Embed Size (px)
Citation preview

Dernières avancées de la chimie organiqueÉtude des applications récentes utilisant la spectroscopie in situ
Adrian Burke, Dominique Hebrault, METTLER TOLEDO
Les transformations catalysées par des métaux représentent un domaine clé de la recherche universitaire autant pour leur intérêt scientifique, qu’en raison du grand nombre de réactions essentielles dans l'industrie. Aujourd’hui, les chercheurs sont principalement confrontés à la nécessité de déterminer avec précision les points de départ et de fin des réactions, et de recueillir suffisamment de données pour bien comprendre, caractériser et optimiser les réactions chimiques. Outre la nécessité de réaliser de nombreuses recherches avec peu de ressources et dans des délais courts, les chercheurs doivent trouver des moyens innovants de recueillir les informations requises pour mener à bien leurs tâches.
La spectroscopie in situ IR est de plus en plus utilisée dans le domaine de la chimie
organique de synthèse, car elle fournit des informations qui aident les chercheurs à clarifier le mécanisme, la cinétique et le déroulement de réactions très diverses.
Le ReactIR™ est un système d'analyse de réaction en temps réel in situ utilisé par les chercheurs pour approfondir leur compréhension de la chimie organique. Le ReactIR™ permet de contrôler la chimie réactive à l'aide de la spectroscopie infrarouge moyen, qui est parfaitement connue. Une sonde ATR résistante est insérée directement dans la cuve pour offrir une vidéo moléculaire de la réaction. Elle permet d'observer les variations de concentration de toutes les principales espèces réactives et intermédiaires pour identifier le déroulement et le mécanisme du procédé. Une analyse cinétique précise est réalisée en collectant de façon
automatisée un ensemble complet de données analysées au moyen du logiciel iC Kinetics™, qui génère un modèle cinétique avec un nombre d'expériences inférieur à celui de l'approche traditionnelle.
Ce livre blanc présente quatre exemples tirés de la recherche universitaire dans lesquels le ReactIR™ a été utilisé pour déterminer les principaux paramètres de chaque étude. Le but n'est pas d'entrer dans des détails scientifiques précis. Ces détails ont été consignés dans les publications concernées, dont la lecture est vivement recommandée. En revanche, les auteurs décrivent ici le contexte d'utilisation du système ReactIR™ et comment il les a aidés à répondre à des questions essentielles.
Transformations catalysées par des métaux
2
+
R CO2R'
N2
catalyseur
CH2Cl2 40°CPh R
CO2R'
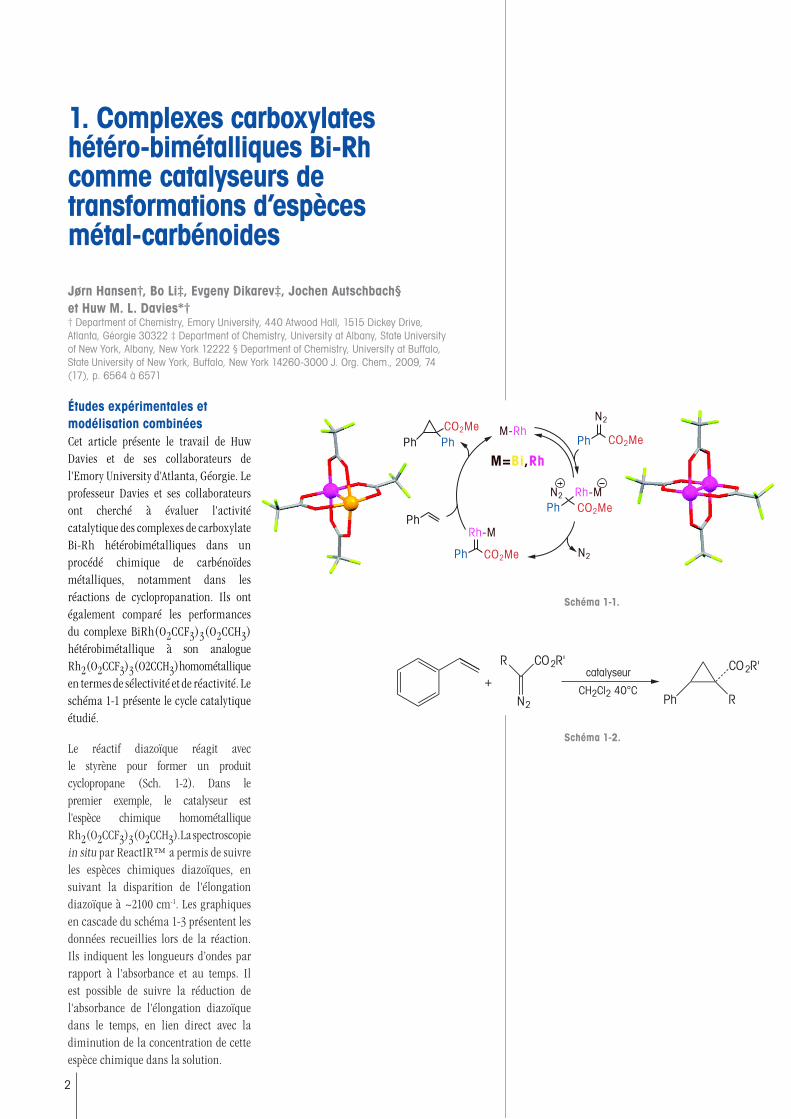
Études expérimentales et modélisation combinéesCet article présente le travail de Huw Davies et de ses collaborateurs de l'Emory University d'Atlanta, Géorgie. Le professeur Davies et ses collaborateurs ont cherché à évaluer l'activité catalytique des complexes de carboxylate Bi-Rh hétérobimétalliques dans un procédé chimique de carbénoïdes métalliques, notamment dans les réactions de cyclopropanation. Ils ont également comparé les performances du complexe BiRh(O2CCF3)3(O2CCH3) hétérobimétallique à son analogue Rh2(O2CCF3)3(O2CCH3) homométallique en termes de sélectivité et de réactivité. Le schéma 1-1 présente le cycle catalytique étudié.
