Embed Size (px)
Citation preview

1
NUCLEAR FACTOR 1 AND METAL TRANSCRIPTION FACTOR-1 SYNERGISTICALLY ACTIVATE THE MOUSE METALLOTHIONEIN-1 GENE IN RESPONSE TO METAL IONS*
Olivier LaRochelle1, Simon Labbé1,2, Jean-François Harrisson1, Carl Simard1, Véronique Tremblay1, Geneviève St-Gelais1 & Carl Séguin1
From Centre de recherche en cancérologie de l’Université Laval1, CHUQ, Hôtel-Dieu de Québec, Québec (Québec) Canada G1R 2J6, and Département d’anatomie et de physiologie, Faculté de médecine,
Université Laval, Québec (Québec) Canada G1K 7P4. Present address2: Département de Biochimie, Faculté de médecine, Université de Sherbrooke, Sherbrooke, Québec (Québec) Canada, J1H 5N4.
Running Title: NF-1 activates the mouse metallothionein-1 promoter Address correspondence to: Carl Séguin, Centre de recherche en cancérologie, Hôtel-Dieu de Québec, 11 côte du Palais, Québec (Québec) Canada G1R 2J6. Tel.: 418-525-4444, ext. 15544 ; Fax : 418-691-5439 ; e-mail : [email protected] Metal activation of metallothionein (MT) gene transcription is dependent on the presence of metal regulatory elements (MREs), which are present in five non-identical copies (MREa through MREe) in the promoter of the mouse MT-1 gene, and on the capacity of metal transcription factor-1 (MTF-1) to bind to the MREs in the presence of zinc. We detected a protein, distinct from MTF-1, specifically binding to the MREc region. DNA binding competition experiments using synthetic oligonucleotides and specific anti-NF1 antibodies showed that this protein binds to an NF1 site overlapping the MREc element as well as to a second site upstream of the Sp1a site, and corresponds to NF1 or a related protein. Transfection experiments showed that loss of the two NF1 sites decreased metal-induced MT promoter activity by 55-70% in transiently transfected cells, and almost completely abrogated metal and tert-butylhydroquinone (tBHQ) induction in stably transfected cells. Similarly, expression of an inactive NF1 protein strongly inhibited MT-1 promoter activity. Using synthetic promoters containing NF1 and MRE sites fused to a minimal MT promoter, we showed that these NF1 sites did not confer metal induction but enhanced metal-induced promoter activity. Chromatin immunoprecipitation assays confirmed that NF1 binds to the mouse MT-1 promoter in vivo and showed that NF1 binding is zinc-inducible. In addition, zinc-induced NF1 DNA binding was MTF-1-dependent. Taken together, these studies show that NF1 acts synergistically with MTF-1 to activate the mouse MT-1 promoter in response to metal
ions and tBHQ, and contributes to maximal activation of the gene. Metallothioneins (MTs) are small metal-binding stress proteins grouped into four classes, MT-1 through MT-4 (1,2). MTs have been identified in a wide range of species and are present in various tissues and cell types from yeast to human. In mice, MT-1 and MT-2 are ubiquitous and coordinately expressed in all tissues, whereas MT-3 is mainly expressed in the brain (3) and in the organs of the reproductive system (4), and MT-4 is restricted to stratified squamous epithelia (5). MTs have no enzymatic function, but appear to play important roles in metal ion homeostasis, as an active donor of zinc to other sites within the cell, in detoxification of toxic metals, and in protection against oxidative damage, ionizing radiation, and xenobiotics (1,2). MT genes are inducible at the transcription level by hormones, cytokines and a variety of stress conditions that include exposure to transition metal ions, UV irradiation, hypoxia, and reactive oxygen species (ROS) (2). Metals are the most general and potent of these inducers. Metal activation of MT gene transcription depends on the presence of regulatory DNA sequences termed metal regulatory elements (MREs), and involves metal-responsive transcription factor-1 (MTF-1) interacting with the MREs in a zinc-dependent manner (6,7). MTF-1 is also involved in the response to hypoxia (8), ROS (9), and amino acid starvation (10). The highly conserved core sequence 5'-TGCRCNC-3' (R, purine; N, any nucleotide) is necessary and sufficient for induction by metals (11-13). MREs are present in five non-identical copies (MREa through MREe) in the 5' flanking region of the mouse MT-1 gene (Fig. 1), and different MREs have different
http://www.jbc.org/cgi/doi/10.1074/jbc.M800640200The latest version is at JBC Papers in Press. Published on January 29, 2008 as Manuscript M800640200
Copyright 2008 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

2
transcriptional efficiencies. MREd is the strongest, MREa and MREc are 50 to 80% weaker, MREb is very weak and MREe is apparently non-functional (14). In addition to MTF-1, several other proteins interact with the mouse MT-1 promoter, including USF-1, USF-2 (15-19), Sp1 (9,19-21), c-fos (19) and c-jun (9). However, the mechanism by which these factors contribute to MT gene expression in not known. MTF-1 gene knock out showed that MTF-1 is essential for basal and metal-induced MT gene transcription (22). Notably, no MRE-binding protein could be detected in MTF-1 null mutant cells. This led to the hypothesis that MTF-1 is the only factor that binds MREs, and the only transcription factor that mediates responsiveness to different metals. We previously identified and purified a mouse nuclear protein, termed metal element protein-1, specifically binding with high affinity to MRE elements in a zinc-dependent manner, and subsequently showed that this protein corresponds to MTF-1 thus supporting the contention that MTF-1 is the only MRE-binding factor (23,24). We also showed that purified MTF-1 binds to MREc, MREd and MREe, as assayed by DNaseI footprinting analysis (23). More recently, in a model depicting the dynamic transcription factor complexes found at the proximal region of the mouse MT-1 promoter, it has been suggested that MTF-1 occupies all MREs, including MREc, under metal-induced conditions (19). However, on the basis of other experiments carried out in vitro and in vivo, it was suggested that NF1 interacts with the MREc element, as well as with two other sites in the MREb region, and inhibits both constitutive and metal-induced MT gene transcription (25,26). However, earlier papers from the same laboratory had concluded that C/EBPδ (or CP-1) (27) and CP-2 (28) bind to the MREc region and activate MT transcription. In the course of our studies on the characterization of the mouse MT-1 gene promoter, DNaseI footprinting analyses revealed the presence of a nuclear protein, distinct from MTF-1, binding to the MREc region. Given that the identity of the MREc-binding protein remains controversial, we further analyzed this region. We show here that the MREc element overlaps with an atypical NF1 binding site and that a NF1-like protein binds in vitro to this region as well as to a second site contiguous with the Sp1a site. The
NF1 protein contributes positively to the constitutive expression of the MT-1 gene and acts cooperatively with MTF-1 to activate MT gene transcription in response to metal ions and the phenolic antioxidant tert-butylhydroquinone (tBHQ). A model is presented in which zinc treatment induces MTF-1-mediated alteration of chromatin structure, which allows the binding of positively acting factors to the MT promoter.
EXPERIMENTAL PROCEDURES Material- Restriction and DNA modifying enzymes were obtained from New England Biolabs (Pickering, Ontario), [α-32P]dCTP was from (NEN, Boston, MA), and synthetic oligonucleotides (oligos) were from Invitrogen (Carlsbad, CA). The polyclonal anti-NF1 antibody used for the supershift was provided by René Saint-Arnaud, Shriners Hospital, Montréal (29). All other chemicals were purchased from Sigma Chemical Company (St-Louis, Mo). Plasmid constructs and mutagenesis- Dr. Nicolas Mermod kindly provided the NF1 RSV expression plasmids p113-CTF-1 and p113-CTF-1∆ (30). Plasmid MT1-LUC (31) contains 1843 bp of the 5' flanking sequence and 68 bp of the 5' untranslated region from the mouse MT-1 gene in the pGL2 basic vector (Promega, Madison, WI). Constructs ∆−590-LUC, ∆−238-LUC and ∆−150-LUC contain mouse MT-1 promoter sequence positions −590 (relative to the transcription start point) to +68, −238 to +68, and −150 to +68, respectively, in pGL2 basic. To construct the reporter plasmids (NF1ab)MT1m-LUC and (MREdd)MT1m-LUC, the double-stranded oligos NF1ab and MREdd (Table 1), respectively, were inserted in front of a minimal mouse MT-1 gene promoter (MT1m; −34 to +68) (31), in pGL2 basic. The NF1ab oligos contains NF1a and NF1b sites organized in tandem, while the MREdd oligo contains two strong mouse MT-1 MREd elements in opposite orientation. The reporter plasmid NF1ab(MREdd)MT1m-LUC was generated by inserting the NF1ab oligo into plasmid (MREdd)MT1m-LUC upstream of the MREdd oligo. Plasmid (MREa)6MT1m-LUC contains six mouse MT-1 MREa elements in front of a minimal mouse MT-1 promoter in pGL2 basic (32). For mutagenesis, a 1843-bp (−1843 to +68) MT1 promoter fragment was subcloned into
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3
the plasmid pAlter-1 (Promega), and mutations were introduced in different sites using specific oligos (Table 1) according to the instructions of the manufacturer. The resulting fragments were subcloned into pGL2 Basic to generate a series of plasmids with mutation(s) in the NF1a (NF1a∆-LUC), NF1b (NF1b∆-LUC), and NF1a and NF1b (NF1ab∆-LUC) sites. For all the constructs and mutants, correct insertions and mutations were confirmed by sequencing. Cell culture and transfection- Metal resistant mouse L50 fibroblast cells were obtained from Dean H. Hamer (NIH, Bethesda, MD). The human HepG2 cells used for transient transfection were from A. Anderson (Centre de recherche, l’Hôtel-Dieu de Québec), while those used to generate stable transfectants were from Jacques Pouysségur (Institute of Signaling, Developmental Biology and Cancer Research, Nice, France). For large scale nuclear extract preparations, L50 cells were grown in suspension in Eagle's minimal essential medium supplemented with 5% fetal calf serum and 5% horse serum in the continuous presence of 50 µM ZnCl2. For small scale analytic experiments, L50, and HepG2 cells were cultured in monolayer’s in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Cells were transfected with the different plasmids by the calcium phosphate method (33), treated or not with CdCl2 (final concentration 2.5 µM), ZnCl2 (100 µM), or tBHQ (100 µM) and harvested after 6 - 8 hours (32). Briefly, cells were seeded (~4 x 105/6 cm plate) 16 h prior to transfection. Cells were transfected with 1 µg of the reporter plasmid MT1-LUC, or the different mutants, or 7.5 µg of the plasmids (NF1ab)MT1m-LUC, (MREdd)MT1m-LUC, or NF1ab(MREdd)MT1m-LUC, shocked for 3 min at 37oC with 15% glycerol in HEPES buffered saline, incubated for 12 h in growth medium, and then treated or not with metals or tBHQ. In some experiments, cells were cotransfected with 1, 5, 10 or 50 ng of the NF1 expression vectors p113-CTF-1 or p113-CTF-1∆, as indicated in the figure legend. The plasmid pTK-rLUC (Promega) was used as internal standard to monitor transfection efficiency. Total amount of DNA added to the cells was adjusted to 10 µg per dish with pGL2 basic DNA. Luciferase (LUC) activities were determined with a Dual-LUC assay kit (Promega) according to the recommendations of the
manufacturer. The transcriptional activity of the reporter plasmids was evaluated in duplicate in two or three different independent transfections.
For stable transfections, HepG2 cells were cotransfected with the MT1-LUC, NF1a∆-LUC, NF1b∆-LUC, NF1ab∆-LUC, or pGL2-basic reporter plasmid, and the amiloride-resistant Na+-H+ exchanger expression vector, pNHE1-R (kindly provided by Dr. J. Pouysségur) (34), with a 20:1 ratio. For the selection of stably transfected cells, three H+-suicide selections were applied (34). Cell lysates were prepared from two different pools of stable population’s transfected with the different plasmids. LUC activity was measured and expressed as relative LUC activity per µg of total cellular protein. Protein concentration was determined by the Bradford method using a Bio-Rad assay kit (Mississauga, Ontario). Nuclear extract preparation, electrophoretic mobility shift assays (EMSAs), DNaseI footprinting analysis, UV cross-linking assay and chromatography- For the NF1 EMSA, 5-6 µg of nuclear extract (24) were mixed in EMSA binding buffer (20 mM Tris pH 7.6, 50 mM KCl, 2 mM MgCl2, 3.3% Ficoll, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride) with 500 ng poly [dI-dC]2 (GE Healthcare, Fairfield, CT) and 20 fmol of 32P-end-labeled probe, and the mixture was incubated for 10 min at room temperature. Protein-DNA complexes were subjected to polyacrylamide gel electrophoresis in Tris/Borate buffer (22 mM Tris base and 22 mM boric acid). Methods for EMSA analysis using the C/EBP oligo as the probe have been described (24). DNaseI footprinting assays were performed (35) using a mouse MT-1 restriction fragment 5' end-labeled at −41 and extending to −200, or a mouse MT-1 PCR-amplified fragment, –348 to +72, as the probe (24). For the UV cross-linking assay (36), the probe was prepared by hybridizing the NF1b oligo (Table 1) to a 9-base complementary primer (5’-GCGTCCTT). This oligo was rendered completely double stranded with he Klenow fragment of DNA polymerase I in presence of [α32P]-dATP. For competition experiments, specific double-stranded competitor oligos (Table 1), as indicated in the figure legends, were added together with the probe. In supershift experiments, 2 µl of an anti-NF1 or an anti-C/EBP-β (∆198, Santa Cruz Biotechnologies, Santa Cruz, CA) polyclonal antibody was added to the binding
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

