Embed Size (px)
Citation preview

ETUDE DE LA REACTIVITE DU 3-AMINO-2-BUTENOATE D'ETHYLE VIS-A-VIS D'AZOLINE-5-ONES.
A. MAQUESTIAU, J.-J. VANDEN EYNDE et R. MANDERLIER.
U n i v e z b i t t ? d e t ' E t a t , L a b o z a t o i z e d e C h i m i e O k g a n i q u e , 7 0 0 0 Monb [ B e t g i q u e ) .
Received : 29/11/1984 - Accepted : 14/12/1984 A B S T R A C T .
Various 1.3-disubstituted pyrazolin-5-ones, 3-phenylisoxazolin-5-one and 2-phenyloxezolin-5-one react with ethyl 3-amino-2-butenoate to yield compounds resulting from the elimination o f ammonia between the precursors ; subsequent intramolecular cyclization is only observed in the pyrazolonic series.
Under the same experimental conditions, ethyl 3-oxobutanoate is less reac- tive than the enaminocarbonyl analogue.
IH NNR data show that the structure of the so obtained products is highly sensitive to solvent effects.
INTRODUCTION.
Nous avons montre precedemment que dans la reaction entre le 3-oxobutanoa- te d'ethyle et la l-ph~nyl-3-m~thylpyrazoline-5-one ( I ) , le 8-cetoester peut avantageusement Stre remplace par son analogue enaminocarbonyle, le 3-amino-2- butenoate d'ethyle''). En effet, l'emploi de ce dernier permet non seulement de preparer la l-phenyl-3,4-dim6thylpyrano [2,3-c1 pyrazole-6(1H)-one (20-fig.1) dans des conditions experimentales plus douces que celles habituellement decri-
, mais surtout d'isoler, avec un excellent rendement, l'intermediaire 1 2 implique dans la synthese du derive bicyclique.
tes (2,3)
Nous generalisons ce resultat par l'etude de la reactivite du 3-amino-2- butenoate d'ethyle vis-a-vis d'autres composes pyrazoloniques ainsi que vis-a- vis d'heterocycles de structure proche.
RESULTATS EXPERIMENTAUX ET DISCUSSION.
1 * Eonstatalions-rxeerlmegtalps. Les reactions entre le 3-amino-2-butPnoate d'ethyle et les pyrazoline-5-
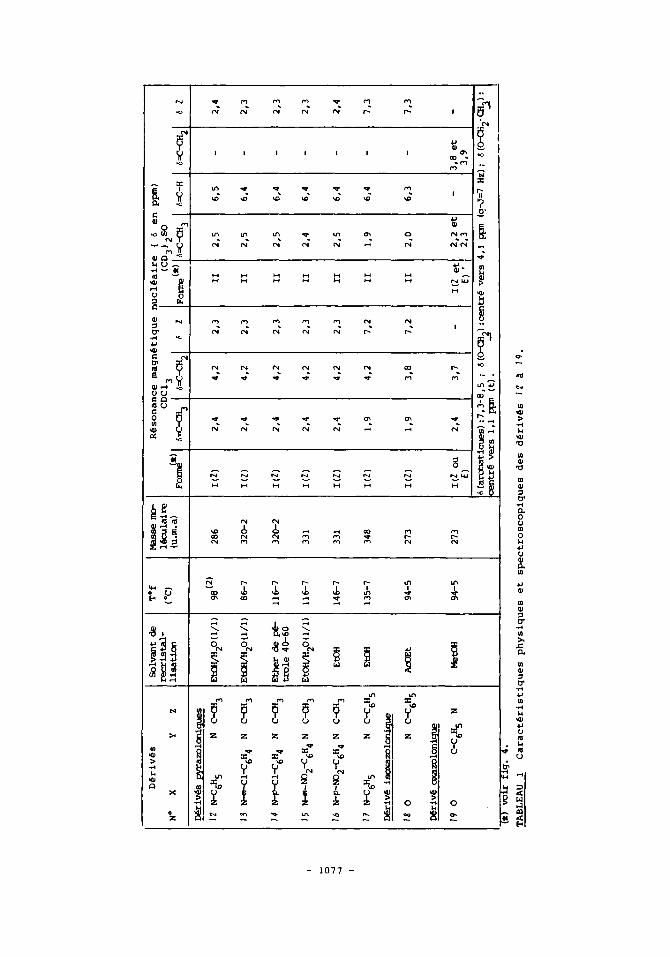
ones I a 6 (fig. 1 ) ont ete realisees dans le benzene a reflux ( 8 heures) .Apr&s evaporation du solvant, on isole les composes 1 2 a 1 7 (fig.1). Ceux-ci resul- tent de l'elimination d'ammoniac entre les precurseurs c o m e en temoigne l'en- semble des caracteristiques spectrales figurant au tableau 1.
