Embed Size (px)
Citation preview

PCEM2 Fiches
Génétique
Médicale
By Puyraimond-Zemmour Jérémy ©

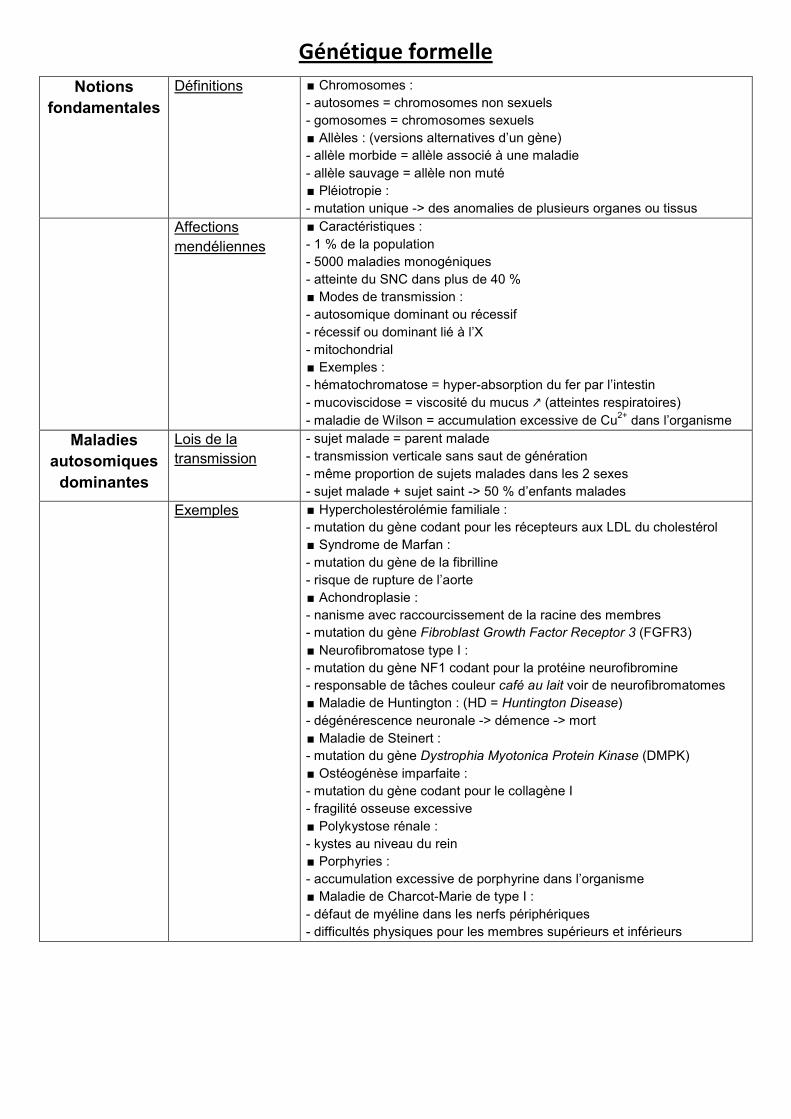
Génétique formelle
Notions
fondamentales
Définitions ∎ Chromosomes :
- autosomes = chromosomes non sexuels
- gomosomes = chromosomes sexuels
∎ Allèles : (versions alternatives d’un gène)
- allèle morbide = allèle associé à une maladie
- allèle sauvage = allèle non muté
∎ Pléiotropie :
- mutation unique -> des anomalies de plusieurs organes ou tissus
Affections
mendéliennes
∎ Caractéristiques :
- 1 % de la population
- 5000 maladies monogéniques
- atteinte du SNC dans plus de 40 %
∎ Modes de transmission :
- autosomique dominant ou récessif
- récessif ou dominant lié à l’X
- mitochondrial
∎ Exemples :
- hématochromatose = hyper-absorption du fer par l’intestin
- mucoviscidose = viscosité du mucus ↗ (atteintes respiratoires)
- maladie de Wilson = accumulation excessive de Cu2+
dans l’organisme
Maladies
autosomiques
dominantes
Lois de la
transmission
- sujet malade = parent malade
- transmission verticale sans saut de génération
- même proportion de sujets malades dans les 2 sexes
- sujet malade + sujet saint -> 50 % d’enfants malades
Exemples ∎ Hypercholestérolémie familiale :
- mutation du gène codant pour les récepteurs aux LDL du cholestérol
∎ Syndrome de Marfan :
- mutation du gène de la fibrilline
- risque de rupture de l’aorte
∎ Achondroplasie :
- nanisme avec raccourcissement de la racine des membres
- mutation du gène Fibroblast Growth Factor Receptor 3 (FGFR3)
∎ Neurofibromatose type I :
- mutation du gène NF1 codant pour la protéine neurofibromine
- responsable de tâches couleur café au lait voir de neurofibromatomes
∎ Maladie de Huntington : (HD = Huntington Disease)
- dégénérescence neuronale -> démence -> mort
∎ Maladie de Steinert :
- mutation du gène Dystrophia Myotonica Protein Kinase (DMPK)
∎ Ostéogénèse imparfaite :
- mutation du gène codant pour le collagène I
- fragilité osseuse excessive
∎ Polykystose rénale :
- kystes au niveau du rein
∎ Porphyries :
- accumulation excessive de porphyrine dans l’organisme
∎ Maladie de Charcot-Marie de type I :
- défaut de myéline dans les nerfs périphériques
- difficultés physiques pour les membres supérieurs et inférieurs
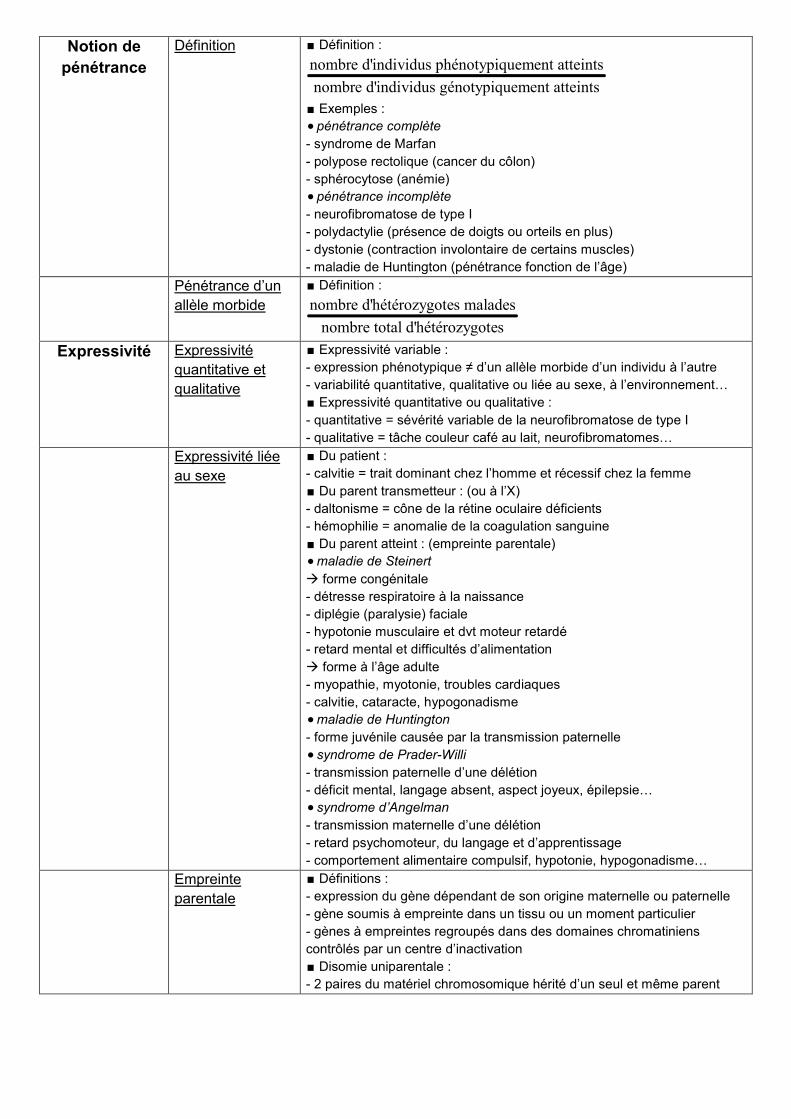
Notion de
pénétrance
Définition ∎ Définition :
nombre d'individus phénotypiquement atteints
nombre d'individus génotypiquement atteints
∎ Exemples :
pénétrance complète
- syndrome de Marfan
- polypose rectolique (cancer du côlon)
- sphérocytose (anémie)
pénétrance incomplète
- neurofibromatose de type I
- polydactylie (présence de doigts ou orteils en plus)
- dystonie (contraction involontaire de certains muscles)
- maladie de Huntington (pénétrance fonction de l’âge)
Pénétrance d’un
allèle morbide
∎ Définition :
nombre d'hétérozygotes malades
nombre total d'hétérozygotes
Expressivité Expressivité
quantitative et
qualitative
∎ Expressivité variable :
- expression phénotypique ≠ d’un allèle morbide d’un individu à l’autre
- variabilité quantitative, qualitative ou liée au sexe, à l’environnement…
∎ Expressivité quantitative ou qualitative :
- quantitative = sévérité variable de la neurofibromatose de type I
- qualitative = tâche couleur café au lait, neurofibromatomes…
Expressivité liée
au sexe
∎ Du patient :
- calvitie = trait dominant chez l’homme et récessif chez la femme
∎ Du parent transmetteur : (ou à l’X)
- daltonisme = cône de la rétine oculaire déficients
- hémophilie = anomalie de la coagulation sanguine
∎ Du parent atteint : (empreinte parentale)
maladie de Steinert
forme congénitale
- détresse respiratoire à la naissance
- diplégie (paralysie) faciale
- hypotonie musculaire et dvt moteur retardé
- retard mental et difficultés d’alimentation
forme à l’âge adulte
- myopathie, myotonie, troubles cardiaques
- calvitie, cataracte, hypogonadisme
maladie de Huntington
- forme juvénile causée par la transmission paternelle
syndrome de Prader-Willi
- transmission paternelle d’une délétion
- déficit mental, langage absent, aspect joyeux, épilepsie…
syndrome d’Angelman
- transmission maternelle d’une délétion
- retard psychomoteur, du langage et d’apprentissage
- comportement alimentaire compulsif, hypotonie, hypogonadisme…
Empreinte
parentale
∎ Définitions :
- expression du gène dépendant de son origine maternelle ou paternelle
- gène soumis à empreinte dans un tissu ou un moment particulier
- gènes à empreintes regroupés dans des domaines chromatiniens
contrôlés par un centre d’inactivation
∎ Disomie uniparentale :
- 2 paires du matériel chromosomique hérité d’un seul et même parent
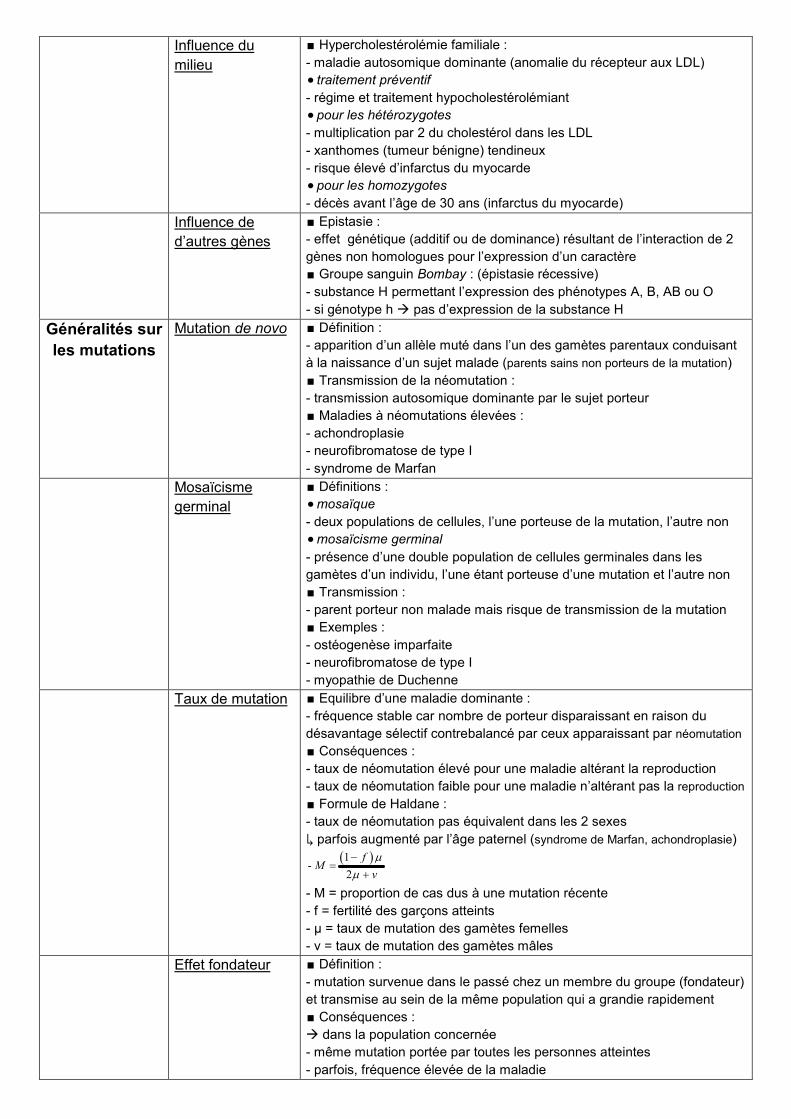
Influence du
milieu
∎ Hypercholestérolémie familiale :