4
reaction, the mixture was incubated for 10 min at 21oC, followed by addition of the labeled oligo probe. Rabbit antiserum to mouse MT-3 (Moffatt, P. and Séguin, C., unpublished results) was used as a negative control. MRE-binding proteins were purified from 60 ml of L50-cell crude nuclear extracts (10 µg protein/µl) by standard chromatography with NaCl gradient elution (24). Chromatin immunoprecipitation (ChIP) experiments- NF1 ChIP assays were performed using the ChIP-it Express kit from Active Motif (Carlsbad, CA) following the manufacturer’s instruction. Cells were treated or not with 100 µM zinc for 3 h and then cross-linked with 1% formaldehyde. The chromatin was immunoprecipitated with an anti-NF1 antibody (N-20X or H-300, Santa-Cruz Biotechnology, Inc.), or normal rabbit serum or IgG (Millipore, Billerica, MA). The MTF-1 ChIP assay was performed as described (37) with some modifications. Protein A Sepharose beads were first coupled to 6 µg of an anti-MTF-1 polyclonal antibody or preimmune serum in presence of 20 µg BSA and 20 µg herring sperm DNA (Invitrogen). The Sepharose-conjugated anti-MTF-1 antibody was then incubated overnight at 4oC with an amount of chromatin corresponding to 1.6 x 106 L cells. The resulting DNA was analyzed by PCR using a pair of primers corresponding to the mouse MT-1 promoter region −230 to −80. As a negative control, each ChIP sample was also subjected to PCR using primers specific to a region located in the coding region of the glucose-6-phosphate-dehydrogenase (G6PD) gene (GenBank accession number X53617), positions +1841 to +1992. In some experiments, MTF-1-null mutant dko7 cells (38) (generously provided by Dr. W. Schaffner, Zurich) were transfected with 500 ng of a CMV-MTF-1 expression vectors (24) 24 h before metal-induction using the ExGen 500 transfection reagent (Fermentas LifeSciences, Burlington, Ontario) following the manufacturers’ instructions. PCR products were separated by agarose gel electrophoresis and visualized by SYBR Gold (Invitrogen) staining. Samples were subjected to PCR for different numbers of cycles to ensure that amplification was in the linear range. These ChIP experiments were performed three time using two different chromatin preparations.
The anti-MTF-1 antibody was raised in rabbit by using purified, bacterially expressed protein representing the C-terminal region of mouse MTF-1 (amino acids 577 to 675) fused to glutathione S-transferase. Anti-MTF-1 antibody was purified on a PROSEP-A column (Millipore, Etobicoke, ON). The anti-MTF-1 antibody specifically recognized a protein of Mr 100 000 that is absent in dko7 cell extracts (data not shown).
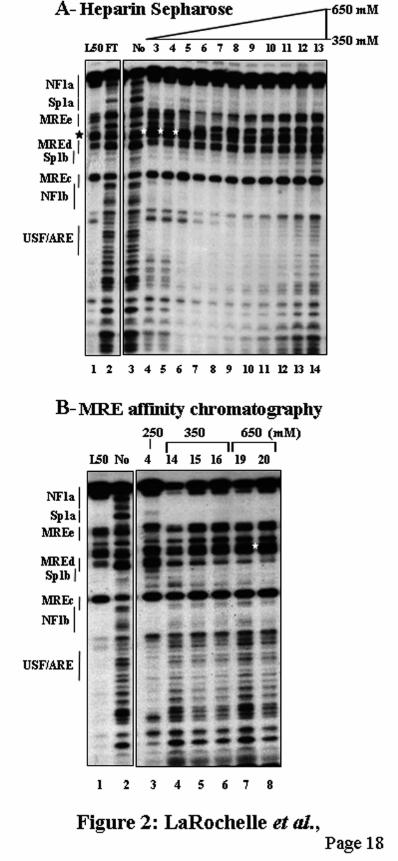
RESULTS Identification of a protein specifically binding to the MREc region. To elucidate the nature of the MREc-binding protein, mouse nuclear extracts were fractionated on a heparin-Sepharose column and analyzed by DNaseI footprinting analysis. In agreement with our previous observations (23,24), footprints were present over the Sp1a, MREd-Sp1b, MREc, and USF/ARE sites (Fig. 2A, lane 1). MTF-1 binds to MREd and induces the formation of a DNaseI-hypersensitive site at −153 (Fig. 2A, lanes 4-6, stars). In heparin-Sepharose chromatography, MTF-1, defined as the MREd-binding protein, mainly eluted in fractions 3-5, as indicated by the hypersensitive site at −153. However, the MREc-binding protein as well as the protein binding to the Sp1a region remained present in all the other fractions, including fractions 11-13 depleted of MTF-1 (Fig. 2A, compare lanes 4 and 14). This suggests the presence of two distinct proteins. To assess the specificity of the MREc-binding protein, fractions 3 to 13 (Fig. 2A) were loaded on an MREa-affinity column and eluted by three salt steps. Two MRE-binding proteins species were eluted from the MRE affinity chromatography. First, an MREc-binding activity present in all the fractions from 250 mM to 650 mM salt and second, MTF-1 found predominantly in the second 650 mM salt fraction (Fig. 2B, lane 8, star). Like the MREc-binding protein, the Sp1a-binding protein was present in all the fractions (Fig. 2B), whereas USF/ARE activity was found in the 250 mM fraction (Fig. 2B, lane 3). Note that the 250 mM salt fraction containing the MREc-binding protein is completely devoid of MREd-binding activity (Fig. 2B, lane 3), clearly indicating that the MREc-binding protein is distinct from the MREd-binding protein MTF-1.
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

