Embed Size (px)
Citation preview

MARIE-THERESE BAWOLAK
ADAPTATION DES RECEPTEURS DES KININES
Thèse présentée à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en Médecine Expérimentale pour l'obtention du grade de Philosophiae Doctor (Ph.D.)
DEPARTEMENT DE MICROBIOLOGIE-IMMUNOLOGIE-INFECTIOLOGIE FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
2011
Marie-Thérèse Bawolak, 201

Résumé
Les kinines sont des peptides exerçant des fonctions régulatrices au niveau cardiovasculaire
et rénal, une activité pro-inflammatoire ainsi qu'un rôle important dans la perception de la
douleur. Leurs effets dépendent de leur liaison à deux récepteurs à sept domaines
transmembranaires couplés aux protéines G, soient les récepteurs Bi et B2 des kinines.
Suivant leur activation, les récepteurs se distinguent grandement l'un de l'autre par les
voies de désensibilisation et les partenaires moléculaires recrutés, et ultimement l'arrêt de
leur signalisation. Les présents travaux de doctorat ont permis une meilleure
compréhension de l'adaptation des récepteurs Bj et B2, grâce à l'utilisation de modèles
cellulaires exprimant les récepteurs recombinants, de type sauvage ou fusionnés à un
epitope myc ou à une protéine fluorescente verte. D'abord, l'utilisation d'agonistes
peptidiques et non peptidique du récepteur B2 résistants au métabolisme permit un
raffinement des connaissances sur les événements essentiels au niveau endosomal pour une
réexpression du récepteur à la membrane, ainsi que sur les caractérisiques des interactions
entre celui-ci et les |3-arrestines. Par ailleurs, ces agonistes ont été identifiés comme
agonistes biaises, et ce, en fonction de leur durée d'action, puisqu'ils généraient une
signalisation qui n'était pas observée avec l'agoniste endogène. Les ligands fluorescents,
dont une paire agoniste-antagoniste a été synthétisée pour chaque type de récepteur, ont
permis de mieux étudier le comportement du récepteur activé, au niveau des phénomènes
de translocation cellulaire, à l'identification des sites où il se retrouve suivant son
activation, ainsi qu'au caractère distinctif de chaque récepteur à cet égard. Les peptides
agonistes du RB2 dont les extrémités N-terminales sont modifiées ou prolongées, dans ce
cas-ci par l'ajout d'un fluorophore, ainsi que la séquence naturelle d'origine amphibienne,
la maximakinine, ont tous mené à une dégradation du RB2-GFP, indiquant ainsi qu'au
niveau endosomal, la principale voie de dégradation de la BK est une activité
aminopeptidase. Finalement, l'emploi d'anticorps anti-récepteur a permis de démontrer que
le RB2, lorsqu'il est une partie intégrante d'un complexe immun, peut toujours être
internalise de manière dépendante de la présence de l'agoniste, ce qui suggère que le
couplage de différentes fonctions aux anticorps employés pourront conférer de nouvelles
propriétés au récepteur.

I l l
Abstract
Kinins are peptides that regulate cardiovascular processes, exhibit pro-inflammatory
activities and are implicated in pain perception. The initiation of their effects is dependent
on their binding to two G protein coupled receptors, possessing seven transmembrane-
spanning domains, namely the bradykinin Bi and the B2 receptors. Following their
activation, the receptors exhibit a very distinct behaviour, in terms of desensitization,
pathway recruitment and molecular partners implicated, which ultimately lead to signal
arrest. The results presented in this thesis provide insights in the relatively unique
adaptation mechanisms to which both receptors are submitted to, allowed by the use of
cellular models expressing both subtypes of recombinant receptors, either their wild type
version, or as fused to epitopes or fluorescent proteins. First, use of inactivation-resistant
agonists of the B2 receptor allowed a more comprehensive understanding of the necessary
events leading to the receptor reexpression at the cell membrane, as well as a clearer view
of the interactions between the receptor and the (3-arrestins. Furthermore, these agonists
were identified as biased agonists as a function of time, as they induced signalling events
that were not echoed by similar treatment by the endogenous agonist bradykinin. Secondly,
a pair of fluorescent agonist-antagonist ligands were synthesized for each receptor subtype,
and permitted visual assessment of the behaviour of the activated receptor, regarding the
phenomenon of subcellular translocation, site of redistribution upon activation and
comparison of the general pattern followed by each receptor. Thirdly, use of many peptide
agonists of the B2 receptor, presenting either a modification of their N-terminal sequence or
the extension of it (in this case exemplified by the addition of a fluorophore) lead to the
degradation of the B2 receptor coupled to a green fluorescent protein. This observation was
further supported by the use of maximakinin, a natural sequence from amphibian source.
This data strongly points to an endosomal aminopeptidase as the major bradykinin
degrading activity at the intracellular level. Lastly, anti-receptor antibodies used to
assemble immune complexes containing the B2 receptor can be internalized in an agonist-
dependant fashion, which lead us to hypothesize that the coupling of various functions to
anti-receptor antibodies could confer new properties to the receptor.

IV
Avant-propos Je tiens à exprimer ma gratitude à mon directeur de recherche, le Docteur François
Marceau. Cela a été un immense honneur que d'effectuer mes études de doctorat, ainsi que
de maîtrise, sous sa direction. Il est un excellent mentor; sa grande connaissance et
l'enthousiasme avec lequel il la partage m'ont permis d'acquérir une formation scientifique
de grande qualité. Mon passage au sein de son laboratoire m'aura enseigné la rigueur et le
jugement critique. Je veux aussi le remercier pour sa compréhension, sa générosité et sa
grande disponibilité. Je le considère comme un homme d'exception, autant au niveau de sa
carrière professionnelle que pour sa personnalité.
Je remercie les membres du jury, les Drs Jacques Huot, Gaétan Guillemette et Robert
Lodge, d'avoir accepté d'évaluer mes travaux de doctorat, tels que présentés par cette thèse.
Je tiens à souligner la participation de tous nos collaborateurs, tout particulièrement les Drs
Lajos Géra, René C.-Gaudreault et Sébastien Fortin, qui ont synthétisé plusieurs ligands des
récepteurs de la bradykinine qui ont permis la réalisation des travaux présentés par cette
thèse, ainsi que M. Maurice Dufour, le responsable de la Plateforme de cytométrie du
CRCHUL. Finalement, je désire remercier le Fonds de Recherche en Santé du Québec pour
le soutien financier accordé durant mon doctorat.
J'aimerais remercier les membres du laboratoire Marceau. Je suis très reconnaissante à
Mme Johanne Bouthillier, qui tient le laboratoire d'une main de maître. Son assiduité, son
sens de l'organisation et l'aide qu'elle apporte à tous les étudiants sont réellement
appréciés. J'aimerais souligner le plaisir que j 'ai eu de travailler avec des collègues qui me
sont particulièrement chers, les Drs Jean-Philippe Fortin et Guillaume Morissette. Je
souhaite à Mlle Caroline Roy beaucoup de succès dans ses études de maîtrise. Finalement,
le Centre de Recherche en Rhumatologie et Immunologie du CHUL offre un
environnement de recherche stimulant qui contribue aux succès des étudiants.
J'aimerais exprimer ma très grande reconnaissance à mes parents pour leur amour et leur
support. Aux gens qui sont le cœur de ma vie, merci.

A mes parents qui m'ont choyée. A Edward Bawolak, mon père, que j 'ai eu le
grand malheur de perdre et à Jadwiga Trojanowska, ma mère, quej 'ai l'immense
bonheur d'avoir encore.

VI
Table des matières Résumé ii Abstract iii Avant-propos iv Table des matières vi Liste des tableaux ix Liste des figures x Liste des abréviations xiv 1. Introduction 1 Le système kinine-kallikréine 1
1.1 Les kinines 1 1.2 La formation des kinines 2
1.2.1 Assemblage du système kinine-kallikréine plasmatique au niveau vasculaire ..3 1.2.2 Assemblage du système kinine-kallikréine tissulaire 5 1.2.3 Voies alternatives menant à la production des kinines 5
1.3 Métabolisme des kinines 6 1.3.1 Carboxypeptidases N et M 7 1.3.2 Enzyme de conversion de l'angiotensine I 7 1.3.3 Endopeptidase neutre (E.C.3.4.24.11) 9 1.3.4 Aminopeptidase P (APP) 10 1.3.5 Aminopeptidase N (APN) 11 1.3.6 Oligopeptidase thimet (EP24.15) 11 1.3.7 Ligands résistants au métabolisme 13
1.4 Les récepteurs des kinines 14 1.4.1 Expression des récepteurs des kinines 14 1.4.2 Structure des récepteurs des kinines 17
1.4.2.1 Glycosylation 18 1.4.2.2 Phosphorylation 19 1.4.2.3 Oligomérisation 20 1.4.2.4 Palmitoylation 22
1.4.3 Relations de structure-activité des récepteurs des kinines 23 1.4.3.1 Fonction de reconnaissance des ligands 23 1.4.3.2 Activation du récepteur et signalisation 26
1.4.4 Adaptation des récepteurs 30 1.4.4.1 La phosphorylation du RB2 par les GRK 32 1.4.4.2 La phosphorylation du RB2 par la PKC 34 1.4.4.3 Le recrutement au RB2 des (3-arrestines 34 1.4.4.4 Endocytose du RB2 37 1.4.4.5 Redistribution membranaire des récepteurs des kinines 39
1.5 Le système kinine-kallikréine en physiologie et en pathologie 40 1.5.1 Système cardiovasculaire et rénal 40 1.5.2 Inflammation 43 1.5.3 Cancer 45 1.5.4 Système nerveux 47
1.6 Objectif des travaux 49

Vil
2. Matériel et Méthodes 53 2.1 Synthèse des analogues des kinines 53
2.1.1 Synthèse peptidique 53 2.1.2 Synthèse du composé 47a 55 2.1.3 Maximakinine 57
2.2 Construction des récepteurs recombinants 57 2.3 Culture cellulaire et transfection 58 2.4 Essais de contractilité 61
2.4.1 Identification de la nature et de la spécificité des nouveaux ligands du RB2 à séquence contrainte 62 2.4.2 Caractérisation approfondie du peptide B-9972, ainsi que du composé 47a, un agoniste non peptidique du RB2, deux agonistes résistants au métabolisme 65 2.4.3 Étude des ligands présentant une extension en N-terminal 66 2.4.4 Étude de l'effet antagoniste du LF16-0687 68
2.5 Essais de liaison 68 2.5.1 Essais de saturation et de compétition 69 2.5.2 Endoyctose de la [3H]BK 72
2.6 Microscopie 73 2.6.1 Microscopie à fluorescence et confocale 73 2.6.2 Immunocytochimie 75
2.7 Immunobuvardages 76 2.7.1 Expression et abondance des protéines d'intérêt 76 2.7.2 Signalisation 77
2.8 Essais de mobilisation calcique 79 2.9 Cytométrie en flux 81
3. Résultats 82 3.1 Caractérisation pharmacologique des analogues de la BK à la structure contrainte et conséquences de leur résistance au métabolisme 82
3.1.1 Propriétés pharmacologiques des analogues de la BK à la structure contrainte.. 82 3.1.2 Caractérisation de la résistance métabolique à l'aide des essais de contractilité. 88 3.1.3 Identification des voies d'inactivation de la BK au niveau de la veine ombilicale humaine à l'aide des essais de contractilité 90 3.1.4 Effets des agonistes résistants au métabolisme au niveau de cellules HEK 293 RB2-GFP 90 3.1.5 Validation du vecteur myc-RB2 97 3.1.6 Désensibilisation fonctionnelle du RB2-GFP par le peptide B-9972 100
3.2 Caractérisation approfondie d'agonistes résistants à l'inactivation 103 3.2.1 Caractérisation pharmacologique du composé 47a 103 3.2.2 Effets des agonistes résistants au métabolisme sur les cellules exprimant le RB2-GFP 108 3.2.3 Effets des agonistes résistants au métabolisme au niveau de l'interaction du RB2 et de la P-arrestine2 118 3.2.4 Dégradation du RB2-GFP secondairement à sa stimulation par les agonistes résistants à l'inactivation 118
3.3 Extension en N-terminal des ligands des récepteurs des kinines 121 3.3.1 Analogues de l'antagoniste B-9430 121

vin
3.3.1.1 Profil pharmacologique d'analogues de l'antagoniste B-9430 prolongés en N-terminal 121 3.3.1.2 Imagerie des RB2 exprimés par des cellules HEK 293a intactes 126
3.3.2 Ligands fluorescents du RBi 126 3.3.2.1 Caractérisation pharmacologique des nouveaux ligands par la réalisation d'essais de liaison 126 3.3.2.2 Évaluation fonctionnelle de nouveaux ligands fluorescents par les essais de contractilité 128 3.3.2.3 Évaluation fonctionnelle des nouveaux ligands fluorescents par les essais de mobilisation calcique 132 3.3.2.4 Imagerie du RBi exprimé par des cellules HEK 293a 134 3.3.2.5 Exploitation de l'antagoniste B-10376 en cytométrie de flux 144
3.3.3 Analogues fluorescents de la BK 147 3.3.3.1 Caractérisation du myc-RB2TRUi\ic 147 3.3.3.2 Pharmacologie des analogues fluorescents de la BK 149 3.3.3.3 Caractérisation et endocytose de l'analogue fluorescent CF-eACA-BK.... 152 3.3.3.4 Imagerie de l'ECA 162
3.3.4 Études de l'analogue naturel maximakinine 165 3.3.4.1 Pharmacologie de la maximakinine 165 3.3.4.2 Propriétés de la maximakinine 167
3.4 Cargos anticorps anti-récepteur 170 4. Discussion 182
4.1 Profil pharmacologique de l'agoniste résistant au métabolisme B-9972 et induction efficace de la dégradation du RB2 par son action 182
4.1.1 Profil pharmacologique de la triade de peptides isomères 182 4.1.2 Dégradation du RB2 par un traitement prolongé avec l'agoniste B-9972 185
4.2 Effets des agonistes résistants à l'inactivation sur la signalisation, la désensibilisation et la dégradation des RB2 189
4.2.1 Caractérisation pharmacologique et effets découlant de la résistance métabolique du composé non peptide 47a au niveau de modèles ex vivo 189 4.2.2 Caractérisation pharmacologique et effets découlant de la résistance métabolique du composé non peptide 47a au niveau de modèles cellulaires 190 4.3 Ligands des récepteurs des kinines possédant une extension en N-terminale 196
4.3.1 Caractérisation de l'antagoniste B-10380 196 4.3.2 Ligands fluorescents du RBi 197 4.3.3 Agonistes fluorescents du RB2 201 4.3.4 La maximakinine 205 4.4 Transport de cargos anticorps anti-récepteur 208
4.5 Perspectives 212 4.5.1 Agonistes résistants à l'inactivation 212 4.5.2 Imagerie des récepteurs des kinines 215 4.5.3 Cargos internalises par le RB2 217
5. Conclusion 220 6. Bibliographie 222 7. ANNEXE 263

IX
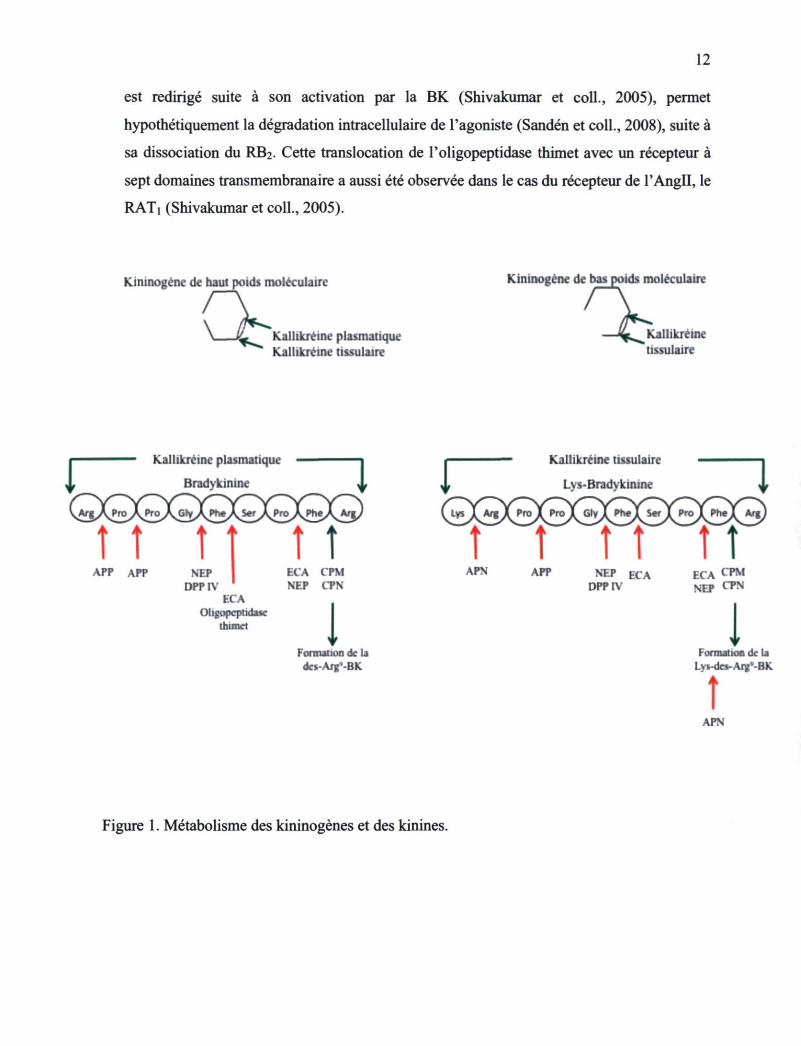
Liste des tableaux Tableau 1. Séquences des nouveaux analogues des kinines possédant des extensions en N-
terminal 55 Tableau 2. Résumé de la pharmacologie des peptides liant le RBi utilisés lors des présentes
études 130

Liste des figures Figure 1. Métabolisme des kininogènes et des kinines 12 Figure 2. Structure et stéréochimie de la région C-terminale des trois ligands des récepteurs
des kinines, les isomères B-9430, B-9972 et B-10344 53 Figure 3. Structures du composé 47a, un agoniste du récepteur B2 de la bradykinine, et du
LF160687, un antagoniste compétitif de ce récepteur 56 Figure 4. Compétition de la liaison du radioligand [3H]BK (3 nM) au RB2 par les nouveaux
antagonistes peptidiques non radiomarqués 83 Figure 5. Effet de la triade d'isomères sur l'activité contractile de la veine jugulaire de
lapin, un essai fonctionnel pour le RB2 84 Figure 6. Effet antagoniste du peptide B-9430 au niveau de l'essai de contractilité de l'aorte
de lapin, stimulée par la des-Arg9-BK. Il s'agit d'un essai fonctionnel du RBi 86 Figure 7. Effets de la triade d'isomères au niveau d'essais de contractilité de la veine
ombilicale humaine, un essai fonctionnel du RB2 87 Figure 8. Paramètres additionnels dérivés des courbes concentration-réponse cumulatives,
construites pour des agonistes résistants au métabolisme du RB2 89 Figure 9. Effet de l'énalaprilat, du phsophoramidon et de l'apstatine sur la relation
contraction-effet de la BK comme agent contractile au niveau de la veine ombilicale humaine 91
Figure 10. Études par microscopie en épifluorescence de cellules intactes HEK 293 exprimant de manière stable le RB2-GFP et stimulées pendant 30 min (en A) ou 12 h (en B) avec différents ligands du RB2 92
Figure 11. Effet des agonistes sur le RB2-GFP exprimé de manière stable par des cellules HEK 293 : immunobuvardage de lysats cellulaires totaux basés sur les anticorps anti-GFP 95
Figure 12. Valeurs moyennes de densitométrie obtenues à partir d'immunobuvardages pour le RB2-GFP et pour le myc-RB2 afin de quantifier la dégradation du récepteur 96
Figure 13. Caractérisation pharmacologique du myc-RB2 recombinant, exprimé de manière transitoire par les cellules COS-1 98
Figure 14. Immunofluorescence des cellules HEK 293a exprimant le myc-RB2, basée sur l'utilisation de l'anticorps anti-myc 99
Figure 15. Effet des agonistes sur l'abondance du myc-RB2 exprimé de manière transitoire par des cellules HEK 293a : immunobuvardages d'extraits cellulaires totaux basés sur l'anticorps anti-myc (clone 4A6) 101
Figure 16. Désensibilisation fonctionnelle du RB2-GFP exprimé par les cellules HEK 293 soumises à des prétraitements de 12 h avec les agonistes BK et B-9972 102
Figure 17. Effet de l'antagoniste LF 16-0687 sur la contraction induite par la BK et de la 5-HT dans la veine ombilicale humaine 104
Figure 18. Effet antagoniste du LF 16-0687 au niveau d'un essai fonctionnel du RB2 au niveau de la veine ombilicale humaine stimulée avec le composé 47a 106
Figure 19. Stimulation séquentielle de la préparation de veine ombilicale humaine afin de déterminer la désensibilisation spécifique à chaque agent 107
Figure 20. En A, temps pour atteindre la demi-relaxation à partir de la réponse contractile maximale pour chacun des agonistes du RB2 après le lavage des tissus suivant l'ajout de la concentration la plus importante 109

XI
Figure 21. Déplacement de la liaison de [3H]BK (à une concentration de 3 nM) à des cellules de HEK 293 exprimant de manière stable RB2-GFP par des agonistes non radiomarqués 110
Figure 22. Études de microscopie en épifluorescence de cellules HEK 293 intactes exprimant de manière stable le RB2-GFP et stimulées pour des périodes de 30 min, 3 h ou 12 h avec des ligands du RB2 aux concentrations indiquées 112
Figure 23. La mobilisation calcique induite au niveau de cellules HEK 293 exprimant de manière stable le RB2-GFP 114
Figure 24. Réponses de signalisation différentielles induites par les agonistes présentant différentes résistances à l'inactivation au niveau de cellules HEK 293 exprimant de manière stable le RB2-GFP ou de cellules HEK 293a exprimant de manière transitoire lemyc-RB2 115
Figure 25. Études de microscopie en épifluorescence de cellules HEK 293a exprimant de manière transitoire la construction codant pour la |3-arrestine2-GFP, et optionnellement le myc-RB2, stimulées pour différentes durées de traitement (de 10 min à 12 h) avec des agonistes du RB2 119
Figure 26. Études en microscopie en épifluorescence (au grossissement de 1000 X) de la translocation de la (B-arrestinei.-Cherry FP induite par un traitement de 12 h avec des agonistes du RB2 120
Figure 27. Effets des agonistes sur les cellules HEK 293 exprimant le RB2-GFP de manière stable tel qu'évalués par l'immunobuvardage d'extraits cellulaires totaux basés sur un anticorps anti-GFP 122
Figure 28. Profil pharmacologique du composé B-10380 au niveau d'essais contractiles pour le RB2 et les RBi et des essais de liaison du radioligand du RB2 123
Figure 29. Imagerie de cellules HEK 293a intactes exprimant un récepteur des kinines recombinant, basée sur la fluorescence verte de l'antagoniste du RB2 B-10380 127
Figure 30. Compétition de la liaison du radioligand [3H]Lys-des-Arg9-BK au RBi-FLAG ou au RBi de type sauvage de lapin par des analogues des kinines non radiomarqués.
129 Figure 31. Effets des nouveaux ligands du RBi au niveau de la contraction de l'aorte de
lapin fraîchement isolée, qui constitue un essai fonctionnel du RBi, 131 Figure 32. Mobilisation calcique au niveau de cellules de muscle lisse d'artère ombilicale
humaine, exprimant des RBi endogènes 133 Figure 33. Études de microscopie en épifluorescence basées sur la fluorescence verte des
antagonistes du RB] B-10372 et B-10376 marquant les cellules HEK 293a intactes exprimant de manière transitoire le RBi-FLAG, le RBi de type sauvage de lapin ou le vecteur FLAG vide 135
Figure 34. Études de microscopie en épifluorescence du marquage de cellules HEK 293a intactes exprimant optionnellement le RBi-FLAG, le RB] de type sauvage de lapin ou le vecteur FLAG vide, avec l'agoniste du RB| B-10378 136
Figure 35. Études de microscopie en épifluorescence présentant l'effet de la concentration des ligands fluorescents sur l'intensité du marquage de cellules HEK 293a intactes exprimant le RBi 137
Figure 36. Caractérisation de l'apparence du marquage de cellules HEK 293a non transfectées ou exprimant l'une ou l'autre construction codant le RBi tel qu'obtenu soit par l'antagoniste B-10376 ou par l'agoniste B-10378 139

Xll
Figure 37. Reconstitutions tridimensionnelles de cellules HEK 293a exprimant de manière transitoire des RBi recombinants à partir de séries de coupes confocales basées sur les ligands fluorescents, l'antagoniste B-10376 ou l'agoniste B-10378 141
Figure 38. Études de microscopie confocale; plans confocaux équatoriaux de cellules exprimant de manière transitoire le RBj de type sauvage de lapin et la cavéoline-1-CherryFP, et optionnellement incubées en présence des ligands antagoniste B-10376 ou agoniste B-10378 143
Figure 39. Cytométrie en flux de cellules HEK 293a marquées avec l'antagoniste fluorescent B-10376 145
Figure 40. Cytofluorométrie de cellules de muscle lisse d'artère ombilicale humaine soumises à un prétraitement avec l'IL-1(3 avant leur marquage avec l'antagoniste fluorescent B-10376 146
Figure 41. Validation de la construction myc-RB2TRUNC 148 Figure 42. Études de microscopie en épifluorescence portant sur la distribution cytosolique
de la p-arrestine2-GFP au niveau de cellules HEK 293a transfectées avec cette seule construction ou co-exprimant le myc-RB2 ou le myc-RB2TRUNC 150
Figure 43. Caractérisation pharmacologique des analogues fluorescents de la BK 151 Figure 44. Études de microscopie en épifluorescence de cellules HEK 293a non
transfectées ou exprimant de manière transitoire le myc-RB2 ou le myc-RB2TRUNc-153 Figure 45. Reconstitutions tridimensionnelles de cellules HEK 293a exprimant le RB2 de
type sauvage de lapin ou exprimant l'ECA humaine, générées à partir d'une série de coupes confocales pour chaque champ 155
Figure 46. Études de microscopie en épifluorescence de cellules exprimant le myc-RB2 soumises à une incubation en présence de l'agoniste fluorescent CF-eACA-BK 157
Figure 47. Études de microscopie en épifluorescence de cellules HEK 293a exprimant le myc-RB2, de concert avec d'autres transgènes 159
Figure 48. Compétition de la liaison de 2 nM [3H]énalaprilat à l'ECA endogène exprimée par les cellules endothéliales de la veine ombilicale humaine par la BK et des analogues de la BK fluorescents 163
Figure 49. Études de microscopie en épifluorescence de cellules HEK 293a exprimant de manière transitoire l'ECA recombinante 164
Figure 50. Caractérisation pharmacologique de la maximakinine 166 Figure 51. Effets de la MK sur l'adaptation du RB2-GFP exprimé de manière stable par des
cellules HEK 293 168 Figure 52. Études de microscopie confocale montrant l'endocytose médiée par le myc-RB2
d'un anticorps monoclonal par des cellules intactes HEK 293 171 Figure 53. Proportion de la fluorescence corticale (associée à la membrane plasmique) au
niveau de cellules HEK 293 exprimant de manière transitoire le myc-RB2 et soumises à l'internalisation du complexe ligand-récepteur 172
Figure 54. Mobilisation calcique aiguë induite par l'injection de ligands du myc-RB2, exprimé de manière transitoire par des cellules HEK 293a 174
Figure 55. Études de microscopie en épifluorescence de l'endocytose d'un complexe immun 175
Figure 56. Études de microscopie en épifluorescence de l'endocytose d'un complexe immun 176

Xlll
Figure 57. Proportion de fluorescence corticale (associée à la membrane) au niveau de cellules HEK 293a intactes exprimant de manière transitoire le myc-RB2 177
Figure 58. Proportion de fluorescence corticale (associée à la membrane) au niveau de cellules HEK 293a intactes exprimant de manière transitoire le myc-RB2 ou la version tronquée du récepteur 178
Figure 59. Études de microscopie en épifluorescence 180 Figure 60. Études de microscopie en épifluorescence 181 Figure 61. Schéma présentant le cycle d'endocytose-recyclage du RB2-GFP en fonction de
l'agoniste 188 Figure 62. Représentation schématique des mécanismes recrutés en réponse à la stimulation
du RB2 de la BK par ses agonistes au niveau de cellules HEK 293 195 Figure 63. Représentation schématique des mécanismes d'endocytose de cargos de diverses
tailles liés au RB2 en réponse à la stimulation de la BK, par des cellules HEK 293a intactes 211

XIV
Liste des abréviations
JH 4,5-PIP2 A AcSDKP ADN AMPC Angl Ang II ANOVA APM APN APP AP-2 Arg ARNm ASIS Asn RAT, RB, RB2 BK " m a x
BSA C C1INH CF CGRP CherryFP CHX CML C02 COX-2 CpG CPM CPN cm C-terminale Cys DAG des-Arg9-BK des-Arg10-KD DMEM DMSO
Tritium Phosphatidylinositol 4,5-bisphosphate Adenine N-acétyl-séryl-aspartyl-lysyl-proline Acide déoxyribonucléique Adenosine monophosphate cyclique Angiotensine I Angiotensine II Analyse de la variance Aminopeptidase M Aminopeptidase N Aminopeptidase P Protéine Adaptatrice-2 Arginine Acide ribonucléique messager Active site-inhibited seven Asparagine Récepteur de l'angiotensine II de type Récepteur B, de la bradykinine Récepteur B2 de la bradykinine Bradykinine Nombre maximal de sites de liaison Bovine serum albumin Cytidine Cl Inhibitor Carboxyfluorescéine Calcitonin-related gene protein Cherry Fluorescent Protein Cycloheximide Cellules de muscle lisse Dioxyde de carbone Cyclooxygénase-2 a-cyclopentylglycine Carboxypeptidase M Carboxypeptidase N Centimètre Carboxy terminale Cysteine Diacylglycérol des-Arg9-bradykinine des-Arg ' °-kal lidine Dttbelcco 's Modified Eagle Medium Dimethyl sulfoxyde

XV
E-ACA E-aminocaproyl ECA Enzyme de conversion de I'angiotensine I EDTA Acide ethylene diamine-tétraacétique eNOS Endothelial nitric oxide synthase EP24.15 Thimet oligopeptidase ERK Extracellular signal regulated kinase FBS Fetal bovine serum F(ab')2 Deux fragments Fab reliés par la région charnière fmol Femto mole FXII Facteur de Hageman g Gramme nG Guanidine GFP Green Fluorescent Protein Gly Glycine GMPC Guanosine monophosphate cyclique GRK G-protein coupled receptor kinase h Heure hRBi-FLAG Protéine de fusion conjuguant le RB, humain et l'épitope
FLAG HBSS Hank's buffered salt solution KFM Kininogène de bas poids moléculaire KHM Kininogène de haut poids moléculaire HRP Horseradish peroxydase HS Human serum IECA Inhibiteur de l'enzyme de conversion de I'angiotensine I IFN-y Interféron-gamma Igl o>(2-indanyl)glycine IL-1 (3 Interleukine-1 bêta iNOS Inducible nitric oxide synthase IP3 Inositol 1,4,5-triphosphate KD Kallidine kDa Kilodalton KD Constante de dissociation à l'équilibre kg Kilogramme Leu Leucine LPS Lipopolysaccharide Lys Lysine Lys-BK Lys-bradykinine Lys-des-Arg9-BK Lys-des-Arg -bradykinine M Molaire M 199 Medium 199 MK Maximakinine mg Milligramme min Minute ml Millilitre mM Millimolaire

XVI
mmol MAP MAPK NaOH Na3V04 NEP NF-KB N-terminale ng nM NO 0 2 Oic pb PBS PG PGI2 PCR PE Phe PKC PMA pmol Pro RCPG s Sar SDS Ser siRNA SP T Thr Tic TM TNBS TNF-a tPA Tyr uCi ug ul uM
Millimole Mitogen activated protein Mitogen activated protein kinase Hydroxyde de sodium Orthovanadate de sodium Endopeptidase neutre Nuclear factor- KB Amino terminale Nanogramme Nanomolaire Monoxyde d'azote Oxygène (3as, 7as) octahydroindol-2-yl-carbonyl Paire de base Phosphate buffered saline Prostanglandine Prostacycline Polymerase chain reaction Phenylephrine Phenylalanine Protéine kinase C Phorbol 12-myristate 13-acétate Picomole Proline Récepteur couplé aux protéines G Seconde Sarcosine Sodium dodécyl sulfate Serine Small interfering RNA Substance P Thymidine Thr 1,2,3,4-tétrahydroisoquinoline-3-acide carboxylique Domaine transmembranaire 2,3,6- acide trinitrobenzènosulfonique Tumor necrosis factor alpha Tissue plasminogen activator Tyrosine Microcurie Microgramme Microlitre Micromolaire

1. Introduction
Le système kinine-kallikréine
Le système kinine-kallikréine repose sur le clivage de précurseurs protéiques par des
enzymes de type serine protease afin de générer les kinines. Les précurseurs protéiques,
dont il existe deux types qui diffèrent par leur poids moléculaire, sont nommés les
kininogènes et sont principalement synthétisés par le foie. Leur clivage par les kallikréines,
dont il existe également deux types, soit la kallikréine plasmatique (KP) et la kallikréine
tissulaire (KT), permet la libération des kinines. Les kinines sont de petits peptides à
activité locale qui engagent la signalisation de deux types de récepteurs à sept domaines
transmembranaires couplés aux protéines G, les récepteurs B, et B2 de la bradykinine, afin
d'exercer leurs fonctions.
1.1 Les kinines
Le terme général kinine se rapporte à un ensemble de peptides, dont le nonapeptide
bradykinine (BK; Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg) et la kallidine (KD, aussi connue
sous le nom de lysyl-bradykinine, Lys-BK), ainsi qu'à leurs metabolites. La BK et la Lys-
BK sont libérées suite à la dégradation protéolytique des kininogènes, et sont donc aussi
identifiées comme étant les kinines natives, toutes deux des ligands préférentiels du RB2.
Par ailleurs, les metabolites des kinines natives générées par les carboxypeptidases N et M
(qui seront décrites plus bas), respectivement la des-Arg9-BK et la Lys-des-Arg9-BK, sont
les ligands préférentiels du RB, (Marceau et coll., 1998; Leeb-Lundberg et coll., 2005). La
Lys-BK conserve une certaine affinité pour le RB,, mais de manière dépendante de
l'espèce, puisque si l'affinité est appréciable pour le RB, humain et de lapin (K de 2,54 et
de 19,0 nM, respectivement (Bastien et coll., 1997; MacNeil et coll., 1995)), elle est

négligeable pour le RB, murin (K de 510 nM; Hess et coll., 1996). Pour le RB2, l'ordre
décroissant de puissance pour les kinines endogènes peut être ainsi illustré : BK ~ Lys-BK
» -9 T W „* T , , „ , ! „ „ A _ „ 9 des-Arg -BK et Lys-des-Arg -BK, alors que celui du RB| est plutôt comme suit : Lys-
des-Arg9-BK > Lys-BK a des-Arg9-BK » BK (Leeb-Lundberg et coll., 2005). Il est
possible de considérer que le RB, fait partie d'une sorte de sous-système du système
kinine-kallikréine, sur la base des peptides qu'il lie, qui dépendrait plutôt de l'action de la
kallikréine tissulaire sur les kininogènes.
Il existe un nombre important d'analogues des kinines chez les animaux non mammifères,
comme les oiseaux (Shroeder et coll., 1997), les amphibiens (Colon et Aronsson, 1997;
Yasuhara et coll., 1979) et les reptiles (Kwok et coll., 2008). La peau d'amphibien est une
source particulièrement riche en peptides régulateurs et vasoactifs des vertébrés, constituant
un poison servant à la défense de l'animal. Le venin contenu dans les pustules sécrétoires
de l'espèce Bombina Maxima, un crapaud de la Chine, présente un peptide de dix-neuf
acides aminés, dont les neuf derniers reproduisent la séquence de la BK (Chen et coll.,
2003; O'Rourke et coll., 2004; Chen et coll., 2005). Ce peptide, connu sous le nom de
maximakinine, possède une activité vasorelaxante plus persistante et plus profonde que
celle observée suite à l'ajout de la BK, dans des essais exploitant divers tissus composés de
muscle lisse (O'Rourke et coll., 2004). Les auteurs de cette étude posent l'hypothèse que
les modifications présentées par la maximakinine comparativement à la BK sont
stabilisantes et constituent un exemple d'arme défensive provenant de l'évolution.
1.2 La formation des kinines
Le kininogène de haut poids moléculaire (KHM), une a-globuline circulante comportant 6
domaines, est métabolisée par la KP et libère ainsi la bradykinine (BK), dont la séquence
est contenue au niveau de son quatrième domaine. Chaque domaine de ce kininogène
comporte des fonctions distinctes; il est à souligner que ces fonctions se rapportent à la
liaison de cofacteurs (domaine 1 : liaison du calcium), d'adhésion à divers types cellulaires

(domaine 3 : liaison aux plaquettes et aux cellules endothéliales), ainsi qu'un site de liaison
pour le zymogène de la kallikréine (prékallikréine) et un site d'activation du système de
contact. De fait, la prékallikréine se trouve majoritairement complexée au KHM au niveau
plasmatique (Mandle et coll., 1976; Reddigari et Kaplan, 1989). Son activation dépend de
l'activation du système de contact plasmatique. Deux étapes protéolytiques sont nécessaires
à la génération de la BK : le KHM est d'abord clivé entre les résidus Arg389 et Ser390, en C-
terminal de la séquence de la BK, puis il est clivé entre les résidus Lys et Arg , ce qui
libère la BK (Mori et coll., 1981). Le kininogène de faible poids moléculaire (KFM), une |3-
globuline, est quant à lui clivé par la KT, ce qui permet la formation de la lysyl-BK (Lys-
BK). Le KFM ne se distingue du KHM que par son extrémité C-terminale, sa séquence plus
courte ne comporte ni le site de liaison à la prékallikréine plasmatique, ni ne permet
l'activation du système de contact, aussi appelé la voie intrinsèque de coagulation.
1.2.1 Assemblage du système kinine-kallikréine plasmatique au niveau vasculaire
Lorsque le plasma entre en contact avec une surface chargée négativement, il y a initiation
de la voie de coagulation intrinsèque, qui met en scène l'activation séquentielle de plusieurs
enzymes, généralement des serine proteases. Celle-ci repose sur l'autoactivation du facteur
de Hageman (FXII) en FXIIa, qui est complexé à la prékallikréine et au KHM, ce qui mène
à l'activation de la kallikréine et au clivage subséquent du KHM. Le clivage du KHM
permet de libérer la bradykinine, ainsi qu'une forme active du kininogène (KHMa) dont les
fonctions généralement pro-inflammatoires sont secondaires à sa liaison à des récepteurs
exprimés au niveau d'une variété de cellules immunitaires, ainsi qu'au niveau de cellules
endothéliales (Sainz et coll., 2007). Les surfaces activatrices endogènes ne sont pas
complètement identifiées; parmi elles se retrouveraient hypothétiquement les constituants
de la membrane basale exposée suite à une lésion endothéliale, des proteoglycans libérés
par des cellules immunitaires (Brunnee et coll., 1997), ainsi que différents agents
pathogènes comme les lipopolysaccharides bactériens (Kaplan et Silverberg, 1987) et

d'autres déterminants pathologiques comme les cristaux d'urate (Kaplan et coll., 2002). Par
ailleurs, il est intéressant de souligner que l'activation du système de contact et du
complément, qui libère des peptides antibactériens en plus du peptide pro-inflammatoire
BK, est inhibée par la protéine d'origine bactérienne SIC (Streptococcal Inhibitor of
Complement), libérée par Streptococcus pyogenes et se liant aux kininogènes de manière à
empêcher leur interaction avec des structures activatrices (Akesson et coll., 2010). La
liaison du complexe regroupant la prékallikréine, le KHM et le FXII à celles-ci dépend de
la présence d'une séquence riche en résidus histidines et lysines au niveau du domaine 5 du
KHM (Colman, 1994; Colman et Schmaier, 1997). Bien que les événements décrits ci-haut
soient inclus dans la voie de coagulation intrinsèque, l'importance de cette voie au niveau
de la coagulation est mineure, étant donné l'impact relativement négligeable sur la capacité
hémostatique d'une déficience en FXII, observable chez certains patients (Muller et Renne,
2008). L'activation de cette voie initie aussi le déclenchement de la cascade du complément
(Merlini et coll., 2004). L'activation du système kinine-kallikréine plasmatique et la
production de bradykinine résultantes sont soumises à la régulation négative des serpines,
des inhibiteurs de serine proteases à large spectre qui assurent la conservation de l'équilibre
hémostatique dans le réseau vasculaire (Kaplan et coll., 1997). La serpine majoritairement
impliquée dans la régulation négative du système de contact est l'inhibiteur de Cl
(C1INH), puisqu'elle inhibe les activités de la kallikréine plasmatique et du FXIIa (Cugno
et coll., 1993). Avec l'a2-macroglobuline, elle est responsable de la quasi-totalité de
l'inhibition du système kinine-kallikréine plasmatique, bien que des serpines additionnelles,
notamment l'antithrombine III, l'inhibiteur de protéase-a, et l'a2-antiplasmine, soient aussi
impliquées dans la régulation négative du système de formation des kinines au niveau
plasmatique (Pixley et coll., 1985; Kaplan et coll., 1997;).
Les cellules circulantes du sang, ainsi que les cellules endothéliales bordant la lumière des
vaisseaux sanguins permettent elles aussi l'assemblage du complexe prékallikréine-KHM à
leur membrane plasmique afin de générer les kinines (Zhao et coll., 2001). Par exemple, le
KHM se lie aux cellules endothéliales d'une façon dépendante du zinc (Schmaier et coll.,
1988; Van Iwardeen et coll., 1988) grâce à l'interaction avec un site récepteur du KHM
recrutant le récepteur de Cl (gClqR, un membre du système du complément; Joseph et
coll., 1996; Herwald et coll., 1996), la cytokératine 1 (Hasan et coll., 1998) et le récepteur

de l'activateur du plasminogène urokinase (u-PAR; Colman et coll., 1997). Des voies
alternatives de l'activation de la production de BK au niveau des cellules endothéliales font
intervenir soient la protéine de choc thermique HSP-90 (Joseph et coll., 2002) ou une
prolylcarboxypeptidase (Shariat-Madar et coll., 2002), qui remplacent le FXII dans
l'activation de la prékallikréine.
1.2.2 Assemblage du système kinine-kallikréine tissulaire
La kallikréine tissulaire (KT), aussi identifiée sous le terme peptidase apparentée à la
kallikréine-1 (KLK-1), est probablement le seul membre de la famille des KLK générant
efficacement les kinines (Oikonomopoulou et coll., 2010). Les substrats des autres KLK
sont peu connus, mais parmi ceux-ci se retrouvent les récepteurs activés par les proteases
(PAR). La KT est une serine protease dont la présence dans une variété de tissus (rein,
vaisseaux sanguins, système nerveux central, estomac, glandes salivaires et sudoripares,
rate, neutrophiles; Bhoola et coll., 1992; Mahabeer et Bhoola, 2000) suggère l'importance
de son activité localisée et sa fonction paracrine (Marcondes et Antunes, 2005). Elle est
synthétisée sous forme de zymogène et est activée, quoique peu efficacement, par la
plasmine et la kallikréine plasmatique. La KT permet la libération de la kallidine (KD ou
Lys-BK; Mahabeer et Bhoola, 2000), par le clivage entre les résidus Met379 et Lys380 ainsi
qu'entre les résidus Arg389 et Ser390 (Fogaca et coll., 2004).
1.2.3 Voies alternatives menant à la production des kinines
En plus des kallikréines tissulaire et plasmatique, d'autres proteases ont été rapportées
comme possédant une activité kininogénase. D'abord, la plasmine, une enzyme catalysant
la fibrinolyse, permet la libération de la bradykinine ainsi que celle de la des-Arg -BK par
son activité sur le KHM (Molinaro et coll., 2002). À l'instar des kallikréines tissulaire et

plasmatique, la plasmine se retrouve sous forme de zymogène au niveau de la circulation
sanguine. Le plasminogène, le zymogène de la plasmine, est activé notamment par
l'activateur de plasminogène tissulaire (t-PA), sécrété par les cellules endothéliales, très
actif lorsqu'il est lié à la fibrine (Dobrovolsky et Titaeva, 2002). La proteinase 3 dérivée du
neutrophile (PR3), qui se trouve dans les granules azurophiles et spécifiques ainsi qu'au
sein des vésicules sécrétoires, est libérée lors de la dégranulation du neutrophile (van der
Geld et coll., 2000). Récemment, Kahn et collaborateurs (2009) ont décrit la libération
d'une nouvelle kinine vasoactive libérée par la PR3 à partir du KHM, dont la séquence, qui
comporte deux extensions en N-terminal et en C-terminal de la séquence de la bradykinine
de deux acides aminés chacune, est Met-Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-Ser-
Ser. Cette équipe rapporte que la PR3-kinine, le nom identifiant ce composé, est un
agoniste du RB,, mais suivant son métabolisme, peut aussi activer le RB2. Finalement, il est
à noter que certains microorganismes pathogènes (De Bernardis et coll., 1998), dont ceux
de l'espèce Candida libèrent des enzymes protéolytiques de type aspartyl proteinase qui ont
pour susbtrats les kininogènes et qui permettent ainsi la libération de kinines (Rapala-Kozik
et coll., 2010). Le profil pharmacologique des kinines ainsi produites semble dépendre du
pH de l'environnement du microorganisme.
1.3 Métabolisme des kinines
La demi-vie des kinines au niveau plasmatique est très courte (Ferreira et Vane, 1967), ce
qui s'explique par le nombre important de peptidases pouvant métaboliser les kinines, ainsi
que par leur efficacité. Les peptidases principales reconnues comme étant responsables du
métabolisme des kinines sont les carboxypeptidases M et N (CPM et CPN), l'enzyme de
conversion de I'angiotensine I (ECA), l'endopeptidase neutre 24.11 (néprilysine; NEP),
l'aminopeptidase P (APP) et l'aminopeptidase N (APN). Elles partagent la caractéristique
de nécessiter la présence de zinc comme groupe prosthétique pour être catalytiquement
actives (Moreau et coll., 2005). Plus récemment, la thimet oligopeptidase (EP24.15) a aussi

été identifiée comme étant impliquée dans le métabolisme des kinines (Shivakumar et coll.,
2005). La présente section présentera succinctement ces différentes peptidases, leurs
substrats préférentiels ainsi que leurs metabolites.
1.3.1 Carboxypeptidases N et M
Ces carboxypeptidases sont des métallopeptidases qui présentent une homologie de
séquence de 41 %, qui se distinguent au niveau de leur distribution, puisque la
carboxypeptidase N est sécrétée dans le plasma, alors que la carboxypeptidase M est
exprimée par l'épithélium rénal et pulmonaire (Moreau et coll., 2005). Elles sont aussi
connues sous la nomenclature kininases I (Tan et coll., 1989). Par leur activité
carboxypeptidase qui retire le résidu Arg9 terminal des kinines natives, elles permettent la
génération de la des-Arg9-BK et de la Lys-des-Arg9-BK, des agonistes du RB, (Leeb-
Lundberg et coll., 2005). Leur rôle dans l'inactivation des kinines est mineur; toutefois,
l'inhibition de l'ECA accroît l'importance de leur implication dans le métabolisme des
kinines (Erdôs et coll., 1997; Skidgel, 1988).
1.3.2 Enzyme de conversion de I'angiotensine I
L'ECA, aussi connue sous le nom de kininase II, est une dipeptidyl carboxypeptidase
membranaire de type I. Elle existe sous deux formes; la forme somatique présente une
distribution importante (endothelium vasculaire, poumon et rein), alors que la forme
germinale, est exprimée au niveau testiculaire (Turner et Hooper, 2002; Hagaman et coll.,
1998). Cette enzyme hydrolyse I'angiotensine I (Angl) et la BK, qu'elle metabolise
respectivement en angiotensine II (Ang II, la forme active et vasoconstrictrice) et en BK(,_
5j; un metabolite inactif de la BK vasodilatatrice (Marceau et coll., 1998). La dégradation
de la BK et de la Lys-BK par l'ECA se produit en deux étapes : elle retire d'abord les
acides aminés situés à l'extrémité C-terminale Phe -Arg , ce qui produit la BK(,.7). Elle-
même se verra retirer deux acides aminés additionnels Phe -Ser', afin de générer la BK (1-5)

8
(Inokuchi et Nagamatsu, Oshima et coll., 1985). Un substrat additionnel de l'ECA est le
peptide N-Acétyl-Séryl-Aspartyl-Lysyl-Proline (AcSDKP), un inhibiteur naturel de
l'hématopoïèse et un activateur de l'angiogénèse (Liu et coll., 2003). La forme somatique
de l'ECA possède deux sites catalytiques (Williams et coll., 1994), qui présentent le motif
de liaison à un ion zinc HEXXH. Identifiés sous la terminologie domaine N et domaine C,
ces sites actifs présentent une cinétique de dégradation similaire de la BK (Jaspard et coll.,
1993), quoique le métabolisme de F Angl en Angll soit plus efficacement accompli par le
domaine C (Costerousse et coll., 1992). Une peptidase apparentée à l'ECA a été identifiée
sous le nom ECA2 (Tipnis et coli, 2000), se distingue de l'ECA par sa distribution
restreinte au niveau testiculaire, cardiaque et rénal, par la présence d'un site actif unique et
par son activité carboxypeptidase (Tian et coll., 2004). Elle hydrolyse la des-Arg9-BK en
lui retirant un acide aminé; par contre elle n'a aucune action métabolique sur la BK
(Vickers et coll., 2002). Elle est impliquée dans le métabolisme de l'Angl en Ang[,_9] et en
Ang[i_7]. Le premier de ces metabolites est un peptide amplifiant le relâchement d'acide
arachidonique et facilitant la resensibilisation du RB2, suite à la stimulation par la BK de
cellules endothéliales qui expriment le RB2 et l'ECA de manière constitutive (Marcic et
coll., 1999)etenAng[i_7].
Il est pertinent de souligner l'importance de l'ECA comme cible thérapeutique. Étant donné
son activité vasopressive indirecte par l'activation du peptide vasopresseur Angll et la
dégradation du peptide vasodilatateur BK, elle est impliquée dans une variété de désordres
cardiovasculaires et rénaux. Il existe une grande quantité d'inhibiteurs de l'ECA (IECA)
présents sur le marché, et ceux-ci constituent le traitement de première ligne pour plusieurs
pathologies dont l'hypertension, l'insuffisance cardiaque, l'infarctus du myocarde et la
néphropathie diabétique. Les IECA empêchent la transformation de l'Angl en Angll,
atténuant ainsi les effets hypertenseurs de cette hormone, présentent par contre certains
effets secondaires, dont l'apparition d'une toux sèche (Semple, 1995; Lalloo et coll., 1996;
Dicpinigaitis, 2006) et un risque accru d'angio-œdème (Nussberger et coll., 1998; 2002).
Une partie des effets bénéfiques des IECA, ainsi que leurs effets secondaires découlent de
l'accumulation de la BK. Il a été rapporté récemment que la liaison des inhibiteurs de
l'ECA à leur cible induisait une transduction de signal émanant de l'extrémité C-terminale
intracellulaire de l'ECA (Fleming, 2006). Suivant la liaison de l'inhibiteur à l'ECA, le

résidu Ser1270 est phosphoryle, ce qui active le facteur de transcription c-jun et entraîne sa
translocation nucléaire. Ce facteur de transcription initie l'expression de certains gènes pro
inflammatoires dont la cyclooxygénase-2 (COX-2; Kohlstedt et coll., 2005). Il a par ailleurs
été rapporté qu'un traitement chronique avec les IECA réduit la quantité d'ARN messager
(ARNm) du RB2 (Marceau et coll., 1999) et induit l'expression du RB, (Moreau et coll.,
2005), dépendamment des modèles animaux étudiés.
1.3.3 Endopeptidase neutre (E.C.3.4.24.11)
Aussi appelée néprylisine, l'endopeptidase neutre (NEP) est exprimée au niveau des
cellules épithéliales du rein, du système nerveux central, de l'endothélium et des poumons
(Sales et coll., 1991). Cette enzyme clive son substrat préférentiellement en position N-
terminal d'un résidu hydrophobe. Parmi ses substrats se trouvent les peptides
natriurétiques, l'Angl, l'AnglI, les kinines et la substance P (Campbell et coll., 2003). Elle
procède à l'hydrolyse de la BK en clivant d'abord les deux acides aminés à l'extrémité de
la séquence (Phe8-Arg9), pour ensuite retirer trois acides aminés additionnels (Phe5-Ser6-
Pro7), produisant ainsi le peptide inactif BK[,.4] (Erdôs et coll., 1988). De plus, la NEP
génère aussi la BK[,_4] à partir de la des-Arg -BK. Le métabolisme des kinines au niveau
rénal dépend essentiellement de l'expression de la NEP. Son implication au sein de
l'endothélium vasculaire est controversée et semble être dépendante du modèle exploité
(Tom et coll., 2002; Moreau et coll., 2005). Finalement, l'importance de son activité est
mineure dans le métabolisme des kinines dans le plasma (Décarie et coll., 1996).
Il a été postulé que l'inhibition de la NEP et la potentialisation subséquente des effets des
peptides natriurétiques et des kinines auraient un effet bénéfique sur les fonctions
cardiaques. Malgré des effets variables sur de la pression sanguine (Favrat et coll., 1995;
Ando et coll., 1995) et l'augmentation de la résistance périphérique vasculaire (Kentsch et
coll., 1996), des inhibiteurs de la NEP présentaient un effet mitigé sur l'insuffisance
cardiaque de patients (Eisner et coll., 1992). Ces données ont incité certains groupes à se

10
pencher sur la synthèse de molécules présentant une double identité pharmacologique
d'inhibiteurs de l'ECA et de la NEP (Burnett, 1999) qui allieraient l'effet antihypertenseur
de l'ECA avec les effets vasodilatateurs, diurétiques et natriurétiques de peptides
natriurétiques non métabolisés. Ces inhibiteurs de vasopeptidases, dont le membre
prototype est l'omapatrilat (Pickering et coll., 2002), présentaient une efficacité
thérapeutique supérieure à celle des IECA pour l'atténuation de l'hypertension et pour
l'amélioration de l'insuffisance cardiaque (Pham et coll., 1993; Seymour et coll., 1991;
Robl et coll., 1997). Par contre, la prévalence de l'angio-œdème secondaire à leur
administration (Zanchi et coll., 2003), dont l'accumulation de bradykinine est probablement
en partie responsable, a grandement diminué l'enthousiasme pour ces composés (Pickering
et coll., 2002; Campbell et coll., 2003). Deux stratégies ont été proposées pour pallier à cet
effet secondaire sérieux; la synthèse d'inhibiteurs mixtes de la NEP et l'enzyme de
conversion de l'endothéline (Jeng et coll., 2002), étant donné la puissante activité
vasocontractile de l'endothéline-1 (Kohan, 2010) ou l'optimisation, par modélisation in
silico, d'inhibiteurs des vasopeptidases ECA et NEP, dont le pharmacophore inhibiteur de
l'ECA ciblerait spécifiquement un site catalytique de l'ECA (le site C), afin de permettre la
dégradation de la BK par le site N de l'ECA (Dimitropoulos et coll., 2010).
1.3.4 Aminopeptidase P (APP)
L'APP, une métallopeptidase présente sous forme membranaire et cytosolique au niveau de
l'endothélium vasculaire, de l'épithélium en brosse de l'intestin et du tubule proximal
rénal, possède une activité X-prolyl peptidase. La forme membranaire de l'APP agit à
l'extrémité N-terminale des kinines et metabolise les substrats BK et des-Arg9-BK en les
metabolites inactifs BK[2-9] et BKp-8] (Décarie et coll., 1996; Erdôs et Skidgel, 1997; Biais
et coll., 1999), puisqu'elle retire le résidu Arg en position 1 de ces peptides. Une seconde
enzyme membranaire, la dipeptidyl peptidase IV dégrade à son tour le metabolite inactif
BK[2-9] pour générer le produit BK^j (Lambeir et coll., 2003).

11
1.3.5 Aminopeptidase N (APN)
La métallopeptidase APN est une enzyme distribuée de façon ubiquitaire et possède un
éventail de substrats étendu, car en plus métaboliser des peptides bioactifs comme les
kinines et les neuropeptides, elle dégrade la matrice extracellulaire, ce qui facilite l'invasion
métastasique et ce qui explique l'intérêt qui lui est porté dans la thérapie du cancer (Luan et
coll., 2010). Dans le cas précis du système kinine-kallikréine, son activité est très
importante puisqu'elle retire l'acide aminé Lys, présent en N-terminal de la Lys-BK et de la
Lys-des-Arg9-BK. Ce dernier peptide est l'agoniste naturel de plus haute affinité pour le
RBi, et sa biotransformation en des-Arg9-BK, constitue une voie d'inactivation majeure
pour cet agoniste issu du système kinine-kallikréine tissulaire. Bien que la des-Arg9-BK
soit aussi un agoniste du RB,, elle est beaucoup moins puissante que la Lys-des-Arg9-BK
(Marceau et coll., 1998). L'effet de la Lys-des-Arg9-BK est potentialisé lorsque l'APN est
inhibée (Drapeau et coll., 1991; Pelorosso et coll., 2005).
1.3.6 Oligopeptidase thimet (EP24.15)
La métalloendopeptidase EP24.15, est vastement distribuée dans l'organisme puisqu'elle se
trouve au niveau du cerveau, des testicules, de l'hypophyse, ce qui est cohérent avec sa
fonction neuroendocrinienne, mais plusieurs tissus périphériques tels la rate, le foie, le rein,
le poumon, les glandes surrénales et la thyroïde expriment aussi cette enzyme (Shrimpton
et coll., 2002). La EP24.15 clive le lien entre le résidu Phe5 et le résidu Ser6 présentés par la
séquence de la BK, clivage qui résulte en la BK[,_5] (Dahms et Mentlein, 1992; Norman et
coll., 2003). Il a été rapporté que cette enzyme qui dégrade la BK, interagit avec le RB2,
plus spécifiquement avec la queue C-terminale de celui-ci, au niveau de la membrane
plasmique. Leur cotranslocation concomitante dans le système endosomal où le récepteur
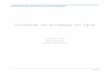
12
est redirigé suite à son activation par la BK (Shivakumar et coll., 2005), permet
hypothétiquement la dégradation intracellulaire de l'agoniste (Sandén et coll., 2008), suite à
sa dissociation du RB2. Cette translocation de l'oligopeptidase thimet avec un récepteur à
sept domaines transmembranaire a aussi été observée dans le cas du récepteur de l'AnglI, le
RATi (Shivakumar et coll., 2005).
Kininogène de haut poids moléculaire
\_C Kallikréine plasmatique Kallikréine tissulaire
Kininogène de bas poids moléculaire
Kallikréine tissulaire
i Kallikréine plasmatique
Bradykinine
î î î î î î i ;
APP APP I NEP ECA CPM DPPIV NEP CPN
ECA Oligopeptidase
thimet 1 Formation dc la dn-ArT-BK
Kallikréine tissulaire
Lys-Bradykinine 1 A ^ ik i r—ik — A j i k
APN APP NEP ECA ECA CPM DPPIV NEP c ? s
I Formation de la
Lys-des-AnT-BK
î APN
Figure 1. Métabolisme des kininogènes et des kinines.

13
1.3.7 Ligands résistants au métabolisme
Le nombre important de peptidases et proteases ayant pour substrats les kinines explique
leur courte demi-vie, de dix-sept à trente secondes pour la BK au niveau plasmatique
(Ferreira et Vane, 1967; Décarie et coll., 1998), et de quelques minutes pour les kinines ne
possédant pas l'arginine en position terminale (des-Arg9 ou des-Arg10, selon la kinine). La
compréhension émergente des fonctions biologiques des kinines a motivé la synthèse de
ligands stables des récepteurs des kinines. La stratégie menant à la synthèse de ligands
peptidiques pour les deux sous-types de récepteurs est essentiellement basée sur la
substitution ou la modification des acides aminés de la séquence naturelle de l'agoniste, par
des acides aminés non naturels (Vavrek et coll., 1992; Drapeau et coll., 1991a; 1991b), ce
qui diminue grandement l'efficacité des diverses peptidases. De plus, le développement de
plusieurs agonistes non peptidiques a contribué à la caractérisation des voies d'inactivation
des kinines, ainsi qu'à l'élucidation des conséquences de cette dégradation sur la
signalisation des récepteurs des kinines. Finalement, les ligands non peptidiques des
récepteurs des kinines présentent une meilleure biodisponibilité orale que les peptides
analogues, ce qui rend ces molécules très attrayantes pour le développement clinique.
La forte activité de dégradation de la BK à proximité des sites récepteurs (Dendorfer et
coll., 2000) a été mise en évidence par l'utilisation de l'agoniste peptidique B-6014, dont la
séquence présente la substitution du résidu proline en position 3 par une hydroxyproline
(Hyp) et celles des résidus phenylalanine en postition 5 et 7 par la thienylalanine (Thi), et
de l'agoniste partiel FR190997 (Asano et coll., 1998), pour mesurer l'importance de la
dégradation de la BK sur la vasodilatation coronarienne. La séquence du peptide B-9972
présente le remplacement de plusieurs acides aminés naturels par des acides aminés non
naturels, ce qui lui confère théoriquement une résistance au métabolisme par diverses
peptidases et proteases, comme les aminopeptidases. In vivo, l'administration du peptide B-
9972 atténue l'hypertension pulmonaire et réduit l'hypertrophie du ventricule droit dans un
modèle de rat présentant une hypertension pulmonaire sévère, en engageant la signalisation
vasodilatatrice classique des RB2 endothéliaux (Taraseviciene-Stewart et coll., 2005).

14
Des analogues de la Lys-des-Arg9-BK, l'agoniste préférentiel du RB,, résistants à l'ECA
(Lys-[D-Phe8]des-Arg9-BK) ou à l'ECA et à l'APM (Sar-[D-Phe8-des-Arg9-BK),
provoquent une hypotension importante et plus persistante chez un modèle de lapin traité
au LPS pendant 5 h, anesthésie et soumis à des bolus intra-artériels de ces agonistes
(Drapeau et coll., 1991a). Des études additionnelles ont aussi mis en évidence la
potentialisation de l'agoniste naturel Lys-des-Arg9-BK au sein d'un essai de contractilité de
l'artère ombilicale humaine, lorsque les courbes concentration-réponse étaient construites
en présence de phosphoramidon, d'amastatine et de captopril, des inhibiteurs de la NEP, de
l'APM et de l'ECA, respectivement (Pelorosso et coll., 2005).
1.4 Les récepteurs des kinines
Il existe deux sous-types de récepteurs des kinines, soient les récepteurs B, (RBi) et B2
(RB2), qui ont été distingués par leur pharmacologie différente (Regoli et Barabé, 1980)
avant d'être caractérisés à l'aide de techniques de biologie moléculaire (Moreau et coll.,
2005b). Les gènes codant chaque récepteur se trouvent en tandem sur un même locus du
chromosome 14. Ce sont des récepteurs à sept domaines hélicoïdaux transmembranaires
couplés aux protéines G (RCPG) de la famille A, couplant préférentiellement les protéines
Gq et Gj, bien qu'il ait été rapporté qu'ils interagissent aussi avec d'autres protéines G,
telles Gs et G12/13 (Leeb-Lundberg et coll., 2005). Cette section présente la régulation de
l'expression des récepteurs, ainsi que les déterminants structuraux des récepteurs qui
expliquent leurs fonctions et leurs partenaires moléculaires.
1.4.1 Expression des récepteurs des kinines
L'expression des RB peut être observée au niveau d'un grand nombre de tissus, comme le
tissu vasculaire (endothelium et muscle lisse; Figueroa et coll., 2001), au sein du système

15
nerveux (Bascands et coll., 2003), ainsi que sur une variété de tissus néoplasiques (Jutras
et coll., 2010; Lu et coll., 2010; Stewart et coll., 2002; Wang et coll., 2001).
Le contexte d'expression des deux récepteurs est très différent. Le RB2 est exprimé de
manière constitutive et peut voir son expression amplifiée, soit de manière dépendante de la
présence de son agoniste, par l'effet de l'AMP cyclique (AMPC) ou par une stimulation des
cellules par les phorbol esters, qui sont des agents mimétiques du DAG (Pesquero et coll.,
1996). Le RB, est très peu exprimé dans un contexte physiologique et dépend de plusieurs
stimuli pour l'induction de son expression (Leeb-Lundberg et coll., 2005), sauf au niveau
de la moelle épinière de souris, où une quantité basale d'ARNm du RB| a été détectée
(Leeb-Lundberg et coll., 2005). Il a été rapporté que l'expression du RB, pouvait être
secondaire à des dommages tissulaires (Regoli et coll., 1978), tels que mimés par l'isolation
de vaisseaux sanguins et leur incubation subséquente in vitro. Par exemple, l'aorte de lapin
ainsi manipulée acquiert une activité contractile en réponse aux agonistes du RB,
(Bouthillier et coll., 1987), ce qui a été observé pour plusieurs autres types de vaisseaux,
provenant de la même espèce ou d'espèces différentes (Marceau, 1995). Cette induction de
l'expression du RB, est dépendante de la transcription du gène du RB, et de sa traduction,
puisqu'un inhibiteur de la synthèse d'ARNm (actinomycine D) ou de la synthèse protéique
(cycloheximide) l'inhibe (Whalley et coll., 1983). Sa présence à la membrane plasmique,
détectable par l'activité contractile générée en réponse aux kinines ne possédant par l'Arg
en position terminale, dépend additionnellement de certaines modifications post-
traductionnelles et de son transport à la membrane (sa transition reticulum endoplasmique-
appareil de Golgi est inhibée par la brefeldine A; Audet et coll., 1994). Par ailleurs, les
tissus provenant d'animaux prétraités avec des lipopolysaccharides bactériens (LPS)
présentent une activité contractile dépendante du RB, beaucoup plus précoce que ne l'est
celle des vaisseaux provenant d'animaux non traités. Conséquemment, l'un des premiers
modèles in vivo de l'induction de l'expression du RB, a été obtenu par l'injection d'une
dose non létale de LPS chez le lapin. L'aorte de lapin traité isolée et installée dans un bain à
organes isolés permet l'observation d'une réponse contractile à la des-Arg9-BK et à la Lys-
des-Arg9-BK (Regoli et coll., 1981; Drapeau et coll., 1981a). C'est cette observation qui
illustre la relation intime entre l'expression du RB| avec les stimuli pro-inflammatoires.
Plusieurs cytokines, dont l'interleukine (IL)-1 p (DeBlois et coll., 1991), l'interféron-y

16
(IFN-y) et le tumor necrosis factor-a (TNF-a; Bawolak et coll., 2008; Koumbadinga et
coll., 2010), ainsi que des facteurs de croissance, comme l'EGF (Schneck et coll., 1994) ont
été impliqués dans la hausse de l'expression du RB,. Le rôle du facteur de transcription
NF-KB dans l'induction de l'expression du RB,, pour lequel le promoteur du récepteur
contient un site de liaison (Leeb-Lundberg et coll., 2005) est mis en lumière par l'effet
inhibiteur de stratégies comme l'atténuation transcriptionnelle de la sous-unité p65 du
facteur de transcription au niveau du muscle lisse vasculaire (Moreau et coli, 2007) et le
traitement du système tissulaire ou cellulaire à l'étude avec des glucocorticoïdes (deBlois et
coll., 1988; Sabourin et coll., 2002a; Moreau et coll., 2007). Il est à noter que le facteur de
transcription NF-KB est impliqué dans l'induction de l'expression d'un grand nombre de
médiateurs de l'inflammation (Lawrence, 2009), et que, par les stimuli menant à son
expression, le RB, s'apparente à certaines molécules proinflammatoires, comme les
enzymes cyclooxygénase-2 (COX-2; Tsatsanis et coll., 2006) et la synthase de l'oxyde
nitrique inductible (iNOS; Kleinert et coll., 2003). À la régulation transcriptionnelle du
RB,, s'ajoute le phénomène de stabilisation de l'ARNm transcrit, qui a d'abord été
documenté au niveau de la lignée cellulaire IMR-90, dérivée de cellules pulmonaires
embryonnaires, suite à leur traitement par ITL-1(3 (Zhou et coll., 1998). Des travaux plus
récents, exploitant un modèle cellulaire d'artère ombilicale humaine, ont contribué à
distinguer les voies inductrices du RB, selon leur dépendance à l'activité du facteur de
transcription NF-KB (IL-1(3 et le phorbol 12-myristate 13-acétate (PMA)) et leur capacité à
stabiliser l'ARNm du RB, (IL-ip, PMA et les sera humain et bovin; Moreau et coll., 2007).
Finalement, il a été rapporté que l'expression du RB, pouvait être soumise à
l'autorégulation par les kinines, puisque la présence de la BK ou de la des-Arg9-BK
augmentait le niveau d'expression du récepteur, dans la lignée cellulaire IMR-90 (Schanstra
et coll., 1998; Phagoo et coll., 1999; 2001). Remarquons que ce modèle cellulaire libère de
l'IL-1(3 de manière autocrine (Bastian et coll., 1998), ce qui serait suffisant à l'induction de
l'expression du RB,; par ailleurs, ces résultats n'ont pu être reproduits dans des modèles de
muscle lisse vasculaire de lapin ou humain, stimulés avec des agonistes des deux sous-
types de récepteurs (Sabourin et coll., 2001 ; Moreau et coll., 2007).

17
1.4.2 Structure des récepteurs des kinines
Les récepteurs des kinines sont des RCPG de la classe A et sont composés d'une longue
chaîne polypeptidique, dont l'extrémité N-terminale est extracellulaire et l'extrémité C-
terminale est intracellulaire. Entre les deux sous-types de récepteurs, on observe une
homologie de séquence de 36%, les domaines présentant la plus forte homologie étant les
domaines transmembranaires (Leeb-Lundberg et coll., 2005). Cette chaîne polypeptidique
traverse la membrane à sept reprises, formant ainsi trois boucles extracellulaires et autant
de boucles intracellulaires. Les deux récepteurs présentent le motif DRY, un motif
hautement conservé au sein de la famille des RCPG de classe A (Fredriksson et coll., 2003)
au niveau de leur deuxième boucle intracellulaire tout juste sous le troisième domaine
transmembranaire (Leeb-Lundberg et coll., 2005). Ce motif remplit une fonction de
maintien de l'état basai du RCPG (Flanagan, 2005), puisque sa mutation mène
fréquemment à une activation constitutive du récepteur. Plus récemment, il a été suggéré
que cette séquence conservée avait une utilité différente dépendamment du RCPG à l'étude;
un premier phénotype de récepteur est contraint dans un état basai grâce au motif DRY,
alors que le second phénotype met plutôt en lumière une fonction de reconnaissance et de
liaison de la protéine G du motif DRY (Rovati et coll., 2007). Par ailleurs, cette séquence a
été impliquée dans le recrutement de la (3-arrestine au récepteur de la chimiokine 5 (CCR5;
Huttenrauch et coll., 2002). Une mutation de cette séquence au niveau du RB2 entraîne une
perte de l'expression de ce récepteur à la surface membranaire (Faussner et coll., 2009)
rendant toute interprétation de son rôle au sein de ce récepteur difficile. Une séquence
additionnelle hautement conservée au sein des RCPG de classe A est la séquence NPXXY
(Stenkamp et coll., 2005), présente dans le domaine transmembranaire 7 (TM7) et
probablement impliquée dans le maintien de la structure intramoléculaire du RCPG.
Lorsque ce motif est muté au niveau du RB2, il a été rapporté que ce récepteur est soumis à
la phosphorylation et l'internalisation de manière constitutive (Kalatskaya et coll., 2004).
Les RB subissent des modifications post-traductionnelles supplémentaires importantes pour
leur structure et leurs fonctions, qui sont présentées dans cette section.

18
1.4.2.1 Glycosylation
La glycosylation est la modification post-traductionnelle la plus fréquemment observée
chez les RCPG (Duvemay et coll., 2005). L'ajout du groupement oligosaccharide sur un
résidu asparagine (Asn ou N; d'où le terme N-glycosylation) au niveau de la séquence Asn-
Xaa-Ser/Thr est initié dans le reticulum endoplasmique, et peut se poursuivre lors de
l'acheminement du RCPG vers la membrane dans l'appareil de Golgi. Son implication dans
la maturation et l'expression membranaire des différents RCPG est caractérisée par un
penetrance variable. Les deux sous-types des récepteurs des kinines sont N-glycosylés.
L'étude des RB2 humain (Hess et coll., 1992) et de rat (McEachern et coll., 1991) a permis
de prédire la présence de 3 sites de N-glycosylation, dont deux au niveau de l'extrémité N-
terminale et un dans la seconde boucle extracellulaire. Les travaux de Yaqoob et
collaborateurs (1995), portant sur le RB2 de rat, ont permis d'abord de vérifier la présence
de glycosylation à ces sites ainsi qu'à caractériser le type d'oligosaccharide ainsi
complexés, qui dans le cas présent contiennent des résidus terminaux d'acide sialique. Par
contre, cette modification du RB2 de rat ne semble pas être impliquée dans la
reconnaissance et la liaison du ligand. Au niveau du RB2 humain, la mutation de sites de N-
glycosylation n'a aucun effet sur la liaison de l'agoniste au RB2, ni sur son internalisation
subséquente, mais diminue le niveau d'expression du RB2 et affecte négativement le
couplage aux seconds messagers habituellement recrutés par le récepteur (Michineau et
coll., 2004). La glycosylation du RB, présente un patron similaire à celui du RB2 et joue un
rôle déterminant dans l'expression membranaire du récepteur mature. Effectivement,
l'incubation de l'artère ombilicale humaine (Sardi et coll., 1999) ou de l'aorte de lapin
(Audet et coll., 1994) fraîchement isolées en présence de tunicamycine, un inhibiteur de la
glycosylation, empêchait l'apparition de l'activité contractile du vaisseau en réponse à la
des-Arg -BK. Par ailleurs, l'utilisation d'un RB, de lapin couplé à la protéine jaune
fluorescente (RB|-YFP) permet la visualisation de la construction fluorescente fortement
retenue au niveau intracellulaire. Son expression membranaire est amplifiée suite à
l'utilisation d'une chaperonne pharmacologique de type antagoniste non peptidique et cette

19
hausse est parallèle à l'augmentation de l'abondance de RB, hautement glycosylés détectés
par immunobuvardage (Fortin et coll., 2006a).
1.4.2.2 Phosphorylation
La phosphorylation des RCPG est un phénomène dépendant principalement de la liaison
d'un agoniste. Cette phosphorylation réversible est observable au niveau de résidus serine
et threonine présents dans l'extrémité C-terminale et joue un rôle déterminant pour la
désensibilisation des RCPG. Les RB2 d'origine mammifère et non mammifère présentent
tous une région riche en serine et threonine au niveau de leur extrémité C-terminale. En
plus du résidu Ser , qui semble être phosphoryle de manière basale, l'équipe de Blaukat et
collaborateurs ont identifié les résidus Ser339, Ser346, Thr342 et Thr345 comme étant les
résidus phosphorylables du RB2 humain (Blaukat et coll., 1999; 2001) en réponse à
l'agoniste. Ces études ont permis de caractériser l'importance majeure de la
phosphorylation de ces résidus serine dans la désensibilisation du récepteur et de
l'influence mineure de la phosphorylation des résidus threonine identifiés. De plus, les
études de Blaukat et collaborateurs (2001) ont révélé le caractère nécessaire et suffisant de
deux événements de phosphorylation minimaux entre les trois résidus serine (Ser et
Ser348 ou entre Ser339 et Ser346) pour permettre une désensibilisation efficace de la
signalisation induite par la stimulation dépendante de la BK. Le RB, présente des résidus
serine et threonine au niveau de son extrémité C-terminale, mais ces résidus sont peu
conservés à travers les espèces et ne sont pas phosphorylés de manière dépendante de la
liaison de l'agoniste (Blaukat et coll., 1999). Les événements menant à la désensibilisation
des RB, ainsi que les intervenants moléculaires de celle-ci seront présentés plus bas.
La phosphorylation de résidus tyrosine au niveau du RB2 a été rapportée, autant chez le n i "KJJ
RB2 humain (Jong et coll., 1993) que pour l'orthologue de rat (les résidus Tyr et Tyr ;
Soskic et coll., 1999). La phosphorylation de ces résidus n'est pas dépendante de la

20
stimulation du récepteur par l'agoniste, ni ne semble influencer sa signalisation (Prado et
coll., 1997).
1.4.2.3 Oligomérisation
L'oligomérisation des RCPG en des complexes de signalisation plus importants regroupant
deux récepteurs ou plus est un phénomène décrit relativement récemment (Milligan, 2004).
Bien qu'un RCPG fonctionne de manière adéquate en monomère, il a été suggéré qu'il
existe un équilibre entre sa forme monomérique et dimérique ou oligomérique (Milligan,
2004; 2008). La stabilité des oligomères dépend de l'affinité de l'interaction entre les
récepteurs, ainsi que du niveau d'expression du récepteur, et peut être soumise à une
modulation par le ligand (Smith et Milligan, 2010). Il existe deux catégories générales
d'oligomères; soient l'homomère (de 2 récepteurs ou plus du même type) ou l'hétéromère,
qui forment tous deux une macromolécule pharmacologiquement et biochimiquement
distincte de ces composants (Ferré et coli, 2009). L'avantage principal d'une telle
réorganisation des récepteurs est l'accroissement des possibilités de signalisation par
l'activation ou l'inhibition croisée d'un RCPG par un autre activé par son agoniste (Smith
et Milligan, 2010). Il a par ailleurs été observé que l'oligomérisation de certains récepteurs
facilitait leur maturation et leur expression membranaire (Canals et coll., 2009) et pouvait
modifier leur profil de désensibilisation en favorisant ou en empêchant leur internalisation
(Smith et Milligan, 2010).
AbdAlla et collaborateurs (1999) ont mis en lumière une dimérisation dépendante de
l'agoniste du RB2 endogène, tel qu'exprimé par des cellules PC-12, qui impliquerait
l'extrémité N-terminale des récepteurs dans la formation de ces homodimères, essentiels à ■y
une internalisation adéquate d'un agoniste radiomarqué [ H]BK et probablement à la
désensibilisation efficace du RB2. La stabilisation des homodimères du RB2 peut être
attribuable à des modifications post-traductionnelles comme la N-glycosylation et la
formation de ponts disulfure (Michineau et coll., 2006). La présence du RB, au sein

21
d'homodimères a été détectée par plusieurs techniques (immunoprecipitation,
immunobuvardage et microscopie électronique) et a pu permettre d'attribuer leur formation
à l'interaction de domaines transmembranaires (Kang et coll., 2005). Cette
homodimérisation constitutive est apparamment requise pour l'expression optimale du
récepteur à la surface membranaire. L'hypothèse d'un hétérodimère entre le RB, et le RB2
exprimé par la lignée cellulaire PC3 (Barki-Harrington et coll., 2003) est supportée par
l'observation d'une inhibition croisée de chacun des sous-types de récepteurs par l'emploi
d'un antagoniste sélectif de l'autre récepteur, sur la signalisation activant la voie des
MAPK, ainsi que sur la prolifération cellulaire. Par ailleurs, des études de Kang et
collaborateurs (2004) ont caractérisé l'état protéolytiquement altéré du RB2 au sein de
l'hétérodimère le conjuguant avec le RB,. La formation spontanée d'un dimère suivant
l'induction du RB, en parallèle avec la dégradation du RB2 pourrait constituer l'un des
mécanismes d'adaptation du système kinine-kallikréine en ce qui a trait à l'expression des
récepteurs lors d'une inflammation aiguë (dont la réponse dépend du RB2) et lors d'une
chronicisation de celle-ci (qui met en lumière l'importance du RB,, suite à la perte de
l'expression du RB2).
Un hétérodimère regroupant le RAT, et le RB2 (Fior et coll., 1993), hypothétiquement
présent au niveau de systèmes natifs (AbdAlla et coll., 2005) ainsi qu'/« vivo chez des
patientes souffrant de préeclampsie (AbdAlla et coll., 2001) pourrait revêtir d'une
importance physiopathologique. On suppose en effet une amplification de la signalisation
pro-hypertensive découlant de la stimulation du RAT,, lorsque complexé au RB2. La
maturation efficace du RB2, aidée par la chaperonne calréticuline (AbdAlla et coll., 2009),
semble être une condition requise pour son hétérodimérisation avec le RAT|. Par contre,
l'existence d'un tel dimère demeure controversée (Hansen et coll., 2009). Très récemment,
l'identification d'un hétérodimère du RB2 et du récepteur pVadrénergique (RP2A) par
Haack et collaborateurs (2010) a mis en évidence la possibilité que deux RCPG, couplés à
des protéines G différentes, forment un complexe permettant la transactivation du récepteur
conjugué (R(32A) par la stimulation du second récepteur (RB2) par son agoniste (BK). Bien
que ces deux récepteurs présentent au sein de leur patron de distribution tissulaire plusieurs
sites de colocalisation, l'importance clinique de cet hétérodimère reste à évaluer.

22
L'importance du phénomène d'oligomérisation des RCPG, relativement peu documenté et
parfois controversé, devra être étudiée plus avant. Par contre, son influence sur l'intégration
de voies de signalisation concurrentes sur la réponse cellulaire ultime sera indéniable, si sa
prépondérance in vivo est démontrée.
1.4.2.4 Palmitoylation
Au niveau de leur domaine C-terminal intracellulaire, à l'instar de plusieurs RCPG
(O'Dowd et coll., 1989), les récepteurs des kinines présentent des résidus cysteine (Cys)
qui peuvent être modifiés par palmitoylation, ce qui permet l'ancrange du groupement acyl
au niveau de la membrane. Les RB2 de mammifères possèdent deux résidus Cys au niveau
de leur extrémité C-terminale, présents dans le motif CxxxGC; leur modification par l'ajout
d'un groupement palmitoyl diffère selon les espèces. Les deux résidus Cys en position
proximale 324 et distale 329 du RB2 humain peuvent être tous deux palmitoylés (Pizard et
coll., 2001), alors que seul le résidu Cys en position proximale 326 (du RB2 de rat) est
modifié de cette façon (Soskic et coll., 1999). Dans le cas du RB,, qui présente une
importante variabilité interespèce de la longueur de son extrémité C-terminale,
l'importance de l'acylation du domaine intracellulaire peut être négligeable pour sa
fonction (Leeb-Lundberg et coll., 2005). Les RB, de rongeur sont dépourvus de cysteines
au niveau de sa très courte queue C-terminale, alors que les RB, non rongeur présentent en
revanche une à deux cysteines à leur extrémité C-terminale (Marceau et coll., 1998). Ce
domaine est impliqué dans le couplage de la protéine G, l'internalisation du RB2, sa
resensibilisation et son trafic intracellulaire (Leeb-Lundberg et coll., 2005). Par la présence
de ce thioester labile modifiable, la palmitoylation ainsi que la dépalmitoylation peuvent
faciliter la phosphorylation et la désensibilisation du RCPG (Bôhm et coll., 1997). La
palmitoylation est une modification post-traductionnelle impliquée dans l'association des
protéines à des domaines membranaires précurseurs des cavéoles, insolubles aux détergents
et présentant un enrichissment en glycosphingolipides.

