Embed Size (px)
Citation preview

Electrochimieen milieux de sels fondus
Jean Claude Poignet / Pierre Chamelot

Avantages et inconvénientsofferts par l’électrochimie en sels fondus
On peut effectuer le même type d’opérations qu’en
solutions aqueuses ou autres à température
ordinaire, mais avec des cinétiques bien plus
élevées, des équilibres atteints très rapidement,
une gamme de fenêtre électrochimique beaucoup
plus grande, mais aussi des difficultés
technologiques accrues.

Avantages
• Variété exceptionnelle de sels : on peut adapter la constitution d’un mélange en fonction des propriétés recherchées.
• Pas de radiolyse.• Stabilité thermique, faibles pressions de vapeur • Viscosités cinématiques faibles• Volumes réduits, peu d’effluents• Domaine de stabilité électrochimique très étendu : on peut préparer
ou traiter la plupart des éléments, de Li à F, dans ces milieux.• Conductivités électrique et thermique élevées.• Solubilités élevées, rendements élevés• Cinétiques réactionnelles (chimiques et électrochimiques) élevées,
équilibres atteints rapidement.

Inconvénients
• Les températures élevées nécessitent l’emploi de matériaux adaptés pour les conteneurs, électrodes etc.
• Les cinétiques chimiques et électrochimiques entraînent des risques accrus de corrosion, si des gradients (de température, de composition …) existent.
• Les sels fondus dissolvent partiellement des métaux, parmi lesquels alcalins et alcalino-terreux.
• La sélection of matériaux peut être délicate dans le cas de certains sels fondus : par exemple, les fluorures fondus dissolvent les oxydes
solides et les céramiques. Mais il est possible de trouver des solutions industrielles :
-
dans le cas de l’électrolyse de l’alumine , qui utilise des fluorures fondus à environ 1000°C, le sels fondu est séparé de la paroi du la cellule par une couche de ‘bain’ solide. Le carbone est aussi très stable en milieu de fluorures fondus, en l’absence de certains métaux alcalins.
- des composants métalliques (conteneur, diaphragmes) peuvent être protégés cathodiquement, ……

Cinétique rapide et gradient de température = problème pour la réalisation d'une électrode
de référence Ag/AgCl

Solubilité
des métaux dans les sels fondus
Les métaux sont plus ou moins solubles dans les sels fondus. A des températures suffisamment élevées, les métaux alcalins et alcalino-terreux (sauf Mg) sont solubles dans leurs halogénures fondus.
La dissolution provoque une augmentation de conductivité électrique du mélange fondu avec souvent une part ‘électronique’.
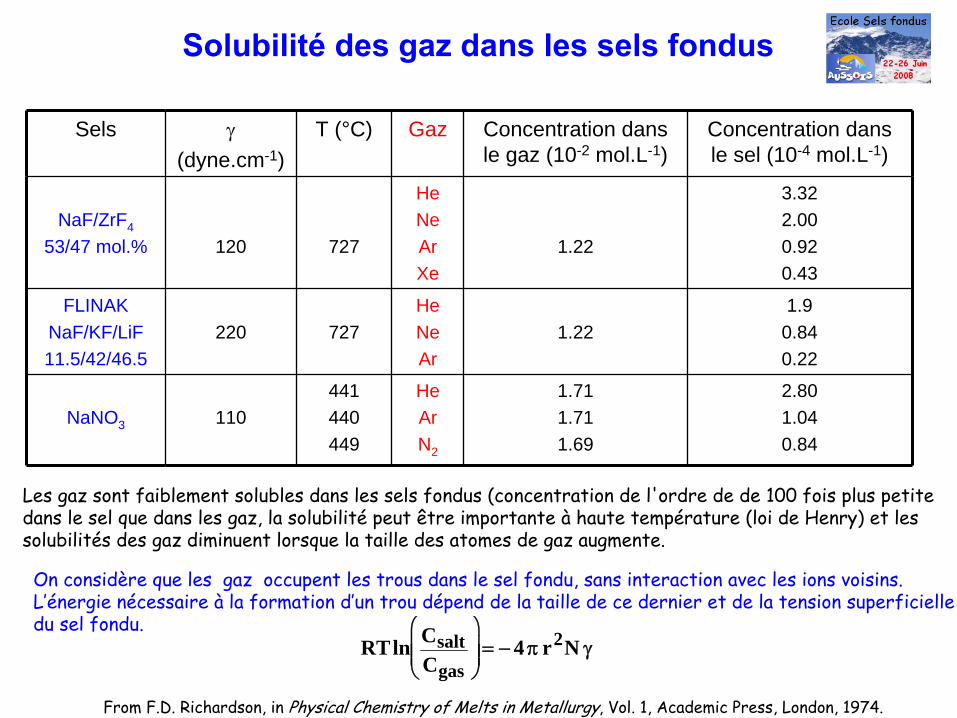
Solubilité
des gaz dans les sels fondus
Sels γ(dyne.cm-1)
T (°C) Gaz Concentration dans le gaz (10-2 mol.L-1)
Concentration dans le sel (10-4 mol.L-1)
NaF/ZrF4
53/47 mol.% 120 727
HeNeArXe
1.22
3.322.000.920.43
FLINAKNaF/KF/LiF11.5/42/46.5
220 727HeNeAr
1.221.9
0.840.22
NaNO3 110441440449
HeArN2
1.711.711.69
2.801.040.84
From F.D. Richardson, in Physical Chemistry of Melts in Metallurgy, Vol. 1, Academic Press, London, 1974.
Les gaz sont faiblement solubles dans les sels fondus (concentration de l'ordre de de 100 fois plus petitedans le sel que dans les gaz, la solubilité
peut être importante à
haute température (loi de Henry) et lessolubilités des gaz diminuent lorsque la taille des atomes de gaz augmente.
On considère que les gaz occupent les trous dans le sel fondu, sans interaction avec les ions voisins.L’énergie nécessaire à
la formation d’un trou dépend de la taille de ce dernier et de la tension superficielledu sel fondu.
RTln CsaltCgas
⎛
⎝ ⎜ ⎜
⎞
⎠ ⎟ ⎟ = − 4 π r2N γ
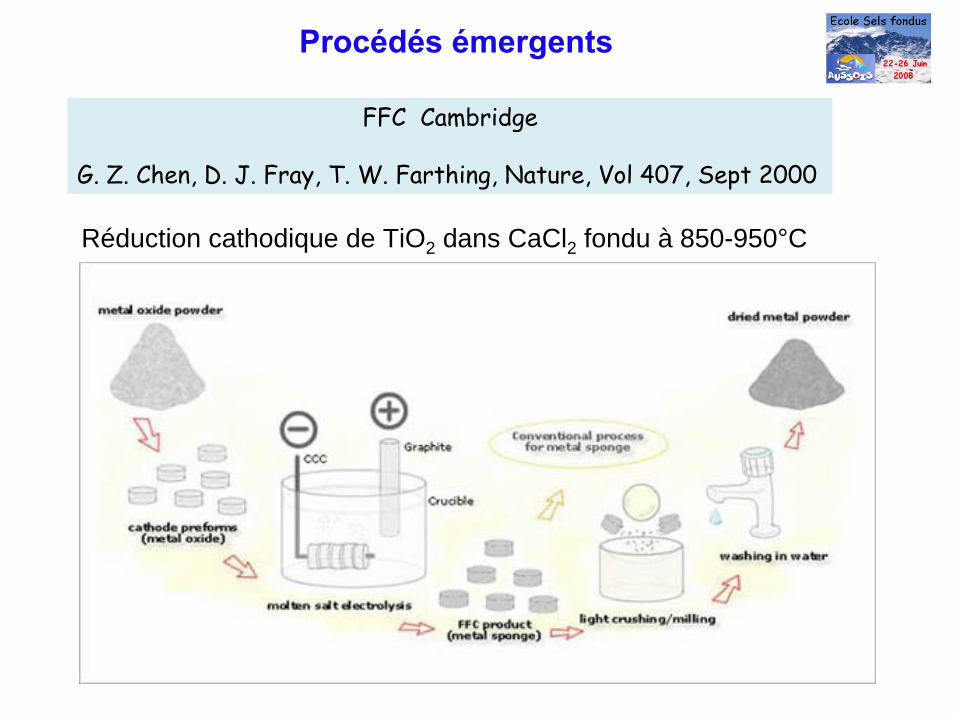
Procédés émergents
FFC Cambridge
G. Z. Chen, D. J. Fray, T. W. Farthing, Nature, Vol 407, Sept 2000
Réduction cathodique de TiO2 dans CaCl2 fondu à 850-950°C
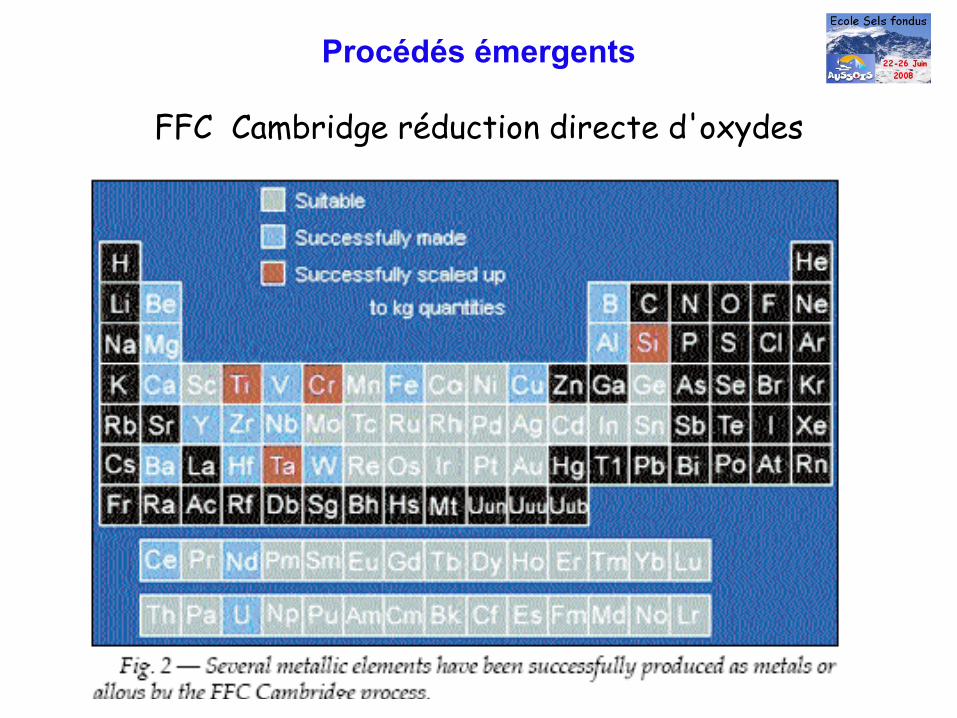
Procédés émergents
FFC Cambridge réduction directe d'oxydes
Procédés émergents
Traitement de déchets :
Voir Farouk Tedjar
Électrolyse de Al2 O3 :
Voir Laurent Cassayre et Airy Pierre Lamaze
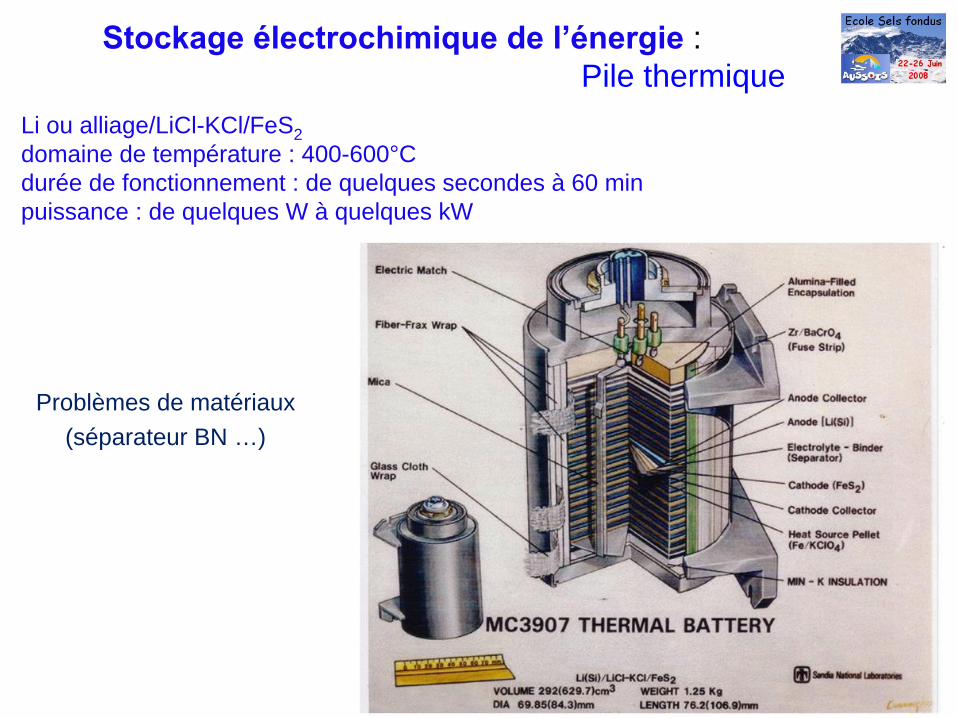
Li ou alliage/LiCl-KCl/FeS2 domaine de température : 400-600°C durée de fonctionnement : de quelques secondes à 60 min puissance : de quelques W à quelques kW
Stockage électrochimique de l’énergie
:Pile thermique
Problèmes de matériaux(séparateur BN …)

Application au transport (automobile)
Na / β
Al2 O3 / S
Énergie massique 100 Wh/kg
température de fonctionnement autour de 350°C
autonomie actuelle 200 km
Problèmes de corrosion, dendrites de Nadurée de vie aléatoire
Stockage électrochimique de l’énergie
:Batteries secondaires (accumulateurs)

