Embed Size (px)
Citation preview

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 1 of 145
CAN 407 – Analyse instrumentale, Travaux pratiques
Protocoles
Date: Automne 2017
Département de Chimie – Division Analytique
Université de Sherbrooke

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 2 of 145
Table des Matières
Table des Matières ..........................................................................................................................2
Expérience 1 : Absorption/Émission Atomique ...........................................................................3
Expérience 2 : Chromatographie liquide 1 ................................................................................10
Expérience 3 : Chromatographie GC-MS ..................................................................................27
Expérience 4 : Voltampérométrie cyclique ................................................................................39
Expérience 5 : Chromatographie liquide 2 & Karl Fischer (KF) ............................................53
Expérience 6 : Analyse MALDI-MS ...........................................................................................65
Expérience 7 : Caractérisation des matériaux ...........................................................................83
Expérience 8: Chromatographie ionique ...................................................................................94
Expérience 9 : Fluorescence et Spectroscopie UV d’absorption ............................................116

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 3 of 145
Expérience 1 : Absorption/Émission Atomique

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 4 of 145
EXPÉRIENCE 1 : ABSORPTION/ÉMISSION ATOMIQUE
Théorie Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire.
Pour la bonne compréhension de la technique de l’absorption et de l’émission
atomique, veuillez lire attentivement votre manuel Principe d’analyse instrumentale de
Skoog concernant ce sujet.
Protocole détaillé Pour l’utilisation de l’appareil, veuillez lire la procédure concernant
l’instrument décrite à la fin du protocole.
Absorption atomique – SAA (EN: AAS)
Analyse du zinc (flamme air/acétylène) – Étalonnage externe
Pour le zinc, la longueur d’onde à utiliser est de λ = 213,9 nm et la courbe est
linéaire jusqu’à 1ppm.
À partir d’une solution mère de 10 ppm, préparer des solutions d’étalonnage de
0 à 4 ppm dans le HCl 1 % dans des ballons volumétriques de 100 mL.
Diluer l’inconnu d’un facteur 100 et à l’aide de ces standards, évaluer la
concentration de zinc dans votre échantillon.
Analyse du sodium (flamme air/acétylène) – Étalonnage par ajouts dosés
La concentration de votre inconnu se situe entre 20 et 40 ppm. Préparer un
étalon dans un ballon de 100 mL contenant 4 ppm de Na pour évaluer votre inconnu.
Pour cette évaluation, diluez votre inconnu d’un facteur 10 dans l’eau déionisée.
Utilisez cette concentration approximative pour préparer une droite
d’étalonnage par additions connues (méthode des ajouts dosés, voir guide de
laboratoire et cours CAN-400) contenant 6 points avec des volumétriques de 100 mL.
Émission atomique SEA (EN: AES)
Analyse du potassium (flamme air/acétylène)
La raie d’émission du Potassium se situe à 𝜆é𝑚𝑖𝑠𝑠𝑖𝑜𝑛= 766,5nm. Préparez les
solutions d’étalonnage selon la table suivante :

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 5 of 145
Table 1. Étalons permettant la quantification du potassium par spectrométrie
d’émission atomique. (valeurs nominales)
Calculs Trouver la teneur en zinc, en sodium et en potassium (en ppm) de vos inconnus.
Effectuer les calculs d’erreur pour chacune des droites d’étalonnage.
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- Bien distinguer les deux différentes techniques, SEA vs SAA (forces, faiblesses,
applications…).
- Commentez les différentes méthodes d’étalonnage.
- Habituellement, les fournisseurs donnent des « recettes » avec l’appareil de
spectrométrie atomique à flamme, vous pouvez les consulter.
Instrument: Utilisation du AAnalyst 200
Ouvrir l’ordinateur en premier, ensuite ouvrir
l’appareil à l’aide du bouton ON/OFF NOTE : Les images dans ce protocole sont à titre indicatif, se
référer au texte pour tous les paramètres.
Choisir : Flame
Appuyer sur OK
Il est conseillé de débuter par l’absorption car
l’appareil a besoin d’un temps de stabilisation pour
ce mode. Ensuite l’émission pourra être aussitôt débutée.
Absorption atomique
S’assurer que la bonne lampe est installée, elle se situe dans le panneau en dessous de
Analyte: Potassium
Solvant: Eau déionisée
Solution: Préparées à partir d'une solution mère de K à 5ppm (Michel Trotier)
Aliquote (mL) Volumétrique (mL) Concentration (ppm)
1 50 0,1
2 50 0,2
3 50 0,3
6 50 0,6
7 50 0,7
9 50 0,9
10 50 1,0
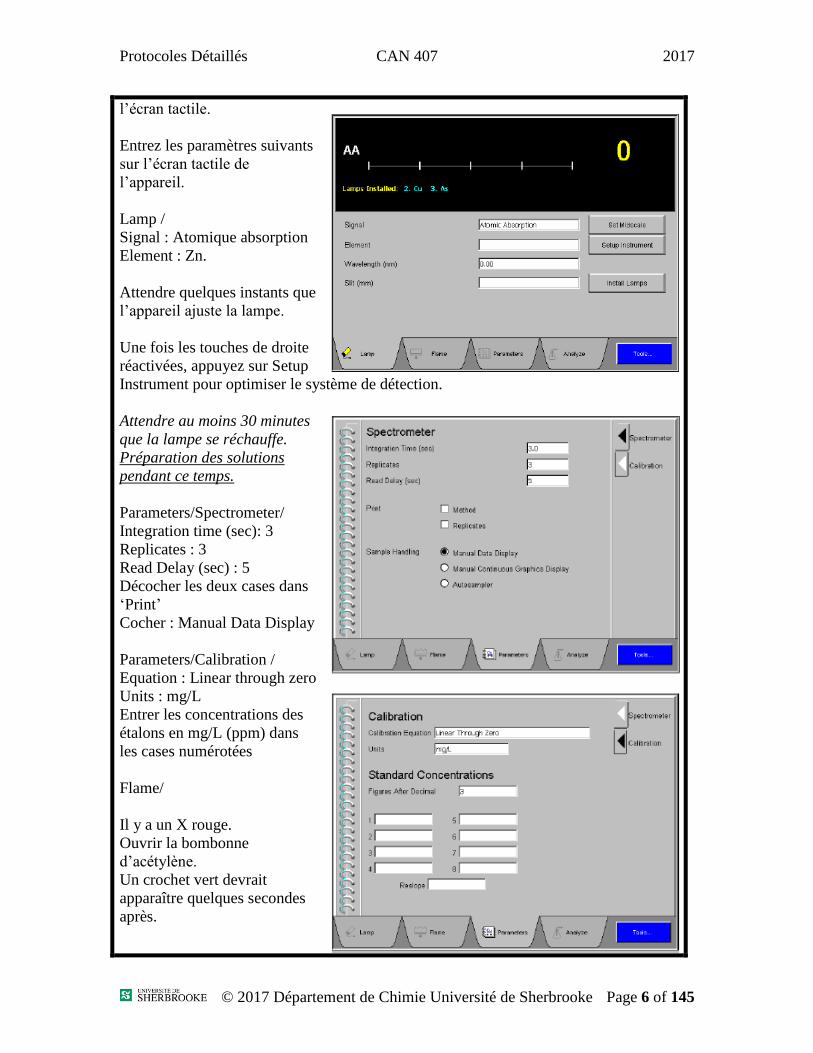
Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 6 of 145
l’écran tactile.
Entrez les paramètres suivants
sur l’écran tactile de
l’appareil.
Lamp /
Signal : Atomique absorption
Element : Zn.
Attendre quelques instants que
l’appareil ajuste la lampe.
Une fois les touches de droite
réactivées, appuyez sur Setup
Instrument pour optimiser le système de détection.
Attendre au moins 30 minutes
que la lampe se réchauffe.
Préparation des solutions
pendant ce temps.
Parameters/Spectrometer/
Integration time (sec): 3
Replicates : 3
Read Delay (sec) : 5
Décocher les deux cases dans
‘Print’
Cocher : Manual Data Display
Parameters/Calibration /
Equation : Linear through zero
Units : mg/L
Entrer les concentrations des
étalons en mg/L (ppm) dans
les cases numérotées
Flame/
Il y a un X rouge.
Ouvrir la bombonne
d’acétylène.
Un crochet vert devrait
apparaître quelques secondes
après.
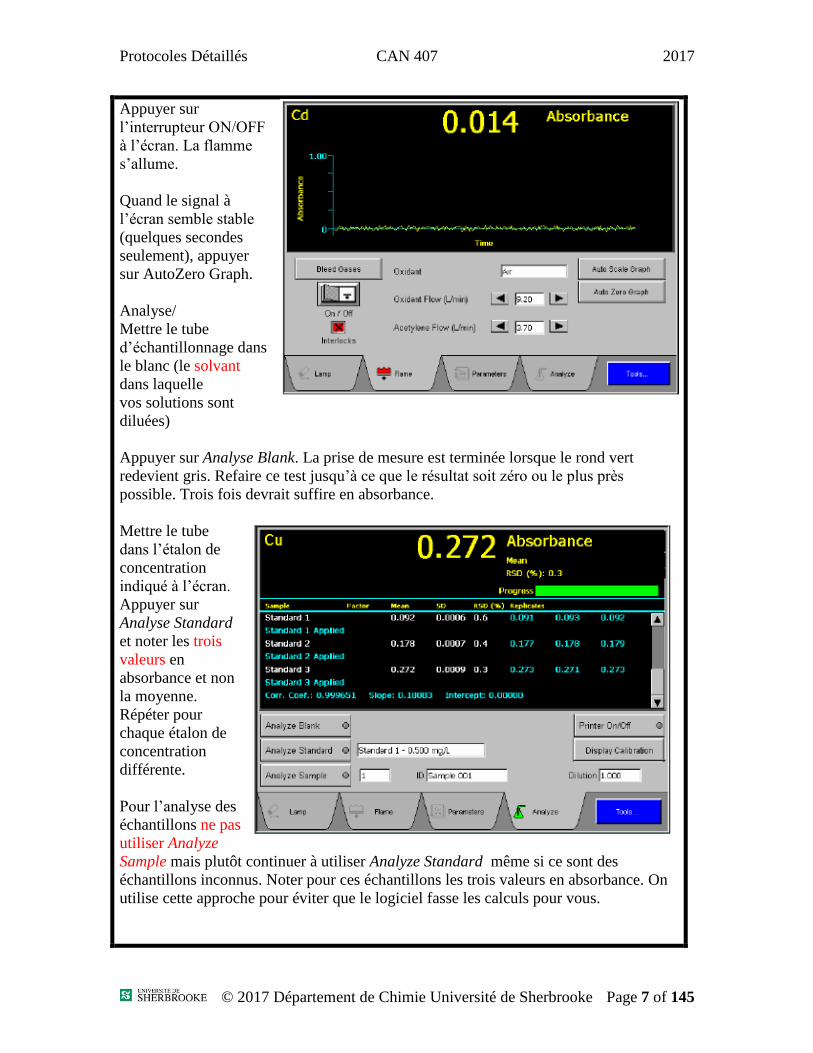
Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 7 of 145
Appuyer sur
l’interrupteur ON/OFF
à l’écran. La flamme
s’allume.
Quand le signal à
l’écran semble stable
(quelques secondes
seulement), appuyer
sur AutoZero Graph.
Analyse/
Mettre le tube
d’échantillonnage dans
le blanc (le solvant
dans laquelle
vos solutions sont
diluées)
Appuyer sur Analyse Blank. La prise de mesure est terminée lorsque le rond vert
redevient gris. Refaire ce test jusqu’à ce que le résultat soit zéro ou le plus près
possible. Trois fois devrait suffire en absorbance.
Mettre le tube
dans l’étalon de
concentration
indiqué à l’écran.
Appuyer sur
Analyse Standard
et noter les trois
valeurs en
absorbance et non
la moyenne.
Répéter pour
chaque étalon de
concentration
différente.
Pour l’analyse des
échantillons ne pas
utiliser Analyze
Sample mais plutôt continuer à utiliser Analyze Standard même si ce sont des
échantillons inconnus. Noter pour ces échantillons les trois valeurs en absorbance. On
utilise cette approche pour éviter que le logiciel fasse les calculs pour vous.

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 8 of 145
Émission atomique
Toutes les solutions doivent être prêtes avant de commencer.
Entrer les paramètres sur l’écran tactile de l’appareil.
Parameters/Spectrometer /
Integration time (sec): 3
Replicates : 3
Read Delay (sec) : 5
Décocher les deux cases dans Print
Cocher : Manual Data Display
Parameters/Calibration /Equation : Linear through zero
Units : mg/L
Entrer les concentrations des étalons en mg/L (ppm)
Si la flamme est fermée faire ce qui suit, si elle est ouverte passer au point Lamp.
Flame/
Il y a un X rouge, ouvrir la bombonne d’acétylène, un crochet vert devrait apparaître
quelques secondes après. Appuyer sur l’interrupteur ON/OFF à l’écran. La flamme
s’allume. Quand le signal à l’écran semble stable (quelques secondes seulement),
appuyer sur AutoZero Graph.
Lamp/
Appuyer sur Install lamp/
Décocher tout. En émission on
n’utilise pas de lampe.
Signal : Flame Emission
Element : K
Une fenêtre s’ouvre.
Mettre le tube dans l’étalon le plus concentré et appuyer sur OK
Lorsque les icones redeviennent en surbrillance, mettre le tube dans le blanc.
Analyse/
Appuyer sur Analyse Blank. La prise de mesure est terminée lorsque le rond vert
redevient gris. Refaire ce test jusqu’à ce que le résultat soit de zéro ou le plus près
possible. 6 fois devrait suffire en émission.
Mettre le tube dans l’étalon de concentration indiqué à l’écran.
Appuyer sur Analyse Standard et noter les trois valeurs.
Répéter pour tous les étalons.
Pour l’analyse des échantillons appuyer sur Analyze Standard même si ce sont des
échantillons inconnus. Noter les trois valeurs.

Protocoles Détaillés CAN 407 2017
© 2017 Département de Chimie Université de Sherbrooke Page 9 of 145
Fermeture de l’appareil
Flame/
Appuyer sur l’interrupteur ON/OFF pour éteindre la flamme.
Fermer la bombonne d’acétylène
Appuyer sur Bleede Gases, cela vide les lignes de gaz pendant quelques secondes.
Éteindre l’appareil avec le bouton d’alimentation par lequel il a été ouvert.

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 10 of 145
Expérience 2 : Chromatographie liquide 1

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 11 of 145
EXPÉRIENCE 2 : CHROMATOGRAPHIE LIQUIDE 1
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
La chromatographie liquide est définie comme toute procédure
chromatographique où la phase mobile est un liquide. Elle est utilisée pour les produits
qui ne sont pas séparables en chromatographie en phase gazeuse parce qu’ils ne sont
pas volatils à basse température ou parce qu’ils se décomposent avec la température.
Dans la pratique, il y a environ 20 % des produits organiques qui sont analysables en
chromatographie en phase gazeuse.
À ses débuts, la chromatographie liquide impliquait des colonnes de
dimensions considérables que traversait la phase mobile par gravité, ce qui pouvait
prendre plusieurs heures. L’ajout d’une pompe pour faire circuler la phase mobile et
d’un détecteur pour détecter les produits à la sortie de la colonne accéléra beaucoup les
analyses. Finalement, le perfectionnement des pompes, la venue d’injecteur à haute
pression et l’immense progrès dans ce qui a trait au support sur les colonnes permet
maintenant de réaliser des séparations très difficiles dans un temps assez court. Ce
type de chromatographie est maintenant désigné sous le terme « chromatographie
liquide à haute performance », plus couramment appelée « HPLC ».
En chromatographie liquide, comme dans toutes les autres méthodes
chromatographiques, les séparations sont fondées sur la différence de distribution des
espèces entre deux phases non miscibles, l’une stationnaire (silice vierge ou greffée,
alumine, résine échangeuse d’ions,…), l’autre mobile (phase liquide constituée par un
solvant pur ou plus souvent par un mélange de solvants). Pour un système
chromatographique donné, on caractérise la distribution de chaque soluté par le
coefficient de distribution (ou coefficient de partage) K défini par la relation :
CS et CM désignant respectivement les concentrations du soluté à l’équilibre
dans les phases stationnaire et mobile.
De la différence d’affinité des divers constituants d’un mélange pour chacune
des deux phases, il résulte une différence de vitesse de migration de chaque composé,
d’où une possibilité de séparation. Expérimentalement, une faible quantité de
l’échantillon à analyser est introduite à l’entrée de la colonne puis, sous l’action de la
phase mobile (éluant), les divers constituants de l’échantillon migrent dans la colonne
d’autant plus lentement qu’ils ont plus d’affinité pour la phase stationnaire. Si les
quantités injectées sont suffisamment faibles, des pics symétriques, plus ou moins
séparés, sont détectés à la sortie de la colonne.
Une bonne séparation en chromatographie en phase liquide implique :

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 12 of 145
• Que les divers constituants du mélange soient retenus dans la colonne, donc
présentent une affinité pour la phase stationnaire suffisante pour qu’ils apparaissent
dans l’effluent après un volume supérieur au volume de la phase mobile contenue dans
la colonne.
• Que les différents pics soient bien séparés, ce qui, pour deux pics consécutifs,
dépend de deux facteurs : la distance séparant les sommets des deux pics et leur
largeur.
• Que l’analyse soit aussi rapide que possible.
Figure 1. Exemple de chromatogramme pour démontrer la notion de résolution en
chromatographie.
• Pour la séparation de deux pics : il faut noter l’influence de la distance entre
les pics et de leur largeur sur la résolution. Sur le chromatogramme (a), il y a
chevauchement des deux pics. Pour les séparer, il y a deux possibilités :
- Soit augmenter la distance entre les pics, en conservant leur largeur
constante (chromatogramme b). On augmente alors la sélectivité.
- Soit diminuer la largeur des pics, en conservant leur écartement
constant (chromatogramme c). On augmente l’efficacité.
Efficacité d’une colonne (nombre de plateaux théoriques)
L’efficacité d’une colonne chromatographique, dont dépend l’étalement des
pics, est mesurée, pour chaque composé, par le nombre de plateaux théoriques N
contenus dans la colonne. La théorie des plateaux établit que, après un certain parcours
dans la colonne, les pics d’élution peuvent être assimilés à des courbes de Gauss, dont
l’écart-type σ (exprimé en unité de temps) est lié au nombre de plateaux théoriques
parcourus par la relation :
Utilisant les caractéristiques géométriques de la courbe de Gauss, on peut
calculer le nombre de plateaux théoriques contenus dans une colonne, pour un soluté
donné, directement à partir du chromatogramme obtenu.

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 13 of 145
Ω = largeur du pic à la base, définie comme la distance entre les points
d’intersections des tangentes d’inflexion avec la ligne de base (ω=4σ), et δ = largeur
du pic à mi-hauteur (δ-σ√5.54 = 2.36 σ).
Figure 2. Calcul du nombre de plateaux théoriques contenu dans une colonne pour un
composé donné selon le profil gaussien
En pratique, on préfère souvent la seconde expression à la première, δ pouvant
être mesuré de façon plus précise que ω, en particulier dans le cas de pics non
parfaitement symétriques. En rapportant la largeur du pic ω à la distance de rétention
mesurée sur le chromatogramme, N constitue bien une mesure de l’élargissement
relatif du pic dans la colonne, ce qui permet, pour un composé donné, de comparer
entre elles les performances de colonnes variées. C’est pourquoi cette notion de
plateaux théoriques, bien qu’elle repose sur un modèle qui ne précise pas l’influence
des différents paramètres, reste d’un usage très général de par sa simplicité.
Pour pouvoir comparer entre elles des colonnes de différentes longueurs (L),
on définit la hauteur équivalente à un plateau théorique (HEPT ou H).
L’équipement en HPLC
L’appareil utilisé en chromatographie liquide haute performance est constitué
des différentes parties suivantes :

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 14 of 145
Figure 3. Schéma d’un chromatographe en phase liquide : (1) réservoir de phase
mobile (solvants); (2) pompe; (3) pré-colonne (filtre à solvant); (4) injecteur; (5)
manomètre (mesure de la pression); (6) colonne; (7) détecteur; (8) acquisition des
données.
La plupart des systèmes sont vendus de façon modulaire. Lorsque l’on monte
un tel système (instrument), on doit éviter le plus possible les volumes morts entre ses
unités, spécialement entre l’injecteur et la colonne ainsi qu’entre cette dernière et le
détecteur car plus le volume de connexion sera grand à ces endroits, plus
l’élargissement des pics sera considérable. Par contre, un volume mort avant
l’injecteur n’affectera pas la qualité de la séparation mais rendra moins pratique les
changements de solvant et occasionnera un délai dans l’application d’un gradient
d’élution. Pour ces raisons, on aura intérêt à utiliser des tuyaux de diamètre interne le
plus petit possible et des raccords ne possédant pas de volume mort; en pratique, on
utilise des tubes en acier inoxydable de diamètre interne de 0,25 à 0,50 mm.
Réservoir de phase mobile
L’extrémité du tuyau d’amené de la phase mobile doit toujours être munie d’un
filtre pour empêcher toute particule de venir boucher les conduits, la pompe ou la
colonne. Généralement, des filtres en acier inoxydable poreux de 20 à 30 µm sont
utilisés (exemple ci-dessous).
Figure 4. Filtre pour phase mobile type
Puisque les solvants utilisés en HPLC sont soumis à de fortes pressions, ils
doivent être exempts de tout gaz dissous. Pour les dégazer, il suffit d’appliquer un

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 15 of 145
vide sur le réservoir pendant 10 à 15 minutes en agitant occasionnellement.
Système de pompage
Le système de pompage doit pouvoir fournir la colonne en solvant de façon
constante et reproductible. Puisque les particules de faible dimension utilisées dans les
colonnes modernes offrent une forte résistance au passage du solvant, on doit pouvoir
disposer de pompes à haute pression.
Les pompes alternatives sont le plus souvent utilisées dans les
chromatographes modernes. Cette popularité est due à une performance globale très
satisfaisante et à ses avantages pratiques d’utilisation. Ces pompes possèdent une
chambre de pompage de faible volume (35-400 µL) et utilisent soit un piston ou un
diaphragme pour pousser le solvant contre la pression de la colonne. Pour varier le
débit, les pompes commerciales font varier soit la fréquence du piston ou le volume de
pompage.
Injecteur
La valve d’échantillonnage est l’injecteur le plus utilisé sur les HPLC. Elle
permet d’introduire de façon reproductible l’échantillon sur la colonne pressurisée
sans interrompre le débit de façon significative.
En position d’introduction, on injecte l’échantillon dans une boucle de volume
prédéterminé alors que la phase mobile s’écoule directement dans la colonne. En
position d’injection, la phase mobile passe dans la boucle et entraîne l’échantillon vers
la colonne. Ce type d’injecteur permet d’introduire de façon rapide, reproductible et
pratiquement indépendante de l’opérateur un large volume d’échantillon à des
pressions jusqu’à 7000 psi avec une erreur inférieure à 0,2 %.
Figure 5. Valve d’échantillonnage à 6 ports type

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 16 of 145
Détecteur
Un des détecteurs les plus utilisés est le spectromètre UV-visible (190-600
nm). Il possède une grande sensibilité pour plusieurs composés; ces derniers doivent
cependant absorber la lumière UV ou visible pour être détectés. Cette absorbance est
reliée à la concentration de l’espèce par la relation de Beer-Lambert :
Où A : absorbance; ε : coefficient d’extinction molaire (L cm-1 mol-1); l :
longueur de la cellule (cm) et c : concentration de l’espèce (mol L-1). Ce mode de
détection possède les avantages suivants :
• Relativement insensible aux changements de débit, de température ou de
composition de la phase mobile.
• Sensibilité absolue de l’ordre 0.002 unité d’absorbance avec un bruit de fond de ±1
%.
• Limite de détection est de l’ordre du nanogramme pour des composés qui absorbent
moyennent.
• Domaine de linéarité est très grand (105 d’ordre de grandeur).
• Très résistant.
• Faible volume mort (petite cellule).
• Très reproductible.
Les colonnes
Caractéristiques physiques
Les colonnes classiques sont des tubes en acier inoxydable dont l’intérieur doit
être le plus lisse possible pour éviter la présence de chemins préférentiels; les
extrémités sont fermées par un filtre en acier dont les pores doivent être plus petits que
les particules dans la colonne. Il en existe de différentes dimensions, le diamètre
intérieur typique allant de 0.2 à 0.8 cm et la longueur variant de 5 à 30 cm.
Différents types de supports solides
Le cœur de la colonne, et probablement le facteur le plus important de la
séparation, demeure le support solide qui s’y trouve et la façon dont il y est introduit.
Comme le remplissage d’une colonne avec des particules plus petites que 20 µm
demande une habileté et un équipement spécial, il est recommandable d’acheter ces
colonnes déjà préparées par le fabricant. Toutefois, pour des particules de dimension
supérieure, le remplissage peut être fait aisément par l’utilisateur. Quant aux
particules elles-mêmes, on peut les classer par les caractéristiques suivantes :
• Solide rigide, gel dur ou gel mou,

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 17 of 145
• Poreuse, superficiellement poreuse ou pelliculaire,
• Particule sphérique ou irrégulière
• Grosseur des particules
Les particules solides rigides sont basées sur une matrice en silice et peuvent
tolérer des pressions élevées (10 000 à 15 000 psi). Ces particules de silice peuvent
être obtenues dans une variété de grosseur, de forme et de porosité. Également, une
variété de groupements fonctionnels ou de couches polymériques peuvent être attachés
à la surface.
Les gels durs sont généralement constitués de particules poreuses de
polystyrène sur lesquelles est greffé du divinylbenzène. Bien que plusieurs de ces gels
puissent être utilisés à des pressions allant jusqu’à 5000 psi, l’usage de certaines de ces
particules est limité à moins de 2000 psi. Les gels durs sont principalement utilisés en
chromatographie d’exclusion ou d’échange d’ion.
Les gels mous sont pratiquement utilisés que pour la séparation de grosses
molécules solubles dans l’eau, comme les protéines. Cependant, ces gels ne peuvent
généralement pas résister aux fortes pressions et leur emploi est ainsi très limité.
Les particules de remplissage des colonnes peuvent également être décrites
comme soit poreuses ou pelliculaires. Ce dernier type de particules est fait à partir de
billes de verre qui sont recouvertes d’une mince couche de phase stationnaire. Dans
une version plus récente de ces particules, une couche poreuse de silice est d’abord
déposée sur la bille de verre, donnant des particules partiellement poreuses. Cette
couche de silice peut à son tour être enrobée avec une phase stationnaire liquide ou
peut réagir chimiquement pour former un lien covalent avec la phase stationnaire. On
peut ainsi greffer différentes phases stationnaires de polarité variée.
Même si en général les petites particules poreuses (5-10 µm) sont les plus
couramment utilisées à cause de leur efficacité élevée et de leur rapidité de séparation,
les colonnes de particules pelliculaires peuvent être adéquates pour des mélanges
simples qui ne requièrent que peu de résolution et lorsque le coût de la colonne est
important, comme pour des analyses de routine. Cependant, la quantité d’échantillon
qui peut être injectée est inférieure aux particules poreuses car il y a moins de phase
stationnaire disponible par unité de volume.
Les particules poreuses peuvent ensuite être décrites comme sphériques ou
irrégulières, de dimension variable (5 µm et plus). Ces deux types de particules
peuvent donner une efficacité (nombre de plateaux N) similaire lorsque les autres
paramètres de la colonne sont les mêmes. Bien qu’une controverse existe quant à la
perte de charge inhérente à ces deux types de particules, l’avantage des particules
sphériques est que l’entassement est plus stable; cela implique que l’efficacité de la
colonne est toujours constante alors des chemins préférentiels peuvent facilement
s’établir au travers de particules irrégulières, ce qui entraîne une perte d’efficacité dans
le temps.

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 18 of 145
Phase normale vs phase inversée
En phase normale, la phase stationnaire est polaire et l’éluant est un solvant pas
ou peu polaire; les composés peu polaires sont alors peu retenus sur la colonne et
sortent en premier. En phase inversée, la phase stationnaire est peu polaire
(généralement de longues chaînes aliphatiques) et l’éluant polaire; les composés
polaires sont peu retenus et sont les premiers détectés. Cette dernière façon de
procéder a très vite gagné en popularité car on a souvent à séparer des composés peu
ou moyennement polaires, séparations jusqu’alors difficile à réaliser en phase normale.
Utilisation des colonnes
Finalement, on doit porter une certaine attention à l’utilisation d’une colonne
particulière. Il peut y avoir une différence notable entre les propriétés
chromatographiques de deux colonnes similaires (même fournisseur, même numéro de
catalogue); on doit donc toujours optimiser les différents paramètres de la séparation
sur une colonne spécifique pour en tirer le maximum de performance.
Enfin, on doit faire très attention de ne pas surcharger la colonne par l’injection
d’une quantité trop grande d’échantillon, ainsi qu’à l’injection de composés pouvant
contaminer la phase stationnaire. Pour éviter de ruiner ainsi une colonne de haute
efficacité, on utilise souvent une précolonne, petite colonne contenant la même phase
stationnaire que la colonne; ces colonnes sont peu coûteuses et faciles à remplir, le cas
échéant.
Protocole détaillé
Objectifs
- Mesurer la quantité de caféine, d’acétaminophène et de benzoate de sodium
dans divers échantillons.
- Séparer un mélange complexe composé de différents HAPs
Appareillage
Un montage HPLC tel que décrit dans la théorie est utilisé. La colonne est du
type C18. Le détecteur est un spectromètre UV-visible; l’utilisation de ce détecteur
permet de sélectionner la longueur d’onde adéquate pour chacun des composés à
analyser.
Préparation
Préparation de la solution-mère
Dans un erlenmeyer de 1 L, préparer environ 800 mL d’un mélange 50/50

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 19 of 145
méthanol/eau. Agiter pour bien mélanger.
Dans un même volumétrique de 500 mL, préparer la solution mère suivante à
partir des composés solides :
• Acétaminophène : entre 500-550 ppm
• Caféine : entre 200-225 ppm
• Benzoate de sodium : entre 500-550 ppm
Utiliser un mélange environ 50/50 méthanol/eau comme solvant.
Optimisation de la méthode
L’optimisation d’une méthode chromatographique implique la variation de
plusieurs paramètres, par exemple : le volume d’injection, le débit et la composition de
l’éluant, etc. Cette optimisation peut prendre une journée, voire plus. Une période de
laboratoire ayant une durée limitée, vous allez déterminer l’effet d’un seul paramètre :
la composition de l’éluant
Conditions opératoires
• Colonne : PoreShell 120 EC-C18 (Agilent) 2.7µm 4,6 x 100 mm
• Éluant : méthanol, eau, acide acétique
• Débit : 1 mL/min
• Volume d’injection : 5 µL
• Détecteur : DAD à 275 nm
Un paramètre non spécifié dans les conditions ci-dessus est la proportion
relative de méthanol et d’acide acétique de l’éluant.
À partir de méthanol (bouteille B), de l’eau (bouteille A) et d’acide acétique
10% dans l’eau (bouteille D), programmer un mélange isocratique de ces trois solvants
pour constituer la phase mobile permettant de séparer les trois composés à doser
sachant que la proportion de la bouteille A devrait être entre 60 et 80%. Faites vos
essais de séparation en injectant de la solution-mère.
Indices : il faut un pourcentage de méthanol appréciable pour pouvoir faire
sortir de la colonne les trois composés mais pas trop pour éviter que les produits se
retrouvent dans le front de solvant. Il faut aussi de l’acide acétique pour aider à
protoner certaines espèces pour obtenir des pics symétriques reproductibles mais il ne
faut pas oublier que cet acide augmente la polarité générale de l’éluant diminuant ainsi
son pouvoir d’élution en phase inverse.
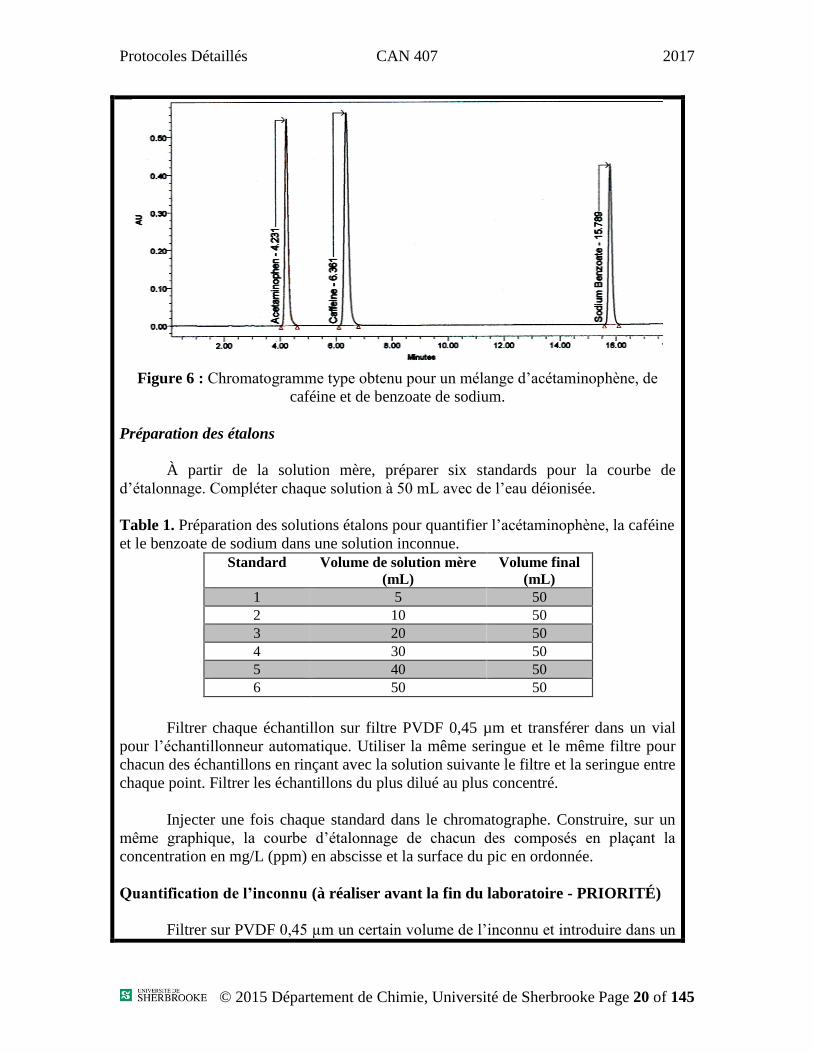
La figure 6 est un exemple de chromatogramme obtenu avec une colonne C18
mais de dimension différente; les temps de rétention sont à titre indicatif seulement.
Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 20 of 145
Figure 6 : Chromatogramme type obtenu pour un mélange d’acétaminophène, de
caféine et de benzoate de sodium.
Préparation des étalons
À partir de la solution mère, préparer six standards pour la courbe de
d’étalonnage. Compléter chaque solution à 50 mL avec de l’eau déionisée.
Table 1. Préparation des solutions étalons pour quantifier l’acétaminophène, la caféine
et le benzoate de sodium dans une solution inconnue.
Filtrer chaque échantillon sur filtre PVDF 0,45 µm et transférer dans un vial
pour l’échantillonneur automatique. Utiliser la même seringue et le même filtre pour
chacun des échantillons en rinçant avec la solution suivante le filtre et la seringue entre
chaque point. Filtrer les échantillons du plus dilué au plus concentré.
Injecter une fois chaque standard dans le chromatographe. Construire, sur un
même graphique, la courbe d’étalonnage de chacun des composés en plaçant la
concentration en mg/L (ppm) en abscisse et la surface du pic en ordonnée.
Quantification de l’inconnu (à réaliser avant la fin du laboratoire - PRIORITÉ)
Filtrer sur PVDF 0,45 µm un certain volume de l’inconnu et introduire dans un
Standard Volume de solution mère
(mL)
Volume final
(mL)
1 5 50
2 10 50
3 20 50
4 30 50
5 40 50
6 50 50

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 21 of 145
vial pour injecteur automatique. Injecter deux fois l’inconnu. À l’aide de votre courbe
d’étalonnage, déterminer la quantité de chacun des produits en ppm.
Quantification du comprimé de Tylénol
• Peser le comprimé de Tylénol; puis le broyer à l’aide d’un mortier et d’un
pilon. Transférer environ 125 mg (pesés précisément) du comprimé dans un
volumétrique de 100 mL. Conserver le reste du comprimé pour l’expérience de GC-
MS. Ajouter environ 50 mL d’un mélange méthanol-eau 1:1, dissoudre (un bain
ultrason peut être utile) et compléter à la marque avec le mélange méthanol eau 1:1
• Diluer avec de l’eau déionisée une partie de l’échantillon 5X précisément.
Filtrer sur PVDF 0,45 µm et introduire dans un vial pour injecteur automatique.
Injecter une fois cet échantillon.
Déterminez la quantité de chacun des produits en milligrammes par comprimé.
Quantification du Diet-Pepsi
Filtrer sur PVDF 0,45 µm un petit volume de Diet-Pepsi® dégazé et introduire
dans un vial pour injecteur automatique. Injecter une fois cet échantillon. Déterminer
la quantité de chacun des produits en ppm.
Quantification du café instantané
Peser précisément entre 350-400 mg de café instantané. Placer dans un ballon
jaugé de 100 mL et diluer avec de l’eau déionisée. Placer dans un bain ultrasonique
durant une à deux minutes pour faciliter la solubilisation du café. Bien mélanger.
Filtrer sur PVDF 0,45 µm et introduire dans un vial pour injecteur automatique.
Injecter une fois cet échantillon. Déterminer la quantité de chacun des produits en
pourcentage poids.
Quantification de la boisson énergétique Red Bull
Pipeter 5 mL de la boisson Red Bull et placer dans un ballon jaugé de 10 mL.
Diluer avec de l’eau déionisée. Filtrer sur PVDF 0,45 µm et introduire dans un vial
pour injecteur automatique. Injecter une fois cet échantillon. Déterminer la quantité de
chacun des produits en milligrammes de composé par 250 mL de boisson.
Séparation d’un mélange contenant plusieurs composés de type HAP (Noter que
cette solution a déjà été préparée par le technicien)
Trouver les conditions expérimentales pour la séparation d’un mélange
contenant sept HAPs, sans toutefois les quantifier. Les solvants à utiliser sont de
l’acétonitrile et de l’eau (normalement bouteille C pour acétonitrile et A pour l’eau).
La colonne chromatographique utilisée est la même que pour les analyses précédentes,
c’est-à-dire une colonne C18. Vous devez changer la longueur d’onde d’analyse du

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 22 of 145
détecteur pour 254 nm. Injecter 1 µL du mélange de standards. La solution standard
de HAPs est relativement concentrée donc il faut en injecter moins afin de ne pas
saturer la colonne et le détecteur.
Calculs Déterminez l’inconnue ainsi que les produits commerciaux analysés. Comparez vos
résultats.
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- Bien démontrer la compréhension de la technique chromatographique utilisée ainsi
que le choix des conditions expérimentales choisies. (colonne, éluant, température,
détecteur…)
- Assurez-vous de savoir ce qu’est un détecteur de type DAD. Quels sont les autres
détecteurs possibles? Quels sont leurs avantages?
- Comparez les résultats obtenus avec les valeurs de référence.
Information Supplémentaire: Utilisation de LC Agilent Series 1100 et
du logiciel HP-Chemstation
Appareil
Figure 7. Chromatographe Agilent Série 1100
Départ du système
Démarrer l’appareil (mettre en marche la pompe) avec la phase mobile
suggérée. Prenez note qu’il faut pomper la phase mobile quelques minutes dans la
colonne chromatographique afin que la pression de stabilise dans cette dernière
(conditionnement de la colonne).
Préparation des phases mobiles (déjà fait dans votre cas!)

