Embed Size (px)
Citation preview

RMN
Travaux pratiques avancés I-II
(PHQ560-660)
Professeur : Je�rey Quilliam
Coordonnateur : Guy Bernier
Département de physique
Faculté des Sciences
Université de Sherbrooke
©Guy Bernier 2019

PHQ560-660 RMN
Table des matières
1 Introduction 3
2 THÉORIE 42.1 Spin et moment magnétique . . . . . . . . . . . . . . . . . . . . . . . . . . . 42.2 Présence d'un champ magnétique : levée de dégénérescence . . . . . . . . . . 52.3 Aimantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.4 Ordres de grandeur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72.5 La précession de Larmor (approche classique) . . . . . . . . . . . . . . . . . 92.6 Référentiel tournant et application d'une induction . . . . . . . . . . . . . . 102.7 Déplacement chimique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.8 Les équations de Bloch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132.9 Remarques générales sur T1 et T2. . . . . . . . . . . . . . . . . . . . . . . . . 14
3 Principe de la mesure 153.1 Montage expérimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153.2 Subtilité de la phase du signal . . . . . . . . . . . . . . . . . . . . . . . . . . 163.3 Di�érence entre et T2 et T ∗2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173.4 Écho de spin et mesure de T2 . . . . . . . . . . . . . . . . . . . . . . . . . . 18
3.4.1 Séquence Carr-Purcell . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.4.2 Séquence de Carr-Purcell-Meiboom-Gill . . . . . . . . . . . . . . . . . 21
3.5 Mesure de T1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
4 Partie expérimentale 234.1 Résonance du proton . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
4.1.1 Impulsion RF (fréquence de résonance du proton) . . . . . . . . . . . 244.1.2 Signal FID . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254.1.3 Impulsion π (180o) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 264.1.4 Transformée de Fourier (LMO) . . . . . . . . . . . . . . . . . . . . . . 264.1.5 Di�érence entre l'huile minérale, la glycérine et l'eau. . . . . . . . . . 264.1.6 Mesures de T 1 et T 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 264.1.7 TMS et chloroforme . . . . . . . . . . . . . . . . . . . . . . . . . . . . 274.1.8 TMS avec et sans gradient de champ . . . . . . . . . . . . . . . . . . . 284.1.9 Imagerie RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284.1.10 Fluorinert FC-43 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
Annexe A : Les transformées de Fourier 30
Annexe B Basculement de l'aimantation[11] 33
Annexe C Hamiltonien nucléaire 34
1.2

PHQ560-660 RMN
Quelques faits marquants :
1896 : Pieter Zeeman (Nobel) observe que des raies spectrales émises par un gaz excitése subdivisent lorsque la source est soumise à un champ magnétique.
1936 : Observation des moments magnétiques nucléaires : Rabi, Alvarez er Ramsey(Nobel).
1946 : Felix Bloch et Edward Mills Purcell (Nobel) réalisent de manière indépen-dante les premières mesures du magnétisme nucléaire par induction magné-tique ; technique à la base de la RMN moderne.
1950 : Découverte des échos de spin par Erwin L. Hahn. Cette technique est largementutilisée aujourd'hui. W. Proctor et W. Dickinson observent indépendammentle phénomène de � déplacement chimique � qui révolutionnera le domaine dela chimie organique.
1959 : Spectres RMN résolus dans les solides grâce à la technique de rotation autourde l'axe magique développée par E. R. Andrew.
1966 : RMN par transformée de Fourier e�ectuée par Richard R. Ernst (Nobel).
Question 1 : Par la suite, deux autres découvertes majeures dans le domaine de laRMN ont mené à l'obtention d'un prix Nobel. Identi�ez-en une.
1 Introduction
Le magnétisme nucléaire est un phénomène très faible, surtout si on le compare au magné-tisme électronique qui peut donner lieu aux phénomènes de ferromagnétisme ou d'antifer-romagnétisme. Nous verrons d'ailleurs que le paramagnétisme nucléaire est pratiquement laseule manifestation du magnétisme nucléaire observable.
Néanmoins, ce magnétisme nucléaire est mesurable et peut, grâce à une technique de réso-nance, la RMN, se révéler une sonde précieuse de la matière et surtout de son environnement.C'est dans le domaine de la chimie organique que la RMN s'est avérée des plus e�caces.Elle permet la détermination de structures complexes. On l'utilise autant pour reconnaitredes structures connues que pour déterminer l'emplacement des atomes d'une nouvelle molé-cule. La RMN des solides est pour sa part beaucoup plus di�cile à réaliser et à interpréter,mais donne quand même beaucoup d'information utile que ce soit du point de vue struc-tural ou électronique. La RMN du solide est particulièrement utile dans les domaines de lasupraconductivité et du magnétisme par exemple.
Dans cette expérience, l'accent sera mis sur la RMN des liquides. L'approche quantique seradélaissée au pro�t d'une approche classique a�n de préserver au maximum le sens physiquedes phénomènes qui seront observés.

PHQ560-660 RMN
2 THÉORIE
2.1 Spin et moment magnétique
Le proton et le neutron possèdent chacun un spin I = 1/2. Pour un noyau donné, la valeurde I dépend du nombre de nucléons qu'il contient. Si le nombre de masse et le numéroatomique du noyau sont pairs, alors I = 0. Pour tous les autres cas, I ≥ 1/2.
Question 2 : Pour quels noyaux (conditions sur le nombre de masse et le numéroatomique) obtient-on une valeur de I entière ?
On peut se représenter un noyau comme une petite sphère uniformément chargée positive-ment tournant autour d'un axe (seulement si S = 1/2 car les spins de plus grande valeuront une distribution de charge non sphérique). Cela donne lieu à l'apparition d'un momentmagnétique µ colinéaire et proportionnel à I :
µ = γI (1)
La constante γ est appelée � rapport gyromagnétique �.
I
(cas: γ > 0) μ
Figure 1: Moment magnétique du noyau.
On détermine expérimentalement γ dont le tableau suivant donne quelques valeurs pour desnoyaux utilisables en RMN.
Noyau Spin γ (106 rad. s-1 T-1) γ/2π(MHz/T) Abondance naturelle (%)11H 1/2 267.513 42.576 99.9921H 1 41.066 6.536 0.016136 C 1/2 67.262 10.705 1.108147 N 1 19.338 3.0777 99.63157 N 1/2 -27.116 -4.316 0.37178 O 5/2 -36.279 -5.774 0.037199 F 1/2 251.662 40.053 100.0
Tableau 1: Rapports gyromagnétiques de quelques noyaux utilisables en RMN.
1.4
PHQ560-660 RMN
Attention aux unités !
Le calcul de γ est fait à partir de la relation ω0 = γB0 (utilisant l'induction magnétique),alors γ s'exprime en rad. s−1T−1.
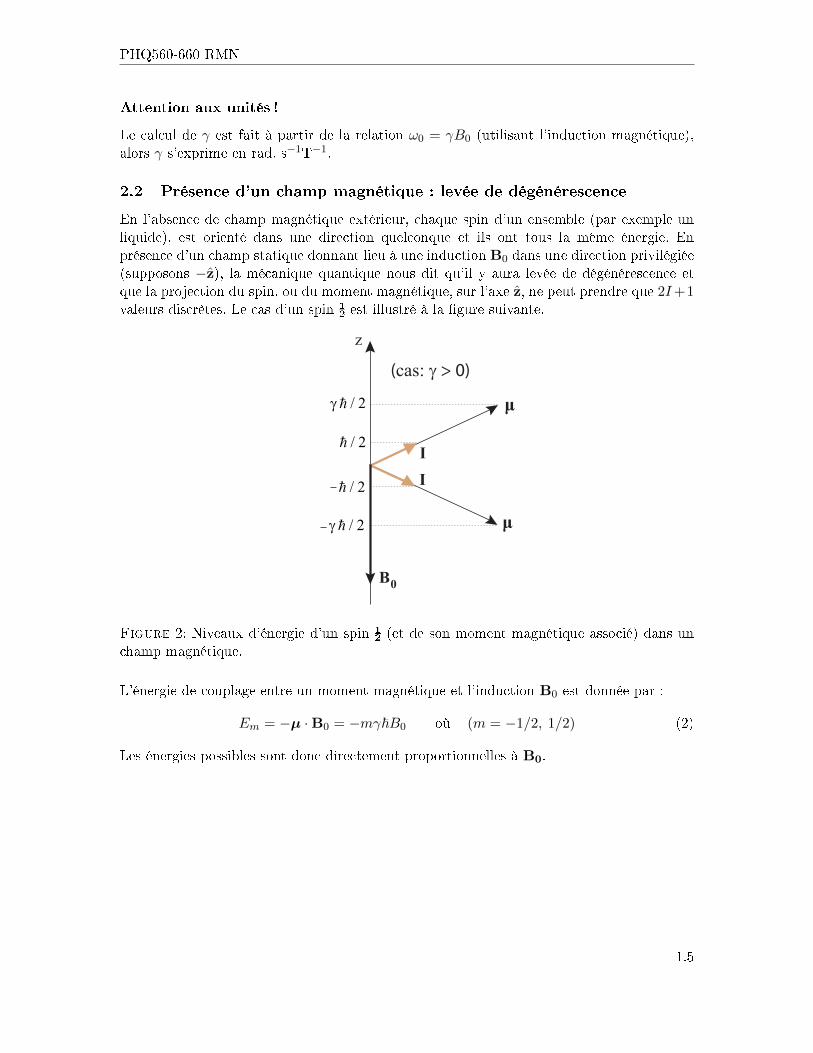
2.2 Présence d'un champ magnétique : levée de dégénérescence
En l'absence de champ magnétique extérieur, chaque spin d'un ensemble (par exemple unliquide), est orienté dans une direction quelconque et ils ont tous la même énergie. Enprésence d'un champ statique donnant lieu à une induction B0 dans une direction privilégiée(supposons −z), la mécanique quantique nous dit qu'il y aura levée de dégénérescence etque la projection du spin, ou du moment magnétique, sur l'axe z, ne peut prendre que 2I+1valeurs discrètes. Le cas d'un spin ½ est illustré à la �gure suivante.
I
z
I
B0
(cas: γ > 0)
/ 2γ
/ 2γ
/ 2
/ 2
μ
μ
Figure 2: Niveaux d'énergie d'un spin ½ (et de son moment magnétique associé) dans unchamp magnétique.
L'énergie de couplage entre un moment magnétique et l'induction B0 est donnée par :
Em = −µ ·B0 = −mγ~B0 où (m = −1/2, 1/2) (2)
Les énergies possibles sont donc directement proportionnelles à B0.
1.5

