Embed Size (px)
Citation preview

TTHHÈÈSSEE
En vue de l'obtention du
DDOOCCTTOORRAATT DDEE LL’’UUNNIIVVEERRSSIITTÉÉ DDEE TTOOUULLOOUUSSEE
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Immunologie et maladies infectieuses
JURY
Elisabeth MENU: Rapporteur Franck HALARY: Rapporteur
Georges HERBEIN: Examinateur Stéphane BERTAGNOLI: Examinateur
Patrice MASSIP: Examinateur Christian DAVRINCHE:Directeur de thèse
Ecole doctorale : Biologie-Santé-Biotechnologies
Unité de recherche : Inserm U563 Directeur(s) de Thèse : Christian DAVRINCHE
Rapporteurs : Elisabeth MENU Franck HALARY
Présentée et soutenue par RAUWEL Benjamin Le 19 Mars 2010
Titre : Activation du récepteur nucléaire PPAR-gamma
par le Cytomégalovirus Humain: Implication dans la réplication virale
et la perturbation de la migration placentaire

TTHHÈÈSSEE
En vue de l'obtention du
DDOOCCTTOORRAATT DDEE LL’’UUNNIIVVEERRSSIITTÉÉ DDEE TTOOUULLOOUUSSEE
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Immunologie et maladies infectieuses
JURY
Elisabeth MENU: Rapporteur Franck HALARY: Rapporteur
Georges HERBEIN: Examinateur Stéphane BERTAGNOLI: Examinateur
Patrice MASSIP: Examinateur Christian DAVRINCHE:Directeur de thèse
Ecole doctorale : Biologie-Santé-Biotechnologies
Unité de recherche : Inserm U563 Directeur(s) de Thèse : Christian DAVRINCHE
Rapporteurs : Elisabeth MENU Franck HALARY
Présentée et soutenue par RAUWEL Benjamin Le 19 Mars 2010
Titre : Activation du récepteur nucléaire PPAR-gamma
par le Cytomégalovirus Humain: Implication dans la réplication virale
et la perturbation de la migration placentaire

1
UNIVERSITE PAUL SABATIER TOULOUSE III
ECOLE DOCTORALE BIOLOGIE-SANTE-BIOTECHNOLOGIE
2010
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Spécialité IMMUNOLOGIE ET MALADIES INFECTIEUSES
Benjamin RAUWEL
Activation du récepteur nucléaire PPARγγγγ par le Cytomégalovirus Humain :
Implication dans la réplication virale
et la perturbation de la migration et de l’invasion placentaire.
Directeur de thèse
Christian DAVRINCHE
Jury
Elisabeth Menu
Franck Halary
Georges HERBEIN
Stéphane BERTAGNOLI
Patrice MASSIP
Christian DAVRINCHE

2
Remerciements
Je tiens à adresser mes remerciements les plus sincères
Au Docteur Christian DAVRINCHE,
Pour m’avoir accueilli au sein de son laboratoire durant toutes ces années, pour m’avoir transmis la passion de la recherche ainsi qu’un esprit scientifique et pour son extrême gentillesse. Je lui exprime toute ma gratitude.
Aux Docteurs Elisabeth MENU, Franck HALARY ainsi qu’aux Professeurs Georges
HERBEIN, Stéphane BERTAGNOLI et Patrice MASSIP,
Pour l’attention qu’ils ont porté à ce travail et pour m’avoir fait l’honneur d’être parmi le jury de cette thèse.
Au Docteur Thierry FOURNIER,
Pour m’avoir accueilli chaleureusement au sein de son laboratoire tout au long de notre collaboration, et pour toute son aide apportée à ce travail ainsi qu’à ma formation.
Au Docteur Danielle EVAIN-BRION,
Pour sa collaboration et son accueil au sein de son laboratoire.
A tous les membres de l’équipe CD/JI (anciens et actuels),
Pour l’excellente ambiance de travail, d’avoir supporté mon humour durant toutes ces années et pour les tennis et squash qui permettent de se détendre après des heures de manips ! Merci à vous tous : Les deux Jérôme, Mélinda, les Charlotte, Bernard, Stéphane (pour toutes ses contrepèteries et ses grands films), Michel, Franck, Hugo, Marie-Émilie, tous les gens de l’IFB. Et tous ceux que j’oublie.
Un remerciement tout particulier à Hélène, qui m’a tout appris, pour sa gentillesse et toute l’aide qu’elle m’a apporté.

3
A Aline DARMANA,
Pour toute son attention, sa gentillesse et tous ses petits gâteaux et gourmandises qui nous ont permis d’égayer nos journées !
Au Docteur Nathalie Vergnolle ainsi que toute son équipe,
Pour la collaboration sur mon projet de thèse et leur sympathie. Un grand merci à Nicolas pour son aide et ses réactifs !!!!
A toute ma famille,
Pour leur soutien durant toutes ces années et bien d’autres encore… Il n’y a pas de mots pour exprimer à quel point ils sont importants pour moi.
A ma belle famille,
Pour m’avoir accueilli au sein de leur famille et soutenu comme leur propre fils.
A tous mes amis,
Pour l’ambiance excellente qui règne au sein du groupe, leur amitié et leur soutien moral.
Et un grand merci à ma petite femme adorée,
Qui m’a soutenu et supporté mon humeur durant tout ce travail et toutes ces années. Sans qui la vie serait différente et nettement moins intéressante. Je t’aime.
Et à notre premier enfant,
Qui est encore dans le ventre de sa mère mais que nous attendons avec impatience pour le mois d’Août.

4
Sommaire
Remerciements ......................................................................................................................... 2�
Sommaire .................................................................................................................................. 4�
Abstract ..................................................................................................................................... 6�
Résumé ...................................................................................................................................... 7�
Abréviations .............................................................................................................................. 8�
INTRODUCTION .................................................................................................................... 9�
LE CYTOMEGALOVIRUS HUMAIN ............................................................................... 10�
I.� Historique ...................................................................................................................... 11�
II.� Généralités ................................................................................................................. 11�
III.� Epidémiologie ............................................................................................................ 12�
IV.� Relevance clinique (Mocarski et al., 2007b) ............................................................. 13�
V.� Réponse immunitaire et vaccination .......................................................................... 15�
a.� Réponse Immunitaire innée ....................................................................................... 15�
b.� Réponse immunitaire adaptative ............................................................................... 16�
c.� Schéma récapitulatif de la réponse immune anti-CMV ............................................. 17�
d.� Vaccination ................................................................................................................ 17�
e.� Mécanismes d’échappement immunitaire du HCMV ............................................... 19�
VI.� Biologie du HCMV ................................................................................................... 21�
a.� Structure de la particule virale et génome ................................................................. 21�
b.� Tropisme et entrée du virus ....................................................................................... 24�
c.� Cycle de réplication ................................................................................................... 25�
d.� Le MIEP et les protéines IE ....................................................................................... 28�
VII.� Peroxisome Proliferator Activated Receptor γ (PPARγ) ........................................... 34�
e.� Généralités sur les PPARs ......................................................................................... 34�
f.� Structure de PPARγ ................................................................................................... 35�
g.� Rôle transcriptionel .................................................................................................... 36�
h.� Rôle de trans-répresseur ............................................................................................ 37�
i.� Les différents rôles de PPARγ ................................................................................... 38�
LE PLACENTA HUMAIN ................................................................................................... 41�
I.� Structure du placenta ..................................................................................................... 42�
II.� Mise en place du placenta au cours de la grossesse................................................... 44�
III.� PPARγ dans la grossesse ........................................................................................... 46�

5
IV.� PAPP-A dans la grossesse ......................................................................................... 49�
V.� MMP et TIMP ........................................................................................................... 50�
VI.� Les récepteurs PAR (Protease Activated Receptor) .................................................. 52�
VII.� HCMV et pathologie de la grossesse ......................................................................... 57�
VIII.� Objectifs du travail ................................................................................................. 59�
TRAVAUX DE RECHERCHE ............................................................................................. 60�
L’activation du récepteur nucléaire PPARγγγγ par le HCMV pour sa réplication perturbe
la migration et les propriétés invasives du cytotrophoblaste. ............................................ 61�
Etude des mécanismes dépendant de l’activation de PPARγγγγ par le HCMV perturbant la
migration et l’invasion du cytotrophoblaste. ....................................................................... 90�
CONCLUSIONS ET PERSPECTIVES ............................................................................. 102�
BIBLIOGRAPHIE ............................................................................................................... 108�
Annexes ................................................................................................................................. 123�

6
Abstract
Activation of nuclear receptor PPARγγγγ by Human Cytomegalovirus: Implication in viral
replication and impairment of placental migration and invasiveness.
Human Cytomegalovirus (HCMV), a member of the betaherpesvirus family contributes to the
development of numerous diseases in immuno-compromised hosts, fetuses and newborns.
Primary infection during the first trimester of pregnancy could lead to miscarriages or
intrauterine growth retardation. It has been demonstrated that the virus benefits from
inflammation by using Cyclooxygenase-2 (COX-2) pathway for the transcription of its
immediate early genes, especially IE2 production which is essential for viral replication and
whose transcription is under the control of the Major Immediate Early Promoter (MIEP). Up
to now, mechanisms were still unknown. Interestingly, COX-2 activation leads to the
production of a natural ligand for PPARγ, a transcription factor belonging to the nuclear
receptors family.
In this study, we have demonstrated for the first time that HCMV induced activation of
PPARγ for its de novo replication and notably for IE2 transcription. Accordingly, we have
identified three new functional DNA binding sequences for PPARγ (PPRE: PPar Response
Element) into the distal part of the MIEP.
Then, we focused on the role of PPARγ in placental migration and invasion, with special care
on consequences of its activation by HCMV. We have demonstrated that HCMV impaired
migration and invasiveness of cytotrophoblast by activating PPARγ. Studies of sustained
mechanisms allowed us to identify potential targets amongst which PAR receptors whose
function was impaired by HCMV.
Further studies should allow to identify new targets useful in prognostic of pregnancy with
high risk for newborns and to develop new therapeutic approaches.�
�
�
�

7
Résumé
Activation du récepteur nucléaire PPARγγγγ par le Cytomégalovirus Humain : Implication
dans la réplication virale et la perturbation de la migration et invasion placentaire.
Le cytomégalovirus Humain, membre de la famille des β-Herpèsvirus, est responsable de
nombreux troubles chez les personnes immuno-déprimées, les fœtus et les nouveau-nés et une
primo-infection de la mère durant le premier trimestre de grossesse peut entraîner des
avortements spontanés ainsi que des retards de croissance intra-utérins. Il a été montré que le
virus tire profit de l’inflammation en utilisant la voie de la cyclooxygénase-2 (COX-2) pour la
transcription des gènes très précoces, aboutissant en particulier à la production de la protéine
IE2, indispensable au cycle viral. Aucun mécanisme n’a été décrit jusqu’à présent. Cette
protéine est sous le contrôle d’un promoteur majeur appelé MIEP (Major Immediate Early
Promoter). En aval de la voie COX-2 se situe le récepteur nucléaire PPARγ, un facteur de
transcription dont un des ligands naturels est un produit de cette voie enzymatique.
Dans cette étude nous avons pu démontrer pour la première fois l’utilisation de ce facteur de
transcription par le HCMV pour sa propre réplication, et notamment pour la transcription de
la protéine très précoce IE2. Nous avons alors identifié trois nouvelles séquences de fixation
de PPARγ à l’ADN (PPRE : PPar Response Element) dans la partie distale du MIEP et la
fonctionnalité de ces séquences a été démontré.
Dans un second temps, nous nous sommes intéressés au rôle de PPARγ dans la migration et
l’invasion placentaire et plus particulièrement aux conséquences de son activation par le
HCMV. Nous avons démontré que le HCMV perturbe la migration et les capacités invasives
du cytotrophoblaste extravilleux en activant PPARγ. L’étude des mécanismes à l’origine de ce
phénomène nous a permis d’identifier des cibles potentielles parmi lesquelles les récepteurs
PAR (Protease Activated Receptor) dont la fonction est perturbée par l’infection.
L’étude approfondie des mécanismes impliqués devrait par ailleurs permettre d’envisager de
nouvelles cibles afin de diagnostiquer les grossesses à risque ainsi que de nouvelles cibles
thérapeutiques.

8
Abréviations
COX-2 : Cyclooxygénase-2
CTEV : Cytotrophoblaste Extravilleux
CTV : Cytotrophoblaste Villeux
GPCMV : Guinea Pig Cytomegalovirus
HCMV : Cytomégalovirus Humain
HETE : Acide hydroxyeicosatétraénoïque
HIPEC : Human Invasive and Proliferative Extravillous Cytotrophoblast
HODE : Acide hydroxyoctadécanoïque
IE : Immediate Early
IGF-II : Insulin-like Growth Factor
IGF-II R : IGF-II receptor
MIEP : Major Immediate Early Promoter
MMP : Matrix Metallo-Proteinase
M.O.I. : Multiplicity Of Infection
PAPP-A : Pregnancy Associated Plasma Protein-A
PAR : Protease-Activated Receptor
PGC-1 : Ppar Gamma Co-factor-1
PPAR : Peroxisome Proliferator Activated Receptor
PPRE : PPar Response Element
RAR : Retinoic Acid Receptor
RXR : Retinoic X Receptor
SCTV : Syncytiotrophoblaste
SUMO : Small Ubiquitin-like Modifier
TIMP : Tissue Inhibitor of Metallo-Proteinase

9
INTRODUCTION

10
PREMIERE PARTIE
LE CYTOMEGALOVIRUS HUMAIN

11
I. Historique
La première description de la pathogénie du cytomégalovirus date du début du XXe siècle.
C’est en 1904 que Ribbert, Jesionek et Kiolemenoglou décrivent pour la première fois la
présence de grandes cellules à inclusion intranucléaire dans les reins, les poumons, le foie
ainsi que la parotide de fœtus et d’enfants mort-nés, caractéristique de ce qu’ils désignent
alors comme « la maladie des inclusions cytomégaliques ». L’origine virale de cette maladie
est démontrée dans les années 20 par un rapprochement de ces lésions avec les cellules de
lésions cutanées de la varicelle ainsi que l’étude histologique de glandes salivaires de cochons
d’Inde infectés. En 1932, Farber publiera une étude démontrant que 12% des enfants mourant
de pathologies variés sans point commun apparent possèdent des inclusions cytomégaliques
dans leurs glandes salivaires. Il désigne alors ce virus comme le « virus des glandes
salivaires ». Son isolement à été réalisé par la suite dans les années 50 par 4 groupes
indépendants : Wyatt suggère la première technique de diagnostic qui consiste en la recherche
de cellules caractéristiques de l’infection dans les urines de bébés. Technique qui sera
appliquée dès 1952 par Fetterman. En 1956, Smith obtient la réplication du virus responsable
de « la maladie des inclusions cytomégaliques » dans des cellules fibroblastiques humaines
cultivées in vitro. Rowe va alors isoler la première souche à partir de tissu adénoïdien d’un
enfant normal. Cette souche sera alors désignée comme AD169. En 1960, Weller et al.
désignent ce virus du terme « cytomégalovirus » en raison de la morphologie des cellules
infectées. Par la suite, des études sérologiques démontreront que l’infection à cytomégalovirus
est largement répandue dans la population mondiale et son rôle étiologique dans les
syndromes mononucléosiques est confirmé. Dans les années 60-70, le lien entre la baisse des
défenses immunitaires et la réactivation du virus est démontré. Des études vont alors établir
que les individus très jeunes, âgés, affaiblis ou immunodéprimés sont plus affectés par
l’infection.
II. Généralités
Le cytomégalovirus appartient à la famille des Herpesviridae dans laquelle il existe 8 virus
infectant l’homme classés en 3 sous-familles, les alpha-, béta- et gamma herpesvirinae. Ces
virus possèdent un large spectre d’hôtes et peuvent induire deux types d’infections : une
infection lytique correspondant à une forte réplication du virus conduisant à la lyse de la
cellule hôte, et une infection latente qui consistera à un contrôle de la réplication du virus en
12
attendant un moment plus approprié tel qu’une immunodéficience ou l’expression de
protéines nécessaires à l’aboutissement du cycle lytique. Ces virus ont un large tropisme mais
un spectre d’hôte très étroit et sont résumés dans le tableau suivant :
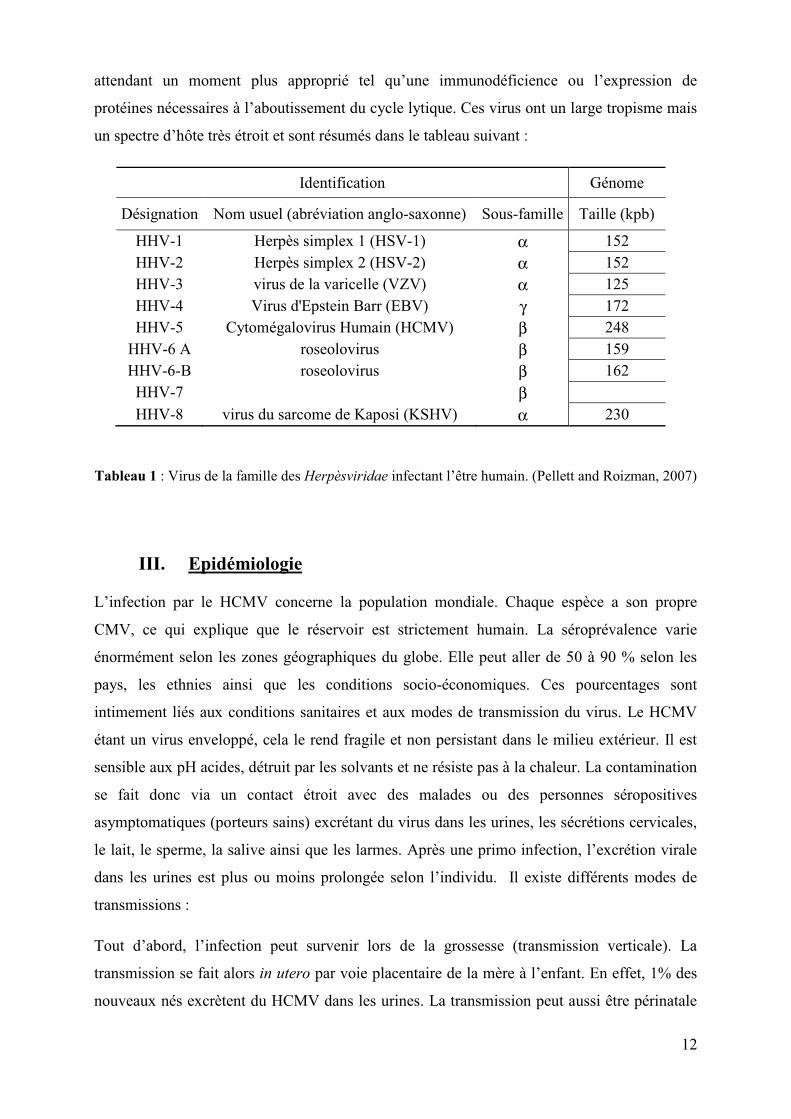
Identification Génome
Désignation Nom usuel (abréviation anglo-saxonne) Sous-famille Taille (kpb)
HHV-1 Herpès simplex 1 (HSV-1) α 152 HHV-2 Herpès simplex 2 (HSV-2) α 152 HHV-3 virus de la varicelle (VZV) α 125 HHV-4 Virus d'Epstein Barr (EBV) γ 172 HHV-5 Cytomégalovirus Humain (HCMV) β 248
HHV-6 A roseolovirus β 159 HHV-6-B roseolovirus β 162
HHV-7 β HHV-8 virus du sarcome de Kaposi (KSHV) α 230
Tableau 1 : Virus de la famille des Herpèsviridae infectant l’être humain. (Pellett and Roizman, 2007)
III. Epidémiologie
L’infection par le HCMV concerne la population mondiale. Chaque espèce a son propre
CMV, ce qui explique que le réservoir est strictement humain. La séroprévalence varie
énormément selon les zones géographiques du globe. Elle peut aller de 50 à 90 % selon les
pays, les ethnies ainsi que les conditions socio-économiques. Ces pourcentages sont
intimement liés aux conditions sanitaires et aux modes de transmission du virus. Le HCMV
étant un virus enveloppé, cela le rend fragile et non persistant dans le milieu extérieur. Il est
sensible aux pH acides, détruit par les solvants et ne résiste pas à la chaleur. La contamination
se fait donc via un contact étroit avec des malades ou des personnes séropositives
asymptomatiques (porteurs sains) excrétant du virus dans les urines, les sécrétions cervicales,
le lait, le sperme, la salive ainsi que les larmes. Après une primo infection, l’excrétion virale
dans les urines est plus ou moins prolongée selon l’individu. Il existe différents modes de
transmissions :
Tout d’abord, l’infection peut survenir lors de la grossesse (transmission verticale). La
transmission se fait alors in utero par voie placentaire de la mère à l’enfant. En effet, 1% des
nouveaux nés excrètent du HCMV dans les urines. La transmission peut aussi être périnatale

13
ou post-natale après acquisition du virus lors du passage des voies génitales ou lors de
l’allaitement. Entre 8 et 60% des enfants sont infectés durant les 6 premiers mois de la vie à
cause de l’alimentation par le lait maternel contaminé par le virus. Ce mode de transmission
reste le plus répandu dans le monde (Reynolds et al., 1973; Stagno et al., 1984). Plus
tardivement, les enfants peuvent se transmettre entre eux le virus par la salive durant la petite
enfance (4 fois plus d’infections chez les enfants mis en crèche). Dans ce cas l’infection se
fait par voie pharyngée.
Chez les adultes, le mode de contamination le plus répandu est la contamination par voie
sexuelle du fait de la présence du HCMV dans les tractus génitaux. La séropositivité est alors
en rapport avec le nombre de partenaires sexuels et l’âge du premier rapport sexuel. De plus le
taux de prévalence des anticorps anti-HCMV chez les adolescents fait plus que doubler durant
la première année d’activité sexuelle.
Enfin, il existe un dernier mode de transmission dans les pays industrialisés, l’infection
iatrogène. Celle-ci se fera en l’occurrence par le sang ou les organes transplantés provenant de
donneurs séropositifs. Pour la transplantation, il peut également y avoir une réactivation du
virus essentiellement due aux traitements immunosuppresseurs (Kotton and Fishman, 2005).
IV. Relevance clinique (Mocarski et al., 2007b)
La primo-infection est en général asymptomatique. Les malades symptomatiques vont
présenter une fièvre, des myalgies ainsi qu’un syndrome mononucléosique identique à celui
causé par le virus d’Epstein-Barr. Environ 8% des mononucléoses sont causées par le HCMV.
Cependant l’atteinte d’un organe cible est exceptionnelle chez le patient immunocompétent :
pneumopathie, myocardite, hépatite, encéphalite ou encore ulcérations coliques. Les
réactivations et les réinfections ne se manifestent que rarement par un syndrome
mononucléosique et sont en général asymptomatiques chez l’adulte non immunodéprimé. Il
peut cependant y avoir des complications et de graves troubles de santé dans certains cas :
Au cours de la grossesse, la primo-infection ainsi qu’une réinfection par une souche différente
de HCMV durant le premier trimestre peut provoquer des avortements spontanés. Au-delà de
ce phénomène, l’infection peut se transmettre au fœtus par le placenta avec une incidence qui
varie de 0.2 à 2%. Elle est rarement symptomatique mais environ 5% des enfants infectés in
utero présentent une infection généralisée se caractérisant par différents symptômes tels qu’un

14
retard de croissance intra-utérin, une microcéphalie ou encore une chorio-rétinite. Tout ceci se
traduit par de graves séquelles telles qu’un retard mental ou des troubles sensoriels (vision,
ouïe) (Cheeran et al., 2009). Les réactivations durant la grossesse ainsi que les infections
périnatales sont pour la plupart asymptomatiques et sans conséquence, cependant dans 5 à
10% des cas elles peuvent conduire plus tardivement à des séquelles neurologiques dont la
plus fréquente est la surdité. Un chapitre sera consacré plus tard aux détails et aux
conséquences de l’infection par le HCMV durant la grossesse.
Dans le cas de greffes, le HCMV occupe un des premiers rangs en termes de complications
infectieuses. Chez les transplantés, les maladies à CMV représentent un risque majeur
d’infection (60 à 100% des transplantés rénaux). Cependant ce pourcentage varie en fonction
de la transplantation (organe solide ou moelle osseuse), de l’organe transplanté et du
traitement utilisé lors de la greffe. Le rejet de greffe est alors très lié à l’infection HCMV dans
de nombreux cas. En effet le traitement immunosuppresseur a une action activatrice sur le
HCMV (réactivation ou primo-infection symptomatique), tandis que la réduction du
traitement immunosuppresseur au cours de l’infection favorise le rejet.
Chez les patients atteint du VIH et au stade SIDA, le HCMV reste une des infections
opportunistes les plus répandues. La prévalence du HCMV ainsi que ses voies de
transmissions font que la majorité des patients infectés par le VIH le sont aussi pour le
HCMV. L’infection reste asymptomatique dans un premier temps et se traduit uniquement par
une excrétion du HCMV dans les urines, la salive et les sécrétions génitales. Cependant,
lorsque le patient devient immunodéprimé, le HCMV se manifestera le plus souvent par une
rétinite. L’atteinte de la rétine se traduit par une baisse de l’acuité visuelle et son extension est
irréversible (Scholz et al., 2003). L’infection peut alors s’aggraver en troubles neuronaux tels
qu’une encéphalite subaigüe. Encore une fois, le HCMV se manifeste de manière opportuniste
et profite de l’immunodépression de son hôte pour se répliquer.
Le HCMV est aussi impliqué dans d’autres phénomènes tels que l’athérosclérose. En effet
l’infection par le HCMV va stimuler la prolifération cellulaire et accélérer l’angiogénèse
(Bentz and Yurochko, 2008). D’autres travaux contradictoires sur le sujet laissent un doute
quant au réel impact de l’infection sur l’athérosclérose.
Enfin, bien que le HCMV ne soit pas répertorié comme un virus oncogène, son infection a été
impliquée dans différents cancers. Un concept « d’oncomodulateur » permet de mieux
expliquer son rôle mais les études sont encore en pleine expansion.(pour revue (Michaelis et
al., 2009)).

15
Afin de contrecarrer le virus, il est important de comprendre la réponse immunitaire
spécifique et de développer une vaccination et de meilleurs outils de diagnostic et
thérapeutiques.
V. Réponse immunitaire et vaccination
a. Réponse Immunitaire innée
La réponse immunitaire innée va jouer un rôle important dans la défense anti-HCMV et
permettra par la suite d’activer la réponse immunitaire adaptative. Les principaux récepteurs
pour cette réponse seront les Toll-like récepteurs (TLR) et plus particulièrement les TLR-3 et
9 pour la reconnaissance de l’ADN viral, mais aussi le TLR-2 au moment de l’entrée du virus
notamment par sa fixation à la glycoprotéine d’enveloppe gB (Boehme et al., 2006; Compton
et al., 2003). Cette activation du système immunitaire inné va induire une sécrétion de
cytokines inflammatoires et engendrer l’activation des cellules dendritiques (DC), des
macrophages ainsi que des cellules NK. Ces cellules pourront alors lyser directement les
cellules infectées (NK) ou activer une réponse immunitaire adaptative (macrophages et DCs).
La réponse immunitaire NK anti-CMV a été décrite pour la première fois chez la souris par
une activité d’élimination ainsi que de protection contre l’infection (Bukowski et al., 1985;
Bukowski et al., 1983; Polic et al., 1998).
Le rôle anti-CMV de ces cellules chez l’Homme est encore assez méconnu. Cependant il a été
démontré que leur activité augmentait durant une infection HCMV et que leur absence était
corrélée avec des primo-infections herpétiques sévères dont le HCMV (Biron et al., 1989;
Venema et al., 1994).
Les récepteurs mis en jeu dans cette reconnaissance sont présentés dans la revue de Guma
(Guma et al., 2006).
16
b. Réponse immunitaire adaptative
Au cours de la réponse immunitaire adaptative, la réponse humorale sera traduite par une
activation des lymphocytes B sécrétant des anticorps neutralisant avec pour cible principale la
glycoprotéine d’enveloppe gB (Britt et al., 1990). Parallèlement à cela, la réponse
immunitaire médiée par les lymphocytes T reste le mécanisme prédominant de contrôle de
l’infection par le HCMV. Il existe un large panel de reconnaissance des antigènes du HCMV
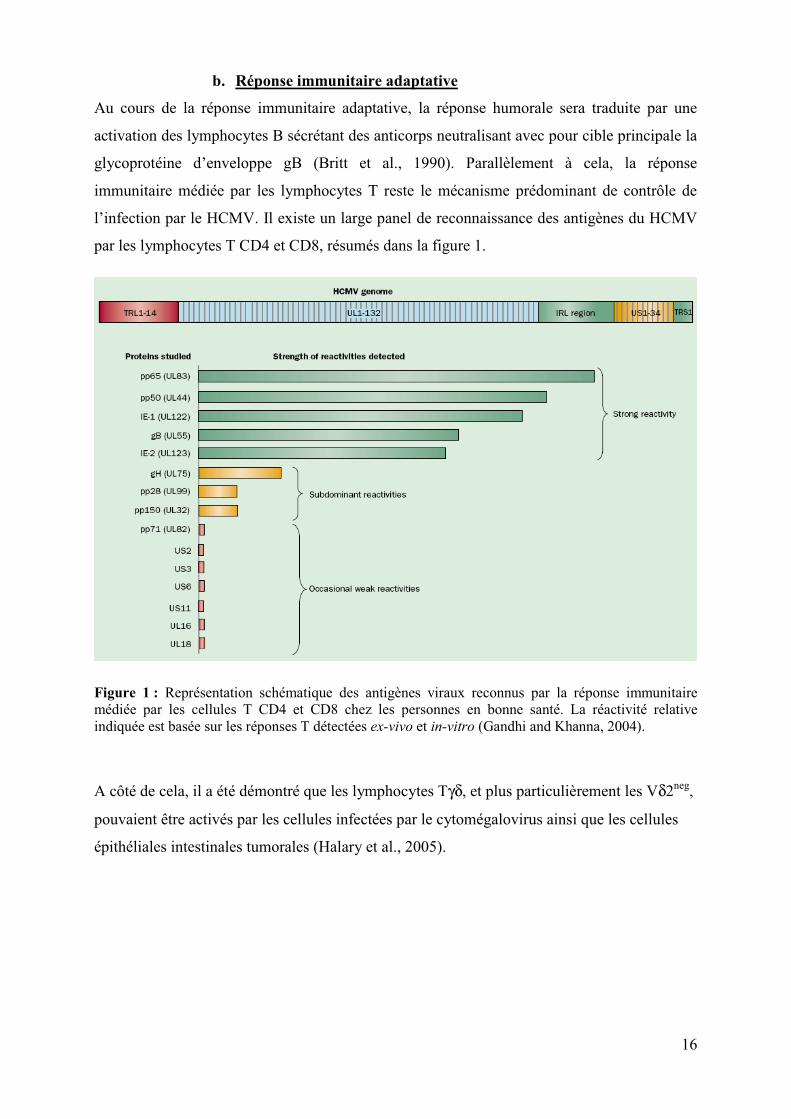
par les lymphocytes T CD4 et CD8, résumés dans la figure 1.
Figure 1 : Représentation schématique des antigènes viraux reconnus par la réponse immunitaire médiée par les cellules T CD4 et CD8 chez les personnes en bonne santé. La réactivité relative indiquée est basée sur les réponses T détectées ex-vivo et in-vitro (Gandhi and Khanna, 2004).
A côté de cela, il a été démontré que les lymphocytes Tγδ, et plus particulièrement les Vδ2neg,
pouvaient être activés par les cellules infectées par le cytomégalovirus ainsi que les cellules
épithéliales intestinales tumorales (Halary et al., 2005).

17
c. Schéma récapitulatif de la réponse immune anti-CMV
Figure 2: Contrôle du HCMV par l’immunité innée et adaptative. L’infection primaire par le HCMV chez les individus en bonne santé commence typiquement dans les épithéliums des muqueuses (A), après quoi le virus se dissémine dans les cellules myéloïdes incluant les monocytes et les cellules CD34+, ou il établira sa latence (B). Une expression restreinte des gènes viraux est observée dans ces cellules infectées de manière latente ce qui limite leur reconnaissance par les cellules effectrices. La différenciation de ces monocytes infectés en macrophage ou cellule dendritique peut initier l’infection productive (C). Les particules virales ou les corps denses (voir chapitre sur le cycle viral plus loin) peuvent être présentés par les cellules dendritiques pouvant alors stimuler les cellules T spécifique de l’antigène (D). Parallèlement à cela, les DCs activées via les TLR peuvent aussi secréter un panel de cytokines activant les cellules effectrices de la réponse innée (D). Les macrophages infectés peuvent aussi activer directement les lymphocytes T spécifique d’antigène (C). Ces lymphocytes T activés (CD8, CD4 et γδ) et les cellules NK peuvent directement lyser les cellules infectées par cytolyse ou bloquer la réplication viral au travers de la sécrétion de cytokines telles que l’IFN-γ ou encore du TNF (E). Une autre réponse importante est médiée par les lymphocytes B activés qui vont contrôler les virions libres dans le milieu extracellulaire par le biais de la production d’anticorps neutralisant (F) (Crough and Khanna, 2009).
d. Vaccination
Actuellement il n’existe aucun vaccin contre le HCMV. Bien que de considérables avancées
thérapeutiques aient été faites, cela reste un véritable challenge. Dans un premier temps,
l’utilisation de virus atténué basée sur le concept de la vaccination jennérienne n’a hélas rien
donné. Par la suite, cette technique a été améliorée et réutilisée en la combinant avec des virus

18
recombinants plus immunogènes que la souche sauvage atténuée. Plus récemment, certains
groupes ont testé de nouveaux vaccins basés sur l’utilisation de protéines virales
recombinantes ou encore de vecteurs viraux tels que les poxvirus ou les adénovirus. Les
principales techniques utilisées en vaccination sont résumées dans la figure 3.
Parmi les modèles animaux actuellement utilisés le modèle cochon d’Inde offre des
avantages uniques en comparaison avec les autres modèles. Un argument clé est que le
GPCMV (CMV du cochon d’Inde) va traverser la barrière placentaire et causer des infections
in utero. C’est pourquoi ce modèle est extrêmement bien adapté pour étudier la vaccination
dont le rôle est de bloquer la transmission du virus (Schleiss, 2008). De plus le génotypage
récent du GPCMV a permis d’améliorer les approches techniques pour l’élaboration des
vaccins, notamment grâce aux recombinaisons génomiques désormais possibles et facilitées
par l’apparition d’une nouvelle technique de modification du génome viral : la technique du
BAC (Bacterial Artificial Chromosome). Nous détaillerons le principe de cette technique par
la suite.
Figure 3 : Les différentes stratégies de vaccination développées contre le HCMV. (Crough and Khanna, 2009)

19
e. Mécanismes d’échappement immunitaire du HCMV
Tout comme la plupart des Herpesviridae, le HCMV a développé de nombreux mécanismes
d’échappement aux acteurs de la réponse immunitaire innée et adaptative. Le virus va utiliser
différents moyens pour bloquer cette réponse, en diminuant l’expression des molécules de
CMH de classe I et II pour limiter la reconnaissance des antigènes viraux par les lymphocytes
T et leur activation. Une protéine homologue du CMH de classe I (UL18) codée par le virus
va limiter la reconnaissance des cellules infectées (Beck and Barrell, 1988). L’expression des
peptides (UL16 et UL40) qui se lieront aux CMH non classiques pour inhiber l’activation des
cellules NK est un autre moyen de limiter la réponse anti-virale. Toutes ces protéines virales
spécifiques sont appelées des immuno-évasines et sont décrites dans la revue de Powers
(Powers et al., 2008). Un dernier exemple est celui de l’homologue viral de la cytokine IL-10
(UL111a) visant à limiter l’inflammation et l’apoptose pour faciliter la dissémination du virus
(Kotenko et al., 2000). Après avoir démontré pour la première fois la présentation croisée
d’antigènes viraux (Arrode et al., 2002; Arrode et al., 2000) pouvant palier le blocage de
l’activation des lymphocytes T, notre équipe s’intéresse désormais aux mécanismes encore
méconnus de la perturbation de la différenciation des monocytes en cellules dendritiques,
autre mécanisme de contrôle de l’inflammation découvert récemment (Gredmark and
Soderberg-Naucler, 2003; Moutaftsi et al., 2004; Moutaftsi et al., 2002).
Très récemment, il a été découvert un système de micro-ARN viraux pouvant inhiber le
système immunitaire (Grey et al., 2005; Stern-Ginossar et al., 2007). Actuellement, 11 micro-
ARNs ont été identifiés dans le génome du HCMV par des moyens bioinformatiques.
Cependant les cibles ainsi que la fonction de la plupart de ces micro-ARN restent méconnus.
Il existe deux scénarii possibles quant à leur fonction, la régulation des gènes cellulaires afin
d’installer un environnement favorable à la réplication virale, ou encore la régulation des
gènes viraux pour optimiser la réplication virale en fonction du tissu ou de la cellule infectée.
L’hypothèse actuelle est la cohabitation de ces deux théories car il a été démontré que certains
micro-ARN viraux pouvaient diminuer l’expression de la protéine très précoce IE1, inhibant
alors l’infection aigüe pour installer une latence, alors que d’autres avaient pour cibles des
gènes cellulaires, tels que MICB (MHC class-I related chain B), responsable de la
reconnaissance suivie de la lyse des cellules infectées par les cellules NK via le récepteur
NKG2D. La diminution de cette protéine par les micro-ARN viraux aurait ici un rôle
d’échappement au système immunitaire. Voir les revues de Dolken et de Grey pour plus
d’informations (Dolken et al., 2009; Grey and Nelson, 2008).

20
Figure 4 : Récapitulatif des différents gènes viraux et cellulaires induits par l’infection, impliqués dans l’échappement à la réponse immunitaire. Chaque gène viral est indiqué avec sa localisation dans le génome viral et sa fonction associée (Gandhi and Khanna, 2004).
Malgré tout, la fonction de ces immuno-évasines n’a été démontrée qu’in vitro. En effet, ces
mécanismes ont été démontrés comme inefficaces in vivo dans des souris
immunocompétentes (Holtappels et al., 2002) et qu’il existait même des lymphocytes T CD8
spécifiques de ces protéines (Elkington et al., 2003). Tous ces mécanismes ne seraient
efficaces que chez un hôte dont le système immunitaire est affaibli, démontrant bien
l’existence d’un équilibre entre la réplication du virus et la réponse immune. Pour bien
comprendre ce phénomène, il est important de connaître la biologie du virus et comment se
déroule son cycle de réplication.
21
VI. Biologie du HCMV
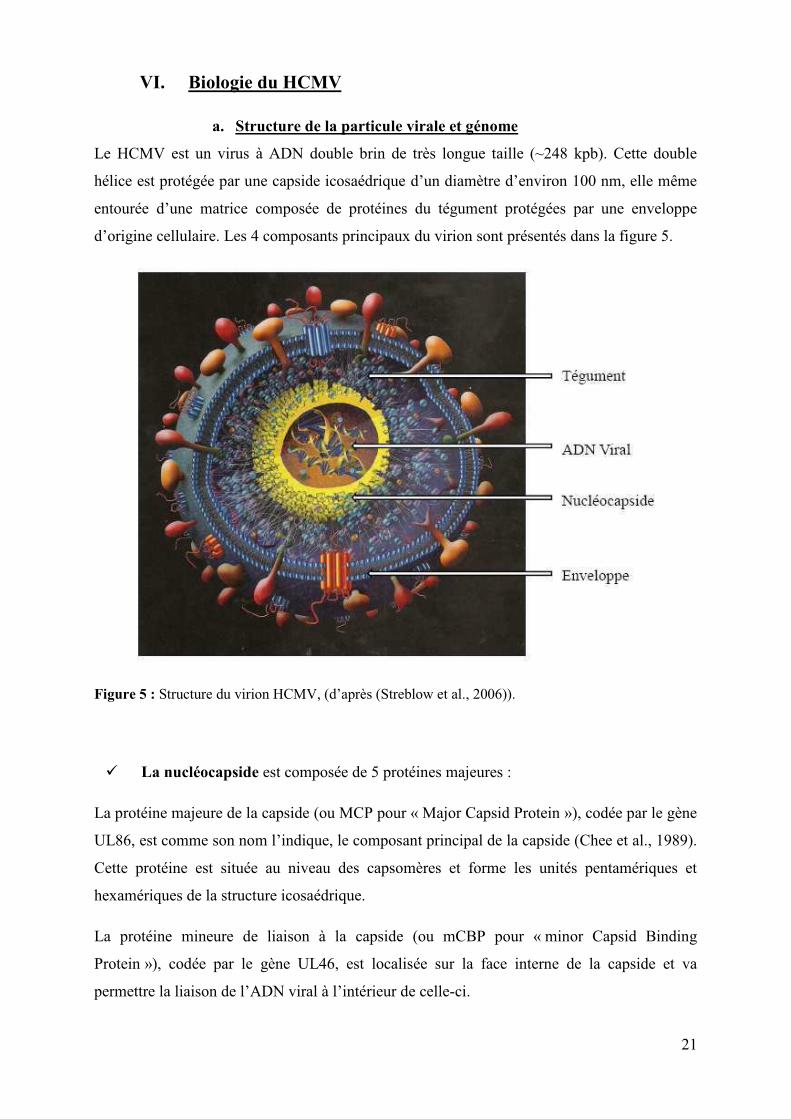
a. Structure de la particule virale et génome
Le HCMV est un virus à ADN double brin de très longue taille (~248 kpb). Cette double
hélice est protégée par une capside icosaédrique d’un diamètre d’environ 100 nm, elle même
entourée d’une matrice composée de protéines du tégument protégées par une enveloppe
d’origine cellulaire. Les 4 composants principaux du virion sont présentés dans la figure 5.
Figure 5 : Structure du virion HCMV, (d’après (Streblow et al., 2006)).
� La nucléocapside est composée de 5 protéines majeures :
La protéine majeure de la capside (ou MCP pour « Major Capsid Protein »), codée par le gène
UL86, est comme son nom l’indique, le composant principal de la capside (Chee et al., 1989).
Cette protéine est située au niveau des capsomères et forme les unités pentamériques et
hexamériques de la structure icosaédrique.
La protéine mineure de liaison à la capside (ou mCBP pour « minor Capsid Binding
Protein »), codée par le gène UL46, est localisée sur la face interne de la capside et va
permettre la liaison de l’ADN viral à l’intérieur de celle-ci.

