Embed Size (px)
Citation preview

N° d’ordre : 2008-ISAL-0056 Année 2008
THESE présentée devant
L’INSTITUT NATIONAL DES SCIENCES APPLIQUEES de LYON
pour l’obtention du
DIPLOME DE DOCTORAT
Ecole Doctorale de Chimie de Lyon
Spécialité Chimie
Soutenue publiquement le 26 septembre 2008
par
Rouba CHEAIB
LES CARBOXYMETHYL GLYCOSIDES LACTONES : SYNTHESE ET APPLICATION A L’IMAGERIE
MEMBRANAIRE
Directeur de thèse : Dr Yves QUENEAU
JURY : M. Benoît JOSEPH, Professeur à l’Université Claude Bernard Lyon 1 Président Mme Amélia RAUTER, Professeur à l’Université de Lisbonne Rapporteur M. Pierre KRAUSZ, Professeur à l’Université de Limoges Rapporteur M. Yann BRETONNIERE, chargé de recherche à l’Ecole Normale Supérieure de Lyon
Examinateur M. Stéphane CHAMBERT, maître de conférences à l’INSA de Lyon Examinateur M. Yves QUENEAU, directeur de recherche au CNRS, INSA de Lyon Examinateur


REMERCIEMENTS
Ce travail de recherche a été réalisé au Laboratoire de Chimie Organique de l’INSA de Lyon sous la
direction du docteur Yves QUENEAU. Je tiens à le remercier pour m’avoir accueillie dans son équipe et
pour m’avoir guidé tout au long de ces trois années. Je remercie le « Cluster Chimie » de la Région Rhône-
Alpes pour avoir financé ce travail de recherche durant ces trois ans.
Je remercie Monsieur Stéphane CHAMBERT, Maître de conférences à l’INSA de Lyon pour son
encadrement durant ces trois ans de thèse.
Je remercie également Monsieur le Professeur Alain DOUTHEAU, directeur du Laboratoire de Chimie
Organique pour sa sympathie, Monsieur Laurent SOULERE, ainsi que Monsieur Arkadiusz
LISTKOWSKI, post-doctorant au laboratoire, et tout le personnel du LCO-INSA.
Je remercie vivement toutes les personnes avec qui j’ai collaboré, Monsieur Yann BRETONNIERE et
Madame le Professeur Chantal ANDRAUD, du Laboratoire de Chimie pour l’Optique à l’Ecole Normale
Supérieure de Lyon. J’adresse mes sincères remerciements à Madame Agnès GIRARD-EGROT et
Mademoiselle Aurélie SANTAFE de m’avoir si chaleureusement accueillie dans le Laboratoire de Génie
Enzymatique et Biomoléculaire, et qui m’ont aidée à réaliser les expériences sur monocouche de Langmuir.
Je tiens à remercier Monsieur Sébastien VIDAL pour sa gentillesse ainsi que tous les chercheurs du
Laboratoire de Chimie Organique II et du Laboratoire de Synthèse Asymétrique que j’ai connus durant ces
quatre ans.
Je remercie Madame le Professeur Amélia RAUTER de l’Université de Lisbonne et Monsieur le
Professeur Pierre KRAUSZ de l’Université de Limoges pour avoir jugé ce travail, ainsi que Monsieur le
Professeur Benoît JOSEPH pour avoir accepté de faire partie du jury.
Je remercie profondément mes parents pour leur amour inconditionnel et pour leur soutien sans faille,
ainsi que Dima ma sœur et la petite princesse Nouna. Je remercie surtout Nader, mon frère, pour nos
souvenirs communs à Lyon.
Je remercie Monsieur Francis MAQUEDA dont la présence était un privilège et un grand soutien
moral.

Je remercie tous les amis que j’ai rencontrés en France et que j’ai appris à apprécier. Je remercie aussi
Aurélie Assalit pour son amitié et son agréable compagnie. Je remercie Momo, Mazen Sleiman et Dina pour
les moments sympathiques que nous avons partagés ensemble, je remercie Jana pour nos discussions
passionnées et épuisantes, Hicham, pour son goût du luxe et de la bonne gastronomie.
Finalement, un grand merci du fond du cœur à Mazen.

Abréviations
Ac : acétyle
ADN : acide désoxyribonucléique
ANEP : Aminonaphthyl éthényl pyridinium
ARN : acide ribonucléique
BAM: Brewster angle microscopy
BHL: Balance hydrophile-lipophile
Bn: benzyle
CCM : chromatographie sur couche mince
CMC : concentration micellaire critique
CMG : carboxyméthyl glycoside
CMGal : carboxyméthyl galactoside
CMGalL : carboxyméthyl galactoside lactone
CMGL : carboxyméthyl glycoside lactone
CMGlc : carboxyméthyl glucoside
CMGlcL : carboxyméthyl glucoside lactone
CMLacL : carboxyméthyl lactoside lactone
CMMan : carboxyméthyl mannoside
1

CMManL : carboxyméthyl mannoside lactone
COSY: Correlated Spectroscopy
DEPT: Distortionless Enhancement by Polarization Transfer
DMF: N,N-diméthylformamide
DMSO: diméthylsulfoxide
DPPC: 2-dipalmitoyl-3-glycéro-3-phosphocholine
EEDQ: N-éthoxycarbonyl-2-éthoxy-1,2-dihydroquinoline
Et3N : triéthylamine
Gal : galactose
Glc : glucose
GSH : génération du second harmonique
HMBC : Heteronuclear Multiple Bond Correlation
IC50 : concentration de l’inhibition à 50% du maximum d’inhibition observée
LC : liquide condensé
LE : liquide expansé
OGT: O-N-acétyl-glucosaminyltransférase
ONL: optique non linéaire
PEG : Polyéthylène glycol
PMB : para-méthoxybenzylidène
Py : pyridine
Rdt : rendement
RMN : résonance magnétique nucléaire
SM: spectrométrie de masse
t-Bu: tert-butyle
THF: tetrahydrofurane
TFA: acide trifluoroacétique
2

TPEF: two-photon excitation fluorescence
UDP-GlcNAc: uridine diphosphate N-acétyl-glucosamine
UV : ultra-violet
VIH : virus de l'immunodéficience humaine
3

4

Sommaire
ABREVIATIONS....................................................................................................................................... 1
SOMMAIRE............................................................................................................................................... 5
INTRODUCTION...................................................................................................................................... 9
PREMIERE PARTIE: BIBLIOGRAPHIE……………………………………………
CHAPITRE I. LES CARBOXYMETHYL GLYCOSIDES (CMGS) ET LES
CARBOXYMETHYL GLYCOSIDES LACTONES (CMGLS) .................................................................. 11
I.1. INTRODUCTION............................................................................................................................ 11 I.1.1. Synthèse de la CMGlcL..................................................................................................... 11 I.1.2. Nomenclature.................................................................................................................... 12
I.2. SYNTHESE DES CARBOXYMETHYL GLYCOSIDES (CMGS) ............................................................ 12 I.2.1. Synthèse des α-CMGs ....................................................................................................... 12 I.2.2. Synthèse des α,β-CMGs.................................................................................................... 13 I.2.3. Synthèse des β-CMGs ....................................................................................................... 16
I.3. EXPLOITATION DES CARBOXYMETHYL GLYCOSIDES (CMGS) ET DES CARBOXYMETHYL
GLYCOSIDES LACTONES (CMGLS) ................................................................................................................. 19 I.3.1. Exploitation des carboxyméthyl glycosides (CMGs)......................................................... 19 I.3.2. Exploitation de la carboxyméthyl glucoside lactone (CMGlcL) ....................................... 23
CHAPITRE II. LES SCAFFOLDS SUCRES.................................................................................... 29
II.1. SCAFFOLD, DEFINITION, HISTORIQUE...................................................................................... 29 II.2. QUELQUES RESULTATS BIOLOGIQUES..................................................................................... 30 II.3. SYNTHESE............................................................................................................................... 35
II.3.1. Scaffolds pyraniques ......................................................................................................... 35 II.3.2. Scaffolds furaniques.......................................................................................................... 41 II.3.3. Scaffolds iminosucres........................................................................................................ 42
5

II.3.4. Scaffolds disaccharidiques................................................................................................ 45
CHAPITRE III. IMAGERIE MEMBRANAIRE PAR OPTIQUE NON LINEAIRE................... 49
III.1. POTENTIEL MEMBRANAIRE EN NEUROBIOLOGIE ..................................................................... 49 III.2. FLUORESCENCE PAR EXCITATION A DEUX PHOTONS (TPEF) ET GENERATION DU SECOND
HARMONIQUE (GSH) POUR LA MESURE DU POTENTIEL MEMBRANAIRE.......................................................... 50 III.2.1. Principe physique......................................................................................................... 50 III.2.2. Avantages de la TPEF et de la GSH en microscopie biologique ................................. 51 III.2.3. Principe de mesure du potentiel membranaire par ONL ............................................. 53 III.2.4. Sondes utilisées pour la mesure du potentiel membranaire ......................................... 54
III.3. INGENIERIE MOLECULAIRE POUR LA SYNTHESE DE NOUVELLES SONDES OPTIQUES
MEMBRANAIRES ............................................................................................................................................. 55 III.3.1. La membrane cellulaire................................................................................................ 55 III.3.2. Les sucres dans l’étude des membranes ....................................................................... 56 III.3.3. Les sucres dans l’optique non linéaire ......................................................................... 57
DEUXIEME PARTIE: RESULTATS ET DISCUSSION…………………………..
CHAPITRE I. SYNTHESE DES CMGS ET DES CMGLS ............................................................ 61
I.1. INTRODUCTION............................................................................................................................ 61 I.2. L’ALKYLATION ANOMERIQUE ..................................................................................................... 62
I.2.1. Synthèse des esters de tert-butyle glycosides .................................................................... 62 I.2.2. Commentaires sur la sélectivité α/β lors de l’alkylation anomérique .............................. 74 I.2.3. Déprotection de l’acide carboxylique ............................................................................... 76 I.2.4. Applications pour la synthèse de glycoclusters persulfurés.............................................. 79 I.2.5. Synthèse des lactones ........................................................................................................ 81
I.3. LACTONES BENZYLEES (GLC/GAL)............................................................................................... 85 I.3.1. Synthèse de l’anomère β (glc/gal)..................................................................................... 85 I.3.2. Synthèse de l’anomère α (glc/gal) .................................................................................... 86
I.4. CONCLUSION ............................................................................................................................... 88
CHAPITRE II. SYNTHESE DE SYSTEMES MULTIFONCTIONNELS .................................... 89
II.1. INTRODUCTION ....................................................................................................................... 89 II.2. L’OUVERTURE DES CARBOXYMETHYL GLYCOSIDES LACTONES .............................................. 90 II.3. FONCTIONNALISATION EN POSITION 2 .................................................................................... 91
II.3.1. Carbamatation .................................................................................................................. 91 II.3.2. Evaluation biologique ....................................................................................................... 95 II.3.3. Ethérification .................................................................................................................... 97 II.3.4. Azidation ........................................................................................................................... 98
II.4. FONCTIONNALISATION EN POSITION 6 .................................................................................. 100
6

II.5. CONCLUSION ........................................................................................................................ 101
CHAPITRE III. SYNTHESE DE SONDES FLUORESCENTES POUR L’IMAGERIE
MEMBRANAIRE NEURONALE ................................................................................................................ 103
III.1. INTRODUCTION ..................................................................................................................... 103 III.2. LE CHROMOPHORE................................................................................................................ 104 III.3. STRUCTURE DES SONDES MEMBRANAIRES ............................................................................ 105 III.4. SYNTHESE DES SONDES GLYCOSYLEES ................................................................................. 106 III.5. PROPRIETES SPECTROSCOPIQUES .......................................................................................... 109
III.5.1. Effet solvatochromique............................................................................................... 109 III.5.2. Le rendement quantique de fluorescence ................................................................... 112
III.6. EVALUATION EN IMAGERIE................................................................................................... 114 III.6.1. Imagerie par TPEF des sondes .................................................................................. 115 III.6.2. GSH de la sonde bis-glucosylée ................................................................................. 120
III.7. CONCLUSION ........................................................................................................................ 122
CHAPITRE IV. ETUDE DE L’INSERTION DANS UNE MONOCOUCHE DE LANGMUIR 125
IV.1. TECHNIQUE DES MONOCOUCHES DE LANGMUIR ................................................................... 125 IV.1.1. Principe de la technique des monocouches de Langmuir........................................... 126
IV.2. MICROSCOPIE A L’ANGLE DE BREWSTER (BAM) ................................................................. 129 IV.2.1. Principe ...................................................................................................................... 129 IV.2.2. Montage expérimental................................................................................................ 130
IV.3. ETUDE DE LA QUALITE D’INSERTION..................................................................................... 130 IV.3.1. Conditions expérimentales ......................................................................................... 131 IV.3.2. Insertion de la sonde à tête lactose et chaîne éthyle (192)......................................... 132 IV.3.3. Insertion de la sonde à tête lactose et chaîne octyle (193)......................................... 137 IV.3.4. Insertion de la sonde à tête glucose et chaîne éthyle (190) ........................................ 138 IV.3.5. Insertion de la sonde à tête glucose et chaîne octyle (191) ........................................ 140
IV.4. CONCLUSION ........................................................................................................................ 141
CONCLUSION GENERALE ET PERSPECTIVES.......................................................................... 143
TROISIEME PARTIE: PARTIE EXPERIMENTALE…………………………………………….145
REFERENCES BIBLIOGRAPHIQUES……………………………………………...…………….203
7

8

Introduction
Grâce à leur diversité structurale et la maîtrise de leur chimie, les sucres sont
abondamment impliqués dans la synthèse d’architectures moléculaires très variées. Leur
importance dans le domaine biologique a entraîné un fort intérêt pour la synthèse de
glycoconjugués et de leurs analogues. D’autre part, la présence de groupements hydroxyles
confère une grande solubilité à des adduits hydrophobes en milieu aqueux, permettant l’accès à
de nouveaux amphiphiles intéressants dans le domaine industriel. Enfin, la rigidité de leur
structure permet l’utilisation des sucres comme châssis moléculaire (scaffolds) sur lesquels des
chaînes latérales peuvent être greffées et orientées d’une façon définie dans l’espace.
Depuis quelques années, il a été montré que la carboxyméthyl 3,4,6-tri-O-acétyl-α-D-
glucopyranoside 2-O-lactone (α-CMGlcL), préparée à partir de l’isomaltulose, est un outil
intéressant pour la synthèse de composés ciblés soit pour leur intérêt biologique soit pour leurs
propriétés physico-chimiques potentielles, tels que des pseudo-oligosaccharides, pseudo-
glycoaminoacides, ou pseudo-glycolipides.
La limitation de cette stratégie est que l’isomaltulose fournit uniquement le lien α-
glucosyle. Dans le but d’élargir l’intérêt de telles lactones, une partie de ce travail a été
consacrée à la mise au point de nouveaux chemins réactionnels pour l’obtention des lactones
synthétisées sur des systèmes mono- ou disaccharidiques, avec les deux configurations
possibles du centre anomérique et présentant des groupements protecteurs différents.
En second lieu, nous avons exploré un autre aspect lié à l’ouverture de ces lactones qui est
la libération de la position 2 alors que les autres groupes hydroxyles restent protégés. Cela nous
a permis de synthétiser des molécules multifonctionnelles par exploitation de l’hydroxyle en
position 2 et de la position hydroxyle primaire en position 6.
9

Enfin, nous avons exploité la stratégie CMGL pour la synthèse de produits amphiphiles
dans un contexte particulier de la conception de sondes membranaires hydrosolubles, dans le
cadre d’une collaboration engagée entre le Laboratoire de Chimie Organique de l’INSA de
Lyon, le Laboratoire de Chimie pour l’Optique de l’Ecole Normale Supérieure de Lyon et le
Laboratoire de Spectrométrie Physique de l’Université Joseph Fourier de Grenoble pour la
synthèse de sondes pour l’imagerie membranaire par microscopie optique non linéaire.
10

PREMIERE PARTIE
BIBLIOGRAPHIE

Bibliographie - Chapitre I
CHAPITRE I. LES CARBOXYMETHYL GLYCOSIDES
(CMGS) ET LES CARBOXYMETHYL GLYCOSIDES
LACTONES (CMGLS)
I.1. INTRODUCTION
I.1.1. SYNTHESE DE LA CMGLCL
L’isomaltulose est un sucre très disponible obtenu à l’échelle industrielle par
bioconversion du saccharose.1 La dégradation de ce dernier par oxydation réalisée par le
peroxyde d’hydrogène en milieu acide, aboutit à la formation du carboxyméthyl α-D-
glucoside (CMGlc). L’acétylation du CMGlc par l’anhydride acétique dans la pyridine a
fourni un nouveau produit qui a été identifié comme étant la lactone triacétylée (1a, schéma
1).2
OAcO
OO
O
OAc
AcOOHOHO
HOO
OH
O
OH
OHO
HOO
OH
HO
O
HO
HOOH
OH
isomaltulose
H2O2, H+
pH=2, 80°C
Ac2O, Py
22-25% sur les2 étapes
α-CMGlc α-CMGlcL, 1a
Schéma 1. Synthèse de la CMGlcL à partir de l’isomaltulose
1 (a) Weidenhagen, R.; Lorenz, S. Angew. Chem., 1957, 69, 641-641; (b) Weidenhagen, R.; Lorenz, S. Ger. Pat. DE 1,049,800, 1957, Chem. Abstr. 1961, 55, 2030b; (c) Schiweck, H.; Munir, M.; Rapp, K. M.; Schneider, B.; Vogel, M. in Carbohydrates as Organic Raw Materials, Lichtenthaler, F. W., Ed., VCH: Weinheim, 1991, Vol. 1, p. 57-94. 2 (a) Trombotto, S.; Bouchu, A; Descotes G.; Queneau, Y. Tetrahedron Lett. 2000, 41, 8273-8277. (b) Queneau, Y.; Chambert, S.; Cheaib, R.; Listkowski, A.; Doutheau, A.; Trombotto, S. Targets in Heterocyclic Systems – Chemistry and Properties, O. A. Attanasi and Spinelli Eds., Italian Society of Chemistry: Rome, 2006, Vol. 10, p.132-151.
11

Bibliographie - Chapitre I
Cette lactone est obtenue après formation d’un anhydride mixte comme intermédiaire
suivie d’une cyclisation par attaque intramoléculaire du groupe hydroxyle en position 2. La
synthèse de cette lactone n’avait jamais été décrite mais un système similaire avait été suggéré
comme intermédiaire dans le cas du β-lactoside.3 En revanche, pour ce qui concerne les
acides caboxyliques du type carboxyméthyl glycoside, plusieurs méthodes décrivent leur
synthèse dans la littérature. L’exposé de ces méthodes fera l’objet de la section I.2.
I.1.2. NOMENCLATURE
Dans ce document, nous avons adopté la nomenclature carboxyméthyl
glycosides/carboxyméthyl glycosides lactones en estimant que la fonctionnalité glycoside est
la fonctionnalité principale. Pour les termes se rapportant aux glycosides acétylés en général,
nous attribuons les termes CMGs et CMGLs. Quand il s’agit d’un sucre particulier, nous
ajouterons un indice se rapportant au sucre (CMGlc et CMGlcL pour le carboxyméthyl
glucoside et sa lactone respectivement, CMGal et CMGalL pour le carboxyméthyl galactoside
et sa lactone respectivement etc…).
Pour ce qui concerne les carboxyméthyl glycosides, ils sont trouvés dans la littérature
sous d’autres noms tels que les O-glycosyl glycolic acid (GGA) ou de 2-(glycosyloxy) acetic
acid (GAA).
I.2. SYNTHESE DES CARBOXYMETHYL GLYCOSIDES (CMGS)
I.2.1. SYNTHESE DES α-CMGS
La seule synthèse dans la littérature qui décrit la formation directe du α-CMGlc est la
voie d’oxydation de l’isomaltulose1 (schéma 2) dans le cadre de l’étude de la dégradation du
sucre par le peroxyde d’hydrogène. Ainsi, l’isomaltulose est traité par un excès de peroxyde
d’hydrogène à pH=4 à 80°C pour donner le CMGlc avec un rendement de 23%. En rajoutant
du tungstate de sodium à la réaction, le rendement est amélioré (38%). Le meilleur rendement
est obtenu quand la réaction se déroule à pH=2 pour donner le CMGlc avec 42% de
rendement. La réaction peut aussi s’effectuer en milieu basique et à température ambiante
mais la formation du α-CMGlc s’est avérée très lente avec un rendement inférieur à 15%.
3 Dean, B.; Oguchi, H.; Cai, S.; Otsuji, E.; Tashiro, K.; Hakomori, S.I.; Toyokuni, T. Carbohydr. Res., 1993, 245, 175-192.
12

Bibliographie - Chapitre I
O
OHOHO
HO
HO
CO2H
Glc
O CO2HOH
OH
OH
Glc
O
HO2C CO2H
O OHOH
Glc
OCO2H
OH
OH
Glc
OCO2HOH
OHOHO
HO
OH
O O
OH
OH OH
OH
i .i ii.
i i.
i i.
i i.
i i.H2O2, H+
pH=2, 80°C
Schéma 2. i. Clivage glycolique, ii. Oxydation-décarboxylation, iii. Hydrolyse
I.2.2. SYNTHESE DES α,β-CMGS
I.2.2.1. Par glycosidation avec un composé présentant une fonction acide
masquée
Petersson et al.4 ont montré une préparation directe du CMGlc par évaporation sous
pression réduite à 35°C d'une solution de glucose et d'acide glycolique, en présence d'acide
chlorhydrique (schéma 3). Le CMGLc est obtenu avec un rendement de 6% sous la forme
d'un mélange d'isomères α et β avec une sélectivité en faveur de l'isomère α (α/β : 7/3).
OHOHO
HO
OH
O
OH
OHOHO
HO
OH
OH O
HOCH2COOH, HCl
35°C, pression réduite,6%
Schéma 3. Synthèse du mélange α,β-CMGLC par glycosylation de l’alcool glycolique
4 Petersson, G.; Pettersson, S.; Samuelson, O. Sv. Papperstidn. 1969, 72, 222-225 (Chem. Abstr. 1969, 71, 23035v).
13

Bibliographie - Chapitre I
Une synthèse décrite par Mandai et al.5 permet l’accès à des dérivés tétracétylés et
tétrabenzylés du CMGlc sous la forme d’un mélange d’anomères par glycosidation du sucre
avec des esters d’acide glycolique suivie d’une hydrolyse en milieu basique comme le montre
le schéma 4. Cette stratégie a été reprise par Nicolaou et al.6 par glycosidation d’un composé
fluoré avec un glycolate d’éthyle en présence de sel d’argent pour donner le produit sous la
forme d’un mélange d’anomères avec un rendement de 55% (α/β : 2/3).
BnOO
BnOBnO
OBn
BnOO
BnOBnO
OBn
O OEt
O
BnOO
BnOBnO
OBn
O OH
OOMe
1. H2SO4, HOAc,100°C, 3h, 85%
2. HOCH2CO2Et,TSOH, toluène
KOH, 75%
Schéma 4. Synthèse de α,β-CMGlc par glycosylation d’un ester d’acide glycolique
Kessler et al.7 ont repris cette méthode en utilisant un alcool comportant une fonction
aldéhyde masquée sous la forme d’un diméthyl acétal. L’aldéhyde est ensuite oxydé pour
conduire au CMMan correspondant.
A partir du thioéthyl mannoside et selon les conditions opératoires, ils ont obtenus des
mélanges α/β avec des proportions différentes. Le clivage des acétals est ensuite effectué en
milieu acide. La fonction acide est enfin obtenue par oxydation de la fonction carbonyle pour
donner le CMMan correspondant (schéma 5).
5 (a) Mandai, T.; Okumoto, H.; Oshitari, T.; Nakanishi, K.; Mikuni, K.; Hara, K. J.; Hara, K. Z.; Iwatani, W.; Amano, T.; Nakamura, K.; Tsuchiya, Y Heterocycles 2001, 54, 561-566. (b) Mandai, T.; Okumoto, H.; Oshitari, T.; Nakanishi, K.; Mikuni, K.; Hara, K. J.; Hara, K. Z. Heterocycles, 2000, 52, 129-132. 6 Nicolaou, K. C.; Trujillo, J. I.; Chibale, K. Tetrahedron, 1997, 53, 8751-8778. 7 (a) Boer, J.; Schuster, A.; Holzmann, B.; Kessler, H. Angew. Chem. Int. Ed. 2001, 40, 3870-387. (b) Locardi, E.; Boer, J.; Modlinger, A.; Schuster, A.; Holzmann, B.; Kessler, H. J. Med. Chem. 2003, 46, 5752-5762.
14

Bibliographie - Chapitre I
OMeO
OR1
OR2
MeO
SEt
OMeO
OR1
OR2
MeOOCH2CH(OMe)2
OMeO
OR1
OR2
MeOOCH2COOH
tBuOH,
OMeO
OR1
OR2
MeO
OCH2CH(OMe)2
1. Br22. HOCH2CH(OMe)2,
Ag2CO3,(α/β : 1/5), 75%
HOCH2CH(OMe)2,NIS, AgOTf,
(α/β: 6/1), 80%
NaClO2
Schéma 5. Synthèse de α,β-CMMan à partir d’un thioéthyl glycoside
I.2.2.2. Par oxydation d’allyl glycoside
L’oxydation de l’allyl glycoside a été une stratégie largement adoptée pour l’accès aux
CMGs. Les allyl glycosides sont obtenus par glycosylation de l’alcool allylique par le sucre
libre par exemple, en présence de l’étherate de trifluorure de bore8 sous la forme d’un
mélange d’anomères α,β. Le passage à l’acide s’effectue par oxydation de la double liaison
soit par RuCl3-NaIO49,10, , ,11 12 13développée par Sharpless et al.14 soit par une bishydroxylation
suivie d’un clivage glycolique par le système OsO4 4-NaIO ,15 KMnO4-NaIO45,16 soit par
ozonolyse.17, 18 Par ces méthodes, les rendements varient de 39 à 99%.
8 Tronchet, J. M. J.; Zsély, M.; Geoffroy, M Carbohydr. Res. 1995, 275, 245-258. 9 Ghosh, M.; Dulina, R. G.; Kalarda. R.; Sofia, M. J. J. Org. Chem. 2000, 65, 8387-8390. 10 Westermann, B.; Dörner, S. Chem. Commun. 2005, 2116-2118. 11 Grandjean, C.; Santraine, V.; Fardel, N.; Polidori, A.; Pucci, B.; Gras-Masse, H.; Bonnet, D. Tetrahedron Lett. 2004, 45, 3451-3454. 12 Virta, P.; Karskela, M.; Lonnberg, H. J. Org. Chem. 2006, 71, 1989-1999. 13 Mogemark, M.; Elofsson, M.; Kihlberg, J. J. Org. Chem. 2003, 68, 7281-7288. 14 Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936-3938. 15 Santacroce, B. V.; Basu, A. Angew. Chem. Int. Ed. 2003, 42, 95-98. 16 Binder, W. H.; Schmid, W. Monatsch. Chem., 1995, 126, 923-931. 17 Lockhoff, O. Angew. Chem. Int. Ed. Engl. 1998, 37, 3436-3439. 18 Santacroce, B. V.; Basu, A. Angew. Chem. Int. Ed. 2003, 42, 95-98.
15

Bibliographie - Chapitre I
ORORO
RO OO
OH
OR
ORORO
OR
OR
O
RuCl3,NaIO4
ou O3ou RuCl3,KMnO4
39-99%
Schéma 6. Synthèse des α,β-CMGs par oxydation de l’allyl glycoside
I.2.3. SYNTHESE DES β-CMGS
I.2.3.1. Par glycosidation avec un composé présentant une fonction acide
masquée
Plusieurs méthodes ont été décrites dans la littérature pour la préparation du β-
CMGlc. La sélectivité de cette réaction vient de la participation du groupement acétate en
position 2 qui favorise l’approche du nucléophile en β.
La réaction de Fischer et Helferich en 1911 (schéma 7) décrit la synthèse du CMGlc par
réaction de l’acétobromoglucose avec le glycolate d’éthyle suivie d’une hydrolyse basique de
l’ester.19 Cette même réaction a été reprise en 2003 pour la synthèse de carboxyméthyl
cellobioside.20
AcOO
AcOAcO
OAc
AcOO
AcOAcO
OAc
Br
OOEt
OAcO
OAcO
AcO
OAc
OOH
OHOCH2COOEt,60%
NaOH,70%
Schéma 7. Synthèse du β-CMGlc par glycosylation d’un alcool glycolique
Lockhoff et al.17 ont proposé la synthèse du dérivé perbenzylé du β-CMGlc à partir du
tétra-O-benzyl D-glucopyranose par formation d’un intermédiaire trichloroacétimidate
(schéma 8). Une glycosidation de ce dernier avec le glycolate d’éthyle suivie d’une
19 Fischer, E.; Helferich, B. Liebigs Ann. Chem. 1911, 383, 68-91. 20 Choudhury, A. K.; Kitaoka, M.; Hayashi, K. Eur. J. Org. Chem. 2003, 2462-2470.
16

Bibliographie - Chapitre I
saponification conduit au β-CMGlc perbenzylé avec un bon rendement. Cette stratégie via
l’intermédiaire trichloroacétimidate a été appliquée pour la synthèse de carboxyméthyl
disaccharides d’anomérie β comme le montrent les travaux de Pieters et al.21,22 et ceux de
Toyokuni.3
BnOO
BnOBnO
OBn
BnOO
BnOBnO
OBn
OOH
OOCCCl3
1. HOCH2CO2Et,BF3.OEt2, THF,
82%2. puis NaOH,MeOH, THF,
72%.
NH
Schéma 8. Synthèse du β-CMGlc à partir d’un trichloroacétimidate glycosyle
I.2.3.2. Par oxydation d’allyl glycoside
Comme précédemment signalé pour le mélange α/β, le β-CMGlc a aussi été synthétisé
par oxydation d’allyl glucoside.
A partir du bromure de glucosyle, Dörner10 a synthétisé l’allyl glucoside de
configuration anomérique β (schéma 9).
AcOO
AcOTFAHN
NHTFA
AcOO
AcOTFAHN
NHTFA
Br
O AcOO
AcOTFAHN
NHTFA
OOH
ONaIO4, RuCl3,70%
AllOH, AgOTf,40%
Schéma 9. Synthèse du β-CMGlc par oxydation de la double liaison de l’allyl glycoside
De plus, Binder et al.17 ont décrit la synthèse de l’allyl glucoside à partir du β-D-glucose
pentaacétylé. Après oxydation, le β-CMGlc est obtenu avec un rendement de 23% (schéma
21 Autar, R.; Liskamp, R. M. J.; Pieters, R. J. Carbohydr. Res. 2005, 340, 2436-2442. 22 Autar, R.; Khan, A. S.; Schad, M.; Hacker, J.; Liskamp, R. M. J.; Pieters, R. J. ChemBioChem. 2003, 4, 1317-1325.
17

Bibliographie - Chapitre I
10). Cette stratégie a été reprise par les équipes d’Overkleeft et van Boom pour la synthèse de
carboxyméthyl β-L-fucoside et carboxyméthyl 2-amino-2-désoxy-β-D-glucoside.23
AcOO
AcOAcO
OAc
AcOO
AcOAcO
OAcNaIO4/KMnO4,
39%O AcO
OAcO
AcO
OAc
OOH
OOAc
AllOHSnCl4, 60%
Schéma 10. Synthèse du β-CMGlc par oxydation de la double liaison de l’allyl glucoside
I.2.3.3. Par dégradation du gentiobiose
Lors de l’étude de la dégradation du gentiobiose par une solution aqueuse de peroxyde
de sodium (schéma 11), Isbell et al.24 ont montré la formation du β-CMGlc. Cependant, aucun
rendement n’est décrit.
HOO
HOOH
OH
HOO
HOOH
OH
OOH
OO
HOO
HOOH OH
Na2O2 aq.4°C, 5 jours
Schéma 11. Synthèse du β-CMGlc par oxydation de la double liaison de l’allyl glucoside
23 Filippov, D. V.; van den Elst, H.; Tromp, C. M.; van der Marel, G. A.; van Boeckel, C. A. A., Overkleeft, H. S.; van Boom, J. H. Synlett. 2004, 773-778. 24 Isbell, H. S.; Frush, H. L. Carbohydr. Res. 1987, 161, 181-193.
18

Bibliographie - Chapitre I
I.3. EXPLOITATION DES CARBOXYMETHYL GLYCOSIDES (CMGS) ET DES
CARBOXYMETHYL GLYCOSIDES LACTONES (CMGLS)
I.3.1. EXPLOITATION DES CARBOXYMETHYL GLYCOSIDES (CMGS)
I.3.1.1. En tant que molécule cible
Le schéma 12 montre différents composés (2-12) possédant une fonction
carboxyméthyle synthétisés pour leurs activités biologiques.
Par exemple, dans le domaine peptidomimétique, Nicolaou et al.6 ont décrit la synthèse
en phase solide d’un dérivé du mannose où la fonction carboxyméthyle est localisée soit en
position 1 (2) soit en position 2 du sucre (3). Plus récemment, l’équipe de Kessler a synthétisé
des scaffolds (4, 5) basés sur une structure β-D-mannopyranose.7 L’équipe de Sofia a
synthétisé des carboxyméthyl glycosides construits sur un squelette N-acétyl-glucosamine9
(6). De même, les groupes de Jiménez-Barbero et Nicotra25 ont proposé des systèmes
furaniques bicycliques (7) à partir de la D-ribonolactone. Billington et al.26 ont synthétisé des
glycosides avec une fonction carboxyméthyle en 4 (8) pour mimer un antagoniste de
l’endothéline. De plus, Chapleur a élaboré une bibliothèque de 126 composés basés sur un
squelette xylose sur lesquelles le carboxyméthyle est localisé en position 2, 3 ou 4 de la
molécule (9, 10, 11),27,28. L’équipe de Sofia a aussi synthétisé une famille de composés
dérivés de sucre où une fonction carboxyméthyle non-anomérique est greffée en position 2 de
la molécule (12). Certains de ces acides sont accrochés à une résine par différents amino-
acides29, , 30 31 dans une synthèse en phase solide.
25 Cipolla, L.; Forni, E.; Jiménéz-Barbéro, J.; Nicotra, F. Chem. Eur. J. 2002, 8, 3976-3983. 26 Le Diguarher, T.; Boudon, A.; Elwell, C.; Paterson, D. E.; Billington, D. C. Bioorg. Med. Chem. Lett. 2004, 16, 1983-1988. 27 Moitessier, N.; Minoux, H.; Maigret, B.; Chrétien, F.; Chapleur, Y. Lett. Pept. Sci. 1998, 8, 75-78. 28 Moitessier, N.; Dufour, H.; Chrétien, F.; Thiery, J. P.; Maigret, B.; Chapleur, Y. Bioorg.Med. Chem. 2001, 9, 511-523. 29 Jain, R.; Kamau, M.; Wang, C.; Ippolito, R.; Wang, H.; Dulina, R.; Anderson, J.; Gange, D.; Sofia, M. J. Bioorg. Med. Chem. Lett. 2003, 13, 2185-2189. 30 Sofia, M. J.; Hunter, R.; Chan, T. Y.; Vaughan, A.; Dulina, R.; Wang, H.; Gange, D. J. Org. Chem. 1998, 63, 2802-2803. 31 Sofia, M. J. Med. Chem. Res. 1998, 8, 362-378.
19

Bibliographie - Chapitre I
O
OOBn
O COOH
5
OO
OMe
OMe
OBnOHO
AcHN
N3
O CO2H6
O
OOH
H
BnOOBn
COOH
N3
7
OOBnO
O
O
O CO2HO
HO
NH
8
OBnOBnO O
OCH2COOHNH
OHOOCCH2OBnO O
OBnNH
9
11
OMeON3
OMe
OCH2COOH12
OH
OMeOBnO
O
O
O
OH
INH NH2
NH
2
OMeOMeO
OO
4
O COOH
OHOMeO
BnO
OOCH2COOH
NH NH2
NH
3
OBnOO O
OBnNH
10COOH
Schéma 12. Différents carboxyméthyl glycosides synthétisés
I.3.1.2. En tant qu’intermédiaire
Vu leur qualité connective, les carboxyméthyl glycosides ont été largement utilisés
comme agents capables de fournir une entité sucre via leur fonction carboxylique, capable
de se lier par une fonction ester ou amide à d’autres adduits.
Par conséquent, cette famille de composés a servi comme élément de construction
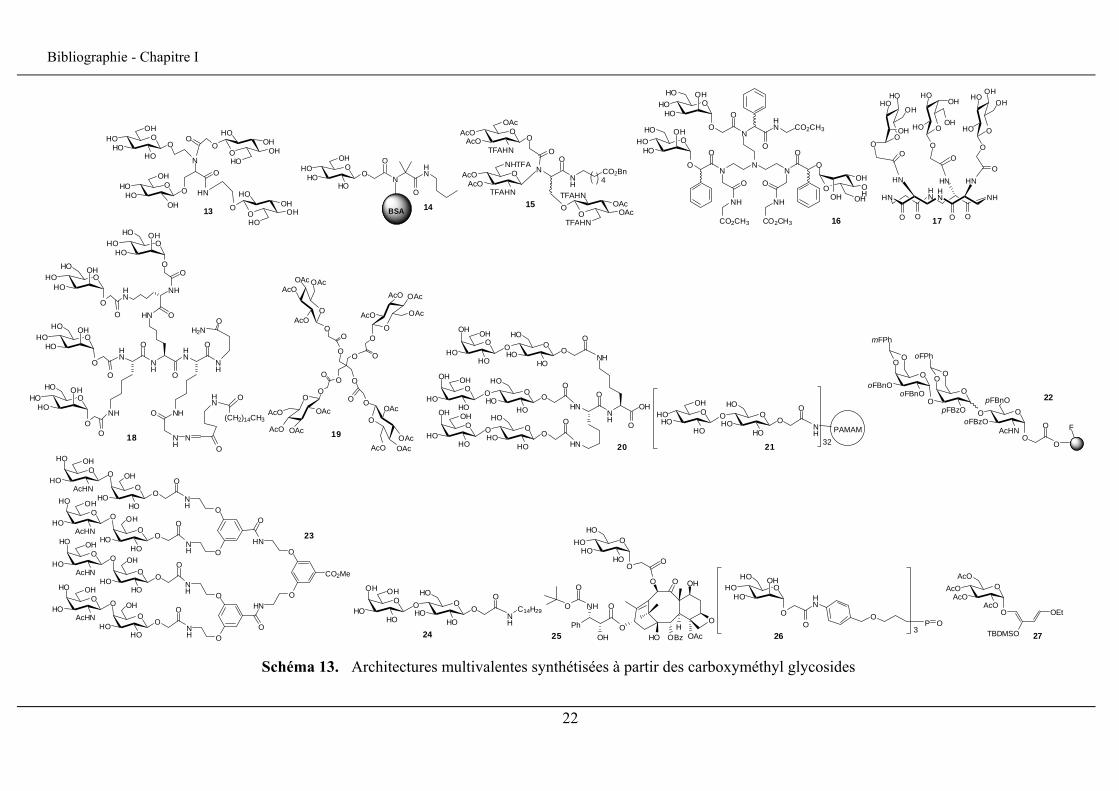
pour la synthèse de nouvelles familles de glycoconjugués (schéma 13).
Dans le contexte de molécules d’intérêt biologique, les sucres ont servi pour la
synthèse de glycodendrimères, macromolécules construites à partir d’unités répétitives,
comme par exemple les glycodendrimères préparés par Lockhoff et al.17 (13), les
20

Bibliographie - Chapitre I
néoglycoprotéines préparées par Ziegler et al.32 (14), les scaffolds cyclopeptidiques pour la
synthèse sur support solide de glycoclusters10 (15, 16) et des clusters cyclopeptidiques
amphiphiles comme ceux synthétisés par Li et al.33 (17) et ceux proposés par Grandjean et
al.11 (18)
Via la stratégie carboxyméthyl glycoside, un cluster contenant quatre unités glucose
sur un pentaérythritol a été synthétisé par Binder et Schmid16 (19), et un autre cluster
lactosyle multivalent3 (20) a aussi servi pour la conception d’un inhibiteur potentiel du
développement des tumeurs métastasiques. Kitaoka et al.20 ont préparé des dendrimères
PAMAM présentant des unités cellobiose (21). D’autres systèmes multivalents présentant
des unités disaccharidiques particulières ont été élaborés pour l’étude de leurs propriétés
antiadhésive à l’E. Coli par Pieters et al.21,22 (22).
De même, Elofsson et Kihlberg se sont servis du lien carboxyméthyle pour connecter
le sucre à une résine dans une approche de synthèse en phase solide13 (23), Basu et
Santacroce pour le lier à une amine à chaîne grasse pour l’obtention des disaccharides
amphiphiles18 (24), Mandai et al.5 pour conférer une solubilité pour des taxoïdes
anticancéreux (25). Il a aussi été utilisé pour la préparation d’oxydes de phosphine
complexes34 (26), et de nouveaux diènes oxygénés (27).35
32 Ziegler, T.; Gerling, S.; Lang M. Angew. Chem. Int. Ed. 2000, 39, 2109-2112. 33 Li, Y.; Zhang, X.; Chu, S.; Yu, K.; Guan, H. Carbohydr. Res. 2004, 339, 873-879. 34 Tamura, J.; Manabu, K. Japanese Patent JP 2002338592 (2002); Chem. Abstr. 2002, 137, 385072. 35 Cousins, R. P. C.; Pritchard, R. G.; Raynor, C. M.; Smith, M.; Stoodley, R. J. Tetrahedron Lett. 2002, 43, 489-492.
21
Bibliographie - Chapitre I
OHOHO
HO
OH
O OHOH
HO
HO
O
HN
O OHOH
HO
HO
OHOHO
OH
OH
O
NO
O
13
OO
OHOHO
HO
OH
ON
HN
O
O
BSA 14
OAcOAcO
TFAHN
OAc
OAcOAcO
TFAHN
NHTFAN
O
O
NH
CO2BnO
O OAcOAc
TFAHN
TFAHN
O
4
15
OHOHO
OHHO
ON
NN
O
N
HN
O
OO
O
OHOHO
OHHO
NH
O
O
CO2CH3
NH
OO O
H
OH
OH OH
CO2CH3 CO2CH3 16
HN
HN
O
HOHO
OH
OH
O
HN NH
O OO O
HN HNHN
O
OHHO
HO
OH
O
O
O
OHHO
HO OH
OO O
17
HN
NH
HN
NH
H2N
O
O
O
HN O
NHHN
NH NHO
NH
O
OHOHO
OHHO
O
OHOHO
OHHO
OO
O
OHOHO
OHHO
OO
OHOHO
OHHO
OO N
O
HN
(CH2)14CH3
O
18
OO
O
O
O
O
O
O
OO
OO
O
OAcAcO
AcO
OAc
O
OAcOAc
OAc
AcO
O
OAcAcO
AcO OAc
O
AcO OAc
OAcAcO
19
OOH
HOHO
OHOO
HOHO
HO
OO
HNNH
OHO
O
NH
OOH
HOHO
OHOO
HOHO
HO
OO
O
OH
HOHO
OHOO
HOHO
HO
OO
HN 20
NH
OHOHO
HO
OHOO
HOHO
HO
OO
PAMAM3221 O
OO
OpFBzO
O
OOoFBzO
AcHN
pFBnO
OO
oFPh
OO
oFBnOoFBnO
O
mFPh
F
22
O
O
CO2Me
HN
HN
O
O
O
O
O
ONH
NH
NH
NH
OHO
HOAcHN
OH
O
O
HOHO
OH
OO
OHO
HOAcHN
OH
O
O
HOHO
OH
OO
OHO
HOAcHN
OH
O
O
HOHO
OH
OO
OHO
HOAcHN
OH
O
O
HOHO
OH
OO
23
NH
OOH
HOHO
OHOO
HOHO
HO
OO
C14H29
24
OO
HO
O
OH
O
O
OHOHO
HO
HO
O
HOAcOBz
O
OHPh
NHO
O
25
HN
OHOHO
OHHO
OO
OP O
326 TBDMSO
OEtO
OAcOAcO
AcO
AcO
27
Schéma 13. Architectures multivalentes synthétisées à partir des carboxyméthyl glycosides
22

Bibliographie - Chapitre I
I.3.2. EXPLOITATION DE LA CARBOXYMETHYL GLUCOSIDE LACTONE (CMGLCL)
La capacité de la α-CMGlcL à s’ouvrir en présence d’espèces nucléophiles a été mise en
évidence par Trombotto et al.2 en 2000 (schéma 14).
L’ouverture de la CMGlcL par des espèces nucléophiles s’effectue souvent sans
catalyseur, dans des conditions douces. Elle donne ainsi accès à de nouveaux glycoconjugués
dans lesquels la structure cyclique du sucre est conservée36 (schéma 14) contrairement à
l’ouverture de la lactone de sucre du type gluconolactone qui donne des conjugués à chaîne
polyols ouverte.
Des structures similaires à cette lactone ont été décrites dans la littérature et utilisées pour
la synthèse de nouveaux glycoconjugués. Une phostone, observée lors de l’étape d’acétylation
du phosphono-C-glycoside a été ouverte pour la formation de dérivés phosphorés
potentiellement actifs.37,38 Un autre type de glucoside bicyclique a aussi été produit à partir
d’une amine39,40. Cette structure a été plus tard utilisée comme un synthon de départ pour la
synthèse de nouveaux conjugués41, , 42 43 (schéma 14).
OAcO
OO
O
OAc
AcOOAcO
AcOHO
O
OAc
O
Nu1a
NuH
O
AcO
AcO
OP O
ORPhostone
Bosco et al.Tetr ahedron Lett. 2003
OHOHO
ONH
OH
S
OHOHO
OH
HN
OH
S
R1
R2NHR1R2 Maya et al.
Tetrahedron Lett. 2001
Trombotto et al.J. Org. Chem. 2000
O
AcO
AcO
OH
PO
OHOR
NaOH
Schéma 14. α-CMGlcL et structures bicycliques similaires
36 Pierre, R.; Chambert, S.; Alirachedi, F.; Danel, M.; Trombotto, S.; Doutheau, A.; Queneau, Y. C. R. Chimie 2008, 11, 61-66. 37 Bosco, M.; Bisseret, P.; Eustache, J. Tetrahedron Lett. 2003, 44, 2347-2349. 38 Bosco, M.; Bisseret, P.; Constant, P.; Eustache, J. Tetrahedron Lett. 2007, 48, 153-157. 39 Steyemark, P. R.; J. Org. Chem. 1962, 27, 1058. 40 Kovacs, J.; Pinter, I.; Messmer, A. Carbohydr. Res.1985, 141, 57. 41 Ichikawa, Y.; Matsukawa, Y.; Minoru, I. J. Am. Chem. Soc. 2006, 128, 3934-3938. 42 Maya, I.; López, Ó.; Fernández-Bolaňos, J. G.; Robina, I.; Fuentes, J. Tetrahedron Lett. 2001, 42, 5413-5416. 43 López, Ó.; Maya, I.; Fuentes, J.; Fernández-Bolaňos, J. G. Tetrahedron 2004, 60, 61-72.
23

Bibliographie - Chapitre I
La α-CMGlcL a été utilisée comme un synthon « donneur de glucoside » dans plusieurs
domaines, forgeant ainsi une nouvelle voie de construction de nouveaux pseudo-
glycoconjugués, alternative aux réactions classiques de glycosylation.
La simplicité de cette stratégie a été exploitée pour la synthèse de nouveaux esters et
amides glycosylés. Par exemple, de nouveaux pseudo-oligosaccharides (28, 29) ont été
synthétisés par ouverture du cycle lactonique par un aminosucre44 (schéma 15).
OHOHO
HO
HO
OO
OHOHO
HO
NH2
OMe
OHO
H2N
OO
OH
OAcOAcO
HO
AcO
OO
OHO
NH
OO
OH
OHOHO
HO
NH
OMe
CMglcL, THF,18h, 90%
1. CMglcL, DMF,15h, ta, 87%;
2. MeONa/MeOH,1h, ta, 83%
28
29
Schéma 15. Synthèse de pseudo-oligosaccharides à partir de la stratégie CMGL
Les glycopeptides ayant suscité l’intérêt de part leur rôle dans de nombreux processus
pathologiques et physiologiques, une famille de glycoaminoacides a été synthétisée grâce à la
stratégie CMGL. Les nucléophiles utilisés sont des aminoacides protégés variés.45 Le schéma
16 en montre un exemple où l’ester méthylique de la glycine est utilisé.
44 Le Chevalier, A.; Pierre, R.; Kanso, R.; Chambert, S.; Doutheau, A.; Queneau, Y. Tetrahedron. Lett. 2006, 47, 2431-2434. 45 Trombotto, S.; Danel, M.; Fitremann, J.; Bouchu, A.; Queneau, Y. J. Org. Chem. 2003, 68, 6672-6678.
24

Bibliographie - Chapitre I
OAcO
OO
O
OAc
AcO
1a
OAcOAcO
AcOOH
OO
NH
NH2CH2COOMeDMAP,CH2Cl2,
3 jours, ta, 60%CO2Me
Schéma 16. Synthèse de glycopeptides à partir de la stratégie CMGL
Dans la perspective de conception de molécules amphiphiles, la α-CMGlcL a été exploitée pour la synthèse de nouveaux glycolipides (schéma 17) connus pour leurs propriétés biologiques et physico-chimiques, comme par exemple leur comportement thermotrope. Des amides aliphatiques ayant une chaîne de 10, 12 et 14 carbones ont été synthétisés dont certains ont montré un comportement lamellaire à haute température (30).
D’autre part, des amino-stéroïdes ont été couplés avec la α-CMGlcL pour donner une série de glycostéroïdes parmi lesquels les composés saturés ont montré un comportement cristal-liquide à des températures moins élevées que leurs homologues insaturés.46,47
OHOHO
HO
OH
ONH
O
n
n = 5, 7, 9, 11, 13, 1530 31
OHOHO
HO
OH
OO
O
Schéma 17. Structures glycolipidiques synthétisées à partir de la stratégie CMGL
Les porphyrines glycosylées ont un intérêt potentiel comme molécules
photosensibilisatrices pour la photochimiothérapie du cancer. La présence des sucres sur les
porphyrines module son caractère amphiphile et permet le ciblage des cellules cancéreuses
46 Chambert, S.; Cowling, S. J.; Mackenzie, G.; Goodby, J. W.; Doutheau, A.; Queneau, Y. J. Carbohydr. Chem. 2007, 26, 27-39. 47 Goodby, J. W.; Görtz, V.; Cowling, S. J.; MacKenzie, G.; Martin, P.; Plusquellec, D.; Benvegnu, T.; Boullanger, P.; Lafont, D.; Queneau, Y.; Chambert, S.; Fitremann, J. Chem.Soc. Rev. 2007, 36, 1971-2032.
25

Bibliographie - Chapitre I
par formation d’interactions spécifiques entre des récepteurs membranaires des cellules
cancéreuses et les groupements glycosyles.
Des porphyrines glucosylées en ortho et en para ont été préparées48 (schéma 18) par la
stratégie CMGL à partir des monohydroxyphényltritolylporphyrines aminopropylées.
La photocytotoxicité de ces porphyrines glucosylées ont été évaluées en utilisant des cellules cancéreuses chroniques K562. La porphyrine en ortho est plus active que celle en para mais reste moins performante que la Photofrin® qui est un médicament commercialisé pour le traitement contre le cancer. La photoactivité par contre, est nettement améliorée par rapport aux porphyrines non glycosylées.
N
NH HN
N
O
OHOHO
HO
HO
OO
NH
N
NH HN
N
O
OHOHO
HO
HO
OO
NH
or tho para
Schéma 18. Les porphyrines glycosylées synthétisées à partir de la stratégie CMGL
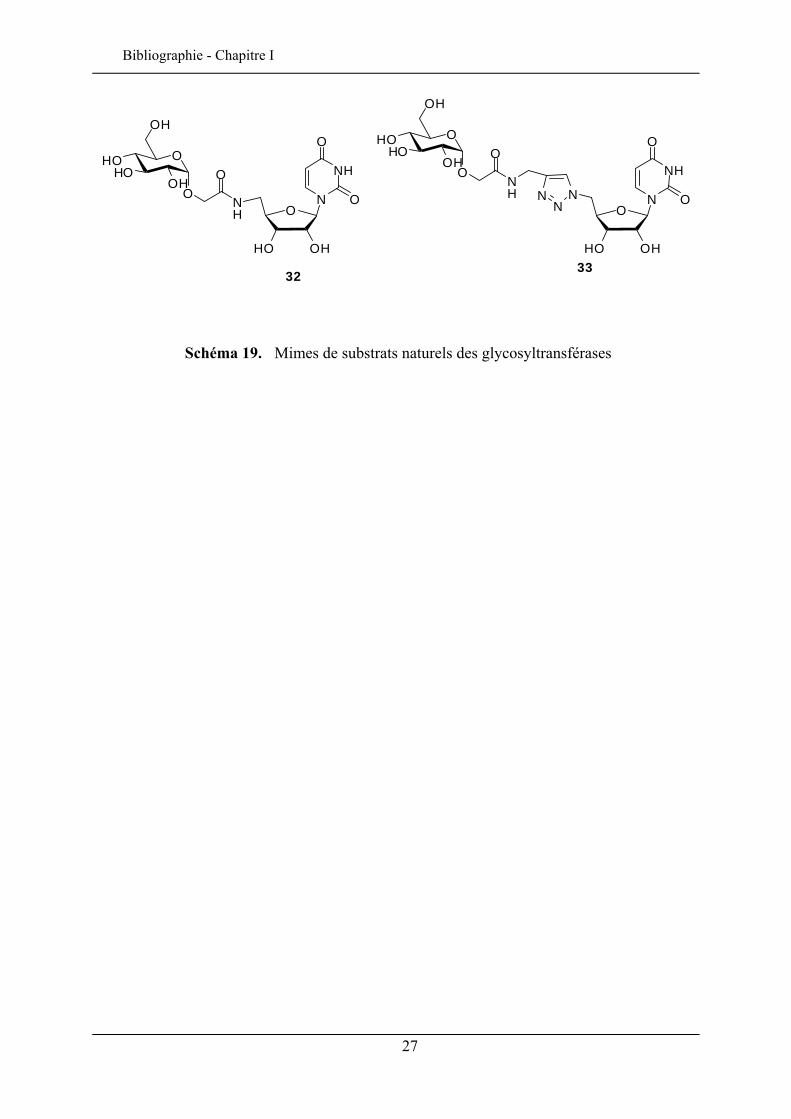
Enfin, une illustration supplémentaire de cette stratégie est donnée lors de la synthèse de
nouveaux analogues de substrats naturels des glycosyltransférases. Les réactions sont
réalisées soit par ouverture de la α-CMGlcL par une azidouridine réduite en amine44 (32) soit
par réaction « Click »49 avec l’azidouridine (schéma 19) dans le but de prolonger l’espace
entre le sucre et l’uridine par un triazole (33).
48 Sol, V., Charmot, A.; Krausz, P.; Trombotto, S.; Queneau, Y. J. Carbohydr. Chem. 2006, 25, 345-360. 49 Listkowski, A.; Ing, P.; Cheaib, R.; Chambert, S.; Doutheau, A.; Queneau, Y. Tetrahedron: asymmetry, 2007, 18, 2201-2210.
26
Bibliographie - Chapitre I
O
HO OH
N
NH
O
ONNN
OHO
OHO
O
OH
NH
HO
O
HO OH
N
NH
O
O
OHO
OHO
O
OH
NH
HO
32 33
Schéma 19. Mimes de substrats naturels des glycosyltransférases
27

Bibliographie - Chapitre II
28

Bibliographie - Chapitre II
CHAPITRE II. LES SCAFFOLDS SUCRES
Dans le premier chapitre, nous avons montré l’essentiel des travaux qui ont visé la
synthèse de scaffolds glucidiques présentant une fonction carboxyméthyle sur un glycoside
ainsi que les scaffolds obtenus par réaction du carboxyméthyle avec une amine via une
fonction amide. Dans le présent chapitre, nous élargissons le propos à la synthèse et
l’utilisation d’autres types de scaffolds à structure glucidique dans une mise au point basée sur
les revues de Meutermans et al.,50 Murphy et al.,51 ,Doores et al.52 mises à jour avec des
références récentes sur le domaine.
II.1. SCAFFOLD, DEFINITION, HISTORIQUE
La structure des sites actifs de protéines étant souvent précisément déterminée par
cristallographie, ainsi que la connaissance des conditions structurales pour les bonnes
interactions protéine-ligand qui en découle, des molécules «scaffolds» ou châssis moléculaires
ont été conçues afin de mimer le positionnement idéal des substituants des substrats naturels.
La conception de molécules scaffolds bioactives consiste à avoir des groupes pharmacophores
capables d’établir des interactions avec la cible et une structure non interactive qui porte la
partie bioactive.
Le scaffold idéal pour une approche biomimétique est une molécule à structure rigide, de
faible poids moléculaire, chimiquement stable, présentant plusieurs groupements fonctionnels
à réactivité orthogonale. L’orientation spatiale des groupements fonctionnels doit être flexible
pour permettre d’établir la plus forte interaction avec le récepteur. De plus, la molécule doit
présenter une stabilité biologique.
50 Meutermans, W.; Le G. T.; Becker, B. ChemMedChem. 2006, 1, 1164-1194. 51 Murphy, P. V. Eur. J. Org. Chem. 2007, 4177-4187. 52 Doores, K. J.; Gramblin, D. P.; Davis, B. G. Chem. Eur. J. 2006, 12, 656-665.
29

Bibliographie - Chapitre II
Les systèmes aromatiques étaient de bons candidats pour la synthèse de scaffolds puisque
l’introduction de différents substituants est relativement facile. Par contre, ce sont en général
des molécules planes qui empêchent la répartition tridimensionnelle des substituants. D’autres
structures, comme les alcaloïdes naturels, sont chimiquement difficiles à manipuler. De
même, les peptides ayant une rigidité faible, présentent une structure très flexible dans un
environnement biologique.
Les sucres, par contre, présentent un squelette rigide avec un bon nombre de groupements
hydroxyle orthogonalement fonctionnalisables ayant une orientation spatiale bien définie, et
servant de points d’attachement entre ces molécules et le récepteur de la protéine qu’on
cherche à inhiber. De plus, ils présentent un grand nombre d’isomères possibles.
Pour ces raisons, des études de molécules sur scaffolds sucre ont été largement menées.
II.2. QUELQUES RESULTATS BIOLOGIQUES
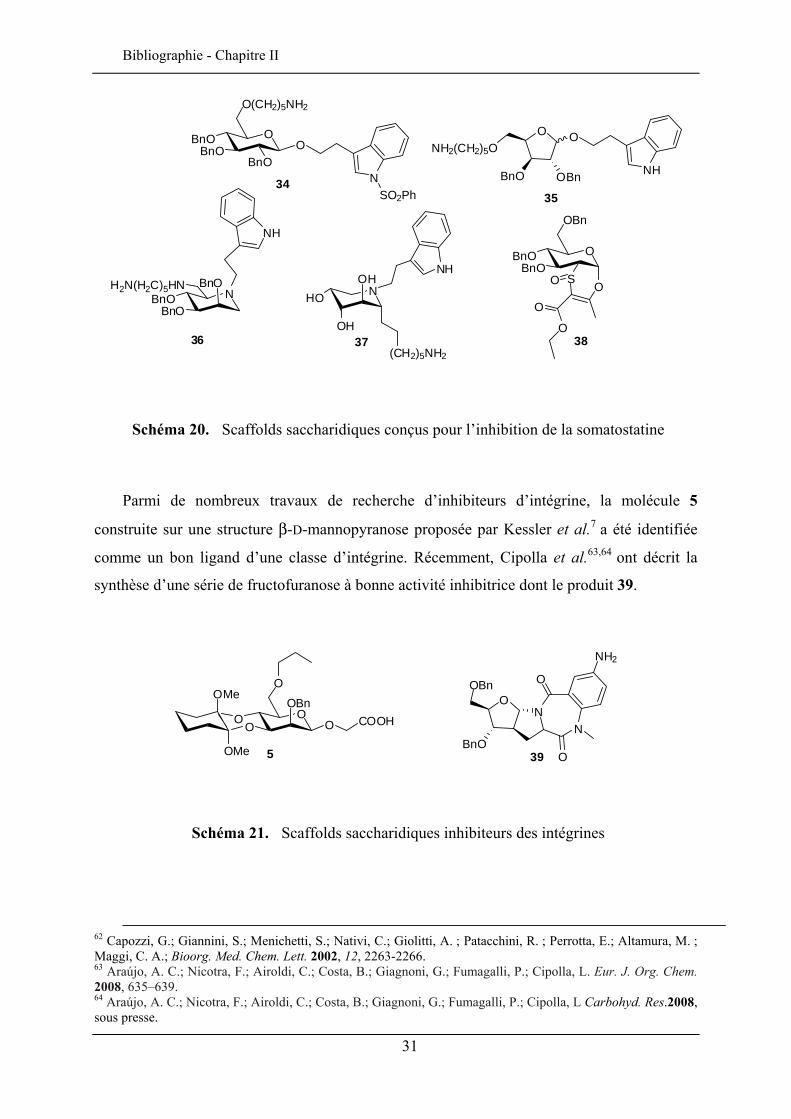
L’hypothèse évoquant que les sucres peuvent être des scaffolds adéquats pour mimer
certains ligands naturels de protéines actives a été proposée par Hirschmann et al.53, , , ,54 55 56 57
en 1990 lors de la recherche de mimes de la somatostatine. Après une modélisation
moléculaire, une collection de molécules a été élaborée sur une structure D-glucose, dont la
molécule 34 qui s’est révélée être un mime du ligand naturel de la protéine. D’autres
synthèses de molécules visant la somatostatine ont été décrites (schéma 20) comme celles
élaborées par l’équipe de Papageorgiou58 (scaffold furanose 35), Depezay et al.,59 Murphy et
al.60,61 (scaffolds iminosucres 36 et 37) ainsi que Nativi, Capozzi et al.62 (dérivé 38).
53 Hirschmann, R.; Nicolaou, K. C.; Pietranico, S.; Salvino, J.; Leahy, E. M.; Sprengeler, P. A.; Furst, G.; Smith III, A. B. J. Am. Chem. Soc. 1992, 114, 9217-9218. 54 Hirschmann, R.; Nicolaou, K. C.; Pietranico, S.; Leahy, E. M.; Salvino, J.; Arison, B.; Cichy, M. A.; Spoors, P. G.; Shakespeare, W. C.; Sprengeler, P. A.; Hamley, P.; Smith III, A. B.; Reisine, T.; Raynor, K.; Maechler, L.; Donaldson, C.; Vale, W.; Freidinger, R. M.; Cascieri, M. R.; Strader, C. D. J. Am. Chem. Soc. 1993, 115, 12550-12568. 55 Hirschmann, R.; Yao, W.; Cascieri, M. R.; Strader, C. D.; Maechler, L.; Cichy, M. A., Jr., J. H.; Sprengler, P. A.; Smith III, A. B.; J. Med. Chem. 1996, 39, 2441. 56 Hirschmann, R.; Hynes, Jr. J.; Cichy-Knight, M. A; van Rijin, R. D.; Sprengeler P. A.; Spoors, P. G.; Shakespeare, W. C.; Pietranico-Cole, S.; Barbosa, J.; Liu, J.; Yao, W.; Rohrer, S.; Smith III, A. B. J. Med. Chem. 1998, 41, 1382-1391. 57 Prasad, V.; Birzin, E. T. ; McVaughn, C. T.; van Rijin, R. D.; Rohrer, S. P.; Chicchi, G.; Underwood, D. J.; Thornton, E. R.; Smith III, A. B.; Hirschmann, R. J. Med. Chem. 2003, 46, 1858-1869. 58 Papageorgiou, C.; Haltiner, R.; Bruns, C.; Petcher, T. J. Bioorg. Med. Chem. Lett. 1992, 2, 135-140. 59 Le Merrer, Y.; Poitout, L.; Depezay, J. Methods in Molecular Medicine, Peptidomimetic Protocols, Vol. 23, (Ed. Kazmierski), Humana Press, Totowa, NJ, 1999, p. 227-257. 60 Gouin, S. G.; Murphy, P. V. J. Org. Chem. 2005, 70, 8527-8523. 61 Chagnault, V.; Lalot, J.; Murphy, P.V ChemMedChem, 2008, sous presse.
30
Bibliographie - Chapitre II
OBnOBnO
BnO
O(CH2)5NH2
O
NSO2Ph
3435
ONH2(CH2)5O
BnO OBn
O
NH
NBnOBnO
BnO
36
NH
H2N(H2C)5HN
OBnOBnO
OBn
S O
O
O38
NOH
HO
OH
(CH2)5NH2
NH
37
O
Schéma 20. Scaffolds saccharidiques conçus pour l’inhibition de la somatostatine
Parmi de nombreux travaux de recherche d’inhibiteurs d’intégrine, la molécule 5
construite sur une structure β-D-mannopyranose proposée par Kessler et al.7 a été identifiée
comme un bon ligand d’une classe d’intégrine. Récemment, Cipolla et al.63, 64 ont décrit la
synthèse d’une série de fructofuranose à bonne activité inhibitrice dont le produit 39.
O
O
OBn
O COOHOO
OMe
OMe 5 39
ON
BnO
OBn
N
O
O
NH2
Schéma 21. Scaffolds saccharidiques inhibiteurs des intégrines
62 Capozzi, G.; Giannini, S.; Menichetti, S.; Nativi, C.; Giolitti, A. ; Patacchini, R. ; Perrotta, E.; Altamura, M. ; Maggi, C. A.; Bioorg. Med. Chem. Lett. 2002, 12, 2263-2266. 63 Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla, L. Eur. J. Org. Chem. 2008, 635–639. 64 Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla, L Carbohyd. Res.2008, sous presse.
31