- maladie autosomique dominante (anomalie du récepteur aux LDL)
traitement préventif
- régime et traitement hypocholestérolémiant
pour les hétérozygotes
- multiplication par 2 du cholestérol dans les LDL
- xanthomes (tumeur bénigne) tendineux
- risque élevé d’infarctus du myocarde
pour les homozygotes
- décès avant l’âge de 30 ans (infarctus du myocarde)
Influence de
d’autres gènes
∎ Epistasie :
- effet génétique (additif ou de dominance) résultant de l’interaction de 2
gènes non homologues pour l’expression d’un caractère
∎ Groupe sanguin Bombay : (épistasie récessive)
- substance H permettant l’expression des phénotypes A, B, AB ou O
- si génotype h pas d’expression de la substance H
Généralités sur
les mutations
Mutation de novo ∎ Définition :
- apparition d’un allèle muté dans l’un des gamètes parentaux conduisant
à la naissance d’un sujet malade (parents sains non porteurs de la mutation)
∎ Transmission de la néomutation :
- transmission autosomique dominante par le sujet porteur
∎ Maladies à néomutations élevées :
- achondroplasie
- neurofibromatose de type I
- syndrome de Marfan
Mosaïcisme
germinal
∎ Définitions :
mosaïque
- deux populations de cellules, l’une porteuse de la mutation, l’autre non
mosaïcisme germinal
- présence d’une double population de cellules germinales dans les
gamètes d’un individu, l’une étant porteuse d’une mutation et l’autre non
∎ Transmission :
- parent porteur non malade mais risque de transmission de la mutation
∎ Exemples :
- ostéogenèse imparfaite
- neurofibromatose de type I
- myopathie de Duchenne
Taux de mutation ∎ Equilibre d’une maladie dominante :
- fréquence stable car nombre de porteur disparaissant en raison du
désavantage sélectif contrebalancé par ceux apparaissant par néomutation
∎ Conséquences :
- taux de néomutation élevé pour une maladie altérant la reproduction
- taux de néomutation faible pour une maladie n’altérant pas la reproduction
∎ Formule de Haldane :
- taux de néomutation pas équivalent dans les 2 sexes
↳ parfois augmenté par l’âge paternel (syndrome de Marfan, achondroplasie)
1-
2
fM
v
- M = proportion de cas dus à une mutation récente
- f = fertilité des garçons atteints
- μ = taux de mutation des gamètes femelles
- ν = taux de mutation des gamètes mâles
Effet fondateur ∎ Définition :
- mutation survenue dans le passé chez un membre du groupe (fondateur)
et transmise au sein de la même population qui a grandie rapidement
∎ Conséquences :
dans la population concernée
- même mutation portée par toutes les personnes atteintes
- parfois, fréquence élevée de la maladie
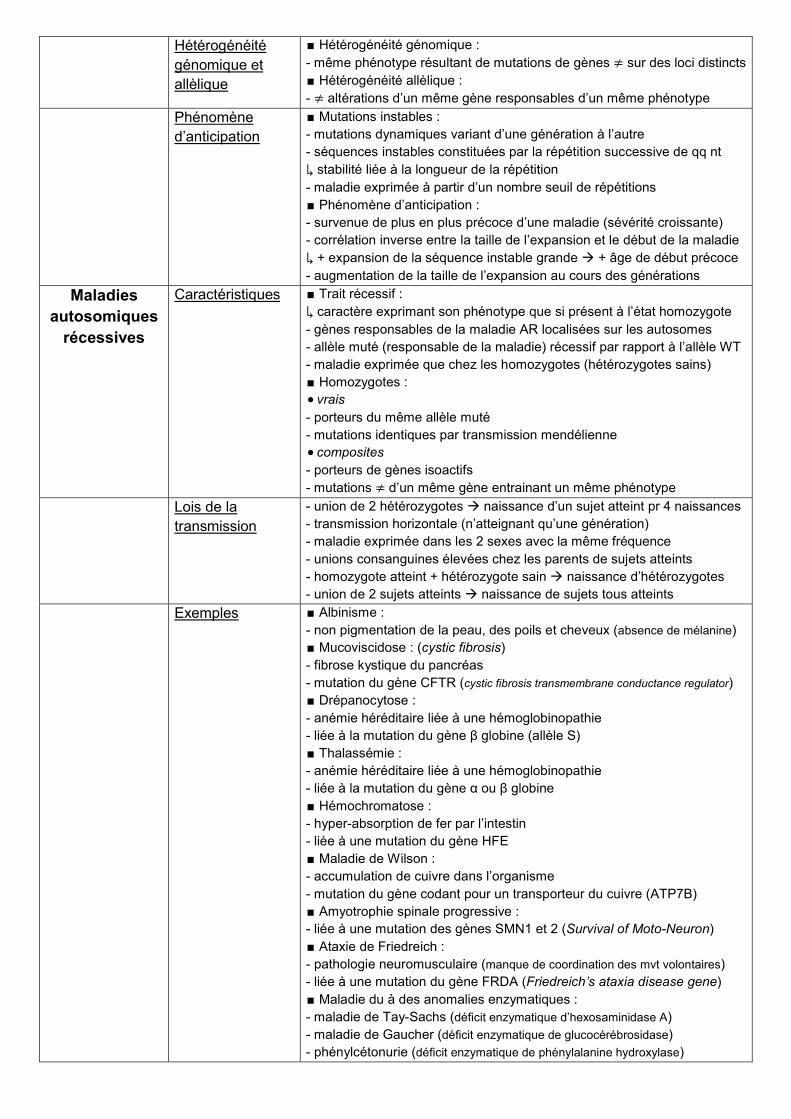
Hétérogénéité
génomique et
allèlique
∎ Hétérogénéité génomique :
- même phénotype résultant de mutations de gènes ≠ sur des loci distincts
∎ Hétérogénéité allèlique :
- ≠ altérations d’un même gène responsables d’un même phénotype
Phénomène
d’anticipation
∎ Mutations instables :
- mutations dynamiques variant d’une génération à l’autre
- séquences instables constituées par la répétition successive de qq nt
↳ stabilité liée à la longueur de la répétition
- maladie exprimée à partir d’un nombre seuil de répétitions
∎ Phénomène d’anticipation :
- survenue de plus en plus précoce d’une maladie (sévérité croissante)
- corrélation inverse entre la taille de l’expansion et le début de la maladie
↳ + expansion de la séquence instable grande + âge de début précoce
- augmentation de la taille de l’expansion au cours des générations
Maladies
autosomiques
récessives
Caractéristiques ∎ Trait récessif :
↳ caractère exprimant son phénotype que si présent à l’état homozygote
- gènes responsables de la maladie AR localisées sur les autosomes
- allèle muté (responsable de la maladie) récessif par rapport à l’allèle WT
- maladie exprimée que chez les homozygotes (hétérozygotes sains)
∎ Homozygotes :
vrais
- porteurs du même allèle muté
- mutations identiques par transmission mendélienne
composites
- porteurs de gènes isoactifs
- mutations ≠ d’un même gène entrainant un même phénotype
Lois de la
transmission
- union de 2 hétérozygotes naissance d’un sujet atteint pr 4 naissances
- transmission horizontale (n’atteignant qu’une génération)
- maladie exprimée dans les 2 sexes avec la même fréquence
- unions consanguines élevées chez les parents de sujets atteints
- homozygote atteint + hétérozygote sain naissance d’hétérozygotes
- union de 2 sujets atteints naissance de sujets tous atteints
Exemples ∎ Albinisme :
- non pigmentation de la peau, des poils et cheveux (absence de mélanine)
∎ Mucoviscidose : (cystic fibrosis)
- fibrose kystique du pancréas
- mutation du gène CFTR (cystic fibrosis transmembrane conductance regulator)
∎ Drépanocytose :
- anémie héréditaire liée à une hémoglobinopathie
- liée à la mutation du gène β globine (allèle S)