23
1.4.3 Relations de s t ructure-act ivi té des récepteurs des kinines
1.4.3.1 Fonction de reconnaissance des ligands
La structure des RCPG doit permettre leur interaction avec minimalement deux molécules;
soient l'agoniste et la protéine G. Le premier modèle d'homologie du RB2, basé sur la
structure de la bactériorhodopsine, a été construit dans le but d'évaluer l'importance de la
longueur de la chaîne carbonée d'une série d'antagonistes dont la structure dérivait du
composé NPC 18352 (Kyle, 1994). Les sites de reconnaissance et de liaison des kinines
avec les deux sous-types de récepteurs ont été élucidés grâce à l'application de la méthode
de reticulation et à la mutagénèse dirigée (Nardone et Hogan, 1994; Herzig et coll., 1996;
Jarnagin et coll., 1996; Meini et coll., 2002; Belluci et coll., 2003; Leeb-Lundberg et coll.,
2005; Ha et coll., 2006). Le site de liaison de l'agoniste au récepteur des kinines se situe
dans la cavité formée par les différents domaines transmembranaires, à l'instar de plusieurs
autres RCPG (Baldwin, 1994; Fredriksson et coll., 2003). Au niveau du RB2, les études de
plusieurs équipes ont permis de dresser le portrait de la liaison de la BK à son récepteur.
Les études de Nardone et Hogan (1994) sur le RB2 de rat ont identifié l'importance de
plusieurs résidus du domaine transmembranaire 6 (TM6) dans la liaison de la BK, puisque
la mutation des résidus F261 (correspondant au résidu F259 de l'orthologue humain) et
T265 réduisait l'affinité de liaison à la BK. Le résidu F259 est essentiel à la liaison de la
BK à son récepteur (Jarnagin et coll., 1996; Marie et coll., 2001). Le TM6 participe
probablement aussi aux contacts intraprotéiques stabilisant la conformation du récepteur
lorsque l'agoniste est lié. Par ailleurs, le pont disulfure présent ente les cysteines 103 (une
cysteine hautement conservée parmi les RCPG de type A au niveau de l'extrémité C-
terminale de la première boucle extracellulaire; Peeters et coll., 2010) et 184 (se trouvant au
niveau de la seconde boucle extracellulaire; Herzig et coll., 1996) permet la formation du
site de liaison de haute affinité pour l'agoniste en rigidifiant les domaines extracellulaires et
donnant une structure adéquate au récepteur (Peeters et coll., 2010). Il a aussi été rapporté
que la seconde boucle extracellulaire présentait une conformation ouverte en l'absence de
ligand (Avlani et coll., 2007) et que la liaison d'un agoniste provoquait sa fermeture et la

24
stabilisation du complexe ligand-récepteur par la formation d'interactions additionnelles
(Peeters et coll., 2010). Des études de Abd Alla et collaborateurs (1996) ont montré, à
l'aide d'anticorps dirigés contre des epitopes présents au niveau des différents domaines
extracellulaire, l'importance de l'accesibilité aux deuxième et troisième boucles
extracellulaires pour une liaison efficace de la BK, comparativement à l'importance de la
deuxième boucle seulement pour la liaison de l'antagoniste HOE-140. Finalement, des
anticorps dirigés contre le domaine N-terminal du récepteur ou la première boucle
extracellulaire n'influençaient pas la liaison des ligands.
L'extrémité C-terminale de la BK pénètre dans la rosette formée par les 7 domaines
transmembranaires à structure en hélice; le résidu arginine en position terminale (position
9) interagit avec le résidu aspartate en position 286 du RB2 de rat (Novotny et coll., 1994;
correspondant au résidu Asp en 287; McEachern et coll., 1991) au niveau du domaine
transmembranaire 7 (TM7), en formant un pont salin (Jarnagin et coll., 1996), ce qui
permet au résidu phenylalanine en position 8 de la BK de pénétrer dans la pochette de
liaison de nature hydrophobe au niveau du TM7. Le résidu proline en position 7 de la BK
interagit quand à lui avec la cavité hydrophobe formée par les résidus phenylalanine 261,
leucine 104, valine 108 et isoleucine 112 au niveau du domaine transmembranaire 3 (TM3;
Chakraborty et coll., 1995). L'extrémité amino-terminale de l'agoniste, dirigée vers le
milieu extracellulaire, se trouve à proximité des résidus cysteine en position 20 à
l'extrémité N-terminale du récepteur et en position 277 au niveau de la troisième boucle
extracellulaire (Herzig et coll., 1996), comme le démontrent des études basées sur la
reticulation entre l'extrémité N-terminale de la BK et un groupement sulfydryle libre
(Herzig et Leeb-Lundberg, 1995). Il est important de noter que les résidus impliqués dans
la liaison des agonistes non peptidiques, telle que présentée par Bellucci et collaborateurs
(2003) pour le composé FR 190997, ainsi que pour les antagonistes peptidiques (Jarnagin et
coll., 1996) et non peptidiques (Meini et coll., 2002) ne sont pas les mêmes.
La transition d'une identité agoniste vers une identité antagoniste, par la substitution de la
proline en position 7 de la séquence naturelle de la BK par le résidu D-phénylalanine a été
rapportée par Vavrek et Stewart (1985). La modification de plusieurs autres peptides a
permis l'identification de l'orientation de l'extrémité C-terminale comme un facteur

25
primordial déterminant la nature des ligands des kinines. Il est important de noter que les
antagonistes dépendent moins de l'interaction avec le TM6, entrant préférentiellement en
contact avec le TM7. L'usage de ligands présentant une conformation contrainte est
intéressant à plusieurs points de vue, puisqu'une telle structure limite l'adoption d'un grand
nombre de conformations par le peptide. Il est alors possible d'observer la plus grande
efficacité d'un composé par la stabilisation de la conformation optimalement active, de
diminuer la dégradation enzymatique du peptide et accroître ainsi sa stabilité. D'un point de
vue du développement clinique, cela permet d'éliminer d'éventuels conformères ayant des
effets biologiques indésirables (Amblard et coll., 1999). Par ailleurs, l'utilisation de tels
peptides à la structure contrainte offre plusieurs renseignements sur les interactions
moléculaires qui unissent le ligand et son récepteur. Le peptide B-9430 est un antagoniste
de haute affinité pour le RB2, présentant une structure contrainte (Stewart et coll., 1996).
Son isomère le peptide B-9972 est un agoniste complet ou partiel possédant une importante
activité intrinsèque (Bironaite et coll., 2004; Taraseviciene-Stewart et coll., 2005). De plus,
une contrainte structurale du squelette du peptide ne semble pas incompatible avec une
identité agoniste.
La quantité importante de ligands des récepteurs des kinines synthétisés au cours des trente
dernières années (Marceau et Regoli, 2004; Fortin et Marceau, 2006) a grandement
contribué à parfaire les connaissances sur la relation structure-activité des ligands
peptidiques au niveau de chacun des sous-types de récepteurs. Succinctement, l'extrémité
C-terminale d'un peptide agoniste ou antagoniste pénètre dans la rosette formée par les sept
domaines transmembranaires, où elle interagit avec certains domaines, alors que l'extrémité
N-terminale sort de la rosette et interagit plutôt avec des résidus chargés négativement des
boucles extracellulaires (Leeb-Lundberg et coll., 2005). Ainsi, le couplage d'un cargo à
l'extrémité N-terminale des peptides pourrait être envisagé, sans craindre d'abolir
complètement la liaison du ligand au récepteur. Cela est d'ailleurs confirmé par plusieurs
publications. À titre d'exemple, la séquence naturelle Met-Lys-BK est un agoniste
légèrement moins actif que la BK au niveau des RB2 recombinants humain et murin
(Marceau et coll., 1998). De plus, l'extension permissive en N-terminal d'agonistes et
d'antagonistes du RB, est illustrée par le développement du radioligand de haute affinité
[' I]-Tyr-Gly-Lys-e-aminocaproyl-Lys-des-Arg -BK (Levesque et coll., 1995) ainsi que

26
par le très puissant antagoniste du RB, B-10324 (2,3,4,5,6-pentafluorocinnamyl-B-9958;
Géra et coll., 2008).
La cristallisation des RCPG est un processus complexe à cause de l'instabilité de ceux-ci
lorsqu'ils se retrouvent hors de la couche bilipidique formant la membrane plasmique et de
leur capacité à adopter une multitude de conformations (Peeters et coll., 2010). Le premier
RCPG dont la structure a été cristallisée est la rhodopsine bovine (Palczewski et coll.,
2000). La connaissance de cette structure a permis la modélisation moléculaire du RB, par
Ha et collaborateurs (2006), augmentant la pertinence de la mise en contexte de
l'élucidation les résidus du RB, impliqués dans la liaison de l'agoniste. Cette méthodologie
a permis la localisation des résidus lysine en position 1 et arginine en position 2 de
l'agoniste Lys-des-Arg9-BK à proximité des résidus aspartate 291 et glutamate 273, alors
que le résidu lysine 118 est nécessaire pour une affinité de liaison optimale avec le résidu
phenylalanine en position terminale (9) de l'agoniste, l'anneau aromatique de cet acide
aminé se trouvant à proximité des résidus alanine 270 et leucine 294. De plus, l'emploi
d'un récepteur chimérique RB1-RB2 a mené à la caractérisation du rôle du TM3 au niveau
de la discrimination entre des ligands sélectifs de chaque récepteur. En effet, une
construction présentant le RB, dont ont été remplacé le TM7 et l'extrémité C-terminale par
ceux du RB2 (construction 6; Bastian et coll., 2000), présente une diminution importante de
l'affinité pour des ligands agonistes et antagonistes, alors que la susbtitution du TM7 du
RB2 accroît l'affinité avec laquelle le RB, chimérique lie le HOE-140, un antagoniste
peptidique du RB2.
1.4.3.2 Activation du récepteur et signalisation
La liaison de l'agoniste au récepteur permet un changement conformationnel, qui dépend
du mouvement des TM3, TM6 et TM7 (Gether, 2000; Meng et Bourne, 2001). Le
complexe agoniste-récepteur activé catalyse l'échange de nucleotides guanidines à la sous-
unité a de la protéine G hétérotrimétique (Ga) liée au GDP, complexée aux sous-unités

27
(3 et y (G(3y)- La liaison du GTP à la sous-unité a provoque la dissociation de la protéine G
hétérotrimérique et permet aux sous-unités d'activer différentes voies de signalisation.
L'activité GTPase intrinsèque de la sous-unité Ga provoque l'hydrolyse de la molécule et
la sous-unité Ga est de nouveau associée au GDP, ce qui diminue son affinité pour ses
effecteurs, mais qui accroît celle pour les sous-unités G|3y, ce qui permet la réassociation de
la protéine G et son couplage à un récepteur activé (Sidorovski et Willard, 2005). La liaison
sélective de la protéines Gq, partenaire principal de la signalisation du RB2, et de la protéine
Gi, dépend grandement de la seconde boucle intracellulaire (Faussner et coll., 2009), en
particulier au niveau du motif LALV (Pal-Ghosh et coll., 2003). Les résidus aux positions
310 à 320 de l'extrémité C-terminale du RB2 forme une structure hélicoïdale (parfois
identifiée comme l'hélice 8 du RB2; Kang et Leeb-Lundberg; 2002; Faussner et coll.,
2005); cette structure participe aussi à l'interaction du récepteur avec la sous-unité a des
protéines Gq et Gj, comme l'indiquent l'importance des régions proximales et médianes
(surtout le motif KS-(R/W)-EVY) pour l'initiation de la signalisation classique de ces types
de protéines G, telle que mesurée par le métabolisme du phosphatidylinositol (PI), et par le
relâchement de l'acide arachidonique (Yu et coll., 2004). En plus de lier les protéines Gq et
Gj, il a été rapporté que le RB2 activé pouvait catalyser l'échange de GDP pour le GTP des
protéines G12/13, qui activent les petites protéines G impliquées dans la modification du
cytosquelette, la morphologie cellulaire et la mobilité (Gohla et coll., 1999), et les protéines
Gs, activatrices des voies dépendantes de l'AMPc et de la protéine kinase A (PKA; Graness
et coll., 1997). Par contre, l'impact physiologique du couplage du RB2 avec d'autres
protéines G que la protéine Gq est peu documenté (Blaukat, 2003).
Le couplage du récepteur B2 lié à son agoniste active la protéine Gq, qui elle-même activera
ses effecteurs, dont la phospholipase C. La phospholipase C (isoformes (3 et y) a pour
substrat un phospholipide membranaire, le phosphatidylinositol 4,5-bisphosphate (4,5-
PIP2). Son clivage entraîne la formation de deux seconds messagers principaux soit le
diacylglycérol (DAG) et l'inositol-3-phosphate (IP3). LTP3 est un agoniste d'un canal
calcique présent au niveau du reticulum endoplasmique, dont la stimulation provoque le
relâchement du calcium (Ca +) qui est mis en réserve au niveau intracellulaire à cet endroit
(Leeb-Lundberg et coll., 2005). La hausse de la concentration cytosolique de Ca2+ stimule

28
l'entrée de Ca2+ présent au niveau extracellulaire par des canaux cationiques non-sélectifs,
des canaux voltage-dépendants ou par le Ca2+ release-activated calcium channel (CRAC)
(Spassova et coll., 2006). Le Ca2+ intracellulaire se lie à plusieurs protéines appartenant à la
superfamille des calcium related signaling proteins (dont la calmoduline fait partie), qui se
caractérisent par la présence dans leur séquence d'un motif de type Hélice-Boucle-Hélice
(d'une taille de 29 acides aminés) qui permet de lier avec une très haute affinité le Ca2+
(Gifford et coll., 2007). Au niveau endothelial, la liaison du Ca2+ à la calmoduline, qui
comporte quatre sites de liaison au Ca fonctionnant de manière coopérative positive, va
mener à l'activation de la calmoduline, afin que celle-ci puisse à son tour activer une
enzyme nommée la nitric oxyde synthase (NOS) dont il existe une isoforme endothéliale
(eNOS). Cette enzyme est responsable de la synthèse de l'oxyde nitrique (NO), un gaz qui
a la capacité de diffuser à travers les membranes plasmiques et activer ainsi la guanylyl
cyclase cytosolique qui se trouve dans le type cellulaire sous-jacent à l'endothélium au
niveau du vaisseau sanguin, le muscle lisse vasculaire (Mombouli et Vanhoutte, 1995). La
guanylyl cyclase activée produit le GMPC. Le GMPC active à son tour la protéine kinase G
(PKG) dont le substrat est la myosine phosphatase (Nakamura et coll., 1999). Cette
phosphorylation étant activatrice, la myosine phosphatase déphosphorylé alors la chaîne
légère de la myosine et inactive ainsi sa fonction ATPase et empêche de cette façon la
contraction du muscle lisse vasculaire. Dans le cas où l'endothélium est abîmé, la
bradykinine peut aussi activer directement le muscle lisse vasculaire en se liant aux
récepteurs B2 présents à sa surface. La même voie de signalisation calcique est recrutée,
mais l'effet est diamétralement opposé. Le Ca + lie toujours la calmoduline, mais ce
complexe active cette fois-ci la kinase de la chaîne légère de la myosine. La
phosphorylation de la chaîne légère de la myosine active sa fonction ATPase et mène à la
contraction du muscle lisse. Ces effets opposés de la stimulation du récepteur B2 au niveau
de types cellulaires différents formant un même tissu constituent ce qu'on appelle l'effet
paradoxal de la bradykinine (Briner et coll., 1993). Le DAG, quant à lui, est un activateur
de la protéine kinase C (PKC). Cette activation de la PKC se traduit par au moins deux
effets du système kallikréine-kinine. D'une part, la PKC activée favorise le maintien de la
contraction vasculaire en phosphorylant (et en inhibant du même coup) la phosphatase de la
chaîne légère de la myosine. D'autre part, la PKC, par la phosphorylation directe de Raf-1,

29
un membre de la famille des mitogenactivated protein kinases (MAPK), active la voie de
MAPK, ce qui permet d'expliquer certains effets mitogènes de la signalisation des kinines
(LeebLundberg et coll., 2005).
Le récepteur B2 de la bradykinine joue aussi un rôle important dans la synthèse de
médiateurs lipidiques. En effet, la hausse de Ca + secondaire à l'activation du récepteur B2
provoque un changement de conformation de la phospholipase A2 cytosolique, ce qui
permet sa translocation à la membrane plasmique ainsi que la liaison à son substrat, l'acide
arachidonique, se trouvant inclus dans les phospholipides membranaires (Burch et Axelrod,
1987; Calder, 2006). L'acide arachidonique ainsi libéré sert ensuite de substrat pour les
cyclooxygénases (COX), qui sont responsables de la formation des prostaglandines. Les
prostaglandines sont des éicosanoïdes, dont l'action dépend de leur liaison à leurs
récepteurs, qui sont des RCPG (Narumiya, 2009). Les actions des prostaglandines sont
multiples : elles sont impliquées dans la prolifération cellulaire, l'angiogénèse, l'apoptose,
la coagulation, le métabolisme osseux, les fonctions hépatiques, la réponse immunitaire,
ainsi que les fonctions reproductrices. Les prostaglandines ont plusieurs effets pro
inflammatoires, mais elles peuvent aussi être antiinflammatoires (Flower et Perretti, 2005).
Le récepteur B2 de la bradykinine exprimé au niveau neuronal déclenche aussi à cet endroit
une signalisation calcique après son activation par la bradykinine. L'effet nociceptif très y ,
aigu de la bradykinine est dû à l'activité du Ca sur deux types de canaux ioniques.
D'abord, il y a inhibition des canaux potassiques de type M et ouverture des canaux ■ y ,
chloriques dépendants du Ca ; la somme de ces effets provoque une dépolarisation de
certains neurones et l'augmentation aiguë de la douleur (Liu et coll., 2010). Une
conséquence additionnelle de la stimulation du récepteur B2 au niveau des neurones
afférents primaires sensitifs est l'amplification du stimulus, d'abord par l'augmentation du
relâchement de médiateurs tels la substance P et le peptide apparenté à la calcitonine
(Calcitoninrelated gene product : CGRP), deux neuropeptides inflammatoires. Il faut
prendre en considération la production des prostaglandines, de la libération de cytokines et
de la signalisation par le NO dans les différents types cellulaires se trouvant à proximité des
neurones sensitifs, afin de comprendre l'importance de la signalisation dépendante de

30
l'activation du récepteur B2 au niveau de sensibilisation des nocicepteurs à des stimuli
douloureux. Ces phénomènes concourent tous à l'état d'hyperalgésie observé après
l'engagement de la signalisation du récepteur B2 (Mizumura et coll., 2009).
La signalisation qui découle du récepteur B2 explique son importance dans le maintien d'un
tonus vasculaire adéquat (en contrebalançant le système rénine-angiotensine-aldostérone,
dont la signalisation mène en généralement à un état hypertendu) autant en permettant la
synthèse du NO, un facteur vasorelaxant, qu'en libérant des prostaglandines qui peuvent
avoir un effet cardioprotecteur (Murphy et coll., 2008). Par ailleurs, ces diverses actions
contribuent à amplifier la réaction inflammatoire au niveau du système vasculaire,
puisqu'une hypotension puissante facilite l'extravasation et l'apparition d'œdème ainsi que
de la douleur par compression des autres tissus.
Les voies de signalisation recrutées par le récepteur B, sont essentiellement les mêmes que
celles stimulées par le récepteur B2, puisqu'à l'instar du récepteur B2, le récepteur B, est
majoritairement couplé à la protéine Gq et induit ainsi la formation des seconds messagers
IP3 et DAG, avec les effets sur la signalisation calcique qui ont été décrits ci-dessus. La
principale différence se situe au niveau de la durée d'action et de l'intensité de la
signalisation au niveau de la mobilisation calcique. En effet, le signal calcique secondaire à
l'activation du récepteur B2 est transitoire, alors qu'il est prolongé lorsqu'initié de manière
secondaire à l'activation du récepteur B, (Bascands et coll., 1993; Smith et coll., 1995).
1.4.4 Adaptation des récepteurs
Le terme adaptation, lorsqu'il s'applique à un récepteur, définit les événements dépendants
de la liaison d'un agoniste à celui-ci qui mènent soit à sa désensibilisation, dans le cas des
RCPG ou à d'autres phénomènes de signalisation, par exemple, l'autophosphorylation des
récepteurs à activité tyrosine kinase (Bôhm et coll., 1997; Blaukat, 2003). Plusieurs
mécanismes participent à l'atténuation du signal engagé par les RCPG, dont le retrait de

31
l'agoniste du fluide extracellulaire, la désensibilisation et la translocation vers des
compartiments distincts où l'agoniste ne peut plus induire la signalisation. Dans le cas du
système kinine-kallikréine, les kinines sont soumises à une forte pression protéolytique au
niveau extracellulaire, étant donné le grand nombre de peptidases responsables de leur
métabolisme à proximité de leur lieu de synthèse, soient le plasma ou à la surface cellulaire
des tissus qui expriment également leurs récepteurs (voir la section 1.3). La dégradation des
kinines avant leur liaison au récepteur est un moyen de contrôler négativement leur
signalisation. La présence de transporteurs de haute affinité pour certaines fonctions
chimiques spécifiques, comme les transporteurs de monoamines à la surface cellulaire des
neurones, constitue un second mécanisme prévenant l'interaction entre l'agoniste et le
récepteur (Bôhm et coll., 1997), mais ne s'applique pas à des peptides comme la BK.
Suivant la liaison de l'agoniste au récepteur, le changement conformationnel de celui-ci et
l'activation de la protéine G, le récepteur est soumis à une désensibilisation qui permet
l'arrêt du signal, protégeant ainsi la cellule des effets délétères d'une surstimulation. La
désensibilisation du RCPG dépend de la dissociation physique de la protéine G qui y est
couplée, et peut être soit dépendante de la liaison de l'agoniste à son récepteur, on la
qualifie alors d'homologue, ou hétérologue, si elle est secondaire à l'activation d'un autre
récepteur. La désensibilisation homologue fait intervenir les GRK (G protein coupled
receptor related kinase), ainsi que des kinases seconds messagers, alors que la
désensibilisation hétérologue dépend essentiellement de l'activité catalytique de kinases
seconds messagers. La désensibilisation est un phénomène qui peut faire intervenir un
processus de phosphorylation séquentielle et hiérarchique (Ohguro et coll., 1993) comme
modification post-traductionnelle des RCPG (Lefkowitz, 2004; Tobin, 2008), bien qu'elle
ne soit pas essentielle à la désensibilisation efficace de tous les RCPG (Ferguson, 2007).
Il a été montré que le RB2 est phosphoryle de manière dépendante de la liaison de la BK
par les GRK (Blaukat et coll., 2001), ainsi que par la PKC au niveau des résidus Ser et Thr
présents au niveau de l'extrémité C-terminale du RB2, comme il a été mentionné à la
section 1.4.2.2. Par ailleurs, sa phosphorylation est importante pour le recrutement des
protéines nommées les (3-arrestines, qui en plus de participer à la désensibilisation du RB2,
sont des protéines d'échaffaudage recrutant la machinerie endocytaire, ce qui permet

32
ultimement l'internalisation du RB2 (Pizard et coll., 1999) et l'arrêt rapide de sa
signalisation. Contrairement au RB2, le RB, n'est ni phosphoryle ni internalise de manière
dépendante de l'agoniste (Blaukat et coll., 1999); par ailleurs, il possède une signalisation
plus persistante que celle du RB2 (Mathis et coll., 1996; Faussner et coll., 1998; Leeb-
Lundberg et coll., 2001). La présente section s'intéressera aux modifications subies par les
récepteurs après l'activation de la protéine Gaq et aux partenaires moléculaires des
récepteurs qui sont impliqués.
1.4.4.1 La phosphorylation du RB2 par les GRK
Il existe sept isoformes de ces serine threonine kinases, membres de la famille des kinases
A, G et C (Boguth et coll., 2010), qui présentent entre elles une structure bien conservée.
Le domaine C-terminal des GRK est nécessaire à la translocation de ces protéines
cytosoliques à la membrane plasmique, puisqu'il possède la capacité de lier les
phospholipides anioniques de celle-ci, alors que le domaine N-terminal est celui qui
interagit avec le RCPG (Penela et coll., 2003). Sa deletion entraîne la perte de la fonction
des GRK (Noble et coll., 2003; Huang et coll., 2009). Malgré les efforts de diverses
équipes à établir la spécificité entre les GRK et leurs substrats, il est maintenant évident que
les GRK présentent un patron de substrats redondants entre elles, quoiqu'elles soient
spécifiquement recrutées à des RCPG activés (Boguth et coll., 2010). Par ailleurs, leur
distribution particulière au niveau des différents types cellulaires permet une certaine
extrapolation de leur impact au niveau de chaque environnement (Penela et coll., 2003).
L'activation du RCPG et le changement de conformation concomitant provoquent un
accroissement de l'aire membranaire occupée par le récepteur et sa réorganisation mène à
la formation d'un site de liaison pour ses différents partenaires cellulaires (Rosenbaum et
coll., 2009), dont la protéine G, qui par l'hélice de son extrémité C-terminale, reconnaît et
lie le RCPG activé. La dissociation des sous-unités Ga, liée à la GTP, et GPy, permet à
cette dernière de lier une GRK, ce qui induit par le fait même sa translocation à la
membrane et amplifie son activité kinase par modulation allostérique. Ce recrutement

33
secondaire à la dissociation de la protéine G activée pourrait conférer une certaine
spécificité au recrutement des GRK aux RCPG activés (Penela et coll., 2003). En plus de
lier le RCPG et la sous-unité GPy, il a été rapporté que les GRK peuvent aussi lier la forme
activée de la sous-unité Gaq et diminuer ainsi l'activation de la PLCf5 (Caïman et coll.,
1999). Ces diverses interactions, qui constituent des mécanismes d'atténuation de la
signalisation dépendants des GRK, sont dissociables de leur activité kinase (Ferguson,
2007). Les GRK sont potentiellement impliquées dans l'internalisation des récepteurs
auxquels elles sont recrutées puisqu'il a été rapporté qu'elles peuvent se trouver aux
endosomes précoces contenant un RCPG récemment internalise (Schulz et coll., 2002) et
que leur interaction avec les clathrines constitue parfois une voie alternative
d'internalisation, indépendante des (3-arrestines (Claing et coll., 2000). Une signalisation
persistante par un RCPG résulte en une diminution de l'expression des GRK (Penela et
coll., 1998). Une variation positive ou négative des niveaux d'expression des GRK peut
mener à une désensibilisation inadéquate de la signalisation des RCPG, ce qui par ailleurs
peut avoir de graves conséquences en pathologie humaine. Par exemple, des niveaux
incorrects de GRK2 jouent un rôle aggravant dans l'hypothyroïdie, l'arthrite rhumatoïde,
plusieurs troubles cardiovasculaires et dans la dépendance aux opiacés (Penela et coll.,
2003; Métayé et coll., 2006). Il est à noter que les GRK sont aussi impliquées dans la
désensibilisation des récepteurs de type tyrosine kinase (Freedman et coll., 2002) et sérine-
thréonine kinase (Ho et coll., 2005), quoique ces fonctions ne dépendent pas de leur activité
enzymatique.
La GRK2 est l'isoforme principalement présente au niveau des cellules endothéliales
vasculaires (Penela et coll., 2003), ce qui pourrait suggérer qu'elle est l'isoforme impliquée
dans la désensibilisation du RB2. Son expression est augmentée en réponse à la
vasoconstriction, à l'hypertrophie ainsi qu'en réponse à une hausse de l'abondance de sous-
unités Gaq activées, alors que des molécules proinflammatoires diminuent son expression.
Il a par ailleurs été rapporté qu'une stimulation prolongée des neurones du ganglion dorsal
par l'IL-1 p provoque la diminution de l'expression de la GRK2, ce qui a comme
conséquence de réduire l'internalisation du RB2 suite à l'injection de la BK (von Banchet et

34
coll., 2010). Ce phénomène pourrait expliquer la sensibilisation des nocicepteurs par la BK
lors des situations inflammatoires persistantes.
1.4.4.2 La phosphorylation du RB2 par la PKC
Il a été rapporté que la PKC, en tant que kinase second messager, pouvait phosphoryler les
RCPG dont son activité dépend et s'inscrire ainsi dans une forme de rétrocontrôle négatif
(Bôhm et coll., 1997), par sa translocation du cytosol vers la membrane en réponse à la
sitmulation d'un RCPG par son agoniste. Le RB2 est phosphoryle au niveau du résidu
Ser de son extrémité C-terminale (Blaukat et coll., 2001) par la PKC, qui participe ainsi à
l'atteinte d'une phosphorylation en tandem des résidus Ser en position 339, 346 et 348,
dont deux sont suffisantes et nécessaires à la désensibilisation du RB2. Par ailleurs, les
protéines kinases seconds messagers peuvent aussi phosphoryler les RCPG non activés (De
Fea,2010).
Les événements de phosphorylation communs à plusieurs RCPG servent des fonctions
communes lors de la stimulation des RCPG, alors que des patrons de signalisation
particuliers sont plutôt liés à la signalisation engagée dans un type cellulaire donné et des
protéines kinases recrutées (Tobin, 2008).
1.4.4.3 Le recrutement au RB2 des p-arrestines
Les arrestines sont des protéines qui se répartissent en 4 familles, dont la première et la
quatrième sont exclusivement présentes au niveau des cellules en cônes et en bâtonnets de
la rétine et sont appelées les arrestines visuelles (Luttrel et Lefkowitz, 2002). Les arrestines
2 et 3, également nommées respectivement |3-arrestinei et |3-arrestine2, sont celles recrutées
aux RCPG phosphorylés dans les autres cellules de l'organisme (Gurevich et Gurevich,

35
2006). En effet, les arrestines constituent le prototype de la protéine dont l'interaction avec
un RCPG dépend de l'état de phosphorylation de celui-ci (Tobin, 2008), et en ce sens, elles
représentent le cofacteur fonctionnel des GRK (Bôhm et coll., 1997). La conformation de la
P-arrestine libre au niveau cytosolique ne présente pas les domaines d'interaction avec les
RCPG, qui sont camouflés par son état de phosphorylation, son oligomérisation ou par des
interactions protéine-protéine. Le dévoilement de ces domaines par son recrutement au
RCPG permet le changement con form ationnel qui confère à la P-arrestine la conformation
optimale d'affinité pour le RCPG. En effet, la reconnaissance séquentielle du récepteur par
les P-arrestines (Gurevich et Benovic, 1993), grâce à ses sites de phosphorylation, participe
à la reconnaissance subséquente de la conformation active de celui-ci, pour laquelle la P-
arrestine possède une meilleure affinité (Hirsh et coll., 1999). Par ailleurs, cette haute
affinité de la liaison de la P-arrestine dépend de la somme des interactions de basse affinité
entre les différents sites de phosphorylation présentés par le RCPG et la P-arrestine. Par
encombrement stérique, l'interaction de la GRK et de la P-arrestine empêche la
réassociation d'une protéine G au récepteur activé. Il est maintenant connu que, malgré
l'importance de la phosphorylation du RCPG pour le recrutement des P-arrestines, elle
n'est pas essentielle à la désensibilisation de tous les récepteurs. En effet, la présence de
plusieurs résidus chargés négativement qui agissent comme phosphomimétiques peuvent
permettre le recrutement des P-arrestines (Tobin, 2008). Ces données indiquent qu'un
ensemble de charges négatives situées à proximité les unes des autres, conférées ou non par
la phosphorylation des résidus, est peut-être plus important dans le recrutement des P-
arrestines que la séquence exacte des résidus. Par ailleurs, de cette observation pourrait
découler une meilleure compréhension de l'importance du changement conformationnel
induit par une phosphorylation différentielle et de ses effets modulateurs des diverses voies
de signalisation et de désensibilisation.
Il est possible de distinguer deux classes de RCPG quant aux caractéristiques qui
définissent leur interaction avec les p-arrestines (Tobin et coll., 2008). Les RCPG de la
classe A recrutent les p-arrestines de façon très transitoire, s'en dissocient alors qu'ils sont
encore présents à la membrane ou à peine internalises et recyclent rapidement à la
membrane plasmique. Les RCPG de la classe B forment plutôt des complexes beaucoup

36
plus stables avec les P-arrestines et présentent un recyclage plus lent que celui observé pour
les RCPG de classe A (Simaan et coll., 2005). Bien que les deux P-arrestines présentent
entre elles une grande homologie de séquence, elles se distinguent au niveau des récepteurs
auxquels elles sont recrutées (DeFea, 2010). Le RB2 a été identifié comme un récepteur de
la classe A étant donné son recyclage rapide après la dissociation et la dégradation du
ligand au niveau endosomal (Simaan et coll., 2005). La déphosphorylation du RB2 par une
protéine phosphatase, suivie de la dissociation de la p-arrestine sont des étapes essentielles
à la réexpression du RB2 internalise à la membrane plasmique.
Les P-arrestines sont des protéines d'échafaudage, c'est-à-dire qu'elles offrent un support
physique facilitant l'interaction de différentes protéines entre elles. Sa liaison au récepteur
activé phosphoryle permet le recrutement de la machinerie endocytaire, dont font partie les
clathrines, le complexe protéique adaptateur 2 (AP-2), différentes petites protéines G, ainsi
que des ubiquitine ligases (Métayé et coll., 2006). Les mécanismes impliqués dans
l'internalisation du RB2 était un sujet relativement controversé, puisqu'elle avait été décrite
comme ne dépendant ni des arrestines, ni de la dynamine (Lamb et coll., 2001) tout en
impliquant la formation de puits de clathrines (Enquist et coll., 2007) qui s'invaginaient
dans la membrane pour devenir des endosomes. Des données plus récentes font état de
l'importance des P-arrestines et des petites GTPases dont la dynamine dans l'internalisation
du RB2 (Simaan et coll., 2005). Par ailleurs, les P-arrestines permettent le regroupement des
divers intervenants moléculaires de voies de signalisation indépendantes de l'activation des
protéines G (Shenoy et coll., 2007; Violin et Lefkowitz, 2007; DeFea, 2010). Récemment,
plusieurs agonistes de diveres RCPG ont été identifiés comme étant des agonistes biaises,
puisqu'ils vont activer préférentiellement une signalisation qui dépendant de l'un des
partenaires de liaison des RCPG, soit la protéine G associée ou la p-arrestine recrutée au
récepteur; comparativement à un agoniste balancé, qui active l'ensemble des voies de
signalisation connues du récepteur (Shenoy et coll., 2007; Violin et Lefkowitz, 2007 Shukla
et coll., 2008; Gesty-Palmer et coll., 2009; Zheng et coll., 2010).

37
1.4.4.4 Endocytose du RB2
La réponse cellulaire adéquate à un agoniste du milieu extracellulaire dépend de la
disponibilité des récepteurs se trouvant à la membrane plasmique. Il existe une grande
variabilité au sein des RCPG au niveau des conditions qui déterminent l'endocytose, des
mécanismes d'endocytose recrutés, ainsi que du destin du ligand internalise (Bôhm et coll.,
1997). Grâce au recrutement des p-arrestines, l'interaction avec les clathrines est facilitée et
permet ainsi l'internalisation du RB2, quoique les clathrines soient des protéines qui
puissent interagir directement avec les récepteurs. Par contre, ces domaines d'interaction ne
sont pas clairement identifiés (Bôhm et coll., 1997). Le domaine NPXXY a été suggéré
comme étant impliqué dans ces interactions (Barak et coll., 1994; Gabilondo et coll., 1996).
Le complexe agoniste-RB2 activé est internalise par l'invagination de la membrane. La
fusion vésiculaire dépend de plusieurs petites protéines G, dont la dynamine et Rab5. La
dynamine, une protéine à activité GTPase, est attachée à la membrane par son domaine
d'homologie à la pleckstrine (PH; Hinshaw, 2000) et présente la capacité de s'assembler en
oligomères formant des anneaux ou des spirales. Ces spirales de dynamine resserrent le col
de l'invagination membranaire et permettent la fission endosomale (Hinshaw, 2000;
Chappie et coll., 2010). Les protéines Rab sont de petites GTPases impliquées dans la
régulation du trafic vésiculaire (Grosshans et coll., 2006). La protéine Rab5 est membre de
la famille des Rab qui compte approximativement 60 membres (Seachrist et Ferguson,
2003) et dont les fonctions sont associées à l'endocytose, au trafic endosomal et à
l'exocytose. La modification post-traductionnelle de l'extrémité C-terminale des Rab,
menant à l'ajout de groupement géranyl-géranyl, permet leur association à la membrane de
différents organelles, pour lesquels les diverses protéines Rab présentent une localisation
préférentielle. De manière générale, il est à noter que les protéines Rab4, 5, 7 et 9 sont
exprimées de manière ubiquitaire, alors que les autres Rab ont un patron d'expression
spécifique du type cellulaire et du tissu. La localisation de la protéine Rab5 est détectable
au niveau de la membrane plasmique, des puits de clathrines et des endosomes précoces
(Bucci et coll., 1992), lorsqu'elle est associée au GTP. Cette protéine est impliquée dans
l'internalisation du RB2 (Houle et Marceau, 2003) et il a été montré que l'inhibition de

38
l'activité de Rab5 menait à une dégradation augmentée du RB2, de manière secondaire à
l'altération de la voie endosomale qu'il emprunte habituellement. Il est à noter que les
protéines Rab4 et Rabl 1, impliquées dans le recyclage du matériel internalise, se trouvent
aux endosomes qui recyclent rapidement ou plus lentement, respectivement. La progression
du matériel internalise vers les endosomes tardifs se fait de manière concomitante à la
conversion du marquage endosomal de la protéine Rab5 à la protéine Rab7. Finalement la
protéine Rab7 est principalement impliquée dans le trafic du matériel contenu dans
l'endosome tardif, où débute sa dégradation, jusqu'aux lysosomes (Lombardi et coll.,
1993).
L'acidification de l'endosome permet la dissociation de l'agoniste, alors que les différentes
peptidases endosomales procèdent à sa dégradation. Le récepteur libéré de son agoniste
retrouve sa conformation inactive, est déphosphorylé par une phosphatase et se dissocie de
la P-arrestine. De ce fait, l'endocytose, qui n'est pas le mécanisme principal de
désensibilisation, est par contre très importante pour la resensibilisation du récepteur. Ces
étapes précèdent le tri du matériel endosomal qui sera recyclé à la membrane ou de ce qui
progressera au niveau de la voie endosomale jusqu'aux lysosomes, sites de dégradation
majeurs des protéines internalisées. Les mécanismes de ce tri sont peu connus.
L'identification d'«étiquettes» dans la structure des RCPG indiquant leur trafic vers les
lysosomes a précédé la découverte de séquences de résidus impliqués dans le recyclage, ce
qui a mené à l'hypothèse que le recyclage était le destin par défaut des RCPG (Bôhm et
coll., 1997). Cette suggestion était, par ailleurs, cohérente avec l'observation d'un rythme
de recyclage similaire des récepteurs et des lipides membranaires. Par contre,
l'identification de plusieurs séquences de recyclage présentes au niveau d'une variété de
protéines membranaires tend à montrer la spécificité de ce mécanisme de réexpression
membranaire (Pandey et coll., 2005). Deux motifs de recyclage ont été mis en évidence
pour les RCPG; soient les motifs DSLL et GTALL. Le motif DSLL, présent dans la queue
cytoplasmique du récepteur P2-adrénergique, force la réexpression membranaire d'un
RCPG qui n'est habituellement pas recyclé, tel le récepteur ô-opioïde (Hislop et von
Zastrow, 2010), lorsqu'introduit par mutagénèse à son extrémité C-terminale. Le motif
GTALL a quant à lui été identifié dans la queue C-terminale du récepteur de l'hormone
lutéinisante. Le fait que ces motifs soient absents du RB2, dont le cycle endocytose-

39
recylcage a été amplement documenté (Munoz et coll., 1993; Bachvarov et coll., 2001;
Lamb et coll., 2002), suggère qu'il existe des motifs additionnels de recyclage pour les
RCPG.
Lors d'une stimulation persistante des RB2 par la BK, une diminution de l'abondance des
RB2 exprimés à la membrane plasmique est observée (Blaukat et coll., 2003). Ce processus,
connu sous le terme régulation à la baisse (downregulation), est basé sur la dégradation
lysosomale des récepteurs internalises, qui est concomitante à une répression de la
transcription et la traduction de ce récepteur. Par ailleurs, le RB2 est un susbtrat pour une
variété de proteases extracellulaires, dont la kallikréine tissulaire et d'autres proteases,
sécrétées par les neutrophiles activés (Marceau et coll., 2002; Houle et coll., 2003), et
l'activité catalytique de ces enzymes provoque la dégradation du récepteur, dont les
fragments sont rapidement internalises, ce qui a pu être observé grâce à l'utilisation d'une
construction fusionnant le RB2 avec la protéine verte fluorescente (green fluorescent
protein; GFP).
1.4.4.5 Redistribution membranaire des récepteurs des kinines
Il a été rapporté que le RB2 était redistribué transitoirement aux cavéoles, suite à sa
stimulation par l'agoniste BK (De Weerd et Leeb-Lundberg, 1997; Haasemen et coll.,
1998). Le RB,, par le fait qu'il ne soit ni phosphoryle, ni internalise, est beaucoup plus
résistant à la désensibilisation fonctionnelle. Sabourin et collaborateurs (2002b) ont mis en
évidence l'agrégation du signal fluorescent associé à la construction fusionnant le RB, de
lapin avec la protéine jaune fluorescente (YFP) suite à l'ajout au milieu de culture de
l'agoniste Lys-des-Arg9-BK, signal contrastant avec le marquage membranaire fluorescent
lisse présenté par des cellules transfectées non stimulées. La translocation du RB, vers ces
microdomaines membranaires caractérisés par le recrutement de plusieurs molécules
signalétiques est inhibée par la P-cyclodextrine.

40
Il est important de noter que le RB,, s'il n'est pas soumis aux mécanismes de
désensibilisation rapides comme l'est le RB2, et présente une signalisation plus persistante
que ce dernier, est en revanche soumis à une dégradation protéolytique assez rapide et
indépendante de sa stimulation par l'agoniste (Fortin et coll., 2003). Le contexte
pathologique de l'induction de l'expression du RB, offre une explication potentielle de
cette observation. L'état inflammatoire qui induit l'expression du RB,, lorsqu'il est résolu,
ne nécessite plus la présence de ce récepteur, qui peut être rapidement dégradé,
parallèlement à l'arrêt de sa transcription, réprimée en l'absence de stimuli inflammatoires.
1.5 Le système kinine-kallikréine en physiologie et en pathologie
Le système kinine-kallikréine, par la vaste distribution de ses composantes, s'illustre en
tant qu'acteur important dans une variété de phénomènes biologiques, primordiaux à
l'homéostasie de l'organisme. La présente section s'intéresse aux fonctions physiologiques
de l'activation des récepteurs des kinines, ainsi qu'aux effets délétères que cette activation
peut entraîner.
1.5.1 Système cardiovasculaire et rénal
L'homéostasie vasculaire dépend de l'équilibre entre les agents vasopresseurs, tels l'Angll
et l'endothéline, et les agents vasodilatateurs, comme les kinines et le NO. En situation
physiologique, l'endothélium et le muscle lisse vasculaire n'exprimant pas le RB| de
manière détectable (Marceau et coll., 1998), les effets des kinines s'expliquent par
l'engagement de la signalisation du RB2. Les kinines formées au niveau plasmatique se
lient au RB2 présent sur l'endothélium constituant la lumière du vaisseau et induisent le
changement de conformation nécessaire à l'activation de la protéine Gq, dont les voies de

41
signalisation qui lui sont associées ont été décrites plus haut. Au niveau endothelial, la
signalisation secondaire à l'activation du RB2 provoque l'activation de la synthase d'oxyde
nitrique (NO) endothéliale (eNOS). Le NO est un puissant vasorelaxant du muscle lisse
vasculaire sous-jacent, qu'il atteint par diffusion et où son activation de la guanylate
cyclase mène à une hausse de l'abondance du GMPC (Schini et coll., 1990), qui empêche la
contraction du muscle lisse vasculaire par son activité au niveau de la myosine. Le NO
libéré par la stimulation du RB2 diminue le rythme cardiaque et la pression sanguine
(Ribuot et coll., 1993). La stimulation du RB2 endothelial mène aussi à la synthèse de la
prostaglandine I2 par l'activation de la phospholipase A2, un médiateur lipidique dont la
liaison à son récepteur exprimé par le muscle lisse vasculaire permet la hausse de la
quantité d'AMPc (Pyne et coll., 1997), qui appuie l'effet vasorelaxant du NO. Un effet
cardioprotecteur additionnel de la signalisation induite par le RB2 est la libération de
l'activateur tissulaire du plasminogène (tPA; Smith et coll., 1985), un agent fibrinolytique
important. Cette serine protease tissulaire induit l'activité fibrinolytique en générant une
plasmine active à partir du plasminogène (Schmaier, 2003). Ainsi, le RB2 endothelial est un
régulateur négatif du tonus vasculaire et de la fonction plaquettaire.
La bradykinine est aussi impliquée au niveau de l'hyperpolarisation du muscle lisse dans le
lit vasculaire coronarien, dont le mécanisme n'est pas entièrement élucidé mais pourrait
impliquer les canaux potassiques du muscle lisse (Félétou et coll., 2003).
Dans le cas où l'endothélium est endommagé, la réponse du système kinine-kallikréine est
altérée. L'effet de la bradykinine peut activer directement le muscle lisse vasculaire en se
liant aux récepteurs B2 présents à sa surface. L'effet vasomoteur observé est la contraction
du muscle lisse (Briner et coll., 1993). Par ailleurs, comme il a été mentionné
précédemment, un dommage tissulaire induit l'expression du RB,, qui recrute les mêmes
voies de signalisation que le RB2 (Marceau et coll., 1998). L'induction de l'expression du
RB, après une infection (Regoli et coll., 1981; Tirapelli et coll., 2007) permet d'observer
une hypotension secondaire à l'activation de ce récepteur.
En situation pathologique, les kinines sont impliquées au niveau de l'hypertension et dans
l'insuffisance de multiples organes (Adam et coll., 2002). On a souvent suggéré que la
signalisation dépendante de la stimulation du RB2 est positive, alors que la signalisation du

42
RB, démontre des effets délétères. Le RB2 est impliqué dans la perfusion cardiaque
(Martorana et coll., 1990) et rénale (Scicli et coll., 1986; Kakoki et coll., 2007), comme le
supporte sa distribution au niveau de zones particulièrement vulnérables à l'ischémie dans
ces organes. L'importance de la signalisation du RB2 au niveau de l'ischémie cardiaque est
supportée par l'observation que les effets protecteurs d'une inhibition de l'ECA sont abolis
par le cotraitement avec un antagoniste sélectif du RB2 (Linz et coll., 1995). Une méthode
reposant sur la livraison du gène de la kallikréine dans un modèle d'infarctus du myocarde
chez le rat montre les effets positifs sur le débit cardiaque et le remodelage, lors d'une
formation accrue des kinines (Agata et coll., 2002). Dans un modèle d'ischémie
myocardique chez la souris, l'infusion continue d'un agoniste sélectif du RB2 a permis une
meilleure préservation du tissu en atténuant le remodelage cardiaque, une meilleure
fonction ventriculaire gauche et la diminution de l'expression de médiateurs inflammatoires
(Marketou et coll., 2010; Manolis et coll., 2010). Par ailleurs, il a été montré que
l'activation du système kinine-kallikréine tissulaire permet le recrutement de cellules
progénitrices cardiaques au site de l'infarctus du myocarde et une néovascularisation plus
efficace, secondaire à la diminution de l'apoptose du tissu (Spillman et coll., 2006). La
fonction du RB, au niveau de l'ischémie rénale est controversée, puisque sa deletion
génétique présentait soit un effet protecteur (Wang et coll., 2006; 2008) ou un effet délétère
(Kakoki et coll., 2007). Il est à remarquer que l'étude identifiant l'effet négatif du RB,
comparait des souris témoins à des souris qui n'exprimaient pas le RB,, alors que l'étude
donnant les résultats opposés utilisait plutôt un modèle murin dont les gènes des deux
récepteurs des kinines étaient inactivés (Duchene et Ahluwalia, 2009).
Dans l'athérosclérose, le profil d'expression très bas du RB2 est dû à sa désensibilisation
(Kuoppala et coll., 2003). Par ailleurs, les vaisseaux athérosclérotiques présentent une
expression très importante du RB, (Raidoo et coll., 1997), ce qui peut être attribuable au
contexte inflammatoire et au dommage endothelial, ou plutôt à titre compensatoire, en
réponse à la perte de l'expression du RB2 (Manolis et coll., 2010). Il a aussi été suggéré que
l'accroissement des forces de cisaillement au niveau endothelial, accéléré par la
dysfonction endothéliale, est un puissant inducteur du RB, (Duchene et coll., 2007). Les
tissus provenant de patients souffrant d'insuffisance cardiaque de stade terminal présentent
une hausse de l'expression du RB,, comme le témoigne l'abondance de l'ARNm et de la

43
protéine (Liesmae et coll., 2005). Le RB| contribue positivement à la réduction de la
formation de la néointima en favorisant la prolifération endothéliale et en inhibant la
prolifération du muscle lisse vasculaire ainsi que sa migration (Agata et coll., 2000;
Morissette et coll., 2007).
Un facteur aggravant des pathologies rénales, dont découle une perte de la fonction de la
filtration, est le stress oxydatif (Forbes et coll., 2008). La stimulation de la signalisation du
RB2 atténue le stress oxydatif au niveau rénal en inhibant l'expression de médiateurs
inflammatoires et de molécules pro-fibrotiques (Bledsoe et coll., 2006).
L'inactivation du gène RB2 chez la souris (Borkowski et coll., 1995) a permis l'observation
d'une pression sanguine plus élevée et une hypertrophie cardiaque lorsque ces souris sont
soumises à un régime riche en sel (Alfie et coll., 1996; Madeddu et coll., 1997; Maestri et
coll., 2003); ces effets semblent par contre dépendre de facteurs additionnels puisqu'ils ne
sont pas reproductibles dans tous les modèles de souris ne possédant pas le gène du RB2
(Milia et coll., 2001; Schanstra et coll., 2002; Trabold et coll., 2002).
1.5.2 Inflammation
L'injection des kinines mène à l'observation des quatre signes cardinaux de l'inflammation,
soient la chaleur, la douleur, la rougeur et l'œdème, qui peut être associé à une perte de
fonction de l'organe atteint. La signalisation dépendant de la stimulation des récepteurs des
kinines permet le développement et le maintien de l'inflammation par la vasodilatation et la
hausse de la perméabilité vasculaire, favorisant la formation d'œdème, l'hyperalgésie et la
douleur (Campos et coll., 2006). Par ailleurs, de nombreuses cellules immunitaires, dont le
neutrophile (Henderson et coll., 1992) expriment les différents composants du système
kinine-kallikréine. Les manifestations aiguës sont dépendantes de l'activation du RB2, alors
que les phénomènes chroniques dépendent de l'activation du RB, (Leeb-Lundberg et coll.,
2005; Costa-Neto et coll., 2008), ce qui s'explique par la désensibilisation rapide du RB2
comparativement à la désensibilisation très lente du RB|. En plus de la durée de sa

44
signalisation, le fait que le RB, soit induit dans un contexte pathologique, qu'il
n'entretienne pas de fonctions dans l'homéostasie, vue son absence au niveau des tissus
sains, fait de ce récepteur une cible thérapeutique intéressante (Campos et coll., 2006;
Duchene et Ahluwalia, 2009). L'induction du RBi est observable en réponse à l'injection
de LPS au niveau de différents modèles animaux, et sa deletion protège les souris de
l'hypotension sévère secondaire au choc endotoxémique, provoqué par l'injection du LPS
(Cayla et coll., 2007). Le RB, est aussi induit dans le cas de l'inflammation des voies
aériennes et dans la rhinite allergique. Il est à noter que la sensibilité au leukotriene D4 dans
un modèle d'hypersensibilité bronchique est abolie par un antagoniste du RB,. Bien que
chez les patients souffrant de maladies inflammatoires de l'intestin comme la maladie de
Crohn et la colite ulcéreuse, une surexpression du RB, a été rapportée, il semble être
exprimé de manière constitutive au niveau de l'épithélium intestinal (Stadnicki et coll.,
2005). Sa présence peut ête due à la stimulation du système kinine-kallikréine tissulaire par
la flore bactérienne environnante. Les macrophages recrutés au côlon lors de la maladie de
Crohn expriment eux aussi le RB, et participent ainsi à l'amplification de l'inflammation
(Stadnicki et coll., 2005). En plus de promouvoir l'aggravation de la pathologie par le
maintien de l'état inflammatoire par la libération de médiateurs pro-inflammatoires, les
deux récepteurs des kinines participent aux symptômes de la maladie, puisqu'ils sont
impliqués dans le transport d'ions Cl" au niveau de l'épithélium intestinal, qui se fait de
concert avec le transport des ions Na+ et de l'eau et provoquent ainsi la diarrhée (Cuthbert
et coll., 2001). Finalement, dans un modèle d'induction de la colite ulcéreuse par injection
de TNBS, les dommages tissulaires et le recrutement de neutrophiles étaient moins
importants lorsque les souris étaient traitées avec un antagoniste du RB,, ou si elles
présentaient une inactivation du RB, (Hara et coll., 2008). Ces données indiquent que le
RB, pourrait s'avérer une cible thérapeutique intéressante au niveau des maladies
inflammatoires de l'intestin (Hara et coll., 2008; Marceau et Regoli; 2008), à l'instar du
RB2.
L'angio-œdème héréditaire est une pathologie rare (Kaplan et Greaves, 2005) qui est
caractérisée par des épisodes spontanés d'œdème au niveau des extrémités, ainsi que des
muqueuses de la bouche et des voies respiratoires et de l'abodmen. Bien que le mécanisme
d'initiation exact des attaques soit inconnu, la pathologie a été associée à un déficit de la

45
serpine Cl-INH (type I) ou à une altération de sa fonction (type II), bien qu'un troisième
type (type III) soit plutôt secondaire à une mutation au niveau du facteur de Hagemann
(Agostini et coll., 1984). La conséquence du déficit quantitatif ou fonctionnel du Cl-INH
est la réduction de l'inhibition de la voie du système de contact et ainsi une production
accrue de la BK, dont une des actions principales est l'accroissement de la perméabilité
vasculaire et l'extravasation plasmatique qui contribuent à la formation de l'œdème
(Nussberger et coll., 1999). L'icatibant, un antagoniste puissant du RB2, a récemment été
approuvé pour cette indication en Europe, mais son approbation tarde en Amérique du
Nord (Epstein et Bernstein, 2008; Berstein, 2008). Un second modulateur négatif du
système kinine-kallikréine utilisé en clinique est l'écallantide, un inhibiteur
biotechnologique de la kallikréine plasmatique, qui par ailleurs présente une activité
inhibitrice plus importante que celle du Cl-INH (Williams et Baird, 2003). Parmi les autres
pathologies associées à une extravasation importante, il est à noter que l'icatibant est
actuellement à l'étude pour l'indication de l'angio-œdème induit par la prise d'IECA (et à
l'accumulation de la BK qui leur est associé). Il a aussi été suggéré que l'icatibant pourrait
prévenir l'œdème secondaire à des brûlures importantes (Jonkam et coll., 2007).
1.5.3 Cancer
Différents constituants du système kinine-kallikréine ont été impliqués dans l'induction et
la promotion de la tumorigenèse, particulièrement grâce à leur stimulation de l'angiogénèse
et leur activation de la migration cellulaire qui caractérisent le processus néoplasique
(Hanahan et Weinberg, 2000). La signalisation induite par les récepteurs des kinines, en
stimulant la prolifération endothéliale pourrait s'avérer très pertinente dans l'accélération
de l'angiogénèse tumorale (Morbidelli et coll., 1998; Parenti et coll., 2001), un facteur de
première importance pour soutenir un réseau métastatique agressif et efficace. Un autre
mécanisme par lequel les récepteurs des kinines pourraient influencer positivement
l'angiogénèse est par la libération de l'endothéline-1 (Andoh et coll., 2010), un facteur pro-
angiogénique, pro-métastatique et pro-prolifératif, des effets qui sont abrogés par

46
l'utilisation d'antagonistes des récepteurs (Harford-Wright et coll., 2010). Le système
kinine-kallikréine participe au processus de métastase en stimulant la migration de cellules
tumorales, comme cela a été démontré dans le cas du gliome (activation du RB,; Lu et coll.,
2010) et du chondrosarcome (Yang et coll., 2010). Cet accroissement de la migration
pourrait être attribuable à l'activation de la COX-2 (Zhang et coll., 2008; Lu et coll., 2010),
dont la surexpression dans un grand nombre de cancers a été associée avec un
développement tumoral agressif (Koki et coll., 1999), la résistance aux traitements
standards et un mauvais pronostic (Koki et coll., 1999; Liao et coll., 2007).
L'accroissement de la perméabilité vasculaire secondaire à la stimulation des récepteurs des
kinines est un facilitateur potentiel de l'invasion métastatique et du recrutement de cellules
immunitaires. La culture de cellules endothéliales en présence de metabolites de cellules
tumorales dérivant de cancers de la prostate ou de cancers du sein a permis l'observation du
relâchement de la kallikréine tissulaire par ces cellules (Wright et coll., 2008). La
kallikréine tissulaire, par son activation des collagénases de type IV, est responsable de la
dégradation de la matrice extracellulaire (Desrivieres et coll., 1993), un effet qui facilite la
migration cellulaire et l'invasion. Par ailleurs, un inhibiteur de la kallikréine tissulaire
empêche l'invasion tumorale et la formation de métastases dans un modèle exploitant la
colonisation du tissu pulmonaire par la lignée cellulaire MDA-MB-231, dérivée d'un
cancer du sein (Wolf et coll., 2001). Finalement, il a été suggéré que les IECA avaient un
effet protecteur contre le cancer (Lever et coll., 1998); l'effet de l'accumulation de la BK,
potentiellement oncogene, secondaire au traitement par les IECA supposément protecteurs
demande à être mieux exploré (Rosenthal et Gavras, 2009).
Plusieurs antagonistes des récepteurs des kinines ont été synthétisés et mis à l'épreuve
comme agents anti-cancéreux. Par exemple, un composé modifié dont la structure dérive de
l'antagoniste de haute affinité du RB, B-9958 (Géra et coll., 2008) inhibe la prolifération
des cellules PC3, une des nombreuses lignées dérivant de tumeurs prostatiques
surexprimant le RB,. Le développement de plusieurs antagonistes du RB2, qu'ils soient des
peptides (Stewart et coll., 2002; 2004) ou des composés non peptidiques (Stewart et coll.,
2002; Jutras et coll., 2010) a permis de caractériser le rôle pro-prolifératif délétère de la BK
suivant l'initiation de la signalisation du récepteur au niveau de tumeurs originant de la
prostate et des ovaires.

47
La barrière hématoencéphalique est constituée d'un ensemble de structures cellulaires et
vasculaires, dont des jonctions serrées présentées par l'endothélium, des enzymes, des
récepteurs et des pompes qui participent à limiter et à contrôler l'accès de molécules au
cerveau (Juillerat-Jeanneret, 2008). La difficulté à obtenir une biodisponibilité acceptable
au niveau des cancers cérébraux a incité un nombre important d'équipes à exploiter la
stimulation du RB2 pour accroître la perméabilité vasculaire qui découle de l'activation du
récepteur (Bartus et coll., 1996; Emerich et coll., 2001; Black et Ningaraj; 2004) dans le but
de faciliter le transport d'agents chimiothérapeutiques au travers cette barrière (Côté et
coll., 2010; Harford-Wright et coll., 2010; Lu et coll., 2010). Les mécanismes de cet
accroissement de la perméabilité vasculaire ne sont pas entièrement élucidés, bien que le
relâchement accéléré du TNF-a (Qin et coll., 2009), le recrutement du calcium
intracellulaire (Wang et Liu, 2009) et le réarragement du cytosquelette (Liu et coll., 2008)
soient tous des phénomènes qui y participent potentiellement, de manière secondaire à la
signalisation du RB2.
1.5.4 Système nerveux
L'intégrité du système nerveux central, qui présente tous les composants du système
kinine-kallikréine (Walker et coll., 1995), dépend d'un contrôle très serré de la perméabilité
vasculaire (Costa-Neto et coll., 2008). La signalisation induite par les kinines joue un rôle
primordial dans le maintien d'une résistance cérébrovasculaire, ainsi qu'au niveau de la
capacitance vasculaire. La hausse de la perméabilité des vaisseaux est secondaire à la
libération de NO (Gôrlach et Whal, 1996). Des perturbations dans le maintien de la
perméabilité vasculaire due à la surstimulation ou la surexpression des composants du
système kallikréine-kinine ont été identifiées comme jouant un rôle aggravant dans
plusieurs pathologies (Thornton et coll., 2010) dont la schizophrénie (Shcherbakova et
coll., 1999) et du lupus neuropsychiatrique (Svenungsson et coll., 2001), dont la
composante clairement inflammatoire peut être amplifiée par la génération des kinines.

48
L'émergence d'un rôle délétère du système kinine-kallikréine dans la physiopathologie de
la maladie d'Alzheimer est très intéressante. Il a été rapporté que le complexe formé par le
KHM et la prékallikréine plasmatique pouvait être activé par sa liaison aux plaques de P-
amyloïde caractéristiques de cette pathologie au niveau cérébral (Joseph et coll., 1999) et
pouvait ainsi générer la production de BK. Par ailleurs, il a été observé que les sites où se
trouvent les lésions précoces de la maladie d'Alzheimer présentent une expression
importante des composantes du système kinine-kallikréine (Corrêa et coll., 1979; Raidoo et
Bhoola, 1997; Chen et coll., 2000; Jong et coll., 2003; Ashby et coll., 2010). Récemment, il
a été rapporté que dans un modèle de maladie d'Alzheimer reproduit par l'administration
intracérébroventriculaire d'un fragment de la protéine P-amyloïde chez la souris, la
fonction neurologique telle que mesurée par le déficit cognitif, est préservée lorsque les
gènes des RB, et RB2 sont absents de ces souris ou lorsque des antagonistes des deux sous-
types de récepteurs sont employés (Prediger et coll., 2008).
La BK est le médiateur endogène le plus algogène qui soit (Mizumura et coll., 2009).
L'expression des récepteurs de kinines par les terminaisons nerveuses sensitives
nociceptives au niveau du système nerveux central, ainsi qu'aux terminaisons afférentes
primaires de neurones non-myélinisés (telles les fibres Aô et C; Dray, 1997) est connue et
leur stimulation mène à l'activation directe des terminaisons nerveuses et à la
sensibilisation de celles-ci (Marceau et Regoli, 2004). La stimulation du RB2 par la BK
cause en effet l'hyperalgésie et l'allodynie (Raidoo et Bhoola, 1998; Couture et coll.,
2001), bien caractérisée pour des stimuli tels que la chaleur, qui dépend de la
phosphorylation des canaux TRPV1 par la PKCe (Huang et coll., 2006), activée par la
libération du DAG par la PLC, un second messager du RB2. Le recrutement d'une autre
voie, dépendant ici de l'accroissement de la concentration du Ca2+ cytosolique secondaire à
l'action de ITP3, a mis en lumière un mécanisme possible qui expliquerait la perception de
douleur aiguë en réponse à la BK. La hausse de Ca + au niveau cytosolique provoque la
dépolarisation de la terminaison nerveuse (dans le cas à l'étude, au niveau du ganglion
spinal) par la fermeture de canaux potassiques de type M coordonnée à l'ouverture des
canaux Cl" de type TMEM16A, ce qui accroît leur excitabilité (Liu et coll., 2010; Brown et
Passmore, 2010).

49
Le RB, est quant à lui impliqué dans l'établissement et le maintien de la nociception par
1'exacerbation inflammatoire à laquelle il participe par la synthèse et le relâchement de
médiateurs pro-inflammatoires, le recrutement de cellules immunitaires au site de la liaison
et par la sensibilisation des terminaisons sensitives, participant ainsi à une hyperalgésie
persistance (Leeb-Lundberg et coll., 2005). La deletion du gène du RB, dans un modèle
murin d'arthrite induite par l'adjuvant de Freund diminue l'allodynie et l'hyperalgésie
thermique (Ferreira et coll., 2001); alors que la deletion du gène du RB2 n'inhibe que la
perception de la douleur secondaire à l'injection de la BK. Le RB,, présent au niveau de la
moelle épinière est aussi impliqué dans la modulation de la plasticité aux stimuli
douloureux de celle-ci, tel que le suggère la diminution du phénomène d'autoamplification
nociceptive (« windup »), lorsque son gène est inactivé (Pesquero et coll., 2000). Ces
données expliquent l'intérêt que suscite le développement d'antagonistes du RB, comme
agents analgésiques (Campos et coll., 2006; Kuduk et Bock, 2008; Huang et Player, 2010).
1.6 Objectif des travaux
Les récepteurs B, et B2 de la bradykinine ont majoritairement une signalisation secondaire
à la liaison d'un agoniste à leur site orthostérique, ce qui provoque le changement de
conformation nécessaire à l'activation de la protéine G. La disponibilité de ces agonistes est
soumise à la régulation par la présence d'une variété de peptidases qui dégradent les kinines
et leurs analogues afin de moduler négativement la signalisation par les récepteurs. Puis,
suivant la liaison de l'agoniste au récepteur, les sous-types de récepteurs subissent une
redistribution subcellulaire, soit intracellulaire pour le récepteur B2, ou membranaire pour
le récepteur B|. L'objectif général de ces travaux de doctorat visait une meilleure
compréhension de l'adaptation des récepteurs, i.e. une vision approfondie des phénomènes
déclenchés par la liaison d'un agoniste à son récepteur, et comment ces phénomènes
peuvent être manipulés ou utilisés, afin de conférer de nouvelles propriétés aux récepteurs.

50
Dans un premier temps, les études se sont intéressées à la caractérisation d'une triade
d'isomères analogues de la bradykinine, soient les peptides B-9430 (D-Arg°-Hyp3-Igl5-D-
Igf-Oic8-BK), B-9972 (D-Arg°-Hyp3-Igl5-Oic7-Igl8-BK) et B-10344 (D-Arg°-Hyp3-Igl5-D-
Oic7-D-Igl8-BK). Le premier objectif de ce volet était de caractériser pharmacologiquement
ces analogues de la bradykinine au niveau des RB2 humain et lapin. Afin de mieux
comprendre l'importance de la stéréochimie en C-terminal du peptide sur la fonction du
récepteur, le peptide B-10344, l'isomère intermédiaire entre le peptide antagoniste B-9430
et le peptide agoniste B-9972 a été synthétisé et évalué. Le second objectif était de vérifier
l'effet hypothétique de la présence en C-terminal de la protéine verte fluorescente (green
fluorescent protein- GFP) au niveau de la construction RB2-GFP sur le recyclage du RB2 de
lapin. Pour ce faire, une protéine recombinante conjuguant le RB2 à un epitope myc à son
extrémité N-terminale a été générée et a pu servir de base de comparaison au niveau du
cycle endocytose-recyclage du RB2-GFP. Le troisième objectif visait à mettre en lumière
des altérations au cycle endocytose-recyclage des deux constructions, suite à leur
stimulation avec le peptide B-9972. En effet, de par sa demi-vie prolongée, prévisible étant
donnée sa résistance au métabolisme, le peptide B-9972 peut exercer des effets particuliers
au niveau du recyclage du récepteur. Cela est d'ailleurs suggéré par l'observation in vivo
d'une réduction marquée de l'expression du RB2 suivant un traitement avec le peptide B-
9972 chez le rat (Taraseviciene-Stewart et coll., 2005), indiquant l'influence potentielle
d'un agent pharmacologique sur les niveaux de dégradation et de recyclage d'un RCPG.
Dans un deuxième temps, l'utilisation d'un agoniste non peptidique du RB2, soit le
composé 47a (Sawada et coll., 2004b) de concert avec celle du peptide B-9972 avait
comme objectif l'approfondissement de nos connaissances sur le cycle endocytose-
recyclage du RB2. Plus spécifiquement, il s'agissait de mettre en évidence les différences
entre l'agoniste naturel BK et les deux agonistes résistants au métabolisme, au niveau de la
cinétique d'association avec les arrestines non visuelles et de la désensibilisation et la
dégradation du RB2. Un dernier objectif de ce volet était de caractériser la cinétique de
signalisation des agonistes résistants au métabolisme.
Grâce à plusieurs études de structure-activité définissant les interactions entre les récepteurs
des kinines et leurs ligands, l'extrémité N-terminale a été identifiée comme plus permissive

51
à l'ajout de groupements fonctionnels. Ces données permettent de poser l'hypothèse que
l'extension en N-terminal de différents analogues avec une variété de molécules peut
permettre la génération de ligands multifonctionnels, ainsi que potentiellement conférer de
nouvelles propriétés au récepteur qui interagira avec le nouvel analogue. Ce troisième volet
portait sur l'atteinte de deux objectifs précis. Le premier était d'obtenir et de caractériser le
jeu complet des ligands fluorescents, agonistes et antagonistes, pour chaque sous-type de
récepteur. Un objectif secondaire à celui-ci était bien entendu d'améliorer ainsi les outils
permettant la visualisation des récepteurs des kinines, en plus d'étudier plus avant leur
expression et leur distribution cellulaire et subcellulaire à l'état basai et activé. A cette fin,
un RB2 inapte à l'internalisation fût généré, et utilisé de concert avec un RB2 recombinant
conservant toutes ses caractéristiques endocytaires. Un aspect additionnel de ce projet
portait plus spécifiquement sur la caractérisation de la séquence naturelle maximakinine, un
analogue naturel de la BK. Cette kinine, isolée du venin de l'amphibien Bombina maxima,
présente la séquence intégrale de la BK en C-terminal de sa structure, avec une extension
de dix acides aminés en N-terminal. Ce peptide est pharmacologiquement actif au sein de
préparations de tissus de mammifères (O'Rourke et coll., 2004). Les études portant sur la
maximakinine visaient à caractériser ses propriétés pharmacologiques et d'évaluer les
conséquences de la longue extension N-terminale présentée par ce peptide par rapport à son
extrémité active, sur la signalisation et le cycle-endocytose du RB2 stimulé par cet
analogue.
Le dernier volet de ces travaux de doctorat s'est attardé à l'exploitation qui peut être faite
de l'endocytose du RB2. Suivant la stimulation du RB2 par son agoniste, le récepteur est
phosphoryle au niveau d'une région riche en résidus serine et threonine dans son extrémité
C-terminale (Blaukat et coll., 1996; 1999), ce qui permet la reconnaissance du site et le
recrutement de la machinerie endocytaire par l'intermédiaire de l'interaction avec les
arrestines non visuelles, ou P-arrestines (Pizard et coll., 1999; Hamdam et coll., 2007).
L'endocytose dépendante de l'agoniste du RB2, comme celle de plusieurs autres RCPG qui
sont aussi des substrats des GRK, implique l'internalisation et la dégradation subséquente
de l'agoniste (Leeb-Lundberg et coll., 2005). Cette adaptation dépendante de l'agoniste du
RB2 peut être exploitée de manière originale par l'application d'une stratégie visant à

52
provoquer l'internalisation de cargos de taille beaucoup plus importante que celle de
l'agoniste. À cette fin, la construction myc-RB2 a été exploitée afin de permettre
l'utilisation d'anticorps dirigés contre l'épitope. Les objectifs étaient de vérifier la
faisabilité de l'internalisation de cargos anticorps dirigés contre une séquence épitopique en
N-terminal du RB2 dépendante de la présence de l'agoniste et caractériser les voies
d'internalisation du myc-RB2 solidaire d'un vaste complexe immun. Afin de démontrer
l'importance de la voie endocytaire normalement recrutée par le RB2 dans le transport des
complexes anticorps-récepteur-agoniste, une construction myc-RB2 tronquée résistante à
l'internalisation fut utilisée.

53
2. Matériel et Méthodes
2.1 Synthèse des analogues des kinines
2.1.1 Synthèse peptidique
Le peptide B9430, un antagoniste du RB2, a été rapporté comme membre d'une série
d'antagonistes comportant l'acide aminé non naturel indanylglycine (Géra et Stewart, 1996;
Stewart et coll., 1996). Les séquences des peptides B9972 et B10344 dérivent de celle du
peptide B9430; les modifications apportées sont l'inversion des acides aminés en position
7 et 8, ainsi que la modification de la stéréochimie de l'indanylglycine. Toutes les
structures sont présentées à la figure 2. Le peptide B9972 a déjà été décrit (Baironite et
coll., 2004; TarasevicieneStewart et coll., 2005). La synthèse en phase solide, la
purification et la caractérisation du peptide B10344 ont été réalisées par l'application de la
même procédure générale décrite pour les peptides B9430 et B9972 (Géra et coll., 1996).
0
H 4 U
/
iMsfoirBWO: PArp llvp^d' l^roic' BK
H
5> t%.»
O ç
ç_ .A
"■""t"" H
H
OiC'lgl»
BW2: DAif Hyp k\ Ok" IgfBK
■■
t N
j ~ H
c
r
OklMgi». BII1.V44: DArç 4Ivp \ë Oic"iM il'BK
Figure 2. Structure et stéréochimie de la région Cterminale des trois ligands des récepteurs des kinines qui sont des isomères, soient B9430, B9972 et B10344. La structure de référence est celle de la BK (HArg'Pro2Pro3Gly4Phe5Ser6Pro7Phe8Arg9OH).

54
Les ligands fluorescents ont été synthétisés et purifiés en suivant le même protocole
général. L'antagoniste fluorescent du RB2, le peptide B-10380, et le peptide B-10330,
présentent une séquence dérivant du peptide B-9430 (tableau 1), respectivement prolongés
en N-terminal par la D-biotine ou la 5(6)-carboxyfluorescéine (CF) avec un espaceur £-
aminocaproyl (e-ACA). Les agonistes fluorescents du RB2 sont formés de la séquence de la
BK, avec l'ajout en N-terminal de la 5(6)-CF avec ou sans un espaceur e-aminocaproyl
(CF-e-ACA-BK et CF-BK). Les séquences des antagonistes fluorescents du RB,, les
peptides B-10372 et B-10376, sont basées sur la séquence de l'antagoniste B-9958 (Géra et
coll., 1996), avec l'extension CF en N-terminal, avec (le peptide B-10376) ou sans (B-
10372) l'espaceur E-ACA, entre la séquence du fluorophore et celle du peptide B-9958.
L'agoniste fluorescent du RB,, le peptide B-10378, contient la séquence de l'agoniste
naturel préférentiel Lys-des-Arg -BK, à laquelle sont ajoutés l'espaceur e-ACA et la CF,
toujours en position N-terminale de la séquence de l'agoniste. Les séquences de ces
peptides sont présentées au tableau 1.
Tous ces peptides ont été synthétisés par notre collaborateur, le Docteur Lajos Géra
(Université du Colorado, Denver).
55
Peptide position Peptide
-3 -2 -1 0 1 2 3 4 5 6 7 8 9
Agonistes du récepteur B2, substrats de l'ECA
CF-BK [5(6)-
CF]
Arg Pro Pro Gly Phe Ser Pro Phe Arg-
COOH
CF-
EACA-
BK
[5(6)-
CF]
£-
ACA
Arg Pro Pro Gly Phe Ser Pro Phe Arg-
COOH
Antagonistes du récepteur B2
B-
10330
D-
biotinyl
D-
Arg
Arg Pro Hyp Gly Igl Ser D-
Igl
Oic Arg-
COOH
B-
10380
[5(6)-
CF]
E-ACA D-
Arg
Arg Pro Hyp Gly Igl Ser D-
Igl
Oic Arg-
COOH
Antagonistes du récepteur B!
B-
10372
[5(6)-
CF]
Lys Lys Arg Pro Hyp Gly CpG Ser D-
Tic
CpG-
COOH
B-
10376
[5(6)-
CF]-
£-
ACA
Lys Lys Arg Pro Hyp Gly CpG Ser D-
Tic
CpG-
COOH
Agoniste du récepteur B(
B-
10378
[5(6)-
CF]
E-ACA Lys Arg Pro Pro Gly Phe Ser Pro Phe-
COOH
Tableau 1. Séquences des nouveaux analogues des kinines possédant des extensions en N-terminal.
Abbreviations: [5(6)-CF]: 5(6)-carboxyfluorescéine; CpG: a-cyclopentylglycine; E-ACA: £-aminocaproyl; Hyp: /nms-4-hydroxyproline; Igl: a-(2-indanyl)glycine; Oic: (3as, 7as)octahydroindol-2-yl-carbonyl; Tic: l,2,3,4-tétrahydroisoquinoline-3-acide carboxylique
2.1.2 Synthèse du composé 47a
Le composé 47a (ethyl {4-[(3-{[({(2 )-3-(6-(acétylamino)-3-pyridinyl]-2-
propenoyl}amino)acétyl](méthyl)amino}-2,6-dichlorobenzyl)oxy]-2-méthoxy-lH-
benzimidazol-1-yl}acétate) a été synthétisé par l'application des procédures préalablement
décrites par Sawada et collaborateurs (2004b), en se référant aussi à des articles plus
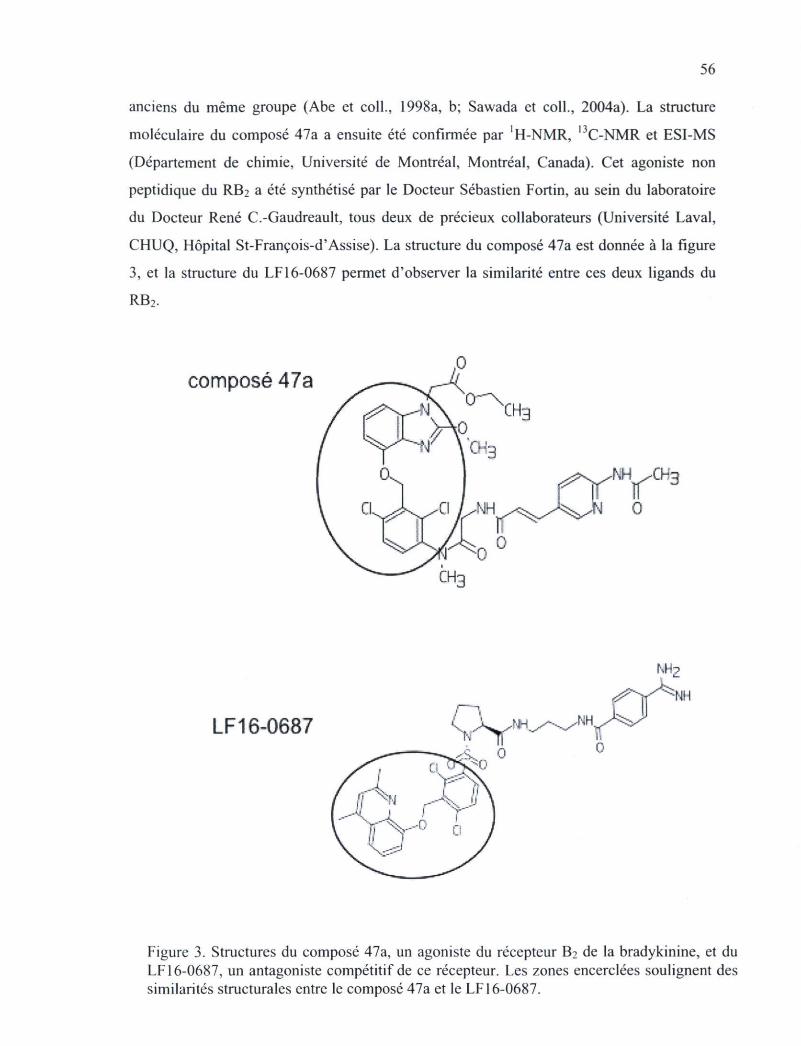
56
anciens du même groupe (Abe et coll., 1998a, b; Sawada et coll., 2004a). La structure
moléculaire du composé 47a a ensuite été confirmée par 'H-NMR, 13C-NMR et ESI-MS
(Département de chimie, Université de Montréal, Montréal, Canada). Cet agoniste non
peptidique du RB2 a été synthétisé par le Docteur Sébastien Fortin, au sein du laboratoire
du Docteur René C.-Gaudreault, tous deux de précieux collaborateurs (Université Laval,
CHUQ, Hôpital St-François-d'Assise). La structure du composé 47a est donnée à la figure
3, et la structure du LF 16-0687 permet d'observer la similarité entre ces deux ligands du
RB2.
composé 47a
NH >-CH3 Y O
LF16-0687 nt/-*^
Figure 3. Structures du composé 47a, un agoniste du récepteur B2 de la bradykinine, et du LF 16-0687, un antagoniste compétitif de ce récepteur. Les zones encerclées soulignent des similarités structurales entre le composé 47a et le LF 16-0687.

57
2.1.3 Maximakinine
La maximakinine, d'origine amphibienne, a été achetée chez Phoenix Pharmaceuticals
(Burlingame, CA). Cet analogue naturel est constitué de dix-neuf acides aminés
(DLPKINRKGPRPPGFSPFR), dont les neuf derniers acides aminés (soulignés) forment la
séquence du peptide prototype de la famille des kinines, soit la BK.
2.2 Construction des récepteurs recombinants
La construction, l'expression stable dans les cellules HEK 293 et les propriétés de la
protéine recombinante RB2 de lapin-GFP ont été décrites précédemment (Houle et coll.,
2000; Bachvarov et coll., 2001). Elle a été comparée à la nouvelle construction myc-RB2
dans laquelle l'épitope myc fut conjugué à la séquence du RB2 de lapin au niveau de
l'extrémité N-terminale (extracellulaire) du récepteur. Pour ce faire, le vecteur pCDNA3
contenant la séquence du RB2 de type sauvage de lapin (Bachvarov et coll., 1995) a été
utilisé comme source d'ADN pour procéder à l'amplification de la séquence codante du
gène du RB2 par réaction en chaîne par polymerase (Polymerase Chain Reaction- PCR).
Les oligonucleotides 5 '-GAACGAATTCGATGCTCAACATCACCTCTCAA-3 ' et 5'-
TGATTCTAGATCACTGTTTGTTCCTCGTCCA-3' servirent d'amorces sens et antisens,
respectivement. Ces amorces dérivent de la séquence du RB2 de lapin (Bachvarov et coll.,
1995). La paire d'amorces permettait l'ajout de sites de restriction additionnels des
enzymes EcoRI et Xbal (soulignés), respectivement, nécessaires au clonage directionnel
adéquat de la région codante du RB2 dans le vecteur d'expression eucaryote pCI-neo
(Promega, Madison, Wl) auquel un epitope myc N-terminal a été ajouté (pCI-neo-myc, don
du Docteur Josée Lavoie, CHUQ, Québec, Canada). Le produit du PCR et le vecteur pCI-
neo-myc ont été digérés avec EcoRI et Xbal (Invitrogen, Carlsbad, CA) et soumis à la

58
ligation à la température de la pièce pendant deux heures. Le vecteur résultant myc-RB2
contient la séquence épitopique myc (MEQKLISEEDLNS) fusionnée à la séquence
codante du RB2 de lapin à son extrémité C-terminale, dans le cadre de lecture approprié.
L'identité du vecteur myc-B2R a été confirmée par séquençage (Plateforme d'analyses
génomiques de l'Université Laval, Pavillon Marchand, Université Laval, Québec).
Une mutation ponctuelle qui détermine une deletion a été introduite dans le vecteur myc-
B2R décrit ci-dessus. Le vecteur myc-RB2 servit de source d'ADN pour l'amplification par
PCR. Les oligonucleotides 5 '-CAGGCGTAGAGCTCCATGGGC-3 ' et 5'-
GCCCATGGAGCTÇTACGCCTG-3', respectivement les amorces sens et anti-sens, ont
permis la mutation du résidu Glu340 en un codon stop (souligné dans la séquence des
amorces). L'identité du vecteur myc-RB2TRUNc fut confirmée par séquençage (Plateforme
de Séquençage et Génotypage, CHUL, Université Laval, Québec).
Le vecteur codant pour la cavéoline-1 (séquence canine) fusionnée à la protéine verte
fluorescente (décrit précédemment par Pelkmans et collaborateurs, (2001)) nous a
gracieusement été donné par le Docteur Ari Helenius. La séquence codante de la cavéoline
a été excisée et insérée à la même position dans un vecteur portant la séquence de la
protéine fluorescente Cherry (CherryFP), afin d'obtenir une protéine recombinante
émettant une fluorescence rouge (travail effectué dans le laboratoire du Dr Michel J.
Tremblay, CHUQ).
Des bactéries TOP 10 (Invitrogen, Carlsbad, CA) ont été transformées avec les vecteurs
produits, ce qui a permis leur surexpression et éventuellement la préparation des plasmides,
à partir d'une colonie isolée.
2.3 Culture cellulaire et transfection
Des cultures primaires de muscle lisse vasculaire provenant de l'artère ombilicale humaine
ont été obtenues par la mise en culture de petits segments de l'artère débarrassée de

59
l'endothélium sur une matrice de gélatine, dans le milieu M199 supplémenté avec du sérum
fœtal bovin (FBS : 20%), de la L-glutamine (2 mM), de la pénicilline (50 U/ml) et de la
streptomycine (50 pg/ml), suivant les procédures rapportées par Angers et collaborateurs,
2000). Un essai conventionnel pour confirmer l'identité de ces cultures est basé sur
l'immunocytochimie de l'a-actine, une protéine fortement exprimée par les cellules
musculaires lisses. Afin de prévenir des effets dus à la dédifférentiation de ces cellules
secondaire à leur culture, ces lignées ne furent utilisées que jusqu'au cinquième passage.
L'obtention de cultures primaires de muscle lisse vasculaire d'aorte de rat nécessite des
étapes très similaires à celles décrites pour les cellules de muscle lisse d'artère ombilicale
humaine; après avoir coupé l'aorte en petits segments, ceux-ci furent mis en culture sur une
matrice de gélatine dans le milieu DMEM supplémenté avec du FBS (10 %), l'acide aminé
essentiel L-glutamine (2 mM) et des antiobiotiques (pénicilline 50 U/ml et streptomycine
50 pg/ml).
Les cellules COS-1 (cellules rénales de singe, transformées avec le virus simien SV40,
inapte à la replication génomique) ont été cultivées dans le milieu DMEM (Dulbecco's
modified Eagle medium) supplémenté avec du FBS (10 %), de la L-glutamine (2 mM), de
la pénicilline (50 U/ml) et de la streptomycine (50 pg/ml). Les cellules HEK 293, obtenues
de FAmerican Type Culture Collection (ATCC; Manassas, VA) ont été cultivées dans le
milieu a-MEM (a-Minimun Essential Medium) supplémenté avec du sérum de cheval (5
%) et du FBS (5 %), avec la L-glutamine (2 mM) et les antibiotiques (pénicilline 50 U/ml
et streptomycine 50 pg/ml). Un clone additionnel des cellules HEK 293, qu'on identifie en
utilisant l'appellation HEK 293a, a été obtenu de Sigma-Aldrich et a été exploité plus
généralement au cours de ces travaux lors des expériences nécessitant des transfections
transitoires. Les cellules HEK 293a étaient cultivées dans le même milieu que celui décrit
ci-dessus pour les cellules COS-1. La lignée cellulaire MG-63, dérivée d'un ostéosarcome
humain, a été cultivée avec le milieu a-MEM (10 % FBS, L-glutamine 2 mM, pénicilline
50 U/ml et streptomycine 50 pg/ml). Cette lignée est un don du Docteur Patrice Poubelle
(CRRI, CRCHUL, Université Laval, QC, Canada).
La génération de lignées de cellules HEK 293 exprimant les protéines recombinantes RB2-
GFP et RBpYFP de façon stable, ainsi que leurs propriétés respectives ont été décrites

60
précédemment (Houle et coll., 2000; Bachvarov et coll., 2001; Fortin et coll., 2006a). Les
cellules HEK 293 non transfectées utilisées lors de certaines expériences servaient de
témoins négatifs. Une lignée de HEK 293a exprimant la construction myc-B2R stablement
a également été produite et utilisée dans quelques expériences. Les lignées transfectées de
manière stable sont cultivées en présence de l'antibiotique généticine (500 pg/ml), qui
permet de sélectionner positivement les cellules exprimant le transfectant.
Les cellules COS-1, HEK 293 et HEK 293a ont été transfectées ou cotransfectées
lorsqu'elles atteignaient 70 % de confluence, pour une durée de 24 à 48 h, dépendamment
des expériences, grâce à l'agent de transfection EX-Gen 500 (MBI-Fermentas Inc.,
Flamborough, ON, Canada), utilisé selon les recommandations du fabricant. En plus des
protéines recombinantes myc-RB2 et RB2-GFP, un troisième vecteur codant pour la
séquence du RB2 de type sauvage du lapin (Bachvarov et coll., 1995) a fréquemment été
employé. Deux vecteurs préalablement décrits codant pour le RB) furent utilisés : le
premier contenait la séquence humaine du RBi et était fusionné avec un epitope FLAG en
C-terminal de sa séquence (Morissette et coll., 2008), alors que le second comportait la
séquence du RBi, de type sauvage de lapin (Larrivée et coll., 2000). Le vecteur codant pour
la (3-arrestine2-GFP (Bernier et coll., 2007) est un don du Docteur Michel Bouvier
(Université de Montréal, QC, Canada) alors que celui codant pour la (3-arrestinei-CherryFP
est un don du Docteur Jean-Martin Beaulieu (Université Laval, QC, Canada). Les vecteurs
C-CherryFP, Rab5-GTP-locked-CherryFP et GFP-Rab7 ont été offerts par le Docteur
Michel J. Tremblay (Université Laval, QC, Canada) et le Docteur Robert Lodge (Université
Laval, QC, Canada). Le vecteur codant pour EEA1-FYVE-GFP est un don du Docteur
Tamas Balla (NICHD, NIH, Bethesda, MD, USA). Le vecteur d'expression de la protéine
de fusion pEGFP-LC3 a été obtenu grâce au Docteur Tamotsu Yoshimori (Université
d'Osaka, Japon). Le vecteur d'expression peACE, contenant la séquence codante de
l'enzyme de conversion de I'angiotensine, est un don du Professeur Pierre Corvol
(INSERM, Paris, France).
Les vecteurs pCI-neo-myc, PCDNA3.1-FLAG (don du Docteur Patrick Provost, CHUQ,
QC, Canada) et C|-CherryFP étaient exprimés dans les cellules servant de témoins négatifs.
Par ailleurs, le vecteur C|-CherryFP était fréquemment utilisé en cotransfection, afin de

61
déterminer l'efficacité de la transfection, en plus de faciliter le repérage de cellules
cotransfectées, lors des expériences de microscopie.
2.4 Essais de contractilité
L'utilisation de vaisseaux sanguins dans la réalisation d'essais fonctionnels peut s'avérer
une source inestimable d'informations, puisque ce sont des cibles importantes d'une variété
de médiateurs inflammatoires et d'hormones (Marceau et coll., 2010). Ces essais peuvent
servir à évaluer de nouveaux composés vasoactifs, à identifier la nature agoniste ou
antagoniste de composés, ainsi qu'à caractériser les intervenants dans le métabolisme des
composés à l'étude et permettre une meilleure compréhension de la distribution des
médicaments. Sans que cette technique ne donne le reflet exact de ce qui se produit in vivo,
elle intégre un niveau d'organisation supérieur et plus complexe que des essais
pharmacologiques moléculaires et cellulaires, très importants eux aussi. La détection de
l'activité biologique en «temps réel» est un facteur très intéressant de ce type d'essais
fonctionnels (Vane, 1983), dont il existe plusieurs variantes.
L'isolation de la veine jugulaire, de l'aorte et de l'artère mésentérique à partir de lapins
(New Zealand White, 1,5-2 kg) a permis la réalisation d'essais fonctionnels bien établis
pour le RB2 (veine jugulaire, Houle et coll., 2000) et RBi (aorte et artère mésentérique,
Petitclerc et coll., 1994; Larrivée et coll., 2000). Les veines étaient coupées en bandelettes
de 3 cm de longueur, alors que l'aorte et l'artère mésentérique étaient plutôt coupées en
anneaux. Tous ces tissus étaient placés dans des bains à organes isolés de 5 ml, retenus au
fond de ceux-ci par un crochet et suspendus à un transducteur isométrique par un fil, sous
une tension de 0,5 g (bandelettes de veine jugulaire), de 2 g (anneaux d'aorte) ou de 1 g
(anneaux d'artère mésentérique). L'obtention de cordons ombilicaux et l'isolation
subséquente de la veine ombilicale humaine a permis le même type d'essai fonctionnel du
RB2 que la veine jugulaire de lapin, à la différence de l'espèce de provenance du récepteur
étudié. Une fois minutieusement disséquée du cordon ombilical et débarrassée du tissu

62
conjonctif excédentaire, la veine était coupée en anneaux d'une largeur de 2 à 3 mm. Ces
anneaux étaient par la suite fixés dans les bains sous une tension basale de 2 g.
Les tissus ont été maintenus à une température de 37 °C, dans une solution de Krebs (117,5
mM NaCl, 4,7 mM KC1, 1,2 mM KH2P04, 1,18 mM MgS04, 2,5 mM CaCl2, 25 mM
NaHC03 et 5,5 mM D-glucose) oxygénée (95 % 0 2 : 5% C02). L'inhibiteur de l'ECA
captopril est continuellement présent dans la solution de Krebs lors des essais réalisés avec
la veine jugulaire de lapin, à raison d'une concentration de 1 pM. Les changements
isométriques du tonus vasculaire secondaires aux traitement avec les différents ligands des
RB] et RB2 sont mesurés par les transducteurs (modèle 52-9545; Harvard Apparatus, South
Natick, MA) couplés à des enregistreurs.
Les manipulations sur les animaux décrites ci-dessus ont été préalablement approuvées par
un comité d'éthique local. L'utilisation anonyme de cordons ombilicaux provenant
d'accouchements par césarienne élective a été approuvée par un comité d'éthique
institutionnel.
2.4.1 Identification de la nature et de la spécificité des nouveaux ligands du RB2 à séquence contrainte
L'expression endogène et abondante du RB2 par la veine jugulaire de lapin et les réponses
contractiles stables qu'elle génère ont rendu possibles l'utilisation de ce tissu dans le
système de bains à organes isolés. Dans un premier temps, après une période
d'équilibration d'une heure dans les bains, les tissus ont été soumis à la construction d'une
première courbe concentration-réponse cumulative, puis d'une seconde quatre heures après
le début de la période d'incubation, pour trois agonistes donnés (BK, B-9972, B-10344),
dont l'ordre a été déterminé au hasard. L'effet maximal obtenu avec l'agoniste naturel BK
servait à déterminer le statut d'agoniste partiel ou complet des autres peptides. Dans un
deuxième temps, d'autres tissus assignés à la caractérisation de l'effet antagoniste du
peptide B-9430, ont servi à la construction de deux courbes concentration-effet à l'aide de

63
la BK. Pour chaque tissu, une première courbe concentration-réponse, constituant la courbe
témoin, a été obtenue au temps 1 h après son montage dans le bain en ajoutant de façon
successive des quantités croissantes de BK. La deuxième courbe construite pour un tissu
donné au temps 4 h, en ajoutant les mêmes quantités croissantes de BK, se faisait en
présence de l'antagoniste B-9430, ajouté au temps 3,5 h. La contraction résultante de la
seconde courbe est exprimée en pourcentage de la contraction maximale obtenue au
moment de la construction de la courbe témoin. La stimulation par l'histamine des
différents tissus à la fin des essais de contractilité portant sur le RB2 servait à démontrer
que la capacité des vaisseaux à répondre à des agents contractiles était intacte.
Les valeurs de CE50 (concentration de l'agoniste à laquelle la moitié de l'effet maximal est
atteint) ont été obtenues à partir de moyennes des courbes concentration-réponse par
l'interpolation de deux points de chaque côté de l'effet à demi maximal sur une échelle
semi-log. Les paramètres de la régression de Schild (le pA2 et la pente) et l'erreur type de la
moyenne pour chacun ont été calculés à partir des points expérimentaux par un programme
informatique (PCS/Pharm; Bawolak et Marceau, 2007). Les valeurs numériques sont
rapportées sous la forme (moyenne ± erreur type). Les groupes de valeurs distribuées de
manière non normale ont été comparés grâce à l'analyse de variance non paramétrique,
avec le test de Kruskal-Wallis, suivie de l'application du test de comparaison multiple de
Dunn. Les ensembles de valeurs suivant une distribution normale ont été comparés en
utilisant le test ANOVA, suivi du test de Dunnett, afin de comparer l'effet des traitements à
un témoin commun.
Une façon de quantifier la puissance d'un antagoniste est en termes de pA2, qui constitue
une mesure d'affinité de l'antagoniste pour son récepteur. Plus spécifiquement, le terme A2
donne la concentration de l'antagoniste qui réduit l'effet d'une dose double de l'agoniste à
celui d'une dose simple, comparativement à celui observé quand l'agoniste est employé
seul. La construction de courbes concentration-réponse d'un agoniste en présence de
différentes concentrations de l'antagoniste étudié permet d'obtenir un ensemble de rapports
de concentration (concentration d'agoniste [A'] nécessaire pour obtenir un effet d'une
intensité déterminée en présence d'une concentration [B] de l'antagoniste / concentration
d'agoniste [A] nécessaire pour obtenir un effet de la même intensité en absence de

64
l'antagoniste). Le calcul de ces rapports sert ensuite à générer la régression de Schild
(Arunlakshana et Schild, 1959), une méthode graphique permettant l'élucidation de la
valeur du pA2. La régression de Schild est donnée par le logarithme des rapports [Log
((AVA)-l)] en fonction du logarithme négatif de la concentration de l'antagoniste [-Log B].
Le prolongement de la droite ainsi tracée mène à son intersection avec l'axe des x ([ -Log
B], point donnant la valeur du pA2. Plus la valeur de pA2 est importante, plus l'antagoniste
est puissant. Il y a toutefois une condition à l'application de ce concept : l'antagoniste doit
être surmontable et déplaçable par des concentrations croissantes de l'agoniste (dans le cas
contraire, l'antagoniste est non compétitif). Dans le cadre des études présentées dans cette
thèse, lorsqu'il a été possible de le faire, les paramètres de la régression de Schild ont été
estimés par une méthode informatique (Tallarida et Murray, 1987) et la valeur de pA2 a été
calculée.
L'effet antagoniste du peptide B-9430 sur la contraction dépendante de la stimulation des
RBi exprimés de manière endogène par l'aorte de lapin par la des-Arg -BK a été évalué en
suivant exactement la procédure décrite par Fortin et collaborateurs (2005a). La possibilité
d'un effet agoniste ou antagoniste du peptide B-9972 dans cette même préparation de
muscle lisse vasculaire a été aussi été étudiée.
Des études contractiles additionnelles portant sur ces nouveaux ligands (B-9972 et B-
10344) du RB2 ont été poursuivies dans le modèle basé sur les anneaux de veine ombilicale
humaine. La [Phe8x|j(CH2NH)Arg9]BK a été ajoutée à l'éventail des agonistes à l'étude (son
effet dans l'essai de contractilité de veine jugulaire isolée de lapin a déjà été rapporté;
Drapeau et coll., 1988). Après une période d'incubation initiale de trois heures après leur
installation dans les bains, les tissus, auxquels ont été assignés aléatoirement les peptides
agonistes, étaient soumis à la construction de courbes concentration-réponse cumulatives.
À la fin de l'expérience, les tissus étaient traités avec la 5-hydroxytryptamine (5-HT; 10
pg/ml). L'effet contractile maximal par la stimulation du RB2 a ensuite été exprimé en
pourcentage de la réponse maximale à la 5-HT. D'autres tissus ont été employés à la
caractérisation de l'effet antagoniste du peptide B-9430; dans le cadre de ces expériences,
le prétraitement des tissus avec soit l'antagoniste B-9430 ou son solvant (une solution

65
saline) se faisait au temps 2,5 h suivant l'installation des tissus dans les bains, soit 0,5 h
avant la construction de la courbe concentration-réponse avec l'agoniste BK.
L'obtention de l'effet contractile maximal suite à la construction d'une courbe
concentration-effet a permis la mesure d'un paramètre additionnel. Après le lavage des
tissus, le temps de relaxation à partir de l'effet contractile maximal a été enregistré et
comparé pour les différents agonistes (la BK, la [Phe8\J>(CH2NH)Arg9]BK et les peptides B-
9972 et B-10344).
Dans le but d'identifier les voies d'inactivation de la BK dans la veine ombilicale humaine,
des courbes concentration-effet ont été construites en présence d'inhibiteurs de l'ECA
(énalaprilat, 1 pM), de la NEP (phosphoramidon, 1 pM) ou de l'APP (apstatine, 20 pM;
Prechel et coll., 1995). Ces molécules étaient appliquées 30 minutes avant la construction
de la courbe concentration-réponse à la BK. De plus, le temps de relaxation à partir de
l'effet contractile maximal a été enregistré.
2.4.2 Caractérisation approfondie du peptide B-9972, ainsi que du composé 47a, un agoniste non peptidique du RB2, deux agonistes résistants au métabolisme
Dans un premier temps, l'agoniste non peptidique 47a a été évalué pour la puissance de son
activité contractile médiée par le RB2 humain exprimé dans la veine ombilicale humaine.
Après une période d'équilibration initiale de trois heures après le montage des tissus dans
les bains, des courbes concentration-réponse ont été construites par l'ajout successif de
quantités croissantes de composé 47a dans la solution de Krebs baignant l'anneau de veine
ombilicale humaine. La construction de courbes concentration-réponse additionnelles à
l'aide d'autres tissus, préalablement traités avec différentes concentrations de l'antagoniste
LF 16-0687 (0-1000 nM), permettait de vérifier la spécificité du composé 47a.
La veine ombilicale humaine peut être contractée par la stimulation du RB2 par son
agoniste de façon répétée, tout en présentant une réponse contractile stable (Marceau et

66
coll., 1994). Dans un deuxième temps et afin de vérifier si les agonistes du RB2 résistants
au métabolisme, le peptide B-9972 et le composé non peptidique 47a, provoquent la
désensibilisation de ce système à la stimulation par l'agoniste BK ou vice versa, le
protocole suivant a été élaboré et appliqué. Chaque tissu servait à la construction de deux
courbes concentration-effet, la première au temps 3 h (toujours par rapport au début de sa
période d'équilibration) avec un agoniste donné et la seconde, au temps 6 h, avec un
agoniste différent.
Troisièmement, le paramètre additionnel de temps de relaxation a pu être mesuré grâce à
ces essais de contractilité. Pour ce faire, les tissus sélectionnés pour cette expérience ont été
stimulés trois heures après leur installation dans le système de bains à organes isolés avec
les différents agonistes (la BK, le peptide B-9972 et le composé 47a) afin qu'ils produisent
leur effet maximal, obtenu par la construction d'une courbe concentration-réponse pour
chaque agoniste. Par la suite, les tissus étaient lavés abondamment et la diminution de la
tension (relaxation) des différents tissus était enregistrée pour calculer la période
correspondant à la relaxation de la moitié de l'effet contractile.
2.4.3 Étude des ligands présentant une extension en N-terminal
Les études présentées dans cette section visaient à évaluer l'effet du prolongement de la
séquence de peptides déjà identifiés comme ligands de l'un des deux sous-types des
récepteurs des kinines au niveau de la conservation de leur identité agoniste ou antagoniste,
leur affinité et leur puissance, comparativement au peptide parent déjà caractérisé. À cet
effet, le peptide B-10380 fut soumis aux mêmes procédures que le peptide dont il dérive,
soit le peptide B-9430. Les propriétés antagonistes du peptide B-10380 ont été quantifiées
par la construction de courbes concentration-effet en présence de cet antagoniste par la
stimulation d'anneaux de veine ombilicale humaine par la BK (essai fonctionnel du RB2
humain) et de bandelettes de veine jugulaire de lapin (essai fonctionnel du RB2 de lapin),

67
ainsi que par la stimulation par la des-Arg9-BK d'anneaux d'aorte de lapin, qui est un essai
fonctionnel du RBi de lapin.
Le peptide CF-eACA-BK a été comparé à l'agoniste naturel préférentiel du RB2 BK, grâce
à la construction de courbes concentration-réponse au niveau du RB2 humain endogène
exprimé par la veine ombilicale humaine. Les courbes ont été construites après une période
d'équilibration de trois heures après l'installation des tissus dans les bains.
La puissance des antagonistes du RBi ayant comme prolongement de leur séquence à
l'extrémité N-terminale la 5(6)-CF, avec ou sans l'espaceur e-ACA, a été évaluée par
l'application exacte de la procédure décrite par Fortin et collaborateurs (2005a), pour le
RB] de lapin. Des courbes concentration-réponse furent construites en ajoutant de manière
cumulative l'agoniste du RBi des-Arg9-BK, au temps 5,5 h compté à partir de la mise en
place des anneaux d'aorte de lapin dans les bains à organes isolés, en présence ou non des
peptides B-10372 et B-10376, ainsi que du peptide B-9958, dont ils dérivent, ajoutés à la
solution physiologique au temps 5 h. Il est important de noter que le respect d'une période
d'incubation plus longue pour les tissus servant à la réalisation d'essais de contractilité du
RBi découle de l'observation que sa synthèse est fortement induite par le dommage
tissulaire secondaire à l'isolation de l'aorte et à sa manipulation subséquente (Leeb-
Lundberg et coll., 2005), ce qui permet un accroissement de la population de RBi
membranaires, et une plus grande réponse contractile durant les essais. L'agoniste
fluorescent du RB|, le peptide B-10378, fut aussi évalué pour la puissance de son activité
contractile dans les préparations d'anneaux d'aorte de lapin. Dans ce cas, les peptides
agonistes des-Arg -BK et Lys-des-Arg -BK ont servi à la réalisation de courbes
concentration-réponse témoins.
Des études additionnelles furent effectuées sur la pharmacologie du peptide maximakinine
(MK) grâce à l'essai de contractilité de la veine ombilicale humaine. La construction de
courbes concentration-réponse cumulatives à l'aide de ce peptide en utilisant la veine
ombilicale humaine comme tissu contractile servit à confirmer son identité
pharmacologique, ainsi que sa puissance. Après un temps d'équilibration de 3 h, une
courbe concentration-réponse cumulative pour la maximakinine fut construite dans des

68
anneaux de veine ombilicale humaine, en parallèle à la construction de courbes
concentration-réponse témoins avec le peptide BK, constituant ici le peptide référence. La
puissance de cet agoniste a ensuite été estimée par la concentration de celui-ci induisant la
moitié de la réponse contractile maximale (CE50) et comparée à celle de l'agoniste naturel
préférentiel pour le RB2, la BK.
2.4.4 Etude de l'effet antagoniste du LF16-0687
Afin d'étudier la puissance du LF 16-0687 à antagoniser l'effet contractile stimulé par la
BK au niveau du RB2 ainsi que pour établir sa sélectivité, des anneaux de veine ombilicale
humaine furent préparés et installés dans des bains à organes isolés, suspendus sous une
tension de 2 g, dans une solution de Krebs oxygénée (95 % O2 : 5 % CO2) et maintenue à
une température de 37°C. Après une période d'équilibration de 3 h des tissus après leur
montage dans les bains, les tissus furent soumis à la construction de deux courbes
concentration-effet successives; la première, réalisée au temps 3 h, en utilisant la BK, et la
seconde, réalisée au temps 5 h, en utilisant la 5-HT, un puissant agent contractile dans ce
tissu (Altura et coll., 1972). L'antagoniste à l'étude, soit le LF 16-0687, a été appliqué au
milieu baignant les tissus trente minutes avant la construction de chaque courbe
concentration-effet. Le solvant de l'antagoniste, une solution saline, a aussi été appliqué au
niveau des tissus servant à la construction de courbes concentration-effet témoins. L'effet
du composé, pour chacune des concentrations à l'essai, a été calculé comme le déplacement
vers la droite de la courbe concentration-effet moyenne obtenue par rapport à la CE50
établie par la courbe témoin.
2.5 Essais de liaison

69
2.5.1 Essais de saturation et de compétition
Les essais de liaison ont été faits sur des cellules adhérentes, intactes et présentant une
confluence quasi maximale, ensemencées dans des plaques de 12 puits (cellules
musculaires lisses d'artère ombilicale humaine et d'aorte de rat, MG-63) ou de 24 puits
(cellules COS-1, HEK 293, HEK 293a transfectées), selon la densité des récepteurs
d'intérêt exprimés par chaque type cellulaire employé.
La version tritiée de l'agoniste naturel BK ([3H]BK ; PerkinElmer Life and Analytical
Sciences, Boston, MA; 90 Ci/ mmol) servit à la réalisation des essais pour le RB2, effectués
sur glace. Comme ces essais visaient à élucider les propriétés des récepteurs à la surface
membranaire, il était impératif d'empêcher le phénomène d'endocytose auquel est soumis
le RB2 activé par la liaison de son agoniste en maintenant la température sous 4 °C. Les
essais de liaison étudiant les diverses propriétés du RBi, qui ne subit pas une internalisation
importante suivant la liaison de son agoniste, ont été effectués à une température de 37 °C,
à l'aide du radioligand [3H]Lys-des-Arg9-BK ([3H]Lys-des-Arg10-KD; 69-88 Ci/mmol;
PerkinElmer Life and Analytical Sciences, Waltham, MA).
Le tampon employé pour la réalisation des essais de liaison au niveau du RB2 (PBS 1 X,
0,02% azoture de sodium, 0,1 % albumine sérique bovine (BSA), 1 mM fluorure
phénylméthylsulfonyle, 1 pM captopril) était conservé à une température de 4 °C alors que
le tampon de liaison utilisé pour les essais de liaison du RBi (milieu M199 pH 7,4, 0,02 %
azoture de sodium, 0,1 % BSA, 1 pM phosphoramidon, 1 pM captopril et 1 pM amastatin,
tel que précédemment décrit par Sabourin et collaborateurs (2002) et Moreau et
collaborateurs (2007)) était préchauffé à 37 °C. Les deux tampons servent de milieux
réactionnels optimisés pour empêcher la dégradation des ligands par la présence
d'inhibiteurs de peptidases et de proteases, en conservant toutefois des caractéristiques
physiologiques, par exemple au niveau de leur force ionique. Avant l'ajout des
compétiteurs non marqués et du radioligand, les cellules furent rincées une fois avec le
tampon de liaison adéquat. Le volume du tampon de liaison employé était de 500 pi pour
les plaques de 24 puits et de 1 ml, pour les plaques de 12 puits.