PHQ560-660 RMN
m = 1/2
(état α)
(état β)m = 1/2
m = -1/2
0E Bγ∆ =
0 0B = 0 0B >
Figure 3: Énergie des spins ½ en fonction de l'induction de module B0.
Dans la littérature, on désigne généralement l'état β comme étant celui ayant l'énergie laplus basse et l'état α comme celui ayant l'énergie la plus haute. On voit donc ici apparaitreun écart d'énergie 4E qui permettra au système d'absorber de l'énergie pour provoquer lephénomène de résonance. À l'équilibre thermodynamique, c'est la statistique de Boltzmannqui permet de calculer la proportion de la population sur le niveau α relative à celle duniveau β :
Nα
Nβ= e− 4EkBT = e
− γ~B0kBT (3)
Dans l'appareil que vous utiliserez, nous aurons des protons en présence d'un champ de 0.5Tesla à 300 K. Si l'on utilise la valeur du rapport gyromagnétique du proton γH (tableau1), on trouve :
Nα
Nβ= e
−267.513·106rad·s−1·T−1·6.626·10−34J·s·0.5 Tesla
2πkBT = 0.9999965 (4)
Le résultat précédent montre à quel point les deux niveaux sont également peuplés. Si le ni-veau supérieur comporte 1 million de spins, alors le niveau inférieur en comptera uniquement4 de plus. C'est très peu, mais su�sant pour être mesuré par une technique de résonance.
2.3 Aimantation
Calculons maintenant l'aimantation pour un système de N spins :
M = Nγ~
I∑m=−I
m exp (γ~mH0/kBT )
I∑m=−I
exp (γ~mH0/kBT )
(5)
Comme le rapport γ~H0/kBT est généralement un nombre très petit, on peut développerau premier ordre l'exponentielle de Boltzmann et obtenir[10] :
M =Nγ2~2H0
kBT
I∑m=−I
m2
2I + 1=Nγ2~2I (I + 1)H0
3kBT= χNH0 (6)
1.6
PHQ560-660 RMN
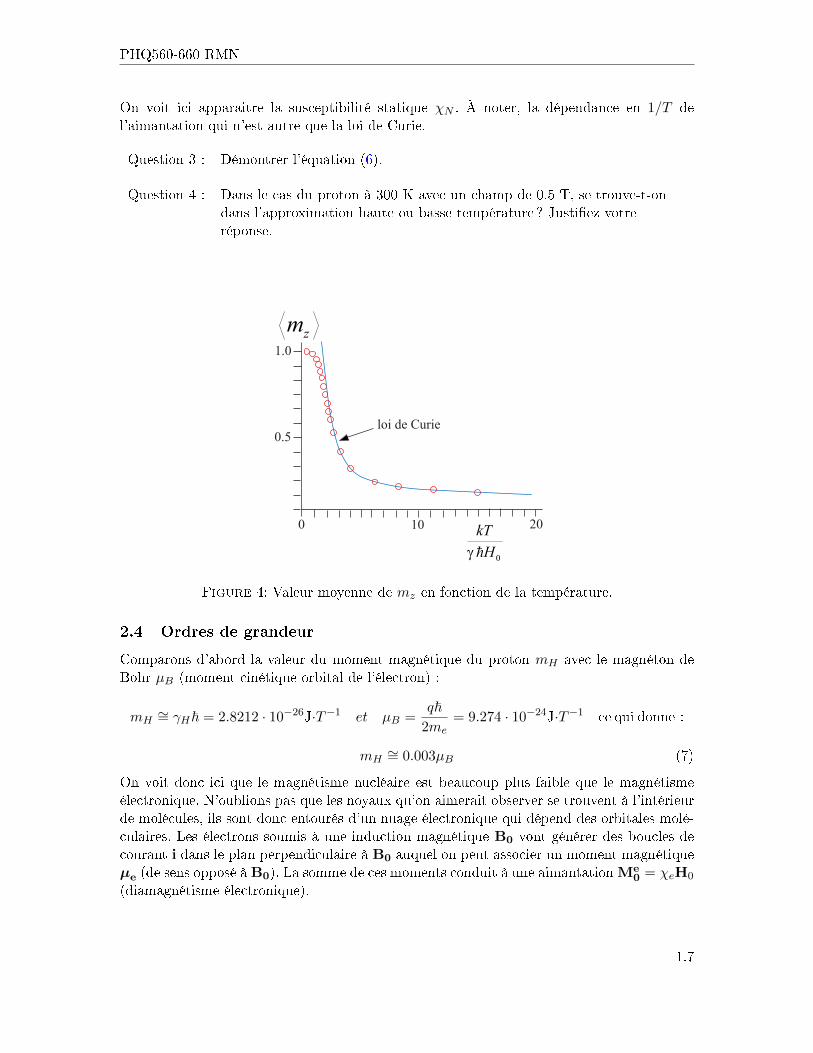
On voit ici apparaitre la susceptibilité statique χN . À noter, la dépendance en 1/T del'aimantation qui n'est autre que la loi de Curie.
Question 3 : Démontrer l'équation (6).
Question 4 : Dans le cas du proton à 300 K avec un champ de 0.5 T, se trouve-t-ondans l'approximation haute ou basse température ? Justi�ez votreréponse.
0
0.5
1.0
loi de Curie
10 20
zm
0
kTHγ
Figure 4: Valeur moyenne de mz en fonction de la température.
2.4 Ordres de grandeur
Comparons d'abord la valeur du moment magnétique du proton mH avec le magnéton deBohr µB (moment cinétique orbital de l'électron) :
mH∼= γH~ = 2.8212 · 10−26J·T−1 et µB =
q~2me
= 9.274 · 10−24J·T−1 ce qui donne :
mH∼= 0.003µB (7)
On voit donc ici que le magnétisme nucléaire est beaucoup plus faible que le magnétismeélectronique. N'oublions pas que les noyaux qu'on aimerait observer se trouvent à l'intérieurde molécules, ils sont donc entourés d'un nuage électronique qui dépend des orbitales molé-culaires. Les électrons soumis à une induction magnétique B0 vont générer des boucles decourant i dans le plan perpendiculaire à B0 auquel on peut associer un moment magnétiqueµe (de sens opposé à B0). La somme de ces moments conduit à une aimantationMe
0 = χeH0
(diamagnétisme électronique).
1.7

PHQ560-660 RMN
e
0I
M
i
/ 2γ−
/ 2−
e00
eeM Hχ=
N Hχ=
0B
0 0
z
μ
μ
Figure 5: Paramagnétisme nucléaireM0 et diamagnétisme électroniqueMe0. La �gure n'est
nullement à l'échelle car χe � χN . La �gure est tirée de la référence [10].
De leur côté, les noyaux plongés dans un champ d'induction B0 = µ0H0 produisent uneaimantation nucléaire macroscopique M0 = χNH0. Il s'agit d'un paramagnétisme qui vaêtre presqu'entièrement masqué par le diamagnétisme électronique Me
0 = χeH0 qui estbeaucoup plus intense[10]. En e�et, on obtient généralement que χe ≈ −105 χN . Il devientdonc tout à fait illusoire de penser appliquer un champ dans une direction donnée et demesurer directement la magnétisation nucléaire dans cette même direction. Celle-ci seraittotalement écrantée par celle produite par les électrons et même si cette dernière était nulle,il faudrait un gaussmètre sensible à 7 chi�res après le point !
L'astuce utilisée pour mesurer le paramagnétisme nucléaire est donc de faire basculer lesspins des noyaux dans le plan perpendiculaire au champ statique appliqué. On va montrerplus loin que cela est possible en soumettant les noyaux à un champ électromagnétique defréquence ν0 tel que hν0 = 4E = γ~B0. Dans le cas des protons soumis à une induction de0.5 Tesla (ce qui est notre cas), on trouve que la fréquence utilisée devrait être :
ν0 =2.675 · 108 × 0.5
2π= 21.28MHz
1.8

PHQ560-660 RMN
2.5 La précession de Larmor (approche classique)
Lorsqu'un moment magnétique µ est en présence d'une induction magnétique B = B0z, ilest animé d'un mouvement de précession autour de cette dernière, comme une toupie dansle champ de gravité. La vitesse angulaire de ce mouvement de précession est :
ω0 = γB0 (8)
Cela provient du fait que le moment magnétique subit un couple de force donné par leproduit vectoriel :
Γ = µ ∧B (9)
Selon le théorème du moment cinétique :
dL
dt= µ ∧B ou
dµ
dt= γµ ∧B (car µ = γ · L) (10)
En considérant maintenant tous les moments magnétiques, l'aimantation nucléaire doit obéiraux relations suivantes (B est selon z) :
dMx
dt= γ ·B ·My
dMy
dt= −γ ·B ·Mx (11)
dMz
dt= 0
À l'équilibre, l'aimantation pointe selon z et a pour grandeurM0 et demeure invariante :Mx(t) =My(t) = 0 et Mz(t) = M0. Maintenant supposons qu'à t = 0, l'aimantation initiale est écar-tée d'un angle θ par rapport à z (suite à une perturbation par exemple). Les conditionsinitiales sont maintenant :
Mx(0) = 0
My(0) = M0 sin θ (12)
Mz(0) = M0 cos θ
L'évolution de l'aimantation est donnée par la solution des équations (11) :
Mx(t) = M0 sin θ sin (ω0t)
My(t) = M0 sin θ cos (ω0t) avec ω0 = γB0 (13)
Mz(t) = M0 cos θ
L'aimantation est donc animée d'un mouvement de précession à la fréquence de Larmor(ω0). Remarquer que pour un noyau ayant un rapport gyromagnétique positif, sa vitesseangulaire est négative. Son moment magnétique possède donc un mouvement de précessiondans le sens des aiguilles d'une montre.
En présence d'ondes de radiofréquence ν0, il est donc possible que les noyaux absorbentde l'énergie (à condition que ω0 = γB0) pour faire passer des spins de l'état β à l'état α,diminuant ainsi l'aimantation selon z. Cette diminution de l'aimantation selon z devraitêtre accompagnée d'une augmentation de l'aimantation dans le plan x-y. Pour étudier lemécanisme par lequel ce basculement d'aimantation est possible, il est préférable de seplacer dans un référentiel tournant.
1.9

PHQ560-660 RMN
2.6 Référentiel tournant et application d'une induction
Regardons maintenant ce qui se passe lorsque notre système de spins, ayant une aimantationM dans un champ d'induction B0 (selon z), est soumis à un champ d'induction B1 oscillantà la fréquence ωr selon x.
Question 5 : En pratique, on utilise une bobine pour générer une onde polarisée li-néairement et qui mathématiquement se décompose en deux polarisa-tions circulaires. Pourquoi l'une des deux polarisations circulaires n'apratiquement aucun e�et sur l'aimantation ?
Dé�nissons premièrement un repère tournant (X, Y, Z) autour de z à la fréquence angulaire−ωr. Dans ce repère, l'induction B1 est stationnaire selon X. Les vecteurs unitaires des axesde ce repère sont notés : i, jetk. L'aimantation y sera donc donnée par : M = MX i+MY j+MZk. Dans le repère �xe on aura :(
dM
dt
)fixe
= MX∂i
∂t+MY
∂j
∂t+MZ
∂k
∂t+
(∂MX
∂ti +
∂MY
∂tj +
∂MZ
δtk
)(14)
L'expression entre parenthèses représente la dérivée temporelle deM dans le repère tournant(∂M∂t
)rot.
. Notons aussi que :
∂i
∂t= ωr ∧ i
∂j
∂t= ωr ∧ j
∂k
∂t= ωr ∧ k
On peut donc réécrire (14) comme étant :(dM
dt
)fixe
=
(dM
dt
)rot.
+ ωr ∧ (MX i +MY j +MZk) (15)
ou : (dM
dt
)rot.
=
(dM
dt
)fixe
− ωr ∧M (16)
Rappelons cependant que dans le repère �xe,(dMdt
)fixe
= γM ∧ (B0 + B1) ce qui permetde réécrire (16) sous la forme :(
dM
dt
)rot.
= γM ∧ (B0 + B1)− γM ∧ ωr
γ
ce qui peut �nalement s'écrire : (dM
dt
)rot.
= γM ∧Beff . (17)
avecBeff . = (B0 − ωr/γ)k +B1i (18)
1.10

PHQ560-660 RMN
Z
X
0rB ωγ
−
1B
θ effB
Figure 6: Induction e�ective vue dans le repère tournant.
On remarque donc que du point de vue du repère tournant, l'aimantation suit un comporte-ment similaire à celle du repère �xe. Elle est encore animée d'un mouvement de précessionmais autour d'un champ e�ectif ~Beff., mouvement qui porte le nom de � nutation � et dontla fréquence vaut :
νnut. =γBeff.
2π=
√γ2B2
1 + (γB0 − ω2r )
2π(19)
L'angle entre la direction de Beff . et l'axe z est donné par :
tan θ =γB1
γB0 − ωr(20)
À la résonance (ωr = ω0),et Beff . se confond avec B1 et la fréquence de nutation devient :
νnut. =γB1
2π(21)
Z
M
1B
X
Y
1 1Bω γ=
Figure 7: Basculement de l'aimantation à la résonance.
Grâce à la résonance, on peut donc manipuler comme on le veut le vecteur d'aimantation. Ene�et, si l'aimantation initiale est selon Z, on peut appliquer un champB1 selon Y d'une durée
1.11

PHQ560-660 RMN
τ1/2 de sorte que γB1τ1/2 = π/2. Ce qui revient à faire basculer l'aimantation en directionY. On nomme une impulsion de cette durée une impulsion π/2. La phase de l'impulsionπ/2 dé�nie la phase de la précession qui suit l'impulsion. Par la suite, on peut appliquer despulses π (avec la bonne phase) pour choisir dans quelle direction on bascule l'aimantation.
Il y a deux paramètres indépendants qui permettent de sélectionner l'angle de basculementde l'aimantation : B1 et τ . Techniquement, il est préférable de restreindre τ à quelquesdizaines de microsecondes. Cela assure que l'angle de basculement est à peu près constantpour toutes les aimantations individuelles d'un spectre moyennent étalé en fréquence[9]. Lacomposition spectrale d'une impulsion courte permet aussi d'avoir accès à une bande defréquence autour de la fréquence centrale.
Supposons maintenant qu'une impulsion π/2 a fait basculer l'aimantation dans le plan X-Y. Que se passe-t-il alors ? L'induction B0 se confond maintenant à Beff . et l'aimantationaura un mouvement de précession autour d'elle. Expérimentalement, on se rend compteque l'aimantation dans le plan X-Y va rapidement disparaitre et que l'aimantation selonZ va graduellement reprendre sa valeur d'origine. Ces relaxations des aimantations trans-verse et longitudinale (par rapport à Z) sont causées par l'interaction des spins avec leurenvironnement.
2.7 Déplacement chimique
Pour un noyau donné, le champ magnétique ressenti dépend non seulement du champ appli-qué mais aussi de la distribution des électrons qui l'entourent de sorte que B = B0 (1− σ).Le mot distribution fait référence à la géométrie des orbitales moléculaires (nature, longueurset angles de liaisons, polarisation, . . .). Les électrons vont produire un e�et d'écrantage duchamp appliqué au niveau du noyau. La RMN tire un grand pro�t de cette réalité car unproton dans un environnement donné aura une fréquence de résonance légèrement di�érentede celle qu'aurait un proton dans un autre environnement. Cela permet aux chimistes d'iden-ti�er les groupements qui sont présents dans des molécules aussi grosses que des protéines.
Le déplacement chimique (δ) est généralement calculé à partir d'un signal de référenceprovenant d'un échantillon dont la distribution électronique est peu déformée. Il est donnéen parties par millions (ppm) selon l'équation suivante :
δ (ppm) =ν − νref.νref.
× 106 =4ν(Hz)
νref. (MHz)(22)
Cette façon de faire permet de s'a�ranchir de la fréquence du spectromètre utilisé. En RMNdes liquides, on utilise souvent le TMS (tétraméthylsilane) comme référence. On �xe sondéplacement chimique à zéro par convention.
Note : Si le liquide de référence n'est pas dilué dans le liquide à mesurer, il faut prendreen considération l'e�et de susceptibilité magnétique qui n'a pas la même valeurpour les deux matériaux (c'est-à-dire que les B0 ne sont pas identiques, même siH0 ne change pas). Si tel est le cas, le champ ressenti par chacun des liquides estdi�érent et il faut appliquer une correction au déplacement chimique mesuré[8].
1.12

