Embed Size (px)
Citation preview

Ronéo n°2 - Cours n°4 Page 1 sur 14
UE1 Biochimie, Biologie moléculaire
Pr. Cavé
Le 04/10/17 de 15h30 à 17h30
Ronéotypeur : Kenza Ait Bouali
Ronéolecteur : Léa Abboud
Cours n°4 : Du gène à la protéine mutante
(Partie 1)
Les diapos avec une icône de stéthoscope en haut à gauche ne sont pas à apprendre.
La prof n’a pas souhaité relire la ronéo ni même nous parler tout simplement…

Ronéo n°2 - Cours n°4 Page 2 sur 14
Sommaire :
I. Les mutations :
A. Caractéristiques générales
B. Mutations constitutionnelles ou somatiques
II. Propriétés générales des cancers :
A. Qu’est-ce qu’une tumeur ?
B. Les propriétés des tumeurs leurs sont conférées par des altérations de l’ADN
C. Etapes clés dans l’étude du génome des cancers
III. Quelles sont les mutations dans les tumeurs ?
A. Aneuploïdies
B. Translocations
C. Amplifications
D. Gains de gènes extérieurs
E. Délétions
F. Mutations ponctuelles

Ronéo n°2 - Cours n°4 Page 3 sur 14
I. Mutation
A. Caractéristiques générales
L’ADN des cellules est exposé en permanence à des agressions pouvant conduire à des mutations.
Ces agressions vont être de différents types :
Agressions exogènes, qui viennent de l’environnement (radiations, agents génotoxiques)
Agressions endogènes (production de radicaux libres toxiques pour la cellule)
Il y a aussi des processus physiologiques qui peuvent conduire à des mutations :
Erreurs de réplication
Accidents de recombinaison
Dans tous les cas la cellule va essayer de réparer les anomalies. Dans la plupart des cas cela
fonctionne mais si ce n’est pas suffisant on va voir s’installer une mutation. Une mutation est une
modification de la séquence de l’ADN (dans une cellule eucaryote) qui va entrainer une modification
de l’information génétique. Mutation est un terme générique qui parle d’une modification du génome,
théoriquement cela ne signifie pas forcément que la modification a des effets délétères. Certaines
mutations vont être pathogènes (la mutation va altérer la fonction d’une protéine de façon pathogène)
et d’autres non pathogènes, c’est-à-dire neutres ou qui peuvent aussi améliorer une fonction (on a
l’habitude de les appeler des polymorphismes). L’avancement des techniques de séquençage permet
de découvrir un nombre important de mutations dont on ne peut statuer sur le caractère pathogène,
d’où le fait que l’on préfère de plus en plus souvent utiliser le terme de variants génétiques pour
désigner ces modifications de séquence.
Les mutations sont le moteur de l’évolution : si le système de réparation corrigeait 100% des
erreurs il y aurait très peu de diversité génétique et sans diversité il y aurait une très faible résistance
aux changements de l’environnement. Mais il y a aussi le risque de développer des maladies
génétiques telle que la drépanocytose (une mutation donne une maladie génétique).
B. Mutations constitutionnelles ou somatiques
Il y a deux types de mutations :
- Les mutations constitutionnelles , présentes avant la fécondation (nouvellement apparues dans la
cellule germinale d’un parent mutation de novo ; ou transmise de génération en génération) ou
survenant lors des premières divisions du zygote (néomutation, qui apparait alors uniquement chez
l’enfant à naître). Ces mutations sont présentes dans toutes les cellules de l’individu (somatiques et
germinales) et vont être transmissibles à la descendance. Mais les mutations survenant lors des
premières divisions du zygote peuvent donner des mosaïques (toutes les cellules ne sont pas
équitables). Les mutations constitutionnelles pathogènes sont à l’origine des maladies génétiques
monogéniques et des maladies génétiques chromosomiques.
- Les mutations somatiques ou « acquises », apparues dans une cellule somatique d’un tissu et qui
n’étaient pas présentes à la naissance dans le génome de l’individu. Elles sont restreintes au tissu
somatique dans lequel elles sont apparues. Elles ne sont pas transmissibles à la descendance.
Lorsqu’une mutation somatique confère un avantage sélectif à la cellule elle peut conduire à la
formation de tumeur.

