Embed Size (px)
Citation preview

Juin 2019
> DMC > DMC avec déficit en mérosine
> DMC avec déficit en collagène VI > DMC avec déficit en collagène XII
> DMC avec déficit en sélénoprotéine N > Alpha-dystroglycanopathies
> DMC de type Fukuyama > Syndrome MEB (muscle-eye-brain)
> Syndrome de Walker-Warburg
Les dystrophies musculaires congénitales (DMC) sont des maladies rares, d’origine génétique. Le terme regroupe plusieurs maladies différentes caractérisées par une atteinte musculaire ("dystrophie") entrainant une faiblesse musculaire - hypotonie et difficultés motrices - se manifestant dès la naissance ou dans les premiers mois de vie ("congénitale"). Le muscle squelettique est le siège d’un processus dystrophique observable au microscope. Cette atteinte musculaire peut se compliquer de rétractions musculo-tendineuses et de difficultés respiratoires. Il peut aussi y avoir des atteintes du système nerveux central. Ce document, publié à l’occasion de l’Assemblée Générale de l’AFM-Téléthon 2019, présente les actualités de la recherche dans les dystrophies musculaires congénitales : colloques internationaux, études ou essais cliniques en cours, publications scientifiques et médicales... Il est téléchargeable sur le site internet de l’AFM-Téléthon où se trouvent aussi d’autres informations concernant les domaines scientifiques, médicaux, psychologiques, sociaux ou techniques dans les dystrophies musculaires congénitales : WEB www.afm-telethon.fr > Concerné par la maladie > Dystrophies
musculaires congénitales
AvanCées
dans les dystrophies musculaires
congénitales

Avancées dans les dystrophies musculaires congénitales
2 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Sommaire
DMC avec anomalies des protéines liées à la matrice extracellulaire ........ 4 Dystroglycanopathies .................................................................................................... 4 Autres formes de DMC ................................................................................................. 6
Des évènements médico-scientifiques................................................ 7 Congrès international de la World Muscle Society (WMS) ............................... 7 Journée Filnemus « Un diagnostic pour chacun » ............................................. 7 Congrès eNMD2019 ....................................................................................................... 8 Myology 2019 ................................................................................................................... 8 Conférence Cure CMD ................................................................................................... 8
Des bases de données internationales ................................................ 9 Une étude américaine pour se préparer aux essais cliniques ........................ 9 Base de données Fukuyama au Japon ................................................................. 10
Des essais cliniques ............................................................................. 11 Omigapil .......................................................................................................................... 11 N-acétylcystéine (NAC) .............................................................................................. 12
Des essais dans des myopathies adultes liées à des gènes impliqués dans des DMC ...................................................................................... 12
Le PF-06252616, un inhibiteur de la myostatine, dans la LGMD R9 liée à FKRP (ex-LGMD2I) ........................................................................................................ 12 Deflazacort (Emflaza®) .............................................................................................. 13
Mieux connaître les dystrophies musculaires congénitales ........... 13 Continuum clinique DMC d’Ulllrich-myopathie de Bethlem ....................... 13 De l’importance d’une évaluation respiratoire combinée ............................ 13 Le gène BET1 impliqué dans une nouvelle forme de DMC avec épilepsie ............................................................................................................................................. 13
Pistes thérapeutiques pharmacologiques ........................................ 14 Le ribitol améliore la souris modèle de DMC liée à la protéine FKRP ..... 14 Effets à cours terme des corticoïdes dans l’ α-dystroglycanopathie liée au gène GMPP ............................................................................................................... 14 Une certaine efficacité de la metformine dans des souris modèles de DMC1A ............................................................................................................................. 14
Sur la piste de la thérapie génique .................................................... 15 Dans une souris FKRP ................................................................................................. 15 Pseudo-exon dans le gène COL6A1 corrigé par saut d’exon ..................... 15
Le saut d’exons ................................................................................................................ 15 Dans des cellules de DMC liée au gène LMNA ................................................. 17 Dans la DMC1A liée à la laminine α2 ................................................................... 17
D’autres avancées médico-scientifiques ........................................... 18 Une stratégie nationale de diagnostic génétique des myopathies .......... 18 Dans les collagénopathies ........................................................................................ 18
Rôle du collagène VI dans la jonction neuromusculaire ................................. 18 Myopathie de Bethlem : pas de corrélation phénotype/génotype. ........... 19 L’adiponectine dans une souris modèle de myopathie de Bethlem .......... 19
Dans les mérosinopathies ......................................................................................... 20 A la recherche de biomarqueur chez la souris .................................................... 20 Élargissement de l’éventail clinique ........................................................................ 20
Dans les dystroglycanopathies ............................................................................... 20
Rédaction Myoinfo, Département d'information sur les maladies neuromusculaires de l'AFM-Téléthon, Évry Validation Dr. Valérie Allamand, Centre de Recherche en Myologie, Sorbonne Université, Inserm, UMRS 974, Institut de Myologie, Hôpital Pitié-Salpêtrière, Paris. Nathalie Loux, Direction Scientifique de l'AFM-Téléthon, Évry

Avancées dans les dystrophies musculaires congénitales
3 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Dystroglycanopathies liées à POMT1 : anomalies génétiques en cause et modèles animaux ............................................................................................................ 20 Anomalies des dystroglycanes selon les anomalies du gène DAG1 .......... 20
Dans la DMC mégaconiale ....................................................................................... 21 Une membrane des mitochondries anormale ..................................................... 21
* * *

Avancées dans les dystrophies musculaires congénitales
4 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Les dystrophies musculaires congénitales (DMC) sont des maladies rares d’origine génétique. Elles sont liées à des anomalies de l'ADN (mutations) qui sont généralement héritées des deux parents (maladies autosomiques récessives) ou parfois d’un seul (maladies autosomiques dominantes). Il existe aussi de nombreuses formes dominantes apparaissant de novo (sans antécédent dans la famille) et qui peuvent représenter jusqu’à la moitié des myopathies liées au collagène VI (COLVI) par exemple. Ces anomalies génétiques à l'origine des DMC conduisent généralement au déficit d'une protéine donnée ou à son dysfonctionnement, alors que cette protéine a une fonction importante pour la cellule musculaire. ▪ Il existe plusieurs formes de dystrophies musculaires congénitales. Elles diffèrent parfois beaucoup les unes des autres, mais peuvent aussi présenter des symptômes communs, ce qui complique le diagnostic. La classification des DMC n’est pas figée et évolue avec les découvertes de nouveaux gènes impliqués dans ces maladies. On distingue à ce jour 3 grands groupes de DMC : - un groupe en lien avec des anomalies des protéines de la matrice extracellulaire ; - un groupe en lien avec des anomalies de l’α-dystroglycane (α-dystroglycanopathies) ; - enfin un troisième groupe constitué par les autres formes de dystrophies musculaires congénitales.
DMC avec anomalies des protéines liées à la matrice extracellulaire ▪ DMC avec déficit en laminine α2 (mérosine) ou mérosinopathie liée à des anomalies dans le gène LAMA2 : les DMC1A. La laminine α2 appartient à un réseau de protéines situées à l’interface entre la membrane de la fibre musculaire et le tissu de soutien du muscle (tissu conjonctif). Ce réseau, appelé lame basale, entoure chaque fibre musculaire.
▪ DMC de type Ullrich (DMCU) liée à un déficit en collagène VI. Elle est due à des anomalies dans l’un des 3 gènes COL6A1, A2 ou A3 qui codent les sous-unités du collagène VI. Le collagène VI est un des constituants du tissu conjonctif qui entoure les fibres musculaires (matrice extracellulaire) et dont le rôle est de les soutenir et de permettre d’être associées les unes aux autres.
▪ DMC due à un déficit en collagène XII, causé par des mutations du gène COL12A1. Comme le collagène VI, le collagène XII participe à la constitution de la matrice extracellulaire.
▪ DMC due à des anomalies du gène de l’intégrine alpha 7, une protéine transmembranaire, en contact avec des protéines de la matrice extracellulaire.
Dystroglycanopathies Les dystroglycanopathies (syndrome de Walker-Warburg ou WWS, syndrome Muscle-œil-cerveau ou MEB, DMC de Fukuyama ou FCMD, dystrophie musculaire précoce avec microcéphalie et retard mental, forme liée à des anomalies du gène DPM3 avec réduction de la N-glycosylation et de la O-mannosylation de l’alpha-dystroglycane) sont dues à une absence ou à une altération de l'alpha-dystroglycane, une protéine qui joue un rôle
Les maladies d'origine génétique sont des maladies
dues à des anomalies de l'ADN, c'est-à-dire de l'information qui
détermine le fonctionnement biologique de notre organisme. Cette information est présente
dans nos cellules sous forme de chromosomes, nous l'héritons de
nos parents et nos enfants héritent de la nôtre. C'est
pourquoi les maladies génétiques sont souvent familiales, c'est-à-dire qu'il peut y avoir plusieurs membres d'une même famille
atteints par la maladie génétique.
La matrice extracellulaire est un réseau complexe de protéines dans lequel baignent les cellules.
Elle assure la cohésion des cellules au sein d’un tissu et joue
un rôle essentiel dans la constitution, le maintien,
l'adhérence, le mouvement et la régulation des cellules. La matrice
extracellulaire du muscle est spécialisée pour répondre aux
contraintes mécaniques inhérentes à l'activité contractile
des fibres musculaires.
La lame basale (ou membrane basale) est une forme particulière
de matrice extracellulaire spécifique à quelques tissus. Elle
est constituée de protéines qui s'enchevêtrent les unes aux autres
pour former un réseau qui entoure une ou plusieurs cellules.

