
M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
TRAVAUX PRATIQUES DE BIOLOGIE CELLULAIRELa séance de TP comprend deux parties distinctes avec la réalisation d’une culture primaire de lymphocytes murins sur laquelle
est réalisée des comptages et une coloration au GIEMSA et de différents tests (effet du SVF) sur les cellules CHO-K1 en
culture. Les cultures de cellules d’ovaire d’hamster chinois sont réalisées en conditions aseptiques. Elles sont cultivées dans
des milieux de culture complexes adaptés aux différents types cellulaires (sels minéraux, vitamines, acides aminés, facteurs de
croissance apportés par le sérum de veau fœtal). Le sérum de veau fœtal est un mélange complexe de molécules dont la
composition fait apparaître des facteurs de croissance, des vitamines, des transferrines….
I. Effet du sérum de veau fœtal (SVF) sur la lignée cellulaire CHO–K1
Elles sont cultivées en présence du milieu de culture MEM supplémenté avec différentes concentrations en SVF. Puis l'effet du
SVF est déterminé par des tests de viabilité.
JOUR 1
A . Lignée cellulaire CHO-K1 et mise en place des cultures
La flasque de 75 cm2 (T75) renferme des cellules CHO-K1 subconfluentes maintenues en culture dans 10 mL de milieu MEM
supplémenté en SVF 10 %, antibiotiques, L glutamine, acides aminés non essentiels (voir annexe 2).
La méthode se déroule en deux étapes (détachement / ensemencement) :
1. Le détachement des cellules de leur support
- Le milieu est aspiré à l’aide d’une pipette de 10 mL pour les deux flasques.
- Faire un lavage du tapis cellulaire de la flasque avec 4 mL de PBS stérile puis éliminer le PBS dans la poubelle en verre
pour les deux flasques.
- Ajouter 2 mL de milieu de dissociation enzymatique à l’aide d’une pipette de 5 mL 10 min dans l’incubateur à CO2 à 37°C..
- Vérifier que les cellules se sont bien détachées de leur support ou qu’elles ont un aspect arrondi à l’aide du microscope
inversé (10X).
- Taper une tranche du flacon contre la paume de la main pour décrocher toutes les cellules (à voir avec l'enseignant).
- A l’aide de la pipette de 5 mL qui a été utilisée pour le prélèvement de la solution de dissociation enzymatique, effectuer
des mouvements de va et vient de façon à dissocier complètement les cellules.
- Pour chacune des flasques deux millilitres de milieu MEM sans SVF sont déposés dans la flasque afin d'arrêter la réaction
de digestion, puis les deux suspensions cellulaires sont transférées dans un seule tube conique stérile de 15 mL (Flacon
A).
- Mélanger l’ensemble et prélever 0,05 mL (2 gouttes) de cette suspension pour le comptage des cellules
- La numération des cellules à l’aide de la cellule de Malassez (voir annexe 4) est réalisée en duplicat selon l’annexe 4.
- Pour réduire les problèmes de mortalité, il faut ensemencer en même temps la boîte 24 puits et les 2 boites de
Pétri pour la coloration de Hoechst (II page 5) (attention au temps).
1 1
M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
- Déterminez le volume contenant les cellules à soustraire pour qu’il y ait 12x105 cellules que vous placez dans un flacon à
prélèvement stérile à bouchon vert en ajoutant par la suite du milieu MEM sans SVF afin d’obtenir un volume final de 2,5
mL (Flacon B).