∎ Thalassémie :
- anémie héréditaire liée à une hémoglobinopathie
- liée à la mutation du gène α ou β globine
∎ Hémochromatose :
- hyper-absorption de fer par l’intestin
- liée à une mutation du gène HFE
∎ Maladie de Wilson :
- accumulation de cuivre dans l’organisme
- mutation du gène codant pour un transporteur du cuivre (ATP7B)
∎ Amyotrophie spinale progressive :
- liée à une mutation des gènes SMN1 et 2 (Survival of Moto-Neuron)
∎ Ataxie de Friedreich :
- pathologie neuromusculaire (manque de coordination des mvt volontaires)
- liée à une mutation du gène FRDA (Friedreich’s ataxia disease gene)
∎ Maladie du à des anomalies enzymatiques :
- maladie de Tay-Sachs (déficit enzymatique d’hexosaminidase A)
- maladie de Gaucher (déficit enzymatique de glucocérébrosidase)
- phénylcétonurie (déficit enzymatique de phénylalanine hydroxylase)
Loi de Hardy-
Weinberg
∎ Définition :
- probabilité de survenue de 2 évènements = produits de probabilité de
chaque évènement : (p + q)² = p² + q² + 2pq
- loi vérifiée dans une population panmictique, sans mutation ni migration
∎ Equilibre de Hardy-Weinberg :
fréquence des gènes constante d’une génération à l’autre si
- population grande avec des unions au hasard (panmixie)
- pas de sélection, pas de néomutation, pas de migration
Consanguinité ∎ Définitions :
- personne consanguine = personne issue de l’union de sujets apparentés
- coeff de consanguinité = prob. que 2 gènes en un locus soient identiques
∎ Conséquence :
- augmentation du risque d’avoir des enfants atteints d’une maladie AR
- proportions d’enfants atteints nés d’union consanguines plus élevée dans
les maladies AR dont le gène est rare
Maladies
récessives
liées à l’X
Caractéristiques - trait déterminé par le chromosome X se manifestant à l’état homozygote
(chez les femmes atteintes) ou hémizygote (chez les hommes atteints)
- femme conductrice = femmes hétérozygotes non atteintes transmettant
la maladie à leur fils (car garçons hémizygotes atteints)
Lois de la
transmission
∎ Femme conductrice avec un homme sain :
- 50 % de garçons atteints et 50 % de filles conductrices
∎ Femme saine avec un homme atteint :
- 100 % de garçons sains et 50 % de filles conductrices
∎ Femme conductrice avec un homme atteint :
- 50 % de garçons atteints et 50 % de garçons sains
- 50 % de filles atteintes et 50 % de filles conductrices
Atteinte fréquente
des hommes
∎ Fréquences : (q = fréquence de l’allèle Xa)
- fréquence des hommes atteints = q (XaY)
- fréquence des femmes atteintes = q² (XaX
a)
∎ Femmes atteintes si :
- homozygotie (XaX
a)
- syndrome de Turner (un chromosome X en moins)
- testicule féminisant (XaY)
- inactivation de l’X normal (phénomène de lyonisation)
∎ Dépistage des femmes conductrices :
- recherche de signes cliniques ou biologiques de l’affection en cause
↳ symptômes de la maladie présents chez quelques femmes conductrices
- analyse génétique moléculaire (quand gène et mutation connus)
Exemples ∎ Myopathie de Duchenne et Becker :
- mutation du gène de la dystrophine
∎ Hémophilies A et B :
- dysfonctionnement d’un des facteurs de coagulation
∎ Syndrome de l’X fragile :
- mutation du gène FMR (Fragility Mental Retardation)
∎ Maladie de Lesh-Nyhan :
- déficit de fonctionnement d’une enzyme spécifique
∎ Choroïdérémie :
- dégénérescence des vaisseaux choroïdiens de l’œil
∎ Favisme :
- déficit en glucose 6 phosphate déshydrogénase
- éviter de manger certain aliments oxydants (fèves)
∎ Adréno-leucodystrophie :
- démyélinisation progressive du système nerveux central
∎ Daltonisme :
- anomalie touchant les cônes de la rétine oculaire
∎ Diabète insipide néphrogénique :
- impossibilité pour les reins de concentrer les urines
Maladies
dominantes
liées à l’X
Caractéristiques - trait déterminé par le chromosome X se manifestant chez les hommes
(H) hémizygotes ou chez les femmes (F) hétérozygotes
- F hétérozygotes moins sévèrement touchées que les H hémizygotes
- transmise par les F atteintes à leurs enfants des 2 sexes (risque 50 %)
- transmise par les H atteints à toutes les filles mais pas les garçons
Lois de la
transmission
∎ Femme atteinte avec un homme sain :
- 50 % de garçons atteints et 50 % de filles atteintes
∎ Femme saine avec un homme atteint :
- 100 % de garçons sains (pas de transmission père-fils)
- 100 % de filles atteintes
Exemples ∎ Rachitisme résistant à la vitamine D :
- mutation du gène codant pour le récepteur de la vitamine D
∎ Maladie de Charcot-Marie-Tooth type X :
- défaut de myéline dans les nerfs périphériques
- difficultés physiques pour les membres supérieurs et inférieurs
∎ Syndrome oro-facio-digital :
- anomalies de la bouche, du visage, des doigts et des orteils
∎ Déficit en ornithine transcarbamylase :
- transport mitochondrial de l’ornithine affecté
