les phospholipides. On distingue selon le site
d'action de l'enzyme
- Phospholipase A1 (EC : 3.1.1.32) enlève l'acide gras lié à la fonction alcool primaire du glycérol
- Phospholipase A2 (EC : 3.1.1.4) enlève l'acide gras lié à la fonction alcool secondaire du glycérol
- Phospholipase C (EC : 3.1.1.4) intervient sur la fonction ester liant le glycérol et le phosphate
- Phospholipase D (EC : 3.1.4.4.) sépare l'acide phosphatidique de l'alcool.
3.3.4 - Hydrolases des peptides et des protéines
Ces hydrolases interviennent sur la liaison peptide des peptides et des protéines. On
distingue les peptidases et les protéinases selon que le produit de la réaction est un acide aminé ou un
peptide.
Les peptidases
- Aminopeptidases libèrent séquentiellement les acides aminés N-terminaux.
- Carboxypeptidases libèrent séquentiellement les acides aminés C-terminaux.
- Dipeptidases hydrolysent les dipeptides.
- Protéinases : hydrolases des protéines

Notes de Cours Prof M. H. DICKO 37
On les appelle des endoprotéinases car elles coupent les liaisons peptidiques situées à l'intérieur
de la protéine loin des acides aminés C et N terminaux. On peut les nommer suivant leur site d'action
ou la structure des protéines elles-mêmes.
- Selon leur lieu d'action on distingue
les protéinases des sucs digestifs
les protéinases du plasma sanguin
les protéinases du tissu conjonctif
les protéinases intracellulaires
Selon la structure, leur nom dérive de l'acide aminé ou du métal situé au niveau de leur site
catalytique . C'est ainsi que nous distinguons :
les sérine-protéinases
la trypsine du pancréas (EC : 3.2.21.4)
la chymotrypsine (EC : 3. 4. 21. 1) du pancréas
la thrombine (EC : 3.4.21.5) du plasma sanguin
les thiol-protéinases
la papaïne (EC : 3.4.22.2) protéine végétale
les métalloprotéinases
collagénase (EC : 3.4.24.3)
3.3.5 - Hydrolases agissant sur les nucléosides, nucléotides et acides nucléiques
On distingue les :
Nucléosidases
- Les Nucléosidases (EC : 3.2.2.1). Elles clivent les liaisons N-osidiques
N-ribosyl-purine + H2O purine + ribose
- AMP-nucléosidase (EC : 3.2.2.4)
AMP + H2O Adénine + ribose phosphate
Nucléotidases
- 5'-Nucléotidases (EC : 3.1.3.5)
Ribonucléoside 5-phosphate Ribonucléoside + H3PO4
Nucléases ou hydrolases des acides nucléiques
Elles clivent les liaisons phosphodiestérs des acides nucléiques. On distingue les endonucléases qui
coupent des liaisons à l'intérieur de la molécule et les exonucléases lorsqu'elles enlèvent les
nucléosides 5'-phosphates les uns après les autres à partir de l'extrémité.

Notes de Cours Prof M. H. DICKO 38
- 3'-Exonucléase ou Phosphodiestérase I (EC : 3.1.4.1), les mononucléotides 5'-phosphates
sont enlevés les uns après les autres à partir de l'extrémité 3'.
- Désoxyribonucléase I (EC : 3.1.4.5) (DNAse 1), c'est une endonucléase qui libère des
oligodésoxynucléotides
- Ribonucléase I (EC : 3.1.4.2.2), endonucléase contenue dans le suc pancréatique. Elle
fragmente aussi les ARN en oligonucléotides. Les enzymes de restriction appartiennent aussi à
cette classe.
3.3.6 - Hydrolases des esters ou anhydrides phosphoriques
- Phosphatase alcaline (EC : 3.1.3.1) : elle n'est active qu'en milieu alcalin, peu spécifique des
monoesters alcalins, active dans toutes les cellules.
- Phosphatase acide (EC : 3.1.3.2) : elle est active dans les cellules osseuses
- ATPases ou adénosine triphosphatase (EC : 3.6.1.3)
- Nucléoside diphosphatase (EC : 3.6.1.1)
ADP (ou GDP) + H2O AMP (ou GMP) + H3PO4
- Pyrophosphatase (EC : 3.6.1.11) transforme le pyrophosphate en 2 orthophosphate
4. - LES LYASES OU SYNTHASES Classe IV. EC :4.X2.X3.X4
On y trouve les enzymes suivantes : décarboxylases, lyases proprement dites, synthases, hydratases,
déshydratases, etc...
3.4.1 - Décarboxylases
Le produit de réaction après le départ de CO2 est un carbonyle ou une amine
- Acétoacétate décarboxylase (EC : 4.1.1.4) : Acétoacétate Acétone + CO2
- Lysine décarboxylase (EC :4.1.1.18) : Lysine cadavérine + CO2
3.4.2 - Aldéhydes-lyases
Les enzymes font apparaître une liaison aldéhyde à la suite du clivage d'une liaison -C-C dont l'un des
carbones porte une fonction alcool secondaire. C'est le cas de
- fructose 1-6 bisphosphate aldolase : fructose 1-6 bis 3-dihydroxyacétone + 3-
glycéraldéhyde
3.4.3 - Acyl-lyases ou Acyl-synthase
- 3-Hydroxy 3-méthyl glytaryl-CoA lyase (EC : 4.1.3.4) :

Notes de Cours Prof M. H. DICKO 39
3-Hydroxy 3-méthyl glytaryl-CoA Acétoacétate + Acétyl-CoA
- Citrate synthase (4.1.3.7) On l'appelle aussi enzyme condensante. C'est une enzyme
importante du cycle de Krebs : Oxaloacétate + acétyl-CoA + H2O Citrate + HSCoA
3.4.4 - Hydratases et déshydratases
Elles fixent ou enlèvent une molécule d'eau, à ne pas confondre avec une hydrolase
R-CHOH-CH2-R' R-CH=CH-R' + H2O
On trouve dans ce groupe :
- la fumarase ou fumarate hydratase (EC : 4.2.1.2)
HOOC-CH=CH-COOH + H2O HOOC-CH2-CHOH-COOH
- l'énolase (4.2.1.11)
2-phosphoglycérate Phosphoénolpyruvate + H2O
5 - LES ISOMERASES Classe V. EC :5.X2.X3.X4
Elles catalysent des changements de structure dans une même molécule sans changer sa
formule globale (isomérisation Cis-trans, épimérisation, déplacement de radicaux, etc.)
3.5.1 - Epimérisation
- Ribulose 5P 3-épimérase (EC : 5.1.3.2) enzyme de la voie des pentoses phosphate
D-ribulose 5- D-xylulose 5-phosphate
- UDP glucose 4-épimérase
UDP glucose 4 UDP galactose
3.5.2 - Oxydoréduction intramoléculaire
- triose phosphate isomérase (EC : 5.3.1.1)
- Glucose 6-phosphate isomérase (EC : 5.3.1.10)
3.5.3 - Transport de radicaux
- Phosphoglycérate mutase (EC : 5.4.2.1)
- Méthyl malonylCoA carboxymutase (EC : 5.4.99.2)
6 - LIGASES ou SYNTHETASES. Classe VI. EC :6.X2.X3.X4
Elles forment des liaisons C-C, C-N, C-S, C-O, O-P grâce à l'utilisation de l'énergie fournie par
l'hydrolyse concomitante d'un groupement phosphate ou pyrophosphate de l'ATP

Notes de Cours Prof M. H. DICKO 40
3.6.1 - LIGASES FORMANT LES LIAISONS C-O
On connaît les ligases des synthèses protéiques et des acides nucléiques.
3.6.2 - LIGASE FORMANT DES LIAISONS C-C
- Pyruvate carboxylase (EC : 6.4.1.1)
Pyruvate + CO2 + ATP Oxaloacétate + ADP + Pi
- Acétyl CoA carboxylase
- Propionyl CoA carboxylase (EC : 6.4.1.3)
3.6.3 - LIGASES DES LIAISONS C-S
- Acétyl CoA synthétase (EC : 6.2.1.1) n'existe pas chez les vertébrés
ATP + Acétate + HSCoA Acétyl CoA + AMP + P-P
- Acyl CoA synthétase (EC : 6.2.1.2) agit sur les acides gras à longue chaîne de carbone.
R-COOH + ATP ° HSCoA R-CO-SCoA + AMP + P-P
3.6.4 - LIGASES DES LIAISONS C-N
- Glutamine synthétase (EC : 6.3.1.2)
ATP + glutamate + NH3 ADP + H3PO4 + Glutamine
- 5-phosphoribosylamine synthétase (EC : 6.3.4.7)
ATP + ribose 5-phosphate + NH3 5-phosphoribosylamine + ADP + H3PO4
Notes de Cours Prof M. H. DICKO 41
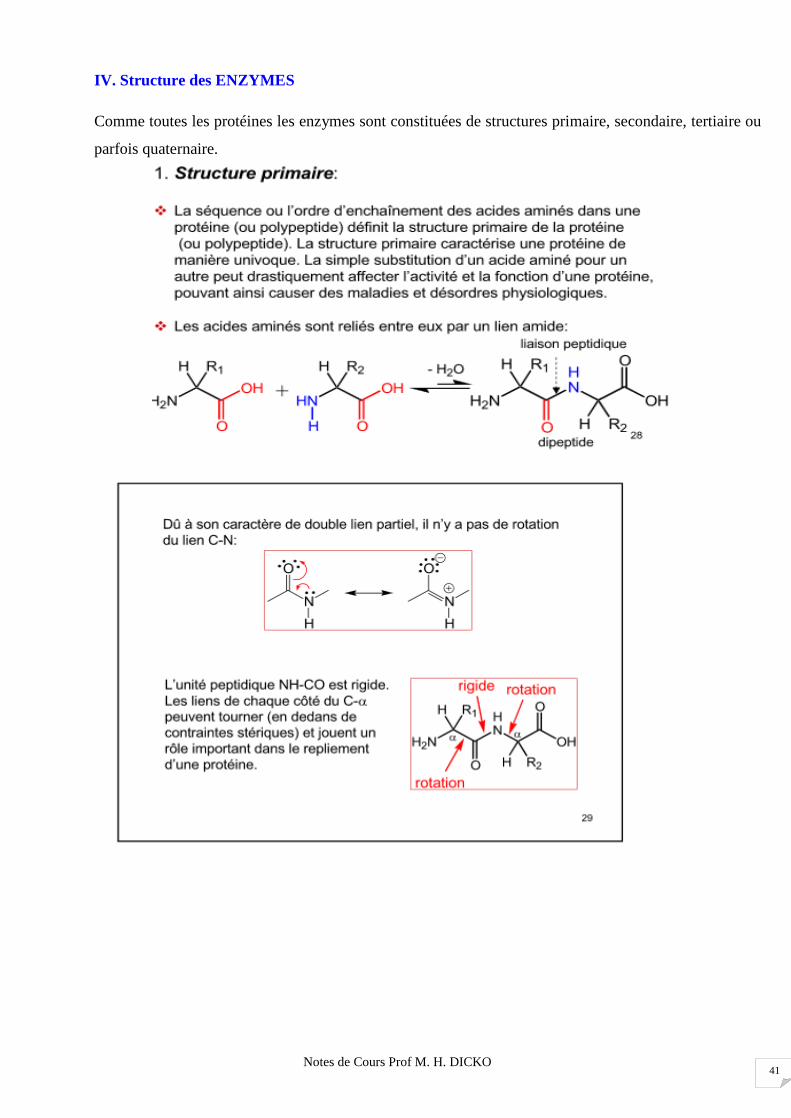
IV. Structure des ENZYMES
Comme toutes les protéines les enzymes sont constituées de structures primaire, secondaire, tertiaire ou
parfois quaternaire.
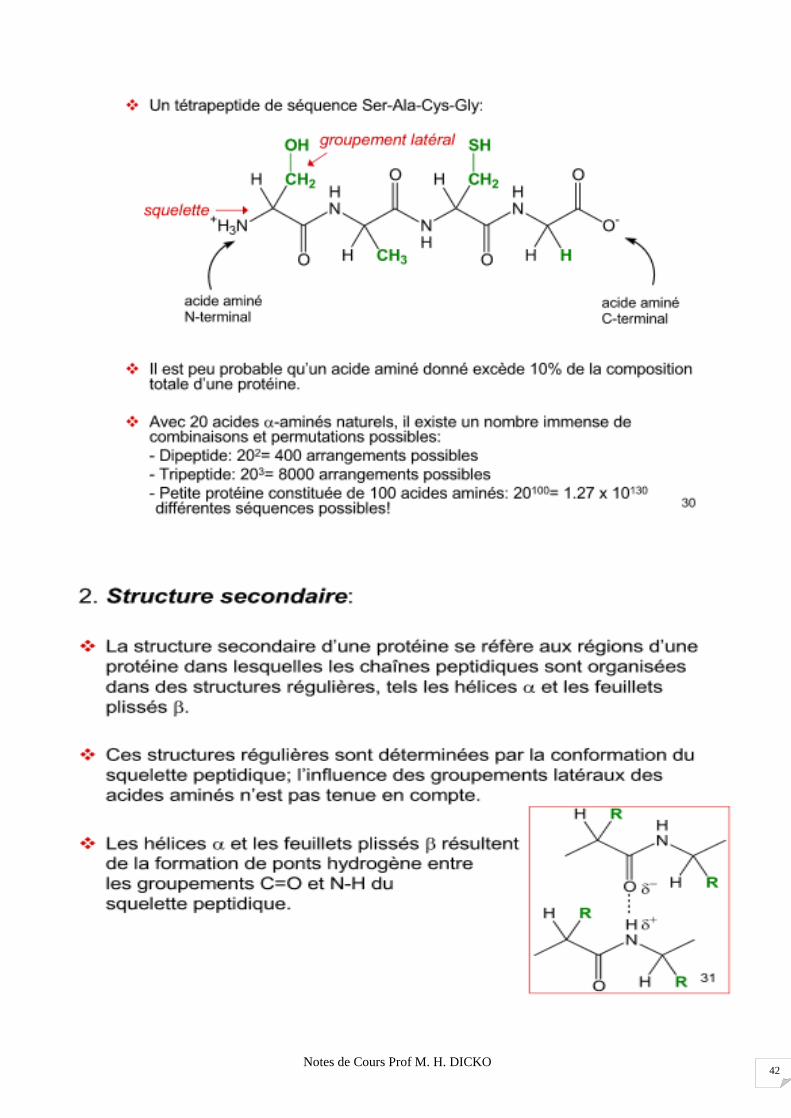
Notes de Cours Prof M. H. DICKO 42
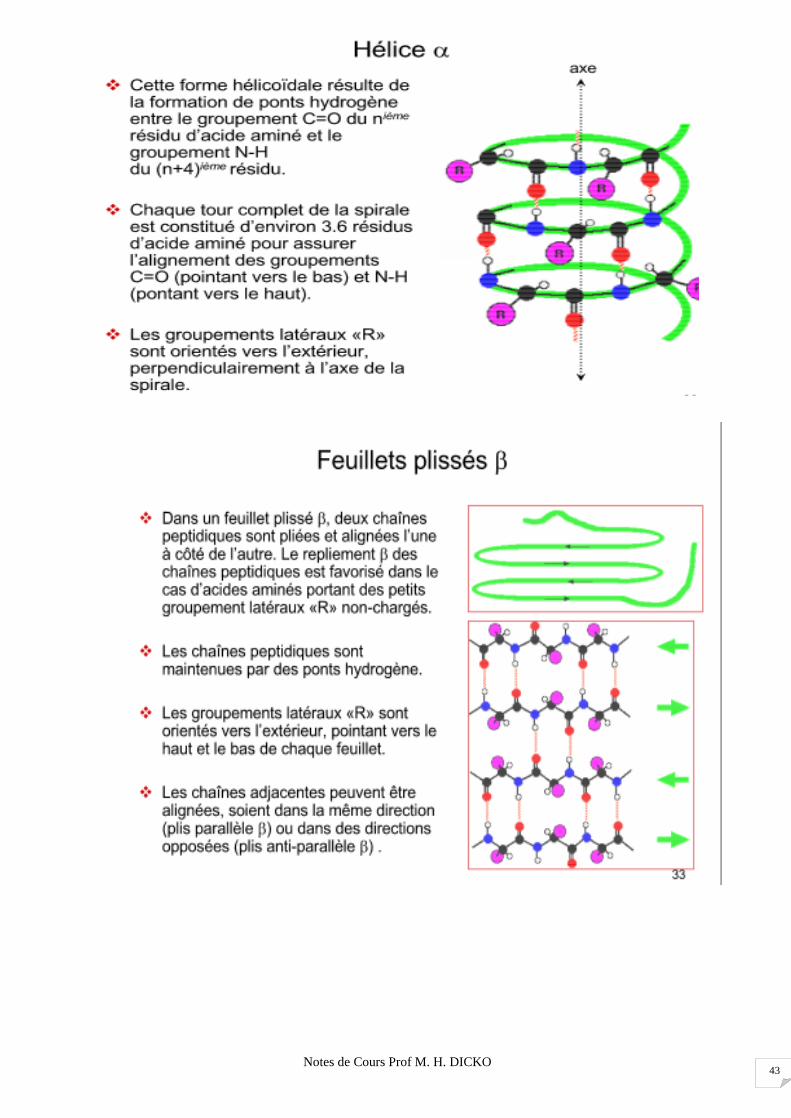
Notes de Cours Prof M. H. DICKO 43
Notes de Cours Prof M. H. DICKO 44
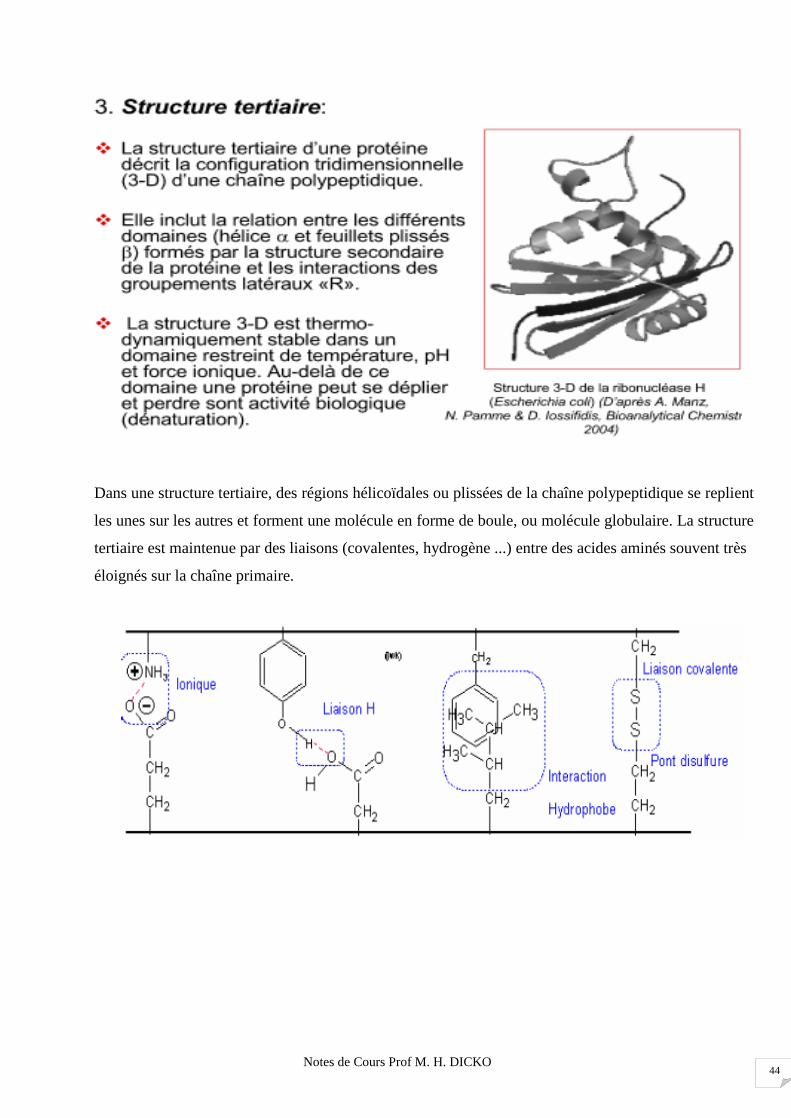
Dans une structure tertiaire, des régions hélicoïdales ou plissées de la chaîne polypeptidique se replient
les unes sur les autres et forment une molécule en forme de boule, ou molécule globulaire. La structure
tertiaire est maintenue par des liaisons (covalentes, hydrogène ...) entre des acides aminés souvent très
éloignés sur la chaîne primaire.

Notes de Cours Prof M. H. DICKO 45

46
V. Nature du site actif
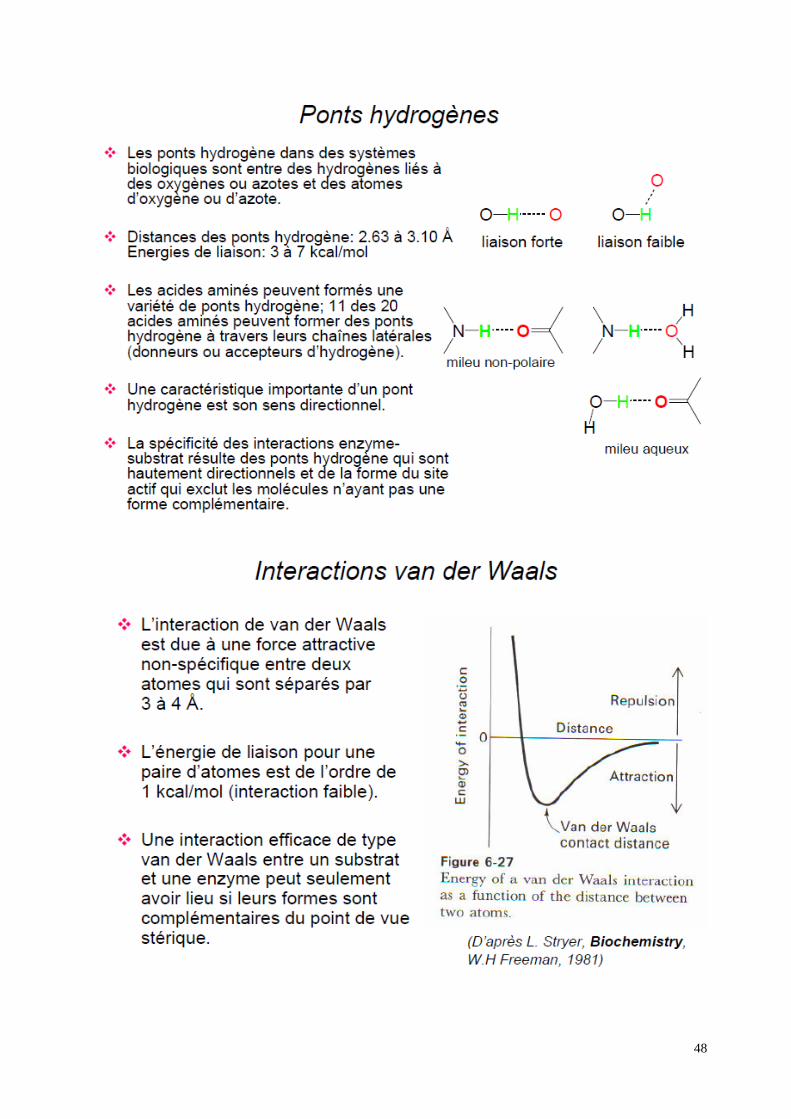
�Le «site actif» d’une enzyme est la région tridimensionnelle qui se lie au substrat.
�Les sites actifs sont des cavités de caractère non polaire et dans lesquelles les substrats
s’insèrent. L’eau est normalement exclut du site actif lorsque le substrat est lié, sauf si elle est
un réactif.
�Le site actif consiste d'au moins deux parties fonctionnelles, qui peuvent ou non être voisines
sur la chaîne polypeptidique (si non, la structure tertiaire les amène près l'une de l'autre):
- le site de reconnaissance du substrat est constitué de certains acides aminés qui sont associés
avec l’orientation du substrat, et donc, avec la spécificité de l’enzyme
- le site catalytique est constitué des résidus qui sont directement impliqués dans la formation et
rupture des liens chimiques. Ces résidus sont souvent localisés dans le fond de la cavité, et dans
la majorité des cas, possèdent des chaînes latérales ioniques ou réactives (ex. His, Lys, Cys,
Ser, Asp, Glu).

47
48

49
CHAPITRE 4 .
DOSAGE DES PROTEINES (ENZYMES)

50
I. Introduction
Les enzymes étant des protéines, sont quantifiées avec toutes les méthodes courantes de
quantification des protéines. Etant donné qu’elles sont de très faibles concentrations, on utilise
rarement les méthodes robustes telles que la HPLC (High performance Liquide
Chromatographie) ou la méthode Kjeldhal (dosage de l’azote totale) pour la quantifier. Les
méthodes les plus couramment utilisées pour la quantification des enzymes sont la
spectrophométrie dans l’UV-visible, la fluorescence ou l’électrophorèse.
II. Principe de la Spectrophotométrie : Loi de Beer Lambert
Principe
L'absorption de la lumière par une molécule est dépendante de:
-la longueur d'onde (transition électronique)
- de la nature chromophore, c’est-à-dire son coefficient d’extinction molaire ()
-la concentration de la molécule (C)
-la longueur du chemin optique parcouru dx (L)
Io It
L

51
Où
A, L’absorbance qui n’a pas d’unité car dérivant d’une fonction logarithmique
ε est le coefficient d’extinction molaire (unités : M-1
cm-1
)
C, la concentration de la molécule (M)
L, le trajet optique (cm).
T, la transmittance (I/Iox 100) exprimé en %.
La loi Beer-Lambert est additive mais il n’est conseillé d’effectuer des mesures à de très fortes
densités optiques (DO>1).
Source polychromatique. Lampe à incandescence
visible Lampe à Xénon (UV)
Monochromateur Permet de
sélectionner la longueur d'onde
Cuve de référence
I
O
Tampon +
échantillon
It
.C.L ε)It
Ilog()log( 0
TDOA
La Densité optique (D.O) ou l'aborsbance A est:

52
III. Quantification des enzymes
II.2. Exemples d'essais de quantification des enzymes
II.2.1 Méthodes spectrophotométriques
(A) Par mesure de l'absorption aux UV.
Ce type d'analyse est très pratique pour suivre la course de protéines sur une série de colonnes
de chromatographie. Tous les systèmes de chromatographie sont équipés d'une cellule
spectrophotométrique pour mesurer l'absorption des rayons UV par le matériel qui sort des
colonnes. C'est cependant une méthode simple dont le type de réponse est "oui, il y a des
protéines" ou "non, il y a eu un os quelque part". On ne peut pas sérieusement s'attendre à
pouvoir faire une relation exacte entre la densité optique d'un mélange protéique en cours de
purification et sa concentration réelle. (On utilise parfois un coefficient d'extinction très
approximatif pour dire qu'une densité optique à 280nm, dans une cuvette de 1 cm, correspond à
1 mg/mL de protéines...Mais c'est comme de dire que la population de la Terre s'élève à
quelques milliards. C'est une idée générale, rien de plus).
En outre, n'oublions pas que l'ADN et l'ARN absorbent dans le coin de 250-260nm, ce qui
risque de nous causer des soucis de précision! (D'habitude, on a plutôt le problème inverse,
celui des protéines qui contaminent notre ADN. Mais tout finit par nous arriver si on vit assez
longtemps).
On peut utiliser le spectrophotomètre pour détecter la présence de protéines parce que le
tryptophane (Trp, W) absorbe la lumière UV avec un pic à 280 nm. La tyrosine (Tyr, Y) le peut
aussi, quoiqu'à un degré moindre, et un troisième acide aminé cyclique, la phénylalanine, le
peut aussi très faiblement. Tous les peptides pourraient absorber l'UV aux environs de 190-200
nm, mais la plupart des tampons absorbent aussi massivement dans cette région qui n'est donc
pas très commode d’utiliser cette longueur d’onde pour les mesures.
Notez que le coefficient d'extinction de chaque protéine variera selon sa composition en W et
Y. Une protéine dépourvue de ces acides aminés passerait devant la cellule optique comme un
fantôme, à peine perçue.
53
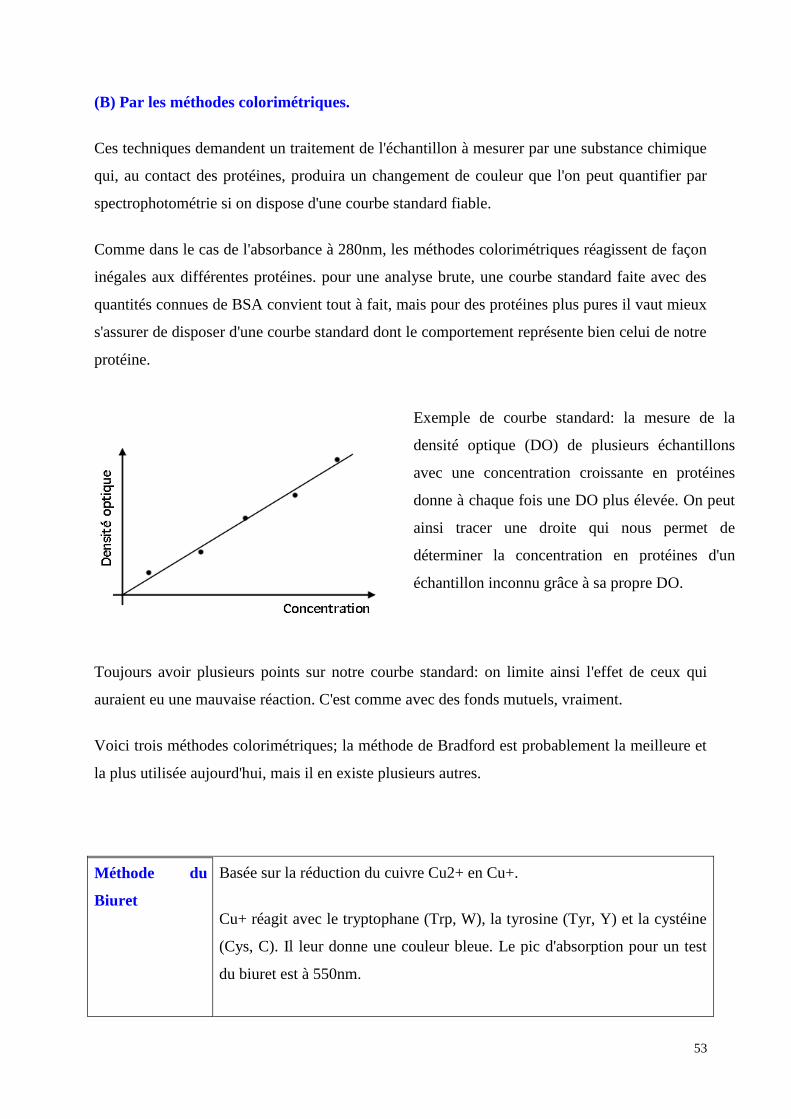
(B) Par les méthodes colorimétriques.
Ces techniques demandent un traitement de l'échantillon à mesurer par une substance chimique
qui, au contact des protéines, produira un changement de couleur que l'on peut quantifier par
spectrophotométrie si on dispose d'une courbe standard fiable.
Comme dans le cas de l'absorbance à 280nm, les méthodes colorimétriques réagissent de façon
inégales aux différentes protéines. pour une analyse brute, une courbe standard faite avec des
quantités connues de BSA convient tout à fait, mais pour des protéines plus pures il vaut mieux
s'assurer de disposer d'une courbe standard dont le comportement représente bien celui de notre
protéine.
Exemple de courbe standard: la mesure de la
densité optique (DO) de plusieurs échantillons
avec une concentration croissante en protéines
donne à chaque fois une DO plus élevée. On peut
ainsi tracer une droite qui nous permet de
déterminer la concentration en protéines d'un
échantillon inconnu grâce à sa propre DO.
Toujours avoir plusieurs points sur notre courbe standard: on limite ainsi l'effet de ceux qui
auraient eu une mauvaise réaction. C'est comme avec des fonds mutuels, vraiment.
Voici trois méthodes colorimétriques; la méthode de Bradford est probablement la meilleure et
la plus utilisée aujourd'hui, mais il en existe plusieurs autres.
Méthode du
Biuret
Basée sur la réduction du cuivre Cu2+ en Cu+.
Cu+ réagit avec le tryptophane (Trp, W), la tyrosine (Tyr, Y) et la cystéine
(Cys, C). Il leur donne une couleur bleue. Le pic d'absorption pour un test
du biuret est à 550nm.