Par chauffage, soit en l'absence de solvant a une temperature proche de celle de leur point de f ~ s i o n ( ~ ' ~ ' ~ ) , soit l'acide acetique a reflux"), 1 2 a I7 sont convertis en pyrano [2,3-cl pyrazole-6(1H)-ones 20 a 2 5 (fig.1) :
la perte d'ethanol, par transesterification intramoleculaire, est confirmee par spectrometrie de masse ainsi que par R.M.N. et, sur les spectres I.R. (KBr) de 20 a 2 5 , on releve, vers 1720 an-', une bande v(C=O) intense caracteristique du cycle o-pyrone(3'6).
dans
- 1073 -
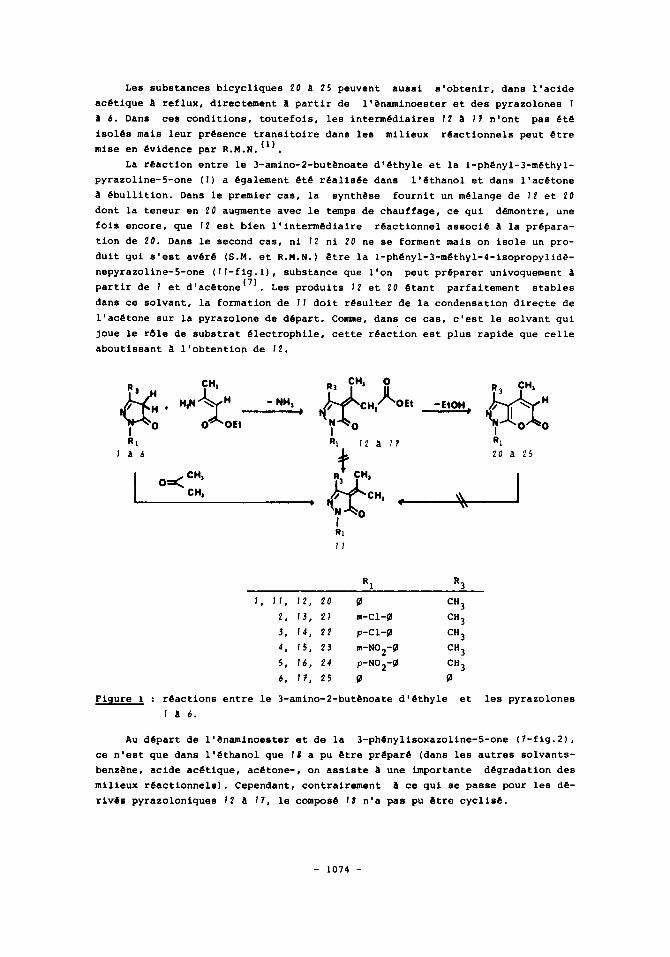
Les substances bicycliques 20 25 peuvent aussi s'obtenir, dans l'acide acetlque a reflux, directement a partir de l'enaminoestar et des pyrazolones 1
a 6. Dans ces conditions, toutefois, lea intermediaires 1 2 a I ? n'ont pas ete isoles mais leur presence transitoire dans les milieux reactionnels peut etre mise en evidence par R.M.N. ( I ) .