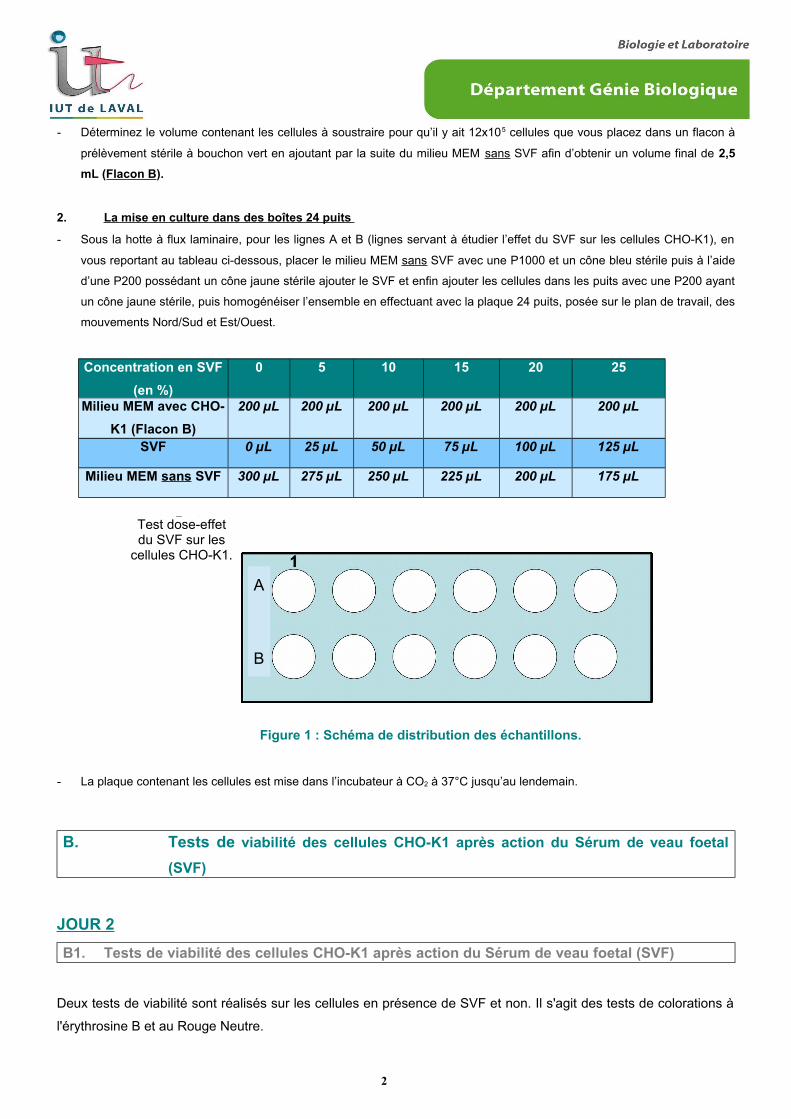
2. La mise en culture dans des boîtes 24 puits
- Sous la hotte à flux laminaire, pour les lignes A et B (lignes servant à étudier l’effet du SVF sur les cellules CHO-K1), en
vous reportant au tableau ci-dessous, placer le milieu MEM sans SVF avec une P1000 et un cône bleu stérile puis à l’aide
d’une P200 possédant un cône jaune stérile ajouter le SVF et enfin ajouter les cellules dans les puits avec une P200 ayant
un cône jaune stérile, puis homogénéiser l’ensemble en effectuant avec la plaque 24 puits, posée sur le plan de travail, des
mouvements Nord/Sud et Est/Ouest.
Concentration en SVF
(en %)
0 5 10 15 20 25
Milieu MEM avec CHO-
K1 (Flacon B)
200 µL 200 µL 200 µL 200 µL 200 µL 200 µL
SVF 0 µL 25 µL 50 µL 75 µL 100 µL 125 µL
Milieu MEM sans SVF 300 µL 275 µL 250 µL 225 µL 200 µL 175 µL
Figure 1 : Schéma de distribution des échantillons.
- La plaque contenant les cellules est mise dans l’incubateur à CO2 à 37°C jusqu’au lendemain.
B. Tests de viabilité des cellules CHO-K1 après action du Sérum de veau foetal
(SVF)
JOUR 2
B1. Tests de viabilité des cellules CHO-K1 après action du Sérum de veau foetal (SVF)
Deux tests de viabilité sont réalisés sur les cellules en présence de SVF et non. Il s'agit des tests de colorations à
l'érythrosine B et au Rouge Neutre.
2 2
A
B
Test dose-effet du SVF sur les
cellules CHO-K1.

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
Test de viabilité par la coloration au Rouge Neutre
Cette méthode est basée sur le fait que les cellules vivantes viables peuvent incorporer certains colorants au contraire des
cellules mortes par un mécanisme de transport actif. L’absorbance de colorant incorporé reflète l’action d’un agent ou d’une
molécule sur les cellules.
Détermination de la viabilité cellulaire : test d’exclusion à l’érythrosine B
Cette méthode est basée sur le fait que les cellules vivantes viables n’incorporent pas certains colorants au contraire des
cellules mortes. La coloration facilite la visualisation de la morphologie des cellules. Toutefois, si les cellules vivantes
n'incorporent pas le colorant par un mécanisme actif, au delà de 10 min cela est moins vrai (risque de fausses cellules mortes).