54
Ce test est peu sensible.
Il résiste assez bien aux différents composés présents dans les tampons mais
est sensible aux sels d'ammonium, ce qui en fait un mauvais choix pour
doser les protéines venant d'être précipitées par ce sel.
Méthode de
Lowry
Basée sur la réaction du biuret, elle utilise aussi la réduction du Cu2+ en
Cu+. Depuis 1951, la méthode de Lowry est la plus citée de toutes dans la
littérature scientifique.
Dans la méthode de lowry, le Cu+ est utilisé pour réduire le réactif de Folin
(une solution phénolique contenant des composés de tungstène et de
molybdène) qui change sa couleur du jaune au bleu. On lit la réaction à 750
nm.
Plus sensible que la réaction du biuret, la méthode de Lowry est utilisée
pour des quantités de 2 — 100 ug.
Elle est sensib -mercapto,
l'Hepes, le Tris, le triton X-100, le NP-40, etc.
Méthode de
Bradford
Le bleu de Coomassie se lie à l'arginine (Arg, R), la tyrosine (Tyr, Y),
le tryptophane (Trp, W), l'histidine (His, H) et la phénylalanine (Phe, F)
(surtout à R; huit fois plus qu'aux autres en fait).
Il est assez insensible aux agents des tampons, mais est sensible aux

55
détergents.
En solution, il a une forme cationique rouge qui absorbe à 470nm. Lié
aux protéines, il a une forme anionique bleue qui absorbe à 595nm.
Parce que les deux spectres d'absorption se chevauchent un peu, une
courbe standard de Bradford n'est pas parfaitement linéaire sur toute sa
distance (quoiqu'en disent les commerçants!), un point souligné par
Bradford lui-même dans son papier original. Elle l'est quand même
suffisamment pour nos besoins.
L’autre variante de la Méthode de Bradford est celle de SEDMAK qui
prend le ratio A620/A450 = f([protéine])
La méthode de Bradford est encore plus sensible que celle de Lowry
(0,2 — 20 ug de protéines).
Structure du bleu de Coomassie G-250
56
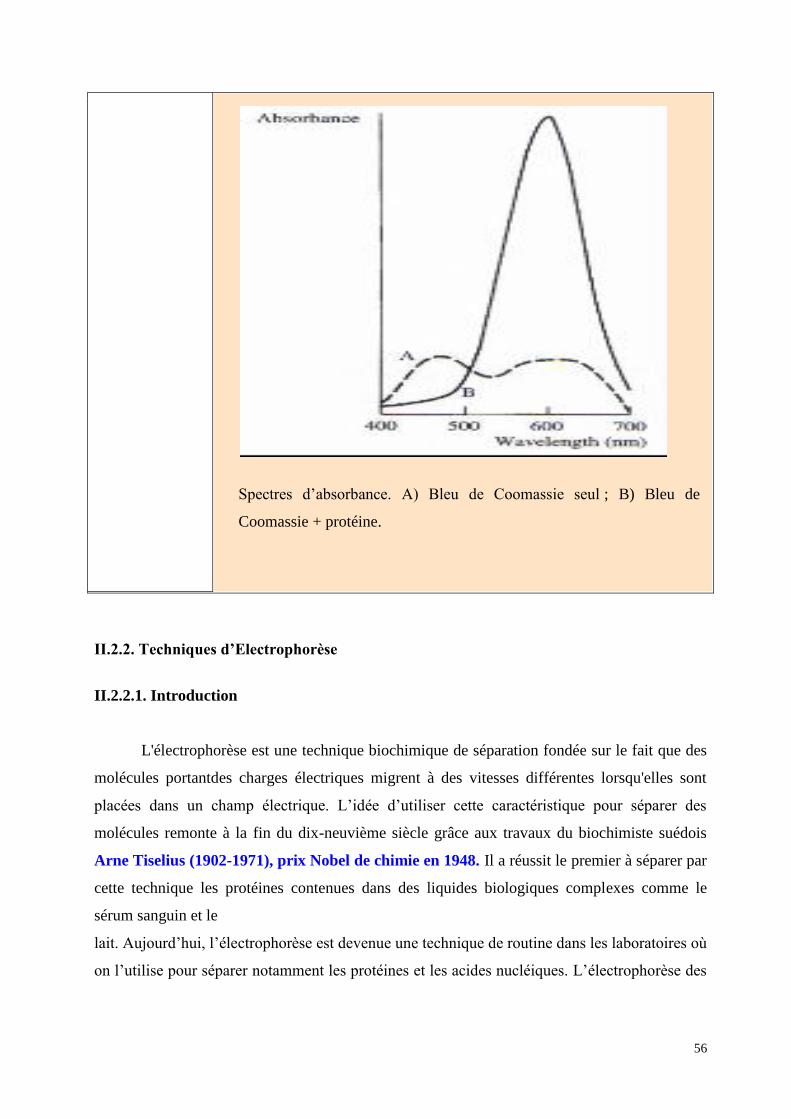
Spectres d’absorbance. A) Bleu de Coomassie seul ; B) Bleu de
Coomassie + protéine.
II.2.2. Techniques d’Electrophorèse
II.2.2.1. Introduction
L'électrophorèse est une technique biochimique de séparation fondée sur le fait que des
molécules portantdes charges électriques migrent à des vitesses différentes lorsqu'elles sont
placées dans un champ électrique. L’idée d’utiliser cette caractéristique pour séparer des
molécules remonte à la fin du dix-neuvième siècle grâce aux travaux du biochimiste suédois
Arne Tiselius (1902-1971), prix Nobel de chimie en 1948. Il a réussit le premier à séparer par
cette technique les protéines contenues dans des liquides biologiques complexes comme le
sérum sanguin et le
lait. Aujourd’hui, l’électrophorèse est devenue une technique de routine dans les laboratoires où
on l’utilise pour séparer notamment les protéines et les acides nucléiques. L’électrophorèse des

57
protéines peut être réalisée sur des supports variés, notamment sur gel de polyacrylamide ou sur
gel d’agarose selon les informations recherchées.
Principe de l’électrophorèse
Selon le principe de l’électrostatique lorsqu’un ion de quantité de charge q est placé dans un
champs électrique E ,une force F s’exerce sur cet ion avec une intensité donnée par l’équation :
Félectrique = qE
L’ion peut être assimilé à une sphère de rayon r en mouvement dans un fluide de viscosité h.
De ce fait une force defriction Ffriction s’oppose à la force électrique F. Par définition
Ffriction= 6πɲrv
Où v = vitesse de migration de l’ion, r = rayon de l’ion, ɲ= viscosité du milieu.
A l’équilibre l’ion migre avec une vitesse constante de sorte que Félectrique = Ffriction
d’où
𝑣 =qE
𝟔 𝛑 ɲ 𝒓
La mobilité électrophorétique μ est donnée par l’équation :
µ =v
𝐄=
q
𝟔 𝛑 ɲ 𝒓
Cette équation nous montre que la mobilité électrophorétique est principalement fonction de la
charge et de la taille de l’ion. Cela est valable pour toutes les techniques d’électrophorèse.
L’électrophorèse est donc une technique de séparation utilisant la migration des molécules
chargées dans un champs électrique. Les protéines possédant de nombreux groupements
ionisables sont des molécules amphotères : leur charge dépend du pH du milieu ainsi que de
leur propre pHi.
Si pH <pHi la charge nette est positive
Si pH = pHi la charge nette est neutre
Si pH >pHi la charge nette est négative

58
La protéine est d’autant plus chargée que la différence entre pH du milieu et pHi est
importante.
De nombreuses méthodes utilisent ce principe, elles peuvent être analytiques (but : séparation
des molécules) ou préparatives (but : obtention d’une quantité plus ou moins importante d’un
mélange). Les principales techniques d’électrophorèse sont mentionnées ci-dessous.
II.2.2.2.Electrophorèse en veine liquide
C’est type d’électrophorèse le plus ancien, mis au point par Tisellius. La migration s’effectue
au sein d’un liquide de composition déterminé. Abandonnée pendant un temps cette technique
est réactualisée pour la technique d’électrophorèse capillaire. Un détecteur optique est placé à
la sortie du tube d’électrophorèse.
La séparation s’effectue selon la charge des protéines.
II.2.2.3 Electrophorèse de zone
La migration est réalisée également dans une phase liquide mais celle ci imprègne un support
solide poreux ou un milieu gélifié; Les supports les plus courants sont : le papier, l’acétate de
cellulose, et les gels (agarose, gélose, amidon, polyacrylamide …)
La séparation s’effectue selon la charge et plus ou moins selon le support utilisé la masse des
protéines.
Après migration les protéines sont fixées par précipitation puis colorées.
Exemple d’utilisation de routine en biochimie clinique : séparation des protéines sériques sur
gel d’amidon.
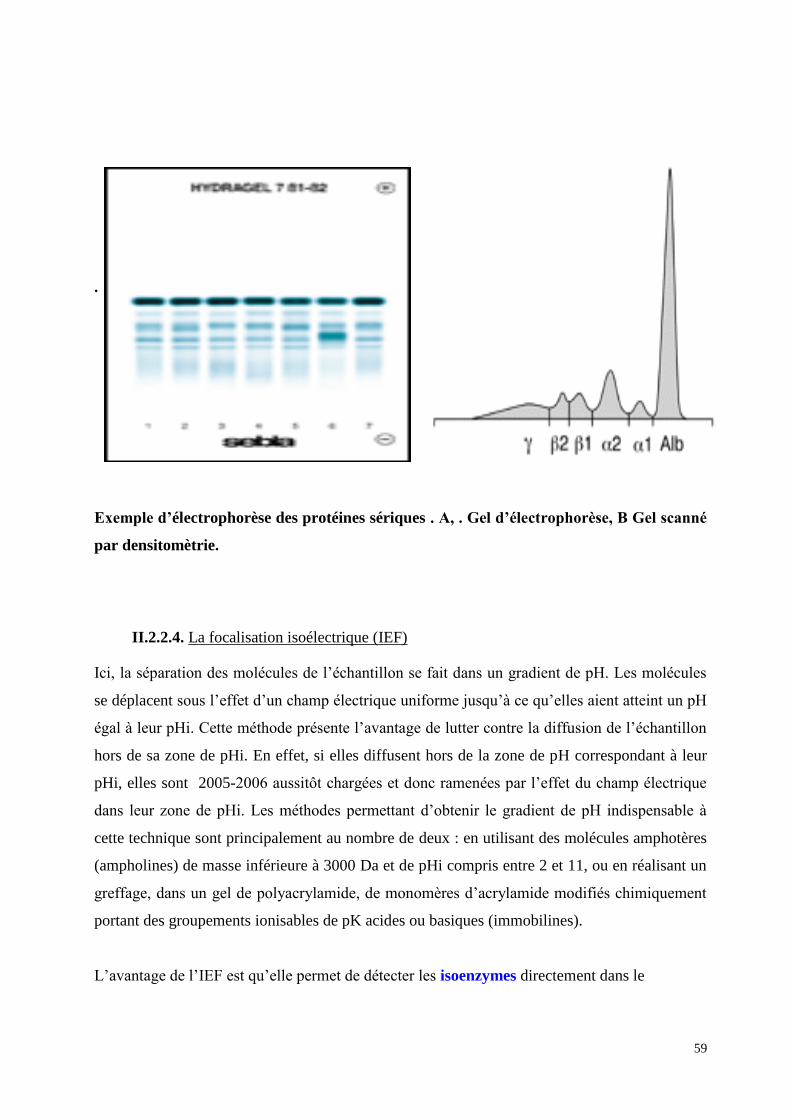
59
.
Exemple d’électrophorèse des protéines sériques . A, . Gel d’électrophorèse, B Gel scanné
par densitomètrie.
II.2.2.4. La focalisation isoélectrique (IEF)
Ici, la séparation des molécules de l’échantillon se fait dans un gradient de pH. Les molécules
se déplacent sous l’effet d’un champ électrique uniforme jusqu’à ce qu’elles aient atteint un pH
égal à leur pHi. Cette méthode présente l’avantage de lutter contre la diffusion de l’échantillon
hors de sa zone de pHi. En effet, si elles diffusent hors de la zone de pH correspondant à leur
pHi, elles sont 2005-2006 aussitôt chargées et donc ramenées par l’effet du champ électrique
dans leur zone de pHi. Les méthodes permettant d’obtenir le gradient de pH indispensable à
cette technique sont principalement au nombre de deux : en utilisant des molécules amphotères
(ampholines) de masse inférieure à 3000 Da et de pHi compris entre 2 et 11, ou en réalisant un
greffage, dans un gel de polyacrylamide, de monomères d’acrylamide modifiés chimiquement
portant des groupements ionisables de pK acides ou basiques (immobilines).
L’avantage de l’IEF est qu’elle permet de détecter les isoenzymes directement dans le

60
Gel par incubation du gel avec le substrat dont le produit est coloré et qui ne diffuse pas
facilement : c’est la technique de ZYMOGRAPHIE.

61
Exemple: Zymogrames des activités de péroxydases dans les grains de variétés de sorghos burkinabè (Sorghum
bicolor) non germés ou germés. Les échantillons ont été deposes au milieu des gels d’EIF (►). Les chiffres
arabes représentent les variétés et la lettre “g” pour la germinstation. Les ronds et les carrés illustrent les
isoenzymes détectés avant et après germination. Les principales isoenzymes anioniques détectées sont A1 (pI
3.1), A2 (pI 4.2), A3 (pI 4.8), A4 (pI 5.2), A5 (pI 6), A6 (pI 6.2) and A7 (pI 7.1). Les principales isoenzymes
cathioniques détectées sont C1 (pI 7.8), C2 (pI 8.1), C3 (pI 8.3), C4 (pI 8.7) and Cx (pI 9). Dicko et al. JSFA.
2006, 86, 953-963.© John Wiley & Sons. Inc. All Rights Reserved.
17 17g 18 18g 19 19g 20 20g
45 45g 46 46g 47 47g 48 48g
29 29g 30 30g 31 31g 32 32g
41 41g 42 42g 43 43g 44 44g
33 33g 34 34g 35 35g 36 36g
5 5g 6 6g 7 7g 8 8g
21 21g 22 22g 23 23g 24 24g
37 37g 38 38g 39 39g 40 40g
pI 3
pI 9
pI 9
pI 3
25 25g 26 26g 27 27g 28 28g
13 13g 14 14g 15 15g 16 16g
pI 9
pI 3 1 1g 2 2g 3 3g 4 4g
pI 3
pI 9
49 49g 50 50g
9 9g 10 10g 11 11g 12 12g
Cx
C1
C3
A4
A6
*Isoenzymes
C2
C4
A2
A1
A3
A5
A7
Zymographie des Enzymes (Péroxydases)

62
II.2.2.5. Electrophorèse bidimensionnelle
Une IEF est effectuée dans un sens sur un boudin de polyacrylamide lâche puis, sans fixer les
protéines, ce boudin est déposé sur un gel polyacrylamide à mailles plus serrées et une
électrophorèse SDS-PAGE est réalisée perpendiculairement à l’IEF. Les protéines sont ainsi
séparées en fonction de leur pHi puis de leur PM. Chaque protéine apparaît comme un spot sur
le gel.
Cette méthode est purement analytique.
Figure. Electrophorèse bidimensionnelle des protéines ribosomales
II.2.2.6. L’isotachophorèse (ITP)
Cette méthode est fondée exclusivement sur la séparation des molécules suivant leur mobilité
électrophorétique. Les anions se déplacent tous vers l’anode, les plus mobiles en tête et les plus
lents en queue. Ceci est possible grâce à un gradient de champ électrique, un champ faible étant
appliqué aux ions lents, alors qu’un champ fort est appliqué aux ions rapides. Ce dispositif
favorise la ségrégation et la répulsion mutuelle des ions de mobilité différente.

63
II.2.2.6. SDS-PAGE
Le système d’électrophorèse le plus utilisé pour l'analyse de la taille des protéines d'un mélange
est le gel de SDS (ou SDS-PAGE, pour sodium docecyl sulfate -polyacrylamide gel
electrophoresis).
La séparation électrophorétique d'une protéine, tout comme celle de n'importe quelle molécule
chargée d'ailleurs, est l'effet de son mouvement sur ou dans une matrice appropriée soumise à
un courant électrique. Plus une molécule est massive ou biscornue, plus elle se déplacera
lentement; plus elle sera petite, plus elle ira vite.
Il serait avantageux, comme ce l'est pour l'ADN, de pouvoir séparer les protéines les unes des
autres par une simple séparation électrophorétique ne dépendant que de la taille des molécules.
Mais alors que l'ADN est uniformément chargé et que sa structure est essentiellement la même
quand il est sous forme linéaire, les protéines, elles, varient énormément non seulement en
poids mais aussi en charge et en forme. Pour les séparer selon leur masse uniquement, il faut
contrecarrer l'effet de leur charge et de leur forme.
Ce tour de passe-passe est accompli en mettant 1% de SDS dans le gel d'électrophorèse et dans
le tampon de migration. On utilise aussi 0,1M β-mercaptoethanol (HO-CH2-CH2-SH) dans le
tampon de largage des échantillons, et ceux-ci sont bouillis de surcroît avant la migration sur
gel.
Le sodium dodecyl sulfate (SDS) est un détergent anionique qui dénature les protéines en
enveloppant la chaîne polypeptidique selon un ratio de masse de 1,4 pour 1. Le SDS confère
aux protéines une charge négative proportionnelle à leur longueur; plus elles sont longues, plus
il y a de SDS. Les polypeptides deviennent donc des chapelets de charges négatives partageant
une même densité de charge par unité de longueur.
Le β-mercaptoethanol, lui, (un agent réducteur qui ne sent pas bon), en combinaison avec la
chaleur, brise les ponts disulfures internes des protéines que la chaleur ont dénaturées. Il brise
aussi les éventuels ponts disulfures inter-protéiques.
Les protéines ainsi dépliées et uniformément chargées en fonction de leur masse peuvent dès
lors être séparées par électrophorèse, de la cathode (-) à l'anode (+). Puisqu'elles sont

64
uniformément chargées par le SDS, leur vitesse de migration sur gel sera proportionnelle à leur
seule masse, les petites protéines étant les plus rapides.
On utilise d'ordinaire un gel discontinu: c'est à dire qu'en haut du gel de séparation
proprement dit, (le "running gel;" en bon franglais) se trouve une bande plus petite d'acrylamide
de composition différente, le gel de concentration ou stackinggel.

65
Alors que le gel de séparation a des pores petits et un pH de 9, le stacking gel a des
pores plus grands et un pH de 6,8. La taille des pores dans un gel de polyacrylamide est
fonction du degré de pontage entre les chaînes d'acrylamide, pontage qui est
proportionnel à la quantité de N,N,N',N',-méthylène bisacrylamide dans le gel; plus il y
en a, plus les chaînes sont pontées et plus petits sont les pores.
De plus, les ions des deux gels au moment du dépôt des protéines sont des ions chlorure alors
que ceux du tampon sont des ions glycine. Les ions glycine qui entrent dans le gel de
concentration à pH 6,8 sont près de leur point isoélectrique et ont une très faible mobilité. Les
ions chlorure, eux, conservent une grande mobilité. Les protéines chargées par le SDS sont plus
lentes que le chlorure mais plus rapides que la glycine.
Quand le courant est appliqué, les ions chlorures filent vers l'anode, laissant derrière elles un
vide ionique temporaire qui n'a qu'une faible conductivité.
Manquant d'électrolytes dans leur entourage immédiat, les protéines dans le gel de
concentration ne peuvent pas migrer vers l'anode. En fait, elles ne peuvent bouger que quand le
front d'ions glycine finit par les rattraper, parce que ces ions remplacent alors les ions chlorure
comme électrolytes. Même alors, parce que les grands pores du gel de concentration permettent
aux protéines d'être aussi rapides que les ions glycine, elles ne se séparent pas les unes des

66
autres mais sont toutes ramassées au passage par le front d'ions.
Ce front d'ions qui pousse et empile les protéines devant lui induit la concentration de toutes les
protéines en une mince bande qui migre avec le front jusqu'au bout du gel de concentration.
Quand le front d'ions atteint la discontinuité de pH et de porosité entre le gel de concentration et
le gel de séparation, le pH soudain plus élevé permet aux ions glycine de prendre leur envol. Ils
deviennent beaucoup plus chargés et rapides et dépassent les protéines.
Maintenant qu'il y a assez d'électrolytes en avant d'elles, les protéines peuvent migrer librement
en fonction de leur masse. (La porosité du gel, qui est plus faible dans le gel de séparation que
dans le gel de concentration, assure que les ions glycine seront plus rapides que les protéines).
Il faut avouer que la mobilité des protéines dans un gel de SDS n'est pas absolument
proportionnelle à leur masse. En effet, on peut observer une variation de vitesse de migration
par rapport à la masse dans une majorité de protéines, allant de très petites variations jusqu'à
des résultats incorrects. La cause peut en être variable: par exemple, certaines protéines ont
naturellement une faible affinité pour le SDS et le ratio masse/charge s'y trouve modifié.
Certaines modifications post-traductionnelles peuvent aussi jouer un rôle: la glycosylation
interfère avec la liaison au SDS. Ces erreurs peuvent être corrigées par des techniques de
spectrométrie de masse.
Les très petites protéines (moins de 15 kDa), en outre, courent de façon très aléatoires dans les
gels de glycine. C'est qu'elles peuvent être capturées dans des micelles de SDS qui se tiennent
près du front de migration. On peut les en libérer en utilisant la tricine à la place de la glycine
dans le tampon de migration. La tricine aide à la libération de ces petites protéines. On offre
commercialement des gels de SDS Tris-tricine pour l'étude des protéines de plus petit poids
moléculaire.
Une fois les protéines dûment séparées selon leur taille, il reste à révéler leur présence. Les
approches les plus courantes sont de les colorer, d'utiliser un anticorps spécifique ou de faire
une zymographie.
On peut colorer les protéines directement sur le gel, ou les colorer après les avoir transféré sur
une membrane par électro-transfert.

67
Les techniques les plus courantes sont la coloration au bleu de Coomassie et au nitrate
d'argent. Le bleu de Coomassie servait jadis à colorer les chandails de laine en bleu; son
nom commémore la prise de la ville Ashanti de Kumassi, au Ghana, pendant l'époque
coloniale. Le bleu de Coomassie colore les protéines de façon à peu près égale mais est de dix
à cent fois moins sensible que la technique de coloration au nitrate d'argent.
Cette dernière repose sur la réduction, facilitée par la présence des protéines, de sels d'argent en
argent métallique (qui précipite). Il existe plusieurs protocoles de coloration au nitrate d'argent
(choisissez la vôtre en fonction de sa facilité d'exécution et du temps qu'elle requiert)! Cette
technique est cependant sensible à plusieurs facteurs et ne colore pas toutes les protéines de
façon égale.
Gel non-dénaturant "Blue native".
Une variante du gel SDS permet de greffer une charge aux protéines ou aux complexes
protéiques sans les dénaturer. Dans ce cas on n’utilise pas le SDS ou le beta-mercapto-éthanol
La distribution des charges ne permet pas de définir un ratio charge/masse aussi constant mais
au moins les protéines natives migrent. Cette technique est appelée "blue native
electrophoresis". (Schagger H, von Jagow G. Blue native electrophoresis for isolation of
membrane protein complexes in enzymatically active form. Anal Biochem. 1991
Dec;199(2):223-31). On peut également effectuer une zymographie après la PAGE en absence
ou en présence du SDS.

68
CHAPITRE 5.
EXTRACTION ET PURIFICATION DES ENZYMES

69
I. Extraction des Enzymes
L’extraction et la purification d'une enzyme sont des processus qui comprennent plusieurs
étapes: extraction initiale, précipitation différentielle (sulphate d’ammonium, solvant
organique, etc.), dialyse, chromatographies de divers types, etc. Les enzymes étant des
protéines, elles se prêtent à toutes les méthodes d’extraction et de purifications des protéines
tout en ayant à l’esprit que les enzymes sont thermolabiles et très sensibles aux substances
chimiques.
1. Broyage
Typiquement, on broie tout d'abord le matériel d'où on veut extraire l’enzyme (tissu
animal, partie de plantes, bactéries, etc). Divers appareils ("Waring Blender", appareil
Potter-Eveljhem, "Polytron", etc.) peuvent être employés à cette fin. Cette homogénéisation se
fait dans un tampon de composition appropriée. Cette étape est évidemment la première
L'homogénat ainsi obtenu est ensuite clarifié, le plus souvent par centrifugation, pour éliminer
les grosses particules peu ou mal broyées ou encore pour obtenir la fraction cellulaire contenant
l’enzyme recherchée. Si la protéine est justement dans un compartiment cellulaire, on utilise
généralement un détergent doux (Triton, Tween, etc., quelquefois déoxycholate) pour la libérer
en dissolvant les membranes de ce compartiment. L'emploi de détergent doit souvent être fait
de façon contrôlée car ils peuvent briser les lysosomes, ce qui libèrent des enzymes
hydrolytiques (protéases, nucléases, etc) qui peuvent attaquer et détruire les enzymes ou autres
molécules qu'on veut isoler. Des précautions particulières doivent être prises si on travaille avec
des enzymes sensibles à la dégradation ou peu nombreuses. Certains tissus comme le pancréas
et de foie sont reconnus pour contenir de grandes quantités de ces enzymes, ce qui pose tout un
défi quand on travaille avec ces tissus. Une solution fréquente à ce problème est l'inclusion
dans les solutions d'inhibiteurs de protéases qui sont soit physiologiques (inhibiteur de trypsine,
antipaïne. leupeptine...) ou artificiels (E64, PMSF...). Pour maximiser leur action, on les
emploie souvent en mélange ("cocktails") à large spectre d'action. Ensuite on utilise diverses
techniques pour séparer l’enzyme recherchée de toutes les autres présentes. Habituellement les
premières étapes sont des techniques peu spécifiques mais bien adaptées à la manipulation de
gros volumes. Ensuite, au fur et à mesure des étapes, on utilise des techniques de plus en plus
spécifiques qui sont souvent applicables à des préparations de volume réduit.

70
Une des méthodes se prêtant le mieux à de gros volumes est la précipitation différentielle au
sulfate d'ammonium. C'est pourquoi on l'utilise très souvent immédiatement après
l'homogénéisation pour se débarrasser du gros des contaminants. Les chromatographies
d'échange ionique ou les chromatographies d`affinité, applicables à de bons volumes
d'échantillon mais ayant un assez bon pouvoir de séparation, constituent de bonnes méthodes
intermédiaires. En fin, on utilise souvent le tamisage moléculaire ou l'isofocalisation qui
permettent de raffiner la pureté, mais nécessitent de très petits volumes d’enzymes concentrées.
Souvent, entre ces étapes, il faut éliminer les sels ou produits utilisés dans ces
chromatographies. On utilise alors une dialyse ou une ultrafiltration.
Figure . Principe de la dialyse (A) et de l’Ultrafiltration (B).
Si on a besoin de concentrer la préparation, on la lyophilise. Si on veut éviter ces procédures,
on doit choisir des méthodes qui ne sont pas affectées négativement par ces produits.

71
Toutes ces étapes doivent être suivies de près pour vérifier si les techniques fonctionnent bien.
Pour cela, il est très courant de doser l’enzyme à purifier après chaque étape. Il faut donc
posséder une méthode de dosage (enzymatique, immunologique, biologique, etc.) pour suivre
le processus. On mesure aussi la quantité totale de protéines. Cette dernière valeur servira à
calculer l'activité spécifique (voir tableau de purification). Dans le cas où il s'agit de la première
fois que l’enzyme est isolée, il faudra développer la méthode de dosage avant même de
développer la méthode de purification.
2. Conservation et rangement des enzymes
Durant toutes les étapes, il faut évidemment éviter de dénaturer l’enzyme qu'on veut isoler. Il
faut donc utiliser des milieux dont les caractéristiques sont compatibles avec la stabilité des
protéines (pH, force ionique, osmolarité, sels, antioxydants, etc.). Pour cela on fabrique certains
milieux plus ou moins "physiologiques". On utilise souvent un tampon destiné à maintenir le
pH (généralement aux alentours de 7.4). Le phosphate buffered saline (PBS) contient un
tampon phosphate pH 7.4. Pour maintenir le pH à un niveau adéquat on inclut généralement un
produit tampon dans les solutions. Le phosphate (en mélange mono- et dibasique) est utilisé
dans le PBS. Le Tris (trishydroxy-méthyl-aminométhane) est très fréquemment utilisé en raison
de son coût peu élevé, même s'il est peu efficace à pH 7.4 et inhibe certaines réactions
physiologiques. L'hepes (hydroxyéthyl-pipérazine-éthane-sulfate) est aussi souvent employé. Il
faut aussi éviter de dénaturer les enzymes en les exposant à l'air ou en les faisant mousser, ce
qui cause une dénaturation et favorise l'oxydation. C'est particulièrement le cas de l'oxydation
de la fonction thiol des cystéines en cystines, i.e. formation d'un lien disulfure. Les enzymes
cytoplasmiques sont particulièrement sensibles car leur milieu naturel, le cytosol, est
légèrement réducteur. Elles supportent donc mal leur solubilisation dans le milieu ambiant qui
est oxydant. L'emploi (à faible concentration et à basse température) d'agents antioxydants
comme le ß-mercapto-éthanol ou un des réactifs de Cleland, dithiothréitol ou
dithiothréitréol, est souvent recommandé pour protéger ces protéines. Les enzymes des
compartiments extracellulaires (sang, liquide céphalo-rachidient, etc.) sont beaucoup moins
susceptibles à ce problème car ce sont des milieux légèrement oxydants. Les enzymes de ces
compartiments ne contiennent pas ou très peu de thiols libres mais plutôt des liens disulfures.
Quand on travaille avec des enzymes en quantités très faibles, il est important d'éviter de
contaminer la préparation avec des protéases. Les sources de protéases peuvent être endogènes,

72
présentes dans la préparation même, par exemple dans les lysosomes qui sont brisés lors du
broyage des cellules ou par la présence de détergents. Il y a également des sources exogènes,
venant de l'extérieur, comme les mains, la salive, etc.
Les enzymes extraites sont souvent peu stables et doivent généralement être conservées
dans des conditions les protégeant de la dégradation. La conservation à basse température
est de rigueur. Certaines enzymes sont beaucoup plus exigeantes que d'autres et doivent
être gardées à -80°C, cependant beaucoup se conservent aux alentours de -20°C. Si les
enzymes sont stables sous forme sèche (en poudre), c'est la meilleure façon de les conserver. Il
est possible aussi de garder des enzymes qui se dénaturent durant la congélation dans une
solution de glycérol. Cette méthode a l'avantage que le glycérol ne dénature pratiquement
jamais les enzymes et ne gèle pas aux températures de l'ordre de -20°C. Certaines
enzymes peuvent aussi être conservées dans une suspension de cristaux de sulfate
d'ammonium. Ces cristaux semblent stabiliser certaines enzymes. Il convient aussi d'éviter de
répéter des cycles de congélation et de décongélation. La façon la plus commune d'éviter ce
problème est de congeler la préparation en petites fractions qui pourront être décongelées une à
la fois. Le rangement dans des solutions de glycérol ou des suspensions de sulfate d'ammonium
permet aussi d'éviter ce problème puisqu'elles restent liquides à -20°C; mais il faudra
probablement se débarrasser de ce glycérol ou de ce sulfate d'ammonium pour travailler avec
ces enzymes. Durant toutes ces étapes, il faut évidemment éviter de dénaturer l’enzyme qu'on
veut isoler. Il faut donc utiliser des milieux dont les caractéristiques sont compatibles avec la
stabilité des enzymes (pH, force ionique, osmolarité, antioxydant, etc.). Pour cela on
fabrique certains milieux plus ou moins "physiologiques", comme le PBS ou le salin, qui
possèdent quelques-unes de ces propriétés. Pour maintenir le pH à un niveau adéquat on inclut
généralement un produit tampon dans les solutions. Il faut aussi éviter de dénaturer les enzymes
en les exposant à l'air ou en les faisant mousser, ce qui cause une dénaturation et favorise
l'oxydation, particulièrement des cystéines en cystine (i.e. formation d'un lien disulfure). Les
enzymes cytoplasmiques sont particulièrement sensibles car leur milieu naturel, le cytosol, est
légèrement réducteur. Elles supportent donc mal une solubilisation dans le milieu ambiant qui
est oxydant. Une oxydation peut non seulement inactiver ou dénaturer les enzymes, mais aussi
causer leur agrégation si la cystéine d’une enzyme forme un lien disulfure avec celle d'une
autre enzyme. L'emploi d'agents antioxydants comme le ß-mercapto-éthanol le dithiothréitol est
recommandé pour protéger ces enzymes particulièrement sensibles. Le dernier est préférable
au ß-mercapto-éthanol, à cause des odeurs désagréables. La présence d'EDTA, pour

73
chélater les ions métalliques, peut être utile car ces derniers accélèrent les réactions d'oxydation
des enzymes. Le travail à basse température, "sur glace", ralentit aussi les processus de
dénaturation. Les opérations d'homogénéisation mécanique et de centrifugation sont
particulièrement à surveiller, car elles causent souvent une surchauffe de l'extrait. Quand on
isole ou on travaille avec de faibles quantités d’enzymes, il est important d'éviter de contaminer
la préparation avec des protéases. Les sources de protéases peuvent être endogènes, présentes
dans la préparation même, par exemple dans les lysosomes} qui sont brisés lors du broyage des
cellules. Certains tissus comme le pancréas en contiennent de grandes quantités. Elles peuvent
aussi être exogènes, provenant de l'extérieur, comme les mains, la salive, etc. Il existe de
nombreuses préparations commerciales d'inhibiteurs ayant des spécificités diverses par rapport
au type de protéase. Ces inhibiteurs peuvent être des produits chimiques simples comme
l'EDTA qui bloque les métalloprotéases. On retrouve aussi des produits chimiques spécialisés,
comme le fluorure de phénylméthylsulfonyle (PMSF) qui inhibe les protéases à sérine. Des
inhibiteurs biologiques sont aussi employés, comme la pepstatine, la a2-macroglobuline, etc.
On peut même se procurer des mélanges contenant un "cocktail" de divers inhibiteurs.
II. TECHNIQUES DE PURIFICATION
La purification d'une protéine est un processus qui prend plusieurs étapes: extraction initiale,
précipitation différentielle, dialyse, chromatographies de divers types, etc.
II.2. Les différentes techniques de chromatographie :
II.2.1. DEFINITION
La chromatographie est une méthode d'analyse physico-chimique, qui sépare les constituants
d'un mélange (les solutés) par entraînement au moyen d'une phase mobile (liquide ou gaz) le
long d'une phase stationnaire (solide ou liquide fixé), grâce à la (ré)partition sélective des
solutés entre ces deux phases. C’est le « phénomène de partage ». Chaque soluté est donc
soumis à une force de rétention (exercée par la phase stationnaire) et une force de mobilité (due
à la phase mobile). La première chromatographie a été réalisée en 1906 par le botaniste russe
Mikhaïl Tswett et consistait à séparer les pigments (en grec : "chromato") d'une feuille
d'épinard (Tswett M. Ber. Dtsch. Chem. Ges., (1906) 24, 316). Tswett avait observé la

74
séparation des colorants végétaux, dont les chlorophylles, lorsqu'il filtrait leur solution dans
l'éther de pétrole, sur une colonne de carbonate de calcium. Dans ces conditions, en effet, des
zones colorées vertes et jaunes se forment. Tswett constatait: "Tout comme les rayons
lumineux d'un spectre, les différentes composantes d'un mélange de colorants se déploient sur
la colonne de carbonate de calcium selon une loi et peuvent être analysées qualitativement et
quantitativement".
Cette invention de la chromatographie d'adsorption par Tswett resta ignorée jusqu'en 1931; où
Kuhn, Winterstein et Lederer (Kuhn R., Lederer E. Ber. Dtsch. Chem. Ges., (1931) 64, 1349 -
Kuhn R., Winterstein A., Lederer E. Hoppe-Seyler's Z. Physiol. Chem. (1931) 197, 141)
l'exploitèrent de nouveau avec succès.
II.2.2. - LES DIFFERENTS TYPES DE TECHNIQUES CHROMATOGRAPHIQUES
Les techniques de chromatographie sont classées en fonction des mécanismes de séparation.
Les facteurs qui interviennent dans le partage des molécules à séparer entre les phases fixe et
mobile sont : la solubilité dans un solvant liquide, la taille (la forme), la polarité, la charge
électrique, la présence de groupements d'atomes formant des sites particuliers. Les différents
types de chromatographie résultent du fait que l'on a privilégié l'effet de l'un de ces facteurs,
mais l'exclusivité d'un mécanisme n'est jamais totale au cours d'une séparation
chromatographique.
- CHROMATOGRAPHIES EN PHASE LIQUIDE A HAUTE PERFORMANCE
(HPLC)
La phase mobile est un liquide sous haute pression. La pression est exercée à l’aide d’une
pompe. Selon la nature de la phase stationnaire, on distingue :
- La chromatographie de partage :C'est une chromatographie liquide-liquide. La phase
stationnaire est un liquide fixé sur un support inerte. Cette chromatographie est ainsi
dénommée car elle est basée sur le partage du soluté dans les deux phases liquides.
La chromatographie d'exclusion :elle est encore appelée chromatographie d'exclusion-
diffusion, tamisage moléculaire, gel-filtration, perméation de gel. La phase stationnaire est un

