Embed Size (px)
Citation preview

REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE
SCIENTIFIQUE
UNIVERSITE MENTOURI CONSTANTINE FACULTE DES SCIENCES EXACTES
DEPARTEMENT DE PHYSIQUE
N° d’ordre : Série :
THESE
PRESENTEE POUR OBTENIR LE DIPLOME DE DOCTORAT D’ETAT EN PHYSIQUE
OPTION
CRISTALLOGRAPHIE
THEME
ETUDE DES PROPRIETES OPTIQUES DE NANOCRISTAUX SEMI-CONDUCTEURS ET DE CENTRES COLORES DISPERSES
DANS LES HALOGENURES ALCALINS KBr, KCl ET NaCl
Par FOUZIA ZEHANI
Soutenue le: 09/12/ 2007
Devant le jury :
Président: A. BOULTIF Prof. Univ. Mentouri-Constantine Rapporteur: M. SEBAIS Prof. Univ. Mentouri -Constantine Examinateurs: B. BOUDINE M.C. Univ. Mentouri -Constantine N. BRIHI Prof. Univ. Jijel Z. TAKKOUK M.C. Univ. Jijel M. S. BOUMAZA M.C. Univ. Guelma

A mes parents
A mes chers frères: Djyad et Ryad
A mes chères sœurs: Souad, Samira, Rofia, Lamis et Rima
A mes beaux frères et ma belle sœur
A mes nièces et neveux
A tous ceux qui me sont chers

REMERCIEMENTS
Ce travail a été réalisé au laboratoire de cristallographie de l’université Mentouri-
Constantine, au laboratoire de Biologie de l’université de Jijel et à l’institut des nanosciences de
Paris (l’INSP).
Mes remerciements vont tout premièrement à Dieu tout puissant pour la volonté, la
santé et la patience qu'il m'a données pour accomplir ce travail.
Je remercie vivement Monsieur Miloud SEBAIS, Professeur à l’université Mentouri-
Constantine, de m’avoir confié et dirigé ce travail avec beaucoup d’efficacité, qu’il soit rassuré
de mon estime et ma reconnaissance. Grâce à son esprit pédagogue, il m’a fait prendre
conscience du côté passionné et exaltant de la recherche scientifique.
J’exprime toute ma reconnaissance à Monsieur Ali BOULTIF, Professeur à l’université
Mentouri-Constantine, qui a bien voulu accepter la présidence du jury de cette thèse.
J’adresse mes respectueux remerciements à Monsieur Boubekeur BOUDINE, maître de
conférence à l’université Mentouri-Constantine, pour avoir accepter de participer au jury de
cette thèse.
Mes remerciements sont aussi adressés à Monsieur Noureddine BRIHI, Professeur à
l’université de Jijel, qui a accepté d’honorer par sa présence le jury de cette thèse.
Je tiens aussi à remercier Monsieur Zahi TAKKOUK, maître de conférence à
l’université de Jijel, qui a bien voulu faire partie du jury de cette thèse.

J’en suis profondément honorée et j’adresse mes remerciements les plus respectueux et le
plus sincères à Monsieur M. S. BOUMAZA, maître de conférence à l’université de Guelma,
d’avoir accepté de jury cette thèse.
J’adresse mes remerciements les plus respectueux au Professeur Tadashi ITOH de
l’université d’Osaka au Japon, au Professeur Phillipe LAVALLARD et au Docteur Carlos
BARTHOU de l’institut des nanosciences de Paris (l’INSP) pour m’avoir aidé au cours de la
préparation de cette thèse et la réalisation des mesures optiques.
Je remercie également les membres du laboratoire de Biologie (Zoologie) pour leur aide
dans la réalisation du traitement thermique des échantillons.
Je tiens à remercier chaleureusement : Melle Samira DIB, Mme Farida MEDJANI, Mr
Abdelhafid BOUNAMIS et Mr Abdnacer TILBI de l’université de Jijel pour l’aide qu’ils
m’ont apportée.
Rien de tout cela n’aurait été possible sans le soutient de mes parents, de toute ma
famille et de toutes mes amies. Qu’ils sachent simplement qu’ils sont au fond de mon coeur
chaque instant et en tout lieu.

SOMMAIRE
INTRODUCTION………………………………………………………………………………1
CHAPITRE I : DEFINITION ET PROPRIETES OPTIQUES DES CENTRES COLORES
DANS LES CRISTAUX D’HALOGENURES ALCALINS
I- 1 INTRODUCTION……………………………………………………………………………3
I- 2 LES CENTRES COLORES…………………………………………………………………..3
I- 2- 1 Les centres colorés à électrons…………………………………………………………..4
I- 2- 1- 1 Centre F……………………………………………………………………………4
I- 2- 1- 2 Centre FA…………………………………………………………………………..5
I- 2- 1- 3 Centre F+2…………………………………………………………………….……5
I- 2- 1- 4 Centre (F+2)A………………………………………………………………...……..5
I- 2- 1- 5 Centre (F+2)H……………………………………………………………………….6
I- 2- 1- 6 Centre (F+2)AH………………………………………………………………….......6
I- 2- 1- 7 Centres R et M …………………………………………………………..…..…….8
I-2 -2 Les centres à trous……………………………….…………………………..……..……..8
CHAPITRE II ABSORPTION ET PHOTOLUMINECSENCE DES CENTRES
COLORES O2- ET (O2- - F+)
II- 1 CENTRES COLORES O2-…………………………………………………………………10
II-1- 1 Introduction…………………………………………………………………...………10
II- 1- 2 Les transitions électroniques dans l’ion moléculaire O2-…………………………….10
II- 1- 3 Types de spectres d’émission des centres O2-…………………...………………......12
II- 2 CENTRES COLORES (O2- - F+)……..…………………………………………………... 16
II- 2- 1 Introduction……………………………………………………………………… ….16
II- 2- 2 Interprétation des bandes d’absorption des centres (O2- - F+)………………………..17
II- 2- 3 Effet du traitement thermique sur les bandes
d’absorption des centres (O2- - F+)…………………………………..…….………...19

CHAPITRE III LES SEMI-CONDUCTEURS II-VI ET LES PROPRIETES
ELECTRONIQUES ET OPTIQUES DES NANOCRISTAUX SEMI-CONDUCTEURS
III- 1 LES SEMICONDUCTEURS II-VI………………………………………………………22
III-1-1 Introduction……………………………..………………………………………….22
III-1-2 Propriétés cristallographiques des semi- conducteurs II-VI……………………….23
III-2 PROPRIETES ELECTRONIQUES DES NANOCRISTAUX
DE SEMI-CONDUCTEURS…………………………………………………………...….25
III-2-1 Introduction…………………………………….………………..…………………...25
III-2-2 L'exciton dans le semi-conducteur massif……..……………………………………..27
III-2-3 L'exciton dans le semi-conducteur nanocristallin…………………………………….28
III-2-4 Régimes du confinement………………………………………………………………28
III-2-4-1 Le confinement fort…………………………………………………………..29
III-2-4-2 Le confinement faible………………………………………………………..29
III-2-4-3 Le confinement intermédiaire………………………………………………..29
III-2-5 Influence du confinement sur les propriétés
optiques des semi-conducteurs………………………………………………………..…30
CHAPITRE IV METHODE DE CZOCHRALSKI ET APPAREILS DE MESURES
UITLISES
IV- 1 INTRODUCTION…………………………………………………………………………31
IV- 2 METHODE DE CZOCHRALSKI………………………………………………………...31
IV- 2- 1 Principe de la méthode……………………………………………………………...31
IV- 2- 2 Appareillage…………….……………………………………………………….…..32
IV- 2- 2- 1 Partie mécanique…………………………………………………….……….32
IV- 2- 2- 2 Partie thermique………………………………………………………….…..32
IV- 3 ELABORATION DES ECHANTILLONS ETUDIES……..……………………….…….34
IV- 3- 1 Elaboration des monocristaux de KBr, KCl et NaCl………………………………..34
IV- 3- 2 Clivage des monocristaux et polissage des pastilles clivées………….……………..35
IV- 3- 3 Traitement thermique des échantillons…………………………….………………..36

IV-3-4 Elaboration des monocristaux de KCl dopés par
Les nanocristaux de CdSe……………………………………………….……………36
IV-4 CARACTERISATION SPECTROSCOPIQUE……………………….…………………..36
IV- 4- 1 Mesure de l’absorption optique UV-visible
à la température ambiante………………………………………………………….….36
IV- 4- 2 Mesure la photoluminescence et l’excitation
de la photoluminescence ……………………………………………………….……38
IV- 5 LE CRYOSTAT………………………………………………………….………………..39
CHAPITRE V CARACTERISATION OPTIQUE
V- 1 CARACTERISATION OPTIQUE DES MONOCRISTAUX
DE KBr, KCl ET NaCl…………………………………………………………………….41
V-1-1 Absorption optique……………………………………………………………………41
V-1-2 LA la photoluminescence et l’excitation de la photoluminescence ……………...….43
V-1-2-1 Cas du KBr………………….….………………..….…………………….………..43
V-1-2-2 Cas du KCl…………………………..……………………………………….….....52
V-1-2-3 Cas du NaCl………………………….…………………………………….……....59
V-2 CARACTERISATION DES MONOCRISTAUX DE KCl DOPES PAR CdSe…………..66
V-3-1 Caractérisation structurale………..……………………………………………...…..66
V -3-2 Caractérisation optique………..……….………………………………………..…..69
V- 3 CONCLUSION……………………………….……………………………………………71
CONCLUSION GENERALE………………………….………………………..…………….72
REFERENCES…………………………………………………………………………….……74

1
INTRODUCTION
Les halogénures alcalins occupent une position particulièrement importante parmi les
matériaux solides car ils présentent des propriétés optiques et électroniques remarquables qui
leur confèrent une place tout à fait importante dans la technologie moderne. Plusieurs de leurs
propriétés sont profondément affectées par les fortes interactions de Coulomb qui donnent lieu à
un caractère ionique fort. Ils sont décrits dans le cadre de la théorie des bandes comme des
isolants avec des bandes interdites très grandes variant entre 6-13 eV [1]. Ils sont caractérisés par
un domaine de transparence exceptionnellement étendu qui va de l’ultraviolet lointain à
l’infrarouge lointain. Il y a donc une très large bande spectrale dans laquelle l’absorption optique
et l'émission des défauts de réseau peuvent être facilement détectées et étudiées.
Vers la fin du dix neuvième siècle, Goldstein a découvert la coloration des cristaux
naturels d’halogénures alcalins qui devraient être transparents. Il a montré qu'il est possible de
reproduire quelques une des couleurs trouvées dans les cristaux naturels si les halogénures
alcalins sont soumis à une irradiation de rayons cathodiques. Cet avantage a fait l’objet des
travaux de recherche de Pohl dans les années trente [2]. Il a trouvé que la coloration de ces
cristaux est due à la présence des défauts ponctuels dans le réseau cristallin qu’il appela centres
colorés; le plus important est connu sous le nom du centre F, nom venant du mot allemand Farbe
signifiant couleur.
Plusieurs types de traitements thermiques ou chimiques ou par irradiation permettent
d’introduire facilement des défauts ponctuels dans le réseau cristallin dans les halogénures
alcalins. Ces derniers sont le siège d’une multitude de centres colorés parmi lesquels les centres
O2- et (O2- - F+) qui n’ont fait l’objet que de quelques rares études de recherche. Ce qui nous a
incités à les étudier et essayer d’ajouter un plus à ce domaine peu exploité.
L’intérêt technologique actuel se concentre sur les nanocristaux élaborés par différentes
méthodes de croissance car les propriétés physico-chimiques de ces matériaux sont généralement
différentes de celles du cristal massif. Pour les semi-conducteurs, les propriétés électroniques et
optiques changent à cause du confinement quantique des excitons dans un volume réduit. C’est
un problème de dimension dans lequel on observe le passage des propriétés du cristal à celles de
la molécule.
L’incorporation des nanocristaux des semi-conducteurs dans les matrices à larges bandes
interdites tel que: les verres et les monocristaux d’halogénures alcalins, a permis la dispersion
des matériaux dont il était impossible d’étudier leurs propriétés optiques.

2
Afin de contribuer à mieux comprendre l’effet de quelques défauts ponctuels sur les propriétés
optiques des halogénures alcalins, nous avons entrepris le présent travail qui consiste à :
1- élaborer des monocristaux d’halogénures alcalins (NaCl, KCl et KBr) sous atmosphère
libre pour les doper avec l’oxygène de l’air et former des défauts ponctuels dans leurs
matrices comme l’ion moléculaire O2- et la paire de défaut (O2- - F+),
2- élaborer des nanocristaux de semi-conducteurs CdSe dispersés dans les monocristaux de
KCl.
Et ce, en vue d’étudier d’une part, l’effet de taille sur les propriétés optiques des semi-
conducteurs; et d’autre part, de montrer de nouvelles bandes d’émission de centres colorés (O2- -
F+) dans le domaine spectral l’UV-visible et de confirmer la transformation des centres O2- en
centres (O2- - F+).
Le travail réalisé dans cette thèse est structuré comme suit:
Le premier chapitre sera consacré à l’étude de quelques centres colorés rencontrés dans
les halogénures alcalins, ainsi que leurs propriétés optiques.
Dans le deuxième chapitre, nous décrirons les propriétés optiques (absorption et
photoluminescence) des centres colorés O2- et (O2- - F+).
L’étude des propriétés électroniques et optiques des nanocristaux de semi-conducteurs
sera présentée dans le troisième chapitre.
La description des méthodes expérimentales qui ont permis l’élaboration et la
caractérisation des échantillons fera l’objet du quatrième chapitre.
Dans le cinquième chapitre, nous exposerons la discussion des résultats concernant la
caractérisation optique des centres O2- et (O2- - F+) dans les matrices cristallines de KBr, KCl et
NaCl et des nanocristaux du semi-conducteur CdSe dans KCl.
Enfin nous terminerons par une conclusion générale où seront regroupés les principaux
résultats avec des suggestions pour travaux futurs.

3
CHAPITRE I DEFINITION ET PROPRIETES OPTIQUES DES CENTRES COLORES
DANS LES CRISTAUX D’HALOGENURES ALCALINS
I-1 INTRODUCTION
Les cristaux ioniques sont formés d’ions positifs et négatifs. La liaison ionique résulte
de l’interaction électrostatique entre ces ions de charges opposées. Elle est sensiblement
supérieure en intensité à la liaison covalente et elle autorise une compacité deux fois plus élevée.
Les cristaux ioniques les plus simples et les plus étudiés sont les halogénures alcalins (formule
A+X-) [3].
Les halogénures alcalins sont des matériaux formés à partir d’atomes appartenant aux
colonnes I (Na, K, Rb) et VII (Cl, Br, F) de la classification périodique. Respectivement, chacun
d’eux comporte un et sept électrons sur sa couche électronique externe. Ils ont donc une valence
de six. De part la grande ionicité qui existe entre les éléments I et VII, leur combinaison donne
des composés à grand gap (ou des isolants) de l’ordre d’une dizaine d’eV. Cette caractéristique
les rend transparents à la lumière visible. Parmi les matériaux de cette famille qui présentent des
propriétés intéressantes, on peut citer: KBr, KCl et NaCl. Ces matériaux ont tous la même
structure, dite structure de type NaCl. Le tableau 1 donne les valeurs de l’énergie du gap (Eg) et
le paramètre de maille (a).
Tableau 1: Valeurs de Eg et a pour quelques halogénures alcalins [3, 5].
I-2 LES CENTRES COLORES
Le cristal parfait est tout à fait hypothétique. L’une des conditions pour qu’il soit parfait
n’est en effet jamais vérifiée. Dans un cristal, on peut distinguer plusieurs types de défauts:
défauts ponctuels (lacune, atome en substitution, atome en insertion…), défauts linéaires
(dislocations), défauts bidimensionnels (joints de grains et macles) et défauts tridimensionnels
précipités et amas ou ségrégations).
I - VII Eg (eV) a )(oA
NaCl 9,00 2,81
KCl 8,50 3,14
KBr 7,50 3,29

4
Durant notre travail, on va étudier quelques défauts ponctuels existant dans les
halogénures alcalins, nommés les centres colorés.
Dans les halogénures alcalins; les défauts sont principalement créés sur le sous réseau
anionique car les ions négatifs ont des niveaux d’excitation électronique plus bas que les cations
[3]. Les défauts ponctuels les mieux connus dans ces matériaux sont les lacunes et les atomes
interstitiels dans le sous réseau des ions halogènes qui constituent respectivement, les centres à
électrons et les centres à trous, et seul la première catégorie est responsable de l’effet laser
découvert dans les halogénures alcalins. Les centres à trous ne sont pas directement liés à l’effet
laser, mais ils contribuent à la formation des centres à électrons [4].
I-2-1 Les centres colorés à électrons
Les centres colorés à électrons sont constitués d’un ou plusieurs électrons piégés dans les
lacunes anioniques du réseau cristallin [6]. Ils sont qualifiés de:
- Centres intrinsèques s’ils sont formés par une combinaison de lacunes anioniques. Ils
sont notés : F, F-, F+….
- Centres extrinsèques s’ils sont formés par une combinaison d'une lacune anionique à côté
des impuretés. Ils sont notés : FA, (F+2) A, (F+
2)AH,…
- Les agrégats du centre F. Ils sont nuisibles pour l’effet laser et ils sont notés : F2, F3,…
I-2-1-1 Centre F
Le centre F est un électron localisé dans une lacune anionique (Fig. 1). C’est un centre
très simple, il occupe une position très particulière parmi les défauts des solides et il fût assimilé
à l’atome d’hydrogène dans la physique atomique [6]. Ce centre ne donne pas l’effet laser, mais
il est à la base de la formation de tous les autres centres. Il est utilisé comme un modèle pour
expliquer les propriétés importantes des centres colorés.
Si la lacune anionique piége deux électrons, on le note par F' ou F-. Mais, si elle ne piége pas
d’électron, on le note par F+ ou a ou par un carré vide ? (Fig. 1). La bande d’absorption du
centre F est déterminée par Mollow, elle est donnée par la relation suivante [7]:
84,1703 a )(oA

5
Fig. 1: Représentation de quelques centres colorés dans les halogénures alcalins [3].
I-2-1-2 Centre FA
Le Centre FA est un centre F à côté d’une impureté cationique alcaline.
I-2-1-3 Centre F+2
Le centre F+2 est constitué par deux lacunes anioniques adjacentes ayant piégé un électron
(Fig. 2). Sa configuration est similaire à celle de l’ion moléculaire H+2 immergé dans un milieu
diélectrique et les deux lacunes jouent le rôle du proton. Ce centre possède deux bandes
d’absorption; l’une dans le visible et l’autre dans l’infrarouge [9].
I-2-1-4 Centre (F+2)A
Le centre (F+2)A représente le centre F+
2 à côté d’une impureté cationique qui
remplace l’ion alcalin. C’est un centre à effet laser et combine les meilleures caractéristiques des

6
centres F+2 et FA. Les bandes d’absorption de ce centre sont légèrement décalées par rapport à
celle du centre F+2. La largeur du déplacement dépend de l’impureté substitutionnelle. Donc, par
un choix adéquat du dopant, les bandes d’absorption du centre F+2 se déplacent vers une nouvelle
gamme de longueurs d’onde [6].
Fig. 2 : Structure du centre F2
+.
I-2-1-5 Centre (F+2)H
Ce centre est formé d’une paire (O2- – F+) (un atome d’oxygène doublement ionisé à côté
d’une lacune anionique vide dans la direction <110> du réseau cristallin) à coté d’un centre F
[10, 11]. Il possède des propriétés identiques à celles du centre F+2. Il peut être considéré comme
un centre F+2 attaché dans différentes configurations possibles à l’ion voisin O2- (Fig. 3) [12]. Il
est noté (F+2)H où l’indice H indique que le centre F+
2 est attaché à l’ion O2- remplaçant un ion
halogène.
I-2-1-6 Centre (F+2)AH
Un autre type de centres colorés similaires aux centres (F+2)H a été développé par
l’association du centre F+2 avec un ion O2- et une impureté cationique d’un métal alcalin (Na+)
[13]. Ces centres ont deux configurations possibles comme le montre la figure 4 [14].
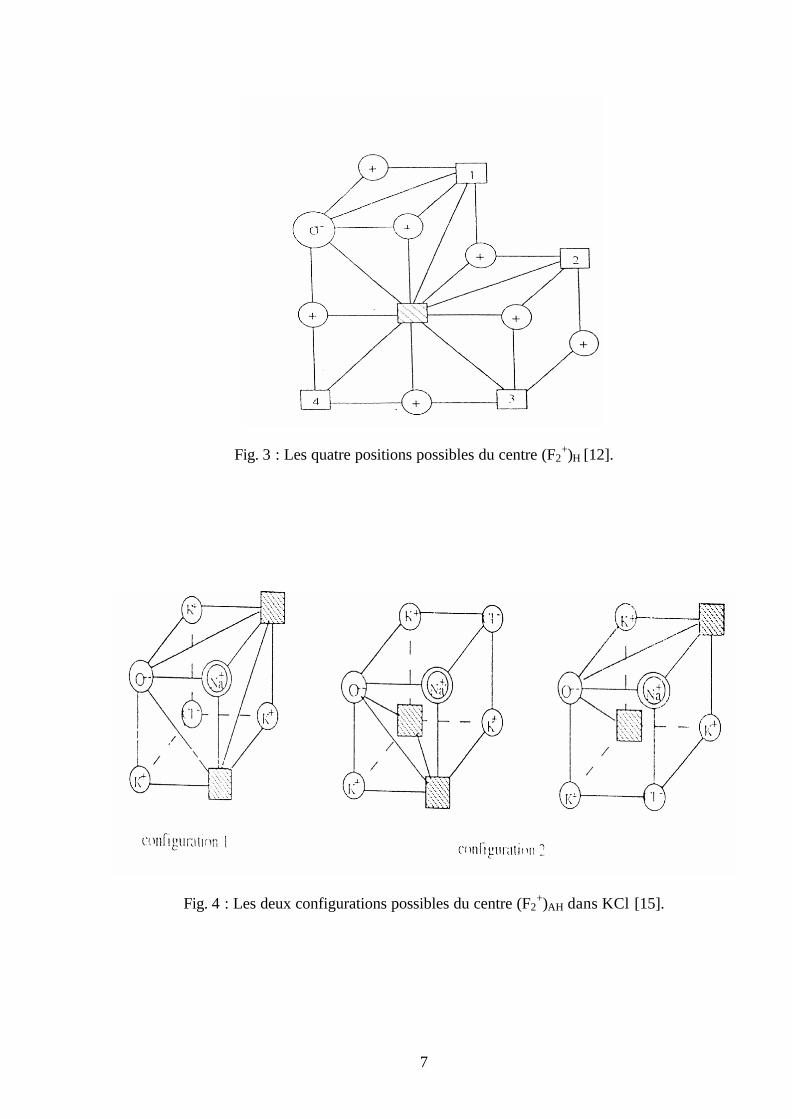
7
Fig. 3 : Les quatre positions possibles du centre (F2+)H [12].
Fig. 4 : Les deux configurations possibles du centre (F2+)AH dans KCl [15].