PHQ560-660 RMN
2.8 Les équations de Bloch
Après avoir fait basculé l'aimantation selon Y avec une impulsion π/2, quelle sera sonévolution dans le temps ? L'équation (17) nous dit qu'une fois queB1 = 0, l'aimantation auraun mouvement de précession perpétuel autour de B0. Cela ne correspond nullement à ce quiest observé expérimentalement. On est donc obligé d'ajouter des termes d'amortissement quivont permettre de re�éter la réalité. Dans le repère tournant, on obtient donc les équationssuivantes :
dMX
dt= (γB0 − ωr)MY −
(1
T2
)MX
dMY
dt= − (γB0 − ωr)MX −
(1
T2
)MY (23)
dMZ
dt=
(1
T1
)(M0 −MZ)
On appelle ces équations phénoménologiques, les équations de Bloch. Pour les résoudre,introduisons l'aimantation transversale complexe MT = MX + iMY et la fréquence de pré-cession dans le repère tournant ν ′ = νr−ν0. Les équations de Bloch peuvent donc se réécrirecomme (avec γB0 = 2πν0) :
dMT
dt= −
(1
T2+ 2πiν ′
)MT (24)
dMZ
dt=
(1
T1
)(M0 −MZ) (25)
Si au départ on a MT (0) = M0 et MZ = 0, on aura :
MT = M0 exp (−t/T2) exp(2πiν ′t
)(26)
d'où :
MX = M0 exp (−t/T2) cos(2πν ′t
)(27)
MY = M0 exp (−t/T2) sin(2πν ′t
)(28)
MZ = M0 [1− exp (−t/T1)] (29)
Après un certain temps, l'aimantation transversale disparait et l'aimantation selon z reprendsa valeur à l'équilibre. Ces deux processus de relaxation s'e�ectuent à des rythmes di�érentsmais on peut montrer que T2 ≤ T1.
Dans la pratique, on est souvent en présence de plusieurs résonances à des énergies rap-prochées. La mesure expérimentale donne donc une superposition de sinusoïdes amorties.Il faut donc e�ectuer une transformée de Fourier pour identi�er chacune des fréquences derésonance. Dans le cas le plus simple où l'on a qu'une seule fréquence de résonance, on a :
TF (MT ) =
∞
0
MT (t) exp (−2iπνt) dt (30)
1.13

PHQ560-660 RMN
TF (MT ) =M0T
∗2
1 + 4π2T ∗2 (ν ′ − ν)2 + iM0T
∗2 2π (ν ′ − ν)
1 + 4π2T ∗2 (ν ′ − ν)2 (31)
Le membre de gauche est relié à l'absorption d'énergie. C'est une raie Lorentzienne ayantune amplitude maximale de M0T
∗2 et une largeur à mi-hauteur 1/πT ∗2 . On verra plus loin
pourquoi on a introduit un T ∗2 plutôt que le T2 des équations de Bloch. Notons aussi que sil'aimantation n'a pas été tournée de π/2 mais plutôt d'un angle α, alors il faut multiplier(27) et (28) respectivement par sinα et cosα.
Note : Normalement, c'est le terme imaginaire qui représente la dissipation d'énergiemais dans notre cas, c'est la partie réelle. Cela ne dépend que de la phaseinitiale de l'aimantation que l'on a choisie.
2.9 Remarques générales sur T1 et T2.
Une fois produite, l'aimantation transversale tend à redevenir nulle. Comme nous venonsde le dire, les inhomogénéités du champ statique vont introduire des fréquences de Larmordi�érentes et cela contribue grandement au déphasage des spins dans le plan x-y. En fait,le temps d'amortissement de l'aimantation transversale en présence d'inhomogénéités dechamp donne une valeur qu'on appelle T ∗2 , souvent beaucoup plus faible que T2, qui luiserait le � vrai � temps de relaxation si le champ était parfaitement homogène. Il y a aussil'interaction dipôle-dipôle entre noyaux de même nature qui cause le déphasage. Commeces noyaux sont de même nature, ils possèdent des fréquences de Larmor identiques et ilsgénèrent donc des champs magnétiques à la bonne fréquence leur permettant ainsi d'échangerde l'énergie facilement. Un noyau peut donc relaxer en passant de l'état excité à l'étatfondamental alors que l'autre noyau absorbera cette énergie pour faire le contraire. Celaconduit à un déphasage dans le plan x-y, sans que de l'énergie ne soit transférée au réseau.Lorsque l'aimantation transversale a totalement disparu, cela ne veut pas dire que le systèmede spins est revenu à son état fondamental.
Les temps de relaxation contrôlent la largeur des raies. De son côté, T2 est toujours pluscourt ou égal à T1. Si on fait basculer l'aimantation, elle reviendra à sa position d'équilibreaprès un certain temps. À ce moment, il ne peut rester d'aimantation transversale. L'énergieacquise par le système de spins en RMN se fait dans le domaine des radiofréquences et nepeut donc pas être réémise par des molécules de façon électromagnétique. Ce sont doncles vibrations et les rotations des molécules qui vont produire des champs oscillants quipourront absorber l'énergie qui a été transmise aux spins. Ces champs devront posséderdes composantes en fréquence leur permettant un échange d'énergie avec les spins de façonà permettre au système de retourner à l'équilibre thermodynamique. L'énergie qui a étéabsorbée par les spins lors de l'impulsion RF sera donc dissipée sous forme de chaleur. Lesmécanismes qui permettent cette dissipation d'énergie régissent la valeur de T1.
1.14

PHQ560-660 RMN
3 Principe de la mesure
3.1 Montage expérimental
La prochaine �gure donne une vue d'ensemble du spectromètre RMN.
090
RF pulsé
fréquence deréférence
détecteurenveloppe
multipl.
multipl.
sortie RF
Filtre P-B
Filtre P-B
Filtre P-B
signalde suppression
sortieenveloppe
sortieen phase (I)
sortieen quadrature(Q)
bobine (L) filtrepasse-bande
suiveur detension
AFB AGV
ligne quartd'onde
C1
C2
Figure 8: Schéma du spectromètre RMN en mode pulsé.
Pour générer des impulsions RF, un synthétiseur fournit un signal sinusoïdal dont la fré-quence est ajustable. Un modulateur transforme ensuite ce signal en un paquet d'onde dontla durée est également ajustable a�n de pouvoir choisir l'angle de basculement de l'aiman-tation. L'impulsion RF est ensuite acheminée vers la bobine d'inductance L qui va générerle champ H1. C'est cette même bobine qui est utilisée pour collecter le signal RMN unefois que la phase d'excitation est terminée. Le mouvement de précession de l'aimantationva donner lieu au phénomène d'induction, qui lui va permettre de récolter une tension auxbornes de la bobine. La capacité C1 permet d'ajuster la fréquence de résonance du cir-cuit
(ωr ≈ 1/
√LC1
), alors que C2 doit être ajustée pour adapter l'impédance de l'ensemble
(LC1, C2) à 50 Ω pour transmettre le maximum d'énergie à la bobine. C1 et C2 sont deuxcapacités variables communément appelées : � Tuning and Matching capacitors �.
Comme le signal RMN est très faible, il faut l'ampli�er à l'aide d'un ampli�cateur à faiblebruit (AFB) combiné à un ampli�cateur à gain variable (AGV). Il faut donc découpler lecircuit d'excitation de celui de réception en utilisant une astuce : l'utilisation de paires dediodes et d'une ligne de transmission quart d'onde. L'amplitude du paquet d'onde pulsé estd'environ 25 V alors que la tension de coude des diodes est de l'ordre de 0.6 V.
Question 6 : Expliquez comment le montage ci-dessus permet d'isoler la source d'ex-citation du signal RMN à mesurer.
Un signal de suppression ajustable permet aussi de mettre le gain du AGV à zéro pendantet juste un peu après la phase d'excitation pour ampli�er uniquement le signal RMN. Un�ltre passe-bande est ensuite utilisé pour enlever le bruit provenant de fréquences autresque celles de la zone d'intérêt. À ce point, on a accès à ce qu'on appelle la � FID � (free
1.15
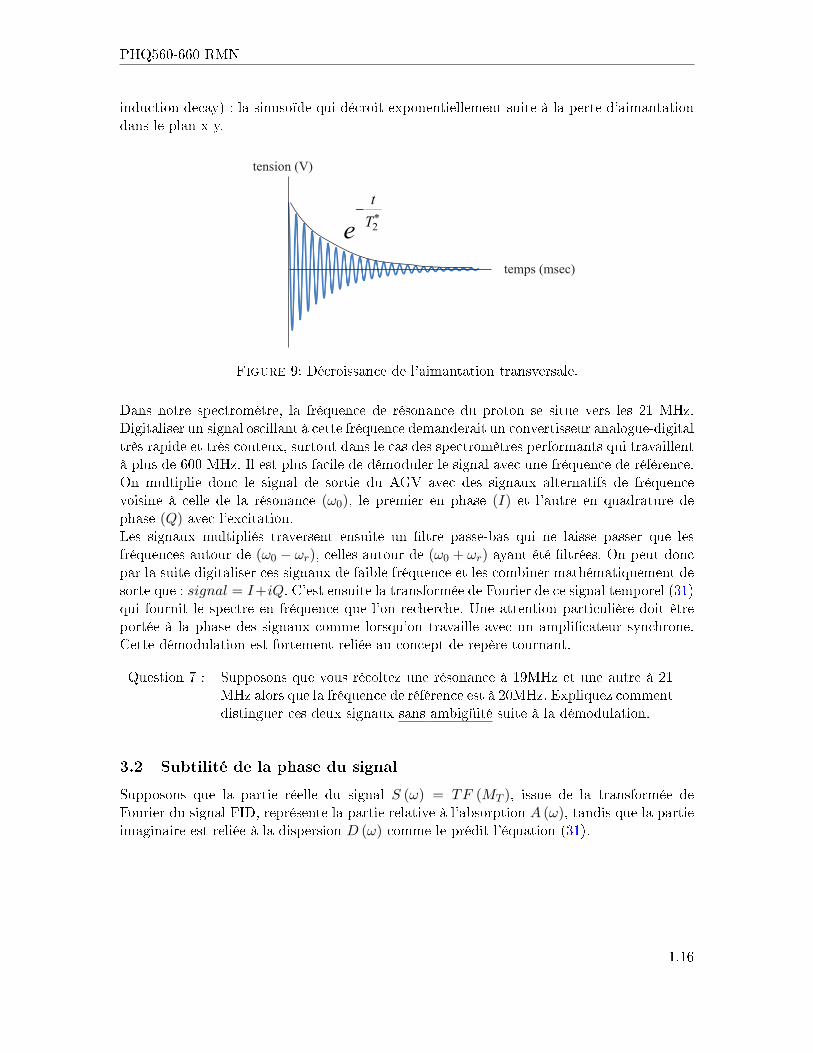
PHQ560-660 RMN
induction decay) : la sinusoïde qui décroit exponentiellement suite à la perte d'aimantationdans le plan x-y.
temps (msec)
tension (V)
2
tTe∗−
Figure 9: Décroissance de l'aimantation transversale.
Dans notre spectromètre, la fréquence de résonance du proton se situe vers les 21 MHz.Digitaliser un signal oscillant à cette fréquence demanderait un convertisseur analogue-digitaltrès rapide et très couteux, surtout dans le cas des spectromètres performants qui travaillentà plus de 600 MHz. Il est plus facile de démoduler le signal avec une fréquence de référence.On multiplie donc le signal de sortie du AGV avec des signaux alternatifs de fréquencevoisine à celle de la résonance (ω0), le premier en phase (I) et l'autre en quadrature dephase (Q) avec l'excitation.Les signaux multipliés traversent ensuite un �ltre passe-bas qui ne laisse passer que lesfréquences autour de (ω0 − ωr), celles autour de (ω0 + ωr) ayant été �ltrées. On peut doncpar la suite digitaliser ces signaux de faible fréquence et les combiner mathématiquement desorte que : signal = I+iQ. C'est ensuite la transformée de Fourier de ce signal temporel (31)qui fournit le spectre en fréquence que l'on recherche. Une attention particulière doit êtreportée à la phase des signaux comme lorsqu'on travaille avec un ampli�cateur synchrone.Cette démodulation est fortement reliée au concept de repère tournant.
Question 7 : Supposons que vous récoltez une résonance à 19MHz et une autre à 21MHz alors que la fréquence de référence est à 20MHz. Expliquez commentdistinguer ces deux signaux sans ambigüité suite à la démodulation.
3.2 Subtilité de la phase du signal
Supposons que la partie réelle du signal S (ω) = TF (MT ), issue de la transformée deFourier du signal FID, représente la partie relative à l'absorption A (ω), tandis que la partieimaginaire est reliée à la dispersion D (ω) comme le prédit l'équation (31).
1.16

