Embed Size (px)
Citation preview

Cours de Cours de PharmacologiePharmacologie
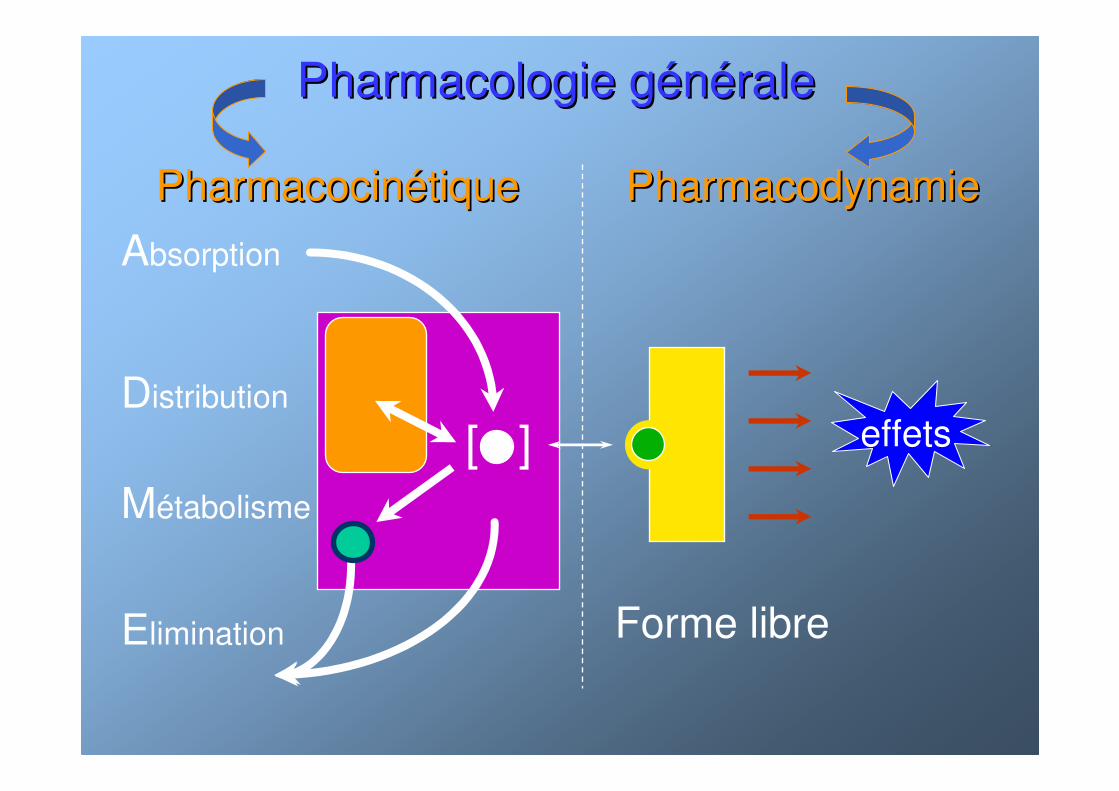
Pharmacologie gPharmacologie géénnééralerale
PharmacocinPharmacocinéétique Pharmacodynamietique Pharmacodynamie
[ ]
Absorption
Elimination
Métabolisme
Distributioneffets
Forme libre
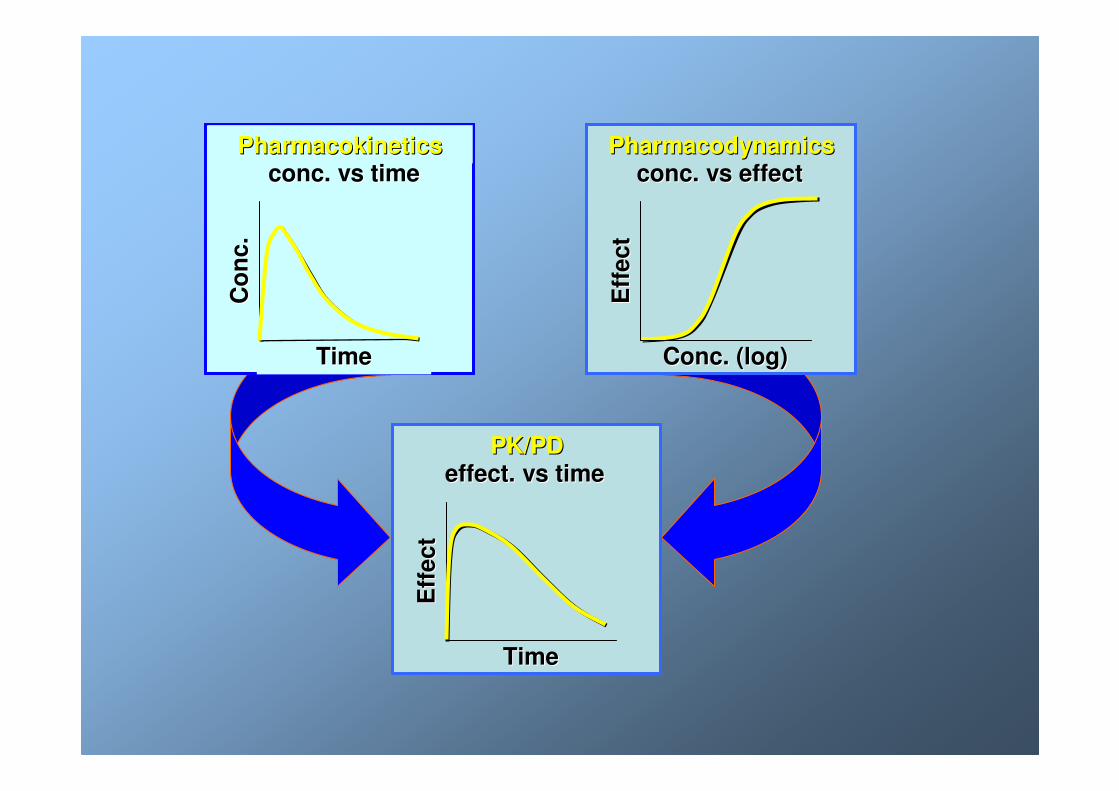
Conc. (log)Conc. (log)
Eff
ect
Eff
ect
conc. vs effectconc. vs effectPharmacodynamicsPharmacodynamics
TimeTime
Co
nc.
Co
nc.
conc. vs timeconc. vs timePharmacokineticsPharmacokinetics
TimeTime
Eff
ect
Eff
ect
effect. vs timeeffect. vs timePK/PDPK/PD
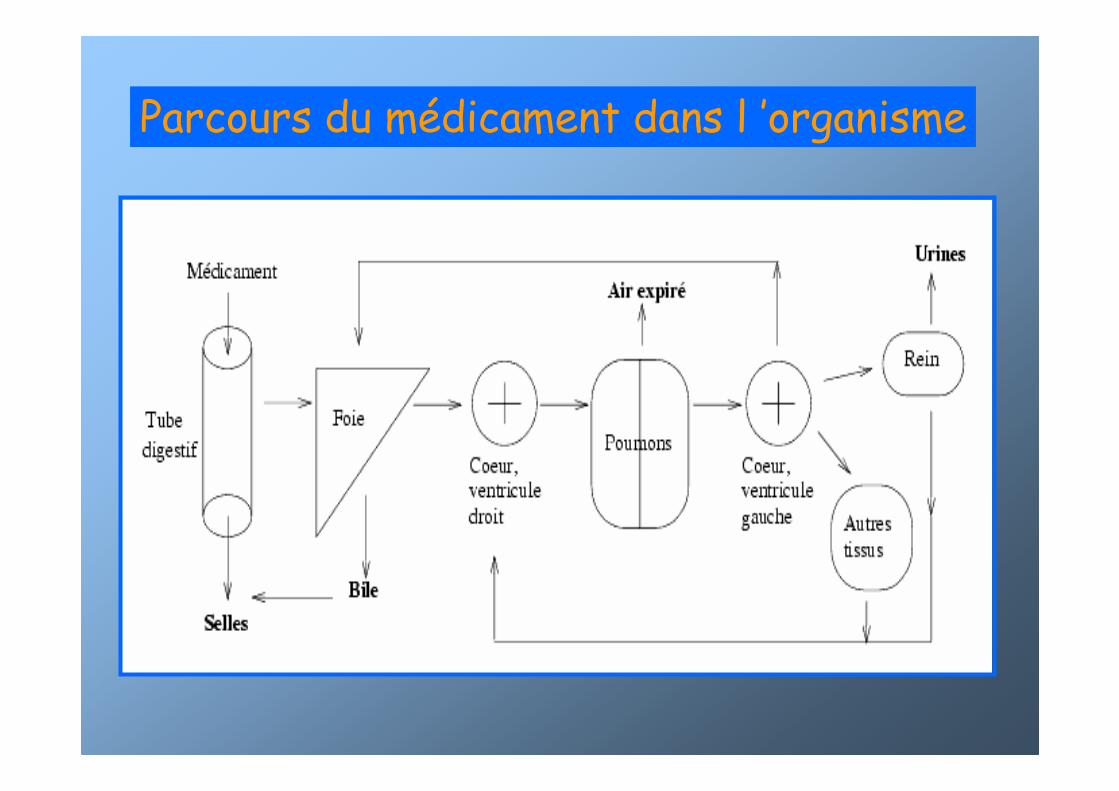
Parcours du médicament dans l ’organisme

RRéésorption/Absorptionsorption/Absorption
��Le mLe méédicament passe dans la circulation gdicament passe dans la circulation géénnééralerale- IV (voie de référence) : veine périphérique ou centrale
- orale ou per os
- sub-linguale : veines linguales et maxillaires vers la veine cave- rectale : veines hémorroïdaires- sous-cutanée : abdomen, bras- cutanée ou trans-dermique
- intra-musculaire : fessier, deltoïde- nasale ou oculaire
- inhalée
- dans un organe ou in-situ : intra-oculaire…

LL’’absorption orale est influencabsorption orale est influencéée pare par::
les caractles caractééristiques du mristiques du méédicamentdicament : physico-chimiques (pka), hydro/liposolubilité, taille des molécules, la forme galénique…
les caractles caractééristiques liristiques liéés s àà ll’’individuindividu : pH digestif, la vitesse de vidange gastrique et la mobilité intestinale, l’alimentation, la prise associé de médicament (pansements digestifs), les pathologies associées…
L’absorption se caractérise par la la biodisponibilitbiodisponibilitéé qui est la fraction de la dose de médicament qui atteint la circulation générale et la vitesse àlaquelle elle l’atteint.
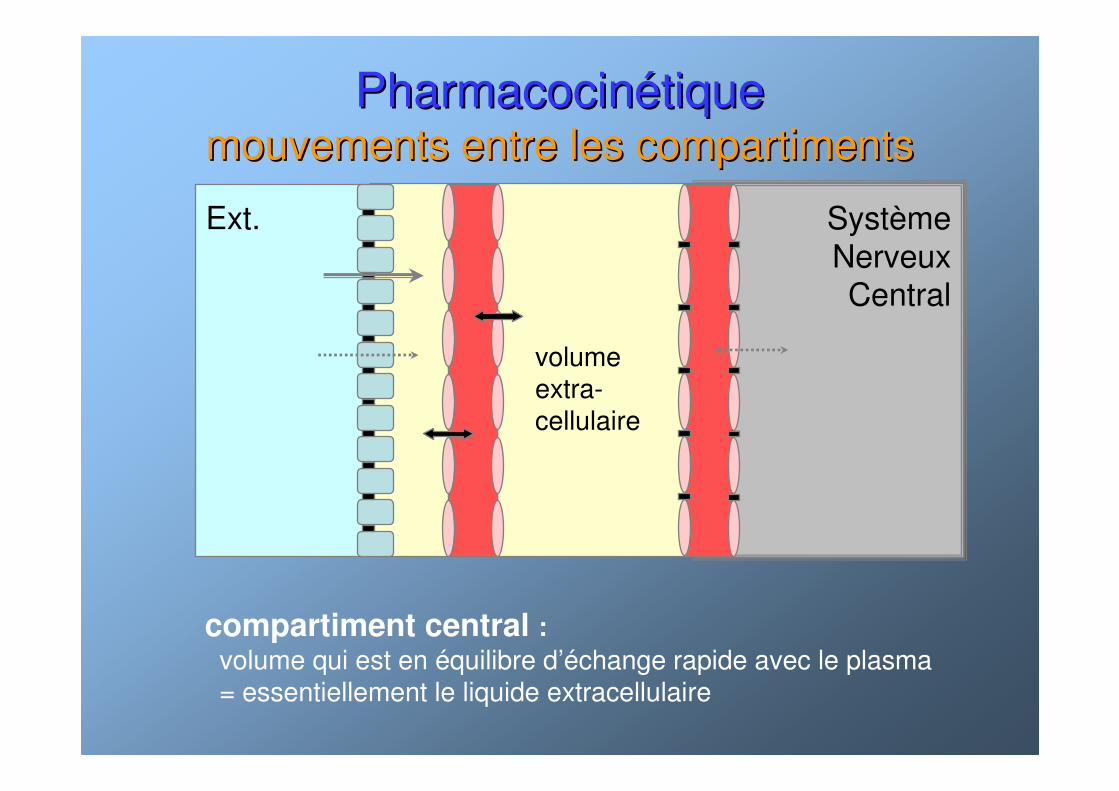
PharmacocinPharmacocinéétiquetiquemouvements entre les compartimentsmouvements entre les compartiments
Ext. SystèmeNerveuxCentral
volumeextra-cellulaire
compartiment central :volume qui est en équilibre d’échange rapide avec le plasma= essentiellement le liquide extracellulaire

Membrane biologique et distribution des Membrane biologique et distribution des principes actifs dans lprincipes actifs dans l’’organismeorganisme
Poids molPoids molééculaire et conformation spatialeculaire et conformation spatiale
DegrDegréé dd’’ionisationionisation
Hydro vs liposolubilitHydro vs liposolubilitéé des formes ionisdes formes ioniséées et es et
non ionisnon ionisééeses
Liaison aux proteines plasmatique vs tissulaires vs Liaison aux proteines plasmatique vs tissulaires vs cible pharmacologiquecible pharmacologique