Accumulateur ZEBRA Constructeur : Rolls Royce
tension aux bornes : 24 –
1000 V puissance : de 2 à
50 kWh
•ZEBRA (Zeolite Research Battery Africa Project)
Na/NaAlCl4 /NiCl2Domaine de température : 160 – 350°C
Véhicules (> 1 million km), navires et sous-marins
Haute densité d’énergie
Stockage électrochimique de l’énergie
:Batteries secondaires (accumulateurs)
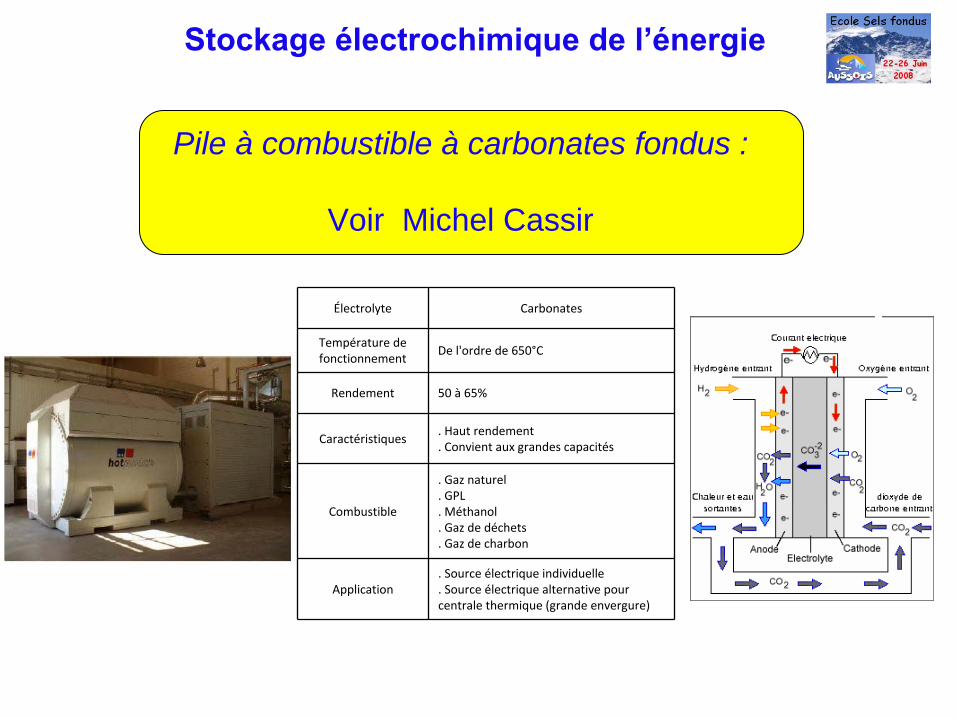
Stockage électrochimique de l’énergie
Pile à combustible à carbonates fondus :
Voir Michel Cassir
Électrolyte Carbonates
Température de
fonctionnement
De l'ordre de 650°C
Rendement 50 à
65%
Caractéristiques. Haut rendement
. Convient aux grandes capacités
Combustible
. Gaz naturel
. GPL
. Méthanol
. Gaz de déchets
. Gaz de charbon
Application. Source électrique individuelle
. Source électrique alternative pour
centrale thermique (grande envergure)

Méthodes électrochimiques d’analyse
• Potentiométrie à courant nul
• Méthodes stationnaires (courant ou potentiel imposé)• Électrode tournante• Spectroscopie d’impédance (voir S. Delpech)
• Méthodes non stationnaires (courant ou potentiel imposé)
- chronopotentiométrie
- voltammétrie
- chronoampérométrie (voir P. Taxil et L. Massot)
- voltammétrie à vague carrée

Le potentiel d’une électrode indicatrice, par rapport à
une référence, est unique.
Potentiomètrie à
courant nul
Il est donné
par la loi de Nernst : E = E°
+ (RT/nF) ln [(Πox/Πred)](en effet, les couples sont rapides en sels fondus)
Si un seul couple est présent :
-Ag+/Ag : Ag+
+ e = Ag
E = E°Ag+/Ag + (RT/F) ln a(Ag+)
-In3+/In+
: In3+
+ 2e = In+
E = E°In3+/In+
+ (RT/2F) ln[(a(In3+)/a(In+)]
La potentiomètrie à
courant nul permet, dans un tel cas, de déterminer une activité, ou une concentration, ou le rapport de deux activités (ou de deux concentrations).
Si plusieurs couples sont présents, ils sont rapidement en équilibre, donc les espèces après réaction sont dans des proportions telles que la loi de Nernst donne la même valeur du potentiel pour tous les
couples.

Choix du matériau de l’électrode indicatrice
C’est une affaire d’expérience : le métal doit conserver son intégrité
dans le milieu considéré, permettre un échange
électronique rapide etc.
Si un seul couple Mn+/M, on choisira souvent M (ex Ag, Ni, etc.)
Mais, dans le cas général, il faudra souvent un autre métal. On tentera souvent d’utiliser W, Mo, Pt , Fe …
(Cf Chamelot)
Choix de l’électrode de référenceEn pratique, les systèmes convenables sont peu nombreux :
Pour les chlorures : Cl2
/Cl‐, Ag+/Ag, Pt2+/PtSystème de prédilection : Ag+/Ag
Pour les hydroxydes : Ag+
/Ag
Pour les fluorures : Ni2+
/ Ni

Données de la littérature :un ouvrage de référence
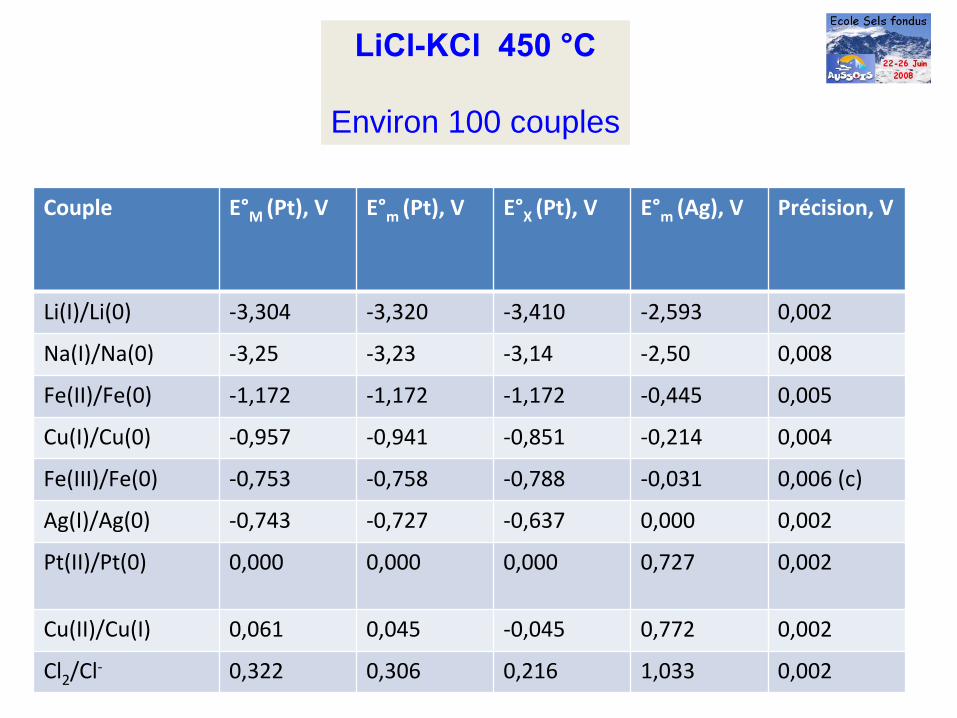
LiCl-KCl
450 °C
Environ 100 couples
Couple E°M
(Pt), V E°m
(Pt), V E°X
(Pt), V E°m
(Ag), V Précision, V
Li(I)/Li(0) ‐3,304 ‐3,320 ‐3,410 ‐2,593 0,002
Na(I)/Na(0) ‐3,25 ‐3,23 ‐3,14 ‐2,50 0,008
Fe(II)/Fe(0) ‐1,172 ‐1,172 ‐1,172 ‐0,445 0,005
Cu(I)/Cu(0) ‐0,957 ‐0,941 ‐0,851 ‐0,214 0,004
Fe(III)/Fe(0) ‐0,753 ‐0,758 ‐0,788 ‐0,031 0,006 (c)
Ag(I)/Ag(0) ‐0,743 ‐0,727 ‐0,637 0,000 0,002
Pt(II)/Pt(0) 0,000 0,000 0,000 0,727 0,002
Cu(II)/Cu(I) 0,061 0,045 ‐0,045 0,772 0,002
Cl2
/Cl‐ 0,322 0,306 0,216 1,033 0,002
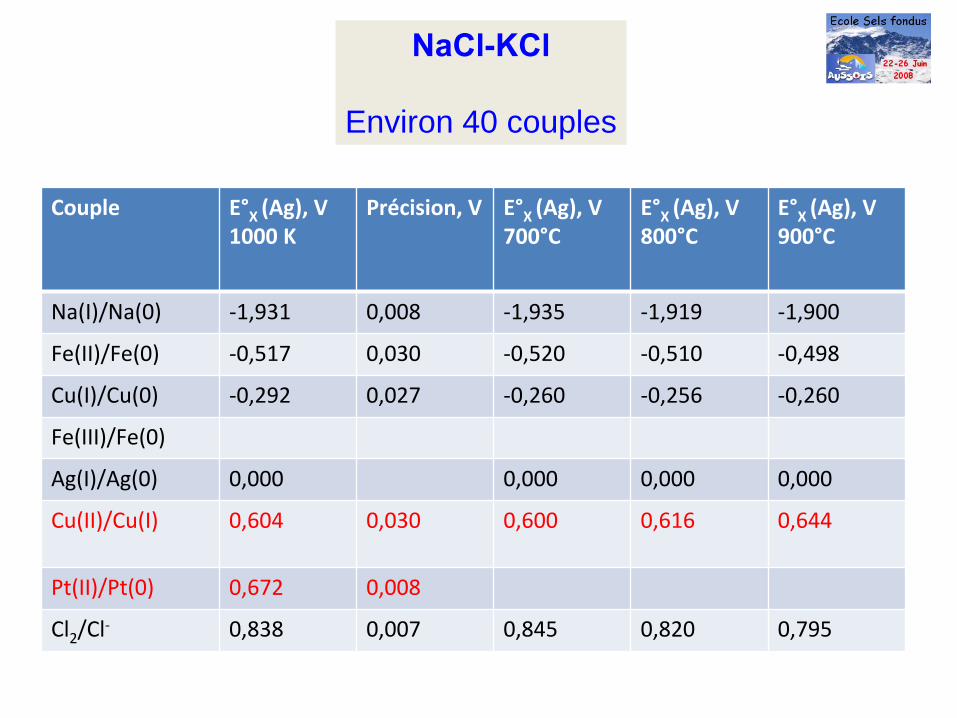
NaCl-KCl
Environ 40 couples
Couple E°X
(Ag), V1000 K
Précision, V E°X
(Ag), V700°C
E°X
(Ag), V800°C
E°X
(Ag), V900°C
Na(I)/Na(0) ‐1,931 0,008 ‐1,935 ‐1,919 ‐1,900
Fe(II)/Fe(0) ‐0,517 0,030 ‐0,520 ‐0,510 ‐0,498
Cu(I)/Cu(0) ‐0,292 0,027 ‐0,260 ‐0,256 ‐0,260
Fe(III)/Fe(0)
Ag(I)/Ag(0) 0,000 0,000 0,000 0,000
Cu(II)/Cu(I) 0,604 0,030 0,600 0,616 0,644
Pt(II)/Pt(0) 0,672 0,008
Cl2
/Cl‐ 0,838 0,007 0,845 0,820 0,795
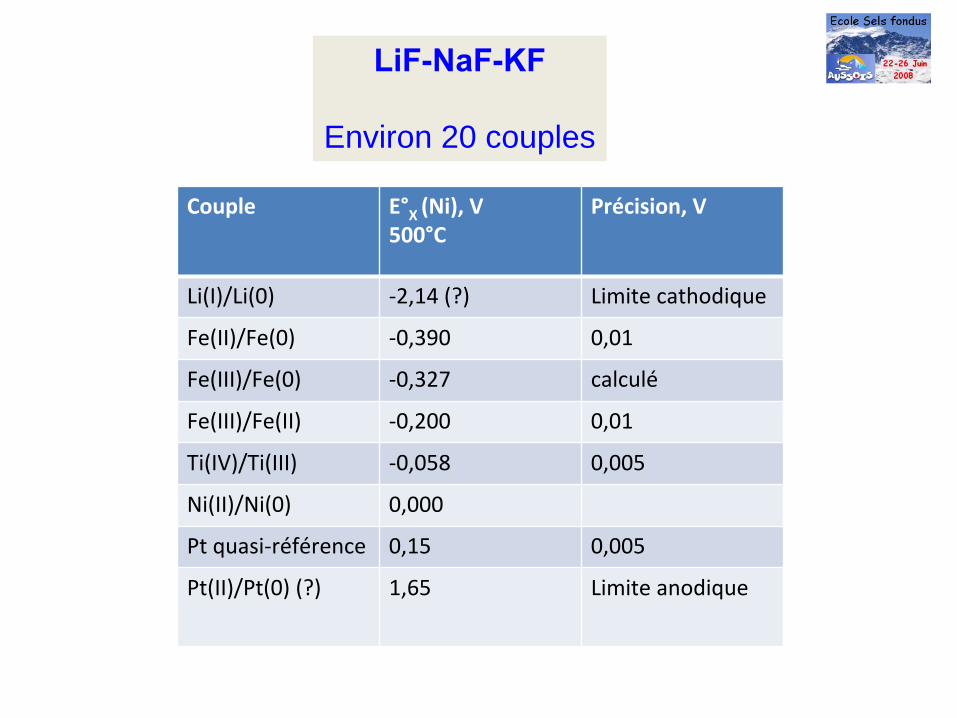
LiF-NaF-KF
Environ 20 couples
Couple E°X
(Ni), V500°C
Précision, V
Li(I)/Li(0) ‐2,14 (?) Limite cathodique
Fe(II)/Fe(0) ‐0,390 0,01
Fe(III)/Fe(0) ‐0,327 calculé
Fe(III)/Fe(II) ‐0,200 0,01
Ti(IV)/Ti(III) ‐0,058 0,005
Ni(II)/Ni(0) 0,000
Pt quasi‐référence 0,15 0,005
Pt(II)/Pt(0) (?) 1,65 Limite anodique