Avancées dans les dystrophies musculaires congénitales
5 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
dans la résistance mécanique du tissu musculaire en faisant le lien entre la cellule musculaire et son environnement.
▪ Déficit de glycosylation de l'alpha-dystroglycane. Ces formes de dystroglycanopathies sont dues à des anomalies dans des gènes codant des protéines impliquées dans le bon fonctionnement de l'alpha-dystroglycane. Pour fonctionner correctement, l'alpha-dystroglycane a besoin de molécules de sucres à sa surface : on appelle glycosylation la fixation de ces sucres. Ces sucres lui permettent d’interagir avec des protéines de la matrice extracellulaire, comme les laminines. L'absence d'ajout de sucres à la surface de l'alpha-dystroglycane empêche sa liaison aux protéines de la matrice extracellulaire. Le lien entre l'intérieur et l'extérieur de la cellule est alors rompu, fragilisant la cellule musculaire.
▪ Déficit en dystroglycane Le déficit en dystroglycane est dû à des anomalies dans le gène DAG1 qui code l’α- et le β-dystroglycane. Chez les trois premiers cas de déficit en dystroglycane rapportés, les anomalies du gène DAG1 altéraient soit l’α-dystroglycane, soit le β-dystroglycane. Plus récemment, il a été décrit une famille concernée par un syndrome de Walker-Warburg, dû à une nouvelle anomalie du gène DAG1 qui entraine une perte complète à la fois de l’α-dystroglycane et du β-dystroglycane.
Dix-neuf gènes impliqués dans les dystroglycanopathies
Les gènes codant une protéine qui transfère une molécule de sucre sur l’alpha-dystroglycane : - gène B3GALNT2 (chromosome 1), - gène B3GNT1 (chromosome 11), - gène FKRP (chromosome 19), - gène FKTN (chromosome 9), - gène GMPPB (chromosome 3), - gène GTDC2 (chromosome 3), - gène ISPD (chromosome 7), - gène LARGE (chromosome 22), - gene POGNT2 (chromosome 3) - gène POMGnT1 (chromosome 1), - gène POMT1 (chromosome 9), - gène POMT2 chromosome 14), - gène SGK196 ou POMK (chromosome 8), - gène TMEM5 (chromosome 12).
Les gènes impliqués dans la fabrication d’un précurseur du sucre : - gène DMP2 (chromosome 9), - gène DMP3 (chromosome 1). - gène DOLK (chromosome 9), - gène DPM1 (chromosome 4), Le gène codant l'α- et le β-dystroglycane : - gène DAG1 (chromosome 3).
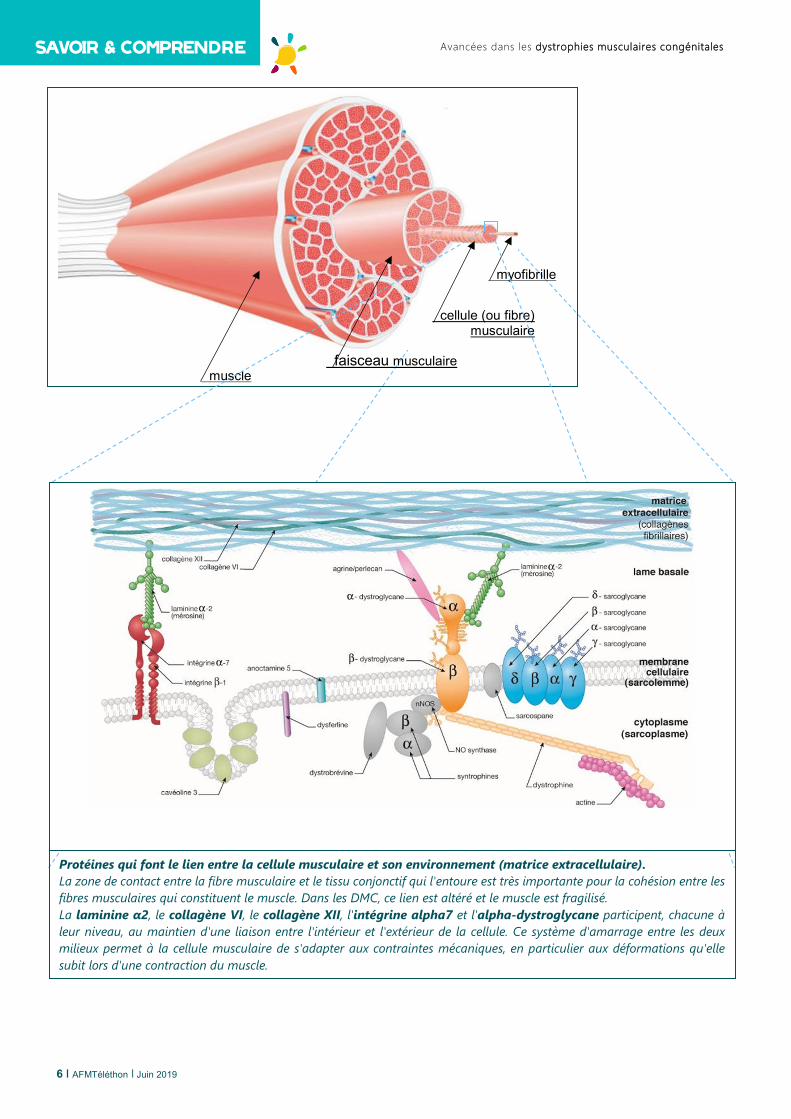
Avancées dans les dystrophies musculaires congénitales
6 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
muscle faisceau musculaire
cellule (ou fibre) musculaire
myofibrille
Protéines qui font le lien entre la cellule musculaire et son environnement (matrice extracellulaire). La zone de contact entre la fibre musculaire et le tissu conjonctif qui l'entoure est très importante pour la cohésion entre les fibres musculaires qui constituent le muscle. Dans les DMC, ce lien est altéré et le muscle est fragilisé. La laminine α2, le collagène VI, le collagène XII, l'intégrine alpha7 et l'alpha-dystroglycane participent, chacune à leur niveau, au maintien d'une liaison entre l'intérieur et l'extérieur de la cellule. Ce système d'amarrage entre les deux milieux permet à la cellule musculaire de s'adapter aux contraintes mécaniques, en particulier aux déformations qu'elle subit lors d'une contraction du muscle.

