Embed Size (px)
Citation preview

Eur J Orthop Surg Traumatol
DOI 10.1007/s00590-007-0221-5ORIGINAL ARTICLE
Development of a molecular methodology to quantify Staphylococcus epidermidis in surgical wash-out samples from prosthetic joint replacement surgery
Fergus J. Byrne · Sinéad M. Waters · Peadar S. Waters · William Curtin · Michael Kerin
Received: 4 December 2006 / Accepted: 11 January 2007© Springer-Verlag 2007
Abstract Prosthetic joint infection is a serious complica-tion of total joint arthroplasty that causes great morbidity inaVected individuals. The most common cause of prosthesisassociated infections are members of Staphylococcus spp.,including Staphylococcus epidermidis. Culture has servedas the gold standard for diagnosis, despite obvious short-comings in terms of sensitivity and time. Bacterial genomicDNA extraction methodologies were evaluated for optimalrecovery of genomic DNA from sterilised wash-out sam-ples, spiked with S. epidermidis. Real time Polymerasechain reaction (PCR) assays targeting the S. epidermidisspeciWc gseA gene were designed to reliably detect andquantify S. epidermidis. Sixty post-operative wash-outsamples from primary hip and knee arthroplasties weretaken aseptically. All were shown to be culture negativeusing the culture-dependent approach. These were sampleswere subjected to S. epidermidis-speciWc real time PCR.Standard curve showed good linearity. Sensitivity limit ofthe assay was <10 CFU S. epidermidis per sample. Repro-ducibility of the assay was conWrmed. S. epidermidis was
not identiWed in any of these samples using the novel spe-cies speciWc SYBR Green real time PCR technique. Resultsindicated that wash-out samples were true negatives anddid not harbour S. epidermidis. To support this, patientsdisplayed no symptoms of infection. To illustrate the fulleVectiveness of the novel real time PCR assay, a largernumber of samples need to be tested (>1,000 patients).
Keywords Staphylococcus epidermidis · Real time PCR · Bacterial infection · Prosthesis · Joint replacement
Mise au point d’une méthode moléculaire pour quantiWer le staphylocoque epidermidis dans les produits de lavage en chirurgie prothétique
Résumé L'infection prothétique est une complicationsérieuse des arthroplasties totales et entraîne une grandemorbidité chez les patients qui en sont atteints La cause laplus commune des infections liées aux prothèses sont duesà diverses espèces de staphylocoques, y compris le staphy-locoque epidermidis. La culture a servi de gold standard audiagnostic, en dépit des imperfections évidentes en termesde sensibilité et de temps. Des méthodologies géniquo-bac-tériennes d'extraction d'ADN ont été évaluées pour le réta-blissement optimal de l'ADN génique des échantillonsstérilisés de lavage, inoculés avec le staphylocoque epide-rmidis. Des analyses en temps réel de PCR visant le gènespéciWque du gseA du staphylocoque epidermidis ont étéconçues pour détecter de façon certaine et pour quantiWer lestaphylocoque epidermidis. Soixante échantillons de produitde lavage pour des arthroplasties primaires de hanche et degenou ont été prélevés aseptiquement. Toutes les culturesont été trouvées stériles en utilisant l'approche culture-dépendante. Des échantillons ont été soumis à une analyse
F. J. Byrne (&) · W. CurtinOrthopaedics Department, Merlin Park Regional Hospital, Galway, Irelande-mail: [email protected]
S. M. WatersTeagasc, Animal Reproduction Research Centre, Mellows Campus, Athenry, Co., Galway, Ireland
P. S. WatersNational University of Ireland, Galway, Irelande-mail: [email protected]
M. KerinDepartment of Surgery, Clinical Science Institute, University College Hospital, Galway, Ireland
123

Eur J Orthop Surg Traumatol
en temps réel de PCR au staphylocoque epidermidis. Lacourbe standard a montré de bonnes linéarités. La limite desensibilité de l'analyse était inférieure à CFU staphylocoqueepidermidis:prélèvement. La reproductibilité de l'analyse aété conWrmée. Le staphylocoque epidermidis n'a pas étéretrouvé dans l'un quelconque d'entre ces échantillons enutilisant la technique nouvelle spéciWque SYBR en tempsréel de PCR. Les résultats ont indiqué que les produits delavage étaient réellement négatifs et ne contenaient pas dustaphylocoque epidermidis.. Les patients n'ont d'ailleursmontré aucun symptôme d'infection, ce qui conforte cetteaYrmation. Pour illustrer la pleine eYcacité de l'analyse entemps réel du nouveau test PCR, un plus grand nombred'échantillons doit être examiné (patients >1000).