Diffusion substances non ionisDiffusion substances non ionisééeses
Diffusion à travers les membranes• la vitesse de diffusion dépend
– de la surface d’absorption S– du coefficient de perméabilité (Kp)
–– du gradient des concentrations de part et ddu gradient des concentrations de part et d’’autre de la autre de la membranemembrane
flux net = flux net = Kp.SKp.S.(C2.(C2--C1)C1)
KpKp ddéépend de la taille de la molpend de la taille de la moléécule et de sa liposolubilitcule et de sa liposolubilitéé
CC11 C2
seules les petites molécules non chargées et peu polaires passent facilement à travers les membranes +++
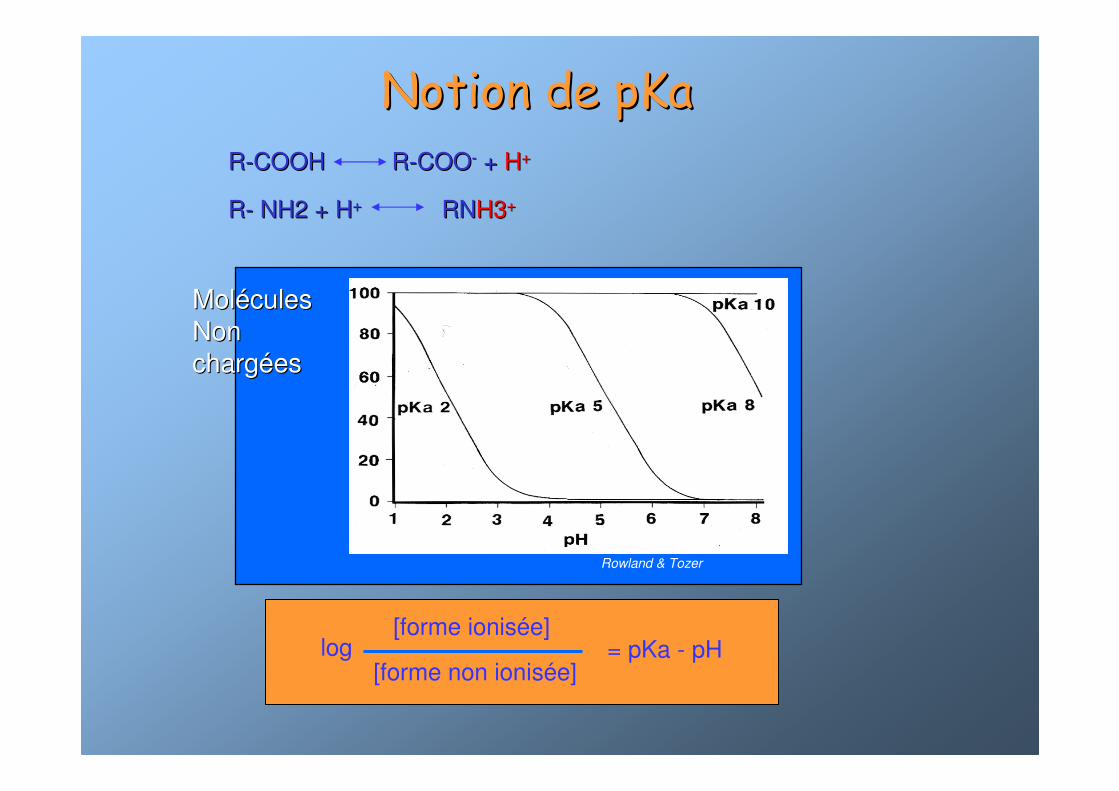
Notion de pKaNotion de pKa
MolMoléécules cules Non Non chargchargééeses
Rowland & Tozer
log = pKa - pH[forme ionisée]
[forme non ionisée]
RR--COOHCOOH RR--COOCOO-- ++ HH++
RR-- NH2 + HNH2 + H++ RNRNH3H3++
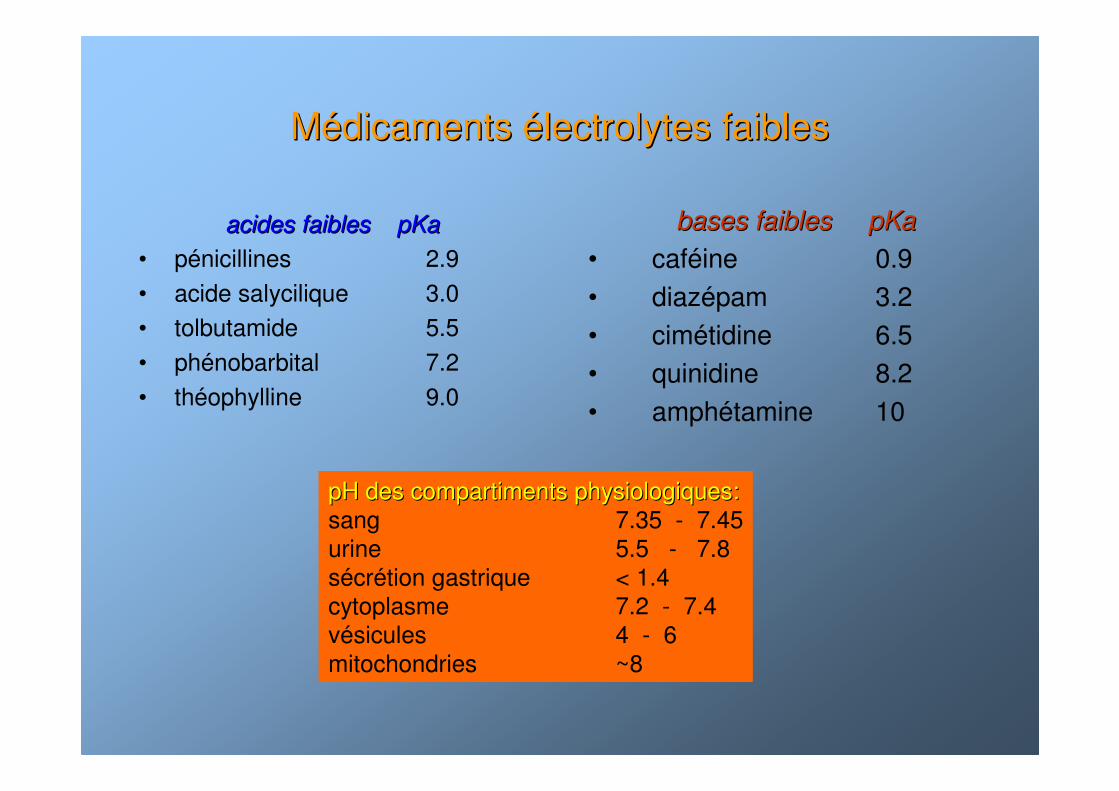
MMéédicaments dicaments éélectrolytes faibleslectrolytes faibles
acides faibles pKaacides faibles pKa
• pénicillines 2.9• acide salycilique 3.0• tolbutamide 5.5• phénobarbital 7.2• théophylline 9.0
bases faibles pKabases faibles pKa
• caféine 0.9• diazépam 3.2• cimétidine 6.5• quinidine 8.2• amphétamine 10
pH des compartiments physiologiques:pH des compartiments physiologiques:sang 7.35 - 7.45urine 5.5 - 7.8sécrétion gastrique < 1.4 cytoplasme 7.2 - 7.4vésicules 4 - 6mitochondries ~8
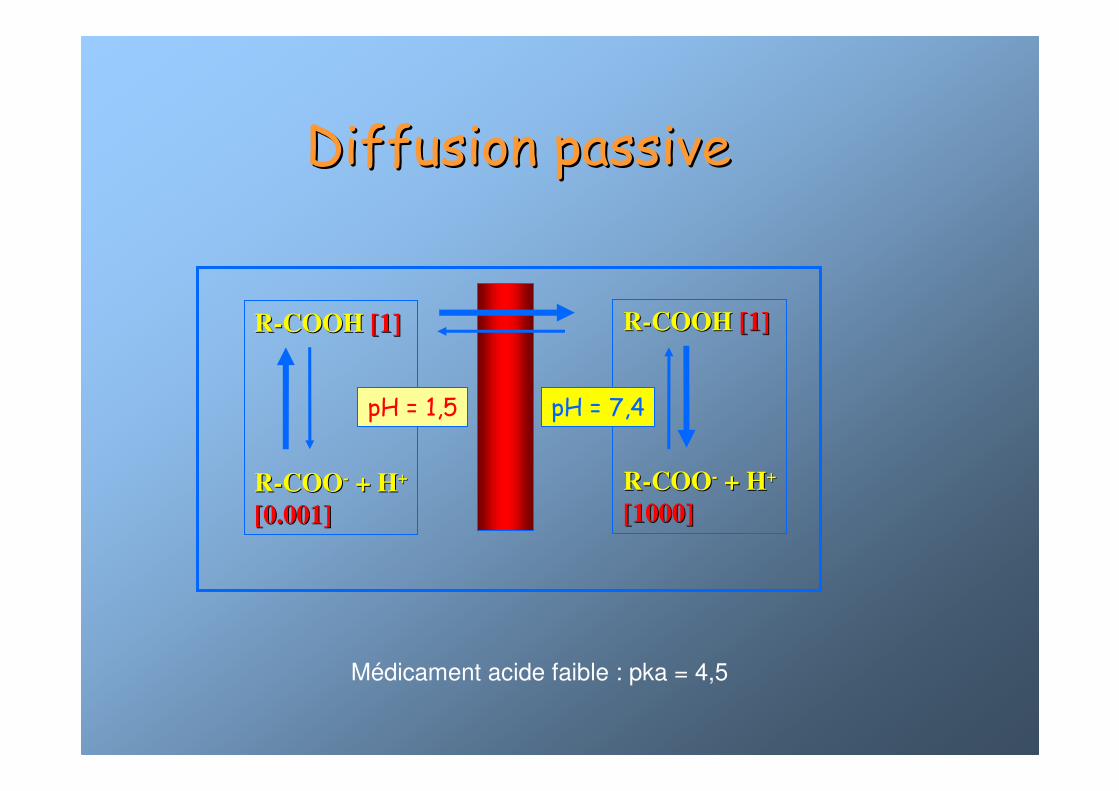
RR--COOH COOH [1][1]
RR--COOCOO-- + H+ H++
[0.001][0.001]
RR--COOH COOH [1][1]
RR--COOCOO-- + H+ H++
[1000][1000]
pH = 7,4pH = 1,5
Diffusion passiveDiffusion passive
Médicament acide faible : pka = 4,5

Application cliniqueApplication clinique
IntoxicationsIntoxications
PhPhéénobarbital, nobarbital, acide faible, pKa = 7,4acide faible, pKa = 7,4
9% non ionis9% non ioniséé pH urinaire = 4,4 pH urinaire = 4,4 100% non ionis100% non ioniséé
alcalinisation urinairealcalinisation urinaire
0,1% non ionis0,1% non ioniséé pH urinaire = 7,5 pH urinaire = 7,5 50% non ionis50% non ioniséé
Acide salicyliqueAcide salicyliqueacide faible, pKa = 3,4acide faible, pKa = 3,4
Seule la fraction libre est filtrSeule la fraction libre est filtréée au niveau re au niveau réénalnal

MMéécanismes de transports actifscanismes de transports actifs
Transport Transport àà ll’’aide de transporteurs membranaires :aide de transporteurs membranaires :
- énergie,- contre gradient électrochimique- sélectif,- saturable- inhibition par compétition