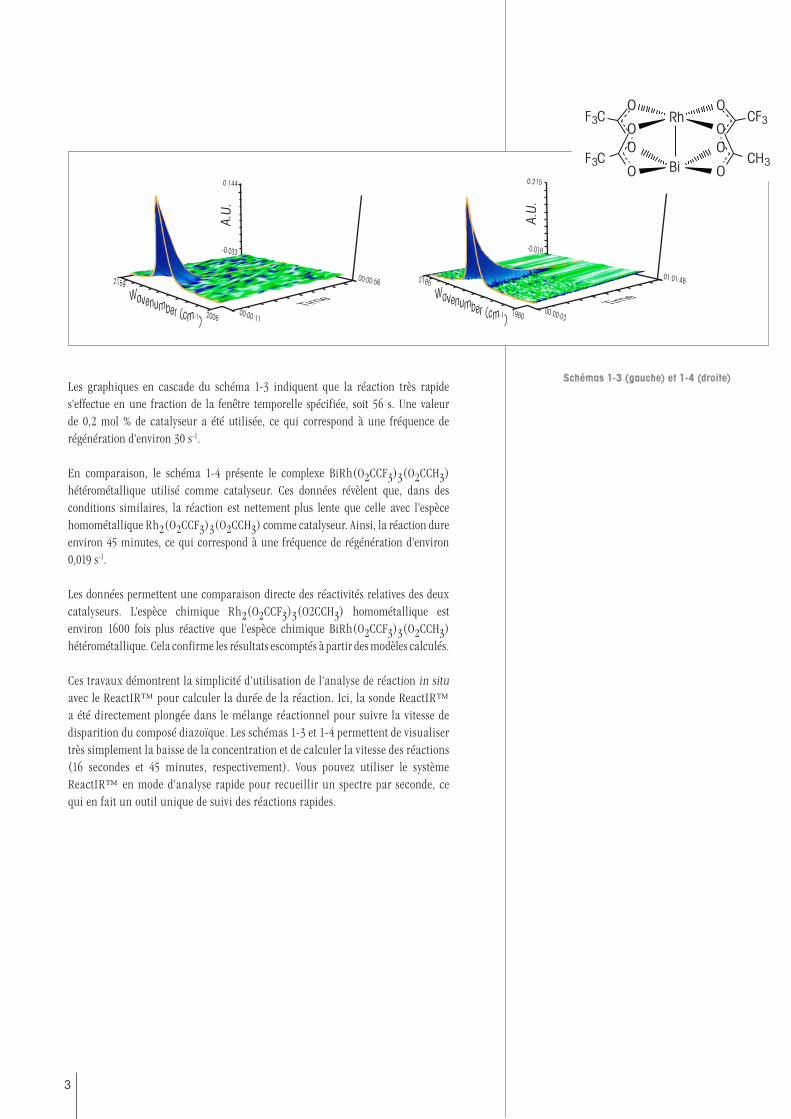
Le réactif diazoïque réagit avec le styrène pour former un produit cyclopropane (Sch. 1-2). Dans le premier exemple, le catalyseur est l'espèce chimique homométallique Rh2(O2CCF3)3(O2CCH3). La spectroscopie in situ par ReactIR™ a permis de suivre les espèces chimiques diazoïques, en suivant la disparition de l'élongation diazoïque à ~2100 cm-1. Les graphiques en cascade du schéma 1-3 présentent les données recueillies lors de la réaction. Ils indiquent les longueurs d’ondes par rapport à l'absorbance et au temps. Il est possible de suivre la réduction de l'absorbance de l'élongation diazoïque dans le temps, en lien direct avec la diminution de la concentration de cette espèce chimique dans la solution.
Jørn Hansen†, Bo Li‡, Evgeny Dikarev‡, Jochen Autschbach§ et Huw M. L. Davies*† † Department of Chemistry, Emory University, 440 Atwood Hall, 1515 Dickey Drive, Atlanta, Géorgie 30322 ‡ Department of Chemistry, University at Albany, State University of New York, Albany, New York 12222 § Department of Chemistry, University at Buffalo, State University of New York, Buffalo, New York 14260-3000 J. Org. Chem., 2009, 74 (17), p. 6564 à 6571
1. Complexes carboxylates hétéro-bimétalliques Bi-Rh comme catalyseurs de transformations d’espèces métal-carbénoides
CO2MeN2
N2
N2
Rh-M
M-Rh
M=Bi,Rh
Rh-M
CO2Me
CO2Me
CO2Me
PhPh
Ph
Ph
Ph
Ph
Schéma 1-1.
Schéma 1-2.
3
Les graphiques en cascade du schéma 1-3 indiquent que la réaction très rapide s'effectue en une fraction de la fenêtre temporelle spécifiée, soit 56 s. Une valeur de 0,2 mol % de catalyseur a été utilisée, ce qui correspond à une fréquence de régénération d'environ 30 s-1.
En comparaison, le schéma 1-4 présente le complexe BiRh(O2CCF3)3(O2CCH3) hétérométallique utilisé comme catalyseur. Ces données révèlent que, dans des conditions similaires, la réaction est nettement plus lente que celle avec l’espèce homométallique Rh2(O2CCF3)3(O2CCH3) comme catalyseur. Ainsi, la réaction dure environ 45 minutes, ce qui correspond à une fréquence de régénération d'environ 0,019 s-1.
Les données permettent une comparaison directe des réactivités relatives des deux catalyseurs. L'espèce chimique Rh2(O2CCF3)3(O2CCH3) homométallique est environ 1600 fois plus réactive que l'espèce chimique BiRh(O2CCF3)3(O2CCH3) hétérométallique. Cela confirme les résultats escomptés à partir des modèles calculés.
