Embed Size (px)
Citation preview

Leucodystrophies de l'adulte [17-066-A-70] - Doi : 10.1016/S0246-0378(15)62629-X
P. Labauge a : MD, PhD, X. Ayrignac a : MD, C. Carra-Dallière a : MD, N. Menjot de Champfleur b : MD, PhD
Sous presse. Épreuves corrigées par l'auteur. Disponible en ligne depuis le mercredi 17 juin 2015
Introduction Les maladies de la substance blanche d'origine génétique sont multiples. Deux grands groupes les opposent : les maladies vasculaires et les maladies démyélinisantes. L'objectif de cette mise au point est d'en définir les principales causes et d'élaborer un arbre diagnostique.
Leucodystrophies vasculaires Les arguments devant faire évoquer au clinicien une maladie de la substance blanche d'origine vasculaire et génétique sont : un âge jeune, moins de 50 ans, l'absence de facteurs de risque vasculaires (hypertension artérielle, diabète, hypercholestérolémie, tabagisme actif) et l'absence de cardiopathie emboligène. En cas de doute, les résultats de l'échographie des vaisseaux du cou, la présence d'une rétinopathie hypertensive vont orienter la démarche étiologique vers une maladie athéroscléreuse plutôt acquise que génétique. À l'inverse, un âge jeune, l'absence de facteurs de risque vasculaires doivent faire évoquer une maladie génétique, même en l'absence d'antécédents familiaux. La maladie la plus connue est le CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) [1].
CADASIL
Il s'agit d'une maladie autosomale dominante, ayant comme symptômes cliniques des accidents lacunaires à répétition, une démence de type sous-cortical, associés à des migraines avec aura, et des états psychiatriques, dépression, voire mélancolie ou état maniaque. Le gène muté à l'origine de cette pathologie est Notch3 [2], localisé sur le bras long du chromosome 19. L'aspect neuroradiologique associe des infarctus de type lacunaire, intéressant la capsule interne, le corps calleux, le thalamus, les noyaux caudés, la partie centrale de la protubérance. Est associée une leucoaraïose, consistant en de vastes plages de démyélinisation, à prédominance périventriculaire et respectant le plus souvent les fibres en U (Figure 1). Ces aspects neuroradiologiques peuvent parfois, lorsque les noyaux gris sont respectés, en imposer pour une sclérose en plaques. Lorsque sont réalisées des séquences en écho de gradient, il est mis en évidence des hyposignaux, rentrant dans le cadre de microbleeds [3, 4]. Leur présence est très importante, car ils ne sont jamais retrouvés dans les leucodystrophies non vasculaires. Un signe neuroradiologique très utile dans ce contexte est la mise en évidence à la phase précoce d'hypersignaux de la pointe des lobes temporaux [5]. Il en est de même de l'atteinte symétrique des capsules externes, réalisant des images dites en « croissant », et de l'atteinte du corps calleux [5].

La cause de cette pathologie est une fragilité des parois des vaisseaux secondaire à une raréfaction des cellules musculaires lisses, appendues aux vaisseaux artériels, qu'ils soient neurologiques ou en dehors du système nerveux central. La mutation du gène Notch3 entraîne une accumulation du récepteur Notch3 sur la membrane de ces vaisseaux. Un aspect caractéristique sur le plan histologique est la mise en évidence de dépôts, appelés granular osmiophilic material (GOM), visibles uniquement en microscopie électronique, sur les membranes basales de ces vaisseaux.
Le gène Notch3 comprend 33 exons, les mutations pathogènes étant clustérisées dans quatre exons principaux (3,4,11,18).
Une forme récessive simulant une maladie CADASIL a été décrite (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy [CARASIL]). Il s'agit d'une affection essentiellement retrouvée dans la population asiatique, associée à des symptômes extraneurologiques, alopécie, atteinte articulaire. Le gène muté, HTRA1, a été récemment identifié [6].
Il est à noter cependant qu'un certain nombre de leucodystrophies vasculaires, remplissant l'ensemble des critères cliniques et neuroradiologiques de CADASIL, n'ont pas de mutation retrouvée dans le gène Notch3. Elles sont appelées Cadasil-like et correspondent à des maladies héréditaires des petits vaisseaux intracrâniens. Il est à souligner que lorsque des biopsies cutanées sont réalisées, il est mis en évidence des dépôts le long des vaisseaux, mais qui n'ont pas les caractéristiques des dépôts observés dans la maladie CADASIL. Il a été ainsi mis en évidence une forme dominante, sans mutation du gène Notch3, partageant certaines caractéristiques neuroradiologiques de CADASIL, mais sans infarctus lacunaire ni hémorragie, et avec une atteinte constante du pont, dont le gène est localisé sur le bras long du chromosome 20 (20q13) [7].
Leucodystrophie vasculaire et mutation du gène COL4A1
Le gène COL4A1 fait partie d'une grande famille de collagène, protéine de soutien des vaisseaux.
Le tableau initial décrit a été celui de sujets présentant une hémiplégie infantile, associée à une leucodystrophie ayant les caractéristiques vasculaires, potentiellement associées à de vastes cavités porencéphaliques, des tortuosités rétiniennes, une cataracte congénitale et d'autres malformations de la chambre antérieure de l'œil [1]. Le tableau s'est ensuite enrichi par la description de patients ayant des crampes musculaires, d'anévrismes intracrâniens et d'insuffisance rénale, tableau appelé HANAC syndrome (hereditary angiopathy with nephropathy, aneurysms and muscle cramps). Ils sont en rapport avec différentes mutations dans le même gène, une douzaine de familles ayant été récemment rapportées.