Mots clés Staphylocoque epidermidis · PCR en temps réel · Infection bactérienne · Prothèse · Arthroplastie
Introduction
Bacterial colonisation or deep infections of the prosthesis orits site are major concerns with a potentially catastrophicoutcome [19]. The most commonly cultured microorgan-isms associated with prosthetic joint infection are coagulase-negative staphylococci (in 30–43% of cases) and Staphylo-coccus aureus (12–23%), followed by mixed Xora (10–11%), streptococci (9–10%), Gram-negative bacilli (3–6%),enterococci (3–7%) and anaerobes (2–4%). However,Staphylococcus epidermidis, a coagulase-negative staphylo-cocci and ubiquitous skin commensal, has emerged as acausative agent in prosthetic joint infection [19]. These bac-teria are a major component of the normal skin Xora andmucous membranes and they are among the most frequentlyisolated bacteria in the clinical microbiology laboratory [15].
Cultures of periprosthetic tissue provide the most reli-able current means of detecting these pathogens and assuch, culture has served as the gold standard for identiWca-tion of these organisms despite obvious shortcomings interms of sensitivity and time. Conventional culture methodsmay require several days to obtain a Wnal result and the useof pre-operative antibiotics may render conventional cul-tures negative. The existence of ‘culture negative’ osteomy-elitis has been reported previously [5].
To overcome the shortcomings associated with the diag-nosis of bacterial infections in orthopaedics by culture andto reduce the number of infected revision arthroplasties,molecular methodologies have been employed [14]. Molec-ular diagnostics relies on the presence of bacterial DNA.Polymerase chain reaction (PCR)-based methods have beenextensively applied in orthopaedics to identify bacterialinfection [14]. Bacterial presence may be detected by tar-
geting the 16S rRNA gene using PCR [9]. The followingspecies have been identiWed using this approach, S. epide-rmidis [1], S. aureus [2] and methicillin-resistant Staphylo-cocci [20], to name but a few. However, the use of primerstargeting the 16S rRNA gene has been associated with ahigh prevalence of false positive results [21]. The broadsensitivity of PCR directed against the 16S rRNA detectseven trace contamination by clinically irrelevant organismsthat occur after specimen acquisition. One way to improvethe speciWcity of PCR is to use primers targeting a fragmentof gene, speciWc to the species of interest.
Another issue of concern in the development of sensitivePCR assays for microbial detection includes the identiWca-tion of an optimal method of DNA isolation from samples.Optimal sample processing protocols for diagnostic bacte-rial PCR should release DNA from an array of target organ-isms with equal eYciencies and washout inhibitory factorsfrom various sample types without introducing bacterialDNA contamination to the ampliWcation reaction. Extrac-tion and PCR ampliWcation of DNA from synovial Xuid isrecognised to be diYcult. The complex macromolecularcomposition of the synovial matrix is claimed to be respon-sible for PCR inhibition and false negative results whenusing bacterial-speciWc PCR for the diagnosis of synovialinfection [13, 16]. Indeed, recent attempts to detect bacte-rial DNA in samples from equine patients with infectioussynovitis were overall not successful [16].
Real-time PCR is based on the detection and quantitationof a Xuorescent reporter during PCR and is extremely sensi-tive [12]. The Wrst and only report in the literature of realtime PCR being applied in the detection of infection inorthopaedics was made by Kobayashi et al. [10]. Thisgroup developed a real time PCR methodology for theidentiWcation of bacteria in culture-negative osteomyelitis,however this assay was not species-speciWc nor fully quan-titative. The aim of the present study was to develop arapid, reliable sensitive and reproducible real time PCRmethod to detect S. epidermidis in surgical wash-out samplesfrom prosthetic joint replacement surgery.
Methods
Sample collection
Wash-out samples consisted of 0.9% saline and were sam-ples of the Wrst wash after reduction of prosthesis. Sixty(2 £ 40 ml2) samples of post-operative wash-out samplesfrom primary hip and knee arthroplasties were taken usingaseptic technique in 50 ml sterilin tubes and stored at¡20°C. One of the duplicate samples was subjected to rou-tine culture-based clinical microbiological analyses whilethe remaining sample was tested using molecular methods.