Ces travaux démontrent la simplicité d'utilisation de l'analyse de réaction in situ avec le ReactIR™ pour calculer la durée de la réaction. Ici, la sonde ReactIR™ a été directement plongée dans le mélange réactionnel pour suivre la vitesse de disparition du composé diazoïque. Les schémas 1-3 et 1-4 permettent de visualiser très simplement la baisse de la concentration et de calculer la vitesse des réactions (16 secondes et 45 minutes, respectivement). Vous pouvez utiliser le système ReactIR™ en mode d'analyse rapide pour recueillir un spectre par seconde, ce qui en fait un outil unique de suivi des réactions rapides.
Schémas 1-3 (gauche) et 1-4 (droite)
Wavenumber (cm-1)
2159
-0.033
A.U.
0.144
200600:00:11
00:00:56
Time
Wavenumber (cm-1)
2185
-0.018
A.U.
0.215
198000:00:02
01:01:48
Time
Wavenumber (cm-1)
2159
-0.033
A.U.
0.144
200600:00:11
00:00:56
Time
Wavenumber (cm-1)
2185
-0.018
A.U.
0.215
198000:00:02
01:01:48
Time
F3C
F3C
CF3
CH 3
O
OO
Rh
BiO
O
OO
O
4
3b. forme hydratée de la carbamazépine
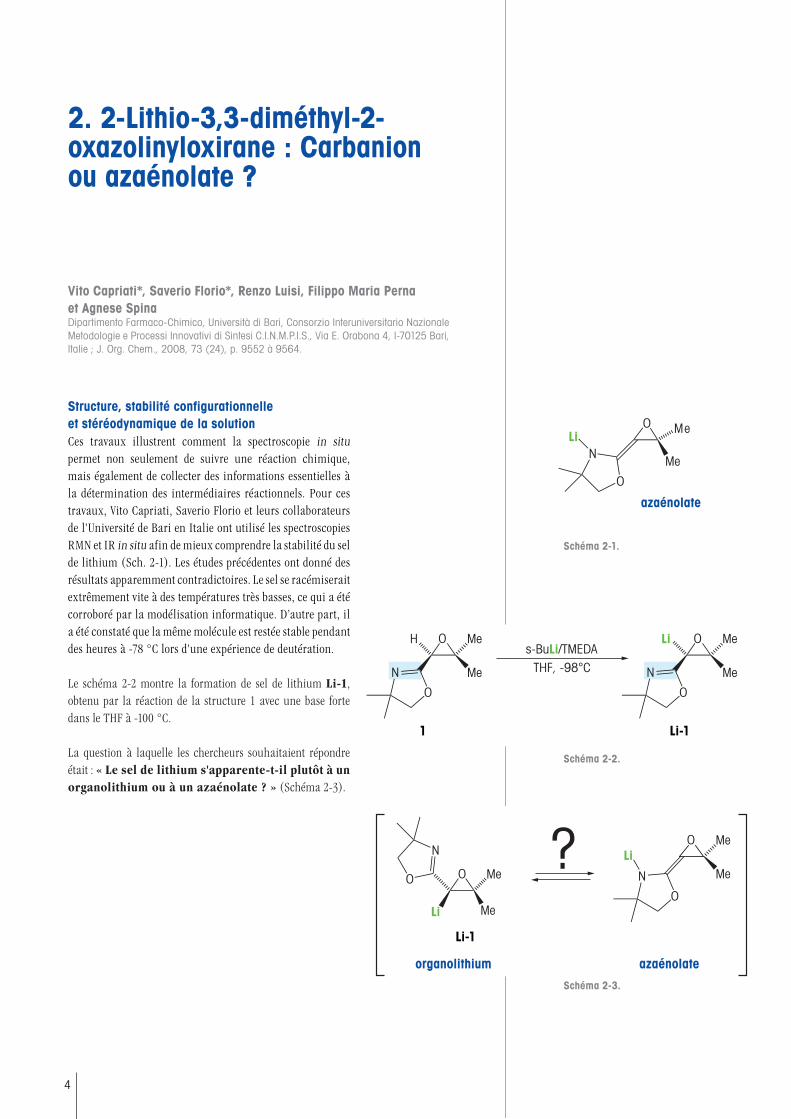
Structure, stabilité configurationnelle et stéréodynamique de la solutionCes travaux illustrent comment la spectroscopie in situ permet non seulement de suivre une réaction chimique, mais également de collecter des informations essentielles à la détermination des intermédiaires réactionnels. Pour ces travaux, Vito Capriati, Saverio Florio et leurs collaborateurs de l'Université de Bari en Italie ont utilisé les spectroscopies RMN et IR in situ afin de mieux comprendre la stabilité du sel de lithium (Sch. 2-1). Les études précédentes ont donné des résultats apparemment contradictoires. Le sel se racémiserait extrêmement vite à des températures très basses, ce qui a été corroboré par la modélisation informatique. D’autre part, il a été constaté que la même molécule est restée stable pendant des heures à -78 °C lors d'une expérience de deutération.
Le schéma 2-2 montre la formation de sel de lithium Li-1, obtenu par la réaction de la structure 1 avec une base forte dans le THF à -100 °C.
La question à laquelle les chercheurs souhaitaient répondre était : « Le sel de lithium s'apparente-t-il plutôt à un organolithium ou à un azaénolate ? » (Schéma 2-3).
Vito Capriati*, Saverio Florio*, Renzo Luisi, Filippo Maria Perna et Agnese Spina Dipartimento Farmaco-Chimico, Università di Bari, Consorzio Interuniversitario Nazionale Metodologie e Processi Innovativi di Sintesi C.I.N.M.P.I.S., Via E. Orabona 4, I-70125 Bari, Italie ; J. Org. Chem., 2008, 73 (24), p. 9552 à 9564.
2. 2-Lithio-3,3-diméthyl-2-oxazolinyloxirane : Carbanion ou azaénolate ?
Schéma 2-1.
Schéma 2-3.
Schéma 2-2.