75
solide poreux : les grosses particules sont exclues de la phase fixe, en revanche les petites
particules incluses diffusent dans les pores du gel.
- les chromatographies d'ADSORPTION :
- La chromatographie d'adsorption : C'est une chromatographie liquide-solide. La phase
stationnaire est un adsorbant solide polaire.
- La chromatographie d'adsorption en phase inverse : C'est une chromatographie liquide-
solide dans laquelle la phase stationnaire est apolaire.
- La chromatographie sur échangeurs d'ions :La phase stationnaire est un échangeur d'ions
constitué par une résine porteuse de groupements ionisés négativement ou positivement,
exerçant des interactions de type électrostatique avec les solutés ioniques du milieu.
- La chromatographie d'affinité : La phase stationnaire est un support macromoléculaire
chimiquement inerte, sur lequel est greffé un effecteur qui présente une affinité biologique
(bio-affinité) pour un soluté de l'échantillon à analyser (affinité enzyme-substrat, ligand-
récepteur, antigène-anticorps).
.1.1 - LA CHROMATOGRAPHIE DE PARTAGE :Principe :
Cette chromatographie liquide-liquide est fondée sur la (ré)partition différentielle de chacun
des solutés entre deux liquides non miscibles, l'un constituant la phase stationnaire, l'autre la
phase mobile. C'est une technique essentiellement qualitative.
Théorie de la partition : La distribution d'un soluté A entre deux solvants non-miscibles, l'un
aqueux ( Saq ), l'autre organique ( Sorg ), relève de l'équilibre :
La constante d'équilibre (qui est le coefficient de partage) est égale à K :

76
C'est l'équation de Nernst pour le partage d'une substance entre 2 phases liquides-non miscibles.
[ASorg] : concentrations en soluté A dans la phase organique
[ASaq] : les concentrations en soluté A dans la phase aqueuse.
Noter qu'ici, nous avons un soluté qui est plus miscible dans le solvant organique que dans le
solvant aqueux (ASorg est le numérateur et ASaq le dénominateur de l'équation). K aura donc une
valeur supérieure à 1. Cette constante d'équilibre à une température donnée est appelée
coefficient de partage ou de distribution. La transposition de cette partition d'un composé entre
deux phases liquides dans un système chromatographique est rendue possible par la fixation de
l'un des « solvants » sur un support inerte, l'autre solvant constituant la phase mobile. Un
soluté très soluble dans la phase fixée migrera lentement, la force de rétention prédominant sur la
force d'entraînement. A l'inverse, un soluté soluble dans la phase mobile migrera rapidement.
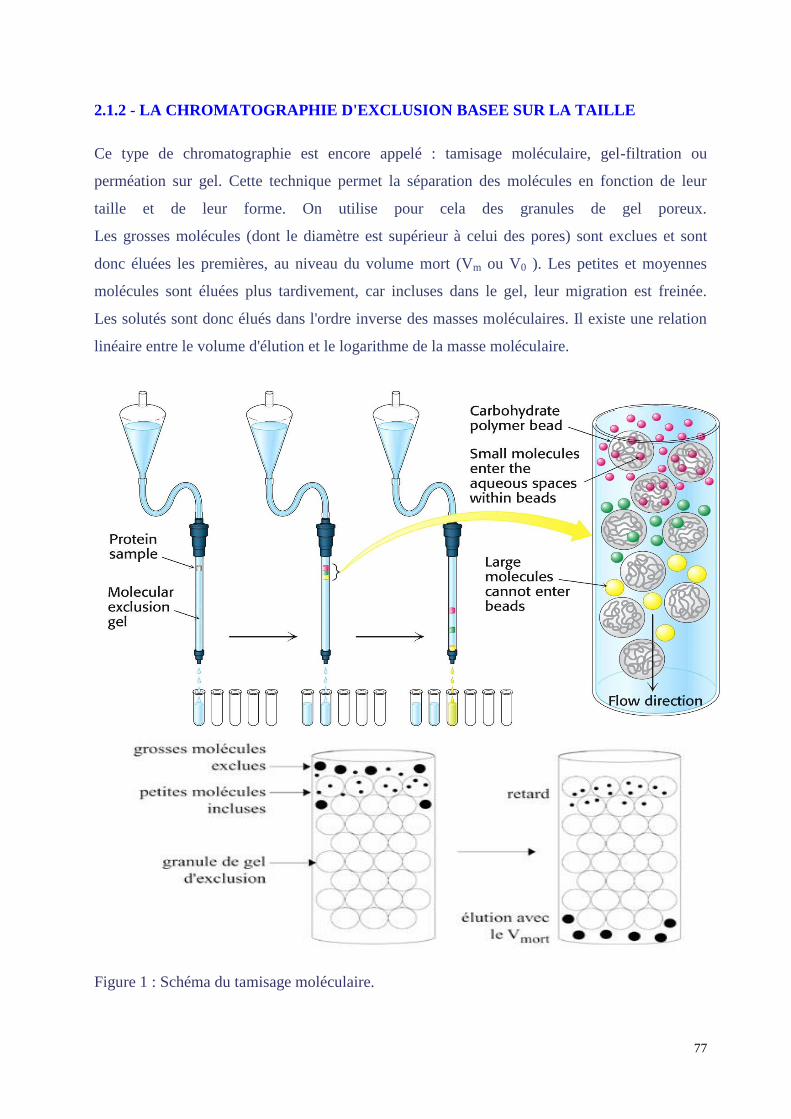
77
2.1.2 - LA CHROMATOGRAPHIE D'EXCLUSION BASEE SUR LA TAILLE
Ce type de chromatographie est encore appelé : tamisage moléculaire, gel-filtration ou
perméation sur gel. Cette technique permet la séparation des molécules en fonction de leur
taille et de leur forme. On utilise pour cela des granules de gel poreux.
Les grosses molécules (dont le diamètre est supérieur à celui des pores) sont exclues et sont
donc éluées les premières, au niveau du volume mort (Vm ou V0 ). Les petites et moyennes
molécules sont éluées plus tardivement, car incluses dans le gel, leur migration est freinée.
Les solutés sont donc élués dans l'ordre inverse des masses moléculaires. Il existe une relation
linéaire entre le volume d'élution et le logarithme de la masse moléculaire.
Figure 1 : Schéma du tamisage moléculaire.

78
Types de gels :
Gels hydratés (les plus utilisés) : le SéphadexTM
(G10 à G200) qui sont des polyosides
bactériens de type dextran (poly D- -> 6), et le SépharoseTM
(agarose). Le gain d'eau correspond au nombre de grammes d'eau fixés par gramme de
substance sêche.
Gels permanents : ce sont des copolymères organiques ou bien des minéraux (silice).
Ici, des effets d'adsorption s'ajoutent au tamisage moléculaire.
Figure 2 : Représentation d'un gel d'exclusion.
Les solutés sont donc élués dans l'ordre inverse des masses moléculaires (voir figure ci-
dessous).
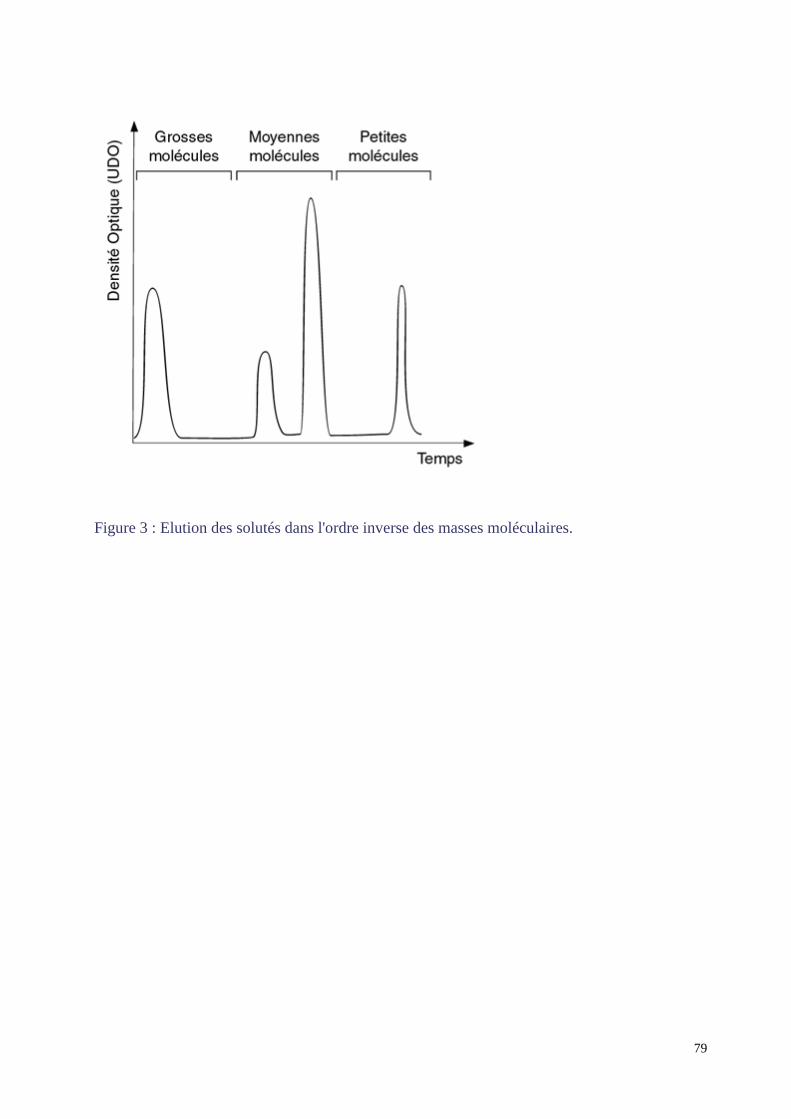
79
Figure 3 : Elution des solutés dans l'ordre inverse des masses moléculaires.
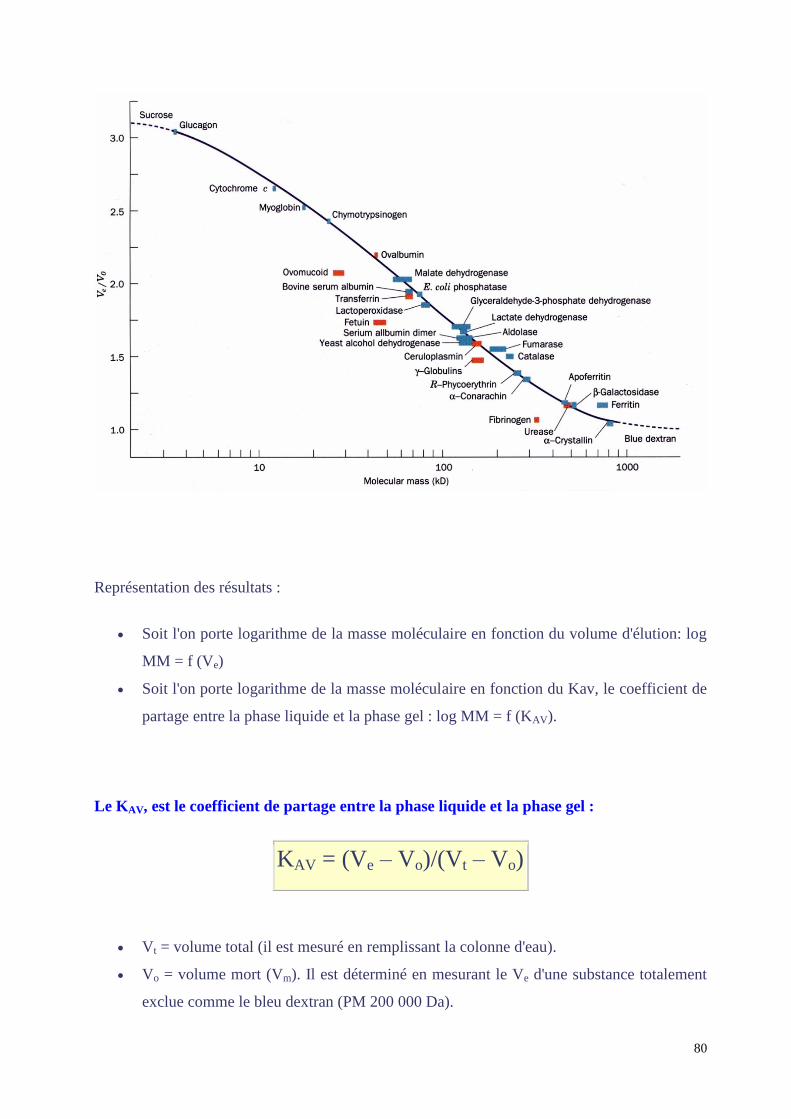
80
Représentation des résultats :
Soit l'on porte logarithme de la masse moléculaire en fonction du volume d'élution: log
MM = f (Ve)
Soit l'on porte logarithme de la masse moléculaire en fonction du Kav, le coefficient de
partage entre la phase liquide et la phase gel : log MM = f (KAV).
Le KAV, est le coefficient de partage entre la phase liquide et la phase gel :
KAV = (Ve – Vo)/(Vt – Vo)
Vt = volume total (il est mesuré en remplissant la colonne d'eau).
Vo = volume mort (Vm). Il est déterminé en mesurant le Ve d'une substance totalement
exclue comme le bleu dextran (PM 200 000 Da).
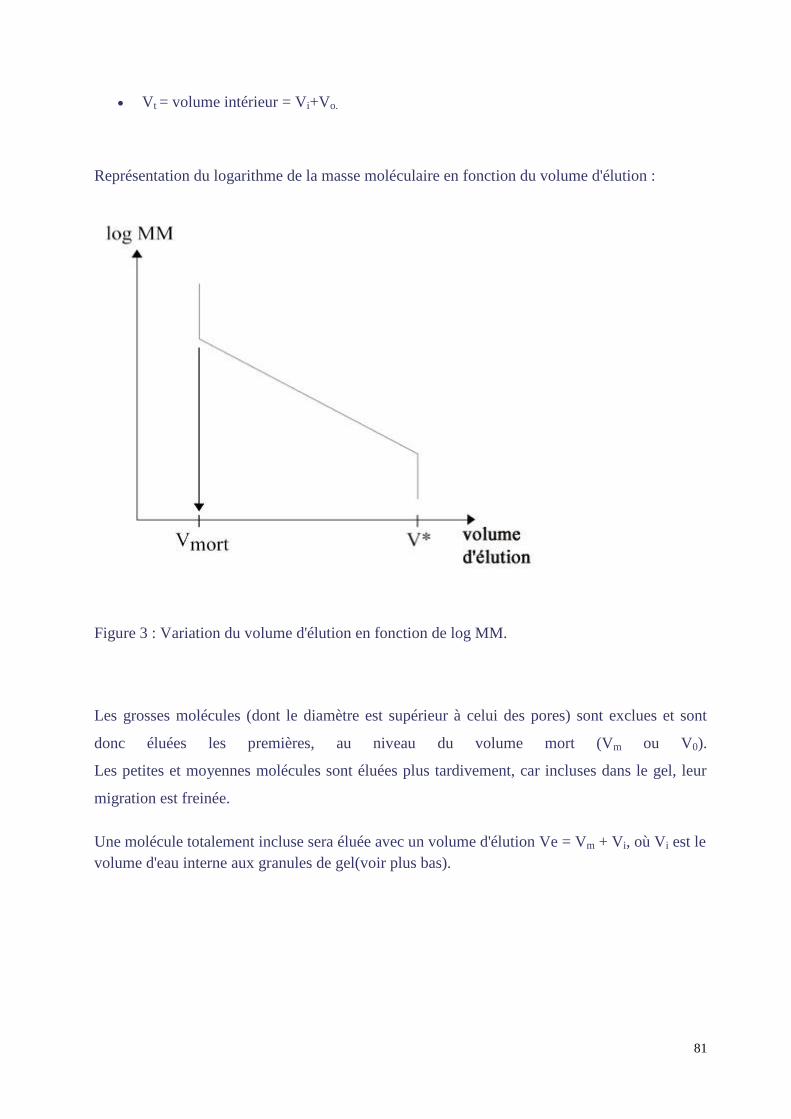
81
Vt = volume intérieur = Vi+Vo.
Représentation du logarithme de la masse moléculaire en fonction du volume d'élution :
Figure 3 : Variation du volume d'élution en fonction de log MM.
Les grosses molécules (dont le diamètre est supérieur à celui des pores) sont exclues et sont
donc éluées les premières, au niveau du volume mort (Vm ou V0).
Les petites et moyennes molécules sont éluées plus tardivement, car incluses dans le gel, leur
migration est freinée.
Une molécule totalement incluse sera éluée avec un volume d'élution Ve = Vm + Vi, où Vi est le
volume d'eau interne aux granules de gel(voir plus bas).

82
2.1.4 - LA CHROMATOGRAPHIE D'ADSORPTION EN PHASE INVERSÉES
Principe :
C'est une chromatographie d'adsorption liquide-solide dans laquelle la phase stationnaire se
distingue par son apolarité. Elle est constituée, la plupart du temps, par des silices apolaires
greffées. Il existe des greffons apolaires de taille différente, de 2 à 18 atomes de carbone (C2 à
C18), donc de polarités différentes. La phase mobile est polaire et hydrophile. Les séparations
sont fondées sur des interactions hydrophobes entre les molécules à séparer et la phase
stationnaire. Ainsi, plus un soluté est apolaire, plus il sera retenu au niveau de la phase solide
stationnaire. A l'inverse, plus un soluté est polaire, plus il sera entraîné par la phase mobile
liquide.

83
Figure 5 : Silices polaire et apolaires greffées.
Technologie : Dans la pratique, cette chromatographie ne s'applique qu'en HPLC, elle porte le
nom de RP-HPLC (ou Reverse Phase HPLC, ou HPLC en phase inverse). La RP-HPLC
s'applique bien à la séparation de petites molécules apolaires : lipides, acides aminés, peptides.

84
2.1.5 - LA CHROMATOGRAPHIE SUR ECHANGEURS D'IONS
Principe :
Les échangeurs d'ions sont des macromolécules insolubles portant des groupements ionisables
qui ont la propriété d'échanger de façon réversible certains de leurs ions, au contact d'autres
ions provenant d'une solution.
La chromatographie par échange d'ions se pratique le plus souvent sur colonne, mais la
méthode peut être transposée sur couche mince. Du papier échangeur d'ions est également
commercialisé. Enfin l'échange d'ions peut être réalisé en batch : la résine est mise en présence
de la solution et agitée mécaniquement.
Propriétés des échangeurs d'ions : Le support peut être minéral, mais il est le plus souvent
organique (résine de copolymérisation, polyosides : dextrans ou celluloses - voir au verso). Les
groupements fonctionnels chargés des résines sont fixés par covalence sur le support.
Capacité de rétention d'un échangeur d'ions : C'est le nombre de millimoles (mmol) d'ions
que la résine peut échanger par unité de masse. On l'exprime par gramme de résine sèche (ou
moins souvent par unité de volume : par ml de résine humide).
Elution : L'élution consiste à déplacer l'ion fixé par un autre, de densité de chargé et de
concentration plus élevée on utilise de petits ions fortement chargés : Cl-, HO
-, Na
+, H
+...
Résine cationique : qui échange réversiblement des cations (ions positifs). Une résine
cationique est chargée négativement.
Résine-G- / X
+ + cation
+ <=> Résine-G
- / cation
+ + X
+
Figure 6 : Résine échangeuse de cations (résine cationique).

85
Les groupements fonctionnels des résines cationiques (résines classées d'après leurs
aptitudes à l'ionisation) :
Résines cationiques fortes :
o sulfoniques (très fortement ionisées, quel que soit le pH) :
Résine sous forme acide : Résine-
SO3- / H
+ (contre-ion : H
+)
Résine sous forme sodique : Résine
SO3- / Na
+ (contre-ion : Na
+)
Résines cationiques intermédiaire :
o à groupement phospho :
Résine-H2PO4- / H
+ Résine-H2PO4
- / Na
+
Résines cationiques faibles :
o carboxyliques (non ionisées en milieu fortement acide) :
Résine-COO- / H
+ Résine-COO
- / Na
+
o à groupement carboxyméthyl :
Résine-CH2-COO- / H
+ Résine-CH2-COO
- / Na
+
Résines cationiques très faibles :
o phénoliques (uniquement ionisées en milieu alcalin) :
Résine-O- / H
+ Résine-O
- / Na
+

86
Exemple de résine échangeuse d'ions
Les résines polyosidiques chargées de type Sephadex : Ces échangeurs sont utilisés pour la
séparation de macromolécules ionisées : protéines, acides nucléiques, etc. La matrice
polyosidique (composée de cellulose ou de dextranes) porte les groupements chargés suivants :
Exemples d’échangeurs cationiques Sephadex :

87
Résines anioniques: qui échangent réversiblement des anions (ions négatifs). Une résine
anionique est chargée positivement.
Résine-G+ / Y
- + anion
- <=> Résine-G
+ / anion
- + Y
-
Figure 7: Résine échangeuse d'anions (résine anionique).
Les Groupements fonctionnels des résines anioniques :
Résines anioniques fortes :
o résines à groupements aminés quaternaires.
o résines à groupements aminés tertiaires.
Résines anioniques faibles :
o résines à groupements aminés secondaires et primaires.
Echangeurs anioniques Sephadex :

88
2.1.6 - LA CHROMATOGRAPHIE D'AFFINITÉ :
Principe :
Dans ce type de chromatographie, la phase stationnaire est un support macromoléculaire
chimiquement inerte sur lequel est greffé un effecteur qui présente une affinité biologique pour
un soluté de l'échantillon à analyser.
Quatre types d'affinités sont utilisées :
affinité enzyme-substrat
affinité enzyme-ligand (pouvant être un inhibiteur)
affinité ligand-récepteur
affinité antigène-anticorps
Très souvent, la molécule fixée sera le substrat, le ligand, ou bien l'anticorps. Ceci permettra de
purifier l'enzyme, le récepteur ou l'antigène, respectivement.

89
Figure 1 : Les trois étapes d'une chromatographie d'affinité.
1 - Etape de FIXATION : Le mélange de molécules contenant le composé à purifié est chargé
sur la colonne d'affinité. Seule la molécule présentant une affinité pour la colonne sera retenue
par l'effecteur greffé sur la phase stationnaire.
2 - Etape de PURIFICATION : En continuant à faire passer du tampon dans la colonne,
toutes les molécules contaminantes sont éliminées et éluées.
3 - Etape d'ELUTION : La molécule purifiée est décrochée de la colonne et est recueillie dans
l'éluant. Souvent, l'un des deux partenaires de l'interaction au moins est une protéine (P), l'autre
sera qualifié de ligand (L) de cette protéine :
P + L <=> PL
L'association de P et de L forme le complexe PL. La constante définissant cet équilibre est :
Ka = (P).(L) / (PL)
NB : C'est une constante d'association à l'équilibre, qui s'écrit donc avec un "K" majuscule.
La phase stationnaire (le gel d'affinité) : elle est constituée d'un effecteur fixé par covalence
à un support (carboxyméthylcellulose, Séphadex, gel de polyacrylamide) par l'intermédiaire

90
d'un bras de fixation ("spacer" en anglais). Exemples de dérivés de la
carboxyméthylcellulose ou CM-cellulose :
La CM-aminohexylique : permet la fixation d'un effecteur à fonction carboxyle.
- O - CH2 - CO - NH - (CH2)6 - NH2 (spacer long)
La CM-hydrazide : permet la fixation d'un effecteur à fonction carboxyle.
- O - CH2 - CO - NH - NH2 (spacer court)
La CM- aminohexylique (ou aminododécylique) succinylée : permet la fixation d'un
effecteur à fonction -NH2 réactive.
- O - CH2 - CO - NH - (CH2)n -NH - CO - CH2 - CH2 - COOH (n = 6 ou 12, spacer
long)
.La longueur du bras est choisie de manière à limiter les contraintes stériques.
Effecteurs :
Affinité enzyme-substrat : substrats, analogues, inhibiteurs réversibles, effecteurs
allostériques, coenzymes.
Affinité ligand-récepteur : haptènes, antigènes, anticorps.
Affinité antigène-anticorps : hormones, peptides, analogues peptidiques.
Elution : elle peut être réalisée de différentes façons :
Tampon de pH différent de celui ayant permis la charge : changement de l'état
d'ionisation d la protéine : désorption
Tampon de force ionique différente de celle ayant permis la charge : changement de
conformation de la protéine.
Compétition avec un ligand libre.
BILAN DE LA PURIFICATION
Tableau de purification
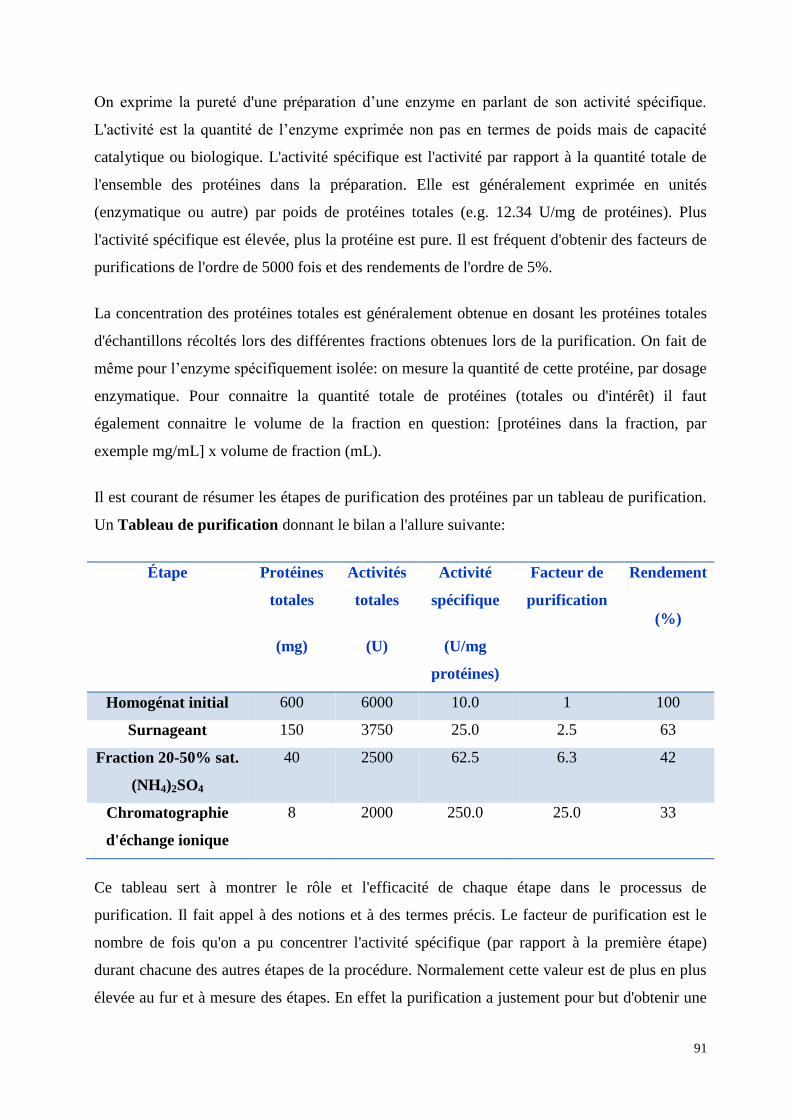
91
On exprime la pureté d'une préparation d’une enzyme en parlant de son activité spécifique.
L'activité est la quantité de l’enzyme exprimée non pas en termes de poids mais de capacité
catalytique ou biologique. L'activité spécifique est l'activité par rapport à la quantité totale de
l'ensemble des protéines dans la préparation. Elle est généralement exprimée en unités
(enzymatique ou autre) par poids de protéines totales (e.g. 12.34 U/mg de protéines). Plus
l'activité spécifique est élevée, plus la protéine est pure. Il est fréquent d'obtenir des facteurs de
purifications de l'ordre de 5000 fois et des rendements de l'ordre de 5%.
La concentration des protéines totales est généralement obtenue en dosant les protéines totales
d'échantillons récoltés lors des différentes fractions obtenues lors de la purification. On fait de
même pour l’enzyme spécifiquement isolée: on mesure la quantité de cette protéine, par dosage
enzymatique. Pour connaitre la quantité totale de protéines (totales ou d'intérêt) il faut
également connaitre le volume de la fraction en question: [protéines dans la fraction, par
exemple mg/mL] x volume de fraction (mL).
Il est courant de résumer les étapes de purification des protéines par un tableau de purification.
Un Tableau de purification donnant le bilan a l'allure suivante:
Étape Protéines
totales
(mg)
Activités
totales
(U)
Activité
spécifique
(U/mg
protéines)
Facteur de
purification
Rendement
(%)
Homogénat initial 600 6000 10.0 1 100
Surnageant 150 3750 25.0 2.5 63
Fraction 20-50% sat.
(NH4)2SO4
40 2500 62.5 6.3 42
Chromatographie
d'échange ionique
8 2000 250.0 25.0 33
Ce tableau sert à montrer le rôle et l'efficacité de chaque étape dans le processus de
purification. Il fait appel à des notions et à des termes précis. Le facteur de purification est le
nombre de fois qu'on a pu concentrer l'activité spécifique (par rapport à la première étape)
durant chacune des autres étapes de la procédure. Normalement cette valeur est de plus en plus
élevée au fur et à mesure des étapes. En effet la purification a justement pour but d'obtenir une

92
protéine de plus en plus pure, ayant donc une préparation dont l'activité spécifique de plus en
plus élevée. Le rendement de la purification est la proportion, en %, de la quantité de la
protéine purifiée qui reste par rapport à la quantité initiale. Le rendement diminue à chaque
étape puisqu'il est inévitable qu'on ait des pertes de matériel à chacune. Une purification est
donc un "compromis" entre le désir d'obtenir une protéine la plus pure possible (facteur de
purification élevé) en quantité maximale (rendement élevé). En effet, chaque étape
supplémentaire, qui permet d'augmenter la pureté, cause des pertes de matériel, donc diminue le
rendement. Au cours d'une purification typique le facteur de purification augmente au fur et à
mesure des étapes tandis que, parallèlement, le rendement diminue. Pour construire un tel
tableau, on dose à chaque étape de purification la quantité de protéines totales et l'activité
volumique de la protéine en question. Généralement il s'agit d'une enzyme et son activité est
exprimée en unités enzymatiques. D'autres unités peuvent servir à définir des protéines sans
activité enzymatique. Sachant le volume de chaque fraction, la concentration des protéines
totales et l'activité volumique, il devient facile de construire le tableau de purification.
On peut obtenir les valeurs du tableau précédent avec les données suivantes. Pour chacune des
fractions qu'on a obtenues lors de la purification, on a mesuré le volume et prélevé des
aliquotes pour doser les protéines totales et l'activité de la protéine qu'on voulait isoler. Par
exemple, l'homogénat du tableau a un volume de 150 mL et contient 4 mg de protéines/mL et
40 U d'enzyme/mL. Cela permet d'obtenir la quantité de protéines totale (150 mL x 4 mg/mL =
600 mg de protéines totales), l'activité totale (40 U/mL x 150 mL = 6000 U) et l'activité
spécifique (6000 U/600 mg = 10 U/mg). De la même façon on obtient les valeurs des autres
fractions. La purification et le rendement se déduisent ensuite facilement. Ainsi pour le
surnageant, on peut calculer le facteur de la purification (25 U/mg/10 U/mg = 2.5X) et le
rendement (3750 U/6000 U = 62.5%).