Bibliographie - Chapitre II
Certains groupes de recherche comme Tsukida, Nishimura et al.65, , ,66 67 68 se sont intéressés
à l’inhibition des métalloprotéinases (schéma 22). Le composé 40 qu’ils proposent a montré
une inhibition de la protéine. Le scaffold bicyclique 41 synthétisé par Nativi et al.69 a été
découvert comme étant le premier inhibiteur d’une métalloprotéinase à base de sucre.
OHOHO
OH
SO
O
OH41
NO
O
OH
40
O2S
NHOHO
OMeHN
COOHO
Schéma 22. Scaffolds saccharidiques pour l’inhibition des métalloprotéinases
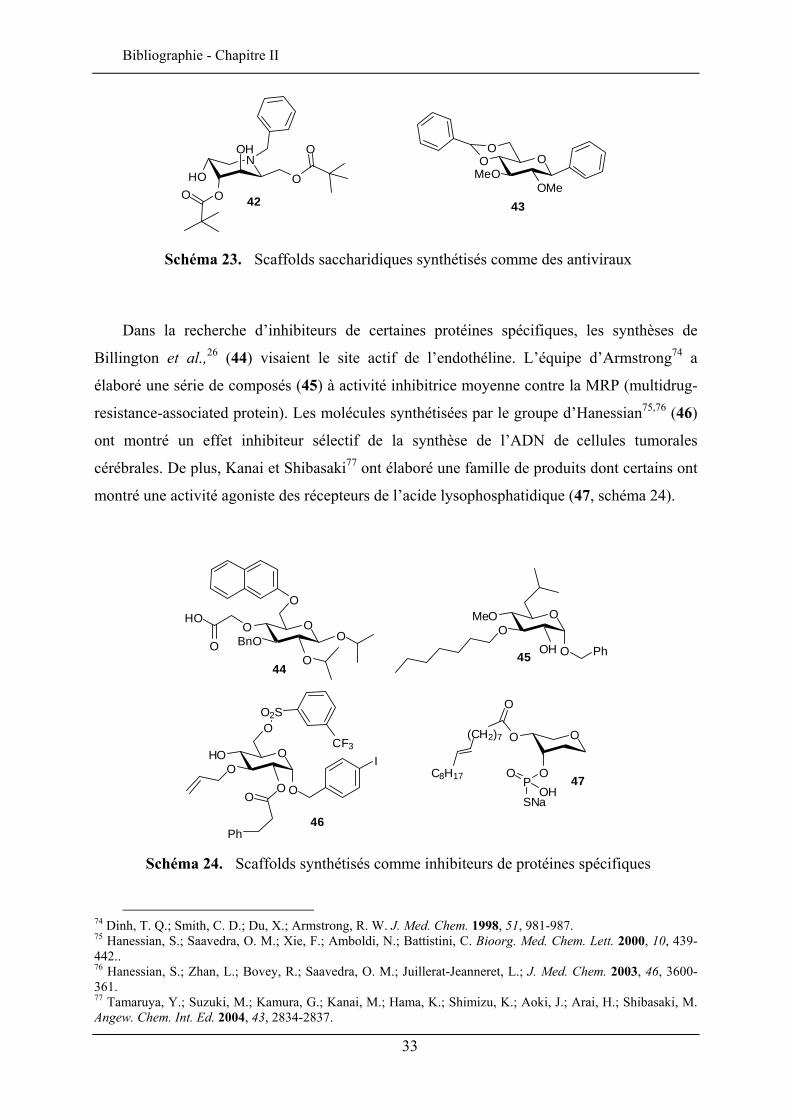
Les antiviraux potentiels ont eux aussi fait l’objet d’étude. Murphy et al.70, ,71 72 visaient
l’inhibition de la protéase du VIH en synthétisant des molécules à base de mannose et glucose
(42) à activité inhibitrice modérée. Van der Eycken et al.73 ont démontré une bonne activité
d’une structure à base de D-glucose (43, schéma 23) contre un virus de la famille des herpès.
65 Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I. Bioorg. Med. Chem. Lett. 2003, 13, 2737-2740. 66 Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I. Bioorg. Med. Chem. Lett. 2003, 13, 2741-2744. 67 Moriyama, H.; Tsukida, T.; Inoue, Y.; Yokota, K. ; Yoshino, K. ; Kondo, H.; Miura, N.; Nishimura, N.-I. J. Med. Chem. 2004, 47, 1930-1938. 68 Tsukida, T.; Moriyama, H.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I. Bioorg. Med. Chem. Lett. 2004, 14, 1569-1572. 69 Fragai, M.; Nativi, C.; Richichi, B.; Venturi, C. ChemBioChem 2005, 6, 1345-1349. 70 Murphy, P. V.; O’Brien, J. L.; Gorey-Feret, L. J.; Smith III, A. B. Tetrahedron 2003, 59, 2259-2271. 71 Chery, F.; Cronin, L.; O’Brien, J. L.; Murphy, P.V. Tetrahedron 2004, 60, 6597-6608. 72 Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla, L. Carbohyd. Res. 2008, sous presse. 73 Van Hoof, S.; Ruttens, B.; Hubrecht, I.; Smans, G.; Blom, P.; Sas, B.; Van Hemel, J.; Vandenkerckhove, J.; Van der Eycken, J. Bioorg. Med. Chem. Lett. 2006, 16, 1495-1498.
32
Bibliographie - Chapitre II
NOH
O
42
O
43O
HO
O
O OMeMeOO
O
Schéma 23. Scaffolds saccharidiques synthétisés comme des antiviraux
Dans la recherche d’inhibiteurs de certaines protéines spécifiques, les synthèses de
Billington et al.,26 (44) visaient le site actif de l’endothéline. L’équipe d’Armstrong74 a
élaboré une série de composés (45) à activité inhibitrice moyenne contre la MRP (multidrug-
resistance-associated protein). Les molécules synthétisées par le groupe d’Hanessian75,76 (46)
ont montré un effet inhibiteur sélectif de la synthèse de l’ADN de cellules tumorales
cérébrales. De plus, Kanai et Shibasaki77 ont élaboré une famille de produits dont certains ont
montré une activité agoniste des récepteurs de l’acide lysophosphatidique (47, schéma 24).
OOBnO
O
O
OO
HO
44
OMeOO
OH O Ph45
OHOO
O
O
O
46
I
Ph
O
O2S
CF3OO
47O
(CH2)7
O
C8H17 PO
SNaOH
Schéma 24. Scaffolds synthétisés comme inhibiteurs de protéines spécifiques
74 Dinh, T. Q.; Smith, C. D.; Du, X.; Armstrong, R. W. J. Med. Chem. 1998, 51, 981-987. 75 Hanessian, S.; Saavedra, O. M.; Xie, F.; Amboldi, N.; Battistini, C. Bioorg. Med. Chem. Lett. 2000, 10, 439-442.. 76 Hanessian, S.; Zhan, L.; Bovey, R.; Saavedra, O. M.; Juillerat-Jeanneret, L.; J. Med. Chem. 2003, 46, 3600-361. 77 Tamaruya, Y.; Suzuki, M.; Kamura, G.; Kanai, M.; Hama, K.; Shimizu, K.; Aoki, J.; Arai, H.; Shibasaki, M. Angew. Chem. Int. Ed. 2004, 43, 2834-2837.
33

Bibliographie - Chapitre II
Dans le domaine des antibactériens, l’équipe de Fleet78 a proposé des molécules basées
sur un iminosucre (48) mais leur évaluation biologique n’a pas été précisée. Spirito et al.79 ont
synthétisé des molécules pyraniques (49) à bonne activité inhibitrice de la neuraminidase. Des
scaffolds disaccharidiques ont été synthétisés par Sofia et al.80 actifs contre la moenomycine
A (50) et par Meutermans et al.81 (51) actifs contre un panel des bactéries Gram-positive. Une
bibliothèque de composés actifs synthétisés par l’équipe de Wong contre des bactéries ARN
(52).82 Enfin, des scaffolds D-glucose (53) ont été préparées par Li et al.83 visant l’acétolactate
synthase et ont montré une bonne capacité à se lier à cette protéine.
O
HN
OBn
ONH
NH
O
SO2NH2
48
OHO
50
HO
OH
NHO O
NH2
OHN
NHO
OH
OP
O
OOH
O
F3C
F3CCl
COOH
O(CH2)11CH3OHOHO
O
NH2
52
ONH
NH2
NH
ONH2
NH2
HN
OHOHO
NH
OH
OO
NH
OHHN
CHNO2S
C C10H21
O
CF3
O
CF3 51
OHO
53
ON
NMeO
SMe
O
O
F3CO
NHO
Ph
OMe
OHO
49
HO
NH
OOH
OHO P
O
ONaONa
Schéma 25. Scaffolds saccharidiques visant des cibles spécifiques
78 Edwards, A. A.; Ichihara, O.; Murfin, S.; Wilkes, R.; Whittaker, M.; Watkin, D. J.; Fleet, G. W. J. J. Comb. Chem. 2004, 6, 230-238. 79 Maione A. M. ; Giordani, E. ; Costa, M.; Spirito A. Arch. Pham Pharm. Med. Chem. 1997, 330, 17-20 80 Sofia, M. J.; Allanson, N.; Hatzenbuhler, N. T.; Jain, R.; Kakarla, R.; Kogan, N.; Liang, R.; Liu, D.; Silva, D. J.; Wang, H.; Gange, D.; Anderson, J.; Chen, A.; Chi, F.; Dulina, R.; Huang, B.; Kamau, M.; Wang, C.; Baizman, E.; Branstrom, A.; Bristol, N.; Goldman, R.; Han, K.; Longley, C.; Midha, S.; Axelrod, H. R. J. Med. Chem. 1999, 42, 3193-3198. 81 Halliday, J.; McKeveney, D.; Muldoon, C.; Rajaratnam, P.; Meutermans, W. Biochem. Pharmacol. 2006, 71, 957-967. 82 Wong, C.-H.; Hendrix, M.; Manning, D. D.; Rosenbohn, C.; Greenberg, W. A. J. Am. Chem. Soc. 1998, 120, 8319-8327. 83 Liao, Y.; Li, Z.; Wong, N. C. Chin. J. Chem. 2001, 19, 1119-1129.
34

Bibliographie - Chapitre II
II.3. SYNTHESE
II.3.1. SCAFFOLDS PYRANIQUES
La première molécule à scaffold sucre est synthétisée par Hirschmann et al.53 à partir
d’un glucopyranose en distinguant, comme dans le produit 54, la position anomérique,
position 6 et les trois autres positions restantes benzylées (schéma 26). Cette stratégie a été
appliquée pour synthétiser plus de 50 produits. Elle a été reprise plus tard par l’équipe de
Papageorgiou qui l’a appliquée à un dérivé furanique.58
OBnOBnO
BnO
OTBDPS
()2
OAcOAcO
AcO
OAc
Br
O NSO2Ph
OBnOBnO
BnO
O(CH2)5NH2
()2O N
SO2Ph
543. HO(CH2)5NHCOCF3,
NaH4. NaOH
3. TBDPSCl,imidazole,
4. BnBr, NaH
1. Tryptophole,AgO
2. NaOMe
1. TBAF2. Tf2O, DMAP
Schéma 26. Synthèse de la première molécule à scaffold sucre
Une autre stratégie développée par le même groupe de chercheurs à partir d’un époxyde
(55)84 pour synthétiser les produits 56. Ces derniers donnent, en changeant les groupements
protecteurs et introduisant des carbamates sur différentes positions, une bibliothèque de
produits de configuration anomérique β dont le composé 57.
OTBDMSOTBDMSO
O
OTBDMS1. EtSH,
TFA OTBDMSOTBDMSO
OH
OTBDMS
SEtOHO
OOBz
OPMB
SEt
NHO
55 56 574. (CH3)3CHNCO,
CuCl5. BH3-Me3N,
AlCl3
1. BzCl2. TBAF
3. PMB(OMe)2,TSA
Schéma 27. Synthèse d’un scaffold pyranique à partir d’un époxyde
84 Hirschmann, R.; Ducry, L.; Smith III, A. B. J. Org. Chem. 2000, 65, 8307-8316.
35
Bibliographie - Chapitre II
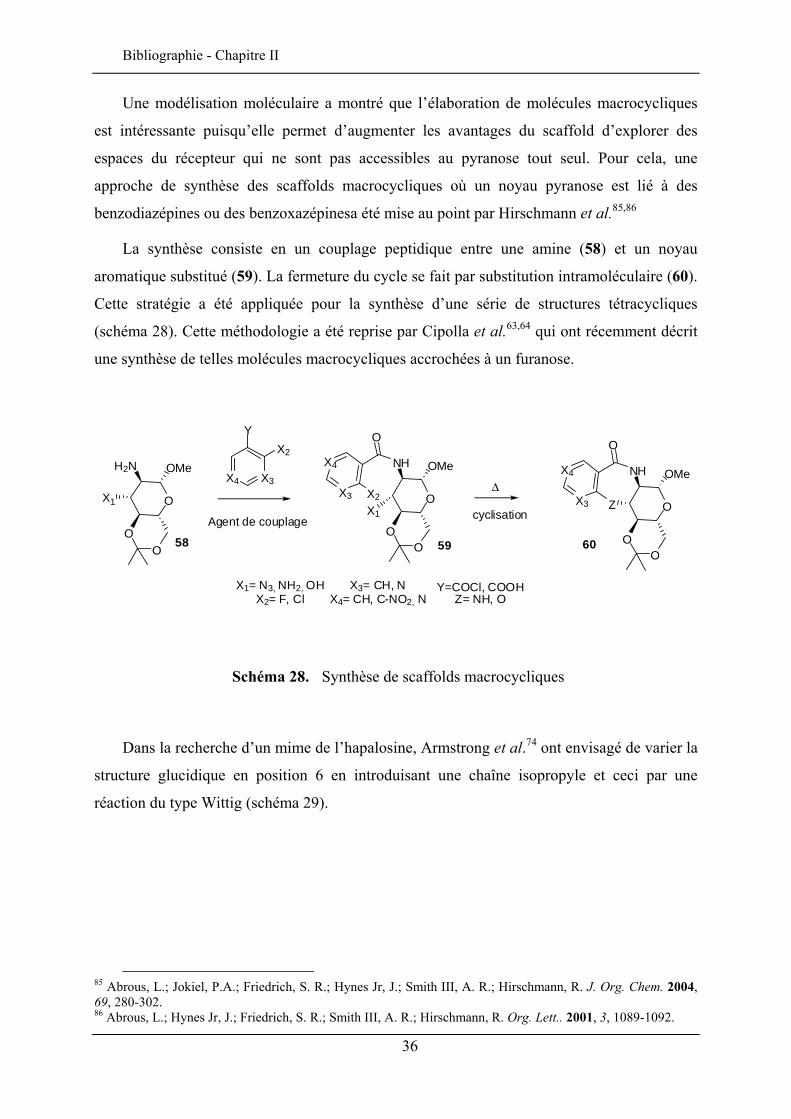
Une modélisation moléculaire a montré que l’élaboration de molécules macrocycliques
est intéressante puisqu’elle permet d’augmenter les avantages du scaffold d’explorer des
espaces du récepteur qui ne sont pas accessibles au pyranose tout seul. Pour cela, une
approche de synthèse des scaffolds macrocycliques où un noyau pyranose est lié à des
benzodiazépines ou des benzoxazépinesa été mise au point par Hirschmann et al.85,86
La synthèse consiste en un couplage peptidique entre une amine (58) et un noyau
aromatique substitué (59). La fermeture du cycle se fait par substitution intramoléculaire (60).
Cette stratégie a été appliquée pour la synthèse d’une série de structures tétracycliques
(schéma 28). Cette méthodologie a été reprise par Cipolla et al.63,64 qui ont récemment décrit
une synthèse de telles molécules macrocycliques accrochées à un furanose.
O
OO
OMe
X1
H2N
O
OO
OMeNH
O
X3
X4X4 X3
YX2
X2X1
O
OO
OMeNH
O
X3
X4
Zcyclisation
∆
Y=COCl, COOHZ= NH, O
58 59 60
Agent de couplage
X1= N3, NH2, OHX2= F, Cl
X3= CH, NX4= CH, C-NO2, N
Schéma 28. Synthèse de scaffolds macrocycliques
Dans la recherche d’un mime de l’hapalosine, Armstrong et al.74 ont envisagé de varier la
structure glucidique en position 6 en introduisant une chaîne isopropyle et ceci par une
réaction du type Wittig (schéma 29).
85 Abrous, L.; Jokiel, P.A.; Friedrich, S. R.; Hynes Jr, J.; Smith III, A. R.; Hirschmann, R. J. Org. Chem. 2004, 69, 280-302. 86 Abrous, L.; Hynes Jr, J.; Friedrich, S. R.; Smith III, A. R.; Hirschmann, R. Org. Lett.. 2001, 3, 1089-1092.
36

Bibliographie - Chapitre II
O
O
O
O
O
HO
OOHepO
BnO
O
OBnMeO
OPMBOHepO
BnOOBn
OMeOHepO
HOOBn61 6462 63 3. MeI, NaH
4. H2, Pd/C4. p-anisaldéhyde,TSA
5. BnBr, NaH
1. DIBAH2. Swern
3. iPrPPh3I,nBuLi
1. n-heptyl-I, NaH2. AcOH
3. H+, BnOH
1. PhS(O)2NHNH2,NaOH
2. DDQ
Schéma 29. Synthèse d’un scaffold sucre à partir du diacétone D-glucose
Utilisant le levoglucosane dérivé du glucose comme scaffold, Tirefort et al.87 ont
synthétisé une bibliothèque de 28 composés à partir de l’époxyde tosylé 65 (schéma 30).
Après ouverture de ce dernier (66), un autre époxyde se forme par substitution nucléophile
(67) qui sera ouvert par des alcools, des nucléophiles azotés ou sulfurés pour conduire aux
produits finaux (68, 69, 70).
O
OTs
OO 1. R1OH,
BF3.OEt2, O
OTs
OHO
R1O O OO
R1O
R2OH,NaH, O
OR2
OHO
R1O
R3NH2, LiClO4,2,6-Lutidine
R4SH, LiPF62,6-Lutidine
O
NHR3
OHO
R1O
O
SR4
OHO
R1O
65 6667 68
70
69
LiOH
Schéma 30. Synthèse de produits à scaffold sucre à partir de levoglucosane
Chapleur et al.27,28 ont utilisé un xylose dans une synthèse parallèle en solution. Une
benzylation aléatoire des allyl xylosides a donné un mélange de composés mono- et di-
benzylés. L’alkylation aléatoire du mélange permet d’avoir tous les isomères possibles de la
87 Brill, W.; K.-D.; Tirefort, D. Tetrahedron Lett. 1998, 39, 787-790.
37
Bibliographie - Chapitre II
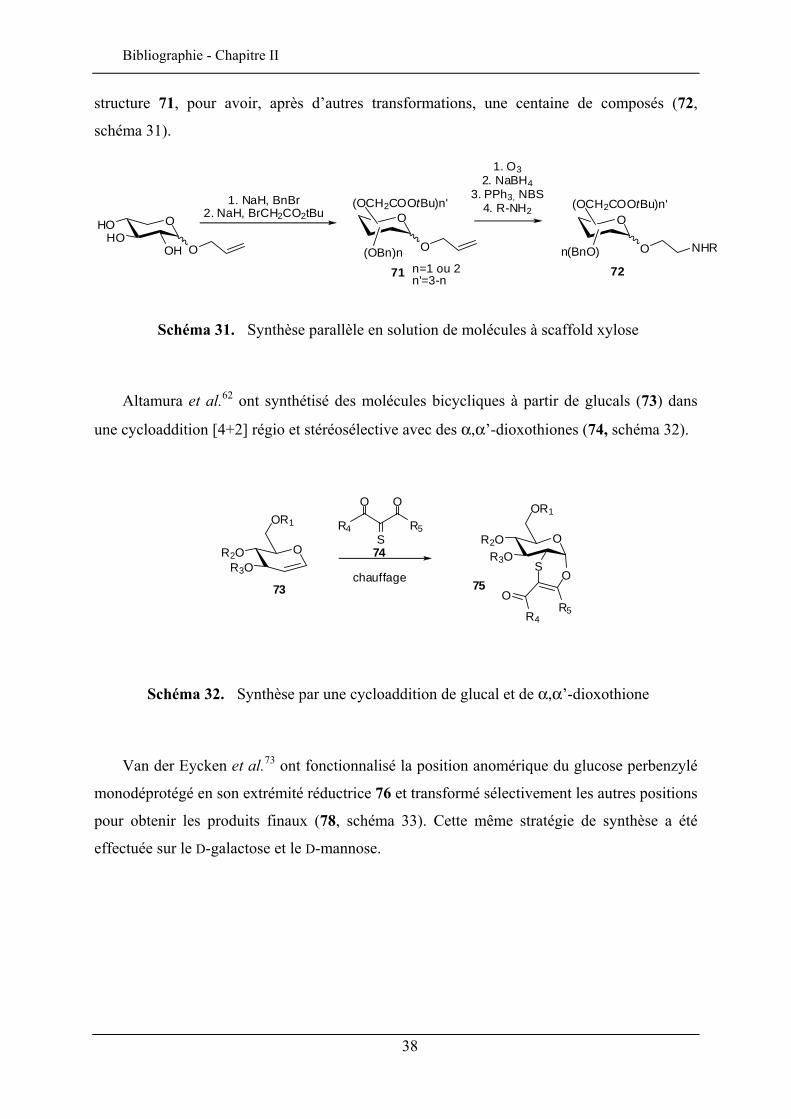
structure 71, pour avoir, après d’autres transformations, une centaine de composés (72,
schéma 31).
OHOHO
OH O
1. NaH, BnBr2. NaH, BrCH2CO2tBu O
O(OBn)n
(OCH2COOtBu)n'
1. O32. NaBH4
3. PPh3, NBS4. R-NH2
O
On(BnO)
(OCH2COOtBu)n'
NHR
71 72n=1 ou 2n'=3-n
Schéma 31. Synthèse parallèle en solution de molécules à scaffold xylose
Altamura et al.62 ont synthétisé des molécules bicycliques à partir de glucals (73) dans
une cycloaddition [4+2] régio et stéréosélective avec des α,α’-dioxothiones (74, schéma 32).
OR2OR3O
OR1 R4
O
S
O
R5OR2O
R3O
OR1
SO
R5O
R4
chauffage73
74
75
Schéma 32. Synthèse par une cycloaddition de glucal et de α,α’-dioxothione
Van der Eycken et al.73 ont fonctionnalisé la position anomérique du glucose perbenzylé
monodéprotégé en son extrémité réductrice 76 et transformé sélectivement les autres positions
pour obtenir les produits finaux (78, schéma 33). Cette même stratégie de synthèse a été
effectuée sur le D-galactose et le D-mannose.
38

Bibliographie - Chapitre II
OBnOBnO
OBn
OBn
OH
OBnOBnO
OBn
OBn1. Bromure
d'oxallyle OO
O
OO
77 7876
1. H2, Pd/C2. PhCH(OMe)2
3. NaH,bromure de propargyle
2. BnMgCl
Schéma 33. Synthèse proposée par van der Eycken et al.
L’équipe de Wong82 a synthétisé une bibliothèque de produits à partir de l’allyle de la N-
acétyl-glucosamine 79 (schéma 34). La double liaison de l’allyle, l’hydroxyle en position 6 et
la position 2 sont transformées en amines dont certaines subissent des modifications
supplémentaires pour avoir les produits finaux (81).
OHOHO
AcHN
OH
OHOHO
H2N
N31. TsCl2. NaN33. Ba(OH)2,
1. acide aminé (R1)2. O3
3. DMS4. Amine (NR2)
5. H2/Pd OHO
R1HN
HO
OO
NH2
ONR2
80 8179
Schéma 34. Synthèse d’une molécule à partir de la N-acétyl-glucosamine
Kessler et al.7 décrivent une stratégie de synthèse à partir du D-glucopyranose en utilisant
les stratégies de protection orthogonales des hydroxyles du sucre (schéma 35). Le clivage
sélectif des groupes protecteurs a permis le greffage de groupements pharmacophores.
39

Bibliographie - Chapitre II
OMeOMeO
OTBDPSOBn
OMeOMeO
O
OR
O ()nCOOH
SEt
OMeOMeO
O
OBn
O()n
COOH
1. TBAF, KN(SiMe3)22. BrCH2CHMe2
3. TFA4. KN(SiMe3)2, MeI
5. NBS, HCl6. SOCl2, Ag2CO3
7. HOCH2CH(OMe)2
1. BrCH2CH2OBn2. HCl
3. tBuOH, NaClO24. H2, Pd/C
Schéma 35. Synthèse de Kessler et al.
Certains glycosides ont été suggérés comme éléments de construction de molécules
scaffolds (schéma 36) parce que les fonctionnalités portées par ces structures sont facilement
manipulables, comme par exemple le produit 81 de Nativi et al.,88 la molécule 6 préparée par
Ghosh et al.,9 les époxydes 82 et 83 de Jensen et al.,89 les diamines 84, 85 et 86 de Chapleur
et al.90 ainsi que le composé bicyclique 87 de Voelter et al.91
OHO
OBn
OH
OTBDMSEtO2C
OBnO
AcHN
N3
HO
OCOOH
O
H2N
OTBDPS
O
OMe
O
H2N
OTBDPSO
OMe
ON3
BocHN
OTBDMS
BnO
OMe
ON3
HNBocOTBDMS
OBn OMe
ON3
HNBocOPhth
OBn OMe
81 6 82 83
84 85 86
OOH
SS O87 S
Schéma 36. Quelques scaffolds synthétisés à base de pyranose
88 Becattini, B.; Capozzi, G.; Falciani, C.; Menichetti, S.; Nativi, C.; Salvini, A. J. Carbohydr. Chem. 2000, 19, 653-657. 89 Svejgaard, L.; Fuglsang, H.; Jensen P. B.; Kelly N. M.; Pederson, H.; Andersen, K.; Ruhland, T.; Jensen, K. J. J. Carbohydr. Chem. 2003, 22, 179-184. 90 Moitessier, N.; Henry, C.; Aubert, N.; Chapleur, Y. Tetrahedron Lett. 2005, 46, 6191-6194. 91 Saeed, M.; Abbas, M.; Raid J. A. J.; Zahid. M.; Voelter, W. Tetrahedron Lett. 2003, 44, 315-317.
40
Bibliographie - Chapitre II
II.3.2. SCAFFOLDS FURANIQUES
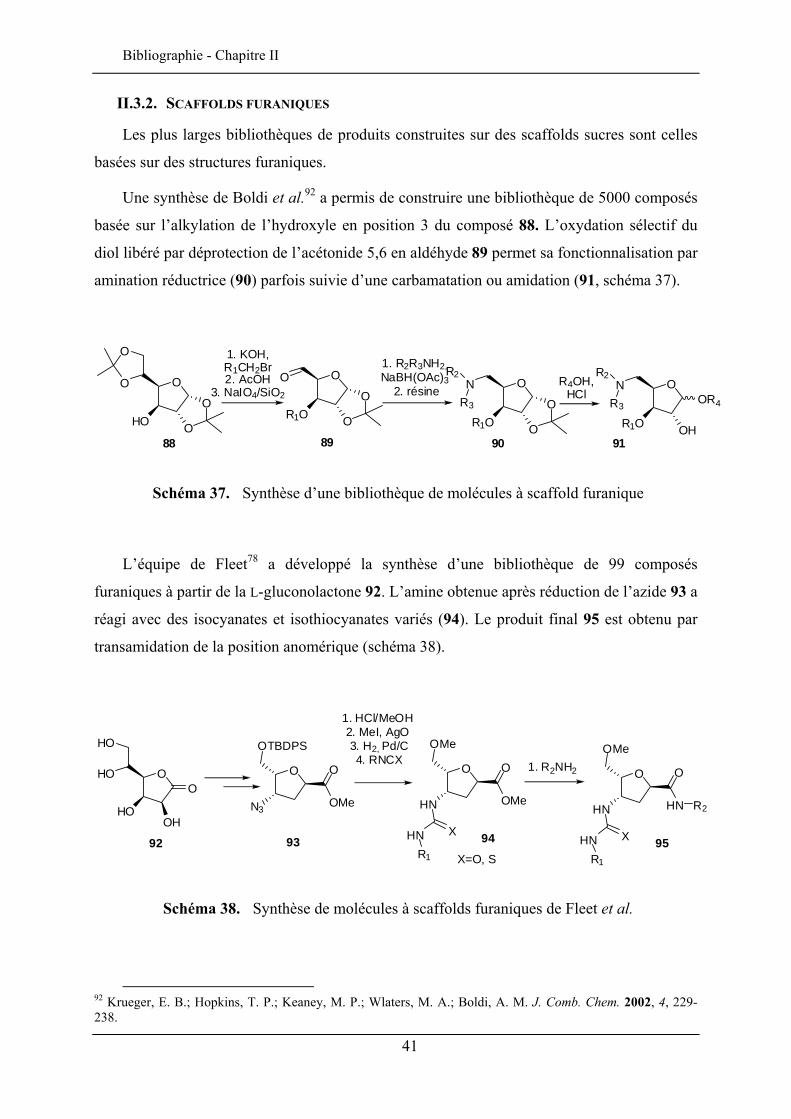
Les plus larges bibliothèques de produits construites sur des scaffolds sucres sont celles
basées sur des structures furaniques.
Une synthèse de Boldi et al.92 a permis de construire une bibliothèque de 5000 composés
basée sur l’alkylation de l’hydroxyle en position 3 du composé 88. L’oxydation sélectif du
diol libéré par déprotection de l’acétonide 5,6 en aldéhyde 89 permet sa fonctionnalisation par
amination réductrice (90) parfois suivie d’une carbamatation ou amidation (91, schéma 37).
O
O
O
O
O
HO
1. KOH,R1CH2Br2. AcOH
3. NaIO4/SiO2
O
O
OO
R1O
O
O
O
N
R1O
1. R2R3NH2,NaBH(OAc)3
2. résineR3
R2O
OH
N
R1O
R3
R2
OR4
88 89 90 91
R4OH,HCl
Schéma 37. Synthèse d’une bibliothèque de molécules à scaffold furanique
L’équipe de Fleet78 a développé la synthèse d’une bibliothèque de 99 composés
furaniques à partir de la L-gluconolactone 92. L’amine obtenue après réduction de l’azide 93 a
réagi avec des isocyanates et isothiocyanates variés (94). Le produit final 95 est obtenu par
transamidation de la position anomérique (schéma 38).
O
HO
HO
HOOH
OO
N3
OTBDPS
OMe
O O
HN
OMe
OMe
O
HNR1
X
O
HN
OMe
HN
O
HNR1
X
X=O, S
R2
92 9593 94
1. R2NH2
1. HCl/MeOH2. MeI, AgO3. H2, Pd/C4. RNCX
Schéma 38. Synthèse de molécules à scaffolds furaniques de Fleet et al.
92 Krueger, E. B.; Hopkins, T. P.; Keaney, M. P.; Wlaters, M. A.; Boldi, A. M. J. Comb. Chem. 2002, 4, 229-238.
41

Bibliographie - Chapitre II
D’autres composés amino-acides furaniques (schéma 39) ont été suggérés comme des
scaffolds potentiels comme ceux de Capozzi, Nativi et al.88 (96), Jiménez-Barbéro, Nicotra et
al.25 (7, 97, 98), ainsi que ceux de Fleet et al.93,94 (99, 100, 101, 102).
O
O
OH
H
BnOOBn
COOH
N3
O
NHCbzO
OCO2Me
O
CO2HBnO
O
BnO
N3
O
OBnBnO
O
BnOHO2C
N3
96 97 987
O
N3
TBDPSOCO2iPr
O
N3
CO2Me
OH
O
CO2Me
N3
HOO
N3
CO2Me
OH
99 100 101 102
Schéma 39. Scaffolds furaniques pour des bibliothèques de scaffolds sucres
II.3.3. SCAFFOLDS IMINOSUCRES
Un iminosucre (molécule ou l’atome d’oxygène intracyclique est remplacé par un azote
sur lequel un groupement fonctionnel peut être greffé) offre des possibilités de scaffold à
géométrie différente.
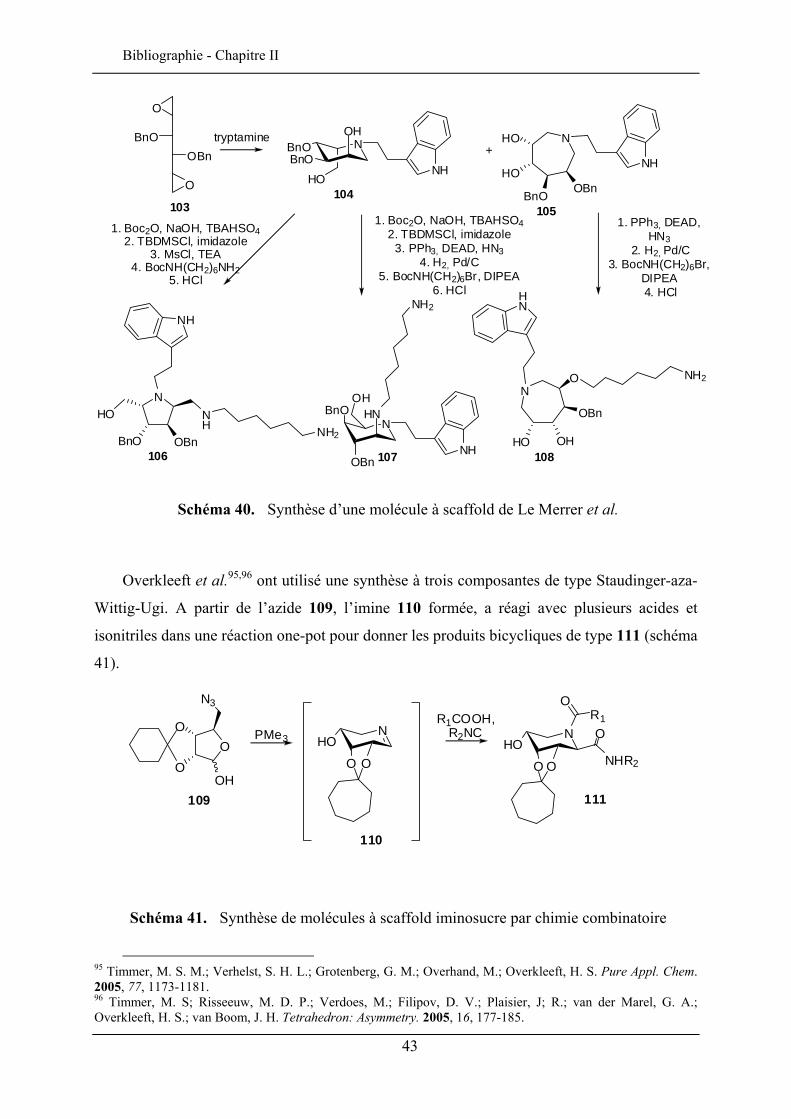
Le Merrer et al.59 ont été les premiers à synthétiser un scaffold iminosucre. La molécule
103 a réagi avec la tryptamine pour donner les produits intermédiaires 104 et 105. Après une
série de protection/déprotection, l’introduction d’une amine sur le cycle donne les 2 amines
106 et 107. Des structures macrocycliques ont aussi été préparées à partir de cette synthèse
(108, schéma 40).
93 Sanjayan, G. J.; Steward, A.; Hachisu, S.; Gonzalez, R.; Watterson, M. P.; Fleet, G. W. J. Tetrahedron Lett. 2003, 44, 5847-5851. 94 Watterson, M. P.; Edwards, A. A.; Leach, J. A.; Smith, M. D.; Ichihara, O.; Fleet, G. W. J. Tetrahedron Lett. 2003, 44, 5853-5857.
42
Bibliographie - Chapitre II
O
BnOOBn
O
NBnOBnO
OH
HONH
NHO
HO
BnO OBn
NH+
NBnO
OBn
HN
NH
OHN
NH
BnO OBn
HO NH NH2
NH2
N
HO OH
OBn
O
HN
NH2
tryptamine
103104
105
106 107 108
1. Boc2O, NaOH, TBAHSO42. TBDMSCl, imidazole
3. MsCl, TEA4. BocNH(CH2)6NH2
5. HCl
1. Boc2O, NaOH, TBAHSO42. TBDMSCl, imidazole
3. PPh3, DEAD, HN34. H2, Pd/C
5. BocNH(CH2)6Br, DIPEA6. HCl
1. PPh3, DEAD,HN3
2. H2, Pd/C3. BocNH(CH2)6Br,
DIPEA4. HCl
Schéma 40. Synthèse d’une molécule à scaffold de Le Merrer et al.
Overkleeft et al.95,96 ont utilisé une synthèse à trois composantes de type Staudinger-aza-
Wittig-Ugi. A partir de l’azide 109, l’imine 110 formée, a réagi avec plusieurs acides et
isonitriles dans une réaction one-pot pour donner les produits bicycliques de type 111 (schéma
41).
OO
O
N3
OH
NHO
O O
R1COOH,R2NC N
HO
O O
OR1
NHR2
O
109
110
111
PMe3
Schéma 41. Synthèse de molécules à scaffold iminosucre par chimie combinatoire
95 Timmer, M. S. M.; Verhelst, S. H. L.; Grotenberg, G. M.; Overhand, M.; Overkleeft, H. S. Pure Appl. Chem. 2005, 77, 1173-1181. 96 Timmer, M. S; Risseeuw, M. D. P.; Verdoes, M.; Filipov, D. V.; Plaisier, J; R.; van der Marel, G. A.; Overkleeft, H. S.; van Boom, J. H. Tetrahedron: Asymmetry. 2005, 16, 177-185.
43
Bibliographie - Chapitre II
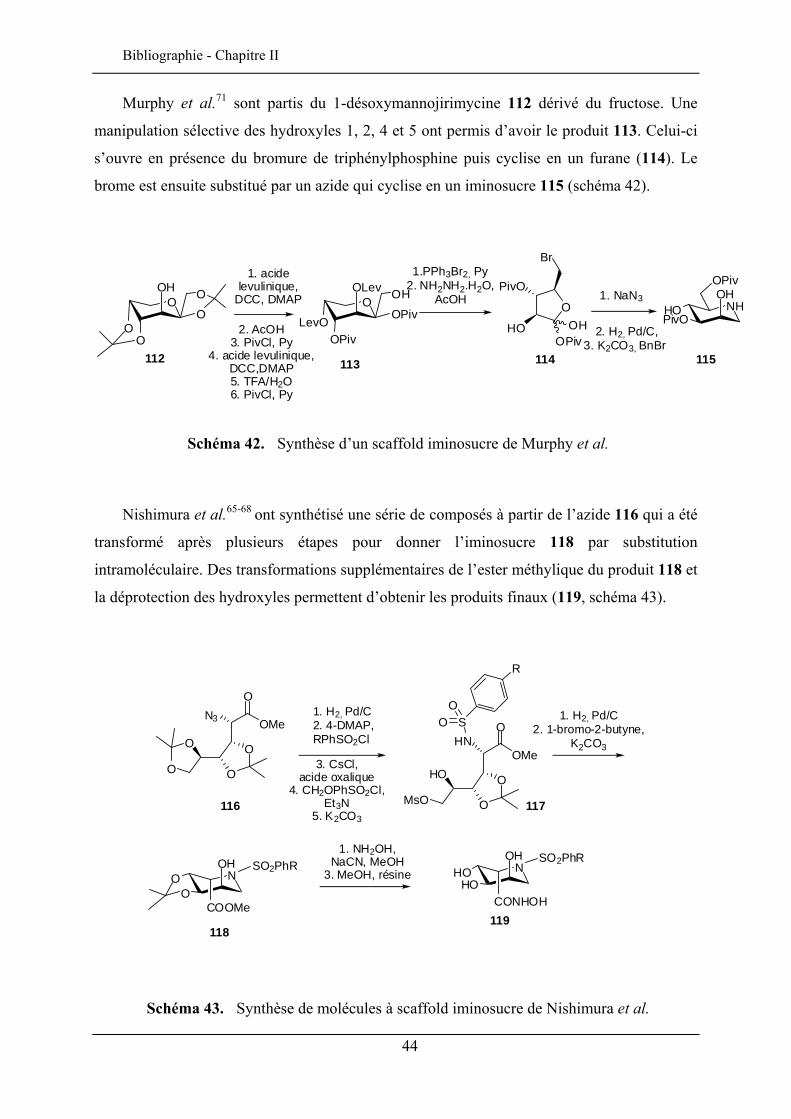
Murphy et al.71 sont partis du 1-désoxymannojirimycine 112 dérivé du fructose. Une
manipulation sélective des hydroxyles 1, 2, 4 et 5 ont permis d’avoir le produit 113. Celui-ci
s’ouvre en présence du bromure de triphénylphosphine puis cyclise en un furane (114). Le
brome est ensuite substitué par un azide qui cyclise en un iminosucre 115 (schéma 42).
OO
OO
OH OO
OPivLevO
OPiv
OLev OHO
HO
PivO
OPivOH
Br
NH
OPiv
PivO
OHHO
2. H2, Pd/C,3. K2CO3, BnBr
112 113 114 115
2. AcOH3. PivCl, Py
4. acide levulinique,DCC,DMAP5. TFA/H2O6. PivCl, Py
1.PPh3Br2, Py2. NH2NH2.H2O,
AcOH 1. NaN3
1. acidelevulinique,
DCC, DMAP
Schéma 42. Synthèse d’un scaffold iminosucre de Murphy et al.
Nishimura et al.65-68 ont synthétisé une série de composés à partir de l’azide 116 qui a été
transformé après plusieurs étapes pour donner l’iminosucre 118 par substitution
intramoléculaire. Des transformations supplémentaires de l’ester méthylique du produit 118 et
la déprotection des hydroxyles permettent d’obtenir les produits finaux (119, schéma 43).
O
O
O
O
OMe
MsO
HO
O
O
OMe
NOH SO2PhR
OO
COOMe
NOH SO2PhR
HOHO
CONHOH
116 117
118119
N3
O
HNS
OO
R
O
3. CsCl,acide oxalique
4. CH2OPhSO2Cl,Et3N
5. K2CO3
1. H2, Pd/C2. 1-bromo-2-butyne,
K2CO3
1. NH2OH,NaCN, MeOH
3. MeOH, résine
1. H2, Pd/C2. 4-DMAP,RPhSO2Cl
Schéma 43. Synthèse de molécules à scaffold iminosucre de Nishimura et al.
44
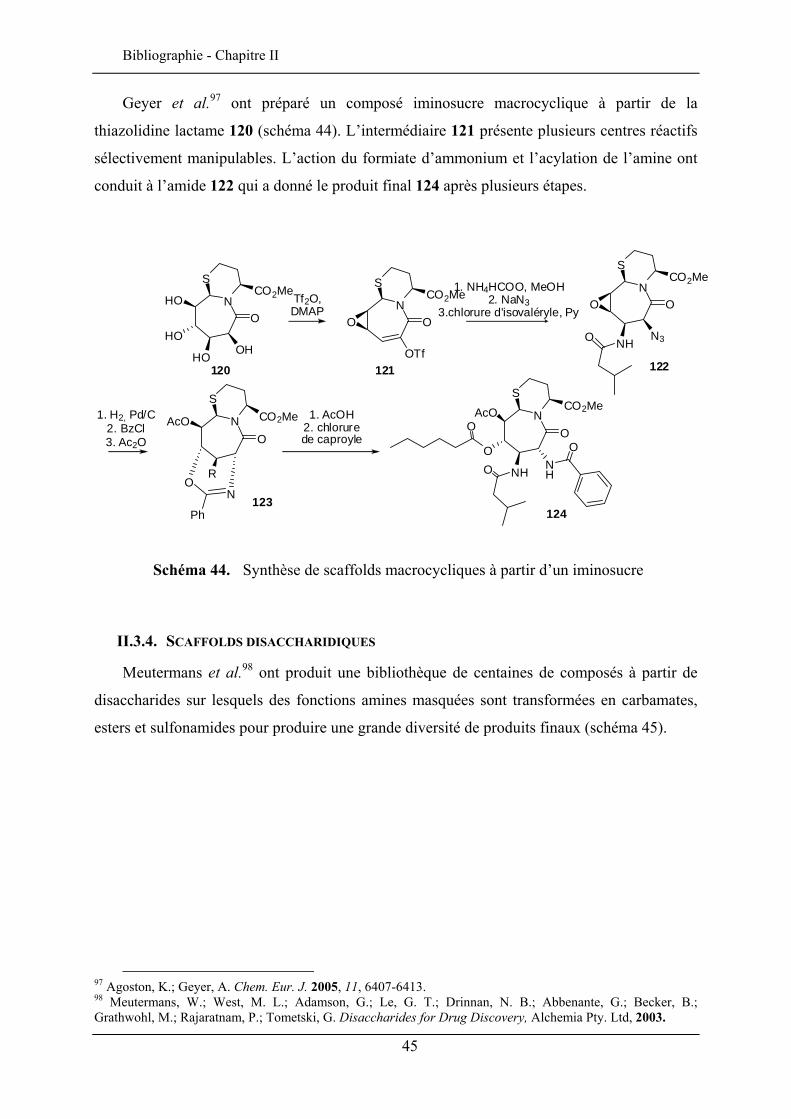
Bibliographie - Chapitre II
Geyer et al.97 ont préparé un composé iminosucre macrocyclique à partir de la
thiazolidine lactame 120 (schéma 44). L’intermédiaire 121 présente plusieurs centres réactifs
sélectivement manipulables. L’action du formiate d’ammonium et l’acylation de l’amine ont
conduit à l’amide 122 qui a donné le produit final 124 après plusieurs étapes.
N
S
HO
HO
HO OH
O
CO2MeN
S
O
CO2Me
O
OTf
N
S
O
CO2Me
O
NHN3O
Tf2O,DMAP
1. NH4HCOO, MeOH2. NaN3
3.chlorure d'isovaléryle, Py
120 121 122
N
S
AcO
OR
O
CO2Me
N
Ph
N
S
AcO
O
NH
CO2Me
O
O NH
OO
1. H2, Pd/C2. BzCl3. Ac2O
124123
1. AcOH2. chlorurede caproyle
Schéma 44. Synthèse de scaffolds macrocycliques à partir d’un iminosucre
II.3.4. SCAFFOLDS DISACCHARIDIQUES
Meutermans et al.98 ont produit une bibliothèque de centaines de composés à partir de
disaccharides sur lesquels des fonctions amines masquées sont transformées en carbamates,
esters et sulfonamides pour produire une grande diversité de produits finaux (schéma 45).
97 Agoston, K.; Geyer, A. Chem. Eur. J. 2005, 11, 6407-6413. 98 Meutermans, W.; West, M. L.; Adamson, G.; Le, G. T.; Drinnan, N. B.; Abbenante, G.; Becker, B.; Grathwohl, M.; Rajaratnam, P.; Tometski, G. Disaccharides for Drug Discovery, Alchemia Pty. Ltd, 2003.
45

Bibliographie - Chapitre II
OPMBOPMBO
H2N
OPMB
OO
NHFmoc
OPMBN3OPMBO
PMBONH
OPMB
OO
NH2
OPMBN3
XR1
1. R3NCOou R3SO2Clou R3COOH
2. TFA
1. R1NCOou R1SO2Clou R1COOH2. Pipéridine
OPMBOPMBO
NH
OPMB
OO
NH
OPMBNH2
XR1
1. R2NCOou R2SO2Clou R2COOH2. Zn, NH4Cl
XR2
OPMBOPMBO
NH
OPMB
OO
NH
OPMBHN
XR1XR2
X R3
Schéma 45. Synthèse d’une famille de molécules à scaffold disaccharide
De nombreux exemples de scaffolds à structure glucidique existent. Certaines structures
se sont révélées biologiquement actives comme par exemple les inhibiteurs de la
somatostatine, d’intégrine, des antiviraux et antibactériens.
C’est dans le cadre de ce concept que ce présent travail se situe. Il vise à valoriser la
stratégie CMGL pour la construction de produits différemment fonctionnalisés.
46

Bibliographie - Chapitre II
47

Bibliographie - Chapitre II
48

Bibliographie – Chapitre III
CHAPITRE III. IMAGERIE MEMBRANAIRE PAR OPTIQUE NON
LINEAIRE
III.1. POTENTIEL MEMBRANAIRE EN NEUROBIOLOGIE
Les microscopies non linéaires combinant la génération de second harmonique (GSH) et
la fluorescence excitée à deux photons (TPEF) connaissent un développement considérable
depuis une dizaine d'années car elles allient à une excellente résolution spatiale, une
profondeur de pénétration du rayonnement lumineux importante et des dommages cellulaires
très réduits.
La technique d’imagerie par détection de l’activité optique de sondes a suscité l’intérêt
des neurobiologistes depuis une dizaine d’années avec la perspective d’imager des neurones
jusqu’à un millimètre de profondeur in vivo pour permettre l’étude de l’activité neuronale à
l’échelle cellulaire avec l’ensemble du réseau neuronal dans un environnement physiologique
préservé. La description de l'activité d'un réseau de neurones dans le cerveau implique la
mesure simultanée de l'activité électrique individuelle d'un grand nombre de neurones in vivo.
Actuellement, ces mesures sont réalisées au moyen de micro-électrodes implantées
directement dans la zone cervicale étudiée. Cette technique très invasive est délicate à mettre
en œuvre et ne permet de mesurer que l'activité moyenne d'un petit nombre de cellules.
La microscopie GSH s'est révélée être une technique très intéressante pour l'imagerie des
membranes cellulaires en permettant d'imager spécifiquement les interfaces et en présentant
une exceptionnelle sensibilité aux variations du potentiel de membrane.99, 100 Elles semblent
particulièrement prometteuses pour évaluer simultanément et instantanément l'activité
individuelle d'une population de neurones101, , , 102 103 104 sans implémentation d’électrodes.
99 Moreaux, L.; Sandre, O.; Charpak, S.; Blanchard-Desce, M.; Mertz, J. Biophys. J. 2001, 80, 1568-1574. 100 Campagnola, P. J.; Wei, M.-D.; Lewis, A.; Loew, L. M. Biophys. J. 1999, 77, 3341-3349. 101 Dombeck, D. A.; Blanchard-Desce M.; Webb, W. W. J. Neurosci. 2004, 24, 999–1003. 102 Nemet, B. A.; Nikolenko, V.; Yuste, R. J. Biomed. Opt. 2004, 9, 873-881. 103 Moreaux, L.; Pons, T.; Dambrin, V.; Blanchard-Desce M.; Mertz, J. Opt. Lett., 2003, 28, 625-627. 104 Bouevitch, O.; Lewis, A.; Pinevsky, I.; Wuskell J. P.; Loew, L. M. Biophys. J. 1993, 65, 672-679.
49

Bibliographie – Chapitre III
En effet, lors de la transmission neuronale des informations, le stimulus se propage d’une
cellule à l’autre en traversant la membrane. Celle-ci se polarise, ce qui induit l’apparition d’un
potentiel membranaire qui modifierait effectivement le signal GSH des sondes synthétiques
associées au niveau de la membrane.
III.2. FLUORESCENCE PAR EXCITATION A DEUX PHOTONS (TPEF) ET
GENERATION DU SECOND HARMONIQUE (GSH) POUR LA MESURE DU
POTENTIEL MEMBRANAIRE
III.2.1. PRINCIPE PHYSIQUE
La fluorescence est un phénomène qui apparaît lorsqu’une molécule absorbe un photon et
passe à un état excité. Une partie de l’énergie absorbée est transmise à l’environnement et
l’autre partie est émise sous la forme d’un photon fluorescent correspondant à la désexcitation
de la molécule.
La fluorescence à deux photons est un phénomène d’optique non linéaire qui diffère du
phénomène de la fluorescence classique par le fait que l’état excité est atteint par absorption
simultanée de deux photons possédant chacun la moitié de l’énergie apportée (figure 1).
La génération du second harmonique (GSH) est un phénomène d’optique non linéaire
dans lequel le faisceau incident de longueur d’onde λ va produire un rayonnement à
exactement la moitié de la longueur d’onde incidente (λ/2). Lors de ce processus, la molécule
n’échange pas d’énergie avec son environnement. C’est un phénomène cohérent puisque la
molécule n’absorbe pas l’énergie incidente mais la diffuse totalement de manière non linéaire
(conversion de 2 photons en un seul, figure 1). La GSH est un phénomène spécifique d’une
molécule non centrosymétrique et qui n’est pas répartie dans le milieu d’une manière
centrosymétrique.
50

Bibliographie – Chapitre III
Figure 1. Conversion de deux photons incidents en un seul par TPEF et GSH
La figure 2 illustre bien la spécificité de la GSH par rapport à la TPEF. L’image obtenue
par TPEF montre un fort signal de fluorescence produit par l’excitation des sondes localisées
dans les membranes des vésicules. La deuxième image, obtenue par GSH, montre que le
signal des sondes localisées dans la zone de contact est éteint. Cette extinction du signal
traduit une distribution symétrique de la sonde dans cette zone.
Figure 2. Images par TPEF (à gauche) et par GSH (à droite) de deux vésicules adhérentes99
III.2.2. AVANTAGES DE LA TPEF ET DE LA GSH EN MICROSCOPIE BIOLOGIQUE
La TPEF et la GSH présentent plusieurs avantages par rapport à la fluorescence
classique.
- Profondeur de pénétration augmentée dans les tissus diffusants :
51

Bibliographie – Chapitre III
La diffusion de la lumière visible par les tissus est le phénomène principal qui limite la
profondeur d’observation en microscopie conventionnelle. Elle a pour effet de diminuer la
fluorescence provenant du point focal105 et d’augmenter le bruit de fond puisque la lumière
émise est générée sur tout le trajet de l’illumination, le signal au point focal se retrouve perdu
dans le bruit du fond de la fluorescence hors le point focal. Par suite, en fluorescence
classique, la faible résolution diminue fortement la profondeur d’imagerie. La microscopie
non linéaire, quant à elle, permet de préserver la résolution à plus grande profondeur et cela
pour les raisons suivantes :
a. Les longueurs d’onde utilisées en ONL, situées dans le proche infra-rouge sont
nettement moins diffusées et absorbées par les tissus que dans le cas de la microscopie
conventionnelle, ce qui permet une meilleure pénétration dans les tissus.
b. Le confinement de l’excitation dans un volume très précis (1µm3) permet d’avoir des
images à grande résolution même en milieu diffusant (figure 3). L’image tridimensionnelle est
ensuite obtenue par balayage du faisceau lumineux à travers tout l’échantillon où la
fluorescence est récoltée point par point.
Ainsi, la possibilité de visualiser des tissus biologiques intacts plus en profondeur, tout en
gardant une grande résolution tridimensionnelle est un des avantages majeurs de la
microscopie par ONL.
Figure 3. Illustration de la dispersion de la lumière sur tout le trajet lumineux dans le cas de la fluorescence classique (a) alors qu’elle est condensée dans un volume
très précis par TPEF (b) (Webb et al., site de l’université Cornell)
105 Centonze, V. E.; White, J. G. Biophys. J. 1998, 75, 2015-2024.
52

Bibliographie – Chapitre III
- Phototoxicité et photoblanchiment réduits :
La cause principale de la phototoxicité est la formation d’espèces oxygénées réactives,
suite à un transfert d’énergie non-radiatif par un chromophore cellulaire. Ces espèces
oxygénées toxiques pour les cellules provoquent rapidement la mort cellulaire, diminuant
ainsi la possibilité de faire une imagerie de longue durée. En microscopie TPEF, le
confinement de l’excitation permet de restreindre les dommages optiques au niveau du
volume focal, donc de réduire la phototoxicité. D’autre part, la fluorescence classique détruit
les colorants par photoblanchiment suite à de nombreux cycles excitation-fluorescence. Ce
phénomène est réduit en ONL pour les mêmes raisons.
Le GSH offre un avantage supplémentaire qui réside dans la possibilité d’effectuer une
imagerie à contraste plus prononcé de la membrane par extinction du signal dans le
cytoplasme et dans le milieu extracellulaire qui sont centrosymétriques (figure 4).
Figure 4. Images simultanées de cellules isolées en culture. Le signal par TPEF est visible partout où il y a du produit. La GSH est générée uniquement dans les
membranes, là où les sondes présentent une direction définie99
La TPEF et la GSH sont deux techniques d’optique complémentaires : la première nous
renseigne sur le nombre et la localisation des sondes, la seconde nous informe en plus sur le
degré d’asymétrie du chromophore qui est fonction de son environnement local.
III.2.3. PRINCIPE DE MESURE DU POTENTIEL MEMBRANAIRE PAR ONL
La membrane cellulaire est polarisée. Cette différence de potentiel due à la séparation de
charge de part et d’autre de la membrane permet les échanges avec le milieu extérieur et le
transfert des signaux d’une cellule à l’autre.
53

Bibliographie – Chapitre III
Le signal de fluorescence d’une sonde dépend de son environnement, un changement de
la polarisation de la membrane modifie donc le signal optique de fluorescence de la sonde
membranaire. Même si cette méthode présente l’inconvénient de ne pas être instantanée, elle
permet de mesurer optiquement la valeur du potentiel membranaire.
La GSH, quant à elle, permet de détecter de façon instantanée un changement de
polarisation selon deux mécanismes :
a. Une réponse électrochromique qui se traduit par une modification des propriétés
optiques intrinsèques du chromophore sous l’effet d’un champ statique.
b. Une réponse orientationnelle de la sonde dans la membrane : le champ électrique est
capable d’orienter les sondes parallèlement les unes aux autres.
Ces deux réponses induisent une exaltation du signal optique de la sonde.
III.2.4. SONDES UTILISEES POUR LA MESURE DU POTENTIEL MEMBRANAIRE
La technique physique choisie (TPEF et GSH simultanées) implique de concevoir des
chromophores dipolaires (associant groupement donneur et accepteur via un système
conjugué). La sonde optimale pour le marquage des membranes doit avoir une bonne réponse
à deux photons à 800 nm. Cette longueur d’onde présente à la fois la zone de transparence
biologique, et la longueur d’onde optimale des lasers à base de Saphire-Titane utilisés
actuellement.
Le schéma 46 présente la structure la plus utilisée pour mesurer le potentiel
membranaire.99,100, , , , 106 107 108 109
N+
N
-O3S
Di-4-ANEPPS
Schéma 46. Structure utilisée pour mesurer le potentiel membranaire par ONL
106 Blanchard-Desce, M., Comptes Rendus Physique 2002, 3, 439-448. 107 Campagnola, P. J.; Wei, M.-D.; Lewis, A.; Loew, L. M. Biophys. J. 1999, 77, 3341-3349. 108 Mertz, J. Curr. Opin. Neurobiol. 2004, 14, 610-616. 109 Zipfel, W. R.; Williams, R. M.; Webb, W. W. Nat. Biotechnol. 2003, 21, 1369-1377.
54

Bibliographie – Chapitre III
Cette sonde synthétique est connue sous le nom de l’ANEP
(AminoNaphtylEthenylPyridinium) développée par l’équipe de Loew.110, 111 C’est une
molécule présentant un donneur et un accepteur d’électrons de part et d’autre d’un pont
électronique (type D-π-A). Le bon rapport signal/bruit et sa haute sensibilité en GSH en
fonction des changements du champ électrique (43% pour 100 mV en 1 ms : donc 4 fois plus
que la sensibilité d’une sonde en fluorescence classique)111 font d’elle une sonde très
intéressante pour les membranes cellulaires.
Les molécules de la famille de l’ANEP ont permis le développement de la microscopie
par TPEF et GSH et son application dans l’étude des membranes. L’inconvénient majeur que
présente cette famille de composés est qu’elle finit par marquer les deux feuillets
membranaires ce qui éteint le signal par GSH à cause de la symétrie obtenue. De là vient
l’intérêt de développer de nouvelles sondes biologiques pour l’optique non linéaire et
respectant les contraintes de forme liées à sa mise en œuvre dans la membrane biologique.
III.3. INGENIERIE MOLECULAIRE POUR LA SYNTHESE DE NOUVELLES
SONDES OPTIQUES MEMBRANAIRES
III.3.1. LA MEMBRANE CELLULAIRE
La membrane cellulaire est essentiellement composée d’une bicouche de phospholipides
amphiphiles (figure 5). Chaque lipide possède une tête polaire hydrophile (phosphates chargés
négativement) orientée vers l’extérieur de la membrane, et une chaîne hydrophobe (chaînes
d’acides gras saturés et insaturés) orientée vers l’intérieur. Certains enchaînements de sucres
sont aussi présents à la surface cellulaire. Ils sont impliqués dans un grand nombre de
phénomènes biologiques liés à la communication intercellulaires (différenciation,
immunologie, métastases…).112
110 Bouevitch, O.; Lewis, A.; Pinevsky, I.; Wuskell, J. P.; Loew, L. M. Biophys. J. 1993, 65, 672-679. 111 Millard, A. C.; Jin, L.; Mei-de, W.; Wuskell, J. P.; Lewis, A.; Loew, L. M. Biophys. J. 2004, 86, 1169-1176. 112 (a) Danishefsky, S. J.; Allen, J. R. Angew. Chem. 2000, 836-863. (b) Galonić, D. P., Gin, D. Y. Nature, 2007, 446, 1000-1007. (c) Seeberger, P. H.; Werz, D. B. Nature, 2007, 446, 1046-1051. (d) Seeberger, P. H. Nature, 2005, 437, 1239.
55

Bibliographie – Chapitre III
Figure 5. Constitution de la membrane plasmique (Biologie moléculaire de la cellule, Médecine-Sciences, Flammarion, p. 503)
III.3.2. LES SUCRES DANS L’ETUDE DES MEMBRANES
La constitution polaire des membranes et la présence des sucres en abondance dans sa
structure nous ont amenées à choisir d’incorporer des sucres comme groupements polaires sur
des sondes fluorescentes pour conférer la solubilité et l’amphiphilie souhaitées afin d’assurer
une bonne insertion dans la bicouche lipidique.
Ces chromophores amphiphiles peuvent s’insérer dans les membranes comme des
lipides. La partie hydrophile polaire restera à l'extérieur de la membrane et la partie lipophile
s'ancrera profondément dans la bicouche lipidique (figure 6).
Cette insertion assure une orientation parallèle de tous les chromophores. Leur sensibilité
au champ électrique permet l’observation instantanée d’un signal par GSH. L’étude
simultanée par TPEF permettra aussi d’étudier la répartition des fluorophores à l’intérieur de
la membrane. Les molécules internalisées dans la cellule manquent d’organisation et ne
produisent pas de signal en GSH.
56

Bibliographie – Chapitre III
O
HO
O
O
A
RO
Figure 6. Insertion d’une sonde amphiphile dans une bicouche lipidique
Dans la littérature, certaines structures portant des sucres comme agents hydrophiles ont
été décrites pour l’étude de la membrane cellulaire. Citons dans ce contexte, les glycolipides
marqués113 qui ont montré une forte insertion dans les membranes et ont permis l’étude de la
distribution des glycolipides dans ces dernières, leur transport et leur métabolisme dans les
cellules, les glycosylphosphatidylinositols greffés sur des protéines marqués ou
fluorescents114 pour le développement des vaccins antitumoraux et des glycopolymères
fluorescents115 pour l’étude des phénomènes à la surface de la cellule.
III.3.3. LES SUCRES DANS L’OPTIQUE NON LINEAIRE
Lors de l’étude du potentiel membranaire par ONL, un phénomène de retournement des
colorants dans les deux feuillets membranaires peut être observé. Ce phénomène de « flip-
flop » qui tend à équilibrer la composition des deux feuillets en colorants conduit à
l’extinction du signal par GSH. L’utilisation des chromophores chiraux a été proposée pour
éviter la diminution du signal même après le flip-flop. Certaines équipes ont utilisé un sucre
pour pallier au problème d’extinction du signal. Il s’agit d’un ribose accroché à un
chromophore104 (schéma 47). La chiralité du sucre aide à garder la non-symétrie dans le cas
ou les sondes sont localisées tête-bêche dans les deux feuillets membranaires, afin de
113 Schwarzmann, G. Semin. Cell. Dev. Biol. 2001, 12, 163-171. 114 (a) Premkumar, D. R.; Fukuoka, Y.; Sevlever, D.; Brunschwig, E.; Rosenberry, T. L.; Tykocinski, M. L.; Medof, M. E. J. Cell Biochem. 2001, 82, 234-245. (b) Paulick, M. G.; Wise, A. R.; Forstner M. B.; Groves, J. T.; Bertozzi, C. R. J. Am. Chem. Soc.2007, 129, 11543-11550. 115 Rabuka; D.; Forstner, M. B.; Groves, J. T.; Bertozzi, C. R. J. Am. Chem. Soc. 2008, sous presse.
57
Bibliographie – Chapitre III
visualiser le signal de la GSH. La solubilité de cette sonde provient de la charge cationique
portée par l’azote.
N
N+O
OH
OHHOH2C Cl-
Schéma 47. Sonde de mesure du potentiel membranaire par ONL présentant un ribose
58

Bibliographie
En résumé, dans ce chapitre bibliographique, nous avons d’abord détaillé les
différentes synthèses décrites dans la littérature pour la synthèse des CMGs, et leurs
utilisations comme éléments de construction de structures plus complexes. La CMGlcL
n’étant jamais décrite, nous avons montré la voie directe de son obtention par oxydation de
l’isomaltulose. Les applications de cette stratégie ont aussi été décrites pour la synthèse de
nouveaux glucoconjugués.
Ensuite, nous avons montré les voies de synthèse qui ont été établies dans la littérature
pour obtenir des scaffolds sucres à base de pyranoses, furanoses, iminosucres et disaccharides.
Plusieurs bibliothèques de molécules ont été construites parmi lesquelles certaines se sont
révélées biologiquement actives comme par exemple, des inhibiteurs de la somatostatine, des
inhibiteurs d’intégrine, des antiviraux et des antibactériens.
Enfin, nous avons rappelé les principes de l’optique non linéaire et décrit les deux
techniques (TPEF et GSH) qui nous intéressent particulièrement. Les structures moléculaires
qui répondent aux exigences de ces deux optiques sont montrées ainsi que leurs applications
pour l’imagerie membranaire.
Le travail qui va être exposé dans les chapitres suivants décrira une nouvelle alternative
plus simple et générale pour l’accès aux CMGLs, et l’utilisation de ces lactones par ouverture
et substitution sélective des différents hydroxyles pour conduire à de nouvelles molécules
fonctionnelles mono- et disaccharidiques.
Du point de vue des applications en optique, notre travail se portera sur l’incorporation de
certains sucres sur des sondes actives à la fois en TPEF et GSH. L’évaluation en imagerie et
les propriétés physico-chimiques de ces sondes seront aussi présentées.
59

60

DEUXIEME PARTIE
RESULTATS ET DISCUSSION

Résultats - Chapitre I
CHAPITRE I. SYNTHESE DES CMGS ET DES CMGLS
I.1. INTRODUCTION
La voie d’oxydation de l’isomaltulose (schéma 48, voie A) donnant exclusivement le α-
CMGlcL (voir commentaire sur la nomenclature section I.1.2, chapitre I de la partie
bibliographique) une autre voie d’accès à cette lactone bicyclique a été mise au point dans le
laboratoire. Elle fait intervenir la glycosidation par l’alcool allylique suivie de l’oxydation de
la double liaison (voie B).49 Cette voie a permis l’obtention d’une série de lactones bâties sur
deux monosaccharides, le glucose et le galactose, avec les deux configurations anomériques
possibles.
Nous avons voulu ensuite explorer la synthèse de ces lactones en utilisant une troisième
voie : l’alkylation anomérique par un bromure présentant une fonction acide masquée, le
bromoacétate de tert-butyle (voie C).
OHOHO
HO
HO
OO OH
OH
OH
OH
voie A
voie Bglycosylation
AllOH
voie CAlkylation
anomérique
ORO
OH
ORO
OO
O
Br O tBuO
ORO
O
ORO
O OtBu
O
ORO
O OH
O
ORO
OO
OH
Schéma 48. Différents chemins d’accès aux CMGs et CMGLs : oxydation de
l’isomaltulose, glycosylation de l’alcool allylique et alkylation anomérique
61