Des phénotypes voisins ont été décrits en rapport avec des mutations du gène COLIVA2, associant hémorragies cérébrales et cavités porencéphaliques.
Autres leucodystrophies vasculaires
Elles sont excessivement rares.
« Cerebroretinal microangiopathy with calcifications and cysts » (CRMCC)

Cette maladie associe une démyélinisation, des calcifications et des kystes intracrâniens (leukoencephalopathy with calcifications and cysts [LCC]). L'association à des signes extraneurologiques (télangiectasies rétiniennes, ostéoporose) la rattache au syndrome de Coats plus (MIM 612199). Un des gènes mutés (CTC1) vient d'être identifié dans les formes pédiatriques de CRMCC.
« Hereditary endotheliopathy with retinopathy, nephropathy and stroke » (HERNS)
L'association de lésions rehaussées par le produit de contraste, notamment dans les régions frontopariétales, entourées d'une réaction œdémateuse, d'une insuffisance rénale dans un contexte de transmission autosomale dominante, fait évoquer le diagnostic de syndrome HERNS. Le gène muté est TREX1 (3p21.11). De façon très intéressante, ce gène est muté dans le syndrome d'Aicardi-Gouttières, décrit essentiellement chez l'enfant sous la forme de leucodystrophie récessive associée à des calcifications intracrâniennes.
Angiopathie amyloïde cérébrale
L'angiopathie amyloïde cérébrale (CAA) associe une démyélinisation, des hémorragies lobaires récidivantes et des microbleeds, prédominant dans les régions lobaires. Les formes familiales sont caractérisées par un âge de début plus précoce, une sévérité clinique plus importante. En dehors d'une notion familiale avérée, la recherche d'une mutation causale, le plus souvent amyloid precursor protein (APP), est peu contributive.
Leucodystrophies cavitaires Un groupe de leucodystrophies doit être dégagé : il s'agit des formes cavitaires. Elles associent de vastes plages d'hyposignaux en fluid attenuated inversion recovery (Flair) au sein de zones de démyélinisation (Figure 2).
Cet aspect particulier oriente vers deux pathologies tout à fait distinctes : une éventuelle sclérose en plaques ou un CACH syndrome (childhood ataxia with central nervous system hypomyelination) ou VWM (vanishing white matter disease). En faveur du premier diagnostic, la notion de poussées cliniques antérieures, de bandes oligoclonales sur la ponction lombaire, d'une myélite sur l'imagerie par résonance magnétique (IRM) médullaire sont autant d'arguments pour une pathologie inflammatoire acquise.
Syndrome CACH/VWM [8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24]
La forme la plus classique et initialement rapportée a été décrite comme se manifestant dans l'enfance, entre 2 et 6 ans. Elle est caractérisée par une atteinte neurologique progressive associant une ataxie cérébelleuse, une spasticité modérée, et une diminution modérée des fonctions cognitives. L'évolution est caractérisée par des épisodes paroxystiques de détérioration majeure et aigus neurologiques, dans les suites de différents facteurs déclenchants, traumatismes crâniens minimes, infections, fièvre, frayeur brutale. Durant ces épisodes paroxystiques, il existe des troubles de conscience, un déficit moteur, une hypotonie, pouvant aboutir à un coma, voire même à un décès. La récupération neurologique est en

général incomplète, avec des séquelles irréversibles et un décès quelques années après ces épisodes paroxystiques.
Variantes phénotypiques
Dépendants de l'âge de début, différents phénotypes ont été décrits :
• formes anténatales : diminution des mouvements du fœtus, oligohydramnios, diminution de la taille fœtale, microcéphalie. Dès la naissance, une détérioration neurologie rapide est notée, comprenant des vomissements, hypotonie axiale, épisodes d'apnées, insuffisance respiratoire, coma et mort en quelques mois ;
• formes infantiles. Dénommée aussi cree leucoencephalopathy, cette forme a été décrite parmi les Indiens de la tribu Cree. Le début se situe entre 3 et 9 mois, et entraîne un décès rapide ;
• forme tardive de l'adolescent et de l'adulte. Ce sont des formes asymptomatiques, à début tardif, commençant en moyenne à 46 ans (extrêmes : 16-62 ans), uniquement psychiatrique, démentielle. Deux caractéristiques sont à retenir dans les formes adultes : facteur environnemental (fièvre, stress, chute) déclenchant ou aggravant les premiers symptômes dans 38 % des cas [24], et une insuffisance ovarienne, extrêmement fréquente chez la femme dans notre série adulte. Cette présence est regroupée sous le terme d'ovarioleukodystrophy. Outre l'aspect cavitaire, retrouvé dans 90 % des formes adultes de notre étude, l'aspect IRM retrouve une atrophie cérébrale, un respect des fibres en U, une atteinte du corps calleux et du cervelet [24].