93
CHAPITRE 6 .
CINETIQUE ENZYMATIQUE

94
I. Définition de la cinétique enzymatique
Introduction
La cinétique enzymatique est l’étude des variations des vitesses de la réaction en fonction
de la concentration du substrat et du temps.
La cinétique sert à la:
- Modélisation de l’activité enzymatique
- L’élaboration de l’Équation de vitesse
- Déterminer les paramètres cinétiques et leur signification
- l’Etudes en présence d’inhibiteurs, conditions optimales d’activité de l’enzyme.
10-15
10-10
10-5
105
1010
1
4 billion years
1 million years
100 years
1 week
1 minute
ADC
STN
FUM
PEP
CMU
CAN
ADC = arginine decarboxylase
STN = staphylococal nuclease
FUM = fumarase
PEP = carboxypeptidase B
CMU = chorismate mutase
CAN = carbonic anhydrase
kcat/KM
(s-1 M-1)
Source: R. Wolfenden 2001
Acc. Chem. Res.
knon
(s-1)
t 1/2 =
Les enzymes sont des catalyseurs
extrêmement efficace
Elle peuvent accélérer une réaction des milliards de milliards de fois

95
La vitesse d'une réaction enzymatique se mesure en dosant le produit apparu ou le substrat
disparu sous l'action de l'enzyme par unité de temps. L'activité moléculaire d'un enzyme est le
nombre de molécules de substrat transformés par minute par une molécule d'enzyme dans les
conditions optimales.
II. Notion d’ordre d’une réaction.
L’ordre d’une réaction tient compte des interactions entre les molécules qui réagissent au cours
de la réaction. L’ordre de la réaction n’est pas obligatoirement la somme des coefficients
stochiométriques de la réaction. On peut dire, grosso modo, que pour un réactant, plus son
ordre partiel est important, plus ce composé influence la cinétique de la réaction. L’ordre global
de la réaction est la somme des ordres partiels.
Considérons une réaction dont l'équation bilan s'écrit :
αA + βB → γC
A et B sont les réactifs et C est l'unique produit et α, β et γ sont les coefficients
stœchiométriques.
La vitesse de réaction dans un réacteur de volumen constan test est définie par :
𝐯 = −𝒅[𝑨]
𝒅𝒕= −
𝒅[𝑩]
𝒅𝒕=
𝒅[𝑪]
𝒅𝒕
Cette vitesse est fonction des concentrations des réactifs. L'étude expérimentale de la réaction
permet d'établir la forme de la loi de vitesse.
Dans certains cas, la loi de vitesse peut se mettre sous la forme d'un monôme des réactifs :
𝐯 = 𝒌[𝑨]𝜶′[𝑩]𝜷′
On dit alors que la réaction admet un ordre et l'on appelle ordre de réaction la somme r des
exposants : r = α’ + β’. L'exposant α’ est l'ordre partiel de la réaction par rapport à l'espèce
reactante A, β’ est l'ordre partiel de la réaction par rapport à l'espèce reactante B.
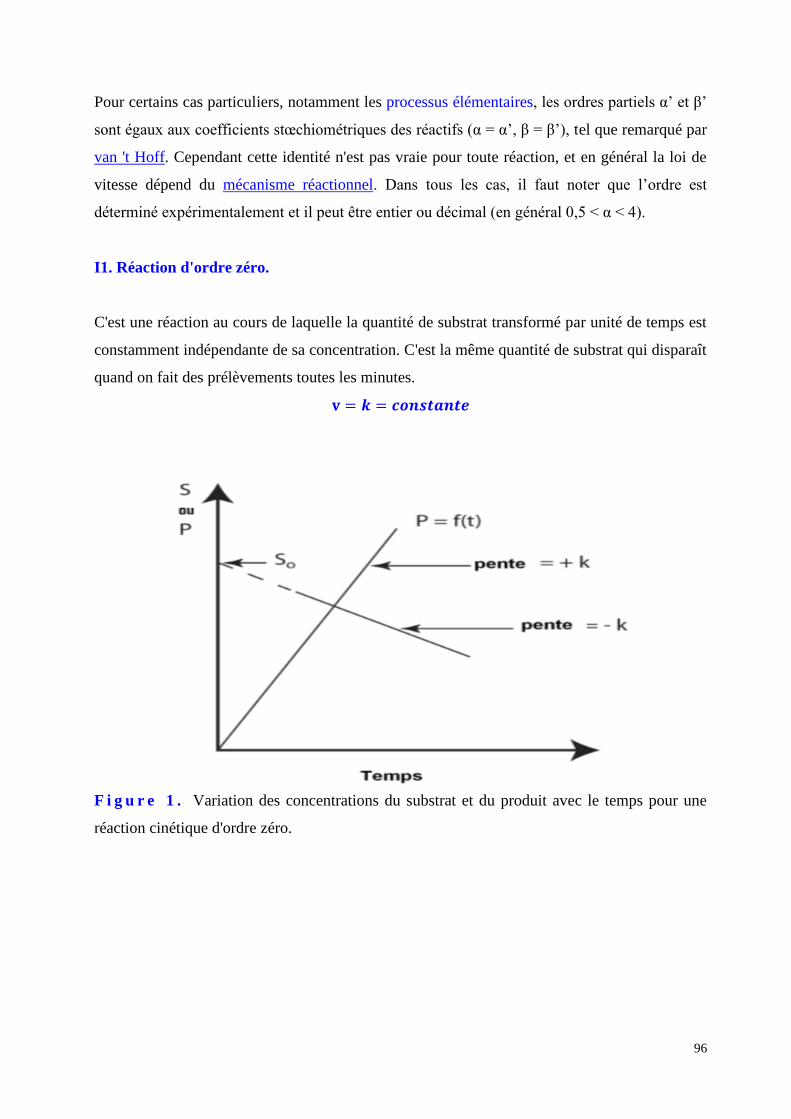
96
Pour certains cas particuliers, notamment les processus élémentaires, les ordres partiels α’ et β’
sont égaux aux coefficients stœchiométriques des réactifs (α = α’, β = β’), tel que remarqué par
van 't Hoff. Cependant cette identité n'est pas vraie pour toute réaction, et en général la loi de
vitesse dépend du mécanisme réactionnel. Dans tous les cas, il faut noter que l’ordre est
déterminé expérimentalement et il peut être entier ou décimal (en général 0,5 < α < 4).
I1. Réaction d'ordre zéro.
C'est une réaction au cours de laquelle la quantité de substrat transformé par unité de temps est
constamment indépendante de sa concentration. C'est la même quantité de substrat qui disparaît
quand on fait des prélèvements toutes les minutes.
𝐯 = 𝒌 = 𝒄𝒐𝒏𝒔𝒕𝒂𝒏𝒕𝒆
F i g u r e 1 . Variation des concentrations du substrat et du produit avec le temps pour une
réaction cinétique d'ordre zéro.
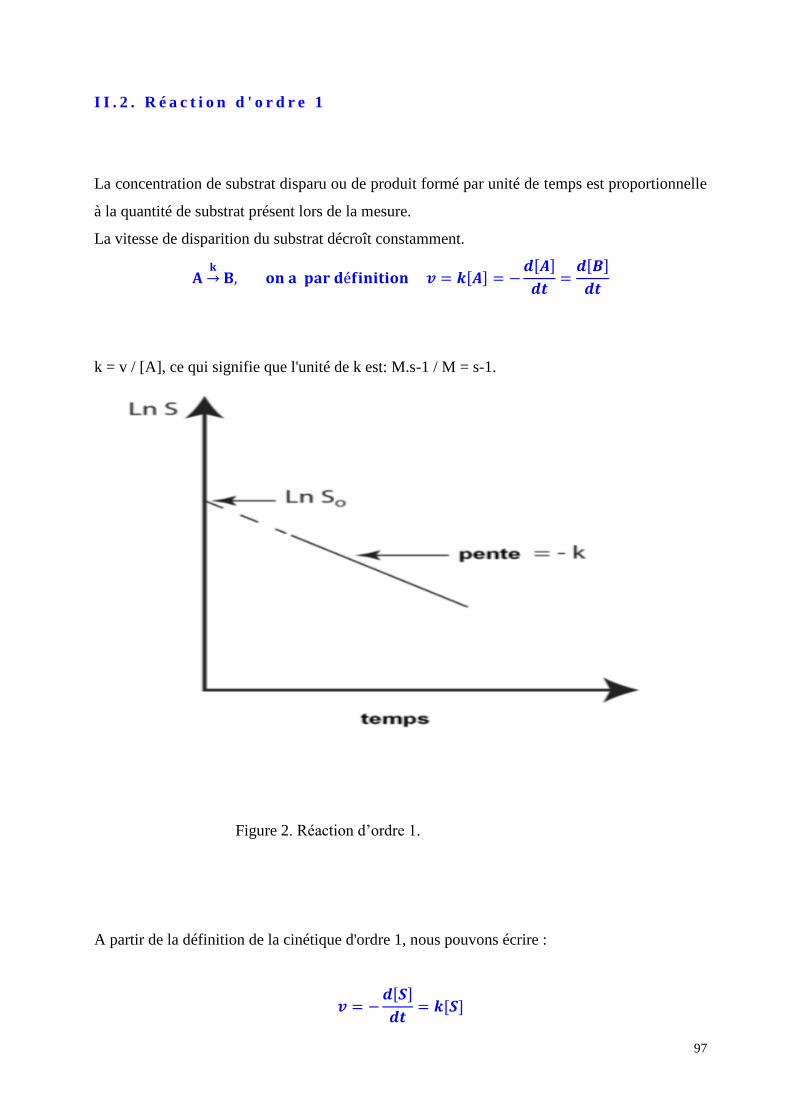
97
I I . 2 . R é a c t i o n d ' o r d r e 1
La concentration de substrat disparu ou de produit formé par unité de temps est proportionnelle
à la quantité de substrat présent lors de la mesure.
La vitesse de disparition du substrat décroît constamment.
𝐀𝐤→ 𝐁, 𝐨𝐧 𝐚 𝐩𝐚𝐫 𝐝é𝐟𝐢𝐧𝐢𝐭𝐢𝐨𝐧 𝒗 = 𝒌[𝑨] = −
𝒅[𝑨]
𝒅𝒕=
𝒅[𝑩]
𝒅𝒕
k = v / [A], ce qui signifie que l'unité de k est: M.s-1 / M = s-1.
Figure 2. Réaction d’ordre 1.
A partir de la définition de la cinétique d'ordre 1, nous pouvons écrire :
𝒗 = −𝒅[𝑺]
𝒅𝒕= 𝒌[𝑺]

98
Au temps t = 0, cte = - Ln[So]. Ainsi, l'équation précédente peut être écrite:
𝐋𝐧[𝐒] = −𝒌𝐭 + 𝐋𝐧[𝐒𝐨]
Le tracé de Ln S en fonction de t, donne une droite de pente égale à – k et de point
d'intersection sur l'axe des Y égal à Ln[So].
L'équation précédente peut être modifiée et écrite sous la forme suivante:
[𝐒] = [𝑺𝒐]𝒆−𝒌𝒕
Donc, S décroît exponentiellement avec le temps.
Pour [S]= [So]/2, nous obtenonst1/2= Ln2
𝒕𝟏/𝟐 =𝑳𝒏𝟐
𝒌= 𝟎, 𝟔𝟗𝟑/𝒌
Où t½ est appelé le temps de la demi-réaction ou demi-vie.
II.3. Réaction du second ordre (ou d'ordre 2)
Soit une réaction élémentaire : A + A P
Si la réaction est d'ordre 2 par rapport à A, la vitesse de réaction s'écrit :
v = -d[A]/dt = k[A]2 , d'où 𝒅
[𝑨]
[𝑨]𝟐=kdt et en intégrant on obtient :
𝟏
[𝑨]= 𝒌𝒕 +
𝟏
[𝑨]𝒐
où [A]0 est la concentration de A à l'instant t0= 0.
La concentration des réactifs diminue hyperboliquement en fonction du temps, et évidemment
celle des produits augmente hyperboliquement en fonction du temps.

99
Une représentation de 1/[A] = f(t) est une droite de pente k Pour une réaction d'ordre deux, la
constante de vitesse k a les dimensions de (M-1 s-1).
II.4. Détermination de l'ordre d'une réaction (méthode différentielle)
Dans les paragraphes précédents, pour chaque ordre de réaction, nous avons déterminé une
représentation particulière f[A]=g(t) qui est linéaire et permet de déterminer la constante de
vitesse. Si nous avons non pas des mesures expérimentales ([A], t) mais des mesures (v, [A])
où v est la vitesse de la réaction, la relation à étudier est v =-d[A]/dt = k[A]n où n est l'ordre de
la réaction :
𝐕 = 𝒌[𝑨]𝒏 ⇒ 𝐥𝐧𝑽 = 𝐥𝐧𝒌 + 𝒏𝒍𝐧[𝑨]
Une représentation de Ln(v) en fonction de Ln([A]) est une droite de pente n (ordre de la
réaction) et d'ordonnée à l'origine Ln(k).
Remarque : si à la place des logarithmes népériens, nous avions utilisé les logarithmes
décimaux (Log), la relation aurait été identique.

100
I I I . C i n é t i q u e e n z y m a t i q u e
Dès 1835, Jöns Jacob Berzelius appela catalyse l'augmentation de la vitesse d'une réaction due
à la présence de substances particulières qui sont inchangées à la fin de la réaction.
En 1895 Wilhelm Ostwald définit un catalyseur comme :
- une substance qui modifie la vitesse d'une réaction sans modifier le bilan énergétique global
- et ajouta en 1902 : une substance que l'on retrouve inchangée à la fin de la réaction. On dit
que la catalyse est homogène si le catalyseur fait partie de la même phase que le système
réactionnel, et dans le cas contraire on parle de catalyse hétérogène.
Un catalyseur est une substance :
1) que l'on retrouve inchangée à la fin de la réaction ;
2) qui modifie la vitesse des réactions thermodynamiquement possibles puisqu'il ne modifie pas
l'énergie du système réactionnel ;
3) qui modifie de la même manière la vitesse de deux réactions opposées selon la règle de
réversibilité, et permet d'atteindre plus rapidement un équilibre chimique.
III. 1. Mécanisme général d'une enzyme
S PE
Une enzyme (E) transforme un substrat (S) en produit (P).
S ES
-Première étape. Le substrat se fixe sur l'enzyme
-La deuxième étape: la catalyse . Le substrat est transformé en produit
EPES
-Troisième étape. Le produit est relâché
EP E+P
K1
K-1
K2
K-2
K3
K-3
Si il y a plusieurs substrats. La réaction se fera étape par étape (et non simultanément)
pour être efficace. C'est une succession de réaction du premier ordre.
On revient toujours à ce système générale
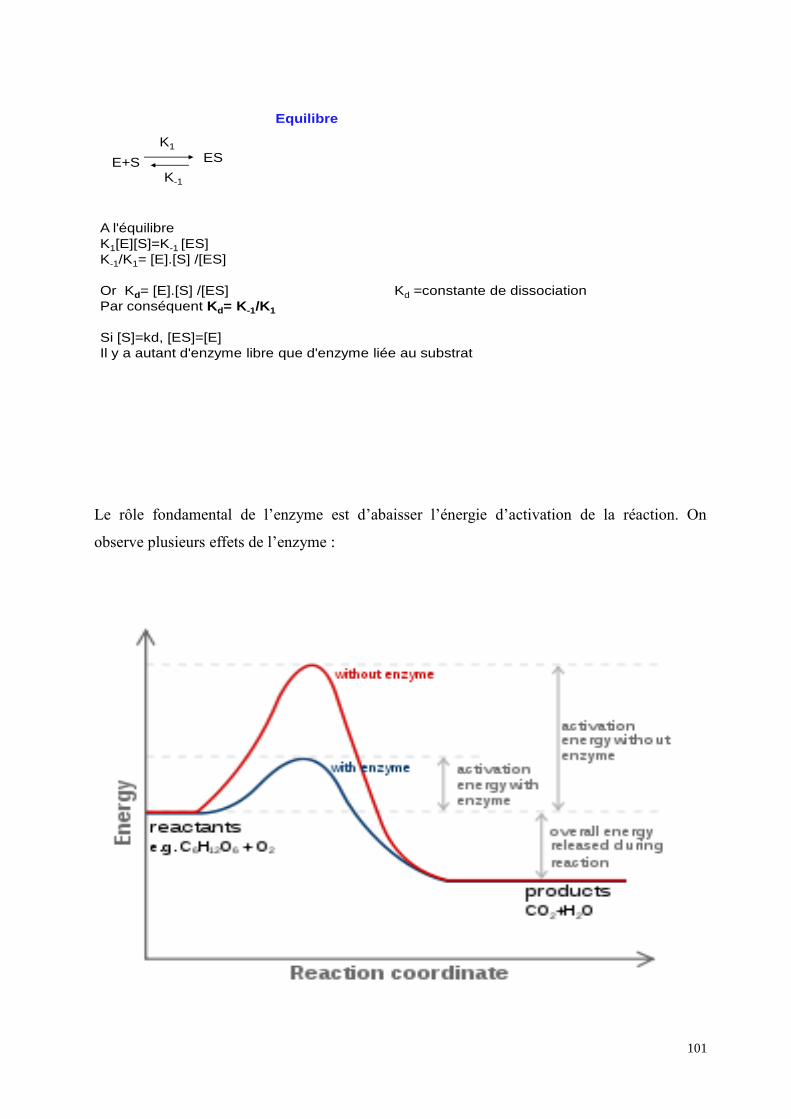
101
Equilibre
A l'équilibre
K1[E][S]=K-1 [ES]
K-1/K1= [E].[S] /[ES]
Or Kd= [E].[S] /[ES] Kd =constante de dissociation
Par conséquent Kd= K-1/K1
Si [S]=kd, [ES]=[E]
Il y a autant d'enzyme libre que d'enzyme liée au substrat
E+SK-1
K1
ES
Le rôle fondamental de l’enzyme est d’abaisser l’énergie d’activation de la réaction. On
observe plusieurs effets de l’enzyme :

102
Effet entropique
Les substrats sont fixés. Il y a augmentation de la concentration locale des réactifs et comme les
substrats sont correctement orientés pour la réaction, il y a plus de chance qu'ils réagissent.
C'est un effet entropique favorable.
Effet énergétique
L'enzyme peut parfois déstabiliser le substrat par déformation géométrique, polarisation d'une
liaison, attaque d'une liaison, etc.
Stabilisation de l'état intermédiaire: charge, liaisons,…
Le produit est formé.
Souvent, le produit a une affinité plus faible que le substrat.
De plus, au début de la réaction comme il n'y a pas encore de produit dans la solution, le
produit sortira plus facilement (effet entropique)
Comme pour l'arrivée du substrat, le produit pour sortir doit franchir une barrière d'énergie.
Déplacement de molécules d'eau, mouvements de la protéine.Quelque soit le chemin, à la fin
S
P
Sans enzyme
Avec Enzyme
ES
EP
DGact, l'énergie d'activation est diminué, la
réaction est fortement accélérée.
L'équilibre n'est pas changé
E+S ESK1
K-1
K2
K-2
EPK2
K-2
E+P

103
on récupère l'énergie de la réaction. Cette énergie peut être couplée à d'autres réactions non
favorables (ex ATP).
Les états ES et EP sont métastable et peuvent être piégé. (concentration P et S, T°, …)
Les autres états intermédiaires sont beaucoup plus difficile à piéger (cryoenzymologie).
Par microcalorimétrie on peut facilement mesurer l'enthalpie (∆H) pour la fixation du substrat,
du produit ou de la réaction).
Par contre il est difficile d'avoir accès à l'enthalpie libre (∆G). Il faudra avoir accès au désordre
((changement de conformation, déplacement des molécules d'eau,…)
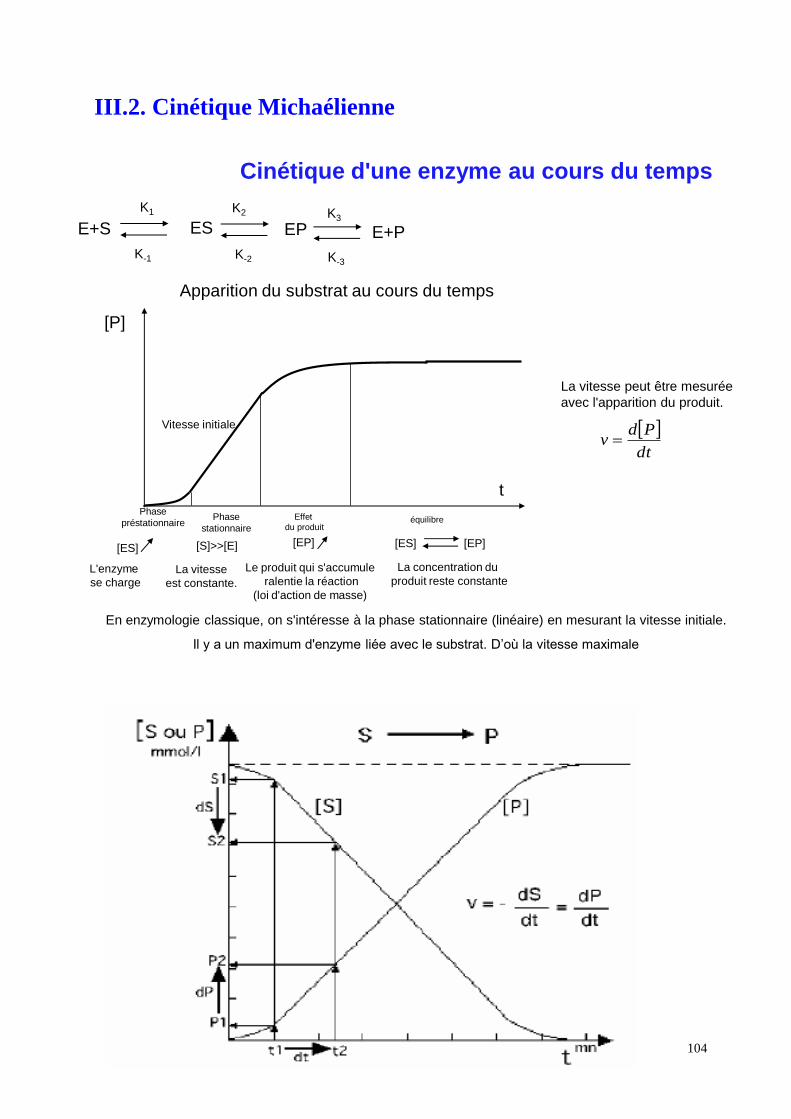
104
III.2. Cinétique Michaélienne
Cinétique d'une enzyme au cours du temps
E+S ES
K-1 K-2
EPK3
K-3
E+P
Phase
préstationnaire
[ES]
[P]
Apparition du substrat au cours du temps
L'enzyme
se charge
Phase
stationnaire
[S]>>[E]
La vitesse
est constante.
Effet
du produit
[EP]
Le produit qui s'accumule
ralentie la réaction
(loi d'action de masse)
équilibre
t
[ES] [EP]
La concentration du
produit reste constante
Vitesse initiale
En enzymologie classique, on s'intéresse à la phase stationnaire (linéaire) en mesurant la vitesse initiale.
Il y a un maximum d'enzyme liée avec le substrat. D’où la vitesse maximale
La vitesse peut être mesurée
avec l'apparition du produit.
dt
Pdv
K1 K2
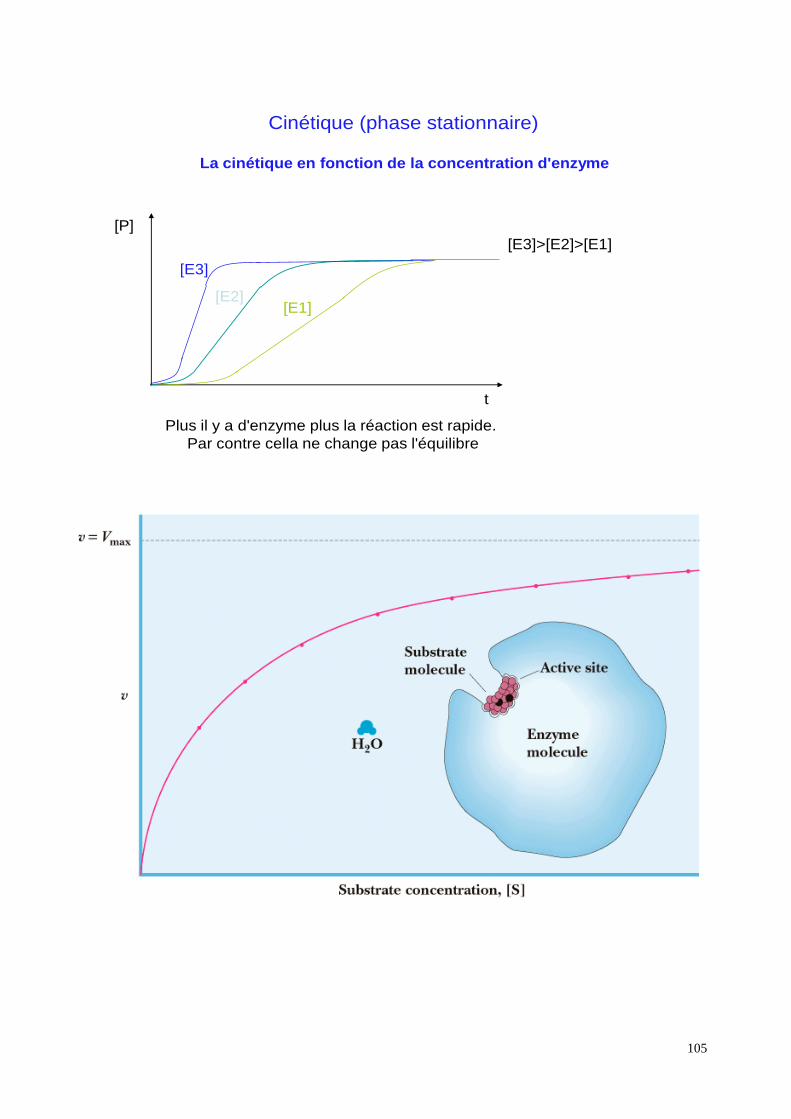
105
Cinétique (phase stationnaire)
en fonction de la concentration du substrat (phase stationnaire)
Courbe cinétique (V en fonction de [S]) classique des enzymes Michaéliennes.
C'est une hyperbole
V
[S]
Plus il y a de substrat, plus l'enzyme est rapide.
C'est la loi d'action de masse.
Néanmoins, c'est augmentation n'est pas infinie
(comme tout processus biologique), et l'enzyme
finie par saturer à la vitesse Vmax. (réaction
d'ordre 0, vitesse constante)
Attention, pour les mesure on atteint Vmax de
façon asymptotique. En réalité, il est difficile de
mesurer Vmax car il faut beaucoup de substrat
pour vraiment atteindre Vmax (en théorie [S]=∞)
Vmax (asymptote)
A faible concentration la vitesse est linéaire en fonction de la concentration de
substrat. C'est un réaction d'ordre 1
La cinétique en fonction de la concentration d'enzyme
[P]
t
[E1][E2]
[E3]
Plus il y a d'enzyme plus la réaction est rapide.
Par contre cella ne change pas l'équilibre
[E3]>[E2]>[E1]

106
INTERPRETATION DE LA SATURATION DE L’ENZYME PAR LE SUBSTRAT
Figure . Saturation de l’enzyme par le substrat. A) Fiable concentration en substrat ; B)
Concentration égale à la Km (50% de saturation) ; C) Saturation de l’enzyme par le substrat
(proche de Vmax)

107
Modèle de Michaelis-Menten (1913)
E+S ES
K-1 K-2
EPK3
K-3
E+P
K1 K2
Conditions (phase stationnaire).
1. On travaille à de très fortes concentrations de substrat [S]>>[P]
2. Il y a beaucoup plus de substrat que d'enzyme que [S]>>[E]
3 . les formes libres de l'enzyme et de l'enzyme lié au substrat sont en
équilibre (rapide).
Avec ces conditions, on peut simplifier la réaction
E+S ES
K-1
E+PComme, il y a peu de produit le retour K-2 est
négligeable
K1 K2
ES
SEKd
K1 K2
E+S ES
K-1
E+P
Par conservation, on doit avoir:
[S]ini=[S] + [ES] + [P] or [S]>>[E] et [S]>>[P]
Donc la concentration de substrat peut être considérée comme constante
[S]=[S]initiale
[E]tot =[E] + [ES], Loi de la conservation de l’enzyme
1
1
k
k
ES
SEKd
L'équilibre étant rapide, la concentration de l'enzyme fixée au substrat est reliée
à la constante de dissociation

108
inid
initot
iniinitotinitot
d
SK
SEES
SES
SE
ES
SESE
ES
SEK
En remplaçant [S] et [E] par leurs valeur
K2ES E+P
La deuxième partie de la réaction est une réaction du premier ordre
KmkdEk
SK
SEkESkV
tot
inid
initot
,Vdéfinit On
][
2max
22
C'est l'équation de Michaelis-Menten (Km =
cte de Michaélis)
V
[S]
Vmax
inim
ini
SK
SV
max
V
On peut définir la constante catalytique
Elle est indépendante de la concentration d'enzyme
tot
catE
VKK
][
max2
][k
ESK
SV
iniM
inicat
1
21
k
kkKM
KM est la constante de Michaelis (M)
Kcat=K2 (s-1)
La vitesse en fonction de la concentration de substrat est donné par l'équation
de Michaelis-Menten
V
Vmax=Kcat [E]
[S]
Si la vitesse de réaction (k2) est faible par rapport
avec la vitesse de dissociation k-1
dM K
k
k
k
kkK
1
1
1
21
On retrouve le modèle précédent

109
Suite du Modèle de Michaelis
K1 K2
E+S ES
K-1
E+P
Dans ce modèle, on tient compte de l'équilibre de ES avec E+S et E+P.
Si on est en phase stationnaire, ES est constant.
Ainsi
iniM
initot
ini
initot
iniinitot
initot
SK
SE
Sk
kk
SEES
kkSkESSEk
ESkkSESEk
ESkkSEkdt
ESd
1
21
2111
211
211
0
0][
1
21
k
kkKM
tot
iniM
initot
Ek
SK
SEkESkV
2max
22
V avec
][
iniM
ini
SK
SV
maxV
De la même façon que précédemment
tot
catE
VKK
][
max2
Analyse des constantes
][k
ESK
SV
iniM
inicat
La vitesse est proportionnelle à la concentration d'enzyme
Unité E,S: Mol
V: Mol/S
Kcat : s-1.Mol-1
KM: Mol
Kcat représente l'efficacité catalytique de l'enzyme, c'est le turnovernumber
Km représente l'affinité du substrat pour l'enzyme
Si [S=]Km
ESE
ESk
kkkE
ESkkkEk
ESkkSEk
m
m
1
21
211
2110
50% des enzyme sont liés au substrat

110
A très grande concentration de substrat, [S]>> KM
max][k][k
VEESK
SV cat
iniM
inicat
Vmax=kcat [E]
KM représente approximativement la constante d'affinité de l'enzyme pour le
substrat. Si on est largement au dessus de cette valeur, l'enzyme est saturé
par le substrat, il y a un maximum d'enzyme en condition pour effectuer la
réaction. C'est la vitesse maximale
Avec la mesure de Vmax on a accès a kcat
Si [S]=KM
2
max
2
][k][
k][
k VEE
KK
KE
SK
SV cat
MM
Mcat
iniM
inicat
Au cas où la concentration du substrat [S]=KM, la moitié des enzymes ont fixé le
substrat. Ainsi la vitesse est Vmax/2.
On peut facilement déduire KM, à partir de la courbe.
KM correspond au point ou la vitesse est Vmax/2
Vmax
Vmax/2
KM
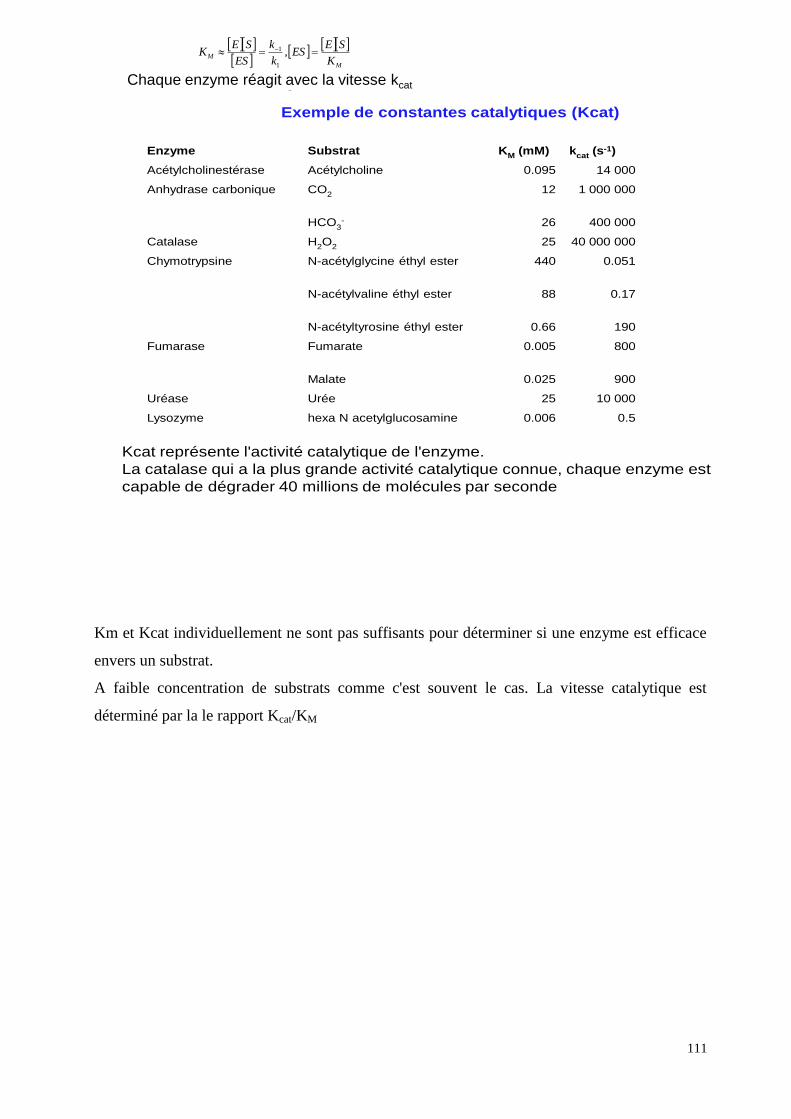
111
Km et Kcat individuellement ne sont pas suffisants pour déterminer si une enzyme est efficace
envers un substrat.
A faible concentration de substrats comme c'est souvent le cas. La vitesse catalytique est
déterminé par la le rapport Kcat/KM
A très faible concentration de substrat, [S]<< KM
][k
][k
ESk
ESk
SV ini
M
cat
iniM
inicat
La vitesse est proportionnelle à la concentration de substrat et d'enzyme
(réaction du premier ordre) avec le rapport
le nombre d'enzymes d'enzyme liées au
substrat approximativement donné par KM
M
MK
SEES
k
k
ES
SEK ,
1
1
Chaque enzyme réagit avec la vitesse kcat
][k
ESk
V ini
M
catD'ou
Pente
M
cat
k
k
Ce rapport mesure la spécificité et l'efficacité de l'enzyme
M
cat
k
k
Exemple de constantes catalytiques (Kcat)
Enzyme Substrat KM
(mM) kcat
(s-1)
Acétylcholinestérase Acétylcholine 0.095 14 000
Anhydrase carbonique CO2
12 1 000 000
HCO3
- 26 400 000
Catalase H2O
225 40 000 000
Chymotrypsine N-acétylglycine éthyl ester 440 0.051
N-acétylvaline éthyl ester 88 0.17
N-acétyltyrosine éthyl ester 0.66 190
Fumarase Fumarate 0.005 800
Malate 0.025 900
Uréase Urée 25 10 000
Lysozyme hexa N acetylglucosamine 0.006 0.5
Kcat représente l'activité catalytique de l'enzyme.
La catalase qui a la plus grande activité catalytique connue, chaque enzyme est
capable de dégrader 40 millions de molécules par seconde
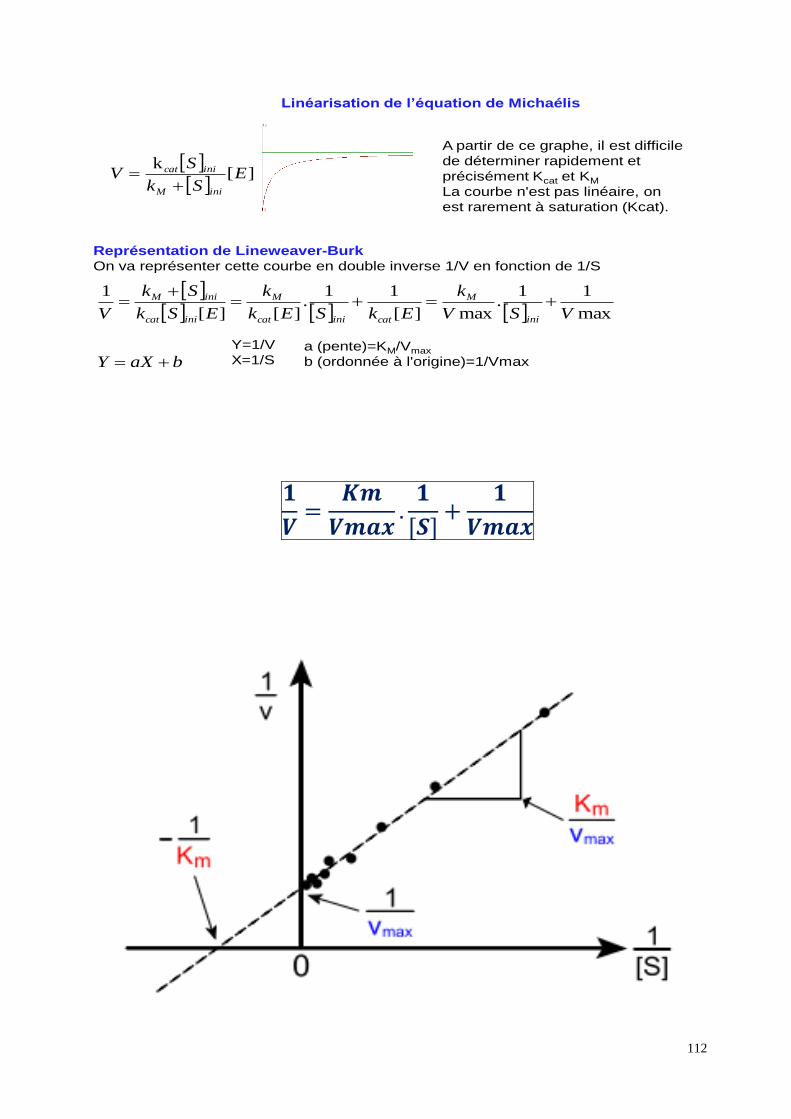
112
Linéarisation de l’équation de Michaélis
][k
ESk
SV
iniM
inicat
A partir de ce graphe, il est difficile
de déterminer rapidement et
précisément Kcat et KM
La courbe n'est pas linéaire, on
est rarement à saturation (Kcat).
Représentation de Lineweaver-Burk
On va représenter cette courbe en double inverse 1/V en fonction de 1/S
baXY
VSV
k
EkSEk
k
ESk
Sk
V ini
M
catinicat
M
inicat
iniM
max
11.
max][
11.
][][
1
Y=1/V
X=1/Sa (pente)=KM/Vmax
b (ordonnée à l'origine)=1/Vmax
𝟏
𝑽=
𝑲𝒎
𝑽𝒎𝒂𝒙.
𝟏
[𝑺]+
𝟏
𝑽𝒎𝒂𝒙

