Embed Size (px)
Citation preview

Crystal structure and spectroscopic investigations of an organicmonophosphate
H. Dhaouadi a, H. Marouani a,*, M. Rzaigui a, A. Madani b
a Laboratoire de Chimie des Matériaux, Faculté des Sciences, 7021 Zarzouna, Bizerte, Tunisiab Laboratoire de Physique des Matériaux, Faculté des Sciences, 7021 Zarzouna, Bizerte, Tunisia
Received 2 December 2007; received in revised form 30 January 2008; accepted 29 February 2008
Available online 16 March 2008
Abstract
Single crystals of ( p-ClC6H4NH3)H2PO4 are synthesized in water by interaction of H3PO4 and ( p-ClC6H4NH2). This compoundcrystallizes in the orthorhombic system with the Pbca space group. Its unit-cell parameters are a = 9.724(3), b = 7.861(1),c = 25.078(6) Å, V = 1917.1(6) Å3 and Z = 8. The crystal structure has been solved and refined to R = 0.039, using 4298independent reflections. The atomic arrangement can be described by inorganic layers parallel to ab plane, between which theorganic cations are located. This compound exhibits a reversible phase transition at 403 K. The electrical conductivitymeasurements show that the ( p-ClC6H4NH3)H2PO4 has a conductivity value which goes from s = 0.88 � 10�6 W�1 cm�1 atroom temperature (293 K) to 3.31 � 10�4 W�1 cm�1 at 433 K. Its characterisation by TA, NMR and IR is reported too.# 2008 Elsevier Ltd. All rights reserved.
Keywords: C. X-ray diffraction; D. Crystal structure; C. Nuclear magnetic spectroscopy (NMR); C. Differential scanning calorimetry (DSC); D.Electrical properties
1. Introduction
Generally, the combination of different organic/inorganic components in a single material can lead to new hybridcompounds, with specific physical and chemical properties. These compounds have attracted great attention becauseof their unique opportunity to combine the remarkable features of organic compounds with those of inorganicmaterials. In particular, hybrid materials based on the phosphates have received much attention in recent years due totheir technological interest in several areas, such as magnetism, electricity, optics and biomaterials research [1,2]. Theproperties of the organic molecule, such as length, geometry and relative position of the donor groups, play a veryimportant role in the production of various hybrid compounds with interesting three-dimensional networks, layers,chains and ribbon structures [3–5]. The cohesion forces in these hybrid compounds are dominated by electrostaticinteractions, Van der Waals contacts, and hydrogen bonds (O–H� � �O, N–H� � �O).
In order to enrich the bibliography in such a kind of hybrid materials and to investigate the influence of hydrogenbonds on the chemical and structural features, we report in this work chemical preparation, crystal structure andphysico-chemical study of a new organic monophosphate ( p-ClC6H4NH3)H2PO4. A correlation attempt of the atomic
www.elsevier.com/locate/matresbu
Materials Research Bulletin 43 (2008) 3234–3244
* Corresponding author. Tel.: +216 72 591 906; fax: +216 72 590 566.E-mail address: [email protected] (H. Marouani).
0025-5408/$ – see front matter # 2008 Elsevier Ltd. All rights reserved.doi:10.1016/j.materresbull.2008.02.027

arrangement and the H-bond scheme with the nature of ring substituents, as well as the properties and the position ofaromatic ring substituents.
2. Experimental
2.1. Crystal chemistry
Crystals of the title compound, ( p-ClC6H4NH3)H2PO4, have been prepared at room temperature by slowly adding0.28 cm3 (5 mmol.) of concentrated monophosphoric acid (85 wt%, d = 1.7) to an aqueous solution containing 0.73 g(5.7 mmol.) of p-chloroaniline. The mixture was fairly heated under constant stirring for 4 h, to obtain a clear solution.Schematically the reaction can be written as follows:
H3PO4þðp-ClC6H4NH2Þ ! ðp-ClC6H4NH3ÞH2PO4
The so obtained solution has been slowly evaporated at room temperature until the formation of short stout prismsof p-ClC6H4NH3)H2PO4 (1.5 g, 61% yield), with suitable dimensions for a crystallographic study. The crystals arestable for months under normal conditions of temperature and humidity.
2.2. Investigation techniques
The intensity data collection was performed with a Mach3 Enraf Nonius diffractometer. The experimentalparameters used during these measurements, the strategy followed for the structure determination, and its final resultsare gathered in Table 1. 31P MAS-NMR and 13C CP MAS-NMR spectra were obtained on a solid state high-resolutionBruker DSX-300 spectrometer operating at 121 MHz for 31P and 75.49 MHz for 13C. 31P NMR chemical shifts aregiven relative to 85% H3PO4 and 13C ones relative to tetramethylsilane as external reference. Thermal analysis wasperformed using the ‘‘multimodule 92 Setaram analyser’’ operating from room temperature up to 723 K at an averageheating rate of 5 K min�1. IR spectrum is recorded in the range 4000–400 cm�1 with a ‘‘PerkinElmer FTIR’’Spectrometer using a sample dispersed in spectroscopically pure KBr pellet. A polycrystalline sample of ( p-ClC6H4NH3)H2PO4 was crushed and pressed at room temperature into a tablet of 13 mm in diameter and 0.92 mm inthickness. Dense pellets suitable for electrophysical measurements were heated at 323 K for 24 h. Metallic silver wasdeposited on both sides which served as electrodes. The pellet is placed between two blocking electrodes in a tubularfurnace, submitted to a temperature regulator.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–3244 3235
Table 1Crystal data and experimental parameters used for the intensity data collection
Empirical Formula C6H9ClNO4PFormula weight 225.57Crystal system OrthorhombicSpace group Pbcaa 9.724(3) Åb 7.861(1) Åc 25.078(6) ÅZ 8V 1917.1(6) Å3
rcal 1.563 g cm�3
Linear absorption factor m(AgK a) = 2.85 cm�1
F(0 0 0) 928.00Index ranges 0 � h � 15, 0 � k � 12, �39 � l � 0Reflections collected 4327Independent reflections 4298Refined parameters 154R [I > 3 s(I)] 0.039R(w) 0.042Goodness-of-fit 1.760
Strategy and final results of the structure determination of ( p-ClC6H4NH3)H2PO4.

