Embed Size (px)
Citation preview

1
AIX-MARSEILLE UNIVERSITÉ
FACULTÉ DE MÉDECINE DE MARSEILLE
ECOLE DOCTORALE : Sciences de la Vie et de la Santé
T H È S E
Présentée et publiquement soutenue
Le 31 octobre 2014
Par M. DEVILLIER Raynier
Né le 5 janvier 1985 à Sainte Foy-lès-Lyon (69)
CARACTÉRISATION MOLÉCULAIRE DES LEUCÉMIES AIGUËS MYÉLOÏDES AVEC DYSMYÉLOPOÏÈSE
Pour obtenir le grade de DOCTORAT d’AIX-MARSEILLE UNIVERSITÉ
SPÉCIALITÉ : Oncologie, Pharmacologie et Thérapeutique
Membres du Jury de la Thèse :
Pr. Norbert VEY Président du Jury
Pr. Michaela FONTENAY Rapporteur
Pr. Christian RECHER Rapporteur
Dr. Marie Joëlle MOZZICONACCI Directeur de thèse
Laboratoire d’Oncologie Moléculaire, Centre de Recherche en Cancérologie de Marseille,
UMR1068, Institut Paoli Calmettes, Marseille, France

2
Je présente tout d’abords mes sincères remerciements aux membres du jury.
Je remercie le Professeur Norbert Vey, qui me fait l’honneur de présider cette
thèse. Merci pour le soutien et les conseils tout au long de ce travail.
Je remercie le Professeur Michaela Fontenay et le Professeur Christian
Recher pour avoir accepté de juger cette thèse. Soyez assurés de ma profonde
reconnaissance.
Je remercie le Docteur Marie-Joëlle Mozziconacci d’avoir dirigé ce travail.
Merci pour votre confiance durant ces dernières années.
Je remercie le Docteur Daniel Birnbaum de m’avoir permis de réaliser ces travaux au
sein de soin équipe.
Je remercie le Professeur Didier Blaise pour son soutien depuis le début de mon
internat.
Je tiens à remercier mes collègues du laboratoire et des unités d’hématologie et
d’oncologie.
Enfin, je dédie cette thèse à ma famille, mes amis et ma compagne Emeline
Tabouret pour leur aide, leur soutien et leur fidélité depuis tant d’années.

3
TABLE DES MATIERES
TABLES ET FIGURES .......................................................................................................... 5
LISTE DES ABREVIATIONS ................................................................................................ 6
INTRODUCTION ................................................................................................................... 7
I) Généralités sur les leucémies aigues myéloïdes ................................................... 7
a. Définition .................................................................................................................... 7
b. Epidémiologie ............................................................................................................ 7
c. Etiologies .................................................................................................................... 8
II) Classification des LAM ............................................................................................11
a. Classification FAB (French-American-British) .................................................... 11
b. Classifications OMS ................................................................................................ 12
III) Anomalies moléculaires dans la leucémogenèse .................................................17
a. Différentes classes d’altérations moléculaires .................................................... 17
b. Anomalies génétiques conférant un avantage prolifératif ................................. 18
c. Anomalies génétiques provoquant un blocage de la différenciation ............... 21
f. Anomalies de gènes impliqués dans la régulation épigénétique ...................... 25
IV) Facteurs pronostiques dans les LAM.....................................................................33
a. Facteurs liés au patient .......................................................................................... 33
b. Facteurs liés à la maladie ....................................................................................... 34
c. Classification de l’European LeukemiaNet (ELN) ............................................... 38
OBJECTIFS ET PRESENTATION DES TRAVAUX .............................................................40
ARTICLE 1 : ........................................................................................................................42
Prognostic significance of myelodysplasia-related changes according to the WHO
classification among ELN-intermediate-risk AML patients .............................................42
ARTICLE 2 : ........................................................................................................................46

4
Acute myeloid leukemia with myelodysplasia-related changes are characterized by a
specific molecular pattern with high frequency of ASXL1 mutations ............................46
ARTICLE 3 : ........................................................................................................................54
Clinical, morphological and molecular characterization of acute myeloid leukemias
with myelodysplasia-related changes...............................................................................54
ARTICLE 4 : ........................................................................................................................91
In vitro multidrug screening and mutational profiles of acute myeloid leukemias with
myelodysplasia-related changes .......................................................................................91
DISCUSSION ET PERSPECTIVES ................................................................................... 107
I) Vers une classification moléculaire des LAM-MRC ? ......................................... 107
a. LAM avec altération de TP53 ................................................................................ 107
b. LAM avec mutation d’ASXL1................................................................................. 109
c. LAM avec mutations de NPM1 ou DNMT3A ........................................................ 110
d. LAM avec d’autres mutations ou sans mutation identifiée ............................... 110
II) Perspectives .......................................................................................................... 111
a. Caractérisation pangénomique des LAM ............................................................ 111
b. Vers une médecine moléculaire personnalisée .................................................. 113
BIBLIOGRAPHIE ............................................................................................................... 115
ANNEXES .......................................................................................................................... 128

5
TABLES ET FIGURES
Table 1 : Maladies congénitales prédisposant au développement d’une LAM, associées ou
non à un syndrome génétique (page 9)
Table 2 : Classification FAB (page 11)
Table 3 : Classification OMS 2008 des LAM (page 13)
Table 4 : Anomalies cytogénétiques suffisantes pour classer une LAM en LAM-MRC selon la
classification OMS 2008 (page 15)
Table 5 : Principales anomalies moléculaires et leur fréquence dans les LAM (page 19)
Table 6 : Caractéristiques des LAM avec mutations d’ASXL1 au sein des principales séries
publiées (page 32)
Table 7 : Groupes de risque cytogénétique selon le CALGB, le MRC et le SWOG/ECOG
(page 36)
Table 8 : Classification pronostique de l’European LeukemiaNet (page 39)
Figure 1 : Modèle de leucémogénèse à 2 évènements (page 17)
Figure 2 : Localisation des mutations d’ASXL1 dans les hémopathies myéloïdes (page 28)
Figure 3 : Rôle d’ASXL1 et du complexe PRC2 dans la régulation des gènes impliqués dans
la leucémogenèse (page 31)
Figure 4 : Proposition de classification moléculaire des LAM-MRC (page 108)
Figure 5 : Différentes classes d’altérations moléculaires dans les LAM (page 112)

6
LISTE DES ABREVIATIONS
CBF : core binding factor
DEP : dysérythropoïèse
DGP : dysgranulopoïèse
DML : dysplasie multilignée
DMP: dysmégacaryopoïèse
ELN : European LeukemiaNet
FAB : French-American-British
ITD : récepteurs à activité tyrosine kinase
LAL : leucémie aiguë lymphoblastique
LAM : leucémie aiguë myéloïde
LAM-MRC : LAM « myelodysplasia-related changes »
LAM-NOS : LAM « not otherwise specified »
LMMC : leucémie myélo-monocytaire chronique
NES : signal d’exportation nucléaire
NMP : néoplasie myéloproliférative
PNN : polynucléaire neutrophile
SMD : syndrome myélodysplasique
TKD : domaine tyrosine kinase
TKR : récepteurs à activité tyrosine kinase

7
INTRODUCTION
I) Généralités sur les leucémies aigues myéloïdes
a. Définition
Les leucémies aigues myéloïdes (LAM) sont des hémopathies malignes
caractérisées par une prolifération clonale de précurseurs hématopoïétiques
myéloïdes (blastes). Suite à des évènements génétiques lors de la leucémogenèse,
les cellules myéloïdes immatures acquièrent des propriétés d’auto-renouvellement et
de prolifération associées à un blocage de maturation entraînant le phénotype
leucémique. L’avantage prolifératif qui en résulte va inhiber l’hématopoïèse
physiologique, et conduire à un envahissement de la moelle osseuse, du sang
périphérique, puis de différents tissus ou organes par les blastes leucémiques. Ce
processus malin va être rapidement identifiable sur le plan clinique et biologique,
notamment par l’apparition d’un syndrome d’insuffisance médullaire et/ou d’un
syndrome tumoral. L’évolution est toujours fatale à court terme en l’absence de
traitement efficace.
b. Epidémiologie
Les LAM représentent environ 80% des leucémies aiguës de l’adulte et 20% de
celles de l’enfant. Ce sont des pathologies rares, représentant environ 0,6% des
cancers dans les pays occidentaux. Elles sont responsables de 1,5% des décès
d’origine tumorale. L’incidence annuelle est de 2 à 4 cas pour 100 000 habitants.
Cette incidence est maximale aux Etats-Unis, en Australie et en Europe occidentale
(Jemal et al., 2010). L’incidence des LAM augmente avec l’âge avec un âge médian
au diagnostic de 65 à 69 ans selon les études (Estey, 2007; Klepin and Balducci,
2009). L’incidence des LAM est actuellement en augmentation, probablement du fait

8
du vieillissement des populations occidentales et de l’utilisation croissante d’agents
cytotoxiques leucémogènes dans le cadre du traitement des cancers (Xie et al.,
2003).
c. Etiologies
Bien que l’étiologie de la LAM reste inconnue dans la majorité des cas, certains
facteurs génétiques ou environnementaux peuvent en favoriser la survenue.
i. Facteurs génétiques constitutionnels
Certaines maladies constitutionnelles augmentent le risque de développer une LAM.
Ces LAM peuvent s’intégrer au sein d’un syndrome génétique bien défini ou être la
seule manifestation clinico-biologique d’une anomalie génétique constitutionnelle.
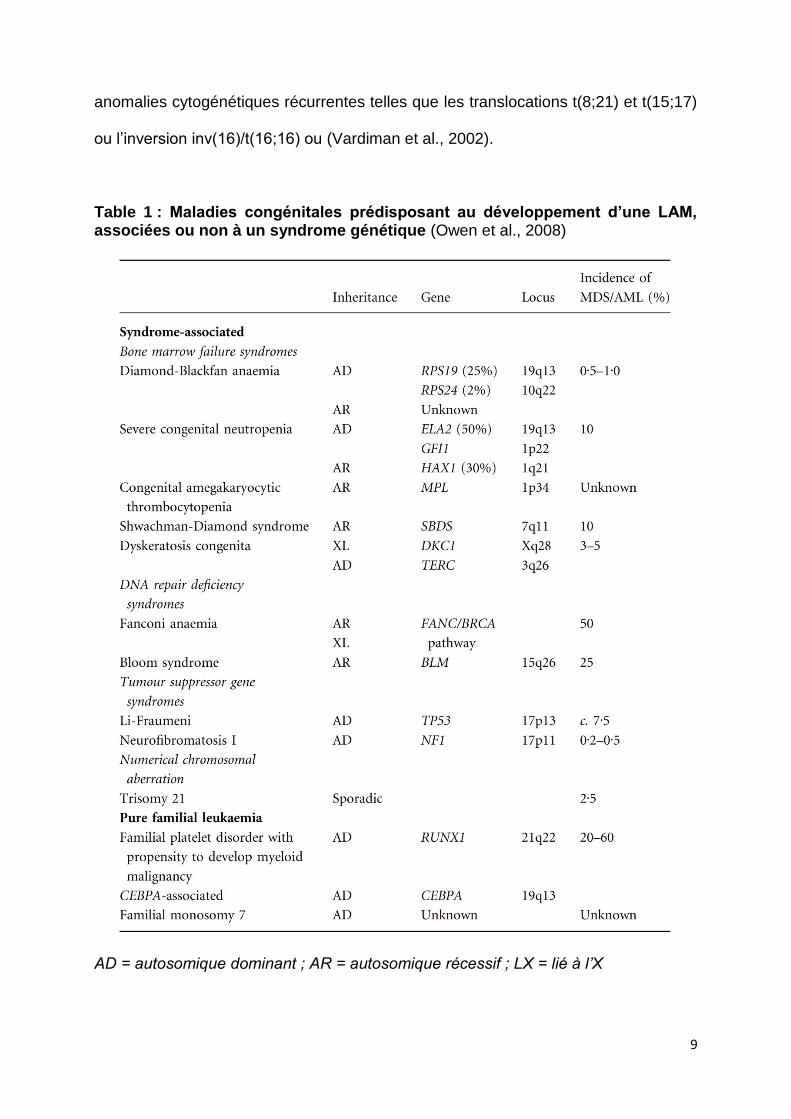
Dans ce dernier cas, elles sont appelées leucémies familiales pures (Table 1).
ii. Facteurs toxiques et environnementaux
Certaines causes toxiques ont été associées au développement d’une LAM. Les plus
fréquemment retrouvées sont les expositions au benzène, aux radiations ionisantes
d’origine professionnelle, accidentelle ou médicale (traitement par radiothérapie) ou à
des chimiothérapies (Belson et al., 2007; Bowen, 2006). Les principaux traitements
identifiés comme leucémogènes sont les agents alkylants (melphalan, cisplatine) et
les inhibiteurs de topo-isomérase II (anthracycline, étoposide) (Bowen, 2006). Les
LAM secondaires aux traitements par agents alkylants surviennent en général après
une latence de 5 ans environ. Elles sont souvent précédées d’une phase de
syndrome myélodysplasique (SMD) et présentent des anomalies cytogénétiques
complexes ou des délétions ou monosomies des chromosomes 5 et 7. Les formes
secondaires aux traitements par inhibiteurs de topo-isomérase II surviennent plus
précocement, environ 2 ans après traitement et sont souvent associées à des
translocations équilibrées impliquant le gène MLL en 11q23, et parfois à des
9
anomalies cytogénétiques récurrentes telles que les translocations t(8;21) et t(15;17)
ou l’inversion inv(16)/t(16;16) ou (Vardiman et al., 2002).
Table 1 : Maladies congénitales prédisposant au développement d’une LAM, associées ou non à un syndrome génétique (Owen et al., 2008)
AD = autosomique dominant ; AR = autosomique récessif ; LX = lié à l’X

10
iii. Evolution à partir d’une autre hémopathie
Certaines hémopathies myéloïdes chroniques acquises peuvent évoluer
naturellement en LAM. Parmi celles-ci, on peut citer :
- les néoplasies myéloprolifératives (NMP) de type leucémie myéloïde chronique
(LMC) ou non LMC, avec une fréquence variable selon les types de NMP.
- les SMD et les syndromes mixtes SMD/NMP, telle que la leucémie myélo-
monocytaire chronique (LMMC) avec également une évolution variable en fonction
du stade de la maladie, essentiellement basé sur les cytopénies, les anomalies
cytogénétiques et la blastose médullaire (Greenberg et al., 2012).
- les aplasies médullaires idiopathiques avec ou sans hémoglobinurie
paroxystique nocturne.

11
II) Classification des LAM
a. Classification FAB (French-American-British)
La classification FAB (Bennett et al., 1976) permettait de distinguer huit entités (de
M0 à M7) selon la lignée d’origine de la population blastique et le degré de
maturation résiduelle à l’aide de critères cyto-morphologiques et cytochimiques
(Table 2). Elle définissait la LAM par une blastose médullaire supérieure à 30%. Bien
que certaines entités de la classification FAB aient un pronostique spécifique, telle
que la LAM3, cette classification n’avait pas pour but de stratifier le pronostic des
LAM.
Table 2 : Classification FAB (Bennett et al., 1976)
Type FAB Nom Caractéristiques cytologiques
M0 LAM sans différenciation Myéloblastes indifférenciés
M1 LAM peu différenciée Myéloblastes peu différenciés
Quelques granulations azurophiles
M2 LAM différenciée Myéloblastes granuleux, corps d’Auer
M3 LA promyélocytaire Promyélocytes anormaux, hyper-
granuleux, avec corps d’Auer en fagots
M4 LA myélo-monocytaire
Monocytose sanguine > 5 G/L ou médullaire > 20 %
Sous type: LAM4 à éosinophile anormaux
M5 LA monoblastique
Cellules monocytaires médullaires > 80 %
M5a : monoblastes (peu différenciés)
M5b : (pro)monocytes (différenciés)
M6 Erythroleucémie > 50 % d'érythroblastes
M7 LA mégacaryocytaire Mégacaryoblastes plus ou moins
différenciés

12
b. Classifications OMS
La classification de l’OMS 2001 identifie des entités distinctes, notamment sur des
critères cytogénétiques (Vardiman et al., 2002). Les LAM avec dysplasie multilignée
apparaissent dans cette classification et sont différenciées des LAM radio ou chimio
induites. Le seuil de blastose médullaire définissant la LAM est abaissé à 20%. La
classification OMS révisée en 2008 (Vardiman et al., 2009) intègre de nouvelles
anomalies cytogénétiques et propose des entités basées sur des anomalies
moléculaires (Table 3).
i. LAM avec anomalie génétique récurrente
La première catégorie de la classification OMS 2008 regroupe les LAM avec
anomalie génétique récurrente. Elle comprend les réarrangements t(8;21)(q22;q22),
inv(16)(p13q22) ou t(16;16)(p13;q22), et t(15;17)(q22;q12) qui étaient déjà présentes
dans la classification de 2001. La translocation t(9;11)(p22;q23) est individualisée
des autres réarrangements de MLL dans la classification 2008. De nouvelles entités
cytogénétiques ont été ajoutées : les LAM avec t(6;9)(p23;q34), les LAM avec
inv(3)(q21q26.2) ou t(3;3)(q21;q26.2) et LAM avec t(1;22)(p13;q13). Enfin, par
rapport à la classification OMS 2001, celle de 2008 intègre 2 entités provisoires
basées sur la présence d’anomalies moléculaires : les LAM avec mutations de NPM1
et celles avec mutations de CEBPA.

13
Table 3 : Classification OMS 2008 des LAM (Vardiman et al., 2009)
Acute myeloid leukemia with recurrent genetic abnormalities
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
APL with t(15;17)(q22;q12); PML-RARA
AML with t(9;11)(p22;q23); MLLT3-MLL
AML with t(6;9)(p23;q34); DEK-NUP214
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
Provisional entity: AML with mutated NPM1
Provisional entity: AML with mutated CEBPA
Acute myeloid leukemia with myelodysplasia-related changes
Therapy-related myeloid neoplasms
Acute myeloid leukemia, not otherwise specified (NOS)
Acute myeloid leukemia with minimal differentiation
Acute myeloid leukemia without maturation
Acute myeloid leukemia with maturation
Acute myelomonocytic leukemia
Acute monoblastic/monocytic leukemia
Acute erythroid leukemia
Pure erythroid leukemia
Erythroleukemia, erythroid/myeloid
Acute megakaryoblastic leukemia
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis (syn.: acute myelofibrosis; acute myelosclerosis)
Myeloid sarcoma (syn.: extramedullary myeloid tumor; granulocytic sarcoma; chloroma)
Myeloid proliferations related to Down syndrome
Transient abnormal myelopoiesis (syn.: transient myeloproliferative disorder)
Myeloid leukemia associated with Down syndrome
Blastic plasmacytoid dendritic cell neoplasm
Acute leukemias of ambiguous lineage
Acute undifferentiated leukemia
Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1
Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged
Mixed phenotype acute leukemia, B/myeloid, NOS
Mixed phenotype acute leukemia, T/myeloid, NOS
Provisional entity: Natural killer (NK)–cell lymphoblastic leukemia/lymphoma

14
ii. LAM-MRC (with myelodysplasia-related changes)
C’est ce sous-groupe spécifique qui sera la base d’étude de notre travail. Apparue en
2001 sous le terme de LAM avec dysplasie multilignée (DML), cette catégorie est
élargie par la classification de l’OMS 2008 en intégrant des critères cytogénétiques.
Elle est actuellement intitulée «AML with myelodysplasia-related changes» (LAM-
MRC). Elle est définie par une blastose médullaire ≥ 20% et la présence d’un
diagnostic préexistant de SMD ou de SMD/NMP ; et/ou de DML définie par au moins
50% de cellules médullaires dysplasiques sur au moins 2 lignées ; et/ou d’anomalies
cytogénétiques associées aux SMD. La présence d’un seul, de deux ou des 3
critères dans une LAM en fait une LAM-MRC . De plus, les patients ne doivent pas
avoir été exposés à un traitement de radiothérapie et/ou de chimiothérapie, sinon ils
sont considérés comme ayant une LAM induite par le traitement (« Therapy-related
AML »).
La DML est définie par la présence d’au moins 50% de cellules dysplasiques sur au
moins 2 lignées. La dysgranulopoïèse (DGP) est caractérisée par la présence
d’anomalies morphologiques au sein des polynucléaires neutrophiles (PNN) : PNN
agranulaire ou hypogranulaire ; PNN avec hypersegmentation nucléaire ; PNN avec
hyposegmentation nucléaire (pseudo Pelger-Huët). Les signes morphologiques
considérés comme des critères de dysérythropoïèse (DEP) sont : la mégaloblastose,
la caryorrhexie, les irrégularités (bourgeonnement) ou les fragmentations nucléaires,
la multinucléation, les vacuoles ou lacunes cytoplasmiques, les sidéroblastes en
couronne et la positivité à l’acide périodique Schiff. Enfin, la dysmégacaryopoïèse
(DMP) est définie par la présence de micro-mégacaryocytes ou d’un noyau hypo ou
non lobulé ou la présence de multinucléation. Certains patients peuvent se
présenter au diagnostic avec des signes de DGP, DEP et/ou de DMP mais sans

15
atteindre les critères nécessaires à la DML (50% de cellules dysplasiques sur au
moins 2 lignées). Cela peut être dû à un nombre insuffisant d’éléments évaluables au
sein d’une ou plusieurs lignées ou à la présence de dysplasie sur moins de 50% des
cellules sur 2 des 3 lignées. Pour ces patients, la présence d’antécédents de SMD ou
de SMD/MNP et/ou d’anomalies cytogénétiques de SMD permet un classement en
LAM-MRC. Ces anomalies cytogénétiques sont listées dans la Table 4. Les
anomalies non équilibrées sont les plus fréquentes, notamment les caryotypes
complexes (≥ 3 anomalies chromosomiques en l’absence d’anomalie cytogénétique
récurrente) et/ou les anomalies de type -5/del(5q) et -7/del(7q). La trisomie 8 et la
del(20q), fréquemment identifiées dans les SMD ne sont pas suffisamment
spécifiques autoriser un diagnostic de LAM-MRC. Les translocations équilibrées sont
plus rares et intéressent principalement la région 5q33. Malgré leur fréquente
association à la présence de DML, les inv(3)(q21;q26.2) et t(3;3)(q21; q26.2) sont à
présent considérées comme des anomalies cytogénétiques récurrentes et sont par
définition exclues des LAM-MRC.
Table 4 : Anomalies cytogénétiques suffisantes pour classer une LAM en LAM-MRC selon la classification OMS 2008 (Vardiman et al., 2009)
Unbalanced changes Balanced changes
del(7q) or -7 t(11;16)(q23;p13.3)
del(5q) or -5 t(3;21)(q26.2;q22.1)
i(17q) or t(17p) t(1;3)(p36.3;q21.1)
del(13q) or -13 t(2;11)(p21;q23)
del(11q) t(5;12)(q33;p12)
del(12p) or t(12p) t(5;7)(q33;q11.2)
del(9q) t(5;17)(q33;p13)
idic(X)(q13) t(5;10)(q33;q21)
Complex karyotype (≥3 abnormalities)
t(3;5)(q25;q34)

16
iii. LAM secondaires à la radio/chimiothérapie
La troisième catégorie de la classification OMS 2008 comprend les LAM induites par
un traitement de chimiothérapie ou de radiothérapie (« Therapy-related AML »). La
présence d’anomalies génétiques récurrentes permet de reclasser certaines de ces
LAM dans la première catégorie.
iv. Autres LAM (LAM-NOS pour « not otherwise specified »)
La quatrième catégorie de la classification OMS 2008 regroupe les LAM qui n’ont pas
de critères pour les catégories précédentes.
v. LAM de lignée ambiguë
Enfin, la classification OMS 2008 identifie une catégorie correspondant aux
leucémies aiguës pour lesquelles l’appartenance à la lignée myéloïde ou lymphoïde
n’est pas bien définie. Pour être assimilée à une leucémie aiguë avec phénotype
mixte, les blastes doivent présenter des caractéristiques d’au moins 2 lignées
(myéloïde, lymphoïde B, lymphoïde T) basés sur des critères phénotypiques.

17
III) Anomalies moléculaires dans la leucémogenèse
a. Différentes classes d’altérations moléculaires
Les LAM sont des hémopathies hétérogènes sur le plan génétique. Les évènements
oncogéniques acquis par la cellule initiatrice sont classiquement divisés en deux
catégories : les anomalies de classe 1, et celles de classe 2 (Gilliland and Griffin,
2002). Les premières confèrent un avantage prolifératif aux cellules. Elles regroupent
par exemple les mutations activatrices de récepteurs à activité tyrosine kinase (TKR)
tels que FLT3, KIT ou RAS. Les anomalies de classe 2 induisent un blocage de
différenciation et une capacité d’auto-renouvellement. Parmi ces anomalies, on
trouve des anomalies de facteurs de transcription avec par exemple les fusions
AML1-ETO, CBFB-MYH11 ou PML-RARA.
Figure 1 : Modèle de leucémogénèse à 2 évènements (Gilliland and Griffin, 2002)

18
D’après ce modèle de leucémogenèse, le phénotype leucémique résulterait de la
combinaison d’une anomalie de classe 1, considérée préférentiellement comme un
évènement secondaire, et d’une anomalie de classe 2, habituellement identifiée
comme une altération initiatrice. Ce modèle se complexifie actuellement avec
l’identification de nouvelles mutations dont l’appartenance à la classe 1 ou la classe
2 n’est pas clairement établie. C’est notamment le cas des mutations portant sur des
régulateurs épigénétiques tels que les gènes ASXL1, TET2, IDH1/2 ou DNMT3A. Il
est possible de regrouper ces mutations pour former de nouvelles classes
d’altérations (Naoe and Kiyoi, 2013). Les principales mutations retrouvées dans les
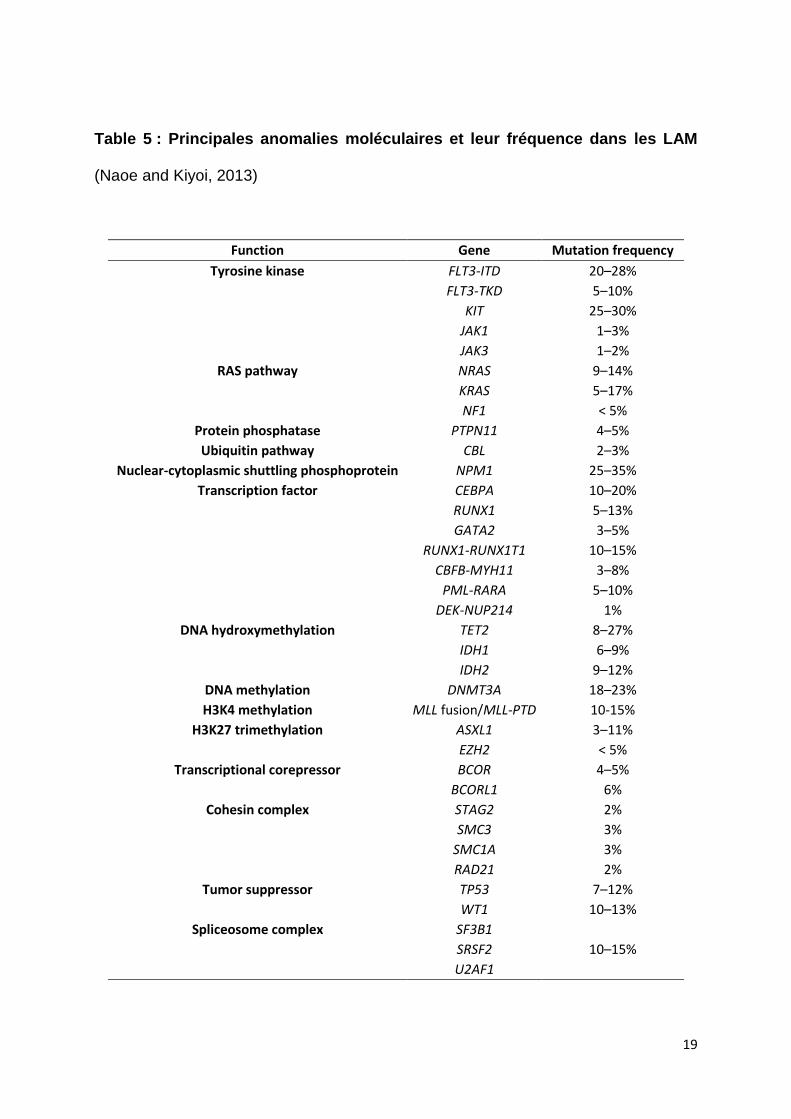
LAM sont récapitulées dans la Table 5.
b. Anomalies génétiques conférant un avantage prolifératif
i. Mutations de FLT3
Le gène codant pour la protéine tyrosine kinase FMS-like (FLT3) est localisé sur le
chromosome 13q12. Il s’agit d’une protéine de la famille des récepteurs à activité
tyrosine kinase de classe III, avec 5 domaines extracellulaires immunoglobuline-like,
un domaine transmembranaire, un domaine juxta-membranaire et deux domaines
tyrosine kinase (TKD) intracellulaires. FLT3 joue un rôle important dans la
prolifération, la différenciation et la survie cellulaire des précurseurs
hématopoïétiques (Gilliland and Griffin, 2002; Stirewalt and Radich, 2003). Des
mutations somatiques de FLT3 sont retrouvées dans 20-25% des LAM (Fröhling et
al., 2002; Schnittger et al., 2002).
19
Table 5 : Principales anomalies moléculaires et leur fréquence dans les LAM
(Naoe and Kiyoi, 2013)
Function Gene Mutation frequency
Tyrosine kinase FLT3-ITD 20–28%
FLT3-TKD 5–10%
KIT 25–30%
JAK1 1–3%
JAK3 1–2%
RAS pathway NRAS 9–14%
KRAS 5–17%
NF1 < 5%
Protein phosphatase PTPN11 4–5%
Ubiquitin pathway CBL 2–3%
Nuclear-cytoplasmic shuttling phosphoprotein NPM1 25–35%
Transcription factor CEBPA 10–20%
RUNX1 5–13%
GATA2 3–5%
RUNX1-RUNX1T1 10–15%
CBFB-MYH11 3–8%
PML-RARA 5–10%
DEK-NUP214 1%
DNA hydroxymethylation TET2 8–27%
IDH1 6–9%
IDH2 9–12%
DNA methylation DNMT3A 18–23%
H3K4 methylation MLL fusion/MLL-PTD 10-15%
H3K27 trimethylation ASXL1 3–11%
EZH2 < 5%
Transcriptional corepressor BCOR 4–5%
BCORL1 6%
Cohesin complex STAG2 2%
SMC3 3%
SMC1A 3%
RAD21 2%
Tumor suppressor TP53 7–12%
WT1 10–13%
Spliceosome complex SF3B1
10–15% SRSF2
U2AF1