123

Eur J Orthop Surg Traumatol
Optimisation of DNA isolation from Staphylococcus epidermidis from orthopaedic surgical wash-out samples
In order to simulate S. epidermidis contaminated wash-outsamples, washout from a surgical operation was sterilisedand was subsequently spiked with known concentrations ofa pure culture of S. epidermidis. In brief, 1 l of wash-outmatrix was obtained from a patient displaying no signs ofinfection and was sterilised by radiation. Sterilisation wasconWrmed by culture-based analyses. S. epidermidis ATCC8558 was purchased from the American Type Culture Col-lection, USA, revitalised and cultured according to manu-facturers instructions. The identity of the strain wasconWrmed following culture by api test analysis (Bio Meri-eux, Marcy-l’Etoile, France). This was performed to ensureno contamination occurred during culture. Serial dilutionsof an overnight culture of S. epidermidis was carried outand a 100 �l aliquot of each dilution was plated in triplicateon nutrient agar at 30°C under aerobic conditions for 24 hfor subsequent quantiWcation. Triplicate 100 �l aliquots ofthe undiluted culture were stored at ¡20°C for spiking ofsterile washout. Tripicate 1 ml samples of sterile wash-outsamples were spiked to contain 1 £ 109, 1 £ 106, 1 £ 103
and 0 CFU S. epidermidis, respectively.Spiked wash-out samples were subjected to the Wve
DNA extraction methods listed below.
1. Standard phenol/chloroform puriWcation method fol-lowed by an ethanol precipitation [17].
2. Protocol as described by Kuipers et al. [11].3. Wizard Bacterial Genomic DNA isolation kit (Pro-
mega, Madison, WI, USA).4. Qiagen DNA mini kit (Qiagen Inc., Valencia, CA,
USA)–a silica-gel column-based method.5. Sigma Bacterial DNA isolation kit (Sigma-Aldrich Ire-
land Ltd., Dublin, Ireland).
For all protocols tested, DNA was eluted in 100 �l ofNuclease-free H2O (Promega). DNA was subsequentlyassessed for optimal DNA recovery. QuantiWcation andpurity assessment of DNA was conducted using a Shima-dzu 160-A ultraviolet (UV) spectrophotometer (ShimadzuScientiWc Instruments Inc., Columbia, MD, USA).
QuantiWcation of S. epidermidis in wash-out samples using real time PCR
Staphylococcus epidermidis ATCC 8558 was culturedovernight according to manufacturers instructions. Closelyrelated bacteria, S. aureus ATCC 29313, Staphylococcusauricularis ATCC 33753, were also cultured overnightaccording to manufacturer’s instructions and were appliedas negative controls. Cells were collected by centrifugation
at 6,000 rpm for 10 min using a MSE Mistral 1000 centri-fuge (Shaw ScientiWc Ltd., Dublin, Ireland).
The extraction procedure using the Wizard BacterialGenomic DNA isolation kit (Promega) according to manu-facturer’s instructions proved most eVective at extractingthe highest quality and quantity of DNA. As such, DNAwas isolated from all cultures using this technique inmolecular studies.
Oligonucleotide primers (Sigma-Genosys, Cambridge,UK), 5�Staph Epi F and 3�Staph Epi R (Table 1) weredesigned using the software programme ‘Primer3’, avail-able online at http://www.gene3.ciat.cgiar.org/PRIMER3/primer3_www.cgi to amplify a 193 bp region of the S. epi-dermidis speciWc gene, which codes for the glutamic acid-speciWc serine protease, GluSE [8]. Sequence analysisusing the Basic Local Alignment Search Tool (BLAST),online at the National Centre for Biotechnology Informa-tion (NCBI) homepage (http://www.ncbi.nlm.nih.gov),conWrmed the species speciWcity of these primers.
DNA from S. epidermidis, S. aureus and S. auriculariswas subjected to S. epidermidis-speciWc PCR using the oli-gonucleotide primers designed in this study and listed inTable 1.
To reduce the possibility of false positive results, PCRreagents were subjected to 5 min exposure to UV irradia-tion prior to this work being carried out and reactions wereformulated in a laminar Xow.
Polymerase chain reaction reactions (100 �l), performedin a DNA thermal cycler (Perkin Elmer Cetus, Wellesley,MA, USA) contained 500 ng genomic DNA, 10£(NH4)2SO4 reaction buVer (10 �l), MgCl2 (3 mM), primers(500 ng each) and deoxynucleotide triphosphates (dNTPs)(500 �M each). Taq DNA polymerase (Bioline™, London,UK) (2 U) was added following the initial denaturationstep.
The PCR ampliWcation programme was: 94°C for 2 min,84°C for 5 min (to allow the addition of Taq DNA poly-merase) and 35 cycles of 93°C for 30 s, 60°C for 30 s, 72°Cfor 30 s, followed by 10 min at 72°C.
Analysis of ampliWed PCR products (10 �l) was carriedout on 1% (w/v) agarose gels using 1£ Tris acetate EDTA(TAE) running buVer, pH 8.3 in a model EC-360-M Maxi-cell gel system (EC apparatus Corporation, St. Petersburg,FL, USA).
