Embed Size (px)
Citation preview

NIZAR CHETOUI
CARACTÉRISATION DU RÔLE DE LA PROTÉINE KINASE MEK1 DANS LES VOIES DE TRANSDUCTION DES MAP KINASES
Thèse présentée à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en biologie cellulaire et moléculaire pour l’obtention du grade de Philosophiae Doctor (Ph.D.)
FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
AVRIL 2005 © Nizar Chetoui, 2005

Résumé
Le développement tumoral nécessite une dérégulation des contrôles normaux de la
prolifération et de la différenciation cellulaires. Il est bien connu que la voie de
signalisation ERK/MAP kinase est impliquée dans ces processus de régulation cellulaire.
De plus, un rôle essentiel dans la transformation cellulaire est attribué à MEK1 qui est un
élément central de cette voie. Une meilleure compréhension de l’implication de MEK1
dans les voies de transduction devrait donc nous permettre de mieux comprendre le
developpement cellulaire et la transformation morphologique. Mes travaux de recherche
tentent d’élucider les mécanismes de régulation des protéines kinases MEK1 et MEK2 dans
le but de mieux comprendre leur divergence fonctionnelle et leur implication dans les
différentes réponses cellulaires. Ainsi, l’étude de la voie ERK/MAPK chez les fibroblastes
embryonnaires mutants pour le gène Mek1 indique que la transduction du signal amorcée
par un neuropeptide, la bombésine, passe spécifiquement par MEK1 et serait indépendant
de MEK2. La région C-terminale de MEK1 semble médier la spécificité de la réponse à la
bombésine. Le domaine MSS, qui est une insertion d’une séquence riche en proline unique
à MEK1 et MEK2 pourrait être la clef de cette réponse spécifique. En outre, nos travaux de
délétion et d’interactions protéiques suggèrent qu’une variation de la conformation de la
région C-terminale de MEK1 pourrait avoir lieu entre l’état inactif et l’état actif de ces
MAPKKs. En absence d’activation, la région en caboxy du domaine kinase semble
interagir avec la boucle d’activation se trouvant au cœur du domaine kinase. Cette
interaction intramoléculaire serait dépendante de l’état de phosphorylation de MEK1. Par
contre, la région en carboxy ne semble pas être un domaine d’autoinhibition ou un pseudo
substrat puisque sa délétion ne met pas la protéine dans un état constitutivement actif.
Ainsi, il est possible que cette région soit essentielle du point de vue structural pour
permettre, en fonction de son activité, la régulation des interactions de MEK1 (ou MEK2)
avec ses activateurs, substrats ou protéines d’échafaudage.

iii
Avant-propos
Je n’avais que 14 ou 15 ans lorsque je suivais, à la télévision, la série de dessins animés Il
était une fois… La vie. J’étais particulièrement émerveillé en découvrant le fonctionnement
du corps humain au niveau cellulaire et moléculaire, la transcription des gènes, la
production des enzymes jusqu'à l’organisation et la mobilisation des différents dispositifs
dans la lutte contre les pathogènes. Cette fascination m’a beaucoup marquée et depuis, je
savais que j’allais faire de la biologie cellulaire, ma carrière.
Voilà que j’en suis arrivé à finir mes études et à franchir une grande étape pour le
couronnement de mes aspirations en décrochant mon doctorat en biologie cellulaire et
moléculaire. Pour en arriver là, j’ai eu tout l’aide et le soutien de professeurs, de collègues,
d’amis et de membres de ma famille. Je tiens ici à les remercier.
Tout d’abord, mon directeur de thèse, le Dr Jean Charron, de m’avoir bien accueilli dans
son laboratoire, de m’avoir soutenue dans mon projet de recherche et d’avoir été un bon
conseil.
Un grand merci à tous les membres de mon comité de thèse, Dr Jacques Huot, Dr Robert
Faure et le Dr Carl Séguin. Le soutien que vous m’avez apporté est inestimable.
Merci à Michel Tremblay, qui a beaucoup participer à l’avancement de mes travaux et qui
m’a transmis tous ces connaissances en culture cellulaire.
Un grand merci à mes chers amis Vick, Seb, Julie et Jérôme. Je suis content de vous avoir
rencontrer au laboratoire Charron/Jeannotte. Je souhaite que notre amitié se prolonge
indéfiniment.
Merci à tous les collègues que j’ai côtoyé au centre de recherche de l’Hôtel-dieu de Québec
tout au long de mes études doctorales.

iv
Merci aux chercheurs du centre de recherche en cancérologie de l’université Laval qui, de
près ou de loin, ont participé à l’avancement de mes travaux, Dr Lucie Jeannotte, Dr Josée
Aubin, Dr Jacques Landry, Dr Josée Lavoie et Dr Luc Bélanger, directeur du centre.
Ma thèse présentée ici représente pour moi l’effort de plusieurs années d’acharnement. Être
patient et très persévérant, c’est ce qu’il faut être pour arriver au bout des difficultés qu’on
rencontre durant les études doctorales. Pour moi, ce n’aurait pas été facile sans le soutien,
l’encouragement, et l’amour de ma femme Kaouther et l’immense joie de vivre que m’ont
procuré mes enfants Walid et Sabrina.
Enfin, c’est grâce à mes parents, Jameleddine et Bärbel Chetoui que je dois tout, ma
réussite scolaire et l’épanouissement de ma vie.
La recherche dans le domaine de la cancérologie est en progression et toute contribution,
aussi modeste qu’elle soit, permettra d’arriver à bout de cette maladie qu’est le cancer. Mes
travaux présentés ici portent sur l’implication de la voie ERK MAP kinase dans la
prolifération et la migration cellulaire, deux processus primordiaux utilisés par les cellules
cancéreuses pour se développer et se propager. Bonne lecture.

À mes parents Jameleddine et Bärbel Chetoui
"Marche avec des sandales jusqu'à ce que la sagesse te procure des souliers"
Avicenne, Ibn Sinà (980-1037)
«La joie de regarder et de comprendre est le plus beau cadeau de la nature»
Albert Einstein (1879-1955)

vi
Table des matières
Résumé................................................................................................................................... ii
Avant-propos ........................................................................................................................ iii
Table des matières .................................................................................................................vi
Liste des figures & des tableaux ............................................................................................ix
Listes des abréviations ............................................................................................................x
1. Introduction générale ......................................................................................................1
1.1. Introduction.....................................................................................................................2
1.2. Activation du sentier ERK par des stimuli extracellulaires............................................4
1.3. Les composantes kinases du module de signalisation ERK/MAPK...............................7 1.3.1 Les isoformes Raf ...............................................................................................7 1.3.2. MEK1 et MEK2................................................................................................12
1.3.2.1 Domaines d’interaction dans MEK1/MEK2.................................................14 1.3.2.1.1. La région MSS......................................................................................15 1.3.2.1.2. Le domaine D........................................................................................17
1.3.2.2. Autres sites importants dans MEK1 .........................................................17 1.3.2.2.1. La séquence NES ..................................................................................17 1.3.2.2.2. Le site d’attache de PD 184352 ............................................................19 1.3.2.2.3. La région C-terminale...........................................................................20
1.3.3. ERK1 et ERK2..................................................................................................21 1.4. Autres voies de signalisation MAP kinases chez les mammifères ...............................23
1.4.1. Le sentier JNK/SAPK.......................................................................................23 1.4.2. Le sentier p38....................................................................................................24 1.4.3. Autres MAP kinases .........................................................................................25
1.4.3.1. ERK3 ........................................................................................................25 1.4.3.2. ERK5 ........................................................................................................26 1.4.3.3. NLK ..........................................................................................................28
1.4.4. MEKKs, la première marche dans la cascade des MAP kinases......................28 1.5. L’organisation, la localisation et la spécificité des cascades des MAP Kinases ..........32
1.5.1. Prédiction de la formation de complexes signalétiques par l’étude de la signalisation intracellulaire chez la levure........................................................................32
1.5.1.1. Ste5p .........................................................................................................33 1.5.1.2. Pbs2p.........................................................................................................35
1.5.2. Association des protéines kinases dans les voies MAP kinases chez les mammifères ......................................................................................................................36

vii
1.5.2.1. Les protéines d’échafaudage du sentier ERK ...........................................36 1.5.2.1.1. MP-1 .....................................................................................................37 1.5.2.1.2. Grb10 ....................................................................................................37 1.5.2.1.3. KSR.......................................................................................................38 1.5.2.1.4. RKIP .....................................................................................................39
1.5.2.2. Les sites d’amarrage ou Docking Sites .....................................................40 1.5.3. L’inactivation des MAP Kinases ......................................................................43 1.5.4. La régulation de la localisation subcellulaire de ERK......................................46 1.5.5. Spécificité et intégrité du signal........................................................................50
1.6. L'organisation différentielle des signaux transmis via la voie ERK/MAPK. ...............54 1.6.1. La différenciation..............................................................................................55 1.6.2. La signalisation entre la prolifération et l'arrêt de la croissance.......................57
1.7. Le projet de recherche...................................................................................................60 1.7.1. Mise en contexte ...............................................................................................60 1.7.2. Hypothèse de travail .........................................................................................63 1.7.3. Objectifs du projet ............................................................................................64
2. Étude de la signalisation ERK/MAPK chez les fibroblastes embryonnaires de souris Mek1-/- ...................................................................................................................................65
2.1. Résumé..................................................................................................................66 2.2. Introduction...........................................................................................................67 2.3. Matériels et méthodes ...........................................................................................72 2.4. Résultats................................................................................................................74 2.5. Discussion.............................................................................................................83 2.6. Références.............................................................................................................86
3. Identification et caractérisation d’un domaine d’interaction phospho-dépendant dans MEK1 et MEK2....................................................................................................................92
3.1. Résumé..................................................................................................................93 3.2. Article ...................................................................................................................94
3.2.1. Abstract.........................................................................................................95 3.2.2. Inroduction....................................................................................................96 3.2.3. Experimental procedures ..............................................................................98 3.2.4. Results.........................................................................................................103 3.2.5. Discussion...................................................................................................123 3.2.6. Acknowledgements.....................................................................................127 3.2.7. References...................................................................................................128
4. Résultats complémentaires .........................................................................................131 4.1. Interaction MEK1-Histones H3 ..........................................................................132 4.2. Interaction MEK1-M96A ...................................................................................133
Discussion générale ............................................................................................................136
Annexe ................................................................................................................................143

viii
A. Criblage double hybride chez la levure ......................................................................144 A.1. Tableaux récapitulatifs des clones isolés lors du criblage double hybride .........144 A.2. Vue d’ensemble des faux positives courrament isolés lors des criblages double hybride chez la levure .....................................................................................................147
Bibliographie ......................................................................................................................148

ix
Liste des figures & des tableaux
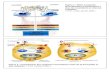
Figure 1-1 : Les composantes kinases de base de la voie ERK/MAP kinase.....................5
Figure 1-2: La transmission du signal extracellulaire via la voie de signalisation ERK/MAPK et les multiples molécules signalétiques qui régulent sa progression. ......8
Figure 1-3 : Les protéines MEK1 et MEK2. Structure et comparaison. ..........................16
Figure 1-4 : Les voies de signalisation des MAP kinases. ...............................................30
Figure 1-5 : Les complexes d'échafaudage de la voie MAP kinase chez la levure saccharomyces cerevisiae.............................................................................................34
Figure 1-6 : MEK1 est impliquée dans la signalisation intracellulaire menant à la migration des fibroblastes embryonnaires de souris.....................................................61
Figure 2-1 : Schéma général de la signalisation induite par la bombésine.......................69
Figure 2-2 : Activation de la voie ERK/MAPK via Gi et Gq...........................................71
Figure 2-3 : Altération de la signalisation induite par la bombésine chez les MEFs mutants pour le gène mek1............................................................................................78
Figure 2-4 : La signalisation induite par la bombésine est spécifique à MEK1. ..............80
Figure 2-5 : Activation de la voie ERK/MAPK en réponse à la bombésine chez les MEFs Mek1 confluents............................................................................................................82
Figure 3-1 : Structure of MEK protein regions recover from yeast two-hybrid screen against MEK1. ............................................................................................................105
Figure 3-2 : Mapping of the MEK1-binding sites by the two-hybrid assay...................109
Table 3-1 : Intra-molecular interaction of the carboxyl terminal with the activation loop is dependent on the phosphorylation state. .................................................................111
Figure 3-3: MEK1 C-terminal domain bind to a truncated form of MEK1 in vivo.......114
Figure 3-4 : Catalytic activity and cellular localisation of MEK1 C362 mutants. .........118
Figure 3-5 : Deletion of the carboxyl-terminal domain of MEK1 does not affect the regulation of MEK1 activity. ......................................................................................120
Figure 3-6 : MEK1 activity is not affected by point mutations in the proline-rich sequence in vitro. ........................................................................................................122
Tableau 4-1 : Essai d’interaction double hybride chez la levure ..................................134
Tableau 4-2 : Les histones H3 et M96A sont des faux positifs ....................................135

x
Listes des abréviations
GÉNÉRALES
ADN Acide désoxyribonucléique
AMPc Adénosine Monophosphate cyclique
ATP Adénosine Triphosphate
CDKs Kinases dépendantes des cyclines (Cyclin-Dependent Kinases)
CRD domaine riche en cystéine (cystein-rich domain)
DAG diacylglycérol
eIF4E Facteur d’initiation de la traduction chez les eucaryotes 4E (eukaryotic
translation initiation factor 4E)
EGF Facteur de croissance épidermique (Epidermal Growth Factor)
EGFR Récepteur du facteur de croissance épidermal (Epidermal Growth Factor
Receptor)
ERK Kinase régulée par un signal extracellulaire (Extracellular signal-Regulated
Kinase)
FAK Kinase d'adhérence focale (Focal Adhesion Kinase)
FGF Facteur de croissance fibroblastique (Fibroblast Growth Factor)
GCK La kinase du centre germinal (germinal center kinase)
GFP Protéine fluorescente verte (Green Fluorescent Protein)
GDP Guanosine di-phosphate
GPCRs récepteurs couplés aux protéines G (G protein-coupled receptors)
Grb2 Protéine qui lie le récepteur des facteurs de croissance 2 (growth-factor-
receptor-bound protein 2)
Grb10 Protéine qui lie le récepteur des facteurs de croissance 10 (growth-factor-
receptor-bound protein10)
GTP Guanosine tri-phosphate
HPK1 (hematopoietic progenitor kinase 1)
HSPs Protéines de choc thermique (Heat Shock Proteins)

xi
HSP27 Protéine de choc thermique 27 (Heat Shock protein of 27 kDa)
IGF-I Facteur de croissance analogue de l’insuline (insulin-like growth factor I)
IL Interleukine
IP3 Inositol- triphosphate
IRS Substrat du récepteur de l'insuline (Insulin Receptor Substrate)
JIP Protéine qui lie JNK (JNK-interacting protein)
JNK Kinase de la partie N-terminale de la protéine c-Jun (c-Jun N-terminal
Kinase)
KSR Protéine kinase suppresseur de RAS (Kinase suppressor of Ras)
MAP Protéine activée par les mitogènes (Mitogen Activated Protein)
MAPK Protéine-kinase activée par les mitogènes (Mitogen Activated Protein
Kinase)
MAPKK Protéine-kinase kinase activée par les mitogènes (Mitogen Activated Protein
Kinase Kinase)
MEC Matrice extracellulaire
MEF Fibroblaste embryonnaire de souris (Mouse Embryonic Fibroblast)
MEK MAP/ERK kinase
MKK MAP kinase kinases
MKKK MAP kinase kinase kinases
MKP MAPK phosphatase
MNK MAP kinase-interacting kinase
MP-1 Partenaire de MEK1 -1 (MEK1-Partner-1)
MSK kinase activée par les mitogènes et par le stress (mitogen- and stress-
activated protein kinase)
MSS Séquence spécifique à MEK (MEK-specific sequence)
NES Séquence d’exclusion nucléaire
NGF Facteur de croissance neuronal
NIK Kinase qui lie Nck (Nck-interacting kinase)
NLS Signal de localisation nucléaire
PAGE Électrophorèse en gel de polyacrylamide (Polyacrylamide gel
electrophoresis)

xii
PAK p21-activated protein kinase
PCR Réaction de polymérisation en chaîne (Polymerase Chain Reaction)
PDGF Facteur de croissance dérivé des plaquettes (Platelet-derived growth factor)
PI-3K Kinase-3 phosphoinositide (Phosphoinositide 3-Kinase)
PIP2 Phosphatidylinositol 4,5-bisphosphate
PKA Protéine kinase A
PKC Protéine kinase C
RKIP Protéine inhibitrice de la kinase RAF (RAF kinase inhibitor protein)
RTK Récepteur à activité tyrosine kinase
SAPK Protéine-kinase activée par le stress (Stress Activated Protein Kinase)
SAPK2 Protéine-kinase 2 activée par le stress (Stress Activated Protein Kinase-2)
Ser Résidu sérine
SOS Facteur d’échange de GTP pour Ras (Son of Sevenless)
ST Syncytiotrophoblastes
TGF-β Facteur de croissance tumorale-béta (Tumor growth factor-β)
Thr Résidu Thréonine
TNF Facteur de nécrose tumorale (Tumor Necrosis Factor)
TNF-α Facteur de nécrose tumorale alpha (Tumor Necrosis Factor Alpha)
TNFR Récepteur du facteur de nécrose tumorale (Tumor Necrosis Factor Receptor)
Tyr Résidu Tyrosine
VEGF Facteur de croissance endothélial vasculaire (Vascular Endothelial Growth
Factor)
WT Type sauvage (Wild Type)
UNITÉS
µ Micro
Da Dalton
g Gramme
h Heure
kb Kilobase
M Molaire

xiii
min Minute
mg Milligramme
sec seconde
RÉACTIFS ET SUBSTANCES CHIMIQUES
3-AT 3-amino-1,2,4-triazole
DMEM Milieu de Eagle modifié par Dulbecco (Dulbecco's modified Eagle's medium)
DMSO Diméthyle sulfoxide
DTT Dithiothréitol
EDTA Acide éthylène-diamine tétra acétique (ethylene diamine tetraacetic acid)
EGTA Acide éthylène glycol-bis-b aminoéthyl éther-N, N, N', N'-tétra acétique
(ethylene glycol tetra acetic acid)
FBS Sérum foetal de bovin (Foetal Bovine Serum)
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
MOPS Acide propane sulfonique 3-(N-morpholino)
PBS Tampon phosphate salin (Phosphate buffer salin)
PMSF Fluorure de phényl-méthyl sulfoxide (Phenyl Methyl Sulfonyl Fluoride)
SDS Dodécyle-sulfate de sodium (Sodium Dodecyl Sulfate)
TRIS Tris (hydroxyméthyle) aminométhane

Chapitre 1
1. Introduction générale

2
1.1. Introduction
Les protéines kinases ainsi que divers systèmes de messagers forment des réseaux
fortement interactifs pour exécuter, au niveau cellulaire, des fonctions intégrées dans
l’organisme. La compréhension des mécanismes de signalisation pour un agent, son
répertoire de transducteurs et les interactions de ceux-ci dans un réseau devrait être définie
dans un contexte cellulaire. Ceci inclut la production de seconds messagers, l’activation de
protéines kinases, et la distribution subcellulaire de ces composants signalétiques pour les
mettre en contact avec les cibles appropriées. La recherche menée depuis la dernière
décennie a permis la découverte de plusieurs réseaux signalétiques intra et extracellulaires
et de faire le lien entre les défaillances possibles de ces réseaux avec plusieurs maladies
comme le cancer [1,2]. Parmi ceux là, on distingue des voies de signalisation formées de
protéines kinases connues sous le nom de MAP kinases pour mitogen-activated protein
kinases. Les voies de transduction des MAP kinases représentent un des systèmes
signalétiques primordiaux que la nature utilise dans plusieurs systèmes cellulaires pour
accomplir une variété étonnante de tâche. Elles existent chez tous les eucaryotes et
contrôlent des processus cellulaires fondamentaux aussi complexes que la prolifération, la
différenciation et l’apoptose.
L'arrangement de base d’une cascade de signalisation MAP kinase inclut une petite GTPase
se situant en amont d'un module central constitué de trois kinases : La MAPK kinase kinase
(MAPKKK), qui phosphoryle et active la MAPK kinase (MAPKK ou MKK), qui à son
tour, active la MAPK (Figure 1-1a). Cette organisation ne permet pas seulement
l'amplification du signal, mais aussi, de façon plus importante, fournit des interfaces
régulées supplémentaires donnant une cinétique, une durée et une amplitude précises à
l’activité [3-9]. Chez les mammifères, nous pouvons distinguer six modules MAP kinases
qui partagent des composantes structuralement apparentées tout en servant de médiateurs
dans des réponses biologiques spécifiques.
En dépit d'une décennie débordante d'intérêt scientifique et révélant d’énormes progrès
dans la compréhension de l'ensemble des circuits de signalisation intracellulaire, la voie

3
ERK/MAPK cache encore beaucoup de secrets. Ceux-ci concernent principalement la
régulation des composantes kinases du module, la compréhension de l’importance, dans
une réponse biologique précise, des changements spatio-temporels de l'activité et de la
distribution subcellulaire des composantes de la voie et enfin l’orchestration de ces
variations au niveau moléculaire [5,10].
Les travaux de plusieurs groupes de recherche pointent les interactions protéine-protéine
comme des moyens puissants de coordination des processus signalétiques. Cette
coordination est identifiée d'une manière claire au niveau de l'assemblage de complexes
protéiques sur les récepteurs activés, ou de complexes de facteurs de transcription sur les
promoteurs des gènes. Cependant, il devient apparent que ce motif de régulation est aussi
largement utilisé pour le contrôle des réseaux de signalisation intracellulaire.
Dans cette introduction, il sera question du rôle des interactions protéine-protéine dans la
régulation de la transduction du signal via les cascades de signalisation des MAP kinases,
en particulier dans la voie ERK/MAPK dont fait partie la protéine kinase MEK1 en étude.
Les points importants qui seront traités sont : 1) Le rôle des protéines d’échafaudage dans
la spécification, la localisation et le maintien de l’intégrité des voies de signalisation MAP
kinases. 2) L’importance des sites d’amarrage (docking sites), qui sont des éléments
d’interaction protéine-protéine puissants, dans la spécification du signal et l’acheminement
de ce dernier via les modules kinases des voies de signalisation. 3) Enfin, une partie de
l’introduction sera consacrée à l’entrecroisement des signaux (cross-talk) entre les
différents modules MAP kinases ainsi qu’entre les modules MAP kinases et les autres voies
de signalisation intracellulaires et leur rôle décisif dans la spécification de la réponse
cellulaire.

4
1.2. Activation du sentier ERK par des stimuli
extracellulaires
Deux composantes de cette voie, Ras et Raf, sont des proto-oncogènes. Il n’est donc pas
surprenant que la fonction majeure de cette voie soit le contrôle de la croissance cellulaire
dans toutes ses facettes, y compris la prolifération cellulaire, la transformation, la
différenciation et l’apoptose. Une grande variété d'hormones, de facteurs de croissance et
de différenciation, ainsi que de substances tumorigènes, utilisent cette voie.
La voie de signalisation la mieux définie depuis la membrane cellulaire jusqu’au MAP
kinases ERK1 et ERK2 est probablement celle qui est initiée par les récepteurs à activité
tyrosine kinase, RTKs [6]. La stimulation de ces récepteurs par le ligand approprié
provoque une augmentation de l’activité catalytique du récepteur et son
autophosphorylation subséquente sur les résidus tyrosines. La phosphorylation du récepteur
induit la formation de multi complexes protéiques dont l'organisation dicte la progression
du signal en aval. En général, l’étape subséquente est l'activation de la protéine
monomérique G, Ras. La transmission du signal des RTKs vers RAS se fait via le
recrutement d’adaptateurs moléculaires, tels que Shc et Grb2 (growth-factor-receptor-
bound protein 2), au récepteur via des interactions entre leurs domaines SH2 et les résidus
phosphotyrosines, puis du facteur d'échange du nucléotide de la guanine (GEF) et Sos (Son
of Sevenless). Lorsque SOS est engagée dans le complexe, elle induit, chez Ras, l’échange
du GDP pour du GTP. Réciproquement, GTP-Ras interagit directement avec plusieurs
effecteurs, y compris les isoformes de Raf. Ras se lie à Raf et provoque un changement
dans sa conformation native, ce qui augmente son activité kinase ou fournit tout
simplement un environnement adéquat pour son activation [11-14]. La localisation de Raf à
la membrane plasmique permettrait aussi à d’autres protéines kinases telles que Src, PKC,
et PAK de modifier Raf pour augmenter son activité [13,15-18]. L'augmentation dans
l’activité Raf est convertie par la suite via le module MEK-ERK. La phosphorylation par
rétroaction de SOS par ERK provoque le démontage du complexe SOS-Grb2 et
l'inactivation de Ras [19].

5
Récepteur
Protéine G
MAPKKK
MAPKK
MAPK
Kinase
P P P
P P
P P
Substrats cytoplasmiques
Substrats nucléaires
Signal extracellulaireMembrane
Noyau
Cytoplasme
Récepteur
Ras
Raf
MEK
ERK
Kinase
P P P
P P
P P
RSK, MLCK
Elk, SAP
Facteurs de croissance et de différenciation
Membrane
Noyau
Cytoplasme
Map kinase kinase kinase
Map kinase kinase
Map kinase
a) b)
Récepteur
Protéine G
MAPKKK
MAPKK
MAPK
Kinase
P P P
P P
P P
Substrats cytoplasmiques
Substrats nucléaires
Signal extracellulaireMembrane
Noyau
Cytoplasme
Récepteur
Ras
Raf
MEK
ERK
Kinase
P P P
P P
P P
RSK, MLCK
Elk, SAP
Facteurs de croissance et de différenciation
Membrane
Noyau
Cytoplasme
Map kinase kinase kinase
Map kinase kinase
Map kinase
a) b)
Récepteur
Protéine G
MAPKKK
MAPKK
MAPK
Kinase
P P P
P P
P P
Substrats cytoplasmiques
Substrats nucléaires
Signal extracellulaireMembrane
Noyau
Cytoplasme
Récepteur
Ras
Raf
MEK
ERK
Kinase
P P P
P P
P P
RSK, MLCK
Elk, SAP
Facteurs de croissance et de différenciation
Membrane
Noyau
Cytoplasme
Map kinase kinase kinase
Map kinase kinase
Map kinase
Récepteur
Protéine G
MAPKKK
MAPKK
MAPK
Kinase
P P P
P P
P P
Substrats cytoplasmiques
Substrats nucléaires
Signal extracellulaireMembrane
Noyau
Cytoplasme
Récepteur
Protéine G
MAPKKK
MAPKK
MAPK
MAPKKK
MAPKK
MAPK
Kinase
P P P
P P
P P
Substrats cytoplasmiques
Substrats nucléaires
Signal extracellulaireMembrane
Noyau
Cytoplasme
Récepteur
Ras
Raf
MEK
ERK
Raf
MEK
ERK
Kinase
P P P
P P
P P
RSK, MLCK
Elk, SAP
Facteurs de croissance et de différenciation
Membrane
Noyau
Cytoplasme
Map kinase kinase kinase
Map kinase kinase
Map kinase
a) b)
Figure 1-1 : Les composantes kinases de base de la voie ERK/MAP kinase.
a). Schéma général d’un sentier MAP kinase. b). la voie ERK/MAPK. Voir texte pour
détails.

6
L’interaction de certaines intégrines avec la matrice extracellulaire peut également recruter
les mêmes complexes et participer ainsi à l’activation de Ras. Les intégrines ne possédant
pas d’activité kinase, plusieurs mécanismes d’activation de la voie ERK/MAPK peuvent
être envisagés. Un mécanisme complexe impliquant les kinases FAK (Focal adhesion
kinase) et src, qui dans un jeu mutuel de phosphorylation, attire une protéine adaptatrice
telle que Grb2, est vraisemblablement impliqué dans la motilité cellulaire. Autrement, la
signalisation mitogène passerait par le recrutement de l’adaptateur Shc via la protéine
membranaire cavéoline (Cav), la phosphorylation s’effectuant par l’un des membres de la
famille Src [20,21].
La signalisation initiée par les récepteurs couplés aux protéines G (G protein-coupled
receptors ; GPCRs) pour l’activation des ERKs implique aussi la modulation de l’activité
Raf. Cependant, les mécanismes de régulation déployés par ces récepteurs sont très variés.
L'existence de multiples classes de protéines G, la capacité de certains récepteurs d'activer
plus qu'une classe de protéine G ainsi que les mécanismes spécifiques aux types cellulaires
contribuent à la diversité [22-24].
Les signaux transmis par les récepteurs via les petites protéines G hétérotrimèriques Gαs
sont particulièrement divers. L’exemple le plus concret de cela serait la variété d'effets sur
l’activité ERK observée suite à l’élévation de la concentration intracellulaire de l’AMP
cyclique (AMPc). La kinase dépendante de l’AMPc, PKA (protein kinase A), a été
rapportée comme étant un inhibiteur de l'activité Raf-1. Dans certains contextes, PKA
phosphoryle directement Raf-1 sur les résidus sérine 43 et sérine 621 [25-27]. Dans
d’autres cas, par exemple chez les cellules d'origine neuronale, PKA phosphoryle la
GTPase Rap1a qui peut influencer positivement ERK via l’activation de B-Raf [28,29].
Dans le cas de l’activation de ERK via Gαi, les sous-unités βγ libres peuvent être les
transducteurs actifs du signal. Cela a été démontré dans la signalisation du mating chez la
levure [30]. Des études de surexpression ont montré que les sous unités βγ seraient
suffisantes pour activer ERKs et que l’inactivation des sous-unités βγ réduirait la
stimulation de ERK par Gαi [31,32]. Dans un modèle proposé, βγ stimulerait l’activité
kinase de Src d’une manière dépendante de PI-3 kinase et de la sous unité γ [33]. Src

7
phosphorylerait alors un récepteur RTK ou une protéine kinase telle que PYK2 ou FAK,
pour créer un motif de liaison SH2 pour un adaptateur moléculaire [34-36]. Dans un autre
modèle, l’activation de la voie ERK/MAPK serait faible chez certaines cellules qui
expriment PI-3 kinase, ce qui suggère une alternative au mécanisme d'activation via Src ou
PYK2 [34,37].
L'activation de ERK2 via Gαq est souvent un processus dépendant de PKC. Elle peut être
dépendante ou indépendante de Ras [35,38]. L’effecteur direct de G αq est le PLCβ qui
produit l'inositol- triphosphate (IP3) et le diacylglycérol (DAG) via le clivage du
phosphatidylinositol 4,5-bisphosphate (PIP2). Quelques isoformes de PKC sont activés par
le DAG et le Calcium intracellulaire libéré à la suite de la production de IP3. PKC
régulerait alors Raf par phosphorylation directe [18]. PKC régulerait aussi MEK, mais le
mécanisme est encore pas clair [39]. Chez les cellules PC12, la stimulation de Gαq par ses
récepteurs induit la phosphorylation de PYK2 et de Shc.
1.3. Les composantes kinases du module de signalisation
ERK/MAPK
1.3.1 Les isoformes Raf
En dépit de la conservation de Ras et autres MAPKKKs, MAPKKs et MAPKs, il n'y a
aucun homologue de Raf dans la levure Saccharomyces cerevisiae. Toutefois, les
homologues de Ras chez la levure ne font pas partie des cascades MAPKs, mais sont
présents dans les voies nutritives qui régulent l’adénylate cyclase. Par ailleurs, la mouche à
fruits Drosophila melanogaster et le ver Caenorhabditis elegans possèdent chacun au
moins un gène raf fonctionnel, dont le produit est présent en aval de Ras dans le sentier
ERK de façon similaire à celle des cellules de mammifères. Chez la Drosophile, Raf est
essentiel pour la prolifération et la différenciation cellulaires pendant le développement
[40].

8
Figure 1-2: La transmission du signal extracellulaire via la voie de signalisation
ERK/MAPK et les multiples molécules signalétiques qui régulent sa progression.
MEK1
Raf-1B - Raf
PKA Ras
Rap
ERK1,2
Akt/PKB
Pak
PI - 3K
cAMP SOS
PKC
Ca2+
He - PTP PTP - SL
Rac
αβ
Facteurs de transcription
MKPNoyau
Membrane
Cytoplasme
RTK PGCRIntegrin
+
-
-
-
+
+
+
+
+
-+
+
+
+ +
+ +++
-+
++
+
+
CavFak - 1
MEK1
B - Raf
PKA Ras
Rap
ERK1,2
Akt/PKB
Pak
PI - 3K
cAMP SOS
PKC
Ca2+
He - PTP PTP - SL
Rac
αβ
Facteurs de transcription
MKPNoyau
Membrane
Cytoplasme
RTK PGCRIntegrin
+
-
-
-
+
+
+
+
+
-+
+
+
+ +
+ +++
-+
++
+
+
CavFak - 1

9
De toutes les MAPKKKs connues, les isoformes Rafs et Mos sont peut-être les seules qui
phosphorylent les MAPKKs dans une cascade unique. En effet, ces protéines semblent
phosphoryler seulement deux membres de famille MEK, MEK1 et MEK2, et se placent
exclusivement dans la cascade ERK/MAPK [41,42]. La famille des protéines kinases Raf
est composée de A-Raf, B-Raf, et Raf-1 (ou c-Raf) [43]. Chaque isoforme contient trois
régions conservées appelées, CR1, CR2, et CR3. Les deux premières régions sont
localisées en amino-terminal de la protéine et sont impliquées dans la régulation du
domaine catalytique. Leur suppression crée un mutant Raf-1 possédant une activité
constitutive élevée ou encore pouvant être activée indépendamment de Ras [44,45]. Le
domaine kinase est localisé dans la région CR3. L’expression de Raf-1 est ubiquitaire. La
plus haute expression de B-Raf se produit dans les tissus neuronaux et dans les testicules.
Enfin A-Raf semble s’exprimer primordialement dans les tissus urogénitaux.
Les phénotypes très différents chez les souris mutantes Raf-1, A-Raf et B-Raf démontrent
d’une façon claire et persuasive que ces protéines ne sont pas redondantes et servent des
fonctions distinctes [46]. Chez l’espèce canine, et selon l'origine génétique, l'élimination de
A-Raf produit des défauts intestinaux et/ou neurologiques, mais les chiots naissent vivants.
Par contre, les souris mutantes pour le gène B-raf présentent des défauts dans la
différenciation des cellules neuro-épitheliales et dans la maturation des cellules
endothéliales, et meurent in utero dû à une hémorragie vasculaire.
La suppression du gène raf-1 chez des souris inbred provoque la mort à la mi-gestation.
Les souris Raf-1 outbred meurent très tôt après la naissance et présentent un retard dans la
croissance et des défauts dans le développement qui sont très apparents dans le placenta, les
poumons et la peau. À noter que les souris sans fonction Raf-1 expriment encore une
protéine Raf-1 tronquée qui est dépourvue de l’activité catalytique, mais qui peut
néanmoins contribuer aux phénotypes anormaux observés en agissant comme un mutant
dominant négatif. En résumé, Raf-1 semble jouer un rôle essentiel dans la prolifération
cellulaire, alors que A-Raf et B-Raf réaliseraient plus de fonctions spécialisées.
La régulation de Raf-1 est complexe. Elle comprend notamment des interactions protéine-
protéine, l’implication de plusieurs sites de phosphorylation dans Raf-1 et une localisation

10
cellulaire variable [43]. Ces multiples modes de régulation permettent à Raf-1 de fluctuer à
travers des états d’activation graduels. Raf fait partie d'un complexe multi protéique
composé de Raf-1 ou B-Raf, de la protéine du choc thermique hsp90, de p50cdc37, et un
nombre indéterminé de protéines 14-3-3 [47-52]. 14-3-3 semble stabiliser Raf-1 dans ses
différentes conformations actives en fonction du degré de phosphorylation de Raf et de ses
interactions avec d’autres protéines régulatrices telles que Ras-GTP. 14-3-3 servirait dans la
spécification du signal transmis via Raf-1 en recrutant ce dernier à des complexes
protéiques hautement agencés [48]. La dissociation du regroupement hsp90-p50 de Raf, par
l'usage d'agents pharmacologiques tels que la geldanamycine et la dexaméthasone ou de
mutants de p50 incapables de lier hsp90, interrompt la signalisation dépendante de Raf
[51,53,54]. La geldanamycine n'affecterait pas la capacité de Raf à former des complexes
avec son activateur Ras et ne réduirait pas son activité qui est stimulée par le facteur
mitogénique EGF. La co-expression de p50 avec Raf chez les cellules Sf9 augmente
l'activité de Raf et potentialise son activation par v-src.
Il y a des différences considérables dans la régulation des divers isoformes de Raf. Raf-1 et
B-Raf sont régulés différemment par les petites protéines G, Ras et Rap1a [28,55-57]. Raf-
1 est activée par H -, K-, et N-Ras [58]. Bien que Raf-1 puisse aussi interagir avec Rap1a,
la fonction de cette interaction n’est pas claire, parce qu'aucune augmentation dans son
activité n’est observée. En revanche, B-Raf est activée aussi bien par Ras que par Rap.
Chez les cellules PC12, l’activation de B-Raf par Rap1 semble être le dispositif dominant.
Cette différence fonctionnelle a été attribuée aux domaines riches en cystéine (CRDs) de
ces protéines. L’échange des domaines CRDs de Raf-1 et B-Raf provoque l'activation de
Raf-1 par Rap1 et inhibe l’activation de B-Raf [59].
L’état de phosphorylation de Raf-1 est influencé par de multiples protéines kinases, y
compris Src, les membres de la famille de la protéine kinase C (PKC), p21 PAK (la
protéine kinase activée par Rac/Cdc42), et Akt (aussi appelé la protéine kinase B ou PKB).
Les sites de phosphorylation par PAK et Src sont localisés au N-terminal du domaine
catalytique, à savoir, la sérine 338 et la tyrosine 340 pour PAK et la tyrosine 341 pour Src
[17,60]. Lorsque phosphorylés, ces sites peuvent potentiellement stimuler l'activité d’une
manière interactive, selon le contexte du signal [13,16,61,62]. Les résidus de la boucle

11
d'activation, les sérines 497 et 499, semblent être des sites de phosphorylation possible par
PKC [18], cependant, la mutation de ces sites n'a aucun impact perceptible sur la
stimulation de Raf par le sérum [41]. Wolfman et ses collègues ont récemment trouvé que
PKCε forme un complexe stable avec Raf-1 et phosphoryle la sérine 338, le même site
modifié par PAK [15,18]. L’inactivation de PKCε bloque l'activation de Raf-1 par l'ester de
phorbol, mais n'a pas d’effet sur l’activation par EGF. Ces lignes d’évidence, parmi
d’autres, démontrent l’existence de divers mécanismes indépendants pour l’activation de
Raf-1.
Akt semble phosphoryler Raf-1 sur le résidu sérine 259 chez les cellules cancéreuses du
sein, MCF-7 [63]. Ce résidu est un site présumé d’interaction de 14-3-3 [64,65]. Lorsque
lié à ce site, 14-3-3 a un effet inhibiteur sur l’activité de Raf-1. In vivo, la mutation de cette
sérine en alanine crée un mutant actif de Raf-1 [60,66]. L’inactivation de ERK1/2 chez les
cellules C2C12 cultivée en présence de sérum peut stimuler les premiers stades de
différenciation du myotube [67]. De même, Akt peut réduire l'activité Raf-1 dans plusieurs
contextes, notament, lors de la différenciation des myoblastes C2C12 [68]. Le site de
phosphorylation de Raf-1 par Akt chez les cellules C2C12 n'a pas encore été cartographié.
Par contre, dans le cas des myotubes différenciées traitées par le facteur de croissance IGF-
I (insulin-like growth factor-I), il y a diminution du niveau de phosphorylation de la sérine
338 lorsque qu’un mutant dominant positif de Akt est exprimé. Ces modes de régulation ne
sont en revanche pas exclusives. La capacité de Akt d'inhiber l'activité Raf-1 peut varier
selon le type cellulaire. Il est aussi intéressant de noter que, chez les cellules C2C12, une
association Akt-Raf-1 se produit seulement pendant la différenciation et est dépendante de
l’activité kinase de Akt. Alors que chez les cellules MCF-7, l'association des deux protéines
paraît être constitutive. Des études futures pourraient déterminer le rôle médiateur de Akt
dans la régulation négative de Raf-1 pendant les processus physiologiques.
Il a été démontré que les inhibiteurs chimiques de l’activité de Raf, ZM336372 et
SB203580, sont incapables de bloquer l'activation de MEK et de ERK par les facteurs de
croissance et les esters de phorbol. Alors que ces inhibiteurs réduisent l'activité Raf in vitro,
ils induisent paradoxalement une activation vigoureuse de Raf-1 lorsque administré aux

12
cellules [69,70]. Une explication possible pour ce phénomène est que cette activité Raf-1
est contrôlée normalement par une autorégulation négative. En empêchant cette régulation,
les inhibiteurs induisent une activation massive de Raf-1 [69,70]. Raf paraît donc être
suspendu dans une balance entre activation et autoinhibition.
La nature de l’autorégulation négative n'est pas encore bien connue, mais elle préconise des
conséquences importantes. Elle jette tout doute sur l'utilité des inhibiteurs de Raf comme
traitement anticancéreux. Elle montre que l'activation de Raf-1 peut être efficacement
accompli en ôtant une contrainte inhibitrice. Il n’est cependant pas clair si ce principe est
utilisé physiologiquement. Enfin, elle suggère que l'attachement de Raf à MEK est un
processus régulé. En dépit de l'hyperactivation de Raf-1, les drogues ne stimulent pas
l’activité de MEK ni celle de ERK, indiquant qu’une association efficace à MEK
nécessiterait plus que l'activité catalytique de Raf. Comme il sera discuté ci-dessous,
l'interface Raf-MEK est en effet utilisée comme moyen de régulation. Raf peut activer
MEK1 et MEK2 avec une efficacité semblable in vitro. Cependant, quelques modèles
génétiques et expériences de transfection suggèrent une interaction préférentielle entre
certains Raf et isoformes de MEK [71,72]. La signification de cela est encore inconnue,
parce que les deux isoformes MEK activent, en aval, les mêmes kinases ERKs.
1.3.2. MEK1 et MEK2
MEK1 et MEK2 font partie de la rare famille de protéines kinases à double spécificité
capables de phosphoryler aussi bien les résidus sérines et thréonines que tyrosines. Bien
que ces deux isoformes de MEK soient fortement homologues dans leur séquence primaire
d'acides aminés, et bien qu'elles génèrent des réponses transcriptionnelles et
morphologiques semblables dans les fibroblastes NIH3T3, elles semblent être
différemment régulés par leurs activateurs ascendants RAF-1, A-RAF, et B-RAF dans
divers types de cellules et en réponse à différents stimuli extracellulaires. A-RAF active
préférentiellement MEK1 [73], tandis que c-Raf-1 active aussi bien MEK1 que MEK2. B-
RAF se lie à MEK1 et à MEK2 mais semble activer MEK1 mieux que MEK2 [74].