PHQ560-660 RMN
( )A ω ( )D ω
Figure 10: Parties réelle et imaginaire de TF (MT ).
Si en plus ce signal possède une phase φ non nulle, alors les parties réelles et imaginairesdeviennent un mélange des fonctions qui décrivent correctement l'absorption et la dispersion.
S (ω) = [A (ω) + iD (ω)] exp (iφ) (32)
S (ω) = R (ω) + iI (ω) (33)
La phase φ conduit donc au mélange suivant :
R (ω) = A (ω) cosφ−D (ω) sinφ (34)
I (ω) = D (ω) cosφ+A (ω) sinφ (35)
Nous sommes donc obligés de faire un changement de base (ce qui revient à ajouter une phaseà notre signal de référence) pour corriger le tout et retrouver les fonctions d'absorption etde dispersion.
A (ω) = R (ω) cosφ+ I (ω) sinφ (36)
D (ω) = I (ω) cosφ−R (ω) sinφ (37)
3.3 Di�érence entre et T2 et T ∗2
Un peu plus tôt, on disait que l'aimantation transversale devrait diminuer avec un tempscaractéristique nommé T2. La valeur T2 provient du déphasage dynamique des spins dûaux interactions entre eux. Lorsqu'on a donné la solution des équations de Bloch pourconfronter la théorie à l'expérience, on a été obligé de substituer T ∗2 à T2 pour tenir comptede la réalité expérimentale. En e�et, l'inhomogénéité du champ H0 et l'e�et du déplacementchimique introduisent un déphase additionnel entre les spins dans le plan x-y car la valeurde la fréquence de Larmor dépend de l'endroit où se trouve le moment magnétique. Bienqu'on essaie d'avoir le champ le plus homogène possible, ce phénomène est toujours présent.Regardons mathématiquement comment cela se traduit.La solution des équations de Bloch pour l'aimantation transversale dans le repère tournantest donnée par l'équation [26] :
MT = M0 exp (−t/T2) exp(2iπν ′t
)où ν ′ = ν − ν0 (38)
1.17

PHQ560-660 RMN
et à la résonance, lorsque ν ′ = 0, on a une exponentielle décroissante pure :
MT = M0 exp (−t/T2) (39)
Dans le cas d'une inhomogénéité du champ qui décale la fréquence de résonance ω0 d'unepetite quantité δ, alors l'aimantation transversale s'écrit :
MT = M0 exp (−t/T2) exp (iδt) (40)
Dans le cas où ce décalage est distribué sur le volume de l'échantillon, le signal provenantde l'ensemble des spins sera donné par :
MT = M0 exp (−t/T2)∑n
exp (iδnt) (41)
Si on suppose une distribution gaussienne des décalages δn alors[1]&[2] :
MT = M0 exp (−t/T2) exp
(−〈δ〉
2 t2
2
)(42)
Expérimentalement, c'est l'inhomogénéité du champ qui domine la relaxation et non le T2.
t
TM
2
tTe
−
Figure 11: E�et de l'inhomogénéité du champ.
Le signal décroit donc avec un temps caractéristique T ∗2 dé�ni par[8] :
1
T ∗2=
1
T2+ γ4H0 (43)
où 4H0 est l'inhomogénéité du champ sur le volume de l'échantillon. Il y a cependant unmoyen de contourner le problème de l'inhomogénéité du champ pour avoir accès à la vraivaleur de T2 ; c'est l'utilisation des échos de spin.
3.4 Écho de spin et mesure de T2
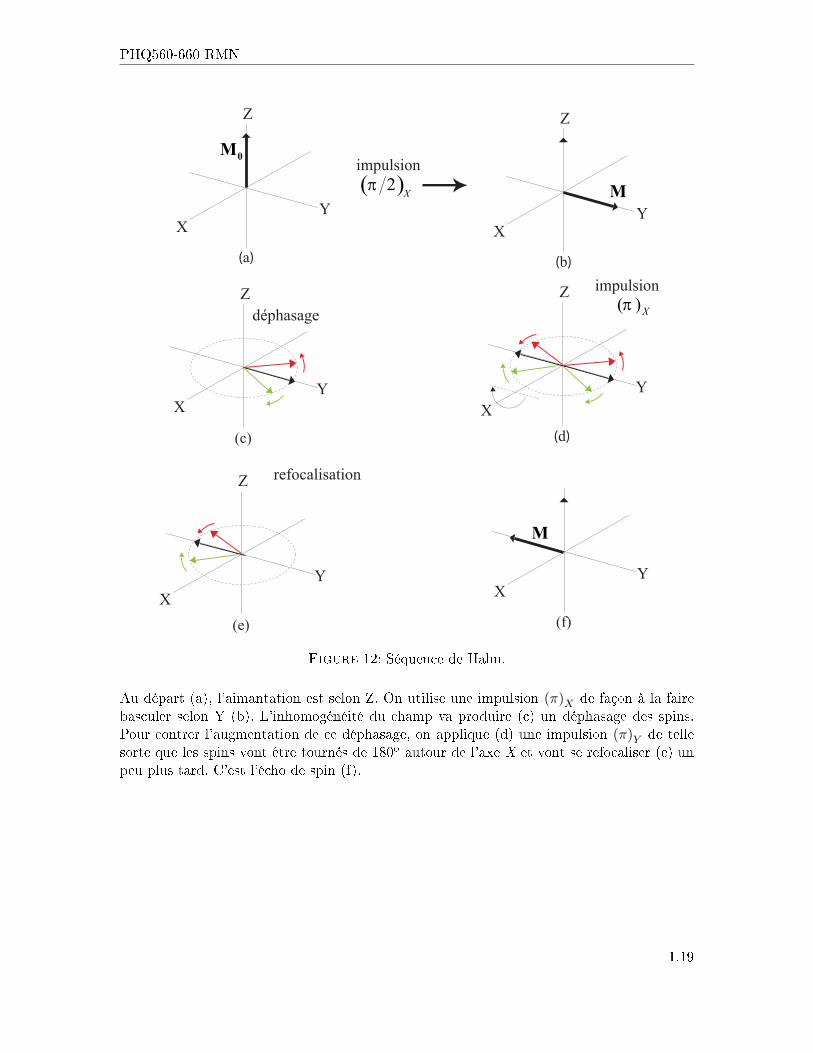
En 1950, Erwin Hahn proposa une explication au phénomène d'écho de spin. Regardons cequi se passe dans le référentiel tournant dans la séquence suivante :
1.18
PHQ560-660 RMN
Z
impulsion
XY
0M
Z
XY
Z
XY
( )2 Xπ
impulsion
déphasage
M
M
(a) (b)
XY
(f)
(c)
Z
XY
(d)
( )Xπ
refocalisationZ
XY
(e)
Figure 12: Séquence de Hahn.
Au départ (a), l'aimantation est selon Z. On utilise une impulsion (π)X de façon à la fairebasculer selon Y (b). L'inhomogénéité du champ va produire (c) un déphasage des spins.Pour contrer l'augmentation de ce déphasage, on applique (d) une impulsion (π)Y de tellesorte que les spins vont être tournés de 180o autour de l'axe X et vont se refocaliser (e) unpeu plus tard. C'est l'écho de spin (f).
1.19

PHQ560-660 RMN
( )2 Xπ
( )Xπt t
TF TF (demi-écho)
*2T 2T
0M22
0 e TM τ−
Figure 13: Production d'un écho de spin.
3.4.1 Séquence Carr-Purcell
On vient de voir que la séquence de Hahn permet de contourner le problème de l'inho-mogénéité du champ mais il en reste un autre. Dans les liquides, on doit aussi faire faceau problème de di�usion lié au mouvement des molécules. Dans la séquence de Hahn, lare-focalisation serait parfaite si la molécule occupait la même position lors de l'applicationde l'impulsion à π que lors de l'impulsion à π/2. On ne s'attardera pas au traitement ma-thématique de cet autre phénomène[9] mais plutôt à une nouvelle séquence qui permet decontourner cet autre problème : la séquence Carr-Purcell.
( )2 Xπ
( )Xπτ τ τ τ τ
...
Figure 14: Séquence de Carr-Purcell
Comme dans la séquence de Hahn, on commence par faire basculer l'aimantation selonY, mais par la suite c'est une série d'impulsions (π)X qui produisent un train d'échos.L'utilisation d'un temps τ assez court permet de s'a�ranchir des e�ets de di�usion (dansun liquide) et de mesurer une bonne valeur de T2. Il y a cependant un problème avec cetteséquence. Si l'impulsion (π)X n'est pas parfaite, c'est-à-dire qu'on a a�aire à une impulsion(π − ε)X , alors la re-focalisation aura lieu en dessous du plan X-Y d'un angle ε.
1.20

PHQ560-660 RMN
impulsionZ
XY
( )Xπ ε−
ε
Figure 15: Re-focalisation sous le plan X-Y due à une impulsion imparfaite.
Dans une séquence de Carr-Purcell, cette erreur est cumulative à chacun des échos. Aprèsn échos, l'aimantation se retrouve avec un angle nε du plan X-Y.
3.4.2 Séquence de Carr-Purcell-Meiboom-Gill
Une variante de la séquence Carr-Purcell proposée par Meiboom et Gill permet d'éviterqu'une erreur d'angle soit cumulative à chaque écho. Il s'agit de déphaser de 90o toutes lesimpulsions π par rapport à l'impulsion initiale à π/2. Cela revient à remplacer les impulsions(π)X par des impulsions (π)Y . Regardons ce que ça donne dans la �gure suivante :
1.21

PHQ560-660 RMN
Z
impulsion
XY
0M
M
Z
XY
( )2 Xπ
(a) (b)
(c) (d)
refocalisationau dessus du plan X-Y
déphasage
Z
XYε
ε
(e)
impulsionZ
XY
( )Yπ ε−Z
XY
Figure 16: Séquence de Carr-Purcell- Meiboom-Gill.
Uniquement les déphasages positifs ont été indiqués sur la �gue pour �n de clarté. Après avoirfait basculer l'aimantation sur Y, le déphasage dû à l'inhomogénéité du champ s'installe. Onapplique ensuite l'impulsion à (π − ε)Y qui bascule les spins autour de Y cette fois. Ensuite,les spins se refocalisent mais à un angle ε au-dessus du plan X-Y. Par contre, si l'on répètele processus, vous vous rendez-compte que le prochain écho sera parfaitement focalisé sur leplan X-Y. Cette célèbre séquence permet donc d'avoir la vrai valeur de l'aimantation unefois sur deux, ce qui est su�sant pour obtenir de bonnes valeurs de T2.
3.5 Mesure de T1
On procède à la mesure de T1 avec ce qu'on appelle une séquence d'inversion-récupération.Il s'agit d'un train de deux impulsions séparé d'un temps mort : π → τ → π/2. .
1.22
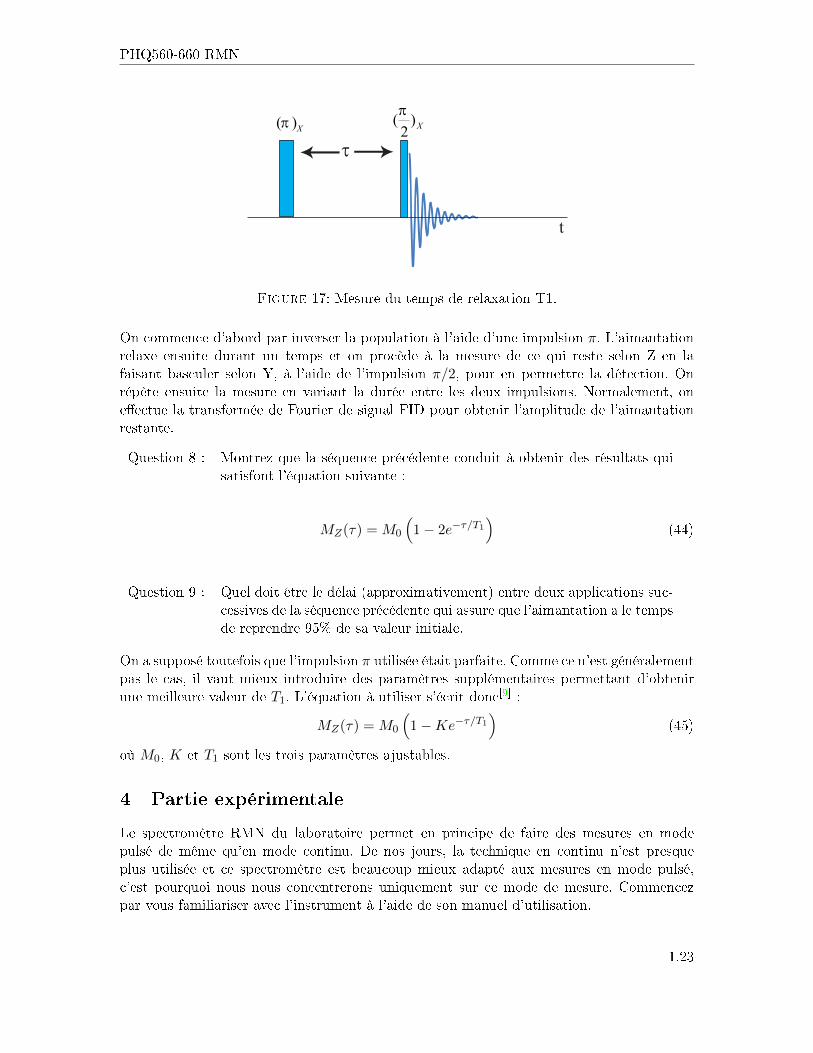
PHQ560-660 RMN
( )2 Xπ
( )Xπ
τ
t
Figure 17: Mesure du temps de relaxation T1.
On commence d'abord par inverser la population à l'aide d'une impulsion π. L'aimantationrelaxe ensuite durant un temps et on procède à la mesure de ce qui reste selon Z en lafaisant basculer selon Y, à l'aide de l'impulsion π/2, pour en permettre la détection. Onrépète ensuite la mesure en variant la durée entre les deux impulsions. Normalement, one�ectue la transformée de Fourier de signal FID pour obtenir l'amplitude de l'aimantationrestante.
Question 8 : Montrez que la séquence précédente conduit à obtenir des résultats quisatisfont l'équation suivante :
MZ(τ) = M0
(1− 2e−τ/T1
)(44)
Question 9 : Quel doit être le délai (approximativement) entre deux applications suc-cessives de la séquence précédente qui assure que l'aimantation a le tempsde reprendre 95% de sa valeur initiale.
On a supposé toutefois que l'impulsion π utilisée était parfaite. Comme ce n'est généralementpas le cas, il vaut mieux introduire des paramètres supplémentaires permettant d'obtenirune meilleure valeur de T1. L'équation à utiliser s'écrit donc[9] :
MZ(τ) = M0
(1−Ke−τ/T1
)(45)
où M0, K et T1 sont les trois paramètres ajustables.
4 Partie expérimentale
Le spectromètre RMN du laboratoire permet en principe de faire des mesures en modepulsé de même qu'en mode continu. De nos jours, la technique en continu n'est presqueplus utilisée et ce spectromètre est beaucoup mieux adapté aux mesures en mode pulsé,c'est pourquoi nous nous concentrerons uniquement sur ce mode de mesure. Commencezpar vous familiariser avec l'instrument à l'aide de son manuel d'utilisation.
1.23