Résultats - Chapitre I
Dans ce chapitre, nous détaillerons la synthèse d’une série de lactones mono- et
disaccharidiques à partir de cette réaction. Dans un second temps, nous décrirons une synthèse
de CMGLs présentant des groupements protecteurs benzyles.
I.2. L’ALKYLATION ANOMERIQUE
La réaction d’alkylation anomérique consiste à déprotoner la position anomérique à l’aide
d’une base et à faire réagir l’anion formé avec un agent électrophile, ici le bromoacétate de
tert-butyle. Cet électrophile présente une fonction acide carboxylique masquée par le
groupement tert-butyle, facilement hydrolysable.
Dans la littérature, l’alkylation en milieu basique du groupement hydroxyle anomérique
par divers agents électrophiles a été largement étudiée par l’équipe de Richard R. Schmidt,116
les agents alkylants étant par exemple des sulphates dialkylés, le bromure de benzyle ou
d’allyle117 et autres O-triflates d’hydroxyles glucidiques pour la synthèse de disaccharides.118
L’alkylation anomérique des sucres libres ou acétylés par le bromoacétate de tert-butyle
n’a, à notre connaissance, jamais été décrite dans la littérature. Les seuls exemples qui s’en
rapprochent sont des brevets de Platzek et al.119, ,120 121 qui décrivent sommairement la réaction
sur des sucres benzylés. En revanche, certaines utilisations de cet électrophile comme
précurseur d’une fonction acide sur différents sucres en position non anomérique26,27,28,122 ou
sur des analogues123 ou sur d’autres structures organiques (négamycine)124 ont été décrites.
I.2.1. SYNTHESE DES ESTERS DE TERT-BUTYLE GLYCOSIDES
I.2.1.1. A partir de sucres mono-déprotégés
Cette réaction a d’abord été étudiée sur les sucres acétylés présentant l’hydroxyle en
position anomérique libre obtenu facilement par déprotection anomérique sélective soit par
116 Schmidt, R. R. Modern Methods in Carbohydrate Synthesis; Kahn, S. H.; O’Neill, R. A.; Eds.; Hardwood academic publishers, Amsterdam, 1996; p. 20-54. 117 Klotz, W.; Schmidt, R. R. Liebigs Ann. Chem. 1993, 683-690. 118 Tsvetkov, Y. E.; Klotz, W.; Schmidt, R. R. Liebigs Ann. Chem. 1992, 371-375. 119 Platzek J.; Graske, K. D.; Niedballa, U. Int Patent WO 2003006474 (2003); Chem. Abstr. 2003, 138, 106941; 120 Platzek J.; Niedballa, U.; Graske, K. D. German patent DE 10129677 (2003); Chem. Abstr. 2003, 138, 56184; 121 Platzek J.; Niedballa, U.; Graske, K. D. Int Patent WO 2002102816 (2002); Chem. Abstr. 2003, 138, 39497. 122 Jarosz, A.; Listkowski, A.; Lewandowski, B.; Ciunik, Z.; Brzuszkiewicz, A. Tetrahedron 2005, 61, 8485-8492. 123 Wang, X.; Migawa, M. T.; Sannes-Lowery, K. A.; Swaze, E. E. Bioorg. Med. Chem. Lett. 2005, 15, 4919-4922. 124 Raju, B.; Mortell, K.; Anandan, S.; O’Dowd, H.; Gao, H.; Gomez, M.; Hackbarth, C.; Wu, C.; Wang, W.; Yuan, Z.; White, R.; Trias, J.; Patel, D. V. Bioorg. Med. Chem. Lett. 2003, 13, 2413-2418.
62

Résultats - Chapitre I
réaction de l'acétate d’hydrazinium125 dans le DMF soit par l’action de l’aminoéthanol dans
l’acétate d’éthyle,126 moins efficace que l’acétate d’hydrazinium.
La réaction a été, en premier lieu, effectuée sur le glucose 125 (schéma 49) à température
ambiante, dans le DMF, avec K2CO3 (2,5 équiv.) comme base, et 2 équivalents du
bromoacétate de tert-butyle (schéma 49).
OAcOAcO
OAc
OAc
OH
OAcOAcO
AcO
OAc
OO
O
i.
125 125a7
82 1
OAcOAcO
OAc
OAc
OO
O
125b
+
Schéma 49. i. DMF, K2CO3 (2,5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 88%,
(α/β = 86:14)
Après 48 heures, la réaction est totale, et l’ester est obtenu avec un rendement de 88%
sous la forme d’un mélange d’anomères 125a et 125b. La séparation des anomères a été
facilement effectuée sur colonne de silice.
Le rapport α:β est déterminé par la RMN 1H sur la base des intégrations des protons
anomériques sur le mélange d’anomères brut. Un doublet avec une constante de couplage de
J1,2 = 3,8 Hz est observé à 5,17 ppm. Il correspond au proton anomérique de l’anomère
α 125a. Celui de l’anomère β (125b) est observé à 4,68 ppm avec une constante de couplage
de J1,2 = 7,8 Hz. Le rapport des intégrations de ces deux protons révèle une sélectivité en
faveur de l’anomère α (α/β : 86/14). La séparation de chaque anomère a été effectuée sur gel
de silice pour avoir les deux glucosides α et β purs avec les rendements respectifs de 72% et
8%.
La figure 7 montre les spectres RMN du glucoside 125b qui a permis son identification
structurale. La RMN du proton montre un singulet à 1,47 ppm qui intègre pour 9 protons. De
plus, un pic intégrant pour deux protons correspondant au groupe carboxyméthyle (H-7)
apparaît à 4,15 ppm. En RMN du carbone, un pic à 28 ppm révélant la présence d’un
125 Excoffier, G.; Gagnaire, D.; Utille J.-P. Carbohydr. Res. 1975, 39, 368-373. 126 Nudelman, A.; Herzig, J.; Gottlieb H. E. Carbohydr. Res. 1987, 162, 145-152.
63
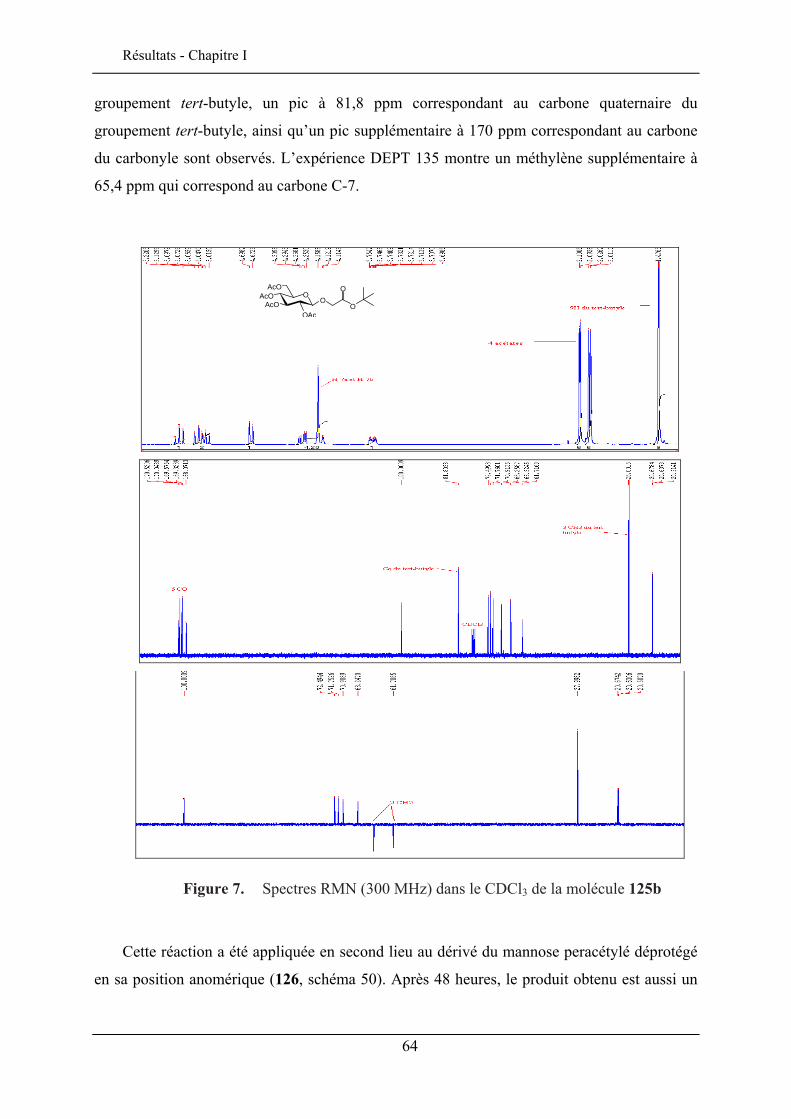
Résultats - Chapitre I
groupement tert-butyle, un pic à 81,8 ppm correspondant au carbone quaternaire du
groupement tert-butyle, ainsi qu’un pic supplémentaire à 170 ppm correspondant au carbone
du carbonyle sont observés. L’expérience DEPT 135 montre un méthylène supplémentaire à
65,4 ppm qui correspond au carbone C-7.
OAcOAcO
OAc
AcO
OO
O
Figure 7. Spectres RMN (300 MHz) dans le CDCl3 de la molécule 125b
Cette réaction a été appliquée en second lieu au dérivé du mannose peracétylé déprotégé
en sa position anomérique (126, schéma 50). Après 48 heures, le produit obtenu est aussi un
64

Résultats - Chapitre I
mélange d’anomères 126a et 126b dont le rapport, calculé par intégration des protons
anomériques du brut de la réaction dans le CDCl3, est de 97/3, en faveur de l’anomère α.
OAcOAcO
OAcOAc
OH
OAcOAcO
OAcOAc
OO
O
i.
126 126a
OAcOAcO
OAcOAc
OO
O
126b
+
Schéma 50. i. DMF, K2CO3 (2,5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 91%,
(α/β = 97:3)
La RMN du proton montre la présence de deux produits avec des constantes de couplage
des protons anomériques très proches : J1,2= 0,6 Hz à 4,96 ppm pour l’un et 1,0 Hz pour
l’autre anomère à 4,84 ppm. L’attribution a été effectuée à partir des spectres de RMN non
découplés du carbone enregistrés sur les produits purs. La constante de couplage du carbone
anomérique obtenue est JC1-H1 = 157,4 Hz ce qui correspond à l’anomère β 126b, elle est de
173,0 Hz dans le cas de l’anomère α 126a. Ces résultats ont été comparés à la littérature et se
sont montrés conformes aux résultats déjà décrits pour des composés similaires.127 La
séparation de l’anomère α a été effectuée sur silice pour donner le produit pur avec 83% de
rendement. L’anomère β n’étant pas en quantité suffisante, il n’a pas pu être purifié à partir de
cette réaction .
Cette réaction au aussi été appliquée au dérivé du galactose peracétylé, déprotégé en sa
position anomérique (127, schéma 51).
127 Ellestad, G. A. ; Martin, J. H. ; Morton, G. O., Sassiver, M. L. ; Lancaster, J. E. J. Antibiotics 1982, 10, 1418-1421.
65

Résultats - Chapitre I
OAcO
AcOOAc
OAc
OH
OAcO
AcOAcO
OAc
OO
O
i .
127 127a
OAcO
AcOAcO
OAc
OO
O
127b
+
Schéma 51. i. DMF, K2CO3 (2,5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 94%,
(α/β = 89:11)
Le mélange d’anomères a été obtenu avec 94% de rendement avec un rapport de α/β de
89/11. Les deux anomères 127a et 127b ont été purifiés sur gel de silice avec des rendements
de 65% et 9% respectivement.
Dans le cas où la réaction a été effectuée sur le dérivé peracétylé du L-fucose déprotégé
en sa position anomérique (128, schéma 52), le mélange d’anomères est obtenu avec 74% de
rendement avec un rapport α/β de 81/19.
OAc
OOAc
OO
OOAc
OAc
OOAc
OHOAc
i .
128 128bOAc
OOAc
OAc
OO
O
128a
+
Schéma 52. i. DMF, K2CO3 (2,5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 74%,
(α/β = 81:19)
Après purification, les deux anomères 128a et 128b sont obtenus avec 45% et 12%
respectivement. Les deux anomères ont été déterminés par des spectres de RMN du proton qui
ont été comparés à ceux de la littérature.23, 128 L’anomère α présente un doublet à 5,11 ppm
correspondant au proton anomérique avec une constante de couplage J1,2 = 3,7 Hz. Pour
128 Flowers, H. M. Carbohydr. Res. 1983, 119, 78-84.
66

Résultats - Chapitre I
l’anomère β, le doublet du H-1 résonne à 4,58 ppm avec une constante de couplage J1,2 = 7,9
Hz.
Cette réaction a été appliquée sur un dernier exemple monosaccharidique, la N-acétyl-
glucosamine peracétylée, déprotégée en sa position anomérique (129, schéma 53).
OAcOAcO
NHAc
OAc
OH
OAcOAcO
AcHN
OAc
OO
O
i.
129 129a
OAcOAcO
NHAc
OAc
OO
O
129b
+
Schéma 53. i. DMF, K2CO3 (2,5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 85%,
(α/β = 87:13)
Le mélange d’anomères est obtenu avec un rendement de 85% dans un rapport de
α/β : 87/13. L’anomère α est purifié sur gel de silice avec un rendement de 65%. L’anomère
β, étant en faible quantité, n'a pas été isolé.
Cette réaction a été étendue aux disaccharides. Elle a nécessité l’utilisation de 5
équivalents de K2CO3, dans le DMF avec 2 équivalents de bromoacétate de tert-butyle. La
séparation des deux anomères étant facile par chromatographie su gel de silice dans le cas des
monosaccharides, elle s’est révélée plus délicate pour les disaccharides à cause de leurs
rapports frontaux très proches.
Le lactose (130, schéma 54), a donné un mélange d’anomères avec un rendement de 68%
dans un rapport α/β de 77/23. L’anomère α 130a est obtenu pur avec un rendement 40%. Une
fraction pure de l’ester β 130b est séparée avec un rendement de 2%.
67

Résultats - Chapitre I
OAcO
AcOOAc
OAc
130
OAcO
OAc
OAc
OH
OO
AcO
AcOOAc
OAc
OAcO
AcO
OAc
O
O
O
O
i .
130a
OAcO
AcOOAc
OAc
OAcO
OAc
OAc
OO O
O
130b
+
Schéma 54. i. DMF, K2CO3 (5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 65%, (α/β =
77:23)
Placé dans les mêmes conditions, le cellobiose (131, schéma 55) a donné un mélange
d’anomères dans 85% de rendement avec un rapport α/β = 77/23. L’anomère α a été séparé
avec un rendement isolé de 72%. L’anomère β 131b n'a pas pu être isolé et caractérisé.
OAcOAcO
AcO
OAc
131
OAcO
OAc
OAc
OH
O OAcOAcO
AcO
OAc
OAcO
AcO
OAc
O
O
O
O
i .
131a
OAcOAcO
AcO
OAc
OAcO
OAc
OAc
OO O
O131b
+
Schéma 55. i. DMF, K2CO3 (5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 85%, (α/β =
77:23)
68
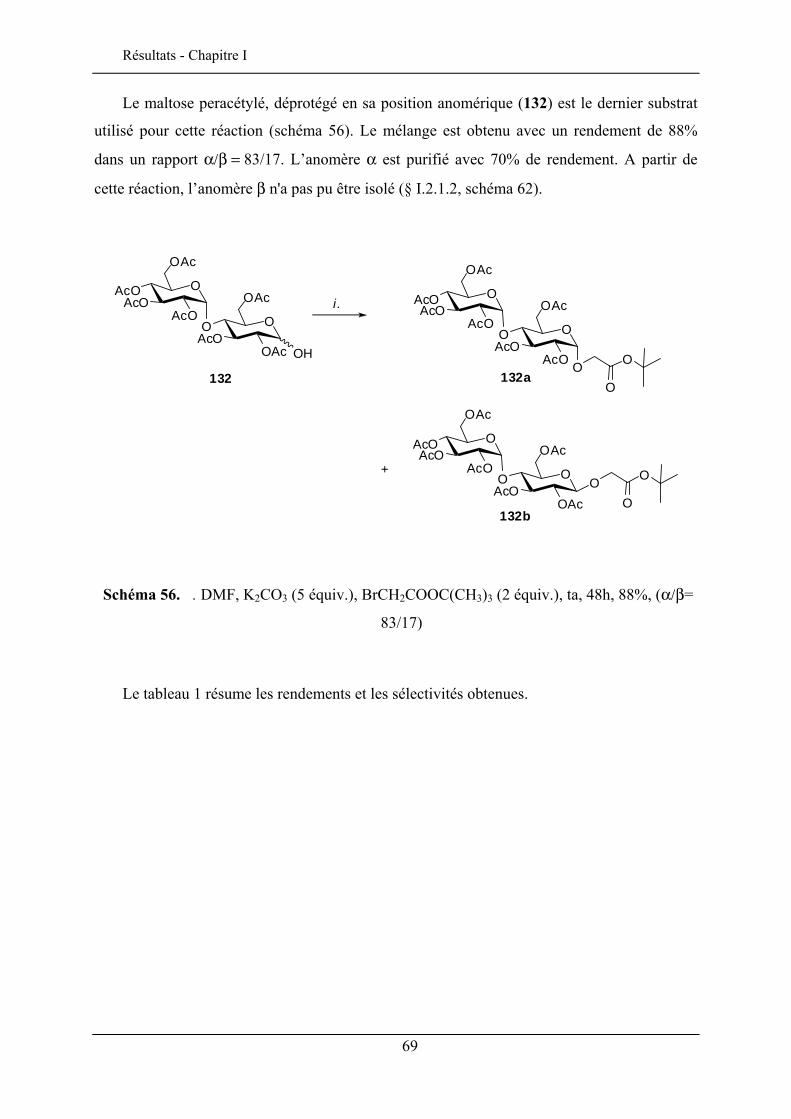
Résultats - Chapitre I
Le maltose peracétylé, déprotégé en sa position anomérique (132) est le dernier substrat
utilisé pour cette réaction (schéma 56). Le mélange est obtenu avec un rendement de 88%
dans un rapport α/β = 83/17. L’anomère α est purifié avec 70% de rendement. A partir de
cette réaction, l’anomère β n'a pas pu être isolé (§ I.2.1.2, schéma 62).
OAcOAcO
AcO
OAc
132
OAcO
OAc
OAc
OH
O
OAcOAcO
AcO
OAc
OAcO
AcO
OAc
O
O
O
i .
132a
OAcOAcO
AcO
OAc
OAcO
OAc
OAc
OO O
O132b
O
+
Schéma 56. . DMF, K2CO3 (5 équiv.), BrCH2COOC(CH3)3 (2 équiv.), ta, 48h, 88%, (α/β=
83/17)
Le tableau 1 résume les rendements et les sélectivités obtenues.
69

Résultats - Chapitre I
Structure Produit Rdt α : β
OAcOAcO
OAc
AcO
OO
O
α = 125a β = 125b
88% 86 : 14
OAcOAcO
OAcAcO
OO
O
α = 126a β = 126b 91% 97 : 3
OAcO
AcOOAc
OAc
OO
O
α = 127a β = 127b 94% 89 : 11
OAc
O
OAc
OO
OOAc
α = 128a β = 128b 74% 81: 19
OAcOAcO
NHAc
AcO
OO
O
α = 129a β = 129b 85% 87 : 13
OOAcO
OAc
AcO
OO
O
OAcO
AcOOAc
OAc
α = 130a β = 130b 68% 77 : 23
OOAcO
OAc
AcO
OO
O
OAcOAcO
OAc
OAc
α = 131a β = 131b 85% 77 : 23
OOAcO
OAc
OAc
OO
O
OAcOAcO
AcO
OAc
α = 132a β = 132b 88% 83 : 17
Tableau 1. Alkylation de sucres partiellement acétylés par le bromoacétate de tert-butyle
I.2.1.2. A partir de sucres libres
La même réaction a été étudiée sur différents sucres libres afin d’éviter des étapes
intermédiaires de protection et de déprotection. L’hydrure de sodium a permis l’obtention des
produits monoalkylés en position anomérique par traitement des sucres dans le DMF avec un
équivalent de base et un équivalent du bromoacétate de tert-butyle en laissant le mélange sous
70

Résultats - Chapitre I
agitation pendant 3 jours à température ambiante. Les produits obtenus sous la forme de
mélanges d’anomères sont séparés par chromatographie sur gel de silice et les rapports
α/β sont déterminés en intégrant les protons anomériques des mélanges dans le méthanol
deutérié.
Cette réaction a été d’abord appliquée au glucose (schéma 57). Le produit obtenu est
obtenu avec un rendement de 50%. La RMN effectuée sur le mélange a montré un mélange
d’anomères avec une sélectivité plus favorable pour l’anomère β (α/β = 26:74).
OHOHO
OH
OH
OH
OHOHO
OH
OH
OO
O
i.
Schéma 57. i. DMF, NaH (1 équiv.), BrCH2COOC(CH3)3 (1 équiv.), ta, 3j, 50%, (α/β =
26:74)
Dans le cas du mannose (schéma 58), le mélange des anomères est obtenu avec 55% de
rendement dans le rapport α/β : 50/50. A partir de cette réaction, nous avons pu séparer
l’anomère β acétylé 126b après acétylation du mélange avec l’anhydride acétique dans la
pyridine avec un rendement global de 11% à partir du mannose.
OHOHO
OHOH
OH
OHOHO
OHOH
OO
O
i.
Schéma 58. i. DMF, NaH (1 équiv.), BrCH2COOC(CH3)3 (1 équiv.), ta, 3j, 55%, (α/β =
50:50)
71

Résultats - Chapitre I
Le galactose, placé dans les mêmes conditions réactionnelles, a donné exclusivement un
produit qui a été identifié comme étant le α-D-furanoside correspondant (schéma 59).
OOH
OHOH
OHO
OOH
HOOH
OH
OHO
Oi .O-
OH
OH
O
OH
HOH
Schéma 59. i. DMF, NaH (1 équiv.), BrCH2COOC(CH3)3 (1 équiv.), ta, 3j, 65%, α
Après acétylation du produit dans la pyridine en présence d’anhydride acétique, le spectre
RMN du proton montre un doublet à 5,25 ppm correspondant au H-1 avec une constante de
couplage de 4,5 Hz. Le H-5 montre un grand déblindage par rapport au dérivé pyranique 127a
(4,04-4,16 ppm) et apparaît à 5,2 ppm sous la forme d’un doublet de doublet de doublet avec
des constantes de couplage J4,5 = 1,5 Hz, J5,6A = 3,4 Hz, J5,6B = 4,5 Hz correspondant à un
cycle furanose. Le dérivé furanique est obtenu par attaque du OH porté par le carbone C-4 sur
l’aldéhyde suivie par l’alkylation avec le bromoacétate de tert-butyle. L’obtention d’un
furanoside pour l’alkylation anomérique du galactose a été déjà observée par Schmidt et al.117
La N-acétyl-glucosamine (schéma 60) a donné un mélange d’anomères avec 77% de
rendement. Contrairement aux cas précédents, la sélectivité obtenue est en faveur de
l’anomère α (α/β : 74/26).
OHOHO
NHAc
OH
OH
OHOHO
NHAc
OH
OO
O
i.
Schéma 60. i. DMF, NaH (1 équiv.), BrCH2COOC(CH3)3 (1 équiv.), ta, 3j, 77%, (α/β =
74:26)
72

Résultats - Chapitre I
Le lactose a donné, dans les conditions de la réaction, un mélange d’anomères avec un
rendement de 57% et un rapport d’anomères α/β = 33 /67 (schéma 61).
OOHO
OH
OH
OH
OHO
HOHO
OH
OOHO
OH
OH
O
OHO
HOHO
OH
O
O
i.
Schéma 61. i. DMF, NaH (1 équiv.), BrCH2COOC(CH3)3 (1 équiv.), ta, 3j, 57%, (α/β =
33:67)
Le mélange d’esters du maltose a été obtenu avec 49% dans un rapport α/β = 36:64
(schéma 62).
OOHO
OH
OH
OH
OHOHO
HO
OH
OOHO
OH
OH
O
OHOHO
HO
OH
O
O
i.
Schéma 62. i. DMF, NaH (1 équiv.), bromoacétate de tert-butyle (1 équiv.), ta, 3j, 49%,
(α/β = 36:64)
A partir de cette réaction, l’anomère β 132b a été isolé avec un rendement de 11% à
partir du maltose libre, après acétylation du mélange dans la pyridine en présence d’anhydride
acétique.
Le tableau 2 résume les résultats obtenus.
73
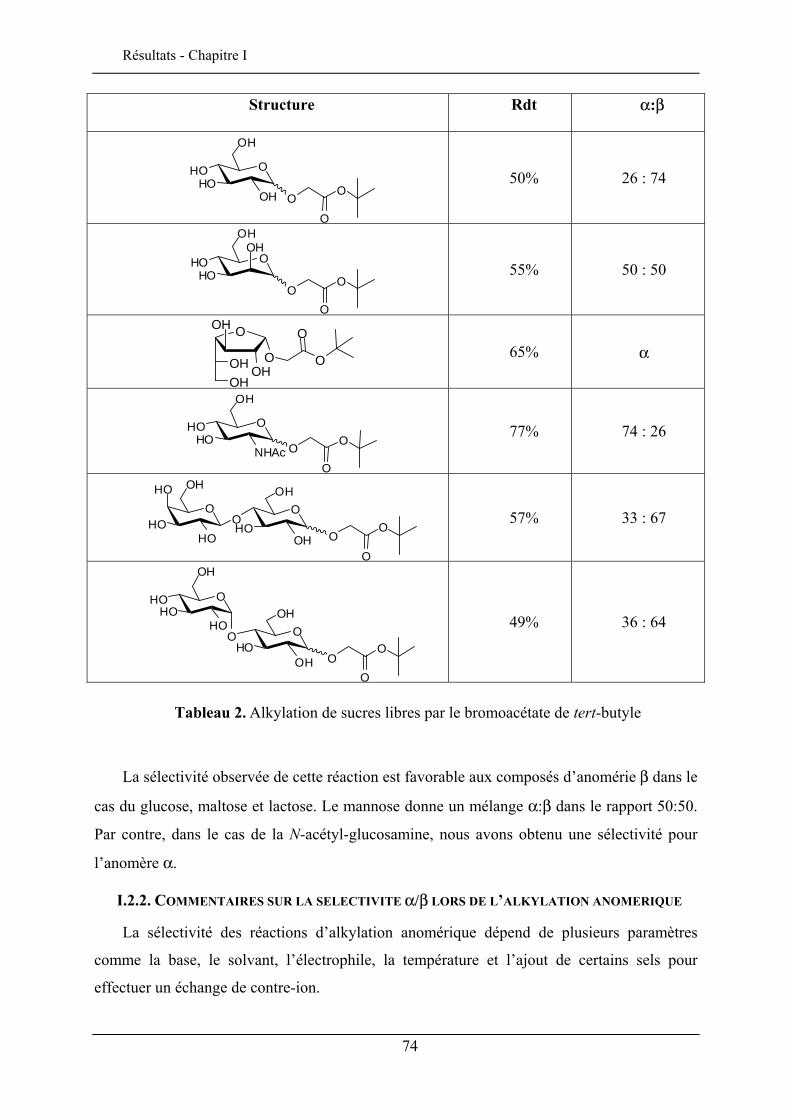
Résultats - Chapitre I
Structure Rdt α:β
OHOHO
OH
OH
OO
O
50% 26 : 74
OHOHO
OHOH
OO
O
55% 50 : 50
OOH
OHOH
OHO O
O
65% α
OHOHO
NHAc
OH
O O
O
77% 74 : 26
OOHO
OH
OH
O
OHO
HOHO
OH
O
O
57% 33 : 67
OOHO
OH
OH
O
OHOHO
HO
OH
O
O
49% 36 : 64
Tableau 2. Alkylation de sucres libres par le bromoacétate de tert-butyle
La sélectivité observée de cette réaction est favorable aux composés d’anomérie β dans le
cas du glucose, maltose et lactose. Le mannose donne un mélange α:β dans le rapport 50:50.
Par contre, dans le cas de la N-acétyl-glucosamine, nous avons obtenu une sélectivité pour
l’anomère α.
I.2.2. COMMENTAIRES SUR LA SELECTIVITE α/β LORS DE L’ALKYLATION ANOMERIQUE
La sélectivité des réactions d’alkylation anomérique dépend de plusieurs paramètres
comme la base, le solvant, l’électrophile, la température et l’ajout de certains sels pour
effectuer un échange de contre-ion.
74

Résultats - Chapitre I
Tous ces paramètres affectent l’équilibre qui s’établit entre les 2 alcoxydes α et β, et leurs
réactivités respectives (schéma 63). En fait, l’anomère α est le produit thermodynamiquement
favorisé alors que l’anomère β réagit plus rapidement (produit cinétique).
O
OH
base
O
O-
OO-O-
O
H
R-X R-X
β-glycosideα-glycoside
Schéma 63. Synthèse de glycosides par alkylation anomérique
A basse température, l’anomère α est favorisé. Quand les réactions sont menées à
température ambiante, l’équilibre entre les deux alcoxydes devient rapide et d’autres facteurs
déterminent la sélectivité. Dans le cas des pyranoses, la nucléophilie plus grande de
l’alcoxyde équatorial (effet cinétique) favorise la formation du produit β.129
Dans la littérature, un solvant apolaire favorise la formation de l’anomère β129, , ,130 131 132
Les solvants polaires favorisent la formation du produit thermodynamique : l’anomère α se
forme majoritairement.118,133
129 (a) Schmidt, R. R. Angew. Chem. Int. Ed. Eng. 1986, 25, 212-235. (b) Schmidt, R. R.; Michel, J. Roos, M. Liebigs Ann. Chem. 1984, 1343-1357. (c) Schmidt, R. R.; Michel, J. Tetrahedron Lett. 1984, 25, 821-824. 130 Lubineau, A.; Escher, S.; Alais, J.; Bonnaffé, D. Tetrahedron Lett. 1997, 38, 4087-4090. 131 Terjung, A.; Jung, K.-H. ; Schmidt, R. R. Carbohydr. Res. 1997, 297, 229-242. 132 Schmidt, R. R. Angew. Chem. Int. Ed. 1986, 25, 212-235. 133 Tamura, J.; Schmidt, R. R. J. Carbohydr. Chem. 1995, 14, 895-911.
75

Résultats - Chapitre I
L’ajout de sels encombrés à la réaction peut aussi affecter la sélectivité. L’échange entre
le cation de la base avec celui du sel plus encombré augmente la réactivité de l’alcoxyde axial
et met la réaction sous contrôle thermodynamique pour donner l’anomère α.130
De même, l’encombrement de l’électrophile agit aussi sur la sélectivité : un agent réactif
réagit avec l’alcoxyde le plus nucléophile (équatorial) et met la réaction sous contrôle
cinétique : c’est le produit β qui se forme. Un agent électrophile encombré n’agira pas
rapidement et favorise la formation de l’anomère α. L’électrophile que nous utilisons étant à
la fois réactif et encombré, il peut favoriser la formation d’un anomère ou de l’autre selon la
réactivité des alcoxydes.
Dans le cas des sucres libres, l’alcoxyde équatorial réagit plus rapidement avec le
bromoacétate de tert-butyle, c’est le produit β qui se forme.
Par contre, dans le cas d’un sucre protégé, les acétates, réduisent considérablement la
réactivité de l’alcoxyde équatorial et encombrent le sucre. L’approche du bromoacétate de
tert-butyle ou d’autres agents alkylants encombrés118, , 134 135 se fait du côté le moins encombré
et place la réaction sous contrôle thermodynamique pour donner le produit α. C’est ce que
nous observons dans nos conditions expérimentales.
Beau et al.136 ont effectué la réaction d’alkylation anomérique avec le bromure d’allyle,
réactif et non encombré, sur la N-acétyl-glucosamine libre. Cette réaction favorise la
formation du glycoside β par contrôle cinétique. La même réaction d’alkylation par le
bromure d’allyle effectuée sur la N-benzoyl-glucosamine menée par Schmidt et al.117 donne
des résultats contraires (sélectivité en faveur de l’anomère α). Dans ce cas, le groupement
benzoyle encombre le sucre et met la réaction sous contrôle thermodynamique.
I.2.3. DEPROTECTION DE L’ACIDE CARBOXYLIQUE
Comme nous avons vu dans la partie bibliographique (§ I.3.1.2, chapitre I de la partie
bibliographie), les carboxyméthyl glycosides sont des synthons intéressants et ont servi à
l’élaboration de structures diverses d’intérêt biologique.
La synthèse des acides acétylés correspondants aux esters tert-butyles isolés ont été
facilement obtenus par déprotection du groupement tert-butyle dans le dichlorométhane par
action de l’acide trifluoroacétique. Cette réaction s’effectue dans un excès de TFA et donne
134 Azoulay, M.; Escriou, V.; Florent, J.-C.; Monneret, C. J. Carbohydr. Chem. 2001, 20, 1093-1101. 135 Klotz, W.; Schmidt, R. R. J. Carbohydr. Chem. 1994, 1093-1101. 136 Vauzeilles, B.; Dausse, B.; Palmier, S.; Beau, J.-M. Tetrahedron Lett. 2001, 42, 7567-7570.
76
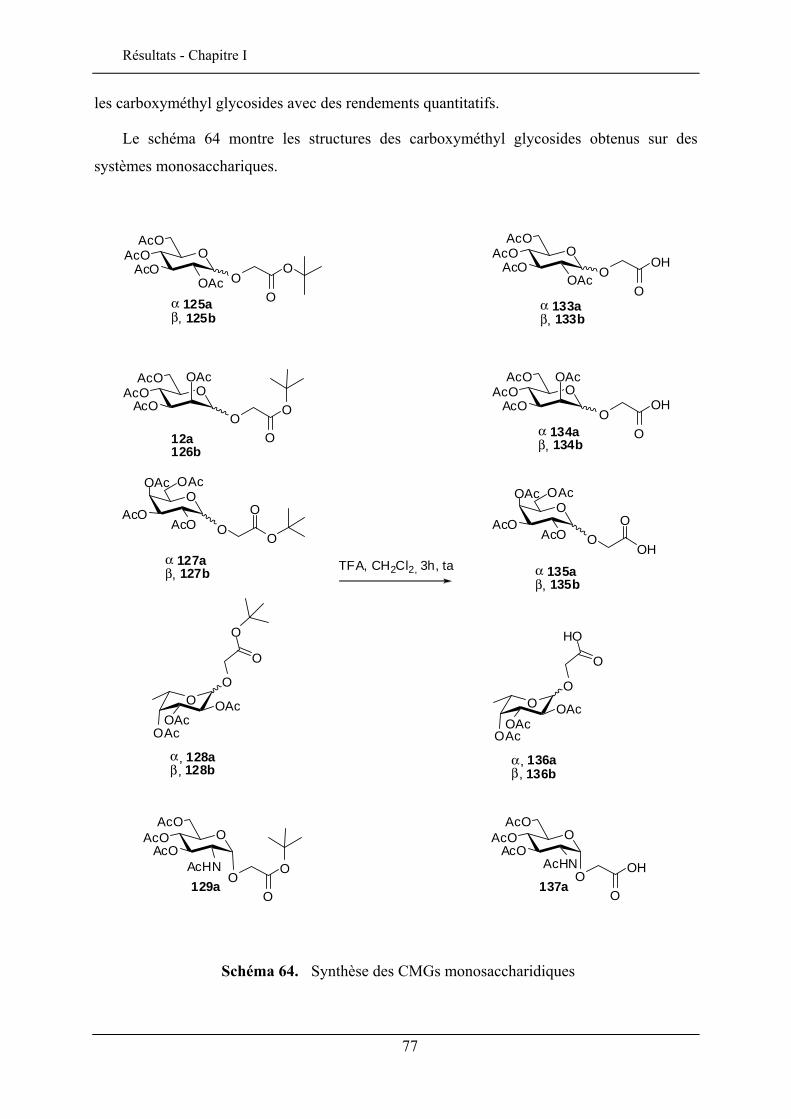
Résultats - Chapitre I
les carboxyméthyl glycosides avec des rendements quantitatifs.
Le schéma 64 montre les structures des carboxyméthyl glycosides obtenus sur des
systèmes monosacchariques.
OAcOAcO
OAcAcO
OOH
O
OOAc
AcOAcO
OAc
OOH
O
α 135aβ, 135b
OAc
OOAc
OAc
O
HO
O
α, 136aβ, 136b
OAcOAcO
OAcAcO
OO
O12a126b
OOAc
AcOAcO
OAc
OO
O
α 127aβ, 127b
OAc
OOAc
OAc
O
O
O
α, 128aβ, 128b
OAcOAcO
AcHN
AcO
OOH
O137a
OAcOAcO
AcHN
AcO
OO
O129a
OAcOAcO
OAc
AcO
OOH
O
OAcOAcO
OAc
AcO
OO
Oα 125aβ, 125b
α 133aβ, 133b
TFA, CH2Cl2, 3h, ta
α 134aβ, 134b
Schéma 64. Synthèse des CMGs monosaccharidiques
77

Résultats - Chapitre I
Les acides 133a,43 133b,16 134a,11 135a,11 135b137 et 136b23 ont déjà été décrits dans la
littérature. Dans les mêmes conditions, des carboxyméthyl glycosides basés sur des
disaccharides ont été obtenus (schéma 65).
OO
AcO
AcOOAc
OAc
OAcO
OAc
OAc
OOH
Oα, 138aβ, 138b
α, 130aβ, 130b
OOAcO
AcOOAc
OAc
OAcO
AcO
OAc
OOH
Oα,139aα,131a
O
OAcOAcO
AcO
OAc
OAcO
OAc
OAc
OOH
Oα, 140aβ, 140b
α, 132aβ, 132b
OO
AcO
AcOOAc
OAc
OAcO
OAc
OAc
OO
O
OOAcO
AcOOAc
OAc
OAcO
AcO
OAc
OO
O
O
OAcOAcO
AcO
OAc
OAcO
OAc
OAc
OO
O
TFA, CH2Cl2, 3h, ta
Schéma 65. Synthèse des CMGs disaccharidiques
Ces acides disaccharidiques n’ont jamais été décrits dans la littérature.
Les spectres de RMN de ces acides ont montré la disparition du singulet à 1,5 ppm
correspondant au groupement tert-butyle. Les protons H-7a et H-7b apparaissent sous la
forme d’un singulet qui intègre pour 2 protons entre 4,30 et 4,60 ppm.
Le spectre de RMN du carbone montre la disparition du pic correspondant au tert-butyle
qui apparaît à 28 ppm dans le cas de l’ester, ainsi que le pic correspondant à carbone
quaternaire du groupement tert-butyle, et le carbone C=O du groupe carboxyméthyle.
137 Sasaki, A.; Naoichi, M. JP Patent 06271597 (1994).
78

Résultats - Chapitre I
A partir d’une réaction simple qui est l’alkylation anomérique, nous avons donc pu
synthétiser une chimiothèque de carboxyméthyl glycosides qui peuvent être greffés sur
d’autres adduits. Une première utilisation de ces acides a été effectuée pour la synthèse de
glycoclusters persulfurés que nous détaillerons dans le paragraphe suivant.
I.2.4. APPLICATIONS POUR LA SYNTHESE DE GLYCOCLUSTERS PERSULFURES
Lors d’une collaboration engagée avec le Laboratoire de Chimie Organique 2 dirigé par
le professeur Peter Goekjian, et dans le cadre de la thèse de Mazen Sleiman,138 certains de ces
acides ont été valorisés pour la synthèse de glyco-astérisques persulfurés (schémas 66 et 67).
Ce sont des molécules fluorescentes, qui représentent une nouvelle classe des clusters
moléculaires à l’interface entre les petits clusters et les dendrimères visant l’étude de «l’effet
cluster glycosidique» impliqué dans plusieurs phénomènes de reconnaissance multivalents des
récepteurs protéiniques. Leurs évaluations biologiques sur des lectines d’origines différentes
ont été effectuées.
En effet, les interactions protéine-saccharide gouvernent plusieurs phénomènes
biologiques importants, à la fois normaux et pathologiques. Ces interactions incluent la
synthèse et la dégradation enzymatique des oligo- et polysaccharides, le transport des sucres
dans les cellules vivantes, les interactions anticorps-antigène, etc. Elles jouent également un
rôle important dans l’adhésion des pathogènes sur les cellules humaines lors de la première
étape d’une infection.
Les acides ont été couplés aux astérisques par liaison amide en utilisant un agent de
couplage, l’EEDQ (1,2 équiv. par couplage) avec les amines correspondantes à chaud (45°C
et 60°C) dans le THF ou le DMF pour obtenir les astérisques finaux avec des rendements
allant de 39 à 65%. La réaction de couplage a été suivie par une désacétylation dans les
conditions de Zemplén pour obtenir les produits polyhydroxylés libres avec de bons
rendements (94-97%, schéma 66-67).
138 Sleiman, M. Thèse de Doctorat de L'Université Claude Bernard Lyon 1, N° d'ordre 291-2007.
79
Résultats - Chapitre I
SS
SS
S
S
HN
HNR
NH
NHRHN
R
HN
R=
OH
OOH
OOOH
OHO
HOHO
OH
OO
OOHO
HO
HO
OO
OHO
HOOH
OH
R
R
R
Schéma 66. Astérisques glycosylés polysulfurés à cœur benzénique
S S
SS
NH
HNHN
HN
R=
OH
OOH
OOOH
OHO
HOHO
OH
OO
R
R
R
R
Schéma 67. Astérisques glycosylés polysulfurés à cœur pyrénique
La plupart des astérisques glycosylés synthétisés ont montré de meilleures propriétés en
ce qui concerne leurs activités biologiques pour inhiber ou agréger certaines lectines par
comparaison à l’action de leurs analogues monovalents (effet multivalent).
80

Résultats - Chapitre I
I.2.5. SYNTHESE DES LACTONES
Le CMGlc 133a est dissous dans le méthanol en présence de soude qui permet la
désacétylation. Après évaporation du solvant, le produit libre est dissous dans la pyridine,
l’action de l’anhydride acétique permet l’acétylation et la fermeture du cycle lactonique
(schéma 68).
L’hypothèse la plus plausible de l’obtention de cette lactone est la formation d’un
anhydride mixte suivie d’une cyclisation par attaque de l’hydroxyle en position 2.
iOAcOAcO
AcO
OAc
OOH
O
α, 83%, 1aβ, 71%, 1b
OAcO
O
O
OAc
AcOOHOHO
HO O
OH
O
α, 133aβ, 133b
O
O O
Schéma 68. i. 1. NaOH, MeOH, t.a., 1h, 2. Ac2O, Pyridine, ta, 12h
La lactone 1a a été caractérisée lors de l’étude de la réaction d’oxydation de
l’isomaltulose avec le peroxyde d’oxygène par Stéphane Trombotto et al.2
Si on désire obtenir une lactone de configuration anomérique parfaitement définie, alors il
est préférable de séparer les anomères au stade de l’ester tert-butylique éventuellement après
acétylation (125ab-132ab). En effet, les lactones α et β se différencient très peu par
chromatographie. Par contre, si on veut se satisfaire d’un mélange de lactones ou si on
suppose qu’à un stade ultérieur (après ouverture), une séparation des deux anomères est
possible, il faut signaler que les lactones peuvent être obtenues directement en traitant le
mélange issu de l’alkylation anomérique des sucres libres pour une déprotection en milieu
basique du groupement tert-butyle (voie d’obtention en trois étapes : a. alkylation
anomérique, b. saponification de l’ester de tert-butyle, c. acétylation et cyclisation).
Le schéma 69 montre les lactones obtenues à partir des monosaccharides dans les mêmes
conditions. Les rendements varient entre 60 et 87%.
81
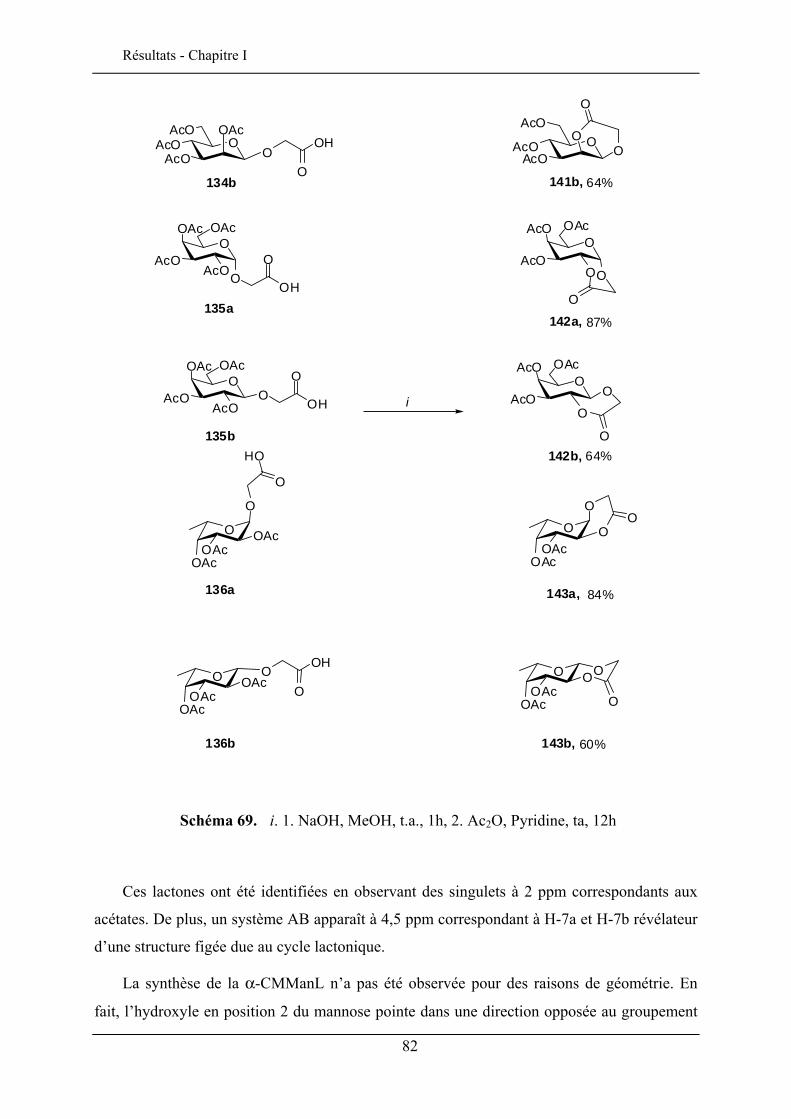
Résultats - Chapitre I
OAcOAcO
AcO
OO
O
OAcOAcO
OAcAcO
OOH
O141b, 64%134b
OOAc
AcOAcO
OAc
OOH
O
142b, 64%
OAcO
O
OAcAcO
135b
OAc
OOAc
OAcO
OH
O
143b, 60%136b
i
OOAc
AcOAcO
OAc
OOH
O
142a, 87%
OAcO
O
O
OAcAcO
135a
O
O
O
OAc
OOAc
OO
O
OAc
OOAc
OAc
O
HO
O
143a, 84%136a
OAc
OOAc
O
OO
Schéma 69. i. 1. NaOH, MeOH, t.a., 1h, 2. Ac2O, Pyridine, ta, 12h
Ces lactones ont été identifiées en observant des singulets à 2 ppm correspondants aux
acétates. De plus, un système AB apparaît à 4,5 ppm correspondant à H-7a et H-7b révélateur
d’une structure figée due au cycle lactonique.
La synthèse de la α-CMManL n’a pas été observée pour des raisons de géométrie. En
fait, l’hydroxyle en position 2 du mannose pointe dans une direction opposée au groupement
82
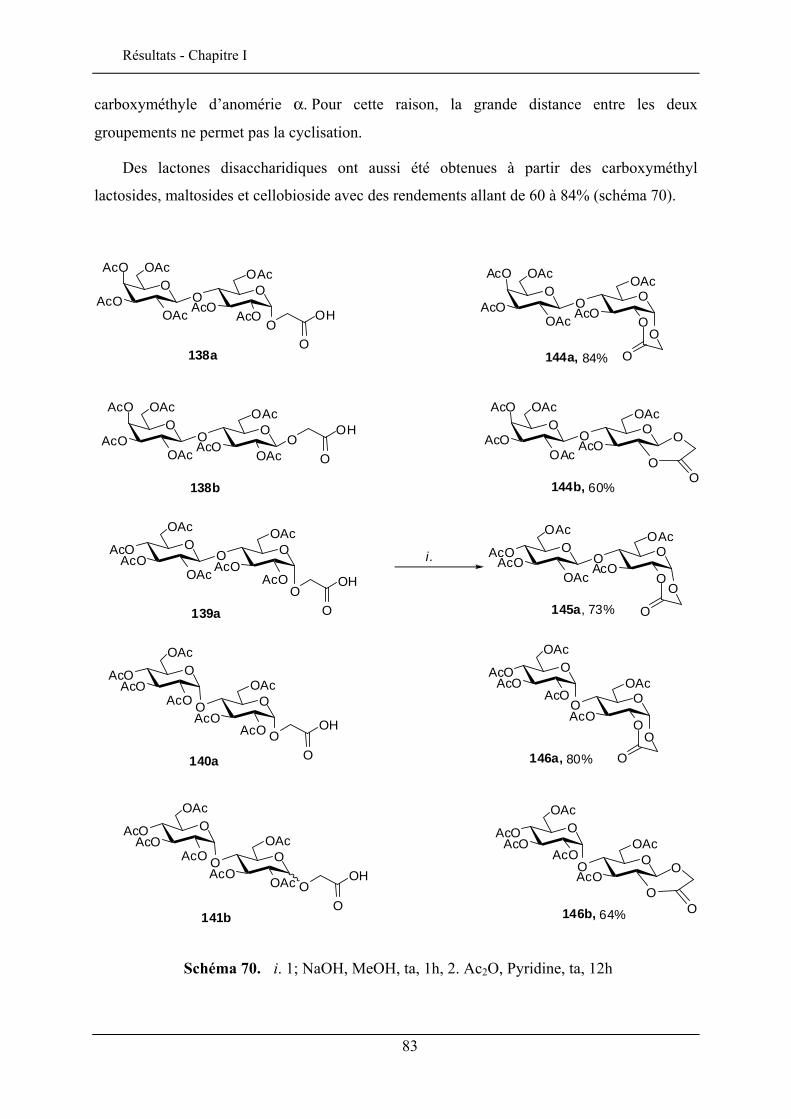
Résultats - Chapitre I
carboxyméthyle d’anomérie α. Pour cette raison, la grande distance entre les deux
groupements ne permet pas la cyclisation.
Des lactones disaccharidiques ont aussi été obtenues à partir des carboxyméthyl
lactosides, maltosides et cellobioside avec des rendements allant de 60 à 84% (schéma 70).
OO
AcO
AcOOAc
OAc
OAcO
AcO
OAc
OOH
O144a, 84%138a
OOAcO
AcOOAc
OAc
OAcO
O
OAc
O
O
OOAcO
AcOOAc
OAc
OAcO
AcO
OAc
OOH
O
i .
145a, 73%139a
O
OAcOAcO
AcO
OAc
OAcO
AcO
OAc
OOH
O 146a, 80%140a
OO
AcO
AcOOAc
OAc
OAcO
OAc
OAc
OOH
O
144b, 60%138b
OO
AcO
AcOOAc
OAc
OAcO
O
OAc
O
O
OO
AcO
AcOOAc
OAc
OAcO
O
OAc
O
O
O
OAcOAcO
AcO
OAc
OAcO
OAc
OAc
OOH
O141b
O
OAcOAcO
AcO
OAc
OAcO
O
OAc
OO
O
OAcOAcO
AcO
OAc
OAcO
O
OAc
O
O146b, 64%
Schéma 70. i. 1; NaOH, MeOH, ta, 1h, 2. Ac2O, Pyridine, ta, 12h
83
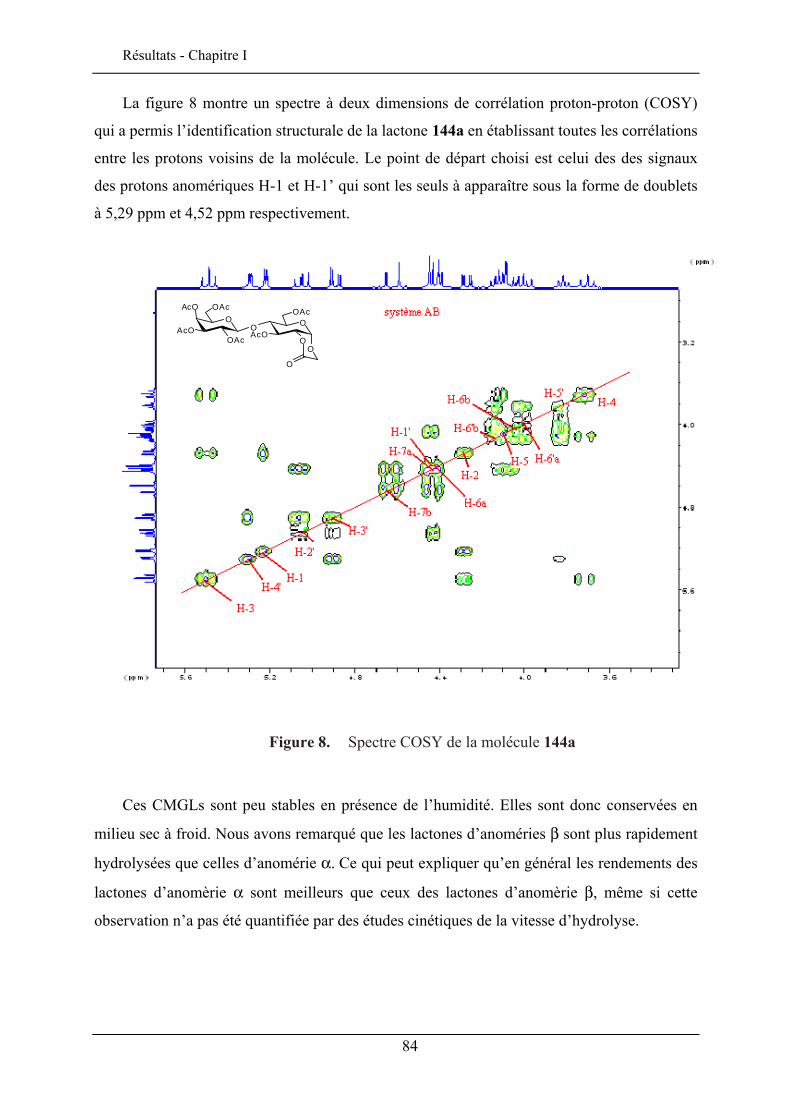
Résultats - Chapitre I
La figure 8 montre un spectre à deux dimensions de corrélation proton-proton (COSY)
qui a permis l’identification structurale de la lactone 144a en établissant toutes les corrélations
entre les protons voisins de la molécule. Le point de départ choisi est celui des des signaux
des protons anomériques H-1 et H-1’ qui sont les seuls à apparaître sous la forme de doublets
à 5,29 ppm et 4,52 ppm respectivement.
OO
AcO
AcOOAc
OAc
OAcO
O
OAc
O
O
Figure 8. Spectre COSY de la molécule 144a
Ces CMGLs sont peu stables en présence de l’humidité. Elles sont donc conservées en
milieu sec à froid. Nous avons remarqué que les lactones d’anoméries β sont plus rapidement
hydrolysées que celles d’anomérie α. Ce qui peut expliquer qu’en général les rendements des
lactones d’anomèrie α sont meilleurs que ceux des lactones d’anomèrie β, même si cette
observation n’a pas été quantifiée par des études cinétiques de la vitesse d’hydrolyse.
84

Résultats - Chapitre I
I.3. LACTONES BENZYLEES (GLC/GAL)
Dans la perspective d’élargir le domaine d’application et la mise en œuvre de la stratégie
carboxyméthyl glycoside lactone (CMGL), nous avons synthétisé ces dernières protégées par
des groupements benzyles.49 Contrairement aux groupements acétyles qui présentent une
sensibilité à la fois aux conditions basiques et acides, les benzyles sont très stables et
permettent d’envisager des possibilités de fonctionnalisations ultérieures des produits.
I.3.1. SYNTHESE DE L’ANOMERE β (GLC/GAL)
La lactone d’anomérie β est obtenue en utilisant la stratégie de l’orthoester développée
par Lemieux et al.139 Cet ester est facilement obtenu en milieu basique à partir du bromure du
glycoside acétylé grâce à la participation du groupement acétate en position 2 en présence
d’un alcool et de sels de mercure ou d’argent. De nombreuses améliorations de cette méthode
ont été proposées dans la littérature. Nous avons suivi le protocole de Bundle et al.140 pour la
synthèse de cet orthoester dans la lutidine en présence d’alcool et du bromure de
tétrabutylammonium (schéma 71).
OR1
AcOO
O
OAcR2
O
OR1
BnOO
O
O
OBnR2
OR1
BnOOH
OBnR2
O
OR1
AcOAcO
Br
OAcR2
i .
i ii iv
149. R1=OAc, R2=H150. R1=H, R2=OAc
151. R1=OAc, R2=H152. R1=H, R2=OAc
153. R1=OBn, R2=H154. R1=H, R2=OBn
155. R1=OBn, R2=H156. R1=H, R2=OBn
OR1
AcOAcO
OAcR2
OAc
i i
147. R1=OAc, R2=H148. R1=H, R2=OAc
Schéma 71. i. HBr/AcOH, ii. Alcool allylique, 2,6-lutidine (2 équiv.), Bu4NBr (1,2 équiv.),
4h; iii. 1. MeONa/MeOH, 2. NaH (1,25 équiv./OH), BnBr (1,25 équiv./OH), DMF; iv.
AllOH, BF3.OEt2 v. O3, MeOH, puis DMS, 2. NaClO2, KH2PO4, H2O, 0 °C; 3. Ac2O, py
139 Lemieux, R. U.; Hayami, J.-I. Can. J. Chem. 1965, 2162-6173. 140 Nitz, M.; Purse, B. W.; Bundle, D. R. Org. Lett. 2000, 2, 2939-2942.
85

Résultats - Chapitre I
Cette stratégie a été appliquée aux glucose et galactose peracétylés (147 et 148). Elle a
été adoptée puisque l’ouverture des deux orthoesters connus 149 et 150 141 libère sélectivement
le groupement hydroxyle en position 2 permettant ainsi la fermeture du cycle lactonique.
L’allyl 3,4,6-tétra-O-benzyl-β-D-glucopyranoside (153) et l’allyl 3,4,6-tétra-O-benzyl-β-D-
galactopyranoside (154) donnent ensuite les lactones correspondantes 155 et 156 via la
réaction d’ozonolyse, suivie par l’oxydation des aldéhydes avec NaClO2 et enfin cyclisation
dans les conditions d’acétylation avec un rendement de 50% et 43%, les intermédiaires
n’étant pas isolés.
I.3.2. SYNTHESE DE L’ANOMERE α (GLC/GAL)
La synthèse des lactones benzylées d’anomérie α a été effectuée à partir des allyl
glycosides correspondants obtenus par la réaction de Fischer réalisée sur les sucres libres. Les
deux anomères ont été séparés après acétylation et ont ensuite été déprotégés avec le mélange
NEt3/MeOH/H2O. La benzylation des sucres est effectuée par le bromure de benzyle et
l’hydrure de sodium dans le DMF pour donner les deux allyles 157 et 158. La déprotection de
la position 2 est nécessaire afin de permettre la cyclisation. Elle est réalisée avec la technique
de la dé-O-alkylation avec le triisobutylammonium (TIBAL) développée par Sinaÿ et al.142, 143
Cette technique, sélective de la position 2 des α-glucosides et galactosides, a été utilisée par
Arek Listkowski pour préparer les lactones benzylées 161 et 162 à partir des allyl glycosides
157 et 158 (schéma 72 et 74).49
OR1
BnOOH
O
OR1
BnOBnO
O
OBn OBnR2 R2
i.
157.R1=OBn, R2=H158.R1=H, R2=OBn
159. R1=OBn, R2=H, 59%160. R1=H, R2=OBn, 56%
Schéma 72. i. TIBAL (5 équiv.), toluène, 50°C, 16 h
141 Bellucci, G.; Chiappe, C.; D'Andrea. Tetrahedron: Asymmetry 1995, 6, 221-230. 142 Lecourt, T.; Herault, A.; Pearce, A. J.; Sollogoub, M.; Sinaÿ, P. Chem. Eur. J. 2004, 10, 2960-2971. 143 Chevalier-du Roizel, B.; Cabianca, E.; Rollin, P.; Sinaÿ, P. Tetrahedron 2002, 58, 9579-9583.
86

Résultats - Chapitre I
La réaction de débenzylation par le TIBAL consiste en une bis-chélation par l’aluminium
des oxygènes orientés en cis suivie d’une hydrolyse de la liaison O-Al (schéma 73).
OBnOBnO
OBn
BnO OMe
OBnOBnO
OBn
BnO OMe
Al
OBnOBnO
OBnOBnO
BnO
OBn
HO OMe
-PhMe H2O
iBuiBu
iBu
Al
O OMePh
TiBAl
HAl
OBnOBnO
OBn
Al
O OMe
Al
Schéma 73. Mécanisme de déprotection par le TIBAL
Dans le cas du galactose, malgré l’orientation cis du benzyloxyde en position 3 par
rapport à celui de la position C-4, celui-ci n’est pas sensible à la débenzylation dans les
conditions de la réaction, ce qui confirme les résultats déjà décrits dans la littérature.143,144
Les CMGlcL et CMGalL benzylées 161 et 162 sont ensuite obtenues selon la séquence
oxydation-acétylation (schéma 74).
OR1
BnOO
OO
OR1
BnOOH
O
OBn OBnR2 R2
i , i i, ii i
159. R1=OBn, R2=H160. R1=H, R2=OBn
161. R1=OBn, R2=H, 56%162. R1=H, R2=OBn, 36%
Schéma 74. i. O3, MeOH, puis DMS, ii. NaClO2, KH2PO4, H2O, 0°C; iii. Ac2O, py
144 Sollogoub, M.; Das, S. K.; Mallet, J-M, Sinaÿ, P. C. R. Acad. Sci. Paris 1999, 2, 441-448.
87

Résultats - Chapitre I
I.4. CONCLUSION
En résumé, une procédure alternative pour la synthèse des carboxyméthyl glycosides
lactones par utilisation du bromoacétate de tert-butyle comme agent électrophile dans
l’alkylation anomérique a été mise au point.
Cette réaction effectuée sur des sucres portant un O- ou N-acétyle en C-2 donne une
sélectivité α alors que la sélectivité β est observée sur des sucres totalement déprotégés. Les
anomères des glycosides tert-butyliques acétylés sont facilement séparables sur une colonne
de silice ce qui a permis la purification et la caractérisation des deux anomères dans la quasi-
totalité des cas.
Les carboxyméthyl glycosides obtenus par cette méthode ont été engagés dans la
synthèse d’astérisques polysulfurés glycosylés pour la reconnaissance de lectines.
D’autre part, les lactones benzylées sont aussi synthétisées par oxydation de double
liaison de l’allyl glycoside lui-même obtenu par glycosidation de Fischer ou par ouverture de
l’orthoester correspondant. Les rendements de ces réactions étant modestes, l’obtention de ces
produits en grande quantité est difficile à réaliser. De plus, les lactones benzylées se sont
révélées très sensibles et facilement hydrolysables ce qui constitue une limitation à leur
utilisation.
Compte tenu de leur stabilité et leur plus grande facilité d’obtention, seules les lactones
acétylées ont été utilisées dans la suite de ce travail.
88

Résultats - Chapitre III
CHAPITRE II. SYNTHESE DE SYSTEMES
MULTIFONCTIONNELS
II.1. INTRODUCTION
Comme nous avons vu dans le chapitre bibliographique, de nombreux travaux ont utilisé
les sucres comme des molécules scaffolds grâce à leur multifonctionnalité et leur diversité
structurale.
Après la synthèse d’une collection de lactones, nous avons donc étudié leur utilité pour la
construction de systèmes 1,2-bis fonctionnalisés en exploitant l’hydroxyle en position 2,
libéré lors de l’étape de l’ouverture de la lactone, pour la seconde fonctionnalisation (schéma
75).
O
OO
O
HOO
O
O
O
OO
ORORORO
Schéma 75. Accès à des systèmes bifonctionnels par ouverture des CMGLs et
fonctionnalisation en position 2
Dans le cadre de la synthèse de dérivés 1-O-carboxyméthylés, cette technique présente
une alternative aux synthèses classiques de composés 1,2-bis-fonctionnalisés145 comme les
acétals 1,2-isopropylidène,146 l’ouverture des 1,2-orthoesters,139,140 les acétals 1,2-O-
145 Garegg, P. J. Adv. Carbohydr. Chem. Biochem. 2004, 59, 69-134. 146 Close, D. M. Chem. Rev. 1979, 79, 491-513.
89

Résultats - Chapitre III
stannylène,147 les glycals,148, 149 ou d’autres stratégies de dé-O-benzylation sélective de la
position 2 avec le TIBAL ou DIBAL-H142,143,144 ou autres acides de Lewis.150
II.2. L’OUVERTURE DES CARBOXYMETHYL GLYCOSIDES LACTONES
Deux lactones ont été choisies comme modèles : la α-CMGlcL 1a et celle d’un système
disaccharidique dérivé du maltose (146a).
La première étape a consisté en une ouverture des lactones par un agent nucléophile.
L’allylamine a été choisie pour sa capacité de conjugaison ultérieure en utilisant la réactivité
des oléfines ou le couplage par métathèse, la propargylamine pour sa capacité à pouvoir être
engagée dans une cycloaddition 1,3 dipolaire de type Huisgen (réaction « Click »).151
L’ouverture du cycle lactonique s’est effectuée dans des conditions douces et les produits
d’ouverture 163, 164 et 165 sont obtenus avec 92%, 96% et 85% de rendement (Schéma 76).
OR1
AcOO
OAc
OO
OR1
AcOHO
OAc
ONH
O
i
163: R1 = OAc, R2 = CH2CH=CH2, 92%164: R1 = OAc, R2 = CH2CCH, 96%165: R1 = α-Glu acétylé, R2= CH2CCH, 85%
R2
1a: R1=OAc146a,R1=α-glu
Schéma 76. i. Amine (1,5 équiv.), CH2Cl2, ta, 12h
Le spectre de RMN du proton a montré un déplacement significatif du H-2 de 4,40 ppm à
3,78 ppm après ouverture de la lactone.
Pour l’amide 165, un triplet apparaît à 2,22 ppm dans le spectre RMN du proton et à 71,7
ppm dans le spectre RMN du carbone correspondant au proton acétylénique. Le méthylène de
147 David, S.; Hanessian, S. Tetrahedron 1985, 41, 643-663. 148 Lemieux, R. U.; Ratcliffe R. M. Can. J. Chem. 1979, 2, 1244-1251. 149 Seeberger, P. H., Danishefsky, S. J. J. Acc Chem. Res. 1998, 31, 685-695. 150 Martin O. R.; Kurz, K. G. ; Rao, S. P. J. Org. Chem. 1987, 52, 2922-2925. 151 Dedola, S., Nepogodiev, S. A., Field, R. A. Org. Biomol. Chem. 2007, 5, 1006-1017.
90