Pathologie
L'examen macroscopique cérébral met en évidence un aspect gélatineux ou cavitaire du cerveau. L'atteinte prédomine au niveau du lobe frontal et pariétal, et des zones périventriculaires sont particulièrement atteintes, tandis que le lobe temporal, les voies optiques, le corps calleux, la capsule interne sont le plus souvent respectés.
L'étude microscopique met en évidence une substance blanche pale, un amincissement des gaines de myéline, des aspects de vacuoles, une perte de fibres myélinisées et des vacuoles et un aspect kystique. La présence de lipophages, macrophages remplis de debris myéliniques, est rarement retrouvée. La substance grise est le plus souvent respectée, comparée à l'atteinte de la substance blanche. Il n'y a jamais de réaction inflammatoire. L'importance de la perte axonale est corrélée à celle des cavités. Les gaines de myéline sont anormales, et variant d'une simple pâleur à un aspect trop fin, voire à des vacuoles.
Il est également noté une augmentation de taille des oligodendrocytes, mise en évidence dans les aires de démyélinisation. Les astrocytes sont quant à eux dysmorphiques et augmentés de taille.
Données génétiques
Cinq gènes mutés EIF2B1-5 sont à l'origine de cette maladie. Ils codent cinq sous-unités d'un facteur de translation protéique (eukaryotic translation initiation factor [eIF2B] [eIF2Bα, β, γ, δ et ]).

Les deux tiers des patients atteints de CACH/VWM sont porteurs de mutations situées sur le gène EIF2B5 qui est la plus grande sous-unité des EIF2B, avec chez l'adulte une mutation récurrente, R113H.
Leucodystrophies métaboliques Elles sont secondaires à des erreurs congénitales du métabolisme (Tableau 1). Leur diagnostic nécessite une confirmation moléculaire.
Les principaux aspects devant faire évoquer une maladie métabolique sont les suivants (Tableau 2) :
• hypersignaux symétriques sur les séquences pondérées T2 ;
• caractère extensif de la démyélinisation ;
• absence de rehaussement des lésions après injection de gadolinium ;
• atteinte sélective de certaines structures de la substance blanche : atteinte préférentielle de la substance blanche du lobe frontal ou pariétal, atteinte élective des fibres corticospinales, du corps calleux ;
• respect ou atteinte des fibres en U ;
• absence de lésions de petite taille ou de forme ovoïde ;
• atteinte du tronc cérébral ou des lobes cérébelleux.
Autres leucodystrophies La généralisation du parc IRM a permis progressivement de discerner quelques aspects IRM de leucodystrophies dont les gènes sont identifiés.
Leucoencéphalopathie mégalencéphale avec kystes sous-corticaux (« megaloencephalic leukoencephalopathy with subcortical cysts » [MLC]) [25, 26, 27, 28, 29, 30, 31, 32, 33, 34]
Décrite pour la première fois en 1995, la MLC est caractérisée par :
• en clinique : macrocéphalie survenant dès la première année, une atteinte modérée ou discrète des fonctions cognitives, une spasticité progressive, entraînant un handicap modéré et d'apparition insidieuse, des crises d'épilepsie dans la moitié des cas ;
• en neuroradiologie (Figure 3) : lésions démyélinisantes diffuses et symétriques périventriculaires et sous-corticales, présence de kystes sous-corticaux dans les régions frontopariétales et les parties antérieures des lobes temporaux.
Les données histologiques consistent en un aspect spongiforme de la substance blanche, dû à des vacuoles situées entre les parties externes des gaines de myéline, respectant les axones.

Ces aspects sont dus à une division des lamelles de myéline le long des lignes intrapériodiques ou une compaction incomplète.
La MLC est observée de manière ubiquitaire dans le monde, excepté une fréquence particulièrement élevée dans le nord de l'Inde (population Agrawals) et en Turquie. De transmission récessive, un premier gène a été identifié. Il est localisé sur le bras long du chromosome 22 (22qtel). Il a été par la suite cloné (KIAA0027), sa dénomination actuelle est MLC1 (MIM 604004), par l'équipe de Leegwater et al. en 2001. Un effet fondateur a été retrouvé dans certaines populations. Il s'agit cependant d'une affection génétiquement hétérogène, puisque 20 % des patients atteints de LMC ne sont pas liés à ce gène.
Leucoencéphalopathie avec atteinte du tronc cérébral et de la moelle avec augmentation de lactate (LBSL syndrome) [35, 36, 37, 38, 39, 40]
Une nouvelle forme de leucoencéphalopathie ataxique a été récemment individualisée par Van der Knaap en 2003. Elle est caractérisée par une paraparésie spastique d'évolution lentement progressive, une ataxie mixte, proprioceptive et cérébelleuse, de début infantile. L'IRM est très particulière avec une démyélinisation extensive, intéressant le corps calleux, la couronne rayonnante, la partie postérieure des capsules internes, le tronc cérébral, et en particulier, les pédoncules cérébelleux, les parties intraparenchymateuses et mésencéphaliques des nerfs trijumeaux. De façon très caractéristique, la moelle épinière est également le siège d'une démyélinisation, et notamment les fibres corticospinales latérales et le faisceau cordonnal postérieur. La démyélinisation peut aussi intéresser la substance blanche du cervelet. La spectro-IRM montre une diminution significative du N-acétylaspartate, une augmentation du myo-inositol, des quantités normales ou modérément élevées de choline, et une élévation des lactates au sein de la substance blanche, donnant le nom de LBSL. Cependant, quelques patients ont été rapportés avec une quantité normale de lactates. La plupart des patients décrits se présentaient de manière sporadique, les rares familles rapportées sont en faveur d'une transmission récessive. Le gène muté a été initialement localisé sur le chromosome 1, et il a été récemment identifié comme étant DARS2, qui code une protéine mitochondriale (aspartyl-ARNt synthétase). L'état muté entraîne une diminution de l'activité enzymatique de l'ARNt synthétase. Plus récemment, une forme hypomyélinisante (HBSL) a été identifiée, dont le gène DARS interagit avec DARS2.