13
Toutefois, cette spécificité est, dans la majorité des cas, une conséquence de variations
quantitatives dans les affinités des interactions activateur/substrat et peut être perdue quand
les associés sont surexprimés [71]. Ceci dit, une étude récente suggère que ces deux
protéines MEK contribuent de manières distinctes à la régulation de l’activité ERK et à la
progression G1/S du cycle cellulaire [75]. Dans une autre étude, il a été suggéré que le
module MEK2-ERK2 inhibe la transition G1/S du cycle celluaire, alors que le module
MEK1-ERK1 inhible celle du G2/M [76]. MEK1 et MEK2 auraient un impact different sur
l’intensité et la durée de la signalisation ERK contribuant distinctivement à l’inhibition ou
l’induction de la prolifération cellulaire. L’activation spécifique de MEK1 ou de MEK2
pourrait donc médier des réponses biologiques différentes dépendamment de la nature du
signal extracellulaire et du contexte cellulaire.
Les analyses histopathologiques d’une lignée de souris mutante dans laquelle le gène mek1
a été perturbé par mutagenèse d'insertion montrent une réduction de la vascularisation du
placenta. Cette anomalie serait due à l’incapacité des cellules endothéliales vasculaires
d’envahir la région du labyrinthe [77]. En outre, les essais de migration cellulaire ont
indiqué que la migration des fibroblastes embryonnaires (MEF) MEK1-/- est altérée lorsque
ces derniers sont étalés sur une matrice de fibronectine. Les niveaux d’expression et
d’activation de MEK2 et de ERK1/2 lors de ces essais semblaient être normaux. La
réexpression de MEK1 dans les MEFs mutants reconstitue leur capacité à migrer (Charron
et collaborateurs, travaux en cours). Ces résultats suggèrent que MEK1 est nécessaire pour
une réponse adéquate aux signaux angiogéniques qui pourraient favoriser la vascularisation
de la région du labyrinthe du placenta [77]. MEK2 ne semble pas être impliquée dans la
maturation placentaire chez la souris. En fait, les souris mutantes pour le gène mek2 sont
viables et fertiles et ne démontrent aucune anomalie placentaire [78].
Les MAPKKKs de la famille Raf ne sont pas les seuls activateurs de MEK1/2. En effet,
MEKK1, Tpl-2 et Mos, semblent phosphoryler et activer MEK1 [79,79-83]. L’activation
du module MEK-ERK par MEKK1 semble jouer un rôle dans la progression du signal
apoptotique médié par les récepteurs Fas. L’activation par Mos a été décrite comme
essentielle à la maturation des ovocytes [84]. Enfin, le rôle de Tpl-2 dans l’activation de la

14
voie ERK n’est pas tout à fait caractérisé. Néanmoins, tout comme Raf et Mos, Tpl-2
semble jouer un rôle dans la différenciation des cellules PC12 [85].
MEK1 et MEK2 sont phosphorylées et activées par phosphorylation directe sur leurs
résidus sérines de la séquence 218SMANS222 présente dans la boucle d’activation. La
phosphorylation de MEK1 sur les deux sites stimulerait son activité par plus que 7,000 fois.
La phosphorylation d’un seul site produit une augmentation moins considérable dans
l’activité [86]. La substitution des deux sites de phosphorylation par des résidus acides
augmente aussi leur activité. De même, la délétion d’une séquence d’acides aminés dans la
région N-terminale accroît encore plus leur activité basale. La combinaison de ces deux
modifications crée des mutants MEK1/2 constitutifs aussi actifs que les protéines de type
sauvage activées [86]. Ces mutants MEK, nommés MEK1R4F, ont été utilisés dans
beaucoup de systèmes pour inférer des événements associés exclusivement à la cascade
ERK [87,88].
MEK1 et MEK2 sont en mesure de phosphoryler et activer ERK1 et ERK2 [89-93]. Ces
MAPKs semblent être les seuls substrats connus de MEK1/2. Les deux MEKs
phosphorylent ERK1/2 sur les résidus tyrosine et thréonine qui se situent dans le segment
d’activation du domaine catalytique (appelé encore boucle d’activation). Cette
phosphorylation est suffisante pour activer ERK1/2 in vitro, comme in vivo [94,95]. À la
suite de la double phosphorylation, l’activité de ERK1/2 augmente par bien plus de 1000
fois avec une activité spécifique de 1-2 mmol/min/mg. Le plus grand effet paraît être dû à
une augmentation du Vmax, la variation du Km pour le substrat étant très faible [96,97]. La
stœchiométrie de phosphorylation de ERK1/2 par MEK2 approche plus aisément les 2 mol
de phosphate/mol de ERK que ne fait la phosphorylation par MEK1.
1.3.2.1 Domaines d’interaction dans MEK1/MEK2
En dehors du domaine catalytique typique conservé chez toutes les protéines kinases des
voies MAP kinases, MEK1 et MEK2 présentent deux éléments nécessaires à leur activité.
Le premier est unique à ces deux MEKs et est communément appelé MSS (MEK specific
sequence). Le deuxième, EDS (ERK docking site), semble être conservé chez tous les

15
membres de la famille MEK. En outre, ces deux éléments représentent aussi le point de
divergence entre les deux kinases. En effet, l’alignement de séquence de MEK1 et MEK2
indique que ces deux protéines sont quasiment identiques dans leurs domaines kinase mais
sont particulièrement différentes au niveau de ces deux éléments (figure 1-3). Cette
variation au niveau de ces deux séquences impliquées dans l’activité suggère une
divergence possible dans le rôle de MEK1 et de MEK2 dans la signalisation chez les
mammifères. En fait, plusieurs observations ont montré que MEK1 et MEK2 peuvent avoir
des fonctions distinctes menant à des réponses physiologiques uniques. MEK1 semble être
activée dans les macrophages et les cellules Swiss 3T3 en réponse au TNF-α et à la
bombésine respectivement [98,99]. D’autre part, MEK2 est spécifiquement activée par le
lactosylcéramide chez les cellules du muscle lisse de l’aorte humaine [100]. MEK2 peut
aussi affecter, d’une façon spécifique, la cinétique du point de contrôle G2/M du cycle
cellulaire [101].
1.3.2.1.1. La région MSS
La région MSS (MEK specific sequence) est un élément unique présent chez les MEKs
dans la voie ERK/MAPK. Cette région riche en proline est insérée entre les domaines
kinases IX et X des protéines MEK1 et MEK2 [71,102,103]. Aucun autre membre connu
de la famille MAPKK ne possède ce domaine spécifique. Du fait de la présence de
plusieurs résidus prolines dans cet élément, certains lui ont proposé tout simplement
l’appellation de séquence riche en proline ou PS (proline-rich sequence). Les études de
délétions ont indiqué que ce domaine est requis pour la stimulation de ERK, bien que son
absence n'ait in vitro aucun effet sur l’activité catalytique de MEK1 [71,103]. L'expression
d'un peptide qui comprend le domaine MSS inhibe l’activation de ERK2 par EGF,
suggérant que cette région est importante pour la progression du signal via cette cascade.
Le domaine MSS semble jouer un rôle essentiel dans la modulation de l’activité de MEK1
ainsi que dans le cross-talk avec d’autres voies de signalisation. En fait, cette région semble
être le site d’interaction de MEK1/2 avec les membres de la famille Raf et est importante à
leur activation subséquente [71,102]. Elle contient de multiples sites d’interaction

16
potentiels pour les domaines SH3 et est phosphorylée par plusieurs protéines kinases
[80,104-106].
a.a 200 300 3931001
EDS NES MSS
EDS NES MSS
66% 90% 36%
S218 / S222
S222 / S226
MEK1
MEK2
a.a 200 300 3931001
EDS NES MSS
EDS NES MSS
66% 90% 36%
S218 / S222
S222 / S226
MEK1
MEK2
Figure 1-3 : Les protéines MEK1 et MEK2. Structure et comparaison.
Les pourcentages représentent l’homologie de séquence de la région indiquée. Par exemple,
il y’a 66% d’homologie au niveau de la région EDS et seulement 36% au niveau de la
région MSS. Les deux protéines sont, dans l’ensemble, homologues à 90%. EDS : Site
d’attachement de ERK. NES : Signal d’exclusion nucléaire. MSS : Séquence spécifique à
MEK1/2. Les sites de phosphorylation indiqués sont ceux qui se retrouvent dans la boucle
d’activation et qui sont donc nécessaires à l’activation de la kinase.

17
1.3.2.1.2. Le domaine D
L’élément qui est conservé chez la famille des MAPK kinases est un site d’interaction
stable pour les MAP kinases, ERK1/2. Il est localisé à l’extrémité N-terminale de MEK1 et
MEK2, dans une courte région riche en acides aminés basiques. Cette séquence possède
toutes les caractéristiques d'un domaine de liaison au substrat. Ce domaine, appelé domaine
D, semble être spécifique aux MAP kinases. Dans MEK1 et MEK2, ce domaine est aussi
nommé EDS pour ERK docking site. Une liste exhaustive des protéines ayant ce domaine D
ont été présentés par Nishida et collaborateurs [107] et par d’autres groupes de recherche
[108,109]. Ce site d’interaction dans MEK1 est requis aussi bien in vitro que chez les
cellules pour l’activation de ERK2. Un mutant de délétion MEK1 dépourvu de la séquence
N-terminale comprenant le domaine D perturbe l’activation de ERK2 par EGF [108].
Lorsque introduit chez les cellules, un peptide dérivé du N-terminal de MEK1 inhibe la
progression du cycle cellulaire [110]. Le facteur mortel de l’Anthrax (ALF) clive le
domaine D de MEK et inhibe l'activation de ERK [111]. En utilisant la mutagenèse et les
études de délétions, le site de liaison au domaine D sur ERK1/2 a été localisé à une paire de
résidu aspartate dans le C-terminale de ERK2, juste à extérieur du domaine catalytique
(discuté plus en détails ultérieurement)[107] et [112].
1.3.2.2. Autres sites importants dans MEK1
1.3.2.2.1. La séquence NES
L’implication de ERK dans la phosphorylation et la régulation d'un grand nombre de
facteur nucléaire suggère que la redistribution de ERK du cytoplasme aux compartiments
nucléaires est nécessaire pour la signalisation. En outre, la localisation nucléaire stable de
ERK a été rapportée pour la différenciation des cellules PC12, un processus nécessitant la
phosphorylation de facteurs nucléaires impliqués dans l’expression de gènes neuronaux
[113].

18
Les premières études faites sur les MEKs ne montraient pas une translocation semblable de
ces protéines losque la voie ERK est activée [114,115]. Ceci a été rectifié plus tard par la
découverte d'une séquence riche en leucine, située en N-terminal de ces protéines. Cette
séquence comprise entre les résidus 32 et 42 dans MEK1 (ALQKKLEELEL) se comporte
comme un signal d'exportation nucléaire (NES) [116]. La délétion ou la mutation du NES
contribuent à la localisation nucléaire constitutive de MEK1. L’ajout d’un peptide
contenant la séquence NES à des protéines hétérologues facilite leur exportation nucléaire.
Les séquences de ce type se sont avérées être des sites de reconnaissance pour lier
Crm1/exportin 1, une protéine nucléaire qui facilite l'exportation des composants cellulaires
y compris PKIα, IκB, Rev, et Dsk1, en plus de MEK1 [117,118]. La leptomycine B, qui est
un antibiotique anti-fongique, lie Crm1 et s’interfère dans son association avec les protéines
ayant le NES et bloque ainsi leur exportation [118-121]. En outre, MEK1 migre vers le
noyau des cellules COS après seulement une heure de traitement avec la leptomycine B
[119], suggérant que, bien que la localisation nucléaire de MEK1 n'est pas stable, son
importation est active avec un taux rapide d'afflux et de reflux.
L’importation nucléaire des mutants MEKs manquant le NES semble se produire en
réponse au traitement par le sérum. Chez les cellules COS ou HEK293, un mutant MEK1
(∆-NES, K97A, et S218E/S222E) est principalement cytosolique mais il est relocalisé dans
le noyau après 10 minutes de traitement au sérum [122]. Ceci suggère que les mécanismes
pour l'importation nucléaire de MEK sont régulés soit par l'importation accrue ou par
l’inhibition de l'exportation. De tels mécanismes exigeraient l'activation des voies de
signalisation, de telle sorte que la phosphorylation et l'activation de MEK pourraient être
les conditions importantes pour stabiliser sa distribution nucléaire.
Deux mécanismes différents pour la translocation nucléaire de ERK ont été proposés. L’un
impliquerait une navette constante et non réglée de MEK entre le noyau et le cytoplasme
aussi bien avant et après la stimulation par des mitogènes. L'autre mécanisme semble exiger
l'activité de MEK et est facilité par la réponse aux stimulations mitogéniques [123].
L’importance de l’interaction ERK avec MEK dans la localisation subcellulaire de ERK a
été adressée en utilisant des mutants MEK1, des peptides dérivés de MEK1, et des

19
protéines chimériques ERK2-MEK1. Plusieurs études de localisation subcellulaire ont
démontré que MEK1 serait essentiellement une protéine cytoplasmique [114,115,124].
MEK1 activée peut se retrouver dans le noyau mais elle y est rapidement exporté grâce à sa
séquence NES [116]. Un peptide MEK1 qui contient le domaine D et le NES cause la
rétention de ERK2 dans le cytoplasme [125]. En outre, une protéine ERK2-MEK1 est
exclue du noyau si le NES de MEK1 est intact, mais s'accumule au noyau si les résidus
leucines dans le NES sont mutés en alanines [126].
1.3.2.2.2. Le site d’attache de PD 184352
Un rôle majeur a été attribué à MEK1 dans le développement des tumeurs. En effet, il a été
rapporté qu’une petite molécule qui inhibe l’activité de MEK, le PD 184352, est capable
d'empêcher la croissance de tumeurs jusqu'à 80% chez les souris porteuses de carcinomes
du colon d'origine humaine ou provenant de souris [127]. Le composé est sélectif et est non
compétitif ni pour la fixation de l’ATP ni pour l’interaction avec la MAP kinase ERK
[127]. PD 184352 peut aussi bien bloquer l'activation de MEK par RAF qu'inhiber la forme
active de MEK (phosphorylée par RAF ou mutée). Cependant, dans une étude récente, le
groupe de Dudley a montré que le PD 184352 ne bloque pas la phosphorylation de MEK et
ce aussi bien in vitro qu’in vivo [128].
Les inhibiteurs rapportés de MEK1 ; PD 98059, PD 184352, et U0126, se sont avérés
particulièrement spécifiques. Ils n’affectent pas les membres relatifs de famille de
MAPKK ; MKK3, MKK4, MKK6, ou MKK7, mais ils inhibent MEK2 [127,129].
Récemment, ces composés ont été rapportés efficaces, quoique plus faibles, contre l'activité
MKK5 [130,131]. Parmi les membres de la famille MAPKK, MKK5 montre l'homologie la
plus étroite avec MEK1 et à MEK2, avec 46% d’identité en acides aminés dans le domaine
catalytique [131].
Le site d’attachement de PD 184352 se trouve entre les domaines kinases III et IV de
MEK1 et MEK2 [128]. Cette région héberge la lysine 97 impliquée dans la fixation de
l’ATP et l’acide glutamique 114 qui est conservé chez tous les protéines kinases et qui est
essentiel pour la stabilité structurale de la protéine [132]. Des études par mutagenèse

20
ponctuée ont permis d’identifier plusieurs résidus importants dans l’attachement de
PD184352 à MEK1. Ces mutations n’affecteraient pas l'activité kinase et n’interféraient pas
dans la capacité de MEK à lier l’ATP. Par contre, tous les mutants MEK1 présentent une
augmentation substantielle de l’activité basale [128].
Une explication possible pour l’augmentation de l’activité de base des mutants est qu'une
conformation plus active serait obtenue par modification de l’emplacement des sous-
domaines dans le domaine kinase. Les structures en trois dimensions de plusieurs protéines
kinases ont indiqué que l'activité catalytique de ces enzymes implique deux changements
primaires dans la conformation [133]. Le premier se produit en raison de la
phosphorylation des résidus qui se trouvent dans la boucle d'activation de la kinase. Cette
phosphorylation aide à stabiliser la kinase sous sa forme active. Le second se produit alors
que les deux lobes que comporte le domaine kinase pivotent l'un par rapport à l'autre pour
fermer la fente catalytique et positionner les résidus essentiels dans une conformation
favorisant la catalyse [133]. Cependant, d'autres études ont indiqué qu’un mouvement
dynamique subtil impliquant diverses structures secondaires peut également être important
pour la catalyse. Précisément, le groupe de Ahn a prouvé par essai d’échange de deutérium
qu’un mouvement et une flexibilité dynamique significative sont observés chez la forme
activée de MEK1, tout particulièrement dans le lobe N-terminal du domaine kinase qui
comprend la région d’attachement du PD 184352 [134].
Enfin, l’inhibiteur PD 184352 pourrait fermer l'enzyme sur une conformation inactive,
empêchant la catalyse tout en permettant la phosphorylation de la Ser218 et de la Ser222 de
la boucle d'activation. Cette conformation pourrait interférer avec la reconnaissance par les
phosphatases spécifiques à MEK1 et provoquer l’hyperphosphorylation de MEK1 qui est
observée chez les cellules KBALB traitées avec le PD184352 [135].
1.3.2.2.3. La région C-terminale
L’extrémité C-terminale de MEK1 se prolonge par environ 40 acides aminés au delà d'une
arginine conservée (Arg349) dans le domaine XI, qui marque la fin du corps kinase [132].
L'importance fonctionnelle de la séquence se trouvant au delà du domaine XI a été explorée

21
en construisant des mutants tronqués C373, C367, C363, C358 ou C343 [86]. L’activité
basale de ces protéines semblait être comparable à celle de la protéine type sauvage,
indiquant que l’activité catalytique est intacte chez ces mutants. Cependant, la
phosphorylation par v-Mos immunoprécipitée n'active pas ces mutants de manière
significative. La troncation C373 dans MEK1R4F qui est constitutivement active produit
une protéine avec seulement 10% de l'activité initiale de MEK1R4F [86]. La région C-
terminale semble être nécessaire pour l'activation de l’enzyme. D'autre part, la région C-
terminale de MEK1 semble être nécessaire pour l’activation de la voie ERK/MAPK et la
survie cellulaire. En effet, Cha et co-auteurs ont indiqué que cette région, et tout
particulièrement la séquence comprise entre les acides aminés 330 et 379, serait importante
pour la localisation adéquate de MEK1 dans le cytoplasme [136]. Sa délétion entraine la
relocalisation de MEK1 dans les compartiments endosomales/lysosomales et provoquer la
mort cellulaire par apoptose. De plus, ces auteurs ont indiqué que la séquence 330-379 est
importante pour l’activation de MEK1 par Raf-1 et qu’elle est donc essentielle pour
l’activation de la voie ERK/MAPK [136].
1.3.3. ERK1 et ERK2
Plusieurs essais biochimiques et expériences de transfection suggèrent que ERK1 et ERK2
sont fonctionnellement équivalentes, à se demander pourquoi existerait-il deux gènes ERK?
Toutefois, le fait que MEK et les isoformes ERK sont habituellement co-exprimées, et la
présence, chez tous les mammifères, de deux gènes chacun pour MEK1/2 et ERK1/2 paraît
indiquer une diversification essentielle. ERK est une sérine/thréonine kinase avec un
portefeuille impressionnant de plus de 50 substrats, ce qui la met clairement à
l’embouchure finale de cette voie. Cependant, cela n'exclut pas l'existence de points
d’embranchement au niveau de Raf ou de MEK.
ERK1 et ERK2 sont des protéines de 43 et 41 kDa qui sont identiques à presque 85%, avec
de plus grande identité dans les régions centrales impliquées dans la liaison des substrats.

22
Les deux sites phosphoaccepteurs, tyrosine et thréonine, dont la phosphorylation est
essentielle pour l’activation, sont séparés par un résidu glutamate dans ERK1 et ERK2 pour
donner motif TEY dans la boucle d'activation [137]. Le remplacement des deux sites de
phosphorylation dans ERK2 par des résidus acides n'élève pas l'activité de la protéine
[138].
Les deux kinases sont exprimées partout, bien que leur abondance relative dans les tissus
soit variable. Par exemple, ERK2 est la forme prédominante chez beaucoup de cellules du
système immunitaire, alors que chez plusieurs cellules d'origine neuroendocrinienne, les
deux formes s’expriment également. Elles sont stimulées dans une certaine mesure par un
vaste nombre de ligands et perturbations cellulaires, avec quelques spécificités liées au type
cellulaire [23]. Chez les fibroblastes (le type cellulaire dans lequel leurs activités et
fonctions ont été développées), elles sont activées par le sérum, les facteurs de croissance,
les cytokines, certains stress mais aussi par les ligands des récepteurs couplés aux protéines
G et agents transformant pour ne citer que quelques-uns. Elles sont fortement exprimées
dans les neurones post-mitotiques et autres cellules hautement différenciées [139]. Chez ces
cellules, elles sont souvent impliquées dans des réponses adaptatives telles que la
potentialisation à long terme [140].
Un variant d’épissage de ERK1, ERK1b, a été identifié comme étant une bande
immunoréactive qui migre plus lentement que la forme ubiquiste de ERK1 [141]. ERK1b
correspondrait originairement à l’isoforme nommée ERK4 [142]. Un épissage alternatif de
erk2, tronquée de quelques résidus en N-terminal, a aussi été rapporté. Des essais de
surexpression ont suggéré que cette forme de ERK2 est localisée essentiellement à la
membrane plasmique [143]. Les structures tridimensionnelles de ERK2 native ou
phosphorylée ont été déterminées.

23
1.4. Autres voies de signalisation MAP kinases chez les
mammifères
1.4.1. Le sentier JNK/SAPK
Une première forme de JNK/SAPK (c-Jun N-terminal kinase/stress-activated protein
kinase), une kinase qui active la protéine MBP, a été purifiée de foies de rats traités à la
cycloheximide [144]. Bientôt, deux formes de JNK/SAPKs de 46 et 54 kDa sont purifiées
par adsorption à une protéine chimérique c-Jun [145]. L'isolement de plusieurs ADNc qui
codent pour ces enzymes et l’analyse subséquente de leur niveau d’expression ont révélé
trois gènes qui codent pour des protéines dont pas moins de 10 formes épissées [146-149].
Les domaines catalytiques de JNK1/SAPKγ, JNK2/SAPKα, et JNK3/SAPKβ sont à 85%
identiques. Les JNK/SAPKs sont activées par phosphorylation sur deux sites, tyrosine et
thréonine, comme tous les autres MAP kinases [149]. Ces résidus sont séparés par un
résidu proline pour donner un motif TPY à l’intérieur de la boucle d'activation. Les
JNK/SAPKs sont activés par les cytokines, certains ligands pour les GPCRs, des agents qui
perturbent la synthèse de l’ADN et des protéines, le stress et dans une certaine mesure par
le sérum, des facteurs de croissance ainsi que des agents transformants [3].
Deux membres de famille MAPKK, MKK4 (SEK1, MEK4, JNKK1, SKK1) et MKK7
(MEK7, JNKK2, SKK4), ont été impliqués dans les voies JNK/SAPK. Ces deux enzymes
ont été identifiées initialement par stratégie de clonage d’ADNc [3,150-157]. L’approche
du système double hybride a identifié MKK7 comme un partenaire de MEK1, bien que la
signification de leur association ne soit pas connue. MKK4 et MKK7 phosphorylent les
membres de la famille de p38 in vitro lorsque surexprimées, bien que JNK/SAPKs soient
les substrats préférés [158]. L’activation de JNK/SAPK est affaiblie chez les cellules
animales dans lesquels l’expression du gène MKK4 a été interrompue, mais les variations
dans l’activation de p38 sont dépendantes du type cellulaire [159]. Le fait que MKK4 agit
exclusivement dans les cascades de JNK/SAPK reste une question ouverte. Les
JNK/SAPKs sont toujours activées par certains stimuli chez les cellules MKK4-/-, indiquant

24
que MKK7 est aussi lié à la cascade JNK/SAPK. In vitro, MKK4 phosphoryle
préférentiellement le résidu tyrosine dans le motif TPY de la boucle d’activation de
JNK/SAPK, alors que MKK7 phosphoryle préférentiellement le résidu thréonine. En se
basant sur cette spécificité, il a été suggéré que ces kinases coopèrent pour activer
JNK/SAPKs, tout en conservant l’intégrité du signal [160,161]. Ces mêmes travaux
indiquent aussi que la phosphorylation du résidu thréonine serait très importante pour
l'activité de JNK3 [161].
1.4.2. Le sentier p38
La MAP kinase p38α a été découverte indépendamment dans trois contextes. C’est une
protéine phosphorylée sur tyrosine qui se retrouve dans des extraits de cellules traités avec
des cytokines inflammatoires. Elle a été identifiée comme étant la cible du SB 203580 de
type pyridinyl imidazole, qui bloque la production du TNFα [162], comme étant la
cytokine- suppressive anti-inflammatory drug-binding protein ou CSBP [163], et comme
étant une kinase pour la MAP kinase activated protein kinase (MAPKAP) kinase-2 [164].
Trois autres gènes qui codent pour des membres de la sous-famille de p38 ont été identifiés
par des stratégies de clonage plutôt que par des approches biologiques: p38β (ou p38-2),
p38γ (ERK6 ou SAPK3), et p38δ (SAPK4) [165-170]. Toutes ces kinases possédent la
séquence TGY dans leur boucle d’activation. p38α et β sont sensibles aux inhibiteurs
dérivés du pyridinyl imidazole, mais les isoformes γ- et δ- sont résistantes à ces drogues
[169,170]. Une variété de facteurs, y compris les cytokines, les hormones, les facteurs
osmotiques, les chocs thermiques ainsi que bien d’autres stress activent les membres de la
famille de p38 [3]. Dans certains contextes, ces protéines kinases peuvent réagir
différemment à un stimulus [171].
Deux protéines MAPKKs, MEK3 et MEK6, active fortement les p38 MAP kinases
[3,150,172,173]. MEK3 paraît favoriser la phosphorylation de p38α et les isoformes de

25
p38β, alors que MEK6 phosphoryle efficacement tous les membres de la famille p38 [172].
Les deux kinases phosphorylent aussi les isoformes de JNK/SAPK. MEK6 phosphoryle les
chimères p38/ERK2, et NLK in vitro, suggérant qu'elle a une spécificité étendue par
rapport aux autres MAPKKs [174,175]. Les implications physiologiques de cette spécificité
ne sont pas encore claires.
1.4.3. Autres MAP kinases
1.4.3.1. ERK3
Un ADNc qui code pour ERK3 chez le rat (ERK3α) a été isolé d'une banque d’ADNc en
utilisant une sonde provenant de ERK1 [139]. Un autre ADNc humain qui code pour une
deuxième protéine kinase ERK3 (ERK3β) a ensuite été isolé. Cette MAP kinase de 63 kDa
est identique à 75% à ERK3α [176]. Ces deux kinases ERK3 sont 50% identiques à ERK1
et ERK2 au niveau du domaine catalytique, et les deux contiennent des extensions en C-
terminal d'approximativement 180 résidus. L’immunodétection, avec des anticorps
spécifiques à ERK3, a révélé des protéines de 63, 95, et 160 kDa présentes dans de
multiples tissus chez le rat et chez plusieurs autres lignées cellulaires [177]. Un clone de
ERK3 codant pour une forme de 100 kDa a été récemment isolé chez la souris par Meloche
et collaborateurs et un seul locus génomique a été cartographié [178]. L'analyse de la base
de données génomiques a suggéré qu'il y a deux gènes fonctionnels, MAPK6 et MAPK4, et
plusieurs pseudogènes [178]. Aucun gène codant pour ERK3 n'a été trouvé dans les
génomes de la levure et des nématodes, suggérant que les isoformes ERK3 proviennent
d'une duplication génomique relativement tardive [179,180].
En dépit de sa ressemblance à ERK1/2, ERK3 a quelques traits qui sont différents des
autres membres de la famille. Le motif des sites de phosphorylation dans la boucle
d'activation chez les isoformes ERK3 a un seul phosphoaccepteur, la serine189 chez
ERK3α. Une glycine remplace le site de phosphorylation tyrosine retrouvé chez la plupart

26
des MAP kinases. Peu est connu sur l’activité catalytique de ERK3α. Cette MAP kinase
s’autophosphoryle mais aucun substrat n’a été clairement identifié [177]. Plusieurs MAP
kinases sont localisées dans le cytoplasme des cellules non stimulées et au noyau quand les
cellules sont stimulées. Par contre, ERK3α est concentrée dans le noyau sous toutes les
conditions examinées [177]. Le mécanisme de la rétention nucléaire est inconnu. En outre,
ERK3 ne possèderait pas la séquence consensus de la localisation nucléaire. Une kinase qui
lie et phosphoryle ERK3α sur la Ser189 a été décrite mais son identité moléculaire est
inconnue [180]. Cette ERK3 kinase ne phosphorylerait pas d’autre MAP kinases.
1.4.3.2. ERK5
ERK5 ou BMK1 (Big MAP kinase-1), a été identifiée indépendamment par deux groupes.
L’un a utilisé un criblage double hybride avec comme l'appât, son activateur potentiel,
MEK5. L'autre a exploité la stratégie de PCR dégénérée pour cloner une nouvelle MAP
kinase [181,182]. Le point marquant chez la protéine ERK5 est sa taille, 816 acides aminés,
due à une extension d'approximativement 400 acides aminés en carboxy du domaine
kinase. Lorsque comparé aux autres MAP kinases de mammifères, la séquence primaire du
domaine catalytique paraît être similaire à celle de ERK2. Cependant, la région C-terminale
de 400 résidus ne présente aucune analogie à d’autres protéines connues et n’a aucune
fonction connue. ERK5 est capable de s’autophosphoryler [183]. Le rôle de cette
autophosphorylation dans la régulation de ERK5 n’a pas été précisé.
Chez les mammifères, ERK5 s’exprime d’une façon ubiquitaire. Comme d'autre MAP
kinases, l'activité ERK5 est régulée par une large variété d’agents prolifératifs et de stress.
Les stimuli prolifératifs incluent le sérum, le EGF, le NGF (nerve growth factor), l’acide
lysophosphatidique (LPA), et l’ester de phorbol [130,184,185]. La capacité de ces agonistes
d'activer ERK5 est dépendante de Ras chez certain type cellulaire. Chez les cellules HeLa,
l'activation par le EGF requière l'activité de MEKK3 [130,184,186]. Les stimuli du stress
incluent le sorbitol, le H2O2, l’irradiation par l’UV, le cisaillement vasculaire, et l’ischemie
[130,185,187-189]. Ces stimuli activeraient la voie ERK5 via Src [190]. Chez les cellules

27
3T3, les formes dominantes négatives de ERK5 peuvent inhiber la prolifération stimulée
par EGF et la formation de foyers de transformation stimulée par RafBXB [184,191].
Anglais et collaborateurs ont examiné la régulation du domaine catalytique de ERK5 après
la délétion de son domaine C-terminal [183]. Le domaine catalytique est activé par V12Ras
et par un mutant actif de MEK5, MEK5DD (les deux sites d'activation sont remplacés par
des résidus acides). In vitro, ERK5 produite chez les bactéries, est phosphorylée sur son
motif TEY par MEK5DD immunoprécipitée et démontre une forte affinité/activité pour son
substrat. Ceci porte à croire que ERK5 se comporte comme la majorité des MAP kinases
dont la taille totale dépasse légèrement celle de leur domaine catalytique. Le domaine
kinase de ERK5 présente la même spécificité d'activation que les autres MAPKs. MEK1,
par exemple, est incapable de phosphoryler ERK5 in vitro ou d’augmenter son activité
lorsque co-exprimées chez les cellules HEK 293. ERK5 phosphoryle des facteurs de
transcription tel que MADS, MEF2A et C, et SAP1a [130,185,192]. Elle active les
isoformes MEF2 et permet la régulation de la concentration intracellulaire de c-Jun [192].
MEK5 est une protéine identifiée à l'origine dans le foie et le cerveau. Il existe des variants
d’épissage qui incluent une isoforme α de 50 kDa et une isoforme β de 40 kDa.
L’expression de l’isoforme β est ubiquitaire. MEK5 est cytoplasmique et son unique
substrat connu est ERK5. Ainsi, le rôle de MEK5 dans la transduction des signaux serait
limité à sa capacité d'activer ERK5 [181,193].
D'après l’alignement de séquences, MEK5 serait principalement reliée à MEK1 et 2. En
fait, MEK5 est aussi inhibée par PD98059 et U0126, deux composés qui sont considérés
comme des inhibiteurs très sélectifs de MEK1 et MEK2 [130,192]. À basses
concentrations, les effets de ces inhibiteurs sont dirigés sur MEK1/2, car le Ki de MEK5 est
considérablement plus élevé. Malgré la ressemblance à MEK1/2, MEK5 n'est pas
phosphorylée par Raf-1 [183]. Bien que Raf-1 soit incapable de réguler l'activité de MEK5,
cette dernière est impliquée intimement à la voie de signalisation Raf-1. MEK5 inactivée,
MEK5KM, peut inhiber la formation de foyers de transformation stimulée par RafBXB
chez les cellules 3T3, alors que la forme constitutivement active de MEK5, MEK5DD,
induit la transformation cellulaire lorsque co-exprimée avec RafBXB [191]. MEK5KM

28
peut inhiber aussi la transformation cellulaire induite par le proto-oncogène Tpl-2/Cot, et la
co-expression de Tpl-2 avec MEK5 augmente les phosphosérines dans MEK5 [194].
L’effet direct de Tpl-2 sur l’activité MEK5 n'a pas été démontré. Les seules MEK5 kinases
identifiées à nos jours sont MEKK2 et 3 [186,195,196].
1.4.3.3. NLK
NLK a été identifiée par le groupe de Erikson comme étant reliée à nemo chez la
Drosophile [197]. Cette kinase a des propriétés qui la placent entre les MAP kinases et les
cdks. Bien qu’elle soit presque 45% identique à ERK2, le motif de double phosphorylation
TXY dans la boucle d'activation est absent, et est remplacé par un motif TQE semblable à
celui des cdks où seul le résidu thréonine est phosphorylé. Néanmoins, NLK paraît prendre
place dans une cascade de MAP kinases qui régule négativement la signalisation par Wnt
[175,198,199]. Des études chez C. elegans ont démontré qu'un homologue à NLK est activé
par la protéine MEKK, Mom-4. Mom-4 est un homologue de TAK1, une MAPKKK dans
la voie p38 MAP kinase. Dans des essais de transfection, TAK1 peut augmenter l'activité
de NLK. Bien qu'aucune MAPKK spécifique à NLK n'a été rapportée, NLK semble être
activée in vitro par MKK6, un substrat de TAK1. Il est donc possible que TAK1 et MKK6
puissent être les régulateurs cellulaires de p38 et de NLK.
1.4.4. MEKKs, la première marche dans la cascade des
MAP kinases
Une enzyme MAPKKK spécifique peut réguler une ou plusieurs MAPKKs dépendamment
de sa spécificité enzymatique, de la distribution cellulaire et subcellulaire des composantes
de la signalisation, de la formation de complexes protéiques et de la nature des stimuli qui
déclanchent la signalisation. Par conséquent, des variations considérables dans l’intensité et
la cinétique d'activation des MAP kinases peuvent se produire sous des circonstances
différentes et en réponse à un agent donné. Plusieurs kinases qui agissent au niveau

29
MAPKKK ont été identifiées, ajoutant de la complexité aux mécanismes de la
signalisation. De plus, il n'y a aucune ressemblance apparente parmi ces protéines en dehors
de leurs domaines catalytiques. La contribution relative de chaque MAPKKK à l'activation
d’une voie MAP kinase unique, avec l'exception possible de Raf dans la voie ERK1/2, est
vague.
Mis à part les isoformes Raf, la première MEK kinase isolée est la protéine à 195 kDa,
nommée MEKK1 [200,201]. Cette kinase est la plus reliée à la protéine kinase de la levure,
Ste11p [202]. Dans leurs domaines catalytiques, MEKK2 et MEKK3, ayant chacune
approximativement 70 kDa et MEKK4, possédant une masse moléculaire proche de 150
kDa, sont presque 50% identiques à MEKK1 [200,203].
Les autres enzymes ayant une activité MAPKKK sont moins analogues et sont de 30 à 40%
identiques à MEKK1. Elles sont, en général, associées à la voie JNKs/SAPKs (figure 1-3);
MAP3kinase (MTK1) [204,205], Tpl-2/Cot [82,206,207], DLK (dual leucine zipper
kinase) [208,209], MLK2/MST (mixed lineage kinase) [210], MLK3/PTK-1/SPRK [211],
TAK1 (transforming growth factor-β (TGFβ)-activated kinase) [212,213],
ASK1/MAPKKK5 [214,215] et ASK2/MAPKKK6 (apoptosis signal-regulating kinases)
[215] et enfin TAOs1 et 2 (thousand and one amino acid kinases) [216,217]. MEKKs 1-3
et Tpl-2 peuvent activer aussi la voie ERK1/2 [82]. MEKK3 et Tpl-2 activent en plus la
voie ERK5 [192,194]. TAK1, ASK1, TAOs1/2, et MTK1 font aussi partie de la voie p38.
Démêler les relations entres ces MAPKKKs et les MAP kinases qu’elles activent a été une
rude tâche. L’identification des spécificités enzymatiques intrinsèques, la distribution, et les
phénotypes d'animaux et de cellules chez lesquels ces gènes MAPKKK sont mutés
devraient commencer à aider à déchiffrer leur diversité et leurs fonctions dans les cascades
de signalisation.

30
MEKK
MEK
MAPK
Rafs, MosMEKK1-4Tpl-2
MLK DLKASK1,2 MTK1MEKK1-4Tpl-2
TAO1,2ASK1 MTK1TAKMEKK1-4
MEKK2,3Tpl-2?TAK ?
MEK1,2 MKK4,7 MKK3,6 ERK3 KinaseMEK5
ERK1,2 JNK1-3
MKK6?
p38α,β,γ ERK5NLK ERK3α,β
Réponse ProliférationDifférenciationdéveloppement
Inflammation Apoptosedéveloppement
Prolifération? ?
Facteurs de croissanceStimulus Cytokines, Stress Sérum
Stress ?
?
?
MEKK
MEK
MAPK
Rafs, MosMEKK1-4Tpl-2
MLK DLKASK1,2 MTK1MEKK1-4Tpl-2
TAO1,2ASK1 MTK1TAKMEKK1-4
MEKK2,3Tpl-2?TAK ?
MEK1,2 MKK4,7 MKK3,6 ERK3 KinaseMEK5
ERK1,2 JNK1-3
MKK6?
p38α,β,γ ERK5NLK ERK3α,β
Réponse ProliférationDifférenciationdéveloppement
Inflammation Apoptosedéveloppement
Prolifération? ?
Facteurs de croissanceStimulus Cytokines, Stress Sérum
Stress ?
?
?
Figure 1-4 : Les voies de signalisation des MAP kinases.
Les différents modules MAP kinases chez les mammifères régulent la croissance cellulaire,
la différenciation, la réponse au stress et le développement.

31
La fonction de MEKK1 est encore obscure. Elle a été impliquée dans l’activation des voies
MAP kinases JNK/SAPK, ERK, et p38. Elle semble aussi jouer un rôle dans l'activation du
facteur nucléaire NF-κB (nuclear factor-κB) [218,219]. In vitro, MEKK1 phosphoryle
MEK 1, 2, 3, 4, 6, et 7 [83,95,201,220]. Cependant, bien que la protéine recombinante
MEKK1 phosphoryle MEK1 et 2 sur les mêmes sites que Raf-1, la phosphorylation de
MEK4 par MEKK1 est relativement plus importante, à l’instar de la découverte que la
signalisation via JNK/SAPKs est la plus affectée chez les cellules sans fonction MEKK1
[221-223].
Bien que le module MAPK classique soit formé de trois niveaux de phosphorylation, une
quatrième kinase peut agir directement en amont comme un activateur des MEKKs. Les
kinases impliquées dans l’activation de JNK/SAPK au niveau MEKK kinase incluent PAK
1-4 [224-226], GCK (germinal center kinase) [227,228], KHS et HGK qui sont apparentées
ou homologues à GCK [229,230], HPK1 (hematopoietic progenitor kinase 1) [231] et NIK
(Nck-interacting kinase) [232].
Les petites protéines G et les protéines hétérotrimèrique G peuvent activer des cascades
MAP kinases (discuté avec plus de détail pour ERK1/2 dans le chapitre précédent) [233].
L’activation de JNK/SAPKs et de p38 en réponse à l’interleukine (IL)-1β, la muscarine, la
bradykinine, et au complexe des sous unités βγ des protéines hétérotrimèrique G peut être
médiées par des membres de la famille des petites protéines G Rho, Rac et Cdc42 [234-
237].

32
1.5. L’organisation, la localisation et la spécificité des
cascades des MAP Kinases
Il y a un haut degré d'homologie entre les différents modules MAPK, dans leur organisation
générale mais aussi au niveau de la composition protéique. Un haut pourcentage de
ressemblance existe dans la séquence primaire des différentes MAPKs (60% entre ERK1/2
et JNK ou p38 MAPK). En outre, les substrats des trois MAPKs principales: ERK, JNK, et
p38 MAPK affichent des motifs de consensus de phosphorylation semblables, (T/S) P.
Deux mécanismes empêchent le cross-talk inapproprié entre les différents modules MAPK.
En premier lieu, les protéines d’échafaudage créent des complexes multi enzymatiques qui
réunissent ensemble des composantes d’une cascade MAP kinase isolée [238]. Ces
complexes préviennent l’activation de la voie par des stimuli inappropriés et favorisent la
transmission rapide du signal à travers la cascade. En second lieu, les sites d’amarrage
spécifiques sur les MAPKs qui sert à l’attachement de substrats, d’activateurs et de
régulateurs, augmentent la fidélité et l'efficacité des réactions enzymatiques.
1.5.1. Prédiction de la formation de complexes signalétiques
par l’étude de la signalisation intracellulaire chez la levure
Les études faites chez la levure ont montré que les protéines d'échafaudage sont
primordiales pour la spécificité et l’intégrité du signal. Ces protéines, qui sont généralement
dépourvues de domaine actif, contrôlent les voies de signalisation en désignant, selon le
contexte cellulaire, la voie qui doit être activée. Chez la levure, et en réponse à la
phéromone, la MAPKKK Ste11p active la MAPKK Ste7, qui a son tour active les MAP
kinases Kss1p et Fus3p. En réponse à un stress osmotique, Ste11p active sélectivement
Pbs2p, la MAPKK de la voie Hog1p (l’équivalent de voie p38 chez les mammifères) [239-
241]. Quel signal activerait Ste11p et quelle MAPKK serait la cible de Ste11p est dictée par

33
les protéines d’échafaudages. En effet, dans le cas d’une réponse à la phéromone, la
protéine d’échafaudage Ste5p rapproche Ste11p à son substrat Ste7p, alors qu’en cas de
réponse à un stress osmotique, Pbs2p crée une interaction stable avec Ste11p et avec le
senseur osmotique Sho1p, ce qui fait transmettre le signal à Hog1p (figure 1-4) [242].
1.5.1.1. Ste5p
Ste5p est la première protéine d'échafaudage identifiée qui est capable de lier les
composantes kinases de la voie MAP kinase chez la levure S. cerevisiae. Les mutants Ste5p
sont stériles, d’où provient le nom de la protéine. Ils ont une déficience dans la progression
du croisement sexué (mating) induite par la phéromone [243]. Des études de double
hybride ont révélé que Ste5p interagit avec les trois kinases du module MAP kinase : la
MAP kinase Fus3p (ou son homologue Kss1p), la protéine MEK, Ste7p et la MEKK,
Ste11p [244-246]. Ces résultats supportent les premières études biochimiques avec la
protéine surexprimée qui montraient que Ste5p est un substrat de Fus3p [247]. Des études
de délétion ont montré que les sites d’interaction de Ste5p avec les MAP kinases sont
distincts, et suggèrent qu'un complexe multi protéique peut être formé [244-246]. La
phosphorylation et l’état d’activation affectent l’interaction des différentes kinases avec
Ste5p [248].
Quatre propriétés de Ste5p ont été soulignées comme clefs possibles de sa fonction. La
première est sa capacité de lier toutes les composantes de la cascade MAP kinase. La
seconde est sa capacité d’interagir avec les transducteurs du signal qui se trouvent en
amont. Ceux-ci incluent la protéine G hétérotrimèrique qui est activée par la phéromone.
En fait, dans cette voie, les sous unités βγ (Ste4p et Ste18p) transmettent le signal avec
l’aide de Ste5p [249] et l’'interaction de Ste5p avec la sous unité Gβ est essentielle pour
l’activation de Ste11p [250,251]. La troisième est que Ste5p forme des oligomères qui
peuvent stimuler l'activation des complexes [251,252]. Finalement, Ste5p peut être aussi un
élément essentiel dans la localisation des kinases dans des complexes. Ste5p est localisée à
la membrane plasmique pour l’activation de la cascade, cependant son entrée et sa sortie du
noyau sont aussi impliquées dans la signalisation induite par la phéromone [253].

34
Figure 1-5 : Les complexes d'échafaudage de la voie MAP kinase chez la levure
saccharomyces cerevisiae.
(a) Le module général de la voie MAP kinase. (b) La voie MAP kinase, en réponse à une
phéromone, comporte la MAP kinase kinase kinase (MKKK) Ste11p, la kinase de la MAP
kinase (MKK) Ste7p, et les map kinases Fus3p et Kss1p. Cette cascade est coordonnée par
la protéine Ste5p d'échafaudage. Ste5p interagit également (directement et indirectement)
avec des composants additionnels de cette voie de signalisation, y compris les sous-unités
Gβ (Ste4p) et Gγ (Ste18p) de la protéine G, Ste20p, Cdc42p, Cdc24p et Bem1p. (c) Une
deuxième protéine d'échafaudage, la MKK Pbs2p, est une protéine kinase qui interagit avec
le senseur osmotique Sho1p, Ste11p et avec la MAP kinase Hog1p pour créer un module
signalétique qui régule la biosynthèse du glycérol en réponse à un stress osmotique [242].
Tiré de la référence [238].