Table 1 Primer sequences for Staphylococcus epidermidis-speciWcPCR
Primer designation Sequence
5�Staph Epi F 5�GGCAAATTTGTGGGTCAAGA3�
3�Staph Epi R 5�TGGCTAATGGTTTGTCACCA3�
123

Eur J Orthop Surg Traumatol
Positive PCR products (»200 bp on the agarose gel)were puriWed using the High Pure PCR product puriWcationkit (Roche, Basel, Switzerland) and DNA sequencing ofPCR products was carried out by Oswel, Southhampton,UK. Analysis of sequence data and homology comparisonswas performed using the BLAST, online at the NCBIhomepage (http://www.ncbi.nlm.nih.gov).
To generate a standard curve for S. epidermidis quantiW-cation, S. epidermidis ATCC 8558 was cultured overnightand quantitated. 50 ml sterile washout was spiked with1013–101 CFU S. epidermidis in triplicate. Contents of steri-lin tubes from samples and standards were collected bycentrifugation. Genomic DNA was extracted from cellsusing the Wizard Bacterial Genomic DNA isolation kit(Promega). S. epidermidis-speciWc primers, 5�Staph Epi Fand 3�Staph Epi R, which were tested for species-speciWc-ity, were used in real time PCR assays.
In SYBR Green real time PCR, the ampliWcation of theDNA target is measured in terms of the increment in thequantity of Xuorescence determined at the end of eachampliWcation cycle. To determine the optimal concentrationof primers, preliminary tests were performed using equimo-lar 300, 50, 25 and 5 nM concentrations of the 5�Staph EpiF and 3�Staph Epi R forward and reverse primers. Opti-mised ampliWcation reactions were performed in a total vol-ume of 50 �l with an ABI prism 7500 sequence detector(Applied Biosystems, Foster City, CA, USA) with 96 wellmicrowell plates (MicroAmp; Applied Biosystems).
In each well, the following were added: 5 �l of puriWedgenomic DNA, 25 �l of SYBR Green I PCR Master Mix(Applied Biosystems), a 50 nM concentration of each S.epidermidis-speciWc primers 5�Staph Epi F and 3�Staph EpiR, in a Wnal volume of 50 �l with Nuclease-free H2O (Pro-mega). Genomic DNA from non-spiked sterile wash-outsamples was applied as a negative control. The reactionmixture was run online at 50°C for 2 min and 95°C for10 min (to activate ampli Taq Gold), followed by 35 cyclesat 95°C for 30 s, 60°C for 30 s and 72°C for 30 s, with anextension phase of 1 cycle at 95°C for 1 min, 60°C for1 min and 95°C (ramp time, 19.59 min).
The results were visualised using the software SequenceDetector 1.7 provided with the ABI Prism 7500 system
(Applied Biosystems) and Cell number values were log10
transformed and plotted against their respective cyclethreshold (CT) values to generate a standard curve for quan-tiWcation of S. epidermidis in wash-out samples. The stan-dard curve was conWrmed by using Microsoft Excel 2000.The melting curve was visualised with the software Disso-ciation Curve 1.0 provided with the ABI Prism 7500 sys-tem (Applied Biosystems). The reproducibility of the assaywas assessed by spiking sterile washout and measuring S.epidermidis using the entire methodology described in thisstudy on three separate occasions.
Results
Sixty surgical wash-out samples from prosthetic jointreplacement surgery were subjected to routine culture-based microbiological analysis and all samples displayedno bacterial growth under both aerobic and anaerobic con-ditions.
To determine the optimal method of DNA isolation fromsterile wash-out samples spiked to contain 1 £ 109,1 £ 106, 1 £ 103 and 0 CFU S. epidermidis, a number ofDNA extraction methodologies reported in the literature[11] including commercial kits [10] were assessed for opti-mal genomic DNA recovery. Table 2 illustrates resultsfrom this evaluation. All DNA isolation generated A260/A280 ratios in region of 1.6–1–8, indicating that the resul-tant DNA was of suitable purity to be used in PCR reac-tions. The Wizard bacterial DNA isolation kit achieved thegreatest recovery of DNA from all dilutions of S. epidermi-dis, while the method employed by Kuipers et al. [11] toisolate DNA from synovial Xuid yielded slightly lowerrecovery rates. The Qiagen commercial kit has previouslybeen used to isolate S. epidermidis from bone and soft tis-sue [10] and to detect bacterial DNA in synovial Xuid formhorses with infectious synovitis [16], however when this kitwas applied in the extraction of S. epidermidis DNA fromwash-out samples, it yielded poor results in comparison toother procedures. The Sigma bacterial DNA isolation kitproved least eVective at recovering high levels of bacterialDNA from surgical wash-out samples. These results dem-
Table 2 QuantiWcation of genomic DNA (mean and standard deviation)
Method of DNA isolation
Weight of DNA recovered
1 £ 109 CFU 1 £ 106 CFU (ng �l¡1)
1 £ 103 CFU (ng �l¡1)
0 CFU
Phenol/chloroform 852 § 0.06 ng �l¡1 521 § 0.04 22 § 0.04 0
Kuipers 1.24 § 0.07 �g �l¡1 654 § 0.07 34 § 0.06 0
Wizard 1.56 § 0.08 �g �l¡1 833 § 0.06 50 § 0.04 0
Qiagen 431 § 0.09 �g �l¡1 266 § 0.04 27 § 0.04 0
Sigma 381 § 0.10 �g �l¡1 184 § 0.03 9 § 0.05 0Results presented as a mean of three replicates
123
Eur J Orthop Surg Traumatol
onstrated that the Wizard kit protocol was most suitable tobe used in further PCR-based studies in this project.