O
NLi
O
Me
Me
azaénolate
O
N
Li
O Me
Me
?N
O
O Me
MeLi
azaénolateorganolithium
N
O
O MeH
Me
s-BuLi/TMEDATHF, -98°C N
O
O MeLi
Me
Li-11
Li-1
5
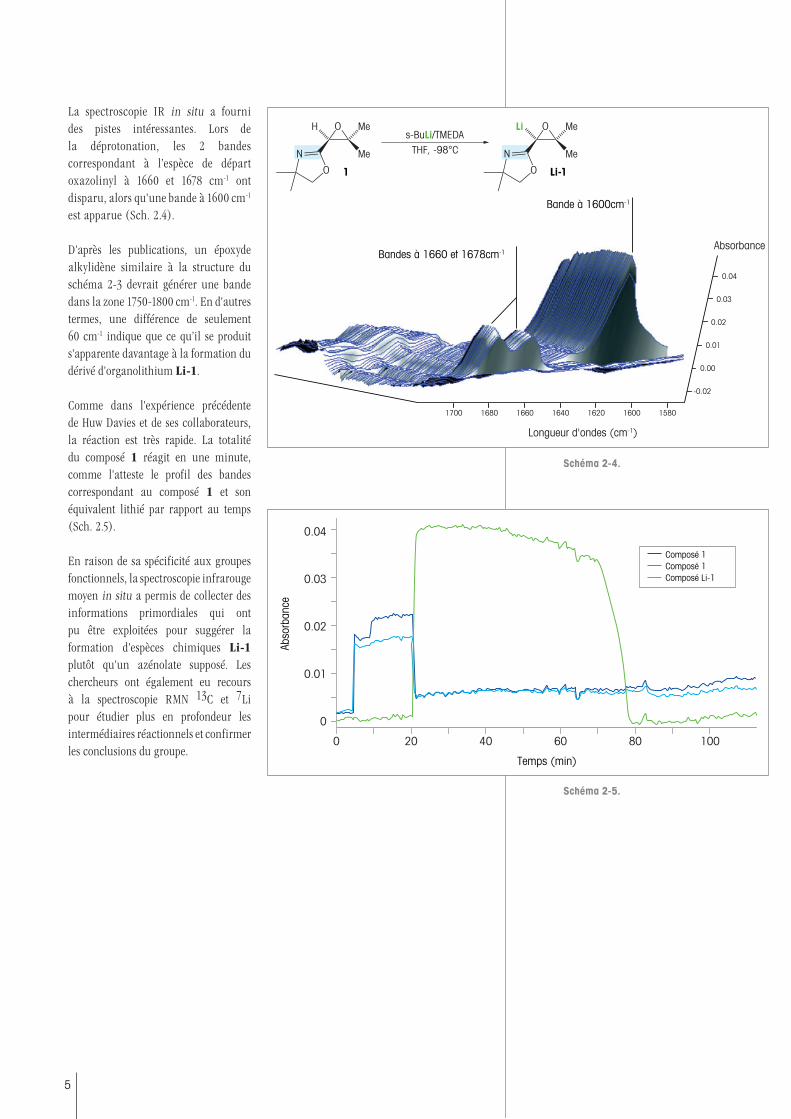
La spectroscopie IR in situ a fourni des pistes intéressantes. Lors de la déprotonation, les 2 bandes correspondant à l’espèce de départ oxazolinyl à 1660 et 1678 cm-1 ont disparu, alors qu'une bande à 1600 cm-1 est apparue (Sch. 2.4).
D'après les publications, un époxyde alkylidène similaire à la structure du schéma 2-3 devrait générer une bande dans la zone 1750-1800 cm-1. En d'autres termes, une différence de seulement 60 cm-1 indique que ce qu’il se produit s'apparente davantage à la formation du dérivé d'organolithium Li-1.
Comme dans l'expérience précédente de Huw Davies et de ses collaborateurs, la réaction est très rapide. La totalité du composé 1 réagit en une minute, comme l'atteste le profil des bandes correspondant au composé 1 et son équivalent lithié par rapport au temps (Sch. 2.5).
En raison de sa spécificité aux groupes fonctionnels, la spectroscopie infrarouge moyen in situ a permis de collecter des informations primordiales qui ont pu être exploitées pour suggérer la formation d'espèces chimiques Li-1 plutôt qu'un azénolate supposé. Les chercheurs ont également eu recours à la spectroscopie RMN 13C et 7Li pour étudier plus en profondeur les intermédiaires réactionnels et confirmer les conclusions du groupe.
0.04
0.03
0.02
0.01
0
0 1004020 8060
Abso
rban
ce
Composé 1Composé 1Composé Li-1
Temps (min)
Schéma 2-5.
Longueur d'ondes (cm-1)
1700 1680 1660 1640 1620 1600 1580
-0.02
0.00
0.01
0.02
0.03
0.04
AbsorbanceBandes à 1660 et 1678cm-1
Bande à 1600cm-1
Schéma 2-4.
N
O
O MeH
Me
s-BuLi/TMEDATHF, -98°C N
O
O MeLi
Me
Li-11

6
0.00
0.05
0.10
0.15
1000 980 960 940
Abso
rban
ce
Longueurs d'ondes (cm-1)
Schémas 3-2. (gauche) et 3-3. (droite)
Wei Shi†, Yingdong Luo†, Xiancai Luo†, Lei Chao†, Heng Zhang†, Jian Wang‡ et Aiwen Lei*† College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. Chine, et Mettler-Toledo AutoChem, 7075 Samuel Morse Drive, Columbia, Maryland 21046; J. Am. Chem. Soc., 2008, 130 (44), p 14713–14720 ; † Wuhan University, ‡ Mettler-Toledo AutoChem, Inc.
3. Réaction de couplage C(sp)-C(sp) efficace avec un catalyseur Palladium contenant un ligand phosphine-oléfine
Application et mécanismesCes travaux, menés par Aiwen Lei de la Wuhan University de Pékin, en Chine, en collaboration avec Mettler-Toledo AutoChem, Inc., portent sur un couplage catalysé par du Pd entre un haloalcyne 1 et un alcyne terminal 2 pour former le diyne 3 (Sch. 3-1). Les chercheurs ont eu recours à la spectroscopie IR in situ pour mesurer la vitesse de réaction dans différentes conditions et comprendre le mécanisme de la réaction. Un article des mêmes auteurs, publié dans Organic Letters, décrit un couplage de Negishi appliquant la même approche mécanistique5.