PHQ560-660 RMN
Stabilisation du champ
� Allumer le spectromètre et stabiliser la température des aimants. Pour ce faire, ilfaut ajuster les potentiomètres du module � Magnet Temperature � à la position oùles LED ne sont ni rouges ni vertes. Ensuite, on place les boutons � Feedback Loop �en position � closed �.
� Dans votre rapport, noter les paramètres utilisés : séquence d'impulsion, période derépétition (pour ne pas saturer), fréquence du spectromètre, longueur des impulsions,etc. pour les di�érents échantillons utilisés. Ces informations peuvent être consignéesdans une annexe.
4.1 Résonance du proton
4.1.1 Impulsion RF (fréquence de résonance du proton)
� Insérer la sonde RF à l'intérieur de la bobine d'excitation et reliez-la au canal 1de l'oscilloscope. Synchroniser l'oscilloscope à l'aide du signal de synchronisation duspectromètre.
� Ajuster le spectromètre à la fréquence de résonance du proton (approximativement21 MHz) et utiliser des impulsions d'excitation d'une durée d'environ 3 μsec avec unepériode de répétition de 400 msec.
� Obtenir le signal produit à la sonde sur l'oscilloscope et le maximiser en ajustant les� tuning capacitors �).
ATTENTION ! Pour l'ajustement des capacités, utilisez uniquement le tournevis en plas-tique pour ne pas perturber les aimants.
� Vous devriez être capables d'obtenir un signal d'environ 40 Volts (crête-à-crête) quiressemble à ceci :
Maintenant que le signal d'excitation est bon, nous pouvons commencer les mesures sur leséchantillons. Un � o-ring � est inséré autour de chaque �acon contenant le liquide à étudier.Ce � o-ring � doit être placé à 39 mm du centre de l'échantillon pour que ce dernier seretrouve au centre de la bobine d'excitation.
1.24
PHQ560-660 RMN
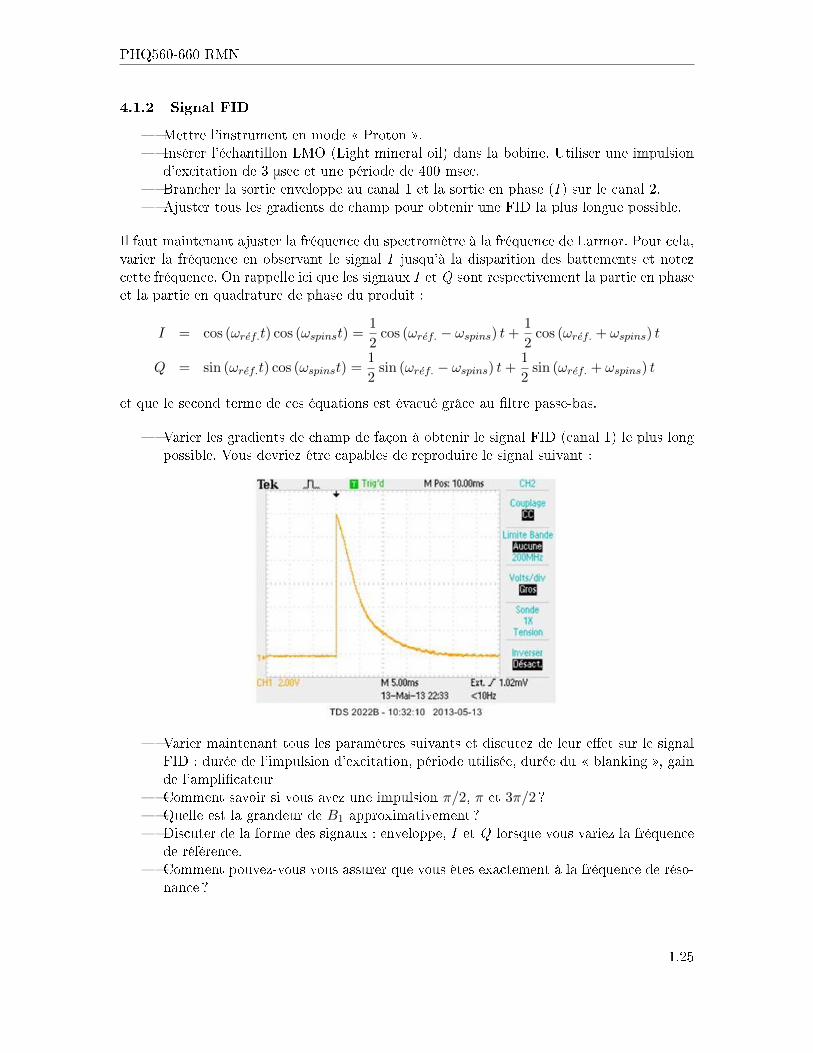
4.1.2 Signal FID
� Mettre l'instrument en mode � Proton �.� Insérer l'échantillon LMO (Light mineral oil) dans la bobine. Utiliser une impulsion
d'excitation de 3 μsec et une période de 400 msec.� Brancher la sortie enveloppe au canal 1 et la sortie en phase (I ) sur le canal 2.� Ajuster tous les gradients de champ pour obtenir une FID la plus longue possible.
Il faut maintenant ajuster la fréquence du spectromètre à la fréquence de Larmor. Pour cela,varier la fréquence en observant le signal I jusqu'à la disparition des battements et notezcette fréquence. On rappelle ici que les signaux I et Q sont respectivement la partie en phaseet la partie en quadrature de phase du produit :
I = cos (ωref.t) cos (ωspinst) =1
2cos (ωref. − ωspins) t+
1
2cos (ωref. + ωspins) t
Q = sin (ωref.t) cos (ωspinst) =1
2sin (ωref. − ωspins) t+
1
2sin (ωref. + ωspins) t
et que le second terme de ces équations est évacué grâce au �ltre passe-bas.
� Varier les gradients de champ de façon à obtenir le signal FID (canal 1) le plus longpossible. Vous devriez être capables de reproduire le signal suivant :
� Varier maintenant tous les paramètres suivants et discutez de leur e�et sur le signalFID : durée de l'impulsion d'excitation, période utilisée, durée du � blanking �, gainde l'ampli�cateur
� Comment savoir si vous avez une impulsion π/2, π et 3π/2 ?� Quelle est la grandeur de B1 approximativement ?� Discuter de la forme des signaux : enveloppe, I et Q lorsque vous variez la fréquence
de référence.� Comment pouvez-vous vous assurer que vous êtes exactement à la fréquence de réso-
nance ?
1.25

PHQ560-660 RMN
4.1.3 Impulsion π (180o)
Il est très di�cile de trouver la durée d'une impulsion π/2 à l'aide du signal FID produit.En revanche, une impulsion π devrait produire un signal FID nul. On détermine donc expé-rimentalement la durée de l'impulsion π et on divise cette dernière par deux pour produireune impulsion π/2. Référez-vous au manuel d'utilisation (page III-8) pour trouver commentdéterminer la durée d'une impulsion π. Expliquer comment on doit s'y prendre.
4.1.4 Transformée de Fourier (LMO)
� Placer la sortie I sur le canal 1 de l'oscilloscope et la sortie Q sur le canal 2. E�ectuerla T.F. avec la référence à 3kHz en-dessous de la résonance en utilisant le signal dupremier canal de l'oscilloscope et ensuite les signaux des deux canaux. Consigner laT.F. dans votre rapport et discuter des résultats.
� Expérimenter l'e�et de tronquer les données, là où les oscillations commencent, avantd'e�ectuer la T.F. Discutez du résultat.
� Quelle information nous donne la détection en quadrature ?� Comment peut-on augmenter la résolution du spectre en fréquence ?� Pourquoi faut-il utiliser le correcteur de phase du logiciel ? Essayer de modi�er la
phase à l'aide du spectromètre de façon à pouvoir vous passer de correcteur de phasedu logiciel.
4.1.5 Di�érence entre l'huile minérale, la glycérine et l'eau.
Obtenir le signal FID suite à une impulsion π/2 pour les échantillons : LMO (light mineraloil), glycérine et eau déminéralisée. Discuter des di�érences observées et de leur comporte-ment en fonction de la période de l'excitation (P ).
4.1.6 Mesures de T1 et T2
En utilisant la méthode � inversion recovery �, obtenez le T1 des liquides : LMO, glycérine eteau. À l'aide de la FID, obtenir T ∗2 . À l'aide des échos de spin (spectromètre ne mode : Carr-Purcell), obtenir le T2 de ces mêmes liquides. Placer maintenant le spectromètre en modeCarr Purcell Meiboom Gill et refaites la mesure de T2. Discuter des di�érences entre les deuxséquences d'échos sur les spectres obtenus. Expérimenter le fait d'augmenter l'inhomogénéitésur les échos obtenus et discuter vos observations.
Note : Pour votre rapport, consigner les graphiques avec les lissages qui ont permisde déterminer les T2, T ∗2 et les T1 pour ces liquides.
Pour les mesures de T1, ajuster la fréquence du spectromètre précisément à la fréquencedu signal pour ces mesures (pour ne plus voir d'oscillations). Il faut regarder le canal Icar vous cherchez des valeurs négatives et positives (impossible à faire avec l'enveloppe).Synchroniser l'oscilloscope avec la deuxième impulsion (B) et noter l'amplitude du signal àla même position en temps pour chaque valeur de τ . Le T1 de l'eau n'est pas facile à mesurer.Ne pas vous inquiéter même si votre mesure n,est qu'approximative.
1.26

PHQ560-660 RMN
Pour les mesures de T2, vous pouvez utiliser l'enveloppe (on n'inversera pas les échos).Essayez d'avoir au moins 15 échos par train CP ou CPMG. Prendre les valeurs maximalesdes échos pour faire un lissage exponentiel. Il y a un logiciel de digitalisation, sur le site desT.P. (sous les expériences de T.P. 1), pour vous aider à entrer les valeurs des maximums deséchos.
Note : Ne pas trop vous attarder sur les valeurs que vous trouverez dans la littératurecar la pureté du liquide peut introduire des di�érences notables aux valeurs deT1 et T2.
Question 10 : Pourquoi est-ce que les valeurs de T1 et T2 changent d'un liquide àl'autre ?
� Pour un échantillon seulement, le LMO, comparer directement des trains d'échos (Ibien phasé) pour les séquences CP et CPMG : démontrer les di�érences entre lesdeux types de séquences sur la phase du signal.
4.1.7 TMS et chloroforme
CH CHClSi 3CH3 3
CH3
CH3(TMS) (chloroforme)
Figure 18: TMS et chloroforme.
� Pour réussir à obtenir deux raies distinctes avec le TMS et le chloroforme, il fautabsolument avoir une très bonne homogénéité du champ au niveau de l'échantillon.On conseille donc au préalable d'utiliser le LMO et d'ajuster les gradients de champpour obtenir une FID la plus longue possible.
� Obtenir ensuite le spectre de Fourier du TMS à 3 kHz en-dessous de la résonance.Il se peut que le signal du TMS soit faible car son T1 est relativement long. Vousdevez donc régler la période des impulsions aux environs d'une seconde. Si la largeurde la raie (FWMH) est de l'ordre de 160 Hz, vous pouvez continuer et ajouter lechloroforme.
� Utiliser maintenant du TMS mélangé à du chloroforme dont le déplacement chimiqueest de 7.26 ppm.
Note : On doit mettre beaucoup plus de chloroforme que de TMS pour avoir dessignaux égaux : donner les proportions à utiliser.
� Obtenir le spectre de la T.F. de ce mélange et le consigner dans votre rapport.� Êtes-vous capable de mesurer le déplacement chimique du chloroforme ? Si oui, quel-
est-il ?
1.27