La reaction entre le 3-amino-2-but6noate d'ethyle et la 1-phenyl-3-methyl- pyrazoline-5-one ( I ) a egalement ete realisee dans l'dthanol et dans l'acetone a ebullition. Dans le premier cas, la synthese fournit un melange de 1 2 et 20 dont la teneur en 20 augmente avec le temps de chauffage, ce q u i dhontre, une fois encore, que 1 2 est bien l'intermediaire reactionnel associe a la prepara- tion de 20. Dans le second cas, ni 1 2 ni 20 ne se forment mais on isole un pro- duit qui s'est aver6 (S.M. et R . M . N . ) Stre la l-ph~nyl-3-m~thyl-4-isopropylid~- nepyrazoline-5-one (ll-fig.l), substance que l'on peut preparer univoquement 1 partir de 1 et d'acetone''). Lea produits 12 et 20 etant stables dans ce solvant, la formation de 1 1 doit resulter de la condensation directe de l'acetone sur la pyrazolone de depart. Comme, dans ce cas, c'est le solvant qui joue le rdle de substrat electrophile, cette reaction eat plus rapide que celle aboutissant a l'obtention de 12.
parfaitement
R . R,
CH3 2, 13, 21 m-C1-0 CH3
CH3 4, 1 5 , 23 m-N02-0 CH3 5, 16, 24 p-N02-0 CH3
I , 1 1 , 12, 20 0
3, 14, 2 2 p-C1-0
6, I ? , 25 0 0
Figure 1 : reactions entre le 3-amino-2-but13noate d'ethyle et les pyrazolones 1 a 6.
A u depart de l'enaminoester et de la 3-phenylisoxazoline-5-one (7-fig.2), ce n'est que dans l'ethanol que 18 a pu &re prepare (dans les autres solvants- benzene, acide acetique, acetone-, on assiste a une importante degradation des milieux reactionnels). Cependant, contrairement a ce qui se passe pour les de- rives pyrazoloniques I2 a 17, le compose 1 8 n'a pas pu &re cyclise.
- 1074 -

I 1 6
Figure 2 : reaction entre le 3-amino-2-butenoate d'ethyle et l'isoxazoline-5- one ? .
Une telle difference de comportement entre substances pyrazoloniques et isoxazoloniques est egalement decrite dans la litterat~re(*'~) pour d'autres reactions intramoleculaires mettant en jeu ces deux types d'heterocycles.
C'est probablement la plus grande electronegativite de l'oxygene (compare a l'azote) en position 1 des heterocycles qui est responsable de l'augmentation sensible de l'energie d'activation associee a l'etape de cyclisation. On releve d'ailleurs que 19 (fig.3), synthetise a partir d'acide hippurique ( 8 ) et de 3- amino-2-butbnoate d'ethyle dans l'anhydride acetique a 100°C, ne reagit pas non plus intramoleculairement.
k 1 9
Figure 3 : preparation du compose 1 9 .
Enfin, il est a souligner que l'utilisation de l'&naminoester a la place du 3-oxobutanoate d'ethyle s'est, a nouveau") , revglee avantageuse car ce der- nier ne reagit ni avec l'isoxazolone ni avec l'oxazolone. Le 6-cetoester n'est consomme que par les pyrazolones 1 a 6, uniquement dans l'acide acetique a re- flux et plus lentement que son analogue enaminocarbonyle.
2. S l r u c ~ u r e - d e s - d e r l v e _ s _ _ 1 2 _ 1 - 1 9 - - e n _ B 0 1 U PimelhYlsulfoxYde -
Les donnees de R.M.N. ( tableau 1 ) indiquent que dans le chloroforme, les substances 1 2 a I9 presentent toutes une double liaison semi-cyclique de la forme I (fig.4). On observe en effet, vers 4 ppm, un singulet dont l'intensite et la position caracterisent les protons du groupe methylene. Par ailleurs,com- me on n'enregistre aucun dedoublement de ce signal ( ni de celui dU aux protons du groupe methyle sur la double liaison semi-cyclique 1 , un seul stereoisomere est, dans chaque cas, predominant en solution.