∎ Incontinentia pigmenti :
- lésions cutanées (incontinence pigmentaire), atteintes viscérales…
- parfois létale chez le garçon
Pathologie
mitochondriale
Caractéristiques - ADN circulaire de 16 000 paires de base, sans introns (500 copies / c)
- 37 gènes codant pour des protéines, des ARNt et des ARNr
- protéines mitochondriales provenant en partie de l’ADN mitochondrial,
mais aussi de l’ADN nucléaire
- multiplication des mitochondries indépendamment du cycle cellulaire
(pendant S et G2) en fonction de la demande en énergie de la cellule
Transmission non
mendélien de
l’ADN
∎ Hérédité cytoplasmique :
- mitochondries maternelles (de l’ovocyte) transmises à l’enfant
- transmission monoparentale (maternelle) de l’ADN mitochondrial
∎ Hétéroplasmie mitochondriale :
- coexistence en proportion variable d’ADN mitochondrial normal et muté
- descendants avec une atteinte non symptomatique, modérée ou sévère
suivant le nombre de mitochondries mutées transmises par la mère
∎ Pathologies mitochondriales :
- myopathie mitochondriale
- atrophie optique de Leber (inflammation du nerf optique)
- syndrome de Leigh (paralysie des muscles oculaires)

Le génome
Structure du
génome
Définitions ∎ Génome :
définition
- ensemble du matériel génétique d’un individu ou d’une espèce
chez l’homme
- 22 paires d’autosomes et 1 paire de gonosomes
- taille = 3400 Mpb avec 25000 gènes
∎ Composition :
séquences codantes (exons)
2 % du génome humain
séquences non codantes
98 % du génome humain
- séquences régulatrices
- introns (90 % des bases des gènes)
- séquences répétées (Long or Short Interspersed Nuclear Elements)
- séquences de fonction structurale de l’architecture chromosomique
Variations du
génome
∎ Polymorphisme :
définition
- toute variation de séquence génomique entrainant l’existence au même
locus d’au moins 2 formes différentes de la séquence dans la population
différents types
- remplacement d’un nucléotide (Single Nucleotide Polymorphism)
- insertion/délétion d’un segment génomique
- séquences répétées en tandem : VNTR > 10 pb, microsatellites < 10 pb
VNTR = Variable Number of Tandem Repeat
∎ Locus :
- emplacement d’un segment d’ADN sur un chromosome
- défini par son contenu informationnel ou sa séquence
∎ Marqueur génétique :
- segment génomique de locus connu et présentant un polymorphisme
- taux d’hétérozygotie : 1 ²H
Recombinaison ∎ Crossing-over :
- segments de chromosomes homologues échangés
- brassage de l’information génétique pour une même paire de K
∎ Taux de recombinaison :
gamètes recombinés
gamètes recombinés gamètes parentaux
Bases
moléculaires
des mutations
Généralités ∎ Mutations somatiques :
- mutations apparues dans les cellules d’un organe ou d’un tissu
∎ Mutations germinales :
- mutations apparues dans la lignée germinale
- transmission à la descendance
∎ Mutation mosaïque :
- mutation apparaissant au cours du développement
- mosaïcisme = présence de 2 types de c chez un individu
∎ Conséquences :
perte de fonction
- mutation ne permettant plus à la protéine d’assurer sa fonction dans les c
gain de fonction
- mutation donnant à la protéine mutée de nouvelles propriétés
- influence délétère sur la physiologie d’au moins un type cellulaire
Mutations simples ∎ Définition :
- mutations pouvant toucher toutes les séquences du génome
∎ Différents types :
- remplacement d’une base
- petits ou grands réarrangements
Remplacement
d’une base
∎ Transitions :
- remplacement d’une base par une autre de même type
∎ Transversions :
- remplacement d’une base purique par une base pyrimidine (ou
inversement) 2 fois moins fréquentes que les transitions
∎ Point chaud de mutation :
- 40 % de toutes les mutations ponctuelles
- dinucléotide CG (transitions vers TG ou CA)
- méthylation associée à la répression transcriptionnelle
transition d’une base C en T
1) méthylation en C5 d’une cytosine
2) désamination et hydratation en C4 de la 5-méthyl-cytosine
3) formation d’une base thymine
transition d’une base G en A
1) désoxydation et amination en C6 d’une base G
2) amination en C2 de la 6-amino-guanine
3) formation d’une base adénine
Réarrangements ∎ Petits réarrangements :
caractéristiques
- petites délétions = perte d’une ou plusieurs bases
- petites insertions = apparition d’une ou plusieurs bases
- si décalage du cadre de lecture -> Premature Termination Stop (PTS)
Gène CFTCR (cystic fibrosis transmembrane conductance regulator)
- mutation ΔF508 = perte du résidu Phe en position 508 du gène CFTCR
- responsable de la mucoviscidose (cystic fibrosis)
∎ Grands réarrangements :
caractéristiques
- délétion ou duplication d’un segment génomique comportant soit un ou
plusieurs exons d’un gène soit le gène en entier
- beaucoup plus de délétions identifiées (létalité des duplications ?)