5
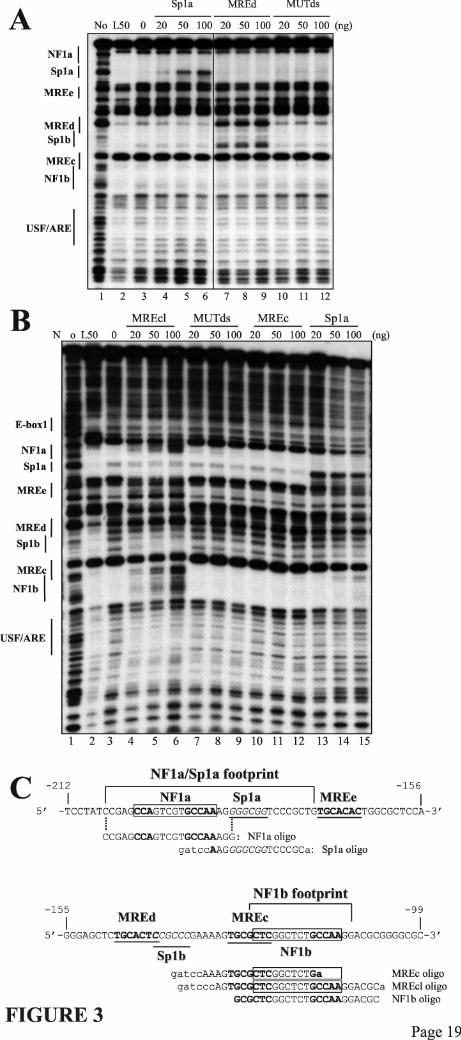
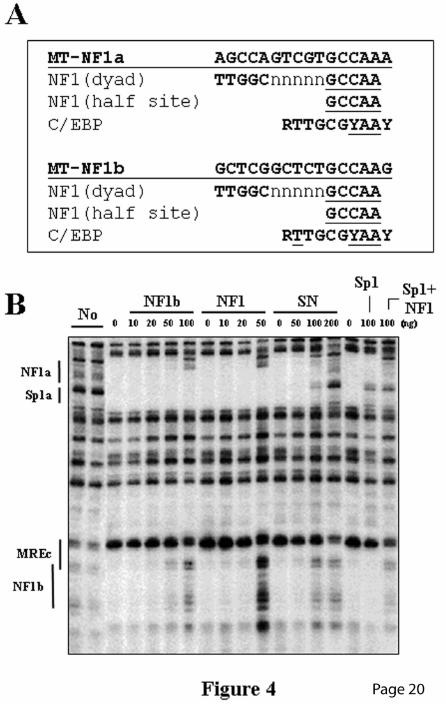
To further characterize the MREc-binding protein, footprinting competition assays were performed using a crude nuclear extract (Fig. 3A) or the 250 mM salt affinity fraction (Fig. 3B) specifically containing the MREc-binding activity and depleted of MTF-1 (See Fig. 2B, lane 3). The oligos MREc and MREd (Table 1), as well as the negative control MUTds (39) did not compete for the MREc-binding protein (Figs. 3, A and B). However, in MTF-1-containing extracts, the MREc and MREd oligos efficiently competed MTF-1 on the MREd element (Fig. 3A, lanes 7-9, and data not shown) (36), thus further indicating that the MREc-binding protein is distinct from MTF-1. Fine mapping of the footprint over the MREc region, performed by comparing with a Maxam and Gilbert sequence ladder, revealed that the protected region extends from −126 to −109 (Fig. 3C). Notably, this excludes the first three nucleotides in the MRE consensus sequence of the MREc element, namely the highly conserved TGC nucleotides. Since each of these three nucleotides is critical for metal induction (40) and MTF-1 DNA binding (41), this virtually excludes the possibility that the MREc-binding protein corresponds to MTF-1. This also suggests that the MREc element is not a bona fide MRE. Close examination of the nucleotide sequence of the protected region using DNA transcription factor binding site prediction programs identified a perfect NF1 half site (GCCAA, NF1b) in the protected region. In addition, a second NF1 half site (NF1a) is present on the mouse MT-1 promoter upstream of the Sp1a site (Figs. 3C and 4A). To further assess the identity of the MREc-binding protein, DNaseI footprinting competition experiments were performed using as competitor DNA an oligo, MREcl, corresponding to the DNaseI protected region in the MREc region, namely the entire MREc consensus sequence and the putative NF1 half site (Fig. 3C). We also used the oligos NF1b, a shorter version of MREcl in which the first nucleotide of the MREc core sequence is excluded (Fig. 3C), MT3-SN (31), an oligo corresponding to the overlapping Sp1/NF1 site of the mouse MT-3 promoter, Sp1a, corresponding to the Sp1a site, and NF1, an oligo containing a generic NF1 site (Santa Cruz Biotechnologies). As shown in Fig. 3B, MREcl efficiently competed the footprint over the MREc region, thus indicating that the
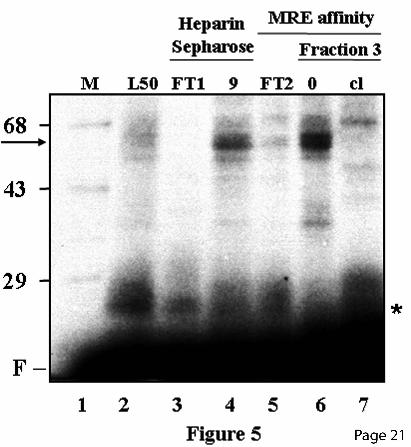
nucleotides required for binding of the MREc-DNA binding protein are distinct from those required for MTF-1, and are located downstream of the MRE consensus sequence in the NF1 half site. Most interestingly, MREcl also competed for a protein binding the NF1 portion of the footprint in the NF1a-Sp1a region (Fig. 3B). In fact, in vivo footprinting assays suggested that an unknown factor(s) interacts with this region (17). This protein is distinct from Sp1 since the Sp1a oligo did not compete the NF1 portion of the Sp1a-NF1a footprint but could efficiently compete its downstream Sp1 portion (Fig. 3, A and B). Similarly, the NF1 and NF1b oligos competed for the protein(s) binding to the MREc region and the NF1 portion of the NF1a-Sp1a footprint (Fig. 4), while the oligo MT3-SN or a mixture of Sp1a and NF1 oligos competed the footprint over the MREc region as well as the entire footprint over the NF1a/Sp1a region (Fig. 4). These results strongly suggest that the MREc-binding protein corresponds to NF1, or a closely related family member, and that this protein interacts with a second site on the mouse MT-1 promoter, namely NF1a, adjacent to the Sp1a site. NF1 binds to the mouse MT-1 promoter. To obtain an indication of the molecular weight of the MREc-binding protein, UV cross-linking experiments were performed with crude L50-cell nuclear extracts and different chromatographic fractions. The major protein species complexed with the NF1b oligo migrated on a denaturating gel with an apparent Mr of approximately 60 000 (Fig. 5, lane 2), and this complex was enriched in the heparin-Sepharose fraction 9 and the affinity chromatography fraction 3, which contains high levels of MREc-binding protein (Fig. 5, lanes 4 and 6). No significant labeled species was generated in chromatographic fractions devoided of MREc-binding activity, namely the heparin-Sepharose and the affinity chromatography column flow through (Fig. 5, lanes 3 and 5). Inclusion of a 250-fold excess of cold MREcl oligo completely abolished complex formation (Fig. 5, lane 7). Since the covalent attachment of short oligos to proteins has only a minor effect on the mobility of these proteins in SDS-polyacrylamide gel electrophoresis, these experiments indicate that a protein of approximately 60 kDa binds to the MREc region.
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

