Embed Size (px)
Citation preview

Université de Genève Faculté de Médecine Section de médecine fondamentale Département de Morphologie
Thèse préparée sous la direction du professeur Paolo Meda
L’EXPRESSION FORCEE DE LA CONNEXINE 32 (Cx32) PAR LES CELLULES ß CAUSE UNE AUGMENTATION POST-NATALE DU
PANCREAS ENDOCRINE
Thèse
Présentée à la Faculté de Médecine de l’Université de Genève pour obtenir le grade de Docteur en médecine
Par
Ambroise Wonkam
De
Bahouan (Cameroun)
Thèse No 10349
Genève, 2003

Dédicace
À la mémoire du professeur Cheick Anta Diop.
« Science alone cannot save Africa, but Africa without science cannot be saved »
Mohamed H.A. Hassan (2001). Science 294, 1609.

Remerciements À Messieurs les professeurs, Paolo Meda, Pour m’avoir donné l’opportunité de m’initier à la recherche fondamentale, pour votre formation pédagogique, scientifique, technique et votre engagement pour l’enseignement au Cameroun. Trouvez ici la marque de mon profond respect. Fritz Baumann, Pour votre implication sans limites, dans le développement de la coopération universitaire entre la Suisse et Cameroun. Alain Perrelet, Pour votre encadrement pédagogique et vos conseils judicieux. Daniel Hoessli, Pour tes enseignements en pathologie générale. Jeremiah Cox, Pour votre précieux coatching. Lelio Orci, Pour m’avoir accueilli au sein du département de morphologie. Aux docteurs Annelise Wohlwend, Pour tes enseignements en histologie et tes techniques pédagogiques. Domenico Bosco, Pour ta collaboration à l’enseignement d’histologie à la Faculté de médecine de l’Université de Yaoundé1. À mon épouse Lilly et à mon fils Ramses, Pour avoir toujours été présents. À tous les membres du laboratoire du professeur Meda Dorothée, Esther, Juliette, Anne, Isabelle, Alessandra, Véronique, Patrick, Thierry Ludovic, David, Igor, Christophe ; Egalement Danielle Ben Nasr et Tutzi Radlgruber Pour votre assistance et votre amitié. À la Commission Fédérale des Bourses pour Etudiants Etrangers, pour son subside. À tous ceux qui de près ou de loin, ont contribué à la bonne marche de mon séjour scientifique au département de morphologie.

Contents Dédicace p.2 Remerciements p.3 Résumé p.6 Chapitre 1 : Introduction p.7 Les Jonctions gap: définition et structure p.9 Les connexons : formation des canaux gap p.9 Les connexines : protéines spécifiques des jonctions gap p.11 Les connexines : structure p.11 Les connexines: gène p.13 Couplage des jonctions gap p.13 Couplage électrique p.13 Couplage métabolique p.14 Régulation des canaux gap p.14 Fonctions des jonctions gap p.15 Connexines et pathologies humaines p.16 Connexines, croissance et prolifération cellulaire p.17 Les communications jonctionnelles sont altérées dans dans beaucoup de cellules néoplasiques p.17 Les facteurs de croissance, les carcinogènes et les oncogènes altèrent les communications jonctionnelles p.17 Les connexines et les communications jonctionnelles varient au cours du cycle cellulaire p.18 Des altérations spécifiques des communications jonctionnelles altèrent le phénotype et la croissance cellulaire : approches antisens, “knock-out” et transfections p.18 Rôle des connexines dans les épithéliums sécrétoires : l’exemple du pancréas p.19 Le développement et la croissance du pancréas endocrine p.21 La problématique de notre étude p.23 Chapitre 2: mon travail original p.24 Methods p.26 Generation of transgenic mice p.26 Tissue sampling p.27 Insulin Radioimmunoassay p.27 Histology p.27 Connexin and E-cadherin expression p.27 Insulin radioimmunoassay p.27 Pancreatic islets and β-cells sorting p.27 Morphometric analysis p.28

The volume density of the pancreatic β-cell p.28 The size of individual islets p.28 The numerical density of islets, dispersed single β-cell, and total β-cells p.28 The size of individual β-cell p.28 Proliferation rate p.28 β-cells apoptosis p.30 Statistical analysis p.30 Results p.32 β-cells of transgenic mice express Cx32 p.32 β-cells coupling was increased in islets of transgenic mice p.33 RIP-Cx32 mice feature a normal phenotype p.34 The four main types of islet cells differentiate in RIP-Cx32 mice p.34 Τhe volume density of β-cells and insulin content was increased in adult, RIP-Cx32 mice p.35 The size of pancreatic islets was increased in adult RIP-Cx32 animals p.36 The number of islets and of single β-cells was decreased in adult RIP-Cx32 mice p.38 β-cells are larger in RIPCx32 mice p.39 RIP-Cx32 adult mice feature increased number of β-cells p.40 β-cells proliferation was normal in adult Rip Cx32 mice p.41 β-cells apoptosis indices was normal in adult RIP-Cx32 mice p.42 E-cadherin was similarly expressed in the islets of transgenic and control adult animals p.42 Analysis of newborn mice p.43 The volume density of β-cells and insulin content of newborn RIP-Cx32 mice is similar to that of controls p.44 The size of β-cell clusters was similar in transgenic RIP-Cx32 and control newborn mice p.45 The proliferation of β-cells was similar in control and RIP-Cx32 newborn mice p.45 Chapitre 3 : Discussion p.46 Conclusion et perspectives p.50 Références p.52
5

Résumé: Nous avons investigué la croissance et la prolifération des cellules β, sécrétrices d’insuline, suite à des changements des communications intercellulaires par les jonctions de type gap. Nous avons utilisé un modèle de souris trangéniques dites RIP-Cx32, dont les cellules β ont été forcées à exprimer la Cx32, une protéine qui n’est pas exprimée dans les îlots pancréatiques natifs. À la naissance, nous n’avons pas observé de différence entre les souris RIP-Cx32 et les souris contrôles. Par contre, à l’âge adulte, les souris transgéniques adultes, montrant une augmentation du niveau de communication cellulaire à travers les cannaux formés de Cx32, montrent une augmentation de 2 à 3 fois de la masse de cellules β, corrélée à une augmentation du contenu en insuline du pancréas. Ces souris montrent aussi une hypertrophie des îlots de Langherhans et des cellules β, une augmentation du nombre de ces cellules, mais pas d’apoptose anormale de ces cellules. Ces résultats indiquent que les connexines influencent in vivo la croissance postnatale des cellules β pancréatiques.
6

Chapitre1 : Introduction

Le fonctionnement normal de tout organisme multicellulaire dépend de la coordination des cellules qui le composent. Cette coordination implique un système de communication grâce auquel chaque cellule est informée de l’activité des cellules voisines et règle, en conséquence, son propre niveau de fonctionnement. Les cellules des vertébrés communiquent entre elles par de nombreux mécanismes. Plusieurs de ceux-ci font intervenir des signaux qui sont transmis à des cellules équipées de récepteurs, canaux, enzymes ou voies métaboliques appropriées. Dans tous ces cas, le transfert d’information d’une cellule à l’autre se fait indirectement via, la diffusion d’ions ou de molécules signal dans l’espace intercellulaire. Cependant, le fonctionnement de nombreux tissus est préservé dans des conditions qui perturbent ou même abolissent le flux de signaux extra-cellulaires, indiquant que d’autres mécanismes de communication intercellulaire sont au moins aussi importants. La perte du fonctionnement normal de certains tissus après isolement de leurs cellules suggère que ces mécanismes dépendent d’interactions directes entre cellules voisines (Meda, 1996a). Ainsi, lorsqu’elles sont en contact, la plupart des cellules échangent des informations par le biais des molécules de surface telles que les molécules d’adhésion, mais aussi par celui des canaux intercellulaires qui peuvent relier deux ou plusieurs cellules et permettre des échanges de molécules hydrosolubles de petite taille (maximum 900 daltons), directement d’une cellule à ses voisines (Beyer, 1990). Ces canaux intercellulaires sont formés de connexines, une famille de protéines intégrales de la membrane plasmique qui sont exprimées par pratiquement tous les types de cellules, quelle que soit la position de l’organisme multicellulaire considéré dans la phylogenèse du monde animal (Bennett et al., 1991).
8

Les jonctions gap: définition et structure L’analyse de coupes fines en microscopie électronique a permis l’identification de jonctions gap dans la plupart des organes et tissus des vertébrés, à l’exception de certaines cellules circulantes du sang, des cellules musculaires striées adultes, de quelques neurones et des spermatozoïdes (Loewenstein and Kanno, 1966 ; Hertzberg et al., 1981; Pitts and Finbow, 1986). Dans les régions de la membrane plasmique où l’on observe des jonctions gap, l’espace extracellulaire est réduit à une mince fente de 2 à 4 nm de large dénommé “gap” en anglais, d’où le terme consacré de jonction gap (fig.1). Ce “gap” a été particulièrement bien mis en évidence par l’emploi de molécules denses aux électrons, qui marquent l’espace extracellulaire et révélent la structure pentalaminaire caractéristique de ces jonctions (Revel and Karnovsky, 1967).
Figure1: Microscopie électronique conventionnelle et en cryofracture des jonctions gap
A: plaque jonctionnelle visualisée par microscopie électronique conventionnelle. B: Plaque jonctionnelle vue par microscopie électronique après cryofracture. D’après Wiszniewski et al., 2000.
B A
Dès 1970, les études par cryofracture ont révélé que les jonctions gap sont formées de particules intra-membranaires, regroupées dans certaines régions de la membrane cellulaire (fig.1). À la même époque, des analyses par cristallographie de jonctions gap isolées à partir de la membrane plasmique d’hépatocytes, ont permis de conclure qu’une jonction gap peut être formée de quelques uns à plusieurs centaines de canaux intercellulaires. Ces canaux sont eux-mêmes formés des 2 hémi-canaux, appelés connexons (fig.2). Les connexons : formation des canaux gap Des analyses par diffraction optique (Caspar et al., 1977) et aux rayons X ont révélé la structure des connexons (Goodenough, 1976). Chacun de ces demi-canaux est composé de 6 protéines nommées connexines. La disposition des six connexines dans chaque connexon détermine un espace hydrophile central de 2 à 4 nm de diamètre (fig.2).

Contrairement à la situation pour la plupart des autres protéines intégrales, l’oligomérisation des six connexines formant chaque connexon survient au niveau du compartiment distal de l’appareil de Golgi (Musil and Goodenough, 1993). Les connexons sont ensuite insérés dans la membrane plasmique. Cette insertion se fait dans des régions membranaires ne participant pas à d’autres contacts intercellulaires tels que les jonctions serrées ou les desmosomes (Musil and Goodenough, 1991). Après son insertion dans la membrane, le connexon est probablement “fermé” jusqu’au moment où les portions extracellulaires de deux connexons sont en “vis-à-vis” et s’arriment pour former un canal gap (Musil and Goodenough, 1993). Cette reconnaissance entre connexons est déterminée par les domaines extracellulaires de chaque connexine. La liaison entre connexons est maintenue par des interactions hydrophobes (Musil and Goodenough, 1991), qui doivent être suffisamment fortes pour isoler le canal de l’espace extracellulaire. Lorsque cette isolation est établie, les canaux jonctionnels s’ouvrent progressivement pour atteindre leur perméabilité maximale (Bennett et al., 1991). Figure 2 : Représentation schématique d’un canal jonctionel et des connexines
A : le canal jonctionnel est formé de deux connexons qui s’assemblent au niveau de zones de contact étroit entre cellules. Chaque connexon correspond à l’assemblage de six protéines transmembranaires appelées connexines. B : Schéma représentant l’insertion dans la membrane plasmique d’une connexine, ainsi que son homologue dans une cellule adjacente. M= segments membranaires ; E= boucles extracellulaires ; BC= boucle cytoplasmique. Schéma adapté d’après Kumar and Gilula, 1996.
A B Cellule 1 NT CT
BC
E1 E2 Zone extracellulaire
M1 M2 M3 M4
Cellule 2
10

Les connexines : protéines spécifiques des jonctions gap Les jonctions gap des hépatocytes de rongeur ont été les premières à être caractérisées biochimiquement, du fait de leur abondance (Benedetti and Emmelot, 1968; Goodenough and Stoeckenius, 1972; Hertzberg, 1984). La première connexine qui a été identifiée a un poids moléculaire de 27 kDa. Elle est composée de 283 acides aminés et son poids moléculaire, calculé à partir de sa séquence ADN, est de 32 kDa, d’où sa dénomination Cx32 (Paul, 1986). Par la suite une autre connexine (Cx43) a été identifiée dans les membranes isolées à partir de cœur de rat (Manjunath et al., 1984; 1987) et clonée en utilisant des conditions d’hybridation de faible stringence et une sonde de rat codant pour la Cx32 (Beyer et al., 1987). Depuis lors, une vingtaine d’autres connexines ont été identifiées en utilisant la même approche, et l’utilisation d’anticorps spécifiques a permis de les mettre en évidence dans de nombreux tissus et types cellulaires. Toutes ces connexines ont été classées selon la même nomenclature (Beyer et al., 1987) (tableau1). Les connexines : structure Les connexines présentent des analogies de structure avec d’autres protéines formant des canaux ioniques. Les connexines traversent la membrane plasmique 4 fois (segments hydrophobes M1 à M4) et leurs deux domaines amino- et carboxy-terminaux sont intra-cytoplasmiques (Beyer et al., 1990; Bennett et al., 1991) (fig.2). Cet arrangement donne deux boucles extracellulaires et une boucle intracellulaire. Les deux boucles extracellulaires sont responsables de l’accrochage entre les connexons de deux cellules voisines (fig.2). Cette topographie a été confirmée par immunomarquage avec des anticorps dirigés contre différent segment des Cx32 et Cx43 (Zimmer et al., 1987; Goodenough et al., 1988; Hertzberg et al., 1988; Milks et al., 1988; Yancey et al., 1989). Les connexines des différentes espèces montrent une structure primaire particulièrement bien conservée. La comparaison de la séquence d’acides aminés révèle que les quatre régions transmembranaires, les deux boucles extracellulaires et le domaine amino-terminal sont hautement conservés entre differentes connexines. Notamment, chacune des deux boucles extracellulaires contient trois résidus cystéines qui ont conservé la même position au cours de l’évolution (Bennett et al., 1991). Inversement, la boucle intracytoplasmique connectant le second et le troisième domaine transmembranaire et la région carboxy-terminale sont très différentes d’une connexine à l’autre. Ainsi, l’extrémité carboxy-terminale comporte 18 acides aminés dans Cx26 et 191 dans Cx46, expliquant les variations de poids des differentes molécules (Tableau1). Cette variabilité de l’extrémité carboxyterminale a permis la production d’anticorps spécifiques qui sont utilisés pour l’immunolocalisation des différentes connexines dans les tissus. Ces régions variables sont impliquées dans la régulation de la perméabilité des canaux intercellulaires (Fishman et al., 1991; Steinberg et al., 1994; Veenstra et al., 1994; Veenstra et al., 1994). Contrairement à de nombreuses autres protéines intégrales de la membrane plasmique, les connexines ont une très courte demi-vie, de l’ordre de 3 à 6 heures (Beyer et al., 1990; Kumar and Gilula, 1992). Elles ne sont pas glycosilées et n’ont pas d’activité enzymatique.
11
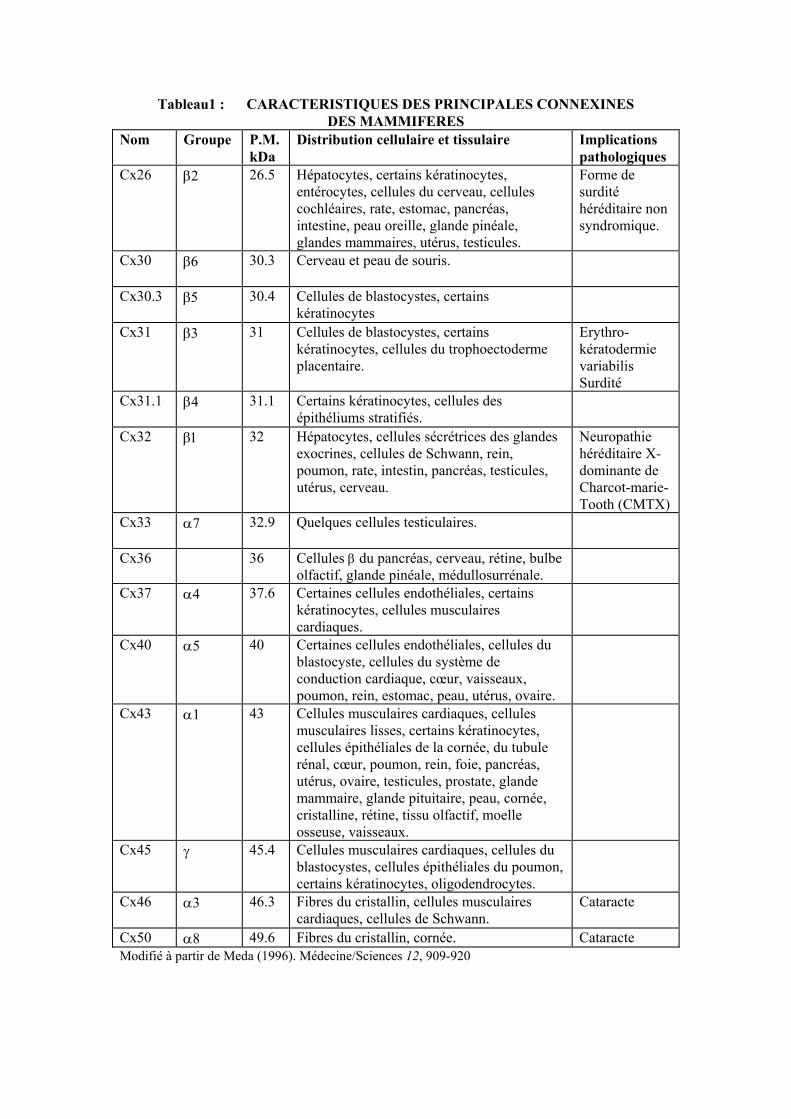
Tableau1 : CARACTERISTIQUES DES PRINCIPALES CONNEXINES DES MAMMIFERES
Nom Groupe P.M. kDa
Distribution cellulaire et tissulaire Implications pathologiques
Cx26 β2
26.5 Hépatocytes, certains kératinocytes, entérocytes, cellules du cerveau, cellules cochléaires, rate, estomac, pancréas, intestine, peau oreille, glande pinéale, glandes mammaires, utérus, testicules.
Forme de surdité héréditaire non syndromique.
Cx30 β6
30.3 Cerveau et peau de souris.
Cx30.3 β5
30.4 Cellules de blastocystes, certains kératinocytes
Cx31 β3
31 Cellules de blastocystes, certains kératinocytes, cellules du trophoectoderme placentaire.
Erythro- kératodermie variabilis Surdité
Cx31.1 β4
31.1 Certains kératinocytes, cellules des épithéliums stratifiés.
Cx32 β1
32 Hépatocytes, cellules sécrétrices des glandes exocrines, cellules de Schwann, rein, poumon, rate, intestin, pancréas, testicules, utérus, cerveau.
Neuropathie héréditaire X-dominante de Charcot-marie-Tooth (CMTX)
Cx33 α7
32.9 Quelques cellules testiculaires.
Cx36 36 Cellules β du pancréas, cerveau, rétine, bulbe olfactif, glande pinéale, médullosurrénale.
Cx37 α4
37.6 Certaines cellules endothéliales, certains kératinocytes, cellules musculaires cardiaques.
Cx40 α5
40 Certaines cellules endothéliales, cellules du blastocyste, cellules du système de conduction cardiaque, cœur, vaisseaux, poumon, rein, estomac, peau, utérus, ovaire.
Cx43 α1
43 Cellules musculaires cardiaques, cellules musculaires lisses, certains kératinocytes, cellules épithéliales de la cornée, du tubule rénal, cœur, poumon, rein, foie, pancréas, utérus, ovaire, testicules, prostate, glande mammaire, glande pituitaire, peau, cornée, cristalline, rétine, tissu olfactif, moelle osseuse, vaisseaux.
Cx45 γ
45.4 Cellules musculaires cardiaques, cellules du blastocystes, cellules épithéliales du poumon, certains kératinocytes, oligodendrocytes.
Cx46 α3
46.3 Fibres du cristallin, cellules musculaires cardiaques, cellules de Schwann.
Cataracte
Cx50 α8 49.6 Fibres du cristallin, cornée. Cataracte Modifié à partir de Meda (1996). Médecine/Sciences 12, 909-920