Résultats - Chapitre III
l’amide apparaît dans la zone 3,80-4,20 ppm en proton et à 28,7 ppm en DEPT 135. Le
carbone quaternaire de la triple liaison apparaît à 76,1 ppm.
En RMN du proton, le spectre du produit 163 comporte aussi à 3,91 ppm un singulet
correspondant aux deux protons du méthylène du groupement NHCH2. Dans le spectre de
RMN du carbone, le carbone de ce groupement est visualisé à 41,5 ppm. Les protons du
méthylène de la double liaison apparaîssent entre 5,07 ppm et 5,11 ppm et à 116,4 ppm en
RMN du carbone, le méthyne de la double liaison à 5,72-5,85 ppm en RMN du proton et
133,7 ppm en RMN du carbone.
Pour ces trois glucosides, le spectre du proton présente un triplet à 7,50-7,60 ppm
correspondant au proton porté par l’azote des fonctions amides.
II.3. FONCTIONNALISATION EN POSITION 2
II.3.1. CARBAMATATION
Les produits obtenus après ouverture présentant un hydroxyle libre en position 2 sont
soumis à des fonctionnalisations ultérieures pour donner les carbamates correspondants.
Les réactions ont été effectuées avec l’isocyanate de propyle et l’isocyanate d’hexadécyle
ajoutés en léger excès dans du dichlorométhane anhydre en présence de triéthylamine. Les
chaînes hydrophobes modulent la polarité des adduits sucres pour conduire à des dérivés
possédant des propriétés tensioactives.
La réaction du composé 164 avec l’isocyanate de propyle a été totale après 12 heures.
Après chromatographie sur gel de silice, le produit acétylé 166 était contaminé par la N,N-
dipropylurée. Une désacétylation dans le système Et3N/MeOH/H2O donne le carbamate
correspondant libre 169 avec un rendement de 72% sur les 2 étapes.
Les amides 164 et 165 ont été soumis à une réaction avec l’isocyanate d’hexadécyle.
Durant la réaction, et contrairement à la N,N-dipropylurée, la précipitation de la N,N-
dihexadécylurée permet son élimination par filtration du mélange réactionnel pour donner
après chromatographie, les carbamates acétylés 167 et 168 avec des rendements respectifs de
74% et 94%. La déprotection de ces produits par action de la triéthylamine a donné les
carbamates 170 et 171 libres avec 77 et 73% de rendement respectivement (schéma 77).
91
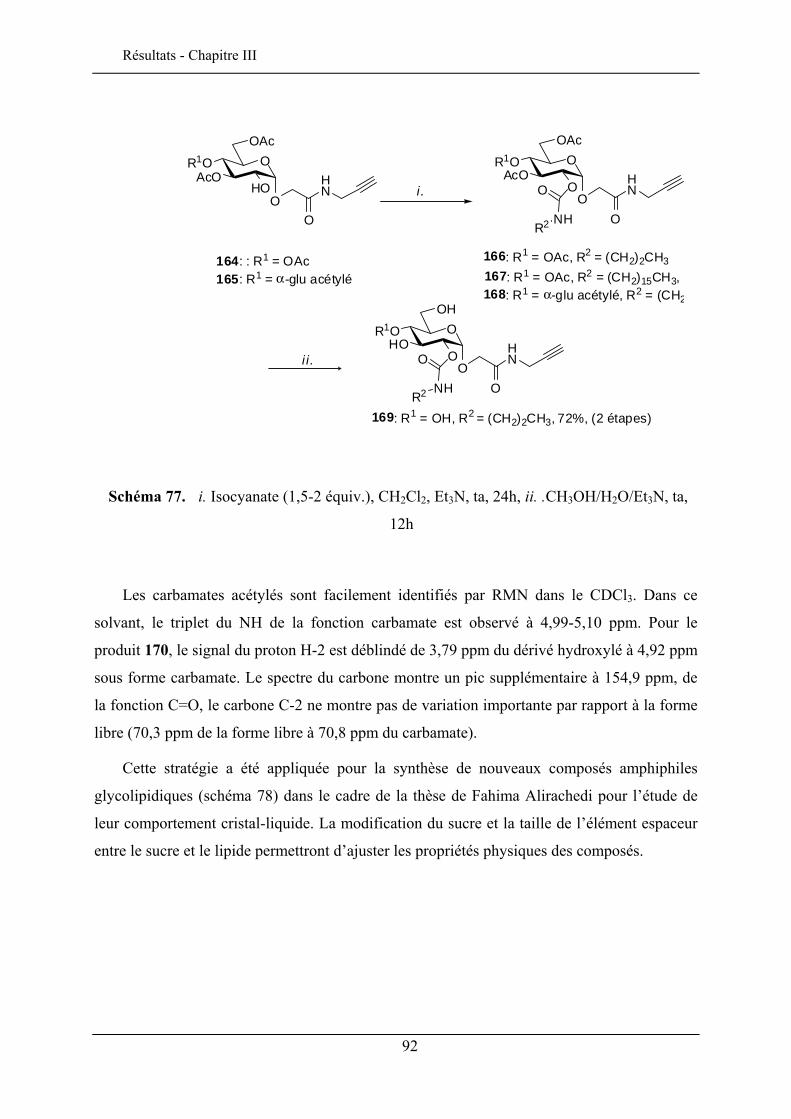
Résultats - Chapitre III
OR1OAcO
HO
OAc
OHN
O
OR1OAcO
O
OAc
OHN
ONH
O
R2
166: R1 = OAc, R2 = (CH2)2CH3167: R1 = OAc, R2 = (CH2)15CH3,168: R1 = α-glu acétylé, R2 = (CH2
OR1OHO
O
OH
OHN
ONH
O
R2
i .
i i .
169: R1 = OH, R2 = (CH2)2CH3, 72%, (2 étapes)
164: : R1 = OAc165: R1 = α-glu acétylé
Schéma 77. i. Isocyanate (1,5-2 équiv.), CH2Cl2, Et3N, ta, 24h, ii. .CH3OH/H2O/Et3N, ta,
12h
Les carbamates acétylés sont facilement identifiés par RMN dans le CDCl3. Dans ce
solvant, le triplet du NH de la fonction carbamate est observé à 4,99-5,10 ppm. Pour le
produit 170, le signal du proton H-2 est déblindé de 3,79 ppm du dérivé hydroxylé à 4,92 ppm
sous forme carbamate. Le spectre du carbone montre un pic supplémentaire à 154,9 ppm, de
la fonction C=O, le carbone C-2 ne montre pas de variation importante par rapport à la forme
libre (70,3 ppm de la forme libre à 70,8 ppm du carbamate).
Cette stratégie a été appliquée pour la synthèse de nouveaux composés amphiphiles
glycolipidiques (schéma 78) dans le cadre de la thèse de Fahima Alirachedi pour l’étude de
leur comportement cristal-liquide. La modification du sucre et la taille de l’élément espaceur
entre le sucre et le lipide permettront d’ajuster les propriétés physiques des composés.
92

Résultats - Chapitre III
OHOHO
OO
OH
O
NH OOHN
OHO
OO
OH
O
NHOO
HN
OHOHO
OHO
OH
Schéma 78. Schéma montrant deux structures mono- et disaccharidiques parmi une famille
de composés amphiphiles synthétisés pour l’étude du comportement cristal-liquide
Une autre transformation de l’hydroxyle libre en position 2 en carbamate a été effectuée
pour former le produit avec une fonction OCONH2 selon une procédure décrite pour la
synthèse d’inhibiteurs de la moenomycine.152,153 Les alcools 163, 164 et 165 sont traités avec
l’isocyanate du trichloroacétyle. Après une heure, la réaction est arrêtée avec du méthanol et
le produit est soumis à une purification sur une colonne de gel de silice. La déprotection du
groupement trichloroacétyle est effectuée par action du zinc dans le méthanol. Les produits
172, 173, 174 sont obtenus avec les rendements respectifs de 88%, 91% et 95%. La
déprotection a ensuite été effectuée dans le système NEt3/H2O/MeOH pour accéder aux
carbamates libres 175, 176, 177 avec des rendements respectifs de 97, 94%, 91% (schéma
79). Ce système de désacétylation a été choisi puisque l’utilisation du méthanolate de sodium
dans le méthanol a montré une élimination du groupement OCONH2.
152 Möller, U.; Hobert, K.; Donnerstag, A.; Wagner, P.; Müller, D.; Fehlhaber H.-W.; Markus. A.; Welzel, P. Tetrahedron 1993, 49, 1635-1648. 153 Luning, J.; Möller, U.; Müller, D.; Welzel, P.; Markus. A.; van Heijenoort, Y.; van Heijenoort, J. Tetrahedron 1993, 49, 10587-10596.
93

Résultats - Chapitre III
OR1OAcO
HO
OAc
OHN
OR2
OR1OAcO
O
OAc
OHN
OR2
H2N
O
OR1OHO
O
OH
OHN
OR2
H2N
O
i .
i i.
173: R1 = OAc, R2 = CH2CCH, 91%174: R1 = α-glu acétylé, R2 = CH2CCH, 95%
172: R1 = OAc, R2 = CH2CH=CH2, 88%
176: R1 = OH, R2 = CH2CCH, 94%177: R1 = α-glu, R2 = CH2CCH, 91%
175: R1 = OH, R2 = CH2CH=CH2, 97%
163: R1 = OAc, R2 = CH2CH=CH2164: R1 = OAc, R2 = CH2CCH165: R1 = α-glu acétylé, R2 = CH2CCH
Schéma 79. i. 1. Trichloroacétyl isocyanate (1,6 équiv.), CH2Cl2, -15°C, 1h, 2. Zn, MeOH,
30 min, ii .CH3OH/H2O/Et3N, ta, 12h
Le spectre du proton du produit acétylé 175 effectué dans le CDCl3 montre à 5,19 ppm un
large singulet qui intègre pour 2 protons correspondant à la fonction CONH2 Pour les
produits libres, ce pic n’est pas observé par échange avec les deutériums de CD3OD. Comme
dans le cas des autres carbamates, H-2 est plus déblindé par rapport au produit de départ (3,79
dans le cas de la molécule hydroxylée à 4,81 ppm), et le carbone C-2 ne montre pas de
changement considérable par rapport à la molécule hydroxylée (de 70,3 ppm pour la molécule
hydroxylée à 71,0 ppm en présence du carbamate). En revanche, nous visualisons un pic
supplémentaire vers 155,2 ppm correspondant au C=O de la fonction carbamoyle.
Dans le contexte de la synthèse d’inhibiteurs potentiels des O-N-acétyl-
glucosaminyltansférases (OGT), les 2 produits 176 et 177 ont été engagés dans une
cycloaddition 1,3-dipolaire de type Huisgen catalysée par du cuivre (I) avec la 5’-azido-5’-
désoxyuridine154 pour obtenir les produits 178 et 179 avec des rendements de 84% et 81%
respectivement (schéma 66).
154 Eager, A. R.; Finney, N. S. J. Org. Chem. 2004, 69, 613-618.
94
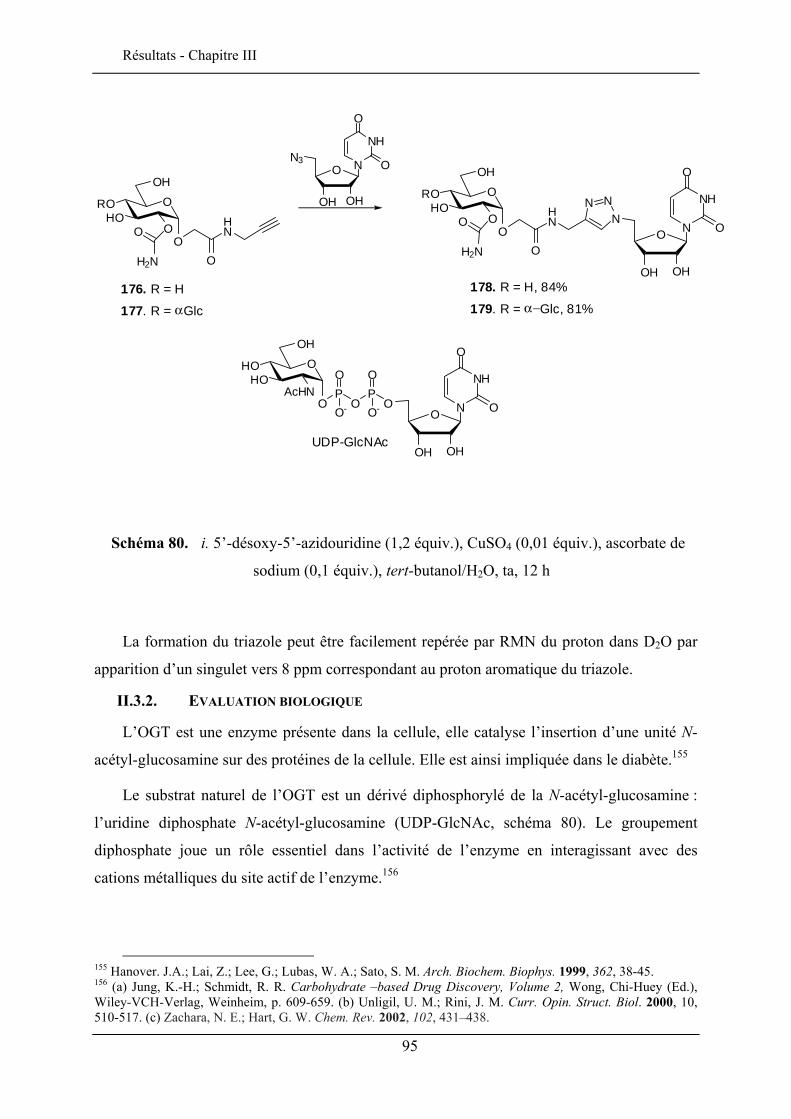
Résultats - Chapitre III
OROHO
O
OH
O
HN
OH2N
O NNN
176. R = H
177. R = αGlc
NH
O
ONO
OHOH
OROHO
O
OH
O
HN
OH2N
O
178. R = H, 84%
179. R = α−Glc, 81%
N3
NH
O
ONO
OHOH
OHOHO
AcHN
OH
O
NH
O
ONO
OHOH
PO
PO
O O
O- O-
UDP-GlcNAc
Schéma 80. i. 5’-désoxy-5’-azidouridine (1,2 équiv.), CuSO4 (0,01 équiv.), ascorbate de
sodium (0,1 équiv.), tert-butanol/H2O, ta, 12 h
La formation du triazole peut être facilement repérée par RMN du proton dans D2O par
apparition d’un singulet vers 8 ppm correspondant au proton aromatique du triazole.
II.3.2. EVALUATION BIOLOGIQUE
L’OGT est une enzyme présente dans la cellule, elle catalyse l’insertion d’une unité N-
acétyl-glucosamine sur des protéines de la cellule. Elle est ainsi impliquée dans le diabète.155
Le substrat naturel de l’OGT est un dérivé diphosphorylé de la N-acétyl-glucosamine :
l’uridine diphosphate N-acétyl-glucosamine (UDP-GlcNAc, schéma 80). Le groupement
diphosphate joue un rôle essentiel dans l’activité de l’enzyme en interagissant avec des
cations métalliques du site actif de l’enzyme.156
155 Hanover. J.A.; Lai, Z.; Lee, G.; Lubas, W. A.; Sato, S. M. Arch. Biochem. Biophys. 1999, 362, 38-45. 156 (a) Jung, K.-H.; Schmidt, R. R. Carbohydrate –based Drug Discovery, Volume 2, Wong, Chi-Huey (Ed.), Wiley-VCH-Verlag, Weinheim, p. 609-659. (b) Unligil, U. M.; Rini, J. M. Curr. Opin. Struct. Biol. 2000, 10, 510-517. (c) Zachara, N. E.; Hart, G. W. Chem. Rev. 2002, 102, 431–438.
95
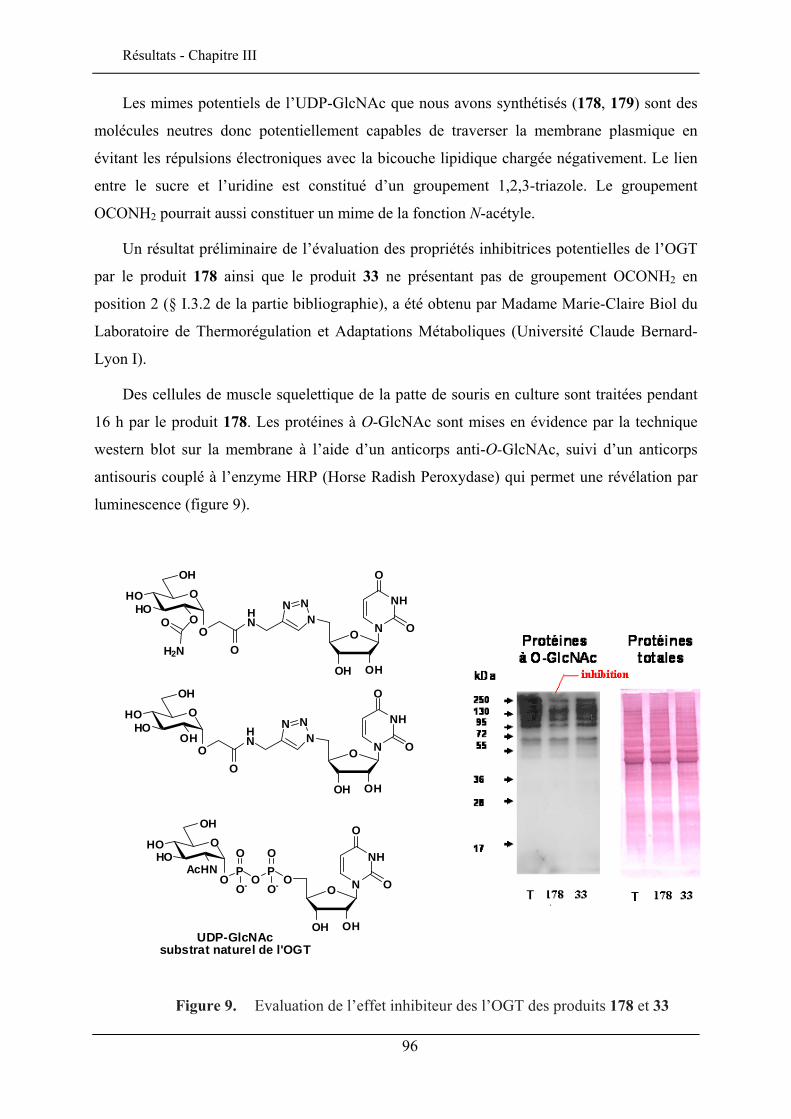
Résultats - Chapitre III
Les mimes potentiels de l’UDP-GlcNAc que nous avons synthétisés (178, 179) sont des
molécules neutres donc potentiellement capables de traverser la membrane plasmique en
évitant les répulsions électroniques avec la bicouche lipidique chargée négativement. Le lien
entre le sucre et l’uridine est constitué d’un groupement 1,2,3-triazole. Le groupement
OCONH2 pourrait aussi constituer un mime de la fonction N-acétyle.
Un résultat préliminaire de l’évaluation des propriétés inhibitrices potentielles de l’OGT
par le produit 178 ainsi que le produit 33 ne présentant pas de groupement OCONH2 en
position 2 (§ I.3.2 de la partie bibliographie), a été obtenu par Madame Marie-Claire Biol du
Laboratoire de Thermorégulation et Adaptations Métaboliques (Université Claude Bernard-
Lyon I).
Des cellules de muscle squelettique de la patte de souris en culture sont traitées pendant
16 h par le produit 178. Les protéines à O-GlcNAc sont mises en évidence par la technique
western blot sur la membrane à l’aide d’un anticorps anti-O-GlcNAc, suivi d’un anticorps
antisouris couplé à l’enzyme HRP (Horse Radish Peroxydase) qui permet une révélation par
luminescence (figure 9).
OHOHO
O
OH
OHN
OH2N
O NNN NH
O
ONO
OHOH
OHOHO
OH
OH
OHN
O
NNN NH
O
ONO
OHOH
OHOHO
AcHN
OH
O
NH
O
ONO
OHOH
PO
PO
O O
O- O-
UDP-GlcNAcsubstrat naturel de l'OGT
Figure 9. Evaluation de l’effet inhibiteur des l’OGT des produits 178 et 33
96

Résultats - Chapitre III
Ainsi, l’intensité relative des taches obtenues montre que le produit 178 utilisé à 250 µM
semble induire une inhibition de la glycosylation des protéines à O-GlcNAc par rapport aux
cellules témoins non traitées (T). Dans les mêmes conditions, le produit 33 semble avoir un
effet moins marqué.
Ces expériences seront répétées afin de confirmer cette première observation. La
quantification de l’inhibition sera aussi effectuée par mesure de l’IC50 de la molécule 178
(Concentration du composé nécessaire pour inhiber 50 % de l'effet observé).
II.3.3. ETHERIFICATION
Des dérivés éthérifiés en position 2 ont été préparés. Une fonction acide, convenable pour
des couplages peptidiques, a été introduite par utilisation du bromoacétate de tert-butyle.
L’amide 164 a ainsi donné accès à l’éther 180 avec un rendement de 72%. Le clivage du
groupement tert-butyle par action du TFA a libéré la fonction acide pour donner le
groupement carboxyméthyle 181 avec un rendement de 90% (schéma 81).
Un lien propargyle a aussi été obtenu par action du bromure de propargyle et de NaH sur
les glucosides 164 et 163 pour donner les éthers 182 et 183 avec des rendements respectifs de
56 et 55% (schéma 81).
OAcOAcO
RO
OAc
OHN
O164: R = H
180: R = COCH2COOC(CH3)3
181: R = COCH2COOH
i
OAcOAcO
RO
OAc
OHN
O
163: R = H
183: R = CH2CCHii
182: R = CH2CCH
iiiiv
Schéma 81. i. Bromoacétate de tert-butyle (1,2 équiv.), K2CO3 (2,5 équiv.), DMF, ta, 16 h,
72%. ii. TFA, CH2Cl2, ta, 2h, 90%; iii. NaH, bromure de propargyle (2,5 équiv.), ta, 30 min,
56%; iv. mêmes conditions, 55%
97

Résultats - Chapitre III
La fonction éther n’induit pas de changements significatifs dans les spectres RMN du
proton par rapport à la forme OH libre. En effet, pour le produit 183 par exemple, le proton H-
2 apparaît à 3,80 ppm, la même zone de déplacement chimique de H-2 dans le cas de la
molécule hydroxylée (3,78 ppm). Par contre, le spectre de RMN du carbone montre un plus
grand déblindage de C-2 qui passe de 70,3 ppm à 76,6 ppm.
Le groupement tert-butyle de l’éther 180 a été identifié par RMN de proton par apparition
d’un singulet à 1,41 ppm intégrant pour 9 protons, le méthylène du groupement
carboxyméthyle en position 2 est observé à 3,96-4,26 ppm. Dans le spectre de RMN du
carbone, un pic à 28,1 ppm du tert-butyle est visualisé ainsi qu’un méthylène à 67,2 ppm
observé en DEPT 135 correspondant au H-7’. Le carbone quaternaire du tert-butyle apparaît à
82,5 ppm et un C=O supplémentaire est observé entre 168,9 et 170,5 ppm. L’amide 181 est
identifié par la disparition des pics relatifs au groupement tert-butyle en RMN du proton et du
carbone.
L’identification des deux éthers 182 et 183 a été possible en observant pour les deux
produits un triplet résonnant à 2,24-2,60 ppm correspondant au proton acétylénique ainsi
qu’un signal correspondant au méthylène de l’éther à 3,93-4,42 ppm. Ce dernier a été identifié
par la corrélation qu’il établit avec le proton acétylénique observé d’après la RMN
bidimensionnelle COSY (H-H).
En RMN du carbone, un méthylène supplémentaire est observé à 59,5 ppm correspondant
à celui de la fonction éther, ainsi qu’un méthyne à 76,0 ppm correspondant au carbone
acétylénique et un carbone quaternaire à 79,0 ppm correspondant au carbone de la triple
liaison.
II.3.4. AZIDATION
Un dernier exemple de fonctionnalisation en position 2 a été l’introduction d’un
groupement azido. Les 2-azido ont déjà été synthétisés soit comme des précurseurs
d’analogues de la N-acétyl-glucosamine et la N-acétyl-mannosamine par azidonitration de
glucals148 ou par une SN2 avec inversion de configuration du carbone C-2,157 soit lors de la
transformation du saccharose en ses manno- et altro-isomères.158
157 (a) Kiso, M.; Ishida, H.; Kawaide, A.; Hasegawa, A. Carbohydr. Res. 1984, 127, 129–135; (b) Knapp, S.; Kukkola, P. J.; Sharma, S.; Dhar, T. G. M.; Naughton, A. B. J. J. Org. Chem. 1990, 55, 5700–5710; (c) Pavliak, V.; Kovac, P. Carbohydr. Res. 1991, 210, 333–337; (d) Sato, K.; Yoshitomo, A. Chem. Lett. 1995, 39–40; (e) Sato, K.; Yoshitomo, A.; Takai, Y. Bull. Chem. Soc. Jpn. 1997, 70, 885–890; (f) Stolz, F.; Reiner, M.; Blume, A.; Reutter, W.; Schmidt, R. R. J. Org. Chem. 2004, 69, 665–679; (g) Popelova, A.; Kefurt, K.; Hlavackova, M.; Moravcova, J. Carbohydr. Res. 2005, 340, 161–166; (h) Teodorović, P.; Slättegård R. ; Oscarson, S. Carbohydr.
98

Résultats - Chapitre III
Après l’ouverture de la lactone du glucose, les dérivés azido-manno sont obtenus par une
substitution SN2 du triflate par l’azoture de sodium. Les 2-azido 184 et 185 sont obtenus avec
96% et 77% de rendements respectifs (schéma 82).
OAcOAcO
N3AcO
OHN
O
164 iOAcOAcO
N3AcO
OHN
O
163 i
184 185
Schéma 82. i. Tf2O (1,5 équiv.), Py (2 équiv.), CH2Cl2, -17°C, 1h, puis NaN3 (20 équiv.),
Bu4NI, DMF, 60°C, 15h, 77%; ii. Mêmes conditions, 96%
Le spectre de RMN du proton du glucoside 184 montre que la fonction azido en position
2 induit un déblindage du proton H-2 de 3,78 ppm à 4,09 ppm avec une faible constante de
couplage J1,3 = 1,7 ppm. La RMN du carbone, par contre, montre un grand blindage du
carbone C-2 qui passe de 70,3 ppm à 60,9 ppm. Les mêmes caractéristiques ont été observées
lors de la formation de l’azido 185.
Des premiers essais de polymérisation du monomère 185 dans des conditions « Click »
ont été effectués dans le cadre de la thèse de Jie Chen au laboratoire (schéma 83). Le but du
projet est de synthétiser et d’étudier les propriétés physico-chimiques de nouveaux polymères
à base de sucre.
Res. 2005, 340, 2675–2676. (i) Zhang, J.; Yan, S.; Liang, X.; Wu, J.; Wang, D. ; Kong, F. Carbohydr. Res. 2007, 342, 2810-2817. 158 Lichtenthaler, F. W.; Mondel, S. Carbohydr. Res. 1997, 303, 293-302.
99

Résultats - Chapitre III
OAcOAcO
AcO
OHN
O
OAcOAcO
N3AcO
OHN
O
OAcOAcO
AcO
OHN
O
N NN
N NN
n
Schéma 83. Un polymère obtenu par polymérisation du monomère 185
II.4. FONCTIONNALISATION EN POSITION 6
En plus de la substitution de la position-2, nous avons voulu étudier la possibilité de
rajouter un groupement fonctionnel en position 6 du sucre. Pour cela, la molécule bis-
propargylée 182 a été engagée dans une réaction d’azidation par déplacement du tosylate
après désacétylation ; le but était de synthétiser des systèmes AB2 intéressants pour la
polymérisation.
En effet, les acétates ont été enlevés par action du méthanolate de sodium dans le
méthanol. Le tosylate est synthétisé par action du chlorure de tosyle dans la pyridine pour
donner le produit attendu 187 avec 54% de rendement. Celui-ci est ensuite substitué par
action de NaN3 dans le DMF à 60°C pour donner le composé 6-azido avec un rendement non
optimisé de 16% (188, schéma 84).
100

Résultats - Chapitre III
OHOHO
O
HO
OHN
O
182 i
186
OHOHO
O
N3
OHN
O
187
OHOHO
O
TsO
OHN
O
188
i i. i ii .
Schéma 84. i. MeOH, MeONa, ta, 3h, 82%, ii. TsCl (2,5 équiv.), Py, 12h, 54% iii. NaN3 (5
équiv.), DMF, 60°C, 15h, 16%.
Le spectre de RMN du proton du produit 186 montre la disparition des acétates vers 2
ppm ainsi qu’un système AB doublé correspondant au méthylène de la position 2 (H-7’) par
couplage avec le proton acétylénique.
Sur le composé 187, le tosyle en position 6 est facilement repérable par apparition de
protons aromatiques à 7,27-7,73 ppm, un singulet à 2,38 ppm correspondant au méthyle du
groupement tosyle ainsi qu’un déblindage de H-6a et H-6b qui passent de 3,69 et 3,83 ppm à
4,12 et 4,31 ppm respectivement. Le spectre de RMN du carbone montre un signal à 21,8 ppm
correspondant au méthyle du groupement tosyle ainsi que 4 carbones aromatiques vers 128,1-
130,1 ppm. Le C-6 qui apparaît à 62,3 ppm sous la forme libre passe à 67,5 ppm en présence
du tosyle.
Le groupement azido du composé 188 induit un blindage des H-6 qui sont observés à
3,44-3,52 ppm en RMN du proton. Le spectre de RMN du carbone montre une variation
considérable du déplacement chimique du carbone C-6 qui passe de 67,5 ppm en présence du
tosyle à 51,5 ppm après substitution par l’azide.
II.5. CONCLUSION
A la suite de l’ouverture de deux lactones mono- et disaccharidique par une amine, la
présence d’une fonction hydroxyle libre a permis la fonctionnalisation de la position 2 pour
donner de nouveaux carbamates, éthers, azides. Cette voie de synthèse a donné accès à de
nouvelles structures glycolipidiques qui seront par la suite étudiées pour leurs propriétés en
phase cristal-liquide. Des inhibiteurs potentiels des glycosyltransférases ont aussi été
101

Résultats - Chapitre III
synthétisés dont la première évaluation a montré une certaine activité inhibitrice de l’OGT.
Par ailleurs, des systèmes comportant à la fois des systèmes propargyles et azido ont pu être
synthétisés pour accéder à une nouvelle famille de glycomonomères. Ces résultats montrent
les potentialités offertes par la stratégie CMGL pour accéder à de nouveaux composés
bifonctionnels.
Enfin, une première approche vers des systèmes trifonctionnels a été réalisée par
substitution sélective de l’hydroxyle en position 6, conduisant, comme premier exemple
choisi, à un monomère de type AB2.
102

Résultats - Chapitre III
CHAPITRE III. SYNTHESE DE SONDES FLUORESCENTES POUR
L’IMAGERIE MEMBRANAIRE NEURONALE
III.1. INTRODUCTION
Les travaux présentés dans ce chapitre s’inscrivent dans le cadre d’une collaboration
entre le Laboratoire de Chimie Organique de l’INSA de Lyon, le Laboratoire de Chimie pour
l’Optique de l’Ecole Normale Supérieure de Lyon et le Laboratoire de Spectrométrie
Physique de l’Université Joseph Fourier de Grenoble. Le but de ce projet est de synthétiser et
d’évaluer les propriétés de nouvelles sondes biologiques optimisées en optique non linéaire en
mettant en avant quelques-uns des principaux inconvénients des fluorophores commerciaux, à
savoir leur faible solubilité en milieu biologique et leurs activités non linéaires modérées.
Du point de vue physique, le but est de mettre au point une imagerie efficace des
variations du potentiel membranaire de cellules au sein de tissus nerveux, plus
particulièrement le cortex cérébral embryonnaire. L’équipe du Laboratoire de Spectrométrie
Physique se charge des cultures cellulaires et de mettre au point l’appareillage pour la
détection du second harmonique généré par des molécules sondes insérées dans les
membranes plasmatiques de ces cellules.
Du point de vue chimique, l’équipe de Chantal Andraud du Laboratoire « Chimie pour
l’Optique » a synthétisé des chromophores qui sont optimisées en GSH et TPEF. En ce qui
nous concerne, nous nous intéressons à rendre ces molécules solubles et amphiphiles et ceci
par greffage des entités sucre sur les chromophores.
103

Résultats - Chapitre III
III.2. LE CHROMOPHORE
Un chromophore modèle159 a été synthétisé et étudié par le Laboratoire de Chimie pour
l’Optique. Sa structure est présentée dans le schéma 85. Cette molécule non centrosymétrique
présente des propriétés optiques et spectroscopiques très intéressantes en TPEF et GSH
simultanément. L’absorption linéaire est entre 350 nm et 450 nm. Par conséquent,
l’absorption à deux photons se trouve entre 700 nm et 900 nm donc optimisée pour travailler
dans la fenêtre de transparence des tissus biologiques.
De plus, son rendement quantique (§ III.5.2 du présent chapitre) est fort dans un solvant
apolaire (89% dans le CH2Cl2). Son solvatochromisme (§ III.5.1 du présent chapitre) est
faible en .absorption et fort en émission, caractéristique des molécules qui ont un faible
moment dipolaire à l’état fondamental et un important dipôle à l’état excité.
N
N
O
N
O
N
63
site C
site A
site B
Schéma 85. Chromophore modèle
159 Barsu, C.; Fortrie, R.; Nowika, K.; Baldeck, P. L.; Vial, J. C.; Barsella, A.; Fort, A.; Hissler, M.; Bretonnière, Y.; Maury, O.; Andraud, C. Chem. Commun. 2006, 4744–4746.
104

Résultats - Chapitre III
Ce composé modèle est très hydrophobe et il ne présente pas l’amphiphilie nécessaire aux
sondes membranaires. Cependant, du point de vue structural, il présente trois sites qui
pourraient être fonctionnalisés pour rendre la molécule amphiphile et contrôler la balance
hydrophile/lipophile :
a. Site A : Un groupement pyridine dicarboxamide accepteur d’électrons avec des
chaînes hydrophobes. La modification de la longueur de ces chaînes permettrait
éventuellement de contrôler la profondeur de sa pénétration dans la membrane.
b. Site B : Un groupement donneur fort de type dialkylamino, auquel des groupements
hydrosolubles pourraient être greffés.
c. Site C : Un groupement éthynyl-fluorène. Le fluorène est connu pour ses propriétés de
fluorescence importante et l’alcyne est un très bon pont de transfert de charge. Les
chaînes latérales hexyles, qui apportent une hydrophobie à la sonde sont difficilement
modifiables pour des problèmes de solubilité d’intermédiaires réactionnels.
III.3. STRUCTURE DES SONDES MEMBRANAIRES
La balance hydrophile/lipophile des sondes membranaires choisies pour ce travail a été
modulée au niveau des sites A et B.
Deux types de chaînes alkyles ont été utilisés sur le site A. Une chaîne éthyle, comme
dans la structure du chromophore modèle et une chaîne octyle pour augmenter la lipophilie de
la sonde (schéma 86).
Trois modifications ont été proposées pour rendre le chromophore hydrosoluble en
fonctionnalisant le site B (schéma 86):
- des fonctions amines libres, qui seront chargées à pH physiologique.
- l’incorporation des chaînes polyéthylène glycols qui grâce à la sphère d’hydratation de
leurs oxygènes, confèrent la solubilité nécessaire. Ces groupements sont connus pour
augmenter l’hydrosolubilité de polymères160 ou de dendrimères,161 de nanoparticules162 et de
chromophores pour la TPEF.163
160 Lottner, C.; Bart, K.-C.; Bernhardt, G.; Brunner, H. J. Med. Chem. 2002, 45, 2079-2089. 161 Oar, M. A.; Serin, J. M.; Dichtel, W. R.; Frechet, J. M. J.; Ohulchanskyy, T. Y.; Prasad, P. N. Chem. Materials. 2005, 17, 2267-2275. 162 Pengo, P.; Polizzi, S.; Battagliarin, M.; Pasquato, L.; Scrimin, P. J. Mat. Chem. 2003, 13, 2471-2478. 163 Abert, M.; Mora, N.; Lacombe, J.-M. Carbohydr. Res. 2002, 337, 11, 997-1006.
105
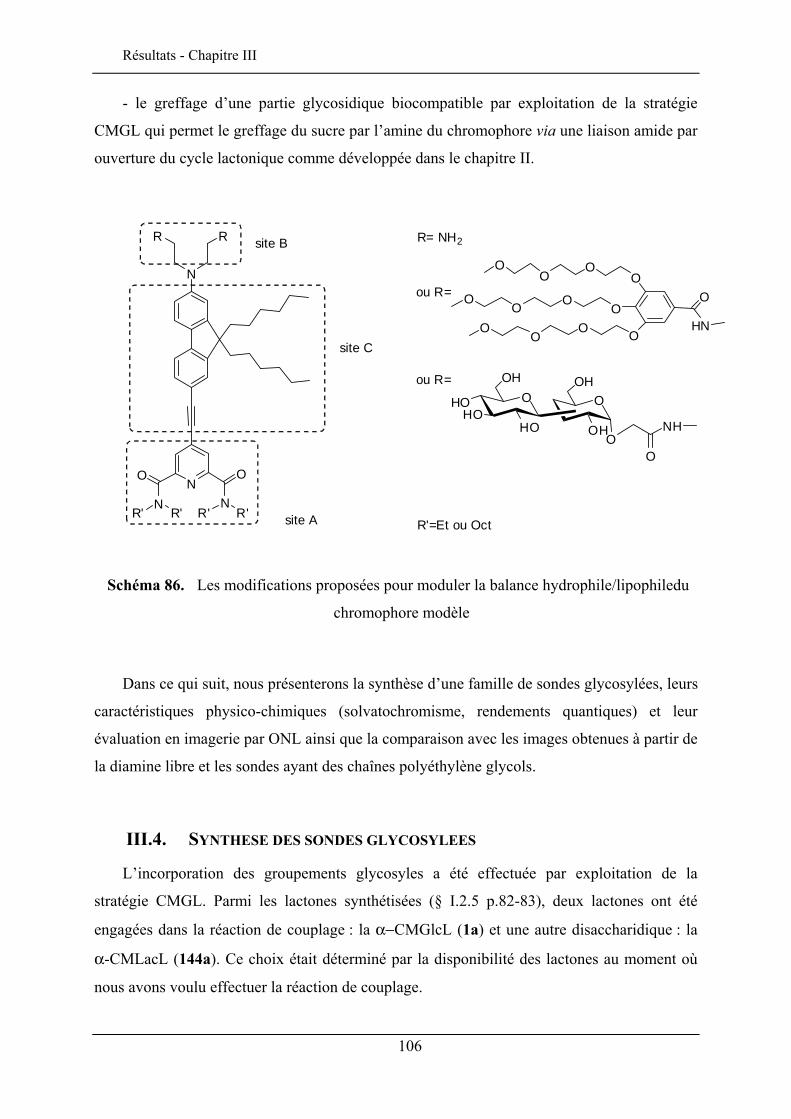
Résultats - Chapitre III
- le greffage d’une partie glycosidique biocompatible par exploitation de la stratégie
CMGL qui permet le greffage du sucre par l’amine du chromophore via une liaison amide par
ouverture du cycle lactonique comme développée dans le chapitre II.
N
N
O
N
O
NR'R' R'R'
site C
site A
site BR R R= NH2
O
HN
O
O
O
OO
O
OOO
OO
O
OOH
OOH
HOHO
HO OOH
NH
O
ou R=
ou R=
R'=Et ou Oct
Schéma 86. Les modifications proposées pour moduler la balance hydrophile/lipophiledu
chromophore modèle
Dans ce qui suit, nous présenterons la synthèse d’une famille de sondes glycosylées, leurs
caractéristiques physico-chimiques (solvatochromisme, rendements quantiques) et leur
évaluation en imagerie par ONL ainsi que la comparaison avec les images obtenues à partir de
la diamine libre et les sondes ayant des chaînes polyéthylène glycols.
III.4. SYNTHESE DES SONDES GLYCOSYLEES
L’incorporation des groupements glycosyles a été effectuée par exploitation de la
stratégie CMGL. Parmi les lactones synthétisées (§ I.2.5 p.82-83), deux lactones ont été
engagées dans la réaction de couplage : la α−CMGlcL (1a) et une autre disaccharidique : la
α-CMLacL (144a). Ce choix était déterminé par la disponibilité des lactones au moment où
nous avons voulu effectuer la réaction de couplage.
106
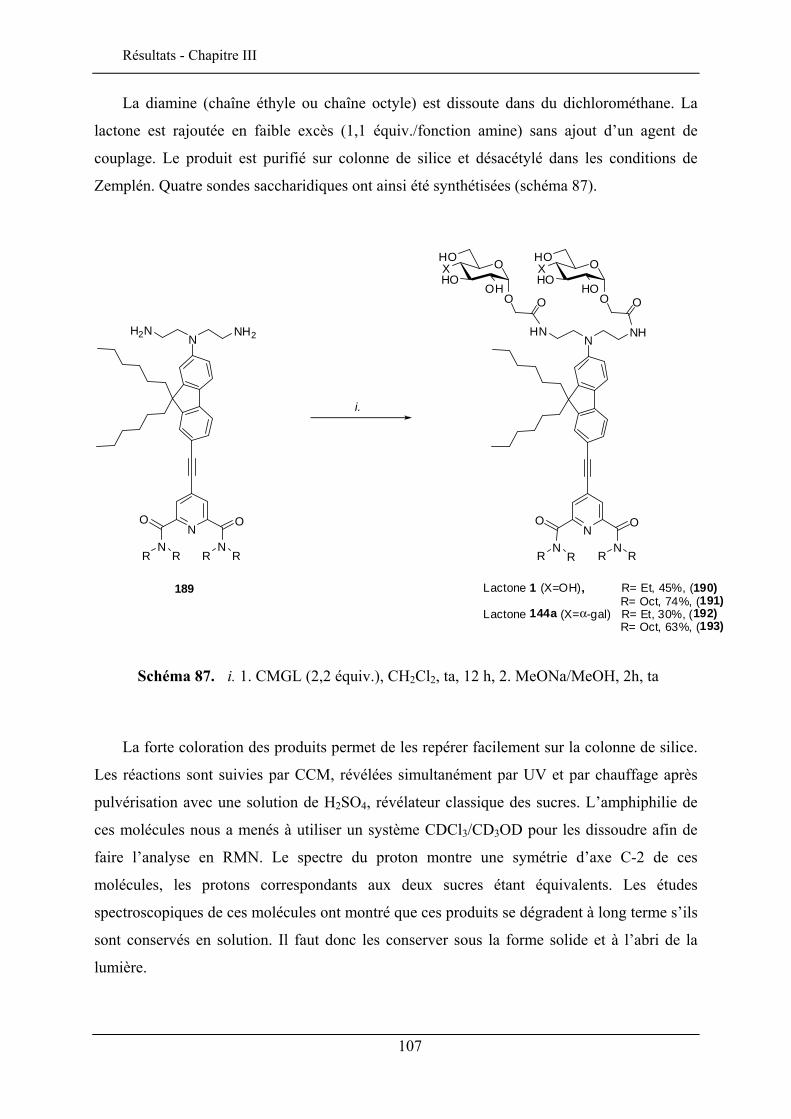
Résultats - Chapitre III
La diamine (chaîne éthyle ou chaîne octyle) est dissoute dans du dichlorométhane. La
lactone est rajoutée en faible excès (1,1 équiv./fonction amine) sans ajout d’un agent de
couplage. Le produit est purifié sur colonne de silice et désacétylé dans les conditions de
Zemplén. Quatre sondes saccharidiques ont ainsi été synthétisées (schéma 87).
N
NN
O
N
O
R R
NH2H2N
RR
Lactone 1 (X=OH), R= Et, 45%, (190)R= Oct, 74%, (191)
Lactone 144a (X=α-gal) R= Et, 30%, (192)R= Oct, 63%, (193)
OXHO
OH
HO
O O
NNHHN
NN
O
N
O
R RRR
OXHO
HO
HO
O O
i.
189
Schéma 87. i. 1. CMGL (2,2 équiv.), CH2Cl2, ta, 12 h, 2. MeONa/MeOH, 2h, ta
La forte coloration des produits permet de les repérer facilement sur la colonne de silice.
Les réactions sont suivies par CCM, révélées simultanément par UV et par chauffage après
pulvérisation avec une solution de H2SO4, révélateur classique des sucres. L’amphiphilie de
ces molécules nous a menés à utiliser un système CDCl3/CD3OD pour les dissoudre afin de
faire l’analyse en RMN. Le spectre du proton montre une symétrie d’axe C-2 de ces
molécules, les protons correspondants aux deux sucres étant équivalents. Les études
spectroscopiques de ces molécules ont montré que ces produits se dégradent à long terme s’ils
sont conservés en solution. Il faut donc les conserver sous la forme solide et à l’abri de la
lumière.
107
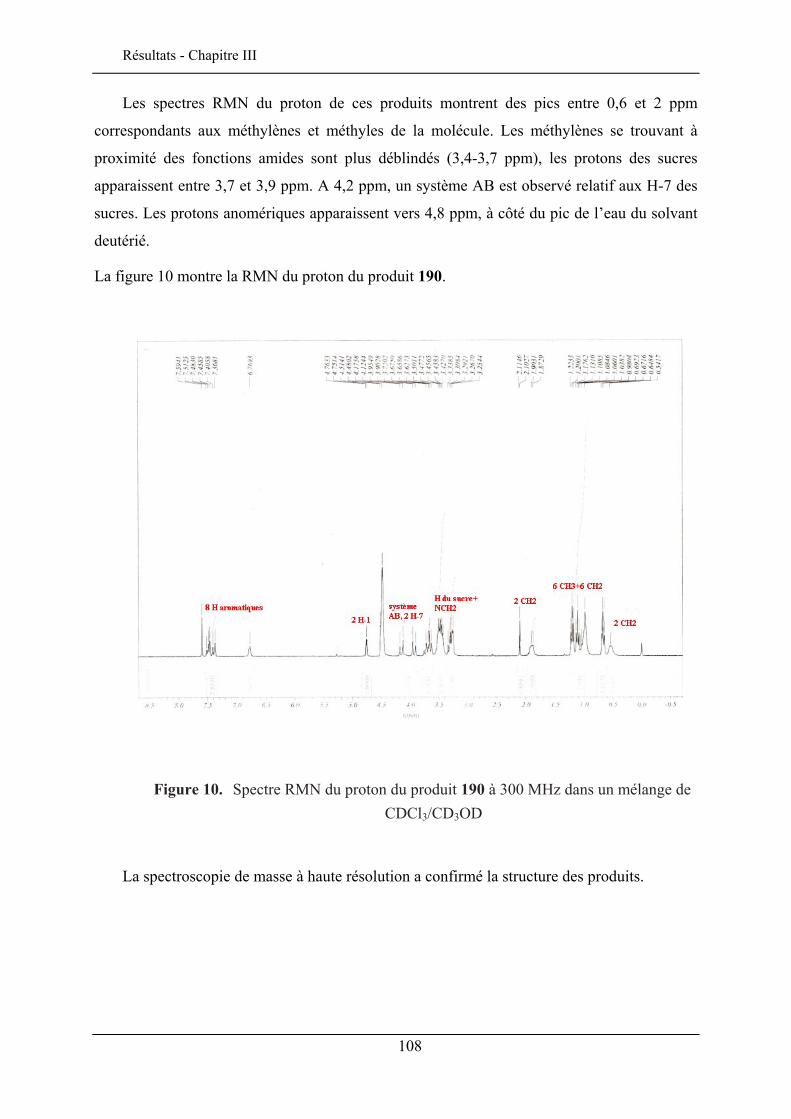
Résultats - Chapitre III
Les spectres RMN du proton de ces produits montrent des pics entre 0,6 et 2 ppm
correspondants aux méthylènes et méthyles de la molécule. Les méthylènes se trouvant à
proximité des fonctions amides sont plus déblindés (3,4-3,7 ppm), les protons des sucres
apparaissent entre 3,7 et 3,9 ppm. A 4,2 ppm, un système AB est observé relatif aux H-7 des
sucres. Les protons anomériques apparaissent vers 4,8 ppm, à côté du pic de l’eau du solvant
deutérié.
La figure 10 montre la RMN du proton du produit 190.
Figure 10. Spectre RMN du proton du produit 190 à 300 MHz dans un mélange de CDCl3/CD3OD
La spectroscopie de masse à haute résolution a confirmé la structure des produits.
108
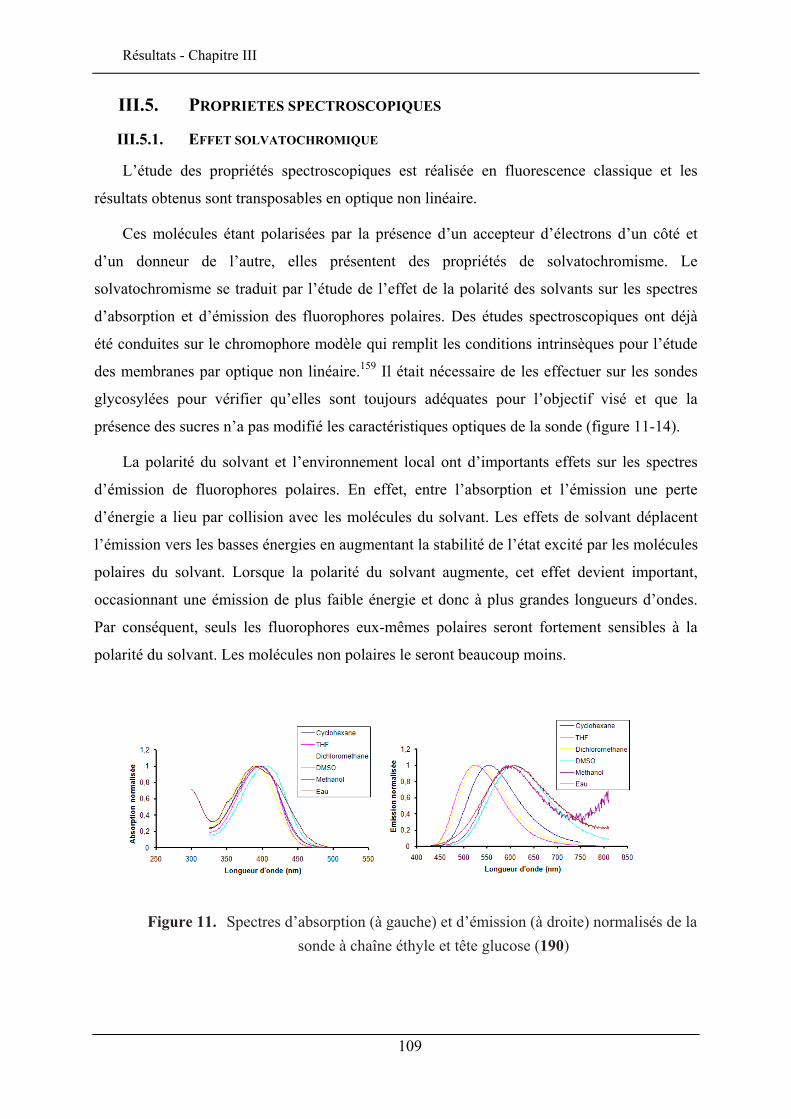
Résultats - Chapitre III
III.5. PROPRIETES SPECTROSCOPIQUES
III.5.1. EFFET SOLVATOCHROMIQUE
L’étude des propriétés spectroscopiques est réalisée en fluorescence classique et les
résultats obtenus sont transposables en optique non linéaire.
Ces molécules étant polarisées par la présence d’un accepteur d’électrons d’un côté et
d’un donneur de l’autre, elles présentent des propriétés de solvatochromisme. Le
solvatochromisme se traduit par l’étude de l’effet de la polarité des solvants sur les spectres
d’absorption et d’émission des fluorophores polaires. Des études spectroscopiques ont déjà
été conduites sur le chromophore modèle qui remplit les conditions intrinsèques pour l’étude
des membranes par optique non linéaire.159 Il était nécessaire de les effectuer sur les sondes
glycosylées pour vérifier qu’elles sont toujours adéquates pour l’objectif visé et que la
présence des sucres n’a pas modifié les caractéristiques optiques de la sonde (figure 11-14).
La polarité du solvant et l’environnement local ont d’importants effets sur les spectres
d’émission de fluorophores polaires. En effet, entre l’absorption et l’émission une perte
d’énergie a lieu par collision avec les molécules du solvant. Les effets de solvant déplacent
l’émission vers les basses énergies en augmentant la stabilité de l’état excité par les molécules
polaires du solvant. Lorsque la polarité du solvant augmente, cet effet devient important,
occasionnant une émission de plus faible énergie et donc à plus grandes longueurs d’ondes.
Par conséquent, seuls les fluorophores eux-mêmes polaires seront fortement sensibles à la
polarité du solvant. Les molécules non polaires le seront beaucoup moins.
Figure 11. Spectres d’absorption (à gauche) et d’émission (à droite) normalisés de la sonde à chaîne éthyle et tête glucose (190)
109
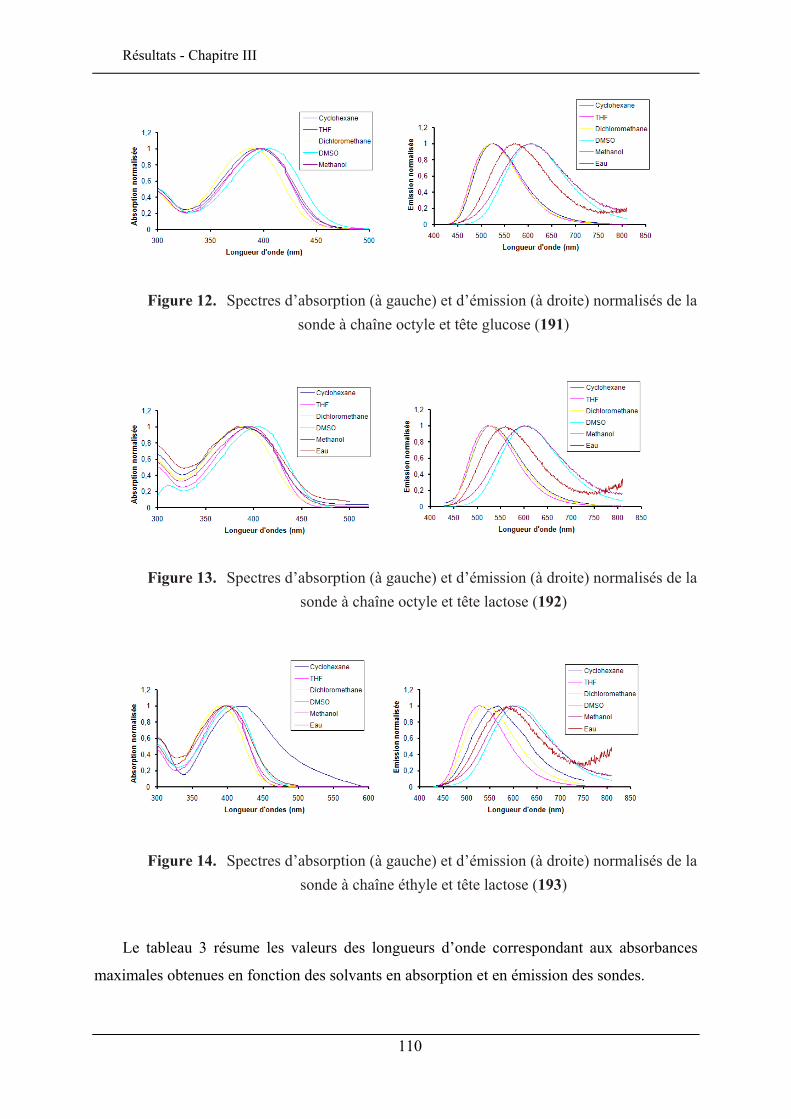
Résultats - Chapitre III
Figure 12. Spectres d’absorption (à gauche) et d’émission (à droite) normalisés de la sonde à chaîne octyle et tête glucose (191)
Figure 13. Spectres d’absorption (à gauche) et d’émission (à droite) normalisés de la sonde à chaîne octyle et tête lactose (192)
Figure 14. Spectres d’absorption (à gauche) et d’émission (à droite) normalisés de la sonde à chaîne éthyle et tête lactose (193)
Le tableau 3 résume les valeurs des longueurs d’onde correspondant aux absorbances
maximales obtenues en fonction des solvants en absorption et en émission des sondes.
110
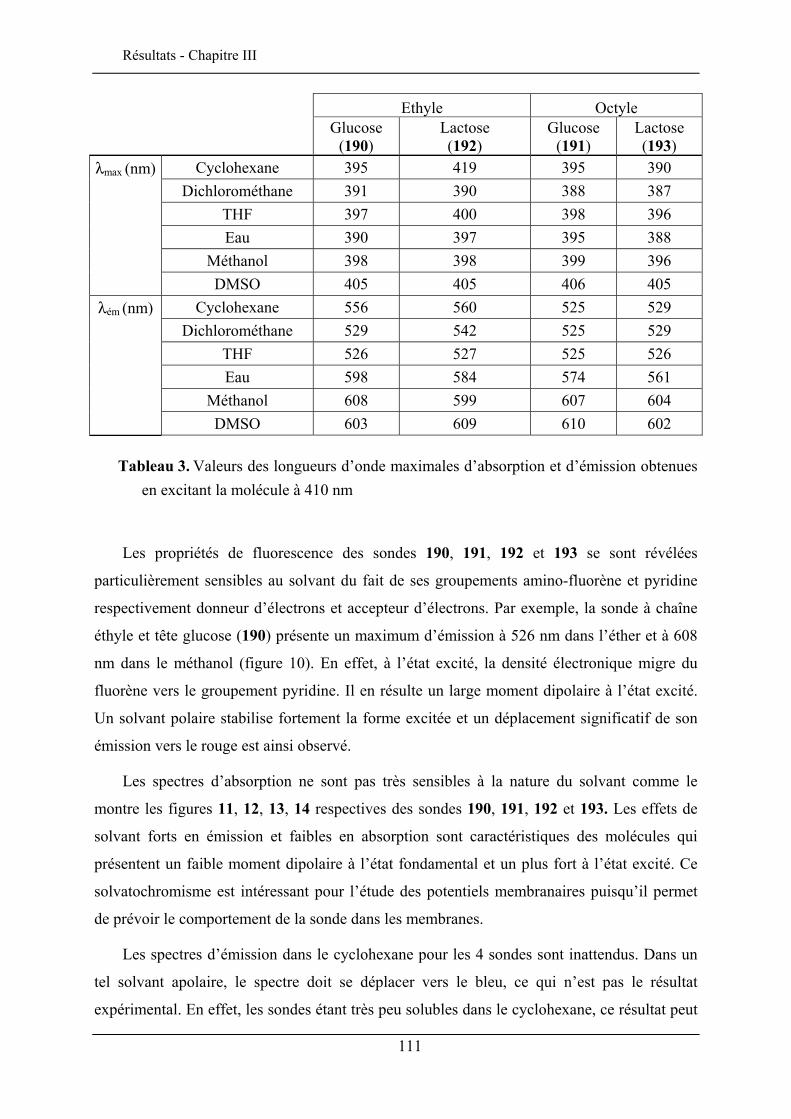
Résultats - Chapitre III
Ethyle Octyle Glucose
(190) Lactose (192)
Glucose (191)
Lactose (193)
λmax (nm) Cyclohexane 395 419 395 390 Dichlorométhane 391 390 388 387 THF 397 400 398 396 Eau 390 397 395 388
Méthanol 398 398 399 396
DMSO 405 405 406 405 λém (nm) Cyclohexane 556 560 525 529
Dichlorométhane 529 542 525 529 THF 526 527 525 526 Eau 598 584 574 561
Méthanol 608 599 607 604
DMSO 603 609 610 602
Tableau 3. Valeurs des longueurs d’onde maximales d’absorption et d’émission obtenues en excitant la molécule à 410 nm
Les propriétés de fluorescence des sondes 190, 191, 192 et 193 se sont révélées
particulièrement sensibles au solvant du fait de ses groupements amino-fluorène et pyridine
respectivement donneur d’électrons et accepteur d’électrons. Par exemple, la sonde à chaîne
éthyle et tête glucose (190) présente un maximum d’émission à 526 nm dans l’éther et à 608
nm dans le méthanol (figure 10). En effet, à l’état excité, la densité électronique migre du
fluorène vers le groupement pyridine. Il en résulte un large moment dipolaire à l’état excité.
Un solvant polaire stabilise fortement la forme excitée et un déplacement significatif de son
émission vers le rouge est ainsi observé.
Les spectres d’absorption ne sont pas très sensibles à la nature du solvant comme le
montre les figures 11, 12, 13, 14 respectives des sondes 190, 191, 192 et 193. Les effets de
solvant forts en émission et faibles en absorption sont caractéristiques des molécules qui
présentent un faible moment dipolaire à l’état fondamental et un plus fort à l’état excité. Ce
solvatochromisme est intéressant pour l’étude des potentiels membranaires puisqu’il permet
de prévoir le comportement de la sonde dans les membranes.
Les spectres d’émission dans le cyclohexane pour les 4 sondes sont inattendus. Dans un
tel solvant apolaire, le spectre doit se déplacer vers le bleu, ce qui n’est pas le résultat
expérimental. En effet, les sondes étant très peu solubles dans le cyclohexane, ce résultat peut
111

Résultats - Chapitre III
être expliqué par le fait que les molécules s’agrègent entre elles (micelle inversée) même à de
très faible concentration (DO inférieure à 0,2), par suite le spectre n’est pas celui d’une
molécule individuelle mais celui de l’agrégat. En fait, une molécule ne verra pas les
molécules du cyclohexane apolaires mais son homologue polaire, par suite, elle se polarise de
la même façon et son spectre se déplace vers le rouge. Le spectre du cyclohexane n’est donc
pas représentatif du comportement de la molécule dans un solvant apolaire.
Le spectre d’émission dans l’eau devrait être le plus déplacé vers le rouge. Ceci n’a pas
été confirmé par l’expérience. Nous pouvons aussi prédire que dans l’eau, nos sondes
amphiphiles s’organisent sous la forme de micelle, ce qui donne un spectre d’émission dans
l’eau inattendu.
III.5.2. LE RENDEMENT QUANTIQUE DE FLUORESCENCE
Toutes les molécules excitées n’émettent pas de photons de fluorescence. Pour un
fluorophore donné, le rendement quantique de fluorescence est le rapport entre le nombre de
photons émis et celui absorbé.
φ = Ném/Nabs
Le rendement quantique est indépendant de la longueur d’onde d’excitation. Un
phénomène qui contribue essentiellement à la diminution du rendement quantique est les
collisions avec les molécules environnantes, en particulier avec les molécules de solvants, qui
pourront inhiber la fluorescence. Par suite, certains corps, fluorescents en milieu apolaire, ne
le sont plus en milieu polaire (dans l’eau).
Une détermination du rendement quantique nécessite des mesures des absorbances et de
spectres d’émission. Il est nécessaire de disposer d’un composé fluorescent de référence (r)
dont le rendement quantique est connu, et dont les spectres d’absorption et d’émission
recouvrent du mieux possible ceux du composé dont on cherche à déterminer le rendement
quantique pour s’affranchir de la réponse du détecteur. La formule suivante s’applique :
(1)
φx est le rendement quantique à déterminer, φr est le rendement quantique connu de la
référence, DO et DOr sont leurs absorbances respectives à la longueur d’onde commune (dans
rrrx A
AxDOr ×⎟⎟⎜⎜××= φφnn
DOx ⎠
⎞
⎝
⎛2
112

Résultats - Chapitre III
la pratique, il est conseillé que ces absorbances soient, sinon égales, aussi proches que
possible), A et Ar leurs intensités de fluorescence respectives mesurées par intégration des
spectres d’émission corrigés correspondants, n et nr les indices de réfraction des solvants
respectifs.
En pratique, nous pouvons aussi trouver φx de chaque échantillon en appliquant la
variation dite de Stern-Volmer. En effet, pour des concentrations diluées (DO inférieure à
0,1), l’aire du spectre d’émission varie linéairement en fonction de DO. En mesurant les
valeurs des aires par intégration des spectres d’émission à des DO différentes (par dilution de
l’échantillon), nous pourrons déduire la pente P de la droite :
et (2)
Cette pente injectée dans l’équation (1), nous permettra de déduire le rendement
quantique du composé :
(3)
La longueur d’onde d’excitation est λexc.=410 nm pour toutes les expériences. Comme
produit de référence, nous avons pris la Coumarine 153 (schéma 87) qui a un rendement
quantique connu de 0,45 dans le méthanol et dont les spectres d’absorption et d’émission
recouvrent le mieux les spectres de nos produits (à λexc.=420 nm, λém entre 500 et 600 nm
avec un maximum d’émission à 550 nm).
r
rr
DOAP =
r
rr
DOAP =
DOxAxPx =
DOxAxPx =
rrrx P
Pxnn
×⎟⎟⎠
⎞⎜⎜⎝
⎛×=
2
φφ
N O O
CF3
Schéma 88. Structure de la Coumarine 153
113
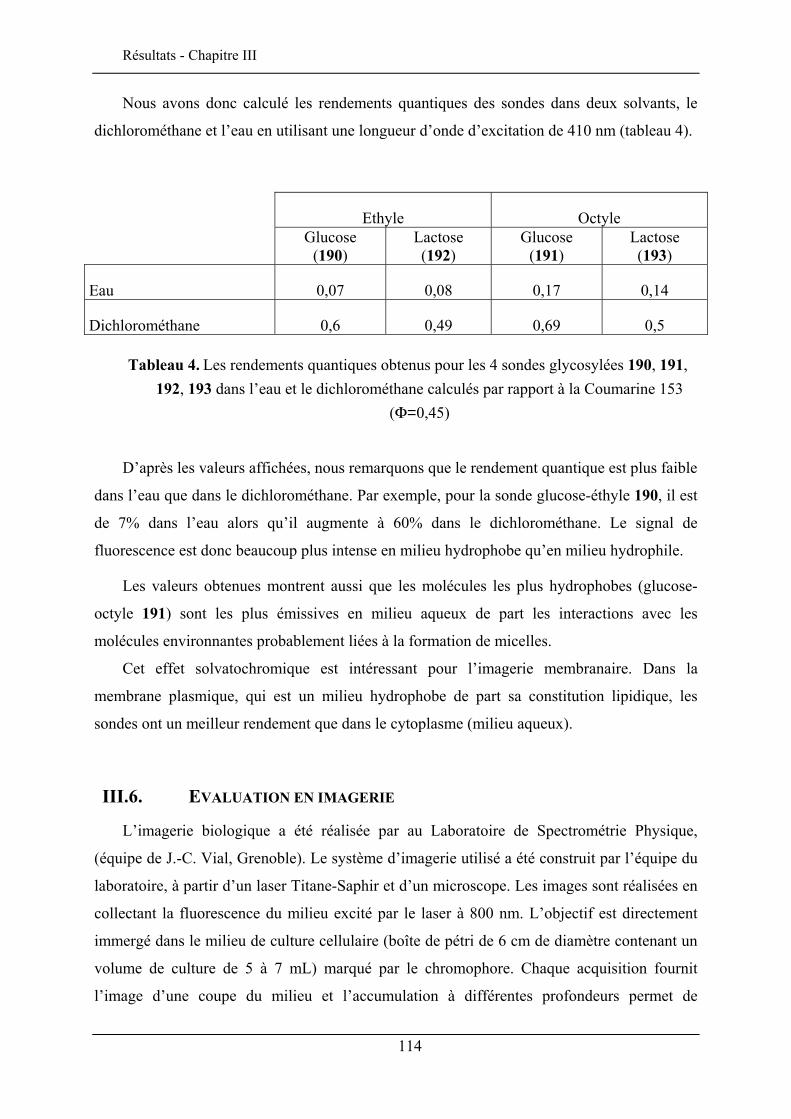
Résultats - Chapitre III
Nous avons donc calculé les rendements quantiques des sondes dans deux solvants, le
dichlorométhane et l’eau en utilisant une longueur d’onde d’excitation de 410 nm (tableau 4).
Ethyle Octyle
Glucose
(190) Lactose (192)
Glucose (191)
Lactose (193)
Eau 0,07 0,08 0,17 0,14
Dichlorométhane 0,6 0,49 0,69 0,5
Tableau 4. Les rendements quantiques obtenus pour les 4 sondes glycosylées 190, 191, 192, 193 dans l’eau et le dichlorométhane calculés par rapport à la Coumarine 153
(Φ=0,45)
D’après les valeurs affichées, nous remarquons que le rendement quantique est plus faible
dans l’eau que dans le dichlorométhane. Par exemple, pour la sonde glucose-éthyle 190, il est
de 7% dans l’eau alors qu’il augmente à 60% dans le dichlorométhane. Le signal de
fluorescence est donc beaucoup plus intense en milieu hydrophobe qu’en milieu hydrophile.
Les valeurs obtenues montrent aussi que les molécules les plus hydrophobes (glucose-
octyle 191) sont les plus émissives en milieu aqueux de part les interactions avec les
molécules environnantes probablement liées à la formation de micelles.
Cet effet solvatochromique est intéressant pour l’imagerie membranaire. Dans la
membrane plasmique, qui est un milieu hydrophobe de part sa constitution lipidique, les
sondes ont un meilleur rendement que dans le cytoplasme (milieu aqueux).
III.6. EVALUATION EN IMAGERIE
L’imagerie biologique a été réalisée par au Laboratoire de Spectrométrie Physique,
(équipe de J.-C. Vial, Grenoble). Le système d’imagerie utilisé a été construit par l’équipe du
laboratoire, à partir d’un laser Titane-Saphir et d’un microscope. Les images sont réalisées en
collectant la fluorescence du milieu excité par le laser à 800 nm. L’objectif est directement
immergé dans le milieu de culture cellulaire (boîte de pétri de 6 cm de diamètre contenant un
volume de culture de 5 à 7 mL) marqué par le chromophore. Chaque acquisition fournit
l’image d’une coupe du milieu et l’accumulation à différentes profondeurs permet de
114