The electrical conductivity measurements were carried out from 293 to 433 K with 5–20 K steps, by checking thecomplex impedance spectroscopy with a Hewlett-Packard 4129A impedance analyzer. The signal frequency rangedfrom 10 to 13 MHz.
3. Results and discussion
3.1. Structure description
An ORTEP view of the asymmetric unit of the title compound is depicted in Fig. 1. The final atomic coordinates andthermal parameters of the ( p-ClC6H4NH3)H2PO4 are listed in Table 2. The main geometrical features of the variousentities involved in the ( p-ClC6H4NH3)H2PO4 structure are reported in Table 3.
The atomic arrangement of the title compound is characterized by the existence of inorganic layers, built byH2PO4
� anions. Each H2PO4� group is connected to its adjacent neighbours by two hydrogen bonds. These layers
cross the unit cell parallel to the ab plane at z = 0 and z = 1/2. Fig. 2 shows a structure projection along the b direction.In each anion, the central phosphorus atom is tetrahedrally coordinated by four distinct oxygen atoms O1, O2, O3 andO4. Among these four oxygen atoms, two of the coordinating oxygen can be attributed to doubly bonded oxygenatoms, while the two other ones (d (P1–O1) = 1.537(7) Å and d (P1–O2) = 1.551(2) Å correspond to the P–OH bonds.The average values for the P–O distances and O–P–O angles are 1.523 Å and 109.4288, respectively. These values arein excellent agreement with those relative to the protonated oxoanions [6].
Regarding the organic cation arrangement, the protonated p-chloroaniline molecule is localized in the interlayerspace, and neutralizes the negative charge of the anionic part. These groups are organized in a similar direction so theyform intermolecular van der Waals interactions between them and particularly strong hydrogen bonds with oxygenatoms of the anionic layers. The arrangement in opposition of p-chloroanilinium groups is probably the meanconstraint which justifies the formation of this structure with local inversion center [7,8]. Each organic entity isbounded to three different (H2PO4
�) groups belonging to the same inorganic layer through three N–H� � �O hydrogenbonds. The organic molecule exhibits a regular spatial configuration with usual distances C–C, C–N, and angles C–C–
C, C–C–N. The aromatic ring of the protonated organic amine is planar, with a mean plane deviation of 0.0047 Å. Thebond lengths of C1–C2, C2–C3, C3–C4, C4–C5, C5–C6 and C6–C1 are 1.369(2) Å, 1.378(3) Å, 1.368(3) Å,1.383(3) Å, 1.382(2) Å, 1.381(2) Å, respectively which are between single bond and double bond and agree with thatin benzene [9]. Furthermore, the distance of N–C(1) [1.457(2) Å] clearly indicates a single bond.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–32443236
Fig. 1. ORTEP view of the configuration of ( p-ClC6H4NH3)H2PO4 (50% thermal ellipsoids).
Hydrogen bonding play a significant role in the linking the organic molecules with the anionic sheets made byH2PO4 moieties. This interaction contributes to the cohesion of the structure. All the D (donor)-H� � �A (acceptor)hydrogen bonds are listed in Table 3, with an upper limit of 2.10(2) Å for H���A distance and a lower limit of 162(2)8for the D–H� � �A bond angles. Thus, this atomic arrangement exhibits two types of intermolecular interaction: O–
H� � �O and N–H� � �O, classified respectively as strong and weak hydrogen bonds. The first type, involving two short
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–3244 3237
Table 2Final atomic coordination and Beq. for the non-hydrogen atoms
Atoms x(s) y(s) z(s) Béq (Å2)
Cl 0.88823(6) 0.13631(8) 0.30583(2) 6.57(1)P 1.11897(3) �0.30318(4) 0.03604(1) 2.351(5)O(1) 1.2399(1) �0.3662(2) 0.07020(4) 5.76(3)O(2) 0.9836(1) �0.3765(2) 0.05933(5) 5.63(3)O(3) 1.10879(9) �0.1154(1) 0.04363(4) 3.04(2)O(4) 1.13588(8) �0.3562(1) �0.02156(3) 2.54(2)N 0.8708(1) 0.0311(1) 0.07209(4) 2.58(2)C(1) 0.8740(1) 0.0543(2) 0.12974(5) 2.45(2)C(2) 0.7708(1) �0.0085(2) 0.16120(5) 3.47(3)C(3) 0.7761(2) 0.0153(2) 0.21562(6) 4.12(3)C(4) 0.8833(2) 0.1037(2) 0.23743(5) 3.82(3)C(5) 0.9874(2) 0.1688(2) 0.20587(6) 3.90(3)C(6) 0.9829(1) 0.1435(2) 0.15135(5) 3.30(3)
Esd are given in parentheses Beq = 4/3P
i
Pj bijaiaj.