La densité
de courant, donc la vitesse de la réaction d’électrode, est proportionnelle au flux interfacial
de l’espèce
Si le transport de matière par diffusion ou par diffusion‐ convection
est largement prédominant, (pas de migration)
0)( =∂∂
−== xxCnFDnFJi
On perturbe l’état d’équilibre en modifiant le potentiel de l’électrode ou en imposant le passage d’un courant . La réponse attendue est une variation du courant, ou du potentiel, en fonction du temps.
Méthodes stationnaires et non stationnaires : potentiel (ou intensité) imposé
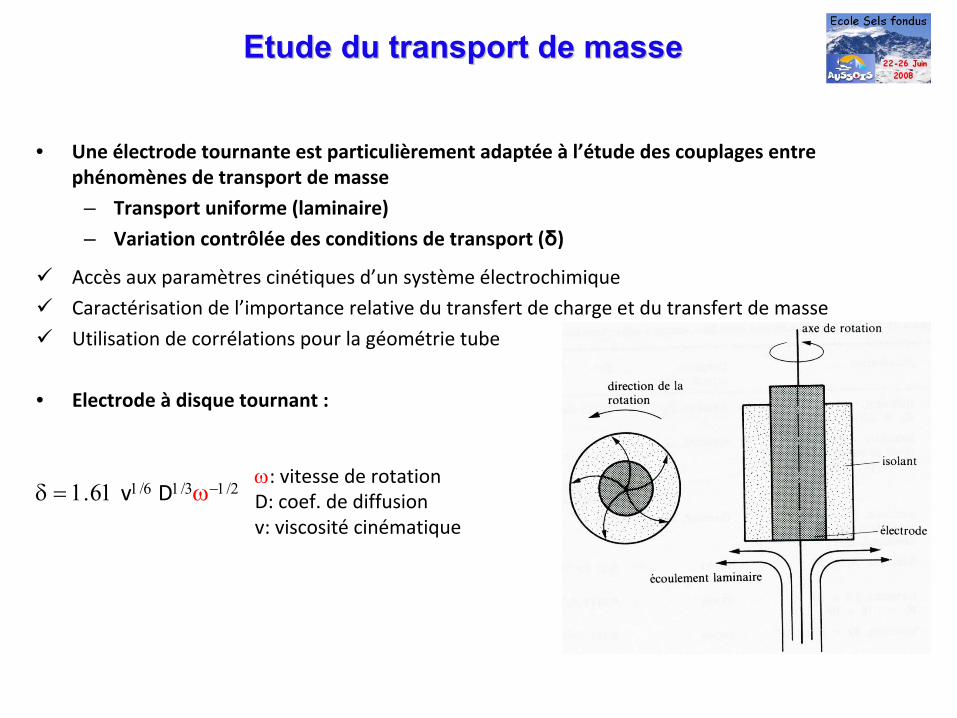
Etude du transport de masseEtude du transport de masse
• Une électrode tournante est particulièrement adaptée à l’étude des couplages entre
phénomènes de transport de masse
– Transport uniforme (laminaire)
– Variation contrôlée des conditions de transport (δ)
Accès aux paramètres cinétiques d’un système électrochimique
Caractérisation de l’importance relative du transfert de charge et du transfert de masse
Utilisation de corrélations pour la géométrie tube
• Electrode à disque tournant :
δ = 1.61 v1/6
D1/3ω−1/2ω: vitesse de rotationD: coef. de diffusionv: viscosité
cinématique
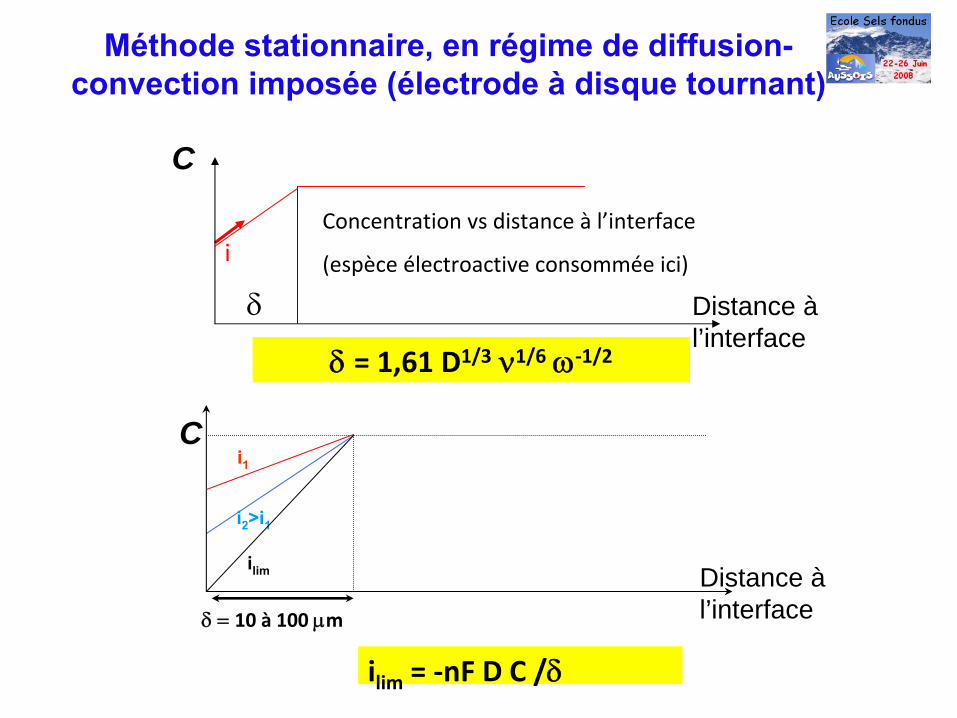
Méthode stationnaire, en régime de diffusion- convection imposée (électrode à
disque tournant)
δ = 1,61 D1/3
ν1/6
ω‐1/2
Distance à l’interface
i2
>i1
ilim
i1
δ = 10 à
100 μm
C
C
δ
iConcentration vs distance à
l’interface
(espèce électroactive consommée ici)
ilim
= ‐nF D C /δ
Distance à l’interface
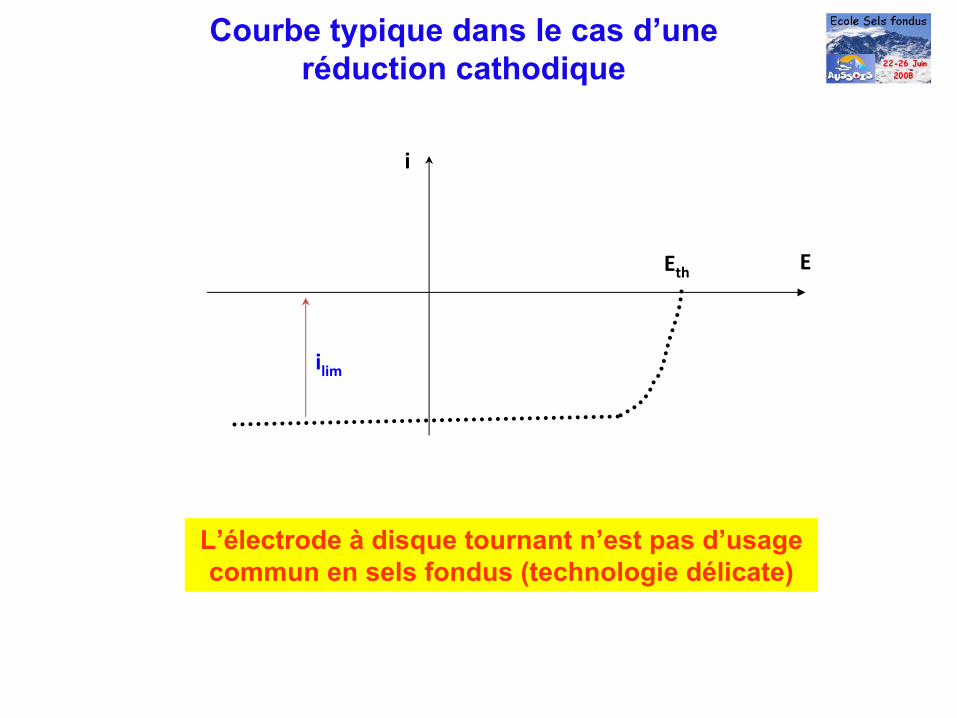
i
Eth
ilim
E
L’électrode à
disque tournant n’est pas d’usage commun en sels fondus (technologie délicate)
Courbe typique dans le cas d’une réduction cathodique
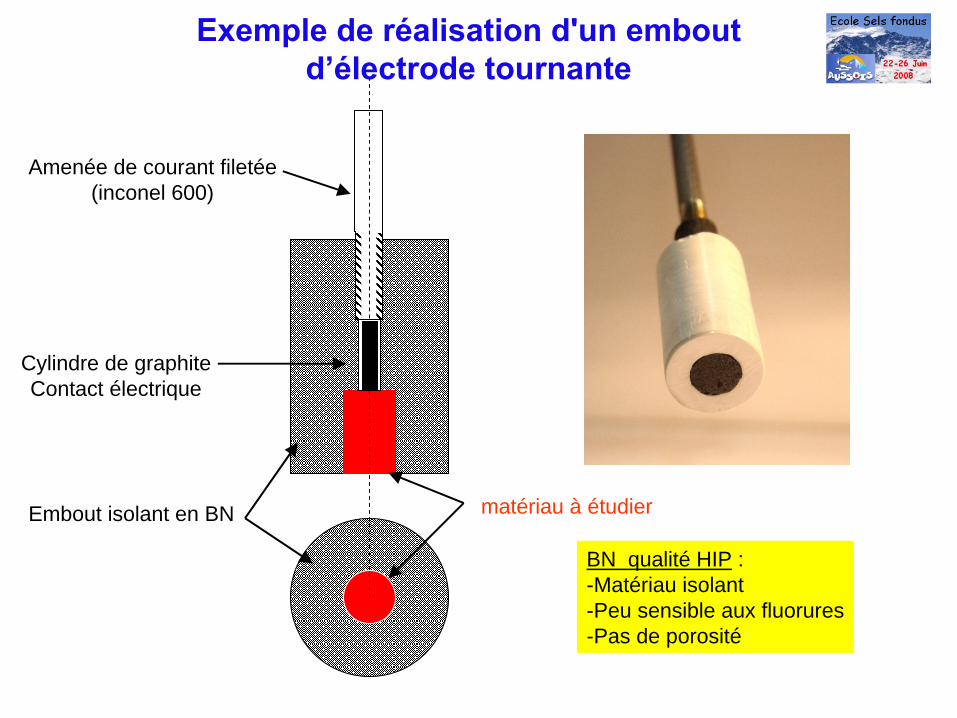
BN qualité HIP : -Matériau isolant-Peu sensible aux fluorures-Pas de porosité
matériau à étudierEmbout isolant en BN
Cylindre de graphiteContact électrique
Amenée de courant filetée(inconel 600)
Exemple de réalisation d'un emboutd’électrode tournante

Méthodes très utilisées en sels fondus
Le transport ionique par diffusion doit être prépondérant
C
xδ
L’épaisseur de la couche de diffusion croît en fonction du temps.
3 Modes de transport en solution :- diffusion- migration - convection
La diffusion de l’espèce électroactive doit régir le transport
Méthodes non-stationnaires

0x)( =
∂∂
−==xCnFDnFJi
Quelques équations importantes dans l’hypothèse du transport régi par la diffusion
Concentrations aux électrodes :
Conditions initiales et aux limites :
Équations différentielles régissant le transport :
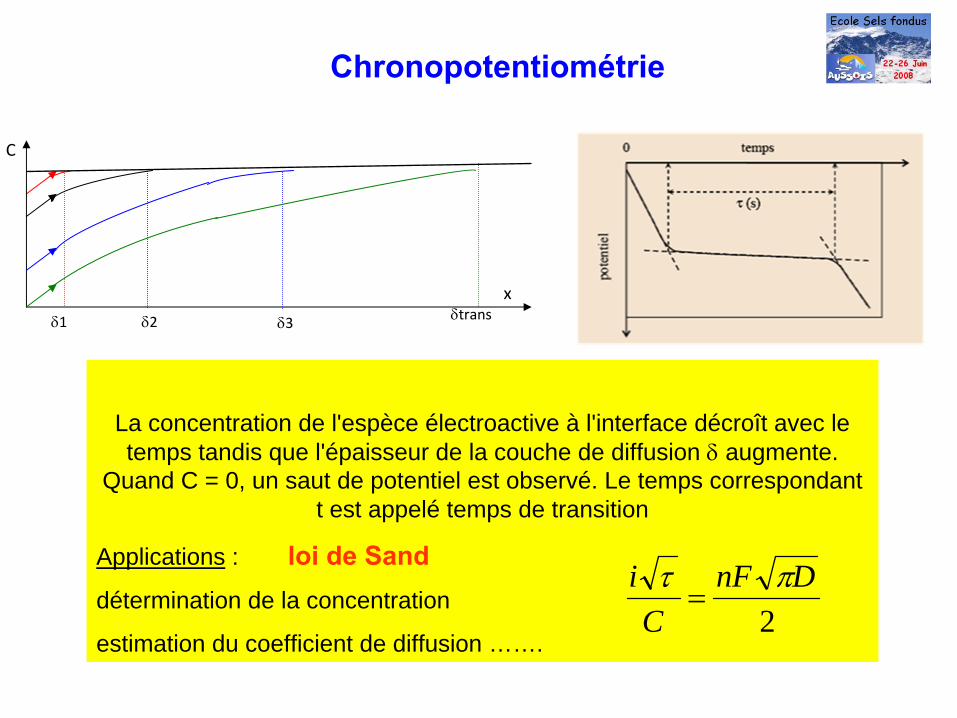
Chronopotentiométrie
δ1
x
C
δ2 δ3 δtrans
La concentration de l'espèce électroactive à l'interface décroît avec le temps tandis que l'épaisseur de la couche de diffusion δ
augmente.
Quand C = 0, un saut de potentiel est observé. Le temps correspondant t est appelé temps de transition
Applications : loi de Sanddétermination de la concentration
estimation du coefficient de diffusion …….2
DnFC
i πτ=
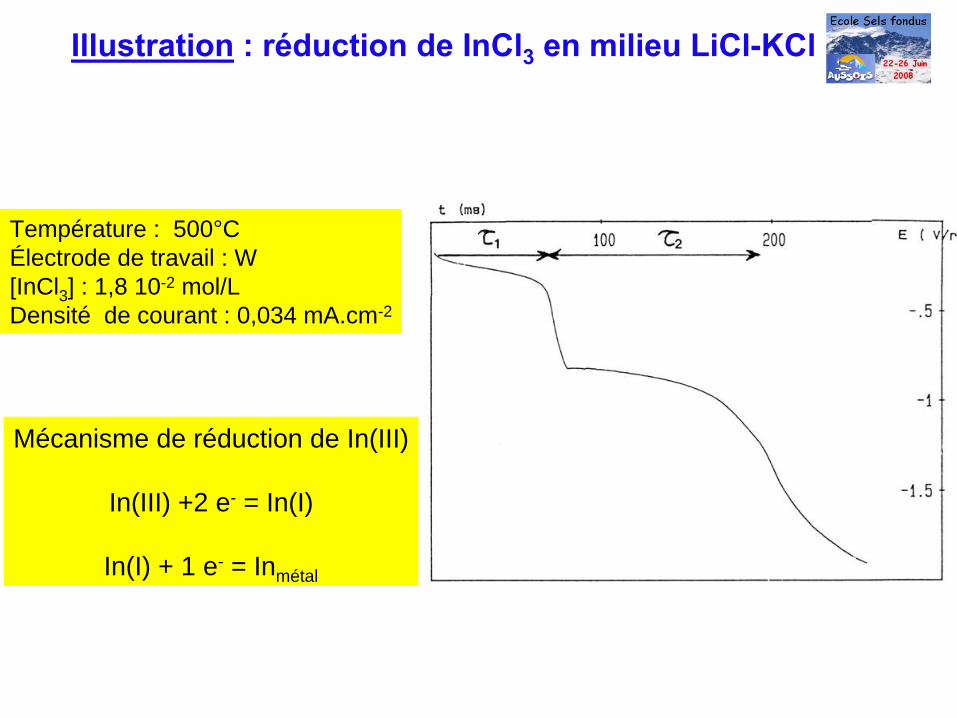
Température : 500°CÉlectrode de travail : W [InCl3 ] : 1,8 10-2 mol/LDensité de courant : 0,034 mA.cm-2
Mécanisme de réduction de In(III)
In(III) +2 e- = In(I)
In(I) + 1 e- = Inmétal
Illustration
: réduction de InCl3
en milieu LiCl-KCl
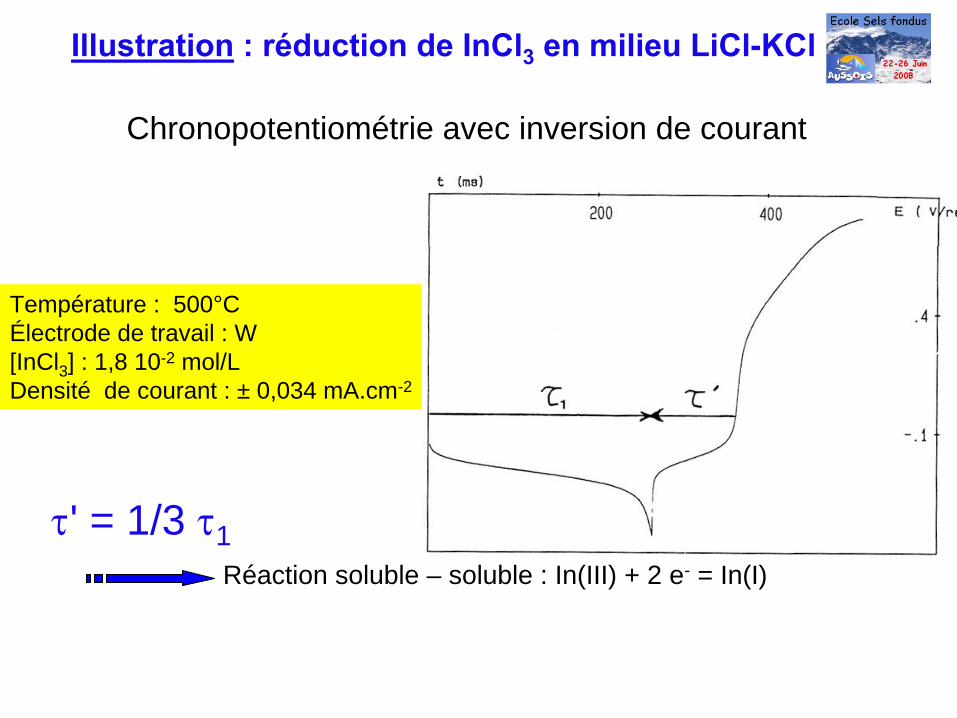
Illustration
: réduction de InCl3
en milieu LiCl-KCl
Chronopotentiométrie avec inversion de courant
Température : 500°CÉlectrode de travail : W [InCl3 ] : 1,8 10-2 mol/LDensité de courant : ± 0,034 mA.cm-2
τ' = 1/3 τ1Réaction soluble – soluble : In(III) + 2 e- = In(I)