Résultats - Chapitre III
reconstruire le milieu en 3 dimensions. Les images biologiques ont été réalisées sur deux
lignées cellulaires différentes :
- La première lignée, F98, est constituée de cellules gliales cancéreuses. Les cellules
gliales, avec les neurones, sont les deux types de cellules dont est constitué le système
nerveux. Ces cellules, très résistantes, ont été choisies cancéreuses pour bénéficier d’une
multiplication rapide.
- La deuxième lignée, CHO (Chinese Hamster Ovary), est constituée de cellules d’ovaires
de hamsters chinois. Ces cellules sont très utilisées pour les études biologiques car elles sont à
la fois faciles à cultiver et très résistantes.
III.6.1. IMAGERIE PAR TPEF DES SONDES
III.6.1.1. Les sondes comportant des groupements polyéthylène glycols
Une solution de la sonde 194 à chaîne éthyle à 10-2 M dans l’eau a été préparée et injectée
dans un milieu cellulaire pour avoir une concentration finale de 10-5 M, la sonde était
totalement soluble dans le milieu cellulaire à cette concentration de part la sphère
d’hydratation des oxygènes (figure 15).
O
NHNHN
NN
O
NO
O
OO
O
O
O
O
O
O
O
OO
O
O
OO
O
O
O
O O O
O
O
O
194
115
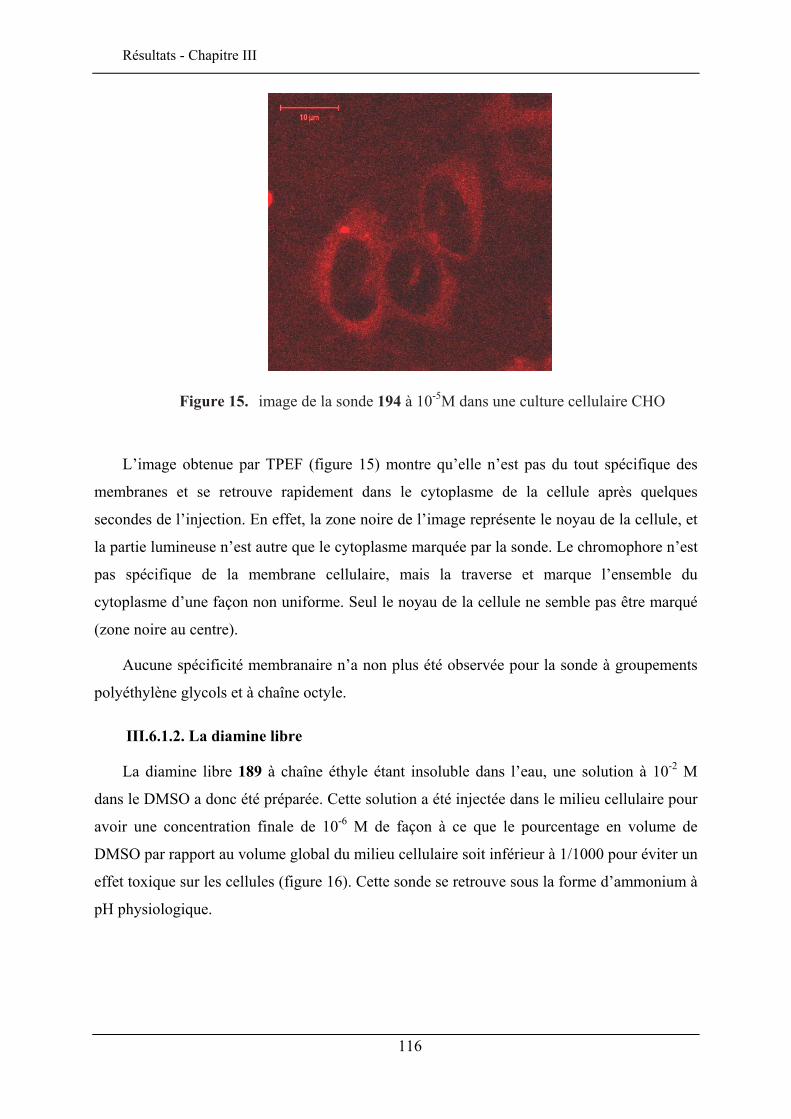
Résultats - Chapitre III
Figure 15. image de la sonde 194 à 10-5M dans une culture cellulaire CHO
L’image obtenue par TPEF (figure 15) montre qu’elle n’est pas du tout spécifique des
membranes et se retrouve rapidement dans le cytoplasme de la cellule après quelques
secondes de l’injection. En effet, la zone noire de l’image représente le noyau de la cellule, et
la partie lumineuse n’est autre que le cytoplasme marquée par la sonde. Le chromophore n’est
pas spécifique de la membrane cellulaire, mais la traverse et marque l’ensemble du
cytoplasme d’une façon non uniforme. Seul le noyau de la cellule ne semble pas être marqué
(zone noire au centre).
Aucune spécificité membranaire n’a non plus été observée pour la sonde à groupements
polyéthylène glycols et à chaîne octyle.
III.6.1.2. La diamine libre
La diamine libre 189 à chaîne éthyle étant insoluble dans l’eau, une solution à 10-2 M
dans le DMSO a donc été préparée. Cette solution a été injectée dans le milieu cellulaire pour
avoir une concentration finale de 10-6 M de façon à ce que le pourcentage en volume de
DMSO par rapport au volume global du milieu cellulaire soit inférieur à 1/1000 pour éviter un
effet toxique sur les cellules (figure 16). Cette sonde se retrouve sous la forme d’ammonium à
pH physiologique.
116

Résultats - Chapitre III
Figure 16. Image de la diamine libre (189) à 10-6 M dans une culture cellulaire CHO
L’image obtenue par TPEF montre que cette sonde marque le cytoplasme et après
quelques temps, traverse la membrane nucléaire et s’accumule dans le noyau.
Aucune spécificité membranaire n’a non plus été observée pour la sonde diamine à
chaîne octyle.
III.6.1.3. Les sondes glycosylées
Des solutions des sondes glycosylées à chaîne éthyle 190 et 192 à 10-2 M dans le DMSO
sont introduites dans un milieu cellulaire du CHO pour avoir une concentration finale de 10-
5M. A cette concentration, les images obtenues par TPEF au moment de l’injection montrent
que les produits s’insèrent bien, d’une façon instantanée dans les membranes cellulaires
(figure 17). Les zones rouges sont des domaines d’agrégats du produit montrant que la
solubilité n’est pas parfaite à cette concentration. Le bruit de fond de luminescence qui est
observé montre que la sonde est partout dans le milieu de culture cellulaire.
117
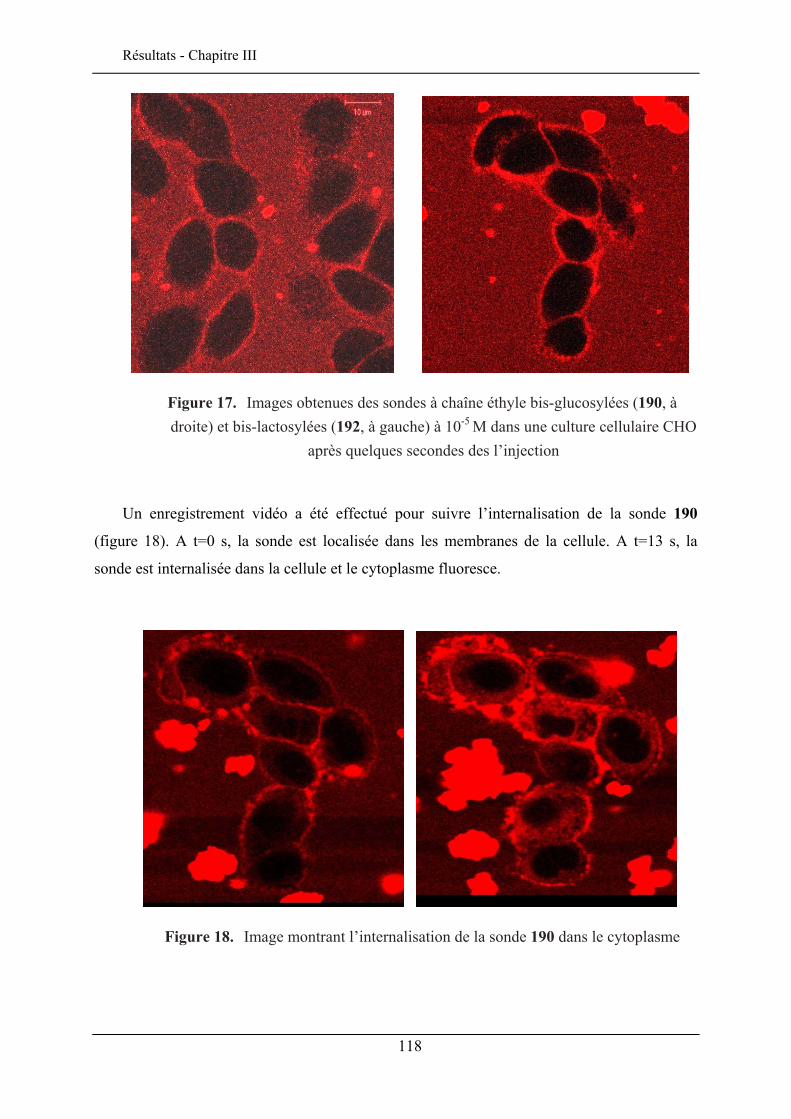
Résultats - Chapitre III
Figure 17. Images obtenues des sondes à chaîne éthyle bis-glucosylées (190, à droite) et bis-lactosylées (192, à gauche) à 10-5 M dans une culture cellulaire CHO
après quelques secondes des l’injection
Un enregistrement vidéo a été effectué pour suivre l’internalisation de la sonde 190
(figure 18). A t=0 s, la sonde est localisée dans les membranes de la cellule. A t=13 s, la
sonde est internalisée dans la cellule et le cytoplasme fluoresce.
Figure 18. Image montrant l’internalisation de la sonde 190 dans le cytoplasme
118
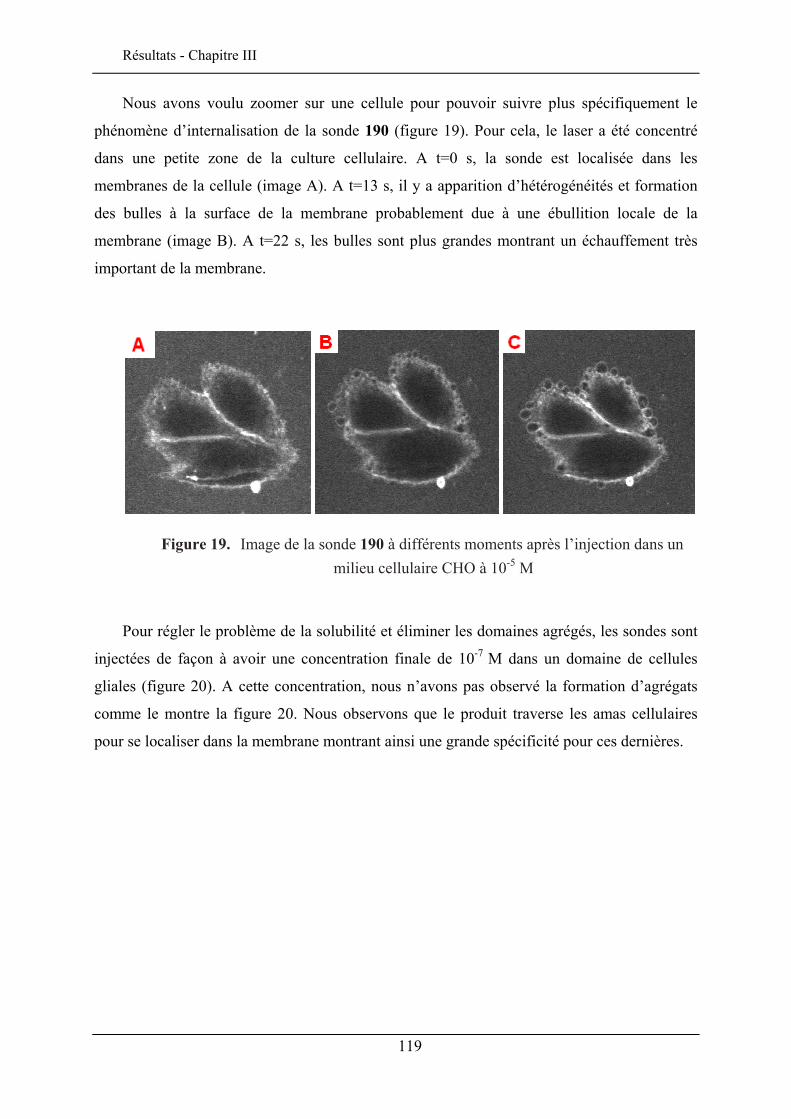
Résultats - Chapitre III
Nous avons voulu zoomer sur une cellule pour pouvoir suivre plus spécifiquement le
phénomène d’internalisation de la sonde 190 (figure 19). Pour cela, le laser a été concentré
dans une petite zone de la culture cellulaire. A t=0 s, la sonde est localisée dans les
membranes de la cellule (image A). A t=13 s, il y a apparition d’hétérogénéités et formation
des bulles à la surface de la membrane probablement due à une ébullition locale de la
membrane (image B). A t=22 s, les bulles sont plus grandes montrant un échauffement très
important de la membrane.
Figure 19. Image de la sonde 190 à différents moments après l’injection dans un milieu cellulaire CHO à 10-5 M
Pour régler le problème de la solubilité et éliminer les domaines agrégés, les sondes sont
injectées de façon à avoir une concentration finale de 10-7 M dans un domaine de cellules
gliales (figure 20). A cette concentration, nous n’avons pas observé la formation d’agrégats
comme le montre la figure 20. Nous observons que le produit traverse les amas cellulaires
pour se localiser dans la membrane montrant ainsi une grande spécificité pour ces dernières.
119
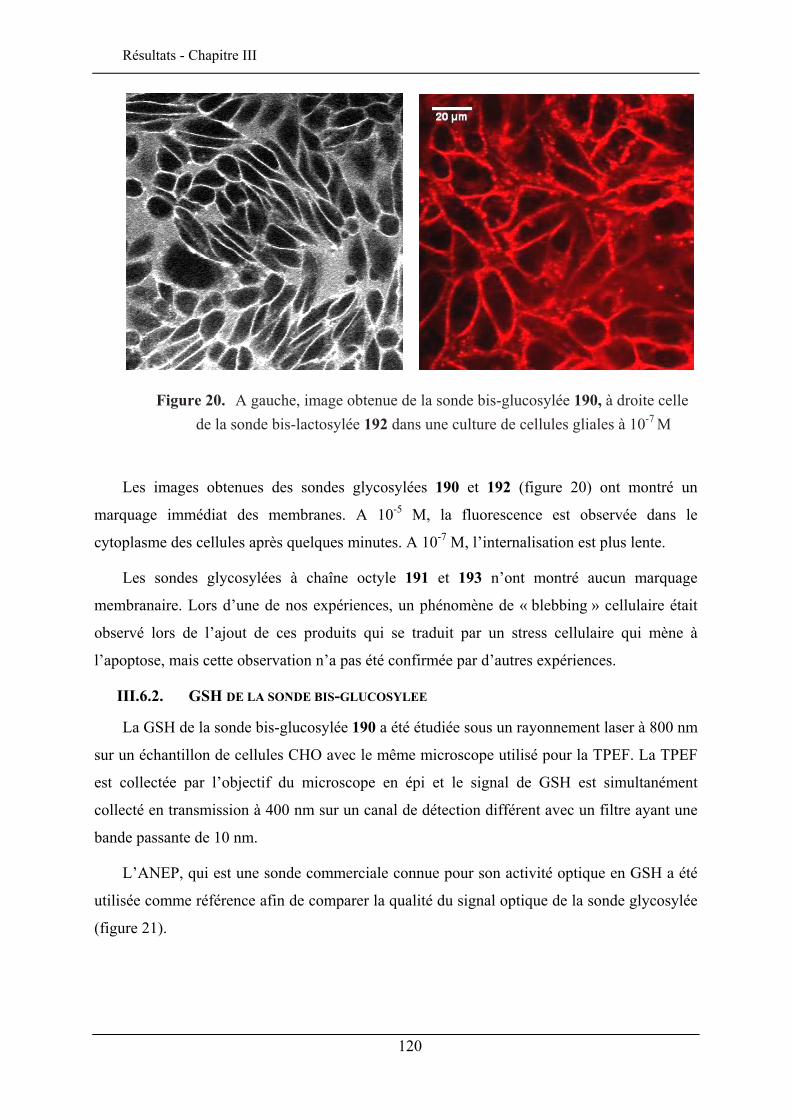
Résultats - Chapitre III
Figure 20. A gauche, image obtenue de la sonde bis-glucosylée 190, à droite celle de la sonde bis-lactosylée 192 dans une culture de cellules gliales à 10-7 M
Les images obtenues des sondes glycosylées 190 et 192 (figure 20) ont montré un
marquage immédiat des membranes. A 10-5 M, la fluorescence est observée dans le
cytoplasme des cellules après quelques minutes. A 10-7 M, l’internalisation est plus lente.
Les sondes glycosylées à chaîne octyle 191 et 193 n’ont montré aucun marquage
membranaire. Lors d’une de nos expériences, un phénomène de « blebbing » cellulaire était
observé lors de l’ajout de ces produits qui se traduit par un stress cellulaire qui mène à
l’apoptose, mais cette observation n’a pas été confirmée par d’autres expériences.
III.6.2. GSH DE LA SONDE BIS-GLUCOSYLEE
La GSH de la sonde bis-glucosylée 190 a été étudiée sous un rayonnement laser à 800 nm
sur un échantillon de cellules CHO avec le même microscope utilisé pour la TPEF. La TPEF
est collectée par l’objectif du microscope en épi et le signal de GSH est simultanément
collecté en transmission à 400 nm sur un canal de détection différent avec un filtre ayant une
bande passante de 10 nm.
L’ANEP, qui est une sonde commerciale connue pour son activité optique en GSH a été
utilisée comme référence afin de comparer la qualité du signal optique de la sonde glycosylée
(figure 21).
120

Résultats - Chapitre III
Figure 21. Image par TPEF (A) et GSH (B) de cellules CHO obtenues de la sonde bis-glucosylée 190 et image par GSH d’une sonde modèle, l’ANEP (C)
Pour la sonde 190, nous avons observé un signal en GSH. Le signal lumineux est obtenu
par balayage du laser et juxtaposition de 50 images, il est obtenu sur fond noir (amélioration
du contraste) et il est spécifique de la membrane contrairement à celui de la TPEF obtenu sur
un fond plus lumineux et émis partout où il y a du produit.
Les qualités optiques du signal restent à améliorer mais la grande importance de ce
résultat vient du fait que c’est la première observation du GSH par une sonde optique non
ionique. De plus, le signal de l’ANEP s’éteint au bout de quelques minutes à cause du
121

Résultats - Chapitre III
phénomène du flip-flop des lipides membranaires. La sonde bis-glucosylée a montré un temps
de résidence relatif plus long mais ce résultat n’a pas pu être quantifié.
III.7. CONCLUSION
Les sondes fluorescentes que nous avons synthétisées sont actives en TPEF et GSH. Elles
permettent d’obtenir des images avec un rapport signal/bruit élevé. Les signaux optiques de
ces sondes renseignent sur les propriétés physico-chimiques de l’environnement de la sonde et
de son organisation dans la cellule. Les résultats obtenus suggèrent que les groupements
polaires glycosyles apportent une amphiphilie ce qui permet l’ancrage à l’interface de la
membrane en gardant une orientation parallèle de la partie hydrophobe aux chaînes d’acides
gras de la membrane, avec les groupements polaires orientés vers l’extérieur de la bicouche.
Leur ancrage à la surface, leur orientation dans la bicouche et la chiralité du sucre
maintiennent un milieu non centrosymétrique et permettent donc l’imagerie par TPEF et
GSH : c’est la première observation de GSH par une sonde neutre. La sonde polyéthylène
glycol et la diamine libre, quant à elles, n’ont montré aucune spécificité membranaire.
Les sondes glycosylées que nous avons synthétisés se sont montrées sensibles à la nature
du solvant de telle sorte que les interactions avec l’environnement induisent une perturbation
de la position de leurs bandes d’émission. Son rendement quantique augmente lorsque la
polarité diminue et le spectre d’émission est déplacé vers le bleu. Cette sensibilité vis-à-vis de
la polarité des solvants rend ces molécules intéressantes pour l’étude des propriétés
biophysiques des membranes. En effet, par solvatochromisme, le signal par TPEF dans le
cytoplasme (milieu aqueux), sera affaibli malgré la présence du produit, et celui des
membranes (milieu apolaire) sera exalté. Ce qui permet un plus fort contraste et une image
mieux résolue.
De point de vue solubilité, les sondes polyéthylène glycols ont la meilleure solubilité
dans l’eau de l’ordre de 10-2 M. Ils sont donc de très bons groupements hydrosolubilisants.
Les diamines et les sondes glycosylées ont une solubilité comparable de 10-6 M. la
substitution du glucose par le lactose (disaccharide plus polaire) n’a pas affecté la solubilité
des sondes mais ce résultat n’a pas été mesuré.
Le temps de résidence des sondes glycosylées dans la membrane est amélioré par rapport
à celui de l’ANEP avant l’internalisation dans le cytoplasme. Cela traduit une très bonne
interaction hydrophobe de ces molécules avec les lipides membranaires.
122

Résultats - Chapitre III
Ce travail a montré la difficulté de l’ingénierie moléculaire pour trouver la bonne balance
hydrophile-lipophile pour une insertion dans la membrane. Son intérêt vient justement d’avoir
optimisé la balance hydrophile-lipophile par les sucres. Les résultats obtenus ne sont pas
évidents quant à l’échec des molécules PEG-ylées pour marquer les membranes. Ce travail a
aussi montré l’importance de la fluorescence pour suivre la dynamique d’internalisation des
chromophores dans la membrane cellulaire.
123

Résultats - Chapitre IV
124

Résultats - Chapitre IV
CHAPITRE IV. ETUDE DE L’INSERTION DANS UNE
MONOCOUCHE DE LANGMUIR
Afin de comprendre les propriétés d’insertion dans la membrane cellulaire, nous avons
effectué une étude d’insertion de ces produits dans une monocouche de Langmuir qui est un
modèle d’étude membranaire constitué d’un demi-feuillet de phospholipides constitutifs de la
membrane plasmique. L’imagerie par microscopie à l’angle de Brewster (BAM) a aussi été
effectuée pour visualiser l’effet de la sonde sur l’agrégation des lipides dans cette membrane
synthétique. Cette étude fait l’objet d’une collaboration avec Dr Agnès Girard-Egrot du
Laboratoire de Génie Enzymatique et Biomoléculaire dirigé par le Professeur Loïc Blum.
IV.1. TECHNIQUE DES MONOCOUCHES DE LANGMUIR
La technique des monocouches de Langmuir est fondée sur la formation de couches
monomoléculaires de molécules amphiphiles qui sont des films insolubles de molécules
organiques confinées à l’interface eau/air.164,165 Le principe de cette technique réside dans la
capacité des espèces amphiphiles à se repartir sur une surface aqueuse (sous-phase) au contact
de l’air et à s’auto-assembler pour former une monocouche sous l’effet de la compression, les
têtes polaires hydrophiles se retrouvent immergées dans la sous-phase, les queues polaires
s’orientent vers la phase gazeuse.
Les monocouches de Langmuir sont largement utilisées pour l’étude des interactions
protéines-lipides.166 Elles permettent de simuler les conditions physico-chimiques de la
membrane.
Dans la littérature, les monocouches de Langmuir ont été utilisées comme un
environnement ordonné représentatif de la membrane cellulaire afin d’étudier les propriétés
164 Vollhardt, D.; Fainerman, V. B. J. Colloid. Interface Sci. 2000, 86, 103-151. 165 Maget-Dana, R. Biochim. Biophys. Acta. 1999, 1462, 109-140. 166 Girard-Egrot, A.; Chauvet, J.-P.; Gillet, G.; Moradi-Améli, M. J. Mol. Biol. 2004, 335, 321-331.
125

Résultats - Chapitre IV
optiques des sondes par fluorescence classique167, 168 ou par optique non
linéaire.169, , , , ,170 171 172 173 174
Les résultats obtenus ont permis de prédire le comportement de ces sondes en milieu
aqueux. Par contre, aucune mesure de la qualité d’insertion membranaire n’a été effectuée sur
les sondes membranaires.
IV.1.1. PRINCIPE DE LA TECHNIQUE DES MONOCOUCHES DE LANGMUIR
Lors de leur dépôt à l’interface air-eau, les lipides se repartissent sur toute la surface
disponible entre les deux barrières mobiles pour former un film organisé. Sous l’effet de
l’avancée symétrique des barrières, la monocouche subit une compression par réduction
progressive de la surface. Suite à cette compression, les molécules se condensent et exercent
une pression plus élevée à la surface de l’eau, mesurable au moyen d’un tensiomètre (balance
de Wilhelmy) partiellement immergé au sein du film. La condensation du film lipidique peut
ainsi être suivie par mesure de la pression superficielle et permettra de tracer une isotherme de
Langmuir (pression de surface en fonction de l’aire moyenne occupée par une molécule à la
surface de l’eau) caractéristique du lipide utilisé.
La pression de surface (π) est définie comme la différence entre la tension superficielle
de l’eau pure (γ0) et celle de l’eau après dépôt des molécules amphiphiles (γ).
Elle représente une force par unité de longueur et s’exprime en milliNewton par mètre
(mN.m-1).
π = γ− γ0
La lame de platine partiellement immergée dans le film est connectée par son autre
extrémité à un capteur de force qui permet de mesurer la tension de surface.
167 Mchedlov-Petrossyan N.O.; Vodolazkaya N.A., Bezkrovnaya O. N.; Yakubovskaya A. G.; Tolmachev A. V.; Grigorovich A. V. Spectrochimica Acta. 2008, 69, 1125–1129. 168 Biadasz, A.; Qabuszewska, K.; Chrzumnicka, E.; Michalowski, E.; Martynski, T.; Bauman D. Dyes and Pigments 2007, 74, 598-607 169 Lenahan, K.M.; Wang; Y.X.; Liu, Y.J.; Claus, R.O.; Heflin, J.R.; Marciu, D.; Figura, C. Adv. Mat. 1998, 10, 853. 170 Li, H.; Zhou, D.J; Huang, C.H.; Xu, J.M.; Li, T.K; Zhao, X.S.; Xia, X.H J. Chem. Soc. Faraday trans. 1996, 92, 2585-2592. 171 Lupo, D.; Ringsdorf, H.; Schuster, A.; Seitz, M. J. Am. Chem. Soc. 1994, 116, 10498-10506. 172 Schoondorp, M. A.; Schouten, M. A.; Hulshof, J. B. E.; Feringa, B. L. Thin Sol. Film 1992, 210, 166-168. 173 Laschewsky, A.; Paulus, W.; Ringsdorf, H.; Lupo, D.; Ottenbreit, P.; Prass, W.; Bubeck, C.; Neher, D.; Wegner, G. Makromol. Chemie, Macromol. Symp. 1991, 46, 205-210. 174 Kato, N.; Saito, K.; Ueso, Y. Thin. Sol. Films. 1999, 338, 5-8.
126
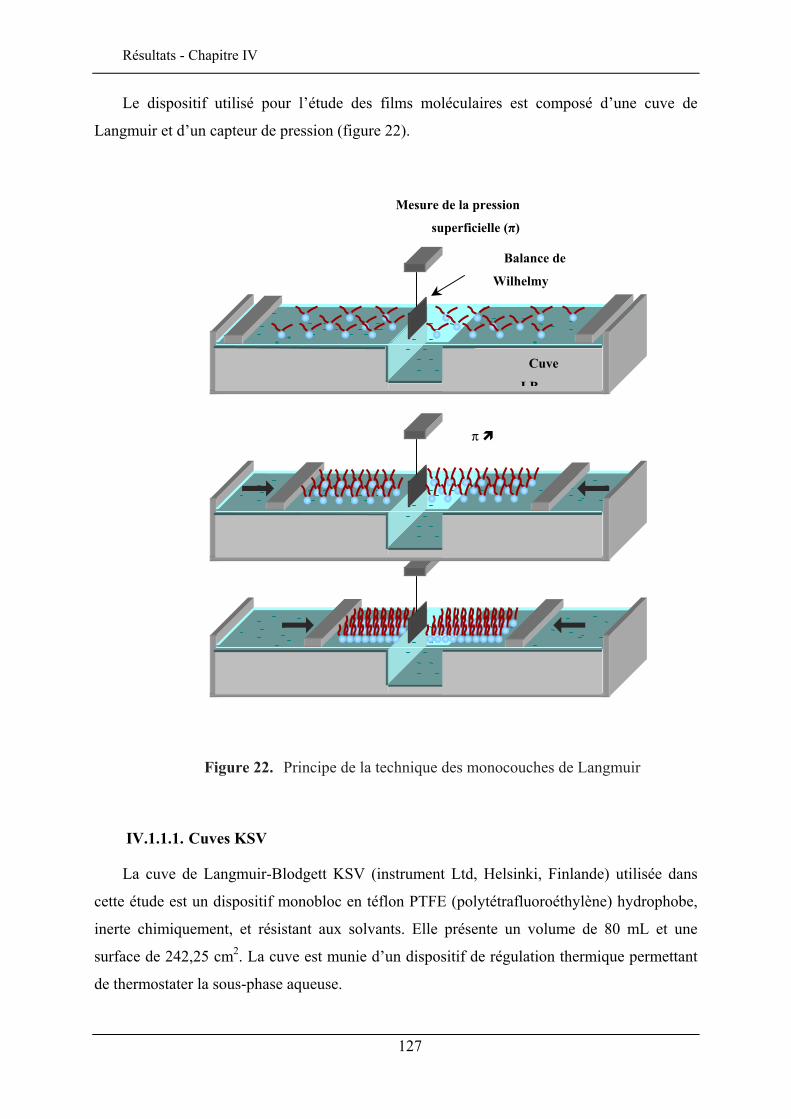
Résultats - Chapitre IV
Le dispositif utilisé pour l’étude des films moléculaires est composé d’une cuve de
Langmuir et d’un capteur de pression (figure 22).
Mesure de la pression
superficielle (π)
π
Cuve
LB
Balance de
Wilhelmy
Figure 22. Principe de la technique des monocouches de Langmuir
IV.1.1.1. Cuves KSV
La cuve de Langmuir-Blodgett KSV (instrument Ltd, Helsinki, Finlande) utilisée dans
cette étude est un dispositif monobloc en téflon PTFE (polytétrafluoroéthylène) hydrophobe,
inerte chimiquement, et résistant aux solvants. Elle présente un volume de 80 mL et une
surface de 242,25 cm2. La cuve est munie d’un dispositif de régulation thermique permettant
de thermostater la sous-phase aqueuse.
127

Résultats - Chapitre IV
IV.1.1.2. Appareillage KSV
La cuve est reliée à un appareillage KSV (instrument Ltd, Helsinki, Finlande)
comprenant un système de mesure de pression constitué d’une balance de Wilhelmy et un
système de compression constitué de deux barrières mobiles (système de compression
bilatérale symétrique) en Delrin.
Cet appareillage est installé sur une plaque de marbre posée sur des coussinets, le tout
monté sur une table anti-vibration afin de limiter les oscillations parasites de la surface
aqueuse. L’appareillage est ensuite relié à une unité informatique qui permet le contrôle du
système de mesure (pression, compression) et la gestion des données, ainsi qu’un boîtier de
contrôle manuel.
La balance de Wilhelmy, placée au niveau du centre de la cuve est constituée d’une sonde
de platine (10x20 mm2) suspendue à un étrier fixé sur un système mobile. La sonde de platine
est immergée dans la sous-phase sur une hauteur de 3 mm environ. Le zéro du capteur est
réglé sur une sous-phase parfaitement nettoyée avant dépôt de la solution lipidique. Ce
système de balance haute sensibilité présente une résolution de 4 µN.m-1.
IV.1.1.3. Préparation de la cuve
Du fait de la grande sensibilité aux poussières et aux contaminants organiques pouvant
modifier considérablement la tension superficielle, la technique d’étude des monocouches
interfaciales nécessite des précautions particulières lors de l’usage et du nettoyage de la
verrerie, ou de la manipulation de la cuve. Le nettoyage préalable de la verrerie avec un
mélange à base de persulfate d’ammonium et d’acide sulfurique puis avant usage, avec du
chloroforme, permet de prévenir la contamination des solutions stockées. Lors du dépôt des
solutions lipidiques, les seringues sont rincées avant chaque utilisation avec du chloroforme
puis du méthanol.
Avant chaque expérience, la cuve et les barrières sont systématiquement nettoyées avec
un chiffon imbibé d’éthanol, puis de chloroforme, et rincée avec de l’eau ultrapure avant de
verser la sous-phase définitive. La sous-phase utilisée dans toutes les expériences est
confectionnée à l’aide d’eau ultrapure. Sa surface est soigneusement nettoyée en surface au
niveau de la zone de compression, à l’aide d’un système d’aspiration par pompe à vide.
L’aspiration est renouvelée plusieurs fois en faisant avancer les barrières jusqu’à l’absence de
variation de la pression superficielle par élimination totale des poussières à la surface.
128

Résultats - Chapitre IV
La température de la sous-phase est maintenue constante à une valeur de 22°C à l’aide
d’un cryothermostat.
IV.1.1.4. Dépôt des molécules lipidiques
La solution chloroformique de lipides est déposée goutte à goutte à la surface de la phase
aqueuse, à l’aide d’une microseringue en faisant fusionner les gouttes de la solution lipidique
avec la surface aqueuse. Une attente de 15 minutes est nécessaire pour l’évaporation du
solvant. Les molécules se repartissent à l’interface eau/air pour former la monocouche.
IV.1.1.5. Isothermes de Langmuir
L’enregistrement des isothermes de compression/décompression de la monocouche de
Langmuir se fait par avancée symétrique des barrières à vitesse continue de 5 mm.min-1
jusqu’à une pression cible, suivie d’un cycle de décompression par écartement des barrières
jusqu’à une pression proche de zéro.
IV.2. MICROSCOPIE A L’ANGLE DE BREWSTER (BAM)
IV.2.1. PRINCIPE
La technique du BAM est une technique qui permet l’étude in situ des films minces à
l’interface air/eau. Cette technique est, avec la microscopie de fluorescence à l’interface, l’une
des rares méthodes permettant l’analyse directe de l’organisation bidimensionnelle des
monocouches lipidiques de Langmuir à l’interface air/eau (figure 23).
α
Faisceau réfracté
α
Faisceau réf léchi
Faisceau réfracté
air
eauFilm minceinterface eau/air
détécté par unecaméra CCDFaisceau incidentFaisceau incident
P P
air
eauinterface eau/air
Figure 23. Principe de la microscopie à l’angle de Brewster
Son principe se base sur la réflexion de la lumière polarisée dans le plan d’incidence à
l’interface de deux milieux diélectriques distincts, ici l’air et l’eau, caractérisés par des indices
129

Résultats - Chapitre IV
de réfraction différents. Lorsqu’un faisceau de lumière polarisée (P) se propage d’un milieu
d’indice n1 à un milieu d’indice n2, il existe un angle d’incidence α pour lequel la totalité du
faisceau incident est réfractée. Cet angle, l’angle de Brewster, dépend de la nature des milieux
diélectriques, et est défini par la relation suivante (loi de Brewster) :
Tan α = nsubstrat/nair.
Où n est l’indice de réfraction du milieu indiqué. La valeur de l’angle de Brewster est de
53° dans le cas de l’interface eau/air.
En absence de film, l’exposition de l’interface eau/air par un faisceau polarisé à l’angle
de Brewster induit une réflexion nulle. L’introduction d’un film mince entre ces deux phases
modifie les propriétés optiques du système de telle manière qu’une petite portion de l’intensité
incidente est réfléchie. Le faisceau réfléchi est alors capté par un détecteur (caméra CDD de
résolution de 2µm et de taille 572x430 µm), permettant ainsi de visualiser le film interfacial.
IV.2.2. MONTAGE EXPERIMENTAL
Le microscope à l’angle de Brewster utilisé dans cette étude est l’Ep3-SW
(NanofilmTchnology, Göttingen, Allemagne) couplée à la cuve de Langmuir.
Le BAM est positionné au-dessus de la cuve au moyen d’une structure métallique fixe à
quelques centimètres de la cuve. Afin d’éliminer le signal parasite dû à la réfraction du
faisceau incident sur le fond de la cuve, une plaque de verre noir en partie biseautée est placée
au fond de la cuve avec le biseau orienté dans la direction d’arrivée du faisceau réfracté.
Avant le dépôt de la solution lipidique, le réglage du BAM est effectué : après allumage du
BAM, les goniomètres porteurs du laser de la caméra CCD s’orientent de telle sorte que le
faisceau incident arrive à l’interface à l’angle de Brewster et que la caméra CCD capte le
faisceau issu de la réflexion. A ce moment précis, en absence de film à l’interface, ce faisceau
réfléchi est d’intensité lumineuse nulle.
IV.3. ETUDE DE LA QUALITE D’INSERTION
La qualité d’insertion d’une molécule dans la monocouche se fait en gardant l’aire de la
surface constante en maintenant les barrières fixes, et en mesurant la variation de la pression
liée à la pénétration dans la monocouche de la molécule préalablement injectée dans la sous-
phase.
130

Résultats - Chapitre IV
IV.3.1. CONDITIONS EXPERIMENTALES
Le lipide utilisé dans notre étude est 2-dipalmitoyl-3-glycéro-3-phosphocholine
(DPPC, schéma 89) qui est un phospholipide abondant dans la membrane cellulaire. Cette
molécule est formée par association d’une molécule de glycérol, de deux molécules d'acide
gras (acide palmitique : C-16), d’une molécule d'acide phosphorique et d’une molécule d'un
alcool : la choline.
)14( O
O
O O
()14
OPO-
O N+O
Schéma 89. Structure chimique du DPPC
Ce sont des molécules amphiphiles présentes sous la forme zwittérionique. L’isotherme
de compression du DPPC s’organise en deux phases, LE (liquide expansé dans lequel les
chaînes hydrophobes des molécules ne sont pas organisées) puis LC (liquide condensé dans
lequel les chaînes hydrocarbonées cristallisent et les molécules passent à un état plus
condensé), séparées par un plateau de transition de phase LE-LC.
Une solution de DPPC à une concentration de 1mM dans un mélange de CHCl3/MeOH
(2/1) est préparée dont 15 µL sont délicatement déposées à la surface de la cuve. Le volume à
déposer est calculé par le logiciel après calcul du nombre total de molécules nécessaires pour
obtenir une aire moléculaire donnée.
La sous-phase utilisée est le PBS qui est un tampon phosphate salin (pH=7,4)
couramment utilisé en biochimie. Il s'agit d'une solution physiologique contenant du chlorure
de sodium (137 mM), du chlorure de potassium (2,7 mM) et du phosphate de sodium (10
mM). En général, la concentration de ces sels est celle du corps humain (solution isotonique).
Les sondes sont injectées dans le DMSO à une solution finale dans la cuve de 10-9 M à
une température de 22°C.
Les isothermes enregistrées présentent une évolution caractéristique des monocouches de
phospholipides avec une augmentation de pression superficielle au fur et à mesure de la
diminution de l’aire disponible.
131

Résultats - Chapitre IV
La qualité d’insertion est ensuite étudiée en enregistrant les cinétiques de pénétration des
molécules d’intérêt dans la monocouche. Ce type d’expérience consiste à enregistrer la
variation de la pression de surface (∆π) au cours du temps et à pression initiale donnée, après
injection de notre sonde dans la sous-phase. La sonde injectée va modifier cette pression de
surface par insertion dans la monocouche. Plus la variation de la pression (∆π) est grande,
plus l’insertion de la sonde dans la monocouche est importante. Cette insertion traduit
l’affinité de la molécule pour le lipide. L’interaction de la tête polaire (sucre) de la sonde et de
la tête polaire du lipide favorise leur approche. Ainsi, la queue hydrophobe pénètre
rapidement dans le feuillet membranaire par interaction hydrophobe et la sonde se retrouve
insérée dans la monocouche.
IV.3.2. INSERTION DE LA SONDE A TETE LACTOSE ET CHAINE ETHYLE (192)
L’isotherme de la pression de surface en fonction de l’aire moléculaire moyenne occupée
par une molécule (exprimée en Å2/molécule) à la surface du tampon a été enregistrée avant et
après l’injection du produit 192 (figure 24). Elle nous permet de vérifier que notre milieu
n’est ou n’est pas contaminé par une quelconque impureté qui viendrait changer le tracé de
l’isotherme du DPPC. Sur cette isotherme, nous retrouvons la phase LE et la phase LC,
séparées par le plateau de transition LE-LC.
132
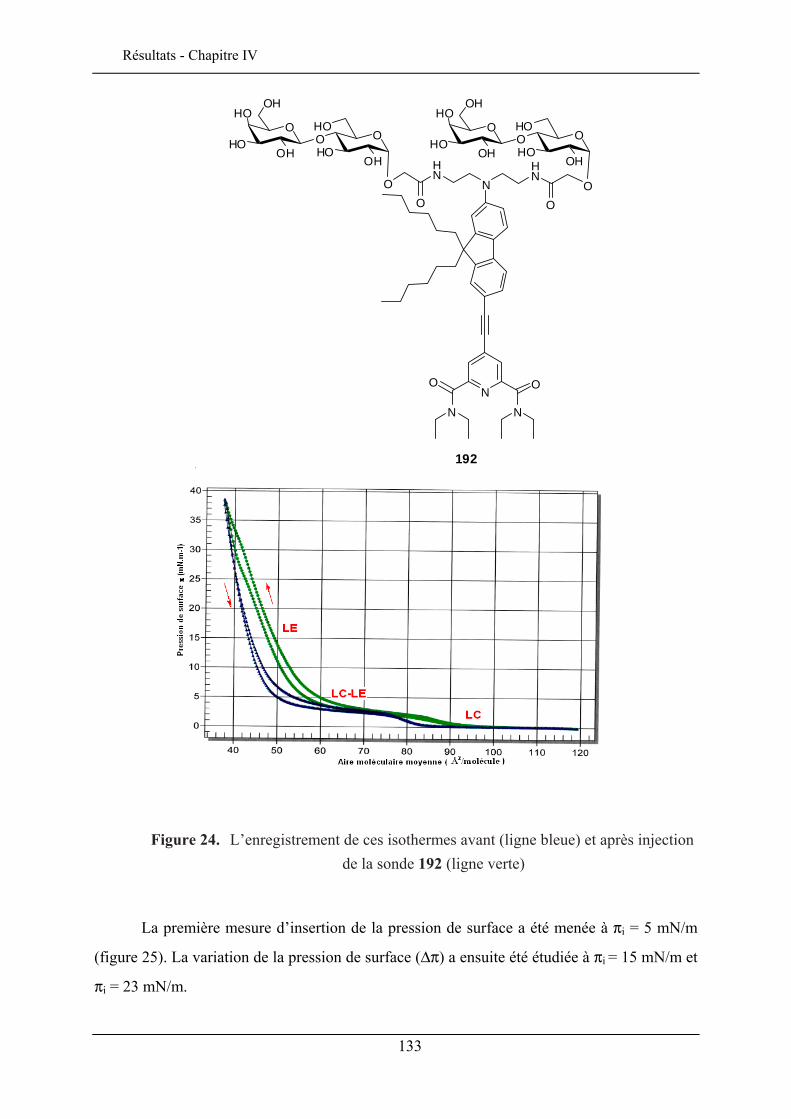
Résultats - Chapitre IV
192
OOHO
OH
HO
NHN
N
N
O
N
O
O
OHO
OH
OHHO
O
HN
OO
OOHO
OH
HOOHO
OH
OHHO
Figure 24. L’enregistrement de ces isothermes avant (ligne bleue) et après injection de la sonde 192 (ligne verte)
La première mesure d’insertion de la pression de surface a été menée à πi = 5 mN/m
(figure 25). La variation de la pression de surface (∆π) a ensuite été étudiée à πi = 15 mN/m et
πi = 23 mN/m.
133
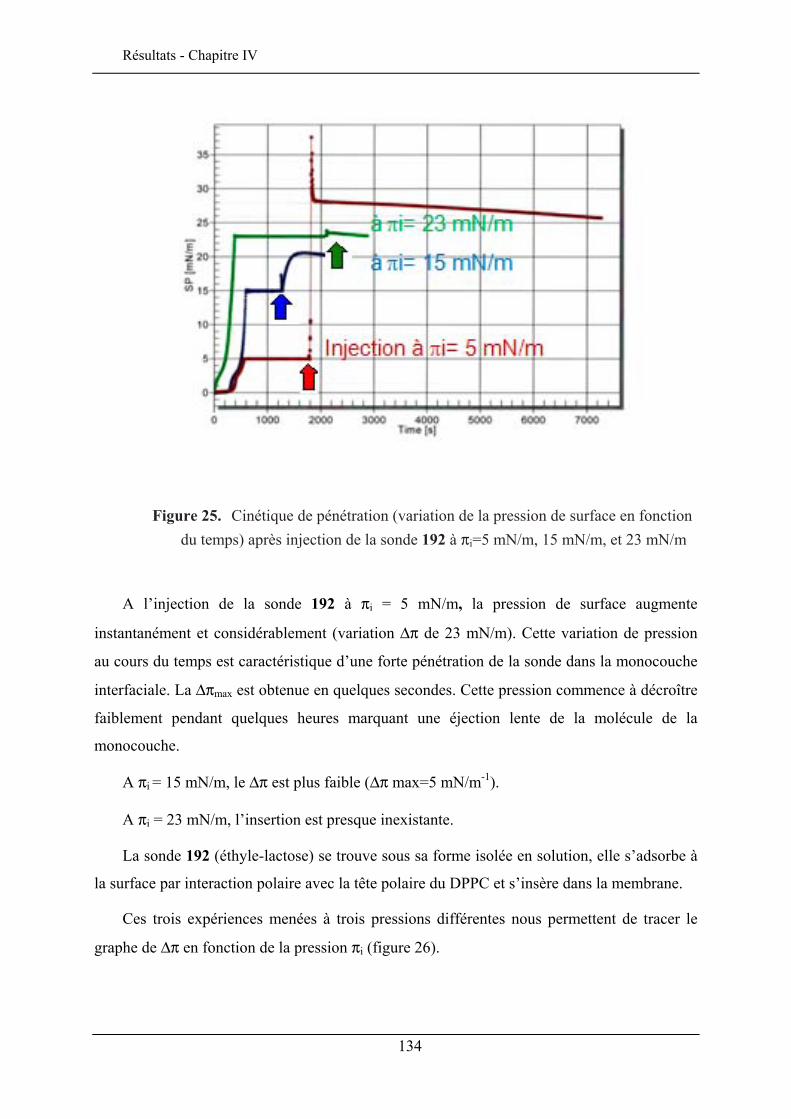
Résultats - Chapitre IV
Figure 25. Cinétique de pénétration (variation de la pression de surface en fonction du temps) après injection de la sonde 192 à πi=5 mN/m, 15 mN/m, et 23 mN/m
A l’injection de la sonde 192 à πi = 5 mN/m, la pression de surface augmente
instantanément et considérablement (variation ∆π de 23 mN/m). Cette variation de pression
au cours du temps est caractéristique d’une forte pénétration de la sonde dans la monocouche
interfaciale. La ∆πmax est obtenue en quelques secondes. Cette pression commence à décroître
faiblement pendant quelques heures marquant une éjection lente de la molécule de la
monocouche.
A πi = 15 mN/m, le ∆π est plus faible (∆π max=5 mN/m-1).
A πi = 23 mN/m, l’insertion est presque inexistante.
La sonde 192 (éthyle-lactose) se trouve sous sa forme isolée en solution, elle s’adsorbe à
la surface par interaction polaire avec la tête polaire du DPPC et s’insère dans la membrane.
Ces trois expériences menées à trois pressions différentes nous permettent de tracer le
graphe de ∆π en fonction de la pression πi (figure 26).
134
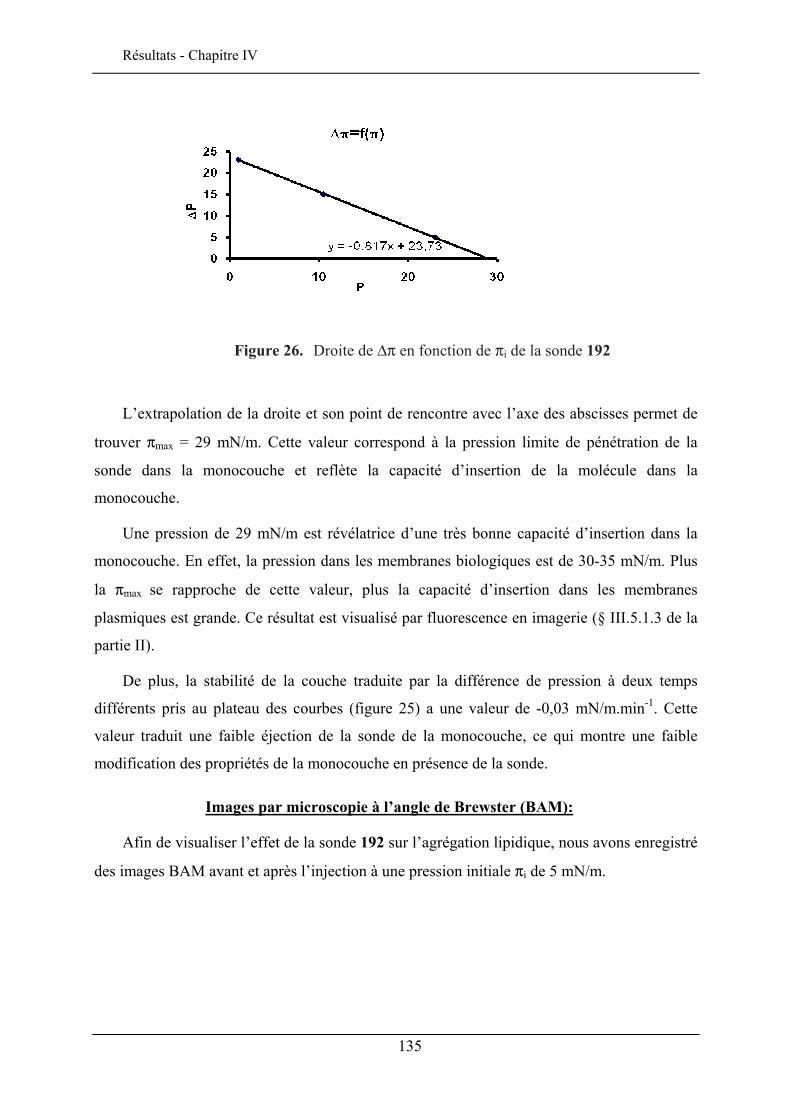
Résultats - Chapitre IV
Figure 26. Droite de ∆π en fonction de πi de la sonde 192
L’extrapolation de la droite et son point de rencontre avec l’axe des abscisses permet de
trouver πmax = 29 mN/m. Cette valeur correspond à la pression limite de pénétration de la
sonde dans la monocouche et reflète la capacité d’insertion de la molécule dans la
monocouche.
Une pression de 29 mN/m est révélatrice d’une très bonne capacité d’insertion dans la
monocouche. En effet, la pression dans les membranes biologiques est de 30-35 mN/m. Plus
la πmax se rapproche de cette valeur, plus la capacité d’insertion dans les membranes
plasmiques est grande. Ce résultat est visualisé par fluorescence en imagerie (§ III.5.1.3 de la
partie II).
De plus, la stabilité de la couche traduite par la différence de pression à deux temps
différents pris au plateau des courbes (figure 25) a une valeur de -0,03 mN/m.min-1. Cette
valeur traduit une faible éjection de la sonde de la monocouche, ce qui montre une faible
modification des propriétés de la monocouche en présence de la sonde.
Images par microscopie à l’angle de Brewster (BAM):
Afin de visualiser l’effet de la sonde 192 sur l’agrégation lipidique, nous avons enregistré
des images BAM avant et après l’injection à une pression initiale πi de 5 mN/m.
135

Résultats - Chapitre IV
Figure 27. BAM d’une monocouche de DPPC sur sous-phase PBS (pH=7,4) à 22°C, à πi= 5 mN/m. Taille de l’image : 572x430 µm
Figure 28. BAM d’une monocouche de DPPC sur sous-phase PBS (pH= 7,4) à 22°C après injection de la sonde 192 à πi= 5 mN.m-1. Image prise à 10 mN.m-1 pendant
l’insertion. Taille de l’image : 572x430 µm
Les images de BAM obtenues en sous-phase de PBS (pH=7,4) (figure 27) montrent que
la morphologie d’une monocouche de DPPC présente des domaines de forte luminosité
contrastant sur un fond plus sombre. Après injection du produit (figure 28), la pression
136

Résultats - Chapitre IV
superficielle augmente et la taille de ces domaines lumineux augmente aussi sans
modification de l’organisation des lipides à la surface.
L’isotherme de la figure 19 traduit bien la stabilité de la monocouche en présence de la
sonde. En effet, en présence du produit, l’isotherme est décalée vers la droite due à la
présence d’une nouvelle structure dans la monocouche. En revanche, la présence de la sonde
ne modifie pas le tracé général de l’istherme quant à la présence des phases LC, LC-LE et LE.
L’organisation lipidique reste donc inchangée.
IV.3.3. INSERTION DE LA SONDE A TETE LACTOSE ET CHAINE OCTYLE (193)
L’étude d’insertion de la sonde 193 à πi =5 mN/m montre une première partie présentant
une pente moyenne suivie d’une brusque augmentation avec une ∆π de 4 mN/m (figure 29).
193
OOHO
OH
HO
NHN
NN
O
N
O
O
OHO
OH
OHHO
O
HN
OO
OOHO
OH
HOOHO
OH
OHHO
137
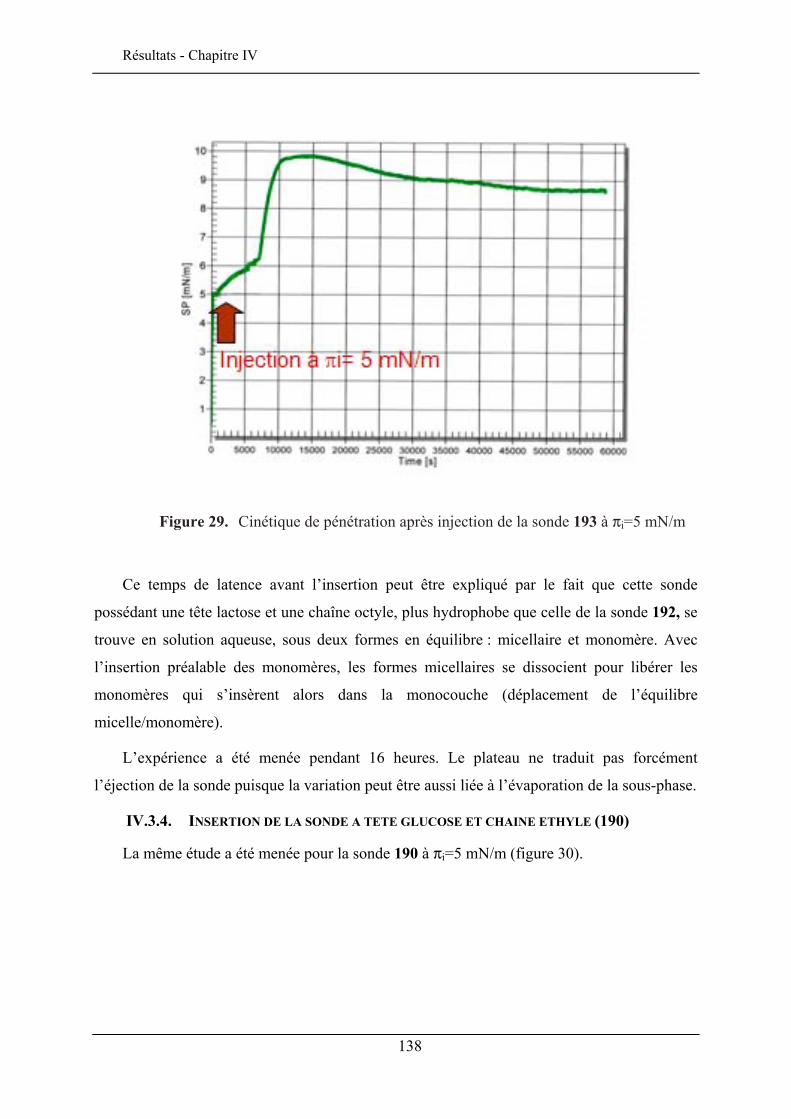
Résultats - Chapitre IV
Figure 29. Cinétique de pénétration après injection de la sonde 193 à πi=5 mN/m
Ce temps de latence avant l’insertion peut être expliqué par le fait que cette sonde
possédant une tête lactose et une chaîne octyle, plus hydrophobe que celle de la sonde 192, se
trouve en solution aqueuse, sous deux formes en équilibre : micellaire et monomère. Avec
l’insertion préalable des monomères, les formes micellaires se dissocient pour libérer les
monomères qui s’insèrent alors dans la monocouche (déplacement de l’équilibre
micelle/monomère).
L’expérience a été menée pendant 16 heures. Le plateau ne traduit pas forcément
l’éjection de la sonde puisque la variation peut être aussi liée à l’évaporation de la sous-phase.
IV.3.4. INSERTION DE LA SONDE A TETE GLUCOSE ET CHAINE ETHYLE (190)
La même étude a été menée pour la sonde 190 à πi=5 mN/m (figure 30).
138
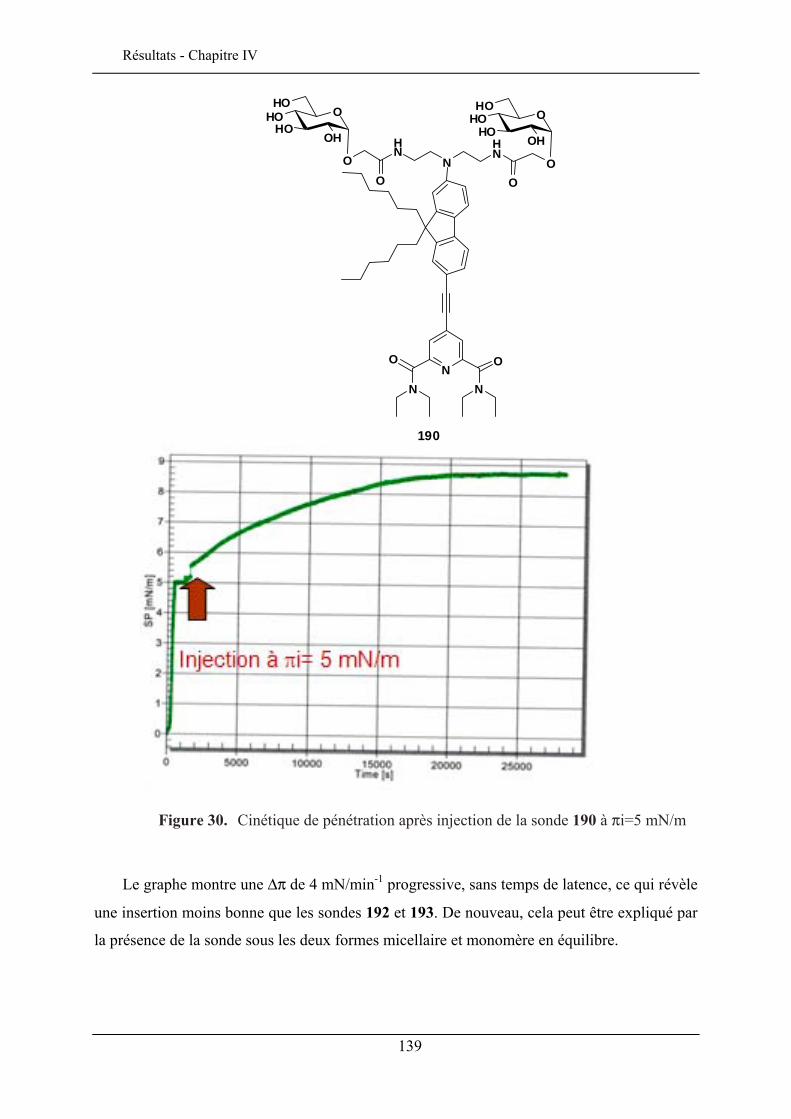
Résultats - Chapitre IV
OHOHO
OH
HO
NHN
NN
O
N
O
OO
HN
OO
OHOHO
OH
HO
190
Figure 30. Cinétique de pénétration après injection de la sonde 190 à πi=5 mN/m
Le graphe montre une ∆π de 4 mN/min-1 progressive, sans temps de latence, ce qui révèle
une insertion moins bonne que les sondes 192 et 193. De nouveau, cela peut être expliqué par
la présence de la sonde sous les deux formes micellaire et monomère en équilibre.
139
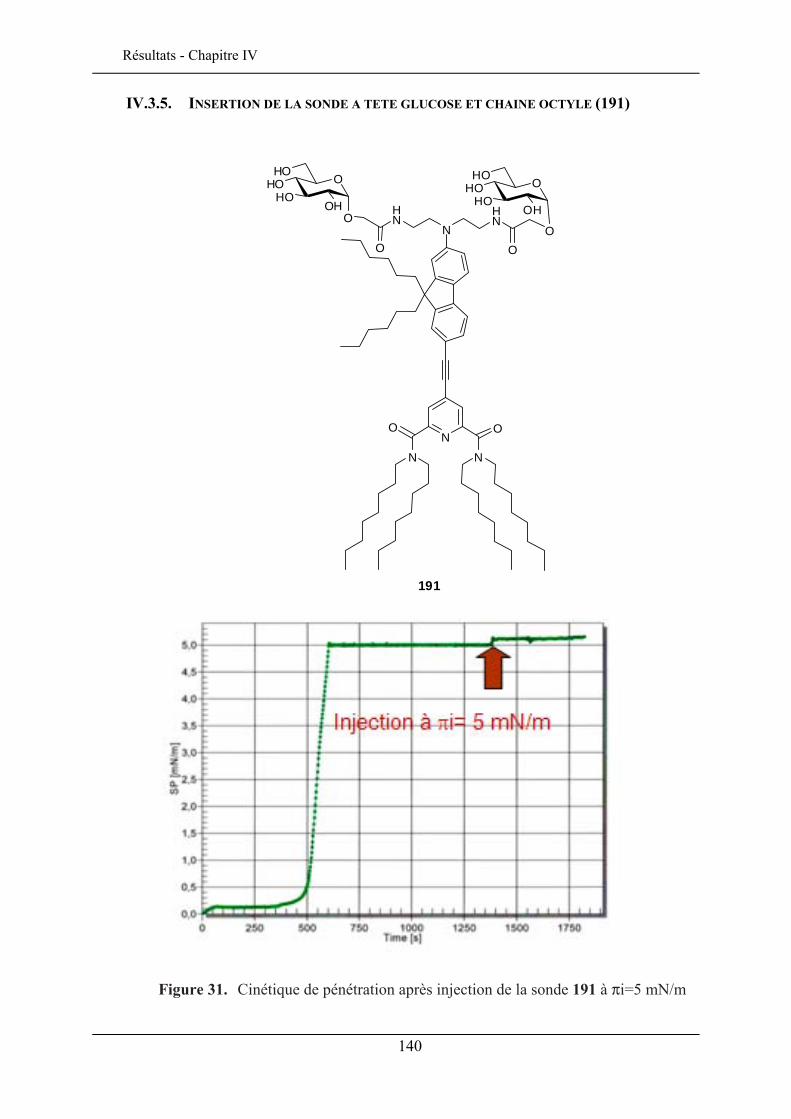
Résultats - Chapitre IV
IV.3.5. INSERTION DE LA SONDE A TETE GLUCOSE ET CHAINE OCTYLE (191)
191
OHOHO
OH
HO
NHN
N
N
O
N
O
OO
HN
O
O
OHOHO
OH
HO
Figure 31. Cinétique de pénétration après injection de la sonde 191 à πi=5 mN/m
140

Résultats - Chapitre IV
La sonde possédant une tête glucose avec une chaîne octyle 191 ne montre aucune
interaction avec la monocouche (figure 31). La variation de pression de surface à son injection
est de l’ordre de 0,1 mN/m-1.
IV.4. CONCLUSION
La sonde éthyle à tête lactose 192 a montré une insertion immédiate et importante dans la
membrane synthétique modèle. La sonde octyle à tête lactose 193 montre une insertion
moyenne et plus lente que la première. L’insertion de la sonde éthyle à tête glucose 190 est
comparable à la sonde 193 mais elle s’effectue à une vitesse plus grande. Par contre,
l’insertion est inexistante dans le cas du produit glucose-octyle 191, le moins hydrophile. Ce
comportement interfacial peut être expliqué par la différence de la balance
hydrophile/hydrophobe de ces molécules.
L’interprétation de ces résultats reste malgré tout délicate puisque l’insertion de ces
molécules dans la monocouche n’est pas uniquement fonction des interactions entre sondes et
monocouche ; elle dépend aussi de leur capacité à se détacher des agrégats qu’elles forment
entre elles. Plus on augmente l’hydrophobie, plus la dissociation de ces micelles paraît
difficile. Il est donc important de mesurer la concentration micellaire critique (CMC) de ces
molécules pour connaître la concentration maximale à laquelle il faut les solubiliser pour les
avoir sous forme de monomères en solution.
Dans la conception de nouveaux chromophores, l’architecture de ces derniers devra
prendre en compte la Balance Hydrophile-Lipophile (BHL) et ceci par augmentation de la
polarité sans augmenter la lipophilie puisque, comme nous l’avons remarqué, plus la sonde est
polaire, plus l’insertion est forte et instantanée.
141