Ronéo n°2 - Cours n°4 Page 4 sur 14
II. Propriétés générales des cancers :
A. Qu’est-ce qu’une tumeur ?
Il existe une grande variabilité des types de tumeurs, et pourtant les cellules tumorales partagent un
certain nombre de propriétés fondamentales qui ont été résumées par les chercheurs Hanathan et
Weinberg :
Les tumeurs présentent une activation constitutive (= qui n’est plus régulée, à distinguer de
constitutionnelle) des circuits de prolifération, il n’y a pas d’extinction du signal donc on ne
va plus pouvoir contrôler les activations ce qui est l’un des principaux problèmes.
De plus elles ont une insensibilité aux inhibiteurs de prolifération, comme si les régulateurs
n’existaient pas.
Et en même temps, elles sont résistantes à la mort cellulaire. Normalement le circuit de
contrôle entraîne la mort cellulaire en cas de prolifération trop importante, mais cette propriété
est absente chez les cellules tumorales.
Les cellules tumorales vont donc soutenir une prolifération chronique incontrôlée mais celle-ci
seule ne suffit pas à conduire à une tumeur, il faut également que la cellule ne puisse « échapper » à
cette prolifération via la mort cellulaire et qu’elle soit indépendante vis-à-vis des signaux de
l’environnement (les inhibiteurs).
De nouvelles capacités vont également apparaître : la capacité d’invasion et de métastases, celle
d’angiogenèse (induction de nouveaux vaisseaux alimentant la prolifération tumorale) et celle
d’immortalité (par perte de la sénescence).

Ronéo n°2 - Cours n°4 Page 5 sur 14
B. Les propriétés des tumeurs leurs sont conférées par des altérations de l’ADN
Les altérations sont de deux grands types :
- génétiques : elles touchent la séquence de l’ADN (par exemple les mutations, les délétions) et vont
modifier la fonction et/ ou l’expression de gènes.
- épigénétiques : elles marquent la séquence d’ADN (par la méthylation par exemple) ce qui va
entraîner une modification de l’expression des gènes.
Le cancer résulte d’altérations de l’ADN. C’est pour cela qu’elles sont considérées comme des
maladies génétiques qui dans la plupart des cas résultent de mutations somatiques , on parle à ce
moment de formes sporadiques : c’est une maladie génétique somatique où les altérations seront
restreintes aux cellules tumorales (exemple de mosaïques avec présence au sein d’un organisme de
tissus génétiquement différents mais provenant du même zygote). C’est pourquoi il faut prélever des
cellules du tissu tumoral (par biopsie par exemple) pour retrouver la mutation à l’origine de cette
tumeur.
Il y a également des formes héréditaires de cancers (tumeurs avec prédisposition) qui résultent de
mutations constitutionnelles (germinales) et somatiques. Elles ont une transmission autosomique
dominante et représentent 1 à 10% des cancers (pénétrance incomplète car tous les individus ne
développeront pas le cancer).
C. Etapes clés dans l’étude du génome des cancers
Dès le 19ème
siècle on se doute que des mutations de l’ADN sont à l’origine des cancers. Il a
toutefois fallu attendre les années 60 pour le confirmer avec l’observation grâce à la cytogénétique du
premier réarrangement chromosomique récurrent dans les cancers ; suivi dans les années 80 des
mutations du premier gène identifié dans le cancer. La recherche autour des tumeurs s’accélère grâce à
un boom des techniques d’exploration du génome, notamment avec l’arrivée dans les années
2000/2010 des technologies de séquençage de nouvelle génération qui rendent aujourd’hui possible de
screener énormément de gènes d’affilée voire un génome ou un exome entier.
III. Quelles sont les mutations dans les tumeurs ?
A. Aneuploïdie
Une aneuploïdie correspond à un nombre anormal de chromosomes (trop ou pas assez
important) qui résulte d’anomalies de disjonction à la mitose. Les aneuploïdies constitutionnelles sont
moins sévères que les aneuploïdies somatiques retrouvées dans les cancers car elles doivent rester
compatibles avec le développement et la vie. Les aneuploïdies sont très répandues dans les cancers.
La technique la plus standard pour observer une aneuploïdie est l’établissement d’un caryotype.
On peut aussi utiliser la FISH (ciblée par chromosome), la CGH et l’index ADN (technique de
cytométrie de flux qui permet de quantifier l’ADN dans la cellule).
(Les techniques d’observation vont être étudiées en ED)