voltammétrie à
balayage linéaire des potentiels
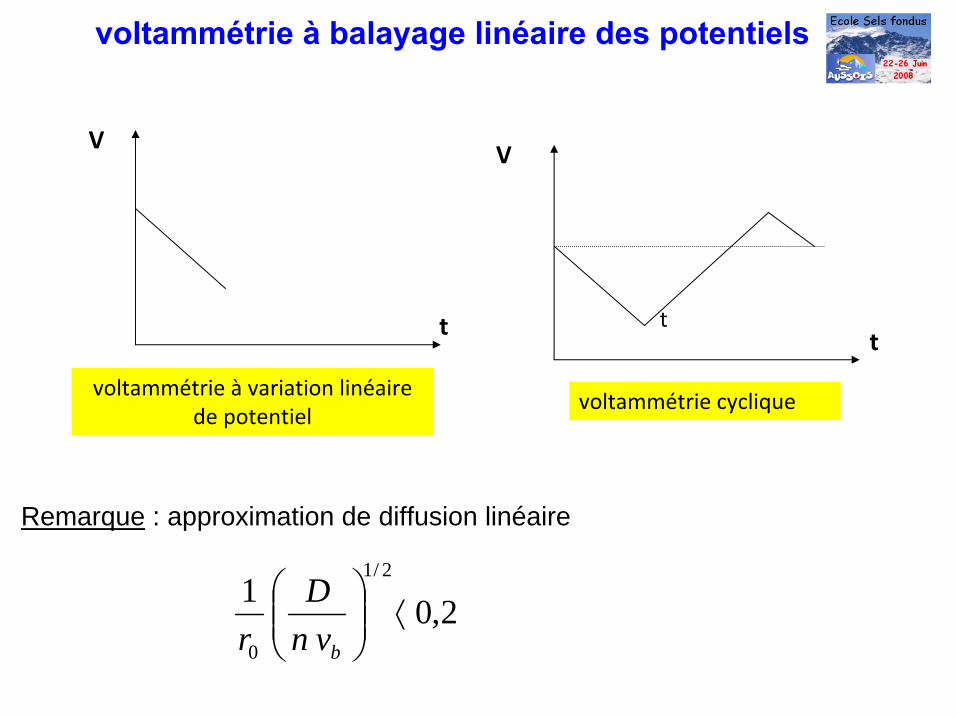
voltammétrie à
balayage linéaire des potentiels
t
V
t
voltammétrie à variation linéaire
de potentielvoltammétrie cyclique
V
t
Remarque : approximation de diffusion linéaire
2,012/1
0
⟨⎟⎟⎠
⎞⎜⎜⎝
⎛
bvnD
r
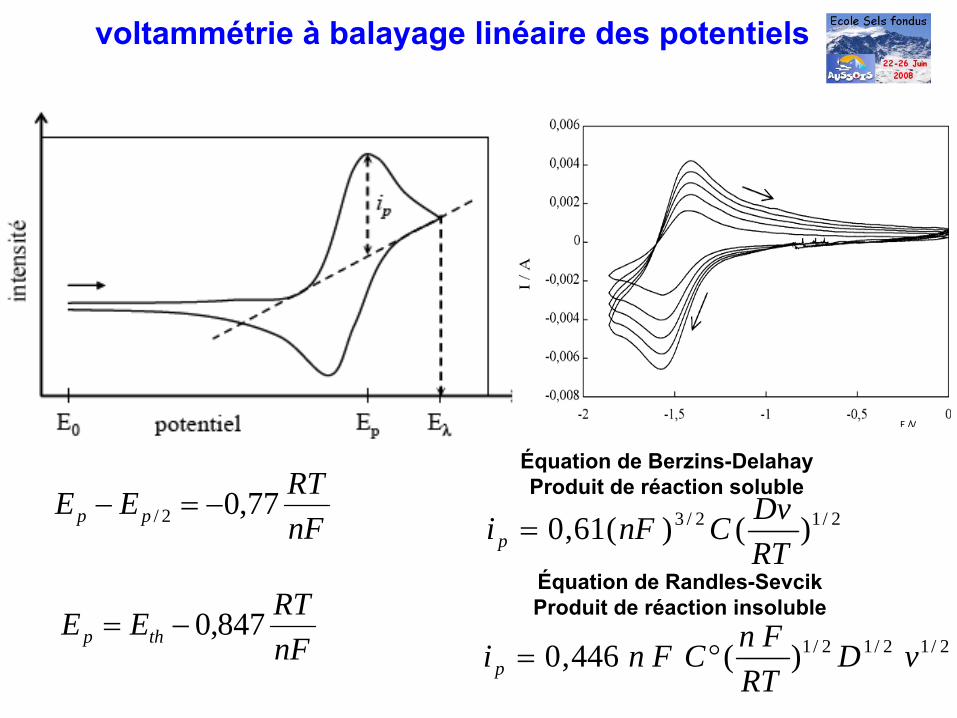
2/12/3 )()(61,0RTDvCnFip =nF
RTEE pp 77,02/ −=−
nFRTEE thp 847,0−=
voltammétrie à
balayage linéaire des potentiels
E /VE /V
Équation de Berzins-DelahayProduit de réaction soluble
2/12/12/1)(446,0 vDRT
FnCFnip °=
Équation de Randles-SevcikProduit de réaction insoluble
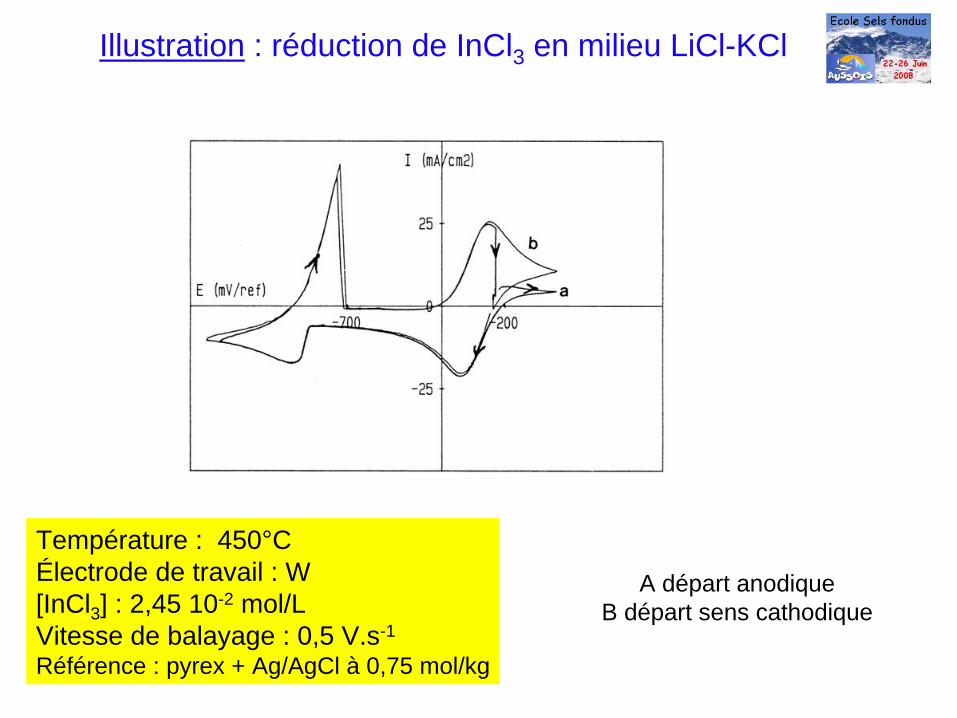
A départ anodiqueB départ sens cathodique
Illustration : réduction de InCl3 en milieu LiCl-KCl
Température : 450°CÉlectrode de travail : W [InCl3 ] : 2,45 10-2 mol/LVitesse de balayage : 0,5 V.s-1
Référence : pyrex + Ag/AgCl à 0,75 mol/kg
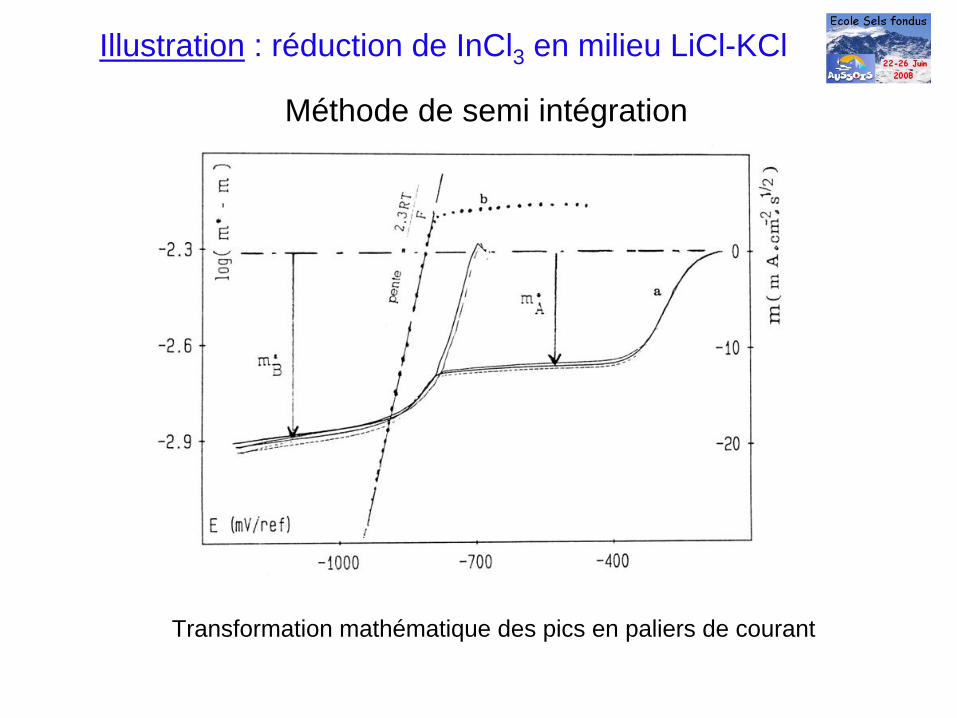
Méthode de semi intégration
Transformation mathématique des pics en paliers de courant
Illustration : réduction de InCl3 en milieu LiCl-KCl
Voltammogramme
cyclique électrode : W (S=0,31 cm2)[ThCl4
] : 0,43mass% T=773 KVitesse de balayage : 100 mV.s‐1.
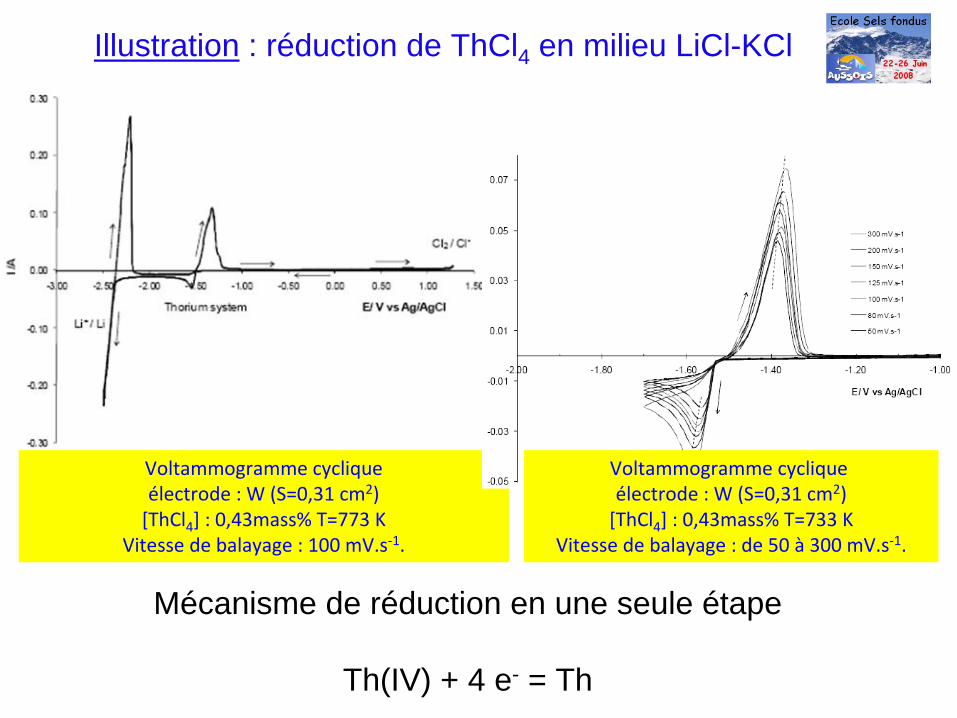
Illustration : réduction de ThCl4 en milieu LiCl-KCl
Voltammogramme
cyclique électrode : W (S=0,31 cm2)[ThCl4
] : 0,43mass% T=733 KVitesse de balayage : de 50 à
300 mV.s‐1.
Mécanisme de réduction en une seule étape
Th(IV) + 4 e- = Th
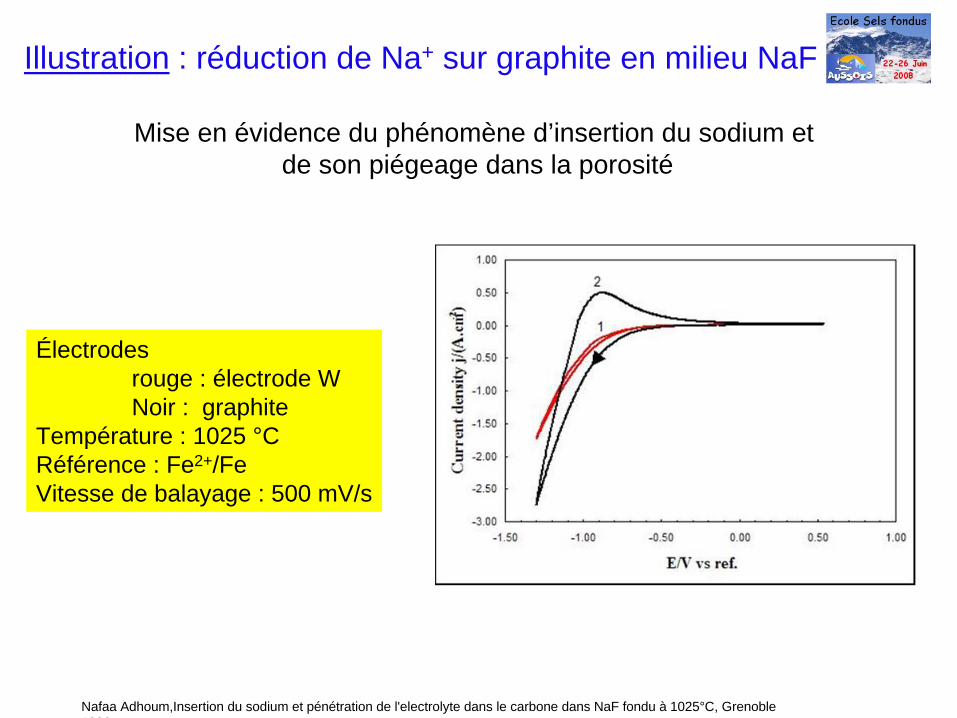
Électrodesrouge : électrode WNoir : graphite
Température : 1025 °CRéférence : Fe2+/FeVitesse de balayage : 500 mV/s
Illustration : réduction de Na+ sur graphite en milieu NaF
Mise en évidence du phénomène d’insertion du sodium etde son piégeage dans la porosité
Nafaa Adhoum,Insertion du sodium et pénétration de l'electrolyte dans le carbone dans NaF fondu à 1025°C, Grenoble 1996
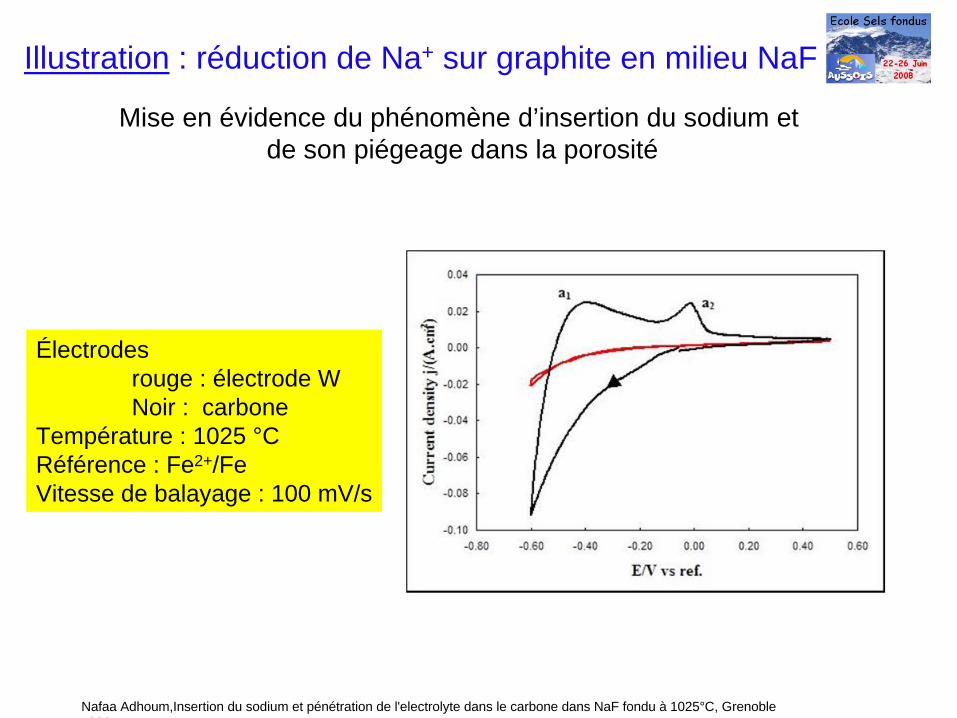
Mise en évidence du phénomène d’insertion du sodium etde son piégeage dans la porosité
Nafaa Adhoum,Insertion du sodium et pénétration de l'electrolyte dans le carbone dans NaF fondu à 1025°C, Grenoble 1996
Illustration : réduction de Na+ sur graphite en milieu NaF
Électrodesrouge : électrode WNoir : carbone
Température : 1025 °CRéférence : Fe2+/FeVitesse de balayage : 100 mV/s
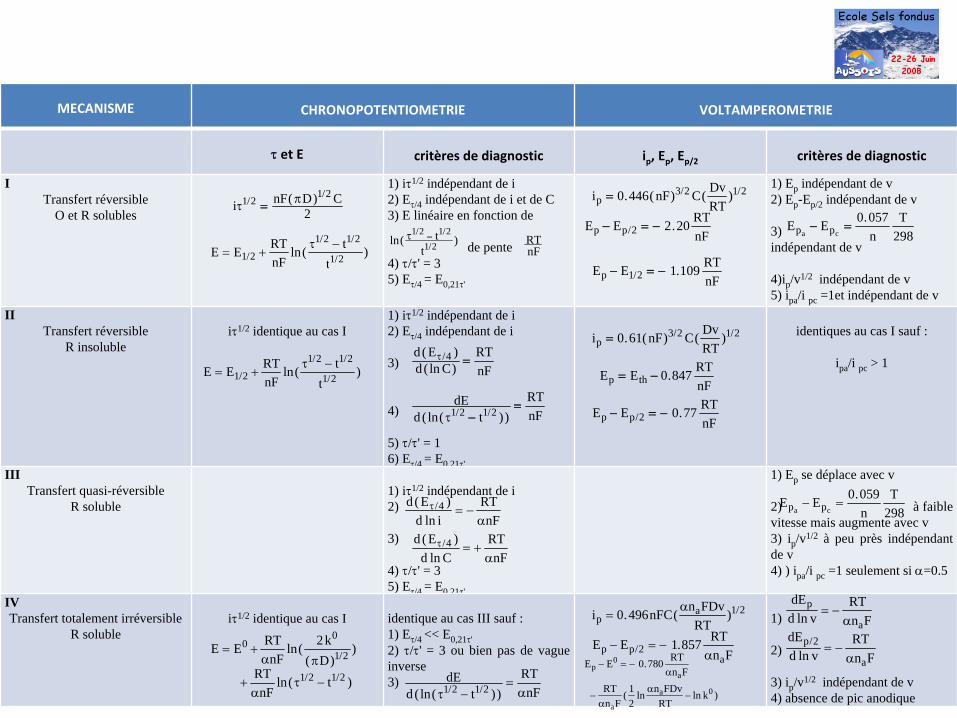
MECANISME CHRONOPOTENTIOMETRIE VOLTAMPEROMETRIE
τ
et E critères de diagnostic ip
, Ep
, Ep/2 critères de diagnostic
ITransfert réversible
O et R solubles
1) iτ1/2 indépendant de i2) Eτ/4 indépendant de i et de C3) E linéaire en fonction de
de pente 4) τ/τ' = 35) Eτ/4 = E0,21τ'
1) Ep indépendant de v2) Ep -Ep/2 indépendant de v
3) indépendant de v
4)ip /v1/2 indépendant de v5) ipa /i pc =1et indépendant de v