- si décalage du cadre de lecture -> codon Stop prématuré
dystrophie musculaire de Duchenne
- délétion d’au moins un exon du gène de la dystrophine (2/3 des cas)
maladies touchant la myéline
- Charcot-Marie-Tooth = duplication du segment génomique
- Neuropathie Tomaculaire = délétion du segment génomique
- duplication ou délétion de 40 gènes dont celui codant la protéine PMP22
- effet dose = quantité de protéine PMP22 dans la myéline proportionnelle
au nombre de copies du gène (délétion des autres gènes non délétère)
Conséquences ∎ En fonction de leur localisation :
mutation en 5’ du gène
- diminution du taux d’ARNm transcrit à partir du gène muté
mutation touchant le codon ATG
- absence de synthèse de la protéine
- protéine de plus petite taille (séquence ATG en aval)
mutation touchant le codon Stop
- production d’une protéine allongée
mutation des sites d’épissages
- rétention de l’intron dans l’ARNm
- épissage d’une séquence ≠ (site cryptique d’épissage)
∎ En fonction de leur nature :
codons Stop prématurés
- dégradation de la protéine incomplète
- destruction précoce de l’ARNm non-sens
dégradation des ARNm non-sens
- déterminée par la relation spatiale entre le premier codon Stop du cadre
de lecture et les limites exon-exon formées par l’épissage de l’ARNm
- mis en place de protéines nucléaires sur l’épissure après l’épissage
dégradation de l’ARNm si codon Stop en amont des limites exon-exon
Mutations
instables
∎ Séquences instables :
- répétitions successives et homogènes d’un motif trinucléotides
- seuil d’instabilité dépendant du nombre de répétitions
- instabilité méiotique (mutation transmise) ou mitotique (mutation mosaïque)
∎ Dans les séquences codantes :
- répétitions du triplet CAG codant pour une Glutamine
- formation de protéines anormales entrainant des pathologies neuro-
dégénératives autosomiques dominantes (par mort des neurones)
- maladie de Huntington (au dessus de 40 CAG -> allèle pathologique)
- syndrome de Kennedy (récessive liée à l’X)
∎ Dans les séquences non codantes :
- répétitions de très grande taille (plus de centaines de répétitions)
maladie de Steinert
- maladie autosomique dominante
- répétitions (GTG)n dans un exon du gène codant pour la myotonine
- effet cytotoxique en altérant l’épissage alternatif des ARNm
ataxie de Friedreich
- maladie autosomique récessive
- répétitions (GAA)n dans un intron du gène codant pour la frataxine
- blocage de la transcription empêchant la synthèse de frataxine
- anomalies mitochondriales (frataxine = protéine mitochondriale)
syndrome du X fragile
- maladie autosomique dominante
- répétitions (CGG)n dans le promoteur du gène codant pour la FMRP
- FMRP = Fragile X Mental Retardation Protein
Transmission
des maladies
génétiques
Autosomique
récessive
∎ Caractéristiques :
- perte de fonction si mutation des 2 copies (allèles) du gène
- pas de perte de fonction si présence d’une seule copie normale du gène
∎ Exemples :
- mucoviscidose
- ataxie de Friedreich
- thalassémies
Autosomique
dominante
∎ Perte de fonction :
- haplo-insuffisance = insuffisance due à la perte d’une copie d’un gène
- mutations dans le site actif de la protéine
- perte d’une région contenant le gène (neuropathie tomaculaire)
- mutations non-sens avec destruction des ARNm mutés
- mutations d’un gène d’une protéine en quantité limitante à l’état normal
∎ Gain de fonction :
augmentation du nombre de copies du gène
- effet dosage génique = produit des gènes trop élevé
- conséquences délétères d’une trisomie ou d’une duplication
- maladie de Charcot-Marie-Tooth (trop de PMP22 dans la myéline)
mutation avec introduction de propriétés nouvelles
- nouvelle propriété à l’origine de la maladie
mutation auto-active
- activation permanente d’une protéine cellulaire (récepteur…)
- achondroplasie (récepteur FGFR3 auto-actif)
mutation avec effet toxique
- expansion de la répétition trinucléotidique
- résistance de la protéine mutée à la protéolyse
- formation d’agrégats puis d’inclusions nucléaires
- interaction avec des facteurs de transcriptions
- maladie de Huntington
mutation dominante négative
- protéine mutée dominante sur la protéine normale
- mutation faux sens de l’une des chaines du collagène

Diagnostic moléculaire
Indications du
diagnostic
moléculaire
Généralités ∎ Objectifs :
- identifier la mutation (diagnostic direct)
- identifier le segment de K porteur de la mutation (diagnostic indirect)
- chez un sujet atteint d’une maladie génétique ou chez un sujet à risque
∎ Etude de l’arbre généalogique :
- pour évaluer un risque chez tous les sujets d’une même famille dans
laquelle ségrége une maladie héréditaire
- pour connaitre le mode de transmission de la maladie (par probabilité)
∎ Deux interlocuteurs :
- médecin clinicien (suivie de la famille)
- médecin biologiste (analyses génétiques)
Conseil génétique ∎ Centres génétiques :
- centres spécialisés pour répondre aux demandes de couple de futurs
parents présentant un risque de transmettre une maladie génétique
- réponse à ce type de demande appelé conseil génétique
↳ si demande des patients : diagnostic prénatal ou présymptomatique
∎ Conseil génétique :
pour évaluer le risque pour le couple d’avoir un enfant atteint
- recherche d’hétérozygotie chez les conjoints pour une maladie AR
- recherche des femmes conductrices dans une MR liée à l’X
diagnostic génétique
- réalisé suite à la demande du patient (consentement et information ++)
Diagnostic
génétique
∎ Diagnostic positif :
définition
- recherche d’une mutation chez un sujet présentant probablement une
maladie génétique (antécédents familiaux, signes cliniques)
demandé
- pour confirmer un diagnostic (affirmer la maladie)
- pour en caractériser le type (formes de début, formes frustres)
- en vue d’un diagnostic prénatal
∎ Diagnostic prénatal :
définition
- recherche de la mutation identifiée chez le parent à risque sur un
prélèvement de tissu fœtal (choriocentèse, amniocentèse, cordocentèse)
pour les maladies graves à début précoce
- mucoviscidose, amyotrophie spinale, dystrophie de Duchenne
∎ Diagnostic présymptomatique :
définition
- déterminer si une personne à risque, encore asymptomatique, est
porteuse de l’anomalie génétique responsable de la maladie
pour les maladies à révélations tardives
- formes familiales de cancer, de cardiopathie, maladie de Huntington
Stratégie
diagnostique
Démarche du
clinicien
∎ Définir le phénotype du cas index :
- à partir des éléments cliniques
- à partir des examens complémentaires
- à partir de l’évolution de la maladie
∎ Etablir un arbre généalogique :
- utiliser les symboles officiels
- mettre le nom et le prénom de tous les individus représentés
- leur date de naissance (ou l’âge)
- indiquer ceux qui ont eu un prélèvement (*)
- indiquer les personnes décédées
∎ Prélèvement veineux envoyé au laboratoire :
- après information au patient avec signature d’un consentement
- toujours associé à un dossier clinique ou une synthèse
- 1 à 2 tubes de 7 mL de sang acheminé au laboratoire en moins de 48 h
Questions guidant
le diagnostic
moléculaire
∎ Quel est le mode de transmission ?
rechercher une seule mutation
- si transmission autosomique dominante (neurofibromatose)
- si transmission récessive liée à l’X (myopathie Duchenne)
rechercher 2 mutations
- si transmission autosomique récessive (mucoviscidose, phénylcétonurie)
∎ Existe-t-il une hétérogénéité génétique ?