35
1.5.1.2. Pbs2p
La formation de complexes protéiques est déterminante pour la régulation des MAP kinases
et leur bon fonctionnement. Cette idée a été fortement suggérée par les conclusions de
l’étude d’un deuxième module MAP kinase présent chez la levure ; la voie de réponse
homéostatique au choc osmotique [241,254,255]. La voie HOG contient la MAP kinase
Hog1p, l’homologue de p38 chez les mammifères [255]. La MAPKK qui se trouve en
amont dans la voie est Pbs2p [241,255]. Deux senseurs osmotiques peuvent activer la voie
HOG via l’un des trois différents MAPKKKs ; Ste11p, Ssk2p et Ssk22p [240]. Un senseur
osmotique transmembranaire, Sho1p, active Ste11p, la même MAPKKK de la voie qui
répond à la phéromone [241]. Donc, les voies du mating et du stress osmotique partagent
une MAPKKK commune. Quand la voie osmotique est activée, Ste11p se lie à Pbs2p. Cette
dernière favorise alors l’assemblage et l’activation des composantes MAP kinases de la
voie HOG. Pbs2p lie Sho1p, Ste11p et Hog1p [241]. Ste5p est absente ou alors présente à
très faible quantité chez les cellules diploïdes. Sa présence peut être exigée pour la
reconnaissance de Ste7p par Ste11p. Par conséquent, son absence serait un facteur
important dans la spécificité de Ste11p pour Pbs2p. Les partenaires d’interaction de Ste11p
paraissent donc déterminer les signaux qu’elle transmet. Ce mode de régulation peut bien
se passer dans les cascades des MAP kinases chez les mammifères.
La spécificité des interactions entre les différentes composantes de la cascade des MAPKs
n'est pas seulement déterminée par leur association avec Ste5p. Par exemple, la MEK Ste7p
interagit avec Fus3p et Kss1p indépendamment de Ste5p [246,256]. L'interaction de Ste7p
avec Fus3p (ou Kss1p) est tout à fait spécifique parce que deux autres MAPKs, Hog1p et
Slt2p, n’interagissent pas avec Ste7p [256]. L'interaction entre Ste7p et Fus3p (ou Kss1p)
pourrait être une interaction enzyme/substrat qui implique la liaison de la MAPK au site
catalytique de la MAPKK. Cependant, parce que Ste7p est un substrat de Fus3p (et de
Kss1p) dans un mécanisme potentiel de rétroaction [256,257], cette interaction pourrait
également refléter l'attachement de la MAPKK au site catalytique de la MAPK. Ni l'un ni
l'autre de ces possibilités n'explique la forte interaction entre la MAPKK et la MAPK. Par
contre, Fus3p (ou Kss1p) lie étroitement une région NH2-terminale de Ste7p qui ne

36
contient aucun site de phosphorylation et ne fait pas partie du domaine kinase qui est en
COOH-terminale de la protéine [256,258].
1.5.2. Association des protéines kinases dans les voies MAP
kinases chez les mammifères
La fonction cellulaire de Ste5p n’est encore pas complètement élucidée. Néanmoins, le fait
que Ste5p est importante pour l’activité de la voie MAP kinase a mis en évidence
l'importance de la formation de complexes dans les cascades de signalisation. En outre, le
contrôle de la spécificité de Ste11p, qui paraît être exercé par son attachement à Ste5p (ou à
Pbs2p,) indique que la réception du signal et sa transmission peuvent être canalisées par la
formation de complexes protéiques. Une autre fonction essentielle de Ste5p est sa capacité
de se déplacer et de se localiser convenablement dans la cellule.
Alors que la spécificité enzymatique inhérente des isoformes Rafs et de MEK1/2 peut être
suffisante pour garantir leur sélectivité pour ERK1/2, quelques-unes des MAPKKKs et
MAPKKs impliquées dans les voies du stress paraissent être moins spécifiques in vitro et
lors d’essais de surexpression chez les cellules en culture. Par exemple, la surexpression de
Tpl-2 a été reliée à l'activation d'au moins cinq voies MAP kinases et MKK6 phosphoryle
au moins sept différentes kinases in vitro. Ce manque apparent de sélectivité enzymatique
suggère que l'assemblage de ces enzymes dans des complexes pourrait restreindre leurs
actions au MAP kinases et autres kinases présentes dans le complexe et permettre une
progression contrôlée du signal.
1.5.2.1. Les protéines d’échafaudage du sentier ERK
Plusieurs études d’interaction, de clonage, et essais d’activité de différents mutants MEKs
suggèrent que les interactions protéine-protéine sont primordiales pour la transmission des
signaux intracellulaires via la voie ERK1/2. Ces interactions régissent la localisation des
kinases pour la réception du signal, le déplacement des kinases aux sites d'action, la

37
reconnaissance et la spécificité envers les substrats et enfin le contrôle temporel de
l’activité kinase. Plusieurs protéines d’échafaudage spécifiques à la voie ERK/MAPK ont
récemment été identifiées. Comme leurs homologues chez la levure, ces protéines de
soutien semblent jouer un rôle important dans l’organisation, la localisation et le maintien
de l’intégrité fonctionnelle des voies de signalisation des MAP kinases [259].
1.5.2.1.1. MP-1
MP-1 est une protéine d’échafaudage d'approximativement 13 kDa qui a été identifiée par
Weber et les collaborateurs dans un criblage double hybride avec MEK1 comme appât
[260]. La suppression de la région MSS de MEK1 empêche son interaction, suggérant que
cette région est le site d'interaction entre MEK1 et MP-1. Il a été suggéré que MP-1 est une
protéine d’échafaudage qui rehausse la formation de complexes protéiques, parce qu'elle se
lie aussi à ERK1. MP-1 lie beaucoup moins bien ERK2, indiquant une sélectivité
inattendue entre ces deux MAP kinases homologues. Des études au niveau cellulaire ont
démontré que MP-1 augmente l'activation de ERK1 [260]. À cause de sa petite taille, MP-1
est loin d'être un équivalent fonctionnel de Ste5p. Cependant, MP-1 peut être une unité d'un
système d’échafaudage modulaire qui peut faciliter la formation d'un regroupement de
complexes protéiques possédant des différences mineures dans la composition en protéines.
Récemment, un nouveau partenaire à MP-1, p14, a été découvert. Le complexe MP-1-p14
entraîne MEK1 et ERK1 à la surface cytoplasmique des endosomes/lysosomes tardifs où
p14 est localisée [261]. La présence de ERK1 au niveau des endosomes/lysosomes pourrait
l’impliquer dans la signalisation par les récepteurs membranaires internalisés ou alors dans
la régulation de la sécrétion lysosomales.
1.5.2.1.2. Grb10
Nantel et collaborateurs [262] ont montré que le MSS de MEK1 lie Grb10. Grb10 a été
isolé originairement dans un criblage de protéines qui lient le domaine C-terminal tyrosine-
phosphorylé du récepteur EGF. Grb10 [263] et ses homologues Grb7 [264] et Grb14 [265]
sont considérés des protéines adaptateurs parce qu'ils interagissent avec plusieurs protéines

38
de signalisation et manquent d’activité enzymatique apparente (voir la référence [266] pour
revue). Ensemble, ils se nomment la famille Grb7 et partagent un domaine SH2 dans la
région C-terminale qui leur permet de se lier à plusieurs récepteurs RTK activés, un
domaine PH (Pleckstrin homologie) central, et une petite séquence riche en proline pouvant
lier les domaines SH3 [267]. Le domaine BPS, situé entre les domaines PH et SH2 dans
Grb10 et dans Grb14, lie les récepteurs de l’insuline et de l’IGF-I [268,269].
Les complexes Grb10-MEK1 ont été identifiés en association avec la mitochondrie et
peuvent être impliqués dans la progression des signaux de la survie cellulaire qui sont
produits au niveau de la mitochondrie [270]. Grb10 interagit aussi avec Raf-1
mitochondriale [262,268], ce qui suggère que Grb10 pourrait être employé dans la
régulation de la mort cellulaire programmée en modulant l'activité de la voie ERK
mitochondriale. Grb10 servirait de lien entre les récepteurs membranaires et le complexe
apoptotique localisé à la membrane externe des mitochondries. Grb10 pourrait entre autres
faire le lien avec la voie du phosphatidylinositol 3-kinase (PI-3K)/Akt, qui joue un rôle
dans la régulation de l'activité anti-apoptotique de l'insuline et des récepteurs de IGF-I
[271-274]. La voie du PI-3K/Akt régulerait en fait la phosphorylation de Grb10 et celle de
Raf-1 mitochondriale [275,276].
1.5.2.1.3. KSR
Les premières études génétiques faites sur le développement de l’oeil chez Drosophile et
sur l’induction vulvaire chez le nématode C. elegans ont permit de discerner les
mécanismes de base de la signalisation par la protéine G Ras et par les autres composantes
de la cascade ERK/MAP kinase. Dans chacun des systèmes d’étude, la cascade MAP
kinase est régulée par un récepteur à activité tyrosine kinase ; le récepteur Sevenless chez la
mouche et le récepteur de EGF chez le nématode. Chacun fonctionne via Ras pour
contrôler un processus cellulaire. Pour identifier des molécules qui sont nécessaires au
fonctionnement de la voie Ras, des mutants Ras sans effet sur Raf et sur les molécules en
aval ont été utilisés [277-279]. KSR (Kinase suppressor of Ras) a résulté de ces criblages et
semble agir dans de nombreuses voies. KSR, comme Raf, a une région N-terminale riche

39
en cystéine et un domaine kinase C-terminal. Des homologues à KSR ont été identifiés
chez de nombreuses espèces animales mais pas chez la levure. Plusieurs évidences
substantielles indiquent que KSR agit comme une molécule d’échafaudage qui lie les
composantes kinases de la voie ERK/MAPK [277,280]. Cependant, KSR possède une forte
homologie de séquence lorsque comparée à d’autres protéines kinases, sa fonction pourrait
donc dépendre de son activité kinase, bien que son domaine kinase interagit à la fois avec
Raf-1 et avec MEK1 [277,280-283]. Récemment, on a démontré que l’activité catalytique
de KSR est importante dans la transmission du signal stimulé par EGF via la voie Ras-
ERK/MAPK [284]. Cette activité serait une propriété intrinsèque de KSR et indépendante
de son attachement à MEK [285]. KSR1 transite du cytoplasme à la membrane cellulaire en
réponse au traitement par des facteurs de croissance. Ce processus est contrôlé par la
sérine/thréonine kinase C-TAK1 qui phosphoryle KSR à un site qui lui confère la fixation à
14-3-3 [286]. KSR est donc séquestrée dans le cytoplasme en absence de stimulation. En
réponse à un facteur de croissance, le site S392 de KSR est déphosphorylé par une
phosphatase inconnue, et KSR est libérée de 14-3-3 et transite à la membrane plasmique où
elle amène MEK et ERK au complexe signalétique actif de Raf. Par conséquent, KSR
paraît agir comme une protéine d’échafaudage pour maintenir la spécificité du signal et
amplifier ce dernier à travers la cascade ERK/MAPK. Comme Ste5p, KSR pourrait
localiser le module des MAP kinases à la membrane pour y être activé. Il semble aussi se
lie à la sous- unité γ de la protéine G hétérotrimèrique, suggérant que KSR peut avoir un
rôle aussi bien dans la signalisation médiée par les récepteurs associés aux protéines
hétérotrimèriques G que par les récepteurs à activité tyrosine kinase [287].
1.5.2.1.4. RKIP
Une protéine qui lie Raf 1, nommé RKIP (RAF kinase inhibitor protein), a été isolée lors
d'un criblage double hybride utilisant Raf-1 comme appât [288]. Tel que suggéré par son
nom, RKIP inhibe la phosphorylation et l’activation de MEK par Raf-1. En fait, RKIP
paraît interrompre la formation du complexe Raf-MEK. Elle se lie directement à Raf-1,
MEK, et ERK in vitro et en essais de co-immunoprécipitation de lysats cellulaires, et
semble ainsi prévenir l’activation de la voie ERK/MAPK [289]. Basé sur des études de

40
surexpression et de l’usage d’ARN anti-sens et d’anticorps inhibiteurs, il a été conclu que
RKIP fonctionne physiologiquement pour empêcher l'activation du module ERK1/2. Il est
possible que RKIP ait d'autres fonctions, par exemple, comme une molécule d’échafaudage
qui soutient l’activation de la cascade dans certaines circonstances ou localise la cascade à
une organelle spécifique. Bien qu'il n'y ait pas actuellement de données qui supportent cette
idée, JIP, la protéine qui lie JNK [290] et PKI, une protéine qui inhibe l’activité de PKA
[291], ont été identifiées originairement comme inhibiteurs et sont maintenant connues
pour avoir d’autres fonctions. JIP est un échafaud relié à Ste5p. PKI précise l'activité
nucléaire de PKA en formant un complexe qui stimule l'exportation de la sous unité
catalytique de PKA du noyau [290,292-294].
De toutes ces études, un nouveau concept émerge: les protéines d'échafaudage ne sont pas
seulement des isolants entre les modules signalétique homologues, mais elles jouent un rôle
important comme régulateurs de l’activité et de la localisation subcellulaire des
composantes de la cascade.
Ces leçons tirées de l’étude des voies MAP kinases, chez la levure au début puis chez les
mammifères, ont permis de caractériser le rôle que peut avoir les protéines d’échafaudage
dans la progression du signal de façon spécifique et précise. Quatre fonctions clés de ces
molécules sont ainsi à considérer: 1) Elles peuvent organiser les cascades MAP kinases
pour une activation efficace des différentes composantes. 2) Elles peuvent restreindre la
réception du signal en reconnaissant des signaux provenant uniquement de certains
récepteurs; 3) Elles peuvent restreindre la spécificité de la transmission du signal en
interagissant avec un répertoire limité de composantes de la cascade. Et 4) elles peuvent
déterminer la destination du signal pas seulement par la sélection des MAP kinases qu’il
faut impliquer, mais aussi en localisant la cascade à des sites d'action présélectionnés, par
exemple, la membrane plasmique, le noyau, la mitochondrie, les microtubules, etc.
1.5.2.2. Les sites d’amarrage ou Docking Sites
La régulation précise des réseaux des protéines kinases est cruciale afin que les cellules
puissent répondre convenablement aux signaux dans leur environnement. En outre, un

41
certain nombre de maladies, en particulier le cancer, serait associé à la dérégulation d’une
voie de transduction. Par conséquent, l’élucidation des mécanismes par lesquels la fidélité
et l'efficacité du signal sont maintenues peut permettre le développement de procédés
thérapeutiques avec une spécificité accrue et des traitements avec des effets secondaires
réduits.
Un des mécanismes qui influence aussi bien la spécificité que l'efficacité du signal est
l'interaction des protéines kinases avec leurs substrats et régulateurs (activateurs) avec une
affinité élevée via les sites d’amarrage [9,258,295,296]. Ces interactions sont distinctes des
interactions transitoires qui se produisent entre le site actif d'une protéine kinase et le site
phospho- accepteur de son substrat. Plusieurs études ont permis d’élucider le rôle des sites
d’amarrage dans l'attachement des MAPKs à leurs activateurs MAPKKs
[107,125,222,256,297-300], aux protéines d'échafaudage [242,301,302], aux facteurs de
transcription [303-309], à des protéines kinases cibles [107,310,311], aux phosphatases
[107,312-315] et à toute autre enzyme [316-318].
Les trois MAPKs majeures phosphorylent leurs substrats sur la séquence consensus (T/S)
P. Plusieurs substrats potentiels contiennent ce motif [23]. Par conséquent, les MAPKs ont
acquis des sites d’amarrage spécifiques pour trier les substrats pertinents avec qui elles
interagissent. Le site d’amarrage principal de ERK est composé d'un groupe d'acides
aminés chargés négativement, conservé de C. elegans à l’être humain (common docking;
CD) [107,258]. Les données sur la structure en trois dimensions de ERK indiquent que le
site CD est localisé sur le côté opposé de la kinase par rapport à la poche catalytique. Les
substrats doivent donc se dissocier du site d’amarrage pour être phosphorylé, ce qui indique
que l'association via le site d’amarrage est très dynamique [319]. Les sites CD dans ERK
lui permettent d’interagir avec une panoplie de protéines, certains sont des substrats,
d’autres des activateurs, des protéines d’échafaudage ou des phosphatases. Le site
d’amarrage dans ces protéines est généralement constitué par un groupe d'acides aminés
chargés positivement (Le domaine D), ce qui permet d’interagir avec le site CD riche en
résidus chargés négativement. Cela implique que l’interaction de ces protéines avec ERK
est mutuellement exclusive, tout en fournissant un mécanisme moléculaire pour l'activation
et l’inactivation séquentielles de ERK.

42
La spécificité d'interaction de ERK avec ses partenaires ne peut pas être déterminée
uniquement par le motif chargé négativement de ERK. En fait, l’échange, par mutation, du
site présent sur ERK par celui présent sur p38 MAPK permet toujours l’attachement de
MEK à ERK et aucune interaction avec MKK6 n’est détectée [107]. Il est donc possible
que la région d'amarrage dans ERK soit contenue dans une zone d'amarrage où plusieurs
motifs d’interaction coopèrent pour conférer un lien fort et spécifique pour chaque
molécule liant la MAPK [319,320]. L'espacement et l’organisation de ces différents motifs
sur les protéines liant les MAPKs seraient à l’origine des spécificités différentielles
observées chez les MAPKs [295].
D'ailleurs, l'état de phosphorylation des protéines associées peut également moduler
l'affinité de l'interaction. Par exemple, l'association de ERK avec son substrat Elk-1 est
renforcée suite à l'activation de ERK [303], tandis que l'interaction de ERK avec son
activateur MEK est réduite après activation du sentier de signalisation [125]. Fait
intéressant, l'interférence avec d'autres voies de signalisation peut être atténuée en régulant
des interactions d'amarrage. Par exemple, dans certains type cellulaire, la tyrosine
phosphatase PTP-SL retient ERK dans le cytoplasme par association via le site d'amarrage
tout en la maintenant sous une forme inactive [321,322]. Après phosphorylation du résidu
Ser231 de PTP-SL par PKA, l'attachement et la déphosphorylation de ERK sont altérés, ce
qui permet son activation et sa translocation nucléaire subséquente.
Il existe deux classes de site d'amarrage sur les protéines substrats ; le domaine D et le
motif FXFP, dont le site d’ancrage sur ERK reste à être déterminée. Une étude
systématique de sites d'amarrage sur Elk-1 indique que le domaine D et le motif FXFP
forment un système modulaire flexible qui a deux fonctions : D’une part, l'affinité d'un
substrat pour ERK peut être régulée par le nombre, le type, la position et l’arrangement de
ces sites. D’autre part, les sites d’amarrage peuvent diriger la phosphorylation vers les
résidus appropriés, comme ceux de la séquence consensus (S/T) P [304,319].
La découverte des sites d’amarrage a fourni de nouveaux outils pour la dérégulation de la
voie ERK. Par exemple, la micro-injection dans le noyau d'un peptide qui comprend le
domaine D de MEK1 interrompt l'association de ERK-MEK dans le noyau et inhibe

43
considérablement l’exportation de ERK hors du noyau [116]. De la même façon, la micro-
injection dans le noyau d'un peptide qui correspond au site d’interaction avec ERK de
p90rsk interrompt l'interaction entre ERK et les phosphatases nucléaires, ce qui augmente le
taux de ERK active dans le noyau [323]. Cette dernière expérience suggère que plusieurs
protéines interagissent avec les MAPKs via des sites d'amarrage très homologues.
La constatation que les sites d’amarrage des MAPKs dans MEKs, Elk-1 et MKPs
concurrencent tous pour la liaison à ERK suggère que chacune des trois classes de
protéines interagit avec ERK sur le(s) même site(s). En accord avec ceci, l’accepteur
commun identifiée dans ERK2 serait en fait la région CD décrites ci-dessus [107,112]. Les
connaissances actuelles sur la localisation subcellulaire de toutes ces protéines indiquent
que MEK1 et MEK2 maintiennent ERK1 et ERK2 dans le cytoplasme en absence
d'activation [125,323-325]. Après activation, ERK se déplace au noyau où elle phosphoryle
Elk-1 et d'autres cibles nucléaires, puis se relocalise au cytoplasme. Comme Elk-1, MKP-1
et MKP-2 sont des protéines nucléaires. Il est donc possible que MKP-1 et MKP-2
concurrencent Elk-1 pour lier ERK activée au noyau. Dans ce cas, les molécules ERK qui
rencontrent Elk-1, ou d'autres substrats physiologiques, seraient temporairement protégées
contre l'action des phosphatases, tandis que les molécules qui ne trouvent pas de cibles
appropriées sont rapidement désactivées. Une étude récente a démontré que la
relocalisation de ERK2 de nouveau au cytoplasme dépend de son attachement à MEK, qui
fait la navette entre le noyau et le cytoplasme [326]. Ainsi, les protéines ERKs qui ont
trouvé une cible nucléaire appropriée peuvent aussi être protégées contre l'exportation
nucléaire médiée par MEK. Ainsi, l'amarrage concurrentiel des substrats et des régulateurs
des MAPKs peut jouer un rôle direct dans la consolidation de la fidélité de signal.
1.5.3. L’inactivation des MAP Kinases
La durée et l'amplitude de l'activation de la MAP kinase représentent l'équilibre entre le
signal et les mécanismes d'inactivation. Tous les deux semblent être influencés par la

44
rétroaction négative déclenchée par le signal activateur en amont de la MAP kinase. Tel
que discuté plutôt, l'activité de ERK est étroitement stimulée par la phosphorylation des
résidus tyrosine et thréonine dans la boucle d'activation. La soustraction d'un ou des deux
phosphates par les tyrosine ou la sérine/thréonine phosphatases ou encore par les
phosphatases à double spécificité diminue nettement l'activité de la MAP kinase. La
spécificité des phosphatases est fortement dictée par la localisation intracellulaire. Ainsi, il
a été difficile de définir clairement leur implication dans la régulation des MAP kinases.
Néanmoins, plusieurs études biochimiques et analyses génétiques ont démontré
l’implication des phosphoprotéine phosphatases de chacune des trois catégories principales
dans l'inactivation des MAP kinases [327,328,328,329].
Schématiquement, l'activation de ERK par les mitogènes lors de la transition G0/G1 du
cycle cellulaire se produit en quatre phases. D'abord une forte activation suivie d’une
inactivation très rapide (quelques minutes). Par la suite, une activation prolongée se produit
avec un maximum qui se situe entre 2 et 4 heures après stimulation. Enfin, l'activation
diminue graduellement et l'activité de ERK est réduite au niveau basal. Considérant que la
déphosphorylation de la thréonine ou de la tyrosine dans le motif TEY de la boucle
d'activation de ERK est suffisante pour l'inactivation enzymatique totale [330], de
nombreuses phosphatases pourraient être impliquées dans les deux phases d'inactivation : la
phase initiale rapide et la phase tardive qui est plus lente.
La sérine/thréonine phosphatase PP2A est impliquée dans la première phase d’inactivation
de ERK observée après quelques minutes de stimulation des cellules NIH 3T3 [331] et des
ovocytes de Xenopus [329]. Le résidu phospho-tyrosine restant semble être modifié par une
phosphatase constitutive. Plusieurs phosphatases spécifiques au résidu tyrosine, telles que
PTP-SL, STEP et He-PTP/LC-PTP démontrent une spécificité envers les MAP kinases
ERKs [320,332-334]. Cependant ces tyrosine-phosphatases cytosoliques semblent présenter
un patron d'expression restreint. Aucune phosphatase exprimée d’une façon ubiquiste et
pouvant jouer le même rôle chez la plupart des cellules n’a été identifiée. Une tyrosine
phosphatase cytosolique de la drosophile, PTP-ER, homologue aux tyrosine phosphatases
mentionnées ci-dessus, joue un rôle important dans la répression de ERK pendant le

45
développement de l’oeil [335]. Toutefois on ne connaît pas si PTP-ER joue un rôle
important dans l'inactivation du premier pic d’activation de ERK.
La phase tardive de l'inactivation dépend de la synthèse de protéines, indiquant que la néo-
synthèse des phosphatases est exigée [331,336]. En plus, ces phosphatases semblent avoir
une spécificité au résidu tyrosine puisqu'elles sont inactivées par le traitement au vanadate
[323,329,331]. Les phosphatases qui possèdent ces critères sont les MAPK phosphatases
(MKPs). Les MKPs appartiennent à la famille des phosphatases ayant une double
spécificité, puisqu'elles sont capables de déphosphoryler les résidus tyrosine et thréonine
des MAPKs [337,338].
Un nombre substantiel de phosphatases à double spécificité est consacré à l'inactivation des
MAP kinases aux temps et aux endroits appropriés [337,339]. Les MKPs sont classées en
deux groupes : Elles sont encodées par des gènes induits par le stress et par des facteurs de
croissance et sont localisées principalement au noyau. Ou alors, elles ne sont pas
ardemment régulées au niveau transcriptionnel et sont situés dans le cytosol. Les deux
classes ont une organisation semblable de leurs domaines : un domaine de régulation N-
terminal et un domaine catalytique C-terminal qui démontre une homologie de séquence
avec la phosphatase à double spécificité VH1 [340]. Les différences dans la localisation et
l'induction impliquent des divergences dans l'inactivation temporelle et spatiale qu'elles
peuvent produire. La spécificité des MKPs semble être différente pour chaque membre de
la famille des MAP kinases [315,341-343].
Certaines évidences indiquent que les MKPs sont de bons candidats pour le contrôle de la
rétroaction négative dans l’activation de ERK. D'abord, on a montré que la production de
MKP1 et MKP2 sont induites au niveau transcriptionnel par l'activation de la voie ERK
[336]. De plus, MKP1 et MKP2 semblent être stabilisées suite à la phosphorylation par
ERK [344]. En fait, MKP1 est phosphorylée sur la Ser359 et la Ser364 par ERK, ce qui ne
modifie en rien son activité phosphatase, mais réduit le taux de sa dégradation par les
protéasomes. Enfin, MKP3 [345,346] et MKP1 [313] sont activées suite à l’attachement de
ERK à leur domaine de régulation via des sites d'amarrage. Par conséquent, la spécificité

46
envers le substrat est assurée par deux moyens : l’interaction protéine-protéine et
l’activation catalytique de la phosphatase.
Le rôle précis de chacune des MKPs in vivo n'est pas encore clair. L'expression de certaines
MKPs est limitée à des compartiments subcellulaires spécifiques, là où elles doivent
couvrir la gamme de substrats disponibles. En plus, certaines phosphatases, telle que MKP1
et MKP2, sont les produits de gènes induits par la voie ERK, alors que d’autres, tel que
MKP3, sont produites passivement à un rythme beaucoup plus lent. Enfin, une MKP peut
être efficace dans l'inactivation de plusieurs MAPKs, fournissant de ce fait une possibilité
de cross-talk avec certaines voies de signalisation alors qu’elle est activée par une voie de
signalisation distincte. En outre, il est probable qu'il y ait redondance entre les MKPs
puisque la suppression du gène MKP1 n'affecte en rien la physiologie de la souris mutante
[347]. De nouvelles approches seraient nécessaires pour évaluer, d’une façon individuelle,
le rôle fonctionnel des MKPs in vivo.
Alors que la déphosphorylation des MAP kinases semble être un moyen d’inactivation
efficace et largement répandu chez tous les membres de la famille des MAPKs, la
phosphorylation peut aussi être un moyen pour arrêter la progression du signal et
l’inactivation des composantes kinases des voies de signalisation. La rétroaction négative
est un phénomène courant dans la voie ERK/MAPK. En effet, ERK semble phosphoryler
plusieurs composants de la voie dont SOS et MEK1/2 ce qui provoquerait la dissociation
des différents modules de la cascade et l’arrêt du signal [348,349].
1.5.4. La régulation de la localisation subcellulaire de ERK
L'organisation spatiale des kinases et de leurs substrats détermine la nature des signaux
transmis et leur destination. En outre, le type et/ou le contexte cellulaire et l’agencement
des protéines de signalisation sont des éléments déterminants qui régissent la distribution et
la localisation intracellulaire adéquates des MAP kinases. Ainsi, une population distincte de
MAP kinases peut répondre, d’une façon spécifique, à un signal extracellulaire.

47
L’agencement des protéines kinase peut se produire grâce à la formation de complexes très
spécialisés, à la concentration d'une MAP kinase à un compartiment unique, tel que la
membrane plasmique, ou encore grâce à la concentration d’une population de MAP kinases
à un compartiment où la diffusion est limitée, tel que les vésicules membranaires ou le
noyau.
Les MAP kinases ERK1 et 2 sont généralement associées au cytosquelette [350,351]. Une
fraction de ERK1 constitutivement active serait associée à la tubuline, alors que ERK2
serait liée aux microtubules via la protéine MAP-2 (microtubule-associated protein 2)
[352]. Les ERKs peuvent aussi se localiser à la membrane plasmique grâce à leur
association avec des composantes membranaires [353,354]. Cependant, leurs formes
actives sont généralement retrouvées dans le noyau [355-357], associées aux kinétochores
et aux asters pendant certaines étapes de la mitose [358,359]. Leur protéine d’ancrage
nucléaire semble être la protéine du centromère CENP-E [359], une découverte
particulièrement intéressante puisque ERKs et p38 MAPKs ont été impliquées dans la
régulation du point de restriction (checkpoint) pendant la phase M du cycle cellulaire
[360,361].
Les mécanismes de ciblage visant les composantes des voies MAPKs sont encore mal
compris et plusieurs stratégies sont possibles. Par exemple, les substrats ou les activateurs
ayant une distribution subcellulaire restreinte peuvent faciliter le recrutement des kinases
par des interactions directes. Par exemple, Ras associée à la membrane plasmique lie RAF
et la recrute à la membrane [44]. D’autre part, la MAP kinase JNK est localisée à des
structures ponctuées le long des microtubules en association avec son activateur, la JNK
kinase kinase MLK2 [362].
La localisation nucléaire semble être essentielle pour la transformation morphologique des
fibroblastes et la différenciation des cellules PC12 [126,363]. Bien que la régulation de la
transcription dépendante de la phosphorylation exige généralement la localisation nucléaire
des kinases appropriées, dans certains cas, les facteurs de transcription peuvent être activés
tandis qu’ils sont encore dans le cytoplasme [364,365].

48
La translocation nucléaire peut se produire via des mécanismes distincts selon la MAP
kinase et le contexte cellulaire. Dans le cas des ERK/MAP kinases, plusieurs événements
peuvent contribuer à la régulation de la quantité de ERK1/2 qui transitent entre le
cytoplasme et le noyau : 1) la séquestration cytoplasmique, 2) l’entrée nucléaire par
diffusion, 3) la phosphorylation et la dimérisation subséquente, 4) le transport actif des
monomères, des dimères, ou des complexes de protéines à travers la membrane nucléaire,
5) l’exportation nucléaire de MAP kinases seules ou dans des complexes et 6)
l’attachement à des sites de rétention au noyau [125,325,326,356,357].
La redistribution intracellulaire des protéines ERKs suite à une stimulation mitogènique se
produit en deux phases. D’abord, il y a entrée rapide mais partielle dans le noyau. Puis,
après plusieurs heures de stimulation, ces ERKs s’accumulent massivement dans le noyau.
Les stimuli non-mitogéniques induisent l'entrée nucléaire initiale de ERK mais ne provoque
pas son accumulation nucléaire [114,323]. Les signaux menant à la différenciation
semblent provoquer une activation prolongée et une accumulation nucléaire de ERK [366].
La rétention artificielle de ERK activée dans le cytoplasme semble être suffisante pour
bloquer la réplication de l’ADN induite par les facteurs de croissance [367] et empêcher la
transformation des cellules [126]. Alternativement, en rétablissant la translocation nucléaire
de ERK dans les fibroblastes sénescents, il est possible de rétablir l'activation de c-fos
[368]. En outre, la rétention cytoplasmique de ERK peut jouer un rôle critique dans le
maintient du phénotype différencié de certains types de cellule. Par exemple, la
surexpression de la protéine PEA15 qui séquestre ERK dans le cytoplasme bloque la
prolifération cellulaire des astrocytes [369]. D’autre part, la β-arrestine semble s’associer
avec ERK dans le cytoplasme et inhiber la transcription [370]. La régulation du module
ERK/MAPK par la séquestration cytoplasmique semble donc être un phénomène fréquent.
La régulation de la translocation nucléaire de ERK serait alors une étape critique pour
générer des réponses biologiques adaptées selon le contexte cellulaire.
Divers groupes de recherche suggèrent que les interactions MEK-ERK jouent un rôle
prédominant dans la redistribution cellulaire de ERK1/2 [112,125,126,326,371]. ERK
s'associe avec MEK dans le cytoplasme des cellules en repos via leurs sites d'amarrage. La
protéine d’échafaudage KSR participerait à cette localisation cytoplasmique du complexe

49
MEK-ERK. La contribution de KSR dans le dégagement de ERK du complexe
cytoplasmique n’est pas claire. Cependant, l'activation de la voie est essentielle, puisque
l’inhibition de l'activité MEK abroge la translocation nucléaire de ERK [357]. D’autre part,
Les mutants ERK constitutivement inactifs migrent au noyau après activation de la cascade
[356,357]. De plus, chez la levure, les mutants inactifs Fus3-GFP migrent au noyau au
même taux que le type sauvage [372]. L’activation des MEKs semble donc suffisante pour
le détachement des ERKs du complexe cytoplasmique.
ERK2 lie également, d’une façon stable, un certain nombre de protéines qui peuvent limiter
son accès au noyau. Par exemple, le détachement, suite à l’activation de la voie
ERK/MAPK, de ERK2 de la phosphatase cytoplasmique PTP-SL serait responsable de
l'accumulation nucléaire de ERK2 [321].
Il est maintenant certain qu'il y a un flux continu de mouvement de ERK entre le
cytoplasme et le noyau. En effet, lorsque des cellules quiescentes sont traitées avec la
Leptomycine B, qui bloque l'exportation nucléaire dépendante de crm-1, ERK apparaît
rapidement dans le noyau [323]. Ceci se produit en absence d'activation de la voie ERK
puisqu'il n'est pas perturbé par le traitement antérieur avec l'inhibiteur U0126 de MEK
[373].
Il semble que plusieurs mécanismes pourraient réguler le taux de transfert de ERK vers le
noyau. Il a été proposé que ERK traverse la membrane nucléaire par diffusion passive car
l’inhibition du trafic nucléo-cytoplasmique actif avec de l'agglutinine n'empêche pas
l'importation nucléaire de ERK [325]. Cependant, chez la levure, le trafic de Fus3-GFP
semble être réduit mais n'est pas complètement inhibé quand le transport nucléo-
cytoplasmique actif est altéré, suggérant l’existence d’un mécanismes de transport actif
dans l’entrée de ERK au noyau. Ce mode de transport n’est pas encore clair car ERK ne
possède pas de motif de localisation nucléaire (NLS) connue. On a également proposé que
ERK se dimèrise et que cette dimérisation favoriserait son entrée au noyau. En effet, les
protéines de fusion ERK-β-galactosidase ne peuvent passer dans le noyau quand les motifs
de dimérisation de ERK sont mutés [325]. Enfin, il a été récemment proposé que le
transport de ERK à travers le pore nucléaire serait propulsé par un mouvement brownien.

50
En effet, chez des cellules perméabilisées, ERK s'associe directement au complexe présent
dans les pores nucléaires et se transfère au noyau indépendamment des facteurs solubles et
de l’ATP. En outre, ERK interagit in vitro avec une région à répétition de FG de la
nucléoporine CAN/Nup214. [374]
L'exportation de ERK hors du noyau n'est pas entièrement comprise non plus. ERK1 et
ERK2 ne présentent aucun motif d'exportation nucléaire et ne devraient pas ainsi être
exportées activement par l'intermédiaire d’un mécanisme dépendant de la protéine Crm-1.
Le blocage de l'exportation nucléaire par Crm-1 avec la Leptomycine B provoque
l'accumulation nucléaire de ERK et de MEK [325,326]. Par conséquent, l'exportation active
de MEK par l'intermédiaire de son NES pourrait être l'un des mécanismes qui conduit ERK
hors du noyau.
Quand l'activation de la voie ERK/MAPK est transitoire, les protéines ERKs sont
rapidement expulsées du noyau [114]. Toutefois lors d’une activation soutenue, elles s'y
accumulent. L'accumulation nucléaire de ERK exigerait une induction rapide et dépendante
de son activité de la transcription de gènes qui codent pour des protéines d’ancrage
nucléaire [357]. L'identité de ces ancres nucléaires demeure inconnue. Toutefois,
l'utilisation d’anticorps anti-phospho-ERK a fourni de nouveaux indices pour expliquer
l’accumulation nucléaire de ERK. En fait, les phosphatases MKP1 et MKP2 semblent être
de bonnes candidates pour la séquestration de ERK dans le noyau car elles possèdent les
caractéristiques d’ancres nucléaires ; leur expression est brève et est stimulée par la voie
ERK/MAPK [336,344], elles sont nucléaires et elles possèdent des sites d'amarrage pour
les MAP kinases ERK1/2. D’autres études devraient dans le futur démontrer le rôle de
MKP1/2 dans l'inactivation et la séquestration nucléaires des MAP kinases ERKs.
1.5.5. Spécificité et intégrité du signal
Les modules MAPKs chez les mammifères sont entrelacés dans un réseau complexe de
cascades signalétiques. L'induction d'une réponse biologique particulière exige souvent plus

51
d'un signal d'entrée. La coordination des voies d'entrée et l'intégration subséquente des
signaux sont requises pour produire un signal de sortie approprié. Pour réaliser ceci, les
modules MAPK utilisent différentes stratégies pour communiquer avec d'autres voies.
Parmis ces stratégies, l’entrecroisement ou cross-talk peut affecter les propriétés
signalétiques et l'effusion de l'information et peut modifier la spécificité des voies
impliquées.
Le cross-talk entre les cascades de signalisation se produit dans de nombreuses voies dans
le but d’intégrer des réponses spécifiques ou des effets modérées. Plusieurs évidences
laissent croire que les MAP kinases ont des spécificités aux substrats qui sont
chevauchantes [8,23]. L’activité résultante des substrats reflète l’enjeu de la
phosphorylation sur les sites régulateurs qui sont partagés par une multitude de protéines
kinases. Tout au long des cascades, les MAP kinases forment des complexes qui facilitent
leur activation et renfoncent leur localisation, leur spécificité, et précise leurs cibles. La
régulation de la formation de ces complexes protéiques fournit un contexte pour le cross-
talk entre les voies de signalisation.
Plusieurs membres de la famille MEK contiennent des sites qui sont phosphorylés par des
kinases d’autres voies. Ces phosphorylation semblent influencer la capacité des MEKs à
interagir dans les complexes [104,375-377]. L'intégration peut se produire aussitôt dans la
voie de signalisation et au sommet du module kinase. Certaines MEKK peuvent réguler
plus qu'une cascade de MAP kinases, de même que plusieurs cascades peuvent être
contrôlées par plusieurs MEKKs distinctes.
Par exemple, un niveau élevé de l’AMPc qui induit l'activation de PKA peut déterminer
quelle isoforme de RAF sera engagée pour stimuler MEK1/2. Chez les fibroblastes Rat-1,
l'activation de c-Raf-1 peut être sélectivement inhibée par des niveaux élevés de l’AMPc
intracellulaire [27,378,379] et la phosphorylation directe de c-Raf-1 par PKA [27]. En
revanche, chez les cellules PC12, B-RAF serait activée par un niveau élevé d’AMPc
indépendamment de Ras [55,380,381]. Chez les cellules LNCaP du cancer de la prostate,
l’AMPc renforce l'activation des MAPKs en réponse à l’EGF [382]. Cette activation est
médiée par la petite GTPase Rap1 qui est sensible à l’AMPc [383] et qui active directement

52
une isoforme B-RAF [28,384]. Ainsi, un niveau élevé d'AMPc et l’activation subséquente
de PKA peuvent changer le cheminement de l’information, résultant dans l'activation
sélective de B-RAF et l'inhibition de c-Raf-1. Ceci suggère que l'AMPc pourrait servir de
commutateur moléculaire pour préciser la spécificité des isoformes dans la cascade des
MAPKs [385,386]. De même, MEK1 peut être sélectivement régulée par la machinerie du
cycle cellulaire. En effet, la phosphorylation de MEK1 (mais pas de MEK2) par la kinase
dépendante de la cycline- 2 (cdk-2) peut réduire son activité kinase [387].
Les modules de MAPK peuvent affecter les propriétés d'autres voies de signalisation. Tel
que mentionné précédemment, JNK peut contrecarrer l'activation du facteur de transcription
NFAT4 par phosphorylation directe [388]. ERK peut répondre à des signaux d'oestrogènes
en phosphorylant son récepteur ER (estrogen receptor) à la sérine 118 [389]. Cette
phosphorylation augmente l’activité transcriptionnelle du récepteur ER induite par
l’œstrogène ou le tamoxifène. Ces observations indiquent que l’attachement de l'oestrogène
et la stimulation de ERK par les facteurs de croissance peuvent coopérer pour une
activation maximale du récepteur de l'oestrogène. ERK interagit également avec la voie de
signalisation JAK-STAT activée par l’interféron alpha/bêta, et la forme dominante négative
de ERK2 peut inhiber l'activation transcriptionnelle d'un gène rapporteur sensible à
l’interféron [390].
Les propriétés signalétiques des modules MAPKs peuvent également être modifiées par des
cascades MAPKs étroitement liées. Par exemple, les petites protéines G, Rac1 et Cdc42,
qui activent normalement le sentier JNK, peuvent coopérer avec RAF pour activer la
cascade ERK [104]. Cette collaboration semble solliciter la protéine kinase activée par Rac
et par Cdc42, PAK1, qui phosphoryle MEK1 sur le résidu Ser298 dans sa région riche en
proline impliquée dans la signalisation [103,391]. Cette phosphorylation semble accroître
l'affinité de MEK1 à Raf-1 et être impliquée dans la migration cellulaire et la réponse aux
signaux mitogéniques qui est dépendante de l’adhérence à la matrice extracellulaire
[104,377,391,392]. PAK3, un membre de la même famille de kinases semble augmenter
l'activité de Raf-1 par phosphorylation directe sur le résidu sérine 338 [62], soutenant
encore le concept de communication par cross-talk. Dans le cas de MEKK1, un effet cross-
talk indirect peut se produire. En fait, MEKK1 semble induire l'expression du gène codant

53
pour la phosphatase MKP-1, atténuant, de ce fait, la signalisation par des cascades MAPKs
hétérologues [389,393].
Il est aussi clair que les modules MAPK en aval peuvent converger. Par exemple, ERK et
p38 MAPK peuvent tous deux activer MNK1 (signal-integrating kinase 1) [394,395] et
MSK1 (mitogen- and stress-activated protein kinase 1) [396]. De même que, la MAPKAP
kinase 3 peut être ciblée par ERK, JNK, et p38 MAPK [397]. En outre, beaucoup de
facteurs de transcription sont régulés par plus qu’une voie MAPK. C’est le cas de Elk-1 qui
peut être activé aussi bien par ERK que par JNK ou p38 MAPK [398-400]. D'autres
facteurs de transcription interagissent avec un sous-ensemble de MAPKs. C’est le cas de
ATF2, qui est régulé tant par JNK que par p38 MAPK [400,401].
Pourquoi les cellules mettraient-elles tant d'efforts dans la génération d'un signal spécifique
si, à l’extrémité des voies, le signal converge sur les mêmes cibles ? Ou comment est-ce
que la spécificité des signaux peut-elle être maintenue quand plusieurs voies utilisent la
même protéine comme cible ultime de la signalisation ? Bien que la compréhension de cette
question soit inachevée, plusieurs mécanismes ont jusqu'ici été identifiés.
D'abord, des modules MAPKs peuvent être liés à certains effecteurs d'une façon spécifique
au type cellulaire. Ceci a été suggéré dans une étude récente qui démontrait que, chez les
cellules PC12, ERK mais pas JNK active c-Jun pour induire le développement neuronal
[113]. Cependant, chez d'autres lignées cellulaires, c-Jun est sélectivement régulée par JNK
[402,402,403,403], suggérant que des facteurs additionnels spécifiques au type cellulaire
peuvent déterminer quel module MAPK activerait c-Jun.
Ensuite, les cibles qui sont communes à tous les modules MAPKs seraient impliquées dans
la régulation de fonctions cellulaires courantes, telles que la régulation de la traduction.
Cette notion est soutenue par l'observation que MNK1, une kinase qui est un substrat
commun de ERK et de p38 MAPK, peut phosphoryler eIF4E (eukaryotic translation
initiation factor 4E) à un site physiologiquement approprié, affectant probablement de ce
fait le déclenchement de la traduction [395].

54
Finalement, les cibles communes qui sont en aval des MAPKs peuvent également
acheminer des signaux d'entrée uniques à un module MAPK pour générer des réponses
biologiques qui sont discrètes.
1.6. L'organisation différentielle des signaux transmis
via la voie ERK/MAPK.
L'amplitude et la durée de l'activation de ERK peuvent être affectées à plusieurs points dans
cette cascade de signalisation et sont régulées par un équilibre découlant de l’activation par
les kinases et l’inactivation par les phosphatases. L'importance et la durée des signaux
dérivés de MEK1-ERK1/2 seraient déterminantes dans l'issue physiologique finale des
cellules.
Le système expérimental qui illustre le mieux ceci est le modèle de
différenciation/prolifération des cellules cultivées du phéochromocytome 12 de rat (PC-12).
Ces cellules prolifèrent en réponse au facteur de croissance épidermique (EGF), alors que
l’exposition au facteur de croissance neuronal (NGF) ou au facteur de croissance
fibroblastique (bFGF) cause la différenciation des cellules marquée par l’immaturation
neuronale et l’arrêt de cycle cellulaire en G1. La réponse différentielle est en grande partie
régie par la prédisposition du NGF, mais pas le EGF, de provoquer l'activation soutenue de
ERK1/2 et leur translocation au noyau. Cette activation prolongée peut durer pendant
plusieurs heures et semble être nécessaire pour maintenir l’état différencié des cellules
[404-408]. À l’encontre des effets observés chez les cellules PC-12, l’activation soutenue
de ERKs chez les fibroblastes mène à la croissance et à la transformation des cellules.
Cependant, dans les deux cas, la translocation nucléaire de ERK se produit seulement en
réponse à l'activation prolongée. Ceci suggère que les événements ultimes responsables de
la différenciation des cellules PC-12 ou la prolifération des fibroblastes impliquent la
phosphorylation de cibles nucléaires.