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 23 of 145
S’assurer que les bouteilles contiennent assez de liquide porteur (au moins
500mL), sinon en préparer. Tout liquide utilisé en chromatographie liquide doit être
de qualité HPLC (grade). Normalement, il est préférable de filtrer les solvants sur un
filtre 0,45µm avant de les utiliser. Sur ce système, il est inutile de dégazer les solvants
car l’appareil est muni d’un dégazeur.
Démarrer l’appareil
Mettre les modules sous tension (si ce n’est pas déjà fait). S’assurer que la
valve noire est ouverte (légèrement dévissée d’environ 2 tours). Cette valve de purge
sert à diriger les phases mobiles soit vers la colonne (en position fermée) ou vers le «
waste » (en position ouverte). On ouvre cette valve au démarrage pour « purger » les
lignes des phases mobiles (éliminer l’air dans la tuyauterie).
Allumez l’ordinateur et démarrer HP Chemstation. Sur la figure suivante, on
voit l’écran d’ouverture du logiciel avec le diagramme de l’instrument.
Figure 8. Saisie d’écran d’une séance dans le logiciel de gestion d’instrumentation
graphique Chemstation
Vous pouvez corriger les volumes des contenants des phases mobiles en
cliquant sur une bouteille (sur le diagramme de l’instrument) et en choisissant «
Solvent bottles filling ». Entrer les nouveaux volumes.
Instrument/acquisition
Si vous cliquer sur l’icône de la pompe (sur le diagramme de l’instrument)
vous pouvez ajuster les paramètres de la pompe dans le menu « Setup pump ».

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 24 of 145
Figure 9. Menu de réglage de la pompe
• Flow : ajustement du débit de la pompe
• Stop time : Temps d’analyse
• Post time : temps d’attente avant le début de la prochaine analyse
• Solvents : Composition des phases mobiles
• Timetable : Table de programmation de la pompe lorsqu’en mode gradient
Entrez le % de composition que vous désirez et régler le débit désiré (1mL/min
pour le moment). S’assurer que la valve de « purge » est en position ouverte. Partir la
pompe en cliquant sur l’icône de la pompe, puis en sélectionnant « Pump ON ». La
pompe devient alors verte sur le diagramme de l’instrument, signe qu’elle est en
fonction. Augmenter le débit à 5mL/min (seulement si la purge est en position
ouverte). Laisser la pompe à ce débit pendant quelques minutes afin de s’assurer que
l’air présent dans les tubes soit évacué.
Remettre le débit dans la colonne à votre débit désiré (environ 1mL/min), puis
fermer la valve noire de purge (sans utiliser une force herculéenne!) pour diriger la
phase mobile dans la colonne. Laisser le temps au système de se stabiliser. Tout
changement de la composition des phases mobiles ou du débit doit être suivi d’une
période d’attente pour la stabilisation de la colonne. La colonne est stable et à
l’équilibre lorsque la pression dans le système est stable pendant 5 à 10 minutes.
Configuration des paramètres d’injection
Cliquer sur l’icône de l’injecteur sur le diagramme de l’instrument pour avoir
accès aux options qui permettent la programmation de l’injecteur.

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 25 of 145
Figure 10. Menu configuration de l’injecteur
Vous verrez 3 modes d’injections différents :
• Standard injection
• Injection with needle wash
• Use injector program
Aujourd’hui nous utiliserons le mode « standard injection ». Vous devez
également régler le volume d’injection à 10µL.
Détecteur de type DAD
Cliquer sur l’icône du détecteur DAD sur le diagramme de l’instrument vous
aurez alors accès aux paramètres du détecteur UV-visible.
Figure 11. Menu de configuration du détecteur.
• Signals : permet de définir les caractéristiques des 5 signaux possibles
d’enregistrer simultanément.
• Spectrum : permet de définir pour quelle partie du signal, le spectre sera
sauvegardé.
Voyons maintenant plus en détails les sections de signals et spectrum.
• Signals :
- Store : en cochant cette case, ce signal sera sauvegardé
- Sample : longueur d’onde de l’échantillon à laquelle l’absorbance sera
mesurée en fonction du temps. L’absorbance de la référence est soustrait de
l’absorbance de l’échantillon.
- Bw (bandwidth) : largeur de la bande (en nm) de la longueur d’onde de
l’échantillon.
- Reference : longueur d’onde de référence à laquelle l’absorbance sera
mesurée en fonction du temps.
- Bw (bandwidth) : largeur de la bande (en nm) de la longueur d’onde de

Protocoles Détaillés CAN 407 2017
© 2015 Département de Chimie, Université de Sherbrooke Page 26 of 145
référence.
• Spectrum
- Store : l’option All in peak permet d’acquérir les spectres entre les pics
- Range : permet de spécifier les limites des longueurs d’onde à être
sauvegardées pour le spectre
Analyses
Préparation de l’échantillon
Filtrer toutes les solutions sur un filtre de 0.45µm. Les vials spécialement
conçues pour aller dans le HPLC doivent être remplis au 2/3.
Injection
Insérer vos vials dans l’échantillonneur de l’injecteur automatique.
Cliquer sur l’icône montrant 3 vials sur le diagramme de l’instrument. Vous
pourrez ainsi accéder aux paramètres vous permettant de créer votre séquence
d’injection.
Cliquer sur start lorsque votre séquence est créée, pour démarrer les injections.
Sur la droite de l’écran, vous verrez le schéma de l’échantillonneur. Vous
pourrez ainsi suivre la progression de votre séquence.
Si vous voulez faire des injections ponctuelles afin de déterminer votre
composition de phase mobile idéale pour la séparation de vos composés d’intérêts,
vous pouvez cliquer sur l’icône du vial seul. Vous pouvez ainsi entrer les paramètres,
puis faire l’injection.
Consulter votre démonstrateur afin de faire approuver votre composition
de phase mobile, avant de débuter vos injections.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 27 of 145
Expérience 3 : Chromatographie GC-MS

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 28 of 145
EXPÉRIENCE 3 : CHROMATOGRAPHIE GC-MS
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
Dilution isotopique
La dilution isotopique consiste à comparer l’intensité d’un ion moléculaire
spécifique (fragment obtenu par MS) du composé d’intérêt à celui d’un autre ion
moléculaire analogue de même structure, marqué par un isotope et ajouté (dopé, EN:
spike) à la matrice (l’échantillon) en quantité connue. Cette méthode est très utilisée
dans l’analyse environnementale, médicale et médicolégale (ex : analyse d’urine dans
le dopage).
Malgré l’élégance de cette méthode, des problèmes pratiques empêche souvent
son utilisation (ex : coûts des standards isotopiques) dans les laboratoires sous-
gradués. Cependant il est possible de synthétiser facilement une forme trideutérée de la
molécule de caféine (7,7,7 caféine-d3) en utilisant deux produits peu dispendieux,
c’est à dire la théophylline (1,3-dimethylxanthine) et l’iodométhane-d3. Le produit de
cette réaction élue comme un seul pic par HPLC et démontre le même temps de
rétention et le même spectre UV que la caféine.
Extraction sur phase solide
L’autre intérêt de cette analyse réside dans la méthode d’extraction de la
caféine des breuvages. Cette extraction se fera par extraction en phase solide (EN :
solid phase extraction, SPE).
L’extraction en phase solide permettra d’extraire la caféine d’un breuvage dont
le composant principal est l’eau, pour la dissoudre dans un solvant organique plus
approprié à l’utilisation d’une colonne capillaire de GC.
L’extraction se fera à l’aide de cartouche contenant une petite quantité de
copolymère. Ce copolymère se comporte comme la phase stationnaire C18. Le C18 est
la phase stationnaire non polaire contenu dans la majorité des colonnes HPLC utilisées
pour la chromatographie liquide en phase inverse. Cette cartouche est en fait une mini-
colonne HPLC. Ces cartouches sont très utiles dans la préparation d’une analyse soit
pour nettoyer un échantillon et/ou pour concentrer un composé d’intérêt. La
préparation de l’échantillon est toujours une des étapes les plus importantes d’une
analyse. Cette étape améliore l’analyse parce que le chromatogramme est plus simple
et la durée de vie des colonnes est augmentée.
Quatre étapes sont nécessaires dans la procédure d’extraction par phase solide :
• le conditionnement de la phase stationnaire
• le chargement de l’échantillon sur la phase stationnaire

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 29 of 145
• l’élution de produits indésirables
• l’isolement du composé d’intérêt
Conditionnement de la cartouche
Cette étape est analogue à celle de conditionnement d’une colonne HPLC.
Premièrement toutes contaminations présentes dues à la fabrication de la cartouche et
à sa manutention lors de la mise en marché sont éliminées. L’élimination de ces
impuretés est essentiel parce qu’ils peuvent éluer et contaminer l’échantillon. Le
conditionnement « mouille » la phase stationnaire et la laisse dans un état compatible
avec la phase mobile initiale et l’échantillon.
Le premier solvant à utiliser pour le conditionnement doit posséder une force
d’élution aussi forte ou plus forte que le solvant qui servira à l’élution du composé
d’intérêt. Ceci assure que toute contamination qui peut éluer avec le composé est
éliminée de la cartouche avant l’introduction de l’échantillon. Le deuxième solvant de
conditionnement doit être un solvant plus faible que le premier, mais qui sera miscible
avec le solvant contenant le composé d’intérêt. Ordinairement, H2O est utilisée avec
les cartouches non polaires comme la C18. Ceci permet d’éliminer les impuretés plus
polaires contenues sur la C18.
Introduction de l’échantillon
L’échantillon est introduit sur la colonne dans un solvant faible (l’eau pour une
cartouche de C18) ce qui assure que le composé d’intérêt, moins polaire que le solvant
dans lequel il est contenu, soit retenu sur la cartouche.
Rinçage ou élution de produits indésirables
Après l’introduction de l’échantillon, une phase mobile légèrement plus forte
ou d’égale force d’élution que la phase initiale (celle qui a servi à l’introduction du
composé) est utilisée pour éliminer des produits qui éluent plus facilement (plus
polaires) que le composé d’intérêt. Un mélange eau/solvant organique peut aussi être
utilisé.
Isolement du composé d’intérêt
Après avoir éliminé les composés faiblement retenus sur la colonne, on élue les
composés d’intérêt. Si un solvant trop fort est utilisé, il y aura co-élution de composés
qui sont retenus plus fortement que le composé d’intérêt. La phase mobile optimale
sera celle qui retiendra sur la colonne ces composés plus fortement retenus au lieu de
co-éluer avec le composé d’intérêt.
L’utilisation d’un solvant trop faible nécessitera un plus grand volume de
solvant et engendrera une dilution du composé d’intérêt, ce qui représente la perte
d’un avantage de la SPE qui est de concentrer le composé d’intérêt. Une fois élué, le

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 30 of 145
composé peut être injecté directement dans un chromatographe ou concentré par
évaporation du solvant et redissout dans un autre solvant plus approprié pour fin
d’analyse. Le volume de solvant d’élution nécessaire est déterminé
expérimentalement. Par exemple pour une cartouche de C18 ayant une masse de 200
mg, le volume est d’environ de 1 à 2 mL.
Chromatographie en phase gazeuse avec couplage d’un spectromètre de masse
Le GC-MS combine le pouvoir séparateur de haute résolution d’un
chromatographe capillaire à la détection sensible et sélective de la spectrométrie de
masse. La spectrométrie de masse est grandement utilisée dans tous les domaines de
la science à cause de l’information spectrale des masses que l’on en retire, de sa
sélectivité, de sa sensibilité et de sa versatilité. D’un instrument de recherche qu’il
était, le spectromètre de masse est vite devenu un instrument de routine indispensable
lorsque couplé à un GC.
Instrumentation d’un GC-MS
À l’exception du système d’acquisition (ordinateur), les parties importantes
d’un GC-MS sont :
• Un chromatographe
• Une interface GC-MS
• Une chambre d’ionisation
• Un analyseur de masse
• Un détecteur
• Un système pour faire le vide dans l’interface GC-MS (située entre
l’extrémité de la colonne capillaire et la chambre d’ionisation)
Dans le spectromètre de masse proprement dit, des ions sont formés (à partir
des molécules qui proviennent de la colonne) dans la chambre d’ionisation, ils sont
séparés dans l’analyseur de masse et mesurés par le détecteur. Le type d’ionisation,
soit l’ionisation par impact d’électrons (I.E) ou par ionisation chimique (I.C),
détermine le type d’ions produits et leur abondance.
Les ions produits peuvent posséder :
• un poids moléculaire égal au poids moléculaire de la molécule parente;
on l’appelle dans ce cas l’ion moléculaire
• un poids moléculaire inférieur à la molécule originale; dans ce cas on
l’appelle fragment
• un poids moléculaire supérieur à la molécule parente dû à des réactions
chimiques entre la molécule parente et la matrice de l’échantillon ou à des réactions
entre un gaz servant dans l’ionisation chimique et la molécule originale.
Le spectre de masse est une présentation du ratio masse/charge des fragments

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 31 of 145
de charge positive (incluant l’ion moléculaire) versus leur concentration relative.
Habituellement, les ions produits ont une charge de 1 et le ratio masse/charge est donc
égal à la masse de l’ion. Le pic le plus intense dans le spectre est appelé le pic de base
et on lui assigne une intensité de 100. Tous les autres pics, incluant l’ion moléculaire,
ont des intensités (hauteur du pic X facteur de sensibilité) qui sont des pourcentages
par rapport à l’intensité du pic de base. Parfois l’ion moléculaire est aussi le pic de
base.
Interface GC-Ms
Étant donné que le GC qui est jumelé avec le MS est de type capillaire et que le
débit du gaz porteur est habituellement entre 1 et 2 mL/min à 1 atm, l’interface entre le
GC et le MS est assez simple. Il est facile pour des pompes à vide communes de
maintenir un vide de 10-5 torr à 500 L/s et d’évacuer ce 1 à 2 mL/min de gaz porteur.
L’interface consiste essentiellement en une pièce cylindrique dans laquelle sont
introduits les derniers 20 cm de la colonne capillaire.
Figure 1. Interface GC-MS
Cette interface sert à chauffer les 20 derniers centimètres de la colonne avant
qu’elle ne soit introduite dans la chambre d’ionisation pour prévenir toutes traces de
condensation. La température de l’interface est maintenue entre 250 et 320°C. Cet
interfaçage direct procure au MS un maximum de sensibilité.
L’ionisation
Par impact d’électrons
L’échantillon est introduit dans la chambre d’ionisation sous forme de gaz. La
température de la chambre d’ionisation est maintenue entre 150-250°C et à une
pression plus basse que 7.5 x 10-4 torr avec une pompe à diffusion pour éviter des
collisions entre les ions et d’autres molécules. Afin de maintenir une pression basse et
constante dans la chambre il faut que l’échantillon introduit dans la chambre soit petit.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 32 of 145
Le mode d’ionisation du GC-MS utilisé est par impact d’électrons (I.E.). Le
gaz sortant de la colonne est introduit perpendiculairement par rapport au faisceau
d’électrons qui ionisera le gaz sortant de la colonne. Les électrons sont produits par un
filament de tungstène ou de rhénium et sont accélérés vers une anode par une
différence de potentiel de 70 volts. Les ions positifs produits par la collision avec le
faisceau d’électrons sont entrainés dans l’analyseur de masse par une différence de
potentiel de 5 à 15 volts.
Figure 2. Spectromètre de masse type
L’efficacité de l’ionisation est faible, une molécule sur 1 million sera ionisée.
Un voltage de 70 volts pour accélérer les électrons est standard mais un voltage plus
bas produira plus d’ions moléculaires et moins de fragments, ce qui permet de
connaître plus facilement la masse moléculaire.
Selon la nature des molécules neutres qui entrent en collisions avec le faisceau
d’électrons (e-) différents ions sont produits tels des ions radicalaires ( , l’ion
moléculaire) accompagné de la perte d’un électron.
ou des ions négatifs par la capture d’un électron,
ou la formation d’un ion à double charge
Notez que le symbole +• à la droite de M signifie qu’il y a un électron non
pairé dans la molécule et ne signifie pas qu’il y a un électron de plus dans la molécule.
Si c’était le cas, ce serait un anion.
M
2M e M e
M e M
2 3M e M e

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 33 of 145
Un spectromètre de masse peut donner des renseignements autant pour les
cations que pour les anions mais ordinairement on favorise la détection des cations par
le choix de la polarité du voltage entre les différentes plaques de la chambre
d’ionisation. La nature du radical formé ainsi que la localisation de la charge positive
sur l’ion moléculaire dépend de l’énergie d’ionisation des électrons formant la
molécule neutre. L’énergie nécessaire pour enlever un électron dans une liaison sigma
σ est plus élevée que pour un électron dans une liaison pi π. Cette dernière nécessitant
une plus grande énergie que des électrons non liants. (Insertion page 48 dans Mc
Lafferty).
L’énergie requise pour ioniser une molécule organique neutre est généralement
en deçà de 20 eV, ce qui fait que l’énergie excessive du faisceau d’électrons
communique beaucoup d’énergie à la molécule, si bien qu’elle reste dans un état
d’excitation très élevé. Après relaxation, il y a production d’ions de fragmentation,
dont le rapport m/z est inférieur à celui de l’ion moléculaire. Cependant, dans le cas
de certaines molécules, la fragmentation est si grande que l’ion moléculaire ne se
retrouve pas dans le spectre. N’ayant pas la masse moléculaire, il est plus difficile
d’identifier le composé d’intérêt. Toutefois, la fragmentation importante et le nombre
élevé de pics agissent comme une empreinte digitale de la molécule et constituent
souvent un avantage pour l’identification.
Par ionisation chimique
La substance à analyser est injectée dans la source avec un large excès de
méthane ou autre hydrocarbure de faible masse molaire. L’hydrocarbure, ionisé en
premier, ionise la substance par transfert de charge.
Figure 3. Exemple d’ionisation chimique
Analyseur de masse du GC-MS : quadripôle
Aujourd’hui le quadripôle produit plus de spectre de masse que tout autre type
d’analyseur de masse. Il excelle surtout dans des applications qui ne requièrent pas une
haute résolution et une mesure exacte de la masse. Dans ce domaine il surpasse
avantageusement les analyseurs de masse traditionnels.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 34 of 145
Le quadripôle, avec sa courte distance entre la chambre d’ionisation et le
détecteur (habituellement moins de 15 cm), combiné avec de très bonnes propriétés de
focalisation (court temps de balayage), est très bien adapté à des applications requérant
des pressions relativement élevées (5 x 10-5 torr) comme dans le cas de la GC-MS et
de la chromatographie liquide couplée avec la spectrométrie de masse (LC-MS). Un
autre avantage du quadripôle est sa simplicité mécanique. Il ne requiert pas de champs
magnétique, ni de fentes de focalisation qui doivent être ajustées mécaniquement.
Dans un appareil à quadripôle, la focalisation se fait électroniquement. En n’ayant pas
recours à des électro-aimants, on réduit considérablement l’espace, le coût, le poids et
le balayage lent des appareils magnétiques.
Les spécifications du spectromètre de masse que nous utiliserons sont les
suivantes :
• Domaine : balayage possible des masses entre 1.6 et 800 uma.
• Résolution : unitaire
• Vitesse de balayage : 8000 amu/sec
• Sensibilité : 1 pg d’octafluoronaphathalene (m/z 272) donne un rapport signal
sur bruit de 60 :1 lorsque balayé de 50 à 300 amu
Détecteur
Dans notre GC-MS le détecteur est un multiplicateur d’électrons à dynode
continue qui se présente comme un dispositif en forme de trompe, façonnée en verre,
fortement dopé en plomb. Une tension de 0 à 3000 volts est appliquée sur toute la
longueur du traducteur. Les ions qui frappent la surface près de l’entrée éjectent des
électrons qui, rebondissant le long de la surface, en éjectent de plus en plus à chaque
impact. Un gain d’au moins 100 000 fois peut être obtenu.
Protocole détaillé
Objectifs
- Les teneurs en caféine dans un breuvage, dans un comprimé d’analgésique et
dans un inconnu seront quantifiées. Pour cette quantification, une méthode appelée «
dilution isotopique » sera utilisé.
- Les résultats de ces dosages seront comparés à ceux obtenus par HPLC.
- Cette expérience est une introduction à l’interprétation des spectres de masse.
Pour l’utilisation de l’appareil, veuillez lire la procédure d’utilisation des
spectrophotomètres dans les sections suivantes.
Préparation des standards de caféine/caféine-d3
Des solutions de 1000 ppm de caféine et de caféine-d3 dans le méthanol sont
fournies.
• Préparer d’abord des solutions de 200 ppm de chacun des deux produits dans

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 35 of 145
des volumétriques de 50 mL. Compléter à la marque avec du méthanol.
• Préparez les autres solutions tel qu’indiqué dans le tableau ci-dessous. Ces
solutions sont ensuite transférées dans des vials adaptés pour le carrousel du GC-MS.
Table 1. Préparation des solutions pour la quantification de la caféine
Préparation des échantillons de Tylénol, de Pepsi et de l’inconnu
Peser le comprimé de Tylénol; le comprimé est ensuite broyé à l’aide d’un
mortier et d’un pilon. Transférer environ 125 mg (pesés précisément) du comprimé
dans un volumétrique de 100 mL. Conserver le reste du comprimé pour l’expérience
de HPLC. Ajouter à ce volumétrique 8 mL de caféine-d3 1000 ppm. Diluer
partiellement avec de l’eau puis soniquer dans un bain ultrason pendant 15 minutes.
Après dissolution, compléter à la marque avec de l’eau.
Le Pepsi et l’inconnu sont utilisés tels quels sans dilution. À un volumétrique
de 100 mL, ajouter 5 mL de caféine-d3 1000 ppm et compléter à la marque avec du
Pepsi dégazé. Pour l’inconnu, ajouter à un volumétrique de 10 mL, 1 mL de caféine-
d3 200 ppm et amener à la marque avec la solution inconnue de caféine qui sera la
même que celle utilisée à l’expérience de HPLC #1.
De ces trois solutions, il faut extraire la caféine de l’eau pour la diluer dans le
méthanol.
Procédure SPE
Les cartouches utilisées sont vendues par Waters sous le nom Oasis HLB. Elles
contiennent 225 mg de phase polymérique.
Conditionnement de la cartouche
• à l’aide d’une seringue faire passer 2 mL de méthanol à travers la cartouche
en poussant le piston lentement
• faire passer ensuite 2 mL d’eau dans la même cartouche
• la cartouche est prête à être utiliser pour l’extraction
Introduction de l’échantillon sur la cartouche
• Enlever le piston au bout d’une seringue de 2 mL.
Ratio
Caf/Caféine-d3
Volume de
Caféine 200 ppm
(mL)
Volume de
caféine-d3
200 ppm (mL)
Concentration
caf/caf-d3
(ppm)
1 :1 5 5 100/100
1.5 :1 6 4 120/80
2.33 :1 7 3 140/60
4 :1 8 2 160/40

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 36 of 145
• Placer une cartouche au bout de la seringue.
• Transférer ~ l mL du Pepsi dans le réservoir de la seringue.
• Remettre le piston sur la seringue en prenant garde de ne pas introduire
subitement le Pepsi sur la cartouche.
• Avec une pression constante et un débit d’environ 1 à 3 mL/min, introduire le
liquide dans la cartouche.
• Enlever la cartouche et ensuite retirer le piston de la seringue.
• Remettre la cartouche au bout de la seringue.
Lavage de la cartouche (élimination des produits polaires)
• Remplir le réservoir de la seringue avec 2 mL d’eau déionisée.
• Replacer avec précautions le piston au bout du réservoir de la seringue.
• Éluer les produits polaires avec un débit de 1 à 3 mL/min.
• Enlever la cartouche, retirer le piston puis replacer la cartouche.
• Remettre le piston et enlever l’excédent d’eau sur la cartouche pour assécher
complètement la cartouche.
Élution de la caféine et des produits moins polaires
• Retirer la cartouche, retirer le piston du réservoir puis replacer la cartouche.
• Remplir le réservoir avec 2 mL de méthanol.
• Avec précautions remettre le piston sur le réservoir
• Placer la cartouche au-dessus d’un vial pour GC-MS et éluer la caféine avec
une pression constante et un débit de 1 mL/min maximum.
Refaire les mêmes étapes avec l’inconnu et le Tylénol.
Analyse par GC-MS et création d’une méthode
Il faut d’abord éditer une méthode d’analyse, c’est-à-dire spécifier les
différents paramètres qui contrôleront le GC-MS. Chaque appareil a ses particularités
de programmation. Votre démonstrateur vous assistera pour la création de la méthode.
Il faut spécifier à la fois les paramètres qui contrôlent la séparation (GC) et ceux qui
contrôlent la détection (MS). Typiquement, pour votre expérience :
• Température de l’injecteur : 275°C
• Volume d’injection : 1 μL
• Programmation de la température du four : de 150°C à 230°C
• Vitesse de balayage : 30°C/min
• Temps du début du balayage : 2 min
• Temps de l’analyse : 13 min
• Mode d’injection (split ou splitless) : split
• Split ratio : 20:1
• Solvent delay : 3 min

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 37 of 145
Temps pendant lequel le détecteur de masse sera fermé en attente de l’élution
du solvant. Ceci rallonge la durée de vie des filaments de la source,
• Mode d’utilisation du spectromètre de masse (SCAN ou SIM) : SIM
Dans ce mode, les ions à quantifier sont spécifiés, ce qui procure une
sensibilité accrue comparativement au mode SCAN où un domaine de balayage des
masses (m/z) (ex : de 50 à 250 m/z) est spécifié. Pour l’analyse de la caféine, ce seront
exclusivement les ions de m/z 194 et 197 qui seront balayés.
• Temps d’échantillonnage d’un ion : 100 ms
Injection des échantillons avec l’injecteur automatique
Placer les vials contenant les standards et les échantillons sur le carrousel de
l’injecteur automatique. Les vials doivent être remplis au tiers (minimum) pour que
l’aiguille puisse atteindre l’échantillon. Quel que soit l’appareil utilisé, il faut bien
noter leur position sur le carrousel et fournir à l’appareil les détails concernant chaque
échantillon.
Traitement des données, résultats et discussion
Tracer un graphique du rapport des surfaces des ions 194/197 en fonction du
rapport des masses de la caféine/caféine-d3. Connaissant la masse de caféine-d3
introduite dans l’inconnu et le rapport des surfaces de l’inconnu, trouver la quantité de
caféine naturelle dans votre échantillon.
Prendre un spectre de masse en mode SCAN pour observer tous les ions et
discuter du spectre de masse de la caféine dans votre discussion. Commenter le spectre
de masse de la caféine. Où est l’ion moléculaire?
Séparation d’un mélange contenant plusieurs composés
Trouver les conditions expérimentales pour la séparation d’un mélange
contenant sept HAPs sans toutefois les quantifier.
Calculs - Déterminez les diverses concentrations d’analytes dans des différentes matrices.
- Quantifiez vos écarts avec vos résultats obtenus pour le laboratoire de
chromatographie liquide 1.
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- La spectrométrie de masse, dans ces conditions, est-elle une méthode analytique,
quantitatif, semi-quantitative ou qualitative?
- Quelles sont les propriétés d’un bon étalon interne?

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie Université de Sherbrooke Page 38 of 145
- Quel sont les caractéristiques et avantages/désavantages d’une méthode reposant sur
un étalonnage interne.
- Bien démontrer votre compréhension de l’extraction sur phase solide.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 39 of 145
Expérience 4 : Voltampérométrie cyclique

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 40 of 145
EXPÉRIENCE 4 : VOLTAMPÉROMÉTRIE CYCLIQUE
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
Introduction à la voltampérométrie
La voltampérométrie est la mesure du courant circulant entre deux électrodes
(électrode de travail et contre-électrode) lorsqu’on applique une différence de potentiel
(relative à une électrode de référence). Cette technique représente donc un système à
trois électrodes. Le passage d’un courant faradique est possible lorsqu’il se produit un
échange d’électron par une réaction d’oxydoréduction des espèces en solution.
L’échange d’électron requière un potentiel minimal caractéristique à chaque espèce
électrochimique.
En voltampérométrie cyclique, la différence de potentiel est augmentée
graduellement jusqu’à un maximum, puis réduite jusqu’à une valeur minimum. Le
nombre de cycle effectué dépend de l’analyse. La mesure effectuée est celle du courant
qui passe dans le circuit extérieur en fonction de la différence de potentiel appliquée. La
figure 1. Représente une généralisation du principe.
Figure 1. Graphiques représentants le principe de la voltampérométrie cyclique 1
Il y a deux sortent de courant observables dans cette méthode. D’abord, le
courant faradique est celui correspondant au transfert d’électron des espèces
électrochimique, c’est celui qu’on utilise pour étudier la réaction d’oxydoréduction.
Ensuite, le courant non-faradique, aussi appelé courant capacitif, est associé aux
phénomènes du système comme l’adsorption/désorption, la polarisation de l’électrode
etc.
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 41 of 145
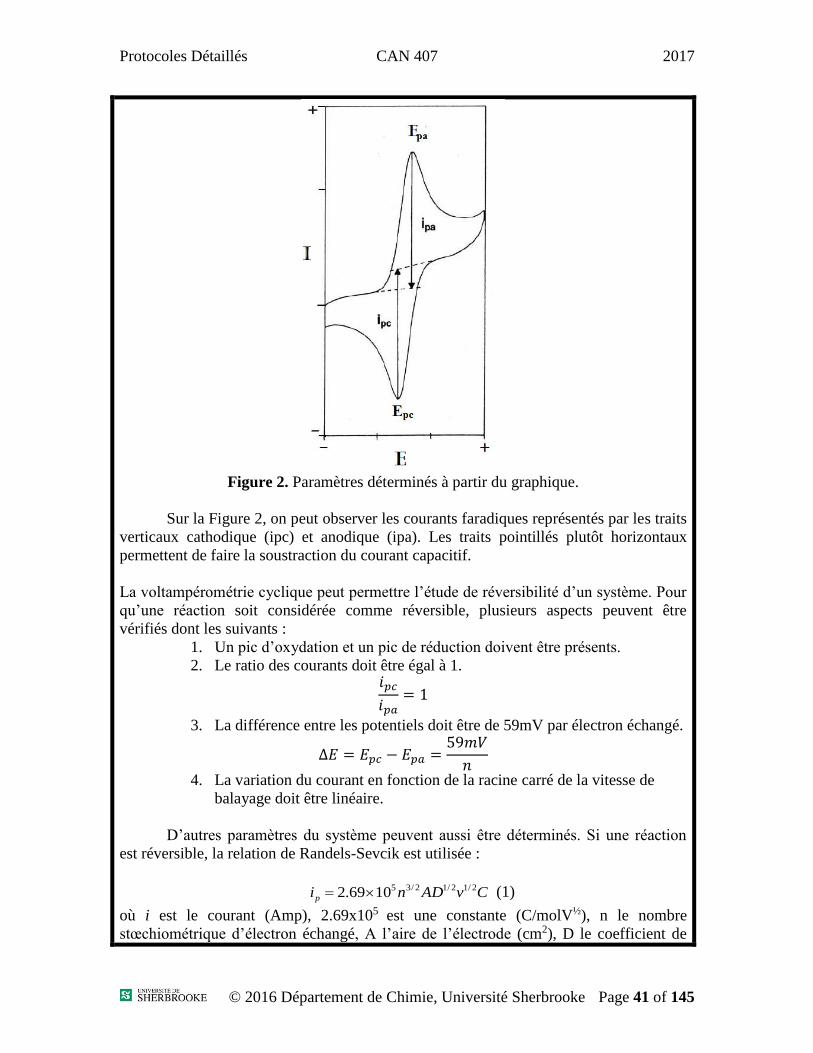
Figure 2. Paramètres déterminés à partir du graphique.
Sur la Figure 2, on peut observer les courants faradiques représentés par les traits
verticaux cathodique (ipc) et anodique (ipa). Les traits pointillés plutôt horizontaux
permettent de faire la soustraction du courant capacitif.
La voltampérométrie cyclique peut permettre l’étude de réversibilité d’un système. Pour
qu’une réaction soit considérée comme réversible, plusieurs aspects peuvent être
vérifiés dont les suivants :
1. Un pic d’oxydation et un pic de réduction doivent être présents.
2. Le ratio des courants doit être égal à 1. 𝑖𝑝𝑐
𝑖𝑝𝑎= 1
3. La différence entre les potentiels doit être de 59mV par électron échangé.
∆𝐸 = 𝐸𝑝𝑐 − 𝐸𝑝𝑎 =59𝑚𝑉
𝑛
4. La variation du courant en fonction de la racine carré de la vitesse de
balayage doit être linéaire.
D’autres paramètres du système peuvent aussi être déterminés. Si une réaction
est réversible, la relation de Randels-Sevcik est utilisée :
CvADnip
2/12/12/351069.2 (1)
où i est le courant (Amp), 2.69x105 est une constante (C/molV½), n le nombre
stœchiométrique d’électron échangé, A l’aire de l’électrode (cm2), D le coefficient de