Avancées dans les dystrophies musculaires congénitales
7 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Autres formes de DMC ▪ Syndrome de la colonne raide (Rigid spine syndrome) lié à des anomalies du gène SEPN1 qui code la sélénoprotéine N. ▪ Forme sévère liée à des anomalies sporadiques du gène LMNA qui code les lamines A/C (LMNA) ou laminopathies. ▪ Forme avec anomalies mitochondriales (myopathie mégaconiale) liée à des anomalies du gène CHKB qui code la choline kinase β. ▪ Forme liée à des anomalies du gène BAG3, que l'on retrouve impliqué aussi dans les myopathies myofibrillaires. ▪ Forme liée à des anomalies du gène INPP5K, qui code une enzyme retrouvée principalement dans les muscles et le cerveau. ▪ Formes avec épilepsie liées à des anomalies du gène BET1 ou du gène GOSR2, qui codent des protéines intervenant dans le transport des vésicules entre le réticulum endoplasmique et l’appareil de Golgi, deux compartiments de la cellule où se déroule la synthèse des protéines. ▪ Forme avec lissencéphalie et neuropathie périphérique. ▪ Forme avec retard mental et cataracte congénitale. ▪ Forme avec myocardiopathie primitive.
Des évènements médico-scientifiques Congrès international de la W orld M uscle Society (W M S) Le Congrès international de la World Muscle Society (WMS) est un congrès annuel de référence sur les maladies neuromusculaires, réunissant chaque année plusieurs centaines de chercheurs, médecins et industriels venus du monde entier. Il a pour but de partager et de faire avancer les connaissances dans le champ des maladies neuromusculaires, de favoriser les collaborations multidisciplinaires, de soutenir les jeunes chercheurs. Lors de sa 23ème édition (WMS 2018), qui s’est déroulée du 2 au 6 octobre 2018 à Mendoza en Argentine, une session de posters a été consacrée aux dystrophies musculaires congénitales (DMC). Elle a été l’occasion de présenter des travaux précisant les manifestations de la maladie, leur évolution et pour certains, les moyens de les mesurer, notamment pour les dystrophies musculaires congénitales liées au collagène VI, à la laminine α2 ou à la sélénoprotéine N1 ; un poster présentait les résultats de pharmacocinétique et de tolérance de l’Omigapil dans l’essai CALLISTO. Une communication orale a présenté une nouvelle forme de dystrophie musculaire congénitale avec épilepsie liée au gène BET1. La prochaine édition, WMS 2019, aura lieu à Copenhague (Danemark) du 1er au 5 octobre 2019.
Journée Filnemus « Un diagnostic pour chacun » Le plan « Un diagnostic pour chacun », conçu en collaboration avec l’AFM-Téléthon et la filière nationale de santé neuromusculaire Filnemus, a été lancé en 2017 pour lutter contre l’errance diagnostique des patients atteints de maladies neuromusculaires et favoriser l’accès à un diagnostic précis de leur maladie. « Un diagnostic pour chacun » s’inscrit dans le cadre du 3ème Plan national maladies rares ; l’accès au diagnostic pour toutes les maladies rares y est un des enjeux majeurs. Il s’appuie aussi sur le plan « Médecine France
La Filière de santé maladies rares neuromusculaires FILNEMUS anime, coordonne et favorise les échanges entre les acteurs participant au diagnostic, à la prise en charge et à la recherche dans les maladies neuromusculaires (centres de références et centres de compétences, laboratoires de diagnostic, équipes de recherche, associations de personnes concernées…). Elle a été créée en février 2014, dans le cadre du deuxième Plan National Maladies Rares 2011-2014. WEB www.filnemus.fr Organisation des soins et maladies neuromusculaires, Repères Savoir & Comprendre, AFM-Téléthon
Les myopathies myofibrillaires sont des myopathies touchant les adultes et caractérisées par une désorganisation du réseau de fibres qui permet aux cellules musculaires de se contracter, les myofibrilles, ainsi qu’une accumulation excessive de diverses protéines.
Le réticulum endoplasmique est le compartiment de la cellule où se déroule la fabrication des protéines et des lipides. Dans la cellule musculaire, il joue en plus un rôle essentiel lors de la contraction musculaire en libérant et en recaptant le calcium qu'il contient.Le réticulum endoplasmique de la cellule musculaire est aussi appelé réticulum sarcoplasmique.

Avancées dans les dystrophies musculaires congénitales
8 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
génomique 2025 » qui finance le déploiement en France des plateformes de séquençage à haut débit (NGS). ▪ Le 5 novembre 2018, la journée Filnemus « Un diagnostic pour chacun » a été l’occasion de faire un état des lieux des outils et moyens existant ou en cours de construction pour l’accès au diagnostic dans les maladies neuromusculaires : outils de repérages et d’accompagnement des personnes en attente de diagnostic au sein de l’AFM-Téléthon, banques de données médicales, listes consensuelles de gènes pour le dépistage génétique des maladies neuromusculaire, circuit d’accès au diagnostic… WEB www.filnemus.fr/menu-filiere/evenements/les-journees-thematiques-de-filnemus/reunion-filnemus-un-diagnostic-pour-chacun WEB www.afm-telethon.fr/actualites/strategie-nationale-diagnostic-genetique-maladies-neuromusculaires-127176
Congrès eN M D 2019 Organisé en 2019 par le CHU de Nice en partenariat avec l'AFM Téléthon et le réseau européen d’excellence neuromusculaire EURO-NMD, le congrès eNMD2019 était le premier grand rassemblement européen de spécialistes de la e-santé travaillant dans le domaine des maladies neuromusculaires. Il a réuni les 22 et 23 mars 2019 des professionnels de santé, des acteurs publiques, des associations de malades et des start-ups. WEB www.afm-telethon.fr/actualites/congres-europeen-sur-e-sante-dans-maladies-neuromusculaires-127197
Myology 2019 Organisé par l’AFM-Téléthon, le 6ème Congrès international de myologie – Myology 2019 – s’est tenu du 25 au 28 mars à Bordeaux. Cet événement a réuni près de 1000 experts de la myologie (chercheurs, médecins-cliniciens) pour faire le point sur la recherche fondamentale et sur les thérapies innovantes développées ces dernières années (thérapie génique, thérapie cellulaire, saut d’exon, pharmacogénétique…) dans les maladies neuromusculaires. Deux présentations orales ont été plus particulièrement consacrées aux stratégies de thérapie génique à l’étude dans les dystrophies musculaires congénitales : l’une présentée par Carsten Bonnemann a porté sur le saut d’un pseudo-exon anormal dans le gène du collagène VI, l’autre de Markus Ruegg a porté sur sur l’ajout de protéines connectrices à la laminine dans les déficits en laminine α2.
Conférence Cure CM D L’association américaine Cure CMD organise une conférence scientifique les 24 et 25 juillet 2019, suivie d’une conférence pour les familles les 27 et 28 juillets 2019 à Chicago (États-Unis). Au programm : le point sur les différentes formes de dystrophies musculaires congénitales, mais aussi les essais cliniques et les priorités pour les 3 années à venir. WEB www.scifam.info/
Cure CMD est une association américaine crée en 2008 dont la mission consiste à faire émerger des recherches, des soins et des
traitements pour les dystrophies musculaires congénitales.
WEB www.curecmd.org
Avancées dans les dystrophies musculaires congénitales
9 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Des bases de données internationales Les bases de données de patients permettent de recenser les personnes atteintes d'une même maladie, de préciser l’histoire naturelle de celle-ci, d'établir des corrélations génotype/phénotype et de faciliter le recrutement de patients dans les essais cliniques.
Base de données internationale des dystrophies musculaires congénitales (CMDIR) (CMDPROS)
Faciliter l’identification des personnes atteintes de DMC pour des essais cliniques, améliorer le diagnostic et la prise en charge
et collecter des données globales sur la DMC [NCT01403402]
(Promoteur : Cure CMD)
Statut Pays Date de création
Recrutement en cours International (y compris en France) 2009
Base de données internationale des DMC liées à un déficit en collagène VI
Collecter des données pour la recherche et faciliter l’identification des personnes atteintes de ces maladies éligibles à de futurs essais cliniques
[NCT03693898] (soutenue par l’AFM-Téléthon)
Statut Pays Date de création
Recrutement en cours International (y compris en France) 2018
Une étude américaine pour se préparer aux essais cliniques Une étude prospective d’histoire naturelle démarrée en 2006 est toujours en cours aux États-Unis.
Cette étude clinique a déjà donné lieu à deux publications : ▪ l’une en 2015, sur la mise en évidence d’une plus grande fréquence de troubles urinaires et de difficultés pour s’alimenter (dysphagie) chez 30 personnes atteintes de dystroglycanopathie que chez les membres de leur famille ; Urologic and gastrointestinal symptoms in the dystroglycanopathies. Crockett CD, Bertrand LA, Cooper CS, Rahhal RM, Liu K, Zimmerman MB, Moore SA, Mathews KD. Neurology. 2015 Feb 3;84(5):532-9.
Étude d’histoire naturelle Décrire les signes précoces et les manifestations des dystroglycanopathies et
rassembler des informations nécessaires à de futurs essais cliniques [NCT00313677]
(Promoteur : National Institute of Neurological Disorders and Stroke (NINDS))
Statut Nombre de participants
(âge)
Pays Durée du suivi
Début - Fin
Recrutement en cours
120 (Tous âges) États-Unis Annuel, durée
non précisée Avril 2006-Mars 2020
Les études de corrélations génotype/phénotype recherchent l'existence de liens entre les caractéristiques génétiques, le génotype, et les caractéristiques s'exprimant de façon apparente, le phénotype (taille, couleur et forme des yeux, couleur des cheveux, manifestation d'une maladie...). On peut ainsi identifier une relation plus ou moins étroite entre la présence d'une anomalie génétique et les manifestations d'une maladie génétique.
Ce que les médecins appellent l'histoire naturelle d'une maladie est la description des différentes manifestations d'une maladie et de leur évolution au cours du temps en l'absence de traitement.