22
Ces deux protéines vont s’organiser autour d’une charpente nécessaire à leur assemblage.
Cette « pré-capside » est composée de fragments de l’Assembly protéin (codée par le gène
UL80) et de la protéine mineure de la capside (ou mCP pour minor Capsid Protein codée par
le gène UL85) Ces protéines vont former des triplexes pour lier les pentanes et hexanes entre
eux.
Enfin il a été récemment démontré que la plus petite protéine de la capside (SCP pour
Smallest Capsid Protein, codée par le gène UL48-49) est essentielle à l’assemblage de la
particule infectieuse, notamment au travers des interactions avec les protéines du tégument
(Borst et al., 2001).
� Les composants du tégument :
La majorité des protéines du tégument sont des protéines phosphorylées. (Mocarski et al.,
2007b). La protéine pp65 (ppUL83) constitue environ 95% du tégument. Cette protéine
comporte des signaux de transfert nucléaires (Gallina et al., 1996) expliquant sa localisation
dans le noyau des cellules quelques minutes seulement après l’infection. Cependant sa
fonction reste inconnue. Parmi les phosphoprotéines de structure du tégument, pp150
(ppUL32) représente à elle seule environ 20% de la masse du virion. A coté de cela, certaines
protéines moins abondantes vont jouer un rôle important dans la réplication, telles que pp71
(ppUL82) qui est un transactivateur de l’expression des gènes viraux. La phosphoprotéine
pp28 (ppUL99) est hautement immunogène et est retrouvée à la périphérie de la capside. Bien
d’autres protéines sont retrouvées dans cette matrice, telles que pp130 (ppUL56 impliqué dans
la maturation du virion), ppUL84, ppUL47, ppUL69 etc. Cependant, bien qu’un rôle dans la
régulation de l’expression des gènes viraux ou la modification de la réponse cellulaire aient
été démontré pour certaines (comme pp28 (ppUL99) (Britt et al., 2004) (Silva et al., 2003)), la
fonction de la plupart de ces protéines reste indéterminée.
Certaines protéines cellulaires spécifiques sont emportées dans ce tégument, telles que la
cPLA-2 (Allal et al., 2004) ou encore la Caséine Kinase II (Nogalski et al., 2007), protéines
qui auront le rôle d’activer plus rapidement certaines voies de signalisation (NF-κB pour la
CK-II) et faciliter ainsi qu’accélérer la réplication virale.
En complément de ces protéines, le tégument contient de nombreux ARN cellulaires et viraux
(Bresnahan and Shenk, 2000; Greijer et al., 2000) dont la quantité incorporée est
proportionnelle à leurs quantités présentes dans la cellule (Terhune et al., 2004). Ces ARN

23
permettraient à certains gènes viraux d’être exprimés directement après l’entrée du virus dans
la cellule, et ceci en absence de transcription du génome viral.
� L’enveloppe virale :
L’enveloppe est constituée d’une bicouche lipidique d’origine cellulaire (Golgi) et possède à
sa surface une multitude de glycoprotéines virales : gB, gH, gL, gO, gM et gN (codées
respectivement par les gènes UL55, UL75, UL115, UL74, UL100 et UL73). La plupart ont
été démontrées comme étant essentielles à la production de particules infectieuses (Hobom et
al., 2000). Ces protéines vont jouer un rôle essentiel à la pénétration du virus dans la cellule
hôte, dans la transmission du virus de cellule à cellule et dans la formation de syncitia (Keay
and Baldwin, 1992; Navarro et al., 1993). Ces protéines portent de nombreux épitopes
reconnus par des anticorps neutralisant et seront les principales cibles de la vaccination.
� Le génome viral :
Le génome du HCMV est le plus grand de tous les Herpesviridae. Il est constitué de deux
régions uniques non répétées : une région longue (UL : Unique long) et une région courte
(US : Unique Short). Ces deux régions sont flanquées de séquences répétitives terminales
(TR : Terminal Repeat) appelées TRL à l’extrémité de la séquence UL et TRS à l’extrémité
de la séquence US. La jonction des 2 segments UL et US est constituée de la répétition
inversée des TRL et TRS appelées respectivement IRL et IRS. Au cours de la réplication, il
apparait 4 isoformes différentes du génome viral selon l’orientation des séquences UL et US,
illustrée dans la figure 6. On dénombre environ 200 cadres ouverts de lecture dont les gènes
sont nommés selon leur localisation au sein des différentes séquences (exemple : US1, US2
… UL1, UL2 …) (Mocarski et al., 2007a).
Après séquençage de la totalité du génome de plusieurs souches, une différence importante à
été remarquée entre les souches dite « de laboratoire » et les souches « cliniques ». En effet,
bien que montrant près de 98% de séquences identiques, les souches cliniques codent au
minimum 19 gènes supplémentaires, surement perdus dans la souche de laboratoire par de
nombreuses délétions et réarrangement lors de ses nombreux passages en culture de
laboratoire. Ces gènes sont impliqués dans la virulence et le tropisme, pouvant expliquer la
plus grande virulence ainsi que le tropisme plus large (endothéliotrope) des souches
cliniques.(Cha et al., 1996).

24
Figure 6 : Structure du génome du cytomégalovirus Humain.
b. Tropisme et entrée du virus
L’infection à CMV est spécifique d’espèce. Aucune lignée d’origine animale ne permet la
réplication du HCMV. In vivo, le HCMV peut infecter une grande variété de cellules comme
les monocytes/macrophages/dendritiques, les cellules endothéliales, épithéliales, les cellules
musculaires lisses, les fibroblastes, les cellules stromales, les neutrophiles, les hépatocytes
(Plachter et al., 1996; Sinzger and Jahn, 1996; Sinzger et al., 1996) ainsi que les cellules
progénitrices neuronales, les neurones et les astrocytes (Luo et al., 2008). Le tropisme diffère
cependant entre les souches cliniques et les souches de laboratoire. Ce tropisme cellulaire est
basé sur un « îlot génétique de tropisme » comprenant trois ORF : UL128, UL130 et
UL131A. Ces trois gènes seraient actifs dans la souche clinique mais inactifs dans les souches
de laboratoire, perdus après de nombreux passages sur fibroblastes (Jarvis and Nelson, 2007).
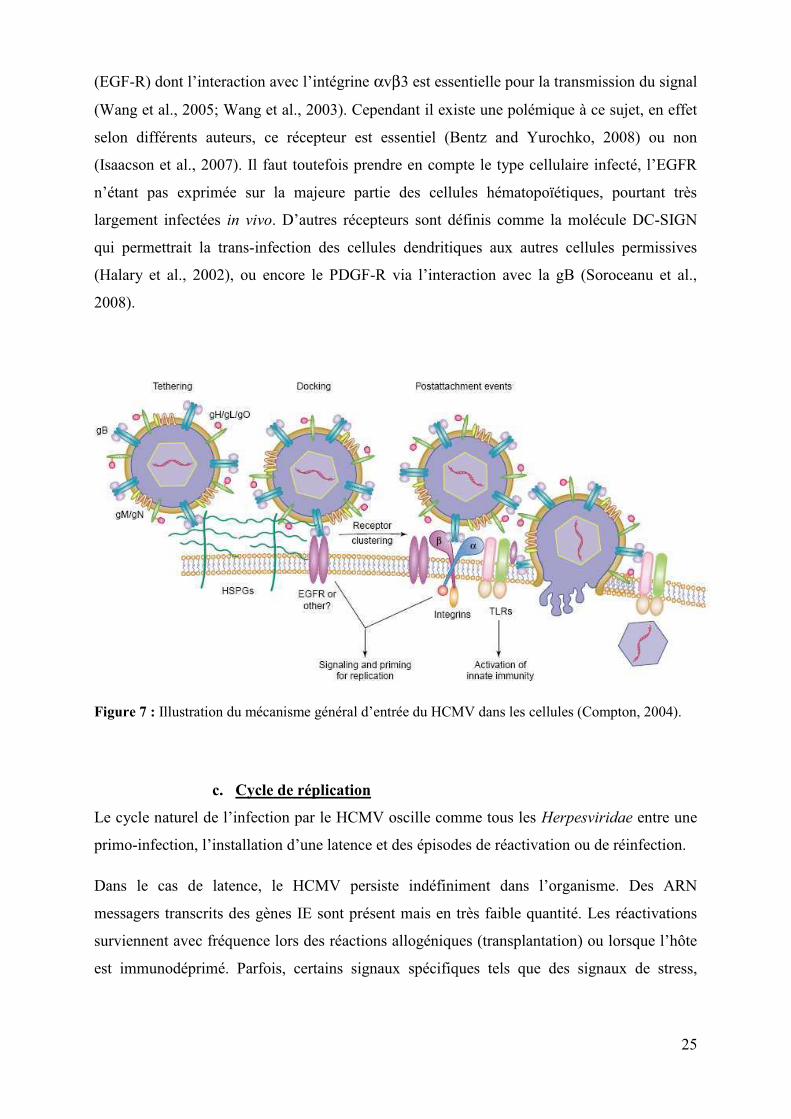
Actuellement l’attachement et l’entrée du virus est celui décrit par Teresa Compton
(Compton, 2004) et illustré dans la figure 7. L’attachement du HCMV commence par des
interactions de basse affinité entre les molécules d’Heparane Sulfates (HSPG) et les
molécules gM/gN et gB de l’enveloppe virale. Cependant, bien que strictement requises, ces
interactions ne sont pas suffisantes pour l’infection. Elles seraient suivies de l’attachement du
virus à ses récepteurs et d’un recrutement de co-récepteurs puis de la fusion (avec ou sans
endocytose selon le type cellulaire infecté) de la membrane virale et de la membrane
cellulaire et libération de la capside dans le cytoplasme. Immédiatement après l’entrée du
virus, les protéines structurales de la capside sont dégradées et l’ADN du virus entre dans le
noyau. A côté de cela, la fixation aux TLRs activerait la réponse immunitaire innée.
Il n’existe pas de récepteur spécifique au HCMV. En effet, les glycoprotéines d’enveloppe
interagissent avec tellement de protéines cellulaires qu’il est difficile de définir un récepteur
type. Le principal récepteur actuellement décrit est le récepteur à l’Epithelial Growth Factor
25
(EGF-R) dont l’interaction avec l’intégrine αvβ3 est essentielle pour la transmission du signal
(Wang et al., 2005; Wang et al., 2003). Cependant il existe une polémique à ce sujet, en effet
selon différents auteurs, ce récepteur est essentiel (Bentz and Yurochko, 2008) ou non
(Isaacson et al., 2007). Il faut toutefois prendre en compte le type cellulaire infecté, l’EGFR
n’étant pas exprimée sur la majeure partie des cellules hématopoïétiques, pourtant très
largement infectées in vivo. D’autres récepteurs sont définis comme la molécule DC-SIGN
qui permettrait la trans-infection des cellules dendritiques aux autres cellules permissives
(Halary et al., 2002), ou encore le PDGF-R via l’interaction avec la gB (Soroceanu et al.,
2008).
Figure 7 : Illustration du mécanisme général d’entrée du HCMV dans les cellules (Compton, 2004).
c. Cycle de réplication
Le cycle naturel de l’infection par le HCMV oscille comme tous les Herpesviridae entre une
primo-infection, l’installation d’une latence et des épisodes de réactivation ou de réinfection.
Dans le cas de latence, le HCMV persiste indéfiniment dans l’organisme. Des ARN
messagers transcrits des gènes IE sont présent mais en très faible quantité. Les réactivations
surviennent avec fréquence lors des réactions allogéniques (transplantation) ou lorsque l’hôte
est immunodéprimé. Parfois, certains signaux spécifiques tels que des signaux de stress,

26
d’inflammation ou simplement l’apport d’une molécule permettant l’activation de certains
facteur de transcription suffisent à réactiver le virus (Meier, 2001).
Les cellules dans lesquelles le virus va installer sa latence sont les cellules progénitrices
CD34+ et leur différenciation en monocyte puis en macrophage ou cellules dendritiques va
alors induire la réactivation du virus (Figure 8). Pour une revue complète sur la latence du
HCMV voir : (Sinclair, 2009; Sinclair and Sissons, 2006).
Figure 8 : Hypothèse actuelle du modèle de latence et de la réactivation du HCMV chez une personne saine. Bien que l’ADN viral puisse être détecté dans toutes les cellules myéloïdes durant l’infection latente, l’expression des protéines IE n’est remarquable que durant la différenciation des cellules en macrophages ou cellules dendritiques, cette expression conduisant alors à la réactivation du virus (d’après (Sinclair and Sissons, 2006)).
Dans le cas d’une infection active, le cycle du HCMV varie de 96 à 120 heures. L’infection
est initiée par la fixation du virus sur la membrane cellulaire, la fusion des membranes virales
et cellulaires ou l’endocytose du virion suivie de la fusion des membranes. Après libération de
la capside, l’ADN viral est directement transloqué au noyau. La circularisation en épisome du
génome viral se fait environ 4 heures après l’infection, ce qui indique que cette étape
nécessite l’assistance de protéines virales ou cellulaires (McVoy and Adler, 1994). La
transcription des gènes viraux s’effectue en 3 phases coordonnées : les phases IE, E et L.

27
� Une phase très précoce IE (Immediate Early)
Durant cette phase, les protéines régulant les transcriptions virales ultérieures vont être
produites. Les deux protéines très précoces principales et essentielles à l’aboutissement du
cycle viral sont les protéines IE1 et IE2, codées respectivement par les gènes UL123 et
UL122, tout deux sous contrôle d’un même promoteur majeur appelé le MIEP (Major
Immediate Early Promoter). Cette région activatrice contient de nombreux éléments de
réponses à différents facteurs de transcription que nous décrirons plus en détails par la suite,
celle-ci étant un des objets principaux de notre étude.
� Une phase précoce E (Early)
Cette phase va aboutir à la production des protéines correspondant aux enzymes de réplication
virale (ADN-polymérase virale codée par le gène UL54) ainsi que le produit du gène UL44
qui s’associera à cette dernière pour permettre sa progression sur le brin matriciel. Une autre
protéine importante sera exprimée durant cette phase, il s’agit de la protéine kinase UL97,
intervenant dans la phosphorylation du ganciclovir, drogue antivirale utilisée actuellement en
clinique.
� Une phase tardive L (Late)
C’est durant cette phase que le génome viral sera dupliqué et les protéines structurales
exprimées, telles que les protéines majeures et mineures de la capside (UL86 et UL46). Les
capsides seront alors assemblées dans le noyau et s’envelopperont dans la membrane
nucléaire en migrant vers le cytoplasme. Elles acquièrent alors les protéines du tégument dont
la synthèse en excès dans les saccules de l’appareil de Golgi forme les « corps denses », très
antigèniques comme indiqué précédemment. Une inclusion cytoplasmique va alors se former,
caractéristique du HCMV et les virions seront libérés après acquisition d’une enveloppe à
partir du reticulum endoplasmique. Un schéma récapitulatif du cycle viral est proposé dans la
figure 9.
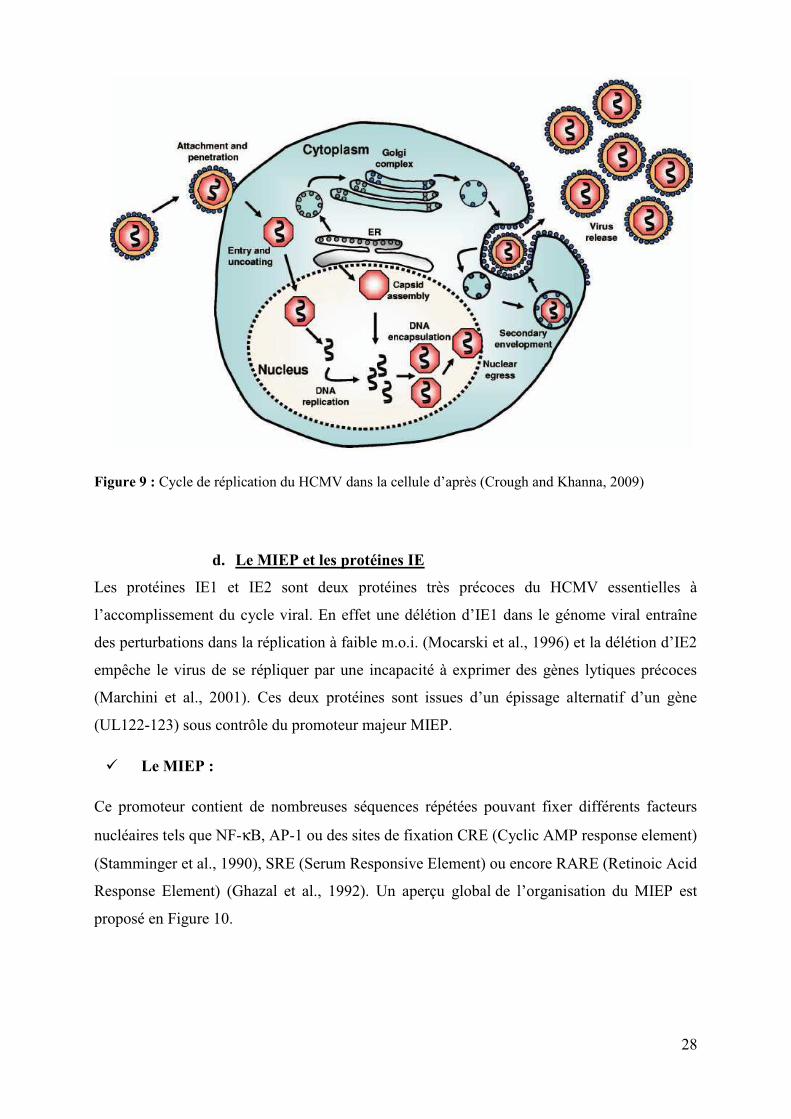
28
Figure 9 : Cycle de réplication du HCMV dans la cellule d’après (Crough and Khanna, 2009)
d. Le MIEP et les protéines IE
Les protéines IE1 et IE2 sont deux protéines très précoces du HCMV essentielles à
l’accomplissement du cycle viral. En effet une délétion d’IE1 dans le génome viral entraîne
des perturbations dans la réplication à faible m.o.i. (Mocarski et al., 1996) et la délétion d’IE2
empêche le virus de se répliquer par une incapacité à exprimer des gènes lytiques précoces
(Marchini et al., 2001). Ces deux protéines sont issues d’un épissage alternatif d’un gène
(UL122-123) sous contrôle du promoteur majeur MIEP.
� Le MIEP :
Ce promoteur contient de nombreuses séquences répétées pouvant fixer différents facteurs
nucléaires tels que NF-κB, AP-1 ou des sites de fixation CRE (Cyclic AMP response element)
(Stamminger et al., 1990), SRE (Serum Responsive Element) ou encore RARE (Retinoic Acid
Response Element) (Ghazal et al., 1992). Un aperçu global de l’organisation du MIEP est
proposé en Figure 10.

29
Figure 10: Organisation générale du MIEP, d’après (Meier and Stinski, 2006).
Le facteur de transcription considéré comme nécessaire à l’activation du MIEP est NF-κB.
Cependant, certaines études démontrent que le virus peut très bien se répliquer sans utilisation
de ce facteur de transcription, notamment dans les cellules épithéliales de la rétine (Cinatl et
al., 2001) durant les premières heures suivant l’infection. Cela dit, d’autres groupes ont
démontré que dans ce type cellulaire, l’activation de NF-κB était nécessaire dans des temps
plus tardifs (Hooks et al., 2006).
D’autres éléments de réponse à différents facteurs de transcription ont été mis en évidence
comme cela à été précisé précédemment, certaines séquences étant plus importantes que
d’autres dans le splicing IE1/IE2 (Meier et al., 2002; Netterwald et al., 2005). La plupart de
ces éléments de réponse ayant été localisés dans la partie distale du MIEP, il a été démontré
que ces séquences jouaient un rôle uniquement à faible m.o.i. (multiplicity of infection) et que
le virus arrivait à palier leur absence à forte m.o.i. dans le cas de délétion complète de cette
partie du génome viral (Keller et al., 2003; Meier and Pruessner, 2000; Meier and Stinski,
1997).
Il a été démontré récemment que les sous-unités de NF-κB utilisées par le virus étaient
différentes selon le type cellulaire et la phase de l’infection (précoce ou tardive). Ainsi le
virus utilisera les sous-unités p50/p65 dans les fibroblastes ou durant les phases précoces de
l’infection dans les macrophages, alors qu’il utilisera les sous-unités p100/Bcl-3 dans la phase
tardive de l’infection de ces mêmes cellules (Khan et al., 2009). Tout ceci montre bien la
complexité de la régulation du MIEP et des acteurs régulant son activation au cours du cycle
viral.
A côté de cela, l’interaction du MIEP avec des protéines de structure de l’ADN cellulaire
telles que les nucléosomes va jouer un rôle important dans la transcription des gènes viraux.
Nous allons assister à une dynamique d’assemblage et de désassemblage des nucléosomes
avec l’ADN viral (Nitzsche et al., 2008) ainsi qu’une véritable régulation de l’acétylation des

30
histones présent sur ce dernier, notamment via l’interaction directe entre les protéines IE et les
histones déacétylase HDAC-1, -2 et -3, paramètre essentiel au contrôle de la transcription des
gènes viraux dans les cycles lytiques ainsi que la latence (Murphy et al., 2002; Reeves et al.,
2005; Woodhall et al., 2006). Tous ces paramètres vont donc réguler l’expression des
protéines IE et pourront être rétro-régulés eux-mêmes par ces dernières.
� Les protéines IE :
Les protéines IE sont issues d’un épissage alternatif. Bien qu’IE1 et IE2 soient les deux
protéines très précoces principales il existe d’autres formes alternatives selon les exons
sélectionnés pouvant jouer un rôle de co-activateurs (Shirakata et al., 2002) mais dont la
fonction principale reste méconnue :
Figure 11 : Les différents produits de l’épissage alternatif du transcrit primaire IE, d’après (Meier and Stinski, 2006).
Ces protéines sont localisées dans le noyau de la cellule à proximité des domaines cellulaires
ND10 et leur fonction principale est de réguler la transcription des gènes viraux précoces et
tardifs, soit en agissant directement comme facteur de transcription soit en interagissant avec
certaines protéines cellulaires impliquées dans la répression ou l’activation des gènes viraux
mais aussi cellulaires.

31
En effet, la protéine IE1 va stimuler de nombreux promoteurs viraux et cellulaires via son
interaction physique avec de nombreuses protéines cellulaires telles que CTF-1, SP1, E2F1 à
5, TAFII130/TAF4, p107, HDAC-2, PML et Daxx (Meier and Stinski, 2006; Nevels et al.,
2004), mais aussi via une activité kinase inhérente sur certains facteurs de transcription
comme Rb, E2F et c-jun (Pajovic et al., 1997). Cette protéine peut également induire
l’expression des sous-unités de NF-κB et disloquer les domaines ND10 cellulaires pour
empêcher leur rôle répressif sur la réplication virale (Muller and Dejean, 1999; Woodhall et
al., 2006). Cette dernière fonction est en particulier permise par la SUMOylation d’IE1, c'est-
à-dire la liaison covalente avec la protéine SUMO-1 (Small Ubiquitin-like Modifier). Il a été
en effet démontré que les protéines IE pouvaient être modifiées de cette manière afin
d’acquérir de nouvelles fonctions (Sadanari et al., 2005). La protéine IE2 SUMOylée va quant
à elle, jouer un rôle essentiel dans la réplication virale (Berndt et al., 2009; Hofmann et al.,
2000; Lee et al., 2003).
Le rôle fondamental de la protéine IE2 a pu être démontré aux moyens de virus dont la
majorité de la séquence du gène UL122 (codant la protéine IE2) a été délétée (Marchini et al.,
2001). Cette construction a été possible grâce à la technique des BAC mise au point par
Martin Messerle (Messerle et al., 1997), consistant à cloner le génome viral entièrement dans
un chromosome bactérien artificiel, insérer ce chromosome dans E.coli permettant alors
d’effectuer de la mutagénèse sur le génome viral. Ce chromosome est alors produit en bactérie
puis extrait pour être ensuite transfecté dans des fibroblastes où son expression et sa
réplication produira de nouveaux virions dont le génome contient les mutations désirées. Le
principe de la technique est résumé dans la figure 12.
IE2 va donc jouer un rôle trans-activateur de promoteurs cellulaires et viraux de la même
manière qu’IE1 (Meier and Stinski, 2006) mais joue également un rôle trans-répresseur sur
son propre promoteur via sa fixation sur la séquence CRS (cis-repression signal) localisée
entre la TATA box et le site de départ du MIEP (Pizzorno and Hayward, 1990).

32
Figure 12 : Principe de la technique BAC. (A) Le clonage du génome HCMV dans le BAC. Le réplicon BAC (en bleu) flanqué de séquences virales est transfecté dans des fibroblastes infectés par la suite par le HCMV. Le génome viral qui a été incorporé dans le BAC par recombinaison homologue est isolé puis transféré dans E.coli. (B) Le principe de la génération de HCMV mutant par la technique BAC. Le génome viral maintenu dans le BAC dans E.coli est utilisé comme substrat pour introduire la mutation désirée (M en rouge). Le BAC contenant le génome viral mutant est alors purifié et transfecté dans des fibroblastes afin de reconstituer des virus mutants. D’après (Brune et al., 2006)
� Régulation des transcripts IE2 par l’activité enzymatique COX-2 :
Les protéines IE1 et IE2 étant essentielles à l’aboutissement du cycle viral, de nombreuses
études se sont portées sur la régulation de leur expression. Comme il a été précisé auparavant,
de nombreux facteurs de transcription jouent un rôle dans cette régulation mais de manière
intéressante, il a été démontré que le virus pouvait tirer profit de l’inflammation, notamment
via l’utilisation de l’activité enzymatique de la cyclooxygénase-2 (COX-2). En effet,
l’expression d’IE2 ainsi que la production de nouveau virions sont très fortement diminuées
lorsque cette enzyme est inhibée (Zhu et al., 2002). Cependant les mécanismes sous-jacents
n’ont jamais été décrits et l’hypothèse d’un rôle de l’AMPc en réponse à la stimulation par les
prostaglandines PGE2, un des produits éventuels de cette activité COX-2, a été émise
(Mocarski, 2002). (Figure 13)

33
Figure 13 : Schéma proposé pour l’implication de COX-2 et de l’AMPc dans la régulation de l’expression d’IE2 et la réplication virale (Mocarski, 2002).
Parmi les différents produits de cette cascade enzymatique se trouve la prostaglandine 15d-
PGJ2, ligand naturel du récepteur nucléaire PPARγ (Peroxisome Proliferator Activated
Receptor γ).
Nous avons émis l’hypothèse que suite à l’activation de la cascade enzymatique COX-2
induite par l’infection, une H- ou L-PGD synthase permettrait la production de la
prostaglandine 15d-PGJ2 à partir de la PGH2, servant alors à l’activation du récepteur
nucléaire PPARγ. Ce récepteur nucléaire étant un facteur de transcription, il pourrait alors
interagir directement avec l’ADN viral ou induire l’expression de certains gènes cellulaires
nécessaires à la transcription des gènes IE. Dans tous les cas, notre hypothèse a été que
l’activation de ce facteur de transcription aurait des conséquences sur la régulation de
l’expression des gènes très précoces IE du HCMV et plus particulièrement sur l’expression de
la protéine IE2.
C’est pourquoi nous nous sommes intéressés au rôle de ce facteur de transcription dans
l’activation du MIEP et la réplication virale.
Notre hypothèse est présentée dans la figure 14, les différents rôles de PPARγ, ainsi que les
protéines interagissant avec ce facteur de transcription et intervenant dans sa fixation à l’ADN
ainsi que dans la régulation de l’expression des gènes seront présentées en détails dans le
chapitre suivant.

34
Figure 14 : Cascade enzymatique conduisant à la synthèse de 15d-PGJ2 dépendante de COX-2, permettant l’activation du facteur de transcription PPARγ. Une fois PPARγ activé par son ligand, une hétérodimérisation avec le récepteur à l’acide rétinoïque RXR, lui-même couplé à son ligand, va permettre la fixation de cet hétérodimère à l’ADN sur des séquences précises appelées PPRE, et le recrutement des complexes co-activateurs induisant la transcription des gènes, d’après (Scher and Pillinger, 2005).
VII. Peroxisome Proliferator Activated Receptor γγγγ (PPARγ)γ)γ)γ)
e. Généralités sur les PPARs
Les PPARs sont des facteurs de transcription appartenant à la super famille des récepteurs
nucléaires aux hormones (famille des récepteurs d’hormones stéroïdes). Ils vont transformer
une grande variété de signaux de l’environnement, nutritionnels et inflammatoires en réponses
cellulaires.
Il existe 3 isoformes de PPARs qui sont les produits de trois gènes différents, couramment
désignés sous les noms de PPARα, PPARγ et PPARδ. Ces protéines présentent une
organisation structurale commune à tous les autres récepteurs nucléaires. Les séquences des
domaines de liaison à l’ADN sont très conservées entre les trois sous-types, tandis que celles
des sites de liaison des ligans le sont moins, signifiant des caractéristiques pharmacologiques
différentes pour chaque sous-type. En effet, chaque isoforme présente des distributions
tissulaires différentes et des spécialités fonctionnelles malgré leurs fortes homologies de
séquence.

35
Ces facteurs de transcription sont donc activés par des ligands spécifiques. Différents types
d’acides gras dont les acides gras insaturés peuvent se fixer et activer les PPARs avec une
certaine spécificité. De plus, les éicosanoïdes produits à partir du métabolisme de l’acide
arachidonique sont aussi des ligands efficaces pour activer spécifiquement les différents
isoformes de PPARs. Ceci inclut les leucotriènes (LTs) produits par la cascade enzymatique
impliquant la 15-lypoxygénase, mais aussi les acides hydroxyeicosatétraénoïques (5-HETE,
12-HETE et 15-HETE) produits respectivement par les 5-, 12- et 15-lipoxygénases. Les
prostaglandines produites par les COX sont aussi impliquées comme précisé précédemment.
La 15d-PGJ2, métabolite provenant de la PGD2 est un puissant ligand de PPARγ in vitro mais
à forte concentration uniquement, tout comme les prostaglandines A2 et D2 pouvant elles
aussi activer ce facteur de transcription dans les mêmes conditions (Vamecq and Latruffe,
1999). Des métabolites oxydés de l’acide linoléique (les acides 9 et 13 hydroxy
octadécanoïques 9- et 13-HODE), dérivés des LDL oxydés sont aussi des ligands de ce
récepteur nucléaire (Nagy et al., 1998; Tontonoz et al., 1998).
A coté de cela, il existe des ligands synthétiques activateurs de PPARγ. Parmi eux se trouvent
des molécules utilisées dans le traitement du diabète de la famille des thiazolidinediones
(troglitazone, rosiglitazone, ciglitazone …) (Lehmann et al., 1995). C’est également le cas des
anti-inflammatoires non-stéroïdiens tels que l’indométacine ou encore l’ibuprofène.
f. Structure de PPARγγγγ
Chez l’homme, il existe 3 ARNs messagers (ARNm) de PPARγ (γ1, γ2 et γ3) (Fajas et al.,
1998; Rocchi and Auwerx, 1999). Les ARN messagers de γ1 et γ2 sont traduits en la même
protéine, à savoir PPARγ1. PPARγ2 contient quant à lui 30 acides aminés de plus à
l’extrémité NH2 terminale. La forme PPARγ1 est prédominante et PPARγ2 sera surtout
exprimée dans les adipocytes (Tontonoz et al., 1998). Les principaux domaines fonctionnels
des PPARs sont communs avec les autres membres de la famille des récepteurs stéroïdiens.
Le domaine C est le domaine de liaison à l’ADN (Chandra et al., 2008). Le domaine E/F
carboy-termianl est le domaine de liaison du ligand (Ligand Binding Domain LBD). Enfin, le
domaine A/B est la région NH2 terminale. La liaison de PPARγ à son ligand est régulé par une
communication intramoléculaire entre le domaine A/B et le LBD (Shao et al., 1998; Vamecq
and Latruffe, 1999).

36
Récemment, il a été développé un inhibiteur spécifique non réversible de PPARγ dont le
mode d’action consiste à modifier une cystéine au niveau du site de fixation du ligand
(Leesnitzer et al., 2002).
g. Rôle transcriptionel
Comme tous les PPARs, PPARγ est activé par la liaison à son ligand. Une fois activé, les
PPARs sont capables de réguler l’expression de gènes cibles possédant un élément de réponse
au PPAR (PPRE pour PPAR response element) dans leur promoteur. Ces séquences PPRE
sont constituées de deux séquences hexanucléotidiques de même sens (AGGTCA) séparées
par un nucléotide aléatoire. Ces séquences sont alors appelées DR1, mais il peut également
exister des séquences allant de DR2 à DR6, dans lesquelles les deux séquences
héxanucléotidiques sont séparées par plusieurs nucléotides aléatoires (de 2 à 6 selon la
séquence PPRE). Cependant ces séquences peuvent varier légèrement et la séquence en 5’
joue un rôle quant à l’intensité de la transcription (Palmer et al., 1995).
Pour la liaison à l’ADN au niveau de ces séquences particulières, une hétérodimérisation avec
le récepteur nucléaire RXR (Retinoic X receptor) couplé lui-même à son propre ligand,
l’acide rétinoïque, est nécessaire (Rocchi and Auwerx, 1999; Vamecq and Latruffe, 1999).
Ainsi ce sont les hétérodimères PPARγ/RXR qui se lient aux séquences PPRE présentes dans
les régions régulatrices des promoteurs des gènes cibles (Figure 15).
Figure 15 : Schéma de l’activation transcriptionnelle de PPARγ, ici activée par la 15d-PGJ2 ( ), le libérant au préalable de son hypothétique co-répresseur ( ). D’après (Scher and Pillinger, 2005).

37
Va s’en suivre un recrutement de protéines co-activatrices pour permettre la transcription des
gènes. Ces protéines comprennent celles modifiant la structure de la chromatine par leur
activité histone acétylase (SRC et CBP/p300), celles du complexe DRIP/TRAP interagissant
avec la machinerie de transcription et enfin celles dont les fonctions sont particulières ou
encore mal définies, comme la protéine PGC-1 (PPAR Gamma Co-factor 1) intervenant dans
l’épissage et « l’exon-skiping » (Kornblihtt, 2005). En effet, l’interaction de PPARγ et de
PGC-1 permettrait d’empêcher ou d’exclure un exon situé entre deux autres lors de l’épissage
de l’ARN. Ce mécanisme interviendrait notamment grâce à l’interaction de la protéine PGC-1
avec la polymérase II ou d’autres protéines du complexe de pré-initiation de la transcription
telles que le facteur d’épissage SRp40.
La fonction transcriptionnelle associée à PPARγ peut alors être limitée à plusieurs facteurs qui
sont :
� la disponibilité de la protéine RXR dans le noyau et qui ne doit pas être déjà associée
en hétérodimère avec une autre protéine comme le récepteur aux œstrogènes par
exemple.
� La disponibilité des cofacteurs de transcription. En effet, ces protéines sont également
impliquées dans l’activité d’autres facteurs de transcription comme NF-κB, AP-1,
STAT1, et un phénomène de compétition peut conduire à la limitation du pool
disponible (Li et al., 2000b; Ricote et al., 1999).
� La phosphorylation de PPARγ peut également inhiber son activité transcriptionnelle
via les MAP kinases (Adams et al., 1997). Cette phosphorylation sur un résidu sérine
112 de la région NH2 terminale va gêner la communication entre les domaines A/Bet
LBD conduisant à une perte d’affinité pour la liaison du ligand et une diminution de
son activité (Shao et al., 1998).
h. Rôle de trans-répresseur
Parallèlement à son rôle trans-activateur, il a été démontré que PPARγ pouvait inhiber
l’expression de gène sous contrôle de certains promoteurs tels que NF-κB, AP1 ou encore
STAT (Welch et al., 2003). Si dans un premier temps l’hypothèse de la compétition au niveau
du pool de cofacteurs comme expliqué précédemment a été émise, un nouveau mécanisme de
trans-répression à été découvert, impliquant la SUMOylation de PPARγ (Pascual et al., 2005).
Ce mécanisme ferait intervenir la liaison covalente de la protéine SUMO-1 à PPARγ sous
l’effet de l’activité enzymatique de PIAS-1. Ce PPARγ SUMOylé interagit directement avec

38
les protéines co-répressives nCOR et HDAC-3 présentes sur les promoteurs cibles, empêchant
leur ubiquitinylation et ainsi leur dégradation par le protéasome. Le complexe co-répresseur
ne pouvant être décroché du promoteur, le complexe co-activateur ne pourra pas venir se fixer
et permettre la transcription du gène (Figure 16). Ce mécanisme a surtout été décrit pour
l’inhibition par PPARγ des gènes pro-inflammatoires.
Figure 16 : Trans-répression par PPARγ. Dans les cellules non stimulées, les promoteurs pro-inflammatoires sont dans un état de répression. Des signaux inflammatoires vont conduire à un état activé avec fixation d’un complexe co-activateur et un recrutement de l’ADN polymerase II, excepté dans le cas d’une co-stimulation avec des ligands de PPARγ où une trans-répression du promoteur aura lieu du fait de la non-dégradation du complexe co-répresseur. D’après (Bailey and Ghosh, 2005).
i. Les différents rôles de PPARγγγγ
Par le biais de ces deux mécanismes bien distincts, PPARγ va jouer de nombreux rôles
physiologiques, agissant comme facteur de transcription dans de nombreuses voies
métaboliques. Dans le cas de l’obésité, son activation va induire l’accumulation de tissus
adipeux dans l’organisme. Certains gènes cibles de PPARγ, comme la lipoprotéine lipase, la
protéine de transport des acides gras et le récepteur au LDL oxydé, vont conduire à
l’accumulation de lipides dans les adipocytes et les autres tissus (Chui et al., 2005; Frohnert et
al., 1999; Schoonjans et al., 1996). Parallèlement à cela, cette activation des adipocytes par
PPARγ va diminuer la résistance à la réponse à l’insuline. Tous ces mécanismes vont
permettre de traiter efficacement les diabètes de type II chez de nombreux patients (Rangwala
and Lazar, 2004).
PPARγ va également jouer un rôle dans le métabolisme des macrophages et sera induit durant
leur différenciation (Ricote et al., 1998). Dans un premier temps, il a été désigné comme
responsable de la capture des lipides ainsi que leur accumulation dans les macrophages
devenant ainsi des cellules spumeuses (foam-cells) conduisant à l’athérosclérose (Nagy et al.,
39
1998). Un peu plus récemment, l’activation de PPARγ par la troglitazone a été démontré
comme ayant un rôle vasoprotecteur et réducteur de l’athérosclérose chez la souris (Li et al.,
2000a) puis chez l’homme (Dormandy et al., 2005; Minamikawa et al., 1998). L’hypothèse
proposée pour le paradoxe de l’accumulation de lipides dans les macrophages sans augmenter
le risque d’athérosclérose serait l’activation de la sécrétion de certains lipides comme
l’excrétion de cholestérol, prévenant ainsi de l’accumulation des lipides impliqués dans
l’athérosclérose (Chawla et al., 2001; Chinetti et al., 2001). Cependant d’autres mécanismes
comme l’inhibition d’AP1 bloquant ainsi la réponse à la thrombine et l’augmentation de la
protéine ET-1 dans les cellules endothéliales vont plutôt pencher en faveur d’une
athérosclérose (Delerive et al., 1999).
Le rôle de PPARγ dans le cancer est encore controversé. En effet, il a été décrit comme un
suppresseur de tumeur mais également comme un oncogène selon le type cellulaire dans
lequel il est activé. Ses capacités anti-prolifératives, anti-migratoires ainsi que promouvant la
différenciation l’ont désigné comme une cible anticancéreuse (Mansure et al., 2009; Ondrey,
2009). Cependant, selon le type cellulaire et les mutations provoquant le cancer, l’activation
de PPARγ peut alors être bénéfique pour la tumeur (Lefebvre et al., 1998; Saez et al., 1998).
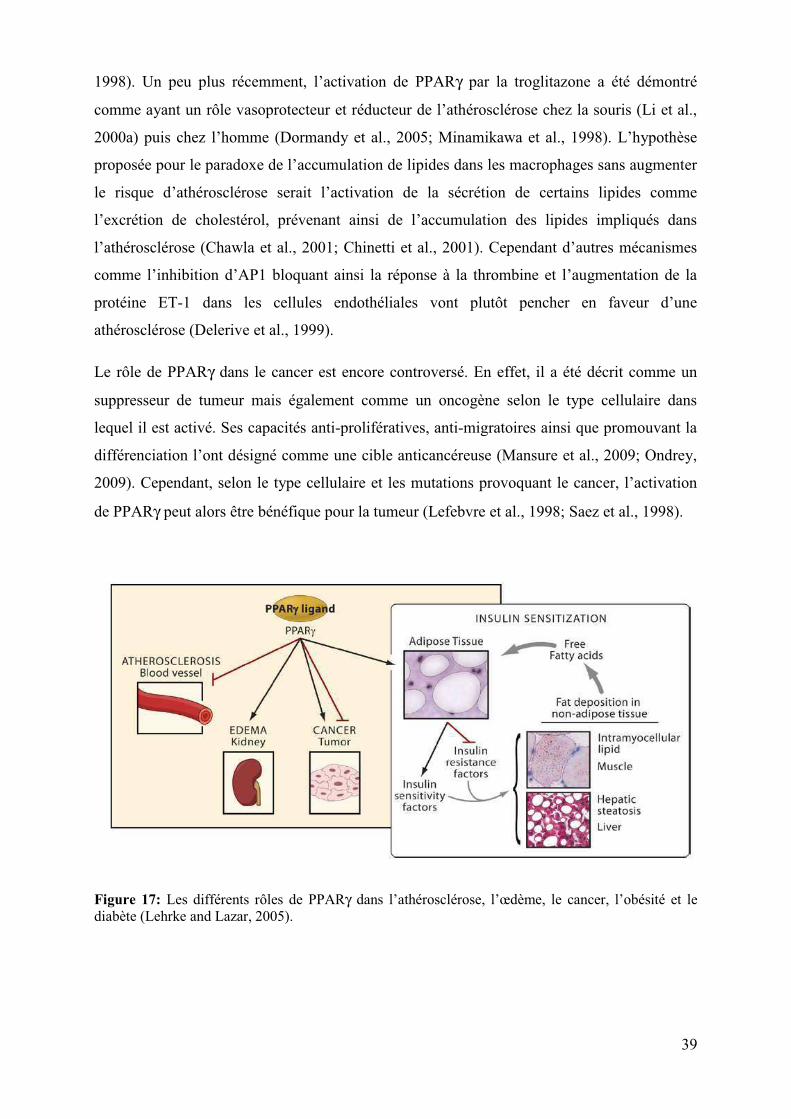
Figure 17: Les différents rôles de PPARγ dans l’athérosclérose, l’œdème, le cancer, l’obésité et le diabète (Lehrke and Lazar, 2005).