55
Au-delà de cela, et sans compter leur organisation temporelle, les interactions spécifiques
enzyme/substrat médiées par les protéines d'échafaudage et d’ancrage peuvent contrôler la
séquestration et l’organisation subcellulaire des composantes signalétiques. Ainsi est née la
notion de la fonction cellulaire à trois dimensions (cellomic fonction) des modules de la
signalisation MAPK. L'organisation différentielle des signaux, qu’elle soit spatiale et/ou
temporelle, pourrait expliquer les effets physiologiques et pathologiques observés chez
différents types cellulaires et chez les organismes mammifères.
1.6.1. La différenciation
Comme chez les cellules PC12, un rôle important a été attribué à la voie ERK/MAPK dans
la différenciation des cellules sanguines. L’expression de MEK1 constitutivement active
(caMEK1) dans deux lignées de cellules humaines d'érythroleucémie, K562 et CMK, mène
à l'inhibition de la croissance des cellules, l’induction de morphologie caractéristique des
mégacaryocytes et l’expression à la surface des cellules de l'intégrine-αIIbβ3, un récepteur
d'adhérence spécifiquement exprimé chez les plaquettes [409,409,410,410,411,411]. En
outre, l'expression des gènes de la globine chez les cellules K562 est bloquée par une
activité élevée du module MEK1-ERK1/2 et est dérégulée par l’inhibition de MEK [412].
Ceci suggère que ce module de signalisation favorise la différenciation des mégacaryocytes
en même temps qu'il supprime la différenciation d'érythrocytes. D'ailleurs, les résultats
obtenus chez des souris sans fonction ERK1 ont démontré que son activité est requise pour
la différenciation des thymocytes double positifs CD4+ CD8+ en simple positif et pour la
prolifération [413].
Ces MAPKs semblent également être impliquées dans la régulation de différentes étapes de
la différenciation du muscle squelettique. Chez les myoblastes C2C12 et en absence de
mitogènes, des gènes spécifiques aux muscles tels que MyoD et myogenin sont exprimés
[414,415]. L'inactivation de la voie ERK/MAPK semble être impliquée dans la myogenèse
des myoblastes C2C12. En fait, la surexpression de la phosphatase MKP1 est suffisante pour

56
interférer avec l'activité ERK2 endogène et la croissance des myoblastes. Cependant,
l’expression de MKP1 endogène décline dans les myotubes différenciés et multi-nucléaires
[67]. Cette diminution dans l’activité phosphatase pourrait être nécessaire pour la formation
de myotube plus tard pendant la myogenèse. Des études de transfection, utilisant caMEK1
et MEK1 dominante négative chez les cardiomyocytes ventriculaires de rat, ont indiqué que
MEK1 peut induire un patron d'expression de gènes caractéristiques des cellules
hypertrophiques [416].
Des approches expérimentales indépendantes utilisant des lignés cellulaires provenant de
rein canin de Madin-Darby (MDCK) suggèrent que l’activation soutenue de ERK2 mène à
la dédifférenciation épithéliale, à l’échec de la morphogenèse et à l’apparition chez ces
cellules d'un phénotype fortement invasif. En effet, chez les cellules MDCK-C7Focus
(C7F) alcali- dédifférenciées, la réduction de l'expression de la protéine ERK1 et la
stimulation de l'activité basique de ERK2 par le sérum sont associés à une conversion
stable du phénotype mésenchymateux des cellules. D’autre part, l’expression stable d'un
mutant caMEK1 chez les cellules MDCK-C7 (cellules C7caMEK1) a comme conséquence
des changements phénotypiques prononcés semblables à ceux obtenus chez les cellules
C7F, y compris l'acquisition d'une morphologie fibroblastique, la réduction de l'expression
des cytokeratines, la surexpression de la vimentine et l’assemblage de fibres de stress
d’actine caractéristiques du muscle lisse α [417,418].
De plus, les cellules dédifférenciées (C7F aussi bien que C7caMEK1) étalées sur collagène
ne peuvent pas reproduire les structures épithéliales et se comportent en tant que cellules
fortement invasives [419]. Par ailleurs, l'inhibition de MEK induit un étalement prononcé
des cellules C7caMEK1 et réduit nettement leur invasion dans la matrice de collagène. En
conclusion, le phénotype trans-différencié et les propriétés invasives de ces cellules
transfectées caMEK1- MDCK-C7 sont reflétées par la la baisse du niveau d’expression de
protéines qui sont cruciales pour les jonctions d’adhérence d’actine comme la E-cadherin et
les α- et β-catenin [420]. L’ensemble de ces résultats suggère que cette activation soutenue
du module MEK1-ERK1/2 représente un mécanisme de signalisation important impliqué
dans la trans-différenciation des cellules épithéliales tubulaires en myofibroblaste. Les

57
cellules épithéliales tubulaires pourraient jouer un rôle pathophysiologique dans les
maladies rénales qui sont associées aux altérations dans la différenciation et/ou la
prolifération des ces cellules, tel la fibrogenèse ou la carcinogenèse rénales. Cependant,
parce l’activation de MEK1 et de ERK1/2 mène à la différenciation de certaines cellules
nerveuses, des mégacaryocytes, et des thymocytes mais induit la dédifférenciation des
cellules épithéliales rénales, les réponses (patho) physiologique lié au module signalétique
MEK1/2-ERK1/2 serait spécifiques au type cellulaire.
1.6.2. La signalisation entre la prolifération et l'arrêt de la
croissance
L’idée, que la divergence dans la fonction du module de signalisation ERK/MAPK est
spécifique au type cellulaire, est encore appuyée par les résultats obtenus à partir
d’expériences faites chez les fibroblastes. L’expression des mutants caMEK1/2, par
exemple, semble transformer les fibroblastes et induire la formation de tumeurs chez les
souris nues [421,422]. La stimulation de ce module de signalisation intracellulaire est non
seulement liée aux altérations de la différenciation cellulaire mais également à la régulation
de la prolifération cellulaire. Pagès et collaborateurs ont été les premiers à prouver que
l'activation de ERK est essentielle pour l’entrée des fibroblastes en phase G0 du cycle
cellulaire [423]. La prolifération est inhibée par l’expression d’une forme dominante
négative de ERK1 ou d'ARN ERK1 anti-sens dans les fibroblastes CCL39. La coexpression
du type sauvage ERK1 renverse ces effets. Plus tard, d’autres études ont rapporté que
l’expression de la forme dominante positive de MEK1 stimule ERKs et accélère la
prolifération. Les inhibiteurs de MEK ou la transfection de phosphatase MKP1 active
empêchent la prolifération cellulaire induite par les mitogènes. L’ensemble des résultats
suggère qu’il existe des liens importants entre la voie ERK/MAPK et le cycle cellulaire. En
fait, les signaux dérivant de cette voie peuvent influencer la croissance cellulaire par
l'intermédiaire de plusieurs mécanismes.

58
D’ailleurs, ERK2 serait essentielle au contrôle de la synthèse de nucléotide, la première
étape dans la production de l'ADN et de l'ARN, en contrôlant directement l'activité de la
carbamoyl-phosphate synthétase II (CPSII) [424]. Cette enzyme est importante dans la
synthèse de novo des nucléotides pyrimidique. ERK2 induit la phosphorylation de CPSII et
son activation subséquente par le phosphoribosyl-pyrophosphate.
De plus, ERK1 et ERK2 semblent accroître la traduction protéique en stimulant la capacité
du facteur de la traduction eIF4E à recruter des ribosomes et autres facteurs d’initiation de
la synthèse protéique aux ARNm [425]. Cet effet peut être médié par la phosphorylation de
facteurs de traduction par des protéines kinases qui sont activées par ERK1/2 [425,426]. En
outre, ERKs favorisent la croissance cellulaire via la phosphorylation de facteurs de
transcription. La phosphorylation de plusieurs facteurs de transcription par ERKs augmente
leur niveau de transcription, ce qui augmente alternativement la production de protéines et
facteurs nécessaires à la croissance cellulaire. D’autre part, les ERKs peuvent également
faciliter la transcription en changeant la structure de la chromatine. Des substrats de
ERK1/2 tels que Rsk-2 et MSK-1 et -2 phosphorylent l'histone H3, l'histone H1, et HMG-
14 (high-mobility group-14) [427-431]. Bien que la fonction exacte de ces événements de
phosphorylations ne soit pas connue, il semble qu’elle améliore l'accessibilité de l'ADN aux
facteurs de transcription ou q’elle permet le recrutement des enzymes de modification.
Finalement, le cycle cellulaire est lui-même influencé par la voie de signalisation
ERK/MAPK. ERK augmente la transcription du gène de la cyclin D1 et facilite également
la formation de complexes actifs cycline D1/ CDK4 [432-434]. Ceux-ci phosphorylent la
protéine rétinoblastoma (rb) [435,436], menant au déplacement de ces protéines et des
histones déacétylases, ce qui a comme conséquence l'activation des gènes régulés par le
facteur de transcription E2F au point de restriction. E2F favorise l'expression de la cyclin A
et de la cyclin E vers la fin G1 [437]. Ces cyclines, à leur tour, s'associent à CDK2 et
favorisent la réplication de l’ADN et la croissance cellulaire. En outre, l'entrée dans la
phase S est favorisée par la dégradation protéolytique de l'inhibiteur p27Kip1 à la fin de la
phase G1, et le dégagement du complexe cycline E-CDK2 de l'inhibition [438-441]. Aussi,
il a été montré que p27 peut être phosphorylée in vitro par ERK et que cet événement de
phosphorylation peut jouer un rôle dans la dégradation de p27 et/ou réguler sa capacité de

59
lier CDK2 [438,442]. Ainsi, la voie MEK/ERK n’agit pas seulement au niveau
trancriptionnel pour induire le gène de la cyclin D mais aussi au niveau posttraductionnel
pour réguler l’assemblage de la cycline D avec CDK4 et empêcher l’effet inhibiteur de
p27kip1 [432].
En plus de fonctionner comme un régulateur positif dans la progression du cycle cellulaire,
la stimulation de la voie ERK/MAPK peut également induire l'arrêt du cycle cellulaire chez
les fibroblastes [443,444]. Ces événements sont associés avec l'induction dépendante de
ERK1/2 de l'inhibiteur de CDK2, p21WAF1/Cip1 [443,445]. L’induction de p21WAF1/Cip1 a été
rapportée pour se produire non seulement par voie transcriptionnelle dépendante de p53,
mais aussi par des mécanismes indépendants de p53 par l'intermédiaire de l’activation de
ERK [444,446,447]. Bien que l'activité ERK soit associée à l'induction de la cyclin D1 et
de p21WAF1/Cip1, plusieurs études ont montré que, contrairement au processus d’activation de
la cycline D1, la décision cellulaire à induire p21WAF1/Cip1 dans la phase G1 du cycle
cellulaire serait en grande partie dicté par l'importance, plutôt que la durée, du signal ERK
[127,443]. Ainsi une activation soutenue de MEK1-ERK1/2 pourrait mener à l’arrêt de la
croissance via l'induction à long terme de p21WAF1/Cip1, alors que l'activation biphasique
mais moins robuste de la voie ERK/MAPK pourrait principalement mener à la prolifération
et à la croissance des cellules. À cet égard, il est intéressant de noter que, chez les cellules
C7caMEK1, l’expression accrue de la cycline D serait liée à une prolifération réduite des
cellules [418,420]. Une forte activation de ERK1/2 chez ces cellules pourrait mener à une
expression forte et persistante de p21WAF1/Cip1, atténuant de ce fait la progression de cycle
cellulaire en dépit de l'expression accrue de la cycline D1.

60
1.7. Le projet de recherche
1.7.1. Mise en contexte
Le laboratoire du Dr Charron du centre de recherche en cancérologie de l’Université Laval
a généré par mutagenèse d’insertion une souris sans fonction MEK1 [77]. L’absence de
protéine MEK1 chez les souris homozygotes pour la mutation cause la mort des embryons
entre les jours 10 et 11 de la gestation. L’analyse histopathologique des embryons a permis
de montrer que les embryons sont normaux. Cependant, MEK1 est essentiel au
développement normal du placenta. Les embryons mutants sont incapables de vasculariser
le labyrinthe, une région du placenta où les sinus maternels (aire de la circulation sanguine
maternelle) se retrouve à proximité des vaisseaux sanguins de l’embryon pour faciliter les
échanges gazeux et nutritionnels. La caractérisation détaillée du phénotype placentaire a
permis de montrer que la néovascularisation n’est pas affectée chez les embryons mutants.
Cependant, les cellules endothéliales des vaisseaux sanguins chez les mutants s’accumulent
à la frontière du labyrinthe suggérant plutôt un problème d’angiogenèse [77].
L’angiogenèse implique la digestion de la matrice extracellulaire pour remodeler les tissus
et permettre la migration, la prolifération et la différenciation des cellules endothéliales
nécessaires à la production de nouveaux vaisseaux [448]. Plusieurs facteurs de croissances
et cytokines stimulant la mobilité et l’angiogenèse ont été identifiés, les FGFs (fibroblast
growth factors), les EGFs (endothelial growth factor) et les VEGFs sont les plus
caractérisés [449]. Des études ont montré que la voie ERK/MAPK est impliquée dans la
migration induite par les matrices extracellulaires. Les récepteurs membranaires à activité
tyrosine kinase et les intégrines sont des éléments essentiels de la cascade intracellulaire
des MAPKs visant la régulation de l’expression génique et de l’organisation
cytosquelletique nécessaires à la migration cellulaire [450].

61
Figure 1-6 : MEK1 est impliquée dans la signalisation intracellulaire menant à la
migration des fibroblastes embryonnaires de souris.
Panneau du haut : Essai le migration en chambre de Boyden : Les MEFs Mek1 type
sauvage ou mutants sont mis au repos pendant 24h puis testés pour leur capacité à migrer à
travers une membrane en polycarbonate recouverte de BSA (contrôle), de collagène ou de
fibronectine. Les photos : Images en contraste de phase des MEFs Mek1 type sauvage ou
mutants 90 min après avoir été mis au repos et étalés sur sur diverses matrices; BSA,
collagène ou fibronectine. Panneau du bas : Niveau de phosphorylation de ERK2 chez les
MEFs Mek1 type sauvage (mek1+/+) ou mutants (mek -/-) en suspension (sus) ou étalés sur
fibronectine (fn). Tiré de la référence [77].

62
L’utilisation de fibroblastes embryonnaires (MEFs) de type sauvage ou mutant pour le gène
Mek1 a permis de montrer que la migration induite par la fibronectine est réduite chez les
MEFs Mek1-/-alors que la migration induite par le collagène n’est pas affectée (figure 1-5)
[77]. La migration induite par la fibronectine peut être restaurée chez les cellules mutantes
par la transfection de vecteur d’expression MEK1 [77]. Ces résultats montrent que
l’induction de la migration par la fibronectine est dépendante de la fonction MEK1.
Cependant ERK1 et ERK2, les substrats de MEK1 et MEK2, sont activés chez les MEFs
Mek1-/- induits par la fibronectine suggérant donc que MEK2, toujours présent dans les
MEFs Mek1-/- pourrait activer ERK1 et ERK2, et que MEK1 serait impliquée dans la
migration via l’activation d’un substrat encore inconnu.
D’autres travaux qui sont en cours dans le laboratoire du Dr Charron suggèrent que la cause
principale de la mort des embryons Mek1-/- serait une morphogenèse anormale de la
barrière de syncytiotrophoblastes (ST) (Bissonauth et Charron, en préparation). Cette
couche de ST constitue la barrière ultime entre le sang fœtal et le sang maternel. Chez les
individus de type sauvage, les précurseurs de ST semblent prendre leur origine dans des
petits amas de cellules au niveau du chorion vers E8.5, lorsque l’allantois fusionne avec le
chorion. Ces précurseurs s’invaginent ensuite dans le chorion pour donner éventuellement
une barrière de cellules entre le sang fœtal et le sang maternel. Chez le placenta Mek1-/- par
contre, bien que les cellules souche ST apparaissent normalement à E8.5, elles n’arrivent
pas à envahir le chorion et se retrouvent plutôt en amas à la jonction chorion-allantois. De
plus, des essais de phosphatase alcaline, d’immunofluorescence et de microscopie
élecronique suggèrent que certains de ces précurseurs Mek1-/- se différencient en ST mais
restent toujours bloqués à la jonction chorion-allantois (Bissonauth et Charron, en
préparation). D’autre part, des expériences d’analyse Western faits à partir d’extraits
protéiques totaux de placentas Mek1-/- montrent que MEK2 n’arrive pas à remplacer MEK1
pour l’activation de la voie ERK/MAPK. En outre, des essais d’immunohistochimie
utilisant des anticorps anti phospho-MEK1/2 et anti phospho-ERK1/2 montrent que MEK2
est phosphorylée de façon ubiquiste au niveau du chorion Mek1-/- mais n’active pas ERK.
(Bissonauth et Charron, en préparation). L’ensemble de ces résultats suggère une fonction
divergente entre MEK1 et MEK2 au niveau des syncytiotrophoblastes placentaires.

63
Récemment, il a été démontré que la voie de signalisation MAPK/ERK peut mener à
l’activation de protéines intracellulaires responsables de la mobilité [450]. Plusieurs
nouvelles protéines d'échafaudage ayant un rôle important dans la voie MAPK ont été
trouvées [260,262,277]. De nouveaux substrats de MEK1, autres que ERK1 et ERK2,
existeraient d’après deux récentes recherches [451,452], indiquant que la voie dépendante
de MEK1 serait plus complexe que celle que l'on connaît aujourd’hui.
1.7.2. Hypothèse de travail
En tenant compte de ces recherches et à la lumière de l’ensemble des résultats obtenus dans
notre laboratoire, notre hypothèse de travail est qu’il y aurait une voie de signalisation
spécifique à MEK1 qui pourrait ne pas impliquer la protéine kinase MEK2 ni les seuls
substrats connus de MEK1 et 2, ERK1 et ERK2. Les protéines kinases MEK1 et MEK2
pourraient être différemment régulées selon le contexte cellulaire et l’issue physiologique
finale imposée par le signal extracellulaire. En d’autres mots, l’activité de MEK1 pourrait
être dépendante de la formation d’un complexe protéique composé de MEK1, de son
activateur (Raf ou autre) et d’un substrat encore inconnu. L’existence de nouveaux
substrats spécifique à MEK1 impliquerait vraisemblablement une nouvelle voie de
signalisation menant à une réponse cellulaire spécifique tel que la migration cellulaire. En
outre, la présence d’un cross-talk entre le sentier ERK et une voie de transduction qui est
médié par MEK1 n’est cependant pas exclue des mécanismes possibles menant à une
réponse cellulaire spécifique. De plus, grâce à des protéines d’échafaudage spécifiques,
MEK1 pourrait être incluse dans un complexe protéique différent de celui impliqué dans la
réponse aux mitogènes et/ou se localiser à des compartiments cellulaires différents.

64
1.7.3. Objectifs du projet
Les MAP kinases de la famille ERK sont impliquées de façon importante dans la
transmission des signaux émis par les facteurs de croissances, les hormones, et divers types
de stress. Elles font partie d’une cascade de signalisation dont leurs activateurs les plus
précoces sont les MAPKKs, MEK1 et MEK2. Mon projet de doctorat portait sur la
caractérisation de la fonction de MEK1.
Les objectifs spécifiques du projet étaient :
1. De démontrer que l’activation de MEK1 et MEK2 pouvait être spécifique à certains
agonistes.
2. De caractériser les mécanismes sous-jacents à cette spécificité.
Dans cette optique, j’ai étudié le comportement de la voie ERK, chez les fibroblastes
embryonnaires de souris mutants pour le gène Mek1, en réponse à divers stimuli,
notamment des facteurs de croissance, des cytokines et des hormones. J’ai observé que
MEK1 mais non pas MEK2 était nécessaire à l’activation de ERK par la bombésine. Ces
résultats vont dans le même sens que ceux des travaux antérieurs faits dans notre
laboratoire et qui démontraient que MEK2 n’active pas ERK chez les trophoblastes
embryonnaires de souris. Afin de caractériser les mécanismes possiblement impliqués dans
ces réponses différentielle, j’ai utilisé l’approche double hybride et fait une étude
approfondie des interactions de MEK1 ou mutants de délétion MEK1 avec d’autres
protéines, notamment Raf, ERK et MEK1 elle-même. Par des études de délétions et
d’interaction protéiques, j’ai pu démonter que MEK1 interagit avec Raf via sa région C-
terminale et que cette même région est impliquée dans une interaction intramoléculaire
MEK1-MEK1. Je me suis, alors, particulièrement intéressé à caractériser le rôle et les
mécanismes de cette interaction intramoléculaire.

Chapitre 2
2. Étude de la signalisation ERK/MAPK chez les fibroblastes embryonnaires de souris Mek1-/-
[MEK1 vs MEK2 est activée par la bombésine chez les MEFs Mek1-/-]
Nizar Chetoui, Michel Tremblay, Sophie Roy et Jean Charron
Les travaux présentés dans ce chapitre font partie d’un projet en cours dans le laboratoire
du Dr Jean Charron au centre de recherche en cancérologie de l’Université Laval.
L’ensemble des résultats obtenus dans ce projet fera l’objet d’une publication. J’ai participé
à la réalisation du projet à plusieurs niveaux, tout particulièrement les essais d’activation de
la voie ERK/MAPK par la bombésine présentés dans la figure 2-5 de ce chapitre. Les
résultats présentés dans les figures 2-3 et 2-4 ont été obtenus par Michel Tremblay, alors
assistant de recherche au laboratoire.

66
2.1. Résumé
La voie de signalisation ERK/MAPK fait partie d’un ensemble de voies de transduction des
signaux à l’intérieur de la cellule qu’on appelle les voies MAP kinases. La voie ERK joue
un rôle majeur dans la prolifération cellulaire, mais elle est aussi impliquée dans la
différenciation, la mobilité et l’apoptose cellulaires. L’issue physiologique de la cellule est
déterminée par la force et la durée de l’activation de la voie ERK/MAPK. Ainsi, elle
dépenderait du type cellulaire, de la nature des agonistes et des récepteurs qui déclenchent
le signal et des caractéristiques des composantes signalétiques qui interviennent dans la
transmission du signal à l’intérieur de la cellule. La voie ERK/MAPK est activée par une
grande panoplie de stimuli extracellulaires qui sont transmis aussi bien via les récepteurs à
activité tyrosine-kinase que via les récepteurs transmembranaires couplés aux protéines G
et les intégrines. Ici, nous montrons que, chez les fibroblastes embryonnaires de souris
(MEF), le neuropeptide bombésine active la voie ERK/MAPK et que cette activation
requiert l’implication de la MAPKK, MEK1, mais pas celle de son homologue MEK2. En
effet, la stimulation, par la bombésine, des MEFs mutants pour le gène Mek1 ne semble pas
induire l’activation de ERK. En outre, la réexpression, chez ces MEFs, de transgènes Mek1
rétablie l’activation de ERK. Par contre, la surexpression de MEK2 n’a aucun effet sur la
voie ERK en réponse à la bombésine. D’autre part, la caractérisation des domaines
protéiques impliqués dans cette réponse spécifique suggère que la séquence riche en
proline, présente dans la région C-terminale de MEK1, est impliquée dans l’activation de
ERK par la bombésine. L’ensemble de nos travaux fournit ainsi un excellent modèle pour
l’étude des divergences fonctionnelles des MAPKKs MEK1 et 2 et pour la caractérisation
du rôle unique de MEK1 dans la voie de signalisation ERK/MAPK.

67
2.2. Introduction
La bombésine fait partie d’un groupe de neuropeptides mitogènes, dit bombesin-like
peptides ou bombésine/GRP, dans lequel on retrouve aussi la gastrine et la
cholécystokinine (CCK) [1]. La bombésine a été décrite comme un facteur de croissance
autocrine dans le cancer des petites cellules du poumon chez l’humain [2]. La bombésine
est un tétradecapeptide qui se lie aux récepteurs transmembranaires couplés à Gq. Chez les
cellules 3T3, qui ont souvent été employées comme système d’étude, elle induit la synthèse
d'ADN et la prolifération cellulaire [3]. Différentes voies de transduction du signal ont été
suggérées pour médier les effets de la bombésine [2,4,5]. La nature de la voie de
signalisation transmettant une réponse à la bombésine semble être dépendant du contexte
cellulaire. Différentes GTPases peuvent être impliquées, mais ces dernières ne semblent pas
être requises chez certains types cellulaires [3,6]. Les premières études menées par
Rozengurt et collaborateurs ont identifié la protéine kinase PKC comment étant un élément
important dans la transmission des signaux intracellulaires médiés par les neuropeptides
bombésine/GRP [7-9]. Des études ultérieures ont indiqué le rôle majeur de la voie
ERK/MAPK dans la prise en charge des signaux provenant de PKC et/ou des GTPases en
réponse à la bombésine/GRP [10-14].
La bombésine lie les récepteurs couplés aux protéines G hétérotrimèriques,
particulièrement ceux du sous-type bombésine/GRP (Figure 2-1) [15]. L’attachement au
récepteur est suivi par l’activation de la phospholipase C et la production des seconds
messagers, l’inositol-1,4,5-trisphosphate et le diacylglycérol, ce qui mène à la mobilisation
du calcium intracellulaire et l’activation de la protéine kinase C [6]. En outre, la
sensibilisation du récepteur de la bombésine peut mener à l'activation des protéines kinases
FAK, JNKs et S6 kinases [16-19]
La bombésine active les MAP kinases ERKs dans divers tissus et cellules telles que les
fibroblastes Swiss 3T3 [3,20,21], les fibroblastes Rat1 [10], les acini pancréatiques de rat
[11,22], les cellules du muscle lisse du rectosigmoïde de lapin [23] et dans la lignée de
cellules STC-1, qui sont des cellules entéroendocrines qui secrètent la CCK

68
(cholecystokinin) [12]. La bombésine semble induire une activation soutenue de ERK1 et 2
chez les cellules Swiss 3T3 [3,5] via une voie de signalisation qui est dépendante de Gq et
de PKC [3] et qui ne semble pas impliquer Ras ou l’activation de Raf-1 [3,24]. Cette voie
se distingue de celle induite par Gi et les récepteurs RTK, qui implique l'activation de Raf-1
via Ras/GTP. Le ou les mécanismes précis par lesquels les isoformes de la famille PKC
activent les MAP kinases ERKs ne sont pas encore clairs [25].
Cependant, Ras peut aussi être impliquée dans l'activation des MAP kinases ERKs par les
récepteurs couplés à Gq [26]. Le contexte cellulaire dans lequel la voie ERK est activée
devient alors un élément important dans la compréhension du rôle de la bombésine dans
divers processus cellulaires [10,27,28]. En effet, chez les cellules Rat-1 dans lesquelles on a
introduit le récepteur de la bombésine/GRP, la bombésine induit l'activation de ERK via
une voie indépendante de PKC mais dépendante de Ras [10,29]. Ces données démontrent
que le même récepteur peut activer la cascade ERK/MAPK via des voies distinctes, et ce
dépendamment du contexte cellulaire (Figure 2-2).
Récemment, on a proposé que, chez certaines cellules (e.g., HEK 293 et PC12), les signaux
via Gβγ et Gα peuvent converger pour favoriser la mobilisation du Ca2+ d’une manière
dépendante du PLC et menant à la stimulation de Pyk2 (proline-rich tyrosine kinase 2) et
l’activation de c-Src (Figure 2-2) [27,30-32]. Src mène alors à l'activation des MAP kinases
via la phosphorylation d’adaptateurs moléculaires, tels que Shc, sur des résidus tyrosines. Il
est donc possible que la transactivation du récepteur de EGF ait lieu via les agonistes des
récepteurs GPCRs et que cela mènerait à l'activation du module Raf-MEK-ERK via
l’activation de la GTPase Ras [33-35]. Ceci dit, les études faites sur cette voie restent
controversées [36]. En effet, on a également montré que l'activation de ERK via certains
GPCRs semble exiger l'endocytose du récepteur [37], reliant ainsi la signalisation du
récepteur à son internalisation subséquente[30].

69
Figure 2-1 : Schéma général de la signalisation induite par la bombésine
La voie ERK/MAPK est une des voies de signalisation induites par la bombésine. Tiré de la
référence [6].

70
Les deux MAPKKs de la voie ERK/MAPK, MEK1 et MEK2, démontrent une forte
homologie de séquence dans leur domaine kinase. Par contre, la région N-terminale ainsi
que la région riche en proline particulière à ces MAPKKs sont très divergentes
(respectivement, 66% et 36% d’identité). Cette divergence ainsi que plusieurs études
récentes suggèrent que MEK1 et MEK2 auraient des fonctions distinctes. Les souris sans
fonction MEK1 meurent in utero suite à une déficience placentaire alors que MEK2 ne
semble pas être importante pour le développement et la croissance [38,39].
Nos travaux indiquent que, chez les fibroblastes embryonnaires de souris, la signalisation
induite par la bombésine emprunte une voie spécifique à MEK1 au dépend de son
homologue MEK2. Alors que la plupart des facteurs de croissance testés ne présentent pas
cette spécificité, l’effet bombésine s’est présenté comme un moyen intéressant pour étudier
la divergence fonctionnelle entre MEK1 et MEK2. Ainsi, nos résultats suggèrent que la
région C-terminale de MEK1 médie l’activation de ERK par la bombésine. Nous proposons
que la séquence riche en proline présente dans cette région soit à l’origine de cette
spcificité.

71
Figure 2-2 : Activation de la voie ERK/MAPK via Gi et Gq.
Importance du contexte cellulaire dans l’acheminement du signal jusqu’au module
RAF/MEK/ERK de la voie de signalisation ERK MAP kinase. Tiré de la référence [6].

72
2.3. Matériels et méthodes
Culture cellulaire et Stimulation
Les MEFs MEK1 sont maintenues à 37°C dans du milieu DMEM (Dulbecco’s modified
Eagle’s medium) avec 10% de sérum foetal bovin (FBS) dans une atmosphère humide
contenant 5 % de CO2. Pour préparer les cellules aux différents essais de stimulation, deux
procédures différentes ont été suivies quant à leur prolifération et leur mise au repos. Dans
la première procédure, les cellules sont étalées à raison de 5 à 7.5 105 cellules par pétri de
35 mm. Après 24 h d’incubation dans du milieu DMEM-FBS 10%, les cellules sont mises
au repos dans du milieu DMEM-FBS 0.1% pendant 24h. Dans la deuxième procédure, les
cellules sont étalées dans des pétri 35 mm à 1-3 105 cellules par pétri et incubées pendant
7–9 jours jusqu’à confluence et quiescence. Les MEFs sont ensuite lavés deux fois avec du
milieu DMEM dépourvu de sérum, conditionnés à 37°C pour environ 10 minute et ensuite
traités avec les agonistes tel qu’indiqué. La stimulation est terminée en plaçant les pétris sur
de la glace, en aspirant le milieu et en solubilisant les cellules dans in 0,3 ml de tampon de
lyse froid (20 mM Tris {pH 7.5}, 10 % glycérol, 5 mM EGTA, 1 mM EDTA, 1 % Triton
X-100, 2 mM ß-glycérolphosphate, 50 mM NaF, 10 µM Na3VO4, 5 mM DTT, 10 µg/ml
leupeptin et 0.2 mM PMSF). Les lysats cellulaires sont ensuite clarifiés par centrifugation à
14,000 rpm pour 10 min à 4°C. Les surnageants sont transférés dans de nouveaux tubes et
conservés à -70 °C.
Immunopurification de ERK2 et essai kinase
Les extraits cellulaires clarifiés sont incubés pendant une heure avec des anticorps anti
ERK2 (Obtenue du laboratoire du Dr. John H. Grose) puis encore une heure en présence de
billes de protéine A-Sépharose. Après immunoprécipitation, les billes sont lavées 3 fois
avec le tampon de lyse puis récupérées dans le tampon kinase (20 mM Tris {pH=7.5}, 10%
glycérol, 120 mM pNPP, 30 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, 1 µM leupeptin, 100
µM ATP, 3 µCi [γ32P] ATP, 1 µg of MBP (Cell Signaling Technology, Pickering, ON)).

73
Les réactions kinase ainsi amorcées sont effectuées à 30oC pendant 20 min et ensuite
arrêtées par ajout de tampon TEX (SDS-loading buffer). Les protéines sont ensuite séparées
par SDS-PAGE et les protéines MBP phosphorylées sont détectées par autoradiogramme.
Immunodétection
Après SDS-PAGE, les protéines sont transférées sur des membranes PVDF (polyvinylidene
difluoride). Pour la détection des protéines, les membranes sont bloquées avec 3% de lait
déshydraté dans du tampon PBS (phosphate buffered saline) {pH 7.5} puis incubées
pendant 1 h avec les anticorps appropriés dilués dans le tampon PBS {pH 7.5} contenant
1% de lait déshydraté. Les anticorps primaires fixées sur les bandes immunoréactives sont
visualisés par la détection de la chemiluminescence (ECL+) des anticorps secondaires
conjugués à la HRP (horseradish peroxidase).
Essais d’activité de ERK1 et ERK2
L’activation de ERK1 et ERK2 se produit par la phosphorylation des résidus thréonine et
tyrosine de la boucle d’activation. Ainsi, ces protéines phosphorylées migrent plus
lentement que les formes non phosphorylées sur un gel dénaturé SDS-PAGE [40]. Ces
formes activées sont détectées soit par l’utilisation d’anticorps anti-ERK2 dans un essai de
retardement ou par l’utilisation d’anticorps antiphospho-p44/p42mapk (New England
Biolabs, Beverly, MA) qui ne reconnaissent que les formes activées de ERK1 et ERK2.

74
2.4. Résultats
La voie ERK n’est pas activée chez les MEFs Mek1-/-traités à la bombésine
Dans le but de mieux comprendre l’implication de MEK1 dans la transmission des signaux
via la voie de signalisation ERK/MAPK, nous avons observé, chez les MEFs Mek1-/-, le
niveau d’activation de cette voie en réponse à différents stimulis et inducteurs connus.
Ainsi, l’absence de MEK1 ne semble pas affecter l’activation de la cascade ERK/MAPK
lorsque les cellules sont traitées avec PDGF, EGF, LIF, HGF (Figure 2-3a et b), la
thrombine ou encore le VEGF (données non présentées). MEK2, qui est toujours présente
chez les MEFs Mek1-/-, phosphoryle et active ERK et semble ainsi compenser la perte de
MEK1. Par contre, lorsque les cellules sont traitées avec le neuropeptide bombésine, la
perte de MEK1 semble empêcher la progression du signal vers le module ERK (Figure 2-
3c). Ainsi, ERK1 et ERK2 ne semblent pas être activées par MEK2 en réponse à la
bombésine. L’ensemble de ces résultats indique que la bombésine génère une cascade de
signalisation spécifique à MEK1 chez les fibroblastes embryonnaires de souris. Nos
résultats corroborent ceux obtenus avec la lignée de fibroblastes Swiss 3T3 et qui
démontraient que l’activation de ERK par la bombésine est spécifiquement médiée par
MEK1 [3].
La bombésine induit la voie ERK via MEK1
Pour confirmer le rôle unique de MEK1 dans la réponse à la bombésine, nous avons
analysé l’activation de la voie ERK/MAPK chez les MEFs Mek1-/- dans lesquels nous
avons réexprimé MEK1 ou surexprimé MEK2 (Figure 2-4). Nos résultats indiquent que la
réintroduction d’un transgène Mek1, qui permet la production de la protéine MEK1 à
seulement 10% du niveau normal présent dans les MEFs, peut rétablir l’activation de la
voie ERK/MAPK en réponse à la bombésine. La surexpression à l’aide d’un transgène
Mek2 à de très hauts niveaux, allant jusqu’à 380% du niveau d’expression de Mek2
endogène, ne peut restaurer l’activation de la voie ERK/MAPK.

75
La région riche en proline de MEK1 médie l’activation de la voie ERK par la bombésine
La région riche en proline qui est unique à MEK1 et MEK2 mais qui est divergente dans sa
composition primaire se trouve dans la région C-terminale de ces protéines et est décrite
comme élément important dans la fonction signalétique de MEK1 et MEK2 (voir la région
MSS dans le chapitre 1 pour détails). Pour déterminer si cette région est essentielle à la
spécificité de MEK1 en réponse à la bombésine, nous avons réexprimé dans les MEFs
Mek1-/- une protéine chimérique MEK2/1 formée de la région N-terminale de MEK2 (a.a 1-
116) et la région C-terminale de MEK1 (a.a 117-393). Tel qu’indiqué dans la figure 2-4, la
présence de MEK2/1 chez les cellules sans fonction Mek1 rétablit l’activation du sentier
ERK par la bombésine. Ces résultats suggèrent que la région C-terminale de MEK1 médie
la spécificité de la réponse cellulaire à la bombésine.
La confluence cellulaire serait importante pour l’induction de la voie ERK par la
bombésine
Nos essais d’activation de la voie ERK par la bombésine ont démontré que la confluence
des MEFs serait importante pour l’activation adéquate de la voie ERK/MAPK. En effet,
nous n’avons pu observé l’activation de la voie ERK/MAPK par la bombésine lorsque nous
avons repris les expériences de stimulation des MEFs Mek1-/-. Les MEFs Mek1-/- aussi bien
que les MEFs de type sauvage ne semblent pas répondre à la bombésine, bien que la
réponse aux facteurs de croissance tel que EGF ou au sérum semblait être normale (données
non présentées). Nous avons alors repris les essais d’induction en utilisant un nouveau
protocole de mise en culture des cellules et de stimulation (voir section Matériels et
méthodes pour comparer les deux protocoles). Alors qu’au premier protocole, les MEFs
cultivés n’atteignent généralement pas la confluence, au deuxième prototole, ils sont
maintenus à confluence pendant plusieurs jours. Nos résultats indiquent que, dans ces
nouvelles conditions de culture, les MEFs de type sauvage répondent bien à l’induction par
la bombésine (Figure 2-5). Ces données suggèrent, que la bombésine est plus susceptible
d’induire l’activation de la voie ERK/MAPK chez les cellules qui atteignent la confluence
que chez celles qui ne sont pas confluentes. Le rôle de la confluence cellulaire dans la

76
réponse à la bombésine n’est pas clair. Cependant, il est possible que, chez les MEFs,
l’expression des récepteurs de la bombésine soit régulée en fonction de la confluence
cellulaire et/ou dépendante de la présence de facteurs mitogéniques qui sont produits par les
MEFs en état de confluence.
Par ailleurs, l’activation de la voie ERK/MAPK par la bombésine semble atteindre son
seuil maximale au bout de 5 min de stimulation et s’estomper rapidement de telle sorte qu’à
15 min, il y a perte totale d’activation (Figure 2-5). La réponse cellulaire à la bombésine
chez les MEFs, tout comme celle de EGF, semble être brève et rapide.
Les MEFs Mek1-/- meurent en condition de confluence prolongée
Plusieurs lignées MEFs Mek1-/- qui ont fait l’objet des essais de stimulation par la
bombésine décollaient du fond du pétri après 4 à 5 jours d’incubation ce qui suggère que
ces cellules ne tolèrent pas des niveaux élevés de confluence ou qu’elles tolèrent moins
l’épuisement de sérum dans le milieu de culture. D’autre part, les lignées MEFs Mek1-/-
stables chez lesquelles nous avons réintroduit un transgène Mek1 présentent la même
défaillance et décollent aussi après une incubation prolongée sans apport de sérum.
L’absence de MEK1 chez les MEFs serait-elle la cause de cette défaillance? L’expression
du transgène Mek1 est sous le contrôle d’un promoteur CMV, ce dernier est un promoteur
fort mais qui est sensible à l’absence de sérum (Aubry et Charron, résultat non publié). Il
est donc possible que l’expression de transgène Mek1 soit faible chez les MEFs cultivés
plusieurs jours sans apport de sérum. Échanger le promoteur CMV par un autre promoteur
insensible à l’absence de sérum, tel que le promoteur RSV, pourrait être une alternative
pour mieux comprendre le rôle de MEK1 dans la prolifération et la survie des MEFs
lorsque cultivés à une forte confluence.

77
Figure 2-3

78
Figure 2-3 : Altération de la signalisation induite par la bombésine chez les MEFs
mutants pour le gène mek1.
Les MEFs Mek1 de type sauvage (Mek1+/+ ; 10126) ou mutants (Mek1-/-, 10124 ou 13187)
sont étalés sur des pétris 35 mm à raison de 5-7.5 x 105 cellules par pétri et maintenus
pendant 24 heures en milieu contenant 10% de FBS. Ils sont ensuite mis au repos en les
incubant dans un milieu ne contenant que 0.1 % de FBS pendant encore 24 heures. (a) Les
fibroblastes sont induits par le sérum foetal de veau FCS (fetal calf serum), ou par les
facteurs de croissance PDGF ou EGF pendant 5 min. Les concentrations utilisées sont
indiquées. L’activation de la voie ERK/MAPK est détectée par le niveau de
phosphorylation de MBP dans un essai kinase in vitro. (b) Les fibroblastes sont stimulés
par la cytokine LIF (leukemia inhibitory factor) ou le facteur de croissance HGF
(hepatocyte growth factor) avec les concentrations ou les dilutions indiquées pendant 5
min. Les niveaux de phosphorylation de ERK1/2 sont détectés en utilisant des anticorps
anti phospho-ERK1/2. (c) Les fibroblastes MEK1 sont induits par la bombésine en utilisant
les concentrations indiquées pendant 5 min. Les niveaux de phospho-ERK1/2 sont détectés
avec les anticorps dirigés contre ERK1/2, du fait que les formes phosphorylées et non
phosphorylés de ERK1/2 migrent différemment sur un gel de polyacrylamide dénaturé. ctl ;
cellules non traitées. L’induction au FCS a servi de contrôle positif pour le niveau
d’activation de la voie ERK/MAPK.

79
Figure 2-4

80
Figure 2-4 : La signalisation induite par la bombésine est spécifique à MEK1.
Les MEFs Mek1 de type sauvage ou mutants (Mek1-/-) sont étalés sur des pétris 35 mm à
raison de 5-7.5 x 105 cellules par pétri et maintenues pendant 24 heures en milieu contenant
10% de FBS. Ils sont ensuite mis au repos dans un milieu ne contenant que 0.1 % de FBS
pendant encore 24 heures jusqu’à quiescence. Les fibroblastes sont ensuite induits par des
concentrations variables de bombésine (tel qu’indiqué, 5 ou 10 ng/ml) ou par 20 % de FBS
qui a servi comme contrôle positif de l’activation de ERK2. ctl ; cellules non induites.
MEK2/1 signifie une protéine chimérique formée de la région N-terminale de MEK2 (a.a
1-116) fusionnée à la région C-terminale de MEK1 (a.a 117-393).

81
Figure 2-5
ERK1-p
ERK2-pα-pp ERK1/2
α- ERK1/2
Bombésine (µM) 0 FBS5 10 50 10 10
ERK2-pERK2
ERK1-pERK1
Induction (min) 5 3 15 5
ERK1-p
ERK2-pα-pp ERK1/2
α- ERK1/2
Bombésine (µM) 0 FBS5 10 50 10 10
ERK2-pERK2
ERK1-pERK1
Induction (min) 5 3 15 5Induction (min) 5 3 15Induction (min) 5 3 15 5

82
Figure 2-5 : Activation de la voie ERK/MAPK en réponse à la bombésine chez les MEFs
Mek1 confluents
Les MEFs Mek1 de type sauvage (10126) sont étalés sur des pétris 35 mm à raison de 105
cellules par pétri puis incubés pendant 7–9 jours jusqu’à confluence et quiescence. Les
fibroblastes sont ensuite induits, tel qu’indiqué, avec des concentrations variables de
bombésine et pour des temps variables d’induction. Le taux d’activation de la voie
ERK/MAPK est détecté de deux façons ; par l’utilisation d’anticorps anti ERK1/2 (panneau
du haut) ou par l’utilisation d’anticorps anti phospho-ERK1/2 (panneau du bas). La
stimulation par le FBS à 20% a servi de contrôle positif.

83
2.5. Discussion
La compréhension des mécanismes permettant des réponses biologiques spécifiques est une
des issues majeures dans la transduction des signaux intracellulaire. Cette compréhension
ne pourrait être parfaite que par l’illustration des voies de transduction utilisée par un
agoniste distinct et par la désignation des composantes impliquées dans une réponse
cellulaire particulière. Plusieurs mécanismes favorisant une telle réponse ont été proposés :
Elle résulterait d’une force et/ou d’une durée spécifique de l'activation de la cascade
conventionnelle. Elle résulterait de la combinaison de signaux ou de cascades multiples tel
que le cross-talk. Enfin, seulement quelques composantes d'une cascade seraient
spécifiquement activées et être ainsi impliquées dans la production d'une réponse
particulière.
Il est ainsi plausible que, chez les mammifères, MEK1 et son homologue MEK2 pourraient
jouer des rôles distincts dans la cascade ERK pour réaliser des fonctions uniques.
D'ailleurs, beaucoup d'observations indiquent des différences fonctionnelles entre MEK1 et
MEK2. Par exemple, seulement MEK1 est activée chez les cellules Suisse 3T3 en réponse
à la bombésine et chez les macrophages stimulés par le TNFα [3,41].
Dans nos travaux, nous avons montré que la signalisation induite par la bombésine
emprunte une voie spécifique à MEK1 au dépend de son homologue MEK2. Alors que
plusieurs facteurs de croissance testées dans notre étude ne présentent pas cette spécificité,
l’effet de la bombésine sur la voie ERK/MAPK s’est présenté comme un moyen intéressant
pour étudier la divergence fonctionnelle entre MEK1 et MEK2 et mieux définir les voies de
signalisation qui mènent à des effet biologiques uniques.
La voie de signalisation conventionnelle Ras-Raf-MEK1/2-ERK1/2 activée par des signaux
mitogéniques tels que EGF ou PDGF ne semble pas présenter une divergence fonctionnelle
entre les modules homologues MEK1 et MEK2 ou ERK1 et ERK2. Cette voie, dite
proliférative, pourrait être considérée comme une voie principale dont l’issue pourrait être

84
modulée par des voies annexes plus spécifiques et qui font appel à d’autres modules de
signalisation et au phénomène de cross-talk entre les différentes voies de signalisation.
La divergence fonctionnelle entre MEK1 et MEK2 telle qu’observée dans le cas de la
l’induction par la bombésine, pourrait être une alternative à la voie de signalisation
conventionnelle Ras-ERK. Elle suggère l’existence de chevauchement entre plusieurs voies
de signalisation via le module MEK. Ce module formé de deux protéines, bien que très
homologue et généralement équivalent de point de vue fonctionnel, permettrait dans un
contexte unique de médier une réponse spécifique.
L’élément ou la séquence protéique dans MEK1 qui semble médier la réponse spécifique à
la bombésine se retrouverait dans la région C-terminale. Nos résultats indiquent que la
réexpression d’une protéine chimérique MEK2/1; (a.a 1-116)/MEK1 (a.a 117-393), dans
les MEFs Mek1-/- rétablit l’activation de ERK. La région C-terminale, tout particulièrement
le domaine riche en proline ou MSS (MEK specific sequence), semble jouer un rôle
essentiel dans la régulation de l’activité catalytique de MEK1 et MEK2. Le MSS qui se situ
approximativement entre les résidus 270 et 310 semble prendre part à la modulation de
l’activité de MEK1 ainsi qu’au cross-talk du sentier ERK avec d’autres voies de
signalisation. En fait, cette région semble être le site d’interaction de MEK1/2 avec les
membres de la famille Raf et être importante pour l’activation de ERK et l’attachement de
certaines protéines d’échafaudage tel que MP-1 [42-45]. Elle contient de multiples sites de
phosphorylation pour plusieurs protéines kinases, dont PAK1, cdk5 et ERK [46-49]. PAK1
phosphoryle spécifiquement MEK1 sur le résidu Ser298, ce qui permet le cross-talk entre
le sentier ERK et la voie Rac-cdc42. Cette phosphorylation serait nécessaire pour générer
des réponses cellulaires particulières telles que la migration et la prolifération cellulaires
dépendantes de l’adhérence à la matrice extracellulaire [50-52]. L’absence d’un site de
phosphorylation de PAK1 dans MEK2 indique que MEK2 ne peut répondre à des signaux
provenant du sentier Rac/cdc42 et qu’il n’est donc pas impliqué dans le cross-talk avec
cette voie de signalisation.
Le rôle de la confluence cellulaire dans la réponse à la bombésine n’est pas clair.
Cependant, il est possible que, chez les MEFs, l’expression des récepteurs de la bombésine

85
soit régulée en fonction de la confluence cellulaire et/ou dépendante de la présence de
facteurs mitogèniques. Alors que les mécanismes d’induction de la voie ERK/MAPK par
les récepteurs RTKs soient largement identifés, ceux des récepteurs GPCR semblent être
très variés et plus complexes. En effet, plusieurs voies de signalisation peuvent coopérer
pour l’activation du module Raf-MEK-ERK de la voie ERK/MAPK. Ces voies
impliqueraient différentes composantes signalétiques dépendamment du contexte cellulaire
(le type cellulaire, l’adhérence à la MEC, etc.) et de la réponse cellulaire recherchée [53-
55].
Nos résultats fournissent de nouvaux arguments quant au rôle unique que pourrait jouer
MEK1 dans la cascade ERK/MAPK et son importance pour accomplir certains processus
cellulaire spécifique. Alors que, dans un organisme, chaque cellule est exposée
physiologiquement aux multiples agonistes tels les facteurs de croissance, les hormones et
les cytokines, il paraît clair que la nature de la voie transmettant le signal soit susceptible de
dépendre des signaux extracellulaires qui rentrent en contact avec la cellule. Ce concept est
propice au développement de drogues qui sont spécifiques à des composantes signalétiques
uniques et qui tiennent compte de la nature du signal et de la réponse cellulaire que ce
dernier engendre. Ainsi, il a été proposé que MEK1 serait une bonne cible pour des drogues
ou d'autres agents capables d'empêcher la transformation cellulaire [56]. L’utilisation de
drogues spécifiques à MEK1 pourrait inhiber des processus cellulaires spécifiques qui
dépendent de MEK1, sans pour autant toucher à la voie proliférative qui elle sera
maintenue par MEK2.

