
1
Large-scale Characterization of Drug Responses of Clinically Relevant Proteins
in Cancer Cell Lines
Wei Zhao1,2,#, Jun Li1,#, Mei-Ju Chen1, Zhenlin Ju1, Nicole K. Nesser3, Katie Johnson-Camacho3,
Christopher T. Boniface3, Yancey Lawrence3, Nupur T. Pande3, Michael A. Davies4, Meenhard
Herlyn5, Taru Muranen6, Ioannis Zervantonakis6, Erika Von Euw7, Andre Schultz1, Shwetha V.
Kumar1, Anil Korkut1, Paul T. Spellman3, Rehan Akbani1, Dennis J. Slamon7, Joe W. Gray8, Joan S.
Brugge6, Yiling Lu2, Gordon B. Mills9*, and Han Liang1,2*
1Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson
Cancer Center, Houston, TX 77030, USA 2Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston,
TX 77030, USA 3Department of Molecular and Medical Genetics, Oregon Health and Science University, Portland,
OR 97201, USA 4Department of Melanoma Medical Oncology, The University of Texas MD Anderson Cancer
Center, Houston, TX 77030, USA. 5Molecular and Cellular Oncogenesis Program, Wistar Institute, Philadelphia, PA 19104, USA 6Department of Cell Biology, Ludwig Center at Harvard, Harvard Medical School, Boston, MA
02115, USA 7Division of Hematology-Oncology, Department of Medicine, David Geffen School of Medicine,
University of California Los Angeles, Los Angeles, CA 90095, USA 8Center for Spatial Systems Biomedicine, Department of Biomedical Engineering, Oregon Health &
Science University, Portland, OR 97201, USA 9Knight Cancer Institute and Cell, Developmental and Cancer Biology, Oregon Health and Science
University, Portland, OR 97201, USA
#These authors contributed equally to this study
*Correspondence: H.L., [email protected] (lead contact) and G.B.M., [email protected]
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

2
Summary
Perturbation biology is a powerful approach to developing quantitative models of cellular behaviors
and gaining mechanistic insights into disease development. In recent years, large-scale resources for
phenotypic and mRNA responses of cancer cell lines to perturbations have been generated. However,
similar large-scale protein response resources are not available, resulting in a critical knowledge gap
for elucidating oncogenic mechanisms and developing effective cancer therapies. Here we generated
and compiled perturbed expression profiles of ~210 clinically relevant proteins in >12,000 cancer
cell-line samples in response to >150 drug compounds using reverse-phase protein arrays. We show
that integrating protein response signals substantially increases the predictive power for drug
sensitivity and aids in gaining insights into mechanisms of drug resistance. We build a systematic
map of protein-drug connectivity and develop an open-access, user-friendly data portal for
community use. Our study provides a valuable information resource for a broad range of quantitative
modeling and biomedical applications.
Highlights
A large collection of cancer cell line protein responses to drug perturbations
Perturbed protein responses greatly increase predictive power for drug sensitivity
Build a systematic map of protein-drug connectivity based on response profiles
Develop a user-friendly, interactive data portal for community use
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

3
Introduction
Cancer is a highly heterogeneous disease encompassing many tissue types and diverse oncogenic
drivers, with treatment responses that are often variable in distinct tumor contexts. Over the last
decade, extensive efforts have been made to characterize the tremendous heterogeneity of human
cancers at the molecular level (Berger et al., 2018; Hutter and Zenklusen, 2018; Jiang et al., 2019;
Liu et al., 2018; Taylor et al., 2018). A real challenge in cancer research, however, is to obtain a
systematic understanding of causality and mechanisms underlying the behaviors of cancer cells with
the eventual goal of improving patient outcomes (Wise and Solit, 2019). To address this challenge,
perturbation experiments provide a powerful approach in which cells are modulated by perturbagens,
and downstream consequences are monitored (Korkut et al., 2015; Molinelli et al., 2013; Ng et al.,
2018). The longitudinal data thus obtained provide considerably greater information content of both
the basal biological network wiring and its associated changes under stress, thereby leading to a
deeper understanding of mechanisms underlying cell survival under stress. Recently, large-scale
compendia of the phenotypic and cellular effects of perturbed cancer cell lines have been established.
For example, large-scale pharmacologic perturbation studies, cell viability measurement upon
different drug treatments across many cell lines, have been published (Barretina et al., 2012; Basu et
al., 2013; Garnett et al., 2012; Iorio et al., 2016); several studies have built genome-wide “cancer
dependency” maps across a large number of cell lines using loss-of-function siRNA, shRNA, or
CRISPR/cas9 screens (McDonald et al., 2017; Tsherniak et al., 2017); a “connectivity map” of
profiled mRNA responses of cancer cell lines to diverse perturbations using an efficient, robust RNA
measurement platform, L1000 has been developed (Subramanian et al., 2017). These studies provide
valuable resources for gaining a systems-level understanding of cancer mechanisms and phenotypes.
However, similar large-scale resources for analysis and integration of protein responses of perturbed
cancer cell lines have yet to be established. This knowledge gap is even more striking, considering
that proteins comprise the basic functional units in biological processes and represent the major
targets for cancer therapy.
To fill this gap, we generated and compiled a large compendium of perturbed protein expression
profiles of cancer cell lines in response to a diverse array of clinically relevant drugs using reverse-
phase protein arrays (RPPAs). As a quantitative antibody-based assay, RPPA can assess a large
number of protein markers in many samples in a cost-effective, sensitive, and reproducible manner
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

4
(Hennessy et al., 2010; Nishizuka et al., 2003; Tibes et al., 2006). We have applied this technology to
quantify protein expression levels of large patient cohorts (e.g., The Cancer Genome Atlas) (Akbani
et al., 2014; Zhang et al., 2017) and cancer cell lines (e.g., MD Anderson Cell Line project and Cancer
Cell Line Encyclopedia) (Ghandi et al., 2019; Li et al., 2017). The current antibody repertoire covers
key oncogenic pathways such as PI3K/AKT, RAS/MAPK, Src/FAK, TGFβ/SMAD, JAK/STAT,
DNA damage repair, Hippo, cell cycle, apoptosis, histone modification, and immune-oncology.
Compared with proteome-wide mass spectrometry approaches, our RPPA-based approach has
several advantages. First, although the number of protein markers in RPPA readout is much smaller
(~200), this highly select protein set is enriched in therapeutic targets and biomarkers, thereby greatly
increasing the ability to generate clinically relevant hypotheses and make translational impacts.
Statistically speaking, this more focused assessment also substantially reduces the burden of multiple
testing, a major challenge in identifying significant hits from unbiased proteomic searches. Second,
one RPPA slide can measure up to 1,000 samples simultaneously. Thus, the high-throughput and
cost-effectiveness make RPPA a practical platform for assessing a large number of samples
(e.g., >10,000), which is simply not feasible for alternative proteomic approaches. Third, protein-
level responses, particularly changes in post-translational modifications, more likely reflect how
cancer cells rewire their signaling pathways to adapt and survive a specific drug treatment, as most targeted
therapies act by modulating protein phosphorylation and activity. The superior ability of RPPA to
quantify some key post-translationally modified proteins has the potential to capture such adaptive
responses and can provide stronger predictors of therapy response or resistance mechanisms (Mertins
et al., 2014). Indeed, our recent studies have demonstrated the value of RPPA-based adaptive
responses in the rational design of combination therapies (Fang et al., 2019; Iavarone et al., 2019; Korkut
et al., 2015; Krepler et al., 2017; Krepler et al., 2016; Kwong et al., 2015; Molinelli et al., 2013; Muranen
et al., 2012; Sun et al., 2017; Sun et al., 2018), with several of these translated to the clinic
(NCT01623349, NCT03586661, NCT02208375, NCT02338622, NCT03162627; NCT03579316,
NCT03565991, NCT03801369, NCT03544125) with patient benefit.
Results
A large, high-quality collection of perturbed protein expression profiles of cancer cell lines
To generate a high-quality resource of perturbed protein responses, we measured RPPA-based protein
expression profiles of cancer cell lines in response to >150 preclinical and clinical therapeutics (often
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

5
across multiple time points), generated normalized RPPA data (including baseline level p0 and post-
treatment level p1) and protein response to perturbation (Δp = p1 - p0) profiles using a standardized
data processing pipeline, and made the data public through a user-friendly data portal (Figure 1A).
In total, this compendium contains RPPA profiles (~210 total and phosphorylated protein markers)
of 15,867 samples (12,222 drug-treated samples and 3,647 control samples). The cancer cell lines
come from several lineages, including breast, ovarian, uterus, skin, blood, and prostate; and the drug
compounds target a broad range of cancer-related processes, including PI3K/mTOR signaling,
ERK/MAPK signaling, RTK signaling, EGFR signaling, TP53 pathway, genome integrity, cell cycle,
antipsychotic drugs, and chromatin remodeling (Figure 1B). Due to time and cost constraints and the
clinical relevance of different drugs, instead of profiling all possible perturbations across all cell-line
and drug combinations, we took a more pragmatic approach in which some cell lineages and drug
groups were more frequently profiled but still represent an extensive survey of drug perturbations
(Figure 1B). Our sample set is highly enriched in responses from a subset of common, well-
characterized cancer cell lines that have rich molecular profiling and drug response data in public
resources (Figure 1C). For example, >1,500 drug-treated samples were from MCF7, and >250 drug-
treated samples were from BT20, SKBR3, MDA-MB-468, BT549, UACC812, BT474, SKOV3, and
HCC1954 (Figure S1A). For drug treatment, 86.2% of the samples were treated with monotherapy,
and ~1,700 samples were treated with double or triple-drug combinations (Figure S1B). Among the
drug compounds used, 23 compounds have >150 treated samples, with lapatinib (485 samples, HER2
inhibitor), GSK690693 (453 samples, AKT inhibitor), and AZD8055 (424 samples, mTOR inhibitor)
being the top three (Figure S1C). Importantly, for many of the therapeutic targets, we profiled
multiple targeting agents, including those that target different members of the same pathway, to
increase our ability to identify on-target activity. To assess overall data quality, we compared protein
response (Δp) correlations of technical replicate samples (n = 2,771 pairs) to those of randomly
selected sample pairs. We found that replicate samples showed much higher correlations across
protein markers (mean R = 0.87) than random pairs (mean R = 0.059) (Figure 1D), indicating high
reproducibility of our RPPA data.
To further confirm the quality of the RPPA data output, we sought to validate our protein response
data using independent mRNA response data from the connectivity map (Subramanian et al., 2017).
Since this analysis is for different molecules (protein vs. RNA) and across different platforms (RPPA
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