8
I-2-1-7 Centres M et R
Parmi les centres qui sont formés par les agrégats du centre F, on peut citer les centres M
et R (Fig. 1). Le premier centre est formé par deux centre F ou F2 et le deuxième est formé par
trois centres F ou F3. La bande d’absorption des centres M est donnée par la relation
suivante [7] : 56,11400 a )(oA
Alors que le centre R a deux bandes d’absorption notées: R1 et R2 dont les positions sont
données par [7]:
Pour R1 : )(816 84,1oAa
Pour R2 : 84,1884 a )(oA
I-2-2 Les centres à trous
Parmi les centres à trous les plus connus actuellement, on peut citer les centres Vk et les centres
H lesquels sont obtenus par irradiation des cristaux d’halogénures alcalins. Le centre Vk a été
mis en évidence par Castner et Känzig [15]. Il est formé par des paires électron-trou. Les trous
libres ont tendance à se piéger formant un centre Vk. Le trou est alors localisé autour de deux
anions (halogène X) et l’ensemble est équivalent à une molécule X-2. Cette configuration est
appelée "exciton auto-piégé centré". Dans certain cas, cette configuration semble pouvoir gagner
de l’énergie en décalant légèrement la molécule X-2 le long d’un axe cristallographique <110> et
en devenant une paire centre F- centre H. L’anion correspondant, lié en position interstitielle à un
autre anion du cristal, forme le centre H également appelé "dumbbell" (Fig.1) [3].
Remarque
Il existe d’autres défauts ponctuels dans les halogénures alcalins qui forment des centres
colorés comme les ions d’hydroxydes OH- et les ions H- (ou les centres U). Ces centres occupent
9
des positions substitutionnels à la place des halogènes. Les bandes d’absorption de ces deux
centres sont données par les deux relations suivantes [7, 16]:
Pour OH- : 95,0681 a )(oA
Pour U (ou H-) : 10,1615 a )(oA
Les valeurs des bandes d’absorption des centres colorés F, R1, R2, M, OH- et U pour
quelques halogénures alcalins sont regroupées dans le tableau 2.
Tableau 2: Longueurs d’onde d’absorption des centres colorés
F, M, R1, R2, U et OH- [7, 8].
I - VII F (nm) M (nm) R1 (nm) R2 (nm) OH-(nm) U (nm)
NaCl 471 701 547 592 190 192
KCl 576 835 669 725 203,5 216
KBr 630 897 732 792 215 228

10
CHAPITRE II ABSORPTION ET PHOTOLUMINESCENCE DES CENTRES
COLORES O2- ET (O2- – F+)
II- 1 CENTRES COLORES O2-
II-1-1 Introduction
La luminescence jaune des halogénures alcalins lorsqu’ils sont excités par la lumière UV
a été connue depuis 1937 par W. Honrath [17]. En 1961, J. Rolfe et al. étaient les premiers à
identifier cette émission et avaient montré qu’elle était due aux centres colorés O2- [18]. En 1962,
W. Sander avait confirmé que les cristaux d’halogénures alcalins élaborés sous atmosphère libre
contiennent de l’oxygène [19]. Dernièrement et à partir de 1994, la luminescence de ce centre
coloré dans les halogénures alcalins a attiré une nouvelle fois l’attention de plusieurs chercheurs
après la découverte de son activité laser [20-25].
L’ion moléculaire O2- est une impureté très soluble dans les cristaux d’halogénures alcalins. Il est
aussi une impureté commune entre tous ces matériaux élaborés par la méthode de fusion à l’air
libre ou sous atmosphère contenant de l’oxygène. Lorsque cette impureté entre dans ces cristaux,
elle occupe une position substitutionnelle dans le réseau cristallin à la place des halogènes et
forme un centre coloré [26]. Les propriétés optiques et électroniques de ce centre coloré sont
étudiées par plusieurs auteurs durant les années soixante et soixante-dix [18, 20-37].
II-1-2 Les transitions électroniques dans l’ion moléculaire O2-
Le spectre d’émission des centres colorés O2- dans les cristaux d’halogénures alcalins
présente une série de bandes d’émission situées entre 400-900 nm. Ces bandes sont en fonction
de la température de mesure. Plus cette dernière diminue plus les bandes sont étroites et on
obtient des bandes d’émission intenses (Fig.5) qui sont nommées pour la première fois par
Rebane, les lignes d’émission [38]. Zeller et Känzig ont montré que ces émissions sont dues aux
transitions électroniques du niveau vibrationnel bas de l’état excité uπ2 aux niveaux vibrationnels
de l’état fondamental gπ2 de la molécule O2- [39].
La figure 6 montre les transitions électroniques de la molécule O2-. On voit que l’absorption de
cette dernière est effectuée à partir de la transition basse de l’état fondamental 0=′′ν à un
niveau vibrationnel supérieur de l’état excité ν ′ [32]. Cette transition absorbe à 5 eV (247 nm ou
40000 cm-1) dans tous les halogénures alcalins [28]. Alors que pour l’émission, chaque bande ou
ligne d’émission correspond à une transition électronique à partir du niveau bas 0=′ν de l’état
11
excité à un niveau vibrationnel de l’état fondamental noté par le nombre quantiqueν ′′ . En effet,
chaque nombre quantique ν ′′ correspond à une émission comme montre la figure 6.
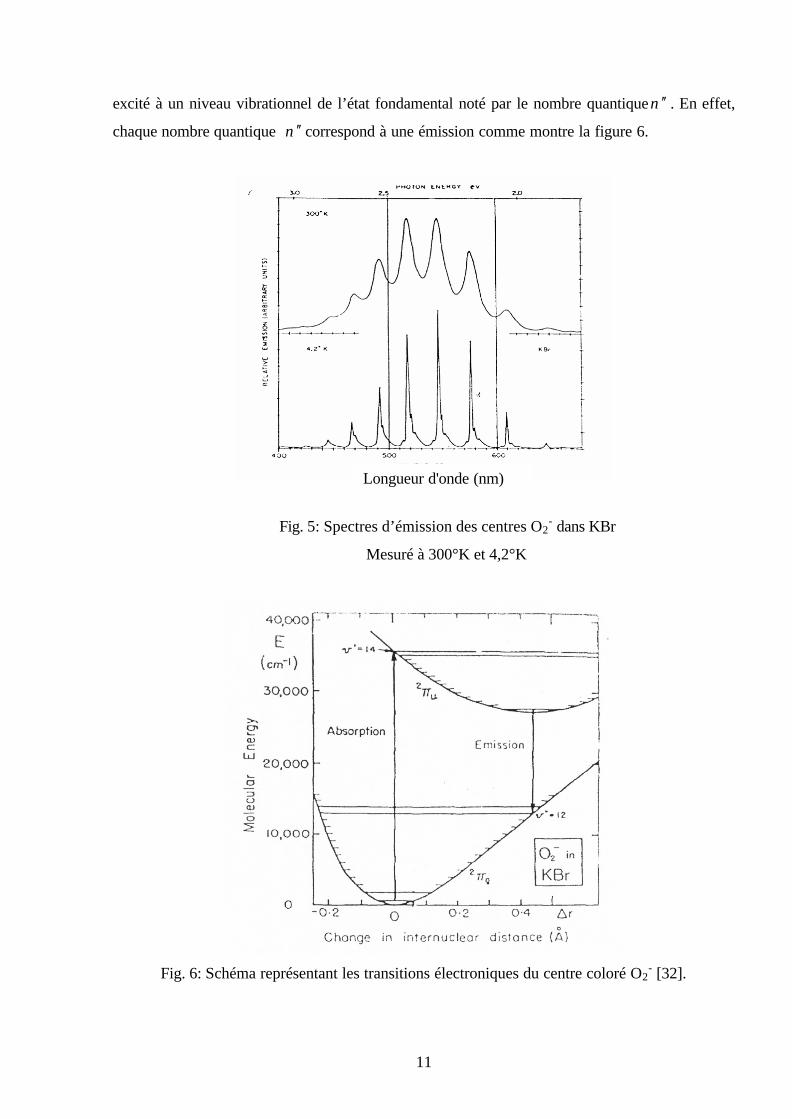
Fig. 5: Spectres d’émission des centres O2- dans KBr
Mesuré à 300°K et 4,2°K
Fig. 6: Schéma représentant les transitions électroniques du centre coloré O2
- [32].
Longueur d'onde (nm)

12
Le nombre d’onde
λ1 de chaque ligne d’émission est déterminé à partir du nombre quantique
ν ′′ selon la relation suivante [32] :
.....1 3
002
00000 +′′″′′+′′″′′+′′′′−= νωνωνωνλ
yx
Où ?00 est un nombre d’onde qui représente la différence entre les deux niveaux 0=′ν et 0=′′ν ,
et 0ω ′′ , ″′′ 00 xω et ″′′ 00 yω sont appelées les constantes vibrationnelles de l’état fondamental gπ2 . Il a
été montré qu’un polynôme de second ordre est suffisant pour déterminer la valeur
λ1 de
chaque ligne d’émission et que la valeur ″′′ 00 yω est négligeable. En effet, à partir de cette
relation, a été calculé le nombre quantique de chaque longueur d’onde d’émission des centres O2-
dans les halogénures alcalins.
Les tableaux 3 et 4 donnent les valeurs des nombres d’onde, nombres quantiques et
longueurs d’onde des lignes ordinaires des émissions de centres O2- dans les trois monocristaux
KBr, KCl et NaCl, mesurées à 300°K et 4,2°K.
II-1-3 Types de spectres d’émission des centres colorés O2-
Les spectres d’émission des centres colorés O2- dans les halogénures alcalins sont classés
en deux groupes [36] :
a- Les spectres constitués d’une série de lignes d’émission formées par une structure simple
dont les lignes sont appelées les lignes ordinaires (Fig. 7-a).
b- Les spectres constitués d’une série de lignes d’émission formées par une double structure
dont les lignes intenses sont appelées les lignes ordinaires alors que les faibles lignes sont
appelées les lignes particulières (Fig. 7-b).
Les spectres de type (a) apparaissent dans KBr, KI, NaCl et NaBr. Les centres O2- dans ces
cristaux ont l’axe moléculaire orienté suivant la direction <111>. Tandis que les spectres de type
(b) apparaissent dans KCl et Rb-halogènes. Les centres O2- dans ces cristaux ont l’axe
moléculaire orienté suivant la direction <110>.

13

14
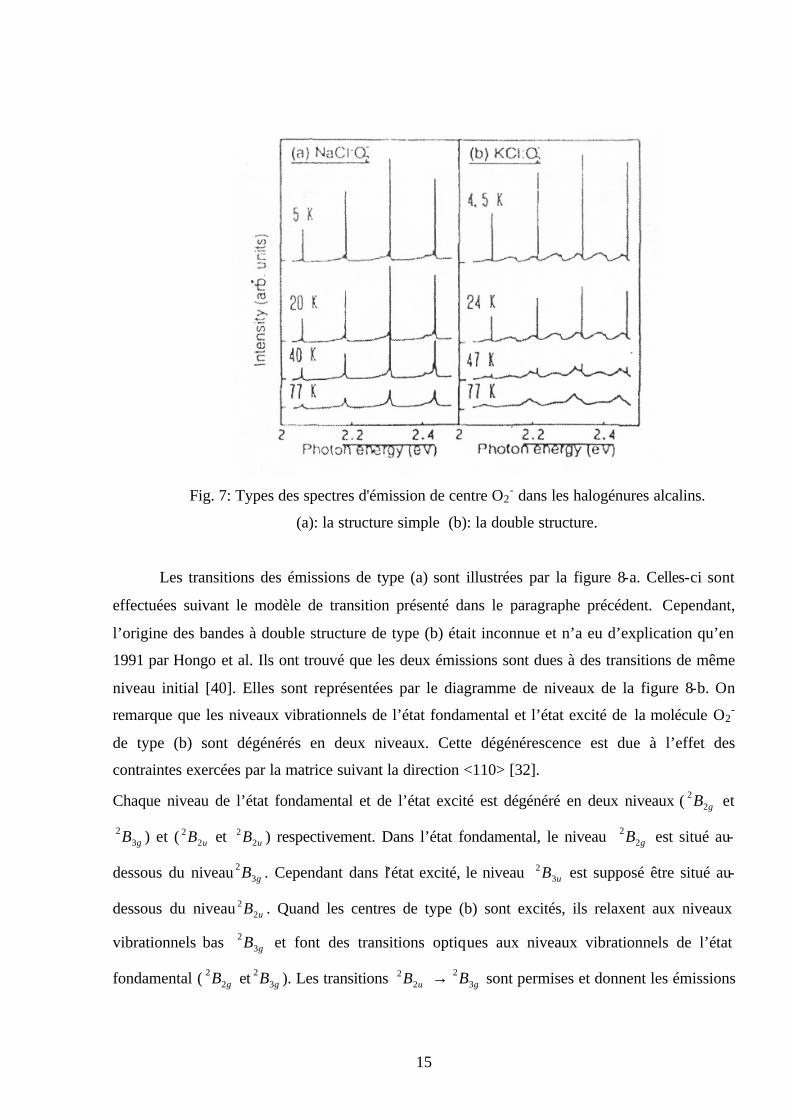
15
Fig. 7: Types des spectres d'émission de centre O2
- dans les halogénures alcalins.
(a): la structure simple (b): la double structure.
Les transitions des émissions de type (a) sont illustrées par la figure 8-a. Celles-ci sont
effectuées suivant le modèle de transition présenté dans le paragraphe précédent. Cependant,
l’origine des bandes à double structure de type (b) était inconnue et n’a eu d’explication qu’en
1991 par Hongo et al. Ils ont trouvé que les deux émissions sont dues à des transitions de même
niveau initial [40]. Elles sont représentées par le diagramme de niveaux de la figure 8-b. On
remarque que les niveaux vibrationnels de l’état fondamental et l’état excité de la molécule O2-
de type (b) sont dégénérés en deux niveaux. Cette dégénérescence est due à l’effet des
contraintes exercées par la matrice suivant la direction <110> [32].
Chaque niveau de l’état fondamental et de l’état excité est dégénéré en deux niveaux ( gB22 et
gB32 ) et ( uB2
2 et uB22 ) respectivement. Dans l’état fondamental, le niveau gB2
2 est situé au-
dessous du niveau gB32 . Cependant dans l’état excité, le niveau uB3
2 est supposé être situé au-
dessous du niveau uB22 . Quand les centres de type (b) sont excités, ils relaxent aux niveaux
vibrationnels bas gB32 et font des transitions optiques aux niveaux vibrationnels de l’état
fondamental ( gB22 et gB3
2 ). Les transitions uB22 → gB3
2 sont permises et donnent les émissions

16
des lignes ordinaires. Alors que les transitions uB22 → gB2
2 sont faiblement permises et donnent
des émissions de faibles intensités. Elles sont appelées les émissions des lignes particulières. Les
deux émissions (ordinaire et particulière) donnent la double structure.
Fig. 8: Schéma représente les transitions électroniques de deux types d'émission.
(a): la structure simple (b): la double structure.
II-2 CENTRES COLORES (O2-– F+)
II-2-1 Introduction
Les centres colorés (O2-– F+) ont été étudiés par F. Fischer, G. Gümmer, H. Gundig et R.
Hilsch dans les années soixante [41-43]. F. Fischer a confirmé, par la mesure du dipôle
électrique sur plusieurs cristaux d’halogénures alcalins dopés par les ions O2-, que les bandes
d’absorption de ces matériaux situées dans l’ultraviolet sont dues aux centres colorés (O2- – F+)
nommés par ce chercheur le dipôle (O2-– F+) [41]. Ce centre présente un atome d’oxygène
doublement ionisé à côté d’une lacune anionique vide et se trouve suivant la direction <110> du
réseau cristallin [44].

17
Dans les années quatre vingt, les centres (O2-– F+) sont utilisés par E. Georgiou et al., D. Wandt
et al. et G. Lifante et al. pour stabiliser les centres colorés (F+2), afin d’obtenir les centres colorés
(F+2)H et (F+
2)AH à effet laser [44-47].
Les centres (O2-– F+) peuvent se former dans les halogénures alcalins à partir des deux centres
OH- et O2- en présence des centres F selon les deux réactions suivantes [41, 47]:
OH- + 2F → (O2- – F+) + U
O2- + 2F → 2 (O2- – F+)
II-2-2 Interprétation des bandes d’absorption des centres (O2- – F+)
Les centres colorés (O2- – F+) ont plusieurs bandes d’absorption situées dans l’UV. Un
autre centre qui possède également plusieurs courtes bandes d’absorption est le centre F. les
courtes bandes d’absorption de ce dernier centre sont nommées les bandes L. Elles sont
attribuées aux transitions de charge à partir des centres F aux ions alcalins voisins. De la même
manière seront décrites les courtes bandes d’absorption de (O2- – F+) car elles sont équivalentes
aux bandes L des centres F [41, 43].
Interprétation des bandes L
En mesurant l’absorption de quelques cristaux colorés, F. Lüty a découvert trois bandes
d’absorption situées dans le domaine des courtes longueurs d’onde. Ces bandes sont appelées les
bandes L1, L2 et L3 (L vient du nom Lüty) [38]. Peu après, M. Hirai et M. Ueta ont découvert la
quatrième bande L4 [49]. C. C. Klick et M. N. Kabler ont prouvé que ces courtes bandes sont
dues aux centres colorés F [50]. Pour interpréter ces bandes, ces deux chercheurs ont utilisé le
modèle de transfert de charge utilisé pour la première fois par Hilsh et Pohl afin d’expliquer les
bandes fondamentales d’absorption des halogénures alcalins [51]. Ce modèle est basé sur le
déplacement des électrons à partir des ions halogènes aux ions alcalins voisins. Zwerling et al.
ont montré que les niveaux des défauts dans les cristaux ioniques n’apparaissent pas seulement
dans la bande interdite mais aussi au-dessus des niveaux d’énergie de la bande de conduction. En
se basant sur le modèle de transfert de charge et le résultat obtenu par Zwerling, C. C. Klick et
M. N. Kabler ont interprété les bandes L du centre F utilisant la figure 9 [50]. Celle-ci illustre la

18
bande de conduction correspondant à l’atome alcalin et les deux états de la bande de valence.
Ces derniers représentent les deux niveaux d’énergie des halogènes. L’état fondamental du
centre F se trouve dans la bande interdite. Les transitions de la bande F se font à un état excité à
côté du minimum le plus bas de la bande de conduction. La bande L1 correspond à une transition
de l’atome alcalin dans son état excité bas, alors que les bandes L2, L3, et L4 correspondent aux
transitions vers les plus hauts états excités de l’atome alcalin. Les énergies des bandes L sont
données approximativement par la somme de l’énergie de la bande F et de l’énergie d’excitation
de l’atome alcalin. En effet, ces transitions se font à partir de l’état fondamental du centre F vers
les états excités existants au-dessus de chaque niveau de la bande de conduction. Ces états
représentent les niveaux de l’atome alcalin. L’existence de ces différents niveaux dans la bande
de conduction permet d’obtenir plusieurs bandes d’absorption dont les énergies sont très petites.
On se basant sur le même principe de la formation de bandes d’absorption L, les bandes
d’absorption des centres (O2- – F+) se forment de la même manière car à la place du centre F dans
la bande interdite on a le centre (O2- – F+). Les bandes d’absorption de ce dernier sont dues aux
transitions électroniques à partir de l’ion O2- vers les états excités situés au-dessus des niveaux
de l’atome alcalin [41].
Fig. 9: Schéma représentant les transitions électroniques des bandes L [50].

19
G. Gümmer a montré que si l’électron dans son état excité peut revenir à son état
fondamental dans l’ion O2- par une transition directe, on obtient des bandes d’émission situées
dans le domaine spectral de l’UV-visible [43].
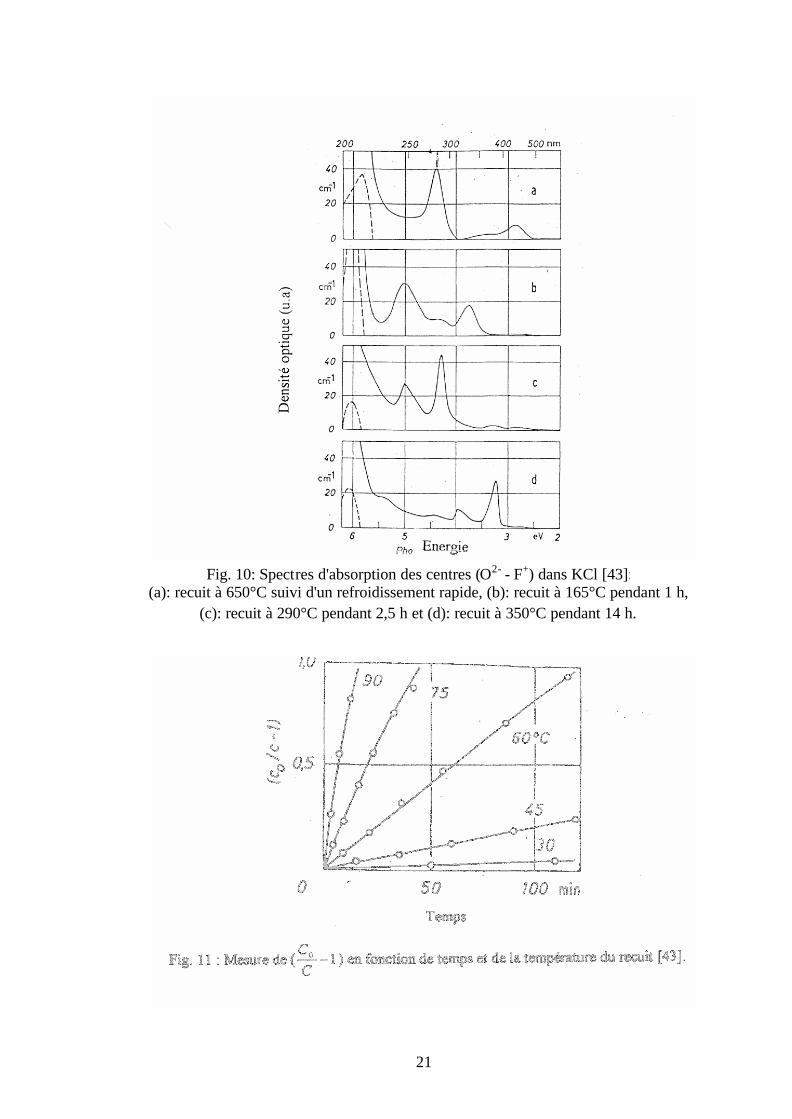
II-2-3 Effet du traitement thermique sur les bandes d’absorption des centres (O2- – F+)
G. Gümmer a étudié l’effet du traitement thermique sur les positions des bandes
d’absorption des centres (O2- – F+) dans les cristaux de KCl. Il a constaté que si les échantillons
sont refroid is rapidement à la température ambiante ou si on fait le recuit avec des températures
moyennes, on obtient des spectres d’absorption tout à fait différents. Les résultats obtenus sont
représentés sur la figure 10. La figure 10-a montre le spectre d’absorption des échantillons qui
ont subi un recuit à 650°C suivi d'un refroidissent rapide à la température 20°C. On remarque
l’apparition de trois bandes situées à 435 nm, 282 nm et 212 nm. Alors que la figure 10-b montre
le spectre après un recuit à 165°C pendant 1h. On note que les trois bandes sont décalées et leurs
maximums sont situés maintenant à 330 nm, 247 nm et 206 nm. Le recuit à 290°C pendant 2,5 h
influe encore sur le spectre et on obtient deux bandes relativement étroites situées à 290 nm et
250 nm (Fig. 10-c). Tandis que le recuit à 330°C pendant 14 h fait apparaître deux bandes situées
à 315 nm et 380 nm (Fig 10-d).
G. Gümmer s’est intéressé au changement du spectre d’absorption des centres colorés (O2- – F+)
pendant le traitement thermique. Il a montré que ce phénomène est dû à la formation des agrégats
ou des complexes de ces centres pendant le recuit. L’association des (O2- – F+) est effectuée
aléatoirement dans les cristaux et peut donner, dans le cas le plus simple, des doubles centres
comme suit [43] :
(O2- – F+) + (O2- – F+) → (O2- – F+)2

20
Afin d’expliquer la formation des agrégats durant le recuit, G. Gümmer a développé la relation
suivante [43] :
tCCC
00 1 α=−
C0 : la concentration initiale des centres (O2- – F+),
C : la concentration momentanée des centres (O2- – F+),
a: une constante dépendant de la température de recuit,
t: le temps.
Les courbes de la figure 11 sont obtenues suite à des mesures de la valeur ( 10 −CC
) en fonction
de plusieurs temps et température de recuit. On remarque que plus le temps et la température de
recuit augmentent plus la valeur ( 10 −CC
) augmente. Ceci explique clairement la diminution de
la concentration C durant le traitement thermique. Cette diminution est due à la formation
progressive des agrégats des centres colorés (O2- – F+).
Dans le cadre de cette thèse, on a proposé de considérer le cas qui n’a pas été étudié par
G. Gümmer; et qui consiste en un recuit à 650°C pendant 24 h suivi d’un refroidissement lent
aux échantillons de KBr, KCl et NaCl élaborés à l’air libre. Ce traitement thermique est effectué
dans le but de montrer les nouveaux spectres d’absorption et les nouvelles bandes d’émission
situées dans l’UV-visible des centres colorés (O2- – F+).
21
Fig. 10: Spectres d'absorption des centres (O2- - F+) dans KCl [43]: (a): recuit à 650°C suivi d'un refroidissement rapide, (b): recuit à 165°C pendant 1 h,
(c): recuit à 290°C pendant 2,5 h et (d): recuit à 350°C pendant 14 h.

22
CHAPITRE III SEMI-CONDUCTEURS II-VI ET PROPRIETES ELECTRONIQUES
ET OPTIQUES DES NANOCRISTAUX DE SEMI-CONDUCTEURS
III- 1 SEMI-CONDUCTEURS II-VI
III-1-1 Introduction
Parmi les éléments du tableau de Mendeleiv douze sont considérés comme semi-
conducteurs: le bore (B), le carbone (C), le silicium (Si), le phosphore (P), le souffre (S), le
germanium (Ge), l’arsenic (As), le séléniure (Se), l’étain (Sn), l'antimoire (Sb), le tellure (Te) et
l'iode (I). De part leurs propriétés physiques, le silicium et le germanium sont les plus importants
pour leurs applications en électronique. Cependant, les progrès de la recherche mettent à la
disposition de la technologie des nouveaux semi-conducteurs de plus en plus performants. Parmi
ces semi-conducteurs, on peut citer: les semi-conducteurs II-VI.
Les semi-conducteurs II-VI sont des matériaux formés à partir d’atomes appartenant aux
colonnes II (Zn, Cd, Hg ) et VI (S, Se, Te) de la classification périodique. Respectivement,
chacun d’eux comporte deux et six électrons sur sa couche électronique externe, ils ont donc une
valence de quatre. La combinaison des élements II-VI donne des composés de semi-conducteurs
à grand gap dont la valeur varie entre 1,5-3,9 eV. Ils sont caractérisés par deux propriétés: gap
direct et large sur quoi repose l’essentiel de l’intérêt porté aux semi-conducteurs II-VI. Parmi
les matériaux de cette famille, qui présentent des propriétés intéressantes, on peut citer ZnS,
ZnSe, ZnTe, ZnO, CdS, CdSe et CdTe, à ceux là s’ajoutent les alliages ternaires (formés de trois
éléments) dont la bande interdite peut être modulable. Par exemple pour Hg1-xCdxTe le gap varie
de 300 meV HgTe jusqu’à 1,5 eV d’où l’origine du développement de HgCdTe et son intérêt
pour les applications militaire IR et communication optique [53, 54].
Les semi-conducteurs II-VI jouent un rôle important dans l'industrie de la micro-
électronique et l'optoélectronique. Des applications de plus en plus nombreuses se confirment tel
que la détection IR et nucléaire, les cellules solaires, les transistors, les diodes
électroluminescentes et les diodes lasers à courtes longueurs d'onde (bleu et proche UV), les
effets non linéaires...Parmi toutes ces applications spécifiques des semi-conducteurs II-VI à
grand gap, la réalisation d'un laser à semi-conducteur émettant dans le bleu est celle qui suscite le
plus grand intérêt. Ces semi-conducteurs sont utilisés sous différentes formes: couches minces,

23
couches épitaxiques, monocristaux et nanocristaux. En effet, si on regarde la figure 12 on
constate que seuls les semi-conducteurs II-VI en particulier les composés du Zn ont un gap
suffisamment élevé pour couvrir le domaine des courtes longueurs d'ondes du spectre visible et
proche ultra violet.
Fig. 12: Gap d'énergie des semi-conducteurs II-VI en fonction du paramètre de maille [53].
III-1-2 Propriétés cristallographiques des semi- conducteurs II-VI
L’étude structurale des composés II-VI pose quelques problèmes du fait du
polymorphisme de ces composés. Ils peuvent se cristalliser dans deux structures différentes:
structure cubique du type sphalérite et structure hexagonale du type wurtzite. Les deux modes se
caractérisent par une disposition tétraédrique des atomes, analogue à celle que l'on observe dans
les semi-conducteurs de la colonne IV (Fig. 13). Ils peuvent former différents polytypes, qui
maintiennent la disposition tétraédrique des atomes, et ne sont en fait que des structures dérivées
de la sphalérite et de la wurtzite. Sous l’action de fortes pressions extérieures certains de ces
composés peuvent acquérir une structure du sel gemme (NaCl) caractérisée par une disposition
octaédrique des atomes, bien que ces phases soient instables dans les conditions usuelles, elles
peuvent subsister aux basses températures. Les paramètres de maille pour quelques semi-
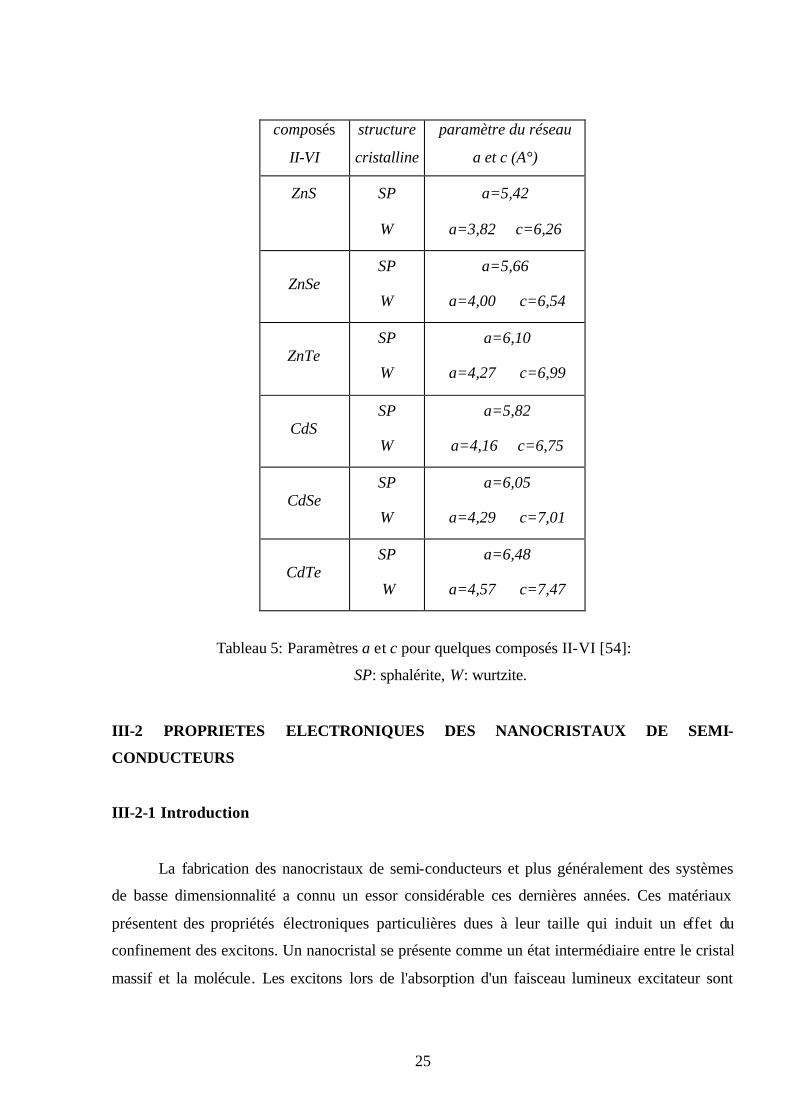
conducteurs II-VI sont représentés dans le tableau 5.