113
La représentation en double inverse : 1/V = f (1/[S]) est la plus utilisée en cinétique
enzymatique. Le problème principal de cette méthode est la prépondérance accordée aux
valeurs les plus faibles de S et de V, pour lesquelles l’incertitude est pourtant la plus grande.
Elle peut conduire à des estimations fausses. Elle ne devient intéressante que pour des valeurs
de [S] élevées, donc dans des conditions « saturantes ».
Méthode de Eadie-Hofstee
[𝑺]
𝑽=
𝟏
𝑽𝒎[𝑺] +
𝑲𝒎
𝑽𝒎𝒂𝒙

114
Le tracé de V en fonction de V/[S] donne une droite. De pente –Km et d’ordonnée à l’origine
Vmax
Représentation de Eisenthal et Cornich-Bowden ou de Schwarzenbach
Cette représentation fût publiée par Robert Eisenthal et AthelstanCornish-Bowden en 1974.
droite, définie par le couple (j) de points [(-[S]0, j, 0) : (0, vj)], est tracée dans le repère
cartésien (v, [S]).
Méthode purement graphique : on reporte sur l’axe des abscisses les valeurs de –[S] et sur celui
des ordonnées celles de V (pour des couples S(i)/V(i) de résultats expérimentaux), et les droites
se coupent en un point d’abscisse Km et d’ordonnée Vm.
L'équation générique est :
v = aj[S] + bj, avec pour chaque droite : b=vj et a = vi/[S]o,j
cela donne
𝐯 =𝐯𝐣
[𝑺]𝒐,𝒋[𝑺] + 𝐯𝐣
pour chacune de ces droites, calculons la valeur de v pour la valeur de [S] égale à KM, en
utilisant la relation de Briggs-Haldane :
𝐯𝒋 =𝑽𝒎𝒂𝒙[𝑺]𝒐,𝒋
𝑲𝒎 + [𝑺]𝒐,𝒋
𝐯 =𝒗𝒋
[𝑺]𝒐,𝒋𝑲𝒎 + 𝒗𝒋
En utilisant la relation de Briggs-Haldane on obtient :
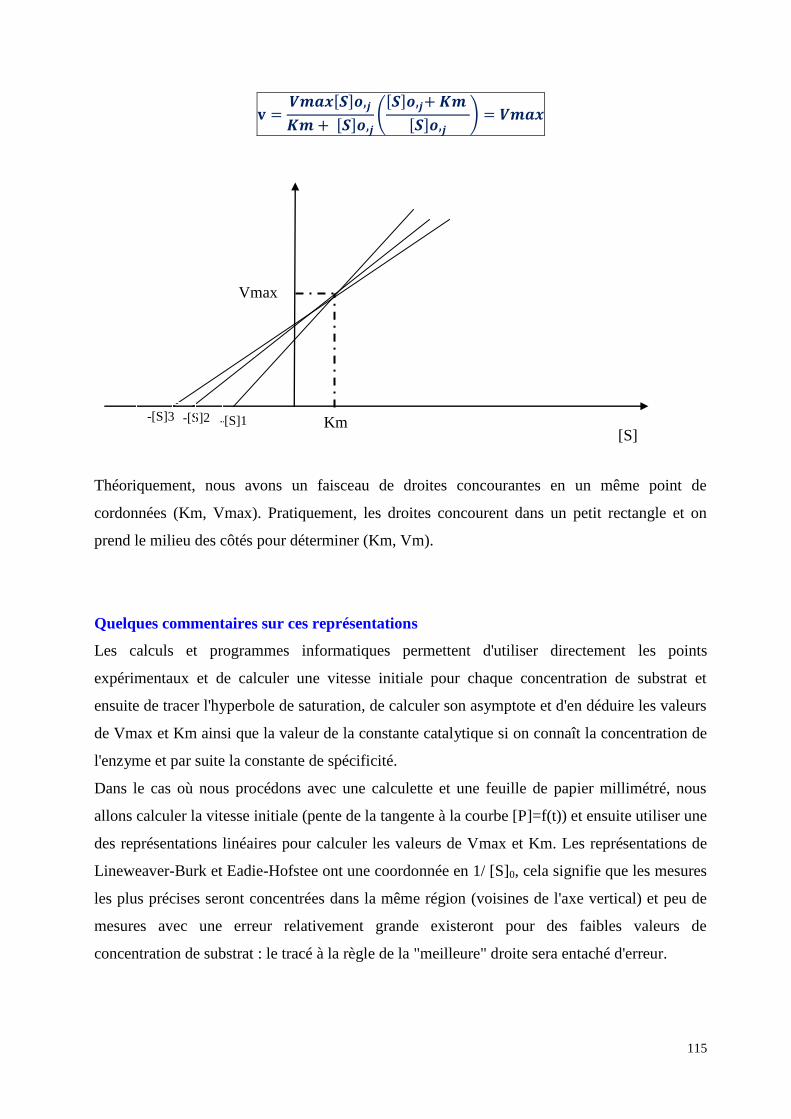
115
𝐯 =𝑽𝒎𝒂𝒙[𝑺]𝒐,𝒋
𝑲𝒎 + [𝑺]𝒐,𝒋(
[𝑺]𝒐,𝒋+ 𝑲𝒎
[𝑺]𝒐,𝒋) = 𝑽𝒎𝒂𝒙
Théoriquement, nous avons un faisceau de droites concourantes en un même point de
cordonnées (Km, Vmax). Pratiquement, les droites concourent dans un petit rectangle et on
prend le milieu des côtés pour déterminer (Km, Vm).
Quelques commentaires sur ces représentations
Les calculs et programmes informatiques permettent d'utiliser directement les points
expérimentaux et de calculer une vitesse initiale pour chaque concentration de substrat et
ensuite de tracer l'hyperbole de saturation, de calculer son asymptote et d'en déduire les valeurs
de Vmax et Km ainsi que la valeur de la constante catalytique si on connaît la concentration de
l'enzyme et par suite la constante de spécificité.
Dans le cas où nous procédons avec une calculette et une feuille de papier millimétré, nous
allons calculer la vitesse initiale (pente de la tangente à la courbe [P]=f(t)) et ensuite utiliser une
des représentations linéaires pour calculer les valeurs de Vmax et Km. Les représentations de
Lineweaver-Burk et Eadie-Hofstee ont une coordonnée en 1/ [S]0, cela signifie que les mesures
les plus précises seront concentrées dans la même région (voisines de l'axe vertical) et peu de
mesures avec une erreur relativement grande existeront pour des faibles valeurs de
concentration de substrat : le tracé à la règle de la "meilleure" droite sera entaché d'erreur.
Km [S]
-[S]1 -[S]2 -[S]3
Vmax

116
La représentation de Hanes-Woolf améliorera la détermination des valeurs expérimentales par
une distribution plus équilibrée des points expérimentaux. Celle de Eisenthal et Cornish-
Bowden donnera les meilleures estimations des valeurs de Vm et Km. Toutefois, malgré ses
Signification de la constante catalytique ou turnover-number
Kcat = nombre de moles de P formées par seconde et par mole d’enzyme. Si l’enzyme possède
plusieurs sites catalytiques, l’unité change et s’exprime par mole de sites. Kcat = Vmax / mole
d’enzyme = Vmax / [Et]. Le rapport Kcat/Km est souvent donné comme mesure de l'efficacité
catalytique, car il correspond à une constante de vitesse pour de basses concentrations de
substrat. (S) << Km, donc (E) = (ET)
Il faut noter qu’il existe Parfois des enzymes non Mikaeliennes telles que les enzymes
allostériques. Leurs courbes n’est pas une hyperbole mais un sigmoïde.Le modèle déterminer
n'est plus valable. On ne peut déterminer directement les paramètres cinétiques. Il faut vérifier
que le modèle "fit" les mesures.
On peut mesurer ces constantes kcat et Km pour caractériser une enzyme envers un substrat.
Spécificité de l'enzyme
En testant différents substrats on peut déterminer les type de substrat reconnus par l'enzyme
(Km) et on peut voir l'efficacité Kcat/Km de la réaction selon la nature chimique du substrat (ex
type de liaison).
Rôle physiologique de l'enzyme
Le rapport Kcat/Km peut nous aider à trouver le ou les substrats naturels de l'enzyme (ère
génomique).
Mutation rationnelle et mécanisme enzymatique.
En testant différents mutants on peut déterminer les acides aminés impliqués dans la
reconnaissance (il modifie le Km) et ceux directement impliqués dans la réaction (il modifie
Kcat).

117
Biotechnologie
La compréhension du mécanisme et la caractérisation de l'enzyme permet d'avoir un intérêt
biotechnologique.
Stabilisation (T°, pH, Sel,..)
Mutants plus actifs, etc.
I I I . 4 . L e s i n h i b i t e u r s
III.4.1 Les Inhibiteurs enzymatiques
Des inhibiteurs enzymatiques sont des espèces qui sont capables de diminuer l'activité
enzymatique.Les inhibiteurs enzymatiques interagissent généralement avec l’enzyme elle-
même, formant ainsi un complexe enzyme-inhibiteur (E·I). Dans certains cas, le mécanisme
d’inhibition implique une réaction avec le substrat.
-L’inhibition enzymatique peut être:
- réversible (activité spécifique et les paramètres de Michaelis-Menten sont affectés
- irréversible (la concentration d’enzyme active est diminuée)
- L’inhibition de réactions par des produits de la réaction catalysée (ou d’une réaction
subséquente) - mécanisme de régulation/« feedback » pour le métabolisme cellulaire.
�Inhibition sélective d’une enzyme par des substances chimiques - à la base de la
pharmacologie et de la chimiothérapie.
Quantification d’inhibiteurs- à partir de mesures de l’activité enzymatique (l’échantillon
d’inhibiteur ne contenant pas d’enzyme). Des standards contenant une quantité fixe d’enzyme,
une concentration saturée de substrat et des concentrations variables d’inhibiteur sont préparés.
Des courbes de l’activité enzymatique vs. [I] sont réalisées à partir des standards.
III.4.2Inhibiteur compétitif
Il y une compétition entre l’inhibiteur et le substrat pour le site actif de l’enzyme. L’inhibiteur
se lie réversiblement à l’enzyme mais n’est pas converti en produit.
Modèle pour un inhibiteur compétitif
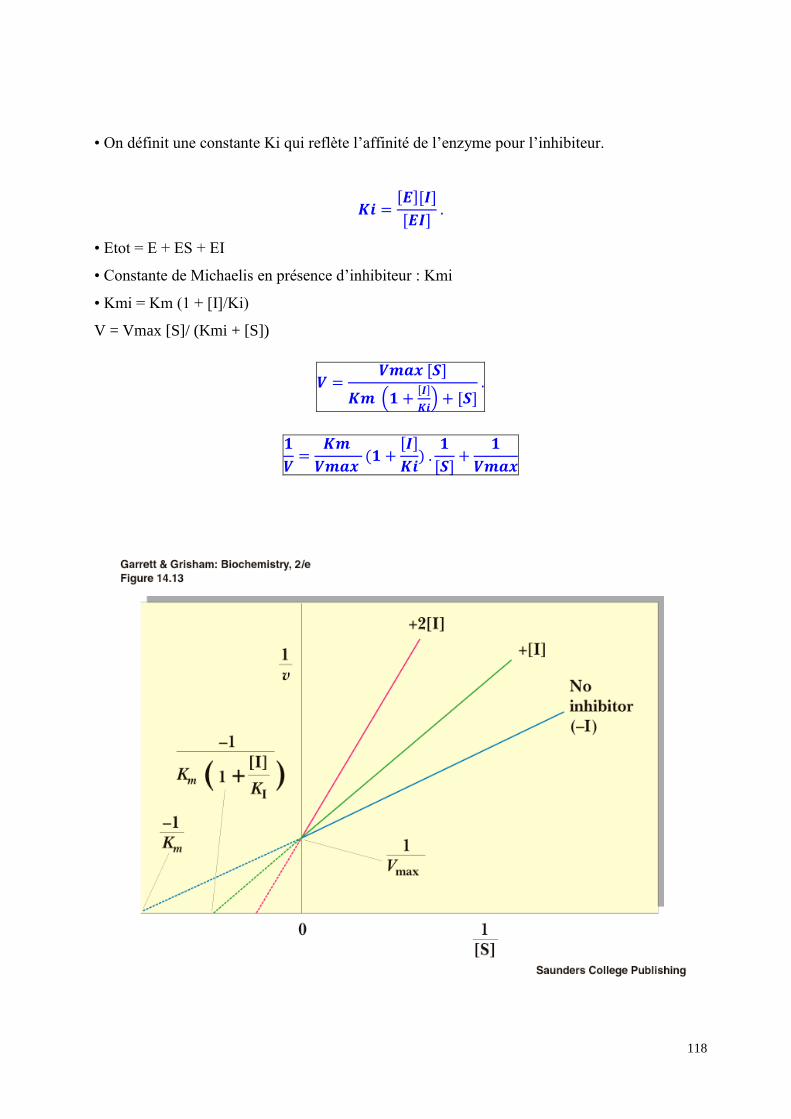
118
• On définit une constante Ki qui reflète l’affinité de l’enzyme pour l’inhibiteur.
𝑲𝒊 =[𝑬][𝑰]
[𝑬𝑰] .
• Etot = E + ES + EI
• Constante de Michaelis en présence d’inhibiteur : Kmi
• Kmi = Km (1 + [I]/Ki)
V = Vmax [S]/ (Kmi + [S])
𝑽 =𝑽𝒎𝒂𝒙 [𝑺]
𝑲𝒎 (𝟏 +[𝑰]
𝑲𝒊) + [𝑺]
.
𝟏
𝑽=
𝑲𝒎
𝑽𝒎𝒂𝒙 (𝟏 +
[𝑰]
𝑲𝒊) .
𝟏
[𝑺]+
𝟏
𝑽𝒎𝒂𝒙
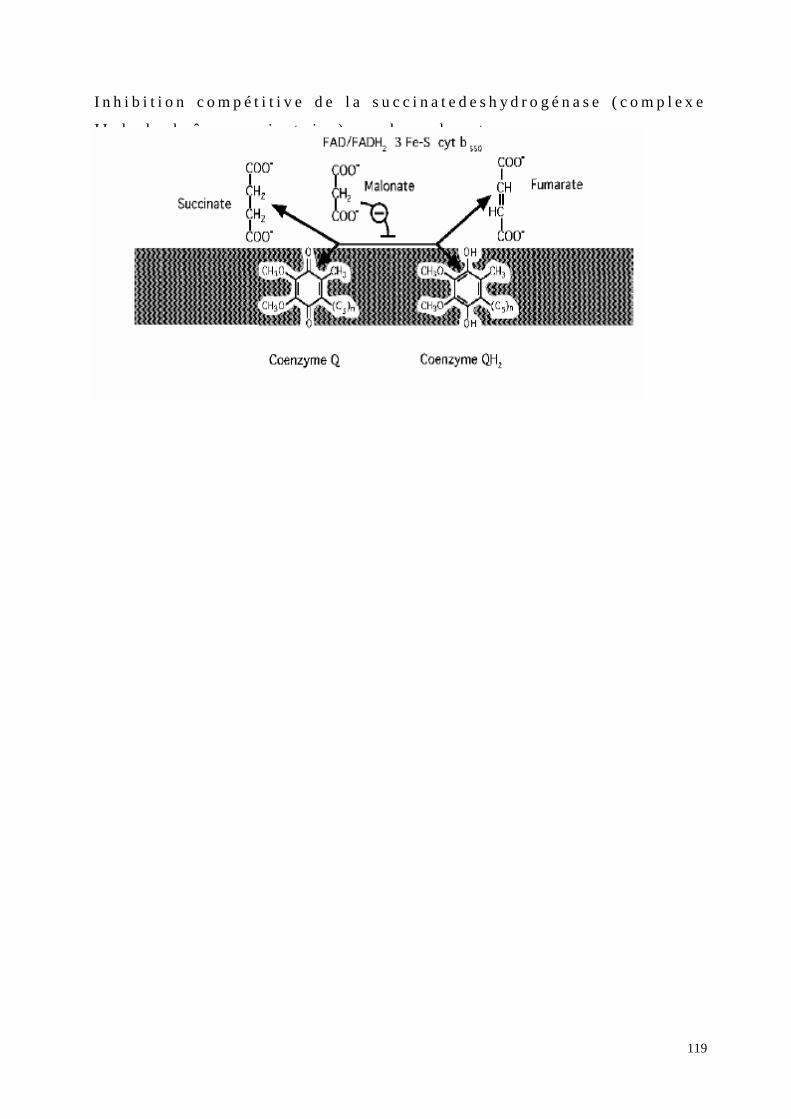
119
I n h i b i t i o n c o m p é t i t i v e d e l a s u c c i n a t e d e s h y d r o g é n a s e ( c o m p l e x e
I I d e l a c h a î n e r e s p i r a t o i r e ) p a r l e m a l o n a t e .

120
III.4.3. Inhibiteur incompétitif
L’inhibiteur incompétitif se lie seulement au complexe ES. De ce fait la Km et la Vmax
changent
–Km devient Km/(1 + [I]/KI’). La Km diminue apparemment. C’est è dire qu’on a besoin de
faible concentration en substrat pour suturer l’enzyme.
–Vmax devient Vmax/(1 + [I]/KI)
Modèle pour un inhibiteur incompétitif
𝑽 =𝑽𝒎𝒂𝒙
(𝟏 +[𝑰]
𝑲𝒊)
.[𝑺]
𝑲𝒎
(𝟏+[𝑰]
𝑲𝒊)
+ [𝑺] .
𝟏
𝑽=
𝑲𝒎
𝑽𝒎𝒂𝒙 .
𝟏
[𝑺]+
𝟏
𝑽𝒎𝒂𝒙 (𝟏 +
[𝑰]
𝑲𝒊)

121
III.4.4. Inhibiteurs non compétitifs
Un inhibiteur non compétitif classique n'a aucune influence sur la fixation du substrat (et
réciproquement) : les sites de fixation du substrat et de l'inhibiteur sont distincts.
En conséquence, l'inhibiteur se fixe à l'enzyme libre E et au complexe ES ; de même, le substrat
se fixe à l'enzyme libre E et au complexe EI. Le complexe EI (enzyme-inhibiteur) et ESI
(enzyme-substrat-inhibiteur) n’ont pas d’activité. Les inhibiteurs non compétitifs n'ont pas
d'homologie structurale avec le substrat. On suppose dans ce modèle que Ki = K’i.
Modèle pour un inhibiteur non-compétitif
EQUATION DE VITESSE
𝑽 =𝑽𝒎𝒂𝒙
(𝟏 +[𝑰]
𝑲𝒊)
. [𝑺]
𝑲𝒎 + [𝑺] .
𝟏
𝑽=
𝑲𝒎
𝑽𝒎𝒂𝒙 (𝟏 +
[𝑰]
𝑲𝒊) .
𝟏
[𝑺]+
𝟏
𝑽𝒎𝒂𝒙 (𝟏 +
[𝑰]
𝑲𝒊)
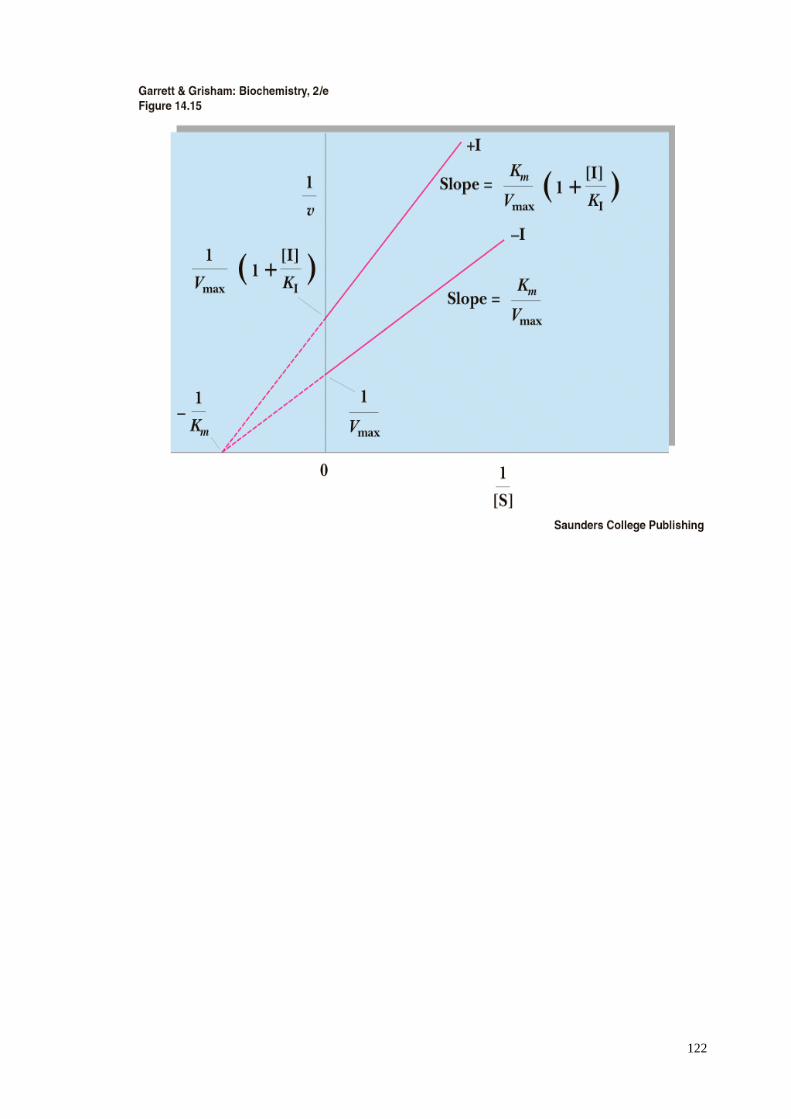
122
Figure . Inhibiteur non- compétitif
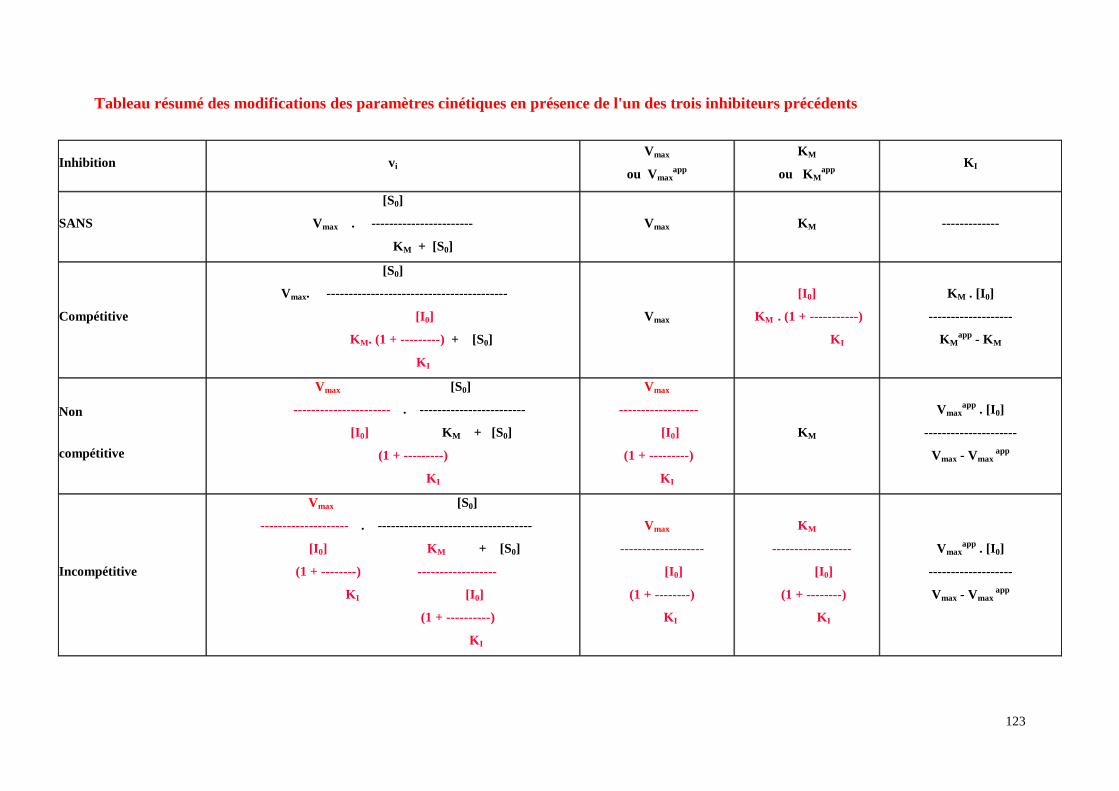
123
Tableau résumé des modifications des paramètres cinétiques en présence de l'un des trois inhibiteurs précédents
Inhibition vi Vmax
ou Vmaxapp
KM
ou KMapp
KI
SANS
[S0]
Vmax . -----------------------
KM + [S0]
Vmax KM -------------
Compétitive
[S0]
Vmax. -----------------------------------------
[I0]
KM. (1 + ---------) + [S0]
KI
Vmax
[I0]
KM . (1 + -----------)
KI
KM . [I0]
-------------------
KMapp
- KM
Non
compétitive
Vmax [S0]
---------------------- . ------------------------
[I0] KM + [S0]
(1 + ---------)
KI
Vmax
------------------
[I0]
(1 + ---------)
KI
KM
Vmaxapp
. [I0]
---------------------
Vmax - Vmax app
Incompétitive
Vmax [S0]
-------------------- . -----------------------------------
[I0] KM + [S0]
(1 + --------) ------------------
KI [I0]
(1 + ----------)
KI
Vmax
-------------------
[I0]
(1 + --------)
KI
KM
------------------
[I0]
(1 + --------)
KI
Vmaxapp
. [I0]
-------------------
Vmax - Vmax app

124
III.4.5. Inhibition par excès de substrat
Soit la réaction 𝐸 + 𝑆 ↔ 𝐸𝑆 → 𝑃 + 𝐸
Supposons qu’une deuxième molécule de substrat puisse se fixer sur le complexe ES tel que
dans le cas de l’inhibiteur incompétitif et le rendre inactif : cette molécule se fixe sur un site
différent du premier, et nous l’appellerons site inhibiteur. Cette molécule se comporte comme
un inhibiteur.
L’équation de la conservation de l’enzyme donne :
[𝐄]𝐨 = [𝐄] + [𝐄𝐒] + [𝐄𝐒𝐒]
A l’état stationnaire on a :
[𝑬] =(𝒌−𝟏 + 𝒌𝟐 )[𝑬𝑺]
𝒌𝟏 [𝑺]= 𝑲𝒎
[𝑬𝑺]
[𝑺]
On a aussi :
[𝑬𝑺𝑺] =[𝑬𝑺][𝑺]
𝒌𝒊
Donc
[𝐄]𝐨 = 𝑲𝒎[𝑬𝑺]
[𝑺]+ [𝐄𝐒] +
[𝑬𝑺][𝑺]
𝒌𝒊
= (𝟏 +𝒌𝒎
[𝑺]+
[𝑺]
𝒌𝒊
)[𝑬𝑺]
[𝑬𝑺] =[𝑬]𝒐
(𝟏 +𝒌𝒎
[𝑺]+
[𝑺]
𝒌𝒊
)
ESS

125
On sait qu’à l’état stationnaire 𝑣 = 𝒌𝟐 [𝑬𝑺]
𝑣 = 𝒌𝟐
[𝑬]𝒐
(𝟏 +𝒌𝒎
[𝑺]+
[𝑺]
𝒌𝒊
)=
𝑽𝒎𝒂𝒙
(𝟏 +𝑲𝒎
[𝑺]+
[𝑺]
𝒌𝒊
)
L’observation de cette équation montre que si [S] est trop grand (tend vers l’infini) 𝑲𝒎
[𝑺] 𝒕𝒆𝒏𝒅 𝒗𝒆𝒓𝒔 𝟎 𝒆𝒕
𝑽𝒎𝒂𝒙
(𝟏+𝟎+[𝑺]
𝒌𝒊
)
𝒆𝒕 𝒍𝒆 𝒓𝒂𝒕𝒊𝒐𝑽𝒎𝒂𝒙
(∞) 𝒕𝒆𝒏𝒅 𝒗𝒆𝒓𝒔 𝒐, 𝒅𝒐𝒏𝒄 𝒗 𝒕𝒆𝒏𝒅 𝒗𝒆𝒓𝒔 𝟎‼‼
Ceci démontre l’inhibition par excès de substrat.
Sur le plan mathématique quand on porte 1/Vi en fonction de 1/[S], on obtient une hyperbole
dont l’asymptote oblique est de pente Km/Vmax, coupe l’axe en un point -1/Km.
Quand on porte 1/Vi en fonction de [S], on obtient une hyperbole dont l’asymptote oblique,
est de pente 1/Vmax. Km coupe l’axe des [S] en un point d’abscisse -Ki.
Dans les deux cas, Vi passe par un maximum pour [𝑺] = √𝑲𝒎. 𝑲𝒊

126
V. Le s e nzy m e s à de ux subs t r a t s
Beaucoup d’enzymes agissent sur deux substrats ou plus. La réaction de Michaélis-Menten est
obtenue en considérant un seul substrat, mais il est toujours possible d’obtenir des équations
de vitesse des enzymes à deux substrats en saturant l’un des substrats et en faisant varier la
concentration du second et vice-versa.
On trois modèles de fixation des substrats :
1 . M o d è l e a l é a t o i r e
Dans ce modèle chacun des substrats se fixe sur l’enzyme de manière aléatoire, aucun ordre
d’arrivé n’est nécessaire mais la réaction finale n’a lieu que si les deux substrats sont présents
dans le centre actif. Pour ces enzymes les deux substrats sont capables de former le complexe
Enzyme-S1 ou Enzyme-S2 en fixant les deux substrats (ou coenzyme libre) l’un après l’autre
mais sans ordre fixe : la probabilité de commencer par Enzyme-S1 ou par Enzyme-S2 ne
dépendant que des affinités respectives de l’enzyme pour ces deux corps chimiques. C’est ce
qui justifie l’appellation bibi aléatoire qu’on donne à ce mécanisme
Exemple : La citrate synthase
La citrate synthase est une enzyme des mitochondries. Elle catalyse le transfert d’un radical
acétyl du premier substrat, l’acétate transporté par le coenzyme A vers l’autre substrat,
l’oxaloacétate. L’affinité de l’enzyme pour ces deux corps chimiques étant voisine, la liaison
de l’enzyme avec chacun d’entre eux se fait dans un ordre qui dépend uniquement des
concentrations.

127
2. Modèle bi-bi ordonné
Dans ce modèle les deux substrats se lient dans un ordre obligatoire ordonné. C’est le substrat
S1 qui de lie pour former un complexe ES1 et ensuite le second substrat se fixe pour donner le
complexe ES1S2. Dans ce contexte aucun produit n’est formé avant que les deux substrats ne
se lient à l’enzyme. L’enzyme libre n’ayant pas d’affinité pour le substrat S2, le complexe ne
peut pas se former dans un ordre différent : c’est ce qui justifie l’appellation bibi ordonné
qu’on donne à ce mécanisme.
Exemple : L’alcool déshydrogénase
Cette enzyme qu’on trouve dans le cytoplasme de la plupart de nos cellules. Elle catalyse
l’oxydation de l’éthanol en acétaldéhyde en réduisant simultanément un coenzyme NAD+ en
NADH.
Cette réaction se déroule selon un mécanisme de type bibi ordonné : l’enzyme n’a pas
d’affinité pour l’alcool si elle n’est pas préalablement associée au coenzyme NAD+ en un

128
premier complexe ; puis le complexe ternaire Enzyme-NAD+-Ethanol se transforme en un
complexe Enzyme-NADH-Acétaldéhyde ; ce dernier complexe se dissocie en libérant
l’acétaldéhyde puis le NAD réduit. De nombreuses autres déshydrogénases utilisant le NAD
comme coenzyme suivent un mécanisme
de type bibi ordonné.
3. Modèle ping pong ordonné
Dans ce mécanisme, il n’y a pas de formation d’un complexe tertiaire ES1S2. Le premier
substrat est converti en produit P1 avant que le second substrat ne se lie à l’enzyme. Mais
dans ce cas il faut nécessairement que l’enzyme soit active (E’) par le premier substrat avant
que le second ne se fixe et l’inverse n’est pas possible. Pour ces enzymes, la réaction sera
catalysée en deux temps. Le complexe formé entre l’enzyme et le substrat S1 est transformé
d’abord en enzyme + produit P1, mais l’enzyme E a été chimiquement modifiée en enzyme E’
au cours de cette première partie de la réaction. L’enzyme E’ ayant une affinité pour le
deuxième substrat, va former un deuxième complexe Enzyme E’-Substrat S2 qui va être
transformé en complexe Enzyme-Produit P2 dans une seconde partie de la réaction où
l’enzyme va retrouver sa forme chimique initiale. Il n’y a jamais de complexe ternaire dans un
tel mécanisme, mais l’enzyme (ou un coenzyme lié à sa structure) subit une transformation

129
réversible et provisoire qui permet le lien entre les deux substrats. C’est ce qui justifie
l’appellation de ping-pong ou de déplacement qu’on donne à ce mécanisme.
Exemple : Alanaine amino-transférase (ALAT)
L’ALAT catalyse le transfert de la fonction amine de l’alanine vers l’α-cétoglutarate qu’elle
transforme en glutamate.
• Dans un premier temps, l’ALAT se lie à l’alanine puis transfère la fonction amine sur un
coenzyme lié : le phosphate de pyridoxal qui devient phosphate de pyridoxamine sans cesser
d’être lié à l’enzyme. L’enzyme se dissocie alors du pyruvate.
• Dans le second temps, l’enzyme liée au phosphate de pyridoxamine, forme un complexe
avec l’α-cétoglutarate, puis transfère la fonction amine du coenzyme qui redevient phosphate
de pyridoxal, vers le second substrat qui est transformé en glutamate. Enfin, le complexe
ALAT-glutamate se dissocie : l’enzyme et son coenzyme lié ont recouvré leurs structures
initiales.