Table 3Main distances (Å) and angles (8) for ( p-ClC6H4NH3)H2PO4 atomic arrangement
Tetrahedron PO4
P O(1) O(2) O(3) O(4)O(1) 1.537(2) 2.509(2) 2.441(2) 2.515(2)O(2) 108.7(1) 1.551(2) 2.418(2) 2.517(2)O(3) 107.38(8) 105.25(8) 1.491(1) 2.514(4)O(4) 111.12(7) 110.46(7) 113.68(7) 1.512(1)
O(1)–H(O1) 0.99(3) P–O(1)–H(O1) 123(2)O(2)–H(O2) 0.95(3) P–O(2)–H(O2) 118(2)
( p–ClC6H4NH3+) group
N–C(1) 1.457(2) N(1)–C(1)–C(2) 120.70(1)C(1)–C(2) 1.369(2) C(1)–C(2)–C(3) 121.3(2)C(2)–C(3) 1.378(3) C(2)–C(3)–C(4) 119.6(2)C(3)–C(4) 1.368(3) C(3)–C(4)–C(5) 121.1(2)C(4)–C(5) 1.383(3) C(4)–C(5)–C(6) 119.3(2)C(5)–C(6) 1.382(2) C(5)–C(6)–C(1) 119.0(2)C(6)–C(1) 1.381(2) C(6)–C(1)–C(2) 121.3(2)C(4)–Cl 1.735(2) C(6)–C(1)–N(1) 118.0(2)
C(3)–C(4)–Cl 119.4(2)C(5)–C(4)–Cl 119.4(2)
D–H� � �A D–H (Å) H� � �A (Å) D� � �A (Å) DHA (8)
Hydrogen-bond schemeO(1)–H(O1)� � �O(3) 0.99(3) 1.55(3) 2.539(2) 177(3)O(2)–H(O2)� � �O(4) 0.95(3) 1.66(3) 2.581(2) 162(2)N–H(1N)� � �O(3) 0.95(2) 1.74(2) 2.682(2) 171(2)N–H(2N)� � �O(4) 0.98(2) 1.87(2) 2.853(2) 177(2)N–H(3N)� � �O(4) 0.88(2) 2.10(2) 2.952(3) 163(2)
Estimated standard deviations are given in parentheses.

contacts with two lengths of 1.55(3) and 1.66(3) Å, ensures the cohesion between PO4 tetrahedron to build inorganiclayers, whereas the second type including three distances with N–H� � �O values in the range [1.74(2)–2.10(2)] Å linksorganic entities to inorganic layers. A three-dimensional frame work is then created.
A comparison of the title compound (I) to p-methylanilinium dihydrogenmonophosphate (II) [10] and to p-ethylanilinium dihydrogenmonophosphate (III) [11] shows that they differ only by the nature of the substituents at thepara-position on the phenyl ring.
However, some similarities can be observed between these structures. In each of the three cases, the structure has alayer organization, which contains the H2PO4
� groups encapsulating the organic entities in the same Pbca space groupof the orthorhombic system, Z = 8. In these structures, the anionic networks contain centrosymmetric pairs ofdihydrogenphosphates where each dihydrogenphosphate is simultaneously donors and acceptors of hydrogen bond. Inaddition, the two-dimensional network of (I) seems to be as cohesive as the two-dimensional anionic network of (II),and (III) as confirmed by the values of hydrogen bonds in the anionic aggregates. The geometrical parameters ofanilinium group have a comparable value in the three compounds and are similar to those observed in other complexescontaining anilinium cation. Similarly as in (I), the asymmetric unit of (II) and (III) consists of the organic cation andthe dihydrogenphosphate anion, joined by medium and strong N–H� � �O hydrogen bonds. As shown in the schematicdiagrams, the organic cation has different substituents which are placed in opposite ends of the aromatic ring. Thenature of different substituents affects weakly the strength of the intermolecular hydrogen bonds and the atomicarrangement.
Recently a similar structure for the (4-chloro-2-methylanilinium dihydrogenphosphate (4-Cl-2-CH3C6H4NH3)H2PO4 has been published [12]. This latter and the title compound differ only by the nature ofsubstituent of the organic cation at the C2 position. This simple difference affects the atomic arrangement, hydrogenbond network and the unit-cell parameters. Thus, the structure (IV) crystallizes in the monoclinic system (P21/C). Thislatter contains the dihydrogenphosphate anions interconnected by O–H� � �O hydrogen bonds which form a thick layer,parallel to (1 0 0) plane. Compare the structure of compound I with that of compound IV, we found that the connectionmode of H2PO4
� anions are different. The difference in connection mode in I and IV induces the differentrearrangement atomic type. In both structures which contain three similar N–H� � �O interactions, each aryl ammonium
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–32443238
Fig. 2. The atomic arrangement of ( p-ClC6H4NH3)H2PO4 in projection along the b-axis. The phosphoric groups are given in tetrahedralrepresentation. The other atoms are indicated by their symbols. Intermolecular H-bonds are denoted by dotted lines.