24
La majorité des composés II-VI présentent des structures cristallines que l’on peut classer
en considérant la distribution spatiale régulière des cations (II) occupant certains interstices d’un
empilement compact d’anions (VI). Dans le cas où les cations occupent les interstices
octaédriques d’un empilement cubique dense d’anions, on obtient la structure du sel gemme.
Dans le cas où ils occupent la moitié des interstices tétraédriques avec une périodicité qui fait
alterner les sites occupés et les sites vacants, on obtient la structure cubique de la sphalérite. Si
les cations occupent dans les mêmes conditions la moitié des interstices tétraédriques d’un réseau
hexagonal compact on obtient la structure hexagonale de la wurtzite. Dans les différents cas où
les cations occupent la moitié des sites tétraédriques d’un empilement mixte en alternance
l’empilement cubique et l’empilement hexagonal dense des anions, on se trouve en présence
d’une multitude de polytypes hexagonaux et orthorhombiques [54].
Sphalérite Wurtzite
Fig. 13: Structure cristalline de semi-conducteurs II-VI.
25
composés
II-VI
structure
cristalline
paramètre du réseau
a et c (A°)
ZnS SP
W
a=5,42
a=3,82 c=6,26
ZnSe SP
W
a=5,66
a=4,00 c=6,54
ZnTe SP
W
a=6,10
a=4,27 c=6,99
CdS SP
W
a=5,82
a=4,16 c=6,75
CdSe SP
W
a=6,05
a=4,29 c=7,01
CdTe SP
W
a=6,48
a=4,57 c=7,47
Tableau 5: Paramètres a et c pour quelques composés II-VI [54]:
SP: sphalérite, W: wurtzite.
III-2 PROPRIETES ELECTRONIQUES DES NANOCRISTAUX DE SEMI-
CONDUCTEURS
III-2-1 Introduction
La fabrication des nanocristaux de semi-conducteurs et plus généralement des systèmes
de basse dimensionnalité a connu un essor considérable ces dernières années. Ces matériaux
présentent des propriétés électroniques particulières dues à leur taille qui induit un effet du
confinement des excitons. Un nanocristal se présente comme un état intermédiaire entre le cristal
massif et la molécule. Les excitons lors de l'absorption d'un faisceau lumineux excitateur sont

26
libres de se déplacer dans tout le cristal semi-conduc teur massif; par contre, dans un nanocristal,
ils se trouvent limités par les parois de la particule. Le confinement des excitons se traduit alors
par deux effets: l'apparition de transitions électroniques d'énergie discrètes au lieu de la structure
de bandes habituelle, et un déplacement du seuil d'absorption vers les hautes énergies, traduisant
ainsi un élargissement du gap, de plus en plus important au fur et à mesure que la taille des
cristaux diminue (Fig. 14) [55].
La principale modification que subit la structure électronique d’un nanocristal concerne la
densité d’états électroniques [56]:
Pour un cristal massif et en ne tenant pas compte de l’interaction colombienne, la densité d’états
varie comme E :
22
21
)(2/3
2,
*
2ghe E
Em
E −
=
hπρ
Pour un nanocristal, elle prend des valeurs discrètes:
∑
−−−−=
zyx
zyxKKK
heK
heK
heK
g EEEE
EE,,
,,,
22)( δρ
E: niveaux d'énergie. K: vecteur d'onde de l'exciton.
Ainsi, le confinement spatial fait évaluer la densité d’états d’un continuum vers une série des
raies discrètes.
Massif Nanocristal
Fig. 14: Diagramme des niveaux d’énergie d'un semi-conducteur
massif et un semi-conducteur nanocristallin [58].

27
III-2 2 L'exciton dans le semi-conducteur massif
Un cristal semi-conducteur possède une bande de valence et une bande de conduction
séparée par une bande interdite, le gap, dont la largeur en énergie est de quelques eV; le gap
correspond donc à l'énergie minimale nécessaire à la création d'une paire électron-trou. Ces deux
porteurs peuvent se déplacer indépendamment dans le cristal, contribuant ainsi à la conductivité
électrique. Ils peuvent également être liés par l'interaction coulombienne; la paire électron-trou
est alors appelée exciton, elle représente une excitation élémentaire du cristal. L’exciton peut
être considéré comme un système hydrogénoïde décrit par le modèle de Bohr, et possédant donc
des niveaux d'énergie quantifiés [57]:
MK
n
EEE R
gn 2
22
2
h+−= (*)
he mmM ** += : masse effective totale.
m*e,h : masse effective de l'électron (e) et du trou (h).
he
he
mmmm
**
**
+=µ : masse effective réduite.
ε: constante diélectrique du matériau.
K : vecteur d'onde de l'exciton.
Eg : gap d'énergie.
RE est l'énergie de Rydberg effective (en analogie avec celle de l'atome d'hydrogène). Elle est
égale à: 22
4
2 εµ
he
ER = est représente l'énergie de liaison de l'exciton dans son niveau fondamental
(n=1). 2e
aB µεh
= est le rayon de Bohr de l'exciton. Il mesure son extension spatiale dans ce
même niveau. Il est d'autant plus petit que l'interaction coulombienne est grande. Le rayon de
Bohr pour quelques composés II-VI est : CdS: 3 nm, ZnO: 1,3 nm, CdSe: 5,4 nm [58].
La relation (*) montre qu'il existe une série de valeurs permises pour l'énergie de l'exciton
dans un semi-conducteur massif, chaque valeur correspondant à un état excité particulier de la
paire électron-trou. Les niveaux d'énergie excitonique convergent vers une valeur limitée de
haute énergie égale à celle de la bande interdite (Fig. 14).

28
III-2-3 L'exciton dans le semi-conducteur nanocristallin
La réduction de l'espace disponible à son évolution spatiale conduit l'exciton à un état de
confinement. Les effets seront plus ou moins importants suivant la taille du nanocristal
considéré, le rayon de Bohr étant la longueur de référence. Un nanocristal de semi-conducteur
encapsulé dans une matrice isolante est habituellement modélisé par un puits de potentiel de
symétrie sphérique, souvent considéré infini. Nous nous plaçons dans le cas d’un potentiel
sphérique U tel que:
U rpour r Rpour r R
( ) =∞
≤≥
0
Le confinement de l’exciton dans un nanocristal, décrit en mécanique quantique comme
le cas d'une ``particule dans une boîte'', conduit alors à une quantification de son énergie
cinétique. L’énergie cinétique de l'exciton ainsi confiné dans une particule de rayon R prend
alors des valeurs discrètes [55, 58]:
2
222
2MRnEn
πh=
L'exciton subit donc deux potentiels :
• L'interaction coulombienne, qui lie les deux particules qui le forment; elle est caractérisée
par l'énergie de liaison.
• Le puits de potentiel qui quantifie son énergie cinétique (∝ 1/R2).
III-2-4 Régimes du confinement
Différents régimes du confinement existent alors selon l'importance relative de ces deux
potentiels, qui dépend bien évidemment de la taille du nanocristal, mais aussi de la constante
diélectrique et des masses effectives que l'on retrouve dans le rayon de Bohr. Les trois régimes
du confinement considérés sont alors définis par comparaison du rayon R de la cristallite et du
rayon de Bohr aB: l'effet du confinement quantique sera d'autant plus important que le rayon du
nanocristal sera petit devant le rayon de Bohr de l'exciton [57, 59].

29
III-2-4-1 Le confinement fort ( BaR⟨ ):
Dans ce cas l’énergie cinétique individuelle de l’exciton est alors beaucoup plus
importante que l’énergie potentielle d’interaction colombienne. L’exciton n’a pas assez de place
pour se former. L’électron et le trou ne sont pas couplés, ils évaluent alors séparément dans un
milieu très confiné. Les énergies des premières transitions sont données par:
Rg ER
eR
EE 248,0786,1
2
2
2
22
+−+=εµ
πh
III-2-4-2 Le confinement faible ( BaR⟩ ):
On retrouve un cas proche de celui du semi-conducteur massif; les effets dus au puits sont
faibles. Dans ce cas le potentiel colombien qui domine, l’exciton peut être considéré comme une
quasi-particule qui subit alors globalement l’effet du confinement. Le mouvement de son centre
de masse est quantifié. Les énergies des premières transitions sont données par :
2
22
2MREEE exg
πh+−=
III-2-4-3 Le confinement intermédiaire
Lorsque la masse effective du trou est très grande par rapport à celle de l’électron et que
le rayon de nanocristallite est plus petit que le rayon de Bohr de l’électron mais beaucoup plus
large que celui du trou, le confinement est dit intermédiaire. L’énergie cinétique minimale de
l’électron est alors plus grande que celle du trou et son mouvement est plus rapide. Il subit peu
l’effet du potentiel colombien. Par contre, le potentiel colombien agit sur le trou et il est
moyenné sur le mouvement rapide de l’électron. Dans ce cas, le trou se positionner au centre de
la nanocristallite. L'énergie de la première transition s'écrit alors:
Re
mmRmEE
heeg ε
π 2
**2
22 8,1118
−
++=
h
30
III-2-5 Influence du confinement sur les propriétés optiques des semi-conducteurs
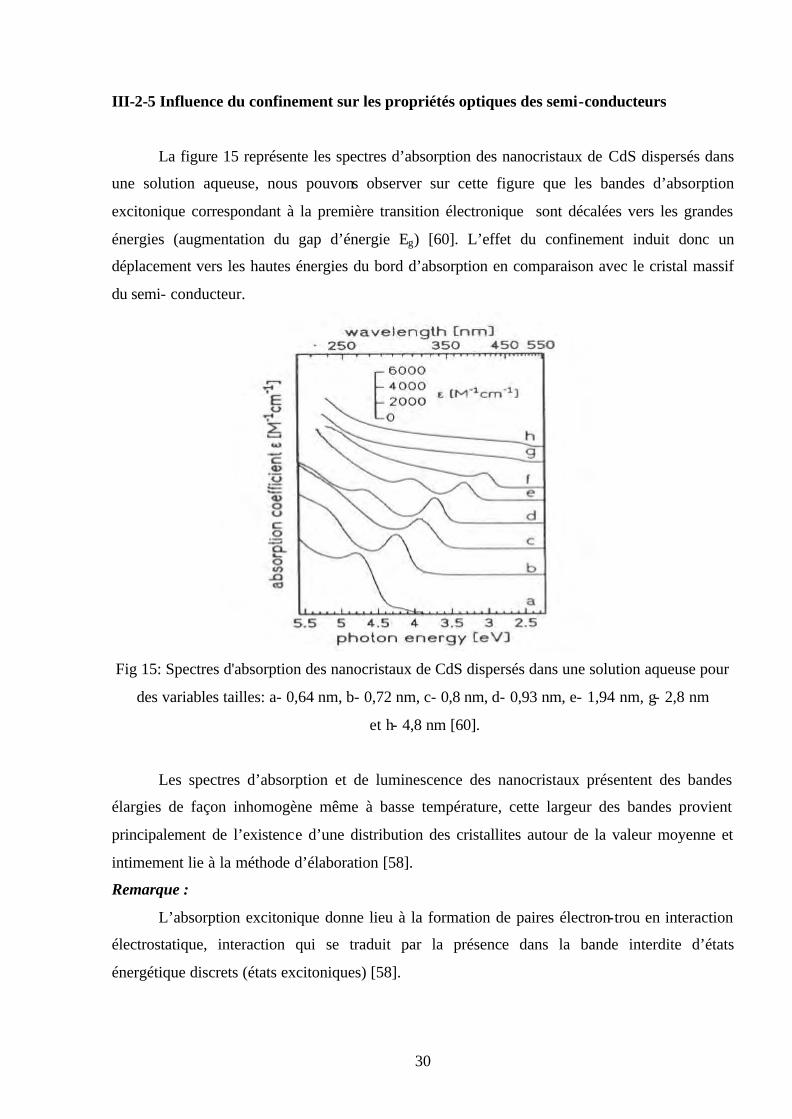
La figure 15 représente les spectres d’absorption des nanocristaux de CdS dispersés dans
une solution aqueuse, nous pouvons observer sur cette figure que les bandes d’absorption
excitonique correspondant à la première transition électronique sont décalées vers les grandes
énergies (augmentation du gap d’énergie Eg) [60]. L’effet du confinement induit donc un
déplacement vers les hautes énergies du bord d’absorption en comparaison avec le cristal massif
du semi- conducteur.
Fig 15: Spectres d'absorption des nanocristaux de CdS dispersés dans une solution aqueuse pour
des variables tailles: a- 0,64 nm, b- 0,72 nm, c- 0,8 nm, d- 0,93 nm, e- 1,94 nm, g- 2,8 nm
et h- 4,8 nm [60].
Les spectres d’absorption et de luminescence des nanocristaux présentent des bandes
élargies de façon inhomogène même à basse température, cette largeur des bandes provient
principalement de l’existence d’une distribution des cristallites autour de la valeur moyenne et
intimement lie à la méthode d’élaboration [58].
Remarque :
L’absorption excitonique donne lieu à la formation de paires électron-trou en interaction
électrostatique, interaction qui se traduit par la présence dans la bande interdite d’états
énergétique discrets (états excitoniques) [58].

31
CHAPITRE IV METHODE DE CZOCHRALSKI ET APPAREILS
DE MESURE UTILISES IV-1 INTRODUCTION
L’élaboration des monocristaux peut être effectuée par plusieurs méthodes (techniques).
Le choix de l’une d’entre elle est dicté par la nature et les propriétés physico-chimiques du
matériau considéré. Pour notre cas précis, les halogénures alcalins (KBr, KCl, NaCl,…) sont peu
solubles en solution aqueuse [61] et ne permettent que de très faibles sursaturations, même à
température relativement haute; d’où no tre orientation vers l’obtention des monocristaux de KBr,
KCl et NaCl par des méthodes utilisant des solutions fondues. La plus adéquate est la méthode
de Czochralski.
IV-2 METHODE DE CZOCHRALSKI
IV-2-1 Principe de la méthode
Le liquide en fusion du matériau est contenu dans un creuset, dans lequel est plongé
partiellement un germe suspendu par un porte germe. Afin d’homogénéiser la fusion et de
contrôler la géométrie du cristal, le germe et le creuset sont animés de deux mouvements de
rotation inverse, une fois l’équilibre thermique (fusion-germe) est atteint, un ménisque se forme
autour du germe. Puis après le germe est tiré vers le haut avec une vitesse appropriée. La matière
en fusion est absorbée par le germe, et ainsi le cristal croît (Fig. 16) [62].
Fig. 16: Principe de la méthode de Czochralski [62].

32
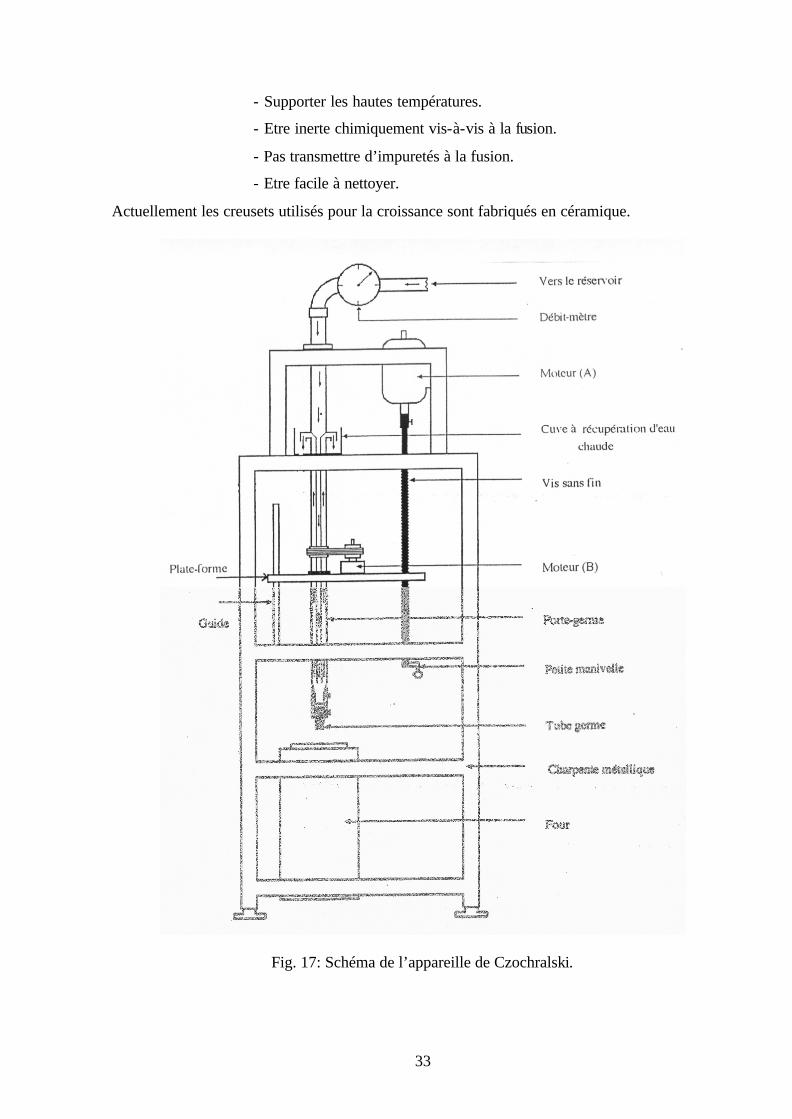
IV-2-2 Appareillage
L’appareil de Czochralski du laboratoire de cristallographie de l’université Mentouri-
Constantine, utilisé pour l’élaboration de nos échantillons est constitué de deux parties dont l’une
est mécanique et l’autre thermique (Fig. 17).
IV-2-2-1 Partie mécanique : elle comporte :
a) Moteur (A): c’est un moteur électrique dont l’arbre est reliée a une vis sans fin,
l’alimentation est réglée par un rhéostat, pour obtenir des vitesses de rotation allant de zéro
jusqu’à 7 tr/min. Ce moteur est fixé en haut de l’appareil à l’aide d’une charpente métallique.
b) Vis sans fin: c’est une tige en acier filetée de pas 1,25 mm. Elle permet la translation
verticale du porte germe et se termine par une petite manivelle qui permet la translation rapide
ainsi que l’ajustement du tube germe au début et à la fin de la croissance de chaque cristal.
c) Moteur B: c’est un petit moteur dont la vitesse est constante et vaux un tour par
minute. Il assure la rotation du porte germe à l’aide de pignons et courroie dentée.
d) Porte germe : c’est une tige en acier inoxydable creuse. Sur le bout inférieur de la tige
est fixé un tube de diamètre 3 mm utilisé comme germe pendant la croissance.
e) Petite plate forme: pour obtenir le mouvement hélicoïdal du porte germe, une
combinaison de deux mouvements de rotation et translation est nécessaire. Pour cela, on a
recours à une petite plate forme à la quelle est solidaire le moteur (B) ainsi que le porte germe à
l’aide d’un roulement. Cette plate forme se déplace verticalement sous l’action de la vis sans fin.
f) Guides: afin de minimiser les vibrations dues au moteur et d’assurer l’alignement de
l’axe du porte germe avec celui du four on utilise des guides fixés à la charpente métallique.
IV-2-2-2 Partie thermique : elle comporte :
a) Four: c’est un four tubulaire a résistance électrique. Les fils généralement de nature
nickel-chrome, kantal ou tungstène.
b) Système de régulation de la température: le contrôle de la température lors de la
croissance à l’intérieur du four s’effectue aux moyens de thermocouples placés à coté des
résistances chauffantes et liés à un contrôleur électrique permettant de connaître la température,
ce dernier actionne un interrupteur, lequel ouvre et ferme le circuit d’alimentation des éléments
chauffants du four.
c) Creuset: il joue un rôle très important dans la croissance des cristaux, le choix de celui-
ci dépend de plusieurs facteurs, il doit :
33
- Supporter les hautes températures.
- Etre inerte chimiquement vis-à-vis à la fusion.
- Pas transmettre d’impuretés à la fusion.
- Etre facile à nettoyer.
Actuellement les creusets utilisés pour la croissance sont fabriqués en céramique.
Fig. 17: Schéma de l’appareille de Czochralski.

34
Remarque :
La technique de Czochralski nécessite pour le tirage des monocristaux l’utilisation des
germes par exemple:
- Germe monocristallin de même nature ou de nature différente, pour obtenir une
croissance orientée.
- Tube en métal ou en céramique ou en quartz pour obtenir une croissance non orientée.
Le tube est généralement utilisé pour la croissance non orientée et pour la préparation des
germes. Il doit être in soluble dans la fusion, avoir une haute température de fusion et inerte
chimiquement avec celle-ci. Lorsque le tube pénètre légèrement dans le liquide en surfusion,
les particules s’y attachent et forment un amas polycristallin autour de celui-ci. Pour avoir un
monocristal, il est nécessaire de surmonter le problème de polycristallisation. Pour cela on a
recours à l’étranglement du cristal qui s’effectue par augmentation de la vitesse du tirage ou
de la température de la fusion. Ce phénomène peut être répéter plusieurs fois jusqu’à avoir un
seul grain.
IV-3 ELABORATION DES ECHANTILLONS ETUDIES
L’élaboration des échantillons s’effectue suivant les étapes suivantes:
- Elaboration des monocristaux purs de KCl, KBr et NaCl par la méthode de Czochralski.
- Clivage des monocristaux et polissage des pastilles clivées.
- Traitement thermique des échantillons (pastilles clivées).
- Caractérisation spectroscopique.
IV-3-1 Elaboration des monocristaux de KCl, KBr et NaCl
L’ordre chronologique des opérations effectuées lors de l’élaboration des monocristaux
est le suivant:
- Le nettoyage de porte germe et du creuset est réalisé avec de l’eau bidistillée et du
méthanol.
- La vitesse de tirage est fixée à la valeur voulue (7 mm/h).

35
- La poudre de KCl ou KBr ou NaCl est mise dans le creuset (la poudre utilisée est d’une
pureté 99,99 %).
- Le creuset est ensuite placé dans le four dont la température est élevée progressivement
jusqu’à la température de fusion de la poudre.
- Après la fusion totale de la poudre, le germe qui se présente sous forme d’un bâtonnet de
longueur 10 à 15 mm, dont l’axe [100] est confondu avec l’axe de rotation de la tige et
qui permet la croissance orientée, est introduit légèrement (2 à 3 mm) dans la solution en
fusion par le biais de la petite manivelle. Lorsque un ménisque se forme (un anneau
circulaire brillant autour du germe), et dés qu’il atteint une taille appréciable, le tirage
automatique commence.
- Au fur et à mesure que la croissance progresse, on diminue la température du four. La
vitesse de diminution varie suivant le diamètre désiré du cristal, le contrôle de la
croissance s’effectue visuellement.
- Quand le cristal atteint une taille convenable la croissance est interrompue par
détachement rapide du cristal en agissant sur la petite manivelle.
- Le cristal est maintenant juste au dessus de la fusion à une température assez élevée puis
il est recouvert par des couvercles en céramique pour éviter le choc thermique et le
claquage du cristal.
- Le refroidissement du cristal s’effectue par abaissement progressif de la température du
four.
IV-3-2 Clivage des monocristaux et polissage des pastilles clivées
Les monocristaux de KCl, KBr et NaCl obtenus sont sous forme de carottes cylindriques.
Pour pouvoir réaliser des mesures optiques sur ces monocristaux, il est nécessaire de les tailler et
de polir les pastilles obtenues.
Les monocristaux de KCl, KBr et NaCl cristallisent dans le système cubique (structure
NaCl). Ils se clivent facilement suivant les plans (100). Leur clivage permet donc d’obtenir des
pastilles avec des faces planes perpendiculaires à la direction [100] et dont l’épaisseur est de 2 à
3 mm. Le clivage est réalisé à l’aide d’une lame tranchante qu’on dispose perpendiculaire à l’axe
de la carotte (axe [100]) et sur laquelle on applique une certaine pression. Après le clivage, les
faces planes des pastilles sont soumises à un polissage mécanique, avec de la poudre d’alumine

36
et finalement à un polissage chimique, où on utilise du méthanol puis de l’acétone ou de
l’éthanol, repartis sur un tissu de soie doux afin d’obtenir des surfaces rigoureusement planes.
IV-3-3 Traitement thermique des échantillons
Le traitement thermique des échantillons (pastilles clivées) est effectué sous atmosphère
libre dans un four thermique modèle Thermolyne 6000 au laboratoire de biologie de l’Université
de Jijel. Le recuit est réalisé à la température 650°C pendant plusieurs temps.
On place les échantillons dans des creusets en céramique ensuite on fixe la température du recuit.
A la fin du chaque recuit, les échantillons sont refroidis lentement jusqu’à la température
ambiante.
IV-3-4 Elaboration des monocristaux de KCl dopés par les nanocristaux de CdSe
Pour l'élaboration des monocristaux de KCl dopés par les nanocristaux du semi-
conducteur CdSe, on a mélangé la poudre de CdSe avec celle de KCl. Le cristal obtenu est
polycristallin à cause de la grande concentration du dopant. Pour garder le cristal monocristallin,
on fait fondre le polycristal et on ajoute de la poudre de KCl ensuite on fait la croissance du
cristal suivant le tirage de Czochralski. Cette opération est répétée jusqu’à ce qu’on obtient un
cristal de KCl monocristallin et dopé. Le clivage de ce cristal permet d’obtenir des échantillons
sous forme des pastilles d’épaisseur de 2 à 3 mm.
IV-4 CARACTERISATION SPECTROSCOPIQUE
IV-4-1 Mesure de l’absorption optique UV-visible à la température ambiante :
Pour la caractérisation optique à la température ambiante de nos échantillons, nous avons
utilisé un spectrophotomètre de type UV.3101 de l’Université Mentouri-Constantine. Ce
spectrophotomètre est constitué de deux monochromateurs dont le premier est constitué de trois
réseaux concaves (G1, G2 et G3) et le deuxième est constitué de trois réseaux plans (G4, G5 et
G6) (Fig. 18).