20
Deux types de mutations de FLT3 ont été identifiés. La plus fréquente est une
duplication interne en tandem (ITD) des exons 14 et 15, dans la région juxta-
membranaire. La taille de la duplication est variable et respecte le cadre de lecture.
L’autre type de mutation est une mutation ponctuelle faux-sens dans l’exon 20, au
niveau du domaine TKD, le plus fréquemment sur les acides aminés D835 ou I836.
Sa fréquence est plus faible, de l’ordre de 5 à 10%. Les 2 types de mutations
entraînent une phosphorylation constitutive du récepteur mais la réponse cellulaire
n’est pas équivalente (Bailey et al., 2013; Stirewalt and Radich, 2003). Sur le plan
clinique, les mutations de type FLT3-ITD sont préférentiellement associées aux LAM
à caryotype normal ou au LAM avec translocation t(15;17) ou t(6;9). Les mutations
de type FLT3-TKD représentent la forme majoritaire au sein des LAM du core
binding factor (CBF) (Allen et al., 2013; Jourdan et al., 2013). Alors que le pronostic
péjoratif des mutations de type FLT3-ITD est bien décrit, l’impact pronostique des
mutations FLT3-TKD semble moindre (Bacher et al., 2008; Mead et al., 2007;
Schnittger et al., 2002).
ii. Mutations de KIT
Le gène KIT localisé en 4q12 code pour un récepteur tyrosine kinase de type III dont
le ligand est le stem cell factor (SCF). Il est impliqué dans l’activation des voies de
signalisation induisant la prolifération, la différenciation, la migration et la survie
cellulaire. L’activation constitutive du récepteur peut être due à plusieurs types de
mutations (insertion in-frame, duplication interne en tandem ou mutation ponctuelle
faux sens). La plus fréquente est la mutation D816V, intéressant le domaine tyrosine
kinase (substitution d’un acide aminé, la plus fréquente étant la mutation D816V).
Les mutations de KIT sont rares dans l’ensemble des LAM, mais sont

21
particulièrement représentées dans le sous-groupe des LAM CBF (20-30% des cas)
(Allen et al., 2013; Jourdan et al., 2013).
iii. Mutations de NRAS et KRAS
Les protéines de la famille RAS participent à la transduction du signal de récepteurs
membranaires tels que FLT3 ou KIT. Elles interviennent dans la régulation de la
prolifération et de la différenciation cellulaire. Il existe 3 gènes RAS fonctionnels :
NRAS, HRAS, et KRAS. Les mutations des gènes NRAS et KRAS sont présentes
dans environ 10% et 5% des LAM, respectivement. Elles sont fréquemment
associées aux LAM avec inv(16)/t(16;16) ou avec inv(3)/t(3;3). Ces mutations
ponctuelles (principalement sur les codons G12, G13 ou Q61) entrainent une perte
de l’activité GTPasique intrinsèque, provoquant ainsi une activation constitutive de la
protéine et de la voie de signalisation d’aval (RAF, MAP kinase, ERK) (Beaupre and
Kurzrock, 1999; Renneville et al., 2008).
c. Anomalies génétiques provoquant un blocage de la différenciation
i. Anomalies des gènes CBFA/RUNX1et CBFB
Le gène RUNX1 est localisé en 21q22 et code pour la sous-unité alpha du CBF. Une
fois associée à la sous-unité béta, elle forme le complexe transcriptionnel CBF qui
joue un rôle majeur dans la régulation de l’hématopoïèse (Cai et al., 2000; Kurokawa
and Hirai, 2003; Tanaka et al., 1995). RUNX1 est un des gènes les plus souvent
dérégulés dans les LAM, à travers différents mécanismes incluant des translocations
du locus 21q22, des mutations ponctuelles et des amplifications géniques. La
translocation t(8;21)(q22;q22) produit une protéine de fusion RUNX1-ETO/RUNX1T1
constituée de la partie N-terminale de RUNX1 (contenant le domaine RUNT
nécessaire à la fixation à l’ADN et à l’hétéro-dimérisation avec CBFB) et de la quasi-
totalité d’ETO, qui conserve ses capacités à recruter des corépresseurs

22
transcriptionnels (en s’associant avec NCoR). Il en résulte, entre autres, une
répression transcriptionnelle de gènes nécessaires à la différenciation myéloïde. Les
fonctions de RUNX1 sont également altérées indirectement en présence de la
protéine de fusion CBFB-MYH11 issue de l’inv(16)/t(16;16), par inhibition compétitive
avec le CBFB sauvage.
Des mutations somatiques de RUNX1, mono- ou bi-alléliques, ont été mises en
évidence dans 5% à 10% des LAM. Elles intéressent soit le domaine RHD (surtout
mono-alléliques), soit le domaine C-terminal (surtout bi-allélique), et sont fortement
associées aux LAM avec trisomie du chromosome 13 (Gaidzik et al., 2011; Osato et
al., 1999; Preudhomme et al., 2000; Roumier et al., 2003). Alors que les
translocations t(8;21) et inv(16)/t(16;16) ont un pronostic relativement favorable, les
mutations ponctuelles de RUNX1 sembleraient présenter un pronostic péjoratif, bien
que leur valeur indépendante reste discutée (Gaidzik et al., 2011).
Des mutations ponctuelles constitutionnelles de RUNX1 ont été décrites dans des
cas de «familial platelet disorder» (FPD), prédisposant au développement de LAM
(Song et al., 1999).
ii. Altérations de CEBPA
Le gène CEBPA (CCAAT/enhancer-binding protein alpha), localisé en 19q13, code
pour un facteur transcriptionnel majeur de la différenciation granulocytaire et
monocytaire et joue un rôle important dans la leucémogenèse (Koschmieder et al.,
2009). Deux types de mutations ponctuelles de CEBPA ont été décrits qui
conduisent à son inactivation : les mutations N-terminales (ponctuelles non-sens ou
avec décalage du cadre de lecture) et les mutations C-terminales (ponctuelles faux-
sens respectant le cadre de lecture) (Leroy et al., 2005). Les mutations sont bi-
alléliques dans 2/3 des mutants CEBPA et généralement de type hétérozygote

23
composite (mutation N-terminale sur un allèle et C-terminale sur l’autre), suggérant
une possible coopération entre ces deux évènements (Marcucci et al., 2011). La
présence d’une double mutation de CEBPA confère un pronostic favorable (Fasan et
al., 2014; Wouters et al., 2009). Le fait que ces mutations soient associées à des
profils phénotypiques et d’expression génique spécifiques supporte la classification
OMS qui considère les LAM mutées CEBPA comme une entité biologiquement
distincte (Lin et al., 2005; Valk et al., 2004).
d. Altérations de TP53
TP53 est un gène suppresseur de tumeur, localisé en 17p13. Il code pour un facteur
de transcription qui régule l’expression de gènes impliqués dans l’arrêt du cycle
cellulaire, la réparation de l’ADN et l’apoptose. La perte de fonction de TP53 induit
une instabilité génomique, associée à une inhibition de l’apoptose et à une
dérégulation du cycle cellulaire, malgré la présence de lésions de l’ADN. Dans les
LAM, TP53 peut être inactivé par des délétions ou des mutations ponctuelles du
gène. Les altérations de TP53 sont souvent bi-alléliques, avec soit une mutation sur
chaque allèle, soit une mutation associée à une délétion sur l’autre allèle. Plus
rarement il peut s’agir d’une double délétion. Les mutations intéressent le plus
souvent le domaine de liaison à l’ADN, de l’exon 4 à 8. Les altérations de TP53 sont
souvent retrouvées chez des patients atteints de LAM à caryotype complexe,
notamment les complexes monosomaux avec perte d’un chromosome 5 (ou délétion
5q) et 7. Les mutations de TP53 sont associées à un pronostic péjoratif,
indépendamment de la complexité du caryotype (Grossmann et al., 2012; Rücker et
al., 2012).

24
e. Mutations de NPM1
Le gène NPM1 est localisé en 5q35. NPM1 est une protéine chaperonne ubiquitaire
se déplaçant entre le noyau et le cytoplasme, avec une localisation
préférentiellement nucléolaire. Elle régule l’assemblage et le transport des
composants pré-ribosomaux à travers la membrane nucléaire. NPM1 joue un rôle
majeur de senseur nucléolaire pour l’activation de TP53. En l’absence de stress,
NPM1 forme un complexe stable intra-nucléolaire avec la protéine suppresseur de
tumeur ARF. En cas de stress (UV, radiation, drogues interférant avec la structure de
l’ADN), le complexe ARF-NPM1 est transféré dans le cytoplasme et entre en
compétition avec HDM2, un inhibiteur de TP53. Les complexes ARF-HDM2 et
NPM1-HDM2 séquestrent HDM2 permettant ainsi l’activation de TP53 en réponse au
stress (Falini et al., 2007). Les travaux de Falini et al. ont montré que les mutations
de NPM1 s’accompagnent d’une délocalisation aberrante de NPM1 dans le
cytoplasme (Falini et al., 2005, 2006). Il s’agit de mutations hétérozygotes
récurrentes dans l’exon 12 qui consistent dans la majorité des cas en une insertion
de type A (4 nucléotides, TCTG, en position 960 sur la séquence codante) (Falini et
al., 2005). Plus de 50 mutations ont été identifiées à ce jour. Toutes provoquent un
décalage du cadre de lecture et entraînent à l’extrémité C-terminale de la protéine la
perte du tryptophane 290, associée ou non à celle du tryptophane 288. Cette
modification se traduit par la création d’un site NES (nuclear exportation signal)
supplémentaire conduisant à la délocalisation cytoplasmique de NPM1 (Falini et al.,
2009). Le mécanisme oncogénique des mutations de NPM1 n’est pas entièrement
identifié. Il est possible que la protéine NPM1 mutée puisse entraîner avec elle la
délocalisation d’autres protéines dont la dérégulation pourrait favoriser le processus
leucémogène. Les mutations de l’exon 12 de NPM1 constituent l’anomalie

25
moléculaire la plus fréquente dans les LAM de l’adulte, avec une fréquence globale
de 35%. Ces mutations sont fréquemment observées dans les LAM à caryotype
normal, au sein desquelles l’incidence atteint 50-60%. Elles sont également souvent
associées aux LAM de novo, hyperleucocytaires au diagnostic, aux types FAB
M4/M5, et à une expression faible ou absente du CD34. Tout comme les LAM
mutées CEBPA, les LAM mutées NPM1 possèdent un profil d’expression génique
spécifique associé à un pronostic favorable, notamment en l’absence de mutations
de FLT3 que l’on retrouve de manière concomitante dans 40% des cas (Becker et
al., 2010; Döhner et al., 2005; Renneville et al., 2008; Schnittger et al., 2005;
Verhaak et al., 2005).
f. Anomalies de gènes impliqués dans la régulation épigénétique
i. Anomalies de MLL
Le gène MLL est localisé en 11q23 et code pour une histone méthyl-transférase qui
a un rôle de coactivateur transcriptionnel. Il intervient lors de l’embryogenèse et de
l’hématopoïèse, notamment par la régulation épigénétique de l’expression des gènes
HOX. Le domaine SET de MLL permet la méthylation de la lysine 4 de l’histone 3
(H3K4), qui représente une marque activatrice de la transcription. Environ 10% des
LAL et des LAM présentent un réarrangement de MLL. Au sein des LAM, les
translocations t(9;11)/(MLL-AF9) et t(6;11)/(MLL-AF6), sont les réarrangements les
plus fréquents (Marschalek, 2011; Meyer et al., 2009). En dehors des
réarrangements cytogénétiques, des mutations de type duplication partielle en
tandem (PTD) ont été identifiés dans 5 à 10% des LAM. Elles sont associées aux
LAM à caryotype normal ou à la trisomie du chromosome 11 (Basecke et al., 2006).
Situées dans la partie 5’ du gène, ces mutations MLL-PTD entraînent des répétitions

26
de plusieurs exons, aboutissant à une protéine de plus grande taille avec une
augmentation d’affinité pour l’ADN (Caligiuri et al., 1998; Marschalek, 2011).
ii. Mutations de TET2
Le gène TET2, situé en 4q24, est une hydroxylase qui intervient dans le processus
de déméthylation de l’ADN en convertissant la méthylcytosine en 5-
hydroxyméthylcytosine (Ko et al., 2010). Les mutations de TET2 ont été décrites
dans diverses hémopathies myéloïdes, dont les LAM. Le plus souvent, il s’agit
d’insertions ou de délétions de petite taille ou de mutations ponctuelles non-sens ou
faux-sens, aboutissant à une perte de fonction de la protéine (Delhommeau et al.,
2009). Les mutations de TET2 sont retrouvées dans environ 15-25% des LAM,
notamment dans les LAM à caryotype normal et peuvent être associées aux
mutations de NPM1. Elles sembleraient avoir un pronostic péjoratif (Chou et al.,
2011; Metzeler et al., 2011; Nibourel et al., 2010; Weissmann et al., 2012).
iii. Mutations d’IDH1 et d’IDH2
Les gènes IDH1 en 2q33 et IDH2 en 15q26 codent respectivement pour les enzymes
isocitrate deshydrogénase 1 et 2 qui convertissent l’isocitrate en alpha-cétoglutarate.
Initialement, les mutations d’IDH ont été mises en évidence grâce aux approches de
séquençage pangénomique dans les glioblastomes puis les LAM (Mardis et al.,
2009; Yan et al., 2009). Dans les LAM, leurs mutations intéressent le codon R132
d’IDH1 et les codons R140 et R172 d’IDH2 et modifient l’activité enzymatique
d’IDH1/2, leur conférant la capacité de réduire l’alpha-cétoglutarate en 2-
hydroxyglutarate. De façon compétitive, ce métabolite va inhiber l’activité de TET2,
aboutissant à une diminution de la synthèse de 5-hydroxyméthylcytosine. Ces
modifications affecteraient la méthylation de l’ADN, aboutissant alors aux mêmes
conséquences que les mutations de TET2 (Figueroa et al., 2010). Au sein des LAM,

27
la fréquence des mutations d’IDH1/2 est de 15-20%. Les mutations des codons R132
d’IDH1 et R140 d’IDH2 sont fréquemment associées aux mutations de NPM1 alors
que celles du codon R172 d’IDH2 sont rarement retrouvées en présence d’autres
mutations (Abbas et al., 2010; Boissel et al., 2010, 2011; Marcucci et al., 2010).
Comme pour les mutations de TET2, il semblerait que ces mutations soient
associées à un pronostic défavorable, notamment au sein des LAM avec mutations
de NPM1.
iv. Mutations de DNMT3A
Le gène DNMT3A est localisé en 2p23 et code pour une DNA méthyltransférase qui
catalyse l’addition d’un groupement méthyl en position 5 de la cytosine (5-
méthylcytosine). Elle permet la méthylation de novo de l’ADN, jouant ainsi un rôle
essentiel dans la différenciation des CSH (Challen et al., 2012). Les mutations de
DNMT3A ont été découvertes en 2010 grâce à l’avènement des techniques de
séquençage à haut débit. La majorité des mutations intéressent le domaine portant
l’activité méthyltransférase, notamment l’exon 23 au niveau du codon R882. Elles
sont responsables d’une diminution de l’activité catalytique de la protéine et d’une
hypométhylation de certaines régions de l’ADN (Ley et al., 2010; Yan et al., 2011).
Les mutations de DNMT3A sont retrouvées dans environ 20% des LAM de l’adulte,
notamment dans les formes à différentiation monocytaire et les LAM avec mutations
de NPM1 et de FLT3. Il semblerait qu’elles soient associées à un pronostic péjoratif
(Hou et al., 2012; Ley et al., 2010; Renneville et al., 2012; Shen et al., 2011; Tie et
al., 2014; Yan et al., 2011).
v. Mutations d’ASXL1
Le gène ASXL1, situé en 20q11, est impliqué dans les modifications post-
traductionnelles des histones, notamment en régulant la désubiquitination de la

28
lysine 119 de l’histone H2A (H2A119KUb, via le complexe de désubiquitination formé
avec BAP1) et la tri-méthylation de la lysine 27 de l’histone 3 (H3K27me3, via EZH2,
effecteur du complexe polycomb répressif 2, PRC2) (Abdel-Wahab and Dey, 2013;
Gelsi-Boyer et al., 2012). Les mutations d’ASXL1 sont situées dans l’exon 12 et sont
majoritairement de type non-sens avec ou sans décalage du cadre de lecture,
aboutissent à la formation d’une protéine dont l’extrémité C-terminale est tronquée
(Gelsi-Boyer et al., 2009). Il en résulte une perte du domaine PHD (« plant homeo
domain ») qui serait nécessaire à la liaison à l’ADN et à la reconnaissance de lysines
méthylées (Gelsi-Boyer et al., 2012; Figure 2).
Figure 2 : Localisation des mutations d’ASXL1 dans les hémopathies
myéloïdes (Gesli-Boyer et al. 2012)

29
Ces mutations empêchent le recrutement et la stabilisation du complexe PRC2,
s’accompagnant ainsi d’une diminution globale des marques répressives H3K27me3,
notamment au niveau des gènes de la famille HOX (HOXA) (Abdel-Wahab et al.,
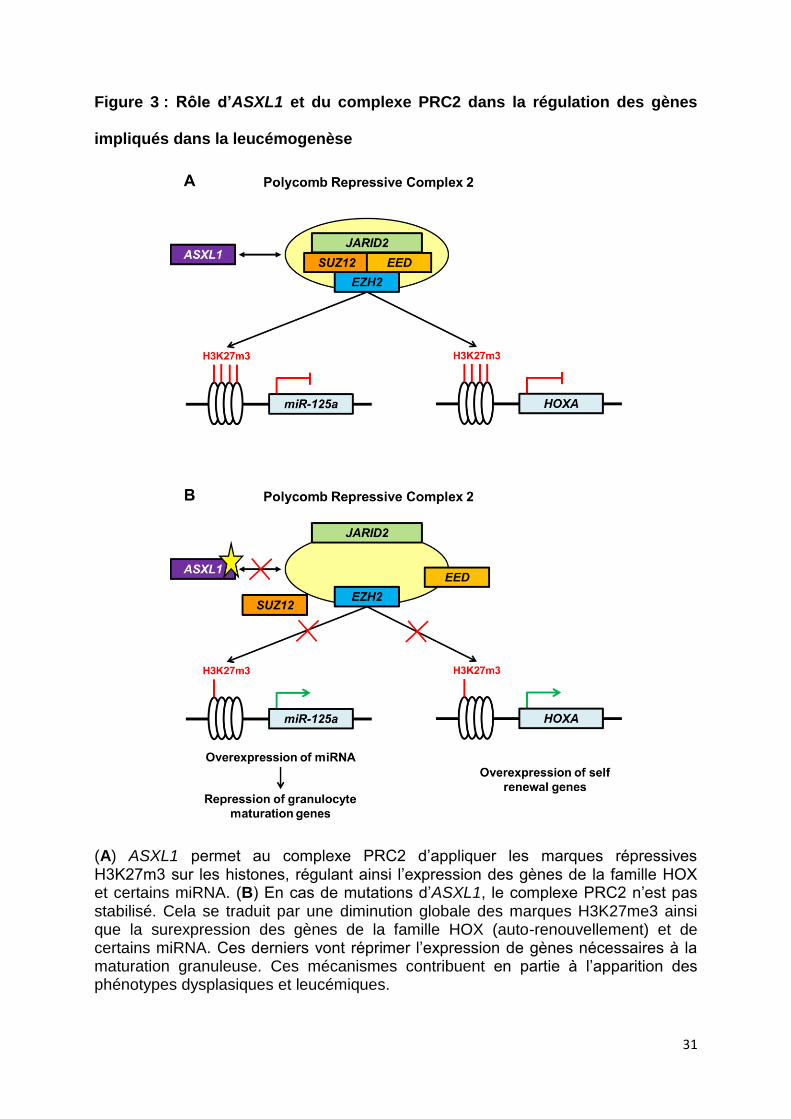
2012). Cela aboutit à une expression aberrante de ces gènes (Figure 3). De façon
similaire, les mutations d’ASXL1 semblent entrainer une surexpression de certains
miRNA, notamment miR-125a. Ce dernier est impliqué dans la régulation des
précurseurs hématopoïétiques et induit des syndromes myéloprolifératifs dans des
modèles de transplantation chez la souris (Guo et al., 2012; Gerrits et al., 2012).
Récemment, il a été montré que la dérégulation de miR-125a par les mutations
d’ASXL1 entraine une diminution de l’expression de Clec5a, protéine membranaire
indispensable à la différenciation granulocytaire, aboutissant ainsi un blocage de
maturation et à l’apparition de dysplasie morphologique (Inoue et al., 2013). Enfin,
grâce à des modèles de coopérations avec les mutations de N-RAS (N-RAS-G12V),
il a été montré que les mutations d’ASXL1 participent à l’apparition de dysplasie et à
la transformation leucémique sur un terrain de syndrome myéloprolifératif engendré
par des mutations de N-RAS (Abdel-Wahab et al., 2012; Inoue et al., 2013). Cela
pourrait expliquer en partie l’association entre la présence des mutations d’ASXL1 et
la transformation aigue des LMMC, lesquelles impliquent fréquemment des
mutations de RAS.
L’altération la plus fréquente d’ASXL1 est la mutation c.1934dupG; p.G646Wfsx12,
qui représente 50 à 60% des mutations d’ASXL1 identifiées dans les hémopathies
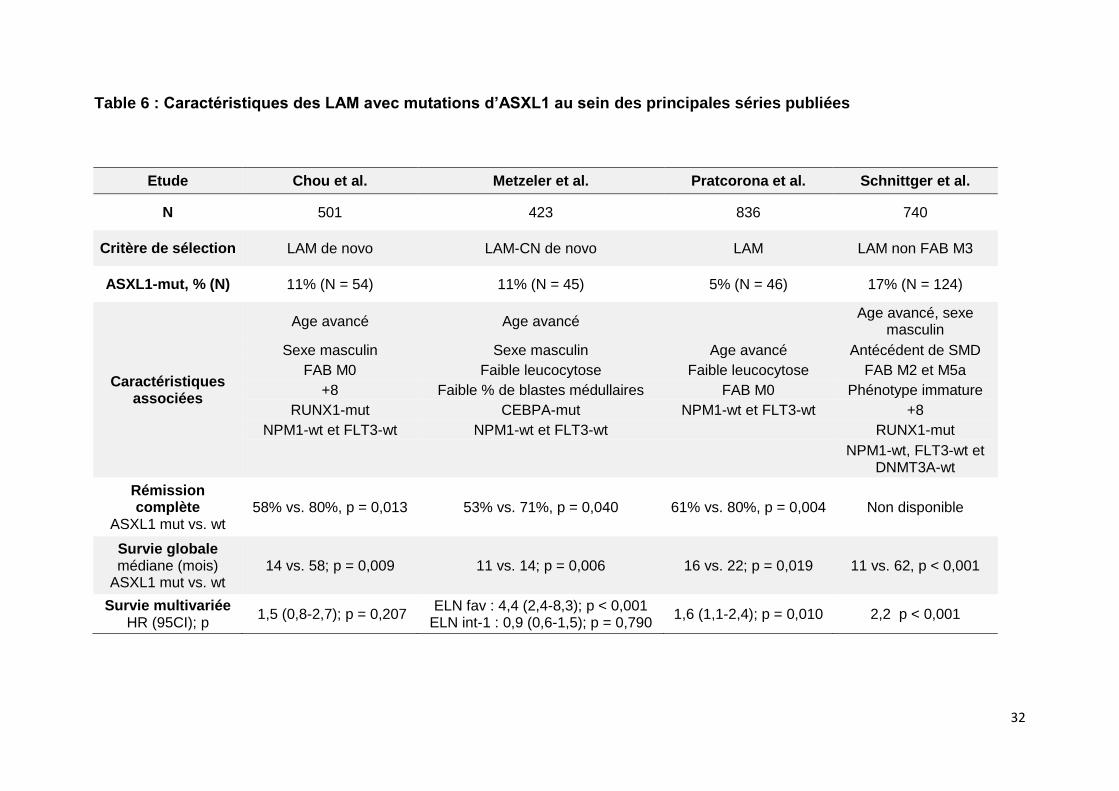
myéloïdes. La table 6 rapporte les plus importantes séries publiées décrivant les
caractéristiques clinicobiologiques des LAM avec mutation d’ASXL1. Elles sont
retrouvées dans 5-10% des LAM et sont plutôt associées au sexe masculin, à un âge
plus élevé au diagnostic, au type M0 de la classification FAB, et à caryotype normal

30
ou avec trisomie 8 isolée (Chou et al., 2010; Schnittger et al., 2013). La présence de
mutations d’ASXL1 semble corrélée à celle de mutations de RUNX1 ainsi qu’aux
formes sauvages de NPM1 et de FLT3 (Chou et al., 2010; Schnittger et al., 2013).
Enfin, il semblerait que ces mutations soient associées à un pronostic péjoratif au
sein des hémopathies myéloïdes (Bejar et al., 2011; Brecqueville et al., 2014; Chou
et al., 2010; Gelsi-Boyer et al., 2010; Schnittger et al., 2013; Thol et al., 2011;
Vannucchi et al., 2013).
31
Figure 3 : Rôle d’ASXL1 et du complexe PRC2 dans la régulation des gènes
impliqués dans la leucémogenèse
(A) ASXL1 permet au complexe PRC2 d’appliquer les marques répressives H3K27m3 sur les histones, régulant ainsi l’expression des gènes de la famille HOX et certains miRNA. (B) En cas de mutations d’ASXL1, le complexe PRC2 n’est pas stabilisé. Cela se traduit par une diminution globale des marques H3K27me3 ainsi que la surexpression des gènes de la famille HOX (auto-renouvellement) et de certains miRNA. Ces derniers vont réprimer l’expression de gènes nécessaires à la maturation granuleuse. Ces mécanismes contribuent en partie à l’apparition des phénotypes dysplasiques et leucémiques.
32
Table 6 : Caractéristiques des LAM avec mutations d’ASXL1 au sein des principales séries publiées
Etude Chou et al. Metzeler et al. Pratcorona et al. Schnittger et al.
N 501 423 836 740
Critère de sélection LAM de novo LAM-CN de novo LAM LAM non FAB M3
ASXL1-mut, % (N) 11% (N = 54) 11% (N = 45) 5% (N = 46) 17% (N = 124)
Caractéristiques associées
Age avancé Age avancé Age avancé, sexe
masculin
Sexe masculin Sexe masculin Age avancé Antécédent de SMD
FAB M0 Faible leucocytose Faible leucocytose FAB M2 et M5a
+8 Faible % de blastes médullaires FAB M0 Phénotype immature
RUNX1-mut CEBPA-mut NPM1-wt et FLT3-wt +8
NPM1-wt et FLT3-wt NPM1-wt et FLT3-wt RUNX1-mut
NPM1-wt, FLT3-wt et
DNMT3A-wt
Rémission complète
ASXL1 mut vs. wt 58% vs. 80%, p = 0,013 53% vs. 71%, p = 0,040 61% vs. 80%, p = 0,004 Non disponible
Survie globale médiane (mois)
ASXL1 mut vs. wt 14 vs. 58; p = 0,009 11 vs. 14; p = 0,006 16 vs. 22; p = 0,019 11 vs. 62, p < 0,001
Survie multivariée HR (95CI); p
1,5 (0,8-2,7); p = 0,207 ELN fav : 4,4 (2,4-8,3); p < 0,001
ELN int-1 : 0,9 (0,6-1,5); p = 0,790 1,6 (1,1-2,4); p = 0,010 2,2 p < 0,001

33
IV) Facteurs pronostiques dans les LAM
a. Facteurs liés au patient
i. Âge
L’âge avancé est un facteur pronostique péjoratif majeur, notamment parce qu’il
représente la principale limite aux traitements intensifs (Appelbaum et al., 2006;
Estey, 2007). Pour la plupart des groupes coopératifs, un patient atteint de LAM est
considéré comme «âgé» au-delà de 60 ans. Bien que les traitements intensifs
puissent être proposés aux patients de plus de 60 ans, ils sont associés à une
mortalité précoce plus importante que chez les sujets jeunes. De plus, les LAM du
sujet âgé présentent souvent des caractéristiques biologiques de plus mauvais
pronostic, telles que les anomalies cytogénétiques ou moléculaires défavorables.
L’âge avancé est aussi associé à des taux plus élevés de chimiorésistance primaire
et de rechute (Appelbaum et al., 2006; Büchner et al., 2009). Le traitement de
patients âgés reste à ce jour un enjeu majeur qui doit intégrer des problématiques
spécifiques comme la polymédication, la pharmacocinétique, la
pharmacodynamique, l’autonomie et la qualité de vie (Elliot et al., 2014; Klepin et al.,
2014; Peyrade et al., 2012; Walter and Estey, 2014). Il doit donc s’intégrer dans une
approche spécifique onco-gériatrique.
ii. Etat général et comorbidités
Ces facteurs ont un impact pronostique majeur en conditionnant la réalisation d’un
traitement curatif. Les scores tels que le « performance status » (PS) ou l’index de
comorbidité pour les greffes allogéniques de cellules souches hématopoïétiques
(HCT-CI) sont des outils importants prédisant la mortalité précoce liée au traitement
et la survie globale (Giles et al., 2007; Sorror et al., 2005). Cependant, il n’existe à ce
jour aucune recommandation précise quant au choix d’un traitement intensif en