6
vs. L1000), we employed the Goodman-Kruskal’s gamma (ɣ) correlation to conduct a robust
assessment. Based on the same cell lines perturbed by the same compounds (n = 46 unique cell-line-
drug perturbations), we first converted the original continuous response scores into categorical
response groups (i.e., upregulated, neutral, and downregulated) and then compared the mRNA-
protein response concordance by calculating mRNA-protein response association and sample-sample
association (Figure 2A). We observed that the matched mRNA-protein responses from the same
condition were highly associated with each other (median ɣ = 0.63), which is significantly higher
than that from the randomly shuffled background distribution (paired Student’s t-test, p = 5.8×10-5,
Figure 2B). Then, we tested whether the sample-sample association inferred from the RPPA-based
protein responses were preserved in the L1000-based mRNA responses. Among the significant
sample-sample associations identified by either platform (FDR < 0.01), the RPPA-based ɣ scores
showed a strong, positive correlation with the L1000-based ɣ scores (Pearson’s correlation, R = 0.65,
p = 5×10-6). Further, categorized RPPA-based associations are highly consistent with L1000-based
associations (Fisher’s exact test, p = 2.3×10-3). These cross-molecule, cross-platform, and cross-study
comparisons strongly support a high quality of the protein response data.
Predictive power and mechanistic insights for drug sensitivity by protein responses
Our previous study demonstrated that RPPA-based baseline protein levels showed considerable
predictive power for drug sensitivity in cancer cell lines (Li et al., 2017). To assess the predictive
power of protein responses for drug sensitivity, we integrated our perturbed RPPA data and drug
sensitivity data available in GDSC (Iorio et al., 2016) and identified seven drugs whose sensitivity
and protein expression data were available in at least six different cell lines. Then, for each drug, we
defined three types of protein markers that may be informative about drug sensitivity: (i) p0: the
baseline expression of a protein shows a significant correlation with the sensitivity to the drug across
cell lines (Pearson correlation, p < 0.05); (ii) Δp only: the protein response shows a significant
correlation with drug sensitivity (Pearson correlation, p < 0.05); and (iii) Δp|p0: given p0, the protein
response shows additional information content in predicting drug sensitivity (see STAR Methods).
Across all the drugs, the numbers of Δp-informative (Δp only + Δp|p0) protein markers were
significantly higher than those of p0-based markers (paired t-test, p = 1.38×10-3, n = 7 drugs, Figure
3A). We next focused on two representative drugs, pictilisib (PI3K inhibitor), and talazoparib (PARP
inhibitor), with markedly different targets, for which a large number of cell lines have drug sensitivity
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

7
data (Iorio et al., 2016; Seashore-Ludlow et al., 2015). We found that across cell lines, baseline
proteins (p0) and protein response (Δp) showed distinct sets of proteins whose levels significantly
correlated with drug sensitivity (Pearson correlation, p < 0.05), respectively; and there were
additional proteins where Δp correlated with drug sensitivity when considering the information
content of p0 (Δp|p0). When combining Δp and Δp|p0, the number of informative protein responses
increased dramatically compared to baseline protein levels: pictilisib, from 17 to 27; and talazoparib,
33 to 52 (Figure 3B, 3C). Considering the potential noise of drug sensitivity data, we further
validated this pattern, either using independent public drug sensitivity data or generating in-house
drug sensitivity data (Figure S2). The correlations of baseline protein levels or protein responses
with drug sensitivity between the different resources are highly correlated, despite the assessment of
independent cell line sets (Figure 3D, Figure S2). These results not only further support the high
quality of the RPPA expression data, but also suggest that changes in protein levels on therapeutic
challenge provide substantial additional information content beyond that provided by baseline protein
levels for predicting treatment responses.
To demonstrate how protein response could help elucidate drug resistance mechanisms and suggest
therapy combinations, we focused on MEK inhibitors (MEKi), using cobimetinib as an illustration
example and considering both baseline (p0) and protein response levels (Δp) (Figure 4). Cell lines
were divided into MEKi-resistant (OVCAR432: RAS pathway WT, OVCAR3: RAS pathway WT,
and OAW28: MAP2K4 mutant) and MEKi-sensitive (OV90: BRAF mutant, CAOV3: RAS pathway
WT, ES2: BRAF and MEK mutant, OVCAR5: KRAS mutant, JHOM1: RAS pathway WT, and
OVCAR8: KRAS mutant) based on response to multiple MEK inhibitors in our and publicly available
data (CTRPv2 and GDSC). As expected, cell lines with aberrations in the RAS/MAPK pathway have
a higher propensity to RAS/MAPK baseline pathway activity and sensitivity to MEKi, as indicated
by low BIM and high EGFR, DUSP4, transglutaminase, pYB1, p90RSK, pMAPK, pMEK, and pJun
(Pohl et al., 2005) (Figure 4A). There was also a suggestion that cell state and, particularly, decreased
epithelial characteristics, or epithelial-mesenchymal transition (EMT) (low E-cadherin, beta-catenin,
RAB25, ERalpha, GATA3, high EPPK1, N-cadherin, AXL, PAI-1, and fibronectin) were associated
with sensitivity to MEKi. The EMT characteristics were likely mediated, at least in part, by effects
of the RAS/MAPK pathway activation noted above (Shao et al., 2014).
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

8
Adaptive responses (Δp) to cobimetinib demonstrated a greater dynamic range in terms of sensitivity
and resistance to MEKi than baseline data. Sensitivity to cobimetinib was associated with evidence
for a greater cobimetinib-induced decrease in RAS/MAPK pathway activity (decreased DUSP4,
transglutaminase, FOXM1, p90RSK, pMAPK, pYB1, pS6, and pJun, and increased BIM), and
decreased cell cycle progression (decreased pRB, cyclinB1 CDK1, PLK1, cdc25c, and CHK1, and
increased p16, p21, and p27), likely as a consequence of RAS/MAPK signaling inhibition (Figure
4B). Further, there was a marked shift to an epithelial phenotype, as indicated by increased EMA,
EPPK1, Claudin1, and beta-catenin (Figure 4B). Many of the associations with sensitivity to
cobimetinib were identifiable in the pre-treatment samples, with the associations markedly
accentuated and extended in cobimetinib-treated samples. The marked increase in BIM in response
to MEKi has been identified previously and provides a biomarker for response to combined inhibition
of MEKi and BCL2 family members (Cragg et al., 2008; Iavarone et al., 2019). We also performed
a similar analysis using trametinib (Figure S3) and observed a marked overlap of potential
biomarkers despite the analysis of different cell lines and different MEK inhibitors. Importantly, the
key adaptive pathway-level changes associated with drug sensitivity include cell cycle inhibition in
sensitive cell lines (t-test, p = 4.1 ×10-4, Figure 4C) and PI3K/Akt signaling activation in resistant
cell lines (t-test, p = 0.015, Figure 4C). Together, the results argue that (i) sensitivity to RAS/MAPK
pathway inhibition is associated with baseline pathway activity and cell state, and (ii) adaptive
responses to RAS/MAPK pathway inhibition in resistant cells could be overcome by PI3K inhibitors
(although the toxicity of combinations of RAS/MAPK and PI3K pathway inhibitors has to be
considered).
A systematic “protein-drug” connectivity map
To systematically evaluate the utility of our protein response data, we built a protein-drug
connectivity map based on the RPPA data. In this map, each node represents a protein or a drug,
protein-drug connections are based on whether the drug treatment caused a significant change of the
protein, and drug-drug connections are based on whether the two drugs caused similar protein
responses (Figure 5A). As expected, drugs for the same target are clustered together: for example,
several MEK inhibitors and mTOR/PI3K inhibitors are highly connected, highlighting their similar
downstream protein responses. This map also identifies novel connections: a PARP inhibitor showed
both similar and opposite relationships with some drugs, suggesting potential additive or agonistic
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

9
effects that could direct the development of rational drug combinations. Indeed, based on assessment
of functional proteomics changes as assessed by RPPA, we have validated synergistic activity of
PARP inhibitors and inhibition of PI3K pathway, MEK, ATR, and WEE1 inhibitors in preclinical
and clinical studies (Fang et al., 2019; Shen et al., 2015; Sun et al., 2017; Sun et al., 2018).
We next studied protein-protein relationships in the map. For any given drug treatment, we classified
proteins into perturbed proteins and other proteins. We found that perturbed proteins are more likely
to interact than other proteins, based on the STRING database (Szklarczyk et al., 2019) (t-test, p =
3.2 ×10-6), suggesting that proteins co-perturbed by a drug tend to be involved in the same biological
processes and to interact as part of a signaling cascade (Figure 5B). This global assessment using
prior protein interaction knowledge supports the utility of the approach to drive biological discoveries.
Using drug-centered protein neighborhoods, we initially focused on signaling through tyrosine
kinases and their downstream networks: selumetinib (target: MEK) (Figure 6A), AZD8055 (target:
mTOR) (Figure 6B), GSK1838705A (target: IGF1R/ALK) (Figure 6C), and sapatinib (target:
EGFR/ERBB2) (Figure 6D), and demonstrated a marked overlap in protein networks in inhibitor-
perturbed cells. Interestingly, the Hsp90 inhibitor (gamitrinib) protein neighborhood (Figure S4A),
demonstrated similarities to that of the tyrosine kinase pathway inhibitors, potentially due to a role
of Hsp90 in stabilizing multiple members of the tyrosine kinase signaling pathway. Indeed, the
similarities in the protein networks argue that the major effects of Hsp90 are likely attributable to its
effects on tyrosine kinase signaling pathways (Lee et al., 2017). In contrast, rabusertib (target: Chk1)
(Figure S4B), and chlorpromazine (target: autophagy) (Figure S4C) demonstrated distinct protein
neighborhoods consistent with markedly different mechanisms of action.
As described above, the MEKi protein neighborhood is strongly associated with signaling through
the MAPK and mTOR pathways, cell cycle progression, and cell state. There is also a strong
association with apoptotic balance (BIM, BAX, and MCL1). Based on extensive validation of the
relationships between these pathways and RAS/MAPK signaling, the association with multiple other
proteins in the neighborhood map in Figure 6A are likely valid. Given that the MAPK pathway is a
key regulator of the TSC1/2 complex that is upstream of mTORC1 signaling, it is not surprising that
the protein neighborhood of mTOR inhibitor AZD8055 (Figure 6B) is highly related to the
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