Facteurs modifiant la rFacteurs modifiant la réésorption sorption digestive des mdigestive des méédicamentsdicaments
�� LiLiéés au ms au méédicament:dicament:-- ddéésagrsagréégation stomacale (enrobage gastrogation stomacale (enrobage gastro--rréésistant,sistant,
forme forme àà liblibéération programmration programméée)e)-- dissolution (solutions, sels, taille particules)dissolution (solutions, sels, taille particules)
�� LiLiéés au patient :s au patient :-- vidange gastriquevidange gastrique-- ddéébit sanguin intestinal (liposolubles)bit sanguin intestinal (liposolubles)-- alimentationalimentation-- association massociation méédicamenteusesdicamenteuses
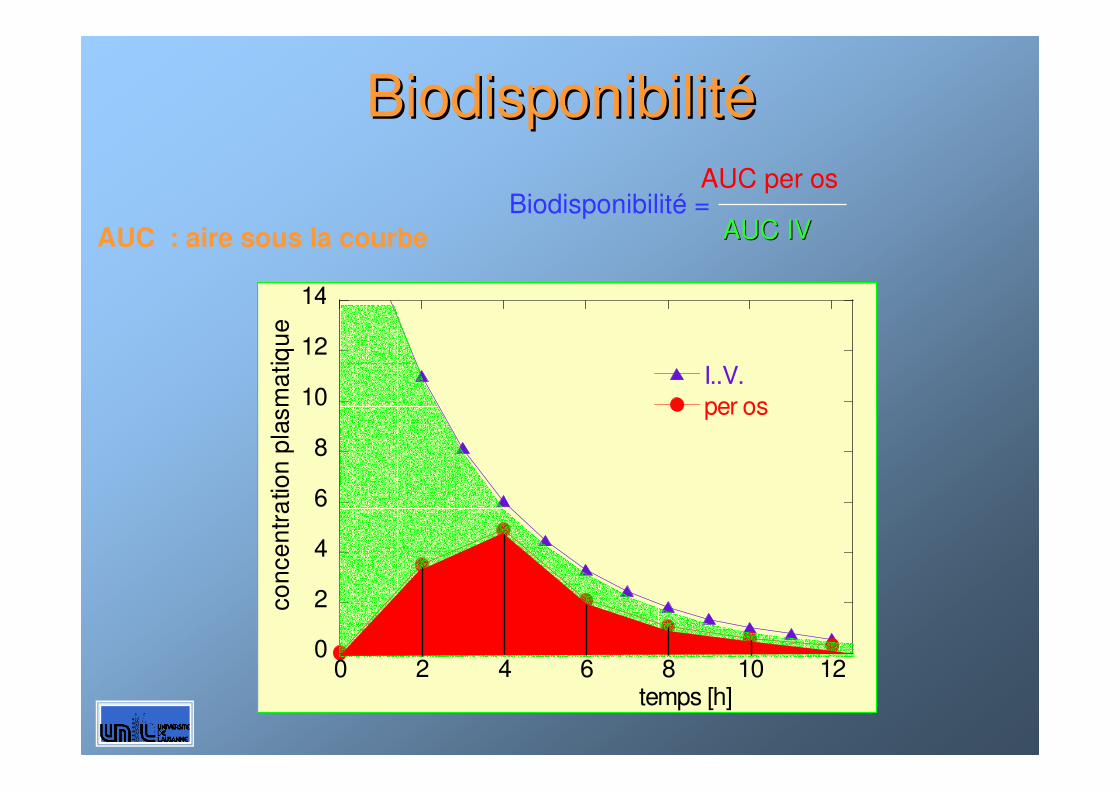
AUC per osBiodisponibilité =
AUC IVAUC IVAUC : aire sous la courbe
BiodisponibilitBiodisponibilitéé
0
2
4
6
8
10
12
14
0 2 4 6 8 10 12
I..V.per os
conc
entr
atio
n pl
asm
atiq
ue
temps [h]
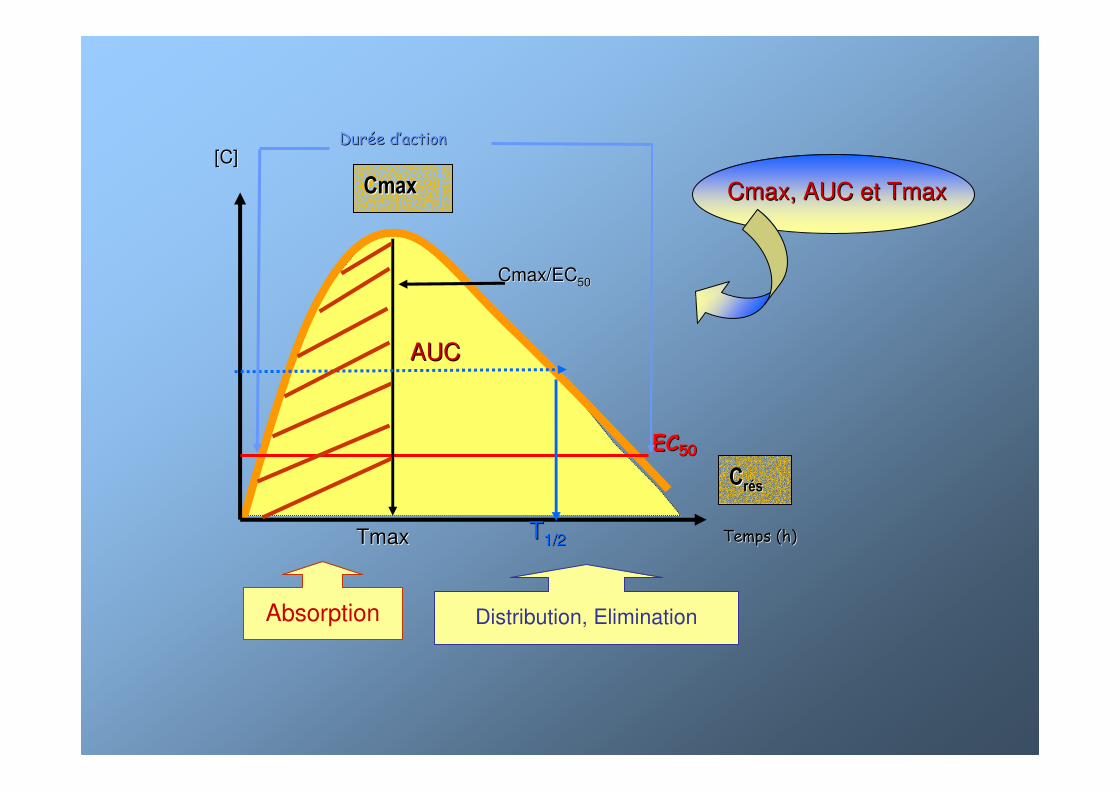
Temps (h)Temps (h)
ECEC5050
CCrrééss
CmaxCmax
Cmax/ECCmax/EC5050
[C][C]
TmaxTmax
AUCAUC
TT1/21/2
DurDuréée de d’’actionaction
Absorption Distribution, Elimination
Cmax, AUC et TmaxCmax, AUC et Tmax

Voies dVoies d’’administration parentadministration parentééralesrales• i.v., intraveineuse = directement dans le compartiment
central
• i.m. intramusculaire , ou s.c. sous-cutanée :
– injection d’un petit volume de solution concentrée
– la vitesse d’absorption dépend de la solubilitéet du débit sanguin dans le tissu concerné
(muscle > tissus sous-cutané)
• voie intra-artérielle : concentration plus élevée dans un territoire pendant la durée de la perfusion
• voie intrarachidienne : directement dans le LCR
• voie intrapéritonéale : absorption par une surface de 1-2 m2 de surface épithéliale

Voies dVoies d’’administration entadministration entééralesralesvoie oralevoie orale :
• résorption sublinguale: pour les substances à haut coefficient de perméabilité(ex. : nitroglycérine)
• résorption faible dans l’estomac (env. 1 m2 de surface muqueuse), épithélium « serré ».
• résorption surtout dans le grêle (env. 200 m2), épithélium « lâche ».
• vu le passage obligé par le système porte et le foie, effet de premier passage : métabolisme hépatique avant que la substance parvienne dans le compartiment central.
voie rectalevoie rectale : passage partiel (env. 30 %) par le système porte.
voie nasalevoie nasale : topique pour la muqueuse nasale systémique pour peptides (mais problème d’immunisation)
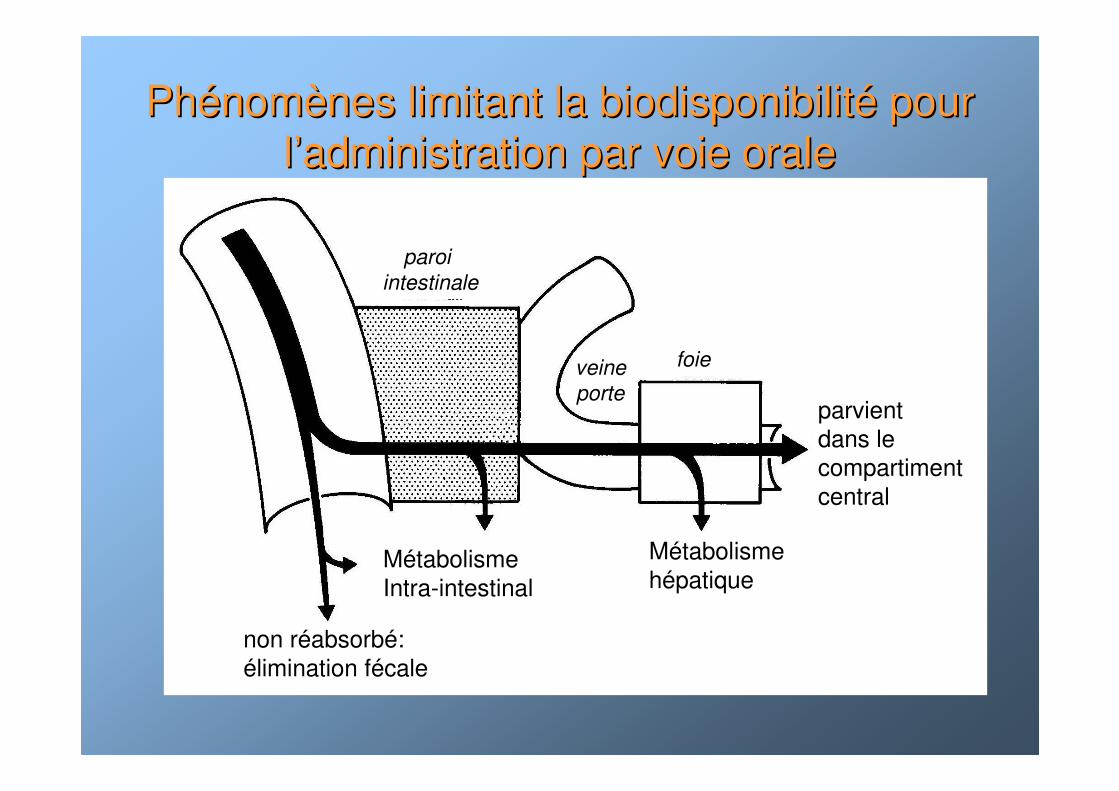
PhPhéénomnomèènes limitant la biodisponibilitnes limitant la biodisponibilitéé pour pour ll’’administration par voie oraleadministration par voie orale
MétabolismeIntra-intestinal
Métabolismehépatique
paroi
intestinale
foieveine
porte
non réabsorbé:élimination fécale
parvient dans le compartiment central

Administration par inhalationAdministration par inhalation
voie bronchiquevoie bronchique : • aérosols ou micro-particules qui se déposent sur les
muqueuses bronchiques• topique pour bronchodilatateurs et vasoconstricteurs• systémique : nicotine !
voie pulmonairevoie pulmonaire (alv(alvééolaire)olaire) ::• très grande surface et perfusion sanguine très
importante ==> absorption potentiellement très rapide des gaz ou substances volatiles
(utilisation clinique : surtout gaz anesthésiques,mais aussi important pour toxiques gazeux !)

Administration par voie cutanAdministration par voie cutanééee
•• topiquetopique : traitements dermatologiques
• ou systsystéémiquemique : quelques substances à haut Kp (petit poids moléculaire, liposoluble) par ex: nitroglycérine, oestrogènes, nicotine et autres toxiques : organophosphorés, DDT, fentanyl, …
• absorption très dépendante de l’état normal ou pathologique de la peau ! (attention par exemple aux effets systémiques des corticostéroïdes topiques par exemple).

DistributionDistribution
Liaison aux protLiaison aux protééinesinesVolume de distributionVolume de distribution
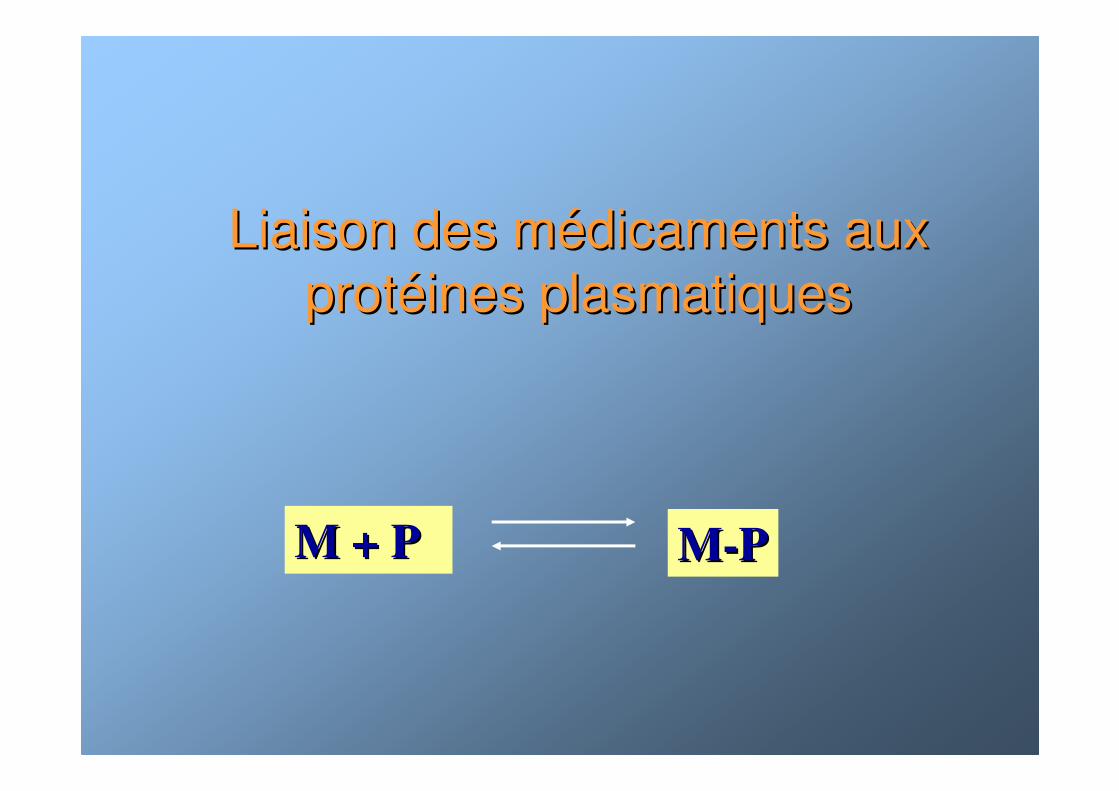
Liaison des mLiaison des méédicaments aux dicaments aux protprotééines plasmatiquesines plasmatiques
M + P M + P MM--PP

Les principaux paramLes principaux paramèètres cintres cinéétiquestiques
• Vd : Volume de distribution• C : concentration dans le compartiment central
• Q : quantité dans l’organisme
C = Q / Vd
Vd = Q / C
Vd
[C]
élimination

On a Vd = dose / C0 (concentration initiale). Par exemple, si
l'on injecte par voie intraveineuse 100 mg d'un médicament
et que sa concentration initiale, C0, dans le plasma est de
10 mg/L, le volume de distribution est de 10 L. Pour un
médicament donné, la connaissance de sa concentration
souhaitée dans le sang et de son volume de distribution
permet d'évaluer la dose à administrer.
Volume (apparent) de distribution (Vd)Volume (apparent) de distribution (Vd)

DDistributionistribution
rappel des volumes liquidiens de l’organisme : (pour un homme de 70 Kg)
• eau totale 60 % (80 - 50 %) 42 l.
• volume intracellulaire 40 % 28 l.
• volume extracellulaire 20 % 14 l.
• volume plasmatique 5 % 3.5 l.
Volume de distribution apparent (Vd)Volume de distribution apparent (Vd)
C = Q / VdC = Q / Vd Vd = Q / CVd = Q / C