130
VI . Not io ns sur l e s e nzy m e s a l l o s tér i que s
1. Définition
L’allostérie désigne une variation de conformation de protéines sous l’effet de la fixation d’un
substrat ou d’une molécule effectrice, d’où l’acquisition de propriétés particulières
(changement d’activité). On décrit cela comme des effets coopératifs. Ceux ci sont
concevables si et seulement si la macromolécule est sous forme oligomérique.
Exemple : un tétramère constitué de 4 sous-unités (monomères) à activité biologique et de 4
sous-unités de liaison.
La fixation d’un substrat entraîne une modification de l’ensemble de la structure.
Toutes les sous-unités reproduisent à l’unisson la variation de conformation subie
par l’une d’elles.
2. Propriétés d’une molécule allostérique
Il existe une structure quaternaire (en pratique, entre un dimère et un tétramère). La variation
de conformation de la structure dépend du taux d’occupation des sites de liaison. Pour toutes
les enzymes allostériques, la cinétique n’est pas michaélienne.
Ces molécules jouent des rôles clef dans la régulation du métabolisme.
Il y a deux types d’enzymes allostériques :
Système K : la régulation se traduit par la variation de la fixation du substrat ou par la
variation de l’affinité du substrat pour l’enzyme, d’où une variation du « Km »
Système V : la régulation porte sur la vitesse maximale. Dans ce cas, cela correspond à une
variation de la constante k2 des enzymes michaéliennes.
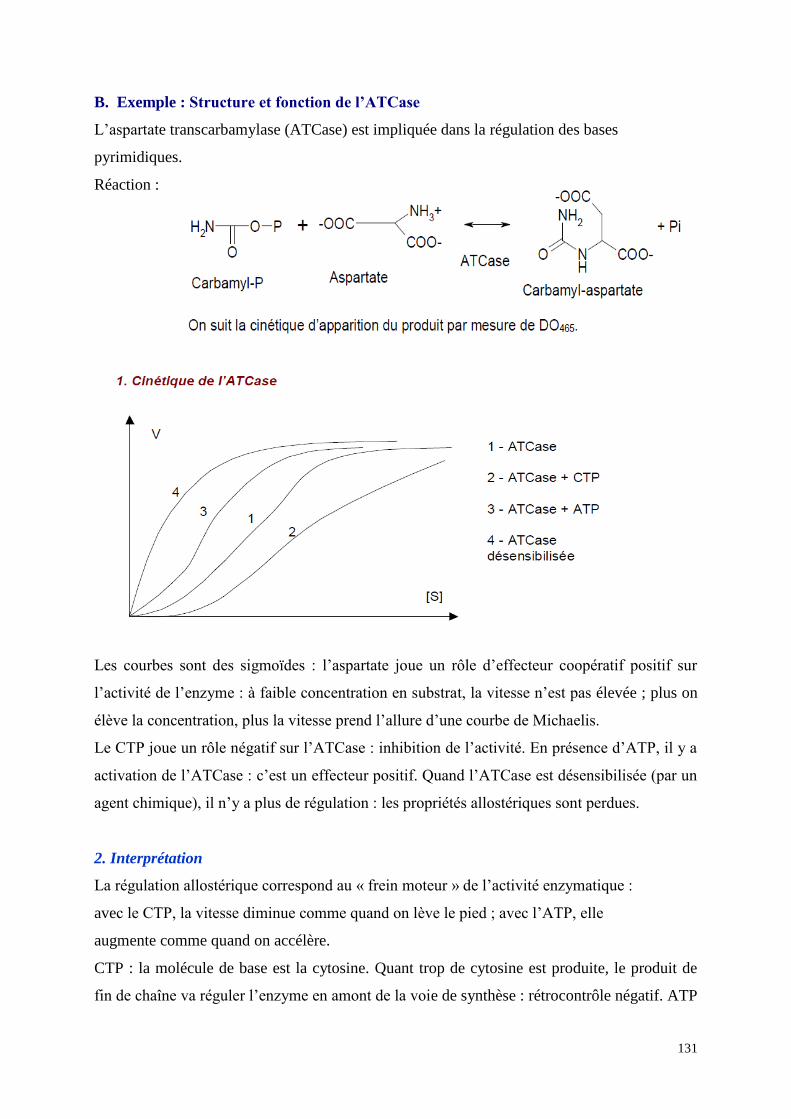
131
B. Exemple : Structure et fonction de l’ATCase
L’aspartate transcarbamylase (ATCase) est impliquée dans la régulation des bases
pyrimidiques.
Réaction :
Les courbes sont des sigmoïdes : l’aspartate joue un rôle d’effecteur coopératif positif sur
l’activité de l’enzyme : à faible concentration en substrat, la vitesse n’est pas élevée ; plus on
élève la concentration, plus la vitesse prend l’allure d’une courbe de Michaelis.
Le CTP joue un rôle négatif sur l’ATCase : inhibition de l’activité. En présence d’ATP, il y a
activation de l’ATCase : c’est un effecteur positif. Quand l’ATCase est désensibilisée (par un
agent chimique), il n’y a plus de régulation : les propriétés allostériques sont perdues.
2. Interprétation
La régulation allostérique correspond au « frein moteur » de l’activité enzymatique :
avec le CTP, la vitesse diminue comme quand on lève le pied ; avec l’ATP, elle
augmente comme quand on accélère.
CTP : la molécule de base est la cytosine. Quant trop de cytosine est produite, le produit de
fin de chaîne va réguler l’enzyme en amont de la voie de synthèse : rétrocontrôle négatif. ATP

132
: quand il y a beaucoup d’adénine (base purique), il y a activation de la synthèse des bases
pyrimidiques : rétrocontrôle positif. L’allostérie est donc un outil de réglage fin, de régulation
directe, instantanée, qui n’est pas du même ordre que, par exemple, l’activation de l’opéron
Lactose qui est un système très lourd.
3. Structure de l’ATCase
La sous-unité régulatrice possède un domaine allostérique et un site de fixation du zinc qui
permet l’interaction avec les trimères.
La sous-unité catalytique possède un domaine équatorial pour la fixation de l’aspartate et un
domaine polaire pour la fixation du carbamyl-phosphate.
L’oligomère présente deux formes, la forme « T » inactive (compacte) et la forme
« R » active (relâchée).
4. Effets homotropes ou hétérotropes
[ T ] Û [ R ] avec Kéq.
En présence de substrat :
R + S Û RS à R + P
Mais, quand on augmente [S], on induit la formation de RS, et donc une diminution de [R]
libre, d’où un passage de la forme T à la forme R, d’où une diminution de T dans la cellule :
Le substrat S joue un rôle homotrope positif.

133
En présence d’un effecteur positif (ATP) :
On a toujours équilibre entre les formes T et R, mais cette fois R se lie aussi à E :
T Û R + E à RE + S à …
Donc [R] diminue, donc transition T à R, donc diminution de [T] libre. Effet hétérotrope
positif.
En présence d’un effecteur négatif (CTP)
L’effecteur se lie à la forme T pour former un complexe TE, d’où une diminution de la forme
T libre, transition de R vers T et diminution de la forme R, ce qui correspond à une
inactivation de l’enzyme. Effet hétérotrope négatif sur l’enzyme.

134
IV. Influence de paramètres physiques sur les réactions enzymatiques
IV.1. Effet de la température
Notion de « température optimale »
Quand la Température augmente, l’énergie cinétique des molécules augmente, ainsi que la
fréquence des collisions. L’apport d’énergie (énergie d’activation) permet une augmentation
de la vitesse initiale.
Loi d’Arrhénius. Arrhénius démontre qu’une réaction chimique a besoin d’un « amorçage ».
Il faut une certaine quantité d’énergie pour préparer les réactifs à réagir. En particulier ; il faut
qu’ils entrent en collision avec une bonne orientation et que la collision permette un
affaiblissement des liaisons chimiques qui doivent se rompre pour former les nouveaux corps
appelés produits. La vitesse des réactions enzymatiques croit avec la température jusqu’à un
certain seuil : Température optimale (To). Après ce seuil, la vitesse n’est plus linéaire à
cause de la dénaturation des protéines enzymatiques. A tout moment on Vmax = kcat [E]o et
kcat est une constante cinétique obéissant à la loi d’Arrhénius selon l’équation :
Où k =constante de Boltzman et h constante de Plank. T tempertaure en °K. En traçant la
courbe ln(kcat) ou ln(Vmax)= f(1/T) on obtient une droite de pente –Ea/R et d’ordonnée à
l’origine lnA selon l’équation :
Dans les réactions enzymatiques on admet que A est une constante. On définit un autre
paramètreQ10 = facteur d’augmentation de .l’activité quand on augmente la température de
:
RT
EA
RT
E
h
kTk aa
cat expexp
21
12
1
2
10 lnTT
TT
R
Ea
k
kQ
cat
cat
.1
.lnTR
EaLnAkcat

135
Une augmentation de la température entraîne:
- augmente la vitesse de la réaction chimique (Loi d’Arrhénius)
- augmente la vitesse de dénaturation de l’enzyme (une protéine), diminuant ainsi son
activité catalytique. La somme de ces deux effets donne une courbe caractéristique de
l’activité enzymatique- température qui passe par un maximum, montrant ainsi
l'existence d'une température optimale.
IV.2. E f f e t d u p H
Les enzymes sont très sensibles aux variations du pH et fonctionnent bien sur une gamme
limitée du pH.
Les mesures de nombreuses activités enzymatiques en fonction du pH donnent des courbes
qui passent par un maximum, montrant ainsi l'existence d'un pH optimum.
Parmi les facteurs qui influencent la catalyse, le pH intervient de deux manières différentes :
soit en modifiant la structure secondaire ou tertiaire de l’enzyme, soit en modifiant les charges
électriques des radicaux des acides aminés du site actif.
• Lorsqu’une enzyme est conservée dans un milieu dont le pH est défavorable au maintien de
sa structure, elle va subir une dénaturation.
Température optimum
Température (°C)
Act
ivit
é en
zym
atiq
ue

136
• On peut examiner l’effet sur un graphe représentant l’activité résiduelle d’une enzyme qui a
été conservée pendant 24 heures à une température de 30°C., en fonction du pH de ce milieu
de conservation.
• A pH 3 cette enzyme a été conservée dans des conditions optimales et son activité résiduelle
est la plus grande : 100%. A pH 6 l’enzyme a subi une dénaturation partielle si bien qu’au
bout de 24 heures son activité n’est plus que de la moitié de ce qu’elle aurait été si on l’avait
conservée à pH 3. A pH 9 ou bien en dessous de pH 3, cet effet dénaturant est encore plus
marqué.
Le milieu dans lequel se produit la réaction enzymatique détermine la charge électrique des
radicaux des acides aminés de la protéine.
• Aux pH très acides la plupart des fonctions ionisables de ces radicaux sont sous la forme
protonée c’est à dire COOH pour la fonction acide carboxylique et NH3+ pour la fonction
amine.
• Aux pH les plus alcalins les fonctions ionisables de ces mêmes radicaux sont sous la forme
déprotonée c’est à dire COO- pour la fonction acide carboxylique, et NH2 pour la fonction
amine.
• A pH voisin de la neutralité, une très grande majorité de ces radicaux à fonctions ionisables
sont chargés ce qui facilite les liaisons enzyme-substrat ou enzyme-coenzyme de type
électrostatique.
• Il existe donc un pH du milieu réactionnel où les charges électriques des radicaux du site
actif de l’enzyme seront les plus favorables à la liaison enzyme-substrat : on appelle ce pH le
pH optimum de la réaction enzymatique.
Le pH peut agir sur plusieurs facteurs :
- l'ionisation des résidus de l’enzyme et du substrat et/ou du produit
- la structure tertiaire des protéines et donc la stabilité de l'enzyme
- la liaison du substrat à l'enzyme
- l'activité catalytique de l'enzyme
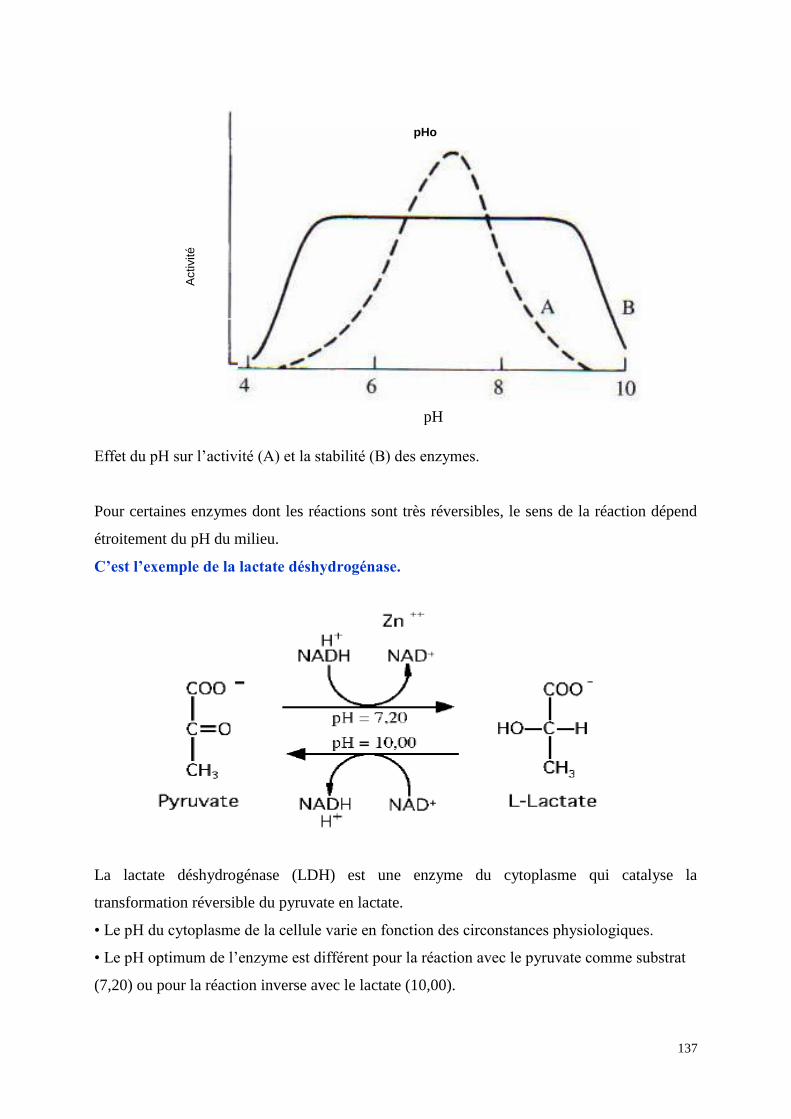
137
Effet du pH sur l’activité (A) et la stabilité (B) des enzymes.
Pour certaines enzymes dont les réactions sont très réversibles, le sens de la réaction dépend
étroitement du pH du milieu.
C’est l’exemple de la lactate déshydrogénase.
La lactate déshydrogénase (LDH) est une enzyme du cytoplasme qui catalyse la
transformation réversible du pyruvate en lactate.
• Le pH du cytoplasme de la cellule varie en fonction des circonstances physiologiques.
• Le pH optimum de l’enzyme est différent pour la réaction avec le pyruvate comme substrat
(7,20) ou pour la réaction inverse avec le lactate (10,00).
Activité
pHo
pH

138
• De sorte que lorsque le pH du cytoplasme est bas (acidose) l’enzyme favorisera la
transformation du pyruvate en lactate.
• Le pH agit donc ici comme un effecteur de la réaction.

139
CHAPITRE 7
IMMOBILISATION DES ENZYMES

140
Introduction
Les enzymes étant difficiles à purifier et à conserver, les biotechnologues ont développé des
méthodes pour immobiliser les enzymes sur des supports inertes non dégradables pour
l’enzyme et résistant aux solvants.
En immobilisation est on cherche généralement à conserver l’activité enzymatique, qui a
tendance:
à diminuer après immobilisation du fait de possibles gênes stériques et de limitations
dans l’accessibilité au site actif.
à augmenter sa stabilité dans le temps.
On peut combiner des méthodes pour assurer une meilleure fixation de l’enzyme sur le
support, par exemple :
• La rétention physique exploite la grande différence de taille entre l’enzyme et le
substrat. Elle peut être un réseau, une capsule ou une membrane.
• Le traitement chimique se réalise par simple interaction ionique, la création d’une
vraie liaison covalente et présente l’avantage de stabiliser la protéine et de permettre une
utilisation prolongée.
LES PRINCIPALES TECHNIQUES D’IMMOBILISATION DES ENZYMES
Plusieurs méthodes d'immobilisation des enzymes ont été décrites dans la littérature.
Cependant, il existe quatre grandes méthodes, couramment utilisées pour immobiliser des
enzymes: l’inclusion, le confinement, la réticulation, la fixation sur un support. Le principe de
ces méthodes est résumé dans le tableau suivant.
Méthode Type Support
Immobilisation par rétention
physique
Inclusion dans une matrice constituée par le réseau
tridimensionnel d’un polymère
Confinement dans une microcapsule réunie par une membrane
semi-perméable dans une fibre creuse
Immobilisation par interaction
physique ou liaison chimique
Réticulation établissement d’un réseau par liaisons covalentes
intermoléculaires
Fixation sur un
support

141
EXEMPLE I.. IMMOBILISATION PAR RETENTION PHYSIQUE
L’enzyme est dispersée dans une solution homogène de monomère ou d’émulsion. La
polymérisation du monomère conduit à la formation d’un réseau au sein duquel l’enzyme est
emprisonnée d’une manière purement physique.Les molécules d’enzyme seront retenues dans
le réseau tridimensionnel du polymère (insoluble dans l’eau) dont les pores sont suffisamment
larges pour permettre le passage des molécules du substrat ou des produits de la réaction.
L’enzyme est incorporée dans un gel insoluble (inclusion en gel). Ce gel peut être constitué
d’une matrice organique (polymère) ou inorganique (argiles). Sont généralement utilisés
comme matrice pour l’inclusion les gelsà partir des polymères naturels (alginate,
Carraghénane, Chitosane…) et les gels à partir des polymères synthétiques (polyacrylamide).
Figure I. Inclusion dans un réseau de polymère insoluble
Exemples d’inclusion : inclusion dans un gel
Figure II : A : Polymérisation en masse d’une solution de monomères contenant une enzyme
B : Polymérisation en bille après suspension de la solution de monomères en phase organique
A
B

142
Figure : Inclusion dans un gel de polyacrylamide.
fibres de polyacétate de cellulose
On réalise une solution de triacétate de cellulose dans le chlorure de méthylène.
On ajoute sous agitation une solution d’enzyme dans l’eau froide ou dans un tampon
contenant de glycérol
L’émulsion obtenue est ensuite extrudée sous pression d’azote dans un bain de coagulation
contenant de toluène.
La fibre obtenue est séchée sous vide pour éliminer les solvants et conserver à 5°C.
Figure IV : Inclusion dans des fibres de polyacétate de cellulose
II.1.2. Confinement ou micro encapsulation
La méthode consiste à inclure l’enzyme à l’intérieur de la membrane semi-perméable obtenue
en copolymérisation un monomère hydrophobe, dissous dans la phase organique, et un
monomère hydrophile, dissous dans la phase aqueuse en émulsion dans la phase organique.
Les microcapsules emprisonnant l’enzyme sont recueillies par centrifugation.Leur diamètre
peut varier de quelques microns à une centaine de microns.

143
Figure V : Inclusion dans des microcapsules
EXEMPLE II. IMMOBILISATION PAR INTERACTION PHYSIQUE OU LIAISON
CHIMIQUE
II.2.1. Réticulation
C’est un procédé d'association des différentes unités biocatalytiques ou de protéines à l'aide
d'un agent réticulant (masse molaire élevée). Elle peut être effectuée après inclusion ou
adsorption afin de stabiliser l’enzyme immobilisée et limiter les phénomènes de fuites ou de
désorption.
L’agent réticulant le plus utilisés est le glutaraldéhyde (OHC-(CH2)3-CHO).
II.2.2. Fixation sur un support
II.2.2.1. Adsorption
Cette méthode est extrêmement simple à mettre en œuvre. Elleconsiste à assurer la rétention
de la molécule enzymatique à la surface d’un support insoluble, par interaction faible entre les
groupes fonctionnels de l’enzyme et du support. Parmi les diverses techniques
d'immobilisation, l'adsorption demeure la méthode la plus simple et la plus rentable.
Figure VI : Réticulation au glutaraldéhyde

144
Différents supports, tels que la silice, les billes de verre poreux, l'alumine, la terre diatomée, la
célite et le charbon actif ont déjà été exploités pour l'immobilisation d'enzymes. Différents
types de liaisons interviennent dans les réactions d'adsorption à savoir interactions de Van der
Waals, liaison hydrogène, transfert de charges (liaison ionique), échanges d’ions, interactions
hydrophobes.
La concentration en enzyme, le temps de contact, le pH, température, composition du milieu
et la quantité du support sont des paramètres qui influencent l'adsorption.
La célite est utilisée comme support d'adsorption dans la plupart des applications industrielles
des lipases et ce, en raison de son caractère poreux.
Le gel de silice compte parmi les principales substances absorbantes et se prête à de
nombreuses applications. C'est une forme d'acide silicique composé de granulés irréguliers et
poreux. Les produits de base sont essentiellement le silicate de sodium et l'acide sulfurique.
Ces substances sont exposées à une réaction chimique et produisent un gel de silice très riche
en SiO2. Le gel de silice est compatible avec tous les matériaux à l'exception des substances
fortement alcalines et l'acide fluorhydrique.
Figure VII : immobilisation par adsorption
II.2.2.2. Liaison covalente
L’immobilisation par liaison covalente a été développée dans le souci d’obtenir des liaisons
très solides entre enzymes et supports. Pour la réalisation de la liaison covalente, il faut une
activation préalable soit du support soit de l’enzyme car les groupes fonctionnels de l’enzyme
ne sont généralement pas suffisamment réactifs. Mais c’est surtout l’activation des supports
d’adsorption qui a fait l’objet de nombreuses études car l’activation des groupes fonctionnels
de l’enzyme est délicate et peut conduire à la dénaturation de l’enzyme.
Un cardodiimide est un groupe fonctionnel de type . En chimie organique
synthétique, les composés contenant la fonctionnalité de carbodiimide sont des agents de
déshydratation et sont souvent employés pour activer les acides carboxyliques vers la
formation d’amide ou d’esters et l’avantage d’avoir une très faible toxicité pour l’enzyme.
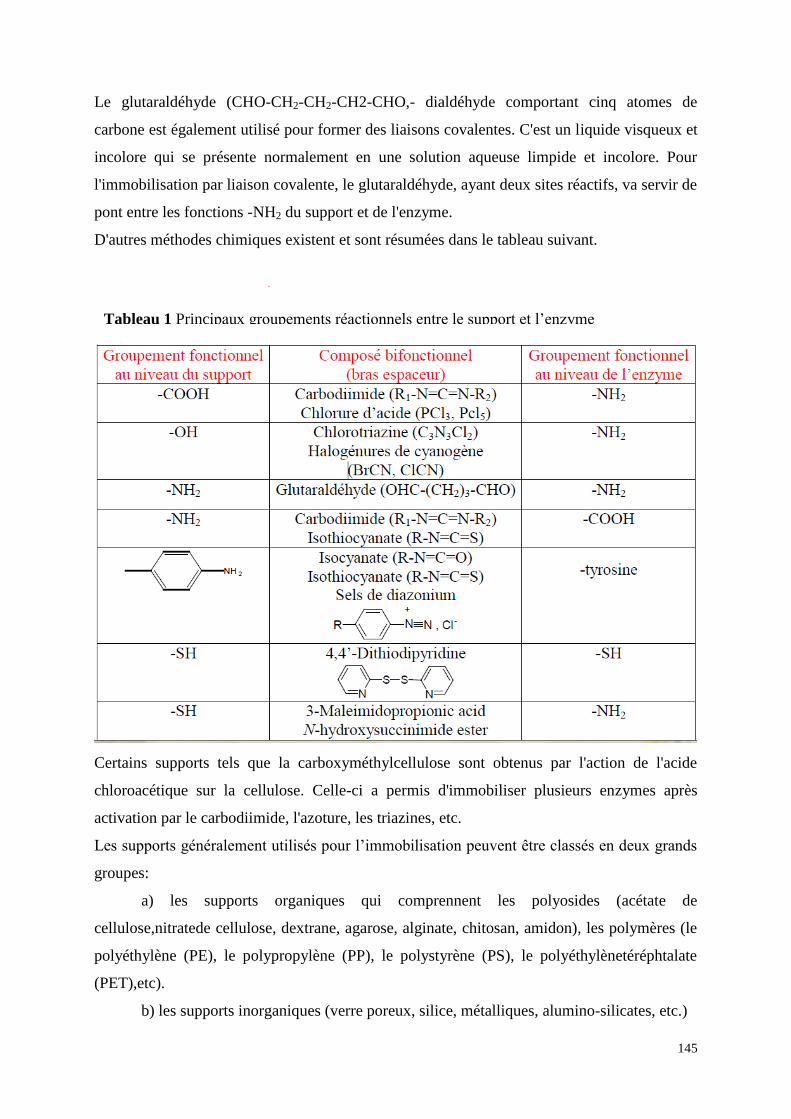
145
Le glutaraldéhyde (CHO-CH2-CH2-CH2-CHO,- dialdéhyde comportant cinq atomes de
carbone est également utilisé pour former des liaisons covalentes. C'est un liquide visqueux et
incolore qui se présente normalement en une solution aqueuse limpide et incolore. Pour
l'immobilisation par liaison covalente, le glutaraldéhyde, ayant deux sites réactifs, va servir de
pont entre les fonctions -NH2 du support et de l'enzyme.
D'autres méthodes chimiques existent et sont résumées dans le tableau suivant.
Certains supports tels que la carboxyméthylcellulose sont obtenus par l'action de l'acide
chloroacétique sur la cellulose. Celle-ci a permis d'immobiliser plusieurs enzymes après
activation par le carbodiimide, l'azoture, les triazines, etc.
Les supports généralement utilisés pour l’immobilisation peuvent être classés en deux grands
groupes:
a) les supports organiques qui comprennent les polyosides (acétate de
cellulose,nitratede cellulose, dextrane, agarose, alginate, chitosan, amidon), les polymères (le
polyéthylène (PE), le polypropylène (PP), le polystyrène (PS), le polyéthylènetéréphtalate
(PET),etc).
b) les supports inorganiques (verre poreux, silice, métalliques, alumino-silicates, etc.)
Tableau 1 Principaux groupements réactionnels entre le support et l’enzyme

146
Les supports organiques peuvent être chimiquement activés par différentes techniques, ce qui
constitue l’un de leurs principaux avantages car ils permettent la fixation d’enzymes par
différentes voies. Les supports inorganiques sont généralement plus stables (résistance à
l’usure, aux agents chimiques et aux bactéries) mais la fixation covalente sur ces supports est
difficile à cause de leur faible réactivité.
III. INFLUENCE DE L’IMMOBILISATION SUR LES ENZYMES
III.1. SUR LES CARACTERISTIQUES PHYSICO-CHIMIQUES
Une enzyme immobilisée est une enzyme liée par des moyens physico-chimiques en
surfaceou à l’intérieur d’un support solide. On cherche généralement à conserver son activité
enzymatique qui a tendance à diminuer après immobilisation du fait de possibles gênes
stériques et de limitations dans l’accessibilité au site actif ; aussi à augmenter sa stabilité dans
le temps (ce qui est souvent le cas)du fait de la structure plus rigide conférée par
l’immobilisation. On distingue normalement trois types de stabilité pour une enzyme
immobilisée:
1. Stabilité de stockage: amélioration de la conservation de l’enzyme par rapport à
l’enzyme libre dans les mêmes conditions.
2. Stabilité opérationnelle: évolution de l’activité de l’enzyme pendant des essais
répétitifs
3. Stabilité thermique: résistance à des températures plus élevées auxquelles l’enzyme
est normalement dénaturée.
Par exemple, la glycéraldéhyde-3-phosphate déhydrogénase (GAPDH) a été greffée à l’aidedu
glutaraldéhyde sur la surface du verre poreux. On observe une augmentation de la stabilité de
laGAPDH fixée sur la surface par liaison covalente par rapport à la stabilité de la même
enzyme enphase homogène. L’enzyme libre perd son activité assez rapidement (dès les 4
premières heures) tandis que l’activité de l’enzyme immobilisée reste stable pendant 30 jours.
La trypsine immobilisée sur des polyesters via différents espaceurs (polyéthylène
glycol(PEG)-diamine, sérum albumine Bovine (BSA), dextran aminé ou porteur de fonctions
aldéhyde),présente une stabilité thermique supérieure après l’immobilisation : l’activité reste
la même pendant environ 350 minutes à la température de 50°C alors qu’à la même
température l’enzyme libre présente une perte de la moitié de son activité en une durée de
moins de 20 min.

147
III.2. SUR LA CINETIQUE
Du fait de son immobilisation dans une structure, l’enzyme et la réaction catalysée
subissent des contraintes nouvelles. Celles-ci modifient à leur tour la cinétique enzymatique.
L’immobilisation des enzymes affecte beaucoup leurs propriétés cinétiques.En effet, à la
vitesse de réaction enzymatique proprement dite il faut ajouter l’effet du microenvironnement
qui conditionne toute l’activité catalytique. Ainsi, les phénomènes de diffusion limitent
l’accès du substrat au niveau du site enzymatique
Les propriétés cinétiques d'une enzyme immobilisée ne sont pas parfaitement corréléesà celles
de l'enzyme libre. La diminution du transfert de masse du substrat dans le support résultant de
l'immobilisation, augmente la valeur du Km de l'enzyme. Il s'ensuit que le domaine
correspondant à la cinétique de premier ordre de l'enzyme vis-à-vis du substrat est plus
étendu. De plus, l'orientation stérique de l'enzyme fait que le site actif ne peut être que
partiellement accessible, voire inaccessible au substrat: en d'autres termes, l'enzyme est
maintenue dans une conformation telle que la fonction catalytique est partiellement ou
totalement bloquée.
Enfin, le microenvironnement de l'enzyme, tel qu'il est déterminé par le support lui-même, a
une influence importante sur les paramètres cinétiques. Une explication de ce type de
comportement serait une interaction du support lui-même avec le site actif.
Toutes les enzymes ont un pH optimum auquel elles présentent une vitesse de réaction
maximale. Quand l'enzyme est immobilisée, le pH optimum peut se décaler suivant la nature
et la charge positive ou négative du support. Par exemple, un support chargé négativement
peut entraîner un pH de la couche enzymatique plus faible que celui du pH du milieu. En
conséquence, on observe un décalage du pH optimum vers une zone plus alcaline pour
compenser l'augmentation locale de la concentration en protons. On constate que l'effet est
contraire pour un support chargé positivement.
AVANTAGES ET INCONVENIENTS DE L’IMMOBILISATION
IV.1. Immobilisation par inclusion
Avantages
• Réaction de polymérisation et de gélification bien connues.
• Réaction chimiques avec l’enzyme limitée (inclusion dans des gels naturels).

148
• Application à toutes les enzymes (utilisation de mélanges).
• Obtention de supports de forma adaptables (films, billes, fibre…)
• Risque de fuite prévenue par réticulation du support après immobilisation.
Inconvénients
• Certaines polymérisations font appel à des agents dénaturants ou à des radicaux.
• Limitation diffusionnelle des substrats, des produits (problème de transfère de masse-
inaccessibilité de certains substrats)
• Risque de fuite de l’enzyme (le réseau formé est presque toujours trop large pour
retenir complètement l’enzyme)
IV.2. IMMOBILISATION PAR ADSORPTION
Avantages
• Très grande facilité de mise en œuvre.
• Possibilité de régénérer les complexes enzyme-support (si l’enzyme perd son activité
en cours de son fonctionnement, il est possible de la désorber et de la remplacer par une
préparation active)
• Fixation rapide du support et immobilisation simple et non dénaturante.
Inconvénients
• Malgré sa simplicité de mise en œuvre, elle demeure peu utiliser pour la conception
des bioréacteurs (risque de désorption)
• La fragilité de la fixation (les enzymes peuvent facilement se désorber sous l’action de
variation de pH, température…
• L’orientation de l’enzyme et mauvaise accessibilité au site actif.
• Les autres désavantages de cette méthode sont les risques de mauvaise accessibilité au
site actif, du fait de l’absence de bras espaceur, les difficultés d’utilisation en réacteurs
continus du fait d’une désorption progressive de la protéine, qui est alors libérée dans le
milieu réactionnel

149
Figure VIII :mauvaise accessibilité au site actif
IV.3. IMMOBILISATION PAR LIAISON COVALENTE
Avantages
• Stabilité accrue à cause de la liaison covalente
• Grand choix de méthodes différentes
• Grande variété de support minéraux (verre, silice, céramique…) et organiques
(cellulose, polymère synthétiques…)
• Possibilité d’effectuer l’immobilisation en présence de substrat pour éviter
l’inactivation (protection du site actif).
Inconvénients
• Mise en œuvre de réactions chimiques souvent complexes
• Longueur des expériences (étape de l’activation du support)
• Rendements de fixation inferieurs à 100%
• Risques de modifications chimiques de l’enzyme (perte d’activité)
• Nécessite de purifier l’enzyme préalablement
• Investissement important
I. APPLICATIONS INDUSTRIELLES D’ENZYMES IMMOBILISEES
L’immobilisation d’une enzyme sur un support est généralement choisie pour
améliorer sa stabilité dans les conditions opératoires employées. Dans certains cas, une
augmentation de l’activité de 700 a pu être obtenue. L’immobilisation permet aussi d’éviter
une oxydation ou une autolyse dans le cas des peptidases, de protéger l’enzyme des effets du
solvant et/ou du pH, de séparer plus facilement la protéine des produits et réactants pour une
nouvelles utilisation.