10
selumetinib protein neighborhood. The most marked differences between the MEK and mTOR
inhibitor protein neighborhoods are represented in the upper components of the PI3K and MAPK
pathway that appear relatively independent of each other. Interestingly, the IGF1R/ALK inhibitor,
GSK1838705A, protein neighborhood encompasses components of both the MEK and mTOR
protein neighborhoods, consistent with the IGF1R having input into both pathways. While the strong
link to the PI3K pathway was expected, the link between the IGF1R and MAPK pathway has been
less studied. The pan-EGFR family inhibitor, sapatinib, neighborhood reflects EGFR family receptors
being the key regulators of the PI3K and MAPK pathways in epithelial cells (Akbani et al., 2014).
The EGFR family has a stronger link than either mTOR or MEK inhibitors to the DNA damage repair
pathway (i.e., 53BP1, Rad50, XRCC1, pChk1/2, and BRCA2), consistent with recent studies (Russo
et al., 2019; Wang et al., 2013).
A user-friendly data portal for protein responses of perturbed cell lines
To facilitate the utilization of our protein response data by a broad biomedical community, we
provided unrestricted access to the data through a user-friendly portal, called “Cancer Perturbed
Proteomics Atlas” for fluent data exploration and analysis, which can be accessed at
http://bioinformatics.mdanderson.org/main/:CPPAOverview. The data portal provides four
interactive modules: “Data Summary,” “My Protein,” “Connectivity Map,” and “Analysis” (Figure
7i). The “Data Summary” module provides detailed information about each sample (including cell
line, compound, dose, time, and culture conditions) (Figure 7ii). The datasets can be easily
downloaded through a tree-view interface. “My protein” module provides annotation of RPPA
protein markers, including the corresponding genes, and antibody information (Figure 7iii). The
“Connectivity Map” provides an interactive approach to exploring the map, through which protein-
drug and drug-drug connectivity can be examined through different visual and layout styles (Figure
7iv). The “Analysis” module provides three common analyses through which users can explore
protein responses associated with a drug/compound, including protein response (Δp) rank (Figure
7v), volcano plots for the correlations between protein responses and drug sensitivity (Figure 7vi),
and box plots for differential protein responses between sensitive and resistant cell lines (Figure
7vii). Collectively, this effort provides a valuable platform that enables researchers to explore,
analyze, and visualize RPPA-based protein response data intuitively and efficiently.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

11
Discussion
Understanding the functional consequences of drug treatment is central to identifying patients who
will likely benefit from specific therapies and to develop effective personalized combination cancer
therapies. Here we present a large collection of protein responses (including total and post-
translationally modified proteins) upon drug treatments (>12,000 treated samples) using RPPA, the
same platform employed for protein profiling of TCGA patient samples and CCLE cell line samples.
Our dataset is several magnitudes larger than previously published studies, and for such a resource,
data quality is key. We validated the quality of our datasets in several ways. First, we demonstrated
the high reproducibility of technical replicate samples using the same platform. Second, we
established a high consistency between RPPA-based protein responses and the independently
generated L1000 mRNA responses to the same perturbation conditions. Third, we validated
observed correlations of protein responses with public drug sensitivity data using newly generated
drug sensitivity data on independent cell line sets. Finally, the quality of our dataset is also
supported by the meaningful patterns observed on the systematic “protein-drug” connectivity map,
such as the clustering of similar drugs and higher node connectivity of perturbed proteins annotated
in the STRING protein interaction database. Our study represents a unique, high-quality
compendium of protein responses of cancer cell lines to a diversity of compound perturbations
available for use by the wider community.
By establishing the connections between a core set of proteins and drug treatment in simplified well-
controlled cell line models, the utility of our protein response dataset is several-fold. First, our dataset
provides a basis for understanding cause-effect relationships, which is complementary to correlation
analyses and associations that can be obtained from patient cohorts. Based on these data, it will be
possible to develop quantitative predictive models of how signaling networks function in intact
cellular systems. Second, we show that while there is information content in biomarkers at baseline,
the information content is markedly increased when baseline and response signals are combined. This
is predicted by systems biology and engineering precepts, wherein, perturbed systems contain more
information than static analysis. Biomarkers designed to select treatment using baseline data
frequently have a limited power to predict benefit, and our results suggest that adaptive protein
responses after initial treatment could be highly informative in terms of treatment response, and clinical
benefit. Further clinical investigations are warranted to assess the potential benefit gains using such a
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

12
strategy. Third, since protein responses reflect how cancer cells critically rewire their signaling pathways to
survive and adapt to the stress of a specific drug treatment, these protein signals provide a strong basis for
rational design of combination therapies as we have demonstrated previously (Iavarone et al., 2019; Krepler
et al., 2017; Krepler et al., 2016; Kwong et al., 2015; Molinelli et al., 2013; Muranen et al., 2012; Sun et al.,
2017; Sun et al., 2018).
We recognize some limitations of this study. First, compared with mass spectrometry-based protein
level or mRNA level readout assays, the number of protein markers that can be effectively monitored
by the RPPA technology is much smaller. However, the increased sensitivity (particularly for some
key proteins and phosphoproteins), and cost considerations, make RPPA a practical platform for
generating such a large resource. In capturing protein responses, RPPA and mass spectrometry are
complementary because of their different scopes and focuses. Second, although many perturbed
protein response profiles were generated, some cell lines and drug treatments (including different
dosages) are still sparsely sampled. Further efforts are required to obtain more compensative sets;
moreover, machine learning approaches may have the potential to fill some of these gaps. Finally, as
with other high-throughput technologies, there can be technical measurement errors for individual
samples, and interesting observations from our study should be followed by further in-depth
investigations.
We have provided an interactive, user-friendly data portal through which biomedical researchers can
explore, visualize, and intuitively analyze these data. With this bioinformatics tool, we expect an
effective translation of the large-scale perturbed protein data into biological knowledge and clinical
utility. Together with recent efforts that have systematically characterized phenotypic and molecular
responses to drug treatment, our study provides a rich resource for the research community to
investigate the behaviors of cancer cells and the dependencies of treatment responses.
Acknowledgements
This study was supported by the National Institutes of Health (U01CA168394, U24CA143883,
U54HG008100, P50CA098258, and P50CA217685 to G.B.M., U24CA209851 and U01CA217842
to H.L. and G.B.M., U24CA210950 and U24CA210949 to R.A. and G.B.M., and Cancer Center
Support Grant P30CA016672), a kind gift from the Sheldon and Miriam Adelson Medical Research
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

13
Foundation, Susan G Komen (SAC110052), Ovarian Cancer Research Foundation (545152), and
Breast Cancer Research Foundation (BCRF-18-110) to G.B.M., a DoD/CDMRP (W81XWH-16-1-
0237) to R.A., and the Lorraine Dell Program in Bioinformatics for Personalization of Cancer
Medicine (to H.L.). We thank Kamalika Mojumdar for editorial assistance. RA is a bioinformatics
consultant for the University of Houston. G.B.M. is on the Scientific Advisory Board or a consultant
for AstraZeneca, ImmunoMet, Lilly, Nuevolution, PDX Pharmaceuticals, Symphogen, and
Tarveda, has stock options with Catena Pharmaceuticals, Immunomet, Signalchem, and Tarveda, and
has licensed technology to Myriad and Nanostring. H.L. is a shareholder and scientific advisor of
Precision Scientific Ltd.
Author Contributions
G.B.M. and H.L. conceived of the project. W.Z., J.L., M.C., J.Z., A.S., S.V.K., A.K., R.A., G.B.M.,
and H.L. contributed to the data analysis. N.K.N., K.J.C., C.T.B, Y. Lawrence, N.T.P., M.A.D., M.H., T.M.,
I.Z., E.V.E., P.T.S., D.J.S., J.S.B., J.W.G., Y. Lu and G.B.M. contributed to the experiments. M.C. and
J.L. implemented the web portal. W.Z., J.L., M.C., G.B.M., and H.L. wrote the manuscript, with input
from other authors. H.L. supervised the whole project.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

14
STAR methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled
by the Lead Contact, Han Liang ([email protected]).
Experimental Model and Subject Details
Cell lines
We collected cancer cell lines through the MD Anderson Cancer Center (MDACC) CCSG supported
Cell Line Characterization Core Facility (Houston, TX, USA) and from several outside collaborations.
All cell lines prepared at MDACC were confirmed by short tandem repeat (STR) analysis in the core
per institutional policy, and the outside collaborators also routinely confirmed cell lines by STR
analysis.
RPPA experiments
Cell line samples were prepared, and antibodies were validated as previously described
(Hennessy et al., 2010; Li et al., 2017). RPPA data were generated by the RPPA core facility at
MDACC. RPPA slides were first quantified using ArrayPro (Media Cybernetics) to generate signal
intensities (level 1), then processed by SuperCurve to estimate the relative protein expression level
(level 2), and were then normalized by median polish (level 3). RPPA slide quality was assessed by
a quality control classifier (Ju et al., 2015), and only slides above 0.8 (range: 0-1) were retained for
further analysis. In total, we generated RPPA data from 15,867 samples, including 12,222 treated cell
line samples and 3,647 baseline samples (e.g., treated by DSMO) from 8 batches. Through the
comparison of RPPA signals between post-treatment (p1) and baseline samples (p0), we generated
8,111 unique protein response (Δp) profiles after combining replicate samples.
Comparison of RPPA-based protein response and L1000-based mRNA response
To validate our RPPA perturbation data, we downloaded the level-5 data of L1000 phase 1 from the
GEO database (GSE92742). For a fair comparison, we collected data from the same cell lines
perturbed by the same compound. In total, 46 “perturbation-cell-line” IDs (60 samples) and 316
genes/proteins (total proteins only) commonly shared by the two platforms were used in the
subsequent analyses. For a perturbation-cell-line ID with multiple concentration and/or time points,
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