Staphylococus epidermidis-speciWc primers were designedusing the software programme ‘Primer3’ and conWrmedto be species-speciWc using the bioinformatic analysistool, BLAST. DNA, isolated from S. epidermidis, S. aureusand S. auricularis was subjected to S. epidermidis-speciWcPCR using the oligonucleotide primers designed in thisstudy. Following agarose gel electrophoresis the gel was vis-ualised (Fig. 1). PCR product of the correct size (194 bp)was generated from S. epidermidis while no ampliWcationwas evident from the negative controls, indicating that theprimers designed in this study were indeed S. epidermidis-speciWc. To further conWrm the species speciWcity of theprimers, the positive PCR product was puriWed and sub-jected to sequence analysis. BLAST analysis of resultantsequence conWrmed that this product was indeed speciWc toS. epidermidis (data not shown). This result in addition tothe inability of the primers to amplify any product from neg-ative controls compounds the speciWcity of the primersdesigned and as such, were applied in real time PCR studies.
During the optimisation of real time PCR conditions, theoptimal concentration of primers chosen for the experimentswas determined as 50 nM. This concentration was chosenbecause it provided the lowest CT and because the CT valuesobtained at lower primer concentrations (5 and 25 nM) weresigniWcantly higher (P < 0.05; data not shown).
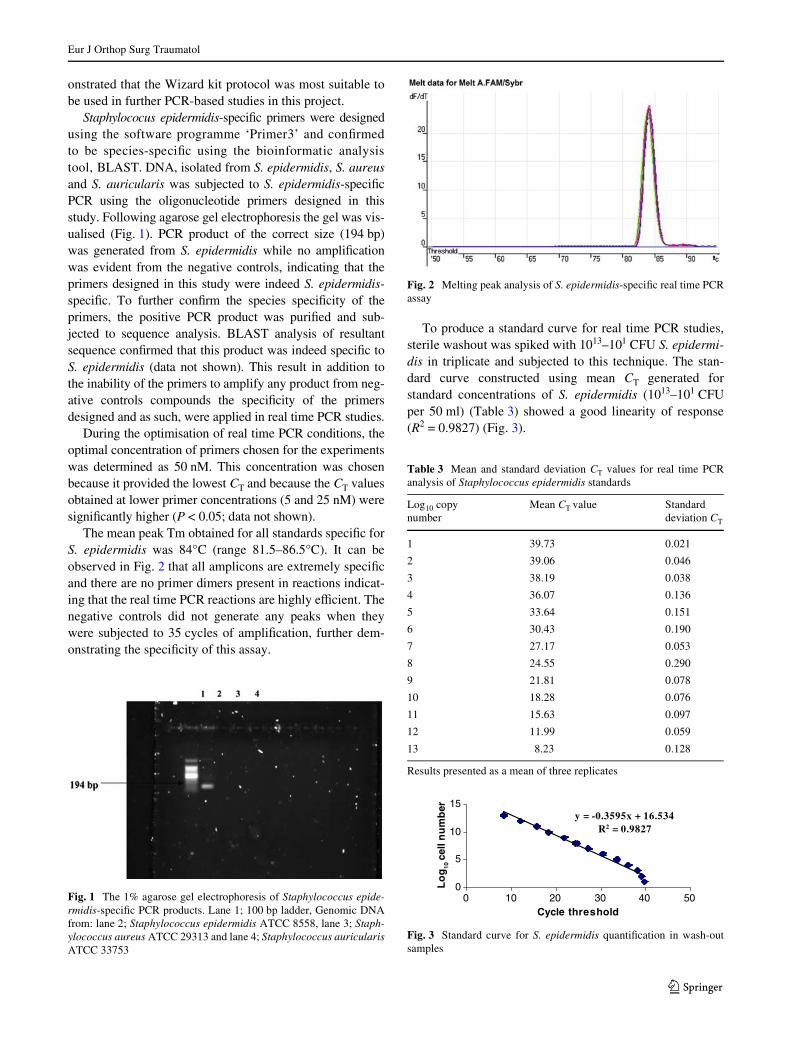
The mean peak Tm obtained for all standards speciWc forS. epidermidis was 84°C (range 81.5–86.5°C). It can beobserved in Fig. 2 that all amplicons are extremely speciWcand there are no primer dimers present in reactions indicat-ing that the real time PCR reactions are highly eYcient. Thenegative controls did not generate any peaks when theywere subjected to 35 cycles of ampliWcation, further dem-onstrating the speciWcity of this assay.