40
PPARγ joue également un rôle anti-inflammatoire important en inhibant l’expression de
nombreuses protéines inflammatoires telles que iNOS, TNFα et la MMP9 dans les
macrophages par exemple (Li et al., 2000b). Son activation va également perturber la
différenciation des cellules dendritiques en inhibant leur maturation, en diminuant
l’expression de protéines de co-stimulation, provoquant une perte d’activation des
lymphocytes T (Szatmari et al., 2006). Son action anti-inflammatoire va également s’exprimer
dans les cellules T (plus particulièrement un rôle de PPARα), mais aussi dans les cellules
endothéliales et épithéliales en diminuant l’expression de cytokines, chimiokines et de
molécules d’adhésion (Straus and Glass, 2007). Il a même été démontré que la protéine Nef
du SIV et du VIH activait PPARγ pour perturber l’hématopoïèse (Prost et al., 2008). Plus
récemment, un rôle de PPARγ dans l’augmentation de la sécrétion de l’IL-10, cytokine anti-
inflammatoire, a été décrit (Are et al., 2008; Ramakers et al., 2007).
Figure 18: Rôle anti-inflammatoire des PPAR (Straus and Glass, 2007).
PPARγ a également été décrit comme un facteur de transcription essentiel à la placentation
(mise en place du placenta, organe essentiel à la grossesse), chez la souris, et chez l’Homme
notamment en régulant l’invasion et la migration trophoblastique et la formation du
syncytiotrophoblaste. Ce rôle étant un élément majeur de notre étude, il sera présenté plus en
détails par la suite car il est essentiel de décrire la structure du placenta et sa mise en place
pour bien comprendre la fonction de PPARγ.

41
DEUXIEME PARTIE
LE PLACENTA HUMAIN

42
I. Structure du placenta
Le placenta se présente comme un organe discoïde comprenant 2 faces: la plaque choriale
relié au fœtus par le cordon ombilical et la plaque basale implantée dans l’endomètre.
Les villosités chorioniques sont formées d’un axe mésenchymateux comprenant des vaisseaux
sanguins fœtaux, des fibroblastes, des myofibroblastes ainsi que des macrophages ou cellules
de Hofbauer présentes dès la troisième semaine de grossesse et dont les fonctions sont la
phagocytose, la capture des anticorps maternels traversant les villosités et la sécrétion de
cytokines. La composition du stroma varie cependant en fonction de l’âge gestationnel et du
degré de développement de l’arbre villeux. Il est entouré d’une membrane basale recouverte
de cellules progénitrices : les cytotrophoblastes. Leur différenciation impliquant un processus
de fusion cellulaire va conduire à la formation du syncytiotrophoblaste, un tissu multinucléé
couvrant la surface des villosités flottantes (floating villi) dont le rôle est d’être une zone
d’échanges en gaz et nutriments entre le sang maternel et la circulation sanguine fœtale. Le
syncytiotrophoblaste constitue également le tissu endocrine du placenta humain par sa
capacité à synthétiser et déverser dans le compartiment maternel de grandes quantités
d’hormones comme la progestérone et la β-HCG.
Au centre des villosités chorioniques se situent donc les capillaires fœtaux, mis en place vers
la 9eme semaine de grossesse. Ils se composent de cellules endothéliales reposant sur une fine
lame basale qui les sépare du stroma.
CPG
Villosité chorionique
Décidua
ASM
Villositéd’ancrage
Stroma
CTV
SCTV
CTEV
Figure 19 : Représentation schématique de la structure placentaire. CTV (cytotrophoblaste villeux) SCTV (Syncytiotrophoblaste villeux) CTEV (cytotrophoblaste extravilleux CPG (cellule placentaire géante) ASM (artère spiralée maternelle). D’après (Alsat et al., 1997).

43
Les villosités ancrées ou crampons (anchoring villi) permettent l’implantation du placenta à
l’endomètre et la mise en place de la vascularisation utéro placentaire. Elles sont constituées à
la base des villosités choriales de colonnes de cellules cytotrophoblastiques dîtes
extravilleuses qui envahissent la décidue et le tiers supérieur du myomètre utérin et colonisent
spécifiquement les artères spiralées utérines. Les CTEV participent ainsi à leur remodelage en
vaisseaux atones de large diamètre facilitant l’arrivée du sang maternel dans la chambre
intervilleuse où flottent les villosités choriales. Ce processus physiologique d’invasion et de
remodelage des artères utérines par les CTEV est indispensable au développement placentaire
et à la croissance fœtale. Ce phénomène est possible grâce aux capacités invasives et
migratoires du cytotrophoblaste extravilleux. Il a lieu essentiellement au premier trimestre de
la grossesse et est dûment contrôlé dans le temps et l’espace mettant en jeu un dialogue étroit
entre trophoblaste et cellules d’origine maternelle. Revue de la structure placentaire (Burton
and Watson, 1997). (Figure 19)
Figure 20 : Organisation de l’interface foeto-maternelle au milieu de grossesse. Zone I : le syncytiotrophoblaste recouvre la surface des villosités flottantes contrôlant les échanges gazeux et nutritionnels ainsi que l’apport d’IgG du sang maternel vers la circulation sanguine fœtale. Zone II :Les villosités ancrées « connectent » le fœtus à la mère. Elles sont formées par des colonnes de cellules cytotrophoblastiques dîtes extravilleuses qui prolifèrent puis envahissent la paroi utérine. A l’extrémité de ces colonnes se trouvent les cellules cytotrophoblastiques extravilleuses invasives pénétrant la décidua. Zone III : Les artères spiralées maternelles sont ouvertes et élargies par l’invasion du cytotrophoblaste pour augmenter le flux sanguin vers le placenta. De 1 à 4: les différents sites possibles d’infections par le HCMV (Tabata et al., 2007).

44
L’interface foeto-maternelle formée par le placenta peut alors se décomposer en trois zones
bien distinctes présentées en figure 20.
Le placenta est un organe évolutif durant les premiers jours de grossesse pour organiser sa
structure et se développer lors d’étapes précises, bien connues aujourd’hui.
II. Mise en place du placenta au cours de la grossesse
Dans les premiers temps de la grossesse, le blastocyte va pénétrer dans le stroma utérin
maternel et déclencher le développement du placenta. Une première étape de nidation du
blastocyte est essentielle à ce processus. Ce blastocyte est composé d’un épithélium formé par
les cellules du trophectoderme, dont un des pôles contient la masse cellulaire précurseur de
l’embryon. Le blastocyte va s’insérer entre les cellules épithéliales de la muqueuse utérine
vers le 6eme jour après l’ovulation. Peu de temps après, le trophoblaste va se différencier en
deux couches : à l’extérieur en syncytiotrophoblaste multinuclée (syncytium primitif) et à
l’intérieur en cytotrophoblaste primitif mononuclée. Ces cellules syncytiales vont alors
s’infiltrer entre les cellules épithéliales et atteindre le stroma utérin maternel. Apparaissent
ensuite dans le syncytium, des vacuoles (ou lacunes), qui grandissent et finissent par
communiquer entre elles. Une mise en place de la circulation sanguine entre l’utérus et le
placenta s’établit alors lorsque les capillaires veineux maternels se retrouvent entourés par le
syncytium. Le sang maternel sera ainsi amené vers le système vacuolaire. Ce système
vacuolaire deviendra l’espace intervilleux du placenta.
Une fois cette étape de nidification accomplie, le trophoblaste va pouvoir se différencier en
cytotrophoblaste villeux (CTV) d’une part et extravilleux (CTEV) d’autre part. Le CTV
entourant les villosités chorioniques du placenta est impliqué dans le transport d’oxygène et
des nutriments de la mère vers le fœtus. Parallèlement à cela, le CTEV va envahir la
muqueuse utérine jusqu’au myomètre permettant ainsi un bon ancrage du placenta et une
relation directe avec plusieurs types cellulaires maternels.
La croissance et la ramification des villosités seront assurées par l’émergence du CTV dans le
syncytium primitif. Ces cellules jouent alors un rôle important dans les échanges mère-enfant
car leur différenciation en syncytiotrophoblaste va permettre de développer les villosités
chorioniques flottantes baignant dans le sang maternel et ainsi mettre en place les échanges
gazeux et nutritionnels.

45
A la base de ces villosités flottantes, les cellules du CTEV forment des structures solides
appelées colonnes. Ces structures vont se fixer à la décidua maternelle et seront désignées
comme villosités d’ancrage. Leur fusion avec les colonnes voisines latérales va former une
plaque englobant le sac embryonnaire. Ces cellules du CTEV vont aussi envahir les parois des
artères maternelles spiralées afin de les agrandir et d’augmenter le flux sanguin, détournant
ainsi la circulation sanguine maternelle au profit du développement placentaire via
l’augmentation des échanges mère-enfant.
Les différentes étapes de la placentation sont résumées dans la figure 21 et la revue suivante
(Burton and Watson, 1997).
Figure 21 : Les différentes étapes de la placentation humaine (Alsat et al., 1997).
Toutes ces étapes sont régulées par l’expression et la régulation de nombreux gènes dans les
cellules placentaires. Le récepteur nucléaire PPARγ, dont les différentes implications dans la
grossesse sont développées par la suite, est un des facteurs de transcription essentiels au
développement du placenta.

46
III. PPARγγγγ dans la grossesse
Au cours de la grossesse, PPARγ va se révéler être un facteur de transcription essentiel à la
placentation ainsi qu’au bon fonctionnement du placenta. Dans cet organe, il est
principalement exprimé dans le cytotrophoblaste et va notamment jouer un rôle important de
régulateur de différents mécanismes comme le développement placentaire, l’invasion, la
différenciation, le métabolisme des acides gras ainsi que la parturition (l’accouchement).
Figure 22.
Figure 22 : La régulation transcriptionnelle de gènes cibles par l’hétérodimère PPARγ-RXR résulte en l’influence cruciale de différents aspects du développement placentaire et de sa fonction. Les ligands de PPARγ ( ) et de RXR ( ) peuvent provenir du sang fœtal, maternel, du liquide amniotique et du placenta (Schaiff et al., 2006).
� PPARγγγγ et le développement placentaire
De nombreuses études ont été effectuées dans des modèles animaux où l’absence de PPARγ
peut être étudiée. Les résultats montrent que son absence provoque une malformation de la
zone labyrinthique avec une imperméabilisation des vaisseaux fœtaux ainsi qu’une rupture
des sinus sanguins maternels. De manière intéressante, il a été observé que l’absence de
PPARγ uniquement dans le placenta provoquait des amincissements sévères du myocarde
chez l’embryon et une perturbation du métabolisme lipidique, démontrant ainsi son rôle
essentiel dans le développement placentaire et métabolique (Barak et al., 1999).

47
De récentes études sur les gènes cibles impliqués dans ce mécanisme ont mis en évidence le
gène placentaire Muc1 comme cible de PPARγ (Shalom-Barak et al., 2004). Ce gène Muc1
code la protéine Mucine 1, protéine transmembranaire impliquée dans la défense de l’hôte
contre les pathogènes mais aussi dans la signalisation cellulaire. Curieusement, ce gène est
régulé par ce facteur de transcription dans le placenta mais pas dans les adipocytes,
démontrant bien encore une fois que la régulation des gènes dépend énormément de la cellule
cible ainsi que de son état et que l’épigénétique reste un mécanisme essentiel dans la
régulation génique.
� PPARγγγγ et le métabolisme lipidique placentaire
La découverte de PPARγ dans les adipocytes a ouvert la voie pour démontrer son rôle dans la
régulation du transport des acides gras et de leur accumulation dans ces cellules (Tontonoz et
al., 1994; Tontonoz et al., 1998). Des études récentes ont montré que ce facteur de
transcription joue le même rôle dans le trophoblaste (Barak et al., 1999). Par la suite, il a été
démontré une accumulation de gouttelettes lipidiques dans le cytotrophoblaste durant sa
différenciation en syncytiotrophoblaste (Tarrade et al., 2001a). Ce phénomène est expliqué
par l’augmentation de l’accumulation d’acides gras dans les trophoblastes primaires lorsque
PPARγ et RXR sont activés, et peut être bloqué par les inhibiteurs de MAPK p38 (Schaiff et
al., 2005; Schild et al., 2006). Les protéines impliquées dans ce métabolisme et régulées par
PPARγ sont les protéines de transport des acides gras (FATP : Fat Acid Transport Protein),
les protéines PAT (Perilipine, Adipophiline et Tip47) mais la régulation par PPARγ des
protéines de fixation des acides gras (FABP : Fat Acid Binding Protein) reste à démontrer.
Toutes ces informations dénotent bien un rôle de PPARγ dans la capture et l’accumulation des
lipides dans le trophoblaste.
� PPARγγγγ et l’accouchement
L’accouchement précoce ou à terme est associé à une accumulation de cytokines pro-
inflammatoires impliquées dans la stimulation des contractions utérines (Andrews et al., 2000;
Keelan et al., 2003). La capacité de PPARγ à inhiber les gènes pro-inflammatoires suggère
qu’il pourrait jouer un rôle dans l’inhibition des contractions utérines durant la grossesse
(Pascual et al., 2005; Ricote et al., 1998; Scher and Pillinger, 2005). De plus, son expression
diminuant dans la membrane amniotique et chorionique au moment de l’accouchement,
laissant place à une augmentation de COX-2, laisse croire en un rôle de PPARγ dans le
contrôle de l’accouchement. Cependant, les études démontrant l’inhibition des gènes pro-

48
inflammatoires dans le placenta ont été effectuées avec de la 15d-PGJ2 et cet agoniste s’est
révélé jouer un rôle indépendamment de PPARγ (Pirianov et al., 2009). Finalement le rôle de
PPARγ dans le contrôle de l’accouchement est plausible mais reste encore à démontrer.
� PPARγγγγ et la différenciation du cytotrophoblaste
La différenciation du cytotrophoblaste humain est caractérisée par la fusion des cellules
cytotrophoblastiques mononuclées en syncytiotrophoblastes multinucléés à la surface des
villosités. Ce processus est accéléré par l’activation de PPARγ dans les cellules primaires du
trophoblaste et est accompagné d’une augmentation de sécrétion de β-hCG (human Chorionic
Gonadotropin), considéré comme un marqueur biochimique de la différenciation
trophoblastique (Schaiff et al., 2000; Tarrade et al., 2001a). Le syncytiotrophoblaste étant plus
résistant à l’hypoxie, il est primordial d’avoir une bonne différenciation trophoblastique pour
la fonction placentaire (Crocker et al., 2003). En effet, l’activation de PPARγ dans des
cultures de cytotrophoblastes les rendent plus résistantes à l’apoptose médiée par l’hypoxie
(Elchalal et al., 2004). Durant ce mécanisme de différenciation, l’expression de PPARγ ne
variant pas, on peut suggérer que c’est l’apport de ligand pour PPAR/RXR qui va réguler ce
phénomène.
� PPARγ γ γ γ dans l’invasion trophoblastique
Comme nous l’avons expliqué précédemment, le cytotrophoblaste extravilleux va envahir la
paroi utérine très tôt dans la grossesse. Ce processus est primordial pour l’accès du
trophoblaste à la circulation sanguine maternelle et l’établissement des échanges foeto-
maternels. L’équipe du docteur Thierry Fournier (Inserm U767) avec qui nous collaborons
dans cette étude, a démontré l’implication de l’hétérodimère PPARγ/RXRα dans la régulation
de l’invasion cytotrophoblastique (Tarrade et al., 2001b). Il a également développé une lignée
de cytotrophoblastes obtenue par immortalisation de CTEV purifiés par les gènes T/t du virus
SV40 : les HIPEC (Human Invasive Proliferative Extravillous Cytotrophoblast) que nous
avons également utilisé au cours de ce travail de thèse (Pavan et al., 2003). Cette régulation
négative de l’invasion ne peut se faire qu’avec l’activation de PPARγ, l’activation de RXR
seul n’ayant aucun effet (Fournier et al., 2002; Tarrade et al., 2001b). Ce rôle de PPARγ dans
l’invasion cytotrophoblastique pourrait être une piste pour la compréhension de la pré-
éclampsie (Evain-Brion et al., 2002). Cependant, les gènes cibles responsable de cette
inhibition de l’invasion restent à déterminer. Très récemment, une cible potentielle a été

49
découverte en plus de la β−ΗCG comme expliqué précédemment. Il s’agit de la protéase
PAPP-A (Pregnancy Associated Plasma Protein-A) (Handschuh et al., 2006).
IV. PAPP-A dans la grossesse
La protéase PAPP-A appartient à la famille des métalloprotéases metzincines. Elle a été isolée
pour la première fois dans le sérum de femmes enceintes dans les années 70 (Bischof, 1979;
Lin et al., 1974). Son expression augmente dans la circulation sanguine maternelle durant la
grossesse et diminue après l’accouchement (Folkersen et al., 1981). La PAPP-A est sécrétée
sous la forme d’un dimère de 400 kDa mais circule dans le sang maternel sous la forme d’un
complexe 2:2 de 500 kDa, relié par des ponts disulfures avec la protéine inhibitrice proMBP
(proform of eosinophil major basic protein) (Overgaard et al., 2000). Elle est exprimée dans le
placenta et plus particulièrement dans le cytotrophoblaste villeux, extravilleux mais aussi dans
la décidua (Bischof, 1989; Bonno et al., 1994). Outre la β-HCG libre, la PAPP-A fait partie
des marqueurs sériques maternels couramment utilisés dans le dépistage du syndrome de
Down (Trisomie 21) au premier trimestre (Gagnon et al., 2008).
Le rôle principal connu à ce jour de cette protéase est de cliver les protéines de liaison des
facteurs de croissance insulino-mimétiques IGF-BP4 et 5 (Insulin-like Growth Factor-
dependent Binding Protein 4) libérant ainsi la protéine IGF-II (Insulin-like Growth Factor II)
de sa ségrégation et lui permettant alors de se fixer à son récepteur IGF-II-R (Byun et al.,
2001). Cette protéine IGF-II va être impliquée dans l’invasion et la migration du
cytotrophoblaste, notamment via sa fixation à son récepteur induisant une phosphorylation
rapide des MAPK ERK 1 et 2 et une activation des protéines Gi et Gs inhibitrices de
l’Adenylate cyclase bloquant alors la synthèse d’AMPc (McKinnon et al., 2001).
L’activité protéasique de la PAPP-A est apparemment liée à sa fixation à la membrane
plasmique, lui suggérant un éventuel récepteur (Sun et al., 2002).
Comme il a été précisé auparavant, PPARγ va contrôler négativement l’expression de la
PAPP-A et inhiber ainsi la migration et l’invasion cytotrophoblastique (Handschuh et al.,
2006).Le mécanisme d’action de la PAPP-A sur l’invasion et sa régulation par PPARγ est
résumé dans la figure 23.
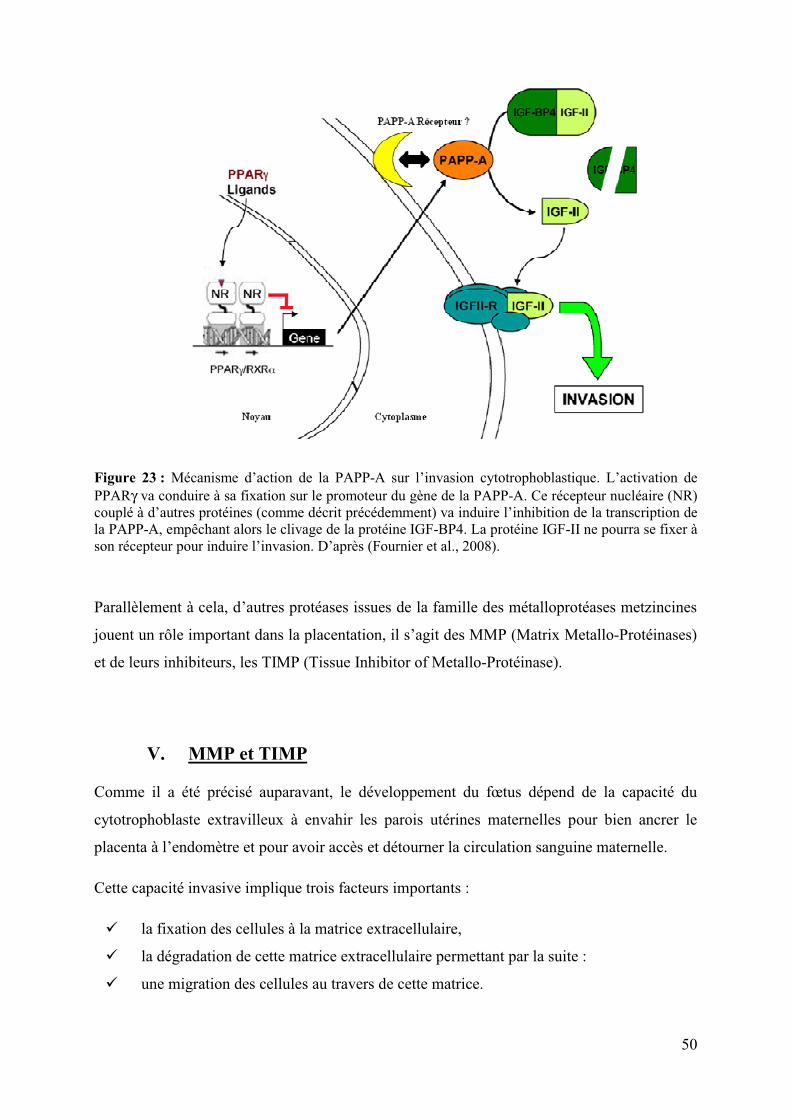
50
Figure 23 : Mécanisme d’action de la PAPP-A sur l’invasion cytotrophoblastique. L’activation de PPARγ va conduire à sa fixation sur le promoteur du gène de la PAPP-A. Ce récepteur nucléaire (NR) couplé à d’autres protéines (comme décrit précédemment) va induire l’inhibition de la transcription de la PAPP-A, empêchant alors le clivage de la protéine IGF-BP4. La protéine IGF-II ne pourra se fixer à son récepteur pour induire l’invasion. D’après (Fournier et al., 2008).
Parallèlement à cela, d’autres protéases issues de la famille des métalloprotéases metzincines
jouent un rôle important dans la placentation, il s’agit des MMP (Matrix Metallo-Protéinases)
et de leurs inhibiteurs, les TIMP (Tissue Inhibitor of Metallo-Protéinase).
V. MMP et TIMP
Comme il a été précisé auparavant, le développement du fœtus dépend de la capacité du
cytotrophoblaste extravilleux à envahir les parois utérines maternelles pour bien ancrer le
placenta à l’endomètre et pour avoir accès et détourner la circulation sanguine maternelle.
Cette capacité invasive implique trois facteurs importants :
� la fixation des cellules à la matrice extracellulaire,
� la dégradation de cette matrice extracellulaire permettant par la suite :
� une migration des cellules au travers de cette matrice.

51
Le premier ainsi que le troisième facteur nécessitent la présence de récepteurs de surface à la
matrice extracellulaire tels que les intégrines. Leur expression va donc varier tout au long de
la différenciation des cellules placentaires durant la grossesse (Damsky et al., 1992).
La dégradation de la matrice extracellulaire dépend de l’expression et de la sécrétion
d’enzymes spécifiques, dont les plus importantes sont les MMPs (Matrix Metallo-
Protéinases). La famille des MMP est toujours en expansion et comporte 25 membres
actuellement divisés en quatre groupes selon la spécificité des substrats et leur
localisation (cités ici avec quelques exemples):
� Les collagénases comprenant la collagènase interstitielle MMP-1 et la collagènase
neutrophile MMP-8, dégradant les collagènes fibrillaires I, II et III.
� Les gélatinases incluant la gélatinase A (MMP-2) et la gélatinase B (MMP-9)
dégradant la plupart des collagènes dénaturés et natifs.
� Les stromélysines incluant la stromélysine-1 (MMP-3), -2 (MMP-10) et -3 (MMP11)
ainsi que la matrilysine (MMP-7) dont le spectre de substrat est le plus large
(fibronectines, laminine, collagène III, IV et V, élastine …).
� Enfin, les MMP de type membranaire (MT-MMP Membrane type Matrix Metallo
Proteinase) seraient plutôt une sous classe comprenant les MT-MMP-1, -2 et -3
localisées à la surface des cellules, et joueraient un rôle d’activateur des MMPs.
Toutes les MMPs sont sécrétées sous une pro-forme inactive et doivent être clivées pour
devenir actives. C’est pourquoi des MMPs sous formes latentes et activées peuvent être
trouvées dans la matrice extracellulaire. Comme de nombreux membres de cette famille
peuvent s’activer entre eux, il est alors parfois possible avec l’activation d’une seule enzyme,
de dégrader complètement une matrice extracellulaire. Tout ceci explique la présence
d’inhibiteurs spécifiques : les TIMPs (Tissue Inhibitor of Metallo-Protéinase). Ces inhibiteurs
spécifiques ont été décrits dans la decidua humaine pour la première fois in vitro (Graham and
Lala, 1991) puis in vivo (Marzusch et al., 1996). A ce jour, il existe 4 TIMP (TIMP-1, TIMP-
2, TIMP-3 et TIMP-4). Ces inhibiteurs interagissent avec les MMPs activées au niveau de
leur site d’activité catalytique. Cependant, il a également été démontré que TIMP-1 pouvait
interagir avec la MMP-2 et la MMP-9 inactives. Ces TIMPs vont alors permettre une
régulation de l’activité enzymatique des MMP durant la grossesse (Huppertz et al., 1998) ou
tout autre phénomène les impliquant, en fonction de leur niveau d’expression et leurs
interactions. Il est également à noter que les PPARs jouent un rôle dans la régulation
d’expression des MMPs ainsi que des TIMPs (Shen et al., 2007; Shu et al., 2000).

52
Il est évident que la placentation s’effectue en grande partie grâce à la sécrétion et la
régulation de protéases spécifiques, c’est pourquoi nous nous sommes intéressés à certains
récepteurs spécifiques, activés par des protéases, les récepteurs PAR (Protease-Activated
Receptor) jouant eux-mêmes un rôle dans l’invasion cytotrophoblastique.
VI. Les récepteurs PAR (Protease Activated Receptor)
La découverte des récepteurs PAR eu lieu dans les années 90. Dans un premier temps, PAR1
fut identifié comme étant le récepteur responsable de l’agrégation cellulaire induite par la
thrombine (Rasmussen et al., 1991; Vu et al., 1991). Puis PAR2 fut identifié comme un
récepteur activé par les mêmes mécanismes que PAR1 mais indépendamment de la thrombine
(Bohm et al., 1996; Nystedt et al., 1995). Vers la fin des années 90, PAR3 et PAR4 furent
identifiés et clonés comme deux nouveaux et derniers récepteurs PAR possibles présents chez
l’Homme (Ishihara et al., 1997; Xu et al., 1998). L’expression de ces 4 récepteurs PARs est
assez ubiquitaire dans les tissus et les différents types cellulaires. Cependant, la signalisation
en aval de leur activation va être différente selon le type cellulaire considéré. En effet, ces
récepteurs son associés à des protéines Gα mais selon la nature de cette dernière (q, 12/13,
i,…) les kinases agissant en second messagers diffèrent (PKC, Tyrosine kinase, PI3kinase …)
(Macfarlane et al., 2001). Les PAR sont donc des récepteurs couplés aux protéines G (RCPG)
avec un système d’activation bien particulier. Comme leur nom l’indique, ces récepteurs
doivent être clivés au niveau de leur domaine N-terminal extracellulaire par des protéases. Il
existe 3 modes d’activation différents des récepteurs PARs résumés dans la figure 23.
� Les protéases clivent au niveau d’un site spécifique du domaine N-terminale
extracellulaire créant ainsi un nouveau domaine N-terminal qui agira comme un ligand
attaché se fixant à la seconde boucle extracellulaire du récepteur pour induire une
signalisation intracellulaire. Le clivage protéolytique spécifique provoque par ailleurs
un relargage d’un petit peptide correspondant à l’ancienne partie N-terminale du
récepteur. Ce peptide est appelé PARstatin et il a été démontré qu’il exerçait des effets
biologiques comme l’inhibition de l’angiogénèse mais les mécanismes impliqués
restent encore à découvrir (Zania et al., 2009) (Figure 24A).
� Certains récepteurs PAR possèdent des séquences d’interaction avec des protéases
avant ou après leur site d’activation. Ces séquences vont permettre d’attirer des
protéases près de leur site de clivage et de faciliter ainsi leur action. Ces séquences
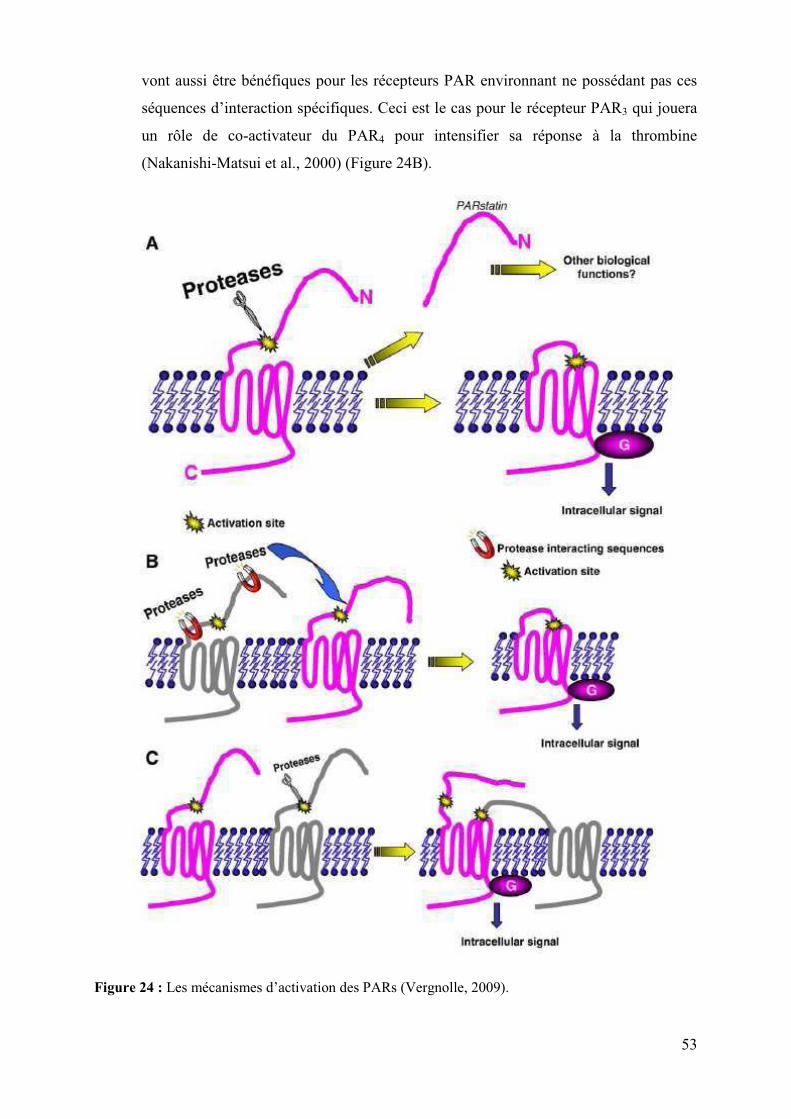
53
vont aussi être bénéfiques pour les récepteurs PAR environnant ne possédant pas ces
séquences d’interaction spécifiques. Ceci est le cas pour le récepteur PAR3 qui jouera
un rôle de co-activateur du PAR4 pour intensifier sa réponse à la thrombine
(Nakanishi-Matsui et al., 2000) (Figure 24B).
Figure 24 : Les mécanismes d’activation des PARs (Vergnolle, 2009).

54
� Enfin le ligand attaché d’un récepteur PAR clivé pourra activer un récepteur PAR
adjacent en se fixant à sa seconde boucle extracellulaire, même si le PAR adjacent est
différent (exemple PAR3 clivé activant PAR1 et PAR2 adjacents) (Hansen et al., 2004)
(Figure 24C).
L’activation de ces récepteurs peut également se faire au moyen de peptide agoniste dont la
séquence en acide aminés correspond à la séquence N-terminale extracellulaire jouant le rôle
de ligand attaché. Il existe également des peptides synthétiques antagonistes de ces récepteurs.
Tous les agonistes et les antagonistes des PAR sont résumés dans les tableaux 2 à 4.
Tableau 2 : Les différentes protéases activant ou désactivant les PAR1 et PAR2 (Vergnolle, 2009).

55
Tableau 3 : Les différentes protéases activant les PAR3 et PAR4 (Vergnolle, 2009).
Le clivage des PARs étant irréversible, le ligand attaché reste fixé à son récepteur jusqu’à une
désensibilisation ou une internalisation du récepteur. En aucun cas le récepteur ne pourra être
réactivé de novo. C’est pourquoi les PARs ne seront pas recyclés et la désensibilisation du
récepteur aura lieu grâce à son endocytose. Ce mécanisme est dépendant de phosphorylation
spécifique ainsi que de fixation de β-arrestine selon les récepteurs et reste à être déterminé
pour d’autres. Ainsi l’endocytose des PAR1 se fera via des puits de clathrine dépendant de la
dynamine (Hoxie et al., 1993; Trejo et al., 2000) alors que l’internalisation de PAR2 sera
médiée par un mécanisme dépendant de l’arrestine (Dery et al., 1999; Kumar et al., 2007).
Pour les récepteurs PAR3 et PAR4, leur mécanisme d’endocytose reste à déterminer.
Tableau 4 : Les différents agonistes et antagonistes synthétiques des PARs (Vergnolle, 2009). Les plus sélectifs et efficaces sont indiqués en rouge.

56
Enfin, des études ont montré que certaines protéases pouvaient inhiber l’activation des
plaquettes induites par la thrombine, ce qui suggéraient que certaines protéases pouvaient
bloquer l’activation des PAR (Renesto et al., 1997). Il a alors été démontré que ces protéases
pouvaient « désarmer » les récepteurs PAR en les clivant entre leur site d’activation et leur
premier domaine transmembranaire (Chignard and Pidard, 2006) (Figure 25). Ces protéases
peuvent provenir aussi bien de la cellule que d’un microorganisme extérieur et sont présentées
dans le tableau 2. Les récepteurs restent alors dans un état inactivé mais peuvent cependant
être activé par les récepteurs PAR adjacent par le mécanisme présenté figure 23C. Ce
mécanisme d’inhibition peut aussi dépendre de l’état de glycosylation du récepteur. Ainsi
certaines protéases vont inhiber les récepteurs PAR dans un type cellulaire alors qu’elles vont
les activer dans d’autres. C’est le cas par exemple de l’elastase neutrophile ou de la cathépsine
G désarmant PAR2 dans les cellules épithéliales de poumons alors qu’elles l’activent dans les
cellules non épithéliales (Chignard and Pidard, 2006; Uehara et al., 2003).
Figure 25 : Mécanisme de désarmage des PAR. Les protéases clivent en amont du site d’activation détachant ainsi la partie N-terminale extracellulaire contenant le ligand attaché. Après une telle protéolyse, le récepteur est considéré comme silencieux et ne peut plus être activé par son propre ligand attaché. Il pourra cependant être activé par les ligands attachés des récepteurs PAR adjacents (Vergnolle, 2009).
Ces récepteurs sont tous impliqués dans les mécanismes d’inflammation et de douleurs. En
effet, ces récepteurs jouent de nombreux rôles pro-inflammatoires dans de nombreuses
maladies telles que l’arthrite, la colite, l’inflammation des poumons et leur blocage ou leur
inhibition fait partie des nombreuses cibles étudiées actuellement en pharmacologie (pour
revue (Vergnolle, 2009)).

57
Parallèlement à cela, ces récepteurs PAR vont être impliqués dans la migration et l’invasion
des cellules cancéreuses. En effet, il a été démontré que leur activation avait lieu dans certains
cancers (cancer du sein, osteosarcome) et promouvait l’invasion tumorale (Arora et al., 2007;
Radjabi et al., 2008). Dans ce type de cellule, leur activation a le plus souvent lieu grâce à
l’action protéolytique de la MMP1 (Boire et al., 2005; Pei, 2005).
Enfin, le point nous intéressant le plus dans cette étude est le rôle récemment découvert de ces
récepteurs dans la placentation et plus particulièrement dans la migration et l’invasion des
cellules trophoblastiques. En effet, il a été démontré que PAR1 jouait un rôle dans la
placentation (O'Brien et al., 2003), tout ceci via une signalisation en aval passant par la voie
WNT et une stabilisation de la β-cathénine (Grisaru-Granovsky et al., 2009). De manière
intéressante, la thrombine est également impliquée dans l’invasion des ostéosarcomes en
augmentant la sécrétion de la MMP9 (Radjabi et al., 2008). Et il a été démontré que PAR1 et
PAR3 pouvaient induire la sécrétion de MMP9 dans les astrocytes et dans les monocytes
(Chang et al., 2009; Choi et al., 2008). La MMP9 jouant un rôle essentiel dans la placentation,
ce mécanisme de régulation n’est pas à négliger quant à l’implication des récepteurs PAR
dans la placentation. Cependant tout ceci reste encore à découvrir.
VII. HCMV et pathologie de la grossesse
Il est temps maintenant de regrouper toutes ces données importantes dans le contexte de
l’infection par le CMV pendant la grossesse. Actuellement, l’infection materno-fœtale à
cytomégalovirus est la principale cause infectieuse de malformation congénitale, de retards
mentaux et de surdité. Les techniques d’aujourd’hui permettent de diagnostiquer les infections
maternelles, fœtales et néonatales. Cependant, l’absence de traitement ainsi que l’observation
que la plupart des fœtus infectés sont pour la plupart asymptomatiques à la naissance ont
poussé l’Anaes à ne pas recommander d’effectuer de sérologie CMV en cours de grossesse
(Anaes, Septembre 2004). A ce jour, la seule intervention possible reste l’interruption
médicale de grossesse (IMG).
Pourtant l’incidence des primo-infections chez la femme enceinte n’en est pas moins non
négligeable et varie selon les conditions socio-économiques, passant d’environ 2 % par an
dans les pays en voie de développement et allant jusqu’à 6 % dans les pays développés
(Benoist et al., 2008). Tout cela sans compter les avortements spontanés causés par l’infection
HCMV. Contrairement à l’infection par le VIH, le premier organe infecté par le CMV est le

58
placenta, véritable centre de réplication du virus. Le placenta agit donc à la fois comme
barrière contre le virus mais aussi, comme un réservoir pouvant libérer ce dernier dans la
circulation fœtale. L’un des premiers signes visibles d’une infection par le HCMV à
l’échographie est l’augmentation de l’épaisseur placentaire associée à un aspect globalement
hétérogène de celui-ci avec des calcifications coexistant avec des zones hypoéchogènes
(Drose et al., 1991). Après l’infection placentaire, le virus va se propager au fœtus et infecter
ses différents organes pouvant alors provoquer de graves troubles neurologiques (voir la revue
de Cheeran (Cheeran et al., 2009)). Cette transmission au travers du placenta se fait
majoritairement par transcytose des virions au travers des syncytiotrophoblastes grâce aux
anticorps IgG maternels et aux récepteurs FcRn placentaires (Maidji et al., 2006) (Figure 26).
Au cours de l’infection materno-fœtale, c’est dans le cytotrophoblaste que le HCMV va se
répliquer. Il a été démontré que le virus pouvait se répliquer in vitro et in vivo dans le
cytotrophoblaste de placenta précoce ou à terme (Fisher et al., 2000; Halwachs-Baumann et
al., 1998). Cette infection va alors conduire à des disfonctionnements du cytotrophoblaste.
Ces disfonctionnements apparaissent par le biais de changement phénotypiques comme la
diminution de l’expression de l’intégrine α1β1 provoquant alors une perte de la cohésion
tissulaire ainsi qu’une perte d’adhérence à la matrice extracellulaire (Pereira et al., 2006).
Cependant, certaines intégrines bloquant l’invasion ne sont quant à elles pas inhibées, ce qui
entraîne la perturbation de l’invasion trophoblastique. En effet, il a été démontré in vitro que
l’infection par le HCMV perturbe les capacités invasives du cytotrophoblaste (Fisher et al.,
2000). Cette perturbation touche la majorité des cellules in vitro même si toutes les cellules ne
sont pas infectées. Ceci suggère que la présence de cellules infectées va influencer la
migration et l’invasion des cellules avoisinantes, et ceci par le biais d’un éventuel facteur viral
sécrété ou un défaut de sécrétion cellulaire naturel. En accord avec cette hypothèse, l’équipe
de Lénore Pereira a démontré que le HCMV perturbait la migration et l’invasion du
cytotrophoblaste en augmentant la sécrétion d’Il-10, induisant par la suite la sécrétion de
l’homologue viral de l’IL-10 (cmvIL-10). Tout ceci a pour conséquence de diminuer la
production de MMP-9 active, enzyme essentielle à la placentation (Yamamoto-Tabata et al.,
2004). Ces études ont donc permis de démontrer que l’exploitation de certains acteurs de la
réponse immunitaire comme l’IL-10 par le virus peut perturber la mise en place du placenta.
Parallèlement à cela, il reste à décrire les bases moléculaires des mécanismes responsables de
cette perturbation de la placentation, notamment les facteurs de transcription mis en jeu et
leurs cibles potentiels dans les cellules placentaires et avoisinantes.

59
Figure 26 : Schéma de la transmission du HCMV dans les villosités chorioniques flottantes. Voie 1 : Dans le syncytiotrophoblaste non infecté, les IgG du sang maternel sont absorbés par pinocytose en se fixant sur les récepteurs FcRn. Les endosomes sont alors recyclés ou traversent le syncytium vers la membrane basale par transcytose. Voie 2 : Suppression de l’infection lorsque le complexe est fortement neutralisant à faible pH dans les vésicules d’endocytose. Les complxes IgG-gB sont stockés dans des cavéosomes à pH neutre (Cav). Les complexes virions-IgG sont alors amenés par transcytose au travers du syncytiotrophoblaste et capturés par les macrophages villeux (M) après avoir été internalisés dans le cytotrophoblaste. Voie 3 : Infection focale du cytotrophoblaste par les complexes IgG-virions avec une faible activité neutralisante. L’infection peut alors se propager vers les fibroblastes stromales, les vaisseaux sanguins, les leucocytes et l’intérieur des villosités. En rouge sont représentés les noyaux des cellules infectées (Maidji et al., 2006).
VIII. Objectifs du travail
Les objectifs de ce travail de thèse ont été dans un premier temps d’approfondir nos
connaissances de la réplication du HCMV, à partir des observations de Zhu sur l’utilisation de
l’activité enzymatique COX-2 par le virus pour sa propre réplication (Zhu et al., 2002). Ceci
nous a amené à découvrir pour la première fois l’activation et l’utilisation d’un récepteur
nucléaire jouant un rôle important dans la placentation par le virus, nous conduisant alors à
étudier les perturbations que cette activation pouvait engendrer durant la grossesse, et enfin à
déterminer les cibles potentielles de ce facteur de transcription, modifiées par l’infection.

60
TRAVAUX DE RECHERCHE

61
PREMIERE PARTIE :
L’activation du récepteur nucléaire PPARγγγγ par
le HCMV pour sa réplication perturbe la
migration et les propriétés invasives du
cytotrophoblaste.