PHQ560-660 RMN
4.1.8 TMS avec et sans gradient de champ
� Obtenir la T.F. du TMS à 3 kHz en-dessous de la résonance avec le champ le plusuniforme possible. Comparer ce spectre à celui que vous obtenez en appliquant unfort gradient de champ selon y.
4.1.9 Imagerie RMN
Sachant que le champ statique est selon z et la bobine selon y, utiliser l'échantillon conte-nant de la glycérine à deux endroits (séparés par un bout de té�on) et réaliser une séried'expériences illustrant le principe d'imagerie RMN. Prenez soin de placer le O-ring à 39mm du centre du morceau de plastique près fond du vial. Discuter l'allure des spectres deT.F. obtenus. Calculer la grandeur du gradient selon y à partir du graphique o�rant lameilleure résolution.
4.1.10 Fluorinert FC-43
� Mettre le spectromètre en mode � Fluor �.� Vous devez recommencer la section sur l'Impulsion RF de façon à obtenir une réso-
nance à la fréquence du Fluor (environ 19.72 MHz). À l'aide de la sonde, obtenir denouveau un signal d'environ 40 volts crête-à-crête en ajustant les � tuning capaci-tors �.
� Insérer maintenant l'échantillon FC-43 et discutez l'allure de la FID obtenue. Consi-gner cette FID dans votre rapport. Discuter du phénomène de battements observé.
� E�ectuer la T.F. avec la référence à 3kHz en-dessous de la résonance. Consigner cespectre dans votre rapport.
� E�ectuer la T.F. à l'aide de l'oscilloscope. Transposer ce spectre (en dB) sur uneéchelle linéaire et le comparer avec la T.F. e�ectuée précédemment dans Labview.
� Discuter des résultats obtenus des transformations de Fourier.� Rechercher la structure chimique du Fluorinert FC-43 et tenter d'identi�er les quatre
sites de �uor dans votre spectre RMN. Typiquement, les sites près du centre d'unemolécule sont plus écrantés par le diamagnétisme des orbitales.
Références
[1] http ://www.tp.physique.usherbrooke.ca/experiences_�chiers/RMN/inhomogeneite.pdf.
[2] http ://www.en.wikipedia.org/wiki/Gaussian_integral.
[3] http ://www.en.wikipedia.org/wiki/Discrete-time_Fourier_transform.
[4] http ://www.users.polytech.unice.fr/ leroux/courssignal/node65.html.
[5] http ://www.en.wikipedia.org/wiki/Spin_echo.
[6] http ://www.2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/Spectrpy/nmr/nmr1.htm.
[7] Kittel C. Introduction to Solid state physics. John Wiley & Sons, 1986.
1.28

PHQ560-660 RMN
[8] Becker E. D. High resolution NMR : theory and chemical applications.Academic Press Inc., 1969.
[9] Canet D. La RMN Concepts et méthodes. Dunod, 1991.
[10] Ducauze C.J. et Rutledge D.N. La résonance magnétique nucléaire (rmn),principes et applications. Notes de cours.
[11] Günther H. La spectroscopie RMN. Masson, 1994.
[12] Levitt M. H. Spin Dunamics. John Wiley & Sons, 2003.
[13] Balci M. Basic 1H- 13C-NMR Spectroscopy. Elsevier Science, 2005.
1.29

PHQ560-660 RMN
Annexe A : Les transformées de Fourier
Avec des données digitalisées, nous ne disposons pas d'un nombre in�ni de points. Cela ades conséquences importantes lorsqu'on fait de l'analyse de Fourier au niveau de la précisiondes fréquences obtenues et de leur poids spectral. Vous pourrez discuter avec le moniteurdes e�ets : de la discrétisation du signal de FID, de la fréquence d'échantillonnage utiliséeet du fenêtrage du signal pour améliorer les résultats résultant d'une TF.
La transformée de Fourier d'une fonction f(t) est dé�nie par :
T.F. [f(t)] = F (ω) =
+∞ˆ
−∞
f(t)e−iωtdt (A-1)
et sa transformée inverse est donnée par :
T.F.−1 [F (ω)] =1
2π
+∞ˆ
−∞
F (ω) eiωtdω (A-2)
En pratique, on doit digitaliser le signal de FID qui est en phase avec le signal de référenceainsi que celui qui est en quadrature de phase avec lui.
Le temps entre deux point ts est appelé � sampling time �. Le temps total d'acquisition estdonc le produit du nombre de points acquisitionnés (N) par ts.
TAcquis. = Nts (A-3)
Dans le domaine des fréquence, après avoir e�ectué la transformée de Fourier, on obtientdonc N points complexes pour des fréquences allant de νf = 1/TAcquis. à νmax. = 1/ts =N/TAcquis.. La transformée de Fourier discrète s'écrit donc [3],[4] :
F (ωk) =1
N
N−1∑j=0
f(tj)e−iωktj (A-4)
Comme on a que tj = jts et que ωk = kωf alors :
F (ωk) =1
N
N−1∑j=0
f(tj)e− 2iπkj
N (A-5)
f (tj) =
N−1∑k=0
F (ωk)e2iπkjN (A-6)
Notons la relation inverse entre l'échelle du temps et celle des fréquences : plus un signalvarie rapidement en temps, plus la bande de fréquences nécessaires est large.
1.30

PHQ560-660 RMN
En RMN, on utilise souvent le fait que l'amplitude de la FID au temps zéro est égale àl'intégrale du spectre en fréquence :
f (0) =1
2π
+∞ˆ
−∞
F (ω) dω (A-7)
Passons maintenant en revue quelques transformées de Fourier utiles dans le domaine de laRMN.
Précession à une seule fréquence :
m (t) = eiω0t ←TF→ F (ω) = δ (ω − ω0) (A-8)
et uniquement un pic apparaitra dans le spectre des fréquences. Il a donc fallu inclurela partie en phase et la partie en quadrature et s'assurer que les deux correspondaientrespectivement à un cosinus et un sinus avant d'e�ectuer la TF. Sinon, il faut corriger laphase comme mentionné précédemment dans le texte.
cosinus :m (t) = cos (ω0t)←TF→ F (ω) =
1
2(δ (ω − ω0) + δ (ω + ω0)) (A-9)
Si on se contente de ne conserver uniquement que la partie en phase du signal, alors onobtiendra un doublet symétrique pour ω = ω0 et ω = −ω0.
sinus :m (t) = sin (ω0t)←TF→ F (ω) =
1
2(δ (ω − ω0)− δ (ω + ω0)) (A-10)
Si on se contente de ne conserver uniquement que la partie en phase du signal, alors onobtiendra un doublet asymétrique pour ω = ω0 et ω = −ω0.
exponentielle :
m (t) = exp
(− t
T2
)←TF→ F (ω) =
T2
1 + ω2T 22
− iωT 22
1 + ω2T 22
(A-11)
La TF d'une exponentielle correspond donc à une Lorentzienne centrée à ω = 0.
Problème de l'échantillonnage
Lorsqu'on digitalise un signal analogique, il faut prendre soin d'utiliser une fréquence d'échan-tillonnage su�samment élevée pour ne pas faire apparaitre de � fausses � composantes enfréquence dans le spectre de la TF . La �gure ci-dessous montre clairement que la fréquenced'échantillonnage (carrés noirs) n'est pas su�sante pour reproduire correctement la courbeen rouge (fréquence élevée), alors qu'elle est tout à fait appropriée pour la courbe en bleu(fréquence faible).
1.31
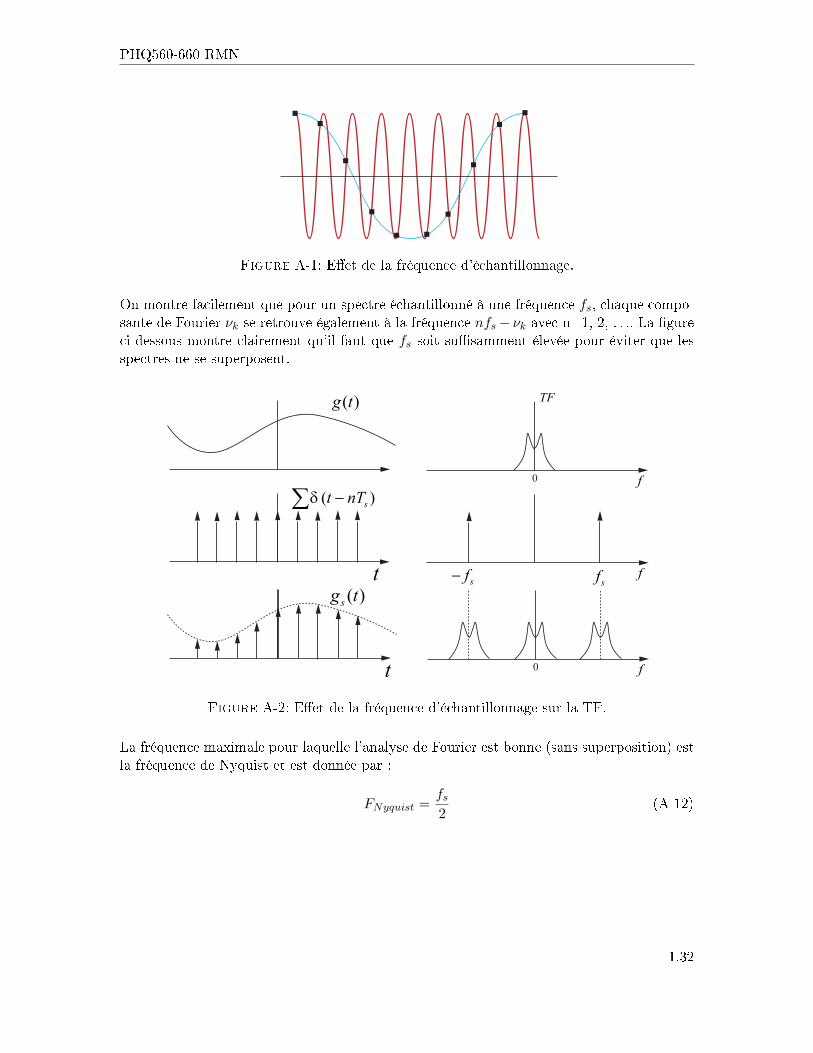
PHQ560-660 RMN
Figure A-1: E�et de la fréquence d'échantillonnage.
On montre facilement que pour un spectre échantillonné à une fréquence fs, chaque compo-sante de Fourier νk se retrouve également à la fréquence nfs− νk avec n=1, 2, . . .. La �gureci-dessous montre clairement qu'il faut que fs soit su�samment élevée pour éviter que lesspectres ne se superposent.
( )g t TF
f0
f0
fsfsf−
( )st nTδ −∑
t
t
( )sg t
Figure A-2: E�et de la fréquence d'échantillonnage sur la TF.
La fréquence maximale pour laquelle l'analyse de Fourier est bonne (sans superposition) estla fréquence de Nyquist et est donnée par :
FNyquist =fs2
(A-12)
1.32
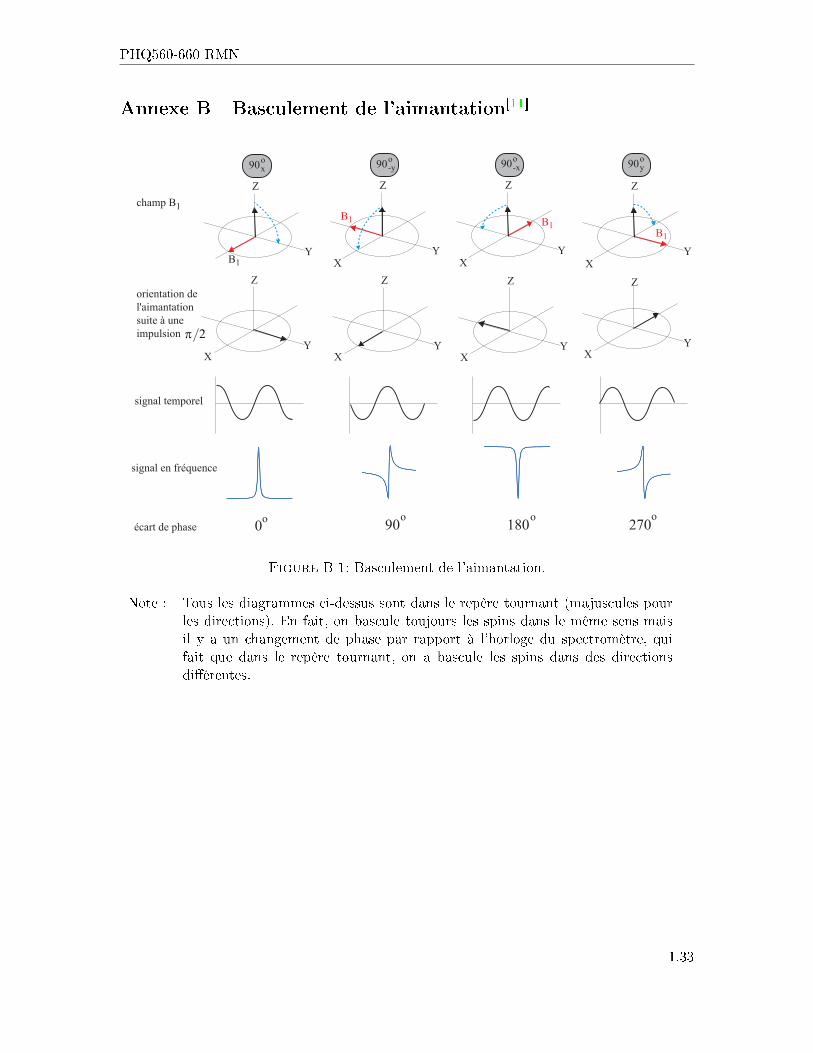
PHQ560-660 RMN
Annexe B Basculement de l'aimantation[11]
Z
1champ B
o o o o
signal temporel
signal en fréquence
écart de phase 0 90 180 270
orientation del'aimantationsuite à uneimpulsion
B1
90xo
Y
Z
YX
Z
YX
Z
YX
Z
YX
Z
X
B1
90-yo
Y
Z
X
B1
90-xo
Y
Z
X
B1
90yo
Y
2π
Figure B-1: Basculement de l'aimantation.
Note : Tous les diagrammes ci-dessus sont dans le repère tournant (majuscules pourles directions). En fait, on bascule toujours les spins dans le même sens maisil y a un changement de phase par rapport à l'horloge du spectromètre, quifait que dans le repère tournant, on a bascule les spins dans des directionsdi�érentes.
1.33