34
fonction des comorbidités, laissant cette décision à l’appréciation des cliniciens. Deux
publications récentes soulignent les limites du score HCT-CI, incitant à reconsidérer
l’évaluation des comorbidités de façon maladie spécifique (Ostgård et al., 2014;
Versluis et al., 2014).
b. Facteurs liés à la maladie
i. Leucocytose
L’hyperleucocytose au diagnostic est considérée comme un facteur de mauvais
pronostic. Le seuil de leucocytes permettant de définir l’hyperleucocytose n’est pas
clairement défini et varie selon les études, de 30 à 100 G/L, avec une exception pour
les LAM promyélocytaires qui sont considérées hyperleucocytaires à partir de 10
G/L. L’hyperleucocytose représente un facteur de risque de mortalité précoce car elle
est associée à la survenue de complications graves telles que la leucostase, le
syndrome de lyse tumoral ou la CIVD (Greenwood et al., 2006). L’admission initiale
systématique en unité de soins intensifs des patients hyperleucocytaires au
diagnostic pourrait permettre une meilleure prise en charge (Lengliné et al., 2012).
ii. Antécédent de SMD ou SMP et dysplasie multilignée
Les LAM issues de la transformation d’un SMD ou d’une NMP sont généralement de
plus mauvais pronostic que les LAM de novo (Larson, 2007). Ces formes se
présentent souvent au diagnostic avec une DML, définie sur le plan cytologique par
la présence de dysmyélopoïèse sur plus de 50% des cellules d’au moins 2 des 3
lignées hématopoïétiques. Cependant, certaines LAM sans antécédent de SMD
présentent également des signes de DML au diagnostic, suggérant un certain degré
de similitude entre ces LAM et les LAM secondaires aux SMD ou NMP. La
classification OMS de 2001 les a regroupées sous le terme « AML with multilineage
dysplasia » (Vardiman et al., 2002). Ces LAM avec DML sont fréquemment

35
associées à un âge avancé ou à des anomalies cytogénétiques défavorables, leur
conférant un pronostic péjoratif. Cependant, l’impact pronostique indépendant de la
présence d’antécédent de SMD et/ou de DML reste controversé (Haferlach et al.,
2003; Miesner et al., 2010; Wandt et al., 2008; Xu et al., 2014).
iii. LAM radio/chimio induites
Le pronostic des LAM induites par des traitements de radiothérapie ou de
chimiothérapie est globalement plus sombre que celui des LAM de novo (Larson,
2007). Comme pour les LAM avec DML, la valeur pronostique indépendante du
caractère induit de la LAM reste discutée. Ces pathologies sont souvent associées à
des anomalies cytogénétiques de risque défavorable (anomalies des chromosomes 5
et 7 ou translocation impliquant MLL). D’autres facteurs extrinsèques à la LAM
peuvent expliquer le pronostic plus sombre de ces pathologies induites : la
persistance de la maladie primitive ou de séquelles organiques des traitements et/ou
des complications antérieures comme la déplétion des CSH normales, l’altération du
stroma médullaire ou l’immunosuppression chronique.
iv. Anomalies cytogénétiques
Le caryotype des cellules leucémiques est le facteur pronostique majeur permettant
de prédire la réponse à la chimiothérapie d’induction et la survie globale des patients.
La cytogénétique représente la base des stratifications pronostiques actuelles des
LAM (Döhner et al., 2010). En fonction du caryotype, les groupes coopératifs
internationaux ont établi différentes classifications pronostiques en séparant les
patients de façon similaire en 3 groupes de risque cytogénétique : favorable,
intermédiaire et défavorable (Byrd et al., 2002; Grimwade et al., 1998, 2010; Slovak
et al., 2000). Toutes ces classifications considèrent que les anomalies t(8;21),
inv(16)/ t(16;16) et t(15;17) sont de bon pronostic. En revanche, elles diffèrent sur les
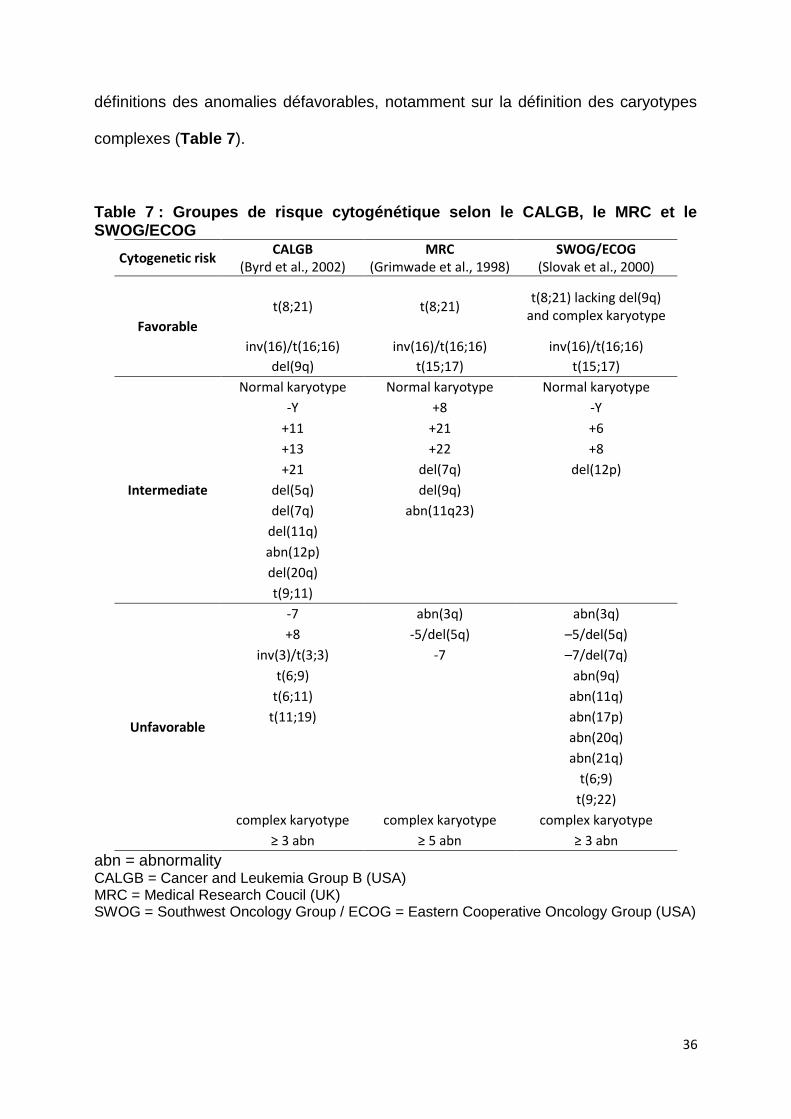
36
définitions des anomalies défavorables, notamment sur la définition des caryotypes
complexes (Table 7).
Table 7 : Groupes de risque cytogénétique selon le CALGB, le MRC et le SWOG/ECOG
Cytogenetic risk CALGB
(Byrd et al., 2002) MRC
(Grimwade et al., 1998) SWOG/ECOG
(Slovak et al., 2000)
Favorable
t(8;21) t(8;21) t(8;21) lacking del(9q)
and complex karyotype
inv(16)/t(16;16) inv(16)/t(16;16) inv(16)/t(16;16)
del(9q) t(15;17) t(15;17)
Intermediate
Normal karyotype Normal karyotype Normal karyotype
-Y +8 -Y
+11 +21 +6
+13 +22 +8
+21 del(7q) del(12p)
del(5q) del(9q)
del(7q) abn(11q23)
del(11q)
abn(12p)
del(20q)
t(9;11)
Unfavorable
-7 abn(3q) abn(3q)
+8 -5/del(5q) –5/del(5q)
inv(3)/t(3;3) -7 –7/del(7q)
t(6;9) abn(9q)
t(6;11) abn(11q)
t(11;19) abn(17p)
abn(20q)
abn(21q)
t(6;9)
t(9;22)
complex karyotype complex karyotype complex karyotype
≥ 3 abn ≥ 5 abn ≥ 3 abn
abn = abnormality CALGB = Cancer and Leukemia Group B (USA) MRC = Medical Research Coucil (UK) SWOG = Southwest Oncology Group / ECOG = Eastern Cooperative Oncology Group (USA)

37
Récemment, une nouvelle entité cytogénétique, appelée caryotype monosomal, a
été définie par la présence d’au moins deux monosomies autosomales ou d’une
monosomie autosomale associée une anomalie de structure (Breems et al., 2008).
Cette entité cytogénétique confère un pronostic très sombre mais n’est pas toujours
utilisée en tant que telle dans les stratifications actuelles car elle est en grande partie
redondante avec le groupe de risque cytogénétique défavorable (Grimwade et al.,
2010). Par défaut, les patients ayant des caryotypes non classés dans les groupes
favorable ou défavorable forment le groupe de risque cytogénétique intermédiaire.
Ce groupe contient les LAM à caryotype normal, les trisomies 8 et les anomalies
cytogénétiques rares dont la valeur pronostique est difficilement identifiable par
manque d’effectif.
v. Anomalies moléculaires utilisées en pratique courante
Les LAM à caryotype normal représentent environ 40 à 50% des LAM de l’adulte,
dont le pronostic est intermédiaire par défaut. Au cours des dernières années,
l’identification de mutations récurrentes a permis d’affiner le pronostic de ce type de
LAM. A ce jour il est possible d’identifier des mutations récurrentes dans la majorité
des LAM à caryotype normal. Ces mutations permettent de mettre en évidence
l’hétérogénéité de ce groupe cytogénétique sur le plan biologique et pronostique. Les
mutations de NPM1, FLT3 et CEBPA sont considérées comme nécessaires pour la
stratification pronostique et la prise en charge thérapeutique des LAM en pratique
courante. Les mutations de NPM1 et de CEBPA sont mutuellement exclusives et
sont considérées comme associées à des entités distinctes au sein de la
classification OMS 2008 (Vardiman et al., 2009). Lorsqu’elles surviennent au sein
d’une LAM à caryotype normal, elles confèrent un pronostic favorable en l’absence
des mutations de FLT3-ITD. Il faut noter que seules les doubles mutations de

38
CEBPA sont associées à ce pronostic favorable (Fasan et al., 2014; Wouters et al.,
2009). Ainsi, la combinaison du statut mutationnel de ces 3 gènes permet d’identifier
2 génotypes favorables au sein des LAM à caryotype normal (NPM1+/FLT-ITD- et
CEBPA+/FLT3-ITD-) pour lesquels la greffe allogénique n’est plus indiquée en
première rémission complète (RC) (Schlenk et al., 2008). Les autres génotypes
(NPM1-/FLT3-ITD+, CEBPA-/FLT3-ITD+ et NPM1-/CEBPA-/FLT3-ITD-) restent de
risque intermédiaire. Au sein des LAM avec mutations de FLT3-ITD, l’analyse du
ratio allèles mutés/sauvages pourrait encore affiner cette stratification moléculaire et
guider les choix thérapeutiques (Linch et al., 2014; Pratcorona et al., 2013;
Schnittger et al., 2011).
c. Classification de l’European LeukemiaNet (ELN)
Cette classification a été publiée 2010 et constitue actuellement la référence pour la
stratification pronostique des patients atteints de LAM et traités en dehors d’essais
thérapeutiques (Döhner et al., 2010). Elle classe ces patients en 4 groupes en se
basant sur les anomalies cytogénétiques et moléculaires : favorable, intermédiaire-1,
intermédiaire-2 et défavorable (Table 8). L’impact pronostique de cette classification
a été validé chez les sujets de moins de 60 ans comme chez les plus âgés (Lazenby
et al., 2014; Mrózek et al., 2012; Röllig et al., 2011; Wetzler et al., 2014). Elle permet
actuellement de guider les traitements de post-rémission, notamment la réalisation
d’une greffe allogénique en première RC. A ce jour, les patients de risque favorable
ne sont pas candidats à l’allogreffe alors que les patients de risque défavorable
pourraient être indiqués pour des approches expérimentales dès l’initiation du
traitement. Les patients des groupes intermédiaires restent quant à eux indiqués
pour une allogreffe en première RC si un donneur compatible est disponible.

39
Table 8 : Classification pronostique de l’European LeukemiaNet
(Döhner et al., 2010)
ELN risk Cytogenetic/molecular abnormality
Favorable
t(8;21)(q22;q22); RUNX1-RUNX1T1
inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
Normal karyotype and: Mutated NPM1 without FLT3-ITD
Mutated CEBPA
Intermediate-1 Normal karyotype and:
Mutated NPM1 and FLT3-ITD
Wild-type NPM1 and FLT3-ITD
Wild-type NPM1 without FLT3-ITD
Intermediate-2 t(9;11)(p22;q23); MLLT3-MLL
Cytogenetic abnormalities not classified as favorable or adverse
Unfavorable
inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
t(6;9)(p23;q34); DEK-NUP214
t(v;11)(v;q23); MLL rearranged
-5 or del(5q)
-7
abn(17p)
complex karyotype (≥ 3 abnormalities)

40
OBJECTIFS ET PRESENTATION DES TRAVAUX
Nos travaux ont porté sur les LAM avec dysplasie, identifiées par la classification
OMS 2008 sous le nom de LAM-MRC (« AML with myelodysplasia-related
changes »). Actuellement définies par la présence de critères cliniques, cytologiques
et cytogénétiques, les LAM-MRC forment un groupe hétérogène tant sur le plan
biologique que pronostique. La complexité de cette définition rend difficile son
utilisation courante. En effet, les termes de LAM de novo et secondaires restent
encore souvent utilisés pour séparer les patients avec ou sans antécédent de SMD.
Cela peut porter à confusion puisque certaines LAM de novo peuvent avoir des
critères morphologiques ou cytogénétiques de SMD, les classant de ce fait comme
des LAM-MRC au même titre que la présence d’antécédent de SMD. De plus, il n’est
pas exclu que certaines LAM aient évolué à partir d’un SMD qui n’a pas été
préalablement diagnostiqué. Cela pourrait se produire en cas de cytopénies
chroniques, peu profondes et cliniquement asymptomatiques. Dans ces cas, le
diagnostic d’hémopathie se ferait uniquement au moment de la transformation en
LAM et celle-ci serait considérée comme de novo. La présence de DML ou
d’anomalies cytogénétiques de SMD pourrait alors permettre de les reclasser en
LAM-MRC, mais ne sont pas d’interprétation facile. Alors qu’un minimum de 10% de
cellules dysplasiques est suffisant pour les diagnostics de SMD, 50% sur au moins 2
lignées sont nécessaires pour affirmer la DML et poser ainsi le diagnostic de LAM-
MRC. Le fait que cette DML ne soit pas toujours retrouvée chez les patients ayant un
antécédent de SMD et/ou des anomalies cytogénétiques de SMD pourrait remettre
en question ce seuil arbitraire de 50%.

41
Nous avons fait l’hypothèse que la caractérisation moléculaire des LAM-MRC
pourrait permettre d’identifier des marqueurs spécifiques associés à ces pathologies.
Ils pourraient alors être considérés comme des critères moléculaires de LAM-MRC
pour affiner les définitions actuelles et stratifier le pronostic de patients (comme c’est
déjà le cas pour les LAM mutées NPM1 ou CEBPA dans le LAM à caryotype
normal). Enfin, ces marqueurs pourraient éventuellement être prédictifs de la
réponse aux traitements ciblés sur des voies de leucémogenèse particulièrement
impliquées dans les LAM-MRC et à terme pourraient être à l’origine du
développement de nouvelles thérapeutiques.
Dans ce mémoire, nous avons posé les questions suivantes :
- Quelle est la valeur pronostique des critères actuels définissant les LAM-
MRC ? (article 1)
- Existe-il des marqueurs moléculaires spécifiques des LAM-MRC ? (article 2)
- La caractérisation moléculaire des LAM-MRC permet-elle la stratification
pronostique de ces patients ? (article 3)
- Les anomalies moléculaires des LAM-MRC peuvent-elles être utilisées pour
prédire la sensibilité aux nouvelles drogues dans une démarche de médecine
personnalisée ? (article 4)

42
ARTICLE 1 :
Prognostic significance of myelodysplasia-related
changes according to the WHO classification among
ELN-intermediate-risk AML patients
Devillier R, Gelsi-Boyer V, Murati A, Prebet T, Rey J, Etienne A, D’Incan E,
Charbonnier A, Blaise D, Mozziconacci MJ, Vey N
American Journal of Hematology 2014 Sep 13 [Epub ahead of print]
43

44

45
Article 1 :
“Prognostic significance of myelodysplasia-related changes according to the WHO
classification among ELN-intermediate-risk AML patients”
Résumé et discussion spécifique
Les LAM-MRC sont classiquement considérées comme des LAM de haut risque du
fait de leur association à un caryotype défavorable et à l’âge avancé des patients
limitant les possibilités de traitements curatifs (Appelbaum et al., 2006; Miesner et al.,
2010). Cependant, l’impact de la dysplasie morphologique, indépendamment de ces
facteurs, reste controversé (Haferlach et al., 2003; Miesner et al., 2010; Wandt et al.,
2008; Xu et al., 2014).
Afin de s’affranchir de l’impact de la cytogénétique, nous avons sélectionné pour
cette étude uniquement des patients atteints de LAM de risque intermédiaire selon la
stratification de l’ELN (Döhner et al., 2010) et comparé le devenir des patients selon
qu’ils aient ou non des critères définissants les LAM-MRC.
Nos résultats ne montrent pas de différence en termes de taux de rémission
complète, de survie sans leucémie et de survie globale entre les patients avec et
sans critères de LAM-MRC. Cela suggère que ces critères n’ont pas d’impact
pronostique au sein des LAM de risque intermédiaire, ce qui pourrait être expliqué
par l’hétérogénéité biologique de cette entité. La caractérisation moléculaire des
LAM-MRC pourrait ainsi mettre en évidence des altérations spécifiques et aider à la
classification diagnostique et pronostique de ces pathologies.

46
ARTICLE 2 :
Acute myeloid leukemia with myelodysplasia-related
changes are characterized by a specific molecular pattern
with high frequency of ASXL1 mutations
Devillier R, Gelsi-Boyer V, Brecqueville M, Carbuccia N, Murati A, Vey N, Birnbaum
D, Mozziconacci MJ.
Am J Hematol. 2012 Jul;87(7):659-62.

47
Acute myeloid leukemia with myelodysplasia-related changes arecharacterized by a specific molecular pattern with high frequency of
ASXL1 mutations
Raynier Devillier,1,2 Veronique Gelsi-Boyer,1,3,4 Mandy Brecqueville,1 Nadine Carbuccia,1 Anne Murati,1,3
Norbert Vey,1,2,4 Daniel Birnbaum,1 and Marie-Joelle Mozziconacci1,3*
To determine whether the distinct and heterogeneous WHO category called ‘‘AML with myelodysplasia-
related changes’’ (MRC-AML), presents speci c molecular alterations we searched for mutations in genes
known to be mutated in malignant myeloid diseases. In 48 MRC-AML patients analyzed, we found 17 muta-tions in ASXL1 (35%), eight in RUNX1 (17%), seven in TET2 (15%), 12 in IDH (n 5 2) or IDH2 (n 5 10) (25%),four in DNMT3A (8%), four in NPM1 (8%), and one in FLT3 (2%). Mutations were more frequent in the inter-
mediate cytogenetic (IC) subgroup of 36 patients than in the unfavorable karyotype subgroup, with an aver-
age ratio mutations/patients of 1.36 [0–3] vs. 0.33 [0–2] (P < 0.001). Then, we compared these 36 patients
with IC MRC-AML with a control panel of 37 no-MRC-AML patients, who had both IC and no dysplasia. ICMRC-AMLs were associated with higher incidence of ASXL1 mutations (47% vs. 0%, P < 0.001) and lower
incidence of DNMT3A (6% vs. 38%, P 5 0.001), NPM1 (11% vs. 62%, P < 0.001) and FLT3 (3% vs. 49%, P <
0.001) mutations. No difference was found in the incidence of IDH1/2 or TET2 mutations according to the
presence of dysplasia. Complete remission rate after intensive treatment was lower in the MRC-AML groupthan in the no-MRC-AML group (48% vs. 78%, P 5 0.023) and in wild type NPM1 patients (50% vs. 84%, P 5
0.009). Our study showed that MRC-AML as de ned in the WHO 2008 classi cation presents a speci c
mutation pattern characterized by a high frequency of ASXL1 mutations and a low rate of NPM1, FLT3, and
DNMT3A mutations. Am. J. Hematol. 87:659–662, 2012. VVC 2012 Wiley Periodicals, Inc.
IntroductionThe 2008 revised WHO classi cation1 recognizes a spe-
ci c category of acute myeloid leukemia (AML) called AMLwith myelodysplasia-related changes (MRC-AML). MRC-AMLs present myelodysplasia-related phenotype and cyto-genetic abnormalities and/or exhibit dysplasia in 50% ormore of the cells in two or more myeloid lineages and/orhistory of myelodysplastic syndrome (MDS). This categoryincludes heterogeneous diseases with distinct clinical andcytogenetic entities. MRC-AMLs are classically consideredas high-risk diseases because they are frequently second-ary and/or unfavorable cytogenetic cases associated witholder age, un t for intensive care.2 To our knowledge, noprevious study has ever reported the predictive value ofdysplasia independently of age, cytogenetic, or previouslydiagnosed MDS. To date, cytogenetic and molecular altera-tions but not dysplasia are considered as independent fac-tors in uencing both prognosis and therapeutic choices.3–5
To determine whether this WHO category, currently de nedsolely upon morphological and cytogenetic criteria,presents speci c molecular alterations, we searched formutations in genes known to be mutated in malignant mye-loid diseases. We report clinical, biological, and molecularcharacteristics of AML according to the presence of criteriafor MRC-AML as described in the WHO classi cation.
MethodsSelection criteria. Patients with the following criteria were included
(1) Marrow blast > 20% at the time of diagnosis except for erythroleuke-
mia according to the FAB classi cation (AML6); (2) No favorable cyto-
genetic abnormalities, i.e., t(8;21), inv(16)/t(16;16), t(15;17); (3) Total
bone marrow sample available at diagnosis; and (4) Signed informed
consent for somatic genetic analyses.
Cases were separated in two groups according to the presence or
not of dysplastic features at diagnosis. Patients with at least one criteria
for AML with MRC according to the WHO classi cation constituted the
MRC-AML group: presence of multilineage dysplasia with (secondary
MRC-AML subgroup) or without (primary MRC-AML subgroup) history
of MDS or myelodysplastic/myeloproliferative syndrome (MDS/MPN);
myelodysplasia-related cytogenetic abnormalities (complex karyotype,
-5 or del(5q), -7, 11q23 abnormalities except t(9;11), t(6;9), 3q26
abnormalities and 17p abnormalities). Cases without any of these crite-
ria formed the no-MRC-AML group.
Treatment modality and response. Patients received intensive
induction therapy (anthracycline and cytarabine), non-intensive treat-
ment (azacytidine, low-dose cytarabine), or supportive care only associ-
ated or not with oral chemotherapy such as 6-mercaptopurine and/or
methotrexate and/or hydroxyurea. Induction treatment modalities were
chosen according to patient’s age, performance status, and cytogenetic
abnormalities. After induction therapy, response was de ned according
to Cheson criteria:6 Bone marrow blast < 5%; no Auer rods; platelet
count > 100 G/L; absolute neutrophil count > 1 G/L and absence of
extramedullary disease de ned complete remission (CR). Patients
reached CR with incomplete recovery when they presented with all cri-
teria for CR except for residual neutropenia < 1 G/L or thrombopenia
< 100 G/L.
Direct gene sequencing. We used direct Sanger gene sequencing
to search for somatic mutations in ASXL1 (Exon 12), RUNX1 (Exons
1–8), TET2 (Exons 3–11), IDH1 (Exon 4) and IDH2 (Exon 4), DNMT3A
(Exons 15–23), FLT3 (internal tandem duplications—ITD—Exons 14
and 15 and point mutation in Exon 20-encoded kinase domain—TKD)
and NPM1 (Exon 12). Methods have been described previously.7
1Centre de Recherche en Cancerologie de Marseille, Laboratoire d’Onco-logie Moleculaire, UMR1068 Inserm, Institut Paoli-Calmettes, Marseille,France; 2Departement d’Hematologie, Institut Paoli-Calmettes, Marseille,France; 3Departement de BioPathologie, Institut Paoli-Calmettes, Marseille,France; 4Faculte de Medecine, Aix-Marseille Universite, Marseille, France
Con ict of interest: Nothing to report.
*Correspondence to: Dr. Marie-Joelle Mozziconacci, Institut Paoli-Calmettes,232 Bd de Sainte-Marguerite, Marseille 13009, France.E-mail: [email protected]
Received for publication 9 March 2012; Accepted 14 March 2012
Am. J. Hematol. 87:659–662, 2012.
Published online 30 March 2012 in Wiley Online Library (wileyonlinelibrary.com).DOI: 10.1002/ajh.23211
Research Article
VVC 2012 Wiley Periodicals, Inc.
American Journal of Hematology 659 http://wileyonlinelibrary.com/cgi-bin/jhome/35105
48
Statistical analyses. We used a Chi-square test to compare patient
characteristics and mutation frequencies in different groups, and to nd
predictive factors associated with CR achievement. Overall survival
(OS) was calculated from date of AML diagnosis to death or last con-
tact using Kaplan Meier8 estimates and Log Rank test.
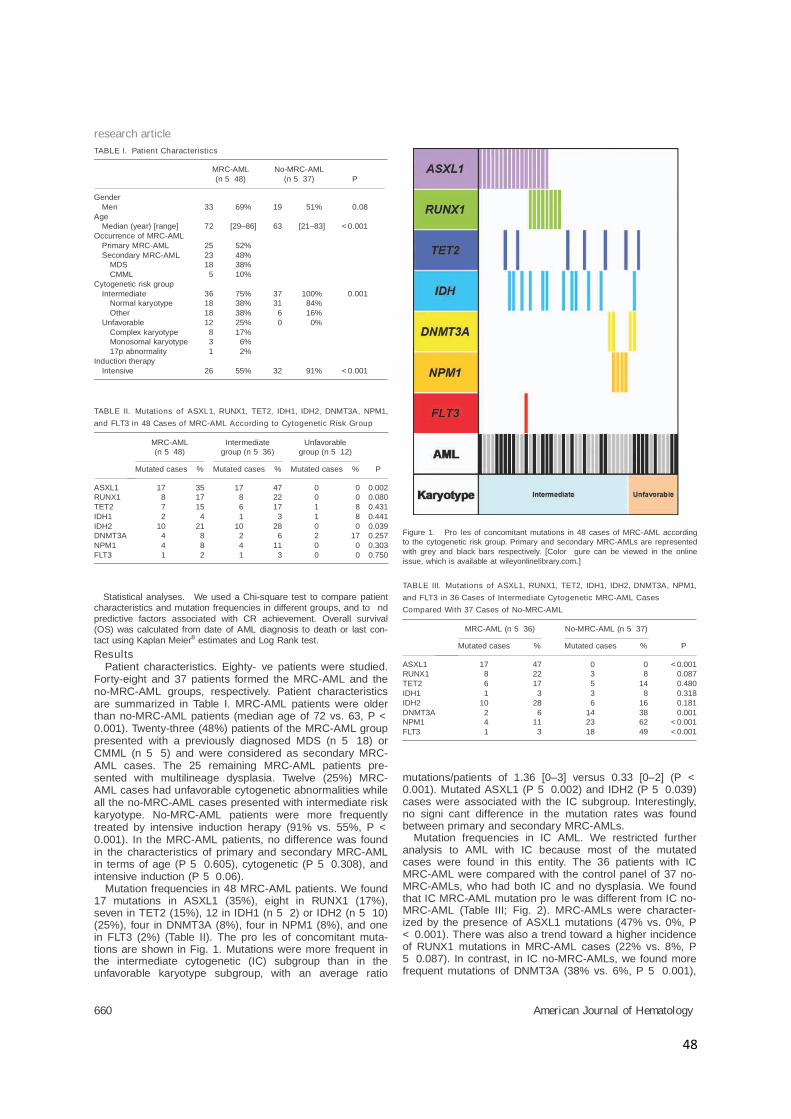
ResultsPatient characteristics. Eighty- ve patients were studied.
Forty-eight and 37 patients formed the MRC-AML and theno-MRC-AML groups, respectively. Patient characteristicsare summarized in Table I. MRC-AML patients were olderthan no-MRC-AML patients (median age of 72 vs. 63, P <0.001). Twenty-three (48%) patients of the MRC-AML grouppresented with a previously diagnosed MDS (n 5 18) orCMML (n 5 5) and were considered as secondary MRC-AML cases. The 25 remaining MRC-AML patients pre-sented with multilineage dysplasia. Twelve (25%) MRC-AML cases had unfavorable cytogenetic abnormalities whileall the no-MRC-AML cases presented with intermediate riskkaryotype. No-MRC-AML patients were more frequentlytreated by intensive induction herapy (91% vs. 55%, P <0.001). In the MRC-AML patients, no difference was foundin the characteristics of primary and secondary MRC-AMLin terms of age (P 5 0.605), cytogenetic (P 5 0.308), andintensive induction (P 5 0.06).
Mutation frequencies in 48 MRC-AML patients. We found17 mutations in ASXL1 (35%), eight in RUNX1 (17%),seven in TET2 (15%), 12 in IDH1 (n 5 2) or IDH2 (n 5 10)(25%), four in DNMT3A (8%), four in NPM1 (8%), and onein FLT3 (2%) (Table II). The pro les of concomitant muta-tions are shown in Fig. 1. Mutations were more frequent inthe intermediate cytogenetic (IC) subgroup than in theunfavorable karyotype subgroup, with an average ratio
mutations/patients of 1.36 [0–3] versus 0.33 [0–2] (P <0.001). Mutated ASXL1 (P 5 0.002) and IDH2 (P 5 0.039)cases were associated with the IC subgroup. Interestingly,no signi cant difference in the mutation rates was foundbetween primary and secondary MRC-AMLs.
Mutation frequencies in IC AML. We restricted furtheranalysis to AML with IC because most of the mutatedcases were found in this entity. The 36 patients with ICMRC-AML were compared with the control panel of 37 no-MRC-AMLs, who had both IC and no dysplasia. We foundthat IC MRC-AML mutation pro le was different from IC no-MRC-AML (Table III; Fig. 2). MRC-AMLs were character-ized by the presence of ASXL1 mutations (47% vs. 0%, P< 0.001). There was also a trend toward a higher incidenceof RUNX1 mutations in MRC-AML cases (22% vs. 8%, P5 0.087). In contrast, in IC no-MRC-AMLs, we found morefrequent mutations of DNMT3A (38% vs. 6%, P 5 0.001),
TABLE I. Patient Characteristics
MRC-AML
(n 5 48)
No-MRC-AML
(n 5 37) P
Gender
Men 33 69% 19 51% 0.08
Age
Median (year) [range] 72 [29–86] 63 [21–83] < 0.001
Occurrence of MRC-AML
Primary MRC-AML 25 52%
Secondary MRC-AML 23 48%
MDS 18 38%
CMML 5 10%
Cytogenetic risk group
Intermediate 36 75% 37 100% 0.001
Normal karyotype 18 38% 31 84%
Other 18 38% 6 16%
Unfavorable 12 25% 0 0%
Complex karyotype 8 17%
Monosomal karyotype 3 6%
17p abnormality 1 2%
Induction therapy
Intensive 26 55% 32 91% < 0.001
TABLE II. Mutations of ASXL1, RUNX1, TET2, IDH1, IDH2, DNMT3A, NPM1,
and FLT3 in 48 Cases of MRC-AML According to Cytogenetic Risk Group
MRC-AML
(n 5 48)
Intermediate
group (n 5 36)
Unfavorable
group (n 5 12)
PMutated cases % Mutated cases % Mutated cases %
ASXL1 17 35 17 47 0 0 0.002
RUNX1 8 17 8 22 0 0 0.080
TET2 7 15 6 17 1 8 0.431
IDH1 2 4 1 3 1 8 0.441
IDH2 10 21 10 28 0 0 0.039
DNMT3A 4 8 2 6 2 17 0.257
NPM1 4 8 4 11 0 0 0.303
FLT3 1 2 1 3 0 0 0.750
Figure 1. Pro les of concomitant mutations in 48 cases of MRC-AML according
to the cytogenetic risk group. Primary and secondary MRC-AMLs are represented
with grey and black bars respectively. [Color gure can be viewed in the online
issue, which is available at wileyonlinelibrary.com.]
TABLE III. Mutations of ASXL1, RUNX1, TET2, IDH1, IDH2, DNMT3A, NPM1,
and FLT3 in 36 Cases of Intermediate Cytogenetic MRC-AML Cases
Compared With 37 Cases of No-MRC-AML
MRC-AML (n 5 36) No-MRC-AML (n 5 37)
PMutated cases % Mutated cases %
ASXL1 17 47 0 0 < 0.001
RUNX1 8 22 3 8 0.087
TET2 6 17 5 14 0.480
IDH1 1 3 3 8 0.318
IDH2 10 28 6 16 0.181
DNMT3A 2 6 14 38 0.001
NPM1 4 11 23 62 < 0.001
FLT3 1 3 18 49 < 0.001
660 American Journal of Hematology
research article
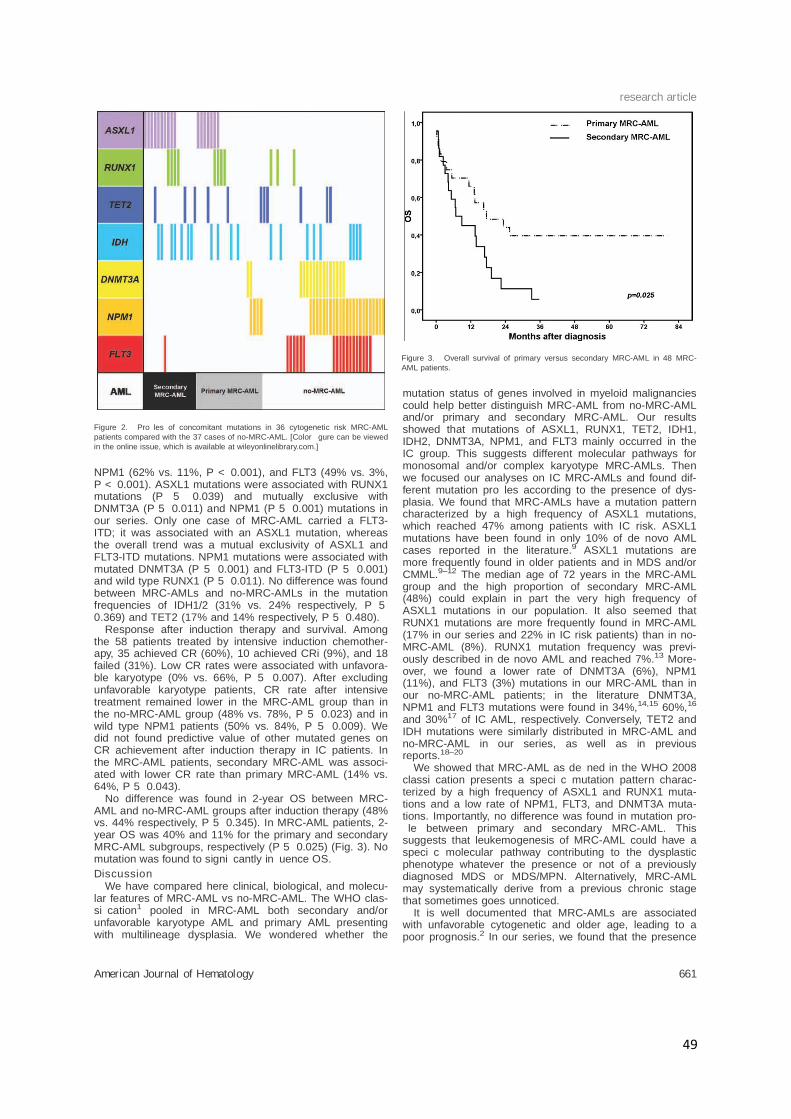
49
NPM1 (62% vs. 11%, P < 0.001), and FLT3 (49% vs. 3%,P < 0.001). ASXL1 mutations were associated with RUNX1mutations (P 5 0.039) and mutually exclusive withDNMT3A (P 5 0.011) and NPM1 (P 5 0.001) mutations inour series. Only one case of MRC-AML carried a FLT3-ITD; it was associated with an ASXL1 mutation, whereasthe overall trend was a mutual exclusivity of ASXL1 andFLT3-ITD mutations. NPM1 mutations were associated withmutated DNMT3A (P 5 0.001) and FLT3-ITD (P 5 0.001)and wild type RUNX1 (P 5 0.011). No difference was foundbetween MRC-AMLs and no-MRC-AMLs in the mutationfrequencies of IDH1/2 (31% vs. 24% respectively, P 50.369) and TET2 (17% and 14% respectively, P 5 0.480).
Response after induction therapy and survival. Amongthe 58 patients treated by intensive induction chemother-apy, 35 achieved CR (60%), 10 achieved CRi (9%), and 18failed (31%). Low CR rates were associated with unfavora-ble karyotype (0% vs. 66%, P 5 0.007). After excludingunfavorable karyotype patients, CR rate after intensivetreatment remained lower in the MRC-AML group than inthe no-MRC-AML group (48% vs. 78%, P 5 0.023) and inwild type NPM1 patients (50% vs. 84%, P 5 0.009). Wedid not found predictive value of other mutated genes onCR achievement after induction therapy in IC patients. Inthe MRC-AML patients, secondary MRC-AML was associ-ated with lower CR rate than primary MRC-AML (14% vs.64%, P 5 0.043).
No difference was found in 2-year OS between MRC-AML and no-MRC-AML groups after induction therapy (48%vs. 44% respectively, P 5 0.345). In MRC-AML patients, 2-year OS was 40% and 11% for the primary and secondaryMRC-AML subgroups, respectively (P 5 0.025) (Fig. 3). Nomutation was found to signi cantly in uence OS.
DiscussionWe have compared here clinical, biological, and molecu-
lar features of MRC-AML vs no-MRC-AML. The WHO clas-si cation1 pooled in MRC-AML both secondary and/orunfavorable karyotype AML and primary AML presentingwith multilineage dysplasia. We wondered whether the
mutation status of genes involved in myeloid malignanciescould help better distinguish MRC-AML from no-MRC-AMLand/or primary and secondary MRC-AML. Our resultsshowed that mutations of ASXL1, RUNX1, TET2, IDH1,IDH2, DNMT3A, NPM1, and FLT3 mainly occurred in theIC group. This suggests different molecular pathways formonosomal and/or complex karyotype MRC-AMLs. Thenwe focused our analyses on IC MRC-AMLs and found dif-ferent mutation pro les according to the presence of dys-plasia. We found that MRC-AMLs have a mutation patterncharacterized by a high frequency of ASXL1 mutations,which reached 47% among patients with IC risk. ASXL1mutations have been found in only 10% of de novo AMLcases reported in the literature.9 ASXL1 mutations aremore frequently found in older patients and in MDS and/orCMML.9–12 The median age of 72 years in the MRC-AMLgroup and the high proportion of secondary MRC-AML(48%) could explain in part the very high frequency ofASXL1 mutations in our population. It also seemed thatRUNX1 mutations are more frequently found in MRC-AML(17% in our series and 22% in IC risk patients) than in no-MRC-AML (8%). RUNX1 mutation frequency was previ-ously described in de novo AML and reached 7%.13 More-over, we found a lower rate of DNMT3A (6%), NPM1(11%), and FLT3 (3%) mutations in our MRC-AML than inour no-MRC-AML patients; in the literature DNMT3A,NPM1 and FLT3 mutations were found in 34%,14,15 60%,16
and 30%17 of IC AML, respectively. Conversely, TET2 andIDH mutations were similarly distributed in MRC-AML andno-MRC-AML in our series, as well as in previousreports.18–20
We showed that MRC-AML as de ned in the WHO 2008classi cation presents a speci c mutation pattern charac-terized by a high frequency of ASXL1 and RUNX1 muta-tions and a low rate of NPM1, FLT3, and DNMT3A muta-tions. Importantly, no difference was found in mutation pro- le between primary and secondary MRC-AML. Thissuggests that leukemogenesis of MRC-AML could have aspeci c molecular pathway contributing to the dysplasticphenotype whatever the presence or not of a previouslydiagnosed MDS or MDS/MPN. Alternatively, MRC-AMLmay systematically derive from a previous chronic stagethat sometimes goes unnoticed.
It is well documented that MRC-AMLs are associatedwith unfavorable cytogenetic and older age, leading to apoor prognosis.2 In our series, we found that the presence
Figure 2. Pro les of concomitant mutations in 36 cytogenetic risk MRC-AML
patients compared with the 37 cases of no-MRC-AML. [Color gure can be viewed
in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3. Overall survival of primary versus secondary MRC-AML in 48 MRC-
AML patients.
American Journal of Hematology 661
research article