Pour I ? et 18, la geometric 7 est proposee. En effet, sur leur spectre, on releve la presence d'un singulet dU a cinq protons magnetiquement equiva- lents a 6 = 7,2 ppm et le signal correspondant aux protons du groupe methyle sur la double liaison n'apparalt qu'a 1,9 ppm alors qu'il est situe a 2,4 ppm pour les substances 12 a 16. C o m e le laissait prevoir l'examen de modeles mo- leculaires, c'est l'interaction sterique entre le groupe methyle et le noyau aromatique qui force ce dernier hors du plan de l'heterocycle. Dans une telle conformation, les protons du groupe methyle sont soumis a un champ diamagneti- que local.
- 1075 -

Pour lee derives pyrazoloniques 1 2 a 17, la deplacement chimique des pro- tons du groupement methylene sur la double liaison semi-cyclique n'est pas af- fecte par la nature du substituant ( methyle ou phenyle 1 en position 3 des he- terocycles. L'environnement de ces protons est donc la m&ne dans 17 et dans 1 2
16. Par consequent, dans ces substances (12 a 1 6 ) , la double liaison semi- cyclique est aussi de configuration 2.
Aucun argument ne permet cependant de determiner la geometrie autour de la double liaison semi-cyclique dans l'oxazolone 19.
Sur les spectres de R.M.N. des composes 1 2 a 18 en solution dans le dime- thylaulfoxyde, on relave un signal echangeable a l'eau lourde vers 9-11 ppm et un pic dO a la resonance d'un proton sur carbone doublement lie vers 6,4 ppm. Ceci signifie, d'une part, que l'heterocycle se trouve sou8 la forme OH ou NH (fig.4) et, d'autre part, que la double liaison est exocyclique. Elle est de geometrie E;: ainsi qu'en t h o i g n e la va1eur(l0) mesuree (1,2 Hz) de la constan- te de couplage allylique 4J (H-C=C-CH3).
Pour 1 2 a 17, l'aspect du massif aromatique du substituant en position 1 des heterocycles est superposable a celui enregistre pour lea pyrano [2,3-cl pyrazole-6(1H)-ones correspondantes (20 a 25-fig.1). Cette observation suggere que toutes ces molecules ( 1 2 a 1 7 et 2 0 I 25) possedent un ensemble structural identiqua, a savoir un noyau de type pyrazole. Aussi, dans les derives 1 2 a 17, les heterocycles existeraient sous la forme hydroxypyrazolique(forme 11-fig.4). Cette proposition est confinnee par la similitude que l'on peut egalement ob- server dans la region aromatique des spectres de R.M.N. de 1 2 et du l-phenyl-3- methyl-5-ethoxypyrazole ( 9 ) .
Forre I Forhie I1 (ou OH) Force I11 (ou CS rom nl sauf l m r 191
Figure 4 : structures des derives 1 5 a 19 (defMtion de X, Y et 2 : voir tableau 1).
I1 resaort de cette etude que la presence de la chaLne exocyclique dans 1 2
I 17 ne modifie pas sensiblement le sens gen€ral('l)du deplacement de l'equili- bre tautomare. Partant, et compte tenu du parallelisme observe entre le compor- tement prototropique des pyrazoline-5-ones et des isoxazoline-5-ones (I2) , on peut raisonnablement supposer que dans f K , l'hbterocycle se presente, dans le dimethylsulfoxyde, sous la forme OH.
Par ailleurs, il est a noter qu'une forme hydroxylee n'a jamais ete mise en evidence avec certitude en aerie oxazolonique (12'13). Dans le cas de 19, il n'est donc paa surprenant que ce soit un melange (1/1) d'isomeres E et 2 autour de la double liaison semi-cyclique q u i coexiste en solution dans le dimbthyl- sulfoxyde.
at Pour 17 et 1 8 , il semble que le noyau aromatique en position 3 des heterocy-
cles soit a nouveau hors du plan ceux-ci ( 5"protons equivalents a 6 = 7,3 ppm) et qu'une telle situation entralne un " gel des molecules dans une con- formation b - c i d autour de la simple liaison -C-heterocycle. On observe, en ef- fet, toujours par rapport a 1 2 a 16, un deblindage important des protons du groupe methyle Bur carbone doublement lie.