PHQ560-660 RMN
Annexe C Hamiltonien nucléaire
Il y a toute une foule de processus qui couplent les spins entre eux et avec le réseau. Parréseau, on entend ici tout l'environnement du liquide ou du solide. Les spins ne perdentpas l'énergie acquise uniquement via un couplage avec les vibrations ou rotations. Certainsde ces processus a�ectent la valeur de T1, d'autres la valeur de T2, et d'autres les valeursde T1 et T2 simultanément. Le but de la présente section est d'en présenter quelques-unssans démonstrations formelles. C'est ici qu'un traitement quantique rigoureux deviendraitnécessaire et cela sort du cadre de cette expérience qui se veut une introduction à la techniqueexpérimentale. On peut approximer l'hamiltonien d'un système de spins portés par desmolécules soumis à un champ extérieur par :
Htot. = HZ +HD +HJ +HQ (C-1)
HZ représente la partie Zeeman, qui contient également l'e�et du champ extérieur et l'e�etdu déplacement chimique (écrantage) produit par l'environnement électronique local.HD estl'interaction dipolaire entre deux moments magnétiques. HJ est l'interaction entre momentsmagnétiques qui se transmet par les électrons de liaison et qui donne lieu à la structure�ne des spectres de RMN. Finalement, HQ est l'interaction quadrupolaire qui est non nulleuniquement pour les noyaux de spin supérieur à 1/2. Nous allons nous intéresser unique-ment à la résonance du proton qui peut être décrite par les trois premièrement interactionscontrairement au 19F.
-Hamiltonien Zeeman
Comme nous l'avons montré, cette interaction donne lieu à la levée de dégénérescence desdeux niveaux d'énergie d'un moment magnétique de spin 1/2. On a aussi mentionné que lesélectrons au voisinage du moment magnétique nucléaire produisent un écrantage du champmagnétique appliqué :
H = H0 − σ (C-2)
Cela diminue donc la fréquence de Larmor d'une certaine fraction (ppm) car σ est propor-tionnel au champ magnétique appliqué. Le déplacement chimique est donc une signature del'environnement d'un moment magnétique nucléaire.
-Interaction dipolaire (ou dipôle-dipôle)
C'est l'interaction entre deux moments magnétique à travers l'espace. Dans les solides, c'estgénéralement l'interaction dominante responsable de l'élargissement des raies.
ije
i
i jj
Figure C-1: Interaction dipôle-dipôle.
1.34

PHQ560-660 RMN
L'hamiltonien de cette interaction s'écrit[12] :
H ijD = −µ0γiγj~
4πr3ij
[3 (Ii · eij) (Ij · eij)− Ii · Ij ] (C-3)
Dans les liquides, cette interaction est moyennée à zéro par les mouvements rapides desmolécules. Cela donne lieu à de la relaxation mais ne conduit pas à un élargissement desraies.
Attention : Lorsque les molécules tournent, la direction des spins ne changent pas !N'oubliez pas que ceux-ci se comportent comme des petits gyroscopes. . .
Pour diminuer l'élargissement causé par cette interaction dans les solides, on utilise l'astucesuivante :
dipB
0B
1
2
54.7oθ =
Figure C-2: Champ produit par un dipôle magnétique à l'angle magique.
Remarquez que dans la �gure précédente, le champ que produit le moment magnétique 1 à laposition du moment magnétique 2 est perpendiculaire à l'orientation de ce dernier. L'angleauquel se produit ce phénomène est appelé � l'angle magique �. L'interaction dipolaire estdonc nulle pour cette con�guration particulière.
On pourra donc réduire considérablement l'élargissement des raies d'un solide (réduit enpoudre) en le plaçant dans un petit cylindre à l'angle magique et en lui appliquant un mou-vement de rotation rapide su�samment rapide pour éliminer les interactions. En pratique,on parle de vitesse de plusieurs kilohertz.
1.35

PHQ560-660 RMN
0B
54.7oθ =
Figure C-3: Rotation de l'échantillon solide à l'angle magique.
Les interactions spin-spin sont donc beaucoup plus faibles dans les liquides que dans lessolides. On peut donc, en première approximation, considérer les spins comme indépendants.Néanmoins, l'ampleur des interactions se manifeste au niveau de la largeur des raies. Àcause des mouvements rapides dans les liquides, les moments magnétiques voient un champlocal qui �uctue rapidement. Sa valeur moyenne, c'est-à-dire sa valeur moyennée sur untemps beaucoup plus long que celui des �uctuations, est beaucoup plus faible que sa valeurinstantanée. C'est ce qui donne lieu au rétrécissement de la raie de résonance. Les raiesobservées dans les solides sont souvent beaucoup plus larges (et asymétriques) que cellesobservées dans les gaz et les liquides.
-Interaction indirecte (ou couplage J)
Cette interaction se manifeste sur les spectres à haute résolution. C'est elle qui permetl'identi�cation des groupements moléculaires et qui rend la RMN si utile en chimie organique.Malheureusement, notre spectromètre ne permet pas de voir les structures �nes que nousdécrivons dans cette section principalement à cause du manque d'inhomogénéité du champet aussi de sa faible valeur.
Les moments magnétiques d'une même molécule occupant des sites magnétiquement dif-férents peuvent se coupler par l'intermédiaire des électrons de liaison. Rappelons que lecouplage direct (dipolaire) s'annule dans un liquide. Appelons ce couplage J, dont l'hamil-tonien s'écrit :
HJ = J IA · IB (C-4)
Remarquez que ce couplage est intrinsèque à la molécule et donc indépendant du champmagnétique appliqué. On le mesure généralement en Hz.
-Cas no1 : 2 spins ½ (protons) sans couplage J.
L'hamiltonien Zeeman de ce système de 2 spins est :
HZ = ωAIZA + ωBIZB (C-5)
1.36

