Embed Size (px)
Citation preview

DIAGRAMMES POTENTIEL-PH (1).
page 1/8
LES DIAGRAMMES POTENTIEL-PH (OU DIAG. DE POURBAIX).
I : Principe d'un diagramme potentiel-pH. 1°) Description d'un diagramme potentiel-pH.
Un diagramme potentiel-pH est relatif à un élément chimique donné, présent en solution aqueuse à divers nombres d'oxydation dans différentes espèces chimiques. Ces diagrammes ont été proposés et établis par le chimiste belge POURBAIX.
On représente, pour les différents couples rédox mis en jeu, les variations du potentiel rédox E en fonction du pH.
Un diagramme potentiel-pH fait apparaître les différents domaines de prédominance ou d'existence de chaque espèce. La superposition de diagrammes relatifs à plusieurs éléments permet, par une méthode graphique simple de prévoir les réactions mises en jeu et leur sens d'évolution pour des concentrations initiales fixées des différents produits.
2°) Les domaines de prédominance (en abrégé D.P.) (ou d'existence). Interprétation graphique.
Soit le couple Ox /Red, caractérisé par la demi-équation rédox: Red = -e n + +aqH q +Ox βα .
dont le potentiel rédox à 25 °C est donné par :⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛° β
α
RedaOxa
log n
0,06 + pH 0,06
nq
- E = E
Lorsque les activités de Ox et red sont fixées (arbitrairement), le potentiel E est une fonction affine du pH, qui sépare le plan E-pH en deux domaines. On note Ef cette droite frontière.
Supposons les espèces Ox et Red placées dans une solution maintenue au potentiel E. On cherche les concentrations à l'équilibre [Ox] et [Red].
Si E > Ef : domaine d'existence ou de prédo-minance de Ox.
Si E < Ef : domaine d'existence ou de prédo-minance de Red.
Pourquoi spécifier prédominance ou existence ? - Entre deux solutés (ions ou complexes), Ox et Red (ou Acide et Base) coexistent toujours, en
proportions variables selon la valeur du potentiel à l'équilibre (ou du pH) : on parle de D.P.
- Entre un soluté et une phase condensée pure (liquide ou solide), la présence de cette phase condensée est conditionnelle: cette phase est présente ou absente, selon que le produit de solubilité est atteint ou pas. La droite frontière délimite donc un domaine d'existence de l'espèce condensée: la frontière peut être horizontale (couple rédox) ou verticale (couple acide-base).
Les conventions sur les droites frontières.
Il faut d'abord fixer la concentration totale atomique Ctot de l'élément étudié.
1- Les deux espèces sont solubles:
pH (ou E) frontière = pH (ou E) pour lequel il y a équiconcentration ato-mique en l'élément X (équations du type : [ ] [ ]p qp X q X= ).
2- Une seule des deux espèces est soluble: pH (ou E) frontière calculé pour [Xdissout]atomique = Ctot.
pH
E
E f = f(pH)
Ox
Red

DIAGRAMMES POTENTIEL-PH (1).
page 2/8
3- Une des deux espèces est un gaz:
On fixe Pgaz = 1 bar et pH (ou E) frontière pour [Xdissout]atomique = Ctot.
3°) Prévision des réactions par lecture d'un diagramme potentiel-pH. L'utilisation d'un diagramme potentiel-pH repose sur le critère suivant:
Dans un système à l'équilibre thermodynamique, tous les couples rédox pré-sents ont le même potentiel.
Soit une solution aqueuse de Ox1, Ox2, Red1, Red2, On trace les droites E1f = f(pH) et E2f = f(pH) dans un diagramme potentiel-pH.
Considérons cette solution à un pH fixé par un mé-lange tampon à pH1 < pH0.
A l'état initial, on a deux valeurs possibles différen-tes pour le potentiel E de la solution, ce qui ne corres-pond pas à un état de stabilité. Il faut donc que le potentiel du couple 2 diminue et que celui du couple 1 augmente. Ainsi, le potentiel à l'équilibre est dans le domaine de prédominance de Ox1 et Red2, seules espèces com-patibles entre elles.
Pour pH > pH0, un raisonnement analogue conduit à la conclusion suivante: Ox1 et Red2 dis-paraissent en faveur de Ox2 et Red1, qui sont compatibles entre eux.