62
Introduction
Le Cytomégalovirus Humain (HCMV) peut causer de graves pathologies chez les personnes
immuno-déprimées, les fœtus et les nouveau-nés et une primo-infection de la mère durant le
premier trimestre de grossesse peut entraîner des fausses couches ainsi que des retards de
croissance intra-utérins. Il a été montré que le virus tire profit de l’inflammation en utilisant la
voie de la cyclooxygénase-2 (COX-2) dépendante des prostaglandines pour la transcription
des gènes très précoces, aboutissant en particulier à la production de la protéine IE2,
indispensable au cycle viral. Aucun mécanisme n’a été décrit jusqu’à présent. En effectuant
des expériences d’activation de « gène rapporteur », de microscopie confocale en présence
d’antagonistes spécifiques, nous avons démontré pour la première fois que l’activation du
récepteur nucléaire PPARγ (Peroxisome Proliferator-Activated Receptor gamma) dans les
cellules infectées est impliquée dans la réplication virale, en aval de l’activation de COX-2.
Nous avons démontré qu’un antagoniste de PPARγ diminue considérablement l’expression
des ARNm de la protéine virale IE2 ainsi que la réplication virale, et nous avons mis en
évidence grâce à des expériences de « gel-retard » et de ChIP que le promoteur majeur des
gènes IE (MIEP) contient des séquences de réponses à PPAR (PPRE) capables de fixer
PPARγ pour activer la transcription de gènes. De plus, l’expression ectopique de PPARγ dans
des cellules non permissives Hek-293 augmente l’expression des ARNm IE2 et la production
de virions, ce qui démontre que l’activation de PPARγ peut être un processus clé pour la
réplication du HCMV. PPARγ jouant un rôle essentiel dans l’invasion cytotrophoblastique,
nous nous sommes intéressés aux conséquences de cette activation dans des cellules
placentaires. En utilisant une technique in vitro d’invasion et de migration de
cytotrophoblastes humains isolés à partir de placentas précoces, nous avons démontré que
l’activation de PPARγ médiée par le HCMV inhibe l’invasion placentaire. Ces résultats
ouvrent une nouvelle voie de recherche et donnent de nouveaux indices pour expliquer
certains troubles de la grossesse pouvant intervenir suite à une primo-infection de la mère
durant le premier trimestre et permettent d’envisager de nouvelles thérapies anti-virales. Ce
travail est présenté dans l’article suivant accepté dans Journal of Virology en décembre 2009.
L’article est ici sous la forme manuscrite. Dans un souci de lisibilité, les figures ainsi que
leurs légendes ont été incorporées dans le texte, certaines ont été fractionnées par rapport au
format de l’article publié. (Version définitive de l’article dans J. Virol disponible en annexes
p123).

63
Activation of PPARγγγγ�by Human Cytomegalovirus for de novo
Replication Impairs Migration and Invasiveness of
Cytotrophoblast from Early Placenta
�
Benjamin Rauwel a,b, Bernard Mariamé a,b, Hélène Martin a,b, Ronni Nielsen c, Sophie Allart a,b, Bernard Pipy d, Susanne Mandrup c, Marie Dominique Devignes e, Danièle Evain-Brionf,g,h, Thierry Fournier f,g,h, and Christian Davrinche a,b,1
a. INSERM, U563, Toulouse, F-31300 France
b. Université Toulouse III Paul-Sabatier, Centre de Physiopathologie de Toulouse
Purpan, Toulouse, F-31300 France
c. Department of Biochemistry and Molecular Biology, University of Southern
Denmark, 5230 Odense M, Denmark
d. EA 2405, IFR 31, BP 84225, 31432, Toulouse Cedex 4, France
e. CNRS-UMR 7503, Laboratoire Lorrain de Recherche en Informatique et Ses
Applications, 54506, Vandoeuvre-les-Nancy, France
f. INSERM, U767, 4 avenue de l’Observatoire, 75006, Paris, France
g. Université Paris Descartes, Paris, France
h. PremUP, Fondation, 75006, Paris, France
Running title : PPARγ�in HCMV replication and placentation
1. Corresponding author: C. Davrinche, Tel: 33 5 62748385, Fax: 33 5 62744558, E-Mail :

64
���������
Human cytomegalovirus (HCMV) contributes to pathogenic processes in immuno-suppressed
individuals, in fetuses and in neonates. In the present report by using reporter gene activation
assays and confocal microscopy in the presence of specific antagonist we show for the first
time that HCMV infection induced PPARγ transcriptional activity in infected cells. We
demonstrated that PPARγ antagonist dramatically impaired virus production and that the
major immediate-early promoter (MIEP) contained PPAR response elements (PPRE) able to
bind PPARγ, as assessed by electrophoretic mobility shift and chromatin immunoprecipitation
assays. Due to the key role of PPARγ in placentation and its specific trophoblast expression
within the human placenta, we then provided evidence that by activating PPARγ human
cytomegalovirus dramatically impaired early human trophoblast migration and invasiveness,
as assessed by using well-established in vitro models of invasive trophoblast i.e. primary
cultures of EVCT isolated from first trimester placentas and the EVCT-derived cell line
HIPEC. Our data provide new clues to explain how early infection during pregnancy could
impair implantation, placentation and therefore embryonic development.

65
Introduction
Human cytomegalovirus (HCMV), a member of the betaherpesvirus family causes
miscarriages, morbidity and mortality in fetuses and newborns and contributes to the
development of numerous diseases in immune-compromised hosts, especially transplant
recipients and AIDS patients (for review see (Mocarski et al., 2007a)). A crucial step in
regulation of HCMV replication and reactivation from latency lies on activation of the major
immediate early promoter (MIEP) which controls production of the immediate-early gene
products IE1 and IE2, the latter being essential for viral replication. Amongst transcription
factors which have been involved in the MIEP regulation (Meier and Stinski, 1996), NF�κB
was the most studied since four cognate recognition sites have been identified into the MIEP
sequence and because it is clearly one of the most important regulators of pro-inflammatory
gene expression including those encoding cytokines, such as TNF- �� IL-���� IL-6, and IL-8,
and cyclooxygenase 2 (Cox-2). Interestingly, it has been demonstrated that in infected
fibroblasts, inhibitors of Cox-2 reduced the yield of HCMV by a factor 100 and blocked the
accumulation of IE2 mRNA and protein (Zhu et al., 2002). These data suggested regulation of
the MIEP downstream of prostaglandin (PG) production, a byproduct of arachidonic acid
(AA) conversion by Cox-2. Up to now, mechanisms underlying regulation of IE2 mRNA by
prostaglandins, either at the transcriptional or post-transcriptional level, were still unknown.
Evidence that induction of Cox-2 and synthesis of prostaglandins including PGE2 were
essential for efficient HCMV replication prompted us to consider a possible role for the
peroxisome proliferator-activated receptor gamma (PPARγ, a nuclear receptor able to induce
transcription of genes involved in lipogenesis and to repress genes involved in inflammatory
processes (Glass and Ogawa, 2006). Indeed, Cox-2 is required for the production of
prostaglandins, some of which are agonists of PPAR����
PPARγ� is an isoform of the PPAR subset of nuclear receptors also including PPARα� and
PPARβ/δ�� that all bind to DNA as heterodimers with retinoid X receptors (RXR).
Heterodimers activate expression of target genes by binding to peroxisome proliferator-
responsive elements (PPREs) composed of direct tandem repeats of a consensus sequence
spaced by a single nucleotide. In addition, PPARs suppress gene expression by interfering
with the activity of transcription factors like NF-�B�� Stats and AP-1. Beside its role as
regulator of lipid metabolism, inflammation and immune response (Glass and Ogawa, 2006),
PPARγ was demonstrated to play a major role for trophoblast differentiation and early
placental development in knock-out mice (Barak et al., 1999; Kubota et al., 1999). In human

66
placenta, PPARγ was shown to control trophoblast invasion and differentiation (Tarrade et al.,
2001b), a process known to be impaired by infection with HCMV during pregnancy (Fisher et
al., 2000). Here we first demonstrate that PPARγ� could be used for HCMV replication
through its binding to the major immediate-early viral promoter and that by this way infection
dramatically impaired trophoblast migration and invasiveness.
�
Materials and Methods
Cells, Viruses and Reagents
U373MG astrocytoma cells, MRC5 fibroblasts, Hek-293 cells (InvivoGen) were propagated
in medium containing 10% fetal calf serum and extravillous cytotrophoblast (EVCT, HIPEC)
were as described previously (Tarrade et al., 2001b) (Pavan et al., 2003). Chorionic villi from
first trimester placentas (8-12 weeks of gestation) were obtained from volunteers who legally
chose to terminate pregnancy after written approbation (Broussais Hospital, Paris, France).
Cultures of trophoblasts were approved by the local ethical committee. After dissection,
EVCTs were isolated by trypsin digestions and purified through a discontinuous percoll
gradient. Cell viability was determined by flow cytometry analysis of annexin V (AV)
positive cells (FITC-AV, Dako). About 90 % viable EVCT stained positive for human
invasive extravillous cytotrophoblast markers after culture on matrigel (CK07, HLA-G, CD9,
hPL, c-erB2, α5 subunit of the fibronectin receptor) and expressed RXRα and PPARγ �in their
nuclei. Immunohistochemistry for PPARγ and Cytokeratin 7 were performed as previously
described (Fournier et al., 2007) on paraffin-embedded sections of an implantation site
(placenta and myometrium at 16 weeks of pregnancy). Immunocytochemistry with anti-
RXRα, -PPARγ and -CK07 on EVCT and HIPEC was performed as in (Pavan et al., 2003).
Virus strains used were AD169 (ATCC), Towne (a gift from S. Michelson, Institut Pasteur,
Paris), and VHLE (a gift from C. Sinzger, Tubingen, Germany). Virus stocks were collected
when cytopathic effects on MRC5 (Biomérieux, France) were > 90%. Supernatants were
clarified of cell debris by centrifugation at 1,500g for 10 min, ultracentrifuged at 100,000g for
30 min at 4° C and stored at -70°C until use. Virus titers were determined by plaque assay on
MRC5 cells using standard methods. UV irradiation of HCMV was performed with a
Spectroline irradiator (EF-140/F) for 20 min.

67
NS-398 (Sigma, 10 µM) and G3335 (Calbiochem, 30 µM) were added to cells 30 min before
infection and cultures were refed with G3335 every 24 h. Gw9662 was from Calbiochem. 15d
PGJ2 and Rosiglitazone were from Cayman Chemicals. Dose-effect experiments were done
for all reagents which were used at the optimal concentrations (peak dose) which did not
affect cell viability as tested by trypan blue exclusion, cell morphology and nuclei
condensation or fragmentation (Dapi staining). The solvent used for each of them (vehicle
control) was assayed as a control in all sets of experiments.
Reporter plasmids and cell transfection.
A PPRE-luciferase (Luc) plasmid provided by W. Wahli (University of Lausanne,
Switzerland) and a home-made construct containing PPRE sequences were used
independently. A reporter vector for�PPAR-�pB3xPPRE-Luc) was constructed� by modifying
the pNF-κB-Luc plasmid which has been described previously (18) Briefly, the NF-κb
binding sites of the pNF-κB-Luc plasmid were removed by digestion with MluI and BglII and
replaced by the phosphorylated, double stranded MluI-BglII oligonucleotide obtained by
annealing 3XPPRE-S (5’-CGCGTAGGTCACAGGTCACAGGTCAGAGGTCA
GAGGTCATAGGTCA-3’) and 3X PPRE-AS (5’-GATCTGACCTATGACCTCTGACCTCT
GACCTGTGACCTGTGACCTA-3’). As a control, the MluI/BglII linearized pNF-κB-Luc
plasmid was also directly recircularized, providing a vector deleted of any responsive element
(p0RE-Luc).
Three reporter vectors containing the putative PPRE sequences of the MIEP (PP1-Luc, PP2-
Luc and PP3-Luc) were similarly constructed by replacing the NF-κb binding sites of the
pNF-κB-Luc plasmid with the phosphorylated, double stranded MluI-BglII oligonucleotides
obtained by annealing PP1-S (5’-CGCGTAGTTCATAGCCCATAGTTCATAGCCCA-3’)
and PP1-AS (5’-GATCTGGGCTATGAACTATGGGCTATGAAC
TA-3’) for PP1, PP2-S (5’-CGCGTAACTTACGGTAAATGGCCCGTAACTTACGGT
AAATGGCCCA-3’) and PP2-AS (5’-GATCTGGGCCATTTACCGTAAGTTACGGGC
CATTTACCGTAAGTTA-3’) for PP2 and PP3-S (5’-CGCGTACGTCAATGACGGTA
AATGGCCCGACGTCAATGACGGTAAATGGCCCA-3’) and PP3-AS : (5’-GATCTG

68
GGCCATTTACCGTCATTGACGTCGGGCCATTTACCGTCATTGACGTA-3’) for PP3.
All constructs were checked by DNA sequencing. For transfection, U373MG cells were
seeded in a 24-well plate at 50,000 cells per well. Sixteen hours later, pPPRE-reporter
plasmids and FuGene 6 reagent (Roche) were added according to manufacturer’s instruction.
After 24h of treatment or infection, cells were lysed with 1X cell culture lysis reagent
(Promega) and luciferase activity was quantified using a Mithras luminometer.
�
Electrophoretic Mobility Shift Assay
Sequences of probes were selected according to previous data demonstrating the importance
of the 5’-flanking sequence (Juge-Aubry et al., 1997; Palmer et al., 1995) of the DR-1
elements. Except for PP1 which contains a RARE sequence in its 5’-flanking region,
nucleotides upstream of the DR-1 motif have been conserved for PP2 and PP3. 200ng of each
double-strand probe PP1, PP2, PP3 as used for reporter plasmids contruction, were labeled
with ATP-γ-32P (Perkin-Elmer) by T4 Polynucleotide kinase (New England Biolabs), for 30
min at 37°C and purified on Mini Quick Spin Oligo Columns (Roche). 3µg of nuclear protein
extract (Q Proteome Nuclear Subfractionnation kit, Qiagen)) from HCMV-infected U373MG
cells (24h p.i) were incubated at room temperature for 30 min with 15 ng of each probe, poly-
I:C and 2µg of either control IgG (AbCam) or PPAR-γ antibody (Santa-Cruz), or 200ng of
cold probe in 10 µl of binding buffer (0.2M KCl, 0.1M K+HEPES, pH7.6, 5mM MgCl2, 5mM
EGTA, 4% Ficoll). Mixes were submitted to electrophoresis on 6% Novex retardation Gel
(InVitroGen) in 0.5X TBE buffer at 100V for 90min. Revelation was made on Kodak BMR
Film at -80°C overnight.
Chomatin Immunoprecipitation (ChIP) Assay
U373MG cells (5x104 cells/cm2) were infected with HCMV (AD169, 3 m.o.i) for 24 hours,
fixed with 1% formaldehyde, lysed (Abcam) and sonicated (Vibracell 72405, Bioblock), in
order to obtain DNA fragments with size <1000bp. DNA-protein complexes were
immunoprecipitated according to Abcam procedure with ChIP grade antibodies (Abcam):
control IgG, anti-Histone-3 (H3), anti-PPARγ, then incubated with Protein A sepharose beads
from Sigma-Aldrich. Primers to detect HCMV MIEP were complementary to positions -272
and +13 relative to the MIEP start site as described in (Reeves et al., 2005): sense primer (5’-

69
ATT ACC ATG GTG ATG CGG TT-3’) and antisense primer (5’-GGC GGA GTT GTT
ACG ACA T-3’). The PCR amplification parameters were as follows: 95°C for 5 min, 50
cycles at 94°C (40s), 50°C (40s), and 72°C (90s).
Generation of purified adenovirus and transduction of Hek-293 cells.
Full-length mouse PPARγ2 as well as transduction in Hek-293 cells were done as follows:
PPAR-γ2 was initially N-terminally HA-tagged and cloned into p.Shuttle-CMV (Stratagene)
as described in (Nielsen et al., 2006). The CMV-HA-PPAR cassette was transferred to the
AdEasy-1 vector by homologous recombination in Escherichia coli to generate the AdHA-
PPAR-γ2 construct (Ad-PPARγ). The plasmid was linearized and transfected into Hek-293
cells for amplification. The amplified adenovirus was purified using CsCl gradients, and the
viral titer was estimated by a plaque assay-based approach as recommended by the AdEasy
protocol (Stratagene). Transduction with adenovirus encoding PPARγ (Ad-PPARγ) or not
(Ad-Vector) were performed at a m.o.i of 0.1 in Hek-293 cells at 80% confluence. After 2
hours of incubation, the medium was replaced by fresh one. The next day, cells were infected
with HCMV (m.o.i. of 3) as indicated. Virus titers were determined on Hek-293 supernatants
by plaque assay on MRC5 cells.
Confocal microscopy.
Cells were cultured on coverglass, washed, fixed and permeabilized in methanol for 5 min at -
20°C. PPARγ was detected with specific MAb (Santa-cruz E-8) followed by PE-conjugated
anti-mouse IgG antibody (Beckmann Coulter). IE1 and IE2 proteins were detected with FITC-
conjugated MAb (Chemicon). Fluorescence was analyzed with a Zeiss LSM 510 confocal
microscope (Zeiss, Jena, Germany). Images were reconstructed by using LSM image browser
software system (Zeiss).
Oil red O staining.Cells were fixed and permeabilized in methanol for 2 min at -20°C and
incubated for 10 min with 0.3% oil Red O in isopropanol (w/v). After 30 sec of incubation in
60% isopropanol, cells were washed in water and nuclei were counter-stained in hematoxylin.

70
RT-PCR.
RNA was extracted by Trizol (Invitrogen), reverse transcribed (RT) with Superscript III
(Invitrogen) and cDNA amplified with Mastermix (Eppendorf). 215-bp and 217-bp fragments
corresponding to IE1 and IE2 cDNA were amplified with the following primers: sense (5’-
AGG TGC CAC GGC CCG AGA CAC C-3’) and antisense (5’-TCT GTT TGA CCG
AGT TCT GCC-3’) for IE1 and sense (5’-AGG TGC CAC GGC CCG AGA CAC C-3’) and
antisense (5’-GGC GAG GAT GTC CGA GTT CTG CC-3’) for IE2. A 300-bp fragment
from actin was amplified with sense primer (5’-TCC CTG GAG AAG AGC TAC GAG-3’)
and antisense primer (5’-CAT CTG CTG GAA GGT GGA CA-3’). PCR was performed as
follows: 95°C for 5 min followed by 35 cycles of 95°C (30s), 58°C (for actin) or 60°C (for
IE1) or 62°C (for IE2) for 1 min, 72°C for 1 min, and 72°C for 2 min.
Western Blot Analyses.
Cells lysates were submitted to SDS/PAGE on 4-12% polyacrylamide gels (InVitroGen).
Blots on Hybond C-extra membrane (Amersham) were treated with anti-IE antibody (mouse
hybridoma E13, a gift from S. Michelson), anti-UL44 (ABCAM), anti-UL99 (ABCAM) anti-
βactin (Santa Cruz), anti-PPARγ (Santa Cruz), then with polyclonal rabbit HRP-conjugated
antibody and protein bands detected by using an ECL-plus kit (Amersham
Scanning CMV sequence with PPREFinder program.
PPREfinder program was developed at Loria to exhaustively list PPRE elements present in
DNA sequences (http://bioinfo.loria.fr/projects/pprefinder/PpreFinder1.1.zip/view). This
three-step program first identifies and localizes all PPRE half-sites (consensus sequence:
AGGTCA) in the input sequence allowing for mismatches (3 in this study) at any place in the
consensus 6-base sequence. The second step consists in localizing pairs of half-sites separated
for this study by only one nucleotide. The third step filters direct repeats (DR-1 motifs) from
the preceding pair list and ranks them according to the total mismatch number over the two
half-sites. A statistical module based on a stationary Markov chain provides an estimation of
the probability that the finding of a given DR-1 motif reflects a particular base composition in
the sequence

71
Human trophoblast wound healing and invasion assays.
Monolayers of HIPEC cells (80% confluence) were infected with HCMV, scratched (three per
well), left for 24 hours in culture, washed with PBS and photographed under a light
microscope. Wound closure reflecting migration of cells was quantified by using Image J
software (NIH). EVCT were cultured on Matrigel-coated Transwell (8 µm diameter pore
membranes) for 48 h in the presence or absence of 1 µM of rosiglitazone, 35 µM of G3335, 3
m.o.i of HCMV. After CK07 immunostaining and counterstaining with Dapi, pseudopodia
crossing the porous membrane were quantified and normalized to the total number of nuclei.
Results of a representative experiment (triplicate) are expressed as % of variation relative to
the control.
�
�
�
�
�
�
�
�
�
�
�
�
�
�
�
�
�
�
�

72
Results
HCMV induces PPARγγγγ transcriptional activity in infected cells.
U373MG astrocytoma cells which constitutively express PPARγ were transfected with
different PPRE-Luc plasmids encoding a luciferase gene under the control of the thymidine
kinase minimal promoter and a multimerized PPRE recognized by PPARγ and infected with
HCMV (VHLE clinical strain). Figure 1aA shows that expression of luciferase increased
throughout infection, which can be assumed to reflect transcription of the Luc gene through
binding of PPARγ to the PPRE sequence. Luciferase activity in uninfected cells may be due to
ligand independent activity of PPAR� (Nielsen et al., 2006) or to low levels of endogenous
agonists. Addition of 15dPG-J2, a natural ligand of PPARγ�to uninfected cells induced Luc
transcription with a quite less efficiency than HCMV (Fig 1aB). This may reflect a better
availability of endogenously synthesized 15dPG-J2 in infected cells and/or that activation of
PPARγ by HCMV could be in part mediated by other metabolites. To provide a direct proof
that PPARγ was involved in the PPRE-driven expression of luciferase, U373MG cells were
infected in the presence of G3335, a cell-permeable dipeptide that acts as a specific and
reversible antagonist of PPARγ through blockade of ligand binding. Figure 1C shows that in
these conditions, luciferase activity was strongly decreased, demonstrating that luciferase
transcription depends on interaction of PPARγ with its ligand. Treatment with NS398, an
inhibitor of Cox-2 activity, substantially reduced luciferase activity in infected cells (Fig
1aC), demonstrating that Cox-2 activation takes part in PPARγ transcriptional activity likely
through its role in prostaglandin synthesis from arachidonic acid. Specificity of PPARγ
activation by HCMV was further assessed by using one shot of Gw9662 (Gw), an irreversible
antagonist known to covalently bind and modify ligand binding site of PPARγ. Figure 1aD
shows the inhibitory effect of Gw on cells transfected with pB3xPPRE-Luc plasmid and
treated with Rosiglitazone as well as on those infected with HCMV. To address whether viral
neosynthesis was required for activation of PPARγ, luciferase activity was quantified from
cells infected with UV-irradiated virus. According to usual consideration, failure of the
irradiated virus (Fig 1aD) to drive luciferase expression suggests a role for newly synthesized
viral proteins in PPARγ activation. Transfection with PPRE-deleted control plasmid (right
histogram) did not show any luciferase activity.

73
Figure 1a. HCMV infection induces PPARγγγγ transcriptional activity
(A-D) ) U373MG cells were transfected with a PPRE-luciferase plasmid, left for 24 hours in culture, then mock-infected for 24 hours (n.i) or with 3 m.o.i of HCMV VHLE strain (cmv) for times as indicated (h p.i) in (A) or for 24 hours in (B) (C) and (D) before treatment or not (NT) with PPARγagonist 15dPGJ2 (15d in DMSO) or Rosiglitazone (Ro in DMSO), or inhibitor of Cox-2 (NS398 in DMSO), or antagonists of PPARγ (G3335 (G33), in distilled water, Gw9662 (Gw) in DMSO) at indicated final concentrations. Alternatively, HCMV was inactivated by UV irradiation (CMV-UV). Luciferase activity (Luc) was quantified by using a Mithras luminometer. Histograms representing means of duplicates are representative of five (A, B) and three (C, D) independent experiments with standard deviations. Right histogram in (D) corresponds to transfection of cells with control plasmid deprived of PPRE sequences (p0RE-Luc).
To further address whether PPARγ activity was restricted to infected cells, either MRC5
fibroblasts or U373MG cells were infected with HCMV. In MRC5 cells, infection strongly
redistributed PPARγ staining from the cytoplasm to the nucleus of infected cells as assessed
by confocal microscopy showing a merge expression of IE proteins and PPARγ in the nucleus
of infected cells (Fig 1E). Identical results were obtained in U373MG cells although with less
intensity (data not shown) and we further assessed the function of PPARγ in these cells
through its ability to induce activation of genes involved in lipogenesis. Staining with Oil red
O revealed accumulation of lipid droplets in infected cells contrary to those treated with the
PPARγ antagonist G3335 (Fig 1F). These experiments provided evidence that PPARγ was
activated in HCMV-infected cells as demonstrated by nuclear translocation and lipids
accumulation.
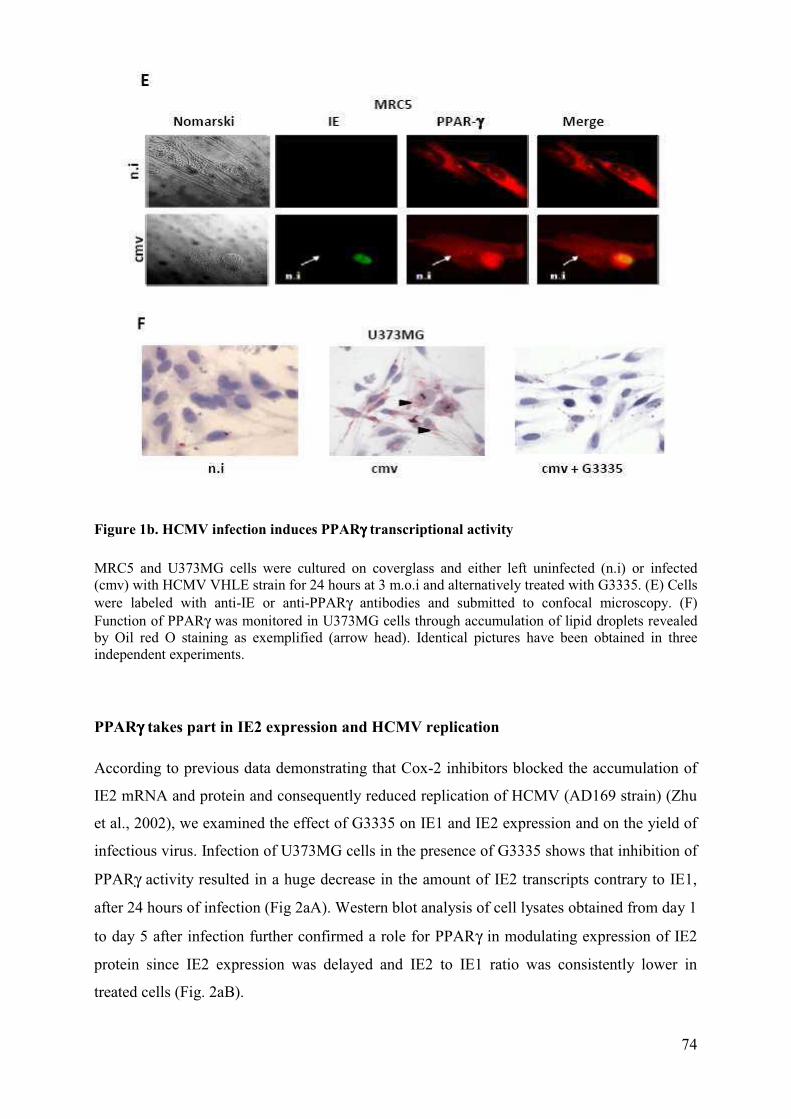
74
Figure 1b. HCMV infection induces PPARγγγγ transcriptional activity
MRC5 and U373MG cells were cultured on coverglass and either left uninfected (n.i) or infected (cmv) with HCMV VHLE strain for 24 hours at 3 m.o.i and alternatively treated with G3335. (E) Cells were labeled with anti-IE or anti-PPARγ antibodies and submitted to confocal microscopy. (F) Function of PPARγ was monitored in U373MG cells through accumulation of lipid droplets revealed by Oil red O staining as exemplified (arrow head). Identical pictures have been obtained in three independent experiments.
�
PPARγ γ γ γ takes part in IE2 expression and HCMV replication
According to previous data demonstrating that Cox-2 inhibitors blocked the accumulation of
IE2 mRNA and protein and consequently reduced replication of HCMV (AD169 strain) (Zhu
et al., 2002), we examined the effect of G3335 on IE1 and IE2 expression and on the yield of
infectious virus. Infection of U373MG cells in the presence of G3335 shows that inhibition of
PPARγ� activity resulted in a huge decrease in the amount of IE2 transcripts contrary to IE1,
after 24 hours of infection (Fig 2aA). Western blot analysis of cell lysates obtained from day 1
to day 5 after infection further confirmed a role for PPARγ in modulating expression of IE2
protein since IE2 expression was delayed and IE2 to IE1 ratio was consistently lower in
treated cells (Fig. 2aB).

75
Figure 2a. PPARγγγγ takes part in regulation of IE2 expression and HCMV replication
U373MG cells were infected (3 m.o.i of VHLE) for times as indicated in the presence (+) or not (-, NT) of PPARγ antagonist G3335 (G33). (A) At times as indicated (hp.i) RT-PCR and western blotting (B) for IE1, IE2 and β-actin were performed on cell extracts. Experiments shown in (A) and (B) are representative of three independent ones. In (C) virus titers were determined every day (dp.i) in cells supernatants of U373MG cells as plaque forming units per ml (PFU/ml/24h) on MRC5 cells. Histograms are presented as means with standard deviations of 3 independent experiments in which virus titers are means of duplicates.
As expected, expression of early (UL44) and late (UL99) proteins was modified in the same
way by treatment with PPARγ antagonist (Fig S1 in the supplemental material). Furthermore,
production of infectious virus, as assessed by plaque assay every 24 hours, was reduced by a
factor 100 in the presence of PPARγ antagonist (Fig 2aC), demonstrating that in these cells
HCMV used PPARγ activity for its replication. Identical results were obtained with MRC5
fibroblasts (data not shown). Altogether our data demonstrated that PPARγ could regulate
expression of IE2 mRNA and viral replication in a way depending on association with its
ligand.
Figure S1. Expression of early (UL44) and late (UL99) HCMV proteins is impaired by viral
activation of PPARγγγγ.
U373MG cells were infected (3 m.o.i of VHLE) for times as indicated in the presence (G3335) or not (NT) of PPARγ antagonist G3335. At times as indicated (days p.i) western blotting for UL44, UL99 and β-actin were performed on cell extracts. Experiments shown are representative of three independent ones. Time of exposure was longer for UL99 blot than for UL44 one.

76
To further characterize the responsiveness of putative PPREs contained in the MIEP and to
provide direct proof for the role of PPARγ in HCMV replication, Hek-293 cells expressing
RXR but no PPARγ were co-infected with adenoviral vectors encoding PPARγ and with
HCMV. Under ectopic expression of PPARγ� HCMV-infected Hek-293 cells expressed a
higher ratio of IE2 to IE1 mRNA compared with those either non transduced or transduced
with control adenovirus and the relative amount of IE2 mRNA with respect to β-actin was
much higher than in control cells (Fig 2bD), suggesting a role for PPARγ in production of
IE2. Quantification of infectious viruses by plaque assay 48 hours after addition of HCMV
shows that ectopic expression of PPARγ improved virus production by a factor of 100 (Fig
2bE). Under infection with Ad-Vector, production of virus could be due to activation of
transcription factors by adenovirus itself. Moreover, no plaque were observed when HEK
cells were infected with HCMV alone (Mock) despite expression of IE mRNA, as well as
with Ad-PPARγ alone (data not shown), the latter ensuring that the presence of plaque
forming units was due to HCMV replication. Altogether, our data provide evidence that
activation of PPARγ in Hek-293 cells improved their permissivity to HCMV which could in
part result from an increased expression of IE2.
Figure 2b. PPARγγγγ takes part in regulation of IE2 expression and HCMV replication
(D) Hek-293 cells were either uninfected (a, mock) or infected with control adenovirus (b, Ad-Vector) or adenovirus encoding PPARγ (c, Ad-PPARγ) at an m.o.i of 0.1 and expression of PPARγ ( ) was revealed by western blotting. 24 hours after infection with adenovirus, RT-PCR was performed on uninfected (n.i) or HCMV-infected cells (3 m.o.i) for times as indicated (hp.i). (E) Supernatants from Hek-293 cells cultures were harvested after 48 hours of infection with HCMV and viral titers determined (pfu/ml) using standard methods. Identical results have been obtained in three independent experiments.
�
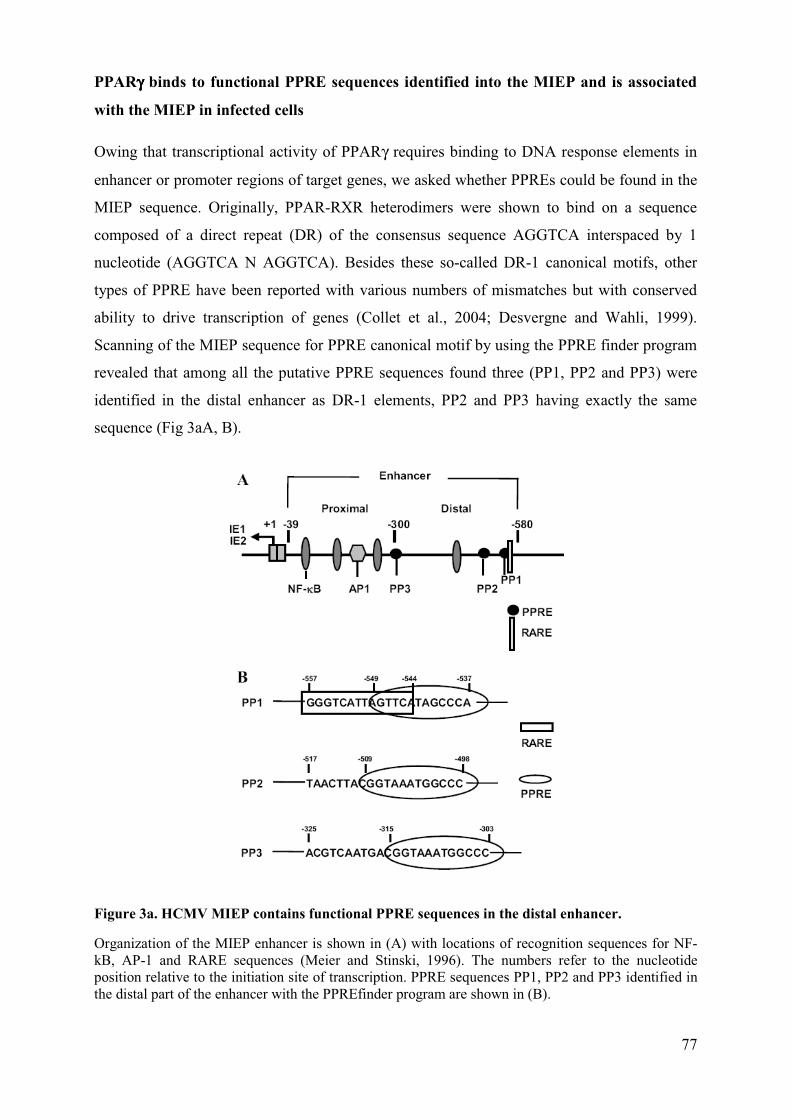
77
PPARγγγγ binds to functional PPRE sequences identified into the MIEP and is associated
with the MIEP in infected cells
Owing that transcriptional activity of PPARγ requires binding to DNA response elements in
enhancer or promoter regions of target genes, we asked whether PPREs could be found in the
MIEP sequence. Originally, PPAR-RXR heterodimers were shown to bind on a sequence
composed of a direct repeat (DR) of the consensus sequence AGGTCA interspaced by 1
nucleotide (AGGTCA N AGGTCA). Besides these so-called DR-1 canonical motifs, other
types of PPRE have been reported with various numbers of mismatches but with conserved
ability to drive transcription of genes (Collet et al., 2004; Desvergne and Wahli, 1999).
Scanning of the MIEP sequence for PPRE canonical motif by using the PPRE finder program
revealed that among all the putative PPRE sequences found three (PP1, PP2 and PP3) were
identified in the distal enhancer as DR-1 elements, PP2 and PP3 having exactly the same
sequence (Fig 3aA, B).
Figure 3a. HCMV MIEP contains functional PPRE sequences in the distal enhancer.
Organization of the MIEP enhancer is shown in (A) with locations of recognition sequences for NF-kB, AP-1 and RARE sequences (Meier and Stinski, 1996). The numbers refer to the nucleotide position relative to the initiation site of transcription. PPRE sequences PP1, PP2 and PP3 identified in the distal part of the enhancer with the PPREfinder program are shown in (B).
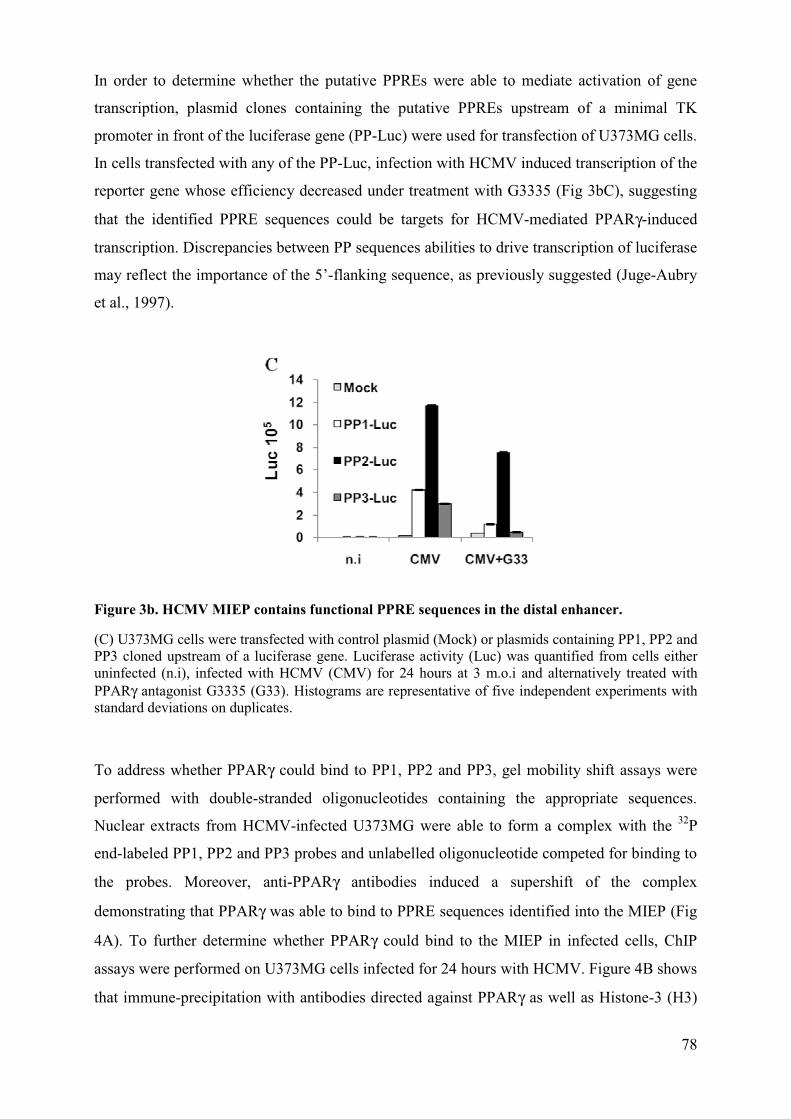
78
In order to determine whether the putative PPREs were able to mediate activation of gene
transcription, plasmid clones containing the putative PPREs upstream of a minimal TK
promoter in front of the luciferase gene (PP-Luc) were used for transfection of U373MG cells.
In cells transfected with any of the PP-Luc, infection with HCMV induced transcription of the
reporter gene whose efficiency decreased under treatment with G3335 (Fig 3bC), suggesting
that the identified PPRE sequences could be targets for HCMV-mediated PPARγ-induced
transcription. Discrepancies between PP sequences abilities to drive transcription of luciferase
may reflect the importance of the 5’-flanking sequence, as previously suggested (Juge-Aubry
et al., 1997).
Figure 3b. HCMV MIEP contains functional PPRE sequences in the distal enhancer.
(C) U373MG cells were transfected with control plasmid (Mock) or plasmids containing PP1, PP2 and PP3 cloned upstream of a luciferase gene. Luciferase activity (Luc) was quantified from cells either uninfected (n.i), infected with HCMV (CMV) for 24 hours at 3 m.o.i and alternatively treated with PPARγ antagonist G3335 (G33). Histograms are representative of five independent experiments with standard deviations on duplicates.
To address whether PPARγ could bind to PP1, PP2 and PP3, gel mobility shift assays were
performed with double-stranded oligonucleotides containing the appropriate sequences.
Nuclear extracts from HCMV-infected U373MG were able to form a complex with the 32P
end-labeled PP1, PP2 and PP3 probes and unlabelled oligonucleotide competed for binding to
the probes. Moreover, anti-PPARγ antibodies induced a supershift of the complex
demonstrating that PPARγ was able to bind to PPRE sequences identified into the MIEP (Fig
4A). To further determine whether PPARγ could bind to the MIEP in infected cells, ChIP
assays were performed on U373MG cells infected for 24 hours with HCMV. Figure 4B shows
that immune-precipitation with antibodies directed against PPARγ as well as Histone-3 (H3)
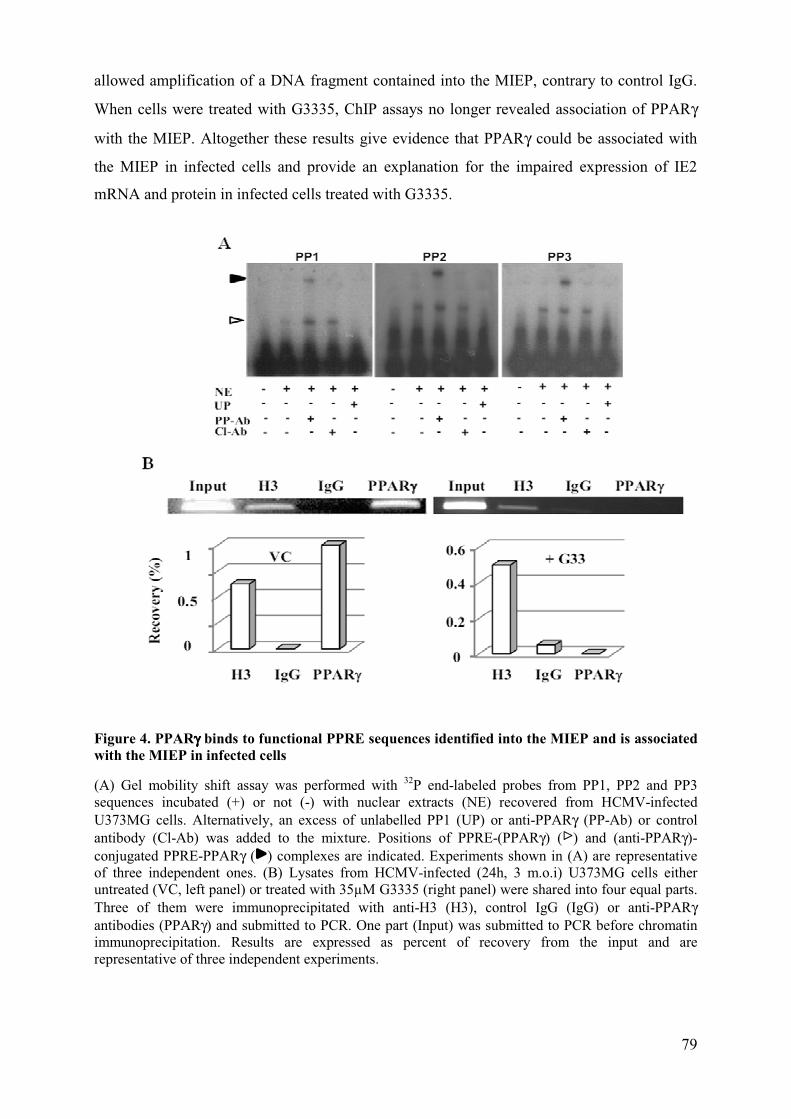
79
allowed amplification of a DNA fragment contained into the MIEP, contrary to control IgG.
When cells were treated with G3335, ChIP assays no longer revealed association of PPARγ
with the MIEP. Altogether these results give evidence that PPARγ could be associated with
the MIEP in infected cells and provide an explanation for the impaired expression of IE2
mRNA and protein in infected cells treated with G3335.
Figure 4. PPARγγγγ binds to functional PPRE sequences identified into the MIEP and is associated
with the MIEP in infected cells
(A) Gel mobility shift assay was performed with 32P end-labeled probes from PP1, PP2 and PP3 sequences incubated (+) or not (-) with nuclear extracts (NE) recovered from HCMV-infected U373MG cells. Alternatively, an excess of unlabelled PP1 (UP) or anti-PPARγ (PP-Ab) or control antibody (Cl-Ab) was added to the mixture. Positions of PPRE-(PPARγ) ( ) and (anti-PPARγ)-conjugated PPRE-PPARγ ( ) complexes are indicated. Experiments shown in (A) are representative of three independent ones. (B) Lysates from HCMV-infected (24h, 3 m.o.i) U373MG cells either untreated (VC, left panel) or treated with 35µM G3335 (right panel) were shared into four equal parts. Three of them were immunoprecipitated with anti-H3 (H3), control IgG (IgG) or anti-PPARγantibodies (PPARγ) and submitted to PCR. One part (Input) was submitted to PCR before chromatin immunoprecipitation. Results are expressed as percent of recovery from the input and are representative of three independent experiments.