142

CONCLUSION GENERALE
ET PERSPECTIVES
Ce travail de recherche décrit la synthèse de carboxyméthyl glycosides lactones, leur
application pour la synthèse de molécules multifonctionnalisées pour la conception de
molécules scaffolds, ainsi que des molécules amphiphiles pour l’imagerie membranaire.
Par la réaction d’alkylation anomérique avec le bromoacétate de tert-butyle, nous avons
eu accès à une collection de lactones de divers mono- et disaccharidiques d’anomérie α et β.
Deux de ces lactones, une monosaccharidique (α-glc) et une disaccharidique (α-malt),
ont été ouvertes par des amines et des fonctionnalisations ont été effectuées en position 2 et 6,
pour donner de nouveaux carbamates, éthers, azido et des inhibiteurs potentiels des
glycosyltransférases dont une a montré une activité inhibitrice de l’OGT. De plus, elles ont
servi pour la conception de nouveaux glycolipides ainsi que des monomères de type AB et
AB2.
La stratégie CMGL a été appliquée pour la synthèse de sondes membranaires par optique
non linéaire. L’introdution de groupements glycosyles par la stratégie CMGL en utilisant les
deux sucres α-glc et α-lact induit une forte polarité à la sonde et la rend suffisamment soluble
en milieu biologique. De plus, l’amphiphilie qu’ils apportent permet l’ancrage à l’interface de
la membrane en gardant une orientation parallèle de la partie hydrophobe aux chaînes
d’acides gras de la membrane, contrairement aux sondes PEG-ylée et diaminiques qui n’ont
montré aucune spécificité membranaire. Cela a permis l’imagerie en TPEF et GSH. Un signal
de GSH a été observé pour la première fois par une sonde optique non ionique.
143

Les caractérisations optiques et les études d’insertion dans une monocouche lipidique de
Langmuir nous ont permis de mesurer la capacité d’insertion de ces sondes amphiphiles dans
une membrane synthétique modèle.
Par la suite, dans la conception de nouvelles sondes, la balance hydrophile-hydrophobe
devrait être prise en compte afin de garder la bonne insertion observée dans les membranes de
ces molécules. Cependant, il faudra augmenter la polarité pour éviter la formation des
agrégats.
Actuellement, l’équipe de Chantal Andraud de l’Ecole Normale Supérieure continue
l’exploration synthétique vers de nouvelles structures dont les propriétés optiques sont
améliorées. L’équipe du Laboratoire de Spectrométrie Physique adapte les systèmes
expérimentaux (lignées cellulaires, appareillage laser et détection) pour effectuer des
expériences sur des neurones. Quant à notre équipe, nous cherchons à varier la nature du sucre
et à comprendre les modifications apportées aux propriétés des sondes quant à leur solubilité,
leur aptitude à l’insertion dans les membranes et leur temps de résidence dans les membranes.
D’autre part, nous voulons exploiter la position 2 libérée après ouverture des lactones pour
synthétiser des composés présentant deux chaînes hydrophobes qui pourraient mieux s’insérer
dans les membranes comme le font les glycosphingolipides. Ces derniers sont présents au
niveau du feuillet externe de la bicouche lipidique et les résidus oligosaccharidiques émergent
de la membrane plasmique et recouvrent partiellement la surface des cellules (schéma 90).
O O
NHHN
NN
O
N
O
R RRR
OHO
OH
HO
OHO
OH
O
O
OH
NHO
glycosphingolipidesonde bicaténaire
HONH
O
O
OHO
OH
OH
HO
Schéma 90. Une sonde bicaténaire similaire à un sphingolipide
144

TROISIEME PARTIE
PARTIE EXPERIMENTALE

Partie Expérimentale
A. GENERALITES
A.1. SOLVANTS
Les solvants techniques sont distillés :
- le pentane et le dichlorométhane sur P2O5.
- l’éther éthylique sur potasse.
- l’acétate d’éthyle sur K2CO3.
Les solvants anhydres sont redistillés :
- le dichlorométhane est distillé sur hydrure de calcium sous atmosphère d’azote.
- le DMF est distillé sur hydrure de calcium.
A.2. REACTIFS
Les réactifs ont été fournis par Aldrich et ont été utilisés généralement sans purification
supplémentaire.
La triéthylamine et la pyridine sont distillées sur potasse.
A.3. CHROMATOGRAPHIES
Les chromatographies sur couche mince ont été réalisées sur des plaques de silice Merck
60 F. Les produits sont révélés par vaporisation d’une solution à 10 % d’acide sulfurique dans
l’éthanol.
Les purifications par chromatographie ont été réalisées sur colonne de gel de silice flash
Merck (60, 40-63 µm).
A.4. CARACTERISATION DES PRODUITS
RMN : Les spectres RMN 1H (300 MHz et 500 MHz) et 13C (75 MHz et 125 MHz) ont
été enregistrés sur des appareils Bruker ALS 300, DRX 300 et DRX 500. Le pic résiduel du
solvant est choisi comme référence pour étalonner le spectre. Les déplacements chimiques
145

Partie Expérimentale
sont exprimés en ppm. Les abréviations utilisées dans la description des spectres sont les
suivantes : s (singulet) ; ls (large singulet) ; d (doublet) ; ddd (doublet de doublet dédoublé) ; t
(triplet) ; dt (doublet de triplet) ; dd (doublet dédoublé) ; q (quadruplet) ; m (multiplet).
SM-HR : Les spectres de masse haute résolution ont été réalisés sur un spectromètre
ThermoFinnigan par ionisation chimique ou électronique.
Points de fusion : Les points de fusion ont été mesurés sur un banc Kofler.
Pouvoirs rotatoires : Les pouvoirs rotatoires ont été mesurés à 22°C grâce à un
polarimètre PERKIN-ELMER 241 à la longueur d’onde de la raie D du sodium, la longueur
de la cellule est de 1 dm. Le pouvoir rotatoire est déterminé par la loi de Biot. Les
concentrations (c) sont données en g/100 mL.
Analyses élémentaires : les analyses élémentaires ont été effectuées par le Service
Central d’Analyse du CNRS à Vernaison.
A.5. REMARQUES GENERALES :
Les phases organiques sont séchées sur sulfate de sodium anhydre.
Les réactions utilisant des solvants anhydres ont été réalisées sous atmosphère d’azote.
Les points de fusion des solides et des cristaux ont été mesurés.
146

Partie Expérimentale
B. SYNTHESE
B.1. PROCEDURES GENERALES POUR LA PREPARATION DES (TERT-
BUTYLOXYCARBONYL)METHYL GLYCOSIDES ACETYLES
1. A partir de sucres déprotégés en position anomérique
A une solution de 75 mM solution du glycoside partiellement acétylé possédant un
groupement hydroxyle libre en position 1 dans du DMF anhydre, K2CO3 (2,5-5 équiv) est
ajouté suivi de 2 équiv. du bromoacétate de tert-butyle. Le mélange réactionnel est agité sous
azote pendant 48 heures à température ambiante. Les sels sont filtrés et rincés au CH2Cl2 et les
solvants sont évaporés sous pression réduite. Le résidu est dissous dans du CH2Cl2 et lavé à
l’eau. La fraction organique est séchée sur Na2SO4, filtrée et évaporée. Le brut est ensuite
soumis à une purification sur silice pour donner les (tert-butyloxycarbonyl)méthyl glycosides
correspondants sous la forme d’un mélange d’anomères. La séparation de chaque anomère est
réalisée ensuite sur gel de silice.
2. A partir de sucres libres
A une solution de 220 mM du glycoside dans du DMF anhydre, NaH (1 equiv) est ajouté
suivi d’un équiv. du bromoacétate de tert-butyle. La réaction est gitée sous azote pendant 3
jours. Ensuite le DMF est évaporé et le résidu est soumis à une purification sur gel de silice
(CH2Cl2: CH3OH, 9:1), pour donner l’ester correspondant sous la forme de mélange
d’anomères. Les deux anomères sont séparés par colonne chromatographique après une
peracétylation dans la pyridine en présence d’anhydride acétique.
147

Partie Expérimentale
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranoside (125a)
OAcOAcO
AcO
OO
OAcO
72%. (Ether/ Pentane : 1/1). Solide blanc.
RMN 1H (300 MHz, CDCl3): δ (ppm)= 1,46 (s ; 9 H) ; 2,02 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,10
(s ; 3 H) ; 2,13 (s ; 3 H) ; 4,07-4,22 (m ; 2 H ; H-5 ; H-6b) ; 4,10 (système AB ; δa 4,05 , δb
4,15 ; 2 H ; J = 16,4 Hz ; H-7) ; 4,27 (dd ; 1 H ; J = 4,0 Hz ; J = 12,1 Hz ; H-6a) ; 4,92 (dd ; 1
H ; J = 3,9 Hz ; J = 9,8 Hz ; H-2) ; 5,09 (d ; 1 H ; J = 9,8 Hz ; H-4) ; 5,17 (d ; 1 H ; J = 3,8 Hz ;
H-1) ; 5,54 (t ; 1 H ; J = 9,8 Hz ; H-3).
RMN 13C (CDCl3, 75 MHz): δ (ppm)= 20,6 ; 20,6 ; 20,7 ; 20,7 (4 CH3) ; 28,0 (3 CH3) ;
61,7 (C-6) ; 64,7 ( C-7) ; 67,8 (C-5) ; 68,4 (C-4) ; 69,7 (C-3) ; 70,4 (C-2) ; 82,0 (Cq) ; 95,7
(C-1) ; 168,2 ; 169,6 ; 169,9 ; 170,3 ; 170,6 (5 CO).
Tf: 88°C.
[α]D +129 (c = 1,0 ; CH2Cl2).
SM (Basse Résolution-ESI) m/z pour C20H30O12Na (M+Na)+ = 485,0.
Anal. Calc. pour C20H30O12: C, 51,94; H, 6,54 ; trouvée: C, 51,82 ; H, 6,45.
(tert-Butyloxycarbonyl)methyl 2,3,4,6-tetra-O-acétyl-β-D-glucopyranoside (125b)
OAcOAcO
AcO
O
O
O
OAc
8%. (Ether/ Pentane : 1/1). Cristaux blancs.
RMN 1H (300 MHz, CDCl3): δ (ppm)= 1,47 (s ; 9 H) ; 2,01 (s ; 3 H) ; 2,02 (s ; 3 H) ; 2,09
(s ; 3 H) ; 2,10 (s ; 3 H) ; 3,72 (ddd ; 1 H ; J = 2,2 Hz ; J = 4,5 Hz ; J = 9,6 Hz ; H-5) ; 4,11-
4,16 (m ; 3 H ; H-7 ; H-6a) ; 4,27 (dd ; 1 H ; J = 4,5 Hz ; J = 12,4 Hz ; H-6b) ; 4,68 (d ; 1 H ; J
148

Partie Expérimentale
= 7,8 Hz ; H-1) ; 5,04 (dd ; 1 H ; J = 7,8 Hz ; J = 9,6 Hz ; H-2) ; 5,10 (t ; 1 H ; J = 9,6 Hz ; H-
4) ; 5,25 (t ; 1 H ; J = 9,6 Hz ; H-3).
RMN 13C (CDCl3, 75 MHz): δ (ppm) 20,6 ; 20,6 ; 20,7 ; 20,7 (4 CH3) ; 28,0 (3 CH3) ;
61,8 (C-6) ; 65,4 (C-7) ; 68,3 (C-4) ; 71,0 (C-2) ; 71,8 (C-5) ; 72,6 (C-3) ; 81,9 (Cq) ; 100,1
(C-1) ; 168,1 ; 169,4 ; 169,6 ; 170,1 ; 170,6 (5 CO).
Tf.: 102°C.
[α]D -29 (c = 1,0 ; CH2Cl2).
SM (Basse Résolution-ESI) m/z pour C20H30O12Na (M+Na)+ = 485,0.
Anal. Calc. pour C20H30O12: C, 51,94; H, 6,54 ; trouvée: C, 51,88 ; H, 6,55.
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tetra-O-acétyl-α-D-mannopyranoside (126a)
OAcOAcO
AcOAcO
OO
O
83%. (Ether/ Pentane : 1/1). Cristaux blancs.
RMN 1H (300 MHz, CDCl3): δ (ppm)= 1,48 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,99 (s ; 3 H) ;
2,05 (s ; 3 H) ; 2,11 (s ; 3 H) ; 2,16 (s ; 3H) ; 4,09 (dd ; 1 H ; J = 2,25 Hz ; J = 12,1 Hz ; H-6a)
; 4,11 (système AB ; δa 4.05 , δb 4.17 ; 2 H ; J = 16,3 Hz ; H-7) ; 4,18 (m ; 1 H ; H-5) ; 4,30
(dd ; 1 H ; J = 4,7 Hz ; J = 12,1 Hz ; H-6b) ; 4,96 (d ; 1 H ; J = 0,6 Hz ; H-1) ; 4,31-4,37 (m ; 3
H ; H-2 ; H-3 ; H-4).
RMN 13C (CDCl3, 75 MHz): δ (ppm)= 20,7 ; 20,8 ; 20,8 ; 20,9 (4 CH3) ; 28,0 (3 CH3 du
t-butyle) ; 62,5 (C-6) ; 65,0 (C-7) ; 66,1 (C-4) ; 69,1 , 69,3 (C-2 ; C-3) ; 69,5 (C-5) ; 82,3 (Cq)
; 97,9 (C-1) ; 168,4 ; 169,8 ; 169,8 ; 169,8 ; 170,6 (5 CO).
Tf: 85° C.
SM (Basse Résolution-ESI) m/z pour C20H30O12Na (M+Na)+= 485,0.
[α]D + 57 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C20H30O12: C, 51,94; H, 6,54 ; trouvée: C, 52,08; H, 6,56.
149

Partie Expérimentale
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tetra-O-acétyl-β-D-mannopyranoside (126b)
OAcOAcO
AcOAcO
O
O
O
11%. Isolé à partir de la réaction sur sucre libre. (Ether/ Pentane : 1/1). Huile jaune.
RMN 1H (300 MHz, CDCl3): δ (ppm)= 1,44 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,95 (s ; 3 H) ;
2,00 (s ; 3 H) ; 2,06 (s ; 3 H) ; 2,15 (s ; 3H) ; 3,64 (m ; 1 H ; H-5) ; 4,11 (dd ; 1 H ; J = 2,6 Hz ;
J = 12,2 Hz ; H-6b) ; 4,21-4,07 (m ; 2 H ; H-7) ; 4,27 (dd ; 1 H ; J = 5,3 Hz ; J = 12,2 Hz ; H-
6a) ; 4,84 (d ; 1 H ; J = 1,0 Hz ; H-1) ; 5,05 (dd ; 1 H ; J = 3,2 Hz ; J = 10,0 Hz ; H-3) ; 5,23
(dd ; 1 H ; J = 9,9 Hz ; J = 10,0 Hz ; H-4) ; 5,55 (dd ; 1 H, J = 1,0 Hz ; J = 3,2 Hz ; H-2).
RMN 13C (CDCl3, 75 MHz): δ (ppm)= 20,4 ; 20,6 ; 20,6 ; 20,7 (4 CH3) ; 28,0 (3 CH3 du
t-butyle) ; 62,3 (C-7) ; 65,4 (C-6) ; 65,9 (C-4) ; 68,6 (C-2) ; 80,8 (C-3) ; 72,4 (C-5) ; 82,1 (Cq)
; 97,5 (C-1) ; 169,3 ; 169,5 ; 169,8 ; 170,1 ; 170,6 (5 CO).
SM (Haute Résolution-ESI) m/z calculée C20H30O12Na (M+Na)+ = 485,1635 ; trouvée =
485,1636.
[α]D +46 (c = 1,0 ; CHCl3).
Anal. Calc. pour C20H30O12: C, 51,94; H, 6,54 ; trouvée: C, 51,53; H, 6,37.
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tetra-O-acétyl-α-D-galactopyranoside (127a)
OAcO
OAc
OAcAcO
OO
O
65%. (Ether/ Pentane : 1/1). Cristaux blancs.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,46 (s ; 9H 3 CH3 du t-butyle) ; 1,99 (s ; 3H) ; 2,05 (s ; 3H) ; 2,14 (s ; 6H) ;
4,10 (système AB: δa 4,06 , δb 4,14 ; J = 16,6 Hz ; H-7) ; 4,04-4,16 (m ; 2 H ; H-5 ; H-6b) ;
4,36 (pseudo t ; 1 H ; J = 6,6 Hz ; H-6a) ; 5,17 (dd ; 1 H ; J = 3,6 Hz ; J = 10,4 Hz ; H-2) ;
150

Partie Expérimentale
5,21 (d ; 1 H ; J = 3,6 Hz ; H-1) ; 5,42 (dd ; 1 H ; J = 3,6 Hz ; J = 10,4 Hz ; H-3) ; 5,48
(pseudo-d ; 1 H ; J = 3,6 Hz ; H-4).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,6 ; 20,6 ; 20,7 ; 20,9 (4 CH3) ; 28,1 (3 CH3 du t-butyle) ; 61,6 (C-7) ; 64,5
(C-6) ; 66,8 (C-5) ; 67,2 (C-3) ; 67,6 (C-2) ; 68,0 (C-4) ; 82,1 (Cq) ; 96,1 (C-1) ; 168,3 ; 169,8
; 170,2 ; 170,4 ; 170,6 (5 CO).
Tf: 102°C.
[α]D +115 (c = 0,5 ; CH2Cl2).
SM (Basse Résolution-ESI) m/z pour C20H30O12Na (M+Na)+ = 485,0.
Anal. Calc. pour C20H30O12: C, 51,94 ; H, 6,54 ; trouvée: C, 51,86 ; H, 6,64.
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranoside (127b)
OAcO
O
OAcO
OOAcAcO
9%. (Ether/ Pentane : 1/1). Cristaux blancs.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,42 (s ; 9 H 3 CH3 du t-butyle) ; 1,93 (s ; 3 H) ; 2,00 (s ; 3 H) ; 2,06 (s ; 3 H) ;
2,10 (s ; 3 H) ; 3,86 (ddd ; 1 H ; J = 1,0 Hz ; J = 6,8 Hz ; J = 13,4 Hz ; H-5) ; 4,08-4,13 (m ; 2
H ; H-6) ; 4,11 (s ; 2 H ; H-7) ; 4,59 (d ; 1 H ; J = 7,9 Hz ; H-1) ; 5,00 (dd ; 1 H ; J = 3,4 Hz ; J
= 10,5 Hz ; H-3) ; 5,19 (dd ; 1 H ; J = 7,9 Hz ; J = 10,5 Hz ; H-2) ; 5,33 (dd ; 1 H ; J = 1,0 Hz ;
J = 3,4 Hz ; H-4).
RMN 13C (CDCl3, 75 MHz):
δ (ppm)= 20,6 ; 20,6 ; 20,7 ; 20,9 (4 CH3) ; 28,1 (3 CH3 du t-butyle) ; 61,2 (C-6) ; 61,2 (
C-6) ; 65,2 (C-4) ; 66,9 (C-2) ; 68,5 (C-5) ; 70,7 (C-3) ; 81,9 (Cq) ; 100,5 (C-1) ; 168,2 ; 169,9
; 170,1 ; 170,2 ; 170,4 (5 CO).
Tf: 98°C.
[α]D –16 (c = 1,0 ; CH2Cl2).
SM (Basse Résolution-ESI) m/z pour C20H30O12Na (M+Na)+ = 484,99.
151

Partie Expérimentale
Anal. Calc. pour C20H30O12: C, 51,94 ; H, 6,54 ; trouvée: C, 51,49 ; H, 6,57.
(tert-Butyloxycarbonyl)méthyl 2,3,4,-tri-O-acétyl-α-L-fucopyranoside (128a)
O
OAc
O
O
OAc
O
OAc
45%. (Ether/ Pentane : 1/1). Solide blanc.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,03 (d ; 3 H ; J = 6,5 Hz ; H-6) ; 1,36 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,88 (s ; 3
H) ; 2,04 (s ; 3 H) ; 2,07 (s ; 3 H) ; 3,98 (système AB: δa 3,94 , δb 4,04 ; J = 16,5 Hz ; H-7) ;
4,19 (dq ; 1 H ; J = 1,0 Hz ; J = 6,5 Hz ; H-5) ; 5,11 (d ; 1 H ; J = 3,7 Hz ; H-1) ; 5,24 (dd ; 1
H ; J = 1,0 Hz ; J = 3,0 Hz ; H-4) ; 5,34 (dd ; 1 H ; J = 3,0 Hz ; J = 10,2 Hz ; H-3) ; 5,35 (dd ;
1 H ; J = 3,7 Hz ; J = 10,2 Hz ; H-2).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 15,6 (C-6) ; 20,5 ; 20,6 ; 20,6 ; 20,8 (3 CH3) ; 28,0 (3 CH3 du t-butyle) ; 64,6
(C-7) ; 64,9 (C-5) ; 67,6 (C-4) ; 67,6 (C-3) ; 71,0 (C-2) ; 81,7 (Cq) ; 96,1 (C-1) ; 168,4 ; 169,8
; 170,4 ; 170,5 (4 CO).
Tf: 120°C
SM (Basse Résolution-ESI) m/z pour (M+Na)+ = 426,9.
[α]D -103 (c = 3,0 ; CH2Cl2).
Anal. Calc. pour C20H30O12: C, 53,46 ; H, 6,98 ; trouvée: C, 53,47 ; H, 6,96.
(tert-Butyloxycarbonyl)méthyl 2,3,4,-tri-O-acétyl-β-L-fucopyranoside (128b)
O
AcOOAc
OAcO
O
O
12%. (AcOEt/ Pentane : 1/1). Cristaux blancs.
152

Partie Expérimentale
RMN 1H (500 MHz, CDCl3):
δ (ppm)= 1,20 (d ; 3 H ; J = 6,4 Hz ; H-6) ; 1,44 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,97 (s ; 3
H) ; 2,09 (s ; 3 H) ; 2,15 (s ; 3 H) ; 3,78 (dq ; 1 H ; J = 0,8 Hz ; J = 6,4 Hz ; H-5) ; 4,15 (s ; 2 H
; H-7) ; 4,58 (d ; 1 H ; J = 7,9 Hz ; H-1) ; 5,03 (dd ; 1 H ; J = 3,4 Hz ; J = 10,5 Hz ; H-3) ; 5,25
(dd ; 1 H ; J = 7,9 Hz ; J = 10,5 Hz ; H-2) ; 5,26 (dd ; 1 H ; J = 0,8 Hz ; J = 3,4 Hz ; H-4).
RMN 13C (CDCl3, 125 MHz):
δ (ppm)=16,0 (C-6) ; 20,5 ; 20,6 ; 20,8 (3 CH3) ; 28,0 (3 CH3 du t-butyle) ; 65,0 (C-7) ;
68,5 (C-2) ; 69,2 (C-5) ; 70,1 (C-4) ; 71,1 (C-3) ; 81,6 (Cq) ; 100,2 (C-1) ; 168,3 ; 169,9 ;
170,0 ; 170,1 (4 CO).
Tf: 114°C.
[α]D + 10 (c = 2,0 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z calculée pour (M+Na)+ = 427,1580, trouvée 427,1577.
Anal. Calc. pour C18H28O10 : C, 53,46 ; H, 6,98 ; trouvée: C, 53,19 ; H, 6,73.
(tert-Butyloxycarbonyl)méthyl 2-acétamido-3,4,6-tri-O-acétyl-2-déoxy-α-D-
glucopyranoside (129a)
OAcO
AcHN
OAc
O O
OAcO
65%. (AcOEt/ Pentane : 4/1). Solide blanc.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,48 (s ; 9 H ; 3 CH3 du t-butyle) ; 2,00 (s ; 3 H) ; 2,02 (s ; 3 H) ; 2,03 (s ; 3 H) ;
2,10 (s ; 3 H) ; 4,07-4,11 (m ; 4 H ; H-5 ; H-6b ; H-7) ; 4,18 (dd ; 1 H ; J = 4,7 Hz ; J = 13,0
Hz ; H-6a) ; 4,37 (dd ; 1 H ; J = 6,6 Hz ; J = 13,0 Hz ; H-6a) ; 4,37 (td ; 1 H ; J = 3,6 Hz ; J =
10,3 Hz ; H-2) ; 4,85 (d ; 1 H ; J = 3,6 Hz ; H-1) ; 5,15 (t ; 1 H ; J = 10,3 Hz ; H-4) ; 5,48 (t ; 1
H ; J = 10,3 Hz, H-3) ; 6,47 (d ; 1 H ; J = 9,2 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
153

Partie Expérimentale
δ (ppm)= 20,4 ; 20,4 ; 20,5 ; 23,0 (4 CH3) ; 27,9 (3 CH3 du t-butyle) ; 51,4 (C-2) ; 61,6
(C-6) ; 65,1 (C-7) ; 67,8 (C-4) ; 68,3 (C-5) ; 71,0 (C-3) ; 82,4 (Cq) ; 98,2 (C-1) ; 168,6 ; 169,1
; 170,3 ; 170,5 ; 170,9 (5 CO).
Tf: 140°C.
[α]D +73 (c = 1,0 ; CH2Cl2).
SM (Basse Résolution-ESI) m/z pour (M+Na)+ = 484,0.
Anal. Calc. pour C20H31NO11 : C, 52,06 ; H, 6,74 ; H, 3,04 ; trouvée: C, 51,90 ; H, 6,69 ;
N, 3,01.
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-
2,3,6-tri-O-acétyl-α-D-glucopyranoside (130a)
40%. (CH2Cl2/ Ether: 1/5). Solide blanc.
RMN 1H (500 MHz, CDCl3) :
δ (ppm)= 1,43 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,96 (s ; 3 H) ; 2,05 (s ; 6 H) ; 2,06 (s ; 3 H) ;
2,11 (s ; 3 H) ; 2,13 (s ; 3 H) ; 2,16 (s ; 3 H) ; 3,76 (t ; 1 H ; J = 10,1 Hz ; H-4) ; 3,86 (t ; 1 H ;
J = 6,4 Hz ; H-5’) ; 4,03-4,13 (m ; 6 H ; H-7 ; H-5 ; H-6’; H-6b) ; 4,47 (dd ; 1 H ; J = 1,9 Hz ;
J = 12,3 Hz ; H-6a) ; 4,47 (d ; 1 H ; J = 8,0 Hz ; H-1’) ; 4,82 (dd ; 1 H ; J = 3,8 Hz ; J = 10,1
Hz ; H-2) ; 4,94 (dd ; 1 H ; J =3,5 Hz ; J = 10,4 Hz ; H-3’) ; 5,09 (d ; 1 H ; J =3,8 Hz ; H-1) ;
5,10 (dd ; 1 H ; J = 8,0 Hz ; 10,4 Hz ; H-2’) ; 5,33 (d ; J = 3,5 Hz ; H-4’) ; 5,52 (t ; 1 H ; J
=10,1 Hz ; H-3).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 20,9 ; 21,0 ; 21,1 ; 21,2 ; 21,3 ; 21,3 ; 21,3 (7 CH3) ; 28,4 (3 CH3 du t-butyle) ;
61,2 (C-6’) ; 62,2 (C-6) ; 64,8 (C-7) ; 67,0 (C-4’) ; 67,0 (C-5) ; 69,2 (C-2’) ; 69,6 (C-3) ; 69,8
(C-2) ; 71,0 (C-5’) ; 71,4 (C-3’) ; 76,7 (C-4) ; 82,4 (Cq) ; 95,8 (C-1) ; 101,4 (C-1’) ; 168,5 ;
169,4 ; 169,8 ; 170,5 ; 170,6 ; 170,7 ; 170,8 ; 171,0 (8 CO).
154

Partie Expérimentale
Tf: 92-97°C.
[α]D +30 (c = 1,0 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z pour calculée pour (M+Na)+ = 773,2480 trouvée
773,2481.
Anal. Calc. pour C32H46O20: C, 51,20 ; H, 6,18 ; trouvée: C, 50,82 ; H, 6,07.
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-
2,3,6tri-O-acétyl-β-D-glucopyranoside (130b)
2%. (CH2Cl2/ Ether: 1/5). Solide blanc.
RMN 1H (300 MHz, CDCl3) :
δ (ppm) 1,48 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,98 (s ; 3 H) ; 2,05 (s ; 3 H) ; 2,07 (s ; 3 H) ;
2,08 (s ; 3 H) ; 2,10 (s ; 3 H) ; 2,14 (s ; 3 H) ; 2,17 (s ; 3 H) ; 3,59 (m ; 1 H ; H-5) ; 3,83 (t ; 1
H, J = 9,2 Hz ; H-4) ; 3,88 (t ; 1 H ; J = 6,8 Hz ; H-5’) ; 4,03-4,12 (m ; 3 H ; H-6’; H-6a) ;
4,12 (s ; 2 H ; H-7) ; 4,48 (d ; 1 H ; J =8,0 Hz ; H-1’) ; 4,49 (dd ; 1 H ; J =11,0 Hz ; H-6b) ;
4,62 (d ; 1 H ; J = 7,7 Hz, H-1) ; 4,92-4,98 (m ; 2 H ; H-1 ; H-3’) ; 5,11 (dd ; 1 H ; J = 7,7 Hz
; J = 10,2 Hz ; H-2’) ; 5,23 (t ; 1 H ; J = 9,2 Hz ; H-3) ; 5,35 (d ; 1 H ; J = 3,1 Hz ; H-4’).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 20,9 ; 21,0 ; 21,0 ; 21,0 ; 21,2 ; 21,2 ; 21,3 (7 CH3) ; 28,4 (3 CH3 du t-butyle) ;
61,2 (C-6’) ; 62,3 (C-6) ; 65,9 (C-7) ; 67,0 (C-4’) ; 69,5 (C-2’) ; 71,1 (C-5’) ; 71,4-71,8 (C-2 ;
C-3’) ; 71,8 (C-3) ; 72,9 (C-5) ; 76,5 (C-4) ; 82,3 (Cq) ; 100,2 (C-1) ; 101,4 (C-1’) ; 168,5 ;
169,4 ; 170,1 ; 170,3 ; 170,5 ; 170,55 ; 170,7 ; 170,7 (8 CO).
Tf.: 93°C
[α]D +5 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C32H46O20: C, 51,20 ; H, 6,18 ; trouvée: C, 51,05 ; H, 6,25.
155

Partie Expérimentale
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tétra-O-acétyl-β-D-glucopyranosyl-(1→4)-
2,3,6-tri-O-acétyl -α-D-glucopyranoside (131a)
O
OAc
O
O
OAcO
AcOOAc
OAc
AcOOAcO
O
72%. (CH2Cl2/ Ether: 1/7). Solide blanc.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,43 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,97 (s ; 3 H) ; 2,00 (s ; 3 H) ; 2,01 (s ; 3 H) ;
2,04 (s ; 3 H) ; 2,08 (s ; 3 H) ; 2,10 (s ; 3 H) ; 2,13 (s ; 3 H) ; 3,64-3,69 (m ; 1 H ; H-5’) ; 3,75
(t ; 1 H ; J = 9,9 Hz ; H-4) ; 3,99-4,17 (m ; 3 H ; H-5 ; H-6’b; H-6b) ; 4,37 (dd ; 1 H ; J = 4,3
Hz ; J = 12,4 Hz ; H-6’a) ; 4,49 (dd ; 1 H ; J = 1,7 Hz ; J = 11,9 Hz ; H-6a) ; 4,53 (dd ; 1 H ; J
= 7,9 Hz ; H-1’) ; 4,84 (dd ; 1 H ; J = 3,7 Hz ; J = 9,9 Hz ; H-2) ; 4,93 (t ; 1 H ; J = 8,6 Hz ;
H-4’) ; 5,04-5,18 (m ; 2 H ; H-2’ ; H-3’) ; 5,10 (d ; J = 3,7 Hz ; H-1) ; 5,50 (t ; 1 H ; J =9,9 Hz
; H-3).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,5 ; 20,5 ; 20,6 ; 20,6 ; 20,7 ; 20,8 ; 20,9 (7 CH3) ; 28,4 (3 CH3 du t-butyle) ;
61,7-61,8 (C-6’ ; C-6) ; 64,4 (C-7) ; 67,7 (C-2’) ; 68,8 (C-5) ; 69,1 (C-3) ; 70,5-71,7 (C-2 ou
C-5’) ; 71,9 (C-4’) ; 73,0 (C-3’) ; 76,5 (C-4) ; 82,4 (Cq) ; 95,4 (C-1) ; 100,7 (C-1’) ; 169,3 ;
169,5 ; 170,0 ; 170,5 ; 170,7 ; 170,8 ; 170,8 ; 178,3 (8 CO).
[α]D +43 (c = 1,0 ; CH2Cl2).
Tf: 160-162°C.
SM (Haute Résolution-ESI) m/z pour calculée pour (M+Na)+ = 773,2480 ; trouvée
773,2478.
Anal. Calc. pour C32H46O20: C, 51,20 ; H, 6,18 ; trouvée: C, 50,81 ; H, 6,24.
156

Partie Expérimentale
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tetra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-
tri-O-acétyl-α-D-glucopyranoside (132a)
O
OAc
O
O
OAcO
AcOOAc
OAc
AcO
O
OAcO
70%. (CH2Cl2/ Acétone: 12/1). Mousse blanche.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,48 (s ; 9 H ; 3 CH3 du t-butyle) ; 2,02 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,04 (s ; 3 H) ;
2,08 (s ; 3 H) ; 2,11 (s ; 3 H) ; 2,12 (s ; 3 H) ; 2,16 (s ; 3 H) ; 3,94 (m ; 1 H ; H-5’) ; 4,05 (t ; 1
H ; J = 9,2 Hz ; H-4) ; 4,08 (dd ; 1 H ; J = 1,5 Hz ; J = 8,3 Hz ; H-6’a) ; 4,15 (système AB: δa
4,08 ; δb 4,17 ; J = 16,5 Hz ; H-7) ; 4,16-4,30 (m ; 3 H ; H-5 ; H-6a ; H-6’b) ; 4,48 (dd ; 1 H ; J
= 2,5 Hz ; J = 12,4 Hz, H-6a) ; 4,80 (dd ; 1 H ; J = 3,6 Hz ; J = 9,8 Hz ; H-2) ; 4,88 (dd ; 1 H ;
J = 3,9 Hz ; J = 10,3 Hz ; H-2’) ; ), 5,09 (t ; 1 H ; J = 9,6 Hz ; H-4’) ; 5,12 (d ; 1 H ; J = 3,6
Hz ; H-1) ; 5,38 (t ; 1 H ; J = 9,8 Hz ; H-3’) ; 5,45 (d ; 1 H ; J = 3,9 Hz ; H-1’) ; 5,60 (t ; 1 H ;
J = 9,8 Hz ; H-3).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,6 ; 20,6 ; 20,7 ; 20,7 ; 20,7 ; 20,8 ; 20,9 (7 CH3) ; 28,1 (3 CH3 du t-butyle) ;
61,4 (C-6’) ; 62,6 (C-6) ; 64,5 (C-7) ; 68,0 (C-4’) ; 68,3 (C-5’) ; 68,4 (C-5) ; 69,3 (C-3’) ; 70,0
(C-2’) ; 71,0 (C-2) ; 72,2 (C-3) ; 72,3 (C-4) ; 82,1 (Cq) ; 95,4 (C-1’) ; 95,5 (C-1) ; 168,2 ;
169,5 ; 169,7 ; 169,9 ; 170,5 ; 170,5 ; 170,5 ; 170,7 (8 CO).
SM (Haute Résolution-ESI) m/z pour calculée pour C32H46O20 Na (M+Na)+ = 773,2480 ;
trouvée 773,2481.
[α]D +69 (c = 0,5 ; CH2Cl2).
Anal. Calc. pour C32H46O20: C, 51,20 ; H, 6,18 ; trouvée: C, 51,17 ; H, 6,21.
157

Partie Expérimentale
(tert-Butyloxycarbonyl)méthyl 2,3,4,6-tetra-O-acétyl-α-D-glucopyranosyl-(1→4)-
2,3,6-tri-O-acétyl-β-D-glucopyranoside (132b)
O
OAcO
AcOOAc
OAc
AcO
AcO
O
OAc
OO
O
11%. Isolé à partir de la réaction du maltose libre avec le bromoacétate de tert-butyle
suivi d’une acétylation. (CH2Cl2/ Acétone: 12/1). Huile incolore.
RMN 1H (500 MHz, CDCl3) :
δ (ppm)= 1,40 (s ; 9 H ; 3 CH3 du t-butyle) ; 1,93 (s ; 3 H) ; 1,94 (s ; 3 H) ; 1,95 (s ; 3 H) ;
1,97 (s ; 3 H) ; 2,00 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,08 (s ; 3 H) ; 3,61 (m ; 1 H ; H-5) ; 3,88 (ddd ;
1 H ; J = 2,4 Hz ; J = 5,1 Hz ; J = 10,2 Hz ; H-5’) ; 3,96 (t ; 1 H ; J = 9,1 Hz ; H-4) ; 3,97 (dd ;
1 H ; J = 2,4 Hz ; J = 12,9 Hz ; H-6’a) ; 4,06 (s ; 2 H ; H-7) ; 4,15 (dd ; 1 H ; J = 5,1 Hz ; J =
12,9 Hz, H-6’b) ; 4,18 (dd ; 1 H ; J = 4,1 Hz ; J = 12,6 Hz ; H-6a) ; 4,42 (dd ; 1 H ; J = 2,5 Hz
; J = 12,6 Hz ; H-6b) ; 4,61 (d ; 1 H ; J = 7,6 Hz ; H-1) ; 4,79 (dd ; 1 H ; J = 4,0 Hz ; J = 10,2
Hz ; H-2’) ; 4,81 (dd ; 1 H ; J = 7,9 Hz ; J = 9,1 Hz ; H-2) ; 4,98 (t ; 1 H ; J = 10,2 Hz ; H-4’) ;
5,21 (t ; 1 H ; J = 9,1 Hz ; H-3) ; 5,29 (t ; 1 H ; J = 10,2 Hz ; H-3’) ; 5,33 (d ; 1 H ; J = 4,0 Hz
; H-1’).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 20,7 ; 20,7 ; 20,8 ; 20,9 ; 20,9 ; 21,0 ; 21,0 (7 CH3) ; 28,2 (3 CH3 du t-butyle) ;
61,1 (C-6’) ; 62,8 (C-6) ; 65,6 (C-7) ; 68,1 (C-4’) ; 68,6 (C-5’) ; 69,4 (C-3’) ; 70,1 (C-2’) ;
72,0 (C-2) ; 72,3 (C-5) ; 72,7 (C-4) ; 75,2 (C-3) ; 82,0 (Cq) ; 95,6 (C-1’) ; 99,7 (C-1) ; 168,2 ;
169,5 ; 170,0 ; 170,0 ; 170,2 ; 170,6 ; 170,6 ; 170,7 (8 CO).
[α]D +59 (c= 0,3 ; CH2Cl2).
Anal. Calc. pour C32H46O20: C, 51,20 ; H, 6,18 ; trouvée: C, 51,15 ; H, 6,26.
B.2. PROCEDURE GENERALE POUR LA PREPARATION DES CARBOXYMETHYL
GLYCOSIDES PERACETYLES
Les (tert-butoxy)carbonylméthyl glycosides sont dissous une solution 50% vol. de TFA
(20 equiv.) dans CH2Cl2. La réaction est agitée pendant 3 heures à température ambiante et les
158

Partie Expérimentale
solvants sont ensuite coévaporés deux fois avec du toluène. Le résidu est chromatographié sur
gel de silice avec un gradient de pentane/AcOEt pour donner les acides correspondants 10-17
avec un rendement quantitatif.
Carboxyméthyl-2,3,4,6-tetra-O-acétyl-β-D-mannopyranoside (134b)
OAcOAcO
AcOAcO
O
OH
O
Huile jaune.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,94 (s ; 3 H) ; 1,99 (s ; 3 H) ; 2,05 (s ; 3 H) ; 2,13 (s ; 3H) ; 3,67 (m ; 1 H ; H-
5) ; 4,11 (dd ; 1 H ; J = 2,6 Hz ; J = 12,6 Hz ; H-6b) ; 4,25 (dd ; 1 H ; J = 4,7 Hz ; J = 12,6 Hz
; H-6a) ; 4,30 (s ; 2 H ; H-7) ; 4,85 (ls ; 1 H ; H-1) ; 5,05 (dd ; 1 H ; J = 1,9 Hz ; J = 9,8 Hz ;
H-3) ; 5,19 (t ; 1 H ; J = 9,8 Hz ; H-4) ; 5,50 (d ; 1 H ; J = 1,9 Hz ; H-2).
RMN 13C (CDCl3, 75 MHz):
δ (ppm) 20,6 ; 20,6 ; 20,7 ; 20,8 (4 CH3) ; 62,5 (C-6) ; 65,0 (C-7) ; 65,9 ( C-4) ; 68,7 (C-
2) ; 71,0 (C-3) ; 72,4 (C-5) ; 97,8 (C-1) ; 170,1 ; 170,5 ; 171,1 ; 171,6 ; 171,6 (5 CO).
[α]D -47 (c = 2,0 ; CH2Cl2).
Anal. Calc. pour C16H22O12: C, 45,29; H, 5,70 ; trouvée: C, 45,61 ; H, 5,58.
Carboxyméthyl 2,3,4,-tri-O-acétyl-α-L-fucopyranoside (136a)
O
OAc
OH
O
OAc
OAc
O
Huile jaune.
RMN 1H (300 MHz, CDCl3):
159

Partie Expérimentale
δ (ppm)= 1,05 (d ; 3 H ; J = 6,0 Hz ; H-6) ; 1,91 (s ; 3 H) ; 2,02 (s ; 3 H) ; 2,08 (s ; 3 H) ;
4,07-4,26 (m ; 3 H ; H-5 ; H-7) ; 5,13 (dd ; 1 H ; J = 3,3 Hz ; J = 10,7 Hz; H-2) ; 5,19 (d ; 1 H
; J = 3,3 Hz ; H-1) ; 5,33 (ls ; 1 H ; H-4) ; 5,41 (dd ; 1 H ; J = 3,3 Hz ; J = 10,7 Hz ; H-3).
RMN 13C (CDCl3, 75 MHz):
δ (ppm)= 15,6 (C-6) ; 20,4 ; 20,5 ; 20,5 (3 CH3) ; 63,7 (C-7) ; 65,0 (C-5) ; 67,6-67,8 (C-2
; C-C-3) ; 71,0 (C-4) ; 96,0 (C-1) ; 125,2 ; 128,1 ; 128,1 ; 128,9 (4 CO).
SM (Basse Résolution-ESI) m/z pour (M+Na)+ = 371,0.
[α]D –135 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C14H20O11 .H2O : C, 45,90 ; H, 6,05 ; trouvée: C, 45,50 ; H, 5,70.
Carboxyméthyl 2-acétamido-3,4,6-tri-O-acétyl-2-déoxy-α-D-glucopyranoside (137a)
OAcO
AcHN
OAc
O OH
OAcO
Solide blanc.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,02 (s ; 3H) ; 2,03 (s ; 6 H) ; 2,10 (s ; 3 H); 4,03-4,13 (m ; 2 H ; H-6b ; H-5) ;
4,19-4,40 (m ; 4 H ; H-2 ; H-7 ; H-6a) ; 4,90 (d ; 1 H ; J = 3,6 Hz ; H-1) ; 5,14 (t ; 1 H ; J =
10,3 Hz ; H-4) ; 5,27 (t ; 1 H ; J = 9,8 Hz ; H-3) ; 6,59 (d ; 1 H ; J = 9,2 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,5 ; 20,7 ; 20,7 ; 22,7 (4 CH3) ; 51,7 (C-2) ; 61,8 (C-6) ; 64,6 (C-7) ; 67,8 (C-
4) ; 68,4 (C-5) ; 70,9 (C-3) ; 97,9 (C-1) ; 169,4 ; 170,8 ; 171,2 ; 171,8 ; 172,2 (5 CO).
Tf: 96°C.
[α]D +73 (c = 1,0 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z calculée pour (M+Na)+ = 428,1169, trouvée 428,1167.
Anal. Calc. pour C16H23O11N.0,5H2O: C, 46,38 ; H, 5,84 ; H, 3,38 ; trouvée: C, 46,39 ; H,
5,61 ; N, 3,30.
160

Partie Expérimentale
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-
acétyl-α-D-glucopyranoside (138a)
Huile jaune.
RMN 1H (300 MHz, CDCl3) :
δ (ppm) 1,97 (s ; 3 H) ; 2,05 (s ; 3 H) ; 2,07 (s ; 6 H) ; 2,09 (s ; 3 H) ; 2,14 (s ; 3 H) ; 2,17
(s ; 3 H) ; 3,78 (t ; 1 H ; J = 9,9 Hz ; H-4) ; 3,89 (t ; 1 H ; J = 7,0 Hz ; H-5’) ; 4,02-4,20 (m ; 4
H ; ; H-5 ; H-6’; H-6b) ; 4,26 (s ; 2 H ; H-7) ; 4.46 (dd ; 1 H ; J =1,7 Hz ; J = 11,5 Hz ; H-6a) ;
4,50 (d ; 1 H ; J =7,9 Hz ; H-1’) ; 4,82 (dd ; 1 H ; J = 3,7 Hz ; J = 9,9 Hz ; H-2) ; 4,97 (dd ; 1
H ; J = 3,4 Hz ; J = 10,4 Hz ; H-3’) ; 5,11 (dd ; 1 H ; J = 7,9 Hz ; J = 10,4 Hz ; H-2’) ; 5,12 (d
; 1 H ; J = 3,7 Hz ; H-1) ; 5,36 (dd ; 1H ; J = 0,9 Hz ; J = 3,4 Hz , H-4’) ; 5,51 (t ; 1 H ; J =9,9
Hz ; H-3).
RMN 13C (75 MHz, CDCl3) :
δ (ppm) 20,9 ; 21,0 ; 21,0 ; 21,0 ; 21,2 ; 21,3 ; 21,8 (7 CH3) ; 61,3 (C-6’) ; 62,2 (C-6) ;
64,3 (C-7) ; 67,1 (C-4’) ; 69,3 (C-5) ; 69,6 (C-2’) ; 69,8 (C-3) ; 71,0 (C-5’) ; 71,1 (C-2) ; 71,4
(C-3’) ; 76,4 (C-4) ; 96,1 (C-1) ; 101,2 (C-1’) ; 169,8 ; 170,5 ; 170,8 ; 170,9 ; 171,1 ; 171,1 ;
171,3 ; 173,6 (8 CO).
SM (Haute Résolution-ESI) m/z calculée pour (M+Na)+ = 717,1854 ; trouvée 717,1858.
[α]D +57 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C28H38O20. H2O: C, 47,19 ; H, 5,66 ; trouvée: C, 47,47 ; H, 5,55.
161

Partie Expérimentale
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-
acétyl-β-D-glucopyranoside (138b)
OO
OAcOH
OOAc
OAcO
OAc
OAcAcO
AcOO
Huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,90 (s ; 3 H) ; 1,97 (s ; 3 H) ; 1,99 (s ; 9 H) ; 2,06 (s ; 3 H) ; 2,08 (s ; 3 H) ;
3,59 (m ; 1 H ; H-5) ; 3,76 (t ; 1 H ; J = 9,3 Hz ; H-4) ; 3,84 (t ; 1 H ; J = 6,7 Hz ; H-5’) ; 3,98-
4,07 (m ; 3 H ; H-6’ ; H-6a) ; 4,22 (s ; 2 H ; H-7) ; 4,41-4,45 (m ; 2 H ; H-1’ ; H-6b) ; 4,56 (d ;
1 H ; J = 7,7 Hz ; H-1) ; 4,86-4,93 (m ; 2 H ; H-2 ; H-3’) ; 5,02 (dd ; 1 H ; J = 7,8 Hz ; J =
10,1 Hz ; H-2’) ; 5,16 (t ; 1 H ; J = 9,2 Hz ; H-3) ; 5,28 (d ; 1 H ; J = 2,8 Hz ; H-4’).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,5 ; 20,6 ; 20,6 ; 20,6 ; 20,7 ; 20,8 ; 20,8 (7 CH3) ; 60,9 (C-6’) ; 61,8 (C-6) ;
65,3 (C-7) ; 66,7 (C-4’) ; 69,2 (C-5) ; 70,7 (C-5’) ; 71,0-71,3 (C-2 ; C-3’) ; 72,4 (C-3) ; 72,9
(C-5) ; 76,0 (C-4) ; 100,1 (C-1) ; 101,0 (C-1’) ; 169,3 ; 169,8 ; 170,0 ; 170,1 ; 170,2 ; 170,3 ;
170,6 ; 170,7 (8 CO).
SM (Haute Résolution-ESI) m/z calculée pour (M+Na)+ = 717,1854, trouvée 717,1853.
[α]D -6 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C28H38O20. H2O: C, 47,19 ; H, 5,66 ; trouvée: C, 47,52 ; H, 5,55.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl-
α-D-glucopyranoside (139a)
O
OAcOH
O
OAcO
AcOOAc
OAc
AcOOAcO
O
Solide blanc.
RMN 1H (300 MHz, CDCl3) :
162

Partie Expérimentale
δ (ppm)= 1,92 (s ; 3 H) ; 1,94 (s ; 3 H) ; 1,96 (s ; 6 H) ; 2,02 (s ; 3 H) ; 2,02 (s ; 3 H) ;
2,07 (s ; 3 H) ; 3,61 (m ; 1 H ; H-5’) ; 3,68 (t ; 1 H ; J = 9,6 Hz ; H-4) ; 3,97-4,01 (m ; 2 H ; H-
5 ; H-6’a) ; 4,06 (dd ; 1 H ; J = 4,5 Hz ; J = 12,3 Hz ; H-6b) ; 4,18 (s ; 2 H ; H-7) ; 4,42-4,49
(m ; 2 H ; H-1’ ; H-6b) ; 4,74 (dd ; 1 H ; J = 3,7 Hz ; J = 10,3 Hz ; H-2) ; 4,86 (t ; 1 H ; J = 8,5
Hz ; H-4’) ; 4,97-5,11 (m ; 3 H ; H-1 ; H-2’ ; H-3’) ; 5,42 (t ; 1 H ; J = 9,8 Hz ; H-3).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,5 ; 20,5 ; 20,5 ; 20,6 ; 20,6 ; 20,8 ; 21,4 (7 CH3) ; 61,6-61,6 (C-6 ; C-6’) ;
64,3 (C-7) ; 67,8 (C-2’) ; 68,9 (C-5) ; 69,1 (C-3) ; 70,6 (C-2) ; 71,7 (C-5’) ; 71,9 (C-4’) ; 73,0
(C-3’) ; 76,4 (C-4) ; 95,6 (C-1) ; 100,6 (C-1’) ; 169,3 ; 169,5 ; 170,0 ; 170,5 ; 170,7 ; 170,8 ;
170,8 ; 178,3 (8 CO).
Tf: 165-167°C.
[α]D +50 (c = 0,5 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z calculée pour C28H38O20 Na (M+Na)+ = 717,1854 ;
trouvée 717,1850.
Anal. Calc. pour C28H38O20. 1,5 H2O: C, 46,60 ; H, 5,73 ; trouvée: C, 46,68 ; H, 5,51.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl
-α-D-glucopyranoside (140a)
O
OAc
OH
O
OAcO
AcOOAc
OAc
AcO
AcO
O
O
Huile jaune.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,97 (s ; 3 H) ; 1,99 (s ; 3 H) ;
2,03 (s ; 3 H) ; 2,08 (s ; 3 H) ; 3,88 (m ; 1 H ; H-5’) ; 3,93 (t ; 1 H ; J = 9,7 Hz ; H-4) ; 3,98
(dd ; 1 H ; J = 1,9 Hz ; J = 12,3 Hz ; H-6’a) ; 4,00-4,10 (m ; 1 H ; H-5) ; 4,15-4,22 (m ; 5 H ;
H-5’; H-6b ; H-6’b ; H-7) ; 4,39 (dd ; 1 H ; J = 2,2 Hz ; J = 12,3 Hz ; H-6a) ; 4,70 (dd ; 1 H ; J
= 3,8 Hz ; J = 9,7 Hz ; H-2) ; 4,79 (dd ; 1 H ; J = 3,8 Hz ; J = 10,1 Hz ; H-2’) ; 5,00 (t ; 1 H ;
163

Partie Expérimentale
J = 10,1 Hz ; H-4’) ; 5,03 (d ; 1 H ; J = 3,8 Hz ; H-1) ; 5,30 (t ; 1 H ; J = 10,4 Hz ; H-3’) ; 5,36
(d ; 1 H ; J = 3,8 Hz ; H-1’) ; 5,51 (t ; 1 H ; J = 9,7 Hz ; H-3).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,6 ; 20,6 ; 20,6 ; 20,7 ; 20,7 ; 20,8 ; 20,9 (7 CH3) ; 67,2 (C-6’) ; 68,0 (C-6) ;
68,0 (C-7) ; 68,4 (C-4’) ; 69,4 (C-5) ; 70,0 (C-5’) ; 70,0 (C-3’) ; 70,9 (C-2’) ; 72,0 (C-2) ; 72,4
(C-3) ; 74,0 (C-4) ; 93,8 (C-1’) ; 95,7 (C-1) ; 169,6 ; 170,0 ; 170,1 ; 170,6 ; 170,8 ; 170,8 ;
170,8 ; 173,1 (8 CO).
SM (Haute Résolution-ESI) m/z calculée pour C28H38O20Na (M+Na)+ = 717,1854 ;
trouvée 717,1858.
[α]D +91 (c = 0,5 ; CH2Cl2).
Anal. Calc. pour C28H38O20: C, 48,42 ; H, 5,51 ; trouvée: C, 48,37 ; H, 5,85.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl-
β-D-glucopyranoside (140b)
O
OAcO
AcOOAc
OAc
AcO
AcO
O
OAc
OOH
O
Huile jaune.
RMN 1H (500 MHz, CDCl3) :
δ (ppm)= 1,93 (s ; 3 H) ; 1,95 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,98 (s ; 3 H) ; 2,03 (s ; 3 H) ;
2,03 (s ; 3 H) ; 2,08 (s ; 3 H) ; 3,65 (m ; 1 H ; H-5) ; 3,88 (br d ; 1 H ; J = 9,8 Hz ; H-5’) ;
3,93-3,99 (m ; 2 H ; H-6’a ; H-4) ; 4,16 (dd ; 1 H ; J = 4,1 Hz ; J = 12,0 Hz ; H-6a) ; 4,18 (dd ;
1 H ; J = 3,4 Hz ; J = 12,9 Hz ; H-6’b) ; 4,24 (s ; 2 H ; H-7) ; 4,44 (br d ; 1 H ; J = 12,0 Hz ;
H-6b) ; 4,62 (d ; 1 H ; J = 7,6 Hz ; H-1) ; 4,78 (dd ; 1 H ; J = 30,6 Hz ; J = 9,8 Hz ; H-2’) ;
4,81 (dd ; 1 H ; J = 7,4 Hz ; J = 9,6 Hz ; H-2) ; 4,98 (t ; 1 H ; J = 9,8 Hz ; H-4’) ; 5,22 (t ; 1 H
; J = 9,6 Hz ; H-3) ; 5,28 (t ; 1 H ; J = 9,8 Hz ; H-3’) ; 5,33 (d ; 1 H ; J = 3,6 Hz ; H-1’).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 20,7 ; 20,7 ; 20,7 ; 20,8 ; 20,9 ; 21,0 ; 21,0 (7 CH3) ; 53,8 (C-6’) ; 61,6 (C-6) ;
62,6 (C-7) ; 68,1 (C-4) ; 68,7 (C-5’) ; 69,4 (C-3’) ; 70,1 (C-2’) ; 71,9 (C-2) ; 72,5 (C-5) ; 72,6
164

Partie Expérimentale
(C-4) ; 75,0 (C-3) ; 95,7 (C-1’) ; 99,9 (C-1) ; 169,6 ; 170,0 ; 170,1 ; 170,3 ; 170,7 ; 170,7 ;
170,7 ; 170,7 (8 CO).
[α]D +61 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C28H38O20: C, 48,42 ; H, 5,51 ; trouvée: C, 48,73 ; H, 5,87.
B.3. PROCEDURE GENERALE POUR LA PREPARATION DES CARBOXYMETHYL
GLYCOSIDE LACTONES
A une solution du carboxyméthyl glycoside (0,1 M) dans le MeOH, NaOH (1M, 10
équiv.) est rajouté. Le mélange est agité pendant 30 minutes à température ambiante et les
solvants sont évaporés. Le résidu est placé dans les conditions classiques d’acétylation (50%
vol anhydride acétique dans la pyridine) pendant une nuit. Après évaporation du solvant, le
résidu est dissous dans CH2Cl2 est lavé avec une solution aqueuse de NH4Cl solution (10%).
La phase organique est séchée sur Na2SO4, filtrée et le solvant est évaporé pour donner, après
purification sur gel de silice, la lactone correspondante.
Carboxyméthyl 3,4,6-tri-O-acétyl-β-D-glucopyranoside-2-O-lactone (1b)
OAcOAcO
AcO
O
OO
71%. (AcOEt/Pentane : 1/1). Mousse blanche.
RMN 1H (300 MHz, CDCl3):
δ (ppm) 2,05 (s ; 3 H) ; 2,09 (s ; 3 H) ; 2,09 (s ; 3 H) ; 3,91 (ddd ; 1 H ; J = 2,3 Hz ; J =
4,5 Hz ; J = 10,2 Hz ; H-5) ; 4,12 (dd ; 1 H ; J = 2,3 Hz ; J = 12,5 Hz ; H-6a) ; 4,21 (dd ; 1 H ;
J = 4,7 Hz ; J = 12,5 Hz ; H-6b) ; 4,30 (dd ; 1 H ; J = 7,9 Hz ; J = 10,2 Hz ; H-2) ; 4,50
(système AB ; δa 4,52 , δb 4,69 ; 2 H ; J = 17,4 Hz ; H-7) ; 4,75 (d ; 1 H ; J = 7,9 Hz ; H-1) ;
5,05 (dd ; 1 H ; J = 9,1 Hz ; J = 10,2 Hz ; H-4) ; 5,30 (dd ; 1 H ; J = 9,1 Hz ; J = 10,2 Hz ; H-
3).
RMN 13C (CDCl3, 75 MHz):
165

Partie Expérimentale
δ (ppm) 20,6 ; 20,7 ; 20,8 (3 CH3) ; 61,6 (C-6) ; 64,5 (C-7) ; 68,2 ; 71,2 ; 73,8 ; 76,5 (C-2
; C-3 ; C-4 ;C-5) ; 95,0 (C-1) ; 164,9 ; 169,6 ; 169,9 ; 170,6 (4 CO).
[α]D +93 (c = 1,0 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z calculée pour (M+H)+ = 347,0978 trouvée 347,0979.
Anal. Calc. pour C14H18O10: C, 48,54 ; H, 5,24 ; trouvée: C, 48,46 ; H, 5,41.
Carboxyméthyl 3,4,6-tri-O-acétyl-β-D-mannopyranoside-2-O-lactone (141b)
OAcOAcO
AcO
OO
O
74%. (AcOEt/Pentane : 1/1). Mousse blanche.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,99 (s ; 3 H) ; 2,01 (s ; 3 H) ; 2,05 (s ; 3 H) ; 3,73 (m ; 1 H ; H-5) ; 4,14 (m ; 2
H ; H-6) ; 4,38 (système AB ; δa 4,29 , δb 4,46 ; 2 H ; J = 17,5 Hz ; H-7) ; 4,85 (dd ; 1 H ; J =
1,1 Hz ; J = 3,0 Hz ; H-2) ; 5,03 (dd ; 1 H ; J = 3,0 Hz ; J = 10,1 Hz ; H-3) ; 5,03 (d ; 1 H ; J =
1,1 Hz ; H-1) ; 5,27 (t ; 1 H ; J = 10,1 Hz ; H-4).
RMN 13C (CDCl3, 75 MHz):
δ (ppm)= 20,6 ; 20,6 ; 20,7 (3 CH3) ; 60,3 (C-7) ; 61,9 (C-6) ; 64,7 (C-4) ; 70,7 (C-3) ;
73,0 (C-5) ; 75,0 (C-2) ; 90,4 (C-1) ; 165,0 ; 169,3 ; 170,4 ; 170,7 (4 CO).
SM (Basse Résolution-ESI) m/z (M+H)+ = 346,88.
[α]D -55 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C14H18O10.0,2 H2O: C, 48,06; H, 5,30 ; trouvée: C, 48,03 ; H, 5,03.
166

Partie Expérimentale
Carboxyméthyl-3,4,6-tri-O-acétyl-α-D-galactopyranoside-2-O-lactone (142a)
OAcO
O
OAcAcO
O
O
87 %. (AcOEt/Pentane : 1/1). Mousse blanche.
RMN 1H (300 MHz) :
δ (ppm)= 2,055 (s ; 3 H), 2,06 (s ; 3 H), 2,15 (s ; 3 H), (3 OAc) ; 4,09-4,15 (m ; 2 H ;
H-6) ; 4,45 (m, ; 1 H ; H-5) ; 4,59 (système AB ; 2 H ; δa 4,52, δb 4,67 ; J = 18,0 Hz ; H-7) ;
4,65 (dd ; 1 H ; J = 3,0 Hz ; J = 3,3 Hz ; H-2) ; 5,39 (d ; 1 H ; J = 3,0 Hz ; H-1) ; 5,43 (dd ; 1 H
; J = 3,3 Hz ; J = 9,9 Hz ; H-3) ; 5,48 (dd ; 1 H, J = 3,3 Hz ; J = 1,2 Hz ; H-4).
RMN 13C (75 MHz) :
δ (ppm)= 20,8 ; 20,9 ; 21,1 (3 CH3) ; 61,7 (C-6) ; 64,9 (C-7) ; 67,3 (C-4) ; 70,5 (C-5) ;
71,9 (C-3) ; 76,4 (C-2) ; 91,5 (C-1) ; 164,2 ; 170,2 ; 170,5 ; 170,8 (4 CO).
SM (Basse Résolution-ESI) m/z pour C15H18O11 (M+H)+ = 346,09.
[α]D +113 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C14H18O10: C, 48,56; H, 5,24 ; trouvée: C, 48,64; H, 5,49.
Carboxyméthyl 3,4,6-tri-O-acétyl-β-D-galactopyranoside-2-O-lactone (142b)
OAcO
O
O
OAcAcO
O
64 %. (AcOEt/Pentane : 1/1). Solide blanc.
RMN 1H (500 MHz) :
δ (ppm) = 2,04 (s ; 6 H) ; 2,14 (s ; 3 H), 4,11 (m ; 1 H ; H-5) ; 4,16-4,20 (m ; 2 H ; H-6) ;
4,53 (dd ; 1 H ; J = 7,6 Hz ; J = 10,5 Hz, H-2) ; 4,60 (système AB ; 2 H ; δa 4,49 ; δb 4,70 ; J
= 17,7 Hz ; H-7) ; 4,77 (d ; 1 H ; J = 7,6 Hz ; H-1) ; 5,16 (dd ; 1 H ; J = 3,5 Hz ; J = 10,5 Hz ;
H-3) ; 5,46 (dd ; 1 H ; J = 1,9 Hz ; J = 3,5 Hz ; H-4).
167

Partie Expérimentale
RMN 13C (125 MHz)
δ (ppm) = 20,4 ; 20,5 ; 20,6 (3 OAc) ; 61,0 (C-6) ; 64,5 (C-7) ; 66,8 (C-4) ; 69,5 (C-3) ;
72,8 (C-5) ; 74,6 (C-2) ; 95,5 (C-1) ; 165,2 ; 169,7 ; 170,3 ; 170,3 (4 CO).
[α]D +85 (c = 1,0 ; CH2Cl2).
SM: Haute résolution (ESI) m/z calculée pour C14H18O10Na (M+Na)+ 369,0798, trouvée
369,0797.
Anal. Calc. pour C14H18O10: C, 48,56 ; H, 5,24 ; trouvée: C, 48.64; H, 5.49.
Carboxyméthyl 3,4-di-O-acétyl-α-L-fucopyranoside-2-O-lactone (143a)
O
OAcOAc
O
OO
84%. (AcOEt/Pentane : 1/2). Solide blanc.
RMN 1H (500 MHz, CDCl3):
δ (ppm)= 1,12 (d ; 3 H ; J = 6,6 Hz ; H-6) ; 2,00 (s ; 3 H) ; 2,11 (s ; 3 H) ; 4,32 (br q ; 1 H
; J = 6,6 Hz ; H-5) ; 4,50 (système AB: δa 4,43 , δb 4,58 ; 2 H ; J = 17,8 Hz ; H-7) ; 4,55 (dd ;
1 H ; J = 3,1 Hz ; J = 8,5 Hz ; H-2) ; 5,25 (d ; 1 H ; J = 3,1 Hz ; H-1) ; 5,27 (dd ; 1 H ; J = 3,1
Hz ; J = 10,2 Hz ; H-4) ; 5,38 (dd ; 1 H ; J = 3,1 Hz ; J = 10,2 Hz ; H-3).
RMN 13C (CDCl3, 125 MHz):
δ (ppm)= 15,9 (C-6) ; 20,7 ; 20,8 (2 CH3) ; 64,8 (C-7) ; 67,6 (C-5) ; 70,0 (C-3) ; 70,7 (C-
4) ; 73,6 (C-2) ; 92,0 (C-1) ; 164,2 ; 170,2 ; 170,3 (3 CO).
Tf: 156-158°C.
[α]D –124 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C18H28O10 : C, 50,0 ; H, 5,59 ; trouvée: C, 49,81 ; H, 5,40.
168