Lorsque deux espèces ayant leurs domaines de prédominance disjoints à un pH donné sont mises en présence, il se produit une réaction en faveur de leur disparition. Deux espèces ne peuvent être compatibles entre elles que si leurs domaines de prédominance (ou d'existence) sont contigus.
Stabilité d'une espèce. Dismutation. Soit une espèce A jouant le rôle de réducteur dans le couple Ox/A et d'oxydant dans le couple A/Red. Considérons un dia-gramme potentiel-pH ayant l'allure ci-contre:
Pour pH > pH0, A est incompatible avec elle-même, elle réagit donc selon la réaction: 2 A → Ox + Red. On dit que A se dismute.
Une dismutation est une réaction d’oxydo-réduction dans laquelle l’oxydant et le réducteur qui réagissent sont une seule et même espèce.
Ainsi dans ce diagramme potentiel-pH, A ne peut plus figurer pour pH > pH0 : Ox et Red ont alors une frontière commune, qu'il faut déterminer. Le diagramme prend l'allure représentée ci-contre.
Il existe aussi les cas de dédismutation (ou rétromutation).
Par exemple : 3 22 3Fe Fe Fe+ ++ → : réaction dite de médiamutationrétromutation
⎧⎨⎩
.
Une médiamutation est une réaction d’oxydo-réduction dans laquelle l’oxydant et le réducteur formés sont une seule et même espèce.
pH
E
pH0 pH1
Ox2
Red2
Ox1
Red1
E 2f
E 1f E2f(pH)
E1f(pH)
A
Red Ox
A
E
pH pH0
A
Red
Ox
E
pH pH0

DIAGRAMMES POTENTIEL-PH (1).
page 3/8
II : Méthode de tracé d'un diagramme potentiel-pH. 1°) Classement des espèces.
Classer les différentes espèces contenant l'élément X du diagramme par nombre d'oxydation croissant de bas en haut. Identifier les couples acide-base et faire apparaître sur un axe horizontal les domaines de pré-dominances des espèces acides et basiques. Déterminer, pour chaque degré d’oxydation, les valeurs de pH limitant les domaines d’existence ou de prédominance. Utiliser pour cela les constantes d’équilibre correspondantes :
- Ka pour les équilibres acido-basiques : [ ][ ]a
base hK
acide= , où 3h H O+⎡ ⎤= ⎣ ⎦ ,
- Ks pour les équilibres de précipitation : ( ) zp n
X OH pX nOH+ −= + , où p nz
SK X OH+ −⎡ ⎤ ⎡ ⎤= ⎣ ⎦ ⎣ ⎦
- βn pour les équilibres de complexation (métal M et ligand L) : nML M nL= + [ ][ ][ ]
n
nn
M LML
β =
2°) Première construction du diagramme. Construire un premier tableau d'espèces prépondérantes à partir duquel se déduisent les frontières nécessaires à la construction du diagramme. Numéroter ces différentes frontières. On aura intérêt à numéroter de bas en haut (sens des n.o. croissants) et de gauche vers la droite (sens des pH croissants).
(Cet ordre permet de corriger rapidement le tableau en cas d'une éventuelle dismutation).
Intervention d'une dismutation (ou d'une dédismutation): Rechercher au brouillon l'équation des droites frontières se trouvant le plus à gauche (Voir § 3°)), et les tracer afin de remarquer s'il apparaît une dismutation (ou une dédismutation). Dans ce cas, modifier le tableau précédent en conséquence, en faisant apparaître les nouvelles frontières. 3°) Tracé du diagramme.
Déterminer les équations des droites frontières à partir des données thermodynamiques (va-leurs des potentiels rédox standards, des pKa ou des pKs) et des conventions sur les frontières.
Remarque: Les droites d'un diagramme potentiel-pH se raccordent continûment.
Ce résultat peut rendre plus rapide le tracé du diagramme, mais cette méthode ne permet pas de remarquer les éventuelles erreurs se produisant lors d'un calcul. En revanche, si la méthode de continuité n'a pas été employée, l'erreur se dévoilera facilement car le diagramme ne sera pas conti-nu.
III Quelques diagrammes potentiel-pH. 1°) Le diagramme potentiel-pH de l'eau.