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 42 of 145
diffusion des espèces (cm2/sec), v la vitesse de balayage (V/s) et C la concentration
(mol/cm3).
Si une réaction est irréversible, la relation suivante est utilisée :
i p 2.99105(na )
1/2nAD
1/2v1/2C (2)
où na est une constante pour chaque réaction.
Pour arriver à déterminer certain paramètre, on utilise une substance dont le couple
réversible d’oxydoréduction bien étudié, le ferrocène.
Figure 3. Couple d’oxydoréduction du ferrocène. 3
Un paramètre intéressant à évaluer est l’aire réelle (A) de l’électrode de travail. Comme
le ferrocène possède un coefficient de diffusion connu, il est possible d’utiliser la
relation de Randels-Sevcik pour déterminer celle-ci après avoir pris des lectures à
différentes vitesses de balayage.
Principes importants
Analyte
L’acide L-ascorbique, de nom commun vitamine C, se retrouve naturellement
dans les agrumes et les légumes. C’est un nutriment nécessaire à l’alimentation. Il est
utile au maintien des tissus conjonctifs et des os, ainsi que dans plusieurs voies
métaboliques grâce à son rôle de coenzyme4. Une carence en vitamine C conduit à
plusieurs problèmes de santé. Cette vitamine s’utilise maintenant partout dans
l’industrie. En majeure partie elle est présente en alimentation (antioxydant dans les
aliments préparés, pour prévenir la décoloration, en remplacement des sulfites, dans la
farine pour améliorer la cuisson, comme ajout dans la nourriture pour poisson), mais
aussi en métallurgie, dans les encres, les explosifs, les cosmétiques, ainsi que comme
catalyseur pour les polymères 5. L’acide L-ascorbique est un agent réducteur et donc, un
bon antioxydant. La figure 4 montre la réaction d’oxydoréduction qui se produit.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 43 of 145
Figure 4. Oxydoréduction de l’acide L-ascorbique 1
Électrodes
L’électrode d’or
Elle agit comme électrode de travail, c’est dont à cette électrode que se produit la
réaction principale. C’est cette électrode qui sera fonctionnalisé pour voir l’effet de
différents composés.
L’électrode de platine
C’est la contre-électrode, elle permet le transfert des électrons sans directement interagir
avec le processus électrochimique.
Ag/AgCl
L’électrode de référence a un potentiel théorique fixe et connu. En mesurant la
différence de potentiel entre notre système et cette électrode, on obtient le potentiel de
notre système.
Fonctionnalisation
Les composés contenant du soufre ont la capacité de se lier fortement sur les
surfaces d’or. Grâce à cette forte interaction, les molécules forment une couche
uniforme lorsqu’on immerge la surface d’or. L’immobilisation de molécules soufrées
permet de modifier l’interaction avec l’espèce électroactive. Les deux substances
servant à la fonctionnalisation sont représentées à la figure 5 ; à gauche l’hydrochlorure
de cystéamine, à droite l’acide α-lipoïque.
Figure 5. Composés utilisés pour la fonctionnalisation1
Protocole détaillé
Objectifs
A. Comparer l’aire théorique et expérimentale d’une électrode d’or.
B. Étudier l’effet de la fonctionnalisation d’une surface d’or sur la détection en
voltampérométrie cyclique.
C. Déterminer la quantité d’acide L-ascorbique dans un comprimé commercial.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 44 of 145
Matériel
Équipement - Électrodes d’or (3)
- Référence Ag/AgCl (1)
- Contre-électrode de platine (1)
- Potentiostat (1)
- Plaque agitatrice (1)
- Barreau magnétique 8mm et 6cm
- Papiers polissage (2)
- Cellule électrochimique (1)
- pH-mètre (1) et solutions étalons
- Tubes de plastique 15mL (2)
- Seringues 5 mL (1)
- Filtres 0,2 µm (1)
- Erlenmeyer 500 mL
- Pipettes de verre 4,8,10 mL
- Volumétrique 25(6), 50, 100 mL
Produits - Ferrocène (1g)
- Poire d’acétone
- Tampon Bu4NPF6 (100mL)
- Tampon phosphate (500mL)
- NaOH 1N (10mL)
- HCl 1M (10mL)
- Acide L-ascorbique (1g)
- Comprimé de vitamine C (1)
- Hydrochlorure de cystéamine (1g)
- Acide alpha-lipoïque (1g)
- Éthanol (50mL)
- Eau déionisée
- Azote
Expérimentation
Préparation des solutions de la PARTIE B
Solution d’hydrochlorure de cystéamine 10mM
- Pesez 0,012 g d’hydrochlorure de cystéamine.
- Transférez le produit dans un tube de plastique de 15 mL.
- Jaugez à 10 mL d’eau déionisée,
Solution d’acide alpha-lipoïque 10mM
- Pesez 0,021 g d’acide alpha-lipoïque.
- Transférez le produit dans un tube de plastique.
- Jaugez à 10 mL d’éthanol 99%.
Préparation des électrodes d’or
Polissage
Demander une démonstration pour effectuer ces étapes.
Faites cette procédure pour les 3 électrodes d’or.
- ATTENTION : Les électrodes sont FRAGILES et DISPENDIEUSES !!!! - Déposez une petite quantité d’eau sur la feuille à polir la moins fine (1µm, rose).
- Polissez l’électrode délicatement en faisant des « 8 » sur la surface pendant 45 à 60
secondes.
- Rincez l’électrode avec de l’eau déionisée.
- Déposez une petite quantité d’eau sur la feuille à polir plus fine (0.3µm, bleu).
- Polissez l’électrode 45 à 60 secondes.
- Rincez l’électrode avec de l’eau déionisée.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 45 of 145
Or - Non fonctionnalisée
Mettre une (1) électrode d’or polie de côté pour les mesures de la PARTIE A et B.
Or - Hydrochlorure de cystéamine
- Ajoutez 1 mL de la solution dans un tube de plastique 1,5 mL identifié.
- Déposez ce tube dans un petit bécher.
- Immergez une électrode d’or polie dans la solution.
- Laissez dans la solution pendant 1h30.
Or - Acide α-lipoïque
- Ajoutez 1 mL de la solution dans un tube de plastique 1,5 mL identifié.
- Déposez ce tube dans un petit bécher.
- Immergez une électrode d’or polie dans la solution.
- Laissez dans la solution pendant 1h30.
Préparation des solutions de la PARTIE A
Solution de ferrocène 1mM
- Pesez 0,0093 g de ferrocène.
- Déposer dans une fiole jaugée de 50 mL et compléter avec le tampon Bu4NPF6
dissout dans l’acétonitrile (fourni par le technicien).
Mesures PARTIE A
Tableau 1. Paramètres des mesures de la partie A. Solution Init E
(V)
High E
(V)
Low E
(V)
Final E
(V)
Scan
Rate
Sweep
Segments
Enable
Final E
Blanc Bu4NPF6 0 1.2 -1.5 0 0.1 3 X
Ferrocène 1mM
0 0.7 0 0 0.02 2 -
0 0.7 0 0 0.05 2 -
0 0.7 0 0 0.10 2 -
0 0.7 0 0 0.25 2 -
0 0.7 0 0 0.50 2 -
1) Ouvrez le module électrochimique.
2) Ouvrez l’ordinateur.
3) Ouvrez le logiciel CHI600D.
4) Set up / Technique / Cyclic voltammetry
5) Installez l’électrode d’or polie comme électrode de travail ainsi que celle de platine
et de référence (enlevez le bouchon bleu).
6) Remplissez le bas de la cellule de mesure avec la solution de blanc de façon à ce
que toutes les électrodes soient dans la solution.
7) Set up / parameters (Voir tableau 1.)

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 46 of 145
Potentiel initial
Potentiel le plus haut
Potentiel le plus bas
Potentiel final
Direction du potentiel
Vitesse de balayage
Nombre de segments
Intervalle des enregistrements
Temps d’attente
Sensibilité
Aller jusqu’au potentiel final
8) Vérifiez que les pinces ne se touchent pas et ne soient pas en contact avec du
métal.
9) Démarrez la mesure : Control / Run experiment
o Si « Link failed » apparaît, rouvrez le logiciel et ajustez les paramètres de
nouveau.
o C’est normal si l’affichage n’est pas complet à l’écran durant la mesure.
o L’ordinateur émet un « bip » lorsque la mesure est finie ou lorsque le
maximum de courant est atteint.
10) File / Save as / Donnez un nom clair, laissez la mesure en .bin.
o Si le graphique ne s’ajuste pas en fonction des échelles: Graphics / Graph
options / cochez Freeze x et y / entrer les valeurs / Ok.
11) Notez les résultats de potentiel et de courant pour les deux segments.
o S’il y a un problème d’identification des pics : Cliquez sur « Data Plot »,
ensuite cliquez sur « Manual Results ». Avec la souris, tracer la tangente de
la base du pic en tenant le clic gauche enfoncé et étirez la droite sous le pic.
De nouveaux résultats devraient s’afficher.
o Si rien n’est affiché après avoir enregistré une mesure, cliquez sur
«Datalist».
12) Videz la cellule.
13) Conditionnez (rinçage) la cellule et les électrodes avec la solution de ferrocène à
analyser.
14) Refaites les étapes 7) à 11) selon le Tableau 1, en agitant légèrement (15 secs) entre
chaque mesure.
15) Lorsque toutes les mesures sont complétées, videz la cellule. Nettoyez la cellule et
les électrodes avec une petite quantité d’acétone (poire).
Enregistrement des graphiques PARTIE A - File / New / Graphics / Overlay Plots / Sélectionner les 6 mesures du Tableau 1.
- Ajustez les axes du graphique.
- Faites une capture d’écran et enregistrez celle-ci dans le logiciel Paint ou Word.
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 47 of 145
Préparation des solutions PARTIE B ET C
Ajustement du pH du tampon phosphate
- Étalonner le pH-mètre avec les solutions de pH 4, 7 et 10. - Mettre 500 mL de tampon dans un erlenmeyer assez grand avec un barreau
magnétique. - Prendre la lecture du pH sous agitation. - Ajuster le pH à 7,4 avec le NaOH 1M ou le HCl 1M.
Bullage du tampon phosphate
- Dans une hotte avec une sortie d’azote, installez une pipette pasteur au bout du tube de la sortie d’azote.
- Mettez la pipette pasteur au fond de l’erlenmeyer et recouvrez partiellement celui-ci de paraffine.
- Assurez-vous que le cylindre d’azote est ouvert. - Ouvrez graduellement le robinet de la hotte jusqu’à ce que de gros bouillons se
forment, mais sans déborder. - Laissez buller pendant 10 minutes. - Assurez-vous de bien refermer l’azote ensuite et de recouvrir votre erlenmeyer
avec la paraffine.
Solution acide L-ascorbique 1mM
- Pesez 0,018 g d’acide L-ascorbique.
- Ajoutez à la solution tampon bullée dans une fiole jaugée de 100 mL.
- Préparer les solutions diluées du tableau ci-dessous.
*NOTE IMPORTANTE !!! Toutes les solutions d’acide L-ascorbique doivent être
utilisées aussi rapidement que possible avant que celles-ci ne soit oxydées par l’oxygène
de l’air.
Solutions d’étalonnage d’acide L -ascorbique
Tableau 2. Dilution de la solution d’acide L-ascorbique pour l’étalonnage.
Concentration en
acide
L-ascorbique (mM)
Aliquote de la solution
d’acide L-ascorbique 1mM
(mL)
Volume final à atteindre
avec la solution tampon (mL)
0,16 4 25
0,32 8 25
0,64 16 25
0,80 20 25

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 48 of 145
Solution de vitamine C - Pesez un comprimé de vitamine C commercial.
- Pulvérisez-le à l’aide d’un pilon et d’un mortier.
- Pesez 0,01 g de la poudre formée.
- Dans un bécher de 50 mL, ajoutez à la poudre 10 mL de la solution tampon afin de
la dissoudre.
- Mettez la solution dans le haut d’une seringue munie d’un filtre.
- Filtrez la solution dans un volumétrique de 25 mL.
- Ajoutez 5 mL de la solution tampon dans le haut de la seringue et filtrez trois fois de
plus celle-ci en récupérant la solution dans le même volumétrique.
- Complétez le volumétrique à 25 mL avec la solution tampon.
- Prélevez 6 mL de la solution de vitamine C concentrée.
- Ajoutez dans un volumétrique de 25 mL et complétez avec la solution tampon.
C’est cette solution de vitamine C diluée qui est utilisée pour l’analyse.
Mesures PARTIE B
Tableau 3. Paramètres des mesures de la partie B.
Solution Électrode de travail Init E
(V)
High
E (V)
Low
E (V)
Final
E (V)
Scan
Rate
Sweep
Segments
Enable
Final E
Acide L-
ascorbique
1mM
Or -0.2 0.8 -0.2 0 0.1 2 -
Hydrochlorure de
cystéamine
-0.2 0.2 -0.2 0 0.1 2 -
Acide α-lipoïque -0.2 0.8 -0.2 0 0.1 2 -
1) Sélectionnez la bonne électrode de travail (Voir tableau 3). Rincez celle-ci avec de
l’eau (ou de l’éthanol pour l’α-lipoïque).
2) Installez l’électrode.
3) Conditionnez la cellule et les électrodes avec la solution à analyser.
4) Remplissez la cellule avec la solution d’acide L-ascorbique 1mM.
5) Set up / parameters (Voir tableau 3.)
6) Vérifiez que les pinces ne se touchent pas et ne soient pas en contact avec du
métal.
7) Démarrez la mesure : Control / Run experiment
8) File / Save as / Donnez un nom clair, laissez la mesure en .bin.
9) Notez les résultats de potentiel et de courant pour les deux segments.
10) Faites 3 mesures pour chaque électrode de travail, sans changer la solution, mais en
agitant légèrement entre chaque mesure.
11) Refaites les étapes 1) à 10) selon le Tableau 3. Remettre l’électrode
fonctionnalisée avec l’hydrochlorure de cystéamine dans sa solution après
utilisation.
12) Videz la cellule.
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 49 of 145
Enregistrement des graphiques PARTIE B - File / New / Graphics / Overlay Plots / Choisir les 3 mesures de l’électrode d’or.
- Ajustez les axes du graphique.
- Faites une capture d’écran et enregistrez celle-ci.
- New / Graphics / Overlay Plots / Choisir les 3 mesures de l’électrode
d’hydrochlorure de cystéamine.
- Ajustez les axes du graphique.
- Faites une capture d’écran et enregistrez celle-ci.
- New / Graphics / Overlay Plots / Choisir les 3 mesures de l’électrode d’acide α-
lipoïque.
- Ajustez les axes du graphique.
- Faites une capture d’écran et enregistrez celle-ci.
- New / Graphics / Overlay Plots / Choisir les 3 premières mesures de chaque
électrode.
- Ajustez les axes du graphique.
- Réfléchissez à qu’elle électrode serait la meilleure pour quantifier la vitamine C…
- Faites une capture d’écran et enregistrez celle-ci.
Mesures PARTIE C
Tableau 4. Paramètres des mesures de la partie C.
Solution Électrode de
travail
Init
E
(V)
High
E
(V)
Low
E
(V)
Final
E
(V)
Scan
Rate
Sweep
Segments
Enable
Final
E
Tampon phosphate
Hydrochlorure
de cystéamine -0.2 0.2 -0.2 0 0.1 2 -
Acide L-asc. 0.16 mM
Acide L-asc. 0.32 mM
Acide L-asc. 0.64 mM
Acide L-asc. 0.80 mM
Acide L-asc. 1 mM
Vitamine C diluée
Procédure 1) Rincez l’électrode de travail avec de l’eau.
2) Installez l’électrode.
3) Set up / parameters (Voir tableau 4.)
4) Conditionnez la cellule et les électrodes avec la solution à analyser.
5) Remplissez la cellule avec la solution à analyser.
6) Vérifiez que les pinces ne se touchent pas et ne soient pas en contact avec du
métal.
7) Démarrez la mesure : Control / Run experiment
8) File / Save as / Donnez un nom clair, laissez la mesure en .bin.
9) Notez les résultats de potentiel et de courant pour les deux segments.
10) Faites 5 mesures pour le tampon et 3 mesure pour chaque autre solution, sans
changer la solution, mais en agitant légèrement enter chaque mesure.
11) Videz la cellule. 12) Refaites les étapes 4) à 11) selon le Tableau 4.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 50 of 145
Enregistrement des graphiques PARTIE C - File / New / Graphics / Overlay Plots / Choisir la première mesure de chaque
solution du Tableau 4.
- Ajustez les axes du graphique.
- Faites une capture d’écran et enregistrez celle-ci.
FIN DU LABORATOIRE - Repolissez, rincez et séchez toutes les électrodes d’or. Rangez-les correctement. - Séchez et récupérez les feuilles de polissage. - Disposez des solutions dans les bons contenants : Organique non-halogéné
(acétonitrile, acétone, éthanol, …) ou aqueux (tampon phosphate, eau, …).
Analyse des résultats Insérez les graphiques enregistrés lors de l’expérience pour votre rapport. Ceux -ci doivent être présentés convenablement, bien identifiés et avec une légende adéquate.
PARTIE A - Déterminez la fenêtre d’analyse électrochimique possible pour le système d’après la
mesure avec le blanc pour le ferrocène.
- Déterminez la réversibilité de la réaction en fonction des 4 conditions présentés
dans la théorie. Vous devez discuter de votre choix entre ipa ou ipc. Vous devez
ajouter les tableaux et les graphiques à l’appui.
- Déterminez l’aire de l’électrode à l’aide de la bonne relation et comparez ce résultat
avec la valeur attendue.
PARTIE B - Pour chaque électrode, discutez de l’effet observé après 3 analyses consécutives.
- En vous basant sur le graphique de la première mesure de chaque électrode, discutez
des différences observées.
- Déterminez qu’elle serait la meilleure électrode à utiliser pour la quantification de la
vitamine C et expliquez pourquoi (principe de fonctionnalisation, nature des
composés à la surface de l’électrode, interaction avec la solution, …).
PARTIE C - Faites une droite d’étalonnage du courant (ip) en fonction de la concentration des
étalons d’acide L-ascorbique. - Déterminez la concentration en acide L-ascorbique dans le comprimé de vitamine
C et comparez avec la valeur attendue. Discutez du résultat. - Effectuez les tests supplémentaires nécessaires pour la détermination des
paramètres analytiques de la technique (LOD, LOQ, domaine linéaire, …).
Notes supplémentaires pour la discussion - Démontrez votre compréhension du fonctionnement et de la raison de l’utilisation
de la voltampérométrie cyclique. - Discutez des causes d’erreurs possibles, des améliorations possibles et des autres
techniques adéquates pouvant être utilisées pour répondre aux objectifs de cet

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 51 of 145
expérience.
Information Supplémentaire
Références
1. Ito, T.; Perera, D. M. N. T.; Nagasaka, S., Lab Documentation. Gold Electrodes
Modified with Self-Assembled Monolayers for Measuring L-Ascorbic
Acid: An Undergraduate Analytical Chemistry Laboratory Experiment. Journal of
Chemical Education 2008, 85 (8), 1112-1115.
2. Ito, T.; Perera, D. M. N. T.; Nagasaka, S., Gold Electrodes Modified with Self-
Assembled Monolayers
for Measuring l-Ascorbic Acid. Journal of Chemical Education 2008, 85 (8), 1112-
1115.
3. Neuse, E. W., Synthetic polymers as drug-delivery vehicles in medicine. Metal-
based drugs 2008, 2008, 469531.
4. Medicine, U. S. N. L. o., Ascorbic acid, [En ligne]
http://chem.sis.nlm.nih.gov/chemidplus/rn/50-81-7. Consulté le 2014-12-12.
5. John Wiley & Sons, I., ASCORBIC ACID. Kirk-Othmer Encyclopedia of
Chemical Technology.
6. Chem 371, A. I. C. L., Cyclic Voltammetry of Ferrocene, [Ru(bpy)3]2+,
[Co(bpy)3]2+ and Iodide. UMass Boston.
Informations diverses – électrochimie analytique
Le potentiostat
Le potentiostat sert à la fois à mesurer et à contrôler la différence de potentiel
entre l’électrode de travail et l’électrode de référence et aussi à mesurer le courant qui
circule entre l’électrode de travail et l’électrode auxiliaire. Le potentiostat permet aussi,
par l’entremise d’un générateur de signaux, d’imposer à l’électrode de travail un signal
variable en fonction du temps (rampe, créneaux, sinusoïde, etc.).
Différentes techniques voltampérométriques
En voltampérométrie, le potentiel E appliqué à l’électrode de travail est varié en
fonction du temps et le courant est mesuré en fonction du potentiel. La variation du
potentiel, linéaire ou modulée, imposée à l’électrode de travail, et la manière dont on
mesure le courant, en continue ou à des temps précis, distinguent les différentes
techniques voltampérométriques. Le courant mesuré est principalement la somme de
deux courants distincts : le courant faradique if et le courant capacitif ic.
Courant capacitif (ic)
Le courant capacitif est dû au chargement du condensateur représenté par
l’interface entre la surface de l’électrode et la couche de la solution adjacente. Le

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 52 of 145
courant capacitif dépend :
• De la surface de l’électrode.
• De la vitesse du changement de potentiel avec le temps.
• De la composition du milieu mais non de la concentration du composé analysé.
Mode de transport de masse et utilisation d’un électrolyte support
Les espèces électroactives disposent de plusieurs modes de transport pour se
rendre sur le lieu du transfert d’électrons, à savoir l’interface électrode-électrolyte. Il en
existe trois principaux :
• Le transport par convection est le déplacement de matière sous l’effet d’un
gradient de température, de pression ou d’agitation mécanique.
• Le transport par diffusion est le déplacement des ions sous l’effet d’un
gradient de potentiel chimique, c’est-à-dire du milieu le plus concentré vers le milieu le
moins concentré.
• Le transport par migration est le déplacement des ions sous l’influence d’un
gradient de potentiel électrique, c’est-à-dire sous l’effet d’un champ électrique.
En analyse électrochimique quantitative, le mode de transport privilégié est le
transport par diffusion. Lorsque le transfert de charges est rapide et que le transport de
matière est uniquement régi par la diffusion, le courant mesuré est directement relié à la
concentration des espèces électroactives en solution.
Expérimentalement, pour s’approcher d’un régime pur de diffusion, il faut
ajouter, à la solution contenant les espèces électroactives, un électrolyte de support
formé d’ions non électroactifs en grande concentration. Les ions de l’électrolyte
subissent entièrement l’effet du gradient électrique (migration), et les ions électroactifs
(les composés d’intérêts), de concentration plus faible, ne sont soumis qu’au gradient de
concentration (diffusion). Par exemple, à une solution d’ions Cu2+ électroactifs, on
ajoute une solution de sulfate de sodium. Parfois, on utilise un tampon comme
électrolyte de support pour fixer le pH et empêcher la précipitation de métaux sous
forme d’hydroxydes. La concentration de l’électrolyte doit être au moins 100 fois plus
élevée que celle du composé d’intérêt. C’est en général la réduction de l’électrolyte de
support qui limite le domaine d’électroactivité du côté des potentiels négatifs (ex. :
réduction du cation K+ ou de l’ion hydronium).
En plus de supprimer la contribution des éléments électroactifs au courant de
migration, ce qui simplifie les relations (équations) entre courant et concentration, cet
ajout :
• Augmente la conductivité de la solution, ce qui a pour effet de diminuer la
chute ohmique et de permettre ainsi de mieux contrôler le potentiel de l’électrode de
travail dans des montages nécessitant trois électrodes.
• Fixe la force ionique du milieu à une valeur élevée, ce qui limite les
phénomènes capacitifs à une zone de très faible épaisseur au voisinage de l’interface
électrode/solution.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 53 of 145
Expérience 5 : Chromatographie liquide 2 & Karl Fischer (KF)

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 54 of 145
EXPÉRIENCE 5 : CHROMATOGRAPHIE LIQUIDE 2 & KARL FISCHER (KF)
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
Pour une bonne compréhension de la technique de chromatographie liquide,
consultez votre manuel Principes d’analyse instrumentale de Skoog au chapitre 28,
intitulé « Chromatographie liquide haute performance ». Vous devez absolument
consulter la section de la théorie de l’expérience de chromatographie liquide #1 de ces
notes de cours.
Théorie HPLC-RID
Type de colonne (phase stationnaire)
La séparation des composés en chromatographie liquide est basée sur la différence
d’affinité des composés entre deux phases non miscibles, l’une stationnaire et l’autre
mobile. Il existe plusieurs types de colonnes en HPLC et il faut choisir celle-ci selon le
type de composés à analyser. Le bon type de phase stationnaire permet d’obtenir une
séparation avec résolution et sensibilité adéquates. En chromatographie liquide, on utilise
souvent les termes « chromatographie en phase normale », où la colonne est constituée de
gel de silice (un matériau très polaire) ou « chromatographie en phase inverse » où la
colonne est constituée de silice greffée par des chaînes hydrocarbonées de 8 ou 18
carbones, C8 ou C18 (un matériau très peu polaire).
Il existe aussi des colonnes de types échangeuses d’ions, constituées de
polystyrène / divinylbenzène, qui peuvent s’apparenter à les colonnes en
chromatographie ionique. Ces colonnes permettent de séparer une vaste gamme de
composés organiques plus solubles dans l’eau que dans certains solvants organiques.
Pour l’analyse des sucres par chromatographie liquide avec détection par indice
de réfraction, la colonne (AMINEX ou semblable) utilisée possède quelques avantages.
• Elle permet d’utiliser une méthode isocratique simple (la phase mobile est
composée d’eau uniquement) ce qui permet de faire la détection par indice de réfraction
(voir la théorie sur le détecteur à indice de réfraction un peu plus loin)
• De plus, comparativement à certaines autres méthodes, l’analyse des sucres par
chromatographie liquide ne nécessite pas de procédure de dérivation afin de favoriser une
bonne séparation et la méthode demande un minimum de préparation des échantillons.
Effet de la température sur la séparation
Le contrôle de la température est souvent critique en chromatographie liquide. Les
temps de rétention et la hauteur des pics peuvent être grandement influencés par la
température. C’est pourquoi sur le système HPLC que nous utiliserons, il y a un chauffe-

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 55 of 145
colonne afin de contrôler précisément la température.
L’avantage d’utiliser de hautes températures en chromatographie liquide est
d’obtenir de meilleures séparations et plus rapidement. L’augmentation de la température
fait en sorte de diminuer la viscosité de la phase mobile, ce qui permet aux sucres une
meilleure pénétration dans les pores de la phase stationnaire (favorise de meilleurs
échanges entre la phase stationnaire et mobile), favorisant ainsi une meilleure résolution
des pics chromatographiques.
Détecteur à indice de réfraction
L’indice de réfraction d’une matière est un nombre qui caractérise le pouvoir que
possède cette matière à ralentir et dévier la lumière. Le détecteur à indice de réfraction
mesure en continu la différence d’indice de réfraction entre la phase mobile pure
(référence) et l’effluent qui sort de la colonne HPLC. Lorsque la phase mobile et
l’effluent sont identiques, les intensités lumineuses de la référence et de l’échantillon sont
identiques et aucun signal n’est détecté. Par contre, lorsque la cellule échantillon contient
une solution avec un indice de réfraction différent, le faisceau lumineux est dévié suivant
la loi de Snell-Descartes.
Le changement de l’intensité lumineuse est proportionnel à la concentration et à
l’indice de réfraction de la solution de l’échantillon.
Le détecteur à indice de réfraction est un détecteur universel car il permet de
détecter quasiment tous les composés. Par contre, il est très sensible aux variations de
température. De plus, ce détecteur ne peut être utilisé qu’en mode isocratique (la
composition de l’éluant ne varie pas au cours du temps). En effet, la cellule de référence
qui contient la phase mobile est remplie avec cette dernière au début de l’expérience mais
c’est la cellule échantillon qui est alimentée par l’effluent de la colonne tout au long des
analyses.
Le détecteur à indice de réfraction possède un désavantage majeur, c’est d’être
moins sensible que la plupart des autres détecteurs.
Théorie Titrimétrie-Karl-Fischer
La réaction de Karl Fischer (voir Skoog … Chimie analytique, traduction de la
8ième édition, pages 580-582
La détermination de la teneur en eau est basée sur la réaction de Bunsen :
42222 22 SOHHIOHSOI

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 56 of 145
Karl Fischer a découvert que cette réaction pouvait être utilisée pour le dosage de
l’eau dans un système non-aqueux contenant un excès de SO2. Le solvant utilisé était du
méthanol. Pour diriger la réaction vers la droite, il est nécessaire de neutraliser les acides
formés (HI et H2SO4); Karl Fischer a utilisé la pyridine, C5H5N à cet effet. Le réactif Karl
Fischer est composé d’iode, de dioxyde de soufre, de pyridine et de méthanol. La
réaction, se fait en deux étapes :
En présence d’une grande quantité de pyridine, tous les réactifs et les produits
existent sous forme de complexes, comme l’indiquent les équations. Notez que seule la
première étape consomme de l’eau lors de l’oxydation du dioxyde de soufre par l’iode
pour donner du trioxyde de soufre et de l’iodure d’hydrogène. La deuxième étape, qui se
produit en présence d’un excès de méthanol, est cruciale pour la réussite du titrage, parce
que le complexe pyridine-trioxyde de soufre est également capable de consommer de
l’eau.
Cette réaction indésirable peut être complètement évitée en présence d’un grand
excès de méthanol. Contrairement à la réaction de Bunsen, la stœchiométrie du titrage
Karl Fischer implique la consommation d’une mole d’iode, d’une mole de SO2, et de trois
moles de pyridine par mole d’eau. En pratique, on emploie un excès de SO2 et de
pyridine pour que la capacité du réactif à se combiner avec l’eau ne soit déterminée que
par sa teneur en iode. Pour les dosages habituels, de 2 à 5 mg d’eau réagissent avec 1 mL
de réactif en présence d’un excès en SO2 d’un facteur 2 et d’un excès en pyridine d’un
facteur 3 à 4.
D’autres études ont démontrées
• que la pyridine ne participait pas directement à la réaction mais servait plutôt
comme un tampon et pouvait être remplacée par une autre base.
• que la vitesse de la réaction dépendait du pH.
La vitesse de réaction est optimale dans le domaine de pH entre 5,5 et 8. La
vitesse augmente encore au-dessus de pH 8,5 mais à ce pH le point de fin de titrage est
moins bon et il y a une plus grande consommation d’iode.
Modification du milieu réactionnel
En 1984 E. Sholtz a développé un réactif Karl Fischer sans pyridine en utilisant
l’imidazole comme base. Ceci a permis non seulement de remplacer la pyridine, un
produit toxique, mais aussi de rendre la réaction de titrage plus rapide et plus précise en
3222 2 SOPyIHPyOHPySOPyIPy
4333 SOCHHPyOHCHSOPy
4333 SOCHHPyOHCHSOPy

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 57 of 145
tamponnant dans un domaine de pH plus favorable. Sholtz a aussi trouvé que d’autres
alcools pouvaient améliorer la stabilité du réactif. Le méthanol peut être remplacé par
l’éthanol, le 2-propanol ou le méthoxyéthanol.
Considérations pratiques
Dépendance de la réaction Karl Fischer sur du pH
Étant donné que la vitesse maximale est atteinte à un pH entre 5,5 et 8, il faut
éviter un pH au-dessus de 8 et inférieur à 4 en pratique. Avec des échantillons acides ou
basiques, on doit ajuster le pH de la solution par l’addition d’imadazole comme tampon
basique et d’acide salicylique comme tampon acide.
Effet du solvant sur la réaction
La stœchiométrie (ratio molaire H2O/I2) dépend du type de solvant. Le méthanol
devrait être toujours présent dans une proportion de 20 % ou plus pour que la réaction
entre l’iode et l’eau ait une stœchiométrie 1:1. Cependant, les cétones et les aldéhydes
forment des acétals avec les alcools en produisant aussi du H2O ce qui fausse les
résultats. Dans ces cas, il faut utiliser un réactif Karl Fischer modifié (voir la section «
réactifs ».
Réactifs
Le réactif de Karl Fischer n’est pas stable à long terme. Pour cette raison, il est
souvent commercialisé sous forme de deux solutions qu’il faut mélanger avant usage. Il y
a sur le marché des réactifs à un seul composant formulés pour être plus stables dans le
temps ainsi que des réactifs spéciaux pour le dosage de l’eau dans les aldéhydes et
cétones.
Réactifs à deux composants
Le titrant contient de l’iode et du méthanol. Le solvant contient le SO2,
l’imidazole (ou la pyridine) et le méthanol. Les composants séparés sont très stables en
entreposage. Par contre, lorsque le réactif et le solvant sont mélangés pour effectuer les
titrages, il y a dégradation et il faut étalonner le titrant régulièrement.
Réactif à un seul composant
Le titrant contient de l’iode, du SO2 et de l’imidazole dissous dans un alcool. Le
solvant est le méthanol. On peut utiliser un mélange à base de méthanol, comme solvant,
spécialement adapté à l’échantillon. Le réactif peut être entreposé pendant deux ans. La
baisse du titre est d’environ 0,5 mg/mL par année dans une bouteille scellée. Avec ce
type de réactif, la vitesse de réaction est de 2 à 3 fois moins rapide qu’avec les réactifs à
deux composants.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 58 of 145
Réactifs spéciaux pour aldéhydes et cétones
La compagnie Riedel-de Haën fabrique un réactif à un composant sans méthanol
pour le titrage en présence des aldéhydes et des cétones :Hydranal® Composite 5K
comme titrant et l’Hydranal® Working medium K comme solvant.
Réactifs avec éthanol
Étant donné que l’éthanol est moins toxique que le méthanol Riedel-de Haën a
mis sur le marché des réactifs à base d’éthanol. Ces réactifs permettent le titrage de
plusieurs cétones qui forment des acétals mais à une vitesse beaucoup moins grande
qu’en présence de méthanol. Un solvant spécial à base d’éthanol est disponible pour le
réactif à un composant.
Contrôle du titrage
L’ajout du titrant durant le titrage doit être contrôlé comme pour tout autre titrage.
Le titrant doit être ajouté aussi rapidement que possible et arrêté exactement au point
d’équivalence. La vitesse du titrage dépend de quatre facteurs :
• La vitesse de réaction entre le titrant et l’échantillon
• Le mélange du titrant avec l’échantillon qui dépend de la vitesse d’agitation
• Des paramètres de contrôle
• La détection du point d’équivalence
Indication du point d’équivalence
Le point d’équivalence peut être détecté visuellement par le changement de
couleur car lorsque toute l’eau a été titrée, un excès d’iode apparait en solution, la
coloration devient alors plus foncée. Pour les appareils automatisés, la détection du point
d’équivalence se fait toujours de façon électronique, soit par mesure potentiométrique ou
ampérométrique.
Détection potentiométrique
Dans le cas d’une détection potentiométrique, le circuit de détection maintient un
courant constant (5 à 10 µA) entre deux électrodes de platine et mesure en même temps
le voltage nécessaire pour maintenir ce courant constant. En absence d’iode en solution,
le potentiel est élevé, mais au point de fin du titrage, il y a apparition d’iode en solution,
ce qui fait chuter le potentiel pour maintenir le courant constant. Cette chute du potentiel
indique le point de fin du titrage.
Détection ampérométrique
Dans le cas d’une détection ampérométrique, une différence de potentiel
constante, suffisante pour réduire l’iode (I2) en iodure (I-), est maintenue entre deux

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 59 of 145
électrodes de platine. Lorsqu’un excès d’iode apparaît en solution, le courant augmente
de façon importante, ce qui signale la fin du titrage.
Vitesse de réaction
La vitesse de réaction dépend : de la concentration d’eau; de la concentration de
SO2; de la concentration d’iode et du pH de la solution. Initialement, lorsque la
concentration de l’eau est élevée, le titrant peut être ajouté rapidement étant donné que
l’iode réagit avec l’eau immédiatement. Vers la fin du titrage, lorsque la concentration
d’eau est plus faible, la vitesse de réaction étant plus lente, le titrant doit être ajouté plus
lentement.
Vitesse d’agitation et dispersion du titrant
Pour un titrage précis il faut un bon mélange des réactifs. Le mélange des réactifs
est influencé par : la vitesse d’agitation; le point d’entrée du titrant et par la forme du
contenant de titrage
La vitesse d’agitation optimale est obtenue lorsqu’il y a apparition d’un petit
vortex. Si la vitesse d’agitation est trop basse le titrage est lent et irrégulier et un
dépassement du point de fin de titrage est possible. Si des bulles se forment en solution,
l’agitation est trop rapide.
Point d’entrée du titrant. Le titrant doit être ajouté à l’endroit où la turbulence est
maximale. L’ajout doit se faire assez loin des électrodes pour laisser assez de temps à
l’iode pour réagir. Si le titrant est ajouté trop près des électrodes le circuit de détection
détecte de l’iode qui n’a pas réagi et le titrant est automatiquement ajouté plus lentement
ce qui entraîne un titrage prolongé.
Figure 1. Considérations expérimentales agitation/point d’entrée
Paramètres de contrôle du titrage
Comme l’appareil que nous utiliserons est un appareil automatique, la précision
du titrage dépend aussi de différents paramètres de contrôle que l’on peut modifier selon
le type d’échantillon à analyser. Voici les différents paramètres suggérés par le
manufacturier pour le type de titrage que nous effectuerons.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 60 of 145
Sources :1. Mode d’emploi du 784 KFP Titrino de Metrohm
2. Publication Metler Toledo pour DL31/DL38 Titrator
Protocole détaillé

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 61 of 145
Objectif
- Mesurer la quantité de sucrose, de fructose et de glucose dans divers échantillons
alimentaires par HPLC-RID.
- Apprendre à utiliser un titrimètre de Karl-Fischer
Expérimentation HPLC-RID
Appareillage
Un montage HPLC, tel que décrit dans la théorie de l’expérience HPLC #1, est
utilisé avec en plus un module de chauffe-colonne afin de contrôler la température. La
colonne utilisée est une colonne Phenomenex RPM-Monosaccharide 300 x 7,8 mm. Le
détecteur est un réfractomètre.
Préparation de la droite d’étalonnage
À partir des trois sucres (solides) fournis, préparer 250 mL de solution-mère
contenant entre 950 et 1050 ppm (précisément) (les 3 sucres dans le même ballon
jaugée); utiliser de l’eau désionisée comme solvant.
À partir de cette solution mère, préparer cinq standards pour la courbe
d’étalonnage: Compléter chaque solution à 50 mL avec de l’eau désionisée.
Table 1. Préparation des solutions d’étalonnage pour la quantification de sucres
Filtrer chaque échantillon sur nylon 0,45 µm et transférer dans un vial pour
échantillonneur automatique. Utiliser la même seringue et le même filtre pour chacun des
échantillons en rinçant avec la solution suivante le filtre et la seringue entre chaque point.
Filtrer les standards du plus dilué au plus concentré.
Injecter chaque standard dans le chromatographe. Construire, sur un même
graphique, la courbe d’étalonnage de chacun des composés en plaçant la concentration en
mg/L (ppm) en abscisse et la surface du pic en ordonnée. Déterminer la limite de
détection de chacun des produits en utilisant les courbes d’étalonnage.
Quantification de l’inconnu
Standard Volume de solution mère
(mL)
Volume final
(mL)
1 10 50
2 20 50
3 30 50
4 40 50
5 50 50