15
we adopted the median value across all conditions as the representative response score. As shown in
Figure 2A, for each platform, we first converted the continuous response to a categorical response:
up-regulated, down-regulated, or neutral. Random events were defined by the global median 35%
quantile, calculated from the full matrix. Next, we excluded the random events and computed
Goodman-Kruskal’s gamma to estimate sample associations across genes. We evaluated the
concordance between RPPA and L1000 platforms through two analyses. (i) Protein-mRNA response
associations: for each sample, a gamma (ɣ) association between the two platforms was computed
across genes/proteins when at least 12 genes showed up-/down-regulation. To generate the
background distribution, we randomly shuffled protein labels and computed the response associations
between the shuffled proteins and mRNAs (the seed used for randomization is “1234”). Then, a paired
Student’s t-test was used to evaluate the statistical significance of the group difference between the
real and matched randomly shuffled responses. (ii) Sample-sample associations: in our RPPA dataset,
a perturbation-cell-line ID might have replicate samples. Here, we only retained the one with the best
protein-mRNA response association from the previous analysis. Next, for samples that showed up-
/down-regulation of >3 genes, gamma associations for every two such samples were computed within
each platform (within the same batch). Then, Pearson’s correlation between the significant gramma
associations (FDR < 0.01 for each platform) was used to evaluate the consistency between the protein
and mRNA responses.
Analysis of predictive protein markers of drug sensitivity
We collected drug sensitivity data from two databases: GDSC and CTRPv2. For validation, an in-
house drug screening data set was generated for selected compounds. RPPA perturbation data of the
same cell line treated with the same compound at different dosages or time points were averaged
using mean values. The baseline level (p0) and protein response levels (Δp) were tested for
associations with drug sensitivity (IC50 or AUC score) in univariate linear models. The joint markers
(Δp|p0) were defined as the predictions of linear regression models, including both baseline and
protein response for specific antibodies. Predictive markers were selected at a significance level of p
= 0.05. To identify the differential protein markers of drug sensitivity and resistance, cell lines were
classified as sensitive or resistant to a specific drug based on the consensus call of CTRPv2, GDSC,
and in-house datasets. Baseline levels (p0) and protein response levels (Δp) with a significant
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

16
difference between sensitive and resistant cell lines were identified by Student’s t-test. Pathway-level
scores (Li et al., 2017) were similarly analyzed.
Construction of a drug-protein connectivity map
The association of each drug-protein pair was assessed by testing the difference of protein expression
between baseline (p0) and post-treatment level (p1) based on the paired t-test across cell lines. For
each drug-drug pair, we used Goodman-Kruskal’s gamma to calculate the associations, as described
in the comparison between RPPA-based protein response and L1000-based mRNA response data.
The significantly correlated drug-protein and drug-drug pairs (FDR < 0.1) were used to construct a
global drug-protein connectivity map. In the connectivity map, proteins were grouped and colored by
their related protein functional pathways, and drugs were grouped and colored by their targeted genes
or pathways. For each drug, the network densities were calculated for the two subsets of proteins: (i)
proteins significantly differentially expressed between p0 and p1 (perturbed proteins), and (ii) other
proteins (neutral proteins). The network density D of a protein subset with size N was defined as a
ratio of the number of protein-protein interactions (E) to the number of all possible protein pairs
(𝐸𝑚𝑎𝑥 = (𝑁2)), i.e., 𝐷 = 𝐸/𝐸𝑚𝑎𝑥 . Protein-protein interaction information was obtained from the
STRING database (Szklarczyk et al., 2019). A paired Wilcoxon test was performed to assess the
difference of the network densities between the perturbed and other proteins of all the drugs. For
Figure 6, the examples of drug-centered connectivity maps were generated separately with colored
edges (red: up-regulated in post-treatment; blue: down-regulated in post-treatment). The edge widths
were proportional to the differential expression between baseline (p0) and post-treatment levels (p1).
All network views were generated by the Rcy3 library and Cytoscape (Otasek et al., 2019; Shannon
et al., 2003).
Data portal development
All RPPA, mRNA expression, and drug sensitivity data accompanying the pre-calculated analytic
results were stored in a CouchDB database. We generated all the analytic results in R before
loading them into the database. We implemented a user-friendly and interactive web interface
in JavaScript. Specifically, tabular results were generated by DataTables, box, and scatter plots were
generated by HighCharts, and interactive network views were implemented by Cytoscape.js library.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

17
References
Akbani, R., Ng, P.K., Werner, H.M., Shahmoradgoli, M., Zhang, F., Ju, Z., Liu, W., Yang, J.Y.,
Yoshihara, K., Li, J., et al. (2014). A pan-cancer proteomic perspective on The Cancer Genome
Atlas. Nat Commun 5, 3887.
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A.A., Kim, S., Wilson, C.J.,
Lehar, J., Kryukov, G.V., Sonkin, D., et al. (2012). The Cancer Cell Line Encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature 492, 290-290.
Basu, A., Bodycombe, N.E., Cheah, J.H., Price, E.V., Liu, K., Schaefer, G.I., Ebright, R.Y.,
Stewart, M.L., Ito, D., Wang, S., et al. (2013). An interactive resource to identify cancer genetic
and lineage dependencies targeted by small molecules. Cell 154, 1151-1161.
Berger, A.C., Korkut, A., Kanchi, R.S., Hegde, A.M., Lenoir, W., Liu, W., Liu, Y., Fan, H., Shen,
H., Ravikumar, V., et al. (2018). A Comprehensive Pan-Cancer Molecular Study of Gynecologic
and Breast Cancers. Cancer Cell 33, 690-705 e699.
Cragg, M.S., Jansen, E.S., Cook, M., Harris, C., Strasser, A., and Scott, C.L. (2008). Treatment of
B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3
mimetic. J Clin Invest 118, 3651-3659.
Fang, Y., McGrail, D.J., Sun, C., Labrie, M., Chen, X., Zhang, D., Ju, Z., Vellano, C.P., Lu, Y.,
Li, Y., et al. (2019). Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity
while Maintaining Efficacy. Cancer Cell 35, 851-867 e857.
Garnett, M.J., Edelman, E.J., Heidorn, S.J., Greenman, C.D., Dastur, A., Lau, K.W., Greninger,
P., Thompson, I.R., Luo, X., Soares, J., et al. (2012). Systematic identification of genomic markers
of drug sensitivity in cancer cells. Nature 483, 570-575.
Ghandi, M., Huang, F.W., Jane-Valbuena, J., Kryukov, G.V., Lo, C.C., McDonald, E.R., Barretina,
J., Gelfand, E.T., Bielski, C.M., Li, H., et al. (2019). Next-generation characterization of the
Cancer Cell Line Encyclopedia. Nature 569, 503-+.
Hennessy, B.T., Lu, Y., Gonzalez-Angulo, A.M., Carey, M.S., Myhre, S., Ju, Z., Davies, M.A.,
Liu, W., Coombes, K., Meric-Bernstam, F., et al. (2010). A Technical Assessment of the Utility
of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected
Human Breast Cancers. Clin Proteomics 6, 129-151.
Hutter, C., and Zenklusen, J.C. (2018). The Cancer Genome Atlas: Creating Lasting Value beyond
Its Data. Cell 173, 283-285.
Iavarone, C., Zervantonakis, I.K., Selfors, L.M., Palakurthi, S., Liu, J.F., Drapkin, R., Matulonis,
U.A., Hallberg, D., Velculescu, V.E., Leverson, J.D., et al. (2019). Combined MEK and BCL-
2/XL Inhibition Is Effective in High-Grade Serous Ovarian Cancer Patient-Derived Xenograft
Models and BIM Levels Are Predictive of Responsiveness. Mol Cancer Ther 18, 642-655.
Iorio, F., Knijnenburg, T.A., Vis, D.J., Bignell, G.R., Menden, M.P., Schubert, M., Aben, N.,
Goncalves, E., Barthorpe, S., Lightfoot, H., et al. (2016). A Landscape of Pharmacogenomic
Interactions in Cancer. Cell 166, 740-754.
Jiang, Y.Z., Ma, D., Suo, C., Shi, J., Xue, M., Hu, X., Xiao, Y., Yu, K.D., Liu, Y.R., Yu, Y., et al.
(2019). Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and
Treatment Strategies. Cancer Cell 35, 428-440 e425.
Korkut, A., Wang, W., Demir, E., Aksoy, B.A., Jing, X., Molinelli, E.J., Babur, O., Bemis, D.L.,
Onur Sumer, S., Solit, D.B., et al. (2015). Perturbation biology nominates upstream-downstream
drug combinations in RAF inhibitor resistant melanoma cells. Elife 4.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