IITransfert réversible
R insolubleiτ1/2 identique au cas I
1) iτ1/2 indépendant de i2) Eτ/4 indépendant de i
3)
4)
5) τ/τ' = 16) Eτ/4 = E0.21τ'
identiques au cas I sauf :
ipa /i pc > 1
IIITransfert quasi-réversible
R soluble1) iτ1/2 indépendant de i 2)
3)
4) τ/τ' = 35) Eτ/4 = E0,21τ'
1) Ep se déplace avec v
2) à faible vitesse mais augmente avec v3) ip /v1/2 à peu près indépendant de v4) ) ipa /i pc =1 seulement si α=0.5
IVTransfert totalement irréversible
R solubleiτ1/2 identique au cas I identique au cas III sauf :
1) Eτ/4 << E0,21τ'2) τ/τ' = 3 ou bien pas de vague inverse3)
1)
2)
3) ip /v1/2 indépendant de v4) absence de pic anodique
i nF D Cτ π1 21 2
2/
/( )=
E E RTnF
t= +
−1 2
1 2 1 2
1 2/
/ /
/ln( )τ
t
E E RTnF
t= +
−1 2
1 2 1 2
1 2/
/ /
/ln( )τ
t
E E RTnF
kD
= +00
1 22
α πln(
( ))/
+ −RTnF
tα
τln( )/ /1 2 1 2
ln( )/ /
/τ1 2 1 2
1 2− t
tRTnF
d Ed C
RTnF
( )( ln )
/τ 4 =
dEd t
RTnF(ln( ))/ /τ1 2 1 2−
=
d Ed i
RTnF
( )ln
/τα
4 = −
d Ed C
RTnF
( )ln
/τα
4 = +
dEd t
RTnF(ln( ))/ /τ α1 2 1 2−
=
i nF C DvRTp = 0 446 3 2 1 2. ( ) ( )/ /
E E RTnFp p− = −/ .2 2 20
E E RTnFp − = −1 2 1 109/ .
E En
Tp pa c
− =0 057
298.
i nF C DvRTp = 0 61 3 2 1 2. ( ) ( )/ /
E E RTnFp th= − 0 847.
E E RTnFp p− = −/ .2 0 77
E En
Tp pa c
− =0 059
298.
i nFC n FDvRTpa= 0 496 1 2. ( ) /α
E E RTn Fp p
a− = −/ .2 1 857
αE E RT
n Fpa
− = −0 0 780.α
− −RTn F
n FDvRT
ka
aα
α( ln ln )12
0
dEd v
RTn F
p
aln = −α
dEd v
RTn F
p
a
/ln
2 = −α
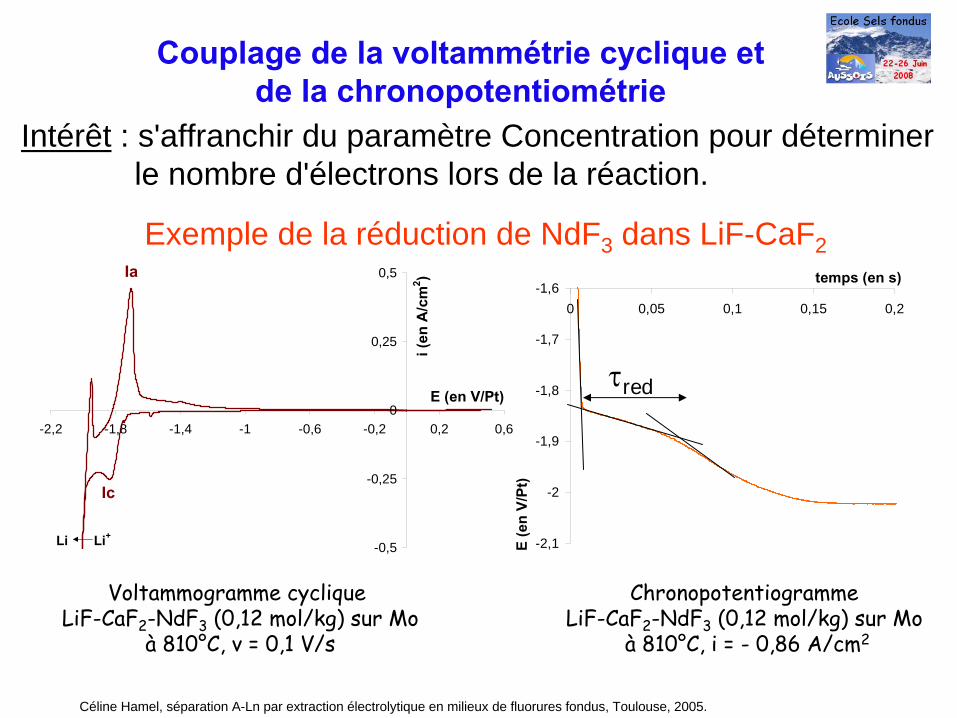
Couplage de la voltammétrie cyclique etde la chronopotentiométrie
Intérêt : s'affranchir du paramètre Concentration pour déterminerle nombre d'électrons lors de la réaction.
Exemple de la réduction de NdF3 dans LiF-CaF2
Voltammogramme cyclique LiF-CaF2
-NdF3
(0,12 mol/kg) sur Mo à
810°C, v = 0,1 V/s
-0,5
-0,25
0
0,25
0,5
-2,2 -1,8 -1,4 -1 -0,6 -0,2 0,2 0,6
E (en V/Pt)
i (en
A/c
m2 )
Ic
Ia
Li Li+
Chronopotentiogramme LiF-CaF2
-NdF3
(0,12 mol/kg) sur Mo à
810°C, i = -
0,86 A/cm2
-2,1
-2
-1,9
-1,8
-1,7
-1,60 0,05 0,1 0,15 0,2
temps (en s)
E (e
n V/
Pt)
τred
Céline Hamel, séparation A-Ln par extraction électrolytique en milieux de fluorures fondus, Toulouse, 2005.
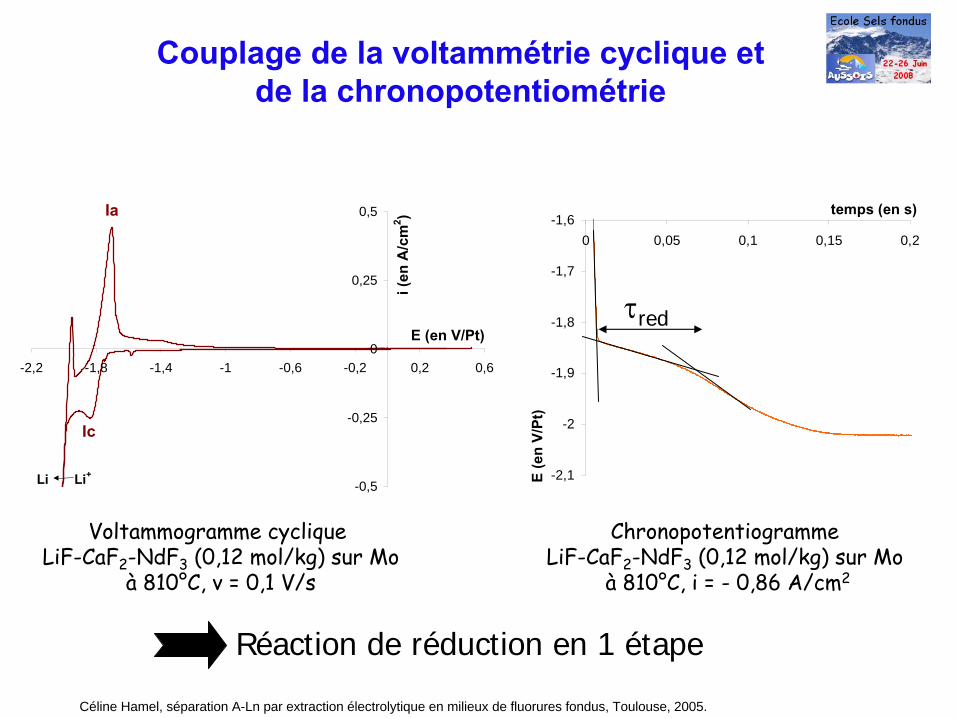
Voltammogramme cyclique LiF-CaF2
-NdF3
(0,12 mol/kg) sur Mo à
810°C, v = 0,1 V/s
Réaction de réduction en 1 étape
-0,5
-0,25
0
0,25
0,5
-2,2 -1,8 -1,4 -1 -0,6 -0,2 0,2 0,6
E (en V/Pt)
i (en
A/c
m2 )
Ic
Ia
Li Li+
Chronopotentiogramme LiF-CaF2
-NdF3
(0,12 mol/kg) sur Mo à 810°C, i = - 0,86 A/cm2
-2,1
-2
-1,9
-1,8
-1,7
-1,60 0,05 0,1 0,15 0,2
temps (en s)
E (e
n V/
Pt)
τred
Céline Hamel, séparation A-Ln par extraction électrolytique en milieux de fluorures fondus, Toulouse, 2005.
Couplage de la voltammétrie cyclique etde la chronopotentiométrie