86
2.6. Références
1. Rozengurt, E. (1995). Convergent signalling in the action of integrins, neuropeptides, growth factors and oncogenes. Cancer Surv. 24:81-96., 81-96.
2. Sethi, T. and Rozengurt, E. (1992). Gastrin stimulates Ca2+ mobilization and clonal growth in small cell lung cancer cells. Cancer Res. 52, 6031-6035.
3. Seufferlein, T., Withers, D. J., and Rozengurt, E. (1996). Reduced requirement of mitogen-activated protein kinase (MAPK) activity for entry into the S phase of the cell cycle in Swiss 3T3 fibroblasts stimulated by bombesin and insulin. J.Biol.Chem. 271, 21471-21477.
4. Casamassima, A. and Rozengurt, E. (1997). Tyrosine phosphorylation of p130(cas) by bombesin, lysophosphatidic acid, phorbol esters, and platelet-derived growth factor. Signaling pathways and formation of a p130(cas)-Crk complex. J.Biol.Chem. 272, 9363-9370.
5. Seufferlein, T., Withers, D. J., Mann, D., and Rozengurt, E. (1996). Dissociation of mitogen-activated protein kinase activation from p125 focal adhesion kinase tyrosine phosphorylation in Swiss 3T3 cells stimulated by bombesin, lysophosphatidic acid, and platelet-derived growth factor. Mol.Biol.Cell 7, 1865-1875.
6. Rozengurt, E. (1998). Signal transduction pathways in the mitogenic response to G protein-coupled neuropeptide receptor agonists. J.Cell Physiol 177, 507-517.
7. Zachary, I., Millar, J., Nanberg, E., Higgins, T., and Rozengurt, E. (1987). Inhibition of bombesin-induced mitogenesis by pertussis toxin: dissociation from phospholipase C pathway. Biochem.Biophys.Res.Commun. 146, 456-463.
8. Zachary, I., Sinnett-Smith, J., and Rozengurt, E. (1991). Stimulation of tyrosine kinase activity in anti-phosphotyrosine immune complexes of Swiss 3T3 cell lysates occurs rapidly after addition of bombesin, vasopressin, and endothelin to intact cells. J.Biol.Chem. 266, 24126-24133.
9. Zurier, R. B., Kozma, M., Sinnett-Smith, J., and Rozengurt, E. (1988). Vasoactive intestinal peptide synergistically stimulates DNA synthesis in mouse 3T3 cells: role of cAMP, Ca2+, and protein kinase C. Exp.Cell Res. 176, 155-161.
10. Charlesworth, A. and Rozengurt, E. (1997). Bombesin and neuromedin B stimulate the activation of p42(mapk) and p74(raf-1) via a protein kinase C-independent pathway in Rat-1 cells. Oncogene 14, 2323-2329.

87
11. Duan, R. D., Zheng, C. F., Guan, K. L., and Williams, J. A. (1995). Activation of MAP kinase kinase (MEK) and Ras by cholecystokinin in rat pancreatic acini. Am.J.Physiol 268, G1060-G1065.
12. Nemoz-Gaillard, E., Cordier-Bussat, M., Filloux, C., Cuber, J. C., Van Obberghen, E., Chayvialle, J. A., and Abello, J. (1998). Bombesin stimulates cholecystokinin secretion through mitogen-activated protein-kinase-dependent and -independent mechanisms in the enteroendocrine STC-1 cell line. Biochem.J. 331, 129-135.
13. Seufferlein, T., Withers, D. J., Broad, S., Herget, T., Walsh, J. H., and Rozengurt, E. (1995). The human CCKB/gastrin receptor transfected into rat1 fibroblasts mediates activation of MAP kinase, p74raf-1 kinase, and mitogenesis. Cell Growth Differ. 6, 383-393.
14. Rozengurt, E. (1989). Signal transduction pathways in mitogenesis. Br.Med.Bull. 45, 515-528.
15. Zachary, I. and Rozengurt, E. (1985). High-affinity receptors for peptides of the bombesin family in Swiss 3T3 cells. Proc.Natl.Acad.Sci.U.S.A 82, 7616-7620.
16. Zachary, I., Sinnett-Smith, J., and Rozengurt, E. (1992). Bombesin, vasopressin, and endothelin stimulation of tyrosine phosphorylation in Swiss 3T3 cells. Identification of a novel tyrosine kinase as a major substrate. J.Biol.Chem. 267, 19031-19034.
17. Dabrowski, A., Grady, T., Logsdon, C. D., and Williams, J. A. (1996). Jun kinases are rapidly activated by cholecystokinin in rat pancreas both in vitro and in vivo. J.Biol.Chem. 271, 5686-5690.
18. Bragado, M. J., Dabrowski, A., Groblewski, G. E., and Williams, J. A. (1997). CCK activates p90rsk in rat pancreatic acini through protein kinase C. Am.J.Physiol 272, G401-G407.
19. Withers, D. J., Seufferlein, T., Mann, D., Garcia, B., Jones, N., and Rozengurt, E. (1997). Rapamycin dissociates p70(S6K) activation from DNA synthesis stimulated by bombesin and insulin in Swiss 3T3 cells. J.Biol.Chem. 272, 2509-2514.
20. Hansson, A. (1994). Map kinase activation in Swiss 3T3 cells stimulated with gastrin-releasing peptide is associated with increased phosphorylation of a 78,000 M(r) protein immunoprecipitated by anti-raf kinase anti-serum. Cell Signal. 6, 423-431.
21. Withers, D. J., Bloom, S. R., and Rozengurt, E. (1995). Dissociation of cAMP-stimulated mitogenesis from activation of the mitogen-activated protein kinase cascade in Swiss 3T3 cells. J.Biol.Chem. 270, 21411-21419.
22. Dabrowski, A., Groblewski, G. E., Schafer, C., Guan, K. L., and Williams, J. A. (1997). Cholecystokinin and EGF activate a MAPK cascade by different mechanisms in rat pancreatic acinar cells. Am.J.Physiol 273, C1472-C1479.

88
23. Yamada, H., Strahler, J., Welsh, M. J., and Bitar, K. N. (1995). Activation of MAP kinase and translocation with HSP27 in bombesin-induced contraction of rectosigmoid smooth muscle. Am.J.Physiol 269, G683-G691.
24. Mitchell, F. M., Heasley, L. E., Qian, N. X., Zamarripa, J., and Johnson, G. L. (1995). Differential modulation of bombesin-stimulated phospholipase C beta and mitogen-activated protein kinase activity by [D-Arg1,D-Phe5,D-Trp7,9,Leu11]substance P. J.Biol.Chem. 270 , 8623-8628.
25. Schonwasser, D. C., Marais, R. M., Marshall, C. J., and Parker, P. J. (1998). Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol.Cell Biol. 18, 790-798.
26. Gutkind, J. S. (1998). Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene 17, 1331-1342.
27. Della Rocca, G. J., van Biesen, T., Daaka, Y., Luttrell, D. K., Luttrell, L. M., and Lefkowitz, R. J. (1997). Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J.Biol.Chem. 272, 19125-19132.
28. Dikic, I., Tokiwa, G., Lev, S., Courtneidge, S. A., and Schlessinger, J. (1996). A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383, 547-550.
29. Charlesworth, A., Broad, S., and Rozengurt, E. (1996). The bombesin/GRP receptor transfected into Rat-1 fibroblasts couples to phospholipase C activation, tyrosine phosphorylation of p125FAK and paxillin and cell proliferation. Oncogene 12, 1337-1345.
30. Luttrell, L. M., Daaka, Y., Della Rocca, G. J., and Lefkowitz, R. J. (1997). G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J.Biol.Chem. 272, 31648-31656.
31. Wan, Y., Kurosaki, T., and Huang, X. Y. (1996). Tyrosine kinases in activation of the MAP kinase cascade by G-protein-coupled receptors. Nature 380, 541-544.
32. Ward, C. W., Gough, K. H., Rashke, M., Wan, S. S., Tribbick, G., and Wang, J. (1996). Systematic mapping of potential binding sites for Shc and Grb2 SH2 domains on insulin receptor substrate-1 and the receptors for insulin, epidermal growth factor, platelet-derived growth factor, and fibroblast growth factor. J.Biol.Chem. 271, 5603-5609.
33. Cunnick, J. M., Dorsey, J. F., Standley, T., Turkson, J., Kraker, A. J., Fry, D. W., Jove, R., and Wu, J. (1998). Role of tyrosine kinase activity of epidermal growth

89
factor receptor in the lysophosphatidic acid-stimulated mitogen-activated protein kinase pathway. J.Biol.Chem. 273, 14468-14475.
34. Daub, H., Weiss, F. U., Wallasch, C., and Ullrich, A. (1996). Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557-560.
35. Daub, H., Wallasch, C., Lankenau, A., Herrlich, A., and Ullrich, A. (1997). Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 16, 7032-7044.
36. Kranenburg, O., Verlaan, I., Hordijk, P. L., and Moolenaar, W. H. (1997). Gi-mediated activation of the Ras/MAP kinase pathway involves a 100 kDa tyrosine-phosphorylated Grb2 SH3 binding protein, but not Src nor Shc. EMBO J. 16, 3097-3105.
37. Lefkowitz, R. J. (1998). G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J.Biol.Chem. 273, 18677-18680.
38. Giroux, S., Tremblay, M., Bernard, D., Cardin-Girard, J. F., Aubry, S., Larouche, L., Rousseau, S., Huot, J., Landry, J., Jeannotte, L., and Charron, J. (1999). Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr.Biol. 9, 369-372.
39. Belanger, L. F., Roy, S., Tremblay, M., Brott, B., Steff, A. M., Mourad, W., Hugo, P., Erikson, R., and Charron, J. (2003). Mek2 is dispensable for mouse growth and development. Mol.Cell Biol. 23, 4778-4787.
40. Seger, R. and Krebs, E. G. (1995). The MAPK signaling cascade. FASEB J. 9, 726-735.
41. Winston, B. W., Remigio, L. K., and Riches, D. W. (1995). Preferential involvement of MEK1 in the tumor necrosis factor-alpha-induced activation of p42mapk/erk2 in mouse macrophages. J.Biol.Chem. 270, 27391-27394.
42. Catling, A. D., Schaeffer, H. J., Reuter, C. W., Reddy, G. R., and Weber, M. J. (1995). A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol.Cell Biol. 15, 5214-5225.
43. Papin, C., Denouel, A., Calothy, G., and Eychene, A. (1996). Identification of signalling proteins interacting with B-Raf in the yeast two-hybrid system. Oncogene 12, 2213-2221.
44. Dang, A., Frost, J. A., and Cobb, M. H. (1998). The MEK1 proline-rich insert is required for efficient activation of the mitogen-activated protein kinases ERK1 and ERK2 in mammalian cells. J.Biol.Chem. 273, 19909-19913.

90
45. Schaeffer, H. J., Catling, A. D., Eblen, S. T., Collier, L. S., Krauss, A., and Weber, M. J. (1998). MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668-1671.
46. Frost, J. A., Steen, H., Shapiro, P., Lewis, T., Ahn, N., Shaw, P. E., and Cobb, M. H. (1997). Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 16, 6426-6438.
47. Mansour, S. J., Resing, K. A., Candi, J. M., Hermann, A. S., Gloor, J. W., Herskind, K. R., Wartmann, M., Davis, R. J., and Ahn, N. G. (1994). Mitogen-activated protein (MAP) kinase phosphorylation of MAP kinase kinase: determination of phosphorylation sites by mass spectrometry and site-directed mutagenesis. J.Biochem.(Tokyo) 116, 304-314.
48. Sharma, P., Veeranna, Sharma, M., Amin, N. D., Sihag, R. K., Grant, P., Ahn, N., Kulkarni, A. B., and Pant, H. C. (2002). Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J.Biol.Chem. 277, 528-534.
49. Gardner, A. M., Vaillancourt, R. R., Lange-Carter, C. A., and Johnson, G. L. (1994). MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Mol.Biol.Cell 5, 193-201.
50. Coles, L. C. and Shaw, P. E. (2002). PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 21, 2236-2244.
51. Eblen, S. T., Slack, J. K., Weber, M. J., and Catling, A. D. (2002). Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol.Cell Biol. 22, 6023-6033.
52. Slack-Davis, J. K., Eblen, S. T., Zecevic, M., Boerner, S. A., Tarcsafalvi, A., Diaz, H. B., Marshall, M. S., Weber, M. J., Parsons, J. T., and Catling, A. D. (2003). PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J.Cell Biol. 162, 281-291.
53. English, J., Pearson, G., Wilsbacher, J., Swantek, J., Karandikar, M., Xu, S., and Cobb, M. H. (1999). New insights into the control of MAP kinase pathways. Exp.Cell Res. 253, 255-270.
54. Lewis, T. S., Shapiro, P. S., and Ahn, N. G. (1998). Signal transduction through MAP kinase cascades. Adv.Cancer Res. 74:49-139., 49-139.
55. Luttrell, L. M., Daaka, Y., and Lefkowitz, R. J. (1999). Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr.Opin.Cell Biol. 11, 177-183.

91
56. Cowley, S., Paterson, H., Kemp, P., and Marshall, C. J. (1994). Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77, 841-852.

Chapitre 3
3. Identification et caractérisation d’un domaine
d’interaction phospho-dépendant dans MEK1 et MEK2
Ce chapitre fait l’objet d’un manuscrit préparé en vue d’une publication. Ce manuscrit a été
écrit avec les résultats découlant de mes travaux. J’ai effectué l’ensemble des figures. J’ai
également écrit la première version du manuscrit et contribué à cette version.
Dans le chapitre précédant, nous avons montré que les protéines MEK1 et son homologue
MEK2 n’ont pas nécessairement des propriétés signalétiques redondantes. Les résultats
obtenus montraient clairement que l’activation de la voie ERK MAP kinase par la
bombésine fait appel à MEK1 et que MEK2 n’est pas impliquée dans ce processus. De
plus, nous avons montré que la région C-terminale de MEK1est à l’origine de la spécificité
de la réponse à la bombésine.
Dans le présent chapitre, nous avons tenté de caractériser les raisons sous-jacentes à cette
spécificité de MEK1. Par le biais du système double hybride, nous avons procédé à une
étude systématique visant à identifier des protéines pouvant intéragir d’une façon
spécifique avec MEK1. L’ensemble de nos résultats suggère un rôle essentiel à la région C-
terminale de MEK1 dans la régulation de son activité et de sa localisation cellulaire.

93
3.1. Résumé
MEK1 et MEK2 sont deux kinases à double spécificité qui sont homologues et qui agissent
au niveau MAPKK dans la voie de signalisation ERK/MAPK. Ces MAPKKs contribuent,
de manière distincte, à la régulation de l’activité de ERK. La région C-terminale de MEK1
et de MEK2 semble jouer un rôle essentiel dans la régulation des interactions protéiques et
la distribution cytoplasmique des protéines MEKs. La région riche en proline typique de
MEK1 et MEK2, présente en C-terminal, semble être importante dans l'interaction avec la
kinase Raf-1 et la régulation de l'activité de ERK. En outre, la région C-terminale de MEK1
contient des sites de phosphorylation unique qui peuvent augmenter ou atténuer son activité
intrinsèque. En particulier, la phosphorylation du résidu Ser298 par PAK-1 semble régler la
formation de complexes MEK1-ERK spécifiques et l'activation de la voie ERK/MAPK par
les intégrines. Par le biais du système double hybride chez la levure, nos travaux
démontrent que la région C-terminale de MEK1 interagit avec la région de la boucle
d'activation. Cette interaction semble être de nature intramoléculaire puisque les protéines
MEKs ne semblent pas former de dimères. Par ailleurs, la phosphorylation de MEK1 sur
les sites Ser218 et Ser222 de la boucle d'activation empêche cette interaction, suggérant que
cette dernière est dépendante de l'état d'activation de MEK1. La perturbation de cette
interaction par la délétion de la région C-terminale n'est pas suffisante pour augmenter
l'activité catalytique de MEK1. En outre, la phosphorylation de la boucle d'activation est
toujours requise pour l’activation de MEK1 indiquant que la région C-terminale n’est pas
un domaine d’autoinhibition. Nous proposons que cette interaction reflète une variation
conformationnelle entre l’état actif et inactif de MEK1 qui peut être importante pour la
régulation des interactions MEK1 et MEK2 avec leurs substrats, activateurs ou leurs
protéines d'échafaudage.

3.2. Article
CHARACTERIZATION OF PHOSPHO-DEPENDENT INTERACTING DOMAIN
OF MEK1 AND MEK2 PROTEINS
Nizar Chetoui and Jean Charron*
Centre de recherche en cancérologie de l’Université Laval, Centre Hospitalier
Universitaire de Québec, L’Hôtel-Dieu de Québec, Québec, QC, Canada, G1R
2J6.
* Corresponding author. Mailing address:
Centre de recherche de L'Hôtel-Dieu de Québec,
9 rue McMahon, Québec, QC, Canada, G1R 2J6.
Phone: (418) 525-4444 ext 15557. Fax (418) 691-5439.
E-mail: [email protected].

95
3.2.1. Abstract
MEK1 and MEK2 are two homologue dual specificity kinases that act at the MAP kinase
kinase level in the ERK/MAPK signaling pathway. They have distinct ways to contribute to
a regulated ERK activity. It was previously shown that the C-terminal region of MEK1 and
MEK2 may function in regulating protein-interaction and were necessary for regulating the
cytoplasmic distribution of MEK proteins. The MEK C-terminal region contains a proline-
rich region that seems to be important in regulating interaction with the Raf-1 kinase and
ERK activities. In addition, the C-terminus of MEK1 contains phosphorylation sites that
may either sustain or attenuate MEK1 activity. Particularly, Ser298 phosphorylation by
PAK-1 kinase has been suggested to regulate the formation of a specific MEK1-ERK
signaling complex and the activation of the ERK/MAPK signaling pathway by integrins.
Here, using the yeast Two-hybrid system, we show that the carboxyl-terminal end of
MEK1 and 2 interact with the activation loop. This interaction seems to be intramolecular
since no interaction was observed between MEK proteins. Furthermore, phosphorylation of
MEK1 at Ser218 and Ser222 in the activation loop prevents this interaction indicating that
this interaction may depend on the activation state of MEK. Elimination of this interaction
by truncation is not sufficient to activate the catalytic activity of MEK. Moreover,
phosphorylation of the activation loop is still required indicating that this interaction does
not act as an intramolecular inhibitory interaction modulated by phosphorylation of the
activation loop. We propose that the intramolecular interaction reflects a conformational
adjust between the active and inactive states that may be important to regulate MEK protein
interactions with substrates, activators or scaffolding proteins.

96
3.2.2. Inroduction
The MAP kinase signaling pathways consist in protein kinase cascades linking extracellular
stimuli to various targets scattered in the cytoplasm, the cytoskeleton, the membrane and
the nucleus [1,2]. There are at least three distinct MAPK signaling pathways in mammals
including the extracellular signal-regulated kinases (ERKs), the c-Jun N-terminal kinases
(JNKs) and the p38 MAPKs [3]. These kinases are activated in cascades by
phosphorylation on both threonine and tyrosine residues in the regulatory TXY loop present
in all MAP kinases. This phosphorylation is carried out via distinct upstream dual-
specificity MAP kinase kinases (MAPKKs). The classical pathway, that appears to be the
major one in growth factor signaling, uses MAP kinase or ERK-activating kinases (MEK;
MAPKK) and ERK isoforms (MAPK) and is also named the ERK/MAP kinase-signaling
pathway.
In mammals, the MAPKKs constitute a small family of related proteins that activate the
MAPKs. However, only MEK1 and MEK2 are known participants in the ERK/MAP kinase
cascade [2]. MEK1 and MEK2 distinguish themselves by the presence of a proline rich
region call MEK specific sequence (MSS) between the kinase domain IX and X [4,5]. This
region is involved with the interaction of MEK1 and MEK2 to its activators and is required
for efficient activation of its substrats [5]. The only identified substrates of MEK1 and
MEK2 are the MAP kinase ERK1 and ERK2 even if another substrate such as a Golgi-
associated ERK was suggested [6,7].
The mechanisms of regulation of MEK1 and MEK2 activity are not completely understood.
RAFs, MOS, PAK1, MEKK1, cyclin B-CDC2, CDK5 and ERKs proteins are able to
phosphorylate MEK but only RAFs and Mos lead to phosphorylation of Ser 218 and 222 in
the activation loop and activation of the ERK/MAP kinase [8-10]. MEKK1 seems to
phosphorylate Ser 218 and Ser 222 without significant activation of the MAP kinase ERK2
[8-10]. Moreover, A-RAF, Mos and PAK phosphorylate only MEK1 suggesting divergent
mechanisms of regulation of the MEK1 and MEK2 activities [11,12]. The site of PAK1
phosphorylation in MEK1 is Ser 298. The phosphorylation of Ser 298 by PAK1 does not

97
regulate MEK1 activity but prime MEK1 for activation by Raf-1. The phosphorylation of
MEK by Cyclin B-CDC2 and CDK5 at Thr 286 represses MEK activity [13,14]. In the case
of cyclin B-CDC2, the regulation of MEK activity also involves a cyclinB-CDC2
dependent proteolytic cleavage of the N-terminal ERK docking site [13]. The major
phosphorylation site of ERK on MEK are Thr 292 and Thr 386 but their role in regulation
of MEK activity are not clear [12,15]. More recently, the phosphorylation of Ser 212 of
MEK has been shown to be involved in the negative regulation of MEK activity [16].
Yeast two-hybrid system, as well as genetic screen, has allowed the identification of other
MEK partners involved in the regulation of its activity or in its activation. These are Grb10,
KSR (kinase suppressor of ras-1) and MP1 (MEK partner 1) [17-22]. The scaffolding
protein MP1 has been reported to facilitate the specific interaction and activation of ERK1
by MEK1 whereas KSR associates independently with RAF and MEK to facilitate the
phosphorylation of MEK by RAF [18,23]. Grb10 can bind RAF1 and MEK1; however the
binding to MEK1 seems to require the phosphorylation of Thr 386 by ERK [17-20,22,24].
Finally, the Rac-PAK pathway has been shown to be involved in the activation of the
MAP/ERK kinase cascade by regulating the formation of a specific MEK1-ERK signaling
complex, which is dependent on phosphorylation of Ser 298 by PAK, a site exclusively
present in MEK1 [25]. The regulation of ERK/MAP kinase signaling involved a cascade of
phosphorylation that is facilitated by specific molecular interaction with activators,
substrates and adaptor or scaffolding proteins.
In the present study we show using the yeast two-hybrid system that two non-contiguous
regions of MEK1 and MEK2 proteins can interact together. One of the regions corresponds
to the activation loop whereas the second is located at the carboxyl-terminal end of MEK.
This interaction has also been observed in mammalian cells and seems to be dependent on
the phosphorylation state of the protein. Our results suggest that this interaction is
intramolecular. However, the elimination of this interaction by truncation is not sufficient
to activate the catalytic activity of MEK. Phosphorylation of the activation loop is still
required indicating that this interaction does not act as an intramolecular inhibitory
interaction modulated by phosphorylation of the activation loop.

98
3.2.3. Experimental procedures
Yeast two-hybrid Assays
The yeast strain used in this study is S. cerevisiae L40 (MATa trp1 leu2 his3 LYS2 : lexA-
HIS3 URA3 : lexA-lacZ). Yeast strain was grown in rich medium (1% yeast extract, 2%
Bacto-Peptone, 2% glucose, and 0.1 mg/ml adenine) or in synthetic minimal medium with
appropriate supplements as described by Gietz et al. [26]. Yeast cells were transformed by
the lithium acetate method as described in [26]. A human MEK1 cDNA BamHI-BglII
fragment of a kinase dead form of MEK1 (S218A/S222A; kindly provided by Dr K.L.
Guan) coding for amino acid 1 to 362 was inserted in frame into the BamHI site of pFBL23
vector in order to generate a fusion protein in amino-terminus of the DNA binding domain
(DBD) of LexA (MEK1 C362 AA-DBD LexA vector; Figure 1; [27]. Two hybrid
screening of 3.5 x 106 primary transformants from a mouse brain cDNA library fused to
the transcriptional activation domain (AD) of GAL-4 (Clonetech #ML4008AH) were
performed as previously described in [27]. Briefly, L40 cells were transformed with the
MEK1 C362 AA-DBD LexA vector. Yeast transfectants were isolated in minimal medium
without Trp then the cDNA libraries were transfected in this population and selected in
minimal medium without Trp, Leu and His but supplemented with 1 mM 3-amino-1,2,4-
triazole (3-AT) for a week at 30oC.
β-galactosidase activity assay was performed on positive clones, essentially as described in
[26]. Briefly, the positive colonies were replicated on minimal medium without Trp and
Leu and grown for 3 to 5 days. The colonies were lift off on nitrocellulose filter and lysed
by freeze/thaw procedure. The filter soaked into the X-gal staining buffer was incubated at
30oC. Blue color in a few hours is indicative of β-galactosidase activity. The strength of
interaction between the two proteins is determined in term of intensity of staining and time
to reveal the staining. His+/LacZ+ clones were grown in minimal medium -Leu for 48 hrs.
The cells were lyzed in 200 µl of lysis buffer (10 mM Tris-Cl pH 8.0, 100 mM NaCl, 1 mM
EDTA, 2% (v/v) Triton X-100 et 1% SDS) containing 200-600 µm glass beads using 3
cycles of freeze (10 sec) / thaw (90 sec at 37oC) / vortex (1 min). The plasmid DNA was

99
recovered from the supernatant by ethanol precipitation and introduced into HB101 LeuB-
allowing selection for the cDNA library vector in minimal medium without Leu. Primary
positive clones analyzed were confirmed for targeted specificity by co-transformation with
different LexA DNA-binding domain fusions (lamine and Hsp27; kindly provided by Steve
Charette and Jacques Landry). Clones containing MEK1 specific interacting proteins were
further analyzed. The length of the cDNA inserted was determined and the sequence
established.
Mammalian expression vectors
Wild-type, constitutively active and inactive MEK1 expression vectors were obtained for
various sources. Recombinant Flag-MEK1 wt (pCMV-Tag2-MEK1 wt; Flag-tagged at
NH2-terminus) was kindly provided by Drs A. Colanzi and V. Malhotra (University of
California, San Diego, CA), whereas constitutively active and inactive MEK1 mutant
S218/S222A, (AA), S218/222D (DD) and S218E/S222D (ED), which are HA or Glutamic
acid-tagged, were kindly provided by Dr R. Erikson (Harvard University, Cambridge, MA),
Dr H.L. Guan (University of Michigan, Ann Arbor, MI) and Tom Moss (Université Laval,
Québec, Canada). The hamster ERK-HA tagged expression vector was provided by Dr J.
Landry (Université Laval, Québec, Canada [28]. pCMV-Tag2-MEK1 AA , DE or DD were
obtained by exchange of restriction enzyme fragment in pCMV-Tag2-MEK1 wt by the
corresponding fragment carrying the constitutively active and inactive mutation. MEK1 C-
terminal deletion mutants C380 wt, AA and DD were generated by PCR using the 5' primer
5'-CAGCGGGCAGCTCAT-3' and the 3' primer 5'-
AAGCTTTTAGCCGATGGTGGAGCA-3'. The 3' primer contains a stop codon after the
triplet encoding the amino acid 380 of MEK1 and a HindIII site. A BsgI-HindIII fragment
encoding the amino acid 242 to 380 was isolated from the PCR product and cloned in
pCMV-Tag2-Mek1 wt, AA and DD vectors between the BsgI-HindIII sites in order to
interrupt the translation of MEK1 protein after amino acid 380 in the pCMV-Tag2-Mek1
C380 wt, AA and DD vectors. The pEGFP-HA-MEK1 N324 expression vector was
generated by the insertion of the HA-N324 encoding sequence, which has been isolated

100
from a yeast two-hybrid cDNA library clones identified during the yeast two-hybrid screen
(Figure 1), in the pEGFP-C2 mammalian expression vector (Clonetech Laboratories Inc.).
The pEGFP-HA control vector was generated by transferring only the HA epitope tag from
the cDNA library vector (pACT11, Clonetech Laboratories Inc.).
Cell culture and transfection
HEK 293 cells were grown in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin, 100 µg of
streptomycin and 2 mM glutamine (Invitrogen, Burlington, ON). Cells were transfected at
70 to 80% confluence. Transfections were performed by the calcium phosphate method in
culture dishes coated with polylysine as described [29] and analyzed 40 to 48 hours after
transfection. When specified the cells were serum starved 24 hours before harvesting by
replacing the medium by DMEM containing only 0.1% of FBS.
Coimmunoprecipitation assays
HEK 293 transfected cells were harvested 40 hours after transfection in 400 µl of hypotonic
lysis buffer ( 20 mM Tris {pH7.5}, 2 mM MgCl2, 2 mM EGTA, 10mg/ml leupeptin and 10
mM PMSF) or MGM lysis buffer (20 mM Tris {pH 7.5}, 10 % glycerol, 150 mM NaCl, 5
mM EGTA, 1 mM EDTA, 1 % Triton X-100, 2 mM MgCl2, 50 mM NaF, supplemented by
10 µg/ml leupeptin and 10 mM PMSF) for ERK or MEK coimmunoprcipitation,
respectively. Extracts were clarified at 14,000 X g for 20 min at 4oC. Protein extract from
one 60mm diameter tissue culture dish were incubate with 8 µg of monoclonal anti-Flag
antiserum M2 (Sigma-Aldrich Inc. # F3165; Oakville, ON) for 2 hours at 4oC. Then 40µl of
protein A-Sepharose (50% vol/vol; Amersham Biosciences, Baie d'Urfé, QC) was added
and the incubation prolonged an additional hour. Immunoprecipitates (IP) were washed
three times with 200 µl of hypotonic lysis buffer, resuspended in SDS-PAGE sample buffer
and resolved by SDS-PAGE. Gels were transferred to nitrocellulose and probe with
monoclonal anti-HA antiserum HA.11 (BabCo, Richmond, CA). Antibody binding was

101
revealed with the ECL-Plus Western blotting detection system (Amersham Biosciences,
Baie d'Urfé, QC).
Cell stimulation, extraction and immunoprecipitation for ERK kinase assay
Serum starved cells were stimulated with 50 ng/ml of epidermal growth factor (EGF; Gibco
Invitrogen Corporation, Burlington ON) for 5 min at 37oC. 60-mm-diameter tissue culture
dishes were washed with iced cold phosphate buffer saline before extraction in 300 µl of
MGM lysis buffer (20 mM Tris {pH 7.5}, 10 % glycerol, 5 mM EGTA, 1 mM EDTA, 1 %
Triton X-100, 2 mM ß-glycerol phosphate, 50 mM NaF, 10 µM Na3VO4, 5 mM
Benzamidine 5 mM DTT, 10 µg/ml leupeptin and 0.2 mM PMSF). Extracts were clarified
at 14,000 X g for 20 min at 4oC. Proteins from a 60-mm-diameter tissue culture dishes were
mixed with 600 µl of MIKI buffer (20 mM Tris {pH=7.5}, 150 mM NaCl, 0.1 mM EDTA,
1 mM EGTA, 1 mM Na3VO4, 1 mM MgCl2, 1% Triton X-100 and 1 mM PMSF)
containing 8 µg of monoclonal anti-Flag antiserum M5 (Sigma # F4042) and incubated for
2 hours at 4oC. Subsequently, 40µl of protein A-Sepharose was added and the incubation
prolonged an additional hour. Immunoprecipitate (IP) were washed three times with 200 µl
of MIKI buffer, resuspended in kinase buffer (20 mM Tris {pH=7.5}, 10% glycerol, 120
mM pNPP, 30 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, 1 µM leupeptin, 100 µM ATP, 3
µCi [γ32P] ATP, 1 µg of inactive ERK2 (K52R) (Cell Signaling Technology, Pickering,
ON)) and incubated 20 min at 30oC. Reactions were terminated with addition of SDS-
PAGE sample buffer and products were resolved by SDS-PAGE. Incorporation into K52R
was determined by autoradiogram.
Immunofluorescence
Cells were grown on 35-mm plates and transfected as described above. Cells were fixed
with 4% paraformaldehyde (Electron Microscopy Sciences) for 5 min and permeabilized
with 0.2% Triton X-100 for 2 min. Localization of MEK1 proteins was identified by

102
immunostaining for the Flag tag followed by ALEXA-594- or ALEXA-488-labeled goat
anti-mouse IgG (Molecular Probes) and counterstained for cellular DNA with 4',6-
diamidino-2-phenylindole (DAPI, 0.2 µg/ml in phosphate-buffered saline). Cells were
identified using a Nikon TE-300 fluorescence microscope and a Micromax 1300YHS
(B/W) cooled CCD camera (-30°C; Princeton Instruments). The Flag-MEK1 pattern of
staining in cells was examined in 3 different assays. Normal cells displayed a diffuse
homogenous Flag pattern throughout the cytoplasm and sometimes in nucleus. The
punctuate pattern of staining was characterized as many distinct spots located throughout
the cytoplasm.

103
3.2.4. Results
Interaction of MEK1 and MEK2 with truncated MEK1 protein in Yeast Two-hybrid
Screen
The COOH-terminal region of MEK1 and MEK2 was isolated from the two-hybrid screen
of a mouse brain cDNA library using a constitutively inactive truncated form of MEK1
(amino acids (aa) 1-362 S218/222A) fused at the amino-terminus of the DNA binding
domain of LexA (Figure 3-1A). This approach keeps the amino terminus end of MEK1 free
of any exogenous a.a sequences and available for interaction with new substrates or any
other interaction. The Screening of 3.2 x 106 transformants from the mouse brain cDNA
library results in the isolation of 3 independent genes. Among the 53 positive clones
identified, 37 correspond to 18 individual Mek1 cDNA clones, 16 correspond to 12
individual Mek2 cDNA clones and one corresponds to catalytic domain of c-Raf. The 5'
ends of all Mek1 and Mek2 cDNA isolated are located between the sequences encoding the
codons 175 and 332 and they all extend to the end of the coding sequences. As shown in
Figure 3-1B, some of the clones has deleted the phosphorylation loop as well as the proline
rich region also known as the MEK specific sequence (MSS).

104
Figure 3-1

105
Figure 3-1 : Structure of MEK protein regions recover from yeast two-hybrid screen
against MEK1.
A, The schematic diagram depicts the MEK1 (1-362 a.a) N-terminal fusion of LexA DNA
binding domain used as bait in the yeast two-hybrid screen. EDS, ERK docking site; MSS,
MEK specific sequence (proline rich sequence); NES, nuclear exclusion sequences; the two
stars represent the phosphorylation site in the activation (or phosphorylation) loop. B,
Schematic representation of the various clones isolated from the two-hybrid study. The N-
terminus of the different clones are grouped according to the specific domain of MEK;
N175 (174 or 175); N195 (195 or 211); N247 (231 to 262); N295 (270 to 296); N324 (319
to 332).

106
COOH-terminal region of MEK interacts with the activation loop-
To identify the interacting region involved in this binding, deletion mutant have been tested
in two-hybrid assay. We used the N324 portions of the MEK1 protein (a.a 324-394) as a
bait in frame with one of the two-hybrid expression vector and various COOH- and NH2-
terminal portions of the MEK protein in frame with the complementary vector. The GAL-4
AD construct and LexA DBD constructs were co-transfected in yeast containing the His3
and LacZ reporter genes. Transformants were then tested for their ability to grow in the
absence of histidine and for β-galactosidase activity.
Results presented in Figure 3-2 show that the full-length MEK1 interacted with the
carboxyl-terminal domain of MEK1 (N324) in the two-hybrid assay. Deletion of 32, 44 or
94 amino acids in the carboxyl terminus of MEK1 (construct C362, C350 and C300,
respectively; Figure 3-2 and Table 2-1) does not have any effect on the interaction with the
carboxyl domain of MEK1. As the fusion protein N324 MEK1 can interact with the fusion
protein C300 MEK1, and since this two proteins do not contain common MEK1 amino
acids sequences we conclude that the interaction take place between two adjacent regions
of MEK1.
Amino terminal deletion of 175 and 195 residues of MEK1 do not also affect the
interaction to N324 MEK1 (construct N175 and N195; Figure 3-2). However, deletion of
247 amino acids or more at the amino terminus of MEK1 abolished interaction with the
carboxyl domain of MEK1 (construct N247, N295 and N324: Figure 3-2). This absence of
interaction with some of the construct tested (N247, N295 and N324) is not an artefact of
the two-hybrid assay, since these construct were able to generate interaction with other
MEK construct (see Table 3-1 and Figure 3-1). Altogether these results suggest that the
region of the phosphorylation loop of MEK can interact with the COOH-terminus of MEK
protein. However, the use of the amino acids 195-247 in the two-hybrid assay does not
show interaction with MEK1 carboxyl-terminal region (wt, N175 and N324; data not
shown). Either, the fusion protein between MEK1 amino acids 195-247 and the LexA DBD
is unstable (rapidly degraded or its solubility is impair) or the interaction domain extends

107
outside of this amino acids sequence. Thus the region of interaction in the activation loop
with the MEK1 carboxyl terminal region might not be restricted to amino acids 195-247.
To confirm the interaction of MEK to the carboxyl-terminal domain (a.a 342-394) of MEK
observed in the two-hybrid assay and to determine whether this interaction also occur in
vivo, we performed co-immunoprecipitation in Triton-soluble protein extracts of HEK 293
cells in which both Flag-C380 MEK1 and HA-GFP-N324 MEK1 or HA-ERK were
transiently expressed. In this experiment Flag- and HA-GFP tagged truncated form of
MEK, Flag-C380 MEK1 and HA-GFP-N324 were used in order to produce soluble protein.
We have shown that the C362 and the N324 are produced but are barely soluble and not
suitable for immunoprecipitation protocol. In HEK 293 cells, HA-ERK and HA-GFP-N324
MEK1 proteins were co-immunoprecipitated by the anti-Flag antibody (Figure 3-3A, lane 2
and 5 to 7, respectively). Thus, this interaction appears to occur in vivo, which is consistent
with the results that have been obtained in two-hybrid assay.

108
Figure 3-2

109
Figure 3-2 : Mapping of the MEK1-binding sites by the two-hybrid assay.
Full-length, COOH-terminal (∆C) or NH2-terminal deletion (∆N) of MEK1 proteins were
fused to the LexA DNA-binding domain or the Gal-4 transcriptional activation domain and
tested in the two-hybrid assay against the COOH-terminal domain of MEK1 N324 (a.a
324-394). Binding was detected by the growth of colonies on histidine-deficient media (+)
and confirmed by the β–galactosidase straining (+ staining in 4 to 8 hours; ++ staining in
less than 4 hours). EDS, ERK docking site; MSS, MEK specific sequence (proline rich
sequence); NES, nuclear exclusion sequences; the two stars represent the phosphorylation
site in the activation (or phosphorylation) loop.

110
Table 3-1
MEK1 C350
MEK1 C362
MEK1 wt
MEK1 N324 c-Raf Lamine Hsp27
MEK1 N324 +/++ +/+ +/+ -/- -/- -/- -/-
MEK1 C350 -/- -/- +/+ +/++ -/- -/- -/-
MEK1 C350 (DD) nd nd nd -/- -/- nd nd
MEK1 C362 -/- -/- +/+ +/++ +/+++ -/- -/-
MEK1 wt +/+ +/+ -/- +/+ +/+++ -/- -/-
MEK1 (DD) nd nd -/- -/- +/+++ nd nd

111
Table 3-1 : Intra-molecular interaction of the carboxyl terminal with the activation loop
is dependent on the phosphorylation state.
Full-length, COOH-terminal (∆C) or NH2-terminal deletion (∆N) of MEK1 proteins were
fused to the LexA DNA-binding domain and tested in the two-hybrid assay against Full-
length, COOH-terminal (∆C) or NH2-terminal deletion (∆N) of MEK1 proteins, c Raf,
lamine or Hsp27 protein fused to the Gal-4 activation domain. Binding was detected by the
growth of colonies on histidine-deficient media (+) and confirmed by the β–galactosidase
straining (+ staining in 4 to 8 hours; ++ staining in less than 4 hours).

112
Internal interaction of MEK1 carboxyl terminal domain with the activation loop-
To decipher whether the MEK1 carboxyl terminal domain interacts with the activation loop
via an intramolecular or intermolecular interaction, we return to the two hybrid system to
determine whether two full-length MEK1 protein can interact together. As shown in Table
3-1, the amino terminal and carboxyl terminal deletion of MEK1 (N320, C350 and C362)
are able to bind to full-length MEK1 protein. However, in the same experiment the full-
length MEK1 is not able of interacting to the full-length MEK1 suggesting that the
interaction was intramolecular. The truncated forms of MEK1, lacking one of the
interacting domains can compete with the interacting domain of the MEK1 full-length to
generate intermolecular interaction whereas two full-length MEK1 proteins cannot most
likely due to the stability of the intramolecular interaction or to limited access in the context
of the full-length protein.
Since one of the domains implicated in this intramolecular interaction involves the
activation loop of MEK1 region that is normally phosphorylated at serine 218 and 222
during MEK1 activation, we have taken advantage of the two-hybrid system to determine if
phosphorylation of the activation loop had any effect on binding. To address this question
we have introduce a charged residue which mimic phosphorylation by changing serine 218
and serine 222 in aspartic acid in the full-length MEK1 and in MEK1C350- LexA DBD
(MEK1 (DD) and MEK1 C350(DD), respectively). As bait the MEK1∆N324- Gal-4 AD
has been used. Both the MEK1 wt and the MEK1 C350 are capable of interacting with the
MEK1 N324 whereas the MEK1 (DD) and MEK1 C350 (DD) forms do not form a stable
interaction with the MEK1 N324 suggesting that the phosphorylation state of the MEK1
can regulate this interaction (Table 3-1).

113
Figure 3-3

114
Figure 3-3: MEK1 C-terminal domain bind to a truncated form of MEK1 in vivo.
HEK 293 cells were transfected with expression vectors for Flag-MEK1-C380 and HA-
GFP, HA-ERK or HA-MEK1-N324. Upper panel: After serum starvation for 16 hrs, cells
were harvested and immunoprecipitations were carried out with α-Flag antibody and
immunoblot were probed with α-HA antibody. Lower panels: Total cell lysates were
resolved on SDS-PAGE and immunoblots were probed with the antibodies indicated to the
left of each panel. The proteins detected are indicated to the left of the panels. Lanes 1-5
and 6-7 are from the same gel but transferred on different membranes.

115
Is there a functional role for the carboxyl-terminal / phosphorylation loop interaction?
Our results suggest that the carboxyl-terminal region of MEK1 interacts with the
phosphorylation loop when this latter is not phosphorylated. This data raises the possibility
that the MEK carboxyl-terminal region can be involved in an intramolecular inhibitory
interaction modulated by phosphorylation of the activation loop. In order to determine the
functional role of the carboxyl terminal region on the regulation of MEK1 activity in intact
cells, we have evaluated the activity of MEK1 truncated mutants.
Various Flag-MEK1 constructs were transiently co-expressed with HA-ERK in HEK 293
cells. The ability of truncated MEK1 protein to phosphorylate ERK in the absence of
activation by EGF was tested. As seen in Figure 3-5A, Flag-MEK1 wt and C380 are unable
to phosphorylate the HA-ERK as well as the endogenous ERKs protein unless the cells
were induced by EGF. In addition, in Flag-MEK1 C380 (AA) transfected cells no
phosphorylation of HA-ERK was detected even after EGF induction. These results indicate
that the truncated MEK1 mutants, Flag-MEK1 C380 and Flag-MEK1 C380 (AA), are
inactive in the absence of induction. Thus the truncation of the carboxyl-terminal region of
MEK1 at position 380 is not sufficient for kinase activation. The catalytic activity is still
dependent on activation by phosphorylation. In order to confirm that Flag-MEK1 C380 was
catalytically inactive in absence of induction by EGF and Flag-MEK1 C380 (AA) was
catalytically even after EGF induction we have performed MEK kinase assay on HEK 293
cell extract expressing various Flag tagged MEK1 mutants. The Flag-MEK1 proteins were
immunoprecipitated with a α-Flag antibody and the catalytic activity tested as described in
Material and Methods. As shown in Figure 3-4B, both mutant Flag-MEK1 C380 and Flag-
MEK1 C380 (AA) are catalytically inactive in the absence of EGF induction and only Flag-
MEK1 C380, which can be phosphorylated, is activated after EGF induction. Use of a
larger carboxyl-terminal deletion such as the C362, in MEK kinase assay has shown that
this deletion lead to a protein that is catalytically inactive (Figure 3-4A). Flag-MEK1 C362,
C362 (AA) and C362 (VV) do not show MEK kinase activity even after EGF induction. In
addition, we have shown that the MEK1 C362 mutant proteins were more difficult to make
soluble and that they were not localize in the cytoplasm like its wild-type counterpart but

116
show an aggregate and punctuated staining (Figure 3-4B). Altogether, these results suggest
that the deletion of the carboxyl-terminal end of MEK1 is not sufficient to activate its
catalytic activity (MEK1 C380) whereas if the deletion includes part of the kinase domain
XI (MEK1 C362) all enzymatic activity is lost.
In order to verify whether phosphorylation of MEK1 at different sites in the C-terminal end
can influence its kinase activity, the ability of some MEK1 mutants to phosphorylate ERK
in absence of activation by EGF was tested in a kinase assay. As seen in Figure 3-6, all
MEK1 mutants S292D, S298D, S292D/S298D (phosphorylation sites by PAK1 that
positively regulate MEK1 activity in vivo), like MEK1 wt, show no activity in the absent of
induction. Active MEK1/S386D seems to be constitutively active. Mutation of S386,
reported to negatively regulate MEK1 activity in vivo, had no effect on the MEK1 DD
mutant activity. Thus phosphorylation at the carboxyl-terminal sites of MEK1 is not
sufficient for kinase activation. The catalytic activity is still dependent on activation by
phosphorylation at the activation loop.