18
Krepler, C., Sproesser, K., Brafford, P., Beqiri, M., Garman, B., Xiao, M., Shannan, B., Watters,
A., Perego, M., Zhang, G., et al. (2017). A Comprehensive Patient-Derived Xenograft Collection
Representing the Heterogeneity of Melanoma. Cell Rep 21, 1953-1967.
Krepler, C., Xiao, M., Sproesser, K., Brafford, P.A., Shannan, B., Beqiri, M., Liu, Q., Xu, W.,
Garman, B., Nathanson, K.L., et al. (2016). Personalized Preclinical Trials in BRAF Inhibitor-
Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin
Cancer Res 22, 1592-1602.
Kwong, L.N., Boland, G.M., Frederick, D.T., Helms, T.L., Akid, A.T., Miller, J.P., Jiang, S.,
Cooper, Z.A., Song, X., Seth, S., et al. (2015). Co-clinical assessment identifies patterns of BRAF
inhibitor resistance in melanoma. J Clin Invest 125, 1459-1470.
Lee, H., Saini, N., Parris, A.B., Zhao, M., and Yang, X. (2017). Ganetespib induces G2/M cell
cycle arrest and apoptosis in gastric cancer cells through targeting of receptor tyrosine kinase
signaling. Int J Oncol 51, 967-974.
Li, J., Zhao, W., Akbani, R., Liu, W.B., Ju, Z.L., Ling, S.Y., Vellano, C.P., Roebuck, P., Yu, Q.H.,
Eterovic, A.K., et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase
Protein Arrays. Cancer Cell 31, 225-239.
Liu, Y., Sethi, N.S., Hinoue, T., Schneider, B.G., Cherniack, A.D., Sanchez-Vega, F., Seoane, J.A.,
Farshidfar, F., Bowlby, R., Islam, M., et al. (2018). Comparative Molecular Analysis of
Gastrointestinal Adenocarcinomas. Cancer Cell 33, 721-735 e728.
McDonald, E.R., 3rd, de Weck, A., Schlabach, M.R., Billy, E., Mavrakis, K.J., Hoffman, G.R.,
Belur, D., Castelletti, D., Frias, E., Gampa, K., et al. (2017). Project DRIVE: A Compendium of
Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi
Screening. Cell 170, 577-592 e510.
Mertins, P., Yang, F., Liu, T., Mani, D.R., Petyuk, V.A., Gillette, M.A., Clauser, K.R., Qiao, J.W.,
Gritsenko, M.A., Moore, R.J., et al. (2014). Ischemia in tumors induces early and sustained
phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol
Cell Proteomics 13, 1690-1704.
Molinelli, E.J., Korkut, A., Wang, W., Miller, M.L., Gauthier, N.P., Jing, X., Kaushik, P., He, Q.,
Mills, G., Solit, D.B., et al. (2013). Perturbation biology: inferring signaling networks in cellular
systems. PLoS Comput Biol 9, e1003290.
Muranen, T., Selfors, L.M., Worster, D.T., Iwanicki, M.P., Song, L., Morales, F.C., Gao, S., Mills,
G.B., and Brugge, J.S. (2012). Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-
attached cancer cells. Cancer Cell 21, 227-239.
Ng, P.K., Li, J., Jeong, K.J., Shao, S., Chen, H., Tsang, Y.H., Sengupta, S., Wang, Z., Bhavana,
V.H., Tran, R., et al. (2018). Systematic Functional Annotation of Somatic Mutations in Cancer.
Cancer Cell 33, 450-462 e410.
Nishizuka, S., Charboneau, L., Young, L., Major, S., Reinhold, W.C., Waltham, M., Kouros-Mehr,
H., Bussey, K.J., Lee, J.K., Espina, V., et al. (2003). Proteomic profiling of the NCI-60 cancer cell
lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A 100,
14229-14234.
Otasek, D., Morris, J.H., Boucas, J., Pico, A.R., and Demchak, B. (2019). Cytoscape Automation:
empowering workflow-based network analysis. Genome Biol 20, 185.
Pohl, G., Ho, C.L., Kurman, R.J., Bristow, R., Wang, T.L., and Shih Ie, M. (2005). Inactivation of
the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous
tumors with KRAS or BRAF mutations. Cancer Res 65, 1994-2000.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

19
Russo, M., Crisafulli, G., Sogari, A., Reilly, N.M., Arena, S., Lamba, S., Bartolini, A., Amodio,
V., Magri, A., Novara, L., et al. (2019). Adaptive mutability of colorectal cancers in response to
targeted therapies. Science 366, 1473-1480.
Seashore-Ludlow, B., Rees, M.G., Cheah, J.H., Cokol, M., Price, E.V., Coletti, M.E., Jones, V.,
Bodycombe, N.E., Soule, C.K., Gould, J., et al. (2015). Harnessing Connectivity in a Large-Scale
Small-Molecule Sensitivity Dataset. Cancer Discov 5, 1210-1223.
Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T., Ramage, D., Amin, N., Schwikowski,
B., and Ideker, T. (2003). Cytoscape: a software environment for integrated models of
biomolecular interaction networks. Genome Res 13, 2498-2504.
Shao, D.D., Xue, W., Krall, E.B., Bhutkar, A., Piccioni, F., Wang, X., Schinzel, A.C., Sood, S.,
Rosenbluh, J., Kim, J.W., et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor
survival. Cell 158, 171-184.
Shen, J., Peng, Y., Wei, L., Zhang, W., Yang, L., Lan, L., Kapoor, P., Ju, Z., Mo, Q., Shih Ie, M.,
et al. (2015). ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to
PARP Inhibitors. Cancer Discov 5, 752-767.
Subramanian, A., Narayan, R., Corsello, S.M., Peck, D.D., Natoli, T.E., Lu, X., Gould, J., Davis,
J.F., Tubelli, A.A., Asiedu, J.K., et al. (2017). A Next Generation Connectivity Map: L1000
Platform and the First 1,000,000 Profiles. Cell 171, 1437-1452 e1417.
Sun, C., Fang, Y., Yin, J., Chen, J., Ju, Z., Zhang, D., Chen, X., Vellano, C.P., Jeong, K.J., Ng,
P.K., et al. (2017). Rational combination therapy with PARP and MEK inhibitors capitalizes on
therapeutic liabilities in RAS mutant cancers. Sci Transl Med 9.
Sun, C., Yin, J., Fang, Y., Chen, J., Jeong, K.J., Chen, X., Vellano, C.P., Ju, Z., Zhao, W., Zhang,
D., et al. (2018). BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction
of Homologous Recombination Deficiency. Cancer Cell 33, 401-416 e408.
Szklarczyk, D., Gable, A.L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., Simonovic, M.,
Doncheva, N.T., Morris, J.H., Bork, P., et al. (2019). STRING v11: protein-protein association
networks with increased coverage, supporting functional discovery in genome-wide experimental
datasets. Nucleic Acids Res 47, D607-D613.
Taylor, A.M., Shih, J., Ha, G., Gao, G.F., Zhang, X., Berger, A.C., Schumacher, S.E., Wang, C.,
Hu, H., Liu, J., et al. (2018). Genomic and Functional Approaches to Understanding Cancer
Aneuploidy. Cancer Cell 33, 676-689 e673.
Tibes, R., Qiu, Y., Lu, Y., Hennessy, B., Andreeff, M., Mills, G.B., and Kornblau, S.M. (2006).
Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of
primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512-2521.
Tsherniak, A., Vazquez, F., Montgomery, P.G., Weir, B.A., Kryukov, G., Cowley, G.S., Gill, S.,
Harrington, W.F., Pantel, S., Krill-Burger, J.M., et al. (2017). Defining a Cancer Dependency Map.
Cell 170, 564-+.
Wang, Y., Yuan, J.L., Zhang, Y.T., Ma, J.J., Xu, P., Shi, C.H., Zhang, W., Li, Y.M., Fu, Q., Zhu,
G.F., et al. (2013). Inhibition of both EGFR and IGF1R sensitized prostate cancer cells to radiation
by synergistic suppression of DNA homologous recombination repair. PLoS One 8, e68784.
Wise, H.C., and Solit, D.B. (2019). Precision Oncology: Three Small Steps Forward. Cancer Cell
35, 825-826.
Zhang, Y., Kwok-Shing Ng, P., Kucherlapati, M., Chen, F., Liu, Y., Tsang, Y.H., de Velasco, G.,
Jeong, K.J., Akbani, R., Hadjipanayis, A., et al. (2017). A Pan-Cancer Proteogenomic Atlas of
PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 31, 820-832 e823.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

20
Figure Legends
Figure 1. Summary of the perturbed RPPA profiling data in this study
(A) Overview of the RPPA profiling experiments and data processing of cell line perturbations.
The pie chart shows the lineage distribution of cancer cell lines (n = 319) profiled. (B) The
distribution of drug-treated samples by cell lineages and drug groups. The bar plots show the
numbers of samples profiled for each lineage or drug group, and the size of the circle is
proportional to the number of samples profiled for each lineage-drug combination. (C) The circles
show the number of cell lines used in this study that were profiled by GDSC and CCLE. (D)
Reproducibility of perturbed RPPA data based on technical replicates (n = 2,771 pairs). See also
Figure S1.
Figure 2. Comparison between the RPPA-based protein responses and the L1000-based
mRNA responses
(A) Overview of the comparison method (see STAR Methods for details). (B) Boxplots of protein-
mRNA response associations between the RPPA and L1000 platforms using the same
perturbations (e.g., the same cell line and the same compound, n = 46). The gamma associations
from the real responses (green box) were compared to those from the randomly shuffled
background distribution (grey box). The P-value is based on a paired Student’s t-test. The vertical
line in the box is the median, the bottom, and top of the box are the first and third quartiles, and
the whiskers extend to 1.5 IQR of the lower quartile and the upper quartile, respectively. (C)
Scatter plot showing the correlation of sample-sample gamma associations from the RPPA (x-axis)
and L1000 (y-axis) platforms. Only significant data points (gamma associations) with FDR < 0.01
in either platform are shown. Pearson’s correlation coefficient and associated P-value are shown.
Figure 3. Comparison of the predictive power of protein markers for drug sensitivity
(A) Summary of predictive markers based on baseline protein expression (p0) and protein response
(Δp) using drug response data from GDSC. Given a specific drug, three types of predictive markers
were identified: (i) proteins whose p0 level is significantly correlated with drug sensitivity; (ii)
proteins whose Δp level is significantly correlated with drug sensitivity; and (iii) proteins whose
Δp level is significantly correlated with drug sensitivity given the p0 contribution. These protein
markers were identified based on both Δp only, and Δp|p0, and are called Δp shared. (B, C)
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

21
Heatmaps showing different types of protein markers for GDC0941/pictilisib (B) and
BMN673/talazoparib (C). In each panel, the heatmap shows Pearson’s correlation coefficients of
different protein levels with sensitivity to the drug of interest; the bar plot shows the number of
predictive protein markers in each category. The protein markers of baseline (p0) and protein
response (Δp) were selected at a significance level of p = 0.05 in univariate correlation. The joint
protein markers, labeled as “Δp|p0,” represent linear regression models, including both p0 and Δp
levels for a specific protein. The coefficients in the heatmap for joint markers indicate the
correlation of the prediction value of the linear model with the real value of drug sensitivity. The
proteins identified by Δp only or “Δp|p0” are marked by Δp total. (D) The scatter plots summarize
the comparisons of Pearson’s correlation coefficients of drug sensitivity and baseline level (p0,
left) as well as protein response (Δp, right) using two independent data sets. See also Figure S2.
Figure 4. Differentially expressed protein markers between cobimetinib-sensitive and
resistant cell lines
(A, B) Heatmaps showing baseline (A) and perturbed protein expressions (B) with significant
differences between sensitive and resistant cell lines (q < 0.05). Each protein marker is annotated
by whether it is a dual marker (i.e., significant both in p0 and Δp), BCL-2 family member, or
belongs to a specific pathway. (C) Cartoon summary of baseline protein levels and adaptive protein
responses to MEK inhibitors between the two cell groups. The difference of pathway scores
between the two groups was assessed based on a Student’s t-test. See also Figure S3.
Figure 5. A “drug-protein” connectivity map based on protein response signals
(A) A global view of the drug-protein connectivity map with highlighted examples of drug-drug
correlation networks (i.e., MEK inhibitors, mTOR, PI3K inhibitors, and neighboring drugs of a
PARP inhibitor). Red/blue edges represent positive/negative drug-drug correlations, respectively.
Proteins were grouped and colored by their related functional pathways. The drugs were grouped
and colored by their targeted genes or pathways. (B) Comparison of node connectivity between
perturbed and neutral proteins in the protein interaction network. P-value was computed based on
a paired Wilcoxon test.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

