Embed Size (px)
Citation preview

Pharmacologie
Devenir du médicament dans l'organisme
IFSI 1ère année ANGERS 2011
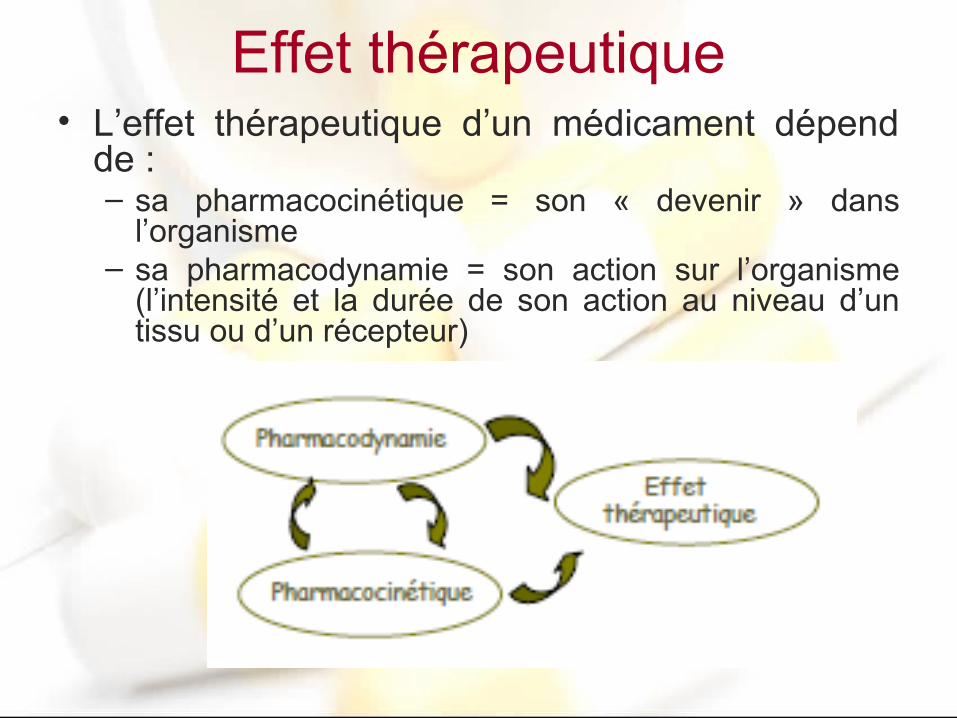
Effet thérapeutique• L’effet thérapeutique d’un médicament dépend
de :– sa pharmacocinétique = son « devenir » dans
l’organisme– sa pharmacodynamie = son action sur l’organisme
(l’intensité et la durée de son action au niveau d’un tissu ou d’un récepteur)