H intervient comme réducteur dans le couple H+(aq) / H2, de demi-équation électronique:
2 H+(aq) + 2 e- = H2.
Sur la frontière, PH2 = 1 bar. D'où )(h log 2
0,06 + )/H(HE = E 2
2+aq1f ° , soit E1f = -0.06 pH.
O intervient comme oxydant dans le couple O2 / H2O, de demi-équation électronique:
O2 + 4 e- + 4 H+(aq) = 2 H2O. Sur la frontière, PO2 = 1 bar. D’où )(h log 4
0,06 + E = 2fE 40
2

DIAGRAMMES POTENTIEL-PH (1).
page 4/8
Soit E2f = 1.23 - 0.06 pH.
Le diagramme potentiel-pH de l'eau: on y distingue trois zones.
1- zone de prédominance de l'eau, d'une largeur de 1,23 V, ∀ pH (domaine compris entre les droites a et b). C'est la :
zone de stabilité thermodynamique de l'eau.
2- Au-dessus, zone de prédominance de O2.
3- Au-dessous, zone de prédominance de H2
Remarque: en réalité, la zone de stabilité couvre une bande plus large, d'environ 2 V, à cause des blocages cinétiques rendant les réactions de dé-composition de l'eau très lentes.
2°) Diagramme potentiel-pH du fer. On cherche à tracer le diagramme (simplifié) du fer à 25 °C pour les espèces suivantes:
Fe, Fe2+, Fe3+ , Fe(OH)2 et Fe(OH)3.
On prendra Ctot = 0.1 mol.L-1.
On donne: E°1(Fe2+/Fe) = - 0.44 V; E°2(Fe3+/Fe2+) = 0.77 V; pKs1(Fe(OH)3) = 36; pKs2(Fe(OH)2) = 15;
a) Construction du diagramme.
Classement des espèces par état d’oxydation : L’étude est limitée ici aux n.o. 0, + II et + III.
Recherche des D.P. acido-basiques. Cherchons le pH de précipitation de chaque hydroxyde :
- Si Fe(OH)2 précipite, on a : 22
1SFe OH K+ −⎡ ⎤ ⎡ ⎤ =⎣ ⎦ ⎣ ⎦ .
Sur la frontière (entre Fe2+ et Fe(OH)2), on fixe : 2totFe C+⎡ ⎤ =⎣ ⎦ .
On en déduit le pH d’apparition du précipité Fe(OH)2 : 7pH =
- Si Fe(OH)3 précipite, on a : 32
2SFe OH K+ −⎡ ⎤ ⎡ ⎤ =⎣ ⎦ ⎣ ⎦ .
Sur la frontière (entre Fe3+ et Fe(OH)3), on fixe : 3totFe C+⎡ ⎤ =⎣ ⎦ .
On en déduit le pH d’apparition du précipité Fe(OH)3 : 2,3pH ≈
Première ébauche du diagramme avec numérotation des frontières : On superpose les D.P. par n.o. croissants avec les D.P. des équilibres acido-basiques pour obtenir une première ébauche du diagramme potentiel-pH et on numérote les différentes frontières du tableau de façon à identifier les différents cou-ples rédox et acido-basiques à prendre en compte. On numérote les couples suivant les potentiels par valeurs de pH (pour repérer d’éventuelles dismutations et pouvoir modifier le tableau).
pH
E (volt)
H2O O2
H2O H2
zone de stabilitéthermodynamique
de l'eau
1,23 V
0 V
1,83 V
-0,2 V
(a)
(b)
zone d'immunité cinétique
Diagramme à 25 °C,sous P = 1 bar.