Ronéo n°2 - Cours n°4 Page 6 sur 14
B. Translocation
Une translocation est une mutation génétique caractérisée par l'échange réciproque de matériel
chromosomique entre des chromosomes non homologues.
Il y a trois types de conséquences moléculaires :
- inhibition de l’expression d’un gène par cassure ;
- modification de l’expression d’un gène par échange de promoteur ;
- création d’un gène hybride avec production d’une protéine de fusion.
Modification de l’expression d’un gène par échange de promoteur :
Dans certaines translocations, la
cassure est située en dehors de la
partie codante du gène : par
exemple on peut observer sur les
gènes 1 et 2 un point de cassure
entre la région promotrice et
l’exon n°1.
Normalement, la cellule exprime
énormément le gène n°1
(symbolisé par la flèche en gras)
alors que le gène n°2 n’est quasiment pas exprimé (la flèche est plus fine).
La cassure au niveau des deux gènes est suivie d’une religation qui aboutit à une séquence d’ADN
contenant la région codante du gène n°2 (normalement peu exprimée dans la cellule) sous la
dépendance du promoteur du gène n°1 (promoteur fort).
Le gène n°2 va alors être très exprimé et cela peut avoir plusieurs conséquences comme la mort
cellulaire ou dans les cas où la dérégulation transcriptionnelle du gène et son expression
inadéquate/ectopique lui procure un avantage sélectif, la constitution d’une première étape vers la
formation d’une tumeur.
Exemple de la translocation (14 ; 18) :
La séquence va être coupée au niveau du promoteur des immunoglobulines (enhancer fort) puis
transposée dans les lymphocytes B rendant l’expression du gène BLC2 dépendante de ce promoteur.
Une cellule mature n’exprime normalement pas
BCL2 (contrairement aux cellules immatures),
gène anti-apoptotique. Dans le lymphocyte B
des centres folliculaires lorsque la translocation
survient et que lymphocyte vieillit, au lieu de
supprimer l’expression de BCL2 et de basculer
vers l’apoptose, celui-ci va continuer à exprimer
BCL2 et avoir une survie anormale . Cette
translocation est initiatrice dans les lymphomes
folliculaires.

Ronéo n°2 - Cours n°4 Page 7 sur 14
Création d’un gène hybride avec production d’une protéine de fusion :
Le gène n°1 subit une cassure entre l’exon 2 et l’exon 3, idem pour le gène n°2 situé sur un autre
chromosome. Le gène de fusion (ou gène « chimère ») obtenu après religation est constitué de la
partie 5’ du gène 1 (avec le promoteur du gène 1, qui est cette fois ci équivaut à celui du gène 2) et la
partie 3’ du gène 2.
La nouvelle séquence va être transcrite (on obtient un « transcrit de fusion ») et si le cadre de
lecture est conservé on peut aboutir à l’expression d’une protéine de fusion qui n’existait pas avant
(néoprotéine avec des domaines issus du gène 1 et des domaines issus du gène 2, chaque domaine
apportant une propriété à la protéine).
Si cette protéine confère un avantage séléctif à la cellule cela entraîne l’apparition d’une tumeur.
Exemple de la protéine BCR-ABL :
Il y a une translocation (9 ; 22) entre
le gène ABL situé sur le chromosome
9 et le gène BCR situé sur le
chromosome 22. Le gène de fusion
BCR-ABL a son cadre de lecture
conservé par rapport aux gènes
initiaux, il aboutit alors à un ARNm
de fusion puis à une protéine de
fusion.
Cette translocation est retrouvée dans
la leucémie myéloïde chronique
(LMC), leucémie survenant
essentiellement chez les patients de
plus de 50 ans.