Le mode opératoire est le suivant :
Le test de viabilité par la coloration au rouge neutre s’applique à la ligne B. (Cf : figure : 1)
Le test de viabilité par exclusion à l’érythrosine B s’applique à la ligne A. (Cf : figure : 1)
1. Test de viabilité par la coloration au Rouge Neutre pour la ligne B
- Dans la ligne B, hors des PSM (hotte à flux laminaire). Après avoir éliminé le milieu, 0,4 mL d’une solution de rouge neutre
à 0,4 % diluée au 1/80 dans du milieu MEM complet est déposé dans les puits de la ligne B.
- Les cellules sont ensuite mises à l’étuve à 37°C pendant 1h45. [Remarque : au bout de 20 min il faut commencer à traiter
la ligne A pour finir dans les temps ].
- La coloration du tapis doit paraître uniforme.
- Le milieu est éliminé au bout de 1h45 min.
- Les puits de la ligne B sont rincés 2 fois par 1 mL de PBS non stérilisé (pour éliminer le Rouge Neutre et le milieu).
Attention, il est important d’agiter doucement pendant 1 min entre chaque rinçage en effectuant, avec la plaque 24 puits
posée sur le plan de travail, des mouvements Nord/Sud et Est/Ouest.
- 1,25 mL de Réactif de Lyse (réactif lyse RN) sont déposés par puits dans la ligne B.
- La plaque est incubée 9 min à l’obscurité puis 20 min à 4°C.
- La lecture de l’absorbance est réalisée à 540 nm :
- l’auto-zéro se fait avec le réactif de lyse RN.
- pour chaque puits, le contenu après homogénéisation est transféré dans une cuve spectrophotométrique
à usage unique de 2,5 mL.
- les valeurs d’absorbance sont converties en pourcentage de viabilité cellulaire :
Viabilité cellulaire (%) = Absorbance essai x 100
Absorbance du témoin
2. Détermination de la viabilité cellulaire : test d’exclusion à l’érythrosine B
Cette méthode est basée sur le fait que les cellules vivantes viables n’incorporent pas certains colorants au contraire des
cellules mortes. La coloration facilite la visualisation de la morphologie des cellules. (Remarque : ici le test doit démarrer 15 min
3 3

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
après le début de l'incubation des cellules dans le Rouge Neutre pour la ligne B) en commençant par le puits 6 vers le 1er). Et
seulement 3 puits à la fois pour éviter d'endommager trop vos cellules.
- Après avoir éliminer le milieu il faut ajouter 0.5 mL de PBS par puits.
- Après le lavage des cellules, le PBS est éliminé.
- Les cellules sont dissociées par 0,1 mL de milieu de dissociation (trypsine-EDTA) pendant 10 min à 37°C. Observer
l’aspect des cellules au microscope inversé, si ces dernières n’ont pas une forme arrondie replacer la plaque dans
l’incubateur. Si elles ont une forme arrondie, à l’aide d’une P1000, décrocher les cellules et homogénéiser en effectuant
des mouvements de flux et de reflux en commençant avec le puits n°6 vers le puits n°1. Quatre-vingt microlitres de
suspension sont ajoutés à 80 µL d’érythrosine B déjà déposés dans une série de 6 microtubes non stériles.
- Après avoir mélangé soigneusement, les échantillons sont incubés 5-10 min à température ambiante (au maximum)
- La numération des cellules colorées et non colorées est réalisée à l’aide d’une cellule de Malassez du puits 6 vers le puits
1 pour chacun des 6 puits et la viabilité cellulaire est déterminée par :
C. Exploitation des résultats
Penser à faire des dessins de cellules CHO-K1 adhérentes en division en apoptose à J1.
Dans un tableau synthétique reportez vos résultats (pour les lignes A et B) et déterminez le nombre de cellules
par puits et également la concentration en cellules par puits (en tenant compte des dilutions) pour la ligne A
seulement.
Tracer un graphe % viabilité cellulaire des CHO-K1= f(pourcentage de SVF) puis déterminer la DL50 (pour
l'érythrosine B) (ligne A)
Tracer un graphe % viabilité cellulaire des CHO-K1= f(pourcentage de SVF) puis déterminer la DL50 (pour le Rouge
neutre)(ligne B)
Comparer les résultats obtenus avec ceux utilisant la méthode à l’érythrosine B et le Rouge neutre. Conclusion.