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 62 of 145
Utiliser l’inconnu tel quel, sans dilution. Filtrer sur nylon 0,45 µm et placer dans
un vial pour échantillonneur automatique. Injecter deux fois cet échantillon. À l’aide de
votre courbe d’étalonnage, déterminer la quantité de chacun des sucres présents en mg/L
(ppm).
Quantification du jus de fruit Oasis
Diluer d’un facteur 100X un échantillon de jus de fruit avec de l’eau désionisée.
Filtrer sur nylon 0,45 µm et placer dans un vial pour échantillonneur automatique.
Injecter l’échantillon. Déterminer la quantité de chacun des sucres en g/250 mL et en
sucres totaux en g/250 mL. Comparer vos résultats avec les valeurs de l’étiquette.
Quantification du miel
Peser précisément environ 1 g de miel et diluer dans un volumétrique de 500 mL
avec de l’eau désionisée. Filtrer sur nylon 0,45µm et placer dans un vial HPLC. Injecter
l’échantillon. Déterminer la quantité de chacun des sucres présents en % (poids/poids).
Comparer vos résultats avec les valeurs trouvées sur Internet.
Quantification du sirop d’érable
Peser précisément environ 0,5 g de sirop d’érable et diluer dans un volumétrique
de 500 mL avec de l’eau désionisée. Filtrer sur nylon 0,45µm et placer dans un vial pour
échantillonneur automatique. Injecter l’échantillon. À l’aide de la courbe d’étalonnage,
déterminer la quantité de chacun des sucres présents en % (poids/poids). Comparer vos
résultats avec les valeurs trouvées sur Internet.
Conditions expérimentales
Colonne : Bio-Rad Aminex HPX-87P 300 x 7.8 mm
Instrument : Agilent 1100 HPLC system
Phase mobile : Eau
Débit : 0,60 mL/min
Volume d’injection : 25 µL
Détecteur : Indice de réfraction
Température de la colonne : 80°C
Expérimentation Karl-Fischer
Utilisation du titrimètre Karl Fischer
Peser sur le bouton avec une icône rouge situé à gauche et au bas de l’écran. Le système
devrait afficher sur l’écran : KFT I (pol) H2O Titer
Si l’écran n’affiche pas cette méthode, il faut la charger en pesant sur le clavier « USER
METHOD » et « ENTER » pour se retrouver à « charger méthode ». On pèse sur «

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 63 of 145
ENTER » et on sélectionne la méthode à charger en pesant sur les flèches <←> et <→>
et en pesant encore sur « ENTER » pour confirmer. La méthode H2OTiter est maintenant
chargée.
Le dosage
Conditionnement
Introduire 20 mL de méthanol dans la cellule, c’est-à-dire jusqu’à la ligne noire. Toute
présence d’eau doit être éliminée de ce méthanol avant de commencer le dosage
proprement dit. On commence ce conditionnement en appuyant sur le bouton « START
». Le conditionnement est terminé lorsque l’écran affiche « dérive OK » et que la lumière
verte située en dessous de « cond » cesse de clignoter.
Titrage (concentration du titrant)
D’abord on veut connaître le titre de la solution Karl Fischer
• Appuyez sur « START » et ajouter 9 μL d’eau avec une seringue en perçant le septum
situé à l’avant de la cellule. L’afficheur vous demandera la quantité d’eau que vous avez
ajoutée et vous confirmerez avec « ENTER ». Dans ce cas, ce sera 0,009 g. Par la suite,
l’appareil titrera l’eau introduite pour ainsi déterminer le titre du titrant Karl Fisher.
• Répétez la même opération lorsque « dérive OK » est affiché en retournant à l’étape a).
Pour bien connaître le titre de la solution Karl Fischer, il faut effectuer au moins trois
dosages de l’eau.
Dosage de l’inconnu
• Appuyez sur « STOP ». Charger la méthode appelée « MIEL » en répétant les mêmes
opérations que lors du chargement de la méthode « H2O Titer ». Peser sur « USER
METH » et « ENTER » etc. Pesez sur « START » l’appareil s’assurera que la dérive est
stable. Pendant ce temps, prendre une seringue de 1 mL et la remplir jusqu’à 0,2 mL de
miel. Peser le miel et la seringue. Lorsque l’afficheur indiquera « Dérive OK » on amorce
le dosage en pesant sur « START ».
• Introduire une goutte de miel (100 mg environ) dans la cellule et on repeser la seringue
pour connaître la quantité introduite. Lorsque l’appareil affichera « prise d’essai », entrer
la quantité pesée par différence et confirmer par « ENTER »et un autre « ENTER » pour
confirmer les unités.
Calculs Déterminez la teneur en différents sucres dans chaque mélange (commerciaux et
inconnu)
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 64 of 145
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- Démontrez votre compréhension du détecteur RID (détecteur à indice de réfraction)
- Quels sont les possibilités pour identifier l’espèce chimique correspondant à un pic?
- Dans votre discussion, faire une comparaison des deux techniques d’analyse des sucres
utilisées au sujet de :
• La préparation des échantillons;
• Les types de séparation (colonnes chromatographiques, phase mobile, etc.);
• Le type de détection
• La sensibilité de la méthode (limites de détection, limites de quantification, …).
- Laquelle des deux techniques utiliseriez-vous pour l’analyse de sucres présents à très
faibles concentrations dans votre échantillon? Laquelle des deux techniques utiliseriez-
vous pour l’analyse de sucres présents à très fortes concentrations dans votre échantillon?
Information Supplémentaire

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 65 of 145
Expérience 6 : Analyse MALDI-MS

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 66 of 145
EXPÉRIENCE 6 : ANALYSE MALDI-MS
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
Introduction
Les cellules des organismes vivants contiennent des milliers de types différents
de protéines. Ces molécules ont une multitude de fonctions dans les organismes vivants
tels que la catalyse de réactions chimiques (p. ex. l’enzyme lactase, protéine
responsable de l’hydrolyse du lactose), la communication cellulaire (p. ex. le récepteur
aux œstrogènes, protéine qui se lie à l’hormone sexuelle estradiol) ou la formation de
structures ou de fibres (p. ex. la kératine, protéine fibreuse qui est le constituant
principal des cheveux et des ongles). Une protéine est un polymère d’acides aminés
(Figure A1, dans l’annexe). Il existe vingt acides aminés naturels qui composent la
plupart des protéines. Les acides aminés sont représentés par des codes à trois ou à une
lettre (p. ex.: alanine = Ala ou A; cystéine= Cys ou C). La séquence peptidique, c’est-à-
dire l’ordre dont les acides aminés sont liés dans une protéine, détermine la structure
tridimensionnelle de celle-ci et donc sa fonction dans les organismes (Figure 1 et
Figure 2).
Figure 1. Composition de l’insuline du porc. Les lignes liant les cystéines sont des
ponts disulfures (liens S-S). Source: Baker E. N., et al. (1988) Philos T Roy Soc B
319(1195):369-456.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 67 of 145
NOTE : Cette expérience est basée sur l’article "Identifying a protein by MALDI-TOF
mass spectrometry: An experiment for the undergraduate laboratory" publiée par
Couterman et al. en 2003 dans le Journal of Chemical Education, 80(2): 177-180.
Figure 2. Structure tridimensionnelle de l’insuline du porc. Source : RSCP Protein
Databank (2001). Molecule of the Month: Insulin. Available at
http://www.rcsb.org/pdb/101/motm.do?momID=14 (accessed on 2015-07-06).
La spectrométrie de masse
D’après l’IUPAC, la spectrométrie de masse (MS) est la science qui « étudie la
matière à travers la formation des ions en phase gazeuse qui sont caractérisés au moyen
des spectromètres de masse par leurs masses, leur charge, leurs structures et leurs
propriétés physico-chimiques ». Le principe de base de la MS est de générer des ions à
partir des composés organiques ou inorganiques et de les séparer par leurs ratios masse
sur charge (m/z). Pour ce faire, les composants de l'échantillon doivent être convertis
en ions et introduits dans un analyseur de masse qui va les séparer en fonction de leurs
m/z. Les analyseurs de masse ont besoin de fonctionner dans un vide élevé (pressions
typiques ~ 109 fois inférieures à la pression atmosphérique) pour éviter les collisions
intermoléculaires. Une fois que les ions sont séparés, un signal est généré à l’aide d’un
système de transduction et d’amplification du courant.
Dans cette expérience, les données seront acquises en utilisant un instrument de
type désorption-ionisation laser assistée par matrice couplée à un spectromètre de type
quadripôle-temps d’envol (MALDI-QTOF). MALDI est la source d’ions, c.-à-d., un
appareil capable de générer des ions à partir de l’échantillon et le QTOF est l’analyseur
de masse qui va séparer les ions en fonction de leurs m/z.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 68 of 145
Pour réaliser une expérience MALDI-QTOF, l'échantillon à analyser est
combiné avec une matrice, souvent constituée d’un acide organique capable d’absorber
la lumière UV. Le ratio molaire entre la matrice et l’analyte est souvent entre 1000 : 1
et 100000 : 1. Ensuite, un volume entre 0.5 et 2 μL du mélange est déposé sur le porte-
échantillons, une plaque de métal (cible MALDI), et on laisse sécher l’échantillon.
Après, la cible est introduite dans l’instrument où elle est frappée par une très courte
impulsion laser (Figure 3). Les molécules de la matrice absorbent l'énergie du laser et
cette énergie est utilisée pour volatiliser l’échantillon et transférer des protons à partir
de la matrice à des molécules de l'échantillon, formant instantanément un panache
d'ions. Quoique des ions de type M•+ ou [M+2H]2+ sont souvent observés, dans les
conditions expérimentales que nous utiliserons, cette méthode d'ionisation favorise la
production d'ions de type [M+H]+.
Figure 3. Schéma de la source MALDI. Source: Gross J. H. (2011) Mass
Spectrometry: A Textbook. 2nd ed. Berlin, Germany: Springer, 716 p.
Ces ions sont ensuite accélérés par une différence de potentiel et sont introduits
dans l’analyseur de masse. Dans ce cas-ci, l’analyseur de masse est un composé d’un
quadripôle (Q) et d’un temps de vol (TOF). Entre les deux, il se trouve une cellule à
collision (q) qui est utilisée pour la fragmentation des ions sélectionnés par le
quadripôle lorsque l’instrument est utilisé dans le mode spectrométrie de masse en
tandem. Dans le cadre de cette expérience, l’instrument sera utilisé en mode « TOF »,
c.-à-d., le quadripôle servira seulement comme guide ionique et la cellule de collision
n’est pas activée. Par conséquent, tous les ions générés par la source MALDI et ayant
un ratio m/z dans la gamme spécifiée par l’expérimentateur seront transmis vers le
TOF.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 69 of 145
Figure 4. Schéma de l’instrument QTOF de la compagnie Waters. Dans cette
expérience la source electrospray est remplacée par la source MALDI. Source : Waters
Corp. (2008) Synapt Mass Spectrometry System : Operators Guide,
71500153502/Revision A.
Avant d’entrer dans le TOF, les ions transmis par le quadripôle sont déviés et
accélérés en direction perpendiculaire à leur trajectoire par le voltage pulsé de 5-10 kV
de l’accélérateur orthogonal (« Pusher » dans la Figure 4). Une fois dans le temps de
vol, les ions sont séparés en fonction du temps qu’ils prennent pour traverser un trajet
de dérive (Figure 4). Puisque la vitesse des ions dépend de leur masse, les ions de
faible masse arriveront plus rapidement au détecteur que les ions de masse élevée.
L’énergie cinétique Ek transféré aux ions par le voltage d’accélération est
exprimée par : 𝐸𝑘 = 𝑒𝑧𝑈
Où z= nombre de charges de l’ion et e=charge de l’électron (1.602x10-19 C). Si l’on
suppose que le voltage d’accélération U est de 20 kV et z=1, alors Ek des ions qui
entrent dans le TOF est de 20 keV (1 électronvolt est l’énergie acquise par une seule
charge électrique accélérée par une différence de potentiel de 1 V, aussi égal à 1.602
x10-19 J).
Cette énergie cinétique dépend de la masse m (en kilogrammes) et la vitesse des
ions v (en mètres par second) selon l’équation :
𝐸𝑘 = 𝑚𝑣2
2⁄
Ces deux équations nous permettent alors de déterminer que le temps t (en
seconds) qu’un ion prend pour se rendre au détecteur placé à une distance s (en mètres)
dans le TOF est déterminé par :
𝑡 =𝑠
√2𝑒𝑈√
𝑚
𝑧
Où U est voltage d’accélération en volts.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 70 of 145
Alors dans un TOF ayant un voltage d’accélération U= 20000 V, une distance s
de 1 m est parcourue par un ion de fullerène (C60+) en :
𝑡 =1𝑚
√2(1.6022 × 10−19 𝐶)(20000 𝑉)
√(
720 𝑔 𝑚𝑜𝑙−1
1000 𝑔 𝑘𝑔−1⁄ )
6.022 × 1023 𝑚𝑜𝑙−1
1
𝑡 = 13.66 𝜇𝑠
Identification des protéines
La protéomique est la science qui s’occupe de la caractérisation globale du
protéome, c.-à-d. l’ensemble des protéines qui peuvent être synthétisées par un
organisme vivant. Elle s’intéresse aussi à l'abondance relative des protéines, leurs
fonctions et aux changements du protéome en fonction des stimuli environnementaux.
Alors, la protéomique nous permet, entre autres, de mieux comprendre les mécanismes
responsables des maladies, du vieillissement et les effets de l’environnement sur le
fonctionnement des organismes vivants.
Une technique largement utilisée pour l’identification des protéines par
spectrométrie de masse est celle de l’empreinte des masses peptidiques (peptide mass
fingerprinting). Cette technique consiste à mesurer les m/z des peptides générés par
digestion enzymatique des protéines d’intérêt. L’enzyme trypsine est couramment
employée pour cette technique. À l’échelle moléculaire, la trypsine agit comme de
« ciseaux » très sélectifs : elle coupe les liens carbonyle-amine entre les acides aminés
liés à la lysine (Lys ou K) ou à l’arginine (Arg ou R) sauf s’ils sont suivis par proline
(Pro ou P). Ceci est illustré à l'aide de la séquence ci-dessous :
AFDEKAWSVARVVAGVANALAHKPEKFVIEI…
Trypsine
AFDEK + AWSVAR + VVAGVANALAHKPEK + FVIEI…Les peptides générés par cette
technique représentent un ensemble unique de m/z (essentiellement une empreinte
digitale) qui est caractéristique de la protéine à partir de laquelle ils proviennent. Les
m/z affichés dans le spectre de masse sont par la suite entrés dans les bases de données
d’empreintes de masses peptidiques pour identifier la protéine d’origine.
Pour calculer la masse en kg d’un
seul ion, nous calculons sa masse
molaire et nous divisons par le
nombre d’Avogadro.
Les unités sont des seconds puisque :
1V=1kgm2
Cs2
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 71 of 145
Figure 5. Spectre de masse obtenu par MALDI-TOF d’un échantillon d’anhydrase
carbonique digéré avec la trypsine. Source de l’image : Protea (2011) Standards for
Protein Mass Spectrometry. Available at :
https://proteabio.com/media/document/Protea_Mass_Spec_Standards_Brochure.pdf
[Accessed on 2015-08-12].
Quoique certaines protéines peuvent avoir un certain degré de similarité entre
leurs séquences peptidiques, des expériences ont démontré qu’une grande partie des
séquences d’une protéine sont uniques et elles peuvent être utilisées pour identifier une
protéine parmi des millions (Pappin et al. 1993; Henzel et al. 2003). En effet, avec
seulement un nombre très petit de peptides (3 à 5), il est possible d’identifier
correctement une protéine.
Protocole détaillé
Objectifs
Identifier une protéine inconnue à l’aide de l’empreinte peptidique déterminée
par MALDI-QTOF et une base de données sur Internet. Cette expérience est divisée en
trois sections :
I. Détermination de l’empreinte de masses peptidiques de la protéine inconnue
par MALDI-QTOF
II. Recherche d’empreintes de masses peptidiques avec une plateforme Web
utilisant des données de la littérature
III. Identification de la protéine inconnue avec une plateforme Web utilisant les
valeurs obtenues expérimentalement.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 72 of 145
Section I : Détermination de l’empreinte de masses peptidiques de la
protéine inconnue par MALDI-QTOF
Matériel et instruments
Solvants et réactifs
• Acétonitrile :Eau (1 :1) 0.1% d’acide trifluoroacétique
• Acide α-cyano-4-hydroxycinnamique : matrice qui devra être mélangée avec
la protéine digérée avant de faire l’analyse par MALDI-QTOF.
• Bicarbonate d’ammonium
• Eau déionisée
• Mélange de calibration du TOF : solution de PEG
• Protéine inconnue
• Trypsine
Pour l’utilisation de l’appareil, veuillez lire la procédure d’utilisation des
spectromètres dans les sections suivantes.
Digestion de la protéine inconnue
(LA PROTÉINE ET LA TRYPSINE DOIVENT ÊTRE CONSERVÉ À -20°C,
ELLES DOIVENT RETOURNER À CETTE TEMPÉRATURE LE PLUS
RAPIDEMENT POSSIBLE)
1- Remplir d’eau et allumer le bain thermostaté puis ajuter la température à 37C.
2- Préparer dans un tube Falcon de 15 mL, 10 mL d’une solution de bicarbonate
d’ammonium 100 mM dans l’eau déionisée.
3- Préparer 10 mL d’une solution de trypsine 0.1 mg/mL dans la solution de
bicarbonate d’ammonium 100 mM.
4- Peser environ 1 mg de la protéine inconnue dans un microtube de 1.5 mL et y
ajouter 500 µL de la solution de trypsine.
5- Placer le tube dans le bain thermostaté maintenu à 37°C pendant environ 3h. Il faut
immerger le tube, mais pas jusqu’au bouchon.
Appareillage
L’instrument utilisé durant l’expérience sera le MALDI-QTOF Synapt de la
compagnie Waters. Cet instrument est équipé d’un quadripôle avec une large gamme
de transmission (environ m/z 200 à 5000), d’un accélérateur orthogonal (« pusher ») et
d’un réflectron (Figure 4). La résolution de cet instrument mesurée à m/z 1400 par la
largeur du pic à la mi-hauteur est de l’ordre de 8000.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 73 of 145
Préparation de la cible MALDI
1- Préparer 1 mL d’une solution de matrice 3.6 mg mL-1 d’acide -cyano-4-
hydroxycinnamique dans 1:1 eau : acétonitrile contenant 0.1 % (v/v) d’acide
trifluoroacétique dans un microtube à centrifugeuse.
2- Mélangez 5 L de solution digérée de la protéine inconnue avec 45 L de la solution
de matrice dans un microtube à centrifugeuse.
3- Déposez 1 L du mélange protéine-matrice sur un des puits de la plaque (cible)
MALDI. Noter sur votre cahier de laboratoire la rangée et colonne des puits utilisés
pour déposer vos échantillons. Répéter cette opération dans deux autres puits.
4- Déposez 1 du mélange de calibration dans un des puits de la plaque (cible) MALDI.
Noter sur votre cahier de laboratoire la rangée et colonne des puits utilisés pour déposer
vos échantillons. Répéter cette opération dans cinq autres puits.
5- Laisser sécher la cible pendant 15-20 minutes à l’air ambiant ou jusqu’à ce que le
puit soit complétement sec.
6- Consulter votre démonstrateur afin de faire approuver votre cible MALDI.
Préparation de la liste d’échantillons et analyse
1- S’il n’est pas déjà ouvert, ouvrez le logiciel « MassLynx V4.1 » en cliquant
sur l’icône suivant dans le Bureau de l’ordinateur :
Sur la fenêtre principale du logiciel Mass Lynx, vous allez voir un menu à
gauche, un menu en haut et la liste d’échantillons au centre. La préparation de votre
liste d’échantillon se fait dans cette dernière. Dans une ligne vide de la liste, taper la
date, le numéro de l’équipe et le code de l’inconnu (p.ex. : AAAAMMJJ-Equipe1-
Inconnu1) dans la colonne « File Name ». Cette information vous aidera à identifier le
fichier de votre échantillon.
2- Dans la colonne « File Text » indiquez la composition de votre échantillon
(p.ex. 10uL-Inconnu1-90uL-matrice). Cette information apparaîtra sur l’entête du
spectre de masse de l’échantillon.
3- Dans la colonne « MS Tune File » tapez « Tune_MALDI_2015 ». Ce fichier
contient l’information sur les paramètres de fonctionnement de la source MALDI
(voltages, gaz de refroidissement, etc.).
4- Dans la colonne « MS Method » tapez « MS-method-Maldi_Highmass ». Ce
fichier contient les paramètres d’acquisition tels que l’intervalle de m/z transmis, le

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 74 of 145
temps et la façon dont le laser frappe la cible ainsi que l’énergie du laser.
5- Dans la colonne « Well location » tapez la position du puit de la cible dont
votre échantillon se trouve. Par exemple pour un échantillon dans la rangée A, colonne
1 de la cible, entrer « A,1 ».
6- Une fois que la cible est sèche, cliquez dans la barre de tâches Windows de
l’ordinateur sur l’icône de la fenêtre de réglage du spectromètre de masse (« MS Tune
») :
7- Dans la fenêtre MS Tune, cliquez sur LOAD dans la section « Sample
Plate » :
8- Introduire la cible MALDI dans l’instrument en suivant les instructions de
votre démonstrateur.
9- Dans la fenêtre MS Tune, cliquez sur l’icône de la caméra pour suivre le
déplacement de la cible MALDI à l’intérieur de l’appareil :
10- Une fois que la cible est visible avec la caméra, tapez la position (p. ex.
« A1 », sans virgule) dans un des puits contenant votre échantillon dans la boîte
« Sample » de la section « Sample Plate ».
11- Minimisez la fenêtre MS Tune (NE JAMAIS FERMER CETTE
FENÊTRE) et retourner à la page principale de Mass Lynx.
12- Sélectionnez les lignes avec vos échantillons et cliquer sur « Start Run »
13- Retournez sur la fenêtre MS Tune et cliquer sur la caméra pour visualiser le
passage du laser sur les échantillons.
Analyse des résultats
1- Une fois que l’acquisition des résultats est finie, sélectionner la ligne d’un de
vos échantillons et cliquer sur « Chromatogram » dans le menu en haut de la
fenêtre principale de Mass Lynx:

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 75 of 145
2- L’image que vous allez voir représente la variation du signal généré par tous
les ions en fonction du temps ; ce n’est pas un vrai chromatogramme, puisqu’il n’y a
pas de séparation chromatographique.
3- Cliquez dois fois sur un des « pics » observés pour visualiser le spectre de
masse au temps donné. Vous allez observer différents pics de m/z 300 à 3000. Fermer
cette fenêtre.
4- Afin de visualiser le spectre de masse combiné, laisser le bouton de droit de
la souris pressé et tracer une ligne du début à la fin du « chromatogramme ». Une
nouvelle fenêtre apparaîtra sur l’écran.
5- Ensuite, vous allez procéder au traitement de votre chromatogramme.
D’abord, aller sur le menu en haut et cliquer sur « Process » et ensuite sélectionner
l’option « Smooth » pour lisser le spectre de masse. Entrer les paramètres suivants :
Smooth windows (channels) = 1
Number of smooths = 2
Smoothing method = Savitzky-Golay
Cliquez sur « OK ». Un nouveau spectre de masse lissée apparaîtra au-dessus de
votre spectre de masse initial.
6- Ensuite, vous allez transformer vos données du mode profile au mode
centroïde (des barres plutôt que des pics) qui sont plus faciles à traiter dans les bases de
données des peptides. Pour faire cela, vous devez d’abord connaître la résolution de
vos pics. Pour mesurer la résolution, lorsque la fenêtre de votre spectre de masse lissé
est active, cliquer sur l’icône de démarrage de Windows et cliquer sur l’icône du
logiciel « Res Cal » :
7- Une fenêtre similaire à celle-ci devra apparaître après quelques secondes :
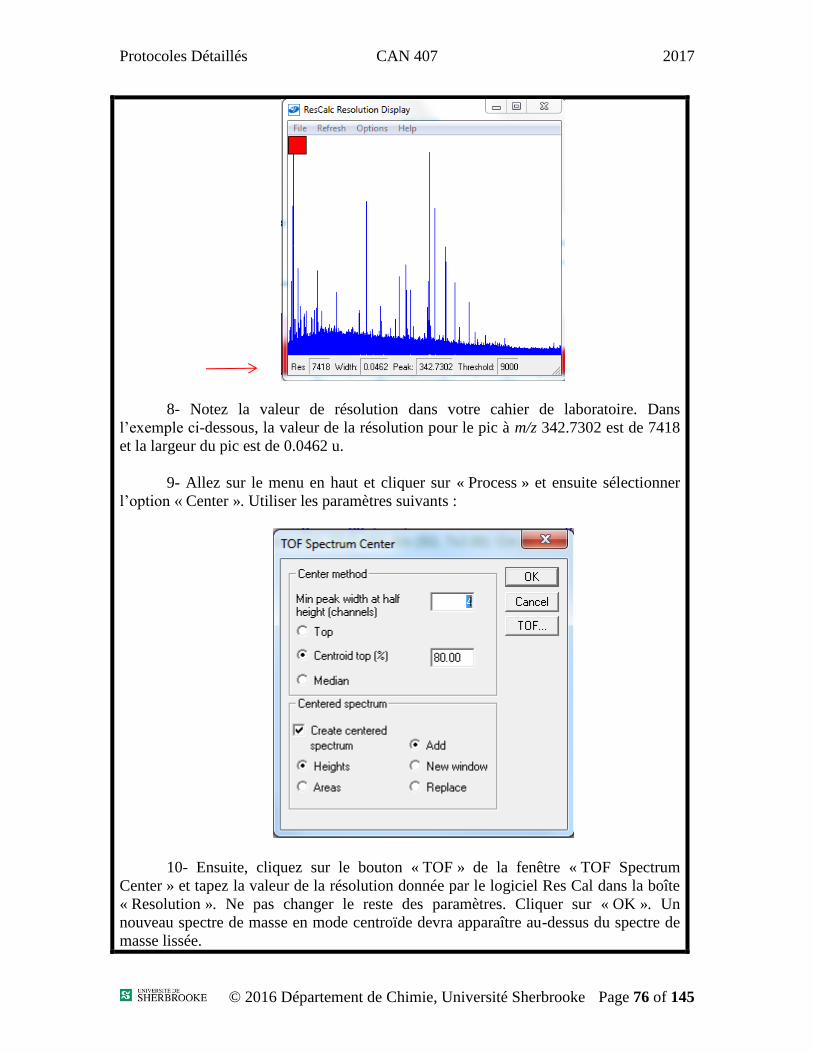
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 76 of 145
8- Notez la valeur de résolution dans votre cahier de laboratoire. Dans
l’exemple ci-dessous, la valeur de la résolution pour le pic à m/z 342.7302 est de 7418
et la largeur du pic est de 0.0462 u.
9- Allez sur le menu en haut et cliquer sur « Process » et ensuite sélectionner
l’option « Center ». Utiliser les paramètres suivants :
10- Ensuite, cliquez sur le bouton « TOF » de la fenêtre « TOF Spectrum
Center » et tapez la valeur de la résolution donnée par le logiciel Res Cal dans la boîte
« Resolution ». Ne pas changer le reste des paramètres. Cliquer sur « OK ». Un
nouveau spectre de masse en mode centroïde devra apparaître au-dessus du spectre de
masse lissée.
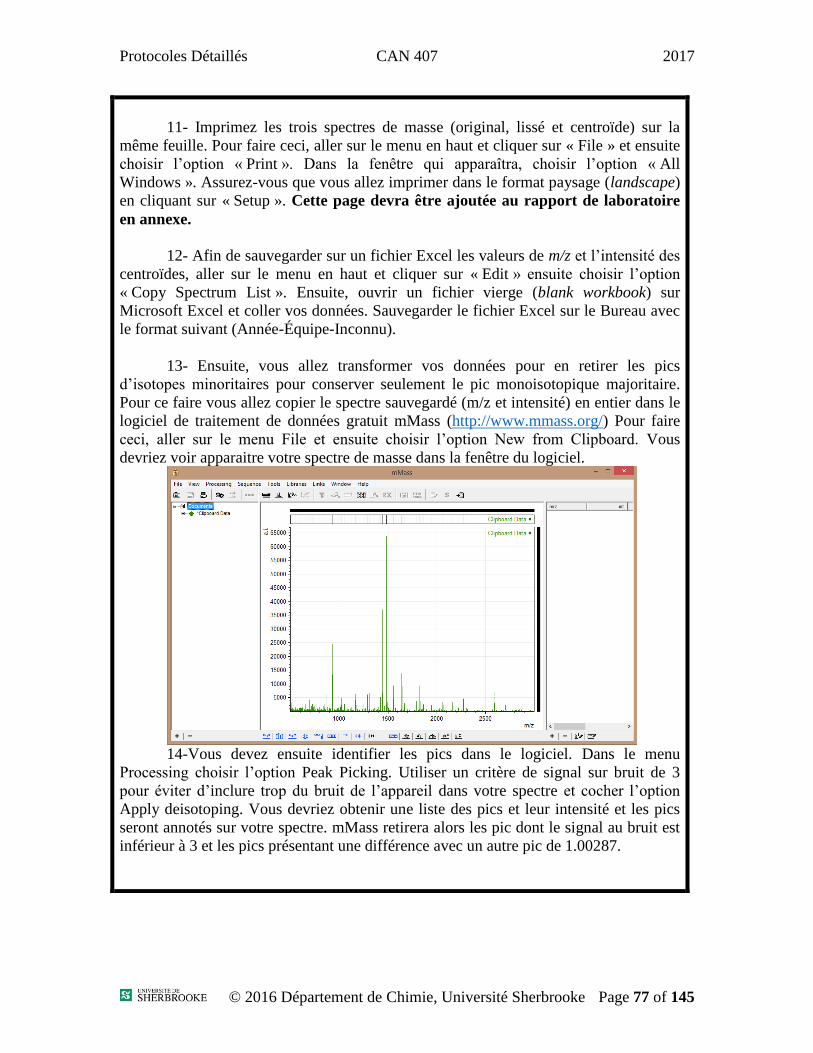
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 77 of 145
11- Imprimez les trois spectres de masse (original, lissé et centroïde) sur la
même feuille. Pour faire ceci, aller sur le menu en haut et cliquer sur « File » et ensuite
choisir l’option « Print ». Dans la fenêtre qui apparaîtra, choisir l’option « All
Windows ». Assurez-vous que vous allez imprimer dans le format paysage (landscape)
en cliquant sur « Setup ». Cette page devra être ajoutée au rapport de laboratoire
en annexe.
12- Afin de sauvegarder sur un fichier Excel les valeurs de m/z et l’intensité des
centroïdes, aller sur le menu en haut et cliquer sur « Edit » ensuite choisir l’option
« Copy Spectrum List ». Ensuite, ouvrir un fichier vierge (blank workbook) sur
Microsoft Excel et coller vos données. Sauvegarder le fichier Excel sur le Bureau avec
le format suivant (Année-Équipe-Inconnu).
13- Ensuite, vous allez transformer vos données pour en retirer les pics
d’isotopes minoritaires pour conserver seulement le pic monoisotopique majoritaire.
Pour ce faire vous allez copier le spectre sauvegardé (m/z et intensité) en entier dans le
logiciel de traitement de données gratuit mMass (http://www.mmass.org/) Pour faire
ceci, aller sur le menu File et ensuite choisir l’option New from Clipboard. Vous
devriez voir apparaitre votre spectre de masse dans la fenêtre du logiciel.
14-Vous devez ensuite identifier les pics dans le logiciel. Dans le menu
Processing choisir l’option Peak Picking. Utiliser un critère de signal sur bruit de 3
pour éviter d’inclure trop du bruit de l’appareil dans votre spectre et cocher l’option
Apply deisotoping. Vous devriez obtenir une liste des pics et leur intensité et les pics
seront annotés sur votre spectre. mMass retirera alors les pic dont le signal au bruit est
inférieur à 3 et les pics présentant une différence avec un autre pic de 1.00287.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 78 of 145
15-Exporter les données : File, Export, Export peak list.
Section II. Recherche d’empreintes de masses peptidiques avec une
plateforme Web utilisant des données de la littérature NOTE: Afin de mieux comprendre l’importance des paramètres lors de la
recherche d’empreintes de masses de peptidiques et faciliter la compréhension des
résultats qui vous seront fournis par la plateforme Web, utilisez les valeurs de m/z des
peptides de l’anhydrase carbonique apparaissant dans la Figure 5.
1- Aller sur le site de ProteinProspector, un site internet intégrant divers outils
de protéomique et développé par l’Université de Californie à San Francisco :
http://prospector.ucsf.edu/prospector/mshome.htm
2- Sur la page principale, dans la section « Database Seach Programs » cliquer
sur « MS-Fit ». MS-Fit est la plateforme Web de recherche de bases de données que
vous allez utiliser pour cette partie de l’expérience.
3- Dans la page de MS-FIT, choisir les paramètres suivants pour effectuer la
recherche d’empreinte de masses peptidiques :
a) Database : SwissProt.2016.9.6
b) Taxonomy : ALL
c) Digest : Trypsin
d) Max. Missed Cleavages : 2
e) Maximum reported hits : 10
f) Tol : 10 mmu
g) Possible modifications : Peptide N-terminal Gln to pyroGlu, Oxidation of M,