Partie Expérimentale
Carboxyméthyl 3,4,6-tri-O-acétyl-β-L-fucopyranoside-2-O-lactone (143b)
O
AcOOAc
OO
O
60 %. (AcOEt/Pentane : 1/2). Solide blanc.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,22 (d ; 3 H ; J = 6,5 Hz ; H-6) ; 1,99 (s ; 3 H) ; 3,98 (s ; 3 H) ; 3,98 (dq ; 1 H ;
J = 1,2 Hz ; J = 6,5 Hz ; H-5) ; 4,44 (dd ; 1 H ; J = 7,7 Hz ; J = 10,7 Hz ; H-2) ; 4,55 (système
AB: δa 4,47 , δb 4,63 ; 2 H ; J = 17,5 Hz ; H-7) ; 4,73 (d ; 1 H ; J = 7,7 Hz ; H-1) ; 5,10 (dd ; 1
H ; J = 3,6 Hz ; J = 10,6 Hz ; H-3) ; 5,23 (dd ; 1 H ; J = 1,2 Hz ; J = 3,6 Hz ; H-4).
RMN 13C (CDCl3, 75 MHz):
δ (ppm)= 15,7 (C-6) ; 20,4 ; 20,5 (2 CH3) ; 64,5 (C-7) ; 69,8-69,9 (C-4 ; C-3) ; 71,3 (C-5)
; 74,8 (C-2) ; 95,2 (C-1) ; 165,6 ; 169,8 ; 170,1 (3 CO).
[α]D –88 (c = 1,0 ; CH2Cl2).
Tf.: 95-96°C.
SM (Haute Résolution-ESI) m/z calculée pour (M+Na)+ = 311,0743, trouvée 311,0745.
Anal. Calc. pour C12H16O8 .1,71 H2O: C, 45,16 ; H, 6,14 ; trouvée: C, 45,21 ; H, 6,00.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-
acétyl-α-D-glucopyranoside-2-O-lactone (144a)
OO
AcO
AcOOAc
OAc
OAcO
O
OAc
O
O
64%. (AcOEt/Pentane : 1/1). Solide blanc.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,97 (s ; 3 H) ; 2,04 (s ; 3 H) ; 2,07 (s ; 3 H) ; 2,13 (s ; 3 H) ; 2,14 (s ; 3 H) ;
2,17 (s ; 3 H) ; 3,77 (t ; 1 H ; J = 9,6 Hz ; H-4) ; 3,89 (t ; 1 H ; J = 6,2 Hz ; H-5’) ; 4,04-4,22
(m ; 4 H ; ; H-5 ; H-6’ ; H-6b) ; 4,35 (d ; 1 H ; J =3,4 Hz ; J = 9,6 Hz ; H-2) ; 4,47-4,52 (m ; 2
169

Partie Expérimentale
H ; H-1’ ; H-6a) ; 4,52 (système AB: δa 4,69 ; δb 4,49 ; J = 17,8 Hz ; H-7) ; 4,96 (dd ; 1 H J =
3,4 Hz ; J = 10,3 Hz ; H-3’) ; 5,12 (dd ; 1 H, J = 7,8 Hz ; J = 10,3 Hz ; H-2’) ; 5,29 (d ; 1 H ; J
= 3,4 Hz ; H-1) ; 5,36 (dd ; 1 H ; J = 1,1 Hz J = 3,4 Hz ; H-4’) ; 5,56 (t ; 1 H ; J = 9,6 Hz ; H-
3).
RMN 13C (125 MHz, CDCl3) :
δ (ppm)= 20,5 ; 20,6 ; 20,7 ; 20,7 ; 20,8 ; 20,9 (6 CH3) ; 60,8 (C-6’) ; 61,5 (C-6) ; 64,4
(C-7) ; 66,6 (C-4’) ; 69,2 (C-2’) ; 70,7 (C-5’) ; 70,8 (C-5) ; 70,9 (C-3’) ; 71,2 (C-3) ; 74,8 (C-
4) ; 74,8 (C-2) ; 90,9 (C-1) ; 100,9 (C-1’) ; 162,9 ; 163,7 ; 169,0 ; 169,7 ; 170,1 ; 170,2 ; 170,4
(7 CO).
SM (Haute Résolution-ESI) m/z calculée pour C26H34O18 Na+ (M+Na)+ = 657,1643,
trouvée 657,1645.
[α]D +68 (c = 1,0 ; CH2Cl2).
Tf: 106-114°C.
Anal. Calc. pour C26H34O18 .H2O: C, 48,75 ; H, 5,46 ; trouvée: C, 48,77 ; H, 5,47.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-
acétyl-β-D-glucopyranoside-2-O-lactone (144b)
OAcO
OAc
OAc
OAcO
O
OAc
O O
O
AcO
60%. (AcOEt/Pentane : 1/1). Mousse blanche.
RMN 1H (500 MHz, CDCl3):
δ (ppm)= 1,90 (s ; 3 H) ; 1,98 (s ; 3 H) ; 2,00 (s ; 3 H) ; 2,06 (s ; 6 H) ; 2,09 (s ; 3 H) ;
3,75-3,76 (m ; 2 H ; H-5, H-4) ; 3,82 (t ; 1 H ; J = 7,2 Hz ; H-5’) ; 4,01 (dd ; 1 H ; J = 7,2 Hz ;
J = 11,0 Hz ; H-6’a) ; 4,08-4,11 (m ; 2 H ; H-6a ; H-6’b) ; 4,12 (dd ; 1 H ; J = 7,7 Hz ; J =
10,4 Hz ; H-2) ; 4,41-4,45 (m ; 2 H ; H-1’ ; H-6b) ; 4,51 (système AB: δa 4,49 ; δb 4,58 ; J =
17,5 Hz ; H-7) ; 4,72 (d ; 1 H ; J = 7,7 Hz ; H-1) ; 4,90 (dd ; 1 H ; J = 3,5 Hz ; J = 10,4 Hz ;
H-3’) ; 5,03 (dd ; 1 H ; J = 7,9 Hz ; J = 10,4 Hz ; H-2’) ; 5,27-5,29 (m ; 2 H ; H-3 ; H-4’).
RMN 13C (125 MHz, CDCl3) :
170

Partie Expérimentale
δ (ppm)= 20,6 ; 20,7 ; 20,7 ; 20,7 ; 20,9 ; 20,9 (6 CH3) ; 60,9 (C-6’) ; 61,7 (C-6) ; 64,4
(C-7) ; 66,6 (C-3 ou C-4’) ; 69,1 (C-2’) ; 70,7 (C-3 ou C-4’) ; 70,9 (C-3’) ; 71,0 (C-5’) ; 74,8
(C-4) ; 76,0 (C-5) ; 76,9 (C-2) ; 94,8 (C-1) ; 101,1 (C-1’) ; 164,9 ; 169,1 ; 169,3 ; 170,1 ;
170,2 ; 170,3 ; 170,4 (7 CO).
SM (Basse Résolution-ESI) m/z pour C26H3’O18 Na (M+Na)+ = 675,1.
[α]D +5 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C26H3’O18: C, 49,21 ; H, 5,40 ; trouvée: C, 48,92 ; H, 5,48.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-β-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl-
α-D-glucopyranoside-2-O-lactone (145a)
O
O
OAcO
AcOOAc
OAc
AcOOAcO
O
O
73 %. (AcOEt/Pentane : 2/1). Solide blanc.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,92 (s ; 3 H) ; 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 2,02 (s ; 3 H) ; 2,04 (s ; 3 H) ;
2,07 (s ; 3 H) ; 3,62 (t ; 1 H ; J = 9,6 Hz ; H-4) ; 4,00 (dd ; 1 H ; J = 1,9 Hz ; J = 12,5 Hz ; H-
6’a) ; 4,01- 4,12 (m ; 2 H ; H-5 ; H-6a) ; 4,28 (dd ; 1 H ; J = 3,0 Hz ; J = 9,6 Hz ; H-2) ; 4,32
(dd ; 1 H ; J = 4,1 Hz ; J = 12,6 Hz ; H-6’b) ; 4,39-4,47 (m ; 2 H ; H-1’ ; H-6b) ; 4,53
(système AB: δa 4,42 ; δb 4,62 ; J = 17,8 Hz ; H-7) ; 4,87 (t ; 1 H ; J = 8,8 Hz ; H-2’) ; 5,01 (t ;
1 H ; J = 8,8 Hz ; H-4’) ; 5,07 (t ; 1 H ; J = 8,8 Hz ; H-3’) ; 5,23 (d ; 1 H ; J = 3,0 Hz ; H-1) ;
5,47 (t ; 1 H ; J = 9,6 Hz ; H-3).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,6 ; 20,6 ; 20,6 ; 20,7 ; 20,7 ; 20,9 (6 CH3) ; 61,4-61,6 (C-6 ; C-6’) ; 64,5 (C-
7) ; 67,8 (C-4’) ; 70,9 (C-5) ; 71,1 (C-3) ; 71,5 (C-5’) ; 72,0 (C-2) ; 72,8 (C-3’) ; 75,2 (C-4) ;
76,2 (C-2) ; 91,0 (C-1) ; 100,7 (C-1’) ; 163,6 ; 169,0 ; 169,3 ; 169,7 ; 170,3 ; 170,3 ; 170,5 (7
CO).
[α]D +45 (c = 1,0 ; CH2Cl2).
Tf: 188°C.
171

Partie Expérimentale
SM (Basse Résolution-ESI) m/z pour C26H34O18 Na (M+Na)+ = 657,1.
Anal. Calc. pour C26H34O18 .0,5 H2O: C, 48,52 ; H, 5,48 trouvée: C, 48,53 ; H, 5,38.
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl -
α-D-glucopyranoside-2-O- lactone (146a)
O
OAcO
AcOOAc
OAc
AcO
AcO
O
OO
O
80%. (AcOEt/Pentane : 1/1). Mousse blanche.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 2,01 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,06 (s ; 3 H) ; 2,10 (s ; 3 H) ; 2,12 (s ; 3 H) ;
2,15 (s ; 3 H) ; 3,94 (m ; 1 H ; H-5) ; 3,99 (t ; 1 H ; J = 9,2 Hz ; H-4) ; 4,07 (dd ; 1 H ; J = 2,3
Hz ; J = 12,4 Hz ; H-6’a) ; 4,20-4,28 (m ; 4 H ; H-6’b ; H-6a ; H-5 ; H-2) ; 4,48-4,74 (m ; 3 H
; H-6b et système AB: δa 4,51 ; δb 4,71 ; J = 17,9 Hz ; H-7) ; 4,85 (dd ; 1 H ; J = 3,9 Hz ; J =
10,3 Hz ; H-2’) ; 5,08 (t ; 1 H ; J = 9,9 Hz ; H-4’) ; 5,30 (d ; 1 H ; J = 2,8 Hz ; H-1) ; 5,36 (t ;
1 H ; J = 10,1 Hz ; H-3’) ; 5,48 (d ; 1 H ; J = 3,9 Hz ; H-1’) ; 5,64 (t ; 1 H ; J = 9,4 Hz ; H-3).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,6 ; 20,6 ; 20,7 ; 20,7 ; 20,9 ; 21,0 (6 CH3) ; 61,4 (C-6’) ; 62,2 (C-6) ; 64,3
(C-7) ; 67,9 (C-4’) ; 68,7 (C-5’) ; 69,3 (C-3’) ; 70,1 (C-2’) ; 70,6 (C-2) ; 71,2 (C-4) ; 73,4 (C-
3) ; 76,4 (C-5) ; 90,8 (C-1) ; 95,7 (C-1’) ; 163,4 ; 169,5 ; 170,0 ; 170,0 ; 170,4 ; 170,6 ; 170,7
(7 CO).
[α]D +109 (c = 1,0 ; CH2Cl2).
SM (Haute Résolution-ESI) m/z calculée pour C26H34O18Na (M+Na)+ = 657,1643 ;
trouvée 657,1640.
Anal. Calc. pour C26H34O18 : C, 49,21 ; H, 5,40 ; trouvée: C, 49,40 ; H, 5,43.
172

Partie Expérimentale
Carboxyméthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-2,3,6-tri-O-acétyl-
β-D-glucopyranoside-2-O-lactone (146b)
OAcO
OAc
OAc
AcO
O OAcO
O
OAc
O
O
64 %. (AcOEt/Pentane : 1/1). Huile incolore.
RMN 1H (500 MHz, CDCl3):
δ (ppm)= 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 2,00 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,04 (s ; 3 H) ;
2,08 (s ; 3 H) ; 3,84 (m ; 1 H ; H-5) ; 3,88 (ddd ; 1 H ; J = 2,5 Hz ; J = 6,0 Hz ; J = 10,4 Hz ;
H-5’) ; 3,98 (t ; 1 H ; J = 9,0 Hz ; H-4) ; 4,00 (dd ; 1 H ; J = 2,5 Hz ; J = 12,3 Hz ; H-6’a) ;
4,05 (dd ; 1 H ; J = 7,9 Hz ; J = 10,4 Hz ; H-2) ; 4,16 (dd ; 1 H ; J = 3,6 Hz ; J = 12,3 Hz ; H-
6a) ; 4,17 (dd ; 1 H ; J = 6,0 Hz ; J = 12,3 Hz ; H-6’b), 4,44 (dd ; 1 H ; J = 2,2 Hz ; J = 12,3
Hz ; H-6b) ; 4,49 (système AB ; δa 4,42 ; δb 4,57 ; 2 H ; J = 17,5 Hz ; H-7) ; 4,75 (d ; 1 H ; J =
7,9 Hz ; H-1) ; 4,81 (dd ; 1 H ; J = 4,2 Hz ; J = 10,3 Hz ; H-2’) ; 5,00 (t ; 1 H ; J = 10,3 Hz ;
H-4’), 5,29 (t ; 1 H ; J = 10,3 Hz ; H-3’), 5,35 (d ; 1 H ; J = 4,2 Hz ; H-1’), 5,36 (dd ; 1 H ; J =
9,0 Hz ; J = 10,4 Hz ; H-3).
RMN 13C (125 MHz, CDCl3):
δ (ppm)= 20,7 ; 20,7 ; 20,7 ; 20,8 ; 20,9 ; 21,0 (6 CH3) ; 61,5 (C-6’) ; 62,6 (C-6) ; 64,4
(C-7) ; 68,0 (C-4’) ; 68,9 (C-5’) ; 69,3 (C-3’) ; 69,4 (C-2’) ; 69,9 (C-4) ; 72,5 (C-3) ; 73,3 (C-
5) ; 74,4 (C-2) ; 94,6 (C-1) ; 95,9 (C-1’) ; 169,5 ; 169,5 ; 169,6 ; 170,0 ; 170,5 ; 170,6 ; 170,7
(7 CO).
[α]D +54 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C26H34O18 0,5 H2O: C, 48,52 ; H, 5,48 ; trouvée: C, 48,38 ; H, 5,19.
173

Partie Expérimentale
(N-Allylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-α-D-glucopyranoside (163)
OAcO
HOO
O
OAc
NH
AcO
A une solution de la lactone 1a (4 g, 11,5 mmol) dans du CH2Cl2 fraîchement distillé (8
mL), l’allylamine (1,30 mL, 0,017 mmol) est rajoutée. La réaction est agitée pendant 12
heures puis, le solvant est évaporé et le produit est purifié sur colonne (CH2Cl2:CH3OH ;
35:1) pour donner le produit (163) pur (4,29 g, 92%) sous la forme d’une huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,99 (s ; 3 H) ; 2,02 (s ; 3 H) ; 2,03 (s ; 3 H) (3 OAc) ; 3,78 (dd ; 1 H ; J = 3,7
Hz ; J = 10,1 Hz ; H-2) ; 3,91 (s ; 2 H ; NHCH2 ) ; 4,00-4,25 (m ; 5 H ; H-5 ; H-6 ; H-7) ; 4,90
(d ; 1 H ; J = 3,7 Hz ; H-1) ; 4,97 (t ; 1 H ; J = 9,8 Hz ; H-4) ; 5,07-5,11 (m ; 2 H ; CH=CH2 ) ;
5,23 (t ; 1 H ; J = 9,6 Hz ; H-3) ; 5,72-5,85 (m ; 1 H ; CH=CH2 ) ; 7,55 (t ; 1 H ; J = 5,6 Hz ;
NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,7 ; 20,8 ; 21,0 (3 CH3) ; 41,5 (NHCH2) ; 61,9 (C-6) ; 67,4 (C-7) ; 68,0 (C-5)
; 68,1 (C-4) ; 70,3 (C-2) ; 73,3 (C-3) ; 99,3 (C-1) ; 116,4 (CH=CH2) ; 133,7 (CH=CH2) ; 169,2
; 169,4 ; 169,4 ; 170,7 (4 CO).
[α]D +118 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C17H25NO10: C, 50,62 ; H, 6,52 ; N, 3,47 ; trouvée: C, 50,28 ; H, 6,31;
N, 3,37.
174

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-
3,6-di-O-acétyl-α-D-glucopyranoside (165)
OAcO
OHO
O
OAc
NH
O
OAcO
AcO
OAc
AcO
Une solution de (146a, 500 mg, 0,78 mmol) et de propargylamine (1,18 mmol, 0,08 mL)
dans du dichlorométhane (3 mL), est agitée à température ambiante pendant 8 heures. Après
évaporation du solvant, le résidu est purifié sur colonne de silice (CH2Cl2/MeOH: 30/1) pour
donner le produit 165 (462 mg, 85%) sous la forme de mousse blanche.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,98 (s ; 3 H) ; 2,04 (s ; 3 H) ; 2,05 (s ; 3 H) ;
2,07 (s ; 3 H) (6 OAc) ; 2,22 (t ; 1 H ; J = 2,4 Hz ; CCH) ; 3,53 (m ; 2H ; H-5 ; H-4) ; 3,80-
4,20 (m ; 9 H ; H-7 ; H-2 ; H-5’ ; H-6b ; H-6’ ; NHCH2) ; 4,35 (dd ; 1 H ; J = 2,2 Hz ; J =
12,0 Hz ; H-6a) ; 4,80 (dd ; 1 H ; J = 4,0 Hz ; J = 10,2 Hz ; H-2’) ; 4,81 (d ; 1 H ; J = 3,4 Hz ;
H-1) ; 5,00 (t ; 1 H ; J = 10,2 Hz ; H-4’) ; 5,24 (t ; 1 H ; J = 10,2 Hz ; H-3’) ; 5,29 (t ; 1 H ; J =
9,6 Hz ; H-3) ; 5,41 (d ; 1 H ; J = 4,0 Hz ; H-1’) ; 7,59 (t ; 1 H ; J = 5,2 Hz ; NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,6 ; 20,6 ; 20,6 ; 20,7 ; 20,9 ; 21,3 (6 OAc) ; 28,7 (NHCH2) ; 61,5 (C-6’) ;
62,8 (C-6) ; 67,2 (C-7) ; 68,0 (C-4’) ; 68,6 (C-5’ou C-2) ; 69,4 (C-3) ; 70,2 (C-2’) ; 71,3 (C-5’
ou C-2) ; 71,7 (CCH) ; 72,4 (C-4) ; 76,1 (C-3’) ; 76,1 (CCH) ; 95,6 (C-1) ; 98,8 (C-1’) ; 168,9
; 169,5 ; 170,0 ; 170,6 ; 170,7 ; 170,7 ; 170,7 ; 172,0 (8 CO).
[α]D +107 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C29H39NO18: C, 50,51 ; H, 5,70 ; N, 2,03 ; trouvée: C, 50,61 ; H, 5,72 ;
N, 2,03.
175

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-hexadecylcarbamoyl-α-D-
glucopyranoside (167)
OAcO
OO
O
OAc
NH
AcO
NHO
( )15
A une solution de (164, 210 mg, 0,523 mmol) dans du dichlorométhane fraîchement
distillé (1 mL), l’isocyanate d’hexadécyle (0,32 mL, 1,04 mmol) est rajouté ainsi q’une
quantité catalytique de triéthylamine. Après 14 heures, la réaction est quenchée avec du
MeOH (0,5 mL), l’urée générée au cours de la réaction est filtrée sur fritté, rincée avec 10 mL
de CH2Cl2 et la solution lavée avec 2x5 mL d’une solution de NH4Cl à 10%. La phase
organique est séchée sur Na2SO4. Après évaporation du solvant, le produit est soumis à une
purification sur colonne de silice (CH2Cl2/ MeOH : 30/1) pour donner le produit 167 pur avec
un rendement de (258 mg, 74%) sous la forme d’une huile transparente.
RMN 1H (500 MHz, CDCl3) :
δ (ppm)= 0,88 (t ; 3 H ; J = 6,7 Hz, CH3) ; 1,25 (m ; 26 H ; 13 CH2) ; 1,49 (t ; 2 H ; J =
6,6 Hz ; CH2) ; 2,04 (s ; 6 H) ; 2,10 (s ; 3 H) ; (3 OAc) ; 2,27 (t ; 1 H ; J = 1,5 Hz ; CCH) ;
3,09 (m, 2 H, NHCH2CH2) ; 4,00-4,27 (m ; 4 H ; H-5 ; H-6a; NHCH2) ; 4,14 (système AB ; 2
H ; δa 4,04 ; δb 4,23 ; J = 15,7 Hz ; H-7) ; 4,25 (dd ; 1 H ; J = 4,7 Hz ; H-6b) ; 4,92 (dd ; 1 H ;
J = 3,6 , J = 10,1 Hz ; H-2) ; 4,99 (t ; 1 H ; J = 5,7 Hz ; NH) ; 5,04 (d ; 1 H ; J = 3,6 Hz ; H-1)
; 5,09 (t ; 1 H ; J = 10,1 Hz ; H-4 ) ; 5,45 (t ; 1 H ; J = 10,1 Hz ; H-3) ; 6,87 (ls ; 1 H ; NH).
RMN 13C (125 MHz, CDCl3) :
14,2 (CH3) ; 20,7 ; 20,8 ; 20,9 (3 CH3) ; 29,4-32,0 (15 CH2) ; 41,4 (NHCH2) ; 61,8 (C-6) ;
67,5 (C-7) ; 68,2 (C-4) ; 68,2 (C-5) ; 70,3 (C-3) ; 70,8 (C-2) ; 70,3 (CCH) ; 79,5 (CCH) ; 97,5
(C-1) ; 154,9 ; 168,2 ; 169,5 ; 170,7 ; 170,7 (5 CO).
[α]D +78 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C34H56N2O11: C, 61,06 ; H, 8,44 ; N, 4,19 ; trouvée: C, 60,91 ; H, 8,41
; N, 4,22.
176

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-
3,6-di-O-acétyl-2-O-hexadécylcarbamoyl-α-D-glucopyranoside (168)
OAcO
OO
O
OAc
NH
O
OAcO
AcO
OAc
AcO
NHO
C16H33
A une solution de (165, 100 mg, 0,145 mmol) dans du CH2Cl2 fraîchement distillé (2 mL)
agitée à température ambiante, l’isocyanate d’hexadécyle (0,09 mL, 0,29 mmol) est rajouté
ainsi qu’une quantité catalytique de triéthylamine. Après 48 heures, la réaction est quenchée
avec du MeOH (0,5 mL), l’urée générée au cours de la réaction est filtrée sur fritté, rincée
avec CH2Cl2 (10 mL). La solution organique est lavée avec 2x5 mL d’une solution de NH4Cl
à 10% et séchée sur Na2SO4. Après évaporation du solvant, le produit est soumis à une
purification sur colonne de silice (CH2Cl2/MeOH : 30/1) donner le carbamate 168 (125 mg,
90%) sous la forme d’une huile transparente.
RMN 1H (300 MHz, CDCl3):
δ (ppm) = 0,81 (t ; 3 H ; J = 6,0 Hz ; CH3) ; 1,20 (m ; 26 H ; 13 CH2) ; 1,40 (m ; 2 H ;
CH2) ; 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,99 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,07 (s ; 3
H) (6 OAc) ; 2,22 (t ; 1 H ; J = 2,4 Hz ; CCH) ; 3,06 (m ; 2 H ; CH2) ; 3,88-4,20 (m ; 8 H ; H-
4 ; H-5 ; H-5’ ; H-6b ; H-6’ ; NHCH2) ; 4,05 (système AB ; 2 H ; δa 3,90 ; δb 4,18 ; J = 15,8
Hz ; H-7) ; 4,38 (dd ; 1 H ; J = 2,2 Hz ; J = 12,2 Hz ; H-6a) ; 4,74 (dd ; 1 H ; J = 3,6 Hz ; J =
9,5 Hz ; H-2) ; 4,79 (dd ; 1 H ; J = 3,8 Hz ; J = 10,0 Hz ; H-2’) ; 4,86 (d ; 1 H ; J = 3,6 Hz ; H-
1) ; 4,99 (t ; 1 H ; J = 10,0 Hz ; H-4’) ; 5,06 (t ; 1 H ; J = 6,6 Hz ; NH) ; 5,29 (t ; 1 H ; J = 10,0
Hz ; H-3’) ; 5,36 (d ; 1 H ; J = 3,8 Hz ; H-1’) ; 5,41 (t ; 1 H ; J = 9,5 Hz ; H-3) ; 6,90 (t ; 1 H ;
J = 5,0 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 14,2 (CH3) ; 20,7 ; 20,7 ; 20,7 ; 20,8 ; 20,8 ; 20,9 (6 OAc) ; 21,1-32,0 (15
CH2) ; 41,4 (NHCH2) ; 61,5 (C-6’) ; 62,7 (C-6) ; 67,4 (C-7) ; 68,0 (C-5 ou C-5’ou C-4) ; 68,6
(C-5 ou C-5’ou C-4) ; 68,7 (C-4’) ; 69,4 (C-3’) ; 70,2 (C-2’) ; 71,1 (C-2) ; 71,9 (C-3) ; 72,7
177

Partie Expérimentale
(CCH) ; 72,8 (C-5 ou C-5’ou C-4) ; 79,0 (CCH) ; 95,8 (C-1) ; 97,3 (C-1’) ; 169,8 ; 170,3 ;
170,7 ; 170,8 ; 170,8 ; 170,9 ; 170,9 ; 170,3 (8 CO).
[α]D +77 (c = 0,5 ; CH2Cl2).
Anal. Calc. pour C46H72N2O19: C, 57,73 ; H, 7,58 ; N, 2,93 ; trouvée C, 57,37 ; H, 7,78 ;
N, 2,86.
(N-Propargylcarbamoyl)méthyl 2-O-propylcarbamoyl-α-D-glucopyranoside (169)
OHO
OO
O
OH
NH
HO
NHO
A une solution de (164, 200 mg, 0,498 mmol) dans du dichlorométhane fraîchement
distillé (3 mL), l’isocyanate de propyle (0,070 mL, 0,747 mmol) y sont rajoutés ainsi qu’une
quantité catalytique de triéthylamine. Après 24 heures, la réaction est quenchée avec du
méthanol (0,1 mL), lavée avec 2x5 mL d’une solution à 10% de NH4Cl et séchée sur Na2SO4.
Après évaporation du solvant, le produit est dissous dans 2 mL d’un mélange
CH3OH/H2O/NEt3 (8:1:1). Après 12 heures, les solvants sont évaporés et le produit est purifié
sur une colonne de silice (CH2Cl2/Acétone/CH3OH/H2O : 78/10/10/2) pour donner le
carbamate (169, 130 mg, 72 %) sous la forme d’un solide blanc.
RMN 1H (300 MHz, CD3OD) :
δ (ppm)= 0,94 (t ; 3 H ; J = 7,0 Hz ; CH2CH2CH3) ; 1,54 (q ; 2 H ; J = 7,0 Hz ;
CH2CH2CH3) ; 2,65 (ls, 1H, CCH) ; 3,10 (td ; 2 H ; J = 2,8 , J = 7,0 Hz ; CH2CH2CH3) ; 3,42
(t ; 1 H ; J = 9,6 Hz ; H-4) ; 3,89 (t ; 1 H ; J = 9,6 Hz ; H-3) ; 4,60-4,73 (m ; 2 H ; H-5 ; H-6a)
; 3,84 (dd ; 1 H ; J = 1,9 ; J = 11,7 Hz ; H-6b) ; 4,02-4,07 (m ; 3 H ; H-7a , NHCH2) ; 4,23 (d ;
1 H ; J = 15,4 Hz ; H-7b) ; 4,56 (dd ; 1 H ; J = 3,6 , J = 9,6 Hz ; H-2) ; 5,01 (d ; 1 H ; J = 3,6
Hz ; H-1).
RMN 13C (75 MHz, CD3OD):
178

Partie Expérimentale
δ (ppm)= 11,6 (CH3) ; 24,0 (CH2CH2CH3) ; 29,1 (NHCH2) ; 43,6 (CH2CH2CH3) ; 62,3
(C-6) ; 67,6 (C-7) ; 71,5 (C-4) ; 72,3 (CCH) ; 72,3 (C-3) ; 74,3 (C-5) ; 74,7 (C-2) ; 80,4 (-
CCH) ; 98,5 (C-1) ; 158,1 ; 171,4 (2 CO).
Tf : 120°C.
[α]D +11 (c = 0,3 ; CH3OH).
Anal. Calc. pour C15H24N2O8.0,25H2O: C, 49,38 ; H, 6,77; N, 7,68 ; trouvée: C, 49,54 ;
H, 6,87 ; N, 7,69.
(N-Propargylcarbamoyl)méthyl 2-hexadécylcarbamoyl-α-D-glucopyranoside (170)
OHO
OO
O
OH
NH
HO
NHO
C16H33
Le produit acétylé (167, 185 mg, 0,277 mmol) est dissous dans 5 mL d’un mélange
CH3OH/H2O/NEt3 (8:1:1). Après 3 heures, les solvants sont évaporés et le produit est purifié
sur une colonne de silice (CH2Cl2/Acétone/CH3OH/H2O : 78/10/10/2) pour donner le
carbamate (170, 125 mg, 77 %) sous la forme d’un solide blanc.
RMN 1H (500 MHz, CD3OD) :
δ (ppm)= 0,89 (t ; 3 H ; J = 6,9 Hz ; CH2CH2CH3) ; 1,28-1,32 (m ; 26 H ; 13 CH2) ; 1,50
(m ; 2 H ; NHCH2CH2) ; 2,60 (t ; 1 H ; J = 2,5 Hz ; CCH) ; 3,10 (m ; 2 H ; NHCH2CH2) ; 3,39
(t ; 1 H ; J = 9,4 Hz ; H-4) ; 3,58-3,61 (m ; 1 H ; H-5) ; 3,70 (dd ; 1 H ; J = 5,6 ; J = 12,0 Hz ;
H-6b) ; 3,85 (dd ; 1 H ; J = 1,9 Hz , J = 12,0 Hz ; H-6a) ; 3,86 (t ; 1 H ; J = 9,4 Hz ; H-3) ;
4,03-4,10 (m ; 3 H ; H-7b, NHCH2) ; 4,23 (d ; 1 H ; J = 15,4 Hz ; H-7b) ; 4,56 (dd ; 1 H ; J =
3,8 , J = 9,4 Hz ; H-2) ; 5,01 (d ; 1 H ; J = 3,8 Hz ; H-1).
RMN 13C (125 MHz, CD3OD) :
δ (ppm)= 14,4 (CH3) ; 23,7-33,0 (15 CH2) ; 41,9 (NHCH2) ; 62,4 (C-6) ; 67,7 (C-7) ; 71,6
(C-4) ; 72,3 (CCH) ; 72,4 (C-3) ; 74,4 (C-5) ; 74,7 (C-2) ; 80,4 (-CCH) ; 98,6 (C-1) ; 158,1 ;
171,4 (2 CO).
179

Partie Expérimentale
Tf: 128 °C.
[α]D +43 (c = 1,0 ; CH3OH).
Anal. Calc. pour C28H50N2O8 H2O: C, 57,51 ; H, 9,42 ; N, 4,79 ; trouvée: C, 57,59 ; H,
9,15 ; N, 4,72.
(N-Propargylcarbamoyl)méthyl α-D-glucopyranosyl-(1→4)-2-O-
hexadécylcarbamoyl-α-D-glucopyranoside (171)
OHO
OO
O
OH
NH
O
OHO
OH
OH
HO
NHH33C16
O
Une solution de (168, 76 mg, 0,079 mmol) dans MeOH/H2O/NEt3 (8/1/1) (2 mL) est
agitée à température ambiante pendant 16 heures. Après évaporation des solvants, le résidu est
purifié sur colonne de silice pour donner le carbamate (171, 41 mg, 73%) sous la forme d’un
solide blanc.
RMN 1H (300 MHz, CD3OD) :
δ (ppm)= 0,97 (t ; 3 H ; J = 6,6 Hz ; CH3) ; 1,35 (m ; 28 H ; 14 CH2) ; 1,59 (t ; 1 H ; J =
6,0 Hz ; CCH) ; 3,19 (dt ; 2 H ; J = 1,5 Hz ; J = 7,0 Hz ; NHCH2CH2) ; 3,67-3,93 (m ; 12 H ;
H-2’ ; H-3’ ; H-4’ ; H-5’ ; H-6’ ; H-6 ; H-4 ; H-5 ; NHCH2) ; 4,10 (m ; 2 H ; H-7a ; H-3) ;
4,24-4,26 (m ; 3 H ; H-7b ; NHCH2) ; 4,68 (dd ; 1 H ; J = 3,7 Hz ; J = 10,3 Hz ; H-2) ; 5,08 (d
; 1 H ; J = 3,7 Hz ; H-1) ; 5,27 (d ; 1 H ; J = 3,9 Hz ; H-1’).
RMN 13C (75 MHz, CD3OD) :
δ (ppm)= 14,4 (CH3) ; 23,4-41,8 (15 CH2) ; 48,4 (NHCH2) ; 61,4 (C-6) ; 62,5 (C-6’) ;
67,6 (C-7) ; 71,2 ; 71,9 ; 72,1 ; 72,5 ; 73,7 ; 73,9 ; 74,4 ; 74,7 (C-2 ; C-3 ; C-4 ; C-5 ; C-2’ ; C-
3’ ; C-4’ ; C-5’) ; 80,0 (CCH) ; 80,7 (CCH) ; 98,3 (C-1) ; 102,6 (C-1’) ; 157,6 ; 170,9 (2 CO).
Tf : 118-120°C.
[α]D +57 (c = 0,5 ; CH3OH).
180

Partie Expérimentale
Anal. Calc. pour C34H60N2O13.H2O: C, 56,49 ; H, 8,65 ; N, 3,88 ; trouvée: C, 56,18 ; H,
8,56 ; N, 3,58.
(N-Allylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-carbamoyl-α-D-glucopyranoside
(172)
OAcO
OO
O
OAc
NH
AcO
NH2
O
A une solution de (163, 200 mg, 0,49 mmol) dans du dichlorométhane anhydre (3 mL),
le trichloroacétyl isocyanate (0,09 mL, 0,79 mmol) est ajouté à -15˚C. Après une heure, la
réaction est quenchée avec du MeOH (2 mL), les solvants sont évaporés et le produit est filtré
sur silice (CH2Cl2/MeOH: 35/1). Ensuite, le produit est redissous dans du MeOH (2 mL) et le
zinc est rajouté (7,35 mmol, 480 mg). Après une heure, la réaction est filtrée sur fritté et le
produit est soumis à une purification sur gel de silice après évaporation du solvant pour
donner le carbamate (172, 195 mg, 88%) sous la forme de cristaux blancs.
RMN 1H (300 MHz, CDCl3):
δ (ppm)= 1,98 (s ; 6 H) ; 2,03 (s ; 3 H) (3 OAc) ; 3,88 (ls ; 2 H ; NHCH2) ; 3,94-4,22 (m
; 5 H ; H-5 ; H-6 ; H-7) ; 4,81 (dd ; 1 H ; J = 3,8 Hz ; J = 10,2 Hz ; H-2) ; 5,02 (t ; 1 H ; J =
10,2 Hz ; H-4) ; 5,03 (d ; 1 H ; J = 3,8 Hz ; H-1) ; 5,09-5,13 (m ; 2 H ; CH=CH2) ; 5,19 (ls ; 2
H ; CONH2) ; 5,40 (t ; 1 H ; J = 10,2 Hz ; H-3) ; 5,72-5,85 (m ; 1 H ; CH=CH2) ; 6,72 (t ; 1 H
; J = 5,6 Hz ; NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,5 ; 20,7 ; 20,7 (3 CH3) ; 41,4 (NHCH2) ; 61,7 (C-6) ; 67,2 (C-7) ; 68,0 (C-5)
; 68,2 (C-4) ; 70,0 (C-3) ; 71,0 (C-2) ; 96,9 (C-1) ; 116,6 (CH=CH2) ; 133,9 (CH=CH2) ; 155,2
; 168,3 ; 169,4 ; 170,4 ; 170,6 (5 CO).
Tf : 116°C
[α]D +107 (c = 1,0, CH2Cl2).
181

Partie Expérimentale
Anal. Calc. pour C18H26N2O11: C, 48,43 ; H, 5,87 ; N, 6,28 ; trouvée: C, 48,27; H, 5,38;
N, 6,10.
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-carbamoyl-α-D-
glucopyranoside (173)
OAcO
OO
O
OAc
NH
AcO
NH2
O
A une solution de (164, 130 mg, 0,32 mmol) dans du dichlorométhane anhydre (3 mL),
le trichloroacétyl isocyanate (0,06 mL, 0,52 mmol) est ajouté à -15˚C. Après une heure, la
réaction est quenchée avec du MeOH (2 mL), les solvants sont évaporés et le produit est filtré
sur silice (CH2Cl2/MeOH: 35/1). Ensuite, le produit est redissous dans du MeOH (2 mL) et le
zinc est rajouté (4,85 mmol, 318 mg). Après 30 min, la réaction est filtrée sur fritté et le
produit est soumis à une purification sur gel de silice après évaporation du solvant pour
donner le carbamate (173, 131 mg, 91%) sous la forme d’huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,97 (s ; 3 H) ; 1,98 (s ; 3 H) ; 2,03 (s ; 3 H) (3 OAc) ; 2,24 (t ; 1 H ; J = 2,4
Hz ; CCH) ; 3,93-4,06 (m ; 4 H ; NHCH2 ; H-5 ; H-6a) ; 4,09 (AB système ; 2 H ; δa 4,00 ; δb
4,18 ; J = 15,6 Hz ; H-7) ; 4,19 (dd ; 1 H ; J = 4,5 Hz ; J = 12,4 Hz ; H-6b) ; 4,84 (dd ; 1 H ; J
= 3,8 Hz ; J = 9,8 Hz ; H-2) ; 5,00 (d ; 1 H ; J = 3,8 Hz ; H-1) ; 5,02 (t ; 1 H ; J = 9,8 Hz ; H-
4) ; 5,16 (br s ; 2 H ; CONH2) ; 5,41 (t ; 1 H ; J = 9,8 Hz ; H-3) ; 6,83 (t ; 1 H ; J = 5,3 Hz ;
NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,6 ; 20,8 ; 20,8 (3 OAc) ; 28,8 (NHCH2) ; 61,7 (C-6) ; 67,4 (C-7) ; 68,2 (C-
5) ; 68,2 (C-4) ; 70,0 (C-3) ; 71,1 (C-2) ; 72,1 (CCH) ; 79,5 (CCH) ; 97,1 (C-1) ; 155,2 ; 168,3
; 169,5 ; 170,6 ; 170,7 (5 CO).
[α]D +98 (c = 1,0 ; CH2Cl2).
Anal. Calc. pour C18H24N2O11.H2O: C, 46,75 ; H, 5,67 ; N, 6,06 ; trouvée: C, 46,71 ; H,
5,38 ; N, 6,10.
182

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 2,3,4,6-tétra-O-acétyl-α-D-glucopyranosyl-(1→4)-
3,6-di-O-acétyl-2-O-carbamoyl-α-D-glucopyranoside (174)
OAcO
OO
O
OAc
NH
O
OAcO
AcO
OAc
AcO
NH2
O
A une solution de (165, 87 mg, 0,13 mmol) dans du dichlorométhane anhydre (2 mL), le
trichloroacétyl isocyanate (0,024 mL, 0,20 mmol) est ajouté à -15˚C. Après deux heures, la
réaction est quenchée avec du MeOH, les solvants sont évaporés et le produit est filtré sur
silice (CH2Cl2/MeOH: 35/1). Après évaporation du solvant, le produit est redissous dans du
MeOH (2 mL) et le zinc est rajouté (124 mg, 1,89 mmol). Après une heure, la réaction est
filtrée sur fritté et le produit est soumis à une purification sur gel de silice pour donner le
carbamate (174, 86 mg, 93%) sous la forme de cristaux blancs.
RMN 1H (300 MHz, CDCl3):
δ (ppm) = 1,94 (s ; 3 H) ; 1,96 (s ; 3 H) ; 1,99 (s ; 6 H) ; 2,04 (s ; 3 H) ; 2,08 (s ; 3 H) (6
OAc) ; 2,22 (t ; J = 2,5 Hz ; CCH) ; 3,89-4,22 (m ; 10 H ; H-4 ; H-5 ; H-6a ; H-4’ ; H-5’; H-6’
; H-7 ; NHCH2) ; 4,03 (dd ; 1 H ; J = 2,0 Hz ; J = 12,1 Hz ; H-6a) ; 4,72 (dd ; 1 H ; J = 3,8 Hz
; J = 10,1 Hz ; H-2) ; 4,79 (dd ; 1 H ; J = 3,9 Hz ; J = 10,2 Hz ; H-2’) ; 4,90 (d ; 1 H ; J = 3,8
Hz ; H-1) ; 5,00 (t ; 1 H ; J = 3,9 Hz ; J = 10,2 Hz ; H-4’) ; 5,29 (dd ; 1 H ; J = 9,6 Hz ; J =
10,2 Hz ; H-3’) ; 5,36 (d ; 1 H ; J = 4,0 Hz ; H-1’) ; 5,45 (dd ; 1 H ; J = 8,3 Hz ; J = 10,1 Hz ;
H-3) ; 6,87 (t ; 1 H ; J = 4,9 Hz ; NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm) = 20,7 ; 20,7 ; 20,7 ; 20,8 ; 20,9 ; 21,1 (6 CH3) ; 28,9 (NHCH2) ; 61,6 (C-6’) ;
62,7 (C-6) ; 67,4 (C-7) ; 68,0 (C-4’) ; 68,7 (C-5 ou C-4 ou C-5’) ; 68,7 (C-5 ou C-4 ou C-
5’) ;69,4 (C-3’) ; 70,2 (C-2’) ; 71,5 (C-2) ; 72,1 (CCH) ; 72,5 (C-3) ; 72,8 (C-5’ ou C-5 ou C-
4) ; 79,5 (CCH) ; 95,1 (C-1) ; 97,0 (C-1’) ; 155,2 ; 163,8 ; 168,4 ; 169,6 ; 170,1 ; 170,6 ;
170,7 ; 170,7 (8 CO).
[α]D +61 (c = 1,0 ; CH2Cl2).
183

Partie Expérimentale
Anal. Calc. pour C30H40N2O19: C, 49,18; H, 5,50; N, 3,82 ; trouvée: C, 49,48; H, 5,74, N,
3,81.
(N-Allylcarbamoyl)méthyl 2-O-carbamoyl-α-D-glucopyranoside (175)
OHO
OO
O
OH
NH
HO
NH2
O
Une solution de (172, 95 mg, 0,21 mmol) dans CH3OH/H2O/NEt3 (8/1/1) (1 mL) est
agitée pendant 12 heures à température ambiante. Après évaporation des solvants sous
pression réduite, le résidu est purifié sur colonne de silice (CH2Cl2/CH3OH: 4/1) pour donner
le produit (175, 63 mg, 94%) sous la forme de cristaux blancs.
RMN 1H (300 MHz, CD3OD) :
δ (ppm)= 3,42 (t ; 1 H ; J = 9,6 Hz ; H-4) ; 3,48-3,90 (m ; 6 H ; H-3 ; H-6 ; H-5 ;
NHCH2) ; 4,10 (système AB ; 2 H ; δa 4,06 ; δb 4,23 ; J = 15,8 Hz ; H-7) ; 4,53 (dd ; 1 H ; J =
3,6 Hz ; J = 10,0 Hz ; H-2) ; 5,03 (d ; 1 H ; J = 3,6 Hz ; H-1) ; 5,11-5,27 (m ; 2 H ; CH=CH2) ;
5,81-5,94 (m ; 1 H ; CH=CH2).
RMN 13C (75 MHz, CD3OD) :
δ (ppm)= 42,3 (NHCH2) ; 62,4 (C-6) ; 67,7 (C-7) ; 71,4 (C-4) ; 71,5 (C-3) ; 72,3 (C-5) ;
73,2 (C-2) ; 100,9 (C-1) ; 116,5 (CH=CH2) ; 135,1 (CH=CH2) ; 159,0 ; 172,0 (2 CO).
Tf: 164°C.
[α]D +51 (c = 0,5 ; CH3OH).
Anal. Calc. pour C12H18N2O8.H2O: C, 44,17 ; H, 6.,38 ; N, 8,59 ; trouvée: C, 44,29 ; H,
6,72; N, 8,33.
184

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl-2-carbamoyl-α-D-glucopyranoside (176)
OHO
OO
O
OH
NH
HO
NH2
O
Une solution de (173, 131 mg, 0,29 mmol) dans MeOH/H2O/NEt3 (8/1/1) (5 mL) est
agitée pendant 12 heures à température ambiante. Après évaporation des solvants sous
pression réduite, le résidu est purifié sur colonne de silice (CH2Cl2/CH3OH: 4/1) pour donner
du carbamate 176 sous la forme de cristaux blancs (90 mg, 97%).
RMN 1H (500 MHz, CD3OD) :
δ (ppm)= 2,62 (t ; 1 H ; J = 2,2 Hz ; CCH) ; 3,42 (t ; 1 H ; J = 9,8 Hz ; H-4) ; 3,64 (ddd ;
1 H ; J = 2,2 Hz ; J = 5,6 Hz ; J = 9,8 Hz ; H-5) ; 3,72 (dd ; 1 H ; J = 5,6 Hz ; J = 12,0 Hz ; H-
6a) ; 3,85 (dd ; 1 H ; J = 2,2 Hz ; J = 12,0 Hz ; H-6b) ; 3,91 (t ; 1 H ; J = 9,8 Hz ; H-3) ; 4,07
(d ; 2 H ; J = 2,2 Hz ; NHCH2) ; 4,15 (système AB ; 2 H ; δa 4,07 ; δb 4,23 ; J = 15,3 Hz ; H-
7) ; 4,54 (dd ; 1 H ; J = 3,8 Hz ; J = 9,6 Hz ; H-2) ; 5,02 (d ; 1 H ; J = 3,8 Hz ; H-1).
RMN 13C (125 MHz, CD3OD) :
δ (ppm)= 29,1 (NHCH2) ; 62,4 (C-6) ; 67,7 (C-7) ; 71,6 (C-4) ; 72,3 (CCH) ; 72,3 (C-3)
; 74,3 (C-5) ; 74,7 (CCH) ; 74,8 (C-2) ; 98,5 (C-1) ; 159,0 ; 171,4 (2 CO).
Tf.: 154°C.
[α]D +32 (c = 0,5 ; CH3OH).
SM : (Haute résolution)-ESI calculée pour C12H18N2O8.Na+ (M+Na+), 341,0961,
trouvée 341,0963.
185

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl α-D-glucopyranosyl-(1→4)-2-O-carbamoyl-α-D-
glucopyranoside (177)
OHO
OO
O
OH
NH
O
NH2
O
OHO
OH
OH
HO
Une solution de (174, 390 mg, 0,53 mmol) dans CH3OH/NEt3/H2O (8/1/1) (2 mL) est
agitée pendant 12 heures à température ambiante. Après évaporation des solvants, le produit
est purifié sur gel de silice pour donner le carbamate (177, 233 mg, 91%) sous la forme d’une
huile incolore.
RMN 1H (500 MHz, CD3OD):
δ (ppm)= 2,5 (t ; J = 2,2 Hz ; CCH) ; 3,58-3,84 (m ; 10 H ; H-4 ; H-5 ; H-6 ; H-2’ ; H-3’
; H-4’ ; H-5’ ; H-6’) ; 4,04 (s ; 2 H ; NHCH2) ; 4,11 (système AB ; 2 H ; δa 4,03 ; δb 4,19 ; J
= 15,3 Hz ; H-7) ; 4,18 (t ; 1 H ; J = 10,1 Hz ; H-3) ; 4,56 (dd ; 1 H ; J = 3,7 Hz ; J = 10,1 Hz ;
H-2) ; 5,00 (d ; 1 H ; J = 3,7 Hz ; H-1) ; 5,21 (d ; 1 H ; J = 3,7 Hz ; H-1’).
RMN 13C (125 MHz, CD3OD):
δ (ppm)= 29,1 (NHCH2) ; 61,8 (C-6) ; 62,7 (C-6’) ; 67,4 (C-7) ; 71,5 ; 72,2 ; 72,2 ; 72,8
; 74,1 ; 74,3 ; 74,7 ; 75,0 ; 79,5 (C-2 ; C-3 ; C-4 ; C-5 ; C-2’ ; C-3’ ; C-4’ ; C-5’ ; CCH) ; 80,8
(CCH) ; 98,4 (C-1) ; 102,7 (C-1’) ; 158.8 ; 171,3 (2 CO).
[α]D +11 (c = 0,3 ; CH3OH).
SM : (Haute résolution)-ESI calculée pour C18H28N2O13.Na (MNa+) 503,1489 ; trouvée
503,1493.
186

Partie Expérimentale
N-méthyl [-4-[1-(5’-déoxyuridin)-1,2,3-triazole]]-carboxyméthyl-2-O-carbamoyl-α-
D-glucopyranoside (178)
O
HO OH
N
NH
O
ONNN
OHO
OO
O
OH
NH
HO
NH2
O
Une solution de (176, 25 mg, 0,078 mmol) et d’azidouridine (16 mg, 0,094 mmol) dans
tert-butanol/H2O (2/1) (1,5 mL) agitée à température ambiante, l’ascorbate de sodium (1 M ;
7,8 µL ; 7.8 µmol) est ajouté suivi par CuSO4 (0,3 M ; 2,6 µL ; 0,78 µmol). Après 12 heures,
la réaction est traitée par résine DOWEX-Na+ 50X8 (50 mg) filtrée et le résidu est purifié sur
colonne de silice (AcOEt/EtOH/H2O: 6/3/1) pour donner le produit (178, 39 mg, 84%) sous la
forme d’une huile incolore.
RMN 1H (500 MHz, CD3OD) :
δ (ppm) = 3,40 (t ; 1 H ; J = 9,6 Hz ; H-4) ; 3,61 (m ; 1 H ; H-5) ; 3,70 (dd ; 1 H ; J = 5,3
Hz ; J = 11,8 Hz ; H-6a) ; 3,83 (dd ; 1 H ; J = 1,6 Hz ; J = 11,8 Hz ; H-6b) ; 3,87 (t ; 1 H ; J =
9,6 Hz ; H-3) ; 4,01 (t ; 1 H ; J = 5,7 Hz ; H-3’) ; 4,15 (système AB ; 2 H ; δa 4,09 ; δb 4,22 ;
J = 15,4 Hz ; H-7) ; 4,19-4,28 (m ; 2 H ; H-2’ ; H-4’) ; 4,53-4,55 (m ; 3 H ; H-2 ; CONH2) ;
4,59 (br s ; 2 H ; NHCH2) ; 4,70 (dd ; 1 H ; J = 6,9 Hz ; J = 14,8 Hz ; H-5’a) ; 4,81 (dd ; 1 H ;
J = 3,1 Hz ; J = 14,8 Hz ; H-5’b) ; 5,00 (d ; 1 H ; J = 3,7 Hz ; H-1) ; 5,72-5,74 (m ; 2 H ;
CHCHCO ; H-1’) ; 7,40 (d ; 1 H ; J = 8,2 Hz ; CHCHCO) ; 7;90 (s ; 1 H ; CCHN).
RMN 13C (125 MHz, CD3OD) :
δ (ppm) = 35,2 (NHCH2) ; 52,5 (C-5’) ; 62,4 (C-6) ; 67,7 (C-7) ; 71,6 (C-4) ; 71,9 (C-3’) ;
72,4 (C-3) ; 74,1 (C-2’) ; 74,4 (C-5) ; 74,7 (C-2) ; 82,9 (C-4’) ; 93,1 (C-1’) ; 98,6 (C-1) ;
103,1 (CHCHCO) ; 125,5 (CCHN) ; 143,3 (CHCHCO) ; 146,0 (CCHN) ; 152,1 ; 158,9 ;
166,1 ; 1719 (4 CO).
[α]D +50 (c = 0,3 ; H2O).
SM: (Haute résolution-ESI) m/z calculée pour C21H29N7O13Na (M+Na)+ 610,1721,
trouvée 610,1715.
187
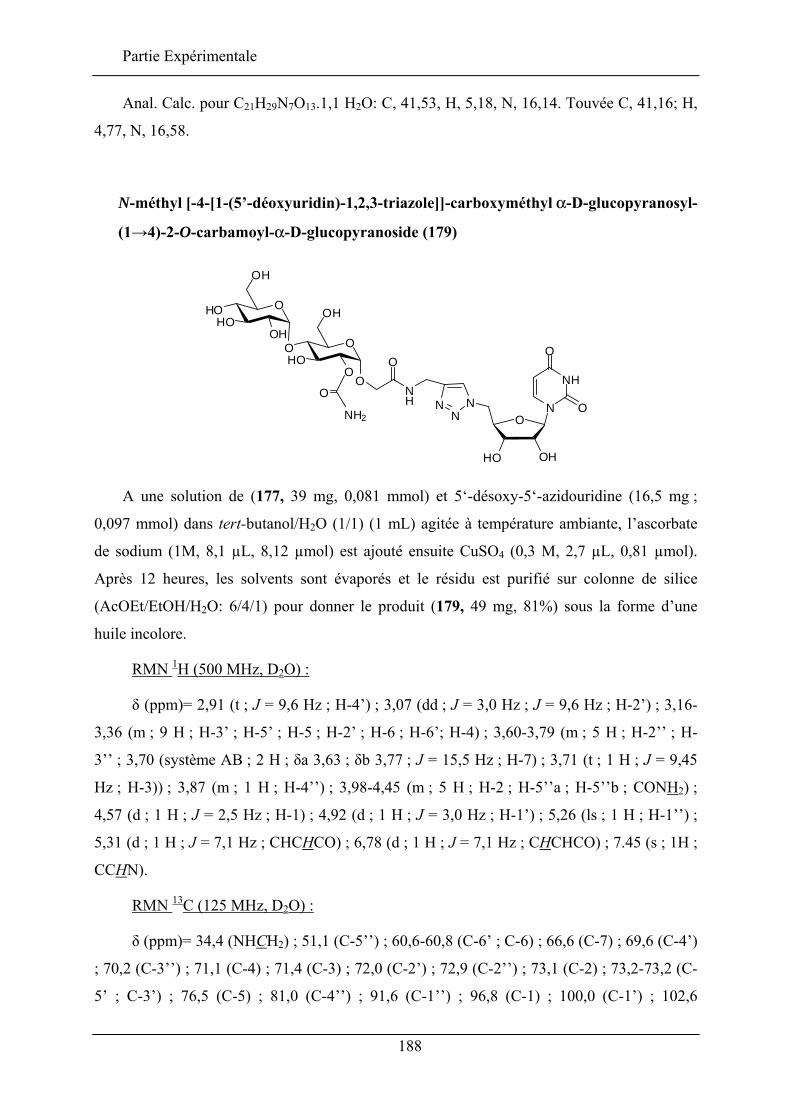
Partie Expérimentale
Anal. Calc. pour C21H29N7O13.1,1 H2O: C, 41,53, H, 5,18, N, 16,14. Touvée C, 41,16; H,
4,77, N, 16,58.
N-méthyl [-4-[1-(5’-déoxyuridin)-1,2,3-triazole]]-carboxyméthyl α-D-glucopyranosyl-
(1→4)-2-O-carbamoyl-α-D-glucopyranoside (179)
O
HO OH
N
NH
O
ONNN
OHO
OO
O
OH
NH
O
NH2
O
OHO
OH
OH
HO
A une solution de (177, 39 mg, 0,081 mmol) et 5‘-désoxy-5‘-azidouridine (16,5 mg ;
0,097 mmol) dans tert-butanol/H2O (1/1) (1 mL) agitée à température ambiante, l’ascorbate
de sodium (1M, 8,1 µL, 8,12 µmol) est ajouté ensuite CuSO4 (0,3 M, 2,7 µL, 0,81 µmol).
Après 12 heures, les solvents sont évaporés et le résidu est purifié sur colonne de silice
(AcOEt/EtOH/H2O: 6/4/1) pour donner le produit (179, 49 mg, 81%) sous la forme d’une
huile incolore.
RMN 1H (500 MHz, D2O) :
δ (ppm)= 2,91 (t ; J = 9,6 Hz ; H-4’) ; 3,07 (dd ; J = 3,0 Hz ; J = 9,6 Hz ; H-2’) ; 3,16-
3,36 (m ; 9 H ; H-3’ ; H-5’ ; H-5 ; H-2’ ; H-6 ; H-6’; H-4) ; 3,60-3,79 (m ; 5 H ; H-2’’ ; H-
3’’ ; 3,70 (système AB ; 2 H ; δa 3,63 ; δb 3,77 ; J = 15,5 Hz ; H-7) ; 3,71 (t ; 1 H ; J = 9,45
Hz ; H-3)) ; 3,87 (m ; 1 H ; H-4’’) ; 3,98-4,45 (m ; 5 H ; H-2 ; H-5’’a ; H-5’’b ; CONH2) ;
4,57 (d ; 1 H ; J = 2,5 Hz ; H-1) ; 4,92 (d ; 1 H ; J = 3,0 Hz ; H-1’) ; 5,26 (ls ; 1 H ; H-1’’) ;
5,31 (d ; 1 H ; J = 7,1 Hz ; CHCHCO) ; 6,78 (d ; 1 H ; J = 7,1 Hz ; CHCHCO) ; 7.45 (s ; 1H ;
CCHN).
RMN 13C (125 MHz, D2O) :
δ (ppm)= 34,4 (NHCH2) ; 51,1 (C-5’’) ; 60,6-60,8 (C-6’ ; C-6) ; 66,6 (C-7) ; 69,6 (C-4’)
; 70,2 (C-3’’) ; 71,1 (C-4) ; 71,4 (C-3) ; 72,0 (C-2’) ; 72,9 (C-2’’) ; 73,1 (C-2) ; 73,2-73,2 (C-
5’ ; C-3’) ; 76,5 (C-5) ; 81,0 (C-4’’) ; 91,6 (C-1’’) ; 96,8 (C-1) ; 100,0 (C-1’) ; 102,6
188

Partie Expérimentale
(CHCHCO) ; 125,4 (CCHN) ; 142,6 (CHCHCO) ; 144,9 (CCHN) ; 151,9 ; 158,3 ; 166,7 ;
172,0 (4 CO).
[α]D +105 (c = 0,3 ; H2O).
SM: (Haute résolution-ESI) calculée pour C27H39N7O18Na (M+Na)+ 772,2249, trouvée
772,2256.
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-[(tert-
butyloxycarbonyl)méthyl]- α-D-glucopyranoside (180)
OAcO
OO
O
OAc
NH
AcO
OO
(260 mg, 0,648 mmol) du produit 164 sont dissous dans 3 mL de DMF anhydre. (223 mg,
1,62 mmol) de K2CO3 y sont rajoutés ainsi que (0,11 mL, 0,78 mmol) du bromoacétate de
tert-butyle. Après 16 heures, la réaction est finie, le K2CO3 est filtré sur fritté, le DMF
évaporé. Ensuite le résidu est dissous dans 10 mL de CH2Cl2 et lavé avec une solution
aqueuse de NH4Cl (10%, 30 mL). La phase organique est séchée sur Na2SO4. Après
évaporation du solvant, le produit est purifié sur colonne (AcOEt/Pentane : 2/1) pour donner
le produit (180, 238 mg, 72%) sous la forme d’un mousse blanche.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,41 (s ; 9 H) ; 1,95 (s ; 3 H) ; 2,01 (s ; 3 H) ; 2,02 (s ; 3 H) (3 OAc) ; 2,12 (t ; 1
H ; J = 2,5 Hz ; CCH) ; 3,54 (dd ; 1 H ; J = 3,7 , J = 9,9 Hz ; H-2) ; 3,96-4,26 (m ; 7 H ; H-5 ;
H-6 ; H-7’ ; NHCH2) ; 4,10 (système AB ; 2 H ; δa 3,99 ; δb 4,22 ; J = 16,8 Hz ; H-7) ; 4,91 (t
; 1 H ; J = 9,9 Hz ; H-4) ; 5,20 (d ; 1H ; J = 3,7 Hz ; H-1); 5,38 (t ; 1 H ; J = 9,8 Hz ; H-3) ;
7,63 (t ; 1 H ; J = 5,1 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
189

Partie Expérimentale
δ (ppm)= 20,6 ; 20,7 ; 20,8 (3 Ac) ; 28,1 (3 CH3) ; 28,8 (NHCH2) ; 61,7 (C-6) ; 67,2 (C-
7’) ; 67,8 (C-5) ; 68,2 (C-4) ; 69,8 (C-7) ; 71,3 (CCH) ; 72,6 (C-3) ; 78,4 (C-2) ; 79,4 (-CCH) ;
82,5 (Cq) ; 98,5 (C-1) ; 168,9 ; 169,1 ; 169,8 ; 170,1 ; 170,5 (5 CO).
[α]D +75 (c = 0,5 ; CH2Cl2).
Anal. Calc. pour C23H33NO2.0,67 H2O : C, 52,37; H, 6,56 ; N, 2,72 ; trouvée: C, 52,02 ;
H, 6,40 ; N, 2,53.
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-[(carboxy)méthyl]- α-D-
glucopyranoside (181)
OAcO
OO
O
OAc
NH
AcO
HOO
(153 mg, 0,297 mmol) de 180 sont dissous dans 1 mL de dichlorométhane fraîchement
distillé et l’acide trifluoroacétique y est rajouté (0,7 mL). Après 3 heures, les solvants sont
coévaporés avec 2x5 mL du toluène, le produit est filtré sur colonne (AcOEt/Pentane: 3/1)
pour donner le produit (181, 123 mg, 90%) sous la forme d’une huile jaune.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,02 (s ; 3 H) ; 2,09 (s ; 3 H) ; 2,1 (s ; 3 H) (3 OAc) ; 2,25 (t ; 1 H ; J = 2,2 Hz ;
CCH) ; 3,65 (dd ; 1 H ; J = 3,4 , J = 9,8 Hz ; H-2) ; 3,96-4,26 (m ; 8 H ; H-5 ; H-6 ; H-7a ; H-
7’ ; NHCH2) ; 4,40 (d ; 1 H ; J = 16,8 Hz ; H-7b) ; 4,99 (t ; 1 H ; J = 9,8 Hz ; H-4) ; 5,30 (d ;
1H ; J = 3,4 Hz ; H-1); 5,46 (t ; 1 H ; J = 9,8 Hz ; H-3) ; 7,85 (t ; 1 H ; J = 4,1 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,7 ; 20,8 ; 21,0 (3 Ac) ; 29,2 (NHCH2) ; 61,8 (C-6) ; 67,6 (C-7’) ; 68,1 (C-5) ;
68,4 (C-4) ; 69,3 (C-7) ; 72,0 (CCH) ; 72,0 (C-3) ; 78,8 (C-2) ; 78,9 (CCH) ; 98,8 (C-1) ;
170,2 ; 170,6 ; 170,6 ; 171,1 ; 172,9 (5 CO).
[α]D +58 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C19H25NO12 0,6H2O: C, 48,53 ; H, 5,62 ; N, 2,98 ; trouvée: C, 48,55 ;
H, 5,47 ; N, 3,52.
190

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-(propargyl)- α-D-
glucopyranoside (182)
OAcO
OO
O
OAc
NH
AcO
Dans un ballon de 5 mL, (100 mg, 0,25 mmol) du produit 164 sont dissous dans 2 mL de
DMF anhydre en présence de tamis moléculaire. (0,03mL, 0,62 mmol) du bromure de
propargyle sont rajoutés ainsi que (12 mg, 0,3 mmol) de NaH. Après 30 min, la réaction est
quenchée avec de l’anhydride acétique (1 mL), le tamis moléculaire est filtré sur fritté, et le
milieu est lavé à l’eau (10 mL). A près séchage de la phase organique sur Na2SO4 et
évaporation des solvants, le résidu est soumis à une purification sur colonne de silice en
utilisant comme éluant le système AcOEt : Pentane (2/1) pour donner le produit 56 (62 mg,
56%) sous la forme d’une huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,02 (s ; 3 H) ; 2,07 (s ; 3 H) ; 2,09 (s ; 3 H) (3 OAc) ; 2,24 (t ; 1 H ; J = 2,5
Hz ; CCH) ; 2,51 (t ; 1 H ; J = 2,2 Hz ; CCH), 3,81 (dd ; 1 H ; J = 3,7 Hz ; J = 9,7 Hz ; H-2) ;
4,00-4,42 (m ; 9 H ; H-5 ; H-6 ; H-7 ; NHCH2 ; OCH2) ; 4,98-5,04 (m ; 2 H ; H-1 ; H-4) ; 5,42
(t ; 1 H ; J = 9,7 Hz ; H-3) ; 7,45 (t ; 1 H ; J = 5,5 Hz ; NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,7 ; 20,8 ; 21,0 (3 OAc) ; 28,8 (NHCH2) ; 59,6 (OCH2) ; 61,8 (C-6) ; 67,6
(C-7) ; 68,2 (C-4) ; 68,8 (C-5) ; 71,8 (C-3) ; 76,3(CCH) ; 76,6 (C-2) ; 77,4 (CCH) ; 78,9
(CCH) ; 79,3 (CCH) ; 98,3 (C-1) ; 168,9 ; 169,9 ; 170,3 ; 170,7 (4 CO).
[α]D +36 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C20H25NO10.0,43 H2O: C, 53,79 ; H, 6,22 ; N, 3,14 ; trouvée: C, 53,47 ;
H, 5,90 ; N, 3,00.
191

Partie Expérimentale
(N-Allylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-O-(propargyl)- α-D-glucopyranoside
(183)
OAcO
OO
O
OAc
NH
AcO
Dans un ballon de 5 mL, (52 mg, 0,129 mmol) du produit (163) sont dissous dans 1 mL
de DMF anhydre en présence de tamis moléculaire. (0,019 mL, 0,258 mmol) du bromure de
propargyle sont rajoutés ainsi que (7,4 mg, 0,155 mmol) de NaH. Après 30 min, la réaction
est quenchée avec de l’anhydride acétique (0,5 mL), le tamis moléculaire est filtré sur fritté, et
le milieu est lavé à l’eau (10 mL). A près séchage de la phase organique sur Na2SO4 et
évaporation des solvants, le résidu est soumis à une purification sur colonne de silice en
utilisant comme éluant le système AcOEt : Pentane (2/1) pour donner le produit (183, 32mg,
55%) sous la forme d’une huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,02 (s ; 3 H) ; 2,07 (s ; 3 H) ; 2,07 (s ; 3 H) (3 OAc) ; 2,60 (t ; 1 H ; J =2,35 Hz
; -CCH) ; 3,80 (dd ; 1 H ; J = 3,6 , J = 9,9 Hz ; H-2) ; 3,93-4,11 (m ; 5 H ; H-5, NHCH2 , -
CH2CCH) ; 4,17-4,35 (m ; 4 H ; H-6 ; H-7) ; 5,04 (d ; 1 H ; J = 3,6 Hz ; H-1) ; 5,08 (t ; 1 H ; J
= 9,5 Hz ; H-4 ) ; 5,14-5,31 (m ; 2 H ; -CH=CH2) ; 5,46 (t ; 1 H ; J = 9,5 Hz ; H-3 ) ; 5,81-5,94
(m ; 1 H ; -CH=CH2) ; 7,55 (t ; 1 H ; J =5,8 Hz ; NH).
RMN 13C (75 MHz, CDCl3):
δ (ppm)= 20,7 ; 20,8 ; 21,0 (3 CH3) ; 41,6 (NHCH2) ; 59,5 (OCH2) ; 61,8 (C-6) ; 67,7 (C-
7) ; 68,1 ,68,4 (C-4 ; C-5) ; 71,9 (C-3) ; 75,9 (CCH) ; 76,6 (C-2) ; 78,8 (CCH) ; 98,3 (C-1) ;
116,9 (CH=CH2) ; 133,8 (CH=CH2) ; 168,8 ; 168,8 ; 170,2 ; 170,7 (4 CO).
[α]D +36 (c = 0,3 ; CH2Cl2).
Anal. Calc. pour C20H27NO10.H2O: C, 52,28 ; H, 6,36 ; N, 3,05 ; trouvée: C, 51,93 ; H,
6,07 ; N, 2,95.
192