Également, à partir des comptages réalisés sur la ligne A, vous déterminerez les concentrations cellulaires, puis vous
tracerez un graphe de la concentration cellulaire des CHO-K1= f (pourcentage de SVF), vous déterminerez l'IC 50 et
conclurez sur l'effet mitogène ou non du SVF ?
II. Recherche d’une activité pro-apoptotique ou non du SVF sur les CHO-K1.
Pour étudier l'effet apoptotique ou non du SVF on va tester des cellules CHO-K1 cultivées en présence de MEM avec
10% de SVF ou 0% de SVF.
Le test de Hoechst permet de colorer l'ADN en bleu fluorescent des cellules en apoptose et normale. Pour les cellules
en apoptose l'ADN se condense et apparaît plus coloré avec le colorant de Hoechst.
4 4
Viabilité cellulaire (%) =nombre total de cellules non colorées et colorées
nombre total de cellules non colorées x 100

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
JOUR 1
A La mise en culture sur lamelle des CHO-K1 en boîte de Pétri
- Deux lamelles sont dégraissées à l’alcool puis elles sont déposées dans deux petites boîtes de Pétri stériles (Figure 2).
- Préparer deux flacons de prélèvement (bouchon vert) pour le premier vous déposez 3 mL de milieu MEM sans SVF et
pour le second 3 mL de milieu avec du SVF 10 % dont il faudra déterminer la quantité. Ensuite pour chaque flacon (C
et D), déposer le volume nécessaire pour avoir 6x105 cellules de la suspension cellulaire contenue dans le flacon A, puis
vous agitez par retournement chaque flacon. Après avoir identifié vos boites et vos lamelles, ajouter la suspension du
flacon C (sans SVF) ou D (avec 10 % SVF). Incuber vos cellules une nuit à 37°C dans l’incubateur sous 5 % de CO2
Figure 2 : Schéma sur la préparation des boites pour les colorations de Hoechst des cellules traitées ou non au SVF.
JOUR 2
B La Détection de condensation nucléaire par la coloration de Hoechst
- le milieu de culture est éliminé.
- Les lamelles sont mises en contact avec 5 mL de méthanol à froid et placées pendant 30 min à -20°C.
- Ces lamelles sont récupérées puis rincées une fois avec 5 mL d'eau ultra-pure et placées dans une chambre humide
(figure 3) .
Figure 3 : modèle de chambre humide.
- Recouvrir les lamelles avec 0,5 mL ou moins de milieu contenant du réactif de Hoechst 5µg.mL-1.
- Incuber 30 min à 37°C.
- Le milieu de Hoechst est éliminé.
5 5
Milieu MEM sans SVF et 6x105 cell. (Flacon C)
Milieu MEM avec SVF 10% et 6x105 cell. (Flacon D)

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
- La lamelle est déposée à l’envers sur une lame en ayant au préalable mis une goutte de glycérol sur la lame.
- La lame est observée au microscope à fluorescence sous UV puis les noyaux sont observés jusqu’à l’objectif à immersion
C. Exploitation des résultats
Faire un comptage des 50 noyaux et déterminer le pourcentage de cellules apoptotiques.
Essayez de déterminer l’impact de l’action du SVF en comparant le contrôle et le test. Pour le compte-rendu,
dessinez un noyau normal et un noyau en phase apoptotique.
A l’aide de l’ensemble des données obtenues dans la partie II, répondez à la question suivante : le SVF est-il
un agent induisant l’apoptose ?
III. Culture primaire de lymphocytes murins
Une culture de lymphocytes isolés du sang de souris est réalisée afin de mettre en évidence ici l'effet du gradient de Ficoll et de
la culture de lymphocytes.
La manipulation est réalisée en conditions non stérile et avec les précautions habituelles relatives aux manipulations de
produits biologiques d'origine humaine.
(exemples d'utilisation des lymphocytes. Ils peuvent être séparés par cytométrie de flux, pour caractériser des populations
ainsi que des sous populations de lymphocytes (B ou T (TH4,TH8…)) ou pour en analyser l’ADN (exemple : caractérisation des
gènes qui codent les chaînes des immunoglobulines), etc...)
JOUR 1
A. Récupération du sang
Le matériel de dissection doit être préparé ainsi que le plan de travail.