70
Les nouvelles constructions myc-RB2 et myc-RB2TRUNc exprimées de manière transitoire
par des cellules COS-1 et HEK 293a, respectivement, ont d'abord été validées pour leur
expression et leur capacité à lier l'agoniste, par la réalisation de courbes de saturation, au
cours desquelles des quantités croissantes de [3H]BK (0,5 à 7 nM) étaient ajoutées aux
puits. Au sein de l'expérience, la moitié des puits contenaient un excès de ligand non
radiomarqué, afin d'évaluer la liaison non spécifique pour une concentration donnée de
[ H]BK. Les valeurs ainsi obtenues pouvaient ensuite être soustraites de la liaison totale
obtenue dans les puits appariés ne contenant que le ligand radiomarqué, afin de permettre le
calcul de la liaison spécifique. Suivant une période d'incubation permettant l'atteinte de
l'équilibre de liaison (90 minutes sur glace), les cellules étaient rapidement lavées à trois
reprises avec du PBS IX pH 7,4 glacé (1 ml pour une plaque 24 puits). L'ajout d'un ml de
NaOH 0,1 M a servi à dissoudre les membranes cellulaires afin de récupérer la radioactivité
encore liée aux récepteurs. Chaque échantillon fut compté par scintillation pour quantifier
la radioactivité présente dans les suspensions cellulaires.
Les essais de liaison reposent sur l'atteinte d'un équilibre entre le ligand lié au récepteur et
les deux membres de ce complexe dissociés : L* + R tt L*R, et c'est l'obtention de cet
équilibre qui permet de dériver les paramètres importants à la caractérisation des récepteurs
et de leurs ligands, soient la constante de dissociation (Ko), qui donne l'affinité et la
saturabilité (Bmax), qui donne la quantité de récepteurs présents dans un système
expérimental donné. Le Ko, donné en unités de concentration, équivaut à la concentration
menant à l'occupation de la moitié des sites spécifiques. La méthode graphique de la
régression de Scatchard permet d'évaluer ces deux paramètres, en générant un graphique
dont l'axe des x est la liaison spécifique (donnée par la soustraction de la liaison non-
spécifique à la liaison totale) et l'axe des y est le rapport entre la liaison spécifique et la
concentration de ligand totale ajoutée à l'essai. La droite résultante (à condition qu'il n'y ait
qu'un seul site de liaison spécifique) a une pente de -1/KD et donne la valeur du Bmax par
l'endroit où elle intercepte l'axe des x.
Lié/Libre Pente=-1/Kd

71
Ici l'emploi d'une méthode informatique (Prism 4.0, GraphPad Software, San Diego, CA,
USA) permit l'estimation des paramètres Ko et Bmax- Par ailleurs, des essais de compétition
furent réalisés en parallèle des courbes de saturation, afin d'évaluer le profil
pharmacologique du myc-RB2, où un ensemble de composés pertinents au système kinine-
kallikréine (agonistes et antagonistes des deux sous-types de récepteurs) servit de
compétiteurs non-radiomarqués. Une concentration fixe de 1 pM fut employée pour
chacun.
La construction des courbes de compétition pour les nouveaux ligands des RB2 et RBi mit
en présence des concentrations croissantes des ligands à l'étude, contre une concentration
constante de radioligand (3 nM de [3H]BK pour le RB2 et 1 nM de [3H]Lys-des-Arg9-BK
pour le RBi). Suivant une période d'incubation permettant l'atteinte de l'équilibre de
liaison (de 90 minutes sur glace ou de 60 minutes à 37 °C), les cellules furent rapidement
rincées et soumises à la dissolution par l'ajout de NaOH. Les suspensions ainsi obtenues
étaient ensuite récoltées et comptées. L'affinité des nouveaux ligands pour un récepteur
donné est exprimée en terme de Ki, une constante de dissociation calculée à partir du Ko
déjà connu du ligand radioactif de référence (dans ce cas-ci la [ H]BK pour le RB2 et la
[3H]Lys-des-Arg9-BK pour le RBi). L'équation servant à calculer cette valeur est Ki =
CI50/O+ L/KD) OÙ le terme CI50 est la concentration du nouveau ligand nécessaire pour
déplacer de la moitié des sites spécifiques le ligand radiomarqué, le terme L est la
concentration du ligand radiomarqué et le terme KD est la constante de dissociation du
ligand radiomarqué et a une valeur connue.
Une variante du protocole décrit ci-dessus a permis de quantifier l'inactivation métabolique
de la BK et du peptide B-9972 par les cellules HEK 293 exprimant le RB2-GFP. Ces
cellules, ensemencées dans des flacons de 75 cm2 et cultivées dans leur milieu de culture
habituel supplémenté de FBS, ont été mises en présence des agonistes BK ou B-9972
(concentration finale pour chacun : 5 pM) pendant 12 h à 37°C. Après la fin de
l'incubation, les milieux ont été récoltés et centrifugés à vitesse maximale dans une

72
centrifugeuse de table, afin d'obtenir des surnageants exempts de cellules décollées et/ou
mortes. Les surnageants ainsi préparés ont ensuite été dilués (facteur de dilution 1:25) dans
le tampon de liaison habituel pour le RB2 et ont été appliqués comme tels à des cellules
HEK 293 RB2-GFP, en tant que compétiteurs froids de la liaison de 3 nM de [3H]BK
(toujours à 0 °C, en présence d'inhibiteurs de proteases et de peptidases; tel que décrit par
Houle et collaborateurs (2000)). La concentration résiduelle des agonistes BK et B-9972 a
pu être estimée en utilisant les courbes de compétition construites au préalable pour chacun,
comme s'il s'agissait de courbes standard. Cet essai est permis par la connaissance des
déterminants de la structure et de l'activité du RB2; en effet, les fragments de la BK ne
retiennent qu'une affinité négligeable pour le RB2.
2.5.2 Endoyctose de la [3H]BK
Une des fonctions du RB2, l'internalisation de la BK, peut être quantifiée par l'utilisation
de la BK tritiée. Des cellules HEK 293 exprimant le RB2-GFP préalablement traitées 12 h
avec les agonistes BK et B-9972 ont été rincées à deux reprises avec du milieu de culture •y
frais, et exposées à 10 nM de [ H]BK pendant 15 minutes. Les cellules ont ensuite été
lysées sur glace en utilisant un tampon sucrose-tricine froid (250 mM sucrose, 20 mM
tricine, 1 mM inhibiteur de la trypsine provenant de la fève de soya, 10 mg/ml leupeptine, 2
mg/ml pepstatine). Les lysats cellulaires ont été brisés mécaniquement (2 min de piston
verre-verre) et soniqués à faible intensité et à vitesse constante pour trente secondes. La
fraction cellulaire contenant les endosomes a pu être extraite grâce à deux centrifugations
successives, la première à 600 g pendant 10 min et une seconde, dont l'échantillon
constituait le surnageant obtenu lors de la première, à 15 000 g pendant 5 min. Les culots
résultant de cette deuxième centrifugation, contenant la fraction endosomale, ont ensuite été
repris dans le même tampon servant à la lyse cellulaire et la radioactivité a été comptée par
luminescence pour chaque échantillon. Cette méthode visait à déterminer si le traitement
prolongé avec l'agoniste résistant au métabolisme provoquait une réduction de la quantité

73
de récepteurs à la membrane cellulaire, et donc une diminution de la radioactivité
internalisée de façon dépendante du récepteur.
2.6 Microscopie
2.6.1 Microscopie à fluorescence et confocale
La localisation subcellulaire du RB2-GFP en réponse à la stimulation avec les agonistes
résistants au métabolisme a été évaluée visuellement par l'utilisation de cellules exprimant
ce récepteur de façon stable, ensemencées dans des pétris de 35 mm. Suite aux traitements
de 30 min, 3 h et 12 h avec la BK, le peptide B-9972 et le composé non peptidique 47a, le
milieu était retiré et une lamelle de verre était délicatement placée sur les cellules. Les
cellules étaient observées à un grossissement de 1000 X (technique avec immersion
d'huile), afin de permettre une plus fine détection de la distribution du récepteur. Ce même
protocole a été appliqué lors des études portant sur la MK. Une condition additionnelle
combinait les effets d'un inhibiteur de l'ECA en concomitance avec la BK. Certaines
expériences portaient sur des cellules HEK 293 RB2-GFP, cotransfectées avec un vecteur
codant pour la (3-arrestinei-CherryFP, traitées avec les différents agonistes (BK, B-9972 et
le composé 47a) afin de déterminer la cinétique de l'interaction entre ces deux protéines.
De façon complémentaire, des cellules HEK 293a étaient transfectées avec la (3-arrestine2-
GFP et le myc-RB2 et soumises aux mêmes conditions pour évaluer la distribution de la |3-
arrestine2-GFP en réponse à la stimulation du récepteur. Certains traitements comportaient
aussi l'ajout du colorant nucléaire Hoechst 33258, facilitant la localisation des protéines
fluorescentes.
Des cellules exprimant des versions incolores du RBi (huRBi-FLAG, RBi de type sauvage
de lapin) et du RB2 (RB2 de type sauvage de lapin, myc-RB2) ou non, optionnellement co
transfectées avec des protéines fusionnées avec des fluorophores rouges ont permis l'étude

74
des ligands fluorescents des deux sous-types de récepteurs. La visualisation des cellules
transfectées avec les différents récepteurs à l'aide des ligands pertinents a d'abord été
effectuée avec un éventail de concentrations (de 5 nM à 100 nM pour les peptides B-10380,
B-10376 et B-10378), avant de sélectionner la concentration de 50 nM pour la suite des
expériences. Dans le cas du peptide CF-EACA-BK, l'éventail des concentrations évaluées
se situaient plutôt entre 1 pM et 10 pM, et la concentration sélectionnée pour les
expériences subséquentes était 5 pM. Les ligands fluorescents étaient ajoutés au milieu de
culture 30 minutes avant l'observation. Un temps additionnel de 3 h de traitement a été
observé pour le peptide CF-EACA-BK. Dans certains pétris, l'incubation avec les ligands
fluorescents était faite en présence d'un ligand antagoniste non fluorescent en excès,
possédant la même sélectivité pour le récepteur (le composé 11, 1 pM, pour le RBi
(Morissette et coll., 2004); le composé LF 16-0687, 1 pM, dans le cas du RB2 (Houle et
coll., 2000). Étant donnée l'internalisation du RB2 avec son agoniste, des études
supplémentaires exploitant le peptide CF-eACA-BK ont été faites pour caractériser sa
localisation subcellulaire. Des cellules exprimant le myc-B2R ont été optionnellement
prétraitées avec un inhibiteur de la V-ATPase, la bafilomycine Al (100 nM, 1 h avant le
traitement avec l'agoniste fluorescent).
Finalement, en tant que substrats de l'ECA, les agonistes fluorescents du RB2 ont été
évalués pour le marquage de cette enzyme membranaire. Le protocole suivi pour le
marquage de cellules transfectées avec un vecteur codant pour l'ECA recombinante est
identique à celui appliqué pour le marquage des RB2 avec le peptide CF-EACA-BK.
Suivant la période d'incubation à 37°C, les cellules ont été lavées avec le tampon Hank's
Balanced Salt Solution (HBSS 1 X, pH 7,4), et observées à un grossissement de 400 X et
de 1000 X (filtres d'excitation bleu et d'émission vert). Les cellules ont aussi été observées
en contraste de phase afin de permettre l'évaluation de l'efficacité de la transfection. Toutes
ces procédures ont été appliquées à l'abri de la lumière, afin de prévenir la dégradation du
fluorophore conjugué au ligand. Les cellules composant l'échantillon de la série
d'expériences exploitant le peptide CF-EACA-BK, ont été cotransfectées avec la protéine
CherryFP, permettant ainsi le repérage des cellules cotransfectées avec myc-RB2.

75
Pour étudier plus avant le comportement des RB suite à la liaison de leur agoniste, des
cellules ensemencées sur des pétris à fond de verre recouvert ou non de poly-L-lysine et
transfectées avec le RB2 de type sauvage de lapin, le myc-RB2, le hRB|-FLAG et le RBi
de type sauvage de lapin ont permis de générer des images présentant des coupes
confocales, à l'aide du microscope confocal à disque tournant Olympus, employé avec le
logiciel Volocity (Improvision; PerkinElmer Life and Analytical Sciences) doté d'une
caméra numérique Hamamatsu EMCCD (Hamamatsu Coloration, Bridgewater, NJ). Les
reconstitutions tridimensionnelles des cellules observées par microscopie confocale ont été
générées à l'aide d'une série d'images confocales traitées par le logiciel FreeSFP (http :
//www.svi.nl).
2.6.2 Immunocytochimie
La possibilité d'utiliser la nouvelle construction myc-RB2 dans le cadre d'essais de
localisation et d'expression basés sur l'utilisation d'anticorps anti-myc a été validée. Des
cellules HEK 293a transfectées avec la construction myc-RB2 de manière stable ou non, ont
été fixées (paraformaldéhye 1%, 20 minutes) et optionnellement perméabilisées (Triton
0,5%, 5 minutes) et soumises à F immunofluorescence indirecte, utilisant l'anticorps anti-
myc monoclonal (clone 4A6; Millipore Corporation, Billerica, MA; dilution 1:1000), et
révélées par un anticorps secondaire de chèvre anti-souris conjugué avec un fluorophore
Alexa-fluor 488 (Molecular Probes-Invitrogen, Eugene, OR; dilution 1:1000).
Une variation de ce protocole visait à évaluer la possibilité de provoquer l'internalisation de
cargos anticorps complexés à l'épitope myc du myc-RB2, se trouvant à l'extrémité N-
terminale du récepteur, donc au niveau du milieu extracellulaire, en réponse à la liaison de
l'agoniste BK. Dans un premier temps, des cellules HEK 293 tranfectées ou non avec le
vecteur myc-RB2, ont été incubées en présence de deux clones d'anticorps anti-myc (clone
4A6, utilisé comme décrit plus haut et clone 9E10; Covance; dilution 1:10) pendant 30 min
à la température de la pièce. Elles ont ensuite été lavées et incubées à 37 °C dans un milieu

76
de culture non supplémenté en FBS (1 h). Quinze min avant la fin de l'incubation, certaines
cellules furent traitées avec l'agoniste BK. À la fin de cette période d'incubation, les
cellules ont été fixées, perméabilisées et marquées avec un anticorps secondaire conjugué
avec le fluorophore Alexa Fluor 488. Par la suite, des cellules HEK 293a intactes exprimant
l'une ou l'autre des constructions myc-RB2, et facultativement des transgènes additionnels,
ont été marquées successivement par l'anticorps anti-myc (clone 4A6) et par l'anticorps
secondaire conjugué avec le fluorophore Alexa Fluor 594. Après la formation des
complexes immuns (le temps d'incubation avec chaque anticorps était identique), les
cellules ont été incubées pendant une heure dans un milieu de culture sans sérum, et
optionnellement stimulées avec l'agoniste BK (100 nM, 15 min ou 3 h). D'autres cellules
HEK 293a exprimant de manière transitoire le myc-RB2 ont été sevrées en FBS 24 h avant
d'être soumises à un traitement avec la BSA-Alexa fluor 594 (20 pg/ml, 5 min) et ensuite
utilisées pour la construction de complexes immuns (anticorps anti-myc clone 4A6 et
anticorps anti-souris couplé au fluorophore Alexa Fluor 488). Après le marquage, certaines
cellules ont été traitées avec la BK pour une période de 3 h, temps auquel la BSA-Alexa
Fluor se retrouve dans les lysosomes (Hurtado-Lorenzo et coll., 2006). Un anticorps
secondaire anti-souris, cette fois-ci couplé à un nanocristal dont les propriétés optiques
dépendent de sa taille, appelé quantum dot (anticorps de chèvre anti-souris conjugué à un
Qdot 705; dilution 1:50) a aussi été utilisé. La visualisation des complexes immuns
exploitant l'anticorps secondaire conjugué aux Qdot comme fluorophore exige une
excitation ultraviolette et l'utilisation d'une caméra électronique qui permet d'observer
toutes les longueurs d'onde du spectre visible.
2.7 Immunobuvardages
2.7.1 Expression et abondance des protéines d'intérêt
L'abondance du RB2-GFP exprimé de manière stable dans les cellules HEK 293 a été
quantifiée par la réalisation d'immunobuvardage reposant sur l'utilisation de l'anticorps
monoclonal anti-GFP JL8 (Clontech, Palo Alto, CA). Les cellules HEK 293 exprimant le

77
■y
RB2GFP ont été ensemencées dans des flacons de 25 cm , cultivées jusqu'à l'atteinte
d'une confluence de 80 à 90 % et traitées avec différents composés pour des périodes de 3
h et de 12 h, dans leur milieu de culture habituel (supplémenté en FBS). Après la période de
traitement, les cellules ont été lavées avec du PBS glacé, et lysées à l'aide d'un tampon
préchauffé à 95°C (1 mM Tris pH 7,4, 1 mM Na3V04, 1 % SDS et des inhibiteurs de
proteases (pastille Cocktail mini, Roche)). Les lysats ont été bouillis et centrifugés à 9000 g
pendant 15 min, et les surnageants ont été recueillis. Un petit volume de ces surnageants a
été prélevé pour doser la concentration des protéines dans chaque échantillon, ce qui a
permis de séparer par electrophorese sur gel SDSpolyacrylamide 9 % une quantité égale de
protéines par condition (entre 5 et 25 pg, dépendamment de la concentration en protéines
par échantillon). Les protéines ont ensuite été transférées sur une membrane de PVDF. Une
fois le transfert complété, la membrane fut incubée dans un tampon de blocage (un tampon
de lavage composé de 10 mM Tris, 100 mM NaCl, 0,1 % Tween20, à pH 7,6 auquel 5 %
de lait écrémé en poudre a été rajouté) pendant une heure avant d'être mise en présence de
l'anticorps antiGFP (dilution 1:1000) pendant 2 h à la température de la pièce. Cette même
procédure a été appliquée aux cellules HEK 293a exprimant transitoirement le mycRB2 ou
le mycRB2TRUNC, dont l'expression et l'abondance peuvent être analysées par l'utilisation
de l'anticorps antimyc (clone 4A6; Upstate Biotechnology, Lake Placid, NY), suite à
F electrophorese sur gel (7,5 %). Afin de vérifier que le dépôt de protéines sur gel était
uniforme, 1'electrophorese sur gel et le transfert ont été faits en double et la membrane
appariée à celle utilisée pour la détection avec l'anticorps d'intérêt fut plutôt analysée avec
un anticorps monoclonal dirigé contre la (3actine (SigmaAldrich, dilution 1:50 000), une
protéine à l'expression constante.
2.7.2 Signalisation
Des essais de phosphorylation des MAP kinases 1/2 (ERK 1/2) ont été réalisés en suivant
une procédure établie précédemment (Morissette et coll., 2007) afin de mieux caractériser
les effets d'un traitement prolongé avec des agonistes (la BK et le peptide B9972, 100 nM,

78
12 h), comme une possible désensibilisation du récepteur à une nouvelle stimulation aiguë.
Les cellules HEK 293 exprimant le RB2-GFP de manière stable ont été maintenues dans un
milieu de culture non supplémenté en FBS pour une durée de 24 h, et soumises à la
stimulation par les agonistes BK et B-9972 ou par un traitement combinant le captopril et la
BK, pour les 12 dernières heures. Après la fin de ce prétraitement, le milieu fut remplacé
par du milieu de culture frais, toujours non supplémenté en FBS, mais contenant
l'inhibiteur de synthèse protéique cycloheximide (CHX, 71 pM) en présence duquel les
cellules furent incubées 30 min à 37 °C, après quoi les cellules furent lavées à nouveau et
soumises à un traitement aigu de 10 min avec l'agoniste BK (10 nM) ou le facteur de
croissance de l'épiderme (EGF; 100 ng/ml). Ces manipulations furent suivies par
l'extraction des protéines totale en appliquant les étapes décrites ci-haut. Les anticorps
utilisés (anti-phospho-ERKl/2, monoclonal, dilution 1:1000; anti-ERKl/2 total, polyclonal,
dilution 1:1000) proviennent de Cell Signaling Technology Inc. (Beverly, MA). Ce même
type d'essai a ensuite été appliqué afin de déterminer l'intensité de la signalisation induite
par le composé 47a. Par ailleurs, cette méthodologie a aussi permis l'étude de la cinétique
de signalisation induite par les agonistes résistants au métabolisme comparativement à celle
induite par la BK et l'identification des seconds messagers recrutés par l'activation du RB2
par ces agonistes, en employant des inhibiteurs spécifiques de différentes voies connues. Le
détail des conditions utilisées est présenté dans la section Résultats. Il avait déjà été
rapporté que la stimulation du RB2 dans certains types cellulaires pouvait entraîner une
augmentation de l'expression de c-Fos (Vidal et coll., 2005). L'induction de l'expression de
du facteur de transcription c-Fos, dépendante de l'activation de la voie des MAP kinases
ERK 1/2 (Glauser et Schlegel, 2007) a donc fait l'objet d'essais d'immunobuvardage, en
analysant les mêmes extraits cellulaires utilisés pour l'étude de l'état de phosphorylation
des MAP kinases avec l'anticorps anti-c-Fos polyclonal K-25 (Santa Cruz Biotechnology,
Santa Cruz, CA, USA; dilution 1:1000). Finalement, des lysats obtenus de cellules HEK
293 exprimant de manière stable le RB2-GFP traitées avec la MK ont été analysés par
l'application de cette méthodologie pour étudier la cinétique d'activation de ERK 1/2
induite par cet agoniste possédant une importante extension N-terminale, ainsi que
l'expression de c-Fos qui peut y être secondaire.

79
Les essais de phosphorylation des MAP kinases ont aussi été employés afin de caractériser
les voies de signalisation recrutées par le myc-RB2TRUNC, afin de déterminer si la deletion
de la queue C-terminale pouvait provoquer des effets délétères sur sa signalisation. Des
cellules non transfectées, exprimant le myc-RB2 ou le myc-RB2TRUNc de manière
transitoire, ont été cultivés dans un milieu non supplémenté en FBS pendant 24 h et ensuite
traitées avec la BK (10 nM, 10 min). Les cellules ont ensuite été lysées, et des échantillons
d'électrophorèse sur gel en ont été dérivés.
2.8 Essais de mobilisation calcique
La mobilisation calcique induite par des agonistes du RB2 a été quantifiée par l'application
de la fluorimétrie exploitant le colorant fluorescent FURA-2AM (Molecular Probes,
Invitrogen Detection Technologies, Carlsbad, CA, USA). Des cellules HEK 293 non
transfectées ou exprimant de manière stable RB2-GFP ont été détachées de leur surface de
culture par un court traitement avec une trypsine supplémentée de EDTA et centrifugées
pendant 5 min à 600 g à la température de la pièce dans du milieu de culture supplémenté
en FBS. Les culots de cellules intactes ont été repris dans une solution saline tamponnée
(HBSS IX, pH 7,4, préparée à partir d'une solution stock de 10 X; Multicell Wisent, St-
Bruno, Canada) contenant 10 mM HEPES et 1,6 mM CaC^, de telle manière que la densité
cellulaire de chaque suspension soit de 2,5 x IO6 cellules/ml. Une concentration finale de 1
pM du colorant fluorescent FURA-2AM a alors été ajouté aux suspensions cellulaires, qui
ont été incubées pendant 30 min dans un bain d'eau à 37 °C, avec agitation. Après la
période d'incubation, les cellules ont été centrifugées et lavées à deux reprises. La
mobilisation calcique induite par l'ajout d'un agoniste (soit la BK, le peptide B-9972 ou le
composé non peptidique 47a; 100 nM pour les 2 premiers et 1 pM pour le dernier, ajoutés
dix secondes après le début de la lecture de fluorescence) dans 2 ml de la suspension
cellulaire a été mesurée à l'aide d'un spectrofluorimètre (FluoroLog-3; HORIBA Jobin-
Yvon, Edison, NJ, USA), dont le thermostat était réglé à 37°C et les longueurs d'onde
d'excitation et d'émission étaient préréglées à 340 nM et à 510 nM, respectivement. Après

so
la prise de lectures, la mobilisation maximale calcique atteignable par la suspension
cellulaire était évaluée par l'ajout d'ionomycine (5 pM), alors que le signal fluorescent
minimal était donné par l'ajout du chélateur MnCh (25 mM). Ces mesures, respectivement
la fluorescence maximale moyenne (Fmax) et la fluorescence mininale moyenne (Fmin), ont
été introduites dans l'équation servant au calcul des concentrations de Ca2+ mobilisées, soit 7+
([Ca ] = 224((y-Fmjn)/(Fmax-y)), où y représente la lecture de fluorescence prise par
l'échantillon. Cette équation a été précédemment décrite par Grynkiewicz, Poenie et Tsien
(1985).
Des cellules de muscle lisse de l'artère ombilicale humaine exprimant de manière endogène
le RB] ont été soumises à une procédure similaire à celle décrite ci-haut, afin d'étudier la
mobilisation calcique induite ou influencée par les ligands fluorescents du RB). Les ligands
fluorescents ont d'abord été injectés 10 sec après le début de la prise de lectures à tour de
rôle afin de vérifier leur capacité à induire une réponse calcique, et ont été comparés avec
leur peptide parent respectif; la Lys-des-Arg9-BK servait de base comparative pour le
composé B-10378 et le peptide B-9958, pour les composés B-10372 et B-10376. Puis,
l'identité des antagonistes B-10372 et B-10376 a été vérifiée en injectant un des
antagonistes au temps 10 sec, ce qui était suivi de l'injection de l'agoniste Lys-des-Arg9-
BK dans la suspension cellulaire au temps 60 sec.
Finalement, cette méthodologie a été appliquée à des cellules HEK 293a exprimant de
manière transitoire le myc-RB2. L'objectif de ces essais était d'identifier un effet agoniste
potentiel de l'anticorps anti-myc (clone 4A6; Upstate Biotechnology, Lake Placid, NY) sur
la signalisation calcique du RB2, suite à sa liaison au site N-terminal du récepteur où
l'épitope myc est fusionné. La stimulation avec la BK des cellules transfectées avec le myc-
RB2 servait à vérifier la transfection des cellules, ainsi que leur viabilité; d'autres cellules
non transfectées ont été soumises aux deux conditions de stimulation et constituaient les
échantillons témoins.

81
2.9 Cytométrie en flux
Afin d'évaluer le potentiel du peptide B-10376, un antagoniste fluorescent du RBi, comme
outil de détection permettant de quantifier les niveaux d'expression du récepteur, la
cytométrie en flux a été appliquée à des cellules HEK 293a exprimant transitoirement la
protéine recombinante de lapin RBi. Certaines cellules ont été soumises à un prétraitement
avec l'anisomycine, un inhibiteur de synthèse protéique, de 2 à 4 h avant la procédure de
marquage, ce qui permettait de mettre en évidence la diminution de la quantité de RB| à la
membrane cellulaire, puisque leur remplacement par la synthèse de nouveaux récepteurs
était empêché (tel que rapporté par Fortin et collaborateurs, 2003). Les cellules ont été
détachées de leur support plastique grâce à un tampon de dissociation sans protease (Cell
Dissociation Buffer, Invitrogen) et incubées dans leur milieu de culture habituel (toutefois
non supplémenté en FBS) à 37°C en présence du peptide B-10376 (100 nM), du composé
11 (1 pM), des deux composés ou en absence de ligand du RBi pendant 30 min sous
agitation à l'abri de la lumière. Après la période d'incubation, les cellules ont été
centrifugées rapidement (30 sec à 15 000 g) et les culots contenant les cellules ont été
resuspendus dans du PBS froid. Les différentes suspensions cellulaires ont ensuite été
analysées grâce à l'appareil de cytofluorimétrie EPICS XL (Beckman Coulter, Fullerton,
CA; fluorescence verte). Le marquage des RBi exprimés par les cellules de muscle lisse
d'artère ombilicale humaine, préalablement traités avec l'IL-ip (5 ng/ml; R&D Systems,
Minneapolis, MN) durant 4 h, a été fait conformément à la technique utilisée pour le
marquage des RBi recombinants.

82
3. Résultats
3.1 Caractérisation pharmacologique des analogues de la BK à la structure contrainte et conséquences de leur résistance au métabolisme
3.1.1 Propriétés pharmacologiques des analogues de la BK à la structure contrainte Les essais de liaison basés sur la liaison spécifique de 3 nM de [3H]BK à des cellules HEK
293 exprimant de manière stable le RB2-GFP, présentés à la figure 4, montrent un
déplacement du ligand marqué par la liaison de la BK non radiomarquée, l'antagoniste B-
9430, ainsi que par ses deux isomères B-9972 et B-10344. Les trois peptides à la structure
contrainte étaient respectivement 2, 9.7 et 36 fois moins puissants que la BK.
L'utilisation de la veine jugulaire de lapin pour générer des essais de contractilité portant
sur le RB2 montre que le peptide B-9972 est un agoniste complet, avec une activité
contractile légèrement inférieure (deux fois moins actif; en A de la figure 5)
comparativement à l'agoniste naturel BK. L'isomère B-10344 présente une faible activité
agoniste, atteignant seulement la moitié de la contraction maximale obtenue par la BK,
pour une concentration cumulative de 6,1 pM. L'identification de ce peptide comme un
agoniste partiel est compatible avec la pente peu élevée de sa courbe concentration-réponse
et l'induction d'une activité contractile 2000 fois inférieure à celle de la BK, quoique
seulement 36 fois moins actif que celle-ci dans l'essai de liaison. L'autre membre de cette
triade d'isomères, le peptide B-9430, n'induit pour sa part aucun effet contractile direct sur
la veine jugulaire de lapin. Il s'y comporte comme un antagoniste puissant et insurmontable
de la BK (en B, figure 5), ce qui n'est pas sans rappeler des observations décrites
précédemment portant sur d'autres antagonistes du RB2 présentant une structure contrainte,
tels le HOE-140 et le NPC 17731 (phénomène décrit par Houle et collaborateurs, 2000).
Les tissus traités avec le
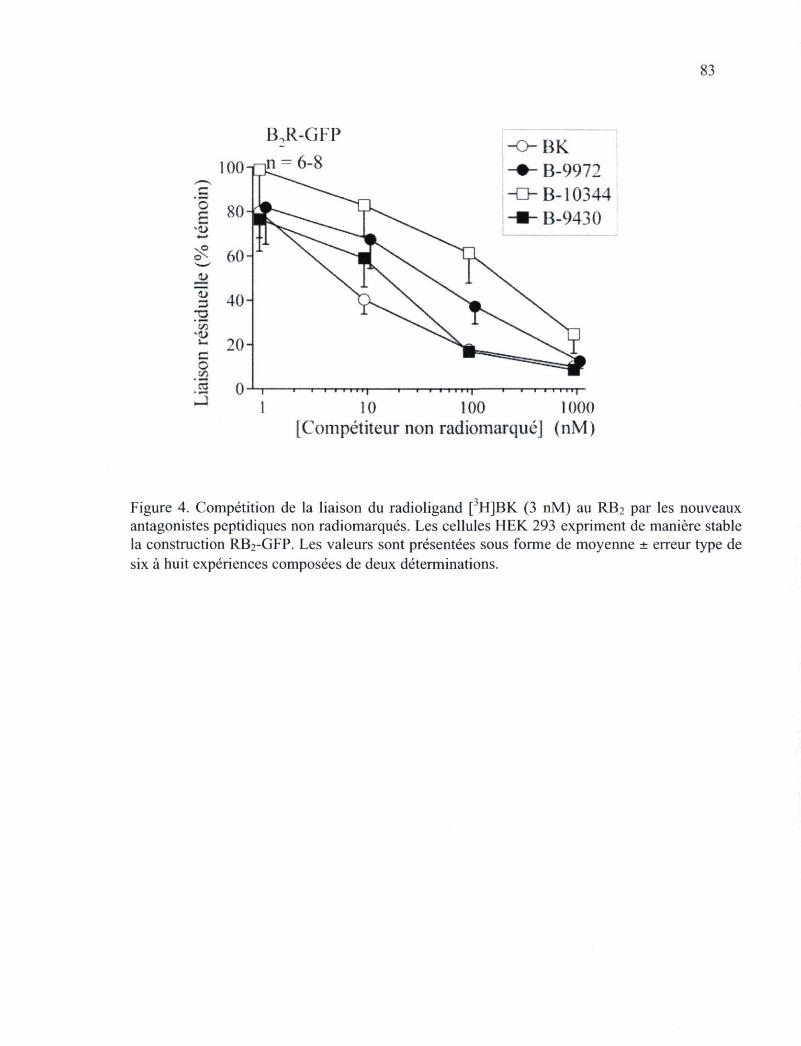
83
c
°
c7
O sa
' s ~ Z
B2RGFP ! 0 0 ^ n 6 8
80
60
3 40
20
K > B K * B9972 O B 1 0 3 4 4 » B9430
■"T— '—i r—i—rrrrrj T ' T T r i ] 1 1—i >'"i" t i i f
10 100 1000 [Compétiteur non radiomarqué] (nM)
Figure 4. Compétition de la liaison du radioligand [3H]BK (3 nM) au RB2 par les nouveaux antagonistes peptidiques non radiomarqués. Les cellules HEK 293 expriment de manière stable la construction RB2GFP. Les valeurs sont présentées sous forme de moyenne ± erreur type de six à huit expériences composées de deux déterminations.
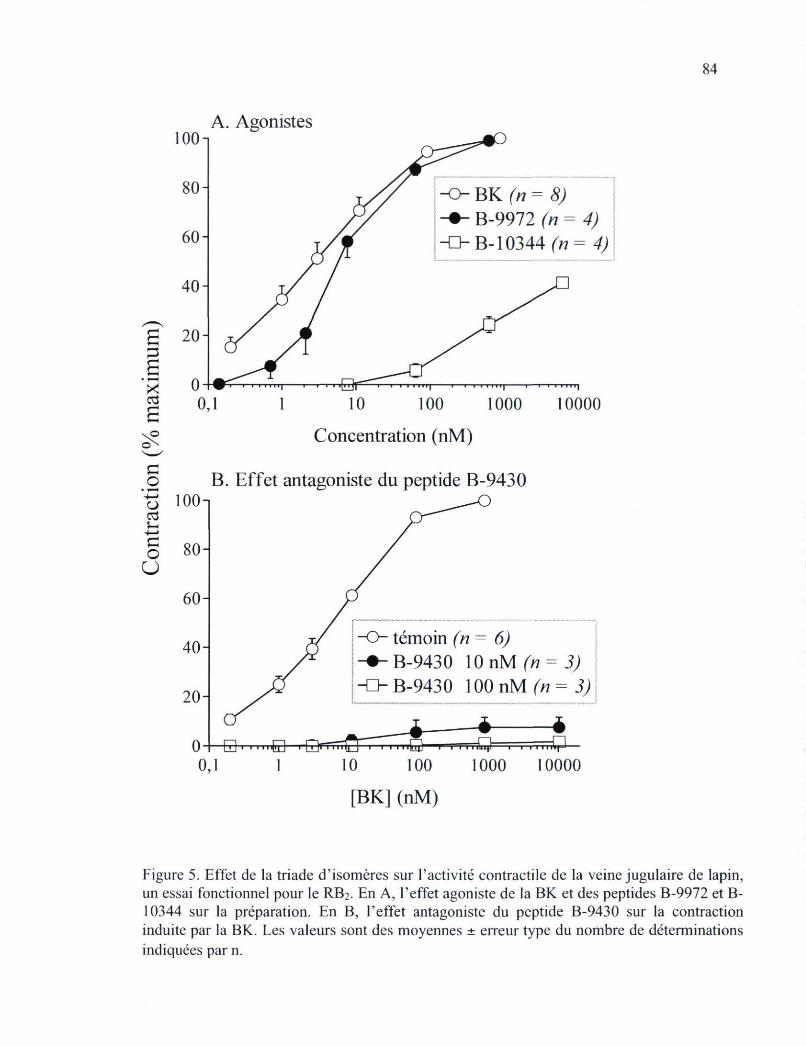
84
A. Agonistes
- O BK (n = 8) - • - B-9972 (n = 4) -O-B-10344(« = 4)
T t I I I I I ! 1 1—I I I I 1 I
1000 10000
10 100 1000 10000
[BK] (nM)
Figure 5. Effet de la triade d'isomères sur l'activité contractile de la veine jugulaire de lapin, un essai fonctionnel pour le RB2. En A, l'effet agoniste de la BK et des peptides B-9972 et B-10344 sur la préparation. En B, l'effet antagoniste du peptide B-9430 sur la contraction induite par la BK. Les valeurs sont des moyennes ± erreur type du nombre de déterminations indiquées par n.

85
peptide B-9430 présentent un effet maximal résiduel très faible de la BK (en B de la figure
5), alors qu'ils répondent fortement à l'histamine (données non présentées), prouvant ainsi
leur préservation fonctionnelle. L'aorte de lapin fraîchement isolée permet la réalisation
d'un essai fonctionnel visant à définir le profil pharmacologique du RBi (Larrivée et coll.,
2000). Comme cela a été rapporté auparavant par Stewart et collaborateurs (1996), le
peptide B-9430 conserve une affinité appréciable pour ce sous-type de récepteur, en
provoquant le déplacement vers la droite de la courbe concentration-effet construite avec
l'agoniste des-Arg9-BK, à des concentrations de 100 nM et 1 pM (figure 6), un
comportement qui est surmontable et qui permet le calcul de la valeur de pA2 de cet
antagoniste de 6,49 (estimée grâce à la génération d'une régression de Schild non présentée
ici). Le protocole de l'essai présent, basé sur la construction de courbes complètes
concentration-réponse avec l'agoniste, établit le caractère surmontable du peptide B-9430
pour le RB] de lapin. Le peptide B-9972 n'avait quant à lui aucun effet significatif de
nature agoniste ou antagoniste au niveau de la contractilité de l'aorte de lapin, et ce jusqu'à
une concentration de 6 pM (résultats non présentés).
L'agoniste [Phe8^(CH2NH)Arg9]BK (dont l'activité sur la veine jugulaire de lapin a été
rapportée ailleurs, Drapeau et coll., 1988) a été ajouté à la triade d'isomères et cet ensemble
de peptides a été mis à l'étude pour les RB2 humains exprimés de la veine ombilicale
humaine isolée; les résultats sont présentés à la figure 7. Le même ordre de puissance
décroissant des agonistes est observé (figure 7A): soit la BK, le B-9972 (activité 4 fois
moindre) et le B-10344 (activité 200 fois moindre). Pour le RB2 humain, le B-10344 est un
agoniste complet puisqu'il permet l'enregistrement d'un effet contractile maximal qui ne
diffère pas significativement des maxima de contraction obtenus avec les autres agonistes,
lorsqu'exprimés en termes de force (unités de poids (g), données non montrées) ou en
pourcentage d'un standard interne de contraction, donné par la contraction maximale du
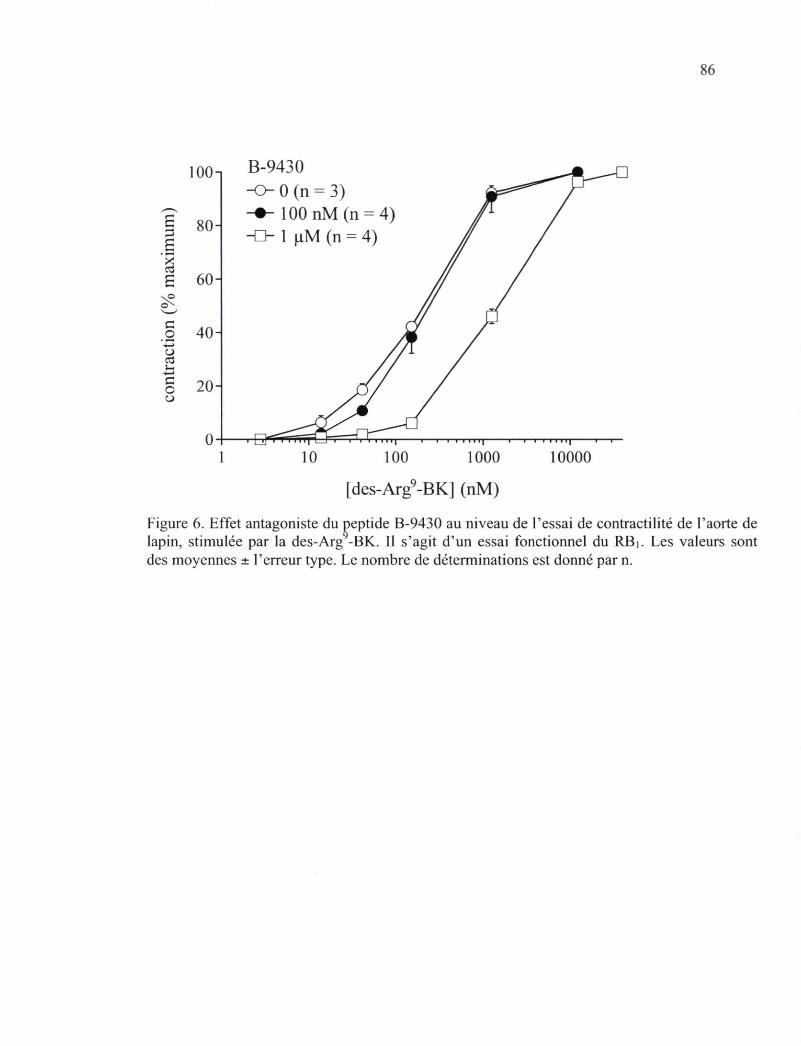
86
E
S
C -.—i
H C C CJ
100 B-9430 K> 0 (n = 3) - • - 1 0 0 n M ( n = 4) ^ > 1 (iM (n = 4)
10000
[des-Ar^-BK] (nM)
Figure 6. Effet antagoniste du peptide B-9430 au niveau de l'essai de contractilité de l'aorte de lapin, stimulée par la des-Arg -BK. Il s'agit d'un essai fonctionnel du RB,. Les valeurs sont des moyennes ± l'erreur type. Le nombre de déterminations est donné par n.
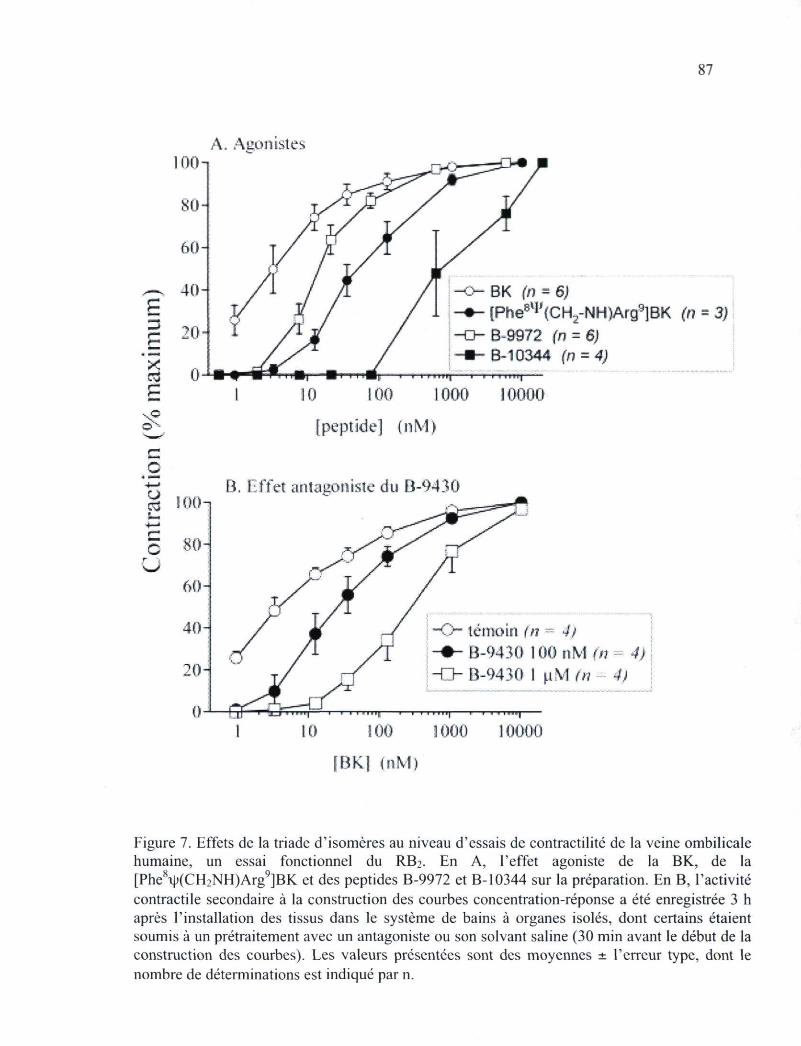
87
A. Agonistes
- c ^BK (n^6) - • - |Phe8U'(CH2-NH)Arg9]BK (n - 3; -O-B-9972 (n = 6) -•-B-10344 (n = 4)
•t t rrjmuj" '" i > i n m\ "'• •
0 100 1000 10000
[peptide] (nM)
^ soo-mm
§ 80H r 'i W
6C
40-
20-
0
B. Effet antagoniste du B-9430
- O - témoin (»= 4) -• -B-9430 100 nM (n= 4) - D - B-9430 I \i M (n - 4)
T—r i i r i n j *—i—i—rrrrn r i « ! i » m i
000 10000
[BK] (nM)
Figure 7. Effets de la triade d'isomères au niveau d'essais de contractilité de la veine ombilicale humaine, un essai fonctionnel du RB2. En A, l'effet agoniste de la BK, de la [Phe8x|)(CH2NH)Arg9]BK et des peptides B-9972 et B-10344 sur la préparation. En B, l'activité contractile secondaire à la construction des courbes concentration-réponse a été enregistrée 3 h après l'installation des tissus dans le système de bains à organes isolés, dont certains étaient soumis à un prétraitement avec un antagoniste ou son solvant saline (30 min avant le début de la construction des courbes). Les valeurs présentées sont des moyennes ± l'erreur type, dont le nombre de déterminations est indiqué par n.

88
tissu en réponse à la 5-HT, injectée à la fin des expériences, tel que présenté à la figure 8A.
Le peptide B-9430 n'a pas d'effet contractile direct sur la veine ombilicale humaine isolée,
mais antagonise compétitivement la contraction induite par la BK (figure 7B), avec une
valeur calculée de pA2 de 7,7 (la pente de la régression de Schild a une valeur de -1,10,
indiquant la nature compétitive de cet antagoniste). L'agoniste [Phe8ip(CH2NH)Arg9]BK,
possédant une résistance au métabolisme par les peptidases hypothétiquement inférieure à
celle du peptide contraint B-9972, montre une puissance 3,5 fois moins importante,
comparativement à celle de ce dernier (figure 7A). Cela suggère que le peptide B-9972 est
plus utile pour la suite de nos études. La BK sert de peptide de référence.
3.1.2 Caractérisation de la résistance métabolique à l'aide des essais de contractilité
Une série d'expériences a mesuré la durée requise pour atteindre un état de demi-relaxation
de tissus maximalement contractés par les différents agonistes, après un changement du
tampon baignant les tissus. Les tissus précontractés avec la concentration cumulative
maximale de BK (10,5 pM) avaient relaxé de moitié après 17,2 ± 2,9 min (figure 8B); le o q
temps de demi-relaxation mesuré pour la [Phe o|>(CH2NH)Arg ]BK (concentration
cumulative maximale: 10,5 pM) n'était pas significativement différent de celui observé
pour la BK (10,5 pM). Par contre, les temps requis pour que les tissus relaxent de moitié
avec les peptides B-9972 et B-10344 (dont les concentrations cumulatives maximales
étaient respectivement de 6,2 pM et de 20,1 pM) étaient beaucoup plus importants et
similaires entre eux (figure 8B). Ainsi la mesure de la vélocité de la relaxation à partir de la
contraction maximale peut dépendre de la résistance prédite aux peptidases, sans dépendre
toutefois de la puissance. Les peptides B-9972 et B-10344 sont les peptides les plus
résistants à la dégradation.
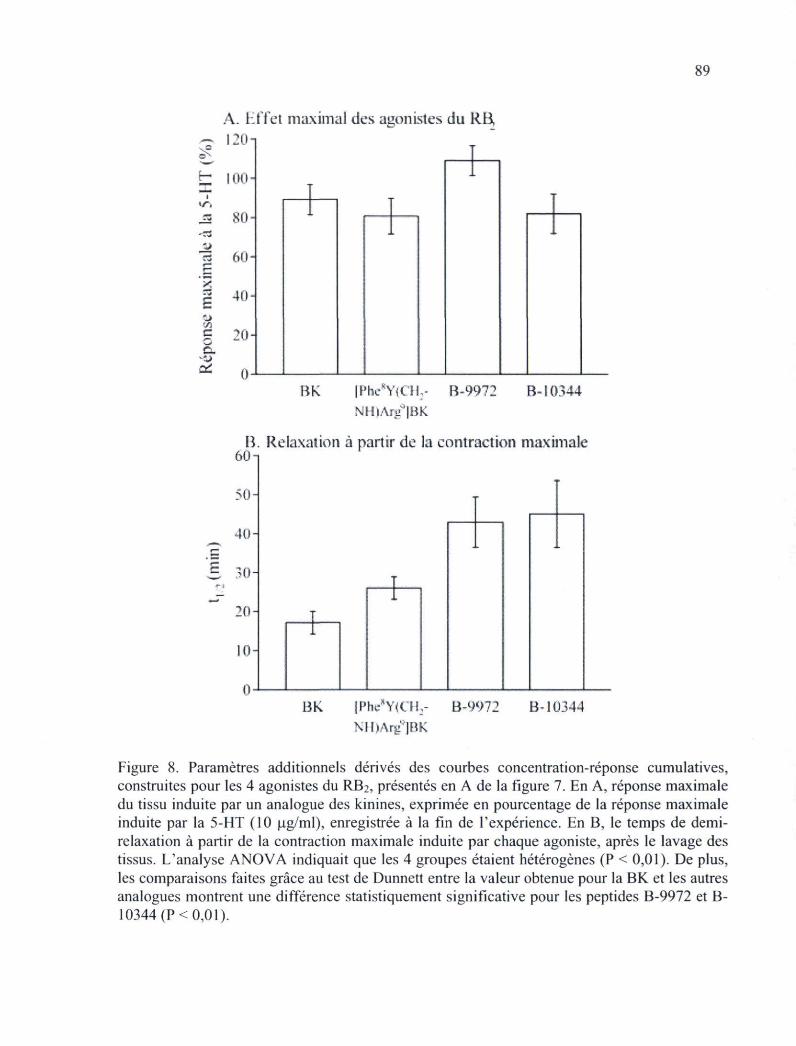
89
A. Effet maximal des agonistes du RB, I20T
b 100
■zss
y
>
80
60
40
20
(i BK |Phe*Y{CH,. B9972
NH)Arg1|BK BI0344
B. Relaxation à partir de la contraction maximale 60 n
50
40
S 30
20
10
BK [Phe YfCH,
NH)Arg*]BK B9972 B10344
Figure 8. Paramètres additionnels dérivés des courbes concentrationréponse cumulatives, construites pour les 4 agonistes du RB2, présentés en A de la figure 7. En A, réponse maximale du tissu induite par un analogue des kinines, exprimée en pourcentage de la réponse maximale induite par la 5HT (10 pg/ml), enregistrée à la fin de l'expérience. En B, le temps de demi
relaxation à partir de la contraction maximale induite par chaque agoniste, après le lavage des tissus. L'analyse ANOVA indiquait que les 4 groupes étaient hétérogènes (P < 0,01). De plus, les comparaisons faites grâce au test de Dunnert entre la valeur obtenue pour la BK et les autres analogues montrent une différence statistiquement significative pour les peptides B9972 et B
10344 (P< 0,01).

90
3.1.3 Identification des voies d'inactivation de la BK au niveau de la veine ombilicale humaine à l'aide des essais de contractilité
Afin d'identifier les voies métaboliques impliquées dans la dégradation de la BK au sein de
la veine ombilicale humaine, la construction de courbes concentration-effet a été effectuée
en présence de l'inhibiteur de l'ECA énalaprilat, l'inhibiteur de l'NEP phosphoramidon ou
l'inhibiteur de l'APP apstatine (Prechel et coll., 1995). Ces inhibiteurs de peptidases n'ont
pas modifié la puissance apparente de la BK dans l'essai de contractilité, comme le montre
la figure 9, en A, ni influencé le temps requis pour atteindre un état de demi-relaxation
après la contraction maximale induite par ce même agoniste (figure 9B). Ces données
suggèrent que les peptidases ciblées par les inhibiteurs ne sont pas celles majoritairement
impliquées dans le métabolisme de la BK dans ce système.
3.1.4 Effets des agonistes résistants au métabolisme au niveau de cellules HEK 293 RB2-GFP
La fluorescence associée à la construction RB2-GFP exprimée par les cellules HEK 293 est
principalement membranaire lorsque l'on soumet ces cellules à l'observation par
épifluorescence (figure 10). L'antagoniste du RB2 B-9430 (5 pM) et l'inhibiteur de l'ECA
captopril (1 pM) n'ont aucun effet important sur la distribution de la fluorescence, alors que
les agonistes du RB2 provoquent la redistribution du récepteur de la membrane plasmique
vers l'intérieur de la cellule (figure 10A). Après une incubation de 30 min, les cellules
exprimant le RB2-GFP exposées à la BK (100 nM), seule ou combinée au captopril, ou au
peptide B-9972 (100 nM) présentaient une perte de la continuité de la fluorescence
membranaire et parfois même la perte de fluorescence membranaire au profit de
l'apparition de structures intracellulaires de tailles variées. Cette redistribution du signal
fluorescent était empêchée par le cotraitement des cellules avec l'antagoniste B-9430 (5
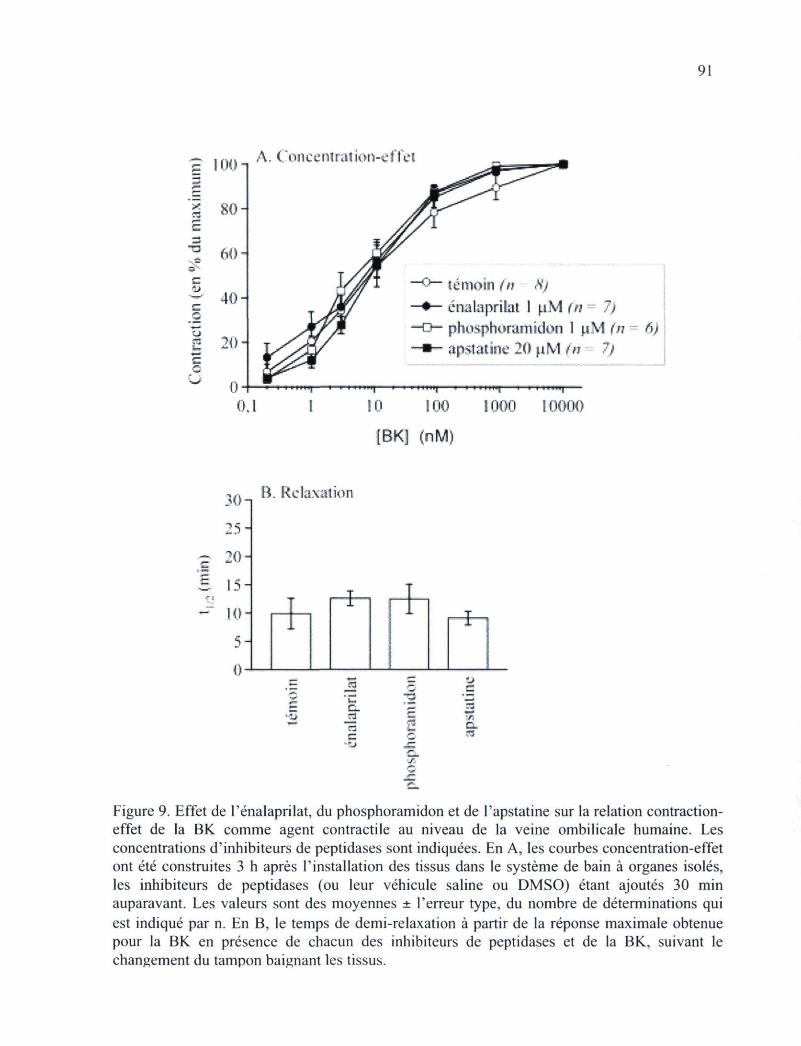
91
s lOOi
x i
ô
c •J
c — —
U
80
60
40
20 4
n
A. Concentrât ioneffet
OJ
témoin (n .S'y énalaprilat 1 \iM (n = 7) phosphoramidon 1 pM (n = 6) apstatine 20 pM fut = 7)
• t ' V ' t t r tm" ' ■> f > #f " i» i f ■■■■■'»'»'*'■ ? r H
IM 100 ■ l ' \ I l l l l I I I ]
1000 toooo
[BK] (nM)
30 n
25
e 20J
p
ç î
10
5
0
B. Relaxation
c c "
c rcj
• o
o
:; .
S
Figure 9. Effet de l'énalaprilat, du phosphoramidon et de l'apstatine sur la relation contraction
effet de la BK comme agent contractile au niveau de la veine ombilicale humaine. Les concentrations d'inhibiteurs de peptidases sont indiquées. En A, les courbes concentrationeffet ont été construites 3 h après l'installation des tissus dans le système de bain à organes isolés, les inhibiteurs de peptidases (ou leur véhicule saline ou DMSO) étant ajoutés 30 min auparavant. Les valeurs sont des moyennes ± l'erreur type, du nombre de déterminations qui est indiqué par n. En B, le temps de demirelaxation à partir de la réponse maximale obtenue pour la BK en présence de chacun des inhibiteurs de peptidases et de la BK, suivant le changement du tampon baignant les tissus.
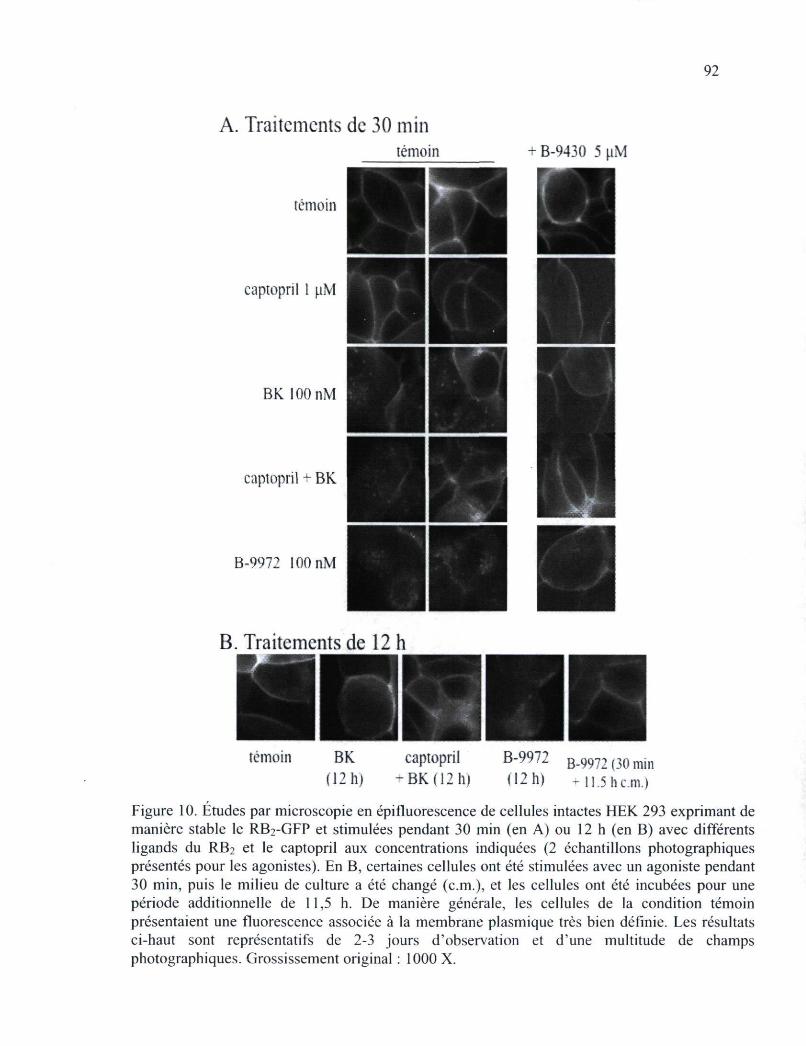
92
A. Traitements dc 30 min témoin
témoin II
captopril 1 pM
BKlOOnM
captopril + BK
-9972 100 nM
+ B-9430 5 pM
B. Traitements de 12 h
témoin BK (12 h)
captopril -BK(12h)
B-9972 112 h)
B-9972 (30 min t11.5 h cm.)
Figure 10. Études par microscopie en épifluorescence de cellules intactes HEK 293 exprimant de manière stable le RB2-GFP et stimulées pendant 30 min (en A) ou 12 h (en B) avec différents ligands du RB2 et le captopril aux concentrations indiquées (2 échantillons photographiques présentés pour les agonistes). En B, certaines cellules ont été stimulées avec un agoniste pendant 30 min, puis le milieu de culture a été changé (cm.), et les cellules ont été incubées pour une période additionnelle de 11,5 h. De manière générale, les cellules de la condition témoin présentaient une fluorescence associée à la membrane plasmique très bien définie. Les résultats ci-haut sont représentatifs de 2-3 jours d'observation et d'une multitude de champs photographiques. Grossissement original : 1000 X.

93
pM), en présence des agonistes. Des traitements plus longs (12 h), tout en soutenant la
réversibilité de l'internalisation du RB2-GFP induite par la BK par la restoration du signal
membranaire, ont révélé qu'un traitement avec le peptide B-9972 retarde ce retour du
récepteur à la membrane plasmique. Les cellules traitées avec une combinaison de BK et de
captopril montraient un retour partiel à la distribution basale du récepteur, i.e. à sa présence
membranaire (figure 10). Cette observation soutient l'implication de l'ECA, présente dans
le milieu de culture, dans la dégradation de la BK et fait écho aux données rapportées par
Bachvarov et collaborateurs (2001) : la présence de captopril dans un milieu de culture
supplémenté de sérum incubé à la température physiologique de 37 °C prolonge la demi-vie
de la BK de 7,7 min (sans captopril) à 102 min (avec captopril). Des cellules stimulées avec
le peptide B-9972 (100 nM) pendant 30 min, lavées après cette première période
d'incubation et remises en présence de milieu de culture complet pendant 11,5 h à 37 °C,
présentaient une fluorescence membranaire indistinguable des cellules constituant
l'échantillon témoin. Cela suggère que le récepteur RB2-GFP lié au peptide B-9972 ne sont
pas condamnées à une séquestration permanente et peuvent être recyclées à la surface
cellulaire. Des données expérimentales obtenues au moyen de l'essai radiorécepteur
montraient que, malgré que ce peptide soit soumis à une importante dégradation, sa
persistance dépasse grandement la demi-vie de la BK; après une période d'incubation de 12
h d'une concentration fixe de 5 pM des agonistes en présence de cellules HEK 293
exprimant de manière stable RB2-GFP cultivées dans un milieu de culture complet, la
concentration résiduelle du peptide B-9972 était d'environ 100 nM; alors que la BK était
indétectable. Il est important que signaler que dans ce cas particulier, la limite de détection
de cette expérience est de 10 nM.
La récepteur RB2-GFP est efficacement internalisée de manière secondaire au traitement
avec un agoniste, mais son abondance totale n'est pas significativement réduite par des
traitements de 3 h avec la BK ou l'agoniste [Phe8i|)(CH2NH)Arg9]BK. Certains de ces
résultats ont été répliqués dans des cellules HEK 293 exprimant le récepteur recombinant
RB2-GFP traitées avec des ligands du récepteur et/ou du captopril en présence de
cycloheximide (71 pM), un inhibiteur de la synthèse protéique qui empêche la synthèse de
nouveaux récepteurs. L'abondance du RB2-GFP était contrôlée grâce à la technique

94
d'immunobuvardage basée sur des anticorps dirigés contre la GFP (figure 11, à gauche;
l'analyse statistique de 4 expériences répliquées dans ces mêmes conditions est présentée à
la figure 12A). La bande possédant un poids moléculaire d'approximativement 101 kDa,
qui correspond au récepteur de fusion RB2-GFP présentait une intensité similaire pour
chaque condition de traitement de 3 h avec la BK (100 nM), le captopril (1 pM, seul ou en
combinaison avec la BK) ou les isomères B-9972 (100 nM) ou B-9430 (5 pM), ce qui
signifie que le récepteur n'était pas dégradé. Un extrait cellulaire total isolé de cellules non
transfectées présentait une très faible réactivité avec l'anticorps anti-GFP. Un traitement
prolongé de 12 h de cellules intactes dans un milieu de culture supplémenté de sérum avec
la BK ne mène pas à la dégradation du récepteur. Par contre, les traitements prolongés avec
la BK combinée au captopril ou avec le peptide B-9972 seul provoquèrent une dégradation
partielle du RB2-GFP, suggérée par la diminution de l'intensité de la bande de 101 kDa et
par l'apparition d'une bande possédant un poids moléculaire approximatif de 27 kDa (poids
de la GFP libre) ainsi que d'autres bandes représentant des metabolites du RB2-GFP (figure
11, centre et droit). Le peptide B-9972 était l'agent le plus actif à ce niveau, puisqu'il
provoquait une diminution significative de l'abondance de la protéine de fusion RB2-GFP
dans les extraits totaux dérivés des cellules traitées avec cet agoniste. L'effet des agonistes
sur l'intensité des bandes de F immunobuvardage a été quantifié en utilisant 4 expériences
(2 expériences représentatives ont été sélectionnées et sont présentées à la figure 11) et les
résultats de cette analyse densitométrique sont présentés à la figure 12, en B. Les
antagonistes du RB2 LF 16-0687 (un antagoniste non peptidique) et le peptide B-9430 ont
empêché la dégradation du RB2-GFP induite par la combinaison de la BK avec le captopril
ou par le B-9972 (résultats présentés par la figure 11, au centre et à droite, respectivement).
Les antagonistes seuls semblent avoir induit une faible dégradation du RB2-GFP, en
particulier le peptide B-9430. Contrairement à la série d'expériences exploitant des
traitments de 3 h, il a été impossible d'utiliser la CHX en cotraitement pour les traitements
de 12 h, étant donnée la cytotoxicité secondaire à son utilisation prolongée.
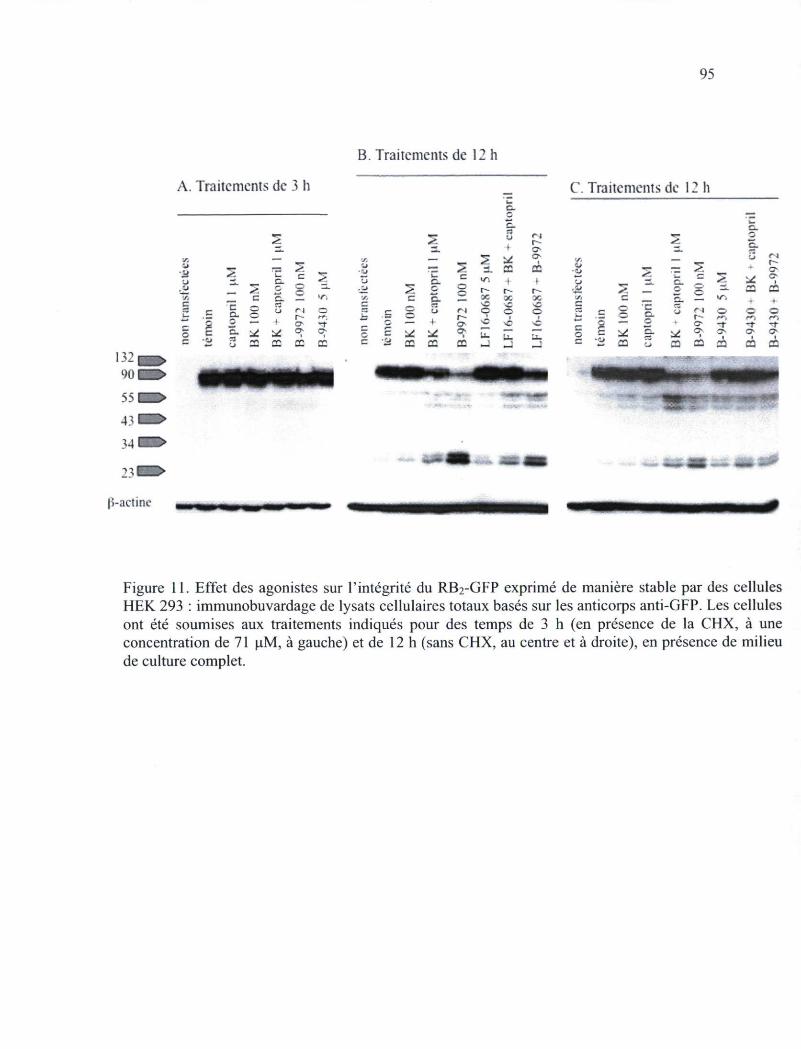
95
B. Traitements dc 12 h
A. Traitement: ;dc 3 h
S
i l cccc 0
U 1
C. Traitements de 12 h A. Traitement: ;dc 3 h
S
i l cccc 0
U 1 5 !> S
i l cccc 0
U 1 5
' —
7 Ê S s
CC tt ■co
x. _ — S ■2
s 2 S S S c
B — r
f r
1 | S
S 5 =
S * 2 CD CS •y. c r i / i Cft B c_ X s. x M cz *"; _* </% 5
5. § u c cr Tt
fc 5 u
r 9 é e TZ c s 5. 5 C
1
r— o O O
a 9 j ^ ' 4 » r C — 1 CC7 w o o _ §
— 0 * CT1
T * r t — ÇL
« F — ^ * cj Cf ~ E u u CO 1 U. tx* [ t Ô s * S. Sx! °T ~ O tt>.
132 » , c •«> Efi nQ CC EC S ■Jj cX ce ce J 3 3 zn ■S C2 u a ca ah eâ CQ
132 » , 90EZ> «■■i *■ HBPBP m m r 55CZ> — "•wumui
43 ■ »
M 1—>
:3CZ> — • ^^^ ^^
jyv*» w « « ^ » #
pactine 0
Figure 11. Effet des agonistes sur l'intégrité du RB2GFP exprimé de manière stable par des cellules HEK 293 : immunobuvardage de lysats cellulaires totaux basés sur les anticorps antiGFP. Les cellules ont été soumises aux traitements indiqués pour des temps de 3 h (en présence de la CHX, à une concentration de 71 pM, à gauche) et de 12 h (sans CHX, au centre et à droite), en présence de milieu de culture complet.
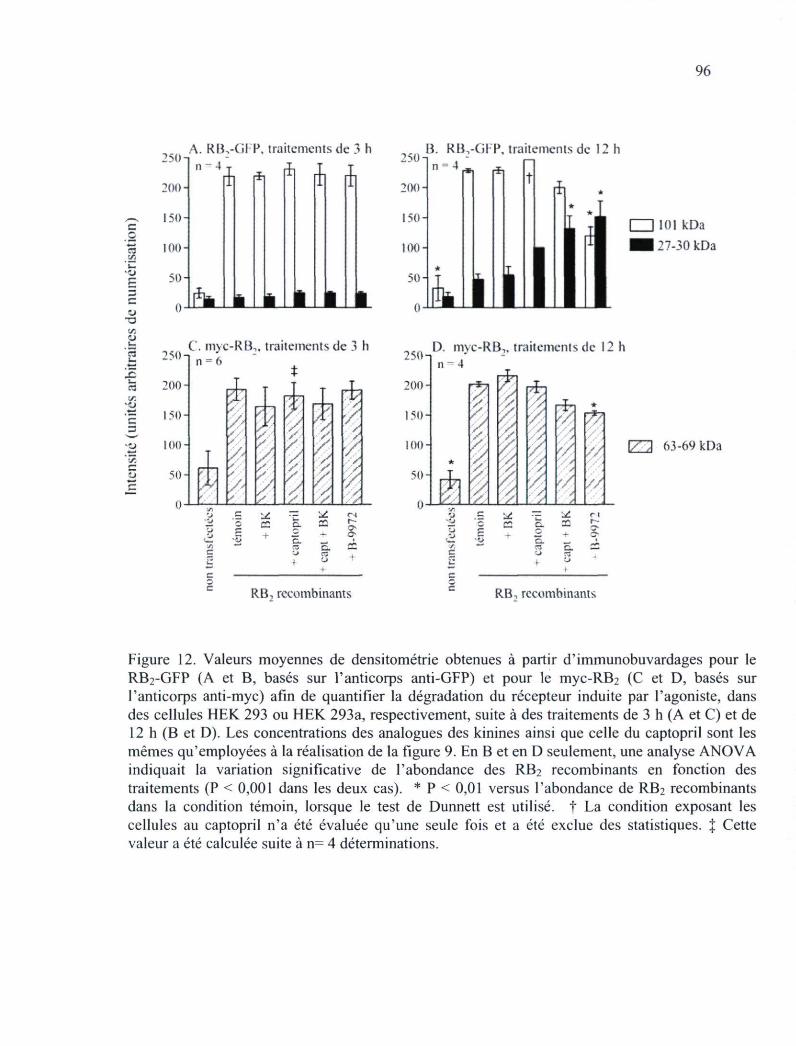
96
rc: s.
cc= J
Ui b ■—
' s j
1 zr.
" J
** ' t e
250i
200
150
100
50
0
A. RB,GI P, traitements de 3 h n 4
A ft A *
250
200
150
100
50 * 7/
/
I _
*
C. mycRB,, traitements de 3 h n = 6
I i f ■
T / ■
V c ^: ccr iui fN v s *r. r—
r— — g.
& + z~t
« EL œ ">
§ c s
f M T
§ c s RB, rccombinants
3. RB.G1 P, traitements de 12 h n = 4
C J 101 kDa ■ I 2730 kDa
250
200
150
100 H
50
0
D. mycRB„ traitements de 12 h n 4
1 J
vy
r/ _
I P. / / IZZ3 6369 kDa
§> £ ^ . ^ « N •2 c es c. su t * P O iX. e> C f « r cy
C. V cx _ » B ss c CS
RB, recombinants
Figure 12. Valeurs moyennes de densitométrie obtenues à partir d'immunobuvardages pour le RB2GFP (A et B, basés sur l'anticorps antiGFP) et pour le mycRB2 (C et D, basés sur l'anticorps antimyc) afin de quantifier la dégradation du récepteur induite par l'agoniste, dans des cellules HEK 293 ou HEK 293a, respectivement, suite à des traitements de 3 h (A et C) et de 12 h (B et D). Les concentrations des analogues des kinines ainsi que celle du captopril sont les mêmes qu'employées à la réalisation de la figure 9. En B et en D seulement, une analyse ANOVA indiquait la variation significative de l'abondance des RB2 recombinants en fonction des traitements (P < 0,001 dans les deux cas). * P < 0,01 versus l'abondance de RB2 recombinants dans la condition témoin, lorsque le test de Dunnett est utilisé, f La condition exposant les cellules au captopril n'a été évaluée qu'une seule fois et a été exclue des statistiques. % Cette valeur a été calculée suite à n= 4 déterminations.