6
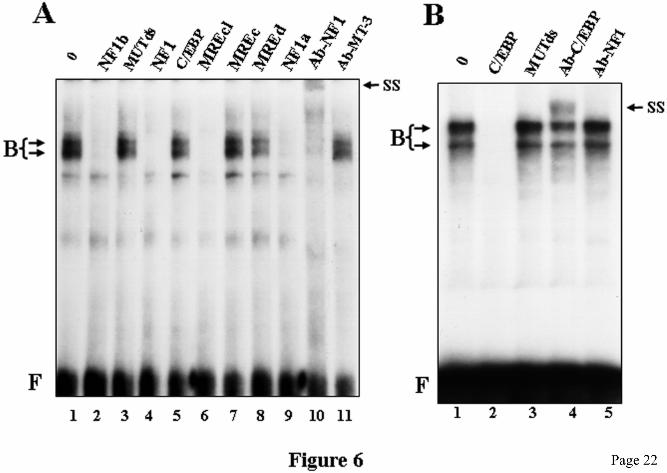
The molecular weight of this protein is consistent with that of NF1 (50-70 kDa). The evidence discussed above strongly suggests that the MREc-binding protein is NF1 or a closely related factor. To further ascertain the identity of the MREc-binding protein, EMSA analyses were performed using 32P-labeled NF1b oligo as the probe. Because a C/EBP-related protein has been reported to bind to the mouse MT-1 promoter regions around the NF1a and NF1b sites (27), EMSA analyses were performed using HepG2 cell nuclear extracts which contain both NF1 and C/EBP protein species (42,43). Incubation of the NF1b oligo with HepG2 cell nuclear extracts led to the formation of two major complexes (Fig. 6A, lane 1). Competition experiments were performed to address the specificity of the binding. Consistent with the DNaseI footprinting data, EMSA analysis showed that the complexes formed with the NF1b oligo and HepG2 cell nuclear proteins were efficiently competed by the NF1, NF1a, NF1b, and MREcl oligos, whereas the MUTds, C/EBP, MREc, MREd oligos (Fig. 6A), as well as oligos corresponding to STAT and NF-κB binding sites (data not shown) did not compete. These competition data further confirm that the MREc-binding protein corresponds to NF1 and not to C/EBP. To obtain more conclusive evidence, we analyzed the MREc-protein by supershift assays using a polyclonal anti-NF1 antibody. HepG2 cell nuclear extracts were incubated with NF1 antiserum or with an anti-C/EBP or an anti-MT3 polyclonal antibody prior to incubation with the NF1b oligo in the EMSA. Incubation with the NF1 antiserum but not with the MT3 or the C/EBP antiserum resulted in the complete elimination of the two major complexes and the formation of a supershifted complex (Fig. 6A, lane 10, and data not shown). The specificity of the supershifted complex was demonstrated by the lack of a similar effect of the same NF1 antibody on complexes formed between oligo C/EBP and HepG2 cell nuclear extracts (Fig. 6B, lane 5). The presence of C/EBP in the HepG2 extract was ascertained by performing an EMSA using the C/EBP oligo as the probe. As shown in Fig 6B the complexes formed with the C/EBP oligo are efficiently competed with cold C/EBP DNA but not with the MUTds oligo. Moreover, treatment with anti-C/EBP antiserum generated a supershift (Fig. 6B, lane 4),
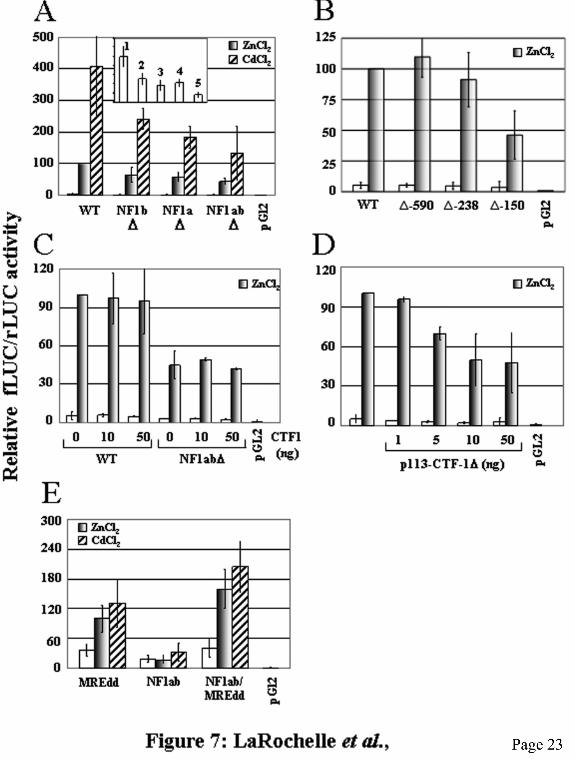
whereas treatment with anti-NF1 antibody did not. This clearly shows that the HepG2 cell extracts contain C/EBP protein and that the binding of NF1 to the NF1b oligo in Fig. 6A was not artificially favored because of the absence of C/EBP protein in the extracts. Taken together, these results strongly support the conclusion that NF1 is the MREc-binding protein. NF1 activates the mouse MT-1 promoter. To determine the functional effect of NF1 on constitutive and metal-induced activity of the mouse MT-1 promoter, HepG2 cells were transfected with the reporter plasmid MT1-LUC or various NF1 mutants. We confirmed that mutation of the NF1 sites abrogated binding of NF1 proteins by DNaseI footprinting analyses using the corresponding mutant promoter fragments as the probe (data not shown). Cells transfected with the wild-type control MT1-LUC reporter plasmid displayed a basal LUC activity that was induced 20- and 80-fold by the addition of ZnCl2 and CdCl2, respectively (Fig. 7A). Mutation of the NF1a site in the context of the intact MT promoter fragment diminished basal MT promoter activity by 60% and zinc- and cadmium-induced levels by approximately 40% and 55%, respectively. However, the mutant promoter remained strongly inducible and was induced 30- and 100-fold in response to zinc and cadmium, respectively. Similarly, inactivation of the NF1b site led to a 50% decrease of basal LUC activity and to a 35% and 40% reduction of zinc- and cadmium-induced promoter activity. The NF1b mutant was still induced 30- and 100-fold in response to zinc and cadmium, respectively. For the double mutant NF1ab, basal levels were inhibited by 55% while zinc and cadmium induction was reduced by 55% and 70%, respectively. Promoter activity of the double mutant was induced 20- and 60-fold by zinc and cadmium, respectively. To further confirm the function of the NF1 sites in stimulating MT promoter activity, we used deletion mutants in transfection experiments. As reported earlier (11,12), deletion of the mouse MT-1 promoter sequences between −1843 and −590, or −238, did not substantially modify either basal or metal-induced expression (Fig. 7B). However, further deletion to −150 produced a 40% decrease in basal level but had only a marginal effect on the capacity of the promoter to be induced by metal ions. Compared to 20-fold for
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7
the −1843 control reporter plasmid, the −150 deletion mutant was induced approximately 15-fold. However, similar to the NF1a or NF1b mutants, the overall activity of the promoter was reduced by approximately 50%. This showed that the region between −238 and −150, which includes the NF1a, E-box1 and Sp1a sites, contains elements required for the basal level of expression of the gene and maximal metal induction. These results are in congruence with those obtained with the NF1 mutants. Overall, these results indicate that while the NF1 sites are not essential, as expected, to confer metal induction to the mouse MT-1 promoter, their presence are required for maximal constitutive and metal-induced promoter activity. To substantiate the stimulating effect of NF1 on MT gene transcription, the MT1-LUC or the double NF1 mutant MT1(NF1ab)∆-LUC reporter plasmids was cotransfected into HepG2 cells along with the NF1 expression plasmid, p113-CTF-1 or the transcriptionally inactive mutant p113-CTF-1∆, in presence or in absence of inducers. Cotransfection of the wild type NF1 expression vector had no effect on either constitutive or metal-induced expression levels of both wild type and mutant MT promoters (Fig. 7C). However, cotransfection of the dominant negative mutant led to a dose-dependent inhibition of metal-induced MT promoter expression (Fig. 7D) but had no effect on basal levels. At the highest doses (10 and 50 ng), transfection of p113-CTFδ reduced zinc-stimulated LUC activity by 50%. These results further support a positive regulatory role of NF1 on MT gene transcription. To determine whether NF1 acts synergistically with MTF-1 in the activation of the MT-1 promoter in response to metal ions, we constructed synthetic promoters containing NF1 and MRE sites isolated from other cis-acting elements. These plasmids contain a minimal mouse MT-1 promoter joined to two MREd (MREdd), the NF1a and NF1b sites (NF1ab), or a combination of both the MREd elements and the NF1 sites (NF1ab/MREdd). HepG2 cells transfected with the NF1ab plasmid showed low constitutive activity that did not significantly increase in presence of metals (Fig. 7E). In contrast, cells transfected with MREdd showed a basal transcription level twice as high as that of NF1ab that was induced 2.7- and 3.6-fold by zinc
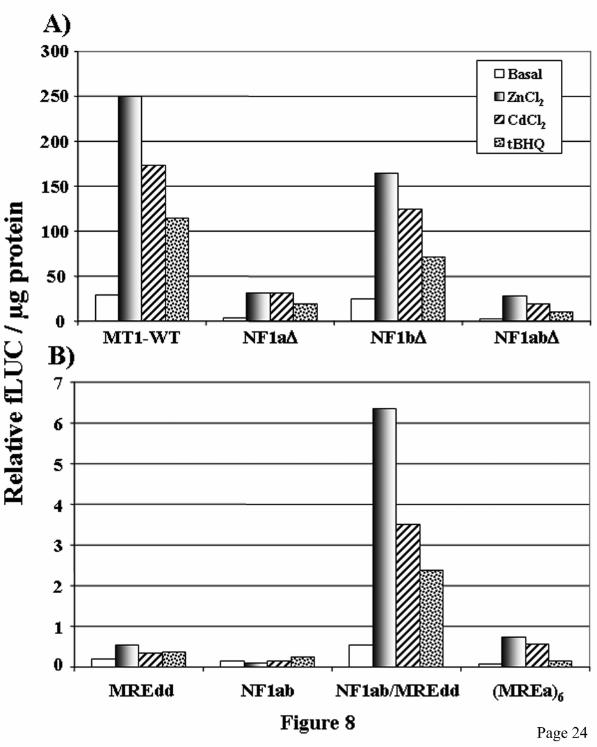
and cadmium, respectively. Interestingly, the fusion promoter NF1ab/MREdd was slightly more active in the non induced state and was induced 4-fold and 5-fold by zinc and cadmium, respectively. These results showed that NF1 acts synergistically with MTF-1 for metal-activation of MT gene transcription. This synergy between these two transcription factors is even more clearly observed in stable transfectants (see below). Because correct chromatin structure, which can not be achieved in small transiently expressed plasmids, may significantly alter gene expression by affecting interactions of transcription factors with the promoter, we confirmed the positive role of NF1 on MT gene expression by generating stable transfectants in HepG2 cells with the same plasmids used in transient transfection experiments. Two pools of clones generated from two distinct transfections for each construct were analyzed. The results obtained with the stable transfectants are in good agreement with those obtained in transient transfections. Indeed, mutation of the NF1 sites reduced basal and metal-induced transcriptional activity of the MT promoter by 40 to 90% (Fig. 8A). As observed in transiently transfected cells, the NF1 mutants were still strongly inducible in response to metals, between 5- and 10-fold. The NF1 mutations also diminished induced transcription levels in response to the phenolic antioxidant tBHQ, a known MT gene inducer (9,44) (Fig. 8A). The induction of MT by tBHQ requires MTF-1 (44). However, the most striking result was obtained with the synthetic promoters. The NF1ab reporter plasmid displayed low basal transcription levels in stable transfected cells and was not metal- nor tBHQ-inducible. The MREdd and (MREa)6 reporters were 3- to 6-fold induced by metals and weakly inducible by tBHQ. Notably, the presence of both MRE and NF1 sites strongly increased basal and metal- and tBHQ-inducible transcriptional activity (Fig. 8B). These results clearly show the importance of NF1 for the optimal activation of MT gene transcription by metal ions and tBHQ. An important question concerns the mechanism by which NF-1 potentialises the activation of MT gene transcription in response to metals and tBHQ. In fact, these results do not address whether NF1 and MTF-1 co-occupy the MT promoter simultaneously in response to metal
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