Ronéo n°2 - Cours n°4 Page 8 sur 14
Dans cette leucémie on peut mettre en évidence un chromosome, « Philadelphie » anormal, ainsi
qu’un ARNm anormal, tous deux absents de la cellule normale et qui résultent de cette translocation.
La partie 5’ de BCR va fusionner avec la
partie 3’ de ABL (donc la partie N-
terminale de la protéine sera un petit bout
de BCR et toute la partie C-terminale sera
ABL).
ABL est une protéine kinase donc par sa
fonction elle a une localisation plutôt
nucléaire, BCR est une protéine
cytoplasmique mais a un rôle mal connu.
ABL comme beaucoup de kinases a un
système d’autoinhibition, elle n’est donc
pas active à l’état basal.
Lorsque ABL va fusionner avec la partie
5’ de BCR la première conséquence va
être une délocalisation d’ABL dans le
cytoplasme . De plus son autoinhibition
va être levée car il y aura possibilité de dimérisation des protéines de fusion grâce à BCR. Les deux
parties tyrosines kinases de ABL vont se retrouver à proximité l’une de l’autre dans une configuration
qui les rend plus actives que dans une protéine ABL normale.
Cela aboutit à une autophosphorylation avec phosphorylation des tyrosines sur la protéine de
fusion (comme le ferait un récepteur à tyrosine kinase). Ces tyrosines phosphorylées vont ensuite
servir de site d’ancrage à la protéine Grb2 qui va pouvoir solliciter deux voies cellulaires : la voie
PI3K/AKT (responsable de la survie cellulaire) et la voie RAS/MAPK-ERK (responsable de la
prolifération). L’activation de ces voies entraîne la survie cellulaire et la prolifération dans des
conditions anormales, à l’origine de la tumeur.
La connaissance de ce mécanisme donne des pistes de recherche pour développer des traitements. Le
but des recherches a été d’inhiber la fonction tyrosine kinase anormale de BCR-ABL en développant
un analogue de l’ATP (substrat de la kinase car donneur de phosphate), l’imatinib. Le développement
de cette classe de médicament a permis un bond thérapeutique dans le traitement de la LMC.
C. Amplifications
Une amplification est un évènement d’insertion consistant en la réplication en tandem d’une
séquence donnée (il s’agit d’une grande séquence de plusieurs centaines de bases, attention à ne pas
confondre l’amplification avec la duplication de 3 ou 4 nucléotides ou l’expansion de triplets). On
parle de duplication s’il y a 2 copies (N=2) et d’amplification lorsqu’il y a plus de 2 copies (N>2).
En conséquence, le niveau d’expression de ces gènes est augmenté. Les copies surnuméraires sont
soit intégrées dans un chromosome ou soit sous forme de mini-chromosomes surnuméraires appelés
les chromosomes double-minute (DM).

Ronéo n°2 - Cours n°4 Page 9 sur 14
Exemple de MDM2 :
Le premier gène dont on a pu mettre en
évidence les amplifications dans les
cancers est le MDM2 (Murine Double
Minute 2). En général c’est toute une
région chromosomique qui est amplifiée
et pas uniquement un seul gène.
Une manière de mettre en évidence les
différents types de duplication est
l’hybridation in situ d’une sonde
fluorescente d’ADN (FISH), technique
adaptée à l’observation de duplications
de taille importante. Il s’agit de choisir
une sonde d’ADN couvrant la région
d’amplification donnée (sonde colorant
notamment MDM2 en rouge), puis de la marquer par un fluorochrome.
D’autres fluorochromes sont également utilisés afin de colorer l’ADN du noyau en bleu (pour se
repérer dans la cellule) ou de colorer la sonde du chromosome 12 en vert (chromosomes contenant
MDM2).
Dans une cellule normale, la sonde du chromosome 12 va s’illuminer deux fois tout comme la sonde
contenant MDM2 : cela correspond à deux chromosomes pour deux allèles de MDM2.
Dans une cellule tumorale par contre, la sonde du chromosome 12 s’illumine toujours deux fois mais
celle de MDM2 s’illumine quant à elle un grand nombre de fois au point de former une « tâche ». Ceci
montre que la région d’ADN contenant MDM2 est énormément dupliquée (on peut aller jusqu'à 50
voire 100 duplications) avec pour conséquence une hyper expression de MDM2.
MDM2 a une action d’ubiquitine ligase dont la cible principale est p53, protéine très importante pour
la stabilité du génome et qui évite que les cellules ne prolifèrent. Cette amplification de MDM2
empêche alors p53 d’assurer ses fonctions de régulateur du génome, du cycle cellulaire et de
l’apoptose.
Autre exemple de HER2/ERBB2:
L’amplification de HER2/ERRB2 est un autre
exemple du rôle de l’amplification dans le cancer
(retrouvée fréquemment dans le cancer du sein).
HER2/ERBB2 (même nom d’une seule et même
protéine) appartient à la famille des récepteurs aux
facteurs de croissance épidermiques (famille EGF).
On peut mettre évidence son amplification par la
FISH sauf que cette fois ci c’est le chromosome 17
qui est concerné et c’est la sonde HER2 qui est en
rouge.