97
3.1.5 Validation du vecteur myc-RB2
Une nouvelle construction fusionnant le RB2 de lapin et l'épitope myc au niveau de
l'extrémité N-terminale du récepteur a été caractérisée pharmacologiquement. Des cellules
COS-1 transfectées de manière transitoire avec le vecteur pCI-neo-myc (vecteur vide) ne
liaient que très peu la [3H]BK, alors que les cellules transfectées avec myc-RB2 exhibaient
une liaison spécifique et saturable (figure 13A). L'affinité dérivée grâce à l'analyse de la
régression de Scatchard (figure 13B) donne une valeur de Kd de 2,38 nM; cette valeur est
très similiaire à celle obtenue pour le récepteur de type sauvage de lapin (exprimé par des
cellules COS-1) par des techniques similaires (Bachvarov et coll., 1995). L'estimation
graphique du Bmax était de 34,3 fmoles/puits (figure 13, en A et en B). Le profil
pharmacologique de la construction myc-RB2 lorsque transitoirement exprimée par les
cellules COS-1 a été étudié plus avant par l'emploi de divers composés apparentés à la
bradykinine et non radiomarqués (à une concentration de 1 pM) servant de compétiteurs à
la liaison de la [3H]BK (en C de la figure 13). La liaison de 1 nM de [3H]BK était abolie en
présence de 1 pM de la BK non radiomarquée, de la Lys-BK et des trois antagonistes du
RB2 soient le HOE-140, la bradyzide ou le LF 16-0687 (figure 13C). Les fragments des-
Arg9 des kinines naturelles BK et Lys-BK, des agonistes sélectifs du RBi, présentaient une
activité marginale, alors que le composé 11, un antagoniste de haute affinité du RBi, était
complètement inactif à compétitionner la liaison de [3H]BK au niveau du myc-RB2.
L'utilisation d'anticorps anti-myc a permis d'évaluer l'abondance du myc-RB2 à la surface
membranaire, indépendamment de techniques basées sur les ligands du RB2- La faisabilité
de cette approche a été montrée par l'exploitation de cellules HEK 293a transfectées
transitoirement, fixées et optionnellement perméabilisées, comme le montre la figure 14.
L'immunofluorescence indirecte, au moyen de l'anticorps anti-myc (clone 4A6) a montré
une expression abondante de la construction myc-RB2 exprimée de manière stable dans des
cellules HEK 293a, mais le signal était majoritairement intracellulaire (figure 14, gauche).
Cette observation concorde avec des données précédemment publiées (Fortin et coll., 2006)
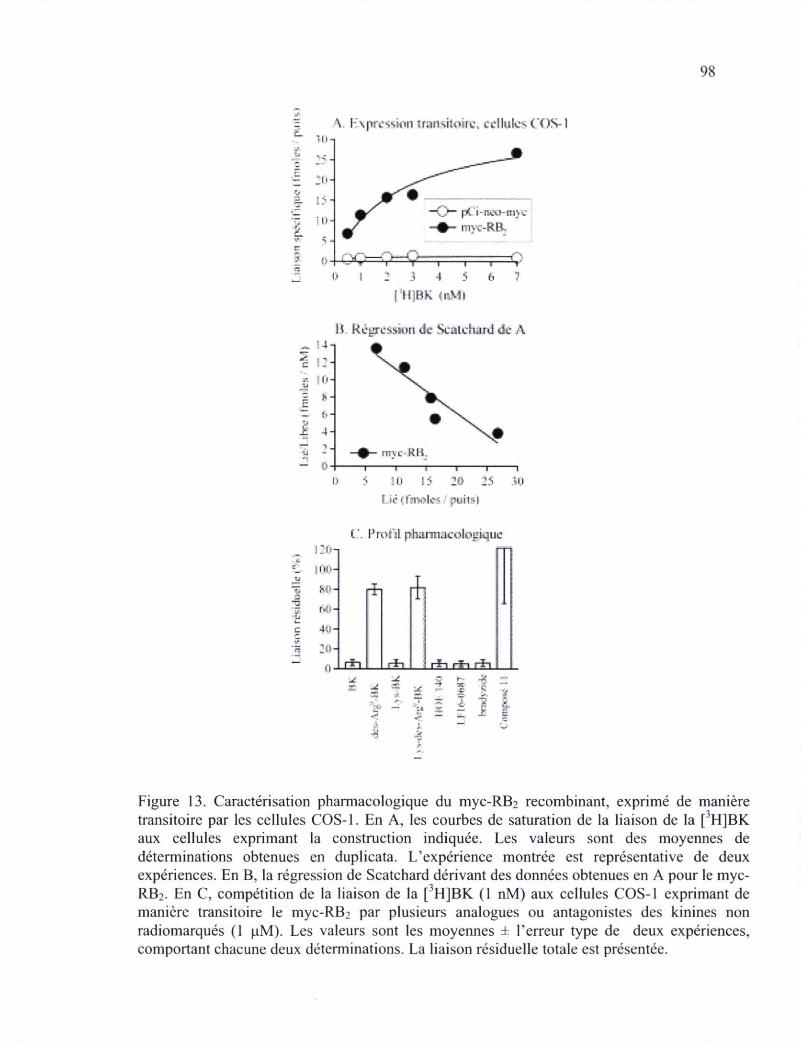
98
I
A. Expression transitoire, cellules C'OSl
■ • :
20
' I I
0 C —i 1 1 r 3 4 5 6 ?
pHWK (lAfl
ï
B. Régression de Scatchard de A I 4
1 * 12
M
8 6 l
0
120 I00Î 80 Wf
Mi
20i n
mvc KU. 1 1 ! 1 1 1
5 10 15 20 25 30 r ië<frooks/puits)
t". Profil pharmacologique
rft, A
six r t i r f i f i t
1 fc I = * U * tf 2 S È > »** ï S J
k *
Figure 13. Caractérisation pharmacologique du mycRB2 recombinant, exprimé de manière transitoire par les cellules COS1. En A, les courbes de saturation de la liaison de la [3H]BK aux cellules exprimant la construction indiquée. Les valeurs sont des moyennes de déterminations obtenues en duplicata. L'expérience montrée est représentative de deux expériences. En B, la régression de Scatchard dérivant des données obtenues en A pour le myc
RB2. En C, compétition de la liaison de la [ H]BK (1 nM) aux cellules COS1 exprimant de manière transitoire le mycRB2 par plusieurs analogues ou antagonistes des kinines non radiomarqués (1 pM). Les valeurs sont les moyennes ± l'erreur type de deux expériences, comportant chacune deux déterminations. La liaison résiduelle totale est présentée.
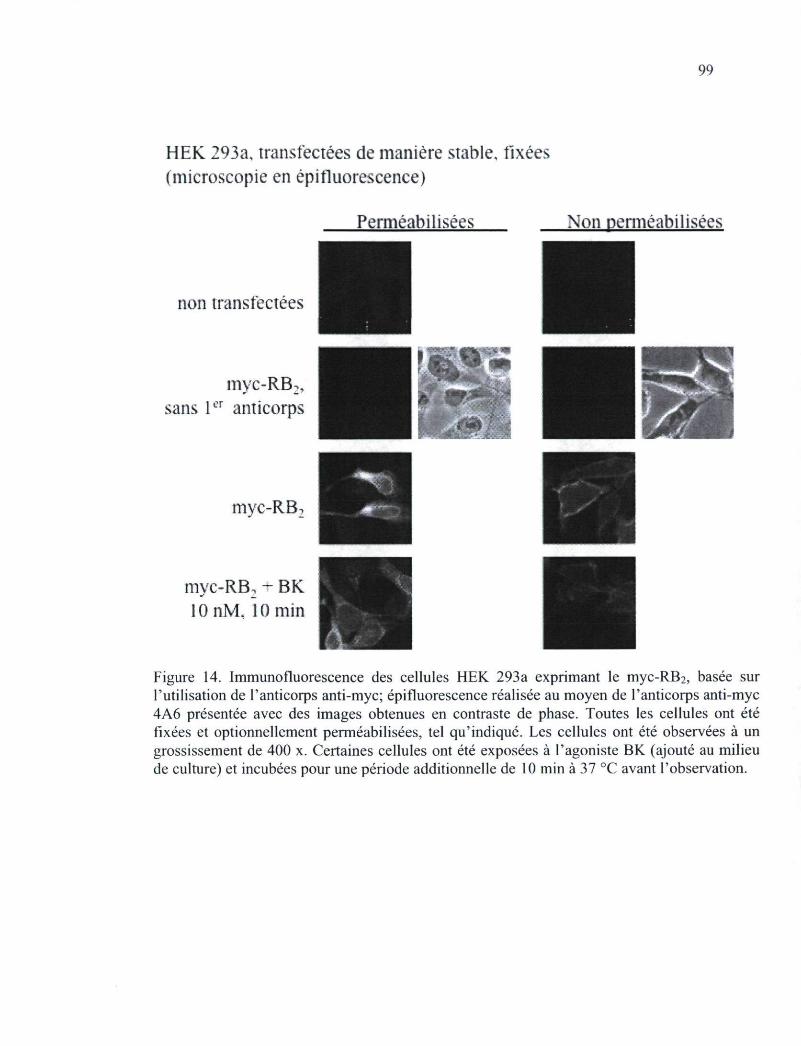
99
HEK 293a, transfectées de manière stable, fixées (microscopie en épifluorescence)
Perméabilisées Non perméabilisées
non transfectées
myc-RB2, sans 1er anticorps
myc-RB,
myc-RB2 + BK 10 nM. 10 min
Figure 14. Immunofluorescence des cellules HEK 293a exprimant le myc-RB2, basée sur l'utilisation de l'anticorps anti-myc; épifluorescence réalisée au moyen de l'anticorps anti-myc 4A6 présentée avec des images obtenues en contraste de phase. Toutes les cellules ont été fixées et optionnellement perméabilisées, tel qu'indiqué. Les cellules ont été observées à un grossissement de 400 x. Certaines cellules ont été exposées à l'agoniste BK (ajouté au milieu de culture) et incubées pour une période additionnelle de 10 min à 37 °C avant l'observation.

100
sur le grand nombre de récepteurs présents dans la voie sécrétoire de cellules exprimant
d'autres constructions. Un traitement avec l'agoniste BK entraînait la translocation de la
fluorescence membranaire vers le cytoplasme, sous forme de matériel granulaire distinct du
signal intracellulaire plus diffus; par ailleurs, des cellules n'ayant pas été perméabilisées
montraient une diminution de l'intensité de la fluorescence moyenne à la surface cellulaire,
ce qui est explicable par l'internalisation du récepteur. Des cellules HEK 293a exprimaient
de manière transitoire une quantité appréciable de myc-RB2, qui était suffisante pour être
détectée par F immunobuvardage réalisé au moyen de l'anticorps anti-myc (clone 4A6;
figure 15). Deux bandes jumelles, possédant des poids moléculaires approximatifs de 63 et
69 kDa, sont considérées comme étant le récepteur mature, ce qui est en accord avec des
données provenant d'autres laboratoires et confirmé par la purification de la forme humaine
du récepteur, présentant un poids moléculaire de 60-70 kDa (Camponova et coll., 2007).
Des bandes de poids moléculaire inférieur ne révélaient aucune information pertinente. Les
cellules exprimant le myc-RB2 ont été soumises aux mêmes traitements avec les agonistes
pour des périodes d'incubation de 3 h et de 12 h, et les expériences résultantes ont permis
une analyse densitométrique (figure 12 C, D). Seul le traitement de 12 h avec l'agoniste
résistant B-9972 (100 nM) provoquait une importante baisse de l'intensité des deux bandes
de 63 et 69 kDa. L'antagoniste B-9430 prévenait la dégradation du récepteur induite par
l'agoniste B-9972 (figure 15). Les puits contenant les lysats de cellules soumises aux
traitements avec les différents agonistes pendant 3 h ne présentaient pas une diminution de
l'intensité des bandes représentant le récepteur mature (figures 12 C et 15).
3.1.6 Désensibilisation fonctionnelle du RB2-GFP par le peptide B-9972
Une fonction bien documentée du RB2-GFP exprimé à la surface des cellules HEK 293 est
l'internalisation rapide de la [3H]BK dans une fraction cellulaire contenant divers
organelles dont les endosomes (Bachvarov et coll., 2001). L'essai permettant la
quantification de l'internalisation de la [3H]BK a été adapté afin de vérifier la fonction
résiduelle des récepteurs dans les cellules prétraitées avec la BK ou le peptide B-9972 (10
nM, 12 h; figure 16A). Des cellules HEK 293 non transfectées n'internalisaient pas la
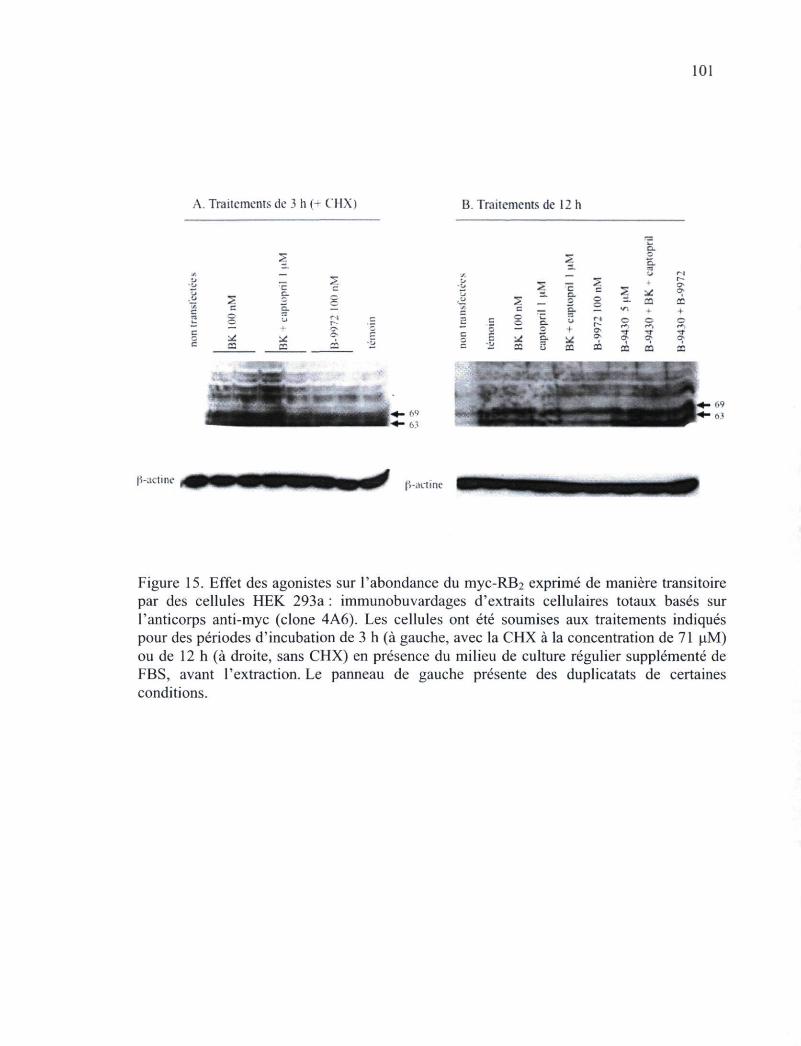
101
A. Traitements dc 3 h (+ CHX) B. Traitements de 12 h
z\W£
HM *
p-actine (5-actïne
Figure 15. Effet des agonistes sur l'abondance du myc-RB2 exprimé de manière transitoire par des cellules HEK 293a : immunobuvardages d'extraits cellulaires totaux basés sur l'anticorps anti-myc (clone 4A6). Les cellules ont été soumises aux traitements indiqués pour des périodes d'incubation de 3 h (à gauche, avec la CHX à la concentration de 71 pM) ou de 12 h (à droite, sans CHX) en présence du milieu de culture régulier supplémenté de FBS, avant l'extraction. Le panneau de gauche présente des duplicatats de certaines conditions.
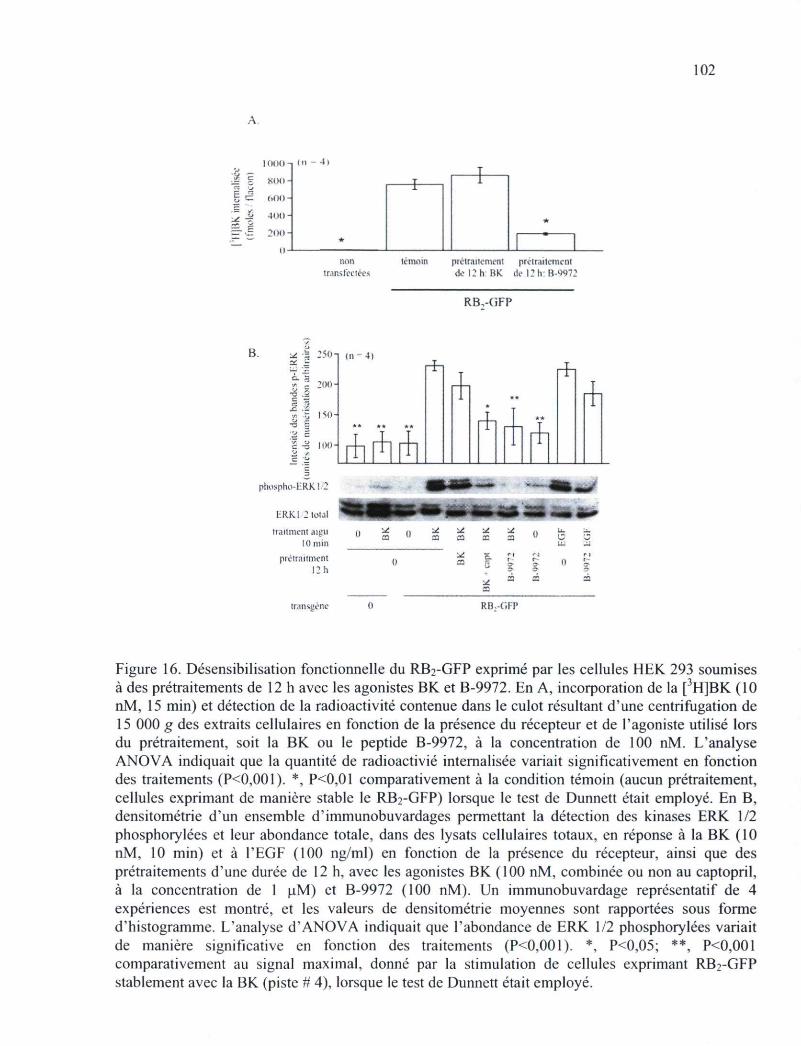
102
"sf. c
IOOOi (n = 4»
800 H
600
400
200
non transfectées
témoin prétraitement prétrai tement d e l 2 h : B K de 12 h : B9972
RB,GFP
u s 250 n 7e. 'é
■r. ^ 200 v — "2
I 1 g .fc 1 5 0 e S 100
( n 4 )
phosphoERK i 2
ERK1/2 total '
n ■P") pw
JC
rl
CCC
mm « W »
trailmcnl aigu 10 min
n ta il SS là, C C =
c^C ca
c^c ■sr:
c ^
JC
rl
CCC
0 3
0
§
prétraitaient 12 h
0
là, C C =
c^C ca
c^c ■sr:
c ^
JC
rl
CCC
0 3
0 r
9
transgéne 0 RB GFP
Figure 16. Désensibilisation fonctionnelle du RB2GFP exprimé par les cellules HEK 293 soumises à des prétraitements de 12 h avec les agonistes BK et B9972. En A, incorporation de la [3H]BK (10 nM, 15 min) et détection de la radioactivité contenue dans le culot résultant d'une centrifugation de 15 000 g des extraits cellulaires en fonction de la présence du récepteur et de l'agoniste utilisé lors du prétraitement, soit la BK ou le peptide B9972, à la concentration de 100 nM. L'analyse ANOVA indiquait que la quantité de radioactivié internalisée variait significativement en fonction des traitements (P<0,001). *, P<0,01 comparativement à la condition témoin (aucun prétraitement, cellules exprimant de manière stable le RB2GFP) lorsque le test de Dunnett était employé. En B, densitométrie d'un ensemble d'immunobuvardages permettant la détection des kinases ERK 1/2 phosphorylées et leur abondance totale, dans des lysats cellulaires totaux, en réponse à la BK (10 nM, 10 min) et à l'EGF (100 ng/ml) en fonction de la présence du récepteur, ainsi que des prétraitements d'une durée de 12 h, avec les agonistes BK (100 nM, combinée ou non au captopril, à la concentration de 1 pM) et B9972 (100 nM). Un immunobuvardage représentatif de 4 expériences est montré, et les valeurs de densitométrie moyennes sont rapportées sous forme d'histogramme. L'analyse d'ANOVA indiquait que l'abondance de ERK 1/2 phosphorylées variait de manière significative en fonction des traitements (P<0,001). *, P<0,05; **, P<0,001 comparativement au signal maximal, donné par la stimulation de cellules exprimant RB2GFP stablement avec la BK (piste # 4), lorsque le test de Dunnett était employé.

103
[3H]BK de manière appréciable, alors que l'expression du RB2-GFP menait à une
importante internalisation de l'agoniste radiomarqué, qui n'était pas modifiée par un
prétraitement de 12 h avec la BK, mais était réduite de 80 % par une exposition prolongée
au peptide B-9972 (figure 16A).
L'activation du RB2-GFP exprimé par des cellules HEK 293 est couplée à la
phosphorylation des MAP kinases ERK 1/2 (Morissette et coli, 2007). Une stimulation
standard de 10 min avec la BK (10 nM) a été appliquée à des cellules non transfectées,
ainsi qu'à des cellules exprimant de manière stable le RB2-GFP qui avaient été prétraitées
avec la BK, une combinaison de BK et de captopril ou le peptide B-9972 pendant une
période de 12 h. La BK n'a pas produit la phosphorylation de ERK 1/2 chez les cellules
non-transfectées. La réponse des cellules exprimant de manière stable le RB2-GFP
dépendait du traitement auquel elles avaient été soumises; les prétraitements avec le B-9972
et celui combinant la BK et le captopril ont complètement aboli une nouvelle stimulation de
la phosphorylation de ERK 1/2, alors que le traitement prolongé avec la BK seule ne
réduisait pas significativement la phosphorylation de ERK 1/2 obtenue suite à une deuxième
stimulation aiguë avec ce même agoniste. L'intense signal de phosphorylation obtenu par
un traitement aigu avec l'EGF de cellules RB2-GFP prétraitées avec le peptide B-9972
pendant 12 h démontre que la désensibilisation subie par le récepteur est homologue.
3.2 Caractérisation approfondie d'agonistes résistants à l'inactivation
3.2.1 Caractérisation pharmacologique du composé 47a
L'antagoniste LF 16-0687 comporte un pharmacophore commun à celui du composé 47a.
La figure 17 présente l'effet de cet antagoniste, dépendant de sa concentration et
surmontable, au niveau de la contraction induite par la BK au niveau de la veine ombilicale
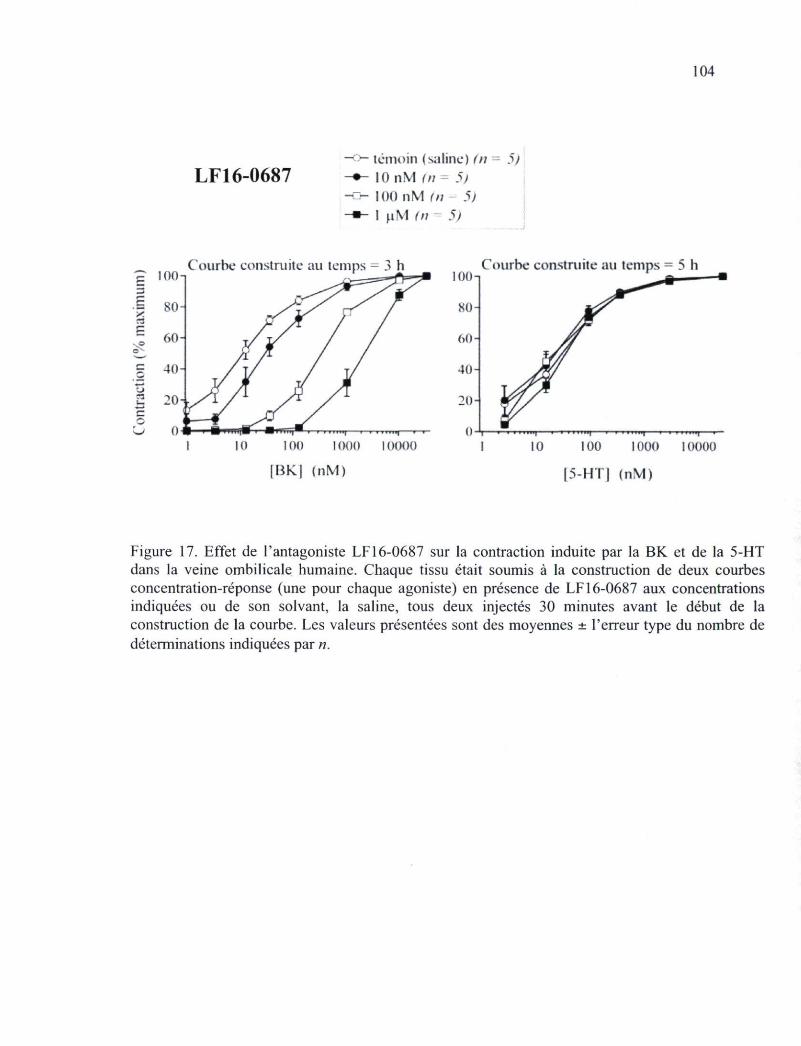
104
LF16-0687 - o - témoin (saline) (n - 5) - • - 10 nM (n= 5) - o - 100 nM f « - 5) H»- | U M (n 5)
Courbe constante au temps = 3 h Courbe construite au temps = 5 h
ta E
CJ
CCZ
O U
10 100 [BK] (nM)
100 1000 10000
[5-HT] (nM)
Figure 17. Effet de l'antagoniste LF 16-0687 sur la contraction induite par la BK et de la 5-HT dans la veine ombilicale humaine. Chaque tissu était soumis à la construction de deux courbes concentration-réponse (une pour chaque agoniste) en présence de LF 16-0687 aux concentrations indiquées ou de son solvant, la saline, tous deux injectés 30 minutes avant le début de la construction de la courbe. Les valeurs présentées sont des moyennes ± l'erreur type du nombre de déterminations indiquées par n.

105
humaine fraîchement isolée. Le pA2 calculé est de 8,27 ± 0,19 et la pente de la régression
de Schild, qui n'est pas montrée, est de -1,04 ± 0,13. La valeur de pA2 rapportée par une
autre équipe était comparable (Pruneau et coll., 1999). Cet antagoniste présente une
excellente sélectivité, puisque l'action contractile de la 5-HT n'était pas altérée par la
présence du LF 16-0687, comme le montre la figure 17. Le composé 47a a d'abord été
caractérisé par des essais de contractilité réalisés avec la veine ombilicale humaine, un essai
bien établi pour le RB2 humain. Le composé 47a stimule la contraction de cette préparation
avec une CE50 de 25,3 nM et la contraction maximale induite par le composé 47a
correspond à 82 % de la contraction maximale induite par la BK, ce qui a été déterminé par
l'ajout de 23,6 pM de BK à la fin de la courbe concentration-réponse construite pour le
composé 47a (figure 18). Cette contraction dépendait de la stimulation du RB2 exprimé par
le vaisseau, étant donné l'inhibition de cet effet par un prétraitement des tissus avec le
LF 16-0687. La méthode graphique de la régression de Schild permet de calculer une valeur
de pA2 de 8,46 ±0,10 (la pente de la régression de Schild= -1,11 ± 0,07) pour cet essai de
contractilité (figure 18, droite). Puisque la veine ombilicale peut être stimulée à répétition
avec la BK et présenter une réponse contractile stable, comme cela a été montré auparavant
(Marceau et coll., 1994), cette préparation a servi à vérifier si le peptide B-9972 et le
composé 47a causaient sa désensibilisation à une stimulation subséquente à la BK. Pour ce
faire, chaque tissu servait à la construction de deux courbes concentration-réponse pour les
différents agonistes au temps 3 h et 6 h après l'installation des tissus dans les bains (figure
19). Lorsqu'utilisée la première, la BK ne provoquait pas la désensibilitation de la veine
ombilicale aux autres agonistes. Les CE50 du B-9972, soit de 69,5 nM et celle du composé
47a, de 68,8 nM ne se distinguent pas significativement des valeurs de CE50 témoins de
40,30 nM et de 53,4 nM pour le B-9972 et le composé 47a, respectivement (figure 19, en A
et en C). Par contre, lorsqu'appliqué le premier, le composé 47a diminuait l'apparente
activité de la BK, dont la CE50 passait de 33,3 nM (lorsque la courbe concentration-
réponse de la BK est construite en premier) à 672 nM. Il est intéressant de remarquer que le
peptide B-9972 était inactif à cet égard, puisque la CE50 de la courbe concentration-réponse
de la BK construite 3 h après celle de la courbe construite à l'aide du B-9972 était de 36,8
nM, alors que celle de la courbe concentration-réponse construite la première est de 41,7
nM (figure 19B et D). De plus, l'effet maximal de la BK était moindre après l'exposition
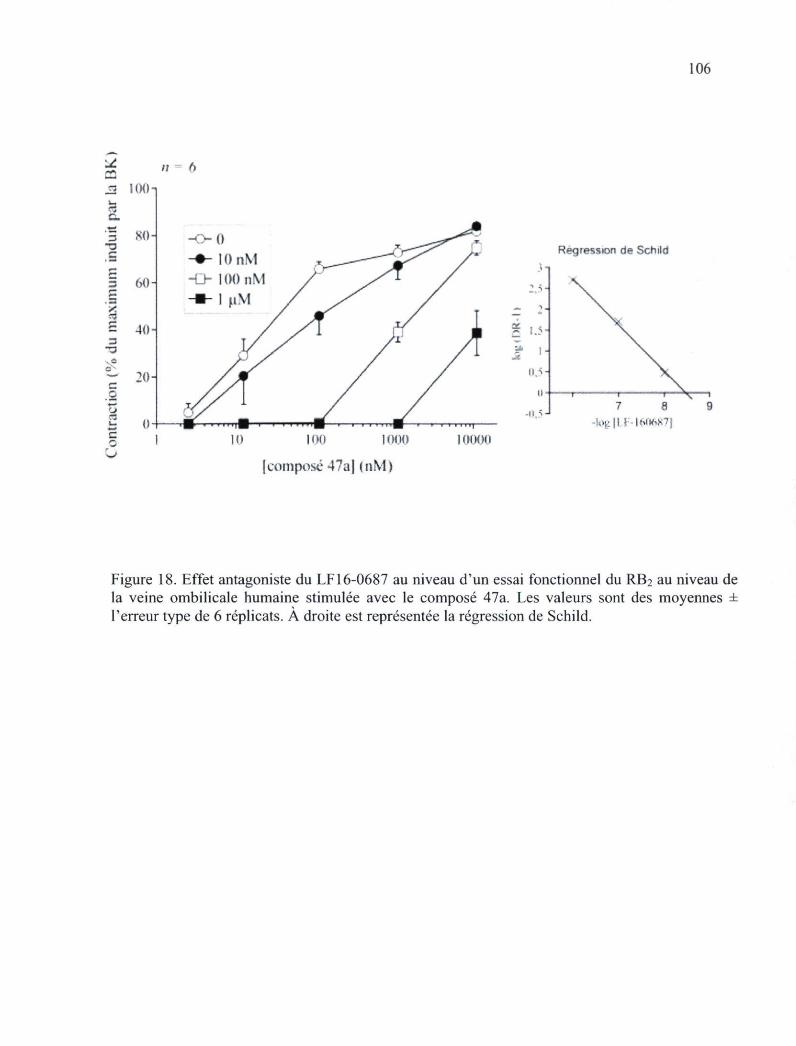
106
S
Regression de Schild
10 100 1000
[composé 47a| (nM) 10000
Figure 18. Effet antagoniste du LF 16-0687 au niveau d'un essai fonctionnel du RB2 au niveau de la veine ombilicale humaine stimulée avec le composé 47a. Les valeurs sont des moyennes ± l'erreur type de 6 réplicats. À droite est représentée la régression de Schild.
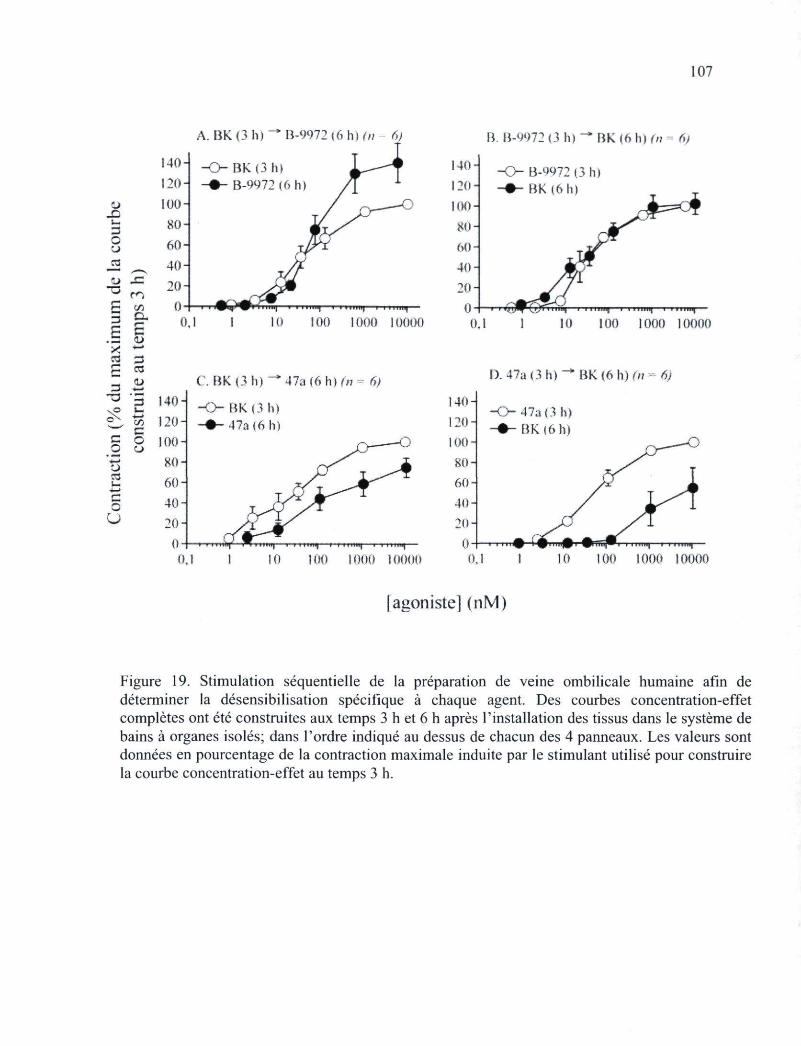
107
A. BK (3 h) "* B-9972 (6 h) (n - 6)
-£ =3 C U
u
E -
C/3
«s 3
- s * t— — - r s— G B 0 O CJ
8 ca H — C C U
000 10000
140 120 100 80 60-40-20
0 o.
C. BK (3 h) ~* 47a (6 h) (n 6)
- O BK (3 h) - • - 4 7 a (6 h)
140 120 100 80 60 40 20 0
B. B-9972 (3 h) "* BK (6 h) (n = 6)
- O B-9972 (3 h) - • - BK(6h)
100 1000 10000
140-120-
-o 100-80-60-40-20-
0- —> rrff i
D.47a<3h>^BK(6h)fw = <5;
47a(3 h) BK(6h)
100 1000 10000 OJ I 00 1000 I0( J00
[agoniste] (nM)
Figure 19. Stimulation séquentielle de la préparation de veine ombilicale humaine afin de déterminer la désensibilisation spécifique à chaque agent. Des courbes concentration-effet complètes ont été construites aux temps 3 h et 6 h après l'installation des tissus dans le système de bains à organes isolés; dans l'ordre indiqué au dessus de chacun des 4 panneaux. Les valeurs sont données en pourcentage de la contraction maximale induite par le stimulant utilisé pour construire la courbe concentration-effet au temps 3 h.

108
initiale des tissus au composé 47a (figure 19D). Le temps de relaxation de la moitié de la
contraction maximale obtenue a été mesuré pour le composé 47a, comme il l'avait été
précédemment pour le peptide B-9972, après le lavage des tissus. Les tissus contractés à la
suite de la construction d'une courbe concentration-réponse avec une concentration
cumulative de 10,5 pM de BK atteignaient la moitié de la contraction en 6,7 ± 0,8 min
(figure 20, en A et en B), comparativement à des temps bien plus élevés pour le peptide B-
9972 et le composé 47a.
L'affinité du composé 47a pour le RB2 de lapin, telle qu'étudiée par les essais de liaison
réalisés à l'aide de cellules HEK 293 exprimant de façon stable le RB2-GFP, est beaucoup
plus faible que celle observée pour la BK, présentant une CI50 de 168 nM, ce qui le rend 10
fois moins actif pour ce récepteur. Au niveau de cette lignée cellulaire, l'antagoniste LF16-
0687 était un ligand puissant, présentant une CI50 de 3,6 nM (figure 21). Afin de déterminer
l'affinité du composé 47a pour le RB2 humain, pour lequel il a d'ailleurs été optimisé, la
lignée cellulaire MG-63 a été employée. Cette lignée dérivant d'un ostéosarcome a
démontré d'importantes réponses fonctionnelles à la BK par la stimulation des RB2 et
l'expression de sites correspondants pour la liaison du radioligand (Wang et coll., 2001;
Brechter et Lerner, 2002). L'utilisation de ces cellules a permis la construction de courbes
de compétition pour la liaison de la [3H]BK; les résultats confirment toutefois une affinité
de composé 47a 10,2 fois moindre que celle de la BK pour le RB2 endogène humain, tout
comme pour le récepteur de lapin. Le LF 16-0687 était 4 fois plus puissant que la BK dans
ce même essai (données non montrées).
3.2.2 Effets des agonistes résistants au métabolisme sur les cellules exprimant le RB2-GFP
Les cellules exprimant le RB2-GFP constituent un modèle approprié pour la réalisation
d'études morphologiques d'endocytose et pour mieux comprendre la dégradation du
récepteur induite par l'agoniste (Bachvarov et coll., 2001).
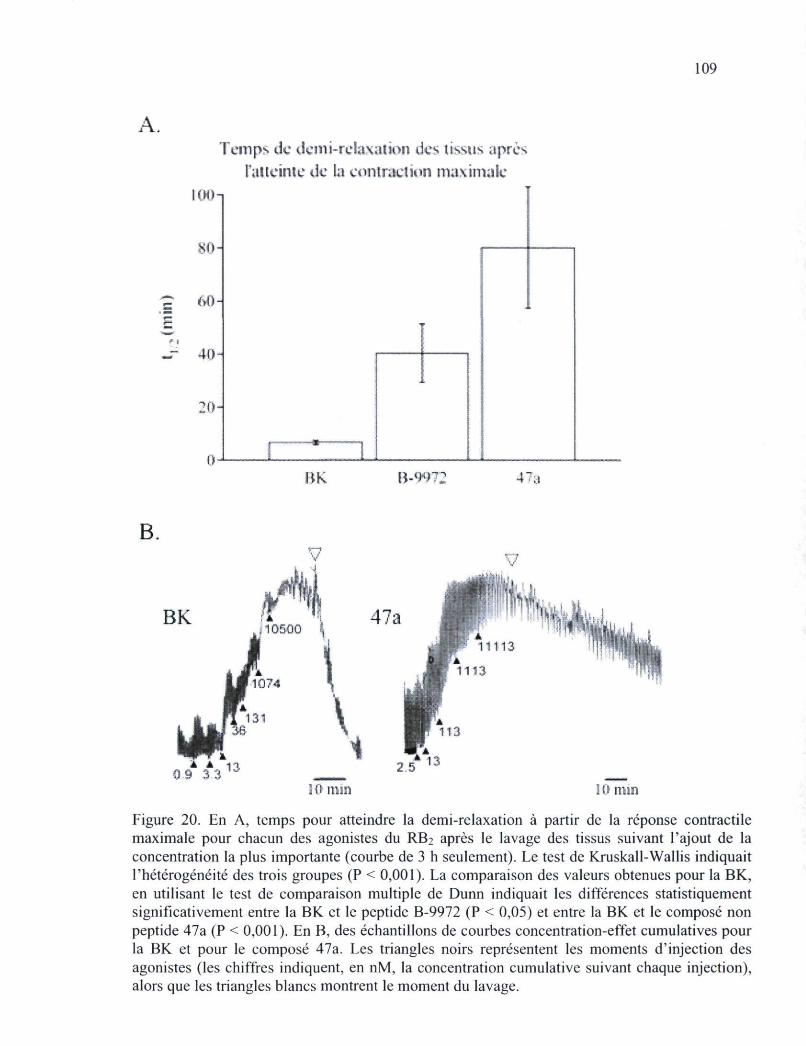
109
A. Temps de demi-relaxation des tissus après
I'alleinlc de la contraction maximale 100-,
80-
2 60-£ _r 40
20-
0 Ik B-9972 47a
B.
47a
2.5
10 mm 10 mm
Figure 20. En A, temps pour atteindre la demi-relaxation à partir de la réponse contractile maximale pour chacun des agonistes du RB2 après le lavage des tissus suivant l'ajout de la concentration la plus importante (courbe de 3 h seulement). Le test de Kruskall-Wallis indiquait l'hétérogénéité des trois groupes (P < 0,001). La comparaison des valeurs obtenues pour la BK, en utilisant le test de comparaison multiple de Dunn indiquait les différences statistiquement significativement entre la BK et le peptide B-9972 (P < 0,05) et entre la BK et le composé non peptide 47a (P < 0,001). En B, des échantillons de courbes concentration-effet cumulatives pour la BK et pour le composé 47a. Les triangles noirs représentent les moments d'injection des agonistes (les chiffres indiquent, en nM, la concentration cumulative suivant chaque injection), alors que les triangles blancs montrent le moment du lavage.
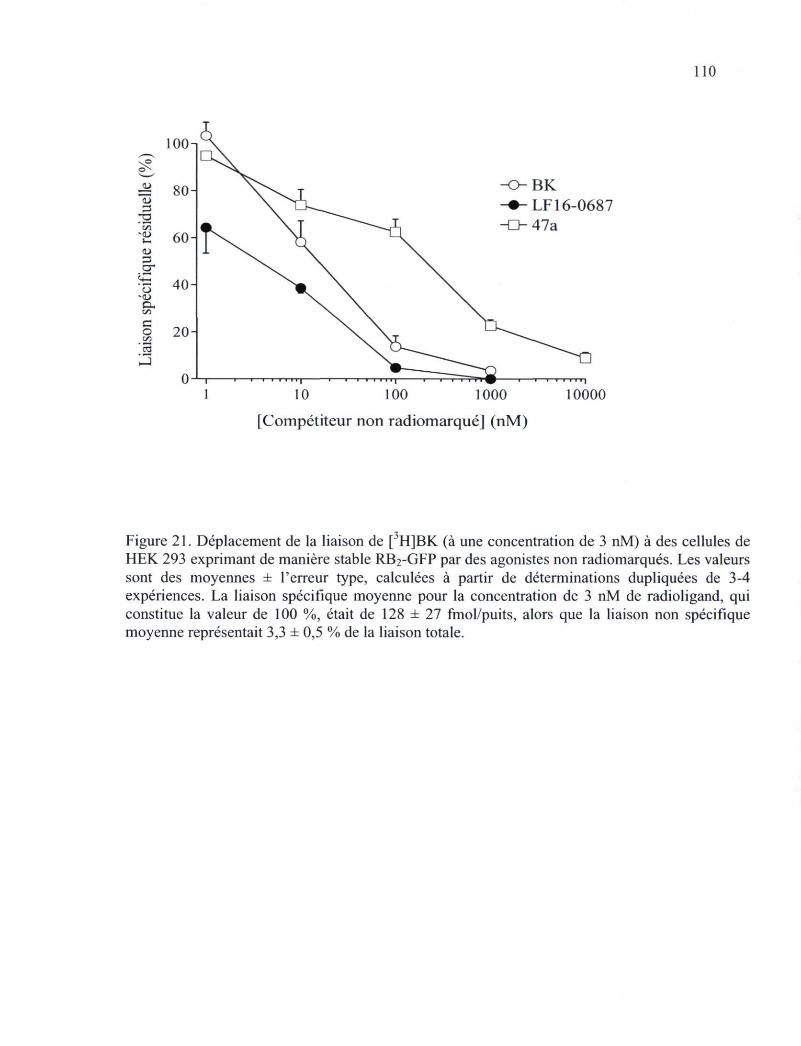
110
O B K - • - L F I 6-0687 ^ > 4 7 a
T 1 1 I I I 1 |
10000 [Compétiteur non radiomarqué] (nM)
Figure 21. Déplacement de la liaison de [3H]BK (à une concentration de 3 nM) à des cellules de HEK 293 exprimant de manière stable RB2-GFP par des agonistes non radiomarqués. Les valeurs sont des moyennes ± l'erreur type, calculées à partir de déterminations dupliquées de 3-4 expériences. La liaison spécifique moyenne pour la concentration de 3 nM de radioligand, qui constitue la valeur de 100 %, était de 128 ± 27 fmol/puits, alors que la liaison non spécifique moyenne représentait 3,3 ± 0,5 % de la liaison totale.

I l l
Pourvu d'un statut d'agoniste partiel, il a été prédit que la liaison du composé 47a au RB2-
GFP mènerait à l'internalisation de ce dernier. Effectivement, un traitement de 30 min avec
cet agoniste non peptidique induit une lente translocation de la fluorescence membranaire,
correspondant au récepteur, vers l'intérieur de la cellule sous forme de particules très fines,
comme en témoigne la figure 22. En revanche, les structures fluorescentes cytosoliques
formées secondairement aux traitements avec des agonistes complets, tels que la BK et le
peptide B-9972, présentent une morphologie endosomale différente, de taille plus
importante et observable plus rapidement (figure 22). L'endocytose induite par le peptide
B-9972 persiste au-delà d'une période de stimulation de 3 h; une baisse de l'intensité de la
fluorescence totale était observable 12 h après la stimulation initiale, alors que la BK induit
une endocytose entièrement réversible après 3 h de traitement et ce, sans que le milieu
réactionnel baignant les cellules ne soit changé.
Lorsque les cellules étaient exposées aux agonistes BK, B-9972 et 47a pendant 30 min,
puis rincées et incubées dans un milieu frais pendant 2,5 h supplémentaires, la distribution
de la fluorescence pour les diverses conditions était différente. Les cellules stimulées par la
BK présentaient une fluorescence membranaire lisse, alors que celles stimulées par le
peptide B-9972 montraient plutôt une persistance des structures endosomales contenant le
récepteur. L'interprétation des résultats obtenus par le traitement du composé 47a est
difficile à cause de l'internalisation modeste stimulée par le composé 47a après un temps
d'incubation de 30 min. L'endocytose du RB2 secondaire au traitement avec le composé
47a persistait après 12 h, et était parallèle à une perte progressive du marquage
membranaire et du signal fluorescent total, suggérant une dégradation partielle du RB2 au
niveau protéique, puisque l'expression du récepteur est sous le contrôle d'un promoteur
viral, qui n'est pas contrôlé par la signalisation des récepteurs des kinines. Les changements
morphologiques induits par le composé 47a sont prévenus par la coincubation des cellules
avec l'antagoniste non peptidique LF 16-0687 (figure 22).
L'évaluation quantitative de la mobilisation calcique, par l'exploitation du colorant
fluorescent FURA-2AM, dans des cellules HEK 293 exprimant de manière stable le RB2-
GFP, permet de mesurer la signalisation calcique induite par les différents agonistes du RB2
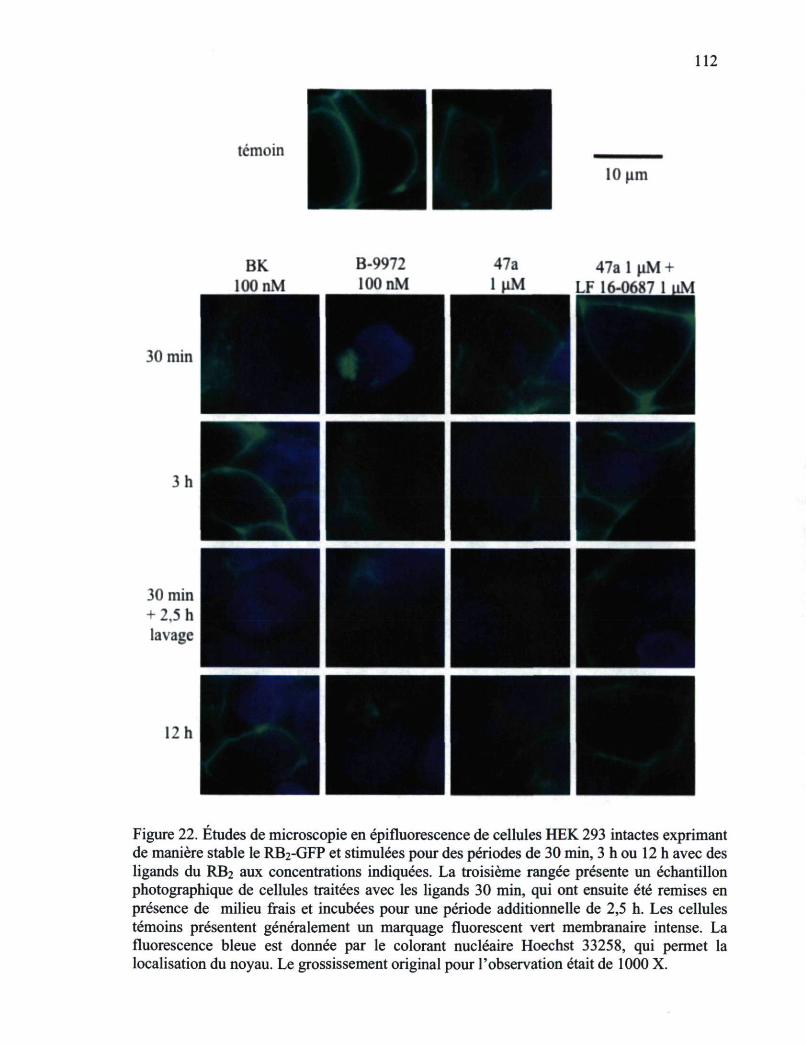
112
témoin 10 pm
30 min
BK 100 nM
B-9972 100 nM
47a lliM
47a 1 nM + LF 16-0687 1 uM
30 min + 2,5 h lavage
12 h
Figure 22. Études de microscopie en épifluorescence de cellules HEK 293 intactes exprimant de manière stable le RB2-GFP et stimulées pour des périodes de 30 min, 3 h ou 12 h avec des ligands du RB2 aux concentrations indiquées. La troisième rangée présente un échantillon photographique de cellules traitées avec les ligands 30 min, qui ont ensuite été remises en présence de milieu frais et incubées pour une période additionnelle de 2,5 h. Les cellules témoins présentent généralement un marquage fluorescent vert membranaire intense. La fluorescence bleue est donnée par le colorant nucléaire Hoechst 33258, qui permet la localisation du noyau. Le grossissement original pour l'observation était de 1000 X.

113
(figure 23). Les cellules HEK 293 non transfectées, constituant la condition témoin,
montraient une réponse faible et brève aux agonistes du RB2 (ces données ne sont pas
présentées). Toutefois, les cellules HEK 293 exprimant le RB2-GFP stablement
présentaient une mobilisation calcique rapide précédant une diminution lente, suivant la
stimulation par la BK (100 nM), le peptide B-9972 (100 nM) et le composé 47a (lpM).
L'effet des trois agonistes déclinait pour atteindre l'état basai 2,5 min après la stimulation
(figure 23).
Les cellules HEK 293 exprimant de manière stable le RB2-GFP permettent la détection de
l'effet stimulant des peptides agonistes ou agonistes partiels de ce récepteur, grâce à
l'identification de réponses spécifiques comme la phosphorylation des MAP kinases ERK
1/2 (Morissette et coll., 2007). Cet essai, lorsqu'appliqué au composé 47a, montrait la
capacité du ligand à induire la phosphorylation des ERK 1/2 à une concentration de 1 pM
lors d'une stimulation de 10 min, quoique cette formation de phospho-ERK ait été moins
intense que celle obtenue par un traitement avec la BK, à une concentration de 10 nM
(figure 24, en A). La phosphorylation de ERK 1/2 secondaire au traitement des cellules
avec le composé 47a était inhibée par le traitement concomitant avec l'antagoniste LF16-
0687, et absente des cellules non transfectées avec le RB2, ce qui démontre que les effets
produits par le traitement au composé 47a dépendent de la stimulation du RB2. L'essai de
phosphorylation des kinases ERK 1/2 a aussi montré que des traitements prolongés de 12 h
avec les agonistes résistants au métabolisme, de nature peptidique ou non, activaient encore
cette voie de signalisation, quoique le niveau de phosphorylation des kinases était déclinant
(Figure 24, en B), ce qui peut être expliqué en partie par la dégradation du récepteur. En
guise de comparaison, l'effet de la BK sur la phosphorylation de ERK 1/2 était à peine
détectable après une stimulation de 3 h et ne l'était plus du tout après une stimulation de 12
h. Des résultats généralement similaires ont été observés dans des cellules HEK 293a qui
exprimaient de manière transitoire le myc-RB2; i.e. que l'effet des agonistes dépendait de
l'expression du récepteur et que les effets du peptide B-9972 et du composé 47a
s'étendaient jusqu'à 12 h après l'ajout des agonistes dans le milieu de culture des cellules.
Toutefois, les cellules transfectées avec la construction myc-RB2 et soumises à un
traitement de 3 h avec 100 nM de BK présentent un niveau de kinases
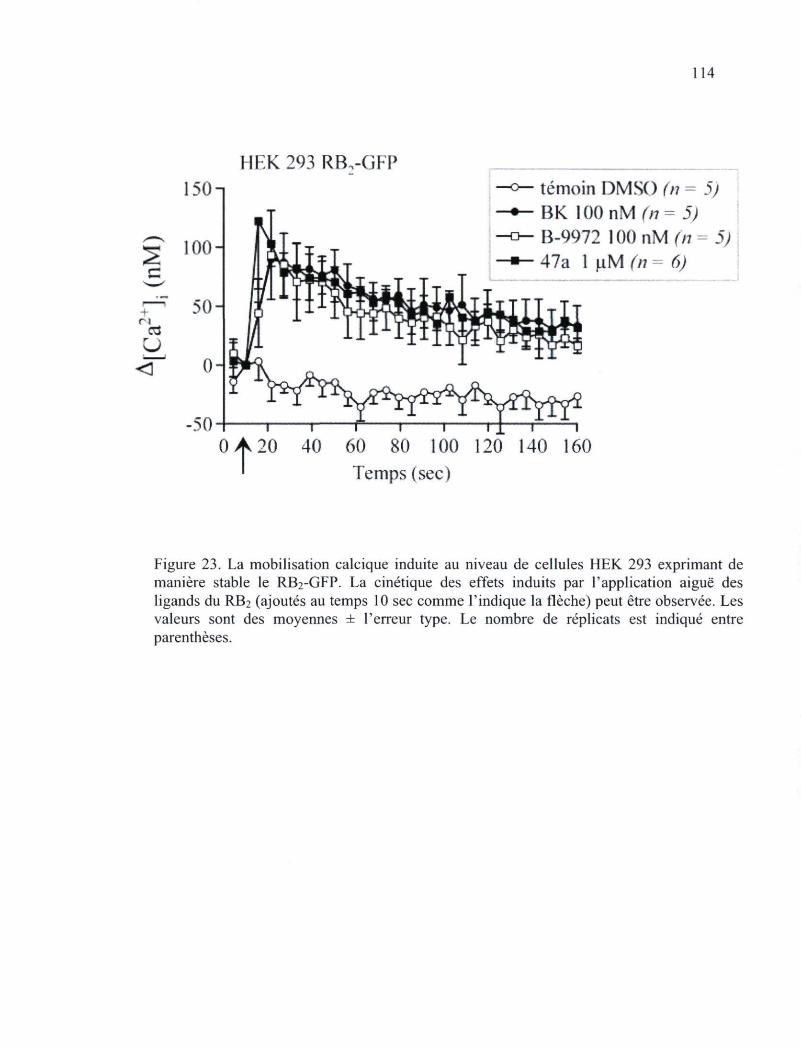
114
HEK 293 RB.-GFP
+ r i
U
témoin DMSO (n = 5) BK 100nMr7i = 5) B-9972 \QQnM(n= 5) 47a 1 p.M (n = 6)
60 80 100 120 Temps (sec)
160
Figure 23. La mobilisation calcique induite au niveau de cellules HEK 293 exprimant de manière stable le RB2-GFP. La cinétique des effets induits par l'application aiguë des ligands du RB2 (ajoutés au temps 10 sec comme l'indique la flèche) peut être observée. Les valeurs sont des moyennes ± l'erreur type. Le nombre de réplicats est indiqué entre parenthèses.
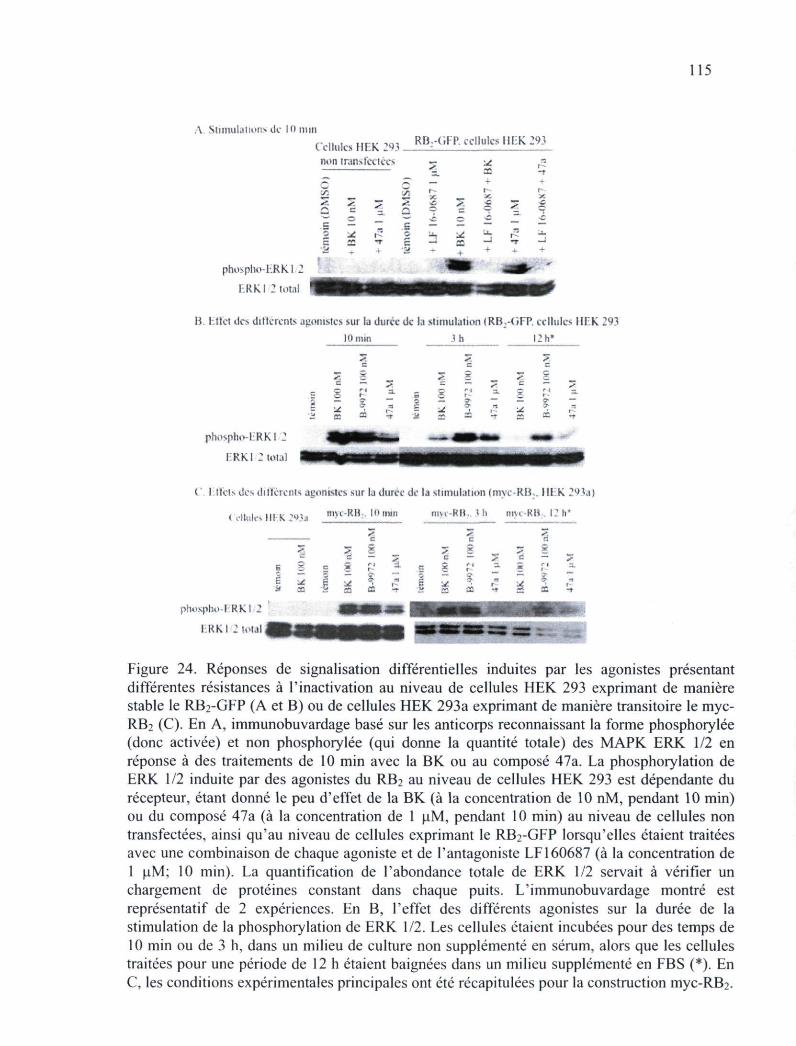
115
A Stimulations de 1 *» mm Cellules HEK 293 non transfectées
RBGEP. cellules HER 293
phosphoERKl/2 ERR I 2 total
B. Effet des différents agonistes sur la durée dc la stimulation (RBOFP. cellules IIEK 29.1 10 min i h 12 h*
phosphoERR 1 2
1RKI 2 total
t ' Effets, des différents agonistes sur la durée de la stimulation (invcRB,, 11ER 293a)
CeliulM HFK "*9Jj m>eRH. KMnm myvRB, 3 Il mycRB,. 12 h*
"K sss
phosphoERKt 2
ERRI 2 total
s * ■
c. c >. * > S ES —, c — Jj
B ■s.
... e
§ r. t m
r i u 2 CCCO
Figure 24. Réponses de signalisation différentielles induites par les agonistes présentant différentes résistances à l'inactivation au niveau de cellules HEK 293 exprimant de manière stable le RB2GFP (A et B) ou de cellules HEK 293a exprimant de manière transitoire le myc
RB2 (C). En A, immunobuvardage basé sur les anticorps reconnaissant la forme phosphorylée (donc activée) et non phosphorylée (qui donne la quantité totale) des MAPK ERK 1/2 en réponse à des traitements de 10 min avec la BK ou au composé 47a. La phosphorylation de ERK 1/2 induite par des agonistes du RB2 au niveau de cellules HEK 293 est dépendante du récepteur, étant donné le peu d'effet de la BK (à la concentration de 10 nM, pendant 10 min) ou du composé 47a (à la concentration de 1 pM, pendant 10 min) au niveau de cellules non transfectées, ainsi qu'au niveau de cellules exprimant le RB2GFP lorsqu'elles étaient traitées avec une combinaison de chaque agoniste et de l'antagoniste LF 160687 (à la concentration de 1 pM; 10 min). La quantification de l'abondance totale de ERK 1/2 servait à vérifier un chargement de protéines constant dans chaque puits. L'immunobuvardage montré est représentatif de 2 expériences. En B, l'effet des différents agonistes sur la durée de la stimulation de la phosphorylation de ERK 1/2. Les cellules étaient incubées pour des temps de 10 min ou de 3 h, dans un milieu de culture non supplémenté en sérum, alors que les cellules traitées pour une période de 12 h étaient baignées dans un milieu supplémenté en FBS (*). En C, les conditions expérimentales principales ont été récapitulées pour la construction mycRB2
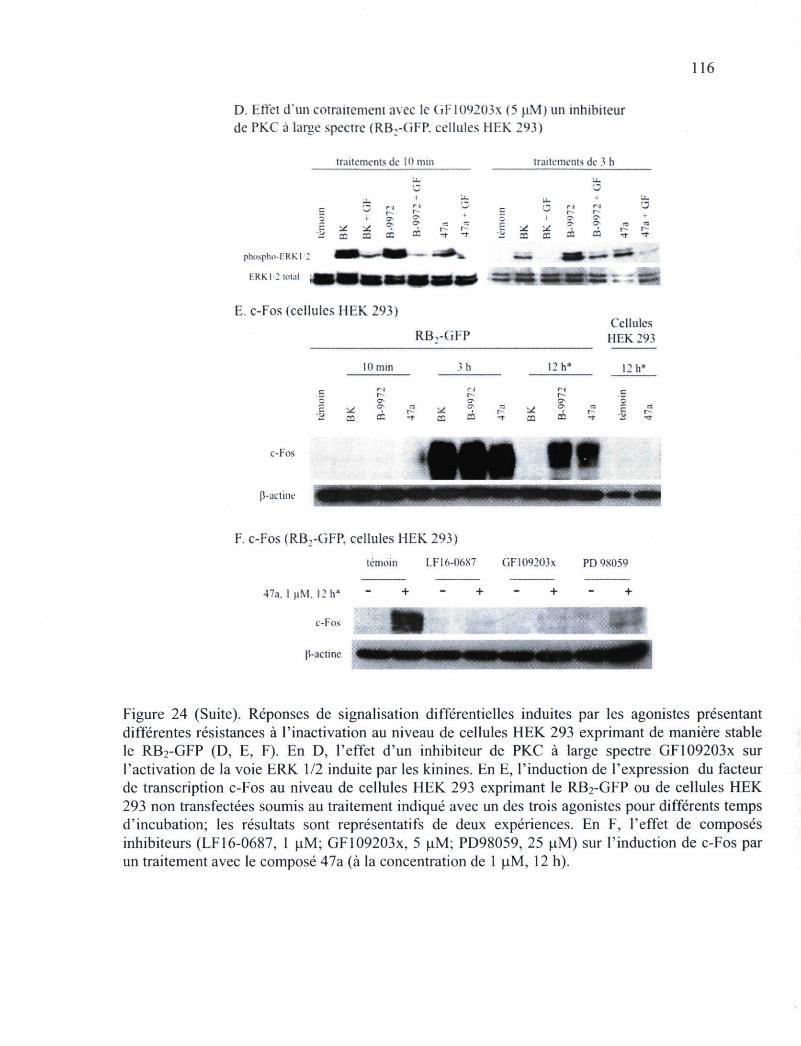
116
D. Effet d'un cotraitement avec le GF109203x (5 u.Vl) un inhibiteur de PKC à large spectre (RB:GFP. cellules HEK 293)
traitements de 10 mm traitements de 3 h
jt Z/. rr<
r~~
■d ce ta a*. » ■*
pbosphoERKI'2
ERK 12 total
E. cFos (cellules HEK 293)
H * 2 rX r r~ 23 PB 33 Πrr r
=2». WÊÊk *
CCJHKK. zwÊÊÊ^ÊÊÊÊ^SSÊÊ& 'SZ Z Smmw :
RB :GFP Cellules HEK 293
10 mm 3 h i : h* 12 h*
S
■ ' j
r t r
r r i
a
cC cq
rr
c
1 s cFos
Bactine
F. cFos (RBGFP. cellules HEK 293) témoin LE 160687 GFI09203x PD 98059
47a. I M M 12 h*
cFos
|lactine
Figure 24 (Suite). Réponses de signalisation différentielles induites par les agonistes présentant différentes résistances à l'inactivation au niveau de cellules HEK 293 exprimant de manière stable le RB2GFP (D, E, F). En D, l'effet d'un inhibiteur de PKC à large spectre GF109203x sur l'activation de la voie ERK 1/2 induite par les kinines. En E, l'induction de l'expression du facteur de transcription cFos au niveau de cellules HEK 293 exprimant le RB2GFP ou de cellules HEK 293 non transfectées soumis au traitement indiqué avec un des trois agonistes pour différents temps d'incubation; les résultats sont représentatifs de deux expériences. En F, l'effet de composés inhibiteurs (LF 160687, 1 pM; GF109203x, 5 pM; PD98059, 25 pM) sur l'induction de cFos par un traitement avec le composé 47a (à la concentration de 1 pM, 12 h).

117
de phosphorylation de ERK 1/2 aussi important que celui présenté par les cellules traitées
avec l'agoniste B-9972. Ces résultats sont présentés à la figure 24, en C. Il est important de
rappeler que les cellules exposées à des traitements de 12 h étaient maintenues dans un
milieu de culture supplémenté en sérum, afin d'assurer leur viabilité; par ailleurs le milieu
n'était pas changé avant le début de la stimulation, pour maintenir un bruit de fond du
signal phospho-ERK 1/2 le plus faible possible.
L'ajout de l'inhibiteur de PKC à large spectre GF109203x, a inhibé l'effet des trois
agonistes sur la phosphorylation de ERK 1/2, aux temps d'analyse de 10 min et de 3 h (en
D de la figure 24). L'inhibiteur du domaine tyrosine kinase du récepteur de l'EGF AG
1478, à une concentration de 1 pM, n'a pas réduit la phosphorylation de ERK 1/2 induite
par les agonistes peptidiques aux temps de 10 min et 3 h (données non montrées).
Les cellules transfectées de manière stable avec la construction RB2-GFP n'expriment pas
le facteur de transcription c-Fos à l'état basai; par contre une lente induction de son
expression a été observée suivant le traitement des cellules avec les agonistes BK, B-9972
et 47a (figure 24, en E). Aucun signal de c-Fos n'était détecté suite à des traitements de 10
min avec les différents agonistes, mais son expression était détectable au temps 3 h. Les
agonistes BK et B-9972 stimulaient une forte expression de c-Fos, alors que le composé
47a stimulait une expression plus faible du facteur de transcription. Par contre, après une
stimulation soutenue de 12 h du RB2-GFP, l'abondance du facteur de transcription c-Fos
était encore très importante au niveau les lysats de cellules exposées avec les agonistes
résistants au métabolisme, le peptide B-9972 et le composé 47a non peptidique, alors qu'il
était complètement absent des cellules traitées avec la BK, pour la même période de temps.
Par ailleurs, le traitement soutenu de 12 h avec soit le B-9972 ou le composé 47a a induit
une augmentation du poids moléculaire moyen de c-Fos. Aucune expression de ce facteur
de transcription n'a été induite dans des cellules HEK 293 n'exprimant pas le RB2-GFP.
Cette induction de l'expression de c-Fos était inhibée dans les cellules HEK 293 RB2-GFP
par l'antagoniste du RB2 LF16-0687, l'inhibiteur de PKC GF109203x ou par l'inhibiteur de
MEK1 PD98059 (résultats présentés à la figure 24, en F).

118
3.2.3 Effets des agonistes résistants au métabolisme au niveau de l'interaction du RB2 et de la p-arrestine2
La (3-arrestine2 est un partenaire moléculaire du RB2 durant son endocytose (Simaan et
coll., 2005). Lorsqu'exprimée dans des cellules HEK 293a non stimulées, la distribution de
la construction fusionnant la GFP et la (3-arrestine2 était essentiellement cytosolique, telle
que présentée à la figure 25. La protéine fluorescente subissait une translocation vers des
particules cytosoliques de 10 à 30 min après la stimulation des cellules exprimant la
construction fluorescente avec le myc-RB2 par l'agoniste naturel BK. Cette condensation
du signal fluorescent de la P-arrestine2-GFP était réversible 3 h ou 12 h après la stimulation
du récepteur avec la BK. Toutefois, un traitement avec le peptide B-9972 ou le composé
47a, à des concentrations de 100 nM ou de 1 pM respectivement, induisait une
translocation soutenue de la |3-arrestine2 à des granules cytosoliques, observable aux temps
de traitement de 3 h et de 12 h (figure 25). D'autres analyses morphologiques de la
distribution subcellulaire des P-arrestines ont été faites et sont résumées à la figure 26.
L'utilisation de la construction (3-arrestinei-CherryFP en combinaison avec le RB2-GFP a
permis l'observation simultanée des deux protéines par des cellules soumises à des
traitements de 12 h avec la BK, le B-9972 ou le composé 47a. Le signal rouge fluorescent
de la 6-arrestinei se localisait à certaines structures intracellulaires vertes où se retrouve le
récepteur. Des cellules exprimant les deux protéines et stimulées par un traitement de 12 h
avec la BK présentaient une distribution lisse et homogène de la P-arrestinei-CherryFP,
tout comme celle montrée par les cellules non traitées.
3.2.4 Dégradation du RB2-GFP secondairement à sa stimulation par les agonistes résistants à l'inactivation
La dégradation du RB2-GFP est facilement détectable par la diminution de l'abondance de
la bande correspondant au récepteur mature, d'un poids moléculaire d'approximativement
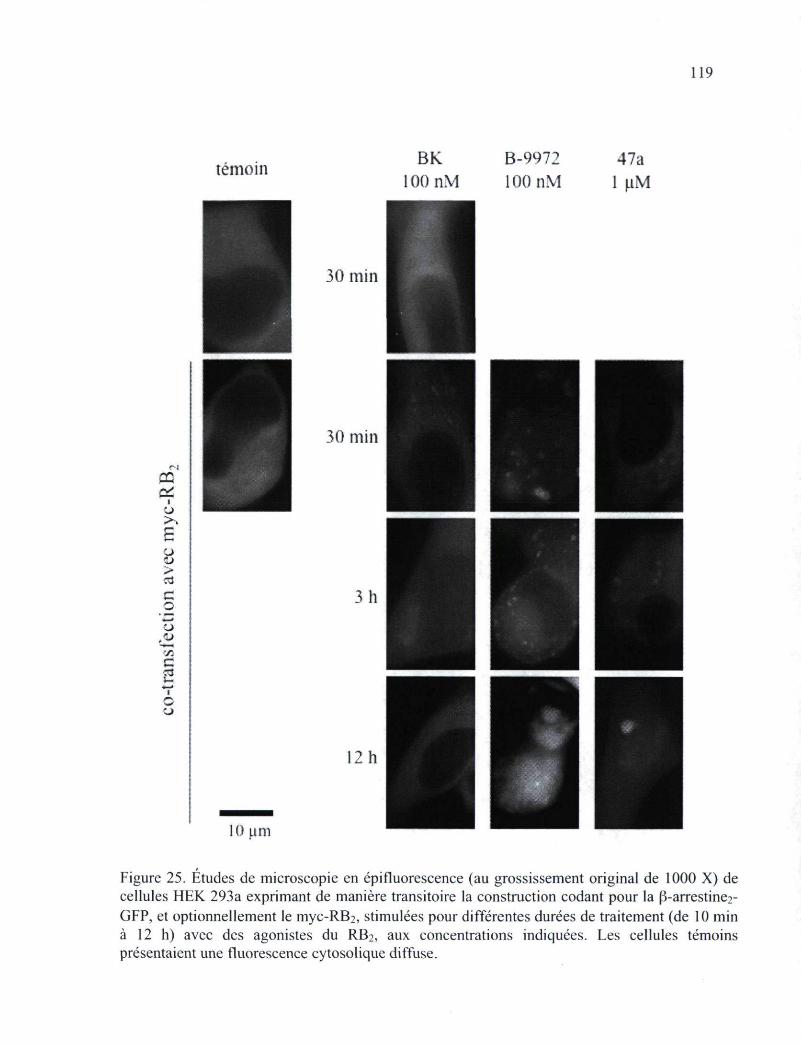
119
témoin
m > z
o >
O
y.
< — ^^
2 ■i
BK .00 nM
B-9972 100 nM
47a 1 ^M
30 min
30 min
3 h
12 h
10 pm
Figure 25. Études de microscopie en épifluorescence (au grossissement original de 1000 X) de cellules HEK 293a exprimant de manière transitoire la construction codant pour la P-arrestine2-
GFP, et optionnellement le myc-RB2, stimulées pour différentes durées de traitement (de 10 min à 12 h) avec des agonistes du RB2, aux concentrations indiquées. Les cellules témoins présentaient une fluorescence cytosolique diffuse.
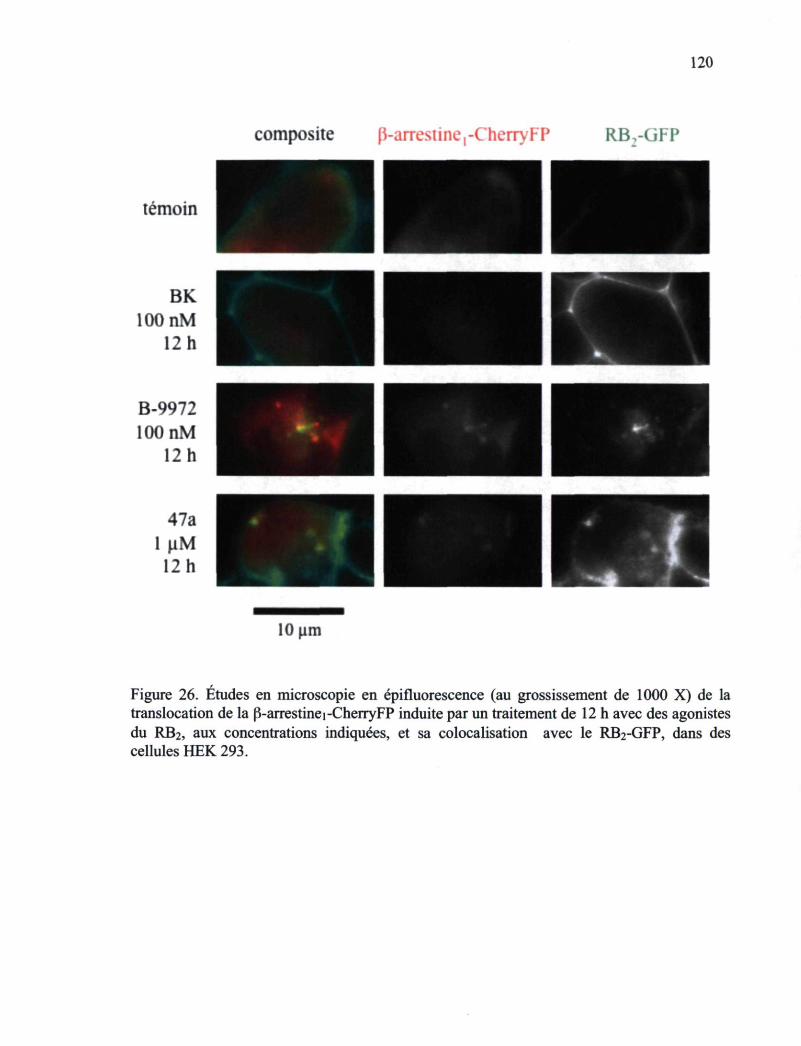
120
composite p-arrestine,-Cherry FP RB.-GFP
témoin
BK 100 nM
12 h
B-9972 100 nM
12 h
47a 1 u M 12h
10 um
Figure 26. Études en microscopie en épifluorescence (au grossissement de 1000 X) de la translocation de la p-arrestine i-CherryFP induite par un traitement de 12 h avec des agonistes du RB2, aux concentrations indiquées, et sa colocalisation avec le RB2-GFP, dans des cellules HEK 293.

121
de 101 kDa et par l'apparition de metabolites du récepteur de la taille de la GFP, une
globuline stable de 27 kDa, grâce à des immunobuvardages exploitant les anticorps anti-
GFP. Tout comme pour l'agoniste peptidique B-9972, la figure 27 montre que le composé
47a, à la concentration de 1 pM, agit comme un agoniste persistant et provoque la
dégradation du récepteur, quoique moins complètement que le peptide B-9972, dont
l'action n'est pas antagonisée par un cotraitement avec le composé 47a. La BK, l'agoniste
naturel du récepteur, est quant à elle inactive à cet égard. Comme pour les autres essais, un
co-traitement avec le LF 16-0687 inhibait l'action du composé 47a, au niveau de la
dégradation du récepteur.
3.3 Extension en N-terminal des ligands des récepteurs des kinines
3.3.1 Analogues de l'antagoniste B-9430
3.3.1.1 Profil pharmacologique d'analogues de l'antagoniste B-9430 prolongés en N-terminal
Le peptide B-10380 ne montrait aucun effet contractile sur la veine ombilicale humaine
fraîchement isolée, mais était un antagoniste compétitif de la contraction induite par la BK
(figure 28A), ce qui permet le calcul d'une valeur de pA2 de 6,83 ± 0,04, grâce à la courbe
de Schild (figure 28), dont la pente était de -1. Ne présentant pas d'activité contractile sur la
veine jugulaire de lapin, le peptide B-10380 se comportait en antagoniste puissant et
insurmontable de la BK (effet présenté en C de la figure 28), ce qui s'apparente à l'activité
de son peptide parent B-9430, ainsi qu'à d'autres antagonistes peptidiques à la structure
contrainte (Houle et coll., 2000, Morissette et coll., 2007) au niveau de RB2 de lapin.
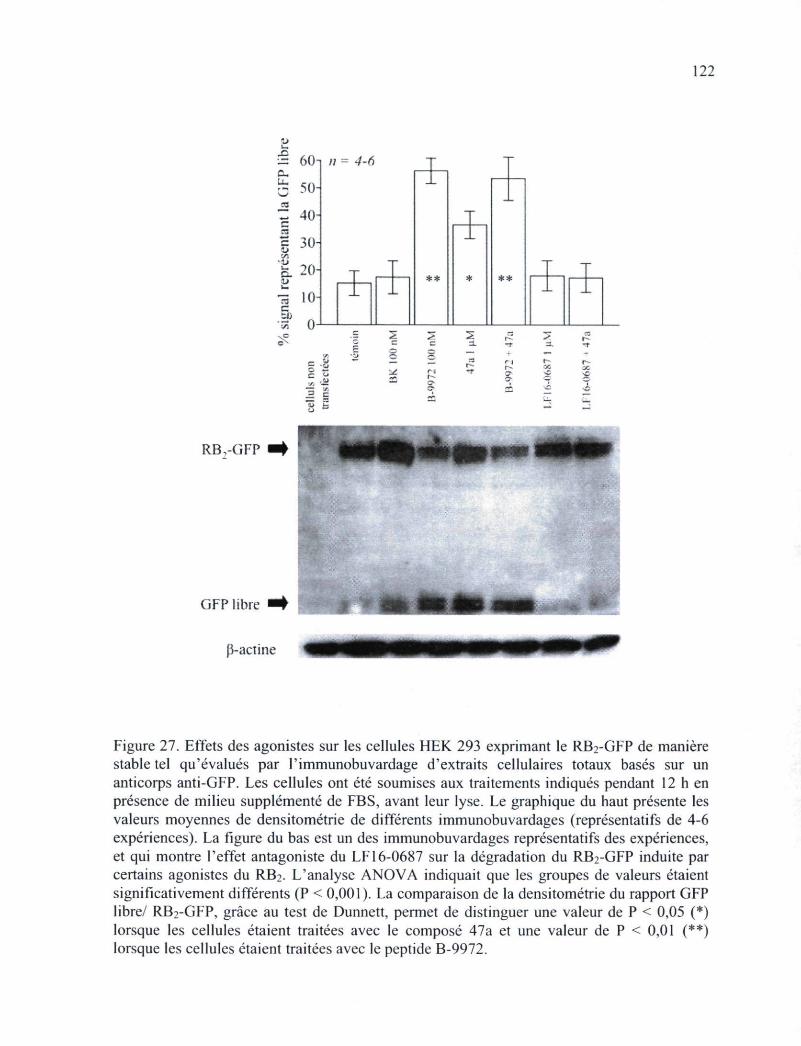
122
•§ 60
S 50i Z 40H
g 30H Cfl
I2
■a 101 5 c
n = 46 i
** i
il 1 ë
RB,GFP .Jk■>
GFP libre
pactine
Figure 27. Effets des agonistes sur les cellules HEK 293 exprimant le RB2GFP de manière stable tel qu'évalués par F immunobuvardage d'extraits cellulaires totaux basés sur un anticorps antiGFP. Les cellules ont été soumises aux traitements indiqués pendant 12 h en présence de milieu supplémenté de FBS, avant leur lyse. Le graphique du haut présente les valeurs moyennes de densitométrie de différents immunobuvardages (représentatifs de 46 expériences). La figure du bas est un des immunobuvardages représentatifs des expériences, et qui montre l'effet antagoniste du LF 160687 sur la dégradation du RB2GFP induite par certains agonistes du RB2. L'analyse ANOVA indiquait que les groupes de valeurs étaient significativement différents (P < 0,001). La comparaison de la densitométrie du rapport GFP libre/ RB2GFP, grâce au test de Dunnett, permet de distinguer une valeur de P < 0,05 (*) lorsque les cellules étaient traitées avec le composé 47a et une valeur de P < 0,01 (**) lorsque les cellules étaient traitées avec le peptide B9972.
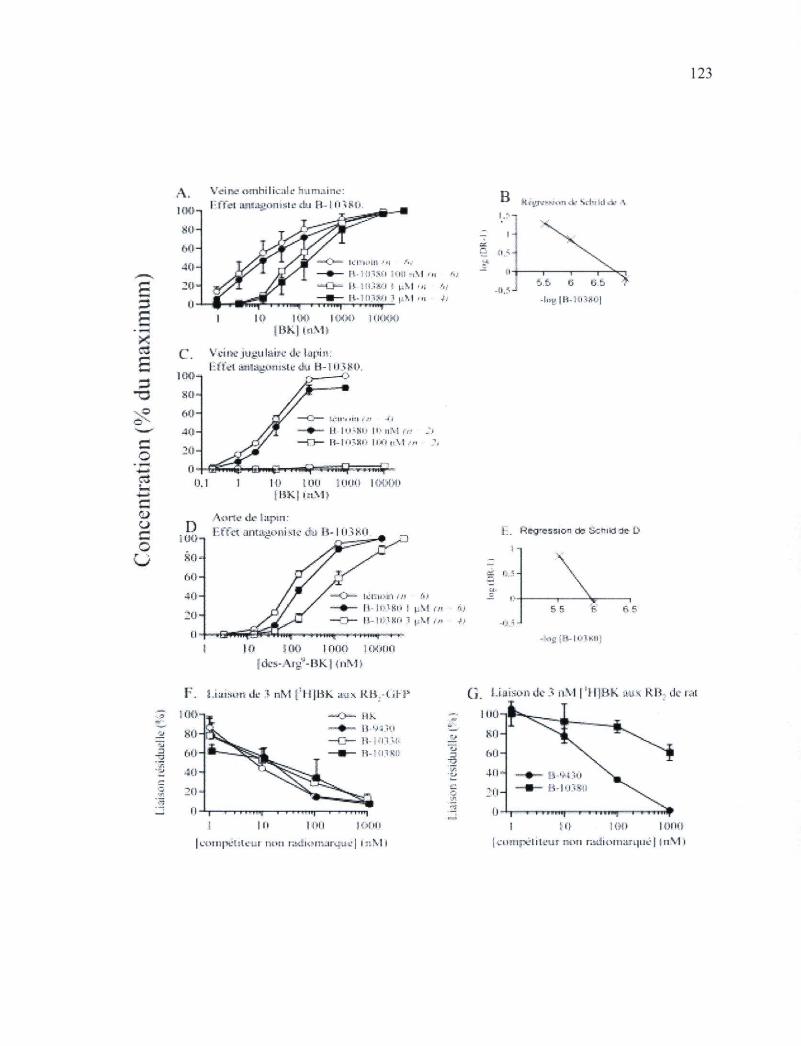
123
!
%
§
!
s O
y^ Veine ombilicale humaine: l'tTet inuagiMiisIedu BlOYRi).
leinmti 'u B K I 3 M I K i t i n M m «rf i l 1«388 î |tM >>: t» B10380 3 vM m *.' I I l>WH ■ I l l l l i j — I I | W ^
HJ 100 IOOO ]0<«>ft |BK| ( t tM>
C . VritcKjugulaire de lapin: Effet «î^os t tk l t ' i k i BUl.ïHO.
U'it'IIH t." H lo.«»0 lOnMrs :« 810380 lOOuMrn ?j
H) t OU 1UOO I<H>SI!,I fBK] («M»
D Aorte d«? t i ipn :
, Vf. Effet antajiom ste eu B1(G KO ,
so
l i Kw.r^**.!^ll vk S i H k i c i ' A
■—O™ Iticîijxsïi i JI fi,"
—o— n)i>.w> 3 nM ,v ^
io too looo KMjiiri fdcsAfg
vBK]0iM)
1.5
ac
— i l
u.: l û j . [ B W 3 S 0 |
s
£. Regression de Schild un D
! «1
.icE!Hli>lKiij
a —
F. tiuàson de 3 nM f HjBK. aux RB,~GPf*
l O O l . O — HK * B.W30 C — BNOifi
T ! 1 I I > IWjj ,—I ' » l " l l |
III 100 I I 'm
(M m
G. Liaison dc 3 tuM | '1 l]BiK twx RB, de rat
100
4 I _
| compétiteur non radiontarqfUÉ] (nM)
i | ' * » f * » « i | f i i f v i K f — — T — n m i ^
I 10 100 1000 [ciimfpt'lileur min rudiomurqué 14riVI i

124
Figure 28. Profil pharmacologique du composé B-10380 au niveau d'essais contractiles pour le RB2 (A-C) et les RBi (D, E) et des essais de liaison du radioligand du RB2 (F, G). En A, l'effet antagoniste du peptide B-10380 au niveau de la contraction induite par la BK, via la stimulation des RB2 exprimés par la veine ombilicale humaine. En B, la régression de Schild dérivée des courbes présentées en A. En C, le caractère insurmontable du B-10380 comme antagoniste au niveau du RB2 de lapin, tel qu'étudié grâce à l'emploi de la veine jugulaire de lapin. En D, effet antagoniste du peptide B-10380 sur la contraction de l'aorte de lapin, un essai pour le RBi, stimulée par la des-Arg -BK. En E, la régression de Schild dérivée des courbes présentées en D. Les valeurs présentées en A, C, D sont des moyennes ± l'erreur type du nombre de déterminations donné par n. En F, la compétition de la liaison de [3H]BK (3 nM) aux RB2-GFP exprimés par les cellules par un ensemble d'analogues de kinines non radiomarqués. Les valeurs présentées sont des moyennes ± l'erreur type de 2-8 expériences répliquées, dont chacune était composée de deux déterminations. Certaines des valeurs répliquées (obtenues de la même façon) et déjà publiées ont été amalgamées aux nouveaux résultats. La liaison spécifique moyenne du radioligand en absence d'un compétiteur (liaison donnant 100 % de liaison) était de 116 ± 20 fmol/ puits. En G, la compétition de la liaison de la [3H]BK (3 nM) aux cellules de muscle lisse d'aorte de rat exprimant des RB2 endogènes, par un ensemble d'analogues des kinines. Les valeurs présentées sont des moyennes ± l'erreur type de deux expériences composées de deux déterminations chacune. La liaison spécifique moyenne en absence de compétiteur (100 % de liaison) est de 12,5 fmol/puits.

125
Les tissus traités avec le B-10380, à une concentration de 100 nM, montraient un effet
maximal résiduel très faible pour la BK, mais répondaient fortement à l'histamine (données
non montrées), un agent contractile puissant et montraient ainsi la préservation de leurs
fonctions contractiles malgré les manipulations subies. Comme le peptide parent B-9430, le
peptide B-10380 retient une certaine affinité pour le RB|, tel que le montre un essai de
contractilité réalisé grâce à l'utilisation de l'aorte de lapin fraîchement isolée, un essai
fonctionnel du RB]. Il agit comme un antagoniste compétitif en provoquant un déplacement
de la courbe concentration-réponse construite par la BK vers la droite, déplacement
proportionnel à la concentration d'antagoniste ajoutée (de 0,31 à 3 pM; données montrées à
la figure 28, en D). Son identité d'antagoniste surmontable permet le calcul d'une valeur de
pA2 égale à 5,95 (la régression de Schild associée est présentée à la figure 28E). Tous ces
essais fonctionnels ont révélé une perte d'activité du peptide B-10380, par rapport à son
parent, le B-9430; la perte de puissance observée était d'un facteur 7.4, 10 et 3.2
comparativement au précédent, au niveau de la veine ombilicale humaine, la veine jugulaire
de lapin et l'aorte de lapin, respectivement.
Dans des cellules HEK 293 exprimant le RB2-GFP stablement, les composés B-9430, B-
10330 et B-10380 déplaçaient la liaison de la [3H]BK avec une puissance appréciable,
quoique les deux peptides comportant une extension N-terminale de la séquence du B-9430
présentent une affinité légèrement moins élevée que celle du peptide parent (figure 28, en
F). La BK produit une variété de réponses fonctionnelles au niveau des cellules de muscle
lisse d'aorte de rat, par la stimulation de RB2 endogènes (Velarde et coll., 1999). Bien que
le peptide B-9430 soit un compétiteur efficace de la liaison de la [3H]BK au niveau des RB2
de rat lors de la liaison d'essai de liaison dans ce système cellulaire (figure 28, en G), le
peptide B-10380 présentait quant à lui une puissance 35 fois inférieure.