de
- 1076 -
a F $
z w
P Y) -
- 1077 -

3 * s _ t r u c _ t u r e _ d e ~ - c r ? l ~ ~ ~ ~ - ~ ~ - ~ - ~ ~ - ~ ~ - ~ ~ ~ u ~ ~ ~ ~ - - ~ ~ ~ ~ - - ~ e - ~ ~ ~ ~ _ t ~ Y ~ ~ u ~ ~ ~ ~ Y ~ ~ - ~ d d ~ ~ -__ tiopne ------------- d'ammoniac.
Les reactions etudi6es (vide supra) procedant avec liberation d'annnoniac, les spectres de R . M . N . de 12 a 17 en solution dans du dimethylsulfoxyde addi- tionne d'ammoniac ont ete releves. En effet, la presence de cette base dans le milieu est, a priori, susceptible de modifier la structure des produits finals qui c o m e vu precedemment est particulierement sensible a w effets de solvant. Si l'on observe bien l'apparition du pic a 6.4 ppm traduisant l'existence dans la molecule d'une double liaison exocyclique, l'aspect du massif aromatique est nettement different de celui correspondant B la forme OH. En effet, il est ca- racterise par un deblindage de deux protons (fig.5) dCI au voisinage d'une fonc- tion carbonyle. Comme on ne releve pas de signal attribuable B un proton en po- sition 4 des heterocycles (forme IV, fig.41, les produits se presentent dans le dimethylsulfoxyde additionne d'ammoniac sous la forme NH (fig.4).
Les modifications de structure que subissent les substances 1 2 a 1 7 sont nearnoins reversibles et elles resultent d'interactions specifiques solvant- solute. En effet, la neutralisation de l'anmoniac par de l'acide chlorhydrique dilue (') conduit 1 la reapparition du tautomere preponderant (hydroxypyrazoli- que) dans le dimethylsulfoxyde pur. De m&e, l'adjonction de volumes croissants de chloroforme B des solutions de 1 2 B I7 dans le dimethylsulfoxyde pur fait reapparartre proqressivement les signaux propres a la forme possedant une dou- ble liaison semi-cyclique (forme I, fig. 4 ) .
1 1 1 1 8 1 3 7
Figure 5 : region aromatique des spectres de R . M . N . de 1 2 dans le dimethylsul- foxyde (a) pur : (b) additionne d'amnoniac.
4 - pxeeriences-cin4tlsuel. Les reactions entre le 3-amino-2-but&noate d'ethyle et les heterocycles I
a 6, 9 et I0 one ete etudiees par R.M.N. selon une methode decrite precedem- merit"). Elle consiste B mesurer, en fonction du temps, la variation d'intensi- te du signal dCI aux protons du groupe methyle sur carbone doublement lie de l'enaminoester.
Experimentalement ( tableau 2 ) , on observe que la vitesse de disparition des reactifs est d'autant plus rapide que le solvant utilise est polaire. Ceci suggere que les syntheses procedent selon un mecanisme dans lequel intervient au moins une espece chargee. Dane le dimethylsulfoxyde, il pourrait s'agir de l'anion des heterocycles. On releve en effet que : a) le l-phenyl-3-methyl-5- ethoxypyrazole ( 9 ) et la l-ph~nyl-2,3-dim~thylpyrazoline-5-one ( 1 0 1 , hetero-
- 1078 -

cycles non ionisables, ne reagissent pas avec le 3-amino-2-but&noate d'ethyle : b) dans la serie des l-aryl-3-m~thylpyrazoline-5-ones, la stabilisation de l'anion par l'introduction d'un groupament electroattracteur sur le noyau aro- matique se traduit par une acceleration des reactions.
En ce qui concerne 1'Btape determinante, elle mettrait en jeu, comme il est rapport6 pour des reactions entre des pyrazoline-5-ones et des sels de dia- zonium(14 a 16! l'anion de l'heterocycle et le substrat electrophile. La vites- se d'apparition des produits finals repond d'ailleurs a une cinetique d'ordre global egal a deux et la reaction est ralentie par la presence d'un substituant volumineux, tel un groupement phenyle, au voisinage du site nucleophile (cfr. I par rapport a 6).