Pharmacocinétique• Définition:
– Étude de l’évolution dans le temps des concentrations des médicaments (PA et métabolites) dans les liquides biologiques…
• Entre l’administration d’un médicament et son action sur le récepteur de l’organe cible permettant d’obtenir la réponse pharmacologique recherché, survient un certain nombre d’évènements regroupés sous le terme de pharmacocinétique
• Classiquement, la pharmacocinétique d’un médicament est divisé en quatre étapes– Résorption– Distribution– Biotransformation (ou métabolisme)– L’élimination
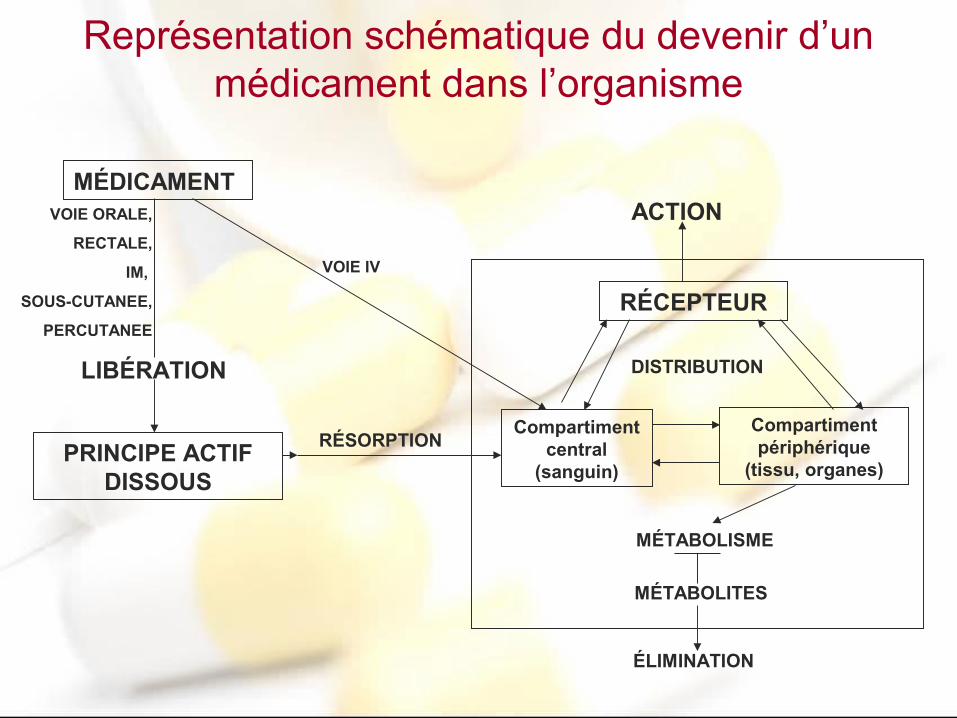
Représentation schématique du devenir d’un médicament dans l’organisme
MÉDICAMENT
PRINCIPE ACTIF DISSOUS
Compartiment central
(sanguin)
Compartiment périphérique
(tissu, organes)
RÉCEPTEUR
RÉSORPTION
DISTRIBUTION
ÉLIMINATION
MÉTABOLISME
MÉTABOLITES
ACTIONVOIE ORALE,
RECTALE,
IM,
SOUS-CUTANEE,
PERCUTANEE
LIBÉRATION
VOIE IV

Résorption (1)• La résorption est le passage d’un médicament dans
la circulation générale à partir de son lieu d’administration.
• Le terme d’absorption doit être réservé à la résorption après administration orale– Dans ce cas, l’absorption se situe nécessairement après les
phénomènes de désagrégation et de dissolution de la forme galénique au niveau du tube digestif, aboutissant à la libération du principe actif
• Cette étape suppose que le médicament franchisse certaines « barrières » cellulaires (membranes cellulaires) afin d’accéder à la circulation sanguine.
• L’étape de résorption n’existe pas lorsque le médicament est introduit directement dans la circulation par voie intraveineuse

Résorption (2)• Différents types de résorption (1)
– Résorption par diffusion passive en phase lipidique• La plupart des médicaments franchissent la membrane
cellulaire par diffusion passive à travers la partie lipidique de cette membrane (double couche lipidique dans laquelle sont insérées des protéines
• Deux caractéristiques physico-chimiques du médicament conditionnent fortement son aptitude à traverser les membranes biologiques
– La liposolubilité du médicament– Le degré d’ionisation (caractère acide ou basique) : une faible
ionisation permet une meilleure résorption• En pratique, l’intestin est le lieu privilégié de résorption en
raison de la grande surface d’échange qu’il présente (villosités intestinales)
• Il faut signaler la possible dégradation de certains médicaments au niveau digestif en raison de la présence des enzymes digestives et de l’acidité gastrique (insuline par exemple)

Résorption (3)• Différents types de résorption (2)
– Résorption par diffusion passive en phase aqueuse• Possible pour les molécules hydrosolubles ionisées de
faible poids moléculaire (ex : lithium) à travers les pores aqueux des membranes biologiques
– Résorption par diffusion facilitée• Processus passif de résorption, ne nécessitant pas de
dépense d’énergie mais la présence d’un « transporteur membranaire »
• Mécanisme saturable et susceptible de phénomène de compétition
– Résorption par transport actif• Mécanisme permettant la résorption de certains
médicaments contre un gradient de concentration (c’est-à-dire passage vers le milieu le plus concentré), et nécessitant un transporteur et de l’énergie
• Mécanisme saturable• Exemple : lévodopa (antiparkinsonien) dont la résorption
est diminuée par compétition avec certains acides aminés de l’alimentation