Ronéo n°2 - Cours n°4 Page 10 sur 14
Pour montrer la surexpression de la protéine il faut employer une autre technique qui cette fois met en
évidence la protéine et non plus l’ADN : c’est l’immunohistochimie . Pour détecter la protéine sur une
coupe cellulaire on se sert d’un anticorps marqué par un fluorochrome ou une molécule permettant la
coloration et spécifique de ce récepteur.
Ici, le marquage coloré délimite les cellules ce qui montre d’une part une surexpression de la
protéine et d’autre part que c’est un récepteur qui se situe bien à la membrane cytoplasmique.
HER2 est un récepteur à activité tyrosine kinase avec activation de la voie RAS (prolifération) ainsi
que de la voie PI3K (survie cellulaire). Ces deux voies sont hyperactives dans le cancer du sein. Le
trastuzumab (herceptine) est un anticorps qui a été développé afin de bloquer la signalisation via
HER2 et qui a changé le pronostic de survie des femmes atteintes d’un cancer du sein avec
surexpression de HER2. A titre indicatif l’herceptine permet aussi de traiter la lèpre.
D. Gains de gènes extérieurs
Un certain nombre de virus est capable de contrôler la prolifération cellulaire et vont être impliqués
dans les cancers par ce que l’on appelle l’oncogenèse virale .
Exemple du papillomavirus (HPV) :
C’est un virus à ADN cancérigène responsables de 100% des cancers du col de l’utérus . Il s’agit de
la plus fréquente des infections sexuellement transmissibles et qui touche essentiellement des femmes
jeunes entre 20 et 30 ans. Le génome viral est capable de s’intégrer dans celui de la cellule et
d’hyper exprimer des oncoprotéines virales (E6 et E7) à l’origine du développement de tumeurs.
E6 et E7 vont bloquer respectivement P53 et RB et interfèrent avec la machinerie cellulaire, ce qui
cause la tumeur.
Exemple de l’infection par HTLV-1 (Human T-Cell Leukemia Virus) :
Ce rétrovirus est associé au développement de l’ATL (Adult T cell Leukemia/Lymphoma), qui
est une leucémie affectant les lymphocytes T.
Tout comme le HPV, il est capable de s’intégrer au génome cellulaire. L’ATL a pour origine la
production d’une protéine virale oncogénique, la protéine TAX qui va stimuler deux voies de
signalisation : la voie NFκB (très impliquée dans la réponse immune et la survie cellulaire) et la voie
PI3K (voie de la survie cellulaire).
On sait que environ 2% des sujets infectés par HTLV-1 lors de la petite enfance vont développer une
ATL avec une latence de 30 à 50 ans. Cela montre qu’une seule mutation ne suffit pas à développer un
cancer et qu’il faut au contraire une accumulation d’évènements : la protéine TAX est indispensable
pour développer la leucémie mais elle ne suffit pas à elle seule à provoquer la leucémie.
De plus ce virus est endémique de régions comme le Japon, les Caraïbes, l’Amérique du Sud et
l’Afrique mais peu présent en Europe.
E. Délétion
Pas de diapos sur cette notion.
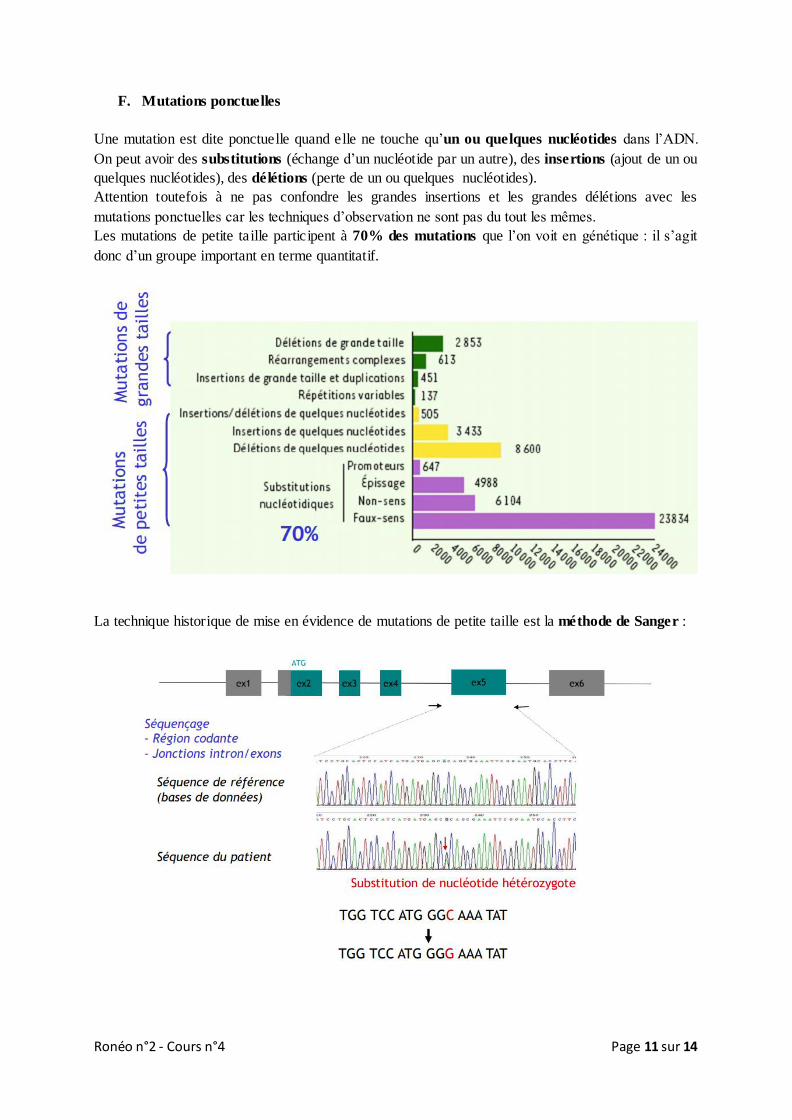
Ronéo n°2 - Cours n°4 Page 11 sur 14
F. Mutations ponctuelles
Une mutation est dite ponctuelle quand elle ne touche qu’un ou quelques nucléotides dans l’ADN.
On peut avoir des substitutions (échange d’un nucléotide par un autre), des insertions (ajout de un ou
quelques nucléotides), des délétions (perte de un ou quelques nucléotides).
Attention toutefois à ne pas confondre les grandes insertions et les grandes délétions avec les
mutations ponctuelles car les techniques d’observation ne sont pas du tout les mêmes.
Les mutations de petite taille participent à 70% des mutations que l’on voit en génétique : il s’agit
donc d’un groupe important en terme quantitatif.
La technique historique de mise en évidence de mutations de petite taille est la méthode de Sanger :