Les connexines : gène À ce jour, environ 20 connexines distinctes ont été identifiées chez les mammifères. Chaque connexine est codée par un gène distinct (Kumar and Gilula, 1992; Willecke et al., 1991). Cependant l’existence d’une liaison génétique entre connexines de structure primaire différente, suggère que les gènes des connexines dérivent d’une même séquence originale encore indéterminée, qui a subi au moins deux duplications (Bennett et al., 1991; Kumar and Gilula, 1992) juste avant, ou au début de l’évolution des vertébrés. Ces duplications ont permis de la divergence de deux groupes de protéines : Le groupe α comprend les connexines comportant une boucle cytoplasmique courte, alors que le groupe β comprend les connexines comportant des boucles cytoplasmiques longue (tableau1). Le groupe γ crée récemment pourrait réunir des connexines ayant une taille intermédiaire de cette boucle. La plupart les gènes de connexines étudiés à ce jour montrent une structure similaire (Fishman et al., 1991; Hennemann et al., 1992; Hennemann et al., 1992) comportant deux exons séparés par un intron. Le premier exon est petit et code pour une séquence 5’ soit non transcrite, soit codant pour une petite partie de la protéine. L’intron varie en taille d’un gène à l’autre, mais, dans tous les cas, contient un site accepteur d’épissage en amont du codon de début de traduction. Le deuxième exon est plus grand et contient en un bloc toute la séquence codante. Les éléments régulateurs des régions promotrices varient d’un gène à l’autre, expliquant sans doute l’expression différentielle de diverses connexines. La plupart des gènes de connexines sont sur des chromosomes différents (Fishman et al., 1991; Hennemann et al., 1992; Hennemann et al., 1992). Par exemple dans l’espèce humaine, les gènes codant pour les Cx26, Cx32 et Cx43 se trouvent respectivement sur les chromosomes 13, X et 6. Il ne semble pas exister de lien entre la localisation et le regroupement de gène sur certains chromosomes, et leur expression dans divers tissus. La raison de la multiplicité des connexines, la diversité de leur distribution tissulaire et leur co-localisation dans certaines cellules restent inexpliquées. Couplage et régulation des jonctions gap Couplage électrique Le couplage électrique a d’abord été détecté en faisant un enregistrement avec des micro-électrodes de haute résistance entre cellules cardiaques (Weidmann, 1952), synapses moteurs d’écrevisses (Furshpan et Potter, 1959) et muscles lisses de mammifères (Dewey et Barr,1962). Cependant, ce n’est qu’en 1963 qu’une corrélation a été établie entre la présence de jonctions gap observées par microscopie électronique et le couplage électrique (Robertson, 1963). Ce couplage qui a d’abord été mis en évidence dans des tissus électriquement excitables a été ensuite observé dans des tissus épithéliaux, tels que les glandes salivaires de drosophile (Loewentein et Kanno, 1964) ou les hépatocytes de souris (Penn, 1966), mais aussi dans la neuroglie (Kuffler et Potter, 1963). À la différence des autres canaux ioniques, les canaux gap ne sont pas sélectifs pour un type donné d’ions. En effet, ils permettent le

passage direct entre le cytoplasme de cellules voisines de presque tous les ions (Bennett et al., 1991). Le couplage électrique peut être évalué indirectement, en mesurant le passage d’un courant électronique d’une cellule à une autre ou, plus directement, en évaluant la conductance des canaux gap par la technique dite de clamp avec double voltage imposé (Spray et al., 1979). Les propriétés électriques des canaux gap varient sensiblement selon le type de connexine et la présence ou non de sites phosphorylés sur la portion carboxy-terminale de connexines spécifiques (Bennett et al., 1991; Veenstra et al., 1992). Couplage métabolique En 1966, Kanno et Lowenstein ont montré que le couplage jonctionnel n’était pas limité aux ions mais intéressait également des molécules hydrophiles. Ainsi, des molécules cytoplasmiques ne pouvant pas traverser la membrane cellulaire peuvent être échangées entre cellules sans passer par le milieu extracellulaire (Kanno and Loewenstein, 1966). Les caractéristiques physico-chimiques du canal de chaque connexon limitent le poids moléculaire des molécules perméables à 900 daltons (Simpson et al., 1977) et leur diamètre maximal à ≤ 1,5 nm. Ce transfert intercellulaire de molécules a été démontré par l’injection de traceurs fluorescents pouvant diffuser par les jonctions gap, tels que le jaune de Lucifer (Stewart, 1978), la fluorescéine (Kanno et Loewenstein, 1964 ; Kanno and Loewenstein, 1966) et plus récemment la neurobiotine (Peinado et al., 1993). Cette approche permet de révéler la perméabilité des jonctions gap entre cellules en culture (Loewenstein, 1979) ou dans des tissus intacts (Meda et al., 1987, 1988; Salomon et al., 1988) et de déterminer le nombre de cellules dans lesquelles le traceur a diffusé par la voie des jonctions gap. Le transfert inter-cellulaire de molécules via les canaux jonctionnels se fait par diffusion le long d’un gradient électrochimique. Ce passage se fait rapidement (quelques fractions de secondes à quelques minutes) et peut concerner un très grand nombre de cellules. Ainsi, environ 106 molécules de la taille de l’AMP cyclique (AMPc) pourraient être échangées en une seconde entre une centaine de cellules couplées. L’utilisation de nucléotides radiomarqués a aussi permis de démontrer, par des techniques d’autoradiographie, l’existence d’échanges intercellulaires de molécules endogènes telles que l’AMPc (Subark-Sharpe et al., 1966, 1969; Meda et al., 1981) ou encore d’acides aminés, de cofacteurs vitaminiques, de métabolites, de et de sucres (Saez et al., 1989). Régulation des canaux gap Dans des conditions physiologiques, la perméabilité des canaux gap est modulée par le Ca++ libre intracellulaire, le pH du cytoplasme (Spray et al., 1982), de très nombreuses hormones (Meda et al., 1984; Stagg and Fletcher, 1990), des neuromédiateurs, des facteurs de croissance, des métabolites de l’acide arachidonique, les proteine-kinases A et C (Kanno, 1985; Loewentein, 1985; Stagg and Fletcher, 1990), des phosphatases, des oncogènes et enfin certains morphogènes. Ainsi la régulation de la perméabilité des jonctions gap et de l’expression des connexines est multifactorielle et diffère selon le type de cellule et leur état d’activité (Bennett et al., 1991).
14

Expérimentalement, il est possible de fermer des canaux gap en employant des molécules de la famille des alcanols tels l’heptanol ou l’octanol (Meda et al., 1986), ou des anesthésiques tel l’halothane (Burt and Spray, 1988). Actuellement, les études les plus prometteuses pour diminuer ou augmenter l’expression d’une connexine, utilisent des techniques de biologie moléculaire basées sur l’emploi d’oligonucléotides antisens (Bevilacqua et al., 1989; Moore and Burt, 1994), ou la transfection de gènes dominant-négatifs (Paul, 1995). Suivant le type de connexine exprimé dans une jonction, on peut envisager des échanges intercellulaires sélectifs pour certains messagers secondaires, ou des échanges se faisant dans une direction préférentielle. Des études in vitro ont confirmé possibilité (Veenstra et al., 1992; White et al., 1995; Goldberg et al., 1999). Au total, la diversité des molécules régulatrices de la perméabilité des canaux gap et le grand nombre de connexines, chacune ayant des caractéristiques propres, compliquent la compréhension des systèmes régulant la fonction des jonctions gap. Fonctions des jonctions gap Bien que durant l’évolution phylogénique, la plupart des types cellulaires forme des jonctions gap, soulignant l’importance physiologique de ces structures, nos connaissances quant à cette fonction restent fragmentaires. Les jonctions gap interviennent dans le développement embryonnaire (Weir and Lo, 1982; Warner et al., 1984; Paul, 1995), le contrôle de la croissance (Mehta et al., 1986; Yamasaki and Naus, 1996 ; Ruch, 2000) et de la différenciation cellulaire (Loewenstein and Rose, 1992). Nous connaissons le rôle majeur joué par le couplage jonctionnel dans la propagation électrique des potentiels d’action au sein de tissus excitables tels que le cœur (Weidmann, 1952; Woodbury et Crill, 1961; Barr et al., 1965), les muscles lisses (Dewey et Barr, 1962) et les synapses électro-toniques (Furshpan et Potter, 1959; Auerbach and Bennett, 1969; Bennett and Goodenough, 1978). Par contre, dans les tissus non excitables, dont les activités ne requièrent pas la transmission de courants ioniques, le rôle des jonctions gap reste en grande partie inconnu. Le couplage par les canaux jonctionnels est unique, en ce qu’il permet d’équilibrer des gradients ioniques et moléculaires entre cellules voisines. Dans un tel système, l’augmentation de la concentration d’un ion ou d’une molécule cytoplasmique dans une cellule est rapidement suivie par son passage dans les cellules adjacentes. Ce passage conduit à un équilibrage des concentrations électrochimiques de part et d’autre du canal. Lorsque la concentration finale ainsi obtenue atteint le seuil permettant d’activer un mécanisme effecteur, un changement d’activité sera observé non seulement dans la cellule où sont survenus initialement les changements ioniques et moléculaires, mais aussi dans toutes les autres cellules qui sont couplées avec elle. Ainsi, le couplage jonctionel peut contrebalancer une hétérogénéité fonctionnelle des cellules au sein d’un tissu, en assurant par exemple le recrutement fonctionnel de cellules qui n’auraient pas été activées individuellement (Meda, 1996a). Des expériences sur des souris transgéniques, présentant un défaut d’expression d’une connexine ont maintenant confirmé in vivo que ces protéines contribuent à
15

l’homéostasie de divers tissus (Kumar and Gilula, 1986). Par exemple, les souris déficientes pour les Cx43 et Cx32 n’ont pas de défaut majeur durant la vie in utero, alors que celles déficientes en Cx26 meurent au onzième jour de la vie embryonnaire, du fait d’un défaut de maturation du placenta. Cependant, les souris “Knock-out” pour le gène de la Cx43 meurent quelques heures après la naissance d’une malformation de l’artère pulmonaire (Reaume et al., 1995). L’intérêt de ce modèle animal est qu’il reproduit en partie des anomalies observées chez des patients porteurs d’une mutation de la Cx43, qui rend cette connexine non fonctionnelle (Britz-Cunningham et al., 1995). Connexines et pathologies humaines L’importance des connexines est soulignée par le fait que certaines de leurs mutations sont génétiquement liées à diverses maladies humaines (Lamartine et al., 2000 ; Maestrini et al., 1999 ; Meda et Spray, 2000 ; Kelley et al., 1999 ; Kelsell et al., 200 ; Krutovskikh; Richard et al., 1998; Richard et al., 1998 ; Richard et al., 2000, ; Solis et al., 2001 and Yamasaki, 2000). Par exemple dans le cas de la Cx32, les malades sont atteints de la maladie de Charcot-Marie-Tooth, la plus commune des neuropathies périphériques héréditaires (Bergoffen et al., 1993; Fairweather et al., 1994; Ionasescu et al., 1994). Dans le type 1 de cette maladie (CMTX1), le locus génétique a été localisé sur une région du chromosome X où se trouve, entre autres le gène codant pour la Cx32. Des mutations ponctuelles de la Cx43 ont été observées chez des enfants atteints d’hétérotaxie viscero-atriale (syndrome d’Ivermark), un ensemble cliniquement et génétiquement hétérogène, associant le plus souvent de sévères malformations cardiaques à l’absence de rate et à des formes plus ou moins complètes de situs inversus thoracique et abdominal (Britz-Cunningham et al., 1995). Ces enfants présentent une sténose ou une atrésie de l’artère pulmonaire, proche de celle observée chez les souris transgéniques qui n’expriment plus de Cx43 (Britz-Cunningham et al., 1995). Des mutations dans les gènes des Cx26, Cx30, Cx30.3 et Cx31 ont été détectées dans plusieurs formes de surdité congénitale et dans certaines pathologies épidermiques. Les mutations récessives sont les plus fréquentes. Elles provoquent une surdité chez les sujets atteints de façon homozygote, et n’induisent pas de phénotype chez les sujets hétérozygotes. Il existe de nombreuses causes à l’origine des surdités, cependant, les mutations dans du gène de la Cx26 sont responsables de la majorité des surdités congénitales. La Cx30 est impliquée dans deux pathologies différentes. Une forme de surdité (Grifa et al., 1999) et le syndrome de Clouston (Lamartine et al., 2000), une dysplasie ectodermale hydrotique. Cette maladie rare, à transmission autosomique dominante, se caractérise par une hypotrichose associée à une dystrophie des ongles, ainsi qu’une hyperkératose de la paume des mains et de la plante des pieds, mais pas de surdité. Des mutations ponctuelles dans le gène des Cx46 et Cx50 ont été détectées dans deux types différents de cataractes (Paul et al., 2000 ; Berry et al., 1999).
16

Connexines, croissance et prolifération cellulaire. Les communications jonctionelles sont altérées dans beaucoup de cellules néoplasiques La comparaison de cellules cancéreuses et de cellules normales d’un même tissu a généralement montré que les cellules cancéreuses présentent une diminution du couplage jonctionnel, de l’expression des connexines et du nombre des jonctions gap (Ruch, 2000). Wilgenbus et al., (1992) ont examiné plusieurs tissus néoplasiques et normaux humains pour l’expression de la Cx26, de la Cx32 et de la Cx43. Ils ont trouvé que la plupart des tumeurs bénignes ont une expression normale de connexines, alors que la plupart de tumeurs malignes montrent une diminution de l’expression des connexines ou les expriment inadéquatement. Oyamada et al. (1990), ont également observé une diminution d’expression de la Cx32 et une expression inadéquate de la Cx43 dans le carcinome hepato-cellulaire humain. La majorité des carcinomes humains et murins exprime moins de Cx43 que les cellules épithéliales pulmonaires normales (Cesen-Cummings et al., 1998). Dans le cas des cellules cancéreuses du sein chez l’homme, la diminution de la transcription du gène de la Cx26 a été attribuée à une expression inappropriée et une fonction anormale de facteurs de transcription (Lee et al., 1992). Dans le cancer du côlon humain, la diminution de l’expression de la Cx32 a été corrélée à une méthylation du promoteur du gène correspondant (Zhu et al., 1998). Cependant dans d’autres formes de cancers, l’expression des connexines est similaire ou même augmentée par rapport aux cellules normales (Ruch, 2000). Ainsi, dans des lignées dérivées de carcinomes pulmonaires humains et murins, l’expression de la Cx43 va de l’indétectable à la surexpression (Cesen-Cummings et al., 1998). Des facteurs de croissance, des carcinogènes et des oncogènes altèrent les communications jonctionnelles De nombreux agents qui stimulent directement ou indirectement la croissance cellulaire, inhibent les communications jonctionnelles. Ces agents incluent des facteurs de croissance, des carcinogènes et des oncogènes. Plusieurs facteurs de croissance, comme le facteur de croissance épidermique (EGF) ou fibroblastique (bFGF) inhibent les communications jonctionnelles. Lorsque qu’ils sont appliquées à des cultures cellulaires, de nombreux carcinogènes non mutagéniques et quelques carcinogènes mutagéniques inhibent les communications jonctionnelles in vivo et in vitro (Trosko et al., 1990; Klaunig and Ruch, 1990; Budunova and Williams, 1994; Yamasaki and Naus, 1996 ; Upham et al., 1998). Ces produits sont chimiquement divers et incluent des pesticides, des extraits végétaux et des hydrocarbones poly-aromatiques. Les oncogènes sont dérivés de gènes cellulaires normaux (proto-oncogènes) qui ont été activés par une mutation et qui fonctionnent dans la transformation de cellules normales en cellules néoplasiques. Les produits protéiques des oncogènes jouent un rôle dans plusieurs voies de signalisation, ainsi que dans la régulation d’autres gènes, le contrôle de la croissance et plusieurs autres facettes de l’homéostasie tissulaire et cellulaire. Plusieurs oncogènes bloquent également les communications jonctionnnelles (Trosko et al., 1990; Yamasaki and Naus, 1996).
17

Les connexines et les communications jonctionnelles varient au cours du cycle cellulaire Les communications jonctionnelles pourraient également réguler la progression des cellules dans le cycle cellulaire. Par exemple, lors de la régénération des cellules hépatiques faisant suite à une hepatectomie partielle, la communication jonctionnelle, le nombre de jonctions gap et l’expression de Cx32 diminuent à la fin de la phase G1 et S puis réapparaissent dans les phases tardives du cycle cellulaire (Meyer et al., 1981; Dermietzel et al., 1987). En culture, une réduction des communications jonctionnelles a été observée dans la phase G1 tardive et la mitose (Ruch, 1994). D’autres expériences ont montré que la croissance des cellules néoplasiques, peut être inhibée lorsque ces cellules sont cultivées ensemble avec des cellules normales et, dans certains cas, cet effet peut être attribué aux communications jonctionnelles (Ruch, 1994). Ainsi, la croissance des hépatocytes transformés par les oncogènes Ras et Neu est inhibée si ces cellules sont cultivées avec des hépatocytes normaux, mais pas avec des hépatocytes mutés, déficients du point de vue de la communication jonctionnelle (Esinduy et al., 1995). La croissance réapparaît aussi lorsque la co-culture est faite avec des hépatocytes transfectées avec la Cx43. Des altérations spécifiques des communications jonctionnelles altèrent le phénotype et la croissance cellulaire : approches anti-sens, “Knock out” et transfections L’inhibition des connexines par des approches anti-sens dans des cellules normales, s’accompagne d’effets sur la croissance cellulaire (Goldberg et al., 1994; Ruch et al., 1995; Oyoyo et al., 1997). En particulier, Oyoyo et al. (1997) ont observé que les cellules adrénocorticales bovines transfectées avec une construction anti-sens de la Cx43 croissaient plus rapidement que les cellules contrôles et perdaient la capacité de répondre à l’effet inhibiteur du dibutyryl cAMP et de l’ACTH sur leur croissance. Les souris déficientes en Cx43 décèdent à la naissance suite à une malformation de l’artère pulmonaire (Reaume et al., 1995). Des lignées cellulaires obtenues de ces souris présentent une croissance anormale (Martyn et al., 1997; Naus et al., 1997). Des souris « knock-out » pour la Cx32 ont été aussi développées. Ces souris vivent normalement (Temme et al., 1997), mais, chez l’adulte, présentent une augmentation de prolifération la hépatocytaire et de la susceptibilité à développer des tumeurs hépatiques, que ces tumeurs soient spontanées ou induites par des carcinogènes (Temme et al., 1997). Les communications jonctionnelles ont été augmentées par transfection des gènes de connexines dans divers types de lignées cellulaires néoplasiques, déficients en connexines ou en communications jonctionnelles (Mehta et al., 1991; Chen et al., 1995; Huang et al., 1998; Ruch et al., 1998). Dans ces cas, la croissance in vitro et /ou la formation de tumeurs était fortement réduite, en parallèle avec l’augmentation des communications jonctionnelles. Les cellules tranfectées pour la Cx43 ont également présenté des changements dans le contenu des protéines régulatrices du cycle cellulaire, comme les cyclines D1 et p27kip (Chen et al., 1995). Leur croissance était inhibée en phase G1 et S, mais pas en phase G2 et M (Chen et al., 1995; Huang et al., 1998). Ces données suggèrent que les communications jonctionnelles peuvent altérer la croissance cellulaire à des stades spécifiques du cycle.
18

Rôle des connexines dans les épithéliums sécrétoires : l’exemple du pancréas. Dans les systèmes qui ne sont pas spécialisés dans la conduction électrique ou mécanique et qui fonctionnent, dans la plupart des situations physiologiques, sans changements de la prolifération et de la différenciation cellulaire, les fonctions du couplage jonctionnel sont moins claires. Dans plusieurs épithéliums sécrétoires, le couplage semble important pour la synthèse, le stockage et la libération de produits de sécrétion spécifiques (Meda et al., 1979; Meda, 1996). Au sein des îlots de Langherhans qui forment le pancréas endocrine, les cellules β, produisant l'insuline sont connectées majoritairement par des jonctions gap constituées de Cx36 (fig. 3) (Serre-Beinier et al., 2000).
Fig.3 : Les cellules β du pancréas expriment préférentiellement la Cx36 :
Le double immunomarquage de la Cx36, (fluorescence jaune) et de l’ insuline(rouge) révèle que la Cx36 est distribuée sur les cellules β qui constituent l’essentie des îlots pancréatiques
Les canaux de ces jonctions permettent la diffusion de potentiels électrotoniques entre les cellules β, la synchronisation des oscillations de leur Ca++ libre et de leurs potentiels membranaires, ainsi que l'échange de nucléotides et d'intermédiaires de la glycolyse (Meda et al., 1984; Meda, 1996a, b et d). Ce type de couplage n'est pas présent entre toutes les cellules β puisque les jonctions «gap» ont été observées dans seulement 30% des interfaces entre ces cellules. Un îlot pancréatique est donc un assemblage de plusieurs territoires de communication. Plusieurs observations suggèrent que le couplage intercellulaire contrôle la réponse sécrétoire des cellules β : premièrement, les cellules β isolées ne répondent pas correctement au glucose, puisqu'elles n'augmentent que peu leur libération d'insuline en présence de ce sucre et montrent une expression basale diminuée du gène de l'insuline (Salomon and Meda, 1986). La restauration de contacts intercellulaires s'accompagne d'un meilleur contrôle de la libération d'insuline, mis en évidence par une diminution de la sécrétion basale, une augmentation de la réponse aux sécrétagogues et une plus grande biosynthèse d'insuline (Bosco and Meda, 1991). Deuxièmement, la stimulation de la sécrétion d'insuline par des sulfonylurées augmente le couplage des cellules β in vivo et in vitro et s'accompagne d'une augmentation de Cx43 (Meda et al., 1991, Meda,
19