Résorption (4)• Notion de biodisponibilité (1)
– Fraction de la dose administrée qui atteint la circulation générale et vitesse à laquelle elle l’atteint
– Directement liée à l’intensité de la résorption du médicament– Ce n’est pas une simple mesure de la résorption digestive
dans le cas de la prise d’un médicament par voie orale. Avant qu’il puisse atteindre la circulation générale, à partir de la circulation porte, le médicament doit franchir le foie, susceptible de capter et de transformer une plus ou moins grande quantité du principe actif résorbé : il s’agit de l’effet de premier passage hépatique (« first pass effect).
– Par définition, la biodisponibilité est de 100% pour un médicament administré par voie intraveineuse
– Il est important de connaître la biodisponibilité, particulièrement pour les formes galéniques destinées à la voie orale : un même PA présenté en gouttes buvable, comprimé,ou en comprimé à libération prolongée ne présentera nécessairement la m^me biodisponibilité
– Si les quantités biodisponibles peuvent être identiques après administration d’une même posologie, c’est le plus souvent la vitesse avec laquelle le principe actif atteint la circulation générale qui est différente
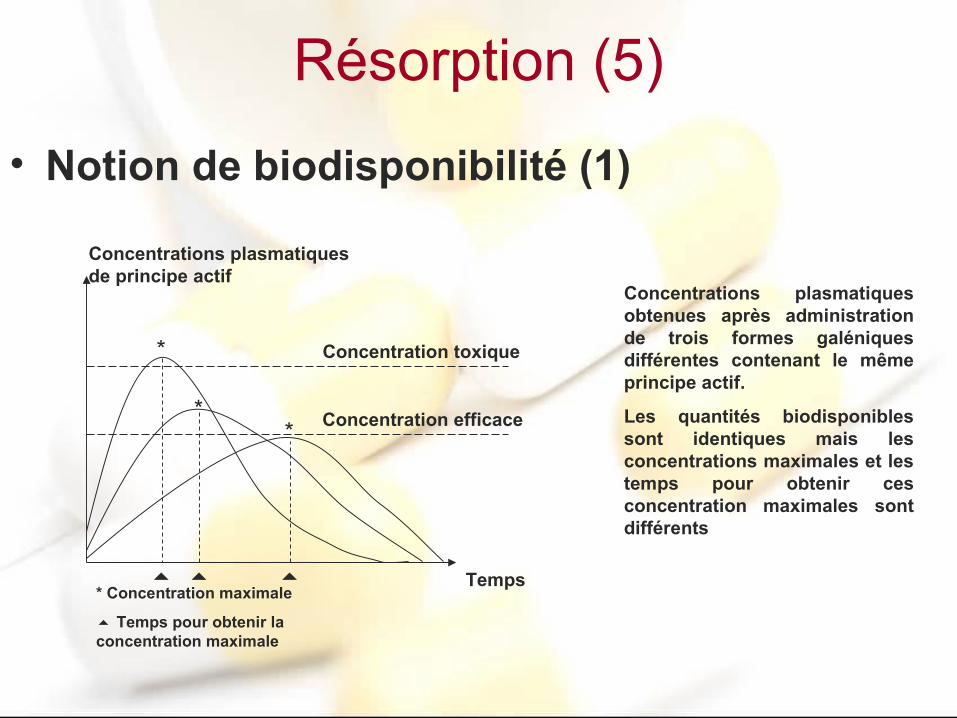
Résorption (5)
• Notion de biodisponibilité (1)
*
**
* Concentration maximale
Temps pour obtenir la concentration maximale
Temps
Concentrations plasmatiques de principe actif
Concentration toxique
Concentration efficace
Concentrations plasmatiques obtenues après administration de trois formes galéniques différentes contenant le même principe actif.
Les quantités biodisponibles sont identiques mais les concentrations maximales et les temps pour obtenir ces concentration maximales sont différents