8
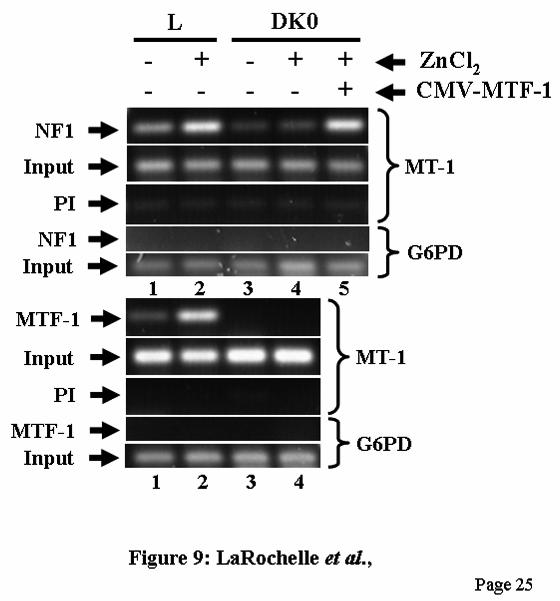
induction or whether the binding of NF1 requires first the binding of MTF-1. Thus, to better understand the mechanisms by which NF1 affects MT gene transcription, we address directly whether binding of NF1 to the mouse MT-1 promoter is modulated by metal and whether the presence of MTF-1 is required for NF-1 DNA binding by using the ChIP assay, a method that allows to study the dynamic in which transcription factors interact with DNA in vivo in the context of the intact chromatin. First, to validate our ChIP assay, we examined the interaction of MTF-1 with the MT-1 promoter in L cells. As previously shown (19), MTF-1 bound to the MT promoter in uninduced cells and this binding is strongly enhanced in presence of zinc (Fig. 9, lower panel, lanes 1 and 2). ChIP assays of chromatin from MTF-1-null dko7 cells confirmed the specificity of the immunoprecipitation (Fig. 9, lower panel, lane 3 and 4). Second, we studied the binding of NF1 to the MT promoter. In agreement with the in vitro studies, the ChIP assay showed that NF1 binds to the MT promoter in vivo in L cells but not to the G6PD gene coding region used as negative control (Fig. 9, upper panel, lane 1). Notably, zinc treatment strongly stimulated NF1 DNA binding to the MT promoter (Fig. 9, upper panel, lane 2). In dko7 cells, constitutive NF-1 DNA-binding activity was lower than in L cells and was not metal-inducible (Fig. 9, upper panel, lanes 3 and 4). Most interestingly, metal-induction of NF-1 DNA binding activity was completely restored in dko7 cells by expressing MTF-1 (Fig. 9, upper panel, lane 5). These results indicate that MTF-1 enhances NF1 DNA binding to the MT promoter in the uninduced state and show that MTF-1 is essential for the induction of NF-1 DNA binding in response to metals.
DISCUSSION
NF1 binds to and activates the mouse metallothionein-1 promoter. We have performed in vitro footprinting analyses and showed that an NF1 family member, or a closely related factor, binds to the MREc region nucleotides −126 to −105 and to a second site, −205 to −187, upstream of the Sp1a site. Five kinds of evidence support this conclusion. First, they contain a perfect NF1 half site (GCCAA). NFI protein binds as a dimer to the dyad symmetric consensus sequence
TTGGC(N5)GCCAA on duplex DNA (45). However, NF1 could also bind specifically to individual half sites (TTGGC or GCCAA). In fact, both protected regions in the footprint experiments include a NF1 half site (Fig. 3C). Moreover, the sequence TCG(N5)GCCAA in the promoter of the α2 (I) collagen promoter is identical to the NF1a site of the mouse MT-1 promoter and binds NF1 (46). Second, that this factor is NF1 is further demonstrated by competition experiments using a fragment of the mouse MT-3 promoter that contains a NF1 binding site as well as a commercial oligo containing a consensus sequence for NF1 binding sites. Third, UV cross-linking experiments indicate that a protein of approximately 60 kDa, consistent with that of NF1 (50-70 kDa) (45), binds to the MREc region. Fourth, an anti-NF1 antiserum recognized the complexes formed in vitro between HepG2 nuclear proteins and the NF1b oligo in supershift experiments (Fig. 6A, and data not shown), and fifth, ChIP assays showed that NF1 binds in vivo to the MT-1 proximal promoter in a metal- and MTF-1-dependent manner. Our results do not support the contention that C/EBPδ (C’BP-1) (27) or C’BP-2/CP2 (28) binds to the NF1b site. First, EMSA competition experiments using an oligo containing a consensus C/EBP site did not prevent the binding of nuclear proteins to the NF1 probe. Second, anti-C/EBP antibody failed to disrupt the formation of DNA/protein complexes (data not shown). The potential role of C/EBP on MT gene expression, if any, appears to be inhibitory rather than stimulatory, as stable expression of C/EBPα in prostate cancer cells down-regulates MT gene expression (47). The molecular mass of C’BP-2/CP2 is 28 kDa while that of the protein interacting with the NF1 oligo is approximately 60 kDa. Hence it is most unlikely that the protein detected in this study corresponds to C’BP-2/CP2. The reason of these apparent discrepancies are not clear but could be related to cell type differences. The NF1 family of site-specific DNA-binding proteins (also known as CTF or CAAT box transcription factor) is composed of four members in vertebrates (NFI-A, NFI-B, NFI-C and NFI-X). NF1 genes are differentially spliced, yielding as many as nine distinct proteins from a single gene. The products of the four NF1 genes
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

9
differ in their abilities to either activate or repress transcription (45). To determine the role of the NF1 sites in MT gene transcription, transfection experiments were performed in HepG2 cells with a MT1-LUC reporter plasmid and specific mutants in which the NF1 sites were inactivated individually or together. Inactivation of either of the NF1 two sites diminished basal and metal-induced transcription rate of the MT-1 promoter by 35 to 60% in transient transfections, while inactivation of both sites led to a 55 to 70 % inhibition. This inhibition was even more pronounced in stable transfectants in which transcriptional activity in NF1 mutants was reduced by up to 90%. However, both in transient and stable transfectants, metal induction, i.e. the ratio of basal over induced transcription levels, was largely unaffected. Deletion mutant studies support the idea that the NF1 sites are positive-regulatory cis-acting elements. Indeed, in agreement with earlier studies (11,12), deletions of the promoter region encompassing the NF1a sequences significantly reduced both basal and metal-induced transcriptional activity of the MT-1 promoter. Moreover, experiments with synthetic promoter reporter plasmids containing NF1 or MRE sites or both regulatory sequences fused to a minimal mouse MT-1 promoter showed that a much higher constitutive and metal-induced activity was observed with the reporter plasmids containing both the MRE and the NF1 sites (Figs. 7E and 8B). Overall, these results clearly show that NF1 is a positive regulator of both basal and metal- and tBHQ-induced MT transcription, acts synergistically with MTF-1 to activate the mouse MT-1 promoter in response to metal ions and tBHQ, and contributes to maximal activation of the gene. However, other studies have indicated a negative effect of NF1 on MTF-1-mediated transactivation (25,48). In those studies, transfection of vectors expressing NF1 proteins in HepG2 cells inhibited transcription from the MT or MTF-1 reporter plasmids. Under our experimental conditions, expression of wild type NF1 did not affect MT1-LUC transcription most likely because NF1 proteins are abundant and present inside the cell in saturating concentration. However, expression of the transcriptionally
inactive mutant p113-CTF-1∆ reduced zinc-induced MT1 promoter activity by up to 50% in a dose dependent manner (Fig. 7D), thus supporting the idea that NF1 is a positive regulator of MT gene transcription. Indeed, if NF1 played a negative regulatory function on MT gene expression, the inactive mutant would have been expected to lead to an increase in the transcriptional activity of the reporter plasmid. The reason for this apparent discrepancy is not clear, but it may reflect the fact that NF1-mediated inhibition of MT promoter transcription was observed in transfection experiments using a strong CMV expression vector and 30 to 1000-fold more vector than in this study, that is 1500 (25) and 5000 (48) ng, compared to 5 - 50 ng. NF1 is an abundant, constitutively expressed, and ubiquitous transcription factor. Increasing too much its concentration could interfere with some cellular components of the transcription machinery and indirectly compromise or quench MTF-1 activity. The strong CMV promoter can drive high levels of transcription leading to potentially non physiological concentrations of the corresponding protein. Consequently, by using a weaker RSV expression vector and by keeping the amount of transfected plasmid DNA in the lower nanogram range, we may have avoided possible non physiological effects. Proposed mechanism. Using the CASTing method to identify MTF-1 binding motifs, several strong consensus sequences for NF1 were pulled out (48). These NF1 sequences did not bind MTF-1, thus suggesting that MTF-1 and NF1 may physically interact. However, co-immunoprecipitation analyses using NF1 and MTF-1 antibodies failed to demonstrate a direct interaction between these two proteins (1). The molecular mechanisms by which metals exert their action on MTF-1 are only partially understood. It has been proposed that MTF-1 act as positive regulator in the presence of zinc ions by undergoing conformational changes that promote DNA binding and transcription, thus facilitating the recruitment of other components of the transcription machinery including other transcription factors or cofactors (6,7). In fact, TFIID and the Mediator complex interact functionally in a MTF-1-dependent manner to modulate transcriptional response to metal ions (49). Phosphorylation is also involved in the activation of MTF-1 (32). Several other proteins
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