1996c). Troisièmement, des agents pharmacologiques tels que l'heptanol ou l' octanol, qui bloquent avec une certaine sélectivité les jonctions gap, abolissent l'insulinosécrétion induite par le glucose aussi bien dans des îlots de Langerhans isolés que dans le pancréas intact (Meda et al., 1990). Quatrièmement, des lignées cellulaires tumorales ou transformées produisant de l'insuline mais ne répondant pas de façon normale au glucose, n'expriment pas de connexine et ne sont pas couplées (Vozzi et al., 1995). Cette déficience ne peut pas être simplement expliquée par une dérégulation in vitro des canaux fonctionnels, puisque les cellules β d’îlots de Langerhans natifs continuent à exprimer la Cx43 en culture (Meda et al., 1981). Suite à la transfection stable du gène codant pour la Cx43, rétablissant dans les lignées un couplage similaire à celui des cellules β primaires, la réponse au glucose et le stockage d'insuline sont considérablement améliorés (Vozzi et al., 1995). Ces observations indiquent que les communications jonctionnelles et la fonction sécrétoire des cellules β sont étroitement liées et que ce lien est impliqué dans la régulation ponctuelle de la sécrétion d'insuline, mais aussi dans le contrôle à plus long terme de l'expression du gène codant cette hormone. Cinquièmement, des conditions qui inhibent la libération d'insuline diminuent ou abolissent le couplage des cellules β in vitro. In vivo, cependant, cette inhibition provoque l'apparition d'hyperglycémie et augmente le couplage intercellulaire, suggérant que le niveau de glucose circulant et la capacité des cellules β à reconnaître ce sucre influencent de façon indépendante la communication jonctionnelle. Des observations analogues ont été faites à propos de la sécrétion d'amylase par les cellules acineuses du pancréas exocrine (Chanson et al., 1991). Dans ce système, cependant, les sécrétagogues naturels induisent une diminution du couplage et le blocage pharmacologique des canaux jonctionnels entraîne rapidement une augmenta-tion de la sécrétion d'amylase (Meda et al., 1987). Ce comportement, opposé à celui des cellules β, pourrait être lié à la synthèse d'une connexine différente. En effet, les cellules insulaires et acineuses du pancréas, comme d'ailleurs celles de nombreuses autres glandes endocrines et exocrines, synthétisent différentiellement deux isoformes distinctes de connexines (Meda et al., 1993), qui sont fonctionnellement incompatibles (White et al., 1995). À ce jour, la démonstration rigoureuse qu'une fonction physiologique est spécifiquement assurée par le couplage jonctionnel n'a pu encore être fournie, entre autres parce que les observations à ce propos n'ont été faites qu'exceptionnellement in vivo (Chanson et al., 1993). À première vue, la grande variété des fonctions proposées pour les connexines est aussi surprenante. Cependant, l'échange intercellulaire de la plupart des seconds messagers au travers des canaux jonctionnels permet de concevoir que ce couplage pourrait compenser aussi bien des déficits ioniques que métaboliques, ou des insuffisances des systèmes effecteurs de quleques cellules, en cas de distribution hétérogène de récepteurs ou de l'innervation au sein d'un tissu. Le récent développement de souris transgéniques, chez lesquelles l'expression génique d'une connexine est altérée, suggère que ces fonctions sont fondamentales, voire vitales pour l'organisme.
20
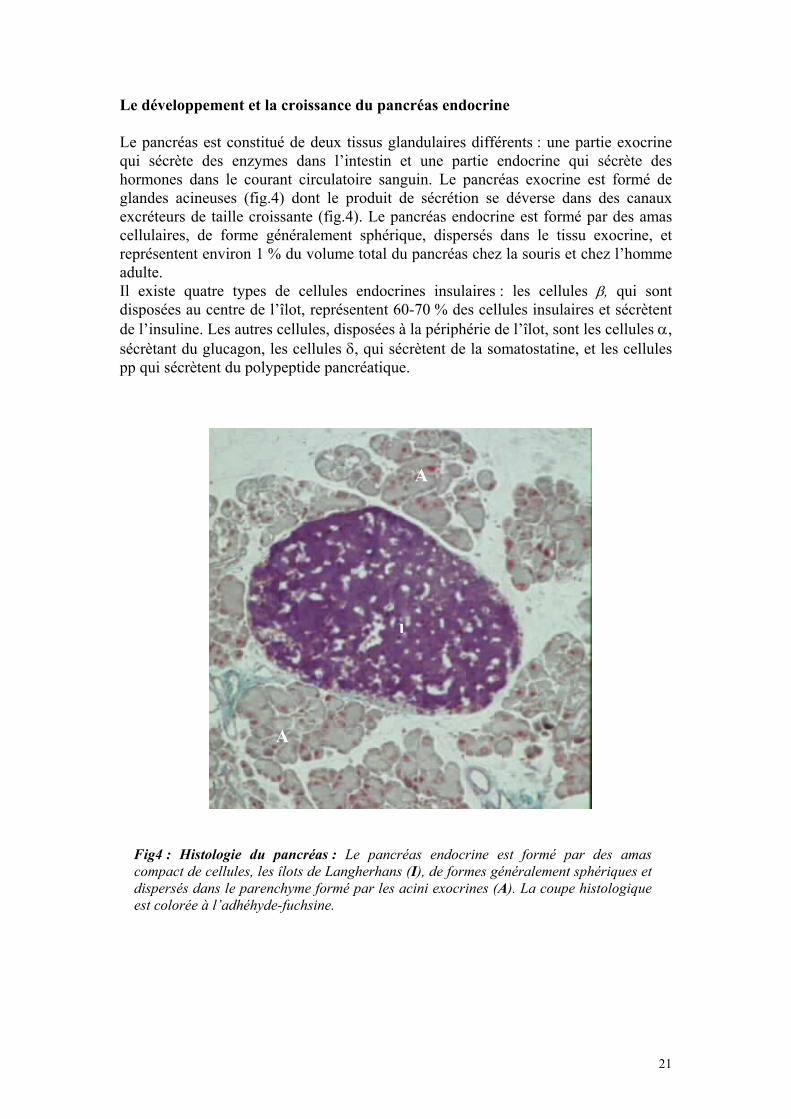
Le développement et la croissance du pancréas endocrine Le pancréas est constitué de deux tissus glandulaires différents : une partie exocrine qui sécrète des enzymes dans l’intestin et une partie endocrine qui sécrète des hormones dans le courant circulatoire sanguin. Le pancréas exocrine est formé de glandes acineuses (fig.4) dont le produit de sécrétion se déverse dans des canaux excréteurs de taille croissante (fig.4). Le pancréas endocrine est formé par des amas cellulaires, de forme généralement sphérique, dispersés dans le tissu exocrine, et représentent environ 1 % du volume total du pancréas chez la souris et chez l’homme adulte. Il existe quatre types de cellules endocrines insulaires : les cellules β, qui sont disposées au centre de l’îlot, représentent 60-70 % des cellules insulaires et sécrètent de l’insuline. Les autres cellules, disposées à la périphérie de l’îlot, sont les cellules α, sécrètant du glucagon, les cellules δ, qui sécrètent de la somatostatine, et les cellules pp qui sécrètent du polypeptide pancréatique.
A
A
I
Fig4 : Histologie du pancréas : Le pancréas endocrine est formé par des amas compact de cellules, les îlots de Langherhans (I), de formes généralement sphériques et dispersés dans le parenchyme formé par les acini exocrines (A). La coupe histologique est colorée à l’adhéhyde-fuchsine.
21

Le pancréas endocrine est un organe particulièrement important en médecine humaine du fait de l’importance du diabète qui affecte 4 à 5 % de la population mondiale. La croissance et le développement de ce tissu ont été largement étudiés, suscitant des questions quant à la régulation de la masse des cellules β (Herrera et al., 1991 ; Mally et al., 1994 ; Slack,1995 ; Sosa-Pineda et al., 1997 ; Edlund et al., 2001). Les cellules pancréatiques exocrines et endocrines sont formées soit par différenciation des cellules des canaux excréteurs embryonnaires (néogenèse), soit par réplication des cellules différenciées préexistantes. Il est admis que, jusqu’à la fin de la gestation, la plupart des cellules β sont le résultat d’une néogenèse et, qu’après la naissance, d’autres cellules β se forment surtout par réplication (Slack,1995; Bonner-Weir et al., 2001). Cependant cette réplication est faible, evaluée chez le rat adulte à environ 3 % par jour (Hellerstrom et al.,1988). Ce faible taux de réplication implique que la masse des cellules β ne change pas considérablement après la naissance. Hellerstrom et al. (1988) ont montré qu’avec un taux de réplication d’environ 3 % par jour, la masse des cellules β doublerait au bout d’un mois s’il n’y avait pas, parallèlement, un fort taux de mort cellulaire. L’apoptose de cellules β est vraisemblablement le mécanisme par lequel des cellules âgées ou défectueuses sont éliminées (Wyllie et al., 1980). L’homéostasie de la masse de la masse des cellules β, résulte donc d’une balance entre la prolifération, la croissance et la mort cellulaire (Finegood, 1995 ; Bonner-Weir et al., 2001). On pourrait dès lors modifier la masse des cellules β en changeant la néoformation, la réplication et la différenciation de nouveaux îlots (néogenèse), mais aussi en changeant la taille/volume des cellules individuelles ou le taux de cellules apoptotiques. La contribution de chacun de ces mécanismes au développement et à la croissance du pancréas endocrine n’est pas bien comprise (Bonner-Weir et al., 2001). Des modèles de pancréatectomie partielle chez le rat démontrent que la néogenèse peut se faire à l’âge adulte, mais il y a peu d’évidence de néogenèse après la période périnatale, du moins en l’absence de stimulus externe (Bonner-Weir et al., 1993 ; Slack,1995). Le peu de capacité de régénération des cellules β à l’âge adulte peut être légèrement augmenté par des molécules de la matrice extracellulaire, qui sont présumées reconnues par des intégrines spécifiques (Lucas-Clerc et al., 1993; Hulinsky et al., 1995, Lefebvre et al., 1998). À l’inverse des cellules β primaires, les cellules productrices d’insuline qui ont été transformées ou qui proviennent de tumeurs, prolifèrent continuellement. Ce changement apparaît être contrôlé par des molécules d’adhésion cellulaire, dans la mesure où des insulinomes se développent chez des souris transgéniques qui n’expriment plus d’E-cadhérine (Perl et al., 1998). Ces données indiquent que les molécules d’adhésion participent au contrôle de la croissance et à la dissémination des cellules tumorales. Il existe des corrélations entre l’expression des molécules d’adhésion et celle des connexines. La transfection d’E-cadherine dans des lignées cellulaires déficientes en couplage, résulte en une induction des communications jonctionnelles (Mege et al., 1988; Jongen et al., 1991; Meyer et al., 1992). La transfection de N-CAM induit également du couplage, suite à un changement de la phosphorylation de la Cx43 endogène et la translocation de la protéine du cytoplasme à la membrane (Musil et al., 1990). L’association entre molécules d’adhésion et communication jonctionnelle, est particulièrement significative dans la transformation cellulaire, et pourrait être impliqué dans les processus de suppression tumorale (Yamasaki, 1990; Birchmeier, 1995).
22

La problématique de notre étude. Comme indiqué précédemment, le rôle spécifique joué par les connexines, n’est pas encore démontré dans de nombreux tissus, même si de nombreuses études indiquent une implication, dans la croissance et la différenciation de cellules adultes hautement spécialisées (Meda et Spray, 2000). Ainsi la croissance adéquate, la différenciation et la prolifération des cellules folliculaires de l’ovaire (Simon et al., 1997), des neurones du cerveau (Dermietzel et al., 1989; Rozental et al., 2000), des fibres du cristallin (Gong et al., 1997; White et al., 1998) et des kératinocytes de la peau (Salomon et al., 1994) dépendent de l’expression adéquate d’un isoforme spécifique de connexine. Certains de ces effets semblent applicables à l’homme. Ainsi les patients souffrant d'une forme de surdité héréditaire non-syndromique, qui est lié à une mutation de la Cx26 (Richard, 2000), ont montré une diminution du taux de prolifération et forment un épiderme anormalement mince (Wiszniewski et al., 2000). En sens inverse, des traitements topiques qui affectent la croissance et la différenciation des kératinocytes, résultent également en une altération de l’expression de leurs connexines (Labarthe et al., 1998; Masgrau-Peya et al., 1997). L’ensemble de ces données montre que l’expression in vivo des connexines joue un rôle significatif dans la croissance et la différenciation de divers types cellulaires hautement spécialisés. Aucune étude n’a jusqu’ici investigué les cellules productrices d’insuline dans ce contexte. Cependant des études récentes sur des cellules tumorales productrices d’insuline ont montré, qu’après transfection stable de la Cx43, ces cellules croissaient in vivo plus lentement, et sécrétaient plus d’insuline que les cellules contrôles, qui n’expriment pas de connexine (Vozzi et al., 1997). Des études parallèles ont également indiqués que la Cx36, la connexine prédominante des cellules β (Serre-Beinier et al., 2000; Meda and Bosco, 2001), change pendant des changements séquentiels des îlots natifs de rats (Chen et al., 1992). Dans cemodèle l’hypotrophie des îlots, causée par une hyperinsulinémie chronique, était associée à une perte de l’expression de Cx36, alors que la croissance des îlots, due à la normalisation de l’insulinémie, entraînait une augmentation de la connexine. Ces données indiquent que la croissance in vivo des cellules β pourrait, être tributaire d’une expression adéquate des connexines. Le but de ce travail a donc été d’étudier la croissance et la prolifération des cellules β dans un modèle de souris transgéniques, chez qui les cellules sécrétrices d’insuline ont été forcées d’exprimer ectopiquement de la Cx32. Nous avons étudié ces souris à la naissance et à lâge adulte, pour déterminer les effets éventuels des connexines sur la croissance et la différenciation du pancréas endocrine.
23

Chapitre 2 : Mon travail original
ECTOPIC EXPRESSION OF Cx32 IN β-CELLS LEADS TO A POST-NATAL INCREASE IN THE GROWTH OF THE ENDOCRINE
PANCREAS
24

The growth capacity of primary islet cells markedly decreases with their postnatal, final differentiation, even though adult, fully differentiated β-cells retain a limited proliferation potential throughout life. This potential appears to be enhanced by molecules of the extracellular matrix, that are presumably recognised by specific integrins (Lucas-Clerc et al., 1993, Hulinsky et al., 1995, Lefebvre et al., 1998). In contrast to primary β-cells, transformed and tumoral insulin-producing cells may growth almost continuously. This change appears to be controlled by cell adhesion molecules (CAMs), inasmuch as insulinomas develop in transgenic mice that cannot express E-cadherins (Perl et al., 1999). Several data indicate that CAMs also play a primordial role in controlling the growth and the spatial dissemination of islet tumours (Perl et al., 1998; Perl et al., 1999). Still other mechanisms may contribute to this control. Communications trough connexin (Cx) channels are an almost ubiquitous mechanism for direct intercellular signalling within animal tissues (Kumar and Gilula, 1996). There is ample evidence for an inverse relationship between the extent of connexin-dependent communication and tumorogenesis (Yamasaki and Naus, 1996, Ruch, 2000). In view of this evidence, connexins have been regarded as tumour suppressors, inasmuch as they allow for the passage of growth regulatory signals that inhibit cell division (Ruch, 2000). Accordingly, enhancement of coupling after connexin transfection has been shown to be associated with a reduction in the in vivo growth of multiple types of tumour cells (Yamasaki and Naus, 1996), including experimental insulinomas (Yamasaki and Naus, 1996, Vozzi et al., 1997). The insulin producing β-cells are interconnected by connexins channels (Meda, 1996; Serre-Beinier et al., 2000). In view of the relationship that likely exists between cell adhesion molecules and connexins, and of the growing evidence implicating junctional communication in the control of cell growth, we have investigated whether the in vivo development of primary β-cells is affected by changes in connexins. To address this question, we have chosen a transgenic approach to change the pattern of connexins expressed by native β-cells. Since cells of pancreatic islets appear to express multiple connexins, whose precise distribution, relative abundance and specific roles remain to be fully ascertained (Serre-Beinier et al., 2000), we have chosen here to selectively force β-cells to express another connexin, referred to as Cx32 (Paul, 1986), which is not expressed in native pancreatic islets (Meda, 1996), still shows most of the structural and functional features of islet cells connexins (Goldberg et al., 1999, Suchyna et al., 1999¸ Bevans et al., 1998). We show that mice featuring increased β-cells coupling, as a result of Cx32 expression (Charollais et al., 2000), have a post-natal increase in the growth of the endocrine pancreas, in insulin content, in the size of islets, and in that of β-cells. These results provide the first clue that proper levels of junctional communication are essential for normal β-cell growth, and open new perspectives for the characterisation of the factors controlling the proliferation and production of primary β-cells.
25

Methods Generation of transgenic mice A transgene was designed by ligating in sequence a 460bp sequence of the rat insulin II promoter (Hanahan, 1985), a cDNA comprising the entire coding sequence of rat Cx32 (Paul, 1986), and the coding region of human growth hormone as a source of introns and polyadenylation signal (Herrera et al., 1994) (fig.5). A Xba1/Xho1 fragment containing the transgene was microinjected into zygotic pronuclei and surviving embryos were transferred into pseudopregnant female mice (Herrera et al., 1994). Transgenic founders were identified by dot blot screening of tail DNA, using as probe for the hGH sequence (probe 1), and by southern blot of EcoR1-digested DNA, using a probe for the sequence of rat Cx32 (probe 2). Two founder mice were successfully bread with C57Bl6 controls to generate two independent lines of transgenic animals, referred to as D6 and B9 (Charollais et al., 2000). Heterozygous transgenic mice were identified in the progeny by PCR amplification of tail DNA, using primers hybridising to the hGH sequence. Homozygous mice, obtained by crossing heterozygous D6 and B9 animals, were identified by dot blot using probe 1. All animals were kept under standard housing conditions and experiments were conducted according to the regulations of our institutional and state committees on animal experiments.
DCB
A
Figure 5. Strategy for expressing Cx32 in pancreatic β-cells. A: A transgene that contained parts of the rat insulin II promoter (RIP), the second exon ofCx32 cDNA (Cx32; coding region between white dots) and the coding region of the humangrowth hormone gene (hGH) was microinjected into zygotic pronuclei and surviving embryoswere transferred into pseudopregnant foster mothers. B: Transgenic mice were identified by southern blot. Compared to wild type controls (W), which featured only the 7.6Kb endogenousgene (CX32e), two positive founders were identified that also expressed the expected 1.5 Kbtransgene (Cx32t). These mice were used to generate two independent lines (lane 1, line B9; lane 2, LineD6) by backcrossing with C56BI6 controls. C: The progeny was screened by PCR using primers along the hGH sequence. Heterozygous mice carrying the transgene (lane 1,3and B) were distinguished from both control littermates (lanes 2 and A) and wild type controls (W) by the amplification of a 390 bp product (lane H =distilled water, lane S = sizestandards). D: heterozygous mice of the of D6 line were crossed to obtain a homozygousprogeny. Mice carrying two alleles of the transgene (lanes1 and 2) were distinguished from heterozygous littermates (lane 3) by hybridising DNA with probe 1. Dot intensity wascompared to that observed with DNA of mice from previous litters whose homozygosity (laneC) or heterozygosity (lane B) had been previously checked by crossing with C57BI6 controls. From Charollais et al., 2000.

Tissue sampling Adult mice were anaesthetised by inhalation of 5% Ethrane® (Abbott laboratories, Cham, Switzerland), sacrificed by cervical dislocation and immediately used for pancreas sampling. Pancreas from newborn mice was sampled within one hour from birth. Histology Pieces of pancreas were fixed in Bouin’s solution and processed according to standard methods for either aldehyde fuchsin staining, or immunofluorescence labelling of the four main islet hormones (Stefan et al., 1987). Briefly, sections were incubated for 2 h at room temperature with one of the following antibodies: mouse monoclonal against insulin, diluted at 1:200; rabbit polyclonal against glucagon, diluted 1.600; rabbit polyclonal against somatostatin, diluted 1:1600; rabbit polyclonal against bovine pancreatic polypeptide, diluted 1:300. After rinsing in PBS, sections were incubated for 1 h at room temperature with a fluorescein-conjugated goat serum against either mouse or rabbit IgS, whichever applicable, diluted 1:400. After a 4 min staining in 0.03% Evan’s blue, sections were coverslipped with a drop of 0.02% paraphenylenediamine in glycerol-PBS (2:1) and photographed with an Axiophot fluorescence microscope. Connexin and E-cadherin expression Immediately after sampling, the pancreas of each mouse was frozen in liquid nitrogen, embedded in Tissue-TeK® and sectioned in a 3000 cryostat (Leica AG, Glattbrugg, Switzerland). Five µm-thick sections were exposed for 3 min to –20°C acetone, air-dried, rinsed in PBS containing 0.5% BSA, and incubated for 2 h at room temperature in the presence of either a mouse monoclonal antibody directed to the C-terminus of Cx32, diluted 1:200, or a rabbit polyclonal antibody against the extracellular domain of E-cadherin, diluted at 1:200. Insulin radioimmunoassay The entire pancreas of adult and one day old (J1) newborn mice was weighted wet, sonicated three times for 10s (output control 2 of a Sonifier 250; branton, Danbury, CT) in acid-ethanol. Extracted insulin was evaluated using a radioimmunoassay with a charcoal separation step, 125I porcine insulin (SB-INS I –1, Sorin Biomedica, Saliggia, Italy) as tracer and rat insulin as standard (Meda et al., 1990). Pancreatic islets and β-cell sorting Islets of Langherhans were isolated from pancreas by collagenase digestion and purification on a histopaque gradient (Giordano et al., 1993). The isolated islets were washed twice in a phosphate-buffered saline (PBS), prepared without adding Mg++ and Ca++, and containing 0.2 mM EDTA. The islets were then exposed for 6-7 min at 37°C to the same medium supplemented with 0.16-mg/ml trypsin (1:250; GIBCO, Grand Island, NY), with periodic aspiration through a pipette tip. The incubation was stopped by the addition of 10 ml ice-cold Krebs-Ringer-Bicarbonate buffer, supplemented with 0.5% bovine serum albumin (BSA), 2.8 mM glucose and 10 mM Hepes, pH 7.4 (KRB buffer). The resulting suspension, which comprised mostly single cells, was centrifuged for 5 min at 130g. The pellet was resuspended in KRB buffer containing 2.8mM glucose, to obtain a final concentration of 3×106 cells/ml.
27