Avancées dans les dystrophies musculaires congénitales
10 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
▪ l’autre, en décembre 2017, sur une aggravation transitoire de la faiblesse musculaire des personnes atteintes de dystroglycanopathies lors d’une maladie intercurrente fébrile (près de 25% des 52 cas étudiés). Illness-associated muscle weakness in dystroglycanopathies. Carlson CR, McGaughey SD, Eskuri JM, Stephan CM, Zimmerman MB, Mathews KD. Neurology. 2017 Dec 5;89(23):2374-2380.
Base de données Fukuyama au Japon La dystrophie musculaire congénitale de Fukuyama est la deuxième forme de dystrophie musculaire la plus fréquente au Japon. C’est une α-dystroglycanopathie due à des anomalies du gène FKTN, qui code la fukutine. La Japan Muscular Dystrophy Association (JMDA) a mis en place en 2011 une base de données nationale des cas génétiquement confirmés de dystrophie musculaire congénitale de Fukuyama. ▪ Un article publié en octobre 2018 fait le point sur cette base de données. Fin septembre 2013, la base contenait les données médicales de 207 patients (104 garçons, 103 filles). Une mutation fondatrice dans le gène FKTN a été retrouvée chez 80% des cas colligés. Soixante-neuf patients (33%) présentaient des convulsions fébriles et/ou une epilepsie. Au niveau oculaire, la myopie était l’atteinte plus fréquente (8,7%), suivie par le strabisme (5,9%). Seize pour cent des patients avaient besoin d’une assistance respiratoire, 16% présentaient une atteinte cardiaque et 22% des difficultés pour avaler (dysphagie). National registry of patients with Fukuyama congenital muscular dystrophy in Japan. Ishigaki K, Ihara C, Nakamura H, Mori-Yoshimura M, Maruo K, Taniguchi-Ikeda M, Kimura E, Murakami T, Sato T, Toda T, Kaiya H, Osawa M. Neuromuscul Disord. 2018 Oct;28(10):885-893. Une étude sur l’histoire naturelle du développement mental dans la dystrophie musculaire congénitale de Fukuyama a été réalisée grâce à un questionnaire adressé à 49 familles référencées dans cette base de données. ▪ Les résultats ont été présentés au cours de la session de posters consacrées aux DMC lors du Congrès international de la World Muscle Society (WMS) en octobre 2018. Ils permettent de mieux cerner les difficultés cognitives des personnes atteintes de dystrophie musculaire congénitale de Fukuyama et d’améliorer ainsi leur prise en charge rééducative et leur prise en compte dans l’éducation des enfants. Review of the natural history of mental development in Fukuyama congenital muscular dystrophy patients, based on a written questionnaire from their families. Shichiji M, Ishigaki K, Sato T, Yamashita A, Nagata S. Neuromuscul Disord. 2018 Oct; 28(10) : S128
Avancées dans les dystrophies musculaires congénitales
11 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Des essais cliniques Les essais cliniques consistent à évaluer les effets d’un traitement potentiel dans une maladie (un candidat-médicament, un dispositif médical…) dans le but de s’assurer qu’il est bien toléré et efficace dans cette maladie.
Omigapil ▪ Des recherches précliniques ont montré que l’omigapil entrainait une diminution de la sévérité des symptômes et une augmentation de la survie de modèles animaux de la DMC1A. Des travaux précliniques de l’omigapil ont été financés en 2009 par l’AFM-Téléthon.
▪ Depuis l’annonce en avril 2018 des résultats démontrant la sécurité d’utilisation de l’Omigapil dans l’essai de phase I (CALLISTO), dans les formes de dystrophies musculaires congénitales liées au collagène VI ou à la laminine α2 (mérosine) par le laboratoire Santhera, ceux-ci ont fait l’objet d’un poster lors de la WMS2018. Cette étude en ouvert [NCT01805024] qui s’est déroulée aux États-Unis de décembre 2014 à janvier 2018, a permis d’établir le profil pharmacocinétique de différentes doses d’omigapil chez 20 enfants atteints de DMC1A ou de DMC de type Ullrich, âgés de 5 à 16 ans. L’administration orale quotidienne de l’omigapil a duré 4 semaines et les participants ont été suivis pendant 8 semaines après l’arrêt de la prise du candidat-médicament. L’omigapil a été bien toléré pendant toute la durée de l’essai. CALLISTO : a phase I open-label, sequential group, cohort study of pharmacokinetics and safety of omigapil in LAMA2 and COL6 -related dystrophy patients. Foley A.R, Leach M, Averion G, Hu Y, Yun P, Neuhaus S, Saade D, Arevalo C, Fink M, DeCoster J, Mendoza C, Mayer O, Hausmann R, Petraki D, Cheung K, Bönnemann C. Neuromuscul Disord. 2018 Oct; 28(10) : p329
▪ Le laboratoire Santhera envisageait en avril 2018 de mettre en place un essai pivot de phase III pour évaluer les propriétés thérapeutiques du candidat-médicament dans les DMC, qui n’a, à ce jour, pas encore été lancé. L’omigapil a reçu de la part des autorités de santé américaines (FDA pour Food and drug administration) la désignation de produit bénéficiant d'une procédure d'évaluation accélérée (« fast track designation ») dans les DMC en mai 2016. Santhera Announces Successful Completion of First Clinical Trial with Omigapil in Patients with Congenital Muscular Dystrophy. Santhera Pharmaceuticals, Communiqué de presse du 5 avril 2018
Essai de phase I dans la DMC1A et la DMC de type Ullrich (CALLISTO) Évaluer le devenir dans l’organisme (pharmacocinétique) et les effets
secondaires à court terme afin de déterminer la dose optimale d’omigapil chez des enfants atteints de DMC1A ou de DMC de type Ullrich
[NCT01805024] (Promoteur : Santhera Pharmaceuticals)
Statut Nombre de participants
(âge)
Pays Durée du suivi
Début - Fin
Essai terminé, données en
cours d’analyse
20 (5 à 16 ans)
États-Unis 8 semaines Décembre 2014 – janvier
2018
Au cours d'un essai clinique de phase I un médicament dont l'intérêt thérapeutique a été montré sur des modèles animaux et/ou cellulaires (essais précliniques) est administré pour la première fois à un petit groupe de volontaires sains, plus rarement à des malades, afin d'évaluer leur tolérance à la substance en fonction de la dose (Comment le futur traitement est-il absorbé et éliminé ? Comment se fait sa répartition dans les organes ? Est-il toxique et à quelles doses ? Existe-t-il des effets secondaires ?). Essais cliniques et maladies neuromusculaires, Repères Savoir & Comprendre, AFM-Téléthon.
La pharmacocinétique étudie le devenir d'un médicament dans l'organisme : comment est-il absorbé (quantité, vitesse...) ? Comment diffuse-t-il dans l'organisme (quantité, vitesse...) ? Comment est-il transformé, puis éliminé (par le foie, par le rein...) ?
Avancées dans les dystrophies musculaires congénitales
12 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
N-acétylcystéine (NAC) La N-acétylcystéine (NAC) est un antioxydant qui pourrait agir sur le stress oxydatif de personnes atteintes de dystrophie musculaire congénitale avec déficit en sélénoprotéine N.
Des essais dans des myopathies adultes liées à des gènes impliqués dans des DMC A l’instar de la myopathie de Bethlem et de la dystrophie musculaire congénitale d’Ullrich, toutes les deux dues à des anomalies du collagène VI, certaines myopathies de l’adulte sont dues à des altérations d’un gène impliqué aussi dans une forme de dystrophie musculaire congénitale. Il s’agit essentiellement de formes de myopathies des ceintures, appelées aussi LGMD, pour limb girdle muscular dystrophy en anglais. Les pistes thérapeutiques étudiées dans ces formes adultes ouvrent la voie à de futurs traitements pour les formes congénitales liées à des anomalies impliquant les mêmes gènes.
Le PF-06252616, un inhibiteur de la myostatine, dans la LGMD R9 liée à FKRP (ex-LGMD2I) La LGMDR9 (ex-LGMD2I) est une myopathie des ceintures liée à des anomalies du gène FKRP. Dans cette dystrophie musculaire des ceintures, une piste thérapeutique à l’étude a pour but d’augmenter la taille des fibres musculaires en bloquant la myostatine, un inhibiteur de la croissance musculaire. Le laboratoire Pfizer mène un essai du PF-06252616, un inhibiteur de la myostatine, chez des personnes atteintes de LGMDR9 (ex-LGMD2I) ambulantes.
Essai SelNac Déterminer si la N-acétylcystéine améliore le stress oxydatif de personnes
atteintes de DMC avec déficit en sélénoprotéine N [NCT02505087] (Promoteur : Assistance Publique - Hôpitaux de Paris)
Statut Nombre de participants
(âge)
Pays Durée du suivi
Début - Fin
Recrutement en cours
48 (18 à 60 ans)
France 1 an Septembre 2015 –
Novembre 2017
Essai de phase I/II du PF-06252616 Évaluer les effets du PF-06252616 chez des personnes atteintes
de LGMD R9 liée à FKRP (ex-LGMD2I) ambulantes [NCT02841267] (Promoteur : Pfizer)
Statut Nombre de participants
(âge)
Pays Durée du suivi
Début - Fin
Recrutement terminé, essai
en cours
19 (18 ans et
plus) États-Unis, 2 ans Juillet 2016 –
Août 2018
Le stress oxydatif correspond à une situation où la cellule ne
contrôle plus la présence excessive de molécules toxiques
issues de la respiration cellulaire : les radicaux libres. En
excès, ces radicaux libres peuvent endommager les
cellules et l’ADN.
Avancées dans les dystrophies musculaires congénitales
13 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Deflazacort (Emflaza®)
La phase initiale de six mois en double aveugle sera suivie d’une phase de six mois en ouvert où tous les participants prendront du déflazacort.
Mieux connaître les dystrophies musculaires congénitales Continuum clinique DMC d’Ulllrich-myopathie de Bethlem Une équipe russe a publié en septembre 2018 l’observation d’un garçon âgé de 8 ans, présentant des signes de sévérité intermédiaire entre ceux de la dystrophie musculaire congénitale d’Ullrich et ceux de la myopathie de Bethlem. Outre des anomalies génétiques nouvellement décrites, cette observation confirme le continuum clinique entre la dystrophie musculaire congénitale d’Ullrich et la myopathie de Bethlem. Two novel COL6A3 mutations disrupt extracellular matrix formation and lead to myopathy from Ullrich congenital muscular dystrophy and Bethlem myopathy spectrum. Marakhonov AV, Tabakov VY, Zernov NV, Dadali EL, Sharkova IV, Skoblov MY. Gene. 2018 Sep 25;672:165-171.
De l’importance d’une évaluation respiratoire combinée Les examens de routine de la fonction respiratoire sont basés sur des manoeuvres volontaires non invasives (mesure des volumes respiratoires, spirométrie, pressions statiques maximales), qui peuvent être difficiles voire impossibles à effectuer chez les jeunes enfants. Des tests utilisant des gestes naturels comme renifler, tousser ou siffler sont plus faciles à réaliser pour les petits. La combinaison de plusieurs tests respiratoires est essentielle aussi bien au diagnostic de l’atteinte respiratoire qu’à la robustesse des données dans les essais cliniques qui évaluent les nouvelles thérapies. Respiratory insight to congenital muscular dystrophies and congenital myopathies and its relation to clinical trial. Fauroux B, Amaddeo A, Quijano-Roy S, Barnerias C, Desguerre I, Khirani S. Neuromuscul Disord. 2018 Sep;28(9):731-740.
Le gène B ET 1 impliqué dans une nouvelle forme de DMC avec épilepsie Une équipe américaine a présenté dans un poster lors de la WMS 2018 la situation d’un garçon de 4 ans présentant des manifestations de dystrophie musculaire congénitale avec épilepsie. L’analyse de son génome entier a permis de retrouver des anomalies dans le gène BET1.
Essai de phase III en double aveugle Évaluer la tolérance et l’efficacité du deflazacort (Emflaza®)
dans la LGMD R9 liée à FKRP (ex-LGMD2I) [NCT03783923] (Promoteur : PTC Therapeutics)
Statut Nombre de participants
(âge)
Pays Durée du suivi
Début - Fin
En préparation
100 (18 ans et
plus)
Allemagne, Danemark, États-Unis
6 mois
Mai 2019-Août 2020