80
HCMV infection impairs human trophoblast migration and invasiveness through a
PPARγγγγ-dependent pathway.
During the first trimester of pregnancy the cytotrophoblasts (extravillous cytotrophoblasts,
EVCT) proliferate to form multilayered columns of cells that rapidly migrate out of the
chorionic villi and invade the decidua and the upper third of the myometrium as illustrated in
Figure 5aA. At the implantion site, EVCT are characterized by CK07 and nuclear PPARγ
expression all along their differentiating pathway (Fig 5aB, C).
Figure.5a. PPARγγγγ is expressed in trophoblasts in situ, in primary (EVCT) and in transformed
(HIPEC) cytotrophoblast.
(A) Schematic representation of a chorionic villous at the implantation site. EVCT (in red) invade the decidua up to the first third of the myometrium and the uterine arteries (vct, villous cytotrophoblast; st, syncytiotrophoblast; evct, extravillous cytotrophoblast; sc, stromal core; is, intervillous space; ua, uterine artery; d, decidua). (B) Immunohistochemical staining for CK07, a specific trophoblast marker of invasive EVCT and (C) for PPARγ in placental sections at the implantation site (16-week of pregnancy).
To study human trophoblast invasion and migration in vitro, we used either primary cultures
of EVCT purified from first trimester placentas (Tarrade et al., 2001b) or a cell line (HIPEC)
established from primary cultures which share the phenotypic and functional characteristics of
primary EVCT (Pavan et al., 2003). In the placenta, trophoblasts constitutively express
PPARγ in their nucleus as shown in situ (Fig 5aA) and in vitro in primary (Fig 5bD) and
transformed EVCT (Fig 5bE).
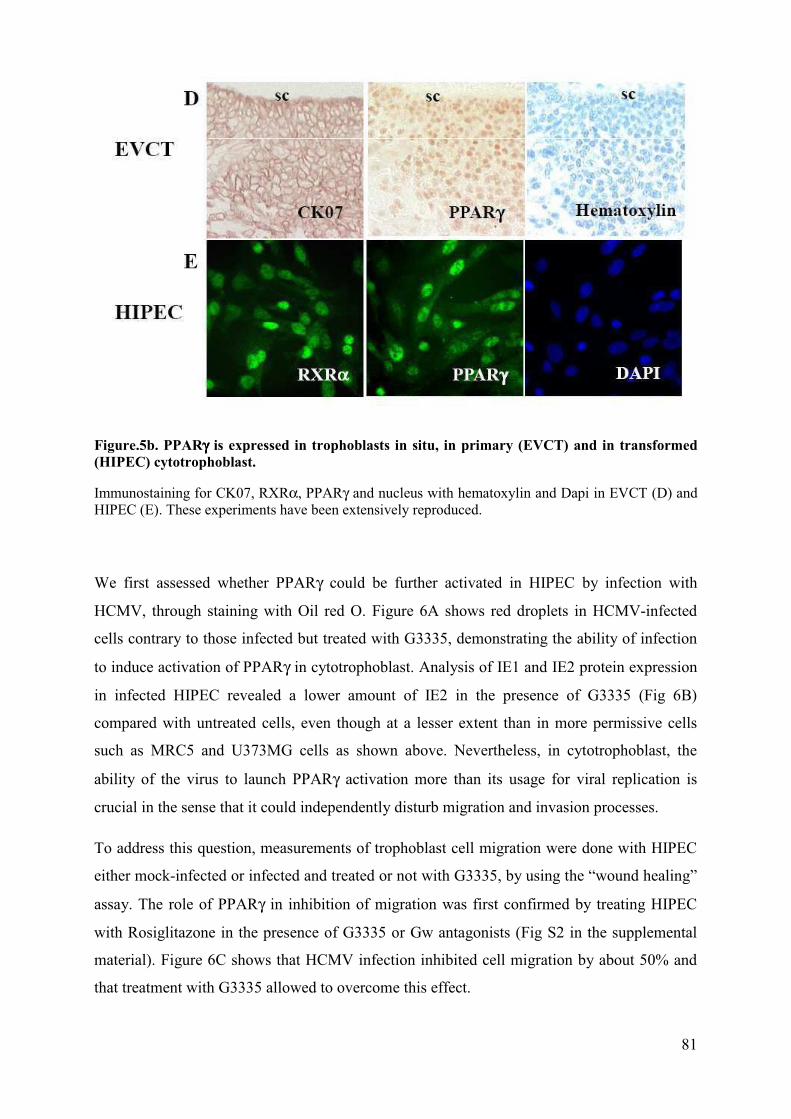
81
Figure.5b. PPARγγγγ is expressed in trophoblasts in situ, in primary (EVCT) and in transformed
(HIPEC) cytotrophoblast.
Immunostaining for CK07, RXRα, PPARγ and nucleus with hematoxylin and Dapi in EVCT (D) and HIPEC (E). These experiments have been extensively reproduced.
We first assessed whether PPARγ could be further activated in HIPEC by infection with
HCMV, through staining with Oil red O. Figure 6A shows red droplets in HCMV-infected
cells contrary to those infected but treated with G3335, demonstrating the ability of infection
to induce activation of PPARγ in cytotrophoblast. Analysis of IE1 and IE2 protein expression
in infected HIPEC revealed a lower amount of IE2 in the presence of G3335 (Fig 6B)
compared with untreated cells, even though at a lesser extent than in more permissive cells
such as MRC5 and U373MG cells as shown above. Nevertheless, in cytotrophoblast, the
ability of the virus to launch PPARγ activation more than its usage for viral replication is
crucial in the sense that it could independently disturb migration and invasion processes.
To address this question, measurements of trophoblast cell migration were done with HIPEC
either mock-infected or infected and treated or not with G3335, by using the “wound healing”
assay. The role of PPARγ in inhibition of migration was first confirmed by treating HIPEC
with Rosiglitazone in the presence of G3335 or Gw antagonists (Fig S2 in the supplemental
material). Figure 6C shows that HCMV infection inhibited cell migration by about 50% and
that treatment with G3335 allowed to overcome this effect.
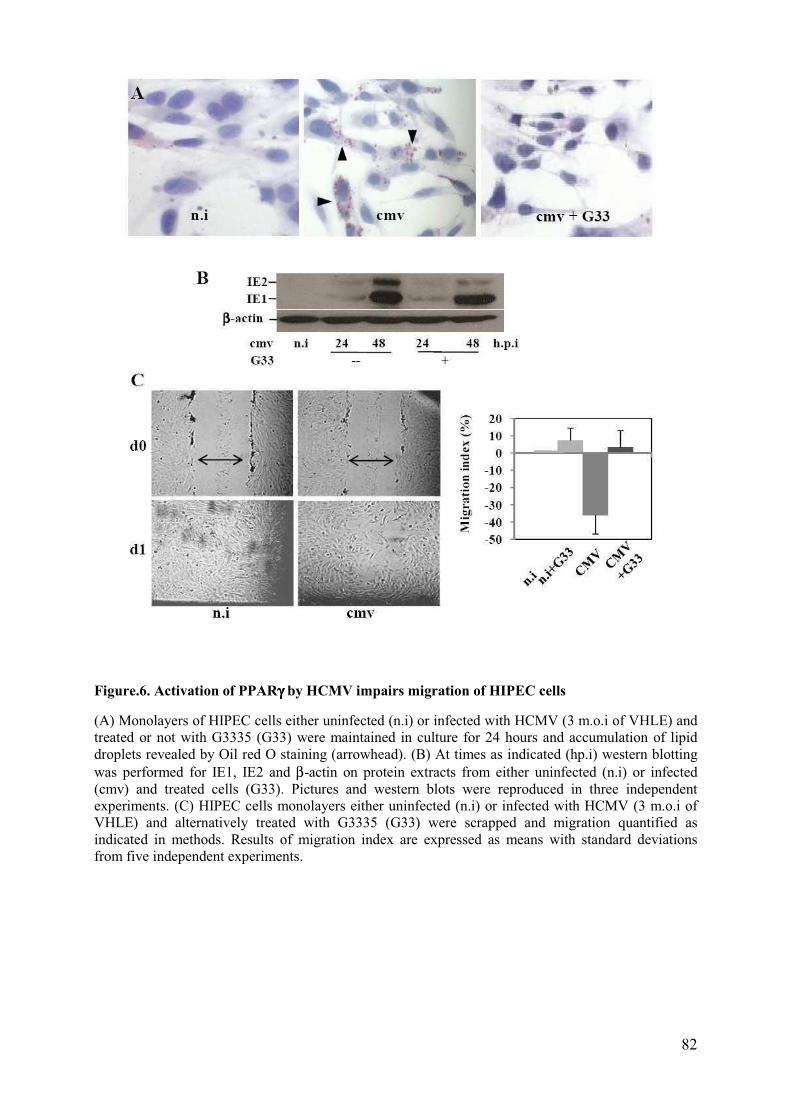
82
Figure.6. Activation of PPARγγγγ by HCMV impairs migration of HIPEC cells
(A) Monolayers of HIPEC cells either uninfected (n.i) or infected with HCMV (3 m.o.i of VHLE) and treated or not with G3335 (G33) were maintained in culture for 24 hours and accumulation of lipid droplets revealed by Oil red O staining (arrowhead). (B) At times as indicated (hp.i) western blotting was performed for IE1, IE2 and β-actin on protein extracts from either uninfected (n.i) or infected (cmv) and treated cells (G33). Pictures and western blots were reproduced in three independent experiments. (C) HIPEC cells monolayers either uninfected (n.i) or infected with HCMV (3 m.o.i of VHLE) and alternatively treated with G3335 (G33) were scrapped and migration quantified as indicated in methods. Results of migration index are expressed as means with standard deviations from five independent experiments.
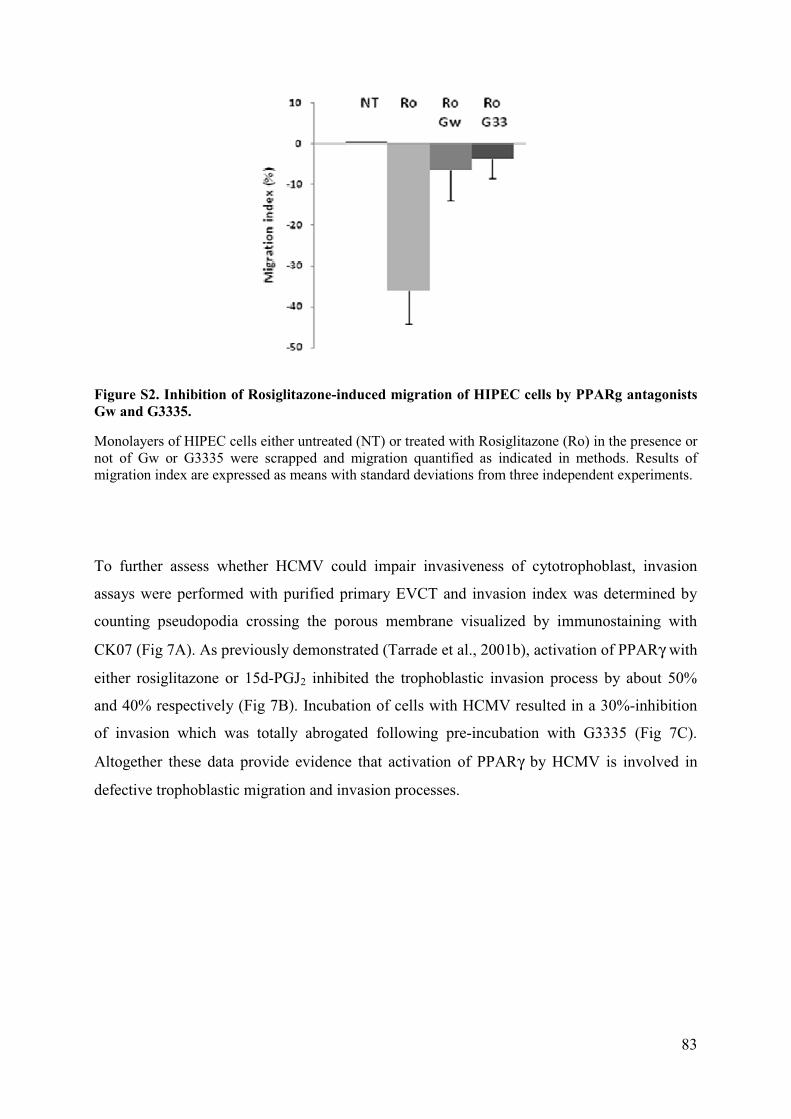
83
Figure S2. Inhibition of Rosiglitazone-induced migration of HIPEC cells by PPARg antagonists
Gw and G3335.
Monolayers of HIPEC cells either untreated (NT) or treated with Rosiglitazone (Ro) in the presence or not of Gw or G3335 were scrapped and migration quantified as indicated in methods. Results of migration index are expressed as means with standard deviations from three independent experiments.
To further assess whether HCMV could impair invasiveness of cytotrophoblast, invasion
assays were performed with purified primary EVCT and invasion index was determined by
counting pseudopodia crossing the porous membrane visualized by immunostaining with
CK07 (Fig 7A). As previously demonstrated (Tarrade et al., 2001b), activation of PPARγ with
either rosiglitazone or 15d-PGJ2 inhibited the trophoblastic invasion process by about 50%
and 40% respectively (Fig 7B). Incubation of cells with HCMV resulted in a 30%-inhibition
of invasion which was totally abrogated following pre-incubation with G3335 (Fig 7C).
Altogether these data provide evidence that activation of PPARγ by HCMV is involved in
defective trophoblastic migration and invasion processes.
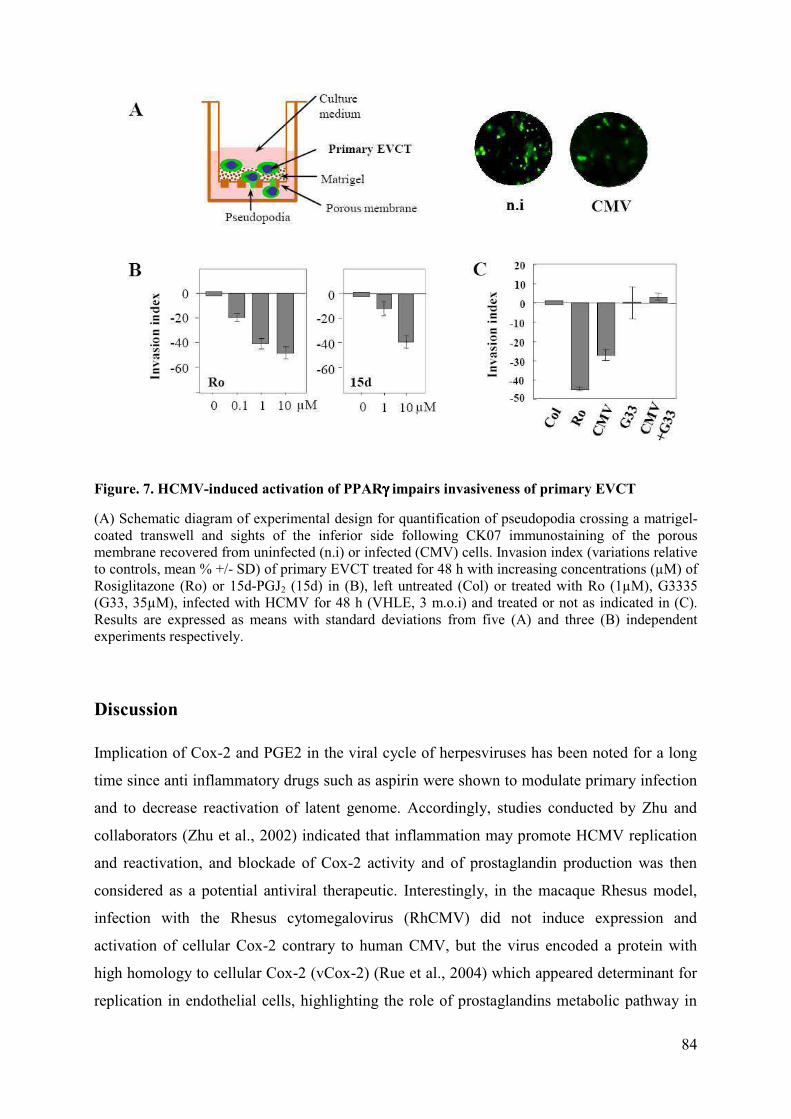
84
Figure. 7. HCMV-induced activation of PPARγγγγ impairs invasiveness of primary EVCT
(A) Schematic diagram of experimental design for quantification of pseudopodia crossing a matrigel-coated transwell and sights of the inferior side following CK07 immunostaining of the porous membrane recovered from uninfected (n.i) or infected (CMV) cells. Invasion index (variations relative to controls, mean % +/- SD) of primary EVCT treated for 48 h with increasing concentrations (µM) of Rosiglitazone (Ro) or 15d-PGJ2 (15d) in (B), left untreated (Col) or treated with Ro (1µM), G3335 (G33, 35µM), infected with HCMV for 48 h (VHLE, 3 m.o.i) and treated or not as indicated in (C). Results are expressed as means with standard deviations from five (A) and three (B) independent experiments respectively.
�
Discussion
Implication of Cox-2 and PGE2 in the viral cycle of herpesviruses has been noted for a long
time since anti inflammatory drugs such as aspirin were shown to modulate primary infection
and to decrease reactivation of latent genome. Accordingly, studies conducted by Zhu and
collaborators (Zhu et al., 2002) indicated that inflammation may promote HCMV replication
and reactivation, and blockade of Cox-2 activity and of prostaglandin production was then
considered as a potential antiviral therapeutic. Interestingly, in the macaque Rhesus model,
infection with the Rhesus cytomegalovirus (RhCMV) did not induce expression and
activation of cellular Cox-2 contrary to human CMV, but the virus encoded a protein with
high homology to cellular Cox-2 (vCox-2) (Rue et al., 2004) which appeared determinant for
replication in endothelial cells, highlighting the role of prostaglandins metabolic pathway in

85
replication of cytomegaloviruses. HCMV replication and reactivation require transcription of
the IE genes UL123 and UL122 encoding IE1 and IE2 proteins respectively, two major
products resulting from a differential splicing. Production of IE2 but not IE1 was shown to be
crucial for productive replication and transcription of IE2 but not IE1 was significantly
affected by Cox-2 inhibitors (Zhu et al., 2002). A role for cAMP response elements contained
into the MIEP was suggested (Mocarski, 2002) with respect to the known ability of
prostaglandins to increase cAMP, but for the moment no direct proof has been obtained to
support this hypothesis. Our results demonstrate for the first time a role for PPARγ in
production of IE2 through binding to PPRE sequences contained into the MIEP and
experiments demonstrating a huge decrease in viral production in the presence of PPARγ
antagonist confirmed the key role of PPARγ in viral replication. Upstream of PPARγ
activation, molecular and cellular events underlying activation of Cox-2 are known to involve
the enzymatic activity of the cellular phospholipase A2 (c-PLA2). It is known for a long time
that c-PLA2 activity could be induced by HCMV infection (AbuBakar et al., 1990), but more
interestingly, a connection between infection and PPARγ activation could be supported by a
study demonstrating that HCMV particles carry a cell-derived PLA2 boarded during the
assembly process, which plays a major role in infectivity of MRC5 fibroblasts (Allal et al.,
2004). PLA2 activity is responsible for the production of arachidonic acid, a substrate for both
Cox-2 and lipoxygenase activities and we can assume that it’s a reason why c-PLA2-specific
inhibitors dramatically impaired virus production (Allal et al., 2004).
Nevertheless, given that cPLA2 is involved in many complex processes including
embryogenesis, inflammation and reproduction, the potential side effects of inhibitors,
provided that highly selective ones are available, might discredit their potential therapeutic
indications. With respect to these observations and to those reported in the present paper, we
can imagine a scenario where virus entry could initiate the release of arachidonic acid through
boarded c-PLA2 activation, a starting point of a process involving Cox-2- as well as
lipoxygenases-mediated production of PPARγ ligands, the latter being considered as a final
requisite for PPARγ-mediated transcription in infected cells. Nevertheless, our data
demonstrating the inability of UV-irradiated virus to induce activation of PPARγ raises the
question of a role for neosynthesized viral proteins in the activation process but also raise the
question whether irradiation could alter structure and function of virions constituents
including boarded cPLA2.

86
Surprisingly, and in agreement with previous data using Cox-2 inhibitors (Zhu et al., 2002),
PPARγ inhibition differentially impaired the expression of IE1 and IE2 mRNA, suggesting a
role for PPARγ in favoring IE2 mRNA production. This was supported by our experiments
demonstrating a huge increase in expression of IE2 but not IE1 mRNA under ectopic
expression of PPARγ in HCMV-infected Hek-293 cells. Since IE1 and IE2 transcripts result
from differential splicing events which are temporally regulated after infection, IE1 being
produced before IE2, we can assume that binding of PPARγ to the MIEP could be coordinated
for instance by temporal chromatin remodeling as reported to explain IE2-mediated auto-
repression of the MIEP at late times of infection (Reeves et al., 2006). Interestingly, several
studies showing that transcription and RNA processing could be spatially and temporally
coupled and that the transcriptional PPARγ co-activator 1 (PGC-1) could play a major role in
pre-mRNA splicing (Kornblihtt, 2005), we can speculate that binding of PPARγ to the MIEP
could recruit PGC-1 which in turn could control exon4 skipping to give rise to IE2 mRNA,
some issues which remain to be extensively explored.
PPARγ was shown to play a major role in human cytotrophoblast differentiation and function.
Implantation of the conceptus involves early invasion of the uterine epithelium and of the
underlying stroma by extravillous trophoblastic cells, and we demonstrated that PPARγ
activation repressed this trophoblastic invasion process (Tarrade et al., 2001b). Since
migration/invasion is essential for implantation and remodeling of uteroplacental
vascularisation necessary for fetal growth, we can speculate that through activation of PPARγ,
infection of the placenta by HCMV, a phenomenon known to precede infection of the fetus,
could take part in miscarriages and low-birthweight babies. In addition to 15d-PGJ2, a
metabolite of cyclooxygenase, the metabolite of 15-lipoxygenase (Lox) 15-HETE was also
shown to inhibit human trophoblastic invasion by about 50 % (Fournier et al., 2008; Pavan et
al., 2004). Whether HCMV infection might lead to production of 15-HETE by activating Lox
in cytotrophoblast is a relevant question since incubation of EVCT with 3H arachidonic acid
(AA) followed by quantitative HPLC analysis of 3H AA-metabolites indicated that 15-HETE
was the main metabolite produced by EVCT (personal data). In the course of the invasion
process, degradation of the basal membrane and of the extracellular matrix of the uterine
stroma is ensured by metallo-proteases (MMP) such as MMP-9, whose secretion is elevated
during the early steps of gestation (Librach et al., 1994). Active MMP-9 is required for
invasion to proceed and, for instance, we can assume that HCMV-induced activation of
PPARγ could lead to a decrease in expression or activity of MMP-9 thus impairing placental

87
differentiation. Remarkably, PPARγ activation was shown to induce trans-repression of the
MMP-9 protein (Marx et al., 1998) whose transcription depends on NF-κB activation, and
HCMV infection is known to decrease MMP activity and to disregulate the cell-cell and/or
cell-matrix interactions of infected cytotrophoblasts (Yamamoto-Tabata et al., 2004).
Experiments are in progress to address this issue. In utero, viral infections have been
associated with an adverse pregnancy outcome and may have a causative role in the
unexplained cause of congenital infections in developed countries and are considered as one
of the most common pathogens implicated in fetal loss cases. Cytomegalovirus accounts for
miscarriages and intrauterine growth retardation and congenital anomalies and our study
provides the first evidence that activation of PPARγ by infection could take part in these
pathological processes through abnormal placentation related to impairment of the trophoblast
migration/invasion mechanisms. Overall, understanding of mechanisms underlying activation
of PPARγ and characterization of its target genes following infection of cytotrophoblast by
HCMV should help to propose new prognostic and therapeutic approaches to prevent
miscarriage and risks of sequelae in new-borns.
Acknowledgments
We thank Claudie Offer and Hélène Brun for sequencing, Michel Baron for helpful advices in
performing gel mobility shift assays, John Sinclair (University of Cambridge, UK) and
Stéphane Chavanas in ChIP assays, Vassilis Tsatsaris for immunochemistry of placental
sections, Ananda Mookerjee and Charlotte Casper for critical reading of the manuscript.
This work was supported by institutional grants from INSERM.
�
�
�
�
�
�
�
�

88
Conclusions
Dans ce travail, nous avons démontré pour la première fois que le HCMV active le récepteur
nucléaire PPARγ. Nous avons par la suite découvert que le virus utilisait ce facteur de
transcription pour sa propre réplication et plus particulièrement pour la transcription de la
protéine très précoce IE2, indispensable à l’accomplissement du cycle viral.
Les bases moléculaires du rôle de PPARγ sont fondées sur l’identification pour la première
fois de 3 séquences PPRE fonctionnelles dans le MIEP, à l’aide du logiciel « PPRE Finder »,
d’expériences de gels retards, de ChIP et de tests fonctionnels au moyen de vecteurs
plasmidiques contenant le gène de la luciférase sous contrôle d’un promoteur minimal associé
à ces séquences.
Il est intéressant de noter que seule l’expression d’IE2 est contrôlée par PPARγ, l’expression
d’IE1 restant inchangée lorsque PPARγ est inhibé. Cela ouvre donc des perspectives de
recherche sur la régulation d’expression d’IE1 et IE2 par ce facteur de transcription, et plus
particulièrement son rôle, s’il en est, dans l’épissage alternatif du gène IE du HCMV. Pour
cela, nous proposons l’hypothèse du rôle de la protéine PGC-1 (PPAR Gamma Co activator-
1), protéine interagissant avec PPARγ décrite dans d’autres modèles comme permettant le
saut d’exon (« exon skipping ») lors de l’épissage de l’ARN (Kornblihtt, 2005).
PPARγ jouant un rôle essentiel dans la placentation, et plus particulièrement dans l’invasion
du cytotrophoblaste, nous nous sommes alors intéressés aux conséquences de son activation
par le HCMV sur la migration et l’invasion de cellules cytotrophoblastiques. Pour cela nous
avons développé une collaboration avec le docteur Thierry Fournier de l’unité Inserm U767 à
Paris, spécialiste de l’étude des fonctions de PPARγ dans le placenta. Cette collaboration a
permis de mettre en évidence une perturbation par le virus de la migration et de l’invasion
cytotrophoblastique totalement dépendante de l’activation de PPARγ.
Ainsi ce travail a permis de démontrer pour la première fois l’utilisation directe du facteur de
transcription PPARγ dans la réplication du HCMV et plus particulièrement dans la
transcription de la protéine IE2.
Cependant, une démonstration de l’utilisation des PPARα et γ pour la transcription et la
réplication du virus de l’Hépatite B avait déjà été publiée, avec des mutations des séquences
PPRE présentes sur le génome viral, appelées ici NRRE pour « Nuclear receptor response
element » et la sur-expression des facteurs de transcription pour observer la modification des

89
transcrits du virus. Aucune fixation directe des récepteurs nucléaires sur l’ADN viral n’avait
cependant été relevé (Yu and Mertz, 2001).
En dehors de l’aspect concernant la réplication virale, ce travail nous a permis de démontrer
en parallèle les effets néfastes de l’activation d’un récepteur nucléaire par un virus sur un
processus physiologique comme ici la migration et l’invasion placentaire. Ces travaux
suggèrent donc de nouvelles pistes pour la compréhension des pathologies de la grossesse lors
d’une infection par le HCMV. Ces résultats nous ont alors amené à développer la deuxième
partie de ce travail de recherche, consistant à étudier les mécanismes impliqués dans la
perturbation de la migration cytotrophoblastique, dépendant de PPARγ et régulés lors de
l’infection par le HCMV.

90
DEUXIEME PARTIE :
Etude des mécanismes dépendant de
l’activation de PPARγγγγ par le HCMV perturbant
la migration et l’invasion du cytotrophoblaste.

91
Introduction
D’après nos premières observations, l’infection du cytotrophoblaste par le HCMV va
conduire à l’activation du récepteur nucléaire PPARγ, perturbant alors l’invasion et la
migration des cellules. La mobilité du cytotrophoblaste ainsi que ses capacités invasives étant
primordiales dans le processus de placentation, nous nous sommes intéressés dans ce travail
aux mécanismes impliquées dans la perturbation de ces capacités, liés à l’activation de PPARγ
par le HCMV. A l’heure actuelle, les mécanismes impliqués sont principalement la sécrétion
d’IL-10 cellulaire et viral (cmvIL-10, UL111a) conduisant à une diminution de la quantité de
MMP-9 active dans le milieu extracellulaire (Yamamoto-Tabata et al., 2004). Certaines
études ont démontré que le HCMV pouvait également diminuer la sécrétion de MMP9 et
augmenter celle de son inhibiteur TIMP-1 dans les macrophages (Straat et al., 2009). Il a
également été démontré un rôle de PPARγ dans le contrôle de la sécrétion de TIMP-1 et de la
MMP-9 conduisant à un défaut de migration dans les monocytes (Kintscher et al., 2000), et
très récemment un rôle dans la production d’IL-10 cellulaire (Are et al., 2008).
Ceci nous a alors amené à étudier la régulation des MMP et de leurs inhibiteurs TIMPs dans
les cellules infectées.
Parallèlement à cela, Thierry Fournier avait mis en évidence une régulation de l’expression de
la PAPP-A par PPARγ dans les cellules cytotrophoblastiques, proposant alors une cible
directe de ce facteur de transcription, impliquée dans la migration et l’invasion placentaire
(Handschuh et al., 2006). Nous nous sommes alors intéressés au rôle de la PPAP-A dans la
perturbation de la migration placentaire liée à l’infection par le HCMV.
Toutes ces protéines cibles connues à ce jour étant des protéases, et la possibilité de collaborer
avec Nathalie Vergnolle spécialiste des récepteurs PAR dans notre centre de recherche, et
dont le rôle dans la placentation est avéré, nous a conduit à étudier les effets de l’infection par
le HCMV en rapport avec le rôle de ces récepteurs PAR dans la migration et l’invasion
placentaire.
Dans le chapitre suivant seront présentés les résultats préliminaires obtenus.

92
Matériels et méthodes
Seules les nouvelles techniques spécifiques de ce chapitre seront décrites.
Antibody Array :
Le principe de l’antibody array (Raybiotech) est assez proche du test ELISA. Une membrane
recouverte avec plusieurs anticorps différents est incubée avec le surnageant à tester. Puis on
incube cette même membrane avec les mêmes anticorps que ceux fixés à la membrane, mais
couplés ici à la biotine. Après cette réaction, la membrane est alors incubée avec une solution
de streptavidine-HRP qui viendra se fixer aux biotines présentes sur les anticorps. A l’aide
d’un kit de détection par luminescence chimique réagissant avec la HRP, les cytokines
présentes dans le surnageant sont alors révélées. Le principe est présentée en figure 27.
Figure 27 : principe du test par Antibody Array.
Zymogramme :
Une solution protéique provenant d’un extrait cellulaire ou d’un surnageant est déposée sur un
gel d’agarose contenant de la gélatine. Les protéines sont alors séparées selon leur poids
moléculaire par électrophorèse. Après migration, les activités enzymatiques des MMP-2 et -9
(gélatinases) sont favorisées par incubation dans des tampons renaturant puis de
développement. Les gels sont ensuite colorés avec du bleu de Coomassie, colorant le

93
collagène et mettant alors en évidence l’activité des collagénases, là ou le gel se trouve être
décoloré. Les gels utilisés dans cette étude sont des gels Novex d’Invitrogen, avec leurs
tampons de migration, renaturant et de développement associés.
Réactifs PAR :
Les anticorps spécifiques des PAR-1 et-4 proviennent de chez AbCAM , PAR-2 (Santa Cruz).
Les peptides agonistes des PAR (TFLLR pour PAR-1, SLIGRL pour PAR-2 et AYP pour
PAR-4) ainsi que leurs peptides reverses (RLLFT, LRGILS et PYA) proviennent du
laboratoire du Docteur Nathalie Vergnolle et sont utilisés à la concentration de 100 µM. La
thrombine provient de chez Sigma-Aldrich et est utilisée à raison de 1 à 2 Unités par puits de
cellules.
Flux Calciques :
Dans cette expérience, nous observons l’activation des récepteurs PAR grâce à la
quantification de l’internalisation du calcium dans les cellules après leur activation. Les
cellules sont traitées avec une solution de Fluo-3 contenant 4 µM de Fluo-3-AcetoxyMethyl
(AM) (Invitrogen), 20 % de pluronic F127 (Sigma) dilué dans du HBSS-BSA-probenecid
Hepes (Hank’s balanced Salt Solution à 0.1% d’albumine de sérum bovin, 2.5 mM
probenecid, 25 mM Hepes à pH 7.2) puis incubées 30 minutes à température ambiante dans le
noir. Les cellules sont ensuite lavées 2 fois avec du HBSS-BSA-probenecid Hepes et incubées
dans cette solution 30 min à 37°C. La fluorescence est ensuite mesurée à 530 nm sur un
lecteur de plaque (Novostar, BMG Labtech) pendant 250 secondes. A 5 sec, l’agoniste à tester
est injecté, à 90 sec la solution Fmin est injectée puis à 200 sec la solution Fmax. La solution
Fmin 5X est composée de 5 µM d’ionomycine (Calbiochem), 50 µM de thapsigargine
(Calbiochem) et 10 nM d’EGTA (Ethylene Glycol Tetraacetic Acid). La solution Fmax est
composée de 125 mM de CaCl2. Les deux solutions sont diluées dans du HBSS-BSA-
probenecid Hepes. Les données sont collectées et analysées avec le programme Novostar et la
mobilisation de calcium est calculée selon la formule :
[Ca2+]Free intracellular nM = Kd Fluo-3 * ((F-Fmin)/(Fmax-F))

94
Résultats
MMP et TIMP
Dans un premier temps, s’appuyant sur l’observation que le HCMV perturbe la production de
MMP-9 (Yamamoto-Tabata et al., 2004) et que l’expression de la MMP-9 pouvait être sous le
contrôle de PPARγ (Are et al., 2008), nous avons décidé d’analyser la régulation des
différentes MMPs et de leurs inhibiteurs spécifiques (les TIMPs) dans notre lignée de
cytotrophoblaste extravilleux (HIPEC pour Human Invasive and Proliferative Extravillous
Cytotrophoblast) (Pavan et al., 2003). Pour cela, nous avons effectué des expériences
d’Antibody Array afin de regarder la variation des différentes MMP et des différents TIMP
dans les cellules infectées par rapport aux cellules non infectées. La figure 28 montre les
résultats d’un des 2 « antibody arrays » effectués sur des HIPEC infectées ou non durant 24 h
par la souche clinique VHLE. Comme on peut le voir, l’infection par le HCMV ne va
augmenter que l’expression des MMP-1 et -3, mais va également augmenter de manière
importante la sécrétion des inhibiteurs TIMP-1 et TIMP-2.
Figure 28 : Variation de l’expression des MMP et TIMP lors de l’infection par le HCMV. (A) Schéma représentant la disposition des anticorps spécifiques sur la membrane dont les contrôles positifs (POS) et négatifs (NEG). En rouge sont indiquées les protéines dont l’expression varie avec l’infection. (B) et (C) Révélation de la membrane avec plus ou moins de temps d’exposition.
Cette augmentation des TIMP-1 et -2 pourrait expliquer la diminution de MMP-9 active dans
les surnageants de cellules infectées (Yamamoto-Tabata et al., 2004).