Islet cell sorting was performed with a Epics-V flow cytometer equipped with an excitation argon laser (innova90, Coherent, Palo Alto, CA) tuned to either 488nm (FAD fluorescence) or 360nm (NADPH fluorescence) at 500-600mW output power, and connected to an MDADS microcomputer (Culter Electronics, Hilalech, FL). β−cells were sorted from the other islets cell types, according to both flavin adenine dinucleotide autofluorescence (510-550nm) and forward light scattering (Giordano et al., 1993). Morphometric analysis Islets were evaluated by on 5-µm-thick sections which were cut serially throughout each pancreas and immunostained with a mouse monoclonal antibody against insulin. 3-5 fields were randomly photographed, at a 16× magnification, in 1 out of every 50 sections (adult mice) or 1 out of 20 sections (newborn mice). A total of 15-20 sections were therefore evaluated per pancreas. The photographs were projected at the final magnification of ×420 on a ACECAD professional graphic tablet connected to a Quantimet Leica 500+ (Leica, Cambridge Ltd, England) programmed for semi-automatic measurement. The volume density of pancreatic β-cells was determined by calculating the ratio of the islet to the area of pancreas (Stefan et al., 1987). The size of individual islets was determined by planimetric evaluation of islet profiles, the shortest islets axis and longest islets axis, which were taken perpendicular to each other. The numerical density of islets, dispersed single β-cells and total β-cells were evaluated by scoring the total number of pancreatic islets, single or total intra- and extra- islet β-cells. Scores were expressed per unit area of pancreas in 10-15 sections randomly selected per animal. Photographs were taken at a magnification of 0.64x, using a video camera Sanyo VC-2512 (Sanyo Electric Co, LTD, Japan). The photographs were computerised at a final magnification of ×7 and analysed on a Quantimet Leica 500+ (Leica, Cambridge Ltd, England) programmed for semi-automatic area measurement. The size of individual β-cells was determined by planimetric evaluation of individual β-cell profiles, in islets photographs, projected at a final magnification of ×2320. At least 200 insulin-immunoreactive cells showing a nucleus, were randomly selected in the four quadrants of each islet, and > 20 randomly selected pancreatic islets were score per animal. Thus a total of 800 β-cells, were measured in control, heterozygous and homozygous RipCx32 adult mice. The size of individual β-cells was also, independently determined by another investigator evaluating FACS purified β-cells by planimetry. Proliferation rate. The proliferation rate of β-cells was determined by studying either the in vivo incorporation of 5-bromo2’-deoxyuridine (BrdU) (Davidson et al., 1989), or the in vitro expression of the Ki67 nuclear antigen (Terada et al., 1998). For BrdU detection, transgenic mice and control littermates were injected intraperitoneally with 2mg/Kg BrdU (BrdU B-5002 Sigma-Aldrich Chemie GmbH, Steinheim, Germany), every 6 hours, and sacrificed after 18 hours. Pancreas were
28

dyingremoved and immediately fixed in 4% paraformaldehyde (PFA) for 24 hours, embedded in paraffin and sectioned. Sections were processed for streptavidin-biotin amplification and immunofluorescence labelling for both BrdU antibody and insulin. Briefly, sections were deparaffinised, boiled in 10mM sodium citrate (pH 6.0) for 25 min (for antigen retrilation), rinsed in PBS, treated with 3% Triton in 2N HCl for 30 min at 37 °C (to linearize DNA), and washed 3 times for 5 min with sodium tetraborate pH 8.5 (to neutralise the acid). After rinsing in PBS, sections were incubated 30 min in 2% bovine serum albumin (BSA) in PBS (to decrease non specific staining). Sections were then incubated overnight at 4°C with monoclonal mouse antibody anti-BrdU, diluted 1:100 (Sigma Saint Louis, Missouri 63103 USA) and, then 2 hours with a guinea pig serum against mouse insulin, diluted 1:400. To visualise the antibodies, sections were incubated sequentially with a biotin-labelled mouse antiserum IgG, diluted 1:100 (Jackson immunoreaseach laboratories, West Grove, PA), streptavidin conjugated to fluorescein, diluted 1:200 (Jackson immunoreaseach laboratories), and a rabbit serum against guinea pig IgG, which was conjugated to rhodamine and used at a 1:200 dilution. Each incubation was performed for 60 min at room temperature in 0.5% BSA/PBS. Sections were rinsed, coverslipped with a drop of 0.02% paraphenylene-diamine in glycerol-PBS (2:1) and photographed with an Axiophot fluorescence microscope (fig.6). For detection of the Ki67 antigen, pancreas of transgenic and normal littermates were removed and immediately fixed in 4% PFA for 24 hours, embedded in paraffin and sectioned. Sections were processed for streptavidin-biotin amplification and immunofluorescence labelling for both Ki67 and insulin. Briefly, sections were deparaffinised, boiled in 10 mM sodium citrate (pH 6.0) for 25 min, rinsed in PBS, exposed 30 min to 2% BSA/PBS ad incubated overnight at 4°C with a rabbit polyclonal antiserum to Ki-67 (Novocastra Laboratories Ltd, Newcastle upon Tyne, United Kingdom), diluted 1:1000 in 0.5% BSA/PBS. Sections were then incubated for 2 hours with a mouse monoclonal antibodies against insulin diluted 1: 400. To visualise the antibodies, sections were incubated sequentially with a biotin-labelled goat against rabbit IgG, diluted 1:200 (Jackson immunoreaseach laboratories, West Grove, PA), a fluorescein-conjugated streptavidin diluted 1:200 (Jackson immunoreaseach laboratories), and rhodamine-conjugated serum against mouse IgG, diluted 1:400. Each incubation step was performed for 60 min at room temperature in 0.5 BSA/PBS. Sections were rinsed, coverslipped with a drop of 0.02% paraphenylenediamine in glycerol-PBS, and photographed with an Axiophot fluorescence microscope (fig.6). In each experiment, a positive control of the BrdU and Ki-67 labelling of proliferating cells was provided by incubating in parallel a section of mouse duodenum (Patel et al., 1993 Burholt et al., 1985). All sections were then scored for the number of islet cells, stained for either Ki-67 or BrdU, and insulin. At least 1000 islets cells were counted per pancreas. The results were expressed as the percentage of Ki-67 and BrdU positive cells per total number of β-cells scored.
29