Ce procédé chimique est très efficace pour synthétiser les 1,3-diynes. Le schéma 3-2 montre que le bromoalcyne 1 et le diyne 3 possèdent des absorbances uniques à 973 et 955 cm-1, respectivement. Le schéma 3-3 révèle que l'étude de ces bandes dans le temps offre une vision claire de l'évolution de la réaction pour les réactifs et les produits formés.
R1 R2R2R1 Br
Pd (dba)2/L1NEt 3, Cul
DMF, r.t., 2-9h+
Schéma 3-1.
PPh2
O
L1=
1c= 3a=
985 975 965
Longueurs d'ondes (cm-1)
Abso
rban
ce
Temps
/min
955 945
0:08:00
0:28:00
0:48:00
0.01
0.00
0.02
0.03
0.04
955cm-1
973cm-1
1c
3a
0.05
Br OHOH
1c 973 cm-11c
973 cm-1
3a 955 cm-1
3a 955 cm-1
1 2 3
77
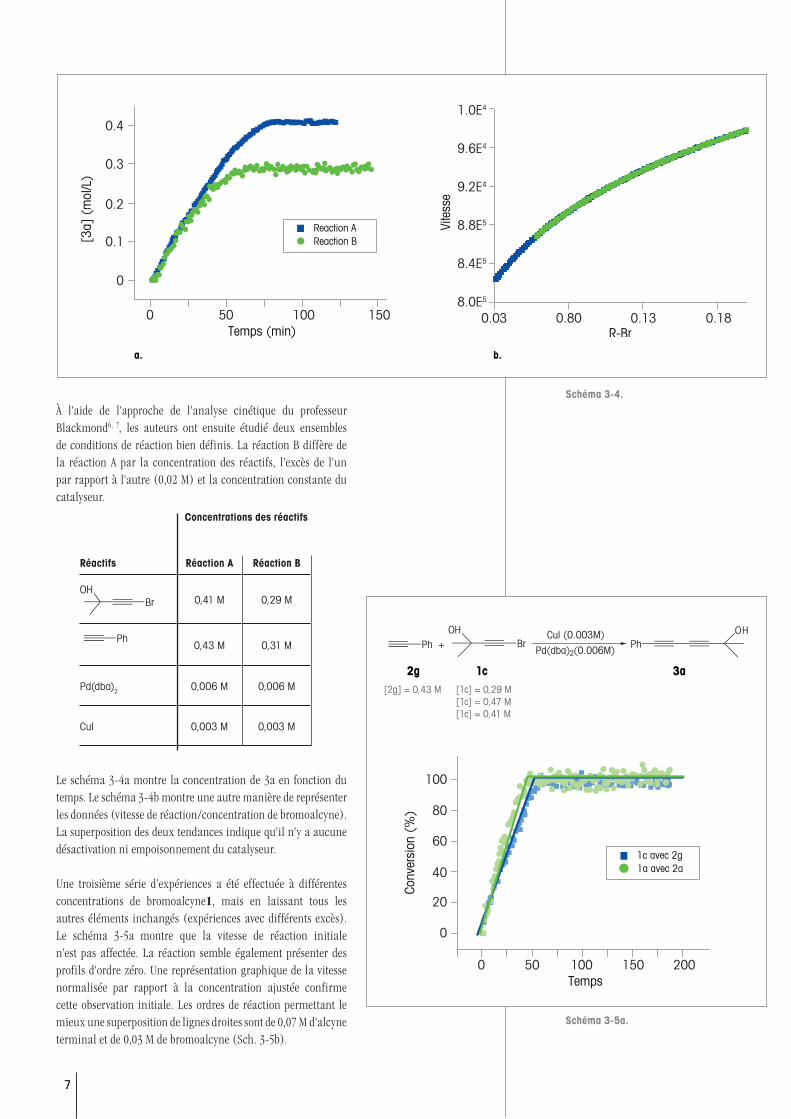
À l'aide de l'approche de l'analyse cinétique du professeur Blackmond6, 7, les auteurs ont ensuite étudié deux ensembles de conditions de réaction bien définis. La réaction B diffère de la réaction A par la concentration des réactifs, l’excès de l'un par rapport à l'autre (0,02 M) et la concentration constante du catalyseur.
Schéma 3-5a.
Ph PhBrOHOH
+Cul (0.003M)
Pd(dba)2(0.006M)
3a2g 1c[1c] = 0,29 M[1c] = 0,47 M[1c] = 0,41 M
[2g] = 0,43 M
Schéma 3-4.
a. b.
Le schéma 3-4a montre la concentration de 3a en fonction du temps. Le schéma 3-4b montre une autre manière de représenter les données (vitesse de réaction/concentration de bromoalcyne). La superposition des deux tendances indique qu'il n'y a aucune désactivation ni empoisonnement du catalyseur.
Une troisième série d’expériences a été effectuée à différentes concentrations de bromoalcyne1, mais en laissant tous les autres éléments inchangés (expériences avec différents excès). Le schéma 3-5a montre que la vitesse de réaction initiale n'est pas affectée. La réaction semble également présenter des profils d'ordre zéro. Une représentation graphique de la vitesse normalisée par rapport à la concentration ajustée confirme cette observation initiale. Les ordres de réaction permettant le mieux une superposition de lignes droites sont de 0,07 M d'alcyne terminal et de 0,03 M de bromoalcyne (Sch. 3-5b).