Tissus rTissus rééservoirsservoirs• tissus adipeux : toutes les substances liposolubles
• os : plomb, fluor, aluminium, tétracyclines
• diverses protéines tissulaires : amiodarone, cadmium
• acides nucléiques : chloroquine
Effets des tissus réservoirs sur la cinétique d’élimination :
• pour une clairance constante, l’élimination est d’autant plus lente que le volume de distribution est grand.
Ke = Cl / Vd
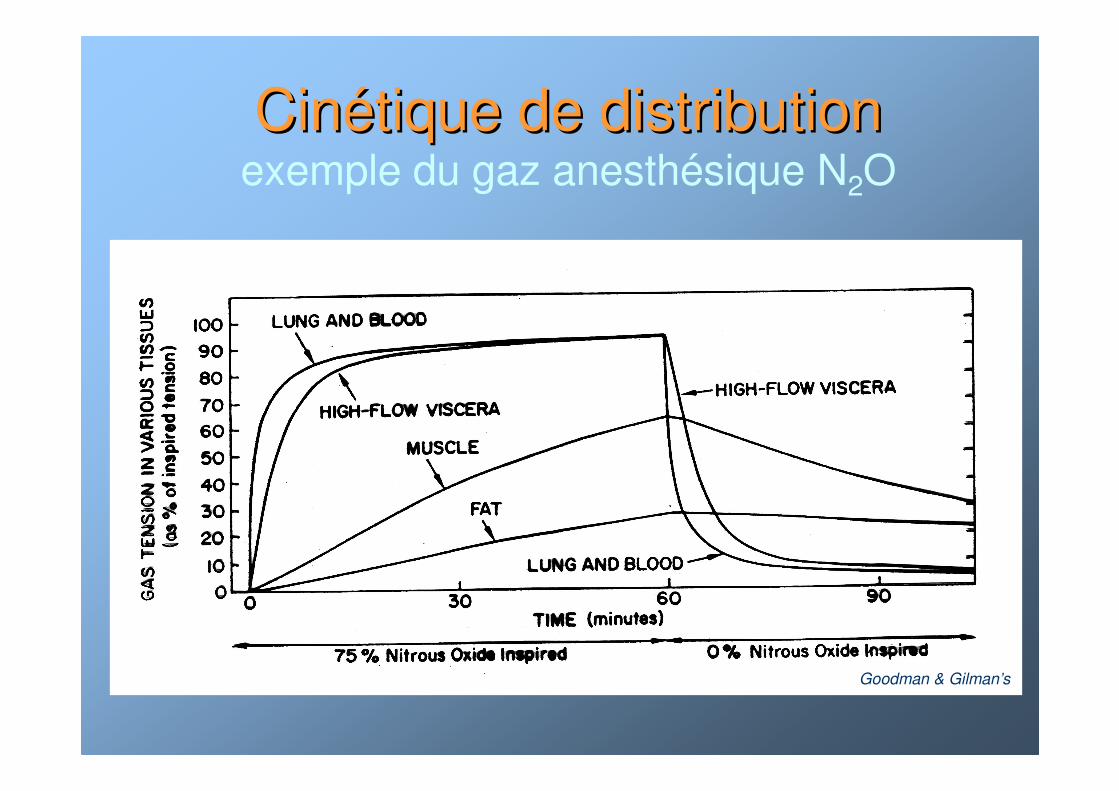
CinCinéétique de distributiontique de distributionexemple du gaz anesthésique N2O
Goodman & Gilman’s

La liaison aux protLa liaison aux protééines (I)ines (I)
☯ La liaison aux protéines est généralement réversible. La fraction liée doit-être considérée comme une forme de stockage
☯ Seule la fraction libre d ’un médicament est diffusible et active. Sa concentration est déterminante pour l ’activitépharmacologique.
☯ Seule la fraction libre est filtrée au niveau rénal.

La liaison aux protLa liaison aux protééines (II)ines (II)
�������� 22èème me cible, les tissus et organes cible, les tissus et organes ::
La fixation se fait en quantité variable selon le principe actif et les organes.
Elle est généralement réversible.
Elle dépend de l ’affinité respective pour les protéines tissulaires et plasmatiques.
Après défixation le médicament quitte le tissu pour rejoindre le sang et y être éliminé..

Liaison aux protLiaison aux protééines plasmatiquesines plasmatiques
Exemples de protéines plasmatiques qui lient les médicaments:• albumine 0.5 - 0.7 mM très nombreuses substances
• glycoprotéine acide 10 µM subst. Basiques
• transcortine ~1 µM cortisol
liaisons : dépendent du pH sanguin � attention aux modifications de fraction libre en cas d’acidose ou d’alcalose
taux de liaison très variables : la fraction libre peut varier entre 0.1 % et 100 % (tableau)

Liaison aux protLiaison aux protééines plasmatiquesines plasmatiques
Attention aux interactions possibles par compétition entre les substances qui se lient aux mêmes sites : �déplacement d’une première substance par une autre qui se lie au même site
p. ex. dicoumarol (anticoagulant oral) et plusieurs anti-inflammatoires, anti-diabétiques oraux, etc.
+

MMéétabolismetabolisme
InductionInduction
InhibitionInhibition
PharmacogPharmacogéénnéétiquetique

MMéétabolisme des mtabolisme des méédicamentsdicamentsprincipaux tissus responsables du métabolisme des xénobiotiques : surtout le foie
mais aussi rein, tube digestif, poumon, peau, enzymes plasmatiques, etc.
Phase IOxydation hépatiqueCytochrome P 450
Introduction ou exposition d’un groupe réactif
Phase II
Réactions de conjugaisonTous les tissus
hydrosolubilité
Résultat du passage par les deux phases : - production d’un dérivé conjugué hautement soluble qui rend l’élimination rénale possible (en particulier par sécrétion tubulaire active pour certains conjugués anioniques)

Sites Importants du MSites Importants du Méétabolisme Mtabolisme Méédicamenteuxdicamenteux
• Ensemble du tractus GI
• Foie
• Rein
• Cerveau
• Sang
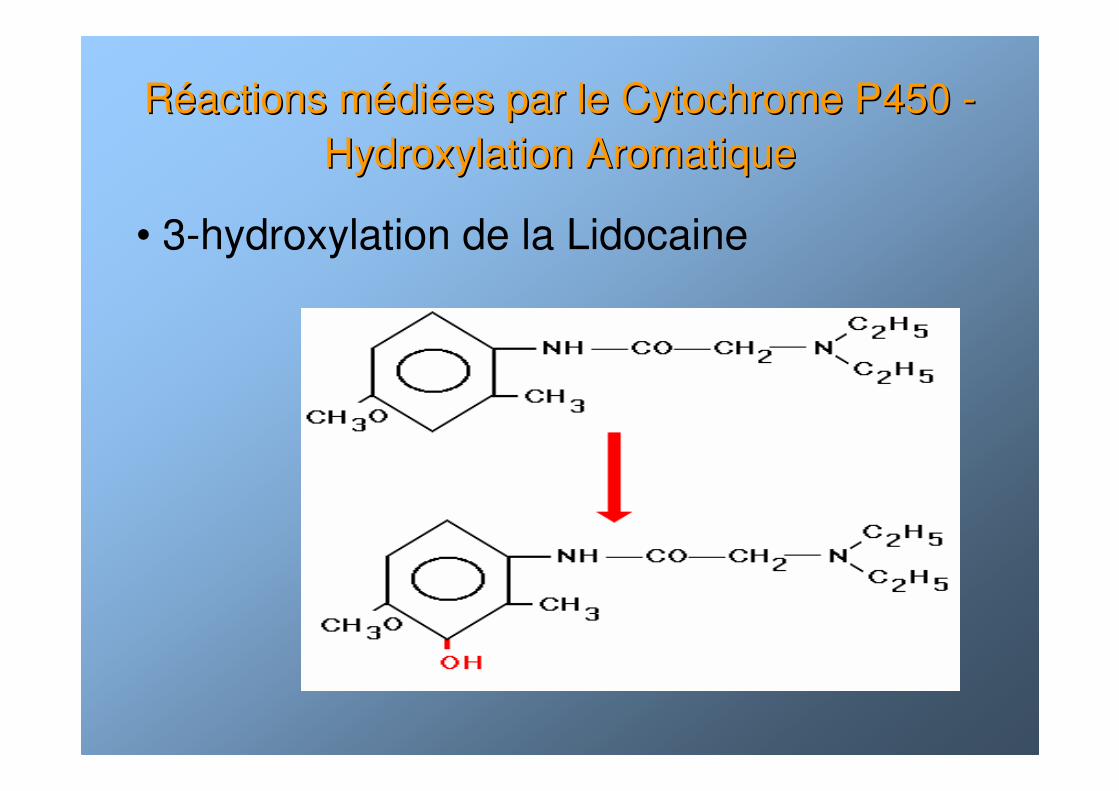
RRééactions mactions méédidiéées par le Cytochrome P450 es par le Cytochrome P450 --Hydroxylation AromatiqueHydroxylation Aromatique
• 3-hydroxylation de la Lidocaine
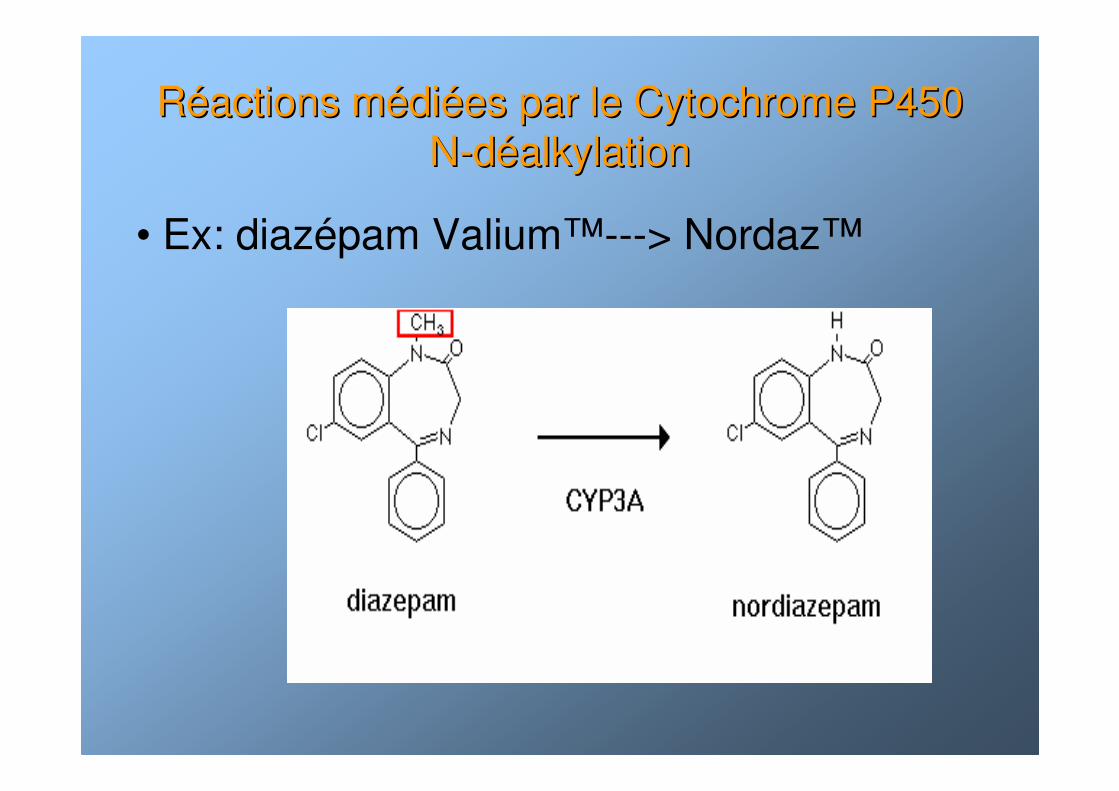
RRééactions mactions méédidiéées par le Cytochrome P450 es par le Cytochrome P450 NN--ddééalkylationalkylation
• Ex: diazépam Valium™---> Nordaz™
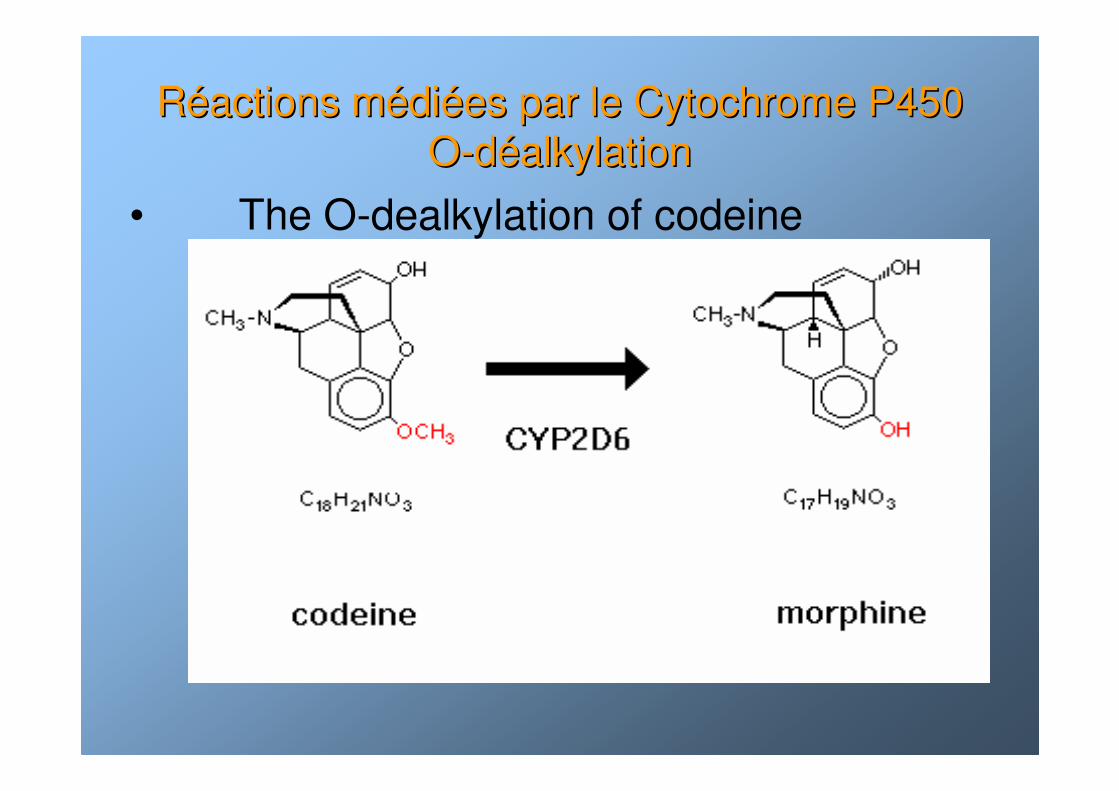
RRééactions mactions méédidiéées par le Cytochrome P450 es par le Cytochrome P450 OO--ddééalkylationalkylation
• The O-dealkylation of codeine