Partie Expérimentale
(N-Allylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-azido-2-déoxy-α-D-mannopyranoside
(184)
OAcO
OO
OAc
NH
AcON3
A une solution de (163, 520 mg, 1,29 mmol) dans du dichlorométhane anhydre (5 mL), et
de pyridine (0,21 mL, 2,58 mmol), en présence de tamis moléculaire, l’anhydride triflique
(0,32 mL, 1,93 mmol) dilué dans du CH2Cl2 (1 mL) est rajouté goutte à goutte à -15°C. Après
une heure, la réaction est filtrée, lavée avec une solution de NaHCO3 (10%), et séchée sur
Na2SO4. Après évaporation du solvant, le résidu est dissous dans 5 mL du DMF anhydre en
présence de tamis moléculaire, NaN3 (1,67g ; 25,8 mmol) est rajoutée ainsi qu’une quantité
catalytique de l’iodure de tétrabutylammonium. La réaction est agitée à 60°C pendant une
nuit. Ensuite, elle est filtrée, lavée à l’eau, et le résidu est purifié sur colonne de silice
(Toluène/AcOEt: 2/1) pour donner l’azide correspondant (184, 527 mg, 96%) sous la forme
d’une huile jaune.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,99 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,05 (s ; 3 H) (3 OAc) ; 3,82-3,91 (m ; 3 H ; H-5
; NHCH2) ; 4,03 (dd ; 1 H ; J = 2,2 Hz ; J = 12,3 Hz ; H-6a) ; 4,05 (système AB ; 2 H ; δa
3,97 ; δb 4,14 ; J = 15,3 Hz ; H-7) ; 4,09 (dd ; 1 H ; J = 1,7 Hz ; J = 3,2 Hz ; H-2) ; 4,18 (dd ;
1 H ; J = 5,3 Hz ; J = 12,3 Hz ; H-6b) ; 4,85 (d ; 1 H ; J = 1,7 Hz ; H-1) ; 5,09-5,21 (m ; 2 H ;
CH=CH2) ; 5,22-5,28 (m ; 2 H ; H-3 ; H-4) ; 5,70-5,90 (m ; 1 H ; CH=CH2) ; 6,47 (t ; 1 H ; J
= 5,3 Hz, NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,4 ; 20,5 ; 20,6 (3 OAc) ; 41,3 (NHCH2) ; 60,9 (C-2) ; 61,9 (C-6) ; 65,5 (C-
4) ; 66,7 (C-7) ; 69,1 (C-5) ; 70,7 (C-3) ; 98,0 (C-1) ; 116,5 (CH=CH2) ; 133,6 (CH=CH2) ;
167,6 ; 169,3 ; 170,0 ; 170,5 (4 CO).
[α]D +69 (c= 0,5 ; CH2Cl2).
SM: (Haute résolution-ESI) calculée pour C17H24N4O9.Na+ (MNa+) 451,1441, trouvée
451.1442.
193

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 3,4,6-tri-O-acétyl-2-azido-2-déoxy-α-D-
mannopyranoside (185)
OAcO
OO
OAc
NH
AcON3
A une solution de (164, 94 mg, 0,23 mmol) dans du dichlorométhane anhydre (2 mL), et
de pyridine (38 µL ; 0,47 mmol), en présence de tamis moléculaire, l’anhydride triflique (88
µL ; 0,35 mmol) dilué dans du dichlorométhane (200 µL) est rajouté goutte à goutte à -17°C.
Après une heure, la réaction est filtrée, rincée au CH2Cl2, lavée avec une solution de NaHCO3
(10%), et séchée sur Na2SO4. Après évaporation du solvant, le résidu est dissous dans 2 mL du
DMF anhydre en présence de tamis moléculaire, NaN3 (304 mg ; 4,68 mmol) est rajoutée
ainsi qu’une quantité catalytique de l’iodure de tetrabutylammonium. La réaction est agitée à
60°C pendant une nuit. Ensuite, elle est filtrée, le solvant est évaporé, et le résidu est purifié
sur colonne de silice (Toluène/AcOEt: 2/1) pour donner l’azide correspondant (185, 70 mg,
77%) sous la forme d’une huile jaune.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 1,99 (s ; 3 H) ; 2,03 (s ; 3 H) ; 2,05 (s ; 3 H) (3 OAc) ; 2,22 (t ; 1 H ; J = 2,5 Hz
; CCH) ; 3,85 (m ; 1 H ; H-5) ; 4,03-4,09 (m ; 4 H ; H-2 ; H-6a ; NHCH2) ; 4,09 (système AB
; 2 H ; δa 4,00 ; δb 4,17 ; J = 15,0 Hz ; H-7) ; 4,18 (dd ; 1 H ; J = 5,2 Hz ; J = 12,2 Hz ; H-6b)
; 4,84 (ls ; 1 H ; H-1) ; 5,21-5,33 (m ; 2 H ; H-3 ; H-4) ; 6,60 (t ; 1 H ; J = 5,5 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 20,6 ; 20,7 ; 20,8 (3 OAc) ; 28,8 (NHCH2) ; 61,0 (C-2) ; 62,0 (C-6) ; 65,7 (C-4)
; 67,0 (C-7) ; 69,4 (C-5) ; 70,9 (C-3) ; 72,0 (CCH) ; 79,1 (CCH) ; 98,4 (C-1) ; 167,6 ; 169,5 ;
170,3 ; 170,7 (4 CO).
[α]D +27 (c = 1,0 ; CH2Cl2).
SM: (Haute résolution-ESI) calculée pour C17H22N4O9.Na+ (MNa+) 449.1284 ; trouvée
449,1291.
194

Partie Expérimentale
(N-Propargylcarbamoyl)méthyl 2-O-(propargyl)- α-D-glucopyranoside (186)
OHO
OO
O
OH
NH
HO
Le produit (182, 346 mg, 78 mmol) est dissous dans MeOH (5 mL). Une quantité
catalytique d’une solution de MeONa/MeOH (1 N) est rajoutée et la solution est agitée
pendant 4 heures à température ambiante. La réaction est ensuite traitée avec DOWEX-H+ et
le produit est purifié sur gel de silice (AcOEt/MeOH : 8/1) pour donner le produit final sous la
forme d’une huile incolore (186, 205 mg, 82%).
RMN 1H (300 MHz, CD3OD) :
δ (ppm)= 2,73 (t ; 1 H ; J =2,5 Hz ; -CCH) ; 2,98 (t ; 1 H ; J =2,4 Hz ; -CCH) ; 3,36 (t ; 1
H ; J = 9,7 Hz ; H-4) ; 3,51 (dd ; 1 H ; J = 3,7 Hz, J = 9,5 Hz ; H-2) ; 3,59 (m ; 1 H ; J = 1,3
Hz, J = 5,5 Hz ; J = 9,7 Hz ; H-5) ; 3,69 (dd ; 1 H ; J = 5,5 Hz ; J = 12,0 Hz ; H-6a) ; 3,81 (t ;
1 H ; J = 9,7 Hz ; H-3) ; 3,83 (dd ; 1 H ; J = 1,3 Hz, J = 12,00 Hz ; H-6b) ; 4,08 (dd ; 2 H ; J =
2,4 Hz ; J = 5,8 Hz ; NHCH2) ; 4,15 (système AB ; 2 H ; J = 16,0 Hz ; H-7) ; 4,49 (système
AB doublé ; J = 2,4 Hz ; J = 15,8 Hz ; H-7’) ; 5,03 (d ; 1 H ; J = 3,7 Hz ; H-1).
RMN 13C (75 MHz, CD3OD) :
δ (ppm)= 29,1 (NHCH2) ; 60,0 (C-7’) ; 62,3 (C-6) ; 67,9 (C-7) ; 71,4 (C-4) ; 72,6 (CCH) ;
74,1 (C-5) ; 74,2 (C-3) ; 76,7 (CCH) ; 80,3 (C-2) ; 80,3 (CCH) ; 80,8 (CCH) ; 99,4 (C-1) ;
171,7 (CO).
(N-Propargylcarbamoyl)méthyl 2-O-(propargyl)-6-O-(paratoluènesulfonyl)-α-D-
glucopyranoside (187)
OHO
OO
O
OTs
NH
HO
195

Partie Expérimentale
A une solution du produit (186, 81 mg, 0,25 mmol) dans de la pyridine distillée (2 mL),
le chlorure de p-toluène sulfonyl est rajouté (121 mg, 0,63 mmol) en présence de tamis
moléculaire 5A. La réaction est agitée à température ambiante. Après 48 heures, le tamis
moléculaire est filtré sur fritté en rinçant avec le CH2Cl2 (5 mL) et les solvants sont évaporés.
Le produit est lavé avec une solution saturée de NH4Cl (5 mL) et extrait au CH2Cl2 (5 mL).
Après évaporation du solvant, le produit est ensuite purifié sur une colonne de silice
(CH2Cl2/MeOH : 20/1) pour avoir au final le tosyle (187, 64 mg, 54%) sous la forme d’une
huile incolore.
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,19 (t ; 1 H ; J =2,4 Hz ; -CCH) ; 2,38 (s ; 3 H ; CH3) ; 2,45 (t ; 1 H ; J =2,4 Hz
; -CCH) ; 3,45-3,53 (m ; 2 H ; H-2 ; H-4) ; 3,72 (m ; 1 H ; H-5) ; 3,85 (t ; 1 H ; J = 9,2 Hz ; H-
3) ; 399 (système AB ; 2 H ; J = 16,2 Hz ; H-7) ; 4,01 (dd ; 2 H ; J = 2,4 Hz, J = 5,3 Hz ;
NHCH2) ; 4,12 (dd ; 2 H ; J = 1,9 Hz ; J = 11,1 Hz ; H-6a) ; 4,31 (dd ; 1 H ; J = 3,9 Hz ; J =
11,1 Hz ; H-6b) ; 4,38 (système AB doublé ; J = 2,4 Hz ; J = 16,0 Hz ; H-7’) ; 4,82 (d ; 1 H ; J
= 3,8 Hz ; H-1) ; 7,27-7,30 (m ; 2 H ; Harom) ; 7,51 (t ; 1 H ; J = 5,3 Hz ; NH) ; 7,71-7,73
(m ; 2 H ; Harom).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 21,8 (CH3) ; 28,8 (NHCH2) ; 59,6 (C-7’) ; 67,5 (C-6) ; 68,7 (C-7) ; 69,6 (C-4) ;
70,0 (C-5) ; 71,8 (CCH) ; 73,0 (C-3) ; 75,8 (CCH) ; 78,4 (C-2) ; 79,3 (CCH) ; 79,5 (CCH) ;
98,6 (C-1) ; 128,1 (C-arom) ; 128,1 (C-arom) ; 130,1 (Cq arom) ; 130,1 (Cq arom) ; 169,5
(CO).
(N-Propargylcarbamoyl)méthyl 2-O-(propargyl)-6-déoxy -6-azido-α-D-
glucopyranoside (188)
OHO
OO
O
N3
NH
HO
A une solution du tosyle (187, 53 mg, 0,11 mmol) dans le DMF distillé (2 mL), l’azoture
de sodium (38 mg, 0,57 mmol) est rajouté en présence due tamis moléculaire 5A. La réaction
196

Partie Expérimentale
est agitée pendant 16 heures à 60°C. Ensuite, le tamis est filtré sur fritté et rincé à l’acétate
d’éthyle (5 mL). Après évaporation du DMF sous pression réduite, le produit est extrait dans
l’acétate d’éthyle (5 mL). La phase organique est séchée sur Na2SO4, le solvant est évaporé et
le résidu est soumis à une chromatographie sur colonne (CH2Cl2/MeOH : 20/1) pour avoir
l’azide (188, 6 mg, 16%).
RMN 1H (300 MHz, CDCl3) :
δ (ppm)= 2,19 (t ; 1 H ; J =2,4 Hz ; -CCH) ; 2,48 (t ; 1 H ; J =2,4 Hz ; -CCH) ; 3,44-3,52
(m ; 4 H ; H-2 ; H-4 ; H-6) ; 3,75 (m ; 1 H ; H-5) ; 3,85 (t ; 1 H ; J = 9,2 Hz ; H-3) ; 4,05 (dd ;
2 H ; J = 2,4 Hz ; J = 5,6 Hz ; NHCH2) ; 4,10 (système AB ; 2 H ; J = 16,0 Hz ; H-7) ; 4;38
(t ; 2 H ; J = 2,4 Hz ; H-7’) ; 4,93 (d ; 1 H ; J = 3,8 Hz ; H-1) ; 7,51 (t ; 1 H ; J = 5,6 Hz ; NH).
RMN 13C (75 MHz, CDCl3) :
δ (ppm)= 29,1 (NHCH2) ; 51,5 (C-6) ; 59,8 (C-7’) ; 68,0 (C-7) ; 71,2 (C-4) ; 71,4 (C-5) ;
72,1 (CCH) ; 73,4 (C-3) ; 76,3 (CCH) ; 77,6 (CCH) ; 79,3 (C-2) ; 79,7 (CCH) ; 98,7 (C-1) ;
169,4 (CO).
B.4. PROCEDURE GENERALE POUR LA PREPARATION DES SONDES OPTIQUES
GLYCOSYLEES
A une solution du chromophore dans du dichlorométhane anhydre (1mL de solvant pour
100 mg de chromophore), 2,2 équiv. de la lactone saccharidique est rajoutée. La réaction est
agitée pendant 12 heures à température ambiante. Ensuite, le solvant est évaporé et le produit
est purifié sur gel de silice (CH2Cl2/MeOH). Le produit est ensuite dissous dans le méthanol
(1mL pour 100 mg de produit) et une quantité catalytique d’une solution de MeONa/MeOH
est rajoutée à la solution. Après 16 heures, le solvant est évaporé et le produit est purifié sur
une colonne de silice (CH2Cl2/(CH3)2CO/MeOH/H2O).
197
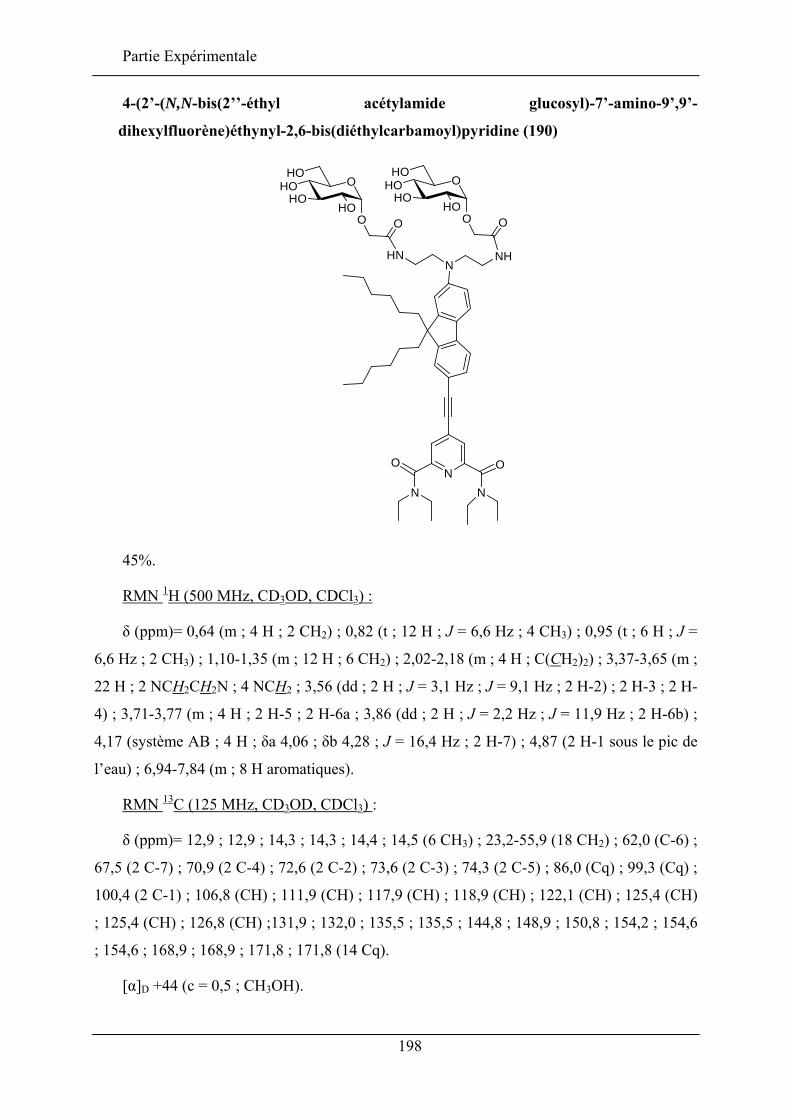
Partie Expérimentale
4-(2’-(N,N-bis(2’’-éthyl acétylamide glucosyl)-7’-amino-9’,9’-
dihexylfluorène)éthynyl-2,6-bis(diéthylcarbamoyl)pyridine (190)
OHOHO
HO
HO
O O
OHOHO
HO
HO
O O
NNHHN
NN
O
N
O
45%.
RMN 1H (500 MHz, CD3OD, CDCl3) :
δ (ppm)= 0,64 (m ; 4 H ; 2 CH2) ; 0,82 (t ; 12 H ; J = 6,6 Hz ; 4 CH3) ; 0,95 (t ; 6 H ; J =
6,6 Hz ; 2 CH3) ; 1,10-1,35 (m ; 12 H ; 6 CH2) ; 2,02-2,18 (m ; 4 H ; C(CH2)2) ; 3,37-3,65 (m ;
22 H ; 2 NCH2CH2N ; 4 NCH2 ; 3,56 (dd ; 2 H ; J = 3,1 Hz ; J = 9,1 Hz ; 2 H-2) ; 2 H-3 ; 2 H-
4) ; 3,71-3,77 (m ; 4 H ; 2 H-5 ; 2 H-6a ; 3,86 (dd ; 2 H ; J = 2,2 Hz ; J = 11,9 Hz ; 2 H-6b) ;
4,17 (système AB ; 4 H ; δa 4,06 ; δb 4,28 ; J = 16,4 Hz ; 2 H-7) ; 4,87 (2 H-1 sous le pic de
l’eau) ; 6,94-7,84 (m ; 8 H aromatiques).
RMN 13C (125 MHz, CD3OD, CDCl3) :
δ (ppm)= 12,9 ; 12,9 ; 14,3 ; 14,3 ; 14,4 ; 14,5 (6 CH3) ; 23,2-55,9 (18 CH2) ; 62,0 (C-6) ;
67,5 (2 C-7) ; 70,9 (2 C-4) ; 72,6 (2 C-2) ; 73,6 (2 C-3) ; 74,3 (2 C-5) ; 86,0 (Cq) ; 99,3 (Cq) ;
100,4 (2 C-1) ; 106,8 (CH) ; 111,9 (CH) ; 117,9 (CH) ; 118,9 (CH) ; 122,1 (CH) ; 125,4 (CH)
; 125,4 (CH) ; 126,8 (CH) ;131,9 ; 132,0 ; 135,5 ; 135,5 ; 144,8 ; 148,9 ; 150,8 ; 154,2 ; 154,6
; 154,6 ; 168,9 ; 168,9 ; 171,8 ; 171,8 (14 Cq).
[α]D +44 (c = 0,5 ; CH3OH).
198
Partie Expérimentale
SM (Haute résolution-ESI) m/z calculée pour C62H90N6O16Na (M+Na)+ = 1197,6311 ;
trouvée = 1197,6303.
4-(2’-(N,N-bis(2’’-éthyl acétylamide glucosyl)-7’-amino-9’,9’-
dihexylfluorène)éthynyl-2,6-bis(dioctylcarbamoyl)pyridine (191)
OHOHO
HO
HO
O O
OHOHO
HO
HO
O O
NNHHN
NN
O
N
O
C8H17 C8H17C8H17 C8H17
74%.
RMN 1H (500 MHz, CD3OD, CDCl3) :
δ (ppm)= 0,63 (m ; 4 H ; 2 CH2) ; 0,80 (t ; 6 H ; J = 7,2 Hz ; 2 CH3) ; 0,89 (t ; 6 H ; J =
7,2 Hz ; 2 CH3) ; 0,95 (t ; 6 H ; J = 7,2 Hz ; 2 CH3) ; 1,09-1,75 (m ; 60 H ; 30 CH2) ; 2,02-
2,18 (m ; 4 H; C(CH2)2) ; 3,35-3,41 (m ; 22 H ; 2 NCH2CH2N ; 4 NCH2 ; 2 H-2 ; 2 H-3 ; 3,39
(t ; 2 H ; J = 9,4 Hz, 2 H-4)) ; 3,68-3,73 (m ; 4 H ; 2 H-5 ; 2 H-6a) ; 3,83 (dd ; 2 H ; J = 2,2
Hz, J = 11,9 Hz ; 2 H-6b) ; 4,14 (système AB ; 4 H ; δa 4,03 ; δb 4,24 ; J = 15,4 Hz ; 2 H-7) ;
4,87 (2 H-1 sous le pic de l’eau) ; 6,61-7,81 (m ; 8 H aromatiques).
RMN 13C (125 MHz, CD3OD, CDCl3) :
δ (ppm)= 14,3 ; 14,3 ; 14,5 ; 14,5 ; 14,5 ; 14,5 (6 CH3) ; 23,4-56,2 (42 CH2) ; 62,3 (C-6) ;
67,8 (2 C-7) ; 71,3 (2 C-4) ; 73,0 (2 C-2) ; 74,0 (2 C-3) ; 74,7 (2 C-5) ; 86,1 (Cq) ; 99,4 (Cq) ;
100,7 (2 C-1) ; 107,1 (CH) ; 112,3 (CH) ; 118,2 (CH) ; 119,1 (CH) ; 122,3 (CH) ; 125,8 (CH)
; 125,8 (CH) ; 127,0 (CH) ; 130,4 ; 132,3 ; 132,3 ; 135,7 ; 145,2 ; 149,4 ; 151,1 ; 154,4 ; 155,0
199
Partie Expérimentale
; 155,0 ; 169,5 ; 169,5 ; 172,2 ; 172,2 (14 Cq).
[α]D +37 (c = 1,0 ; CH3OH).
SM (Haute résolution-ESI) m/z calculée pour C86H138O16N6 H (M+H)+ = 1512,0248 ;
trouvée = 512,02525.
4-(2’-(N,N-bis(2’’-éthyl acétylamide lactosyl)-7’-amino-9’,9’-dihexylfluorène)éthynyl-
2,6-bis(diéthylcarbamoyl)pyridine (192)
O O
NHNHN
NN
O
N
O
OHO
OH
HO
OHO
OH
HO
O
OH
OHO
OH
HO
OHO
OH
HO
O
OH
O
O
30%.
RMN (500 MHz, CD3OD, CDCl3) :
δ (ppm)= 0,68 (m ; 4 H ; 2 CH2) ; 0,83 (t ; 6 H ; J = 6,6 Hz ; 2 CH3) ; 1,09-1,21 (m ; 12 H
; 6 CH2) ; 1,27 (t ; 6 H ; J = 6,6 Hz ; 2 CH3) ; 1,36 (t ; 6 H ; J = 6,6 Hz ; 2 CH3) ; 2,06 (m ; 4
H ; 2 C(CH2)2) ; 3,44 (q ; 4 H ; J = 6,6 Hz ; 2 NCH2) ; 3,55-3,69 (m ; 22 H ; 3,55 (dd ; 2 H ; J
= 3,1 Hz , J = 9,7 Hz, 2 H-2) ; 2 H-2’ ; 2 H-3’ ; 2 H-4 ; 2 H-5’ ; 2 NCH2CH2N ; 2 NCH2) ;
3,68-3,73 (m ; 2 H ; 2 H-5) ; 3,75 (dd ; 2 H ; J = 4,4 Hz ; J = 11,3 Hz ; 2 H-6’a) ; 3,82-3,92 (m
; 10 H ; 2 H-3 ; 2 H-4’ ; 2 H-6’b ; 2 H-6a; 2 H-6b) ; 4,12 (système AB ; 4 H ; δa 4,02 ; δb 4,22
; J = 15,4 Hz ; 2 H-7) ; 4,39 (d ; 2 H ; J =7,9 Hz ; 2 H-1’) ; 4,89 (2 H-1 sous le pic de l’eau) ;
6,90-7,75 (m ; 8 H aromatiques).
200
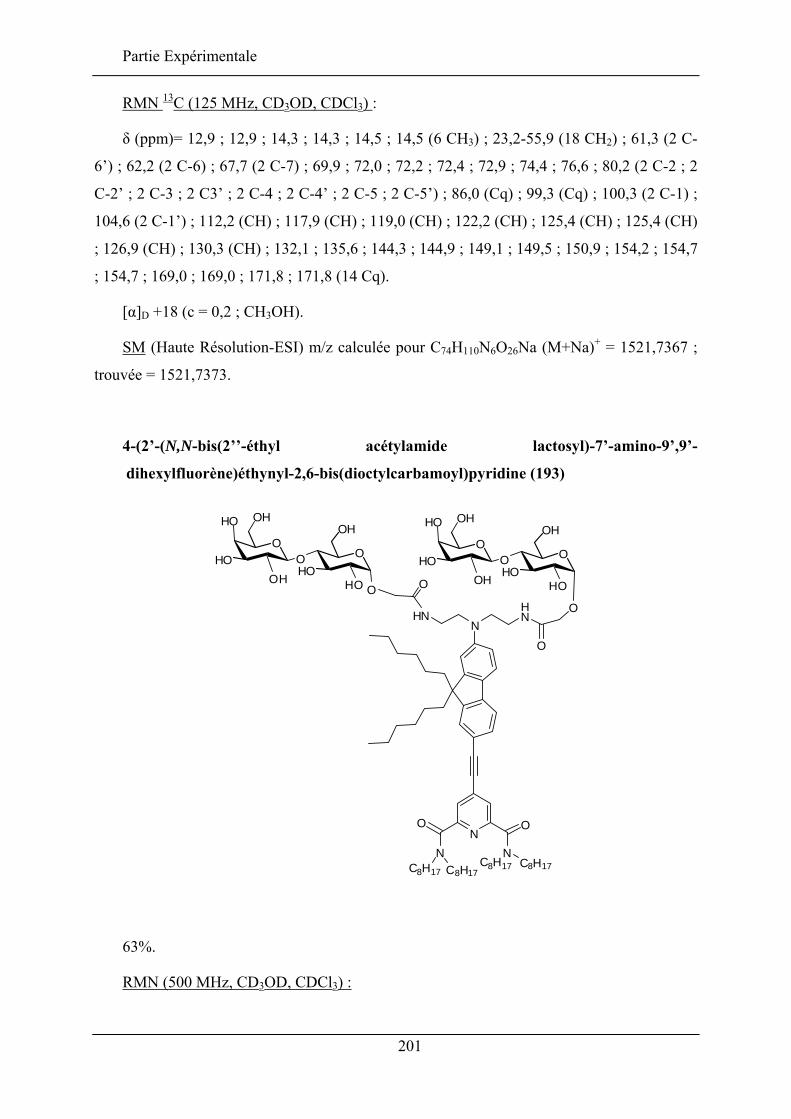
Partie Expérimentale
RMN 13C (125 MHz, CD3OD, CDCl3) :
δ (ppm)= 12,9 ; 12,9 ; 14,3 ; 14,3 ; 14,5 ; 14,5 (6 CH3) ; 23,2-55,9 (18 CH2) ; 61,3 (2 C-
6’) ; 62,2 (2 C-6) ; 67,7 (2 C-7) ; 69,9 ; 72,0 ; 72,2 ; 72,4 ; 72,9 ; 74,4 ; 76,6 ; 80,2 (2 C-2 ; 2
C-2’ ; 2 C-3 ; 2 C3’ ; 2 C-4 ; 2 C-4’ ; 2 C-5 ; 2 C-5’) ; 86,0 (Cq) ; 99,3 (Cq) ; 100,3 (2 C-1) ;
104,6 (2 C-1’) ; 112,2 (CH) ; 117,9 (CH) ; 119,0 (CH) ; 122,2 (CH) ; 125,4 (CH) ; 125,4 (CH)
; 126,9 (CH) ; 130,3 (CH) ; 132,1 ; 135,6 ; 144,3 ; 144,9 ; 149,1 ; 149,5 ; 150,9 ; 154,2 ; 154,7
; 154,7 ; 169,0 ; 169,0 ; 171,8 ; 171,8 (14 Cq).
[α]D +18 (c = 0,2 ; CH3OH).
SM (Haute Résolution-ESI) m/z calculée pour C74H110N6O26Na (M+Na)+ = 1521,7367 ;
trouvée = 1521,7373.
4-(2’-(N,N-bis(2’’-éthyl acétylamide lactosyl)-7’-amino-9’,9’-
dihexylfluorène)éthynyl-2,6-bis(dioctylcarbamoyl)pyridine (193)
O O
NHNHN
NN
O
N
O
C8H17C8H17
OHO
OH
HO
OHO
OH
HO
O
OH
OHO
OH
HO
OHO
OH
HO
O
OH
O
O
C8H17C8H17
63%.
RMN (500 MHz, CD3OD, CDCl3) :
201

Partie Expérimentale
δ (ppm)= 0,62 (m ; 4 H ; 2 CH2) ; 0,78 (t ; 6 H ; J = 6,6 Hz ; 2 CH3) ; 0,87 (t ; 6 H ; J =
6,6 Hz, 2 CH3) ; 0,93 (t ; 6 H ; J = 6,6 Hz ; 2 CH3) ; 1,08-2,03 (m ; 64 H ; 32 CH2) ; 3,50-3,65
(m ; 40 H ; 2 NCH2CH2N ; 4 NCH2 ; 2 H-2 ; 2 H-2’ ; 2 H-3 ; 2 H-3’ ; 2 H-4 ; 2 H-4’ ; 2 H-5 ;
2 H-5’ ; 2 H-6a ; 2 H-6b ; 2 H-6’a ; 2 H-6’b) ; 4,12 (système AB ; 4 H ; δa 4,02 ; δb 4,21 ; J =
15,6 Hz ; 2 H-7) ; 4,41 (d ; 2 H ; J = 7,7 Hz ; 2 H-1’) ; 4,83 (2 H-1 sous le pic de l’eau) ; 6,91-
7,80 (m ; 8 H aromatiques).
RMN 13C (125 MHz, CD3OD, CDCl3) :
δ (ppm)= 14,1 ; 14,1 ; 14,2 ; 14,2 ; 14,2 ; 14,2 (6 CH3) ; 22,8-55,4 (42 CH2) ; 60,9, 61,8
(2 C-6 et 2 C-6’) ; 67,2 (2 C-7) ; 69,4 ; 71,4 ; 71,8 ; 72,2 ; 73,6 ; 75,8 ; 77,7 ; 79,5 (2 C-2 ; 2
C-3 ; 2 C-4 ; 2 C-5 ; 2 C-2’ ; 2 C-3’; 2 C-4’ ; 2 C-5’) ; 85,7 (Cq) ; 98,6 (Cq) ; 99,6 (2 C-1) ;
103,9 (2 C-1’) ; 106,3 (CH) ; 117,5 (CH) ; 118,5 (CH) ; 121,7 (CH) ; 125,4 (CH) ; 125,4 (CH)
; 126,4 (CH) ; 129,8 (CH) ; 134,8 ; 140,0 ; 140,4 ; 140,7 ; 144,0 ; 148,3 ; 150,3 ; 153,7 ; 154,0
; 154,0 ; 168,4 ; 168,4 ; 171,1 ; 171,1 (14 Cq).
[α]D +20 (c = 0,2 ; CH3OH).
SM (Haute résolution-ESI) m/z calculée pour C98H158N6O26Na (M/2+Na)+ = 940,5506 ;
trouvée = 940,5516.
202

REFERENCES
BIBLIOGRAPHIQUES

Partie Expérimentale
1. (a) Weidenhagen, R.; Lorenz, S. Angew. Chem., 1957, 69, 641-641; (b) Weidenhagen,
R.; Lorenz, S. Ger. Pat. DE 1,049,800, 1957, Chem. Abstr. 1961, 55, 2030b; (c)
Schiweck, H.; Munir, M.; Rapp, K. M.; Schneider, B.; Vogel, M. in Carbohydrates as
Organic Raw Materials, Lichtenthaler, F. W., Ed., VCH: Weinheim, 1991, Vol. 1, p.
57-94.
2. (a) Trombotto, S.; Bouchu, A; Descotes G.; Queneau, Y. Tetrahedron Lett. 2000, 41,
8273-8277. (b) Queneau, Y.; Chambert, S.; Cheaib, R.; Listkowski, A.; Doutheau, A.;
Trombotto, S. Targets in Heterocyclic Systems – Chemistry and Properties, O. A.
Attanasi and Spinelli Eds., Italian Society of Chemistry: Rome, 2006, Vol. 10, p.132-
151.
3. Dean, B.; Oguchi, H.; Cai, S.; Otsuji, E.; Tashiro, K.; Hakomori, S.I.; Toyokuni, T.
Carbohydr. Res., 1993, 245, 175-192.
4. Petersson, G.; Pettersson, S.; Samuelson, O. Sv. Papperstidn. 1969, 72, 222-225 (Chem.
Abstr. 1969, 71, 23035v).
5. (a) Mandai, T.; Okumoto, H.; Oshitari, T.; Nakanishi, K.; Mikuni, K.; Hara, K. J.; Hara,
K. Z.; Iwatani, W.; Amano, T.; Nakamura, K.; Tsuchiya, Y Heterocycles 2001, 54,
561-566. (b) Mandai, T.; Okumoto, H.; Oshitari, T.; Nakanishi, K.; Mikuni, K.; Hara,
K. J.; Hara, K. Z. Heterocycles, 2000, 52, 129-132.
6. Nicolaou, K. C.; Trujillo, J. I.; Chibale, K. Tetrahedron, 1997, 53, 8751-8778.
7. (a) Boer, J.; Schuster, A.; Holzmann, B.; Kessler, H. Angew. Chem. Int. Ed. 2001, 40,
3870-387. (b) Locardi, E.; Boer, J.; Modlinger, A.; Schuster, A.; Holzmann, B.;
Kessler, H. J. Med. Chem. 2003, 46, 5752-5762.
8. Tronchet, J. M. J.; Zsély, M.; Geoffroy, M. Carbohydr. Res. 1995, 275, 245-258.
9. Ghosh, M.; Dulina, R. G.; Kalarda. R.; Sofia, M. J. J. Org. Chem. 2000, 65, 8387-8390.
10. Westermann, B.; Dörner, S. Chem. Commun. 2005, 2116-2118.
11. Grandjean, C.; Santraine, V.; Fardel, N.; Polidori, A.; Pucci, B.; Gras-Masse, H.;
Bonnet, D. Tetrahedron Lett. 2004, 45, 3451-3454.
12. Virta, P.; Karskela, M.; Lonnberg, H. J. Org. Chem. 2006, 71, 1989-1999.
13. Mogemark, M.; Elofsson, M.; Kihlberg, J. J. Org. Chem. 2003, 68, 7281-7288.
14. Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46,
3936-3938.
15. Santacroce, B. V.; Basu, A. Angew. Chem. Int. Ed. 2003, 42, 95-98.
203

16. Binder, W. H.; Schmid, W. Monatsch. Chem., 1995, 126, 923-931.
17. Lockhoff, O. Angew. Chem. Int. Ed. Engl. 1998, 37, 3436-3439.
18. Santacroce, B. V.; Basu, A. Angew. Chem. Int. Ed. 2003, 42, 95-98.
19. Fischer, E.; Helferich, B. Liebigs Ann. Chem. 1911, 383, 68-91.
20. Choudhury, A. K.; Kitaoka, M.; Hayashi, K. Eur. J. Org. Chem. 2003, 2462-2470.
21. Autar, R.; Liskamp, R. M. J.; Pieters, R. J. Carbohydr. Res. 2005, 340, 2436-2442.
22. Autar, R.; Khan, A. S.; Schad, M.; Hacker, J.; Liskamp, R. M. J.; Pieters, R. J.
ChemBioChem 2003, 4, 1317-1325.
23. Filippov, D. V.; van den Elst, H.; Tromp, C. M.; van der Marel, G. A.; van Boeckel, C.
A. A., Overkleeft, H. S.; van Boom, J. H. Synlett. 2004, 773-778.
24. Isbell, H. S.; Frush, H. L. Carbohydr. Res. 1987, 161, 181-193.
25. Cipolla, L.; Forni, E.; Jiménéz-Barbéro, J.; Nicotra, F. Chem. Eur. J. 2002, 8, 3976-
3983.
26. Le Diguarher, T.; Boudon, A.; Elwell, C.; Paterson, D. E.; Billington, D. C. Bioorg.
Med. Chem. Lett. 2004, 16, 1983-1988.
27. Moitessier, N.; Minoux, H.; Maigret, B.; Chretien, F.; Chapleur, Y. Lett. Pept. Sci.
1998, 8, 75-78.
28. Moitessier, N.; Dufour, H.; Chrétien, F.; Thiery, J. P.; Maigret, B.; Chapleur, Y.
Bioorg.Med. Chem. 2001, 9, 511-523.
29. Jain, R.; Kamau, M.; Wang, C.; Ippolito, R.; Wang, H.; Dulina, R.; Anderson, J.;
Gange, D.; Sofia, M. J. Bioorg. Med. Chem. Lett. 2003, 13, 2185-2189.
30. Sofia, M. J.; Hunter, R.; Chan, T. Y.; Vaughan, A.; Dulina, R.; Wang, H.; Gange, D. J.
Org. Chem. 1998, 63, 2802-2803.
31. Sofia, M. J. Med. Chem. Res. 1998, 8, 362-378.
32. Ziegler, T.; Gerling, S.; Lang M. Angew. Chem. Int. Ed. 2000, 39, 2109-2112.
33. Li, Y.; Zhang, X.; Chu, S.; Yu, K.; Guan, H. Carbohydr. Res. 2004, 339, 873-879.
34. Tamura, J.; Manabu, K. Japanese Patent JP 2002338592 (2002); Chem. Abstr. 2002,
137, 385072.
35. Cousins, R. P. C.; Pritchard, R. G.; Raynor, C. M.; Smith, M.; Stoodley, R. J.
Tetrahedron Lett. 2002, 43, 489-492.
36. Pierre, R.; Chambert, S.; Alirachedi, F.; Danel, M.; Trombotto, S.; Doutheau, A.;
Queneau, Y. C. R. Chimie 2008, 11, 61-66.
37. Bosco, M.; Bisseret, P.; Eustache, J. Tetrahedron Lett. 2003, 44, 2347-2349.
38. Bosco, M.; Bisseret, P.; Constant, P.; Eustache, J. Tetrahedron Lett. 2007, 48, 153-157.
204

39. Steyemark, P. R.; J. Org. Chem. 1962, 27, 1058.
40. Kovacs, J.; Pinter, I.; Messmer, A. Carbohydr. Res.1985, 141, 57.
41. Ichikawa, Y.; Matsukawa, Y.; Minoru, I. J. Am. Chem. Soc. 2006, 128, 3934-3938.
42. Maya, I.; López, Ó.; Fernández-Bolaňos, J. G.; Robina, I.; Fuentes, J. Tetrahedron Lett.
2001, 42, 5413-5416.
43. López, Ó.; Maya, I.; Fuentes, J.; Fernández-Bolaňos, J. G. Tetrahedron 2004, 60, 61-
72.
44. Le Chevalier, A.; Pierre, R.; Kanso, R.; Chambert, S.; Doutheau, A.; Queneau, Y.
Tetrahedron. Lett. 2006, 47, 2431-2434.
45. Trombotto, S.; Danel, M.; Fitremann, J.; Bouchu, A.; Queneau, Y. J. Org. Chem. 2003,
68, 6672-6678.
46. Chambert, S.; Cowling, S. J.; Mackenzie, G.; Goodby, J. W.; Doutheau, A.; Queneau,
Y. J. Carbohydr. Chem. 2007, 26, 27-39.
47. Goodby, J. W.; Görtz, V.; Cowling, S. J.; MacKenzie, G.; Martin, P.; Plusquellec, D.;
Benvegnu, T.; Boullanger, P.; Lafont, D.; Queneau, Y.; Chambert, S.; Fitremann, J.
Chem. Soc. Rev. 2007, 36, 1971-2032.
48. Sol, V., Charmot, A.; Krausz, P.; Trombotto, S.; Queneau, Y. J. Carbohydr. Chem.
2006, 25, 345-360.
49. Listkowski, A.; Ing, P.; Cheaib, R.; Chambert, S.; Doutheau, A.; Queneau, Y.
Tetrahedron: asymmetry, 2007, 18, 2201-2210.
50. Meutermans, W.; Le G. T.; Becker, B. ChemMedChem. 2006, 1, 1164-1194.
51. Murphy, P. V. Eur. J. Org. Chem. 2007, 4177-4187
52. Doores, K. J.; Gramblin, D. P.; Davis, B. G. Chem. Eur. J. 2006, 12, 656-665.
53. Hirschmann, R.; Nicolaou, K. C.; Pietranico, S.; Salvino, J.; Leahy, E. M.; Sprengeler,
P. A.; Furst, G.; Smith III, A. B. J. Am. Chem. Soc. 1992, 114, 9217-9218.
54. Hirschmann, R.; Nicolaou, K. C.; Pietranico, S.; Leahy, E. M.; Salvino, J.; Arison, B.;
Cichy, M. A.; Spoors, P. G.; Shakespeare, W. C.; Sprengeler, P. A.; Hamley, P.; Smith
III, A. B.; Reisine, T.; Raynor, K.; Maechler, L.; Donaldson, C.; Vale, W.; Freidinger,
R. M.; Cascieri, M. R.; Strader, C. D. J. Am. Chem. Soc. 1993, 115, 12550-12568.
55. Hirschmann, R.; Yao, W.; Cascieri, M. R.; Strader, C. D.; Maechler, L.; Cichy, M. A.,
Jr., J. H.; Sprengler, P. A.; Smith III, A. B. J. Med. Chem. 1996, 39, 2441.
56. Hirschmann, R.; Hynes, Jr. J.; Cichy-Knight, M. A; van Rijin, R. D.; Sprengeler P. A.;
Spoors, P. G.; Shakespeare, W. C.; Pietranico-Cole, S.; Barbosa, J.; Liu, J.; Yao, W.;
Rohrer, S.; Smith III, A. B. J. Med. Chem. 1998, 41, 1382-1391.
205

57. Prasad, V.; Birzin, E. T. ; McVaughn, C. T.; van Rijin, R. D.; Rohrer, S. P.; Chicchi,
G.; Underwood, D. J.; Thornton, E. R.; Smith III, A. B.; Hirschmann, R. J. Med.
Chem. 2003, 46, 1858-1869.
58. Papageorgiou, C.; Haltiner, R.; Bruns, C.; Petcher, T. J. Bioorg. Med. Chem. Lett.
1992, 2, 135-140.
59. Le Merrer, Y.; Poitout, L.; Depezay, J. Methods in Molecular Medicine,
Peptidomimetic Protocols, Vol. 23, (Ed. Kazmierski), Humana Press, Totowa, NJ,
1999, p. 227-257.
60. Gouin, S. G.; Murphy, P. V. J. Org. Chem. 2005, 70, 8527-8523.
61. Chagnault, V.; Lalot, J.; Murphy, P.V. ChemMedChem, 2008, sous presse.
62. Capozzi, G.; Giannini, S.; Menichetti, S.; Nativi, C.; Giolitti, A. ; Patacchini, R. ;
Perrotta, E.; Altamura, M. ; Maggi, C. A.; Bioorg. Med. Chem. Lett. 2002, 12, 2263-
2266.
63. Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla,
L. Eur. J. Org. Chem. 2008, 635–639.
64. Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla,
L. Carbohydr. Res.2008, sous presse.
65. Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I.
Bioorg. Med. Chem. Lett. 2003, 13, 2737-2740.
66. Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I.
Bioorg. Med. Chem. Lett. 2003, 13, 2741-2744.
67. Moriyama, H.; Tsukida, T.; Inoue, Y.; Yokota, K. ; Yoshino, K. ; Kondo, H.; Miura,
N.; Nishimura, N.-I. J. Med. Chem. 2004, 47, 1930-1938.
68. Tsukida, T.; Moriyama, H.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, N.-I.
Bioorg. Med. Chem. Lett. 2004, 14, 1569-1572.
69. Fragai, M.; Nativi, C.; Richichi, B.; Venturi, C. ChemBioChem 2005, 6, 1345-1349.
70. Murphy, P. V.; O’Brien, J. L.; Gorey-Feret, L. J.; Smith III, A. B. Tetrahedron 2003,
59, 2259-2271.
71. Chery, F.; Cronin, L.; O’Brien, J. L.; Murphy, P.V. Tetrahedron 2004, 60, 6597-6608.
72. Araújo, A. C.; Nicotra, F.; Airoldi, C.; Costa, B.; Giagnoni, G.; Fumagalli, P.; Cipolla,
L Carbohydr. Res.2008, sous presse.
73. Van Hoof, S.; Ruttens, B.; Hubrecht, I.; Smans, G.; Blom, P.; Sas, B.; Van Hemel, J.;
Vandenkerckhove, J.; Van der Eycken, J. Bioorg. Med. Chem. Lett. 2006, 16, 1495-
1498.
206

74. Dinh, T. Q.; Smith, C. D.; Du, X.; Armstrong, R. W. J. Med. Chem. 1998, 51, 981-987.
75. Hanessian, S.; Saavedra, O. M.; Xie, F.; Amboldi, N.; Battistini, C. Bioorg. Med.
Chem. Lett. 2000, 10, 439-442..
76. Hanessian, S.; Zhan, L.; Bovey, R.; Saavedra, O. M.; Juillerat-Jeanneret, L.; J. Med.
Chem. 2003, 46, 3600-361.
77. Tamaruya, Y.; Suzuki, M.; Kamura, G.; Kanai, M.; Hama, K.; Shimizu, K.; Aoki, J.;
Arai, H.; Shibasaki, M. Angew. Chem. Int. Ed. 2004, 43, 2834-2837.
78. Edwards, A. A.; Ichihara, O.; Murfin, S.; Wilkes, R.; Whittaker, M.; Watkin, D. J.;
Fleet, G. W. J. J. Comb. Chem. 2004, 6, 230-238.
79. Maione A. M. ; Giordani, E. ; Costa, M.; Spirito A. Arch. Pham Pharm. Med. Chem.
1997, 330, 17-20
80. Sofia, M. J.; Allanson, N.; Hatzenbuhler, N. T.; Jain, R.; Kakarla, R.; Kogan, N.;
Liang, R.; Liu, D.; Silva, D. J.; Wang, H.; Gange, D.; Anderson, J.; Chen, A.; Chi, F.;
Dulina, R.; Huang, B.; Kamau, M.; Wang, C.; Baizman, E.; Branstrom, A.; Bristol, N.;
Goldman, R.; Han, K.; Longley, C.; Midha, S.; Axelrod, H. R. J. Med. Chem. 1999,
42, 3193-3198.
81. Halliday, J.; McKeveney, D.; Muldoon, C.; Rajaratnam, P.; Meutermans, W. Biochem.
Pharmacol. 2006, 71, 957-967.
82. Wong, C.-H.; Hendrix, M.; Manning, D. D.; Rosenbohn, C.; Greenberg, W. A. J. Am.
Chem. Soc. 1998, 120, 8319-8327.
83. Liao, Y.; Li, Z, Wong, N. C. Chin. J. Chem. 2001, 19, 1119-1129.
84. Hirschmann, R.; Ducry, L.; Smith III, A. B. J. Org. Chem. 2000, 65, 8307-8316.
85. Abrous, L.; Jokiel, P.A.; Friedrich, S. R.; Hynes Jr, J.; Smith III, A. R.; Hirschmann, R.
J. Org. Chem. 2004, 69, 280-302.
86. Abrous, L.; Hynes Jr, J.; Friedrich, S. R.; Smith III, A. R.; Hirschmann, R. Org. Lett..
2001, 3, 1089-1092.
87. Brill, W.; K.-D.; Tirefort, D. Tetrahedron Lett. 1998, 39, 787-790.
88. Becattini, B.; Capozzi, G.; Falciani, C.; Menichetti, S.; Nativi, C.; Salvini, A. J.
Carbohydr. Chem. 2000, 19, 653-657.
89. Svejgaard, L.; Fuglsang, H.; Jensen P. B.; Kelly N. M.; Pederson, H.; Andersen, K.;
Ruhland, T.; Jensen, K. J. J. Carbohydr. Chem. 2003, 22, 179-184.
90. Moitessier, N.; Henry, C.; Aubert, N.; Chapleur, Y. Tetrahedron Lett. 2005, 46, 6191-
6194.
207

91. Saeed, M.; Abbas, M.; Raid, J. A. J.; Zahid, M.; Voelter, W. Tetrahedron Lett. 2003,
44, 315-317.
92. Krueger, E. B.; Hopkins, T. P.; Keaney, M. P.; Wlaters, M. A.; Boldi, A. M. J. Comb.
Chem. 2002, 4, 229-238.
93. Sanjayan, G. J.; Steward, A.; Hachisu, S.; Gonzalez, R.; Watterson, M. P.; Fleet, G. W.
J. Tetrahedron Lett. 2003, 44, 5847-5851.
94. Watterson, M. P.; Edwards, A. A.; Leach, J. A.; Smith, M. D.; Ichihara, O.; Fleet, G.
W. J. Tetrahedron Lett. 2003, 44, 5853-5857.
95. Timmer, M. S. M.; Verhelst, S. H. L.; Grotenberg, G. M.; Overhand, M.; Overkleeft,
H. S. Pure Appl. Chem. 2005, 77, 1173-1181.
96. Timmer, M. S; Risseeuw, M. D. P.; Verdoes, M.; Filipov, D. V.; Plaisier, J; R.; van der
Marel, G. A.; Overkleeft, H. S.; van Boom, J. H. Tetrahedron: Asymmetry. 2005, 16,
177-185.
97. Agoston, K.; Geyer, A. Chem. Eur. J. 2005, 11, 6407-6413.
98. Meutermans, W.; West, M. L.; Adamson, G.; Le, G. T.; Drinnan, N. B.; Abbenante, G.;
Becker, B.; Grathwohl, M.; Rajaratnam, P.; Tometski, G. Disaccharides for Drug
Discovery, Alchemia Pty. Ltd, 2003.
99. Moreaux, L.; Sandre, O.; Charpak, S.; Blanchard-Desce, M.; Mertz, J. Biophys. J.
2001, 80, 1568-1574.
100. Campagnola, P. J.; Wei, M.-D.; Lewis, A.; Loew, L. M. Biophys. J. 1999, 77, 3341-
3349.
101. Dombeck, D. A.; Blanchard-Desce M.; Webb, W. W. J. Neurosci. 2004, 24, 999–
1003.
102. Nemet, B. A.; Nikolenko, V.; Yuste, R. J. Biomed. Opt. 2004, 9, 873–881.
103. Moreaux, L. ; Pons, T. ; Dambrin, V. ; Blanchard-Desce M. ; Mertz, J. Opt. Lett.,
2003, 28, 625–627.
104. Bouevitch, O.; Lewis, A.; Pinevsky, I.; Wuskell J. P.; Loew, L. M. Biophys. J. 1993,
65, 672–679.
105. Centonze, V. E.; White, J. G. Biophys. J. 1998, 75, 2015-2024.
106. Blanchard-Desce, M. Comptes Rendus Physique 2002, 3, 439-448.
107. Campagnola, P. J.; Wei, M.-D.; Lewis, A.; Loew, L. M. Biophys. J. 1999, 77, 3341-
3349.
108. Mertz, J. Curr. Opin. Neurobiol. 2004, 14, 610-616.
109. Zipfel, W. R.; Williams, R. M.; Webb, W. W. Nat. Biotechnol. 2003, 21, 1369-1377.
208

110. Bouevitch, O.; Lewis, A.; Pinevsky, I.; Wuskell, J. P.; Loew, L. M. Biophys. J. 1993,
65, 672-9.
111. Millard, A. C.; Jin, L.; Mei-de, W.; Wuskell, J. P.; Lewis, A.; Loew, L. M. Biophys. J.
2004, 86, 1169-1176.
112. (a) Danishefsky, S. J.; Allen, J. R. Angew. Chem. 2000, 836-863. (b) Galonić, D. P.,
Gin, D. Y. Nature, 2007, 446, 1000-1007. (c) Seeberger, P. H.; Werz, D. B. Nature,
2007, 446, 1046-1051. (d) Seeberger, P. H. Nature, 2005, 437, 1239.
113. Schwarzmann, G. Semin. Cell. Dev. Biol. 2001, 12, 163-171.
114. (a) Premkumar, D. R. ; Fukuoka, Y. ; Sevlever, D. ; Brunschwig, E.; Rosenberry, T.
L.; Tykocinski, M. L.; Medof, M. E. J. Cell Biochem. 2001, 82, 234-245. (b) Paulick,
M. G. ; Wise, A. R. ; Forstner M. B.; Groves, J. T.; Bertozzi, C. R. J. Am. Chem. Soc.
2007, 129, 11543-11550.
115. Rabuka ; D. ; Forstner, M. B. ; Groves, J. T.; Bertozzi, C. R. J. Am. Chem. Soc. 2008,
sous presse.
116. Schmidt, R. R. Modern Methods in Carbohydrate Synthesis; Kahn, S. H.; O’Neill, R.
A.; Eds.; Hardwood academic publishers, Amsterdam, 1996; p. 20-54.
117. Klotz, W.; Schmidt, R. R. Liebigs Ann. Chem. 1993, 683-690.
118. Tsvetkov, Y. E.; Klotz, W.; Schmidt, R. R. Liebigs Ann. Chem. 1992, 371-375.
119. Platzek J.; Graske, K. D.; Niedballa, U. Int Patent WO 2003006474 (2003); Chem.
Abstr. 2003, 138, 106941.
120. Platzek J.; Niedballa, U.; Graske, K. D. German patent DE 10129677 (2003); Chem.
Abstr. 2003, 138, 56184.
121. Platzek J.; Niedballa, U.; Graske, K. D. Int Patent WO 2002102816 (2002); Chem.
Abstr. 2003, 138, 39497.
122. Jarosz, A.; Listkowski, A.; Lewandowski, B.; Ciunik, Z.; Brzuszkiewicz, A.
Tetrahedron 2005, 61, 8485-8492.
123. Wang, X.; Migawa, M. T.; Sannes-Lowery, K. A.; Swaze, E. E. Bioorg. Med. Chem.
Lett. 2005, 15, 4919-4922.
124. Raju, B.; Mortell, K.; Anandan, S.; O’Dowd, H.; Gao, H.; Gomez, M.; Hackbarth, C.;
Wu, C.; Wang, W.; Yuan, Z.; White, R.; Trias, J.; Patel, D. V. Bioorg. Med. Chem.
Lett. 2003, 13, 2413-2418.
125. Excoffier, G.; Gagnaire, D.; Utille J.-P. Carbohydr. Res. 1975, 39, 368-373.
126. Nudelman, A.; Herzig, J.; Gottlieb H. E. Carbohydr. Res. 1987, 162, 145-152.
209

127. Ellestad, G. A. ; Martin, J. H. ; Morton, G. O., Sassiver, M. L. ; Lancaster, J. E. J.
Antibiotics 1982, 10, 1418-1421.
128. Flowers, H. M. Carbohydr. Res. 1983, 119, 78-84.
129. (a) Schmidt, R. R. Angew. Chem. Int. Ed. Eng. 1986, 25, 212-235. (b) Schmidt, R. R.;
Michel, J. Roos, M. Liebigs Ann. Chem. 1984, 1343-1357. (c) Schmidt, R. R.; Michel,
J. Tetrahedron Lett. 1984, 25, 821-824.
130. Lubineau, A.; Escher, S.; Alais, J.; Bonnaffé, D. Tetrahedron Lett. 1997, 38, 4087-
4090.
131. Terjung, A.; Jung, K.-H. ; Schmidt, R. R. Carbohydr. Res. 1997, 297, 229-242.
132. Schmidt, R. R. Angew. Chem. Int. Ed. 1986, 25, 212-235.
133. Tamura, J.; Schmidt, R. R. J. Carbohydr. Chem. 1995, 14, 895-911.
134. Azoulay, M.; Escriou, V.; Florent, J.-C.; Monneret, C. J. Carbohydr. Chem. 2001, 20,
1093-1101.
135. Klotz, W.; Schmidt, R. R. J. Carbohydr. Chem. 1994, 1093-1101.
136. Vauzeilles, B.; Dausse, B.; Palmier, S.; Beau, J.-M. Tetrahedron Lett. 2001, 42, 7567-
7570.
137. Sasaki, A.; Naoichi, M. JP Patent 06271597, (1994).
138. Sleiman, M. Thèse de Doctorat de L'Université Claude Bernard Lyon 1, N° d'ordre
291-2007.
139. Lemieux, R. U.; Hayami, J.-I. Can. J. Chem. 1965, 2162-6173.
140. Nitz, M.; Purse, B. W.; Bundle, D. R. Org. Lett. 2000, 2, 2939-2942.
141. Bellucci, G.; Chiappe, C.; D'Andrea. Tetrahedron: Asymmetry 1995, 6, 221-230.
142. Lecourt, T.; Herault, A.; Pearce, A. J.; Sollogoub, M.; Sinaÿ, P. Chem. Eur. J. 2004,
10, 2960-2971.
143. Chevalier-du Roizel, B.; Cabianca, E.; Rollin, P.; Sinaÿ, P. Tetrahedron 2002, 58,
9579-9583.
144. Sollogoub, M.; Das, S. K.; Mallet, J-M, Sinaÿ, P. C. R. Acad. Sci. Paris 1999, 2, 441-
448.
145. Garegg, P. J. Adv. Carbohydr. Chem. Biochem. 2004, 59, 69-134.
146. Close, D. M. Chem. Rev. 1979, 79, 491-513.
147. David, S.; Hanessian, S. Tetrahedron 1985, 41, 643-663.
148. Lemieux, R. U.; Ratcliffe R. M. Can. J. Chem. 1979, 2, 1244-1251.
149. Seeberger, P. H., Danishefsky, S. J. J. Acc Chem. Res. 1998, 31, 685-695.
150. Martin O. R.; Kurz, K. G. ; Rao, S. P. J. Org. Chem. 1987, 52, 2922-2925.
210

151. Dedola, S., Nepogodiev, S. A., Field, R. A. Org. Biomol. Chem. 2007, 5, 1006-1017.
152. Möller, U.; Hobert, K.; Donnerstag, A.; Wagner, P.; Müller, D.; Fehlhaber H.-W.;
Markus. A.; Welzel, P. Tetrahedron 1993, 49, 1635-1648.
153. Luning, J.; Möller, U.; Müller, D.; Welzel, P.; Markus. A.; van Heijenoort, Y.; van
Heijenoort, J. Tetrahedron 1993, 49, 10587-10596.
154. Eager, A. R.; Finney, N. S. J. Org. Chem. 2004, 69, 613-618.
155. Hanover. J.A.; Lai, Z.; Lee, G.; Lubas, W. A.; Sato, S. M. Arch. Biochem. Biophys.
1999, 362, 38-45.
156. (a) Jung, K.-H.; Schmidt, R. R. Carbohydrate –based Drug Discovery, Volume 2,
Wong, Chi-Huey (Ed.), Wiley-VCH- Verlag, Weinheim, p.609-659. (b) Unligil, U.
M.; Rini, J. M. Curr. Opin. Struct. Biol. 2000, 10, 510-517.(c) Zachara, N. E.; Hart, G.
W. Chem. Rev., 2002, 102, 431–438.
157. (a) Kiso, M.; Ishida, H.; Kawaide, A.; Hasegawa, A. Carbohydr. Res. 1984, 127, 129–
135; (b) Knapp, S.; Kukkola, P. J.; Sharma, S.; Dhar, T. G. M.; Naughton, A. B. J. J.
Org. Chem. 1990, 55, 5700–5710; (c) Pavliak, V.; Kovac, P. Carbohydr. Res. 1991,
210, 333–337; (d) Sato, K.; Yoshitomo, A. Chem. Lett. 1995, 39–40; (e) Sato, K.;
Yoshitomo, A.; Takai, Y. Bull. Chem. Soc. Jpn. 1997, 70, 885–890; (f) Stolz, F.;
Reiner, M.; Blume, A.; Reutter, W.; Schmidt, R. R. J. Org. Chem. 2004, 69, 665–679;
(g) Popelova, A.; Kefurt, K.; Hlavackova, M.; Moravcova, J. Carbohydr. Res. 2005,
340, 161–166; (h) Teodorović, P.; Slättegård R. ; Oscarson, S. Carbohydr. Res. 2005,
340, 2675–2676. (i) Zhang, J.; Yan, S.; Liang, X.; Wu, J.; Wang, D.; Kong, F.
Carbohydr. Res. 2007, 342, 2810-2817.
158. Lichtenthaler, F. W.; Mondel, S. Carbohydr. Res. 1997, 303, 293-302.
159. Barsu, C.; Fortrie, R.; Nowika, K.; Baldeck, P. L.; Vial, J. C.; Barsella, A.; Fort, A.;
Hissler, M.; Bretonnière, Y.; Maury, O.; Andraud, C. Chem. Commun., 2006, 4744–
4746.
160. Lottner, C.; Bart, K.-C.; Bernhardt, G.; Brunner, H. J. Med. Chem. 2002, 45, 2079-
2089.
161. Oar, M. A.; Serin, J. M.; Dichtel, W. R.; Frechet, J. M. J.; Ohulchanskyy, T. Y.;
Prasad, P. N. Chem. Materials 2005, 17, 2267-2275.
162. Pengo, P.; Polizzi, S.; Battagliarin, M.; Pasquato, L.; Scrimin, P. J. Mat. Chem. 2003,
13, 2471-2478.
163. Abert, M.; Mora, N.; Lacombe, J.-M., Carbohydr. Res. 2002, 337, 11, 997-1006.
164. Vollhardt, D.; Fainerman, V. B. J. Colloid. Interface Sci. 2000, 86, 103-151.
211

165. Maget-Dana, R. Biochim. Biophys. Acta. 1999, 1462, 109-140.
166. Girard-Egrot, A.; Chauvet, J.-P.; Gillet, G.; Moradi-Améli, M. J. Mol. Biol. 2004,
335, 321-331.
167. Mchedlov-Petrossyan N.O.; Vodolazkaya N.A., Bezkrovnaya O. N.; Yakubovskaya
A. G.; Tolmachev A. V.; Grigorovich A. V. Spectrochimica Acta. 2008, 69, 1125–
1129.
168. Biadasz, A.; Qabuszewska, K.; Chrzumnicka, E.; Michalowski, E.; Martynski, T.;
Bauman D. Dyes and Pigments 2007, 74, 598-607
169. Lenahan, K.M.; Wang; Y.X.; Liu, Y.J.; Claus, R.O.; Heflin, J.R.; Marciu, D.; Figura,
C. Adv. Mat. 1998, 10, 853.
170. Li, H.; Zhou, D.J; Huang, C.H.; Xu, J.M.; Li, T.K; Zhao, X.S.; Xia, X.H J. Chem.
Soc. Faraday trans. 1996, 92, 2585-2592.
171. Lupo, D.; Ringsdorf, H.; Schuster, A.; Seitz, M. J. Am. Chem. Soc. 1994, 116, 10498-
10506.
172. Schoondorp, M. A.; Schouten, M. A.; Hulshof, J. B. E.; Feringa, B. L. Thin Sol. Film
1992, 210, 166-168.
173. Laschewsky, A.; Paulus, W.; Ringsdorf, H.; Lupo, D.; Ottenbreit, P.; Prass, W.;
Bubeck, C.; Neher, D.; Wegner, G. Makromol. Chemie, Macromol. Symp. 1991, 46,
205-210.
174. Kato, N.; Saito, K.; Ueso, Y. Thin. Sol. Films. 1999, 338, 5-8.
212

RESUME La carboxyméthyl 3,4,6-tri-O-acétyl-α-D-glucopyranoside 2-O-lactone, préparée à partir de l’isomaltulose, est un bon « donneur de glycoside » pour la synthèse de composés ciblés soit pour leur intérêt biologique soit pour leurs propriétés physico-chimiques potentielles tels que des pseudo-oligosaccharides, pseudo-glycoaminoacides, et pseudo-glycolipides. Dans ce contexte, de nouvelles stratégies de synthèse ont été mises au point pour l’obtention de ces lactones construites sur des systèmes mono- et disaccharidiques avec des groupements protecteurs variés. D’autre part, la fonctionnalisation de la position 2 sélectivement libérée après ouverture de cette lactone par des agents nucléophiles, a été effectuée pour la synthèse de systèmes multifonctionnels. Une application de cette stratégie a aussi été étudiée pour la synthèse de produits amphiphiles pour la conception de sondes membranaires hydrosolubles pour l’imagerie membranaire par microscopie optique non linéaire. TITLE CARBOXYMETHYL GLYCOSIDE LACTONES : SYNTHESIS AND APPLICATION FOR MEMBRANE IMAGERY ABSTRACT The carboxymethyl 3,4,6-tri-O-acetyl-α-D-glucopyranoside 2-O-lactone, prepared starting from isomaltulose, has shown to be a good “glycoside donor” for the synthesis of compounds targeted either for their biological interest or for their potential physicochemical properties such as pseudo-oligosaccharides, pseudo-glycoaminoacids, and pseudo-glycolipids. New strategies of synthesis for obtaining carboxymethyl glycoside lactones based on mono- and disaccharidic systems with varied protective groups are now presented. In addition, the fonctionnalisation of position 2 selectively released after opening of these lactones by nucleophilic agents allowed the synthesis of multifunctionnalised systems. An application of this strategy was also studied for the synthesis of amphiphilic products for the design of water-soluble membrane probes for the membrane imagery by non linear optical microscopy. DISCIPLINE Chimie MOTS-CLES Carboxyméthyl glycosides, carboxyméthyl glycosides lactones, scaffolds saccharidiques, alkylation anomérique, sondes membranaires, optique non linéaire, fluorescence à deux photons, génération du second harmonique. KEYWORDS Carboxymethyl glycosides, carboxymethyl glycoside lactones, saccharidic scaffolds, anomeric alkylation, membrane probes, non linear optics, two-photon excitation fluorescence, second harmonic generation. INTITULE ET ADRESSE DE DU LABORATOIRE Laboratoire de Chimie Organique de l’INSA de Lyon Institut de Chimie et Biochimie Moléculaire et Supramoléculaire (ICBMS) UMR CNRS-UCBL-CPE-INSA 5246 Domaine Scientifique de la DOUA, bâtiment Jules Verne 20 avenue Albert Einstein, 69621 Villeurbanne Cedex, France






![Grenadille...Grenadille 3 Composition D'après la revue bibliographique de Dhawan et als[5] (2004), les phyto-constituants de Passiflora edulis sont constitués par • des glycosides](https://img.pdfslide.fr/doc/110x75/5e80415cc5b8852f6f0db725/-grenadille-3-composition-daprs-la-revue-bibliographique-de-dhawan-et-als5.jpg)






