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 79 of 145
Protein N-terminus Acetylated
h) Instrument : MALDI-Q-TOF
g) Data Format : PP M/Z Intensity Charge
NOTE : L’identification de protéines peut être très sensible aux paramètres
utilisés pour la recherche, surtout le nombre minimum de peptides requis pour
l’identification (Min. # peptides required to match), le nombre maximal de clivages
ratés (Max. Missed Cleavages) et la tolérance de l’erreur sur la masse (Tol).
4- Une fois que les paramètres sont entrés, copier les valeurs des m/z affichées
dans la Figure 5 (utiliser seulement les pics d’intensité supérieure ou égale à celle de
m/z 1255.5) dans la boîte « Data Paste Area » et cliquer sur « Start Search ».
5- Après quelques seconds, un sommaire des résultats sera affiché à l’écran
contenant l’information suivante :
Protein Hit Number Classement des protéines qui correspondent
potentiellement à la protéine dans l’échantillon en
fonction du pointage MOWSE obtenu.
MOWSE Score Pointage calculé par un algorithme. Cet algorithme est
basé sur la comparaison des m/z des ions obtenus
expérimentalement et les valeurs calculées des m/z des
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 80 of 145
peptides de la protéine identifiée en fonction de la
tolérance de l’erreur sur la masse.
# pep
Nombre de peptides identifiés.
# mat
Nombre d’ions identifiés.
% mat
Pourcentage d’ions identifiés sur le nombre total d’ions
soumis.
# pks Nombre total d’ions soumis.
% Cov Recouvrement de la séquence, c.-à-d. le pourcentage des
acides aminés identifiés sur le nombre total composant la
protéine.
% TIC Pourcentage de la somme des intensités des ions
identifiés sur les la somme des intensités de tous les ions
soumis (cette valeur est identique au % mat si les
intensités des ions ne sont pas soumises).
MeanErr ppm Erreur moyenne sur la masse des ions identifiés en ppm.
L’erreur relative sur la masse d’un ion se calcule de la
façon suivante :
δ𝑚/𝑚(𝑝𝑝𝑚) =𝑚/𝑧𝑒𝑥𝑝 − 𝑚/𝑧𝑐𝑎𝑙
𝑚/𝑧𝑐𝑎𝑙× 106𝑝𝑝𝑚
DataTol ppm 2 écart-type de l’erreur sur les m/z des ions identifiés
en ppm.
# Hom Prot Nombre de protéines homologues (c.-à-d. ayant au
moins un peptide en commun)
MS-Digest Index # Code de la protéine dans la base de données MS-Digest
(digestion simulée par ordinateur).
Protein MW(Da)/pI Poids moléculaire de la protéine en Da/ point
isoélectrique.
Accession # Code d’identification de la protéine dans la base de
données UniProt.
Species Code à cinq lettres de l’organisme vivant (espèce) où la
protéine a été trouvée.
Protein Name : Nom de la protéine.
6- Cliquer sur le symbole « + » de la section « Detailed Results » qui se trouve
en bas de la page. Une série de données apparaîtra pour chaque identité possible de la
protéine dans l’échantillon. Dans cette section, la composition des peptides
correspondante aux valeurs m/z soumises est affichée (« Sequence »). En cliquant sur la
carte peptidique de la protéine (« Coverage Map for This Hit ») vous pouvez observer
dans quel endroit précis de la protéine les peptides identifiés se trouvent.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 81 of 145
Section III. Identification de la protéine inconnue avec une plateforme
Web utilisant les valeurs obtenues expérimentalement.
1- Ouvrez les fichiers Excel contenant vos données expérimentales.
2- Répétez les étapes 1-4 de la section II, mais cette fois-ci en utilisant les
données expérimentales sélectionnées. Faites cette même opération pour les deux
autres échantillons de protéine inconnue.
Références
Counterman A. E., Thompson M. S., Clemmer D. E. 2003 Identifying a protein by
MALDI-TOF mass spectrometry: An experiment for the undergraduate laboratory.
Journal of Chemical Education 80(2), 177-180.
Henzel W. J., Watanabe C., Stults J. T. 2003 Protein identification: the origins of
peptide mass fingerprinting. Journal of the American Society for Mass Spectrometry
14(9), 931-942.
Pappin D. J., Hojrup P., Bleasby A. J. 1993 Rapid identification of proteins by peptide-
mass fingerprinting. Current Biology 3(6), 327-332.
Calculs a. Calculez le temps qui prend les ions m/z 927.50 et 927.85 à se rendre au
détecteur si le voltage d’accélération utilisé dans le QTOF est de 15 kV et le trajet de
dérive est de 1 m. Quel serait le temps de vol de ces ions si le trajet de dérive est
doublé ?
b. Quel est l’avantage d’un trajet de dérive plus grand ?
Notes supplémentaires pour la discussion
- Discussion des résultats (spectres de masse, tableaux) en intégrant les réponses
aux questions suivantes :
a. Pourquoi utilise-t-on l’acide α-cyano-4-hydroxycinnamique comme matrice
en MALDI ?
b. Pourquoi l’identification des protéines par la méthode d’empreinte de masses
peptidiques est sensible au minimum de peptides requis pour l’identification, au
nombre maximal de clivages ratés et à la tolérance de l’erreur sur la masse ?
c. Selon les résultats du tableau sommaire, est-ce que vous pouvez être certain
que la protéine inconnue a été identifiée correctement ? Expliquez.
- Conclusion
- En Annexe :
a. Le spectre de masse de la protéine inconnue (section II, numéro 12).
b. Le tableau sommaire des résultats obtenus avec la plateforme Web MS-FIT
pour la protéine inconnue.
c. Le tableau des résultats détaillés pour la protéine ayant le pointage le plus
élevé.
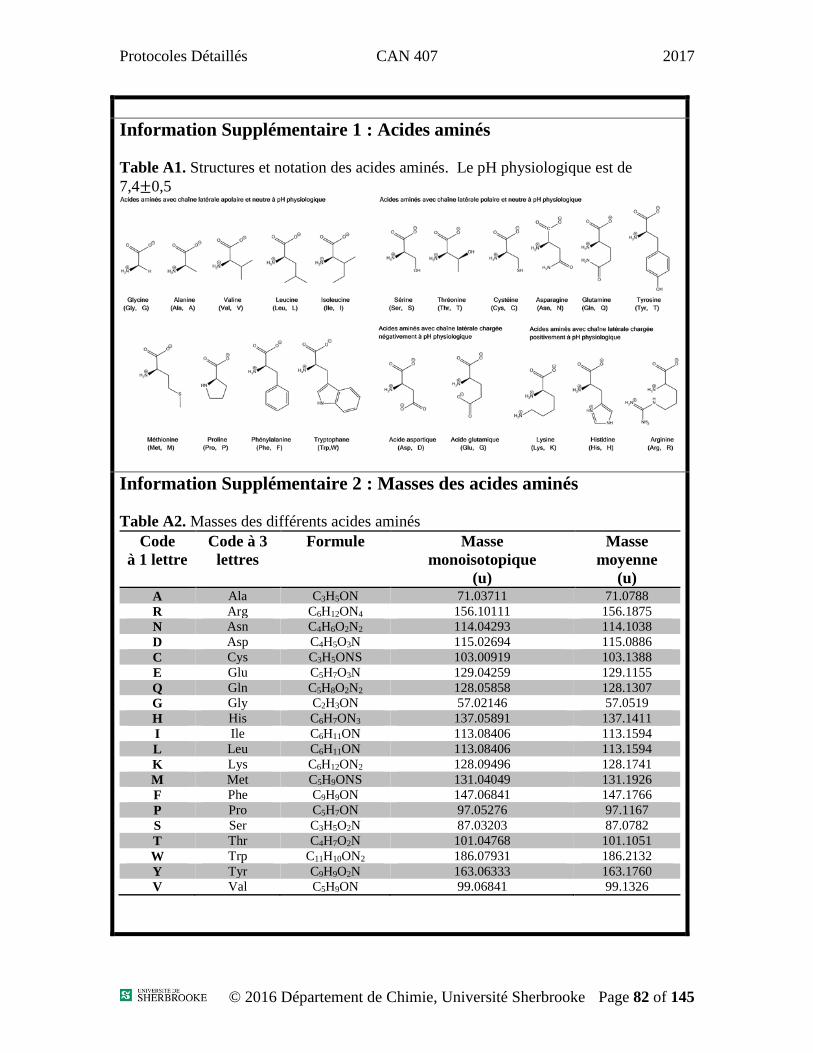
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 82 of 145
Information Supplémentaire 1 : Acides aminés
Table A1. Structures et notation des acides aminés. Le pH physiologique est de
7,4±0,5
Information Supplémentaire 2 : Masses des acides aminés
Table A2. Masses des différents acides aminés
Code
à 1 lettre
Code à 3
lettres
Formule Masse
monoisotopique
(u)
Masse
moyenne
(u) A Ala C3H5ON 71.03711 71.0788 R Arg C6H12ON4 156.10111 156.1875 N Asn C4H6O2N2 114.04293 114.1038 D Asp C4H5O3N 115.02694 115.0886 C Cys C3H5ONS 103.00919 103.1388 E Glu C5H7O3N 129.04259 129.1155 Q Gln C5H8O2N2 128.05858 128.1307 G Gly C2H3ON 57.02146 57.0519 H His C6H7ON3 137.05891 137.1411 I Ile C6H11ON 113.08406 113.1594 L Leu C6H11ON 113.08406 113.1594 K Lys C6H12ON2 128.09496 128.1741 M Met C5H9ONS 131.04049 131.1926 F Phe C9H9ON 147.06841 147.1766 P Pro C5H7ON 97.05276 97.1167 S Ser C3H5O2N 87.03203 87.0782 T Thr C4H7O2N 101.04768 101.1051 W Trp C11H10ON2 186.07931 186.2132 Y Tyr C9H9O2N 163.06333 163.1760 V Val C5H9ON 99.06841 99.1326

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 83 of 145
Expérience 7 : Caractérisation des matériaux

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 84 of 145
EXPÉRIENCE 7 : CARACTÉRISATION DES MATÉRIAUX
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
Introduction
La pierre à chaux (souvent simplement appelée « chaux ») est composée de
carbonate de calcium (CaCO3) avec une teneur plus ou moins élevée en carbonate de
magnésium (MgCO3). Elle peut contenir plusieurs composés mineurs, par exemple de
la silice, des oxydes de fer, etc. Elle est classée en trois variétés :
- La chaux calcique
- La chaux magnésienne
- La chaux dolomitique
Après broyage, la pierre à chaux est utilisée pour neutraliser l’acidité des sols.
Pour cette application, le pouvoir neutralisant de la chaux est une caractéristique
importante. Le choix du type de chaux à appliquer dépend des besoins du sol en
calcium et en magnésium.
Avant d’utiliser la chaux dans l’industrie chimique, on lui fait souvent subir un
traitement :
Soit par chauffage, on la transforme en oxyde puis en hydroxyde de calcium :
CaCO3 → CaO + CO2 et CaO + H2O → Ca(OH)2
L’hydroxyde de calcium est une base forte mais peu soluble utilisée pour
ajuster le pH, neutraliser les acides, capter le SO2, etc.
Soit on la purifie par précipitation : mêmes étapes que ci-dessus, puis :
Ca(OH)2 + CO2 → CaCO3 + H2O
Le carbonate de calcium précipité est utilisé à grande échelle comme agent de
remplissage dans les industries du plastique, du papier, de la peinture, etc.
Note concernant les normes
Les composés d’importance industrielle sont régis par des normes. En
Amérique du Nord, ce sont généralement les normes ASTM qui s’appliquent. La
caractérisation de la chaux et des composés semblables est décrite dans la norme
ASTM C25, « C25-11 Standard Test Methods for Chemical Analysis of Limestone,
Quicklime, and Hydrated Lime » que vous trouverez à la bibliothèque, à la section
« Normes », dans le volume ASTM 04.01. Notez que :

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 85 of 145
- les normes contiennent des procédures d’analyse très détaillées. Ce sont de
bonnes sources de renseignements pour les chimistes analystes.
- S’il y a un différend entre deux sociétés concernant la qualité d’un produit,
les analyses doivent être faites conformément aux normes, et souvent, par les
méthodes les moins instrumentales.
Les normes ASTM couvrent plus de 80 volumes répartis en 15 sections. Vous
pouvez consulter ces sections à partir de la bibliothèque, BANQUES de données,
ASTM.
http://libguides.biblio.usherbrooke.ca/az.php?a=a
Pour vous familiariser avec les normes ASTM,
- consultez l’index du volume 04.01 :
(Recherchez : ASTM C25-11)
- Vous devriez pouvoir répondre aux deux questions suivantes :
- Dans la norme ASTM C25-11 (note : 11 signifie l’année de la dernière
révision), par quelle méthode est analysé le phosphate dans la chaux?
- Comment le résultat de l’analyse du phosphate est-il exprimé?
Protocole détaillé
Objectifs
Le but de cette expérience est de caractériser un échantillon de pierre à chaux
ou autre échantillon de carbonate de calcium. Cette caractérisation se déroulera en
deux parties.
La première partie se déroulera dans les laboratoires du Département de
chimie :
- Préparation de l’échantillon
- Tamisage, séchage
- Perte au feu
- Détermination des phases et de la composition chimique approximative par
diffraction des rayons-X (DRX).
- Détermination du pouvoir neutralisant par titrage potentiométrique.
La deuxième partie se déroulera au Centre de caractérisation des matériaux de
l’Université :
- Analyse thermique différentielle (ATD) et analyse thermogravimétrique

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 86 of 145
couplée à la spectroscopie de masse (ATG-MS).
- Analyse élémentaire par fluorescence des rayons-X (FRF).
- Observation de la composition chimique en surface par microscopie
électronique à balayage (MEB) couplée à une microsonde.
Par cette caractérisation, les étudiants seront mis en contact avec une variété de
méthodes d’analyse modernes.
PREMIÈRE PARTIE : Préparation, DRX et Détermination du
pouvoir neutralisant (Au département de chimie)
Préparation de l’échantillon
Séchage
Normalement, les résultats d’analyses chimiques d’un solide sont exprimés sur
la base de la masse sèche de l’échantillon.
- Dans un pèse-tare en aluminium, peser précisément environ 30 g de chaux.
(Fait par le technicien avant le jour du laboratoire)
- Mettre à l’étuve à 120°C pendant trois heures et laisser refroidir dans un
dessiccateur. (Fait par le technicien avant le jour du laboratoire)
- Peser de nouveau puis remettre l’échantillon dans le dessiccateur. (Vous
devrez faire cette étape vous-même, votre échantillon de chaux vous attendra au
dessiccateur accompagné de la masse initiale)
- Exprimer le résultat en termes de pourcentage d’humidité de votre échantillon
de départ.
Note : Normalement, il faudrait remettre l’échantillon à l’étuve pendant une
autre période de trois heures pour vérifier si la masse est constante (dans le cadre d’un
laboratoire, nous n’avons pas le temps).
Tamisage
La granulométrie d’une chaux est une caractéristique importante directement
reliée à sa réactivité : plus elle est fine, plus elle est réactive.
Le but du tamisage dans cette expérience n’est pas de déterminer la
granulométrie de l’échantillon de chaux mais plutôt d’obtenir un échantillon le plus
homogène possible en y retirant les particules grossières. On peut noter que cette
opération peut modifier légèrement la composition chimique globale de l’échantillon.
En effet, le carbonate de calcium est un matériau relativement facile à broyer; des
impuretés difficiles à broyer, tel le quartz, un matériau dur, sont susceptibles de se

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 87 of 145
retrouver plus concentrées dans la fraction grossière de l’échantillon.
NOTE : Dès que vous aurez suffisamment de poudre tamisée, débutez le
titrage!
- Pesez le tamis micrométrique et le contenant récepteur. (Notez bien toutes les
informations)
- Transférez l’échantillon séché sur le tamis et peser de nouveau.
- Secouez (vigoureusement) le tamis pour que l’échantillon passe au-travers.
- Si nécessaire, utilisez mortier et pilon pour re-broyer votre chaux.
- Retirer le tamis du contenant récepteur. Frappez légèrement le tamis pour en
retirer toutes les particules fines. Pesez
- Enlevez les particules grossières et peser de nouveau.
- Exprimez votre résultat en termes du pourcentage de l’échantillon passant le
tamis micrométrique que vous avez utilisé.
Transférez l’échantillon tamisé dans un vial de 20 mL avec bouchon
hermétique. Cet échantillon sera utilisé pour toutes les autres expériences. Conservez
la partie de l’échantillon retenue sur le tamis pour un titrage acido-basique.
Perte au feu (PAF)
La perte au feu (PAF) (en anglais « loss on ignition » (LOI)) consiste à
chauffer à haute température un échantillon qui a normalement été séché auparavant.
Selon le type d’échantillon, la perte au feu peut comprendre :
- L’eau d’hydratation (ex. : CaSO4.2H2O → CaSO4 + 2 H2O)
- L’eau de structure (ex. : Ca(OH)2 → CaO + H2O)
- La matière organique qui se transforme en CO2 lors de la combustion
- La décomposition des carbonates (ex. : CaCO3 → CaO + CO2). Etc.
Pour un échantillon de chaux, la perte au feu est principalement due à la
décomposition des carbonates.
- Préchauffez le four à 400°C. (Utilisez le four avec délicatesse puisque ce type
de four est très dispendieux).
- Mettez deux creusets de porcelaine propres au four pendant ~15 minutes; les
laisser refroidir dans un dessiccateur.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 88 of 145
- Pesez les creusets et transférer environ 4 g (pesés précisément) de
l’échantillon dans chacun.
- Couvrir (en laissant un espace pour laisser s’échapper les gaz de
décomposition) et insérer dans le four à 400°C pendant 30 minutes.
- Une fois la période de 30 minutes écoulée, augmentez la température du four
à 1000°C.
- Chauffez pendant 20 minutes à 1000°C puis retirer du four et transférer dans
un dessiccateur.
- Pesez. Transférez l’échantillon dans un vial de 5 mL avec bouchon
hermétique (il servira à l’analyse par fluorescence X)
- Exprimer le résultat en termes de pourcentage de perte au feu.
- En supposant que la perte au feu soit due uniquement à la décomposition des
carbonates, exprimer le résultat en termes de pourcentage de CaCO3 équivalent dans
l’échantillon.
Note : Normalement, on remettrait l’échantillon au four pour une autre période
de 20 minutes pour s’assurer que la masse est constante.
Diffraction des rayons-X
Remettre à Daniel Fortin environ 200 mg de votre échantillon original (avant la
perte au feu). Il va enregistrer le DRX et vous remettre les résultats. Dans le cadre du
cours CAN 400, Daniel vous présentera en détail les principes de l’analyse par rayon-
X, mais ces concepts seront aussi expliqué au cours de la séance s’information se
tenant à la première rencontre du cours CAN407.
Le diffractomètre est couplé à une banque de données qui permet d’identifier
directement les composés présents dans l’échantillon ou encore leurs formes
cristallines. Par exemple, le carbonate de calcium existe sous deux formes cristallines :
la calcite et l’aragonite. La DRX permet de distinguer ces deux formes.
Pouvoir neutralisant
Pour déterminer le pouvoir neutralisant de la chaux, une quantité connue est
dissoute dans un excès de HCl, puis l’excès est titré par NaOH. Vous avez déjà fait
beaucoup de titrages de HCl par NaOH. Cette fois, vous allez utiliser la technique
suivante:
• Titrage à l’aide d’un titrateur automatique

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 89 of 145
Titrateur automatique
Un titrateur automatique est extrêmement utile pour effectuer des titrages en
série, surtout s’il est couplé à un module échantillonneur. Le fonctionnement du
titrateur vous sera expliqué au laboratoire; vous pouvez consulter la documentation sur
le titrateur « Titrino751 GPD » sur le site de Metrohm :
http://www.metrohm.com/en/support-and-service/
Recherchez : Titrino751 GPD
--> Vous pouvez consulter « Short Instructions for Use 751 GPD Titrino » ou
« Quick References 751 GPD Titrino ». Le mode DET sera utilisé.
Vous devez d’abord vous familiariser avec le fonctionnement du titrateur
automatique en effectuant quelques titrages de HCl par NaOH.
*** Pour l’échantillon de chaux tamisé dans le tamis micrométrique ***
- Dans un bécher de 250 mL. pesez précisément environ 2,1 g de chaux.
- Ajoutez 50 mL de HCl 1 M et bouillir légèrement pendant 5 minutes pour
accélérer la dissolution et permettre l’expulsion du CO2 de la solution.
- Titrez l’excès de HCl par NaOH 1 M en utilisant le titrateur automatique.
- Exprimez le pouvoir neutralisant de la chaux en termes de pourcentage de
CaCO3 équivalent dans l’échantillon.
- Une fois le point d’équivalence de HCl atteint, poursuivre le titrage jusqu’à
pH ~ 11. S’il y a une quantité appréciable d’ions Mg2+ dans l’échantillon, un deuxième
point d’équivalence sera observé. Il devrait correspondre à la fin de la précipitation de
Mg(OH)2 en présence de NaOH.
- Calculez la concentration en magnésium dans votre échantillon; exprimer le
résultat en termes de pourcentage de MgO.
Pour déterminer si le tamisage a modifié la composition chimique de
l’échantillon, effectuez la même procédure de titrage, mais cette fois pour la partie
retenue sur le tamis. (Inclure à votre discussion)
SECONDE PARTIE : ATG/ATD, Fluorescence-X, MEB (Au CCM)
Note : Cette deuxième partie de la caractérisation d’un échantillon de chaux
sera effectuée au Centre de caractérisation des matériaux (CCM) situé à la faculté de
Génie. Le personnel du CCM vous expliquera en détail le fonctionnement des
instruments et les procédures d’analyse. LA PONCTUALITÉ EST OBLIGATOIRE!!!
Tout retard sera considéré comme injustifié et entrainera des manquements au niveau

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 90 of 145
de la formation offerte sa traduisant par des faiblesses aux rapports. RAPPEL :
Toutes les règles décrites au plan de cours CAN407 s’appliquent dans les locaux du
CCM.
Analyse thermogravimétrique (ATG) et analyse thermique différentielle (ATD)
Le document suivant a été préparé par un professionnel du Centre de
caractérisation des matériaux. En prendre connaissance avant d’effectuer les
expériences.
http://www.ccm.usherbrooke.ca/fr/services/ccm/LCG/pdf/analyse_thermique.pdf
Contrairement à la perte au feu, qui fournit un résultat global de la perte (ou
gain) de masse à une température donnée (ex. 1000°C), l’analyse thermogravimétrique
mesure la perte (ou le gain) de masse en fonction de la température. Connaissant la
température de décomposition de composés purs, ces données peuvent permettre
d’identifier des composés présents dans un échantillon, ou encore d’en exclure la
présence. Par exemple, l’hydroxyde de calcium, Ca(OH)2, se décompose en CaO et
H2O à 580°C. Une perte de masse d’un échantillon aux environs de cette température
indique la possibilité de présence de Ca(OH)2 dans l’échantillon, alors que l’absence
de perte de masse à cette température exclut la présence de Ca(OH)2 dans
l’échantillon.
L’analyse thermique différentielle mesure la différence de température entre
une cellule contenant l’échantillon et une cellule de référence, en fonction de la
température. Par exemple, la décomposition de l’hydroxyde de calcium est une
réaction endothermique; pendant la décomposition de ce composé, la température de la
cellule échantillon va être plus basse que celle de la cellule de référence. Dans ce cas,
l’ATG indique une perte de masse, et l’ATD un refroidissement de la cellule.
Autour de 580°C, le quartz β se transforme en quartz α. Cette transition
endothermique n’implique pas de perte de masse, donc aucun signal ne serait détecté
en ATG. Par contre, l’ATD indiquerait un refroidissement de la cellule échantillon.
• Avec l’appareil ATG couplé à l’ATD, enregistrer le thermogramme de votre
échantillon à partir de la température de la pièce jusqu’à 1000°C.
• S’il y a plusieurs plages de pertes de masse en fonction de la température,
essayer d’identifier et de quantifier les produits responsables de ces pertes de masse
(en vous aidant des résultats de l’analyse chimique par fluorescence X).
• Exprimer la perte de masse totale en termes du pourcentage de CaCO3
équivalent de votre échantillon.
• En comparant les signaux ATG et ATD, vérifier s’il y a des transitions
thermiques qui n’impliquent pas de perte de masse. Pour ce faire, vous devez amplifier

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 91 of 145
le signal ATD, car les transitions d’une forme cristalline à une autre sont en général
beaucoup moins endothermiques que les décompositions.
Analyse élémentaire par fluorescence - X
Le document suivant présente les bases de la fluorescence X. L’appareil que
vous allez utiliser est un spectromètre à dispersion de longueur d’onde.
https://www.youtube.com/watch?v=bPNV3jZW0ds --> Anxios Advanced
Fusion de l’échantillon
L’analyse par fluorescence X peut être effectuée directement sur un échantillon
solide, sans traitements préalables. Par contre, la précision des résultats est de
beaucoup supérieure si l’échantillon est d’abord solubilisé dans un fondant, ce qui
rend la matrice très homogène. Une fois l’échantillon solubilisé dans le fondant, il peut
être transféré dans un petit moule pour former un verre solide. Ce verre s’appelle une «
perle ». L’échantillon peut aussi être transféré dans une solution acide pour le
solubiliser et être ensuite analysé par absorption atomique, ICP, etc.
Le tétraborate de lithium est souvent utilisé comme fondant. Les masses
molaires très faibles du lithium et du bore font qu’ils émettent peu de fluorescence X.
Vous pouvez visualiser la vidéo suivante qui illustre le processus de fusion. (Note : le
fondant utilisé pour l’expérience est composé de 66.67 % de tétraborate de lithium
(Li2B4O7), 32,83 % de métaborate de lithium (Li3BO3) et de 0,5 % de bromure de
lithium (LiBr) comme agent pour aider au démoulage).
http://www.katanax.com/cgi/show.cgi?video.l=fr
L’étalonnage et mesure
L’appareil de fluorescence X est étalonné avec des standards préparés dans le
même fondant que l’échantillon de façon à minimiser les effets de matrice. Les
courbes d’étalonnage pour les différents éléments sont déjà intégrées dans la méthode
d’analyse.
Les résultats d’analyse sont souvent exprimés en termes de pourcentage
d’oxyde des différents éléments. Lors du traitement d’un l’échantillon à haute
température (perte au feu) dans une atmosphère oxydante (l’air), les composés volatils
(H2O; CO2; etc.) sont éliminés, et les métaux, si présents, sont transformés en leur
oxyde (stable à 1000°C). Pour cette raison, la somme des pourcentages de tous les
éléments présents dans un échantillon, exprimés sous la forme de leur oxyde stable à
1000°C, devrait égaler 100 % ou s’en approcher.
Microscopie électronique à balayage

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 92 of 145
Vous allez observer un échantillon de chaux (pas nécessairement le vôtre) par
microscopie électronique à balayage (MEB). Le fort grossissement de ce microscope
permet d’observer la taille des grains, leur structure, etc. De plus, une microsonde
couplée au MEB permet de connaitre la composition chimique de surface d’un grain
(ou une partie d’un grain) de l’échantillon par rapport à un autre.
Calculs
- Déterminez le % d’eau dans la chaux avant le séchage
- Déterminez le pouvoir neutralisant de votre échantillon de chaux
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- Mettez bien en évidence l’apport de chaque technique dans le cadre de cette
caractérisation.
- Aurait-on pu utiliser d’autres techniques analytiques pour obtenir les mêmes ou
davantage d’information sur l’échantillon étudié?
- Mettez TOUJOURS vos résultats en contexte!
- Vous devrez répondre aux deux questions suivantes :
* Dans la norme ASTM C25-11 (note : 11 signifie l’année de la dernière
révision), par quelle méthode est analysé le phosphate dans la chaux?
* Comment le résultat de l’analyse du phosphate est-il exprimé?
Voici quelques pistes supplémentaires pour votre discussion :
Les suggestions ci-dessous ne sont pas exclusives :
• Comparer les résultats des différentes analyses, leur niveau de précision, etc.
• D’après les résultats de fluorescence X, de perte au feu, de titrages et d’ATG,
est-ce que d’autres composés que CaCO3 et MgCO3 contribuent au pouvoir
neutralisant de la chaux?
• Définissez clairement ce qu’on entend par « pouvoir neutralisant », quels
sont les applications, ces applications sont-elles exclusives à la chaux?
• La sommation des pourcentages de tous les composés présents dans
l’échantillon, déterminés par fluorescence X se rapproche-t-elle de 100 %?
Commenter.
• Commenter l’utilité des différentes méthodes d’analyse que vous avez mises
en œuvre.
• Après la relâche, le module ATD sera remplacé par un module DSC, qui
permettra de déterminer la variation d’enthalpie des transitions de phase et des

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 93 of 145
réactions de décomposition lors de l’analyse thermique. À partir des enthalpies de
formation, calculer la variation d’enthalpie de la décomposition des carbonates de
votre échantillon, de façon à pouvoir comparer cette enthalpie à la valeur obtenue par
DSC.
Information Supplémentaire Plusieurs ouvrages sont disponibles à la bibliothèque de la Faculté des Sciences
sur la caractérisation des matériaux ainsi que sur chacune des techniques employées.
Faites en bon usage.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 94 of 145
Expérience 8: Chromatographie ionique

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 95 of 145
EXPÉRIENCE 8 : CHROMATOGRAPHIE IONIQUE
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
La chromatographie ionique (CI en français et IC en anglais) est une version haute
performance de la chromatographie par échange d’ions. Elle trouve beaucoup
d’applications pour l’analyse des ions (ou des composés pouvant être ionisés) dans des
domaines tels que de l’eau, l’alimentation, les cosmétiques, le pharmaceutique, les semi-
conducteurs (pour suivre les traces d’anions ou de cations dans l’eau désionisée), etc.
Chromatographie par échange d’ions
Dans la chromatographie par échange d’ions, la rétention est basée sur l’attraction
électrostatique entre les ions du soluté et des sites chargés attachés à une phase
stationnaire. Sur une colonne échangeuse d’anions, des groupements chargés
positivement sont liés par un lien covalent à la phase stationnaire et attirent les anions du
soluté. A l’inverse, une colonne échangeuse de cations contient des groupements négatifs
fixes qui attirent les cations.
Les colonnes échangeuses d’ions
Les colonnes échangeuses d’ions consistent en un support (polymère, particules
de silice, etc.) sur lequel on peut greffer sur un noyau aromatique, par exemple, des
groupements fonctionnels acides ou basiques qui attirent soit les cations soit les anions.
Les groupements les plus fréquents sont les groupements ammonium NR3+ pour les
colonnes échangeuses d’anions (anex est une abréviation pour colonne échangeuse
d’anions) et le groupement sulfonate –SO3- pour les colonnes échangeuses de cations
(catex).
Figure 1. Illustration de la constitution de la résine ou support

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 96 of 145
Figure 2. (gauche) Phase stationnaire échangeuse de cation avec des groupements
sulfonates. (droite) Phase stationnaire échangeuse d’anions avec des groupements
ammonium.
Si on utilise l’acide méthacrylique au lieu du styrène, on fabrique un polymère
avec un groupement carboxylique
Une très grande variété de substrats peut être utilisée pour la fabrication de
colonnes échangeuses d’ions incluant les polymères d’esters, d’amides et des halogénures
d’alkyl. Les résines basées sur un copolymère de polystyrène et de divinylbenzène sont
les plus utilisées. Tel qu’illustré plus haut, la résine est essentiellement constituée de
polystyrène auquel on ajoute une quantité de divinylbenzène (DVB) pour la réticulation.
Cette réticulation confère à la résine sa stabilité mécanique et réduit considérablement sa
solubilité en augmentant le poids moléculaire de la chaîne du polymère. Dans bien des
cas, le nom de la résine indique le pourcentage de réticulation. Par exemple une colonne
Dowex 50X4 contient 4% de divinylbenzène.
Résines microporeuses et résines macroporeuses
Il y a deux types de résine utilisés pour la fabrication de colonnes échangeuses
d’ions : les résines microporeuses et les résines macroporeuses. Leurs propriétés
différentes proviennent du degré de réticulation et de leur procédé de fabrication. Pour les
résines microporeuses, on utilise de 2 à 25 % (typiquement 8 %) en poids de composé
réticulaire (ex : divinylbenzène) tandis que pour les résines macroporeuses, on utilise
jusqu’à 55 % de composé réticulaire.
Résine microporeuse
Après sa fabrication, la résine microporeuse est sous forme de billes uniformes et
solides mais microporeuses. La grosseur des billes dépend de la vitesse d’agitation lors de
la fabrication; une agitation plus rapide produit des billes plus petites. Après introduction
des groupements fonctionnels sur la résine, ces billes deviendront plus polaires. Un
désavantage de ce type de résine est que des solvants polaires comme l’eau la feront

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 97 of 145
gonfler alors que des solvants non polaires déshydrateront la résine et la feront rétrécir.
Ces résines microporeuses, souvent appelés « gels » lorsqu’elles ont un degré de
réticulation élevé, ont tendance à exclure les gros ions et la diffusion des ions de
dimensions ordinaires à travers le gel laisse à désirer. Les résines microporeuses sont
surtout utilisées comme support pour des colonnes constituées d’une pellicule de
gouttelettes de latex. Ce type de colonne est efficace et versatile. Nous verrons ces
colonnes plus loin.
Résine macroporeuse
La structure des billes de résine macroporeuse est formée d’une multitude de
microsphères interconnectées par des pores et des canaux. Parce que chaque sphère de
résine est constituée de milliers d'autres petites sphères (comme du maïs soufflé), la
surface des résines macroporeuses est beaucoup plus importante que la surface des
résines microporeuses. La surface spécifique d’un gel est de l’ordre de 1 m2/g tandis que
celle d’une résine macroporeuse peut être entre 25 et 800 m2/g.
Les résines macroporeuses sont remarquablement rigides à cause du haut degré de
réticulation. Elles sont particulièrement avantageuses lorsqu’on les utilise avec des
solvants organiques car elles ne gonfleront ni ne rétréciront avec la polarité du solvant.
Aussi elles ne font pas obstruction à la diffusion ou au procédé d’échange ionique comme
le font les gels car les ions peuvent pénétrer facilement leurs pores.
Le tableau ci-dessous présente un certain nombre de colonnes avec leur groupe
fonctionnel, la constitution de la résine, leurs noms commerciaux et la sélectivité de
différents ions
Les colonnes de résine de polystyrène possèdent une densité de charges tellement
importante que des macromolécules hautement chargées comme les protéines peuvent
être irréversiblement liées à la résine. Pour cela on a créé des colonnes dont les polymères
sont la cellulose ou le dextran, deux polymères du glucose. Le dextran réticulé avec la
glycérine (illustré ci-dessous) est vendu sous le nom commercial Sephadex. Parce que les
polymères de dextran et leurs proches parents sont beaucoup moins rigides que des
résines de polystyrène, on les appelle aussi des gels.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 98 of 145
Figure 3. Colonne de type Sephadex
D’autres échangeurs d’ions macro poreux sont basés sur des polysaccharides
d’agarose et des polyacrylamide. Des groupements fonctionnels sont liés au groupement
hydroxyle du polysaccharide. Pour les échangeurs de cations sur gel on utilise les
groupements acides suivants :
Sulfopropyl (SP) --OCH2CH2CH2SO3H acide fort
Sulfoéthyl (SE) --OCH2CH2SO3H
Phosphate (P) --OPO3H2 acide moy. fort
Carboxyméthyle (CM) --OCH2CO2H acide faible
Pour les échangeurs d’anions sur gel, on utilise les bases suivantes :
Triéthylaminoéthyl (TEAE) --OCH2CH2N+(CH2CH3)3 base forte
Diethylaminoethyl (DEAE) --OCH2CH2N(CH2CH3)2 moyennement forte
p-aminobenzyl (PAB) --OCH2—Benzyl—NH2 base faible
Ex : DEAE-Sephadex réfère à un échangeur d’anions Sephadex avec des
groupements diéthylaminoéthyl
Colonnes utilisées pour la séparation des sucres
Les groupements –OH des sucres se dissocient partiellement en anions O- en
milieu basique; on peut les considérer comme des acides faibles. Le pKa de plusieurs
sucres est de l’ordre de 12 à 12,5. Comme ces sucres sont ionisés à pH élevé, on peut les