95
Parallèlement à cela, nous avons analysé l’activité gélatinase dans les surnageants d’HIPEC
infectées ou non avec le HCMV ainsi que traitées ou non avec l’inhibiteur non réversible de
PPARγ (Gw9662) grâce à des expériences de Zymogramme. La figure 29 nous montre qu’une
gélatinase est inhibée dans les cellules infectées, mais que cette inhibition est levée lorsque
PPARγ est lui-même inhibé. Malheureusement ici, nous n’avons pas encore identifié la MMP
concernée. Mais ceci nous démontre bien que l’activation de PPARγ par le HCMV peut
diminuer l’expression de certaines MMP et augmenter l’expression de leurs inhibiteurs.
Figure 29 : L’infection HCMV diminue l’activité enzymatique de certaines MMP de manière
dépendante de PPARγγγγ.
Zymogramme de surnageants de cellules HIPEC infectées pendant 24 h (CMV) ou non (Ni) par le VHLE (m.o.i. 3) traitées ou non avec l’inhibiteur spécifique de PPARγ (Gw) 30 minutes avant l’infection.
Cependant, ces résultats ne sont encore qu’à un stade préliminaire, et le rôle de PPARγ dans la
régulation de ces protéines au cours de l’infection reste à démontrer en approfondissant ces
expériences et en les complétant notamment avec des expériences de RT-PCR ou de Western-
blot afin de regarder directement la régulation de ces protéines.
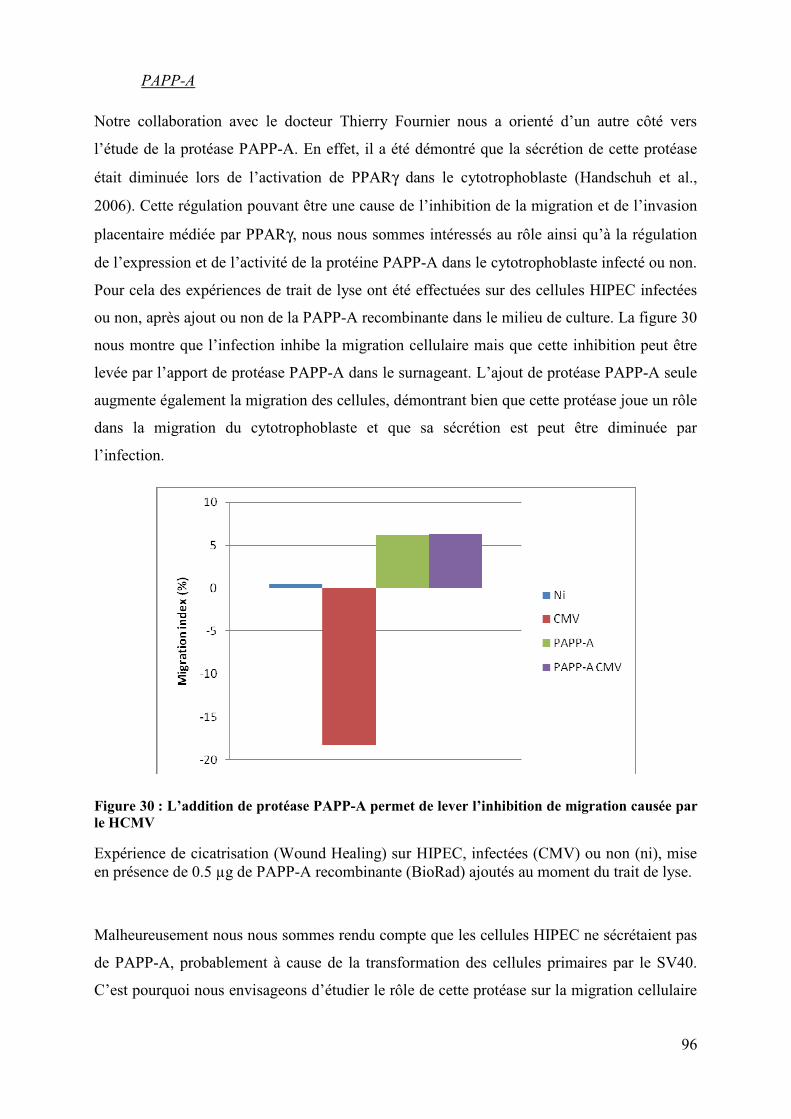
96
PAPP-A
Notre collaboration avec le docteur Thierry Fournier nous a orienté d’un autre côté vers
l’étude de la protéase PAPP-A. En effet, il a été démontré que la sécrétion de cette protéase
était diminuée lors de l’activation de PPARγ dans le cytotrophoblaste (Handschuh et al.,
2006). Cette régulation pouvant être une cause de l’inhibition de la migration et de l’invasion
placentaire médiée par PPARγ, nous nous sommes intéressés au rôle ainsi qu’à la régulation
de l’expression et de l’activité de la protéine PAPP-A dans le cytotrophoblaste infecté ou non.
Pour cela des expériences de trait de lyse ont été effectuées sur des cellules HIPEC infectées
ou non, après ajout ou non de la PAPP-A recombinante dans le milieu de culture. La figure 30
nous montre que l’infection inhibe la migration cellulaire mais que cette inhibition peut être
levée par l’apport de protéase PAPP-A dans le surnageant. L’ajout de protéase PAPP-A seule
augmente également la migration des cellules, démontrant bien que cette protéase joue un rôle
dans la migration du cytotrophoblaste et que sa sécrétion est peut être diminuée par
l’infection.
Figure 30 : L’addition de protéase PAPP-A permet de lever l’inhibition de migration causée par
le HCMV
Expérience de cicatrisation (Wound Healing) sur HIPEC, infectées (CMV) ou non (ni), mise en présence de 0.5 µg de PAPP-A recombinante (BioRad) ajoutés au moment du trait de lyse.
Malheureusement nous nous sommes rendu compte que les cellules HIPEC ne sécrétaient pas
de PAPP-A, probablement à cause de la transformation des cellules primaires par le SV40.
C’est pourquoi nous envisageons d’étudier le rôle de cette protéase sur la migration cellulaire
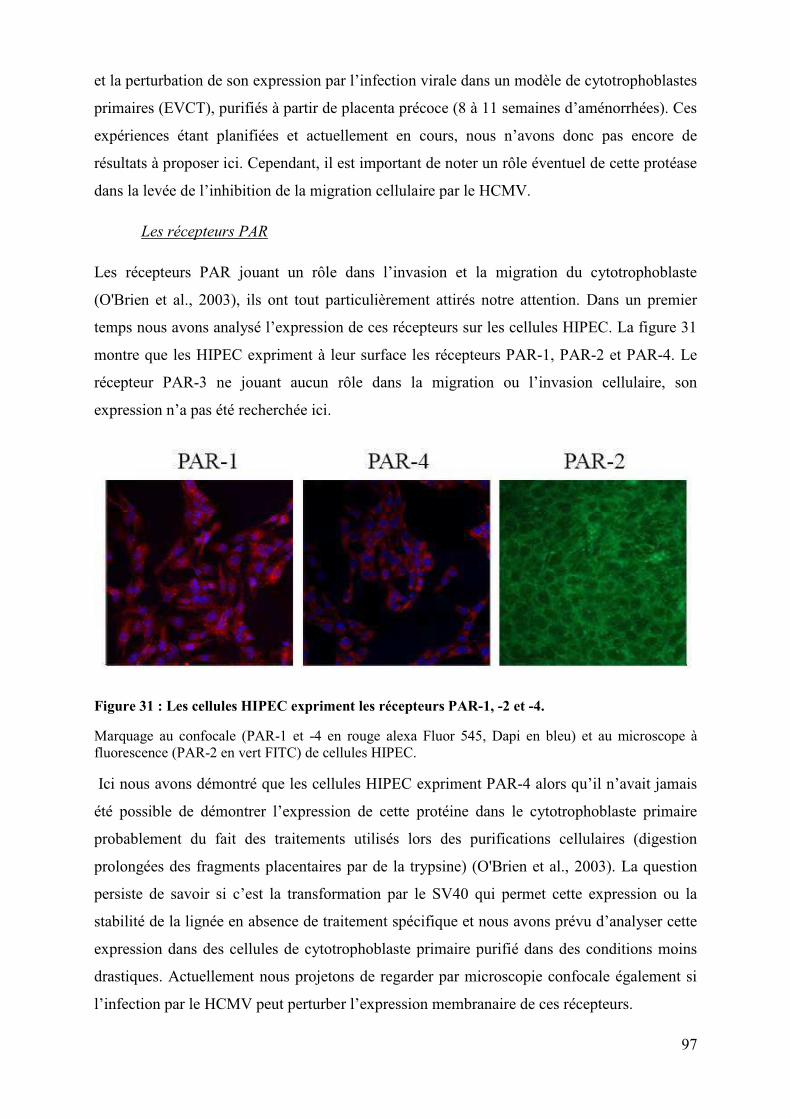
97
et la perturbation de son expression par l’infection virale dans un modèle de cytotrophoblastes
primaires (EVCT), purifiés à partir de placenta précoce (8 à 11 semaines d’aménorrhées). Ces
expériences étant planifiées et actuellement en cours, nous n’avons donc pas encore de
résultats à proposer ici. Cependant, il est important de noter un rôle éventuel de cette protéase
dans la levée de l’inhibition de la migration cellulaire par le HCMV.
Les récepteurs PAR
Les récepteurs PAR jouant un rôle dans l’invasion et la migration du cytotrophoblaste
(O'Brien et al., 2003), ils ont tout particulièrement attirés notre attention. Dans un premier
temps nous avons analysé l’expression de ces récepteurs sur les cellules HIPEC. La figure 31
montre que les HIPEC expriment à leur surface les récepteurs PAR-1, PAR-2 et PAR-4. Le
récepteur PAR-3 ne jouant aucun rôle dans la migration ou l’invasion cellulaire, son
expression n’a pas été recherchée ici.
Figure 31 : Les cellules HIPEC expriment les récepteurs PAR-1, -2 et -4.
Marquage au confocale (PAR-1 et -4 en rouge alexa Fluor 545, Dapi en bleu) et au microscope à fluorescence (PAR-2 en vert FITC) de cellules HIPEC.
Ici nous avons démontré que les cellules HIPEC expriment PAR-4 alors qu’il n’avait jamais
été possible de démontrer l’expression de cette protéine dans le cytotrophoblaste primaire
probablement du fait des traitements utilisés lors des purifications cellulaires (digestion
prolongées des fragments placentaires par de la trypsine) (O'Brien et al., 2003). La question
persiste de savoir si c’est la transformation par le SV40 qui permet cette expression ou la
stabilité de la lignée en absence de traitement spécifique et nous avons prévu d’analyser cette
expression dans des cellules de cytotrophoblaste primaire purifié dans des conditions moins
drastiques. Actuellement nous projetons de regarder par microscopie confocale également si
l’infection par le HCMV peut perturber l’expression membranaire de ces récepteurs.
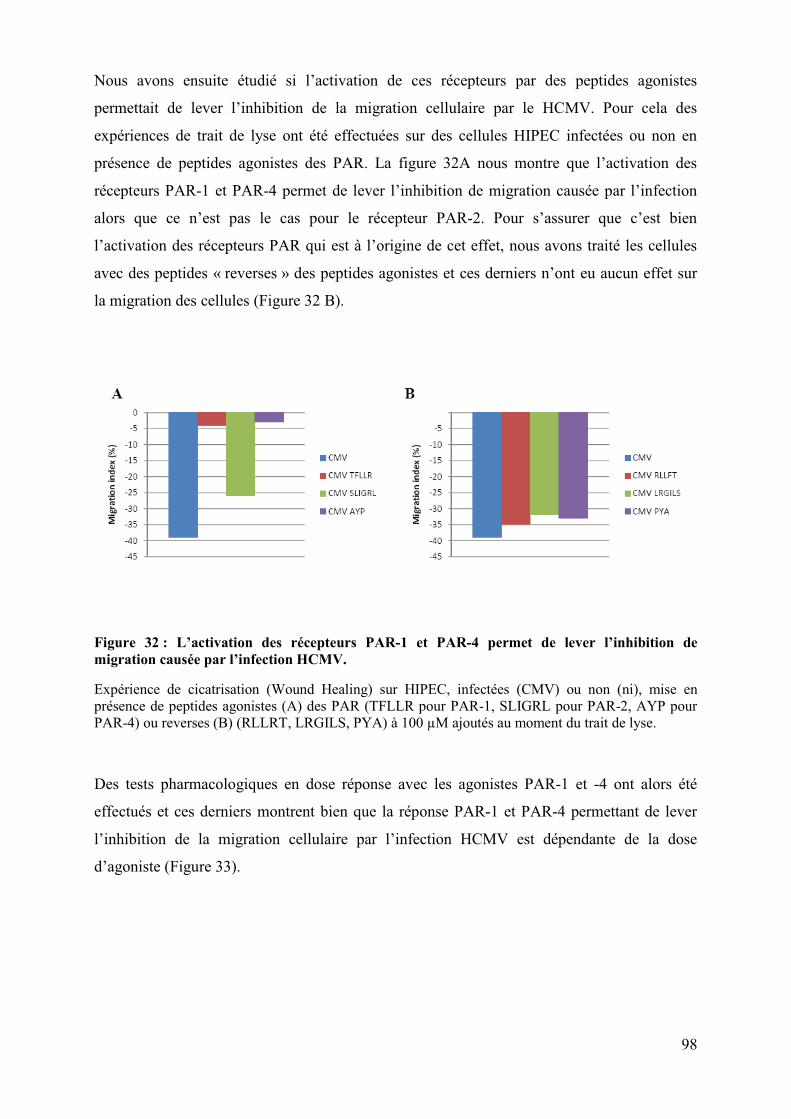
98
Nous avons ensuite étudié si l’activation de ces récepteurs par des peptides agonistes
permettait de lever l’inhibition de la migration cellulaire par le HCMV. Pour cela des
expériences de trait de lyse ont été effectuées sur des cellules HIPEC infectées ou non en
présence de peptides agonistes des PAR. La figure 32A nous montre que l’activation des
récepteurs PAR-1 et PAR-4 permet de lever l’inhibition de migration causée par l’infection
alors que ce n’est pas le cas pour le récepteur PAR-2. Pour s’assurer que c’est bien
l’activation des récepteurs PAR qui est à l’origine de cet effet, nous avons traité les cellules
avec des peptides « reverses » des peptides agonistes et ces derniers n’ont eu aucun effet sur
la migration des cellules (Figure 32 B).
Figure 32 : L’activation des récepteurs PAR-1 et PAR-4 permet de lever l’inhibition de
migration causée par l’infection HCMV.
Expérience de cicatrisation (Wound Healing) sur HIPEC, infectées (CMV) ou non (ni), mise en présence de peptides agonistes (A) des PAR (TFLLR pour PAR-1, SLIGRL pour PAR-2, AYP pour PAR-4) ou reverses (B) (RLLRT, LRGILS, PYA) à 100 µM ajoutés au moment du trait de lyse.
Des tests pharmacologiques en dose réponse avec les agonistes PAR-1 et -4 ont alors été
effectués et ces derniers montrent bien que la réponse PAR-1 et PAR-4 permettant de lever
l’inhibition de la migration cellulaire par l’infection HCMV est dépendante de la dose
d’agoniste (Figure 33).
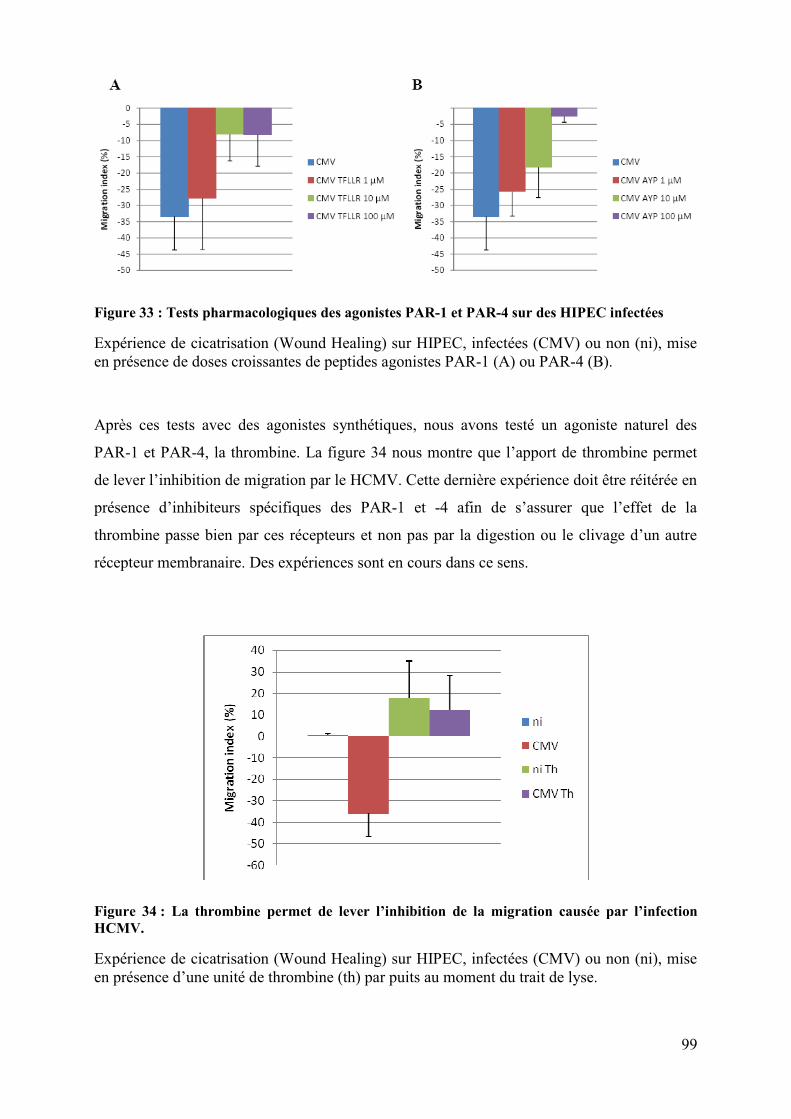
99
Figure 33 : Tests pharmacologiques des agonistes PAR-1 et PAR-4 sur des HIPEC infectées
Expérience de cicatrisation (Wound Healing) sur HIPEC, infectées (CMV) ou non (ni), mise en présence de doses croissantes de peptides agonistes PAR-1 (A) ou PAR-4 (B).
Après ces tests avec des agonistes synthétiques, nous avons testé un agoniste naturel des
PAR-1 et PAR-4, la thrombine. La figure 34 nous montre que l’apport de thrombine permet
de lever l’inhibition de migration par le HCMV. Cette dernière expérience doit être réitérée en
présence d’inhibiteurs spécifiques des PAR-1 et -4 afin de s’assurer que l’effet de la
thrombine passe bien par ces récepteurs et non pas par la digestion ou le clivage d’un autre
récepteur membranaire. Des expériences sont en cours dans ce sens.
Figure 34 : La thrombine permet de lever l’inhibition de la migration causée par l’infection
HCMV.
Expérience de cicatrisation (Wound Healing) sur HIPEC, infectées (CMV) ou non (ni), mise en présence d’une unité de thrombine (th) par puits au moment du trait de lyse.
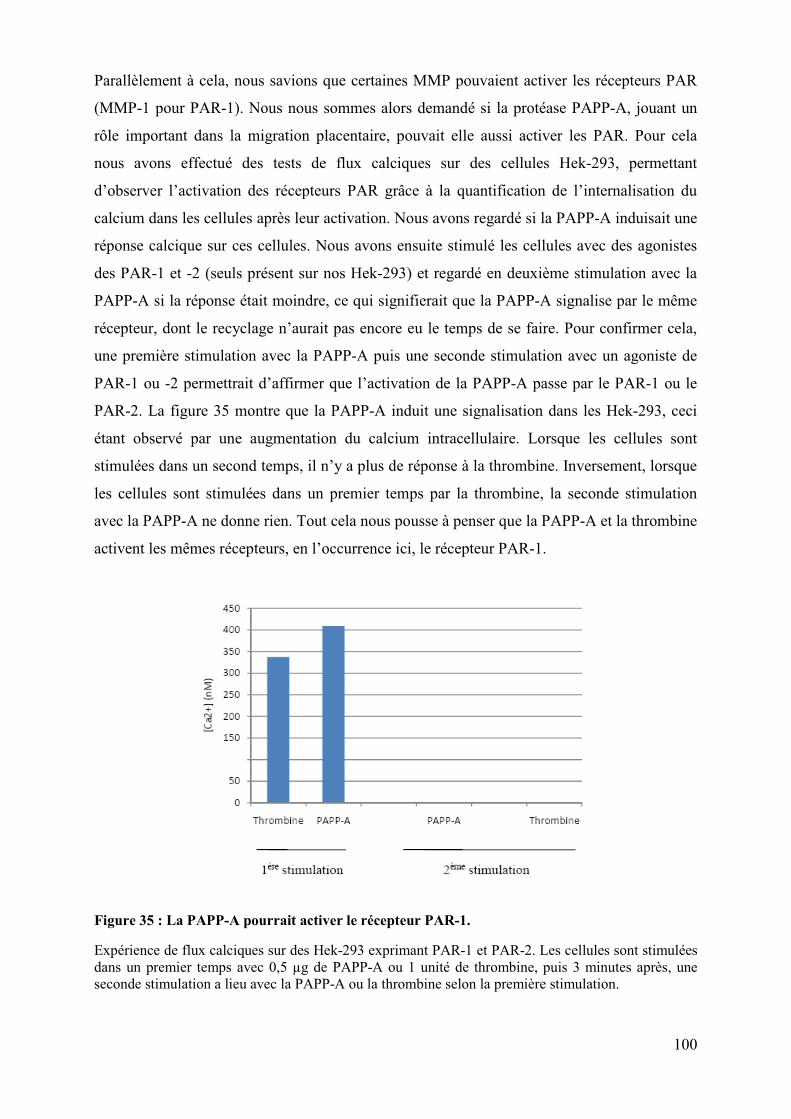
100
Parallèlement à cela, nous savions que certaines MMP pouvaient activer les récepteurs PAR
(MMP-1 pour PAR-1). Nous nous sommes alors demandé si la protéase PAPP-A, jouant un
rôle important dans la migration placentaire, pouvait elle aussi activer les PAR. Pour cela
nous avons effectué des tests de flux calciques sur des cellules Hek-293, permettant
d’observer l’activation des récepteurs PAR grâce à la quantification de l’internalisation du
calcium dans les cellules après leur activation. Nous avons regardé si la PAPP-A induisait une
réponse calcique sur ces cellules. Nous avons ensuite stimulé les cellules avec des agonistes
des PAR-1 et -2 (seuls présent sur nos Hek-293) et regardé en deuxième stimulation avec la
PAPP-A si la réponse était moindre, ce qui signifierait que la PAPP-A signalise par le même
récepteur, dont le recyclage n’aurait pas encore eu le temps de se faire. Pour confirmer cela,
une première stimulation avec la PAPP-A puis une seconde stimulation avec un agoniste de
PAR-1 ou -2 permettrait d’affirmer que l’activation de la PAPP-A passe par le PAR-1 ou le
PAR-2. La figure 35 montre que la PAPP-A induit une signalisation dans les Hek-293, ceci
étant observé par une augmentation du calcium intracellulaire. Lorsque les cellules sont
stimulées dans un second temps, il n’y a plus de réponse à la thrombine. Inversement, lorsque
les cellules sont stimulées dans un premier temps par la thrombine, la seconde stimulation
avec la PAPP-A ne donne rien. Tout cela nous pousse à penser que la PAPP-A et la thrombine
activent les mêmes récepteurs, en l’occurrence ici, le récepteur PAR-1.
Figure 35 : La PAPP-A pourrait activer le récepteur PAR-1.
Expérience de flux calciques sur des Hek-293 exprimant PAR-1 et PAR-2. Les cellules sont stimulées dans un premier temps avec 0,5 µg de PAPP-A ou 1 unité de thrombine, puis 3 minutes après, une seconde stimulation a lieu avec la PAPP-A ou la thrombine selon la première stimulation.
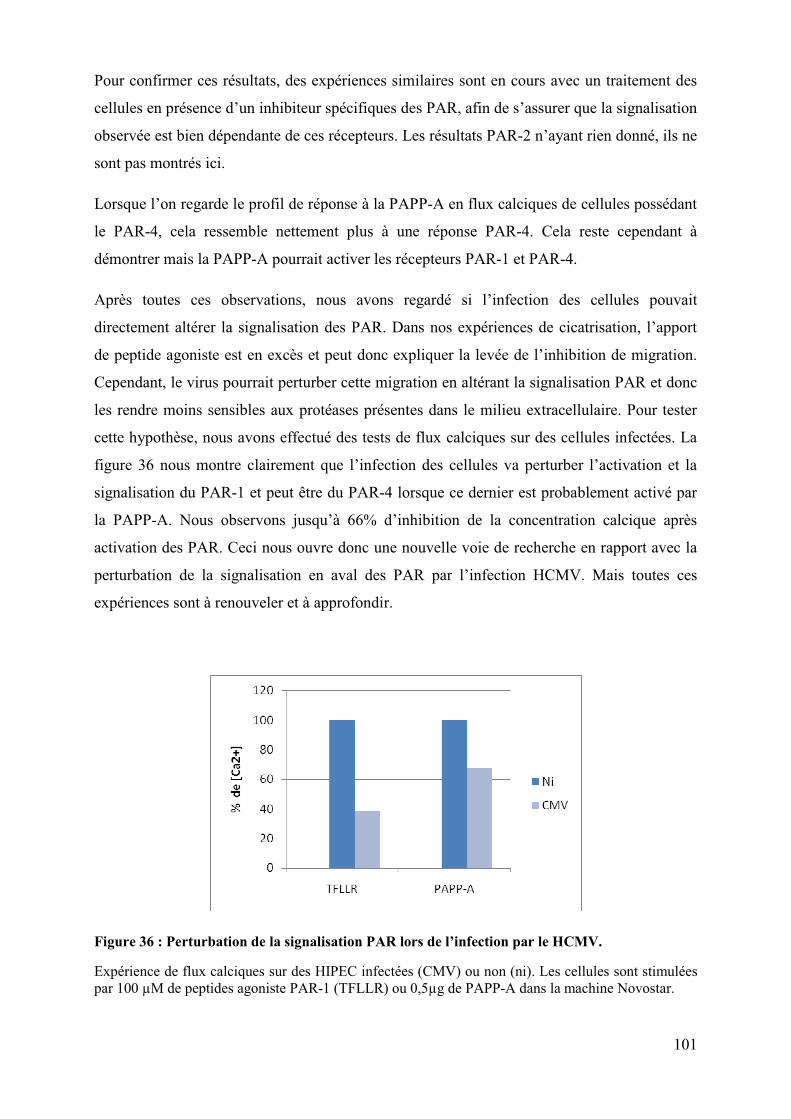
101
Pour confirmer ces résultats, des expériences similaires sont en cours avec un traitement des
cellules en présence d’un inhibiteur spécifiques des PAR, afin de s’assurer que la signalisation
observée est bien dépendante de ces récepteurs. Les résultats PAR-2 n’ayant rien donné, ils ne
sont pas montrés ici.
Lorsque l’on regarde le profil de réponse à la PAPP-A en flux calciques de cellules possédant
le PAR-4, cela ressemble nettement plus à une réponse PAR-4. Cela reste cependant à
démontrer mais la PAPP-A pourrait activer les récepteurs PAR-1 et PAR-4.
Après toutes ces observations, nous avons regardé si l’infection des cellules pouvait
directement altérer la signalisation des PAR. Dans nos expériences de cicatrisation, l’apport
de peptide agoniste est en excès et peut donc expliquer la levée de l’inhibition de migration.
Cependant, le virus pourrait perturber cette migration en altérant la signalisation PAR et donc
les rendre moins sensibles aux protéases présentes dans le milieu extracellulaire. Pour tester
cette hypothèse, nous avons effectué des tests de flux calciques sur des cellules infectées. La
figure 36 nous montre clairement que l’infection des cellules va perturber l’activation et la
signalisation du PAR-1 et peut être du PAR-4 lorsque ce dernier est probablement activé par
la PAPP-A. Nous observons jusqu’à 66% d’inhibition de la concentration calcique après
activation des PAR. Ceci nous ouvre donc une nouvelle voie de recherche en rapport avec la
perturbation de la signalisation en aval des PAR par l’infection HCMV. Mais toutes ces
expériences sont à renouveler et à approfondir.
Figure 36 : Perturbation de la signalisation PAR lors de l’infection par le HCMV.
Expérience de flux calciques sur des HIPEC infectées (CMV) ou non (ni). Les cellules sont stimulées par 100 µM de peptides agoniste PAR-1 (TFLLR) ou 0,5µg de PAPP-A dans la machine Novostar.

102
CONCLUSIONS ET PERSPECTIVES

103
Conclusions et perspectives
Le HCMV peut causer de graves pathologies chez les personnes immunodéprimées, les fœtus
et les nouveau-nés et une primo-infection de la mère durant le premier trimestre de grossesse
peut entraîner des fausses couches ainsi que des retards de croissance intra-utérins.
Dans ce travail, nous avons pu mettre en évidence pour la première fois l’utilisation du
récepteur nucléaire PPARγ par le HCMV pour sa propre réplication. En effet, nous avons pu
identifier des séquences de fixation PPRE sur le génome viral et plus particulièrement dans le
promoteur majeur des protéines IE (MIEP), protéines indispensables à l’aboutissement du
cycle viral. Nous avons donc identifié trois nouvelles séquences PPRE dans le génome du
HCMV, localisées dans la partie distale du MIEP. Ceci a permis de découvrir que
l’expression de la protéine IE2 peut dépendre de l’activation de PPARγ contrairement à celle
d’IE1. IE1 et IE2 étant issues d’un épissage alternatif, nos résultats que PPARγ, en se fixant
au génome viral, va réguler l’épissage du transcrit primaire des protéines IE. Un nouveau
projet de recherche dans notre laboratoire consistera à étudier la régulation de l’expression des
protéines IE1 et IE2, de l’épissage alternatif en amont et surtout à identifier les acteurs
majeurs de cette régulation par PPARγ. Parmi les cibles potentielles envisagées, figure la
protéine PGC-1, protéine co-activatrice de PPARγ dont le rôle dans le saut d’exon au moment
de la transcription a été découvert récemment (Kornblihtt, 2005). Des expériences de ChIP
permettront de savoir si la protéine PGC-1 peut se fixer à l’ADN viral et plus particulièrement
au niveau des séquences PPRE du MIEP. L’interaction entre PPARγ et PGC-1 durant
l’infection pourra être révélée par des expériences de co-immuno-précipitation. Des
constructions de plasmides contenant le gène IE sous contrôle d’un MIEP délété des
séquences PPRE permettraient d’approfondir la compréhension du rôle de PPARγ dans la
régulation de l’expression des protéines IE. La technologie BAC est actuellement développée
dans le laboratoire afin d’obtenir des virus mutants délétés des séquences PPRE que nous
avons identifiées dans le MIEP. L’utilisation de cette technique permettrait de voir si
l’utilisation de PPARγ est essentielle pour l’expression de la protéine IE2 et pour la
réplication du virus. En effet, la production de virions délétés des PPRE devraient être
fortement diminuée dans des fibroblastes infectées par le BAC.

104
A côté de cela, les mécanismes à l’origine de l’activation de PPARγ par le HCMV restent à
élucider. Une hypothèse serait que le virus en délivrant une cPLA-2 cellulaire intégrée dans le
tégument, la relarguerait dans le cytosol au moment de sa fusion avec la membrane plasmique
(Allal et al., 2004). Cette enzyme permettrait alors de transformer les acides gras
membranaires en acide arachidonique, molécule transformée par la suite par une cascade
enzymatique en ligand naturels de PPARγ tels que la 15d-PGJ2, la 15-HETE ou encore la 9-
HODE. Ces deux derniers ligands sont produits en grande quantité dans les cellules
placentaires et leur production dépend de l’activité lipoxygénase, elle-même activée lors de
l’infection par le HCMV (Qiu et al., 2008). Cependant, la réplication virale semble nécessaire
à l’activation de PPARγ, comme nous le montrent nos expériences avec le virus irradié aux
UV. En effet, l’infection par un virus traité aux UV n’active pas PPARγ. Il n’est cependant
pas exclu que le traitement aux UV altère également la structure et/ou la fonction de la
cPLA-2 contenue dans les virions et ne puisse donc pas activer PPARγ. Tout ceci reste à
démontrer par le biais d’expériences avec des inhibiteurs de cPLA-2 ou des lipoxygénases.
Ces expériences sont actuellement en cours dans notre laboratoire afin de déterminer
précisément comment le virus active PPARγ dans les cellules (Figure 37).
Figure 37 : Hypothèse de l’activation de PPARγγγγ par le HCMV pour sa propre réplication

105
Dans un second temps, PPARγ jouant un rôle important dans la régulation de l’invasion et de
la migration des cellules placentaires, nous nous sommes intéressés aux effets de son
activation par le HCMV sur la placentation. Nous avons observé et démontré que son
activation par le HCMV perturbe la migration ainsi que l’invasion des cellules
cytotrophoblastiques provenant de placentas précoces. Par la suite, nous avons cherché à
identifier les mécanismes dépendant de PPARγ activés par le HCMV et perturbant la
migration placentaire. A ce niveau, nous avons pu observer une augmentation des inhibiteurs
de metalloprotéinases TIMP-1 et TIMP-2 dont l’expression peut être modulée par
PPARγ (Shen et al., 2007). Parallèlement à cela, une étude à démontré que la signalisation en
aval de l’intégrine αvβ3 pouvait réguler la transcription de TIMP-1 et de MMP-9 dans les
cellules cancéreuse ovariennes (Kim et al., 2008). Le HCMV pouvant se fixer à cette intégrine
et même induire la signalisation dépendante de ce récepteur, il n’est pas à exclure que la
régulation de la protéine TIMP-1 lors de l’infection par le HCMV se fasse par cette voie.
La diminution de la MMP-9 n’a pas été observée dans notre modèle mais des expériences
supplémentaires sont nécessaires, sachant qu’il a été démontré que l’activation de PPARγ peut
diminuer son expression (Kintscher et al., 2000). Actuellement nous cherchons à identifier de
nouvelles cibles de PPARγ par des expériences de puce ADN. Pour cela, nous récupérerons
des ARN provenant de cellules infectées ou non et en présence ou non d’inhibiteurs de
PPARγ, et uniquement dans le cas où l’infection a causé un défaut de migration, révélé grâce
à un test de trait de lyse en premier lieu. Ces expériences vont nous permettre de mettre en
évidence des gènes régulés par PPARγ lors de l’infection par le HCMV et plus
particulièrement dans le cas où la migration cellulaire est altérée. Un contrôle positif d’ARN
de cellules traitées avec un ligand synthétique de PPARγ (la rosiglitazone) nous permettra de
différencier et de bien mettre en évidence les gènes spécifiquement régulés par PPARγ
uniquement dans le cas de l’infection par le HCMV. En effet, l’état d’acetylation des histones
ainsi que la condensation de la chromatine cellulaire pourrait être totalement différente entre
les cellules infectées et les cellules saines du fait de l’interaction des protéines virales IE avec
les protéines histones déacetylases par exemple (Reeves et al., 2006).
Il a été observé dans notre laboratoire dans des études histologiques de placenta à terme
infectés ou non in vivo que l’infection HCMV diminuait la vascularisation placentaire (M.
Bénard, C. Casper). Ce défaut de vascularisation pourrait s’expliquer par un défaut de
migration des cellules endothéliales qui pourrait être dû à l’activation de PPARγ.

106
Nous avons pu mettre en évidence que la protéase PAPP-A était capable de lever l’inhibition
de la migration cellulaire lors de l’infection par le HCMV. Ceci nous permet de croire que la
diminution eventuelle de l’expression de cette protéase dans le cytotrophoblaste infectée
pourrait être une des causes de perturbation de la migration et de l’invasion. Des expériences
complémentaires doivent être effectuées sur des cultures de cellules cytotrophoblastiques
primaires.
Notre intérêt pour les récepteurs PAR a permis de découvrir que la PAPP-A pouvait être une
nouvelle protéase activatrice de PAR-1 et PAR-4. Ceci reste cependant à confirmer par des
expériences complémentaires.
Nous avons pu mettre en évidence une perturbation de la signalisation des récepteurs PAR par
l’infection. Nous avons également montré ici que l’apport en agoniste de ces récepteurs
permettait de lever l’inhibition de migration des cellules. Il nous reste cependant à confirmer
le rôle de ces récepteurs dans la migration placentaire et surtout si la perturbation de la
signalisation de ces récepteurs est la cause de la diminution de la migration placentaire lors de
l’infection par le HCMV. Certaines études ont montré que la migration placentaire impliquait
ces récepteurs notamment via la signalisation en aval par la voie WNT et la stabilisation de la
β-caténine (Grisaru-Granovsky et al., 2009). Parallèlement à cela, il a été démontré que
PPARγ pouvait inhiber la voie des WNT (Katoh and Katoh, 2007). Nous pouvons alors
émettre l’hypothèse que le virus en activant PPARγ perturberait la signalisation PAR en
inhibant la voie WNT. Certaines études ont montré également que la thrombine en activant les
PAR-1 et -3 pouvait augmenter la sécrétion de MMP-9 dans les monocytes et les
ostéosarcomes, permettant alors une meilleure invasion et migration des cellules (Chang et al.,
2009; Radjabi et al., 2008). Ces observations nous permettent alors d’envisager l’hypothèse
suivante : le virus en perturbant la signalisation des PAR, diminuerait la sécrétion de MMP-9
entrainant le défaut de migration déjà observé par Lénore Pereira (Yamamoto-Tabata et al.,
2004). Mais ceci reste également à prouver par des expériences. Il a également été démontré
que l’activation de PAR-1 pouvait induire la sécrétion de MMP-9 dans les astrocytes de rat
(Choi et al., 2008). Cette observation suggère une hypothèse supplémentaire et un nouveau
projet de recherche est développé dans ce sens dans le laboratoire. En effet, il est actuellement
connu que l’infection du placenta et du fœtus par le HCMV peut entraîner de graves lésions
neuronales ainsi que de graves troubles sensoriels (Cheeran et al., 2009). Parallèlement à cela,
il a été démontré que PPARγ jouait un rôle dans la différenciation des cellules souches
neuronales (Cimini and Ceru, 2008; Wang et al., 2009). C’est pourquoi dans le laboratoire, un

107
projet a été initié et consiste à étudier l’impact de l’activation de PPARγ par le HCMV sur la
différenciation de ces cellules. Ceci permettra de mieux comprendre l’origine des lésions et
des troubles neuronaux observés chez les enfants infectés in utero.
En conclusion, ces travaux ont permis d’approfondir les connaissances sur la réplication du
HCMV grâce à l’identification d’un nouveau facteur de transcription utilisé par le virus pour
sa réplication. Ceux-ci ont également permis de mieux comprendre comment l’invasion et la
migration placentaire pouvaient être perturbées par le HCMV et a ouvert de nouvelles voies
pour identifier des protéines utiles pour diagnostiquer les grossesses à risque et pour
développer de nouveaux traitements.

108
BIBLIOGRAPHIE

109
AbuBakar, S., I. Boldogh, and T. Albrecht. 1990. Human cytomegalovirus stimulates arachidonic acid metabolism through pathways that are affected by inhibitors of phospholipase A2 and protein kinase C. Biochem Biophys Res Commun. 166:953-9.
Adams, M., M.J. Reginato, D. Shao, M.A. Lazar, and V.K. Chatterjee. 1997. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 272:5128-32.
Allal, C., C. Buisson-Brenac, V. Marion, C. Claudel-Renard, T. Faraut, P. Dal Monte, D. Streblow, M. Record, and J.L. Davignon. 2004. Human cytomegalovirus carries a cell-derived phospholipase A2 required for infectivity. J Virol. 78:7717-26.
Alsat, E., J. Guibourdenche, D. Luton, F. Frankenne, and D. Evain-Brion. 1997. Human placental growth hormone. Am J Obstet Gynecol. 177:1526-34.
Anaes. Septembre 2004. Evaluation de l’intérêt du dépistage de l’infection à Cytomégalovirus chez la femme enceinte en France.
Andrews, W.W., J.C. Hauth, and R.L. Goldenberg. 2000. Infection and preterm birth. Am J
Perinatol. 17:357-65. Are, A., L. Aronsson, S. Wang, G. Greicius, Y.K. Lee, J.A. Gustafsson, S. Pettersson, and V.
Arulampalam. 2008. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc Natl Acad Sci
U S A. 105:1943-8. Arora, P., T.K. Ricks, and J. Trejo. 2007. Protease-activated receptor signalling, endocytic
sorting and dysregulation in cancer. J Cell Sci. 120:921-8. Arrode, G., C. Boccaccio, J.P. Abastado, and C. Davrinche. 2002. Cross-presentation of
human cytomegalovirus pp65 (UL83) to CD8+ T cells is regulated by virus-induced, soluble-mediator-dependent maturation of dendritic cells. J Virol. 76:142-50.
Arrode, G., C. Boccaccio, J. Lule, S. Allart, N. Moinard, J.P. Abastado, A. Alam, and C. Davrinche. 2000. Incoming human cytomegalovirus pp65 (UL83) contained in apoptotic infected fibroblasts is cross-presented to CD8(+) T cells by dendritic cells. J
Virol. 74:10018-24. Bailey, S.T., and S. Ghosh. 2005. 'PPAR'ting ways with inflammation. Nat Immunol. 6:966-7. Barak, Y., M.C. Nelson, E.S. Ong, Y.Z. Jones, P. Ruiz-Lozano, K.R. Chien, A. Koder, and
R.M. Evans. 1999. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 4:585-95.
Beck, S., and B.G. Barrell. 1988. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature. 331:269-72.
Benoist, G., F. Jacquemard, M. Leruez-Ville, and Y. Ville. 2008. Infection congénitale à Cytomégalovirus (CMV). Gynécologie Obstétrique & Fertilité. 36:248–260.
Bentz, G.L., and A.D. Yurochko. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and beta1 and beta3 integrins. Proc Natl Acad Sci U S A. 105:5531-6.
Berndt, A., H. Hofmann-Winkler, N. Tavalai, G. Hahn, and T. Stamminger. 2009. Importance of covalent and non-covalent SUMO interactions of the major human cytomegalovirus transactivator IE2p86 for viral infection. J Virol.
Biron, C.A., K.S. Byron, and J.L. Sullivan. 1989. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 320:1731-5.
Bischof, P. 1979. Purification and characterization of pregnancy associated plasma protein A (PAPP-A). Arch Gynecol. 227:315-26.
Bischof, P. 1989. Three pregnancy proteins (PP12, PP14, and PAPP-A): their biological and clinical relevance. Am J Perinatol. 6:110-6.

110
Boehme, K.W., M. Guerrero, and T. Compton. 2006. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J
Immunol. 177:7094-102. Bohm, S.K., W. Kong, D. Bromme, S.P. Smeekens, D.C. Anderson, A. Connolly, M. Kahn,
N.A. Nelken, S.R. Coughlin, D.G. Payan, and N.W. Bunnett. 1996. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 314 ( Pt 3):1009-16.
Boire, A., L. Covic, A. Agarwal, S. Jacques, S. Sherifi, and A. Kuliopulos. 2005. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 120:303-13.
Bonno, M., C. Oxvig, G.M. Kephart, J.M. Wagner, T. Kristensen, L. Sottrup-Jensen, and G.J. Gleich. 1994. Localization of pregnancy-associated plasma protein-A and colocalization of pregnancy-associated plasma protein-A messenger ribonucleic acid and eosinophil granule major basic protein messenger ribonucleic acid in placenta. Lab Invest. 71:560-6.
Borst, E.M., S. Mathys, M. Wagner, W. Muranyi, and M. Messerle. 2001. Genetic evidence of an essential role for cytomegalovirus small capsid protein in viral growth. J Virol. 75:1450-8.
Bresnahan, W.A., and T. Shenk. 2000. A subset of viral transcripts packaged within human cytomegalovirus particles. Science. 288:2373-6.
Britt, W.J., M. Jarvis, J.Y. Seo, D. Drummond, and J. Nelson. 2004. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J Virol. 78:539-43.
Britt, W.J., L. Vugler, E.J. Butfiloski, and E.B. Stephens. 1990. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J
Virol. 64:1079-85. Brune, W., M. Wagner, and M. Messerle. 2006. Manipulating Cytomegalovirus Genomes by
BAC Mutagenesis: Strategies and Applications. Cytomegaloviruses, molecular
biology and immunology. Chapter 4:63-89. Bukowski, J.F., J.F. Warner, G. Dennert, and R.M. Welsh. 1985. Adoptive transfer studies
demonstrating the antiviral effect of natural killer cells in vivo. J Exp Med. 161:40-52. Bukowski, J.F., B.A. Woda, S. Habu, K. Okumura, and R.M. Welsh. 1983. Natural killer cell
depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol. 131:1531-8.
Burton, G.J., and A.L. Watson. 1997. The Structure of the Human Placenta: Implications for Initiating and Defending Against Virus Infections. Rev Med Virol. 7:219-228.
Byun, D., S. Mohan, M. Yoo, C. Sexton, D.J. Baylink, and X. Qin. 2001. Pregnancy-associated plasma protein-A accounts for the insulin-like growth factor (IGF)-binding protein-4 (IGFBP-4) proteolytic activity in human pregnancy serum and enhances the mitogenic activity of IGF by degrading IGFBP-4 in vitro. J Clin Endocrinol Metab. 86:847-54.
Cha, T.A., E. Tom, G.W. Kemble, G.M. Duke, E.S. Mocarski, and R.R. Spaete. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol. 70:78-83.
Chandra, V., P. Huang, Y. Hamuro, S. Raghuram, Y. Wang, T.P. Burris, and F. Rastinejad. 2008. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature:350-356.

111
Chang, C.J., L.A. Hsu, Y.H. Ko, P.L. Chen, Y.T. Chuang, C.Y. Lin, C.H. Liao, and J.H. Pang. 2009. Thrombin regulates matrix metalloproteinase-9 expression in human monocytes. Biochem Biophys Res Commun. 385:241-6.
Chawla, A., W.A. Boisvert, C.H. Lee, B.A. Laffitte, Y. Barak, S.B. Joseph, D. Liao, L. Nagy, P.A. Edwards, L.K. Curtiss, R.M. Evans, and P. Tontonoz. 2001. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 7:161-71.
Chee, M., S.A. Rudolph, B. Plachter, B. Barrell, and G. Jahn. 1989. Identification of the major capsid protein gene of human cytomegalovirus. J Virol. 63:1345-53.
Cheeran, M.C., J.R. Lokensgard, and M.R. Schleiss. 2009. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin
Microbiol Rev. 22:99-126, Table of Contents. Chignard, M., and D. Pidard. 2006. Neutrophil and pathogen proteinases versus proteinase-
activated receptor-2 lung epithelial cells: more terminators than activators. Am J
Respir Cell Mol Biol. 34:394-8. Chinetti, G., S. Lestavel, V. Bocher, A.T. Remaley, B. Neve, I.P. Torra, E. Teissier, A.
Minnich, M. Jaye, N. Duverger, H.B. Brewer, J.C. Fruchart, V. Clavey, and B. Staels. 2001. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 7:53-8.
Choi, M.S., Y.E. Kim, W.J. Lee, J.W. Choi, G.H. Park, S.D. Kim, S.J. Jeon, H.S. Go, S.M. Shin, W.K. Kim, C.Y. Shin, and K.H. Ko. 2008. Activation of protease-activated receptor1 mediates induction of matrix metalloproteinase-9 by thrombin in rat primary astrocytes. Brain Res Bull. 76:368-75.
Chui, P.C., H.P. Guan, M. Lehrke, and M.A. Lazar. 2005. PPARgamma regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J Clin Invest. 115:2244-56.
Cimini, A., and M.P. Ceru. 2008. Emerging roles of peroxisome proliferator-activated receptors (PPARs) in the regulation of neural stem cells proliferation and differentiation. Stem Cell Rev. 4:293-303.
Cinatl, J., Jr., S. Margraf, J.U. Vogel, M. Scholz, J. Cinatl, and H.W. Doerr. 2001. Human cytomegalovirus circumvents NF-kappa B dependence in retinal pigment epithelial cells. J Immunol. 167:1900-8.
Collet, P., L. Domenjoud, M.D. Devignes, H. Murad, H. Schohn, and M. Dauca. 2004. The human semaphorin 6B gene is down regulated by PPARs. Genomics. 83:1141-50.
Compton, T. 2004. Receptors and immune sensors: the complex entry path of human cytomegalovirus. Trends Cell Biol. 14:5-8.
Compton, T., E.A. Kurt-Jones, K.W. Boehme, J. Belko, E. Latz, D.T. Golenbock, and R.W. Finberg. 2003. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 77:4588-96.
Crocker, I.P., S. Cooper, S.C. Ong, and P.N. Baker. 2003. Differences in apoptotic susceptibility of cytotrophoblasts and syncytiotrophoblasts in normal pregnancy to those complicated with preeclampsia and intrauterine growth restriction. Am J Pathol. 162:637-43.
Crough, T., and R. Khanna. 2009. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev. 22:76-98, Table of Contents.
Damsky, C.H., M.L. Fitzgerald, and S.J. Fisher. 1992. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J Clin
Invest. 89:210-22.