22
Figure 6. Examples of drug-centered protein connectivity maps
(A-D) Drug-protein connectivity maps for individual drugs: selumetinib (A), AZD8055 (B),
GSK1838705A (C), and sapitinib (D). In the network view, the edge color indicates the response
direction of protein markers (red/blue: up/down-regulated in post-treatment). The proteins (nodes)
of the same functional pathways are highlighted in the same colors. See also Figure S4.
Figure 7. A snapshot of the data portal for the perturbed RPPA data
The CPPA portal (i) contains four modules: the “Summary” module (ii); the “My protein” module
(iii); the “Network Visualization” module (iv); and the “Analysis” module, which offers protein
perturbation analysis (v), correlation analysis of drug and protein response (vi), and differential
expression analysis of protein response in sensitive and resistant cell lines (vii).
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

23
Supplementary Figures
Figure S1. Summary of the perturbed RPPA profiling data in this study, related to Figure 1.
(A) Cell lines associated with the largest sample sizes. (B) Sample numbers by different types of
compound perturbations. (C) Compounds associated with the largest sample sizes.
Figure S2. Confirmation of predictive protein markers for selected drugs, related to Figure
3
(A-C) Pictilisib (drug response data from CTRPv2) and (D) talazoparib (drug response data from
in-house experiments). The analysis was based on drug sensitivity data independent from Figure
3. (A, D) The heatmaps show Pearson’s correlation coefficients of protein level and drug
sensitivity. The bar plots show the number of predictive protein markers in each category. The
protein markers of baseline (p0) and protein response (Δp) were selected at the significance level
of p = 0.05 in the univariate correlation test. The joint protein markers, labeled as “Δp|p0,” are the
linear regression models of baseline and protein response levels for specific proteins. The
coefficients in the heatmap for joint markers indicate the correlation between the prediction of the
linear model and the drug sensitivity. Δp total is defined as the union of “Δp only” and “Δp given
p0.” (B, C) The scatter plots summarize the comparison of the Pearson’s correlation coefficients
of drug sensitivity and (B) baseline level (p0) as well as (C) protein response (Δp) in the two
independent data sets.
Figure S3. Differentially expressed proteins between trametinib-sensitive and -resistant cell
lines, related to Figure 4
(A, B) Heatmaps showing baseline protein expression (A), and perturbed protein response (B) with
a significant difference between the sensitive and resistant cell lines (q < 0.05).
Figure S4. Examples of drug-centered protein connectivity maps, related to Figure 6
(A-C) Drug-protein connectivity maps for individual drugs: gamitrinib (A), rabusertib (B), and
chlorpromazine (C). In the network views, the edge color indicates the response direction of
protein markers (red/blue: up/down-regulated in post-treatment). The proteins (nodes) of the same
functional pathways are highlighted in the same colors.
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

AFigure 1
C
B
D
150+ compounds
Cell cultureRPPA profiling
Level 1 Level 2 Level 3
L3 p1 p0
∆p
-
This study(N=319)
GDSC(N=78)
CCLE(N=92)
771 15
Breast
Skin
Ovary
Blood
Other
Prostate
Uterus
PI3K/M
TOR si
gnali
ngOthe
r
ERK MAPK si
gnali
ng
RTK sign
aling
Genom
e inte
grity
Chromati
n
EGFR sign
aling
Antips
ycho
tic
Cell cy
cleHsp
90
Other, k
inase
s
Mitosis
p53 p
athway
Num
ber o
f sa
mpl
es
032102432768
Number of samples
Number of samples
0
32
1024
0 32 1024
Breast(56%)
Skin(23%)
Ovary(12%)
Blood(5%)
Other(2%)
Prostate(1%)
Uterus(1%)
Molecularly characterizedcell lines
CPPA portal
PI3K/MTORERK MAPKRTK
Genome integrity...
−1.0 −0.5 0.0 0.5 1.0Pearson's R
Freq
uenc
y
0.871
−1.0 −0.5 0.0 0.5 1.0
0.0592
ReplicatesRandom pairs
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

B C
RPPA (protein reponse)
L1000(mRNA reponse)
Protein-mRNA response association Sample-sample association
Platform A
Figure 2
Down-regulation Neutral Up-regulation
Down-regulation
Neutral
Up-regulation
Categorical responseGoodman-Kruskal's gamma association
RPPA
L100
0
Protein-mRNA response association
Sample-sample association
RPPA L1000
Samples Samples
Pro
tein
s
Gen
es
GammaRPPA L1000
Samples Samples
Pro
tein
s
Gen
es
Gamma Gamma
Signi�cant in both
p = 5.8 x 10-5 R = 0.65, p = 5 x 10-6
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

Figure 3
A
Talazoparib (PARP1/2)Cp0 only
∆p only
∆p | p0
∆p total
p21
Myosin
−IIa_
pS19
43
ATM_p
S1981
Myosin
−11
p38−
MAPK_pT18
0_Y18
2TFRC
E−Cad
herin
b−Cate
nin_p
T41_S
45CD31
Rad50
E2F1PMS2
ATRJA
B1Syk
Chk1_
pS34
5
FoxM
1
XRCC1Gys
Rb_pS
807_
S811ARHI
Heregu
lin
PDGFR−b
GSK3_pS
9
Cox−IVPA
L1PCNA
Caveo
lin−1
Caspa
se−8
Cyclin
−E1
PDK1_pS
241CD29
14−3
−3−e
psilo
ncIA
P
14−3
−3−b
eta DJ1
UBAC1
Cyclin
−D1
TIGAR Lc
kPDK1
PRAS40_p
T246
Rictor_
pT11
35
Paxillin
FoxO3a
_pS31
8_S32
1
4E−B
P1_pT
37_T
46
Stathm
in p27
Annex
in−VII
Akt_pT
308 Rb
PKC−alph
a_pS
657TSC1
PKC−alph
a
4E−B
P1_pS
65IR
S1
Akt_pS
473
0 20 40
33
24
29
52
P−Cad
herin
FoxM1
Annex
in-1
Bad_p
S112
Src_pY
416
Glutam
inase
Tyro3
p21
Notch1
UBAC1
FRA−1p3
8−a
ACC1_PS79
Rictor
G6PD
SF2
Myosin
−11
Cyclop
hilin−
FeIF
4E
Myosin
−IIa_
pS19
43
Elk1_p
S383SCD1
Transg
lutam
inase
AMPK−alph
a
DM−Hist
one−
H3
PDK1_pS
241PDK1
HER3
ER−alph
a Rb
Annex
in-VII
Rad50
53BP1
ATM_pS19
81Smac
0 10 20
Number of predictors
17
10
18
27
p0 only
∆p only
∆p | p0
∆p total
Pictilisib (PI3K)B
Correlationcoefficient
00.20.40.60.81
Correlationcoefficient
−1−0.500.51
PathwaysApoptosisCell cycleCore reactiveDNA damage response
PI3K/AktRas/MAPKRTKTSC/mTOR
Hormone a
EMT
∆p markers
GSK3−alp
ha−b
eta_p
S21_S
9
DTalazoparib p0 Talazoparib ∆p
Pearson’s correlation (GDSC) Pearson’s correlation (GDSC)
Pea
rson
’s c
orre
latio
n (M
DA
CC
)
R=0.725p=1.12x10-36
Pea
rson
’s c
orre
latio
n (M
DA
CC
)
-1.0 -0.5 0 0.5 1.0 -1.0 -0.5 0 0.5 1.0
1.0
0.5
0
-0.5
-1.0
1.0
0.5
0
-0.5
-1.0
R=0.616p=4.75x10-24
Number of predictors
GSK2118436 GSK690693 MK2206
p0 ∆p p0 ∆p p0 ∆p p0 ∆p p0 ∆p p0 ∆p p0 ∆p0
10
20
30
40
50N
umbe
r of m
arke
rsProtein markers
p0 only∆p only∆p | p0∆p shared
Dactolisib Olarparib Pictilisib Talazoparib
p = 1.38x10-3
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