Résorption (6)• Facteurs pouvant modifier la résorption
(1)– Facteurs physiopathologiques
• Âge, activité physique, position du corps, grossesse, diarrhée, constipation, insuffisance cardiaque…peuvent modifier la résorption
– Facteurs exogènes (1)• Alimentation
– médicament mieux résorbés lorsque pris en dehors des repas (antituberculeux)
– Médicament avec meilleure biodisponibilité lors d’une prise au cours des repas (nitrofurantoïne, griséofulvine)
– Les laitage, en raison de leur forte teneur en calcium, peuvent former des complexes insolubles non résorbables avec certains médicaments (tétracyclines)

Résorption (7)• Facteurs pouvant modifier la résorption (2)
– Facteurs exogènes (2)• Médicaments associés
– Des interactions médicamenteuses peuvent se produire lors de l’administration simultanée de plusieurs médicaments
» antiacides utilisés dans le traitement de l’ulcère gastroduodénal peuvent réduire l’absorption d’autres médicaments pris au même moment
» D’autres médicaments peuvent accélérer la vidange gastrique et le transit digestif, ou à l’inverse, les ralentir avec des conséquences variables sur la résorption de certains médicaments associés
» Exemple : un ralentisseur de vidange gastrique (anticholinergique)peut augmenter la résorption de certains médicaments peu solubles par prolongation de leur séjour dans l’estomac (digoxine), ou à l’inverse, la réduire en majorant leur dégradation par exposition prolongée aux enzymes gastriques (lévodopa)

Distribution (1)• Étape correspondant à la diffusion du
médicament dans l’organisme au niveau plasmatique et tissulaire
• Le volume de distribution est un volume théorique exprimé en litres qui traduit la répartition du médicament dans l’ensemble des tissus et organes, en particulier dans ceux où il peut atteindre ses récepteurs et donc exercer son action pharmacologique

Distribution (2)
• Fixation protéique (1)– Le médicament une fois résorbé parvient dans
le plasma où il se trouve sous deux formes• Forme liée aux protéines plasmatiques (albumine) :
fraction inactive, non diffusible et constituant une réserve de principe actif qui est progressivement libéré
• Forme libre : active, diffusible, pouvant exercer son action pharmacologique

Distribution (3)• Fixation protéique (2)
– La fixation protéique des médicaments sur les protéines plasmatiques est un phénomène réversible, variable en intensité en fonction de la nature du PA (jusqu'à 99% de fixation de la dose administrée)
– Dès qu’une fraction de médicament sous forme libre quitte la circulation générale (diffusion tissulaire ou élimination), une fraction équivalente se libère du complexe protéine-médicament. La fixation protéique est un phénomène saturable, c’est-à-dire que la capacité de fixation protéique n’est pas inépuisable

Distribution (4)• Fixation protéique (3)
– Toute variation dans les concentrations en protéines plasmatiques, toute modification de leur structure ou encore un déplacement possible des médicaments par des substances endogènes (bilirubine par exemple) s’y fixant également, va entraîner des modifications de l’intensité de fixation des médicaments, avec dans certains cas une augmentation de la fraction libre, active et/ou toxique du médicament
– Il peut exister également des phénomènes de compétition entre deux ou plusieurs médicament se fixant habituellement sur les m^mes sites protéiques, susceptibles d’entraîner des interactions médicamenteuses
• Exemple : défixation protéique des antivitamines K (anticoagulants par voie orale) par administration d’anti-inflammatoires non stéroïdiens (rhumatologie), pouvant dans certains cas aboutir à un accident hémorragique (augmentation de l’activité de l’anticoagulant, dont la fraction libre est majorée)

Distribution (5)• Fixation protéique (4)
– Des facteurs physiopathologiques peuvent également modifier la fixation protéique des médicaments :
• Fixation le plus souvent faible chez les jeunes enfants et sujet âgés. Il faut être très prudent lors de l’utilisation des médicaments à forte fixation protéiques (sulfamides, par exemple), particulièrement chez le nourrisson présentant une hyperbilirubinémie
• L’insuffisance rénale ou hépatique, une baisse du taux d’albumine plasmatique, une fuite protéique d’origine rénale