hétérogénéité génétique
- maladie génétique résultant de mutations dans des gènes différents
situés sur des segments chromosomiques (locus) différents
- exemples : hémophilie (A et B), neurofibromatose (NF1, NF2), formes
familiales de cancer du sein (BRCA1, BRCA2)
homogénéité génétique
- pour les maladies héréditaires fréquentes
↳ mucoviscidose, myopathie de Duchenne, phénylcétonurie
∎ Existe-t-il une hétérogénéité allèlique ?
hétérogénéité allèlique
- ≠ mutations d’1 même gène responsable de la même maladie génétique
- exemples : mucoviscidose, myopathie de Duchenne, phénylcétonurie
↳ mais existence de mutations récurrentes (ΔF508 -> mucoviscidose)
homogénéité allèlique (très rare)
- exemple : drépanocytose
∎ Type de mutation ?
mutations ponctuelles dans la séquence codante
- mutations récurrentes identifiées par des Kits commerciaux
- autres mutations identifiées par séquençage des exons et des régions
flanquantes (ADN interagissant avec des protéines pour réguler la transcription)
réarrangements chromosomiques
- technique de microsatellites
- méthode quantitative basée sur un PCR multicomplexe de courts
fragments fluorescents (Quantitative Multicomplex PCR of Shorts Fluorescents Fragments)
- méthode d’amplification multicomplexe de sondes dépendantes d’une
liaison (Multiplex Ligation-dependent Probe Amplification)
mutation par expansion de triplets
- détectée par amplification PCR (ou Southern blot parfois)
Techniques
d’identification
des mutations
La PCR ∎ Caractéristiques :
- (RT)-PCR = (reverse transcription) polymerase chain reaction
- permet d’amplifier de l’ADN
∎ Mécanisme :
1) dénaturation en simple brin de l’ADN à 95°C
2) hybridation d’amorces aux 2 extrémités de la région à amplifier à 60°C
-> amorce sens (en 5’ du brin sens) s’hybridant avec le brin anti-sens
-> amorce anti-sens (en 3’ du brin sens) s’hybridant avec le brin sens
3) synthèse d’un brin complémentaire à partir de l’extrémité 3’OH de
l’amorce par la Taq polymérase à 70°C
4) formation de 4 brins d’ADN
5) à partir du 3ème
cycle -> formation de 2 fragments d’ADN de la région
amplifiée (les autres n’ont pas la bonne taille)
6) après 35 cycles -> formation de 34 milliards brins d’ADN
Séquençage ∎ Réaction de séquence :
1) dénaturation en simple brin de l’ADN à 95°C
2) hybridation d’une amorce en 3’ de l’ADN simple brin
3) ADN simple brin dans un milieu avec des ddNTP fluorescents
4) synthèse du brin complémentaire par l’ADN polymérase
(avec incorporation de dNTP normaux et parfois de ddNTP fluorescents)
5) si incorporation de ddNTP -> arrêt de la synthèse
6) séparation des molécules par électrophorèse en fonction de leur taille
7) lecture de la séquence de l’ADN sur l’électrophorégramme
8) fragments proches du pôle (–) = extrémité 3’ de l’ADN séquencé
9) fragments proches du pôle (+) = extrémité 5’ de l’ADN séquencé
∎ Gel de séquence :
1) séparation par électrophorèse des fragments d’ADN
2) migration des fragments du plus petit au plus grand
3) détection par un faisceau laser -> excitation de la fluorescence
4) lumière émise reçue par des cellules photo-électriques
5) analyse par l’ordinateur des résultats expérimentaux
Applications Responsabilité
d’une mutation
∎ Argument bibliographique :
- mutation identifiée chez le patient déjà connue
∎ Ségrégation du variant avec la maladie de la famille :
- variant responsable de la maladie = variant présent chez tous les
individus atteints de la famille (certitude ↗ si nb de sujets atteint avec variant ↗)
- variant non responsable de la maladie = au moins un individu atteint de
la famille de possède pas le variant en question
∎ Absence du variant dans une population contrôle :
variant non responsable de la pathologie
- pour une maladie AD : variant présent chez les hétérozygotes sains
- pour une maladie AR : variant présent chez les homozygotes sains
∎ Nature délétère du variant :
se baser sur le type de mutation
- pour maladies AD = présence de mutations non-sens peu significatif
- pour maladies AR = présence de mutations non-sens sur les 2 variants
↳ variants responsables de la pathologie autosomique récessive
aa très conservé au cours de l’évolution
- aa conservé au cours de l’évolution = aa très important (structure…)
- variant responsable de la maladie si aa conservé touché dans ce variant
Application au
conseil génétique
∎ Conseil génétique :
pour évaluer le risque pour le couple d’avoir un enfant atteint
- recherche d’hétérozygotie chez les conjoints pour une maladie AR
- recherche des femmes conductrices dans une MR liée à l’X
diagnostic génétique
- réalisé suite à la demande du patient (consentement et information ++)
∎ Allele Specific Oligonucleotide hybridization : (ASO)
1) dénaturation de l’ADN et dépôt sur une membrane
2) hybridation avec une sonde mutée radiomarquée
↳ complémentarité parfaite si ADN porteur de la mutation
3) déshybridation de la sonde mutée (lavage)
↳ ADN muté toujours fixé à la sonde (ADN double brin très stable)
4) hybridation (de l’ADN non muté) avec une sonde normale
Application au
diagnostic
prénatal
∎ Définitions :
- diagnostic porté sur l’embryon ou le fœtus in utero pour déceler une
anomalie morphologique, une maladie génétique ou chromosomique
actuelle ou une prédisposition à développer une maladie dans le futur
- pratiques médicales ayant pour but de détecter in utero chez l’embryon
ou le fœtus une affection d’une particulière gravité
∎ Principes :
- connaitre la mutation responsable de la maladie dans la famille et le
statut moléculaire du sujet à risque du couple
- donner une information au couple sur le déroulement, les risques et les
conséquences d’un DPN lors d’une consultation de conseil génétique
- avoir l’autorisation d’interrompre la grossesse pour des raisons
médicales (maladie sévère pour laquelle il n’existe pas de traitement)
∎ En pratique :
1) prélèvement de villosités choriales (8 à 12 semaines après les règles)
2) élimination sous loupe binoculaire du tissu maternel contaminant
3) caryotype : recherche d’une anomalie du nombre de chromosomes
4) extraction de l’ADN et amplification par PCR
5) recherche ciblée de mutations connues
Application au
diagnostic pré-
symptomatique
∎ Principes :
- connaitre la mutation responsable de la maladie dans la famille
- être dans le cadre d’un protocole de diagnostic présymptomatique
- sujet réellement à risque, asymptomatique et majeur
∎ Formes familiales de cancer du sein :
caractéristiques
- maladie autosomique dominante
- hétérogénéité génétique (BRCA1, BRCA2) et allèlique
- mutations variées (ponctuelle ou réarrangement génomique)
suivi médical proposé
- examen direct par palpation 2 à 3 fois par an
- mammographie annuelle à partir de 30 ans
- dépistage par IRM mammaire tous les ans
- chirurgie prophylactique ovarienne ou mastectomie (après 35 ans)
- chimio-prévention (tamoxifène) à partir de 50 ans (sur 5 ans)
Diagnostic
indirect
Définition et
principes
∎ Définition :
- déterminer si un sujet à risque à reçu le segment chromosomique porteur
de la mutation qui ségrége dans la famille
∎ Quand est-il utilisé ?