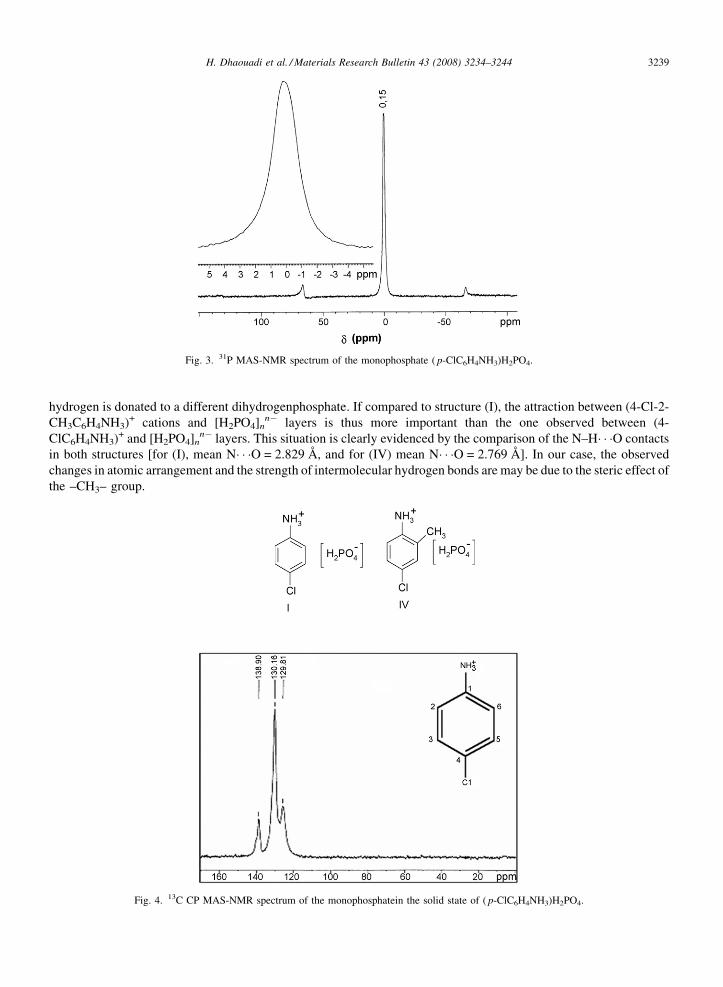
hydrogen is donated to a different dihydrogenphosphate. If compared to structure (I), the attraction between (4-Cl-2-CH3C6H4NH3)+ cations and [H2PO4]n
n� layers is thus more important than the one observed between (4-ClC6H4NH3)+ and [H2PO4]n
n� layers. This situation is clearly evidenced by the comparison of the N–H� � �O contactsin both structures [for (I), mean N� � �O = 2.829 Å, and for (IV) mean N� � �O = 2.769 Å]. In our case, the observedchanges in atomic arrangement and the strength of intermolecular hydrogen bonds are may be due to the steric effect ofthe –CH3– group.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–3244 3239
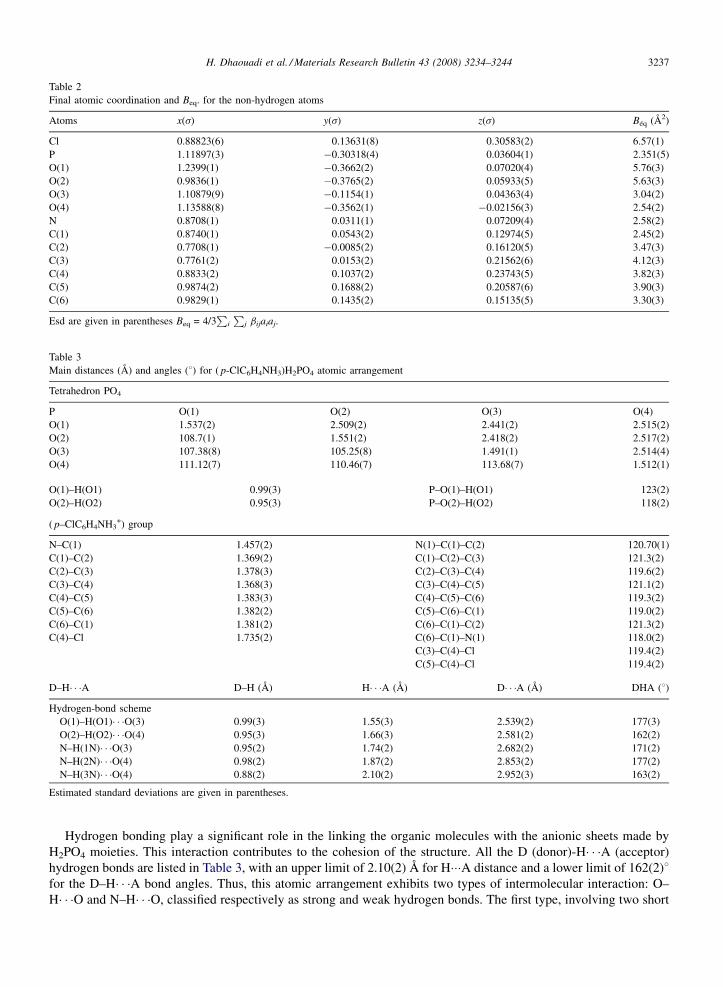
Fig. 3. 31P MAS-NMR spectrum of the monophosphate ( p-ClC6H4NH3)H2PO4.
Fig. 4. 13C CP MAS-NMR spectrum of the monophosphatein the solid state of ( p-ClC6H4NH3)H2PO4.
The results of the comparative study have shown that the atomic arrangement is strongly affected by two factors: thepresence of various substituents attached to the aryl ring, and the strength of H-bonds.
3.2. NMR results
The high resolution NMR spectroscopy is a powerful technique for the characterization of phosphates. From theisotropic chemical shift values of NMR components, structural aspects have been studied. Proton decoupled 31P MAS-NMR spectrum of crystalline monophosphate is presented in Fig. 3. It exhibits a unique sharp peak. The correspondingchemical shift value (0.15 ppm) is recorded with respect to 85% H3PO4 aqueous solution. This chemical shift valueagrees with those of monophosphate (between �10 and +5 ppm), depending on the compound [13–18]. The singlepeak is related to the number of phosphorus sites, which exist in this atomic arrangement.