To produce a standard curve for real time PCR studies,sterile washout was spiked with 1013–101 CFU S. epidermi-dis in triplicate and subjected to this technique. The stan-dard curve constructed using mean CT generated forstandard concentrations of S. epidermidis (1013–101 CFUper 50 ml) (Table 3) showed a good linearity of response(R2 = 0.9827) (Fig. 3).
Fig. 1 The 1% agarose gel electrophoresis of Staphylococcus epide-rmidis-speciWc PCR products. Lane 1; 100 bp ladder, Genomic DNAfrom: lane 2; Staphylococcus epidermidis ATCC 8558, lane 3; Staph-ylococcus aureus ATCC 29313 and lane 4; Staphylococcus auricularisATCC 33753
Fig. 2 Melting peak analysis of S. epidermidis-speciWc real time PCRassay
Table 3 Mean and standard deviation CT values for real time PCRanalysis of Staphylococcus epidermidis standards
Results presented as a mean of three replicates
Log10 copy number
Mean CT value Standard deviation CT
1 39.73 0.021
2 39.06 0.046
3 38.19 0.038
4 36.07 0.136
5 33.64 0.151
6 30.43 0.190
7 27.17 0.053
8 24.55 0.290
9 21.81 0.078
10 18.28 0.076
11 15.63 0.097
12 11.99 0.059
13 8.23 0.128
Fig. 3 Standard curve for S. epidermidis quantiWcation in wash-outsamples
y = -0.3595x + 16.534R2 = 0.9827
0
5
10
15
0 10 20 30 40Cycle threshold
g10
oL
50
lecln
um
bre
123

Eur J Orthop Surg Traumatol
The sensitivity limit of the reaction was <10 CFU per50 ml (Fig. 3).
The reproducibility of the assay was assessed. When S.epidermidis was measured in standards employing theentire real time PCR procedure on three separate occasions,results were identical, ensuring the reproducibility of thetechnique.
To recall, the sixty wash-out samples taken from opera-tions were shown to be culture negative using microbiolog-ical culture-dependent techniques. S. epidermidis was alsonot identiWed in any of these samples using our novel spe-cies speciWc SYBR Green real time PCR assay, indicatingthat these samples are indeed true negatives and do not har-bour this species. To support this result, none of thesepatients have displayed any symptoms of infection (datanot shown).
Discussion
A novel, rapid sensitive and reproducible real time PCRmethodology has been developed for the speciWc detectionand quantiWcation of S. epidermidis in surgical wash-outsamples from prosthetic joint replacement procedures. Thisis the Wrst report of a molecular procedure, which optimallyisolates bacterial genomic DNA from orthopaedic wash-outsamples and speciWcally quantiWes this species using realtime PCR technology. The methodology is culture-indepen-dent and extremely sensitive, capable of detecting S. epide-rmidis at levels as low as 10 CFU per reaction.
At present, S. epidermidis is routinely quantiWed in hos-pital laboratories by culture. The main disadvantage associ-ated with culturing is the turnabout time that may extend toseveral days. The use of real time however can circumventthis problem. The DNA isolation is completed in <2 h andthe subsequent real time PCR step only takes 2 h, a lowturnabout time compared to days with culture. In addition,the present of Staphylococcus spp. has been detected in cul-ture negative osteomyelitis clinical samples using real timePCR [10], indicating its lack of sensitivity.
It has been reported in previous studies that the methodof DNA preparation in PCR-based bacterial detection andquantiWcation methodologies is of crucial importance inimproving the sensitivity of the procedure [4, 22]. Kuiperset al. [11], in a study describing the optimisation of samplepreparation of synovial Xuid for detection of Chlamydiatrachomatis DNA by PCR, concluded that the method ofsample preparation signiWcantly inXuences the sensitivityof subsequent PCR. Daly et al. [4], noted appreciable diVer-ences in the sensitivity of a PCR-ELISA method to detectEscherichia coli depending on the DNA extraction tech-nique employed. Other groups found that in using certainDNA isolation methods, PCR inhibitors co-puriWed with
the target DNA, thereby requiring extensive sample prepa-ration to remove, dilute or inactivate inhibitors prior toPCR ampliWcation [6, 7, 11]. As a consequence of thesereports, a variety of DNA extraction methods were evalu-ated in this study. It was established that diVerent DNAisolation procedures yielded varying recovery of DNA fromS. epidermidis spiked washout and that the Wizard bacterialgenomic DNA preparation kit (Promega) isolated optimalconcentrations of DNA in the most pure form from sam-ples. This method of DNA preparation was thus utilised inall real time PCR quantiWcation procedures.