β-cells apoptosis. The rate of β-cell apoptosis was measured by the DNA fragmentation (TUNEL) method, using a commercially available kit (Boehringer-Mannheim, Germany) with modifications. Briefly, pancreas of transgenic RipCx32 and control adult mice were fixed in 4% PFA for 24 hours at 4° C, embedded in paraffin and sectioned. Sections were deparaffinised, treated with 3µg /ml proteinase K for 15 min at room temperature, rinsed in water, treated with 0.5% H2O2 for 10 min and again rinsed in water. The sections were then incubated in TdT buffer containing 0.05% BSA, incubated with a dNTP mixture for 1 hour at 37°, washed in PBS, treated with 5% BSA in PBS for 15 min, and eventually incubated with a streptavidin-biotin-complex-pox for 30 min. After rinsing in PBS (pH 7.4), sections were exposed to di-amino-benzidine (DAB) for 1 min, rinsed with water, counterstained with haematoxylin, cover-slipped and photographed. Positive controls were given by staining apoptotic cells in a section of the small mouse intestine (fig.6) or lymphoid follicles (Potten, 1992; Hall et al., 1994). Sections were scored for at least 1000 islet nuclei per pancreas. The results were expressed as the percentage of islet cells showing an apoptotic nucleus. Statistical analysis Data were compared by the median test or the Kolmogorov-Smirnov test, as provided by the 06.01.02 Windows version of Statistical Package for Social Sciences (SPSS Inc., Chicago,IL)
30
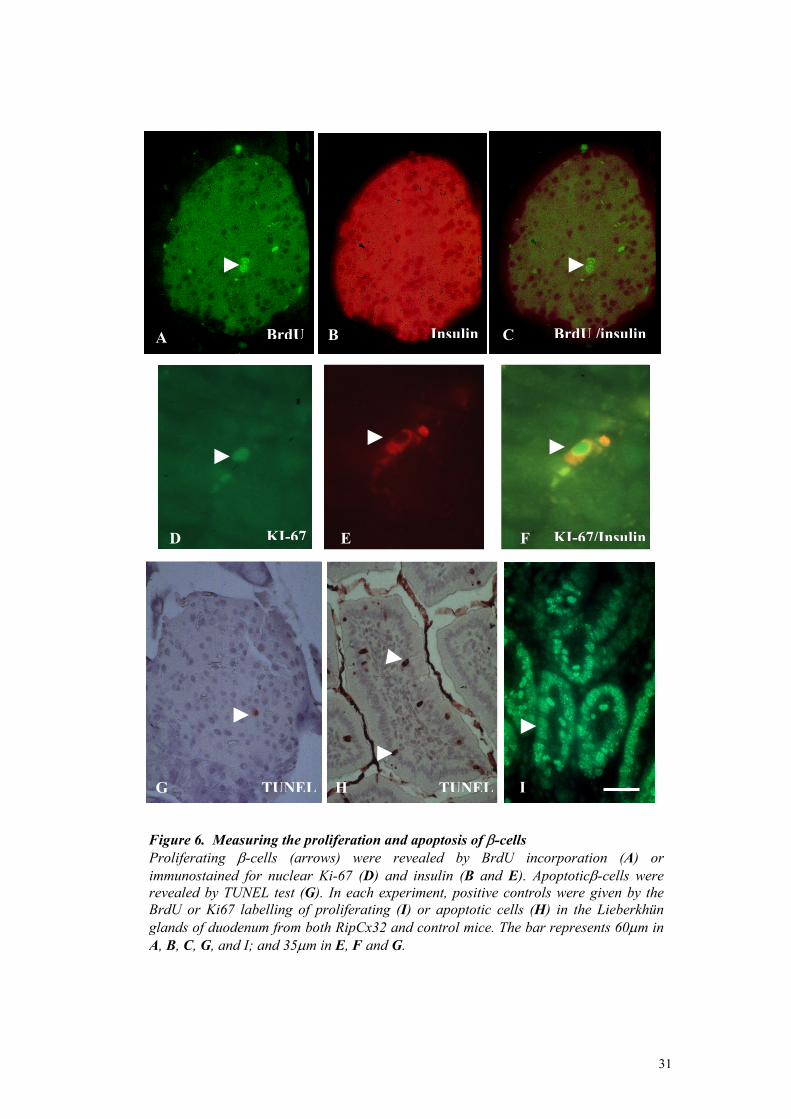
Insulin BrdU /insulinBrdU BA C
KI-67D E
A G TUNEL H
Figure 6. Measuring the proliferation and aProliferating β-cells (arrows) were reveimmunostained for nuclear Ki-67 (D) and irevealed by TUNEL test (G). In each experiBrdU or Ki67 labelling of proliferating (I) glands of duodenum from both RipCx32 and A, B, C, G, and I; and 35µm in E, F and G.
Insulin
F KI-67/Insulin
ITUNEL
31
poptosis of β-cells aled by BrdU incorporation (A) or nsulin (B and E). Apoptoticβ-cells were ment, positive controls were given by theor apoptotic cells (H) in the Lieberkhün control mice. The bar represents 60µm in
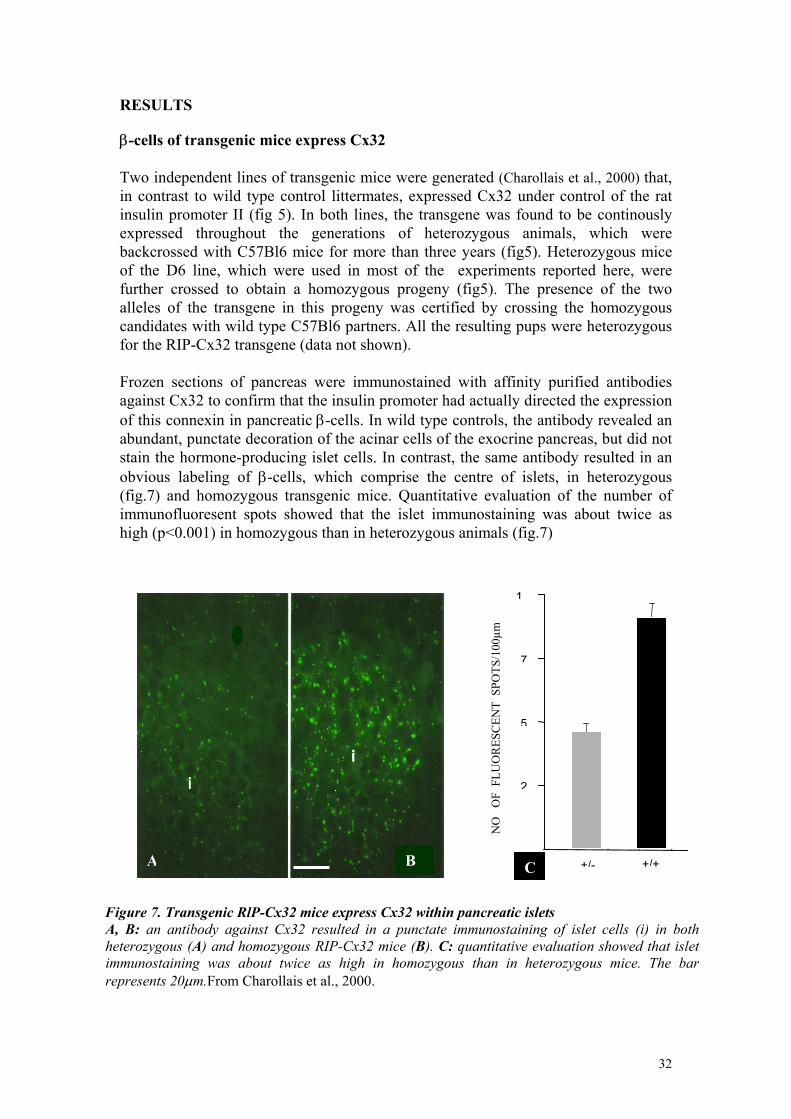
RESULTS β-cells of transgenic mice express Cx32 Two independent lines of transgenic mice were generated (Charollais et al., 2000) that, in contrast to wild type control littermates, expressed Cx32 under control of the rat insulin promoter II (fig 5). In both lines, the transgene was found to be continously expressed throughout the generations of heterozygous animals, which were backcrossed with C57Bl6 mice for more than three years (fig5). Heterozygous mice of the D6 line, which were used in most of the experiments reported here, were further crossed to obtain a homozygous progeny (fig5). The presence of the two alleles of the transgene in this progeny was certified by crossing the homozygous candidates with wild type C57Bl6 partners. All the resulting pups were heterozygous for the RIP-Cx32 transgene (data not shown). Frozen sections of pancreas were immunostained with affinity purified antibodies against Cx32 to confirm that the insulin promoter had actually directed the expression of this connexin in pancreatic β-cells. In wild type controls, the antibody revealed an abundant, punctate decoration of the acinar cells of the exocrine pancreas, but did not stain the hormone-producing islet cells. In contrast, the same antibody resulted in an obvious labeling of β-cells, which comprise the centre of islets, in heterozygous (fig.7) and homozygous transgenic mice. Quantitative evaluation of the number of immunofluoresent spots showed that the islet immunostaining was about twice as high (p<0.001) in homozygous than in heterozygous animals (fig.7)
2
5
7
1
+/++/-
1
CB A
NO
O
F F
LUO
RES
CEN
T S
POTS
/100
µm
Figure 7. Transgenic RlP-Cx32 mice express Cx32 within pancreatic islets A, B: an antibody against Cx32 resulted in a punctate immunostaining of islet cells (i) in bothheterozygous (A) and homozygous RIP-Cx32 mice (B). C: quantitative evaluation showed that islet immunostaining was about twice as high in homozygous than in heterozygous mice. The barrepresents 20µm.From Charollais et al., 2000.
32
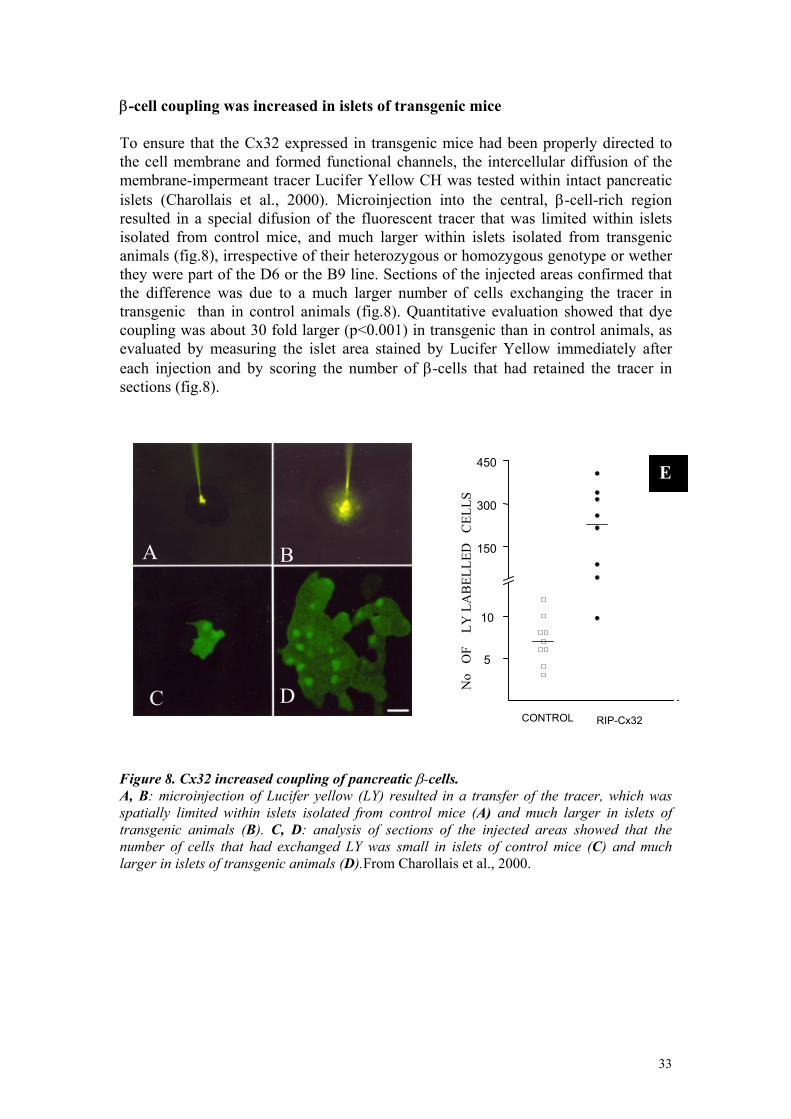
β-cell coupling was increased in islets of transgenic mice To ensure that the Cx32 expressed in transgenic mice had been properly directed to the cell membrane and formed functional channels, the intercellular diffusion of the membrane-impermeant tracer Lucifer Yellow CH was tested within intact pancreatic islets (Charollais et al., 2000). Microinjection into the central, β-cell-rich region resulted in a special difusion of the fluorescent tracer that was limited within islets isolated from control mice, and much larger within islets isolated from transgenic animals (fig.8), irrespective of their heterozygous or homozygous genotype or wether they were part of the D6 or the B9 line. Sections of the injected areas confirmed that the difference was due to a much larger number of cells exchanging the tracer in transgenic than in control animals (fig.8). Quantitative evaluation showed that dye coupling was about 30 fold larger (p<0.001) in transgenic than in control animals, as evaluated by measuring the islet area stained by Lucifer Yellow immediately after each injection and by scoring the number of β-cells that had retained the tracer in sections (fig.8).
D C
B A
5
10
150
300
450N
o O
F
LY L
AB
ELLE
D
CEL
LS
E
CONTROL RIP-Cx32
Figure 8. Cx32 increased coupling of pancreatic β-cells. A, B: microinjection of Lucifer yellow (LY) resulted in a transfer of the tracer, which was spatially limited within islets isolated from control mice (A) and much larger in islets of transgenic animals (B). C, D: analysis of sections of the injected areas showed that the number of cells that had exchanged LY was small in islets of control mice (C) and much larger in islets of transgenic animals (D).From Charollais et al., 2000.
33
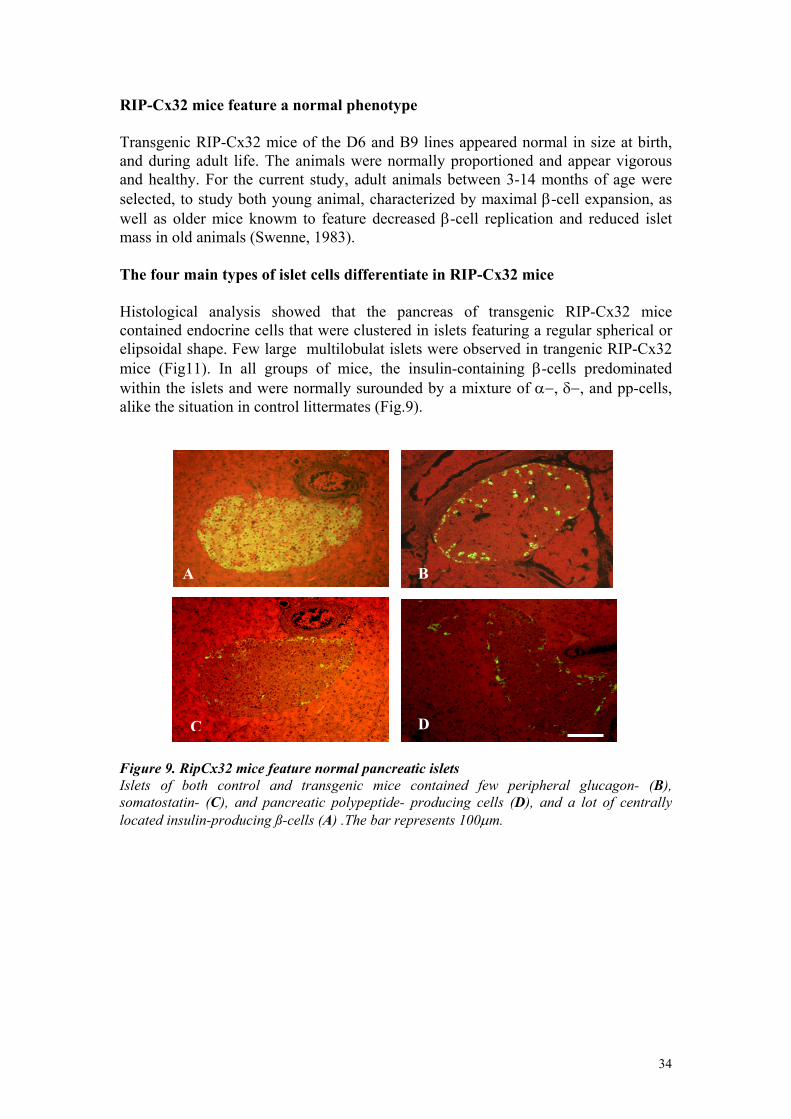
RIP-Cx32 mice feature a normal phenotype Transgenic RIP-Cx32 mice of the D6 and B9 lines appeared normal in size at birth, and during adult life. The animals were normally proportioned and appear vigorous and healthy. For the current study, adult animals between 3-14 months of age were selected, to study both young animal, characterized by maximal β-cell expansion, as well as older mice knowm to feature decreased β-cell replication and reduced islet mass in old animals (Swenne, 1983). The four main types of islet cells differentiate in RIP-Cx32 mice Histological analysis showed that the pancreas of transgenic RIP-Cx32 mice contained endocrine cells that were clustered in islets featuring a regular spherical or elipsoidal shape. Few large multilobulat islets were observed in trangenic RIP-Cx32 mice (Fig11). In all groups of mice, the insulin-containing β-cells predominated within the islets and were normally surounded by a mixture of α−, δ−, and pp-cells, alike the situation in control littermates (Fig.9).
DC
A B
Figure 9. RipCx32 mice feature normal pancreatic islets Islets of both control and transgenic mice contained few peripheral glucagon- (B), somatostatin- (C), and pancreatic polypeptide- producing cells (D), and a lot of centrally located insulin-producing ß-cells (A) .The bar represents 100µm.
34
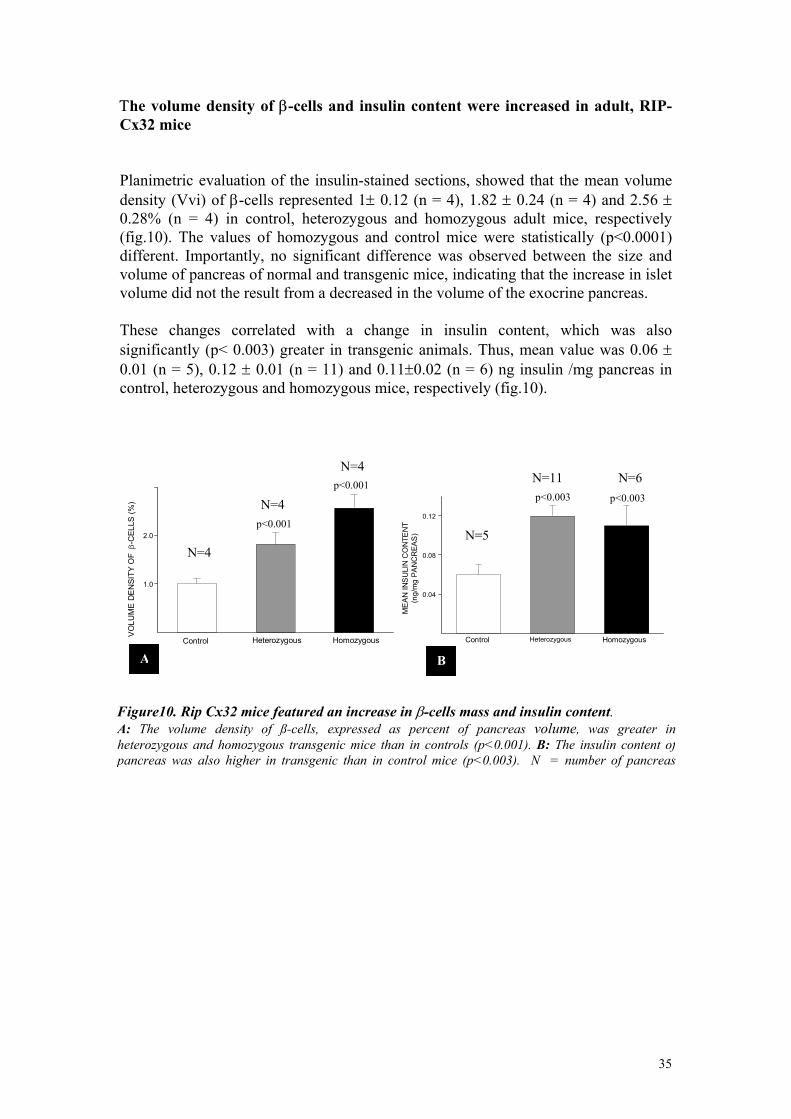
Τhe volume density of β-cells and insulin content were increased in adult, RIP-Cx32 mice Planimetric evaluation of the insulin-stained sections, showed that the mean volume density (Vvi) of β-cells represented 1± 0.12 (n = 4), 1.82 ± 0.24 (n = 4) and 2.56 ± 0.28% (n = 4) in control, heterozygous and homozygous adult mice, respectively (fig.10). The values of homozygous and control mice were statistically (p<0.0001) different. Importantly, no significant difference was observed between the size and volume of pancreas of normal and transgenic mice, indicating that the increase in islet volume did not the result from a decreased in the volume of the exocrine pancreas. These changes correlated with a change in insulin content, which was also significantly (p< 0.003) greater in transgenic animals. Thus, mean value was 0.06 ± 0.01 (n = 5), 0.12 ± 0.01 (n = 11) and 0.11±0.02 (n = 6) ng insulin /mg pancreas in control, heterozygous and homozygous mice, respectively (fig.10).
Control Heterozygous Homozygous
1.0
2.0
VO
LUM
E D
EN
SITY
OF
β-C
ELLS
(%)
A
p<0.001
N=4
p<0.001
N=4
N=4
Control Heterozygous Homozygous
0.04
0.08
0.12
ME
AN
INS
ULI
N C
ON
TEN
T (n
g/m
g PA
NC
REA
S)
p<0.003
N=6 p<0.003
N=11
N=5
B
Figure10. Rip Cx32 mice featured an increase in β-cells mass and insulin content. A: The volume density of ß-cells, expressed as percent of pancreas volume, was greater in heterozygous and homozygous transgenic mice than in controls (p<0.001). B: The insulin content of pancreas was also higher in transgenic than in control mice (p<0.003). N = number of pancreas
35
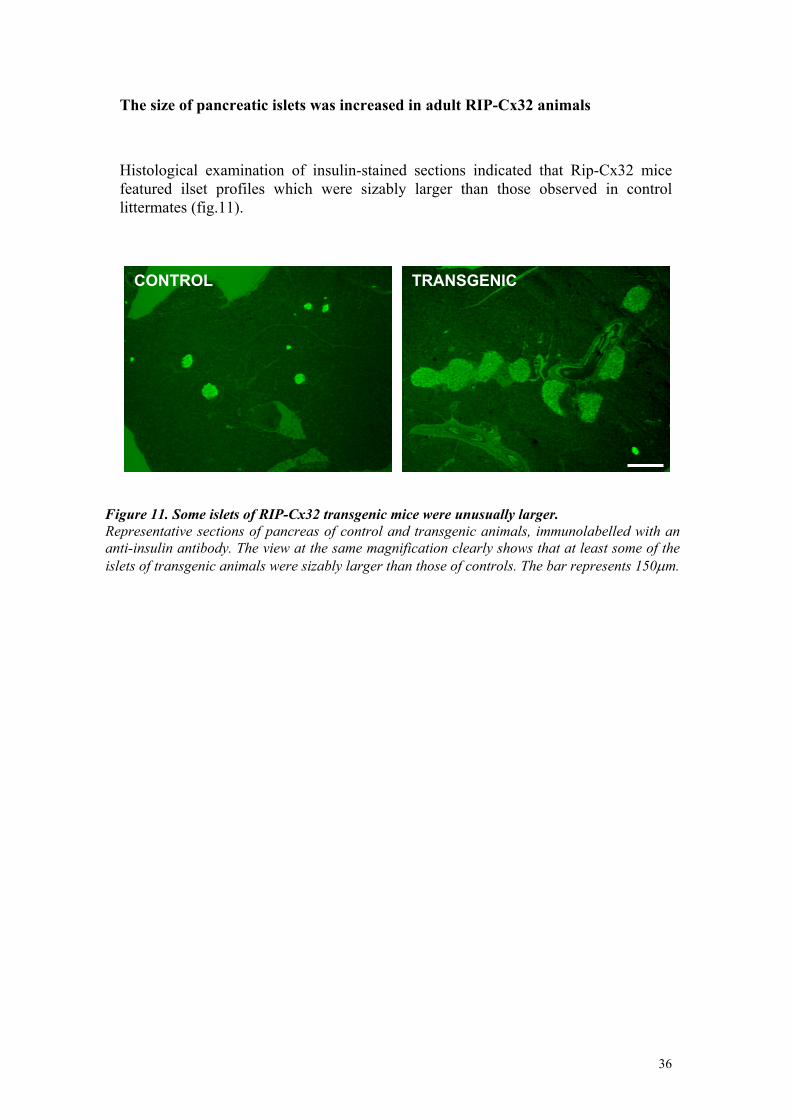
The size of pancreatic islets was increased in adult RIP-Cx32 animals Histological examination of insulin-stained sections indicated that Rip-Cx32 mice featured ilset profiles which were sizably larger than those observed in control littermates (fig.11).
TRANSGENICCONTROL
Figure 11. Some islets of RIP-Cx32 transgenic mice were unusually larger. Representative sections of pancreas of control and transgenic animals, immunolabelled with ananti-insulin antibody. The view at the same magnification clearly shows that at least some of theislets of transgenic animals were sizably larger than those of controls. The bar represents 150µm.
36
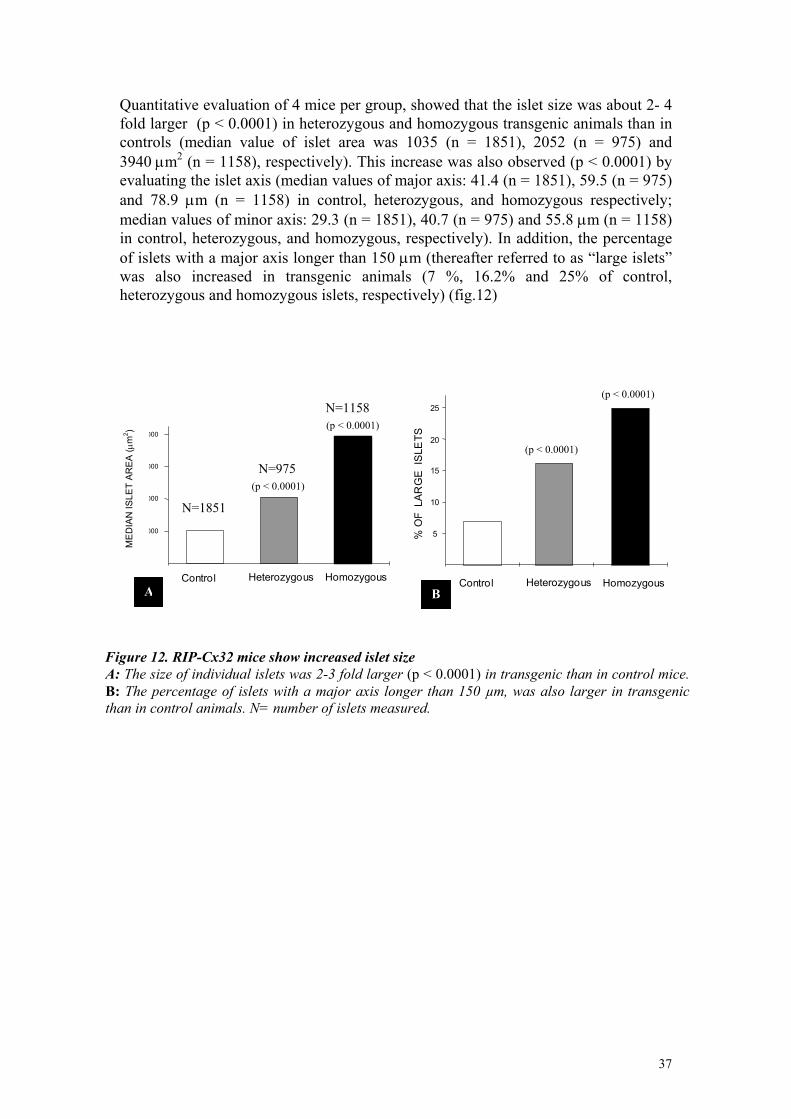
Quantitative evaluation of 4 mice per group, showed that the islet size was about 2- 4 fold larger (p < 0.0001) in heterozygous and homozygous transgenic animals than in controls (median value of islet area was 1035 (n = 1851), 2052 (n = 975) and 3940 µm2 (n = 1158), respectively). This increase was also observed (p < 0.0001) by evaluating the islet axis (median values of major axis: 41.4 (n = 1851), 59.5 (n = 975) and 78.9 µm (n = 1158) in control, heterozygous, and homozygous respectively; median values of minor axis: 29.3 (n = 1851), 40.7 (n = 975) and 55.8 µm (n = 1158) in control, heterozygous, and homozygous, respectively). In addition, the percentage of islets with a major axis longer than 150 µm (thereafter referred to as “large islets” was also increased in transgenic animals (7 %, 16.2% and 25% of control, heterozygous and homozygous islets, respectively) (fig.12)
Control Heterozygous Homozygous
5
10
15
20
25%
OF
LA
RG
E I
SLE
TS
(p < 0.0001)
(p < 0.0001)
BA
N=1851
N=975
N=1158
(p < 0.0001)
(p < 0.0001)
Control Heterozygous Homozygous
1000
2000
3000
4000
ME
DIA
N IS
LET
AR
EA
(µm
2 )
Figure 12. RIP-Cx32 mice show increased islet size A: The size of individual islets was 2-3 fold larger (p < 0.0001) in transgenic than in control mice. B: The percentage of islets with a major axis longer than 150 µm, was also larger in transgenicthan in control animals. N= number of islets measured.
37
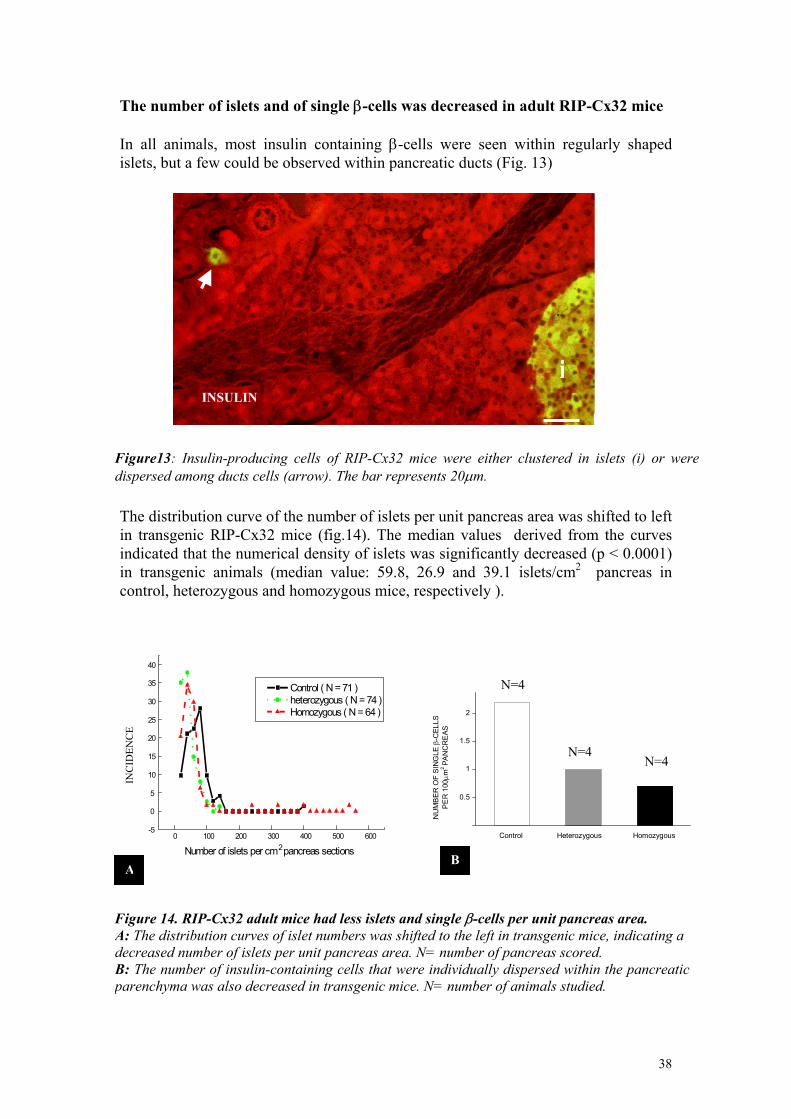
The number of islets and of single β-cells was decreased in adult RIP-Cx32 mice In all animals, most insulin containing β-cells were seen within regularly shaped islets, but a few could be observed within pancreatic ducts (Fig. 13)
iINSULIN
Figure13: Insulin-producing cells of RIP-Cx32 mice were either clustered in islets (i) or were dispersed among ducts cells (arrow). The bar represents 20µm.
The distribution curve of the number of islets per unit pancreas area was shifted to left in transgenic RIP-Cx32 mice (fig.14). The median values derived from the curves indicated that the numerical density of islets was significantly decreased (p < 0.0001) in transgenic animals (median value: 59.8, 26.9 and 39.1 islets/cm2 pancreas in control, heterozygous and homozygous mice, respectively ).
0 100 200 300 400 500 600-5
0
5
10
15
20
25
30
35
40
Control ( N = 71 ) heterozygous ( N = 74 ) Homozygous ( N = 64 )
Number of islets per cm 2 pancreas sections
INC
IDEN
CE
A
Control Heterozygous Homozygous
0.5
1
1.5
2
NU
MB
ER
OF
SIN
GLE
β-C
ELLS
P
ER
100
µ m2 PA
NC
RE
AS
N=4N=4
N=4
B
Figure 14. RIP-Cx32 adult mice had less islets and single β-cells per unit pancreas area. A: The distribution curves of islet numbers was shifted to the left in transgenic mice, indicating a decreased number of islets per unit pancreas area. N= number of pancreas scored. B: The number of insulin-containing cells that were individually dispersed within the pancreaticparenchyma was also decreased in transgenic mice. N= number of animals studied.
38
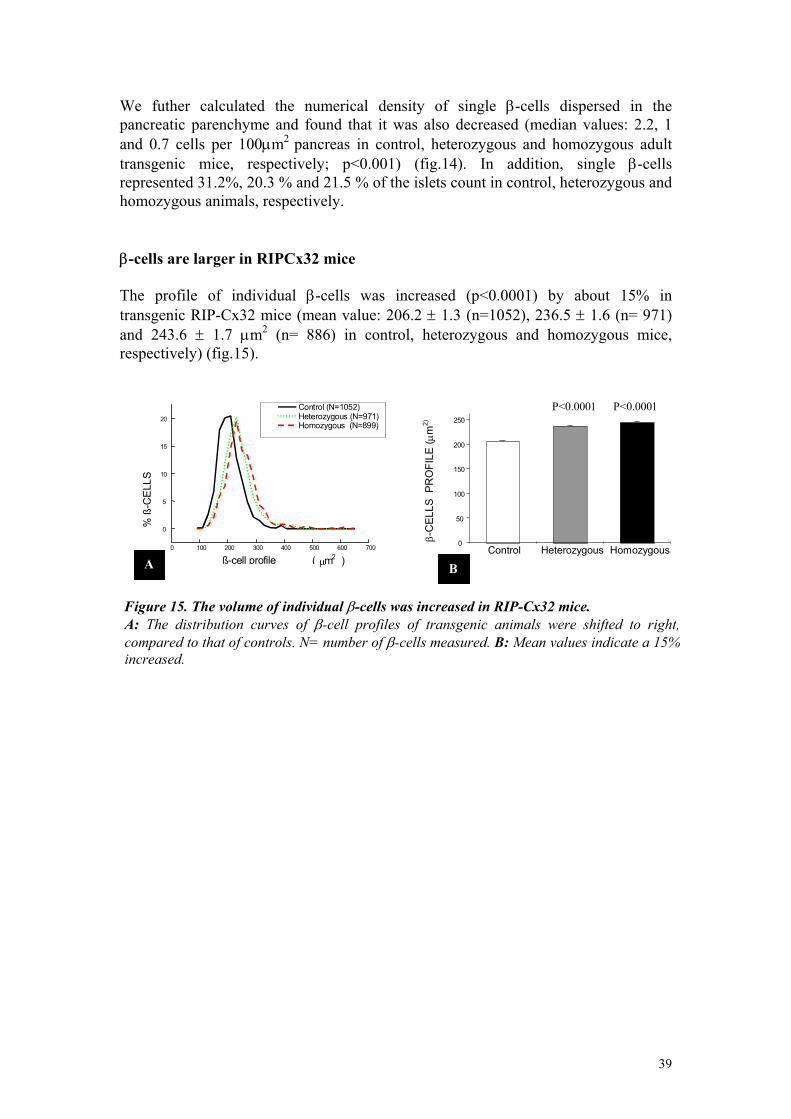
We futher calculated the numerical density of single β-cells dispersed in the pancreatic parenchyme and found that it was also decreased (median values: 2.2, 1 and 0.7 cells per 100µm2 pancreas in control, heterozygous and homozygous adult transgenic mice, respectively; p<0.001) (fig.14). In addition, single β-cells represented 31.2%, 20.3 % and 21.5 % of the islets count in control, heterozygous and homozygous animals, respectively. β-cells are larger in RIPCx32 mice The profile of individual β-cells was increased (p<0.0001) by about 15% in transgenic RIP-Cx32 mice (mean value: 206.2 ± 1.3 (n=1052), 236.5 ± 1.6 (n= 971) and 243.6 ± 1.7 µm2 (n= 886) in control, heterozygous and homozygous mice, respectively) (fig.15).
0 100 200 300 400 500 600 700
0
5
10
15
20 Control (N=1052) Heterozygous (N=971) Homozygous (N=899)
% ß
-CE
LLS
ß-cell profile ( µ m2 ) A
Control Heterozygous Homozygous0
50
100
150
200
250
P<0.0001 P<0.0001
β-C
ELL
S P
RO
FILE
(µm
2)
B
Figure 15. The volume of individual β-cells was increased in RIP-Cx32 mice. A: The distribution curves of β-cell profiles of transgenic animals were shifted to right, compared to that of controls. N= number of β-cells measured. B: Mean values indicate a 15% increased.
39
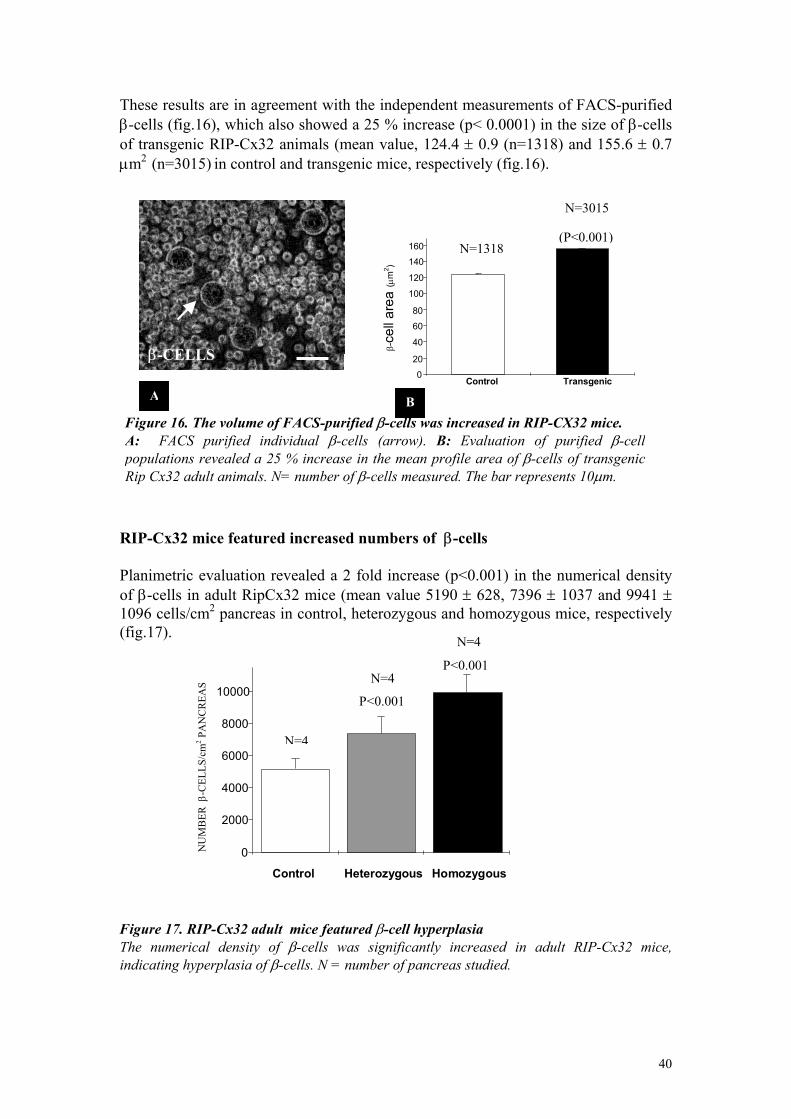
These results are in agreement with the independent measurements of FACS-purified β-cells (fig.16), which also showed a 25 % increase (p< 0.0001) in the size of β-cells of transgenic RIP-Cx32 animals (mean value, 124.4 ± 0.9 (n=1318) and 155.6 ± 0.7 µm2 (n=3015) in control and transgenic mice, respectively (fig.16). N=3015 RIP-Cx32 mice featured increased numbers of β-cells Planimetric evaluation revealed a 2 fold increase (p<0.001) in the numerical density of β-cells in adult RipCx32 mice (mean value 5190 ± 628, 7396 ± 1037 and 9941 ± 1096 cells/cm2 pancreas in control, heterozygous and homozygous mice, respectively (fig.17). Figure 17. RIP-Cx32 adult mice featured β-cell hyperplasia The numerical density of β-cells was significantly increased in adult RIP-Cx32 mice, indicating hyperplasia of β-cells. N = number of pancreas studied.
Figure 16. The volume of FACS-purified β-cells was increased in RIP-CX32 mice. A: FACS purified individual β-cells (arrow). B: Evaluation of purified β-cellpopulations revealed a 25 % increase in the mean profile area of β-cells of transgenicRip Cx32 adult animals. N= number of β-cells measured. The bar represents 10µm.
β-CELLS
AControl Transgenic 0
20
40
60
80
100
120
140
160
β -ce
ll ar
ea (µ
m2 )
(P<0.001) N=1318
B
N=4
Control Heterozygous Homozygous
0
2000
4000
6000
8000
10000 P<0.001
P<0.001
N=4
N=4
NU
MB
ER β
-CEL
LS/c
m2 P
AN
CR
EAS
40
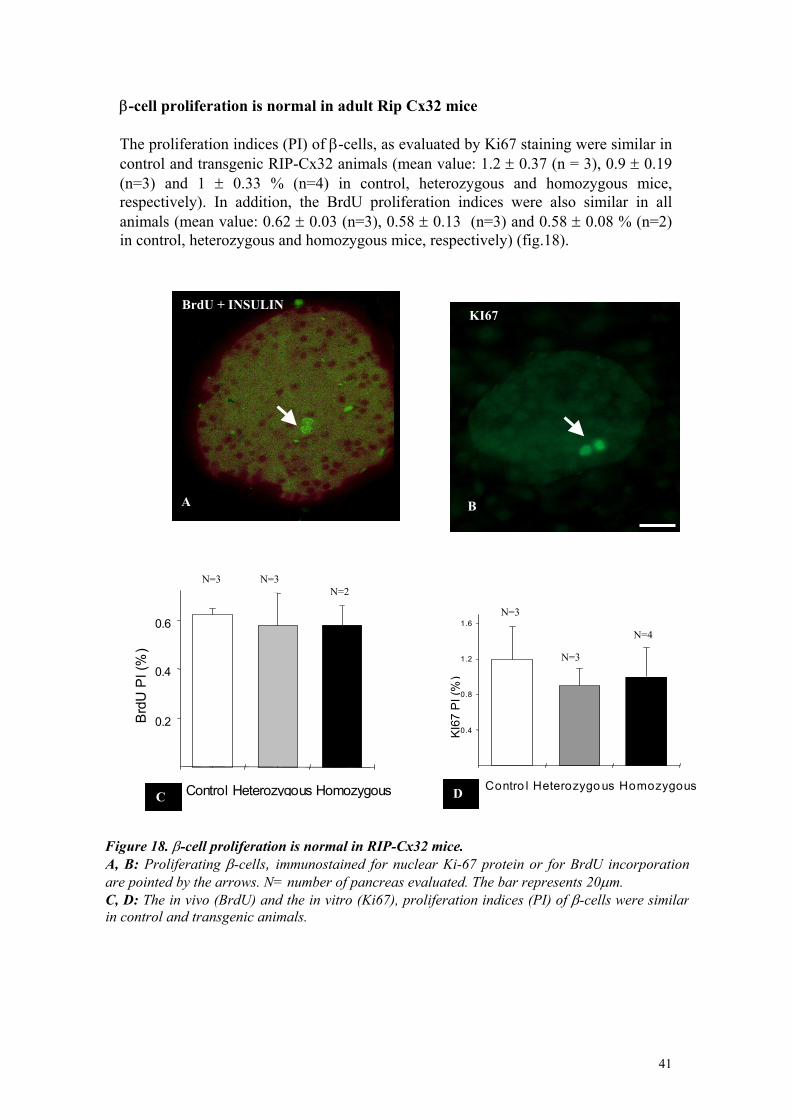
β-cell proliferation is normal in adult Rip Cx32 mice The proliferation indices (PI) of β-cells, as evaluated by Ki67 staining were similar in control and transgenic RIP-Cx32 animals (mean value: 1.2 ± 0.37 (n = 3), 0.9 ± 0.19 (n=3) and 1 ± 0.33 % (n=4) in control, heterozygous and homozygous mice, respectively). In addition, the BrdU proliferation indices were also similar in all animals (mean value: 0.62 ± 0.03 (n=3), 0.58 ± 0.13 (n=3) and 0.58 ± 0.08 % (n=2) in control, heterozygous and homozygous mice, respectively) (fig.18). BrdU + INSULIN
A
B
KI67
Control Heterozygous Homozygous 0.2 0.4 0.6
Brd
U P
I (%
)
C
N=2 N=3 N=3
C
0.4
0.8
1.2
1.6
KI6
7 P
I (%
)
D
Figure 18. β-cell proliferation is normal in RIP-Cx32 mice. A, B: Proliferating β-cells, immunostained for nuclear Ki-67 prare pointed by the arrows. N= number of pancreas evaluated. TheC, D: The in vivo (BrdU) and the in vitro (Ki67), proliferation inin control and transgenic animals.
N=3
ontro l Het
otein or fo bar repredices (PI)
N=3
erozygo us H
r BrdU incsents 20µm. of β-cells w
N=4
omozygous
orporation ere similar
41
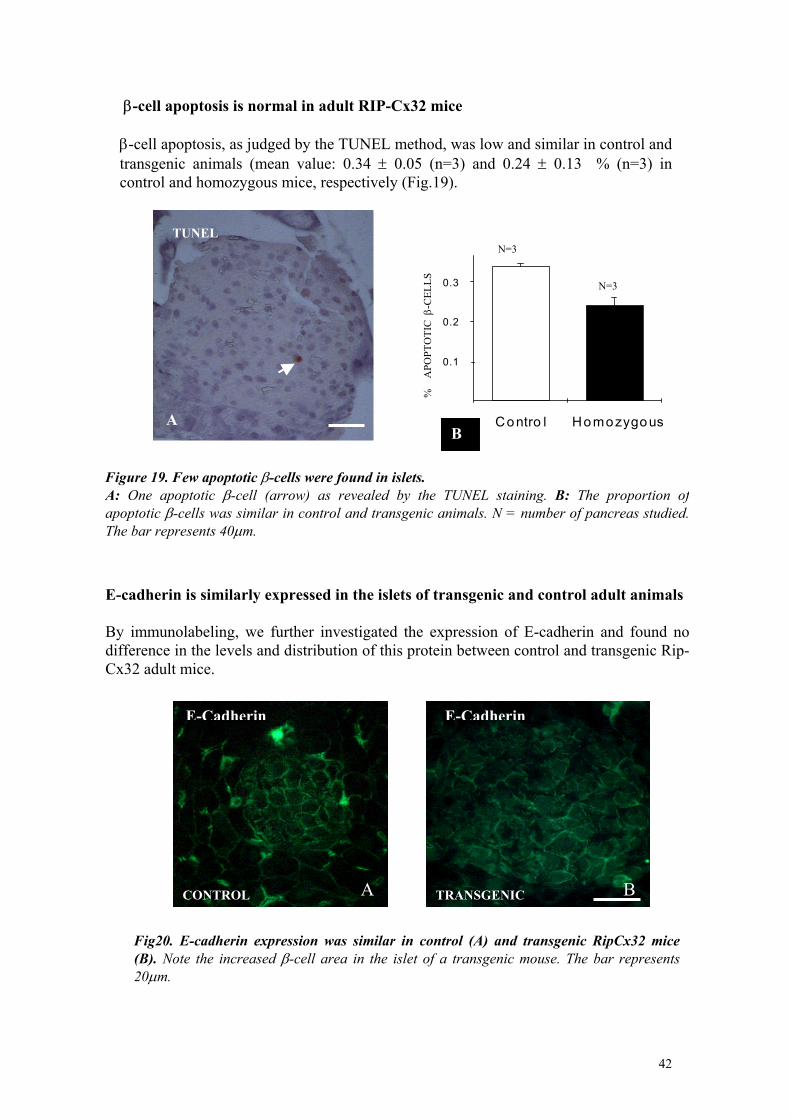
β-cell apoptosis is normal in adult RIP-Cx32 mice β-cell apoptosis, as judged by the TUNEL method, was low and similar in control and transgenic animals (mean value: 0.34 ± 0.05 (n=3) and 0.24 ± 0.13 % (n=3) in control and homozygous mice, respectively (Fig.19).
TUNEL
A
N=3
N=3
BContro l Homozygous
0.1
0.2
0.3
%
APO
PTO
TIC
β-C
ELLS
Figure 19. Few apoptotic β-cells were found in islets. A: One apoptotic β-cell (arrow) as revealed by the TUNEL staining. B: The proportion of apoptotic β-cells was similar in control and transgenic animals. N = number of pancreas studied.The bar represents 40µm.
E-cadherin is similarly expressed in the islets of transgenic and control adult animals By immunolabeling, we further investigated the expression of E-cadherin and found no difference in the levels and distribution of this protein between control and transgenic Rip-Cx32 adult mice.
CONTROL
ACONTROL
E-Cadherin
TRANGENIC RIPCX32
B TRANSGENIC
E-Cadherin
Fig20. E-cadherin expression was similar in control (A) and transgenic RipCx32 mice (B). Note the increased β-cell area in the islet of a transgenic mouse. The bar represents20µm.
42
Analysis of newborn mice To assess, whether the endocrine tissue of RIP-Cx32 transgenic increased in volume place during the embryonic life, we investigated newborn mice, within one hour from birth. Histological analysis of newborn transgenic animal showed that the distribution of islet cells, was similar to that observed in control littermates (fig.21).
Insulin
CONTROL
C
A
CONTROL
Glucagon
Insulin B
TRANSGENIC
Insulin
DGlucagon
SomaSomatostatin
ESomatostatin
GPancreatic polypeptide
Pancpolyp
Figure 21. Newborn Rip-Cx32 mice feature islePancreas sections of newborn RIP-Cx32 traimmunostained with specific antibodies against pancreatic polypeptide and show a distributionobserved in controls (left panels). The bar represeD, E, F, G, H, and I.
A
Ftostatin
Hreatic eptide
ts alike those of control littermates.nsgenic mice (right panels) were insulin, glucagon, somatostatin and of endocrine cells similar to thatnts 100µm A and B; and 40µm in C,
43
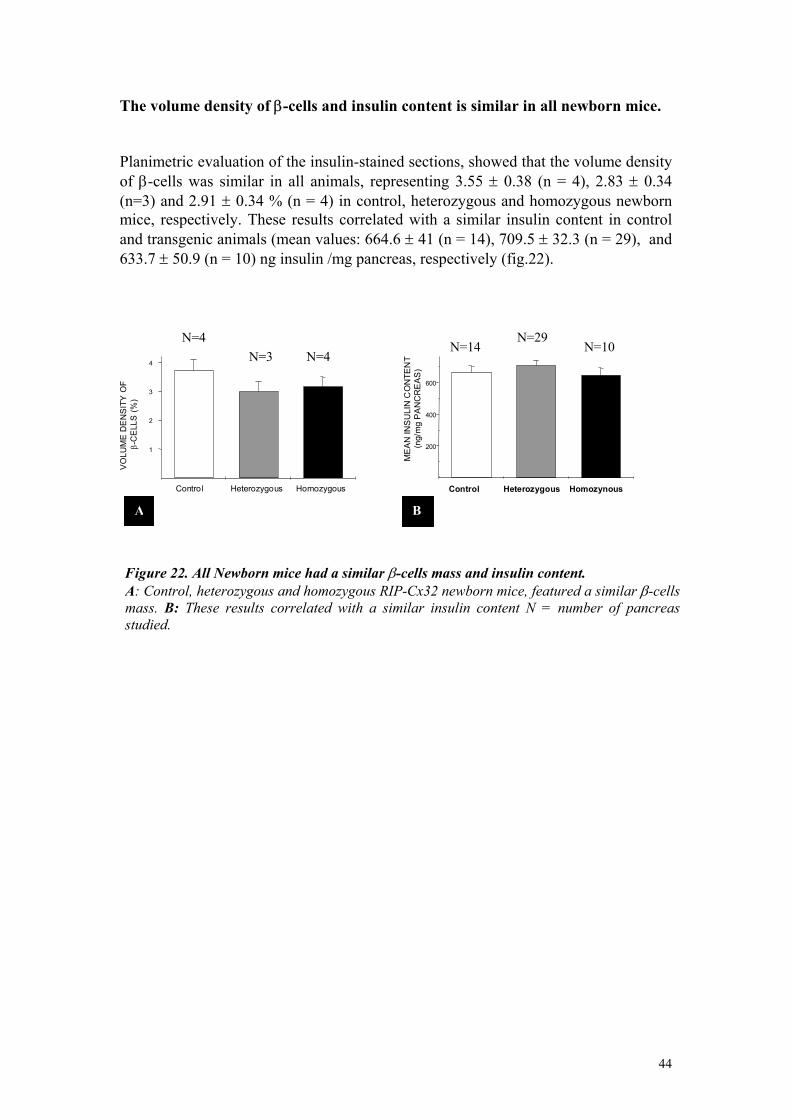
The volume density of β-cells and insulin content is similar in all newborn mice. Planimetric evaluation of the insulin-stained sections, showed that the volume density of β-cells was similar in all animals, representing 3.55 ± 0.38 (n = 4), 2.83 ± 0.34 (n=3) and 2.91 ± 0.34 % (n = 4) in control, heterozygous and homozygous newborn mice, respectively. These results correlated with a similar insulin content in control and transgenic animals (mean values: 664.6 ± 41 (n = 14), 709.5 ± 32.3 (n = 29), and 633.7 ± 50.9 (n = 10) ng insulin /mg pancreas, respectively (fig.22).
Control Heterozygous Homozynous
200
400
600
ME
AN
INS
ULI
N C
ON
TEN
T (n
g/m
g P
AN
CR
EA
S)
N=10 N=29
N=14
Control Heterozygous Homozygous
1
2
3
4
VO
LUM
E D
EN
SIT
Y O
F β
-CE
LLS
(%)
N=4N=3 N=4
A B
Figure 22. All Newborn mice had a similar β-cells mass and insulin content. A: Control, heterozygous and homozygous RIP-Cx32 newborn mice, featured a similar β-cellsmass. B: These results correlated with a similar insulin content N = number of pancreasstudied.
44
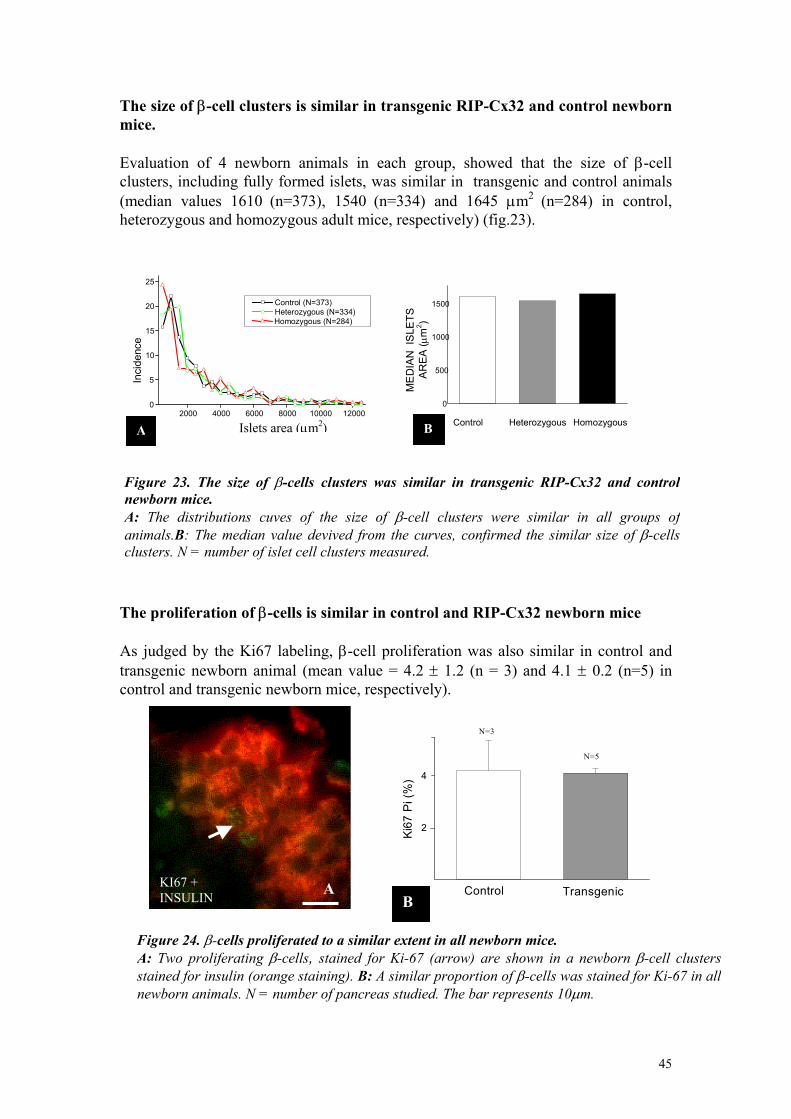
The size of β-cell clusters is similar in transgenic RIP-Cx32 and control newborn mice. Evaluation of 4 newborn animals in each group, showed that the size of β-cell clusters, including fully formed islets, was similar in transgenic and control animals (median values 1610 (n=373), 1540 (n=334) and 1645 µm2 (n=284) in control, heterozygous and homozygous adult mice, respectively) (fig.23).
2000 4000 6000 8000 10000 120000
5
10
15
20
25
Control (N=373) Heterozygous (N=334)Homozygous (N=284)
Inci
denc
e
Islets area (µm2)A
Control Heterozygous Homozygous
0
500
1000
1500
ME
DIA
N I
SLE
TSA
RE
A ( µ
m2 )
B
Figure 23. The size of β-cells clusters was similar in transgenic RIP-Cx32 and control newborn mice. A: The distributions cuves of the size of β-cell clusters were similar in all groups of animals.B: The median value devived from the curves, confirmed the similar size of β-cellsclusters. N = number of islet cell clusters measured.
The proliferation of β-cells is similar in control and RIP-Cx32 newborn mice As judged by the Ki67 labeling, β-cell proliferation was also similar in control and transgenic newborn animal (mean value = 4.2 ± 1.2 (n = 3) and 4.1 ± 0.2 (n=5) in control and transgenic newborn mice, respectively).
KI67 + INSULIN A
Control Transgenic
2
4
Ki67
Pi (
%)
N=3
N=5
B
KI67 + INSULIN
Figure 24. β-cells proliferated to a similar extent in all newborn mice. A: Two proliferating β-cells, stained for Ki-67 (arrow) are shown in a newborn β-cell clusters stained for insulin (orange staining). B: A similar proportion of β-cells was stained for Ki-67 in all newborn animals. N = number of pancreas studied. The bar represents 10µm.
45