50
of dysplasia was signi cantly associated with less CR afterinduction therapy (48% vs. 78%, P 5 0.023). However,these results were not translated into an OS bene t. Thelow numbers and the bias related to patient selection for in-tensive induction could explain in part this result. It seemsthat secondary MRC-AML reached lower CR rate (14% vs.64%, P 5 0.043) and had unfavorable outcome comparedwith primary MRC-AML (2-year OS: 11% vs. 25%, P 50.025).
Beyond the speci c morphologic and cytogenetic fea-tures that de ne MRC-AML, molecular abnormalities suchas ASXL1 and RUNX1 mutations, although not speci c,could help delineate this recent distinct entity. The high fre-quency of ASXL1 mutations in MRC-AML suggests epige-netic deregulation in this kind of myeloid malignancies. Thiscould open perspectives through molecular characterizationof MRC-AML and adapted treatment strategies with newdrugs such as demethylating agents or HDAC inhibitors.
References1. Vardiman JW, Thiele J, Arber DA, Brunning RD, et al. The 2008 revision of the
World Health Organization (WHO) classi cation of myeloid neoplasms and
acute leukemia: Rationale and important changes. Blood 2009;114:937–951.
2. Appelbaum FR, Gundacker H, Head DR, Slovak ML, et al. Age and acute
myeloid leukemia. Blood 2006;107:3481–3485.
3. Schlenk RF, Dohner K, Krauter J, Frohling S, et al. Mutations and treatment
outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med
2008;358:1909–1918.
4. Byrd JC, Mrozek K, Dodge RK, Carroll AJ, et al. Pretreatment cytogenetic
abnormalities are predictive of induction success, cumulative incidence of
relapse, and overall survival in adult patients with de novo acute myeloid leu-
kemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood
2002;100:4325–4336.
5. Grimwade D, Walker H, Oliver F, Wheatley K, et al. The importance of diag-
nostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered
into the MRC AML 10 trial. The Medical Research Council Adult and Child-
ren’s Leukaemia Working Parties. Blood 1998;92:2322–2333.
6. Cheson BD, Bennett JM, Kopecky KJ, Buchner T, et al. Revised recommen-
dations of the International working group for diagnosis, standardization of
response criteria, treatment outcomes, and reporting standards for therapeu-
tic trials in acute myeloid leukemia. J Clin Oncol 2003;21:4642–4649.
7. Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, et al. Mutations of poly-
comb-associated gene ASXL1 in myelodysplastic syndromes and chronic my-
elomonocytic leukaemia. Br J Haematol 2009;145:788–800.
8. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations.
J Am Stat Assoc 1958;53:457–481.
9. Chou WC, Huang HH, Hou HA, Chen CY, et al. Distinct clinical and biological
features of de novo acute myeloid leukemia with additional sex comb-like 1
(ASXL1) mutations. Blood 2010;116:4086–4094.
10. Gelsi-Boyer V, Trouplin V, Roquain J, Adelaide J, et al. ASXL1 mutation is
associated with poor prognosis and acute transformation in chronic myelo-
monocytic leukaemia. Br J Haematol 2010;151:365–375.
11. Metzeler KH, Becker H, Maharry K, Radmacher MD, et al. ASXL1 mutations
identify a high-risk subgroup of older patients with primary cytogenetically nor-
mal AML within the ELN Favorable genetic category. Blood 2011;118:6920–
6929.
12. Thol F, Friesen I, Damm F, Yun H, et al. Prognostic signi cance of ASXL1
mutations in patients with myelodysplastic syndromes. J Clin Oncol
2011;29:2499–2506.
13. Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, et al. RUNX1 muta-
tions in acute myeloid leukemia: Results from a comprehensive genetic and
clinical analysis from the AML study group. J Clin Oncol 2011;29:1364–
1372.
14. Ley TJ, Ding L, Walter MJ, McLellan MD, et al. DNMT3A mutations in acute
myeloid leukemia. N Engl J Med 2010;363:2424–2433.
15. Thol F, Damm F, Ludeking A, Winschel C, et al. Incidence and prognostic
in uence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol
2011;29:2889–2896.
16. Falini B, Mecucci C, Tiacci E, Alcalay M, et al. Cytoplasmic nucleophosmin in
acute myelogenous leukemia with a normal karyotype. N Engl J Med
2005;352:254–266.
17. Frohling S, Schlenk RF, Breitruck J, Benner A, et al. Prognostic signi cance
of activating FLT3 mutations in younger adults (16 to 60 years) with acute my-
eloid leukemia and normal cytogenetics: A study of the AML Study Group
Ulm. Blood 2002;100:4372–4380.
18. Abbas S, Lugthart S, Kavelaars FG, Schelen A, et al. Acquired mutations in
the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute
myeloid leukemia: Prevalence and prognostic value. Blood 2010;116:2122–
2126.
19. Boissel N, Nibourel O, Renneville A, Gardin C, et al. Prognostic impact of iso-
citrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid
leukemia: A study by the Acute Leukemia French Association group. J Clin
Oncol 2010;28:3717–3723.
20. Metzeler KH, Maharry K, Radmacher MD, Mrozek K, et al. TET2 mutations
improve the new European LeukemiaNet risk classi cation of acute myeloid
leukemia: A cancer and leukemia group B study. J Clin Oncol 2011;29:1373–
1381.
662 American Journal of Hematology
research article

51
Article 2 :
“Acute myeloid leukemia with myelodysplasia-related changes are characterized by a
specific molecular pattern with high frequency of ASXL1 mutations”
Résumé et discussion spécifique
Nous avons montré précédemment que les critères de l’OMS 2008 définissant les
LAM-MRC ne semblent pas présenter de valeur pronostique propre mais que cette
évaluation reste délicate du fait d’une importante hétérogénéité. L’identification de
sous-groupes au sein des LAM-MRC en tant qu’entités distinctes sur le plan
biologique et pronostique semble primordiale pour améliorer les stratifications
actuelles et les orientations thérapeutiques. Afin de déterminer si les LAM-MRC
présentent des altérations moléculaires spécifiques, le statut mutationnel de gènes
impliqués dans les hémopathies myéloïdes a été évalué au sein d’une cohorte de 48
patients atteints de LAM-MRC. Nous avons sélectionné des gènes dont les mutations
sont majoritairement rapportées dans les LAM : DNMT3A (Ley et al., 2010), NPM1
(Falini et al., 2005), FLT3 (Fröhling et al., 2002), IDH (Boissel et al., 2010), RUNX1
(Gaidzik et al., 2011) ou dans les SMD ou NMP1 : ASXL1(Gelsi-Boyer et al., 2009),
TET2 (Delhommeau et al., 2009). Nous avons fait l’hypothèse que les LAM-MRC, qui
partagent des caractéristiques de LAM et de SMD, pourraient présenter une
combinaison spécifique de ces mutations.
Nous avons retrouvé 17 mutations sur ASXL1 (35%), 8 sur RUNX1 (17%), 7 sur
TET2 (15%), 12 sur IDH1 (n = 2) ou IDH2 (n = 10) (25%), 4 sur DNMT3A (8%), 4 sur
NPM1 (8%), et une sur FLT3 (2%). Les mutations d’ASXL1 étaient spécifiquement
identifiées au sein des 36 LAM-MRC de risque cytogénétique intermédiaire, où leur
incidence était de 50%.

52
Nous avons ensuite comparé ces profils mutationnels à ceux de 36 patients sans
critère de LAM-MRC (LAM-non-MRC). Les mutations d’ASXL1 ont été exclusivement
retrouvées chez les patients du groupe LAM-MRC (47% vs. 0%, p < 0,001) tandis
que celles de RUNX1 semblaient plus fréquentes dans ce même groupe (22% vs.
8%, p = 0,087). Inversement, ces patients présentaient moins fréquemment de
mutations de DNMT3A (6% vs. 38%, p = 0,001), de NPM1 (11% vs. 62%, p < 0,001)
et de FLT3 (3% vs. 49%, p < 0,001). Les mutations d’ASXL1 semblaient associées à
celles de RUNX1 (p = 0,059) et étaient exclusives de celles de DNMT3A (p = 0,016),
de NPM1 (p < 0,001) et de FLT3 (p = 0,032). Inversement, les mutations de NPM1
étaient associées à celles de DNMT3A (p = 0,001) et de FLT3 (p = 0,012), et
exclusives de celles de RUNX1 (p = 0,005). Les mutations des gènes IDH et TET2
étaient mutuellement exclusives, sans différence de fréquence entre les groupes
LAM-MRC et LAM-non-MRC.
Ces résultats semblent indiquer la présence de 2 profils mutationnels exclusifs,
associant les mutations d’ASXL1 et de RUNX1 aux LAM-MRC et les mutations de
DNMT3A, NPM1 et FLT3 aux LAM-non-MRC. Dans cette étude, les mutations
d’ASXL1 n’étaient observées que dans les LAM-MRC (47% vs. 0%). Dans des
cohortes aspécifiques de LAM dont les critères de LAM-MRC n’étaient pas rapportés,
les taux de mutations d’ASXL1 variaient de 11 à 18% (Chou et al., 2010; Schnittger
et al., 2013). Ces études comparant des LAM avec et sans mutations d’ASXL1
montrent que les mutations sont associées à l’âge avancé, le sexe masculin, les
antécédents de SMD, les mutations de RUNX1, et aux formes sauvages de
DNMT3A, NPM1 et FLT3. Elles semblent également corrélées à un pronostic
péjoratif. Ces résultats sont proches de ceux que nous avons mis en évidence lors
de notre analyse comparative LAM-MRC vs LAM-non-MRC et suggèrent que les

53
mutations d’ASXL1 pourraient être un marqueur moléculaire potentiel de LAM-MRC.
Cependant, mutations d’ASXL1 et critères de LAM-MRC ne sont pas entièrement
superposables, puisque 1 patient sur 2 dans notre série avait une LAM-MRC sans
mutation d’ASXL1. Parmi ces patients non mutés pour ASXL1, d’autres altérations
moléculaires équivalentes aux mutations d’ASXL1 pourraient aboutir au même
phénotype leucémique. Les études pangénomiques pourraient en partie répondre à
cette problématique. En parallèle, les patients présentant des mutations de type
« LAM-non-MRC » telles que celles de NPM1 ou de DNMT3A pourraient
éventuellement être reclassés hors du groupe des LAM-MRC. Cette hétérogénéité
moléculaire au sein même des LAM-MRC participe à la difficulté d’établir une valeur
pronostique spécifique (Wandt et al., 2008; Xu et al., 2014). Du fait du faible effectif
et de l’hétérogénéité des traitements reçus, nous n’avons pas pu mettre en évidence
l’impact pronostique péjoratif des mutations d’ASXL1 tel que rapporté dans les
hémopathies myéloïdes (Bejar et al., 2011; Brecqueville et al., 2014; Chou et al.,
2010; Gelsi-Boyer et al., 2010; Schnittger et al., 2013; Thol et al., 2011; Vannucchi et
al., 2013).
Au-delà des aspects clinico-morphologiques et cytogénétiques qui définissent
actuellement les LAM-MRC, l’identification de marqueurs moléculaires associés à la
dysplasie telles que les mutations d’ASXL1 pourraient être utile pour affiner les
classifications actuelles, permettant de les différencier en entités biologiquement
distinctes, associées à des pronostics spécifiques.

54
ARTICLE 3 :
Clinical, morphological and molecular
characterization of acute myeloid leukemias with
myelodysplasia-related changes
Devillier R, De Mas V, Gelsi-Boyer V, Demur C, Murati A, Corre J, Prebet T, Bertoli
S, Brecqueville M, Recher C, Vey N, Mozziconacci MJ, Delabesse R and Birnbaum D
Manuscrit soumis à Oncotarget, revisions majeures en cours

55
Clinical, morphological and molecular characterization of acute myeloid
leukemias with myelodysplasia-related changes
Raynier Devillier1,2,3, Véronique De Mas5*, Veronique Gelsi-Boyer2,3,4*, Cecile
Demur5*, Anne Murati3,4*, Jill Corre5*, Thomas Prebet1,3, Sarah Bertoli5, Mandy
Brecqueville2,3, Christian Recher5,6, Norbert Vey1,2,3, Marie-Joelle Mozziconacci3,4,
Eric Delabesse5,6 and Daniel Birnbaum3
1Hematology department, Institut Paoli Calmettes, Marseille, France
2Aix-Marseille University, Marseille, France
3Département d'Oncologie Moléculaire ; Centre de Recherche en Cancérologie de
Marseille (CRCM) ; Institut Paoli-Calmettes; UMR1068 Inserm; Marseille, France
4Biopathology department, Institut Paoli Calmettes, Marseille, France
5Hematology department, Institut Universitaire du Cancer Toulouse – Oncopole,
Toulouse, France
6Toulouse University, Toulouse, France
* These authors contributed equally to this work
Running title: Characterization of AML with myelodysplasia-related changes
Corresponding author:
Marie-Joelle Mozziconacci
Institut Paoli Calmettes, Biopathology department
232 boulevard Sainte Marguerite
13009 Marseille, France
Tel: +33 4 91223377
E-mail: [email protected]
Keywords: acute myeloid leukemia; myelodysplasia-related changes; mutational
status; ASXL1; TP53

56
Abstract
Acute myeloid leukemias (AML) with myelodysplasia-related changes (AML-MRC)
are defined by the presence of multilineage dysplasia (MLD), and/or myelodysplastic
syndrome (MDS)-related cytogenetics, and/or previous MDS. To determine if
molecular characterization could identify distinct biological and prognostic subgroups
we analyzed the mutational status of ASXL1, RUNX1, DNMT3A, NPM1, FLT3 and
TP53 in 125 AML-MRC patients according to the presence of MLD, cytogenetics and
outcome. ASXL1 mutations (n=26, 21%) were associated with a higher proportion of
marrow dysgranulopoiesis (mutant vs. wild-type: 75% vs. 55%, p=0.030) and were
mostly found in intermediate cytogenetic AML (23/26) in which they predicted inferior
2-year overall survival (OS, mutant vs. wild-type: 14% vs. 37%, p=0.030). TP53
mutations (n=28, 22%) were mostly found in complex karyotype AML (26/28) and
predicted poor outcome within unfavorable cytogenetic risk AML (mutant vs. wild-
type: 9% vs. 40%, p=0.040). In multivariate analysis, the presence of either ASXL1 or
TP53 mutation was the only independent factor associated with shorter OS (HR,
95%CI: 2.53, 1.40-4.60, p=0.002) while MLD, MDS-related cytogenetics and previous
MDS history did not influence OS. We conclude that ASXL1 and TP53 mutations
identify two molecular subgroups among AML-MRCs, with specific poor prognosis.
This could be useful for future diagnostic and prognostic classifications.

57
Introduction
In the WHO 2008 classification, acute myeloid leukemia (AML) with myelodysplasia-
related changes (AML-MRC) is defined as a distinct entity by the presence of
multilineage dysplasia (MLD), and/or myelodysplastic syndrome (MDS)-related
cytogenetics, and/or previously diagnosed MDS or MDS/Myeloproliferative neoplasm
(MDS/MPN)[1]. The prognostic value of these three criteria is not established. The
independent prognostic value of MLD is controversial and varies among different
subsets of AML[2–7]. AML with MDS-related cytogenetics or previously diagnosed
MDS or MDS/MPN have often unfavorable cytogenetics, and are associated with
poorer outcome than AML without criteria of AML-MRC[2,8,9].
Although gene mutations are now a major tool for AML classification into distinct
entities with specific prognosis[10–12], no molecular pattern is currently associated
with AML-MRC. We hypothesized that molecular characterization of AML-MRC could
help identify subgroups of AML-MRC with distinct biological features and specific
outcome. We had previously reported that AML-MRC have a specific mutation
pattern sharing mutations found in both AML and high risk MDS and a particularly
high frequency of ASXL1 mutation[13]. We report here a cohort of patients with AML-
MRC for whom we analyzed the presence of mutational events according to AML-
MRC criteria (MLD, cytogenetics and patient history) and identified mutation-based
subgroups with specific poor outcome.

58
Patients and Methods
Selection criteria
We conducted a retrospective analysis of patients from two French centers. Selection
criteria for analyses were: (i) Diagnosis of AML with criteria for AML-MRC according
to the WHO classification[1]: previously diagnosed MDS or MPN/MDS; and/or
multilineage dysplasia (MLD); and/or MDS-related cytogenetic abnormalities
(complex karyotype (CK) defined by three or more chromosomal abnormalities, -7 or
del(7q); -5 or del(5q); i(17q) or t(17p); -13 or del(13q); del(11q); del(12p) or t(12p);
del(9q); idic(X)(q13); t(11;16)(q23;p13.3); t(3;21)(q26.2;q22.1); t(1;3)(p36.3;q21.1);
t(2;11)(p21;q23); t(5;12)(q33;p12); t(5;7)(q33;q11.2); t(5;17)(q33;p13); t(5;
10)(q33;q21); t(3;5)(q25;q34); (ii) Genomic DNA available for mutational analyses.
AML with inv(3)/t(3;3), t(6;9) and t(v;11q23) or with favorable risk according to the
ELN classification (inv(16)/t(16;16), t(8;21), t(15;17), normal karyotype AML with
NPM1 or CEBPA mutations) were excluded as well as therapy-related AML[14].
Morphological analyses
Smears of bone marrow aspirates made at diagnosis were collected and stained by
May-Grünwald-Giemsa. Each smear was retrospectively analyzed by two expert
cytologists in both centers. We assessed the percentage of dysplastic cells in each
lineage. MLD was assessed according to the WHO classification criteria: at least
50% of dysplastic cells in at least 2 lineages. We established the percentage of
dysgranulopoiesis (DGP), dyserythropoiesis (DEP) and dysmegakaryopoiesis (DMP)
for each smear.
Mutational analyses

59
Direct sequencing was done using the Sanger method as previously described[15].
We searched for mutation of ASXL1 (exon 12), RUNX1 (exon 1-8), DNMT3A (exon
15-23), NPM1 (exon 12), FLT3 (internal tandem duplication ITD, exon 14-15) and
TP53 (exon 4-10).
Statistical analyses
Chi-square or Fischer tests were used to compare categorical variables. We used
non-parametric test (U Mann Whitney) to compare the median percentage of
dysplastic cells in each lineage according to mutational status for each gene. As
multivariate model, linear regression was used to find correlation between
morphologic dysplasia and the presence of mutation. Survivals were calculated using
the Kaplan Meier estimator[16]. Time to event started from the date of diagnosis. We
compared survivals using the Log-Rank test. Cox regression was used for
multivariate of OS[17]. Statistics were computed using the R.3.1.0 software.

60
Results
Patient, disease and treatment characteristics
We studied 149 patients. After morphological review, 24 patients were excluded from
the main analysis because of not enough dysplasia to reach the MLD criteria. These
patients were analyzed separately (Supplemental Table 1). The remaining 125
fitting the AML-MRC criteria were considered for the main analysis and their
characteristics are reported in Table 1. Median age was 71 years (range: 18-90).
Fifty-nine patients (47%) had a previously diagnosed MDS. Seventy-one patients
(57%) had MDS-related cytogenetics, including 42 patients (34%) with complex
karyotype AML (CK-AML). Ninety-four patients were evaluable for morphological
dysplasia by the double centralized review. Multilineage dysplasia (MLD) was found
in 38/94 patients (40%). Sixty-seven patients (54%) received intensive induction
chemotherapy. Mutations were found in ASXL1 (n = 26, 21%), RUNX1 (n = 15, 12%),
DNMT3A (n = 11, 9%), NPM1 (n = 4, 3%), FLT3 (n = 9, 7%) and TP53 (n = 28, 22%).
No mutation was found in 47 (38%) patients (31 with non-complex karyotype NCK-
AML and 16 with CK-AML).
Mutation profiles according to previous history of MDS, cytogenetics and MLD
TP53 mutations were exclusive from all other mutations and all but two (26/28, 93%)
were found in CK-AML. Mutations in the other genes were almost exclusively found
in NCK-AMLs (Table 2A, Figure 1). In NCK-AMLs, ASXL1 was the most frequently
mutated gene (26/83, 31%). These mutations were associated with the absence of
MDS-related cytogenetics (MDS-related cytogenetics: yes vs. no: 4/29, 14% vs.