Causes de variation du mCauses de variation du méétabolisme tabolisme des xdes xéénobiotiquesnobiotiques
1.1. InductionInduction(m(méédicaments, composants alimentaires,dicaments, composants alimentaires,contaminants de lcontaminants de l’’environnement)environnement)
2.2. InhibitionInhibition3.3. Polymorphisme gPolymorphisme géénnéétiquetique4.4. ÂgeÂge5.5. PathologiePathologie
}

Induction: exemples•• phphéénobarbitalnobarbital : induction du CYP3A mais aussi nombreux autres enzymes
hépatiques, amplification du RE. Activation peu spécifique du métabolisme hépatique de nombreuses substances.
•• benzpyrbenzpyrèènene : stimulation très rapide et très importante du CYP1A2 (epoxydation du benzpyrène et d’autres hydrocarbures aromatiques polycycliques).
•• PCBPCB (polychlorinated biphenyl) : induction du métabolisme des stéroïdes
Conséquences de l’induction :
Accélération considérable de la vitesse d’élimination de toutes les substances métabolisées par le système induit (demi-vie diminuée), médicaments mais aussi hormones endogènes
mais aussi
formation accélérée de métabolites toxiques ou carcinogènes
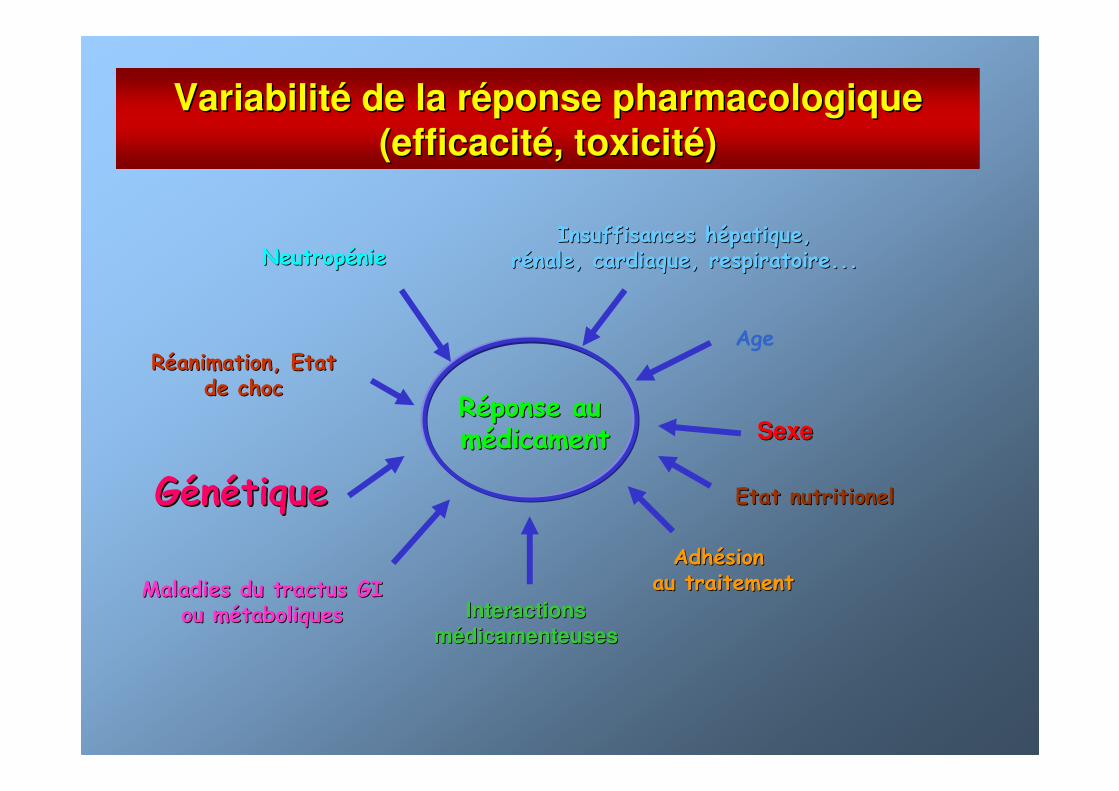
VariabilitVariabilitéé de la rde la rééponse pharmacologiqueponse pharmacologique
(efficacit(efficacitéé, toxicit, toxicitéé))
RRééponse au ponse au mméédicamentdicament
NeutropNeutropéénienieInsuffisances hInsuffisances héépatique,patique,
rréénale, cardiaque, respiratoire...nale, cardiaque, respiratoire...
AgeRRééanimation, Etat animation, Etat
de chocde choc
Maladies du tractus GIMaladies du tractus GIou mou méétaboliquestaboliques
GGéénnéétiquetique
AdhAdhéésion sion au traitementau traitement
InteractionsInteractions
mméédicamenteusesdicamenteuses
SexeSexe
Etat nutritionelEtat nutritionel

DDééfinitionsfinitions
PharmacogPharmacogéénnéétiquetique : étude de l'influence du
polymorphisme génétique au niveau d'un gène
spécifique sur la réponse au traitement. C'est un
élément important de la variabilité interindividuelle
de cette réponse.
Les gènes qui codent pour les protéines impliquées
dans la pharmacocinétique des médicaments peuvent
présenter des anomalies de séquence

Polymorphisme génétique de la N-Acétylation NAT-2
• Années1940 : Neuropathie Périphérique notée chez les patients traités pour la tuberculose.
• 1959 : Facteurs Génétiques influençant les taux sanguins d’INH chez l’homme.
• Trans Conf Chemother Tuberc 1959: 8, 52–56.• Molécule mère : active, neurotoxique• métabolites acétylés non actifs, hépatotoxiques

EEliminationlimination
Principaux organes d’élimination
•• ReinsReins : élimination urinaires des substances hydrosolubles (ou rendues hydrosolubles par le métabolisme hépatique)
•• FoieFoie : élimination biliaire, puis fécale d’autres susbtances
•• PoumonPoumon : élimination de substances gazeuses ou volatiles
•• Peau, phanPeau, phanééresres…
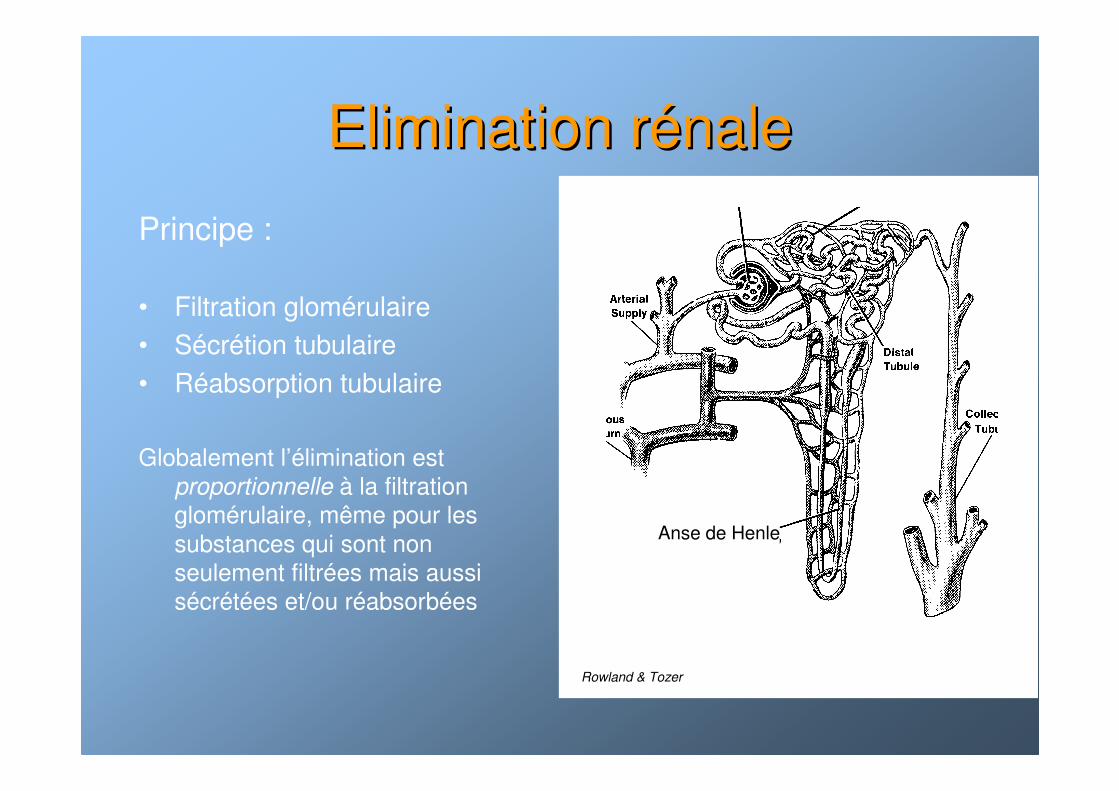
Elimination rElimination réénalenale
Principe :
• Filtration glomérulaire• Sécrétion tubulaire• Réabsorption tubulaire
Globalement l’élimination est proportionnelle à la filtration glomérulaire, même pour les substances qui sont non seulement filtrées mais aussi sécrétées et/ou réabsorbées
Schéma rein
Anse de Henle
Rowland & Tozer

Filtration glomFiltration gloméérulairerulaire
• Filtration glomérulaire mesurée par la clairance à la créatinine
• Pour une substance seulement filtrée (ni sécrétée, ni réabsorbée) : clairance = environ 100 ml/min (adulte de 70 kg)
Ce qui correspond, pour une volume de distribution de 20 l par exemple, à une demi-vie de 200 minutes
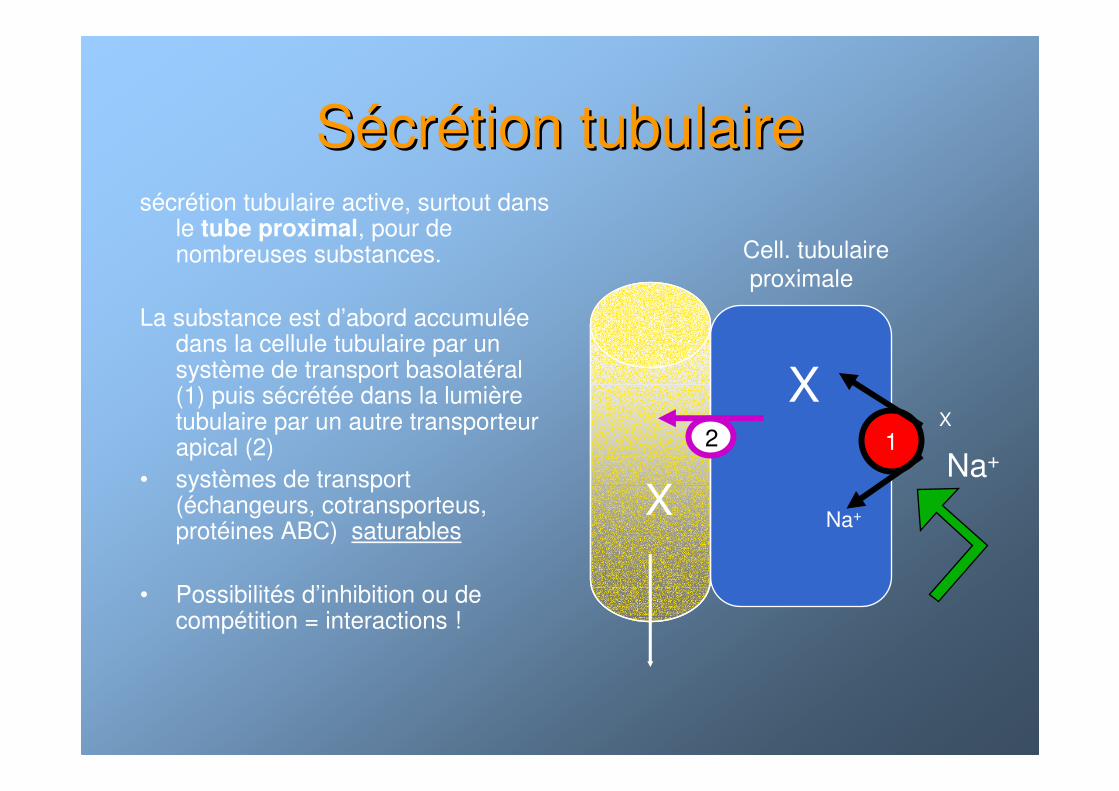
SSéécrcréétion tubulairetion tubulairesécrétion tubulaire active, surtout dans
le tube proximal, pour de nombreuses substances.
La substance est d’abord accumulée dans la cellule tubulaire par un système de transport basolatéral (1) puis sécrétée dans la lumière tubulaire par un autre transporteur apical (2)
• systèmes de transport (échangeurs, cotransporteus, protéines ABC) saturables
• Possibilités d’inhibition ou de compétition = interactions !
1X
X
Na+
Na+
X
Cell. tubulaireproximale
22
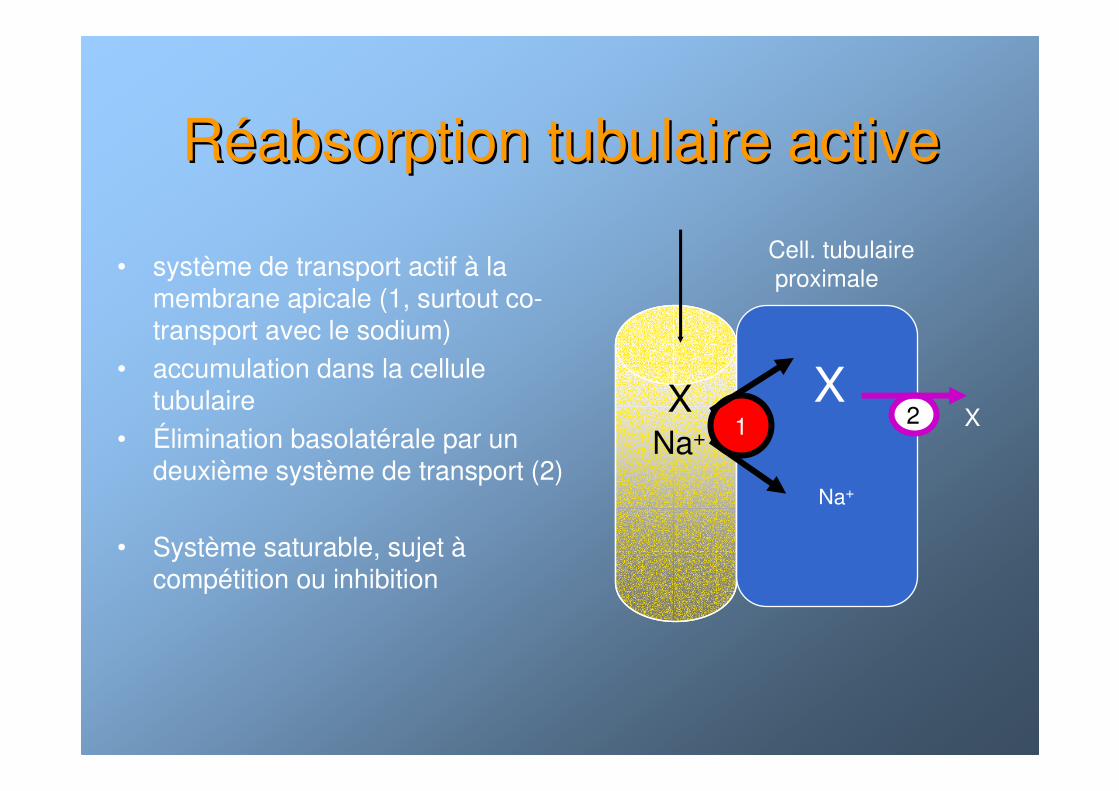
RRééabsorption tubulaire activeabsorption tubulaire active
• système de transport actif à la membrane apicale (1, surtout co-transport avec le sodium)
• accumulation dans la cellule tubulaire
• Élimination basolatérale par un deuxième système de transport (2)
• Système saturable, sujet àcompétition ou inhibition
XX
Na+
X
Cell. tubulaireproximale
2Na+ 1