PHQ560-660 RMN
Notez que les deux protons ont des fréquences de résonance di�érentes car ils ont des dé-placements chimiques di�érents. Nous allons maintenant regarder le diagramme d'énergiede ce système. Chaque spin a deux états possible, |↑〉ou |↓〉 , avec comme valeurs propresassociées +~/2 et −~/2. Les quatre états possibles de ce système sont donc : |↑↑〉, |↑↓〉, |↓↑〉,|↓↓〉. Nous avons donc le diagramme d'énergie et le spectre RMN associé :
↑↑
↑↓↓↑
↓↓
( ) 2A Bω ω+
( ) 2A Bω ω− +
( ) 2A Bω ω−( ) 2B Aω ω−
Aω
AωAωBω
Bω
Bω
spectre RMN
Figure C-4: Niveaux d'énergie de deux spins ayant des déplacements chimiques di�érentsen l'absence de couplage J.
-Cas no2 : 2 spins ½ (protons) avec faible couplage J.
Un couplage faible signi�e que : J � |ωA − ωB|.
En faisant un calcul de perturbation au premier ordre, seul le terme HJ = JIZAIZB del'équation (C-4) intervient. Notez que HJ commute avec HZ .
On obtient donc des variations d'énergie données par :
〈↑↑| JIZAIZB |↑↑〉 = 〈↓↓| JIZAIZB |↓↓〉 = +J/4 (C-6)
〈↑↓| JIZAIZB |↓↑〉 = 〈↓↑| JIZAIZB |↑↓〉 = −J/4 (C-7)
Ce qui conduit au diagramme d'énergie suivant :
1.37
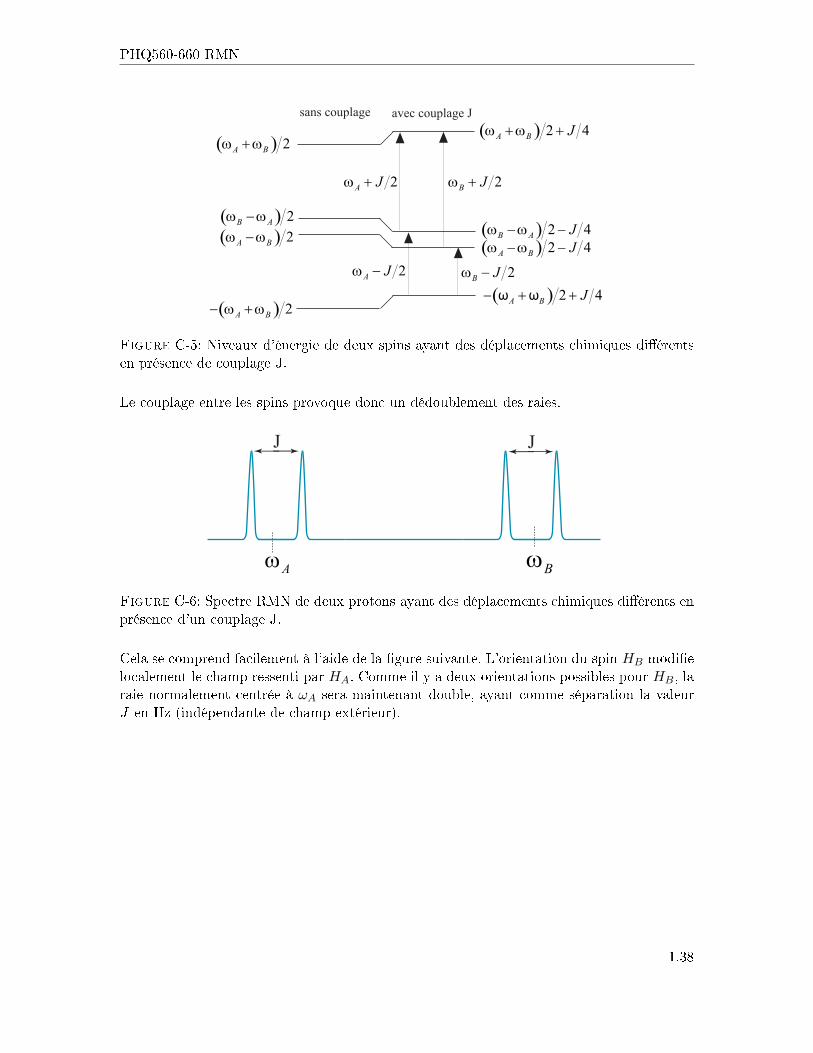
PHQ560-660 RMN
( ) 2A Bω ω+
( ) 2A Bω ω− +
( ) 2 4A B Jω ω+ +
( ) 2 4A B Jω ω− + +
( ) 2 4A B Jω ω− −( ) 2A Bω ω−( ) 2B Aω ω−
( ) 2 4B A Jω ω− −
2A Jω + 2B Jω +
2B Jω −2A Jω −
sans couplage avec couplage J
Figure C-5: Niveaux d'énergie de deux spins ayant des déplacements chimiques di�érentsen présence de couplage J.
Le couplage entre les spins provoque donc un dédoublement des raies.
Aω Bω
J J
Figure C-6: Spectre RMN de deux protons ayant des déplacements chimiques di�érents enprésence d'un couplage J.
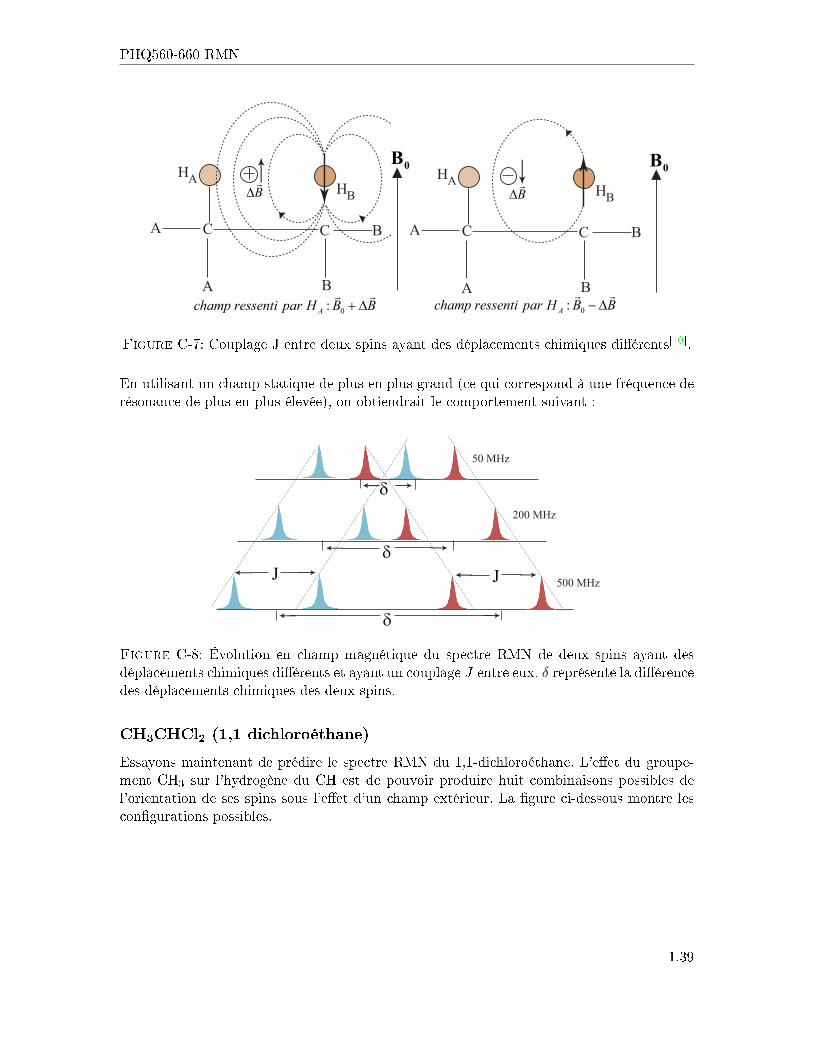
Cela se comprend facilement à l'aide de la �gure suivante. L'orientation du spin HB modi�elocalement le champ ressenti par HA. Comme il y a deux orientations possibles pour HB, laraie normalement centrée à ωA sera maintenant double, ayant comme séparation la valeurJ en Hz (indépendante de champ extérieur).
1.38
PHQ560-660 RMN
A
A
H
C C B
B
AHB
0B0B
B∆
0:Achamp ressenti par H B B+ ∆
A
A
H
C C B
B
AHBB∆
0:Achamp ressenti par H B B− ∆
Figure C-7: Couplage J entre deux spins ayant des déplacements chimiques di�érents[10].
En utilisant un champ statique de plus en plus grand (ce qui correspond à une fréquence derésonance de plus en plus élevée), on obtiendrait le comportement suivant :
δ
δJ J
δ
50 MHz
200 MHz
500 MHz
Figure C-8: Évolution en champ magnétique du spectre RMN de deux spins ayant desdéplacements chimiques di�érents et ayant un couplage J entre eux. δ représente la di�érencedes déplacements chimiques des deux spins.
CH3CHCl2 (1,1 dichloroéthane)
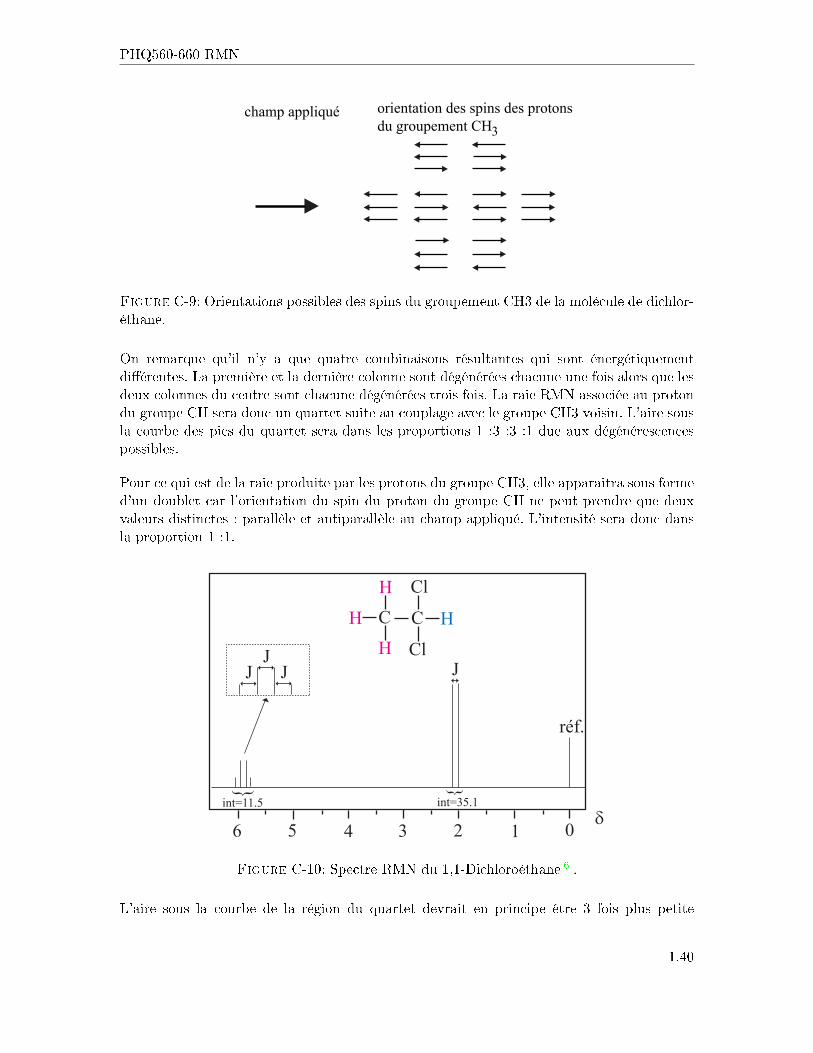
Essayons maintenant de prédire le spectre RMN du 1,1-dichloroéthane. L'e�et du groupe-ment CH3 sur l'hydrogène du CH est de pouvoir produire huit combinaisons possibles del'orientation de ses spins sous l'e�et d'un champ extérieur. La �gure ci-dessous montre lescon�gurations possibles.
1.39
PHQ560-660 RMN
champ appliqué3
orientation des spins des protonsdu groupement CH
Figure C-9: Orientations possibles des spins du groupement CH3 de la molécule de dichlor-éthane.
On remarque qu'il n'y a que quatre combinaisons résultantes qui sont énergétiquementdi�érentes. La première et la dernière colonne sont dégénérées chacune une fois alors que lesdeux colonnes du centre sont chacune dégénérées trois fois. La raie RMN associée au protondu groupe CH sera donc un quartet suite au couplage avec le groupe CH3 voisin. L'aire sousla courbe des pics du quartet sera dans les proportions 1 :3 :3 :1 due aux dégénérescencespossibles.
Pour ce qui est de la raie produite par les protons du groupe CH3, elle apparaitra sous formed'un doublet car l'orientation du spin du proton du groupe CH ne peut prendre que deuxvaleurs distinctes : parallèle et antiparallèle au champ appliqué. L'intensité sera donc dansla proportion 1 :1.
C C
Cl
Cl
H
δ
réf.
J
1 0int=35.1int=11.5
3 2
}}
46 5
H
H
HJ J
J
Figure C-10: Spectre RMN du 1,1-Dichloroéthane[6].
L'aire sous la courbe de la région du quartet devrait en principe être 3 fois plus petite
1.40

PHQ560-660 RMN
que celle du doublet car il y a 3 fois plus de protons dans le groupement CH3 que dans legroupement CH. C'est sensiblement ce qui a été mesuré dans le graphique précédent.
Nous venons de prédire le spectre � idéal � au premier ordre qu'on obtient lorsque le couplagese fait entre des noyaux ayant des déplacements chimiques très di�érents. Cela revient aussià dire : ∆δ � J .
-Sommaire des règles entourant les noyaux de spin ½
1. Les noyaux possédant des déplacements chimiques identiques ne montrent pas delevée de dégénérescence, bien qu'ils puissent être réellement couplés.
2. Les noyaux qui sont su�samment rapprochés (3 liaisons ou moins) sont généralementcouplés et leurs résonances seront mutuellement divisées en multiplets. Ils auront lemême J à condition d'avoir des déplacements chimiques di�érents.
3. Le niveau de couplage entre les spins nucléaires est caractérisé par la constante decouplage J qui est indépendante du champ magnétique appliqué.
4. On peut prédire l'allure du spectre RMN à l'aide de la règle du n + 1, où n est lenombre de noyaux voisins qui possèdent un couplage de spin similaire J. Par exemple,si un noyau est couplé à deux voisins identiques, cela donnera un triplet dans lespectre. Les raies centrales d'un multiplet sont toujours les plus intenses. L'intensitédes raies d'un multiplet est proportionnelle à sa dégénérescence et sont données parle � Triangle de Pascal �. Pour un doublet (1 :1), un triplet (1 :2 :1), un quartet(1 :3 :3 :1), etc..
1.41

PHQ560-660 RMN
C C
singulet
Triangle de Pascal
doublet
triplet
quartet
C
C
C H
C C
J1
2
33
4 4
55 10 10
6
1 1
1
1
1
11
1
1
1J J
JJJ
C
C
H
H
C CH
H
H
C C
C
H
H
H
H
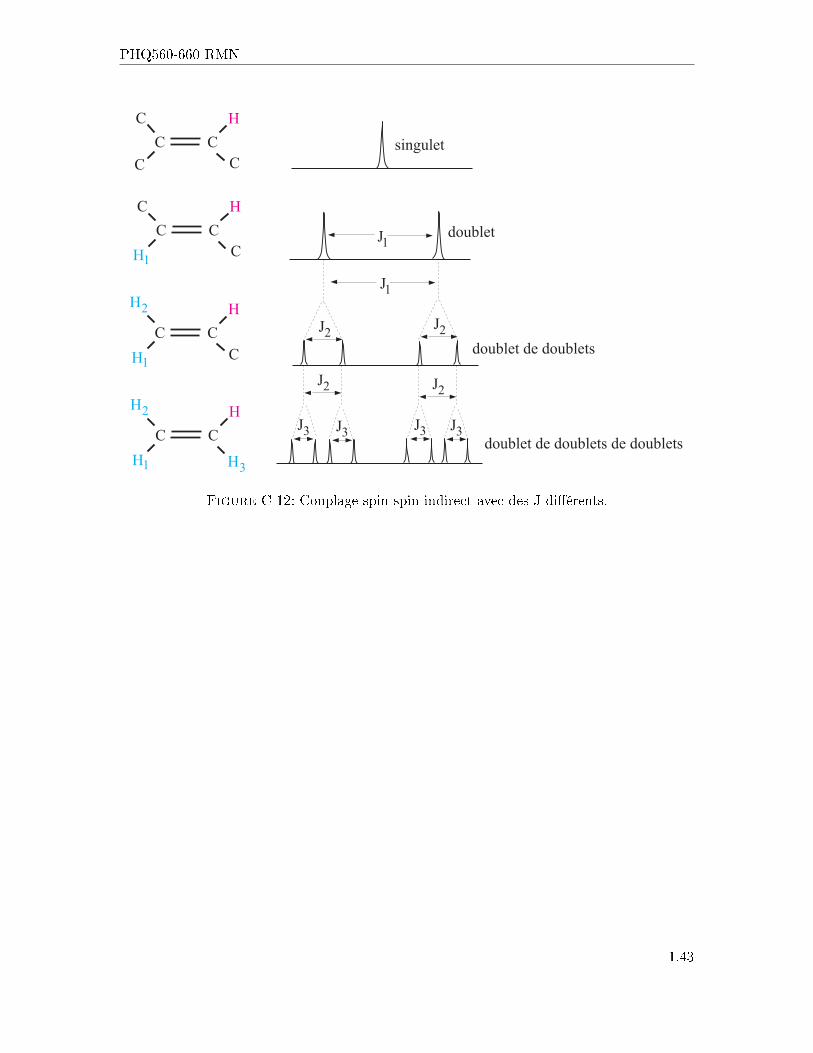
Figure C-11: Structure des multiplets avec J constant.
Pour ce qui est maintenant des noyaux ayant un couplage de spin avec deux voisins ou plusayant des J di�érents, et bien la règle du n+ 1 ne prédit pas entièrement le spectre. Il fautalors prendre en compte que la séparation due à un groupe de J doit être ajoutée à celled'un autre groupe. La �gure ci-dessous montre le phénomène.
1.42
PHQ560-660 RMN
CC
C
Csingulet
doublet
doublet de doublets
doublet de doublets de doublets
CH
CC
C
C
CH
J1H1
CCH
H1
H2
H3
CCH
H1
H2
J1
J3J3J3J3
J2J2
J2 J2
Figure C-12: Couplage spin-spin indirect avec des J di�érents.
1.43





