Le sang est prélevé le plus rapidement possible sur une souris extemporanément sacrifiée. Le matériel (seringue, tube Falcon
de 15 mL) doit être préalablement hépariné avec une solution d’héparine 250 UI.mL-1.
B. Isolement des lymphocytes du sang
1- Recueillir l’anneau lymphocytaire
- Diluer dans un tube stérile le sang au 1/2 en tampon PBS : (exemple : 1 mL de sang + 1 mL de PBS)
- Dans le tube Falcon stérile de 15 mL contenant les 5 mL de solution de Ficoll (ramené à température ambiante), vous
introduirez délicatement le sang dilué au 1/2 goutte à goutte le long de la paroi du tube incliné à 45° en évitant tout
6 6

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
mélange avec la solution de Ficoll.
- Centrifuger à 400 g pendant 30 min à 18°C (vérifier que le frein de la centrifugeuse a été arrêté).
- Eliminer l'essentiel surnageant (plasma + PBS)
- Recueillir avec précautions l'anneau mono-lymphocytaire (transparent) du tube à l'aide d'une pipette stérile.
- Transférer la suspension cellulaire lymphocytaire recueillie dans un tube conique stérile de 15 mL contenant 3 mL de milieu
de culture RPMI non supplémenté. Penser à conserver également le culot sanguin.
2- Lavage des cellules de l'anneau mono-lymphocytaire ( pour éliminer le Ficoll qui est toxique pour lescellules)
- Mélanger le contenu du tube avec précautions (par retournement)
- Centrifuger à 400 g, pendant 5 -10 min (avec le frein)
- Eliminer le surnageant.
3- . Mise en culture des lymphocytes (une plaque multipuits dédiée pour deux Xnomes).
- Mettre en suspension les lymphocytes du culot précédent dans 1,7 mL de milieu RPMI–SVF10 %.
- Répartir la suspension à cultiver dans 2 puits d'une plaque multipuits: introduire 1 mL et 0,5 mL de suspension de
lymphocytes enrichis dans chaque puits, puis ajuster à 1 mL de RPMI –SVF10 %.
- de 0,1 mL à partir d'une dilution du culot au 1/100ème (dans le puits 3), puis ajuster à 1 mL de RPMI–SVF10 % (voir figure
4 ci-dessous).
Figure 4 : Plan de distribution dans la boite (une boite pour deux Xnomes du groupe TP)
* : 0,1 mL d'une solution à diluer au 1/100 dans le RPMI complet.
C. Numération et Contrôle de viabilité (à faire à J1 et J2)
JOUR 1 et JOUR 2 Ce test concerne les puits 1,2, 3 (les lymphocytes et le culot dilué))
- Après remise en suspension
100 µL de suspension cellulaire + 100 µL de solution d’érythrosine B
- Homogénéiser et attendre 5 min (au delà risque de coloration des cellules viables).
- Effectuer le comptage des cellules vivantes et mortes à l’aide de la cellule de Malassez.
JOUR 2
D. Suivi de la culture primaire
7 7
1 mL 0,5 mL * 0,1mL au 1/100è
Puits 1 2 3
Lymphocytes Culot sanguin
Groupe X

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
Le lendemain, vous effectuez un contrôle de viabilité des lymphocytes des 2 puits (remettre les lymphocytes en suspension)
pour chaque série et du puits 3 (culot dilué au 1/100) . (cf. C page 7)
E. Test Giemsa (pour les trois puits contenant les lymphocytes et le culot)
- Après homogénéisation de la suspension cellulaire, prélever 0,5 mL. La suspension cellulaire est centrifugée à 600g
pendant 5 min. Le culot cellulaire sera repris dans 20 µL de PBS. L'ensemble est déposé sur une lame. Réaliser un frottis
en utilisant une lamelle d’étalement et en l’inclinant à 45° pour les puits
- déposer la lame avec frottis sur le bac à coloration
- déposer 10 à 15 gouttes de May-Grünwald pour recouvrir le frottis ==> attendre 3 min
- déposer autant de gouttes d'eau distillée sur le frottis ==> attendre 2 à 3 min
- préparer le Giemsa : 10 mL d'eau distillée + 5 gouttes de Giemsa
- rejeter le colorant sans rinçage
- déposer le Giemsa préparé ==> attendre 20 à 30 min
- rejeter le colorant
- rincer avec de l'eau distillée puis avec l'eau du robinet
- sécher les lames
Vous pouvez déposer une goutte de glycérol, puis une lamelle avant d'observer vos lames. Pour chacune des troislames vos comptages se feront seulement sur 50 cellules.