0
0.2
0.1
0.3
0.4
0 50 100 150
[3a]
(m
ol/L
)
Temps (min)
Reaction AReaction B
8.0E5
8.4E5
8.8E5
9.2E4
9.6E4
1.0E4
0.03 0.80 0.13 0.18
Vite
sse
R-Br
Réactifs Réaction A Réaction B
BrOH
0,41 M 0,29 M
Ph0,43 M 0,31 M
Pd(dba)2 0,006 M 0,006 M
CuI 0,003 M 0,003 M
Concentrations des réactifs
0
40
60
20
80
100
0 50 100 150 200
Conv
ersi
on (
%)
Temps
1c avec 2g1a avec 2a
8
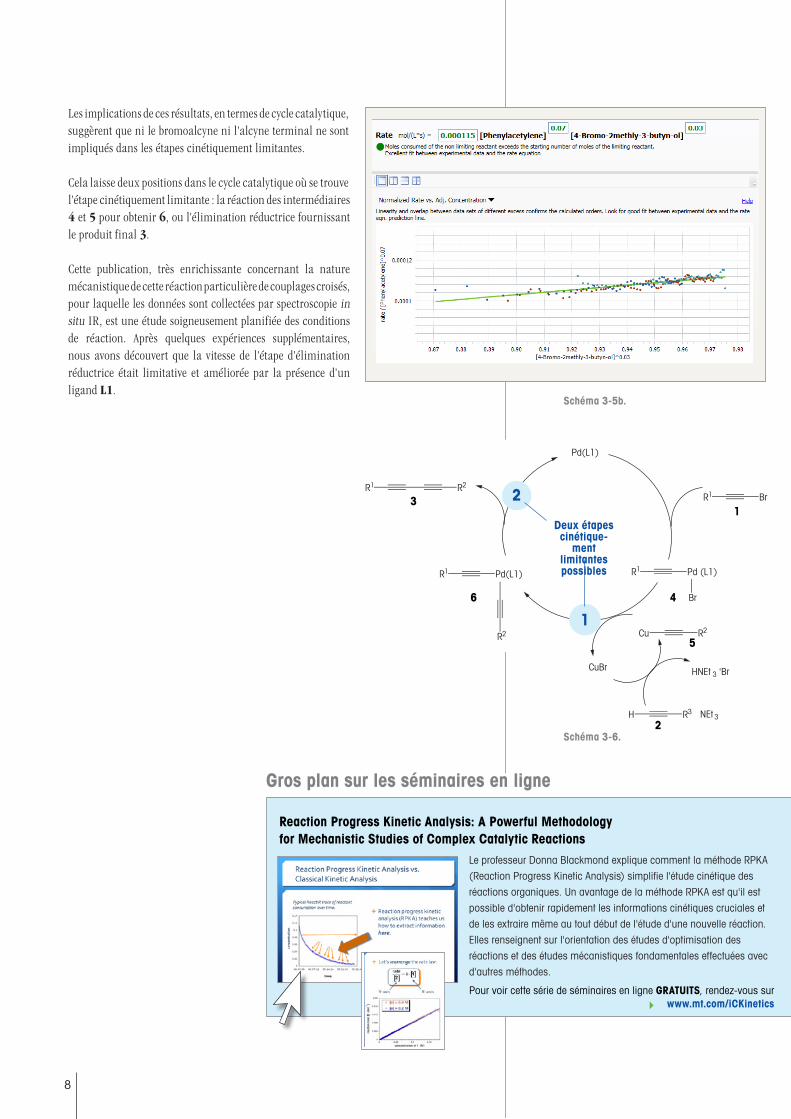
Les implications de ces résultats, en termes de cycle catalytique, suggèrent que ni le bromoalcyne ni l'alcyne terminal ne sont impliqués dans les étapes cinétiquement limitantes.
Cela laisse deux positions dans le cycle catalytique où se trouve l'étape cinétiquement limitante : la réaction des intermédiaires 4 et 5 pour obtenir 6, ou l'élimination réductrice fournissant le produit final 3.
Cette publication, très enrichissante concernant la nature mécanistique de cette réaction particulière de couplages croisés, pour laquelle les données sont collectées par spectroscopie in situ IR, est une étude soigneusement planifiée des conditions de réaction. Après quelques expériences supplémentaires, nous avons découvert que la vitesse de l'étape d'élimination réductrice était limitative et améliorée par la présence d'un ligand L1.
Schéma 3-5b.
Schéma 3-6.
R1
R1
R1
R2
Pd(L1)
Cu R2
BrR2
Pd(L1)
R1 Pd (L1)
Br
CuBr HNEt 3 'Br
H R3 NEt 3
1
4
5
2
6
3 2
1
Deux étapes cinétique-
ment limitantes possibles
Gros plan sur les séminaires en ligne
Le professeur Donna Blackmond explique comment la méthode RPKA
(Reaction Progress Kinetic Analysis) simplifie l'étude cinétique des
réactions organiques. Un avantage de la méthode RPKA est qu'il est
possible d'obtenir rapidement les informations cinétiques cruciales et
de les extraire même au tout début de l'étude d'une nouvelle réaction.
Elles renseignent sur l'orientation des études d'optimisation des
réactions et des études mécanistiques fondamentales effectuées avec
d'autres méthodes.
Pour voir cette série de séminaires en ligne GRATUITS, rendez-vous sur www.mt.com/iCKinetics
Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions
9
Jenny B. Åberg, Madeleine C. Warner et Jan-E. Bäckvall*Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Suède ; J. Am. Chem. Soc., 2009, 131 (38), p. 13622 ) à 13624
4. Formation inattendue d’un complexe ruthénium cyclopentadiènyle alcoxycarbonyle avec une liaison covalente C=C
Une résolution cinétique dynamique catalysée par des complexes de ruthéniumMême si les catalyseurs au ruthénium ont été utilisés avec succès associés à des lipases pour convertir les alcools secondaires racémiques 2 en acétates énantiopurs 3 (« résolution cinétique dynamique »), la même réaction ne fonctionne pas aussi bien avec l'alcool oléfinique 4. Il semble que l'alcool 4 se racémise très lentement, probablement à cause de la coordination de sa double liaison avec le ruthénium. L'article décrit l'utilisation des spectroscopies RMN et in situ IR pour montrer la présence d'un complexe de Ru-oléfine.