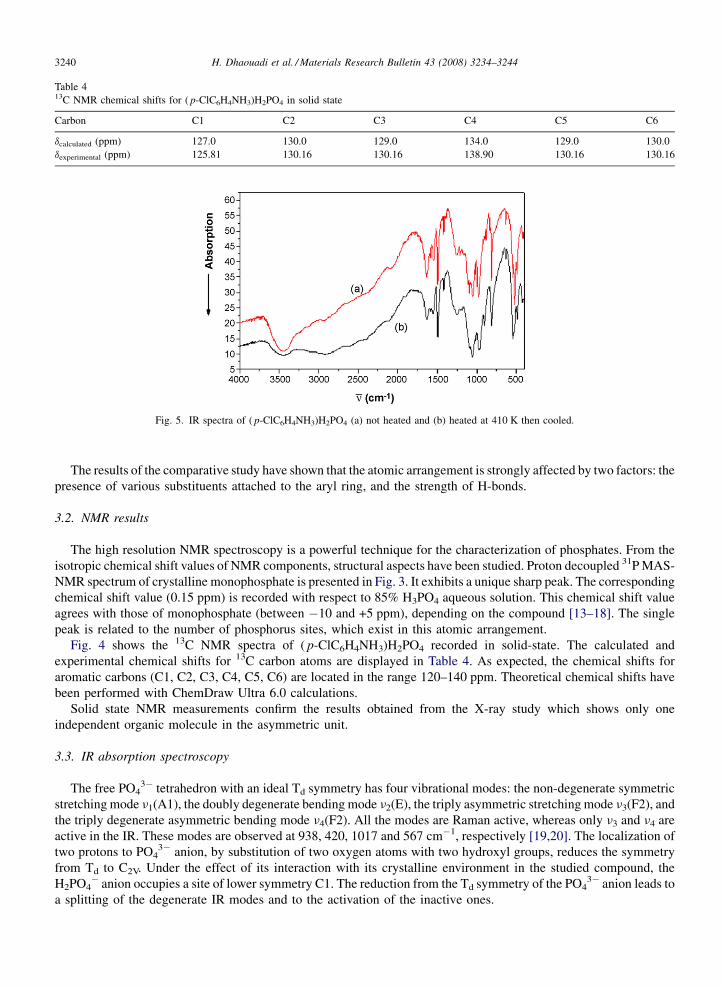
Fig. 4 shows the 13C NMR spectra of ( p-ClC6H4NH3)H2PO4 recorded in solid-state. The calculated andexperimental chemical shifts for 13C carbon atoms are displayed in Table 4. As expected, the chemical shifts foraromatic carbons (C1, C2, C3, C4, C5, C6) are located in the range 120–140 ppm. Theoretical chemical shifts havebeen performed with ChemDraw Ultra 6.0 calculations.
Solid state NMR measurements confirm the results obtained from the X-ray study which shows only oneindependent organic molecule in the asymmetric unit.
3.3. IR absorption spectroscopy
The free PO43� tetrahedron with an ideal Td symmetry has four vibrational modes: the non-degenerate symmetric
stretching mode n1(A1), the doubly degenerate bending mode n2(E), the triply asymmetric stretching mode n3(F2), andthe triply degenerate asymmetric bending mode n4(F2). All the modes are Raman active, whereas only n3 and n4 areactive in the IR. These modes are observed at 938, 420, 1017 and 567 cm�1, respectively [19,20]. The localization oftwo protons to PO4
3� anion, by substitution of two oxygen atoms with two hydroxyl groups, reduces the symmetryfrom Td to C2V. Under the effect of its interaction with its crystalline environment in the studied compound, theH2PO4
� anion occupies a site of lower symmetry C1. The reduction from the Td symmetry of the PO43� anion leads to
a splitting of the degenerate IR modes and to the activation of the inactive ones.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–32443240
Table 413C NMR chemical shifts for ( p-ClC6H4NH3)H2PO4 in solid state
Carbon C1 C2 C3 C4 C5 C6
dcalculated (ppm) 127.0 130.0 129.0 134.0 129.0 130.0dexperimental (ppm) 125.81 130.16 130.16 138.90 130.16 130.16
Fig. 5. IR spectra of ( p-ClC6H4NH3)H2PO4 (a) not heated and (b) heated at 410 K then cooled.
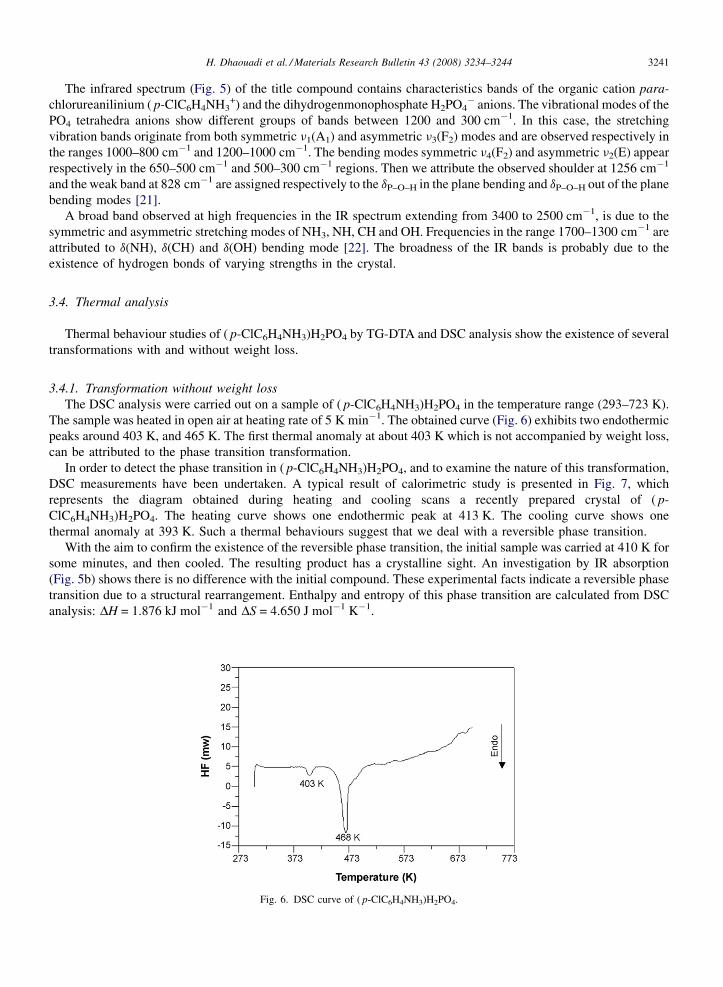
The infrared spectrum (Fig. 5) of the title compound contains characteristics bands of the organic cation para-chlorureanilinium ( p-ClC6H4NH3
+) and the dihydrogenmonophosphate H2PO4� anions. The vibrational modes of the
PO4 tetrahedra anions show different groups of bands between 1200 and 300 cm�1. In this case, the stretchingvibration bands originate from both symmetric n1(A1) and asymmetric n3(F2) modes and are observed respectively inthe ranges 1000–800 cm�1 and 1200–1000 cm�1. The bending modes symmetric n4(F2) and asymmetric n2(E) appearrespectively in the 650–500 cm�1 and 500–300 cm�1 regions. Then we attribute the observed shoulder at 1256 cm�1
and the weak band at 828 cm�1 are assigned respectively to the dP–O–H in the plane bending and dP–O–H out of the planebending modes [21].