10
interact with the mouse MT-1 promoter, including USF-1, USF-2 (15-19), Sp1 (9,19-21), c-fos (19) and c-jun (9). These transcription factors may be essential for maintenance of adequate basal promoter activities and for maximum induction in response to inducers (8). For example, MTF-1 and USF1 cooperate to regulate mouse MT-1 expression in response to zinc, and loss of USF-1 attenuates MT gene expression (18). An important question then concerns the mechanism by which basal elements amplify MTF-1-mediated metal treatment. One possible mechanism is that MTF-1 stabilizes or allows the binding of one or more factors to an adjacent enhancer element. We propose a model based on the induction of the MMTV promoter by glucocorticoids and of cytochrome P450 1A1 by polycyclic aromatic hydrocarbons (50,51). In both cases, activation of the respective genes by the ligand results in binding of NF1 to its site, presumably because of changes in chromatin structure. Notably, the affinity of NF1 for its DNA site is greatly affected by specific chromatin organization (52). We speculate that, in addition to recruit TFIID, MTF-1 controls metal-mediated MT gene induction in part through the modification of chromatin structure and the subsequent recruitment of NF1 to the MT-1 promoter. Consistent with this model, we showed here using ChIP assays that NF1 binds in vivo to the MT-1 promoter in a metal- and MTF-1-dependent manner. Thus, NF1 binding to DNA would largely be inhibited due to a “closed” chromatin structure. Upon metal induction, binding of MTF-1 to MREd and MREa would induce a more “open”, accessible, chromatin structure, thus allowing the binding of NF1 to the MT promoter. Consistent with this hypothesis, the MREc-region encompassing the NF1b site of the mouse MT-1 promoter shows metal-induced protection in in vivo footprinting assays (9,21). While the interaction of c-jun, USF-1, USF-2 and Sp1 with the MT-1 promoter is metal- and MTF1-independent, the recruitment of c-fos requires MTF-1 (19) and could also be recruited to the promoter with NF1. The formation of a DNaseI-hypersensitive site at the level of MREd in presence of nuclear extract (Fig. 2, A and B, and data not shown) provides some evidence of a conformational change in the DNA. In fact, it has been shown that the binding of MTF-1 induces
conformational changes in the MREd (53). Changes in chromatin structure of the MT1 gene after metal induction was also detected by general DNaseI I sensitivity (54,55). This model is also consistent with MTF-1 acting as a chromatin insulator shielding specific transcriptionally active regions from the repressive effects of flanking chromatin (56). Mutation of the NF1 sites did not only impair metal-induction but also reduced constitutive expression thus suggesting that NF1 binds to DNA in the absence of inducers. If, as suggested here, NF1 DNA binding is MTF-1-dependent, it follows that MTF-1 also binds to DNA in basal conditions. In fact, MTF-1 is absolutely required for basal level transcription of MT genes (22). Serum-supplemented medium contains 3-4 µM zinc and this concentration is sufficient to drive MTF-1-dependent expression of the MT-1 gene (19). A significant amount of MTF-1 is located in the nucleus in the noninduced state, as assayed by Western (1) (57) and EMSA (24) analyses, and in vivo footprinting studies show a detectable footprint at MREd in the absence of added metal (21), presumably due to the binding of MTF-1. In addition, ChIP assays confirmed the interaction of some MTF-1 with the MT-1 promoter in uninduced cells (Fig. 9) and (19,49). It is thus possible that MREd-bound MTF-1 allows NF1 to bind to one of the NF1 sites and, with other general transcription factors, control basal transcription. Interestingly, the NF1a/Sp1a site is occupied both in the basal and induced state (17,21), and we showed by in vitro DNA binding competition experiments that the NF1a site has an apparent higher affinity for NF1 that the NF1b site (data not shown). In metal-induced cells, MTF-1 would strongly bind to MREd and MREa, thus causing further changes in chromatin structure and allowing NF1 to interact with the other NF1 site and to induce transcription. Alternatively, binding of NF1 to one NF1 site is MTF-1-dependent while binding to the other site is not. Gene promoter induction is often associated with histone acetylation and recruitment of chromatin remodeling complexes (58). It is likely that following zinc-induced MTF-1 DNA-binding, an ATP-dependent remodeling complex and a histone acetyltransferase (HAT) are recruited to the MT promoter leading to local changes in
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

11
chromatin structure and the subsequent binding of NF1, cofactors and mediator proteins. In fact, MTF-1 DNA-binding activity is sensitive to histone modifications as DNA-binding and expression of MTF-1 increase in lymphosarcoma cells treated with inhibitors of histone deacetylase (59). However, the ATPase chromatin remodeling complex SWI/SNF does not appear to be required for the activation of the MT promoter in response
to metals (60). The identification of the putative chromatin remodeling complexes and HAT recruited to the MT promoter in response to metal induction and the elucidation of the mechanisms of action of these factors will be a challenging task in the future and may reveal new insights on metal-regulated transcription.
REFERENCES
1. Palmiter, R. D. (1998) Proc. Natl. Acad. Sci. USA 95, 8428-8430 2. Coyle, P., Philcox, J. C., Carey, L. C., and Rofe, A. M. (2002) Cell Mol Life Sci. 59, 627-647 3. Palmiter, R. D., Findley, S. D., Whitmore, T. E., and Durnam, D. M. (1992) Proc. Natl. Acad. Sci.
USA 89, 6333-6337 4. Moffatt, P. and Séguin, C. (1998) DNA Cell Biol. 17, 501-510 5. Quaife, C. J., Findley, S. D., Erickson, J. C., Froelick, G. J., Kelly, E. J., Zambrowicz, B. P., and
Palmiter, R. D. (1994) Biochemistry 33, 7250-7259 6. Giedroc, D. P., Chen, X., and Apuy, J. (2001) Antioxid. Redox. Signal. 3, 577-596 7. Laity, J. H. and Andrews, G. K. (2007) Arch. Biochem. Biophys. 463, 201-210 8. Murphy, B. J., Andrews, G. K., Bittel, D., Discher, D. J., McCue, J., Green, C. J., Yanovsky, M.,
Giaccia, A., Sutherland, R. M., Laderoute, K. R., and Webster, K. A. (1999) Cancer Res. 59, 1315-1322
9. Dalton, T. P., Li, Q., Bittel, D., Liang, L., and Andrews, G. K. (1996) J. Biol. Chem. 271, 26233-26241
10. Adilakshmi, T. and Laine, R. O. (2002) J. Biol. Chem. 277, 4147-4151 11. Carter, A. D., Felber, B. K., Walling, M. J., Jubier, M. F., Schmidt, C. J., and Hamer, D. H.
(1984) Proc. Natl. Acad. Sci. USA 81, 7392-7396 12. Stuart, G. W., Searle, P. F., Chen, H. Y., Brinster, R. L., and Palmiter, R. D. (1984) Proc. Natl.
Acad. Sci. USA 81, 7318-7322 13. Stuart, G. W., Searle, P. F., and Palmiter, R. D. (1985) Nature 317, 828-831 14. Searle, P. F., Stuart, G. W., and Palmiter, R. D. (1985) Mol. Cell. Biol. 5, 1480-1489 15. Carthew, R. W., Chodosh, L. A., and Sharp, P. A. (1987) Genes Dev. 1, 973-980 16. Datta, P. K. and Jacob, S. T. (1997) Biochem. Biophys. Res. Commun. 230, 159-163 17. Li, Q. W., Hu, N. M., Daggett, M. A. F., Chu, W. A., Bittel, D., Johnson, J. A., and Andrews, G.
K. (1998) Nucleic Acids Res. 26, 5182-5189 18. Andrews, G. K., Lee, D. K., Ravindra, R., Lichtlen, P., Sirito, M., Sawadogo, M., and Schaffner,
W. (2001) EMBO J. 20, 1114-1122 19. Daniels, P. J. and Andrews, G. K. (2003) Nucleic Acids Res. 31, 6710-6721 20. Westin, G. and Schaffner, W. (1988) EMBO J. 7, 3763-3770 21. Mueller, P. R., Salser, S. J., and Wold, B. (1988) Genes Dev. 2, 412-427 22. Heuchel, R., Radtke, F., Georgiev, O., Stark, G., Aguet, M., and Schaffner, W. (1994) EMBO J.
13, 2870-2875 23. Labbé, S., Larouche, L., Mailhot, D., and Séguin, C. (1993) Nucleic Acids Res. 21, 1549-1554 24. LaRochelle, O., Stewart, G., Moffatt, P., Tremblay, V., and Séguin, C. (2001) Biochem. J. 353,
591-601 25. Majumder, S., Ghoshal, K., Gronostajski, R. M., and Jacob, S. T. (2001) Gene Expr. 9, 203-215 26. Jacob, S. T., Majumder, S., and Ghoshal, K. (2002) Environ. Health Perspect. 110 Suppl 5, 827-
830 27. Aniskovitch, L. P. and Jacob, S. T. (1997) Arch. Biochem. Biophys. 341, 337-346
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