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 99 of 145
séparer par un mécanisme d’échange d’anions. La compagnie Dionex a développé une
colonne dont la résine sulfonée consiste en des billes de 10 µm de diamètre
(éthylvinylbenzène réticulé avec 55 % de divinylbenzène) sur lesquelles on a associé une
couche de particules de latex de 460 nm qui contiennent plusieurs groupements
ammonium quaternaire. Ces dernières particules sont réticulées à 5 %. Pour une particule
de résine de 5 µm on a estimé qu’il y avait ~28 000 particules ou billes de latex. La
structure de cette résine est illustrée ci-dessous.
Figure 4. Schéma d’une colonne de type Dionex
Cette résine agglomérée de latex est très stable chimiquement. Même une solution
de NaOH 4 M ne parvient pas à scinder le lien entre la résine et la pellicule de latex. De
plus, le support donne une stabilité mécanique et une pression de retour (backpressure)
modérée. Le grand nombre de billes de latex et leur location sur le substrat permettent un
processus d’échange rapide et ainsi une bonne efficacité. La compressibilité est réduite au
minimum.
La sélectivité des colonnes échangeuses d’ions
Si on considère la compétition des ions Na+ et Li+ pour les sites sur une colonne
échangeuse de cations de résine R-, le coefficient de sélectivité est donné par :
La sélectivité de la résine de polystyrène tend à augmenter avec l’augmentation du
degré de réticulation, comme l’indique le tableau ci-dessous, parce que le diamètre des
pores de la résine diminue. Les ions comme Li+, avec un rayon hydraté assez large, n’ont
pas un accès aussi facile sur la résine que des ions hydratés ayant un plus petit rayon.
Table 1. Sélectivité selon la nature de la colonne et des ions
R-Na+ + Li+ → R-Li+ + Na+
LiNaR
NaLiRK
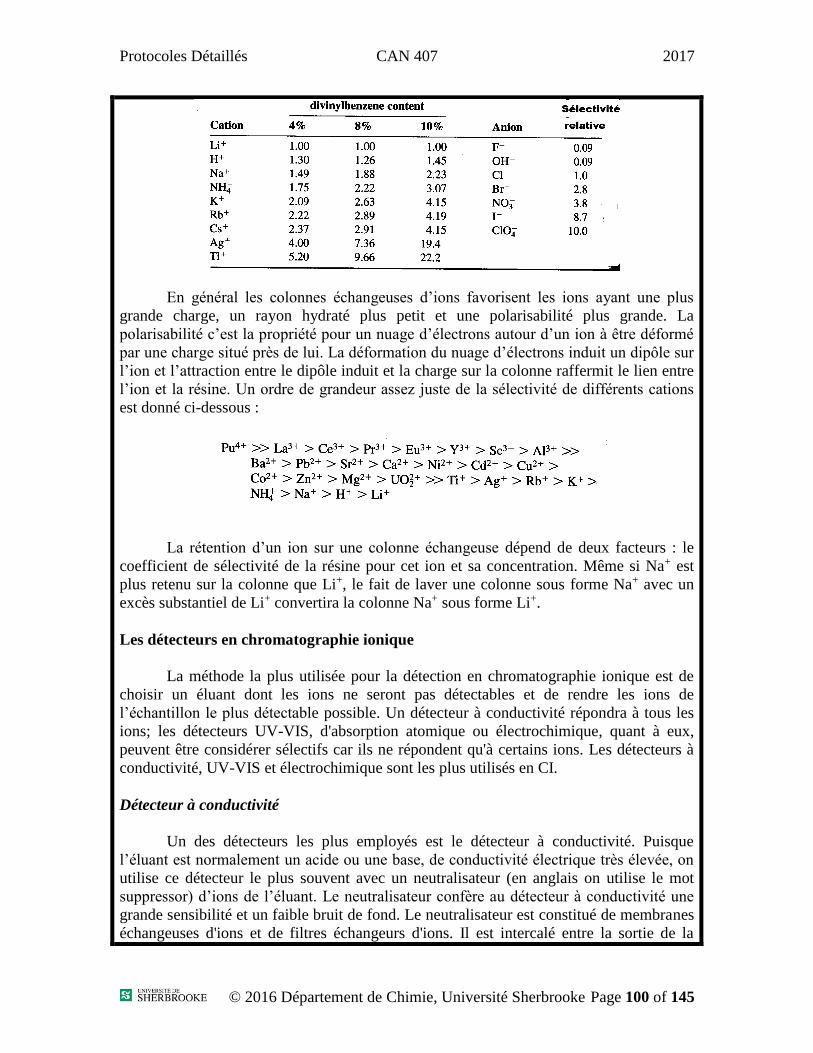
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 100 of 145
En général les colonnes échangeuses d’ions favorisent les ions ayant une plus
grande charge, un rayon hydraté plus petit et une polarisabilité plus grande. La
polarisabilité c’est la propriété pour un nuage d’électrons autour d’un ion à être déformé
par une charge situé près de lui. La déformation du nuage d’électrons induit un dipôle sur
l’ion et l’attraction entre le dipôle induit et la charge sur la colonne raffermit le lien entre
l’ion et la résine. Un ordre de grandeur assez juste de la sélectivité de différents cations
est donné ci-dessous :
La rétention d’un ion sur une colonne échangeuse dépend de deux facteurs : le
coefficient de sélectivité de la résine pour cet ion et sa concentration. Même si Na+ est
plus retenu sur la colonne que Li+, le fait de laver une colonne sous forme Na+ avec un
excès substantiel de Li+ convertira la colonne Na+ sous forme Li+.
Les détecteurs en chromatographie ionique
La méthode la plus utilisée pour la détection en chromatographie ionique est de
choisir un éluant dont les ions ne seront pas détectables et de rendre les ions de
l’échantillon le plus détectable possible. Un détecteur à conductivité répondra à tous les
ions; les détecteurs UV-VIS, d'absorption atomique ou électrochimique, quant à eux,
peuvent être considérer sélectifs car ils ne répondent qu'à certains ions. Les détecteurs à
conductivité, UV-VIS et électrochimique sont les plus utilisés en CI.
Détecteur à conductivité
Un des détecteurs les plus employés est le détecteur à conductivité. Puisque
l’éluant est normalement un acide ou une base, de conductivité électrique très élevée, on
utilise ce détecteur le plus souvent avec un neutralisateur (en anglais on utilise le mot
suppressor) d’ions de l’éluant. Le neutralisateur confère au détecteur à conductivité une
grande sensibilité et un faible bruit de fond. Le neutralisateur est constitué de membranes
échangeuses d'ions et de filtres échangeurs d'ions. Il est intercalé entre la sortie de la
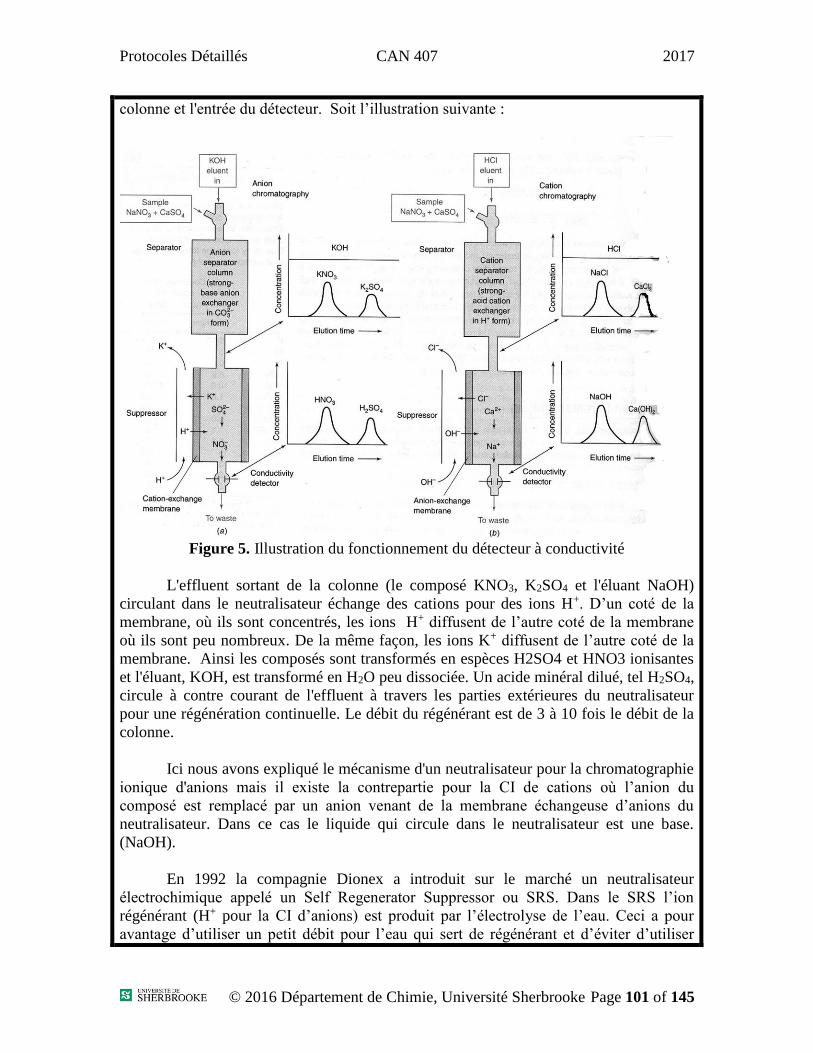
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 101 of 145
colonne et l'entrée du détecteur. Soit l’illustration suivante :
Figure 5. Illustration du fonctionnement du détecteur à conductivité
L'effluent sortant de la colonne (le composé KNO3, K2SO4 et l'éluant NaOH)
circulant dans le neutralisateur échange des cations pour des ions H+. D’un coté de la
membrane, où ils sont concentrés, les ions H+ diffusent de l’autre coté de la membrane
où ils sont peu nombreux. De la même façon, les ions K+ diffusent de l’autre coté de la
membrane. Ainsi les composés sont transformés en espèces H2SO4 et HNO3 ionisantes
et l'éluant, KOH, est transformé en H2O peu dissociée. Un acide minéral dilué, tel H2SO4,
circule à contre courant de l'effluent à travers les parties extérieures du neutralisateur
pour une régénération continuelle. Le débit du régénérant est de 3 à 10 fois le débit de la
colonne.
Ici nous avons expliqué le mécanisme d'un neutralisateur pour la chromatographie
ionique d'anions mais il existe la contrepartie pour la CI de cations où l’anion du
composé est remplacé par un anion venant de la membrane échangeuse d’anions du
neutralisateur. Dans ce cas le liquide qui circule dans le neutralisateur est une base.
(NaOH).
En 1992 la compagnie Dionex a introduit sur le marché un neutralisateur
électrochimique appelé un Self Regenerator Suppressor ou SRS. Dans le SRS l’ion
régénérant (H+ pour la CI d’anions) est produit par l’électrolyse de l’eau. Ceci a pour
avantage d’utiliser un petit débit pour l’eau qui sert de régénérant et d’éviter d’utiliser

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 102 of 145
d’autres sources chimiques comme les anciens systèmes. C’est ce système que nous
utiliserons dans nos expériences.
L’illustration ci-dessous décrit le mécanisme SRS en mode d’analyse des anions.
Les ions H+ générés par l’anode traversent la membrane échangeuse de cations pour
neutraliser l’éluant basique. Les contre ions Na+ sont attirés par la charge négative de la
cathode et traversent la membrane, pour maintenir l’électro-neutralité de la chambre
cathodique, en se pairant aux ions hydroxydes. L’oxygène du côté de l’anode et
l’hydrogène du côté de la cathode sont éliminés avec une solution aqueuse de NaOH.
Figure 6. Mécanisme SRS en mode d’analyse des ions
La chromatographie ionique sans neutralisation de l'anion ou du cation de l'éluent
(non-suppressed ion chromatography; abréviation NSIC) est possible à trois conditions :
1. L’utilisation d’une colonne échangeuse de cations ou d’anions à faible capacité
est obligatoire (0.007 à 0.04méq/g).
2. L’utilisation d’un éluant à faible concentration ionique et par le fait même de
faible conductivité.
3. les ions de l’éluant ont, d’une façon significative, une conductivité équivalente
plus faible que celle des ions analysés.
Les anions d’acide forts peuvent être séparés en solution acide ou basique. Les
anions d’acide faible, quant à eux, ne peuvent exister sous forme anionique qu’en
solution basique. La séparation des anions borate, silicate, sulfite ne peut se faire avec un
suppresseur car ces anions sont convertis en espèces faiblement conductrices. Cependant
ces anions peuvent être séparés par NSIC.
Même si un grand nombre d’anions organiques peuvent être séparé par « NSIC »,
cette méthode n’est pas toujours viable. Les anions d’acides organiques de petite taille
peuvent être détectés par conductivité mais il en est autrement lorsque la taille devient
plus grande. Dans ce cas, la conductivité approche celle de l’éluant et la sensibilité à la
détection disparaît.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 103 of 145
Détecteur UV-Visible
Le détecteur spectrophotométrique est très utile en chromatographie ionique. C'est
un détecteur sélectif et sa sélectivité peut être facilement changée en sélectionnant la
longueur d'onde. Les métaux alcalins ne sont pas détectés en UV. Cependant beaucoup
d'anions absorbent à de basses longueurs d'onde; en voici une liste.
Table 2. Solutés par détection spectrophotométrique directe après une séparation
par chromatographie ionique
Les composés aromatiques ont une bonne absorption dans l'UV et des méthodes
très puissantes existent pour détecter leurs ions.
L'utilité de la détection UV directe peut paraître limitée si on considère que les
sulfates ne sont pas détectés par UV et que les chlorures, phosphates et autres sont
difficiles à détecter (ces anions, par contre, peuvent facilement être détectés avec un
détecteur à conductivité). D'autre part les anions qui sont difficiles à détecter peuvent être
d'excellentes phases mobiles anioniques.
Détection spectrophotométrique indirecte
Dans cette méthode, on utilise une phase mobile qui absorbe dans l’UV et on
mesure la diminution de l’absorbance lorsque les ions transparents à l’UV de
l’échantillon éluent de la colonne et remplacent dans le détecteur les anions de l’éluant.
Cette méthode permet d’analyser les anions difficiles à détecter par spectrophotométrie
directe (chlorure, phosphate, sulfate, etc.). La figure ci-contre illustre la détection
spectrophotométrique d’anions transparents à l’UV (phase mobile le phtalate de sodium

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 104 of 145
2,0 mM à pH 6 et une détection indirecte à 280nm.
Dans la détection spectrophotométrique indirecte, la concentration de l’anion de
l’éluant doit être assez élevé pour éluer le composé d’intérêt, et doit excéder la
concentration de ce dernier au maximum du pic sans pour cela être trop élevé pour
augmenter considérablement le bruit de fond. En fait l’anion choisi pour la détection
indirecte doit avoir une grande affinité pour la colonne échangeuse d’ions pour pouvoir
l’utiliser à une concentration relativement basse.
L'absorbance des complexes métalliques de chlorure est utilisée pour détecter des
ions métalliques en CI. Les longueurs d'onde maximum d'absorption de ces complexes
métalliques de chlorure sont données ci-dessous :
Table 3. Formation de complexes d'anions inorganiques avec un réactif colorant
de perchlorate ferrique (0,8 M HClO4, 0,05 M Fe(ClO4)3
Ces anions sont détectés par une méthode post-colonne. Le réactif de perchlorate
ferrique est ajouté avec l’effluent, dans un réacteur, à la sortie de la colonne. Le complexe
de perchlorate ferrique est incolore mais beaucoup d'anions forment des complexes avec
le fer et forment des espèces colorées qui sont détectées.
Les détecteurs électrochimiques
Les détecteurs électrochimiques comprennent les détecteurs potentiométriques,
ampérométriques et conductométriques. Les détecteurs potentiométriques mesurent le
voltage, les détecteurs ampérométriques, le courant, et les détecteurs conductométriques,
la résistance (vus plus haut).
Les détecteurs potentiométriques opèrent sur le même principe que les électrodes
sélectives d’ions. Une électrode indicatrice mesure un changement de potentiel en
présence de certains ions. Une électrode d’argent recouverte d’un sel (ex. : AgCl,) peut
utiliser pour l’analyse des halogénures et les pseudos halogénures (CN-, SCN-, S2O3-,
etc.). Une électrode d’argent répond rapidement et d’une façon reproductible à l’activité
d’ions Ag+ en solution. À 25°C :

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 105 of 145
Si l’électrode d’argent est recouverte d’un solide légèrement soluble (AgX), aAg+
est déterminé par le produit de solubilité de AgX :
Ks = aAg+ . aX- aAg+ = Ks/aX-
E = E° + 0,0592 log Ks/aX-
Les détecteurs ampérométriques sont des détecteurs très sélectifs et très sensibles
utilisés dans la séparation par CI. Ils sont sélectifs parce qu’ils opèrent sur les principes
de l’oxydation et de la réduction des substances à l’électrode. Le potentiel nécessaire est
différent pour chaque ion, la sélectivité est contrôlée aussi par la grandeur du potentiel
appliquée à la cellule, le matériel utilisé pour l’électrode (ex. : or, platine) et le pH de la
solution
Ces détecteurs peuvent mesurer des nano ampères ce qui peut correspondre à des
concentrations de pico-équivalents. Ils ont un large domaine de réponse (3 à 4 ordre de
grandeur), un petit volume mort (aussi bas que 1 µL), ils sont simples, peu dispendieux et
fiables. Ils ont cependant certains désavantages : Ils sont sensibles au débit d’éluant et au
pH. Des traces de substances, tel l’oxygène, peuvent réagir dépendant du type d’électrode
et du potentiel appliqué. Les électrodes d’or sont moins sensibles que les électrodes de
platine à l’oxygène.
En principe n’importe laquelle espèce chimique peut être détecté avec le voltage
approprié. En pratique, le domaine de potentiel utilisable est limité entre –1,5V à +1,5V.
Dans la partie négative du domaine, le potentiel est limité par la réduction de l’oxygène
dissous dans la phase mobile et dans l’échantillon. Si l’oxygène a été retiré, on est quand
même limité par la réduction de l’eau en hydrogène. Dans la région positive du domaine
on est limité par l’hydrolyse de l’eau et la production d’oxygène.
Le détecteur à indice de réfraction
Ce détecteur peut être considéré comme un détecteur universel parce que
n’importe laquelle substance ajoutée à l’eau provoquera un changement de l’indice de
réfraction. Il a été utilisé autant pour la séparation des anions que pour les cations. Le
détecteur à indice de réfraction permet un grand éventail dans la sélection de l’éluant, du
pH, et de la force ionique. En principe ce détecteur peut remplacer un détecteur à
conductivité ou un détecteur UV dans plusieurs séparations. On a rapporté un seuil de
détection de 20 à 50 ng pour des anions communs comme les nitrates, les chlorures et les
sulfates, comparativement à un seuil de 1 à 5 ng avec un détecteur à conductivité.
Conditions opératoires pour la chromatographie ionique d’anions
Plusieurs paramètres peuvent être variés pour obtenir une bonne séparation dans
E = E° + 0,0592 log aAg+

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 106 of 145
un temps minimum.
Le choix de la résine Des changements dans la composition chimique peuvent
altérer la sélectivité. Par exemple un groupement éthanolamine au lieu d’un groupe
méthyl dans la fonction d’ammonium quaternaire augmente l’affinité de OH- pour la
colonne et fait en sorte que NaOH et KOH sont des phases mobiles plus fortes.
La capacité de la colonne Une capacité de colonne plus petite donnera une élution
plus rapide et permettra un éluant de plus faible concentration. Les ions divalents sont
plus affectés que les ions monovalents par ce changement. La capacité d’une résine peut
être définie comme étant le nombre de milliéquivalents de sites chargés par gramme de
résine sèche.
Le diamètre de la colonne Pour un même débit, l’utilisation d’une colonne de
diamètre plus petit donnera une élution plus rapide.
Force de la phase mobile
Les phases mobiles anioniques varient beaucoup dans leur habilité à éluer les
anions. Un anion avec une charge plus élevé tel que le carbonate possède un plus grand
pouvoir éluant qu’un ion monovalent. Voici une liste d’éluant utilisé avec la CI à
neutralisation d’anions.
Table 4. Liste d’éluant utilisé avec la CI à neutralisation d’anions
Un mélange de bicarbonate de sodium et de carbonate de sodium est souvent
utilisé. Le carbonate, avec une charge de –2, est un éluant plus fort que le bicarbonate.
En variant la proportion des deux on peut ajuster la force de l’éluant. Avec l’apparition de
colonne ayant une grande affinité pour l’anion OH- et des neutralisateurs tolérant une
plus grande concentration de l’éluant, l’utilisation de KOH ou NaOH devient plus
populaire.
La concentration de la phase mobile Une plus grande concentration de la phase
mobile anionique amène des temps de rétention plus courts.
L’utilisation de solvants organiques (ex. : méthanol, acétonitrile) dans la phase
mobile à un pourcentage de 10 à 20% peut assurer une plus grande solubilisation de
certains composants de l’échantillon. Une autre fonction des solvants organiques est
d’assurer la compatibilité entre les ions de l’échantillon et la colonne constituée souvent
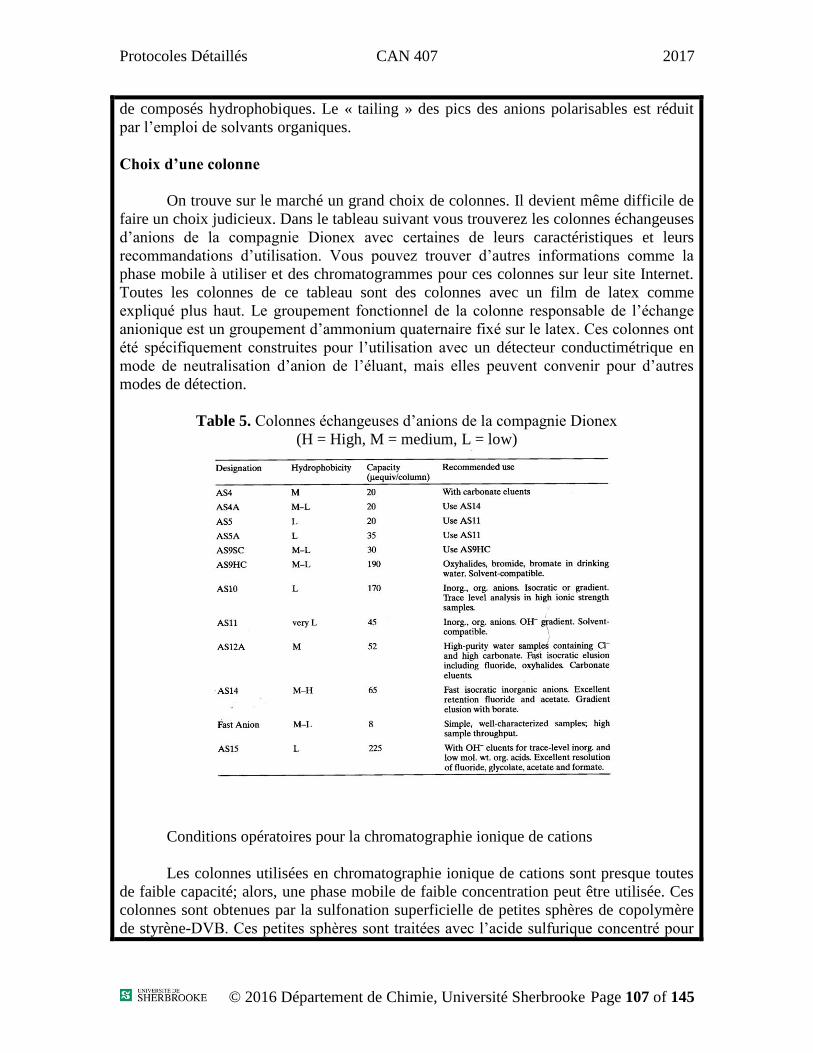
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 107 of 145
de composés hydrophobiques. Le « tailing » des pics des anions polarisables est réduit
par l’emploi de solvants organiques.
Choix d’une colonne
On trouve sur le marché un grand choix de colonnes. Il devient même difficile de
faire un choix judicieux. Dans le tableau suivant vous trouverez les colonnes échangeuses
d’anions de la compagnie Dionex avec certaines de leurs caractéristiques et leurs
recommandations d’utilisation. Vous pouvez trouver d’autres informations comme la
phase mobile à utiliser et des chromatogrammes pour ces colonnes sur leur site Internet.
Toutes les colonnes de ce tableau sont des colonnes avec un film de latex comme
expliqué plus haut. Le groupement fonctionnel de la colonne responsable de l’échange
anionique est un groupement d’ammonium quaternaire fixé sur le latex. Ces colonnes ont
été spécifiquement construites pour l’utilisation avec un détecteur conductimétrique en
mode de neutralisation d’anion de l’éluant, mais elles peuvent convenir pour d’autres
modes de détection.
Table 5. Colonnes échangeuses d’anions de la compagnie Dionex
(H = High, M = medium, L = low)
Conditions opératoires pour la chromatographie ionique de cations
Les colonnes utilisées en chromatographie ionique de cations sont presque toutes
de faible capacité; alors, une phase mobile de faible concentration peut être utilisée. Ces
colonnes sont obtenues par la sulfonation superficielle de petites sphères de copolymère
de styrène-DVB. Ces petites sphères sont traitées avec l’acide sulfurique concentré pour

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 108 of 145
former une mince couche de groupements d’acide sulfonique à la surface. La capacité
finale de la résine dépend de l’épaisseur de la couche, du type de résine, du diamètre des
sphères, de la température et du temps de contact avec l’acide sulfurique.
Table 6. Liste des différentes colonnes disponibles commercialement est présentée ici.
Depuis peu, la tendance dans la chromatographie de cations est d’utiliser des
résines avec des fonctions d’acides faibles. Un groupement carboxylique sera
complètement sous la forme anionique à des valeurs de pH plus élevés que son pKa. En
travaillant à des pH plus acides, l’ionisation du groupement carboxylique est diminuée et
l’affinité de la colonne pour l’échange d’ions est diminuée. Ainsi le contrôle du pH de
l’éluant ainsi que sa force ionique permet d’obtenir le degré de séparation voulu. La
colonne de Alltech « universal cation » contient de petites sphères enrobées d’un
copolymère de butadiène et d’acide maléique qui lui donne sa fonction d’échangeuse d
‘ions. L’acide maléique possède deux groupements carboxyliques par molécule pK1 =
2.0, pK2 = 6.3 et retient son caractère anionique à un des valeurs de pH plus acides qu’un
simple acide carboxylique.
Dans le tableau précédant il y a des colonnes avec des groupements fonctionnels
mixtes. Un groupement d’acide phosphorique possède un pKa intermédiaire entre un
acide carboxylique faible (pKa 4 à 5) et un groupement fortement acide tel l’acide
sulfonique. En incorporant un groupement carboxylate et un groupe phosphate, cela
permet de moduler l’efficacité de la colonne échangeuse d’ions en variant le pH sur un
plus grand domaine. En commençant à un pH de 5, une réduction du pH de l’éluant
permet de protonner d’abord l’acide carboxylique mais le groupement phosphate
demeure sous la forme anionique. En diminuant encore le pH le groupe phosphate se
protonne graduellement et l’efficacité de la colonne échangeuse d’ions est réduite.
Séparation des cations avec des phases mobiles ioniques
Dans ce type de séparation, les différents cations des composés compétitionnent
avec le cation de la phase mobile pour les sites de la colonne échangeuse d’ions et
avancent dans la colonne à différentes vitesses. L’éluant choisi dépend des cations à
séparer, du type de colonne et du détecteur. Dans plusieurs cas, l’emploi d’une solution
d’un acide fort tel l’acide chlorhydrique, sulfurique ou méthanesulfonique est satisfaisant.
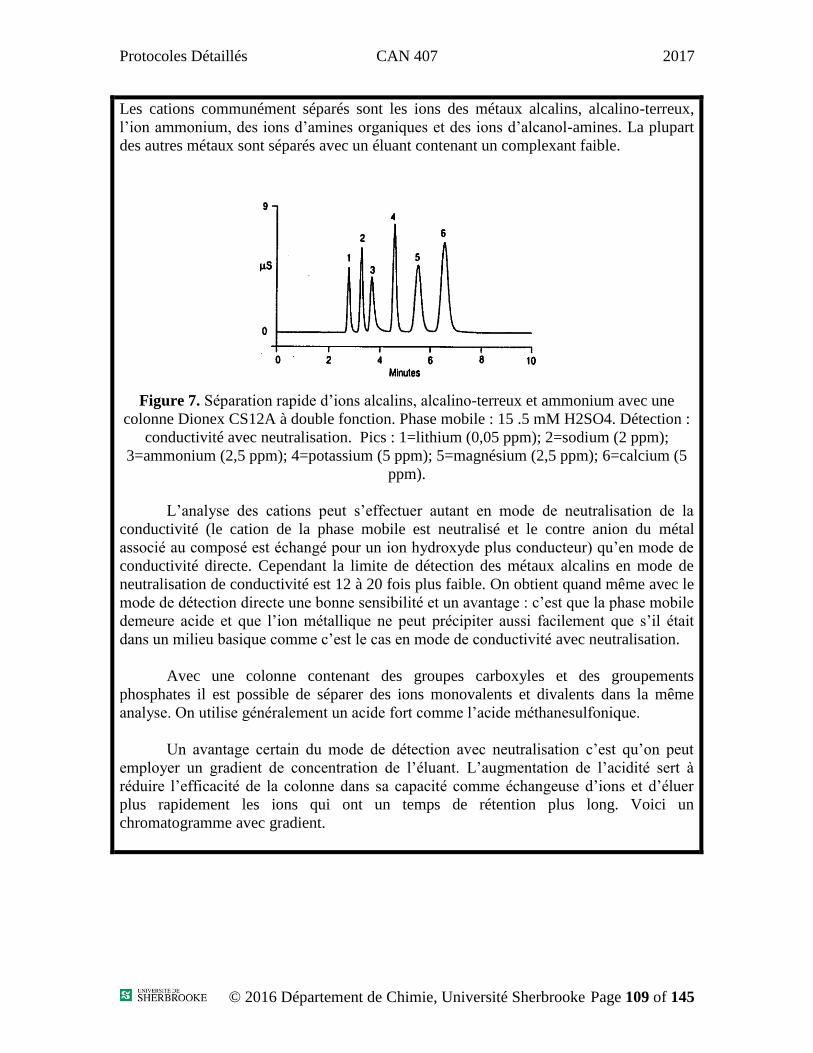
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 109 of 145
Les cations communément séparés sont les ions des métaux alcalins, alcalino-terreux,
l’ion ammonium, des ions d’amines organiques et des ions d’alcanol-amines. La plupart
des autres métaux sont séparés avec un éluant contenant un complexant faible.
Figure 7. Séparation rapide d’ions alcalins, alcalino-terreux et ammonium avec une
colonne Dionex CS12A à double fonction. Phase mobile : 15 .5 mM H2SO4. Détection :
conductivité avec neutralisation. Pics : 1=lithium (0,05 ppm); 2=sodium (2 ppm);
3=ammonium (2,5 ppm); 4=potassium (5 ppm); 5=magnésium (2,5 ppm); 6=calcium (5
ppm).
L’analyse des cations peut s’effectuer autant en mode de neutralisation de la
conductivité (le cation de la phase mobile est neutralisé et le contre anion du métal
associé au composé est échangé pour un ion hydroxyde plus conducteur) qu’en mode de
conductivité directe. Cependant la limite de détection des métaux alcalins en mode de
neutralisation de conductivité est 12 à 20 fois plus faible. On obtient quand même avec le
mode de détection directe une bonne sensibilité et un avantage : c’est que la phase mobile
demeure acide et que l’ion métallique ne peut précipiter aussi facilement que s’il était
dans un milieu basique comme c’est le cas en mode de conductivité avec neutralisation.
Avec une colonne contenant des groupes carboxyles et des groupements
phosphates il est possible de séparer des ions monovalents et divalents dans la même
analyse. On utilise généralement un acide fort comme l’acide méthanesulfonique.
Un avantage certain du mode de détection avec neutralisation c’est qu’on peut
employer un gradient de concentration de l’éluant. L’augmentation de l’acidité sert à
réduire l’efficacité de la colonne dans sa capacité comme échangeuse d’ions et d’éluer
plus rapidement les ions qui ont un temps de rétention plus long. Voici un
chromatogramme avec gradient.
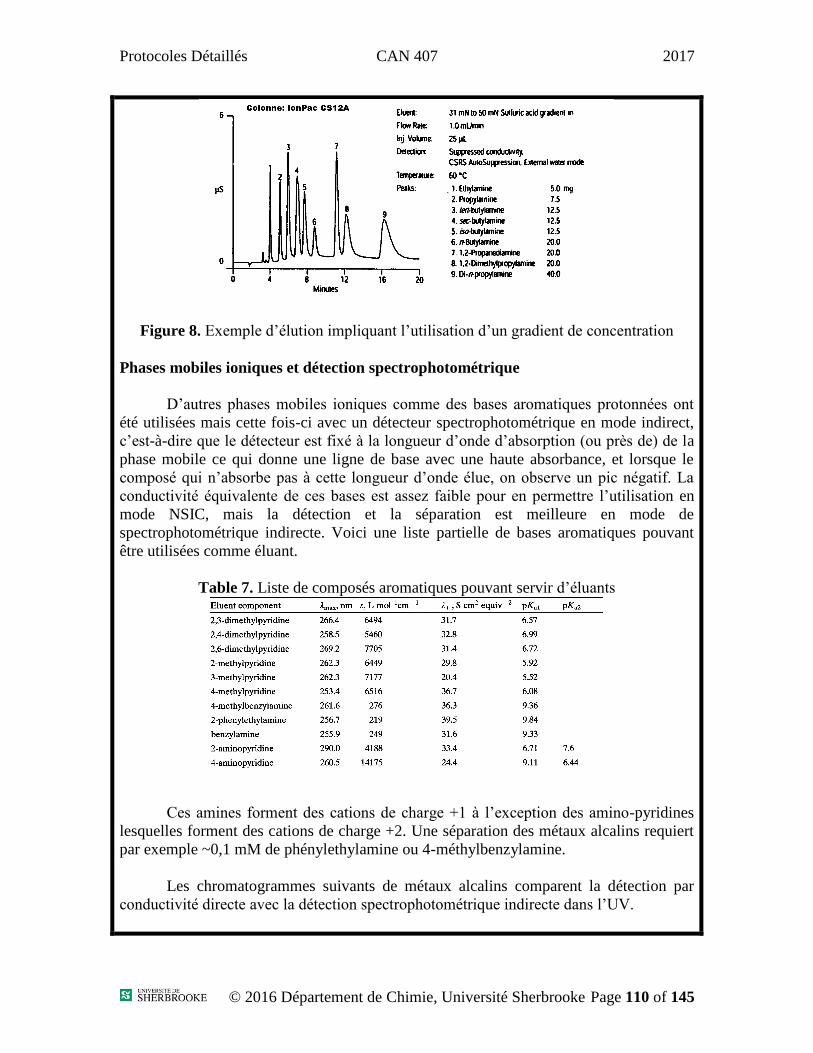
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 110 of 145
Figure 8. Exemple d’élution impliquant l’utilisation d’un gradient de concentration
Phases mobiles ioniques et détection spectrophotométrique
D’autres phases mobiles ioniques comme des bases aromatiques protonnées ont
été utilisées mais cette fois-ci avec un détecteur spectrophotométrique en mode indirect,
c’est-à-dire que le détecteur est fixé à la longueur d’onde d’absorption (ou près de) de la
phase mobile ce qui donne une ligne de base avec une haute absorbance, et lorsque le
composé qui n’absorbe pas à cette longueur d’onde élue, on observe un pic négatif. La
conductivité équivalente de ces bases est assez faible pour en permettre l’utilisation en
mode NSIC, mais la détection et la séparation est meilleure en mode de
spectrophotométrique indirecte. Voici une liste partielle de bases aromatiques pouvant
être utilisées comme éluant.
Table 7. Liste de composés aromatiques pouvant servir d’éluants
Ces amines forment des cations de charge +1 à l’exception des amino-pyridines
lesquelles forment des cations de charge +2. Une séparation des métaux alcalins requiert
par exemple ~0,1 mM de phénylethylamine ou 4-méthylbenzylamine.
Les chromatogrammes suivants de métaux alcalins comparent la détection par
conductivité directe avec la détection spectrophotométrique indirecte dans l’UV.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 111 of 145
Figure 9. Comparaison entre les modes de détection par conductivité et en mode
spectrophotométrique UV indirect.
Voici la limite de détection (LOD/(ppb)) calculée pour certaines phases mobiles
avec une détection par UV indirecte :
Table 8. LOD en ppb pour certaines phases mobiles pour détection UV indirecte.
Les solvants organiques comme phase mobile
En chromatographie ionique, il n’y a pas seulement un simple mécanisme
d’échange d’ions. Il y a aussi des interactions hydrophobes entre les cations de
l’échantillon et la résine qui entrent en jeu. Une bonne séparation dépend aussi bien des
différences dans l’attraction hydrophobes entre les ions du soluté et la colonne
échangeuse que des différences dans l’attraction électrostatique.
L’utilisation d’une phase organique permet à l’ion inorganique d’être solvaté par
des molécules organiques au lieu d’être solvaté par des molécules d’eau, ce qui rendrait
la colonne et le soluté plus compatible. En incorporant des solvants organiques dans
l’éluant, ceci a pour effet aussi d’augmenter la solubilité des solutés organiques. Il est
préférable de d’utiliser un mélange eau solvant organique comme éluant que d’utiliser
seulement un solvant organique. L’utilisation de solvants organiques tels le méthanol,
l’éthanol, le 2-propanol, et l’acétonitrile avec des résines macroporeuses permet des
séparations qui sont très difficiles à effectuer avec des solvants aqueux.
Séparation de cations avec des phases mobiles complexantes
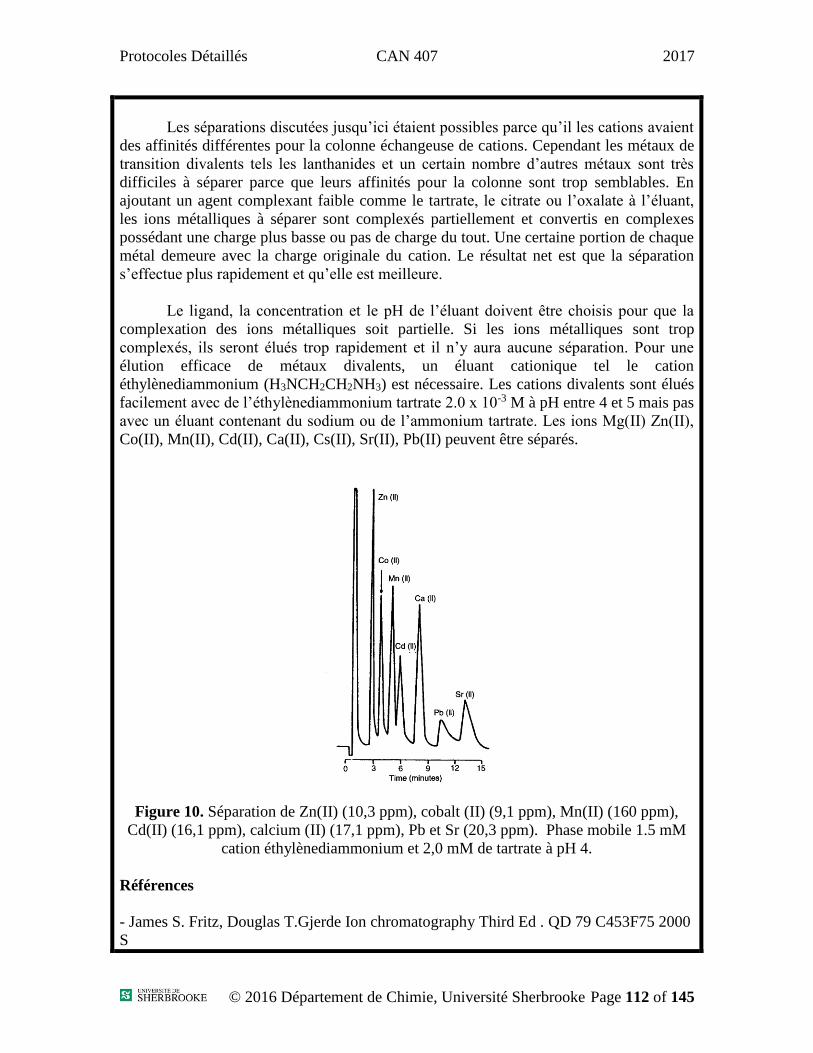
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 112 of 145
Les séparations discutées jusqu’ici étaient possibles parce qu’il les cations avaient
des affinités différentes pour la colonne échangeuse de cations. Cependant les métaux de
transition divalents tels les lanthanides et un certain nombre d’autres métaux sont très
difficiles à séparer parce que leurs affinités pour la colonne sont trop semblables. En
ajoutant un agent complexant faible comme le tartrate, le citrate ou l’oxalate à l’éluant,
les ions métalliques à séparer sont complexés partiellement et convertis en complexes
possédant une charge plus basse ou pas de charge du tout. Une certaine portion de chaque
métal demeure avec la charge originale du cation. Le résultat net est que la séparation
s’effectue plus rapidement et qu’elle est meilleure.
Le ligand, la concentration et le pH de l’éluant doivent être choisis pour que la
complexation des ions métalliques soit partielle. Si les ions métalliques sont trop
complexés, ils seront élués trop rapidement et il n’y aura aucune séparation. Pour une
élution efficace de métaux divalents, un éluant cationique tel le cation
éthylènediammonium (H3NCH2CH2NH3) est nécessaire. Les cations divalents sont élués
facilement avec de l’éthylènediammonium tartrate 2.0 x 10-3 M à pH entre 4 et 5 mais pas
avec un éluant contenant du sodium ou de l’ammonium tartrate. Les ions Mg(II) Zn(II),
Co(II), Mn(II), Cd(II), Ca(II), Cs(II), Sr(II), Pb(II) peuvent être séparés.
Figure 10. Séparation de Zn(II) (10,3 ppm), cobalt (II) (9,1 ppm), Mn(II) (160 ppm),
Cd(II) (16,1 ppm), calcium (II) (17,1 ppm), Pb et Sr (20,3 ppm). Phase mobile 1.5 mM
cation éthylènediammonium et 2,0 mM de tartrate à pH 4.
Références
- James S. Fritz, Douglas T.Gjerde Ion chromatography Third Ed . QD 79 C453F75 2000
S
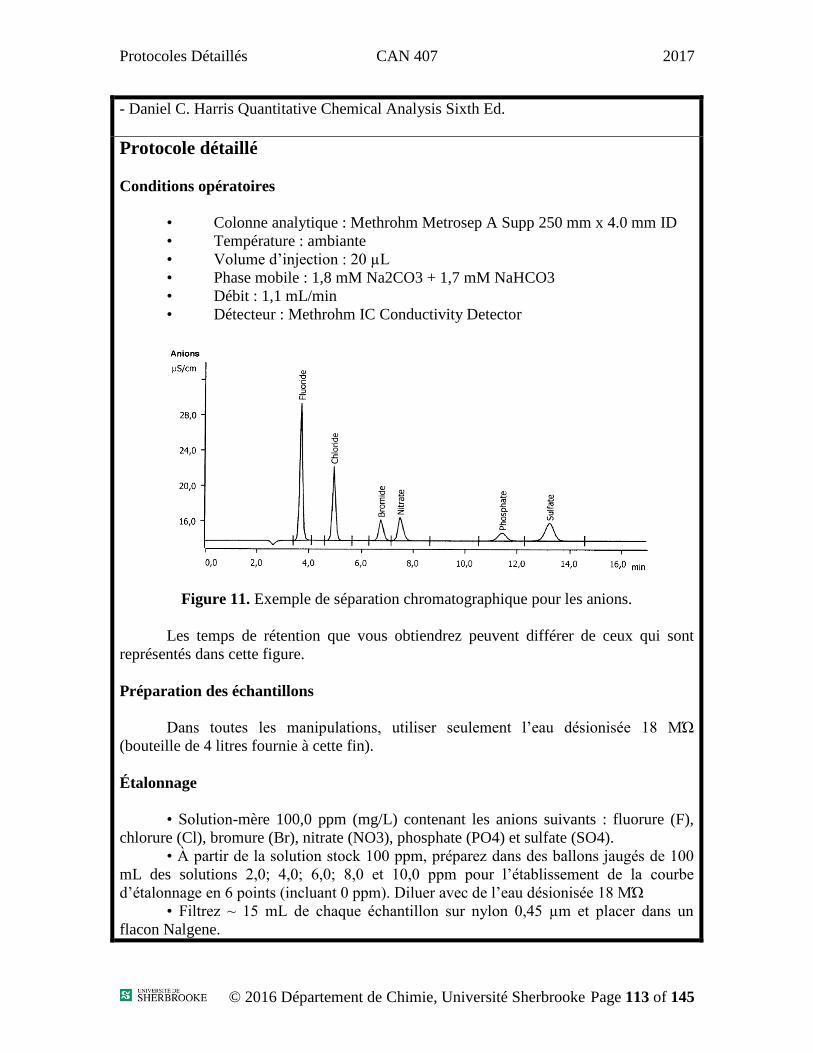
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 113 of 145
- Daniel C. Harris Quantitative Chemical Analysis Sixth Ed.
Protocole détaillé
Conditions opératoires
• Colonne analytique : Methrohm Metrosep A Supp 250 mm x 4.0 mm ID
• Température : ambiante
• Volume d’injection : 20 µL
• Phase mobile : 1,8 mM Na2CO3 + 1,7 mM NaHCO3
• Débit : 1,1 mL/min
• Détecteur : Methrohm IC Conductivity Detector
Figure 11. Exemple de séparation chromatographique pour les anions.
Les temps de rétention que vous obtiendrez peuvent différer de ceux qui sont
représentés dans cette figure.
Préparation des échantillons
Dans toutes les manipulations, utiliser seulement l’eau désionisée 18 MΏ
(bouteille de 4 litres fournie à cette fin).
Étalonnage
• Solution-mère 100,0 ppm (mg/L) contenant les anions suivants : fluorure (F),
chlorure (Cl), bromure (Br), nitrate (NO3), phosphate (PO4) et sulfate (SO4).
• À partir de la solution stock 100 ppm, préparez dans des ballons jaugés de 100
mL des solutions 2,0; 4,0; 6,0; 8,0 et 10,0 ppm pour l’établissement de la courbe
d’étalonnage en 6 points (incluant 0 ppm). Diluer avec de l’eau désionisée 18 MΏ
• Filtrez ~ 15 mL de chaque échantillon sur nylon 0,45 µm et placer dans un
flacon Nalgene.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 114 of 145
Échantillon inconnu (PRIORITÉ)
• Filtrez ~ 15 mL de l’inconnu sur nylon 0,45 µm et placer dans un flacon
Nalgene.
• Exprimez vos résultats en ppm pour chaque anion présent.
Eau potable de la ville de Sherbrooke
• Laissez couler l’eau froide du robinet environ 2 minutes avant d’en recueillir un
peu dans un bécher de 100 mL.
• Filtrez ~ 15 mL sur nylon 0,45 µm et placer dans un flacon Nalgene.
• Exprimez vos résultats en ppm pour chaque anion présent.
Eau provenant d’un lac ou d’une rivière (2 échantillons)
• Filtrez environ ~ 15 mL de chaque échantillon sur nylon 0,45 µm et placer dans
un flacon Nalgene.
• Exprimez vos résultats en ppm pour chaque anion présent.
Eau minérale embouteillée
• À partir des concentrations en anions indiquées sur l’étiquette, choisir les
dilutions pour que chaque anion à doser soit dans les mêmes concentrations que votre
courbe d’étalonnage. Si nécessaire, diluer avec l’eau désionisée 18 MΩ.
• Filtrer ~ 15 mL de chaque solution sur nylon 0,45 µm dans un flacon Nalgene.
• Exprimer vos résultats en ppm pour chaque cation présent.
Rince-bouche Listerine
• Bien agiter la bouteille avant les dilutions.
• Dans un ballon jaugé, préparer une dilution de 20X avec de l’eau désionisée 18
MΩ pour le dosage du fluorure et 2000X pour le dosage du nitrate.
• Filtrez ~ 15 mL de chaque solution sur nylon 0,45 µm et placer dans un flacon
Nalgene.
• Exprimez vos résultats en pourcentage (% p/v) de fluorure de sodium (NaF) et
de nitrate de potassium (KNO3). Comparer vos résultats avec les valeurs de l’étiquette.
Note : Le mode opératoire de l’instrument vous sera fourni par le démonstrateur
Calculs - Déterminez la teneur en ion de chaque solution. Assurez-vous au minimum d’avoir le
résultat en triplicatas pour l’inconnue, afin de faire une étude statistique.
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 115 of 145
Notes supplémentaires pour la discussion
- Démontrez que vous comprenez bien le principe de la chromatographie ionique.
- Proposez d’autres approches analytiques pour déterminer ou contrevérifier les valeurs
déterminées.
- Comparez vos valeurs avec la littérature disponible.
Information Supplémentaire