Avancées dans les dystrophies musculaires congénitales
14 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
L’étude des cellules du garçon a montré une diminution importante de la quantité de la protéine BET1. Cette protéine joue un rôle dans le transport de molécules via des vésicules (trafic membranaire) entre le réticulum endoplasmique et l’appareil de Golgi, deux compartiments de la cellule essentiels à la fabrication de protéines fonctionnelles. Recessive mutations in BET1 and GOSR2 establish Q-SNARE Golgi vesicle-transport genes as a cause for congenital muscular dystrophy with epilepsy. Donkervoort S, Hu Y, Shieh P, Koliwer J, Tsai L, Cummings B, Snyder M, Chao K, Kaur R, Bharucha-Goebel D, Iannaccone S, MacArthur D, Foley A.R, Schwake M, Bönnemann C. Neuromuscul Disord. 2018 Oct; 28(10) : S30
Pistes thérapeutiques pharmacologiques Le ribitol améliore la souris modèle de DMC liée à la protéine FKRP Le ribitol, une molécule dont on ne connaît pas la fonction exacte dans les cellules de mammifères, a restauré partiellement une O-mannolysation (l’ajout de sucre déficitaire dans la dystrophie musculaire congénitale liée à la protéine FKRP) de l’α-dystroglycane dans un modèle cellulaire. Administré avant ou après l’apparition des premiers signes de la maladie chez une souris modèle de dystrophie musculaire congénitale liée à FKRP, le ribitol a diminué la fibrose cardiaque et a amélioré les fonctions respiratoires et motrices de ces souris. Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Cataldi MP, Lu P, Blaeser A, Lu QL. Nat Commun. 2018 Aug 27;9(1):3448.
Effets à cours terme des corticoïdes dans l’ α-dystroglycanopathie liée au gène G M PP Une équipe italienne rappporte dans un article publié en novembre 2018, le cas d’un enfant atteint d’une α-dystroglycanopathie due à des anomalies du gène GMPP, traité pendant 3 mois par des corticoïdes (0,75mg/kg/j de prednisone). Ce traitement a apporté une amélioration de la force musculaire et des scores fonctionnels, associée à une diminution du taux de créatine kinase, qui ne se sont pas poursuivies après l’arrêt du traitement. Les auteurs concluent à l’intérêt de considérer ce type de traitement dans l’α-dystroglycanopathie liée au gène GMPP Steroid therapy in an alpha-dystroglycanopathy due to GMPPB gene mutations: A case report. Fecarotta S, Gragnaniello V, Della Casa R, Romano A, Raiano E, Torella A, Savarese M, Nigro V, Strisciuglio P, Andria G, Parenti G. Neuromuscul Disord. 2018 Nov;28(11):956-960.
Une certaine efficacité de la metformine dans des souris modèles de DMC1A Compte tenu des altérations du métabolisme musculaire retrouvées dans des cellules musculaires de patients atteints de dystrophie musculaire congénitale liées à la laminine α2 (mérosinopathie) et dans des souris modèles de cette maladie, une équipe suédoise a exploré les effets de la metformine, un médicament déjà utilisé dans le diabète et plus récemment dans la dystrophie myotonique de type 1.
L'appareil de Golgi est le compartiment de la cellule où les
protéines nouvellement synthétisées subissent les
dernières modifications nécessaires à leur bon
fonctionnement (ajout d’un sucre (glycosylation), ajout de sulfates
ou de phosphates...), sont stockées puis envoyées à des
destinations différentes, dans ou hors de la cellule, suivant leur
fonction.