3" qN 0
I Ph
Ph 6h
I 0 9
R 1 R3
1 0 CH3 2 m-Cl-0 CH3
I 4 m-N02-0 CH3 & 3 p-c1-0 CH3
R1 5 p-N02-0 CH3 I a 6 6 0 0
~ ~
Reactions menees a 293 K avec des concentra- tions initiales de 0,25 mole par litre
Heterocycle Solvant Rendement apres 24 heures
10 % I 'sD6
I CgDsN 40 %
1 DCON (CD3) 50 %
I (CD3) 2S0 70 %
9 (CD3) 2S0 pas de reactioi 1 0 (CD3) 2S0 pas de reactioi
Reactions menees dans (CD3l2SO 1 315 K
I CDC13 20 %
0,50 100
0,25 2 00 1 0 , 4 0 125 0,02
3 0.50 65 0,03 2 0,50 50 0,04
4 0.25 80 0.05 5 0.25 20 0.14 6 0.50 200 0.01
TABLEAU 2. Etude quantitative par R.M.N. des reactions entre le 3-amino-2-bute- noate d'ethyle et les heterocycles I a 6, 9 ainsi que 1 0 .
CONCLUSIONS.
Des pyrazoline-5-ones 1,3-disubstituees reagissent avec le 3-oxobutanoate d'ethyle pour fournir des pyrano [2,3-c] pyrazole-6(1H)-ones.
L'utilisation du 3-amino-2-but&noate d'ethyle 1 la place du 8-cetoester correspondant donne lieu a une diminution sensible de l'energie d'activation de l'etape intermoleculaire, ce qui permet d'isoler, avec d'excellents rendements, les intermediaires associes a la preparation de ces derives bicycliques. Les
- 1079 -

produits ainsi obtenus existent, en solution dans le chloroforme, sous une for- me presentant une double liaison semi-cyclique de geometric 2, mais il est pos- sible, par effet de solvant, d'induire, de facon reversible, une migration de la double liaison vers la chafne exocyclique et de stabiliser l'une ou l'autre structure prototropique (OH ou NH) de l'heterocycle.
En ce qui concerne la 3-ph~nyllsoxazoline-5-one et la 2-phenyloxazoline- s-one, elles ne reagissent pas avec le 3-oxobutanoate d'ethyle. Par contre, a partir de 3-amino-2-but6noate d'ethyle, on forme des substances resultant de l'elimination d'ammoniac entre les pr6curseure. De telles substances ne cycli- sent toutefois pas en pyrano [3,2-d] (is)oxazole-6-ones.
PARTIE EXPERIMENTALE.
1 - APeaEpilla9E. - Spectroscopie de R.M.N. : Varian EM 360-L ou Varian XL-100-12 equip6 d'un
Les deplacements chimiques (6) sont exprimes en ppm par rapport au TMS utili- se comme reference interne.
VFT- 100.
- Spectroscopie I.R. : Perkin-Elmer 577.
- Spectrometrie de masse : Varian Mat 311 A. - Les points de fusion ( non corriges ) sont releves sur un microscope a plaque
chauffante.
2. S.Yn_the_seE, - Les syntheses sont realides au depart de solutions molaires de reactifs dans
le solvant indique ( benzene, acide acetique, ethanol ou acetone 1 . Apres 8 heures de chauffage a reflux, le solvant est evapore et le residu est recris- tallise. Les rendements sont toujours superieurs a 50 0. Le produit 19 est prepare selon la ref. 17 (page 30) en remplapant le benzaldehyde par du 3- amino-2-butenoate d'ethyle.
- Produits c m e r c i a u x : le 3-amino-2-butenoate d'ethyle et l'acide hippurique (Aldrich), le 3-oxobutanoate d'ethyle ( Merck ) ainsi que les heterocycles 1 ,
10 (Aldrich) et 2, 3, 4 (Schuchardt).