37
Le faisceau des lampes de déterium (D2) ou de tungstène (W1) est focalisé par un miroir
inverseur M1. Ce dernier peut changer sa position automatiquement pour choisir la lampe: la
lampe de déterium balaye le domaine 190-360 nm; l’autre de tungstène balaye le domaine 360-
3200 nm. Avant de passer par la fonte S1, le faisceau va traverser le filtre (F) et passer ensuite
par le premier monochromateur. Chaque réseau est équivalent à un domaine de longueur d’onde
qu’on peut sélectionner automatiquement (G1 pour le visible et proche UV et G2 et G3 pour
l’infrarouge). Ce réseau réfléchit le faisceau qui va traverser la deuxième fente (S2) et passer par
le deuxième monochromateur après avoir été réfléchi par le miroir M2. Comme dans le premier
cas, le réseau est choisi suivant le domaine de la longueur d’onde (G4) pour le visible et proche
UV et G5, G6 pour l’infrarouge. Le fa isceau est réfléchi par le même miroir M2 et va passer par
la fente S3 et sera ensuite réfléchi par les miroirs M3, M4 et M5 pour pouvoir traverser le
miroir CH qui le divise en deux faisceaux : le premier est réfléchi par le miroir M7 vers la
référence et le deuxième est réfléchi par le miroir M8 vers l’échantillon à étudier et il sera
détecté ensuite par un détecteur photomultipier PM (pour visible et proche UV) ou par le PbS
dans le cas de l’infrarouge.
Fig. 18: Schéma du principe de spectrophotomètre UV.3101.

38
IV-4-2 Mesure de la photoluminescence et l’excitation de la photoluminescence
La mesure des spectres d’émission a été réalisée avec un spectromètre Jobin-Yvon HR
460 muni d’un monochromateur blazé à 500 nm (150 traits/mm, dispersion 14 nm/mm) de
l’institut des nanosciences de Paris (l’INSP). Le détecteur est un CCD (Charged Coupled
Device) composé de 2000×800 pixels, dont la sensibilité est de 1.7 photons par électron et qui
couvre une plage spectrale de 200 à 1000 nm.
La résolution de l’ensemble est de 0.3 nm/pixel pour une fente d’entrée inférieur à 30 µm. La
source d’excitation dans le domaine UV-visible est une lampe de xénon (450 W). Le faisceau
incident est perpendiculaire à la surface de l’échantillon et l’émission est collectée du côte de
l’excitation avec un angle de 30° environ par rapport à la normale, par une fibre optique d’angle
de collection de 20° et située à environ 10 mm de l’échantillon. La section du faisceau incident
au niveau de l’échantillon est de 1mm2 environ.
Pour l’enregistrement des spectres d’excitation, nous avons utilisé un spectrofluoromètre
Fluorlog-3 de la société Jobin-Yvon à (l’INSP). Il est composé d’une lampe de Xénon (450 W)
pour la source d’excitation couvrant un domaine de longueur d’onde de 240 à 850 nm, d’un
spectromètre d’excitation équipé d’un double monochromateur (blazé à 330 nm, 1200 traits/mm,
dispersion: un niveau de l’échantillon est de 1mm2 environ 2,1 nm/mm), d’un compartiment où
le faisceau incident est focalisé sur l’échantillon, d’un spectromètre Triax 320 muni d’un
monochromateur (blazé à 750 nm, 600 traits/mm) dont la fonction est d’analyser l’émission et
d’un photomultiplicateur pour la détection des photons émis. La variation de l’intensité
d’émission à longueur d’onde fixe (?analyse) est mesurée en fonction de la longueur d’onde
d’excitation. Le spectre de la lampe est mesuré par un détecteur à base de photodiode, ce qui
permet de corriger les spectres des variations de l’émission d la lampe ainsi que de ses
fluctuations en fonction de la longueur d’onde; ce qui revient à travailler à intensité d’excitation
constante. Les spectres sont corrigés de la réponse du détecteur à photodiodes. Un schéma
illustrant le principe de l’appareillage est donné sur la figure 19.
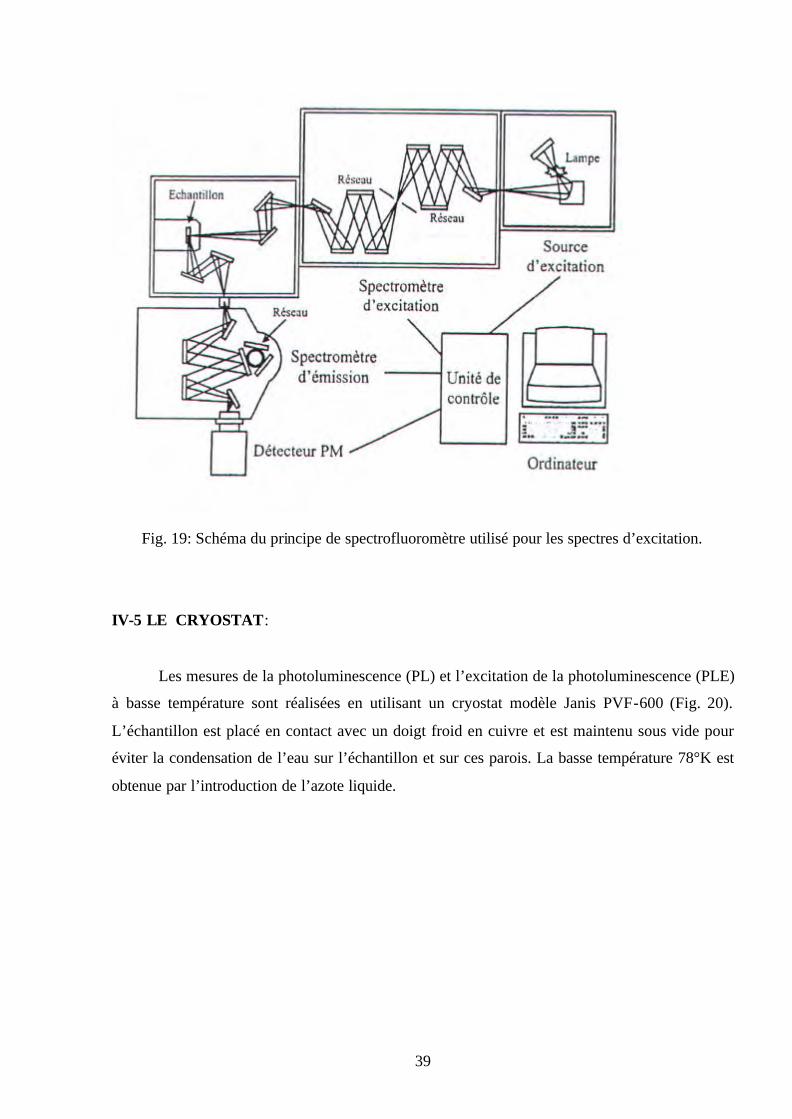
39
Fig. 19: Schéma du principe de spectrofluoromètre utilisé pour les spectres d’excitation.
IV-5 LE CRYOSTAT:
Les mesures de la photoluminescence (PL) et l’excitation de la photoluminescence (PLE)
à basse température sont réalisées en utilisant un cryostat modèle Janis PVF-600 (Fig. 20).
L’échantillon est placé en contact avec un doigt froid en cuivre et est maintenu sous vide pour
éviter la condensation de l’eau sur l’échantillon et sur ces parois. La basse température 78°K est
obtenue par l’introduction de l’azote liquide.
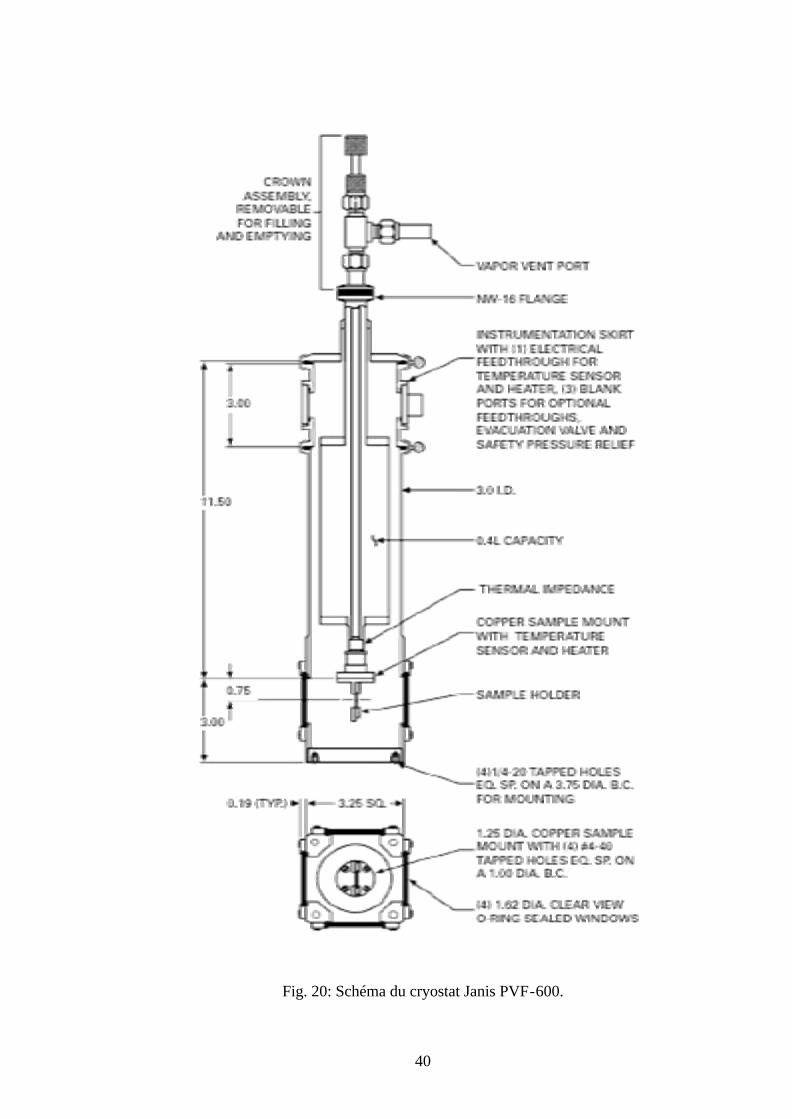
40
Fig. 20: Schéma du cryostat Janis PVF-600.

41
CHAPITRE V CARACTERISATION OPTIQUE
V- 1 CARACTERISATION OPTIQUE DES MONOCRISTAUX DE KBr, KCl ET NaCl
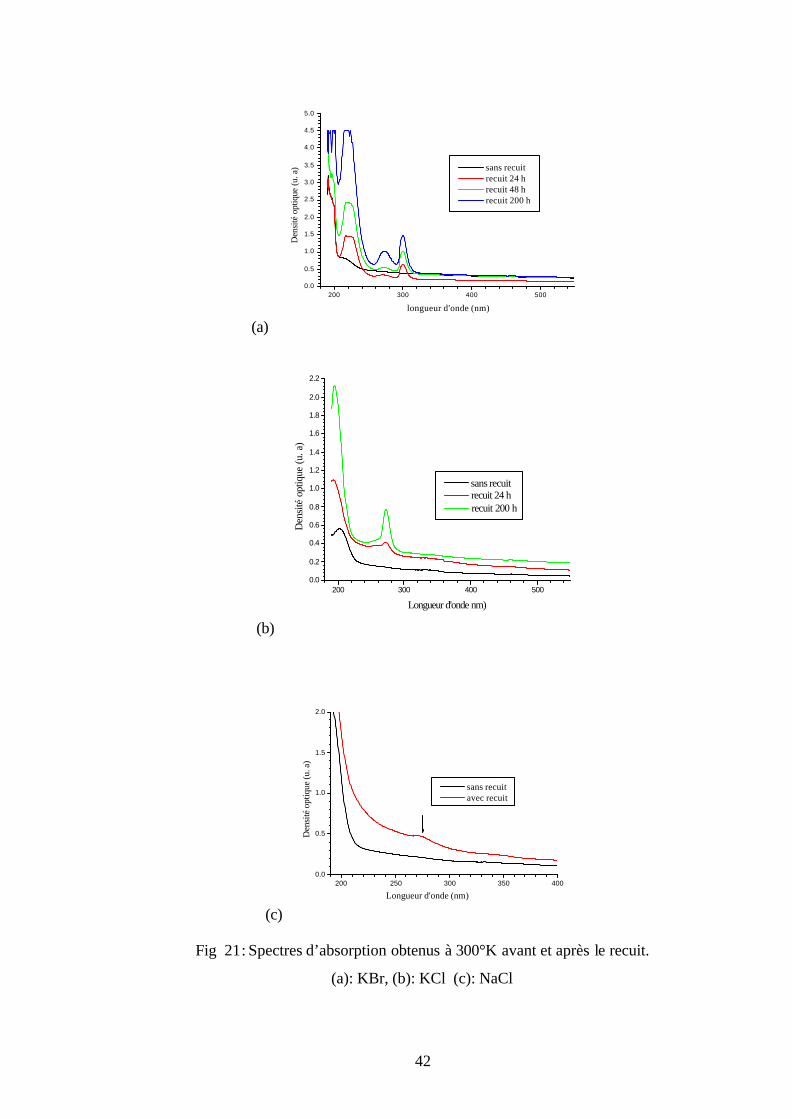
V- 1- 1 Absorption optique
Pour montrer les nouvelles bandes d’émission des centres colorés (O2- – F+) dans les
cristaux de KBr, KCl et NaCl et de confirmer la transformation des centres O2- aux centres (O2- –
F+) par étude optique, nous avons effectué des mesures optiques sur ces échantillons avant et
après le recuit. Ces mesures sont: l’absorption optique, la photoluminescence (la PL) et
l’excitation de la photoluminescence (la PLE).
La figure 21 représente les spectres d’absorption UV-visible des monocristaux de KBr,
KCl et NaCl avant et après recuit à 650°C pendant plusieurs temps. Avant le recuit, le spectre de
KBr présente une seule bande d’absorption à 215 nm. Alors qu’après le recuit, cette bande est
devenue intense et large et de plus il y a deux bandes situées à 272 nm et 301 nm (Fig. 21-a).
Pour les spectres d’absorption de KCl, ils sont montrés sur la figure 21-b. Avant le recuit, on
remarque qu’il y a une seule bande à 203 nm. Après le recuit, cette bande a disparu et il apparaît
deux bandes à 193 nm et 272 nm. Quant aux spectres de NaCl, ils sont représentés sur la figure
21-c. Avant le recuit, les spectres ne présentent aucune bande d’absorption alors qu’après le
recuit, on voit apparition d’une seule faible bande à 272 nm.
Les bandes d’absorption 215 nm dans KBr et 203 nm dans KCl sont attribuées aux
centres colorés OH- [8]. Ces ions sont introduits durant l’élaboration des monocristaux parce
qu’elle est effectuée sous atmosphère libre et donc il y a la contamination des cristaux par
l’humidité de l’air. On remarque que l’intensité de la bande d’absorption des centres OH- dans
KBr augmente pendant le recuit parce que ce dernier est réalisé à l’air libre. Tandis que dans KCl
cette bande a disparu. Cette disparition est probablement due à la transformation à haute
température des centres colorés OH- en centres (O2- – F+) selon la réaction suivante [10]:
OH- + 2F → (O2- – F+) + U
42
(a)
200 300 400 5000.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
Den
sité
opt
ique
(u. a
)
longueur d'onde (nm)
sans recuit recuit 24 h recuit 48 h recuit 200 h
(b)
200 300 400 5000.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
2.2
Den
sité
opt
ique
(u. a
)
Longueur d'onde nm)
sans recuit recuit 24 h recuit 200 h
(c)
200 250 300 350 4000.0
0.5
1.0
1.5
2.0
Den
sité
opt
ique
(u. a
)
Longueur d'onde (nm)
sans recuit avec recuit
Fig 21: Spectres d’absorption obtenus à 300°K avant et après le recuit.
(a): KBr, (b): KCl (c): NaCl
43
Dans [43], G. Gümmer a montré que les centres colorés (O2- – F+) dans les halogénures
alcalins ont plusieurs bandes d’absorption situées dans l’UV dont les positions sont variables et
dépendent du traitement thermique. Par conséquent, les bandes d’absorption 193 nm, 272 nm et
301 nm obtenues dans les trois matrices peuvent être causées par les centres colorés (O2- – F+).
On remarque que la bande 272 nm apparaît dans les trois cristaux et son intensité est très faible
dans NaCl. Ceci est peut être dû à la faible concentration des centres (O2- – F+) dans cette
matrice.
Une étude comparative entre nos résultats et ceux obtenus par G. Gümmer, montre que le
recuit à 650°C pendant 24 h mais avec un refroidissement lent des échantillons permettent
d'obtenir un spectre d’absorption complètement différent de celui trouvé dans [43]. En effet, il y
a apparition d'autres bandes situées à 193 nm, 272 nm et 301 nm.
V-1-2 La photoluminescence (la PL) et l'excitation de la photoluminescence (La PLE)
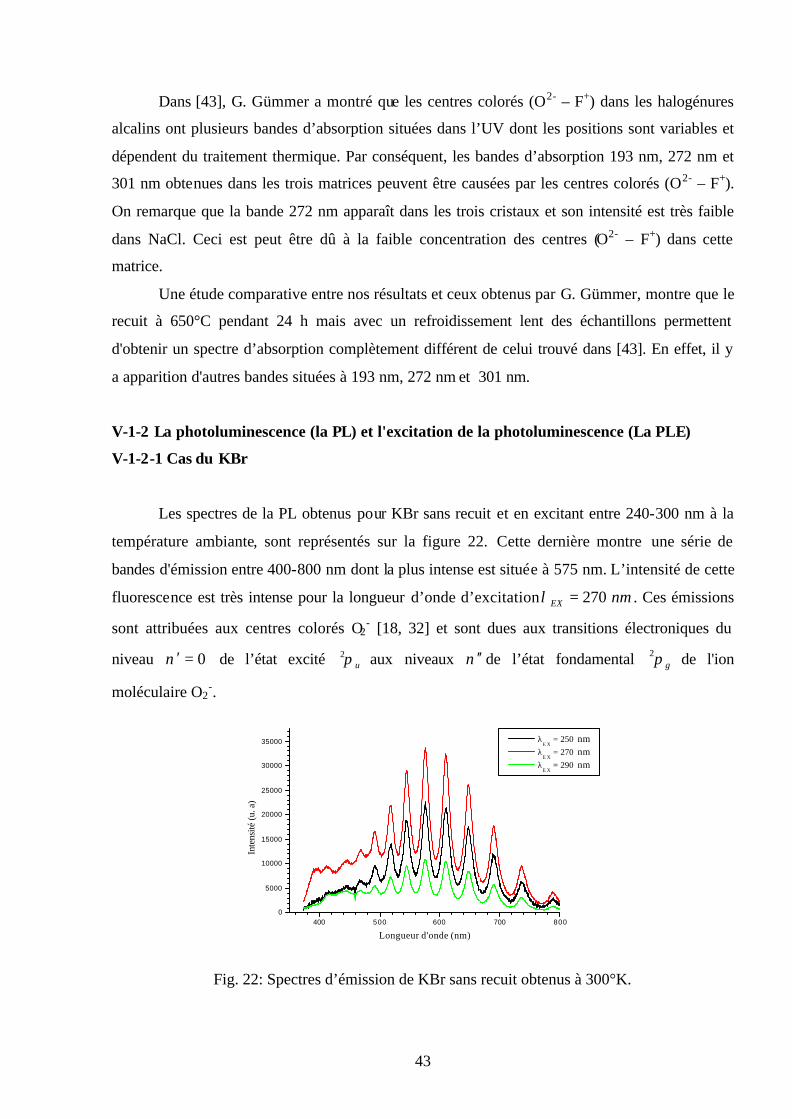
V-1-2-1 Cas du KBr
Les spectres de la PL obtenus pour KBr sans recuit et en excitant entre 240-300 nm à la
température ambiante, sont représentés sur la figure 22. Cette dernière montre une série de
bandes d'émission entre 400-800 nm dont la plus intense est située à 575 nm. L’intensité de cette
fluorescence est très intense pour la longueur d’onde d’excitation nmEX 270=λ . Ces émissions
sont attribuées aux centres colorés O2- [18, 32] et sont dues aux transitions électroniques du
niveau 0=′ν de l’état excité uπ2 aux niveaux ν ′′ de l’état fondamental gπ2 de l'ion
moléculaire O2-.
400 500 600 700 8000
5000
10000
15000
20000
25000
30000
35000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕ Χ
= 250 nm λ
Ε Χ = 270 nm
λΕ Χ
= 290 nm
Fig. 22: Spectres d’émission de KBr sans recuit obtenus à 300°K.
44
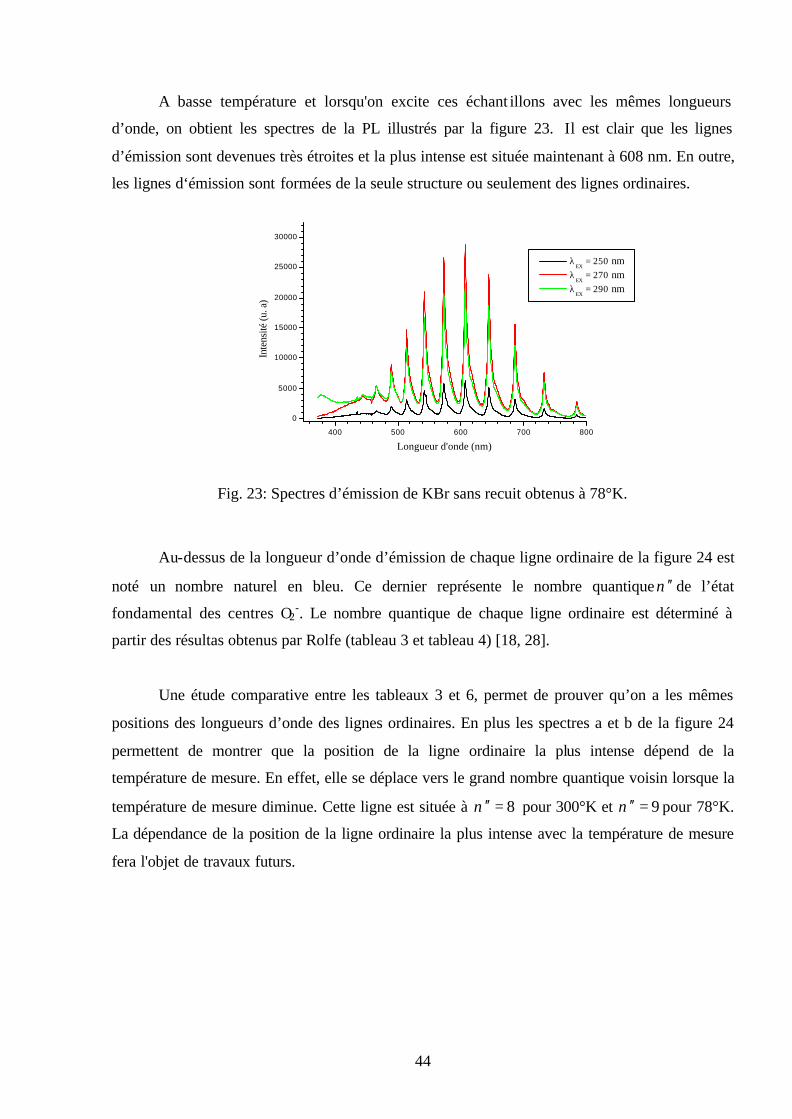
A basse température et lorsqu'on excite ces échant illons avec les mêmes longueurs
d’onde, on obtient les spectres de la PL illustrés par la figure 23. Il est clair que les lignes
d’émission sont devenues très étroites et la plus intense est située maintenant à 608 nm. En outre,
les lignes d‘émission sont formées de la seule structure ou seulement des lignes ordinaires.
400 500 600 700 800
0
5000
10000
15000
20000
25000
30000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 250 nm λ
ΕΧ = 270 nm
λΕΧ
= 290 nm
Fig. 23: Spectres d’émission de KBr sans recuit obtenus à 78°K.
Au-dessus de la longueur d’onde d’émission de chaque ligne ordinaire de la figure 24 est
noté un nombre naturel en bleu. Ce dernier représente le nombre quantiqueν ′′ de l’état
fondamental des centres O2-. Le nombre quantique de chaque ligne ordinaire est déterminé à
partir des résultas obtenus par Rolfe (tableau 3 et tableau 4) [18, 28].
Une étude comparative entre les tableaux 3 et 6, permet de prouver qu’on a les mêmes
positions des longueurs d’onde des lignes ordinaires. En plus les spectres a et b de la figure 24
permettent de montrer que la position de la ligne ordinaire la plus intense dépend de la
température de mesure. En effet, elle se déplace vers le grand nombre quantique voisin lorsque la
température de mesure diminue. Cette ligne est située à 8=′′ν pour 300°K et 9=′′ν pour 78°K.
La dépendance de la position de la ligne ordinaire la plus intense avec la température de mesure
fera l'objet de travaux futurs.
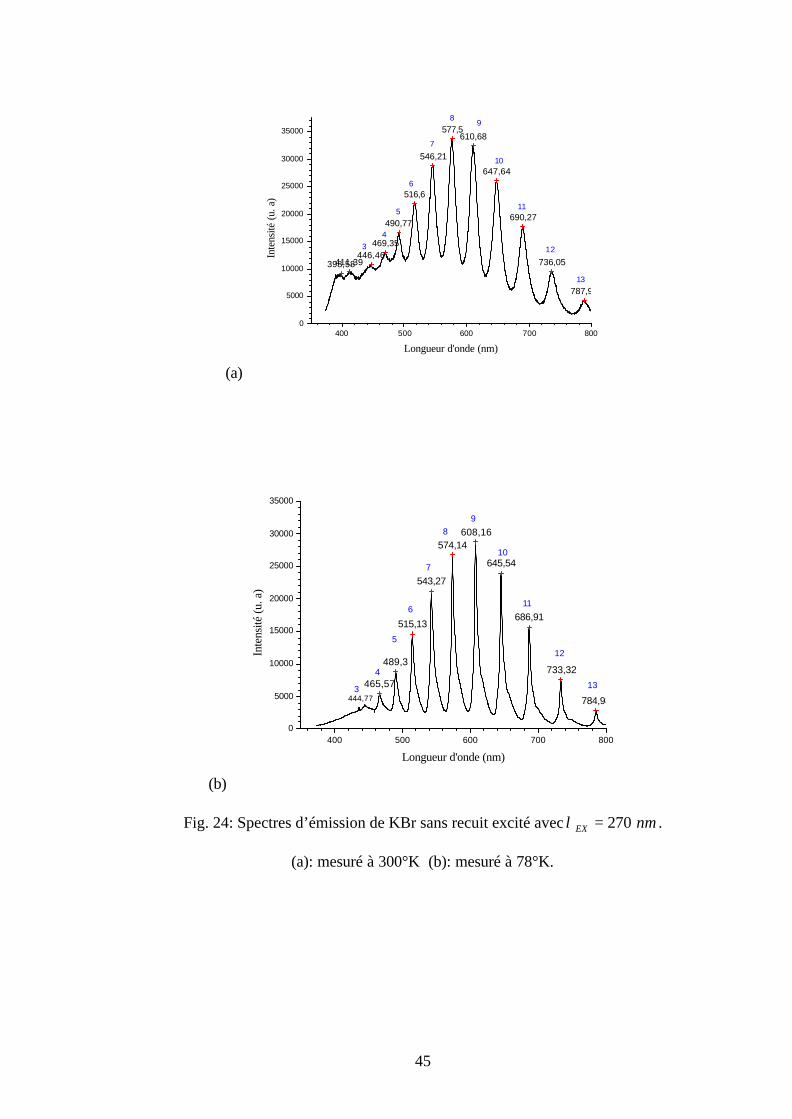
45
(a)
400 500 600 700 8000
5000
10000
15000
20000
25000
30000
35000
398,58411,39446,46
469,35
490,77
516,6
546,21
577,5610,68
647,64
690,27
736,05
787,92
34
5
6
7
13
12
11
10
98
Inte
nsité
(u. a
)
Longueur d'onde (nm)
(b)
400 500 600 700 8000
5000
10000
15000
20000
25000
30000
35000
465,57
489,3
515,13
543,27
574,14608,16
645,54
686,91
733,32
784,983
444,77
4
5
6
7
8
13
12
11
10
9
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 24: Spectres d’émission de KBr sans recuit excité avec nmEX 270=λ .
(a): mesuré à 300°K (b): mesuré à 78°K.