61
22/54, 41%, p = 0.013). Other mutations were equally distributed whether MDS-
related cytogenetics was present or not (Figure 1).
We analyzed the presence of mutations according to the cytogenetic risk group
(Table 2B). In the 94 patients evaluable for MLD, patients with criteria for MLD had
more ASXL1 mutations (MLD: 15/38, 39% vs. no MLD: 8/48, 14%, p = 0.007). We did
not find any correlation between the presence of MLD and other mutations. Median
percentages of bone marrow DGP, DEP and DMP in the 94 patients were 64%, 24%
and 45% respectively. ASXL1 mutations were associated with higher DGP (ASXL1-
mut: 75% vs. ASXL1-wt: 55%, p = 0.030) but similar DEP (ASXL1-mut: 20% vs.
ASXL1-wt: 25%, p = 0.933) and DMP (ASXL1-mut: 53% vs. ASXL1-wt: 40%, p =
0.139). There was no difference in percent of morphologic dysplasia according to any
other genes (Supplemental Table 2). In a linear regression analysis including the
mutation status of the 6 genes, mutation of ASXL1 remained associated with higher
DGP (Coefficient beta = 20, p = 0.023, Supplemental Table 3). The frequencies of
these mutations were not different according to the presence or not of prior MDS or
MDS/MPN.
Outcome after intensive chemotherapy
Only the 67 patients who received intensive induction chemotherapy were considered
for the outcome analyses. Among them, 42 achieved CR (63%). Cytogenetic risk
group, the presence of MDS-related cytogenetics, previous history of MDS or
MDS/MPN, and MLD did not influence the CR rate (data not shown). The presence of
an ASXL1 mutation was associated with a lower CR rate (ASXL1-mut vs. ASXL1-wt:
40% vs. 69%, p = 0.039). Other gene mutations did not influence the CR rate.
The 2-year OS was 24% in the 67 intensively treated patients. Cytogenetics did not
predict outcome, with a 2-year OS of 27% and 20% in the intermediate and

62
unfavorable groups, respectively (p=0.351, Table 3). Similarly, MDS-related
cytogenetics, previous history of MDS or MDS/MPN, and MLD did not significantly
influence OS (data not shown). Among the intermediate cytogenetic (IC-AML)
patients, the presence of an ASXL1 mutation was associated with worse 2-year OS
(14%) compared to patients without ASXL1 mutation (37%, p = 0.030, Figure 2A,
Table 3).
In the unfavorable cytogenetic (UC-AML) group, TP53-mutated patients had a lower
2-year OS (9%) than TP53-wild type patients (26%, p = 0.040, Figure 2B, Table 3).
In multivariate analyses adjusted for age and WBC, IC-AML with mutated ASXL1 (HR
= 2.67, 95%CI = [1.15-6.24], p = 0.023) and UC-AML with mutated TP53 (HR = 5.44,
95%CI = [2.16-13.65], p < 0.001) had shorter OS than IC-AML with wild type ASXL1
(considered as reference, HR = 1). Of note, patients with UC-AML with wild type
TP53 (HR = 1.14, 95%CI = [0.52-2.50], p = 0.743) had similar OS compared to those
with intermediate cytogenetic and no ASXL1 mutation (considered as reference, HR
= 1).
Using only the mutational status of ASXL1 and TP53 to stratify patient outcome, we
found that patients who presented with either ASXL1 or TP53 mutation had worse 2-
year OS (15%) than those with both ASXL1 and TP53 wild type (29%, p = 0.005,
Table 3, Figure 3). In multivariate analysis including age, WBC and cytogenetics, the
presence of either ASXL1 or TP53 mutation remained the only independent
predictive factor associated with both lower CR rate (HR = 0.29, 95%CI = [0.09-0.89],
p = 0.031) and shorter OS (HR = 2.53, 95%CI = [1.40-4.60] , p = 0.002).
Patients with dysplasia but without criteria for MLD
These 24 patients were separately analyzed because they did not reach AML-MRC
criteria after morphological review. Indeed, they had no previous history of MDS or

63
MDS/MPN and no MDS-related cytogenetics. Morphological review showed
dysplasia that did not reach criteria for MLD. All of them had intermediate
cytogenetics and 15 (63%) had normal karyotype (Supplemental table 1). Among
these 24 patients, median percentage of DGP, DEP and DMP was 50% (range: 7-
97%), 20% (range: 0-45%) et 26% (range: 0-83%), respectively. We found 4
mutations in ASXL1 (17%), 5 in RUNX1 (21%), 1 in DNMT3A (4%), 2 in NPM1 (8%),
4 in FLT3 (17%) and no TP53 mutation. ASXL1 mutated patients had a median
percent of DGP of 62% (range: 35-90%) vs. 50% (range: 7-97%) in those with wild
type ASXL1.

64
Discussion
We previously reported that AMLs with MRC harbor a specific mutational profile with
a high proportion of ASXL1 and RUNX1 mutations and less DNMT3A, FLT3 and
NPM1 mutations than AMLs without criteria of AML-MRC[13]. In this series including
only patients with AML-MRC, we have evaluated the correlation between these
mutations and the different criteria defining AML-MRC (previous history of MDS or
MDS/MPN, MDS-related cytogenetics, and MLD). We found that none of the three
MRC criteria could help identify a specific mutational profile within AML-MRC,
suggesting that these criteria do not have a molecular basis, at least within the limit of
the genes studied. This heterogeneity in terms of biological features as well as in
prognostic significance suggests that AML-MRC is not a true distinct entity. Although
they are defined as AML-MRC because of MDS-related cytogenetics, CK-AML
should be considered separately because of the presence of a specific mutational
profile consisting in a high frequency of TP53 mutations and the absence of other
mutations. As expected, TP53 mutations predicted a worse outcome among patients
with unfavorable cytogenetics, supporting that genetic stratification is useful to predict
differential patient outcome although classified in the same cytogenetic risk group.
This is in line with previous reports showing the close correlation between TP53
mutation, CK-AML and poor outcome[18,19]. In NCK-AML, we found that ASXL1 was
the most frequently mutated gene (31%). It was the only mutation associated with the
presence of MLD and a higher proportion of DGP in bone marrow. This is in
agreement with previous biological reports showing the role of ASXL1 in the
appearance of dysplasia and myeloid transformation[20–23]. However, the overlap

65
between ASXL1 mutations and morphological MLD was not complete, because 39%
of patients with MLD had ASXL1 mutation and 14% patients without MLD had ASXL1
mutation. Thus, there is no strong association between morphological analyses and
the presence of an ASXL1 mutation. Interestingly, ASXL1 mutations were associated
with the absence of MDS-related cytogenetics among the NCK-AML, as if ASXL1
mutations and karyotype with MDS-related abnormalities were mutually exclusive.
Finally, there was no correlation between ASXL1 mutation and previous history of
MDS or MDS/MPN.
Taken together, these results suggest that the presence of an ASXL1 mutation could
be considered as an independent molecular marker of dysplasia in AML that is not
redundant with the criteria defining AML-MRC. This could be helpful in the
perspective of developing a molecular classification of AML-MRC. This statement is
also supported by the presence of a specific gene expression profile associated with
ASXL1-mutated AML[24]. Like TP53 stratification of outcome in UC-AML, ASXL1
mutations were associated with a shorter OS in intermediate risk patients, with a 2-
year OS (14%) close to that observed in unfavorable cytogenetic patients (20%).
Different studies that did not specifically focus on AML-MRC also reported this poor
outcome associated to ASXL1 mutations[25,26]. Thus, stratification upon ASXL1 and
TP53 mutation, which identified two distinct biological subgroups among AML-MRC,
was the only significant predictor of outcome while usual cytogenetic risk
classification or the different criteria defining AML-MRC (prior MDS or MDS/MPN,
MDS-related cytogenetics and MLD) failed to predict patient outcome in our series.
Although ASXL1 and TP53 mutations could classify distinct subgroups of AML-MRC,
patients without any of these mutations still represent a heterogeneous group of
AML-MRC harboring different morphological, cytogenetic and molecular features.

66
The mutations of DNMT3A and/or NPM1, which are usually found in de novo AML
and are mutually exclusive with ASXL1 and TP53 mutations, could identify patients
for whom the definition of AML-MRC should be questioned. Falini et al. reported that
MLD did not identify distinct biological and clinical entities among NPM1-mutated
AML, with overlapping gene expression profiling and similar outcome[4]. In contrast,
some patients had morphological dysplasia but not enough to reach MLD criteria,
leading to their exclusion from the AML-MRC category in the absence of other
criteria. Among these patients, those who presented with ASXL1 mutations might be
included in the same subgroup as ASXL1-mutated AML-MRC. This supports the
need of redefining AML-MRC upon molecular abnormalities. We propose a model in
which the presence of these molecular abnormalities could help reclassify AML-MRC
(Figure 4).
Finally, we did not find any mutation in the 6 genes studied in 47 patients, suggesting
the need to identify other molecular markers to stratify these patients. Because
ASXL1 is a key regulator of the polycomb repressive complex 2 (PRC2), it is possible
that AML carrying alterations of other genes involved in methylation marks via
PRC2 and myeloid transformation such as EZH2[27,28], JARID2[29] or BAP1[30,31]
could share characteristics of ASXL1-mutated AML. This could help identify AML-
MRC through a common altered molecular pathway and could help develop future
targeted treatments.
We conclude that the criteria defining AML-MRC do not identify distinct clinical and
biological subgroups and do not predict outcome of patients with AML-MRC. In
contrast, ASXL1 and TP53-mutated AML identify two distinct biological subgroups of
AML-MRC with very poor outcome. This molecular characterization could be useful to

67
redefine AML-MRC in a future classification aiming at merging biological
characterization and specific prognostic value.

68
Acknowledgements
This work was supported by Inserm, Institut Paoli-Calmettes and grants from the
Fondation ARC pour la Recherche sur le Cancer (DB) and INCa-DGOS-Inserm 6038.

69
Conflict of interest
The authors have nothing to disclose.

70
References
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris
NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision
of the World Health Organization (WHO) classification of myeloid neoplasms and
acute leukemia: rationale and important changes. Blood. 2009;114(5):937-51.
2. Haferlach T, Schoch C, Löffler H, Gassmann W, Kern W, Schnittger S,
Fonatsch C, Ludwig W-D, Wuchter C, Schlegelberger B, Staib P, Reichle A, et al.
Morphologic Dysplasia in De Novo Acute Myeloid Leukemia (AML) Is Related to
Unfavorable Cytogenetics but Has No Independent Prognostic Relevance Under the
Conditions of Intensive Induction Therapy: Results of a Multiparameter Analysis From
the German AML Cooperative Group Studies. J Clin Oncol. 2003;21(2):256-65.
3. Miesner M, Haferlach C, Bacher U, Weiss T, Macijewski K, Kohlmann A, Klein
H-U, Dugas M, Kern W, Schnittger S, Haferlach T. Multilineage dysplasia (MLD) in
acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities
and a prior history of MDS or MDS/MPN but has no independent prognostic
relevance: a comparison of 408 cases classified as « AML not otherwise specified »
(AML-NOS) or « AML with myelodysplasia-related changes » (AML-MRC). Blood.
2010;116(15):2742-51.
4. Falini B, Macijewski K, Weiss T, Bacher U, Schnittger S, Kern W, Kohlmann A,
Klein H-U, Vignetti M, Piciocchi A, Fazi P, Martelli MP, et al. Multilineage dysplasia
has no impact on biologic, clinicopathologic, and prognostic features of AML with
mutated nucleophosmin (NPM1). Blood. 2010;115(18):3776-86.
5. Bacher U, Schnittger S, Macijewski K, Grossmann V, Kohlmann A, Alpermann
T, Kowarsch A, Nadarajah N, Kern W, Haferlach C, Haferlach T. Multilineage
dysplasia does not influence prognosis in CEBPA-mutated AML, supporting the WHO
proposal to classify these patients as a unique entity. Blood. 2012;119(20):4719-22.

71
6. Rozman M, Navarro J-T, Arenillas L, Aventín A, Giménez T, Alonso E, Perea
G, Camós M, Navarrete M, Tuset E, Florensa L, Millá F, et al. Multilineage dysplasia
is associated with a poorer prognosis in patients with de novo acute myeloid
leukemia with intermediate-risk cytogenetics and wild-type NPM1. Ann Hematol.
2014;
7. Devillier R, Gelsi-Boyer V, Murati A, Prebet T, Rey J, Etienne A, D’Incan E,
Charbonnier A, Blaise D, Mozziconacci M-J, Vey N. Prognostic significance of
myelodysplasia-related changes according to the WHO classification among ELN-
intermediate-risk AML patients. Am J Hematol. 2014;
8. Wandt H, Schäkel U, Kroschinsky F, Prange-Krex G, Mohr B, Thiede C,
Pascheberg U, Soucek S, Schaich M, Ehninger G. MLD according to the WHO
classification in AML has no correlation with age and no independent prognostic
relevance as analyzed in 1766 patients. Blood. 2008;111(4):1855-61.
9. Xu X-Q, Wang J-M, Gao L, Qiu H-Y, Chen L, Jia L, Hu X-X, Yang J-M, Ni X,
Chen J, Lü S-Q, Zhang W-P, et al. Characteristics of acute myeloid leukemia with
myelodysplasia-related changes: A retrospective analysis in a cohort of Chinese
patients. Am J Hematol. 2014;
10. Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, Lai YJ, Tseng MH,
Huang CF, Chiang YC, Lee FY, Kuo YY, et al. Integration of cytogenetic and
molecular alterations in risk stratification of 318 patients with de novo non-M3 acute
myeloid leukemia. Leukemia. 2014;28(1):50-8.
11. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van
Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, et al.
Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl
J Med. 2012;366(12):1079-89.
12. Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, Schmid
C, Wendtner C-M, Staib P, Serve H, Kreuzer K-A, Kern W, et al. A novel hierarchical
prognostic model of AML solely based on molecular mutations. Blood.
2012;120(15):2963-72.

72
13. Devillier R, Gelsi-Boyer V, Brecqueville M, Carbuccia N, Murati A, Vey N,
Birnbaum D, Mozziconacci M-J. Acute myeloid leukemia with myelodysplasia-related
changes are characterized by a specific molecular pattern with high frequency of
ASXL1 mutations. Am J Hematol. 2012;87(7):659-62.
14. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK,
Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, et al. Diagnosis
and management of acute myeloid leukemia in adults: recommendations from an
international expert panel, on behalf of the European LeukemiaNet. Blood.
2010;115(3):453-74.
15. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N,
Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, et al. Mutations of
polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic
myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788-800.
16. Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J
Am Stat Assoc. 1958;53(282):457-81.
17. Cox D. Regression models and life tables. J R Stat Soc Ser B Methodol.
1972;34(2):187-220.
18. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M,
Kugler C-M, Holzmann K, Gaidzik VI, Paschka P, Held G, et al. TP53 alterations in
acute myeloid leukemia with complex karyotype correlate with specific copy number
alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114-21.
19. Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green C, Gale RE, Hills R,
Linch DC. TP53 gene mutation is frequent in patients with acute myeloid leukemia
and complex karyotype, and is associated with very poor prognosis. Leukemia.
2009;23(1):203-6.
20. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S,
Patel JP, Chung YR, Koche R, Perna F, Zhao X, et al. ASXL1 mutations promote
myeloid transformation through loss of PRC2-mediated gene repression. Cancer
Cell. 2012;22(2):180-93.

73
21. Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C,
Hricik T, Ndiaye-Lobry D, Lafave LM, Koche R, Shih AH, et al. Deletion of Asxl1
results in myelodysplasia and severe developmental defects in vivo. J Exp Med.
2013;210(12):2641-59.
22. West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired
ASXL1 mutations are common in patients with inherited GATA2 mutations and
correlate with myeloid transformation. Haematologica. 2014;99(2):276-81.
23. Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, Kagiyama
Y, Kawabata KC, Nakahara F, Izawa K, Oki T, Maehara A, et al. Myelodysplastic
syndromes are induced by histone methylation–altering ASXL1 mutations. J Clin
Invest. 2013;123(11):4627-40.
24. Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K,
Nicolet D, Whitman SP, Wu Y-Z, Schwind S, Powell BL, Carter TH, et al. ASXL1
mutations identify a high-risk subgroup of older patients with primary cytogenetically
normal AML within the ELN Favorable genetic category. Blood. 2011;118(26):6920-9.
25. Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V,
Kohlmann A, Illig T, Klopp N, Wichmann H-E, Kreuzer K-A, Schmid C, et al. ASXL1
exon 12 mutations are frequent in AML with intermediate risk karyotype and are
independently associated with an adverse outcome. Leukemia. 2013;27(1):82-91.
26. Chou W-C, Huang H-H, Hou H-A, Chen C-Y, Tang J-L, Yao M, Tsay W, Ko B-
S, Wu S-J, Huang S-Y, Hsu S-C, Chen Y-C, et al. Distinct clinical and biological
features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1)
mutations. Blood. 2010;116(20):4086-94.
27. Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, Sun A, Ruan J, Chen S, Wu
D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -
7/del(7q) in De Novo Acute Myeloid Leukemia. Richards KL, éditeur. PLoS ONE.
2013;8(4):e61341.
28. Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, Zhang Z, He Q, Chang C.
Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic

74
syndromes: relation to adverse epigenetic alteration and poor prognostic scoring.
Ann Hematol. 2011;90(6):643-53.
29. Puda A, Milosevic JD, Berg T, Klampfl T, Harutyunyan AS, Gisslinger B, Rumi
E, Pietra D, Malcovati L, Elena C, Doubek M, Steurer M, et al. Frequent deletions of
JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol.
2012;87(3):245-50.
30. Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new factors in myelopoiesis,
cancer and epigenetics. Leukemia. 2013;27(1):10-5.
31. Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS,
Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, Wu J, Phu L, et al. Loss of the
tumor suppressor BAP1 causes myeloid transformation. Science.
2012;337(6101):1541-6.

75
Table legends
Table 1: patient and disease characteristics
Table 2: Frequencies of mutation in ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD and
TP53 in the 125 patients and according to the karyotypes (A) and to the cytogenetic
risk groups (B)
Table 3: Univariate analyses of overall survival
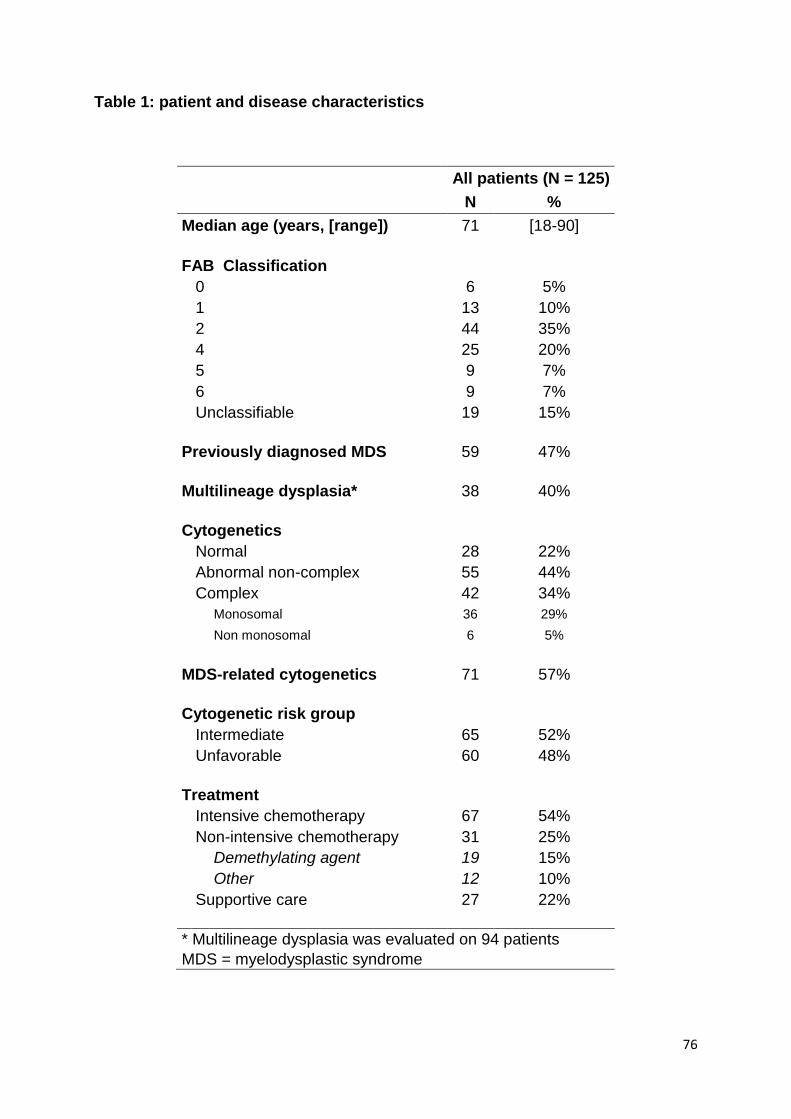
76
Table 1: patient and disease characteristics
All patients (N = 125)
N %
Median age (years, [range]) 71 [18-90]
FAB Classification
0 6 5%
1 13 10%
2 44 35%
4 25 20%
5 9 7%
6 9 7%
Unclassifiable 19 15%
Previously diagnosed MDS 59 47%
Multilineage dysplasia* 38 40%
Cytogenetics
Normal 28 22%
Abnormal non-complex 55 44%
Complex 42 34%
Monosomal 36 29%
Non monosomal 6 5%
MDS-related cytogenetics 71 57%
Cytogenetic risk group
Intermediate 65 52%
Unfavorable 60 48%
Treatment
Intensive chemotherapy 67 54%
Non-intensive chemotherapy 31 25%
Demethylating agent 19 15%
Other 12 10%
Supportive care 27 22%
* Multilineage dysplasia was evaluated on 94 patients
MDS = myelodysplastic syndrome
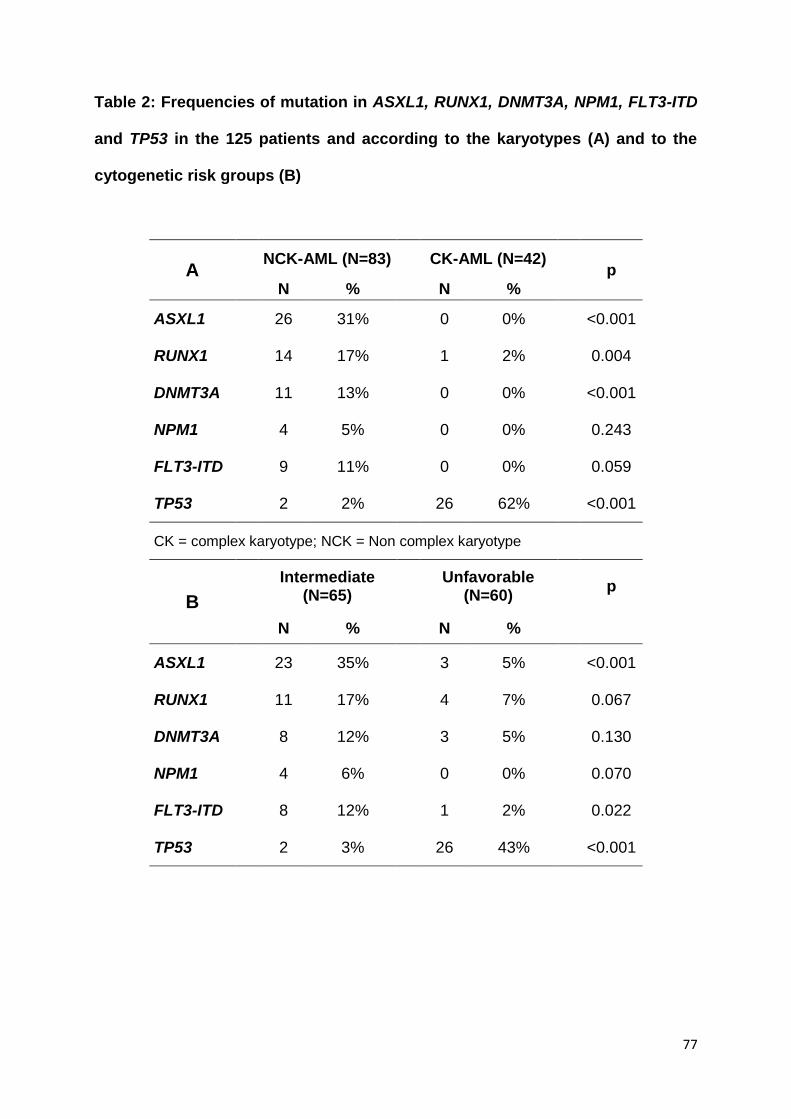
77
Table 2: Frequencies of mutation in ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD
and TP53 in the 125 patients and according to the karyotypes (A) and to the
cytogenetic risk groups (B)
A NCK-AML (N=83) CK-AML (N=42)
p
N % N %
ASXL1 26 31% 0 0% <0.001
RUNX1 14 17% 1 2% 0.004
DNMT3A 11 13% 0 0% <0.001
NPM1 4 5% 0 0% 0.243
FLT3-ITD 9 11% 0 0% 0.059
TP53 2 2% 26 62% <0.001
CK = complex karyotype; NCK = Non complex karyotype
B
Intermediate (N=65)
Unfavorable
(N=60) p
N % N %
ASXL1 23 35% 3 5% <0.001
RUNX1 11 17% 4 7% 0.067
DNMT3A 8 12% 3 5% 0.130
NPM1 4 6% 0 0% 0.070
FLT3-ITD 8 12% 1 2% 0.022
TP53 2 3% 26 43% <0.001
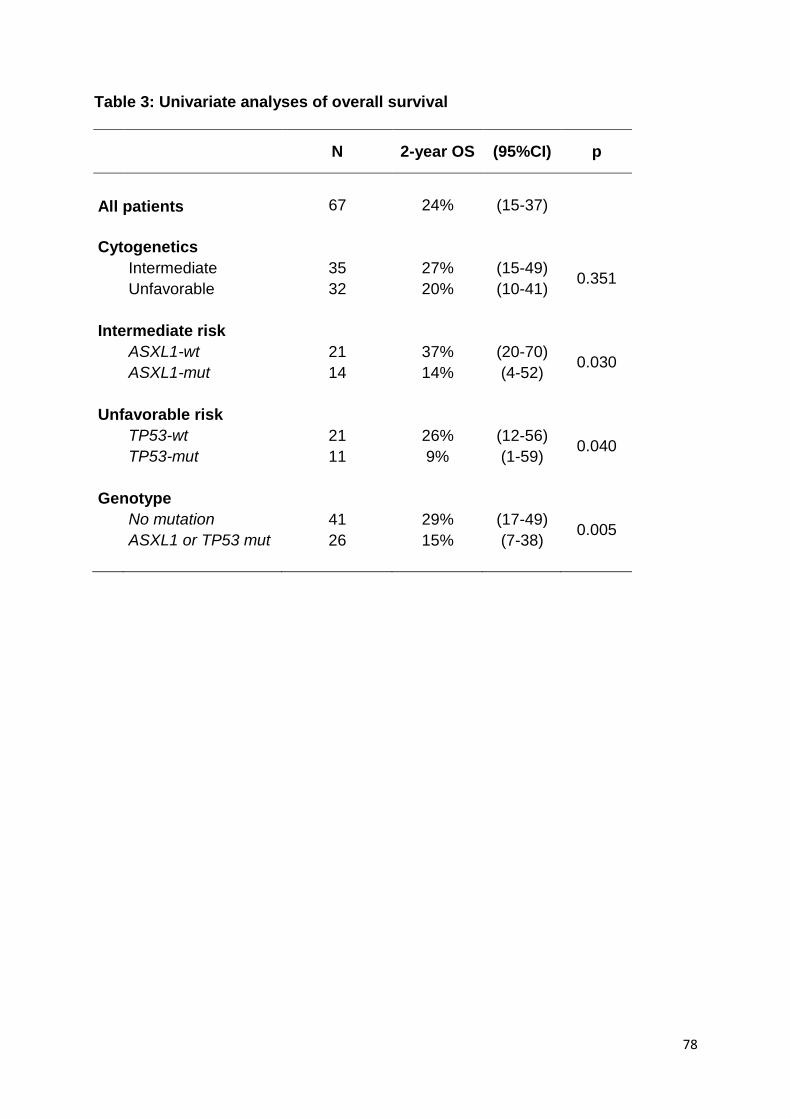
78
Table 3: Univariate analyses of overall survival
N 2-year OS (95%CI) p
All patients 67 24% (15-37)
Cytogenetics
Intermediate 35 27% (15-49) 0.351
Unfavorable 32 20% (10-41)
Intermediate risk
ASXL1-wt 21 37% (20-70) 0.030
ASXL1-mut 14 14% (4-52)
Unfavorable risk
TP53-wt 21 26% (12-56) 0.040
TP53-mut 11 9% (1-59)
Genotype
No mutation 41 29% (17-49) 0.005
ASXL1 or TP53 mut 26 15% (7-38)