150
Exemple du bioréacteur
Les techniques d’immobilisation, d’inclusion ou d’encapsulation d’enzymes ont
permis d’optimiser, dans des conditions opératoires constantes et répétables, les réactions
utilisant des volumes importants de milieu réactionnel. Différents types de réacteurs sont
employés, certains sont constituées d’une membrane semi-perméable retenant l’enzyme mais
laissant diffuser substrats et produits, d’autres comprennent un support sur lequel l’enzyme est
immobilisée. Selon un principe analogue à la filtration tangentielle, le support est parcouru
par un milieu contenant le substrat.
L’utilisation de réacteurs enzymatiques est très diversifiée, par exemple le dosage d’un
inhibiteur difficile à quantifier par les techniques usuelles, peut être réalisé par le passage d’un
mélange de substrat et d’inhibiteur dans un réacteur enzymatique. L’analyse du milieu
réactionnel par chromatographie permet la détermination du rapport produit/substrat mais
aussi celui obtenu sans inhibiteur et de calculer la concentration de l’inhibiteur connaissant
son IC50 (fig. V.9).
L’agro-alimentaire
• Production d’acides aminés (pénicilline amidase )
• Production du glucose, sirop
• Production de fructose en utilisant glucose isomérase
• Amélioration des propriétés de boissons alimentaires : viscosité, clarification,
digestibilité, goût, etc
• Amélioration de procédé de panification

151
• Amélioration de la production de produits lactés.
•
Exemple d’application : Lactase immobilisé par inclusion
C’est une enzyme utilisée pour l’hydrolyse du lactose du lait. Elle est produite par
Saccharomyceslactis. L’immobilisation se fait par inclusion dans des fibres de triacétate de
cellulose.Les fibres ainsi obtenues sont mise en œuvre dans un réacteur fonctionnant en batch
alimenté avec du lait écrémé stérilisé.
La chimie industrielle et pharmaceutique
• Production d’acides divers (lipase, pénicilline amidase)
• Modification de la structure d’antibiotique.
Autres secteurs
• Textile
• Blanchissage
• Tannerie

152
CHAPITRE 8 .
INTRO DUCTIO N A l ’ ENZY M O LOG IE
INDUSTRIELLE

153
I. Historique de l’enzymologie Industrielle
C'est à la fin du XIXème siècle que les fondements de la technologie enzymatique moderne
sont apparus, principalement grâce aux travaux du chimiste danois C. Hansen, en 1864 qui fût
le premier à extraire la rennet à partir d'estomacs de veaux et d'en faire une préparation de
pureté encore relative à des fins industrielles alors que, en 1897, Eduard Buchner, en montrant
qu'un extrait cellulaire de levures seul était capable de propriétés catalytiques similaires à
celles des cellules vivantes entières, démontra que les enzymes étaient capables de
fonctionner seules pour convertir le glucose en éthanol et dioxide de carbone. Cette première
purification d'enzymes liait le concept à la terminologie apparue 20 ans plus tôt (W. Kühne,
1876/1978) qui, éthymologiquement, signifie "dans la levure".
Outre l'établissement des bases théoriques sur la spécificité vis-à-vis du substrat (Fisher), sur
la cinétique (Michaélis&Menten) et sur la nature macromoléculaire et protéique des enzymes,
c'est aussi pendant la période allant de 1900 à 1930 que naissent les premiers développements
de l'enzymologie appliquée à l'industrie. Ainsi, le scientifique japonais Takamine développe
un procédé de fermentation pour la production industrielle d'une amylase fongique, procédé
basé sur la culture d'Aspergillus oryzaesur riz humidifié ou traité à la vapeur. Cette
technologie de fermentation appelée culture "en surface" ou culture "semi-solide" n'est plus
aujourd'hui que très peu utilisée. Pendant cette période, l'enzymologie trouva également son
application dans l'industrie textile. Etant donné les conditions thermiques particulières
requises dans ce processus, l'utilisation d'une enzyme thermo-resistante était indispensable.
Une telle amylase fût isolée pour la première fois par Boidin&Effront en 1917 à partir de
Bacillus subtilis mais ne put être produite à large échelle, la technologie de fermentation "en
surface" étant mal adaptée à la culture de bactéries. C'est aussi à cette époque que sont
rapportés les premiers exemples de limitation du développement de produits enzymatiques par
les conditions dans lesquelles ces catalyseurs doivent travailler, ce qui demeure à l'heure
actuelle le principal souci des producteurs. En attestent les recherches menées par
l'industriel allemand O. Röhm menant en 1913 au dépôt d'un brevet pour une
préparation enzymatique à base de trypsine (extraite de glandes pancréatiques)
utilisable dans les détergents. Cette préparation, appliquée seule, s'est révélée efficace
pour l'élimination de taches de nature protéique sur les vêtements mais lorsqu'elle fût
incorporée au produit détergent complet, l'activité de la trypsine n'était que très limitée,
étant donné la forte alcalinité du milieu, une fois le produit dissous.
Durant l'entre-deux guerres, d'énormes progrès ont été réalisés sur les méthodes de
purification et de cristallisation des enzymes, mais peu de développements sont apparus dans

154
les technologies de fermentation permettant de produire les micro-organismes à l'échelle
industrielle. Le véritable essor des cultures dites "submergées", dans lesquelles la biomasse
est produite en milieu liquide agité et aéré dans des réacteurs fermés stérilisables, n'a eu lieu
qu'après la seconde guerre. Il fût concomitant avec le développement de plusieurs
préparations enzymatiques destinées, dans les années 50, essentiellement à l'industrie des
détergents. En alliant la sélection de protéases dites alcalines à une capacité de production
accrue, l'utilisation des enzymes dans l'industrie des détergents a progressé rapidement après
1965. Ce secteur est encore à l'heure actuelle le principal demandeur d'enzymes mais d'autres
domaines tel que la transformation de l'amidon ont également eu de plus en plus recours aux
enzymes lorsque sont apparus les avantages d'un traitement biochimique par rapport à
l'hydrolyse acide classiquement employée pour dépolymériser cette matière première. En
traitant les principaux types ou familles d'enzymes au cas par cas, les prochains chapitres ont
pour but de mettre en lumière la multitude et la variété des secteurs industriels intéressés par
leur exploitation.
II. LES ENZYMES DANS LES DIFFERENTS SECTEURS
L'utilisation des bioconversions à la production industrielle est souvent appelée
biotechnologie blanche ("white biotechnology"). Les bioconversions enzymatiques,
représentent une partie de la biotechnologie blanche. Elles font aussi partie de ce que
l'on appelle la chimie verte. Les applications commerciales les plus importantes des
enzymes sont: lessive (protéases, lipases et amylases), production de sirop de fructose à
partir d'amidon de maïs (8.5x10 tonnes/an aux USA, amylases et glucose isomérase),
production d’éthanol (amylase ou cellulase). Par contre, il existe de plus en plus
d'applications en chimie fine. Le travail de R&D (Research/development) se fait à
l’intersection entre la chimie, la biochimie, et le génie chimique.
Avantages des enzymes en industrie
variés; donc le site actif peut être chimio-, régio- et stéréosélectif.
000 000 fois plus rapide que les
réactions non-catalysées et peuvent catalyser jusqu'à 10 évènements par seconde (par
exemple, l'anhydrase carbonique est limitée par la vitesse de diffusion des substrats).
: insuline,
aspartame, etc.)

155
Désavantages des enzymes
solubles
-facteurs et co-enzymes
tabilité (même en
conditions réactionnelles relativement douces, par rapport aux conditions de catalyse
chimique
- dénaturation (température)
- oxydation de certains résidus (Met, Cys, Trp)
- hydrolyse (acides, bases, protéases)
- mauvais repliement (perte d'activité, insolubilité).
Le marché mondial des enzymes est en croissance rapide depuis une plusieurs
d'années et représente environ 1,5 milliards de dollars US (Laretta-Garde, 1997).
Le champ d'application des enzymes ne cesse de s'élargir. En 2007, le marché mondial
des enzymes industrielles a été estimé à 2,3 milliards de dollars. Une croissance de 4 % était,
par ailleurs, obtenue pour l’année 2012. Avec un taux d’utilisation d’enzymes industrielles de
41%, le domaine pharmaceutique se place en tête du classement suivi par l’industrie des
détergents (17%), l’industrie agroalimentaire (17%), la papeterie (17%) et le textile (8%)
Comme le montre le graphique de la figure 1, leur incorporation dans les produits détergents
représente près de la moitié de la part de ce marché si l'on considère le volume commercialisé.
Le secteur textile est, avec plus de 10 % de parts du marché en volume, l'autre demandeur
principal d'enzymes dites "techniques" par opposition aux enzymes utilisées en agro-
alimentaire. D'autres applications des enzymes techniques restant plus marginales à l'heure
actuelle en terme d'importance de marché se trouvent dans le domaine pharmaceutique, dans
la fabrication du papier, dans l'industrie des huiles et graisses ou encore dans la fabrication du
cuir.
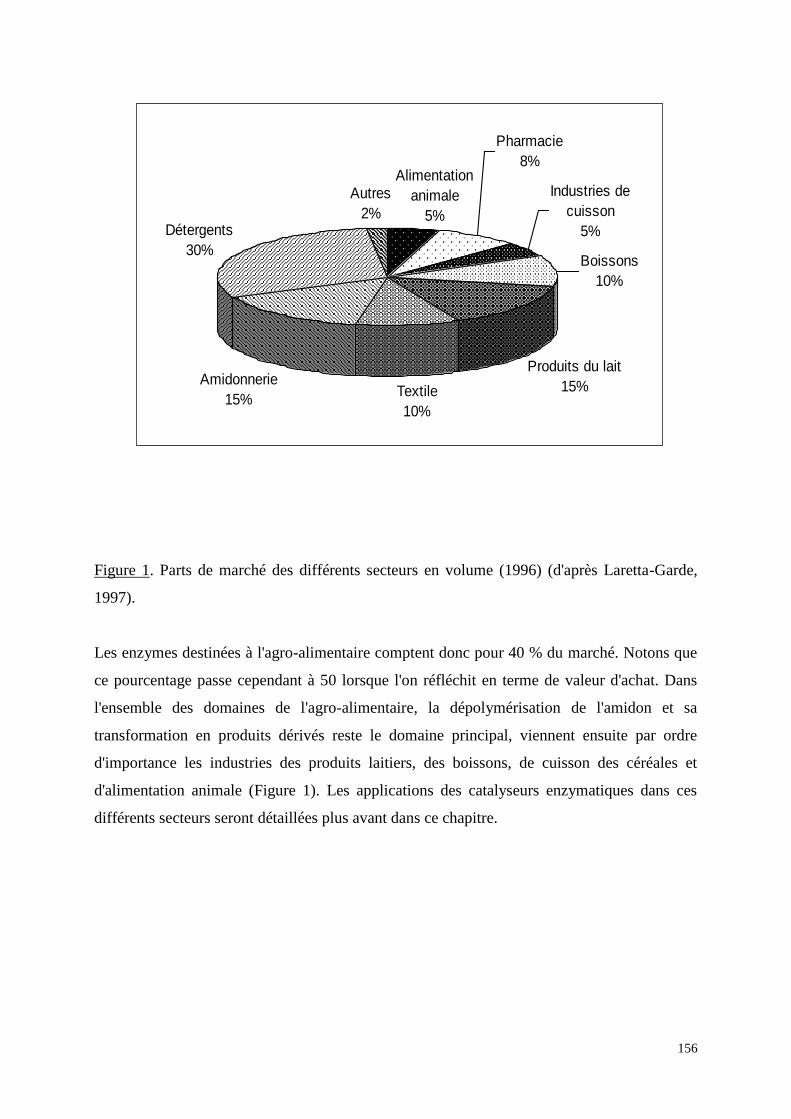
156
Figure 1. Parts de marché des différents secteurs en volume (1996) (d'après Laretta-Garde,
1997).
Les enzymes destinées à l'agro-alimentaire comptent donc pour 40 % du marché. Notons que
ce pourcentage passe cependant à 50 lorsque l'on réfléchit en terme de valeur d'achat. Dans
l'ensemble des domaines de l'agro-alimentaire, la dépolymérisation de l'amidon et sa
transformation en produits dérivés reste le domaine principal, viennent ensuite par ordre
d'importance les industries des produits laitiers, des boissons, de cuisson des céréales et
d'alimentation animale (Figure 1). Les applications des catalyseurs enzymatiques dans ces
différents secteurs seront détaillées plus avant dans ce chapitre.
Alimentation
animale
5%
Pharmacie
8%
Industries de
cuisson
5%
Boissons
10%
Produits du lait
15%Textile
10%
Amidonnerie
15%
Détergents
30%
Autres
2%

157
IV.4.10. Applications des enzymes
IV.4.10.1. En boulangerie
Les peroxydases tout comme les polyphénoloxydases (ou laccases responsables de l'oxydation
des composés phénoliques en quinones), de part leur effet oxydant sont susceptibles d'induire
des liaisons covalentes entre les pentosanes substitués par l'acide férulique (subissant
l'oxydation) et les protéines présents dans le gluten. Macroscopiquement cela résulterait en
une amélioration de la consistance de la pâte et un accroissement du volume du pain car la
structure renforcée du réseau protéique permettrait de mieux piéger le CO2 produit lors de la
fermentation.
IV.4.10.2. A usages analytiques
La peroxydase isolée de raifort est une des deux enzymes les plus employées dans les kits de
diagnostiques basés sur des tests immunologiques. Alors que dans les applications
industrielles les préparations enzymatiques sont peu pures, il en va différemment pour les
catalyseurs appelés à être utilisés à des fins de diagnostique. Dans ce dernier cas, le test est
basé sur la haute spécificité du catalyseur vis-à-vis du substrat et la présence d'activités
secondaires est inacceptable. C'est pourquoi les enzymes de diagnostique doivent être d'une
grande pureté, cela impliquant un traitement approfondi du jus de fermentation ou des tissus
faisant bien souvent appel à des étapes de chromatographie.
Les analyseurs automatiques permettant de doser le glucose ou le cholestérol sont basés sur
l'utilisation des oxydases respectives et de peroxydase qui permet de quantifier l'H2O2 produit
dans la première réaction. Dans ce cas, la peroxydase catalyse souvent la réaction de l'H2O2
avec un composé de type o-dianisidine qui forme une molécule colorée dont la concentration
est mesurée par spectrophotométrie.
L'utilisation de kits de diagnostique faisant appel à un test immunologique (ELISA, enzyme-
linked immunosorbent assay) s'est considérablement développée dans le secteur médical mais
aussi en agro-alimentaire pour la détection de molécules aussi diverses que les aflatoxines, les
résidus d'antibiotiques, de pesticides ou pour la détection de microorganismes tels les
Salmonelles ou Listeria. Les anticorps utilisés pour la reconnaissance de la molécule
recherchée sont attachés à une enzyme qui permettra de développer une réaction
colorimétrique proportionnelle à la quantité d'anticorps ayant fixé leur substance cible

158
(exemple dans la figure IV.5.). Dans ce contexte les peroxydases sont les enzymes les plus
utilisées car elles peuvent catalyser l'oxydation de multiples composés chromogéniques
permettant des détections très sensibles.
Figure IV.5. Utilisation de la peroxydase dans les tests immunologiques.
Enfin, avec un principe similaire de fonctionnement basé sur le dosage colorimétrique,
signalons également l'utilisation du couple glucose oxydase - peroxydase dans la fabrication
des bandelettes pour le dosage du glucose dans les urines et dans le sang à usage notamment
des personnes diabétiques.
L'exploitation de l'activité des peroxydases pourrait s'étendre à d'autres secteurs tels que
l'industrie papetière ou dans le prétraitement de matériaux cellulosiques. Mais le potentiel de
certaines peroxydases à dégrader des composés organochlorés phénoliques ou non tels que le
DDT, la dioxine, le pentachlorophénol ou des composés hydrocarbonés polycycliques
aromatiques tels que l'anthracène, le fluorène, le phénanthrène a également été rapportée.
Leur utilisation pourrait donc être efficace pour la dépollution ou la biotransformation de
composés organochlorés (pesticides ou herbicides) dans les sols ou dans l'eau.
La production de peroxydases (d'une lignine et d'une manganèse peroxydase avec Mn2+
en
tant que premier substrat pour l'oxydation) synthétisées par le champignon Phanerochaete
chrysosporium a été étudiée au Centre Wallon de Biologie Industrielle en vue de son
utilisation dans l'industrie du papier. Le pouvoir ligninolytique de cette enzyme pourrait être
exploité pour générer une délignification plus sélective des pulpes Kraft avec une altération
des fibres de cellulose limitée comparativement à celle engendrée par le procédé chimique par
alcalinisation à haute température. Cela permettrait d'obtenir du papier plus résistant et donc
de meilleure qualité.
AnticorpsSubstance
cible
Peroxydase
attachée
Peroxydase
attachée
AnticorpsSubstance
cible liée
2H2O + 4-aminoantipyrine + p-OH-benzène sulfonate Colorant quinoneimine + 4H2O
H2O2
AnticorpsSubstance
cible
Peroxydase
attachée
Peroxydase
attachée
AnticorpsSubstance
cible liée
Peroxydase
attachée
AnticorpsSubstance
cible liée
2H2O + 4-aminoantipyrine + p-OH-benzène sulfonate Colorant quinoneimine + 4H2O2H2O + 4-aminoantipyrine + p-OH-benzène sulfonate Colorant quinoneimine + 4H2O
H2O2

159
Ces peroxydases pourraient aussi intervenir dans le blanchiment de la pâte à papier en se
substituant à des composés chimiques oxydants normalement utilisés pour éliminer la lignine
responsable de la coloration. Le traitement chimique entraînant le rejet de produits toxiques,
l'utilisation d'un procédé enzymatique permettrait de réduire la pollution environnementale.
V. Autres applications des enzymes
L’utilisation d’enzymes pour réaliser des travaux de recherche scientifique ne peut
s’envisager que si la pureté de ces catalyseurs biologiques est suffisamment poussée. Le
clonage des gènes a révolutionné les méthodes de purification classiques des enzymes en
permettant un isolement rapide, une forte concentration et une simplification des méthodes de
fractionnement.
V.1. Protéolyse
La protéolyse facilite généralement l’utilisation des protéines, elle peut aussi permettre
la quantification des résidus aminoacides.
Traditionnellement, l’hydrolyse totale des protéines qui a pour but de libérer tous les
acides aminés en vu d’établir la proportion de chacun d’eux est effectuée en solution d’acide
chlorhydrique 6M chauffée à 110°C pendant 24 à 48 heures. L’un des désavantages de cette
méthode est que sous ces conditions, le tryptophane est totalement détruit, la sérine et la
thréonine le sont partiellement, asparagine et l’acide glutamique sont transformés
respectivement acide aspartique et glutamique. La digestion enzymatique de protéine et de
peptides permet d’éviter ces inconvénients, en particulier, lorsque la composition en
asparagine et glutamine est recherchée.
Pour des peptides contenant moins de 35 résidus aminoacides, la digestion complète
s’effectue avec l’aminopeptidase M et la prolidase. Pour des peptidases de taille supérieure ou
des protéines, une première digestion obtenue avec des peptidases non spécifiques telles que
la pronase est suivie de celle utilisant l’aminopeptidase M et la prolidase. Les digestions sont
effectuées à 37°C pendant 24 heures environ.

160
V.1.1. La Pronase
Le nom pronase (EC 3.4.24.4) correspond à un groupe de dix enzymes protéolytiques
obtenues à partir du microorganisme Streptomyces griseus. De ce fait, la pronase ayant peu de
spécificité est capable d’hydrolyser de nombreuses liaisons peptidiques. En présence de
calcium, l’activité de cette enzyme est maximale entre pH 7 et 8. Etant active dans les milieux
dénaturants utilisés pour déployer et/ou dissocier les structures protéiques tels que SDS à 1%,
chlorure de guanidium à 6 M ou urée à 8 M, la pronase permet une obtention rapide de petits
fragments de peptides.
V.1.2. L’Aminopeptidases M
L’aminopeptidase M (EC3.4.11.2) est une métalloprotéase à zinc trouvée en particulier
dans les membranes plasmiques de cellules entérocytaires et les microsomes de rein. Elle
libère séquentiellement les résidus aminoacides à partir des l’extrémité N-terminale de
peptides ou de protéine. Toute fois, lorsque les peptides contiennent soit une extrémité N-
terminale bloquée, soit une séquence X-Pro, où X correspond à un résidu à chaîne la térale
encombrante telle que Leu, Trp, Tyr, Met-sulfone, le clivage ne s’effectue pas. C’est pour
cette raison que la prolidase est utilisée conjointement à l’aminipeptidase M pour obtenir une
hydrolyse totale.
V.1.3. Prolidase
La prolidase (EC 3.4.13.9) possède une haute spécificité en hydrolysant à partir de
l’extrémité C-terminale les peptides contenant un résidu de proline ou d’hydroxyproline
(HyP). L’enzyme en ne clivant pas les tripeptides montre la meilleure affinité pour Ala-Pro et
Gly-Pro. La présence d’ions manganèse dans le milieu est réactionnel est indispensable pour
une activité optimale se situant entre pH 6 et 8.

161
V.2. Réaction en chaîne par l’ADN polymérase (PCR)
La technique de réaction en chaîne par l’ADN polymérase appelée PCR (Polymerase
Chain Réaction) a été mise au point par Mullis et collaborateurs en 1986. La PCR a
révolutionné la biologie moléculaire en permettant d’amplifier selon un facteur très important
le l’ordre de 2.109 le nombre de copie d’une ADN isolé. Cette technique emploi en particulier
une ADN polymérase thermostable dans un milieu réactionnel soumis à des transitions
cycliques de température.
V.2.1 Principe
En exploitant les propriétés de l’ADN polymérase, la PCR utilise un ADN double
brin, appelé amplicon, qui est dénaturé par une température de 95°C en mono brin. En
diminuant la température à 55°C une hybridation de chaque brin d’ADN est effectuée avec
une courte séquence nucléotidique servant d’amorce. L’extension de l’ADN est mise en
œuvre grâce à une plolymérase obtenue de microorganismes thermophile comme Thermus
aquaticus (Taq polymérase) et qui présente une activité optimale à 72°C. Un nouveau cycle
de température permet de séparer les brins d’ADN nouvellement dupliqués et d’effectuer une
seconde hybridation avec les mêmes séquences amorces suivie d’une autre extension. Il s’en
suit que le nombre de d’exemplaires d’ADN croit de manière exponentielle. La séquence
nucléotidique comprise entre les positions des amorces est majoritaire (V.2).
La longueur des séquences amorces influence l’optimisation de la PCR : une longueur
de 25 à 30 bases constitue un bon compromis afin d’éviter des différences de leur température
de demi-dénaturation (Tm) supérieure à 2°C. Pour la même raison, la teneur en GC des
amorces ne doit pas être supérieure à 40 à 50%. En outre, la valeur du Tm doit être la plus
haute possible pour garantir la spécificité des produits.
L’activité de l’ADN polymérase nécessite la présence d’ions magnésium dans le
milieu réactionnel. En outre l’ion magnésium améliore la fiabilité de cette technique ne
contribuant à limiter les erreurs de copie des produits amplifiées. Toute fois cette propriété
peut être utilisée en évolution dirigée par l’addition de manganèse dans le milieu réactionnel,
cette technique est appelée error-prone PRC. Des polymérases isolées d’organismes et bien
adaptés à la PCR sont commercialement disponibles (tableau V.1).
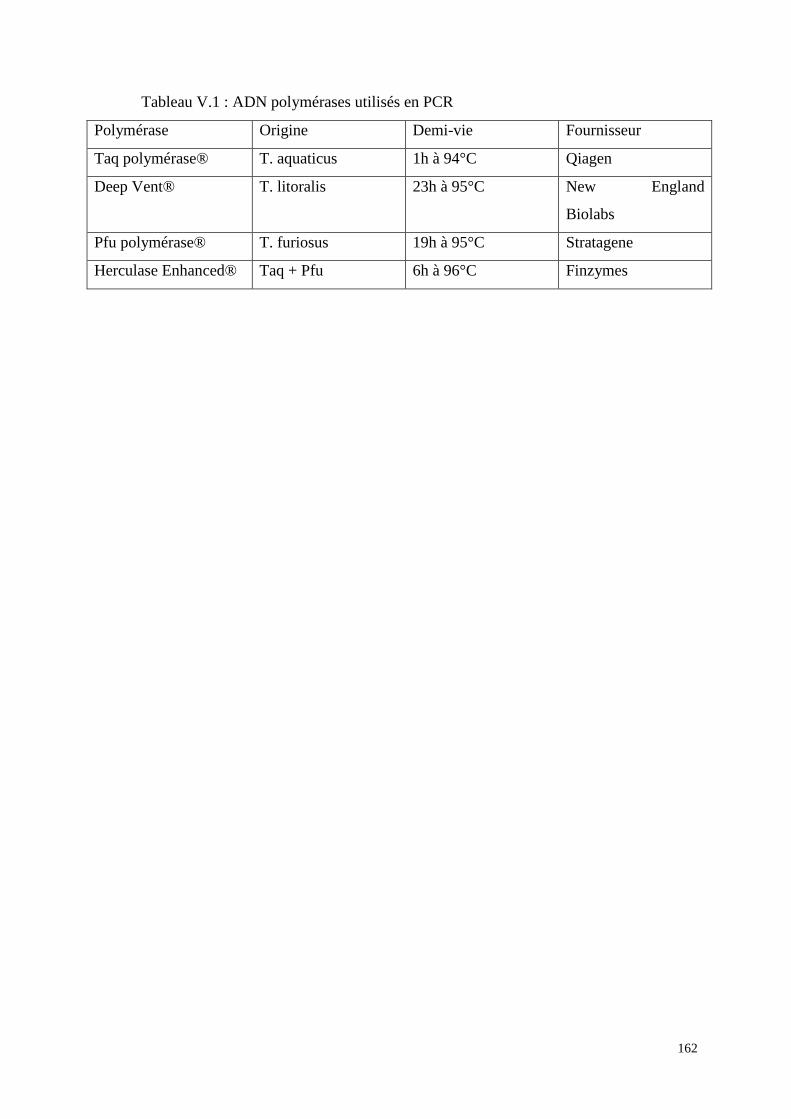
162
Tableau V.1 : ADN polymérases utilisés en PCR
Polymérase Origine Demi-vie Fournisseur
Taq polymérase® T. aquaticus 1h à 94°C Qiagen
Deep Vent® T. litoralis 23h à 95°C New England
Biolabs
Pfu polymérase® T. furiosus 19h à 95°C Stratagene
Herculase Enhanced® Taq + Pfu 6h à 96°C Finzymes

163

164
V.2.2 ADN polymérase I
L’ADN polymérase I catalyse aussi un processus chargé de respecter la fidélité de
l’élongation. Une erreur occasionnelle dans l’addition d’un nucléotide s’accompagne d’une
déformation de l’ADN due à un mauvais appariement des bases. La reconnaissance de cette
déformation par un domaine contenant une activité 3’,5’-exonucléase permet grace à une
« fonction d’édition » de remplacer le nucléotide incorrect par celui respectant la
complémentarité des bases. La structure tridimensionnelle de la polymérase I a la particularité
de présenter plusieurs protubérances ressemblant à une sorte de main (doigts, pouce, paume)
permetant ainsi à l’enzyme d’avoir un contact étroit avec la chaîne d’ADN (fig.V.3)
V.3. Endonucléases
Pour incorporer un fragment d’ADN dans un vecteur plasmidique il est indispensable
de dispose de méthodes permettant de couper mais aussi d’assembler un ADN double brin. La

165
découverte d’endonucléases spécifiques appelées aussi enzymes de restriction au cours des
années 60 a constitué une étape décisive pour le clonage des gènes.
Les enzymes de restriction sont classées en trois types (I,II,III). Les enzymes des types
I et III nécessitent la présence d’ATP pour effectuer l’hydrolyse de l’ADN. Toute fois, les
enzymes de type I effectuent un clivage de manière aléatoire alors que celles de type III sont
capables d’hydrolyse l’ADN double brin au niveau d’un site formé par une séquence
nucléotidique précise. Les endonucléases de type II sont largement utilisées pour cloner et
séquencer les molécules d’ADN. Leur activité qui n’est pas ATP-dépendante se manifeste
spécifiquement au niveau de séquences nucléotidiques particulières. La reconnaissance d’une
séquence de quatre à six nucléotides peut s’effectuer aussi bien sur l’un des brins que sur
l’autre (brin complémentaire). Toutes les endonucléases de type II, après clivage, libèrent une
extrémité 3’-OH et une extrémité 5’-PO42-
(fig V.4).
V.3.1. Exemple : EcoRI
EcoRI correspond à une endonucléase de type II isolée de E. coli reconnaissant la
séquence GAATTC. Cette séquence lue de 5’ vers 3’ est la même sur chaque brin. Deux
clivages, effectués en quinconce, forment deux extrémités 5’ caractérisées par le depassement
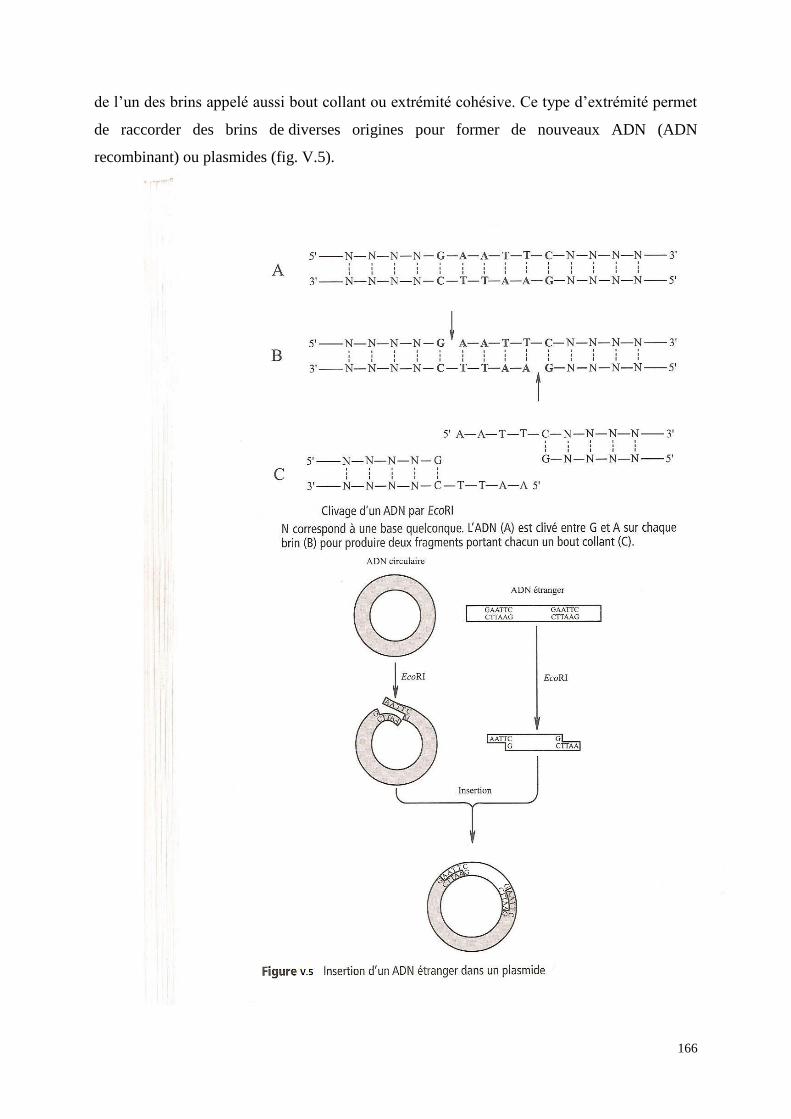
166
de l’un des brins appelé aussi bout collant ou extrémité cohésive. Ce type d’extrémité permet
de raccorder des brins de diverses origines pour former de nouveaux ADN (ADN
recombinant) ou plasmides (fig. V.5).

167
V.3.2. Carte restrictive
Les enzymes de restriction sont principalement utilisées pour cartographier les
molécules d’ADN sous forme de carte de restriction. Le nombre de paires de bases de certains
ADN peut être relativement grand, leur clivage aboutit généralement à un mélange de
fragments de restriction. L’action simultanée de plusieurs endonucléases à spécificité
différente permet d’augmenter la précision de la cartographie en fournissant un plus grand
nombre de repères correspondant aux sites de coupure. La séparation et l’identification de ces
fragments par électrophorèse sur gel d’agarose facilite l’établissement des cartes de
restriction. Le marquage de l’une des extrémités de l’ADN initial permet généralement de
sélectionner l’une des deux propositions du positionnement des sites de coupure.
Obtenues à partir d’ADN recombinants, beaucoup d’enzymes de restriction sont
désormais disponible en quantité significative afin de satisfaire les besoins de la recherche
scientifique.
V.4. Marquage enzymatique
La fixation covalente de certaines enzymes à des molécules biologiques permet
notamment de réaliser des dosages.
V.4.1. Phosphatase alcaline
La phosphatase alcaline (EC 3.1.3.1) de la muqueuse intestinale bovine est une
enzyme stable, de taille modérée (140 kDA) au tur over number élevé qui peut agir sur des
substrats chromogène ou fluorescents. Le marquage des immunoglobulines (IgG) constitue
l’une des applications de la phosphatase alcaline.
Le couplage de l’enzyme et de l’anticorps d’effectue généralement avec le
glutaradéhyde qui réagit principalement avec des groupes aminés sans former de base de
Schiff. Cette méthode de marquage a le mérite d’être simple, de présenter peu d’autocouplage
de l’enzyme ou de l’anticorps et permet de conserver à la fois l’activité enzymatique et
l’immunoréactivité des conjuguées obtenus.
V.4.2. Test ELISA
Ce test dont le nom provient de « enzyme-linked immunoadsorbant assay » emploi des
anticorps pour révéler et doser une très faible quantité d’antigène. Les protéines contenues
dans l’échantillon sont adsorbées sur la paroi en plastique des puits d’une plaque de
microtitration. La plaque est lavée avec une solution protéique (albumine, gélatine, caséine,

168
sérum…) qui a pour but de saturer tous les sites de fixation disponibles (fig. V.6). La surface
est ensuite traitée avec une solution contenant un anticorps dirigé contre la protéine d’intérêt,
c’est-à-dire l’antigène.
Ces anticorps portent le nom d’anticorps primaires. Après une période d’incubation et
élimination par lavage, la plaque est traitée avec une solution d’anticorps (anticorps
secondaires) reconnaissant les anticorps primaire. Les anticorps couplés à une enzyme
(peroxydase,β-galactosidase ou phosphatase alcaline) permettent de quantifier l’antigène
grâce à une réaction colorée.
Selon l’enzyme couplée aux anticorps secondaires, la réaction emploi, par exemple
comme substrat, l’o-nitrophénylgalactoside pour la β-galactosidase , p-nitrophénylphosphate
pour la phosphatase alcaline ou o-phénylènediamine + H2O2 pour la peroxydase (fig V.7).

169
V.5. Dosage par voie enzymatique
Divers composés biologiques peuvent être dosés par voie enzymatique, souvent grâce
au couplage de plusieurs réactions du métabolisme. Beaucoup de ces méthodes utilisent des
réactants chromogènes qui permettent l’emploi de spectrophotomètres (visble et/ou
ultraviolet). Un temps d’incubation généralement long est nécessaire pour obtenir la
conversion complète du composé à doser. Certains de ces dosages sont commercialisés sous
forme de kit pour faciliter la mise en œuvre et la répétabilité.
V.5.1. Glucose et fructose
Le D-glucose en présence d’ATP et d’hexokinase est d’abord converti en glucose-6-
phospohate et en ADP. Le glucose-6-phosphate libéré est ensuite transformé en 6-phospho-D-
δ-lactone par la glucose-6-phosphate déshydrogénase en présence de NAD+. La
transformation de NAD+en NADH permet de mesurer une variation de densité optique (DO) à
340 nm puisque seul NADH absorbe à cette longueur d’onde.
Le D-fructose est dosé grâce à la phosphoglucoisomérase qui le converti en glucose-6-
phosphate. Ce dernier est à son tour utilisé comme substrat par la glucose-6-phosphate
déhydrogénase pour produire le dérivé gluconolactone et du NADH. Le dosage du saccharose

170
peut être aussi réalisé puisque son hydrolyse par la β-fructosidase fournit du D-glucose et du
D-fructose. Le fructose est ensuite transformé en glucose-6-phosphate puis en dérivé lactone
avec production de NADH. Pour le dosage du saccharose, une alternative est possible avec la
transformation du glucose seul en glucose-6-phosphate puis en dérivé lactone.
V.5.2. Cholestérol
Le cholestérol et les esters de cholestérol peuvent être dosés par méthode
chimique mais plus récemment par voie enzymatique employant la cholestérol estérase
(CE), la cholestérol oxydase (CO) et la péroxydase (POX). Cette méthode utilise en
outre, l’Amplex Red (10-acétyl-3,7-phénoxazine) qui par oxydation se transforme en
résorufine. La haute fluorescence de la résorufine permet d’augmenter très
significativement la sensibilité du dosage. La méthode disponible sous forme de kit,
emploi trois réactions couplées.
Le dosage du cholestérol est effectué par spectrofluorimétrie dont les longueurs d’onde
d’excitation et d’émission de la résorufine sont respectivement à 563 et 587 nm. La variation
moyenne de la fluorescence en fonction du temps permet d’obtenir une réponse
proportionnelle à la concentration de cholestérol (fig V.8).