46
Tabl
eau
6:
47
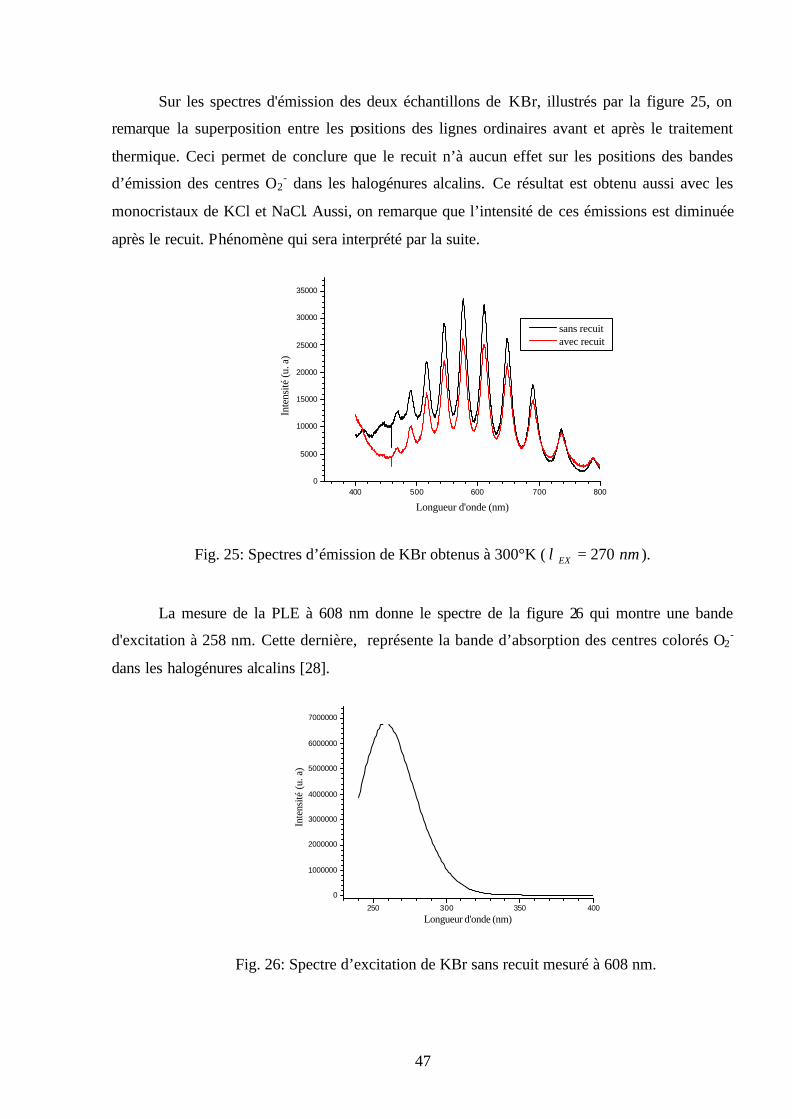
Sur les spectres d'émission des deux échantillons de KBr, illustrés par la figure 25, on
remarque la superposition entre les positions des lignes ordinaires avant et après le traitement
thermique. Ceci permet de conclure que le recuit n’à aucun effet sur les positions des bandes
d’émission des centres O2- dans les halogénures alcalins. Ce résultat est obtenu aussi avec les
monocristaux de KCl et NaCl. Aussi, on remarque que l’intensité de ces émissions est diminuée
après le recuit. Phénomène qui sera interprété par la suite.
400 500 600 700 8000
5000
10000
15000
20000
25000
30000
35000In
tens
ité (u
. a)
Longueur d'onde (nm)
sans recuit avec recuit
Fig. 25: Spectres d’émission de KBr obtenus à 300°K ( nmEX 270=λ ).
La mesure de la PLE à 608 nm donne le spectre de la figure 26 qui montre une bande
d'excitation à 258 nm. Cette dernière, représente la bande d’absorption des centres colorés O2-
dans les halogénures alcalins [28].
250 300 350 400
0
1000000
2000000
3000000
4000000
5000000
6000000
7000000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 26: Spectre d’excitation de KBr sans recuit mesuré à 608 nm.
48
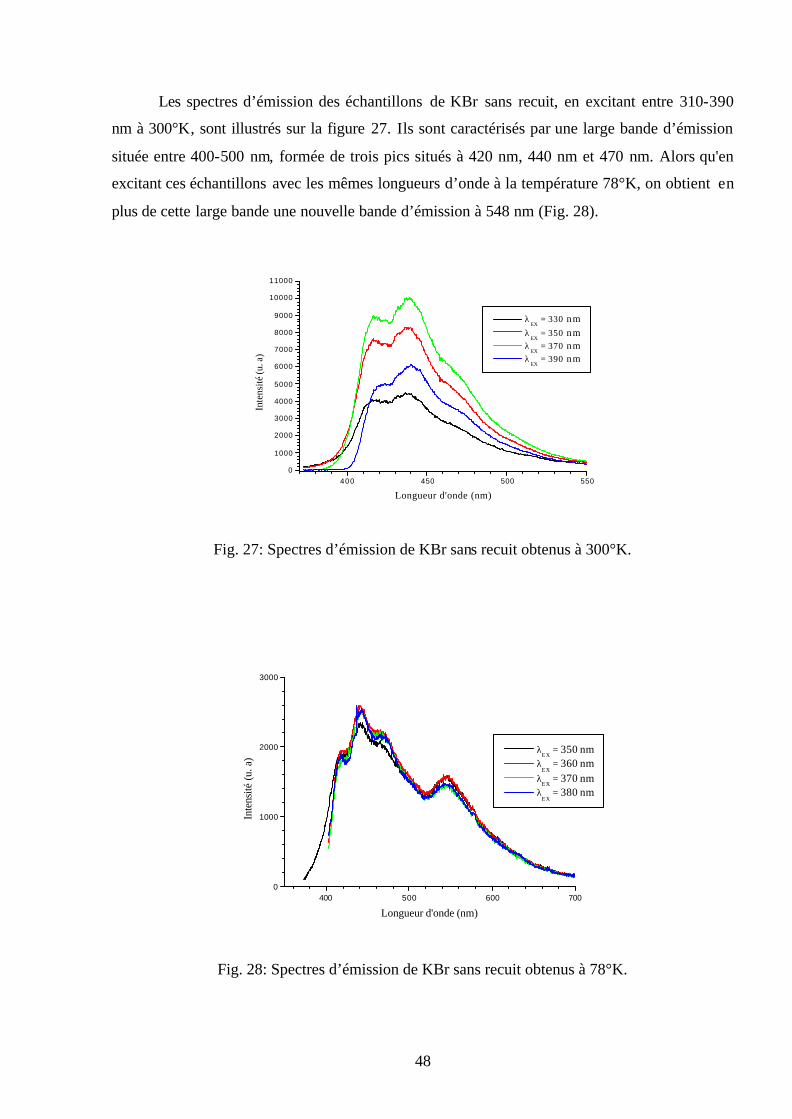
Les spectres d’émission des échantillons de KBr sans recuit, en excitant entre 310-390
nm à 300°K, sont illustrés sur la figure 27. Ils sont caractérisés par une large bande d’émission
située entre 400-500 nm, formée de trois pics situés à 420 nm, 440 nm et 470 nm. Alors qu'en
excitant ces échantillons avec les mêmes longueurs d’onde à la température 78°K, on obtient en
plus de cette large bande une nouvelle bande d’émission à 548 nm (Fig. 28).
400 450 500 5500
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 330 nm λ
ΕΧ = 350 nm
λΕΧ
= 370 nm λ
ΕΧ = 390 nm
Fig. 27: Spectres d’émission de KBr sans recuit obtenus à 300°K.
400 500 600 7000
1000
2000
3000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 350 nm λ
ΕΧ = 360 nm
λΕΧ
= 370 nm λ
ΕΧ = 380 nm
Fig. 28: Spectres d’émission de KBr sans recuit obtenus à 78°K.
49
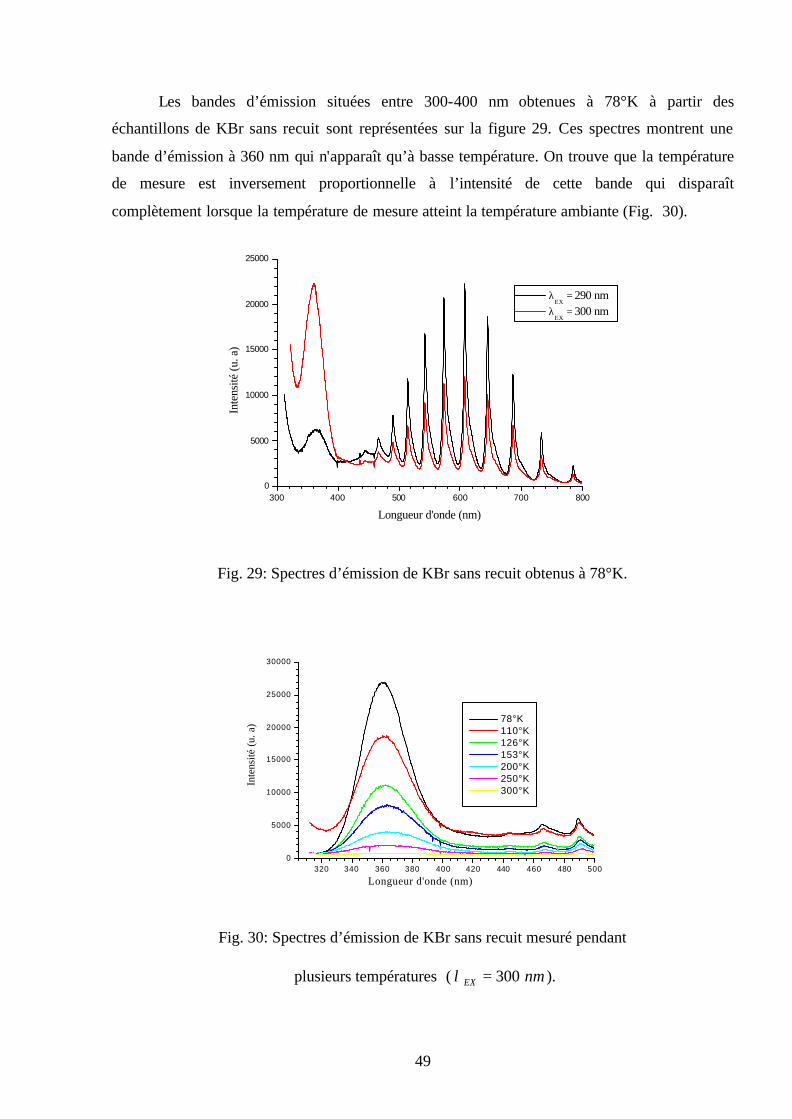
Les bandes d’émission situées entre 300-400 nm obtenues à 78°K à partir des
échantillons de KBr sans recuit sont représentées sur la figure 29. Ces spectres montrent une
bande d’émission à 360 nm qui n'apparaît qu’à basse température. On trouve que la température
de mesure est inversement proportionnelle à l’intensité de cette bande qui disparaît
complètement lorsque la température de mesure atteint la température ambiante (Fig. 30).
300 400 500 600 700 8000
5000
10000
15000
20000
25000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 290 nm λ
ΕΧ = 300 nm
Fig. 29: Spectres d’émission de KBr sans recuit obtenus à 78°K.
320 340 360 380 400 420 440 460 480 5000
5000
10000
15000
20000
25000
30000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
78°K 110°K 126°K 153°K 200°K 250°K 300°K
Fig. 30: Spectres d’émission de KBr sans recuit mesuré pendant
plusieurs températures ( nmEX 300=λ ).
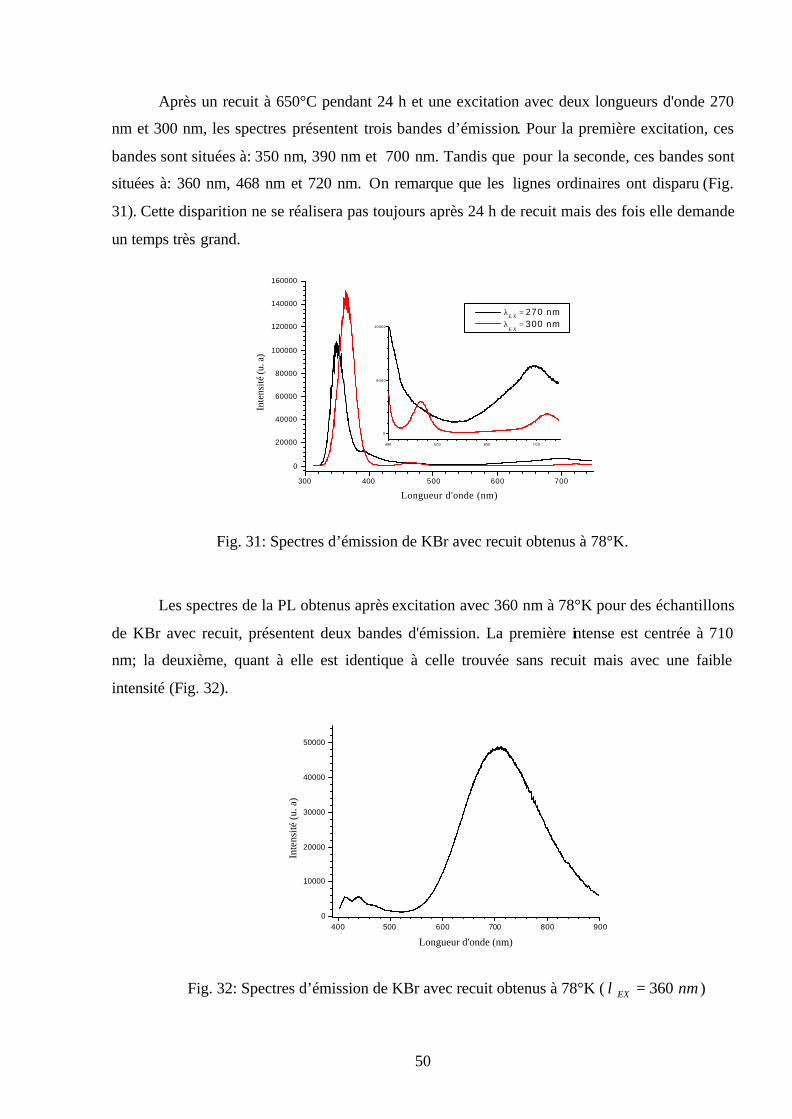
50
Après un recuit à 650°C pendant 24 h et une excitation avec deux longueurs d'onde 270
nm et 300 nm, les spectres présentent trois bandes d’émission. Pour la première excitation, ces
bandes sont situées à: 350 nm, 390 nm et 700 nm. Tandis que pour la seconde, ces bandes sont
situées à: 360 nm, 468 nm et 720 nm. On remarque que les lignes ordinaires ont disparu (Fig.
31). Cette disparition ne se réalisera pas toujours après 24 h de recuit mais des fois elle demande
un temps très grand.
400 500 600 7 0 0
0
5000
10000
300 400 500 600 700
0
20000
40000
60000
80000
100000
120000
140000
160000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕ Χ = 270 nm λ
Ε Χ = 300 nm
Fig. 31: Spectres d’émission de KBr avec recuit obtenus à 78°K.
Les spectres de la PL obtenus après excitation avec 360 nm à 78°K pour des échantillons
de KBr avec recuit, présentent deux bandes d'émission. La première intense est centrée à 710
nm; la deuxième, quant à elle est identique à celle trouvée sans recuit mais avec une faible
intensité (Fig. 32).
400 500 600 700 800 9000
10000
20000
30000
40000
50000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 32: Spectres d’émission de KBr avec recuit obtenus à 78°K ( nmEX 360=λ )
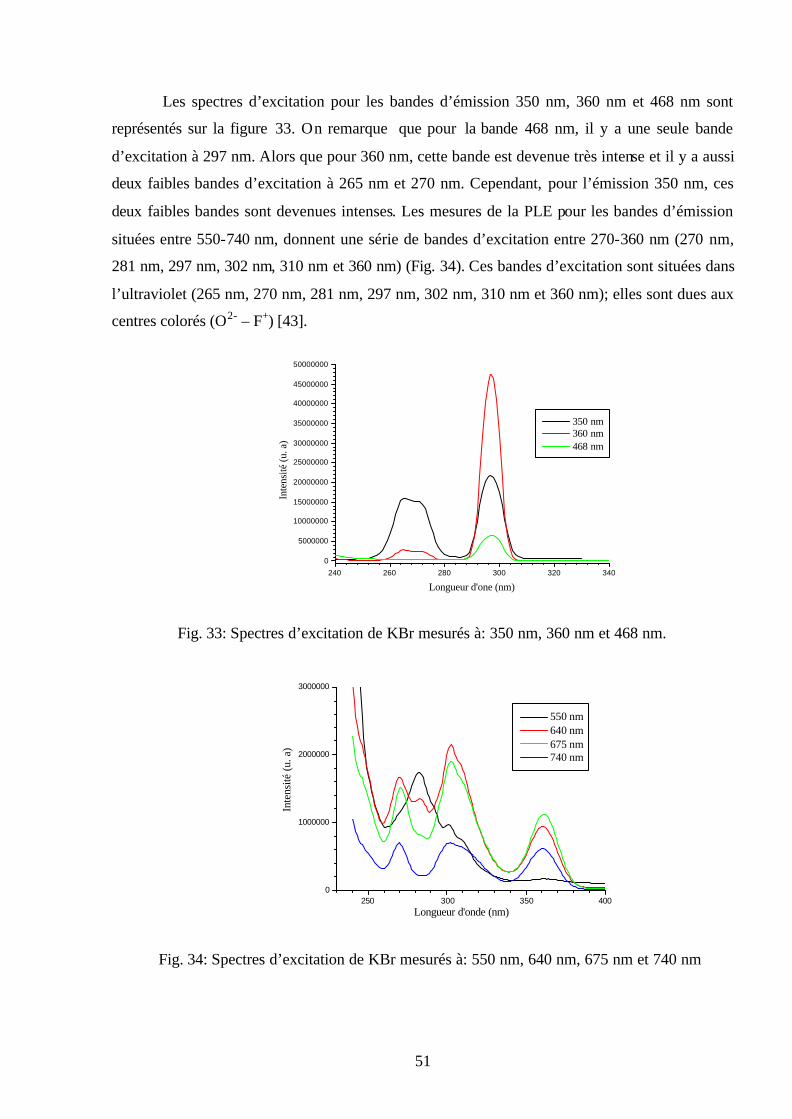
51
Les spectres d’excitation pour les bandes d’émission 350 nm, 360 nm et 468 nm sont
représentés sur la figure 33. On remarque que pour la bande 468 nm, il y a une seule bande
d’excitation à 297 nm. Alors que pour 360 nm, cette bande est devenue très intense et il y a aussi
deux faibles bandes d’excitation à 265 nm et 270 nm. Cependant, pour l’émission 350 nm, ces
deux faibles bandes sont devenues intenses. Les mesures de la PLE pour les bandes d’émission
situées entre 550-740 nm, donnent une série de bandes d’excitation entre 270-360 nm (270 nm,
281 nm, 297 nm, 302 nm, 310 nm et 360 nm) (Fig. 34). Ces bandes d’excitation sont situées dans
l’ultraviolet (265 nm, 270 nm, 281 nm, 297 nm, 302 nm, 310 nm et 360 nm); elles sont dues aux
centres colorés (O2- – F+) [43].
240 260 280 300 320 340
0
5000000
10000000
15000000
20000000
25000000
30000000
35000000
40000000
45000000
50000000
Inte
nsité
(u. a
)
Longueur d'one (nm)
350 nm 360 nm 468 nm
Fig. 33: Spectres d’excitation de KBr mesurés à: 350 nm, 360 nm et 468 nm.
250 300 350 4000
1000000
2000000
3000000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
550 nm 640 nm 675 nm 740 nm
Fig. 34: Spectres d’excitation de KBr mesurés à: 550 nm, 640 nm, 675 nm et 740 nm
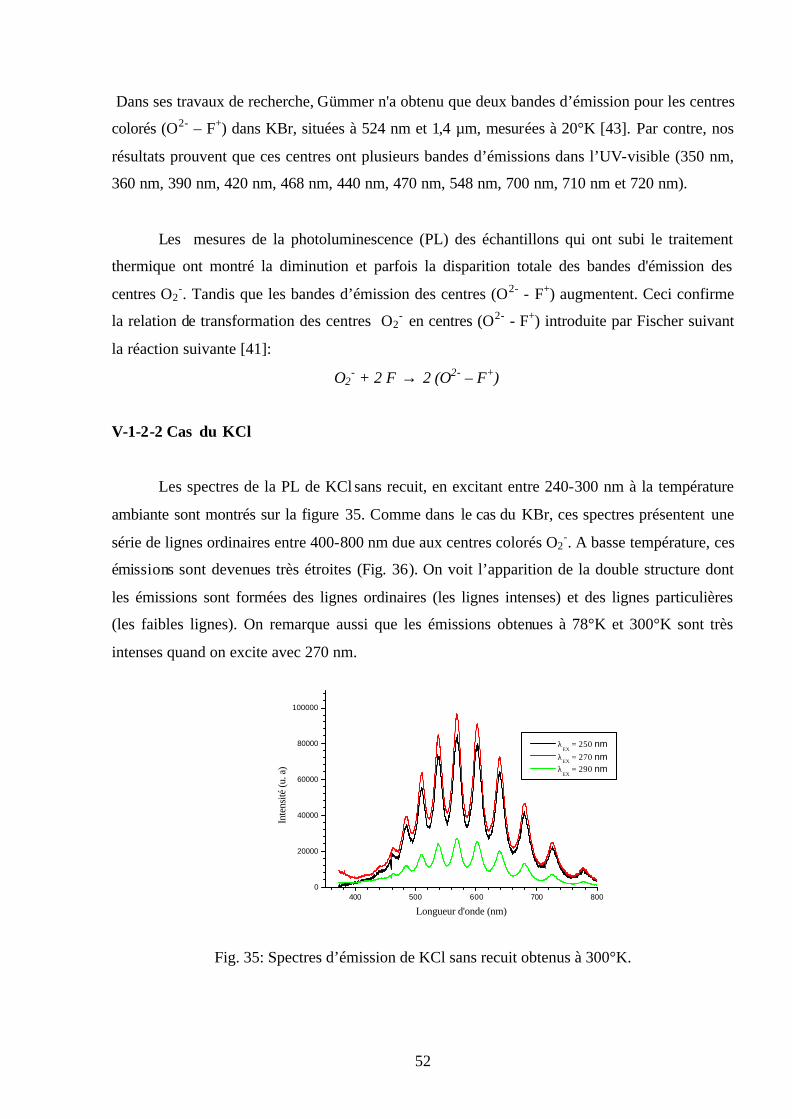
52
Dans ses travaux de recherche, Gümmer n'a obtenu que deux bandes d’émission pour les centres
colorés (O2- – F+) dans KBr, situées à 524 nm et 1,4 µm, mesurées à 20°K [43]. Par contre, nos
résultats prouvent que ces centres ont plusieurs bandes d’émissions dans l’UV-visible (350 nm,
360 nm, 390 nm, 420 nm, 468 nm, 440 nm, 470 nm, 548 nm, 700 nm, 710 nm et 720 nm).
Les mesures de la photoluminescence (PL) des échantillons qui ont subi le traitement
thermique ont montré la diminution et parfois la disparition totale des bandes d'émission des
centres O2-. Tandis que les bandes d’émission des centres (O2- - F+) augmentent. Ceci confirme
la relation de transformation des centres O2- en centres (O2- - F+) introduite par Fischer suivant
la réaction suivante [41]:
O2- + 2 F → 2 (O2- – F+)
V-1-2-2 Cas du KCl
Les spectres de la PL de KCl sans recuit, en excitant entre 240-300 nm à la température
ambiante sont montrés sur la figure 35. Comme dans le cas du KBr, ces spectres présentent une
série de lignes ordinaires entre 400-800 nm due aux centres colorés O2-. A basse température, ces
émissions sont devenues très étroites (Fig. 36). On voit l’apparition de la double structure dont
les émissions sont formées des lignes ordinaires (les lignes intenses) et des lignes particulières
(les faibles lignes). On remarque aussi que les émissions obtenues à 78°K et 300°K sont très
intenses quand on excite avec 270 nm.
400 500 600 700 8000
20000
40000
60000
80000
100000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 250 nm λΕΧ = 270 nm λ
ΕΧ = 290 nm
Fig. 35: Spectres d’émission de KCl sans recuit obtenus à 300°K.
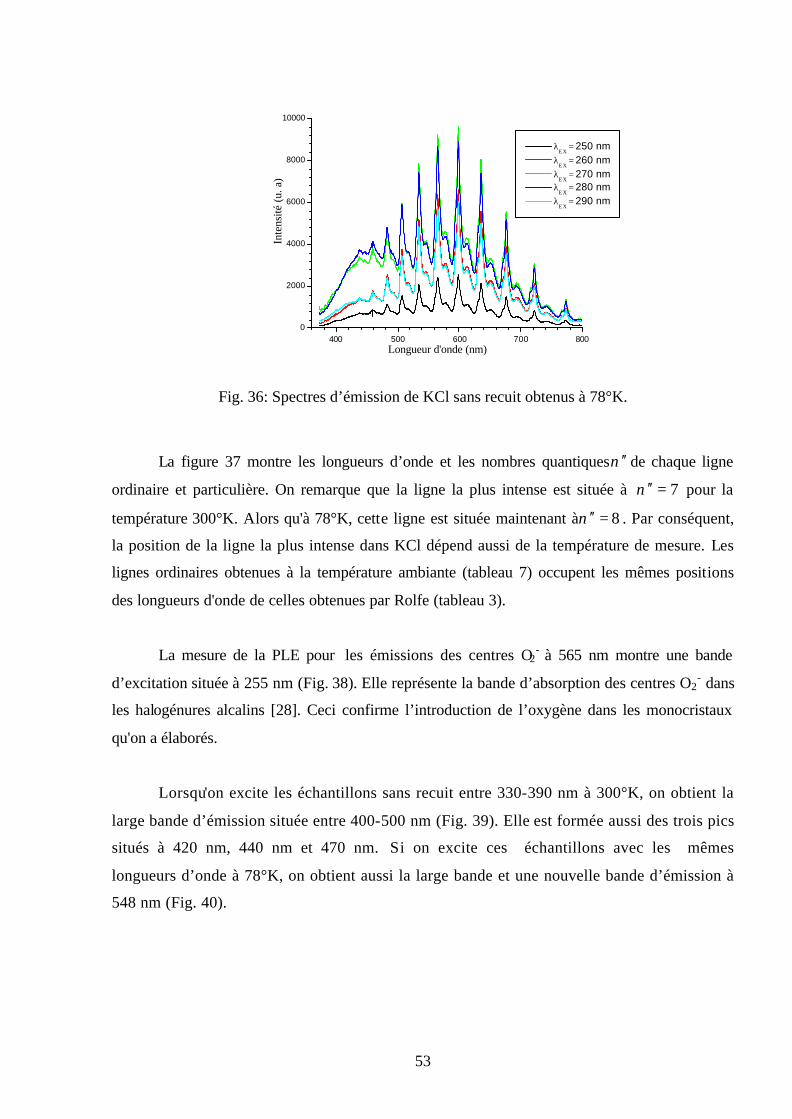
53
400 500 600 700 8000
2000
4000
6000
8000
10000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕ Χ
= 250 nm λ
Ε Χ = 260 nm
λΕ Χ
= 270 nm λ
Ε Χ = 280 nm
λΕ Χ
= 290 nm
Fig. 36: Spectres d’émission de KCl sans recuit obtenus à 78°K.
La figure 37 montre les longueurs d’onde et les nombres quantiquesν ′′ de chaque ligne
ordinaire et particulière. On remarque que la ligne la plus intense est située à 7=′′ν pour la
température 300°K. Alors qu'à 78°K, cette ligne est située maintenant à 8=′′ν . Par conséquent,
la position de la ligne la plus intense dans KCl dépend aussi de la température de mesure. Les
lignes ordinaires obtenues à la température ambiante (tableau 7) occupent les mêmes positions
des longueurs d'onde de celles obtenues par Rolfe (tableau 3).
La mesure de la PLE pour les émissions des centres O2- à 565 nm montre une bande
d’excitation située à 255 nm (Fig. 38). Elle représente la bande d’absorption des centres O2- dans
les halogénures alcalins [28]. Ceci confirme l’introduction de l’oxygène dans les monocristaux
qu'on a élaborés.
Lorsqu'on excite les échantillons sans recuit entre 330-390 nm à 300°K, on obtient la
large bande d’émission située entre 400-500 nm (Fig. 39). Elle est formée aussi des trois pics
situés à 420 nm, 440 nm et 470 nm. Si on excite ces échantillons avec les mêmes
longueurs d’onde à 78°K, on obtient aussi la large bande et une nouvelle bande d’émission à
548 nm (Fig. 40).
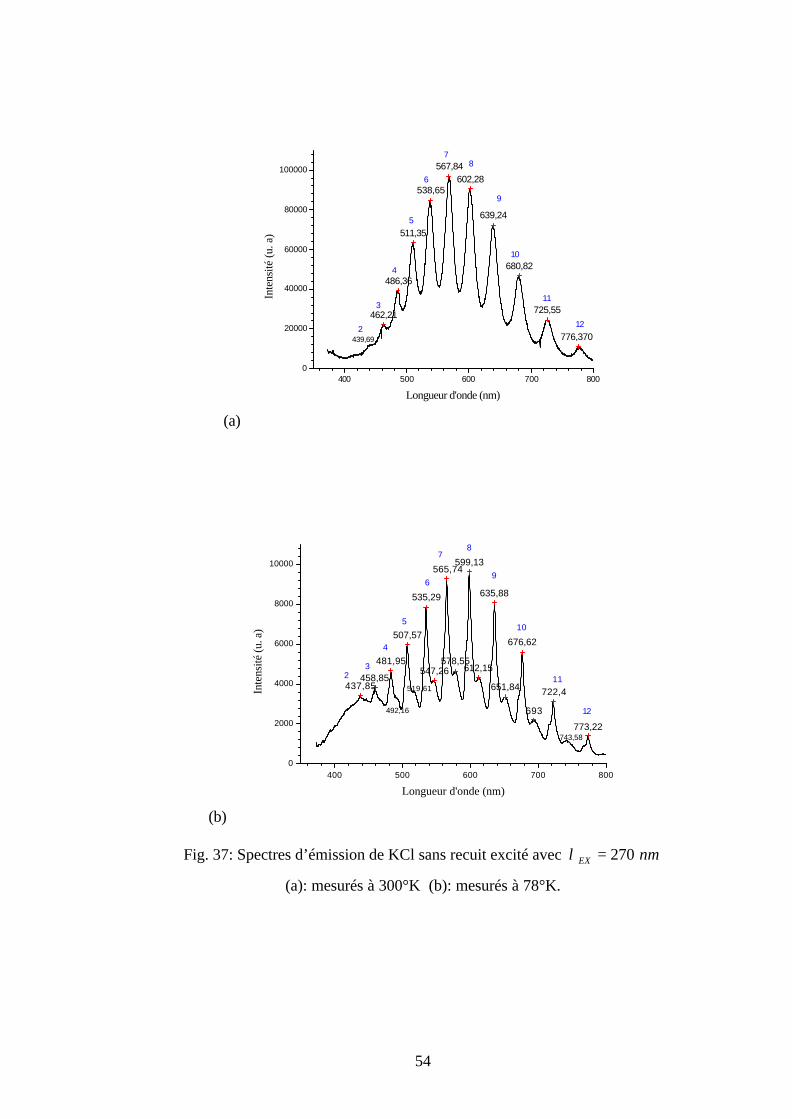
54
(a)
400 500 600 700 8000
20000
40000
60000
80000
100000
462,21
486,36
511,35
538,65
567,84602,28
639,24
680,82
725,55
776,37012
3
4
5
6
12
11
10
9
87
439,69
Inte
nsité
(u. a
)
Longueur d'onde (nm)
(b)
400 500 600 700 8000
2000
4000
6000
8000
10000
437,85458,85
481,95
507,57
535,29
547,26
565,74
578,55
599,13
612,15
635,88
651,84
676,62
693
722,4
773,22
23
4
5
6
7
12
11
10
9
8
492,16
519,61
743,58
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 37: Spectres d’émission de KCl sans recuit excité avec nmEX 270=λ
(a): mesurés à 300°K (b): mesurés à 78°K.
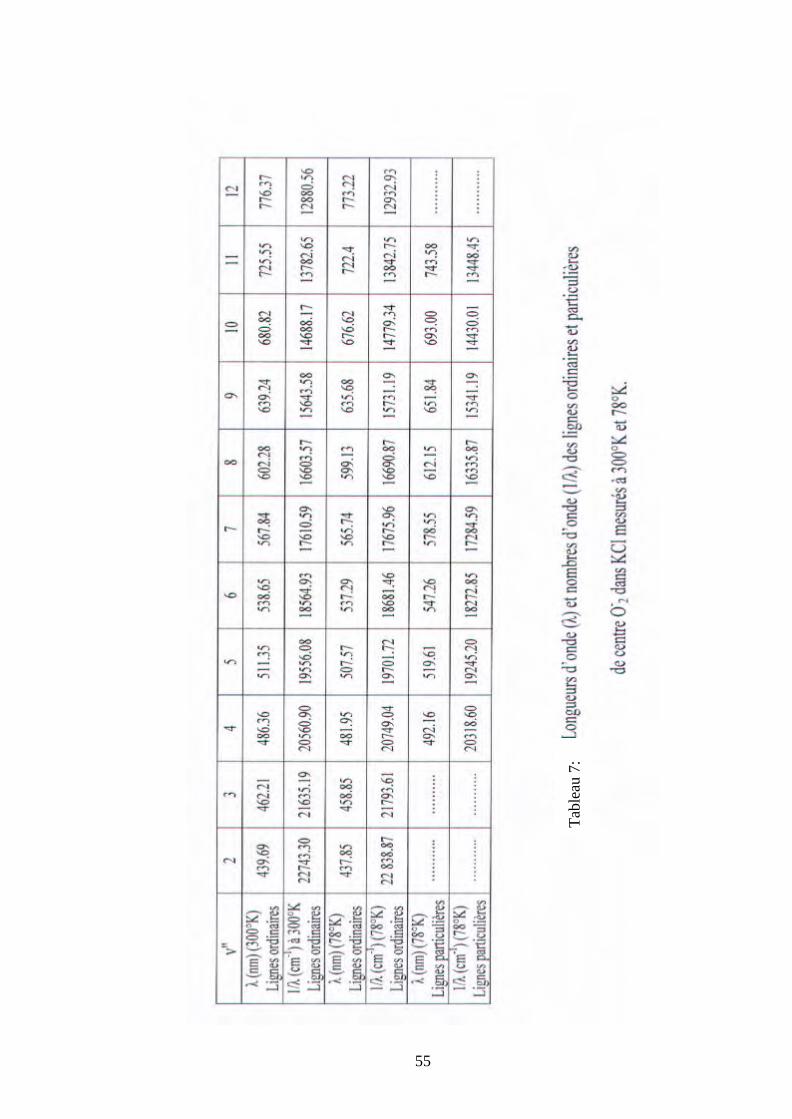
55
Tabl
eau
7:
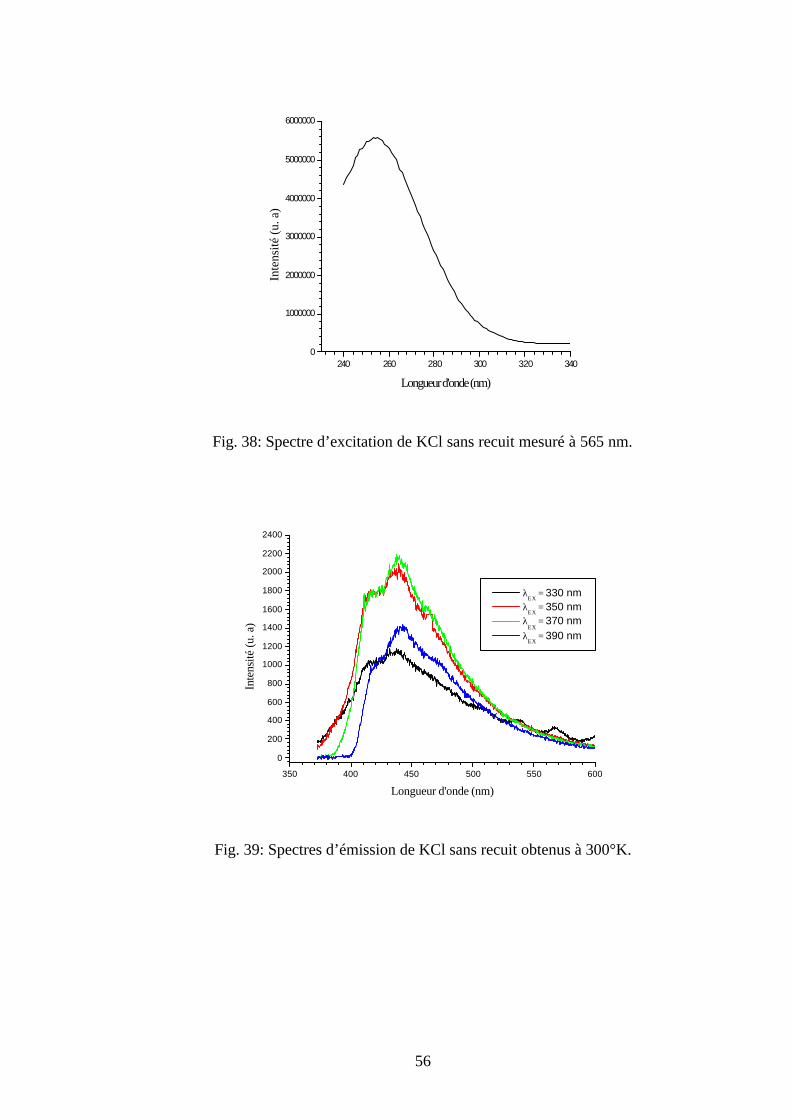
56
240 260 280 300 320 3400
1000000
2000000
3000000
4000000
5000000
6000000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 38: Spectre d’excitation de KCl sans recuit mesuré à 565 nm.
350 400 450 500 550 600
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 330 nm λ
ΕΧ = 350 nm
λΕΧ
= 370 nm λ
ΕΧ = 390 nm
Fig. 39: Spectres d’émission de KCl sans recuit obtenus à 300°K.
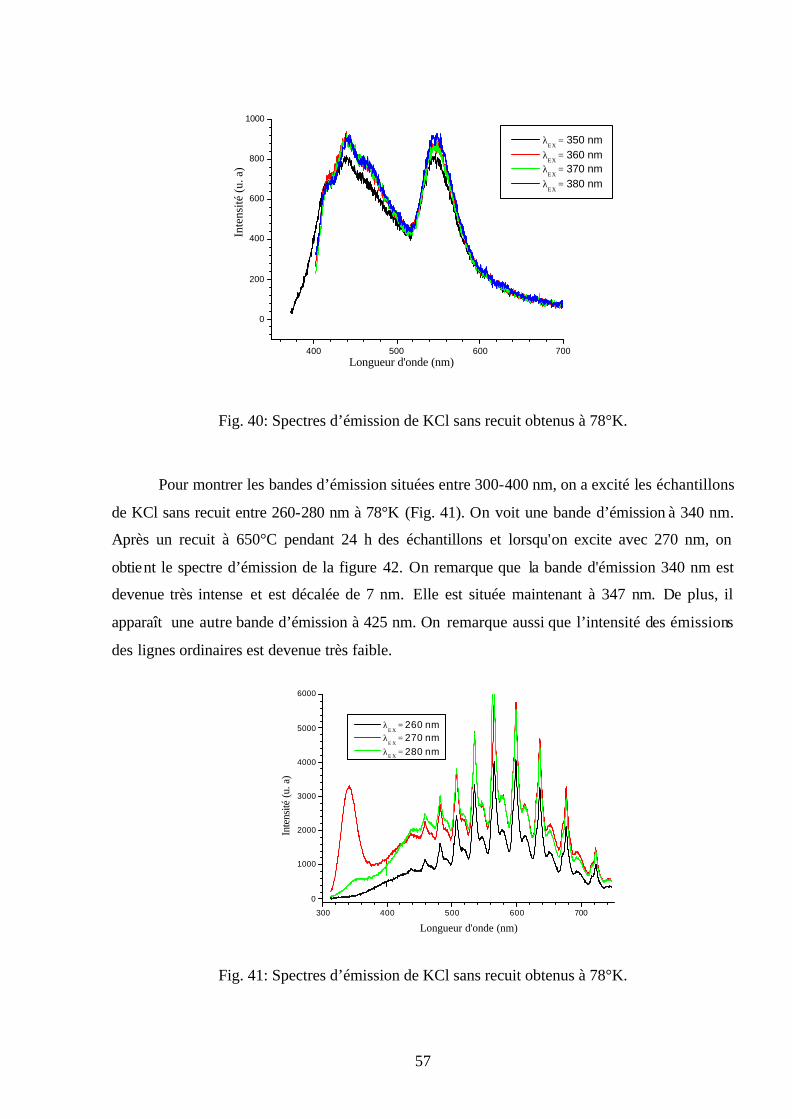
57
400 500 600 700
0
200
400
600
800
1000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 350 nm λ
ΕΧ = 360 nm
λΕΧ
= 370 nm λ
ΕΧ = 380 nm
Fig. 40: Spectres d’émission de KCl sans recuit obtenus à 78°K.
Pour montrer les bandes d’émission situées entre 300-400 nm, on a excité les échantillons
de KCl sans recuit entre 260-280 nm à 78°K (Fig. 41). On voit une bande d’émission à 340 nm.
Après un recuit à 650°C pendant 24 h des échantillons et lorsqu'on excite avec 270 nm, on
obtient le spectre d’émission de la figure 42. On remarque que la bande d'émission 340 nm est
devenue très intense et est décalée de 7 nm. Elle est située maintenant à 347 nm. De plus, il
apparaît une autre bande d’émission à 425 nm. On remarque aussi que l’intensité des émissions
des lignes ordinaires est devenue très faible.
300 400 500 600 700
0
1000
2000
3000
4000
5000
6000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕ Χ
= 260 nm λ
Ε Χ = 270 nm
λΕ Χ = 280 nm
Fig. 41: Spectres d’émission de KCl sans recuit obtenus à 78°K.
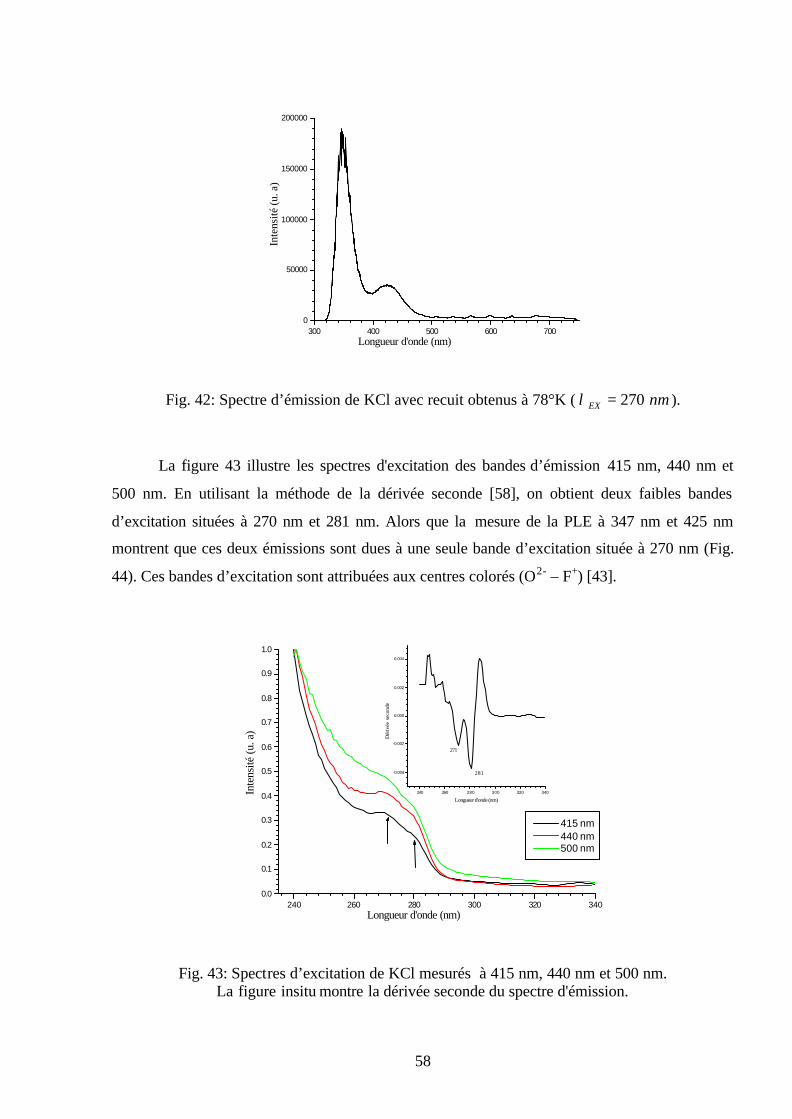
58
300 400 500 600 7000
50000
100000
150000
200000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 42: Spectre d’émission de KCl avec recuit obtenus à 78°K ( nmEX 270=λ ).
La figure 43 illustre les spectres d'excitation des bandes d’émission 415 nm, 440 nm et
500 nm. En utilisant la méthode de la dérivée seconde [58], on obtient deux faibles bandes
d’excitation situées à 270 nm et 281 nm. Alors que la mesure de la PLE à 347 nm et 425 nm
montrent que ces deux émissions sont dues à une seule bande d’excitation située à 270 nm (Fig.
44). Ces bandes d’excitation sont attribuées aux centres colorés (O2- – F+) [43].
240 260 280 300 320 3400.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Inte
nsité
(u. a
)
Longueur d'onde (nm)
415 nm 440 nm 500 nm
240 260 280 300 320 340
-0.004
-0.002
0.000
0.002
0.004
281
271
Dér
ivée
sec
onde
Longueur d'onde (nm)
Fig. 43: Spectres d’excitation de KCl mesurés à 415 nm, 440 nm et 500 nm. La figure insitu montre la dérivée seconde du spectre d'émission.
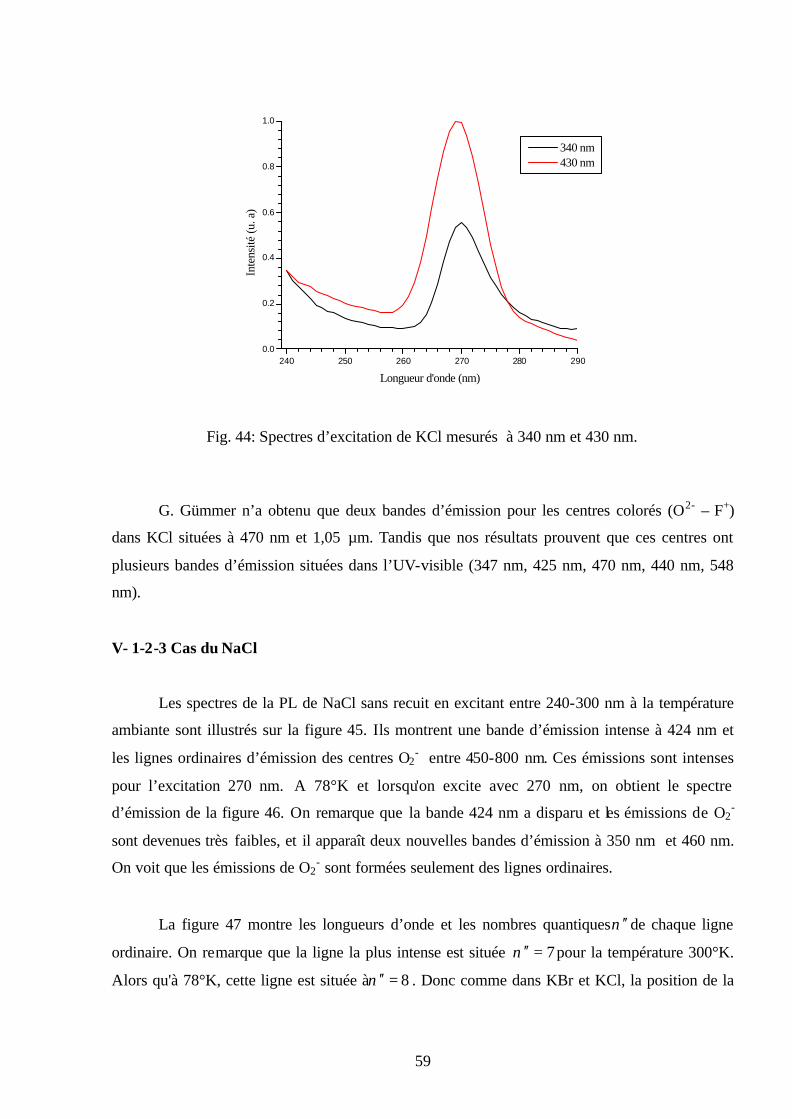
59
240 250 260 270 280 2900.0
0.2
0.4
0.6
0.8
1.0
Inte
nsité
(u. a
)
Longueur d'onde (nm)
340 nm 430 nm
Fig. 44: Spectres d’excitation de KCl mesurés à 340 nm et 430 nm.
G. Gümmer n’a obtenu que deux bandes d’émission pour les centres colorés (O2- – F+)
dans KCl situées à 470 nm et 1,05 µm. Tandis que nos résultats prouvent que ces centres ont
plusieurs bandes d’émission situées dans l’UV-visible (347 nm, 425 nm, 470 nm, 440 nm, 548
nm).
V- 1-2-3 Cas du NaCl
Les spectres de la PL de NaCl sans recuit en excitant entre 240-300 nm à la température
ambiante sont illustrés sur la figure 45. Ils montrent une bande d’émission intense à 424 nm et
les lignes ordinaires d’émission des centres O2- entre 450-800 nm. Ces émissions sont intenses
pour l’excitation 270 nm. A 78°K et lorsqu'on excite avec 270 nm, on obtient le spectre
d’émission de la figure 46. On remarque que la bande 424 nm a disparu et les émissions de O2-
sont devenues très faibles, et il apparaît deux nouvelles bandes d’émission à 350 nm et 460 nm.
On voit que les émissions de O2- sont formées seulement des lignes ordinaires.
La figure 47 montre les longueurs d’onde et les nombres quantiquesν ′′ de chaque ligne
ordinaire. On remarque que la ligne la plus intense est située 7=′′ν pour la température 300°K.
Alors qu'à 78°K, cette ligne est située à 8=′′ν . Donc comme dans KBr et KCl, la position de la
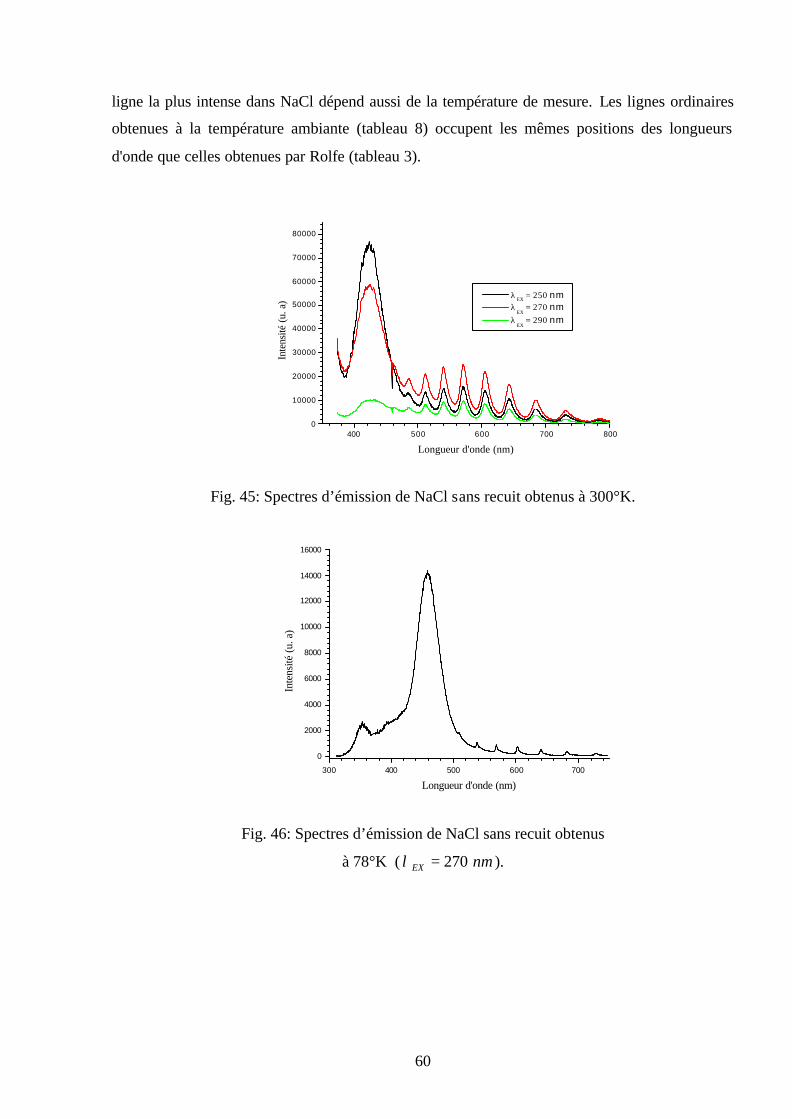
60
ligne la plus intense dans NaCl dépend aussi de la température de mesure. Les lignes ordinaires
obtenues à la température ambiante (tableau 8) occupent les mêmes positions des longueurs
d'onde que celles obtenues par Rolfe (tableau 3).
400 500 600 700 8000
10000
20000
30000
40000
50000
60000
70000
80000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λ ΕΧ = 250 nm λ
ΕΧ = 270 nm
λΕΧ
= 290 nm
Fig. 45: Spectres d’émission de NaCl sans recuit obtenus à 300°K.
300 400 500 600 700
0
2000
4000
6000
8000
10000
12000
14000
16000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 46: Spectres d’émission de NaCl sans recuit obtenus
à 78°K ( nmEX 270=λ ).
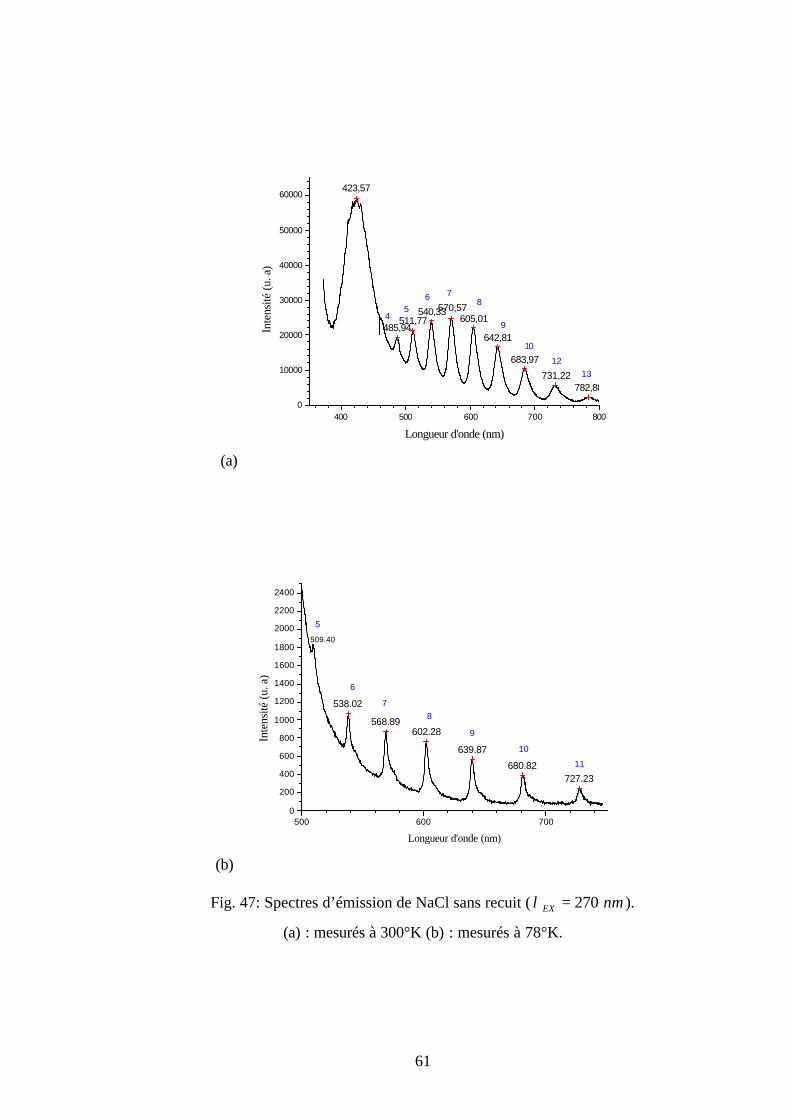
61
(a)
400 500 600 700 8000
10000
20000
30000
40000
50000
60000423,57
485,94511,77
540,33570,57605,01
642,81
683,97
731,22782,88
1312
10
9
876
54
Inte
nsité
(u. a
)
Longueur d'onde (nm)
(b)
500 600 7000
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
538.02
568.89602.28
639.87
680.82727.23
5
6
7
11
10
9
8
509.40
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 47: Spectres d’émission de NaCl sans recuit ( nmEX 270=λ ).
(a) : mesurés à 300°K (b) : mesurés à 78°K.
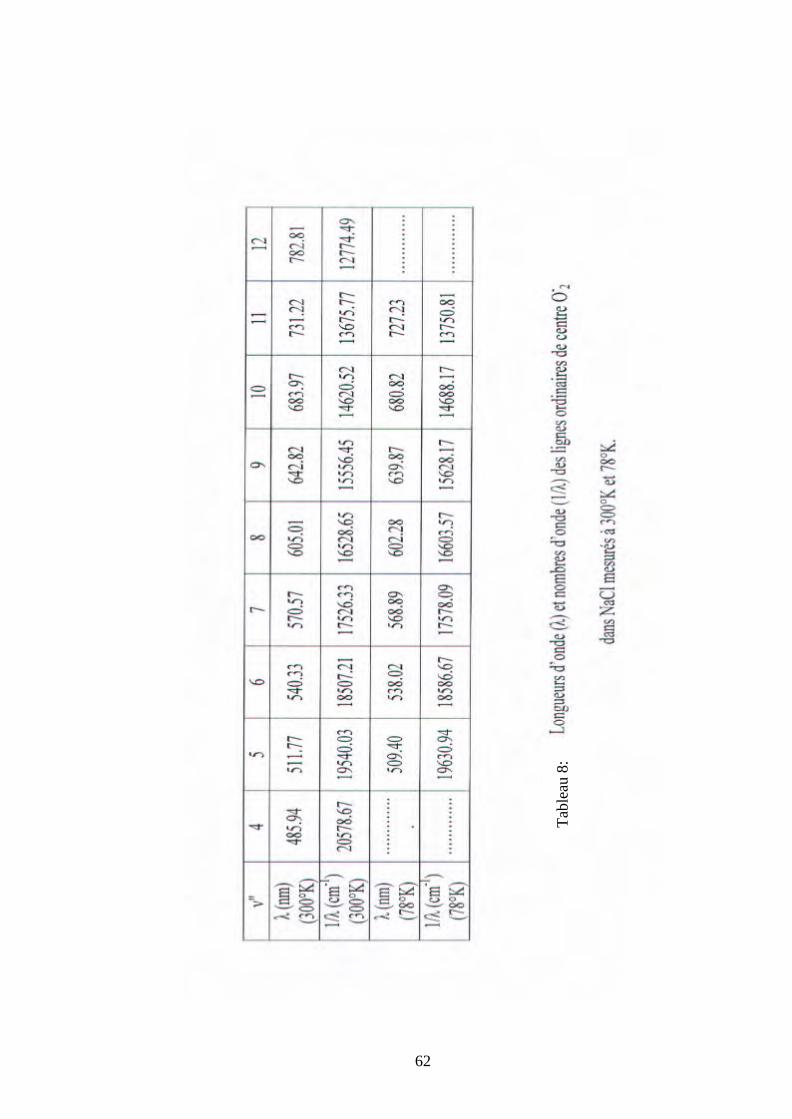
62
Tabl
eau
8:
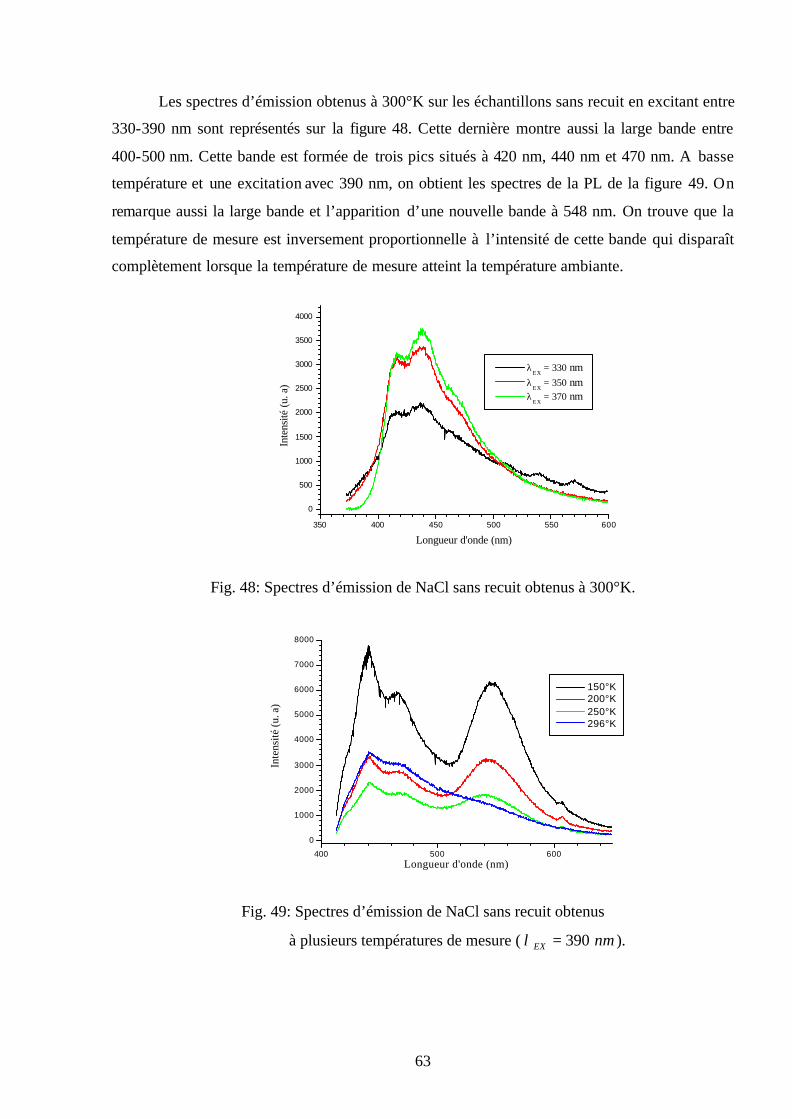
63
Les spectres d’émission obtenus à 300°K sur les échantillons sans recuit en excitant entre
330-390 nm sont représentés sur la figure 48. Cette dernière montre aussi la large bande entre
400-500 nm. Cette bande est formée de trois pics situés à 420 nm, 440 nm et 470 nm. A basse
température et une excitation avec 390 nm, on obtient les spectres de la PL de la figure 49. On
remarque aussi la large bande et l’apparition d’une nouvelle bande à 548 nm. On trouve que la
température de mesure est inversement proportionnelle à l’intensité de cette bande qui disparaît
complètement lorsque la température de mesure atteint la température ambiante.
350 400 450 500 550 600
0
500
1000
1500
2000
2500
3000
3500
4000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 330 nm λ
ΕΧ = 350 nm
λΕΧ
= 370 nm
Fig. 48: Spectres d’émission de NaCl sans recuit obtenus à 300°K.
400 500 600
0
1000
2000
3000
4000
5000
6000
7000
8000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
150°K 200°K 250°K 296°K
Fig. 49: Spectres d’émission de NaCl sans recuit obtenus
à plusieurs températures de mesure ( nmEX 390=λ ).
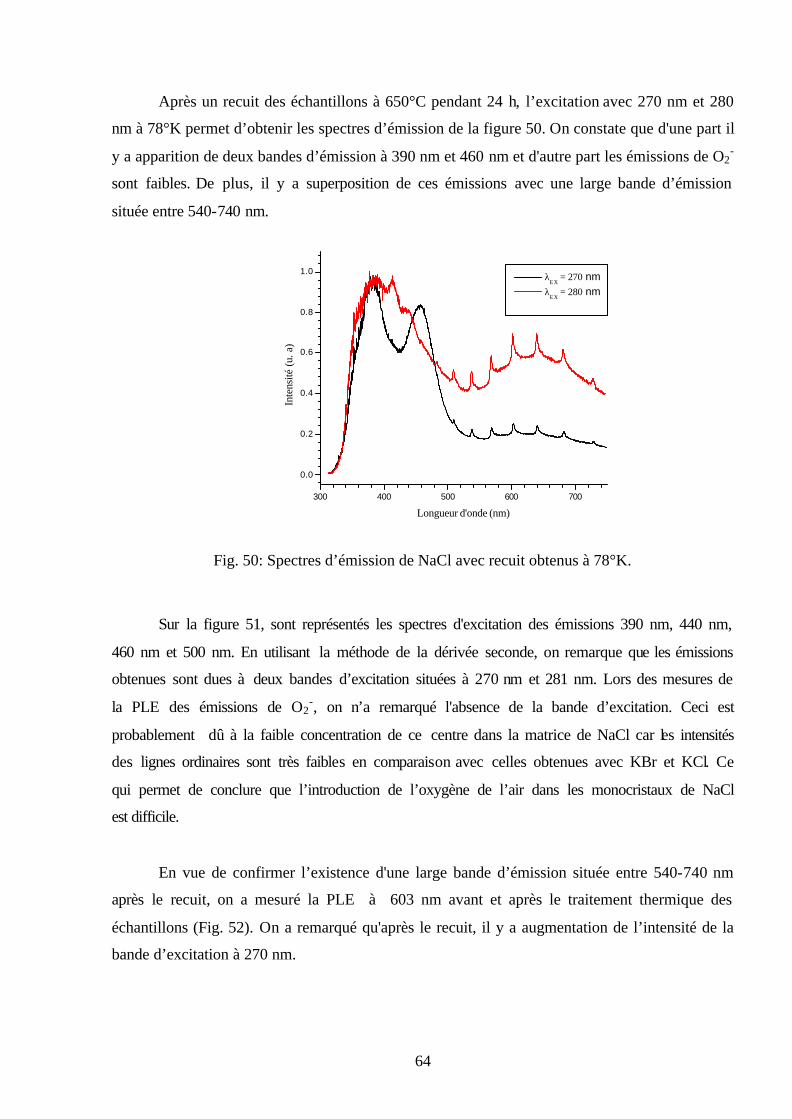
64
Après un recuit des échantillons à 650°C pendant 24 h, l’excitation avec 270 nm et 280
nm à 78°K permet d’obtenir les spectres d’émission de la figure 50. On constate que d'une part il
y a apparition de deux bandes d’émission à 390 nm et 460 nm et d'autre part les émissions de O2-
sont faibles. De plus, il y a superposition de ces émissions avec une large bande d’émission
située entre 540-740 nm.
300 400 500 600 700
0.0
0.2
0.4
0.6
0.8
1.0
Inte
nsité
(u. a
)
Longueur d'onde (nm)
λΕΧ
= 270 nm λ
ΕΧ = 280 nm
Fig. 50: Spectres d’émission de NaCl avec recuit obtenus à 78°K.
Sur la figure 51, sont représentés les spectres d'excitation des émissions 390 nm, 440 nm,
460 nm et 500 nm. En utilisant la méthode de la dérivée seconde, on remarque que les émissions
obtenues sont dues à deux bandes d’excitation situées à 270 nm et 281 nm. Lors des mesures de
la PLE des émissions de O2-, on n’a remarqué l'absence de la bande d’excitation. Ceci est
probablement dû à la faible concentration de ce centre dans la matrice de NaCl car les intensités
des lignes ordinaires sont très faibles en comparaison avec celles obtenues avec KBr et KCl. Ce
qui permet de conclure que l’introduction de l’oxygène de l’air dans les monocristaux de NaCl
est difficile.
En vue de confirmer l’existence d'une large bande d’émission située entre 540-740 nm
après le recuit, on a mesuré la PLE à 603 nm avant et après le traitement thermique des
échantillons (Fig. 52). On a remarqué qu'après le recuit, il y a augmentation de l’intensité de la
bande d’excitation à 270 nm.
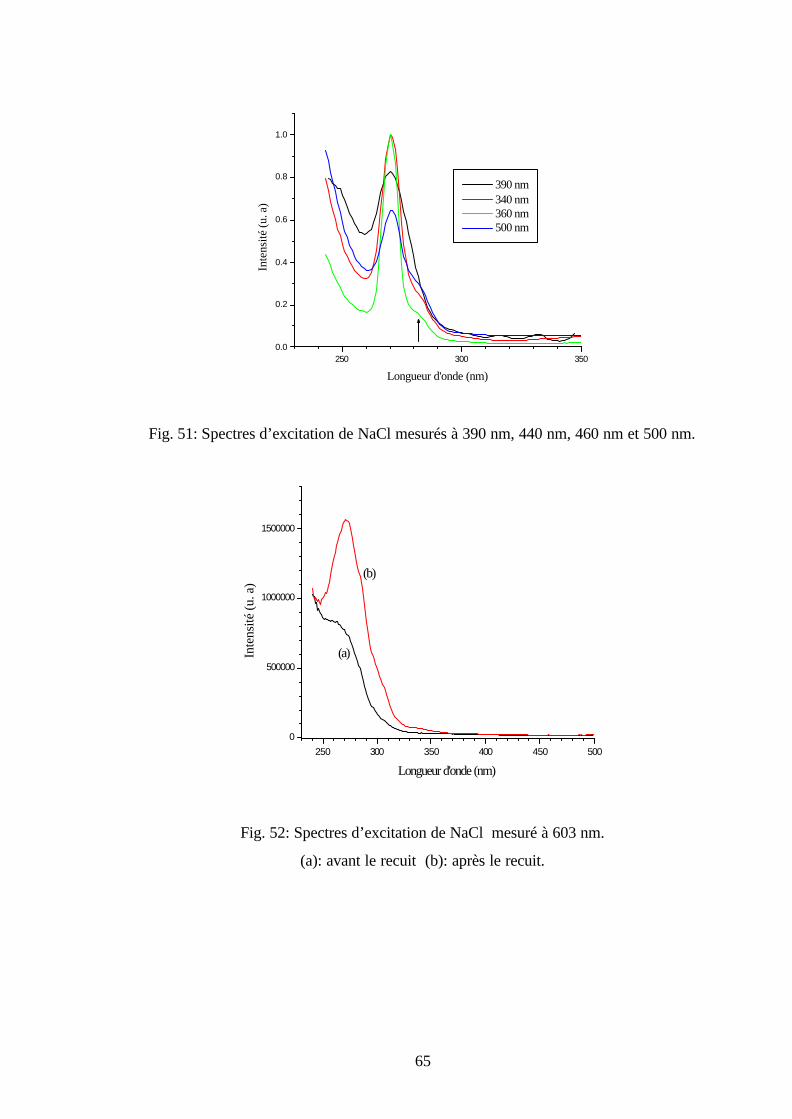
65
250 300 3500.0
0.2
0.4
0.6
0.8
1.0
Inte
nsité
(u. a
)
Longueur d'onde (nm)
390 nm 340 nm 360 nm 500 nm
Fig. 51: Spectres d’excitation de NaCl mesurés à 390 nm, 440 nm, 460 nm et 500 nm.
250 300 350 400 450 500
0
500000
1000000
1500000
(b)
(a)Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 52: Spectres d’excitation de NaCl mesuré à 603 nm.
(a): avant le recuit (b): après le recuit.
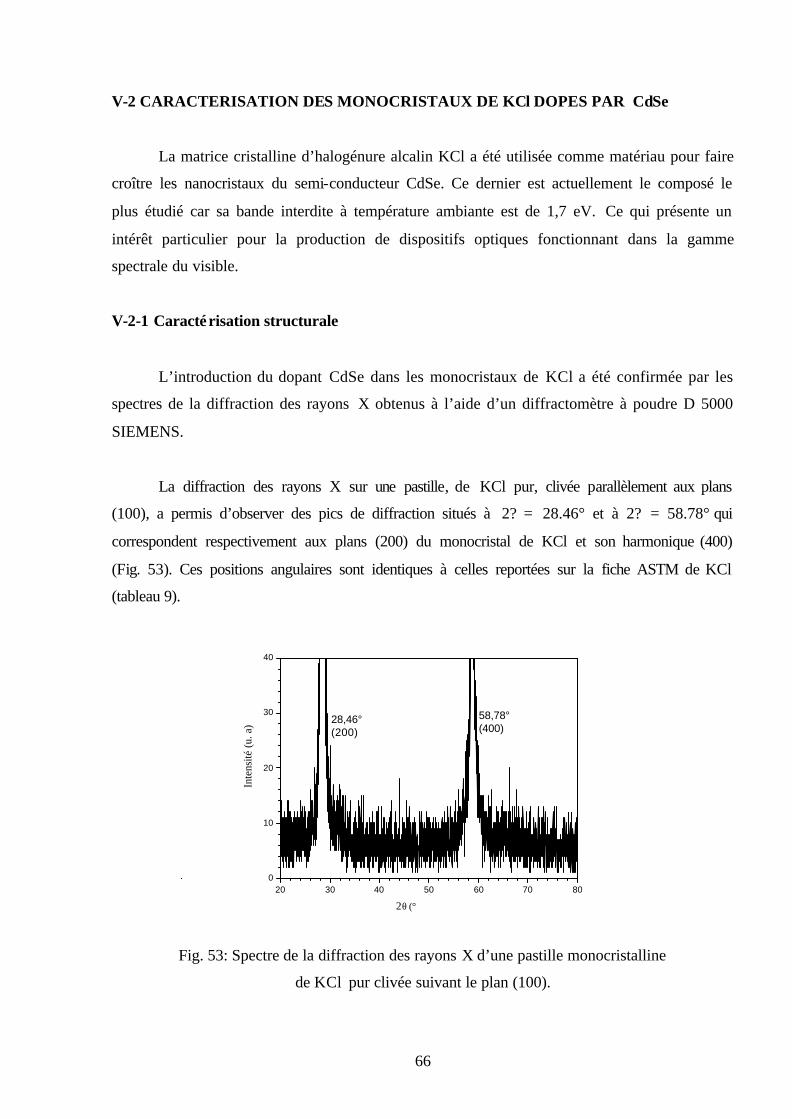
66
V-2 CARACTERISATION DES MONOCRISTAUX DE KCl DOPES PAR CdSe
La matrice cristalline d’halogénure alcalin KCl a été utilisée comme matériau pour faire
croître les nanocristaux du semi-conducteur CdSe. Ce dernier est actuellement le composé le
plus étudié car sa bande interdite à température ambiante est de 1,7 eV. Ce qui présente un
intérêt particulier pour la production de dispositifs optiques fonctionnant dans la gamme
spectrale du visible.
V-2-1 Caractérisation structurale
L’introduction du dopant CdSe dans les monocristaux de KCl a été confirmée par les
spectres de la diffraction des rayons X obtenus à l’aide d’un diffractomètre à poudre D 5000
SIEMENS.
La diffraction des rayons X sur une pastille, de KCl pur, clivée parallèlement aux plans
(100), a permis d’observer des pics de diffraction situés à 2? = 28.46° et à 2? = 58.78° qui
correspondent respectivement aux plans (200) du monocristal de KCl et son harmonique (400)
(Fig. 53). Ces positions angulaires sont identiques à celles reportées sur la fiche ASTM de KCl
(tableau 9).
20 30 40 50 60 70 800
10
20
30
40
Inte
nsité
(u. a
)
2θ (°)
28,46°(200)
58,78°(400)
Fig. 53: Spectre de la diffraction des rayons X d’une pastille monocristalline
de KCl pur clivée suivant le plan (100).
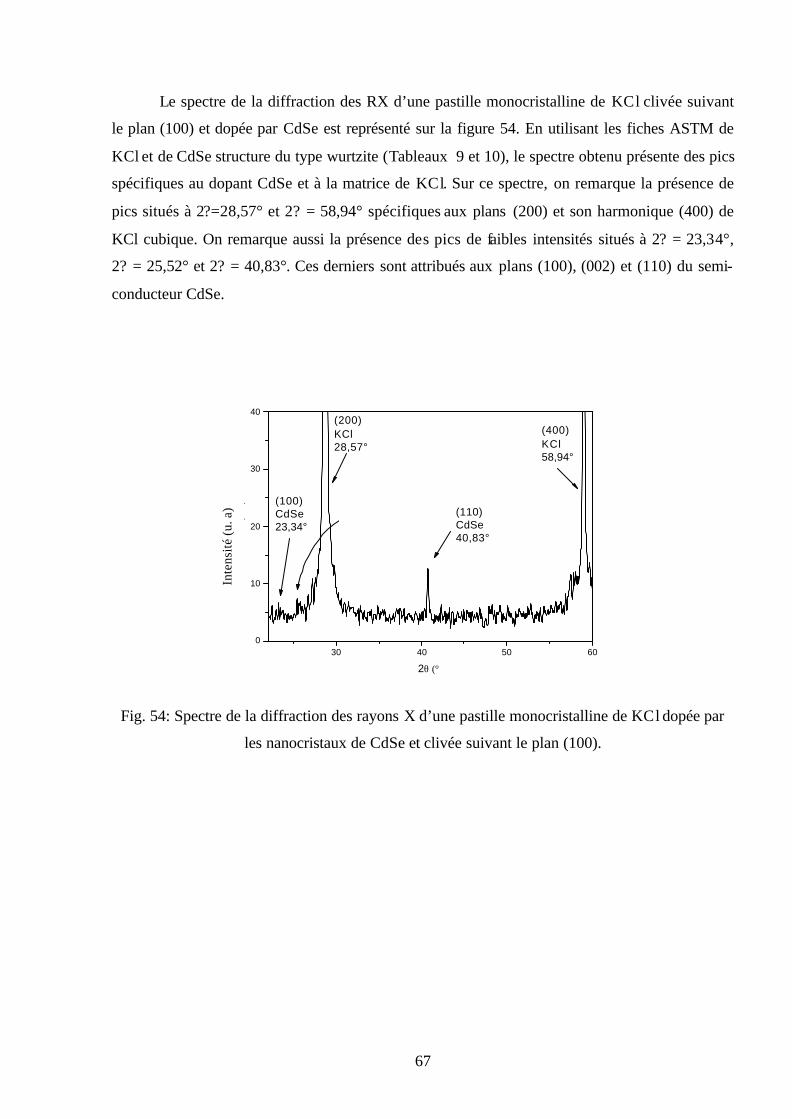
67
Le spectre de la diffraction des RX d’une pastille monocristalline de KCl clivée suivant
le plan (100) et dopée par CdSe est représenté sur la figure 54. En utilisant les fiches ASTM de
KCl et de CdSe structure du type wurtzite (Tableaux 9 et 10), le spectre obtenu présente des pics
spécifiques au dopant CdSe et à la matrice de KCl. Sur ce spectre, on remarque la présence de
pics situés à 2?=28,57° et 2? = 58,94° spécifiques aux plans (200) et son harmonique (400) de
KCl cubique. On remarque aussi la présence des pics de faibles intensités situés à 2? = 23,34°,
2? = 25,52° et 2? = 40,83°. Ces derniers sont attribués aux plans (100), (002) et (110) du semi-
conducteur CdSe.
30 40 50 600
10
20
30
40
Inte
sité
(c/
s)
2θ (°)
(100)CdSe23,34°
(002)CdSe25,52°
(110)CdSe40,83°
(200)KCl28,57°
(400)KCl58,94°
Fig. 54: Spectre de la diffraction des rayons X d’une pastille monocristalline de KCl dopée par
les nanocristaux de CdSe et clivée suivant le plan (100).
Inte
nsité
(u. a
)
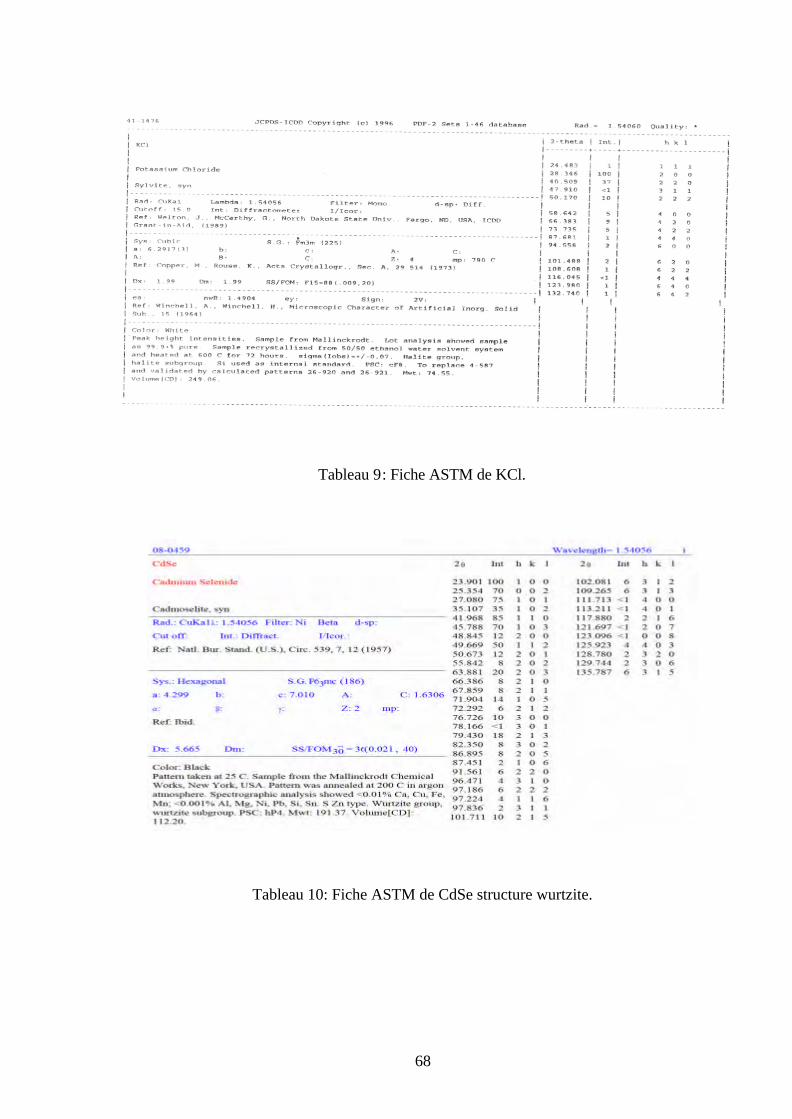
68
Tableau 9: Fiche ASTM de KCl.
Tableau 10: Fiche ASTM de CdSe structure wurtzite.
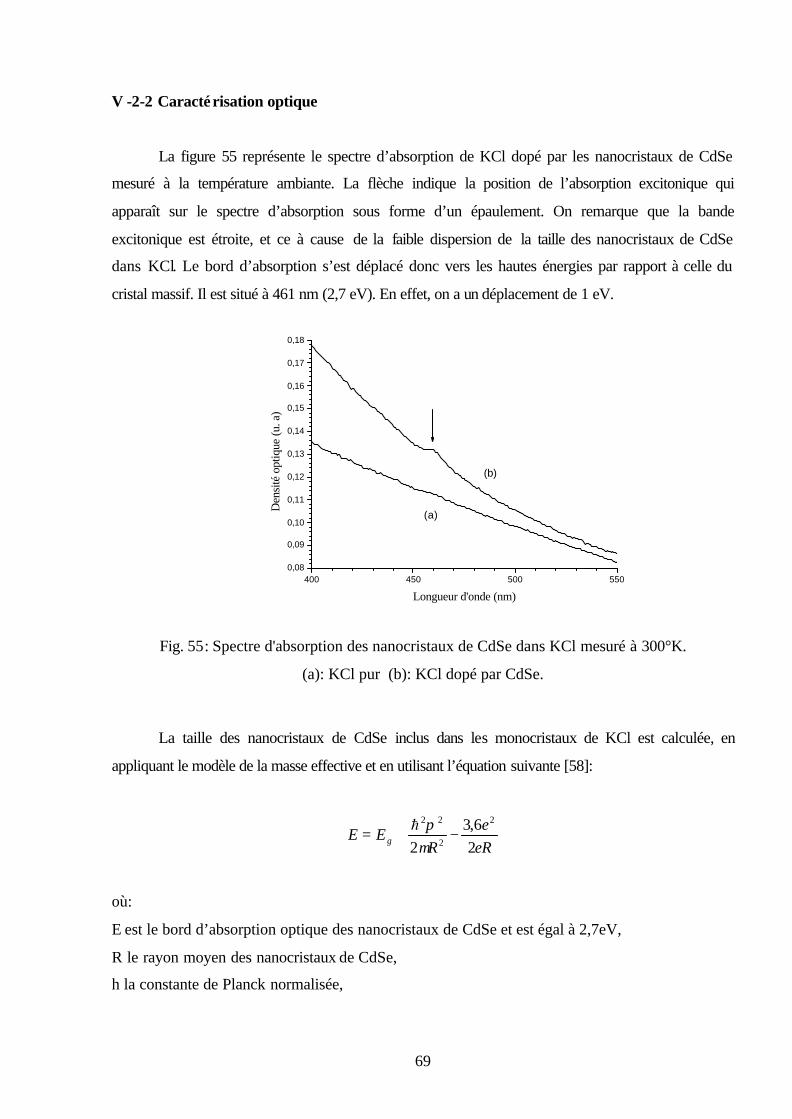
69
V -2-2 Caractérisation optique
La figure 55 représente le spectre d’absorption de KCl dopé par les nanocristaux de CdSe
mesuré à la température ambiante. La flèche indique la position de l’absorption excitonique qui
apparaît sur le spectre d’absorption sous forme d’un épaulement. On remarque que la bande
excitonique est étroite, et ce à cause de la faible dispersion de la taille des nanocristaux de CdSe
dans KCl. Le bord d’absorption s’est déplacé donc vers les hautes énergies par rapport à celle du
cristal massif. Il est situé à 461 nm (2,7 eV). En effet, on a un déplacement de 1 eV.
400 450 500 5500,08
0,09
0,10
0,11
0,12
0,13
0,14
0,15
0,16
0,17
0,18
(b)
(a)
Den
sité
opt
ique
(u. a
)
Longueur d'onde (nm)
Fig. 55: Spectre d'absorption des nanocristaux de CdSe dans KCl mesuré à 300°K.
(a): KCl pur (b): KCl dopé par CdSe.
La taille des nanocristaux de CdSe inclus dans les monocristaux de KCl est calculée, en
appliquant le modèle de la masse effective et en utilisant l’équation suivante [58]:
Re
REE g εµ
π26,3
2
2
2
22
−+=h
où:
E est le bord d’absorption optique des nanocristaux de CdSe et est égal à 2,7eV,
R le rayon moyen des nanocristaux de CdSe,
h la constante de Planck normalisée,
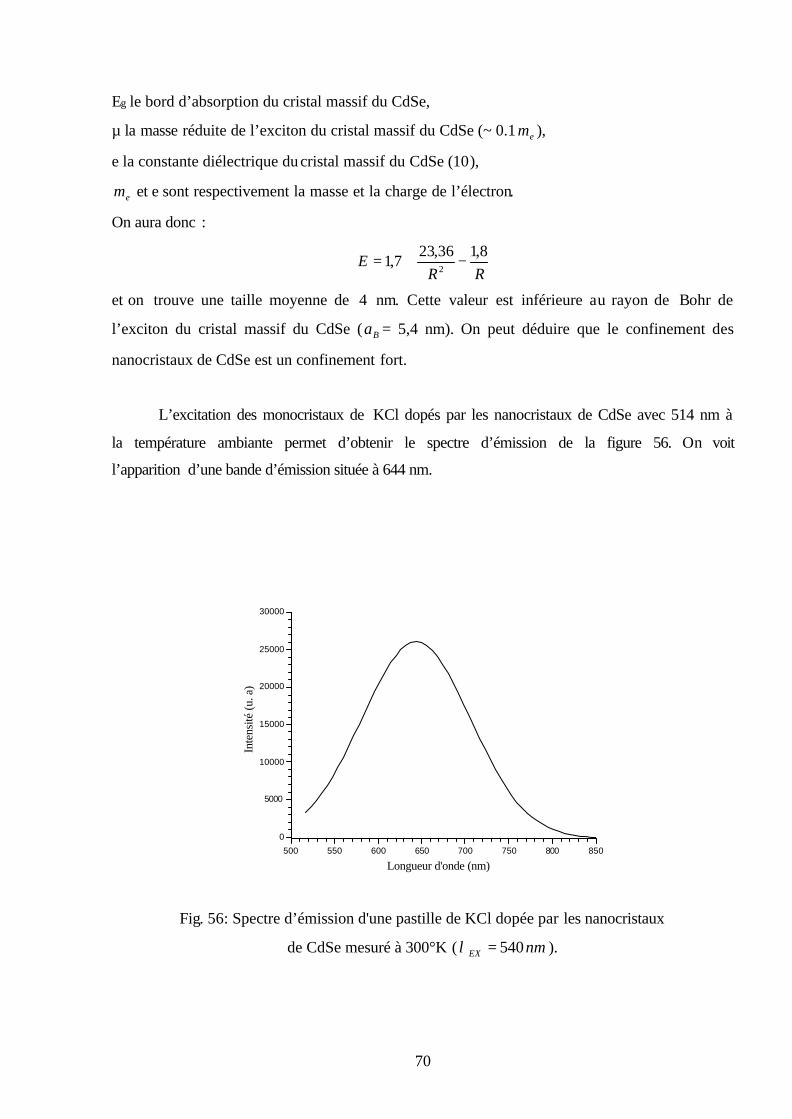
70
Eg le bord d’absorption du cristal massif du CdSe,
µ la masse réduite de l’exciton du cristal massif du CdSe (~ 0.1 em ),
e la constante diélectrique du cristal massif du CdSe (10),
em et e sont respectivement la masse et la charge de l’électron.
On aura donc :
RRE
8,136,237,1
2−+=
et on trouve une taille moyenne de 4 nm. Cette valeur est inférieure au rayon de Bohr de
l’exciton du cristal massif du CdSe ( Ba = 5,4 nm). On peut déduire que le confinement des
nanocristaux de CdSe est un confinement fort.
L’excitation des monocristaux de KCl dopés par les nanocristaux de CdSe avec 514 nm à
la température ambiante permet d’obtenir le spectre d’émission de la figure 56. On voit
l’apparition d’une bande d’émission située à 644 nm.
500 550 600 650 700 750 800 850
0
5000
10000
15000
20000
25000
30000
Inte
nsité
(u. a
)
Longueur d'onde (nm)
Fig. 56: Spectre d’émission d'une pastille de KCl dopée par les nanocristaux
de CdSe mesuré à 300°K ( nmEX 540=λ ).