126
3.3.1.2 Imagerie des RB2 exprimés par des cellules HEK 293a intactes
Des cellules transfectées avec le RB2 de type sauvage de lapin soumises au traitement
séquentiel du B-10330 (conjugué biotine) et de la streptavidine-Alexa fluor 594 n'ont pas
été marquées par la fluorescence de cette deuxième protéine (non montré). Pourtant, un
peptide ayant pratiquement une affinité identique, le B-10380, a permis l'imagerie directe
de cellules transfectées soit avec le RB2 de type sauvage de lapin ou le myc-RB2 (figure
29A). Le traitement avec le B-10380, à la concentration de 50 nM, était fait dans le milieu
de culture habituel des cellules. Suite à une incubation de 30 min, les cellules étaient
rincées et observées en épifluorescence. Le peptide fluorescent marquait la surface
cellulaire et possédait une distribution lisse chez les cellules qui étaient transfectées. Les
images obtenues en microscopie se trouvent à la figure 29, en A. Comme cela peut être
attendu lors d'une transfection transitoire, l'intensité du marquage varie largement d'une
cellule à une autre, puisque toutes les cellules n'étaient pas marquées, ni marquées
également. Le marquage des cellules exprimant des récepteurs recombinants de manière
transitoire était observable avec une concentration de l'antagoniste B-10380 aussi faible
que 5 nM. Le marquage de ces cellules était fortement réduit par un cotraitement avec
l'antagoniste LF16-0687 (Houle et coll., 2000). Les cellules non transfectées ne
présentaient aucun marquage non spécifique par le B-10380. Par ailleurs,
l'autofluorescence de ces cellules était faible, et l'antagoniste fluorescent B-10380 (à la
concentration de 50 nM) ne marquait pas les cellules exprimant de manière transitoire ni la
construction RBi. de type sauvage de lapin, ni la construction hRBi-FLAG (figure 29, B).
3.3.2 Ligands fluorescents du RB]
3.3.2.1 Caractérisation pharmacologique des nouveaux ligands par la réalisation d'essais de liaison
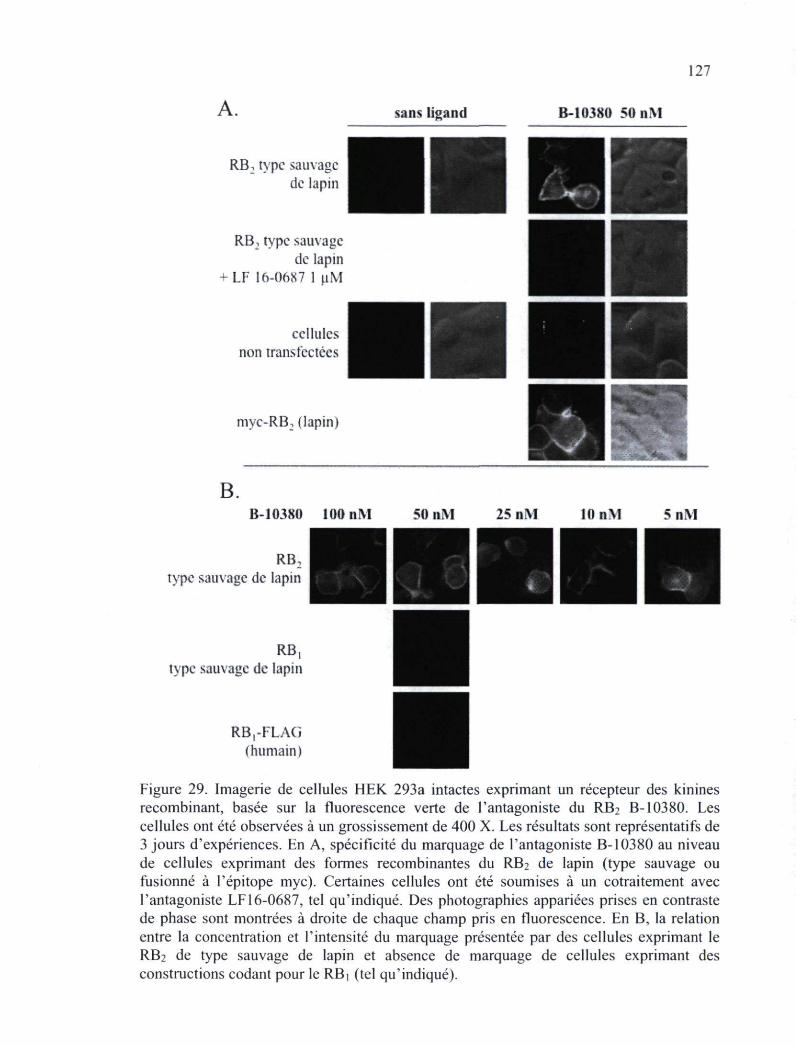
127
A. sans ligand B-10380 50 nM
RB: type sauvage de lapin
RB, type sauvage de lapin
+ LF 16-0687 1 pM
cellules non transfectées
myc-RB, (lapin)
, ^ 1
#
\j/r 1
B. B-10380 100 nM 50 nM 25 nM 10 nM 5 nM
RB2 type sauvage de lapin
RB, type sauvage de lapin
RBrFLAG (humain)
Figure 29. Imagerie de cellules HEK 293a intactes exprimant un récepteur des kinines recombinant, basée sur la fluorescence verte de l'antagoniste du RB2 B-10380. Les cellules ont été observées à un grossissement de 400 X. Les résultats sont représentatifs de 3 jours d'expériences. En A, spécificité du marquage de l'antagoniste B-10380 au niveau de cellules exprimant des formes recombinantes du RB2 de lapin (type sauvage ou fusionné à l'épitope myc). Certaines cellules ont été soumises à un cotraitement avec l'antagoniste LF 16-0687, tel qu'indiqué. Des photographies appariées prises en contraste de phase sont montrées à droite de chaque champ pris en fluorescence. En B, la relation entre la concentration et l'intensité du marquage présentée par des cellules exprimant le RB2 de type sauvage de lapin et absence de marquage de cellules exprimant des constructions codant pour le RBi (tel qu'indiqué).

128
Le déplacement de la [ H]Lys-des-Arg -BK aux récepteurs recombinants humain (hRBi-
FLAG; Morissette et coll., 2008) et de lapin (RB| de type sauvage de lapin) exprimés de
manière transitoire a servi à évaluer les affinités des ligands fluorescents pour ce sous-type
de récepteur dans chaque espèce. L'agoniste de référence, la Lys-des-Arg9-BK, dont dérive
le peptide B-10378, et l'antagoniste B-9958, le peptide parent des analogues fluorescents
B-10372 et B-10376, sont des compétiteurs très puissants du radioligand, possédant des
valeurs de Kj d'ordre nanomolaire, selon les données présentées dans le tableau 2. Les
analogues fluorescents qui en dérivent sont beaucoup moins puissants à cet égard, ce qui
indique que l'ajout du fluorophore, avec ou sans l'espaceur e-ACA, a un effet négatif sur
l'affinité du ligand pour le récepteur. Ainsi, les antagonistes B-10372 et B-10376, qui
possèdent des puissances très similaires pour le RBi de chaque espèce, présentaient une
puissance de 5 à 6 fois moindre que le peptide B-9958 pour le récepteur de lapin, mais une
diminution de 32 fois par rapport au peptide parent au niveau du récepteur humain.
L'agoniste B-10378 était 24 fois moins puissant que la Lys-des-Arg9-BK au récepteur
lapin, mais présente une perte d'affinité d'un facteur de 200, au niveau du récepteur humain
(figure 30; tableau 2).
3.3.2.2 Évaluation fonctionnelle de nouveaux ligands fluorescents par les essais de contractilité
Des anneaux d'aorte de lapin préincubés in vitro montrent une réponse contractile aux
kinines sélectives pour le RB|, ce qui fait que ce système expérimental se prête bien à la
caractérisation d'antagonistes et d'agonistes de ce sous-type de récepteur (résultats
présentés à la figure 31). Les dérivés fluorescents de l'antagoniste B-9958, les peptides B-
10372 et B-10376, ne présentaient aucun effet stimulant direct sur la préparation vasculaire
exploitée, mais antagonisaient de manière compétitive l'agoniste conventionnel des-Arg -
BK avec une puissance similaire, puisque les valeurs de pA2 estimées (grâce à la
construction de courbes de régression de Schild montrées à la figure 31, en E, dont les
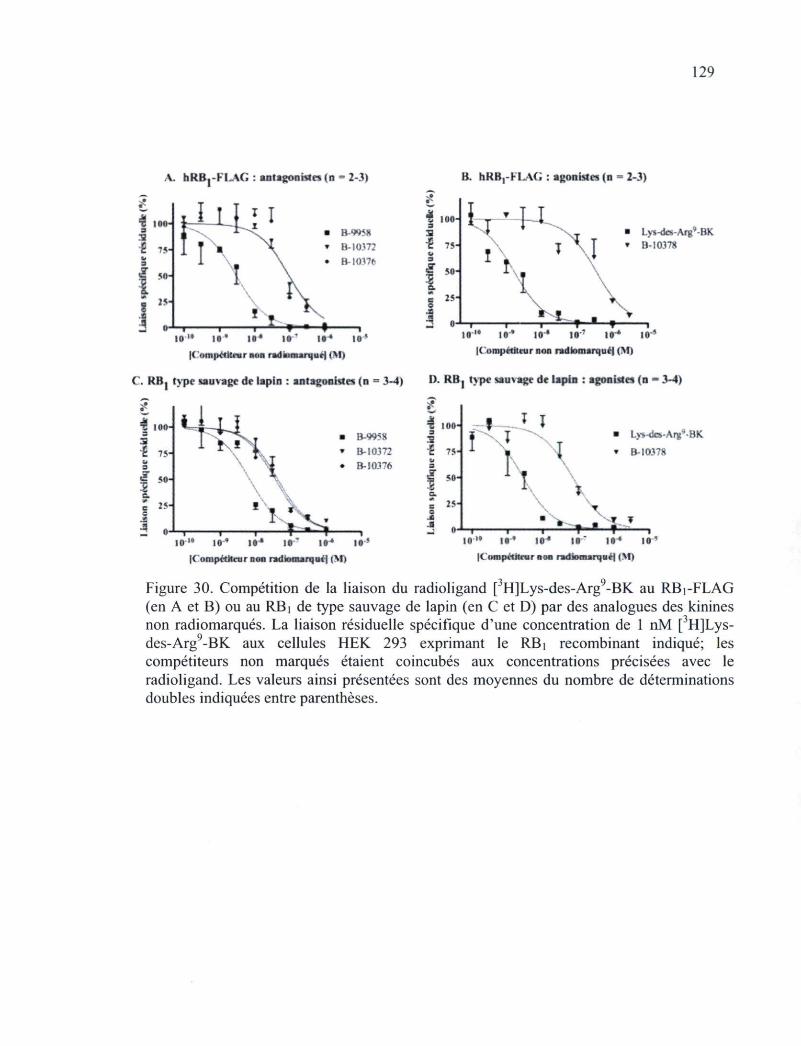
129
A. h R B , H _ \ < ; : antagonivliMn = 23)
I t i n
|( ompétHeor non r»il»im*rnur| Of)
C. RB. type sauvage de lapin : antagonistes (n = 34)
i f 100s S l 75u a i soi 5 &
S o
K1
\
■ BW58 » B10372 ♦ 010376
tS
h nr" \ f i f i f tr* io!
[Compétiteur non radiununjut j (M)
^
B. hKB,KLA<; : agonistes (n = 23)
% mues
t »■
50
Î5
trM. ■ Lys<JesArg''BK » B10378
t r
■ T I
H ) ' T « t
t a " i o ' \ f i f i f |( ompititiur non radtomari|Uii| (M)
1 i n '
D. RBj type sauvage de lapin : agonistes (n = 34)
? i io»' a 3 'Ê 75'
50
25
■ Lytv<ksArg*BK » B10378
\ i ■ M r w r n p i M i i ' i
ie* tr" ir* |t ompctitnir nan m<J>oman)in'| (M)
— I
l i . l f
Figure 30. Compétition de la liaison du radioligand [3H]LysdesArg9BK au RBiFLAG (en A et B) ou au RBi de type sauvage de lapin (en C et D) par des analogues des kinines non radiomarqués. La liaison résiduelle spécifique d'une concentration de 1 nM [3H]Lys
desArg BK aux cellules HEK 293 exprimant le RBi recombinant indiqué; les compétiteurs non marqués étaient coincubés aux concentrations précisées avec le radioligand. Les valeurs ainsi présentées sont des moyennes du nombre de déterminations doubles indiquées entre parenthèses.
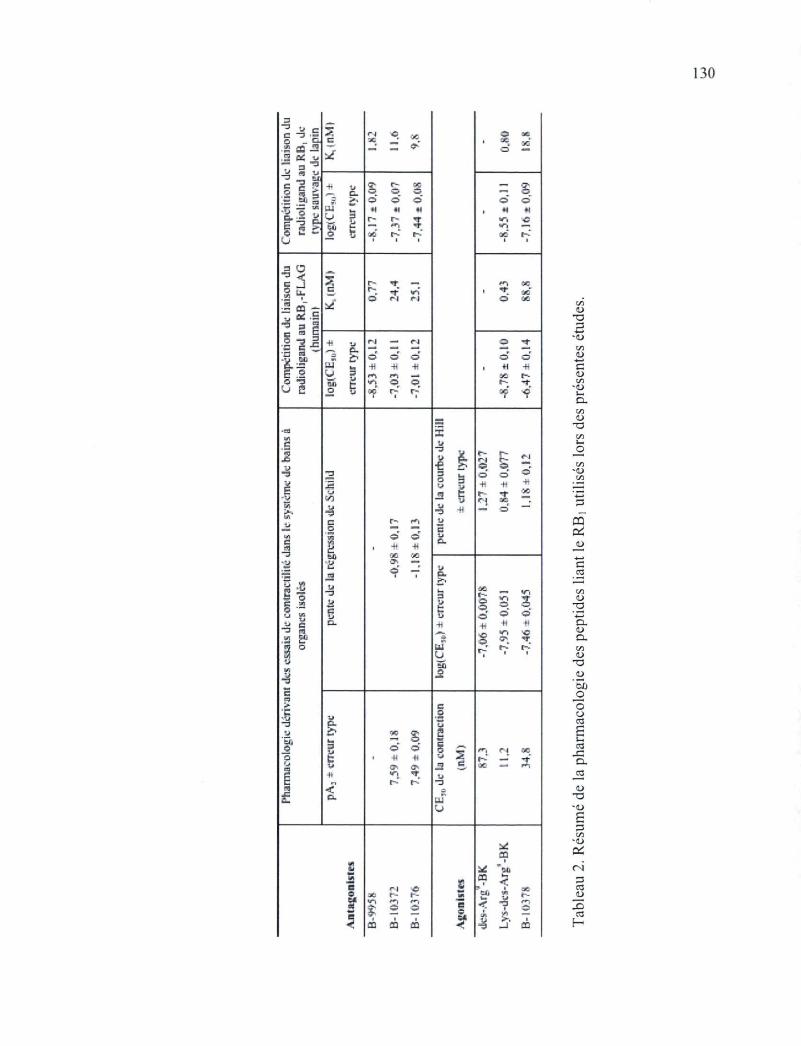
130
a ■3 y c
«N « £ 0 c= a c p _ C l s as —
c >C| i ^ '
■ JS.
cr n ,3 d a
c
û a f J I y ™ —. fi c £ r> 1 II * S —' SE S c £ r> 1 II S >. 9
; o O o
I l S ill
14 U i • i
r• i
r« i • • i
zr, ♦ i
I l S ill 1 t i
z r .
t" U i ■ i
5 ° 7, n
ri
T
<rt • T ■0 mi
. 2 1 . =>' 1 M ssssl M
B «j c
*
B «j c
■B 'S c 1H iL
1 — C J • t
il H c ' d o' d o' il pu ta + H H , ♦1 H
Ë S u 3 ". 1*1 y c 1
o s CCI.
J5 Ë ■ r , es
r3 t
KJ M x c • J
J SS!
I i s. rf 4
rr C 4
S •^ •E ccc; 5
o D © '
i 1 S?
3 o •y
5 «■ H H
i 1 S?
u»
w J t ■i
r.. 1 X i i
1 S? « 4j M
r..
o ~" D C r r , u n H «a I i
* ■H
© '
S. t i j S OC
•JJ
J 3
A « H *
J 3 * —' 1 i .fi Q Mî
3 U
5 u
M
•' «n
o .8 ^ D. H + •H •H "3 « a s
1 ^Z s; l p * .
j ° ui U r r"
i
u r j . w>
j C C 15 j r C
i " 3
.2
S U W
i S o
.2
S i S
r ; SC
y ta
v ■
rp.. I I i
S 1 — X
H r j *r Z)
fc ., t' r 3 H < z
ë c
Mi ap
1 ^ 'at •S 1
m s <
s a i JS
1 m B? i
i r
1 2
i S
f i
e m
i <
i
CA,
CA. CU
C CU Cfl
' 5 J
«a eu
o
zr, ■eu c/5
X ZC
.5 zr eu
72 D eu Q. «5 eu o
c eu C3
rej
= 2. eu
3 •y:
•eu Cri ci 3 r3
JU J ^ c : H
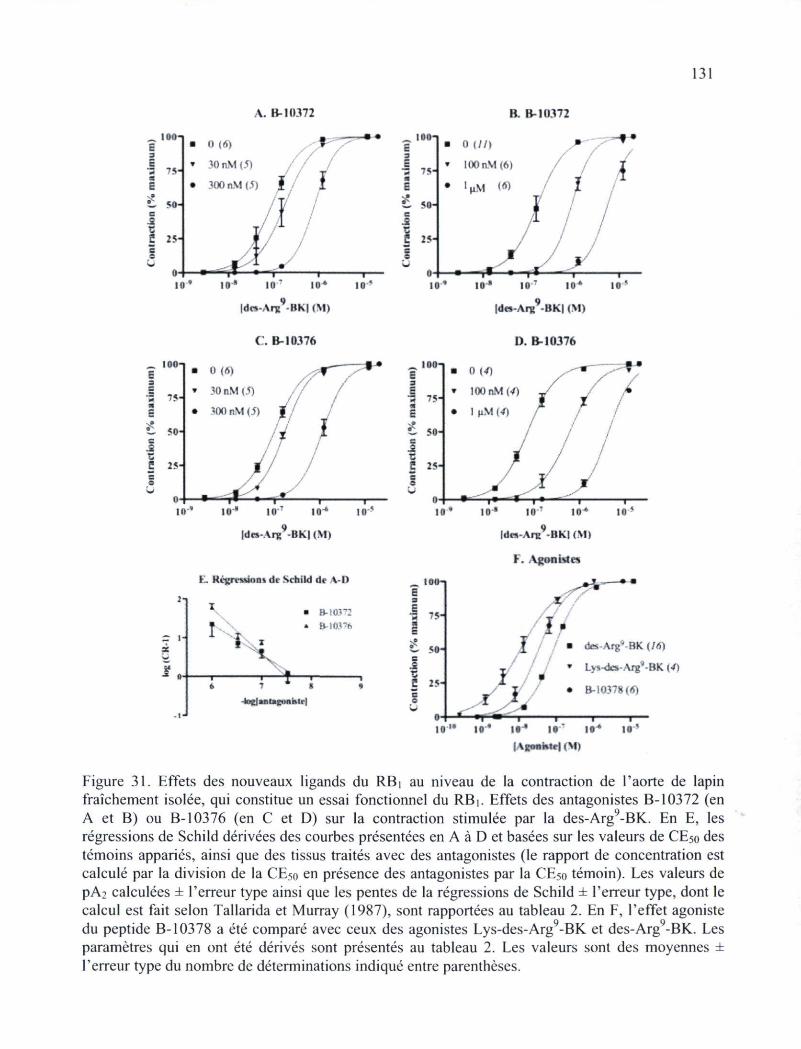
131
A. B10372
i9»n = .1 =
TS
0 (6)
» 30nM{5)
300 nM (5)
!_ S0=
25
IdesArg BK| (M)
G B10376
|d*sArjj BK| (M)
D. B10376
_ 1001
g 1
I I i i a
■ 0(6)
» 30nM(5)
• .300 DM (5) J' /
100'
13 75 i
£ 50H i 2 2SH
■ 0 {4)
» lOOniMK)
• I nM (/) / / •
X I » llll %
|d«s Are BK| (M)
1 KtviiNMi.tiN iti S c h i l d d c X I )
■ B10372
HI
_ io<N
■ 3
1 HH
C 50e j 9
r * — * — i — 10J IO7 10*
|d<sAre9BK| (M)
F. Agonistes
IO'»
6 T i loglantagonhtr]
25
2k
j m <k*Arg*BK (16)
♦ Lvsdes Arg*BK (4)
B10378 (IS)
i a«» io* tr* t r* i r * I \goafete) (Vf)
I i '
Figure 31. Effets des nouveaux ligands du RBi au niveau de la contraction de l'aorte de lapin fraîchement isolée, qui constitue un essai fonctionnel du RBi.. Effets des antagonistes B10372 (en A et B) ou B10376 (en C et D) sur la contraction stimulée par la desArg9BK. En E, les régressions de Schild dérivées des courbes présentées en A à D et basées sur les valeurs de CE50 des témoins appariés, ainsi que des tissus traités avec des antagonistes (le rapport de concentration est calculé par la division de la CE50 en présence des antagonistes par la CE50 témoin). Les valeurs de PA2 calculées ± l'erreur type ainsi que les pentes de la régressions de Schild ± l'erreur type, dont le calcul est fait selon Tallarida et Murray (1987), sont rapportées au tableau 2. En F, l'effet agoniste du peptide B10378 a été comparé avec ceux des agonistes LysdesArg BK et desArg BK. Les paramètres qui en ont été dérivés sont présentés au tableau 2. Les valeurs sont des moyennes ± l'erreur type du nombre de déterminations indiqué entre parenthèses.

132
pentes s'approchent de l'unité) pour chacun sont de 7,59 pour le B-10372 et de 7,49 pour le
B-10376. Le peptide parent B-9958, un antagoniste compétitif, est plus puissant que les
deux analogues fluorescents, présentant une valeur de pA2 de 8,27 (la valeur de pA2 s'élève
à 9,11 en présence de l'amasatine, un inhibiteur de l'aminopeptidase N, telle que décrit par
Géra et collaborateurs, 2006). Le peptide B-10378 est un agoniste complet 3 fois moins
puissant que le peptide parent, Lys-des-Arg -BK, mais 2,5 fois plus puissant que le peptide
naturel des-Arg -BK, comme le montrent les courbes concentration-réponse présentées en
F de la figure 31. Ces données suggèrent que l'extension de la séquence de l'agoniste Lys-
des-Arg9-BK, permettant l'introduction d'un fluorophore, ne nuit que partiellement au gain
d'affinité conféré par la présence du résidu lysine en position 0 de l'agoniste, pour le RBi
de lapin. Dans les essais de contractilité, la pente de Hill expérimentale de la courbe
concentration-effet construite à l'aide de des-Arg9-BK, de 1,27 est similaire à celle de B-
10378, qui se chiffre à 1,18, mais celle de la Lys-des-Arg9-BK est inférieure, se chiffrant à
0,84. À titre d'hypothèse, cela peut s'expliquer par le fait que ce dernier peptide est
susceptible à la dégradation par l'aminopeptidase N, exprimée par l'aorte de lapin (Fortin et
coll., 2005), contrairement à la des-Arg9-BK et aux ligands protégés à leur extrémité N-
terminale.
3.3.2.3 Évaluation fonctionnelle des nouveaux ligands fluorescents par les essais de mobilisation calcique
Les effets des peptides fluorescents ont été évalués au niveau de la mobilisation calcique
secondaire à la stimulation du RB| exprimé par des cellules de muscle lisse d'artère
ombilicale humaine sont présentés à la figure 32. L'agoniste B-10378, à la concentration de
1 pM, reproduit l'effet d'une concentration de 10 nM de l'agoniste naturel Lys-des-Arg9-
BK. L'injection des peptides B-10372 et B-10376 n'induisait pas de mobilisation calcique,
à la concentration de 100 nM. Les concentrations utilisées pour chaque ligand ont été
choisies en fonction des concentrations efficaces à déplacer la liaison du radioligand des
RBi humains. Par contre, lorsque les peptides dérivés de l'antagoniste B-9958 étaient
ajoutés à la suspension cellulaire avant l'injection de 10 nM de l'agoniste naturel, ils
prévenaient l'effet stimulant de ce dernier. Ces expériences confirment l'identité
pharmacologique des trois nouveaux ligands conjugués à la CF du RB|.
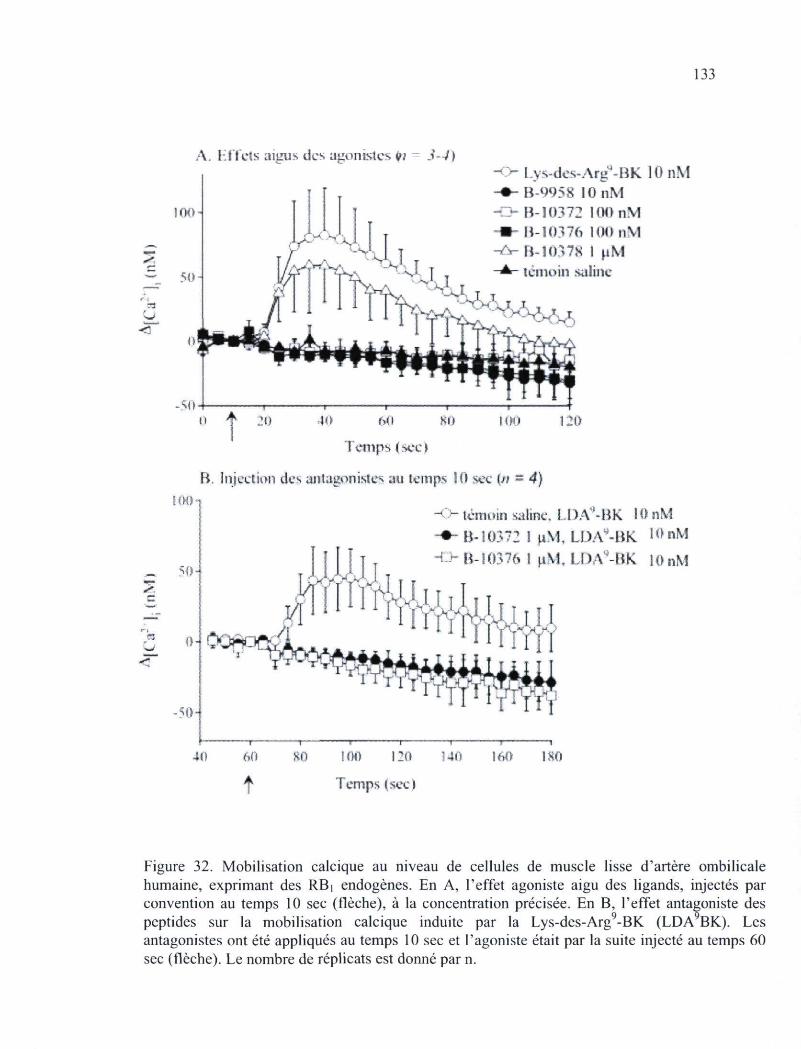
133
"
—
A. Effets aigus des agonistes ifi = 34) LysdesArg'BK 10 nM
• B9958 10 nM Z B10372 lOOnM ■B10376 lOOnM A~ B10378 1 pM * témoin saline
40 60 KO
Temps (S©c) loo 120
B. Injection des antagonistes au temps 10 sec (// = 4)
« j
I0CH
,. 50
SZ
50
O témoin saline, LDA^BK 10 nM • B10372 1 p\L LDÂ*BK 10 «M O B10376 1 pM, LDA*BK 10 nM
™%4to 40 60 S0 100 120 140 160 180
f Temps (sec)
Figure 32. Mobilisation calcique au niveau de cellules de muscle lisse d'artère ombilicale humaine, exprimant des RBi endogènes. En A, l'effet agoniste aigu des ligands, injectés par convention au temps 10 sec (flèche), à la concentration précisée. En B, l'effet antagoniste des peptides sur la mobilisation calcique induite par la LysdesArg BK (LDA BK). Les antagonistes ont été appliqués au temps 10 sec et l'agoniste était par la suite injecté au temps 60 sec (flèche). Le nombre de réplicats est donné par n.

134
3.3.2.4 Imagerie du RB( exprimé par des cellules HEK 293a
Des cellules HEK 293a exprimant de manière transitoire des RBi recombinants, hRB]-
FLAG ou RBi de type sauvage de lapin, ont été incubées en présence d'une concentration
uniforme de 50 nM de chacun des peptides fluorescents pour une période de 30 min à 37°C.
Après un lavage, elles ont été observées en microscopie à épifluorescence. Les antagonistes
B-10372 et B-10376 marquaient la membrane plasmique des cellules exprimant l'une ou
l'autre des constructions comportant le RBi. Un traitement concomitant avec l'antagoniste
de haute affinité non peptidique, le composé 11 (Morissette et coll., 2004), abolissait le
marquage des cellules exprimant les RBi recombinants. Les photographies sont présentées
à la figure 33. La transfection transitoire explique la grande variabilité de l'intensité du
marquage, puisque certaines cellules étaient fortement marquées, d'autres plus faiblement,
alors que d'autres encore, pas du tout. Les cellules transfectées avec le vecteur FLAG ne
contenant pas le récepteur ne présentaient aucune fluorescence. L'agoniste du RBi B-10378
permettait aussi l'observation de cellules exprimant soit le hRBi-FLAG, soit le RBi de type
sauvage de lapin, lorsqu'il était utilisé à la concentration de 50 nM, et incubé en présence
de cellules pendant 30 min. Un échantillon photographique des résultats obtenus est
présenté à la figure 34.
Ici encore, le co-traitement avec le composé 11 inhibait le marquage des cellules exprimant
les récepteurs. La fluorescence présentée par les cellules transfectées mais non traitées avec
les ligands fluorescents ou encore par des cellules non transfectées soumises au traitement
avec l'un ou l'autre des ligands fluorescents était négligeable.
L'antagoniste fluorescent B-10376 permettait l'observation de cellules exprimant l'une des
deux constructions de RB| aux concentrations minimales de 5 à 10 nM (figure 35); ces
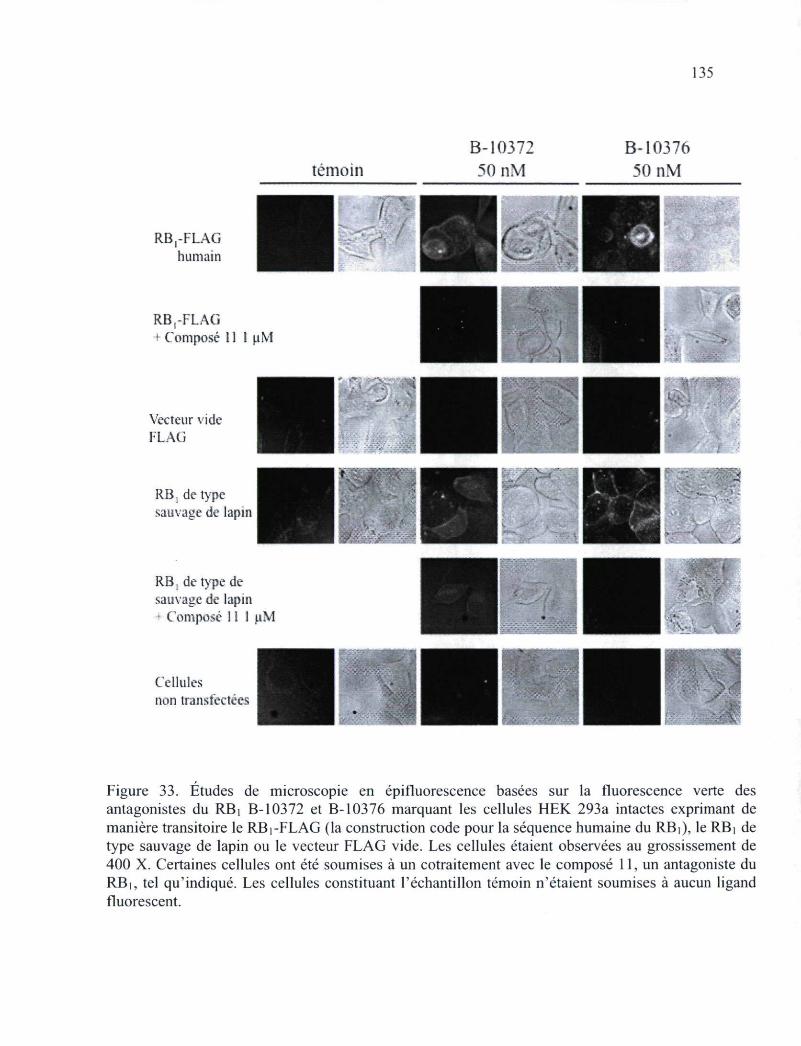
135
témoin B-10372 50 nM
B-10376 50 nM
RB.-FLAG humain
RB,-FLAG i Composé 11 1 pM
Vecteur vide FLAG
RB; de type sauvage de lapin
RB, de type de sauvage de lapin * Composé 11 1 uM
Cellules non transfectées
Figure 33. Études de microscopie en épifluorescence basées sur la fluorescence verte des antagonistes du RBi B-10372 et B-10376 marquant les cellules HEK 293a intactes exprimant de manière transitoire le RBi-FLAG (la construction code pour la séquence humaine du RBi), le RB| de type sauvage de lapin ou le vecteur FLAG vide. Les cellules étaient observées au grossissement de 400 X. Certaines cellules ont été soumises à un cotraitement avec le composé 11, un antagoniste du RBi, tel qu'indiqué. Les cellules constituant l'échantillon témoin n'étaient soumises à aucun ligand fluorescent.
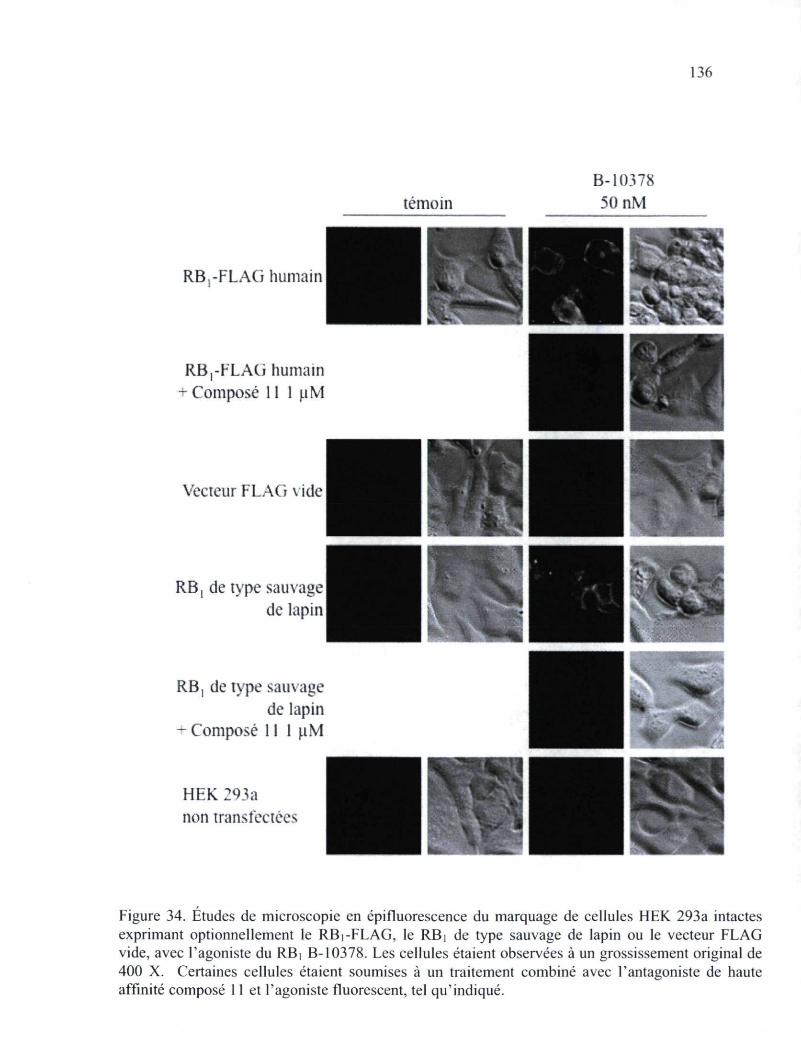
136
RB,-FLAG humain
RB,-FLAG humain -Compose 11 1 uM
Vecteur FLAG vide
RB, de type sauvage de lapin
RB, de type sauvage de lapin
+ Composé 11 1 uM
HEK 293a non transfectées
témoin B-10378 50 nM
:
ion
Figure 34. Études de microscopie en épifluorescence du marquage de cellules HEK 293a intactes exprimant optionnellement le RB|-FLAG, le RB| de type sauvage de lapin ou le vecteur FLAG vide, avec l'agoniste du RBi B-10378. Les cellules étaient observées à un grossissement original de 400 X. Certaines cellules étaient soumises à un traitement combiné avec l'antagoniste de haute affinité composé 11 et l'agoniste fluorescent, tel qu'indiqué.
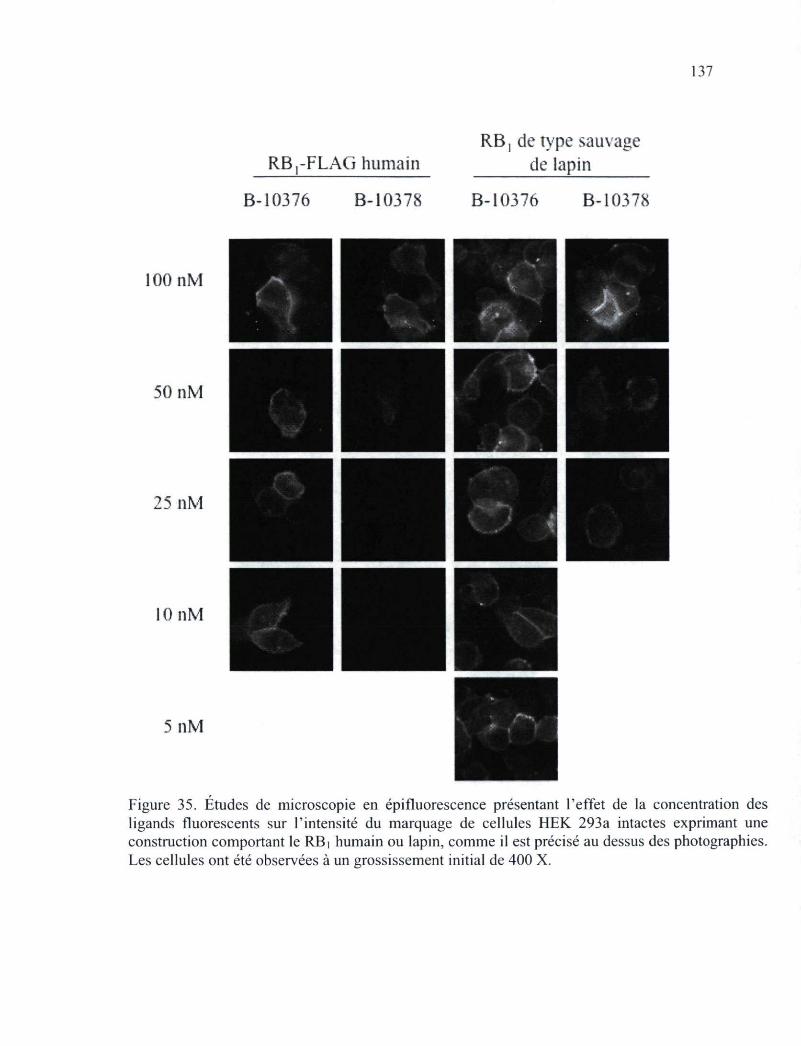
137
RB,-FLAG humain RB, de type sauvage
de lapin
B-10376 B-10378 B-10376 B-10378
100 nM
50 nM
25 nM
lOnM
5 nM
Figure 35. Études de microscopie en épifluorescence présentant l'effet de la concentration des ligands fluorescents sur l'intensité du marquage de cellules HEK 293a intactes exprimant une construction comportant le RB| humain ou lapin, comme il est précisé au dessus des photographies. Les cellules ont été observées à un grossissement initial de 400 X.

138
données sont cohérentes avec sa valeur de Ki pour ces récepteurs. À titre de comparaison, le
peptide B-10378 ne s'avérait pas aussi efficace, à des concentrations aussi basses, à
marquer les cellules exprimant les RBt recombinants, principalement le hRBi-FLAG.
Deux facteurs peuvent expliquer ce phénomène; d'abord la faible affinité de ce ligand pour
le RB) humain (se référer au tableau 2), puis, le fait qu'un changement de distribution du
RBi au niveau de la membrane plasmatique par la liaison de l'agoniste fluorescent rende
plus difficile l'observation du récepteur.
Afin d'estimer la validité de cette dernière hypothèse, des cellules HEK 293a ont été
transfectées de manière transitoire avec le RBi. de type sauvage de lapin ou le hRBi-FLAG,
et traitées avec l'un des ligands fluorescents. Elles ont ensuite été observées en microscopie
confocale afin d'étudier les différences qualitatives des patrons de marquage entre celui
présenté par l'agoniste B-10378 et celui obtenu par l'antagoniste B-10376. Un échantillon
des coupes confocales est montré à la figure 36. Les cellules non transfectées ont montré
une liaison très faible des peptides. Les cellules exprimant l'une ou l'autre des
constructions portant le RBi de l'espèce humaine ou de lapin, montraient une fluorescence
associée à leur membrane d'une intensité plus élevée que l'autofluorescence. Les cellules
exprimant le RBi de type sauvage de lapin ou le hRBi-FLAG et traitées avec l'antagoniste
B-10376, à une concentration de 20 nM, ont été observées à proximité du plan equatorial.
Elles offraient à l'observation un signal membranaire lisse et continu chez 58,6 % des
cellules marquées exprimant le récepteur lapin et chez 34,0 % des cellules marquées
exprimant le récepteur humain. La proportion de cellules présentant un signal membranaire
continu était significativement plus faible, tel qu'évalué par le test du %2, lorsque les
cellules exprimant un ou l'autre des RBi. recombinants exploités dans ces expériences
étaient exposées à l'agoniste B-10378 (20 nM ou 50 nM, indifféremment); le signal
fluorescent se présentait sous forme d'une condensation plus ponctuée, et plusieurs de ces
points fluorescents se situaient juste sous la membrane plasmatique (les données
photographiques sont présentées à la figure 36). Cette redistribution du signal fluorescent
rappelle la translocation du RB|-YFP, en réponse à son agoniste Lys-des-Arg9-BK,
observée par Sabourin et collaborateurs (2002). Ces différences dans l'apparence du
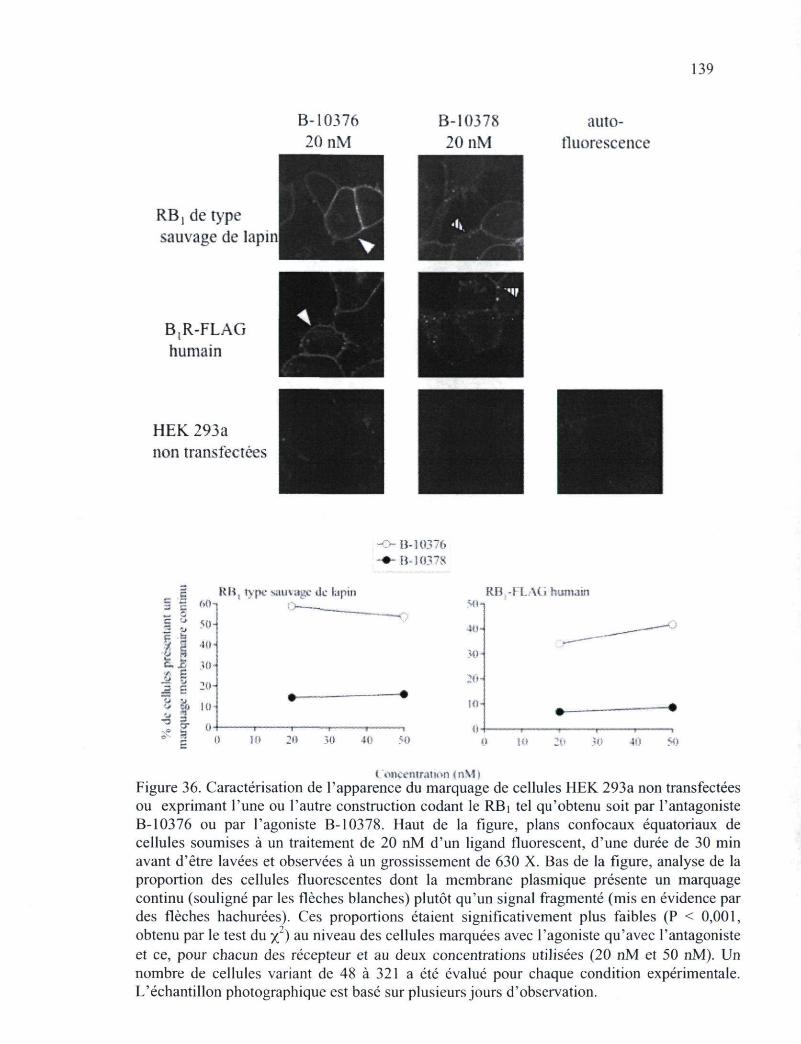
139
B10376 20 nM
B10378 20 nM
auto
fluorescence
RB | de type sauvage de lapin
BjRFLAG humain
HEK 293a non transfectées
OB10376 "♦•B1037B
RR, type sauvage dc lapin RB.4L Ad hiwi.icn
m ■ »
tfl
0 2» \n u 50 H ll'l M l
CoiKi?niraiJi»n (nM i Figure 36. Caractérisation de l'apparence du marquage de cellules HEK 293a non transfectées ou exprimant l'une ou l'autre construction codant le RBi. tel qu'obtenu soit par l'antagoniste B10376 ou par l'agoniste B10378. Haut de la figure, plans confocaux équatoriaux de cellules soumises à un traitement de 20 nM d'un ligand fluorescent, d'une durée de 30 min avant d'être lavées et observées à un grossissement de 630 X. Bas de la figure, analyse de la proportion des cellules fluorescentes dont la membrane plasmique présente un marquage continu (souligné par les flèches blanches) plutôt qu'un signal fragmenté (mis en évidence par des flèches hachurées). Ces proportions étaient significativement plus faibles (P < 0,001, obtenu par le test du x2) au niveau des cellules marquées avec l'agoniste qu'avec l'antagoniste et ce, pour chacun des récepteur et au deux concentrations utilisées (20 nM et 50 nM). Un nombre de cellules variant de 48 à 321 a été évalué pour chaque condition expérimentale. L'échantillon photographique est basé sur plusieurs jours d'observation.

140
marquage obtenu avec l'antagoniste et l'agoniste demeurent importantes et significatives
lorsque la concentration des ligands est élevée à 50 nM, l'agoniste B-10378 menant
toujours à une distribution moins continue; ces données ne sont pas montrées. Les coupes
confocales des conditions témoins montrent une potentielle contribution de
l'autofluorescence des cellules au signal fluorescent total observé aux réglages employés, à
laquelle s'ajoute une certaine internalisation indépendante du récepteur des peptides
fluorescents.
Des reconstitutions tridimensionnelles ont été réalisées à partir de séries de coupes
confocales de cellules exprimant soit le FLAG-RB| humain ou le RBi de type sauvage de
lapin et traitées avec l'un ou l'autre des ligands fluorescents. Elles sont présentées à la
figure 37; il faut remarquer l'ajout d'une fausse coloration jaune ou bleue, indiquant
respectivement une fluorescence verte d'intensité faible ou élevée, respectivement. Les
cellules intactes exprimant les récepteurs humain et de lapin et marquées grâce à
l'antagoniste B-10376 présentaient une fluorescence membranaire lisse et homogène, alors
que le marquage associé à l'agoniste était significativement moins continu et présentait des
agrégats fluorescents à la membrane des cellules. Ces observations sont potentiellement
compatibles avec la migration du récepteur aux cavéoles.
Afin d'investiguer l'identité des structures où se condensent la fluorescence associée au
peptide B-10378, les cellules HEK 293a ont été cotransfectées de manière transitoire avec
une protéine de fusion conjuguant la cavéoline-1 avec une protéine rouge fluorescente, et le
RBi de type sauvage de lapin. Un échantillon photographique des résultats obtenus est
présenté à la figure 38. Le signal fluorescent rouge présentait une distribution variée au
niveau subcellulaire, étant présent à la membrane plasmique, ainsi qu'au niveau
cytosolique, toujours sous forme particulaire et granulaire, au sein des cellules témoins.
L'antagoniste B-10376, à la concentration de 20 nM, marquait la membrane plasmique de
manière généralement lisse dans les coupes confocales successives, alors que la
fluorescence verte associée à l'agoniste B-10378 était fréquemment localisée avec la
fluorescence rouge, au niveau de granules de taille irrégulière, à proximité de la membrane
plasmique des cellules traitées avec cet agoniste. Des flèches ajoutées sur les photographies
pointent des exemples de telles structures à la figure 38. Il est aussi possible que la
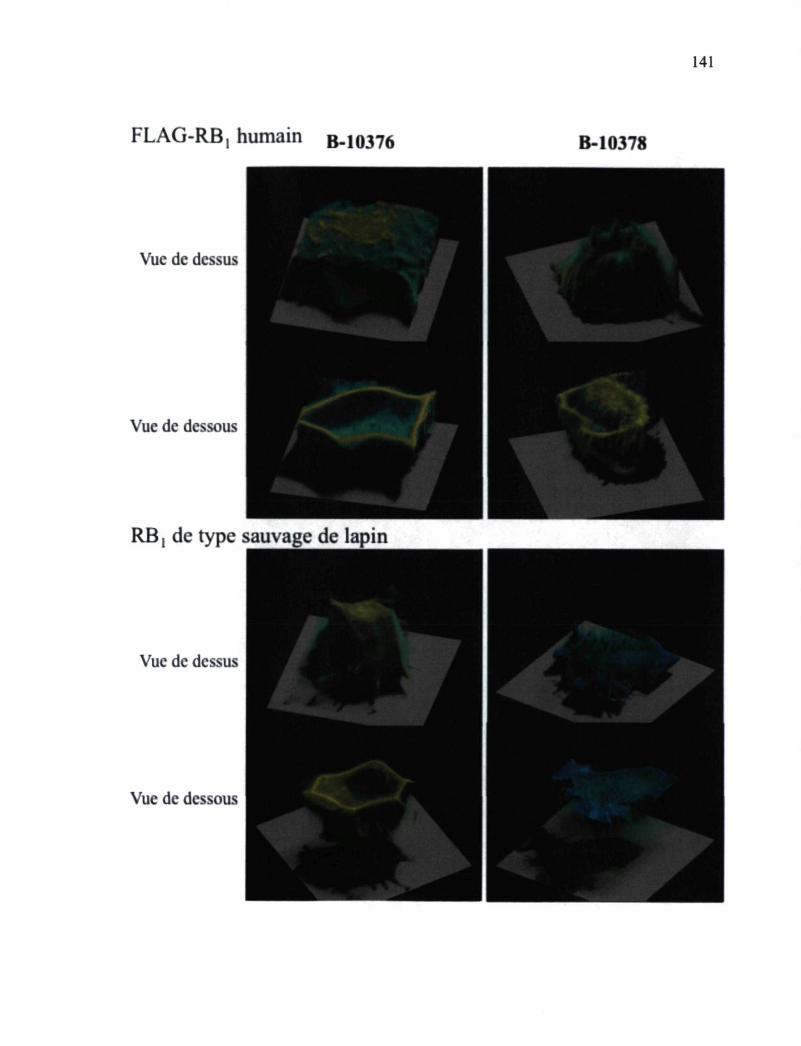
141
FLAG-RB, humain B-10376 B-10378
Vue dc dessus Ef
Vue dc dessous "*^^flfl / » 1
RB, de type sauvage de lapin
Vue de dessus
Vue dc dessous

142
Figure 37. Reconstitutions tridimensionnelles de cellules HEK 293a exprimant de manière transitoire des RBi recombinants à partir de séries de coupes confocales basées sur les ligands fluorescents, l'antagoniste B-10376 ou l'agoniste B-10378 (à une concentration de 50 nM; réaction de marquage de 30 minutes à 37°C, suivie de rinçages). L'échantillon photographique montré, présentant une seule cellule, est représentatif de l'ensemble des cellules photographiées, cultivées en monocouche parmi lesquelles certaines n'étaient pas transfectées. Chaque cellule est présentée du dessus et du dessous, par rapport à la surface de la cellule qui est adhère au substrat de culture. La coloration jaune ou bleue est ajoutée par ordinateur et sert à illustrer les niveaux d'intensité variables de la fluorescence verte, plus faible ou plus élevée, respectivement. L'ajout d'un effet d'ombre sous la cellule aide à la compréhension de l'image. Les cellules ont été observées à un grossissement original de 630 X.
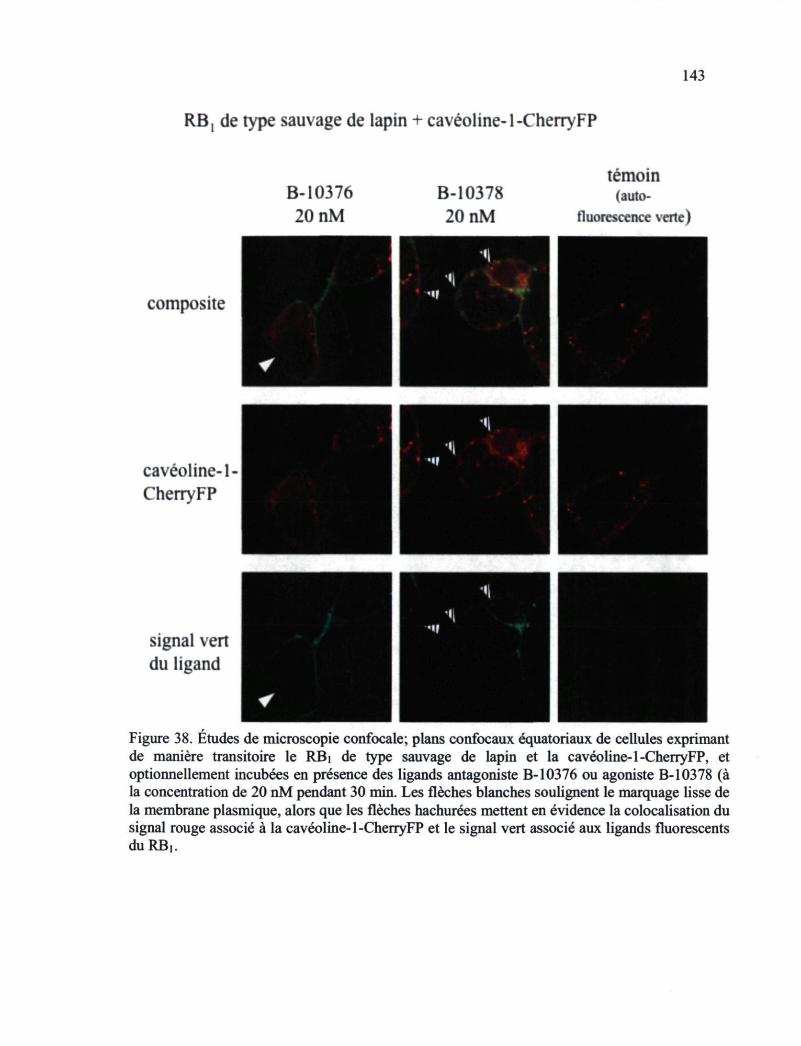
143
RB, de type sauvage de lapin + cavéoline-1-CherryFP
B-10376 B-10378 témoin
(auto20 nM 20 nM fluorescence verte)
composite
cavéoline-1-CherryFP
signal vert du ligand
Figure 38. Études de microscopie confocale; plans confocaux équatoriaux de cellules exprimant de manière transitoire le RBi de type sauvage de lapin et la cavéoline-1-CherryFP, et optionnellement incubées en présence des ligands antagoniste B-10376 ou agoniste B-10378 (à la concentration de 20 nM pendant 30 min. Les flèches blanches soulignent le marquage lisse de la membrane plasmique, alors que les flèches hachurées mettent en évidence la colocalisation du signal rouge associé à la cavéoline-1-CherryFP et le signal vert associé aux ligands fluorescents duRBi.

144
stimulation par l'agoniste provoquait la translocation une partie du signal granulaire
cytosolique de la cavéoline-1 à la membrane plasmique (ces données ne sont pas montrées).
3.3.2.5 Exploitation de l'antagoniste B-10376 en cytométrie de flux
Des cellules HEK 293a marquées avec l'antagoniste fluorescent ne présentaient pas une
fluorescence verte associée aux cellules différente de l'autofluorescence de ces cellules
(figure 39). Par contre, près de la moitié des cellules soumises à la transfection avec la
construction RBj de type sauvage de lapin présentaient une intense fluorescence en
présence du B-10376 (figure 39, en D). Cette distribution était complètement renversée si le
marquage était fait en présence du composé 11, un antagoniste non peptidique de haute
affinité du RBi (à une concentration de 1 pM), comme le montre la figure 39, en E.
L'utilité de l'antagoniste fluorescent à détecter l'abondance du RB] a été mise à l'essai dans
une étude plus poussée, où certaines cellules exprimant le RBt de type sauvage de lapin ont
été soumises à des prétraitements de 2 à 4 h avec l'anisomycine, un inhibiteur de la
synthèse protéique, avant d'être mises en présence du B-10376. Le prétraitement avec
l'anisomycine réduisit la proportion de cellules intensément fluorescentes, ainsi que
l'intensité médiane du marquage, comme le présente la figure 39 (en G et en F,
respectivement). Ces données supportent par ailleurs les observations rapportées par Fortin
et collaborateurs (2003), montrant que les RBi exprimés à la membrane plasmique ont une
courte demi-vie et sont rapidement remplacés par la synthèse de novo.
L'expression du RBi par les cellules de muscle lisse ainsi que les réponses fonctionnelles
qui découlent de sa stimulation ont déjà fait l'objet de plusieurs études, en exploitant des
cellules musculaires lisses dérivant de l'artère ombilicale humaine, dont celles décrites plus
haut, et d'autres encore (Morissette et coll., 2004; Moreau et coll., 2007). La fluorescence
de ces cellules, prétraitées avec l'IL-l(3, et ensuite incubées avec l'antagoniste B-10376, à
la concentration de 100 nM, était systématiquement plus haute que l'autofluorescence,
comme il est d'ailleurs illustré à la figure 40 (la valeur de P < 0,05, telle qu'obtenue par le
test t de Student apparié). Il y a par contre un chevauchement entre ces deux distributions. Il
est à noter que l'abondance des RBi endogènes tels qu'exprimés par ce tissu représente
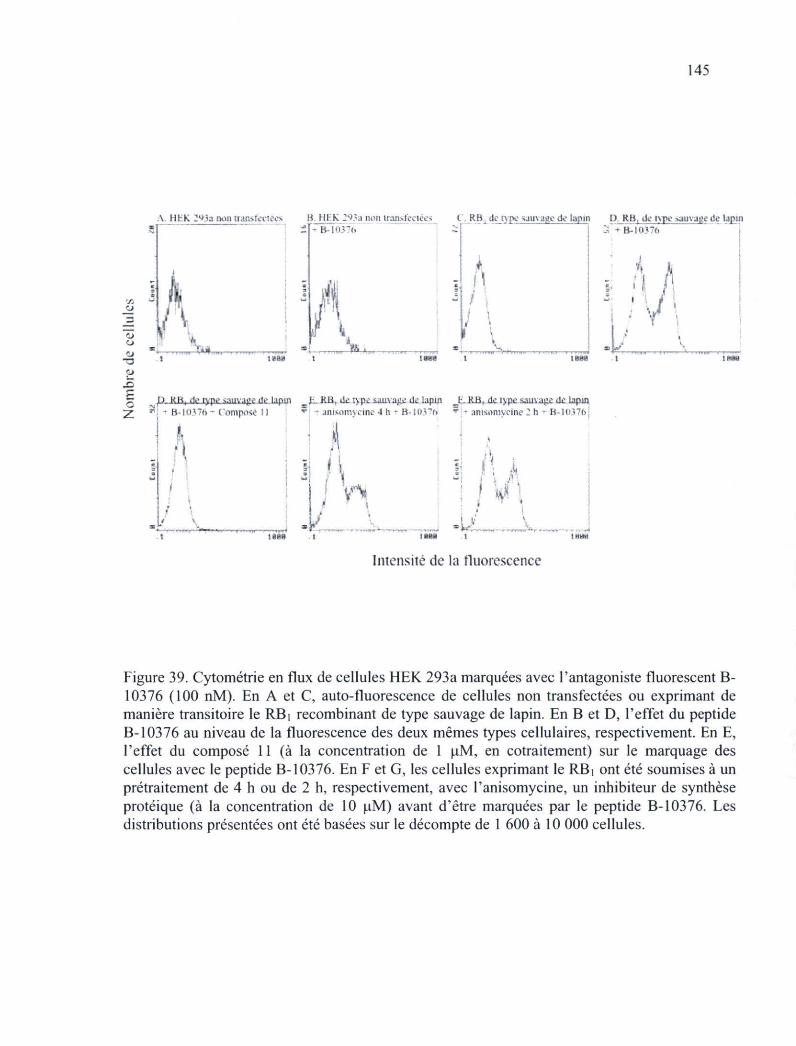
145
■V HEK 293a non iransfcciees B.HEK 293a non Iransleclèes C. RB. de type sauvage do lapin DJtB^ de lype sauvage de lapin S
;+ B10376
A
IHM 1 IHW i
ia%r de Upin p . RR, dp lyrtf sauvagf «talapip JL RBt deiype Minage dc lipin ■"• t anisomycine 4 Ii • B10378 *H anisomveine 2 h iB*10376
t f i a
. =LJ.,
1 IH
Intensité de la fluorescence
Figure 39. Cytométrie en flux de cellules HEK 293a marquées avec l'antagoniste fluorescent B
10376 (100 nM). En A et C, autofluorescence de cellules non transfectées ou exprimant de manière transitoire le RBi recombinant de type sauvage de lapin. En B et D, l'effet du peptide B10376 au niveau de la fluorescence des deux mêmes types cellulaires, respectivement. En E, l'effet du composé 11 (à la concentration de 1 pM, en cotraitement) sur le marquage des cellules avec le peptide B10376. En F et G, les cellules exprimant le RBi ont été soumises à un prétraitement de 4 h ou de 2 h, respectivement, avec l'anisomycine, un inhibiteur de synthèse protéique (à la concentration de 10 pM) avant d'être marquées par le peptide B10376. Les distributions présentées ont été basées sur le décompte de 1 600 à 10 000 cellules.
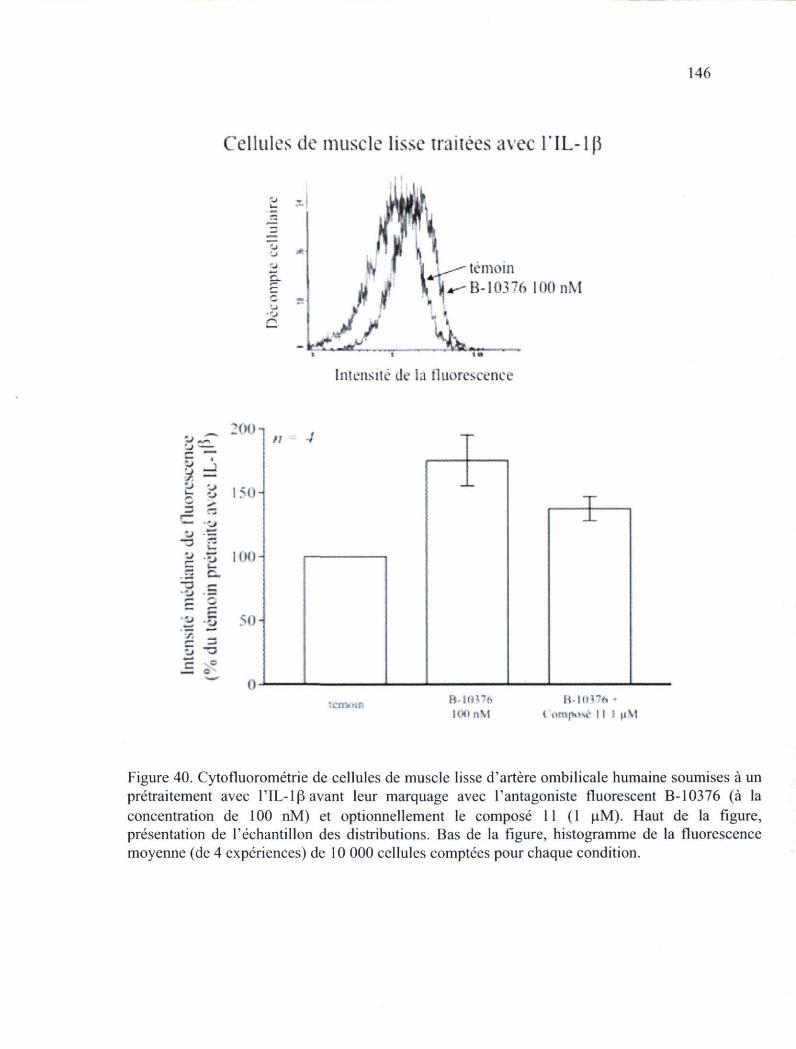
146
Cellules de muscle lisse traitées avec l'ILlp
témoin B10376 100 n M
Intensité de la fluorescence
V fc^* u
■"'"■■■■■
c i i _ 0 a
8 ** r i
J tece*
. J L~
g *— u . 1 C—
3 .> n ^ r—
»— o ZJ
M r —> t j *o
_ç
2001
150
100
so
0
II
tntviin BlOVJf, 100 nM
B10176 • t ompùât Il i JIVI
Figure 40. Cytofluorométrie de cellules de muscle lisse d'artère ombilicale humaine soumises à un prétraitement avec l'IL1 p avant leur marquage avec l'antagoniste fluorescent B10376 (à la concentration de 100 nM) et optionnellement le composé 11 (1 pM). Haut de la figure, présentation de l'échantillon des distributions. Bas de la figure, histogramme de la fluorescence moyenne (de 4 expériences) de 10 000 cellules comptées pour chaque condition.

147
approximativement 1 % de l'expression de la construction huBiR-FLAG en transfection
transitoire, par les cellules HEK 293a (Moreau et coll., 2007; Morissette et coll., 2008).
Le composé 11, un antagoniste du RBi, réduisait la fluorescence des cellules prétraitées
avec des cytokines, lorsqu'il était introduit en présence de l'antagoniste B-10376 lors de la
procédure du marquage. Cela permet de confirmer que la fluorescence mesurée est associée
aux RBi endogènes.
3.3.3 Analogues fluorescents de la BK
3.3.3.1 Caractérisation du myc-RB2TRUNC
Le myc-RB2 représente un RB2 de lapin fonctionnel qui est fusionné avec un epitope myc
en N-terminal de la séquence du récepteur. Malgré cette modification dans la séquence du
récepteur, il conserve une affinité identique pour le radioligand [3H]BK à celle du récepteur
de type sauvage de lapin. Le myc-RB2TRUNC est une version tronquée du myc-RB2, dont les
28 derniers acides aminés C-terminaux ont été supprimés; cette région contient le site
substrat des GRK (Leeb-Lundberg et coll., 2005). L'expression transitoire de ce récepteur
par des cellules HEK 293a a permis la réalisation d'essais de liaison qui ont montrés une
liaison de la [3H]BK avec une affinité similaire à celle du myc-RB2 (un Ko de 2,97 nM
pour le myc-RB2TRUNc, comparativement à un Ko de 2,42 nM, pour le récepteur non
tronqué). L'expression du myc-RB2TRUNC (Bmax de 120,1 fmol/puits) était légèrement
moindre que celle du myc-RB2 (Bmax de 148,8 fmol/puits); ces paramètres ont été dérivés
des courbes de saturation construites pour la [3H]BK (figure 41, en A). La liaison
spécifique d'une faible quantité de radioligand par les cellules HEK 293a non transfectées
est cohérente avec la présence d'une petite population endogène de RB2 rapportée par
Kramarenko et collaborateurs (2009). L'utilisation d'anticorps anti-myc a permis la
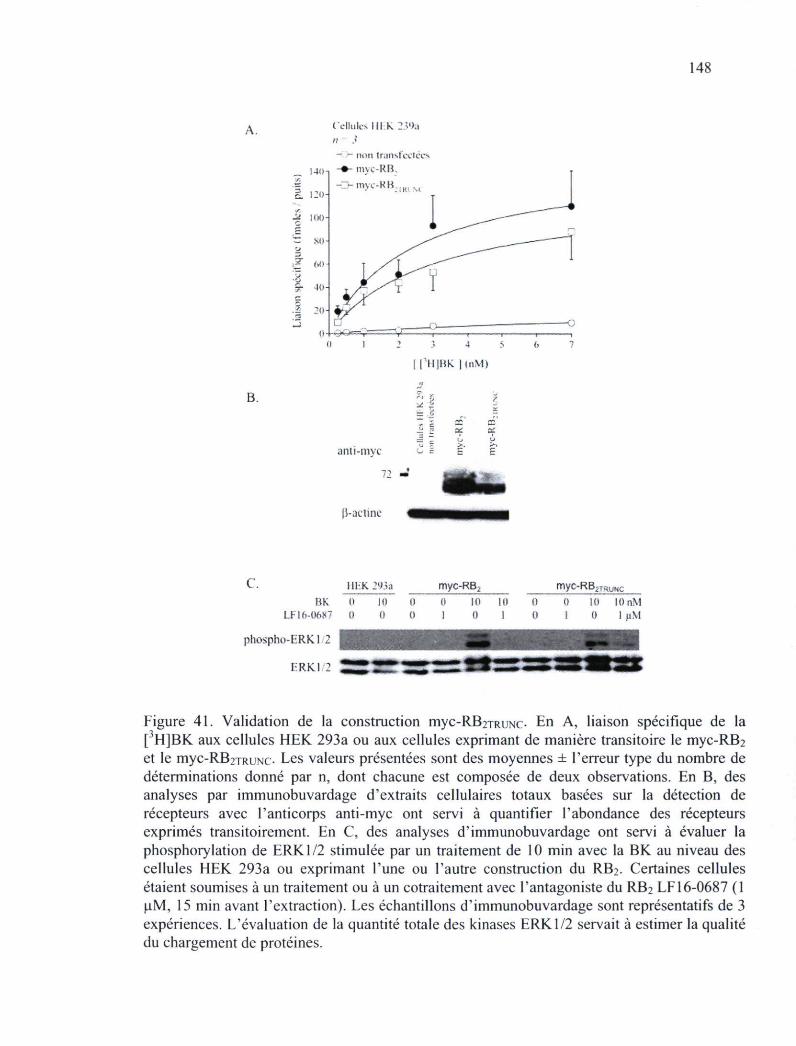
148
Cellules HF.K 239a Il s
>■ non Ircinsfcclécs 140, • mycRB,
['HJBK |(iiM)
B. cr cet
aniimvc
[5actine
C. BK
LF 1606X7
phosphoERKl/2
lîRK.1 2
HEK 293a mycRB2 mycRBiTSUNC
0 10 0 0 10 10 0 0 10 10 nM 0 II 0 1 0 1 0 1 0 l f iM
Figure 41. Validation de la construction mycRB2TRUNC En A, liaison spécifique de la [ H]BK aux cellules HEK 293a ou aux cellules exprimant de manière transitoire le mycRB2 et le mycRB2TRUNC Les valeurs présentées sont des moyennes ± l'erreur type du nombre de déterminations donné par n, dont chacune est composée de deux observations. En B, des analyses par immunobuvardage d'extraits cellulaires totaux basées sur la détection de récepteurs avec l'anticorps antimyc ont servi à quantifier l'abondance des récepteurs exprimés transitoirement. En C, des analyses d'immunobuvardage ont servi à évaluer la phosphorylation de ERK 1/2 stimulée par un traitement de 10 min avec la BK au niveau des cellules HEK 293a ou exprimant l'une ou l'autre construction du RB2. Certaines cellules étaient soumises à un traitement ou à un cotraitement avec l'antagoniste du RB2 LF 160687 (1 pM, 15 min avant l'extraction). Les échantillons d'immunobuvardage sont représentatifs de 3 expériences. L'évaluation de la quantité totale des kinases ERK1/2 servait à estimer la qualité du chargement de protéines.

149
détection de la construction myc-RB2 sous sa forme glycosylée dans des essais
d'immunobuvardage, dont la bande représentative était assez large, d'un poids moléculaire
variant entre 63 à 68 kDa, alors que la bande représentative de la construction myc-
RB2TRUNC présentait une perte de poids moléculaire d'approximativement 3 kDa, ce qui
était prévisible. Un échantillon représentatif des données d'immunobuvardage est présenté
à la figure 41, en B. L'expression transitoire du récepteur tronqué permet la
phosphorylation des kinases ERK 1/2 par la stimulation aiguë avec la BK avec une
intensité similaire à celle découlant du récepteur complet, signalisation qui est par ailleurs
inhibée par un cotraitement avec l'antagoniste du RB2 LF 16-0687 pour les deux
constructions, comme permet de le constater la figure 41, en C.
Le signal fluorescent associé à la (3-arrestine2-GFP, apparaît généralement très homogène
dans le cytosol de cellules HEK 293a non stimulées cotransfectés avec le myc-RB2, alors
qu'il se condense au niveau de structures endosomales intracellulaires lorsque les cellules
sont soumises à la stimulation par la BK. Par contre, sous ces mêmes conditions, on le
retrouve très peu au niveau de la membrane plasmique. Cette translocation vers des
particules intracellulaires n'est pas observable dans les cellules transfectées uniquement
avec la f>-arrestine2-GFP, ni dans les cellules exprimant le myc-RB2TRUNc, ce qui confirme
l'incompétence de cette construction à recruter les (3-arrestines, comme cela est montré à la
figure 42.
3.3.3.2 Pharmacologie des analogues fluorescents de la BK
Des essais de liaison par l'exploitation du radioligand [3H]BK ont servi à évaluer l'affinité
du nouvel analogue CF-eACA-BK pour le myc-RB2 exprimé transitoirement par les
cellules HEK 293a. La CF-EACA-BK déplace la liaison spécifique de la [3H]BK avec une
CI50 de 800 nM, ce qui la rend 400 fois moins puissante que son peptide parent, dont la CI50
est de 2 nM. Les courbes de compétition construites pour chaque peptide sont illustrées à la
figure 43.
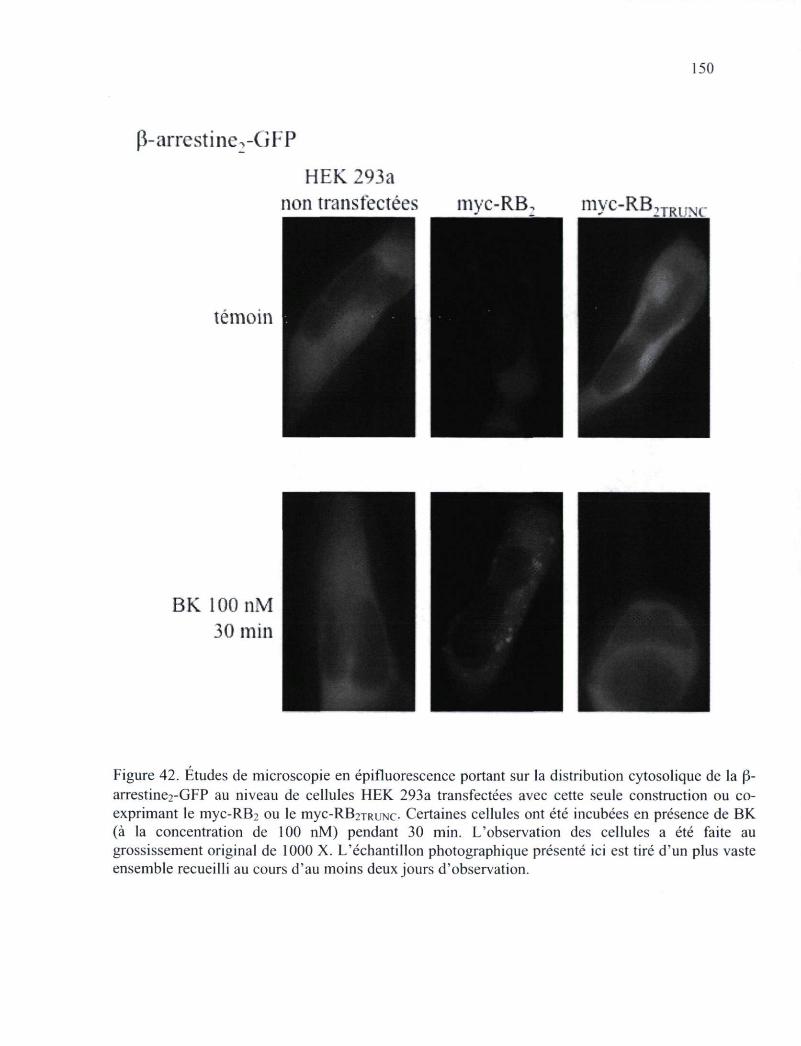
150
(J-arrcstincvGFP
HEK 293a non transfectées
témoin
mvc-RB- myc-RB-, TRUNC
BK 100 nM 30 min
Figure 42. Études de microscopie en épifluorescence portant sur la distribution cytosolique de la |3-arrestine2-GFP au niveau de cellules HEK 293a transfectées avec cette seule construction ou co-exprimant le myc-RB2 ou le myc-RB2TRUNC- Certaines cellules ont été incubées en présence de BK (à la concentration de 100 nM) pendant 30 min. L'observation des cellules a été faite au grossissement original de 1000 X. L'échantillon photographique présenté ici est tiré d'un plus vaste ensemble recueilli au cours d'au moins deux jours d'observation.
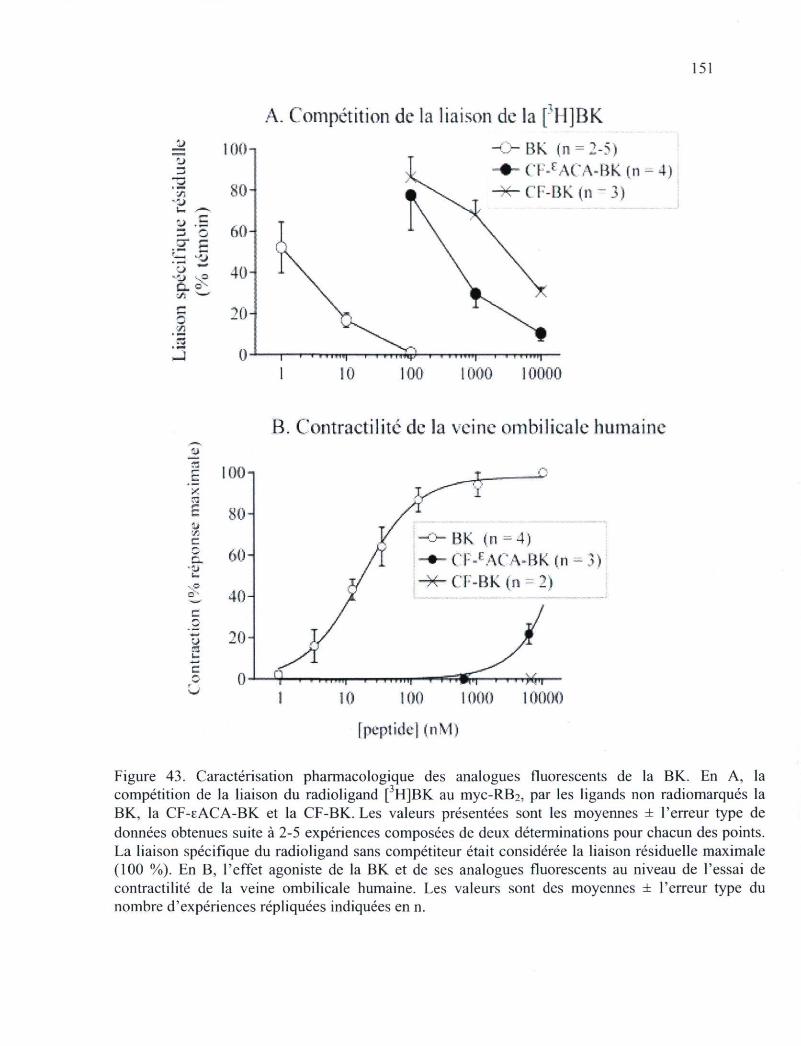
151
41
c/c
-t> -s? Û, c--cA ' > - '
0
lOO-i
80-
3 o 60-
40
20-
0
A. Compétition de la liaison de la [3H]BK
- BK (n = 2-5) EACA-BK (n - 4) BK(n-3)
p T T TTflWy" T T f T T I * t f i »T—■T»nT.yTTfy;|-...-.—r. ■.■■r.,r■rTT,f ...
M) 100 .000 10000
î !
s
■ *
■'-i
u ■ -
—
u
00
80
60-
40-
Z 20
B. Contractilité de la veine ombilicale humaine
- O - B K ( n - 4 ) •-CF-€ACA-BK (n = 3) H- CF-BK (n - 2)
11
1 i i 1111 l l | I I ! I M U !
10 100 [peptide] (nM)
oooo
Figure 43. Caractérisation pharmacologique des analogues fluorescents de la BK. En A, la compétition de la liaison du radioligand [3H]BK au myc-RB2, par les ligands non radiomarqués la BK, la C F - E A C A - B K et la CF-BK. Les valeurs présentées sont les moyennes ± l'erreur type de données obtenues suite à 2-5 expériences composées de deux déterminations pour chacun des points. La liaison spécifique du radioligand sans compétiteur était considérée la liaison résiduelle maximale (100 %). En B, l'effet agoniste de la BK et de ses analogues fluorescents au niveau de l'essai de contractilité de la veine ombilicale humaine. Les valeurs sont des moyennes ± l'erreur type du nombre d'expériences répliquées indiquées en n.

152
La veine ombilicale humaine permet la réalisation d'essais fonctionnels pour le RB2, et a
servi à la caractérisation approfondie des analogues fluorescents de la BK, ce qui a d'abord
mené à la confirmation de l'identité d'agoniste de la CF-EACA-BK pour le RB2. Par contre,
sa CE50 au niveau du récepteur humain était supérieure à 10 000 nM, et ne produisait que
21,8 % de la réponse maximale contractile à une concentration de 6,3 pM, et la rend ainsi
1000 fois moins active que la BK pour cet essai (figure 43B). À cette concentration, la
CF-BK était inactive. Ainsi les essais de liaison et de contractilité suggèrent tous deux une
importante perte d'affinité des analogues fluorescents de la BK, qui est probablement due à
leur extension en N-terminal, quoique l'affinité soit partiellement préservée par la présence
d'un motif espaceur entre la BK et le fluorophore. Les études de microscopie effectuées
suite à la caractérisation pharmacologique de l'agoniste C F - E A C A - B K ont été basées sur
des concentrations pharmacologiquement actives de cet agoniste.
3.3.3.3 Caractérisation et endocytose de l'analogue fluorescent C F - E A C A - B K
La figure 44 présente le marquage fluorescent de cellules HEK 293a non transfectées,
transfectées avec la construction myc-RB2 ou avec le myc-RB2TRUNC soumises à des
concentrations croissantes de CF-EACA-BK, variant de 1 à 10 pM. Les cellules non
transfectées ou encore les cellules non traitées avec l'agoniste fluorescent ne présentent
aucune autofluorescence verte, sous les réglages employés en microscopie en
épifluorescence. Le peptide fluorescent ne marquait ni les cellules non transfectées, ni les
cellules exprimant le récepteur tronqué. La distribution du signal fluorescent, au sein de
cellules exprimant le myc-RB2, était double; d'abord les cellules montraient une
fluorescence diffuse intracellulaire, mais présentaient aussi des petites structures denses
hautement fluorescentes. Cette fluorescence condensée au niveau intracellulaire des cellules
exprimant le myc-RB2 suggérait l'internalisation de l'agoniste fluorescent vers les
endosomes d'une façon dépendante du récepteur, comme l'est l'agoniste naturel, suivant sa
liaison au récepteur. L'intensité du marquage fluorescent dépendait de la concentration
employée de C F - E A C A - B K et était d'ailleurs cohérente avec l'occupation du récepteur telle
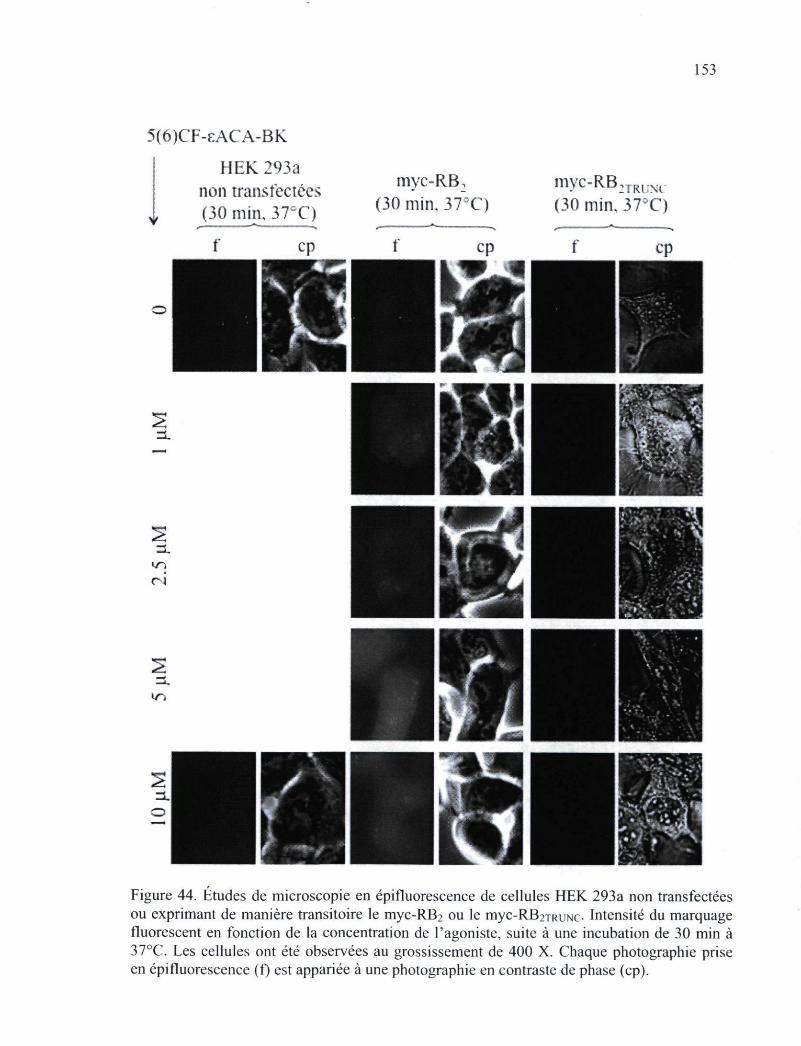
153
5 ( 6 ) C F - E A C A - B K
HEK 293a non transfectées (30 min. 37°C)
f cp
myc-RB2
(30 min, 37°C)
t
H
myc-RB2TRUNC
(30 min, 37°C)
Figure 44. Études de microscopie en épifluorescence de cellules HEK 293a non transfectées ou exprimant de manière transitoire le myc-RB2 ou le myc-RB2TRUNC- Intensité du marquage fluorescent en fonction de la concentration de l'agoniste, suite à une incubation de 30 min à 37°C. Les cellules ont été observées au grossissement de 400 X. Chaque photographie prise en épifluorescence (f) est appariée à une photographie en contraste de phase (cp).