Observation au microscope photonique et déterminer les % de cellules lymphocytaires par rapport au nombre
total de cellules sanguines.
F. Exploitation des résultats
1- Suivi de la culture lymphocytaire: l'action du gradient de Ficoll.
Présenter et interpréter les résultats obtenus à partir des colorations à l'érythrosine B et au Giemsa pour
chacun des 3 puits.
Dessiner les lymphocytes.
8 8

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
Objectifs rédactionnels pour le compte rendu de TP :
Choisir un titre précis sur la première page Un sommaire paginé Une introduction présentant les objectifs du TP de culture cellulaire Une partie avec le principe et les méthodes utilisées. (En prenant compte de votre manip.) Une partie présentant les résultats (données brutes et les résultats analysés) et leurs
interprétations (cette partie sera impérativement) rédigée à l'ordinateur) Une partie discussion sur les résultats et les méthodes. Une conclusion
Ne pas mettre vos données en annexe !Attention à la présentation des figures et des graphesVous devez utiliser la forme passive
Attention à la présentation des documents, soyez le plus rigoureux possible ainsi qu'à la forme !
Le nombre de page est inférieur ou égale à 10 pour le TP.
Annexe 1
Milieux et solutions prêts à l’emploi pour la lignée cellulaire
Le milieu de culture utilisé pour les cellules CHO-K1 est le MEM qui contient déjà des antibiotiques (Pénicilline/Streptomycine40 UI), de la L-glutamine (2mM) et des acides aminés non essentiels (1 %) et le sérum de veau fœtal 10 %.
Milieux,Tampons et solutions (prêts à l’emploi) :
- Milieu de culture MEM (annexe 2)
- Tampon phosphate salin 20 mM pH 7,2
Milieux et réactifs prêt à l’emploi pour la culture primaire
- Milieu RPMI 1640 avec tampon HEPES (modification de Dutch) (annexe 3)Le Milieu RPMI 1640 sera, pour ce TP déjà supplémenté en L-glutamine, et en pénicilline/streptomycine.Ce milieu a été élaboré au "Roswell Park Memorial Institute". Ce milieu s'est avéré utile pour une large gamme d'applications(croissance de nombreux types cellulaires en particulier lymphocytes dans le test de stimulation par la PHA). Le milieu utilisé (modification de Dutch) contient un tampon organique : tampon HEPES (acide N-2hydroxyéthylpipérazine-N'-2éthanesulfonique) qui permet de mieux stabiliser le pH que le système CO2/HCO3
-
- Milieu RPMI 1640 avec tampon HEPES (modification de Dutch) (annexe 3)- Solution d’érythrosine B non stérile (1 g.L-1 dans du tampon PBS).- Colorant Grünwald- Colorant Giemsa