Syndrome Nasu-Hakola (NHD) [41, 42, 43, 44, 45]
Le syndrome Nasu-Hakola, également connu sous le terme de polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL ; MIM221770), est une affection autosomale récessive, caractérisée par une démence présénile, et une raréfaction osseuse. Initialement décrit au début des années 1970, plus de 150 cas ont été rapportés, essentiellement au Japon, Finlande, mais aussi dans d'autres pays. Les symptômes cliniques débutent dans la troisième décennie, et sont représentés avant tout par des douleurs et des gonflements des articulations, poignets, genoux, chevilles, des fractures distales dans les suites de traumatismes souvent mineurs. Les radiographies osseuses mettent en évidence une perte osseuse dans les extrémités distales des os longs, et la présence de kystes dans les doigts et les orteils. En général, le crâne et le rachis sont respectés. Ces cavités kystiques contiennent des cellules lipidiques et sont entourées de membranes lipidiques de 1 à 2 μm d'épaisseur. Les symptômes neurologiques surviennent dix ans après, comprenant des crises d'épilepsie, une démence de type frontal, et des mouvements choréiques. Le caractère frontal de la démence est supporté par les données cliniques mais également par les études en imagerie fonctionnelle

(positon emission tomography [PET]-scan). Le décès survient en général 20ans plus tard, soit à un âge moyen de 50ans. L'évolution neuroradiologique est caractérisée par l'aggravation progressive des lésions neuroradiologiques : atrophie cérébrale, avec au départ un respect de la substance blanche, une augmentation progressive de l'espace intercaudé, apparition de calcifications des noyaux gris. En fin d'évolution, l'atrophie cérébrale est diffuse, et la démyélinisation intéresse toute la substance blanche. Les études neuropathologiques ont montré une perte des fibres myéliniques du lobe frontal et un aspect de gonflement axonal, dit « sphéroïde », la présence de macrophages avec inclusions lipidiques et une réaction étendue astrocytaire et gliale. De plus, il est noté une réduction du volume des ganglions de la base, essentiellement des noyaux caudés. Des lésions vasculaires sont également notées, il s'agit d'épaississement des parois des vaisseaux avec diminution de la lumière et occlusion au sein des petites artérioles et capillaires. Les études en immunomarquage ont montré une augmentation de la quantité de collagène de type IV sur les lames basales des vaisseaux. Sur ces données, la physiopathologie de cette pathologie repose sur des anomalies du métabolisme lipidique ou une hypoplasie vasculaire.
Le gène muté (DAP12) est localisé sur le bras long du chromosome 19 (19q13.1). Il a été mis en évidence au sein de la population finlandaise un effet fondateur. Différents types de mutations ont été identifiés : délétion d'une simple base chez un patient japonais, délétion de plusieurs exons (d'un à quatre).
Cette pathologie est elle aussi hétérogène sur le plan génétique, depuis la publication de familles suédoises et norvégiennes remplissant tous les critères mais sans mutation retrouvée dans le gène DAP12. Un deuxième gène a été identifié, TREM2.
La pathogénie de cette pathologie n'est pas clairement démontrée. Plusieurs hypothèses ont été proposées :
• un mécanisme vasculaire, avec interruption de la barrière hématoencéphalique et ischémie secondaire, entraînant une atteinte axonale et oligodendrocytaire, expliquant la formation d'axones sphéroïdaux, et la perte axonale et des gaines myéliniques ;
• des anomalies du métabolisme lipidique systémique, aboutissant à des lésions des gaines de myéline.
Leucodystrophies de l'adulte à transmission dominante
Leucodystrophie de l'adulte autosomale dominante (ADLD) [46, 47, 48]
La leucodystrophie autosomale dominante de l'adulte (OMIM 169500) a été décrite pour la première fois dans une grande famille irlando-américaine en 1984. Le début des symptômes se situe entre 50 et 60ans ; ils associent :
• une atteinte végétative (troubles urinaires, intestinaux, hypotension orthostatique) ;
• un syndrome cérébelleux et pyramidal.
L'IRM est caractérisée par une démyélinisation de la substance blanche à prédominance frontopariétale et cérébelleuse, prédominant dans les pédoncules cérébelleux. Dans les formes

avancées, les anomalies peuvent être également observées au niveau occipital et à un moindre degré dans les lobes temporaux. Le faisceau corticospinal est intéressé dans son ensemble, ainsi que le corps calleux.
Cette démyélinisation respecte les régions périventriculaires, et prédomine au niveau des lobes. Cette démyélinisation est également mise en évidence chez des sujets asymptomatiques, allant de modifications minimes à une démyélinisation extensive.