71
V- 3 CONCLUSION
Les résultats des mesures optiques obtenus à partir des échantillons de KBr, KCl et NaCl
élaborés sous atmosphère libre ont montré l’introduction de l’oxygène dans les monocristaux et
la formation des centres colorés O2- et (O2- – F+).
Ces mesures ont montré les émissions optiques des centres colorés O2- dans les trois
matrices et la variation de la position de la ligne ordinaire la plus intense avec la température de
mesure.
Nous avons montré aussi par étude de la photoluminescence (PL) et l’excitation de la
photoluminescence (PLE), les nouvelles bandes d’absorption des centres colorés (O2- – F+)
situées dans l’UV et leurs nouvelles émissions situées dans l’UV-visible.
Le recuit thermique des échantillons à la température 650°C pendant 24 h montre la
transformation à haute température des centres O2- en centres (O2- – F+) à cause de la présence
des centres F. Ceci était confirmé par l’étude des spectres de la PL avant et après le recuit des
échantillons et justifie la relation de transformation obtenue par F. Fischer et al.
La mesure de l'absorption optique des monocristaux de KCl dopés par les nanocristaux
du semi-conducteur CdSe a montré l’effet du confinement sur leurs propriétés optiques dont le
bord d’absorption s’est déplacé vers les grandes énergies par comparaison avec le cristal massif.