A broad band observed at high frequencies in the IR spectrum extending from 3400 to 2500 cm�1, is due to thesymmetric and asymmetric stretching modes of NH3, NH, CH and OH. Frequencies in the range 1700–1300 cm�1 areattributed to d(NH), d(CH) and d(OH) bending mode [22]. The broadness of the IR bands is probably due to theexistence of hydrogen bonds of varying strengths in the crystal.
3.4. Thermal analysis
Thermal behaviour studies of ( p-ClC6H4NH3)H2PO4 by TG-DTA and DSC analysis show the existence of severaltransformations with and without weight loss.
3.4.1. Transformation without weight lossThe DSC analysis were carried out on a sample of ( p-ClC6H4NH3)H2PO4 in the temperature range (293–723 K).
The sample was heated in open air at heating rate of 5 K min�1. The obtained curve (Fig. 6) exhibits two endothermicpeaks around 403 K, and 465 K. The first thermal anomaly at about 403 K which is not accompanied by weight loss,can be attributed to the phase transition transformation.
In order to detect the phase transition in ( p-ClC6H4NH3)H2PO4, and to examine the nature of this transformation,DSC measurements have been undertaken. A typical result of calorimetric study is presented in Fig. 7, whichrepresents the diagram obtained during heating and cooling scans a recently prepared crystal of ( p-ClC6H4NH3)H2PO4. The heating curve shows one endothermic peak at 413 K. The cooling curve shows onethermal anomaly at 393 K. Such a thermal behaviours suggest that we deal with a reversible phase transition.
With the aim to confirm the existence of the reversible phase transition, the initial sample was carried at 410 K forsome minutes, and then cooled. The resulting product has a crystalline sight. An investigation by IR absorption(Fig. 5b) shows there is no difference with the initial compound. These experimental facts indicate a reversible phasetransition due to a structural rearrangement. Enthalpy and entropy of this phase transition are calculated from DSCanalysis: DH = 1.876 kJ mol�1 and DS = 4.650 J mol�1 K�1.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–3244 3241
Fig. 6. DSC curve of ( p-ClC6H4NH3)H2PO4.
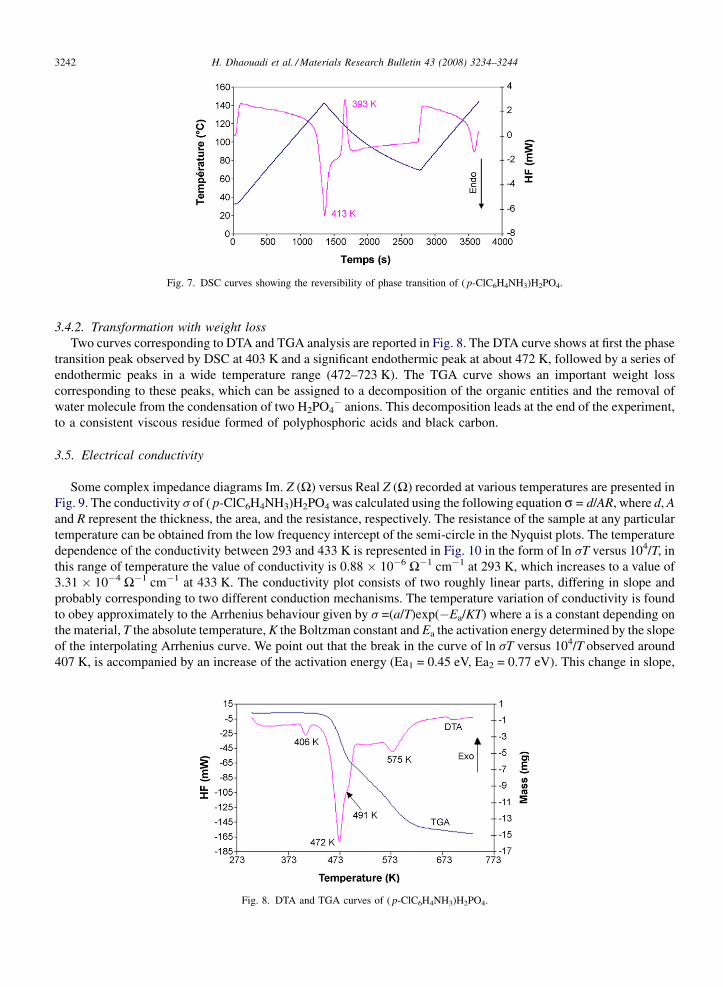
3.4.2. Transformation with weight lossTwo curves corresponding to DTA and TGA analysis are reported in Fig. 8. The DTA curve shows at first the phase
transition peak observed by DSC at 403 K and a significant endothermic peak at about 472 K, followed by a series ofendothermic peaks in a wide temperature range (472–723 K). The TGA curve shows an important weight losscorresponding to these peaks, which can be assigned to a decomposition of the organic entities and the removal ofwater molecule from the condensation of two H2PO4
� anions. This decomposition leads at the end of the experiment,to a consistent viscous residue formed of polyphosphoric acids and black carbon.