Res
ista
ntS
ensi
tive
dual markerspathway
BCL2 family
Baseline level
−4−2024
Protein response
−2−1012
PathwaysApoptosisCell cycleDNA damage response
PI3K/AktRas/MAPKRTK
Dual markersBaseline and response
BCL2 familyBCL−2 family
Figure 4A
C
Baseline level (p0)
BProtein response (∆p)
Res
ista
ntS
ensi
tive
dual markerspathway
BCL2 family
OVCA432OVCAR3OAW28OV90CAOV3ES2OVCAR5JHOM1OVCAR8
OVCA432OVCAR3OAW28OV90CAOV3ES2OVCAR5JHOM1OVCAR8
Syk
IGFB
P2
Bim
ATR
XE
−Cad
herin
Sta
t3R
ab25
HE
R2
Chk
1G
ysG
ATA
3S
tat3
_pY
705
VH
L−E
PP
K1
IR−b
Gys
_pS
641
ER
−alp
haD
M−H
isto
ne−H
3W
IPI2
Wee
1Fo
xO3a p2
1C
hk2
Sm
ad1
Pdc
d4be
ta−C
aten
inm
TOR
Cyc
lin−D
3N
otch
1C
reb
CA
SPA
SE
7_C
LEAV
ED
HE
R2_
pY12
48P
MS
2IG
F1R
_pY
1135
_Y11
36c−
IAP
2LC
3A−B
Myt
1P
RE
X1
CD
K1_
pY15
Vim
entin
TSC
2TF
AM
RS
KP
RA
S40
_pT2
46D
−a−T
ubul
inS
nail
4−O
cteE
F2K
STA
T5−a
lpha
Cyc
lin−D
1S
CD
1AC
C1
PK
C−d
elta
_pS
664
Cox
−IV
CD
26R
ab11
GC
LMC
ox2
Cla
udin
−7E
NY
2C
ME
T_P
Y12
34_Y
1235
PLK
1E
lk1_
pS38
3C
aspa
se−8
MS
H6
WIP
I1Ja
gged
1B
AP
1A
RH
ES
1A
nnex
in−V
IIS
ox2
Bax
Myo
sin−
11X
RC
C1
PE
A−1
5A
tg7
b−A
ctin
Sm
ad3
RIP
ER
CC
514
−3−3
−zet
aP
KC
BE
TAII_
PS
660
Akt
MM
P2
LRP
6_pS
1490
Ric
tor
PK
A−a
Glu
tam
inas
eeI
F4G
CD
49b
Cyc
lin−B
1c−
Myc
Rad
50U
GT1
AN
DR
G1_
pT34
6D
J1VA
SP
p53
XPA
CD
29A
MP
K−a
lpha
c−Ju
n_pS
73M
EK
1_pS
217_
S22
1N
−Cad
herin
PD
−L1
NF−
kB−p
65_p
S53
6M
IG6
PD
GFR
−bFo
xM1
BR
D4
cdc2
5CIG
F1R
−bM
AP
K_p
T202
_Y20
4C
yclo
phili
n−F
IRS
1p9
0RS
K_p
T573
YB
1_pS
102
Glu
tam
ate−
D1−
2M
ER
LIN
Myo
sin−
IIa_p
S19
43E
GFR
Rb_
pS80
7_S
811
FRA
−1M
CT4
Fibr
onec
tinP
KC
−alp
ha_p
S65
7D
US
P4
AM
PK
a_pT
172
CD
44Tr
ansg
luta
min
ase
PAI−
1A
xl
DU
SP
4C
DK
1R
b_pS
807_
S81
1P
LK1
p90R
SK
_pT5
73C
yclin
−B1
FoxM
1M
AP
K_p
T202
_Y20
4Y
B1_
pS10
2FR
A−1
Tran
sglu
tam
inas
ecd
c25C
Chk
14E
−BP
1H
ES
1PA
RP
1TG
IF2
eIF4
ELR
P6_
pS14
90S
6_pS
235_
S23
6C
DK
1_pY
15eI
F4E
_pS
209
Elk
1_pS
383
C−R
afAT
R_p
S42
8E
GFR
ND
UFB
4Au
rora
−BP
RA
S40
_pT2
46C
ox2
MS
H6
c−Ju
n_pS
73A
RID
1AB
−Raf
_pS
445
Bad
_pS
112
AM
PK
−alp
haJN
K2
BR
D4
RPA
32U
BAC
1P
CN
AeE
F2K
YAP
_pS
127
Sta
thm
inE
RC
C5
IRS
1eI
F4G
p27_
pT19
8W
ee1
TRIM
25B
AP
1IN
PP
4bIG
FBP
2P
I3K
−p11
0−al
pha
mTO
RC
D29
D−a
−Tub
ulin
Ric
tor_
pT11
35TS
C1
MC
T4C
reb
53B
P1
Mnk
1E
R−a
lpha
WIP
I2YA
Pp1
6IN
K4a
Gra
nzym
e−B
GAT
A3
Ann
exin
−1G
6PD
TSC
2be
ta−C
aten
inC
yclin
−E1
VE
GFR
−2 p27
Her
egul
inIR
−b p21
ULK
1_pS
757
Cla
udin
−7FA
K_p
Y39
7S
HP
−2_p
Y54
2C
D17
1S
OD
2V
HL−
EP
PK
1P
DG
FR−b
EM
AB
im
EMT
�
���
��
�
�
�
�
�
�
�
�
�
��
�
�
�
�
��
�
�
�
�
−0.5
0.0
0.5
1.0
resistant
sensitive
Ras
/MA
PK
pa
thw
ay s
core
p=3.79x10−6
��
�
��
�
�
�
�
�
�
�
��
�
�
�
�
��
�
�
�
�
�
�
�
−1.0
−0.5
0.0
0.5
1.0
sensitive
EM
T p
athw
ay s
core
Drug responseresistantsensitive
p=9.56x10−9
Cell cycle inhibition in sensitive lines
Sensitive
Resistant
Ras/MAPK high
EMT high(Mesenchymal)
Ras/MAPK low
EMT low(Epithelial)
MEKi treatment
Ras/MAPK
�
�
�
�
��
�
�
�
�
�
�
�
�
�
��
�
�
�
�
�
�
�
��
�
−0.4
0.0
0.4
resistant
sensitive
Ada
ptiv
e P
I3K
/Akt
pa
thw
ay s
core
(∆p)
p = 1.50x10−2
�
�
�
��
��
�
�
�
�
�
�
�
�
�
�
�
�
�
��
�
�
�
�
�
−1.5
−1.0
−0.5
0.0
0.5
resistant
sensitive
Ada
ptiv
e ce
ll cy
cle
path
way
sco
re (∆
p)
p=4.10x10−4
PI3K/Akt activation in resistant lines
p0 (base level) ∆p (adaptive response)
resistant
+
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

PTCH
Tau
PD-L1
Src_pY416
Gab2
Jak2
Porin
GCN5L2
IGF1R_pY1135_Y1136
Src_pY527
Notch1
Mre11
Syk
BRCA2
NF2
Chk1_pS345
ATM
IGF1RXPF
TFF1C-Raf
RPA32_pS4_S8
Cyclophilin-DRPA32
PI3K-p110-alphaGranzyme-B
CD26 EMA
Connexin-43Cyclophilin-FLck
RSK
Cytokeratin-19PKC-delta_pS664
S6
Chk2_pT68
XRCC1
XBP1IGF1R-b
Rad51
AIB1
53BP1
Rad50
p53
eIF4E
4E-BP1
b-Act in
ERCC1
eIF4G
P-Cadherin
PI3K-p85
E-Cadherin
Claudin-7
Caveolin-1
beta-Catenin
SHP-2_pY542
Stat3_pY705
Stat3
RBM15
PI3K-p110-b
Stathmin
Hif-1-alpha
p38_MAPKATRXChk1
Aurora-B
SF2
H2AX_pS140
p38-MAPK_pT180_Y182c-Met_pY1235
PDK1
P38
PDK1_pS241
Chk1_pS296
p38-a
E2F1 WIPI1
PARP-cleaved
14-3-3-ze ta
PARP1
eEF2
eIF4E_pS209
ERCC5
PAR
c-Jun_pS73
MEK1_pS217_S221
Shc_pY317 YB1_pS102
p90RSK_pT359_S363
MAPK_pT202_Y204
JNK_pT183_Y185
C-Raf_pS338
ACVRL1
TSC2Myosin-11
Rb
Rb_pS807_S811
TSC2_PT1462
CD29
S6_pS240_S244
mTOR_pS2448
S6_pS235_S236
TAZ_PS89
4E-BP1_pT70
Rictor_pT1135
TAZ
4E-BP1_pT37_T46
4E-BP1_pS65
p70-S6K_pT389
CD31
MEK2
MEK1
b-Catenin_pT41_S45
Src
IRS1
SCD1
FASN
Smac
HER3
PAK1
CASPASE7_CLEAVEDWee1
FAK_pY397AIF
Pdcd4
CASPASE9_CLEAVED FAKComplex-II-SubunitPDGFR-b
Bim
Bid
cIAP
Bax
Bak Bcl-xL
Bad_pS112
c-Ki t
PDH
Cox-IV
Wee1_pS642
CASPASE3_ACTIVE
MGMT
Caspase-3SLC1A5
SOD1
A-Raf
YAP
YAP_pS127
UQCRC2 ATP5A
ATP_SYNTHASE_SUBUNIT_D NDUFB4
Glutamate-D1-2
CD49b Glutaminase Tyro3
TUFMp90RSKHER2_pY1248
PAI-1
Jagged1
NFKB-P65
HES1
CD44
FRA-1
c-IAP2
cdc25C LC3A-BLRP6_pS1490
TTF1
p90RSK_pT573PKM2
MERIT40_pS29 Puma
PLC-gamma2_pY759
PDHK1
c-Abl
RIP
SETD2
PKA-a
Twist
Ubq-Histone-H2B
Sox2
YB1
KRAS
Rab11
VEGFR-2
N-Ras
PKCBETAII_PS660
TFAM
TIGAR
Smad3
SOD2
Mcl-1
NF-kB-p65_pS536
STAT5-alpha
Cox2
c-Myc
DUSP4
Smad4
Rab25Snail
UGT1A
P70S6K
MSH6
TGIF2
Slfn11
UBIQUITIN_CLONE_P4D1-A11 eEF2K
MSI2
WIPI2
VHL-EPPK1
IR-b
Mitochondria
Rheb
Myt1
NAPSIN-A
ULK1_pS757
XPA
Annexin-1 FoxO3a
PYGM
p27
MIF
BiP-GRP78
HER2MIG6
NDRG1_pT346
Akt_pT308
PTEN
Akt_pS473
PRAS40_pT246GSK3_pS9
GSK3-alpha-beta_pS21_S9
AMPK-a2_pS345
PAK4
Raptor
HER3_pY1289
GSK-3B
PKC-pan_BetaII_pS660
Mnk1
GSK-3b_pS9
EGFR_pY1173
PRAS40
CXCR4
EGFRPEA-15_pS116
Gys_pS641MMP2
PEA-15
Ets-1GAPDH
PKC-alpha_pS657
Gys
Hexokinase-II
EGFR_PY992
Transglutaminase
LDHA
FoxO3a_pS318_S321
Myosin-IIa_pS1943
PKC-alpha
14-3-3-be ta
ACC1_PS79
AMPKa_pT172
ACC1
JNK2
GSK3-alpha-beta
HYDIN Beclin
DJ1
Annexin-VII
HMGB1
HIAPFoxM1
Chk2
PAICS
PMS2
TRIM25
MSH2
p16INK4a
BMX
Creb
Coup-TFII
ATP5H
ARID1A
Atg3
4-Oct
ENY2
ZAP-70
CD171p21 GPBB
Cyclin-D3p44-42-MAPK
ATM_pS1981
AMPK-alpha
DM-K9-Histone-H3
BAP1
COG3
Dvl3
Elk1_pS383
Histone-H3
ER-alpha_pS118
DM-Histone-H3ARHIAtg7
GATA3
ARB7-H4BRD4
Heregulin
AktUBAC1
SDHA
MDM2_pS166MCT4
Notch3
GCLM
Pilaralisib
GSK2118436
KA2237
PI3Kinhibitor-05
PI3Kinhibitor-04
PDK1inhibitor-03
Vemurafenib
Gamitrinib
GSK690693
MEKinhibitor-06
AKTinhibitor-04
MK2206
MEKinhibitor-07
Trametinib PD0325901
MEKinhibitor-01
Fulvestrant
Cobimetinib
MEKinhibitor-08
BETinhibitor-01
Selumetinib
Paclitaxel
AZD8055
PARPinhibitor-02
Dactolisib
MTORinhibitor-01
Rabusertib
MTORinhibitor-02
RAPAMYCINOlaparib
Talazoparib
DasatinibChlorpromazine
KA1463
HA130
Carboplatin
Gleevec
Lapatinib
Gefitinib
6-THIO-DG
Sapitinib
Smad1
XIAP
mTORBcl2
B-Raf
B-Raf_pS445
INPP4b
FAKinhibitor-02
Vimentin
METinhibitor-01
IGFBP2
PD173074
Perphenazine
FASinhibitor-01
COMPLEX_II_SUBUNIT_RIESKE
GNE493
Adavosertib
Pictilisib
GSK1838705A
IGFR
Omipalisib
COMPLEX_II_SUBUNIT_CORE2
Voxtalisib
WEE1inhibitor-01
CDK1Cyclin-D1
Cyclin-E1 p27_pT198
p27_pT157
Cyclin-B1
PCNA
TRFC
HSP27_pS82
ATR
ATR_pS428
TFRC
HSP27
JAB1
HSP70
Fibronectin
alpha-Catenin
N-Cadherin
Collagen-VI
VASP
MERLIN
14-3-3-epsi lon
TSC1
Rock-1
PREX1
CDK1_pY15
D-a-Tubul in
Rictor
Paxillin
PLK1ER-alpha
PR
ADAR1
IRF-1
Caspase-8
c-Met
CMET_PY1234_Y1235 Axl
IGFBP5 G6PD
Figure 5A
Selumetinib
MEKinhibitor-01
Trametinib
MEKinhibitor-06
PD0325901
MEKinhibitor-08
MEKinhibitor-07 Cobimetinib
Voxtalisib
GNE493
Pictilisib
Omipalisib
DactolisibRAPAMYCIN
KA2237
PI3Kinhibitor-04
MTORinhibitor-01
MTORinhibitor-02
Pilaralisib
AZD8055
Trametinib
Paclitaxel
Talazoparib
WEE1inhibitor-01
Gleevec
Chlorpromazine
GSK1838705A
Cobimetinib
Carboplatin
FAKinhibitor-02
PARPinhibitor-02
Adavosertib
Dactolisib
PARPi and neighboring compoundsmTOR, PI3K inhibitorsMEK inhibitors
B
+
Perturbedproteins
Other proteins
0.2
0.4
0.6
0.8
1.0
p−value: 3.2x10−6
Perturbed proteins Other proteins
Nod
e co
nnec
tivity
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