72
CONCLUSION GENERALE
Au cours de ce travail, nous nous sommes intéressés à l’étude des centres colorés O2- et
(O2- - F+) dans les cristaux de KBr, KCl et NaCl, et des nanocristaux du semi-conducteur CdSe
dispersés dans KCl. Ceci nous a permis une meilleure compréhension de certaines propriétés
optiques de ces matériaux.
L’étude des centres colorés a nécessité l’élaboration des cristaux de KBr, KCl et NaCl.
Ces derniers ont été élaborés par la méthode de croissance de Czochralski sous atmosphère libre
qui nous a permis aussi l’introduction de l’oxygène aux cours de la croissance à partir de
l’atmosphère. Ceci a été confirmé par les mesures de la photoluminescence et l’excitation de la
photoluminescence.
Les mesures de l’absorption optique à la température ambiante montrent des bandes
situées à 215 nm dans KBr et à 193 nm dans KCl spécifique aux centres OH-. Ces mesures ont
montré aussi l’existence de quelques bandes d’absorption situées dans l’UV dans les trois
matrices. Elles sont attribuées aux centres colorés (O2- - F+).
L’étude des spectres d’émission dans le domaine spectral 400-800 nm à 300°K et 78°K a
montré les lignes d’émission des centres O2- dans les trois cristaux. A basse température ces
émissions sont formées de la structure simple ou seulement des lignes ordinaires dans KBr et
NaCl. Alors que dans KCl, elles sont formées de la double structure ou des lignes ordinaires et
des lignes particulières. L’étude de la ligne ordinaire la plus intense a montré que sa position
dépend de la température de mesure dans les trois matrices. En effet, elle se déplace vers le grand
nombre quantique voisin lorsque la température de mesure diminue.
Une étude comparative entre les émissions du centre O2-, obtenues dans les trois cristaux,
a montré que les intensités des lignes ordinaires dans NaCl sont très faibles par rapport à celles
obtenues dans KBr et KCl. Ce qui explique la faible concentration d’O2- dans cette matrice. Par
conséquent, l’introduction de l’oxygène de l’air dans NaCl est difficile par rapport à KBr et KCl.