En raison de l'âge de début tardif, de la lenteur évolutive, un diagnostic de sclérose en plaques peut être initialement évoqué.
Une étude neuropathologique a été possible chez trois patients décédés. Elle met en évidence une perte extensive de fibres myéliniques, survenant de manière isolée ou en nappes, intéressant la substance blanche dans son ensemble, cérébrale et cérébelleuse. Le respect des cellules oligodendrogliales et l'absence relative de gliose dans les zones de démyélinisation et l'absence de réaction inflammatoire sont les signes histologiques caractéristiques.
Le gène a été localisé sur le bras long du chromosome 5 (5q31.6). Il s'agit d'une duplication du gène codant la lamine B1, protéine de structure nucléaire.
Maladie d'Alexander [49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59]
La maladie d'Alexander est une maladie progressive, considérée comme fatale, débutant en général dans l'enfance. Le début de la maladie est généralement avant 2 ans (plus de trois quarts des cas publiés) ; les symptômes associent un retard mental, un syndrome bulbaire, des crises d'épilepsie, une macrocéphalie et une spasticité. Le décès survient en général vers l'âge de 10 ans. Les études neuropathologiques ont mis en évidence une perte de la myéline dans les lobes frontaux. Des formes à début plus tardif ont été décrites : forme juvénile, à début entre 2 et 12 ans, caractérisée par un syndrome bulbaire et une évolution plus lente ; forme à début tardif de l'adulte : évolution particulièrement lente, ataxie, myoclonies du voile, absence d'atteinte cognitive et de macrocéphalie. D'autres particularités phénotypiques ont été rapportées : insuffisance ovarienne primaire, oscillopsie, dysthyroïdie, hypothermie, dysautonomie, évolution aiguë avec décès en moins de deux mois.
Le pronostic de la maladie d'Alexander est corrélé à l'âge de début : le délai moyen de survie dans les formes infantiles est de 3,6 ans, 8,1 ans dans les formes juvéniles et 15,0 ans dans les formes de l'adulte.
Des critères neuroradiologiques ont été établis en 2001 par Van Der Knaap et al. :
• atteinte de la substance blanche à prédominance frontale ;
• hypersignal périventriculaire en T1 visible en hyposignal en T2 ;
• atteinte des ganglions de la base et des thalamus ;
• atteinte du tronc cérébral ;
• possibilité de rehaussement par l'injection de produit de contraste.

Dans les formes tardives de l'adulte, les anomalies en IRM sont mises en évidence sous la forme d'hyposignaux en T2 dans la substance grise, le tronc cérébral et la moelle cervicale avec une atrophie marquée. De plus, les lobes frontaux sont souvent respectés dans ces formes de l'adulte.
Les données histologiques mettent en évidence une accumulation massive de fibres de Rosenthal, communes à toutes ces formes évolutives. Ces fibres sont particulièrement mises en évidence dans les régions piales et sous-épendymaires, dans les formes juvéniles, et dans les formes tardives de l'adulte dans le cervelet et le tronc cérébral.
La plupart des observations rapportées sont secondaires à des mutations du gène codant la glial fibrillary acidic protein (GFAP), dont la mutation entraîne le dépôt de fibres de Rosenthal dans les astrocytes.
Nouvelles leucodystrophies à transmission autosomale dominante
Il a été ainsi décrit récemment deux nouvelles leucodystrophies à transmission autosomale dominante :
• une atteinte clinique et neuroradiologique à prédominance frontale oriente vers une dystrophie neuroaxonale, dont le gène identifié est CSF1R [60] ;
• la présence d'une syndactylie associée à une hypomyélinisation évoque le syndrome oculo-dento-digital et doit faire rechercher une mutation du gène de la connexine 43 (GJA1).
Conclusion Le diagnostic d'une leucoencéphalopathie de l'adulte constitue une problématique régulière pour les neurologues. Certains arguments neuroradiologiques plaident pour une origine vasculaire (atteinte centropontique, des noyaux gris, présence de microbleeds). Deux éléments permettent de guider le clinicien : un aspect cavitaire sur l'IRM, orientant vers le diagnostic de CACH syndrome, et une transmission dominante, suggérant une mutation de la lamine B1, une maladie d'Alexander à révélation tardive et une dystrophie neuroaxonale. En dehors de ces éléments d'orientation, une maladie métabolique doit être recherchée par le biais d'un bilan enzymatique standardisé. L'exploration en IRM doit être rigoureuse et les séquences requises sont le T1 sans et avec injection de gadolinium, le T2 deuxième écho et les séquences Flair et l'écho de gradient.