Lors de la réaction d’un alcool allylique avec un complexe Ru, on constate la diminution au cours du temps des deux bandes IR à 2 021 cm-1 et 1 964 cm-1. Celles-ci correspondent au complexede ruthénium 1. La formation d'une nouvelle bande à 1 982 cm-1 est visible dans la zone des carbonyles. Un nouveau pic se forme dans la zone des acyles à 1 644 cm-1 (Sch. 4-1).
0.03
0.02
0.01
0
2040 164018801960 17201800
Spec
tre (
A.U.
)
T= 29minT= 0 min
Longueurs d'ondes (cm-1)
2021cm-1
(Ru-O -Bu6)t
1982cm-1
1644cm-1
1964cm-1
(Ru-O -Bu6)t
CO R
egio
n
Acyl
Reg
ion
Schéma 4-1.
PhPh
PhPh
Ph
RuCO
CO CO
R
OH
R
OAc
OAc
cat. 1, cat. t-BuOK
CALB,
racémique toluène R-acétate
1
2 R = grand groupe (alkyle, aryle)
4 R = H2C = CHCH2CH25 R = H2C = CHCH2CH2 <50 %; pas de résolution cinétique dynamique
3 R = grand groupe (alkyle, aryle)
rendement chimique élevéexcès énantiométrique élevé
10
Ph Ph
Ph
Ph
Ph
RuOC C
O O
1
65
4
32
0.03
0.02
0.01
0
00:20:00 00:40:0000:30:00 00:50:00
Spec
tre (
A.U.
)
1644 cm-1
1982 cm-1
1964 cm-1
2021 cm-1
Temps relatif (min)
Addition Alcool 4
Schéma 4-2.
Schéma 4-3.
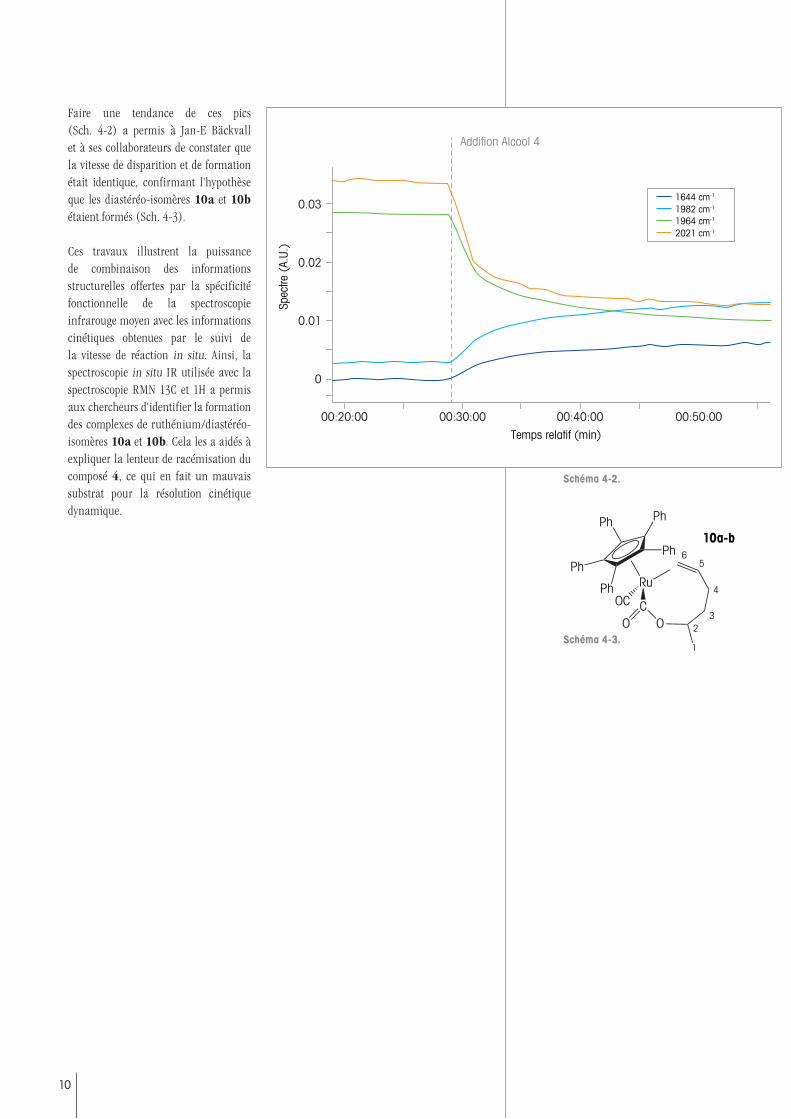
Faire une tendance de ces pics (Sch. 4-2) a permis à Jan-E Bäckvall et à ses collaborateurs de constater que la vitesse de disparition et de formation était identique, confirmant l'hypothèse que les diastéréo-isomères 10a et 10b étaient formés (Sch. 4-3).
Ces travaux illustrent la puissance de combinaison des informations structurelles offertes par la spécificité fonctionnelle de la spectroscopie infrarouge moyen avec les informations cinétiques obtenues par le suivi de la vitesse de réaction in situ. Ainsi, la spectroscopie in situ IR utilisée avec la spectroscopie RMN 13C et 1H a permis aux chercheurs d'identifier la formation des complexes de ruthénium/diastéréo-isomères 10a et 10b. Cela les a aidés à expliquer la lenteur de racémisation du composé 4, ce qui en fait un mauvais substrat pour la résolution cinétique dynamique.
10a-b

11
Nos conseillersMETTLER TOLEDO dispose d'un réseau mondial de conseillers en technologies et en applications, forts d'une expérience approfondie du secteur et de la recherche, et qui vous aideront dans les domaines de la chimie organique, et du développement et de l'extrapolation de procédés chimiques.
E-mail : [email protected]él. : 410-910-8500 01 30 97 17 17 (MT-France)
Site WebConsultez notre site Web METTLER TOLEDO (www.mt.com/autochem),où vous trouverez des informations détaillées supplémentaires sur nos produits et applications, notamment la liste complète des prochains séminaires en ligne à la demande.