79
Figure legends
Figure 1: Co mutation profile of ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD and
TP53 genes in the 125 patients:
Karyotypes: normal (light grey), abnormal non-complex (dark grey) and complex (black). Cytogenetics risk group: intermediate (light blue) and unfavorable (dark blue). MLD: green bars indicate the presence of criteria for MLD and grey bars are samples that were not evaluable for MLD. These latter had at least one other criteria for AML-MRC to enrolled them in this study. MDS = myelodysplastic syndrome; MLD = multilineage dysplasia.
Figure 2: Overall survival according to the presence ASXL1 mutation in the
intermediate cytogenetic patients (A) and according to the presence of TP53
mutation in the unfavorable cytogenetic patients (B)
Figure 3: Overall survival according to the presence of ASXL1 or TP53 mutation
Figure 4: Proposition of reclassification of AML-MRC according to molecular
alterations
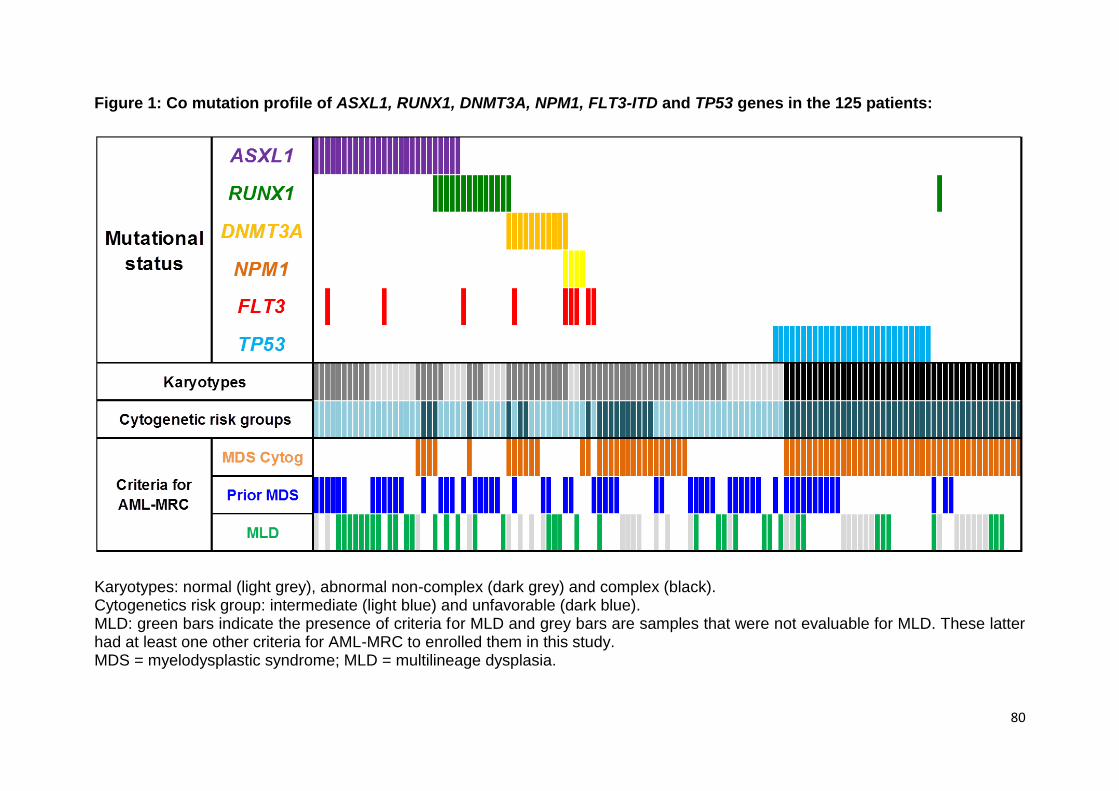
80
Figure 1: Co mutation profile of ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD and TP53 genes in the 125 patients:
Karyotypes: normal (light grey), abnormal non-complex (dark grey) and complex (black). Cytogenetics risk group: intermediate (light blue) and unfavorable (dark blue). MLD: green bars indicate the presence of criteria for MLD and grey bars are samples that were not evaluable for MLD. These latter had at least one other criteria for AML-MRC to enrolled them in this study. MDS = myelodysplastic syndrome; MLD = multilineage dysplasia.
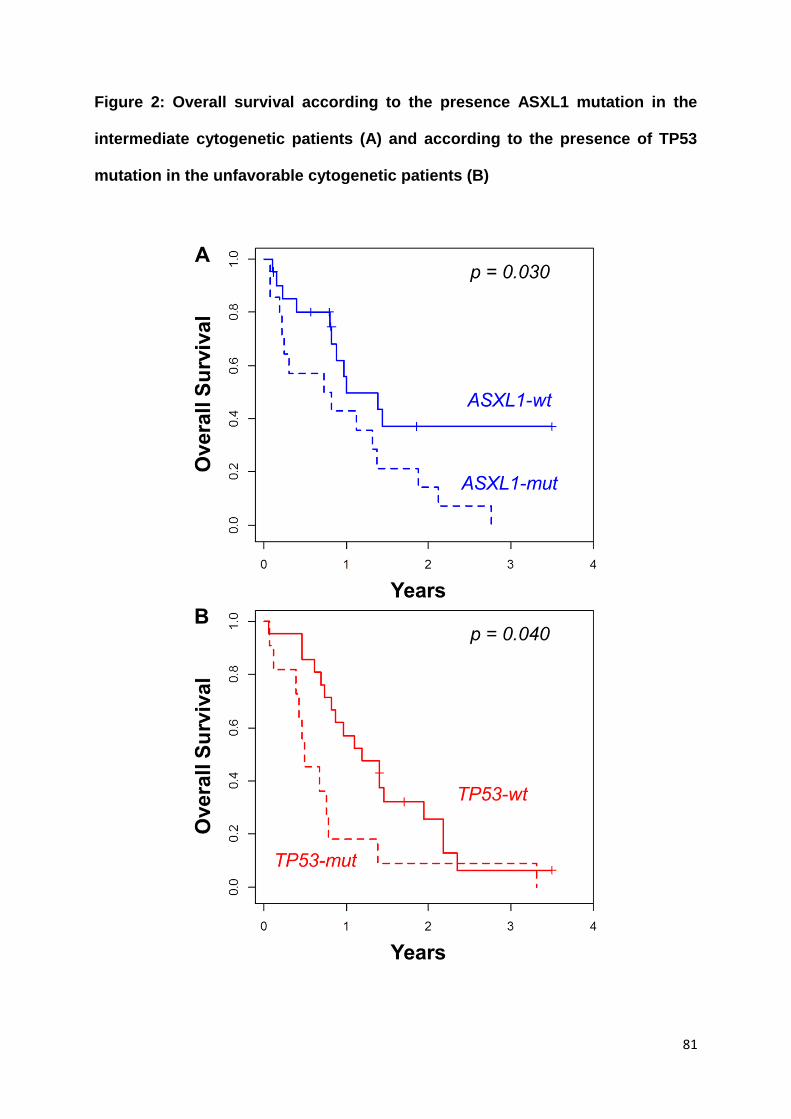
81
Figure 2: Overall survival according to the presence ASXL1 mutation in the
intermediate cytogenetic patients (A) and according to the presence of TP53
mutation in the unfavorable cytogenetic patients (B)
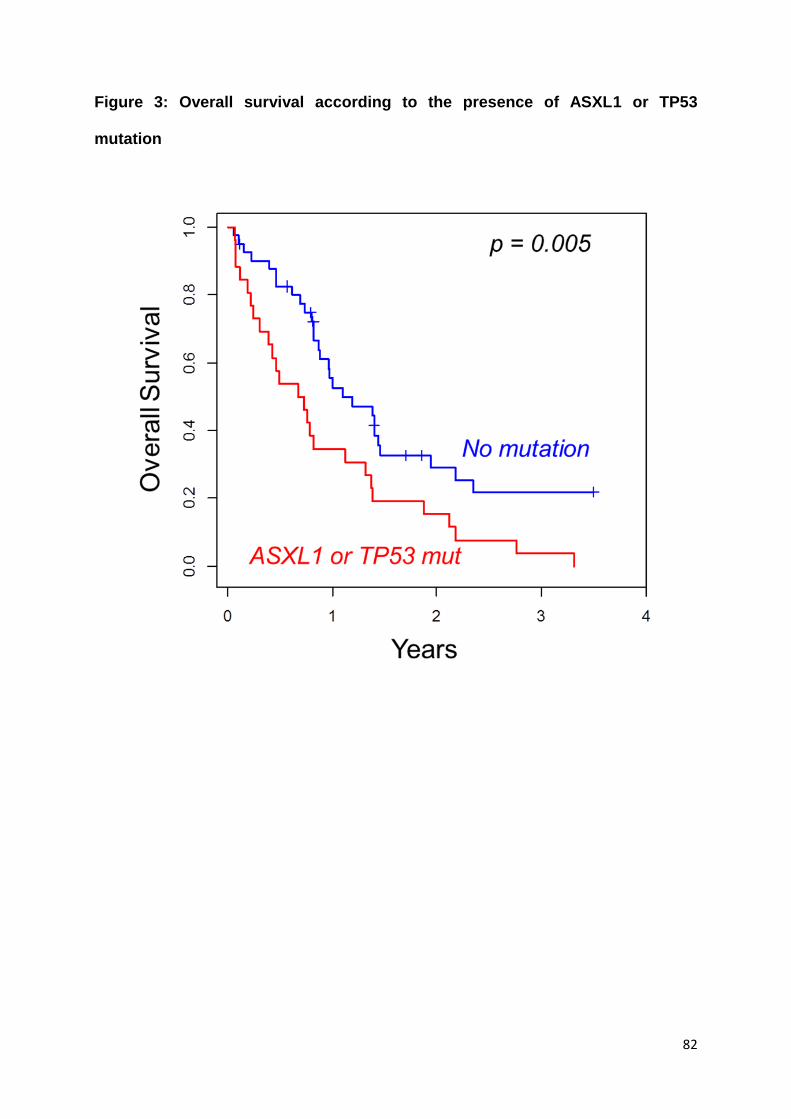
82
Figure 3: Overall survival according to the presence of ASXL1 or TP53
mutation
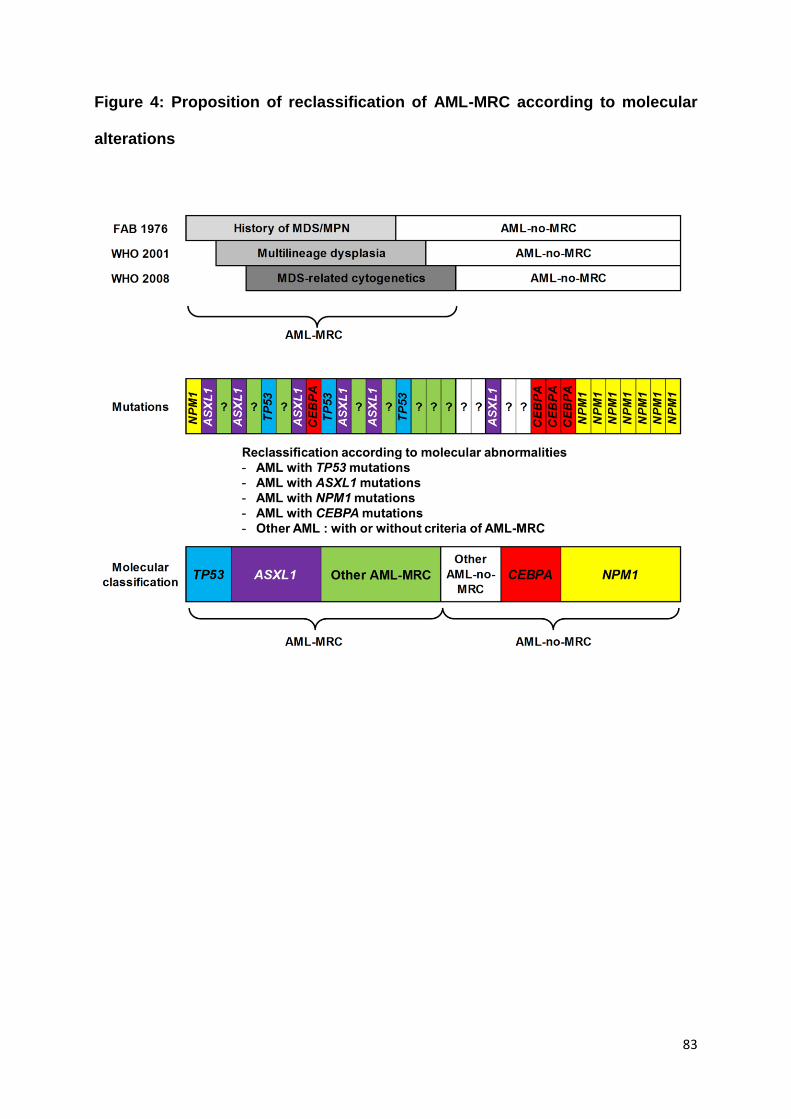
83
Figure 4: Proposition of reclassification of AML-MRC according to molecular
alterations
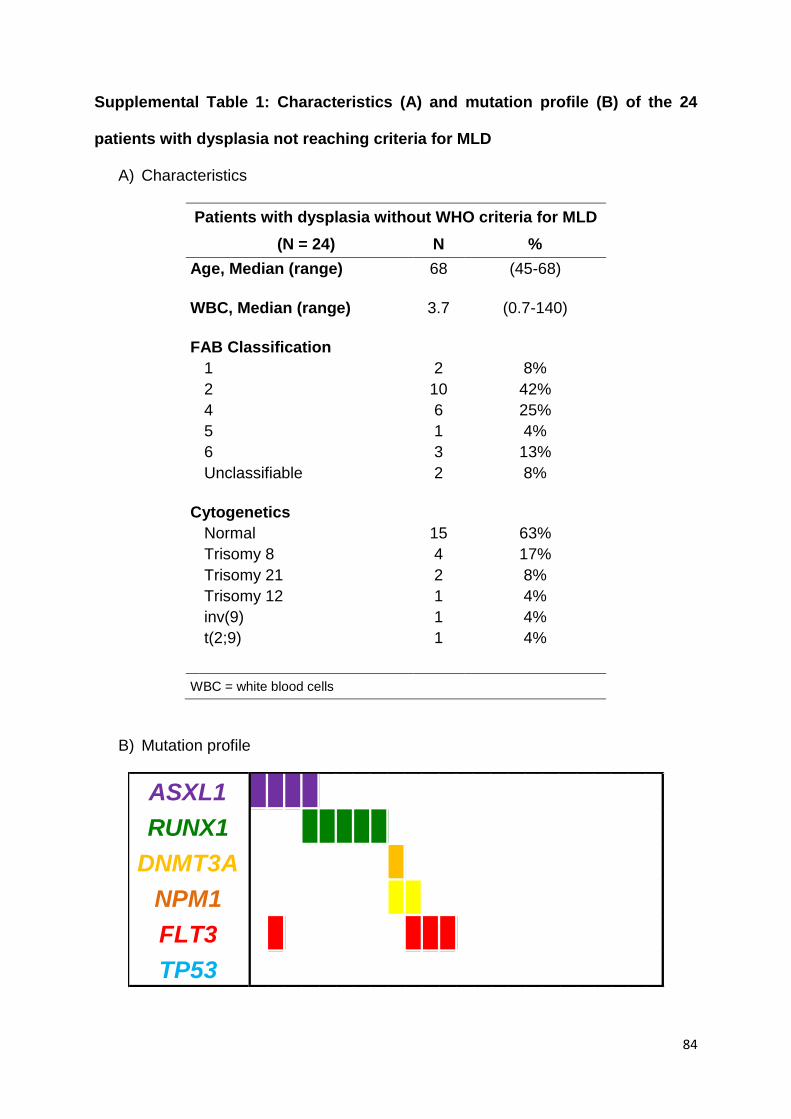
84
Supplemental Table 1: Characteristics (A) and mutation profile (B) of the 24
patients with dysplasia not reaching criteria for MLD
A) Characteristics
Patients with dysplasia without WHO criteria for MLD
(N = 24) N %
Age, Median (range) 68 (45-68)
WBC, Median (range) 3.7 (0.7-140)
FAB Classification
1 2 8%
2 10 42%
4 6 25%
5 1 4%
6 3 13%
Unclassifiable 2 8%
Cytogenetics
Normal 15 63%
Trisomy 8 4 17%
Trisomy 21 2 8%
Trisomy 12 1 4%
inv(9) 1 4%
t(2;9) 1 4%
WBC = white blood cells
B) Mutation profile
ASXL1
RUNX1
DNMT3A
NPM1
FLT3
TP53
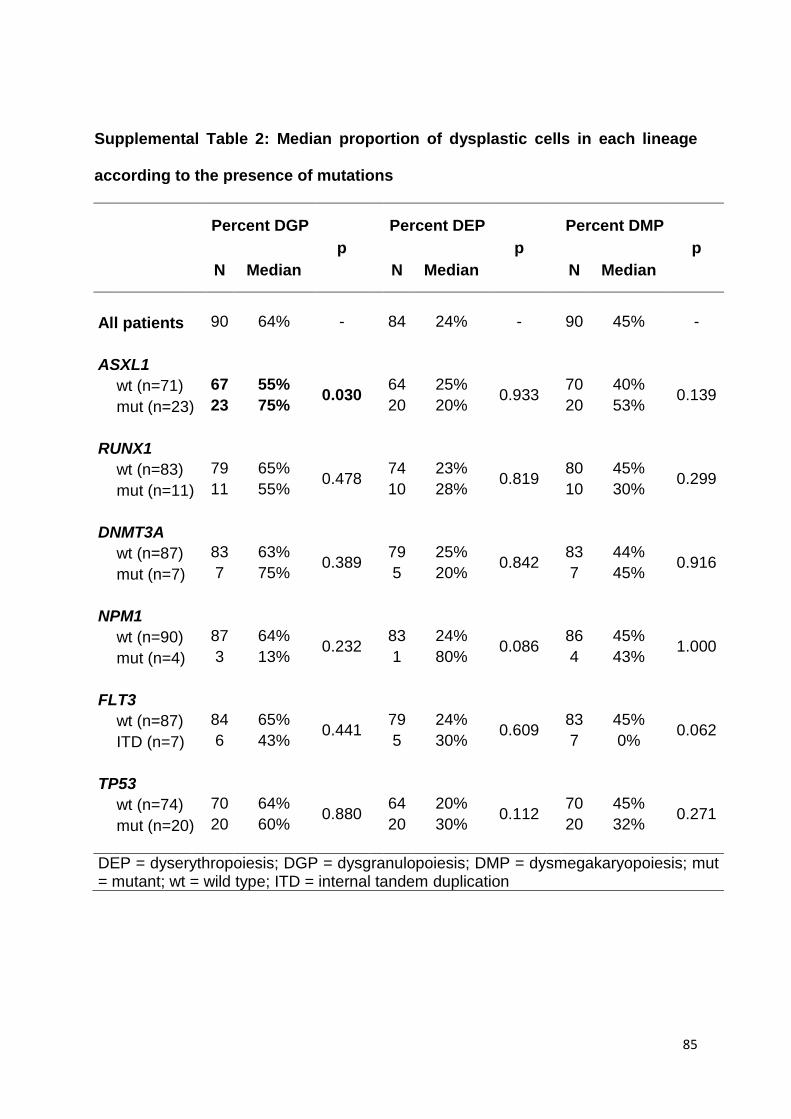
85
Supplemental Table 2: Median proportion of dysplastic cells in each lineage
according to the presence of mutations
Percent DGP
p
Percent DEP
p
Percent DMP
p
N Median N Median N Median
All patients 90 64% - 84 24% - 90 45% -
ASXL1
wt (n=71) 67 55% 0.030
64 25% 0.933
70 40% 0.139
mut (n=23) 23 75% 20 20% 20 53%
RUNX1
wt (n=83) 79 65% 0.478
74 23% 0.819
80 45% 0.299
mut (n=11) 11 55% 10 28% 10 30%
DNMT3A
wt (n=87) 83 63% 0.389
79 25% 0.842
83 44% 0.916
mut (n=7) 7 75% 5 20% 7 45%
NPM1
wt (n=90) 87 64% 0.232
83 24% 0.086
86 45% 1.000
mut (n=4) 3 13% 1 80% 4 43%
FLT3
wt (n=87) 84 65% 0.441
79 24% 0.609
83 45% 0.062
ITD (n=7) 6 43% 5 30% 7 0%
TP53
wt (n=74) 70 64% 0.880
64 20% 0.112
70 45% 0.271
mut (n=20) 20 60% 20 30% 20 32%
DEP = dyserythropoiesis; DGP = dysgranulopoiesis; DMP = dysmegakaryopoiesis; mut = mutant; wt = wild type; ITD = internal tandem duplication
86
Supplemental Table 3: Linear regression for proportion of dysgranulopoeisis
according to the presence of gene mutations
Coefficient
beta Standard deviation
p
ASXL1 20.389 8.824 0.023
RUNX1 -8.026 11.148 0.474
DNMT3A 17.904 13.879 0.201
NPM1 -26.058 22.304 0.246
FLT3 -0.846 15.894 0.958
TP53 2.346 9.405 0.804

87
Article 3 :
“Clinical, morphological and molecular characterization of acute myeloid leukemias
with myelodysplasia-related changes”
Résumé et discussion spécifique
A ce jour, aucun marqueur moléculaire n’est associé spécifiquement aux LAM-MRC,
bien que les mutations de certains gènes soient considérées comme des outils
majeurs dans les classifications diagnostiques et les stratifications pronostiques des
LAM (Grossmann et al., 2012; Hou et al., 2014; Patel et al., 2012). Nous avons
précédemment rapporté que les LAM-MRC présentaient un profil mutationnel
spécifique caractérisé par un taux élevé de mutation d’ASXL1 et une faible
proportion de mutations de DNMT3A, NPM1 et FLT3 (cf article 2). Dans cette série,
nous avons évalué les corrélations entre ces mutations et les différents critères
définissant les LAM-MRC (antécédent de SMD et/ou cytogénétiques de SMD et/ou
dysplasie multilignée). Il semblerait qu’aucun de ces critères ne permettent
d’identifier un profil moléculaire spécifique, suggérant qu’ils ne reposent pas sur un
rationnel/substrat moléculaire. Par définition, les LAM à caryotype complexe sont
rattachées aux LAM-MRC par analogie aux anomalies cytogénétiques de SMD.
Cependant, leur forte association aux mutations de TP53 et l’absence d’autres
anomalies devraient conduire à les classer séparément. De plus, ces mutations
semblaient associées à un pronostic péjoratif au sein des LAM à cytogénétique
défavorable, soulignant l’intérêt d’une sous-stratification pronostique moléculaire au
sein d’un même groupe de risque cytogénétique. Ces résultats sont concordants
avec ceux rapportés dans la littérature (Bowen et al., 2009; Rücker et al., 2012).

88
Après exclusion des LAM à caryotype complexe, le gène ASXL1 était le gène le plus
fréquemment muté dans notre série (31%), essentiellement dans les cytogénétiques
intermédiaires. ASXL1 était le seul gène dont les mutations étaient associées à la
présence de dysplasie multilignée selon les critères OMS et à un pourcentage plus
élevé de cellules dysgranulopoïétiques. Ces résultats semblent concorder avec
certaines études biologiques rapportant le rôle d’ASXL1 dans l’apparition du
phénotype dysplasique et la transformation leucémique (Abdel-Wahab et al., 2012,
2013; Inoue et al., 2013; West et al., 2014). Bien que significativement associées, les
mutations d’ASXL1 et la dysplasie multilignée ne sont pas strictement
superposables. Nous avons également identifié 16% de mutations d’ASXL1 dans la
cohorte analysée séparément, constituée de 24 patients sans critère de LAM-MRC
pour cause de dysplasie médullaire présente mais insuffisante. De plus, les
mutations d’ASXL1 ont été principalement identifiées dans les cas sans anomalies
cytogénétiques de SMD, alors même que les caryotypes complexes étaient exclus
de l’analyse. Enfin, il n’y avait pas de corrélation entre les mutations d’ASXL1 et la
présence d’antécédent de SMD ou de SMD/NMP. Ces résultats suggèrent que les
mutations d’ASXL1 pourraient représenter un marqueur moléculaire de dysplasie
dans les LAM, indépendamment des critères de LAM-MRC utilisés actuellement,
soulignant son potentiel intérêt et la nécessité de son intégration lors d’une révision
de la classification actuelle des LAM. Il a aussi été rapporté que les LAM mutées
ASXL1 présentaient un profil d’expression génique spécifique, suggérant qu’elles
forment une entité biologiquement distincte (Boultwood et al., 2010; Metzeler et al.,
2011).
Dans notre étude, si les mutations de TP53 stratifiaient le devenir des patients
porteurs de cytogénétiques défavorables, celles d’ASXL1 étaient associées à une

89
survie plus courte chez les patients à cytogénétiques intermédiaires. Différentes
études non restreintes aux LAM-MRC retrouvaient également ce pronostic péjoratif
associé aux mutations d’ASXL1 (Chou et al., 2010; Pratcorona et al., 2012;
Schnittger et al., 2013). Après analyse multivariée, la stratification pronostique selon
les mutations d’ASXL1 et de TP53 était le seul facteur prédictif de la survie alors que
la classification de risque cytogénétique usuelle ainsi que les différents critères des
LAM-MRC ne permettaient pas d’identifier des sous-groupes de pronostics différents.
En revanche, les patients pour lesquels aucune de ces deux mutations n’est
retrouvées forment un groupe de LAM-MRC présentant des caractéristiques
morphologiques, cytogénétiques et moléculaires hétérogènes. Les mutations de
DNMT3A et/ou de NPM1, fréquemment associées aux LAM de novo et exclusives
des mutations d’ASXL1 et de TP53, pourraient permettre d’identifier des patients
pour lesquels l’appartenance au groupe LAM-MRC pourrait être remise en cause.
Falini et al. ont rapporté que la présence de dysplasie multilignée ne permettait pas
d’identifier d’entité biologiquement et cliniquement distincte au sein des LAM mutées
NPM1, avec des profils d’expression génique superposables et une survie similaire
(Falini et al., 2010). A nouveau, ces résultats soulignent la nécessité de redéfinir les
LAM-MRC selon les anomalies moléculaires.
Enfin, pour les 47 patients qui n’avaient aucune mutation parmi les 6 gènes étudiés,
il nous semble nécessaire d’identifier d’autres marqueurs moléculaires. ASXL1 étant
un élément régulateur majeur du complexe polycomb répressif 2 (PRC2), il serait
possible que d’autres altérations de la régulation de ce complexe puissent aboutir au
même phénotype. Ainsi, les LAM avec des altérations d’autres gènes impliqués dans
la transformation myéloïdes via le PRC2, comme EZH2 (Wang et al., 2013; Xu et al.,
2011), JARID2 (Puda et al., 2012) ou BAP1 (Abdel-Wahab and Dey, 2013; Dey et

90
al., 2012) pourraient partager des caractéristiques avec les LAM mutées ASXL1.
L’analyse de ces différentes anomalies impliquées dans une même voie cellulaire
pourrait permettre d’identifier un sous-groupe homogène de LAM-MRC, guidant le
développement de futurs traitements ciblés.
Ainsi, les critères définissant actuellement les LAM-MRC ne semblent pas distinguer
des entités biologiques différentes et ne permettent pas de prédire la survie des
patients. Inversement, les mutations d’ASXL1 et de TP53 pourraient identifier des
entités biologiquement distinctes et aider à la stratification pronostique des LAM-
MRC. Cette caractérisation moléculaire pourrait être utile pour affiner la définition des
LAM-MRC dans les futures classifications, qui tendent à faire converger entités
biologiques et impact pronostique.

91
ARTICLE 4 :
In vitro multidrug screening and mutational profiles
of acute myeloid leukemias with myelodysplasia-
related changes
Devillier R et al.
Statut : Manuscrit en préparation

92
Introduction
Nous avons précédemment montré que certaines anomalies moléculaires pouvaient
permettre d’affiner la classification des LAM-MRC. Nous avons fait l’hypothèse que
les mutations d’ASXL1 et de TP53 permettraient d’identifier des sous-entités des
LAM-MRC distinctes des LAM non MRC qui sont majoritairement caractérisées par la
présence de mutations de NPM1 et dans une moindre mesure de DNMT3A1. Cette
reclassification pourrait permettre d’améliorer la stratification pronostique des LAM-
MRC et d’identifier de nouvelles cibles thérapeutiques, dans un contexte de
médecine personnalisée. Ainsi, de nombreuses molécules sont apparues récemment
dans l’éventail thérapeutique comme les agents déméthylants ou les inhibiteurs
d’histone déacétylases (HDACi). L’identification de marqueurs prédictifs de réponse
à ces traitements spécifiques restent alors indispensable2,3. Dans cette optique, nous
avons cherché à déterminer si les différents sous-groupes moléculaires de LAM-
MRC présentaient des profils de sensibilité à certaines drogues spécifiques. Nous
avons réalisé un criblage de drogues sur des cellules leucémiques primaires de
patients atteints de LAM-MRC et tenté de corréler les concentrations inhibitrices
(IC50) obtenues avec les anomalies mutationnelles observées dans cette population.

93
Méthodes
Critères de sélection
Les critères d’inclusion des patients étaient : 1) LAM-MRC selon la classification de
l’OMS 20084 ; 2) Echantillons cellulaires leucémiques au diagnostic congelés en
DMSO disponibles pour les analyses moléculaires et le criblage de drogues ; 3)
patients ayant signé un consentement éclairé autorisant l’étude ces échantillons
congelés dans un but de recherche. Le comité d’orientation stratégique (COS) de
l’Institut Paoli Calmettes a approuvé cette étude, incluse dans le projet translationnel
du SIRIC leucémie.
Séquençage
A l’aide d’un séquenceur à haut débit (Ion Torrent, Life Tech.), nous avons recherché
des mutations sur les régions codantes des gènes sur ASXL1 (sauf exon 12),
RUNX1, TET2, IDH1, IDH2, DNMT3A et TP53. L’exon 12 d’ASXL1 a été séquencé
par la méthode classique de Sanger. Les mutations de NPM1 (exon 12) et FLT3
(ITD, exon 13-14) ont été identifiées par les techniques courantes de génotypage par
analyse de taille de fragments et par séquençage.
Criblage de drogues
Le criblage a été réalisé grâce à des tests de viabilité basés sur la quantité cellulaire
d’ATP (Cell Titer Glo assay). Pour chaque patient et pour chaque drogue, nous
avons défini une valeur IC50 ajustée sur la mortalité des cellules contrôles provenant
du même patient, cultivées dans les mêmes conditions mais sans drogue. Nous
avons séparé les patients en deux groupes de sensibilité à chaque drogue :
sensibilité élevée (IC50 < médiane) et faible (IC50 > médiane). Les drogues testées
ainsi que leur mécanisme d’action sont listées dans la Table 1.

94
Analyses statistiques
Nous avons comparé les groupes de sensibilité aux drogues et le statut mutationnel
pour chaque gène et chaque drogue en utilisant les tests du Chi-2 ou de Fisher.
Nous avons effectué un test non paramétrique de Mann-Whitney pour tenter
d’identifier une association entre un échec de première cure de chimiothérapie
d’induction et les valeurs d’IC50 pour la cytarabine. Les survies ont été calculées à
partir de la date de la chimiothérapie intensive à l’aide de la méthode de Kaplan
Meier et du test du Log Rank5. Pour la survie globale (OS), la survenue du décès a
été considérée comme un évènement. Pour la survie sans évènement (EFS), nous
avons pris en compte l’échec primaire d’induction, la rechute et le décès comme
évènements.