The most common target gene for bacterial identiWcationis the 16S rRNA gene that is conserved in nearly all speciesof bacteria. For example, Tunney et al. [21] used PCR totest for evidence of bacteria in Xuids obtained by sonicationof 120 hip implants retrieved at revision arthroplasty. Withthe use of primers targeting the 16S rRNA gene, this groupfound that this technique was related to a high prevalenceof false positive results. The broad sensitivity of PCRdirected against the 16S rRNA detects even trace contami-nation by clinically irrelevant organisms that occur afterspecimen acquisition. One way to improve the speciWcity ofPCR is to use primers targeting a speciWc organism, orgroups of organisms most likely to be involved in clinicallyimportant orthopaedic infections, as was performed in thecurrent study.
Other investigators have used this approach. Sakai et al.developed a PCR assay for staphylococci, in which post-ampliWcation melting curve analysis allows distinctionbetween S. aureus and coagulase negative staphylococci[18]. Kobayashi et al. used a combination of a modiWeduniversal PCR and sequencing technology to identify bac-teria on the basis of DNA sequences that determine Gram-positive versus Gram-negative staining [9]. Thus, combina-tions of speciWc PCR assays may ultimately prove to bemore useful than broad-spectrum, so-called ‘universal’ bac-terial assays. While there is a report in the literature of thedetection of S. epidermidis in orthopaedic clinical samplesusing real time PCR, this method was not quantitative [10].
The problem of false positivity in PCR-based studies hasfrustrated researchers and clinicians due to unreliable inter-pretation of positive molecular results. To eliminate theseproblems, some precautions were taken in this study. First,PCR reagents were treated with UV irradiation and sec-ondly to avoid contamination from DNA in the air the PCRreactions were formulated carefully in a laminar Xow hood.In addition, intra-operative sampling by the surgeon is ofutmost importance to avoid contamination of wash-outsamples. Care was taken to employ aseptic technique at allstages of sampling.
In order to overcome the likely limitation of this meth-odology of DNA ampliWcation from dead cells, it may bepossible in some instances to incorporate a reverse
123

Eur J Orthop Surg Traumatol
transcription step in conjunction with real time PCR todistinguish between viable and non-viable cells. Other newtechniques, which may have a role in the diagnosis of via-ble infecting bacteria in orthopaedic clinical samplesinclude the use of microarray technology [3]. A microarrayallows the isolation and evaluation of numerous mRNAgenes with a single test. The premise of this technique is toidentify organism-speciWc genes and as mRNA is targeted,only live bacteria are detected.
To conclude, a novel, rapid sensitive and reproduciblereal time PCR methodology has been developed for thespeciWc detection and quantiWcation of S. epidermidis insurgical wash-out samples from prosthetic joint replace-ment procedures. However, to illustrate the full eVective-ness of this real time PCR assay a larger number of samplesneed to be tested (>1,000 patients).