Points essentiels
• En faveur d'une origine vasculaire
○ Atteinte des noyaux gris
○ Atteinte des capsules externes
○ Atteinte de la partie médiane du tronc cérébral
○ Présence de microsaignements sur les séquences en écho de gradient
• En faveur de certaines leucodystrophies avec gènes connus
○ CACH : leucodystrophie cavitaire (Flair indispensable)
○ MLC : kystes bitemporaux
○ LBSL : atteinte du tronc cérébral et de la moelle épinière
○ Alexander : hyposignaux des noyaux gris, du bulbe et atrophie bulbaire
○ Leucodystrophies dominantes : duplication de la lamine B1, mutation du gène CSF1R
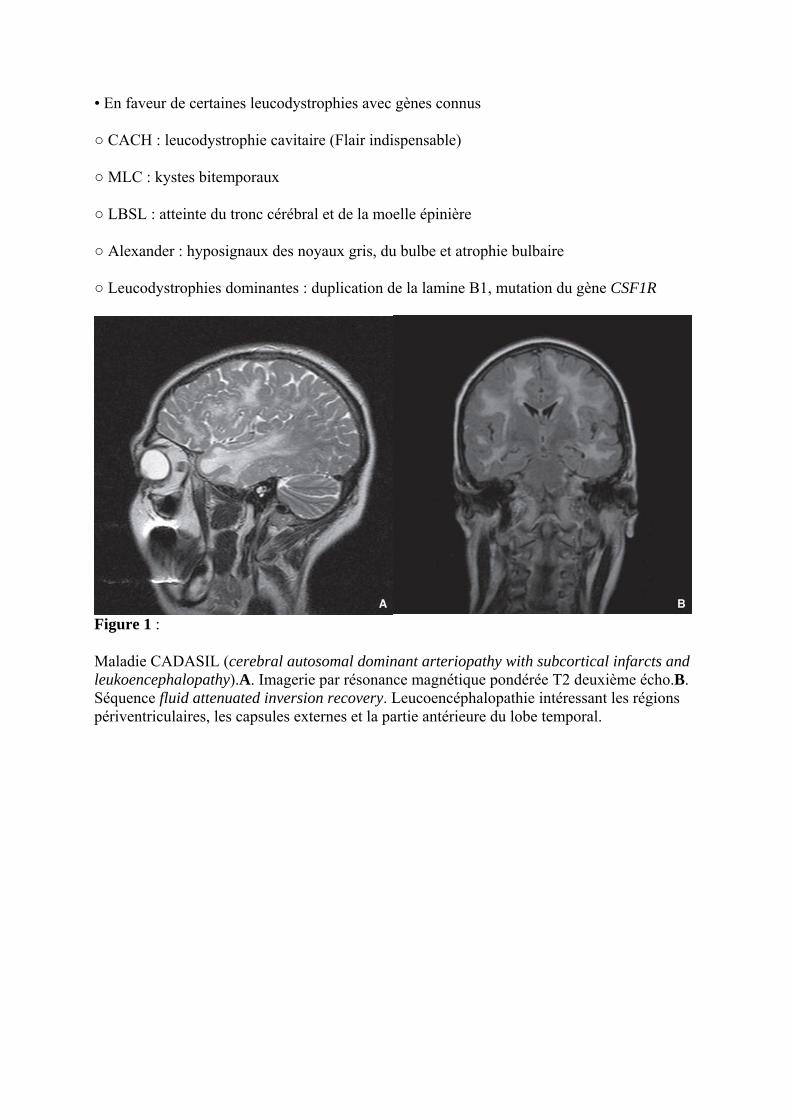
Figure 1 :
Maladie CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy).A. Imagerie par résonance magnétique pondérée T2 deuxième écho.B. Séquence fluid attenuated inversion recovery. Leucoencéphalopathie intéressant les régions périventriculaires, les capsules externes et la partie antérieure du lobe temporal.
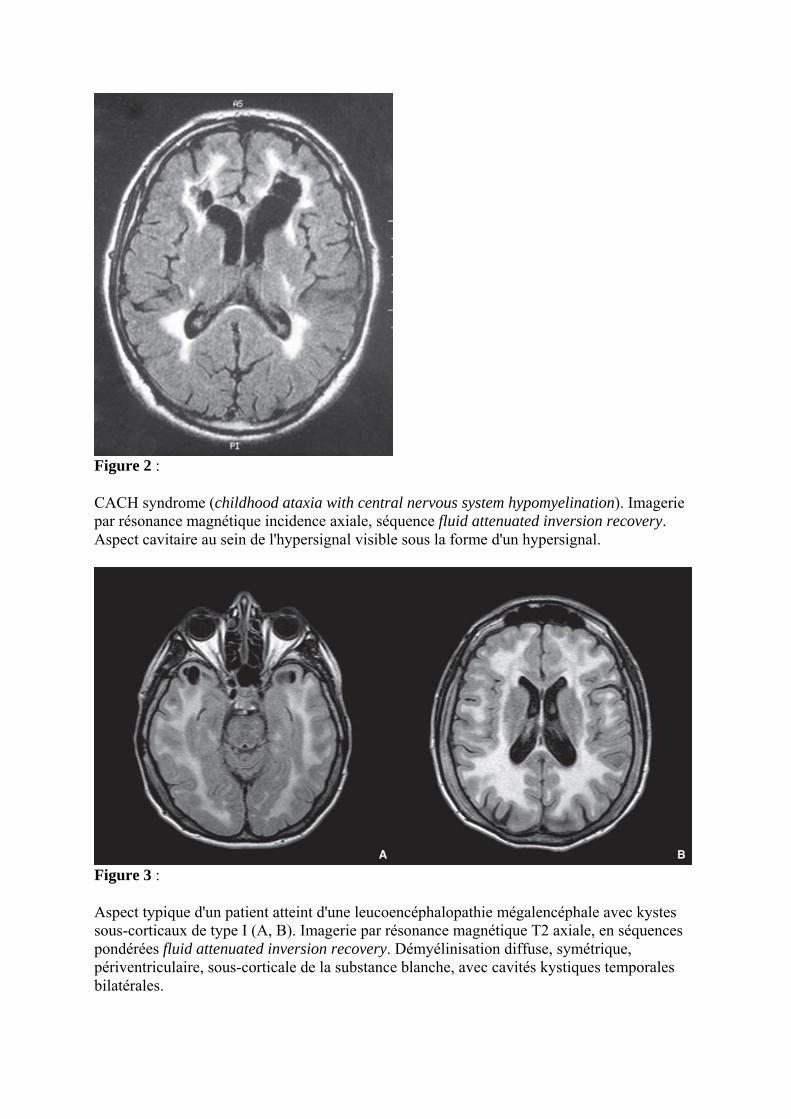
Figure 2 :
CACH syndrome (childhood ataxia with central nervous system hypomyelination). Imagerie par résonance magnétique incidence axiale, séquence fluid attenuated inversion recovery. Aspect cavitaire au sein de l'hypersignal visible sous la forme d'un hypersignal.