3.5. Electrical conductivity
Some complex impedance diagrams Im. Z (W) versus Real Z (W) recorded at various temperatures are presented inFig. 9. The conductivity s of ( p-ClC6H4NH3)H2PO4 was calculated using the following equation s = d/AR, where d, Aand R represent the thickness, the area, and the resistance, respectively. The resistance of the sample at any particulartemperature can be obtained from the low frequency intercept of the semi-circle in the Nyquist plots. The temperaturedependence of the conductivity between 293 and 433 K is represented in Fig. 10 in the form of ln sT versus 104/T, inthis range of temperature the value of conductivity is 0.88 � 10�6 W�1 cm�1 at 293 K, which increases to a value of3.31 � 10�4 W�1 cm�1 at 433 K. The conductivity plot consists of two roughly linear parts, differing in slope andprobably corresponding to two different conduction mechanisms. The temperature variation of conductivity is foundto obey approximately to the Arrhenius behaviour given by s =(a/T)exp(�Ea/KT) where a is a constant depending onthe material, T the absolute temperature, K the Boltzman constant and Ea the activation energy determined by the slopeof the interpolating Arrhenius curve. We point out that the break in the curve of ln sT versus 104/T observed around407 K, is accompanied by an increase of the activation energy (Ea1 = 0.45 eV, Ea2 = 0.77 eV). This change in slope,
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–32443242
Fig. 8. DTA and TGA curves of ( p-ClC6H4NH3)H2PO4.
Fig. 7. DSC curves showing the reversibility of phase transition of ( p-ClC6H4NH3)H2PO4.
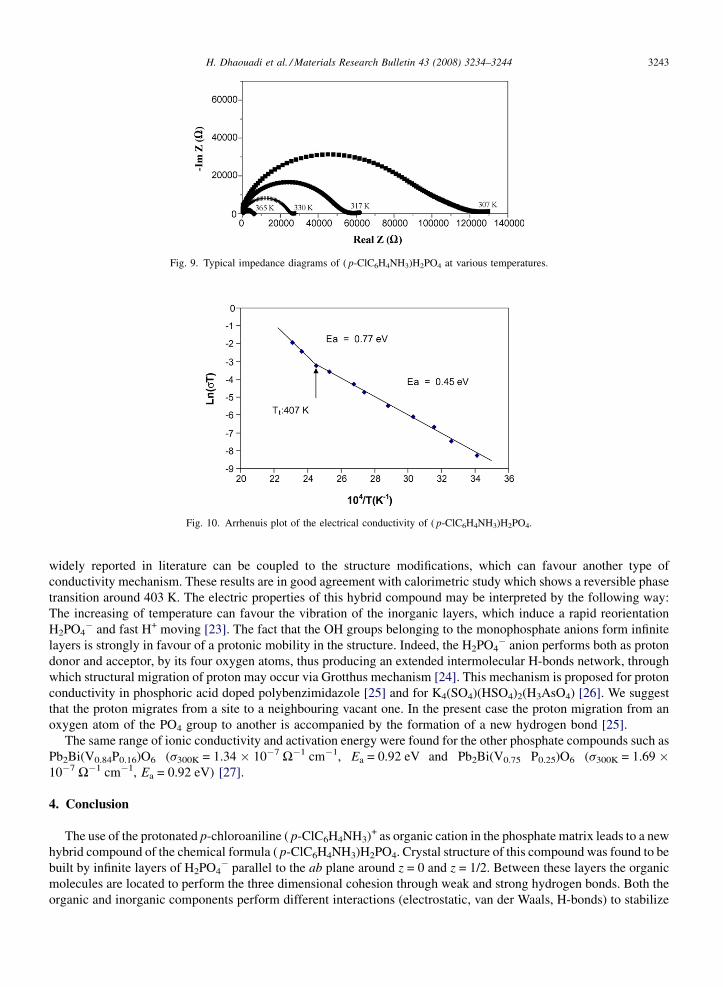
widely reported in literature can be coupled to the structure modifications, which can favour another type ofconductivity mechanism. These results are in good agreement with calorimetric study which shows a reversible phasetransition around 403 K. The electric properties of this hybrid compound may be interpreted by the following way:The increasing of temperature can favour the vibration of the inorganic layers, which induce a rapid reorientationH2PO4
� and fast H+ moving [23]. The fact that the OH groups belonging to the monophosphate anions form infinitelayers is strongly in favour of a protonic mobility in the structure. Indeed, the H2PO4
� anion performs both as protondonor and acceptor, by its four oxygen atoms, thus producing an extended intermolecular H-bonds network, throughwhich structural migration of proton may occur via Grotthus mechanism [24]. This mechanism is proposed for protonconductivity in phosphoric acid doped polybenzimidazole [25] and for K4(SO4)(HSO4)2(H3AsO4) [26]. We suggestthat the proton migrates from a site to a neighbouring vacant one. In the present case the proton migration from anoxygen atom of the PO4 group to another is accompanied by the formation of a new hydrogen bond [25].