-0,7
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
00 0,2 0,4 0,6 0,8 1
v1/2 (en V1/2/s1/2)
i p (e
n A
/cm
2 )
Voltammétrie cyclique ip
=f(v1/2) Chronopotentiométrie i.τ1/2= cste
réaction limitée par la diffusion des ions Nd(III)
n = 3
-0,35
-0,3
-0,25
-0,2
-0,15
-0,1
-0,05
0-2,5 -2 -1,5 -1 -0,5 0
i (en A/cm2)
i. τ1/
2 (en
A.s
1/2 /c
m2 )
2/12/12/1
61,0 vDRTnFFCni o
p ⎟⎠⎞
⎜⎝⎛××= oCnFDi 2/12/12/1 5,0 πτ ×=×
2
1/ 2 1/ 2 2
14 0,61
pi Rn Tv i F
πτ
⎛ ⎞ ×= × × ×⎜ ⎟× × ×⎝ ⎠
Céline Hamel, séparation A-Ln par extraction électrolytique en milieux de fluorures fondus, Toulouse, 2005.
Couplage de la voltammétrie cyclique etde la chronopotentiométrie
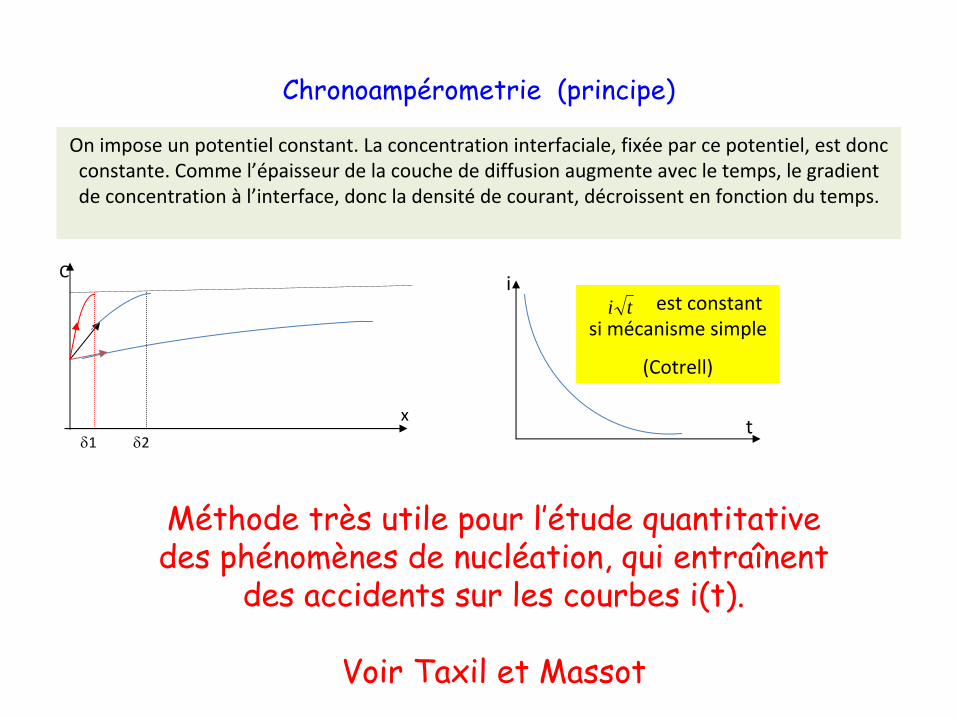
Chronoampérometrie
(principe)
On impose un potentiel constant. La concentration interfaciale, fixée par ce potentiel, est donc
constante. Comme l’épaisseur de la couche de diffusion augmente avec le temps, le gradient
de concentration à
l’interface, donc la densité
de courant, décroissent en fonction du temps.
est constant
si mécanisme simple
(Cotrell)
i
t
ti
Méthode très utile pour l’étude quantitative des phénomènes de nucléation, qui entraînent
des accidents sur les courbes i(t).
Voir Taxil
et Massot
C
x
δ2δ1
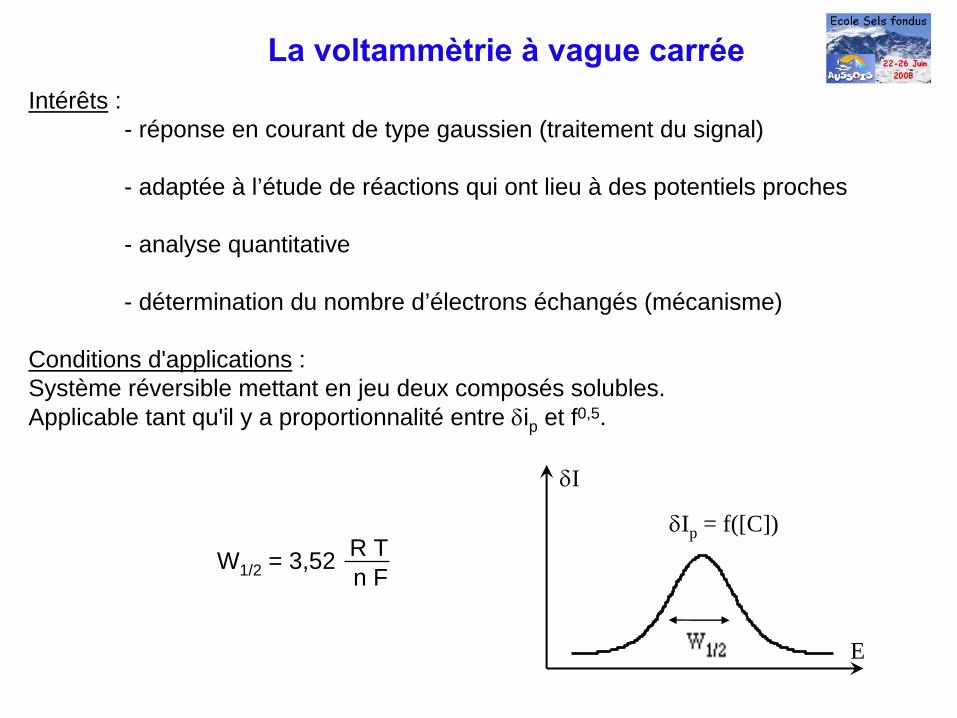
Intérêts : - réponse en courant de type gaussien (traitement du signal)
- adaptée à l’étude de réactions qui ont lieu à des potentiels proches
- analyse quantitative
- détermination du nombre d’électrons échangés (mécanisme)
Conditions d'applications : Système réversible mettant en jeu deux composés solubles.Applicable tant qu'il y a proportionnalité entre δip et f0,5.
δI
E
W1/2 = 3,52 R Tn F
δIp = f([C])
La voltammètrie à
vague carrée
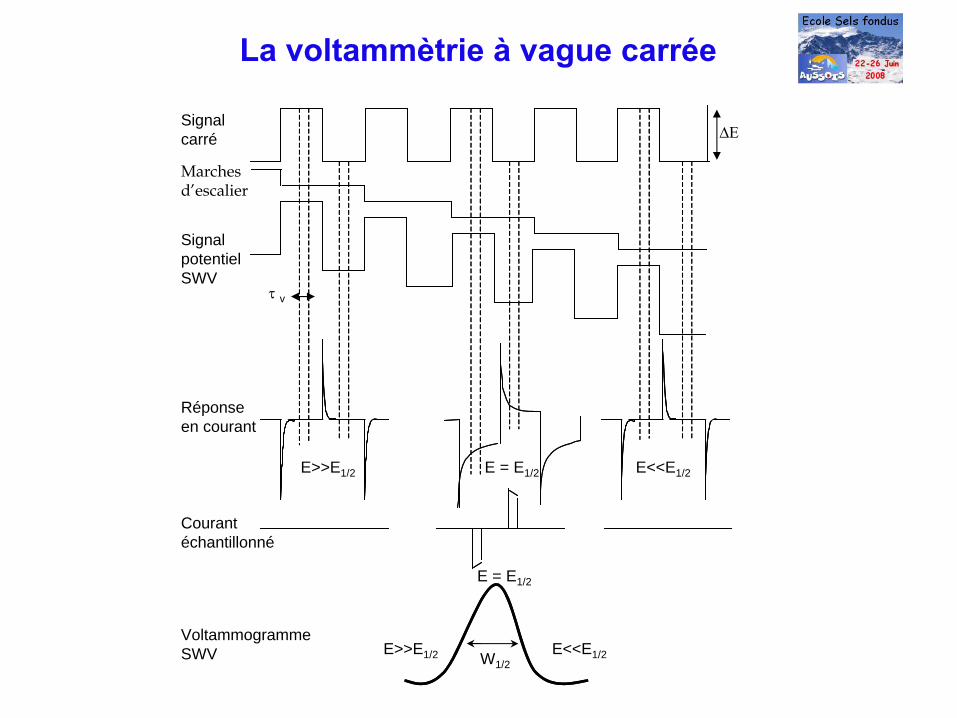
La voltammètrie à
vague carrée
W1/2
E>>E1/2 E = E1/2 E<<E1/2
ΔΕ
τ v
E>>E1/2 E<<E1/2
E = E1/2
Signal carré
Marchesd’escalier
Signal potentiel SWV
Réponse en courant
Courant échantillonné
Voltammogramme SWV
-300
-200
-100
0
100
200
300
400
500
-0.2 -0.1 0 0.1 0.2 0.3 0.4 0.5 0.6
E(V/Pt)
i (m
A.c
m-2
)
-250
-200
-150
-100
-50
0-0.55 -0.45 -0.35 -0.25 -0.15 -0.05 0.05 0.15 0.25
expérience modèle5 e échangés3 e échangés
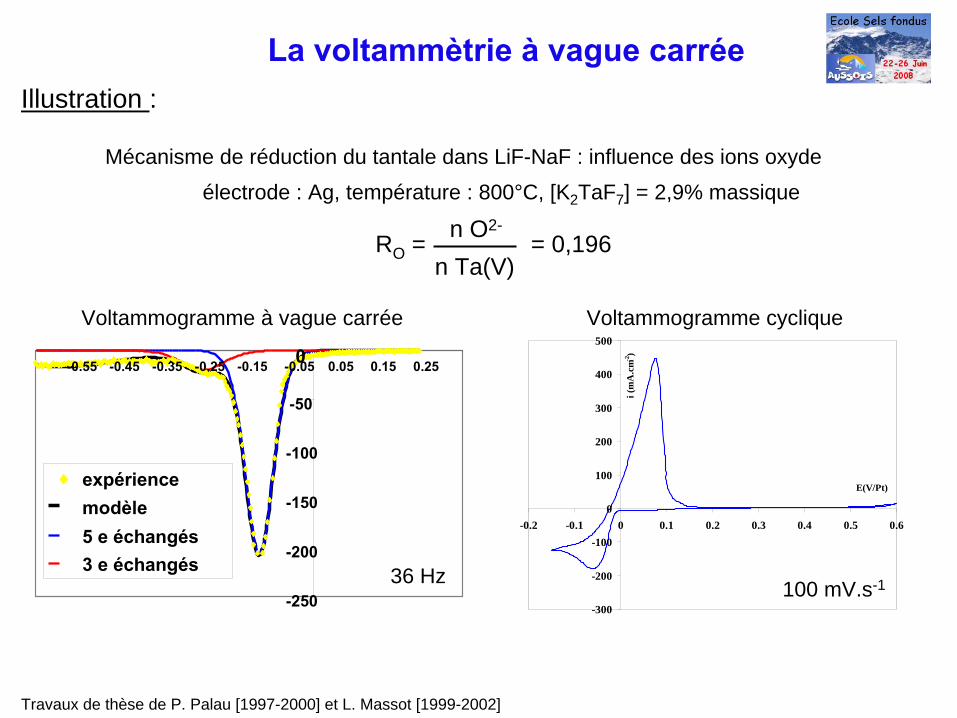
Mécanisme de réduction du tantale dans LiF-NaF : influence des ions oxyde
Travaux de thèse de P. Palau [1997-2000] et L. Massot [1999-2002]
électrode : Ag, température : 800°C, [K2 TaF7 ] = 2,9% massique
100 mV.s-136 Hz
Voltammogramme à vague carrée Voltammogramme cyclique
RO = = 0,196n O2-
n Ta(V)
La voltammètrie à
vague carréeIllustration :
-200
-160
-120
-80
-40
0
-0.55 -0.45 -0.35 -0.25 -0.15 -0.05 0.05 0.15
expérience5 e échangés3 e échangés2 e échangés
-400
-200
0
200
400
600
800
-500 -400 -300 -200 -100 0 100 200 300 400
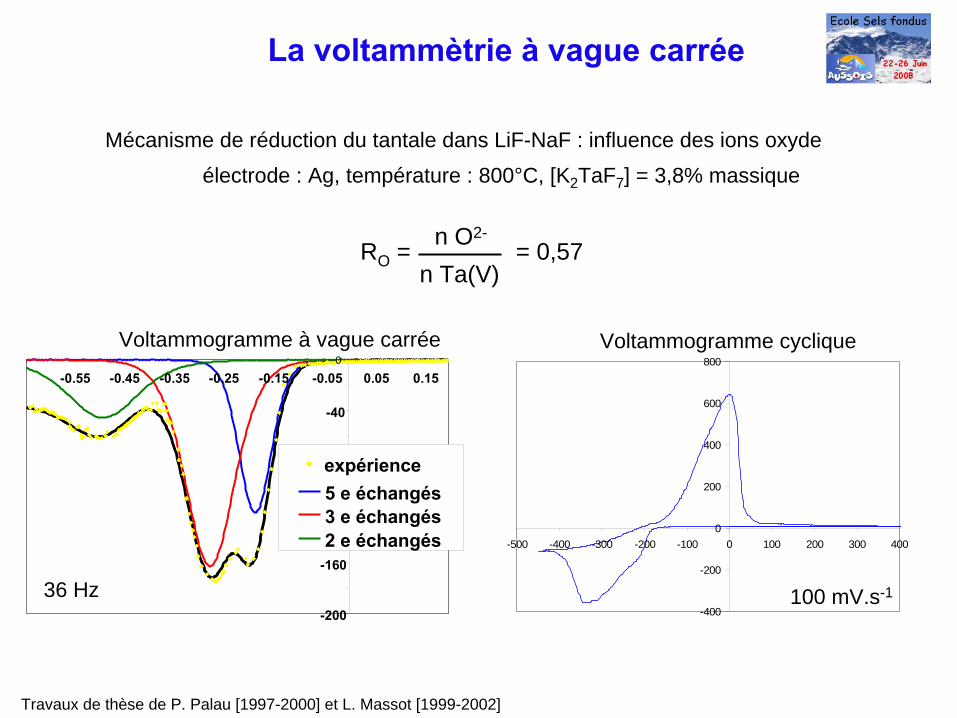
Travaux de thèse de P. Palau [1997-2000] et L. Massot [1999-2002]
Mécanisme de réduction du tantale dans LiF-NaF : influence des ions oxydeélectrode : Ag, température : 800°C, [K2 TaF7 ] = 3,8% massique
36 Hz
Voltammogramme à vague carrée Voltammogramme cyclique
100 mV.s-1
RO = = 0,57n O2-
n Ta(V)
La voltammètrie à
vague carrée
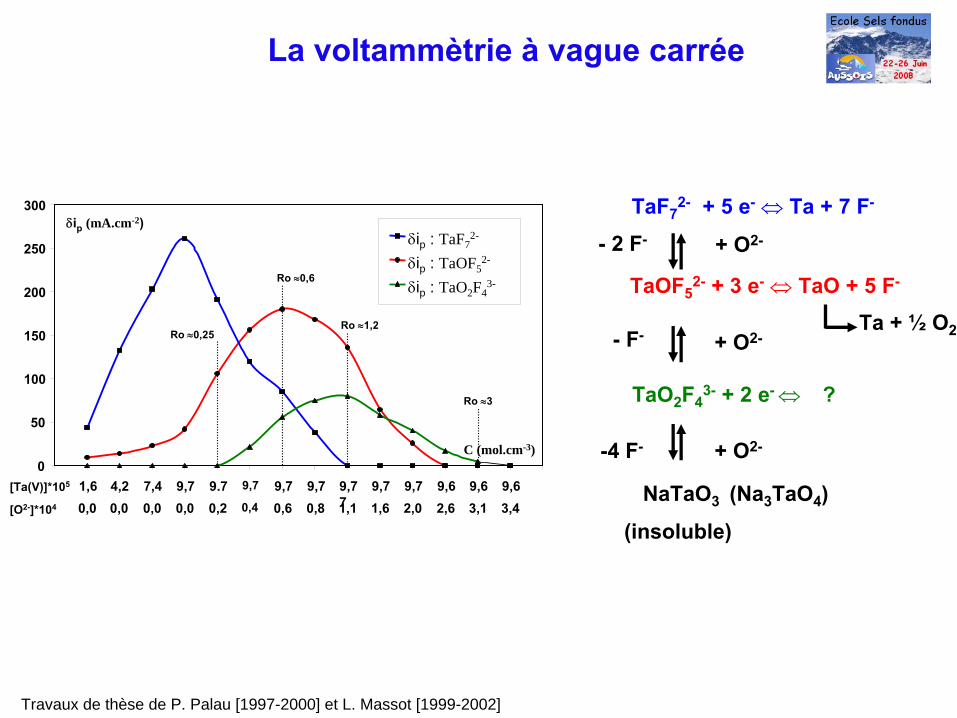
TaF72-
+ 5 e-
⇔ Ta + 7 F-
TaOF52-
+ 3 e-
⇔ TaO + 5 F-
Ta + ½
O2
TaO2
F43-
+ 2 e-
⇔
?
- 2 F-
- F-
+ O2-
+ O2-
NaTaO3 (Na3
TaO4
)
-4 F- + O2-
(insoluble)
[Ta(V)]*105
0
50
100
150
200
250
300
1,6 4,2 7,4 9,7 9.7 9,7 9,7 9,7 9,7
7
9,7 9,7 9,6 9,6 9,6
δip : TaOF52-
δip : TaF72-
δip : TaO2 F43-
[O2-]*104
C (mol.cm-3)
0,0 0,0 0,0 0,0 0,2 0,4 0,6 0,8 1,1 1,6 2,0 2,6 3,1 3,4
δip
(mA.cm-2)
Ro ≈0,25Ro ≈1,2
Ro ≈0,6
Ro ≈3
La voltammètrie à
vague carrée
Travaux de thèse de P. Palau [1997-2000] et L. Massot [1999-2002]
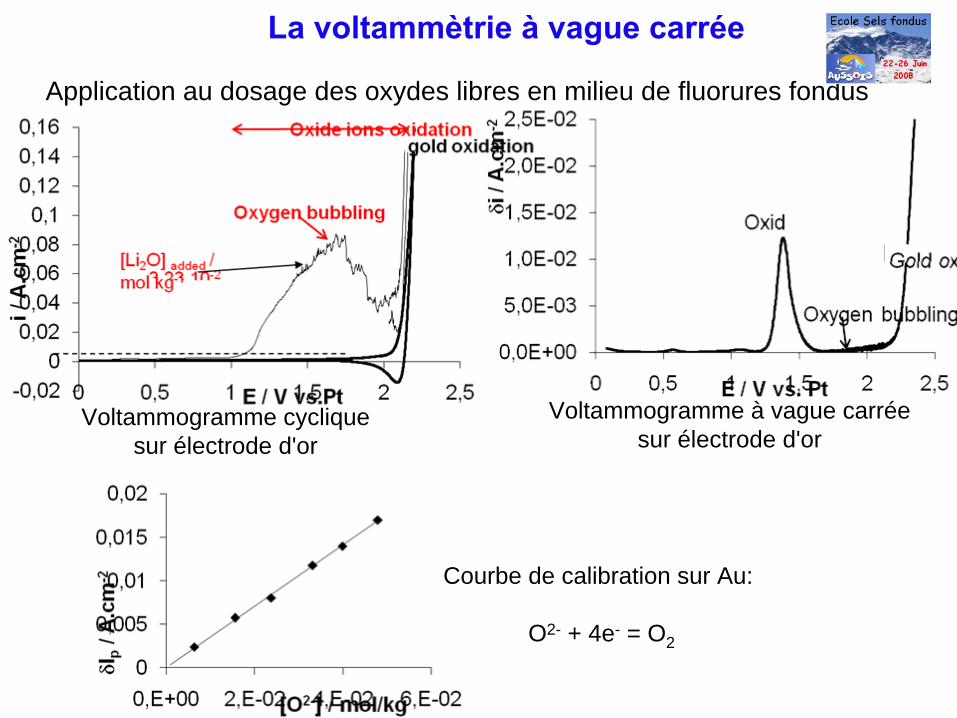
Application au dosage des oxydes libres en milieu de fluorures fondus
La voltammètrie à
vague carrée
Voltammogramme cycliquesur électrode d'or
Voltammogramme à vague carréesur électrode d'or
Courbe de calibration sur Au:
O2- + 4e- = O2