Ronéo n°2 - Cours n°4 Page 12 sur 14
Des amorces nucléotidiques amplifiées par PCR sont placées de part et d’autre d’une région d’intérêt
(généralement un exome car la plupart des mutations que l’on sait interpréter sont dans la région
codante) puis on la séquence pour voir l’enchainement des nucléotides. Ensuite on compare la
séquence du patient à la séquence de référence présente dans les bases de données.
Ici à chaque base de la séquence de référence on a un « pic » tandis que chez le patient on retrouve à
un moment sur la séquence deux pics, cela signifie qu’au lieu d’avoir uniquement un C on a sur un
allèle un C et sur l’autre allèle un G (substitution du C par un G sur l’un des allèles avec pour
conséquence un éventuel changement d’acide aminé).
Depuis quelques années s’est mis en place un type de séquençage beaucoup plus puissant, le NGS
(séquençage de nouvelle génération) qui permet le séquençage parallèle de masse : énormément de
gènes sont séquencés en parallèle.
Cette technique a décuplé les capacités de séquençage et changé les techniques d’étude des maladies :
on partait d’hypothèses et on séquençait un gène à partir de ces hypothèses, maintenant on peut
séquencer sans apriori, de façon très large on séquence l’ensemble des régions codantes (exome)
voire le génome complet puis on réfléchit.
Cela a révolutionné l’étude des cancers via le séquençage des exomes (ensemble des régions codantes)
par cette approche, donnant naissance à une compréhension beaucoup plus fine de cette pathologie.
Article sur le panorama des altérations somatiques dans les ATL :
Les chercheurs ont séquencé tout le génome, l’exome, le transcriptome (séquençage des ARN) et ont
réalisé un séquençage ciblé de 50 gènes sur la cohorte totale ainsi que des puces à ADN leurs
permettant de voir les anomalies de nombre et les anomalies de méthylation (épigénétique). Le but
recherché était de déceler toutes les anomalies afin de mieux comprendre l’ATL et les cancers.
Les résultats montrent un grand nombre de mutations somatiques de type mutation ponctuelle. Celles-
ci touchent de nombreux gènes avec pour chaque gène des fréquences de mutation variable. Un gène
peut être atteint par plusieurs types de mutations, la plus fréquente étant le variant non-synonyme.
Cela montre la diversité des mutations que l’on peut retrouver dans un même cancer, toutes les
tumeurs ne vont pas avoir le même profil génétique (d’où les différents pronostics ou la diversité des
réponses au traitement).
Les mutations synonymes :
Du fait que le code soit dégénéré, la mutation synonyme ne va pas entrainer de changement d’acide
aminé au niveau de la protéine. On a l’habitude de considérer que les variants synonymes ne sont
pas importants en pathologie.