Avancées dans les dystrophies musculaires congénitales
15 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
▪ Les chercheurs ont, dans un premier temps, constaté que les manifestations de la maladie étaient différentes dans les souris modèles de mérosinopathie mâles et dans les souris femelles. ▪ L’administration de metformine entraine un gain de poids, une amélioration de la fonction musculaire et de l’aspect histologique du tissu musculaire des souris femelles. Ces signes d’amélioration ont été moins marqués dans les souris mâles. Effects of metformin on congenital muscular dystrophy type 1A disease progression in mice: a gender impact study. Fontes-Oliveira CC, M Soares Oliveira B, Körner Z, M Harandi V, Durbeej M. Sci Rep. 2018 Nov 2;8(1):16302.
Sur la piste de la thérapie génique À ses débuts, la thérapie génique consistait uniquement à remplacer un gène défectueux en apportant à l’organisme le gène normal. Avec le développement de nouvelles techniques, le terme de thérapie génique désigne plus largement toutes les techniques qui introduisent dans l’organisme du matériel génétique sous forme d’ADN ou d’ARN (gène médicament, oligonucléotides antisens…) à des fins thérapeutiques.
Dans une souris FKRP Des chercheurs américains ont administré par voie intraveineuse, différentes doses (une basse, une moyenne et une forte dose) d’un produit de thérapie génique apportant le gène normal de la FKRP à l’aide d’un vecteur AAV9, à des souris modèles de la dystrophie musculaire congénitale liée à FKRP qui commencaient à avoir des signes de la maladie. ▪ Les anomalies de glycosylation de l’α-dystroglycanes et celles du tissu musculaire, dues au manque de FKRP, ont été améliorées de façon proportionnelle à la dose injectée. ▪ Ces améliorations se sont accompagnées d’une amélioration des performances physiques des souris qui s’est prolongée au delà de 35 semaines après l’injection pour la plus forte dose. Dose-Dependent Effects of FKRP Gene-Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice. Vannoy CH, Leroy V, Lu QL. Mol Ther Methods Clin Dev. 2018 Oct 13;11:106-120.
Pseudo-exon dans le gène COL6A1 corrigé par saut d’exon Le développement des techniques de séquençage nouvelles générations a entrainé la découverte de nouveaux types d’anomalies génétiques. L’une de ces anomalies génétiques dans le gène COL6A1 entraine l’apparition d’un pseudo-exon anormal qui conduit à la fabrication d’une partie du collagène VI produit, qui empêche l’assemblage de celui-ci dans la matrice extracellulaire. ▪ En utilisant une technique de saut d’exon pour que la machinerie cellulaire ignore ce pseudo-exon, dans des fibroblastes de patient en culture, une équipe de chercheurs internationale a pu restaurer une matrice extra-cellulaire normale.
Le saut d’exons Le saut d’exon consiste à intervenir au cours d’une étape de la fabrication des protéines : l’épissage de l’ARN messager.
Un vecteur viral est un virus modifié, dit sécurisé, dont on a éliminé les éléments qui rendent malades (éléments pathogènes), en ne conservant que les éléments indispensables au virus pour atteindre le noyau des cellules. Le génome du virus est reconstruit pour y intégrer les séquences du gène médicament.
Le virus adéno-associé (AAV pour adeno-associated virus) est un virus à ADN, qui peut infecter l'être humain. Toutefois, il ne provoque pas de maladie et n'entraine qu'une réponse de défense immunitaire modérée. Une fois à l'intérieur des cellules, l’AAV exprime ses gènes (et ceux que l’on aurait introduits dans son génome). Il est utilisé en génie génétique comme vecteur pour la thérapie génique.

Avancées dans les dystrophies musculaires congénitales
16 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Pour effectuer un saut d'exon, les chercheurs utilisent de petits ADN artificiels antisens, appelés aussi oligonucléotides antisens et capables de tromper la machinerie d'épissage. ▪ Les chercheurs ont aussi utilisé une nouvelle technique d’édition du génome, le système CRIPR/Cas9, pour détruire précisément la séquence du gène COL6A1 qui permet de sauter le pseudo-exon et de conserver ainsi une matrice extra-cellulaire de constitution normale.
Le système CRISPR/Cas9 est un système rapide et efficace pour couper l’ADN à un endroit précis du génome, dans n’importe quelle cellule. Il est constitué d’un « ARN guide », qui cible une séquence d’ADN particulière, associé à l’enzyme Cas9, qui, comme des ciseaux moléculaires, coupe l’ADN.
▪ Ce type d’anomalie de l’épissage du gène COL6A1 étant l’une des plus fréquentes à l’origine d’une myopathie liée au collagène VI autosomique dominante, les auteurs concluent que ce type de technique de modification spécifique de l’épissage est une voie prometteuse pour une application clinique. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. Bolduc V, Foley AR, Solomon-Degefa H, Sarathy A, Donkervoort S, Hu Y, Chen GS, Sizov K, Nalls M, Zhou H, Aguti S, Cummings BB, Lek M, Tukiainen T, Marshall JL, Regev O, Marek-Yagel D, Sarkozy A, Butterfield RJ, Jou C, Jimenez-Mallebrera C, Li Y, Gartioux C,
De l’ARN pré-messager à l’ARN messager. La transcription est la première étape de la synthèse d’une protéine : le message du gène est "transcrit" en ARN pré-messager (un peu comme une photocopie ou une empreinte de la région d'ADN qui porte le gène). Dans la seconde étape, l'épissage, l'ARN pré-messager devient l’ARN messager : certaines parties (les introns) sont coupées et les morceaux restants (les exons) sont réunis en un seul brin d'ARN messager mature qui ne contient que les informations nécessaires pour guider la synthèse de la protéine.
ARN pré-messager
ARN messager
Le saut d’exon est une technique de thérapie génique Cette technique utilise des oligonucléotides qui se lient à l’ARN messager pendant l’épissage pour empêcher l’inclusion d’un ou de plusieurs exons. La protéine sera alors raccourcie des exons sautés.
ARN pré-messager
ARN messager
Les techniques d’édition du génome (« genome editing » en
anglais) sont des techniques utilisées pour modifier les
séquences d’ADN qui constituent le génome, comme les nucléases
à doigt de zinc, les méganucléases, les TALENs
(nucléases effectrices de type activateur de transcription) ou
plus récemment le système CRISPR/Cas9.