Chapitre 3: discussion
46

So far few studies have examined the possible role of connexins on the development and differentiation of pancreatic islets (Charollais et al., 1999, 2000). We show here, for the first time, that β-cell growth may be promoted in vivo by a selective change in Cx32, a connexin which is not natively expressed in the endocrine pancreas. Previous studies have documented islet neogenesis from uncommitted precursors such as the epithelial cells of pancreatic ducts (Nielsen et al., 1999; Wilkin, 1998 Rosenberg et al., 1996). However it is unlikely that this process plays a major role in the increase of the β-cells mass in RIP-Cx32 adult mice, since disseminated single β-cells (which are thought to indicate neogenesis), as well as the numerical density of β-cells, were decreased in RIP-Cx32 transgenic mice. Thus, this increase may results from hypertrophy of individual islets, secondary to increased number and size of individual β-cells. Moreover, the decrease numerical density of β-cells single may reflect, an increase the replication capacity of β-cells. Using proliferation indices, we failed to demonstrate such a significant increase, even though the numerical density of β-cells was larger in transgenic animals. However, the basal level of proliferation of β-cells is quite low. Assuming that the daily proliferation rate is increased by only 1%, which may not be detectable by the approaches we used, the β-cells mass may increase by about 365% in one year time. This is about the increase we observed in RIP-Cx32 transgenic mice. In addition both the BrdU and the Ki67 labelling indices were evaluated in mice older than 3 months, a stage when β-cell proliferation likely reached steady state. It is unlikely that the random insertion of the RIP promoter can account for our findings, since increased insulin content and β-cells mass were observed in two independent lines of RIP-Cx32 mice. Rather, the results appear to be a specific consequence of the over-expression of Cx32 in pancreatic β-cells, inasmuch as they was not observed in the majority of RIP driven transgenic models (Epstein et al., 1992, Kato et al., 1994, Efrat et al., 1988, Hanahan, 1985). This possibility is further supported by the findings that increase in β-cell mass, islet size and β-cell numbers, was larger in homozygous than heterozygous RIP-Cx32 mice, suggesting a gene dosage effect. CAMs play a primordial role in controlling the growth and spatial distribution of islet cells, inasmuch as insulinomas develop in transgenic mice that no more express E-cadherin (Perl et al., 1998). Since the expression of CAMs and gapjunction proteins is interdependent (Jongen et al., 1991; Meyer et al., 1992), the alteration we observed in RIP-Cx32 mice may result from altered CAMs expression. However, this possibility is not substantiated by our failure to observe a difference in the expression of E-cadherin between control and transgenic Rip Cx32 adult mice. Connexins have been implicated in the intercellular propagation of signals during metabolic stress, which exacerbate cell injury, e.g. during cerebral ischemia (Lin et al., 1998). Since apoptosis plays an essential role in the control of the β-cells mass (Pick et al., 1998; Finegood et al., 1995, Scaglia et al., 1995), we evaluated whether it may be altered as result of Cx32 expression. We found that apoptosis was very low in the 3 animal genotypes we studied, and not different in control and transgenic Rip Cx32 animals.
47