Electrochemistry
of
thorium in LiCl‐KCl
eutectic
melts
Electrochimica
Acta 51(2006) 4024‐4032
L. Cassayre, J. Serp, P. Soucek, R. Malmbeck*, J. Rebizant, J.‐P. Glatz
European
Commission, JRC, Institute
for Transuranium
Elements,
P.O. 2340, 76125 Karlsruhe, Germany
Previous
work
performed
in ITU concerned
the
study
of
the
electrochemical
properties
of
U,Pu, Np
and
Am cations in a LiCl‐KCl
eutectic
molten
salt
[1‐3]. Transient
electrochemicaltechniques were
used
to determine
the
diffusion coefficient of
the
dissolved
actinide ions, aswell
as their
redox potentials. Basic thermochemical
properties
(Gibbs energy
and
activitycoefficient) were
also
determined
in the
673‐823 K temperature
range.The
electrochemical
behaviour
of
Th ions dissolved
in the
same
chloride
melt
is
reported
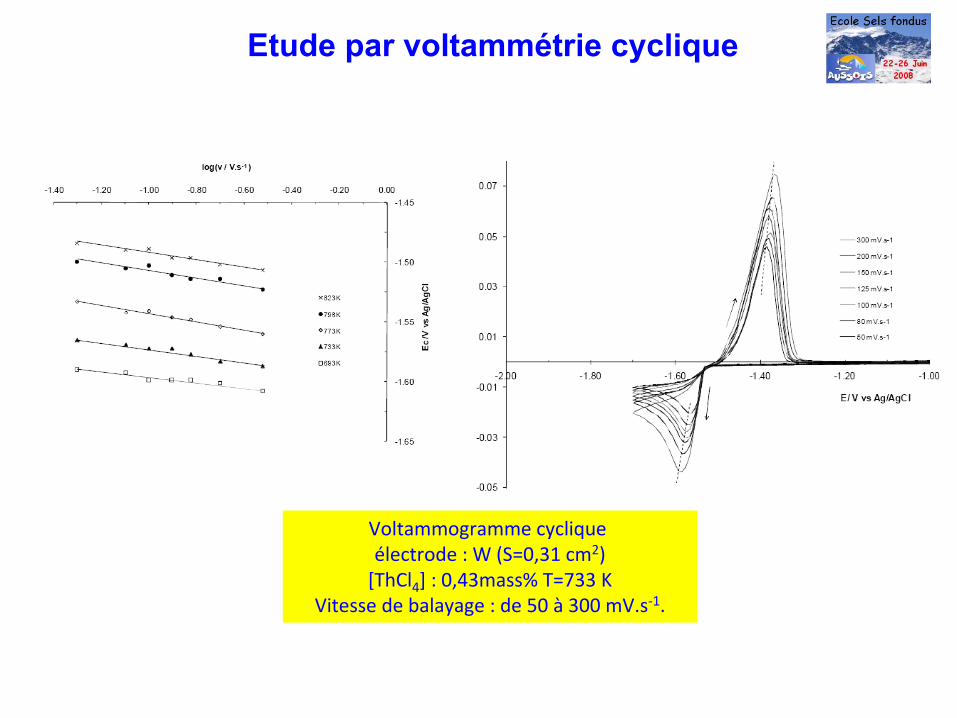
Voltammogramme
cyclique électrode : W (S=0,31 cm2)[ThCl4
] : 0,43mass% T=733 KVitesse de balayage : de 50 à
300 mV.s‐1.
Etude par voltammétrie cyclique
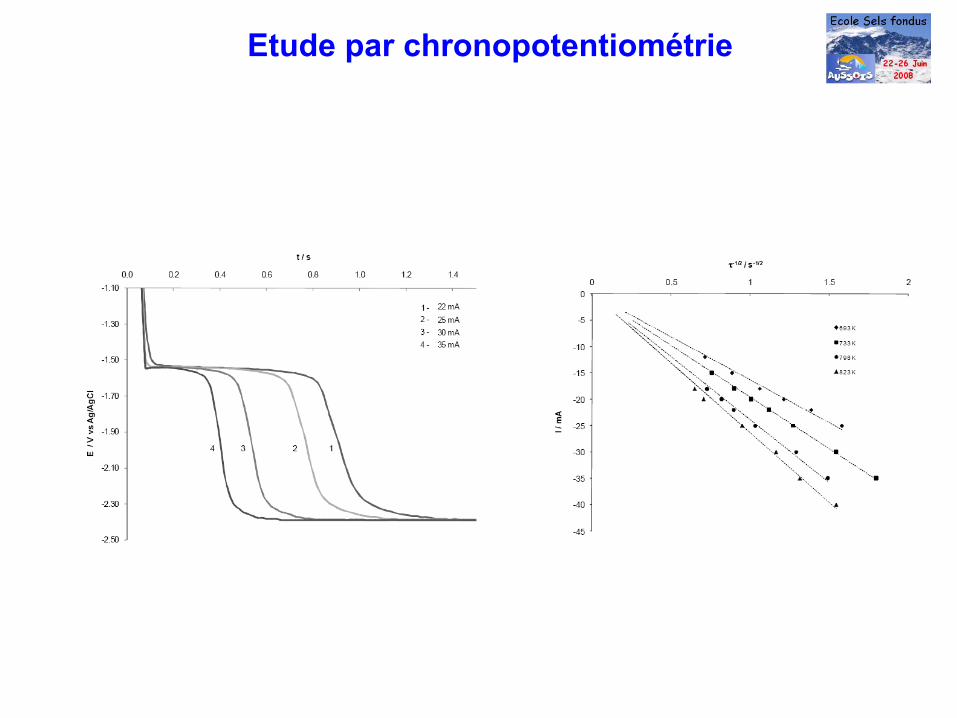
Etude par chronopotentiométrie
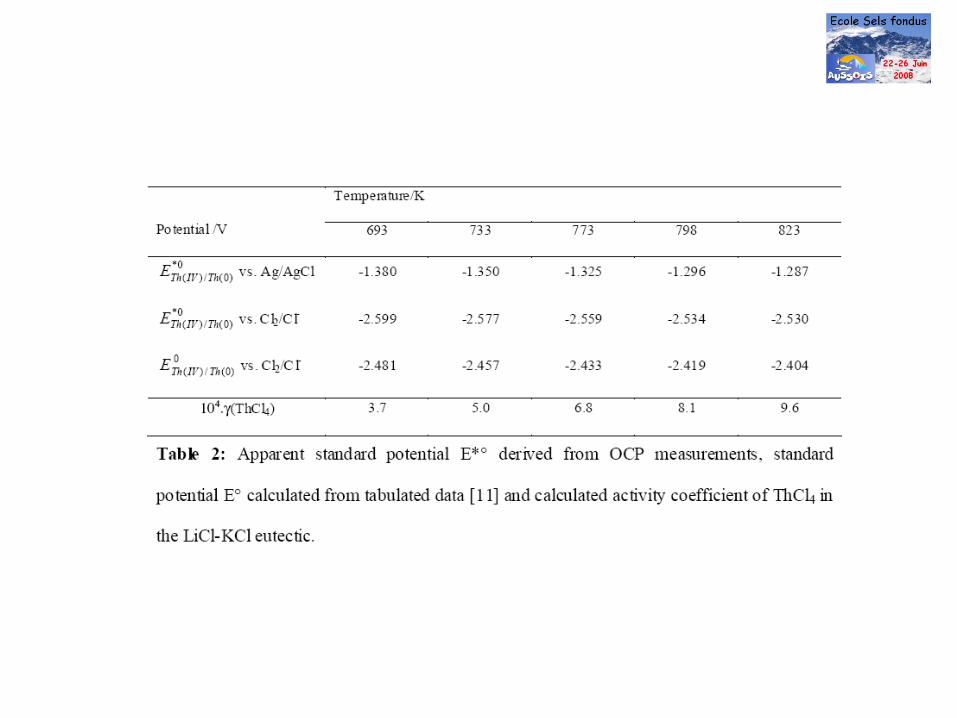