117
Figure 3-4

118
Figure 3-4 : Catalytic activity and cellular localisation of MEK1 C362 mutants.
A. MEK1 C362 mutants are inactive even after EGF stimulation. HEK 293 cells were
transfected with expression vectors for Flag-MEK1 and truncated, constitutively active or
inactive mutants as indicated. After serum starvation for 16 hrs, cells were harvested and
immunoprecipitation were carried out with α-Flag antibody and a kinase reaction was
performed as described in Material and Methods. An autoradiogram of the electrophoresed
kinase reaction is shown. B. Cellular localisation of MEK1 and truncated forms, C380 and
C362. HEK 293 cells were transfected with expression vectors for Flag-MEK1 and
truncated. Experiment was done as described in Material and methods.

119
Figure 3-5

120
Figure 3-5 : Deletion of the carboxyl-terminal domain of MEK1 does not affect the
regulation of MEK1 activity.
A, Activation of MEK1 C380 mutant is still required for kinase activity. HEK 293 cells
were transfected with expression vectors for HA-ERK and, Flag-MEK1, truncated,
constitutively active or inactive MEK1 mutants as indicated. After serum starvation for 16
hrs, cells were harvested and activation of the MAP kinase cascade assess by immunoblot
using α-P-ERK antibody. B, MEK1 C380 mutant is still catalytically active after induction
with EGF. HEK 293 cells were transfected with expression vectors for Flag-MEK1 and
truncated, constitutively active or inactive mutants as indicated. After serum starvation for
16 hrs, cells were harvested and immunoprecipitation were carried out with α-Flag
antibody and a kinase reaction was performed as described in Material and Methods. An
autoradiogram of the electrophoresed kinase reaction is shown. Lanes 1-5 and 6-7 are non-
adjacent lanes from the same blot whereas lanes 1-3, 4-5 and 6 are non-adjacent lanes from
another experiment.

121
Figure 3-6
WTS292D DD/386D
EGF 50ng/ml - - +- - - + + + +
S298D
S292D/S298DWT
S292DS298D
S292D/S298D

122
Figure 3-6 : MEK1 activity is not affected by point mutations in the proline-rich
sequence in vitro.
A, Activation of MEK1 C380 mutant is still required for kinase activity. HEK 293 cells
were transfected with expression vectors for HA-MEK1; WT, WT with point mutations in
the proline-rich sequence; either S292D, S298D or both, constitutively active MEK1 DD-
S386D mutants as indicated. After serum starvation for 16 hrs, cells were treated with EGF
as indicated and harvested and immunoprecipitation were carried out with α-HA antibody.
A kinase reaction was performed as described in Material and Methods. An autoradiograph
of the electrophoresed kinase reaction is shown.

123
3.2.5. Discussion
There is increasing interest in the regulation mechanisms and functions of the MEK/ERK
cascade. There are several molecules that can modulate the localisation of MEK 1 and its
interactions with the MEK/ERK pathway. In this study, we have used the yeast double
hybrid system to identify new MEK interacting molecules that might be involved in the
regulatory function of MEK1 in the ERK MAPK pathway. Unlike several investigators
who have used, as a bait, MEK 1 fused at the C-terminal end of the DNA binding domain
(DBD) of LexA (or Gal4), we have used a MEK1 fused at the N-terminal of LexA-DBD
(Figure 3-1). This construction allowed us to obtain new interacting proteins (probably new
substrates of MEK1), that, just like ERK1/2, interact specifically with the N-terminal of
MEK1. However, it is important to note that the MEK1 bait used was devoided of the last
amino acids at the C-terminal (a.a 362 to 393). These amino acids were deleted in order to
allow the correct fusion with the DNA binding domain of LexA. To our surprise, the
majority of the molecules that were able to interact with MEK1 correspond to peptides of
various length from the C-terminal end of MEK1 and MEK2 themselves (Figure 3-1).
Furthermore, we showed by several deletion mutations that the intermolecular interactions
take place between two specific regions of MEK1 and MEK2 (figure 3-2). The carboxy-
terminal sequences of MEK1 between residues 324 and 393 can interact with the
phosphorylation loop of MEK1, residues 195 to 247. Our data suggest that this interaction
is intramolecular. We and other investigators have shown that MEK1, 2 are unable to form
dimers in double hybrid assays (Table 3-1 and [30]). In addition, the intramolecular
interaction seems to be dependent on the phosphorylation state of MEK1/2. The interaction
observed between the N324 and the C362 SS (Ser 218 and Ser 222) regions is lost when the
serine residues of the activation loop are replaced by aspartate residues which mimic the
phosphorylation in the C362 DD mutant.
These results indicate that there is an intramolecular interaction between the C-terminal
region and the activation loop in MEK1 and MEK2 and that this interaction is dependent on
the phosphorylation state. These results suggest that the electrostatic variations induced by

124
the phosphorylation of the activation loop can modulate a conformational change at the C-
terminal region of MEK1/2. This conformational change could play a role in the transition
between the active and inactive states of MEK1/2 proteins. In order to verify whether the
C-terminal could play a direct role in the activation of the protein, deletion mutants have
been generated and tested for kinase activity. The deletion of the carboxy region from aa
362 to 393 lead to lost of kinase activity. Furthermore, immunoprecipation experiments
using protein extracts from HEK 293 or CCL39 cell lines overexpressing the MEK1 C362
mutant showed that these proteins was trapped in insoluble fractions of the extracts. More
over, immuncytolocalisation experiment showed that the expression of this truncated form
of MEK1 is localized in restricted cytosolic area and specific cytoplasmic compartments.
Altogether, these results indicate that the C362 mutants might be localized in these cellular
comparments due to an altered conformation. Cha et al (2001) showed that similar mutants
MEK1 C296, C330, and ∆330-379 are localised in lysosomes [31]. The C-terminal region
of MEK1 could be important for a stable configuration allowing its proper expression and
localisation in the cytoplasm.
We have also observed a stable interaction between the deleted mutant C362 and ERK
(data not shown). However, the C362 mutant was unable to phosphorylate ERK even in its
active form, C362DD, suggesting that this portion of C terminal of MEK1 is implicated in
the enzymatic activity. The last kinase domain (XI), characterised by its structure and its
Arg349 residue (conserved in protein kinases) [32], seems to be intact in the C362 mutant.
In order to verify this hypothesis, we have performed another MEK1 deletion mutant,
C380. Our results show that this mutant behave as wild type MEK1 protein. Hence we
observed a cytoplasmic localisation of the C380 mutant (Fig 2-5b). Furthermore, the
enzymatic activity of the MEK1 DD C380 mutant is comparable to that of the full length
protein MEK1 DD. This suggests that the C380 mutant activity is dependent on the
phosphorylation of the 218 and 222 serine residues in the activation loop. Deletion of the
last 13 amino acid residues of MEK1 does not seem to alter the basal activity of the latter.
Our data are not in accord with those of Cha et al (2001) who have shown that in vivo the
MEK1 mutant d379 could activate ERK1 and ERK2 by phosphorylation even in absence of
induction [31].

125
Hence our results suggest that the amino acid sequence between the 362 and 380 residues
of MEK1 contains important elements for MEK1 activity. Structural and conformational
integrity of the kinase domain seems to be perturbed in the C362 mutants. In order to
elucidate this issue, further studies need to be done.
Immunoprecipitation experiments indicate that there is an in vivo interaction between the
MEK1 C380 SS and the C-terminal fragment N-324 (figure 3-3). We have not been able to
confirm, in vivo, the role of the C-terminal region on MEK1 activity. Nevertheless, all these
results suggest that the residues between the C362 and C380 region may be important for
the activation of the MEK1 kinase. The stability of the inactive form of MEK1 does not
seem to involve any intra-steric competition by an autoinhibition domain or a pseudo
substrate, since deletion of the C-terminal region does not result in a constitutively active
MEK1 protein. However, it is probable that structural rearrangements in different regions
of MEK1 contribute to the latter’s activity. Several studies have shown that there is a
dynamic movement involving diverse secondary structures of MEK1. This could in turn be
important for MEK1 activity [33-35].
Conformational variations in the C-terminal region could be primordial for the stability of
the interactions of MEK1 with its substrates, its activators and also with scaffold proteins.
Lastly, these variations could also be important for the proper localisation of the MEK1
protein in the cytoplasm. Our Two-hybrid experiments have shown that there is a problem
in the interaction of MEK1C350 and RAF-1 (table 2-1). There have been some reports
suggesting that the MEK Specific Sequence (MSS) could be the site of interaction between
RAF-1 and MEK1/2 [4]. Our results indicate that the portion of MEK1 between the C350
and C362 residues could, on one hand, be directly involved in the attachment of RAF to
MEK. On the other hand, the deletion of the C350 region could result in a conformational
change of the MEK1 protein and this could in turn alter the stability of the MEK1-RAF
complex. It would therefore be extremely useful to elucidate the crystal structure of the
MEK1 protein given its highly conserved sequence between the different species. This
would allow us to better understand the activation and inactivation of the MAPKK family.
Could the change in conformation induced by the deletion of the C-terminal region be
directly linked to the phosphorylation/activation state of MEK1? Could there be other

126
phosphorylation events that control such conformational changes? Several phosphorylation
sites are present in the C-terminal region, in particular in the proline rich sequence (PRS),
which seems to play an important role in stabilising the interaction of MEK1 with RAF
[36], with ERK [25] and with several scaffold proteins such as Grb10 and MP-1 [17].
Lastly, there are sites such as Ser298 that regulate positively MEK1 activity [36].
Phosphorylation of MEK1 on the Ser 298 residue by PAK1 could stimulate a
conformational change which stabilise the MEK-ERK interaction [37]. Other residues such
as Ser212, Thr 292 and Thr386 seem to inhibit MEK1 activity in vivo [17,37].
Phosphorylation of the Ser212 residue could perturb the activation loop thereby blocking
the substrate’s access to the active site of MEK1 [16].

127
3.2.6. Acknowledgements
We thank Drs. Robert Faure and Jacques Huot for critical reading of the manuscript, and Dr
Grosse for ERK2 antibody. This work was supported by grants from the Canadian
Institutes of Health Research (MOP-42523) and the Cancer Research Society Inc. to J. C. J.
C. is a Senior Scholar of the Fonds de la Recherche en Santé du Québec.

128
3.2.7. References
1. Guan, K. L. (1994). The mitogen activated protein kinase signal transduction pathway: from the cell surface to the nucleus. Cell Signal. 6, 581-589.
2. Seger, R. and Krebs, E. G. (1995). The MAPK signaling cascade. FASEB J. 9, 726-735.
3. Robinson, M. J. and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr.Opin.Cell Biol. 9, 180-186.
4. Catling, A. D., Schaeffer, H. J., Reuter, C. W., Reddy, G. R., and Weber, M. J. (1995). A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol.Cell Biol. 15, 5214-5225.
5. Dang, A., Frost, J. A., and Cobb, M. H. (1998). The MEK1 proline-rich insert is required for efficient activation of the mitogen-activated protein kinases ERK1 and ERK2 in mammalian cells. J.Biol.Chem. 273, 19909-19913.
6. Xu, B., Wilsbacher, J. L., Collisson, T., and Cobb, M. H. (1999). The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J.Biol.Chem. 274, 34029-34035.
7. Acharya, U., Mallabiabarrena, A., Acharya, J. K., and Malhotra, V. (1998). Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 92 , 183-192.
8. Pham, C. D., Arlinghaus, R. B., Zheng, C. F., Guan, K. L., and Singh, B. (1995). Characterization of MEK1 phosphorylation by the v-Mos protein. Oncogene %20;10, 1683-1688.
9. Reuter, C. W., Catling, A. D., Jelinek, T., and Weber, M. J. (1995). Biochemical analysis of MEK activation in NIH3T3 fibroblasts. Identification of B-Raf and other activators. J.Biol.Chem. 270, 7644-7655.
10. Xu, S., Robbins, D., Frost, J., Dang, A., Lange-Carter, C., and Cobb, M. H. (1995). MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc.Natl.Acad.Sci.U.S.A 92, 6808-6812.
11. Yashar, B., Irie, K., Printen, J. A., Stevenson, B. J., Sprague, G. F., Jr., Matsumoto, K., and Errede, B. (1995). Yeast MEK-dependent signal transduction: response thresholds and parameters affecting fidelity. Mol.Cell Biol. 15, 6545-6553.
12. Matsuda, S., Gotoh, Y., and Nishida, E. (1993). Phosphorylation of Xenopus mitogen-activated protein (MAP) kinase kinase by MAP kinase kinase kinase and MAP kinase. J.Biol.Chem. 268, 3277-3281.

129
13. Harding, A., Giles, N., Burgess, A., Hancock, J. F., and Gabrielli, B. G. (2003). Mechanism of mitosis-specific activation of MEK1. J.Biol.Chem. 278, 16747-16754.
14. Sharma, P., Veeranna, Sharma, M., Amin, N. D., Sihag, R. K., Grant, P., Ahn, N., Kulkarni, A. B., and Pant, H. C. (2002). Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J.Biol.Chem. 277, 528-534.
15. Gardner, A. M., Vaillancourt, R. R., Lange-Carter, C. A., and Johnson, G. L. (1994). MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Mol.Biol.Cell 5, 193-201.
16. Gopalbhai, K., Jansen, G., Beauregard, G., Whiteway, M., Dumas, F., Wu, C., and Meloche, S. (2002). Negative regulation of MAP kinase kinases by phosphorylation of a conserved serine residue equivalent to Ser212 of MEK1. J.Biol.Chem. ..
17. Nantel, A., Mohammad-Ali, K., Sherk, J., Posner, B. I., and Thomas, D. Y. (1998). Interaction of the Grb10 adapter protein with the Raf1 and MEK1 kinases. J.Biol.Chem. 273, 10475-10484.
18. Schaeffer, H. J., Catling, A. D., Eblen, S. T., Collier, L. S., Krauss, A., and Weber, M. J. (1998). MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668-1671.
19. Stewart, S., Sundaram, M., Zhang, Y., Lee, J., Han, M., and Guan, K. L. (1999). Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol.Cell Biol. 19, 5523-5534.
20. Therrien, M., Michaud, N. R., Rubin, G. M., and Morrison, D. K. (1996). KSR modulates signal propagation within the MAPK cascade. Genes Dev. 10, 2684-2695.
21. Raabe, T. and Rapp, U. R. (2002). KSR--a regulator and scaffold protein of the MAPK pathway. Sci.STKE. 2002, E28.
22. Roy, F. and Therrien, M. (2002). MAP kinase module: the Ksr connection. Curr.Biol. 12, R325-R327.
23. Catling, A. D., Eblen, S. T., Schaeffer, H. J., and Weber, M. J. (2001). Scaffold protein regulation of mitogen-activated protein kinase cascade. Methods Enzymol. 332:368-87., 368-387.
24. Downward, J. (1995). KSR: a novel player in the RAS pathway. Cell 83, 831-834.
25. Eblen, S. T., Slack, J. K., Weber, M. J., and Catling, A. D. (2002). Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol.Cell Biol. 22, 6023-6033.

130
26. Gietz, R. D., Triggs-Raine, B., Robbins, A., Graham, K. C., and Woods, R. A. (1997). Identification of proteins that interact with a protein of interest: applications of the yeast two-hybrid system. Mol.Cell Biochem. 172, 67-79.
27. Beranger, F., Aresta, S., de Gunzburg, J., and Camonis, J. (1997). Getting more from the two-hybrid system: N-terminal fusions to LexA are efficient and sensitive baits for two-hybrid studies. Nucleic Acids Res. 25, 2035-2036.
28. Meloche, S., Pages, G., and Pouyssegur, J. (1992). Functional expression and growth factor activation of an epitope-tagged p44 mitogen-activated protein kinase, p44mapk. Mol.Biol.Cell 3, 63-71.
29. Frost, J. A., Steen, H., Shapiro, P., Lewis, T., Ahn, N., Shaw, P. E., and Cobb, M. H. (1997). Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 16, 6426-6438.
30. Papin, C., Denouel, A., Calothy, G., and Eychene, A. (1996). Identification of signalling proteins interacting with B-Raf in the yeast two-hybrid system. Oncogene 12, 2213-2221.
31. Cha, H., Lee, E. K., and Shapiro, P. (2001). Identification of a C-terminal region that regulates mitogen-activated protein kinase kinase-1 cytoplasmic localization and ERK activation. J.Biol.Chem. 276, 48494-48501.
32. Mansour, S. J., Candia, J. M., Matsuura, J. E., Manning, M. C., and Ahn, N. G. (1996). Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry 35, 15529-15536.
33. Cox, S., Radzio-Andzelm, E., and Taylor, S. S. (1994). Domain movements in protein kinases. Curr.Opin.Struct.Biol. 4, 893-901.
34. Delaney, A. M., Printen, J. A., Chen, H., Fauman, E. B., and Dudley, D. T. (2002). Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol.Cell Biol. 22, 7593-7602.
35. Resing, K. A. and Ahn, N. G. (1998). Deuterium exchange mass spectrometry as a probe of protein kinase activation. Analysis of wild-type and constitutively active mutants of MAP kinase kinase-1. Biochemistry 37, 463-475.
36. Coles, L. C. and Shaw, P. E. (2002). PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 21, 2236-2244.
37. Eblen, S. T., Slack-Davis, J. K., Tarcsafalvi, A., Parsons, J. T., Weber, M. J., and Catling, A. D. (2004). Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol.Cell Biol. 24, 2308-2317.

Chapitre 4
4. Résultats complémentaires
Ce chapitre renferme des résultats complémentaires obtenus lors du criblage double hybride
qui a mené à la préparation du l’article présenté au chapitre 3 de la thèse.

132
Au cours de notre criblage double hybride chez la levure, nous avons isolé plusieurs
protéines qui paraissaient intéragir avec MEK1 (Tableau A.1). La plupart des protéines
repêchées se sont avérés des faux positifs ; elles n’interagissent plus avec MEK1 dans les
essais de double hybride ou alors elles intéragissent d’une façon non spécifique avec MEK1
et avec d’autres protéines testées tel que la lamine ou Hsp27. Par contre, certaines
protéines, dont les histones H3 et M96A, ne semblaient pas être des faux positifs et
semblaient intéragir avec MEK1 d’une façon spécifique lors des essais double hybride.
4.1. Interaction MEK1-Histones H3
Le criblage de deux banques d’ADNc, provenant de cerveaux de souris et de placenta
humain (produit par Clonetech.), ne nous a pas permis d’identifier de nouvelles protéines
agissant au sein de la voie de signalisation ERK (Tableau A.1). Ceci dit, nous avons isolé
des variants de remplacement de l’histone H3 (v.r H3) comme protéine pouvant interagir
avec MEK1 (Tableau 4-1). Les histones H3 semblent jouer un rôle important dans la
progression du cycle cellulaire et sont des substrats légitimes de ERK2 et de RSK1, deux
protéines kinases qui agissent en aval de MEK1 dans le sentier ERK/MAPK [430,453,454].
D’autre part, les histones H3 et H4 ne semblaient pas être des artéfacts courants du système
double hybride chez la levure (Tableau A-2). Ainsi, les histones H3 se sont avérées des
candidats potentiels et fort intéressants pouvant élaborer une fonction nucléaire de MEK1
ou tout simplement justifier sa présence au noyau. Malheureusement, nous n’avons pas pu
confirmer, in vitro, l’interaction de MEK1 avec ces histones. Nos tentatives pour produire,
chez la bactérie, la protéine GST-v.r H3 ont échoué à cause de l’instabilité et l’insolubilité
de la protéine recombinante (données non présentées). Par contre, d’autres essais double
hybride ont été effectués en couplant MEK1 au domaine d’activation de Gal4 et les
histones H3 au domaine de liaison à l’ADN de LexA. Alors que les histones peuvent lier
l’ADN d’une façon non spécifique, cette nouvelle combinaison visait à éliminer
l’activation non spécifique de la transcription des gènes rapporteurs par les histones couplés
au domaine d’activation de Gal4. Les résultats obtenus lors de ces essais ont montré que

133
l’interaction H3-MEK1 n’était pas spécifique et que les histones isolées du criblage sont de
faux positifs (Tableau 4-2).
4.2. Interaction MEK1-M96A
Un autre clone isolé lors du criblage double hybride de la banque d’ADNc provenant du
cerveau de souris contient des séquences d’un gène homologue à M96A. M96A est un
facteur de transcription isolé chez l’humain (Holen, T. et Aasland, R., données non
publiées) et qui est homologue à M96 isolé chez la souris [455]. Aucune donnée n’est
présentée pour M96A, mais son homologue M96, isolé en utilisant le système un hybride
chez la levure, semble lier des séquences MREs (metallotheonin response element) du gène
de la métallothéonine-1 [455]. L’analyse de la séquence protéique de M96A a révélé la
présence de site consensus de phosphorylation par MEK1 et 2. Alors que nos premiers
essais de double hybride indiquaient que l’interaction MEK1-M96A est réelle (Tableau 4-
1), le clone M96A s’est avéré, comme ceux des histones H3, un faux positif (Tableau 4-2).
Les gènes rapporteurs His3 et LacZ sont inactifs lorsque MEK1 est fusionnée au domaine
d’activation de Gal4 et M96A est fusionné au domaine de liaison à l’ADN de LexA. Tout
comme les histones, M96A, qui est un facteur de transcription capable de lier l’ADN,
semble avoir stimulé l’activation non spécifique de la transcription des gènes rapporteurs
lorsque il été fusionné au domaine d’activation de Gal4. La présence d’un site du type
MRE sur les promoteurs des gènes rapporteurs pourrait expliquer l’activation non
spécifique observée lors de nos premiers essais.

134
Gal4 -AD-------------
LexA-DBDMEK1 MEK2 H3.3A H3.3B H4 M96A c-RAF PLZF
MEK1 C362 AA (Amino) + + + + + + + +
hsp27 - - - - - - - +
Lamine - - - - - - - +
Gal4 -AD-------------
LexA-DBDMEK1 MEK2 H3.3A H3.3B H4 M96A c-RAF PLZF
Gal4 -AD-------------
LexA-DBDMEK1 MEK2 H3.3A H3.3B H4 M96A c-RAF PLZF
MEK1 C362 AA (Amino) + + + + + + + +MEK1 C362 AA (Amino) + + + + + + + +
hsp27 - - - - - - - +hsp27 - - - - - - - +
Lamine - - - - - - - +Lamine - - - - - - - +
Tableau 4-1 : Essai d’interaction double hybride chez la levure
Les protéines MEK1, hsp27 et lamine ont été fusionnées au domaine de liaison à l’ADN de
LexA et testées dans un essai double hybride avec les différentes protéines isolées lors du
criblage, MEK1, MEK2, les variants de remplacement de l’histone H3 (H3.3A et H3.3B),
Histone H4, les facteurs de transcription M96A et PLZF et c Raf, fusionnées au domaine
d’activation de Gal-4. L’interaction est déterminée par l’activation des gènes rapporteurs :
His3, qui permet aux levures de pousser sur un milieu sans histidine, et LacZ qui permet de
confirmer l’interaction par un essai β–galactosidase.

135
Gal4 AD------------
LexA DBD
MEK1wt
MEK2wt
Gal4-AD seul
MEK1 D.N (A) + + -
H3.3A - - -
M96A - - -
Gal4 AD------------
LexA DBD
MEK1wt
MEK2wt
Gal4-AD seul
MEK1 D.N (A) + + -
Gal4 AD------------
LexA DBD
MEK1wt
MEK2wt
Gal4-AD seul
MEK1 D.N (A) + + -
H3.3A - - -H3.3A - - -
M96A - - -M96A - - -
Tableau 4-2 : Les histones H3 et M96A sont des faux positifs
Les histones H3.3, M96A et la forme dominante négative de MEK1 C362 ont été fusionnées
au domaine de liaison à l’ADN de LexA et testées dans un essai double hybride avec MEK1
et MEK2 fusionnées au domaine d’activation de Gal-4. L’interaction est déterminée par
l’activation des gènes rapporteurs : His3, qui permet aux levures de pousser sur un milieu
sans histidine, et LacZ qui permet de confirmer l’interaction par un essai β–galactosidase.

Discussion générale

137
Le développement tumoral nécessite la dérégulation des contrôles normaux de la
prolifération et de la différenciation cellulaires. Il est bien connu que la voie de
signalisation ERK/MAP kinase est impliquée dans ces processus. De plus, le rôle de
MEK1, qui est un élément central de la voie, dans la transformation cellulaire a été mis en
évidence par l’utilisation de protéines MEK1 constitutivement actives. L’expression des
formes actives de MEK1 dans les fibroblastes est suffisante pour induire la production de
foyers de transformation, la croissance des fibroblastes en agar et la production de tumeurs
chez les souris nues [421,456]. De plus, la croissance des tumeurs et leur progression vers
l’état métastatique nécessite la vascularisation de la tumeur primaire par angiogenèse
[2,456]. La tumeur ainsi vascularisée n’est pas restreinte dans sa croissance. En outre, si les
cellules acquièrent une certaine mobilité, elles peuvent utiliser les vaisseaux formés pour
coloniser des sites secondaires. Les travaux antérieurs faits au laboratoire du Dr. Charron
ont permis de montrer le rôle essentiel et unique de MEK1 lors du développement
embryonnaire [77]. L’analyse du phénotype placentaire en cours dans le laboratoire tente
de mettre en évidence son rôle dans la morphogenèse du placenta et la migration cellulaire
(Bissonauth et Charron, en préparation). Une meilleure compréhension du rôle de MEK1
dans des voies de transduction devrait donc nous permettre de mieux contrôler ces
processus lors de la transformation cancéreuse.
La voie de signalisation ERK/MAPK est impliquée dans une panoplie de processus
cellulaires complexes permettant aux cellules de proliférer, de survivre, de se différencier,
ou encore de migrer. Les recherches récentes visent à éclaircir l’importance des variations
spatio-temporelle dans l’activité et la distribution subcellulaire des composantes
signalétiques. Les travaux de plusieurs groupes de recherche soulignent l’importance des
interactions protéiques comme un moyen puissant de coordination des processus
signalétiques.
Dans ce sens, mon projet de recherche effectué dans le cadre de mes études de doctorat
tente d’identifier et comprendre les divergences fonctionnelles et structurales qui existent
entre MEK1 et MEK2 et qui permettent de répondre de façon spécifique à des signaux
uniques.

138
Mes premiers travaux, dont les résultats les plus pertinents sont décrits dans le chapitre 2,
ont montré que la signalisation induite par la bombésine emprunte une voie spécifique à
MEK1 au dépend de son homologue MEK2. Alors que la plupart des facteurs de croissance
testés ne présentent pas cette spécificité, l’effet de la bombésine s’est présenté comme un
moyen intéressant pour étudier la divergence fonctionnelle entre MEK1 et MEK2 et mieux
comprendre la spécificité de la réponse.
La voie de signalisation conventionnelle Ras-Raf-MEK1/2-ERK1/2, activée par des
signaux mitogénique via des récepteurs RTKs, ne semble pas présenter une divergence
fonctionnelle entre les modules homologues MEK1 et MEK2 ou ERK1 et ERK2 [462,463].
Cette voie, dite proliférative pourrait être considérée comme une voie principale dont
l’issue pourrait être modulée par des signaux provenant d’autres récepteurs; tels que les
récepteurs couplés aux protéines G ou des intégrines, ou par des voies annexes qui font
appel à d’autres modules signalétiques et au phénomène de cross-talk.
La divergence fonctionnelle entre MEK1 et MEK2 telle qu’observée lors de l’induction par
la bombésine pourrait provenir d’alternative à la voie de signalisation conventionnelle Ras-
ERK et préconiserait l’existence de chevauchement entre plusieurs voies de signalisation
via le module MEK. Ce module formé de deux protéines bien que très homologue et
généralement équivalent de point de vue fonctionnel permettrait, dans un contexte unique,
de médier une réponse spécifique.
L’élément ou la séquence protéique dans MEK1 qui semble médier la réponse spécifique à
la bombésine se retrouverait dans la région C-terminale. Nos résultats indiquent que la
réexpression d’une protéine chimérique MEK2/1; (a.a 1-116)/MEK1 (a.a 117-393), dans
les MEFs Mek1-/- rétablit l’activation de ERK. La région C-terminale, tout particulièrement
la région MSS, semble jouer un rôle essentiel dans la régulation de l’activité catalytique de
MEK1 et MEK2. La MSS semble prendre part dans la modulation de l’activité de MEK1
ainsi que dans le cross-talk du sentier ERK avec d’autres voies de signalisation [391].
En somme, ces résultats fournissent de nouveaux éléments démontrant le rôle unique que
peut jouer MEK1 dans la cascade ERK/MAPK pour accomplir certains processus

139
cellulaires spécifiques. Ces résultats suggèrent que seul MEK1 et non pas MEK2 peut
répondre à des signaux provenant de l’induction par la bombésine.
Dans le but de comprendre cette divergence dans la fonction de MEK1 et MEK2, nous
avons tenté d’identifier de nouvelles protéines interagissant spécifiquement avec MEK1. En
fait, la découverte de substrats spécifiques à MEK1 et/ou une meilleure compréhension du
mode de régulation de MEK1 pourraient bien expliquer cette divergence. Pour atteindre
notre objectif, nous avons utilisé le criblage et l’étude des intéraction protéine-protéine du
système double hybride chez la levure.
Bien de recherches faites avant ou en même temps que la nôtre ont utilisé, comme appât,
MEK1 fusionné en carboxy terminal du domaine de liaison à l’ADN de LexA (ou de Gal4).
L’originalité de nos travaux était de démasquer la région N-terminale de MEK1, clairement
identifiée comme importante et suffisante pour lier ERK, en mettant MEK1 en amino
terminal du domaine de liaison à l’ADN de LexA.
Le criblage de deux banques d’ADNc, provenant de cerveaux de souris et de placenta
humain (produit par Clonetech.), ne nous a pas permis d’identifier de nouveaux éléments
agissant au sein de la voie de signalisation ERK (Annexe A). En outre, presque 70 % des
clones isolés lors de notre criblage double hybride contiennent des séquences de longueurs
variables tous provenant de la région C-terminale de Mek1 et de Mek2 (Annexe A.1 et
chapitre 3). La région repêchée contient le dernier domaine kinase de MEK1 et 2 ainsi que
la région C-terminale dont la fonction est encore inconnue. La séquence la plus courte qui a
été isolée code pour le peptide N324 (résidu 324-393 de MEK1). Des essais double hybride
nous ont permis d’identifier la région entourant la boucle d’activation (a.a 195-241) comme
étant l’autre région impliquée dans l’interaction.
Parce que les protéines MEKs ne forment pas de multimères (Tableau 3-1), l’interaction
observée dans notre étude serait de type intramoléculaire. De plus, cette interaction serait
dépendante de l’état de phosphorylation/activation de MEK1 et MEK2, puisque dans la
présente étude, il y a perte d’interaction entre la région minimale isolée, N324 et la forme
dominante positive de MEK1, MEK1 C350 DD (Tableau 3-1).

140
En somme, ces résultats indiquent qu’il existe une interaction intramoléculaire entre la
région C-terminale et la région de la boucle d’activation de MEK1 et MEK2 et que cette
interaction serait dépendante de l’état de phosphorylation de ces protéines kinases. Elles
suggèrent que les variations électrostatiques induites par la phosphorylation de la boucle
d’activation peuvent contribuer à un changement de la conformation de la région C-
terminale de MEK1 qui, par conséquence, pourrait jouer un rôle dans la transition de l’état
inactif à l’état actif.
À ce stade des travaux, nous avons émis l’hypothèse que la région C-terminale serait un
domaine d’autoinhibition ou un pseudo substrats de MEK1/2 dont le rôle serait de réguler
leur l’activité enzymatique. Lorsque la voie ERK/MAPK est au repos, ce domaine garderait
la protéine kinase dans sa conformation inactive. Cette inhibition serait levée grâce à
l’activation/phosphorylation de MEK1 (ou MEK2) par son activateur. Plusieurs
mécanismes d’autorégulation par l’intermédiaire d’un domaine d’autoinhibition ont été
décrits, entre autres, chez les protéines kinases Raf, Mos, MAPKAPK-2 et WNK1 [457-
460]. Cette inhibition est levée soit par phosphorylation directe par un activateur soit par
autophosphorylation. MEK1 est capable de s’autophosphoryler mais peu est connu sur le
rôle de cette phosphorylation dans la régulation de son activité [461].
Pour vérifier si la région C-terminale pourrait jouer un rôle direct dans l’activation de la
protéine, nous avons entamé des essais d’activation en utilisant des mutants de délétions
MEK1 dépourvus de cette région. Nos résultats ont indiqué que MEK1 perd complètement
son activité kinase lorsque écourtée au résidu 362. Ces mutants MEK1 semblent aussi se
retrouver dans une fraction insoluble lorsque surexprimés chez les cellules HEK 293
(Figure 3-4) ou CCL39 (données non présentées). Des expériences d’immunocytochimie
ont indiqué que ces mutants s’expriment dans des compartiments cellulaires spécifiques (en
occurrence dans les lysosomes) comparativement à l’expression cytoplasmique du type
sauvage. Les délétants C362 seraient produits chez les cellules avec des conformations
altérées qui les mènent à se localiser dans les compartiments endosomales/lysosomaux. Les
travaux de Shapiro et ces collaborateurs ont indiqué que des mutants semblables, MEK1
C296, C330 et ∆330-379 se retrouveraient dans des compartiments
endosomales/lysosomaux [136]. Ces résultats suggèrent que la région C-terminale pourrait

141
être importante pour que la protéine MEK1 adopte une conformation physiologique et
stable lui permettant de s’exprimer convenablement et de se localiser dans la fraction
soluble du cytoplasme.
Alors qu’il était possible d’observer une interaction stable entre les mutants de délétion
C362 et ERK (données non présentées), ces mutants MEK1 semblent être incapables de
phosphoryler ERK même dans leur forme active, C362DD. L’altération de l’activité des
mutants C362 pourrait s’expliquer par la perte d'une région ou d'une structure importante
dans MEK1 qui serait impliquée dans l'activité enzymatique. Le dernier domaine kinase, le
domaine XI qui est caractérisé par sa structure et sa séquence conservée, principalement, le
résidu Arg 349 conservé chez les différentes protéines kinases [86,132], semble être intact
chez les mutants de délétion C362. Ces résultats suggèrent que la région comprise entre les
résidus 362 et 393 est importante pour l’activité de MEK1.
Pour vérifier cette hypothèse, nous avons repris nos expériences en utilisant de nouveaux
mutants de délétion, les MEK1 C380. Nos résultats ont montré que ces délétants se
comportaient comme les protéines de type sauvage. Ainsi, nous avons observé une
localisation cellulaire des mutants C380 identique à celle de la protéine de type sauvage
(Figure 3-5b). De plus, l’activité enzymatique du mutant MEK1 DD C380 est comparable à
l’activité du mutant pleine longueur MEK1 DD (données non présentées), ce qui suggère
que l’activation des mutants C380 est dépendante de la phosphorylation des résidus sérines
218 et 222 de la boucle d’activation. La délétion des 13 deniers résidus de MEK1 ne
semble donc pas avoir d’effet sur son activité basale. Nos résultats vont à l’encontre de
ceux obtenus par le groupe de Shapiro qui démontrait in vivo, qu’un mutant MEK1 d379
stimulait la phosphorylation de ERK1 et ERK2 en absence d’induction [136].
La perte d’activité des mutants de délétion C362 comparée aux mutants C380 laisse croire
que la séquence d’acides aminés comprise entre 362 et 380 dans MEK1 contient des
éléments importants pour l’activité enzymatique de MEK1. L’intégrité ou la conformation
adéquate du domaine kinase semble être perturbée chez les mutants C362 ce qui même à
leur inactivation. Une étude plus poussée sur l’activité des mutants de délétion MEK1 qui

142
sont tronqués à des résidus entre 362 et 380 pourrait nous éclairer quant à l’importance de
cette région dans l’activité de MEK1.
L’ensemble des résultats d’activité et d’interaction suggèrent que la région C-terminale est
impliquée dans une variation conformationnelle du domaine kinase qui se produit entre
l’état inactif et l’état actif de la protéine MEK1. La stabilité de la conformation inactive ne
semble pas impliquer une compétition intra-stérique par un domaine d’autoinhibition ou de
pseudo substrat, puisque la délétion de la région C-terminale ne met pas la protéine dans
une forme active ou constitutivement active. Par contre, il est possible que des
réarrangements structuraux dans différentes régions de MEK1 coopèrent pour mettre
l'enzyme dans un état actif. D’autres études ont indiqué qu’un mouvement dynamique
subtil impliquant diverses structures secondaires peut également être important pour
l’activation complète de MEK1 [128,133,134].
Les variations conformationnelles de la région C-terminale pourraient aussi être
primordiales pour la stabilité des interactions de MEK1 avec ses substrats, activateurs et
protéines d’échafaudage et/ou nécessaires pour la localisation intracellulaire adéquate de
MEK1. Nos expériences de double hybride ont indiqué un défaut d’interaction de MEK1
C350 et RAF-1 (tableau 3-1), Alors que la région MSS semble être désigné comme le site
d’interaction RAF-MEK1/2 [71,102], nos résultats suggèrent une implication directe du
fragment MEK1 compris entre 350 et 362 dans l’attachement à Raf ou une déformation
conformationnelle du mutant C350 qui altère la stabilité de l’interaction MEK1-Raf.
Sachant la nature conservée de la famille de MAPKK des protozoaires jusqu’aux
mammifères, l'élucidation de la structure en cristal de MEK1 dans ses conformations
actives et inactives permettra de mieux comprendre les mécanismes contrôlant l'activation
de cette famille d’enzymes.
D’une façon générale, l’identification et la caractérisation d’une voie de signalisation qui
serait dépendante de MEK1 mais indépendante de MEK2 permettront éventuellement de
développer de nouveaux inhibiteurs spécifiques qui pourront servir dans la lutte contre
certaines maladies, notamment le cancer.

Annexe

144
A. Criblage double hybride chez la levure
A.1. Tableaux récapitulatifs des clones isolés lors du criblage
double hybride
N° Clone Gène/protéine Région identifiée Remarques DNC1 1 Mek2 a.a 296--> Confirmé 2 hybride
2 Mek1 Confirmé 2 hybride 3 Mek1 Confirmé 2 hybride 4 Mek1 a.a 253--> Confirmé 2 hybride 5 Mek1 a.a 253--> Confirmé 2 hybride 6 Mek2 a.a 313--> Confirmé 2 hybride 7 Mek1 a.a 323--> Confirmé 2 hybride 8 Mek1 a.a 322--> Confirmé 2 hybride 9 Mek1 a.a 322--> Confirmé 2 hybride 10 Mek1 a.a 322--> Confirmé 2 hybride 11 Mek2 a.a 296--> Confirmé 2 hybride 12 Mek1 Confirmé 2 hybride 13 Mek1 a.a 319--> Non confirmé 14 Mek1 a.a 279--> Confirmé 2 hybride 15 Mek1 a.a 323--> Confirmé 2 hybride 16 Mek1 a.a 247--> Confirmé 2 hybride 19 Mek2 a.a 296--> Confirmé 2 hybride 20 alpha-2 globin
(HBA1) Négatif 2 hybride
22 Mek1 a.a 291--> Confirmé 2 hybride 23 Mek2 a.a 211--> Confirmé 2 hybride 28 N/D Négatif 2 hybride 30 N/D Négatif 2 hybride
Mauvaise séquence 33 elongation factor
1-alpha Négatif 2 hybride
35 Mek1 a.a 247--> Confirmé 2 hybride

145
38 ? Négatif 2 hybride 39 N/D Négatif 2 hybride 42 Mitochondrial DNA Négatif 2 hybride 45 Replacement variant
of histone H3.3 Essai 2 hybride
séquencé 2 côtés 46 Replacement variant
of histone H3.3 Confirmé 2 hybride
56 Histone H3.3A Confirmé 2 hybride séquencé 2 côtés
58 Mek1 a.a 179--> Confirmé 2 hybride 60 Mek1 a.a 244--> Confirmé 2 hybride 61 H. Sapiens BTF3 Négatif 2 hybride
séquencé 2 côtés 63 prostaglandin
D synthase Négatif 2 hybride
65 ? Négatif 2 hybride 66 Mek2 a.a 332--> Confirmé 2 hybride
Placenta 23 Mek2 a.a 329--> Confirmé 2 hybride
N° Clone Gène/protéine Région identifiée Remarques DNC2 1 Mek1 a.a 323--> Confirmé 2 hybride
2 Mek1 Non confirmé 3 c-Raf Domaine catalytique Confirmé 2 hybride 4 Mek2 a.a 272--> Confirmé 2 hybride 5 Mek1 a.a 211--> Confirmé 2 hybride 6 Mek1 a.a 322--> Confirmé 2 hybride 7 Mek1 a.a 324--> Confirmé 2 hybride 8 Mek2 a.a 270--> Confirmé 2 hybride 9 Mek2 a.a 246--> Confirmé 2 hybride 10 Mek1 a.a 215--> Confirmé 2 hybride 11 Mek1 a.a 259--> Confirmé 2 hybride 12 Mek2 a.a 211--> Confirmé 2 hybride 13 Mek2 a.a 295--> Confirmé 2 hybride 14 Mek2 a.a 273--> Confirmé 2 hybride 15 ? Négatif 2 hybride 16 Mek1 a.a 324--> Confirmé 2 hybride
séquencé 2 côtés

146
17 Mek2 a.a 289--> Confirmé 2 hybride 18 Mek1 a.a 253--> Confirmé 2 hybride 19 Mek1 a.a 327--> Confirmé 2 hybride 20 ? Négatif 2 hybride 22 Mek1 a.a 253--> Confirmé 2 hybride 25 Mek1 a.a 324--> Confirmé 2 hybride 33 Mek1 a.a 324--> Confirmé 2 hybride 35 Mek2 a.a 274--> Confirmé 2 hybride 36 PLZF a.a 416--> Négatif 2 hybride 37 Mek1 a.a 231--> Confirmé 2 hybride 41 Mek1 a.a 231--> Non confirmé 43 Mek1 a.a 231--> Non confirmé 44 Histone H3.3A Essai 2 hybride
séquencé 2 côtés 46 Replacement variant
of histone H3.3 Essai2 hybride
séquencé 2 côtés 50 Gène homologue à
M96A humain a.a 65 –> prot : 594 a.a.
homologue à M96 de souris. 2X séquencé. 2 côtés
55 Mek1 a.a 231--> Non confirmé 60 ? mauvaise séquence 62 Mek1 a.a 323--> Confirmé 2 hybride
séquencé 2 côtés 65 ? Négatif 2 hybride 66 Mek1 a.a 252--> Non confirmé 67 Mouse histone 4 Essai double hybride 69 Mek1 séquencé. 2 côtés 75 ? Négatif 2 hybride 84 EST MDB1180 Mouse
brain, Stratagene Mus musculus cDNA
3'end
Négatif 2 hybride
90 ? Négatif 2 hybride

147
A.2. Vue d’ensemble des faux positives courrament isolés lors
des criblages double hybride chez la levure
Protéines Identifiées comme faux positives
Interactions protéiques réelles
Protéines hsp 16 5 Protéines ribosomales 14 1 Cytochrome oxidase 5 - Protéines mitochondriales 3 1 Sous-unités protéasomales 4 3 ferritine 4 - ARNt synthase 3 - Protéines reliées au collagène 3 - Protéines doigt de Zn 3 4 vimentine 2 -
Adapté du site internet de Golemis E., [464].

Bibliographie

149
1. Lee, J. T., Jr. and McCubrey, J. A. (2002). The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia 16, 486-507.
2. Rak, J., Yu, J. L., Klement, G., and Kerbel, R. S. (2000). Oncogenes and angiogenesis: signaling three-dimensional tumor growth. J.Investig.Dermatol.Symp.Proc. 5, 24-33.
3. Tibbles, L. A. and Woodgett, J. R. (1999). The stress-activated protein kinase pathways. Cell Mol.Life Sci. 55, 1230-1254.
4. Kyriakis, J. M. (1999). Making the connection: coupling of stress-activated ERK/MAPK (extracellular-signal-regulated kinase/mitogen-activated protein kinase) core signalling modules to extracellular stimuli and biological responses. Biochem.Soc.Symp. 64:29-48., 29-48.
5. Robinson, M. J. and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr.Opin.Cell Biol. 9 , 180-186.
6. Denhardt, D. T. (1996). Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem.J. 318, 729-747.
7. Zanke, B. W., Rubie, E. A., Winnett, E., Chan, J., Randall, S., Parsons, M., Boudreau, K., McInnis, M., Yan, M., Templeton, D. J., and Woodgett, J. R. (1996). Mammalian mitogen-activated protein kinase pathways are regulated through formation of specific kinase-activator complexes. J.Biol.Chem. 271, 29876-29881.
8. Schaeffer, H. J. and Weber, M. J. (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol.Cell Biol. 19, 2435-2444.
9. Enslen, H. and Davis, R. J. (2001). Regulation of MAP kinases by docking domains. Biol.Cell 93, 5-14.
10. Pouyssegur, J., Volmat, V., and Lenormand, P. (2002). Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem.Pharmacol. 64, 755-763.
11. Vojtek, A. B., Hollenberg, S. M., and Cooper, J. A. (1993). Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74, 205-214.
12. Zhang, X. F., Settleman, J., Kyriakis, J. M., Takeuchi-Suzuki, E., Elledge, S. J., Marshall, M. S., Bruder, J. T., Rapp, U. R., and Avruch, J. (1993). Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364, 308-313.