- gène de la maladie localisé sur le génome mais non encore identifié
- gène identifié mais mutation familiale difficilement détectable
- maladie très génétiquement hétérogène (identifier le gène en cause)
∎ Principes :
- posséder les marqueurs polymorphes intragéniques ou flanquant pour le
gène étudié, à forte hétérozygotie
- réaliser une étude familiale sur des individus clés de la famille afin de
pouvoir établir la phase des marqueurs sans ambiguïté
- réunir les prélèvements des membres de la famille et établir leur statut
∎ Risques et limites :
- manque d’informativité d’un marqueur
- survenue d’une recombinaison entre 2 marqueurs
↳ utiliser des marqueurs flanquants le gène pour détecter une
recombinaison intragénique

Myopathie de
Duchenne
∎ Aspects cliniques :
- début dans la petite enfance
- fonte et déficit musculaire (d’abord au niveau des membres inférieurs)
- handicap fonctionnel entrainant une mise au fauteuil vers 10 ans
- insuffisance cardio-respiratoire
∎ Aspects génétiques :
- maladie gonosomique récessive liée à l’X
- mutations dans le gène de la dystrophine (réarrangements K)
↳ délétions au niveau de points chauds (60 % des cas)
∎ Recherche :
- de délétion par la méthode de PCR multicomplexe
- d’une délétion de novo par la technique de microsatellites
- de femmes conductrices de la myopathie de Duchenne

Génétique des populations
Introduction Définitions ∎ Population :
- groupe de reproduction, tel que la majorité des individus choisis son
conjoint à l’intérieur du groupe
Objet et finalité de
la génétique des
populations
∎ Objet principal de la génétique des populations :
- étude la fréquence des gènes et des génotypes dans une population
- son évolution dans le temps au cours des générations successives
∎ Finalité de la génétique des populations :
- compréhension des facteurs régissant l’évolution d’un gène
- compréhension de l’origine de l’homme (analyse phylogénétique)
- guider la stratégie de dépistage des maladies génétiques à la naissance
- analyse du risque de survenue de maladie génétique
- identification de facteurs génétiques impliqués dans les maladies
Relations entre
phénotype et
génotype
Phénotype ∎ Définition :
- manifestation apparente du génome sous la forme d’un trait
morphologique, d’un syndrome clinique, d’une variation quantitative ou
qualitative du produit final d’expression d’un gène (protéine)
- phénotype = interactions entre divers gènes, l’environnement et l’âge
∎ Exemple : la phénylcétonurie
- maladie autosomique récessive
- déficience de la phénylalanine hydroxylase (Phe -> Tyr)
- excès d’acide phényl pyruvique très toxique pour les neurones
- arriération mentale profonde chez les enfants non traités
- traitement = régime carencé en Phe
Polymorphisme ∎ Définition :
- toute variation de séquence génomique entrainant l’existence au même
locus d’au moins 2 formes différentes de la séquence dans la population
- fréquence du variant allèlique ≥ 1 %
∎ Différents types :
- remplacement d’un nucléotide (Single Nucleotide Polymorphism)
- insertion/délétion d’un segment génomique
- séquences répétées en tandem : VNTR > 10 pb, microsatellites < 10 pb
VNTR = Variable Number of Tandem Repeat
∎ Conséquences sur le produit du gène :
quantitative
- duplication du gène
- modification de la région régulatrice
- protéine tronquée non stable, non traduite
qualitative
- modification de la séquence d’acide aminé
pas de conséquence
∎ Degré de polymorphisme :

Génétique chromosomique
Chromosome
normal et ses
variants
Structure
et fonctions
chromosomiques
∎ Les centromères :
- constriction primaire du chromosome métaphasique
- région au niveau de laquelle sont associées les chromatides sœurs
- ADN α satellite = courte séquence répétitive spécifique au centromère
- dernière séquence d’ADN à être répliquée
∎ Les télomères :
structure
- séquence nucléotidique TTAGGG répétée plusieurs centaines de fois
- structures adjacentes aux télomères riches en gènes
fonctions
- maintenir l’intégrité structurale du chromosome
- assurer la réplication complète de la partie terminale extrême du K
- participer à l’architecture tridimensionnelle du noyau
∎ Les origines de réplication :
- situées au centre des boucles fonctionnelles de l’euchromatine
- nécessaires pour initier la réplication et maintenir le nb de copies K
∎ L’activité transcriptionnelle :
euchromatine
- chromatine décondensée et dispersée dans le nucléoplasme
- forme active, accessible aux polymérases et transcrite
hétérochromatine
- chromatine dense (sombre) à la périphérie du noyau
- forme inactive, pas associée aux polymérases et non transcrite
- facultative = génétiquement active ou inactive (inactivation de l’X)
- constitutive = tjs inactive (régions répétées autour des centromères)
Variants
chromosomiques
(polymorphisme)
∎ Définition :
- variations individuelles de segments d’hétérochromatine révélées par
une taille, un aspect et des propriétés de coloration ≠ selon les individus
- pas d’effet sur le phénotype
- transmis comme un caractère dominant
∎ Localisation :
- dans des régions très variables des chromosomes 1, 9 et 16
- bras long du chromosome Y, bras court et satellite des K acrocentriques
Inactivation du
chromosome X
∎ Caractéristiques :
- inactivation aléatoire de l’un des X chez la femme (entre J14 et J16)
- transmission de cette inactivation au cours des divisions cellulaires
- mosaïcisme fonctionnel des gènes liés à l’X
∎ Régions pseudo-autosomiques :
- PAP = Pseudo Autosomal Region
- gènes actifs au niveau des extrémités de l’X
- régions homologues au chromosome Y (appariement lors de la méiose)
∎ Hétérochromatine facultative :
- hyper-méthylation du chromosome X entrainant une inactivation
- corpuscule de Barr = condensation de l’X dans le noyau interphasique
- réplication tardive de l’X au cours du cycle cellulaire
Techniques
d’étude des
chromosomes
Un peu d’histoire ∎ Génétique au 20ème
siècle :
- en 1956 : 1er
caryotype humain réalisé par Tjio et Levan
- en 1959 : découverte de la trisomie 21 par Lejeune, Turpin et Gautier
- en 1970 : techniques de marquage des chromosomes (-> cytogénétique)
- en 1990 : techniques d’hybridation in situ fluorescente (FISH)
∎ Génétique au 21ème
siècle :
- techniques utilisées pour le diagnostic des maladies chromosomiques
Caryotype sur
sang
∎ Principes :
1) prélèvement sanguin de quelques mL
2) mise en culture des lymphocytes (milieu nutritif, mitogène, 37 °C)
3) traitement des c à la colchicine (fuseau mitotique bloqué en métaphase)
4) choc hypotonique (meilleur séparation des chromosomes)
5) fixations pour une bonne préservation morphologique des K
6) centrifugation et récupération du culot cellulaire
7) étalement des chromosomes sur lame (coloration au Giemsa)
8) observation au microscope photonique
∎ Classification des chromosomes
- chromosomes classés en fonction de leur taille, leur morphologie, de la
position des centromères et des techniques de marquages
- nomenclature = nombre de chromosomes + sexe (46,XX ou 46,XY)
Marquage en
bandes des
chromosomes
∎ Caractéristiques :
- pour reconnaitre spécifiquement les différents chromosomes
- pour identifier les anomalies de structure
- série de bandes claires et sombres reflétant les variations de structure
des chromosomes (composition en bases, densité en gènes…)
∎ Marquage de l’euchromatine :
bandes Q (quinacrine)
- coloration des chromosomes par la moutarde de quinacrine
- marquage des régions riches en A, T et de réplication tardive
bandes G (Giemsa)
- coloration des chromosomes par le Giemsa
- marquage des régions riches en A, T et de réplication tardive
bandes R (reverse of G)
- obtenues après dénaturation thermique
- complémentaires et inverses des bandes G
- riches en C et G et en gènes actifs, de réplication précoce
∎ Marquage de l’hétérochromatine :
- pour l’identification des variants polymorphiques
bandes C (centromère)
- traitement des chromosomes par l’hydroxyde de Barium