Avancées dans les dystrophies musculaires congénitales
17 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Mamchaoui K, Allamand V, Gualandi F, Ferlini A, Hanssen E; COL6A1 Intron 11 Study Group, Wilton SD, Lamandé SR, MacArthur DG, Wagener R, Muntoni F, Bönnemann CG. JCI Insight. 2019 Mar 21;4(6). Mutation Specific Precision Therapy: The COL6-Related Dystrophies and Beyond Bönnemann C.G. for the “Collagen VI Precision Diagnostics and Therapeutics Collaborative Research Group” Communication orale Myology 2019 In vivo-ready lipid-conjugated siRNAs for allele-specific silencing of a dominant-negative mutation causing collagen VI-related muscular dystrophy Bolduc V, Sarathy A, Biscans A, Chen G.S, Khvorova A, Bönnemann C.G. Poster #433, Myology 2019
Dans des cellules de DMC liée au gène LM N A La dystrophie musculaire congénitale liée aux lamines A/C (L-DMC) est due à des anomalies du gène LMNA. Les lamines A/C sont des filaments intermédiaires, qui constituent une des composantes de la lamina nucléaire, un réseau fibreux qui double la face interne du noyau. Les lamines A/C jouent un rôle dans la résistance du noyau aux contraintes mécaniques, contribuant au maintien de l’intégrité cellulaire. Elles agissent sur l’expression des gènes via leurs interactions avec la chromatine. ▪ Les anomalies en cause dans la L-DMC entraine la production d’une protéine lamine A/C toxique. Par ailleurs, on sait qu’une insuffisance de production de lamine A/C entraine des pathologies cardiaques isolées. Une thérapie génique empêchant seulement la production de la protéine lamine A/C toxique risque de provoquer une production de lamine A/C restante insuffisante. ▪ C’est pourquoi, une équipe française a expérimenté avec succès dans des modèles cellulaires une technique de thérapie génique qui non seulement empêche l’expresssion de la lamine A/C mutée toxique, mais permet d’augmenter l’expression de la lamine A/C restante. Gene therapy for LMNA-related Congenital Muscular Dystrophy (L-CMD). Brull A, Azibani F, Nelson I, Bonne G, Bertrand A.T. Poster #400, Myology 2019
Dans la DMC1A liée à la laminine α2 La société Santhera Pharmaceuticals a annoncé en mai 2019 sa collaboration avec le Biozentrum de l’Université de Bazel (Suisse) dans un projet de thérapie génique pour la dystrophie musculaire congénitale liée à la lamine α (DMC1A ou mérosinopathie) co-financé par Innosuisse. ▪ L’approche mise au point par le Pr Rüegg du Biozentrum, soutenue par l’AFM-Téléthon, consiste à utiliser deux petites protéines dites « connectrices » qui, en se connectant à la laminine, permettent de restaurer le lien avec la matrice extra-cellulaire. L’expression de ces protéines dans des souris modèles déficientes en laminine α2 a permis de restaurer la structure et la fonction du muscle de ces souris modèle de DMC1A. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Reinhard JR, Lin S, McKee KK, Meinen S, Crosson SC, Sury M, Hobbs S, Maier G, Yurchenco PD, Rüegg MA. Sci Transl Med. 2017 Jun 28;9(396). Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Yurchenco PD, McKee KK, Reinhard JR, Rüegg MA. Matrix Biol. 2018 Oct;71-72:174-187.
La chromatine est une substance contenue dans le noyau des cellules. Elle est constituée à la fois d'ADN (matériel génétique de la cellule) et de protéines qui organisent et protègent cet ADN. Lorsque la cellule se divise, la chromatine se condense en petits bâtonnets : les chromosomes.

Avancées dans les dystrophies musculaires congénitales
18 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
▪ La collaboration de Santhera et du Biozentrum a pour but de réaliser des travaux de recherche préclinique explorant la faisabilité d’une thérapie génique apportant les gènes codant les molécules connectrices pour pallier au dysfonctionnement de la laminine α2. Santhera Starts Collaboration in Gene Therapy Research for Congenital Muscular Dystrophy with the Biozentrum, University of Basel, Co-Financed by Innosuisse. Santhera Pharmaceuticals, Communiqué de presse du 21 mai 2019.
D’autres avancées médico-scientifiques Une stratégie nationale de diagnostic génétique des myopathies En France, avec la création de la « Filière de Santé des Maladies Rares Neuromusculaires » (Filnemus) en 2014 et le second Plan National Maladies Rares, « Structurer le diagnostic génétique » est devenu un enjeu majeur dans les myopathies. La standardisation et l’harmonisation des tests génétiques entre les différents laboratoires de biologie moléculaire permettent d’assurer l'équité des examens pour l’ensemble des patients des différentes régions. ▪ À l’initiative de la Commission « Outils Diagnostiques » de Filnemus, les neuf laboratoires français, spécialisés dans l’analyse génétique par séquençage à haut débit, ont élaboré des listes homogènes de gènes pour le diagnostic génétique des myopathies à l’échelle nationale. Cette liste de 199 gènes a été définie en se basant sur les 13 principaux groupes de myopathies. Pour chaque groupe de maladies, il existe 3 types de listes : la « liste de gènes principale » en première intention, puis la « liste de gènes exhaustive » et enfin la « liste de gènes uniques exhaustive», qui sont à utiliser successivement jusqu’à l’obtention d’un résultat positif. Cette stratégie séquentielle de diagnostic moléculaire est en cours d'adoption par tous les laboratoires participants en France. A National French consensus on gene lists for the diagnosis of myopathies using next-generation sequencing. Krahn M, Biancalana V, Cerino M, Perrin A, Michel-Calemard L, Nectoux J, Leturcq F, Bouchet-Séraphin C, Acquaviva-Bourdain C, Campana-Salort E, Molon A, Urtizberea JA, Audic F, Chabrol B, Pouget J, Froissart R, Melki J, Rendu J, Petit F, Métay C, Seta N, Sternberg D, Fauré J, Cossée M. Eur J Hum Genet. 2019 Mar;27(3):349-352.
Dans les collagénopathies
Rôle du collagène VI dans la jonction neuromusculaire Des anomalies dans l'un des gènes COL6A1, 2 ou 3 sont à l’origine de la dystrophie musculaire congénitale de type Ullrich et de la myopathie de Bethlem. Ces anomalies entraînent une absence ou une fabrication anormale de collagène VI, un des constituants de la matrice extracellulaire qui entoure les fibres musculaires. La fente synaptique de la jonction neuromusculaire (l’espace entre la fin du motoneurone et la membrane de la cellule musculaire) est constituée par une matrice extra-cellulaire hautement spécialisée, qui joue un rôle dans la maturation, la mise en place et la transmission synaptique de la jonction neuromusculaire. ▪ Une équipe italienne a mis en évidence pour la première fois, dans des souris modèles et des cellules de patients atteints de dystrophie musculaire congénitale d’Ullrich, que le collagène VI, lui-même, est un composant de
La matrice extracellulaire est un réseau complexe de protéines dans lequel baignent les cellules.
Elle assure la cohésion des cellules au sein d’un tissu et joue
un rôle essentiel dans la constitution, le maintien,
l'adhérence, le mouvement et la régulation des cellules. La
matrice extracellulaire du muscle est spécialisée pour répondre aux
contraintes mécaniques inhérentes à l'activité contractile
La jonction neuromusculaire est la zone de communication
entre le nerf par qui le signal de contraction (influx nerveux)
arrive et le muscle qui se contracte sous l'impulsion de
l'influx nerveux.
La synapse est la zone de contact entre deux cellules nerveuses ou
entre une cellule nerveuse et une autre cellule (musculaire,
récepteur sensoriel...), par laquelle la cellule en amont (pré-
synaptique) transmet l’influx nerveux à la cellule en aval (post-
synaptique).
Séquencer l'ADN permet de déterminer l'ordre (la séquence)
des nucléotides successifs constituants l'ADN. En comparant
les séquences d'une personne atteinte d'une maladie génétique
et d'une personne indemne, on peut mettre en évidence une
anomalie génétique.