Figure 3 :
Aspect typique d'un patient atteint d'une leucoencéphalopathie mégalencéphale avec kystes sous-corticaux de type I (A, B). Imagerie par résonance magnétique T2 axiale, en séquences pondérées fluid attenuated inversion recovery. Démyélinisation diffuse, symétrique, périventriculaire, sous-corticale de la substance blanche, avec cavités kystiques temporales bilatérales.
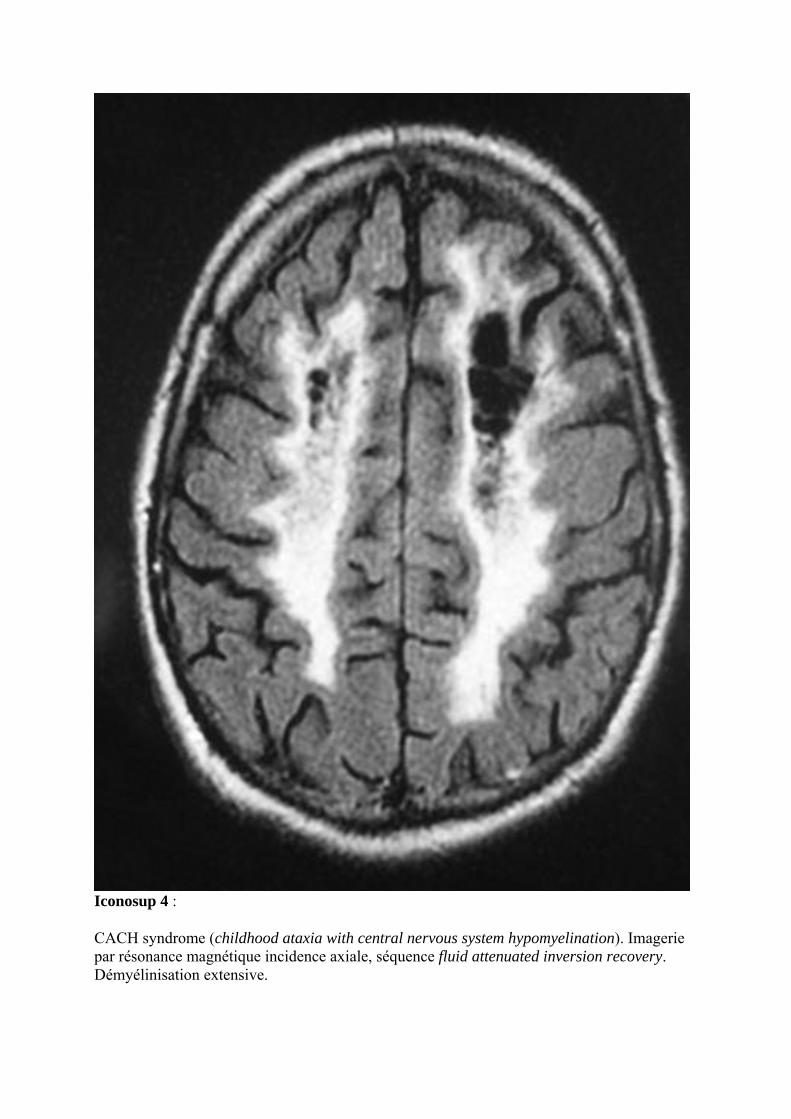
Iconosup 4 :
CACH syndrome (childhood ataxia with central nervous system hypomyelination). Imagerie par résonance magnétique incidence axiale, séquence fluid attenuated inversion recovery. Démyélinisation extensive.