Ronéo n°2 - Cours n°4 Page 13 sur 14
Les mutations faux-sens :
La mutation entraîne le remplacement d’un
acide aminé par un autre, avec pour
conséquence un changement de fonction au
niveau de la protéine.
Ici on la substitution d’un G par un A au niveau
du 661ème nucléotide (noté c.661C>A) et donc
la substitution d’une glycine par une sérine au
niveau du codon 221 (noté p.Gly221Ser ou
encore p.G221S).
Exemple : Mutations ponctuelles activant la
PKC la phosphorylation de IKK (une des
cibles de la PKC) est beaucoup plus importante
que chez un sujet ne portant pas la mutation.
Les mutations non sens :
Mutation qui entraîne le remplacement d’un acide aminé par un codon STOP.
Insertions et délétions de quelques nucléotides :
Il y a deux cas de figure :
insertion ou délétion d’un multiple de 3 nucléotides
insertion ou délétion d’un nombre de nucléotides non-multiple de 3

Ronéo n°2 - Cours n°4 Page 14 sur 14
Dans le cas où c’est un non-multiple de
3, le cadre de lecture des codons se
retrouve décalé et on voit très souvent
apparaitre un codon STOP de façon
précoce.
Les conséquences sont le plus souvent
l’absence de la protéine (équivaut à
une perte de fonction) ou l’apparition
d’une protéine tronquée (avec perte
ou gain de fonction).
Lorsqu’il y a insertion ou délétion d’un
multiple de 3, on aboutit à une
protéine anormale avec gain ou perte
de fonction.
Ces anomalies s’observent grâce au séquençage.
Exemple de CCR4 (récepteur couplé aux protéines G) : mutation entraînant un décalage du cadre de
lecture ou mutation STOP Troncation de la partie C-terminale intracytoplasmique ce qui empêche
l’internalisation du récepteur après stimulation du ligand Augmente la quantité de récepteur en
surface.
Mutation dans une région non codante :
Ces mutations vont jouer sur l’épissage d’un gène et changer l’ARNm ce qui va modifier la protéine.
Elles touchent des régions 5’ non codantes et altèrent la transcription. Si la mutation affecte un
silencer la protéine va gagner une fonction tandis que si elle affecte un enhancer ou un promoteur
elle va perdre sa fonction.
Elles atteignent aussi les sites d’épissage . Si on a une mutation sur la première ou dernière base d’un
exon cela va perturber la maturation de l’ARNm, on parle d’altération d’épissage.
Les conséquences sont diverses. On peut avoir
une perte d’un exon (avec décalage ou non
du cadre de lecture), l’incorporation d’une
séquence intronique qui sera alors reconnue
comme un exon (alors que ça n’en est pas un).
On peut également avoir la création d’un site
d’épissage alternatif au niveau des régions
exoniques alors qu’il n’y en avait pas : les
exons seront donc tronqués.
Ces mutations entrainent une perte de
fonction avec soit absence de la protéine
(par dégradation d’ARNm qui reconnait les
ARN messagers anormaux), soit une protéine non fonctionnelle , soit production d’un dominant
négatif (protéine mutée inactivant une protéine normale). Il peut aussi y avoir gain de fonction avec
soit la production d’une protéine « toxique » ou d’une protéine hyperactive.
RTPCR : technique permettant de comparer le transcrit du gène à un transcrit normal lorsque
l’on suspecte une mutation d’épissage.





