We eventually documented that the increase in islet mass, observed in RIP-Cx32 mice does not take place during embryonic development, as judge by a similar β-cell mass of control and transgenic newborn mice. This finding is consistent with a role of connexins in regulating the islet cell mass during postnatal life. This effect may be due to the cell-to-cell transfer of endogenous metabolites, some of which may act as growth signals, resulting in an increase in the size of the proliferation compartment which is though to be a major limiting factor for the regenerative capacity of the islets (Hellerstrom et al., 1991). A change in connexins may also withdraw increasing numbers of β-cells into G0. Consistent with this possibility, growth alterations have been associated with altered expression of cyclin D1 and p27kip-1 after forced connexin expression in lung and liver carcinoma cells (Koffler et al., 2000). In neonatal β-cells, cycline-dependent kinase-4 (Cdk4) and cycline D1 are associated to the mitogenic stimulation by lysophosphatidic acid (Dunlop et al., 1996), and Cdk4 activation results in β-cell hyperplasia (Rane et al., 1999). There is also ample evidence for an inverse correlation between the extent of connexin-dependent communication and tumorogenesis, including in an experimental insulinoma (Vozzi et al., 1997). Our study indicates that an increase in intercellular gap junctional communication is linked to an increase in the growth pattern of β-cells in RipCx32 mice. Cx32 makes channels with conductance and permeability characteristics different from those of Cx36, which natively connects β-cells (Serre-Beinier et al., 2002). It is an exciting prospective to test whether use this connexin may be use to drive the formation of the large β-cell numbers, which would be required in the perspective of a cell therapy for diabetic patients. There is a clear specificity in the role of different connexins in cell growth (Goldberg et al., Meda and Spray, 2000). In this perspective, it is worthnoting that the increase in β-cells mass and islet size of RIP-Cx32 adult mice, as well as their spontaneous hypoglycaemia, are comparable to those reported in mice over-expressing Hepatocyte Growth Factor (HGF), or the Parathyroid Related Protein (PTHrP). HGF and PtHrP functions include the regulation of cell cycle, differentiation and apoptosis (Garcia-Ocana et al., 2000, Porter et al., 1998, Vasavada et al., 1996). The transfer through gap junctions of putative islet growth signals is worthy of exploration, to determine the mechanism whereby islet neogenesis, differentiation and proliferation, as well as normal islet mass are controlled throughout life. It will be interesting to study β-cell growth and proliferation, after over-expression of other connexins, such as Cx43 or Cx36, and possibly also after knocking out these proteins. In summary, our study shows that abnormal levels of β-cell communication through Cx32 channels, is associated with an increase in the growth pattern of the endocrine pancreas, which results in increased insulin contents. The data provide the first clue that proper connexin expression is required for the adequate growth pattern of the endocrine pancreas. Increased islet mass, due to hypertrophy and hyperplasia of β-cells, appears to be responsible for the increased insulin content. The potential role of proper expression of connexins in islet growth is significant, given the reduced number of β-cells in type II diabetes mellitus, and the current paucity of well-defined islet growth factors. The primary rationale for studying the mechanism responsible for
48

islet development, growth and differentiation is to facilitate the development of effective strategies for treatment of diabetes mellitus. Our data suggest that connexins are worth additional studies in this context.
49

Conclusion et perspectives
50

Notre étude a démontré qu’un taux anormal de communication entre cellules β, au travers de canaux formés de Cx32, est associé à une augmentation de la croissance post-natale des cellules produisant l’insuline, et à une augmentation concomittante du contenu du pancréas en cette hormone. Nous avons montré que cette augmentation post-natale est due à une hypertrophie des îlots de Langherhans, laquelle résulte d’une hyperplasie et d’une hypertrophie des cellules β. Il s’agit du premier indice indiquant qu’une expression adéquate de connexines est nécessaire pour une croissance normale du pancréas endocrine in vivo. Il est donc indispensable de préciser le rôle de divers isoformes de connexines et d’étudier leur mécanisme d’action dans différents modèles de souris transgéniques et de lignées cellulaires. D’autres modèles de souris transgéniques sont déjà disponibles à cet effet notamment pour étudier, l’effet de la surexpression de la Cx36, la principale connexine des cellules β, ou de la Cx43, une connexine également présente dans les îlots pancréatiques. L’analyse des effets de ces proteines sur la dynamique de la masse des cellules β, devrait permettre d’identifier les mécanismes par lesquels les connexines influencent la néogenèse, la différenciation et la prolifération des cellules β. La raison principale d’étudier ces mécanismes est de faciliter le développement de stratégies thérapeutiques effectives pour le traitement du diabète. Eu égard au dysfonctionnement des cellules β dans le diabète de type II et à la problématique du petit nombre de donneurs disponibles, une thérapie cellulaire doit être envisagée. Celle-ci est actuellement confrontée à des problèmes immunologiques (échecs à long terme des thérapies immunosuppressives dans l’éventualité des transplantations du pancréas et des îlots) et quantitatifs (difficulté d’obtenir in vitro suffisamment de cellules β differenciés). Notre travail participe à la recherche des facteurs qui pourraient apporter une solution à ce dernier problème, en montrant que les connexines peuvent influer favorablement sur la croissance postnatale des cellules productrices d’insuline.
51

Références Auerbach, A. A., and Bennett, M. V. (1969). A rectifying electrotonic synapse in the central nervous system of a vertebrate. J Gen Physiol 53, 211-37. Barr L., Dewey M.M. and Berger W.(1965).propagation of action potentials and the structure of the nexus in cardiac muscle. J Gen physiol 48, 797-823 Benedetti, E. L. and Emmelot, P. (1968). Hexagonal array of subunits in tight junctions separated from isolated rat liver plasma membranes. J Cell Biol 38, 15-24. Bennett, M. V., Barrio, L. C., Bargiello, T. A., Spray, D. C., Hertzberg, E., and Saez, J. C. (1991). Gap junctions: new tools, new answers, new questions. Neuron 6, 305-20. Bennett, M. V., and Goodenough, D. A. (1978). Gap junctions, electrotonic coupling, and intercellular communication. Neurosci Res Program Bull 16, 1-486. Bergoffen, J., Scherer, S. S., Wang, S., Scott, M. O., Bone, L. J., Paul, D. L., Chen, K., Lensch, M. W., Chance, P. F., and Fischbeck, K. H. (1993). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039-42. Berry, V., Mackay, D., Khaliq, S., Francis, P. J., Hameed, A., Anwar, K., Mehdi, S. Q., Newbold, R. J., Ionides, A., Shiels, A., Moore, T., and Bhattacharya, S. S. (1999). Connexin 50 mutation in a family with congenital "zonular nuclear" pulverulent cataract of Pakistani origin. Hum Genet 105, 168-70. Bevans, C. G., Kordel, M., Rhee, S. K., and Harris, A. L. (1998). Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem 273, 2808-16. Bevilacqua, A., Loch-Caruso, R., and Erickson, R. P. (1989). Abnormal development and dye coupling produced by antisense RNA to gap junction protein in mouse preimplantation embryos. Proc Natl Acad Sci U S A 86, 5444-8. Beyer, E. C. (1990). Molecular cloning and developmental expression of two chick embryo gap junction proteins. J Biol Chem 265, 14439-43. Beyer, E. C., Paul, D. L., and Goodenough, D. A. (1987). Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J Cell Biol 105, 2621-9. Beyer, E. C., Paul, D. L., and Goodenough, D. A. (1990). Connexin family of gap junction proteins. J Membr Biol 116, 187-94. Birchmeier, W. (1995). E-cadherin as a tumor (invasion) suppressor gene. Bioessays 17, 97-9. Bonner-Weir, S., Baxter, L. A., Schuppin, G. T., and Smith, F. E. (1993). A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 42, 1715-20. Bonner-Weir, S. (2001). B-cell turnover: its assessment and implications. Diabetes 50, Suppl1, S20-S24. Bosco, D., and Meda, P. (1991). Actively synthesizing beta-cells secrete preferentially after glucose stimulation. Endocrinology 129, 3157-66.
52

Britz-Cunningham, S. H., Shah, M. M., Zuppan, C. W., and Fletcher, W. H. (1995). Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med 332, 1323-9. Budunova, I. V., and Williams, G. M. (1994). Cell culture assays for chemicals with tumor-promoting or tumor- inhibiting activity based on the modulation of intercellular communication. Cell Biol Toxicol 10, 71-116. Burholt, D. R., Etzel, S. L., Schenken, L. L., and Kovacs, C. J. (1985). Digestive tract cell proliferation and food consumption patterns of Ha/ICR mice. Cell Tissue Kinet 18, 369-86. Burt, J. M., and Spray, D. C. (1988). Inotropic agents modulate gap junctional conductance between cardiac myocytes. Am J Physiol 254, H1206-10. Caspar, D. L., Goodenough, D. A., Makowski, L., and Phillips, W. C. (1977). Gap junction structures. I. Correlated electron microscopy and x-ray diffraction. J Cell Biol 74, 605-28. Cesen-Cummings, K., Fernstrom, M. J., Malkinson, A. M., and Ruch, R. J. (1998). Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis 19, 61-7. Chanson, M., Chandross, K. J., Rook, M. B., Kessler, J. A., and Spray, D. C. (1993). Gating characteristics of a steeply voltage-dependent gap junction channel in rat Schwann cells. J Gen Physiol 102, 925-46. Chanson, M., Orci, L., and Meda, P. (1991). Extent and modulation of junctional communication between pancreatic acinar cells in vivo. Am J Physiol 261, G28-36. Charollais A, Gjinovci A, Huarte J, Bauquis J, Nadal A, Martin F, Andreu E, Sanchez-Andres JV, Calabrese A, Bosco D, Soria B, Wollheim CB, Herrera PL, Meda P. (2000). Junctional communication of pancreatic beta cells contributes to the control of insulin secretion and glucose tolerance.J Clin Invest. 106, 235-43. Charollais A, Serre V, Mock C, Cogne F, Bosco D, Meda P. (1999). Loss of alpha 1 connexin does not alter the prenatal differentiation of pancreatic beta cells and leads to the identification of another islet cell connexin. Dev Genet. 24, 13-26. Chen, L., Appel, M. C., Alam, T., Miyaura, C., Sestak, A., O'Neil, J., Unger, R. H., and Newgard, C. B. (1992). Factors regulating islet regeneration in the post-insulinoma NEDH rat. Adv Exp Med Biol 321, 71-80. Chen, S. C., Pelletier, D. B., Ao, P., and Boynton, A. L. (1995). Connexin43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin-dependent kinases. Cell Growth Differ 6, 681-90. Davidson, P. M., Campbell, I. L., Oxbrow, L., Hutson, J. M., and Harrison, L. C. (1989). Pancreatic beta cell proliferation in rabbits demonstrated by bromodeoxyuridine labeling. Pancreas 4, 594-600. Dermietzel, R., Traub, O., Hwang, T. K., Beyer, E., Bennett, M. V., Spray, D. C., and Willecke, K. (1989). Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci U S A 86, 10148-52.
53

Dermietzel, R., Yancey, S. B., Traub, O., Willecke, K., and Revel, J. P. (1987). Major loss of the 28-kD protein of gap junction in proliferating hepatocytes. J Cell Biol 105, 1925-34. Dewer M.M. and Barr L. (1962). Intercellular connection between smooth muscle cells by the nexus. Science, 137: 670-672. Dunlop, M., Muggli, E., and Clark, S. (1996). Association of cyclin-dependent kinase-4 and cyclin D1 in neonatal beta cells after mitogenic stimulation by lysophosphatidic acid. Biochem Biophys Res Commun 218, 132-6. Edlund H. (2001) Factors controlling pancreatic cell differentiation and function. Diabetologia 44, 1071-1079. Efrat, S., Linde, S., Kofod, H., Spector, D., Delannoy, M., Grant, S., Hanahan, D., and Baekkeskov, S. (1988). Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc Natl Acad Sci U S A 85, 9037-41. Epstein, P. N., Boschero, A. C., Atwater, I., Cai, X., and Overbeek, P. A. (1992). Expression of yeast hexokinase in pancreatic beta cells of transgenic mice reduces blood glucose, enhances insulin secretion, and decreases diabetes. Proc Natl Acad Sci U S A 89, 12038-42. Esinduy, C. B., Chang, C. C., Trosko, J. E., and Ruch, R. J. (1995). In vitro growth inhibition of neoplastically transformed cells by non- transformed cells: requirement for gap junctional intercellular communication. Carcinogenesis 16, 915-21. Fairweather, N., Bell, C., Cochrane, S., Chelly, J., Wang, S., Mostacciuolo, M. L., Monaco, A. P., and Haites, N. E. (1994). Mutations in the connexin 32 gene in X-linked dominant Charcot-Marie- Tooth disease (CMTX1). Hum Mol Genet 3, 29-34. Finegood, D. T., Scaglia, L., and Bonner-Weir, S. (1995). Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 44, 249-56. Fishman, G. I., Eddy, R. L., Shows, T. B., Rosenthal, L., and Leinwand, L. A. (1991). The human connexin gene family of gap junction proteins: distinct chromosomal locations but similar structures. Genomics 10, 250-6. Furshpan E.J. and Potter D.D. (1959). Transmission at the giant motor synapses of the crayfish. J physiol 145, 289-325. Garcia-Ocana, A., Takane, K. K., Syed, M. A., Philbrick, W. M., Vasavada, R. C., and Stewart, A. F. (2000). Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem 275, 1226-32. Giordano, E., Cirulli, V., Bosco, D., Rouiller, D., Halban, P., and Meda, P. (1993). B-cell size influences glucose-stimulated insulin secretion. Am J Physiol 265, C358-64. Goldberg, G. S., Lampe, P. D., and Nicholson, B. J. (1999). Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol 1, 457-9. Goldberg, G. S., Lampe, P. D., and Nicholson, B. J. (1999). Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol 1, 457-9.
54

Goldberg, G. S., Martyn, K. D., and Lau, A. F. (1994). A connexin 43 antisense vector reduces the ability of normal cells to inhibit the foci formation of transformed cells. Mol Carcinog 11, 106-14. Gong, X., Li, E., Klier, G., Huang, Q., Wu, Y., Lei, H., Kumar, N. M., Horwitz, J., and Gilula, N. B. (1997). Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell 91, 833-43. Goodenough, D. A. (1976). The structure and permeability of isolated hepatocyte gap junctions. Cold Spring Harb Symp Quant Biol 40, 37-43. Goodenough, D. A., Paul, D. L., and Jesaitis, L. (1988). Topological distribution of two connexin32 antigenic sites in intact and split rodent hepatocyte gap junctions. J Cell Biol 107, 1817-24. Goodenough, D. A., and Stoeckenius, W. (1972). The isolation of mouse hepatocyte gap junctions. Preliminary chemical characterization and x-ray diffraction. J Cell Biol 54, 646-56. Grifa, A., Wagner, C. A., D'Ambrosio, L., Melchionda, S., Bernardi, F., Lopez-Bigas, N., Rabionet, R., Arbones, M., Monica, M. D., Estivill, X., Zelante, L., Lang, F., and Gasparini, P. (1999). Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet 23, 16-8. Hall, P. A., Coates, P. J., Ansari, B., and Hopwood, D. (1994). Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci 107, 3569-77. Hanahan, D. (1985). Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115-22. Hellerstrom, C., Sjoholm, A., and Swenne, I. (1991). Effects of growth hormone and related growth factors on DNA replication and insulin production in pancreatic islet beta-cells. Acta Paediatr Scand Suppl 377, 55-62. Hennemann, H., Kozjek, G., Dahl, E., Nicholson, B., and Willecke, K. (1992). Molecular cloning of mouse connexins26 and -32: similar genomic organization but distinct promoter sequences of two gap junction genes. Eur J Cell Biol 58, 81-9. Hennemann, H., Suchyna, T., Lichtenberg-Frate, H., Jungbluth, S., Dahl, E., Schwarz, J., Nicholson, B. J., and Willecke, K. (1992). Molecular cloning and functional expression of mouse connexin40, a second gap junction gene preferentially expressed in lung. J Cell Biol 117, 1299-310. Herrera, P. L., Huarte, J., Zufferey, R., Sanvito, F., Meda P., Orci, L., and Vassalli, J. D. (1991). Embryogenesis of the murine endocrine pancreas; early expression of pancreatic poly peptide gene. Development 113, 1257-1265. Herrera, P. L., Huarte, J., Zufferey, R., Nichols, A., Mermillod, B., Philippe, J., Muniesa, P., Sanvito, F., Orci, L., and Vassalli, J. D. (1994). Ablation of islet endocrine cells by targeted expression of hormone- promoter-driven toxigenes. Proc Natl Acad Sci U S A 91, 12999-3003. Hertzberg, E. L. (1984). A detergent-independent procedure for the isolation of gap junctions from rat liver. J Biol Chem 259, 9936-43.
55

Hertzberg, E. L., Disher, R. M., Tiller, A. A., Zhou, Y., and Cook, R. G. (1988). Topology of the Mr 27,000 liver gap junction protein. Cytoplasmic localization of amino- and carboxyl termini and a hydrophilic domain which is protease-hypersensitive. J Biol Chem 263, 19105-11. Hertzberg, E. L., Lawrence, T. S., and Gilula, N. B. (1981). Gap junctional communication. Annu Rev Physiol 43, 479-91. Huang, R. P., Fan, Y., Hossain, M. Z., Peng, A., Zeng, Z. L., and Boynton, A. L. (1998). Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43). Cancer Res 58, 5089-96. Hulinsky, I., Cooney, S., Harrington, J., and Silink, M. (1995). In vitro growth of neonatal rat islet cells is stimulated by adhesion to matrix. Horm Metab Res 27, 209-15. Ionasescu, V., Searby, C., and Ionasescu, R. (1994). Point mutations of the connexin32 (GJB1) gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Hum Mol Genet 3, 355-8. Jongen, W. M., Fitzgerald, D. J., Asamoto, M., Piccoli, C., Slaga, T. J., Gros, D., Takeichi, M., and Yamasaki, H. (1991). Regulation of connexin 43-mediated gap junctional intercellular communication by Ca2+ in mouse epidermal cells is controlled by E- cadherin. J Cell Biol 114, 545-55. Kanno, Y. (1985). Modulation of cell communication and carcinogenesis. Jpn J Physiol 35, 693-707. Kanno, Y., and Loewenstein, W. R. (1966). Cell-to-cell passage of large molecules. Nature 212, 629-30. Kato, I., Suzuki, Y., Akabane, A., Yonekura, H., Tanaka, O., Kondo, H., Takasawa, S., Yoshimoto, T., and Okamoto, H. (1994). Transgenic mice overexpressing human vasoactive intestinal peptide (VIP) gene in pancreatic beta cells. Evidence for improved glucose tolerance and enhanced insulin secretion by VIP and PHM-27 in vivo. J Biol Chem 269, 21223-8. Kelley, P. M., Abe, S., Askew, J. W., Smith, S. D., Usami, S., and Kimberling, W. J. (1999). Human connexin 30 (GJB6), a candidate gene for nonsyndromic hearing loss: molecular cloning, tissue-specific expression, and assignment to chromosome 13q12. Genomics 62, 172-6. Kelsell, D. P., Di, W. L., and Houseman, M. J. (2001). Connexin mutations in skin disease and hearing loss. Am J Hum Genet 68, 559-68. Kelsell, D. P., Dunlop, J., and Hodgins, M. B. (2001). Human diseases: clues to cracking the connexin code? Trends Cell Biol 11, 2-6. Klaunig, J. E., and Ruch, R. J. (1990). Role of inhibition of intercellular communication in carcinogenesis. Lab Invest 62, 135-46. Koffler, L., Roshong, S., Kyu Park, I., Cesen-Cummings, K., Thompson, D. C., Dwyer-Nield, L. D., Rice, P., Mamay, C., Malkinson, A. M., and Ruch, R. J. (2000). Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1 )after forced connexin expression in lung and liver carcinoma cells. J Cell Biochem 79, 347-54.
56

Krutovskikh, V., and Yamasaki, H. (2000). Connexin gene mutations in human genetic diseases. Mutat Res 462, 197-207. Kuffler R.C. and Potter D.D. (1963). Glia in the leech central nervous system: physiological prpperties and neuron-glia relationship. J neurophysiol 27,290-320. Kumar, N. M., and Gilula, N. B. (1986). Cloning and characterization of human and rat liver cDNAs coding for a gap junction protein. J Cell Biol 103, 767-76. Kumar, N. M., and Gilula, N. B. (1996). The gap junction communication channel. Cell 84, 381-8. Kumar, N. M., and Gilula, N. B. (1992). Molecular biology and genetics of gap junction channels. Semin Cell Biol 3, 3-16. Labarthe, M. P., Bosco, D., Saurat, J. H., Meda, P., and Salomon, D. (1998). Upregulation of connexin 26 between keratinocytes of psoriatic lesions. J Invest Dermatol 111, 72-6. Lamartine, J., Munhoz Essenfelder, G., Kibar, Z., Lanneluc, I., Callouet, E., Laoudj, D., Lemaitre, G., Hand, C., Hayflick, S. J., Zonana, J., Antonarakis, S., Radhakrishna, U., Kelsell, D. P., Christianson, A. L., Pitaval, A., Der Kaloustian, V., Fraser, C., Blanchet-Bardon, C., Rouleau, G. A., and Waksman, G. (2000). Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 26, 142-4. Lee, S. W., Tomasetto, C., Paul, D., Keyomarsi, K., and Sager, R. (1992). Transcriptional downregulation of gap-junction proteins blocks junctional communication in human mammary tumor cell lines. J Cell Biol 118, 1213-21. Lefebvre, V. H., Otonkoski, T., Ustinov, J., Huotari, M. A., Pipeleers, D. G., and Bouwens, L. (1998). Culture of adult human islet preparations with hepatocyte growth factor and 804G matrix is mitogenic for duct cells but not for beta-cells. Diabetes 47, 134-7. Lefebvre, V. H., Otonkoski, T., Ustinov, J., Huotari, M. A., Pipeleers, D. G., and Bouwens, L. (1998). Culture of adult human islet preparations with hepatocyte growth factor and 804G matrix is mitogenic for duct cells but not for beta-cells. Diabetes 47, 134-7. Lin, J. H., Weigel, H., Cotrina, M. L., Liu, S., Bueno, E., Hansen, A. J., Hansen, T. W., Goldman, S., and Nedergaard, M. (1998). Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci 1, 494-500. Loewenstein, W. R. (1979). Junctional intercellular communication and the control of growth. Biochim Biophys Acta 560, 1-65. Loewenstein, W. R. (1985). Regulation of cell-to-cell communication by phospholilation. Biochem soc Symp Lond 50, 43-58. Loewenstein, W. R., and Kanno, Y. (1964). Studies on an epithelial 8Gland) cell junction J cell biol 22, 565-585. Loewenstein, W. R., and Kanno, Y. (1966). Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature 209, 1248-9.
57