BlogNotre blog Chemical Research, Development and Scale-up (Recherche et Développement Chimiques, Extrapolation) reprend les dernières publications et les commentaires de nos experts, ainsi que d'universitaires et de professionnels du secteur.
Communauté de clientsNotre site réservé à la communauté de clients offre aux utilisateurs de nos technologies un accès gratuit à des listes de publications archivées, rapports d'application, études de cas et supports de formation complets, et l'accès immédiat à tous nos séminaires en ligne à la demande.
Réseaux sociauxObtenez, en temps réel via Facebook et Twitter, l'actu sur les derniers développements en synthèse chimique, génie chimique et extrapolation.
ALR
FBRM®
PVM®
ReactIR™
iC Software
Comme indiqué précédemment, le but de ce livre blanc n'est pas d'entrer dans les détails des conclusions scientifiques des auteurs. Il s'agit plutôt de démontrer que la spectroscopie in situ ReactIR™, utilisée seule ou avec d'autres techniques, fournit aux chercheurs des indices pour comprendre le mécanisme, la cinétique et le déroulement de réactions chimiques.
Conclusions
Les exemples ci-dessus démontrent notamment ce qui suit :
- La spectroscopie in situ ReactIR™ offre aux chercheurs de précieuses informations complémentaires leur permettant de déterminer le mécanisme et le déroulement d'un certain nombre de différents types de réactions chimiques.
- Les informations recueillies à l'aide de la spectroscopie in situ complètent d'autres données structurelles comme la spectroscopie par résonance magnétique nucléaire (RMN).
- L'association des données recueillies par spectroscopie in situ ReactIR™ et du logiciel iC Kinetics™ offre une méthode puissante pour étudier la cinétique d'une réaction chimique en moins d’expériences que les approches classiques.

12
Annexe : Liste des publications
1. Jørn Hansen, Bo Li, Evgeny Dikarev, Jochen Autschbach et Huw M. L. Davies*, Combined Experimental and Computational Studies of Heterobimetallic Bi−Rh Paddlewheel Carboxylates as Catalysts for Metal Carbenoid Transformations, J. Org. Chem., 2009, 74 (17), p. 6564 à 6571.
2. Vito Capriati*, Saverio Florio*, Renzo Luisi, Filippo Maria Perna et Agnese Spina, 2-Lithio-3,3-dimethyl-2-oxazolinyloxirane: Carbanion or Azaenolate? Structure, Configurational Stability, and Stereodynamics in Solution, J. Org. Chem., 2008, 73 (24), p. 9552 à 9564.
3. Wei Shi, Yingdong Luo, Xiancai Luo, Lei Chao, Heng Zhang, Jian Wang et Aiwen Lei*, Investigation of an Efficient Palladium-Catalyzed C(sp)−C(sp) Cross-Coupling Reaction Using Phosphine−Olefin Ligand: Application and Mechanistic Aspects, J. Am. Chem. Soc., 2008, 130 (44), p. 14713 à 14720.
4. Jenny B. Åberg, Madeleine C. Warner et Jan-E. Bäckvall*, Unexpected Formation of a Cyclopentadienylruthenium Alkoxycarbonyl Complex with a Coordinated C═C Bond, J. Am. Chem. Soc., 2009, 131 (38), p. 13622 à 13624.
5. Aiwen Lei et al: Org. Lett. 2008, 10, (13), p. 2661 à 2664.
6. Donna G. Blackmond, Angew. Chem. Int. Éd. 2005, 44, p. 4302 à 4320.
7. Reaction Progress Kinetic Analysis: A Powerful Methodology for Streamlining the Study of Complex Organic Reactions, Donna G. Blackmond, Webinar, www.mt.com/ac-webinars
Internet : http://www.mt.com/autochem
Sous réserve de modifications techniques 51725293©03/2011 Mettler-Toledo AutoChem, Inc.7075 Samuel Morse DriveColumbia, MD 21046, États-UnisTéléphone : +1 410 910 8500Fax : +1 410 910 8600 E-mail : [email protected]
www.mt.com/autochem
iC Kinetics™ offre un moyen graphique rapide
de décrire les caractéristiques d'une réaction
chimique et d'optimiser les procédés chimiques.
Le modèle cinétique créé permet de simuler l'effet
des paramètres de concentration et de température
sur les performances de la réaction.
Les données sont générées en moins
d'étapes qu'une approche classique et
les réactions chimiques sont optimisées
plus rapidement.
www.mt.com/iCKinetics
iC Kinetics™
Le système ReactIR™ analyse la composition en
temps réel, vous permettant de suivre les principales
espèces réactionnelles sans avoir à prélever des
échantillons. Grâce au suivi des réactions in situ en
temps réel, les scientifiques recueillent un maximum
d'informations sur leurs réactions en un minimum
d'effort pour une meilleure compréhension de leurs
procédés chimiques.
www.mt.com/ReactIR
Analyse de réactions FTIR In Situ avec ReactIR™
Contactez-nous pour savoir comment ces outils vous aideront dans vos recherches.
EasyMax™ permet aux chimistes organiciens et
de synthèse d'évaluer rapidement les différentes
voies de synthèse, les réactifs, et les conditions
de réactions pour générer la quantité souhaitée de
composés cibles. Ils peuvent mener deux expériences
entièrement indépendantes avec des réacteurs de
différents volumes, pour une synthèse plus rapide et
plus fiable.
www.mt.com/EasyMax
EasyMax™
En savoir plus...
Adrian Burke, BSc (Hons)[email protected]









![Présentation1 [Mode de compatibilité] · L’ACCORD PPCR APPORTE DES AVANCEES MAJEURES •Larestructurationetlarevalorisationdesgrillesindiciaires,pourtouteslescatégoriesA,BetC,dans](https://img.pdfslide.fr/doc/110x75/5edecec9ad6a402d666a2902/prsentation1-mode-de-compatibilit-laaccord-ppcr-apporte-des-avancees-majeures.jpg)



