RRééabsorption tubulaire passiveabsorption tubulaire passive
Diffusion due à la haute concentration tubulaire suite à la réabsorption d’eau le long du tubule.
• dépend de la perméabilité membranaire de la substance • réabsorption passive très importante pour les substances liposolubles de faible
poids moléculaire• Dépend du débit urinaire (réabsorption d’eau plus ou moins importante)
Pour les acides et les bases faibles la réabsorption dépend de la fraction non-ionique• acide faible fortement ionisé (= peu réabsorbé) dans une urine alcaline
alcalinisation des urines ==> élimination plus rapide• L’inverse pour une base faible : acidification ==> élimination plus rapide
d’où modification du pH urinaire pour élimination plus rapide d’un toxiqueex : acidification urinaire lors d’une intoxication aux amphétamines
G

Adaptation des doses en cas de capacité d’élimination diminuée
Insuffisance hépatique : adaptation difficile (pas de mesures faciles de la capacité métabolique hépatique) :
• en cas d’insuffisance hépatique, on cherche à limiter l’utilisation de médicaments éliminés totalement ou principalement par le foie.
Insuffisance rénale : adaptation possible et nécessaire, grâce à la disponibilitéd’une bonne mesure de la fonction rénale par la clairance à la créatinine.
Adaptation nécessaire si
• Clcréat < 60 ml/min et
• élimination principalement par voir rénale

Elimination hépatique et digestive
élimination par sécrétion biliaire de substances plus ou moins lipophiles
• ex : stéroïdes, divers produits du métabolisme hépatique
• calcul de la clairance hépatique :
Clhep = débit sanguin hep X fraction d’excrétion
fraction d’excretion : (Cart-Cvein)/Cart
• réabsorption intestinale possible : cycle entéro-hépatique
ce qui ralentit considérablement l’éliminationde la substance ou de ses métabolites
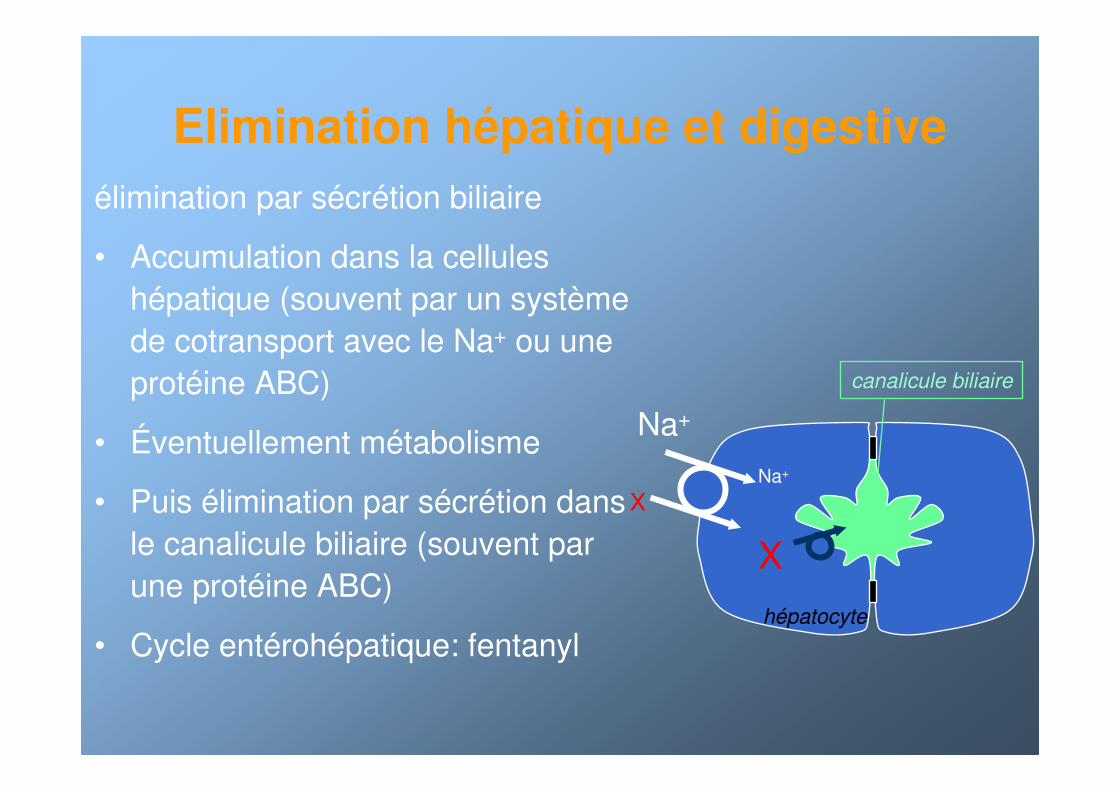
Elimination hépatique et digestive
élimination par sécrétion biliaire
• Accumulation dans la cellules hépatique (souvent par un système de cotransport avec le Na+ ou une protéine ABC)
• Éventuellement métabolisme
• Puis élimination par sécrétion dans le canalicule biliaire (souvent par une protéine ABC)
• Cycle entérohépatique: fentanyl
X
X
Na+
Na+
hépatocyte
canalicule biliaire

E < 0.3
Cl dépend surtout du débit sanguin
E > 0.7
Cl dépend essentiellement de la fraction libre
ClairancesClairances