95
Résultats
Nous avons analysé 52 patients dont les caractéristiques sont présentées dans la
Table 2. Parmi ces patients, 41 étaient analysables pour les tests de sensibilité aux
drogues. La Table 3 présente la fréquence des mutations des gènes ASXL1, RUNX1
TET2, IDH1, IDH2, DNMT3A, NPM1, FLT3 et TP53. La liste des mutations des
gènes RUNX1, TET2, IDH1, IDH2, DNMT3A et TP53 identifiées à l’aide du Ion
Torrent est rapportée dans la Table 4.
Nous avons mis en évidence des profils de sensibilité différents pour le prima-1 selon
la présence des mutations des gènes DNMT3A, NPM1, FLT3 et TP53. Cette drogue
permet de rétablir l’apoptose TP53-dépendante en restaurant la conformation et la
liaison à l’ADN des mutants TP536,7. Les LAM mutées TP53 avaient une sensibilité
élevée pour le prima-1 dans 85% des cas (6/7). Inversement, 100% (4/4) des
mutants DNMT3A, 78% (7/9) des mutants NPM1 et 100% (4/4) des mutants FLT3
avaient une sensibilité faible pour le prima-1 (p = 0,006, Figure 1A).
Le profil de sensibilité à la daunorubicine (Figure 1B) montre que les mutations de
NPM1 tendent à être associées à une IC50 faible pour la daunorubicine (p = 0,073).
Nous n’avons pas identifié d’autre association significative entre présence de
mutations et sensibilité relative aux drogues. La Figure 2 illustre les profils
mutationnels des 41 échantillons analysables pour les tests de sensibilité aux
drogues ainsi que les IC50 pour quelques une de ces drogues.
Sur les 41 patients, 33 avaient reçu une induction par chimiothérapie intensive
comprenant une anthracycline et de la cytarabine. Une sensibilité in vitro à la
cytarabine semblerait être associée à une meilleure EFS à 2 ans (sensibilité élevée
vs. faible : 42% vs. 22%, p = 0.146, Figure 3). En revanche, nous n’avons pas

96
identifié de différence en termes de taux de rémission complète ou de survie globale
selon la sensibilité in vitro à la cytarabine.

97
Discussion
Nous avons fait l’hypothèse que les anomalies moléculaires décrites précédemment
pourraient prédire des sensibilités spécifiques à différentes drogues. Nous avons
donc testé la sensibilité à certaines drogues utilisées en pratique courante comme la
daunorubicine, la cytarabine ou la 5-azacytidine. De plus nous avons testé des
drogues expérimentales supposées actives sur différentes voies cellulaires
potentiellement impliquées dans les LAM (inhibiteurs d’histone deacétylases ou de
tyrosine kinases, activateurs de la voie TP53…). Nous avons mis en évidence que
les mutations de TP53 prédisaient la réponse in vitro au prima-1 alors que celles de
DNMT3A et de NPM1 semblaient être associées à une résistance à cette drogue.
Nous n’avons pas réussi à identifier des profils de sensibilité spécifiquement
associées aux mutations d’ASXL1 car seulement 2 mutants ASXL1 sur 5 ont été
analysables. Le fait que nous n’ayons pas mis en évidence plus de corrélations entre
réponse aux drogues et présence de mutations pourrait être dû à différentes limites
de notre étude. La première est l’effectif réduit dans chaque sous-groupe
moléculaire, limitant ainsi la puissance des tests statistiques. Ensuite, nous avons
testé les drogues sur des échantillons de moelle totale. La réalisation des mêmes
tests de viabilité sur des sous-populations cellulaires triées permettrait peut-être d’en
améliorer la sensibilité et la spécificité. Enfin, nous n’avons étudié que la viabilité
cellulaire, sans analyse de la différenciation, de la prolifération ou d’apoptose, qui
sont également de potentielles cibles thérapeutiques. Cela pourrait être le cas
notamment des épidrogues. Pour celles-ci, l’évaluation des modifications
morphologiques, phénotypiques ou des profils d’expression et de méthylation
seraient probablement plus adaptée.

98
Nous avons trouvé que la sensibilité in vitro à la cytarabine pourrait être un facteur
prédictif de meilleure EFS chez les patients traités intensivement. Ce résultat pourrait
encourager le développement de stratégies de médecine personnalisée basée sur
des chimiogrammes. Cela permettrait d’identifier précocement les patients à haut
risque de maladie réfractaire et de reconsidérer la stratégie thérapeutique de ces
patients. L’absence de significativité pourrait être expliquée par le faible effectif et par
l’influence d’autres facteurs intrinsèques aux patients comme les aspects
pharmacocinétiques ou pharmacogénomiques.
Malgré les limites de notre étude, ces premiers résultats sont encourageants. La
double analyse des profils mutationnels et de réponse aux drogues pourrait être une
approche intéressante dans l’optique d’une médecine personnalisée guidée par les
anomalies moléculaires. Cependant, ces tests nécessitent d’être affinés en intégrant
d’autres paramètres, drogues spécifiques. Cette démarche pourrait ainsi permettre
de mieux orienter les développements thérapeutiques, adaptés à des sous-groupes
moléculaires d’intérêts.

99
Références
1. Devillier R, Gelsi-Boyer V, Brecqueville M, et al. Acute myeloid leukemia with
myelodysplasia-related changes are characterized by a specific molecular pattern
with high frequency of ASXL1 mutations. Am. J. Hematol.. 2012;87(7):659–662.
2. Pemovska T, Kontro M, Yadav B, et al. Individualized systems medicine
strategy to tailor treatments for patients with chemorefractory acute myeloid
leukemia. Cancer Discov.. 2013;3(12):1416–1429.
3. Yadav B, Pemovska T, Szwajda A, et al. Quantitative scoring of differential
drug sensitivity for individually optimized anticancer therapies. Sci. Rep..
2014;4:5193.
4. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health
Organization (WHO) classification of myeloid neoplasms and acute leukemia:
rationale and important changes. Blood. 2009;114(5):937–951.
5. Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J.
Am. Stat. Assoc.. 1958;53(282):457–481.
6. Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor
function to mutant p53 by a low-molecular-weight compound. Nat. Med..
2002;8(3):282–288.
7. Lambert JMR, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant
p53 by covalent binding to the core domain. Cancer Cell. 2009;15(5):376–388.

100
Table 1 : Liste des drogues évaluées
Molécules Cibles / Mécanisme d'action
Chimiothérapies
Clofarabine Analogue nucléosidique
Cytarabine Analogue nucléosidique
Daunorubicine Inhibiteur de topoisomérase II
Inhibiteurs de kinases
Quizartinib FLT3
Rigosertib Polo-like kinase 1 et PI3K
BKM-120 PI3K
MK-2206 AKT
Ruxolitinib JAK1/JAK2
Temsirolimus mTOR
Epidrogues
Décitabine Agent déméthylant
5-Azacitidine Agent déméthylant
Entinostat HDACi
S78454 HDACi
GSK-J1 Inhibiteur de la déméthylation H3K27
Inducteur d'apoptose
Prima-1 Inducteur d'apoptose p53 dépendante
NSC-207895 Inhibiteur de MDMX
Nutlin-3 Inhibiteur de MDM2
Autres
TMI-1 Inhibiteur de la métalloprotéase ADAM-17
Lenalidomide Activateur d'ubiquitination
Bortezomib Inhibiteur du protéasome
101
Table 2 : Caractéristiques des patients
All patients (N = 52)
N %
Median age (years, [range]) 66 [18-83]
FAB Classification
0 3 6%
1 11 21%
2 15 29%
4 9 17%
5 1 2%
6 7 13%
Unclassifiable 6 12%
Previously diagnosed MDS 8 15%
MDS-related cytogenetics 27 66%
Cytogenetics
Normal 16 31%
Complex monosomal 15 29%
Abnormal non-complex 21 40%
monosomy 7 4 7,7%
trisomy 8 5 9,6%
del(5q) 3 5,8%
del(9q) 3 5,8%
del(3p) 1 1,9%
del(20q) 1 1,9%
del(11q) 1 1,9%
trisomy 13 1 1,9%
i(17) 1 1,9%
t(3;12)(q23;p13) 1 1,9%
Cytogenetic risk group
Intermediate 29 56%
Unfavorable 23 44%
La somme des pourcentages peut être différente de 100% à cause des arrondis

102
Table 3 : Fréquence des mutations des gènes ASXL1, RUNX1, TET2, IDH1/2,
DNMT3A, NPM1, FLT3 et TP53
All patients (N = 52)
N %
ASXL1 5 10%
RUNX1 7 13%
TET2 10 19%
IDH1 5 10%
IDH2 4 8%
DNMT3A 6 12%
NPM1 9 17%
FLT3-ITD 4 8%
TP53 8 15%
103
Table 4 : Mutations identifiées par séquençage sur Ion Torrent dans 52 cas
Mutations Ratio allélique (mut / wt+mut)
RUNX1
R201Q 0.47
R201Q 0.41
R201Q 0.67
F330Sfs*265 0.38
R162K 0.68
R204X 0.21
F163fs / R166Q 0.38 / 0.36
TET2
H800fs 0.76
S327X / D1376G 0.40 / 0.42
E1247X 0.38
P1723S 0.38
H1868R 0.27
C1378R 0.96
V1718L 0.44
V1718L 0.43
C1273Y / exon4:c.3500+1G>A 0.43 / 0.23
exon8:c.4044+2T>A 0.73
IDH1
R132H 0.28
R132S 0.37
R132C 0.18
R132C 0.08
R132C 0.17
IDH2
R140Q 0.45
R140Q 0.27
R140Q 0.16
R140Q 0.35
DNMT3A
R882H 0.40
R882H 0.45
R882C 0.29
C497Y 0.44
TP53
R175H / R110H 0.33 / 0.08
Y220C / Q167X 0.13 / 0.15
Y220C 0.73
Y236H / H178fs 0.43
R248G 0.44
R273H 0.72
R273H 0.84
S241Y 0.84
mut = mutant ; wt = wild type

104
Figure 1 : Sensibilité in vitro au prima-1 (A) et à la daunorubicine (B) en
fonction des mutations des gènes ASXL1, RUNX1 TET2, IDH1/2, DNMT3A,
NPM1, FLT3 et TP53
A- Prima-1
B- Daunorubicine

105
Figure 2 : Profil mutationnel et sensibilité à 6 drogues de 41 patients
ASXL1
RUNX1
TET2
IDH
DNMT3A
NPM1
FLT3
TP53
Monosomal
karyotype
prima-1
daunorubicin
cytarabine
S78454
entinostat
decitabine
.
Les barres grises, noires et blanches représentent une sensibilité faible, élevée et
non évaluable, respectivement.
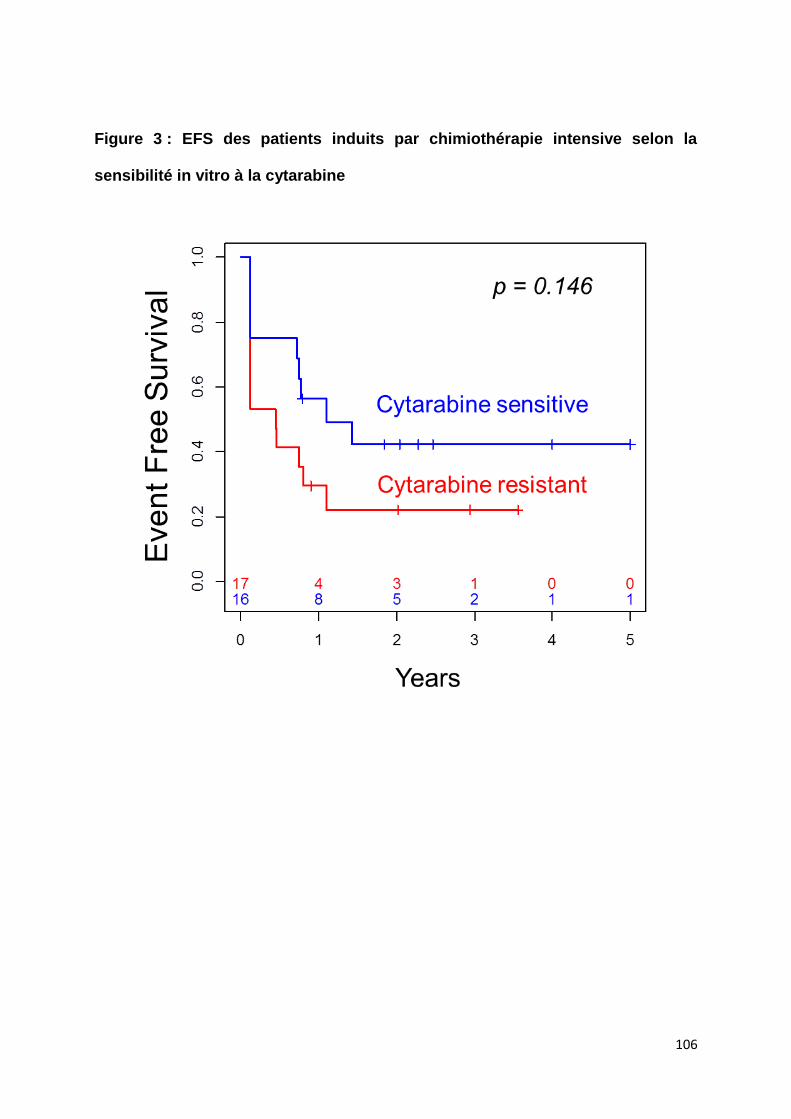
106
Figure 3 : EFS des patients induits par chimiothérapie intensive selon la
sensibilité in vitro à la cytarabine

107
DISCUSSION ET PERSPECTIVES
Nos résultats ont montré que les LAM-MRC définies selon les critères de l’OMS 2008
regroupent des pathologies très hétérogènes tant sur le plan biologique que
pronostique. Ceci pourrait être en partie expliqué par la relative complexité de cette
définition, qui intègre à la fois des critères cliniques (antécédents de SMD ou
SMD/NMP), morphologiques (DML) et cytogénétiques. A l’exception de certaines
anomalies cytogénétiques complexes et/ou celles des chromosomes 5, 7 et 17, les
critères OMS ne se semblent pas avoir d’impact pronostique. Nos analyses
moléculaires ont montré que les LAM-MRC étaient hétérogènes sur le plan
mutationnel. Cette caractérisation moléculaire pourrait permettre d’affiner la définition
des LAM-MRC et de mieux en stratifier le pronostic.
I) Vers une classification moléculaire des LAM-MRC ?
Nous proposons ici une classification des LAM-MRC basée sur le statut mutationnel
qui permettrait de reclasser certains patients du groupe LAM-MRC dans le groupe
LAM-non-MRC et inversement (Figure 4).
a. LAM avec altération de TP53
Au sein des LAM-MRC, la majorité des anomalies cytogénétiques défavorables sont
des caryotypes complexes et/ou monosomaux (70%). Dans ces sous-groupes, les
mutations de TP53 sont retrouvées dans plus de 60% des cas et sont exclusives des
autres mutations. Cela suggère un mécanisme de leucémogenèse spécifique, au
sein duquel l’instabilité chromosomique joue un rôle majeur. Ces LAM ont un
pronostic très sombre quel que soit le traitement. L’identification des mutations de
TP53, associées à ces anomalies cytogénétiques, pourrait permettre le
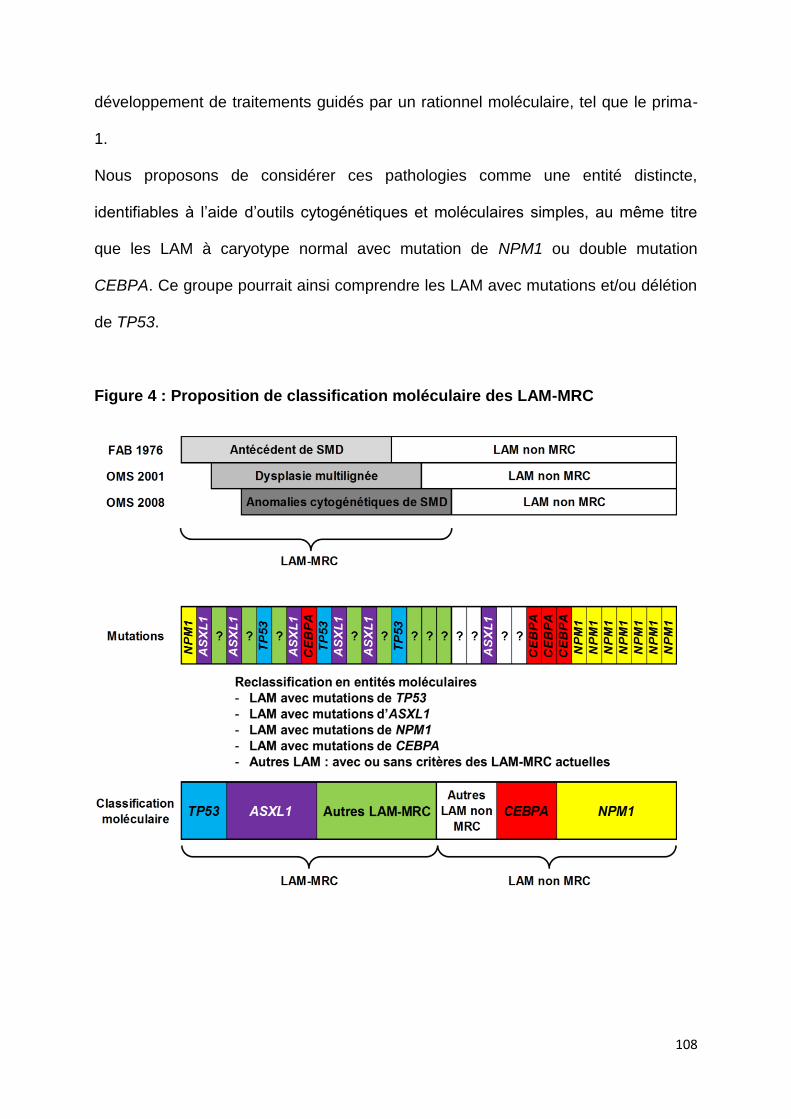
108
développement de traitements guidés par un rationnel moléculaire, tel que le prima-
1.
Nous proposons de considérer ces pathologies comme une entité distincte,
identifiables à l’aide d’outils cytogénétiques et moléculaires simples, au même titre
que les LAM à caryotype normal avec mutation de NPM1 ou double mutation
CEBPA. Ce groupe pourrait ainsi comprendre les LAM avec mutations et/ou délétion
de TP53.
Figure 4 : Proposition de classification moléculaire des LAM-MRC

109
b. LAM avec mutation d’ASXL1
Nous avons montré que les mutations d’ASXL1 pourraient constituer un marqueur de
dysplasie et être ainsi un critère moléculaire définissant les LAM-MRC. Plusieurs
études biologiques confirment que les mutations d’ASXL1 induisent des aspects
myélodysplasiques morphologiques chez la souris et favorisent la transformation en
LAM (Abdel-Wahab et al., 2012, 2013; Inoue et al., 2013; Wang et al., 2014; West et
al., 2014). Chez l’homme, les mutations d’ASXL1 ne sont pas distribuées
aléatoirement au sein des hémopathies myéloïdes chroniques. Alors qu’elles sont
rares dans les SMD de bas risque, la maladie de Vaquez et la thrombocytémie
essentielle (<10%), leur fréquence augmente dans les formes plus agressives : leur
fréquence est de 15-25% dans les SMD de haut risque, 20-25% dans les
myélofibroses et 40-45% dans les LMMC (Bejar et al., 2011; Brecqueville et al.,
2012, 2014; Gelsi-Boyer et al., 2009; Tefferi and Vainchenker, 2011; Thol et al.,
2011; Vannucchi et al., 2013). Dans l’ensemble de ces pathologies, ces mutations
caractérisent les formes à haut risque de transformation leucémique. Dans les LAM-
MRC, elles sont retrouvées dans environ 35-45% des cas et sont toujours exclusives
de celles de NPM1 et de DNMT3A (Carbuccia et al., 2010; Devillier et al., 2012; Hou
et al., 2014; Renneville et al., 2014; Schnittger et al., 2013). Elles semblent être
associées à un pronostic défavorable, notamment par une chimiorésistance primaire.
Dans notre étude, nous avons montré qu’au sein des LAM-MRC, les mutations
d’ASXL1 sont associées à une dysgranulopoïèse plus importante mais variable.
Nous avons également retrouvé des mutations d’ASXL1 chez des patients ayant des
LAM avec dysplasie sans critère de DML. Bien que n’appartenant pas aux LAM-MRC
selon les critères actuels, la présence des mutations d’ASXL1 permettrait de les
intégrer dans une classification moléculaire. L’ensemble de ces résultats suggère

110
que la seule présence d’une mutation d’ASXL1 pourrait être considérée comme un
critère suffisant pour définir une LAM-MRC.
c. LAM avec mutations de NPM1 ou DNMT3A
Dans notre étude, environ 15% des patients atteints de LAM-MRC présentent des
mutations de NPM1 et/ou de DNMT3A. Elles sont fréquemment associées aux
mutations de FLT3, constituant ainsi un profil qui se rapproche de celui des LAM-
non-MRC. Dans l’optique d’une classification moléculaire, nous proposons de ne pas
considérer ces pathologies comme des LAM-MRC malgré la présence de certains
critères actuels tels que la DML ou un antécédent de SMD. Le fait que la DML
n’influence ni les caractéristiques biologiques ni la survie des patients atteints de
LAM mutées NPM1 supporte en partie notre hypothèse.
d. LAM avec d’autres mutations ou sans mutation identifiée
Environ 40% des patients ne présentent aucune des mutations citées ci-dessus. Ils
peuvent avoir des mutations de RUNX1, TET2, d’IDH1/2 ou de FLT3 ou aucune
d’entre elles. Ces mutations ne permettent ni d’identifier des entités distinctes ni
d’aider à une classification moléculaire des LAM-MRC. En effet, les mutations de
TET2 et d’IDH1/2 sont retrouvées dans les LAM-MRC aussi bien que dans les LAM-
non-MRC, avec une fréquence comparable. Les mutations de FLT3 sont
préférentiellement retrouvées dans les LAM-non-MRC et sont associées à celles de
NPM1 et de DNMT3A, mais il n’est pas exceptionnel de les observer en présence de
celles d’ASXL1. Les mutations de FLT3 ont déjà été identifiées au sein de différentes
entités comme les LAM promyélocytaires ou les LAM-CBF, suggérant le caractère
additionnel et non classant de cette altération. Enfin, il semblerait que les mutations

111
de RUNX1 soient plus fréquemment retrouvées au sein des LAM-MRC et soient
associées à celles d’ASXL1. Cependant, nous avons trouvé quelques cas de LAM
mutées RUNX1 en l’absence de tout critère de LAM-MRC et il n’est pas exceptionnel
d’observer des co-mutations RUNX1/DNMT3A ou RUNX1/NPM1 (plus rarement).
Ainsi, nous n’avons pas considéré les mutations de RUNX1 suffisamment
spécifiques pour intégrer une classification moléculaire. Enfin, pour l’ensemble de
ces patients, nous proposons d’utiliser les critères actuels de classification OMS
2008 des LAM-MRC.
II) Perspectives
a. Caractérisation pangénomique des LAM
Ce travail, réalisé majoritairement avec des techniques de séquençage direct
standard, a permis d’identifier des mutations revêtant une particulière importance
pour les classifications futures. Cependant, la majorité des LAM restent classées par
la cytogénétique, le phénotype et/ou la cytologie. L’avènement des techniques de
biologie moléculaire à haut débit a permis d’identifier de nouvelles altérations
moléculaires, dont certaines peuvent jouer un rôle majeur sur le plan diagnostique,
comme par exemple la découverte des mutations de CALR dans la thrombocytémie
essentielle et la myélofibrose primitive (Nangalia et al., 2013). Une étude récente du
« Cancer Genome Atlas Research Network » a analysé 200 cas de LAM de novo par
séquençage de l’ensemble du génome ou des exons (Cancer Genome Atlas
Research Network, 2013). Pour chaque patient, elle identifie au moins une mutation
associée à l’une des voies majeures de leucémogenèse. Ce modèle complexifie
celui de Gilliland et Griffin (mutations de classe I et de classe II) (Gilliland and Griffin,
2002), en considérant 9 catégories de gènes : les transcrits de fusion (anciennement

112
classe II), les suppresseurs de tumeur, les gènes de la méthylation de l’ADN, les
modificateurs de la chromatine, les facteurs de transcription myéloïdes, les
activateurs de la signalisation (anciennement classe I), les gènes du complexe
cohésine et les gènes du spliceosome (Figure 5).
Figure 5 : Différentes classes d’altérations moléculaires dans les LAM
TSG : tumor supressor gene
Les approches pangénomiques permettent une analyse exhaustive du génome et la
réalisation d’analyses intégrées en couplant ces résultats à ceux issus de l’analyse
de l’ARN (profil d’expression génique ou séquençage haut débit de l’ARN codant ou
des micro-ARN) et de la méthylation de l’ADN (méthylome). L’inconvénient principal
est la quantité importante de données générées et la difficulté d’application des
résultats en pratique clinique courante, au moins dans l’immédiat. Pour les

113
altérations rares (<10%), un regroupement par voies cellulaires communes semble
préférable afin d’en évaluer le pronostic. A terme, cela pourrait permettre une
classification basée sur des groupes d’anomalies moléculaires, reposant sur des
arguments physiopathologiques, pronostiques et thérapeutiques.
b. Vers une médecine moléculaire personnalisée
Les approches pangénomiques ont révélé l’hétérogénéité des LAM sur le plan
moléculaire. Cette caractérisation génomique globale pourrait permettre d’orienter le
développement de nouveaux traitements ciblés sur des anomalies moléculaires.
Certaines mutations pourraient devenir des marqueurs prédictifs de réponse ou de
résistance à certains traitements, avec à terme un impact direct sur les stratégies
thérapeutiques. Enfin, une approche combinant un diagnostic moléculaire extensif et
un criblage thérapeutique in vitro guidé par les anomalies identifiées permettrait
d’appliquer un traitement spécifique à chaque patient.

114
CONCLUSIONS GENERALES
Les LAM avec dysmyélopoïèse regroupent des pathologies hétérogènes sur le plan
biologique et pronostique. Une reclassification basée sur la présence d’altérations
moléculaires exclusives entre elles, comme les mutations d’ASXL1 et de TP53,
permettrait d’affiner le diagnostic et la stratification pronostique de ces maladies. A
ce jour, les statuts mutationnels de FLT3, NPM1 et CEBPA sont nécessaires à la
prise en charge diagnostique et thérapeutique des LAM. Nos résultats suggèrent que
la recherche systématique des mutations d’ASXL1 et de TP53 permettrait optimiser
cette prise en charge. Ces travaux pourraient servir de point de départ aux
classifications futures, basées majoritairement sur la présence d’altérations
moléculaires. Enfin, nous avons montré que les tests de sensibilité in vitro peuvent
avoir un intérêt en tant que facteurs prédictifs de réponse et guider les choix
thérapeutiques ainsi que le développement de nouveaux traitements ciblés.