Figure 6
A
B
C
D
GSK1838705A (IGF1R/ALK)
Sapitinib (EGFR, ERBB2)
Selumetinib (MEK)Stathmin
MMP2CASPASE9_CLEAVED
G6PD
PTEN
A-Raf
TSC2
MEK2
D-a-Tubul inHER2_pY1248
beta-Catenin
HER3_pY1289
Caveolin-1
Puma
Porin
14-3-3-ze ta
SOD2
HER2
PDGFR-b
RSK
Stat3_pY705
Bax
Rad51
PKCBETAII_PS660
Chk1_pS296
NDRG1_pT346
Chk1
MIG6
4E-BP1
Hexokinase-II
GCN5L2
PLK1
FRA-1
Mcl-1MDM2_pS166
AMPKa_pT172
SELUMETINIBN-Ras
ATR
MSH6
P70S6K
p27_pT198c-Myc
FoxM1
STAT5-alpha
PKC-alpha
HSP27
SDHA
p70-S6K_pT389
4E-BP1_pS65
CDK1
Rictor_pT1135
Cyclin-B1
S6_pS235_S236
S6_pS240_S244PKC-alpha_pS657SF2
VEGFR-2
DJ1
CD31
Complex-II-Subunit
p27
eIF4G
DUSP4Rb_pS807_S811
MEK1_pS217_S221
YB1_pS102
c-Jun_pS73MAPK_pT202_Y204
Aurora-B
p90RSK_pT573
Hif-1-alpha
Gys_pS641
ATP5A
Annexin-1
c-MeteEF2K
AZD8055 (mTOR)
alpha-Catenin
Src
Transglutaminase
Chk2
N-Ras
MEK1
TSC2
Caveolin-1
SF2ACC1
CASPASE7_CLEAVEDACVRL1
IGFBP2
Rb_pS807_S811
4E-BP1
Annexin-VII
PKC-alpha_pS657
EGFR_pY1173
cIAP
Bid
HER3_pY1289
Bax
HER2_pY1248
Bcl2
Bad_pS112
JNK_pT183_Y185
C-Raf_pS338
MEK1_pS217_S221
MAPK_pT202_Y204
p90RSK_pT359_S363
YB1_pS102
HER2
Syk
STAT5-alpha
eIF4G
Rictor
TTF1
Beclin
Stat3_pY705
Snail
c-Ki t
MSH6
DJ1
Gab2
PTEN
GSK3-alpha-beta_pS21_S9
PRAS40_pT246
GSK3_pS9
Akt_pS473
4E-BP1_pT37_T46
Rictor_pT1135
4E-BP1_pS65
S6_pS235_S236
S6_pS240_S244
p70-S6K_pT389
mTOR_pS2448
Cyclin-B1
Cyclin-D1
p27_pT157
Cyclin-E1
PCNA
Collagen-VI
Rad50
FibronectineEF2
XRCC1
NDRG1_pT346
P70S6K
Smac Smad3CD49b
TRFC
YAP
Claudin-7
PEA-15_pS116
eIF4E
p27PDK1_pS241
KRASINPP4b
14-3-3-be ta
Rab11
CD31GATA3
ER-alpha_pS118
p27_pT198NF2
AZD8055c-Met
SCD1
NF-kB-p65_pS536Chk1
HER3
Src_pY416
Pdcd4
EGFR_pY1173
HER2_pY1248
CD31
Smac
HER3_pY1289
Src_pY527
P70S6KYB1
BAP1HER3
p27
SETD2
PKC-delta_pS664
eEF2K
14-3-3-epsi lon
Chk1
c-Myc
Rb_pS807_S811
RBM15
Rab2514-3-3-be ta
FoxM1eIF4G NDRG1_pT346
BidC-RafBcl-xLmTORPEA-15_pS116
PEA-15
Rad50PKC-alpha
p38_MAPK
53BP1
XRCC1
Rictor
LckSAPITINIB
AMPKa_pT172GSK3-alpha-betaACVRL1
c-Met_pY1235Smad1PKC-pan_BetaII_pS660
BaxBim
eEF2 Bcl2
p27_pT198p27_pT157
Cyclin-B1
Akt_pS473
CDK1PCNA
N-Ras
JNK2
PARP-cleaved
MSH6
FoxO3a
INPP4bAR
ARHISF2
Annexin-VII
c-MetSCD1
GSK3_pS9
p70-S6K_pT389
S6_pS235_S236
PRAS40_pT246
4E-BP1_pS654E-BP1_pT37_T46
GSK3-alpha-beta_pS21_S9
S6_pS240_S244
mTOR_pS2448
CASPASE7_CLEAVEDMIG6
GAPDH c-Ki t
Rab11
ACC1
TSC2_PT1462
MEK1
FASNNF-kB-p65_pS536
Dvl3
Chk2_pT68
ER-alpha_pS118
N-CadherinCollagen-VI
Chk1_pS345
ER-alphaBRCA2
PRTSC1
StathminFoxO3a_pS318_S321
NF2
Smad4
Gab2
JNK_pT183_Y185
TFRC
Chk2Myosin-IIa_pS1943
C-Raf_pS338MAPK_pT202_Y204
c-Jun_pS73
YB1_pS102
MAPK_pT202_Y204
GATA3
JNK_pT183_Y185
MEK1_pS217_S221
PEA-15
Collagen-VIS6_pS240_S244
S6_pS235_S236
Rictor_pT1135
4E-BP1_pS65
NDRG1_pT346
mTOR_pS2448
GSK1838705A
STAT5-alpha
14-3-3-ze ta
p53RBM15
Bad_pS112
Rb_pS807_S811eEF2K
SF2
Bcl2
PCNA
Apoptosis
Cell cycle
Core reactive
DNA damage response
EMT
Hormone_a
Hormone_b
PI3K/Akt
Ras/MAPK
RTK
TSC/mTOR
Pathways
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint

∆P
Proteins
iii
iv
v vii
Spearman's Rho
p-va
lue
(log1
0)
-1 -0.5 0 0.5 1
-5
-4
-3
-2
-1
0
iii
Cancer Perturbed Proteomics Atlas
Cancer Perturbed Proteomics Atlas
vi
-7.5
-5
-2.5
0
2.5
5
∆P
ResistentSensitive0 22 44 66 88 11
013
215
417
619
822
024
226
428
6-1.5
-1
-0.5
0
0.5
1
SkinBreast
OvaryOther
BloodUterusProstate
ERKMAPKsignaling
PI3K/MTORsignaling
Other
Hsp90Chromatin
Genomeintegrity
Cellcycle
RTKsignaling
EGFRsignaling
PI3K/MTOR signaling
RTKsignaling
Other
ERKMAPKsignaling
EGFRsignaling
Chromatin
Antipsychotic
Genomeintegrity
Other,kinases
Mitosis
PI3K/MTORsignaling
Genomeintegrity
Cellcycle
Other
ERKMAPKsignaling
Chromatin
Other
Genomeintegrity
PI3K/MTORsignaling
ERKMAPKsignaling
Other
Genomeintegrity
OtherPI3K/MTORsignaling
was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The copyright holder for this preprint (whichthis version posted July 4, 2020. . https://doi.org/10.1101/2020.07.03.186908doi: bioRxiv preprint
Recommended


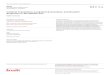







![Reciprocal Responses in the Interaction between ... · Reciprocal Responses in the Interaction between Arabidopsis and the Cell-Content-Feeding Chelicerate Herbivore Spider Mite1[W][OPEN]](https://img.pdfslide.fr/doc/110x75/5f07b53e7e708231d41e56c6/reciprocal-responses-in-the-interaction-between-reciprocal-responses-in-the.jpg)