150
13. Dent, P., Reardon, D. B., Morrison, D. K., and Sturgill, T. W. (1995). Regulation of Raf-1 and Raf-1 mutants by Ras-dependent and Ras-independent mechanisms in vitro. Mol.Cell Biol. 15 , 4125-4135.
14. Warne, P. H., Viciana, P. R., and Downward, J. (1993). Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364, 352-355.
15. Chaudhary, A., King, W. G., Mattaliano, M. D., Frost, J. A., Diaz, B., Morrison, D. K., Cobb, M. H., Marshall, M. S., and Brugge, J. S. (2000). Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr.Biol. 10, 551-554.
16. Diaz, B., Barnard, D., Filson, A., MacDonald, S., King, A., and Marshall, M. (1997). Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol.Cell Biol. 17, 4509-4516.
17. Fabian, J. R., Daar, I. O., and Morrison, D. K. (1993). Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol.Cell Biol. 13, 7170-7179.
18. Kolch, W., Heidecker, G., Kochs, G., Hummel, R., Vahidi, H., Mischak, H., Finkenzeller, G., Marme, D., and Rapp, U. R. (1993). Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature 364, 249-252.
19. Dong, C., Waters, S. B., Holt, K. H., and Pessin, J. E. (1996). SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J.Biol.Chem. 271, 6328-6332.
20. Schwartz, M. A. and Assoian, R. K. (2001). Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J.Cell Sci. 114, 2553-2560.
21. Cary, L. A., Han, D. C., and Guan, J. L. (1999). Integrin-mediated signal transduction pathways. Histol.Histopathol. 14, 1001-1009.
22. English, J., Pearson, G., Wilsbacher, J., Swantek, J., Karandikar, M., Xu, S., and Cobb, M. H. (1999). New insights into the control of MAP kinase pathways. Exp.Cell Res. 253, 255-270.
23. Lewis, T. S., Shapiro, P. S., and Ahn, N. G. (1998). Signal transduction through MAP kinase cascades. Adv.Cancer Res. 74:49-139., 49-139.
24. Luttrell, L. M., Daaka, Y., and Lefkowitz, R. J. (1999). Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr.Opin.Cell Biol. 11, 177-183.

151
25. Hafner, S., Adler, H. S., Mischak, H., Janosch, P., Heidecker, G., Wolfman, A., Pippig, S., Lohse, M., Ueffing, M., and Kolch, W. (1994). Mechanism of inhibition of Raf-1 by protein kinase A. Mol.Cell Biol. 14, 6696-6703.
26. Mischak, H., Seitz, T., Janosch, P., Eulitz, M., Steen, H., Schellerer, M., Philipp, A., and Kolch, W. (1996). Negative regulation of Raf-1 by phosphorylation of serine 621. Mol.Cell Biol. 16, 5409-5418.
27. Wu, J., Dent, P., Jelinek, T., Wolfman, A., Weber, M. J., and Sturgill, T. W. (1993). Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3',5'-monophosphate. Science 262, 1065-1069.
28. Vossler, M. R., Yao, H., York, R. D., Pan, M. G., Rim, C. S., and Stork, P. J. (1997). cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89, 73-82.
29. Grewal, S. S., Horgan, A. M., York, R. D., Withers, G. S., Banker, G. A., and Stork, P. J. (2000). Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J.Biol.Chem. 275, 3722-3728.
30. Wan, Y., Bence, K., Hata, A., Kurosaki, T., Veillette, A., and Huang, X. Y. (1997). Genetic evidence for a tyrosine kinase cascade preceding the mitogen-activated protein kinase cascade in vertebrate G protein signaling. J.Biol.Chem. 272, 17209-17215.
31. Hedin, K. E., Bell, M. P., Huntoon, C. J., Karnitz, L. M., and McKean, D. J. (1999). Gi proteins use a novel beta gamma- and Ras-independent pathway to activate extracellular signal-regulated kinase and mobilize AP-1 transcription factors in Jurkat T lymphocytes. J.Biol.Chem. 274, 19992-20001.
32. Luttrell, L. M., Hawes, B. E., van Biesen, T., Luttrell, D. K., Lansing, T. J., and Lefkowitz, R. J. (1996). Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J.Biol.Chem. 271, 19443-19450.
33. Lopez-Ilasaca, M., Crespo, P., Pellici, P. G., Gutkind, J. S., and Wetzker, R. (1997). Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science 275, 394-397.
34. Dikic, I., Tokiwa, G., Lev, S., Courtneidge, S. A., and Schlessinger, J. (1996). A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383, 547-550.
35. Della Rocca, G. J., Maudsley, S., Daaka, Y., Lefkowitz, R. J., and Luttrell, L. M. (1999). Pleiotropic coupling of G protein-coupled receptors to the mitogen-

152
activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J.Biol.Chem. 274, 13978-13984.
36. Lev, S., Moreno, H., Martinez, R., Canoll, P., Peles, E., Musacchio, J. M., Plowman, G. D., Rudy, B., and Schlessinger, J. (1995). Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature 376, 737-745.
37. Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoyanova, S., Vanhaesebroeck, B., Dhand, R., Nurnberg, B., and . (1995). Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690-693.
38. Daub, H., Weiss, F. U., Wallasch, C., and Ullrich, A. (1996). Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557-560.
39. Schonwasser, D. C., Marais, R. M., Marshall, C. J., and Parker, P. J. (1998). Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol.Cell Biol. 18, 790-798.
40. Nishida, Y., Inoue, Y. H., Tsuda, L., Adachi-Yamada, T., Lim, Y. M., Hata, M., Ha, H. Y., and Sugiyama, S. (1996). The Raf/MAP kinase cascade in cell cycle regulation and differentiation in Drosophila. Cell Struct.Funct. 21, 437-444.
41. Force, T., Bonventre, J. V., Heidecker, G., Rapp, U., Avruch, J., and Kyriakis, J. M. (1994). Enzymatic characteristics of the c-Raf-1 protein kinase. Proc.Natl.Acad.Sci.U.S.A 91, 1270-1274.
42. Kyriakis, J. M., App, H., Zhang, X. F., Banerjee, P., Brautigan, D. L., Rapp, U. R., and Avruch, J. (1992). Raf-1 activates MAP kinase-kinase. Nature 358, 417-421.
43. Morrison, D. K. and Cutler, R. E. (1997). The complexity of Raf-1 regulation. Curr.Opin.Cell Biol. 9, 174-179.
44. Leevers, S. J., Paterson, H. F., and Marshall, C. J. (1994). Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369, 411-414.
45. Whitehurst, C. E., Owaki, H., Bruder, J. T., Rapp, U. R., and Geppert, T. D. (1995). The MEK kinase activity of the catalytic domain of RAF-1 is regulated independently of Ras binding in T cells. J.Biol.Chem. 270, 5594-5599.
46. Mikula, M., Schreiber, M., Husak, Z., Kucerova, L., Ruth, J., Wieser, R., Zatloukal, K., Beug, H., Wagner, E. F., and Baccarini, M. (2001). Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 20, 1952-1962.

153
47. Fantl, W. J., Muslin, A. J., Kikuchi, A., Martin, J. A., MacNicol, A. M., Gross, R. W., and Williams, L. T. (1994). Activation of Raf-1 by 14-3-3 proteins. Nature 371, 612-614.
48. Freed, E., Symons, M., Macdonald, S. G., McCormick, F., and Ruggieri, R. (1994). Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 265, 1713-1716.
49. Jaiswal, R. K., Weissinger, E., Kolch, W., and Landreth, G. E. (1996). Nerve growth factor-mediated activation of the mitogen-activated protein (MAP) kinase cascade involves a signaling complex containing B-Raf and HSP90. J.Biol.Chem. 271, 23626-23629.
50. Stancato, L. F., Chow, Y. H., Hutchison, K. A., Perdew, G. H., Jove, R., and Pratt, W. B. (1993). Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J.Biol.Chem. 268, 21711-21716.
51. Stancato, L. F., Silverstein, A. M., Owens-Grillo, J. K., Chow, Y. H., Jove, R., and Pratt, W. B. (1997). The hsp90-binding antibiotic geldanamycin decreases Raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J.Biol.Chem. 272, 4013-4020.
52. Kimura, Y., Rutherford, S. L., Miyata, Y., Yahara, I., Freeman, B. C., Yue, L., Morimoto, R. I., and Lindquist, S. (1997). Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 11, 1775-1785.
53. Schulte, T. W., Blagosklonny, M. V., Ingui, C., and Neckers, L. (1995). Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J.Biol.Chem. 270, 24585-24588.
54. Schulte, T. W., Blagosklonny, M. V., Romanova, L., Mushinski, J. F., Monia, B. P., Johnston, J. F., Nguyen, P., Trepel, J., and Neckers, L. M. (1996). Destabilization of Raf-1 by geldanamycin leads to disruption of the Raf-1-MEK-mitogen-activated protein kinase signalling pathway. Mol.Cell Biol. 16, 5839-5845.
55. Erhardt, P., Troppmair, J., Rapp, U. R., and Cooper, G. M. (1995). Differential regulation of Raf-1 and B-Raf and Ras-dependent activation of mitogen-activated protein kinase by cyclic AMP in PC12 cells. Mol.Cell Biol. 15, 5524-5530.
56. Seidel, M. G., Klinger, M., Freissmuth, M., and Holler, C. (1999). Activation of mitogen-activated protein kinase by the A(2A)-adenosine receptor via a rap1-dependent and via a p21(ras)-dependent pathway. J.Biol.Chem. 274, 25833-25841.
57. Zwartkruis, F. J., Wolthuis, R. M., Nabben, N. M., Franke, B., and Bos, J. L. (1998). Extracellular signal-regulated activation of Rap1 fails to interfere in Ras effector signalling. EMBO J. 17, 5905-5912.

154
58. Hamilton, M. and Wolfman, A. (1998). Ha-ras and N-ras regulate MAPK activity by distinct mechanisms in vivo. Oncogene 16, 1417-1428.
59. Okada, T., Hu, C. D., Jin, T. G., Kariya, K., Yamawaki-Kataoka, Y., and Kataoka, T. (1999). The strength of interaction at the Raf cysteine-rich domain is a critical determinant of response of Raf to Ras family small GTPases. Mol.Cell Biol. 19, 6057-6064.
60. Morrison, D. K., Heidecker, G., Rapp, U. R., and Copeland, T. D. (1993). Identification of the major phosphorylation sites of the Raf-1 kinase. J.Biol.Chem. 268, 17309-17316.
61. Dent, P., Jelinek, T., Morrison, D. K., Weber, M. J., and Sturgill, T. W. (1995). Reversal of Raf-1 activation by purified and membrane-associated protein phosphatases. Science 268, 1902-1906.
62. King, A. J., Sun, H., Diaz, B., Barnard, D., Miao, W., Bagrodia, S., and Marshall, M. S. (1998). The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 396, 180-183.
63. Zimmermann, S. and Moelling, K. (1999). Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286, 1741-1744.
64. Muslin, A. J., Tanner, J. W., Allen, P. M., and Shaw, A. S. (1996). Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889-897.
65. Thorson, J. A., Yu, L. W., Hsu, A. L., Shih, N. Y., Graves, P. R., Tanner, J. W., Allen, P. M., Piwnica-Worms, H., and Shaw, A. S. (1998). 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol.Cell Biol. 18, 5229-5238.
66. Rommel, C., Radziwill, G., Moelling, K., and Hafen, E. (1997). Negative regulation of Raf activity by binding of 14-3-3 to the amino terminus of Raf in vivo. Mech.Dev. 64, 95-104.
67. Bennett, A. M. and Tonks, N. K. (1997). Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278, 1288-1291.
68. Rommel, C., Clarke, B. A., Zimmermann, S., Nunez, L., Rossman, R., Reid, K., Moelling, K., Yancopoulos, G. D., and Glass, D. J. (1999). Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286, 1738-1741.
69. Hall-Jackson, C. A., Eyers, P. A., Cohen, P., Goedert, M., Boyle, F. T., Hewitt, N., Plant, H., and Hedge, P. (1999). Paradoxical activation of Raf by a novel Raf inhibitor. Chem.Biol. 6, 559-568.

155
70. Hall-Jackson, C. A., Goedert, M., Hedge, P., and Cohen, P. (1999). Effect of SB 203580 on the activity of c-Raf in vitro and in vivo. Oncogene 18, 2047-2054.
71. Catling, A. D., Schaeffer, H. J., Reuter, C. W., Reddy, G. R., and Weber, M. J. (1995). A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol.Cell Biol. 15, 5214-5225.
72. Jelinek, T., Catling, A. D., Reuter, C. W., Moodie, S. A., Wolfman, A., and Weber, M. J. (1994). RAS and RAF-1 form a signalling complex with MEK-1 but not MEK-2. Mol.Cell Biol. 14, 8212-8218.
73. Wu, X., Noh, S. J., Zhou, G., Dixon, J. E., and Guan, K. L. (1996). Selective activation of MEK1 but not MEK2 by A-Raf from epidermal growth factor-stimulated Hela cells. J.Biol.Chem. 271, 3265-3271.
74. Reuter, C. W., Catling, A. D., Jelinek, T., and Weber, M. J. (1995). Biochemical analysis of MEK activation in NIH3T3 fibroblasts. Identification of B-Raf and other activators. J.Biol.Chem. 270, 7644-7655.
75. Ussar, S. and Voss, T. (2004). MEK1 and MEK2: different regulators of the G1/S transition. J.Biol.Chem. ..
76. Liu, X., Yan, S., Zhou, T., Terada, Y., and Erikson, R. L. (2004). The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene 23, 763-776.
77. Giroux, S., Tremblay, M., Bernard, D., Cardin-Girard, J. F., Aubry, S., Larouche, L., Rousseau, S., Huot, J., Landry, J., Jeannotte, L., and Charron, J. (1999). Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr.Biol. 9, 369-372.
78. Belanger, L. F., Roy, S., Tremblay, M., Brott, B., Steff, A. M., Mourad, W., Hugo, P., Erikson, R., and Charron, J. (2003). Mek2 is dispensable for mouse growth and development. Mol.Cell Biol. 23, 4778-4787.
79. Pham, C. D., Arlinghaus, R. B., Zheng, C. F., Guan, K. L., and Singh, B. (1995). Characterization of MEK1 phosphorylation by the v-Mos protein. Oncogene %20;10, 1683-1688.
80. Gardner, A. M., Vaillancourt, R. R., Lange-Carter, C. A., and Johnson, G. L. (1994). MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Mol.Biol.Cell 5, 193-201.
81. Karandikar, M., Xu, S., and Cobb, M. H. (2000). MEKK1 binds raf-1 and the ERK2 cascade components. J.Biol.Chem. 275, 40120-40127.

156
82. Salmeron, A., Ahmad, T. B., Carlile, G. W., Pappin, D., Narsimhan, R. P., and Ley, S. C. (1996). Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 15, 817-826.
83. Xu, S., Robbins, D., Frost, J., Dang, A., Lange-Carter, C., and Cobb, M. H. (1995). MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc.Natl.Acad.Sci.U.S.A 92, 6808-6812.
84. Verlhac, M. H., Lefebvre, C., Kubiak, J. Z., Umbhauer, M., Rassinier, P., Colledge, W., and Maro, B. (2000). Mos activates MAP kinase in mouse oocytes through two opposite pathways. EMBO J. 19, 6065-6074.
85. Hagemann, D., Troppmair, J., and Rapp, U. R. (1999). Cot protooncoprotein activates the dual specificity kinases MEK-1 and SEK-1 and induces differentiation of PC12 cells. Oncogene 18, 1391-1400.
86. Mansour, S. J., Candia, J. M., Matsuura, J. E., Manning, M. C., and Ahn, N. G. (1996). Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry 35, 15529-15536.
87. Mansour, S. J., Matten, W. T., Hermann, A. S., Candia, J. M., Rong, S., Fukasawa, K., Vande Woude, G. F., and Ahn, N. G. (1994). Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966-970.
88. Whalen, A. M., Galasinski, S. C., Shapiro, P. S., Nahreini, T. S., and Ahn, N. G. (1997). Megakaryocytic differentiation induced by constitutive activation of mitogen-activated protein kinase kinase. Mol.Cell Biol. 17, 1947-1958.
89. Crews, C. M., Alessandrini, A., and Erikson, R. L. (1992). The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 258, 478-480.
90. Wu, J., Harrison, J. K., Vincent, L. A., Haystead, C., Haystead, T. A., Michel, H., Hunt, D. F., Lynch, K. R., and Sturgill, T. W. (1993). Molecular structure of a protein-tyrosine/threonine kinase activating p42 mitogen-activated protein (MAP) kinase: MAP kinase kinase. Proc.Natl.Acad.Sci.U.S.A 90, 173-177.
91. Wu, J., Harrison, J. K., Dent, P., Lynch, K. R., Weber, M. J., and Sturgill, T. W. (1993). Identification and characterization of a new mammalian mitogen-activated protein kinase kinase, MKK2. Mol.Cell Biol. 13, 4539-4548.
92. Zheng, C. F. and Guan, K. L. (1993). Dephosphorylation and inactivation of the mitogen-activated protein kinase by a mitogen-induced Thr/Tyr protein phosphatase. J.Biol.Chem. 268, 16116-16119.
93. Ahn, N. G., Seger, R., Bratlien, R. L., Diltz, C. D., Tonks, N. K., and Krebs, E. G. (1991). Multiple components in an epidermal growth factor-stimulated protein

157
kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J.Biol.Chem. 266, 4220-4227.
94. Zheng, C. F. and Guan, K. L. (1993). Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J.Biol.Chem. 268, 11435-11439.
95. Robinson, M. J., Cheng, M., Khokhlatchev, A., Ebert, D., Ahn, N., Guan, K. L., Stein, B., Goldsmith, E., and Cobb, M. H. (1996). Contributions of the mitogen-activated protein (MAP) kinase backbone and phosphorylation loop to MEK specificity. J.Biol.Chem. 271, 29734-29739.
96. Prowse, C. N., Hagopian, J. C., Cobb, M. H., Ahn, N. G., and Lew, J. (2000). Catalytic reaction pathway for the mitogen-activated protein kinase ERK2. Biochemistry 39, 14002.
97. Robinson, M. J., Harkins, P. C., Zhang, J., Baer, R., Haycock, J. W., Cobb, M. H., and Goldsmith, E. J. (1996). Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5'-triphosphate. Biochemistry 35, 5641-5646.
98. Winston, B. W., Remigio, L. K., and Riches, D. W. (1995). Preferential involvement of MEK1 in the tumor necrosis factor-alpha-induced activation of p42mapk/erk2 in mouse macrophages. J.Biol.Chem. 270, 27391-27394.
99. Seufferlein, T., Withers, D. J., and Rozengurt, E. (1996). Reduced requirement of mitogen-activated protein kinase (MAPK) activity for entry into the S phase of the cell cycle in Swiss 3T3 fibroblasts stimulated by bombesin and insulin. J.Biol.Chem. 271, 21471-21477.
100. Bhunia, A. K., Han, H., Snowden, A., and Chatterjee, S. (1996). Lactosylceramide stimulates Ras-GTP loading, kinases (MEK, Raf), p44 mitogen-activated protein kinase, and c-fos expression in human aortic smooth muscle cells. J.Biol.Chem. 271, 10660-10666.
101. Abbott, D. W. and Holt, J. T. (1999). Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J.Biol.Chem. 274, 2732-2742.
102. Papin, C., Denouel, A., Calothy, G., and Eychene, A. (1996). Identification of signalling proteins interacting with B-Raf in the yeast two-hybrid system. Oncogene 12, 2213-2221.
103. Dang, A., Frost, J. A., and Cobb, M. H. (1998). The MEK1 proline-rich insert is required for efficient activation of the mitogen-activated protein kinases ERK1 and ERK2 in mammalian cells. J.Biol.Chem. 273, 19909-19913.

158
104. Frost, J. A., Steen, H., Shapiro, P., Lewis, T., Ahn, N., Shaw, P. E., and Cobb, M. H. (1997). Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 16, 6426-6438.
105. Mansour, S. J., Resing, K. A., Candi, J. M., Hermann, A. S., Gloor, J. W., Herskind, K. R., Wartmann, M., Davis, R. J., and Ahn, N. G. (1994). Mitogen-activated protein (MAP) kinase phosphorylation of MAP kinase kinase: determination of phosphorylation sites by mass spectrometry and site-directed mutagenesis. J.Biochem.(Tokyo) 116, 304-314.
106. Cobb, M. H., Xu, S., Cheng, M., Ebert, D., Robbins, D., Goldsmith, E., and Robinson, M. (1996). Structural analysis of the MAP kinase ERK2 and studies of MAP kinase regulatory pathways. Adv.Pharmacol. 36:49-65., 49-65.
107. Tanoue, T., Adachi, M., Moriguchi, T., and Nishida, E. (2000). A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat.Cell Biol. 2, 110-116.
108. Xu, B., Wilsbacher, J. L., Collisson, T., and Cobb, M. H. (1999). The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J.Biol.Chem. 274, 34029-34035.
109. Yang, S. H., Whitmarsh, A. J., Davis, R. J., and Sharrocks, A. D. (1998). Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 17, 1740-1749.
110. Gotoh, I., Fukuda, M., Adachi, M., and Nishida, E. (1999). Control of the cell morphology and the S phase entry by mitogen-activated protein kinase kinase. A regulatory role of its n-terminal region. J.Biol.Chem. 274, 11874-11880.
111. Duesbery, N. S., Webb, C. P., Leppla, S. H., Gordon, V. M., Klimpel, K. R., Copeland, T. D., Ahn, N. G., Oskarsson, M. K., Fukasawa, K., Paull, K. D., and Vande Woude, G. F. (1998). Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734-737.
112. Rubinfeld, H., Hanoch, T., and Seger, R. (1999). Identification of a cytoplasmic-retention sequence in ERK2. J.Biol.Chem. 274, 30349-30352.
113. Leppa, S., Saffrich, R., Ansorge, W., and Bohmann, D. (1998). Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 17, 4404-4413.
114. Lenormand, P., Sardet, C., Pages, G., L'Allemain, G., Brunet, A., and Pouyssegur, J. (1993). Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J.Cell Biol. 122, 1079-1088.

159
115. Zheng, C. F. and Guan, K. L. (1994). Cytoplasmic localization of the mitogen-activated protein kinase activator MEK. J.Biol.Chem. 269, 19947-19952.
116. Fukuda, M., Gotoh, I., Gotoh, Y., and Nishida, E. (1996). Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J.Biol.Chem. 271, 20024-20028.
117. Kudo, N., Khochbin, S., Nishi, K., Kitano, K., Yanagida, M., Yoshida, M., and Horinouchi, S. (1997). Molecular cloning and cell cycle-dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J.Biol.Chem. 272, 29742-29751.
118. Fornerod, M., Ohno, M., Yoshida, M., and Mattaj, I. W. (1997). CRM1 is an export receptor for leucine-rich nuclear export signals. Cell %19;90, 1051-1060.
119. Fukuda, M., Asano, S., Nakamura, T., Adachi, M., Yoshida, M., Yanagida, M., and Nishida, E. (1997). CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature %20;390, 308-311.
120. Ossareh-Nazari, B., Bachelerie, F., and Dargemont, C. (1997). Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 278, 141-144.
121. Kudo, N., Wolff, B., Sekimoto, T., Schreiner, E. P., Yoneda, Y., Yanagida, M., Horinouchi, S., and Yoshida, M. (1998). Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp.Cell Res. 242, 540-547.
122. Jaaro, H., Rubinfeld, H., Hanoch, T., and Seger, R. (1997). Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc.Natl.Acad.Sci.U.S.A 94, 3742-3747.
123. Yao, Z., Flash, I., Raviv, Z., Yung, Y., Asscher, Y., Pleban, S., and Seger, R. (2001). Non-regulated and stimulated mechanisms cooperate in the nuclear accumulation of MEK1. Oncogene 20, 7588-7596.
124. Tolwinski, N. S., Shapiro, P. S., Goueli, S., and Ahn, N. G. (1999). Nuclear localization of mitogen-activated protein kinase kinase 1 (MKK1) is promoted by serum stimulation and G2-M progression. Requirement for phosphorylation at the activation lip and signaling downstream of MKK. J.Biol.Chem. 274, 6168-6174.
125. Fukuda, M., Gotoh, Y., and Nishida, E. (1997). Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 16, 1901-1908.
126. Robinson, M. J., Stippec, S. A., Goldsmith, E., White, M. A., and Cobb, M. H. (1998). A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr.Biol. 8, 1141-1150.

160
127. Sebolt-Leopold, J. S., Dudley, D. T., Herrera, R., Van Becelaere, K., Wiland, A., Gowan, R. C., Tecle, H., Barrett, S. D., Bridges, A., Przybranowski, S., Leopold, W. R., and Saltiel, A. R. (1999). Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat.Med. 5, 810-816.
128. Delaney, A. M., Printen, J. A., Chen, H., Fauman, E. B., and Dudley, D. T. (2002). Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol.Cell Biol. 22, 7593-7602.
129. Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem.J. 351, 95-105.
130. Kamakura, S., Moriguchi, T., and Nishida, E. (1999). Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J.Biol.Chem. 274, 26563-26571.
131. Mody, N., Leitch, J., Armstrong, C., Dixon, J., and Cohen, P. (2001). Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 502, 21-24.
132. Hanks, S. K. and Hunter, T. (1995). Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576-596.
133. Cox, S., Radzio-Andzelm, E., and Taylor, S. S. (1994). Domain movements in protein kinases. Curr.Opin.Struct.Biol. 4, 893-901.
134. Resing, K. A. and Ahn, N. G. (1998). Deuterium exchange mass spectrometry as a probe of protein kinase activation. Analysis of wild-type and constitutively active mutants of MAP kinase kinase-1. Biochemistry 37, 463-475.
135. Shapiro, P. S. and Ahn, N. G. (1998). Feedback regulation of Raf-1 and mitogen-activated protein kinase (MAP) kinase kinases 1 and 2 by MAP kinase phosphatase-1 (MKP-1). J.Biol.Chem. 273, 1788-1793.
136. Cha, H., Lee, E. K., and Shapiro, P. (2001). Identification of a C-terminal region that regulates mitogen-activated protein kinase kinase-1 cytoplasmic localization and ERK activation. J.Biol.Chem. 276, 48494-48501.
137. Payne, D. M., Rossomando, A. J., Martino, P., Erickson, A. K., Her, J. H., Shabanowitz, J., Hunt, D. F., Weber, M. J., and Sturgill, T. W. (1991). Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 10, 885-892.

161
138. Zhang, J., Zhang, F., Ebert, D., Cobb, M. H., and Goldsmith, E. J. (1995). Activity of the MAP kinase ERK2 is controlled by a flexible surface loop. Structure. 3, 299-307.
139. Boulton, T. G., Nye, S. H., Robbins, D. J., Ip, N. Y., Radziejewska, E., Morgenbesser, S. D., DePinho, R. A., Panayotatos, N., Cobb, M. H., and Yancopoulos, G. D. (1991). ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65, 663-675.
140. English, J. D. and Sweatt, J. D. (1996). Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J.Biol.Chem. 271, 24329-24332.
141. Yung, Y., Yao, Z., Hanoch, T., and Seger, R. (2000). ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J.Biol.Chem. 275, 15799-15808.
142. Boulton, T. G. and Cobb, M. H. (1991). Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 2, 357-371.
143. Gonzalez, F. A., Seth, A., Raden, D. L., Bowman, D. S., Fay, F. S., and Davis, R. J. (1993). Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J.Cell Biol. 122, 1089-1101.
144. Kyriakis, J. M., Brautigan, D. L., Ingebritsen, T. S., and Avruch, J. (1991). pp54 microtubule-associated protein-2 kinase requires both tyrosine and serine/threonine phosphorylation for activity. J.Biol.Chem. 266, 10043-10046.
145. Hibi, M., Lin, A., Smeal, T., Minden, A., and Karin, M. (1993). Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 7, 2135-2148.
146. Gupta, S., Barrett, T., Whitmarsh, A. J., Cavanagh, J., Sluss, H. K., Derijard, B., and Davis, R. J. (1996). Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760-2770.
147. Kyriakis, J. M., Banerjee, P., Nikolakaki, E., Dai, T., Rubie, E. A., Ahmad, M. F., Avruch, J., and Woodgett, J. R. (1994). The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369, 156-160.
148. Kyriakis, J. M. and Avruch, J. (2001). Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 81, 807-869.
149. Derijard, B., Hibi, M., Wu, I. H., Barrett, T., Su, B., Deng, T., Karin, M., and Davis, R. J. (1994). JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025-1037.

162
150. Derijard, B., Raingeaud, J., Barrett, T., Wu, I. H., Han, J., Ulevitch, R. J., and Davis, R. J. (1995). Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267, 682-685.
151. Sanchez, I., Hughes, R. T., Mayer, B. J., Yee, K., Woodgett, J. R., Avruch, J., Kyriakis, J. M., and Zon, L. I. (1994). Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature 372, 794-798.
152. Lin, A., Minden, A., Martinetto, H., Claret, F. X., Lange-Carter, C., Mercurio, F., Johnson, G. L., and Karin, M. (1995). Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 268, 286-290.
153. Holland, P. M., Suzanne, M., Campbell, J. S., Noselli, S., and Cooper, J. A. (1997). MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J.Biol.Chem. 272, 24994-24998.
154. Tournier, C., Whitmarsh, A. J., Cavanagh, J., Barrett, T., and Davis, R. J. (1997). Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc.Natl.Acad.Sci.U.S.A 94, 7337-7342.
155. Yao, Z., Diener, K., Wang, X. S., Zukowski, M., Matsumoto, G., Zhou, G., Mo, R., Sasaki, T., Nishina, H., Hui, C. C., Tan, T. H., Woodgett, J. P., and Penninger, J. M. (1997). Activation of stress-activated protein kinases/c-Jun N-terminal protein kinases (SAPKs/JNKs) by a novel mitogen-activated protein kinase kinase. J.Biol.Chem. %19;272, 32378-32383.
156. Yashar, B. M., Kelley, C., Yee, K., Errede, B., and Zon, L. I. (1993). Novel members of the mitogen-activated protein kinase activator family in Xenopus laevis. Mol.Cell Biol. 13, 5738-5748.
157. Yamauchi, J., Kaziro, Y., and Itoh, H. (1999). Differential regulation of mitogen-activated protein kinase kinase 4 (MKK4) and 7 (MKK7) by signaling from G protein beta gamma subunit in human embryonal kidney 293 cells. J.Biol.Chem. 274, 1957-1965.
158. Meier, R., Rouse, J., Cuenda, A., Nebreda, A. R., and Cohen, P. (1996). Cellular stresses and cytokines activate multiple mitogen-activated-protein kinase kinase homologues in PC12 and KB cells. Eur.J.Biochem. 236, 796-805.
159. Ganiatsas, S., Kwee, L., Fujiwara, Y., Perkins, A., Ikeda, T., Labow, M. A., and Zon, L. I. (1998). SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc.Natl.Acad.Sci.U.S.A 95, 6881-6886.
160. Lawler, S., Fleming, Y., Goedert, M., and Cohen, P. (1998). Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr.Biol. 8, 1387-1390.

163
161. Lisnock, J., Griffin, P., Calaycay, J., Frantz, B., Parsons, J., O'Keefe, S. J., and LoGrasso, P. (2000). Activation of JNK3 alpha 1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry 39, 3141-3148.
162. Han, J., Lee, J. D., Bibbs, L., and Ulevitch, R. J. (1994). A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808-811.
163. Lee, J. C., Laydon, J. T., McDonnell, P. C., Gallagher, T. F., Kumar, S., Green, D., McNulty, D., Blumenthal, M. J., Heys, J. R., Landvatter, S. W., and . (1994). A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739-746.
164. Rouse, J., Cohen, P., Trigon, S., Morange, M., Alonso-Llamazares, A., Zamanillo, D., Hunt, T., and Nebreda, A. R. (1994). A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78, 1027-1037.
165. Stein, B., Yang, M. X., Young, D. B., Janknecht, R., Hunter, T., Murray, B. W., and Barbosa, M. S. (1997). p38-2, a novel mitogen-activated protein kinase with distinct properties. J.Biol.Chem. 272, 19509-19517.
166. Li, Z., Jiang, Y., Ulevitch, R. J., and Han, J. (1996). The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem.Biophys.Res.Commun. 228, 334-340.
167. Jiang, Y., Chen, C., Li, Z., Guo, W., Gegner, J. A., Lin, S., and Han, J. (1996). Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J.Biol.Chem. 271, 17920-17926.
168. Jiang, Y., Gram, H., Zhao, M., New, L., Gu, J., Feng, L., Di Padova, F., Ulevitch, R. J., and Han, J. (1997). Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J.Biol.Chem. 272, 30122-30128.
169. Goedert, M., Cuenda, A., Craxton, M., Jakes, R., and Cohen, P. (1997). Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J. 16, 3563-3571.
170. Kumar, S., McDonnell, P. C., Gum, R. J., Hand, A. T., Lee, J. C., and Young, P. R. (1997). Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem.Biophys.Res.Commun. 235, 533-538.
171. Wang, Y., Huang, S., Sah, V. P., Ross, J., Jr., Brown, J. H., Han, J., and Chien, K. R. (1998). Cardiac muscle cell hypertrophy and apoptosis induced by distinct

164
members of the p38 mitogen-activated protein kinase family. J.Biol.Chem. 273, 2161-2168.
172. Enslen, H., Brancho, D. M., and Davis, R. J. (2000). Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 19, 1301-1311.
173. Stein, B., Brady, H., Yang, M. X., Young, D. B., and Barbosa, M. S. (1996). Cloning and characterization of MEK6, a novel member of the mitogen-activated protein kinase kinase cascade. J.Biol.Chem. 271, 11427-11433.
174. Wilsbacher, J. L., Goldsmith, E. J., and Cobb, M. H. (1999). Phosphorylation of MAP kinases by MAP/ERK involves multiple regions of MAP kinases. J.Biol.Chem. 274, 16988-16994.
175. Ishitani, T., Ninomiya-Tsuji, J., Nagai, S., Nishita, M., Meneghini, M., Barker, N., Waterman, M., Bowerman, B., Clevers, H., Shibuya, H., and Matsumoto, K. (1999). The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature 399, 798-802.
176. Gonzalez, F. A., Raden, D. L., Rigby, M. R., and Davis, R. J. (1992). Heterogeneous expression of four MAP kinase isoforms in human tissues. FEBS Lett. 304, 170-178.
177. Cheng, M., Boulton, T. G., and Cobb, M. H. (1996). ERK3 is a constitutively nuclear protein kinase. J.Biol.Chem. 271, 8951-8958.
178. Turgeon, B., Saba-El-Leil, M. K., and Meloche, S. (2000). Cloning and characterization of mouse extracellular-signal-regulated protein kinase 3 as a unique gene product of 100 kDa. Biochem.J. 346 Pt 1:169-75., 169-175.
179. Plowman, G. D., Sudarsanam, S., Bingham, J., Whyte, D., and Hunter, T. (1999). The protein kinases of Caenorhabditis elegans: a model for signal transduction in multicellular organisms. Proc.Natl.Acad.Sci.U.S.A 96, 13603-13610.
180. Cheng, M., Zhen, E., Robinson, M. J., Ebert, D., Goldsmith, E., and Cobb, M. H. (1996). Characterization of a protein kinase that phosphorylates serine 189 of the mitogen-activated protein kinase homolog ERK3. J.Biol.Chem. 271, 12057-12062.
181. Zhou, G., Bao, Z. Q., and Dixon, J. E. (1995). Components of a new human protein kinase signal transduction pathway. J.Biol.Chem. 270, 12665-12669.
182. Lee, J. D., Ulevitch, R. J., and Han, J. (1995). Primary structure of BMK1: a new mammalian map kinase. Biochem.Biophys.Res.Commun. 213, 715-724.
183. English, J. M., Pearson, G., Baer, R., and Cobb, M. H. (1998). Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J.Biol.Chem. 273, 3854-3860.

165
184. Kato, Y., Tapping, R. I., Huang, S., Watson, M. H., Ulevitch, R. J., and Lee, J. D. (1998). Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 395 , 713-716.
185. Kato, Y., Kravchenko, V. V., Tapping, R. I., Han, J., Ulevitch, R. J., and Lee, J. D. (1997). BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 16, 7054-7066.
186. Chao, T. H., Hayashi, M., Tapping, R. I., Kato, Y., and Lee, J. D. (1999). MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J.Biol.Chem. 274, 36035-36038.
187. Abe, J., Kusuhara, M., Ulevitch, R. J., Berk, B. C., and Lee, J. D. (1996). Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J.Biol.Chem. 271, 16586-16590.
188. Takeishi, Y., Abe, J., Lee, J. D., Kawakatsu, H., Walsh, R. A., and Berk, B. C. (1999). Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ.Res. 85, 1164-1172.
189. Yan, C., Takahashi, M., Okuda, M., Lee, J. D., and Berk, B. C. (1999). Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J.Biol.Chem. 274, 143-150.
190. Abe, J., Takahashi, M., Ishida, M., Lee, J. D., and Berk, B. C. (1997). c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J.Biol.Chem. 272, 20389-20394.
191. English, J. M., Pearson, G., Hockenberry, T., Shivakumar, L., White, M. A., and Cobb, M. H. (1999). Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J.Biol.Chem. 274, 31588-31592.
192. Marinissen, M. J., Chiariello, M., Pallante, M., and Gutkind, J. S. (1999). A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol.Cell Biol. 19, 4289-4301.
193. English, J. M., Vanderbilt, C. A., Xu, S., Marcus, S., and Cobb, M. H. (1995). Isolation of MEK5 and differential expression of alternatively spliced forms. J.Biol.Chem. 270, 28897-28902.
194. Chiariello, M., Marinissen, M. J., and Gutkind, J. S. (2000). Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol.Cell Biol. 20, 1747-1758.

166
195. Nakamura, K. and Johnson, G. L. (2003). PB1 Domains of MEKK2 and MEKK3 Interact with the MEK5 PB1 Domain for Activation of the ERK5 Pathway. J.Biol.Chem. 278, 36989-36992.
196. Sun, W., Kesavan, K., Schaefer, B. C., Garrington, T. P., Ware, M., Johnson, N. L., Gelfand, E. W., and Johnson, G. L. (2001). MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J.Biol.Chem. 276, 5093-5100.
197. Brott, B. K., Pinsky, B. A., and Erikson, R. L. (1998). Nlk is a murine protein kinase related to Erk/MAP kinases and localized in the nucleus. Proc.Natl.Acad.Sci.U.S.A 95, 963-968.
198. Meneghini, M. D., Ishitani, T., Carter, J. C., Hisamoto, N., Ninomiya-Tsuji, J., Thorpe, C. J., Hamill, D. R., Matsumoto, K., and Bowerman, B. (1999). MAP kinase and Wnt pathways converge to downregulate an HMG-domain repressor in Caenorhabditis elegans. Nature 399, 793-797.
199. Behrens, J. (2000). Cross-regulation of the Wnt signalling pathway: a role of MAP kinases. J.Cell Sci. 113, 911-919.
200. Xu, S., Robbins, D. J., Christerson, L. B., English, J. M., Vanderbilt, C. A., and Cobb, M. H. (1996). Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc.Natl.Acad.Sci.U.S.A 93, 5291-5295.
201. Yan, M., Dai, T., Deak, J. C., Kyriakis, J. M., Zon, L. I., Woodgett, J. R., and Templeton, D. J. (1994). Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature 372, 798-800.
202. Rhodes, N., Connell, L., and Errede, B. (1990). STE11 is a protein kinase required for cell-type-specific transcription and signal transduction in yeast. Genes Dev. 4, 1862-1874.
203. Blank, J. L., Gerwins, P., Elliott, E. M., Sather, S., and Johnson, G. L. (1996). Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J.Biol.Chem. 271, 5361-5368.
204. Graves, J. D., Gotoh, Y., Draves, K. E., Ambrose, D., Han, D. K., Wright, M., Chernoff, J., Clark, E. A., and Krebs, E. G. (1998). Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 17, 2224-2234.
205. Takekawa, M., Posas, F., and Saito, H. (1997). A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. EMBO J. 16, 4973-4982.

167
206. Patriotis, C., Makris, A., Chernoff, J., and Tsichlis, P. N. (1994). Tpl-2 acts in concert with Ras and Raf-1 to activate mitogen-activated protein kinase. Proc.Natl.Acad.Sci.U.S.A 91 , 9755-9759.
207. Wang, W., Zhou, G., Hu, M. C., Yao, Z., and Tan, T. H. (1997). Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J.Biol.Chem. 272, 22771-22775.
208. Fan, G., Merritt, S. E., Kortenjann, M., Shaw, P. E., and Holzman, L. B. (1996). Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J.Biol.Chem. 271, 24788-24793.
209. Holzman, L. B., Merritt, S. E., and Fan, G. (1994). Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J.Biol.Chem. 269, 30808-30817.
210. Hirai, S., Katoh, M., Terada, M., Kyriakis, J. M., Zon, L. I., Rana, A., Avruch, J., and Ohno, S. (1997). MST/MLK2, a member of the mixed lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J.Biol.Chem. 272, 15167-15173.
211. Teramoto, H., Coso, O. A., Miyata, H., Igishi, T., Miki, T., and Gutkind, J. S. (1996). Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J.Biol.Chem. 271, 27225-27228.
212. Hirose, T., Fujimoto, W., Tamaai, T., Kim, K. H., Matsuura, H., and Jetten, A. M. (1994). TAK1: molecular cloning and characterization of a new member of the nuclear receptor superfamily. Mol.Endocrinol. 8, 1667-1680.
213. Yamaguchi, K., Shirakabe, K., Shibuya, H., Irie, K., Oishi, I., Ueno, N., Taniguchi, T., Nishida, E., and Matsumoto, K. (1995). Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008-2011.
214. Ichijo, H., Nishida, E., Irie, K., ten Dijke, P., Saitoh, M., Moriguchi, T., Takagi, M., Matsumoto, K., Miyazono, K., and Gotoh, Y. (1997). Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90-94.
215. Wang, X. S., Diener, K., Tan, T. H., and Yao, Z. (1998). MAPKKK6, a novel mitogen-activated protein kinase kinase kinase, that associates with MAPKKK5. Biochem.Biophys.Res.Commun. 253 , 33-37.

168
216. Hutchison, M., Berman, K. S., and Cobb, M. H. (1998). Isolation of TAO1, a protein kinase that activates MEKs in stress-activated protein kinase cascades. J.Biol.Chem. 273, 28625-28632.
217. Chen, Z., Hutchison, M., and Cobb, M. H. (1999). Isolation of the protein kinase TAO2 and identification of its mitogen-activated protein kinase/extracellular signal-regulated kinase kinase binding domain. J.Biol.Chem. 274, 28803-28807.
218. Lee, F. S., Hagler, J., Chen, Z. J., and Maniatis, T. (1997). Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell 88, 213-222.
219. Yin, M. J., Christerson, L. B., Yamamoto, Y., Kwak, Y. T., Xu, S., Mercurio, F., Barbosa, M., Cobb, M. H., and Gaynor, R. B. (1998). HTLV-I Tax protein binds to MEKK1 to stimulate IkappaB kinase activity and NF-kappaB activation. Cell 93, 875-884.
220. Gardner, A. M., Vaillancourt, R. R., Lange-Carter, C. A., and Johnson, G. L. (1994). MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Mol.Biol.Cell 5, 193-201.
221. Yujiri, T., Sather, S., Fanger, G. R., and Johnson, G. L. (1998). Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science 282, 1911-1914.
222. Xia, Y., Wu, Z., Su, B., Murray, B., and Karin, M. (1998). JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 12, 3369-3381.
223. Xia, Y., Makris, C., Su, B., Li, E., Yang, J., Nemerow, G. R., and Karin, M. (2000). MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc.Natl.Acad.Sci.U.S.A 97, 5243-5248.
224. Zhang, S., Han, J., Sells, M. A., Chernoff, J., Knaus, U. G., Ulevitch, R. J., and Bokoch, G. M. (1995). Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J.Biol.Chem. 270, 23934-23936.
225. Tang, Y., Yu, J., and Field, J. (1999). Signals from the Ras, Rac, and Rho GTPases converge on the Pak protein kinase in Rat-1 fibroblasts. Mol.Cell Biol. 19, 1881-1891.
226. Polverino, A., Frost, J., Yang, P., Hutchison, M., Neiman, A. M., Cobb, M. H., and Marcus, S. (1995). Activation of mitogen-activated protein kinase cascades by p21-

169
activated protein kinases in cell-free extracts of Xenopus oocytes. J.Biol.Chem. 270, 26067-26070.
227. Kyriakis, J. M. (1999). Signaling by the germinal center kinase family of protein kinases. J.Biol.Chem. 274, 5259-5262.
228. Yuasa, T., Ohno, S., Kehrl, J. H., and Kyriakis, J. M. (1998). Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J.Biol.Chem. 273, 22681-22692.
229. Yao, Z., Zhou, G., Wang, X. S., Brown, A., Diener, K., Gan, H., and Tan, T. H. (1999). A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J.Biol.Chem. 274, 2118-2125.
230. Tung, R. M. and Blenis, J. (1997). A novel human SPS1/STE20 homologue, KHS, activates Jun N-terminal kinase. Oncogene 14, 653-659.
231. Hu, M. C., Qiu, W. R., Wang, X., Meyer, C. F., and Tan, T. H. (1996). Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 10, 2251-2264.
232. Su, Y. C., Han, J., Xu, S., Cobb, M., and Skolnik, E. Y. (1997). NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 16, 1279-1290.
233. Vojtek, A. B. and Cooper, J. A. (1995). Rho family members: activators of MAP kinase cascades. Cell 82, 527-529.
234. Bagrodia, S., Derijard, B., Davis, R. J., and Cerione, R. A. (1995). Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J.Biol.Chem. 270, 27995-27998.
235. Coso, O. A., Chiariello, M., Kalinec, G., Kyriakis, J. M., Woodgett, J., and Gutkind, J. S. (1995). Transforming G protein-coupled receptors potently activate JNK (SAPK). Evidence for a divergence from the tyrosine kinase signaling pathway. J.Biol.Chem. 270, 5620-5624.
236. Coso, O. A., Chiariello, M., Yu, J. C., Teramoto, H., Crespo, P., Xu, N., Miki, T., and Gutkind, J. S. (1995). The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81, 1137-1146.