Loewenstein, W. R., and Rose, B. (1992). The cell-cell channel in the control of growth. Semin Cell Biol 3, 59-79. Lucas-Clerc, C., Massart, C., Campion, J. P., Launois, B., and Nicol, M. (1993). Long-term culture of human pancreatic islets in an extracellular matrix: morphological and metabolic effects. Mol Cell Endocrinol 94, 9-20. Maestrini, E., Lai, C., Marlow, A., Matthews, N., Wallace, S., Bailey, A., Cook, E. H., Weeks, D. E., and Monaco, A. P. (1999). Serotonin transporter (5-HTT) and gamma-aminobutyric acid receptor subunit beta3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. The International Molecular Genetic Study of Autism Consortium. Am J Med Genet 88, 492-6. Mally I.M., Otonkoski T., Lopez A. D., Hayek A. (1994). Developmental gene expression in the human fetal Pancreas. Pediatric Reaseach 36, 4, 537-544. Martyn, K. D., Kurata, W. E., Warn-Cramer, B. J., Burt, J. M., TenBroek, E., and Lau, A. F. (1997). Immortalized connexin43 knockout cell lines display a subset of biological properties associated with the transformed phenotype. Cell Growth Differ 8, 1015-27. Masgrau-Peya, E., Salomon, D., Saurat, J. H., and Meda, P. (1997). In vivo modulation of connexins 43 and 26 of human epidermis by topical retinoic acid treatment. J Histochem Cytochem 45, 1207-15. Meda, P. (1996) a. Connexines, canaux jonctionnels et communications cellulaires (1996). Médecine/sciences12, 909-20 Meda, P. (1996) b. Gap junction involvement in secretion: the pancreas experience. Clin Exp Pharmacol Physiol 23, 1053-7. Meda, P. (1996) c. Molecular biology of gap junction proteins. Mol biol diabetes; 14:333-56. Meda, P. (1996) d. The role of gap junction membrane channels in secretion and hormonal action. J Bioenerg Biomembr 28, 369-77. Meda, P., Amherdt, M., Perrelet, A., and Orci, L. (1981). Metabolic coupling between cultured pancreatic b-cells. Exp Cell Res 133, 421-30. Meda, P., Atwater, I., Goncalves, A., Bangham, A., Orci, L., and Rojas, E. (1984). The topography of electrical synchrony among beta-cells in the mouse islet of Langerhans. Q J Exp Physiol 69, 719-35. Meda, P. and Bosco, D. (2001). Communication of islet cells : Molecules and functions. In “molecular basis of endocrine pancreas development and function”, ed. By J. F. Habener & M. Husein, Kluwer academic publishers, 143-163. Meda, P., Bosco, D., Chanson, M., Giordano, E., Vallar, L., Wollheim, C., and Orci, L. (1990). Rapid and reversible secretion changes during uncoupling of rat insulin- producing cells. J Clin Invest 86, 759-68. Meda, P., Bruzzone, R., Chanson, M., Bosco, D. (1988). Junctional coupling and secretion of acinar cells. In Gap junction (Hertzberg E.L. and Johnson R.G., eds), Modern cell biology 7, Alan R. Liss, inc, NY, pp353-364, 1988.
58

Meda, P., Bruzzone, R., Chanson, M. and Bosco, D., Orci, L. (1987). Gap junctional coupling modulates secretion of exocrine pancreas. Proc Natl Acad Sci U S A 84, 4901-4. Meda, P., Bruzzone, R., Knodel, S., and Orci, L. (1986). Blockage of cell-to-cell communication within pancreatic acini is associated with increased basal release of amylase. J Cell Biol 103, 475-83. Meda, P., Chanson, M., Pepper, M., Giordano, E., Bosco, D., Traub, O., Willecke, K., el Aoumari, A., Gros, D., Beyer, E. C., and et al. (1991). In vivo modulation of connexin 43 gene expression and junctional coupling of pancreatic B-cells. Exp Cell Res 192, 469-80. Meda, P., Pepper, M. S., Traub, O., Willecke, K., Gros, D., Beyer, E., Nicholson, B., Paul, D., and Orci, L. (1993). Differential expression of gap junction connexins in endocrine and exocrine glands. Endocrinology 133, 2371-8. Meda, P., Perrelet, A., and Orci, L. (1979). Increase of gap junctions between pancreatic B-cells during stimulation of insulin secretion. J Cell Biol 82, 441-48. Meda, P., Perrelet, A., and Orci, L. (1984). Gap junctions and cell-to-cell coupling in endocrine glands. Modern cell biol 3, 131-196. Meda, P., Spray (2000). Gap junctions functions. Adv. Mol. Cell Biol 30, 263-322. Mege, R. M., Matsuzaki, F., Gallin, W. J., Goldberg, J. I., Cunningham, B. A., and Edelman, G. M. (1988). Construction of epithelioid sheets by transfection of mouse sarcoma cells with cDNAs for chicken cell adhesion molecules. Proc Natl Acad Sci U S A 85, 7274-8. Mehta, P. P., Bertram, J. S., and Loewenstein, W. R. (1986). Growth inhibition of transformed cells correlates with their junctional communication with normal cells. Cell 44, 187-96. Mehta, P. P., Hotz-Wagenblatt, A., Rose, B., Shalloway, D., and Loewenstein, W. R. (1991). Incorporation of the gene for a cell-cell channel protein into transformed cells leads to normalization of growth. J Membr Biol 124, 207-25. Meyer, D. J., Yancey, S. B., and Revel, J. P. (1981). Intercellular communication in normal and regenerating rat liver: a quantitative analysis. J Cell Biol 91, 505-23. Meyer, R. A., Laird, D. W., Revel, J. P., and Johnson, R. G. (1992). Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol 119, 179-89. Milks, L. C., Kumar, N. M., Houghten, R., Unwin, N., and Gilula, N. B. (1988). Topology of the 32-kd liver gap junction protein determined by site- directed antibody localizations. Embo J 7, 2967-75. Moore, L. K., and Burt, J. M. (1994). Selective block of gap junction channel expression with connexin- specific antisense oligodeoxynucleotides. Am J Physiol 267, C1371-80. Musil, L. S., Cunningham, B. A., Edelman, G. M., and Goodenough, D. A. (1990). Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol 111, 2077-88.
59

Musil, L. S., and Goodenough, D. A. (1991). Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 115, 1357-74. Musil, L. S., and Goodenough, D. A. (1993). Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 74, 1065-77. Naus, C. C., Bechberger, J. F., Zhang, Y., Venance, L., Yamasaki, H., Juneja, S. C., Kidder, G. M., and Giaume, C. (1997). Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. J Neurosci Res 49, 528-40. Nielsen, J. H., Svensson, C., Galsgaard, E. D., Moldrup, A., and Billestrup, N. (1999). Beta cell proliferation and growth factors. J Mol Med 77, 62-6. Oyamada, M., Krutovskikh, V. A., Mesnil, M., Partensky, C., Berger, F., and Yamasaki, H. (1990). Aberrant expression of gap junction gene in primary human hepatocellular carcinomas: increased expression of cardiac-type gap junction gene connexin 43. Mol Carcinog 3, 273-8. Oyoyo, U. A., Shah, U. S., and Murray, S. A. (1997). The role of alpha1 (connexin-43) gap junction expression in adrenal cortical cell function. Endocrinology 138, 5385-97. Pal, J. D., Liu, X., Mackay, D., Shiels, A., Berthoud, V. M., Beyer, E. C., and Ebihara, L. (2000). Connexin46 mutations linked to congenital cataract show loss of gap junction channel function. Am J Physiol Cell Physiol 279, C596-602. Patel, S., Rew, D. A., Taylor, I., Potten, C. S., Owen, C., and Roberts, S. A. (1993). Study of the proliferation in human gastric mucosa after in vivo bromodeoxyuridine labelling. Gut 34, 893-6. Paul, D. L. (1986). Molecular cloning of cDNA for rat liver gap junction protein. J Cell Biol 103, 123-34. Paul, D. L. (1995). New functions for gap junctions. Curr Opin Cell Biol 7, 665-72. Peinado, A., Yuste, R., and Katz, L. C. (1993). Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron 10, 103-14. Penn, R. D. (1966). Ionic communication between liver cells. J Cell Biol 29, 171-4. Perl, A. K., Dahl, U., Wilgenbus, P., Cremer, H., Semb, H., and Christofori, G. (1999). Reduced expression of neural cell adhesion molecule induces metastatic dissemination of pancreatic beta tumor cells. Nat Med 5, 286-91. Perl, A. K., Wilgenbus, P., Dahl, U., Semb, H., and Christofori, G. (1998). A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392, 190-3. Pick, A., Clark, J., Kubstrup, C., Levisetti, M., Pugh, W., Bonner-Weir, S., and Polonsky, K. S. (1998). Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 47, 358-64. Pitts, J. D., and Finbow, M. E. (1986). The gap junction. J Cell Sci Suppl 4, 239-66.
60

Porter, S. E., Sorenson, R. L., Dann, P., Garcia-Ocana, A., Stewart, A. F., and Vasavada, R. C. (1998). Progressive pancreatic islet hyperplasia in the islet-targeted, parathyroid hormone-related protein-overexpressing mouse. Endocrinology 139, 3743-51. Potten, C. S. (1992). The significance of spontaneous and induced apoptosis in the gastrointestinal tract of mice. Cancer Metastasis Rev 11, 179-95. Rane, S. G., Dubus, P., Mettus, R. V., Galbreath, E. J., Boden, G., Reddy, E. P., and Barbacid, M. (1999). Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 22, 44-52. Reaume, A. G., de Sousa, P. A., Kulkarni, S., Langille, B. L., Zhu, D., Davies, T. C., Juneja, S. C., Kidder, G. M., and Rossant, J. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831-4. Revel, J. P., and Karnovsky, M. J. (1967). Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol 33, C7-C12. Richard, G. (2000). Connexins: a connection with the skin. Exp Dermatol 9, 77-96. Richard, G., Brown, N., Smith, L. E., Terrinoni, A., Melino, G., Mackie, R. M., Bale, S. J., and Uitto, J. (2000). The spectrum of mutations in erythrokeratodermias--novel and de novo mutations in GJB3. Hum Genet 106, 321-9. Richard, G., Smith, L. E., Bailey, R. A., Itin, P., Hohl, D., Epstein, E. H., Jr., DiGiovanna, J. J., Compton, J. G., and Bale, S. J. (1998). Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet 20, 366-9. Richard, G., White, T. W., Smith, L. E., Bailey, R. A., Compton, J. G., Paul, D. L., and Bale, S. J. (1998). Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 103, 393-9. Robertson J.D.(1963). The occurrence of a subunit pattern in the unit membranes of club ending in mauthner cell synapses in goldfish brains. J cell Biol 19, 201-221. Rosenberg, L., Rafaeloff, R., Clas, D., Kakugawa, Y., Pittenger, G., Vinik, A. I., and Duguid, W. P. (1996). Induction of islet cell differentiation and new islet formation in the hamster--further support for a ductular origin. Pancreas 13, 38-46. Rozental, R., Srinivas, M., Gokhan, S., Urban, M., Dermietzel, R., Kessler, J. A., Spray, D. C., and Mehler, M. F. (2000). Temporal expression of neuronal connexins during hippocampal ontogeny. Brain Res Brain Res Rev 32, 57-71. Ruch, R. J. (1994). The role of gap junctional intercellular communication in neoplasia. Ann Clin Lab Sci 24, 216-31. Ruch, R. J. (2000). Role of gap junctions in cellular growth and neoplasia: evidence and mechanisms. Current Topics in membranes 49, 535-54. Ruch, R. J., Cesen-Cummings, K., and Malkinson, A. M. (1998). Role of gap junctions in lung neoplasia. Exp Lung Res 24, 523-39.
61

Ruch, R. J., Guan, X., and Sigler, K. (1995). Inhibition of gap junctional intercellular communication and enhancement of growth in BALB/c 3T3 cells treated with connexin43 antisense oligonucleotides. Mol Carcinog 14, 269-74. Saez, J. C., Gregory, W. A., Watanabe, T., Dermietzel, R., Hertzberg, E. L., Reid, L., Bennett, M. V., and Spray, D. C. (1989). cAMP delays disappearance of gap junctions between pairs of rat hepatocytes in primary culture. Am J Physiol 257, C1-11. Salomon, D., Masgrau, E., Vischer, S., Ullrich, S., Dupont, E., Sappino, P., Saurat, J. H., and Meda, P. (1994). Topography of mammalian connexins in human skin. J Invest Dermatol 103, 240-7. Salomon, D., and Meda, P. (1986). Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp Cell Res 162, 507-20. Salomon, D., Saurat, J. H., and Meda, P. (1988). Cell-to-cell communication within intact human skin. J Clin Invest 82, 248-54. Scaglia, L., Smith, F. E., and Bonner-Weir, S. (1995). Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 136, 5461-8. Serre-Beinier, V., Le Gurun, S., Belluardo, N., Trovato-Salinaro, A., Charollais, A., Haefliger, J. A., Condorelli, D. F., and Meda, P. (2000). Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 49, 727-34. Serre-Beinier V, Mas C, Calabrese A, Caton D, Bauquis J, Caille D, Charollais A, Cirulli V, Meda (2002). Connexins and secretion.Biol Cell. 94 , 477-92. Simon, A. M., Goodenough, D. A., Li, E., and Paul, D. L. (1997). Female infertility in mice lacking connexin 37. Nature 385, 525-9. Simpson, I., Rose, B., and Loewenstein, W. R. (1977). Size limit of molecules permeating the junctional membrane channels. Science 195, 294-6. Slack J.M.W.(1995) Developmental biology of the pancreas. Development 121, 1569-1580. Sosa-pineda B., Chowdhury K., Torres M., Oliver G., Gruss P.(1997). The pax4 gene is essential for differentiation of insulin-produccing β-cells in the mammalian pancreas.Nature 386, 399-401. Solis, R. R., Diven, D. G., and Trizna, Z. (2001). Vohwinkel's syndrome in three generations. J Am Acad Dermatol 44, 376-8. Spray, D. C., Harris, A. L., and Bennett, M. V. (1979). Voltage dependence of junctional conductance in early amphibian embryos. Science 204, 432-4. Spray, D. C., Stern, J. H., Harris, A. L., and Bennett, M. V. (1982). Gap junctional conductance: comparison of sensitivities to H and Ca ions. Proc Natl Acad Sci U S A 79, 441-5. Stagg, R. B., and Fletcher, W. H. (1990). The hormone-induced regulation of contact-dependent cell-cell communication by phosphorylation. Endocr Rev 11, 302-25.
62

Stefan, Y., Meda, P., Neufeld, M., and Orci, L. (1987). Stimulation of insulin secretion reveals heterogeneity of pancreatic B cells in vivo. J Clin Invest 80, 175-83. Steinberg, T. H., Civitelli, R., Geist, S. T., Robertson, A. J., Hick, E., Veenstra, R. D., Wang, H. Z., Warlow, P. M., Westphale, E. M., Laing, J. G., and et al. (1994). Connexin43 and connexin45 form gap junctions with different molecular permeabilities in osteoblastic cells. Embo J 13, 744-50. Stewart, W. W. (1978). Functional connections between cells as revealed by dye-coupling with a highly fluorescent naphthalimide tracer. Cell 14, 741-59. Subark-Sharpe J.H., Burk R.R. and Pitts J.D. Metabolic cooperation between Biochemically marked cells in tissue culture. (1969). J Cell Sci 4, 353-367. Subark-Sharpe J.H., Burk R.R. and Pitts J.D. Metabolic cooperation by cell-cell transfer between genetically different mammalian cells in tissue culture. (1966). Heredity 21, 342. Suchyna, T. M., Nitsche, J. M., Chilton, M., Harris, A. L., Veenstra, R. D., and Nicholson, B. J. (1999). Different ionic selectivities for connexins 26 and 32 produce rectifying gap junction channels. Biophys J 77, 2968-87. Swenne, I. (1983). Effects of aging on the regenerative capacity of the pancreatic B-cell of the rat. Diabetes 32, 14-9. Temme, A., Buchmann, A., Gabriel, H. D., Nelles, E., Schwarz, M., and Willecke, K. (1997). High incidence of spontaneous and chemically induced liver tumors in mice deficient for connexin32. Curr Biol 7, 713-6. Terada, T., Ohta, T., Kitamura, Y., Ashida, K., and Matsunaga, Y. (1998). Cell proliferative activity in intraductal papillary-mucinous neoplasms and invasive ductal adenocarcinomas of the pancreas: an immunohistochemical study. Arch Pathol Lab Med 122, 42-6. Trosko, J. E., Chang, C. C., Madhukar, B. V., and Klaunig, J. E. (1990). Chemical, oncogene and growth factor inhibition gap junctional intercellular communication: an integrative hypothesis of carcinogenesis. Pathobiology 58, 265-78. Upham, B. L., Weis, L. M., and Trosko, J. E. (1998). Modulated gap junctional intercellular communication as a biomarker of PAH epigenetic toxicity: structure-function relationship. Environ Health Perspect 106 Suppl 4, 975-81. Vasavada, R. C., Cavaliere, C., D'Ercole, A. J., Dann, P., Burtis, W. J., Madlener, A. L., Zawalich, K., Zawalich, W., Philbrick, W., and Stewart, A. F. (1996). Overexpression of parathyroid hormone-related protein in the pancreatic islets of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. J Biol Chem 271, 1200-8. Veenstra, R. D., Wang, H. Z., Beyer, E. C., and Brink, P. R. (1994). Selective dye and ionic permeability of gap junction channels formed by connexin45. Circ Res 75, 483-90. Veenstra, R. D., Wang, H. Z., Beyer, E. C., Ramanan, S. V., and Brink, P. R. (1994). Connexin37 forms high conductance gap junction channels with subconductance state activity and selective dye and ionic permeabilities. Biophys J 66, 1915-28.
63

Veenstra, R. D., Wang, H. Z., Westphale, E. M., and Beyer, E. C. (1992). Multiple connexins confer distinct regulatory and conductance properties of gap junctions in developing heart. Circ Res 71, 1277-83. Veenstra, R. D., Wang, H. Z., Westphale, E. M., and Beyer, E. C. (1992). Multiple connexins confer distinct regulatory and conductance properties of gap junctions in developing heart. Circ Res 71, 1277-83. Vozzi, C., Bosco, D., Dupont, E., Charollais, A., and Meda, P. (1997). Hyperinsulinemia-induced hypoglycemia is enhanced by overexpression of connexin 43. Endocrinology 138, 2879-85. Vozzi, C., Ullrich, S., Charollais, A., Philippe, J., Orci, L., and Meda, P. (1995). Adequate connexin-mediated coupling is required for proper insulin production. J Cell Biol 131, 1561-72. Vozzi, C. R., Rodriguez, A. O., Paolantonio, D., Smith, J. A., and Wholey, M. H. (1997). Extracranial carotid angioplasty and stenting. Initial results and short-term follow-up. Tex Heart Inst J 24, 167-72. Warner, A. E., Guthrie, S. C., and Gilula, N. B. (1984). Antibodies to gap-junctional protein selectively disrupt junctional communication in the early amphibian embryo. Nature 311, 127-31. Weidmann S. (1952). The electrical constants of purkinje fibers. J physiol 118:348-360. Weir, M. P., and Lo, C. W. (1982). Gap junctional communication compartments in the Drosophila wing disk. Proc Natl Acad Sci U S A 79, 3232-5. White, T. W. (2000). Functional analysis of human Cx26 mutations associated with deafness. Brain Res Brain Res Rev 32, 181-3. White, T. W., Goodenough, D. A., and Paul, D. L. (1998). Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol 143, 815-25. White, T. W., Paul, D. L., Goodenough, D. A., and Bruzzone, R. (1995). Functional analysis of selective interactions among rodent connexins. Mol Biol Cell 6, 459-70. Wilgenbus, K. K., Kirkpatrick, C. J., Knuechel, R., Willecke, K., and Traub, O. (1992). Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer 51, 522-9. Wilkin, T. J. (1998). Neogenesis of islet cells. Diabetes Metab Rev 14, 331-3. Willecke, K., Hennemann, H., Dahl, E., Jungbluth, S., and Heynkes, R. (1991). The diversity of connexin genes encoding gap junctional proteins. Eur J Cell Biol 56, 1-7. Wiszniewski, L., Limat, A., Saurat, J. H., Meda, P., and Salomon, D. (2000). Differential expression of connexins during stratification of human keratinocytes. J Invest Dermatol 115, 278-85. Woodbury J.W. , Crill W.E.(1961). On the problems of impulse conduction in the atrium. In the Nervous Inhibitions (E. Florey, Ed), Oxford, Pergamon Press, 124, 1961.
64

Wyllie, A. H., Kerr, J. F., and Currie, A. R. (1980). Cell death: the significance of apoptosis. Int Rev Cytol 68, 251-306. Yamasaki, H. (1990). Gap junctional intercellular communication and carcinogenesis. Carcinogenesis 11, 1051-8. Yamasaki, H., and Naus, C. C. (1996). Role of connexin genes in growth control. Carcinogenesis 17, 1199-213. Yamasaki, H., and Naus, C. C. (1996). Role of connexin genes in growth control. Carcinogenesis 17, 1199-213. Yancey, S. B., John, S. A., Lal, R., Austin, B. J., and Revel, J. P. (1989). The 43-kD polypeptide of heart gap junctions: immunolocalization, topology, and functional domains. J Cell Biol 108, 2241-54. Zimmer, D. B., Green, C. R., Evans, W. H., and Gilula, N. B. (1987). Topological analysis of the major protein in isolated intact rat liver gap junctions and gap junction-derived single membrane structures. J Biol Chem 262, 7751-63. Zhu D., Caveney S., Kidder I.G.M. and Nauss C.C.G.(1991). Transfection of C6 gliomacells with connexins43 cDNA : analysis of expression, intercellular coupling, and cell proliferation. Proc Natl Acad Sci USA 88,1883-1887.
65





