Electrochemical behaviour of americium ions in LiCl–KCl eutectic melt
Electrochimica Acta 51(2006) 4024-4032
J. Serpa, P. Chamelot b, S. Fourcaudot a, R.J.M. Konings a, R. Malmbeck a,C. Pernel a,c, J.C. Poignet d, J. Rebizant a, J.-P. Glatz a
a
European Commission, JRC, Institute for Transuranium Elements,
Postfach 2340, 76125 Karlsruhe, Germanyb
Laboratoire de Génie Chimique, Toulouse, Francec
Alliance Materials Development Center, CEA, LETI, 17 Rue des martyrs, 38054 Grenoble Cedex 9, Franced
Laboratoire d’Electrochimie et de Physicochimie des Mat´eriaux et Interfaces, LEPMI-ENSEEG/INP Grenoble,UMR 5631, 1130 rue de la Piscine, BP 75, 38402 Saint Martin d’H`eres, France
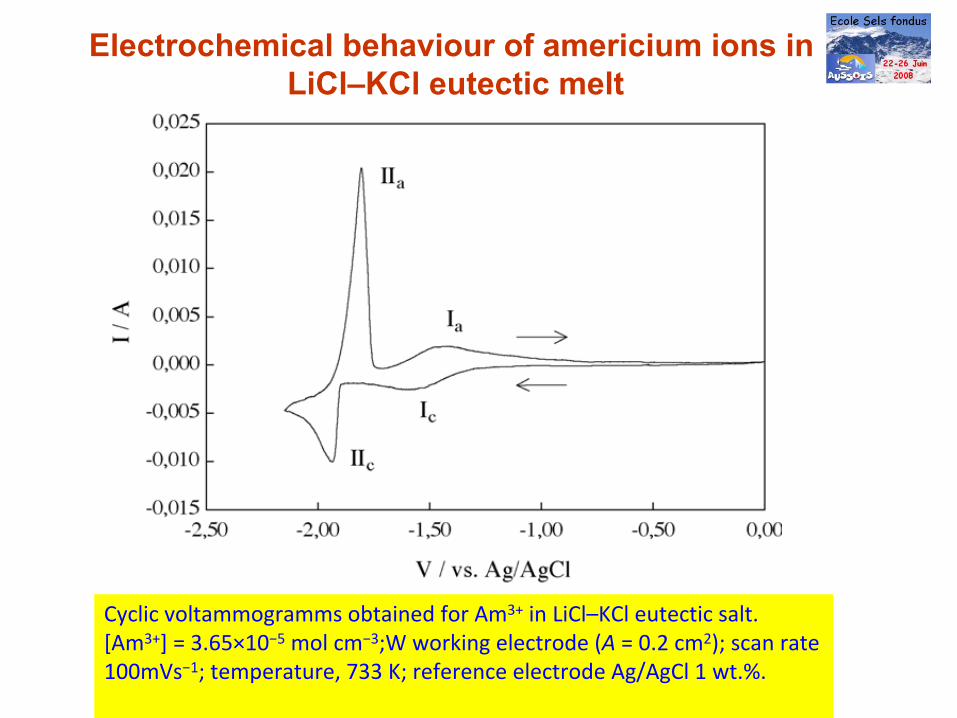
Cyclic voltammogramms obtained for Am3+
in LiCl–KCl eutectic salt.
[Am3+] = 3.65×10−5
mol cm−3;W working electrode (A = 0.2 cm2); scan rate
100mVs−1; temperature, 733 K; reference electrode Ag/AgCl 1 wt.%.
Electrochemical behaviour of americium ions in LiCl–KCl eutectic melt
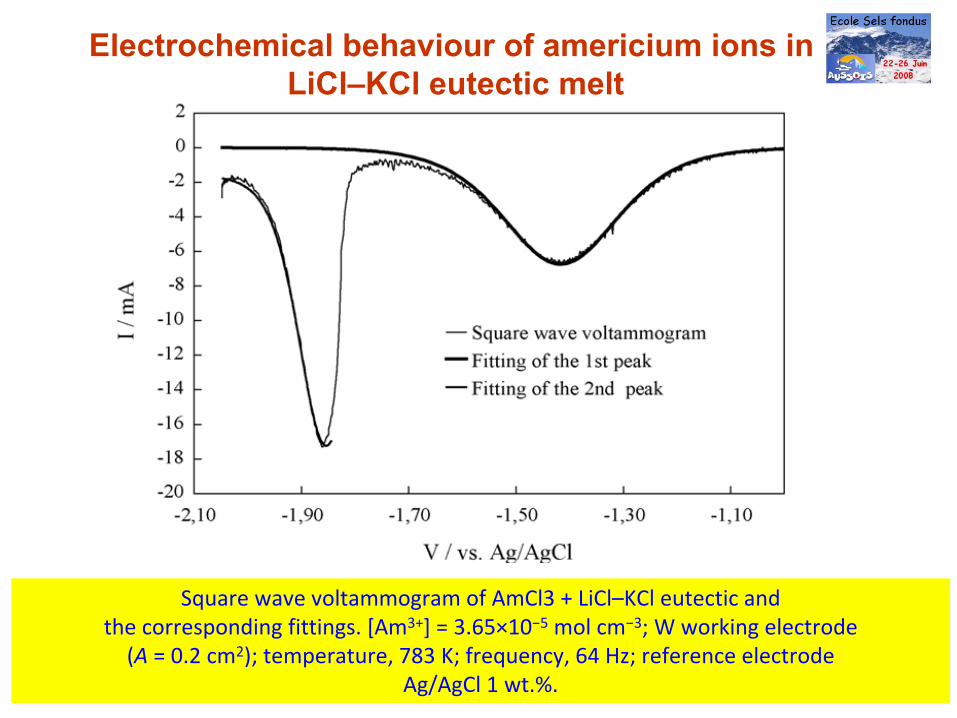
Square wave voltammogram of AmCl3 + LiCl–KCl eutectic andthe corresponding fittings. [Am3+] = 3.65×10−5
mol cm−3; W working electrode(A = 0.2 cm2); temperature, 783 K; frequency, 64 Hz; reference electrode
Ag/AgCl 1 wt.%.
Electrochemical behaviour of americium ions in LiCl–KCl eutectic melt
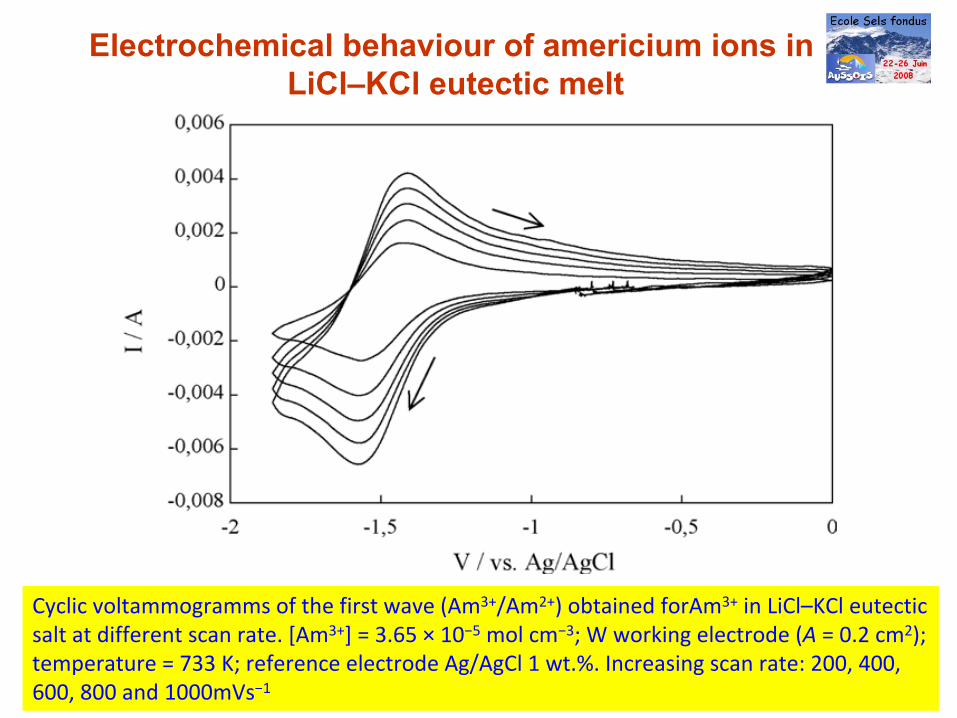
Cyclic voltammogramms of the first wave (Am3+/Am2+) obtained forAm3+
in LiCl–KCl eutectic
salt at different scan rate. [Am3+] = 3.65 × 10−5
mol cm−3; W working electrode (A = 0.2 cm2);
temperature = 733 K; reference electrode Ag/AgCl 1 wt.%. Increasing scan rate: 200, 400,
600, 800 and 1000mVs−1
Electrochemical behaviour of americium ions in LiCl–KCl eutectic melt
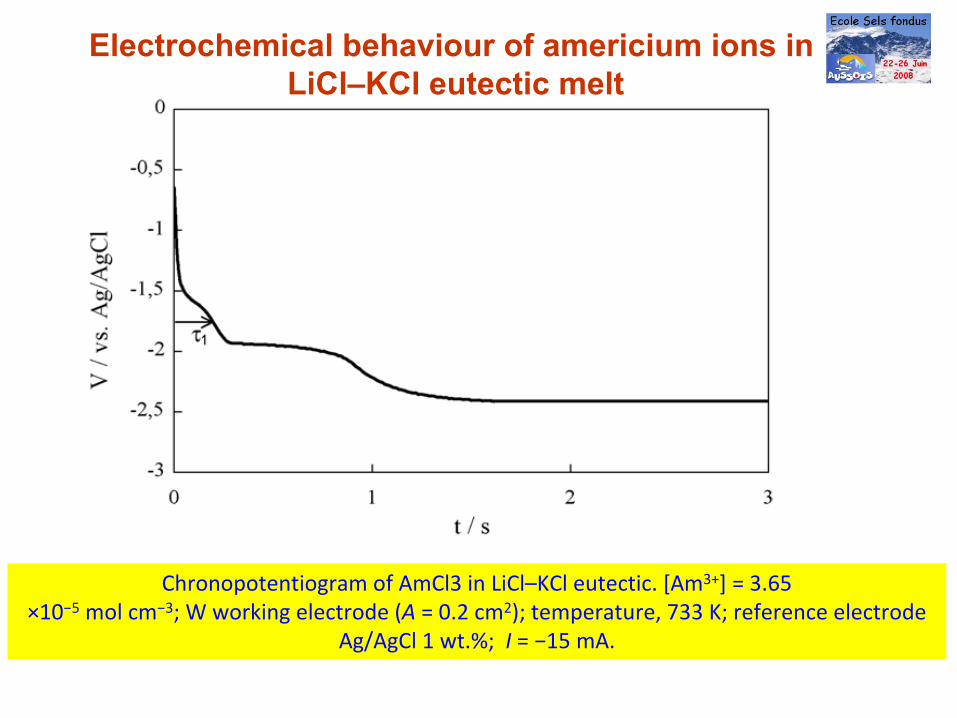
Chronopotentiogram of AmCl3 in LiCl–KCl eutectic. [Am3+] = 3.65×10−5
mol cm−3; W working electrode (A = 0.2 cm2); temperature, 733 K; reference electrode
Ag/AgCl 1 wt.%; I = −15 mA.
Electrochemical
behaviour
of americium
ions in LiCl–KCl
eutectic
melt





