154
que mesurée grâce aux essais de compétition reposant sur l'utilisation du radioligand.
L'emploi de concentrations plus élevées de CF-EACA-BK (entre 5 et 10 pM) menait à un
marquage plus intense et à une meilleure définition des structures fluorescentes
intracellulaires. Les expériences de microscopie subséquentes ont été effectuées à la
concentration de 5 pM, une concentration considérée optimale pour la suite de l'imagerie.
Les reconstitutions tridimensionnelles, présentées à la figure 45 et qui modélisent la liaison
des ligands fluorescents des RB2 recombinants de lapin, ont permis d'observer les
différences prononcées entre les distributions de l'agoniste C F - E A C A - B K et de
l'antagoniste B-10380 par des cellules intactes. Alors que l'antagoniste marquait la surface
cellulaire d'une manière lisse, sans indiquer une distribution apicale polarisée des RB2
comme celle qui a été rapportée au sein d'autres lignées cellulaires d'origine épithéliale
(Gardia-Perez et Smith, 1984), la séquestration endosomale de l'agoniste fluorescent a été
clairement mise en évidence par la perte de la perception de la surface cellulaire, menant
plutôt à un signal fragmenté au niveau du cytosol. Les structures ainsi indentifiées étaient
l'intérieur des endosomes contenant l'agoniste fluorescent, sans marquage significatif de la
membrane plasmique, un phénomène qui peut s'expliquer par la dissociation rapide du
ligand de basse affinité suivant le lavage des cellules et/ou la translocation intracellulaire de
tous les récepteurs exprimés à la surface cellulaire.
Des études additionnelles s'intéressaient à la progression de la C F - E A C A - B K liée au
récepteur au niveau de la voie endosomale. Les cellules HEK 293a intactes utilisées pour
cette série d'expériences étaient cotransfectées de manière systématique avec la
construction myc-RB2 en combinaison avec une protéine fluorescente rouge (CherryFP) et
subséquemment traitées avec le colorant nucléaire Hoechst 33258. Des échantillons
photographiques de chacune des conditions sont présentés aux figures 46 et 47, alors que
l'analyse statistique de la fluorescence cellulaire totale se trouve à la figure 46, à droite. La
fluorescence rouge émise par la CherryFP était utilisée pour prédire l'expression du myc-
RB2, et seules les cellules rouges ont été incluses dans les analyses statistiques de la figure
46, portant sur l'intensité de la fluorescence verte. Cette approche est validée par la
corrélation positive et significative (non montrée) entre l'intensité de la fluorescence rouge
de la CherryFP et celle de la fluorescence verte de la CF-EACA-BK internalisée dans un

155
RB, de type sauvage de lapin CF-cACA-BK
Vue de dessus
Vue de dessous
B-10380
ECA
Vue de dessus
Vue de dessous
CF-cACA-BK.
"N
>
- y Coupe confocale

156
Figure 45. Reconstitutions tridimensionnelles de cellules HEK 293a exprimant le RB2 de type sauvage de lapin (haut de la figure) ou exprimant l'ECA humaine, générées à partir d'une série de coupes confocales pour chaque champ. Haut de la figure, la distribution du RB2 de type sauvage de lapin révélée par l'agoniste fluorescent C F - E A C A - B K OU l'antagoniste fluorescent B-10380, à des concentrations de 5 pM et de 50 nM, respectivement. La réaction de marquage, d'une durée de 30 min à 37°C, était suivie de rinçage et d'une observation au grossissement de 630 X. Bas de la figure, reconstitutions tridimensionnelles d'une cellule HEK 293a exprimant de manière transitoire l'ECA recombinante utilisant une série de coupes confocales basées sur la CF-EACA-BK, à la concentration de 5 pM. Les cellules étaient soumises à une incubation de 30 min en présence de l'agoniste fluorescent du RB2. Une coupe confocale est montrée pour documenter la localisation de la fluorescence, essentiellement membranaire. Chaque champ sélectionné présentait une cellule isolée, dont la fluorescence était représentative de celle observée généralement pour chaque condition; ces cellules isolées faisaient partie d'une monocouche cellulaire et les cellules avoisinantes n'étaient pas transfectées. Chaque cellule est présentée du dessus et du dessous (à partir de la surface du substrat plastique sur lequel la cellule adhérait). Certains plans ont été retirés. Les couleurs jaune et bleu ont été attribuées de manière arbitraire aux deux extrémités d'une échelle d'intensité de fluorescence et sont de fausses couleurs au niveau de l'image qui indiquent les niveaux d'intensité moindre et supérieur, respectivement, de la seule fluorescence enregistrée, la verte. Une fausse ombre a été ajoutée aux cellules afin de faciliter leur visualisation.
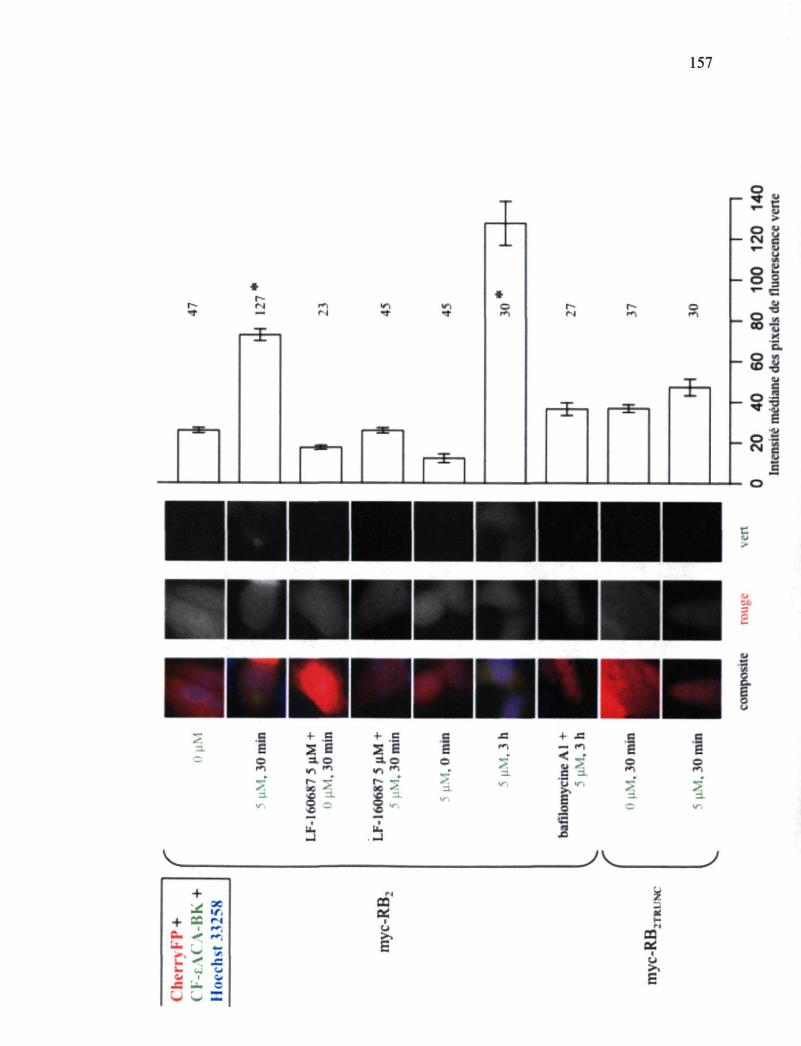
157
r
r i vr. t
g rf i
î i r*n
* 8 O V
o E 1 8 o ■ coS
' J
o ]
C ■J
H a
i £ c
3.
+ ■§ S E a o
3
t c
II a.© S ^
c I e
» :
ccccc un
et sss\
<r.
4 JS
< J
| I I
1
V 2.
JK. J + É X
•/•, + aa — ^ " i u. r
t •^ ** JB
— LJ o «« I fc JB — c u w S
I

158
Figure 46. Études de microscopie en épifluorescence de cellules exprimant le myc-RB2 soumises à une incubation en présence de l'agoniste fluorescent CF-EACA-BK. À gauche, marquage de cellules exprimant le myc-RB2 ou le myc-RB2TRUNC par la C F - E A C A - B K (concentration de 5 pM) soumises à des incubations de différentes durées, afin de déterminer la distribution subcellulaire de la fluorescence verte associée au ligand. Certaines cellules exprimant le myc-RB2 ont été prétraitées avec un inhibiteur de la V-ATPase bafilomycine Al (à la concentration de 100 nM, 1 h avant de procéder au marquage avec l'agoniste fluorescent) ou avec l'antagoniste du RB2 LF 16-0687 (à la concentration de 5 pM, 30 min avant de procéder au marquage avec l'agoniste fluorescent). La protéine CherryFP, émettant une fluorescence rouge, était cotransfectée systématiquement avec le myc-RB2 et le myc-RB2jRUNC afin de vérifier l'efficacité de la transfection. Le colorant Hoechst 33258, ajouté 15 min avant l'observation à la concentration de 25 pM, permettait la visualisation du noyau. Les cellules ont été observées à un grossissement de 1000 X. À droite, intensité de la fluorescence cellulaire médiane associée à la CF-EACA-BK, calculée à partir de réplicats de chaque condition expérimentale, présentées sous forme moyenne ± erreur type. Les chiffres se trouvant à proximité des barres de l'histogramme indiquent le nombre de cellules évaluées. Le test de Kruskal-Wallis indique l'hétérogénéité des valeurs (P < 0,001). L'autofluorescence verte des cellules exprimant le myc-RB2 sans agoniste fluorescent (barre du haut) a servi de base de comparaison pour les autres groupes (test de comparaison multiple de Dunn, * P < 0,001). Des conditions témoins additionnelles ont été incluses dans les statistiques, mais ne sont pas présentées par cette figure: il s'agit de cellules exprimant le RBi de concert avec la CherryFP incubées seules ou en présence de la C F - E A C A - B K (5 pM, 30 min), pour lesquelles les intensités de fluorescence sont de 12,9 ± 1,0 (n= 14) et de 11,4 ± 0,4 (n= 29), elles ne présentent donc pas un signal fluorescent supérieur à l'autofluorescence. Cette observation s'applique aussi aux cellules exprimant le myc-RB2 et traitées avec la bafilomycine seule (13,8 ± 0,8; n= 5) ou à des cellules n'exprimant que la protéine CherryFP, incubées en présence de la C F - E A C A - B K (5 pM, 30 min) ou non traitées, qui par ailleurs ne montraient pas une fluorescence supérieure à l'autofluorescence (22,3 ± 2,4 et 17,2 ± 0,4, n= 44 et 72, respectivement).
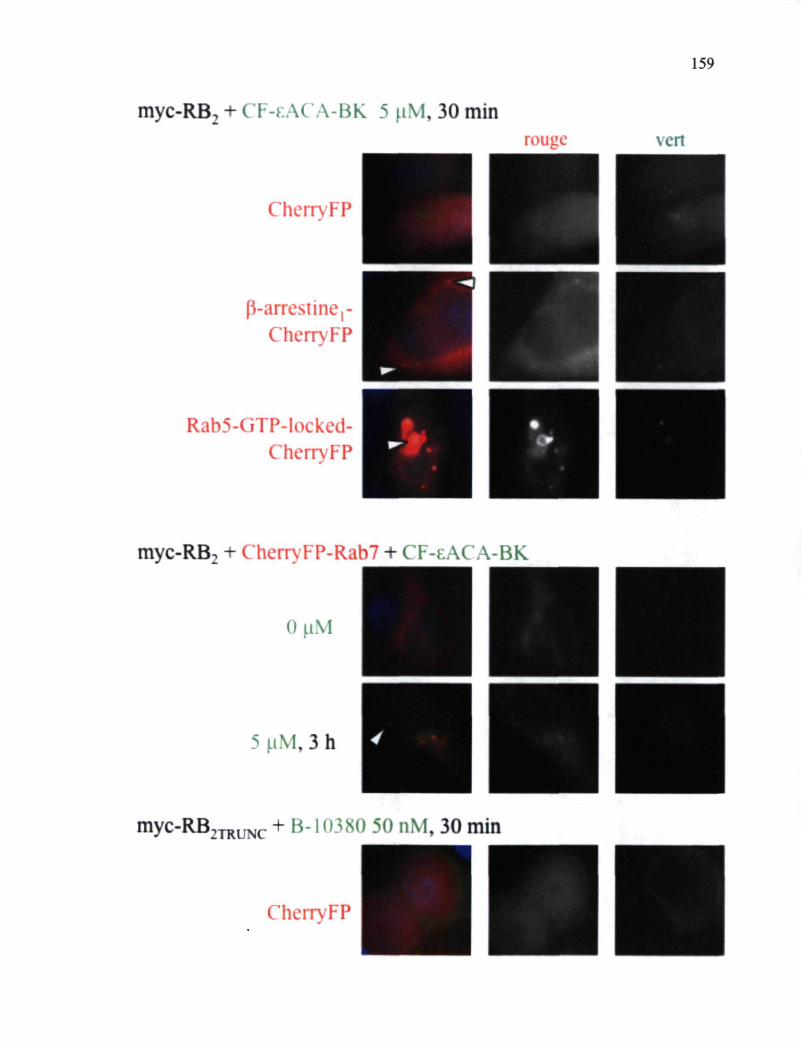
159
myc-RB2 + CF-eACA-BK 5 iM, 30 min
CherryFP
P-arrestine,-CherryFP
Rab5-GTP-locked-CherryFP
rouge
jjflH
vert
myc-RB2 + CherryFP-Rab7 + CF-EACA-BK
O^iM
5 pM, 3 h
myc-RB2TRUNC + B-10380 50 nM, 30 min
CherryFP

160
Figure 47. Études de microscopie en épifluorescence de cellules HEK 293a exprimant le myc-RB2, de concert avec d'autres transgènes. Les 3 premières lignes de la figure présentent des cellules exprimant le myc-RB2 soumises à un traitement de 30 min avec la CF-EACA-BK, à la concentration de 5 pM, dont certaines étaient en plus transfectées avec la construction (3-arrestinei-CherryFP, la construction CherryFP-GTP-locked-Rab5 ou la construction CherryFP. Les flèches blanches soulignent les sites de colocalisation entre la fluorescence verte associée au ligand et la fluorescence rouge émise par les différentes constructions. Les 4e et 5e lignes présentaient des images obtenues par l'observation de cellules HEK 293a exprimant le myc-RB2 de concert avec la construction CherryFP-Rab7, soumises au marquage avec l'agoniste fluorescent CF-EACA-BK durant 3 h. La 6e ligne présente le signal obtenu avec l'antagoniste fluorescent B-10380 (à la concentration de 50 nM, d'une durée de 30 min), lors de son incubation en présence de cellules exprimant le myc-RB2TRUNC- L'expression des différentes protéines fusionnées à la CherryFP servait aussi de témoin de transfection. Les cellules ont été observées à un grossissement de 1000 X.

161
ensemble de cellules cotransfectées avec la protéine fluorescente et le myc-RB2. Les
cellules HEK 293a exprimant le myc-RB2 étaient traitées avec la C F - E A C A - B K et
observées après des périodes variables d'incubation à 37°C.
Lorsque l'observation suivait immédiatement l'ajout de l'agoniste fluorescent au milieu de
culture, un marquage fluorescent très faible était apparent et les endosomes contenant cette
fluorescence se situaient directement sous la membrane, alors qu'un traitement de 30 min
permettait une intensification du marquage du cytosol et l'observation d'une fluorescence
vive au niveau endosomal. Une incubation de 3 h menait à une diminution du signal
fluorescent granulaire, à laquelle était secondaire une augmentation de la fluorescence
cytosolique. Un prétraitement de 1 h avec la bafilomycine Al à une concentration de 100
nM réduisait la diffusion de la fluorescence des endosomes vers le cytosol. Les statistiques
portant sur l'intensité de la fluorescence indiquent une forte réduction de la fluorescence
cellulaire médiane des pixels en présence de la bafilomycine Al; cela s'explique par le fait
que la méthode de mesure employée (intensité médiane dans le périmètre cellulaire) est très
sensible au bruit de fond cytosolique représentant la majorité des pixels et moins au signal
maximal de fluorescence concentré dans les vésicules denses qui n'occupent qu'une petite
surface. Afin de vérifier si un traitement de 3 h provoquait la progression de l'agoniste CF-
EACA-BK dans la voie endosomale jusqu'à sa localisation aux endosomes tardifs et aux
lysosomes, des cellules HEK 293a, servant à la réalisation de ces expériences, ont été
cotransfectées avec l'ADN codant pour une GTPase associée aux endosomes tardifs
CherryFP-Rab7. Un échantillon photographique des résultats obtenus en microscopie en
épifluorescence est présenté au bas de la figure 47. Malgré la quantité restreinte
d'endosomes apparents au temps 3 h après l'addition de l'agoniste CF-EACA-BK, il
semblait y avoir une colocalisation entre ces structures et la fluorescence rouge associée à
celle de la protéine Rab7.
Des expériences additionnelles, présentées à la figure 46, sur les cellules exprimant le myc-
RB2 ont été réalisées en présence de l'antagoniste du RB2 LF16-0687, à une concentration
équimolaire à celle utilisée pour la CF-EACA-BK, soit de 5 pM. Ce traitement avec
l'antagoniste a aboli l'internalisation de l'agoniste fluorescent. La coexpression de la (3-

162
arrestine i-CherryFP avec le myc-RB2 a mis en évidence la translocation considérable de la
fluorescence rouge associée à la (3-arrestinei aux endosomes contenant la CF-EACA-BK,
(figure 47). Les cellules exprimant le myc-RB2 de concert avec la protéine associée aux
endosomes précoces Rab5-GTP-locked-CherryFP et traitées avec l'agoniste fluorescent,
présentaient des endosomes élargis marqués en rouge, contenant une fluorescence verte,
associée à l'agoniste CF-EACA-BK. Les cellules exprimant ces deux constructions, mais
non traitées avec l'agoniste fluorescent, montraient une distribution de la CherryFP-Rab5-
Q79L similaire à celle des cellules traitées, sous forme d'une quantité d'endosomes vides
géants, d'ailleurs typiques de l'expression de la construction Rab5 à activité dominante
positive (Ceresa et coll., 2001). Au grossissement important de 1000 X, l'agoniste CF-
EACA-BK ne présentait aucun marquage significatif des cellules coexprimant le myc-
RB2TRUNC et la CherryFP. De manière contrastante, l'antagoniste fluorescent du RB2 B-
10380 marquait la membrane plasmique des cellules HEK 293a exprimant le myc-
RB2TRUNC, comme le présente la figure 47.
3.3.3.4 Imagerie de l'ECA
La CF-BK a retenu approximativement 20 % de l'affinité que présente la BK pour l'ECA,
alors que la CF-EACA-BK possède une affinité similaire pour l'ECA que la BK, selon les
essais de compétition réalisés à l'aide de l'énalaprilat tritié à l'ECA naturelle (figure 48).
Ces considérations supportent la possibilité d'utiliser les analogues fluorescents de la BK
comme outils servant à l'imagerie de l'ECA, si la liaison du substrat fluorescent au site
catalytique de l'enzyme se fait dans des conditions qui ne permettent pas la réaction
d'hydrolyse trop rapidement. La figure 49 montre le marquage de cellules HEK 293a
exprimant l'ECA recombinante humaine par la CF-EACA-BK, à la concentration de 5 pM,
qui était par ailleurs moins efficace à des concentrations moindres (données non montrées).
Un cotraitement des cellules avec le peptide fluorescent et l'énalaprilat empêchait le
marquage de l'ECA par la CF-EACA-BK, comme le présente la figure 49. Les cellules
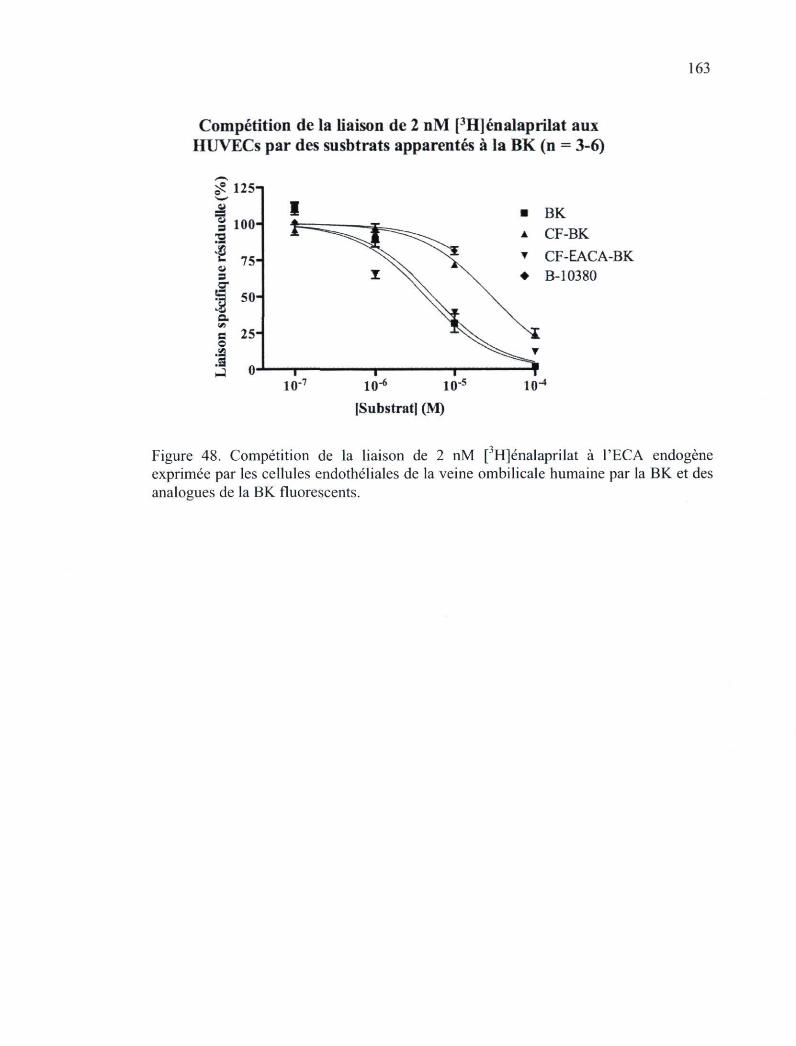
Compétition de la liaison de 2 nM [31I)énalaprilat aux HUVECs par des susbtrats apparentés à la BK (n = 36)
163
^ 1251
I 3 a
i u u 3 3"
I a. <n c o
100
T IO"7 IO"6 10s
[Substrat] (M)
■ BK A CFBK ▼ CFEACABK ♦ B10380
Figure 48. Compétition de la liaison de 2 nM [3H]énalaprilat à l'ECA endogène exprimée par les cellules endothéliales de la veine ombilicale humaine par la BK et des analogues de la BK fluorescents.
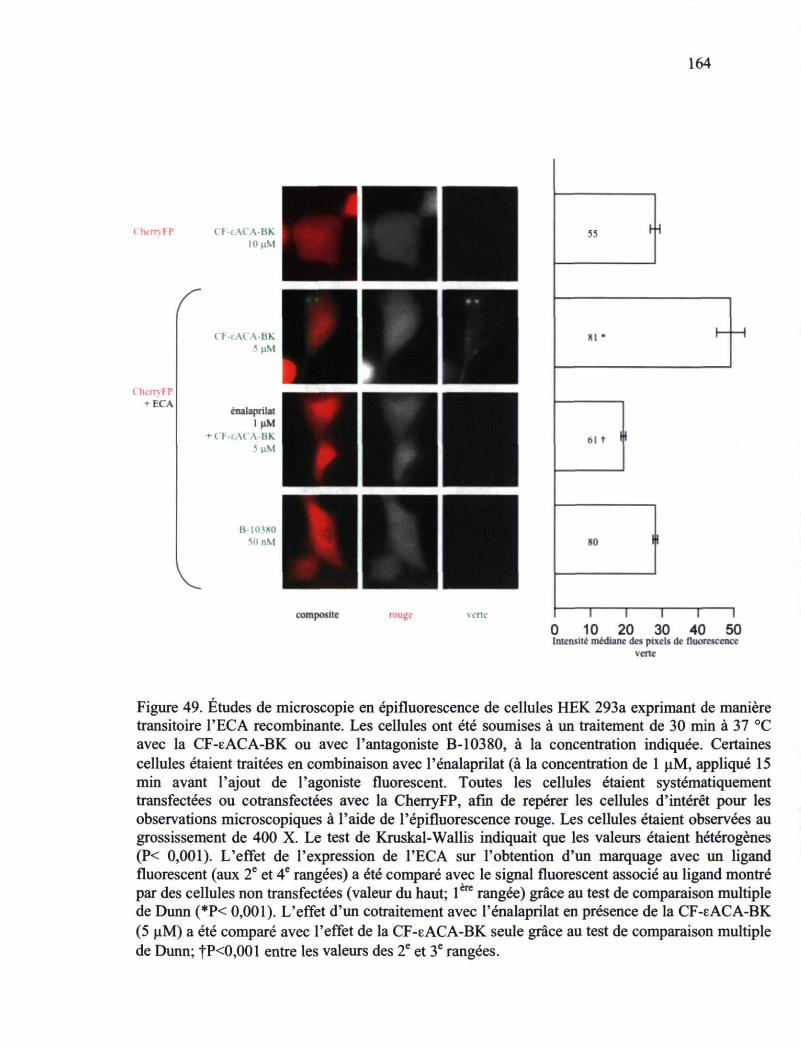
CherryFP IF-cACA-BK 10 MM
r
(.herrvFP + ECA
("F-cACA-BK 5(iM
énalaprilat I (iM
+ CF-CACA-BK 5 jiM
B-10380 50 nM
V
164
composile rouge
611
su
— I 1 1 I 1 0 10 20 30 40 50 Intensité médiane des pixels de fluorescence
verte
Figure 49. Études de microscopie en épifluorescence de cellules HEK 293a exprimant de manière transitoire l'ECA recombinante. Les cellules ont été soumises à un traitement de 30 min à 37 °C avec la CF-eACA-BK ou avec l'antagoniste B-10380, à la concentration indiquée. Certaines cellules étaient traitées en combinaison avec l'énalaprilat (à la concentration de 1 pM, appliqué 15 min avant l'ajout de l'agoniste fluorescent. Toutes les cellules étaient systématiquement transfectées ou cotransfectées avec la CherryFP, afin de repérer les cellules d'intérêt pour les observations microscopiques à l'aide de l'épifluorescence rouge. Les cellules étaient observées au grossissement de 400 X. Le test de Kruskal-Wallis indiquait que les valeurs étaient hétérogènes (P< 0,001). L'effet de l'expression de l'ECA sur l'obtention d'un marquage avec un ligand fluorescent (aux 2e et 4e rangées) a été comparé avec le signal fluorescent associé au ligand montré par des cellules non transfectées (valeur du haut; lère rangée) grâce au test de comparaison multiple de Dunn (*P< 0,001). L'effet d'un cotraitement avec l'énalaprilat en présence de la CF-eACA-BK (5 pM) a été comparé avec l'effet de la CF-eACA-BK seule grâce au test de comparaison multiple de Dunn; fP<0,001 entre les valeurs des 2e et 3e rangées.

165
exprimant seulement le gène marqueur de transfection CherryFP ne présentaient aucun
marquage par la CF-eACA-BK. Les reconstitutions tridimensionnelles de la fluorescence
verte associée à des cellules intactes exprimant l'ECA qui ont été générées à partir d'une
série de plans confocaux suivant le traitement de 30 min (incubation de 37°C) avec la CF-
eACA-BK, à la concentration de 5 pM, sont présentées au bas de la figure 45. Le marquage
secondaire à la liaison de la C F - E A C A - B K se situait à la surface cellulaire d'une manière
uniforme sans la présence significative d'endocytose. L'analogue fluorescent de la BK ne
permettait qu'un faible marquage de l'ECA recombinante lorsque l'incubation se faisait à la
température de la pièce (ces données ne sont pas présentées), ce qui indique que la réaction
d'hydrolyse probablement plus rapide à 37°C ne nuit pas à la production d'une imagerie
optimale à cette température. L'antagoniste fluorescent du RB2 B-10380, qui présente un Kj
en excès de 1 pM en compétition de l'[3H]énalaprilat (figure 48), ne permettait pas
l'imagerie de l'ECA recombinante exprimée dans les cellules HEK 293a, à la concentration
de 50 nM (données présentées à la figure 49), alors que cet antagoniste fluorescent
marquait les RB2 présents à la membrane cellulaire à cette même concentration, comme le
présente le haut de la figure 45.
3.3.4 Études de l'analogue naturel maximakinine
3.3.4.1 Pharmacologie de la maximakinine
La maximakinine (MK) montre une affinité quelque peu inférieure à celle de la BK au
niveau des RB2. L'affinité de la maximakinine dépend de l'espèce d'origine du récepteur.
L'essai de compétition contre le ligand tritié, effectué pour le RB2 de lapin permet
d'évaluer une affinité 20 fois moindre pour la MK par rapport à la BK, alors que l'essai de
contractilité de la veine ombilicale humaine permet d'estimer une affinité 7 fois moindre de
la MK, toujours par rapport à la BK. Les résultats des essais de liaison et de contractilité
sont présentés à la figure 50. La CI50 permettant le déplacement de la liaison de la [3H]BK
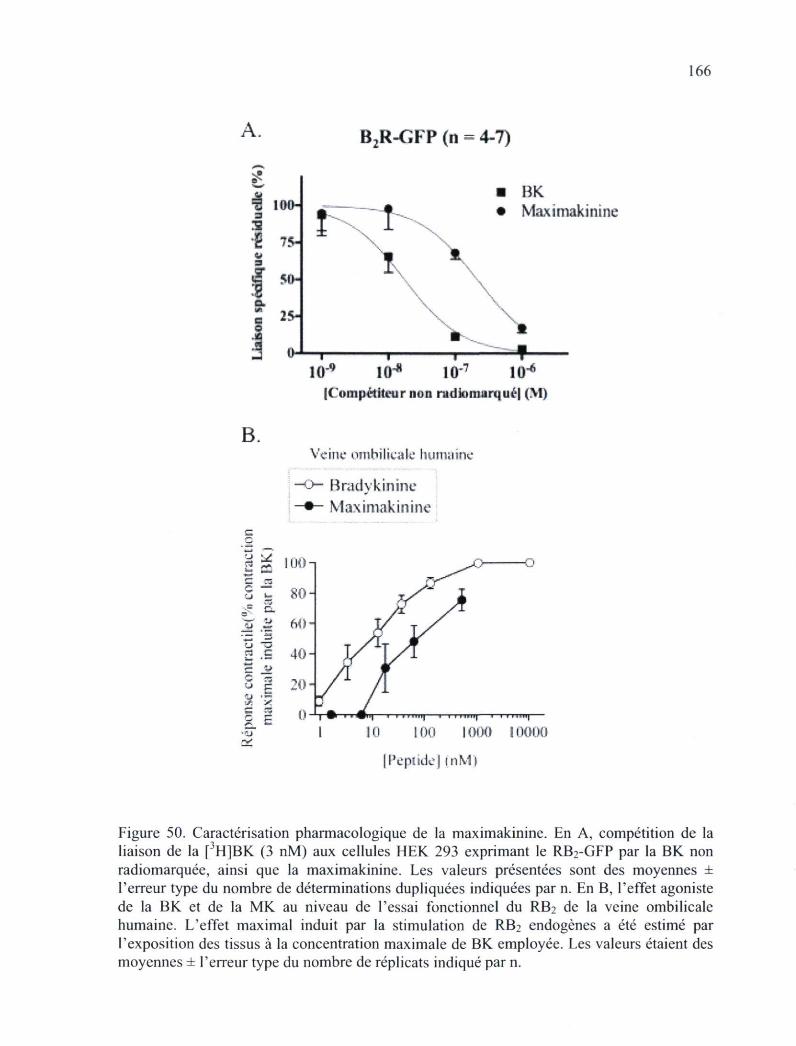
166
B2R-GFP (n = 4-7)
■ BK • Maximakinine
[Compétiteur non mdk>marqué| (M)
B.
T. 3
Veine ombilicale humaine
Bradykinine Maximakinine
■ty
tt, 10 100 1000 10000
[Peptide] (nM)
Figure 50. Caractérisation pharmacologique de la maximakinine. En A, compétition de la liaison de la [3H]BK (3 nM) aux cellules HEK 293 exprimant le RB2-GFP par la BK non radiomarquée, ainsi que la maximakinine. Les valeurs présentées sont des moyennes ± l'erreur type du nombre de déterminations dupliquées indiquées par n. En B, l'effet agoniste de la BK et de la MK au niveau de l'essai fonctionnel du RB2 de la veine ombilicale humaine. L'effet maximal induit par la stimulation de RB2 endogènes a été estimé par l'exposition des tissus à la concentration maximale de BK employée. Les valeurs étaient des moyennes ± l'erreur type du nombre de réplicats indiqué par n.

167
au RB2-GFP se situait entre 7 et 11,1 nM pour la BK, alors qu'elle se situait à 216 nM pour
la MK. La CE5o estimée grâce aux essais de contractilité se trouve entre 10 à 19 nM pour la
BK, comparativement à la CE50 de la MK qui est de 75 nM. Cela suggère une assez bonne
tolérance à l'extension considérable de la séquence N-terminale de cet agoniste du RB2.
3.3.4.2 Propriétés de la maximakinine
Les cellules HEK 293 exprimant de manière stable le RB2-GFP ont été soumises à un
traitement de 12 h avec la MK, à la concentration de 1 pM, afin de déterminer si la
prolongation de la séquence de cet agoniste naturel menait à la dégradation du récepteur.
Effectivement, la séparation par SDS-PAGE de lysats extraits des cellules traitées avec la
MK et suivie d'un immunobuvardage analysé avec l'anticorps anti-GFP révélait une
diminution de l'abondance du récepteur mature et une augmentation de l'abondance de la
GFP libre, à l'instar du patron de dégradation observé par le peptide résistant au
métabolisme B-9972, comme le montre la figure 51. En revanche, l'abondance du récepteur
mature demeurait inchangée dans les cellules exprimant le RB2-GFP traitées avec la BK,
comparativement aux cellules constituant le groupe témoin. De plus, la MK induisait une
activation persistante de la voie ERK 1/2, comme en témoigne l'état de phosphorylation
durable de ces kinases (données présentés en B de la figure 51), suite à un traitement
prolongé avec la MK, qui rappelle les résultats obtenus avec les agonistes résistants au
métabolisme. Par ailleurs, on observe l'induction de l'expression du facteur de transcription
c-Fos, suite à un traitement de 12 h avec la MK et le B-9972, comme le montre la figure
51B. Finalement, des études de microscopie en épifluorescence (dont un échantillon
photographique est présenté en C de la figure 51) permettent d'observer une internalisation
persistante du signal fluorescent associé au RB2-GFP, suite à un traitement prolongé avec la
MK. Le peptide B-9972, à la concentration de 100 nM, était utilisé comme témoin positif
d'un agoniste persistant du RB2-GFP.
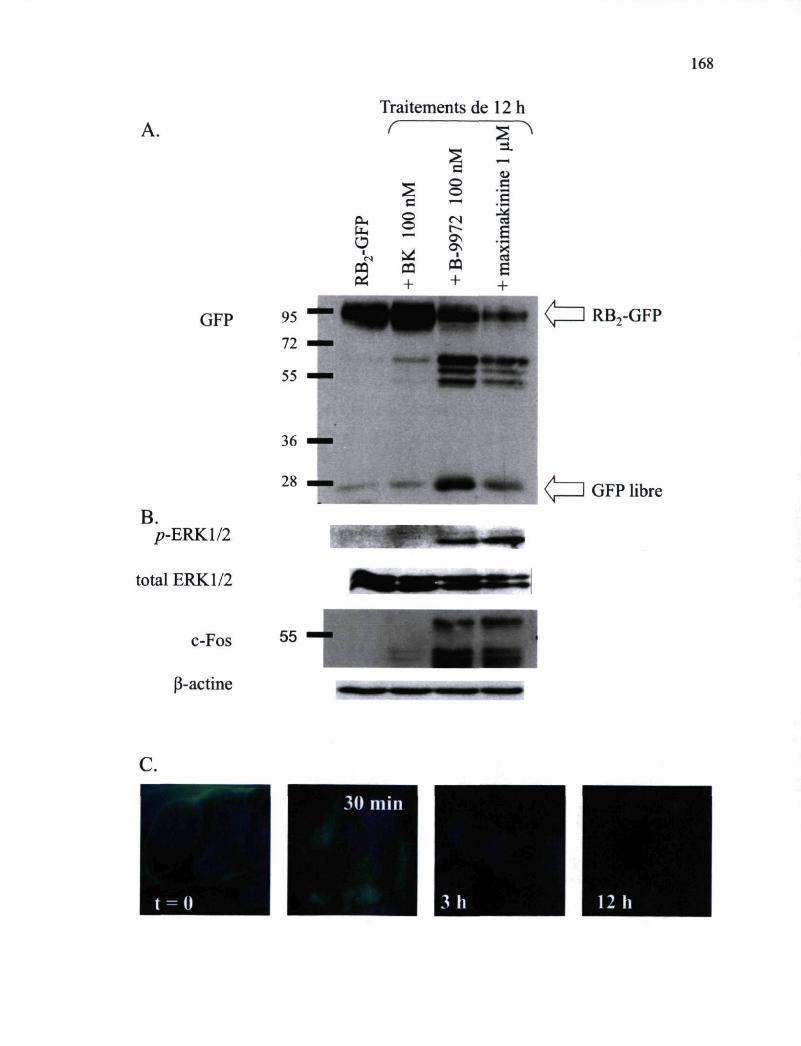
168
Traitements de 12 h
eu
o
GFP 95 72
55
s ^ s zi.
c <u S o o n
c c -^ n c
o CM i o P* E "~ Os E X 1
CÛ CQ £ + + +
RB.-GFP
36
28
B. />-ERKl/2
total ERK1/2
c-Fos 55
P-actine
GFP libre
C.

169
Figure 51. Effets de la MK sur l'adaptation du RB2-GFP exprimé de manière stable par des cellules HEK 293. En A, abondance du RB2-GFP: electrophorese sur gel et immunobuvardage d'extraits cellulaires totaux basés sur l'anticorps anti-GFP (JL-8). Les cellules étaient soumises aux traitements indiqués pour une période d'incubation de 12 h, en présence de milieu de culture supplémenté en FBS, avant leur lyse. La diminution de l'abondance de la bande d'un poids moléculaire d'approximativement 101 kDa, correspondant à la protéine de fusion, parallèle à l'augmentation de la quantité de GFP libre (bande de 27 kDa) est interprétée comme montrant la dégradation induite par l'agoniste. En B, réponses de signalisation différentielles au niveau de l'activation de la voie ERK 1/2 induite par les différents agonistes du RB2 et de l'induction du facteur de transcription c-Fos, suivant la stimulation de cellules HEK 293 exprimant de manière stable le RB2-GFP pendant une période de 12 h avec la BK (100 nM), le peptide B-9972 (100 nM) et la MK (1 pM). Les essais étaient basés sur l'utilisation d'anticorps anti-phospho-ERK 1/2 et anti-c-Fos; l'utilisation d'anticorps anti-ERK et anti-(3-actine permettait de vérifier le chargement uniforme de protéines sur gel. En C, études de microscopie en épifluorescence (grossissement de 1000 X) sur la localisation de la fluorescence associée au RB2-GFP, suite à des traitements de 30 min, 3 h et 12 h avec la MK, à la même concentration que celle employée pour les essais de quantification du RB2-GFP et de signalisation (soit 1 pM). L'utilisation du colorant Hoechst 33258 permettait la visualisation du noyau (en bleu).

170
3.4 Cargos anticorps anti-récepteur
La construction pharmacologiquement active myc-RB2 possède un epitope myc fusionné au
niveau de l'extrémité N-terminale du récepteur, donc extracellulaire, qui permet la liaison
de l'anticorps monoclonal anti-myc (clone 4A6) et le marquage du récepteur exprimé par
des cellules intactes non perméabilisées. Afin de documenter l'endocytose d'un cargo
anticorps, des cellules HEK 293a intactes exprimant de manière transitoire le myc-RB2
étaient préalablement soumises à un traitement avec un anticorps anti-myc (clone 4A6 ou
clone 9E10), lavées et incubées pour une période additionnelle de 45 min et
optionnellement stimulées avec la BK, à une concentration de 10 nM, ajoutée durant les 15
dernières minutes de cette incubation. À la fin de cette incubation, les cellules étaient
fixées, perméabilisées et soumises au marquage avec un anticorps secondaire conjugué à un
fluorophore. Les images obtenues suite à ces expériences sont présentées à la figure 52. La
fluorescence de l'anticorps secondaire employé ne marquait pas la surface des cellules non
stimulées par la BK d'une manière aussi lisse que celle observée avec l'antagoniste
fluorescent B-10380, ce qui peut indiquer une certaine agrégation de complexes immuns au
niveau de cellules vivantes. Des coupes confocales ont montré que les anticorps anti-myc
se trouvaient principalement au niveau de la surface membranaire des cellules non
stimulées, mais se localisaient plutôt dans des granules intracellulaires de taille variable
lorsque les cellules avaient été traitées avec la BK. Le logiciel Photoshop a été utilisé afin
de mesurer la fluorescence associée à une zone manuellement délimitée et incluant la
membrane plasmique, donc identifiée comme la région corticale, versus la région
intracellulaire, à partir d'expériences répliquées. Les résultats de ces analyses se trouvent à
la figure 53. Bien qu'une certaine internalisation non spécifique des anticoqîs soit
observable, cette méthode d'analyse démontrait une endocytose statistiquement
significative des deux types d'anticorps anti-myc suivant le traitement avec l'agoniste BK;
la disparition du signal fluorescent cortical était le critère morphologique le plus
reproductible. Une manière additionnelle de contrôler la dépendance de l'internalisation de
l'anticorps sur la présence de l'agoniste était de vérifier si l'anticorps anti-myc possédait
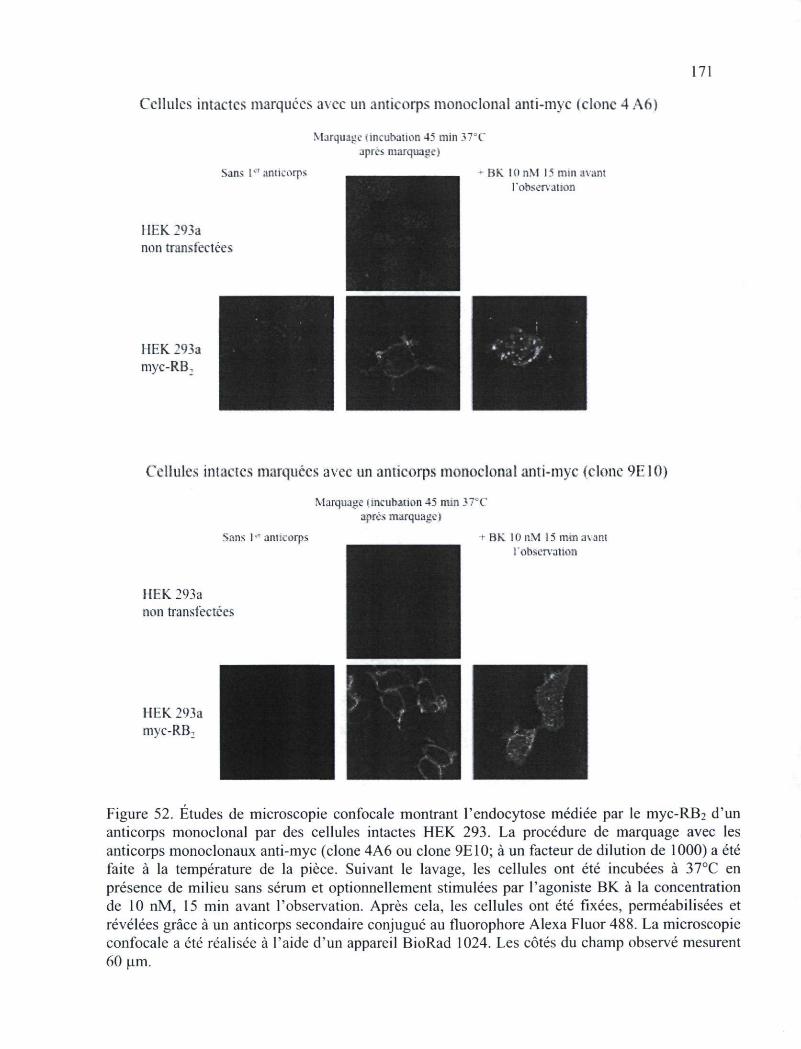
171
Cellules intactes marquées avec un anticorps monoclonal anti-myc (clone 4 A6)
Marquage (incubation 45 min 37T après marquage)
Sans ["anticorps
HEK 293a non transfectées
+ BK lOuM 15 min avant l'observation
HEK 293a mvc-RB,
Cellules intactes marquées avec un anticorps monoclonal anti-myc (clone 9F10)
Marquage (incubation 45 min 37°C après marquage)
Sans 1i* anticorps
HEK 293a non transfectées
r BK 10 nM 15 mita avant }'observation
HEK 293a mvc-RB,
Figure 52. Études de microscopie confocale montrant l'endocytose médiée par le myc-RB2 d'un anticorps monoclonal par des cellules intactes HEK 293. La procédure de marquage avec les anticorps monoclonaux anti-myc (clone 4A6 ou clone 9E10; à un facteur de dilution de 1000) a été faite à la température de la pièce. Suivant le lavage, les cellules ont été incubées à 37°C en présence de milieu sans sérum et optionnellement stimulées par l'agoniste BK à la concentration de 10 nM, 15 min avant l'observation. Après cela, les cellules ont été fixées, perméabilisées et révélées grâce à un anticorps secondaire conjugué au fluorophore Alexa Fluor 488. La microscopie confocale a été réalisée à l'aide d'un appareil BioRad 1024. Les côtés du champ observé mesurent 60 pm.
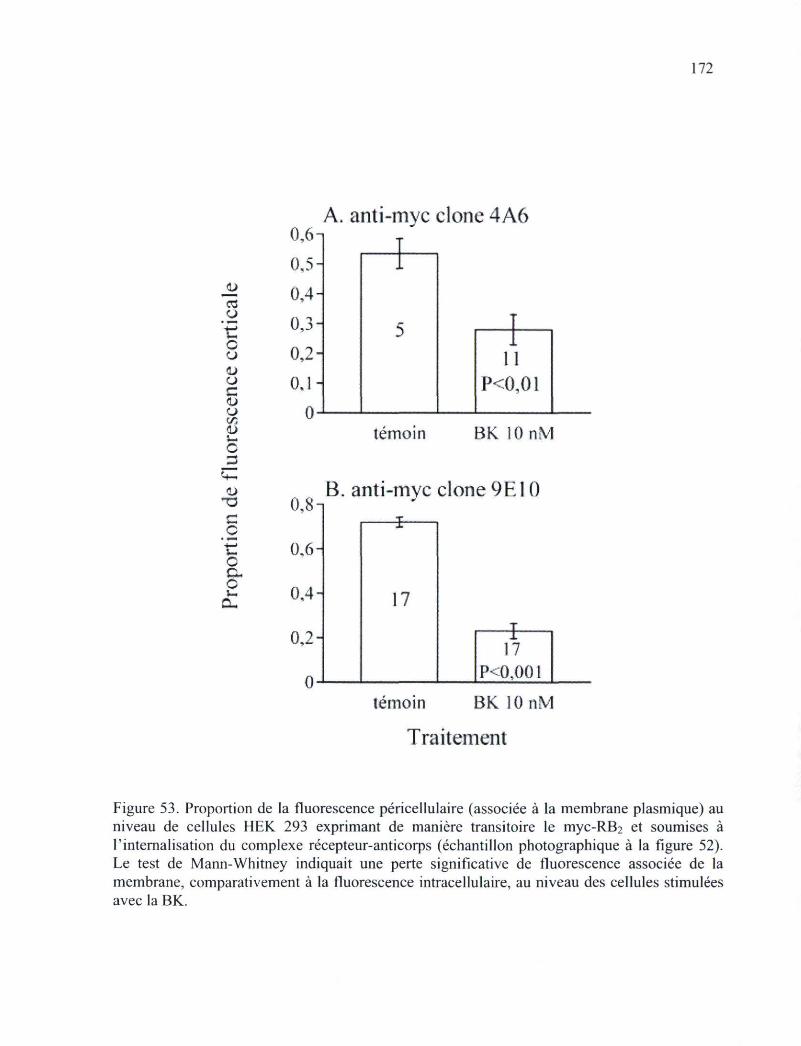
172
03
O c o o O q o « C
•a
c V «nH
c
0,6-1
0,5-0,4
0,3
0,2
0 ,H
0
A. anti-myc clone 4A6
témoin BK 10 nM
0,8-
0,6-
0,4-
0,2-
0-
B. anti-myc clone 9E10 s-
témoin BK 10 nM
Traitement
Figure 53. Proportion de la fluorescence péricellulaire (associée à la membrane plasmique) au niveau de cellules HEK 293 exprimant de manière transitoire le myc-RB2 et soumises à l'internalisation du complexe récepteur-anticorps (échantillon photographique à la figure 52). Le test de Mann-Whitney indiquait une perte significative de fluorescence associée de la membrane, comparativement à la fluorescence intracellulaire, au niveau des cellules stimulées avec la BK.

173
une activité agoniste intrinsèque à la concentration utilisée dans l'essai de marquage.
L'anticorps anti-myc (clone 4A6) s'est révélé incapable d'induire une mobilisation calcique
au niveau de cellules HEK 293a qui exprimaient transitoirement le myc-RB2, comme le
montre la figure 54. La BK induisait quant à elle une intense mobilisation calcique.
Afin de vérifier la capacité des récepteurs à transporter des cargos de taille encore plus
importante, des cellules intactes exprimant le myc-RB2 ont été soumises aux traitements
séquentiels avec l'anticorps anti-myc et avec un anticorps secondaire conjugué à un
fluorophore, qui était soit de type Alexa Fluor (généralement Alexa Fluor 594) ou de type
Qdot. Après une période d'équilibration à 37°C, les cellules ont optionnellement été traitées
avec la BK. Un échantillon photographique des résultats découlant de ce double marquage
est présenté à la figure 55 (pour les anticorps secondaires couplés à un fluorophore de type
Alexa Fluor), ainsi qu'à la figure 56 (pour les anticorps secondaire couplés aux Qdots). Les
photographies des cellules ont été analysées sur la base de la quantification de la
fluorescence corticale, ce qui a permis l'évaluation statistique de l'internalisation
dépendante de l'agoniste. Ces données statistiques sont présentées aux figures 57 (anticorps
couplés à des fluorophores Alexa Fluor) et 58 (anticorps couplés aux Qdots), selon le type
de fluorophore mis en valeur. Les complexes immuns formés à la surface cellulaire étaient
internalises 15 min après la stimulation des récepteurs par l'agoniste; par ailleurs une
préincubation avec l'antagoniste LF 16-0687, à la concentration de 1 pM, inhibait en grande
partie l'endocytose du complexe immun induite par la BK. Il est à noter que la fluorescence
associée aux Qdots demeurait intracellulaire 3 h après le traitement des cellules par la BK,
comme le montre la figure 56. Des cellules non transfectées ne présentaient aucune
fluorescence associée à la construction de complexes immuns à leur surface, telle que
démontré par les figures 55 et 56. La version tronquée du myc-RB2, myc-RB2jRUNC,
lorsqu'exprimée par des cellules HEK 293a, permettait la construction de complexes
immuns contenant les Qdots à la surface cellulaire, mais ceux-ci n'étaient pas transloqués
vers l'intérieur des cellules stimulées avec la BK, comme cela peut être observé à la figure
56 (les analyses statistiques sont présentées à la figure 58). L'agoniste fluorescent CF-
eACA-BK, employé à la concentration de 5 pM pendant 15 min, induisait aussi
l'endocytose de complexes immuns présentant les Qdots, vers des endosomes qui
présentaient un double marquage de fluorescence rouge et verte. Ces expériences sont
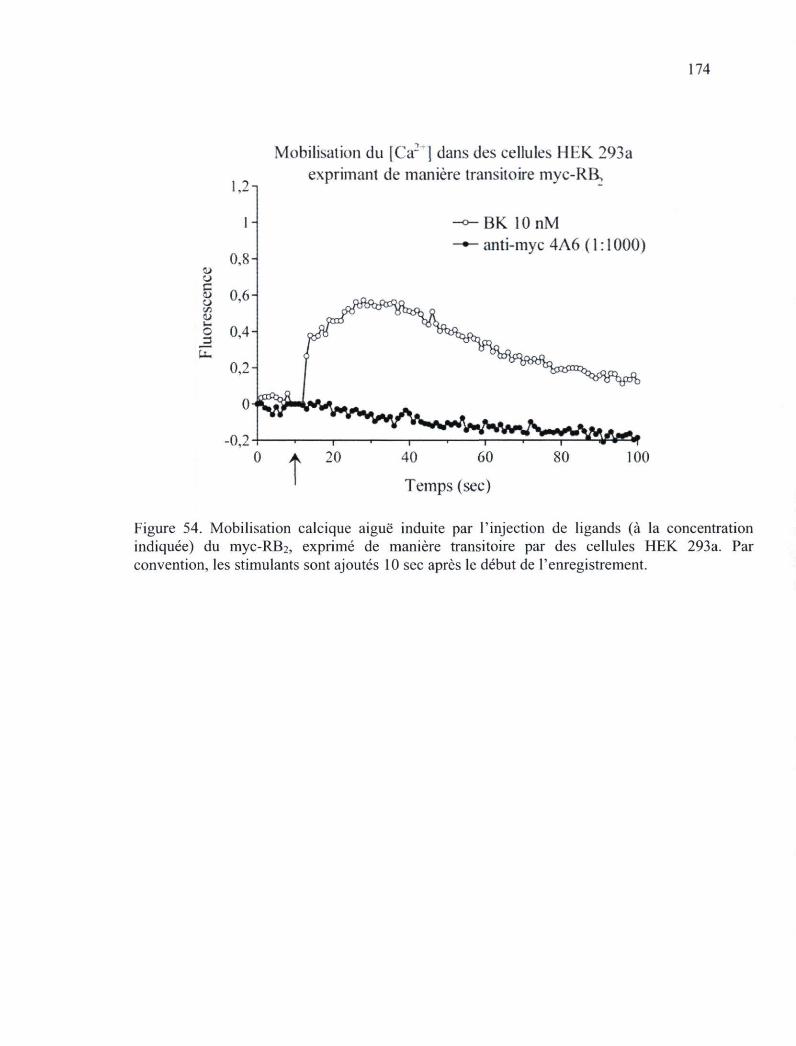
174
Mobilisation du [Ca2+] dans des cellules HEK 293a exprimant de manière transitoire myc-RB,
-<^-BK 10 nM — anti-myc 4A6( 1:1000)
Temps (sec)
Figure 54. Mobilisation calcique aiguë induite par l'injection de ligands (à la concentration indiquée) du myc-RB2, exprimé de manière transitoire par des cellules HEK 293a. Par convention, les stimulants sont ajoutés 10 sec après le début de l'enregistrement.
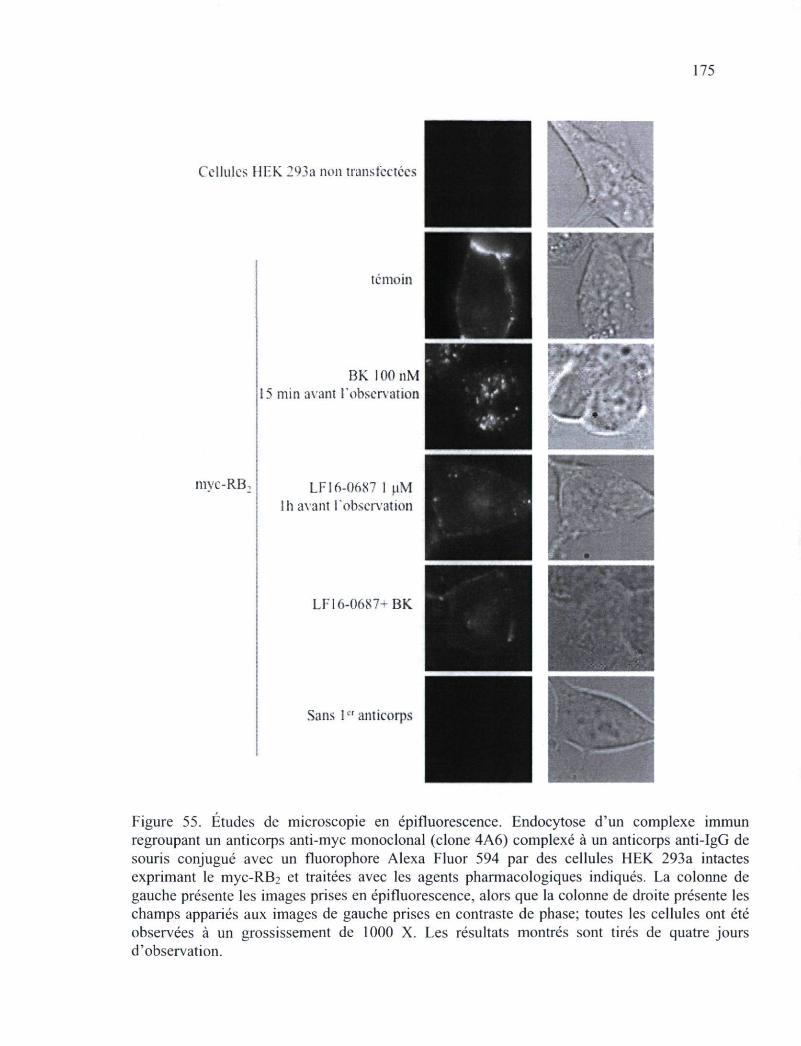
175
Cellules HEK 293a non transfectées
mvc-RB,
témoin
BK 100 nM 5 min avant F observation
LF 16-0687 1 uM lh avant l'observation
LF 16-0687-BK
Sans 1er anticorps
Figure 55. Études de microscopie en épifluorescence. Endocytose d'un complexe immun regroupant un anticorps anti-myc monoclonal (clone 4A6) complexé à un anticorps anti-IgG de souris conjugué avec un fluorophore Alexa Fluor 594 par des cellules HEK 293a intactes exprimant le myc-RB2 et traitées avec les agents pharmacologiques indiqués. La colonne de gauche présente les images prises en épifluorescence, alors que la colonne de droite présente les champs appariés aux images de gauche prises en contraste de phase; toutes les cellules ont été observées à un grossissement de 1000 X. Les résultats montrés sont tirés de quatre jours d'observation.
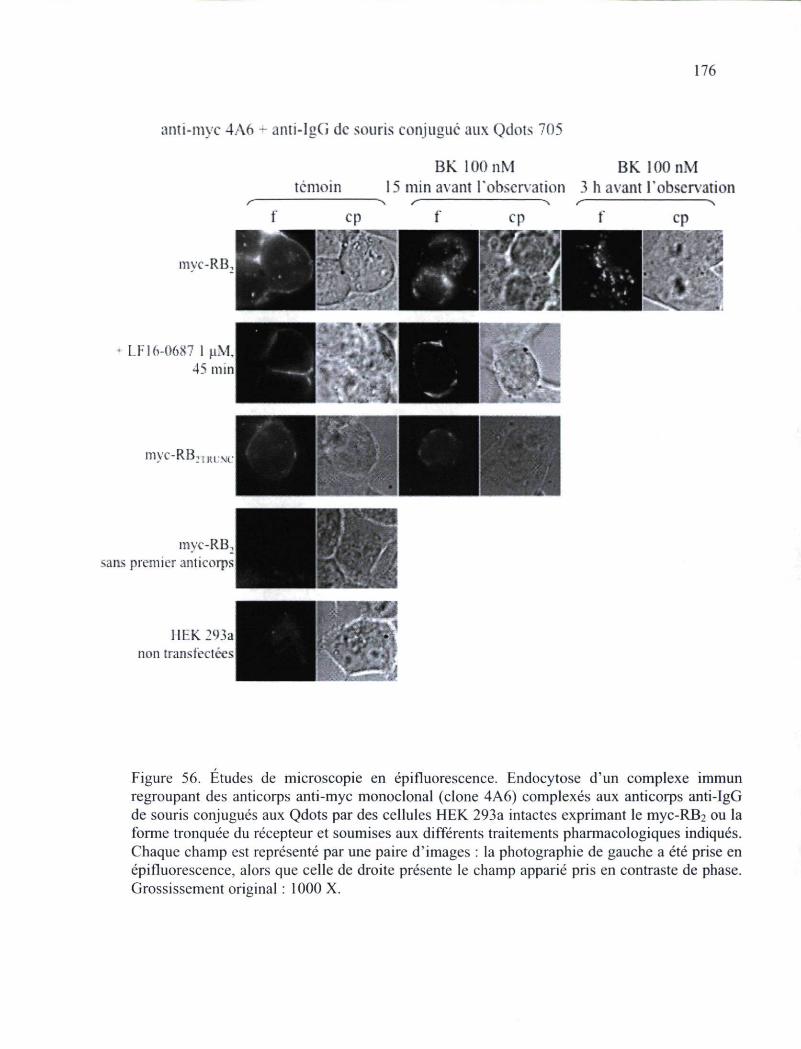
176
anti-myc 4A6 - anti-IgG de souris conjugue aux Qdots 705
BKlOOnM BKlOOnM témoin 15 min avant l'observation 3 h avant l'observation
mvc-RB,
• LF 16-0687 1 p\1, 45 min
mvc-RB tRUNC
myc-RB, sans premier anticorps
HEK 293a non transfectées
Figure 56. Études de microscopie en épifluorescence. Endocytose d'un complexe immun regroupant des anticorps anti-myc monoclonal (clone 4A6) complexés aux anticorps anti-IgG de souris conjugués aux Qdots par des cellules HEK 293a intactes exprimant le myc-RB2 ou la forme tronquée du récepteur et soumises aux différents traitements pharmacologiques indiqués. Chaque champ est représenté par une paire d'images : la photographie de gauche a été prise en épifluorescence, alors que celle de droite présente le champ apparié pris en contraste de phase. Grossissement original : 1000 X.
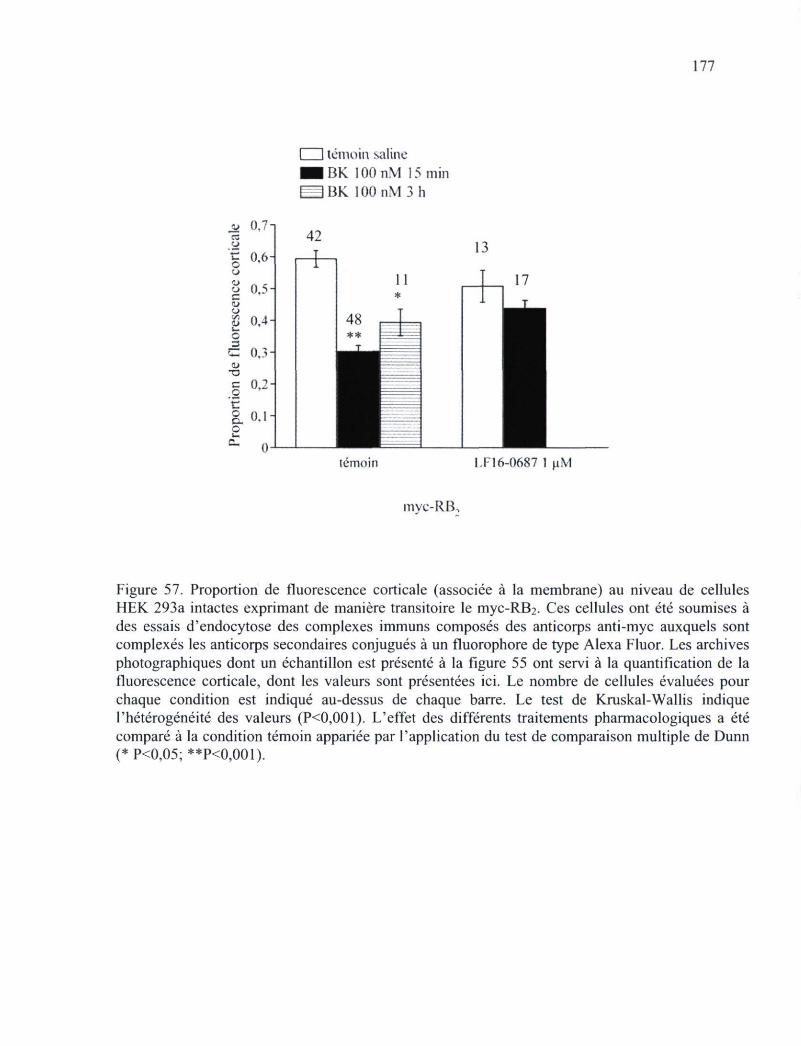
177
I I témoin saline ■ BK lOOnM 15 min Ë H B K 100 nM 3 h
a 0,7
témoin I. F160687 1 pM
mycRB,
Figure 57. Proportion de fluorescence corticale (associée à la membrane) au niveau de cellules HEK 293a intactes exprimant de manière transitoire le mycRB2. Ces cellules ont été soumises à des essais d'endocytose des complexes immuns composés des anticonps antimyc auxquels sont complexés les anticorps secondaires conjugués à un fluorophore de type Alexa Fluor. Les archives photographiques dont un échantillon est présenté à la figure 55 ont servi à la quantification de la fluorescence corticale, dont les valeurs sont présentées ici. Le nombre de cellules évaluées pour chaque condition est indiqué audessus de chaque barre. Le test de KruskalWallis indique l'hétérogénéité des valeurs (P<0,001). L'effet des différents traitements pharmacologiques a été comparé à la condition témoin appariée par l'application du test de comparaison multiple de Dunn (* P<0,05; **P<0,001).
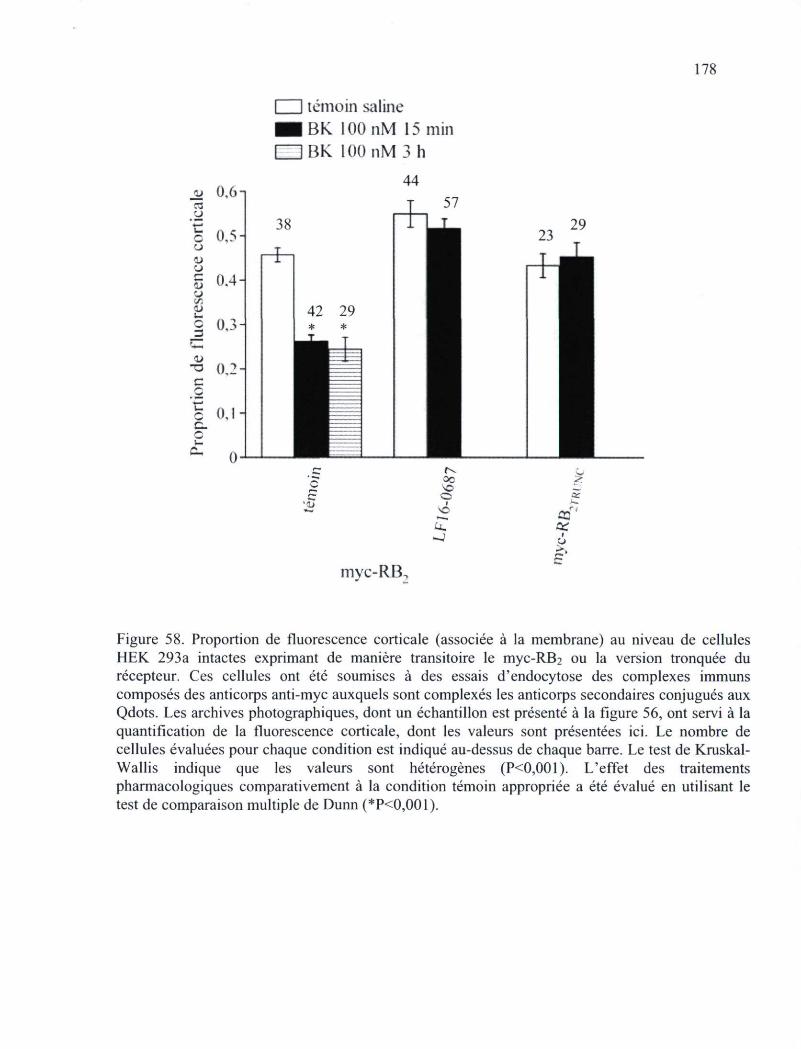
178
I 1 témoin saline ■ 1 B K 100 nM 15 min □ BK 100nM3h
« 0,6
23 29
ea
... j?
mycRB,
Figure 58. Proportion de fluorescence corticale (associée à la membrane) au niveau de cellules HEK 293a intactes exprimant de manière transitoire le mycRB2 ou la version tronquée du récepteur. Ces cellules ont été soumises à des essais d'endocytose des complexes immuns composés des anticorps antimyc auxquels sont complexés les anticorps secondaires conjugués aux Qdots. Les archives photographiques, dont un échantillon est présenté à la figure 56, ont servi à la quantification de la fluorescence corticale, dont les valeurs sont présentées ici. Le nombre de cellules évaluées pour chaque condition est indiqué audessus de chaque barre. Le test de Kruskal
Wallis indique que les valeurs sont hétérogènes (P<0,001). L'effet des traitements pharmacologiques comparativement à la condition témoin appropriée a été évalué en utilisant le test de comparaison multiple de Dunn (*P<0,001).

179
présentées au haut de la figure 59. Les systèmes exploitant les deux types de fluorophores
et menant à la construction de complexes immuns à la surface cellulaire et à leur
internalisation suivant l'ajout de l'agoniste ont été exploités plus avant au sein de cellules
exprimant le myc-RB2 et un autre transgène. Les résultats de cette étude approfondie se
trouvent aux figures 59 et 60. La |3-arrestine2-GFP, qui est généralement cytosolique dans
les cellules au repos, subit une translocation vers les endosomes dans les cellules traitées
avec l'agoniste, comme le montre la figure 42. La (3-arrestine2-GFP présentait une
importante colocalisation avec les complexes immuns internalises, autant lorsque ceux-ci
étaient composés d'anticorps secondaires couplés à un fluorophore Alexa Fluor (les
résultats sont présentés à la figure 60) ou d'anticorps conjugués aux nanocristaux Qdots, au
niveau de cellules co-exprimant le myc-RB2 stimulées par la BK, ce qui indiquait que les
complexes immuns étaient liés à des récepteurs phosphorylés internalises après une
stimulation de 15 min. Les complexes immuns contenant les Qdots se trouvaient aussi
localisés à proximité d'une protéine associée aux endosomes précoces EEA1-FYVE-GFP,
suivant le traitement des cellules marquées avec la BK. La coexpression du myc-RB2 et de
la construction GFP-Rab7, qui marque les endosomes tardifs (Marceau et coll., 2009) par
des cellules HEK 293a, a mené à l'observation d'une faible colocalisation en fonction du
temps d'incubation, avec les deux types d'anticorps secondaires. Cette colocalisation qui
s'intensifie au fil du temps montre la progression des cargos dans la voie endosomale
(figures 59 et 60). De plus, les complexes immuns formés par le récepteur, l'anticorps anti-
myc et l'anticorps secondaire anti-IgG de souris conjugué avec un fluorophore Alexa Fluor
488 étaient partiellement colocalisés avec la BSA marquée avec le fluorophore Alexa Fluor
594 3 h après la stimulation avec la BK. Par contre, il n'y avait aucune colocalisation entre
une protéine se trouvant sur les autophagosomes, le GFP-LC3 (Marceau et coll., 2009) et
l'un ou l'autre de complexes immuns formés à la surface cellulaire et internalises suite à un
traitement de 3 h avec la BK, ce qui indique que l'accumulation des vésicules contenant les
cargos n'induisait pas l'autophagie, comme le montrent les figures 59 et 60.
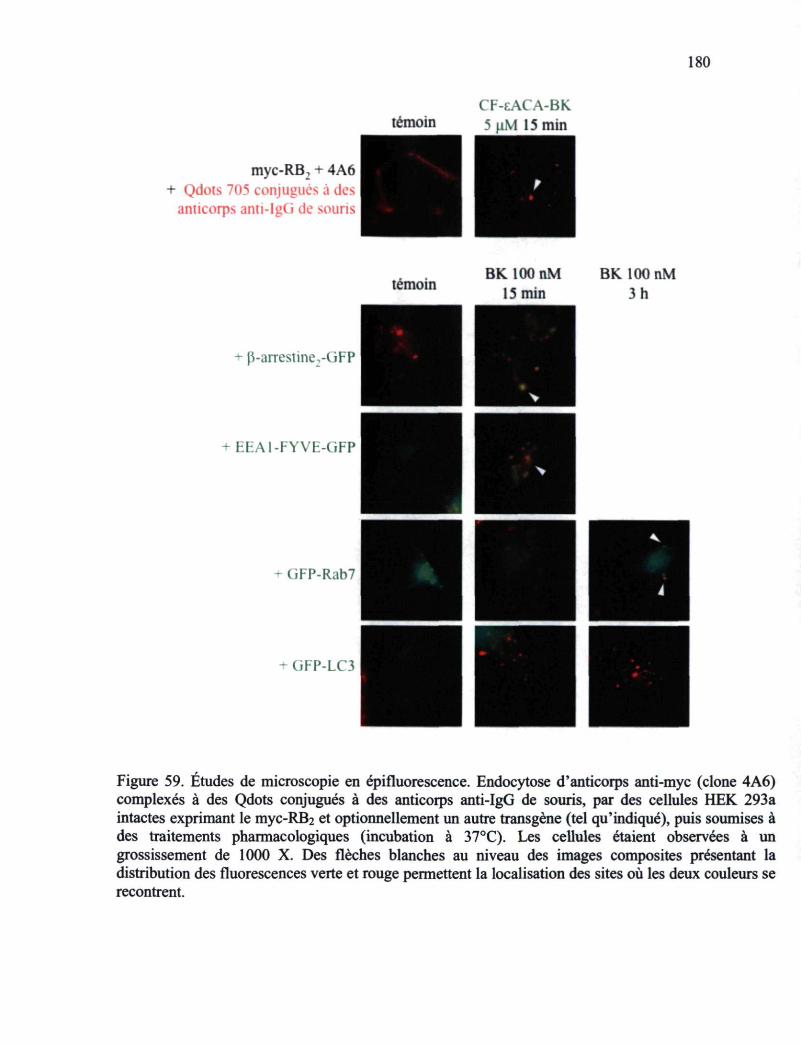
180
témoin C F - E A C A - B K 5 pM 15 min
myc-RB2 + 4A6 + Qdots 705 conjugués à des
anticorps anti-IgG de souris
témoin BKlOOnM
15 min BK 100 nM
3h
[i-arrestine,-GFP
+ EEA1-FYVE-GFP
GFP-Rab7
- GFP-LC3
Figure 59. Études de microscopie en épifluorescence. Endocytose d'anticorps anti-myc (clone 4A6) complexés à des Qdots conjugués à des anticorps anti-IgG de souris, par des cellules HEK 293a intactes exprimant le myc-RB2 et optionnellement un autre transgène (tel qu'indiqué), puis soumises à des traitements pharmacologiques (incubation à 37°C). Les cellules étaient observées à un grossissement de 1000 X. Des flèches blanches au niveau des images composites présentant la distribution des fluorescences verte et rouge permettent la localisation des sites où les deux couleurs se recontrent.
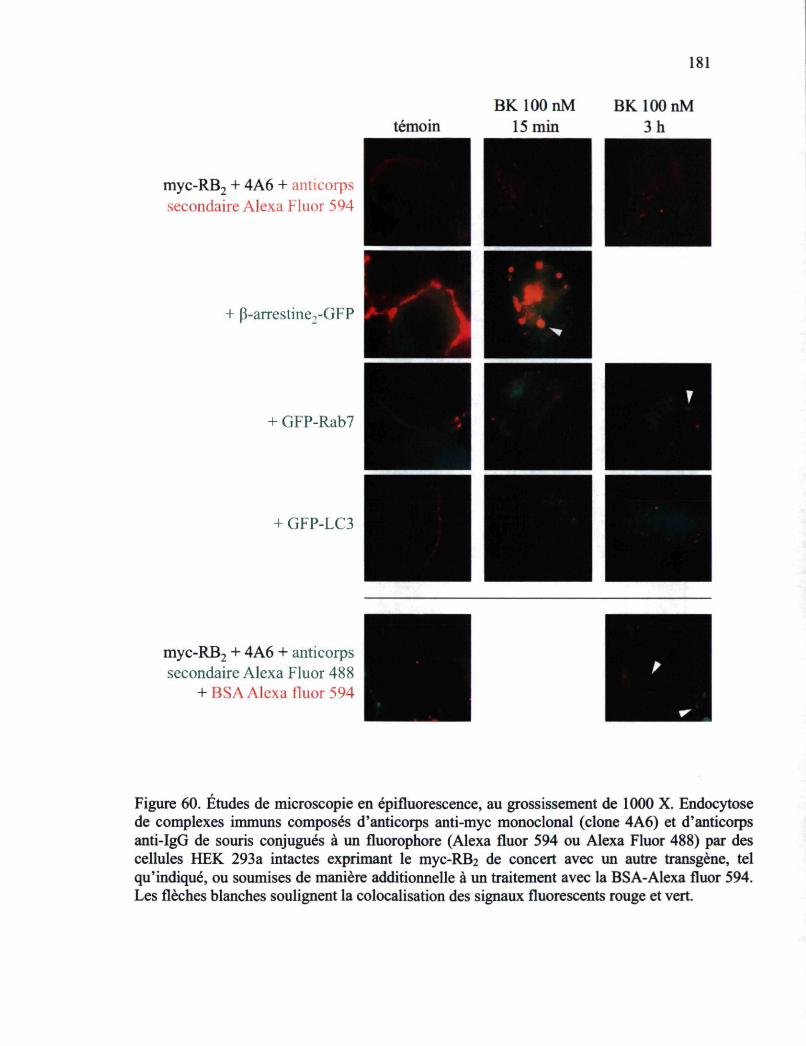
181
témoin BK 100 nM
15 min BKlOOnM
3h
myc-RB2 + 4A6 + anticorps secondaire Alexa Fluor 594
+ P-arrestine2-GFP
+ GFP-Rab7
+ GFP-LC3
myc-RB2 + 4A6 + anticorps secondaire Alexa Fluor 488
+ BSA Alexa fluor 594
Figure 60. Études de microscopie en épifluorescence, au grossissement de 1000 X. Endocytose de complexes immuns composés d'anticorps anti-myc monoclonal (clone 4A6) et d'anticorps anti-IgG de souris conjugués à un fluorophore (Alexa fluor 594 ou Alexa Fluor 488) par des cellules HEK 293 a intactes exprimant le myc-RB2 de concert avec un autre transgène, tel qu'indiqué, ou soumises de manière additionnelle à un traitement avec la BSA-Alexa fluor 594. Les flèches blanches soulignent la colocalisation des signaux fluorescents rouge et vert.

182
4. Discussion
4.1 Profil pharmacologique de l'agoniste résistant au métabolisme B-9972 et induction efficace de la dégradation du RB2 par son action
4.1.1 Profil pharmacologique de la triade de peptides isomères L'antagoniste du RB2 à structure contrainte B-9430 a déjà fait l'objet d'études (Leeb-
Lundberg et coll., 2005), mais n'a pas été caractérisé au niveau d'essais fonctionnels de ce
sous-type de récepteur, comme la contractilité de la veine jugulaire de lapin et de la veine
ombilicale humaine (Marceau et coll., 1994, 2003; Rizzi et coll., 1997; Pruneau et coll.,
1999; Houle et coll., 2000). La combinaison d'un effet insurmontable du B-9430 au niveau
du RB2 de lapin avec un effet compétitif et réversible du même antagoniste au niveau du
RB2 humain est le même profil particulier aussi observé pour l'icatibant, un autre peptide à
structure contrainte (Marceau et coll., 1994; Houle et coll., 2000). Le comportement
insurmontable de l'icatibant, un effet dépendant de l'espèce d'origine du récepteur, a été
vérifié au niveau du RB2 de lapin recombinant (Bachvarov et coll., 1995). Le peptide B-
9430 est peut-être, comme l'icatibant, un antagoniste faiblement réversible au RB2 de lapin,
étant donné le fait qu'un bolus intraveineux de ce composé inhibe l'effet hypotenseur de la
BK pendant 24 h chez le lapin (Stewart et coll., 1996). L'antagonisme non compétitif
exercé par le peptide B-9430 et l'icatibant au RB2 de lapin peut être expliqué par les
différences au niveau de la séquence d'acides aminés (16 %) qu'il présente
comparativement à son orthologue humain (Bachvarov et coll., 1995) pour lequel les deux
composés sont compétitifs. D'autres antagonistes, tel le composé non peptididique LF16-
0687 présente un caractère compétitif pour les RB2 humain et lapin (Pruneau et coll., 1999;
Houle et coll., 2000; Bawolak et Marceau, 2007).
Le peptide B-9870 (CU201) consiste en un dimère de deux molécules du peptide B-9430
liées par leur extrémité N-terminale par le groupement caractéristique suberimidyl (Géra et
coll., 1996). Ce peptide de taille importante exerce, à des concentrations micromolaires, des

183
effets antiprolifératifs qui ne sont pas partagés par le monomère B-9430 sur certaines
lignées cellulaires tumorales (Chan et coll., 2002; Thomas et coll., 2006) et sont
dissociables de sa liaison aux récepteurs des kinines (Morissette et coll., 2007). Par contre,
le peptide B-9870 a plusieurs points en commun avec le peptide B-9430, en tant
qu'antagoniste des récepteurs des kinines possédant une affinité nanomolaire. Ainsi, le
peptide B-9870 est un antagoniste insurmontable de la BK au niveau de la veine jugulaire
de lapin, alors qu'il présente un comportement d'antagoniste compétitif de relativement
basse affinité (pA2, 7,4) pour le RBi exprimé par l'aorte de lapin (Morissette et coll., 2007).
Le peptide B-9430 est aussi un antagoniste surmontable du RB] (pA2, 6,5; les résultats sont
présentés à la figure 6). Des évidences récentes indiquent que le comportement
insurmontable des peptides à structure contrainte au RB2 de lapin découle de l'activité
agoniste partielle détectable dans les cellules qui surexpriment des récepteurs recombinants
de ce sous-type, telle que déterminée par l'activation des MAP kinases, la mobilisation
calcique et l'endocytose lente et partielle des récepteurs membranaires (Morissette et coll.,
2007). Ces données ont été confirmées par les résultats obtenus ici, comme l'indiquent la
dégradation minime du RB2-GFP suite à un traitement prolongé en sa présence (figure 11)
et le comportement d'agoniste partiel dans les expériences de mobilisation calcique (dont
les résultats ne sont pas montrés).
L'isomère obtenu par l'échange des résidus aux positions 7 et 8 du peptide B-9430, soit le
peptide B-10344, est un agoniste de basse affinité, tel que le démontre les essais de
contractilité effectués grâce aux deux systèmes veineux. Il est un agoniste partiel au niveau
de la veine jugulaire de lapin, dont la courbe concentration-effet présente une pente
amoindrie, comparativement à celle obtenue par la BK (figure 5, en A) et dont la puissance
résiduelle est beaucoup plus basse que celle prédite par l'essai de liaison, toujours par
comparaison avec la BK (figure 4). Par contre, il est un agoniste complet dans le même
essai réalisé cette fois avec la veine ombilicale humaine, puisqu'il présente un effet
maximal similaire à celui obtenu avec la BK. L'isomérisation en L du résidu D-Igl , une
modification additionnelle retrouvée chez le peptide B-9972, lui confère un comportement
d'agoniste complet au sein des RB2 des deux espèces. Les modifications séquentielles des
propriétés pharmacologiques de la triade d'isomères présentés ici illustrent l'importance de
l'orientation spatiale de l'extrémité C-terminale dans la détermination de la transition d'un

184
agoniste RB2 en un antagoniste (Vavrek et Stewart, 1985; Fortin et Marceau, 2006). Ces
données sont cohérentes avec des études de modélisation basées sur la technique de
mutagénèse sur les sites d'interaction entre le RB2 de rat et l'icatibant, qui montraient
l'interaction préférentielle de l'extrémité C-terminale avec des résidus du TM7, alors que la
BK interagit davantage avec le TM6 (Jarnagin et coll., 1996). L'agoniste B-9972 présente
une activité supérieure au niveau du RB2 à l'analogue plus ancien [Phe8\|t(CH2NH)Arg9]BK
et intègre théoriquement une résistance plus complète aux différentes peptidases
(aminopeptidases, endopeptidases). L'application de la méthode radiorécepteur a montré
l'inactivation importante du peptide B-9972 lorsqu'il est incubé en présence de cellules
pendant une période de 12 h, quoique sa dégradation soit moins complète que celle de la
BK. La résistance à l'inactivation peut expliquer l'importante puissance du peptide B-9972
comparativement à celle de la BK au niveau de l'essai de contractilité de la veine
ombilicale humaine (rapport de puissance 1:2), alors qu'il retient une faible fraction de
l'affinité de la BK, comme le montre l'essai de liaison exploitant le RB2-GFP (1:7; montrée
à la figure 4). L'essai de contractilité de la veine jugulaire de lapin est généralement réalisé
en présence de l'IECA captopril, un agent qui accroît l'activité apparente de la BK au sein
de cette préparation (Gaudreau et coll., 1981), mais non au sein de la veine ombilicale
humaine (Marceau et coll., 1994). La lente relaxation suivant le lavage de la veine
ombilicale humaine préstimulée avec soit le peptide B-9972, soit le peptide B-10344,
comparativement à la relaxation plus rapide aux tissus stimulés avec la BK ou la
[Phe8iJt(CH2NH)Arg9]BK, supporte plus avant le fait que ces derniers sont moins résistants
à plusieurs voies de dégradation extracellulaires et intracellulaires. Ces voies peuvent
recruter des enzymes différentes de l'ECA, de l'endopeptidase neutre et de
l'aminopeptidase P, une conclusion suggérée par l'absence d'effet des inhibiteurs de ces
peptidases sur la puissance apparente de la BK et sur la cinétique de relaxation, suivant le
retrait de la BK de l'environnement du RB2 par un lavage, comme le montre la figure 9.

185
4.1.2 Dégradation du RB2 par un traitement prolongé avec l'agoniste B-9972
La protéine de fusion RB2-GFP, dont le poids moléculaire est approximativement de 101
kDa, est un récepteur fonctionnel de haute affinité, présentant un profil pharmacologique
intact. La GFP conjuguée au récepteur présente le triple avantage de permettre l'imagerie
du récepteur dans des cellules intactes, d'être un excellent site antigénique pour la
réalisation d'immunobuvardages et d'être un produit de réaction stable après la dégradation
du récepteur (Houle et coll., 2003; Houle et Marceau, 2003). La génération de la GFP libre
ou sa variante, la YFP, a aussi été observée suivant le métabolisme du récepteur B de
l'endothéline (ETB), un récepteur qui est sujet à la dégradation (Oksche et coll., 2000), ou
du RB,, qui présente une courte demi-vie (Fortin et coll., 2003). Les agonistes BK et B-
9972 provoquent une endocytose rapide du RB2-GFP, comme le montrent la figure 10 et les
flèches 1 et 2 du schéma récapitulatif constituant la figure 61. L'abondance de la protéine
de fusion au niveau d'extraits cellulaires totaux restait constante suivant 3 h de stimulation,
comme le présentent les figures 11 et 12; par contre lors de longues stimulations de 12 h, le
peptide B-9972 provoquait la baisse de la quantité du RB2-GFP mature et la hausse de
l'abondance de la GFP libre, comme le présentent les immunobuvardages à la figure 11 et
les statistiques présentées à la figure 12. Cette voie est identifiée par la flèche 4 dans le
schéma présenté à la figure 61. Cet effet spécifique était aboli par l'antagoniste peptide B-
9430 ou l'antagoniste non peptide LF 16-0687. La BK seule était inactive à cet égard; par
contre, lorsque le traitement avec la BK était supplémenté avec 1TECA captopril, cet
agoniste pouvait entraîner une petite fraction de la dégradation du RB2 obtenue par le
peptide B-9972, comme le démontre la génération de la GFP libre. Des effets similaires en
fonction de la durée des traitements ont été observés lors l'utilisation alternative de la
construction myc-RB2, comme le présentent les figures 12 et 15; par contre, la position N-
terminale de l'épitope antigénique était moins favorable à la détection des produits de
réaction du myc-RB2. Ainsi, en l'absence de cycloheximide pendant les traitements de 12
h, il est possible de supposer que les récepteurs sont toujours synthétisés à un rythme

186
constant puisque soumis à la régulation du promoteur viral des vecteurs d'expression, et
que le déclin lent de la quantité de récepteurs s'explique par un taux de dégradation
supérieur des récepteurs au sein des cellules traitées par le peptide B-9972. La diminution
de la fonction du RB2-GFP à la surface cellulaire suite à un traitement prolongé avec le B-
9972 a été corroborée grâce à la réalisation d'essais d'internalisation de la [3H]BK ainsi que
d'essais de phosphorylation des kinases ERK1/2, dont les résultats sont présentés à la
figure 16. Ces observations sont compatibles avec la dégradation des RB2, sans exclure une
possible internalisation du récepteur dépendante de l'agoniste. Les conditions pauvres en
sérum mises en place lors des essais de phosphorylation amélioraient l'efficacité de la
combinaison BK-captopril à désensibiliser fonctionnellement le récepteur. L'ECA est une
enzyme de dégradation majeure dans le milieu de culture supplémenté en sérum des
cellules HEK 293 (Bachvarov et coll., 2001; le schéma à la figure 61 en fait état), mais elle
n'est probablement pas impliquée dans la dégradation de l'agoniste au niveau
intracellulaire. La résistance du peptide B-9972 à de multiples aminopeptidases et
endopeptidases non identifiées, qui lui est conférée par sa structure, peut être cruciale pour
la capacité de cet agoniste à promouvoir la dégradation du récepteur lorsque l'expérience
est réalisée en présence de sérum, comparativement à l'effet obtenu par la combinaison
BK-captopril. Seuls des traitements prolongés par un agoniste synthétique de cellules
exprimant les récepteurs (32-adrénergiques provoquaient la dégradation de ces récepteurs
habituellement recyclés (Moore et coll., 1999); les présents résultats suggèrent un
comportement similaire du RB2 si l'agoniste, de nature fragile, est stabilisé. Par contre, les
cellules HEK 293 exprimant le RB2-GFP ont été utilisées par le passé pour démontrer des
mécanismes de dégradation plus efficaces et rapides en réponse à des proteases
extracellulaires, dont celles sécrétées par les neutrophiles (Marceau et coll., 2002; Houle et
coll., 2003). La suggestion que la conjugaison et la présence de la GFP présente à
l'extrémité C-terminale du RB2-GFP redirige celui-ci vers le recyclage plutôt que vers la
dégradation (Kalatskaya et coll., 2006) n'est pas supportée par les résultats obtenus par les
présentes études, puisque les immunobuvardages montrés à la figure 11 suggèrent une
proportion inférieure de la protéine de fusion myc-RB2 dégradée, comparativement à celle
des RB2-GFP. Par contre, étant donné l'expression de myc-RB2 immunoréactifs au sein de
granules cytosoliques, comme le montre la figure 14, la proportion de récepteurs matures

187
vulnérables à la dégradation est incertaine dans ce système d'expression transitoire. De
plus, l'espèce du RB2, la technique employée pour sa détection et les niveaux d'expression
étaient différents de ceux décrits par Kalatskaya et collaborateurs (2006), rendant une
comparaison détaillée impossible. Néanmoins, la nature de la stimulation qui mène à la
dégradation est similaire pour les deux constructions et le traitement le plus efficace à cet
égard est une incubation de 12 h en présence du peptide B-9972.
En résumé, les résultats illustrent la transition agoniste-antagoniste des ligands peptidiques
présentant une extrémité C-terminale contrainte, la variabilité entre les espèces du profil
pharmacologique du RB2 et la possibilité de provoquer la dégradation de ce récepteur d'une
manière sélective en utilisant un agoniste résistant aux peptidases.
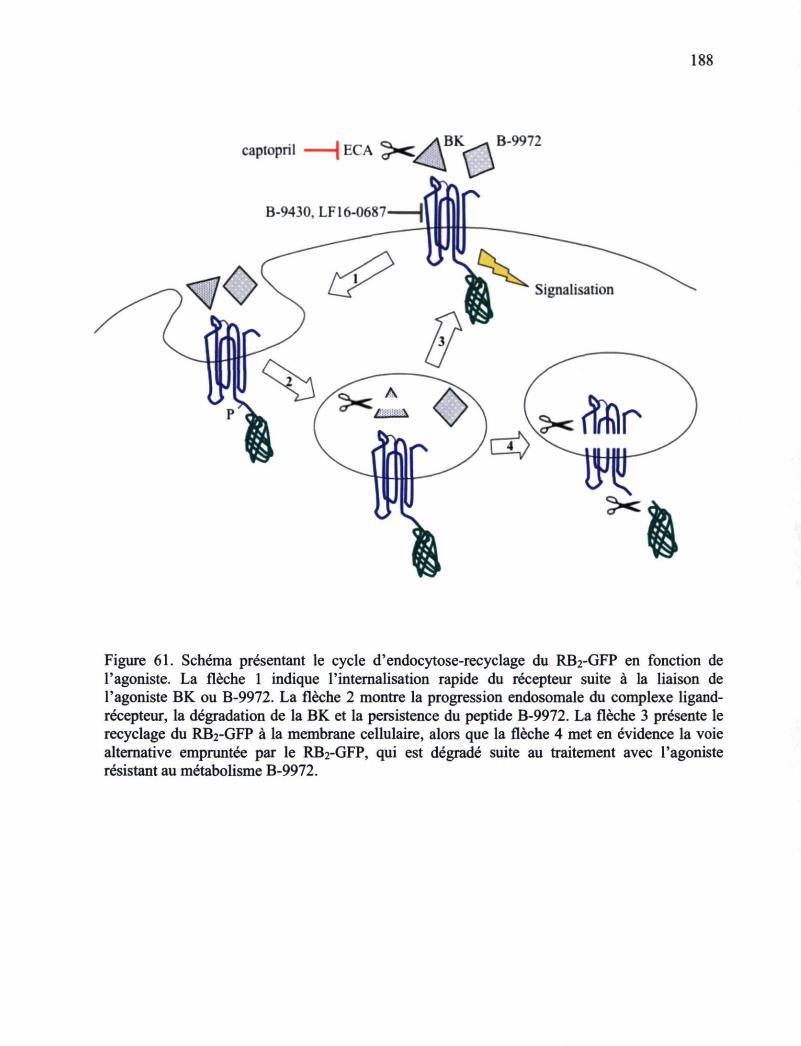
188
captopril ECA BK ^ B-9972 MO B-9430, LF 16-0687
Figure 61. Schéma présentant le cycle d'endocytose-recyclage du RB2-GFP en fonction de l'agoniste. La flèche 1 indique l'internalisation rapide du récepteur suite à la liaison de l'agoniste BK ou B-9972. La flèche 2 montre la progression endosomale du complexe ligand-récepteur, la dégradation de la BK et la persistence du peptide B-9972. La flèche 3 présente le recyclage du RB2-GFP à la membrane cellulaire, alors que la flèche 4 met en évidence la voie alternative empruntée par le RB2-GFP, qui est dégradé suite au traitement avec l'agoniste résistant au métabolisme B-9972.