112
Delerive, P., F. Martin-Nizard, G. Chinetti, F. Trottein, J.C. Fruchart, J. Najib, P. Duriez, and B. Staels. 1999. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 85:394-402.
Dery, O., M.S. Thoma, H. Wong, E.F. Grady, and N.W. Bunnett. 1999. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-Arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem. 274:18524-35.
Desvergne, B., and W. Wahli. 1999. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 20:649-88.
Dolken, L., S. Pfeffer, and U.H. Koszinowski. 2009. Cytomegalovirus microRNAs. Virus
Genes. 38:355-64. Dormandy, J.A., B. Charbonnel, D.J. Eckland, E. Erdmann, M. Massi-Benedetti, I.K. Moules,
A.M. Skene, M.H. Tan, P.J. Lefebvre, G.D. Murray, E. Standl, R.G. Wilcox, L. Wilhelmsen, J. Betteridge, K. Birkeland, A. Golay, R.J. Heine, L. Koranyi, M. Laakso, M. Mokan, A. Norkus, V. Pirags, T. Podar, A. Scheen, W. Scherbaum, G. Schernthaner, O. Schmitz, J. Skrha, U. Smith, and J. Taton. 2005. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 366:1279-89.
Drose, J.A., M.A. Dennis, and D. Thickman. 1991. Infection in utero: US findings in 19 cases. Radiology. 178:369-74.
Elchalal, U., R.G. Humphrey, S.D. Smith, C. Hu, Y. Sadovsky, and D.M. Nelson. 2004. Troglitazone attenuates hypoxia-induced injury in cultured term human trophoblasts. Am J Obstet Gynecol. 191:2154-9.
Elkington, R., S. Walker, T. Crough, M. Menzies, J. Tellam, M. Bharadwaj, and R. Khanna. 2003. Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multispecific reactivities in healthy virus carriers. J Virol. 77:5226-40.
Evain-Brion, D., T. Fournier, P. Therond, A. Tarrade, and L. Pavan. 2002. Pathogénie de la pré-éclampsie: rôle du PPARgamma dans l'invasion trophoblastique. Bull. Acad. Natle
Méd. 186:409-419. Fajas, L., J.C. Fruchart, and J. Auwerx. 1998. PPARgamma3 mRNA: a distinct PPARgamma
mRNA subtype transcribed from an independent promoter. FEBS Lett. 438:55-60. Fisher, S., O. Genbacev, E. Maidji, and L. Pereira. 2000. Human cytomegalovirus infection of
placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol. 74:6808-20.
Folkersen, J., J.G. Grudzinskas, P. Hindersson, B. Teisner, and J.G. Westergaard. 1981. Pregnancy-associated plasma protein A: circulating levels during normal pregnancy. Am J Obstet Gynecol. 139:910-4.
Fournier, T., K. Handschuh, V. Tsatsaris, J. Guibourdenche, and D. Evain-Brion. 2008. Role of nuclear receptors and their ligands in human trophoblast invasion. J Reprod
Immunol. 77:161-70. Fournier, T., L. Pavan, A. Tarrade, K. Schoonjans, J. Auwerx, C. Rochette-Egly, and D.
Evain-Brion. 2002. The role of PPAR-gamma/RXR-alpha heterodimers in the regulation of human trophoblast invasion. Ann N Y Acad Sci. 973:26-30.
Fournier, T., V. Tsatsaris, K. Handschuh, and D. Evain-Brion. 2007. PPARs and the placenta. Placenta. 28:65-76.
Frohnert, B.I., T.Y. Hui, and D.A. Bernlohr. 1999. Identification of a functional peroxisome proliferator-responsive element in the murine fatty acid transport protein gene. J Biol
Chem. 274:3970-7.

113
Gagnon, A., R.D. Wilson, F. Audibert, V.M. Allen, C. Blight, J.A. Brock, V.A. Desilets, J.A. Johnson, S. Langlois, A. Summers, and P. Wyatt. 2008. Obstetrical complications associated with abnormal maternal serum markers analytes. J Obstet Gynaecol Can. 30:918-49.
Gallina, A., E. Percivalle, L. Simoncini, M.G. Revello, G. Gerna, and G. Milanesi. 1996. Human cytomegalovirus pp65 lower matrix phosphoprotein harbours two transplantable nuclear localization signals. J Gen Virol. 77 ( Pt 6):1151-7.
Gandhi, M.K., and R. Khanna. 2004. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis. 4:725-38.
Ghazal, P., C. DeMattei, E. Giulietti, S.A. Kliewer, K. Umesono, and R.M. Evans. 1992. Retinoic acid receptors initiate induction of the cytomegalovirus enhancer in embryonal cells. Proc Natl Acad Sci U S A. 89:7630-4.
Glass, C.K., and S. Ogawa. 2006. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 6:44-55.
Graham, C.H., and P.K. Lala. 1991. Mechanism of control of trophoblast invasion in situ. J
Cell Physiol. 148:228-34. Gredmark, S., and C. Soderberg-Naucler. 2003. Human cytomegalovirus inhibits
differentiation of monocytes into dendritic cells with the consequence of depressed immunological functions. J Virol. 77:10943-56.
Greijer, A.E., C.A. Dekkers, and J.M. Middeldorp. 2000. Human cytomegalovirus virions differentially incorporate viral and host cell RNA during the assembly process. J
Virol. 74:9078-82. Grey, F., A. Antoniewicz, E. Allen, J. Saugstad, A. McShea, J.C. Carrington, and J. Nelson.
2005. Identification and characterization of human cytomegalovirus-encoded microRNAs. J Virol. 79:12095-9.
Grey, F., and J. Nelson. 2008. Identification and function of human cytomegalovirus microRNAs. J Clin Virol. 41:186-91.
Grisaru-Granovsky, S., M. Maoz, O. Barzilay, Y.J. Yin, D. Prus, and R. Bar-Shavit. 2009. Protease activated receptor-1, PAR1, promotes placenta trophoblast invasion and beta-catenin stabilization. J Cell Physiol. 218:512-21.
Guma, M., A. Angulo, and M. Lopez-Botet. 2006. NK cell receptors involved in the response to human cytomegalovirus infection. Curr Top Microbiol Immunol. 298:207-23.
Halary, F., A. Amara, H. Lortat-Jacob, M. Messerle, T. Delaunay, C. Houles, F. Fieschi, F. Arenzana-Seisdedos, J.F. Moreau, and J. Dechanet-Merville. 2002. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 17:653-64.
Halary, F., V. Pitard, D. Dlubek, R. Krzysiek, H. de la Salle, P. Merville, C. Dromer, D. Emilie, J.F. Moreau, and J. Dechanet-Merville. 2005. Shared reactivity of V{delta}2(neg) {gamma}{delta} T cells against cytomegalovirus-infected cells and tumor intestinal epithelial cells. J Exp Med. 201:1567-78.
Halwachs-Baumann, G., M. Wilders-Truschnig, G. Desoye, T. Hahn, L. Kiesel, K. Klingel, P. Rieger, G. Jahn, and C. Sinzger. 1998. Human trophoblast cells are permissive to the complete replicative cycle of human cytomegalovirus. J Virol. 72:7598-602.
Handschuh, K., J. Guibourdenche, M. Guesnon, I. Laurendeau, D. Evain-Brion, and T. Fournier. 2006. Modulation of PAPP-A expression by PPARgamma in human first trimester trophoblast. Placenta. 27 Suppl A:S127-34.
Hansen, K.K., M. Saifeddine, and M.D. Hollenberg. 2004. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology. 112:183-90.

114
Hobom, U., W. Brune, M. Messerle, G. Hahn, and U.H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol. 74:7720-9.
Hofmann, H., S. Floss, and T. Stamminger. 2000. Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J Virol. 74:2510-24.
Holtappels, R., N.K. Grzimek, C.O. Simon, D. Thomas, D. Dreis, and M.J. Reddehase. 2002. Processing and presentation of murine cytomegalovirus pORFm164-derived peptide in fibroblasts in the face of all viral immunosubversive early gene functions. J Virol. 76:6044-53.
Hooks, J.J., M.S. Chin, K. Srinivasan, Y. Momma, L.C. Hooper, C.N. Nagineni, C.C. Chan, and B. Detrick. 2006. Human cytomegalovirus induced cyclooxygenase-2 in human retinal pigment epithelial cells augments viral replication through a prostaglandin pathway. Microbes Infect. 8:2236-44.
Hoxie, J.A., M. Ahuja, E. Belmonte, S. Pizarro, R. Parton, and L.F. Brass. 1993. Internalization and recycling of activated thrombin receptors. J Biol Chem. 268:13756-63.
Huppertz, B., S. Kertschanska, A.Y. Demir, H.G. Frank, and P. Kaufmann. 1998. Immunohistochemistry of matrix metalloproteinases (MMP), their substrates, and their inhibitors (TIMP) during trophoblast invasion in the human placenta. Cell Tissue Res. 291:133-48.
Isaacson, M.K., A.L. Feire, and T. Compton. 2007. Epidermal growth factor receptor is not required for human cytomegalovirus entry or signaling. J Virol. 81:6241-7.
Ishihara, H., A.J. Connolly, D. Zeng, M.L. Kahn, Y.W. Zheng, C. Timmons, T. Tram, and S.R. Coughlin. 1997. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 386:502-6.
Jarvis, M.A., and J.A. Nelson. 2007. Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J Virol. 81:2095-101.
Juge-Aubry, C., A. Pernin, T. Favez, A.G. Burger, W. Wahli, C.A. Meier, and B. Desvergne. 1997. DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5'-flanking region. J Biol Chem. 272:25252-9.
Katoh, M., and M. Katoh. 2007. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 13:4042-5.
Keay, S., and B. Baldwin. 1992. The human fibroblast receptor for gp86 of human cytomegalovirus is a phosphorylated glycoprotein. J Virol. 66:4834-8.
Keelan, J.A., M. Blumenstein, R.J. Helliwell, T.A. Sato, K.W. Marvin, and M.D. Mitchell. 2003. Cytokines, prostaglandins and parturition--a review. Placenta. 24 Suppl A:S33-46.
Keller, M.J., D.G. Wheeler, E. Cooper, and J.L. Meier. 2003. Role of the human cytomegalovirus major immediate-early promoter's 19-base-pair-repeat cyclic AMP-response element in acutely infected cells. J Virol. 77:6666-75.
Khan, K.A., A. Coaquette, C. Davrinche, and G. Herbein. 2009. Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J Immunol. 182:7784-94.
Kim, D.S., O.H. Jeon, H.D. Lee, K.H. Yoo, and D.S. Kim. 2008. Integrin alphavbeta3-mediated transcriptional regulation of TIMP-1 in a human ovarian cancer cell line. Biochem Biophys Res Commun. 377:479-83.

115
Kintscher, U., S. Goetze, S. Wakino, S. Kim, S. Nagpal, R.A. Chandraratna, K. Graf, E. Fleck, W.A. Hsueh, and R.E. Law. 2000. Peroxisome proliferator-activated receptor and retinoid X receptor ligands inhibit monocyte chemotactic protein-1-directed migration of monocytes. Eur J Pharmacol. 401:259-70.
Kornblihtt, A.R. 2005. Promoter usage and alternative splicing. Curr Opin Cell Biol. 17:262-8.
Kotenko, S.V., S. Saccani, L.S. Izotova, O.V. Mirochnitchenko, and S. Pestka. 2000. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad
Sci U S A. 97:1695-700. Kotton, C.N., and J.A. Fishman. 2005. Viral infection in the renal transplant recipient. J Am
Soc Nephrol. 16:1758-74. Kubota, N., Y. Terauchi, H. Miki, H. Tamemoto, T. Yamauchi, K. Komeda, S. Satoh, R.
Nakano, C. Ishii, T. Sugiyama, K. Eto, Y. Tsubamoto, A. Okuno, K. Murakami, H. Sekihara, G. Hasegawa, M. Naito, Y. Toyoshima, S. Tanaka, K. Shiota, T. Kitamura, T. Fujita, O. Ezaki, S. Aizawa, T. Kadowaki, and et al. 1999. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 4:597-609.
Kumar, P., C.S. Lau, M. Mathur, P. Wang, and K.A. DeFea. 2007. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 293:C346-57.
Lee, J.M., H.J. Kang, H.R. Lee, C.Y. Choi, W.J. Jang, and J.H. Ahn. 2003. PIAS1 enhances SUMO-1 modification and the transactivation activity of the major immediate-early IE2 protein of human cytomegalovirus. FEBS Lett. 555:322-8.
Leesnitzer, L.M., D.J. Parks, R.K. Bledsoe, J.E. Cobb, J.L. Collins, T.G. Consler, R.G. Davis, E.A. Hull-Ryde, J.M. Lenhard, L. Patel, K.D. Plunket, J.L. Shenk, J.B. Stimmel, C. Therapontos, T.M. Willson, and S.G. Blanchard. 2002. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 41:6640-50.
Lefebvre, A.M., I. Chen, P. Desreumaux, J. Najib, J.C. Fruchart, K. Geboes, M. Briggs, R. Heyman, and J. Auwerx. 1998. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 4:1053-7.
Lehmann, J.M., L.B. Moore, T.A. Smith-Oliver, W.O. Wilkison, T.M. Willson, and S.A. Kliewer. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 270:12953-6.
Lehrke, M., and M.A. Lazar. 2005. The many faces of PPARgamma. Cell. 123:993-9. Li, A.C., K.K. Brown, M.J. Silvestre, T.M. Willson, W. Palinski, and C.K. Glass. 2000a.
Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 106:523-31.
Li, M., G. Pascual, and C.K. Glass. 2000b. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell
Biol. 20:4699-707. Librach, C.L., S.L. Feigenbaum, K.E. Bass, T.Y. Cui, N. Verastas, Y. Sadovsky, J.P. Quigley,
D.L. French, and S.J. Fisher. 1994. Interleukin-1 beta regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem. 269:17125-31.
Lin, T.M., S.P. Galbert, D. Kiefer, W.N. Spellacy, and S. Gall. 1974. Characterization of four human pregnancy-associated plasma proteins. Am J Obstet Gynecol. 118:223-36.
Luo, M.H., P.H. Schwartz, and E.A. Fortunato. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol. 82:9994-10007.

116
Macfarlane, S.R., M.J. Seatter, T. Kanke, G.D. Hunter, and R. Plevin. 2001. Proteinase-activated receptors. Pharmacol Rev. 53:245-82.
Maidji, E., S. McDonagh, O. Genbacev, T. Tabata, and L. Pereira. 2006. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol. 168:1210-26.
Mansure, J.J., R. Nassim, and W. Kassouf. 2009. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 8:6-15.
Marchini, A., H. Liu, and H. Zhu. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol. 75:1870-8.
Marx, N., G. Sukhova, C. Murphy, P. Libby, and J. Plutzky. 1998. Macrophages in human atheroma contain PPARgamma: differentiation-dependent peroxisomal proliferator-activated receptor gamma(PPARgamma) expression and reduction of MMP-9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 153:17-23.
Marzusch, K., P. Ruck, J.A. Dietl, H.P. Horny, and E. Kaiserling. 1996. Immunohistochemical localization of tissue inhibitor of metalloproteinases-2 (TIMP-2) in first trimester human placental decidua. Eur J Obstet Gynecol Reprod Biol. 68:105-7.
McKinnon, T., C. Chakraborty, L.M. Gleeson, P. Chidiac, and P.K. Lala. 2001. Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory G protein(s) and phosphorylation of MAPK. J Clin
Endocrinol Metab. 86:3665-74. McVoy, M.A., and S.P. Adler. 1994. Human cytomegalovirus DNA replicates after early
circularization by concatemer formation, and inversion occurs within the concatemer. J Virol. 68:1040-51.
Meier, J.L. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J Virol. 75:1581-93.
Meier, J.L., M.J. Keller, and J.J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J Virol. 76:313-26.
Meier, J.L., and J.A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J Virol. 74:1602-13.
Meier, J.L., and M.F. Stinski. 1996. Regulation of human cytomegalovirus immediate-early gene expression. Intervirology. 39:331-42.
Meier, J.L., and M.F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J Virol. 71:1246-55.
Meier, J.L., and M.F. Stinski. 2006. Major Immediate early enhancer and its gene products. Cytomegaloviruses, molecular biology and immunology. Chapter 8:151-166.
Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U.H. Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A. 94:14759-63.
Michaelis, M., H.W. Doerr, and J. Cinatl. 2009. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia. 11:1-9.
Minamikawa, J., S. Tanaka, M. Yamauchi, D. Inoue, and H. Koshiyama. 1998. Potent inhibitory effect of troglitazone on carotid arterial wall thickness in type 2 diabetes. J
Clin Endocrinol Metab. 83:1818-20.

117
Mocarski, E., T. Shenk, and R. Pass. 2007a. Cytomegaloviruses. In Fields Virology. Vol. 2. D.M. Knipe and P.M. Howley, editors. Lippincott, Williams and Wilkins. 2739-2746.
Mocarski, E.S., Jr. 2002. Virus self-improvement through inflammation: no pain, no gain. Proc Natl Acad Sci U S A. 99:3362-4.
Mocarski, E.S., Jr., T. Shenk, and R.F. Pass. 2007b. Cytomegaloviruses. Fields virology. Volume II:2701-2772.
Mocarski, E.S., G.W. Kemble, J.M. Lyle, and R.F. Greaves. 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc Natl Acad Sci U S A. 93:11321-6.
Moutaftsi, M., P. Brennan, S.A. Spector, and Z. Tabi. 2004. Impaired lymphoid chemokine-mediated migration due to a block on the chemokine receptor switch in human cytomegalovirus-infected dendritic cells. J Virol. 78:3046-54.
Moutaftsi, M., A.M. Mehl, L.K. Borysiewicz, and Z. Tabi. 2002. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood. 99:2913-21.
Muller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J
Virol. 73:5137-43. Murphy, J.C., W. Fischle, E. Verdin, and J.H. Sinclair. 2002. Control of cytomegalovirus lytic
gene expression by histone acetylation. Embo J. 21:1112-20. Nagy, L., P. Tontonoz, J.G. Alvarez, H. Chen, and R.M. Evans. 1998. Oxidized LDL
regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 93:229-40.
Nakanishi-Matsui, M., Y.W. Zheng, D.J. Sulciner, E.J. Weiss, M.J. Ludeman, and S.R. Coughlin. 2000. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 404:609-13.
Navarro, D., P. Paz, S. Tugizov, K. Topp, J. La Vail, and L. Pereira. 1993. Glycoprotein B of human cytomegalovirus promotes virion penetration into cells, transmission of infection from cell to cell, and fusion of infected cells. Virology. 197:143-58.
Netterwald, J., S. Yang, W. Wang, S. Ghanny, M. Cody, P. Soteropoulos, B. Tian, W. Dunn, F. Liu, and H. Zhu. 2005. Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J Virol. 79:5035-46.
Nevels, M., C. Paulus, and T. Shenk. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl
Acad Sci U S A. 101:17234-9. Nielsen, R., L. Grontved, H.G. Stunnenberg, and S. Mandrup. 2006. Peroxisome proliferator-
activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol Cell Biol. 26:5698-714.
Nitzsche, A., C. Paulus, and M. Nevels. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol. 82:11167-80.
Nogalski, M.T., J.P. Podduturi, I.B. DeMeritt, L.E. Milford, and A.D. Yurochko. 2007. The human cytomegalovirus virion possesses an activated casein kinase II that allows for the rapid phosphorylation of the inhibitor of NF-kappaB, IkappaBalpha. J Virol. 81:5305-14.
Nystedt, S., K. Emilsson, A.K. Larsson, B. Strombeck, and J. Sundelin. 1995. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem. 232:84-9.

118
O'Brien, P.J., H. Koi, S. Parry, L.F. Brass, J.F. Strauss, 3rd, L.P. Wang, J.E. Tomaszewski, and L.K. Christenson. 2003. Thrombin receptors and protease-activated receptor-2 in human placentation: receptor activation mediates extravillous trophoblast invasion in vitro. Am J Pathol. 163:1245-54.
Ondrey, F. 2009. Peroxisome proliferator-activated receptor gamma pathway targeting in carcinogenesis: implications for chemoprevention. Clin Cancer Res. 15:2-8.
Overgaard, M.T., J. Haaning, H.B. Boldt, I.M. Olsen, L.S. Laursen, M. Christiansen, G.J. Gleich, L. Sottrup-Jensen, C.A. Conover, and C. Oxvig. 2000. Expression of recombinant human pregnancy-associated plasma protein-A and identification of the proform of eosinophil major basic protein as its physiological inhibitor. J Biol Chem. 275:31128-33.
Pajovic, S., E.L. Wong, A.R. Black, and J.C. Azizkhan. 1997. Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol Cell Biol. 17:6459-64.
Palmer, C.N., M.H. Hsu, H.J. Griffin, and E.F. Johnson. 1995. Novel sequence determinants in peroxisome proliferator signaling. J Biol Chem. 270:16114-21.
Pascual, G., A.L. Fong, S. Ogawa, A. Gamliel, A.C. Li, V. Perissi, D.W. Rose, T.M. Willson, M.G. Rosenfeld, and C.K. Glass. 2005. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 437:759-63.
Pavan, L., A. Hermouet, V. Tsatsaris, P. Therond, T. Sawamura, D. Evain-Brion, and T. Fournier. 2004. Lipids from oxidized low-density lipoprotein modulate human trophoblast invasion: involvement of nuclear liver X receptors. Endocrinology. 145:4583-91.
Pavan, L., A. Tarrade, A. Hermouet, C. Delouis, M. Titeux, M. Vidaud, P. Therond, D. Evain-Brion, and T. Fournier. 2003. Human invasive trophoblasts transformed with simian virus 40 provide a new tool to study the role of PPARgamma in cell invasion process. Carcinogenesis. 24:1325-36.
Pei, D. 2005. Matrix metalloproteinases target protease-activated receptors on the tumor cell surface. Cancer Cell. 7:207-8.
Pellett, P.E., and B. Roizman. 2007. The family Herpesviridae: a brief introduction. Fields
Virology. Volume II:2479-2499. Pereira, L., E. Maidji, S. McDonagh, and T. Tabata. 2006. Routes of Human CMV
Transmission and Infection at the Uterine-Placental interface. Cytomegaloviruses,
molecular biology and immunology. 2:29-48. Pirianov, G., S.N. Waddington, T.M. Lindstrom, V. Terzidou, H. Mehmet, and P.R. Bennett.
2009. The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J(2) delays lipopolysaccharide-induced preterm delivery and reduces mortality in the newborn mouse. Endocrinology. 150:699-706.
Pizzorno, M.C., and G.S. Hayward. 1990. The IE2 gene products of human cytomegalovirus specifically down-regulate expression from the major immediate-early promoter through a target sequence located near the cap site. J Virol. 64:6154-65.
Plachter, B., C. Sinzger, and G. Jahn. 1996. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res. 46:195-261.
Polic, B., H. Hengel, A. Krmpotic, J. Trgovcich, I. Pavic, P. Luccaronin, S. Jonjic, and U.H. Koszinowski. 1998. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med. 188:1047-54.
Powers, C., V. DeFilippis, D. Malouli, and K. Fruh. 2008. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol. 325:333-59.

119
Prost, S., M. Le Dantec, S. Auge, R. Le Grand, S. Derdouch, G. Auregan, N. Deglon, F. Relouzat, A.M. Aubertin, B. Maillere, I. Dusanter-Fourt, and M. Kirszenbaum. 2008. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest. 118:1765-75.
Qiu, H., K. Straat, A. Rahbar, M. Wan, C. Soderberg-Naucler, and J.Z. Haeggstrom. 2008. Human CMV infection induces 5-lipoxygenase expression and leukotriene B4 production in vascular smooth muscle cells. J Exp Med. 205:19-24.
Radjabi, A.R., K. Sawada, S. Jagadeeswaran, A. Eichbichler, H.A. Kenny, A. Montag, K. Bruno, and E. Lengyel. 2008. Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and beta1-integrin on the cell surface. J
Biol Chem. 283:2822-34. Ramakers, J.D., M.I. Verstege, G. Thuijls, A.A. Te Velde, R.P. Mensink, and J. Plat. 2007.
The PPARgamma agonist rosiglitazone impairs colonic inflammation in mice with experimental colitis. J Clin Immunol. 27:275-83.
Rangwala, S.M., and M.A. Lazar. 2004. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci. 25:331-6.
Rasmussen, U.B., V. Vouret-Craviari, S. Jallat, Y. Schlesinger, G. Pages, A. Pavirani, J.P. Lecocq, J. Pouyssegur, and E. Van Obberghen-Schilling. 1991. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS
Lett. 288:123-8. Reeves, M., J. Murphy, R. Greaves, J. Fairley, A. Brehm, and J. Sinclair. 2006.
Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J Virol. 80:9998-10009.
Reeves, M.B., P.A. MacAry, P.J. Lehner, J.G. Sissons, and J.H. Sinclair. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A. 102:4140-5.
Renesto, P., M. Si-Tahar, M. Moniatte, V. Balloy, A. Van Dorsselaer, D. Pidard, and M. Chignard. 1997. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood. 89:1944-53.
Reynolds, D.W., S. Stagno, T.S. Hosty, M. Tiller, and C.A. Alford, Jr. 1973. Maternal cytomegalovirus excretion and perinatal infection. N Engl J Med. 289:1-5.
Ricote, M., J.T. Huang, J.S. Welch, and C.K. Glass. 1999. The peroxisome proliferator-activated receptor(PPARgamma) as a regulator of monocyte/macrophage function. J
Leukoc Biol. 66:733-9. Ricote, M., A.C. Li, T.M. Willson, C.J. Kelly, and C.K. Glass. 1998. The peroxisome
proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 391:79-82.
Rocchi, S., and J. Auwerx. 1999. Peroxisome proliferator-activated receptor-gamma: a versatile metabolic regulator. Ann Med. 31:342-51.
Rue, C.A., M.A. Jarvis, A.J. Knoche, H.L. Meyers, V.R. DeFilippis, S.G. Hansen, M. Wagner, K. Fruh, D.G. Anders, S.W. Wong, P.A. Barry, and J.A. Nelson. 2004. A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J Virol. 78:12529-36.
Sadanari, H., R. Yamada, K. Ohnishi, K. Matsubara, and J. Tanaka. 2005. SUMO-1 modification of the major immediate-early (IE) 1 and 2 proteins of human cytomegalovirus is regulated by different mechanisms and modulates the intracellular localization of the IE1, but not IE2, protein. Arch Virol. 150:1763-82.

120
Saez, E., P. Tontonoz, M.C. Nelson, J.G. Alvarez, U.T. Ming, S.M. Baird, V.A. Thomazy, and R.M. Evans. 1998. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 4:1058-61.
Schaiff, W.T., Y. Barak, and Y. Sadovsky. 2006. The pleiotropic function of PPAR gamma in the placenta. Mol Cell Endocrinol. 249:10-5.
Schaiff, W.T., I. Bildirici, M. Cheong, P.L. Chern, D.M. Nelson, and Y. Sadovsky. 2005. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor signaling regulate fatty acid uptake by primary human placental trophoblasts. J Clin Endocrinol
Metab. 90:4267-75. Schaiff, W.T., M.G. Carlson, S.D. Smith, R. Levy, D.M. Nelson, and Y. Sadovsky. 2000.
Peroxisome proliferator-activated receptor-gamma modulates differentiation of human trophoblast in a ligand-specific manner. J Clin Endocrinol Metab. 85:3874-81.
Scher, J.U., and M.H. Pillinger. 2005. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin
Immunol. 114:100-9. Schild, R.L., C.M. Sonnenberg-Hirche, W.T. Schaiff, I. Bildirici, D.M. Nelson, and Y.
Sadovsky. 2006. The kinase p38 regulates peroxisome proliferator activated receptor-gamma in human trophoblasts. Placenta. 27:191-9.
Schleiss, M.R. 2008. Comparison of vaccine strategies against congenital CMV infection in the guinea pig model. J Clin Virol. 41:224-30.
Scholz, M., H.W. Doerr, and J. Cinatl. 2003. Human cytomegalovirus retinitis: pathogenicity, immune evasion and persistence. Trends Microbiol. 11:171-8.
Schoonjans, K., J. Peinado-Onsurbe, A.M. Lefebvre, R.A. Heyman, M. Briggs, S. Deeb, B. Staels, and J. Auwerx. 1996. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. Embo J. 15:5336-48.
Shalom-Barak, T., J.M. Nicholas, Y. Wang, X. Zhang, E.S. Ong, T.H. Young, S.J. Gendler, R.M. Evans, and Y. Barak. 2004. Peroxisome proliferator-activated receptor gamma controls Muc1 transcription in trophoblasts. Mol Cell Biol. 24:10661-9.
Shao, D., S.M. Rangwala, S.T. Bailey, S.L. Krakow, M.J. Reginato, and M.A. Lazar. 1998. Interdomain communication regulating ligand binding by PPAR-gamma. Nature. 396:377-80.
Shen, D., C. Deng, and M. Zhang. 2007. Peroxisome proliferator-activated receptor gamma agonists inhibit the proliferation and invasion of human colon cancer cells. Postgrad
Med J. 83:414-9. Shirakata, M., M. Terauchi, M. Ablikim, K. Imadome, K. Hirai, T. Aso, and Y. Yamanashi.
2002. Novel immediate-early protein IE19 of human cytomegalovirus activates the origin recognition complex I promoter in a cooperative manner with IE72. J Virol. 76:3158-67.
Shu, H., B. Wong, G. Zhou, Y. Li, J. Berger, J.W. Woods, S.D. Wright, and T.Q. Cai. 2000. Activation of PPARalpha or gamma reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res
Commun. 267:345-9. Silva, M.C., Q.C. Yu, L. Enquist, and T. Shenk. 2003. Human cytomegalovirus UL99-
encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J Virol. 77:10594-605.
Sinclair, J. 2009. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta.
Sinclair, J., and P. Sissons. 2006. Latency and reactivation of human cytomegalovirus. J Gen
Virol. 87:1763-79. Sinzger, C., and G. Jahn. 1996. Human cytomegalovirus cell tropism and pathogenesis.
Intervirology. 39:302-19.

121
Sinzger, C., B. Plachter, A. Grefte, T.H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus in vivo. J Infect Dis. 173:240-5.
Soroceanu, L., A. Akhavan, and C.S. Cobbs. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 455:391-5.
Stagno, S., G. Cloud, R.F. Pass, W.J. Britt, and C.A. Alford. 1984. Factors associated with primary cytomegalovirus infection during pregnancy. J Med Virol. 13:347-53.
Stamminger, T., H. Fickenscher, and B. Fleckenstein. 1990. Cell type-specific induction of the major immediate early enhancer of human cytomegalovirus by cyclic AMP. J Gen Virol. 71 ( Pt 1):105-13.
Stern-Ginossar, N., N. Elefant, A. Zimmermann, D.G. Wolf, N. Saleh, M. Biton, E. Horwitz, Z. Prokocimer, M. Prichard, G. Hahn, D. Goldman-Wohl, C. Greenfield, S. Yagel, H. Hengel, Y. Altuvia, H. Margalit, and O. Mandelboim. 2007. Host immune system gene targeting by a viral miRNA. Science. 317:376-81.
Straat, K., R. de Klark, S. Gredmark-Russ, P. Eriksson, and C. Soderberg-Naucler. 2009. Infection with human cytomegalovirus alters the MMP-9/TIMP-1 balance in human macrophages. J Virol. 83:830-5.
Straus, D.S., and C.K. Glass. 2007. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 28:551-8.
Streblow, D.N., S.M. Varnum, R.D. Smith, and J.A. Nelson. 2006. A proteomics analysis of Human Cytomegalovirus particules. Cytomegaloviruses, molecular biology and
immunology Chapter 5:91-110. Sun, I.Y., M.T. Overgaard, C. Oxvig, and L.C. Giudice. 2002. Pregnancy-associated plasma
protein A proteolytic activity is associated with the human placental trophoblast cell membrane. J Clin Endocrinol Metab. 87:5235-40.
Szatmari, I., E. Rajnavolgyi, and L. Nagy. 2006. PPARgamma, a lipid-activated transcription factor as a regulator of dendritic cell function. Ann N Y Acad Sci. 1088:207-18.
Tabata, T., S. McDonagh, H. Kawakatsu, and L. Pereira. 2007. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: a quantitative analysis. Placenta. 28:527-37.
Tarrade, A., K. Schoonjans, J. Guibourdenche, J.M. Bidart, M. Vidaud, J. Auwerx, C. Rochette-Egly, and D. Evain-Brion. 2001a. PPAR gamma/RXR alpha heterodimers are involved in human CG beta synthesis and human trophoblast differentiation. Endocrinology. 142:4504-14.
Tarrade, A., K. Schoonjans, L. Pavan, J. Auwerx, C. Rochette-Egly, D. Evain-Brion, and T. Fournier. 2001b. PPARgamma/RXRalpha heterodimers control human trophoblast invasion. J Clin Endocrinol Metab. 86:5017-24.
Terhune, S.S., J. Schroer, and T. Shenk. 2004. RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J Virol. 78:10390-8.
Tontonoz, P., E. Hu, and B.M. Spiegelman. 1994. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 79:1147-56.
Tontonoz, P., L. Nagy, J.G. Alvarez, V.A. Thomazy, and R.M. Evans. 1998. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 93:241-52.
Trejo, J., Y. Altschuler, H.W. Fu, K.E. Mostov, and S.R. Coughlin. 2000. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem. 275:31255-65.

122
Uehara, A., K. Muramoto, H. Takada, and S. Sugawara. 2003. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. J Immunol. 170:5690-6.
Vamecq, J., and N. Latruffe. 1999. Medical significance of peroxisome proliferator-activated receptors. Lancet. 354:141-8.
Venema, H., A.P. van den Berg, C. van Zanten, W.J. van Son, M. van der Giessen, and T.H. The. 1994. Natural killer cell responses in renal transplant patients with cytomegalovirus infection. J Med Virol. 42:188-92.
Vergnolle, N. 2009. Protease-activated receptors as drug targets in inflammation and pain. Pharmacol Ther. 123:292-309.
Vu, T.K., D.T. Hung, V.I. Wheaton, and S.R. Coughlin. 1991. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 64:1057-68.
Wang, S.H., Z.L. Sun, Y.J. Guo, Y. Yuan, and L. Li. 2009. PPARgamma-mediated advanced glycation end products regulation of neural stem cells. Mol Cell Endocrinol. 307:176-84.
Wang, X., D.Y. Huang, S.M. Huong, and E.S. Huang. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med. 11:515-21.
Wang, X., S.M. Huong, M.L. Chiu, N. Raab-Traub, and E.S. Huang. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 424:456-61.
Welch, J.S., M. Ricote, T.E. Akiyama, F.J. Gonzalez, and C.K. Glass. 2003. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci U S A. 100:6712-7.
Woodhall, D.L., I.J. Groves, M.B. Reeves, G. Wilkinson, and J.H. Sinclair. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol
Chem. 281:37652-60. Xu, W.F., H. Andersen, T.E. Whitmore, S.R. Presnell, D.P. Yee, A. Ching, T. Gilbert, E.W.
Davie, and D.C. Foster. 1998. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 95:6642-6.
Yamamoto-Tabata, T., S. McDonagh, H.T. Chang, S. Fisher, and L. Pereira. 2004. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol. 78:2831-40.
Yu, X., and J.E. Mertz. 2001. Critical roles of nuclear receptor response elements in replication of hepatitis B virus. J Virol. 75:11354-64.
Zania, P., D. Gourni, A.C. Aplin, R.F. Nicosia, C.S. Flordellis, M.E. Maragoudakis, and N.E. Tsopanoglou. 2009. Parstatin, the cleaved peptide on proteinase-activated receptor 1 activation, is a potent inhibitor of angiogenesis. J Pharmacol Exp Ther. 328:378-89.
Zhu, H., J.P. Cong, D. Yu, W.A. Bresnahan, and T.E. Shenk. 2002. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci U S
A. 99:3932-7.