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 116 of 145
Expérience 9 : Fluorescence et Spectroscopie UV d’absorption

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 117 of 145
EXPÉRIENCE 9 : FLUORESCENCE ET SPECTROSCOPIE UV D’ABSORPTION
Théorie (Cette section n’est qu’un bref résumé et la consultation d’ouvrages de référence
supplémentaire est nécessaire)
La fluorescence est l’émission d’un rayonnement à une longueur d’onde plus
élevée, donc moins énergétique que celle du rayon incident absorbé par une molécule. La
majorité des molécules possèdent un nombre pair d’électrons et à l’état fondamental, ils
sont regroupés par paires et chaque paire se retrouve sur une orbitale moléculaire. Deux
électrons ne peuvent occuper une même orbitale que s’ils sont pairés, c’est-à-dire que
leurs champs magnétiques, associés à leur rotation, sont de sens inverse. Lorsque dans
une molécule les spins des électrons sont pairés, ils sont dans un état singulet dénommé
S0, S1, etc.; alors que si les électrons ne sont pas pairés, on dit qu’ils sont dans un état
triplet. Ces états sont appelés triplets parce que lorsqu’ils sont soumis à un champ
magnétique, ils se séparent en 3 sous-états d’énergie légèrement différente. Le plus bas
état triplet s’appelle T1. Pour chaque état singulet, il y a un état triplet.
La figure suivante (Skoog), appelé diagramme de Jablonski, illustre des
transitions de niveaux d’énergie électroniques et de vibration lorsqu’il y a absorption
d’un photon. Les lignes foncées représentent les niveaux vibrationnels fondamentaux des
niveaux électroniques, alors que les lignes plus fines représentent les niveaux d’énergie
vibrationnelle.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 118 of 145
Figure 1. Diagramme de Jablonski résumant les différents processus en spectroscopie
moléculaire. (NOTE : Comprendre et assimiler cette figure est essentiel à votre
apprentissage en chimie)
À la température de la pièce, la grande majorité des molécules se trouvent sur le
plus bas niveau d’énergie électronique et vibrationnel, habituellement un état singulet S0
(note : l’état fondamental de O2 est un état triplet). Lorsqu’une molécule absorbe un
photon, un électron d’une de ses orbitales moléculaires est promu à un niveau
électronique et vibrationnel supérieur, tel qu’illustré sur la figure ci-haut, où la longueur
des flèches est proportionnelle à l’énergie. On dit alors que la molécule est dans un état
excité. L’électron promu peut posséder un spin opposé (comme sur le niveau d’où il est
parti) ou un spin parallèle (dans le même sens) dans lequel cas nous avons un état triplet.
L’excitation de molécules, à partir du niveau fondamental, qui implique des
transitions S0→S1, S0→S2, S0→S3 ne peut s’effectuer que par l’absorption de radiation de
longueur d’onde progressivement plus courte (plus énergétique). La transition d’un
niveau électronique fondamental à un état excité triplet est très peu probable; c’est la
raison pour laquelle cette transition n’est pas représentée sur le diagramme. Les
molécules ne peuvent accéder à un état triplet, à partir d’un état singulet, qu’en passant
par un procédé qu’on appelle conversion externe (inter-system crossing) ou dans certains
cas, en passant par un autre processus telle une réaction chimique. Une transition triplet

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 119 of 145
→ singulet, même si elle est beaucoup moins probable qu’une transition singulet →
singulet, va quand même se produire pour abaisser l’énergie de la molécule. C’est ce
qu’on appelle de la phosphorescence. Les temps caractéristiques des phénomènes sont
intéressants : transition par absorption d’un photon ~ 10-15 s; émission par fluorescence ~
10-8 s et émission par phosphorescence, beaucoup moins probable, de 10-4 s à plusieurs
heures.
Illustrons les différences entre les niveaux d’états électroniques à l’aide de
diagrammes représentant les orbitales moléculaires et la molécule d’acétone. La
molécule d’acétone possède un système π dû à la présence d’un lien double C=O et d’une
paire d’électrons libres sur l’atome d’oxygène. Le premier diagramme illustre la
configuration pour le niveau fondamental S0. Les orbitales occupées (2 électrons) sont
l’orbitale σ (liante), l’orbitale π (liante) et l’orbitale n (non-liante) en ordre croissant
d’énergie. Les orbitales inoccupées sont les orbitales π* et σ* (deux orbitales anti-
liantes).
Figure 2. Illustration des orbitales moléculaires pour la molécule d’acétone
À l’état excité, les deux électrons occupant des orbitales différentes peuvent
posséder des spins parallèles, c’est-à-dire s’aligner dans la même direction.
Figure 3. Illustration de l’état excité avec des spins parallèles
Les configurations des états triplets T1 et T2 correspondant aux états S1 et S2 sont
les suivantes :

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 120 of 145
Figure 4. Configurations triplet, T1 etT2 correspondant à S1 et S2
Chez les molécules organiques, il y a deux types de transitions importantes. Ce
sont les transitions n→π* et π→π*. Pour l’acétone, la transition n→π* implique une des
paires d’électrons libres sur l’atome d’oxygène situé sur une orbitale non-liante. La
transition π→π* implique une des paires d’électrons de l’orbitale π du groupement C=O.
Pour les molécules simples, comme l’acétone, la transition n→π* est de plus
basse énergie mais pour les molécules plus complexes comme dans les cétones
aromatiques, qui possèdent plusieurs orbitales π liantes et anti-liantes, ce n’est pas
toujours le cas.
D’habitude, dans l’ultraviolet, le mode d’absorption, nous observons pour les
liquides une bande large avec un contour régulier. La perte de la structure fine du spectre
est causée surtout par les interactions entre les molécules de la phase liquide. Ceci cause
un petit décalage dans les niveaux d’énergie des molécules individuelles, tout
particulièrement si la molécule est polaire et qu’elle est dissoute dans un solvant polaire.
Cependant, avec certaines molécules, il est possible d’observer les bandes
d’absorption pour chacune des transitions comme l’illustre le spectre du benzène sur la
figure ci-dessous. L’intensité des pics est directement proportionnelle à la probabilité de
la transition.
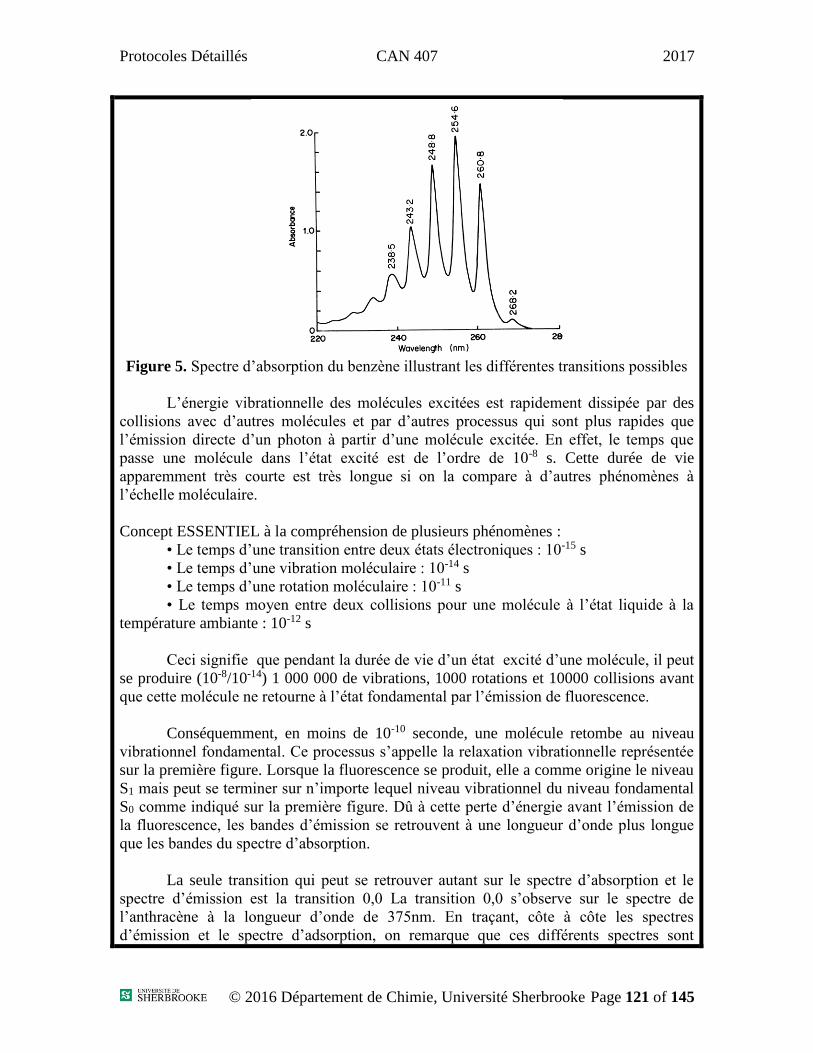
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 121 of 145
Figure 5. Spectre d’absorption du benzène illustrant les différentes transitions possibles
L’énergie vibrationnelle des molécules excitées est rapidement dissipée par des
collisions avec d’autres molécules et par d’autres processus qui sont plus rapides que
l’émission directe d’un photon à partir d’une molécule excitée. En effet, le temps que
passe une molécule dans l’état excité est de l’ordre de 10-8 s. Cette durée de vie
apparemment très courte est très longue si on la compare à d’autres phénomènes à
l’échelle moléculaire.
Concept ESSENTIEL à la compréhension de plusieurs phénomènes :
• Le temps d’une transition entre deux états électroniques : 10-15 s
• Le temps d’une vibration moléculaire : 10-14 s
• Le temps d’une rotation moléculaire : 10-11 s
• Le temps moyen entre deux collisions pour une molécule à l’état liquide à la
température ambiante : 10-12 s
Ceci signifie que pendant la durée de vie d’un état excité d’une molécule, il peut
se produire (10-8/10-14) 1 000 000 de vibrations, 1000 rotations et 10000 collisions avant
que cette molécule ne retourne à l’état fondamental par l’émission de fluorescence.
Conséquemment, en moins de 10-10 seconde, une molécule retombe au niveau
vibrationnel fondamental. Ce processus s’appelle la relaxation vibrationnelle représentée
sur la première figure. Lorsque la fluorescence se produit, elle a comme origine le niveau
S1 mais peut se terminer sur n’importe lequel niveau vibrationnel du niveau fondamental
S0 comme indiqué sur la première figure. Dû à cette perte d’énergie avant l’émission de
la fluorescence, les bandes d’émission se retrouvent à une longueur d’onde plus longue
que les bandes du spectre d’absorption.
La seule transition qui peut se retrouver autant sur le spectre d’absorption et le
spectre d’émission est la transition 0,0 La transition 0,0 s’observe sur le spectre de
l’anthracène à la longueur d’onde de 375nm. En traçant, côte à côte les spectres
d’émission et le spectre d’adsorption, on remarque que ces différents spectres sont
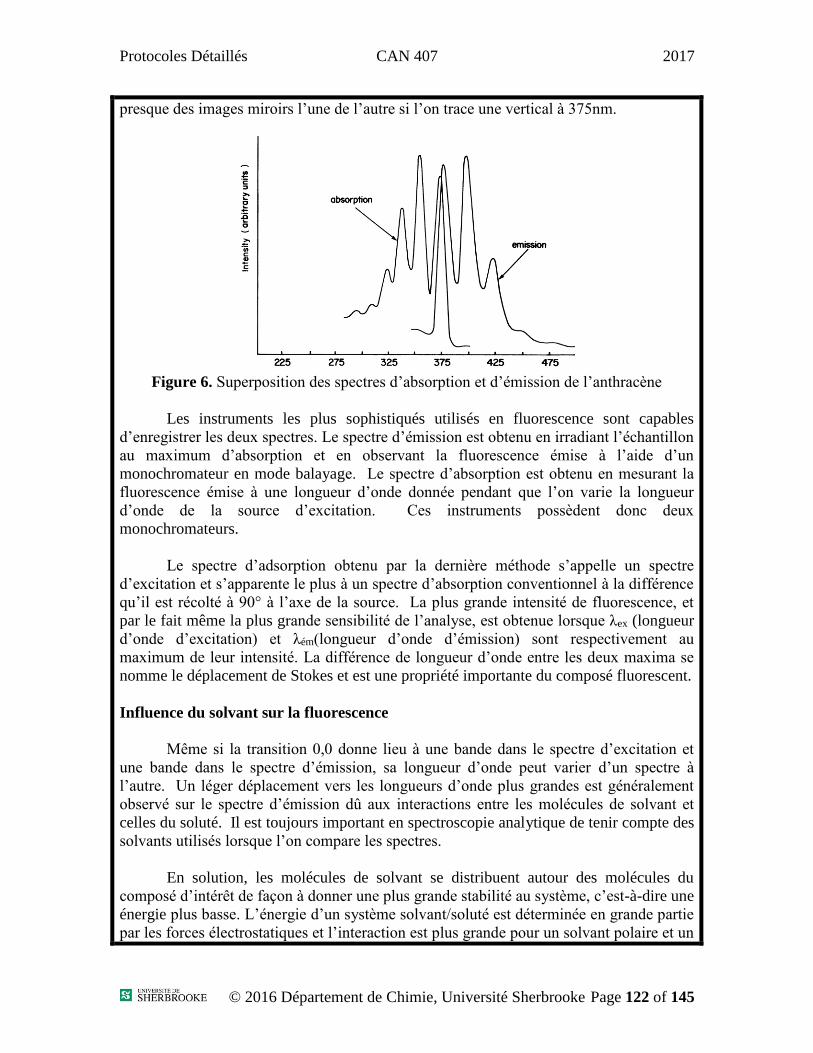
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 122 of 145
presque des images miroirs l’une de l’autre si l’on trace une vertical à 375nm.
Figure 6. Superposition des spectres d’absorption et d’émission de l’anthracène
Les instruments les plus sophistiqués utilisés en fluorescence sont capables
d’enregistrer les deux spectres. Le spectre d’émission est obtenu en irradiant l’échantillon
au maximum d’absorption et en observant la fluorescence émise à l’aide d’un
monochromateur en mode balayage. Le spectre d’absorption est obtenu en mesurant la
fluorescence émise à une longueur d’onde donnée pendant que l’on varie la longueur
d’onde de la source d’excitation. Ces instruments possèdent donc deux
monochromateurs.
Le spectre d’adsorption obtenu par la dernière méthode s’appelle un spectre
d’excitation et s’apparente le plus à un spectre d’absorption conventionnel à la différence
qu’il est récolté à 90° à l’axe de la source. La plus grande intensité de fluorescence, et
par le fait même la plus grande sensibilité de l’analyse, est obtenue lorsque λex (longueur
d’onde d’excitation) et λém(longueur d’onde d’émission) sont respectivement au
maximum de leur intensité. La différence de longueur d’onde entre les deux maxima se
nomme le déplacement de Stokes et est une propriété importante du composé fluorescent.
Influence du solvant sur la fluorescence
Même si la transition 0,0 donne lieu à une bande dans le spectre d’excitation et
une bande dans le spectre d’émission, sa longueur d’onde peut varier d’un spectre à
l’autre. Un léger déplacement vers les longueurs d’onde plus grandes est généralement
observé sur le spectre d’émission dû aux interactions entre les molécules de solvant et
celles du soluté. Il est toujours important en spectroscopie analytique de tenir compte des
solvants utilisés lorsque l’on compare les spectres.
En solution, les molécules de solvant se distribuent autour des molécules du
composé d’intérêt de façon à donner une plus grande stabilité au système, c’est-à-dire une
énergie plus basse. L’énergie d’un système solvant/soluté est déterminée en grande partie
par les forces électrostatiques et l’interaction est plus grande pour un solvant polaire et un

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 123 of 145
composé polaire. Ceci conduit à une plus grande différence entre la configuration la plus
stable et la moins stable.
Lorsqu’une molécule subit un changement de niveau électronique suite à une
excitation, elle subit aussi un changement significatif dans sa polarité. Conséquemment,
la distribution des molécules du solvant, qui donne la configuration la plus stable lorsque
la molécule est au niveau fondamental S0, n’est pas nécessairement celle qui minimise
l’énergie de la molécule lorsqu’elle est dans un état excité. Le processus d’absorption est
virtuellement instantané si on le compare au mouvement moléculaire et les molécules de
solvant s’ajustent par la suite pour fournir la configuration la plus stable à l’état excité,
avec une légère réduction de l’énergie. C’est à partir de ce niveau électronique que la
molécule retourne à l’état fondamental et où l’énergie du système est au-dessus du
minimum car la distribution des molécules du solvant correspond à la situation d’un état
excité. Par la suite, un ajustement dans la distribution des molécules permet de réduire
l’énergie à la valeur à laquelle l’excitation originale s’est produite, ce qui fait que le
photon émis est de plus basse énergie que le photon excitant et que la transition apparaît à
une longueur d’onde supérieure sur le spectre d’émission. Le diagramme suivant illustre
ce phénomène.
Figure 7. Illustration de l’effet du solvant
Fluorescence et structure moléculaire
Avant qu’une molécule puisse émettre une radiation par fluorescence, elle doit
être capable d’absorber la radiation. Par contre, ce ne sont pas toutes les molécules qui
absorbent la radiation qui peuvent émettre de la fluorescence. Dans un premier temps,
établissons une méthode qui quantifiera jusqu’à quel point une molécule sera
fluorescente. On définit Φf, le rendement quantique de fluorescence comme étant :
Les valeurs de Φf sont comprises entre 0 et 1; Φf est largement déterminée par la

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 124 of 145
structure de la molécule. Une valeur élevée de Φf est généralement associée à des
molécules possédant un système délocalisé de liens doubles conjugués, ce qui confère
une rigidité à la molécule. Exemple : fluorescéine ou anthracène.
Si une molécule absorbe de la radiation et n’émet pas de fluorescence, c’est
qu’elle a disposée de l’excès d’énergie d’une autre façon en retournant à l’état
fondamental. Elle peut se décomposer en brisant ses liens chimiques parce qu’elle a reçu
trop de radiation ou elle a disposé de cette énergie par un autre mécanisme sans émission
de radiation.
Transfert d’énergie sans émission de rayonnement
La fluorescence et la relaxation vibrationnelle sont deux mécanismes de
désactivation de l’état excité. L’un est rayonnant et l’autre non. Le chemin de retour à
l’état fondamental le plus favorable est celui qui minimise la durée de vie de l’état excité.
Le mécanisme de relaxation vibrationnelle (par collision entre les molécules excitées et
celle du solvant) est si efficace que la durée de vie d’un état vibrationnel excité est de 10-
12 seconde. C’est une durée nettement plus courte que celle d’un état électronique excité.
L’efficacité de la relaxation vibrationnelle est telle que la bande de fluorescence
correspondant à une transition électronique donnée est déplacée vers des longueurs
d’ondes plus élevées.
Il y a d’autres mécanismes non rayonnants de désactivation de l’état excité
comme la conversion interne, la conversion externe et le changement de multiplicité. Ces
mécanismes sont illustrés sur la première figure. Ces processus de désactivation ne sont
encore bien compris et bien définis mais ils expliquent certains phénomènes observés en
fluorescence.
La conversion interne
La conversion interne, comme mécanisme de désactivation, semble
particulièrement efficace quand deux niveaux énergétiques électroniques sont
suffisamment voisins pour que les niveaux d’énergie vibrationnelle se recouvrent comme
illustré sur la première figure. Sur cette figure, les niveaux vibrationnels de S1 sont
voisins de l’autre état singulet S2 ce qui permet une transition efficace.
Un exemple de cette conversion interne est celle de la quinine où deux longueurs
d’onde d’excitation, celle de 250 nm et celle de 350 nm résultent en une seule longueur
d’onde d’émission, celle de 450 nm. C’est comme si après l’absorption du rayonnement
à 250 nm, le plus énergétique des deux rayonnements d’excitation, la molécule se
retrouvant au niveau S2 (comme sur la figure) empruntait le chemin de la conversion
interne (indiqué par la flèche ondulée) et par la suite, après relaxation vibrationnelle, se
retrouvait au niveau S1 pour enfin émettre la fluorescence. Cette absorption à 250 nm se
termine par la même émission à 450 nm qu’une absorption à 350 nm qui exciterait la
molécule à un niveau vibrationnel excité de S1 et qu’après relaxation vibrationnelle se
retrouve au premier niveau de S1.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 125 of 145
Le mécanisme de la conversion interne peut aussi se produire dans le sens inverse,
c’est-à-dire de S1 à S0 (de la première figure). Une illustration est donnée sur la figure
suivante :
Figure 8. Principe de la conversion interne
Ce dernier processus est encore mal compris mais expliquerait le fait que les
composés aliphatiques sont rarement fluorescents.
La conversion externe
La conversion externe, ou désactivation par collision, résulterait d’une interaction
et d’un transfert d’énergie entre la molécule excitée et le solvant ou autres molécules. Ce
mécanisme est appuyé par l’effet marqué du solvant sur l’intensité de la fluorescence
ainsi que par des changements dans les conditions physico-chimiques, telles la baisse de
température ou l’augmentation de la viscosité du solvant, qui tendent à renforcer la
fluorescence en diminuant le nombre de collisions. Les détails sont encore ici mal
compris.
Changement de multiplicité
« Le changement de multiplicité est un processus par lequel le spin d’un électron
excité est inversé. Comme pour la conversion interne, la probabilité de cette transition est
renforcée par le recouvrement des niveaux de vibration des deux états », tel qu’illustré
sur le dernier diagramme. « Dans ce cas, le niveau vibrationnel le plus bas de l’état
singulet recouvre l’un des niveaux vibrationnels supérieurs de l’état triplet, et un
changement de spin est donc plus probable. Le changement de multiplicité se rencontre
surtout dans les molécules qui contiennent des atomes lourds, l’iode ou le brome (effet
d’atome lourd). En présence de tels atomes, les interactions spin-orbitale deviennent
importantes ce qui favorise un changement de spin. La présence d’espèces
paramagnétiques en solution, telles que l’oxygène moléculaire, favorise aussi le
changement de multiplicité et diminue donc la fluorescence. » (Skoog p.360)

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 126 of 145
Lire le manuel de Skoog à la page 360-361-362 les paragraphes dont les titres
sont :
« Fluorescence et structure » et « Effet de la rigidité structurale ».
Effets de la température et du solvant (important!!!)
Voir Skoog, Principes d’analyse instrumentale, page 363 (traduction 5e édition)
Effet du pH sur la fluorescence (important!!!)
Voir Skoog, Principes d’analyse instrumentale, page 363 (traduction 5e édition).
Relation entre la fluorescence et la concentration
On peut démontrer que l’intensité de la fluorescence pour des solutions diluées est
donnée par :
où If réfère à l’intensité totale dans toutes les directions ce qui signifie que la
partie de droite doit être multipliée par un facteur pour tenir compte qu’il n’y a qu’une
fraction du rayonnement fluorescent qui est détectée. Si la concentration est élevée, cette
relation ne tient plus et on arrive plutôt à une équation qui inclut un terme quadratique et
un terme cubique :
Le terme au carré possède un signe négatif et va réduire If à mesure que la
concentration augmente. Le terme au cube a un signe positif et réduira alors la courbure
mais seulement à des concentrations beaucoup plus élevées. À condition que 2,303 ε cd =
A soit inférieur à 0,05, on peut négliger les termes de puissance supérieure à 1. Dans ce
cas, l’erreur relative ne dépasse pas 0,13 %.
Si on prend un produit fluorescent dont l’absorptivité molaire ε est de 10000 L
mol-1cm-1 (ex :anthracène) et que l’on utilise une cellule dont le chemin optique est de 1
cm et que l’on met en graphique la fluorescence relative If/I0 avec I0 et Φf = 1 (permis
pour un produit donné, mesuré sur le même instrument et à une longueur d’onde fixe) en
fonction de la concentration, en utilisant la dernière équation, nous obtenons pour des
concentrations de 0 à 30 µmol/L le graphique ci-dessous. Nous constatons que la droite
s’éloigne de la linéarité à partir de~3 µmol/L et que le terme cubique devient significatif
à partir de 14 µmol/L. Si le poids moléculaire de ce produit est de 300 nous obtenons
0,720 mg/L comme concentration limite pour la linéarité. Donc, si on veut effectuer une
droite d’étalonnage pour un tel produit, nous devons travailler avec des solutions très
diluées et en-deçà de 3 µmol/L.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 127 of 145
Figure 9. Déviation de la linéarité à fortes concentrations
Peut-on observer la fluorescence à ces concentrations? Ceci dépend de la
sensibilité de notre instrument qui en retour dépend de I0, de l’efficacité optique
d’émission et du détecteur. Heureusement, pour des composés ayant des efficacités
raisonnables de fluorescence cette concentration est de plusieurs ordres au-dessus de la
limite de détection.
Comparaison de la sensibilité et du domaine dynamique entre spectroscopie
d’absorption et la fluorescence
Sensibilité
En spectroscopie d’absorption, la limite de détection est souvent définie comme
étant la concentration qui donne une valeur d’absorbance égale au double du bruit de
fond. La limite supérieure de linéarité, pour une courbe d’étalonnage en fluorescence, est
d’environ 10 fois supérieure à la limite de détection dans l’UV/visible mais la limite de
détection en fluorescence est de 1000 plus basse.
Le domaine dynamique
Le domaine dynamique d’une technique analytique est la concentration la plus
élevée que nous pouvons mesurer, divisée par la concentration la plus basse, sous des
conditions de routine (10 fois la limite de détection est une valeur raisonnable). En
général, pour les techniques d’absorption, ce ratio se limite à 100. La fluorescence, en
commun avec d’autres techniques d’émission, a un domaine dynamique de l’ordre de 10
000.
L’auto-désactivation et l’auto-absorption
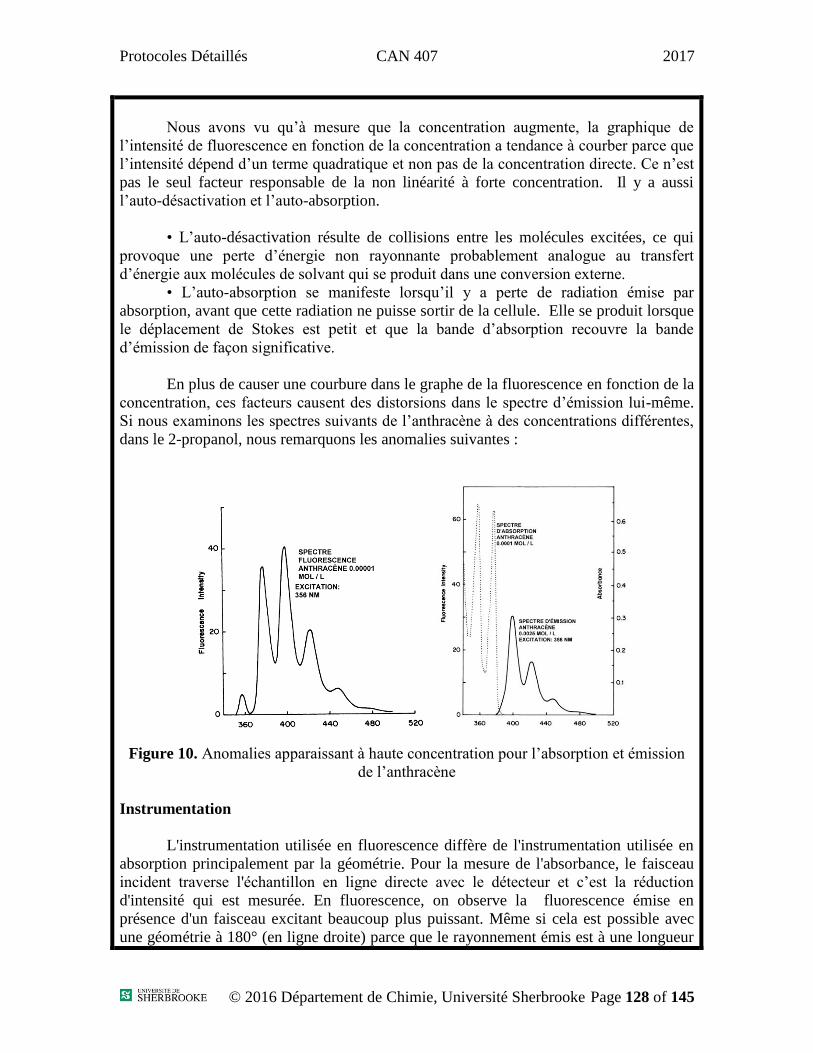
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 128 of 145
Nous avons vu qu’à mesure que la concentration augmente, la graphique de
l’intensité de fluorescence en fonction de la concentration a tendance à courber parce que
l’intensité dépend d’un terme quadratique et non pas de la concentration directe. Ce n’est
pas le seul facteur responsable de la non linéarité à forte concentration. Il y a aussi
l’auto-désactivation et l’auto-absorption.
• L’auto-désactivation résulte de collisions entre les molécules excitées, ce qui
provoque une perte d’énergie non rayonnante probablement analogue au transfert
d’énergie aux molécules de solvant qui se produit dans une conversion externe.
• L’auto-absorption se manifeste lorsqu’il y a perte de radiation émise par
absorption, avant que cette radiation ne puisse sortir de la cellule. Elle se produit lorsque
le déplacement de Stokes est petit et que la bande d’absorption recouvre la bande
d’émission de façon significative.
En plus de causer une courbure dans le graphe de la fluorescence en fonction de la
concentration, ces facteurs causent des distorsions dans le spectre d’émission lui-même.
Si nous examinons les spectres suivants de l’anthracène à des concentrations différentes,
dans le 2-propanol, nous remarquons les anomalies suivantes :
Figure 10. Anomalies apparaissant à haute concentration pour l’absorption et émission
de l’anthracène
Instrumentation
L'instrumentation utilisée en fluorescence diffère de l'instrumentation utilisée en
absorption principalement par la géométrie. Pour la mesure de l'absorbance, le faisceau
incident traverse l'échantillon en ligne directe avec le détecteur et c’est la réduction
d'intensité qui est mesurée. En fluorescence, on observe la fluorescence émise en
présence d'un faisceau excitant beaucoup plus puissant. Même si cela est possible avec
une géométrie à 180° (en ligne droite) parce que le rayonnement émis est à une longueur