9 9
M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
Annexe 2 – MILIEUX ET REACTIFS
MILIEUX ET REACTIFS A PREPARER LE 1er JOUR
Milieux et réactifs pour la culture primaire
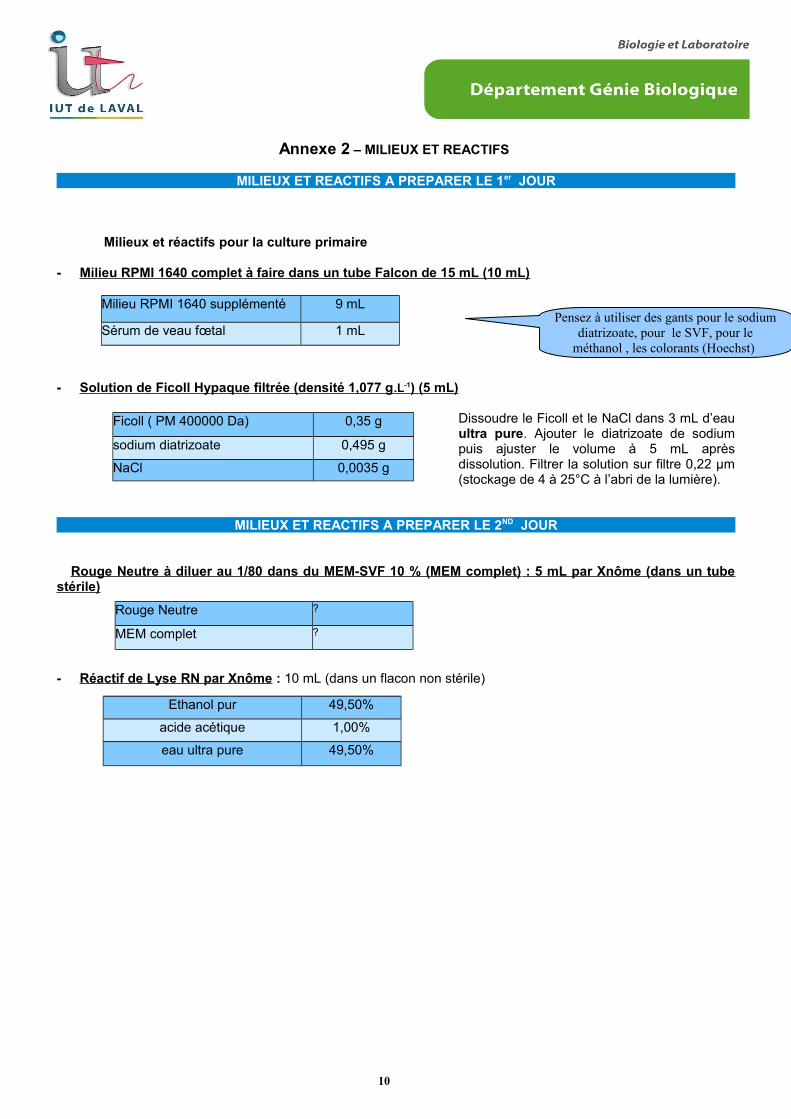
- Milieu RPMI 1640 complet à faire dans un tube Falcon de 15 mL (10 mL)
- Solution de Ficoll Hypaque filtrée (densité 1,077 g .L -1 ) (5 mL)
Dissoudre le Ficoll et le NaCl dans 3 mL d’eauultra pure. Ajouter le diatrizoate de sodiumpuis ajuster le volume à 5 mL aprèsdissolution. Filtrer la solution sur filtre 0,22 µm(stockage de 4 à 25°C à l’abri de la lumière).
MILIEUX ET REACTIFS A PREPARER LE 2ND JOUR
Rouge Neutre à diluer au 1/80 dans du MEM-SVF 10 % (MEM complet) : 5 mL par Xnôme (dans un tubestérile)
- Réactif de Lyse RN par Xnôme : 10 mL (dans un flacon non stérile)
10 1
Milieu RPMI 1640 supplémenté 9 mL
Sérum de veau fœtal 1 mL
Ficoll ( PM 400000 Da) 0,35 g
sodium diatrizoate 0,495 g
NaCl 0,0035 g
Rouge Neutre ?
MEM complet ?
Ethanol pur 49,50%
acide acétique 1,00%
eau ultra pure 49,50%
Pensez à utiliser des gants pour le sodium diatrizoate, pour le SVF, pour le
méthanol , les colorants (Hoechst)

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
ANNEXE 3 : Fiche technique du milieu RPMI-1640
Annexe 3 : Les comptages à l’aide de la cellule de Malassez
ANNEXE 4 : Fiche technique sur la numération cellulaire
11 1
Pensez à utiliser des gants pour le sodium diatrizoate, pour le SVF, pour le
méthanol , les colorants (Hoechst)

M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
ANNEXE 4 : Comptage avec une cellule de Malassez
Le comptage de la concentration d’une culture grâce à la cellule de Malassez (haemacytomètre). Cette méthode est la plus simple pour dénombrer une culture decellules animales, micro-algues, etc .. Le principe peut être appliqué à d’autres types de cellules. Il faut alors reprendre le calcul pour l’adapter à la profondeur età la surface de la cellule utilisée.
Coupe de la cellule de Malassez
Le gravage vertical de la cellule de Malassez est composé de 5 groupes de 6 lignes, plus une ligne seule. Les groupes de lignes sont espacésde 0,25 mm. Les lignes des groupes sont espacées de 0,05 mm. Le gravage horizontal est constitué de 5 groupes de 5 lignes, plus une ligneseule. Les groupes de lignes sont espacés de 0,2 mm. Les lignes des groupes sont espacés de 0,05 mm. La profondeur de la cellule est de0,200 mm.