189
4.2 Effets des agonistes résistants à l'inactivation sur la signalisation, la désensibilisation et la dégradation des RB2
La synthèse d'un agoniste non peptidique pour des récepteurs d'hormones peptidiques est
un objectif intéressant, qui a été accompli pour le RATi (avec le composé L-162,313;
Perlman et coll., 1995) ainsi que pour le récepteur V2 de la vasopressine (avec le composé
OPC-51803; Nakamura et coll., 2004). Le développement clinique de modulateurs non
peptidiques de récepteurs d'hormones peptidiques permettrait la génération de composés
présentant une meilleure biodisponibilité et une plus longue demi-vie.
4.2.1 Caractérisation pharmacologique et effets découlant de la résistance métabolique du composé non peptide 47a au niveau de modèles ex vivo
Pour le RB2, une étude précédente avait montré que le composé FR 190007, qui est
apparenté au composé 47a, produisait 70 % de la contraction maximale obtenue par la BK
au niveau de la veine ombilicale humaine et 30 % de la contraction maximale obtenue par
la BK au niveau de la veine jugulaire de lapin (Rizzi et coll., 1999). Ainsi, en dépit de
l'identification du composé 47a en tant qu'agoniste complet de RB2 recombinants (Sawada
et coll., 2004), celui-ci ne possède pas une activité intrinsèque très importante,
particulièrement au RB2 de lapin, au niveau duquel il agit comme un agoniste partiel dans
plusieurs essais in vitro (phosphorylation de ERK 1/2, induction de c-Fos, dégradation du
RB2-GFP). L'effet du composé 47a dépend de sa liaison au RB2 exprimé par la veine
ombilicale humaine, comme le montrent les paramètres semblables dérivés de la régression
de Schild obtenus suite à la construction de courbes concentration-réponse à la BK ou au
composé 47a en présence de l'antagoniste LF 16-0687 (figures 17 et 18). Bien que
possédant une affinité pour le récepteur inférieure à celle du peptide B-9972, le composé

190
47a cause une relaxation particulièrement lente après son retrait par rinçage des tissus
(tracé type présenté à la figure 20), ce qui est cohérent avec l'utilisation du paramètre de
demi-relaxation pour évaluer la résistance d'un ligand à l'inactivation. Par ailleurs ce
paramètre est indépendant de l'affinité pour le récepteur. De plus, le composé 47a
désensibilise la réponse de la veine ombilicale humaine à la BK durant une période de 3 h,
un effet qui n'est pas partagé par le peptide B-9972, partiellement résistant au métabolisme,
comme le démontre la figure 19. Ainsi, les résultats des essais de contractilité étaient
compatibles avec une signalisation prolongée et la désensibilisation du RB2 après la
stimulation avec le composé 47a. Les mécanismes moléculaires détaillés du processus de
désensibilisation, qui peuvent ou non inclure la dégradation du RB2, ne peuvent être dérivés
à partir de l'essai de contractilité.
4.2.2 Caractérisation pharmacologique et effets découlant de la résistance métabolique du composé non peptide 47a au niveau de modèles cellulaires
Les systèmes cellulaires exploités afin de caractériser les agonistes résistants au
métabolisme exprimaient les constructions RB2-GFP ou myc-RB2. Le RB2-GFP présente la
même affinité que le RB2 de type sauvage de lapin, telle qu'évaluée par des essais de
liaison de la [3H]BK (Bachvarov et coll., 2001) et active les mêmes voies de signalisation
(activation de la PLA2, mobilisation calcique, phosphorylation ERK 1/2; Houle et coll.,
2000; Morissette et coll., 2007). L'affinité du radioligand pour le myc-RB2 est identique à
celle pour le récepteur de type sauvage. Le fait que la cinétique de l'activation de la voie
des kinases ERK 1/2, telle que détectée par la phosphorylation des ERK 1/2, soit similaire
dans les expériences présentées à la figure 24, en B et en C, suggère que ces deux
récepteurs ont une fonction comparable. Il a été rapporté que la conjugaison de la GFP à
l'extrémité C-terminale aux RCPG peut ralentir leur cycle d'endocytose-recyclage
(McLean et Milligan, 2000) et favorise potentiellement le recyclage des RB2 plutôt que leur
dégradation, suite à une stimulation par la BK (Kalatskaya et coll., 2006). Par contre, les
immunobuvardages basés sur les anticorps anti-GFP et anti-myc montrent que les deux

191
protéines de fusion sont dégradées après un traitement de 12 h avec le peptide B-9972, un
effet qui est indétectable suite à un traitement de 3 h, et que le niveau de dégradation est
comparable, lorsqu'est examinée l'abondance de la protéine sur une longue période; et cela,
en dépit de la localisation différentielle des séquences antigéniques au sein des
constructions.
Le comportement de l'agoniste non peptidique 47a au niveau des modèles cellulaires
partage plusieurs similarités avec celui du peptide B-9972. Une demi-vie de 6 à 8 minutes
avait été rapportée pour la BK dans le même système expérimental, grâce à l'emploi d'un
immunoessai enzymatique (Bachvarov et coll., 2001); en conséquence, la réversibilité des
effets induits par la BK peut être attribuée à l'inactivation de l'agoniste et la réexpression
des récepteurs à la surface membranaire. Les trois agonistes utilisés dans la présente série
d'expériences, soient la BK, le B-9972 et le composé 47a, présentent des profils temporaux
similaires au niveau de la mobilisation calcique (figure 23), telle que médiée par des RB2
recombinants (RB2-GFP), à laquelle peut s'ajouter une faible réponse découlant d'une
petite population de RB2 endogènes exprimée par ce type cellulaire (Kramarenko et coll.,
2009). L'antagoniste LF 16-0687, qui n'a aucun effet sur la mobilisation calcique, prévient
l'effet de la BK au niveau du même modèle cellulaire, tel que rapporté précédemment
(Morissette et coll., 2007). Les aspects nouveaux de cette étude des agonistes B-9972 et 47a
incluent la persistence du signal fluorescent associé au RB2-GFP au niveau de structures
endosomales suivant une période d'incubation de 2,5 h suivant le retrait de l'agoniste, une
translocation prolongée des (3-arrestinesi/2 à ces structures ainsi qu'une signalisation par la
voie de MEK/ERK et c-Fos, suivant la stimulation du RB2-GFP avec l'un ou l'autre des
agonistes résistants au métabolisme. Ces observations suggèrent d'abord la persistence du
complexe agoniste-récepteur-|3-arrestine aux endosomes. Par ailleurs, elles indiquent aussi
un comportement de type agoniste biaisé des agonistes résistants à la dégradation,
puisqu'ils induisent un recrutement déséquilibré des voies de signalisation en fonction du
temps, en comparaison de la signalisation induite par la BK. Ces effets sont surtout
remarquables au point de vue de la persistance de l'activation des voies des MAP kinases et
de c-Fos, alors que la mobilisation calcique secondaire à la stimulation des RB2 présentait
une cinétique comparable pour les trois agonistes, comme le montrent les figures 23 et 24.
Les effets différentiels au niveau de l'induction de l'expression du facteur de transcription

192
c-Fos sont une découverte particulièrement intéressante, étant donné que cette réponse
intègre différents mécanismes découlant de l'activation de ERK1/2 en fonction du temps,
tels la stimulation transcriptionnelle, l'inhibition de la dégradation de c-Fos par le
protéasome, de multiples événements de phosphorylation compatibles avec une
augmentation du poids moléculaire apparent (telle que présentée à la figure 24, en E;
Glauser et Schlegel, 2007). L'expression de c-Fos dépendante de la voie des MAP kinases
stimulée par un agoniste du RB2, telle qu'observée ici dans un modèle exploitant les
cellules HEK 293 (les résultats sont présentés à la figure 24), a précédemment été observée
dans les keratinocytes humains stimulés avec un agoniste du RB2 (Vidal et coll., 2005).
Plus récemment, il a été rapporté que les kinines, en tant que modulateurs inflammatoires,
stimulaient la résorption osseuse et avaient un effet synergique sur celle-ci en présence de
cytokines (Brechter et coll., 2008), ce qui pourrait expliquer le rôle aggravant du système
kinine-kallikréine dans les désordres inflammatoires osseux, comme l'arthrite rhumatoïde.
Puisque la phosphorylation stabilisatrice de c-Fos est secondaire à la stimulation du RB2
avec les agonistes résistants au métabolisme, il est possible de penser qu'une signalisation
prolongée in vivo par le RB2, observée dans certaines pathologies inflammatoires, peut
exacerber l'inflammation qui s'installe. Par ailleurs, certains gènes cibles de c-Fos sont
impliqués dans le mécanisme d'invasion cellulaire, en facilitant le réarrangement du
cytosquelette, en augmentant la motilité ainsi qu'en accroissant la sécrétion d'enzymes
protéolytiques (Durchdewald et coll., 2009); des phénomènes qui pourraient être amplifiés
dans un environnement pro-inflammatoire présentant une surstimulation des récepteurs des
kinines.
L'activation de la signalisation de la voie MEK1/ERK dépendante de la PKC a aussi été
observée pour le RATi, principalement couplé à la protéine Gq (Violin et Lefkowitz, 2007;
DeWire et coll., 2008). Il a été postulé qu'une composante de cette signalisation est
dépendante de la (3-arrestine, dépendance identifiable par sa résistance à un inhibiteur
pharmacologique de la PKC. L'utilisation de l'inhibiteur de PKC à large spectre le
GF109203x dans les présentes expériences réduisait de manière marquée la
phosphorylation de ERK 1/2, ce qui excluerait une signalisation des MAP kinases
dépendante des |3-arrestines dans ce système, malgré la translocation prolongée des deux
types d'arrestines aux endosomes contenant le RB2 stimulé par les agonistes résistants au

193
métabolisme. Il est possible que le statut d'agoniste biaisé engageant préférentiellement les
P-arrestines soit attribué à l'analogue de I'angiotensine II [Sar1, Ile4, Ile8]angiotensine II
(Violin et Lefkowitz, 2000) de manière secondaire à une potentielle et imprévue résistance
au métabolisme intracellulaire, due à la présence du résidu sarcosine à l'extrémité N-
terminale. Ce récepteur, apparenté au RB2, est lui aussi soumis à un cycle de
phosphorylation, endocytose et réexpression à la membrane plasmique. Puisque que la
signalisation persistante du RB2 stimulé par un agoniste résistant au métabolisme ne semble
pas dépendre de l'assemblage d'un complexe de signalisation grâce à l'interaction directe
entre le RB2, la (3-arrestine et certains membres de la famille des MAP kinases, cela peut
suggérer que la signalisation du RB2 internalise dépend de son couplage à la protéine G.
Bien que l'internalisation et le recrutement de la (3-arrestine soient des phénomènes qui sont
impliqués dans la désensibilisation de la réponse du récepteur, ainsi que dans l'inhibition de
l'interaction de celui-ci avec la protéine G, il a récemment été rapporté que certaines voies
de signalisation identifiées comme étant initiées par des RCPG membranaires avant leur
internalisation étaient en fait recrutables par l'action d'une protéine G à partir de sites
intracellulaires (Calebiro et coll., 2009; 2010a; 2010b). Les auteurs de ces études identifient
la présence concertée du récepteur de la thyréostimuline (TSHR), un RCPG couplé à la
protéine Gs, de la protéine Gs et de l'adénylyl cyclase, un des seconds messagers activés par
la protéine Gs aux endosomes dont l'apparition est secondaire à la stimulation par
l'agoniste. Par ailleurs, leurs études au niveau du récepteur de la parathormone (PTHR),
montrent une spécificité de l'induction du signal à partir du récepteur internalise qui dépend
de l'agoniste employé. En effet, le PTHR possède deux agonistes naturels soient la PTH et
le peptide apparenté à la PTH (PTHrP) et les auteurs attribuent la différence de durée
d'action de chaque agoniste à la capacité de la PTH de générer un signal du récepteur
émanant du système endosomal (Calebiro et coll., 2010b). Par contre, il est à noter que,
jusqu'à présent, l'activation de la voie de signalisation de la PLC et du Ca2+ par la protéine
Gq (celle couplée préférentiellement au RB2) est principalement considérée comme un
phénomène membranaire.
Les données concernant la persistance de la translocation des (3-arrestines aux granules
endosomaux induits par le peptide B-9972 ou le composé 47a règlent une controverse quant

194
à la classification du RB2 en ce qui a trait à son interaction en tant que RCPG avec la P-
arrestine. Les caractéristiques déterminant la classe du récepteur à ce niveau sont la durée et
le site cellulaire de l'interaction; un récepteur de type A lie les P-arrestines de manière
transitoire et à la membrane plasmique, alors qu'un récepteur de type B présente une
interaction plus longue et intracellulaire (Hamdam, et coll., 2007). Cette interaction, étudiée
en présence d'un agoniste résistant au métabolisme tel le peptide B-9972, montre une
translocation persistante des arrestines et du RB2 au niveau de structures endosomales,
même lorsque l'agoniste est retiré du milieu réactionnel après une incubation initiale de 30
minutes. Cela permet d'affirmer sans équivoque que le RB2 appartient à la classe B, ce qui
est cohérent avec la susceptibilité de celui-ci à la dégradation lorsqu'il est stimulé avec un
agoniste résistant au métabolisme. De plus, les récepteurs de la classe A présentent une
affinité supérieure pour la P-arrestine2, alors que les récepteurs de la classe B lient avec une
haute affinité comparable les deux p-arrestines (Oakley et coll., 2000). Les données de
microscopie obtenues montrant la translocation des deux types d'arrestines aux structures
endosomales contenant le récepteur supportent la classification du RB2 en tant que RCPG
de classe B.
L'utilisation d'agonistes résistants à l'inactivation a révélé un assemblage durable du
complexe agoniste- RB2- p-arrestine. De plus, ces agonistes présentent une signalisation
biaisée en fonction du temps, telle que le démontre l'activation persistante des voies des
MAP kinases ERK 1/2 ainsi que du facteur de transcription c-Fos, comparativement à
l'activation transitoire de ces voies par la stimulation du RB2 par son agoniste naturel, la
BK. Finalement, les agonistes résistants au métabolisme provoquaient la dégradation du
RB2, un effet qui n'est pas observé avec la BK. Ces différentes observations sont
représentées sous forme schématique à la figure 62.
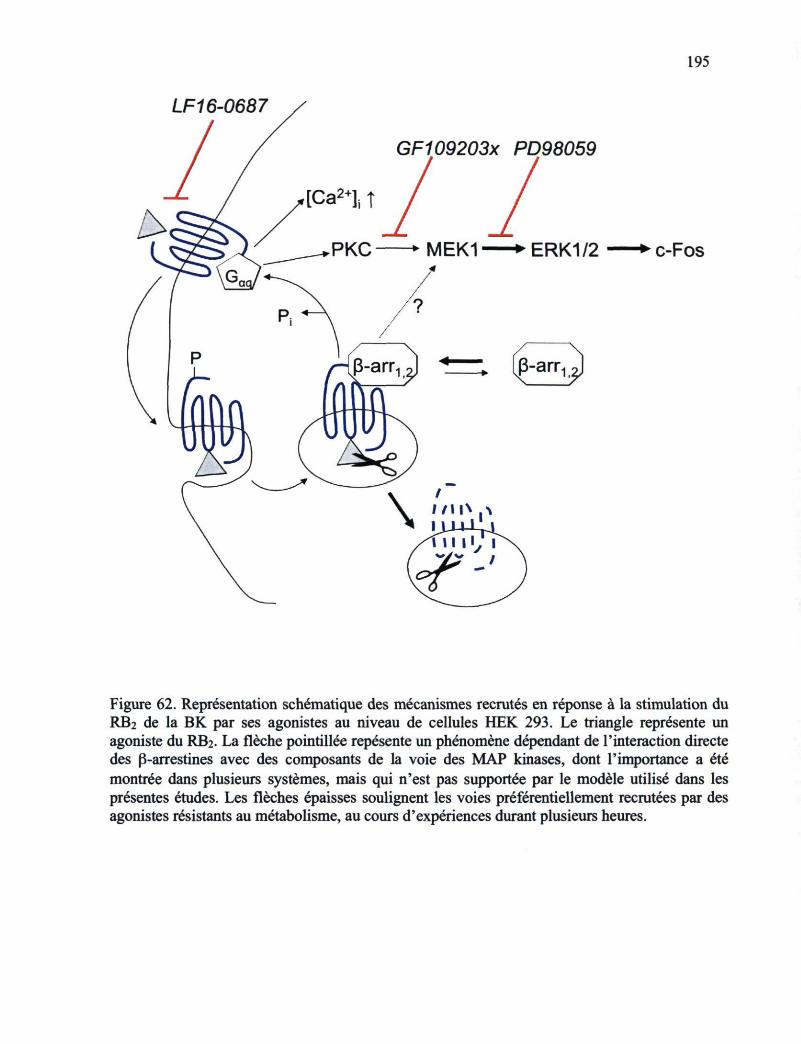
195
LF16-0687
[Ca2+]i î
GF109203X PD98059
-MEK1 ERK1/2 c-Fos
B-aiO M ; (^a[0
Figure 62. Représentation schématique des mécanismes recrutés en réponse à la stimulation du RB2 de la BK par ses agonistes au niveau de cellules HEK 293. Le triangle représente un agoniste du RB2. La flèche pointillée repésente un phénomène dépendant de l'interaction directe des P-arrestines avec des composants de la voie des MAP kinases, dont l'importance a été montrée dans plusieurs systèmes, mais qui n'est pas supportée par le modèle utilisé dans les présentes études. Les flèches épaisses soulignent les voies préférentiellement recrutées par des agonistes résistants au métabolisme, au cours d'expériences durant plusieurs heures.

196
4.3 Ligands des récepteurs des kinines possédant une extension en N-terminale
4.3.1 Caractérisation de l'antagoniste B-10380
La faisabilité de générer des ligands du RB2 comportant de nouvelles propriétés a été
étudiée par l'ajout d'une extension à l'extrémité N-terminale de la séquence de
l'antagoniste B-9430, puisque la modification de cette position est potentiellement moins
délétère du point de vue de l'affinité pour le récepteur. Par ailleurs, la séquence de
l'antagoniste B-9430 est déjà prolongée par l'ajout d'un résidu D-Arg en position N-
terminale, lorsqu'on compare sa structure à celle de la BK. En se basant sur des études de
modélisation de la liaison de la BK, ainsi que de l'icatibant et du composé NPC 17731
(Leeb-Lundberg et coll., 2005), des peptides antagonistes à la structure contrainte, il est
possible de prédire que l'extrémité C-terminale du peptide B-10380 interagit avec des
résidus se trouvant au niveau des domaines transmembranaires du RB2. L'extrémité N-
terminale de l'antagoniste B-10380, présentant un fluorophore carboxyfluorescéine
conventionnel séparé du peptide par l'acide e-aminocaproïque, sort quant à elle du plan
membranaire et se trouve au niveau du milieu extracellulaire. Cette situation présente
l'avantage de permettre la détection de la fluorescence associée à l'antagoniste lié au
récepteur, en évitant autant que possible l'interaction du fluorophore avec la protéine. En
revanche, le groupement caractéristique biotinyl en position N-terminale du peptide B-
10330 ne sort peut-être pas assez de la rosette formée par les TM, ce qui empêche la liaison
de la streptavidine. Un tel encombrement stérique pourrait expliquer l'inefficacité de la
formation du complexe biotine-streptavidine, puisque l'affinité du conjugué biotinyl B-
10330 pour le RB2 est acceptable.
Le peptide B-10380 présente une pharmacologie similaire à celle de son peptide parent à
plusieurs points de vue. En effet, l'antagoniste fluorescent du RB2 est compétitif pour le
RB2 humain, alors que son effet antagoniste est insurmontable et presque irréversible pour

197
l'orthologue de lapin. Au niveau du RB] de lapin, il présente plutôt un profil d'antagoniste
compétitif faible, tel qu'évalué par la réalisation d'essais de contractilité à l'aide de l'aorte
de lapin. Le peptide B-9430 présente une puissance 3 à 10 fois supérieure pour chacun de
ces essais. L'espèce d'origine du RB2 semble être une variable importante dans la
détermination des propriétés pharmacologiques du peptide B-10380, puisque par
comparaison, la différence d'affinité entre ce composé et le peptide parent, le B-9430, est
supérieure au niveau du RB2 de rat, selon les données obtenues par la réalisation d'essais de
liaison.
La perte d'affinité relativement mineure que présente le B-10380 au niveau du RB2 de lapin
comparativement au peptide B-9430 ne nuit pas à l'utilisation de cet agent comme
marqueur fluorescent spécifique de RB2 recombinants de cette espèce exprimés par des
cellules en culture. La sélectivité relativement faible du peptide B-10380 pour le RB2
contre le RBi. permet malgré tout le marquage du RB2 puisqu'à la concentration testée de
50 nM, des cellules exprimant une quantité comparable de RBi recombinants humains ou
de lapin ne présentaient pas une fluorescence significative. Le marquage spécifique des
RB2 par le peptide B-10380 était compétitionné par le composé LF 16-0687, un antagoniste
du RB2 de lapin et humain ne présentant pas de parenté structurale avec le peptide
fluorescent (Houle et coll., 2000). Les études de microscopie portant sur les cellules HEK
293a faisaient la preuve de concept qu'il est possible de produire un ligand fluorescent du
RB2, qui pourrait s'avérer utile dans plusieurs applications telles la cytofluorométrie, la
microscopie confocale, l'identification du RB2 dans des coupes de tissus congelés et
l'imagerie de cellules intactes.
4.3.2 Ligands fluorescents du RBi
L'objectif des études était d'évaluer la possibilité de la conjugaison de peptides ligands du
RB] avec un fluorophore afin de produire des marqueurs fluorescents exploitables. Les
relations de structure-activité connues pour les agonistes et les antagonistes du RB, ont été

198
considérées afin de sélectionner une stratégie basée sur l'extension N-terminale de
l'agoniste naturel préférentiel du RBi, la Lys-des-Arg9-BK, et de l'antagoniste de haute
affinité B-9958. Les peptides résultants ont conservé les identités pharmacologiques de
leurs peptides parents, ce qui a été déterminé par la réalisation d'essais fonctionnels basés
sur les RBi humains et de lapin naturellement exprimés. Le peptide B-10378 est un
agoniste complet, alors que les peptides B-10372 et B-10376 retiennent les propriétés
d'antagoniste du B-9958, une classification qui est compatible avec les résultats obtenus
par la réalisation d'essais de contractilité de l'aorte de lapin et la quantification de la
mobilisation calcique induite par les différents ligands au sein de cellules de muscle lisse
d'artère ombilicale humaine. Par contre, l'extension en N-terminal des ligands sélectionnés
pour l'ajout de la 5(6)-carboxyfluorescéine a mené à une perte d'affinité significative qui
est plus importante pour l'agoniste B-10378 que pour les antagonistes B-10372 et B-10376.
De plus, cette baisse d'affinité est plus marquée pour le RBt humain que pour l'orthologue
lapin. L'affinité des ligands prolongés au niveau de leur extrémité N-terminale était
conservée nonobstant la présence ou l'absence de l'espaceur consitué de l'acide E-
aminocaproïque qui était inséré entre le fluorophore et le peptide, une structure se trouvant
dans l'antagoniste B-10376, qui présente des propriétés pharmacologiques très similaires au
niveau des RB| de lapin et humains. Les affinités des différents ligands pour les deux
récepteurs, mesurées lors d'essais de laison dont dérivent les valeurs de Kj présentées au
tableau 2, permettaient de prédire les concentrations minimales pouvant être utilisées pour
le marquage de cellules intactes et leur visualisation subséquente. Un échantillon
photographique de cette expérience est présenté aux figures 34 et 35. L'affinité plus basse
des nouveaux ligands comparativement à celle des peptides parents, ainsi que l'efficacité
limitée et l'atténuation rapide du signal du fluorophore sélectionné sous une excitation
continue montrent que ces marqueurs fluorescents du RBi ne constituent que la première
génération de ces peptides et qu'ils devront être optimisés.
Un problème pratique qui complexifié le développement d'un marqueur de récepteur
fluorescent destiné à l'usage au niveau de cellules intactes est l'internalisation non-
spécifique de celui-ci. Par exemple, à des concentrations d'ordre micromolaire,
l'internalisation de peptides cationiques présentant une extension en N-terminal composée
de la 5(6)-carboxyfluorescéine peut dépendre de plusieurs voies, mais recrute généralement

199
les clathrines, ainsi que les cavéoles ou autres radeaux lipidiques (Duchart et coll., 2007).
De plus, l'agoniste B-10378, dont la séquence est principalement constituée d'acides
aminés naturels, peut être dégradé et les fragments N-terminaux fluorescents peuvent être
inernalisés. En dépit de ces limites, certaines expériences témoins qui étaient basées sur la
compétition de la liaison au RB| entre le ligand fluorescent et un antagoniste non
peptidique dont la structure n'est pas apparentée à celles des ligands fluorescents ou sur le
marquage des cellules n'exprimant pas de RBi recombinant permettent d'affirmer que la
majorité du signal fluorescent présenté par des cellules exprimant l'une ou l'autre des
constructions du RBi est spécifique, tout au moins pour l'éventail de concentrations d'ordre
nanomolaire exploitées. Ainsi, les affinités acceptables des différents ligands fluorescents
pour le RBi permettent de suggérer plusieurs applications originales soutenues par l'emploi
de ligands fluorescents. Parmi celles-ci, deux applications ont surtout été caractérisées :
l'effet différentiel de la liaison de l'agoniste ou de l'antagoniste sur la distribution du RBi
et l'identification d'une population de ces récepteurs grâce à la cytofluorométrie.
Comme le montre l'échantillon photographique présenté à la figure 36, l'agoniste
fluorescent B-10378 semble reproduire la translocation latérale du RBi., qui avait été
observée précédemment pour la construction codant pour un RBi fluorescent grâce à la
microscopie confocale (Sabourin et coll., 2002). Le signal fluorescent associé à l'agoniste
est particulaire et situé juste au dessous du plan de la membrane plasmique, suggérant une
localisation cavéolaire (figure 38). En effet, les radeaux lipidiques non cavéolaires ne
peuvent pas être observés à l'aide de la microscopie confocale optique (Fortin et coll.,
2005b). Les cellules exprimant un RBi recombinant et incubées avec l'antagoniste B-10376
présentaient quant à elles un marquage membranaire plus lisse, malgré une certaine
variabilité de l'expression du RBt qui est caractéristique de l'expression transistoire des
récepteurs. La démonstration est particulièrement convaincante au niveau du RB] de lapin,
étant donné les affinités similaires en terme de valeurs de Kj absolues de l'agoniste et de
l'antagoniste, malgré que les données obtenues pour le RBi humain appuient aussi la
redistribution du récepteur dépendante de l'agoniste, utilisé à différentes concentrations.
L'emploi de ligands fluorescents du RBi est une méthode complémentaire à l'utilisation de
la construction fluorescente RBi-YFP, puisque les ligands vont influencer uniquement les
récepteurs exprimés à la surface membranaire, sans que la présence de récepteurs au niveau

200
intracellulaire n'interfère avec le marquage fluorescent. Cette approche, basée sur le
marquage de cellules vivantes et intactes, confirme essentiellement la proposition d'une
forme unique d'adaptation induite par l'agoniste du RBi non phosphorylable, par Sabourin
et collaborateurs (2002), qui est une translocation cavéolaire sans internalisation
significative. La colocalisation fréquente de la protéine recombinante cavéoline-1 -
CherryFP, émettant une fluorescence rouge, avec la fluorescence verte associée à l'agoniste
B-10378 (figure 38), supporte cette hypothèse. La surexpression de la cavéoline-1 -
CherryFP comporte ses limites, puisqu'elle peut entraîner l'augmentation du nombre de
cavéoles au niveau des cellules HEK 293a. Ces cavéoles peuvent former des tubes
contournés qui s'étendent profondément au niveau intracellulaire, expliquant ainsi la
colocalisation occasionnelle des signaux rouge et vert présentés par les granules
cytosoliques.
Une seconde application permise par l'utilisation des ligands fluorescents était la détection
de la population de RBi exprimée à la surface cellulaire, par cytofluorométrie.
L'antagoniste B-10376 permettait la mise en évidence d'une densité variable de RB| à la
surface des cellules HEK 293a, d'un niveau nul par les cellules non transfectées à élevé par
les cellules exprimant de manière transitoire le RBi de type sauvage de lapin, montrant par
ailleurs l'efficacité de la transfection. Tel qu'observé au sein d'autres systèmes cellulaires,
la population de RBi. exprimés à la membrane plasmique dépend de l'effet combiné de sa
synthèse de novo et de sa dégradation indépendante de la présence de l'agoniste (Fortin et
coll., 2003). L'antagoniste B-10376 a aussi été employé à la détection de niveaux
endogènes, assez bas, de RBi exprimés par les cellules de muscle lisse vasculaire. Il reste à
déterminer si la quantification de l'expression du RB| par des cellules leucocytaires, par
exemple au niveau des cellules mononucléés du sang périphérique provenant de patients
atteints de sclérose en plaque (Prat et coll., 2005), peut se révéler une application
diagnostique pertinente des ligands fluorescents du RBt. D'autres applications potentielles
de ces ligands pourraient être la détection des récepteurs présents dans des coupes
tissulaires congelées, ainsi que de permettre le tri de cellules intactes exprimant le récepteur
correspondant afin de sélectionner un transfectant stable d'une population de cellules
exprimant une construction codant un récepteur non fluorescent.

201
L'introduction d'un fluorophore dans la séquence de ligands du RBi. a permis la génération
d'un agoniste et de deux antagonistes, qui peuvent tous être employés à la détection du
récepteur. Malgré une perte d'affinité de ces ligands comparativement à leurs peptides
parents, les nouveaux agents permettent des applications originales qui appuient par ailleurs
les études s'intéressant à l'adaptation du RBi, comme la translocation cavéolaire nécessaire
à sa signalisation, secondaire à son activation, et sa quantification par cytofluorométrie.
4.3.3 Agonistes fluorescents du RB2
La connaissance des relations structure-activité des ligands peptidiques des deux sous-types
de récepteurs de la BK, ainsi que des interactions entre le ligand et le récepteur peut être
résumée ainsi : l'extrémité C-terminale des peptides agonistes ou antagonistes liés au
récepteur se situe profondément dans la rosette formée par les domaines transmembranaires
où elle interagit avec plusieurs résidus de ces domaines, alors que l'extrémité N-terminale
interagit plutôt avec certains résidus chargés négativement présents au niveau des boucles
extracellulaires (Leeb-Lundberg et coll., 2005). Ainsi, le couplage d'un cargo à cette
extrémité des peptides peut s'avérer possible, étant donné une certaine tolérance pour des
extensions chimiques sortant du plan membranaire du récepteur vers le milieu
extracellulaire. Par ailleurs, cette prédiction a été validée pour chaque sous-type des
récepteurs des kinines, grâce à la synthèse d'antagonistes fluorescents des deux récepteurs,
ainsi que de l'agoniste du RBi, le peptide B-10378, qui ont tous été décrits plus haut. Par
ailleurs, la séquence naturelle Met-Lys-BK présente une puissance légèrement inférieure
que celle de la BK, en tant qu'agoniste au niveau des RB2 recombinants humains et murins
(Hess et coll., 1994).
L'analogue fluorescent de la BK, soit la CF-EACA-BK, complète l'ensemble des agonistes
et antagonistes des deux sous-types de récepteurs conjugués à la CF. Cette extension se
révèle toujours préjudiciable à l'affinité de ces ligands, mais l'importance de la perte
d'affinité est variable et dépend aussi de l'espèce d'origine du récepteur. Les nouveaux

202
agonistes du RB2 rapportés ici sont les ligands qui ont subi la diminution de puissance la
plus importante en raison de l'extension de leur séquence en N-terminal avec la CF,
comparativement au peptide parent, la BK. Ainsi, la CF-BK lie peu le RB2, comme le
montre l'essai de compétition réalisée avec la [3H]BK (figure 43). La présence de
l'espaceur E-ACA atténue ce problème d'affinité, puisque son ajout permet à l'agoniste C F -
EACA-BK de lier les RB2 humain et de lapin, tout en demeurant 400 fois moins puissant
que la BK au niveau de l'essai de liaison basé sur le radioligand et 1000 fois moins puissant
que la BK comme agent contractile de la veine ombilicale humaine, comme le montre la
figure 43. Un hypothétique encombrement stérique entre la face extérieure du récepteur et
l'extension N-terminale de la séquence du ligand pourrait être à l'origine de l'importante
baisse d'affinité que présente l'agoniste fluorescent. La présence des trois sites de N-
glycosylation du RB2 pourrait être la cause de l'encombrement stérique. Par ailleurs, la
présence de différents états de glycosylation sous lesquels peut exister le récepteur est un
facteur qui expliquerait son apparence hétérogène, sous forme d'une bande épaisse ou de
multiples bandes, lorsque détecté par immunobuvardage (Camponova et coll., 2007). En
dépit de son affinité modeste, l'agoniste fluorescent CF-EACA-BK, utilisé à la
concentration de 5 pM, s'avère un outil de visualisation performant de la progression
endosomale de l'agoniste suivant l'internalisation du complexe ligand-RB2 par des cellules
intactes et permet la réalisation d'expériences de colocalisation avec plusieurs protéines
couplées à un fluorophore rouge, comme les Rab7 et 5, ainsi que la P-arrestine i. Les
représentations tridimensionnelles du marquage obtenu suite à l'incubation de cellules
exprimant le RB2 recombinant de lapin en présence de l'un ou l'autre des ligands
fluorescents ont permis d'observer la profonde différence qu'il y a entre la distribution de
l'agoniste fluorescent C F - E A C A - B K et de l'antagoniste fluorescent B-10380. Un
échantillon photographique de chaque condition est présenté à la figure 45. Le marquage
membranaire lisse obtenu grâce à l'antagoniste se distingue grandement du signal
fluorescent principalement endosomal, où l'agoniste est séquestré après une incubation de
30 min à 37°C (suivie de multiples rinçages), d'un faible signal cytosolique et de la perte de
la définition de la membrane plasmique. Les structures marquées constituent présumément
l'intérieur des endosomes, présentant une colocalisation significative avec la p-arrestinei
fluorescente et Rab5-CherryFP; toutefois, il ne demeure aucun signal membranaire. Cela

203
peut s'expliquer par la dissociation rapide du ligand de basse affinité suivant le rinçage des
cellules, ainsi que par la translocation de tous les récepteurs membranaires liés à l'agoniste
fluorescent. La première option est plus plausible, puisque la construction myc-RB2TRUNC,
dont l'expression membranaire est confirmée par les essais de liaison de la [3H]BK et le
marquage avec l'antagoniste B-10380, ne permet pas le marquage de cellules qui expriment
cette construction par l'agoniste CF-EACA-BK.
Le patron de marquage des RB2 par la C F - E A C A - B K peut être comparé à la fluorescence
associée aux récepteurs au niveau de cellules HEK 293 exprimant de manière stable la
protéine de fusion B2R-GFP, au repos ou stimulées par la BK, respectivement (Bachvarov
et coll., 2001). Le traitement avec l'agoniste naturel non fluorescent BK fragmente la
distribution lisse de la fluorescence associée au RB2-GFP, et permet l'observation d'un
marquage endosomal contenant le récepteur fluorescent. Par contre, un signal résiduel
variable demeure présent à la membrane cellulaire, ce qui est probablement dû à
l'expression élevée du RB2-GFP. De plus, des approches variées indiquent que le recyclage
secondaire à l'endocytose du récepteur fluorescent induite par la BK à la membrane
plasmique est complet après un temps variant de 1 h à 3 h (Bachvarov et coll., 2001), ce qui
est également le cas de RB2 endogènes. Cette cinétique de recyclage est parallèle et dépend
de la déphosphorylation du récepteur et la dissociation des p-arrestines (Blaukat et coll.,
1996; Simaan et coll., 2005). La C F - E A C A - B K modélise le phénomène de dégradation de
l'agoniste : il est connu que la BK internalisée est entièrement métabolisée dans le système
endosomal (Munoz and Leeb-Lundberg, 1992); l'inhibition du relâchement cytosolique de
la fluorescence de la C F - E A C A - B K par un traitement concomitant de bafilomycine Al
supporte l'implication de l'acidification du milieu endosomal par la pompe à protons V-
ATPase dans le recyclage des RB2. Ainsi la C F - E A C A - B K est relativement instable et sa
présence prolongée dans certaines endosomes tardifs positifs pour la protéine Rab7, peut
démontrer le routage différentiel de l'agoniste et du myc-RB2.
La stimulation du RB2 par son agoniste entraîne la phosphorylation de celui-ci au niveau de
certains résidus serine conservés (Ser339, Ser346, Ser34x) par les GRK, ce qui permet le
recrutement des P-arrestines, les protéines d'échafaudage qui constituent en partie la
plateforme de liaison à la machinerie endocytaire. Malgré la publication de données

204
contradictoires (Leeb-Lundberg et coll., 2005), Hamdam et collaborateurs (2007) ont
conclu que l'endocytose du RB2 dépendait de la formation de puits de clathrines, recrutées
par l'interaction moléculaire centrale de la p-arrestine avec la chaîne lourde de la clathrine,
ainsi qu'avec la sous-unité P2 de la protéine adaptatrice AP-2 (Hislop et von Zastrow,
2010), durant la stimulation du récepteur. L'élimination du domaine phosphorylable au
niveau de l'extrémité C-terminale intracellulaire du myc-RB2 a rendu cette construction
incapable de recruter la P-arrestine2, comme le montre la figure 42, en dépit de son profil
d'affinité identique et d'une signalisation inchangée, dont la caractérisation est présentée à
la figure 4L Ces données sont par ailleurs compatibles avec des travaux antérieurs portant
sur le RB2 humain. La transfection de la construction myc-RB2TRUNc ne permet pas
l'internalisation de la CF-EACA-BK, établissant ainsi la dépendance de son transport sur la
phosphorylation du RB2 suite à sa stimulation et sur son interaction avec les P-arrestines.
Ainsi, le marquage des cellules par la C F - E A C A - B K dépend d'événements métaboliques.
L'ECA est une ectoenzyme qui inactive la bradykinine au niveau extracellulaire qui
constitue une molécule hautement impliquée en physiopathologie et est une cible
thérapeutique d'importance en pharmacothérapie humaine. Il a été rapporté que l'ECA est
plus densément exprimée que le RB2 au niveau des cellules endothéliales dérivées de la
veine ombilicale humaine, selon la quantification des sites de liaison de l'énalaprilat tritié
([ HJénalaprilat) comparativement au nombre de sites de liaison de la [ H]BK
(Koumbadinga et coll., 2010a; 2010b). La BK possède l'affinité la plus haute pour cette
enzyme, avec une valeur de Kj de 525 nM, comparativement à ses autres substrats, tels
I'angiotensine I et l'AcSDKP, comme l'indiquent des essais de compétition de la liaison de
1TECA [3H]énalaprilat à l'ECA naturellement exprimée par des cellules endothéliales
humaines en culture (Koumbadinga et coll., 2010a). Pour l'ECA, la CF-BK présentait
approximativement 20 % de l'affinité de la BK, alors que la C F - E A C A - B K démontrait une
affinité similaire à celle de la BK, ce qui souligne l'importance de l'espaceur entre la
séquence de la BK et le groupement CF à l'extrémité N-terminale. Ces considérations
suggèrent la possibilité d'utiliser des analogues fluorescents de la BK comme outils de
détection de l'ECA en imagerie dans des conditions où le peptide fluorescent se lie au site
catalytique avant que ne se produise la réaction d'hydrolyse, une hypothèse qui a été

205
vérifiée, et dont les résultats sont présentés aux figures 45 et 49. La C F - E A C A - B K marquait
la surface des cellules exprimant l'enzyme recombinante d'une manière uniforme, sans
qu'un signal intracellulaire significatif n'indique son endocytose, ce qui est remarquable
étant donné l'endocytose importante de ce peptide dans des conditions identiques par des
cellules exprimant un RB2 recombinant. Par conséquent, l'agoniste du RB2 CF-EACA-BK
permet le marquage du récepteur ainsi que celui de l'ECA par l'emploi d'une échelle
comparable de concentration d'ordre micromolaire. Dans une situation où l'objectif serait
d'identifier les RB2 naturellement exprimés au niveau d'un contexte cellulaire où l'ECA
pourrait être présente, l'antagoniste fluorescent B-10380, présentant une valeur de K; en
excès de 1 pM lors d'une compétition de la liaison de l'[3H]énalaprilat, serait
théoriquement supérieur, étant sélectif pour le RB2 à la concentration de 50 nM. En effet,
l'antagoniste B-10380 ne soutient nullement l'imagerie de l'ECA recombinante exprimée
par des cellules HEK 293a à cette concentration, comme le montre la figure 49, alors qu'il
permet la visualisation de RB? membranaires à la même concentration de 50 nM, comme le
présente la figure 45.
Ainsi, le nouvel analogue de la BK fluorescent CF-EACA-BK, employé à des
concentrations micromolaires, est un marqueur pour deux cibles physiologiques majeures
de la BK, soient le RB2 et l'ECA. Le marquage des cellules exprimant le récepteur est basé
sur l'endocytose du peptide, elle-même dépendante des P-arrestines. La CF-EACA-BK
permet aussi la modélisation de la dégradation subséquente de l'agoniste internalise.
4.3.4 La maximakinine
La perte d'affinité de la séquence amphibienne MK, comportant un important
prolongement de sa séquence en N-terminal, est modeste et quelque peu dépendante de
l'espèce d'origine du RB2. En tant que molécule hypothétiquement optimisée de manière
évolutive, il est intéressant de noter le caractère hydrophile de la séquence en N-terminal de

206
la structure de la BK, ainsi que la présence d'une potentielle région charnière entre ces deux
domaines, formée par les résidus Gly"1 et Pro0.
La dégradation du RB2-GFP, suite à sa stimulation prolongée par des agonistes
synthétiques résistants au métabolisme, qui a été présentée plus haut, montre que
l'inactivation de l'agoniste est un facteur déterminant dans le cycle d'endocytose-recyclage
du RB2. La persistance du RB2 au niveau endosomal et sa dégradation subséquente ont
d'abord été découvertes grâce à l'agoniste peptidique B-9972 et confirmées par la suite
grâce à l'utilisation du composé 47a, un agoniste partiel non peptidique. Les structures de
ces deux agents intègrent une résistance pour un grand nombre de peptidases impliquées
dans le métabolisme de la BK, telles les amino-, carboxy- et endopeptidases. De plus, ces
agonistes provoquent la formation de complexes agoniste-récepteur-P-arrestine durables. Il
est très intéressant de noter que la MK est la première séquence naturelle à se comporter
comme un agoniste persistant du RB2, en produisant une dégradation aussi marquée du
RB2-GFP que celle observée suite à un traitement prolongé avec le peptide B-9972, toutes
deux parallèles à l'apparition de fragments de plus faible poids moléculaire, ainsi que la
génération de GFP libre, comme le montre F immunobuvardage présenté en A à la figure
51. La BK est inactive à ce niveau, lorsqu'elle est employée à une concentration générant
une réponse également efficace. De plus, des essais de signalisation portant sur l'activation
de la voie des MAP kinases ERK 1/2 et de l'induction de c-Fos montrent que la MK se
comporte comme un agoniste résistant au métabolisme, puisqu'elle produit une activation
prolongée de la voie de la voie ERK 1/2, ainsi que l'induction de c-Fos, qui est secondaire à
la stimulation persistante de la voie ERK 1/2 (figure 51B). Finalement, des études de
microscopie en épifluorescence présentent une translocation durable du RB2-GFP au niveau
intracellulaire, lors de traitements prolongés avec la MK, à l'instar des agonistes résistants
au métabolisme (figure 51C). Ces données semblent suggérer qu'au niveau endosomal, la
dégradation de la BK dépend prioritairement d'une aminopeptidase et que la modification
de la séquence N-terminale, par son extension comme dans le cas de la MK et de la C F -
E A C A - B K (l'effet de ce dernier peptide n'est pas montré) ou par l'échange du résidu Arg
par le résidu D-Arg de B-9972, nuit à l'activité optimale de l'aminopeptidase. La
persistance d'un agoniste fonctionnel au niveau endosomal, qui peut toujours lier le
récepteur, inhibera le changement de conformation nécessaire à sa déphosphorylation ainsi

207
qu'à la dissociation de la P-arrestine, empêchant ultimement le recyclage du récepteur. Les
voies menant à la dégradation du RB2, ainsi séquestré au niveau endosomal n'ont pas fait
l'objet d'une caractérisation très poussée, malgré l'observation d'une diminution de
l'expression du RB2 dans certaines situations pathologiques. Par ailleurs, il ne s'agit pas de
la voie préférentiellement suivie par le récepteur.
Il est possible que chaque cycle d'endocytose-recyclage du récepteur soit associé à un
mauvais routage d'une fraction des récepteurs, qui sont dirigés vers la dégradation
lysosomale. Les déterminants moléculaires qui cibleraient le RB2 à la dégradation sont
inconnus, quoique l'ubiquitination pourrait être un des mécanismes recrutés.
L'ubiquitination est une de nombreuses modifications post-traductionnelles réversibles que
peuvent subir les RCPG, qui peut être impliquée dans leur endocytose, leur trafic
endosomal et leur dégradation, puisqu'elle redirige le contenu internalise présent au niveau
des endosomes précoces vers les endosomes tardifs, qui sont appelés à fusionner avec les
lysosomes (Hislop et von Zastrow, 2010). Il a été rapporté que la dégradation lysosomale
du récepteur K-opioïde secondaire à sa stimulation par l'agoniste dépend de sa
polyubiquitination basée sur la construction d'une chaîne sur le résidu Lysine 3 de
l'ubiquitine (Li et coll., 2008). La régulation des RCPG par l'ubiquitination peut se faire à
deux niveaux; d'abord par l'ubiquitination directe de l'extrémité C-terminale intracellulaire
du récepteur, ainsi qu'au niveau des protéines adaptatrices qu'il recrute, par exemple les P-
arrestines. En effet, il a été montré que la cinétique d'interaction de la P-arrestine dépend
non seulement de l'état de phosphorylation de l'extrémité C-terminale du récepteur, mais
aussi de l'état d'ubiquitination de la P-arrestine. Le nombre élevé de ligases de l'ubiquitine
ainsi que de déubiquitinases confère une grande complexité à ce phénomène, qui est par
ailleurs impliqué à de multiples étapes de la voie endocytaire.
Un intervenant moléculaire additionnel potentiellement impliqué dans la redirection des
RB2 stimulés avec des agonistes résistants au métabolisme pourrait être un membre de la
famille des GASP (GPCR-Associated Sorting Proteins). Comme leur nom l'indique, ces
protéines sont impliquées dans le trafic des RCPG, quoique que leur mécanisme d'action
précis soit inconnu (Hislop et von Zastrow, 2010). Il a été suggéré qu'elles lient la
dysbindine, une protéine qui participe à la biogénèse des lysosomes et dont la depletion

208
inhibe la dégradation de certains RCPG, dont le récepteur D2 de la dopamine et le récepteur
ô-opioïde (Marley et von Zastrow. 2010). Il a été rapporté que les GASP lient
préférentiellement les RCPG devant être soumis à la dégradation, en l'absence d'une
séquence de recyclage au niveau de leur extrémité C-terminale (Thompson et coll., 2007).
Le changement conformationnel persistant du RB2 induit par les agonistes résistants au
métabolisme pourrait dissimuler le signal de recyclage du RB2, ou à l'inverse, exposer un
site de liaison potentiel pour les GASP, ce qui l'entraînerait vers la voie de dégradation
lysosomale.
4.4 Transport de cargos anticorps anti-récepteur
Les travaux décrits précédemment portant sur les ligands fluorescents avaient d'abord
comme objectif de vérifier la tolérance de la séquence de la BK à être prolongée à son
extrémité N-terminale afin de générer des ligands conservant une affinité appréciable pour
le récepteur. Bien entendu, l'ajout du groupement CF en N-terminal permet la visualisation
indirecte du récepteur, ce qui est particulièrement pertinent dans le cas d'études portant sur
l'adaptation du RB2. Une méthode additionnelle permettant la visualisation des récepteurs
repose sur l'immunocytochimie, basée sur des anticorps spécifiques contre un site
épitopique présenté par le récepteur. Il s'agissait de déterminer si divers anticorps couplés à
des cargos variés pouvaient être internalises à la suite du récepteur auquel ils étaient liés, de
manière subséquente à sa stimulation. Cela a d'abord été vérifié par l'endocytose d'un seul
anticorps anti-myc lié au myc-RB2; deux anticorps anti-myc différents ont été utilisés,
soient les clones 4A6 et 9E10. L'internalisation d'un complexe composé du récepteur, d'un
anticorps anti-récepteur (anti-myc clone 4A6) ainsi que d'un anticorps secondaire, couplé
soit à un fluorophore Alexa Fluor 594, ou à un nanomatériau de type Qdot fluorescent a
ensuite été observée, de manière subséquente à la stimulation du récepteur par l'agoniste.
Cette approche a permis de confirmer l'hypothèse suggérant qu'un RCPG peut transporter
des molécules de taille beaucoup plus importante que son ligand à l'intérieur de la cellule
lorsque lui-même est internalise. Les mécanismes recrutés pour l'internalisation de ce

209
complexe de taille importante sont identiques à ceux généralement mobilisés par l'agoniste,
comme l'illustre la rapide colocalisation de la P-arrestine2 aux endosomes contenant les
complexes immuns. La taille d'un tel complexe assemblé à la surface cellulaire est
théoriquement assez importante et peut être approximée à 370 kDa, si le rapport pour
chaque constituant est 1; un récepteur pour un anticorps anti-récepteur pour un anticorps
secondaire. Par contre, il est possible qu'il y ait formation de complexes multivalents au
sein desquels, par exemple, un anticorps couplerait deux récepteurs et ainsi de suite. Les
complexes immuns composés des nanomatériaux de type Qdot peuvent présenter une taille
supérieure encore, étant donné qu'une nanoparticule de ce type se situe à un ordre de
grandeur de 1,5 MDa (Lin et coll., 2008). Cette estimation ne tient pas compte de la couche
de polymères entourant le cristal et permettant la liaison des fragments F(ab')2 qui
enveloppent la molécule. Selon le manufacturier, chaque particule Qdot comporte en
moyenne trois molécules d'anticorps. Il est intéressant de remarquer que malgré une
endocytose potentiellement peu affectée par la taille des cargos qu'elle accomode, le trafic
endosomal des deux types de complexes immuns est distinct de celui du myc-RB2 stimulé
par la BK. En effet, les complexes immuns persistent au niveau de structures
intracellulaires partiellement marquées avec la protéine de fusion GFP-Rab7 après une
incubation de 3 h en présence de la BK, comme le montrent les figures 59 et 60. Un
traitement d'une telle durée permet d'observer la dégradation de la BK au niveau
extracellulaire et le recyclage du RB2 à la surface cellulaire (Bachvarov et coll., 2001). Ces
données sont comparables à celles faisant état de la rétention des Qdots couplés à la
transferrine au niveau intracellulaire, alors que les récepteurs de la transferrine sont
généralement recyclés rapidement à la membrane plasmique (Teckle et coll., 2008).
La figure 54 montre l'absence d'un effet agoniste de l'anticorps anti-myc (clone 4A6) au
niveau de cellules HEK 293a exprimant le myc-RB2, comme en témoigne la mobilisation
calcique nulle secondaire à son ajout dans le milieu réactionnel. Abd Alla et collaborateurs
ont cartographie les sites de liaison des agonistes et des antagonistes au RB2, grâce à
l'utilisation d'anticorps dirigés contre plusieurs régions de ses domaines extracellulaires et
ont trouvé que l'extrémité N-terminale du récepteur ne comporte aucun site déterminant à
la liaison des ligands. La modélisation de la position et de l'orientation du composé LF16-
0687 lorsqu'il est lié au RB2 démontre qu'il n'y a pas d'interaction entre l'antagoniste et la

210
région N-terminale du récepteur (Marie et coll., 2001). Ainsi l'anticorps anti-myc n'active
pas le récepteur et n'empêche pas la liaison de l'agoniste, ce qui permet à ce dernier
d'induire l'internalisation du RB2 suite à son ajout dans le milieu réactionnel. Par ailleurs,
la liaison de l'anticorps ne provoque pas un encombrement qui nuit à la liaison de
l'antagoniste LF16-0687, puisque ce composé prévient toujours l'internalisation des
complexes immuns, lorsque les cellules sont coincubées avec la BK et cet antagoniste.
Un certain niveau d'agrégation des anticorps anti-myc à la surface cellulaire indépendante
de l'agoniste, ainsi qu'un bruit de fond dénotant une faible endocytose en l'absence de
l'agoniste, sont des phénomènes qui ont été observés pour tous les systèmes de détection
employés, dont l'importance variable peut être appréciée aux figures 52, 55 et 56. Il reste à
voir si ces effets non spécifiques peuvent être réduits, en privilégiant l'utilisation
d'anticorps anti-récepteur monovalents, par exemple. Des cargos basés sur des anticorps
anti-récepteur pourraient mener à une multitude d'applications. Dans les présentes études,
un des principaux objectifs poursuivis était l'amélioration de l'imagerie du RB2 par
l'emploi de nanocristaux fluorescents, les Qdot, qui sont des fluorophores supérieurs à
plusieurs autres molécules abondamment utilisées, en raison de leur fluorescence intense et
de leur résistance à l'atténuation du signal. La capacité du RB2 à être internalise de manière
secondaire à son activation par la BK, même lorsqu'il se retrouve inclus dans un complexe
de taille importante constitué d'anticorps, a pu être validée. L'ajout de groupements
fonctionnels aux anticorps liés au RB2 internalise pourrait conférer de nouvelles propriétés
au récepteur, ainsi que de générer de nouveaux types d'événements de signalisation
dérivant de l'activation de celui-ci, qui seraient par contre toujours dépendants de la
présence de l'agoniste.
En plus de pouvoir accommoder le transport de petits cargos conjugués à l'agoniste, dont
un exemple est la CF-EACA-BK, des cargos de taille importante constitués de complexes
immuns peuvent être transportés à la suite du RB2 stimulé par l'agoniste, au niveau
intracellulaire. Les deux types de cargos dépendent d'un récepteur apte à interagir avec les
P-arrestines et progressent au niveau de la voie endosomale, comme le prouve leur présence
dans les endosomes tardifs, reconnaissables par leur association avec le marqueur GFP-
Rab7 après une période de 3 h.
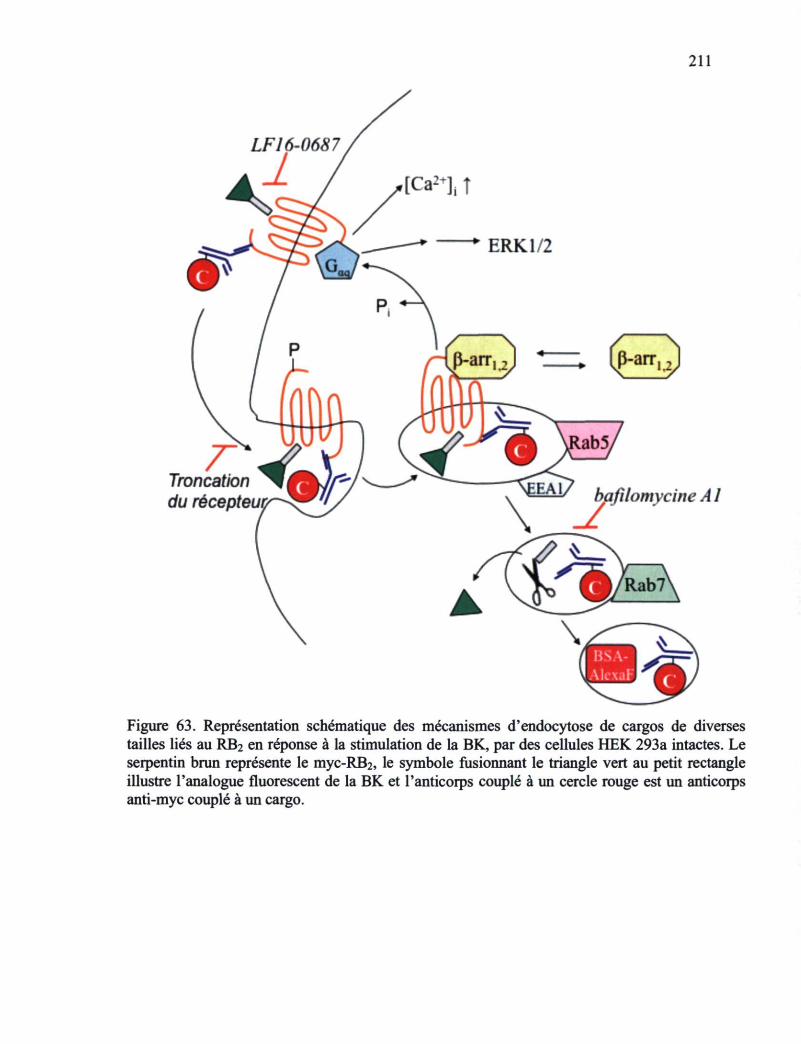
211
LF16-0687
* ERK1/2
P-arr,j t = i (MT U )
7" Troncation
du récepteu bafilomycine Al
Figure 63. Représentation schématique des mécanismes d'endocytose de cargos de diverses tailles liés au RB2 en réponse à la stimulation de la BK, par des cellules HEK 293a intactes. Le serpentin brun représente le myc-RB2, le symbole fusionnant le triangle vert au petit rectangle illustre l'analogue fluorescent de la BK et l'anticorps couplé à un cercle rouge est un anticorps anti-myc couplé à un cargo.

212
4.5 Perspectives
Ces travaux de doctorat ont permis de mieux comprendre l'adaptation des récepteurs des
kinines suivant leur stimulation avec leur agoniste, tout en soulevant un grand nombre de
questions.
4.5.1 Agonistes résistants à l'inactivation
Grâce aux agonistes résistants au métabolisme, on a pu démontrer que la dégradation de
l'agoniste au niveau endosomal est absolument essentielle afin que le RB2 puisse être
déphosphorylé et se dissocie de la p-arrestine afin d'être recyclé à la membrane plasmique.
Le complexe durable qu'il forme avec la P-arrestine suite à un traitement avec un agoniste
résistant au métabolisme permet de raffiner la caractérisation du RB2 quant à son
interaction avec cette protéine d'échafaudage en soulignant qu'elle peut se définir aussi
comme dépendante de l'agoniste utilisé. Il a été rapporté que certains RCPG de la classe B,
possédant une affinité supérieure pour la P-arrestine, étaient aussi plus sujets à la
dégradation (Simaan et coll., 2005). Le fait de persister sous sa conformation active au
niveau de la voie endosomale et lié à la P-arrestine semblent constituer des facteurs
suffisants pour le ciblage du RB2 à la dégradation et est intrinsèquement associé au
ralentissement de l'inactivation des agonistes. Le terme ralentissement est utilisé ici pour
souligner que la résistance métabolique des différents agonistes causant la dégradation du
récepteur n'est pas complète; la quantification de l'agoniste peptidique B-9972 montre qu'il
est éventuellement dégradé, puisque la concentration résiduelle telle que mesurée par
l'essai radiorécepteur ne représente que 2 % de la concentration initiale utilisée, après un
traitement de 12 h. Par ailleurs, l'analogue fluorescent de la BK, qui par son extension en
N-terminal s'oppose à l'activité aminopeptidase potentiellement dominante au niveau

213
endosomal, est lui-même dégradé en fonction du temps, comme en témoigne la diffusion de
la fluorescence des endosomes vers le cytosol, après une incubation de 3 h. Finalement, la
maximakinine, ne présentant pas de résistance intrinsèque aux peptidases comparable aux
artifices présentés par le peptide B-9972, doit éventuellement être dégradée. L'importance
du maintien d'un certain rythme dans le phénomène d'endocytose et de recyclage a
d'ailleurs déjà été montré par Houle et Marceau (2003). Ces auteurs ont mis en évidence
que lorsque la progression du RB2 au niveau endosomal était altérée, celui-ci était plutôt
redirigé vers la voie de la dégradation lysosomale. Comme ces expériences étaient
principalement basées sur la protéine Rab5, dont une version dominante négative
(Rab5S34N) et une version constitutivement active (Rab5Q79L) ont été employées, il serait
intéressant de vérifier si l'altération des fonctions des protéines Rab importantes pour les
fonctions des endosomes recyclants, soient les Rab4 et Rabll, peut aussi mener à la
dégradation lysosomale.
Un phénomène relativement récemment introduit dans la compréhension de la signalisation
des RCPG est la signalisation non conventionnelle qu'ils peuvent induire indépendamment
de la protéine G. En effet, plusieurs équipes ont observé une signalisation émanant de
l'association directe de la P-arrestine, en sa qualité de protéine d'échafaudage, toujours liée
à la queue C-terminale du RCPG, avec des intervenants de plusieurs voies de signalisation,
dont les MAP kinases ainsi que c-src. Plusieurs études ont rapporté l'utilisation d'agonistes
biaises, c'est-à-dire des agonistes qui privilégient l'activation d'une voie de signalisation
particulière parmi celles que recrute habituellement le récepteur. Un exemple de ce type
d'agoniste est le composé [Sar1, Ile4, Ile ]Ang II, qui n'induit pas une signalisation
dépendante de la protéine Gq habituellement couplée au RATi. (Wei et coll., 2003), mais
permet le recrutement de la p-arrestine et l'activation de la voie des MAP kinases. Parmi
les autres RCPG pour lesquels de tels ligands ont été identifiés se retrouve le récepteur 1 de
la parathormone (PTH1R; Gesty-Palmer et coll., 2006; 2009); le récepteur activé par une
protéinase-2 (PAR2; Ramachandran et coll., 2009) et les récepteurs P-adrénergiques
(Tzingounis et coll., 2010). Le PTH1R, qui peut engager une signalisation menant à la
synthèse de l'os trabéculaire s'il est stimulé avec l'agoniste synthétique [D-Trp12,
Tyr34]PTH,7.34), un agoniste biaisé sélectif de la signalisation dépendante de la p-arrestine

214
(Gesty-Palmer et coll., 2009), mène en revanche à la résorption osseuse, dans le cas où il
est stimulé par la PTH(i-34).. Pourtant, dans les travaux présentés ici, le maintien prolongé
d'un complexe RB2-P-arrestine ne semble pas favoriser une signalisation indépendante de
la protéine G. Il est intéressant de s'attarder aux raisons potentielles qui pourraient
expliquer ceci. Shukla et collaborateurs (2008) ont mis en évidence la capacité des P-
arrestines d'adopter de multiples conformations actives, dont dépendent leurs fonctions de
désensibilisation du signal secondaire à l'activation de la protéine G et/ou de facilitation
d'une signalisation indépendante de la protéine G. Ces auteurs suggèrent que l'agonisme
biaisé démontré par certains composés dépend de leur capacité à induire ou à stabiliser une
conformation qui elle-même influencera la conformation de la P-arrestine. Des études de
Marion et collaborateurs (2006) ont identifié un résidu Proline ou Alanine hautement
conservé au niveau de la seconde boucle intracellulaire de la majorité des RCPG, qui est un
site additionnel d'interaction entre le récepteur et la P-arrestine, puisqu'il s'ajoute au
domaine riche en résidus serines et threonines qui se trouve dans la queue C-terminale du
RCPG. Ce site de reconnaissance et d'interaction de la P-arrestine est absent du RB2
(Faussner et coll., 2009). Bien que son absence du RB2 ne nuise pas à la désensibilisation
initiée par le recrutement de la P-arrestine, il est à noter que la variété des sites d'interaction
entre ces protéines peut très certainement dicter leur conformation. En introduisant ce site
d'interaction avec la P-arrestine dans la séquence du RB2, il pourrait être possible
d'observer différents effets de la liaison de la P-arrestine au récepteur. Il est aussi possible
que les agonistes employés ne parviennent pas à induire au niveau du RB2 le changement
de conformation nécessaire qui induirait une conformation permissive de la P-arrestine afin
qu'elle stabilise les interactions entre différentes composantes de voies de signalisation.
Des études de modélisation de la liaison des agonistes biaises sauront renseigner sur les
interactions intermoléculaires entre l'agoniste et le récepteur qui mènent à une
conformation activant préférentiellement une signalisation dépendante soit de la protéine G,
soit de la p-arrestine (Rosenkilde et coll., 2010). Par exemple, l'identification d'un résidu
proline du TM2 se trouvant au niveau de la pochette mineure de liaison de l'agoniste à son
RCPG (qui est par ailleurs présent aussi chez le RB2) a été impliquée dans la sélectivité
pour une voie dépendante de la protéine G, suite à la stimulation du RATi par son agoniste
Angll (Reis et coll., 2007). Finalement, il faut considérer la découverte assez récente du

215
phénomène et le point de vue réducteur avec lequel il est parfois approché; pour l'instant, la
voie de signalisation la plus fréquemment associée à l'activité d'échaffaudage de la P-
arrestine est la voie des MAP kinases. Par contre, une étude portant sur le
phosphoprotéome quantitatif induit par la stimulation du RATi par l'agoniste biaisé [Sar1,
Ile4, Ile8]Ang II semble indiquer une influence beaucoup plus importante de cette
signalisation, vue la quantité de kinases et phosphatases dont le profil de phosphorylation
est modulé par l'agoniste biaisé (Xiao et coll., 2010).
4.5.2 Imagerie des récepteurs des kinines
Les travaux décrits dans la présente thèse montrent la caractérisation pharmacologique et
l'utilisation en imagerie de ligands fluorescents des récepteurs des kinines. L'objectif de
ces études était d'évaluer la faisabilité de créer un analogue conjugant le fluorophore CF
avec un ligand des récepteurs des kinines. La génération d'une paire agoniste-antagoniste
fluorescents pour chaque récepteur a permis la visualisation de récepteurs recombinants
exprimés à haute densité par des cellules HEK 293a, ainsi que celle de l'ECA
recombinante. De plus, leur utilisation a servi à caractériser ainsi qu'à mesurer les
processus biologiques au niveau cellulaire et moléculaire. Ces peptides fluorescents ne
constituent pourtant pas des sondes performantes pour la détection des récepteurs
endogènes exprimés en faible quantité, que ce soit en microscopie ou grâce à la cytométrie
de flux. Une solution qui pourrait être apportée à ce problème serait l'utilisation de
fluorophores beaucoup plus pluissants que ne l'est la CF, comme des fluorophores de type
Alexa Fluor ou même des nanocristaux de type Qdots. Comme la perte d'affinité
démontrée par ces ligands, assez sévère pour les agonistes, est atténuée par l'ajout d'un
espaceur, cela suggère que l'optimisation de la taille de celui-ci et de sa structure pourrait
grandement réduire la perte d'affinité. Les données présentées dans cette thèse portant sur
la maximakinine supportent tout à fait cette hypothèse. En effet, cette molécule présentant à
son extrémité C-terminale la séquence complète de la BK fusionnée à une séquence de dix
résidus en N-terminal présente une perte d'affinité modeste, telle que mesurée par des

216
essais de liaison (RB2 de lapin) et des essais de contractilité (RB2 humain). Ainsi, en plus
d'avoir identifié une séquence naturelle résistante au métabolisme endosomal auquel est
habituellement soumise la BK, il est fort probable que la MK constitue un excellent point
de départ à la synthèse d'une seconde génération d'agonistes fluorescents du RB2.
L'introduction de sa séquence dans un vecteur codant pour une protéine fluorescente est
une technique relativement simple à appliquer qui pourrait permettre l'obtention d'un
analogue fluorescent très prometteur pour son application en imagerie. Il serait fort
intéressant d'utiliser ce type de ligand original pour visualiser les tissus exprimant le RB2,
comme l'endothélium vasculaire, ou encore détecter la présence du RB2 au niveau de tissus
tumoraux et inflammés. Finalement l'optimisation des ligands fluorescents du RBi pourrait
s'avérer fort importante pour la visualisation de tissus inflammés où son expression est
particulièrement élevée.
L'emploi de la technique basée sur l'assemblage de complexes immuns comprenant le RB2
a permis elle aussi l'imagerie du récepteur. Les fluorophores couplés aux différents
anticorps secondaires présentaient une brillance hautement supérieure à celle de la CF, avec
l'avantage évident d'être minimalement soumis au pâlissement du signal, toujours par
comparaison avec la CF. De plus, les Qdots se distinguent des fluorophores de type Alexa
Fluor par leur résistance supérieure à l'atténuation du signal, un spectre d'émission étroit
permettant l'usage de plusieurs fluorophores de manière concomitante et leur importante
stabilité chimique (Pong et coll., 2008). L'internalisation de ces complexes par la
stimulation du RB2 par son agoniste BK a permis de caractériser les voies impliquées dans
son endocytose et d'effectuer une comparaison détaillée entre celles-ci et celles
généralement recrutées par le RB2 activé mais non complexé. Le problème de l'application
de cette technique à des systèmes endogènes dérive du manque d'anticorps fiables pour les
domaines extracellulaires du RB2 dans sa conformation native, bien que certaines équipes
aient décrits l'utilisation de tels anticorps (Haaseman et coll., 1998; Abd Alla et coll., 1996;
Figueroa et coll., 1996). Cette difficulté à générer des anticorps efficaces est vraie pour les
RCPG en général, comme le suggère l'application d'une technique très similaire à celle
présentée ici, pour un récepteur-1 de la sérotonine recombinant, qui présente un epitope HA
au niveau de son extrémité N-terminale, contre lequel est dirigé un anticorps anti-HA
couplé à un Qdot (Ficther et coll., 2010). Par contre, les résultats obtenus par cette équipe

217
diffèrent de ceux présentés ici, puisqu'ils rapportent un cycle d'endocytose-recyclage
inchangé du récepteur ainsi complexé.
Une stratégie additionnelle qui permet de marquer le RB2 a été décrite par Nonaka et
collaborateurs (2010), et repose sur un RB2 de fusion présentant une étiquette CA6D4x2
(CAAAAAADDDDGDDD) qui réagit de manière covalente avec un composé
tétranucléaire Zn(II)Dpa-Tyr, pouvant être optionnellement couplé à un fluorophore ou à la
biotine, dans le but d'étudier l'internalisation et le trafic endosomal du récepteur.
4.5.3 Cargos internalises par le RB2
La démonstration que le RB2 permet la liaison et l'internalisation d'un agoniste comportant
une extension N-terminale ou d'un cargo de taille importante basé sur des anticorps anti
récepteur de manière dépendante à son activité constitue tout de même un élément de
nouveauté qui permet d'émettre l'hypothèse qu'une variété de cargos conjugués médiant
différentes fonctions pourraient être internalises grâce au récepteur. Bien que les résultats
présentés se veulent une preuve de concept de l'internalisation du RB2 au sein d'un
complexe immun suite à la stimulation de son agoniste, puisque leur application est limitée
aux systèmes recombinants, le développement d'un anticorps spécifique contre l'extrémité
N-terminale du RB2 permettra plusieurs applications. Les récepteurs des kinines par leur
expression au sein de plusieurs tissus néoplasiques, inflammatoires ou au niveau de lésions
neuropathiques douloureuses pourraient servir de cibles afin de diriger spécifiquement un
médicament à son site d'action.
Un courant de recherche intensif dans le domaine pharmaceutique s'intéresse aux
conjugués médicament-anticorps et agent toxique-anticorps (immunotoxines) pour
plusieurs applications dont la plus étudiée est la thérapie du cancer. Ces composés
présentent l'avantage d'avoir un index thérapeutique élevé, en facilitant la livraison directe
de l'agent chimiothérapeutique au site désiré, ce qui en théorie garantit une toxicité plus
faible au niveau des tissus sains (Hughes, 2010). Le développement des immunotoxines

218
dépend de l'optimisation de la spécificité de l'anticorps monoclonal, des caractéristiques de
l'espaceur et de la puissance de l'agent cytotoxique employé (Ricart et Tolcher, 2007). Les
molécules toxiques pouvant être couplées aux anticorps sont des radioisotopes, des agents
chimiothérapeutiques ainsi que des toxines peptidiques. Le plus prometteur de ces
composés est présentement le trastuzumab-DM 1 (Lewis Phillips et coll., 2008; Hughes,
2010), un anticorps monoclonal déjà commercialisé dirigé contre HER2 (Chiu et coll.,
2004), le récepteur-2 du facteur de croissance épidermique dont l'expression est fortement
augmentée dans plusieurs formes de cancers du sein, ainsi que des cancers ovariens, qui est
conjugué avec l'agent cytotoxique emtansine (DMI). L'avantage que présentent le HER2 et
le RB2 comme cibles antigéniques idéales pour les immunotoxines est leur internalisation
rapide (Hughes, 2010). En revanche, le HER2 se distingue par son expression très
importante au niveau des tissus néoplasiques et par le fait qu'il n'est pas dégradé suivant
une stimulation prolongée, alors que l'inverse est observé dans le cas du RB2.
Au lieu de recourir à la reconnaissance d'un anticorps pour un antigène, il a été démontré
que la production de ligands bifonctionnels pour plusieurs types de récepteurs, dont les
RCPG, est une stratégie attrayante afin de générer des médicaments ciblés. La modulation
de leur expression dans certaines situations pathologiques présuppose un effet tissu-
spécifique (Shiota et coll., 2007). Un exemple intéressant d'un agoniste portant une
fonction cytotoxique est basé sur la bombésine, un analogue amphibien du peptide
stimulant le relâchement de gastrine (gastrin-releasing peptide), dont les trois sous-types de
récepteurs sont fréquemment surexprimés dans une variété de cancers, dont ceux de la
prostate (Markwalder et Reubi, 1999), du rein (Heuser et coll., 2005), de l'utérus (Cornelio
et coll., 2007) et des ovaires (Sun et coll., 2000). La bombésine a été couplée à un peptide
possédant une activité perturbatrice de la fonction mitochondriale, ce qui accrut la mort par
apoptose des cellules dérivées de tumeurs solides ou de lignées cellulaires leucémiques, en
facilitant l'accès des peptides à leur site d'action intracellulaire (Cai et coll., 2010). Un
second exemple intéressant est la molécule fusionnant la susbtance P, un neuropeptide,
avec la saporine, une enzyme inactivant catalytiquement le ribosome (Wiley et Lappi,
2003) qui pourrait être appliquée dans le cadre de la neurochirugie moléculaire, ainsi que
dans la compréhension de la perception de la douleur. Une telle approche serait
envisageable de manière préférentielle avec des agonistes du RB2, qui est internalise,

219
contrairement au RB|. Dans le cas d'agents thérapeutiques non toxiques, des ligands ou des
anticorps liant les récepteurs des kinines permettraient une livraison adéquate de susbtances
au niveau du système vasculaire, par exemple, où sa conjugaison avec un IECA pourrait
avoir un double effet vasodilatateur. Les deux récepteurs des kinines pourraient être des
cibles exploitables par des ligands ou des anticorps, en tant que nanotransporteurs d'un
composé anti-inflammatoire topique, dont la libération dépendrait de l'acidité du milieu,
afin de cibler les maladies inflammatoires de l'intestin, étant donné l'implication de ces
récepteurs dans F exacerbation du contexte inflammatoire (Marceau et Regoli, 2008). Ici,
les agonistes ou anticorps ciblant le RB2 présenteraient bien entendu l'avantage d'être
internalises par les cellules.
Puisque l'internalisation du RB2 repose sur la présence de son ligand, l'injection et
l'internalisation subséquente d'un anticorps dirigé contre l'extrémité N-terminale du
récepteur fusionné à un agent cytotoxique auraient l'avantage de n'avoir un effet qu'au site
où l'agoniste est présent et produit de manière endogène. Cette approche pourrait se révéler
autrement plus spécifique qu'une stratégie exploitant un agoniste couplé à une toxine, qui
encourerait automatiquement l'internalisation du récepteur. Par ailleurs, une façon de
valoriser l'internalisation du récepteur et son trafic endosomal est d'employer des toxines
qui sont dépendantes du pH, comme la toxine diphtérique ou des toxines dérivées de B.
anthracis (Cabiaux, 2004).

220
5. Conclusion
Les travaux de doctorat présentés par cette thèse ont permis une meilleure caractérisation
de l'adaptation des récepteurs des kinines, en fonction de leur sous-type, grâce à des
agonistes résistants au métabolisme dans le cas du RB2 et à des ligands fluorescents qui ont
permis de suivre leur progression subcellulaire suite à la liaison de l'agoniste, alors que
l'antagoniste permettait l'évaluation de la situation basale. Les ligands fluorescents de
première génération pour les récepteurs des kinines démontrent la faisabilité de prolonger
les séquences des ligands au niveau de leur extrémité N-terminale afin de générer une
variété d'analogues des kinines qui présenteront potentiellement des fonctions variées.
Finalement, l'internalisation de complexes immuns de taille importante par le RB2 stimulé
par son agoniste est une observation qui suggère que le transport que peut supporter le RB2
n'est pas très limité au niveau de la taille des cargos et qu'il pourrait accommoder des
cargos ayant des fonctions différentes de ceux exploités ici, c'est-à-dire des fluorophores de
type Alexa Fluor et d'autres basés sur des nanocristaux semiconducteurs Qdot.
Malgré les fonctions primordiales remplies par les kinines au niveau de la physiologie
humaine au niveau cardiovasculaire et rénal et leur participation néfaste à des phénomènes
pathologiques, la validation des récepteurs comme cibles thérapeutiques au sein de
plusieurs maladies n'est pas entière. En effet, du nombre important d'antagonistes et
d'agonistes des récepteurs, de nature peptidique ou non (Marceau et Regoli, 2004),
synthétisés au cours des trente dernières années, seul l'icatibant, un antagoniste du RB2, est
présentement en usage clinique, pour l'indication de l'angio-œdème héréditaire (Cicardi et
coll., 2010) et sera fort probablement bientôt accepté pour l'angio-œdème induit par la prise
d'IECA (Bas et coll., 2010). En effet, jusqu'à présent la pharmacothérapie du système
kinine-kallikréine enregistre des résultats mitigés pour plusieurs applications qui semblaient
fort prometteuses. La grande difficulté rencontrée avec les kinines découle de la dualité de
leurs fonctions, à la fois protectrices et délétères ; cette dualité fait écho aux deux sous-
types de récepteurs (Gabra et coll., 2003; Frick et coll., 2007), dont l'un serait bon (RB2) et

221
l'autre mauvais (RBi). Les agonistes du RB2 devaient présenter un potentiel inouï dans la
protection contre les désordres cardiovasculaires et la néphropathie diabétique. Un
antagoniste du RB2, l'anatibant (ou LF 16-0687), en essais cliniques pour l'œdème cérébral
post-traumatique (Zausinger et coll., 2003; Marmarou et coll., 2005), n'a malheureusement
révélé que peu d'efficacité réelle et un certain nombre d'effets adverses (Shakur et coll.,
2009). Des antagonistes du RBi. devaient être fort efficaces pour le traitement du choc
septique (Weikert et Bernard, 1996), de la douleur neuropathique (Levy et Zochodne, 2000;
Yamaguchi-Sase et coll., 2003; Gougat et coll., 2003). Par contre, les effets secondaires
observés suite à l'antagonisme systémique du RB] présentent une réalité plus nuancée,
étant donné son recrutement par des phénomènes compensateurs dans certaines situations
pathologiques, auquel cas son inhibition serait particulièrement délétère (Xu et coll., 2005).
Il sera important d'identifier de nouvelles applications pathologiques qui pourraient
bénéficier d'une modulation des récepteurs des kinines en tant que cibles thérapeutiques,
ainsi que de revisiter certaines applications à la lumière des données obtenues. Par exemple,
l'avantage théorique qu'un agoniste du RB2 résistant au métabolisme pourrait avoir en tant
qu'agent hypotenseur, comparativement à un agoniste vulnérable aux multiples peptidases
à activité kininase, ne sera pas nécessairement bénéfique, puisque le puissant effet
vasorelaxant est accompagné d'une stimulation typiquement pro-inflammatoire, tel que le
suggère la surexpression du facteur de transcription c-Fos. L'optimisation des ligands
fluorescents permettra certainement la valorisation des récepteurs des kinines en tant que
marqueurs d'une surexpression des récepteurs au niveau du système nerveux dans les
phases précoces de la maladie d'Alzheimer (Prediger et coll., 2008; Wiel et Buck, 2010),
ainsi que dans la sclérose en plaques (Prat et coll., 2005; Lùhder et coll., 2009).

6. Bibliographie Abd Alla S, Quitterer U, Grigoriev S, Maidhof A, Haasemann M, Jarnagin K, Muller-Esterl W (1996) Extracellular domains of the bradykinin B2 receptor involved in ligand binding and agonist sensing defined by anti-peptide antibodies. J Biol Chem 271: 1748-1755.
AbdAlla S, Zaki E, Lother H, Quitterer U (1999) Involvement of the amino terminus of the B(2) receptor in agonist-induced receptor dimerization. J Biol Chem 274:26079-84.
AbdAlla S, Lother H, el Massiery A, Quitterer U (2001) Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin I responsiveness. Nat Med 7:1003-9.
AbdAlla S, Abdel-Baset A, Lother H, el Massiery A, Quitterer U (2005) Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J Mol Neurosci 26:185-92.
Abe Y, Kayakiri H, Satoh S, Inoue T, Sawada Y, Imai K, Inamura N, Asano M, Hatori, Katayama A, Oku T, Tanaka H (1998) A novel class of orally active non-peptide bradykinin B2 receptor antagonists. 1. Construction of the Basic Framework. J Med Chem 41:564-578.
Abe Y, Kayakiri H, Satoh S, Inoue T, Sawada Y, Inamura N, Asano M, Aramori I, Hatori C, Sawai H, Oku T, Tanaka H (1998) A novel class of orally active non-peptide bradykinin B2 receptor antagonists. 3. Discovering bioisosteres of the imidazo[l,2-a]pyridine moiety. J Med Chem 41: 4062-4079.
Adam A, Cugno M, Molinaro G, Perez M, Lepage Y, Agostoni A (2002) Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet 359:2088-9.
Agata J, Miao RQ, Yayama K, Chao L, Chao J (2000) Bradykinin B| receptor mediates inhibition of neointima formation in rat artery after balloon angioplasty. Hypertension 36: 364-370.
Agata J, Chao L, Chao J (2002) Kallikrein gene delivery improves cardiac reserve and attenuates remodeling after myocardial infarction. Hypertension 40:653-9.
Akesson P, Herwald H, Rasmussen M, Hâkansson K, Abrahamson M, Hasan AA, Schmaier AH, Muller-Esterl W, Bjôrck L (2010) Streptococcal inhibitor of complement-mediated lysis (SIC): an anti-inflammatory virulence determinant. Microbiology 156:3660-8.
Alexander SPH, Mathie A, Peters JA (2008) Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision). Br J Pharmacol 153: S1-S209.

223
Alexander SPH, Mathie A, Peters JA (2009) Guide to Receptors and Channels (GRAC), 4th edition (2009). Br J Pharmacol 158: S1-S254.
Alfie ME, Yang XP, Hess F, Carretero OA (1996) Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem Biophys Res Commun 224:625-30.
Altura BM, Malaviya D, Reich CF, Orkin LR (1972) Effects of vasoactive agents on isolated human umbilical arteries and veins. Am J Physiol 222:345-55.
Amblard M, Daffix I, Berge G, Calmés M, Dodey P, Pruneau D, Paquet JL, Luccarini JM, Bélichard P, Martinez J ( 1999) Synthesis and characterization of bradykinin B(2) receptor agonists containing constrained dipeptide mimics. J Med Chem 42:4193-201.
Ando S, Rahman MA, Butler GC, Senn BL, Floras JS (1995) Comparison of candoxatril and atrial natriuretic factor in healthy men. Effects on hemodynamics, sympathetic activity, heart rate variability, and endothelin. Hypertension 26:1160-6.
Andoh T, Akira A, Saiki I, Kuraishi Y (2010) Bradykinin increases the secretion and expression of endothelin-1 through kinin B2 receptors in melanoma cells. Peptides 31:238-41.
Aramori I, Zenkoh J, Morikawa N, Asano M, Hatori C, Sawai H, Kayakiri H, Satoh S, Inoue T, Abe Y, Sawada Y, Mizutani T, Inamura N, Nakahara K, Kojo H, Oku T, Notsu Y ( 1997) Nonpeptide mimic of bradykinin with long-acting properties at the bradykinin B2 receptor. Mol Pharmacol 52:16-20.
Arunlakshana, Schild (1959) Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 14:48-58.
Asano M, Hatori C, Sawai H, Johki S, Inamura N, Kayakiri H, Satoh S, Abe Y, Inoue T, Sawada Y, Mizutani T, Oku T, Nakahara K (1998) Pharmacological characterization of a nonpeptide bradykinin B2 receptor antagonist, FR 165649, and agonist, FR 190997. Br J Pharmacol 124:441-6.
Ashby EL, Love S, Kehoe PG (2010) Assessment of activation of the plasma kallikrein-kinin system in frontal and temporal cortex in Alzheimer's disease and vascular dementia. Neurobiol Aging [Sous presse].
Audet R, Petitclerc E, Drapeau G, Rioux F, Marceau F (1994) Further analysis of the upregulation of bradykinin BI receptors in isolated rabbit aorta by using metabolic inhibitors. Eur J Pharmacol 271:551 -5.
Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, Christopoulos A (2007) Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem 282:25677-86.

224
Bachvarov DR, Saint-Jacques E, Larrivée JF, Levesque L, Rioux F, Drapeau G, Marceau F (1995) Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J Pharmacol Exp Ther 275:1623-1630.
Bachvarov DR, Houle S, Bachvarova M, Bouthillier J, Adam A, Marceau F (2001) Bradykinin B2 receptor endocytosis, recycling, and down-regulation assessed using green fluorescent protein conjugates. J Pharmacol Exp Ther 297:19-26. Baldwin JM (1994) Structure and function of receptors coupled to G proteins. Curr Opin Cell Biol 6:180-90.
Barak LS, Tiberi M, Freedman NJ, Kwatra MM, Lefkowitz RJ, Caron MG (1994) A highly conserved tyrosine residue in G protein-coupled receptors is required for agonist-mediated beta 2-adrenergic receptor sequestration. J Biol Chem 269:2790-5.
Barki-Harrington L, Bookout AL, Wang G, Lamb ME, Leeb-Lundberg LM, Daaka Y (2003) Requirement for direct cross-talk between BI and B2 kinin receptors for the proliferation of androgen-insensitive prostate cancer PC3 cells. Biochem J 371:581-7.
Bartus RT, Elliott PJ, Dean RL, Hayward NJ, Nagle TL, Huff MR, Snodgrass PA, Blunt DG (1996) Controlled modulation of BBB permeability using the bradykinin agonist, RMP-7. Exp Neurol 142:14-28.
Bas M, Grève J, Stelter K, Bier H, Stark T, Hoffmann TK, Kojda G (2010) Therapeutic efficacy of icatibant in angioedema induced by angiotensin-converting enzyme inhibitors: a case series. Ann Emerg Med 56: 278-282.
Bascands JL, Pécher C, Rouaud S, Emond C, Tack JL, Bastie MJ, Burch R, Regoli D, Girolami JP (1993) Evidence for existence of two distinct bradykinin receptors on rat mesangial cells. Am J Physiol 264:F548-56.
Bascands JL, Schanstra JP, Couture R, Girolami JP (2003) Bradykinin receptors: towards new pathophysiological roles. Med Sci (Paris) 19:1093-100.
Bastian S, Loillier B, Paquet JL, Pruneau D (1997) Stable expression of human kinin BI receptor in 293 cells: pharmacological and functional characterization. Br J Pharmacol 122:393-9.
Bastian S, Paquet JL, Robert C, Cremers B, Loillier B, Larrivée JF, Bachvarov DR, Marceau F, and Pruneau D (1998) Interleukin 8 (IL-8) induces the expression of kinin Bi receptor in human lung fibroblasts. Biochim Biophys Res Commun 253:750-5.
Bastian S, Pruneau D, Loillier B, Robert C, Bonnafous JC, Paquet JL (2000) Identification of a key region of kinin B(l) receptor for high affinity binding of peptide antagonists. J Biol Chem 275:6107-13.

225
Bawolak MT, Marceau F (2007) Does zaltoprofen antagonize the bradykinin receptors? Regul Pept U0:\25-\30.
Bawolak MT, Touzin K, Moreau ME, Désormeaux A, Adam A, Marceau F (2008) Cardiovascular expression of inflammatory signaling molecules, the kinin Bi receptor and COX2, in the rabbit: effect of LPS, anti-inflammatory and anti-hypertensive drugs. Regul. Pept. 146: 157-168.
Bellucci F, Meini S, Cucchi P, Catalani C, Reichert W, Zappitelli S, Rotondaro L, Quartara L, Giolitti A, Maggi CA (2003) A different molecular interaction of bradykinin and the synthetic agonist FR 190997 with the human B2 receptor: evidence from mutational analysis. Br J Pharmacol 140:500-6.
Bernier V, Lagacé M, Lonergan M, Arthus MF, Bichet DG, Bouvier M (2004) Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol Endocrinol 18: 2074-2084.
Bhoola KD, Figueroa CD, Worthy K (1992) Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 44:1-80.
Bironaite D, Gera L, Stewart JM (2004) Characterization of the B2 receptor and activity of bradykinin analogs in SHP-77 cell line by Cytosensor microphysiometer. Chem Biol Interact 150:283-293.
Black KL, Ningaraj NS (2004) Modulation of brain tumor capillaries for enhanced drug delivery selectively to brain tumor. Cancer Control 11:165-73.
Biais C Jr, Marceau F, Rouleau JL, Adam A (2000) The kallikrein-kininogen-kinin system: lessons from the quantification of endogenous kinins. Peptides 21:1903-40.
Blaukat A, Abd Alia S, Lohse MJ, Muller-Esterl W (1996) Ligand-induced phosphorylation/dephosphorylation of the endogenous bradykinin B2 receptor from human fibroblasts. J Biol Chem 271:32366-32374.
Blaukat A, Herzer K, Schroeder C, Bachmann M, Nash N, Muller-Esterl W (1999) Overexpression and functional characterization of kinin receptors reveal subtype-specific phosphorylation. Biochemistry 38: 1300-1309.
Blaukat A, Pizard A, Breit A, Wernstedt C, Alhenc-Gelas F, Muller-Esterl W, Dikic I (2001) Determination of bradykinin B2 receptor in vivo phosphorylation sites and their role in receptor function. J Biol Chem 276:40431-40440.
Blaukat A (2003) Structure and signalling pathways of kinin receptors. Andrologia 35:17-23.

226
Blaukat A, Mieke P, Kalatskaya I, Faussner A, Muller-Esterl W (2003) Downregulation of bradykinin B2 receptor in human fibroblasts during prolonged agonist exposure. Am J Physiol Heart Circ Physiol 284:H 1909-16.
Bledsoe G, Shen B, Yao Y, Zhang JJ, Chao L, Chao J (2006a) Reversal of renal fibrosis, inflammation, and glomerular hypertrophy by kallikrein gene delivery. Hum Gene Ther 17:545-55.
Bledsoe G, Crickman S, Mao J, Xia CF, Murakami H, Chao L, Chao J (2006b) Kallikrein/kinin protects against gentamicin-induced nephrotoxicity by inhibition of inflammation and apoptosis. Nephrol Dial Transplant 21:624-33.
Boguth CA, Singh P, Huang CC, Tesmer JJ (2010) Molecular basis for activation of G protein-coupled receptor kinases. EMBO J 29:3249-59.
Bôhm SK, Grady EF, Bunnett NW ( 1997) Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J 322:1-18.
Borkowski JA, Ransom RW, Seabrook GR, Trumbauer M, Chen H, Hill RG, Strader CD, Hess JF (1995) Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J Biol Chem 270:13706-10.
Bouthillier J, Deblois D, and Marceau F (1987) Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vivo and in vitro. Br J Pharmacol 92:257-64.
Brechter AB, Lerner UH (2002) Characterization of bradykinin receptors in a human osteoblastic cell line. Regul Peptides 103: 39-51.
Brechter AB, Persson E, Lundgren I, Lerner UH (2008) Kinin BI and B2 receptor expression in osteoblasts and fibroblasts is enhanced by interleukin-1 and tumour necrosis factor-alpha. Effects dependent on activation of NF-kappaB and MAP kinases. Bone 43:72-83.
Briner VA, TSAI P, Shrier RW (1993) Bradykinin: potential for vascular constriction in the presence of endothelial injury. Am J Physiol 264: F 322-7'.
Brown DA, Passmore GM (2010) Some new insights into the molecular mechanisms of pain perception. J Clin Invest 120:1380-3.
Brunnée T, Reddigari SR, Shibayama Y, Kaplan AP, Silverberg M (1997) Mast cell derived heparin activates the contact system: a link to kinin generation in allergic reactions. Clin Exp Allergy 27:653-63.