Le diagramme potentiel-pH de l'eau.
immunité cinétique
Fe
Fe2+ Fe(OH)2
Fe(OH)3 Fe3+
n.o
0
+II
+III
pH
E
1
2
3
4
5
6
7
Fe
Fe2+
Fe3+ Fe(OH)3
Fe(OH)2

DIAGRAMMES POTENTIEL-PH (1).
page 5/8
Mise en équation des droites frontières pour chaque couple :
Frontière n° Couple bilan
Équation sur la fron-tière
1 Fe2+ / Fe ( )2 sFe e Fe−+ =
( )0 21 0,03logE E Fe +⎡ ⎤= + ⎣ ⎦
2totFe C+⎡ ⎤ =⎣ ⎦
1 0, 47E V= −
2 Fe3+ / Fe2+
3 2Fe e Fe+ − ++ =
302 2
0,06 logFe
E EFe
+
+
⎛ ⎞⎡ ⎤⎣ ⎦⎜ ⎟= +⎜ ⎟⎡ ⎤⎣ ⎦⎝ ⎠
2 3Fe Fe+ +⎡ ⎤ ⎡ ⎤=⎣ ⎦ ⎣ ⎦
2 0,77E V=
3 Fe(OH)3 / Fe3+ 333 ( )Fe OH Fe OH+ −+ =
3totFe C+⎡ ⎤ =⎣ ⎦
2,3pH =
4 Fe(OH)3 / Fe2+
23 2( ) 3 3aqFe OH H e Fe H O+ − ++ + = +
304 2
10,06 log1
hE EFe +
⎛ ⎞×⎜ ⎟= +⎜ ⎟⎡ ⎤×⎣ ⎦⎝ ⎠
2totFe C+⎡ ⎤ =⎣ ⎦
04 4 0,06 0,18E E pH= + −
5 Fe(OH)2 / Fe2+ 222 ( )Fe OH Fe OH+ −+ =
2totFe C+⎡ ⎤ =⎣ ⎦
7pH =
6 Fe(OH)2 / Fe
2 ( ) 2( ) 2 2 2aq sFe OH H e Fe H O+ −+ + = +
206
0,06 1log2 1 1
hE E⎛ ⎞×
= + ⎜ ⎟×⎝ ⎠
06 6 0,06E E pH= −
7 Fe(OH)3 / Fe(OH)2
3 2 2( ) ( )aqFe OH H e Fe OH H O+ −+ + = +
07
10,06log1 1hE E ×⎛ ⎞= + ⎜ ⎟×⎝ ⎠
07 7 0,06E E pH= −
Il n’est pas besoin de déterminer les valeurs des potentiels standard inconnus : il suffit d’exploiter la continuité du diagramme potentiel-pH. Ainsi, coupe ∩ . On en déduit 0
4E avec : 040,77 0,06 0,18 2,3E= + − × . D’où 0
4 1,12E V= .
De même, coupe ∩ . On en déduit 06 0,06E V= −
Et coupe ∩ , qui donne : 07 0,30E V= −
Il est toutefois possible de déterminer les potentiels standard inconnus à partir de combinaisons linéaires d’équilibres chimiques et de demi-équations rédox (Loi de Hess appliquée aux ΔrG° de réaction). Par exemple, pour le calcul de 0
4E , on forme le cycle de bilans de matière suivant :
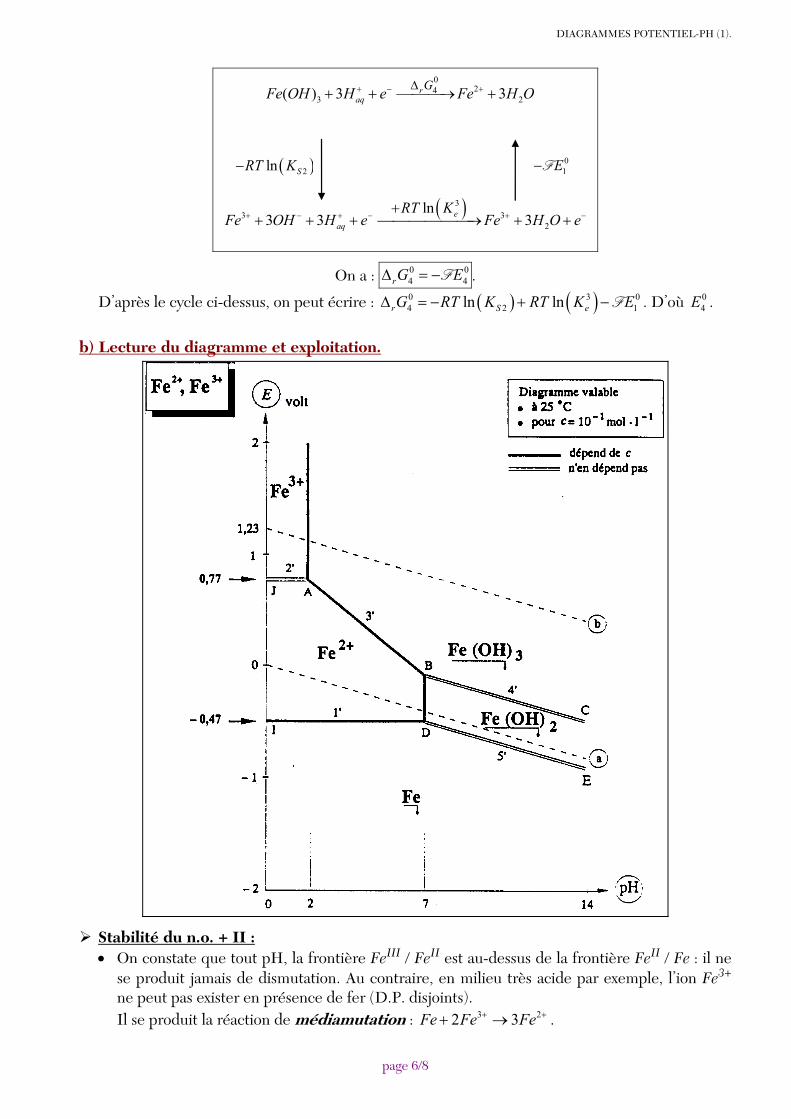
DIAGRAMMES POTENTIEL-PH (1).
page 6/8
0
243 2( ) 3 3r
aqGFe OH H e Fe H O+ − +Δ+ + ⎯⎯⎯→ +
( )33 3
2
ln3 3 3e
aq
RT KFe OH H e Fe H O e+ − + − + −
++ + + ⎯⎯⎯⎯⎯⎯→ + +