1212), 2 0 ' ~ ) et 22 a 25'3).
2'izQicethYleY r e n n - L 2 ' 2 - n l - e Y r a - E o l ~ ~ ~ ~ ~ ~ ~ ~ n ~ e - ~ ~ ~ ~ ~
- Produits decrits dans la litterature : p 3 ) , 6119), #I171 91201, 1117),
- Produits nouveaux : 1 3 a 19 (voir tableau 1) ainsi que la A:Jj=chlgyophe_nyll:
F(CH3CO2H) : 154-6OC - M" : 278-280 u.m.a.
R.M.N. (CDC13-6) : 7,9-7,l (4H) : 5,8 (1H) : 2,4 (3H) : 2,3 (3H).
;( :C=O)-KBr : 1720 an-'.
- 1080 -

BIBLIOGRAPHIE.
1. A. MAQUESTIAU, Y. VAN HAVERBEKE et J . 4 . VANDEN EYNDE, Bull. SOC. Chim. Belges, 92, 451 (1983).
2. R. STOLLE, Chem. Ber., 2, 3023 (1905). 3. M.A. KHAN, A.G. COSENZA et G.P. ELLIS, J . Heterocyclic Chem., 2, 4. A. MUSTAFA, A.M. FLEIFEL, M.I. ALI et N.M. HASSAN, Ann., 9, 75
5. A. MAQUESTIAU et J . 4 . VANDEN EYNDE, Bull. SOC. Chim. Belges, 93, 6. K. NAKANISHI dans "Infrared Absorption Spectroscopy", Holden-Day
San Francisco (1962).
7. L. KNORR, Ann., 238, 137 (1887).
1077
1970
451
nc ,
(1982).
1984).
8. G. TACCONI, G. GATTI, G. DE SIMON1 et V. MESSORI, J. Prakt. Chem., 322, 831 (1980).
9. A. HARHASH, M. ELNAGDI, N. KASSAB et A. NEGM. J. Chem. Eng. data, z, 120
(1975).
10. L.M. JACKMAN et S. STERNHELL dans " Applications of Nuclear Magnetic Resonan- ce Spectroscopy in Organic Chemistry ", Supp. I, Academic Press, Paris (1969).
11. A. MAQUESTIAU, Y. VAN HAVERBEKE et R. JACQUERYE, Bull. SOC. Chim. Belges, g , 215 (1973).
12. J. ELGUERO, C. MARZIN, A.R. KATRITZKY et P. LINDA, " The Tautomerism of He- terocycles ", dans ,, Advances in Heterocyclic Chemistry ", supp. I, Academic Press, London ( 1 976) .
13. W. STEGLICH, Fortschr. Chem. Forsch., 77 (1969).
14. I. DOBAS, V. STERBA et M. VECERA, Collect. Czech. Chem. Commun., 34, 3895 (1969).
15. I. DOBAS, V. STERBA et M. VECERA, Collect. Czech. Chem. Commun., 2, 3905 (1969).
16. Y. HASHIDA, M. KOBAYASHI et K. MATSUI, Bull. Chem. SOC. Japan, 4-4. 2506 (1971).
17. A.O. FITTON et R.K. SMALLEY dans " Practical Heterocyclic Chemistry ", Aca- demic Press, London (1968).
18. W.C. SUMPTER et P.H. WILKEN, J. Am. Chem. SOC., 70, 1980 (1948). 19. L. KNORR et C. KLOTZ, Chem. Ber., 20, 2545 (1887). 20. P.C. FREER, J. Prakt. Chem., 47, 246 (1893).
- 1081 -





























![vis-à-vis de l’examen gynécologique - chups.jussieu.fr€¦ · vis-à-vis de l’examen gynécologique ... [11]. La simulation basse fidélité, développée dès les années](https://img.pdfslide.fr/doc/110x75/5b995ded09d3f29c338c27d4/vis-a-vis-de-lexamen-gynecologique-chups-vis-a-vis-de-lexamen-gynecologique.jpg)