- spécifique des régions centromériques et de l’hétérochromatine
constitutive des chromosomes 1, 9, 16 et Y
∎ Marquage de la région des organisateurs nucléolaires : (NOR)
- traitement des chromosomes au nitrate d’argent (NAR)
- marquage au niveau des organisateurs nucléolaires (bras cours de
chromosomes acrocentriques 13, 14, 15, 21, 22)
- riches en gènes codant pour les ARNr
Technique de
haute résolution
∎ Méthode d’élongation des chromosomes :
- blocage des cellules en phase S (en interrompant la voie des purines)
- reprise de la synthèse après élimination du produit de blocage
- synchronisation des cellules au cours du cycle cellulaire
↳ pour augmenter la proportion de mitoses en pro-métaphase
Cytogénétique
moléculaire
∎ Hybridation in situ fluorescente : (FISH)
- pour visualiser et localiser un fragment d’ADN sur les chromosomes par
hybridation de la séquence d’ADN complémentaire (sonde fluorescente)
- sondes spécifiques du centromère, des télomères, d’un locus particulier
ou de tout le chromosome (peinture chromosomique)
- utilisée pour le diagnostic de microremaniements, de remaniements
complexes ou d’anomalies du nombre de chromosomes (DPN)
∎ Hybridation génomique comparative : (CGH)
- technique d’hybridation in situ modifiée permettant la détection et la
cartographie du ratio d’hybridation entre 2 génomes
- pour repérer des anomalies déséquilibrées (délétions, duplications)
- utilisation de puces d’ADN (CGH-array) pour analyser le génome entier
de manière plus sensible, rapide et avec une meilleure résolution
Mécanique des
chromosomes
Généralité ∎ Tableau clinique :
- retard mentale, de croissance pré- et post-natale
∎ Anomalies des chromosomes :
- en nombre ou en structure
- sur les autosomes ou les gonosomes
- survenant au cours de la formation des c germinales (méiose)
- survenant après la formation de l’œuf, au stade post-zygotique (mitose)
↳ anomalies mitotiques responsables d’un mosaïcisme cellulaire
Méiose et mitose ∎ Méiose normale :
- réduction de l’état diploïde (c germinales) vers l’état haploïde (gamètes)
- assurer la diversité génétique dans les gamètes
- chez la fille : début de la méiose dès le stade embryonnaire
- chez le garçon : début de la méiose dès la puberté
non-disjonction ou mal-ségrégation = pathologie du nombre de K
∎ Mitose normale :
- transmettre une copie intacte et complète du génome aux cellules filles
- erreur mitotique entrainant des anomalies phénotypiques en mosaïque
↳ formation d’une lignée cellulaire anormale au cours de l’embryogenèse
- mosaïcisme chromosomique = coexistence, chez un même individu, de
plusieurs lignées cellulaires ≠ avec un nombre de chromosomes différent
- chimèrisme = coexistence, chez un même individu, de plusieurs lignées
cellulaires différentes liée à l’union de zygotes initialement séparés
Non-
disjonction
En méiose ∎ Caractéristiques :
- échec pour les chromosomes homologues de se séparer (ségrégation)
symétriquement au cours de la division cellulaire (surtout en méiose I)
↳ une c trisomique et une c nullosomique pour un chromosome homologue
- ovogenèse responsable de la plupart des ND autosomiques
- spermatogenèse responsable de la plupart des ND gonosomiques
majorité des aneuploïdies liée à une ND au cours de l’ovogenèse
∎ Causes : âge maternel dégradation avec le temps
- de facteurs de contrôle de la méiose (-> cohésion des chromatides)
- de facteurs extra-ovariens (-> atrésie cellulaire)
En mitose ∎ Sur un zygote normal :
- mosaïcisme pour une lignée trisomique et une lignée monosomique
associée à une lignée normale
- pour les ND autosomiques : élimination de la lignée monosomique
- pour les ND gonosomiques : survie de la lignée monosomique
∎ Sur un zygote aneuploïde :
- non-disjonction chez un œuf initialement trisomique pour un autosome
↳ transformation de la cellule trisomique en cellule normale
- correction post-zygotique d’une aneuploïdie en disomie biparentale ou
monoparentale (si lignée normale à l’origine de tout l’embryon)
Aneuploïdies
et polyploïdies
Définitions ∎ Aneuploïdie = présence d’un chromosome en plus ou en moins
∎ Polyploïdie = lot additionnel entier de chromosomes (triploïdie, tétraploïdie)
Trisomie 21 ∎ Caractéristiques :
- anomalie chromosomique la plus fréquente à la naissance
- phénotype = dysmorphie faciale spécifique + déficit mental
- 3 chromosomes 21 -> effet de dosage génique de gènes en triple dose
∎ Trisomie 21 libre : (95 %)
- présence d’un chromosome 21 supplémentaire dans toutes les cellules
- due à une erreur méiotique maternelle en méiose I (ou en méiose II)
↳ absence de recombinaison entre les chromatides du chromosome 21
∎ Trisomie 21 en mosaïque : (2 %)
- présence d’un chromosome 21 en plus dans une lignée cellulaire
- due à une mal-ségrégation des homologues au stade post-zygotique
∎ Trisomie 21 par translocation Robertsonienne : (4 %)
- fusion de deux chromosomes 21 par translocation
- translocation d’origine familiale (25 %) ou de novo (75 %)
Autres trisomies
autosomiques
∎ Trisomies 13 et 18 :
- beaucoup moins fréquentes que la trisomie 21
- favorisées par l’âge maternel
∎ Autres trisomies autosomiques :
- très rares à terme car peu viable (fréquentes en fausse couche)
Aneuploïdies des
gonosomes
∎ Trisomies sexuelles :
- syndrome du triple X = 47,XXX
- syndrome de Klinefelter = 47,XXY
- due à une erreur méiotique maternelle, paternelle
- due à une erreur mitotique post-zygotique (mosaïcisme)
∎ Monosomie sexuelle :
- syndrome de Turner = 45,X
- due à la perte d’un X paternel au cours de la méiose
- due à la perte d’un X au stade post-zygotique (mosaïcisme)
Polyploïdie ∎ Triploïdies : (3 n = 69 K)
caractéristiques
- fausse couche spontanée premier semestre
- mort fœtale in utero au deuxième semestre
diandrique monogynique
- 2 n chromosomes paternels et 1 n chromosomes maternels
- fécondation de l’ovocyte par 2 spz haploïdes ou par 1 spz diploïde
digynique monoandrique
- 2 n chromosomes maternels et 1 n chromosomes paternels
- due à un ovocyte diploïde (ND de tous les K ou persistance du GP)
∎ Tétraploïdie : (4 n = 92 K)
- très rares
- division anormale des chromosomes sans division cytoplasmique au
cours de la première division du zygote
Remaniements
de structure
Délétions ∎ Grandes délétions :
- visibles en cytogénétique classique
- délétion d’une partie d’un chromosome (perte de gènes adjacents)
↳ syndrome de gènes contigus = chaque trait caractérisant le syndrome du
à la perte d’un gène ou plutôt d’un allèle
monosomie ou haploinsuffisance pour la région du chromosome perdue
∎ Micro-délétions :
- détectée par les techniques de cytogénétiques moléculaires (FISH)
- rôle des microdélétions subtélomériques dans l’étiologie des retards
mentaux idiopathiques (parfois présents chez parents -> polymorphisme)
Translocations
chromosomiques
∎ Translocations réciproques :
- échange mutuel de segments chromosomiques entre 2 chromosomes ≠
↳ chromosomes résultant appelés chromosomes dérivés
- phénotype normal du porteur de translocation réciproque mais risque
d’avoir un enfant avec un chromosome déséquilibré
∎ Translocations Robertsoniennes :
- fusion de deux bras longs de deux chromosomes acrocentriques (13, 14,
15, 21, 22) et non homologues dans la majorité des cas
- perte du bras court génétiquement inactif -> pas d’effet sur le phénotype
- plus de risque pour les femmes de transmettre un déséquilibre de la
translocation que les hommes (sélection négative des spz disomiques)
- responsables de fausses couches spontanées ou d’infertilité masculine
Micro-
remaniements
∎ Caractéristiques :
- remaniements chromosomiques dans une région de taille < à 3 Mb
- pas détectés par la cytogénétique classique (remaniements cryptiques)
- visibles en cytogénétique moléculaire (FISH)
∎ Exemples :
- syndromes de DiGeorge et vélo-cardio-facial (micro-délétion 22q11.2)
- syndrome de Prader-Willi et Angelman (micro-délétion 15q11q13)





