Adaptation des doses Adaptation des doses àà une fonction une fonction rréénale diminunale diminuééee
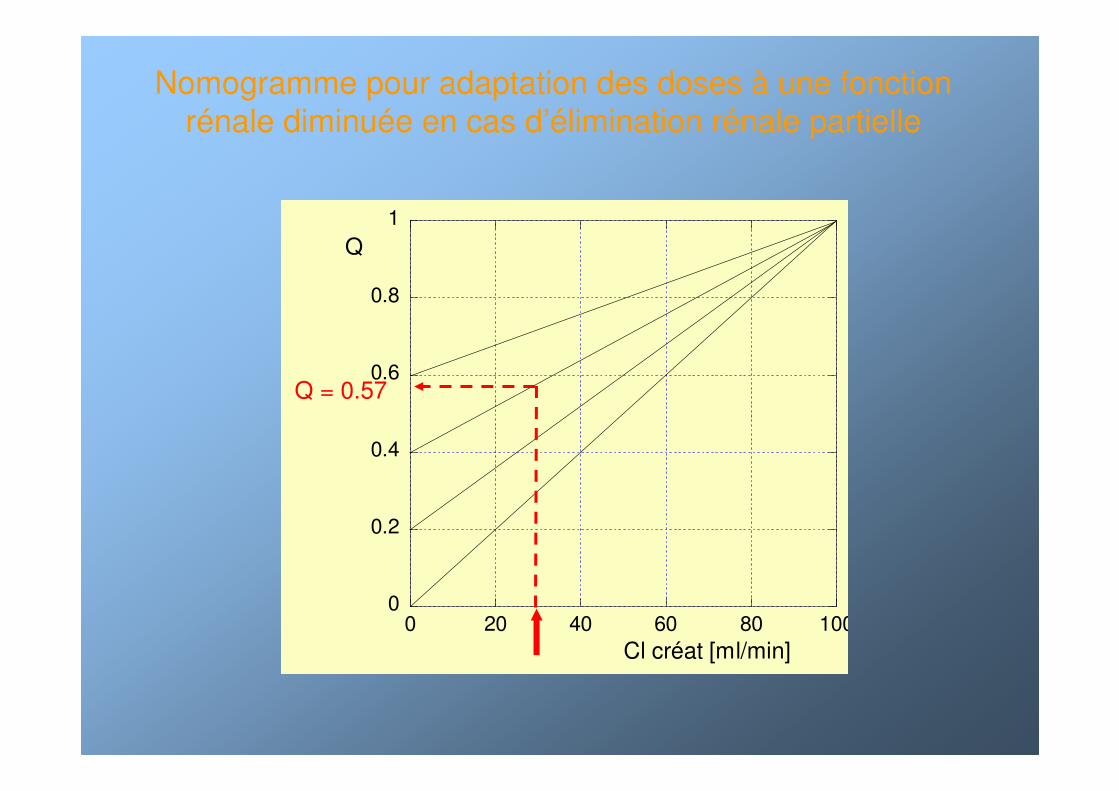
Nomogramme pour adaptation des doses à une fonction rénale diminuée en cas d’élimination rénale partielle
0
0.2
0.4
0.6
0.8
1
0 20 40 60 80 100
Q
Cl créat [ml/min]
Q = 0.57
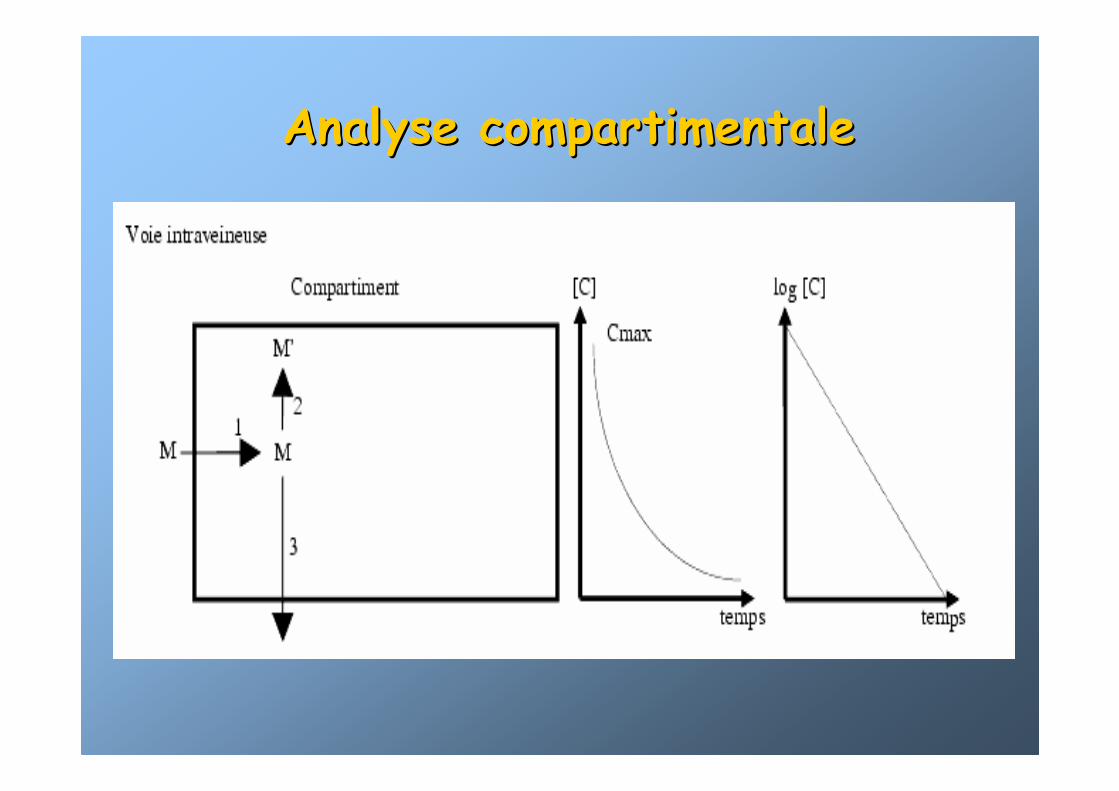
Analyse compartimentaleAnalyse compartimentale
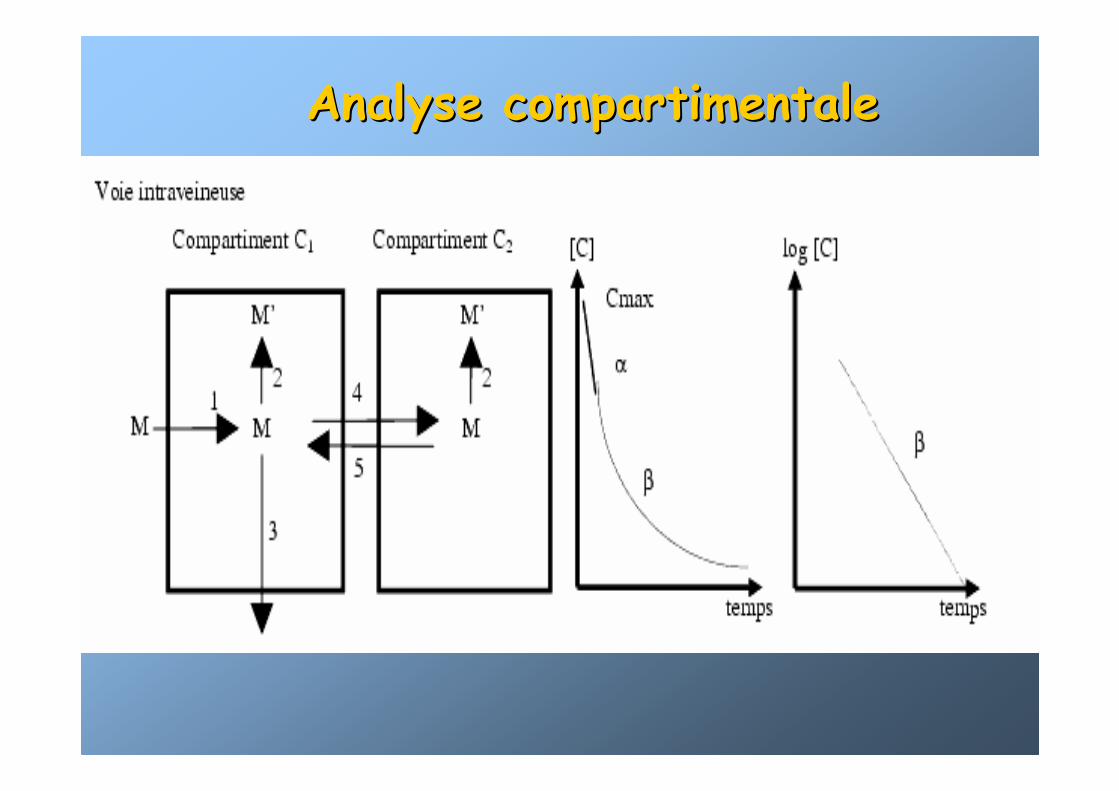
Analyse compartimentaleAnalyse compartimentale
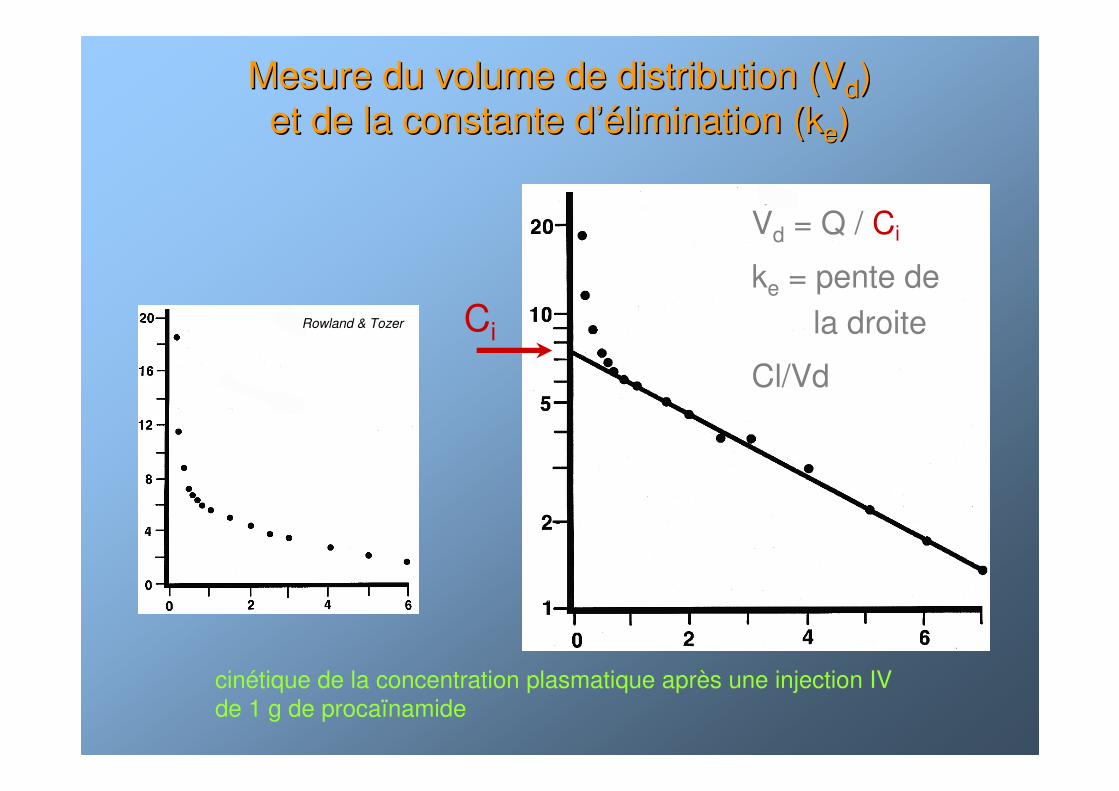
Mesure du volume de distribution (VMesure du volume de distribution (Vdd) ) et de la constante det de la constante d’é’élimination (klimination (kee))
cinétique de la concentration plasmatique après une injection IV de 1 g de procaïnamide
Ci
Vd = Q / Ci
ke = pente de la droite
Cl/Vd
Rowland & Tozer
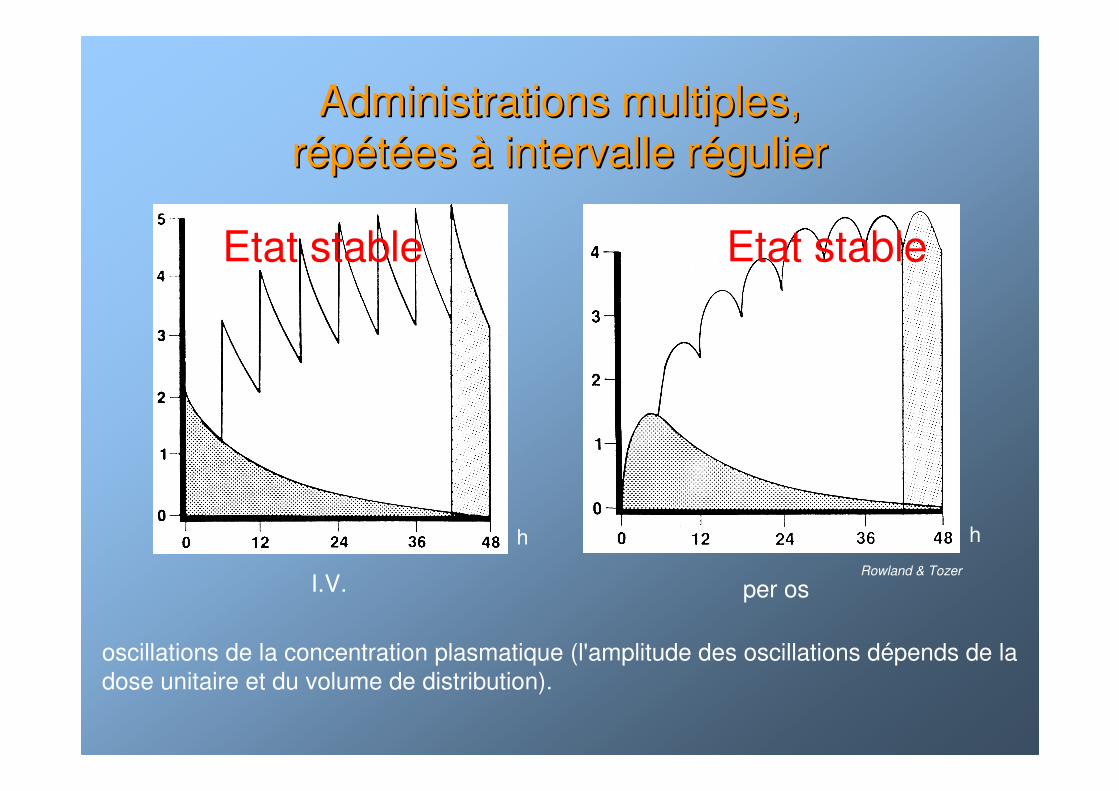
Administrations multiples, Administrations multiples, rrééppééttéées es àà intervalle rintervalle rééguliergulier
oscillations de la concentration plasmatique (l'amplitude des oscillations dépends de ladose unitaire et du volume de distribution).
I.V. per os
hhRowland & Tozer
Etat stableEtat stable
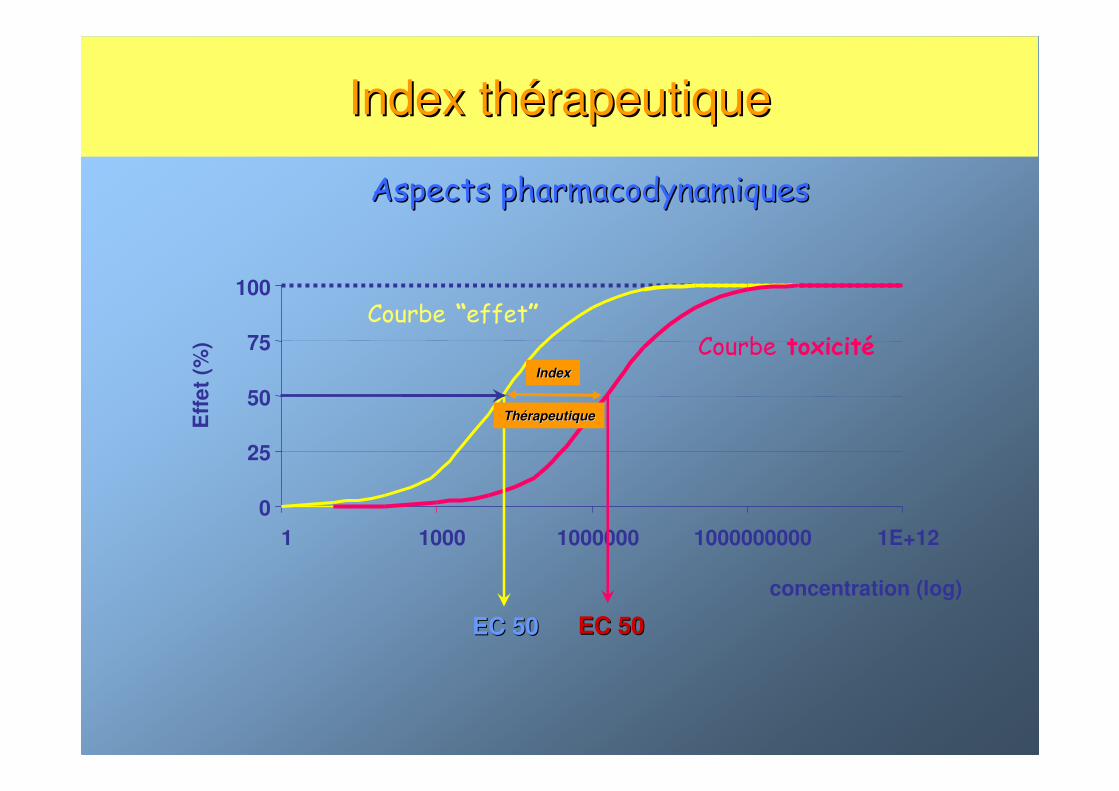
Index thIndex théérapeutiquerapeutique
0
25
50
75
100
1 1000 1000000 1000000000 1E+12
concentration (log)
Eff
et
(%) Courbe toxicité
Courbe “effet”
EC 50EC 50 EC 50EC 50
IndexIndex
ThThéérapeutiquerapeutique
Aspects pharmacodynamiquesAspects pharmacodynamiques
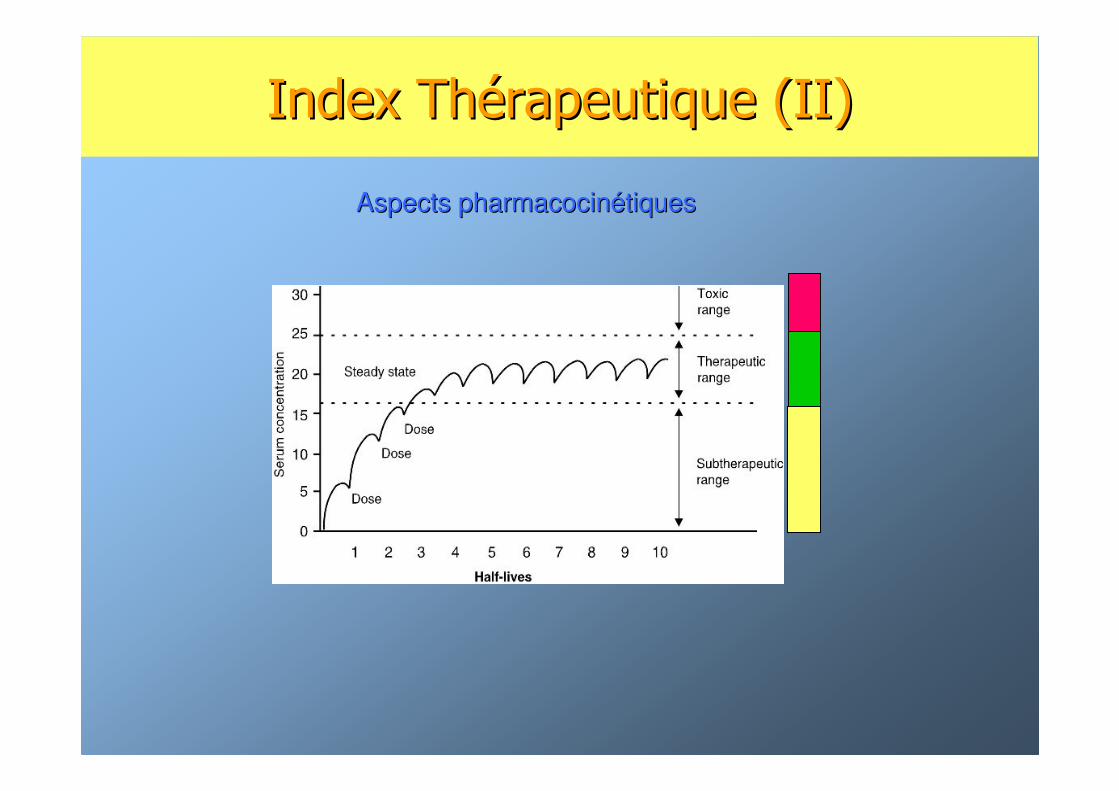
Index ThIndex Théérapeutique (II)rapeutique (II)
Aspects pharmacocinAspects pharmacocinéétiquestiques