170
237. Wilk-Blaszczak, M. A., Stein, B., Xu, S., Barbosa, M. S., Cobb, M. H., and Belardetti, F. (1998). The mitogen-activated protein kinase p38-2 is necessary for the inhibition of N-type calcium current by bradykinin. J.Neurosci. 18, 112-118.
238. Whitmarsh, A. J. and Davis, R. J. (1998). Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem.Sci. 23, 481-485.
239. Elion, E. A. (2000). Pheromone response, mating and cell biology. Curr.Opin.Microbiol. 3, 573-581.
240. Posas, F., Wurgler-Murphy, S. M., Maeda, T., Witten, E. A., Thai, T. C., and Saito, H. (1996). Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 "two-component" osmosensor. Cell %20;86, 865-875.
241. Posas, F. and Saito, H. (1997). Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276, 1702-1705.
242. Whitmarsh, A. J., Cavanagh, J., Tournier, C., Yasuda, J., and Davis, R. J. (1998). A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281, 1671-1674.
243. Elion, E. A. (2001). The Ste5p scaffold. J.Cell Sci. 114, 3967-3978.
244. Choi, K. Y., Satterberg, B., Lyons, D. M., and Elion, E. A. (1994). Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell 78, 499-512.
245. Marcus, S., Polverino, A., Barr, M., and Wigler, M. (1994). Complexes between STE5 and components of the pheromone-responsive mitogen-activated protein kinase module. Proc.Natl.Acad.Sci.U.S.A 91, 7762-7766.
246. Printen, J. A. and Sprague, G. F., Jr. (1994). Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics 138, 609-619.
247. Kranz, J. E., Satterberg, B., and Elion, E. A. (1994). The MAP kinase Fus3 associates with and phosphorylates the upstream signaling component Ste5. Genes Dev. 8, 313-327.
248. Choi, K. Y., Kranz, J. E., Mahanty, S. K., Park, K. S., and Elion, E. A. (1999). Characterization of Fus3 localization: active Fus3 localizes in complexes of varying size and specific activity. Mol.Biol.Cell 10, 1553-1568.

171
249. Whiteway, M. S., Wu, C., Leeuw, T., Clark, K., Fourest-Lieuvin, A., Thomas, D. Y., and Leberer, E. (1995). Association of the yeast pheromone response G protein beta gamma subunits with the MAP kinase scaffold Ste5p. Science 269, 1572-1575.
250. Feng, Y., Song, L. Y., Kincaid, E., Mahanty, S. K., and Elion, E. A. (1998). Functional binding between Gbeta and the LIM domain of Ste5 is required to activate the MEKK Ste11. Curr.Biol. 8, 267-278.
251. Inouye, C., Dhillon, N., and Thorner, J. (1997). Ste5 RING-H2 domain: role in Ste4-promoted oligomerization for yeast pheromone signaling. Science 278, 103-106.
252. Yablonski, D., Marbach, I., and Levitzki, A. (1996). Dimerization of Ste5, a mitogen-activated protein kinase cascade scaffold protein, is required for signal transduction. Proc.Natl.Acad.Sci.U.S.A 93, 13864-13869.
253. Mahanty, S. K., Wang, Y., Farley, F. W., and Elion, E. A. (1999). Nuclear shuttling of yeast scaffold Ste5 is required for its recruitment to the plasma membrane and activation of the mating MAPK cascade. Cell %20;98, 501-512.
254. Hall, J. P., Cherkasova, V., Elion, E., Gustin, M. C., and Winter, E. (1996). The osmoregulatory pathway represses mating pathway activity in Saccharomyces cerevisiae: isolation of a FUS3 mutant that is insensitive to the repression mechanism. Mol.Cell Biol. 16, 6715-6723.
255. Brewster, J. L., de Valoir, T., Dwyer, N. D., Winter, E., and Gustin, M. C. (1993). An osmosensing signal transduction pathway in yeast. Science %19;259, 1760-1763.
256. Bardwell, L., Cook, J. G., Chang, E. C., Cairns, B. R., and Thorner, J. (1996). Signaling in the yeast pheromone response pathway: specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol.Cell Biol. 16, 3637-3650.
257. Errede, B., Gartner, A., Zhou, Z., Nasmyth, K., and Ammerer, G. (1993). MAP kinase-related FUS3 from S. cerevisiae is activated by STE7 in vitro. Nature 362, 261-264.
258. Bardwell, L. and Thorner, J. (1996). A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem.Sci. 21, 373-374.
259. van Drogen, F. and Peter, M. (2002). MAP kinase cascades: scaffolding signal specificity. Curr.Biol. 12 , R53-R55.

172
260. Schaeffer, H. J., Catling, A. D., Eblen, S. T., Collier, L. S., Krauss, A., and Weber, M. J. (1998). MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668-1671.
261. Wunderlich, W., Fialka, I., Teis, D., Alpi, A., Pfeifer, A., Parton, R. G., Lottspeich, F., and Huber, L. A. (2001). A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J.Cell Biol. %19;152, 765-776.
262. Nantel, A., Mohammad-Ali, K., Sherk, J., Posner, B. I., and Thomas, D. Y. (1998). Interaction of the Grb10 adapter protein with the Raf1 and MEK1 kinases. J.Biol.Chem. 273, 10475-10484.
263. Ooi, J., Yajnik, V., Immanuel, D., Gordon, M., Moskow, J. J., Buchberg, A. M., and Margolis, B. (1995). The cloning of Grb10 reveals a new family of SH2 domain proteins. Oncogene %20;10, 1621-1630.
264. Margolis, B., Silvennoinen, O., Comoglio, F., Roonprapunt, C., Skolnik, E., Ullrich, A., and Schlessinger, J. (1992). High-efficiency expression/cloning of epidermal growth factor-receptor-binding proteins with Src homology 2 domains. Proc.Natl.Acad.Sci.U.S.A 89, 8894-8898.
265. Daly, R. J., Sanderson, G. M., Janes, P. W., and Sutherland, R. L. (1996). Cloning and characterization of GRB14, a novel member of the GRB7 gene family. J.Biol.Chem. 271, 12502-12510.
266. Daly, R. J. (1998). The Grb7 family of signalling proteins. Cell Signal. 10, 613-618.
267. Frantz, J. D., Giorgetti-Peraldi, S., Ottinger, E. A., and Shoelson, S. E. (1997). Human GRB-IRbeta/GRB10. Splice variants of an insulin and growth factor receptor-binding protein with PH and SH2 domains. J.Biol.Chem. 272, 2659-2667.
268. He, W., Rose, D. W., Olefsky, J. M., and Gustafson, T. A. (1998). Grb10 interacts differentially with the insulin receptor, insulin-like growth factor I receptor, and epidermal growth factor receptor via the Grb10 Src homology 2 (SH2) domain and a second novel domain located between the pleckstrin homology and SH2 domains. J.Biol.Chem. %20;273, 6860-6867.
269. Kasus-Jacobi, A., Perdereau, D., Auzan, C., Clauser, E., Van Obberghen, E., Mauvais-Jarvis, F., Girard, J., and Burnol, A. F. (1998). Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J.Biol.Chem. 273, 26026-26035.
270. Nantel, A., Huber, M., and Thomas, D. Y. (1999). Localization of endogenous Grb10 to the mitochondria and its interaction with the mitochondrial-associated Raf-1 pool. J.Biol.Chem. 274, 35719-35724.

173
271. O'Connor, R., Kauffmann-Zeh, A., Liu, Y., Lehar, S., Evan, G. I., Baserga, R., and Blattler, W. A. (1997). Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Mol.Cell Biol. 17, 427-435.
272. Kulik, G., Klippel, A., and Weber, M. J. (1997). Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol.Cell Biol. 17, 1595-1606.
273. Peruzzi, F., Prisco, M., Dews, M., Salomoni, P., Grassilli, E., Romano, G., Calabretta, B., and Baserga, R. (1999). Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol.Cell Biol. 19, 7203-7215.
274. Chen, H., Yan, G. C., and Gishizky, M. L. (1998). Identification of structural characteristics that contribute to a difference in antiapoptotic function between human insulin and insulin-like growth factor 1 receptors. Cell Growth Differ. 9, 939-947.
275. Majewski, M., Nieborowska-Skorska, M., Salomoni, P., Slupianek, A., Reiss, K., Trotta, R., Calabretta, B., and Skorski, T. (1999). Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 59, 2815-2819.
276. Dong, L. Q., Du, H., Porter, S. G., Kolakowski, L. F., Jr., Lee, A. V., Mandarino, L. J., Fan, J., Yee, D., Liu, F., and Mandarino, J. (1997). Cloning, chromosome localization, expression, and characterization of an Src homology 2 and pleckstrin homology domain-containing insulin receptor binding protein hGrb10gamma. J.Biol.Chem. 272, 29104-29112.
277. Therrien, M., Michaud, N. R., Rubin, G. M., and Morrison, D. K. (1996). KSR modulates signal propagation within the MAPK cascade. Genes Dev. 10, 2684-2695.
278. Kornfeld, K., Hom, D. B., and Horvitz, H. R. (1995). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83, 903-913.
279. Sundaram, M. and Han, M. (1995). The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83, 889-901.
280. Stewart, S., Sundaram, M., Zhang, Y., Lee, J., Han, M., and Guan, K. L. (1999). Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol.Cell Biol. 19, 5523-5534.
281. Morrison, D. K. (2001). KSR: a MAPK scaffold of the Ras pathway? J.Cell Sci. 114, 1609-1612.

174
282. Cacace, A. M., Michaud, N. R., Therrien, M., Mathes, K., Copeland, T., Rubin, G. M., and Morrison, D. K. (1999). Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol.Cell Biol. 19, 229-240.
283. Yu, W., Fantl, W. J., Harrowe, G., and Williams, L. T. (1998). Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr.Biol. 8, 56-64.
284. Xing, H. R., Lozano, J., and Kolesnick, R. (2000). Epidermal growth factor treatment enhances the kinase activity of kinase suppressor of Ras. J.Biol.Chem. 275, 17276-17280.
285. Xing, H. R., Campodonico, L., and Kolesnick, R. N. (2004). The kinase activity of kinase suppressor of Ras1 (KSR1) is independent of bound MEK. J.Biol.Chem. ..
286. Muller, J., Ory, S., Copeland, T., Piwnica-Worms, H., and Morrison, D. K. (2001). C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol.Cell 8, 983-993.
287. Bell, B., Xing, H., Yan, K., Gautam, N., and Muslin, A. J. (1999). KSR-1 binds to G-protein betagamma subunits and inhibits beta gamma-induced mitogen-activated protein kinase activation. J.Biol.Chem. %19;274, 7982-7986.
288. Yeung, K., Seitz, T., Li, S., Janosch, P., McFerran, B., Kaiser, C., Fee, F., Katsanakis, K. D., Rose, D. W., Mischak, H., Sedivy, J. M., and Kolch, W. (1999). Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 401, 173-177.
289. Yeung, K., Janosch, P., McFerran, B., Rose, D. W., Mischak, H., Sedivy, J. M., and Kolch, W. (2000). Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol.Cell Biol. 20, 3079-3085.
290. Yasuda, J., Whitmarsh, A. J., Cavanagh, J., Sharma, M., and Davis, R. J. (1999). The JIP group of mitogen-activated protein kinase scaffold proteins. Mol.Cell Biol. 19, 7245-7254.
291. Tash, J. S., Welsh, M. J., and Means, A. R. (1980). Protein inhibitor of cAMP-dependent protein kinase: production and characterization of antibodies and intracellular localization. Cell 21, 57-65.
292. Pedram, A., Razandi, M., and Levin, E. R. (1998). Extracellular signal-regulated protein kinase/Jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. J.Biol.Chem. 273, 26722-26728.

175
293. Wiley, J. C., Wailes, L. A., Idzerda, R. L., and McKnight, G. S. (1999). Role of regulatory subunits and protein kinase inhibitor (PKI) in determining nuclear localization and activity of the catalytic subunit of protein kinase A. J.Biol.Chem. 274, 6381-6387.
294. Wen, W., Meinkoth, J. L., Tsien, R. Y., and Taylor, S. S. (1995). Identification of a signal for rapid export of proteins from the nucleus. Cell 82, 463-473.
295. Sharrocks, A. D., Yang, S. H., and Galanis, A. (2000). Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem.Sci. 25, 448-453.
296. Holland, P. M. and Cooper, J. A. (1999). Protein modification: docking sites for kinases. Curr.Biol. 9, R329-R331.
297. Kieran, M. W., Katz, S., Vail, B., Zon, L. I., and Mayer, B. J. (1999). Concentration-dependent positive and negative regulation of a MAP kinase by a MAP kinase kinase. Oncogene 18, 6647-6657.
298. Bardwell, A. J., Flatauer, L. J., Matsukuma, K., Thorner, J., and Bardwell, L. (2001). A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J.Biol.Chem. 276, 10374-10386.
299. Xu, B., Stippec, S., Robinson, F. L., and Cobb, M. H. (2001). Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. J.Biol.Chem. 276, 26509-26515.
300. Ho, D. T., Bardwell, A. J., Abdollahi, M., and Bardwell, L. (2003). A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J.Biol.Chem. 278, 32662-32672.
301. Miller, W. E., McDonald, P. H., Cai, S. F., Field, M. E., Davis, R. J., and Lefkowitz, R. J. (2001). Identification of a motif in the carboxyl terminus of beta -arrestin2 responsible for activation of JNK3. J.Biol.Chem. 276, 27770-27777.
302. Luttrell, L. M., Roudabush, F. L., Choy, E. W., Miller, W. E., Field, M. E., Pierce, K. L., and Lefkowitz, R. J. (2001). Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc.Natl.Acad.Sci.U.S.A 98, 2449-2454.
303. Yang, S. H., Yates, P. R., Whitmarsh, A. J., Davis, R. J., and Sharrocks, A. D. (1998). The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol.Cell Biol. 18, 710-720.

176
304. Fantz, D. A., Jacobs, D., Glossip, D., and Kornfeld, K. (2001). Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J.Biol.Chem. %20;276, 27256-27265.
305. Kallunki, T., Su, B., Tsigelny, I., Sluss, H. K., Derijard, B., Moore, G., Davis, R., and Karin, M. (1994). JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 8, 2996-3007.
306. Jacobs, D., Glossip, D., Xing, H., Muslin, A. J., and Kornfeld, K. (1999). Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163-175.
307. Yang, S. H., Galanis, A., and Sharrocks, A. D. (1999). Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol.Cell Biol. 19, 4028-4038.
308. Kim, D. W. and Cochran, B. H. (2000). Extracellular signal-regulated kinase binds to TFII-I and regulates its activation of the c-fos promoter. Mol.Cell Biol. 20, 1140-1148.
309. Galanis, A., Yang, S. H., and Sharrocks, A. D. (2001). Selective targeting of MAPKs to the ETS domain transcription factor SAP-1. J.Biol.Chem. 276, 965-973.
310. Smith, J. A., Poteet-Smith, C. E., Malarkey, K., and Sturgill, T. W. (1999). Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J.Biol.Chem. 274, 2893-2898.
311. Gavin, A. C. and Nebreda, A. R. (1999). A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr.Biol. 9, 281-284.
312. Chen, P., Hutter, D., Yang, X., Gorospe, M., Davis, R. J., and Liu, Y. (2001). Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J.Biol.Chem. 276, 29440-29449.
313. Slack, D. N., Seternes, O. M., Gabrielsen, M., and Keyse, S. M. (2001). Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J.Biol.Chem. 276, 16491-16500.
314. Zhou, B., Wu, L., Shen, K., Zhang, J., Lawrence, D. S., and Zhang, Z. Y. (2001). Multiple regions of MAP kinase phosphatase 3 are involved in its recognition and activation by ERK2. J.Biol.Chem. 276, 6506-6515.

177
315. Tanoue, T., Yamamoto, T., and Nishida, E. (2002). Modular structure of a docking surface on MAPK phosphatases. J.Biol.Chem. 277, 22942-22949.
316. MacKenzie, S. J., Baillie, G. S., McPhee, I., Bolger, G. B., and Houslay, M. D. (2000). ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. The involvement of COOH-terminal docking sites and NH2-terminal UCR regions. J.Biol.Chem. 275, 16609-16617.
317. Metodiev, M. V., Matheos, D., Rose, M. D., and Stone, D. E. (2002). Regulation of MAPK function by direct interaction with the mating-specific Galpha in yeast. Science 296, 1483-1486.
318. Hill, J. M., Vaidyanathan, H., Ramos, J. W., Ginsberg, M. H., and Werner, M. H. (2002). Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. EMBO J. 21, 6494-6504.
319. Tanoue, T., Maeda, R., Adachi, M., and Nishida, E. (2001). Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 20, 466-479.
320. Tarrega, C., Blanco-Aparicio, C., Munoz, J. J., and Pulido, R. (2002). Two clusters of residues at the docking groove of mitogen-activated protein kinases differentially mediate their functional interaction with the tyrosine phosphatases PTP-SL and STEP. J.Biol.Chem. 277, 2629-2636.
321. Blanco-Aparicio, C., Torres, J., and Pulido, R. (1999). A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J.Cell Biol. 147, 1129-1136.
322. Zuniga, A., Torres, J., Ubeda, J., and Pulido, R. (1999). Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J.Biol.Chem. 274, 21900-21907.
323. Volmat, V., Camps, M., Arkinstall, S., Pouyssegur, J., and Lenormand, P. (2001). The nucleus, a site for signal termination by sequestration and inactivation of p42/p44 MAP kinases. J.Cell Sci. 114, 3433-3443.
324. Wolf, I., Rubinfeld, H., Yoon, S., Marmor, G., Hanoch, T., and Seger, R. (2001). Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J.Biol.Chem. 276 , 24490-24497.
325. Adachi, M., Fukuda, M., and Nishida, E. (1999). Two co-existing mechanisms for nuclear import of MAP kinase: passive diffusion of a monomer and active transport of a dimer. EMBO J. 18 , 5347-5358.

178
326. Adachi, M., Fukuda, M., and Nishida, E. (2000). Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J.Cell Biol. 148, 849-856.
327. Sontag, E., Fedorov, S., Kamibayashi, C., Robbins, D., Cobb, M., and Mumby, M. (1993). The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75, 887-897.
328. Todd, J. L., Tanner, K. G., and Denu, J. M. (1999). Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J.Biol.Chem. 274, 13271-13280.
329. Sohaskey, M. L. and Ferrell, J. E., Jr. (1999). Distinct, constitutively active MAPK phosphatases function in Xenopus oocytes: implications for p42 MAPK regulation In vivo. Mol.Biol.Cell 10, 3729-3743.
330. Posada, J. and Cooper, J. A. (1992). Requirements for phosphorylation of MAP kinase during meiosis in Xenopus oocytes. Science 255, 212-215.
331. Alessi, D. R., Gomez, N., Moorhead, G., Lewis, T., Keyse, S. M., and Cohen, P. (1995). Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr.Biol. 5, 283-295.
332. Pettiford, S. M. and Herbst, R. (2000). The MAP-kinase ERK2 is a specific substrate of the protein tyrosine phosphatase HePTP. Oncogene 19, 858-869.
333. Oh-hora, M., Ogata, M., Mori, Y., Adachi, M., Imai, K., Kosugi, A., and Hamaoka, T. (1999). Direct suppression of TCR-mediated activation of extracellular signal-regulated kinase by leukocyte protein tyrosine phosphatase, a tyrosine-specific phosphatase. J.Immunol. 163, 1282-1288.
334. Ogata, M., Oh-hora, M., Kosugi, A., and Hamaoka, T. (1999). Inactivation of mitogen-activated protein kinases by a mammalian tyrosine-specific phosphatase, PTPBR7. Biochem.Biophys.Res.Commun. 256, 52-56.
335. Karim, F. D. and Rubin, G. M. (1999). PTP-ER, a novel tyrosine phosphatase, functions downstream of Ras1 to downregulate MAP kinase during Drosophila eye development. Mol.Cell 3, 741-750.
336. Brondello, J. M., Brunet, A., Pouyssegur, J., and McKenzie, F. R. (1997). The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J.Biol.Chem. 272, 1368-1376.
337. Camps, M., Nichols, A., and Arkinstall, S. (2000). Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 14, 6-16.

179
338. Keyse, S. M. (2000). Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr.Opin.Cell Biol. 12, 186-192.
339. Theodosiou, A. and Ashworth, A. (2002). MAP kinase phosphatases. Genome Biol. 3, REVIEWS3009.
340. Guan, K., Hakes, D. J., Wang, Y., Park, H. D., Cooper, T. G., and Dixon, J. E. (1992). A yeast protein phosphatase related to the vaccinia virus VH1 phosphatase is induced by nitrogen starvation. Proc.Natl.Acad.Sci.U.S.A 89, 12175-12179.
341. Chu, Y., Solski, P. A., Khosravi-Far, R., Der, C. J., and Kelly, K. (1996). The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J.Biol.Chem. 271, 6497-6501.
342. Groom, L. A., Sneddon, A. A., Alessi, D. R., Dowd, S., and Keyse, S. M. (1996). Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 15, 3621-3632.
343. Muda, M., Theodosiou, A., Rodrigues, N., Boschert, U., Camps, M., Gillieron, C., Davies, K., Ashworth, A., and Arkinstall, S. (1996). The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J.Biol.Chem. 271, 27205-27208.
344. Brondello, J. M., Pouyssegur, J., and McKenzie, F. R. (1999). Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 286, 2514-2517.
345. Camps, M., Nichols, A., Gillieron, C., Antonsson, B., Muda, M., Chabert, C., Boschert, U., and Arkinstall, S. (1998). Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 280, 1262-1265.
346. Muda, M., Theodosiou, A., Gillieron, C., Smith, A., Chabert, C., Camps, M., Boschert, U., Rodrigues, N., Davies, K., Ashworth, A., and Arkinstall, S. (1998). The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J.Biol.Chem. 273, 9323-9329.
347. Dorfman, K., Carrasco, D., Gruda, M., Ryan, C., Lira, S. A., and Bravo, R. (1996). Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene 13, 925-931.
348. Dong, C., Waters, S. B., Holt, K. H., and Pessin, J. E. (1996). SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J.Biol.Chem. 271, 6328-6332.

180
349. Eblen, S. T., Slack-Davis, J. K., Tarcsafalvi, A., Parsons, J. T., Weber, M. J., and Catling, A. D. (2004). Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol.Cell Biol. 24, 2308-2317.
350. Reszka, A. A., Seger, R., Diltz, C. D., Krebs, E. G., and Fischer, E. H. (1995). Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc.Natl.Acad.Sci.U.S.A 92, 8881-8885.
351. Reszka, A. A., Bulinski, J. C., Krebs, E. G., and Fischer, E. H. (1997). Mitogen-activated protein kinase/extracellular signal-regulated kinase 2 regulates cytoskeletal organization and chemotaxis via catalytic and microtubule-specific interactions. Mol.Biol.Cell 8, 1219-1232.
352. Morishima-Kawashima, M. and Kosik, K. S. (1996). The pool of map kinase associated with microtubules is small but constitutively active. Mol.Biol.Cell 7, 893-905.
353. Mineo, C., Anderson, R. G., and White, M. A. (1997). Physical association with ras enhances activation of membrane-bound raf (RafCAAX). J.Biol.Chem. 272, 10345-10348.
354. Furuchi, T. and Anderson, R. G. (1998). Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J.Biol.Chem. 273, 21099-21104.
355. Chen, R. H., Sarnecki, C., and Blenis, J. (1992). Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol.Cell Biol. 12, 915-927.
356. Khokhlatchev, A. V., Canagarajah, B., Wilsbacher, J., Robinson, M., Atkinson, M., Goldsmith, E., and Cobb, M. H. (1998). Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93, 605-615.
357. Lenormand, P., Brondello, J. M., Brunet, A., and Pouyssegur, J. (1998). Growth factor-induced p42/p44 MAPK nuclear translocation and retention requires both MAPK activation and neosynthesis of nuclear anchoring proteins. J.Cell Biol. 142, 625-633.
358. Shapiro, P. S., Vaisberg, E., Hunt, A. J., Tolwinski, N. S., Whalen, A. M., McIntosh, J. R., and Ahn, N. G. (1998). Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J.Cell Biol. 142, 1533-1545.
359. Zecevic, M., Catling, A. D., Eblen, S. T., Renzi, L., Hittle, J. C., Yen, T. J., Gorbsky, G. J., and Weber, M. J. (1998). Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP-E. J.Cell Biol. 142, 1547-1558.

181
360. Wang, X. M., Zhai, Y., and Ferrell, J. E., Jr. (1997). A role for mitogen-activated protein kinase in the spindle assembly checkpoint in XTC cells. J.Cell Biol. 137, 433-443.
361. Takenaka, K., Moriguchi, T., and Nishida, E. (1998). Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science 280, 599-602.
362. Nagata, K., Puls, A., Futter, C., Aspenstrom, P., Schaefer, E., Nakata, T., Hirokawa, N., and Hall, A. (1998). The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 17, 149-158.
363. Cowley, S., Paterson, H., Kemp, P., and Marshall, C. J. (1994). Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77, 841-852.
364. Yordy, J. S. and Muise-Helmericks, R. C. (2000). Signal transduction and the Ets family of transcription factors. Oncogene 19, 6503-6513.
365. Macfarlane, W. M., McKinnon, C. M., Felton-Edkins, Z. A., Cragg, H., James, R. F., and Docherty, K. (1999). Glucose stimulates translocation of the homeodomain transcription factor PDX1 from the cytoplasm to the nucleus in pancreatic beta-cells. J.Biol.Chem. 274, 1011-1016.
366. Traverse, S., Seedorf, K., Paterson, H., Marshall, C. J., Cohen, P., and Ullrich, A. (1994). EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr.Biol. 4, 694-701.
367. Brunet, A., Roux, D., Lenormand, P., Dowd, S., Keyse, S., and Pouyssegur, J. (1999). Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 18, 664-674.
368. Kim, K., Nose, K., and Shibanuma, M. (2000). Significance of nuclear relocalization of ERK1/2 in reactivation of c-fos transcription and DNA synthesis in senescent fibroblasts. J.Biol.Chem. 275, 20685-20692.
369. Formstecher, E., Ramos, J. W., Fauquet, M., Calderwood, D. A., Hsieh, J. C., Canton, B., Nguyen, X. T., Barnier, J. V., Camonis, J., Ginsberg, M. H., and Chneiweiss, H. (2001). PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev.Cell 1, 239-250.
370. Tohgo, A., Pierce, K. L., Choy, E. W., Lefkowitz, R. J., and Luttrell, L. M. (2002). beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J.Biol.Chem. 277, 9429-9436.

182
371. Fukuda, M., Gotoh, I., Adachi, M., Gotoh, Y., and Nishida, E. (1997). A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J.Biol.Chem. %19;272, 32642-32648.
372. van Drogen, F., Stucke, V. M., Jorritsma, G., and Peter, M. (2001). MAP kinase dynamics in response to pheromones in budding yeast. Nat.Cell Biol. 3, 1051-1059.
373. Volmat, V. and Pouyssegur, J. (2001). Spatiotemporal regulation of the p42/p44 MAPK pathway. Biol.Cell 93, 71-79.
374. Matsubayashi, Y., Fukuda, M., and Nishida, E. (2001). Evidence for existence of a nuclear pore complex-mediated, cytosol-independent pathway of nuclear translocation of ERK MAP kinase in permeabilized cells. J.Biol.Chem. 276, 41755-41760.
375. Sharma, P., Veeranna, Sharma, M., Amin, N. D., Sihag, R. K., Grant, P., Ahn, N., Kulkarni, A. B., and Pant, H. C. (2002). Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J.Biol.Chem. 277, 528-534.
376. Siow, Y. L., Kalmar, G. B., Sanghera, J. S., Tai, G., Oh, S. S., and Pelech, S. L. (1997). Identification of two essential phosphorylated threonine residues in the catalytic domain of Mekk1. Indirect activation by Pak3 and protein kinase C. J.Biol.Chem. 272, 7586-7594.
377. Slack-Davis, J. K., Eblen, S. T., Zecevic, M., Boerner, S. A., Tarcsafalvi, A., Diaz, H. B., Marshall, M. S., Weber, M. J., Parsons, J. T., and Catling, A. D. (2003). PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J.Cell Biol. 162, 281-291.
378. Cook, S. J. and McCormick, F. (1993). Inhibition by cAMP of Ras-dependent activation of Raf. Science 262, 1069-1072.
379. Sevetson, B. R., Kong, X., and Lawrence, J. C., Jr. (1993). Increasing cAMP attenuates activation of mitogen-activated protein kinase. Proc.Natl.Acad.Sci.U.S.A 90, 10305-10309.
380. Frodin, M., Peraldi, P., and Van Obberghen, E. (1994). Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J.Biol.Chem. 269, 6207-6214.
381. Young, S. W., Dickens, M., and Tavare, J. M. (1994). Differentiation of PC12 cells in response to a cAMP analogue is accompanied by sustained activation of mitogen-activated protein kinase. Comparison with the effects of insulin, growth factors and phorbol esters. FEBS Lett. 338, 212-216.

183
382. Chen, T., Cho, R. W., Stork, P. J., and Weber, M. J. (1999). Elevation of cyclic adenosine 3',5'-monophosphate potentiates activation of mitogen-activated protein kinase by growth factors in LNCaP prostate cancer cells. Cancer Res. 59, 213-218.
383. de Rooij, J., Zwartkruis, F. J., Verheijen, M. H., Cool, R. H., Nijman, S. M., Wittinghofer, A., and Bos, J. L. (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474-477.
384. York, R. D., Yao, H., Dillon, T., Ellig, C. L., Eckert, S. P., McCleskey, E. W., and Stork, P. J. (1998). Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622-626.
385. Stork, P. J. and Schmitt, J. M. (2002). Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 12, 258-266.
386. Ohba, Y., Kurokawa, K., and Matsuda, M. (2003). Mechanism of the spatio-temporal regulation of Ras and Rap1. EMBO J. 22, 859-869.
387. Rossomando, A. J., Dent, P., Sturgill, T. W., and Marshak, D. R. (1994). Mitogen-activated protein kinase kinase 1 (MKK1) is negatively regulated by threonine phosphorylation. Mol.Cell Biol. 14, 1594-1602.
388. Chow, C. W., Rincon, M., Cavanagh, J., Dickens, M., and Davis, R. J. (1997). Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 278, 1638-1641.
389. Kato, S., Endoh, H., Masuhiro, Y., Kitamoto, T., Uchiyama, S., Sasaki, H., Masushige, S., Gotoh, Y., Nishida, E., Kawashima, H., and . (1995). Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270, 1491-1494.
390. David, M., Petricoin, E., III, Benjamin, C., Pine, R., Weber, M. J., and Larner, A. C. (1995). Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science 269, 1721-1723.
391. Eblen, S. T., Slack, J. K., Weber, M. J., and Catling, A. D. (2002). Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol.Cell Biol. 22, 6023-6033.
392. Coles, L. C. and Shaw, P. E. (2002). PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 21, 2236-2244.
393. Bokemeyer, D., Sorokin, A., Yan, M., Ahn, N. G., Templeton, D. J., and Dunn, M. J. (1996). Induction of mitogen-activated protein kinase phosphatase 1 by the stress-

184
activated protein kinase signaling pathway but not by extracellular signal-regulated kinase in fibroblasts. J.Biol.Chem. 271, 639-642.
394. Fukunaga, R. and Hunter, T. (1997). MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 16, 1921-1933.
395. Waskiewicz, A. J., Flynn, A., Proud, C. G., and Cooper, J. A. (1997). Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16, 1909-1920.
396. Deak, M., Clifton, A. D., Lucocq, L. M., and Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426-4441.
397. Ludwig, S., Engel, K., Hoffmeyer, A., Sithanandam, G., Neufeld, B., Palm, D., Gaestel, M., and Rapp, U. R. (1996). 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol.Cell Biol. 16, 6687-6697.
398. Gille, H., Kortenjann, M., Thomae, O., Moomaw, C., Slaughter, C., Cobb, M. H., and Shaw, P. E. (1995). ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 14, 951-962.
399. Gille, H., Strahl, T., and Shaw, P. E. (1995). Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr.Biol. 5, 1191-1200.
400. Raingeaud, J., Whitmarsh, A. J., Barrett, T., Derijard, B., and Davis, R. J. (1996). MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol.Cell Biol. 16, 1247-1255.
401. Gupta, S., Campbell, D., Derijard, B., and Davis, R. J. (1995). Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science %20;267, 389-393.
402. Minden, A., Lin, A., McMahon, M., Lange-Carter, C., Derijard, B., Davis, R. J., Johnson, G. L., and Karin, M. (1994). Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science 266, 1719-1723.
403. Minden, A., Lin, A., Smeal, T., Derijard, B., Cobb, M., Davis, R., and Karin, M. (1994). c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol.Cell Biol. 14, 6683-6688.
404. Kao, S., Jaiswal, R. K., Kolch, W., and Landreth, G. E. (2001). Identification of the mechanisms regulating the differential activation of the mapk cascade by epidermal

185
growth factor and nerve growth factor in PC12 cells. J.Biol.Chem. 276, 18169-18177.
405. Traverse, S., Gomez, N., Paterson, H., Marshall, C., and Cohen, P. (1992). Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem.J. 288, 351-355.
406. Yan, G. Z. and Ziff, E. B. (1995). NGF regulates the PC12 cell cycle machinery through specific inhibition of the Cdk kinases and induction of cyclin D1. J.Neurosci. 15, 6200-6212.
407. Yasui, H., Katoh, H., Yamaguchi, Y., Aoki, J., Fujita, H., Mori, K., and Negishi, M. (2001). Differential responses to nerve growth factor and epidermal growth factor in neurite outgrowth of PC12 cells are determined by Rac1 activation systems. J.Biol.Chem. 276, 15298-15305.
408. Marshall, C. J. (1995). Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179-185.
409. Melemed, A. S., Ryder, J. W., and Vik, T. A. (1997). Activation of the mitogen-activated protein kinase pathway is involved in and sufficient for megakaryocytic differentiation of CMK cells. Blood 90, 3462-3470.
410. Herrera, R., Hubbell, S., Decker, S., and Petruzzelli, L. (1998). A role for the MEK/MAPK pathway in PMA-induced cell cycle arrest: modulation of megakaryocytic differentiation of K562 cells. Exp.Cell Res. 238, 407-414.
411. Racke, F. K., Lewandowska, K., Goueli, S., and Goldfarb, A. N. (1997). Sustained activation of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway is required for megakaryocytic differentiation of K562 cells. J.Biol.Chem. 272, 23366-23370.
412. Kang, C. D., Do, I. R., Kim, K. W., Ahn, B. K., Kim, S. H., Chung, B. S., Jhun, B. H., and Yoo, M. A. (1999). Role of Ras/ERK-dependent pathway in the erythroid differentiation of K562 cells. Exp.Mol.Med. 31, 76-82.
413. Pages, G., Guerin, S., Grall, D., Bonino, F., Smith, A., Anjuere, F., Auberger, P., and Pouyssegur, J. (1999). Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286, 1374-1377.
414. Tortorella, L. L., Milasincic, D. J., and Pilch, P. F. (2001). Critical proliferation-independent window for basic fibroblast growth factor repression of myogenesis via the p42/p44 MAPK signaling pathway. J.Biol.Chem. 276, 13709-13717.

186
415. Sarbassov, D. D., Jones, L. G., and Peterson, C. A. (1997). Extracellular signal-regulated kinase-1 and -2 respond differently to mitogenic and differentiative signaling pathways in myoblasts. Mol.Endocrinol. 11, 2038-2047.
416. Ueyama, T., Kawashima, S., Sakoda, T., Rikitake, Y., Ishida, T., Kawai, M., Yamashita, T., Ishido, S., Hotta, H., and Yokoyama, M. (2000). Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J.Mol.Cell Cardiol. 32, 947-960.
417. Schramek, H., Schumacher, M., Wilflingseder, D., Oberleithner, H., and Pfaller, W. (1997). Differential expression and activation of MAP kinases in dedifferentiated MDCK-focus cells. Am.J.Physiol 272, C383-C391.
418. Schramek, H., Feifel, E., Healy, E., and Pollack, V. (1997). Constitutively active mutant of the mitogen-activated protein kinase kinase MEK1 induces epithelial dedifferentiation and growth inhibition in madin-darby canine kidney-C7 cells. J.Biol.Chem. 272, 11426-11433.
419. Montesano, R., Soriano, J. V., Hosseini, G., Pepper, M. S., and Schramek, H. (1999). Constitutively active mitogen-activated protein kinase kinase MEK1 disrupts morphogenesis and induces an invasive phenotype in Madin-Darby canine kidney epithelial cells. Cell Growth Differ. 10, 317-332.
420. Marschitz, I., Lechner, J., Mosser, I., Dander, M., Montesano, R., and Schramek, H. (2000). Differential expression of cell-cell adhesion proteins and cyclin D in MEK1-transdifferentiated MDCK cells. Am.J.Physiol Cell Physiol 279, C1472-C1482.
421. Brunet, A., Pages, G., and Pouyssegur, J. (1994). Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene 9, 3379-3387.
422. Greulich, H. and Erikson, R. L. (1998). An analysis of Mek1 signaling in cell proliferation and transformation. J.Biol.Chem. 273, 13280-13288.
423. Pages, G., Lenormand, P., L'Allemain, G., Chambard, J. C., Meloche, S., and Pouyssegur, J. (1993). Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc.Natl.Acad.Sci.U.S.A 90, 8319-8323.
424. Graves, L. M., Guy, H. I., Kozlowski, P., Huang, M., Lazarowski, E., Pope, R. M., Collins, M. A., Dahlstrand, E. N., Earp, H. S., III, and Evans, D. R. (2000). Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature %20;403, 328-332.
425. Knauf, U., Tschopp, C., and Gram, H. (2001). Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol.Cell Biol. 21, 5500-5511.

187
426. Gavin, A. C. and Schorderet-Slatkine, S. (1997). Ribosomal S6 kinase p90rsk and mRNA cap-binding protein eIF4E phosphorylations correlate with MAP kinase activation during meiotic reinitiation of mouse oocytes. Mol.Reprod.Dev. 46, 383-391.
427. Chadee, D. N., Taylor, W. R., Hurta, R. A., Allis, C. D., Wright, J. A., and Davie, J. R. (1995). Increased phosphorylation of histone H1 in mouse fibroblasts transformed with oncogenes or constitutively active mitogen-activated protein kinase kinase. J.Biol.Chem. 270, 20098-20105.
428. Soloaga, A., Thomson, S., Wiggin, G. R., Rampersaud, N., Dyson, M. H., Hazzalin, C. A., Mahadevan, L. C., and Arthur, J. S. (2003). MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 22, 2788-2797.
429. Zhong, S., Zhang, Y., Jansen, C., Goto, H., Inagaki, M., and Dong, Z. (2001). MAP kinases mediate UVB-induced phosphorylation of histone H3 at serine 28. J.Biol.Chem. %20;276, 12932-12937.
430. Schmitt, A., Gutierrez, G. J., Lenart, P., Ellenberg, J., and Nebreda, A. R. (2002). Histone H3 phosphorylation during Xenopus oocyte maturation: regulation by the MAP kinase/p90Rsk pathway and uncoupling from DNA condensation. FEBS Lett. 518, 23-28.
431. Clayton, A. L. and Mahadevan, L. C. (2003). MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 546, 51-58.
432. Cheng, M., Sexl, V., Sherr, C. J., and Roussel, M. F. (1998). Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc.Natl.Acad.Sci.U.S.A 95, 1091-1096.
433. Lavoie, J. N., L'Allemain, G., Brunet, A., Muller, R., and Pouyssegur, J. (1996). Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J.Biol.Chem. 271, 20608-20616.
434. Matsushime, H., Quelle, D. E., Shurtleff, S. A., Shibuya, M., Sherr, C. J., and Kato, J. Y. (1994). D-type cyclin-dependent kinase activity in mammalian cells. Mol.Cell Biol. 14, 2066-2076.
435. Kato, J., Matsushime, H., Hiebert, S. W., Ewen, M. E., and Sherr, C. J. (1993). Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 7, 331-342.
436. Connell-Crowley, L., Harper, J. W., and Goodrich, D. W. (1997). Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol.Biol.Cell 8, 287-301.

188
437. Zerfass-Thome, K., Schulze, A., Zwerschke, W., Vogt, B., Helin, K., Bartek, J., Henglein, B., and Jansen-Durr, P. (1997). p27KIP1 blocks cyclin E-dependent transactivation of cyclin A gene expression. Mol.Cell Biol. 17, 407-415.
438. Kawada, M., Yamagoe, S., Murakami, Y., Suzuki, K., Mizuno, S., and Uehara, Y. (1997). Induction of p27Kip1 degradation and anchorage independence by Ras through the MAP kinase signaling pathway. Oncogene 15, 629-637.
439. Nguyen, H., Gitig, D. M., and Koff, A. (1999). Cell-free degradation of p27(kip1), a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol.Cell Biol. 19, 1190-1201.
440. Yin, M. B., Guo, B., Panadero, A., Frank, C., Wrzosek, C., Slocum, H. K., and Rustum, Y. M. (1999). Cyclin E-cdk2 activation is associated with cell cycle arrest and inhibition of DNA replication induced by the thymidylate synthase inhibitor Tomudex. Exp.Cell Res. 247, 189-199.
441. Bastians, H., Townsley, F. M., and Ruderman, J. V. (1998). The cyclin-dependent kinase inhibitor p27(Kip1) induces N-terminal proteolytic cleavage of cyclin A. Proc.Natl.Acad.Sci.U.S.A 95, 15374-15381.
442. Alessandrini, A., Chiaur, D. S., and Pagano, M. (1997). Regulation of the cyclin-dependent kinase inhibitor p27 by degradation and phosphorylation. Leukemia 11, 342-345.
443. Pumiglia, K. M. and Decker, S. J. (1997). Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc.Natl.Acad.Sci.U.S.A 94, 448-452.
444. Roovers, K. and Assoian, R. K. (2000). Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays 22, 818-826.
445. Kivinen, L. and Laiho, M. (1999). Ras- and mitogen-activated protein kinase kinase-dependent and -independent pathways in p21Cip1/Waf1 induction by fibroblast growth factor-2, platelet-derived growth factor, and transforming growth factor-beta1. Cell Growth Differ. 10, 621-628.
446. Chung, Y. W., Jeong, D. W., Won, J. Y., Choi, E. J., Choi, Y. H., and Kim, I. Y. (2002). H(2)O(2)-induced AP-1 activation and its effect on p21(WAF1/CIP1)-mediated G2/M arrest in a p53-deficient human lung cancer cell. Biochem.Biophys.Res.Commun. 293, 1248-1253.
447. Tang, D., Wu, D., Hirao, A., Lahti, J. M., Liu, L., Mazza, B., Kidd, V. J., Mak, T. W., and Ingram, A. J. (2002). ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J.Biol.Chem. 277, 12710-12717.

189
448. Bischoff, J. (1997). Cell adhesion and angiogenesis. J.Clin.Invest 99, 373-376.
449. Rousseau, S., Houle, F., Landry, J., and Huot, J. (1997). p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15, 2169-2177.
450. Klemke, R. L., Cai, S., Giannini, A. L., Gallagher, P. J., de Lanerolle, P., and Cheresh, D. A. (1997). Regulation of cell motility by mitogen-activated protein kinase. J.Cell Biol. 137, 481-492.
451. Holt, K. H., Kasson, B. G., and Pessin, J. E. (1996). Insulin stimulation of a MEK-dependent but ERK-independent SOS protein kinase. Mol.Cell Biol. 16, 577-583.
452. Haraguchi, S., Naito, K., and Sato, E. (1998). MAP kinase cascade, but not ERKs, activated during early cleavage of mouse embryos. Mol.Reprod.Dev. 51, 148-155.
453. Cheung, P., Tanner, K. G., Cheung, W. L., Sassone-Corsi, P., Denu, J. M., and Allis, C. D. (2000). Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol.Cell 5, 905-915.
454. Zhong, S. P., Ma, W. Y., and Dong, Z. (2000). ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10. J.Biol.Chem. 275, 20980-20984.
455. Inouye, C., Remondelli, P., Karin, M., and Elledge, S. (1994). Isolation of a cDNA encoding a metal response element binding protein using a novel expression cloning procedure: the one hybrid system. DNA Cell Biol. 13, 731-742.
456. Welch, D. R., Sakamaki, T., Pioquinto, R., Leonard, T. O., Goldberg, S. F., Hon, Q., Erikson, R. L., Rieber, M., Rieber, M. S., Hicks, D. J., Bonventre, J. V., and Alessandrini, A. (2000). Transfection of constitutively active mitogen-activated protein/extracellular signal-regulated kinase kinase confers tumorigenic and metastatic potentials to NIH3T3 cells. Cancer Res. 60, 1552-1556.
457. Cutler, R. E., Jr., Stephens, R. M., Saracino, M. R., and Morrison, D. K. (1998). Autoregulation of the Raf-1 serine/threonine kinase. Proc.Natl.Acad.Sci.U.S.A 95, 9214-9219.
458. Puls, A., Proikas-Cezanne, T., Marquardt, B., Propst, F., and Stabel, S. (1995). Kinase activities of c-Mos and v-Mos proteins: a single amino acid exchange is responsible for constitutive activation of the 124 v-Mos kinase. Oncogene 10, 623-630.
459. Zu, Y. L., Ai, Y., and Huang, C. K. (1995). Characterization of an autoinhibitory domain in human mitogen-activated protein kinase-activated protein kinase 2. J.Biol.Chem. 270, 202-206.

190
460. Xu, B. E., Min, X., Stippec, S., Lee, B. H., Goldsmith, E. J., and Cobb, M. H. (2002). Regulation of WNK1 by an Autoinhibitory Domain and Autophosphorylation. J.Biol.Chem. 277, 48456-48462.
461. Resing, K. A., Mansour, S. J., Hermann, A. S., Johnson, R. S., Candia, J. M., Fukasawa, K., Vande Woude, G. F., and Ahn, N. G. (1995). Determination of v-Mos-catalyzed phosphorylation sites and autophosphorylation sites on MAP kinase kinase by ESI/MS. Biochemistry 34, 2610-2620.
462. Guan, K. L. (1994). The mitogen activated protein kinase signal transduction pathway: from the cell surface to the nucleus. Cell Signal. 6, 581-589.
463. Pearson, G., Robinson, F., Beers, G. T., Xu, B. E., Karandikar, M., Berman, K., and Cobb, M. H. (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr.Rev. 22, 153-183.
464. Golemis E. Lab of Erica Golemis. 1997. Available at: http://chaos.fccc.edu/research/labs/golemis



















![MEMOIRE - Tlemcendspace.univ-tlemcen.dz/bitstream/112/15396/1/Modelisation-des... · Table Des Figures Figure 1 : Spectre donnant les dimensions des colloïdes [9]. 3 Figure (1.1)](https://img.pdfslide.fr/doc/110x75/5fae4f446cd5ab65fb77f3b4/memoire-table-des-figures-figure-1-spectre-donnant-les-dimensions-des-collodes.jpg)