123
Annexes


to be impaired by infection with HCMV during pregnancy (6).Here we first demonstrate that PPARg can be used for HCMVreplication through its binding to the major immediate-earlyviral promoter and that by this way infection dramatically im-pairs trophoblast migration and invasiveness.
MATERIALS AND METHODS
Cells, viruses, and reagents. U373MG astrocytoma cells, MRC5 fibroblasts,
and Hek-293 cells (InvivoGen) were propagated in medium containing 10% fetal
calf serum, and extravillous cytotrophoblasts (EVCT; also HIPEC) were prop-
agated as described previously (22, 26). Chorionic villi from first-trimester pla-
centas (8 to 12 weeks of gestation) were obtained from volunteers who legally
chose to terminate pregnancy after written approbation (Broussais Hospital,
Paris, France). Cultures of trophoblasts were approved by the local ethical
committee. After dissection, EVCT were isolated by trypsin digestion and puri-
fied through a discontinuous percoll gradient. Cell viability was determined by
flow cytometry analysis of annexin V (AV)-positive cells (fluorescein isothiocya-
nate [FITC]-AV; Dako). About 90% of viable EVCT stained positive for human
invasive extravillous cytotrophoblast markers after culture on matrigel (CK07,
HLA-G, CD9, hPL, c-erB2, or the a5 subunit of the fibronectin receptor) and
expressed RXRa and PPARg in their nuclei. Immunohistochemistry for PPARg
and cytokeratin 7 was performed as previously described (8) on paraffin-embed-
ded sections of an implantation site (placenta and myometrium at 16 weeks of
pregnancy). Immunocytochemistry with anti-RXRa, -PPARg, and -CK07 anti-
bodies on EVCT and HIPEC was performed as described in reference 22.
Virus strains used were AD169 (ATCC), Towne (a gift from S. Michelson,
Institut Pasteur, Paris, France), and VHLE (a gift from C. Sinzger, Tubingen,
Germany). Virus stocks were collected when cytopathic effects on MRC5 cells
(bioMerieux, France) were .90%. Supernatants were clarified of cell debris by
centrifugation at 1,500 3 g for 10 min, ultracentrifuged at 100,000 3 g for 30 min
at 4°C, and stored at 270°C until use. Virus titers were determined by plaque
assay on MRC5 cells using standard methods. UV irradiation of HCMV was
performed with a Spectroline irradiator (EF-140/F) for 20 min.
NS-398 (10 mM; Sigma) and G3335 (30 mM; Calbiochem) were added to cells
30 min before infection and cultures were refed with G3335 every 24 h. Gw9662
was from Calbiochem. 15-Deoxy-D12,14-prostaglandin J2 (15d-PGJ2) and ros-
iglitazone were from Cayman Chemicals. Dose-effect experiments were done for
all reagents, which were each used at the optimal concentration (peak dose) that
did not affect cell viability as tested by trypan blue exclusion, cell morphology,
and nuclei condensation or fragmentation (496-diamino-2-phenylindole [DAPI]
staining). The solvent used for each of them (vehicle control) was assayed as a
control in all sets of experiments.
Reporter plasmids and cell transfection. A PPRE-luciferase (Luc) plasmid
provided by W. Wahli (University of Lausanne, Lausanne, Switzerland) and a
home-made construct containing PPRE sequences were used independently. A
reporter vector for PPARg (pB3xPPRE-Luc) was constructed by modifying the
pNF-kB-Luc plasmid which was described previously (18). Briefly, the NF-kB
binding sites of the pNF-kB-Luc plasmid were removed by digestion with MluI
and BglII and replaced by the phosphorylated, double-stranded MluI-BglII oli-
gonucleotide obtained by annealing 3XPPRE-S (59-CGCGTAGGTCACAGGT
CACAGGTCAGAGGTCAGAGGTCATAGGTCA-39) and 3X PPRE-AS (59-
GATCTGACCTATGACCTCTGACCTCTGACCTGTGACCTGTGACCTA-
39). As a control, the MluI/BglII-linearized pNF-kB-Luc plasmid was also di-
rectly recircularized, providing a vector deleted of any responsive element
(p0RE-Luc).
Three reporter vectors containing the putative PPRE sequences of the MIEP
(PP1-Luc, PP2-Luc, and PP3-Luc) were similarly constructed by replacing the
NF-kB binding sites of the pNF-kB-Luc plasmid with the phosphorylated, dou-
ble-stranded MluI-BglII oligonucleotides obtained by annealing PP1-S (59-CGC
GTAGTTCATAGCCCATAGTTCATAGCCCA-39) and PP1-AS (59-GATCTG
GGCTATGAACTATGGGCTATGAACTA-39) for PP1, PP2-S (59-CGCGTA
ACTTACGGTAAATGGCCCGTAACTTACGGTAAATGGCCCA-39) and
PP2-AS (59-GATCTGGGCCATTTACCGTAAGTTACGGGCCATTTACCG
TAAGTTA-39) for PP2, and PP3-S (59-CGCGTACGTCAATGACGGTAAAT
GGCCCGACGTCAATGACGGTAAATGGCCCA-39) and PP3-AS (59-GATC
TGGGCCATTTACCGTCATTGACGTCGGGCCATTTACCGTCATTGACG
TA-39) for PP3. All constructs were checked by DNA sequencing. For
transfection, U373MG cells were seeded in a 24-well plate at 50,000 cells per
well. Sixteen hours later, pPPRE-reporter plasmids and FuGene 6 reagent
(Roche) were added according to the manufacturer’s instructions. After 24 h of
treatment or infection, cells were lysed with 13 cell culture lysis reagent (Pro-
mega), and luciferase activity was quantified using a Mithras luminometer.
Electrophoretic mobility shift assay. Sequences of probes were selected ac-
cording to previous data that demonstrated the importance of the 59-flanking
sequence (10, 20) of the DR-1 elements. Except for PP1, which contains a RARE
sequence in its 59-flanking region, nucleotides upstream of the DR-1 motif are
conserved for PP2 and PP3. Two hundred nanograms of each double-stranded
probe, PP1, PP2, or PP3, as used for reporter plasmids construction, was labeled
with [g-32P]ATP (Perkin-Elmer) using T4 polynucleotide kinase (New England
Biolabs) for 30 min at 37°C and purified on MiniQuick spin oligo columns
(Roche). Three micrograms of nuclear protein extract (Q proteome nuclear
subfractionation kit; Qiagen) from HCMV-infected U373MG cells (24 h postin-
fection [p.i.]) were incubated at room temperature for 30 min with 15 ng of each
probe, poly(I z C), and 2 mg of either control IgG (Abcam) or PPARg antibody
(Santa Cruz Biotechnology) or 200 ng of cold probe in 10 ml of binding buffer
(0.2 M KCl, 0.1 M K1 HEPES, pH 7.6, 5 mM MgCl2, 5 mM EGTA, 4% Ficoll).
Mixes were submitted to electrophoresis on a 6% Novex retardation gel (In-
vitrogen) in 0.53 Tris-borate-EDTA buffer at 100 V for 90 min. Revelation was
made on Kodak BMR film at 280°C overnight.
ChIP assay. U373MG cells (5 3 104 cells/cm2) were infected with HCMV
(AD169; multiplicity of infection [MOI], 3) for 24 h, fixed with 1% formaldehyde,
lysed (Abcam), and sonicated (Vibracell 72405; Bioblock), in order to obtain
DNA fragments of sizes of ,1,000 bp. DNA-protein complexes were immuno-
precipitated according to the Abcam procedure with chromatin immunoprecipi-
tation (ChIP)-grade antibodies (Abcam), control IgG, anti-histone-3 (H3), or
anti-PPARg, and then incubated with protein A-Sepharose beads (Sigma-Al-
drich). Primers to detect HCMV MIEP were complementary to positions 2272
and 113 relative to the MIEP start site as described in reference 24, with the
sense primer 59-ATT ACC ATG GTG ATG CGG TT-39 and antisense primer
59-GGC GGA GTT GTT ACG ACA T-39. The PCR amplification parameters
were as follows: 95°C for 5 min, and then 50 cycles at 94°C (40 s), 50°C (40 s), and
72°C (90 s).
Generation of purified adenovirus and transduction of Hek-293 cells. Full-
length mouse PPARg2 as well as transduction in Hek-293 cells were done as
follows: PPARg2 was initially N-terminally hemagglutinin (HA) tagged and
cloned into p.Shuttle-CMV (Stratagene) as described in reference 19. The CMV-
HA-PPAR cassette was transferred to the AdEasy-1 vector by homologous
recombination in Escherichia coli to generate the AdHA-PPARg2 construct
(Ad-PPARg). The plasmid was linearized and transfected into Hek-293 cells for
amplification. The amplified adenovirus was purified using CsCl gradients, and
the viral titer was estimated by a plaque assay-based approach as recommended
in the AdEasy protocol (Stratagene). Transduction with adenovirus encoding
PPARg (Ad-PPARg) or not (Ad-vector) was performed at an MOI of 0.1 in
Hek-293 cells at 80% confluence. After 2 h of incubation, the medium was
replaced with fresh medium. The next day, cells were infected with HCMV
(MOI, 3) as indicated. Virus titers were determined on Hek-293 supernatants by
plaque assay on MRC5 cells.
Confocal microscopy. Cells were cultured on coverglasses, washed, fixed, and
permeabilized in methanol for 5 min at 220°C. PPARg was detected with
specific monoclonal antibody (MAb E-8; Santa Cruz Biotechnology) followed by
phycoerythrin-conjugated anti-mouse IgG antibody (Beckmann Coulter). IE1
and IE2 proteins were detected with FITC-conjugated MAb (Chemicon). Fluo-
rescence was analyzed with an LSM 510 confocal microscope (Zeiss, Jena,
Germany). Images were reconstructed by using the LSM image browser software
system (Zeiss).
Oil Red O staining. Cells were fixed and permeabilized in methanol for 2 min
at 220°C and incubated for 10 min with 0.3% (wt/vol) Oil Red O in isopropanol.
After 30 s of incubation in 60% isopropanol, cells were washed in water and
nuclei were counterstained with hematoxylin.
RT-PCR. RNA was extracted by using Trizol (Invitrogen), reverse transcribed
(RT) with Superscript III (Invitrogen), and cDNA amplified with Mastermix
(Eppendorf). Fragments of 215 bp and 217 bp, corresponding to IE1 and IE2
cDNA, were amplified with the following primers: sense (59-AGG TGC CAC
GGC CCG AGA CAC C-39) and antisense (59-TCT GTT TGA CCG AGT TCT
GCC-39) for IE1 and sense (59-AGG TGC CAC GGC CCG AGA CAC C-39)
and antisense (59-GGC GAG GAT GTC CGA GTT CTG CC-39) for IE2. A
300-bp fragment from actin was amplified with the sense primer 59-TCC CTG
GAG AAG AGC TAC GAG-39 and antisense primer 59-CAT CTG CTG GAA
GGT GGA CA-39. PCR was performed as follows: 95°C for 5 min followed by
35 cycles of 95°C (30 s), 58°C (for actin) or 60°C (for IE1) or 62°C (for IE2) for
1 min, 72°C for 1 min, and 72°C for 2 min.
Western blot analyses. Cells lysates were submitted to SDS-PAGE on 4 to
12% polyacrylamide gels (Invitrogen). Blots on Hybond C-extra membrane (Am-
VOL. 84, 2010 PPARg IN HCMV REPLICATION AND PLACENTATION 2947
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m

ersham) were treated with anti-IE antibody (mouse hybridoma E13; a gift from
S. Michelson), anti-UL44 (Abcam), anti-UL99 (Abcam), anti-b-actin (Santa
Cruz Biotechnology), or anti-PPARg (Santa Cruz Biotechnology), and then with
polyclonal rabbit horseradish peroxidase-conjugated antibody, and protein bands
were detected by using an ECL-plus kit (Amersham).
Scanning the CMV sequence with the PPREFinder program. The PPREfinder
program was developed at Loria to exhaustively list PPRE present in DNA
sequences (http://bioinfo.loria.fr/projects/pprefinder/PpreFinder1.1.zip/view).
This three-step program first identifies and localizes all PPRE half-sites (con-
sensus sequence, AGGTCA) in the input sequence, allowing for mismatches
(three in this study) at any place in the consensus 6-base sequence. The second
step consists of localizing pairs of half-sites separated for this study by only one
nucleotide. The third step filters direct repeats (DR-1 motifs) from the preceding
pair list and ranks them according to the total mismatch number over the two
half-sites. A statistical module based on a stationary Markov chain provides an
estimation of the probability that the finding of a given DR-1 motif reflects a
particular base composition in the sequence.
Human trophoblast wound healing and invasion assays. Monolayers of
HIPEC cells (80% confluent) were infected with HCMV, scratched (three per
well), left for 24 h in culture, washed with phosphate-buffered saline (PBS), and
photographed under a light microscope. Wound closure reflecting migration of
cells was quantified by using ImageJ software (NIH). EVCT were cultured on
matrigel-coated transwell membranes (8-mm-diameter pore membranes) for 48 h
in the presence or absence of 1 mM rosiglitazone, 35 mM G3335, and 3 MOI of
HCMV. After CK07 immunostaining and counterstaining with DAPI, pseudop-
odia crossing the porous membrane were quantified and normalized to the total
number of nuclei. Results of a representative experiment (performed in tripli-
cate) are expressed as the percent variation relative to the control.
RESULTS
HCMV induces PPARg transcriptional activity in infected
cells. U373MG astrocytoma cells, which constitutively expressPPARg, were transfected with different PPRE-Luc plasmidsencoding a luciferase gene under the control of the thymidinekinase minimal promoter and a multimerized PPRE recog-nized by PPARg and infected with HCMV (VHLE clinicalstrain). Figure 1A shows that expression of luciferase increasedthroughout infection, which can be assumed to reflect tran-scription of the Luc gene through binding of PPARg to thePPRE sequence. Luciferase activity in uninfected cells may bedue to ligand-independent activity of PPARg (19) or to lowlevels of endogenous agonists. Addition of 15d-PGJ2, a naturalligand of PPARg, to uninfected cells induced Luc transcriptionwith a much lower efficiency than HCMV (Fig. 1B). This mayreflect a better availability of endogenously synthesized 15d-PGJ2 in infected cells and/or that activation of PPARg byHCMV could be in part mediated by other metabolites. Toprovide direct proof that PPARg was involved in the PPRE-driven expression of luciferase, U373MG cells were infected inthe presence of G3335, a cell-permeable dipeptide that acts asa specific and reversible antagonist of PPARg through block-ade of ligand binding. Figure 1C shows that under these con-ditions, luciferase activity was strongly decreased, demonstrat-ing that luciferase transcription depends on the interaction ofPPARg with its ligand. Treatment with NS398, an inhibitor ofCox-2 activity, substantially reduced luciferase activity in in-fected cells (Fig. 1C), demonstrating that Cox-2 activationtakes part in PPARg transcriptional activity, likely through itsrole in prostaglandin synthesis from arachidonic acid. Speci-ficity of PPARg activation by HCMV was further assessed byusing one shot of Gw9662 (Gw), an irreversible antagonistknown to covalently bind and modify the ligand binding site ofPPARg. Figure 1D shows the inhibitory effect of Gw on cellstransfected with the pB3xPPRE-Luc plasmid and treated with
rosiglitazone as well as on those infected with HCMV. Toaddress whether viral neosynthesis was required for activationof PPARg, luciferase activity was quantified from cells infectedwith UV-irradiated virus. According to the usual consider-ations, failure of the irradiated virus (Fig. 1D) to drive lucif-erase expression suggests a role for newly synthesized viralproteins in PPARg activation. Transfection with PPRE-de-leted control plasmid (Fig. 1D, right histogram) did not showany luciferase activity.
To further address whether PPARg activity was restricted toinfected cells, either MRC5 fibroblasts or U373MG cells wereinfected with HCMV. In MRC5 cells, infection strongly redis-tributed PPARg staining from the cytoplasm to the nucleus ofinfected cells as assessed by confocal microscopy with mergedexpression of IE proteins and PPARg in the nucleus of in-fected cells (Fig. 1E). Identical results were obtained inU373MG cells although with less intensity (data not shown),and we further assessed the function of PPARg in these cellsthrough its ability to induce activation of genes involved inlipogenesis. Staining with Oil Red O revealed accumulation oflipid droplets in infected cells, contrary to those treated withthe PPARg antagonist G3335 (Fig. 1F). These experimentsprovided evidence that PPARg is activated in HCMV-infectedcells, as demonstrated by nuclear translocation and lipid accu-mulation.
PPARg takes part in IE2 expression and HCMV replication.
According to previous data demonstrating that Cox-2 inhibi-tors block the accumulation of IE2 mRNA and protein andconsequently reduce replication of HCMV (AD169 strain)(28), we examined the effects of G3335 on IE1 and IE2 ex-pression and on the yield of infectious virus. Infection ofU373MG cells in the presence of G3335 showed that inhibitionof PPARg activity resulted in a huge decrease in the amount ofIE2 transcripts, compared to IE1, after 24 h of infection (Fig.2A). Western blot analysis of cell lysates obtained from day 1to day 5 after infection further confirmed a role for PPARg inmodulating expression of IE2 protein, as IE2 expression wasdelayed and the IE2/IE1 ratio was consistently lower in treatedcells (Fig. 2B). As expected, expression of early (UL44) andlate (UL99) proteins was modified in the same way by treat-ment with PPARg antagonist (data not shown). Furthermore,production of infectious virus, as assessed by plaque assayevery 24 h, was reduced by a factor of 100 in the presence of aPPARg antagonist (Fig. 2C), demonstrating that in these cellsHCMV uses PPARg activity for its replication. Identical re-sults were obtained with MRC5 fibroblasts (data not shown).Altogether, our data demonstrate that PPARg can regulateexpression of IE2 mRNA and viral replication in a way thatdepends on association with its ligand.
To further characterize the responsiveness of putativePPREs contained in the MIEP and to provide direct proof forthe role of PPARg in HCMV replication, Hek-293 cells ex-pressing RXR but not PPARg were coinfected with adenoviralvectors encoding PPARg and with HCMV. Under ectopicexpression of PPARg, HCMV-infected Hek-293 cells ex-pressed a higher ratio of IE2 to IE1 mRNA compared withthose either nontransduced or transduced with control adeno-virus, and the relative amount of IE2 mRNA with respect tob-actin was much higher than in control cells (Fig. 2D), sug-gesting a role for PPARg in production of IE2. Quantification
2948 RAUWEL ET AL. J. VIROL.
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m
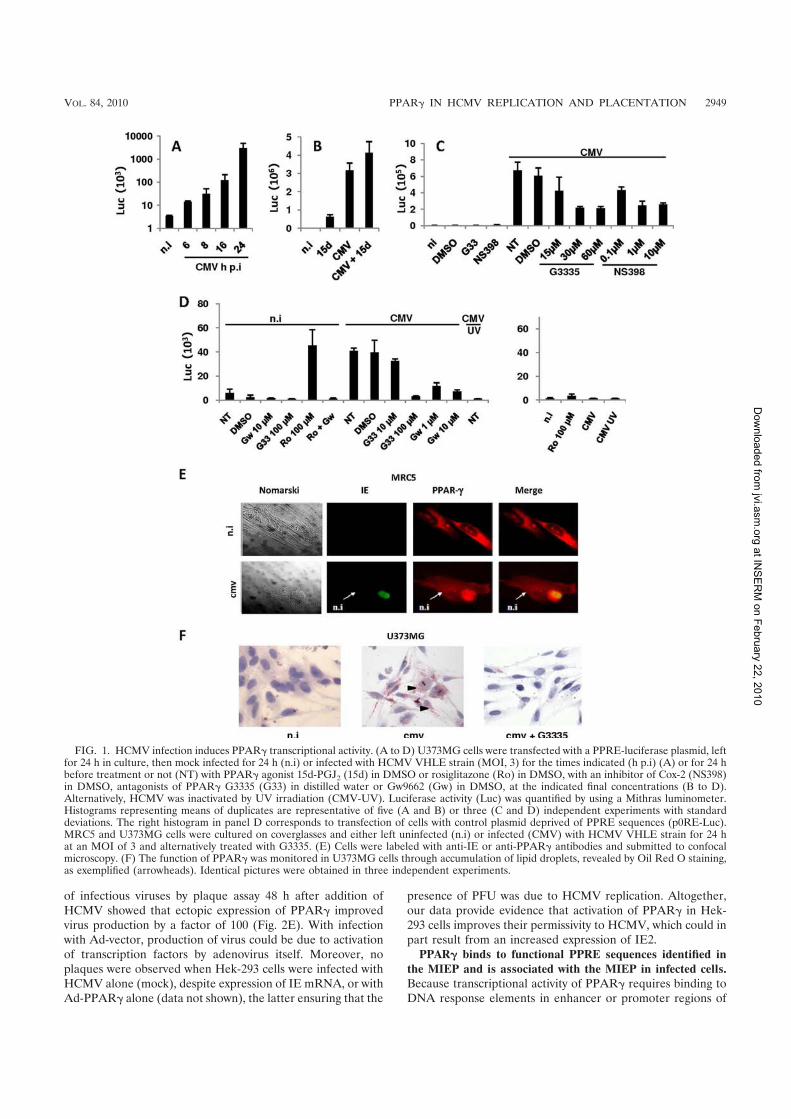
of infectious viruses by plaque assay 48 h after addition ofHCMV showed that ectopic expression of PPARg improvedvirus production by a factor of 100 (Fig. 2E). With infectionwith Ad-vector, production of virus could be due to activationof transcription factors by adenovirus itself. Moreover, noplaques were observed when Hek-293 cells were infected withHCMV alone (mock), despite expression of IE mRNA, or withAd-PPARg alone (data not shown), the latter ensuring that the
presence of PFU was due to HCMV replication. Altogether,our data provide evidence that activation of PPARg in Hek-293 cells improves their permissivity to HCMV, which could inpart result from an increased expression of IE2.
PPARg binds to functional PPRE sequences identified in
the MIEP and is associated with the MIEP in infected cells.
Because transcriptional activity of PPARg requires binding toDNA response elements in enhancer or promoter regions of
FIG. 1. HCMV infection induces PPARg transcriptional activity. (A to D) U373MG cells were transfected with a PPRE-luciferase plasmid, leftfor 24 h in culture, then mock infected for 24 h (n.i) or infected with HCMV VHLE strain (MOI, 3) for the times indicated (h p.i) (A) or for 24 hbefore treatment or not (NT) with PPARg agonist 15d-PGJ2 (15d) in DMSO or rosiglitazone (Ro) in DMSO, with an inhibitor of Cox-2 (NS398)in DMSO, antagonists of PPARg G3335 (G33) in distilled water or Gw9662 (Gw) in DMSO, at the indicated final concentrations (B to D).Alternatively, HCMV was inactivated by UV irradiation (CMV-UV). Luciferase activity (Luc) was quantified by using a Mithras luminometer.Histograms representing means of duplicates are representative of five (A and B) or three (C and D) independent experiments with standarddeviations. The right histogram in panel D corresponds to transfection of cells with control plasmid deprived of PPRE sequences (p0RE-Luc).MRC5 and U373MG cells were cultured on coverglasses and either left uninfected (n.i) or infected (CMV) with HCMV VHLE strain for 24 hat an MOI of 3 and alternatively treated with G3335. (E) Cells were labeled with anti-IE or anti-PPARg antibodies and submitted to confocalmicroscopy. (F) The function of PPARg was monitored in U373MG cells through accumulation of lipid droplets, revealed by Oil Red O staining,as exemplified (arrowheads). Identical pictures were obtained in three independent experiments.
VOL. 84, 2010 PPARg IN HCMV REPLICATION AND PLACENTATION 2949
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m
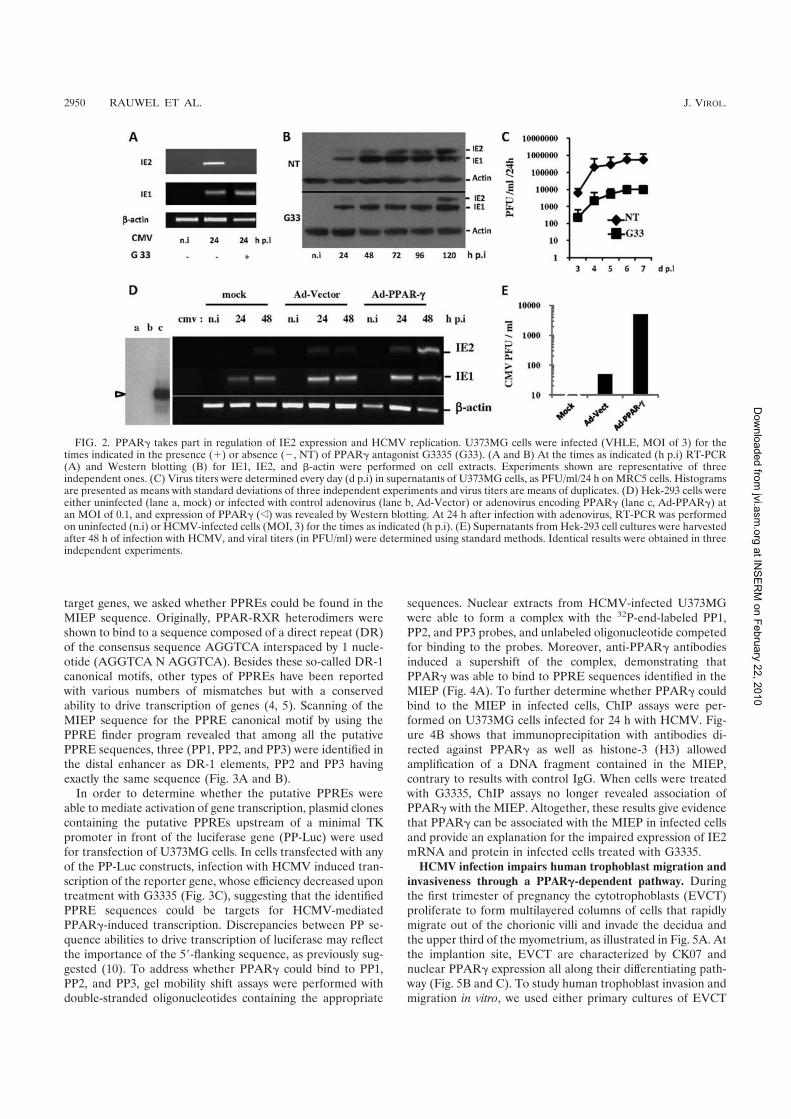
target genes, we asked whether PPREs could be found in theMIEP sequence. Originally, PPAR-RXR heterodimers wereshown to bind to a sequence composed of a direct repeat (DR)of the consensus sequence AGGTCA interspaced by 1 nucle-otide (AGGTCA N AGGTCA). Besides these so-called DR-1canonical motifs, other types of PPREs have been reportedwith various numbers of mismatches but with a conservedability to drive transcription of genes (4, 5). Scanning of theMIEP sequence for the PPRE canonical motif by using thePPRE finder program revealed that among all the putativePPRE sequences, three (PP1, PP2, and PP3) were identified inthe distal enhancer as DR-1 elements, PP2 and PP3 havingexactly the same sequence (Fig. 3A and B).
In order to determine whether the putative PPREs wereable to mediate activation of gene transcription, plasmid clonescontaining the putative PPREs upstream of a minimal TKpromoter in front of the luciferase gene (PP-Luc) were usedfor transfection of U373MG cells. In cells transfected with anyof the PP-Luc constructs, infection with HCMV induced tran-scription of the reporter gene, whose efficiency decreased upontreatment with G3335 (Fig. 3C), suggesting that the identifiedPPRE sequences could be targets for HCMV-mediatedPPARg-induced transcription. Discrepancies between PP se-quence abilities to drive transcription of luciferase may reflectthe importance of the 59-flanking sequence, as previously sug-gested (10). To address whether PPARg could bind to PP1,PP2, and PP3, gel mobility shift assays were performed withdouble-stranded oligonucleotides containing the appropriate
sequences. Nuclear extracts from HCMV-infected U373MGwere able to form a complex with the 32P-end-labeled PP1,PP2, and PP3 probes, and unlabeled oligonucleotide competedfor binding to the probes. Moreover, anti-PPARg antibodiesinduced a supershift of the complex, demonstrating thatPPARg was able to bind to PPRE sequences identified in theMIEP (Fig. 4A). To further determine whether PPARg couldbind to the MIEP in infected cells, ChIP assays were per-formed on U373MG cells infected for 24 h with HCMV. Fig-ure 4B shows that immunoprecipitation with antibodies di-rected against PPARg as well as histone-3 (H3) allowedamplification of a DNA fragment contained in the MIEP,contrary to results with control IgG. When cells were treatedwith G3335, ChIP assays no longer revealed association ofPPARg with the MIEP. Altogether, these results give evidencethat PPARg can be associated with the MIEP in infected cellsand provide an explanation for the impaired expression of IE2mRNA and protein in infected cells treated with G3335.
HCMV infection impairs human trophoblast migration and
invasiveness through a PPARg-dependent pathway. Duringthe first trimester of pregnancy the cytotrophoblasts (EVCT)proliferate to form multilayered columns of cells that rapidlymigrate out of the chorionic villi and invade the decidua andthe upper third of the myometrium, as illustrated in Fig. 5A. Atthe implantion site, EVCT are characterized by CK07 andnuclear PPARg expression all along their differentiating path-way (Fig. 5B and C). To study human trophoblast invasion andmigration in vitro, we used either primary cultures of EVCT
FIG. 2. PPARg takes part in regulation of IE2 expression and HCMV replication. U373MG cells were infected (VHLE, MOI of 3) for thetimes indicated in the presence (1) or absence (2, NT) of PPARg antagonist G3335 (G33). (A and B) At the times as indicated (h p.i) RT-PCR(A) and Western blotting (B) for IE1, IE2, and b-actin were performed on cell extracts. Experiments shown are representative of threeindependent ones. (C) Virus titers were determined every day (d p.i) in supernatants of U373MG cells, as PFU/ml/24 h on MRC5 cells. Histogramsare presented as means with standard deviations of three independent experiments and virus titers are means of duplicates. (D) Hek-293 cells wereeither uninfected (lane a, mock) or infected with control adenovirus (lane b, Ad-Vector) or adenovirus encoding PPARg (lane c, Ad-PPARg) atan MOI of 0.1, and expression of PPARg (�) was revealed by Western blotting. At 24 h after infection with adenovirus, RT-PCR was performedon uninfected (n.i) or HCMV-infected cells (MOI, 3) for the times as indicated (h p.i). (E) Supernatants from Hek-293 cell cultures were harvestedafter 48 h of infection with HCMV, and viral titers (in PFU/ml) were determined using standard methods. Identical results were obtained in threeindependent experiments.
2950 RAUWEL ET AL. J. VIROL.
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m
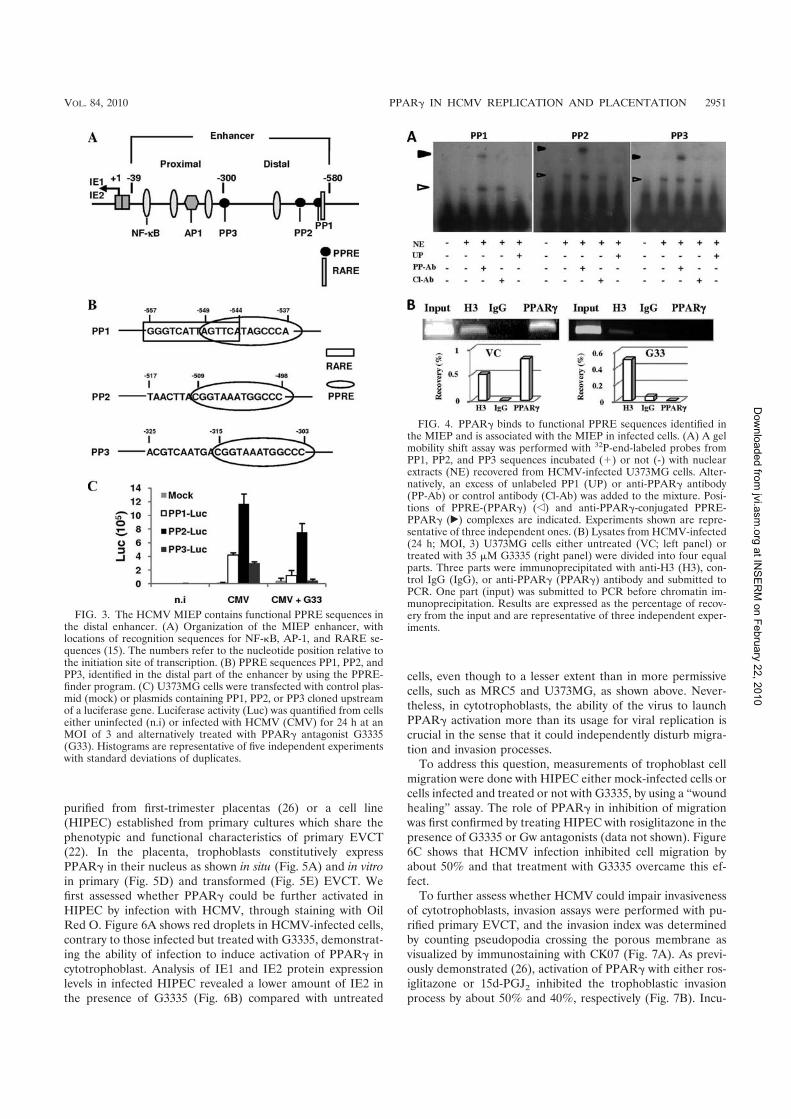
purified from first-trimester placentas (26) or a cell line(HIPEC) established from primary cultures which share thephenotypic and functional characteristics of primary EVCT(22). In the placenta, trophoblasts constitutively expressPPARg in their nucleus as shown in situ (Fig. 5A) and in vitro
in primary (Fig. 5D) and transformed (Fig. 5E) EVCT. Wefirst assessed whether PPARg could be further activated inHIPEC by infection with HCMV, through staining with OilRed O. Figure 6A shows red droplets in HCMV-infected cells,contrary to those infected but treated with G3335, demonstrat-ing the ability of infection to induce activation of PPARg incytotrophoblast. Analysis of IE1 and IE2 protein expressionlevels in infected HIPEC revealed a lower amount of IE2 inthe presence of G3335 (Fig. 6B) compared with untreated
cells, even though to a lesser extent than in more permissivecells, such as MRC5 and U373MG, as shown above. Never-theless, in cytotrophoblasts, the ability of the virus to launchPPARg activation more than its usage for viral replication iscrucial in the sense that it could independently disturb migra-tion and invasion processes.
To address this question, measurements of trophoblast cellmigration were done with HIPEC either mock-infected cells orcells infected and treated or not with G3335, by using a “woundhealing” assay. The role of PPARg in inhibition of migrationwas first confirmed by treating HIPEC with rosiglitazone in thepresence of G3335 or Gw antagonists (data not shown). Figure6C shows that HCMV infection inhibited cell migration byabout 50% and that treatment with G3335 overcame this ef-fect.
To further assess whether HCMV could impair invasivenessof cytotrophoblasts, invasion assays were performed with pu-rified primary EVCT, and the invasion index was determinedby counting pseudopodia crossing the porous membrane asvisualized by immunostaining with CK07 (Fig. 7A). As previ-ously demonstrated (26), activation of PPARg with either ros-iglitazone or 15d-PGJ2 inhibited the trophoblastic invasionprocess by about 50% and 40%, respectively (Fig. 7B). Incu-
FIG. 3. The HCMV MIEP contains functional PPRE sequences inthe distal enhancer. (A) Organization of the MIEP enhancer, withlocations of recognition sequences for NF-kB, AP-1, and RARE se-quences (15). The numbers refer to the nucleotide position relative tothe initiation site of transcription. (B) PPRE sequences PP1, PP2, andPP3, identified in the distal part of the enhancer by using the PPRE-finder program. (C) U373MG cells were transfected with control plas-mid (mock) or plasmids containing PP1, PP2, or PP3 cloned upstreamof a luciferase gene. Luciferase activity (Luc) was quantified from cellseither uninfected (n.i) or infected with HCMV (CMV) for 24 h at anMOI of 3 and alternatively treated with PPARg antagonist G3335(G33). Histograms are representative of five independent experimentswith standard deviations of duplicates.
FIG. 4. PPARg binds to functional PPRE sequences identified inthe MIEP and is associated with the MIEP in infected cells. (A) A gelmobility shift assay was performed with 32P-end-labeled probes fromPP1, PP2, and PP3 sequences incubated (1) or not (-) with nuclearextracts (NE) recovered from HCMV-infected U373MG cells. Alter-natively, an excess of unlabeled PP1 (UP) or anti-PPARg antibody(PP-Ab) or control antibody (Cl-Ab) was added to the mixture. Posi-tions of PPRE-(PPARg) (�) and anti-PPARg-conjugated PPRE-PPARg (‹) complexes are indicated. Experiments shown are repre-sentative of three independent ones. (B) Lysates from HCMV-infected(24 h; MOI, 3) U373MG cells either untreated (VC; left panel) ortreated with 35 mM G3335 (right panel) were divided into four equalparts. Three parts were immunoprecipitated with anti-H3 (H3), con-trol IgG (IgG), or anti-PPARg (PPARg) antibody and submitted toPCR. One part (input) was submitted to PCR before chromatin im-munoprecipitation. Results are expressed as the percentage of recov-ery from the input and are representative of three independent exper-iments.
VOL. 84, 2010 PPARg IN HCMV REPLICATION AND PLACENTATION 2951
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m

bation of cells with HCMV resulted in a 30% inhibition ofinvasion, which was totally abrogated following preincubationwith G3335 (Fig. 7C). Altogether, these data provide evidencethat activation of PPARg by HCMV is involved in defectivetrophoblastic migration and invasion processes.
DISCUSSION
Implication of Cox-2 and PGE2 in the viral cycle of herpes-viruses has been noted for a long time, as anti-inflammatorydrugs such as aspirin were shown to modulate primary infec-tion and to decrease reactivation of latent genome. Accord-ingly, studies conducted by Zhu and collaborators (28) indi-cated that inflammation may promote HCMV replication andreactivation, and blockades of Cox-2 activity and of prostaglan-din production were then considered as potential antiviraltherapeutics. Interestingly, in the macaque rhesus model, in-fection with the rhesus cytomegalovirus (RhCMV) did notinduce expression and activation of cellular Cox-2, contrary tohuman CMV, but the virus encoded a protein with high ho-
mology to cellular Cox-2 (vCox-2) (25) which appeared deter-minant for replication in endothelial cells, highlighting the roleof the prostaglandin metabolic pathway in replication of cyto-megaloviruses. HCMV replication and reactivation requiretranscription of the IE genes UL123 and UL122, encoding IE1and IE2 proteins, respectively, two major products resultingfrom a differential splicing. Production of IE2 but not IE1 wasshown to be crucial for productive replication, and transcrip-tion of IE2 but not IE1 was significantly affected by Cox-2inhibitors (28). A role for cyclic AMP (cAMP) response ele-ments contained in the MIEP was suggested (17) with respectto the known ability of prostaglandins to increase cAMP, butfor the moment no direct proof has been obtained to supportthis hypothesis. Our results demonstrate for the first time arole for PPARg in production of IE2 through binding to PPREsequences contained in the MIEP, and experiments demon-strating a huge decrease in viral production in the presence ofPPARg antagonist confirmed the key role of PPARg in viralreplication. Upstream of PPARg activation, molecular andcellular events underlying activation of Cox-2 are known toinvolve the enzymatic activity of the cellular phospholipase A2(c-PLA2). It has been known for a long time that c-PLA2activity can be induced by HCMV infection (1), but moreinterestingly, a connection between infection and PPARg ac-tivation was supported by a study demonstrating that HCMVparticles carry a cell-derived PLA2 that is boarded during theassembly process, which plays a major role in infectivity ofMRC5 fibroblasts (2). PLA2 activity is responsible for theproduction of arachidonic acid, a substrate for both Cox-2 andlipoxygenase activities, and we assume that it is a reason whyc-PLA2-specific inhibitors dramatically impair virus produc-tion (2).
Nevertheless, given that cPLA2 is involved in many complexprocesses, including embryogenesis, inflammation, and repro-duction, the potential side effects of inhibitors, provided thathighly selective ones are available, might discredit their poten-tial therapeutic indications. With respect to these observationsand to those reported in the present paper, we can imagine ascenario where virus entry could initiate the release of arachi-donic acid through boarded c-PLA2 activation, a starting pointof a process involving Cox-2- as well as lipoxygenase-mediatedproduction of PPARg ligands, the latter being considered afinal requisite for PPARg-mediated transcription in infectedcells. Nevertheless, our data demonstrating the inability ofUV-irradiated viruses to induce activation of PPARg raise thequestion of a role for neosynthesized viral proteins in theactivation process, and they also raise the question of whetherirradiation could alter the structure and function of virionsconstituents including boarded cPLA2.
Surprisingly, and in agreement with previous data usingCox-2 inhibitors (28), PPARg inhibition differentially impairedthe expression of IE1 and IE2 mRNA, suggesting a role forPPARg in favoring IE2 mRNA production. This was sup-ported by our experiments demonstrating a huge increase inexpression of IE2 but not IE1 mRNA under ectopic expressionof PPARg in HCMV-infected Hek-293 cells. Since IE1 andIE2 transcripts result from differential splicing events whichare temporally regulated after infection, IE1 being producedbefore IE2, we can assume that binding of PPARg to theMIEP could be coordinated for instance by temporal chroma-
FIG. 5. PPARg is expressed in trophoblasts in situ, in primary(EVCT) and in transformed (HIPEC) cytotrophoblasts. (A) Schematicrepresentation of a chorionic villous at the implantation site. EVCT (inred) invade the decidua up to the first third of the myometrium and theuterine arteries (vct, villous cytotrophoblast; st, syncytiotrophoblast;evct, extravillous cytotrophoblast; sc, stromal core; is, intervillousspace; ua, uterine artery; d, decidua). (B and C) Immunohistochemicalstaining for CK07, a specific trophoblast marker of invasive EVCT(B) and for PPARg (C) in placental sections at the implantation site(week 16 of pregnancy). (D and E) Immunostaining for CK07, RXRa,PPARg, and nucleus with hematoxylin and DAPI in EVCT (D) andHIPEC (E). These experiments were extensively reproduced.
2952 RAUWEL ET AL. J. VIROL.
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m
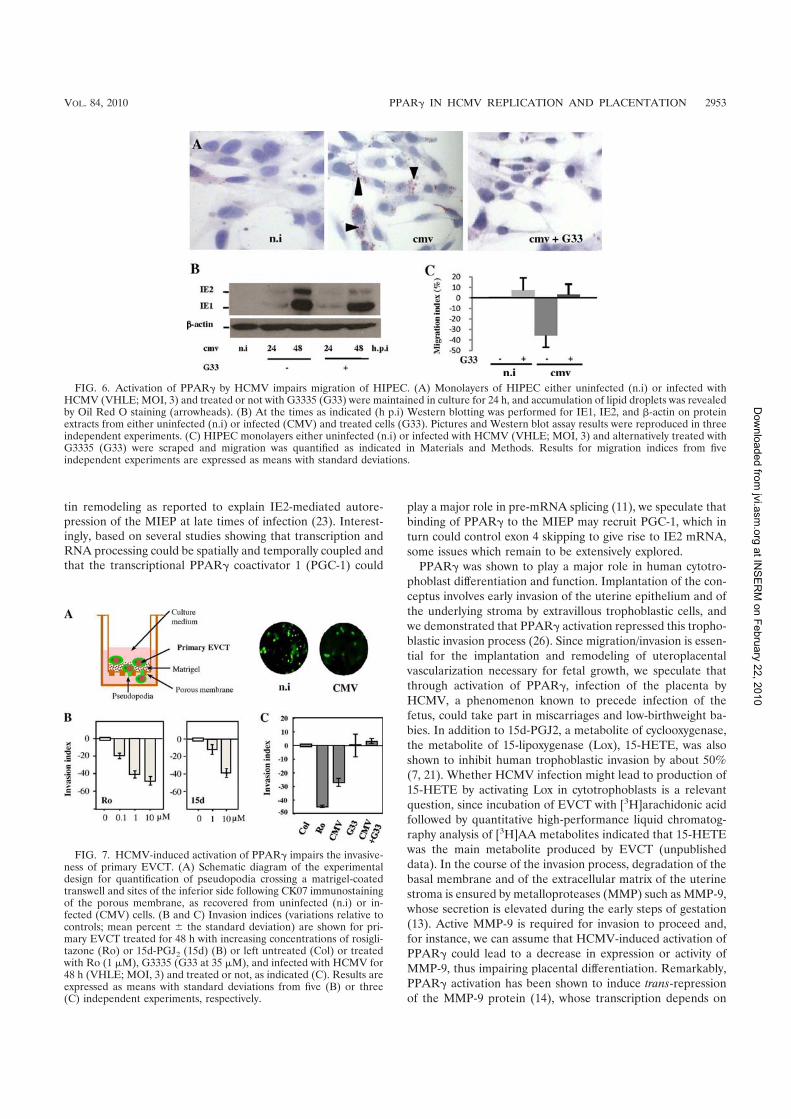
tin remodeling as reported to explain IE2-mediated autore-pression of the MIEP at late times of infection (23). Interest-ingly, based on several studies showing that transcription andRNA processing could be spatially and temporally coupled andthat the transcriptional PPARg coactivator 1 (PGC-1) could
play a major role in pre-mRNA splicing (11), we speculate thatbinding of PPARg to the MIEP may recruit PGC-1, which inturn could control exon 4 skipping to give rise to IE2 mRNA,some issues which remain to be extensively explored.
PPARg was shown to play a major role in human cytotro-phoblast differentiation and function. Implantation of the con-ceptus involves early invasion of the uterine epithelium and ofthe underlying stroma by extravillous trophoblastic cells, andwe demonstrated that PPARg activation repressed this tropho-blastic invasion process (26). Since migration/invasion is essen-tial for the implantation and remodeling of uteroplacentalvascularization necessary for fetal growth, we speculate thatthrough activation of PPARg, infection of the placenta byHCMV, a phenomenon known to precede infection of thefetus, could take part in miscarriages and low-birthweight ba-bies. In addition to 15d-PGJ2, a metabolite of cyclooxygenase,the metabolite of 15-lipoxygenase (Lox), 15-HETE, was alsoshown to inhibit human trophoblastic invasion by about 50%(7, 21). Whether HCMV infection might lead to production of15-HETE by activating Lox in cytotrophoblasts is a relevantquestion, since incubation of EVCT with [3H]arachidonic acidfollowed by quantitative high-performance liquid chromatog-raphy analysis of [3H]AA metabolites indicated that 15-HETEwas the main metabolite produced by EVCT (unpublisheddata). In the course of the invasion process, degradation of thebasal membrane and of the extracellular matrix of the uterinestroma is ensured by metalloproteases (MMP) such as MMP-9,whose secretion is elevated during the early steps of gestation(13). Active MMP-9 is required for invasion to proceed and,for instance, we can assume that HCMV-induced activation ofPPARg could lead to a decrease in expression or activity ofMMP-9, thus impairing placental differentiation. Remarkably,PPARg activation has been shown to induce trans-repressionof the MMP-9 protein (14), whose transcription depends on
FIG. 6. Activation of PPARg by HCMV impairs migration of HIPEC. (A) Monolayers of HIPEC either uninfected (n.i) or infected withHCMV (VHLE; MOI, 3) and treated or not with G3335 (G33) were maintained in culture for 24 h, and accumulation of lipid droplets was revealedby Oil Red O staining (arrowheads). (B) At the times as indicated (h p.i) Western blotting was performed for IE1, IE2, and b-actin on proteinextracts from either uninfected (n.i) or infected (CMV) and treated cells (G33). Pictures and Western blot assay results were reproduced in threeindependent experiments. (C) HIPEC monolayers either uninfected (n.i) or infected with HCMV (VHLE; MOI, 3) and alternatively treated withG3335 (G33) were scraped and migration was quantified as indicated in Materials and Methods. Results for migration indices from fiveindependent experiments are expressed as means with standard deviations.
FIG. 7. HCMV-induced activation of PPARg impairs the invasive-ness of primary EVCT. (A) Schematic diagram of the experimentaldesign for quantification of pseudopodia crossing a matrigel-coatedtranswell and sites of the inferior side following CK07 immunostainingof the porous membrane, as recovered from uninfected (n.i) or in-fected (CMV) cells. (B and C) Invasion indices (variations relative tocontrols; mean percent 6 the standard deviation) are shown for pri-mary EVCT treated for 48 h with increasing concentrations of rosigli-tazone (Ro) or 15d-PGJ2 (15d) (B) or left untreated (Col) or treatedwith Ro (1 mM), G3335 (G33 at 35 mM), and infected with HCMV for48 h (VHLE; MOI, 3) and treated or not, as indicated (C). Results areexpressed as means with standard deviations from five (B) or three(C) independent experiments, respectively.
VOL. 84, 2010 PPARg IN HCMV REPLICATION AND PLACENTATION 2953
at IN
SE
RM
on F
ebru
ary
22, 2
010
jvi.a
sm
.org
Dow
nlo
aded fro
m





