12
28. Aniskovitch, L. P. and Jacob, S. T. (1998) Oncogene 16, 1475-1486 29. Candeliere, G. A., Jurutka, P. W., Haussler, M. R., and St-Arnaud, R. (1996) Mol. Cell. Biol. 16,
584-592 30. Martinez, E., Dusserre, Y., Wahli, W., and Mermod, N. (1991) Mol. Cell. Biol. 11, 2937-2945 31. Faraonio, R., Moffatt, P., LaRochelle, O., St-Arnaud, R., Schipper, H. M., and Séguin, C. (2000)
Eur. J. Biochem. 267, 1743-1753 32. LaRochelle, O., Gagné, V., Charron, J., Soh, J. W., and Séguin, C. (2001) J. Biol. Chem. 276,
41879-41888 33. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular cloning. A laboratory manual,
2nd Ed, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY 34. Pouysségur, J. and Roux, D. (1999) in Methods in Molecular Medecine, Vascular Disease:
Molecular Biology and Gene Therapy Protocols (Baker, A. H. ed) pp. 315-321, Humana Press Inc., Totowa, NJ
35. Thomassin, H., Hamel, D., Bernier, D., Guertin, M., and Belanger, L. (1992) Nucleic Acids Res. 20, 3091-3098
36. Labbé, S., Prévost, J., Remondelli, P., Leone, A., and Séguin, C. (1991) Nucleic Acids Res. 19, 4225-4231
37. Aparicio, O., Geisberg, J. V., Sekinger, E., Yang, A., Moqtaderi, Z., and Struhl, K. (2005) in Current protocols in molecular biology (Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K. eds) pp. 21.3.8-21.3.11, Greene/Wiley Interscience, New York
38. Radtke, F., Georgiev, O., Muller, H. P., Brugnera, E., and Schaffner, W. (1995) Nucleic Acids Res. 23, 2277-2286
39. Séguin, C. and Prévost, J. (1988) Nucleic Acids Res. 16, 10547-10560 40. Culotta, V. C. and Hamer, D. H. (1989) Mol. Cell. Biol. 9, 1376-1380 41. Radtke, F., Heuchel, R., Georgiev, O., Hergersberg, M., Gariglio, M., Dembic, Z., and Schaffner,
W. (1993) EMBO J. 12, 1355-1362 42. Park, J. S., Qiao, L., Gilfor, D., Yang, M. Y., Hylemon, P. B., Benz, C., Darlington, G., Firestone,
G., Fisher, P. B., and Dent, P. (2000) Mol Biol Cell 11, 2915-2932 43. Hung, C. F. and Penning, T. M. (1999) Mol Endocrinol 13, 1704-1717 44. Bi, Y., Palmiter, R. D., Wood, K. M., and Ma, Q. (2004) Biochem. J. 380, 695-703 45. Gronostajski, R. M. (2000) Gene 249, 31-45 46. Oikarinen, J., Hatamochi, A., and de, C. B. (1987) J Biol Chem. 262, 11064-11070 47. Yin, H., Smith, M., and Glass, J. (2005) Prostate 62, 209-216 48. Wang, Y., Lorenzi, I., Georgiev, O., and Schaffner, W. (2004) Biol. Chem. 385, 623-632 49. Marr, M. T., Isogai, Y., Wright, K. J., and Tjian, R. (2006) Genes Dev. 20, 1458-1469 50. Smith, C. L. and Hager, G. L. (1997) J. Biol. Chem. 272, 27493-27496 51. Wu, L. and Whitlock, J. P., Jr. (1992) Proc. Natl. Acad. Sci. U. S. A 89, 4811-4815 52. Blomquist, P., Li, Q., and Wrange, O. (1996) J. Biol. Chem. 271, 153-159 53. Chen, X. H., Chu, M. H., and Giedroc, D. P. (1999) Biochemistry 38, 12915-12925 54. Senear, A. W. and Palmiter, R. D. (1983) Cold Spring Harbor Symp. Quant. Biol. 47, 539-547 55. MacArthur, C. A. and Lieberman, M. W. (1987) J. Biol. Chem. 262, 2161-2165 56. Sutter, N. B., Scalzo, D., Fiering, S., Groudine, M., and Martin, D. I. (2003) Proc. Natl. Acad. Sci.
USA 100, 1105-1110 57. Smirnova, I. V., Bittel, D. C., Ravindra, R., Jiang, H., and Andrews, G. K. (2000) J. Biol. Chem.
275, 9377-9384 58. Mellor, J. (2005) Mol. Cell 19, 147-157 59. Ghoshal, K., Datta, J., Majumder, S., Bai, S., Dong, X., Parthun, M., and Jacob, S. T. (2002) Mol
Cell Biol. 22, 8302-8319 60. De La Serna, I. L., Carlson, K. A., Hill, D. A., Guidi, C. J., Stephenson, R. O., Sif, S., Kingston,
R. E., and Imbalzano, A. N. (2000) Mol. Cell. Biol. 20, 2839-2851
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

13
61. Ramji, D. P. and Foka, P. (2002) Biochem. J. 365, 561-575
FOOTNOTES *We thank Drs. Nicolas Mermod for providing the NF1 expression vectors, and Jacques Pouysségur for the pNHE1-R-1 vector. We are grateful to Alan Anderson for critical reading of the manuscript and to Jacques Côté, Amine Nourani and Manjapra Govindan for helpful suggestion and discussion on chromatin structure and ChIP experiments. This research was supported by a grant from the Conseil de recherches en sciences naturelles et en génie du Canada to C.S. The abbreviation used are, ChIP, chromatin immunoprecipitation; EMSA, electrophoretic mobility shift assay; HAT, histone acetyltransferase; LUC, luciferase; MT, metallothionein; MRE, metal regulatory elements; MTF-1, metal-responsive transcription factor-1; oligo, oligonucleotide; ROS, reactive oxygen species; tBHQ, tert-butylhydroquinone. (1) St-Gelais G. and Séguin, C., unpublished results.
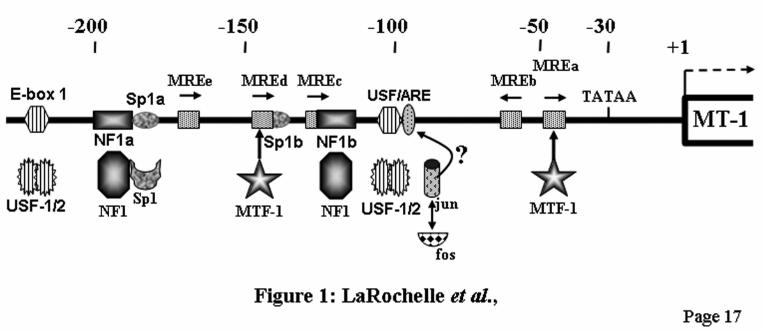
FIGURE LEGENDS Fig. 1. Map of the mouse MT-1 gene promoter. Schematic representation of the mouse MT-1 gene proximal promoter. Arrangement of the five MREs (arrows) (11,12), the E-box 1 and the USF/ARE elements (19), the binding sites for the transcription factor Sp1 and NF1, and the TATA box. Below the line, the corresponding transcription factors interacting with the different elements are shown. The numbers at the top refer to the positions relative to the transcription start point. Fig. 2. Identification of two distinct MRE-binding proteins A) DNaseI footprinting analysis performed with chromatographic fractions from the heparin-Sepharose column eluted with a NaCl gradient, as indicated schematically over the lanes. Note that the MREc-binding activity is present in all the fractions while the MREd-binding activity is mainly present in fractions 3-5, as evidenced by the DNaseI hypersensitive site (stars). This hypersensitive site is generated by the binding of MTF-1 on MREd. B) DNaseI footprinting analysis performed with fractions from the MRE affinity column eluted with a NaCl step gradient, as indicated. Fraction 4 (lane 3) from the 250 mM salt fraction contains a specific MREc-binding activity, whereas MTF-1 mainly eluted in the second 650 mM salt fraction (lane 8). The probe was a mouse MT-1 gene promoter DNA fragment extending from –200 to −41. The positions of the different cis-acting elements are indicated on the left, as determined by Maxam-Gilbert sequencing. Numbers above the lanes indicate the fraction numbers while those below the gel correspond to the lanes. Lanes 2 and 3 on each panel are non-adjacent lanes from the same gel. Lanes: L50, L50-cell nuclear extract; FT, flow through; No, no extract. Fig. 3. Competition experiments in DNaseI footprinting assays. A) Footprinting reaction was carried out with L50-cell crude nuclear extracts, and competition was performed with double-stranded unlabeled oligos (Table 1) corresponding to the mouse MT-1 promoter Sp1a site (Sp1a), the mouse MT-1 MREd (MREd) and the non specific oligo MUTds (39). The probe was the same as in Fig. 2. Lanes 6 and 7 are non-adjacent lanes from the same gel. B) Footprinting reaction was carried out with aliquots of fraction 4 (Fig. 2B, lane 3) of the heparin-Sepharose column containing the specific MREc-binding activity. Competition was performed with double-stranded unlabeled oligos corresponding to an extended region of the mouse MT-1 promoter around MREc (MREcl), the mouse MT-1 MREc (MREc), and the oligos MUTds and Sp1a. The probe was a mouse MT-1 gene promoter DNA fragment extending from –348 to +72. Twenty to 100 ng of competitors, as indicated above the lanes, were added together with the probe and binding was allowed to proceed for 10-15 min at 24OC, before adding the DNaseI. Lanes: 0, no competitor; L50, L50-cell nuclear extract; No, no extract. The position of the different cis-acting elements
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