73
L’excitation des échantillons entre 330-390 nm à 300°K et 78°K a permis d’obtenir les mêmes
émissions dans les trois cristaux. A 300°K, ces émissions présentent une large bande située entre
400-500 nm. Alors qu’à 78°K, il apparaît de plus une nouvelle bande située à 548 nm.
L’étude des bandes d’émission situées entre 300-450 nm à 78°K a montré plusieurs
bandes d’émission dans les trois cristaux. La mesure de l’excitation de la photoluminescence
(PLE) a montré que ces émissions sont dues aux centres (O2- - F+).
Les mesures de la photoluminescence (PL) des échantillons qui ont subi le traitement
thermique ont montré la diminution et parfois la disparition totale des bandes d'émission des
centres O2-. Tandis que les bandes d’émission des centres (O2- - F+) augmentent. Ceci confirme
la relation de transformation des centres O2- en centres (O2- - F+) introduite par Fischer.
Une étude comparative avec les résultats de G. Gümmer nous a permis de montrer de
nouvelles bandes d’émission de centre (O2- - F+) dans KBr, KCl et NaCl situées dans l’UV-
visible. Ces émissions sont obtenues à 300°K et 78°K.
L’élaboration des cristaux de KCl dopés par les nanocristaux du semi-conducteur CdSe
est effectuée par la méthode de croissance de Czochralski. L’incorporation de CdSe dans KCl a
été confirmée par la diffraction des rayons X dont le spectre a présenté des pics spécifiques au
semi-conducteur CdSe et à la matrice cristalline de KCl.
Les mesures de l’absorption optique dans le domaine spectral UV-visible à température
ambiante des cristaux de KCl après dopage, ont montré le déplacement du bord d’absorption des
nanocristaux de CdSe vers les courtes longueurs d’ondes en comparaison avec le cristal massif,
alors que l’excitation de ces échantillons à la température ambiante, a permis l’obtention d’une
bande d’émission située à 644 nm.
Etudier l’effet laser du centre coloré pourra constituer un volet de recherche important vu
l’utilité croissante des lasers dans la technologie moderne. Ce qui fera l’objet de travaux de
recherche dans le cadre de projets effectués au sein du laboratoire LEM de l’université de Jijel et
le laboratoire de cristallographie de l’université Mentouri Constantine

74
REFERENCES
[1] A. Ejiri, A. Hatano, Inst. Phys. Conf. Ser. 193, p. 77 (1992).
[2] Frederick Seltz, Rev. Mod. Phys. 18, No. 3, p. 384 (1946).
[3] Marc Boccanfuso, Thèse de doctorat d’état, Université de Caen (2001).
[4] A. Akrimi, Thèse de doctorat d’état, Université d’Annaba (1994).
[5] C. Z. Vandoorn, Philips. Rpts. Suppl. N° 4 (1962).
[6] C. Pollock, J. Lum. 35, p. 65 (1986).
[7] Henry F. Ivey, Phys. Rev. 72, p. 341 (1947).
[8] J. Rolfe, Phys. Rev. Letters. 1, p. 56 (1958).
[9] M. Aegerter, F. Lüty, Phys. Stat. Solidi. 48, p. 227 (1971).
[10] D. Wandt, W. Gellermann, F. Lüty, H. Welling, ‘’Conference on Tunable Solid Stat
Lasers’’, Zigzog. Oregon (1986).
[11] J. Pinto, E. Efstrastios, C. Pollock, Opt. Lett. 11, p. 519 (1986).
[12] W. Gellermann, J. Phys. Chem. Sol. 52, p. 249 (1991).
[13] D. Wandt, W. Gellermann, Opt. Com. 61, p. 405 (1987).
[14] K. Mollmann, F. Mitschke, W. Gellermann, Opt. Com. 83, p. 177 (1991).
[15] T. Castner, W. Känzig, J. Phys. Chem. Solid. 3, p. 178 (1957).
[16] H. W. Etzel and D. A. Patterson, Phys. Rev. 112, p. 1112 (1958).
[17] W. Honrath, Ann. Physik. 29, p. 421 (1937).
[18] J. Rolfe, F. R. Lipsett and W. J. King, Phys. Rev. 123, p. 447 (1961).
[19] W. Sander, Z. Physik. 169, p. 353 (1962).
[20] R. Florian, L. O. Schwan and D. Schmid, Sol. Stat. Com. 42, p. 55 (1982).
[21] R. Florian, L. O. Schwan and D. Schmid, Phys. Rev A. 29, p. 2709 (1984).
[22] R. Florian, L. O. Schwan and D. Schmid, J. Lum. 31 & 32, p. 169 (1984).

75
[23] M. S. Malcuit, Jeffery J. Maki, D. J. Simkin and R. W. Boyd, Phys. Rev. Letters. 59,
p. 1189 (1986).
[24] M. Ashida, O. Morikawa, H. Arai and R. Kato, Prog. Crystal Growth and Charact. 33,
p. 105 (1996).
[25] M. Ashida, H. Arai, O. Morikawa and R. Kato, J. Lum. 72-74, p. 624 (1997).
[26] David A. Patteron, Phys. Rev. 127, p. 1564 (1962).
[27] W. Känzig and M. H. Cohen, Phys. Rev. Letters. 3, p. 509 (1959).
[28] J. Rolfe, J. Chem. Phys. 40, p. 1664 (1963).
[29] R. Pirc, B. Zeks and P. Gasar, J. Phys. Chem. Solids. 27, p. 1219 (1966).
[30] J. Muggli and H. U. Beyeler, Solid. Stat. Com. 8, p. 217 (1970).
[31] E. Lawther, Chem. Phys. Letters. 35, p. 136 (1975).
[32] M. Ikezawa and J. Rolfe. J. Chem. Phys. 58, p. 2024 (1973).
[33] J. Rolfe, M. Ikezawa and T. Timusk, Phys. Rev B. 7, p. 3913 (1973).
[34] R. Cywinski and J. Z. Damm, J. Lum. 27, p.327 (1982).
[35] W. H. Knox and K. J. Teegarden, J. Lum. 31 & 32, p. 39 (1984).
[36] O. Morikawa and R. Kato, J. Lum. 59, p. 19 (1994).
[37] S. Sogo, O. Morikawa, A. Kato and R. Kato, J. Lum. 65, p. 77 (1995).
[38] K. Rebane and L. Rebane, Sov. Phys. Solid. Stat. 12, p. 154 (1971).
[39] H. R. Zeller and W. Känzig, Helv. Phys. Acta. 40, p. 845 (1965).
[40] S. Hongo, H. Murata and R. Kato, J. Lum. 48 & 49, p. 807 (1991).
[41] F. Fischer, H. Grundig und R. Hilsch, Z. Physik. 189, p. 79 (1966).
[42] F. Fischer und G. Gümmer, Z. Physik. 189, p. 97 (1966).
[43] Günter Gümmer, Z. Physik. 215, p. 256 (1968).
[44] E. Georgiou, J. F. Pinto and C. R. Pollock, Phys. Rev B. 35, p. 7636 (1987).
[45] E. Georgiou, C. R. Pollock, Phys. Rev B. 39, p. 10412 (1981).

76
[46] G. Liffante, P. Silfsten and F. Cusso, Phys. Rev B. 40, p. 9925 (1989).
[47] D. Wandt, W. Gellerman, F. Lüty and H. Wellin, J. Appl. Phys. 61, p. 869 (1987).
[48] F. Lüty, Z. Physik. 160, p. 1 (1960).
[49] H. Hirai and M. Ueta, J. Phys. Soc. Japan. 17, p. 566 (1962).
[50] C. C. Klick and M. Kabler, Phys. Rev. 131, p. 1075 (1963).
[51] R. Hilsch and R. W. Pohl, Z. Physik. 57, p. 145 (1929).
[52] T. Timusk and W. Martienssen, Phys. Rev. 128, p. 1656 (1962).
[53] Thierry Cloitre, Thèse de doctorat d’Etat, Université de Montpellier II (1992).
[54] P.Baranski, V. Klotehkov, I. Potykeritch, ’’Electronique des semi-conducteurs’’,
ED. Mr. Moscou (1978).
[55] Françoise Paille, Thèse de doctorat , Université de Lyon 1 (1997).
[56] S. Schmitt-Rink, D. S. Chemla, D. A. B. Miller, Adv. in Phys. 38, p. 89 (1989).
[57] Philippe Riblet, Thèse de doctorat d’Etat, Université de Strasbourg I (1995).
[58] Boubekeur Boudine, Thèse de doctorat d'état, Université de Mentouri Constantine (2005).
[59] Paul Faller, Thèse de doctorat d’Etat, Université de Strasbourg I (1994).
[60] Horst Weller, Tobias Vossmeyer, Alexander Egchmüller, Alf Mews, Lynne Katsikas,
Günter Reek, Mat. Res. Soc. Symp. Proc. 385, p. 213 (1995).
[61] E. Voronkova, B. Gretsouchnikov, G. Distter, ‘’Matériaux optiques pour les techniques
d’infrarouge’’, Ed. Moscou (1962).
[62] W. Bardsely, D. T. J. Burle, L. B. Mullin, ‘’Crystal growth’’, North- Hollond (1979).










![chapitre -5- PROPORTIONNALITE [A] RECHERCHE (fiche n°150) jeudi 2 avril 2015 exemples recherche dans un tableau recherche sur un graphique recherche](https://img.pdfslide.fr/doc/110x75/551d9de0497959293b8ea3c0/chapitre-5-proportionnalite-a-recherche-fiche-n150-jeudi-2-avril-2015-exemples-recherche-dans-un-tableau-recherche-sur-un-graphique-recherche.jpg)


