227
Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M (1992) The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70:715-28.
Burch RM, Axelrod J (1987) Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci USA 84:6374-8.
Burnett JC Jr (1999) Vasopeptidase inhibition: a new concept in blood pressure management. JHypertens Suppl 17:S37-43.
Cabiaux V (2004) pH-sensitive toxins: interactions with membrane bilayers and application to drug delivery. Adv Drug Deliv Rev 56:987-97.
Cai H, Yang H, Xiang B, Li S, Liu S, Wan L, Zhang J, Li Y, Cheng J, Lu X (2010) Selective apoptotic killing of solid and hematologic tumor cells by bombesin-targeted delivery of mitochondria-disrupting peptides. Mol Pharm 7:586-96
Calder PC (2006) Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. 75:197-202.
Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7:e 1000172.
Calebiro D, Nikolaev VO, Persani L, Lohse MJ (2010a) Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci 31:221-8.
Calebiro D, Nikolaev VO, Lohse MJ (2010b) Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J Mol Endocrinol 45:1 -8.
Campbell DJ (2003) The renin-angiotensin and the kallikrein-kinin systems. Int J Biochem Cell Biol 35:784-91.
Camponova P, Baud S, Mattras H, Duroux-Richard I, Bonnafous JC, Marie J (2007) High-level expression and purification of the human bradykinin B2 receptor in a tetracycline-inducible stable HEK293S cell line. Protein Expr Purif in press (published online, doi : 10.1016/j .pep.2007.04.020).
Campos MM, Leal PC, Yunes RA, Calixto JB (2006) Non-peptide antagonists for kinin BI receptors: new insights into their therapeutic potential for the management of inflammation and pain. Trends Pharmacol Sci 27:646-51.
Canals M, Lopez-Gimenez JF, Milligan G (2009) Cell surface delivery and structural reorganization by pharmacological chaperones of an oligomerization-defective alpha(lb)-

228
adrenoceptor mutant demonstrates membrane targeting of GPCR oligomers. Biochem J 417:161-72.
Ceresa BP, Lotscher M, Schmid SL. (2001) Receptor and membrane recycling can occur with unaltered efficiency despite dramatic Rb5(Q79L)-induced changes in endosome geometry. J Biol Chem 276 : 9649-9654.
Chan D, Gera L, Stewart J, Helfrich B, Verella-Garcia M, Johnson G, Baron A, Yang J, Puck T, Bunn P Jr (2002) Bradykinin antagonist dimer, CU201, inhibits the growth of human lung cancer cell lines by a "biased agonist" mechanism. Proc Natl Acad Sci USA 99:4608-4613.
Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F (2010) G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 465:435-40.
Chen EY, Emerich DF, Bartus RT, Kordower JH (2000) B2 bradykinin receptor immunoreactivity in rat brain. J Comp Neurol 427 :1 -18.
Chen T, O'Rourke M, Orr DF, Coulter DJ, Hirst DG, Rao P, Shaw C (2003) Kinestatin: a novel bradykinin B2 receptor antagonist peptide from the skin secretion of the Chinese toad, Bombina maxima. Regul Pept 116:147-54.
Chen T, O'Rourke M, McKenna J, Hirst DG, Shaw C (2005) Biotransformation of maximakinin, a bradykinin-related nonadecapeptide from toad venom, by mammalian kallikrein and salivary proteases. J Pept Res 66 Suppl 1:106-13.
Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (Kj) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22: 3099-3108.
Chiu SJ, Ueno NT, Lee RJ (2004) Tumor-targeted gene delivery via anti-HER2 antibody (trastuzumab, Herceptin) conjugated polyethylenimine. J Control Release 97:357-69.
Cicardi M, Baneiji A, Bracho F, Malbrân A, Rosenkranz B, Riedl M, Bork K, Lumry W, Aberer W, Bier H, Bas M, Grève J, Hoffmann TK, Farkas H, Reshef A, Ritchie B, Yang W, Grabbe J, Kivity S, Kreuz W, Levy RJ, Luger T, Obtulowicz K, Schmid-Grendelmeier P, Bull C, Sitkauskiene B, Smith WB, Toubi E, Werner S, Anne S, Bjôrkander J, Bouillet L, Cillari E, Hurewitz D, Jacobson KW, Katelaris CH, Maurer M, Merk H, Bernstein JA, Feighery C, Floccard B, Gleich G, Hébert J, Kaatz M, Keith P, Kirkpatrick CH, Langton D, Martin L, Pichler C, Resnick D, Wombolt D, Fernandez Romero DS, Zanichelli A, Arcoleo F, Knolle J, Kravec I, Claing A, Perry SJ, Achiriloaie M, Walker JK, Albanesi JP, Lefkowitz RJ, Premont RT (2000) Multiple endocytic pathways of G protein-coupled receptors delineated by GIT 1 sensitivity. Proc Natl Acad Sci USA 97:1119-24.
Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T (1999) Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem 274:34483-92.

229
Cayla C, Todiras M, Iliescu R, Saul VV, Gross V, Pilz B, Chai G, Merino VF, Pesquero JB, Baltatu OC, Bader M (2007) Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J 21:1689-98.
Colman RW (1994) Structure-function correlates of human high molecular weight kininogen. Braz J Med Biol Res 27:1839-53.
Colman RW, Pixley RA, Najamunnisa S, Yan W, Wang J, Mazar A, McCrae KR (1997) Binding of high molecular weight kininogen to human endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J Clin Invest 100:1481-7.
Colman RW, Schmaier AH (1997) Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90:3819-43.
Conlon JM, Aronsson U (1997) Multiple bradykinin-related peptides from the skin of the frog, Rana temporaria. Peptides 18:361-5.
Comelio DB, Meurer L, Roesler R, Schwartsmann G (2007) Gastrin-releasing peptide receptor expression in cervical cancer. Oncology 73:340-5
Corrêa FM, Innis RB, Uhl GR, Snyder SH (1979) Bradykinin-like immunoreactive neuronal systems localized histochemically in rat brain. Proc Natl Acad Sci USA 76:1489-93.
Costa-Neto CM, Dillenburg-Pilla P, Heinrich TA, Parreiras-e-Silva LT, Pereira MG, Reis RJ, Souza PP (2008) Participation of kallikrein-kinin system in different pathologies. Int Immunopharmacol 8:135-42.
Costerousse O, Jaspard E, Allegrini J, Wei L, Alhenc-Gelas F (1992) Angiotensin converting enzyme (kininase II). Molecular and physiological aspects. C R Seances Soc Biol Fil 186:586-98.
Côté J, Savard M, Bovenzi V, Dubuc C, Tremblay L, Tsanaclis AM, Fortin D, Lepage M, Gobeil F Jr (2010) Selective tumor blood-brain barrier opening with the kinin B2 receptor agonist [Phe(8)psi(CH(2)NH)Arg(9)]-BK in a F98 glioma rat model: an MRI study. Neuropeptides 44:177-85.
Couture R, Harrisson M, Vianna RM, Cloutier F (2001) Kinin receptors in pain and inflammation. Eur J Pharmacol 429:161-76.
Cugno M, Hack CE, de Boer JP, Eerenberg AJ, Agostoni A, Cicardi M (1993) Generation of plasmin during acute attacks of hereditary angioedema. J Lab Clin Med 121:38-43.

230
Cuthbert AW (1999) Anion secretory effects of a non-peptide mimic of bradykinin (FR 190997) on mouse colon epithelium. Immunopharmacology 45: 191-198.
Cuthbert AW (2001) Kinins and epithelial ion transport in the alimentary tract. Biol Chem 382:57-60.
de Agostini A, Lijnen HR, Pixley RA, Colman RW, Schapira M (1984) Inactivation of factor XII active fragment in normal plasma. Predominant role of C-1-inhibitor. J Clin Invest 73:1542-9.
Dahms P, Mentlein R (1992) Purification of the main somatostatin-degrading proteases from rat and pig brains, their action on other neuropeptides, and their identification as endopeptidases 24.15 and 24.16. Eur J Biochem 208:145-54.
De Bernardis F, Muhlschlegel FA, Cassone A, Fonzi WA (1998) The pH of the host niche controls gene expression in and virulence of Candida albicans. Infect Immun 66:3317-25.
deBlois D, Bouthillier J, and Marceau F (1988) Effect of glucocorticoids, monokines and growth factors on the spontaneously developing responses of the rabbit aorta to des-Arg9 -bradykinin. Br J Pharmacol 93: 969-77'.
deBlois D, Bouthillier J, Marceau F (1991) Pulse exposure to protein synthesis inhibitors enhances vascular responses to des-Arg9-bradykinin: possible role of interleukin-1. Br J Pharmacol 103:1057-66.
Décarie A, Raymond P, Gervais N, Couture R, Adam A (1996) Serum interspecies differences in metabolic pathways of bradykinin and [des-Arg9]BK: influence of énalaprilat. Am J Physiol 271 :H 1340-7.
Defea KA (2010) Beta-arrestins as regulators of signal termination and transduction: How do they determine what to scaffold? Cell Signal [Sous presse].
Dendorfer A, Wolfrum S, Schàfer U, Stewart JM, Inamura N, Dominiak P (2000) Potentiation of the vascular response to kinins by inhibition of myocardial kininases. Hypertension 35:32-7.
Desrivières S, Lu H, Peyri N, Soria C, Legrand Y, Ménashi S (1993) Activation of the 92 kDa type IV collagénase by tissue kallikrein. J Cell Physiol 157:587-93.
de Weerd WF, Leeb-Lundberg LM (1997) Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem 272:17858-66.
DeWire SM, Kim J, Whalen EJ, Ahn S, Chen M, Lefkowitz RJ (2008) P-arrestin-mediated signaling regulates protein synthesis. J Biol Chem 283: 10611-10620.

231
Diagaradjane P, Orenstein-Cardona JM, Colôn-Casasnovas NE, Deorukhkar A, Shentu S, Kuno N, Schwartz DL, Gelovani JG, Krishnan S (2008) Imaging epidermal growth factor receptor expression in vivo: pharmacokinetic and biodistribution characterization of a bioconjugated quantum dot nanoprobe. Clin Cancer Res 14: 731-741.
Dicpinigaitis PV (2006) Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest 129T69S-173S.
Dimitropoulos N, Papakyriakou A, Dalkas GA, Sturrock ED, Spyroulias GA (2010) A computational approach to the study of the binding mode of dual ACE/NEP inhibitors. J Chem InfModel 50:388-96.
Dobrovolsky AB, Titaeva EV (2002) The fibrinolysis system: regulation of activity and physiologic functions of its main components. Biochemistry 67:99-108.
Dong L, Zimmermann J, Rosen K, Fan WT (2010) Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N EnglJ Med 363:532-41.
Drapeau G, Rhaleb NE, Dion S, Jukic D, Regoli D (1988) [Phe8W(CH2NH)Arg9]bradykinin, a B2 receptor selective agonist which is not broken down by either kininase I or kininase II. Eur J Pharmacol 155:193- 195.
Drapeau G, deBlois D, Marceau F (1991a) Hypotensive effects of Lys-des-Arg9-bradykinin and metabolically protected agonists of BI receptors for kinins. J Pharmacol Exp Ther 259:997-1003.
Drapeau G, Chow A, Ward PE (1991b) Metabolism of bradykinin analogs by angiotensin I converting enzyme and carboxypeptidase N. Peptides 12:631-8.
Dray A (1997) Kinins and their receptors in hyperalgesia. Can J Physiol Pharmacol 75:704-12.
Duchardt F, Forin-Mleczek M, Schwarz H, Fischer R, Brock R (2007) A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8: 848-866.
Durchdewald M, Angel P, Hess J (2009) The transcription factor Fos: a Janus-type regulator in health and disease. Histol Histopathol 24:1451-61.
Duchene J, Cayla C, Vessillier S, Scotland R, Yamashiro K, Lecomte F, Syed I, Vo P, Marrelli A, Pitzalis C, Cipollone F, Schanstra J, Bascands JL, Hobbs AJ, Perretti M, Ahluwalia A (2009) Laminar shear stress regulates endothelial kinin BI receptor expression and function: potential implication in atherogenesis. Arterioscler Thromb Vase Biol 29:1757-63.
Duchene J, Ahluwalia A (2009) The kinin B( 1 ) receptor and inflammation: new therapeutic target for cardiovascular disease. Curr Opin Pharmacol 9:125-31.

232
Duvemay MT, Filipeanu CM, Wu G (2005) The regulatory mechanisms of export trafficking of G protein-coupled receptors. Cell Signal 17:1457-65.
Emanueli C, Bonaria Salis M, Stacca T, Pintus G, Kirchmair R, Isner JM, Pinna A, Gaspa L, Regoli D, Cayla C, Pesquero JB, Bader M, Madeddu P (2002) Targeting kinin Bj receptor for therapeutic neovascularization. Circulation 105: 360-366.
Emerich DF, Dean RL, Osborn C, Bartus RT (2001 ) The development of the bradykinin agonist labradimil as a means to increase the permeability of the blood-brain barrier: from concept to clinical evaluation. Clin Pharmacokinet 40:105-23.
Eisner D, Miintze A, Kromer EP, Riegger GA (1992) Effectiveness of endopeptidase inhibition (candoxatril) in congestive heart failure. Am J Cardiol 70:494-8.
Enquist J ,Skroder C ,Whistler JL ,Leeb-Lundberg LM (2007) Kinins promote B2 receptor endocytosis and delay constitutive BI receptor endocytosis. Mol Pharmacol 71:494-507.
Epstein TG, Bernstein JA (2008) Current and emerging management options for hereditary angioedema in the US. Drugs 68:2561-73.
Faussner A, Proud D, Towns M, Bathon JM (1998) Influence of the cytosolic carboxyl termini of human BI and B2 kinin receptors on receptor sequestration, ligand internalization, and signal transduction. J Biol Chem 273: 2617-2623.
Faussner A, Bathon JM, Proud D (1999) Comparison of the responses of BI and B2 kinin receptors to agonist stimulation, lmmunopharmacology 45:13-20.
Faussner A, Wennerberg G, Schussler S, Feierler J, Seidl C, Jochum M, Proud D (2009) Alanine screening of the intracellular loops of the human bradykinin B receptor—effects on receptor maintenance, G protein activation and internalization. FEBS J 276:3491-503.
Favrat B, Burnier M, Nussberger J, Lecomte JM, Brouard R, Waeber B, Brunner HR (1995) Neutral endopeptidase versus angiotensin converting enzyme inhibition in essential hypertension. J Hypertens 13:797-804.
Félétou M, Busse R, Edwards G, Fleming I, Weston AH, Vanhoutte PM (2003) Communication between endothelial and smooth muscle cells. Med Sci 19:1242-50.
Ferguson SS (2007) Phosphorylation-independent attenuation of GPCR signalling. Trends Pharmacol Sci 28:173-9.
Fernandez N, Monczor F, Baldi A, Davio C, Shayo C (2008) Histamine H2 receptor trafficking: role of arrestin, dynamin, and clathrin in histamine H2 receptor internalization. Mol Pharmacol 74: 1109-1118.

233
Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R (2009) Building a new conceptual framework for receptor heteromers. Nat Chem Bio 5:131-4.
Ferreira SH, Vane JR (1967) The detection and estimation of bradykinin in the circulating blood. Br J Pharmacol Chemother 29:367-77'.
Ferreira J, Campos MM, Pesquero JB, Araùjo RC, Bader M, Calixto JB (2001) Evidence for the participation of kinins in Freund's adjuvant-induced inflammatory and nociceptive responses in kinin BI and B2 receptor knockout mice. Neuropharmacology 41:1006-12.
Fichter KM, Flajolet M, Greengard P, Vu TQ (2010) Kinetics of G-protein-coupled receptor endosomal trafficking pathways revealed by single quantum dots. Proc Natl Acad Sci 107:18658-63.
Figueroa CD, Dietze G, Muller-Esterl W(1996) Immunolocalization of bradykinin B2 receptors on skeletal muscle cells. Diabetes 45:S24-8.
Figueroa CD, Marchant A, Novoa U, Fôrstermann U, Jarnagin K, Schôlkens B, Muller-Esterl W (2001) Differential Distribution of Bradykinin B(2) Receptors in the Rat and Human Cardiovascular System. Hypertension 37:110-120.
Fior DR, Hedlund PB, Fuxe K (1993) Autoradiographic evidence for a bradykinin/angiotensin II receptor-receptor interaction in the rat brain. Neurosci Lett 163:58-62.
Flanagan CA (2005) A GPCR that is not "DRY". Mol Pharmacol 68:1 -3.
Fleming I (2006) Signaling by the angiotensin-converting enzyme Circ Res 98:887-96.
Flower RJ, Perretti M (2005) Controling inflammation : a fat chance? J Exp Med 201 : 671-4.
Fogaça SE, Melo RL, Pimenta DC, Hosoi K, Juliano L, Juliano MA (2004) Differences in substrate and inhibitor sequence specificity of human, mouse and rat tissue kallikreins. Biochem .7380:775-81.
Forbes JM, Coughlan MT, Cooper ME (2008) Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 57:1446-54.
Fortin JP, Bouthillier J, Marceau F (2003) High agonist-independent clearance of rabbit kinin Bt receptors in cultured cells. Am J Physiol Heart Circ Physiol 284:YH647-H\654.

234
Fortin JP, Géra L, Bouthillier J, Stewart JM, Adam A, Marceau F (2005a) Endogenous aminopeptidase N decreases the potency of peptide agonists and antagonists of the kinin Bi receptors in the rabbit aorta. J Pharmacol Exp Ther 314:1169-1174.
Fortin JP, Rivard GE, Adam A, Marceau F (2005b) Studies on rabbit natural and recombinant tissue factors: intracellular retention and regulation of surface expression in cultured cells. Am J Physiol Heart Circ Physiol 288: H2192-H2202.
Fortin JP, Dziadulewicz EK, Géra L, Marceau F (2006a). A nonpeptide antagonist reveals a highly glycosylated state of the rabbit kinin Bj receptor. Mol Pharmacol 69:1146-1157.
Fortin JP, Marceau F (2006b) Advances in the development of bradykinin receptor ligands. Curr Topics Med Chem 6:1353-1363.
Fredriksson R, Lagerstrôm MC, Lundin LG, Schiôth HB (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63:1256-72.
Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, Peppel K (2002) Phosphorylation of the platelet-derived growth factor receptor-beta and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277:48261-9.
Frick IM, Bjôrck L, Herwald H (2007) The dual role of the contact system in bacterial infectious disease. Thromb Haemost 98:497-502.
Gabilondo AM, Krasel C, Lohse MJ (1996) Mutations of Tyr326 in the beta 2-adrenoceptor disrupt multiple receptor functions. Eur J Pharmacol 307:243-50.
Gabra BH, Couture R, Sirois P (2003) Functional duality of kinin receptors in pathophysiology. Med Sci 19:1101-10.
Gainer JV, Morrow JD, Loveland A, King DJ, Brown NJ ( 1998) Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med 339: 1285-1292.
Garcia-Perez A, Smith WL (1984) Apical-basolateral membrane asymmetry in canine cortical collecting tubule cells. Bradykinin, arginine vasopressin, prostaglandin E2 interrelationships. J Clin Invest 74: 63-74.
Gaudreau P, Barabé J, St-Pierre S, Regoli D (1981) Pharmacological studies of kinins in venous smooth muscle. Can J Physiol Pharmacol 59:371-379.
Géra L, Stewart JM (1996) A new class of bradykinin antagonists containing indanylglycine. Immunopharmacology 33:174-177.

235
Géra L, Stewart JM, Whalley E, Burkard M, Zuzack JS (1996) A new class of potent bradykinin antagonist dimers. Immunopharmacology 33:178-182.
Géra L, Fortin JP, Adam A, Stewart JM, Marceau F (2006) Discovery of a dual-function peptide that combines aminopeptidase N inhibition and kinin B| receptor antagonism. J Pharmacol Exp Ther 317: 300-308.
Gera L, Stewart JM, Fortin JP, Morissette G, Marceau F (2008) Structural modification of the highly potent peptide bradykinin Bj receptor antagonist B9958. Int Immunopharmacol 8: 289-292.
Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ (2006) Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK 1/2 activation. J Biol Chem 281:10856-64.
Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, Luttrell LM (2009) A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transi Me Lirai .
Gether U (2000) Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev 21:90-113.
Gifford JL, Walsh MP, Vogel HJ (2007) Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J 405:199-221.
Glauser DA, Schlegel W (2007) Sequential actions of ERK 1/2 on the AP-1 transcription factor allow temporal integration of metabolic signals in pancreatic p cells. FASEB J 21: 3240-3249.
Gobeil F Jr, Montagne M, Inamura N, Regoli D (1999) Characterization of non-peptide bradykinin B2 receptor agonist (FR 190997) and antagonist (FR 173657). Immunopharmacology 43: 179-185.
Gohla A, Offermanns S, Wilkie TM, Schultz G (1999) Differential involvement of Galphal2 and Galphal3 in receptor-mediated stress fiber formation. J Biol Chem 274:17901-7.
Golias Ch, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A (2007) The kinin system-bradykinin: biological effects and clinical implications. Multiple role of the kinin system-bradykinin. Hippokratia 11:124-8.
Gôrlach C, Wahl M (1996) Bradykinin dilates rat middle cerebral artery and its large branches via endothelial B2 receptors and release of nitric oxide. Peptides 17:1373-8.

236
Gougat J, Ferrari B, Sarran L, Planchenault C, Poncelet M, Maruani J, Alonso R, Cudennec A, Croci T, Guagnini F, Urban-Szabo K, Martinolle JP, Soubrié P, Finance O, Le Fur G (2004) SSR240612 [(2R)-2-[((3R)-3-( 1,3-benzodioxol-5-yl)-3-[[(6-methoxy-2-naphthyl)sulfonyl]amino]propanoyl)amino]-3-(4-[[2R,6S)-2,6-dimethylpiperidinyl]methyl]phenyl)-N-isopropyl-N-methylpropanamide hydrochloride], a new nonpeptide antagonist of the bradykinin BI receptor: biochemical and pharmacological characterization. J Pharmacol Exp Ther 309:661-9.
Graness A, Adomeit A, Ludwig B, Muller WD, Kaufmann R, Liebmann C (1997) Novel bradykinin signalling events in PC-12 cells: stimulation of the cAMP pathway leads to cAMP-mediated translocation of protein kinase Cepsilon. Biochem J 327:147-54.
Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: Achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103: 11821 -11827.
Grynkiewizc G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440-50.
Gurevich VV, Benovic JL (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem 268:11628-38.
Gurevich EV, Gurevich VV (2006) Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol 7:236.
Ha SN, Hey PJ, Ransom RW, Bock MG, Su DS, Murphy KL, Chang R, Chen TB, Pettibone D, Hess JF (2006) Identification of the critical residues of bradykinin receptor BI for interaction with the kinins guided by site-directed mutagenesis and molecular modeling. Biochemistry 45:14355-61.
Haack KK, Tougas MR, Jones KT, El-Dahr SS, Radhakrishna H, McCarty NA (2010) A novel bioassay for detecting GPCR heterodimerization: transactivation of beta 2 adrenergic receptor by bradykinin receptor. JBiomol Screen 15:251-60.
Haasemann M, Cartaud J, Muller-Esterl W, Dunia I ( 1998) Agonist-induced redistribution of bradykinin B2 receptor in caveolae. J Cell Sci 111:917-28.
Hagaman JR, Moyer JS, Bachman ES, Sibony M, Magyar PL, Welch JE, Smithies O, Krege JH, O'Brien DA (1998) Angiotensin-converting enzyme and male fertility. Proc Natl Acad Sci U SA 95:2552-7.
Hamdan FF, Rochdi MD, Breton B, Fessart D, Michaud DE, Charest PG, Laporte SA, Bouvier M (2007) Unraveling G protein-coupled receptor endocytosis pathways using realtime monitoring of agonist-promoted interaction between P-arrestins and AP-2. J Biol Chem 282: 29089-290100.

237
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70.
Hansen JL, Hansen JT, Speerschneider T, Lyngso C, Erikstrup N, Burstein ES, Weiner DM, Walther T, Makita N, Iiri T, Merten N, Kostenis E, Sheikh SP (2009) Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: how common is the AT1R/B2R heterodimer? J Biol Chem 284:1831-9.
Hara DB, Leite DF, Fernandes ES, Passos GF, Guimarâes AO, Pesquero JB, Campos MM, Calixto JB (2008) The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br J Pharmacol 154:1276-86.
Harford-Wright E, Lewis KM, Vink R (2010) Towards Drug Discovery for Brain Tumours: Interaction of Kinins and Tumours at the Blood Brain Barrier Interface. Recent Pat CNS DrugDiscov [Sous presse].
Hasan AA, Zisman T, Schmaier AH ( 1998) Identification of cytokeratin 1 as a binding protein and presentation receptor for kininogens on endothelial cells. Proc Natl Acad Sci U S A 95:3615-20.
Hayashi I, Ishihara K, Kumagai Y, Majima M (2001) Proinflammatory characteristics of a nonpeptide bradykinin mimic, FR190997, in vivo. Br J Pharmacol 133: 1296-1306.
Herwald H, Dedio J, Kellner R, Loos M, Muller-Esterl W (1996) Isolation and characterization of the kininogen-binding protein p33 from endothelial cells. Identity with the gClq receptor. J Biol Chem 271:13040-7.
Herzig MC, Leeb-Lundberg LM (1995) The agonist binding site on the bovine bradykinin B2 receptor is adjacent to a sulfhydryl and is differentiated from the antagonist binding site by chemical cross-linking. J Biol Chem 270:20591-8.
Herzig MC, Nash NR, Connolly M, Kyle DJ, Leeb-Lundberg LM (1996) The N terminus of bradykinin when bound to the human bradykinin B2 receptor is adjacent to extracellular Cys20 and Cys277 in the receptor. J Biol Chem 271:29746-51.
Hess JF, Borkowski J A, Young GS, Strader CD, Ransom RW (1992) Cloning and pharmacological characterization of a human bradykinin (BK-2) receptor. Biochem Biophys Res Commun 184:260-8.
Hess JF, Borkowski JA, MacNeil T, Stonesifer GY, Fraher J, Strader CD (1994) Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol Pharmacol 45: 1-8.
Hess JF, Derrick AW, MacNeil T, Borkowski JA (1996) The agonist selectivity of a mouse BI bradykinin receptor differs from human and rabbit BI receptors. Immunopharmacology 33:1-8.

238
Henderson LM, Figueroa CD, Muller-Esterl W, Stain A, Bhoola KD (1992) Immunovisualisation of plasma prekallikrein and H-kininogen on human neutrophils and in human hepatocytes. Agents Actions Suppl 38:590-4.
Heuser M, Schlott T, Schally AV, Kahler E, Schliephake R, Laabs SO, Hemmerlein B (2005) Expression of gastrin releasing Peptide receptor in renal cell carcinomas: a potential function for the regulation of neoangiogenesis and microvascular perfusion. J Urol 173:2154-9.
Hinshaw JE (2000) Dynamin and its role in membrane fission. Annu Rev Cell Dev Biol 16:483-519.
Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97:257-69.
Hislop JN, Von Zastrow M (2010) Role of Ubiquitination in Endocytic Trafficking of G-Protein-Coupled Receptors. Traffic [Sous presse].
Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ (2005)The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J 24:3247-58.
Houle S, Larrivée JF, Bachvarova M, Bouthillier J, Bachvarov DR, Marceau F (2000) Antagonist-induced intracellular sequestration of rabbit bradykinin B2 receptor. Hypertension 35:1319-1325.
Houle S, Marceau F (2003) Wortmannin alters intracellular trafficking of the bradykinin B2 receptor: role of phosphatidylinositol-3 kinase and Rab5. Biochem J 315:151-158.
Houle S, Molinaro G, Adam A, Marceau F (2003) Tissue kallikrein actions at the rabbit natural or recombinant kinin B2 receptors. Hypertension 41:611-617.
Huang Z, Taylor L, Liu B, Yu J, Polgar P (2006) Modulation by bradykinin of angiotensin type 1 receptor-evoked RhoA activation of connective tissue growth factor expression in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 290:L 1291-9.
Huang CC, Yoshino-Koh K, Tesmer JJ (2009) A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J Biol Chem 284:17206-15.
Huang H, Player MR (2010) Bradykinin BI receptor antagonists as potential therapeutic agents for pain. J Med Chem 53:5383-99.
Hughes B (2010) Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov 9:665-7.

239
Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V (2006) V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 8: 124-136.
Huttenrauch F, Nitzki A, Lin FT, Hôning S, Oppermann M (2002) Beta-arrestin binding to CC chemokine receptor 5 requires multiple C-terminal receptor phosphorylation sites and involves a conserved Asp-Arg-Tyr sequence motif. J Biol Chem 217:30169-71.
Inokuchi J, Nagamatsu A (1981) Tripeptidyl carboxypeptidase activity of kininase II (angiotensin-converting enzyme). Biochim Biophys Acta 662:300-7.
Jarnagin K, Bhakta S, Zuppan P, Yee C, Ho T, Phan T, Tahilramani R, Pease JH, Miller A, Freedman R (1996) Mutations in the B2 bradykinin receptor reveal a different pattern of contacts for peptidic agonists and peptidic antagonists. J Biol Chem 271:28277-28286.
Jaspard E, Wei L, Alhenc-Gelas F (1993) Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem 268:9496-503.
Jeng AY, Savage P, Beil ME, Bruseo CW, Hoyer D, Fink CA, Trapani AJ (2002) CGS 34226, a thiol-based dual inhibitor of endothelin converting enzyme-1 and neutral endopeptidase 24.11. Clin Sci (Lond) 103:98S-101S.
Jong YJ, Dalemar LR, Wilhelm B, Baenziger NL (1993) Human bradykinin B2 receptors isolated by receptor-specific monoclonal antibodies are tyrosine phosphorylated. Proc Natl Acad Sci USA 90:10994-8.
Jong YJ, Ford SR, Seehra K, Malave VB, Baenziger NL (2003) Alzheimer's disease skin fibroblasts selectively express a bradykinin signaling pathway mediating tau protein Ser phosphorylation. FASEB J 17:2319-21.
Jonkam CC, Enkhbaatar P, Nakano Y, Boehm T, Wang J, Nussberger J, Esechie A, Traber LD, Herndon D, Traber DL (2007) Effects of the bradykinin B2 receptor antagonist icatibant on microvascular permeability after thermal injury in sheep. Shock 28:704-9.
Joseph K, Ghebrehiwet B, Peerschke EI, Reid KB, Kaplan AP (1996) Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor that binds to the globular "heads" of Clq (gClq-R). Proc Natl Acad Sci USA 93:8552-7.
Joseph K, Shibayama Y, Nakazawa Y, Peerschke EI, Ghebrehiwet B, Kaplan AP (1999) Interaction of factor XII and high molecular weight kininogen with cytokeratin 1 and gClqR of vascular endothelial cells and with aggregated Abeta protein of Alzheimer's disease. Immunopharmacology 43:203-10.

240
Joseph K, Tholanikunnel BG, Kaplan AP (2002) Activation of the bradykinin-forming cascade on endothelial cells: a role for heat shock protein 90. Int Immunopharmacol 2:1851-9.
Juillerat-Jeanneret L (2008) The targeted delivery of cancer drugs across the blood-brain barrier: chemical modifications of drugs or drug-nanoparticles? Drug Discov Today 13:1099-106.
Jutras S, Bachvarova M, Keita M, Bascands JL, Mes-Masson AM, Stewart JM, Bachvarov D (2010) Strong cytotoxic effect of the bradykinin antagonist BKM-570 in ovarian cancer cells - analysis of the molecular mechanisms of its antiproliferative action. FEBS J 277:5146-5160.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720-5728.
Kahn R, Hellmark T, Leeb-Lundberg LM, Akbari N, Todiras M, Olofsson T, Wieslander J, Christensson A, Westman K, Bader M, Muller-Esterl W, Karpman D (2009) Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol 182:7906-15.
Kakoki M, Kizer CM, Yi X, Takahashi N, Kim HS, Bagnell CR, Edgell CJ, Maeda N, Jennette JC, Smithies O (2006) Senescence-associated phénotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J Clin Invest 116:1302-9.
Kakoki M, McGarrah RW, Kim HS, Smithies O (2007) Bradykinin BI and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc Natl Acad Sci USA 104:7576-81.
Kalatskaya I, Schussler S, Blaukat A, Muller-Esterl W, Jochum M, Proud D, Faussner A (2004) Mutation of tyrosine in the conserved NPXXY sequence leads to constitutive phosphorylation and internalization, but not signaling, of the human B2 bradykinin receptor. J Biol Chem 279:31268-76.
Kalatskaya I, Schussler S, Seidl C, Jochum M, Faussner A (2006) C-Terminal fusion of eGFP to the bradykinin B2 receptor strongly affects down-regulation but not receptor internalization or signaling. Biol Chem 387:603-610.
Kang DS, Leeb-Lundberg LM (2002) Negative and positive regulatory epitopes in the C-terminal domains of the human B1 and B2 bradykinin receptor subtypes determine receptor coupling efficacy to G(q/1 l)-mediated [correction of G(9/l l)-mediated] phospholipase Cbeta activity. Mol Pharmacol 62:281-8.
Kang DS, Ryberg K, Môrgelin M, Leeb-Lundberg LM (2004) Spontaneous formation of a proteolytic BI and B2 bradykinin receptor complex with enhanced signaling capacity. J Biol Chem 279:22102-7'.

241
Kang DS, Gustafsson C, Môrgelin M, Leeb-Lundberg LM (2005) BI bradykinin receptor homo-oligomers in receptor cell surface expression and signaling: effects of receptor fragments. Mol Pharmacol 67:309-18.
Kaplan AP, Silverberg M (1987) The coagulation-kinin pathway of human plasma. Blood 70:1-15.
Kaplan AP, Joseph K, Shibayama Y, Reddigari S, Ghebrehiwet B, Silverberg M (1997) The intrinsic coagulation/kinin-forming cascade: assembly in plasma and cell surfaces in inflammation. Adv Immunol 66:225-72.
Kaplan AP, Greaves MW (2005) Angioedema. J Am Acad Dermatol 53(3):373-88; quiz 389-92.
Kentsch M, Otter W, Drummer C, Nôtges A, Gerzer R, Muller-Esch G (1996) Neutral endopeptidase 24.11 inhibition may not exhibit beneficial haemodynamic effects in patients with congestive heart failure. Eur J Clin Pharmacol 51:269-72.
Kleinert H, Schwarz PM, Fôrstermann U (2003) Regulation of the expression of inducible nitric oxide synthase. Biol Chem 384:1343-64.
Kohan DE (2010) Endothelin, hypertension and chronic kidney disease: new insights. Curr Opin Nephrol Hypertens 19:134-9
Kohlstedt K, Busse R, Fleming I (2005) Signaling via the angiotensin-converting enzyme enhances the expression of cyclooxygenase-2 in endothelial cells. Hypertension 45:126-32.
Koki AT, Leahy KM, Masferrer JL (1999) Potential utility of COX-2 inhibitors in chemoprevention and chemotherapy. Expert Opin Investig Drugs 8:1623-1638.
Koumbadinga GA, Bawolak MT, Marceau E, Adam A, Géra L, Marceau F (2010a) Ligand-based approaches to investigate the expression of angiotensin converting enzyme in intact human umbilical vein endothelial cells. Peptides 31:1546-1554.
Koumbadinga GA, Désormeaux A, Adam A, Marceau F (2010b) Effect of interferon-y on inflammatory cytokine-induced bradykinin Bj receptor expression in human vascular cells. Eur J Pharmacol 647: 117-125.
Kramarenko II, Bunni MA, Morinelli TA, Raymond JR, Garnovskaya MN (2009) Identification of functional bradykinin B2 receptors endogenously expressed in HEK293 cells. Biochem Pharmacol 77: 269-276.
Kuduk SD, Bock MG (2008) Bradykinin BI receptor antagonists as novel analgesics: a retrospective of selected medicinal chemistry developments. Curr Top Med Chem 8:1420-30.

242
Kuoppala A, Shiota N, Lindstedt KA, Rysâ J, Leskinen HK, Luodonpàà M, Liesmaa I, Ruskoaho H, Kaaja R, Kovanen PT, Kokkonen JO (2003) Expression of bradykinin receptors in the left ventricles of rats with pressure overload hypertrophy and heart failure. J Hypertens 21:1729-36.
Kutateladze TG, Ogburn KD, Watson WT, de Beer T, Emr SD, Burd CG, Overduin M (1999) Phosphatidylinositol 3-phosphate recognition by the FYVE domain. Mol Cell 3: 805-811.
Kwok HF, Chen T, O'Rourke M, Ivanyi C, Hirst D, Shaw C (2008) Helokinestatin: a new bradykinin B2 receptor antagonist decapeptide from lizard venom. Peptides 29(l):65-72. Lalloo UG, Barnes PJ, Chung KF (1996) Pathophysiology and clinical presentations of cough. J Allergy Clin Immunol 98:S91-6; discussion S96-7.
Kyle DJ (1994) Structural features of the bradykinin receptor as determined by computer simulations, mutagenesis experiments, and conformational ly constrained ligands: establishing the framework for the design of new antagonists. Braz J Med Biol Res 27:1757-79.
Lamb ME, De Weerd WF, Leeb-Lundberg LM (2001) Agonist-promoted trafficking of human bradykinin receptors: arrestin- and dynamin-independent sequestration of the B2 receptor and bradykinin in HEK293 cells. Biochem J 355:741 -50.
Lambeir AM, Durinx C, Scharpé S, De Meester I (2003) Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci 40:209-94.
Larrivée JF, Géra L, Houle S, Bouthillier J, Bachvarov DR, Stewart J, Marceau F (2000) Non-competitive pharmacological antagonism at the rabbit Bj receptor. Br J Pharmacol 131:885-92.
Leeb-Lundberg LM, Kang DS, Lamb ME, Fathy DB (2001) The human BI bradykinin receptor exhibits high ligand-independent, constitutive activity. Roles of residues in the fourth intracellular and third transmembrane domains. J Biol Chem 276:8785-92.
Leeb-Lundberg LMF, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL (2005) International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev 57:27-77.
Lefkowitz RJ (2004) Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci 25:413-22.
Lever AF, Hole DJ, Gillis CR, McCallum IR, Mclnnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW (1998) Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 352:179-84.

243
Levesque L, Harvey N, Rioux F, Drapeau G, Marceau F (1995) Development of a binding assay for the B| receptors for kinins. Immunopharmacology 29: 141-147.
Levy D, Zochodne DW (2000) Increased mRNA expression of the BI and B2 bradykinin receptors and antinociceptive effects of their antagonists in an animal model of neuropathic pain. Pain 86:265-71.
Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blàttler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX (2008) Targeting HER2-positive breast cancer with trastuzumab-DM 1, an antibody-cytotoxic drug conjugate. Cancer Res 68:9280-90.
Li JG, Haines DS, Liu-Chen LY (2008) Agonist-promoted Lys63-linked polyubiquitination of the human kappa-opioid receptor is involved in receptor down-regulation. Mol Pharmacol 73:1319-30.
Liao Z, Mason KA, Milas L (2007) Cyclo-oxygenase-2 and its inhibition in cancer: is there a role? Drugs 67:821-45.
Liesmaa I, Kuoppala A, Shiota N, Kokkonen JO, Kostner K, Mâyrànpàà M, Kovanen PT, Lindstedt KA (2005) Increased expression of bradykinin type-1 receptors in endothelium of intramyocardial coronary vessels in human failing hearts. Am J Physiol Heart Circ Physiol 288:H2317-22.
Lin P, Chen JW, Chang LW, Wu JP, Redding L, Chang H, Yeh TK, Yang CS, Tsai MH, Wang HJ, Kuo YC, Yang RSH (2008) Computational and ultrastructural toxicology of a nanoparticle, s(1994) Effects of peptide and nonpeptide antagonists of the bradykinin B2 receptors on the venoconstrictor action of bradykinin. J Pharmacol Exp Ther 269:1136-1143.
Liu JM, Lawrence F, Kovacevic M, Bignon J, Papadimitriou E, Lallemand JY, Katsoris P, Potier P, Fromes Y, Wdzieczak-Bakala J (2003) The tetrapeptide AcSDKP, an inhibitor of primitive hematopoietic cell proliferation, induces angiogenesis in vitro and in vivo. Blood 101:3014-20.
Liu LB, Xue YX, Liu YH, Wang YB (2008) Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton. J Neurosci Res 86:1153-68.
Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N (2010) The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated CX channels. J Clin Invest 120 : 1240-52.

244
Linz W, Wiemer G, Gohlke P, Unger T, Schôlkens BA (1995) Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev 47:25-49.
Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR (1993) Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J 12:677-82.
Lu DY, Leung YM, Huang SM, Wong KL (2010) Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin BI receptor in glioma cells. J Cell Biochem 110:141-50.
Luan Y, Wang Q, Liu N, Mou J, Jiao X, Fang H, Li M, Xu W (2011) Synthesis and activity evaluation of a new bestatin derivative LYP2 as an aminopeptidase N inhibitor. Anticancer Drugs 22:99-103.
Luttrell LM, Lefkowitz RJ (2002) The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115:455-65.
Madeddu P, Varoni MV, Palomba D, Emanueli C, Demontis MP, Glorioso N, Dessi-Fulgheri P, Sarzani R, Anania V (1997) Cardiovascular phenotype of a mouse strain with disruption of bradykinin B2-receptor gene. Circulation 96:3570-8.
Mahabeer R, Bhoola KD (2000) Kallikrein and kinin receptor genes. Pharmacol Ther 88:77-89.
Mandle RJ, Colman RW, Kaplan AP (1976) Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc Natl Acad Sci U S A 73:4179-83.
Manolis AJ, Marketou ME, Gavras I, Gavras H (2010) Cardioprotective properties of bradykinin: role of the B(2) receptor. Hypertens Res 33:112-1.
Marceau F (1995) Kinin BI receptors: a review, lmmunopharmacology 30:1-26.
Marceau F, Hess JF, Bachvarov DR (1998) The Bj receptors for kinins. Pharmacol Rev 50: 357-86.
Marceau F, Houle S, Bouthillier J, Said NB, Garratt PJ, Dziadulewicz EK (2001) Effects of two novel non-peptide antagonists at the rabbit bradykinin B2 receptor. Peptides 22:1397-1402.
Marceau F, Sabourin T, Houle S, Fortin JP, Petitclerc E, Molinaro G, Adam A (2002) Kinin receptors: Functional aspects. Int lmmunopharmacol 2:1729-1739.

245
Marceau F, Fortin JP, Morissette G, Dziadulewicz EK (2003) A non-peptide antagonist unusually selective for the human form of the bradykinin B2 receptor. Int Immunopharmacol 3:1529-1536.
Marceau F, Regoli D (2004) Bradykinin receptor ligands: therapeutic perspectives. Nat Rev DrugDiscov 3:845-52.
Marceau F, Regoli D (2008) Therapeutic options in inflammatory bowel disease: experimental evidence of a beneficial effect of kinin B1 receptor blockade. Br J Pharmacol 154:1163-5.
Marceau F, Bawolak MT, Bouthillier J, Morisette G (2009) Vacuolar ATPase-mediated cellular concentration and retention of quinacrine: a model for the distribution of lipophilic cationic drugs to autophagic vacuoles. Drug Metab Dispos 37: 2271-2274.
Marcic B, Deddish PA, Jackman HL, Erdos EG (1999) Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension 33:835- 43.
Marcondes S, Antunes E (2005) The plasma and tissue kininogen-kallikrein-kinin system: role in the cardiovascular system. Curr Med Chem Cardiovasc Hematol Agents 3:33-44.
Marie J, Richard E, Pruneau D, Paquet JL, Siatka C, Larguier R, Poncé C, Vassault P, Groblewski T, Maigret B, Bonnafous JC (2001) Control of conformational equilibria in the human B2 bradykinin receptor. Modeling of nonpeptidic ligand action and comparison to the rhodopsin structure. J Biol Chem 276: 41100-41 111.
Marketou M, Kintsurashvili E, Papanicolaou KN, Lucero HA, Gavras I, Gavras H (2010) Cardioprotective effects of a selective B(2) receptor agonist of bradykinin post-acute myocardial infarct. Am J Hypertens 23:562-8.
Markwalder R, Reubi JC (1999) Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res 59:1152-9.
Marley A, von Zastrow M (2010) Dysbindin promotes the post-endocytic sorting of G protein-coupled receptors to lysosomes. PLoS One 5:e9325.
Marmarou A, Guy M, Murphey L, Roy F, Layani L, Combal JP, Marquer C (2005) A single dose, three-arm, placebo-controlled, phase I study of the bradykinin B2 receptor antagonist Anatibant (LF16-0687Ms) in patients with severe traumatic brain injury. American Brain Injury Consortium. J Neurotrauma. 22:1444-55.
Martorana PA, Kettenbach B, Breipohl G, Linz W, Schôlkens BA (1990) Reduction of infarct size by local angiotensin-converting enzyme inhibition is abolished by a bradykinin antagonist. Eur J Pharmacol 182:395-6.

246
Mathis SA, Criscimagna NL, Leeb-Lundberg LM (1996) BI and B2 kinin receptors mediate distinct patterns of intracellular Ca2+ signaling in single cultured vascular smooth muscle cells. Mol Pharmacol 50:128-39.
McEachern AE, Shelton ER, Bhakta S, Obernolte R, Bach C, Zuppan P, Fujisaki J, Aldrich RW, Jarnagin K (1991) Expression cloning of a rat B2 bradykinin receptor. Proc Natl Acad Sci US A 88:7724-8.
McLean AJ, Milligan G (2000) Ligand regulation of green fluorescent protein-tagged forms of the human pi- and p2-adrenoceptors; comparison with the unmodified receptor. Br J Pharmacol 130: 1825-1832.
Meini S, Cucchi P, Zappitelli S, Rotondaro L, Quartara L, Giolitti A, Maggi CA (2002) Preliminary mutational analysis of the human kinin B2 receptor for nonpeptide antagonist ligands recognition. Can J Physiol Pharmacol 80:303-9.
Meng EC, Bourne HR (2001) Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol Sci 22:587-93.
Merlini PA, Cugno M, Rossi ML, Agricola P, Repetto A, Fetiveau R, Diotallevi P, Canosi U, Mannucci PM, Ardissino D (2004) Activation of the contact system and inflammation after thrombolytic therapy in patients with acute myocardial infarction. Am J Cardiol 93:822-5.
Métayé T, Perdrisot R, Kraimps JL (2006) GRKs and arrestins: the therapeutic pathway?. Med Sci 22:537-43
Michineau S, Muller L, Pizard A, Alhenc-Gélas F, Rajerison RM (2004) N-linked glycosylation of the human bradykinin B2 receptor is required for optimal cell-surface expression and coupling. Biol Chem 385:49-57.
Milia AF, Gross V, Plehm R, De Silva JA Jr, Bader M, Luft FC (2001) Normal blood pressure and renal function in mice lacking the bradykinin B(2) receptor. Hypertension 37:1473-9.
Milligan G (2004) G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol 66:1-7.
Milligan G (2008) A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol 153:S216-29.
Mizumura K, Sugiura T, Katanosaka K, Banik RK, Kozaki Y (2009) Excitation and sensitization of nociceptors by bradykinin : what do we know? Exp Brain Res 196 : 53-65.

247
Molinaro G ,Rouleau JL ,Adam A (2002) Vasopeptidase inhibitors: a new class of dual zinc metallopeptidase inhibitors for cardiorenal therapeutics. Curr Opin Pharmacol 2:131-41.
Mombouli JV, Vanhoutte PM (1995) Kinins and endothelial control of vascular smooth muscle. Annu Rev Pharmacol Toxicol 35:679-705
Moore RH, Tuffaha A, Millman EE, Dai W, Hall HS, Dickey BF (1999) Agonist-induced sorting of human p2-adrenergic receptors to lysosomes during downregulation. J Cell Sci 112:329-339.
Morbidelli L, Parenti A, Giovannelli L, Granger H J, Ledda F, Ziche M (1998) BI receptor involvement in the effect of bradykinin on venular endothelial cell proliferation and potentiation of FGF-2 effects. Br J Pharmacol 124:1286-92.
Moreau ME, Dubreuil P, Molinaro G, Chagnon M, Muller-Esterl W, Lepage Y, Marceau F, and Adam A (2005a) Expression of metallopeptidases and kinin receptors in swine oropharyngeal tissue: effect of ACE inhibition and inflammation. J Pharmacol Exp Ther 315:1065-74.
Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, and Adam A. (2005b) The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci 99:6-38.
Moreau ME, Bawolak MT, Morissette G, Adam A, Marceau F (2007) Role of nuclear factor-KB and protein kinase C signaling in the expression of the kinin B] receptor in human vascular smooth muscle cells. Mol Pharmacol 71: 949-956.
Mori K, Sakamoto W, Nagasawa S (1981) Studies on human high molecular weight (HMW) kininogen. III. Cleavage of HMW kininogen by the action of human salivary kallikrein. J Biochem 90:503-9.
Morissette G, Fortin JP, Otis S, Bouthillier J, Marceau F (2004) A novel nonpeptide antagonist of the kinin Bj receptor: effects at the rabbit receptor. J Pharmacol Exp Ther 311: 1121-1130.
Morissette G, Sabourin T, Adam A, Marceau F (2006) Inhibition of human and rabbit arterial smooth muscle cell migration mediated by the kinin Bj receptor: role of receptor density and released mediators. Can J Physiol Pharmacol 84: 1107-1119.
Morissette G, Houle S, Géra L, Stewart JM, Marceau F (2007) Antagonist, partial agonist and anti-proliferative actions of B-9870 (CU201) as a function of the expression and density of the bradykinin Bj and B2 receptors. Br J Pharmacol 150:369-379.
Morissette G, Couture JP, Désormeaux A, Adam A, Marceau F (2008a) Lack of direct interaction between énalaprilat and the kinin Bj receptors. Peptides 29:606-612.

248
Morissette G, Lodge R, Marceau F (2008b) Intense pseudotransport of a cationic drug mediated by vacuolar ATPase: procainamide-induced autophagic cell vacuolization. Toxicol Appl Pharmacol 228: 364-377.
Muller F, Renne T (2008) Novel roles for factor XH-driven plasma contact activation system. Curr Opin Hematol 15:516-21.
Munoz CM, and Leeb-Lundberg LMF (1992) Receptor-mediated internalization of bradykinin. DDT1 MF-2 smooth muscle cells process internalized bradykinin via multiple degradation pathways. J Biol Chem 267:303-309.
Murphy E, Wong R, Steenbergen C (2008) Signalosomes : delivering cardioprotective signals form GPCRs to mitochondria. Am J hysiol Circ Physiol 295 : H920-H922.
Nakamura M, Ichikawa K, Ito M, Yamamori B, Okinaka T, Isaka N, Yoshida Y, Fukita S, Nakano T (1999) Effects of the phosphorylation of myosin phosphatase by cyclic GMP-dependent protein kinase. Cell Signal 11:671-6.
Nakamura S, Hirano T, Onogawa T, Itoh S, Hashimoto A, Yamamura Y, Kondo K, Mori T, Kambe T (2004) Antidiuretic effects of a novel nonpeptide vasopressin V2-receptor agonist, OPC-51803, administered orally to dogs. J Pharmacol Sci 94: 426-433.
Nardone J, Hogan PG (1994) Delineation of a region in the B2 bradykinin receptor that is essential for high-affinity agonist binding. Proc Natl Acad Sci USA 91:4417-21.
Narumiya S (2009) Prostanoids and inflammation: a new concept arising from receptor knockout mice. J Mol Med 87:1015-22.
Noble B, Kallal LA, Pausch MH, Benovic JL (2003) Development of a yeast bioassay to characterize G protein-coupled receptor kinases. Identification of an NH2-terminal region essential for receptor phosphorylation. J Biol Chem 278:47466-76.
Nonaka H, Fujishima SH, Uchinomiya SH, Ojida A, Hamachi I (2010) Selective covalent labeling of tag-fused GPCR proteins on live cell surface with a synthetic probe for their functional analysis. J Am Chem Soc 132:9301-9.
Norman MU, Lew RA, Smith AI, Hickey MJ (2003) Metalloendopeptidases EC 3.4.24.15/16 regulate bradykinin activity in the cerebral microvasculature. Am J Physiol Heart Circ Physiol 284:H 1942-8.
Novotny EA, Bednar DL, Connolly MA, Connor JR, Stormann TM (1994) Mutation of aspartate residues in the third extracellular loop of the rat B2 bradykinin receptor decreases affinity for bradykinin. Biochem Biophys Res Commun 201:523-30.
Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A (1998) Plasma bradykinin in angio-oedema. Lancet 351:1693-7.

249
Nussberger J, Cugno M, Cicardi M, Agostoni A (1999) Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol 104:1321 -2.
Nussberger J, Cugno M, Cicardi M (2002) Bradykinin-mediated angioedema. TV Engl J Med 347:621-2.
Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS (2000) Differential affinities of visual arrestin, Parrestini and Parrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275: 17201-17210.
O'Dowd BF, Lefkowitz RJ, Caron MG (1989) Structure of the adrenergic and related receptors. Annu Rev Neurosci 12:67-83.
Ohguro H, Palczewski K, Ericsson LH, Walsh KA, Johnson RS (1993) Sequential phosphorylation of rhodopsin at multiple sites. Biochemistry 32:5718-24.
Oikonomopoulou K, Diamandis EP, Hollenberg MD (2010) Kallikrein-related peptidases: proteolysis and signaling in cancer, the new frontier. Biol Chem 391:299-310.
Oksche A, Boese G, Horstmeyer A, Furkert J, Beyermann M, Bienert M, Rosenthal W (2000) Late endosomal/lysosomal targeting and lack of recycling of the ligand-occupied endothelin B receptor. Mol Pharmacol 51:1104-1113.
O'Rourke M, Chen T, Hirst DG, Rao P, Shaw C (2004) The smooth muscle pharmacology of maximakinin, a receptor-selective bradykinin-related nonadecapeptide from the venom of the Chinese toad, Bombina maxima. Reg Peptides 121: 65-72.
Oshima G, Hiraga Y, Shirono K, Oh-ishi S, Sakakibara S, Kinoshita T (1985) Cleavage of des-Arg9-bradykinin by angiotensin I-converting enzyme from pig kidney cortex. Experientia 41:325-8.
Ostrom RS (2002) New determinants of receptor-effector coupling: trafficking and compartmentation in membrane microdomains. Mol Pharmacol 61: 473-476.
Pal-Ghosh R, Yu J, Prado GN, Taylor L, Mierke DF, Polgar P (2003) Chimeric exchanges within the bradykinin B2 receptor intracellular face with the prostaglandin EP2 receptor as the donor: importance of the second intracellular loop for cAMP synthesis. Arch Biochem Biophys 415:54-62.
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289:739-45.

250
Pan W, Kastin AJ, Géra L, Stewart JM (2001) Bradykinin antagonist decreases early disruption of the blood-spinal cord barrier after spinal cord injury in mice. Neurosci Lett 307:25-28.
Pandey KN, Nguyen HT, Garg R, Khurana ML, Fink J (2005) Internalization and trafficking of guanylyl (guanylate) cyclase/natriuretic peptide receptor A is regulated by an acidic tyrosine-based cytoplasmic motif GDAY. Biochem J 388:103-13.
Parenti A, Morbidelli L, Ledda F, Granger HJ, Ziche M (2001) The bradykinin/Bl receptor promotes angiogenesis by up-regulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. FASEB J 15:1487-9.
Peeters MC, van Westen GJ, Li Q, Ijzerman AP (2010) Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci [Sous presse]
Penela P, Ribas C, Mayor F Jr (2003) Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 15:973-81.
Pelkmans L, Kartenbeck J, Helenius A (2001) Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol 3: 473-483.
Pelorosso FG, Brodsky PT, Zold CL, and Rothlin RP (2005) Potentiation of des-Arg9-kallidin induced vasoconstrictor responses by matallopeptidase inhibition in isolated human umbilical artery. J Pharmacol Exp Ther 313:1355-60.
Penela P, Ruiz-Gômez A, Castano JG, Mayor F Jr (1998) Degradation of the G protein-coupled receptor kinase 2 by the protéasome pathway. J Biol Chem 273:35238-44.
Perlman S, Schambye HT, Rivero RA, Greenlee WJ, Hjorth SA, Schwartz TW (1995) Non-peptide angiotensin agonist. Functional and molecular interaction with the ATj receptor. J Biol Chem 270: 1493-1496.
Pesquero JB, Pesquero JL, Oliveira SM, Roscher AA, Metzger R, Ganten D, Bader M (1996) Molecular cloning and functional characterization of a mouse bradykinin BI receptor gene. Biochem Biophys Res Commun 220:219-25.
Pesquero JB, Araujo RC, Heppenstall PA, Stucky CL, Silva JA Jr, Walther T, Oliveira SM, Pesquero JL, Paiva AC, Calixto JB, Lewin GR, Bader M (2000) Hypoalgesia and altered inflammatory responses in mice lacking kinin BI receptors. Proc Natl Acad Sci U S A 97:8140-5. '
Phagoo SB, Poole S, and Leeb-Lundberg LMF (1999) Autoregulation of bradykinin receptors: agonists in the presence of interleukin-1 P shift the repertoire of receptor subtypes from B2 to Bj in human lung fibroblasts. Mol Pharmacol 56:325-333.

251
Phagoo SB, Reddi K, Anderson KD, Leeb-Lundberg LM, and Warburton D (2001) Bradykinin BI receptor up-regulation by interleukin-lb and BI agonist occurs through independent and synergistic intracellular signaling mechanisms in human lung fibroblasts. J Pharmacol Exp Ther 298:77-85.
Pham I, Gonzalez W, el Amrani AI, Fournié-Zaluski MC, Philippe M, Laboulandine I, Roques BP, Michel JB (1993) Effects of converting enzyme inhibitor and neutral endopeptidase inhibitor on blood pressure and renal function in experimental hypertension. J Pharmacol Exp Ther 265:1339-47.
Pickering TG (2002) Effects of stress and behavioral interventions in hypertension: the rise and fall of omapatrilat. J Clin Hypertens 4:371-3.
Pixley RA, Schapira M, Colman RW (1985) The regulation of human factor Xlla by plasma proteinase inhibitors. J Biol Chem 260:1723-9.
Pizard A, Blaukat A, Muller-Esterl W, Alhenc-Gelas F, Rajerison RM (1999) Bradykinin-induced internalization of the human B2 receptor requires phosphorylation of three serine and two threonine residues at its carboxyl tail. J Biol Chem 274: 12738-12747.
Pizard A, Blaukat A, Michineau S, Dikic I, Muller-Esterl W, Alhenc-Gelas F, Rajerison RM (2001 ) Palmitoylation of the human bradykinin B2 receptor influences ligand efficacy. Biochemistry 40:15743-51.
Prado GN, Taylor L, Polgar P (1997) Effects of intracellular tyrosine residue mutation and carboxyl terminus truncation on signal transduction and internalization of the rat bradykinin B2 receptor. J Biol Chem 272:14638-42.
Prat A, Biernacki K, Saroli T, Orav JE, Guttmann CR, Weiner HL, Khoury SJ, Antel JP (2005) Kinin Bj receptor expression on multiple sclerosis mononuclear cells: correlation with magnetic resonance imaging T2-weighted lesion volume and clinical disability. Arch Neurol 62: 795-800.
Prechel MM, Orawski AT, Maggiora LL, Simmons WH (1995) Effect of a new aminopeptidase P inhibitor, apstatin, on bradykinin degradation in the rat lung. J Pharmacol Exp Ther 275:1136-1142.
Prediger RD, Medeiros R, Pandolfo P, Duarte FS, Passos GF, Pesquero JB, Campos MM, Calixto JB, Takahashi RN (2008) Genetic deletion or antagonism of kinin B(l) and B(2) receptors improves cognitive deficits in a mouse model of Alzheimer's disease. Neuroscience 151:631-43.
Pruneau D, Paquet JL, Luccarini JM, Defrêne E, Fouchet C, Franck RM, Loillier B, Robert C, Bélichard P, Duclos H, Cremers B, and Dodey P (1999) Pharmacological profile of LF 16-0687, a new potent non peptide bradykinin B2 receptor antagonist. Immunopharmacology 43:187-194.

252
Pong BK, Trout BL, Lee JY (2008) Modified ligand-exchange for efficient solubilization of CdSe/ZnS quantum dots in water: a procedure guided by computational studies. Langmuir 24:5270-6
Pyne NJ, Tolan D, Pyne S (1997) Bradykinin stimulates cAMP synthesis via mitogen-activated protein kinase-dependent regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle. Biochem J 328:689-94.
Qin LJ, Gu YT, Zhang H, Xue YX (2009) Bradykinin-induced blood-tumor barrier opening is mediated by tumor necrosis factor-alpha. Neurosci Lett 450:172-5.
Raidoo DM, Bhoola KD (1997) Kinin receptors on human neurones. J Neuroimmunol 77:39-44.
Raidoo DM, Bhoola KD (1998) Pathophysiology of the kallikrein-kinin system in mammalian nervous tissue. Pharmacol Ther 79:105-27.
Raidoo DM, Ramsaroop R, Naidoo S, Muller-Esterl W, Bhoola KD (1997) Kinin receptors in human vascular tissue: their role in atheromatous disease. Immunopharmacology 36:153-60.
Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, Hollenberg MD (2009) Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol 76:791-801.
Rapala-Kozik M, Bras G, Chruscicka B, Karkowska-Kuleta J, Sroka A, Herwald H, Nguyen KA, Eick S, Potempa J, Kozik A (2010) Adsorption of Components of the Plasma Kinin-forming System on the Surface of Porphyromonas gingivalis Involves Gingipains as the Major Docking Platforms. Infect Immun. [Sous presse]
Reddigari SR, Kaplan AP (1989) Monoclonal antibody to human high-molecular-weight kininogen recognizes its prekallikrein binding site and inhibits its coagulant activity. Blood 74:695-702.
Regoli D, Marceau F, Barabé J (1978) De novo formation of vascular receptors for bradykinin. Can J Physiol Pharmacol 56:674-7.
Regoli D, Barabé J (1980) Pharmacology of bradykinin and of related kinins. Pharmacol Rev32:\-46.
Regoli D, Marceau F, Lavigne J (1981) Induction of the B|-receptor for kinins in the rabbit by a bacterial lipopolysaccharide. Eur J Pharmacol 71:105-15.
Reis RJ, Santos EL, Pesquero JB, Oliveira L, Schanstra JP, Bascands JL, Pécher C, Paiva AC, Costa-Neto CM (2007) Participation of transmembrane proline 82 in angiotensin II ATI receptor signal transduction. Regul Pept 140:32-6.

253
Ribuot C, Godin D, Couture R, Regoli D, Nadeau R (1993) In vivo B2-receptor-mediated negative chronotropic effect of bradykinin in canine sinus node. Am J Physiol 265:H876-9.
Ricart AD, Tolcher AW (2007) Technology insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Clin Pract Oncol 4:245-55.
Rizk SS, Luchniak A, Uysal S, Brawley CM, Rock RS, Kossiakoff AA (2009) An engineered substance P variant for receptor-mediated delivery of synthetic antibodies into tumor cells. Proc Natl Acad Sci USA 106: 11011 -11015.
Rizzi A, Gobeil F, Calo G, Inamura N, Regoli D (1997) FR 173657: a new, potent, nonpeptide kinin B2 receptor antagonist. An in vitro study. Hypertension 29:951-956.
Rizzi A, Rizzi C, Amadesi S, Calo G, Varani K, Inamura N, Regoli D (1999) Pharmacological characterization of the first non-peptide bradykinin B2 receptor agonist FR 190997: an in vitro study on human, rabbit and pig vascular B2 receptors. Naunyn-Schmied Arch Pharmacol 360: 361-367.
Robl JA, Sun CQ, Stevenson J, Ryono DE, Simpkins LM, Cimarusti MP, Dejneka T, Slusarchyk WA, Chao S, Stratton L, Misra RN, Bednarz MS, Asaad MM, Cheung HS, Abboa-Offei BE, Smith PL, Mathers PD, Fox M, Schaeffer TR, Seymour AA, Trippodo NC (1997) Dual metalloprotease inhibitors: mercaptoacetyl-based fused heterocyclic dipeptide mimetics as inhibitors of angiotensin-converting enzyme and neutral endopeptidase. J Med Chem 40:1570-7.
Rosenbaum DM, Rasmussen SG, Kobilka BK (2009) The structure and function of G-protein-coupled receptors. Nature 459:356-63.
Rosenkilde MM, Benned-Jensen T, Frimurer TM, Schwartz TW (2010) The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol Sci 31:567-74.
Rosenthal T, Gavras I (2009) Angiotensin inhibition and malignancies: a review. J Hum Hypertens 23:623-35.
Rovati GE, Capra V, Neubig RR (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol 71:959-64
Sabourin T, Guay K, Houle S, Bouthillier J, Bachvarov DR, Adam A, et al (2001) Absence of ligand-induced regulation of kinin receptor expression in the rabbit. Br J Pharmacol 133:1154-62.
Sabourin T, Morissette G, Bouthillier J, Levesque L, and Marceau F (2002a) Expression of kinin Bj receptor in fresh or cultured rabbit aortic smooth muscle cells: role of NF-KB. Am J Physiol Heart Circ Physiol 283:H227-37.

254
Sabourin T, Bastien L, Bachvarov DR, Marceau F (2002b) Agonist-induced translocation of the kinin Bj receptor to caveolae-related rafts. Mol Pharmacol 61: 546-553.
Sainz IM, Pixley RA, Colman RW (2007) Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost 98:77-83.
Sales N, Dutriez I, Maziere B, Ottaviani M, Roques BP (1991) Neutral endopeptidase 24.11 in rat peripheral tissues: comparative localization by 'ex vivo' and 'in vitro' autoradiography. Regul Pept 33:209-22.
Sandén C, Enquist J, Bengtson SH, Herwald H, Leeb-Lundberg LM (2008) Kinin B2 receptor-mediated bradykinin internalization and metalloendopeptidase EP24.15-dependent intracellular bradykinin degradation. J Pharmacol Exp Ther 326:24-32.
Sardi SP, Daray FM, Errasti AE, Pelorosso FG, Pujol-Lereis VA, Rey-Ares V, Rogines-Velo MP, Rothlin RP (1999) Further pharmacological characterization of bradykinin BI receptor up-regulation in human umbilical vein. J Pharmacol Exp Ther 290:1019-25.
Sato K, Nagai J, Mitsui N, Ryoko Y, Takano M. (2009) Effects of endocytosis inhibitors on internalization of human IgG by Caco-2 human intestinal epithelial cells. Life Sci 85: 800-7.
Sawada Y, Kayakiri H, Abe Y, Mizutani T, Inamura N, Asano M, Aramori I, Hatori C, Oku T, Tanaka H (2004a) A new class of nonpeptide bradykinin B2 receptor ligand, incorporating a 4-aminoquinoline framework. Identification of a key pharmacophore to determine species difference and agonist/antagonist profile. J Med Chem 47: 2667-2677'.
Sawada Y, Kayakiri H, Abe Y, Mizutani T, Inamura N, Asano M, Hatori C, Aramori I, Oku T, Tanaka H (2004b) Discovery of the first non-peptide full agonists for the human bradykinin B2 receptor incorporating 4-(2-picolyloxy)quinoline and l-(2-picolyl)benzimidazole frameworks. J Med Chem 47: 2853-2863.
Schanstra JP, Bataille E, Marin Castano ME, Barascud Y, Hirtz C, Pesquero JB, Pêcher C, Gauthier F, Girolami JP, and Bascands JL (1998) The B|-agonist [des-Argl0]-kallidin activates transcription factor NF-KB and induces homologous upregulation of the bradykinin B|-receptor in cultured human lung fibroblasts. J Clin Invest 101:2080-91.
Schanstra JP, Neau E, Drogoz P, Arevalo Gomez MA, Lopez Novoa JM, Calise D, Pécher C, Bader M, Girolami JP, Bascands JL (2002) In vivo bradykinin B2 receptor activation reduces renal fibrosis. J Clin Invest 110:371 -9.
Schini VB, Boulanger C, Regoli D, Vanhoutte PM (1990) Bradykinin stimulates the production of cyclic GMP via activation of B2 kinin receptors in cultured porcine aortic endothelial cells. J Pharmacol Exp Ther 252:581-5.

255
Schmaier AH, Kuo A, Lundberg D, Murray S, Cines DB (1988) The expression of high molecular weight kininogen on human umbilical vein endothelial cells. J Biol Chem 263:16327-33.
Schmaier AH (2003) The kallikrein-kinin and the renin-angiotensin systems have a multi layered interaction. Am J Physiol Regul Integr Comp Physiol 285:R1-13.
Schmidt PW, Hirschi MM, Trautinger F (2010) Treatment of angiotensin-converting enzyme inhibitor-related angioedema with the bradykinin B2 receptor antagonist icatibant. J Am Acad Dermatol 63: 913-914.
Schneck KA, Hess JF, Stonesifer GY, Ransom RW (1994) Bradykinin BI receptors in rabbit aorta smooth muscle cells in culture. Eur J Pharmacol 266:277-82.
Schroeder C, Beug H, Muller-Esterl W (1997) Cloning and functional characterization of the omithokinin receptor. Recognition of the major kinin receptor antagonist, HOE 140, as a full agonist. J Biol Chem 272:12475-81.
Schulz R, Wehmeyer A, Schulz K (2002) Opioid receptor types selectively cointernalize with G protein-coupled receptor kinases 2 and 3. J Pharmacol Exp Ther 300:376-84.
Scicli AG, Carretero OA (1986) Renal kallikrein-kinin system. Kidney In 29:120-30.
Seachrist JL, Ferguson SS (2003) Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci 74:225-35.
Semple PF (1995) Putative mechanisms of cough after treatment with angiotensin converting enzyme inhibitors. JHypertens Suppl 13:S 17-21.
Seymour AA, Swerdel JN, Abboa-Offei B (1991) Antihypertensive activity during inhibition of neutral endopeptidase and angiotensin converting enzyme. J Cardiovasc Pharmacol 17:456-65.
Shakur H, Andrews P, Asser T, Balica L, Boeriu C, Quintero JD, Dewan Y, Druwé P, Fletcher O, Frost C, Hartzenberg B, Mantilla JM, Murillo-Cabezas F, Pachl J, Ravi RR, Râtsep I, Sampaio C, Singh M, Svoboda P, Roberts I (2009) The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 10:109.
Shariat-Madar Z, Mahdi F, Schmaier AH (2002) Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem 277:17962-9.

256
Shcherbakova I, Neshkova E, Dotsenko V, Platonova T, Shcherbakova E, Yarovaya G (1999) The possible role of plasma kallikrein-kinin system and leukocyte elastase in pathogenesis of schizophrenia. Immunopharmacology 43:273-9.
Shenoy SK, Barak LS, Xiao K, Ahn S, Berthouze M, Shukla AK, Luttrell LM, Lefkowitz RJ (2007) Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J Biol Chem 282:29549-62.
Shiota M, Ikeda Y, Kaul Z, Itadani J, Kaul SC, Wadhwa R (2007) Internalizing antibody-based targeted gene delivery for human cancer cells. Hum Gene Ther 18:1153-60.
Shivakumar BR, Wang Z, Hammond TG, Harris RC (2005) EP24.15 interacts with the angiotensin II type I receptor and bradykinin B2 receptor. Cell Biochem Funct 23:195-204.
Shrimpton CN, Smith AI, Lew RA (2002) Soluble metalloendopeptidases and neuroendocrine signaling. Endocr Rev 23:647-64.
Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ (2008) Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci U S A 105:9988-93.
Siderovski DP, Willard FS (2005) The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci 1:51 -66.
Simaan M, Bédard-Goulet S, Fessait D, Gratton JP, Laporte SA (2005) Dissociation of beta-arrestin from internalized bradykinin B2 receptor is necessary for receptor recycling and resensitization. Cell Signal 17:1074-1083.
Skidgel RA, Erdos EG. (1998) Cellular carboxypeptidases. Immunol Rev 161:129-41.
Smith D, Gilbert M, Owen WG (1985) Tissue plasminogen activator release in vivo in response to vasoactive agents. Blood66:835-9.
Smith JA, Webb C, Holford J, Burgess GM (1995) Signal transduction pathways for BI and B2 bradykinin receptors in bovine pulmonary artery endothelial cells. Mol Pharmacol 47:525-34.
Smith NJ, Milligan G (2010) Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev 62:701-25.
Soskic V, Nyakatura E, Roos M, Muller-Esterl W, Godovac-Zimmermann J (1999) Correlations in palmitoylation and multiple phosphorylation of rat bradykinin B2 receptor in Chinese hamster ovary cells. J Biol Chem 274:8539-45.

257
Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL (2006) STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci USA 103:4040-5.
Spillmann F, Graiani G, Van Linthout S, Meloni M, Campesi I, Lagrasta C, Westermann D, Tschôpe C, Quaini F, Emanueli C, Madeddu P (2006) Regional and global protective effects of tissue kallikrein gene delivery to the peri-infarct myocardium. Regen Med 1:235-54.
Stadnicki A, Pastucha E, Nowaczyk G, Mazurek U, Plewka D, Machnik G, Wilczok T, Colman RW (2005) Immunolocalization and expression of kinin BIR and B2R receptors in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol 289:G361 -6.
Stenkamp RE, Teller DC, Palczewski K (2005) Rhodopsin: a structural primer for G-protein coupled receptors. Arch Pharm (Weinheim) 338:209-16.
Stewart JM, Gera L, Hanson W, Zuzack JS, Burkard M, McCullough R, Whalley ET (1996) A new generation of bradykinin antagonists. Immunopharmacology 33:51-60.
Stewart JM, Gera L, Chan DC, Bunn PA, York EJ, Simkeviciene V, Helfrich B (2002a) Bradykinin-related compounds as new drugs for cancer and inflammation. Can J Physiol Pharmacol 80:275-280.
Stewart JM, Chan DC, Simkeviciene V, Bunn PA Jr, Helfrich B, York EJ, Taraseviciene-Stewart L, Bironaite D, Gera L (2002b) Bradykinin antagonists as new drugs for prostate cancer. Int Immunopharmacol 2:1781-6.
Stewart JM, Gera L, Chan DC, York EJ, Stewart LT, Simkeviciene V, Helfrich B (2004) New lung cancer drugs from bradykinin antagonists. Chest 125:148S.
Sun B, Schally AV, Halmos G (2000) The presence of receptors for bombesin/GRP and mRNA for three receptor subtypes in human ovarian epithelial cancers. Regul Pept 90:77-84.
Svenungsson E, Andersson M, Brundin L, van Vollenhoven R, Khademi M, Tarkowski A, Greitz D, Dahlstrom M, Lundberg I, Klareskog L, Olsson T (2001) Increased levels of proinflammatory cytokines and nitric oxide metabolites in neuropsychiatrie lupus erythematosus. Ann Rheum Dis 60:372-9.
Tallarida RJ, Murray RB (1987) Manual of Pharmacologic Calculations with Computer Programs. Springer, New York.
Tan F, Jackman H, Skidgel RA, Zsigmond EK, Erdôs EG (1989) Protamine inhibits plasma carboxypeptidase N, the inactivator of anaphylatoxins and kinins. Anesthesiology 70:267-75.

258
Taraseviciene-Stewart L, Scerbavicius R, Stewart JM, Gera L, Demura Y, Cool C, Kasper M, Voelkel NF (2005) Treatment of severe pulmonary hypertension: a bradykinin receptor 2 agonist B9972 causes reduction of pulmonary artery pressure and right ventricular hypertrophy. Peptides 26:1292-1300.
Tekle C, van Deurs B, Sandvig K, Iversen TG (2008) Cellular trafficking of quantum dot-ligand bioconjugates and their induction of changes in normal routing of unconjugated ligands. Nano Lett 8: 1858-1865.
Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino M, Gooding WE, Siegfried JM, Chan DC, Grandis JR (2006) Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cances Res 66:11831-11839.
Thompson D, Pusch M, Whistler JL (2007) Changes in G protein-coupled receptor sorting protein affinity regulate postendocytic targeting of G protein-coupled receptors. J Biol Chem 282:29178-85.
Thornton E, Ziebell JM, Leonard AV, Vink R (2010) Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules 15:6598-618.
Tian XL, Pinto YM, Costerousse O, Franz WM, Lippoldt A, Hoffmann S, Unger T, Paul M (2004) Over-expression of angiotensin converting enzyme-1 augments cardiac hypertrophy in transgenic rats. Hum Mol Genet 13:1441-50.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275:33238-43.
Tirapelli CR, Bonaventura D, de Oliveira AM (2007) Functional characterization of the mechanisms underlying bradykinin-induced relaxation in the isolated rat carotid artery. Life Sci 80:1799-805.
Tobin AB (2008) G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol 153: S167-76.
Tobin AB, Butcher AJ, Kong KC (2008) Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci 29:413-20.
Tom B, Dendorfer A, de Vries R, Saxena PR, Jan Danser AH (2002) Bradykinin potentiation by ACE inhibitors: a matter of metabolism. Br J Pharmacol 137:276-84.
Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN (2006) Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol 38:1654-61.

259
Tschope C, Schultheiss HP, Walther T (2002) Multiple interactions between the renin-angiotensin and the kallikrein-kinin systems: role of ACE inhibition and ATI receptor blockade. J Cardiovasc Pharmacol 39:478-87.
Turner AJ, Hooper NM (2002) The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci 23:177-83.
Tzingounis AV, von Zastrow M, Yudowski GA (2010) {beta}-Blocker drugs mediate calcium signaling in native central nervous system neurons by {beta}-arrestin-biased agonism. Proc Natl Acad Sci USA 107:21028-33.
Ueno A, Naraba H, Kojima F, Morita E, Oh-ishi S (1999) FR 190997, a novel bradykinin B2 agonist, expresses longer action than bradykinin in paw edema formation and hypotensive response. Immunopharmacology 45: 89-93.
Uknis AB, de la Cadena RA, Janardham R, Sartor RB, Whalley ET, Colman RW (2001) Bradykinin receptor antagonists type 2 attenuate the inflammatory changes in peptidoglycan-induced acute arthritis in the Lewis rat. Inflamm Res 50:140-155.
van der Geld YM, Huitema MG, Franssen CF, van der Zee R, Limburg PC, Kallenberg CG (2000) In vitro T lymphocyte responses to proteinase 3 (PR3) and linear peptides of PR3 in patients with Wegener's granulomatosis (WG). Clin Exp Immunol 122:504-13.
van Iwaarden F, de Groot PG, Bouma BN (1988) The binding of high molecular weight kininogen to cultured human endothelial cells. J Biol Chem 263:4698-703.
Vavrek RJ, Stewart JM (1985) Competitive antagonists of bradykinin. Peptides 6:161-164.
Vavrek RJ, Gera L, Stewart JM (1992) Pseudopeptide analogs of bradykinin and bradykinin antagonists. Agents Actions Suppl 38:565-71.
Velarde V, Ullian ME, Morinelli TA, Mayfield RK, Jaffa AA (1999) Mechanisms of MAPK activation by bradykinin in vascular smooth muscle cells. Am J Physiol 277:C253-61.
Vickers C ,Hales P ,Kaushik V ,Dick L ,Gavin J ,Tang J ,Godbout K ,Parsons T, Baronas E, Hsieh F ,Acton S ,Patane M ,Nichols A ,Tummino P (2002) Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277:14838-43.
Vidal MA, Astroza A, Matus CE, Ehrenfeld P, Pavicic F, Sanchez T, Salem C, Figueroa J, Concha M, Gonzales CB, Figueroa CD (2005) Kinin B2 receptor-coupled signa; transduction in human cultured keratinocytes. J Invest Dermatol 124:178-186.
Viel TA, Buck HS (2010) Kallikrein-Kinin System Mediated Inflammation in Alzheimer's Disease In Vivo. Curr Alzheimer Res [Sous presse]

260
Violin JD, Lefkowitz RJ (2007) P-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci 28: 416-422.
von Banchet GS, Fischer N, Uhlig B, Hensellek S, Eitner A, Schaible HG (2010) Molecular effects of interleukin-ip on dorsal root ganglion neurons: Prevention of ligand-induced internalization of the bradykinin 2 receptor and downregulation of G protein-coupled receptor kinase 2. Mol Cell Neurosci [Sous presse].
Walker K, Perkins M, Dray A (1995) Kinins and kinin receptors in the nervous system. Neurochem 7/7/26:1-16; discussion 17-26.
Wang JW, Su W, Law YP, Lu CH, Chen YC, Wang JL, Chang HJ, Chen WC, Jan CR (2001) Mechanisms of bradykinin-induced Ca2+ mobilization in MG63 human osteosarcome cells. Horm Res 55: 265-270.
Wang PH, Cenedeze MA, Pesquero JB, Pacheco-Silva A, Câmara NO (2006) Influence of bradykinin BI and B2 receptors in the immune response triggered by renal ischemia-reperfusion injury. Int Immunopharmacol 6:1960-5.
Wang PH, Cenedeze MA, Campanholle G, Malheiros DM, Torres HA, Pesquero JB, Pacheco-Silva A, Câmara NO (2009) Deletion of bradykinin B1 receptor reduces renal fibrosis. Int Immunopharmacol 9:653-7.
Wang YB, Liu YH (2009) Bradykinin selectively modulates the blood-tumor barrier via calcium-induced calcium release. J Neurosci Res 87:660-7.
Wei L, Alhenc-Gelas F, Soubrier F, Michaud A, Corvol P, Clauser E (1991) Expression and characterization of recombinant human angiotensin I-converting enzyme. Evidence for a C-terminal transmembrane anchor and for a proteolytic processing of the secreted recombinant and plasma enzymes. J Biol Chem 266:5540-5546.
Weikert LF, Bernard GR (1996) Pharmacotherapy of sepsis. Clin Chest Med 17:289-305.
Whalley ET, Wahl M, Sampaio CA (1983) Angiotensin-converting enzyme, bradykinin, angiotensin, and cerebral vessel reactivity. Hypertension 5:V34-7.
Wiley RG, Lappi DA (2003) Targeted toxins in pain. Adv Drug Deliv Rev 55:1043-54.
Williams TA, Corvol P, Soubrier F (1994) Identification of two active site residues in human angiotensin I-converting enzyme. J Biol Chem 269:29430-4.
Williams A, Baird LG (2003) DX-88 and HAE: a developmental perspective. Transfus Apher Sci 29:255-8.

261
Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ (2003) Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A 100:10782-7.
Wolf WC, Evans DM, Chao L, Chao J (2001) A synthetic tissue kallikrein inhibitor suppresses cancer cell invasiveness. Am J Pathol 159:1797-805.
Wright JK, Botha JH, Naidoo S (2008) Influence of the kallikrein-kinin system on prostate and breast tumour angiogenesis. Tumour Biol 29:130-6.
Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villén J, Haas W, Kovacs JJ, Shukla AK, Hara MR, Hernandez M, Lachmann A, Zhao S, Lin Y, Cheng Y, Mizuno K, Ma'ayan A, Gygi SP, Lefkowitz RJ (2010) Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci USA 107:15299-304.
Xu J, Carretero OA, Sun Y, Shesely EG, Rhaleb NE, Liu YH, Liao TD, Yang JJ, Bader M, Yang XP (2005) Role of the BI kinin receptor in the regulation of cardiac function and remodeling after myocardial infarction. Hypertension 45:747-53.
Yamaguchi-Sase S, Hayashi I, Okamoto H, Nara Y, Matsuzaki S, Hoka S, Majima M (2003) Amelioration of hyperalgesia by kinin receptor antagonists or kininogen deficiency in chronic constriction nerve injury in rats. Inflamm Res 52:164-9.
Yang WH, Chang JT, Hsu SF, Li TM, Cho DY, Huang CY, Fong YC, Tang CH (2010) Bradykinin enhances cell migration in human chondrosarcoma cells through BK receptor signaling pathways. J Cell Biochem 109:82-92.
Yasuhara T, Ishikawa O, Nakajima T, Araki K, Tachibana S (1979) The studies on the active peptide on smooth muscle in the skin of Rana rugosa, Thr6-Bradykinin and its analogous peptide, ranakinin-R. Chem Pharm Bull 27:486-91.
Yaqoob M, Snell CR, Burgess GM (1995) Carbohydrate analysis of the B2 bradykinin receptor from rat uterus. J Neurochem 65:1290-6.
Yu J, Liu B, Eramian D, Mierke D, Taylor L, Polgar P (2004) K317, R319, and E320 within the proximal C-terminus of the bradykinin B2 receptor form a motif important for phospholipase C and phospholipase A2 but not connective tissue growth factor related signaling. J Cell Biochem 92:547-59.
Zanchi A, Maillard M, Burnier M (2003) Recent clinical trials with omapatrilat: new developments. Curr Hypertens Rep 5:346-52.
Zausinger S, Lumenta DB, Pruneau D, Schmid-Elsaesser R, Plesnila N, Baethmann (2003) A Therapeutical efficacy of a novel non-peptide bradykinin B2 receptor antagonist on brain

262
edema formation and ischemic tissue damage in focal cerebral ischemia. Acta Neurochir Suppl. 86:205-7.
Zhang W, Bhola N, Kalyankrishna S, Gooding W, Hunt J, Seethala R, Grandis JR, Siegfried JM (2008) Kinin b2 receptor mediates induction of cyclooxygenase-2 and is overexpressed in head and neck squamous cell carcinomas. Mol Cancer Res 6:1946-56.
Zhao Y, Qiu Q, Mahdi F, Shariat-Madar Z, Rojkjaer R, Schmaier AH (2001) Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. Am J Physiol Heart Circ Physiol 280:H 1821 -9.
Zheng H, Loh HH, Law PY (2010) Agonist-selective signaling of G protein-coupled receptor: mechanisms and implications. IUBMB Life 62:112-9.
Zhou X, Prado GN, Taylor L, Yang X, Polgar P (2000) Regulation of inducible bradykinin BI receptor gene expression through absence of internalization and resensitization. J Cell Biochem 78: 351-362.

263
7. ANNEXE
Articles couverts par la thèse.
Bawolak MT, Gera L, Morissette G, Stewart JM, Marceau F (2007) B-9972 (D-Arg-■> c 7 8
[Hyp ,Igl ,Oic ,Igl ]-bradykinin) is an inactivation-resistant agonist of the bradykinin B2 receptor derived from the peptide antagonist B-9430 (D-Arg-[Hyp3,Igl5,D-Igl7,Oic8]-
bradykinin): pharmacologic profile and effective induction of receptor degradation. J Pharmacol Exp Ther 323: 534-46.
Bawolak MT, Gera L, Bouthillier J, Stewart JM, Adam A, Marceau F (2008) A fluorescent version of the bradykinin B2 receptor antagonist B-9430: pharmacological characterization and use in live cell imaging. Peptides 29: 1626-1630.
Bawolak MT, Fortin S, Bouthillier J, Adam A, Gera L, C-Gaudreault R, and Marceau F (2009a) Effects of inactivation-resistant agonists on the signalling, desensitization and down-regulation of bradykinin B2 receptors. Br J Pharmacol 158: 1375-1386.
Bawolak MT, Gera L, Morissette G, Bouthillier J, Stewart JM, Gobeil LA, Lodge R, Adam A, and Marceau F (2009b) Fluorescent ligands of the bradykinin Bj receptors: pharmacologic characterization and application to the study of agonist-induced receptor translocation and cell surface receptor expression. J Pharmacol Exp Ther 329: 159-168.
Gera L, Bawolak MT, Roy C, Lodge R, Marceau F (2011) Design of fluorescent bradykinin analogs: application to imaging of B2 receptor-mediated agonist endocytosis and trafficking and of angiotensin converting enzyme. J Pharmacol Exp Ther 337: 1-9.
Bawolak MT, Lodge R, Morissette G, Marceau F (2011) Bradykinin receptor-mediated transport into intact cells: anti-receptor antibody-based cargoes. Eur J Pharmacol. Soumis.
Autres articles produits durant les études de 2e et 3e cycles.
Bawolak MT, Fortin JP, Vogel LK, Adam A, Marceau F (2006) The bradykinin B2 receptor antagonist icatibant (Hoe 140) blocks aminopeptidase N at micromolar concentrations: off-target alterations of signaling mediated by the bradykinin BI and angiotensin receptors. Eur J Pharmacol 551:108-11.
Bawolak MT, Marceau F (2007) Does zaltoprofen antagonize the bradykinin receptors? Regul Pept 140:125-30.
Moreau ME, Bawolak MT, Morissette G, Adam A, Marceau F (2007) Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin BI receptor in human vascular smooth muscle cells. Mol Pharmacol 71:949-56.

264
Bawolak MT, Touzin K, Moreau ME, Désormeaux A, Adam A, Marceau F (2008) Cardiovascular expression of inflammatory signaling molecules, the kinin BI receptor and COX2, in the rabbit: effects of LPS, anti-inflammatory and anti-hypertensive drugs. Regul Pept 146:157-68.
Marceau F, Bawolak MT, Bouthillier J, Morissette G (2009) Vacuolar ATPase-mediated cellular concentration and retention of quinacrine: a model for the distribution of lipophilic cationic drugs to autophagic vacuoles. Drug Metab Dispos 37:2271-4.
Bawolak MT, Morissette G, Marceau F (2010) Vacuolar ATPase-mediated sequestration of local anesthetics in swollen macroautophagosomes. Can J Anaesth 57:195-200.
Marceau F, deBlois D, Petitclerc E, Levesque L, Drapeau G, Audet R, Godin D, Larrivée JF, Houle S, Sabourin T, Fortin JP, Morissette G, Gera L, Bawolak MT, Koumbadinga GA, Bouthillier J (2010) Vascular smooth muscle contractility assays for inflammatory and immunological mediators. Int Immunopharmacol 10: 1344-53.
Koumbadinga GA, Bawolak MT, Marceau E, Adam A, Gera L, Marceau F (2010) A ligand-based approach to investigate the expression and function of angiotensin converting enzyme in intact human umbilical vein endothelial cells. Peptides 31:1546-54.