Vue du gravage de la cellule de Malassez
On utilise donc pour compter :
25 rectangles de 0,2 x 0,25 mm divisés en 20 carrés de 0,05 mm de côté (ces rectangles sont appelés "périmètres de comptage" -en bleu sur le schéma-). Chaque
périmètre a un volume de 0,2x0,2x0,25=0,01 mm3
soit 10-5 mL. La totalité de la cellule, soit 100 rectangles de 0,2x0,25 mm, dont certains sont divisés horizontalement, verticalement, les deux ou pas du tout. Le volume de la
cellule est de 0,2x2x2,5 = 1 mm3 = 1 µL = 10-3 mL.
Détail de la cellule de Malassez
Exemple : on compte une culture de cellules. Ce genre de culture compte environ 5 à 10.106
cellules par mL choisit de compter 3 périmètres de comptage. Sur ces
trois périmètres, on dénombre 251 cellules. 251/3=83,6 cellules par périmètres, soit 8,36.106
cellules par mL
Discussion sur la méthode :
La valeur du résultat dépend en partie du soin que l’on aura apporté à la préparation de l’échantillon et de la cellule. On prendra donc bien soin durant cette étape ane pas trop faire déborder l’échantillon dans les rainures. Si on utilise du formol, les cellules mortes décantent très vite, il faut donc bien homogénéiser l’échantillonavant de le placer sur la lame.
Une autre source d’erreur est le choix du nombre de périmètres que l’on compte, surtout pour de faibles concentrations. Un nombre de 150 cellules comptéessemble être un minimum. Pour ne pas oublier de compter un périmètre, on compte méthodiquement ! Le plus simple lorsqu’on ne compte que 4 ou 5 périmètresest de suivre une diagonale.
Quel intérêt y-a-t-il à compter les rectangles en diagonale lorsque l’on n’en compte que 4 ou 5 ? (Mickaël Guéret 2002)
12 1
Utilisation :On commence tout d’abord par humecter les bords de la lamelle avec de l'eau, puis elle est déposée sur les supports. Celle-ci doit rester collée à la lame. Ensuite l’échantillon est introduit à l’aide d’une pipette pasteur dans l’espace crée entre la lame et la lamelle par capillarité.
La lame est placée sous un microscope, un grossissement de 400 est pratique. Les cellules peuvent alors être comptées. Les cellules à cheval sur un bord ne sont comptées que pour une moitié. Selon la concentration et la taille des cellules, on choisit la surface sur laquelle on va dénombrer : 3, 4 ou 5 périmètres de comptage pour des cultures très denses (de l’ordre de 2 à 20 millions de cellules par ml). Ensuite, on rapporte le nombre que l’on trouve à un seul périmètre de comptage. Il suffit ensuite
de multiplier ce résultat par 105 et l’on obtient un résultat en nombre de cellules par mL. 25 périmètres, ou la totalité des 100 rectangles pour des cultures beaucoup moins denses (de l’ordre de 500 000 cellules par ml). Si on ne compte que 25 périmètres, on multiplie le résultat par 4 pour rapporter le nombre à la surface totale de la cellule. Il suffit ensuite de multiplier le chiffre que l’on
obtient par 103 et l’on obtient un résultat en nombre de cellules par mL.
Zone de dépôt par capillarité et de comptage
M 3202 : Cultures cellulaires Méthodes alternatives à l’expérimentation animaleNovembre 2016
Feuille de résultats du TP :
13 1
ABS
en %
en %
1 mL 0.5mL
V/M
Résultats colorations érythrosine B :
Résultats à J1(comptages) ….............. …........... ….................... (pourcentages) ….............. …........... …....................
résultats à J2 (comptages) ….............. …........... ….................... (pourcentages) ….............. …........... ….................…
Résultats colorations Giemsa : (comptages) ….............. …........... ….................... (pourcentages) ….............. …........... …....................
1/100
Numéro du Xnôme : Noms des étudiants :
Erythrosine B (J1 et J2)Giemsa (J2)
Résultats du Test “effet dose du SVF sur les CHO-K1
Résultats de la cultureprimaire
neutre
Lymphocytes Culot
Rouge neutre
Résultats des colorations de Hoechst des cellules CHO-K1 :
SVF 0% SVF 10%
comptages .......…….. comptages .....…….. pourcentage …....…… pourcentage …..……...
Erythrosine B
B
Erythrosine B C
D
A1 62 53 4
Recommended

