References
1. Arciola CR, Campoccia D, Gamberini S, Donati ME, MontanaroL (2004) Presence of Wbrinogen-binding adhesin gene in Staphy-lococcus epidermidis isolates from central venous catheters-asso-ciated and orthopaedic implant-associated infections. Biomaterials25(19):4825–4829
2. Arciola CR, Alvi FI, An YH, Campoccia D, Montanaro L (2005)Implant infection and infection resistant materials: a mini review.Int J Artif Organs 28(11):1119–1125
3. Bauer TW, Parvizi J, Kobayashi N, Krebs V (2006) Diagnosis ofperiprosthetic infection. J Bone Joint Surg 88(4):869–882
4. Daly P, Collier T, Doyle S (2002) PCR-ELISA detection of Esc-herichia coli in milk. Lett Appl Microbiol 34:222–226
5. Floyed RL, Steele RW (2003) Culture-negative osteomyelitis. Pe-diatr Infect Dis J 8:731
6. Fratamico PM, Bagi LK, Pepe T (2000) A multiplex polymerasechain assay for rapid detection and identiWcation of Escherichiacoli 0157:H7 in foods and bovine feces. J Food Prot 63:1032–1037
7. González I, Garcia T, Fernandez A, Sanz B, Hernandez PE, MartinR (1999) Rapid enumeration of Escherichia coli in oysters by aquantitative PCR-ELISA. J Appl Microbiol 86:231–236
8. Ikeda Y, Ohara-Nemoto Y, Kimura S, Ishibashi K, Kikuchi K(2004) PCR-based identiWcation of Staphylococcus epidermidistargeting gseA endocing the glutamic-acid speciWc protease. Can JMicrobiol 50(7):493–498
9. Kobayashi N, Bauer TW, Togawa D, Lieberman IH, Sakai H, Fu-jishiro T, Tuohy MJ, Procop GW (2005) A molecular gram stainusing broad range PCR and pyrosequencing technology: a poten-tially useful tool for diagnosing orthopaedic infections. Diagn MolPathol 14(2):83–89
10. Kobayashi N, Bauer TW, Tuohy MJ, Lieberman IH, Krebs V,Togawa D, Fujishiro T, Procop GW (2006) The comparison of py-rosequencing molecular Gram stain, culture, and conventionalGram stain for diagnosing orthopaedic infections. J Orthop Res24(8):1641–1649
11. Kuipers JG, Nietfeld L, Dreses-Werringloer U, Koehler L, Wol-lenhaupt J, Zeidler H, Hammer M (1999) Optimised sample prep-aration of synovial Xuid for detection of Chlamydia trachomatisDNA by polymerase chain reaction. Ann Rheum Dis 58(2):103–108
12. Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K (1995) Oligo-nucleotides with Xuorescent dyes at opposite ends provide aquenched probe system useful for detecting PCR product and nu-cleic acid hybridization. PCR Methods Appl 4(6):357–362
13. Mariani BD, Levine MJ, Booth RE Jr, Tuan RS (1995) Develop-ment of a novel, rapid processing protocol for polymerase chainreaction-based detection of bacterial infections in synovial Xuids.Mol Biotechnol 4(3):227–237
14. Mariani BD, Tuan RS (1998) Advances in the diagnosis of infec-tion in prosthetic joint implants. Mol Med Today 4(5):207–213
15. Martineau F, Picard FJ, Roy PH, Ouellette M, Bergeron MG(1996) Species-speciWc and ubiquitous DNA-based assays for rap-id identiWcation of Staphylococcus epidermidis. J Clin Microbiol34(12):2888–2893
16. Pille F, Martens A, Schouls LM, Peelman L, Gasthuys F, SchotCS, De Baere C, Desmet P, Vandenberghe F (2005) Detection ofbacterial DNA in synovial Xuid from horses with infectious syno-vitis. Res Vet Sci 77:198–195
17. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: alaboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press,Cold Spring Harbor, New York
18. Sakai H, Procop GW, Kobayashi N, Togawa D, Wilson DA, Bor-den L, Krebs V, Bauer TW (2004) Simultaneous detection ofStaphylococcus aureus and coagulase-negative staphylococci inpositive blood cultures by real-time PCR with two Xuorescenceresonance energy transfer probe sets. J Clin Microbiol 42(12):5739–5744
19. Steckelberg JM, Osmon DR (2000) Prosthetic joint infections.American Society for Microbiology, Washington DC, pp 173–209
20. Tarkin IS, Henry TJ, Fey PI, Iwen PC, Hinrichs SH, Garvin KL(2003) PCR rapidly detects methicillin-resistant staphylococciperiprosthetic infection. Clin Orthop Relat Res 414:89–94
21. Tunney MM, Patrick S, Curran MD, Ramage G, Hanna D, NixonJR, Gorman SP, Davis RI, Anderson N (1999) Detection of pros-thetic hip infection at revision arthroplasty by immunoXuores-cence microscopy and PCR ampliWcation of the bacterial 16SrRNA gene. J Clin Microbiol 37(10):3281–3290
22. Waters SM, Doyle S, Murphy RA, Power RF (2005) Developmentof solution phase hybridisation PCR-ELISA for the detection andquantiWcation of Enterococcus faecalis and Pediococcus pento-saceus in Nurmi-type cultures. J Microbiol Methods 63(3):264–275
123






![Pilot Validation Study of the European Association of ... · of trainees, shorten their learning curves for different procedures, and improve surgical safety [9,10]. Nevertheless,](https://img.pdfslide.fr/doc/110x75/601067db42f457177466cf3d/pilot-validation-study-of-the-european-association-of-of-trainees-shorten-their.jpg)





![AESCULAP Surgical InstrumentsAESCULAP® Surgical Instruments エースクラップ 鋼製小物 [ 標準品] ビー・ブラウン エースクラップ(株)は製造・販売だけでなく](https://img.pdfslide.fr/doc/110x75/602222f31846b17d034e948a/aesculap-surgical-instruments-aesculap-surgical-instruments-fffff.jpg)
