01E−F ( )2ln SRT K−
On a : 0 04 4rG EΔ = −F .
D’après le cycle ci-dessus, on peut écrire : ( ) ( )0 3 04 2 1ln lnr S eG RT K RT K EΔ = − + − F . D’où 0
4E .
b) Lecture du diagramme et exploitation.
Stabilité du n.o. + II : • On constate que tout pH, la frontière FeIII / FeII est au-dessus de la frontière FeII / Fe : il ne
se produit jamais de dismutation. Au contraire, en milieu très acide par exemple, l’ion Fe3+ ne peut pas exister en présence de fer (D.P. disjoints).
Il se produit la réaction de médiamutation : 3 22 3Fe Fe Fe+ ++ → .

DIAGRAMMES POTENTIEL-PH (1).
page 7/8
• Les métaux présentant un E° très négatif (Zn, Al) réduisent les ions Fe2+ en fer.
Stabilité de FeII et FeIII : • FeII et FeIII ont des D.P. recouvrant partiellement le domaine de stabilité de l’eau : ils sont
donc stables dans l’eau. • Le D.P. des ions Fe3+ est très limité du côté des pH acides : les solutions ferriques devront
donc être acidifiées, et le choix de l’acide utilisé n’est pas neutre, pouvant modifier de façon importante, tant la couleur de la solution ferrique que le potentiel standard E° !
• Ainsi, en milieu perchlorique (HC O4), on mesure E°(Fe3+ / Fe2+ ) =0,75 V (proche de la va-
leur « théorique de 0,77 V) ; avec l’acide chlorhydrique à 1 mol.L-1 E° chute à 0,70 V et en milieu sulfurique (H2SO4 à 0,5 mol.L-1) il n’est que de 0,67 V.
Ceci montre que les chloro-, sulfato- et aquacomplexes du FeIII sont plus stables que les complexes correspondants du FeII.
• On peut oxyder les solutions ferreuses (FeII) par : o le permanganate de potassium : 2 2 3
4 25 8 5 4aqFe MnO H Mn Fe H O+ − + + ++ + → + + ,
o le dichromate de potassium : 2 2 3 37 26 14 2 6 7aqFe CrO H Cr Fe H O+ − + + ++ + → + + ,
o les ions nitrates : 2 33 23 4 3 2aqFe NO H NO Fe H O+ − + ++ + → + +
Ces réactions consomment des ions H3O+ et si le milieu n’a pas été assez acidifié, c’est Fe(OH)3 qui précipite.