171
V.6. Préparation et extrait cellulaires
V.6.1. obtention de cellules isolées
La préparation de cellules à partir de tissus peut être réalisée par traitement mécanique,
toute fois, avec ce moyen, nombre de cellules sont lysées et les rendements sont généralement
moyens. Les méthodes enzymatiques permettent au contraire d’éviter la lyse cellulaire et
garantissent généralement des rendements satisfaisants. Selon le type de tissu, le choix de
l’enzyme comme agent de dispersion peut varier puisque l’action digestive des protéases n’a
pas la même efficacité :
Dispase<collagènase<hyaluronidase<élastase<papaïne<trypsine
Provoquant la libération des cellules, une digestion des tissus par la trypsine est aussi
susceptible d’altérer les protéines de surface ancrées dans la membrane plasmique. La
substitution de la trypsine par la collagénase permet d’agir essentiellement sur la matrice
intercellulaire et peu à la surface des cellules. En effet, ces enzymes clivent spécifiquement
les liens X-Gly (X représentant un résidu aminoacide neutre) trouvés dans les séquences Pro-
X-Gly-Pro du collagène et rarement dans celles des autres protéines. L’emploi de la
collagénase d’origine bactérienne, disponible commercialement est très répandu en biologie,
embryologie ou histologie ; une bonne pureté de l’enzyme doit être choisie pour obtenir
d’excellents résultats. En effet, certaines préparation de collagénase peuvent contenir d’autres
protéases susceptibles d’altérer la cellule.
V.6.2. Repiquage cellulaire
Le repiquage ou transfert de cellules à partir de cultures monocouche est facilité par
l’emploi d’enzymes. Une exposition à la trypsine interrompt les fibres unissant les cellules à
la paroi des flacons. Cette adhésion généralement observée pour les cellules monocouches se
traduit par une forme allongée des cellules. L’action de la trypsine menée à 4°C pour éviter
toute altération au lyse cellulaire s’accompagne d’un changement de forme (aspect arrondi).
L’activité trypsique peut être arrêtée par ajout d’un inhibiteur comme l’inhibiteur trypsique du
pancréas de bœuf (BPTI) ou par lavage de cellules. Après lavage, les cellules transférées sont
employées pour la culture de cellules souches.

172
V.6.3. Lyse cellulaire
Pour purifier des macromolécules biologiques, il est souvent nécessaire d’effectuer
une lyse cellulaire. Les bactéries par exemple possèdent une paroi rigide constituée de
peptidoglycanes qui doit être rompue pour libérer leur contenu. Traditionnellement, cette
opération est accomplie par des moyens mécaniques (presse de French, appareils à
ultrasons…). Toute fois, ces techniques laborieuses et consommatrices de temps aboutissent
souvent à une lyse incomplète. L’utilisation d’enzymes capables de provoquer la lyse
cellulaire constitue une alternative simple et efficace qui se dispense d’équipement spécifique.
Le lysosyme est enzyme globulaire de 129 résidus aminoacide (140 kDa)
principalement trouvé dans le blanc d’œuf et les sécrétions biologiques comme les
larmes. Cette enzyme a la propriété d’hydrolyser les liaisons osidiques β 1→4 des
polysaccharides constituant la paroi bactérienne, en particulier celles des résidus N-
acétyl muramique (NAM) et N-acetylglucosamine (NAG). Le clivage de ces liens
s’effectue dans une poche où les nombreuses interaction hydrogènes assurent la
reconnaissance spécifique du substrat. Cette hydrolyse suit un mécanisme catalytique
acide-base.
La lyse cellulaire est obtenue par exemple, en plaçant les bactéries dans un milieu
tamponné contenant du lysozyme incubé à 37°C pendant 1 heure. La centrifugation du milieu
à 15000 g pendant quelques minutes permet de séparer l’extrait cellulaire des bactéries lysées.
V.7. Synthèse organique
La synthèse de molécules par voie enzymatique est de plus en plus utilisée bien que la
synthèse chimique soit économique, rapide et conduise généralement à de bons rendements.
Les enzymes ont l’avantage d’effectuer des réactions douces, d’obtenir un produit défini, de
porter un label naturel et d’éviter notamment les solvants et/ou réactifs aux effets toxiques.
Les réactions de synthèse utilisant des enzymes sont nombreuses.
V.7.1. Formation d’une liaison C-C
Les aldolases sont capables de catalyser la formation d’une liaison C-C hautement
stéréospécifique par une réaction de condensation, encore appelée addition aldol. La
formation de liaison C-C peut être réalisée par d’autres voies enzymatiques comme celles
mise en œuvre par la solanapyrone synthétase isolée du champignon Alternaria solana qui
catalyse une réaction de type Diels-Adler.

173
V.7.2. Greffage d’oligosaccharides
Le greffage de résidus osidiques permet par exemple de glycosyler une protéine en
changeant ses propriétés physicochimiques.
In vivo le greffage s’effectue grâce aux glycosyltransférases mais l’utilisation in vitro
de ces enzymes se heurte au coût élevé de la réaction et souvent à des difficultés dues à
l’instabilité des sucres activés utilisés comme donneurs.
Les glycosidases peu onéreuses peuvent être substitués aux glycosyltransférases. Les
α-fucosidases ou α-sialosides, difficiles à synthétiser par voie chimique, peuvent être obtenu
grâce à l’utilisation de donneurs portant un groupe nitrophényle et dont le greffage est
effectué par α-fucosidase ou la sialidase. Le site actif des glycosidases comporte deux résidus
aminoacides à groupe carboxylique dont l’un fixe le donneur. Ce dernier est greffé sur
l’accepteur en position anomérique. A forte concentration de donneur ou d’accepteur, le
rendement de tranglycosylaation est augmenté au détriment de celui de la réaction
d’hydrolyse. Toute fois cette réaction conduit à plusieurs produits qu’il est nécessaire de
séparer.
VI. Applications médicales et pharmaceutiques
En médecine, les enzymes sont employées essentiellement :
- Comme indicateurs de la concentration (ou activité) d’enzymes dans les fluides
biologiques (sérum, urine, liquide céphalo-rachidien…) pour déceler une pathologie ;
- Comme réactifs pour l’analyse de composés non enzymatiques (substrats, molécules
biologique, médicaments…) ou d’enzymes.
- Comme agents thérapeutiques.
VI.1. Aide au diagnostique
Dans la recherche de pathologies, les enzymes sont fréquemment pratiquées par les
laboratoires d’analyse médicale, le sang constituant la cible principale des prélèvements. Les
enzymes circulant sont classées comme étant plasma spécifiques ou non plasma spécifiques.
Les enzymes plasma spécifiques ont habituellement une activité supérieure comparée à celle
trouvée dans les autres tissus. La thrombine, plasmine, cholinestérase et céruloplasmine
constituant des exemples, sont principalement synthétisés par le foie. Des maladies
héréditaires affectant le foie comme l’hémophilie s’accompagnent généralement de taux
anormaux d’activité enzymatique. Une activité nulle ou très faible de cholinestérase peut

174
provoquer la paralysie des muscles lors de l’administration de d’anesthésiques comme la
succinylcholine. Une mesure d’activité de la chlolinestérase permet de prendre compte de
cette éventualité.
Les enzymes non plasma spécifiques sont intracellulaires, mais dans le cas de maladies, elles
peuvent se retrouver libres dans le plasma à de fortes concentrations. C’est le cas pour
l’infarctus du myocarde, l’hépatite, la pancréatite ou le cancer de la prostate. La nature des
enzymes libérés dans le plasma reflète aussi l’importance des lésions : une inflammation sans
gravité peut provoquer l’apparition d’enzymes cytoplasmiques. Par contre l’apparition
d’enzymes mitochondriales peut résulter d’une nécrose tissulaire comme l’hépatite. L’aide au
diagnostique est souvent facilité par la provenance de l’enzyme retrouvée en quantité
anormale (tableau VII.1).
Enzyme Source
Alanine aminotransférase Foie
Amylase Glande salivaire, pancréas
Aspartate aminotransférase Cœur, muscle squelettique
Céruloplasmine Foie
Créatinine kinase Muscle, cœur, cerveau
γ-Glutamyltranférase Rein, prostate, pancréas
Ornithine carbamoyl-transférase foie
Phosphatase acide prostate
Phosphatase alcaline Muqueuse intestinale, rein, os
VI.2. Mesure de la concentration, test biologique
Lorsque le composé est un substrat intervenant dans une réaction enzymatique, sa
concentration peut être déterminée. Le dosage nécessite qu’au moins 99% de substrat soit
transformé par l’enzyme. Deux méthodes sont applicables :
- La méthode du point final employant une période d’incubation suffisamment longue
pour que la catalyse soit complète. Le dosage correspond à la différence entre les
concentrations initiale et finale de produit.
- La mesure des vitesses de réaction de substrat.

175
Le dosage par voie enzymatique est souvent basée sur l’absorption optique dont
l’augmentation ou la diminution est respectivement proportionnelle à l’apparition ou à la
disparition d’un composé chromophore.
Moyen de surveillance
Les services hospitaliers ainsi que les particuliers ont souvent recours à des tests généralement
effectués à partir de l’urine qui permet d’y contrôler la présence d’un métabolite. Le plus
utilise concerne la détection et la quantification du glucose. Ce système se présente sous la
forme d’un kit renfermant des bandes de cellulose préalablement imprégnés de réactifs
comprenant généralement deux enzymes : la glucose oxydase et la péroxydase. La glucose
oxydase convertit le glucose en acide gluconique et peroxyde d’hydrogène, ce dernier en
présence de peroxydase réagit sur un chromogène comme l’o-toluidine en donnant une
couleur bleue à la band de papier. L’intensité de la coloration (chromogène oxydé) est
proportionnelle à la concentration du glucose. Un étalonnage de la couleur fourni avec le kit
permet d’évaluer la concentration en glucose. La réaction est rapide puisqu’une minute suffit
pour lire le résultat.
Glucose + O2 + H2O + (glucose oxydase) → Acide gluconique + H2O2
H2O2 + chromogène réduit + (peroxydase) → Chromogène oxydé + H2O
VI.3. Enzyme thérapeutique
La recherche et le développement de médicaments sont pour la plupart focalisés sur les
anticorps, cytokines ou hormones. Toute fois les enzymes sont aussi employées avec succès
pour obtenir un effet biologique spécifique et bénéfique. Elles peuvent remplacer une enzyme
défectueuse voire manquante, ou compléter une quantité insuffisante d’enzyme secrétée par
un organe. Pour un usage thérapeutique, une enzyme doit posséder une bonne stabilité, une
faible immunogénicité, une action ciblée sur un constituant cellulaire donné et une pénétration
optimale dans les tissus. Depuis les années 1980, les enzymes humaines pour la thérapie sont
obtenues par la technologie de l’ADN recombinant. Les enzymes thérapeutiques actuellement
disponibles sont employées pour soigner de nombreuses pathologies :
- Aide digestive
(pancréatine, cellulase, α-galactosidase, lacase, lipase...). Ces enzymes sont utilisables
comme aide digestive. Dans de nombreux cas, une seule enzyme peur être
sélectionnée pour améliorer l’activité digestive. Toute fois, certaines préparations

176
pharmaceutiques en contenant plusieurs procurent au patient une capacité digestive
plus accrue.
- Agent de coagulation
Pour arrêter un saignement provoqué par une blessure superficielle, des préparations
pharmaceutiques contenant de la thrombine humaine sont souvent utilisées comme
activateurs de la coagulation.
- Agent de débridement
Le débridement se réfère au nettoyage d’une plaie ou d’une brûlure pour retirer un
corps étranger ou un tissu mort. Le nettoyage des tissus facilite leur cicatrisation
minimise les risques d’infections bactérienne notamment lorsque les germes sont
recouverts par des croûtes fibrines. A côté des moyens physiques pour ôter les tissus
morts, le recours à certaines protéases est fréquent. Ces protéases sont contenues
dans des crèmes hydrosolubles ou imprègnent certains pansements. Trypsine,
papaïne, collagénase et autres enzymes provenant de micro-organismes sont
employés.
- Cancer
L’asparaginase est une enzyme capable de catalyser l’hydrolyse de la L-asparagine en
acide aspartique et ammoniac. L’asparaginase a été identifiée et isolée en 1970 grâce à
ses propriétés inhibitrice de la prolifération des cellules leucémiques chez l’animale.
La plupart des cellules de mammifère synthétisent directement l’asparagine à partir de
la glutamine indiquant ainsi que l’asparagine n’est pas un aminoacide essentiel. Toute
fois, de nombreuses cellules cancéreuses perdent leur capacité à synthétiser
l’asparagine qui devient alors essentielle.En hydrolysant l’asparagine, l’asparaginase
prive les cellules cancéreuses de cet aminoacide. L’asparaginase est obtenu à partir de
microorganismes comme E. coli, Erwinia carotovora. L’asparaginase traitée avec du
PEG est administré dans le cas de la leucémie lymphoblastique aigüe de l’enfant.
Comme en général la concentration d’asparagine du plasma est relativement faible (40
µM), l’asparaginase thérapeutique doit avoir une haute affinité pour le substrat. Les
asparaginases de microorganismes tels que E. coli, Erwinia, pseudomonas ou
acinetobacter se sont montrées efficaces à inhiber la croissance de différentes cellules

177
leucémiques et de lignées cellulaires transformées. En outre, le traitement au PEG de
l’asparaginase permet d’accroitre significativement sa demi-vie. Bien que
l’asparaginase ait une action bénéfique, des effets secondaires peuvent être associés à
cette thérapie comme les nausées, vomissements, diarrhées et un fonctionnement
ralenti du foie et des reins.
- Exemple d’une enzyme à thérapie multiple
la bromélaïne est une protéase isolée de l’Ananas comosus qui est utilisée comme
médicament depuis 1957. Cette enzyme non toxique peut être utilisée directement
comme médicament pour les fonctions suivantes : anti-inflammatoire (réduction des
oedèmes), amélioration de la circulation cardiovasculaire, débridement des brûlures,
activité fibrinolytique et mucolytique, inhibition réversible de l’agrégation
palquettaire, soulagement de la douleur (arthrite rhumatoïde), aide digestive,
modulation des cytokines et immunité, activité anti-tumorale..
VI.4. Enzyme pour la synthèse de médicaments
Comparée à la préparation par synthèse chimique, l’usage d’enzymes permet, en évitant la
protection de groupements, de diminuer le nombre d’étapes nécessaires à la production d’un
médicament. Cet avantage est dû à la haute spécificité des réactions enzymatiques. En outre la
plupart des enzymes possèdent des propriétés régioselectives comme c’est le cas avec la
synthèse d’un médicament administré dans le cas de leucémie. La lipase de Candida
antarticaréagit sur une purine en l’acétylant. Le produit anticancéreux acétylé en position 5’
est obtenu à 99%. Les acylases et les estérases sont les enzymes les plus stéréo-spécifiques
Ainsi le Naproxène médicament anti-inflammatoire qui correspond à la forme S est obtenu à
partir d’un mélange racémique d’esters. Une estérase recombinante permet d’obtenir plus de
98% de Naproxène.
VI.5. Enzymes de nettoiement
VI.5.1. Verres correcteurs
Les larmes contiennes différentes protéines dont les mucines (glycoprotéines) capables
d’adhérer aux verres et lentilles de contact qui à l’usage peuvent constituer un dépôt
susceptible de voiler la vision et de provoquer inconfort, irritation voire même d’endommager
la cornée. En outre les corps gras secrétés par les glandes sébacées peuvent aussi s’associer à
ce dépôt.

178
Le nettoyage des lentilles doit être effectué de manière simple, efficace et se faire ne toute
sécurité. C’est pourquoi les solutions d’entretiens vendus en pharmacie contiennent des agents
bien tolérés comme des enzymes et des détergents neutres. La part enzymatique est
généralement constituée par des protéases comme α-chymotripsine, papaïne, pronase P,
subtilisine A, pancréatine, bromélaine ou carboxypeptidase. N’altérant pas la surface des
lentilles, ces enzymes minimisent les problèmes de friction. Certains fabricants
commercialisent leurs produits sous formes de comprimés à dissoudre. Le kit de nettoyage
comprend souvent une solution de désinfection contenant des oxydants. Les lentilles sont
placées en présence des enzymes, l’élimination des dépôts constitue la première étape du
nettoyage. La seconde consiste à porter le milieu à une température comprise entre 60 et
100°C afin d’inactiver irréversiblement les enzymes. Un rinçage des lentilles avec une
solution saline isotonique achève le traitement.
VI.5.2. Matériel médical
Le nettoyage du matériel médical utilisé en chirurgie, endoscopie doit répondre à des normes
précises permettant d’éviter la transmission de maladies infectieuses comme l’hépatite B,
SIDA ou maladies nosocomiales. Les produits de nettoyage et décontaminant contiennent
généralement des poudre enzymatiques capables d’agir sur différents types de molécules
biologiques. En outre la combinaison des enzymes et des détergents renforce l’efficacité des
du produit notamment dans les cas difficiles.
Les activités enzymatiques des protéases, amylases et lipases sont le plus souvent mises à
contribution. La température d’utilisation s’étend généralement de 4 à 22°C. en outre la
capacité des produits nettoyant est fonction de la concentration des enzymes : plus cette
concentration est élevée, et meilleure est l’efficacité du produit. Comme les enzymes ont un
certain coût, leur concentration reste dans une limite acceptable pour garantir un rapport
qualité/prix compétitif.
La désinfection des instruments contaminés par le prion (maladie de Creutzfeldt-jakob)
s’effectue habituellement par des lavages avec de la soude et de l’eau de javel suivi d’un
autoclavage. Les produits à base de protéases offrent une alternative intéressante pour la
destruction du prion. Certains produits nettoyant ont une spécificité qui permet d’éliminer
toute trace de protéine, ils sont souvent utilisés sous forme de spray pour faciliter leur emploi
et minimiser le contact direct avec les instruments contaminés.

179
VI.5.3. Nettoyage de la peau
Dans la culture japonaise, certaines bactéries et/ou enzymes sont été et sont encore utilisés
dans les bains pour rendre la peau lisse. Toute fois le domaine d’application des enzymes
n’est pas limité aux seuls produits de bains. Les protéases d’origine végétale comme la
papaïne ou la bromelaïne capables de peler la peau de manière douce sont employées pour les
soins du visage. Ces enzymes font désormais partie des produits cosmétiques de cosommation
courante.
VI.5.4. Soins des dents
L’addition d’enzymes aux pâtes dentifrices est courante depuis de nombreuses années.
En éliminant les bactéries pathogènes, les enzymes oxydatives permettent en effet de
combattre la plaque dentaire, d’éviter les gingivites ou les caries. Les pâtes dentifrices
associent aussi des enzymes hydrolysant les restes d’aliments (protéines, amidon). Certains
produits dentaires contiennent de la lactoferrine (protéine bactérienne) en association à la
lactoperoxydase et au lysozyme. Ces enzymes trouvées normalement dans la salive, contribue
à augmenter l’effet antibactérien dirigé notamment contre Streptococcus mutans et les
bactéries gram-positives. D’autres enzymes sont aussi utilisées dans les pâtes dentifrices
comme la papaïne pour une action blanchissante de l’émail. En effet, la papaïne a la capacité
d’éliminer les tâches en présence d’acide citrique.
VI.5.5. Soins des cheveux
Les oxydases, peroxydases ou tyrosidases permettent de rendre le procédé de coloration des
cheveux plus doux et mieux adapté à la nature des cheveux. La glucose oxydase comme
uricase sont capable de produire du peroxyde d’hydrogène en présence d’un substrat. Ces
enzymessont utilisées pour générer H2O2 directement au niveau du cheveu ce qui permet
d’en abaisser la concentration comparé aux traitements sans enzymes. En outre les dommages
causés aux cheveux sont minimisés puisque le peroxyde d’hydrogène produit ne s’accumule
pas. Il est continuellement consommé. La coloration avec les oxydases utilise comme substrat
des amines aromatiques telles que la p-phénilènediamine.
Les polyphénol oxydases sont des enzymes contenant du cuivre capable de réduire l’oxygène
en eau et d’oxyder un substrat polyphénolique par une mécanisme radicalaire en donnant une
quionone. Cette famille d’enzyme se subdivise en quatre classes : laccases, catécol oxydase,
ascorbate oxydase et tyrosinases. Parmi les polyphenol oxydases, les tyrosinases sont
considérés depuis de nombreuses années comme les enzymes les plus appropriées pour les

180
traitements colorants, toute fois, les laccases isolées pour la première fois du champignon
Myceliophtora ont été récemment utilisés. Ces enzymes agissent selon un mécanismes
effectué par un transfert de quatre électrons comprenant deux cycles d’oxydation. Au cours de
cette action, l’oxygène moléculaire est réduit en eau. Contrairement aux oxydases et
peroxydases, les tyrosinases et laccases utilisent les précurseurs chromogènes comme
substrats, ce qui constitue un avantage pour leur efficacité.
VI.5.6. Antibiotiques
Dans l'industrie pharmaceutique, la synthèse de certains antibiotiques utilise des enzymes.
La penicillinase (β-lactamase - EC 3.5.2.6) immobilisée est utilisée pour la conversion de la
penicilline-G ou V en acide 6-amino penicillanique (6-APA).
L'ampicilline est produite à partir du 6-APA avec la penicilline G amidase (EC 3.5.1.11)
immobilisée (CLEA).
La rifampicine est l’un des inducteurs les plus puissants des CYP.
Rôles des enzymes dans les antibiorésistances
Les bactéries peuvent résister à un agent antimicrobien en produisant des enzymes qui
détruisent ou qui désactivent l'antibiotique. Comme exemple, nous avons les pénicillines,
carbapenemes, et cephalosporines. Beaucoup de bactéries deviennent résistantes à ces
antibiotiques en produisant plusieurs beta-lactamases qui sont capable de désactiver quelques
formes de ces drogues.
Alors pour palier a cela, d’autres antibiotiques tels l’amoxilline+ acide clavulaniques permet
d’inhiber la beta- lactamase.
VII. Enzyme et industrie de l’environnement et du développement durable

181
S’effectuant généralement à pH neutre et nécessitant une énergie inférieure à celle utilisée en
synthèse chimique, les réactions enzymatiques sont de plus en plus appliquées pour
l’obtention dans des conditions douces, de composés sans rejet toxique pour l’environnement.
Les enzymes peuvent aussi être employées pour traiter ou transformer biologiques ou
recycler, par exemple les eaux usées. A plus de 2 milliards d’euros en 2005, le marché des
enzymes s’est accéléré au cours de ces dernières années. L’ingénierie enzymatique et/ou la
recherche d’espèces extremophiles a permis l’émergence de nouveaux catalyseurs biologiques
ayant des propriétés bien adaptées aux tâches pour lesquelles ils sont destinés.
VII.1. Biocatalyse des nitriles
Les méthodes traditionnelles chimiques pour la conversion, par exemple, de nitriles en acides
ou amides possèdent plusieurs inconvénients. La réaction nécessite des conditions très acides
comme la présence d’HCl 6M ou très alcaline (NaOH 2 M), une haute température et produit
des composés toxiques comme HCN ainsi qu’une grande quantité de sels. L’hydrolyse
biocatalytique des nitriles constitue une voie attractive pour la « synthèse verte ». Les
nitrilases convertissant sélectivement un groupe cyano en groupe amide ou acide offrent des
approches innovantes notamment pour la synthèse de composés chimiques chiraux.
Appartenant à une superfamille, les nitrilases sont représentés chez les plantes, animaux et
bactéries, elles agissent aussi bien sur les nitriles aliphatiques qu’aromatiques. L’hydrolyse du
groupe nitrile produit l’amide correspondant avec l’addition d’une molécule ou d’acide avec
une seconde molécule d’eau. Le mécanisme réactionnel implique l’attaque nucléophile sur le
carbone du groupe nitrile par une fonction sulhydryle d’un résidu de cystéine de l’enzyme
pour donner un intermédiaire tétraédrique. L’intermédiaire tétraédrique peut aussi se dissocier
pour donner l’amide à la place de l’acide. Les nitrilases peuvent être employés
industriellement pour la conversion sélective d’un group cyano d’un polynitrile virtuellement
impossible à obtenir par les méthodes chimiques conventionnelles. Ainsi, la conversion de
l’adiponitrile en 5-cyanovalérique qui est intermédiaire de synthèse du nylon-6 est effectué en
présence des ces enzymes. La nitrilase est employée pour la production de l’acide mandélique
R (-), utilisées par l’industrie pharmaceutique, dont la réaction ne nécessite aucun solvant
organique.
VII.2. Blanchiment du papier
Le recours à des produits chlorés pour des opérations de blanchiment est relativement
restreint. La protection de l’environnement a été améliorée grâce à l’intégration d’enzymes

182
notamment dans le blanchiment de la pâte à papier. Les xylanases jouent un rôle important
dans les opérations de blanchiment pour recycler les fibres de cellulose. Elles permettent
d’aboutir à un éclat final du papier, supérieur à celui obtenu par l’utilisation de composés
chlorés (Cl2, CL2O2) et H2O2. En dégradant les xylan à a surface des fibres de la pulpe de
bois, les xylanases augmentent la pénétration chimique et améliorent l’extraction de la
lignine.
Les xylanases d’organismes alcalophiles et thermophiles comme Bacillus steraro-
thermophilus ont été employés à pH 9 et à 65°C pour une plus grande efficacité. Pulsyme HA
a été la première xylanse commercialisée par Novozymes.
VII.3. Extraction du pétrole
Certaines bactéries extremophiles ont été testées pour améliorer l’extraction mais leur
utilisation s’est révélée complexe puisqu’un milieu nutritif devait être présent. En outre peu
de bactéries peuvent survivre dans un milieu constitué de pétrole. Les enzymes des
microorganismes thermophiles sont capables de supporter des températures allant jusqu’à
100°C ce qui facilite leur mise en œuvre dans les conditions particulières régnant dans les
exploitations pétrolières. Les enzymes sont généralement confinées ou encapsulées pour
minimiser la dénaturation. En modifiant la tension de surface, la peroxydase et la catalase
permettent de relarguer le pétrole adhérant aux grains de sables et aux roches meubles. Des
préparations enzymatiques comme Greenzyme sont adaptées à la récupération du pétrole. Les
propriétés de ces enzymes permettent aussi de dépolluer plus efficacement les surfaces et sols
contaminés. Lors du forage, la viscosité des polysaccharides constitue un facteur important.
Des enzymes stables à des températures atteignant 100°C telles que les cellulase et amylases
ont été employées pour une dégradation partielle de la cellulose polyanionique et de l’amidon
respectivement afin d’assurer un contrôle de la viscosité. L’utilisation des mannases
thermostables commercialisées sous le nom de Pyrolase a permis d’accroitre le rendement et
de diminuer les coûts de production.
III- Quelques enzymes particulières utilisées dans certaines industries au Burkina FASO
2) Cas des Brasseries du Burkina Faso (BRAKINA)
Pour des raisons d’optimisation de la fabrication de la bière, plusieurs enzymes sont utilisées
pendant le brassage et aussi lors de la garde de bière après la fermentation.
a) Hitempase 2XL

183
Source : C’est une alpha amylase bactérienne thermostable produite par une souche de
Bacillus licheniformis. Elle est commercialisé par la firme BIOCON et ou la firme KERRY.
Et existe sous la forme micro granules comme en liquide.
Activité : L’optimum de température de HITEMPASE 2XL est de 90-95°C avec une activité
supérieure à 80% pour un pH compris entre 4,0-8,0.
Usage : HITEMPASE 2XL convient pour des grains crus ou des céréales non maltées, qui ont
une température de liquéfaction élevée tels que le riz, le maïs, sorgho. A la Brakina, elle est
utilisée seulement pour l’empattage du gritz de maïs.
b) La FILTRASE .BR
Source : La FILTRASE BR est une formulation standardisée sur les activités beta-glucanase,
protéase neutre et hémicellulase, présentée sous formes granulée et liquide.
Les activités beta-glucanases et protéases sont issues de souches génétiquement modifiées de
Bacillus amyloliquefaciens.
Activité : L’action maximum de la FILTRASE BR se situe entre une température de 40°C à
65°C avec un pH situé entre 4,0 et 8,0. Elle est spécialement utilisée à la brakina lors de la
liquéfaction du malt afin de permettre d’améliorer la filtrabilité, le rendement en extrait, la
stabilité colloïdale de la bière finie.
Usage : La FILTRASE BR est en générale utilisée pour des céréales comme le malt qui a une
grande viscosité après son empattage et également un pH et une température optimum
envoisinant ceux de la FILTRASE BR.
c) La COLLUPILINE
Source : La papaïne est l’élément actif de la COLLUPILINE. La papaïne est une protéase
cystéine qui dégrade les fractions protéiques de la bière lesquelles, en s’associant aux
polyphénols, sont responsables du trouble au froid.
Activité : Elle agit sur les seuls composants protéiques responsables du trouble au froid et n’a
pas d’influence sur les composés protéiques qui conditionnent la tenue de mousse

184
Usage : La COLLUPILINE est ajoutée en tank de garde ou immédiatement avant la
clarification finale. L’addition en tank de garde semble la plus adaptée car elle offre à
l’enzyme un temps de contact maximal, garant d’une parfaite efficacité.
Les enzymes utilisées au Burkina Faso sont généralement dans le tissu industriel en particulier
dans le domaine alimentaire. D’autres enzymes sont utilisées dans la composition des
médicaments vendus en pharmacie. Cependant nous pouvons retenir que beaucoup d’enzymes
utilisées dans le tissu industriel au Burkina Faso restent inconnues.
D’où il est nécessaire de prévoir une étude exhaustive qui va faire le récapitulatif des enzymes
utilisées au Burkina Faso.
PERSPECTIVES
L’exploration du potentiel de nouvelles sources comme les microorganismes extremophiles
pourrait conduire à isoler des enzymes plus performantes pour opérer dans des conditions
difficiles (pH, température…). Les méthodes génétiques sont aussi capables de concevoir de
nouvelles enzymes à potentiel étendu pour traiter les effluents industriels. L’optimisation de la
production de ces enzymes permettra aussi de diminuer les coûts des traitements. Très
souvent, un coût trop élevé fait préférer les traitements traditionnels au procédé enzymatique.
Les microorganismes très employés pour la transformation des substances ont été utilisés
pendant longtemps de manières empirique dans les techniques traditionnelles. Toutefois, dans
le cas d’une utilisation ciblée, les microorganismes en amenant diverses contraintes
environnementales ne peuvent pas toujours représenter une solution attrayante. Dans ce cas,
les enzymes peuvent constituer des outils efficaces dépourvus de ces contraintes.
BIBLIOGRAPHIE
- Yon-Kahn, Jeannine, Hervé, Guy, Enzymologie moléculaire et cellulaire, Tome 1, 2005
- Bernhard, Sydney A., Structure et fonction des enzymes, Édition française dirigée par
Labeyrie, Françoise,1969
- Branden, Carl, Tooze, John, Introduction à la structure des protéines, traduit de l'anglais par
Lubochinsky Bernard,1996
- Voet, Donald, Voet, Judith G., Biochimie, traduction de la 3e édition américaine par Rousseau Guy
et Domenjoud Lionel, 2005
Cours d’enzymologie. Pr. A. Raisonnier. Université Pierre et Marie-Curie, France, 2005-2006
Cours d’enzymologie. Pr Philippe Collas
Cours d’enzymologie. Université de Montréal, Canada, 2012

185
“Industrial Enzymes and Their Applications” Uhlig H. (Ed.), John Wiley & Sons, Inc., New York (1999).
« Enzymes en Agroalimentaire » Larreta-Garde V. (Ed.), Technique & Documentation Lavoisier, Paris (1997).
« Industrial Enzymology » Godfrey T. and West S. (Eds), MacMillan Press Ltd, London (1996).
RF Boyer (1986) Modern experimenta lbiochemistry, Addison-Wesley Publishing Co, Reading (Mass.,
USA), p. 243-52 (notions de base sur la purification: choix des méthodes, étapes, préparation d'un
extrait brut, etc), 252-64 (purification de la lactalbumine).
RL Dryer, GF Lata (1989) Experimental Biochemistry, Oxford University Press, New York.
P Kamoun(1975) Appareils et méthodes en biochimie, Flammarion Médecine-Sciences, Paris,.
J Kling (1997) Scientists Use Various Methods To Tackle Protein Purification, The Scientist
11[14]:14-.[méthodes de purification de protéines, recombinantes ou non, exprimées dans des systèmes
in vitro]
M le Maire, R Chabaud, G Hervé (1990) Un modèle d'étude: l'aspartate transcarbamylase, Masson,
Paris.
DT Plummer (1987) An Introduction to Practical Biochemistry (3° éd.) McGraw-Hill, London.
NC Price (1992) Techniques for enzymes extraction in R Eisenthal, MJ Danson (éditeurs), Enzymes
assays, IRL Press, Oxford, pp. 255-76.[méthodes de purification des protéines avec accent sur les
enzymes]
B Osterlund, JC Janson (1997) A strategic approach to protein purification: Part 1, Pharmacia
Biotech. 2(3):8-10. [description des stratégies de purification, méthodes de rangement]
K Wilson, J Walker (1994) Principles and techniques of practical biochemistry (4e éd) Cambridge
University Press. Oxford.
Sites web :
http://www.webbioch.net/modules/mydownloads/cache/files/specificite.pdf
- Académie de Nancy-Metz :
http://www.ac-nancy-metz.fr/enseign/Physique/CHIM/Jumber/GLUCIDES/cadrglucides_fichiers
/GLUCIDES.htm
- http://www.chem.qmul.ac.uk/iubmb/enzyme/
- http://fr.wikipedia.org/wiki/Catalyseur
Pour les illustrations :
- http://www.ac-orleans-tours.fr/svt/mol3d/3d/module3/html/3page.htm
- http://membres.lycos.fr/carbonicanhydrase/
- http://www.kuhnle.co.uk/phd/pub/text/html/node17.htm
- http://pedagogie.ac-montpellier.fr:8080/disciplines/scphysiques/academie/ABCDORGA/
Famille/Produit/LESUCRE.html
- http://www.curie.u-psud.fr/U350/1999/french/Biochimie.html
- http://svt.ac-creteil.fr/spip.php?article4012
- http://www.ekcsk12.org/faculty/jbuckley/regbio/practicemidterm5.htm





