The same range of ionic conductivity and activation energy were found for the other phosphate compounds such asPb2Bi(V0.84P0.16)O6 (s300K = 1.34 � 10�7 W�1 cm�1, Ea = 0.92 eV and Pb2Bi(V0.75 P0.25)O6 (s300K = 1.69 �10�7 W�1 cm�1, Ea = 0.92 eV) [27].
4. Conclusion
The use of the protonated p-chloroaniline ( p-ClC6H4NH3)+ as organic cation in the phosphate matrix leads to a newhybrid compound of the chemical formula ( p-ClC6H4NH3)H2PO4. Crystal structure of this compound was found to bebuilt by infinite layers of H2PO4
� parallel to the ab plane around z = 0 and z = 1/2. Between these layers the organicmolecules are located to perform the three dimensional cohesion through weak and strong hydrogen bonds. Both theorganic and inorganic components perform different interactions (electrostatic, van der Waals, H-bonds) to stabilize
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–3244 3243
Fig. 10. Arrhenuis plot of the electrical conductivity of ( p-ClC6H4NH3)H2PO4.
Fig. 9. Typical impedance diagrams of ( p-ClC6H4NH3)H2PO4 at various temperatures.

the three-dimensional network. Solid-state 31P and 13C MAS-NMR spectroscopies are in accordance with the X-raystructure.
Acknowledgement
We would like to thank the Tunisian Secretariat State for Scientific Research and Technology for its support.
References
[1] R.A. Hearn, C.E. Bugg, Acta Cryst. B28 (1977) 1513.[2] J.M. Adams, Acta Cryst. B33 (1972) 3662.[3] L. Baouab, A. Jouini, J. Solid State Chem. 141 (1988) 343.[4] M.T. Averbuch-Pouchot, A. Durif, J.C. Guitel, Acta Cryst. C44 (1988) 99.[5] M.T. Averbuch-Pouchot, A. Durif, J.C. Guitel, Acta Cryst. C45 (1989) 421.[6] G. Ferraris, G. Ivaldi, Acta Cryst. B40 (1984) 1.[7] B.C. Aakerby, B.P. Hitchcook, D.B. Moyle, R.K. Seddon, J. Chem. Soc. Chem. Commun. 161 (1989) 1856.[8] J. Pecaut, R. Masse, Z. Kristallogr. 208 (1993) 241.[9] Z.J. Li, Chen S X.M., Ren S Z.X., Y. Li, X.A. Chen, Z.T. Huang, Chin. J. Struct. Chem. 16 (1997) 311.
[10] W. Smirani, A. Ben Slimene, M. Rzaigui, Z. Kristallogr NCS 219 (2004) 189.[11] K. Kaabi, A. Rayes, C. Ben Nasr, M. Rzaigui, F. Lefebvre, Mater. Res. Bull. 38 (2003) 741.[12] I. Fabry, R. Krupkova, V. Studnicka, Acta Cryst. E38 (2002) 103.[13] A.R. Grimmer, U. Haubenreisser, Chem. Phys. Lett. 99 (1983) 487.[14] D. Mûller, E. Jahn, G. Ladwig, U. Haubenreisser, Chem. Phys. Lett. 109 (1984) 332.[15] A.K. Cheetham, N.J. Clayden, C.M. Dobson, R.J.B. Jakemen, J. Chem. Soc. Chem. Commun. 4 (1986) 195.[16] L. Mudracovskii, V.P. Shmochkova, N.S. Kentsarenko, V.M. Mastikhin, Phys. Chem. Solids 47 (1986) 335.[17] G.L. Turner, K.A. Smith, R.J. Kirkpatrick, E.J. Oldfield, J. Magn. Reson. 70 (1986) 408.[18] S. Prabhakar, K.J. Rao, C.N.R. Rao, Chem. Phys. Lett. 139 (1987) 96.[19] A. Gharbi, A. Jouini, M.T. Averbuch-Pouchot, A. Durif, J. Solid State Chem. 111 (1994) 330.[20] D. Dolphin, A.E. Wick, Tabulation of Infrared Spectra Data, John Wiley and sons, New York, 1977.[21] S. Kammoun, M. Kammoun, A. Daouad, F. Romain, Spectrochim. Acta Cryst. 47A (1991) 1051.[22] A.C. Chapman, L.E. Thirlwell, Spectrochim. Acta Cryst. 20 (1964) 937.[23] A. Schechter, R.F. Savinell, Solid State Ionics 147 (2002) 181.[24] O. Labidi, P. Roussel, M. Huve, M. Drache, P. Conflant, J.P. Wignacourt, J. Solid State Chem. 178 (2005) 2247.[25] N. Zouari, H. Khemakhem, T. Mhiri, A. Douad, J. Phys. Chem. Solid 60 (1999) 1779.[26] M. Amri, N. Zouari, T. Mhiri, S. Pechev, P. Gravereau, R. Von Der Muhll, J. Phys. Chem. Solids 68 (2007) 1281.[27] R. Bouchet, S. Miller, M. Duclot, J.L. Souquet, Solid State Ionics 145 (2001) 69.
H. Dhaouadi et al. / Materials Research Bulletin 43 (2008) 3234–32443244


![Garanties 2009-2010 Crystal Studies€¦ · Garanties 2009-2010 Crystal Studies [ La Mobilité] Particuliers Imprimé sur papier recyclé [ des solutions ] pour les étudiants et](https://img.pdfslide.fr/doc/110x75/5f638ec8469e1048172b0e7d/garanties-2009-2010-crystal-garanties-2009-2010-crystal-studies-la-mobilit.jpg)


