Avancées dans les dystrophies musculaires congénitales
19 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
cette matrice extra-cellulaire et qu’il est nécessaire à l’intégrité structurelle et fonctionnelle de la jonction neuromusculaire. Ces travaux soulèvent la question du rôle des anomalies de la jonction neuromusculaire dans les myopathies liées au collagène VI. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Cescon M, Gregorio I, Eiber N, Borgia D, Fusto A, Sabatelli P, Scorzeto M, Megighian A, Pegoraro E, Hashemolhosseini S, Bonaldo P. Acta Neuropathol. 2018 Sep;136(3):483-499. WEB www.afm-telethon.fr/actualites/dystrophie-musculaire-congenitale-role-collagene-vi-sur-jonction-neuromusculaire-114372
Myopathie de Bethlem : pas de corrélation phénotype/génotype. La myopathie de Bethlem est une forme de myopathie liée au collagène VI de moindre sévérité que la dystrophie musculaire congénitale d’Ullrich. ▪ Une équipe médicale espagnole a fait une étude rétrospective des dossiers de 16 personnes atteintes de myopathie de Bethlem : manifestations de la maladie (signes cliniques, taux de créatine kinase, biopsie musculaire, IRM musculaire) et anomalies génétiques correspondantes. Les signes de la maladie les plus fréquents, mais pas systématiquement présents, ont été une faiblesse musculaire proximale associée à des rétractions des poignets et des doigts. Le taux de CK n’était pas corrélé à la sévérité de la maladie. Parmi les anomalies génétiques retrouvées, la plus fréquente de celles en cause dans la dystrophie musculaire congénitale d’Ullrich (forme congénitale) a été retrouvée dans une myopathie de Bethlem (forme adulte) confirmant l’absence de corrélation pénotype/génotype dans les anomalies du collagène VI. Bethlem myopathy: a series of 16 patients and description of seven new associated mutations. Panadés-de Oliveira L, Rodríguez-López C, Cantero Montenegro D, Marcos Toledano MDM, Fernández-Marmiesse A, Esteban Pérez J, Hernández Lain A, Domínguez-González C. J Neurol. 2019 Apr;266(4):934-941.
L’adiponectine dans une souris modèle de myopathie de Bethlem L’adiponectine est une hormone qui augmente la sensibilité à l’insuline du foie et du muscle squelettique, favorisant la pénétration du glucose dans les cellules hépatiques et musculaires où il est métabolisé. ▪ Les souris modèles de myopathie de Bethlem/dystrophie musculaire congénitale Ullrich ont un taux d’adiponectine plasmatique diminué. La sécretion d’adiponectine par leurs cellules musculaires en cours de développement, les myoblastes, est altérée. ▪ Ces myoblastes Col6a1-/- présentent des anomalies métaboliques (diminution de la captation de glucose, anomalie du potentiel de membrane des mitochondries, diminution de la consommation d’oxygène) qui sont améliorées par l’apport d’adiponectine Role of adiponectin in the metabolism of skeletal muscles in collagen VI-related myopathies. Gamberi T, Magherini F, Mannelli M, Chrisam M, Cescon M, Castagnaro S, Modesti A, Braghetta P, Fiaschi T. J Mol Med (Berl). 2019 Jun;97(6):793-801.
La créatine kinase (CK ou créatine phosphokinase, CPK) est une enzyme musculaire qui joue un rôle dans la production d'énergie directement utilisable par les cellules. Abondamment présente dans les cellules musculaires, elle est libérée dans la circulation sanguine en cas d'atteinte musculaire. Son dosage dans le sang est utile au diagnostic de certaines myopathies.

Avancées dans les dystrophies musculaires congénitales
20 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Dans les mérosinopathies
A la recherche de biomarqueur chez la souris Des chercheurs suédois ont démontré l’existence d’un groupe de miARN urinaires qui sont exprimés de façon spécifique à différents stades de la maladie (avant l’apparition des signes, au début de la maladie et au cours de celle-ci) dans une souris modèle de dystrophie musculaire congénitale liée à la laminine α2 (mérosine). Exploratory Profiling of Urine MicroRNAs in the dy2J/dy2J Mouse Model of LAMA2-CMD: Relation to Disease Progression. Moreira Soares Oliveira B, Gawlik KI, Durbeej M, Holmberg J. PLoS Curr. 2018 Aug 27;10.
Élargissement de l’éventail clinique Une équipe chinoise rapporte l’observation d’une personne présentant des crises d’épilepsie, une faiblesse des muscles proximaux des membres inférieurs peu sévère, une atteinte cognitive modérée et des anomalies marquée à l’imagerie par résonance magnétique (IRM) cérébrale, dues à des anomalies du gène LAMA2. Les auteurs concluent que ces anomalies génétiques pourraient être plus particulièrement en cause dans les atteintes cognitives des mérosinopathies. Missense mutations in LAMA2 causing a new phenotype of mild cognitive impairment, proximal myopathy, seizure, and severe leukoencephalopathy: A case report and protein analysis. Ding M, Wang X, Zeng Y, Lu Z, Cai S, Gao M, Zhu W, Luo S, Zhao C, Xiao Z. Clin Neuropathol. 2019 May/Jun;38(3):100-108.
Dans les dystroglycanopathies
Dystroglycanopathies liées à POMT1 : anomalies génétiques en cause et modèles animaux Afin de mieux comprendre, de mieux classer et d’améliorer le diagnostic des tableaux cliniques complexes des dystroglycanopathies dues à des anomalies du gène POMT1 (formes congénitales, formes adultes des ceintures, avec ou sans atteinte cérébrale ou oculaire...), des chercheurs chinois ont réalisé et publié une synthèse des connaissances sur les mutations en cause et les différents modèles animaux de ces maladies. Molecular genetics of the POMT1-related muscular dystrophy-dystroglycanopathies. Hu P, Yuan L, Deng H. Mutat Res. 2018 Oct - Dec;778:45-50.
Anomalies des dystroglycanes selon les anomalies du gène DAG1 Comparée aux fréquentes anomalies de la glycosylation de l’α-dystroglycane, les anomalies génétiques touchant directement le gène DAG1, qui code cette protéine, sont rarement en cause dans les α-dystroglycanopathies. On parle alors de dystroglycanopathies primaires. ▪ Un chercheur italien a étudié les conséquences des différentes anomalies du gène connues sur la protéine et son fonctionnement : certaines anomalies génétiques entrainent une déstabilisation de la protéine produite mais ne l’empêche pas d’ête bien localisée ; d’autres empêchent la maturation de la protéine conduisant à la destruction totale ou partielle de la protéine immature instable. A molecular overview of the primary dystroglycanopathies. Brancaccio A. J Cell Mol Med. 2019 May;23(5):3058-3062.
Les micro-ARN (miARN) sont des petits ARN produits par la cellule qui ne sont pas traduits
en protéine. Leur rôle est de réguler l’expression de gènes en bloquant la traduction de l'ARN
messager de ces derniers en protéine. L'expression de ces miARN varie en fonction des situations. Dans les maladies
neuromusculaires, certains miARN sont exprimés et pas
d'autres, et la combinaison des miARN exprimés est différente
d'une maladie neuromusculaire à l'autre et spécifique de
chacune.
Les muscles proximaux sont les muscles qui sont proches de l'axe
du corps. Ils sont situés aux racines des membres : muscles
des épaules et des bras pour les membres supérieurs, muscles des
hanches et des cuisses pour les membres inférieurs.

Avancées dans les dystrophies musculaires congénitales
21 ǀ AFMTéléthon ǀ Juin 2019
Savoir & Comprendre
Dans la DMC mégaconiale
Une membrane des mitochondries anormale Une équipe italienne décrit dans un article paru en avril 2019 des anomalies de la membrane interne des mitochondries dans des cellules issues de personnes atteintes de dystrophie musculaire congénitale liée à des anomalies du gène CHKB non décrites jusqu’à présent. Ces anomalies de la membrane interne des mitochondries seraient à l’origine de l’aspect anormal particulier des mitochondries dans la dystrophie musculaire congénitale mégaconiale. Alteration of mitochondrial membrane inner potential in three Italian patients with megaconial congenital muscular dystrophy carrying new mutations in CHKB gene. Marchet S, Invernizzi F, Blasevich F, Bruno V, Dusi S, Venco P, Fiorillo C, Baranello G, Pallotti F, Lamantea E, Mora M, Tiranti V, Lamperti C. Mitochondrion. 2019 Apr 12;47:24-29.
* * *
▪ Tout au long de l'année, suivez l'actualité de la recherche dans les maladies neuromusculaires sur le site de l’AFM-Téléthon : WEB www.afm-telethon.fr > Voir toutes les Actus > Maladies





