MMéédicaments dicaments àà faible index thfaible index théérapeutiquerapeutique
☯ ImmunosuppresseursImmunosuppresseurs
☯☯ AntiAnti--éépileptiquespileptiques
☯☯ AntirAntiréétrovirauxtroviraux
☯☯ Certains antibiotiquesCertains antibiotiques
☯☯ DigitaliquesDigitaliques
☯ AntiAnti--canccancééreuxreux
☯ AntiAnti--coagulantscoagulants
☯☯ Certains psychotropesCertains psychotropes
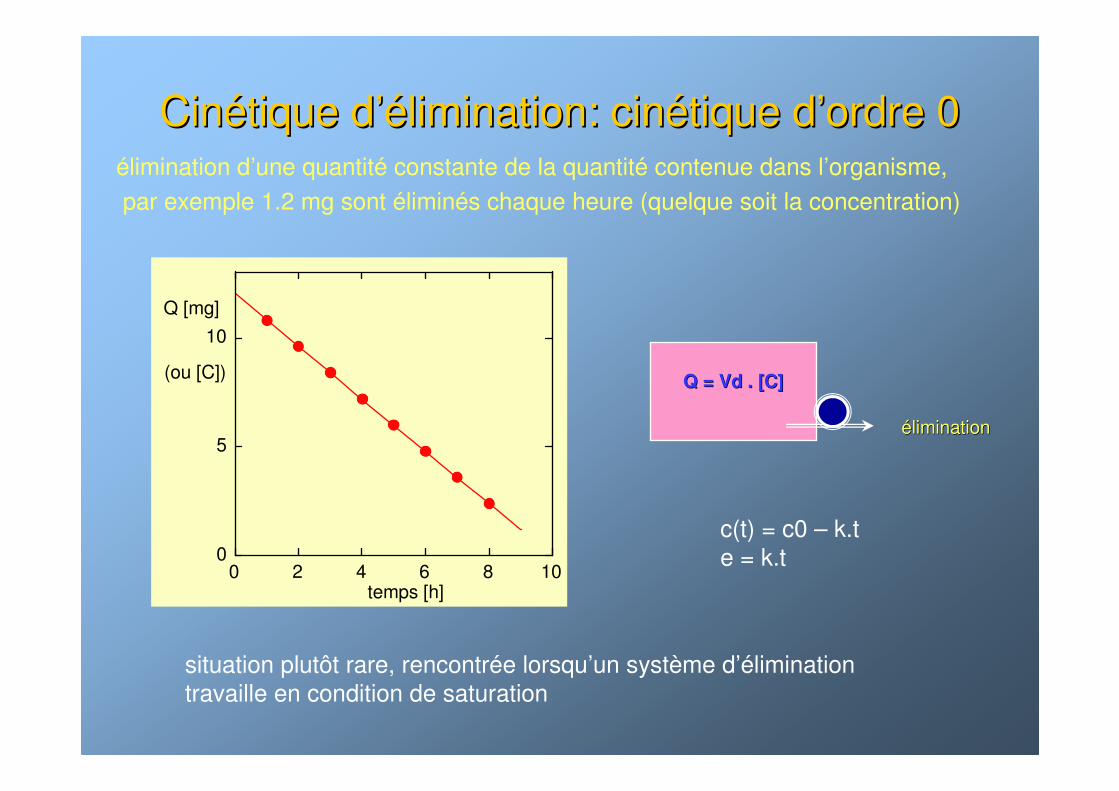
CinCinéétique dtique d’é’élimination: cinlimination: cinéétique dtique d’’ordre 0ordre 0élimination d’une quantité constante de la quantité contenue dans l’organisme, par exemple 1.2 mg sont éliminés chaque heure (quelque soit la concentration)
Q = Vd . [C]Q = Vd . [C]
ééliminationlimination
0
5
10
0 2 4 6 8 10
Q [mg]
(ou [C])
temps [h]
situation plutôt rare, rencontrée lorsqu’un système d’élimination travaille en condition de saturation
c(t) = c0 – k.te = k.t
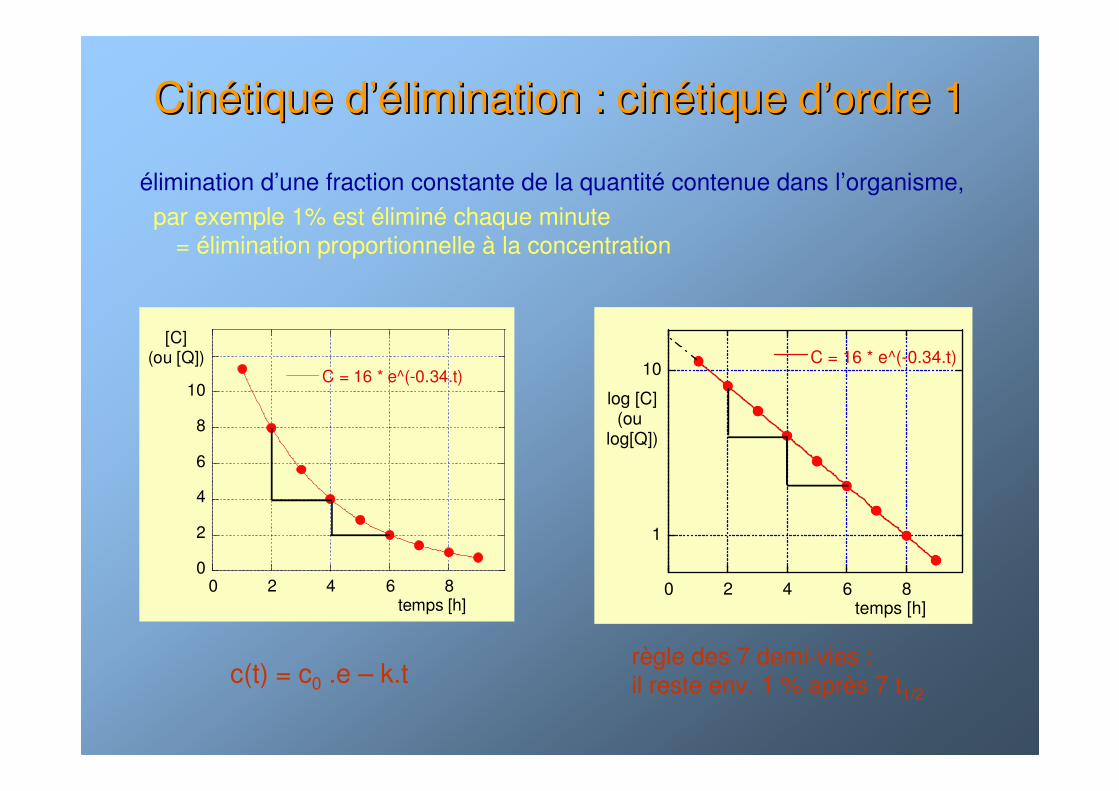
CinCinéétique dtique d’é’élimination : cinlimination : cinéétique dtique d’’ordre 1ordre 1
élimination d’une fraction constante de la quantité contenue dans l’organisme,par exemple 1% est éliminé chaque minute
= élimination proportionnelle à la concentration
0
2
4
6
8
10
0 2 4 6 8
C = 16 * e^(-0.34.t)
temps [h]
[C](ou [Q])
1
10
0 2 4 6 8
C = 16 * e^(-0.34.t)
temps [h]
log [C](ou
log[Q])
règle des 7 demi-vies : il reste env. 1 % après 7 t1/2
c(t) = c0 .e – k.t

Constante dConstante d’é’élimination, demilimination, demi--vie et clairancevie et clairance
• ke [1/s] : constante d’élimination
ke = Cl / Vd
• demi-vie (t1/2) [s] : temps nécessaire àl’élimination de la moitié de la substance
• Clairance (Cl) totale (ou métabolique) [ml/min] = volume épuré par minute
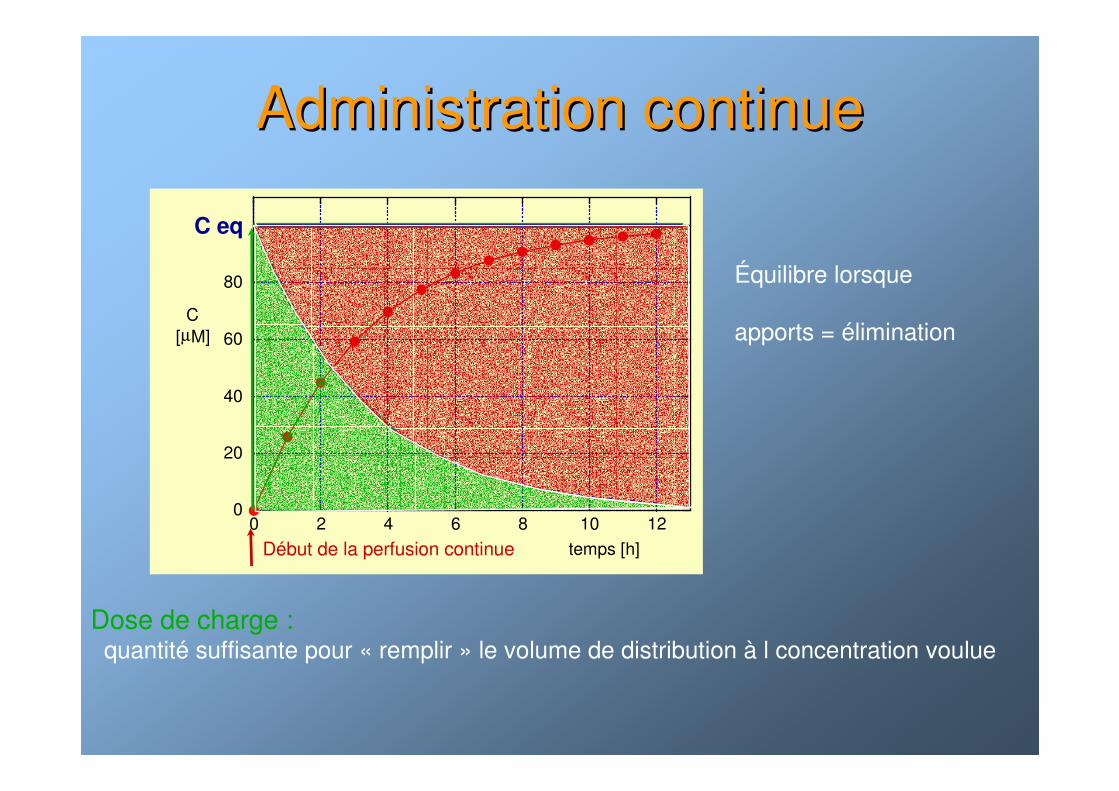
Administration continueAdministration continue
Équilibre lorsque
apports = élimination
0
20
40
60
80
0 2 4 6 8 10 12
C[µM]
temps [h]
C eq
Dose de charge :quantité suffisante pour « remplir » le volume de distribution à l concentration voulue
Début de la perfusion continue
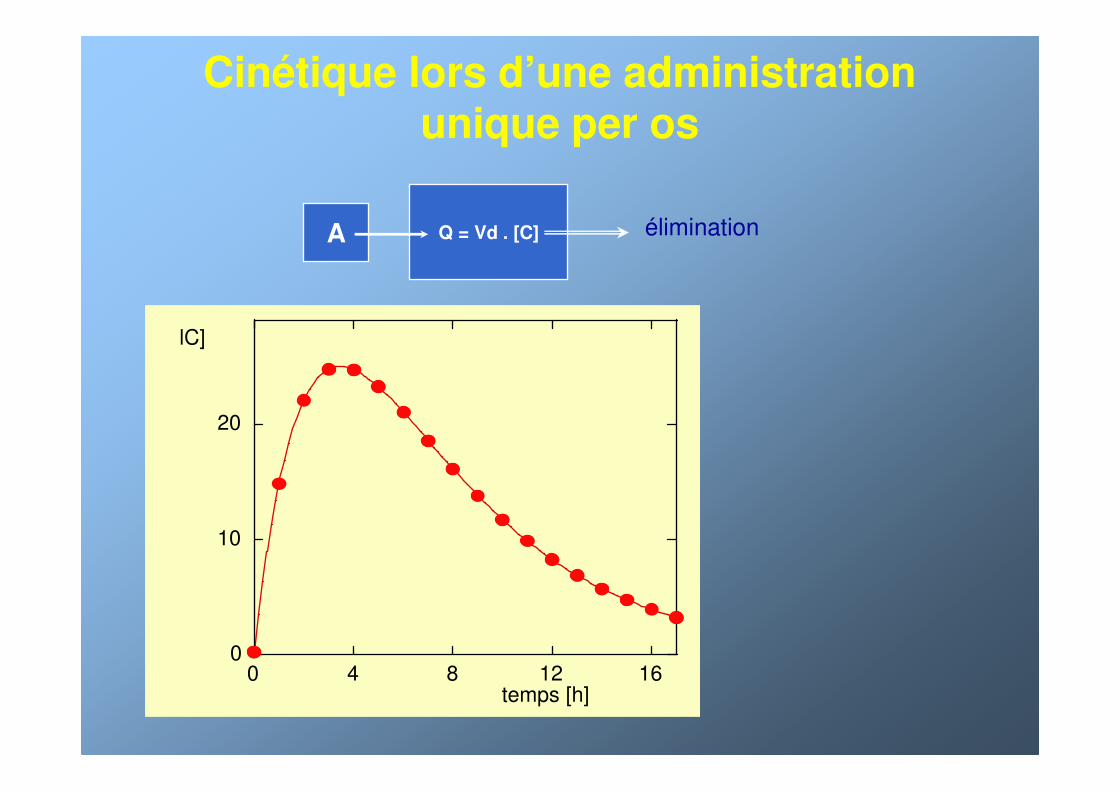
Cinétique lors d’une administration unique per os
Q = Vd . [C] éliminationA
0
10
20
0 4 8 12 16temps [h]
lC]





