Attaque du fer par l’eau ou les acides : • Le domaine d’immunité du fer solide (D.P. de Fe) est extérieur au domaine de stabilité
de l’eau. Le fer est donc attaqué : il n’est pas un métal noble. • On observe que le potentiel rédox du couple O2 / H2O est très supérieur à celui du couple
Fe2+ / Fe, quel que soit le pH. Aussi, faudra-t-il toujours envisager la participation de l’oxygène dissous dans l’eau à la réaction d’oxydation du métal.
On distingue les zones de corrosion (domaines de Fe2+ et Fe3+ ), et de passivation (domaines de Fe(OH)2 et Fe(OH)3).
- Action de l’eau supposée non aérée en milieu neutre : L’eau oxyde le métal de manière peu quantitative en Fe(OH)2 selon :
( ) 2 2( ) 2( )2 ( )s s gFe H O Fe OH H+ +
- Action d’un acide à anion inerte (non oxydant) (type HC ): En milieu acide, les ions H3O+ (couple H2O / H2) oxydent le fer en Fe2+ selon :
2( ) 2( )2s aq gFe H Fe H+ ++ → + .
- Action d’un acide à anion oxydant (type HNO3) : En général, le couple rédox associé à l’anion est un meilleur oxydant que le couple H+/H2. Ainsi, l’anion transforme le fer en Fe3+.
- Action de O2 de l’air (ou dissous dans l’eau) : L’oxydation de Fe(OH)2 est aisée (la droite frontière entre Fe(OH)3 et Fe(OH)2 est très en dessous de celle du couple O2 / H2O) selon : 2( ) 2 2 3( )4 ( ) 2 4 ( )s sFe OH O H O Fe OH+ + → . En revanche, on observe qu’en milieu nettement acide, O2 (et donc l’air) sont sans effet notable sur le fer et les solutions ferreuses (même si thermodynamiquement, la réaction d’oxydation est possible).

DIAGRAMMES POTENTIEL-PH (1).
page 8/8
c) Influence de Ctot sur les équations des droites frontières.
• Les frontières des couples Fe3+ / Fe2+ , Fe(OH)2 / Fe, ou, Fe(OH)3 / Fe(OH)2. sont indépen-dantes de Ctot.
• Si Ctot : les pH d’apparition des précipités Fe(OH)2 et Fe(OH)3 L’ordonnée à l’origine de la frontière entre Fe2+ et Fe L’ordonnée à l’origine de la frontière entre Fe(OH)3 / Fe3+ .
• Fixons un seuil limite : 6 110 .totC mol L− −= (choix usuel dans le domaine dit de la corrosion humide).
En effet, la corrosion est un phé-nomène cinétiquement lent et les concentrations des ions produits res-tent très faibles. En outre, on utilise désormais non plus les hydroxydes, mais les oxydes qui sont plus stables thermodynami-quement mais qui se forment moins rapidement.
Dans le cas du fer, les oxydes stables à la température de 25°C sont l’oxyde ferrique Fe2O3 et l’oxyde magnétique Fe3O4 (FeO n’existe qu’au-delà de 570 °C).
Ainsi, pour Ctot = 1 μmol.L-1, on a changé la nature du système d’étude :
2 3( ) 2 3( ) 3 4( )s s sFe Fe Fe Fe O Fe O+ +
On obtient le diagramme notable-ment modifié représenté figure ci-contre, faisant trois domaines dits d’immnuité, de corrosion et de passi-vation.
Immunité, corrosion et passivation :
Zone d’immunité. = zone de stabilité thermodynamique du métal
Zone de corrosion. = zone d’attaque du métal avec formation d’ions.
On distingue la corrosion en milieu acide ou en milieu basique.
Zone de passivation. = zone de protection du métal par la formation d’oxydes ou
d’hydroxydes à sa surface
Remarque : Ces différentes zones font appel à un raisonnement purement thermodynamique et ne prennent pas en compte les aspects cinétiques ou cristallographiques. En réalité, la passivation du fer n’est pas efficace, la couche de Fe2O3 n’étant pas totalement imperméable.
La zone de corrosion (acide ou basique) est d’autant plus étendue que Ctot . On fixe une limite de 6 110 .totC mol L− −= pour pouvoir parler de corrosion.