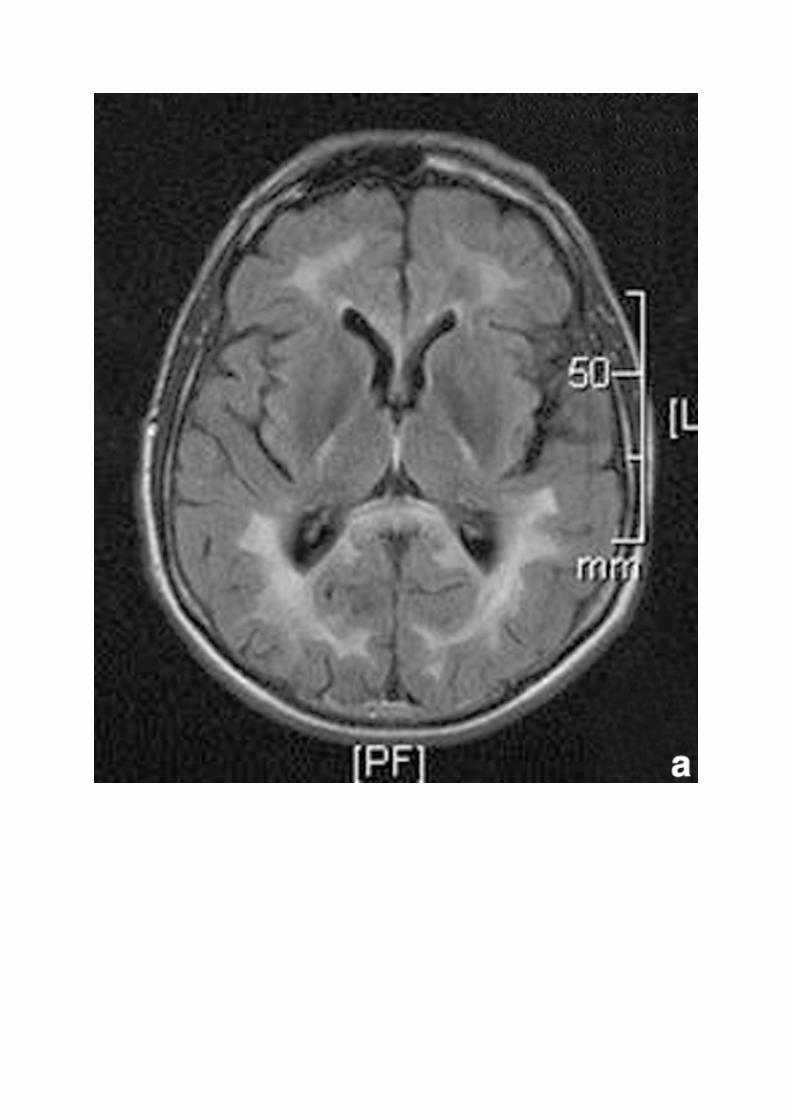
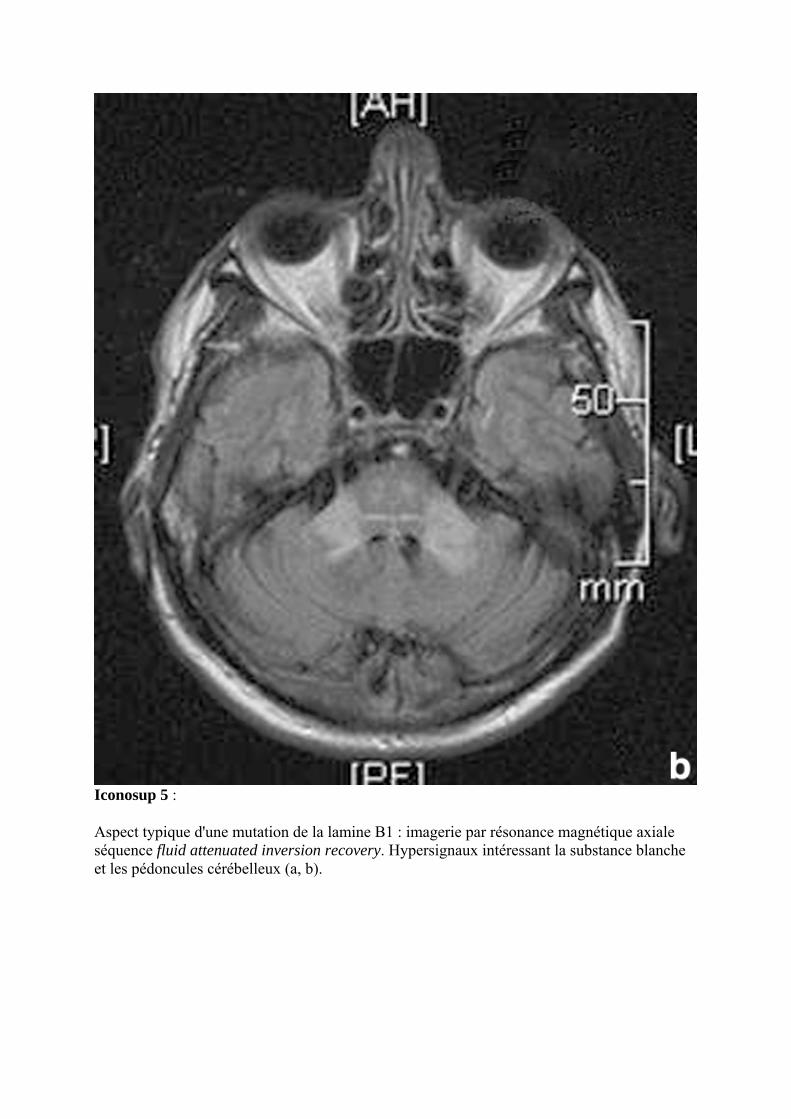
Iconosup 5 :
Aspect typique d'une mutation de la lamine B1 : imagerie par résonance magnétique axiale séquence fluid attenuated inversion recovery. Hypersignaux intéressant la substance blanche et les pédoncules cérébelleux (a, b).