14
is indicated on the right as determined by Maxam-Gilbert sequencing. Numbers below the gel correspond to the lanes. C) Sequence of the mouse MT-1 promoter, nucleotides −212 to −99, encompassing the two NF1 sites. MREs and Sp1 sites are underlined and the NF1 sites are boxed. The footprints present in this region are indicated over the sequences and the oligos used as competitors are listed under the sequence. Fig. 4. Identification of two NF1 half sites in the mouse MT-1 promoter. A) Alignment of the two MT-1 NF1 sites with NF1 (45) and C/EBP (61) consensus sequences. Underlined nucleotides correspond to nucleotides conserved in the mouse MT-1 NF1 sites. B) Competition experiments in DNaseI footprinting assays. The footprinting reaction was carried out as described in Fig. 3B, using as competitors double-stranded unlabeled oligos corresponding to the mouse MT-1 NF1b site (NF1b), a generic NF1 site (NF1), the mouse MT-3 promoter NF1/Sp1 site (SN), and the oligo Sp1a alone (Sp1) or in combination with the oligo NF1b (Sp1+NF1). Fig. 5. Identification of the NF1b DNA-binding activity as a 60-kDa polypeptide by UV crosslinking assay. DNA affinity labeling of NF1b-binding factors in a L50-cell crude nuclear extract (L50), the flow through of the heparin-Sepharose column (FT1), fraction 9 (9) of the heparin-Sepharose column enriched with the MREc-binding protein, the flow trough (FT2) of the MRE affinity column (MRE affinity), and fraction 3 of the affinity column specifically containing the MREc-binding activity. The probe was the NF1b oligo (Table 1). Addition of cold MREcl oligo (cl) to the reaction completely inhibited formation of the approximately 60 kDa DNA-protein complex (Arrow). Numbers below the gel indicate the lanes. The asterisk indicates non specific binding. Lanes: M, molecular weight markers; 0, no competitor. F, free probe. Fig. 6. The NF1 transcription factor present in nuclear extracts specifically binds to the NF1b oligo. A) EMSA reactions were performed by incubating 32P-labeled NF1b oligo (20 fmol, approximately 0.5 ng), with HepG2-cell nuclear extracts. For competition reactions, 100 ng of double-stranded unlabeled oligos (Table 1) were used as indicated above the lanes. B) Reactions were performed using the 32P-labeled C/EBP oligo as the probe and a HepG2-cell nuclear extract. Competition was performed with double-stranded unlabeled C/EBP and MUTds oligos. Anti-NF1 (Ab-NF1), anti-C/EBP (Ab-C/EBP) antibodies or non-specific anti-MT-3 antibody (Ab-MT-3) were added to the EMSA reactions and the mixtures preincubated for 5 min at 21oC before the addition of [32P]-labeled oligo probes. Arrows indicate the DNA-protein complexes. SS refers to supershifted complexes. F, free probe; B, bound DNA. Fig. 7. Transient transfection studies in HepG2 cells. A) Cells transfected with a plasmid mixture including the reporter wild-type MT1-LUC (WT, 1), containing 1843 bp of mouse MT-1 gene 5’ flanking sequence, or the NF1 mutant reporter plasmids NF1a∆-LUC (NF1a∆, 3), NF1b∆-LUC (NF1b∆, 2) or NF1ab∆-LUC (NF1ab∆, 4), and pTK-rLUC, as internal standard, were treated or not with 100 µM ZnCl2 or 2.5 µM CdCl2 for 6-8 h. Cell extracts were prepared and LUC activity was measured with a dual LUC kit. Results are expressed as percentage of firefly LUC (fLUC) activity relative to the level directed by the renilla LUC (rLUC) construct, and as a percentage relative to that of the WT DNA induced by zinc, which is taken as 100. Inset: Basal levels plotted on a different scale. Data represent the average ± S.D. of three independent experiments performed in duplicate or in triplicate. B) Cells were transfected as described in panel A with a plasmid mixture containing MT1-LUC (WT) or 5’ deletion mutant reporter plasmids, as indicated, and pTK-rLUC. C) Cells were transfected as described in panel A with a plasmid mixture of MT1-LUC (WT) or the NF1 double mutant NF1ab∆-LUC (NF1ab∆), the internal standard pTK-rLUC, and increasing amount of the NF1 expression vector p113-CTF-1 (CTF1), as indicated. D) Cells were transfected as described in panel A with a plasmid mixture containing MT1-LUC, the internal standard pTK-rLUC, and increasing amount of the NF1 mutant expression vector p113-CTF-1∆, as indicated. E) Cells were transfected as described in panel A with LUC reporter plasmids containing the mouse MT-1 minimal promoter (−35 to +68) fused to two MREd elements (MREdd), the NF1a and NF1b sites
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

15
(NF1ab), or a combination of both the NF1 sites and the double MREd element (NF1ab/MREdd). Results are expressed as percentage of firefly LUC (fLUC) activity relative to the level directed by the renilla LUC (rLUC) construct, and as a percentage relative to that of MREdd plasmid induced by zinc, which is taken as 100. pGL2 (5 in inset), pGL2-Basic plasmid. Fig. 8. Stable transfection studies in HepG2 cells. A) Cells stably transfected with the MT1-LUC (WT), NF1a∆-LUC (NF1a∆), NF1b∆-LUC (NF1b∆), NF1ab∆-LUC (NF1ab∆), or pGL2-basic (not shown) reporter plasmids, were treated or not with 100 µM ZnCl2, 2.5 µM CdCl2 or 100 µM tBHQ for 6-8 h. Cells lysates were prepared from two different pools of stable transfectants, and LUC activity was measured and expressed as relative fLUC activity per µg of total protein. The results of one pool are shown. B) Cells were stably transfected with synthetic promoters containing the mouse MT-1 minimal promoter fused to two MREd elements (MREdd), six MREa elements [(MREa)6], the NF1a and NF1b sites (NF1ab), or a combination of both the NF1 sites and the double MREd element (NF1ab/MREdd). Cell lysates were prepared and LUC activity was measured as described in panel A. Fig. 9. NF1 binds the mouse MT-1 promoter in vivo in a zinc-inducible and MTF-1-dependent manner. ChIP assays were performed using chromatin isolated from L cells or dko7 mouse embryonic fibroblasts (MTF-1 null) treated or not with 100 µM ZnCl2 for 3 h prior to formaldehyde cross-linking. Immunoprecipitation of cross-linked chromatin was done with NF1 (upper panel), MTF-1 (lower panel) antibodies or a pre-immune normal rabbit serum (PI), as indicated. DNA from both the IP input and the IP bound fractions was amplified by PCR with primer pairs for the mouse MT-1 promoter (MT-1) and the coding region of the G6PD gene. Some dko cells were transfected with a MTF-1 expression vector (CMV-MTF-1), grown for 24 h and then treated with zinc. Input: amplification of DNA prior to immunoprecipitation. The input sample contained 0.4% of the supernatant used for NF1 immunoprecipitation and 0.8% for MTF-1. The PCR products were analyzed by agarose gel electrophoresis. These ChIP assays were performed three times with similar results using two different chromatin preparations.
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

16
Table 1 Sequences of the synthetic oligos used in this study
Nucleotides in bold designate the core MRE sequence, those in bold underlined the NF1 core conserved sequences, and those in italic underlined the Sp1 site. Nucleotides in lower case letters at the extremities of the oligos correspond to restriction sites added to the oligos, while those in the middle of the oligos correspond to mutations introduced in a given site. In NF1ab, the NF1a (bold) and NF1b (underlined) sites are shown. MREdd contains two strong mouse MT-1 MREd in opposite orientation (bold, underlined). The NF1 oligo is from Santa Cruz Biotechnologies.
Name Sequence
NF1 5’-TTTTGGATTGAAGCCAATATGATAA
NF1a 5’-CCGAGCCAGTCGTGCCAAAGG
NF1b 5’-GCGCTCGGCTCTGCCAAGGACGC
NF1ab (5'-tcgagATCCGAGCCAGTCGTGCCAAAGGGCGCTC GGCTCTGCCAAGGACGC)
MREc 5’-gatccAAAGTGCGCTCGGCTCa
MREcl 5’-gatcccAGTGCGCTCGGCTCTGCCAAGGACGCa
MREd 5’-cgatCTCTGCACTCCGCCCGA
MREdd 5'-tcgAAGATCTCGGGCGGAGTGCAGAGCTAGCTCTGC ACTCCGCCCGAc
Sp1a 5’-gatccAAGGGGCGGTCCCGCa
C/EBP 5’-TGCAGATTGCGCAATCTGCA
NFκB 5’-CAACGGCAGGGGAATCTCCCTCTCCTT
NF1a∆ 5’-CGAGCCAGTCGTtaaAAAGGGGCGGTC
NF1b∆ 5’-GCGCTCGGCTCTtaaAAGGACGCGGGG
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Tremblay, Geneviève St-Gelais and Séguin CarlOlivier LaRochelle, Simon Labbé, Jean-François Harrisson, Simard Carl, Véronique
metallothionein-1 gene in response to metal ionsNuclear factor 1 and metal transcription factor-1 synergistically activate the mouse
published online January 29, 2008J. Biol. Chem.
10.1074/jbc.M800640200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on August 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

![New Function of astrocyte MyD88 in high-fat-diet-induced … · 2020. 6. 19. · kinase and nuclear factor kappa B (NF-κB) pathways [19, 20]. A recent study reported that interaction](https://img.pdfslide.fr/doc/110x75/6128bbb7febc6e13b44cc1d9/new-function-of-astrocyte-myd88-in-high-fat-diet-induced-2020-6-19-kinase-and.jpg)