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 129 of 145
d'onde différente du faisceau incident, la partie non absorbée du faisceau incident passe
dans le monochromateur et donne lieu à un rayonnement parasite important.
La configuration la plus utilisée en fluorescence est la configuration à 90° (angle
droit entre la source et le détecteur) illustrée sur la figure ci-contre. Cette géométrie
permet d’amplifier le signal de fluorescence sans pour autant amplifier le signal du
faisceau incident. On peut également augmenter la puissance Po du rayon incident et ainsi
augmenter le signal de fluorescence. Cette possibilité n’existe pas en spectrophotométrie
d’absorption étant donné que le résultat de la mesure est l’absorbance (log Po/P). Une
augmentation de Po conduit à une augmentation proportionnelle de P et n’a aucun effet
sur A, l’absorbance.
L’instrument le plus versatile est sans contredit le spectrofluorimètre qui donne le
choix de sélectionner et la longueur d’onde d’excitation et la longueur d’onde d’émission
avec ses deux monochromateurs.
Fluorimètres avec filtres et monochromateur
Une économie considérable peut-être effectuée si l’un des monochromateurs est
remplacé par un filtre passe-bande. Les filtres interférentiels modernes ont une
transmission de 60 % et ont une largeur de bande comparable à un petit monochromateur
(5 nm). Ils ont même un avantage considérable en termes de pouvoir de collection de la
lumière et peuvent dans certains cas surclasser des instruments plus dispendieux pour
détecter de bas niveaux de fluorescence.
Une source au mercure peut être utilisée en remplacement d’un monochromateur
d’excitation. La source au mercure émet un rayonnement intense à certaines longueurs
d’onde et des filtres interférentiels peuvent être fabriqués pour isoler les principales
bandes du mercure. Avec ce type d’appareil, on peut obtenir un spectre d’émission mais
le spectre d’excitation se limite aux six bandes du mercure. En pratique, ceci permet
d’exciter la plupart des composés parce que les bandes sont assez larges mais dans
certains cas l’efficacité serait faible. En utilisant une source au xénon, il n’y aurait aucune
restriction quant à la longueur d’onde, en autant que l’on puisse trouver sur le marché le
filtre interférentiel qui corresponde à nos besoins.
Instrument exclusivement avec filtres
Étant donné que dans les analyses de routine on connaît les conditions
d’excitation et d’émission, on peut utiliser un appareil avec filtres exclusivement.
• Désavantages : différents filtres à chaque analyse et on ne peut prendre un
spectre d’excitation ou d’émission.
• Avantages : peu coûteux, simple et facile à transporter.
Entre l’échantillon et le détecteur, on introduit un filtre passe-haut qui transmet le
rayonnement au-dessus d’une certaine longueur d’onde mais qui absorbe complètement

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 130 of 145
au-dessous de cette valeur. Ces filtres sont nécessaires pour enrayer les rayonnements
parasites.
Note : C’est ce type d’appareil qui sera utilisé pour l’expérience et vous trouverez,
à la dernière page de votre protocole, les spectres des différents filtres interférentiels
passe-bande (partie supérieure de la page) servant à l’excitation et les filtres passe-haut
(cut off, partie inférieure de la page) utilisés pour isoler la longueur d’onde d’émission.
Pour des informations supplémentaires concernant ces appareils, consulter aussi votre
livre de chimie instrumentale Skoog.
Interférences dues à la diffusion de la lumière
La lumière directe ne peut atteindre le monochromateur car la source et le
monochromateur sont à 90° par rapport à l’échantillon. Une partie du rayonnement
incident atteint quand même le monochromateur à cause du phénomène de diffusion de la
lumière : Dans un faisceau lumineux, les petites particules (ex. : fumée) sont visibles
parce que chaque particule agit comme un émetteur; cette émission de lumière se fait
dans toute les directions. De la même manière, les molécules dans un faisceau lumineux
agissent comme des émetteurs, mais avec une intensité beaucoup plus faible que les
particules. Ce phénomène est appelé « diffusion de Raleigh » et est dû principalement
aux molécules du solvant. L’intensité de cette diffusion peut être comparable à la
fluorescence du composé d’intérêt mais la longueur d’onde de cette lumière diffusée est
la même que celle du faisceau incident, donc plus courte que l’émission par fluorescence.
Un autre type de diffusion, qui peut interférer avec la fluorescence, est la diffusion
Raman Stokes, diffusion plus faible se produisant à une longueur d’onde plus élevée que
le faisceau incident et qui peut interférer avec la bande d’émission de fluorescence du
composé d’intérêt.
Il y a aussi la diffusion Raman anti-Stokes, plus difficile à détecter parce que plus
faible que la diffusion Raman Stokes, dont la longueur d’onde est plus faible que le
faisceau incident. Si l’échantillon contient des particules en suspension, il faut ajouter, à
la diffusion déjà existante, la diffusion de Tyndall; cette dernière est beaucoup plus
intense que la diffusion de Raleigh.
La diffusion de Raleigh se produisant à la même longueur d’onde que le faisceau
incident, même si elle est plus intense que la diffusion Raman, est plus facile à éliminer et
n’atteint pas le détecteur directement car le monochromateur d’émission est réglé à une
longueur d’onde plus élevée. Il en est autrement de la diffusion Raman car se produisant
à une longueur d’onde plus élevée, elle peut être associée à la fluorescence d’une
impureté fluorescente.
La figure suivante montre le spectre d’émission d’une solution aqueuse de
dichloro-fluorescéine (tracé supérieur) et le spectre d’émission de l’eau pure (tracé
inférieur).
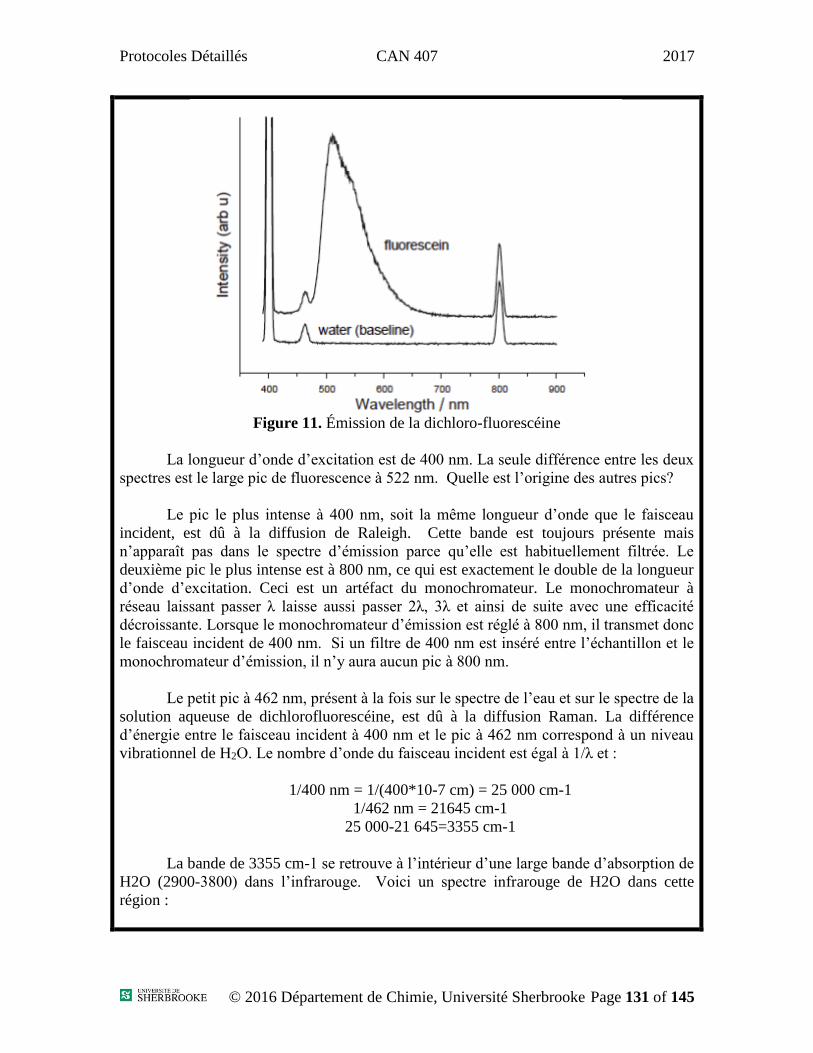
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 131 of 145
Figure 11. Émission de la dichloro-fluorescéine
La longueur d’onde d’excitation est de 400 nm. La seule différence entre les deux
spectres est le large pic de fluorescence à 522 nm. Quelle est l’origine des autres pics?
Le pic le plus intense à 400 nm, soit la même longueur d’onde que le faisceau
incident, est dû à la diffusion de Raleigh. Cette bande est toujours présente mais
n’apparaît pas dans le spectre d’émission parce qu’elle est habituellement filtrée. Le
deuxième pic le plus intense est à 800 nm, ce qui est exactement le double de la longueur
d’onde d’excitation. Ceci est un artéfact du monochromateur. Le monochromateur à
réseau laissant passer λ laisse aussi passer 2λ, 3λ et ainsi de suite avec une efficacité
décroissante. Lorsque le monochromateur d’émission est réglé à 800 nm, il transmet donc
le faisceau incident de 400 nm. Si un filtre de 400 nm est inséré entre l’échantillon et le
monochromateur d’émission, il n’y aura aucun pic à 800 nm.
Le petit pic à 462 nm, présent à la fois sur le spectre de l’eau et sur le spectre de la
solution aqueuse de dichlorofluorescéine, est dû à la diffusion Raman. La différence
d’énergie entre le faisceau incident à 400 nm et le pic à 462 nm correspond à un niveau
vibrationnel de H2O. Le nombre d’onde du faisceau incident est égal à 1/λ et :
1/400 nm = 1/(400*10-7 cm) = 25 000 cm-1
1/462 nm = 21645 cm-1
25 000-21 645=3355 cm-1
La bande de 3355 cm-1 se retrouve à l’intérieur d’une large bande d’absorption de
H2O (2900-3800) dans l’infrarouge. Voici un spectre infrarouge de H2O dans cette
région :

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 132 of 145
Figure 12. Spectre IR de l’eau
Les bandes Raman dans les spectres d’émission se déplacent avec la longueur
d’onde du faisceau incident et l’écart avec le faisceau incident est constant. Les pics dus
à la fluorescence ne changent pas avec la longueur d’onde du faisceau incident. On peut
confirmer le pic de diffusion Raman en prenant plusieurs spectres avec un faisceau
incident différent pour chaque spectre, ensuite on trouve la moyenne des écarts en cm-1
et cet écart devrait être compris à l’intérieur de la large bande d’absorption de l’eau. Un
exemple est donné dans le tableau ci-dessous :
Table 1. Confirmation du pic de diffusion Raman
Bibliographie
• Fluorescence and Phosphorescence; Analytical chemistry by open learning,
David Redell
• Chemistry Experiments for Instrumental Methods : Ed. Wiley, Sawyer,
Heineman, Beebe
• Quantitative Chemical Analysis; Ed. W.H.Freeman; 8ième édition, D.C. Harris
• Skoog
Protocole détaillé
Objectifs
• Utiliser la fluorescence pour quantifier la quinine, un composé fluorescent, dans
λex (nm) λdiffusé (nm) Différence de nombre d’onde (cm-1)
400 462 3352
410 478 3472
420 491 3445
430 503 3374
440 515 3312
450 532 3423

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 133 of 145
un « tonic water » et dans un inconnu.
• Utiliser la spectroscopie UV pour quantifier la quinine dans le même « tonic
water » et dans le même inconnu.
• Étudier l’effet du pH sur la fluorescence de la quinine.
• Étudier de l’extinction (quenching) de la fluorescence de la quinine causée par
les halogénures.
• Doser le bromure dans un inconnu par extinction de la fluorescence.
Étalonnage et mesure par fluorescence
Préparation d’une solution de quinine 200 ppm dans H2SO4 0,05 M :
Dissoudre environ 0,1207 g de sulfate de quinine (Fisher Cat. No. Q39-25
((C20H24N2O2)2.H2SO4 • 2H2O; M = 782,96 g/mol) pesé précisément) dans un
volumétrique de 500 mL contenant environ 250 mL d’eau et 12,5 mL de H2SO4 2M
(ajouté avec un cylindre gradué); compléter au trait de jauge avec de l’eau désionisée.
(Note : Assurez-vous que la quinine soit bien dissoute).
Préparation de la solution mère de quinine 10 ppm dans H2SO4 0,05 M :
Pipeter 5 mL de quinine 200 ppm dans un volumétrique de 100 mL et compléter
avec la solution de H2SO4 0,05 M. Note : Une solution de H2SO4 0,05 M a déjà été
préparée.
Préparation des solutions d’étalonnage dans H2SO4 0,05 M
(Note : Calculez les concentrations réelles de quinine à partir de la masse pesée).
Table 2. Préparation des solutions d’étalonnage
Manipulations et analyse des données
Une cellule carrée en quartz est utilisée pour mesurer la fluorescence. Il est
important de toujours mettre cette cellule avec la même orientation dans le porte
échantillon.
• Ajuster la sensibilité du fluorimètre en utilisant le mode « mutli optionnel
fluorescence brute » (voir la notice d’utilisation de l’appareil en annexe).
- Entrer la valeur de 800 FSU (fluorescence unit) pour le standard le plus
Concentration
(ppb)
Volume de solution
de quinine 10 ppm
Volume final
(mL)
50 500 µL 100
100 1 mL 100
200 1 mL 50
400 1 mL 25
500 5 mL 100

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 134 of 145
concentré (500 ppb).
- Pour faire le zéro (blank), couper totalement les faisceaux d’excitation et
d’émission à l’aide de l’adapteur pour cellules rondes, mais installé à l’envers (vous ferez
régulièrement le 0 à l’aide de cet adapteur).
• Mesurer la fluorescence des solutions standard et de la solution de H2SO4 0,05
M (blanc).
• Mesurer la fluorescence de l’inconnu après dilution 5 dans 100 avec H2SO4
0,05 M.
• Dégazer 100 mL de Tonic Water et en pipeter 5 mL dans un volumétrique de
100 mL et compléter avec H2SO4 0,05 M.
• Après agitation, pipeter 5 mL de cette dernière solution et la diluer dans un
ballon volumétrique de 50 mL avec H2SO4 0,05 M.
• Mesurer la fluorescence de cette solution de Tonic Water.
• Tracer la droite d’étalonnage de l’intensité de la fluorescence en fonction de la
concentration de quinine.
• Dans un tableau, mettre en évidence les résultats et l’incertitude obtenus pour la
concentration en ppm de l’inconnu et celle du Tonic Water.
• Pour le Tonic Water, comparer la valeur obtenue à celle publiée.
Influence du pH sur la fluorescence de la quinine
• À partir de la solution de 200 ppm de quinine dans H2SO4 0,05 M, préparez 100
mL de solution de quinine 10 ppm, mais cette fois en diluant avec de l’eau désionisée.
• À partir de cette solution 10 ppm, préparez six solutions de 50 mL contenant 500
ppb de quinine dans les tampons suivants : 2.0; 2.9; 4.1; 5.0; 5.5 et 6.2. (Note : les
différents tampons ont déjà été préparés).
• Mesurez d’abord la fluorescence de la solution 500 ppb de quinine à pH 2 en
mode « mutli optionnel fluorescence brute »; attribuer une valeur de 800 FSU à cette
solution. Faire le 0 de l’appareil comme dans la première partie.
• Mesurez la fluorescence de chacun des tampons et de chacune des solutions
tampons + 500 ppb de quinine. Pour s’assurer d’obtenir un graphique valable de la
fluorescence de la quinine en fonction du pH (parce que les tampons proviennent de
plusieurs sources différentes : tampons phosphate et acétate), il faut soustraire la
fluorescence du tampon de la valeur de fluorescence obtenue pour chacune des solutions
quinine + tampon.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 135 of 145
• Portez en graphique la fluorescence de la quinine en fonction du pH.
Étude de l’extinction de la fluorescence de la quinine causée par les halogénures et
l’équation de Stern-Volmer
Note : Voir note explicative de l’équation de Stern-Volmer en annexe.
Préparation des solutions pour obtenir la droite de Stern-Volmer
À partir de la solution mère de 10 ppm (10 000 ppb) de quinine dans H2SO4 0,05
M préparée dans la première partie, préparez six volumétriques de 50 mL dans lesquels
vous ajouterez des volumes de la solution mère de quinine et des volumes d’une solution
déjà préparée de NaBr 0,05 M dans H2SO4 0,05 M. Complétez avec H2SO4 0,05 M. La
concentration de quinine de ces solution sera de 400 ppb.
Table 3. Solutions à préparer pour obtenir la droite de Stern-Volmer
Préparation de la solution inconnue
Préparez trois autres volumétriques de 50 mL, mais cette fois ajoutez des volumes
de la solution inconnue de NaBr.
Table 4. Préparation des solutions inconnues
Mesure et analyse des données
• Mesurez d’abord la fluorescence de la solution 1 sans ajout de NaBr en mode «
mutli optionnel fluorescence brute »; attribuer une valeur de 800 FSU à cette solution.
Faire le 0 de l’appareil comme dans la première partie.
• Mesurez la fluorescence de toutes les solutions standard.
Solution Solution de quinine 10 000
ppb dans H2SO4 0,05 M (mL)
Solution de NaBr 0,05 M
dans H2SO4 0,05 M (mL)
1 2 0
2 2 2
3 2 4
4 2 8
5 2 16
6 2 32
Solution Solution de quinine 10 000
ppb dans H2SO4 0,05 M (mL)
Solution de NaBr
inconnue (mL)
7 2 2
8 2 4
9 2 8

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 136 of 145
• Mesurez la fluorescence des trois solutions de l’inconnu. Assurez-vous qu’au
moins deux valeurs de fluorescence pour votre inconnu de NaBr soient comprises dans le
domaine des valeurs de fluorescence des solutions standard.
• Tracez le graphique de l’intensité de la fluorescence en fonction de la
concentration de NaBr.
• Tracez une droite en vous servant la relation de Stern-Volmer
𝜙0
𝜙𝑓= 𝐾𝑐 + 1
Φ°est le rendement quantique de l’espèce fluorescente en absence de l’extincteur,
Φf est le rendement quantique de l’espèce fluorescente lorsque la concentration de
l’extincteur est c. K étant connu comme la constante de Stern-Volmer. La pente K de la
droite est différente pour chacun des halogénures, augmentant de Cl- jusqu’à I-.
• Déterminez la concentration millimolaire de NaBr dans votre inconnu en
utilisant comme courbe standard la droite tracée avec l’équation de Stern-Volmer.
Calculer une moyenne des valeurs de concentration obtenues pour votre inconnu de
NaBr.
Spectroscopie UV d’absorption
Les dosages par fluorimétrie sont très sensibles mais limités aux molécules, peu
nombreuses, qui ont un rendement quantique de fluorescence acceptable. Par contre, un
grand nombre de molécules absorbent dans l’UV, et la spectroscopie UV est souvent la
méthode la plus simple et la plus précise de les doser.
Dosage de la quinine par spectroscopie UV d’absorption
La cellule de quartz utilisée pour les mesures de fluorescence ne convient pas
pour les mesures d’absorption UV (voir section suivante). Vous devez utiliser une cellule
de quartz.
• À partir de la solution de quinine 200 ppm dans H2SO4 0,05 M, préparez des
solutions standard entre 2 et 10 ppm en quinine dans H2SO4 0,05 M.
• Enregistrez le spectre d’absorption UV de H2SO4 0,05 M comme blanc, puis
enregistrez le spectre d’absorption de chaque standard.
• Enregistrez le spectre d’absorption de l’inconnu; diluez si nécessaire.
• Tracez la droite d’étalonnage de l’absorbance en fonction de la concentration et
déterminer la concentration de l’inconnu. (Note : choisissez la longueur d’onde la plus
appropriée pour effectuer vos lectures…. Quelle est-elle et pourquoi???).

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 137 of 145
Absorbance UV des filtres et de la cellule du fluorimètre
Afin de bien comprendre le fonctionnement du fluorimètre et le choix des filtres
pour la mesure de la fluorescence de la quinine :
• Enregistrez le spectre d’absorption UV de la cellule de quartz (remplie de H2SO4
0,05 M).
• Enregistrez les spectres d’absorption UV des filtres d’excitation et d’émission
que vous avez utilisés pour les expériences de fluorescence.
Références supplémentaires
Voir les articles suivants, disponibles en ligne:
http://www.sciencedirect.com/science/article/pii/S0022231302002946
http://onlinelibrary.wiley.com/doi/10.1002/jps.2600630615/pdf
Calculs Trouver la teneur en zinc, en sodium et en potassium (en ppm) de vos inconnus. Effectuer
les calculs d’erreur pour chacune des droites d’étalonnage.
- Comparez la teneur en zinc de l’inconnue avec celle obtenue en voltampérométrie
(expérience 4, si vous l’avez fait, sinon, vous comparerez ce résultat dans votre
discussion de l’expérience 4)
- N’oubliez pas d’effectuer les tests supplémentaires nécessaires pour la détermination
des paramètres analytiques de la technique.
Notes supplémentaires pour la discussion
- Démontrez que vous distinguez bien la spectroscopie d’absorbance et de fluorescence
en termes de fonctionnement, d’instrumentation, de performances analytiques…
- La quelle des technique permet généralement d’obtenir la plus faible limite de
détection? Expliquez pourquoi en vos mots.
- Aussi à inclure à votre discussion :
• Est-ce que l’on aurait pu utiliser HCl pour diluer la solution standard de quinine au lieu
de H2SO4 0,05 M? Expliquer
• En étudiant la courbe obtenue lors de l’étude de la fluorescence en fonction du pH,
discuter de l’importance du pH dans les mesures de fluorescence.
• Pourquoi l’augmentation du pH fait-elle diminuer la fluorescence observée à 450 nm?
Sous quelle forme retrouve-t-on la quinine à pH 2?
• Comparer la méthode de dosage de la quinine par fluorescence à celle par absorption
UV.
• Commenter les spectres d’absorption UV des filtres d’excitation et d’émission du

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 138 of 145
fluorimètre et de la cellule utilisée pour mesurer la fluorescence
Information Supplémentaire 1: Mécanismes d’extinction et l’équation
de Stern-Volmer (Il peut y avoir plusieurs types de mécanismes ou processus
d’extinction (quenching))
L’extinction (raccourcissement de la durée de vie de l’état excité) de la quinine à
l’état singulet en présence d’halogénures est de type dynamique, c’est-à-dire qu’il y a
collisions entre les molécules de quinine excitées et l’halogénure. Ces collisions se
produisent en parallèle avec les autres mécanismes expliqués dans le diagramme de
Jablonski. Ces collisions conduisent à une perte d’énergie du fluorophore et par le fait
même à une diminution du temps de vie de l’état excité fluorescent.
On peut parler aussi d’extinction statique où le mécanisme est la formation, à
l’état fondamental, d’un complexe fluorophore-agent d’extinction non fluorescent.
Lorsque ce complexe absorbe la lumière, il retourne à l’état fondamental sans émission
de photons. Un autre type de mécanisme statique peut se produire lorsque la
concentration de l’agent d’extinction est assez élevée ce qui entraîne la formation d’un
complexe fluorophore-agent d’extinction qui stoppe instantanément sont état excité. Ce
dernier type d’extinction statique ne produit pas de changement dans le spectre
d’absorption du fluorophore tandis que dans le premier, il y a déplacements dans le
spectre d’absorption du fluorophore avec l’addition de l’agent d’extinction.
L’équation de Stern-Volmer modélise le mécanisme d’extinction dynamique
suivant : Supposons qu’une molécule A, à l’état fondamental, absorbe un photon hν et est
excité à l’état singulet A*
Absorption
La vitesse à laquelle A* est formée d[A*]/dt est proportionnelle à la concentration
de A. La constante ka dépend de l’intensité du faisceau incident et de l’absorptivité de A.
À la suite de l’absorption d’énergie, la molécule A* peut émettre un photon et retourner à
l’état fondamental. Ce processus est représenté par :
Émission
ke étant la constante de vitesse du processus de désactivation par voir radiative.
Aussi la molécule excitée A* peut perdre de l’énergie sous forme de chaleur ou par un
processus non rayonnant tel que la conversion interne.
Désactivation

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 139 of 145
Lorsque l’extincteur (quencher) (Q) est ajouté, un autre mode ou mécanisme de
désactivation est maintenant possible, où la molécule excitée (A*) transfère son énergie à
l’extinction (Q) qui est promu à un état excité (Q*) et qui perdra cette énergie par une
multitude de processus. kq est la constante de vitesse de second ordre pour le processus
d’extinction.
Extinction (quenching)
L’extincteur excité pouvant perdre son énergie par une multitude de processus.
Sous une intensité constante d’un faisceau lumineux, le système atteint l’état
stationnaire où les concentrations de A* et de A restent constantes. À ce stade, la vitesse
de formation de A* est égale à sa vitesse de destruction, de sorte que :
Le rendement quantique pour un processus chimique est la fraction de photons
absorbés produisant un certain résultat. Si ce résultat se produit à chaque fois qu’un
photon est absorbé alors le rendement quantique est l’unité. Le rendement est un nombre
entre 0 et 1. Le rendement quantique pour l’émission de A* est la vitesse d’émission
divisée par la vitesse d’absorption.
En l’absence de l’extincteur Q nous désignons le rendement quantique de
fluorescence :
𝜙0 =𝑝ℎ𝑜𝑡𝑜𝑛𝑠 é𝑚𝑖𝑠 𝑝𝑎𝑟 𝑠𝑒𝑐𝑜𝑛𝑑𝑒
𝑝ℎ𝑜𝑡𝑜𝑛𝑠 𝑎𝑏𝑠𝑜𝑟𝑏é𝑠 𝑝𝑎𝑟 𝑠𝑒𝑐𝑜𝑛𝑑𝑒=
𝑣𝑖𝑡𝑒𝑠𝑠𝑒 𝑑′é𝑚𝑖𝑠𝑠𝑖𝑜𝑛
𝑣𝑖𝑡𝑒𝑠𝑠𝑒 𝑑′𝑎𝑏𝑠𝑜𝑟𝑝𝑡𝑖𝑜𝑛=
𝑘𝑒[𝐴∗]
𝑘𝑎[𝐴]
En substituant ka[A] dans l’avant dernière équation et en mettant [Q] = 0 nous
obtenons une expression pour le rendement quantique de fluorescence à l’état
stationnaire :
𝜙0 =𝑘𝑒[𝐴∗]
𝑘𝑒[𝐴∗] + 𝑘𝑑[𝐴∗] + 𝑘𝑞[𝐴∗][0]=
𝑘𝑒
𝑘𝑒 + 𝑘𝑑
Si [Q] ≠ 0 le rendement quantique de fluorescence (ΦQ) est :
𝜙0 =𝑘𝑒[𝐴∗]
𝑘𝑒[𝐴∗] + 𝑘𝑑[𝐴∗] + 𝑘𝑞[𝐴∗][𝑄]=
𝑘𝑒
𝑘𝑒 + 𝑘𝑑 + 𝑘𝑞[𝑄]
En faisant le rapport Φo/ΦQ, nous obtenons l’équation de Stern-Volmer :
𝜙0 =𝑘𝑒[𝐴∗]
𝑘𝑒[𝐴∗] + 𝑘𝑑[𝐴∗] + 𝑘𝑞[𝐴∗][𝑄]=
𝑘𝑒
𝑘𝑒 + 𝑘𝑑 + 𝑘𝑞[𝑄]

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 140 of 145
L’équation de Stern-Volmer stipule que si nous mesurons l’émission relative
Φo/ΦQ en fonction de la concentration de l’extincteur (Q) nous obtiendrons une droite de
pente (kq
ke+kd).
La quantité Φo/ΦQ est équivalente à I0/Iq où I0 est l’intensité de fluorescence en
l’absence d’extincteur et IQ est l’intensité de fluorescence en présence d’extincteur. KSV
étant la constante de Stern-Volmer.
Références
Principles and applications of fluorescence spectroscopy, J.R. Albany;
BlackwellPublishing; QP 519 F56AR42 2007
Méthodes instrumentales d’analyse chimique et applications; Gwenola Burgot, Jean-
Louis Burgot; QD 79 15B87 2006
Lee K. Fraiji, David M. Hayes, T.C. Werner., J. Chem. Educ, 1992, 69, 424-428.
Information Supplémentaire 2 : Fonctionnement du fluorimètre à
cuvette
Opération de l’appareil (TURNER TD-700)
Cet appareil peut être relié à un ordinateur pour faire l’acquisition de données.
Figure A1. Fluorimètre à cuvette TURNER TD-700

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 141 of 145
Figure A2. Illustration du support à cuvette du TURNER TD-700
Figure A3. Fonctionnement du porte cuvette du TURNER TD-700
Utilisation de l’interface du TURNER TD-700
Note : Une procédure en format papier sera disponible avec l’instrument au
laboratoire. Les documents qui suivent sont toutefois plus complets.
Fluorimètre TD-700 Manuel d’utilisation
Page 7
Fluorimètre TD-700
Cylindre porte-filtre et adaptateur
pour tube 13 mm
Adaptateur pour tube 25 mm Adaptateur pour cuve carrée
10 mm
Repère d’alignement Repère d’alignement
Adaptateur échantillon
Cylindre porte-filtre
Repère d’alignement argent
Affichage
Clavier
Compartiment échantillon
Fluorimètre TD-700 Manuel d’utilisation
Page 7
Fluorimètre TD-700
Cylindre porte-filtre et adaptateurpour tube 13 mm
Adaptateur pour tube 25 mm Adaptateur pour cuve carrée10 mm
Repère d’alignement Repère d’alignement
Adaptateur échantillon
Cylindre porte-filtre
Repère d’alignement argent
Affichage
Clavier
Compartiment échantillon

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 142 of 145
Fluorimètre TD-700 Manuel d’utilisation
Page 21
Calibrage: mode multi-
optionnel - fluorescence brute
1. Setup2. Calibration
1. Mode2. Cal Procedure
Simple
<Multi-Optional>
1. Mode2. Cal Procedure
<Raw Fluor.>Direct Conc.
1. Mode
2. Cal Procedure
1. Setup2. Calibration
Insert TypicalSample <ENT>
SETTING SensSens Factor: XX
Sensitivity Set Sens Factor: XX
Set Sample = XXX1. OK 9. Change
Sample = XXX
<ENT>
Read & Subtract
Blank? 1=Yes 9=No
1.
2.
3.
4.
5.
VIII. Calibrage : mode multi-optionnel -
fluorescence brute
La procédure de calibrage en mode multi-optionnel - fluorescence brute consiste
en un point de calibrage unique dans lequel un standard et un blanc optionnelsont lus afin de fixer la gamme et la sensibilité maximales de l’appareil.
L’échantillon choisi sera fixé à 80% de la valeur maximale lisible avec précisionpar cet appareil, sauf en cas de changement de la valeur de cet échantillon. La
gamme et la sensibilité de l’appareil peuvent être ajustées en changeant la valeurde l’échantillon.
1. Pour choisir le mode multi-optionnel - fluorescence brute, appuyer sur
<ENT> à partir de l’écran de départ, appuyer sur <1> pour le réglage,puis sur <1> à nouveau pour le mode. Utiliser <> pour choisir le mode
multi-optionnel. Appuyer sur <ESC> pour revenir à l’écran précédent,puis sur <2> pour choisir la procédure de calibrage. Utiliser la touche <>
pour choisir «Raw Fluor.» (fluorescence brute) pour la procédure decalibrage en fluorescence brute. Appuyer sur <ESC> pour revenir à
l’écran Setup/Cal (réglage/calibrage).
2. Pour accéder à la séquence de calibrage, appuyer sur <2> à partir del’écran Setup/Cal (réglage/calibrage). La séquence de calibrage multi-
optionnel - fluorescence brute apparaît.
3. Remplir un tube à essai ou une cuve propre avec un échantillon ayantenviron 80% de la concentration maximum qui doit être lue. Il n’est pas
nécessaire de connaître la concentration exacte; elle est utilisée pourfixée la gamme et la sensibilité optimales de l’appareil. Essuyer et sécher
l’extérieur du tube à essai ou de la cuve, et l’insérer dans l’adaptateurd’échantillon dans le compartiment échantillon. Appuyer sur <ENT> pour
accéder à l’écran suivant.
4. Si l’échantillon a 80% de la concentration maximum qui doit être lue,accepter la valeur prédéfinie de 800 en appuyant sur <1>. Le TD-700 fixe
la sensibilité de façon à ce que le standard soit lu à environ 80% de lagamme maximale. Si une lecture égale à 800 n’est pas acceptable,
appuyer sur <9> pour changer la valeur. Si une valeur plus élevée estattribuée à l’échantillon, la concentration maximale des échantillons
pouvant être lue diminue, et la sensibilité et la résolution de l’appareilaugmentent. Si une valeur inférieure à 500 est attribuée à l’échantillon
choisi, la concentration maximale des échantillons pouvant être lueaugmente, et la sensibilité et la résolution de l’appareil diminuent. Saisir
le nombre désiré et appuyer sur <ENT>, puis sur <1>.
5. Le TD-700 fixe maintenant la sensibilité, comme indiqué par le SENSFACTOR (facteur de sensibilité), basée sur la valeur de l’échantillon final
accepté. Lorsque l’échantillon est fixé, le TD-700 demande s’il faut lire unblanc. Pour qu’un blanc soit soustrait, appuyer sur <1>. Sinon, appuyer
sur <9> et la séquence de calibrage s’arrête là, et l’écran de départapparaît. A ce stade, la valeur du calibrage est envoyée directement vers
une imprimante ou un ordinateur. Voir l’annexe 2 pour plus de détailsconcernant la sortie du calibrage sur imprimante.

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 143 of 145
Performance du TURNER TD-700
Fluorimètre TD-700 Manuel d’utilisation
Page 22
Blank: X.X
Cal Std: XXX.X
Reading BlankWAIT
Insert Blankand press <ENT>
Press <0> when
value stable XXX
Blank = XXX
8.
6.
<1> - Abort Cal<ESC> - Resume
10.
6. Pour lire un blanc, remplir un tube à essai ou une cuve propre avec lasolution de blanc, essuyer et sécher l’extérieur du tube à essai ou de la
cuve, l’insérer dans l’adaptateur d’échantillon dans le compartimentéchantillon, et appuyer sur <ENT>. Laisser la lecture se stabiliser et
appuyer sur <0>. L’appareil lit le blanc, puis retourne automatiquement àl’écran de départ. A ce stade, la valeur du calibrage est envoyée
directement vers une imprimante ou un ordinateur. Voir l’annexe 2 pourplus de détails concernant la sortie du calibrage sur imprimante.
7. Erreurs: Après la lecture de l’échantillon, le TD-700 s’ajuste
automatiquement sur la gamme optimale pour la mesure de l’échantillon.Si l’échantillon utilisé est trop concentré ou trop dilué, l’appareil ne sera
pas capable d’atteindre la sensibilité voulue. Dans ces cas-là, unmessage apparaît indiquant que, en se basant sur le calibrage, l’appareil
a atteint son maximum ou son minimum de sensibilité. Appuyer sur<ENT> pour dire à l’appareil d’accepter la valeur de sensibilité maximale
ou minimale. Il est cependant conseillé d’ajuster la concentration del’échantillon de façon à tomber dans la gamme, puis de recalibrer.
8. Pour visualiser le dernier calibrage fixé, appuyer sur <ENT> à partir de
l’écran de départ, puis sur <9> pour voir la valeur du blanc et du standardde calibrage. Appuyer sur <H> pour retourner à l’écran de départ.
9. Pour imprimer le dernier calibrage fixé, appuyer sur <ENT> à partir de
l’écran de départ, puis sur <D>. Appuyer sur <H> pour retourner à l’écrande départ.
10. Pour annuler le calibrage, appuyer sur <ESC> n’importe quand pendant
la séquence de calibrage. Appuyer sur <1> pour annuler ou sur <ESC>pour reprendre.
Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 144 of 145

Protocoles Détaillés CAN 407 2017
© 2016 Département de Chimie, Université Sherbrooke Page 145 of 145





