115
BIBLIOGRAPHIE
Abbas, S., Lugthart, S., Kavelaars, F.G., Schelen, A., Koenders, J.E., Zeilemaker, A., van Putten, W.J.L., Rijneveld, A.W., Löwenberg, B., and Valk, P.J.M. (2010). Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116, 2122–2126.
Abdel-Wahab, O., and Dey, A. (2013). The ASXL-BAP1 axis: new factors in myelopoiesis, cancer and epigenetics. Leukemia 27, 10–15.
Abdel-Wahab, O., Adli, M., LaFave, L.M., Gao, J., Hricik, T., Shih, A.H., Pandey, S., Patel, J.P., Chung, Y.R., Koche, R., et al. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193.
Abdel-Wahab, O., Gao, J., Adli, M., Dey, A., Trimarchi, T., Chung, Y.R., Kuscu, C., Hricik, T., Ndiaye-Lobry, D., Lafave, L.M., et al. (2013). Deletion of ASXL1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 210, 2641–2659.
Allen, C., Hills, R.K., Lamb, K., Evans, C., Tinsley, S., Sellar, R., O’Brien, M., Yin, J.L., Burnett, A.K., Linch, D.C., et al. (2013). The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia 27, 1891–1901.
Appelbaum, F.R., Gundacker, H., Head, D.R., Slovak, M.L., Willman, C.L., Godwin, J.E., Anderson, J.E., and Petersdorf, S.H. (2006). Age and acute myeloid leukemia. Blood 107, 3481–3485.
Bacher, U., Haferlach, C., Kern, W., Haferlach, T., and Schnittger, S. (2008). Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood 111, 2527–2537.
Bailey, E., Li, L., Duffield, A.S., Ma, H.S., Huso, D.L., and Small, D. (2013). FLT3/D835Y mutation knock-in mice display less aggressive disease compared with FLT3/internal tandem duplication (ITD) mice. Proc. Natl. Acad. Sci. U. S. A. 110, 21113–21118.
Basecke, J., Whelan, J.T., Griesinger, F., and Bertrand, F.E. (2006). The MLL partial tandem duplication in acute myeloid leukaemia. Br. J. Haematol. 135, 438–449.
Beaupre, D.M., and Kurzrock, R. (1999). RAS and leukemia: from basic mechanisms to gene-directed therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 17, 1071–1079.
Becker, H., Marcucci, G., Maharry, K., Radmacher, M.D., Mrózek, K., Margeson, D., Whitman, S.P., Wu, Y.-Z., Schwind, S., Paschka, P., et al. (2010). Favorable

116
prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 596–604.
Bejar, R., Stevenson, K., Abdel-Wahab, O., Galili, N., Nilsson, B., Garcia-Manero, G., Kantarjian, H., Raza, A., Levine, R.L., Neuberg, D., et al. (2011). Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 364, 2496–2506.
Belson, M., Kingsley, B., and Holmes, A. (2007). Risk factors for acute leukemia in children: a review. Environ. Health Perspect. 115, 138–145.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R., and Sultan, C. (1976). Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 33, 451–458.
Boissel, N., Nibourel, O., Renneville, A., Gardin, C., Reman, O., Contentin, N., Bordessoule, D., Pautas, C., de Revel, T., Quesnel, B., et al. (2010). Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 3717–3723.
Boissel, N., Nibourel, O., Renneville, A., Huchette, P., Dombret, H., and Preudhomme, C. (2011). Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 117, 3696–3697.
Boultwood, J., Perry, J., Pellagatti, A., Fernandez-Mercado, M., Fernandez-Santamaria, C., Calasanz, M.J., Larrayoz, M.J., Garcia-Delgado, M., Giagounidis, A., Malcovati, L., et al. (2010). Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 24, 1062–1065.
Bowen, D.T. (2006). Etiology of acute myeloid leukemia in the elderly. Semin. Hematol. 43, 82–88.
Bowen, D., Groves, M.J., Burnett, A.K., Patel, Y., Allen, C., Green, C., Gale, R.E., Hills, R., and Linch, D.C. (2009). TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 23, 203–206.
Brecqueville, M., Rey, J., Bertucci, F., Coppin, E., Finetti, P., Carbuccia, N., Cervera, N., Gelsi-Boyer, V., Arnoulet, C., Gisserot, O., et al. (2012). Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes. Chromosomes Cancer 51, 743–755.
Brecqueville, M., Rey, J., Devillier, R., Guille, A., Gillet, R., Adélaide, J., Gelsi-Boyer, V., Arnoulet, C., Chaffanet, M., Mozziconacci, M.-J., et al. (2014). Array comparative genomic hybridization and sequencing of 23 genes in 80 patients with myelofibrosis at chronic or acute phase. Haematologica 99, 37–45.
Breems, D.A., Van Putten, W.L.J., De Greef, G.E., Van Zelderen-Bhola, S.L., Gerssen-Schoorl, K.B.J., Mellink, C.H.M., Nieuwint, A., Jotterand, M., Hagemeijer, A.,

117
Beverloo, H.B., et al. (2008). Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 26, 4791–4797.
Büchner, T., Berdel, W.E., Haferlach, C., Haferlach, T., Schnittger, S., Müller-Tidow, C., Braess, J., Spiekermann, K., Kienast, J., Staib, P., et al. (2009). Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: a study by the German Acute Myeloid Leukemia Cooperative Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 27, 61–69.
Byrd, J.C., Mrózek, K., Dodge, R.K., Carroll, A.J., Edwards, C.G., Arthur, D.C., Pettenati, M.J., Patil, S.R., Rao, K.W., Watson, M.S., et al. (2002). Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 100, 4325–4336.
Cai, Z., de Bruijn, M., Ma, X., Dortland, B., Luteijn, T., Downing, R.J., and Dzierzak, E. (2000). Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity 13, 423–431.
Caligiuri, M.A., Strout, M.P., Lawrence, D., Arthur, D.C., Baer, M.R., Yu, F., Knuutila, S., Mrózek, K., Oberkircher, A.R., Marcucci, G., et al. (1998). Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 58, 55–59.
Cancer Genome Atlas Research Network (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074.
Carbuccia, N., Trouplin, V., Gelsi-Boyer, V., Murati, A., Rocquain, J., Adélaïde, J., Olschwang, S., Xerri, L., Vey, N., Chaffanet, M., et al. (2010). Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia 24, 469–473.
Challen, G.A., Sun, D., Jeong, M., Luo, M., Jelinek, J., Berg, J.S., Bock, C., Vasanthakumar, A., Gu, H., Xi, Y., et al. (2012). DNMT3A is essential for hematopoietic stem cell differentiation. Nat. Genet. 44, 23–31.
Chou, W.-C., Huang, H.-H., Hou, H.-A., Chen, C.-Y., Tang, J.-L., Yao, M., Tsay, W., Ko, B.-S., Wu, S.-J., Huang, S.-Y., et al. (2010). Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 116, 4086–4094.
Chou, W.-C., Chou, S.-C., Liu, C.-Y., Chen, C.-Y., Hou, H.-A., Kuo, Y.-Y., Lee, M.-C., Ko, B.-S., Tang, J.-L., Yao, M., et al. (2011). TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 118, 3803–3810.
Delhommeau, F., Dupont, S., Della Valle, V., James, C., Trannoy, S., Massé, A., Kosmider, O., Le Couedic, J.-P., Robert, F., Alberdi, A., et al. (2009). Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301.

118
Devillier, R., Gelsi-Boyer, V., Brecqueville, M., Carbuccia, N., Murati, A., Vey, N., Birnbaum, D., and Mozziconacci, M.-J. (2012). Acute myeloid leukemia with myelodysplasia-related changes are characterized by a specific molecular pattern with high frequency of ASXL1 mutations. Am. J. Hematol. 87, 659–662.
Dey, A., Seshasayee, D., Noubade, R., French, D.M., Liu, J., Chaurushiya, M.S., Kirkpatrick, D.S., Pham, V.C., Lill, J.R., Bakalarski, C.E., et al. (2012). Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 337, 1541–1546.
Döhner, H., Estey, E.H., Amadori, S., Appelbaum, F.R., Büchner, T., Burnett, A.K., Dombret, H., Fenaux, P., Grimwade, D., Larson, R.A., et al. (2010). Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115, 453–474.
Döhner, K., Schlenk, R.F., Habdank, M., Scholl, C., Rücker, F.G., Corbacioglu, A., Bullinger, L., Fröhling, S., and Döhner, H. (2005). Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106, 3740–3746.
Elliot, K., Tooze, J.A., Geller, R., Powell, B.L., Pardee, T.S., Ritchie, E., Kennedy, L., Callahan, K.E., and Klepin, H.D. (2014). The prognostic importance of polypharmacy in older adults treated for acute myelogenous leukemia (AML). Leuk. Res.
Estey, E. (2007). Acute myeloid leukemia and myelodysplastic syndromes in older patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 25, 1908–1915.
Falini, B., Mecucci, C., Tiacci, E., Alcalay, M., Rosati, R., Pasqualucci, L., La Starza, R., Diverio, D., Colombo, E., Santucci, A., et al. (2005). Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352, 254–266.
Falini, B., Martelli, M.P., Bolli, N., Bonasso, R., Ghia, E., Pallotta, M.T., Diverio, D., Nicoletti, I., Pacini, R., Tabarrini, A., et al. (2006). Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 108, 1999–2005.
Falini, B., Nicoletti, I., Martelli, M.F., and Mecucci, C. (2007). Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood 109, 874–885.
Falini, B., Bolli, N., Liso, A., Martelli, M.P., Mannucci, R., Pileri, S., and Nicoletti, I. (2009). Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 23, 1731–1743.
Falini, B., Macijewski, K., Weiss, T., Bacher, U., Schnittger, S., Kern, W., Kohlmann, A., Klein, H.-U., Vignetti, M., Piciocchi, A., et al. (2010). Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 115, 3776–3786.
Fasan, A., Haferlach, C., Alpermann, T., Jeromin, S., Grossmann, V., Eder, C., Weissmann, S., Dicker, F., Kohlmann, A., Schindela, S., et al. (2014). The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28, 794–803.

119
Figueroa, M.E., Abdel-Wahab, O., Lu, C., Ward, P.S., Patel, J., Shih, A., Li, Y., Bhagwat, N., Vasanthakumar, A., Fernandez, H.F., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567.
Fröhling, S., Schlenk, R.F., Breitruck, J., Benner, A., Kreitmeier, S., Tobis, K., Döhner, H., Döhner, K., and AML Study Group Ulm. Acute myeloid leukemia (2002). Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 100, 4372–4380.
Gaidzik, V.I., Bullinger, L., Schlenk, R.F., Zimmermann, A.S., Röck, J., Paschka, P., Corbacioglu, A., Krauter, J., Schlegelberger, B., Ganser, A., et al. (2011). RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 1364–1372.
Gelsi-Boyer, V., Trouplin, V., Adélaïde, J., Bonansea, J., Cervera, N., Carbuccia, N., Lagarde, A., Prebet, T., Nezri, M., Sainty, D., et al. (2009). Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 145, 788–800.
Gelsi-Boyer, V., Trouplin, V., Roquain, J., Adélaïde, J., Carbuccia, N., Esterni, B., Finetti, P., Murati, A., Arnoulet, C., Zerazhi, H., et al. (2010). ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br. J. Haematol. 151, 365–375.
Gelsi-Boyer, V., Brecqueville, M., Devillier, R., Murati, A., Mozziconacci, M.-J., and Birnbaum, D. (2012). Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol.J Hematol Oncol 5, 12.
Gerrits, A., Walasek, M.A., Olthof, S., Weersing, E., Ritsema, M., Zwart, E., van Os, R., Bystrykh, L.V., and de Haan, G. (2012). Genetic screen identifies microRNA cluster 99b/let-7e/125a as a regulator of primitive hematopoietic cells. Blood 119, 377–387.
Giles, F.J., Borthakur, G., Ravandi, F., Faderl, S., Verstovsek, S., Thomas, D., Wierda, W., Ferrajoli, A., Kornblau, S., Pierce, S., et al. (2007). The haematopoietic cell transplantation comorbidity index score is predictive of early death and survival in patients over 60 years of age receiving induction therapy for acute myeloid leukaemia. Br. J. Haematol. 136, 624–627.
Gilliland, D.G., and Griffin, J.D. (2002). The roles of FLT3 in hematopoiesis and leukemia. Blood 100, 1532–1542.
Greenberg, P.L., Tuechler, H., Schanz, J., Sanz, G., Garcia-Manero, G., Solé, F., Bennett, J.M., Bowen, D., Fenaux, P., Dreyfus, F., et al. (2012). Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120, 2454–2465.
Greenwood, M.J., Seftel, M.D., Richardson, C., Barbaric, D., Barnett, M.J., Bruyere, H., Forrest, D.L., Horsman, D.E., Smith, C., Song, K., et al. (2006). Leukocyte count

120
as a predictor of death during remission induction in acute myeloid leukemia. Leuk. Lymphoma 47, 1245–1252.
Grimwade, D., Walker, H., Oliver, F., Wheatley, K., Harrison, C., Harrison, G., Rees, J., Hann, I., Stevens, R., Burnett, A., et al. (1998). The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 92, 2322–2333.
Grimwade, D., Hills, R.K., Moorman, A.V., Walker, H., Chatters, S., Goldstone, A.H., Wheatley, K., Harrison, C.J., Burnett, A.K., and National Cancer Research Institute Adult Leukaemia Working Group (2010). Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116, 354–365.
Grossmann, V., Schnittger, S., Kohlmann, A., Eder, C., Roller, A., Dicker, F., Schmid, C., Wendtner, C.-M., Staib, P., Serve, H., et al. (2012). A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood 120, 2963–2972.
Guo, S., Bai, H., Megyola, C.M., Halene, S., Krause, D.S., Scadden, D.T., and Lu, J. (2012). Complex oncogene dependence in microRNA-125a-induced myeloproliferative neoplasms. Proc. Natl. Acad. Sci. U. S. A. 109, 16636–16641.
Haferlach, T., Schoch, C., Löffler, H., Gassmann, W., Kern, W., Schnittger, S., Fonatsch, C., Ludwig, W.-D., Wuchter, C., Schlegelberger, B., et al. (2003). Morphologic Dysplasia in De Novo Acute Myeloid Leukemia (AML) Is Related to Unfavorable Cytogenetics but Has No Independent Prognostic Relevance Under the Conditions of Intensive Induction Therapy: Results of a Multiparameter Analysis From the German AML Cooperative Group Studies. J. Clin. Oncol. 21, 256–265.
Hou, H.-A., Kuo, Y.-Y., Liu, C.-Y., Chou, W.-C., Lee, M.C., Chen, C.-Y., Lin, L.-I., Tseng, M.-H., Huang, C.-F., Chiang, Y.-C., et al. (2012). DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood 119, 559–568.
Hou, H.A., Lin, C.C., Chou, W.C., Liu, C.Y., Chen, C.Y., Tang, J.L., Lai, Y.J., Tseng, M.H., Huang, C.F., Chiang, Y.C., et al. (2014). Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia 28, 50–58.
Inoue, D., Kitaura, J., Togami, K., Nishimura, K., Enomoto, Y., Uchida, T., Kagiyama, Y., Kawabata, K.C., Nakahara, F., Izawa, K., et al. (2013). Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Invest. 123, 4627–4640.
Jemal, A., Siegel, R., Xu, J., and Ward, E. (2010). Cancer statistics, 2010. CA. Cancer J. Clin. 60, 277–300.
Jourdan, E., Boissel, N., Chevret, S., Delabesse, E., Renneville, A., Cornillet, P., Blanchet, O., Cayuela, J.-M., Recher, C., Raffoux, E., et al. (2013). Prospective

121
evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 121, 2213–2223.
Klepin, H.D., and Balducci, L. (2009). Acute myelogenous leukemia in older adults. The Oncologist 14, 222–232.
Klepin, H.D., Rao, A.V., and Pardee, T.S. (2014). Acute Myeloid Leukemia and Myelodysplastic Syndromes in Older Adults. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol.
Ko, M., Huang, Y., Jankowska, A.M., Pape, U.J., Tahiliani, M., Bandukwala, H.S., An, J., Lamperti, E.D., Koh, K.P., Ganetzky, R., et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843.
Koschmieder, S., Halmos, B., Levantini, E., and Tenen, D.G. (2009). Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 27, 619–628.
Kurokawa, M., and Hirai, H. (2003). Role of AML1/RUNX1 in the pathogenesis of hematological malignancies. Cancer Sci. 94, 841–846.
Larson, R.A. (2007). Is secondary leukemia an independent poor prognostic factor in acute myeloid leukemia? Best Pract. Res. Clin. Haematol. 20, 29–37.
Lazenby, M., Gilkes, A.F., Marrin, C., Evans, A., Hills, R.K., and Burnett, A.K. (2014). The prognostic relevance of FLT3 and NPM1 mutations on older patients treated intensively or non-intensively: a study of 1312 patients in the UK NCRI AML16 trial. Leukemia.
Lengliné, E., Raffoux, E., Lemiale, V., Darmon, M., Canet, E., Boissel, N., Schlemmer, B., Dombret, H., and Azoulay, E. (2012). Intensive care unit management of patients with newly diagnosed acute myeloid leukemia with no organ failure. Leuk. Lymphoma 53, 1352–1359.
Leroy, H., Roumier, C., Huyghe, P., Biggio, V., Fenaux, P., and Preudhomme, C. (2005). CEBPA point mutations in hematological malignancies. Leukemia 19, 329–334.
Ley, T.J., Ding, L., Walter, M.J., McLellan, M.D., Lamprecht, T., Larson, D.E., Kandoth, C., Payton, J.E., Baty, J., Welch, J., et al. (2010). DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433.
Lin, L.-I., Chen, C.-Y., Lin, D.-T., Tsay, W., Tang, J.-L., Yeh, Y.-C., Shen, H.-L., Su, F.-H., Yao, M., Huang, S.-Y., et al. (2005). Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 11, 1372–1379.
Linch, D.C., Hills, R.K., Burnett, A.K., Khwaja, A., and Gale, R.E. (2014). Impact of FLT3(ITD) mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood 124, 273–276.

122
Marcucci, G., Maharry, K., Wu, Y.-Z., Radmacher, M.D., Mrózek, K., Margeson, D., Holland, K.B., Whitman, S.P., Becker, H., Schwind, S., et al. (2010). IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 2348–2355.
Marcucci, G., Haferlach, T., and Döhner, H. (2011). Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 475–486.
Mardis, E.R., Ding, L., Dooling, D.J., Larson, D.E., McLellan, M.D., Chen, K., Koboldt, D.C., Fulton, R.S., Delehaunty, K.D., McGrath, S.D., et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066.
Marschalek, R. (2011). Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 152, 141–154.
Mead, A.J., Linch, D.C., Hills, R.K., Wheatley, K., Burnett, A.K., and Gale, R.E. (2007). FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 110, 1262–1270.
Metzeler, K.H., Maharry, K., Radmacher, M.D., Mrózek, K., Margeson, D., Becker, H., Curfman, J., Holland, K.B., Schwind, S., Whitman, S.P., et al. (2011). TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 1373–1381.
Metzeler, K.H., Becker, H., Maharry, K., Radmacher, M.D., Kohlschmidt, J., Mrózek, K., Nicolet, D., Whitman, S.P., Wu, Y.-Z., Schwind, S., et al. (2011). ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood 118, 6920–6929.
Meyer, C., Kowarz, E., Hofmann, J., Renneville, A., Zuna, J., Trka, J., Ben Abdelali, R., Macintyre, E., De Braekeleer, E., De Braekeleer, M., et al. (2009). New insights to the MLL recombinome of acute leukemias. Leukemia 23, 1490–1499.
Miesner, M., Haferlach, C., Bacher, U., Weiss, T., Macijewski, K., Kohlmann, A., Klein, H.-U., Dugas, M., Kern, W., Schnittger, S., et al. (2010). Multilineage dysplasia (MLD) in acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities and a prior history of MDS or MDS/MPN but has no independent prognostic relevance: a comparison of 408 cases classified as “AML not otherwise specified” (AML-NOS) or “AML with myelodysplasia-related changes” (AML-MRC). Blood 116, 2742–2751.
Mrózek, K., Marcucci, G., Nicolet, D., Maharry, K.S., Becker, H., Whitman, S.P., Metzeler, K.H., Schwind, S., Wu, Y.-Z., Kohlschmidt, J., et al. (2012). Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 30, 4515–4523.

123
Nangalia, J., Massie, C.E., Baxter, E.J., Nice, F.L., Gundem, G., Wedge, D.C., Avezov, E., Li, J., Kollmann, K., Kent, D.G., et al. (2013). Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405.
Naoe, T., and Kiyoi, H. (2013). Gene mutations of acute myeloid leukemia in the genome era. Int. J. Hematol. 97, 165–174.
Nibourel, O., Kosmider, O., Cheok, M., Boissel, N., Renneville, A., Philippe, N., Dombret, H., Dreyfus, F., Quesnel, B., Geffroy, S., et al. (2010). Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood 116, 1132–1135.
Osato, M., Asou, N., Abdalla, E., Hoshino, K., Yamasaki, H., Okubo, T., Suzushima, H., Takatsuki, K., Kanno, T., Shigesada, K., et al. (1999). Biallelic and heterozygous point mutations in the runt domain of the AML1/PEBP2alphaB gene associated with myeloblastic leukemias. Blood 93, 1817–1824.
Ostgård, L.S.G., Nørgaard, J.M., Sengeløv, H., Severinsen, M., Smidstrup Friis, L., Werenberg Marcher, C., Høgh Dufva, I., and Nørgaard, M. (2014). Comorbidity and performance status in acute myeloid leukemia patients: a nation-wide population-based cohort study. Leukemia.
Owen, C., Barnett, M., and Fitzgibbon, J. (2008). Familial myelodysplasia and acute myeloid leukaemia--a review. Br. J. Haematol. 140, 123–132.
Patel, J.P., Gönen, M., Figueroa, M.E., Fernandez, H., Sun, Z., Racevskis, J., Van Vlierberghe, P., Dolgalev, I., Thomas, S., Aminova, O., et al. (2012). Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089.
Peyrade, F., Gastaud, L., Ré, D., Pacquelet-Cheli, S., and Thyss, A. (2012). Treatment decisions for elderly patients with haematological malignancies: a dilemma. Lancet Oncol. 13, e344–e352.
Pratcorona, M., Abbas, S., Sanders, M.A., Koenders, J.E., Kavelaars, F.G., Erpelinck-Verschueren, C.A.J., Zeilemakers, A., Löwenberg, B., and Valk, P.J.M. (2012). Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica 97, 388–392.
Pratcorona, M., Brunet, S., Nomdedéu, J., Ribera, J.M., Tormo, M., Duarte, R., Escoda, L., Guàrdia, R., Queipo de Llano, M.P., Salamero, O., et al. (2013). Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood 121, 2734–2738.
Preudhomme, C., Warot-Loze, D., Roumier, C., Grardel-Duflos, N., Garand, R., Lai, J.L., Dastugue, N., Macintyre, E., Denis, C., Bauters, F., et al. (2000). High incidence of biallelic point mutations in the Runt domain of the AML1/PEBP2 alpha B gene in Mo acute myeloid leukemia and in myeloid malignancies with acquired trisomy 21. Blood 96, 2862–2869.

124
Puda, A., Milosevic, J.D., Berg, T., Klampfl, T., Harutyunyan, A.S., Gisslinger, B., Rumi, E., Pietra, D., Malcovati, L., Elena, C., et al. (2012). Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am. J. Hematol. 87, 245–250.
Renneville, A., Roumier, C., Biggio, V., Nibourel, O., Boissel, N., Fenaux, P., and Preudhomme, C. (2008). Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 22, 915–931.
Renneville, A., Boissel, N., Nibourel, O., Berthon, C., Helevaut, N., Gardin, C., Cayuela, J.-M., Hayette, S., Reman, O., Contentin, N., et al. (2012). Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia 26, 1247–1254.
Renneville, A., Abdelali, R.B., Chevret, S., Nibourel, O., Cheok, M., Pautas, C., Duléry, R., Boyer, T., Cayuela, J.-M., Hayette, S., et al. (2014). Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: results of the ALFA-0701 trial. Oncotarget 5, 916–932.
Röllig, C., Bornhäuser, M., Thiede, C., Taube, F., Kramer, M., Mohr, B., Aulitzky, W., Bodenstein, H., Tischler, H.-J., Stuhlmann, R., et al. (2011). Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 2758–2765.
Roumier, C., Fenaux, P., Lafage, M., Imbert, M., Eclache, V., and Preudhomme, C. (2003). New mechanisms of AML1 gene alteration in hematological malignancies. Leukemia 17, 9–16.
Rücker, F.G., Schlenk, R.F., Bullinger, L., Kayser, S., Teleanu, V., Kett, H., Habdank, M., Kugler, C.-M., Holzmann, K., Gaidzik, V.I., et al. (2012). TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 119, 2114–2121.
Schlenk, R.F., Döhner, K., Krauter, J., Fröhling, S., Corbacioglu, A., Bullinger, L., Habdank, M., Späth, D., Morgan, M., Benner, A., et al. (2008). Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 358, 1909–1918.
Schnittger, S., Schoch, C., Dugas, M., Kern, W., Staib, P., Wuchter, C., Löffler, H., Sauerland, C.M., Serve, H., Büchner, T., et al. (2002). Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100, 59–66.
Schnittger, S., Schoch, C., Kern, W., Mecucci, C., Tschulik, C., Martelli, M.F., Haferlach, T., Hiddemann, W., and Falini, B. (2005). Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106, 3733–3739.

125
Schnittger, S., Bacher, U., Kern, W., Alpermann, T., Haferlach, C., and Haferlach, T. (2011). Prognostic impact of FLT3-ITD load in NPM1 mutated acute myeloid leukemia. Leukemia 25, 1297–1304.
Schnittger, S., Eder, C., Jeromin, S., Alpermann, T., Fasan, A., Grossmann, V., Kohlmann, A., Illig, T., Klopp, N., Wichmann, H.-E., et al. (2013). ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 27, 82–91.
Shen, Y., Zhu, Y.-M., Fan, X., Shi, J.-Y., Wang, Q.-R., Yan, X.-J., Gu, Z.-H., Wang, Y.-Y., Chen, B., Jiang, C.-L., et al. (2011). Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood 118, 5593–5603.
Slovak, M.L., Kopecky, K.J., Cassileth, P.A., Harrington, D.H., Theil, K.S., Mohamed, A., Paietta, E., Willman, C.L., Head, D.R., Rowe, J.M., et al. (2000). Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 96, 4075–4083.
Song, W.J., Sullivan, M.G., Legare, R.D., Hutchings, S., Tan, X., Kufrin, D., Ratajczak, J., Resende, I.C., Haworth, C., Hock, R., et al. (1999). Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 23, 166–175.
Sorror, M.L., Maris, M.B., Storb, R., Baron, F., Sandmaier, B.M., Maloney, D.G., and Storer, B. (2005). Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 106, 2912–2919.
Stirewalt, D.L., and Radich, J.P. (2003). The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 3, 650–665.
Tanaka, T., Tanaka, K., Ogawa, S., Kurokawa, M., Mitani, K., Nishida, J., Shibata, Y., Yazaki, Y., and Hirai, H. (1995). An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 14, 341–350.
Tefferi, A., and Vainchenker, W. (2011). Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 573–582.
Thol, F., Friesen, I., Damm, F., Yun, H., Weissinger, E.M., Krauter, J., Wagner, K., Chaturvedi, A., Sharma, A., Wichmann, M., et al. (2011). Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 29, 2499–2506.
Tie, R., Zhang, T., Fu, H., Wang, L., Wang, Y., He, Y., Wang, B., Zhu, N., Fu, S., Lai, X., et al. (2014). Association between DNMT3A mutations and prognosis of adults with de novo acute myeloid leukemia: a systematic review and meta-analysis. PloS One 9, e93353.

126
Valk, P.J.M., Verhaak, R.G.W., Beijen, M.A., Erpelinck, C.A.J., Barjesteh van Waalwijk van Doorn-Khosrovani, S., Boer, J.M., Beverloo, H.B., Moorhouse, M.J., van der Spek, P.J., Löwenberg, B., et al. (2004). Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 350, 1617–1628.
Vannucchi, A.M., Lasho, T.L., Guglielmelli, P., Biamonte, F., Pardanani, A., Pereira, A., Finke, C., Score, J., Gangat, N., Mannarelli, C., et al. (2013). Mutations and prognosis in primary myelofibrosis. Leukemia 27, 1861–1869.
Vardiman, J.W., Harris, N.L., and Brunning, R.D. (2002). The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100, 2292–2302.
Vardiman, J.W., Thiele, J., Arber, D.A., Brunning, R.D., Borowitz, M.J., Porwit, A., Harris, N.L., Le Beau, M.M., Hellström-Lindberg, E., Tefferi, A., et al. (2009). The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951.
Verhaak, R.G.W., Goudswaard, C.S., van Putten, W., Bijl, M.A., Sanders, M.A., Hugens, W., Uitterlinden, A.G., Erpelinck, C.A.J., Delwel, R., Löwenberg, B., et al. (2005). Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106, 3747–3754.
Versluis, J., Labopin, M., Niederwieser, D., Socie, G., Schlenk, R.F., Milpied, N., Nagler, A., Blaise, D., Rocha, V., Cornelissen, J.J., et al. (2014). Prediction of non-relapse mortality in recipients of reduced intensity conditioning allogeneic stem cell transplantation with AML in first complete remission. Leukemia.
Walter, R.B., and Estey, E.H. (2014). Management of older or unfit patients with acute myeloid leukemia. Leukemia.
Wandt, H., Schäkel, U., Kroschinsky, F., Prange-Krex, G., Mohr, B., Thiede, C., Pascheberg, U., Soucek, S., Schaich, M., and Ehninger, G. (2008). MLD according to the WHO classification in AML has no correlation with age and no independent prognostic relevance as analyzed in 1766 patients. Blood 111, 1855–1861.
Wang, J., Li, Z., He, Y., Pan, F., Chen, S., Rhodes, S., Nguyen, L., Yuan, J., Jiang, L., Yang, X., et al. (2014). Loss of ASXL1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553.
Wang, X., Dai, H., Wang, Q., Wang, Q., Xu, Y., Wang, Y., Sun, A., Ruan, J., Chen, S., and Wu, D. (2013). EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. PLoS ONE 8, e61341.
Weissmann, S., Alpermann, T., Grossmann, V., Kowarsch, A., Nadarajah, N., Eder, C., Dicker, F., Fasan, A., Haferlach, C., Haferlach, T., et al. (2012). Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 26, 934–942.

127
West, R.R., Hsu, A.P., Holland, S.M., Cuellar-Rodriguez, J., and Hickstein, D.D. (2014). Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica 99, 276–281.
Wetzler, M., Mrózek, K., Kohlschmidt, J., Dombret, H., Döhner, H., Pilorge, S., Krug, U., Carroll, A.J., Larson, R.A., Marcucci, G., et al. (2014). Intensive induction is effective in selected octogenarian acute myeloid leukemia patients: prognostic significance of karyotype and selected molecular markers used in the European LeukemiaNet classification. Haematologica 99, 308–313.
Wouters, B.J., Löwenberg, B., Erpelinck-Verschueren, C.A.J., van Putten, W.L.J., Valk, P.J.M., and Delwel, R. (2009). Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113, 3088–3091.
Xie, Y., Davies, S.M., Xiang, Y., Robison, L.L., and Ross, J.A. (2003). Trends in leukemia incidence and survival in the United States (1973-1998). Cancer 97, 2229–2235.
Xu, F., Li, X., Wu, L., Zhang, Q., Yang, R., Yang, Y., Zhang, Z., He, Q., and Chang, C. (2011). Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann. Hematol. 90, 643–653.
Xu, X.-Q., Wang, J.-M., Gao, L., Qiu, H.-Y., Chen, L., Jia, L., Hu, X.-X., Yang, J.-M., Ni, X., Chen, J., et al. (2014). Characteristics of acute myeloid leukemia with myelodysplasia-related changes: A retrospective analysis in a cohort of Chinese patients. Am. J. Hematol.
Yan, H., Parsons, D.W., Jin, G., McLendon, R., Rasheed, B.A., Yuan, W., Kos, I., Batinic-Haberle, I., Jones, S., Riggins, G.J., et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773.
Yan, X.-J., Xu, J., Gu, Z.-H., Pan, C.-M., Lu, G., Shen, Y., Shi, J.-Y., Zhu, Y.-M., Tang, L., Zhang, X.-W., et al. (2011). Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 43, 309–315.

128
ANNEXES
Annexe 1 :
Mutations in ASXL1 are associated with poor prognosis across the spectrum of
malignant myeloid diseases
Véronique Gelsi-Boyer, Mandy Brecqueville, Raynier Devillier, Anne Murati, Marie-
Joelle Mozziconacci and Daniel Birnbaum
Journal of Hematology & Oncology 2012, 5:12
Annexe 2 :
Myeloid malignancies: mutations, models and management
Anne Murati, Mandy Brecqueville, Raynier Devillier, Marie-Joelle Mozziconacci,
Véronique Gelsi-Boyer and Daniel Birnbaum*
BMC Cancer 2012, 12:304





