Distribution (5)• Diffusion tissulaire
– La fraction libre du médicament diffuse vers les tissus et passe ainsi du « compartiment » plasmatique (ou central) vers le « compartiment » tissulaire (ou périphérique) après traversées des membranes tissulaires, par les m^mes mécanismes d’action que ceux décrits pour la phase de résorption
– Cette diffusion dépend de l’importance de la vascularisation du tissu considéré; certains tissus sont richement vascularisés (coeur, cerveau, foie, reins…) alors que d’autres le sont beaucoup moins (os, dents, phanères…) et seront difficilement atteint par les médicaments
– L’obésité, des modifications de l’importance relative des secteurs liquidiens, protéiques ou graisseux sont susceptibles de modifier la diffusion des médicaments
• Chez la femme enceinte : le poids en eau augmente de 50%– Après diffusion tissulaire, le médicament est susceptible de se
fixer sur son récepteur spécifique et d’exercer ainsi son action pharmacologique. Il peut aussi y être stocké (dans les tissus de nature lipidique en particulier) ou être transformé par des mécanismes enzymatiques (biotransformation)

Biotransformation (métabolisme) (1)• Un médicament sera d’autant plus facilement éliminé par l’organisme (par
le rein essentiellement) qu’il sera plus hydrosoluble.• Les biotransformations sont réalisées grâce à des processus
enzymatiques.• Le foie, en raison de sa vascularisation et de sa richesse en enzymes
microsomiaux), joue un rôle primordial, bien que d’autres organes ou tissus (tube digestif, poumons, reins…) contribuent aussi, de façon moins importante, au métabolisme des médicaments
• Les médicaments biotransformés par l’organisme sont appelés métabolites
• Ces métabolites correspondent à des transformations chimiques (oxydation, réduction, hydrolyse…) des principes actifs ou à des réactions de conjugaison (glucuroconjugaison), aboutissant à des composés plus polaire et plus hydrosolubles
• Les métabolites ne présentent généralement plus de propriétés pharmacologiques; on parle de métabolites inactifs
• Pour certains méicaments appelés bioprécurseurs, le métabolisme aboutit à un métabolite actif dont l’activité est similaire (exemple : oxazépam, métabolite du diazépam) voire supérieur (canrénone, métabolite de la spironolactone) à celle du produit administré
• Dans certains cas, le métabolisme peut entraîner la formation de métabolites toxiques (acétylisoniazide, métabolite hépatotoxique de l’isoniazide [antituberculeux])

Biotransformation (métabolisme) (2)• Facteurs pouvant modifier la biotransformation (1)
– Certains facteurs physiologiques ou exogènes peuvent modifier la biotransformation des médicaments
– Facteurs physiologiques• L’insuffisance hépatocellulaire entraîne une diminution du
métabolisme des médicaments (cirrhose, hépatite chronique et virale). Il peut –être nécessaire dans ce cas de réduire la posologie de certains médicaments
• Il existe des variations individuelles, ethnique et génétiques des biotransformations médicamenteuses. Il est possible par exemple de distinguer deux types de « populations » pour le métabolisme de l’isoniazide : les acetyleurs lents et les acetyleurs rapides. La connaissance de la vitesse d’acetylation de cet antituberculeux peut être utile à connaître pour mieux en ajuster la posologie et pour prévoir la nature des effets indésirables

Biotransformation (métabolisme) (3)• Facteurs pouvant modifier la
biotransformation (2)– Facteurs exogènes
• alimentation : la capacité métabolique du foie peut être augmentée (on parle d’ «induction enzymatique ») sous l’influence de certains types d’aliments (choux, brocolis..)
• Alcool : chez le buveur occasionnel, , l’absorption aigue d’alcool provoque un ralentissement (inhibition) du métabolisme de certains médicaments. A l’inverse la prise répétée et excessive d’alcool (alcoolisme chronique) est responsable d’induction enzymatique et de l’accélération du métabolisme des médicaments

Biotransformation (métabolisme) (4)
• Facteurs pouvant modifier la biotransformation (3)– Facteurs exogènes
• Médicaments associés : le métabolisme hépatique d’un médicament peut être modifié par d’autres médicaments administrés simultanément. Il s’agit d’interactions médicamenteuses pouvant entraîner une augmentation ou une diminution d’activité de l’un des médicaments associés
– Inducteurs enzymatiques : phenobarbital, phénytoïne, rifampicine
– Inhibiteurs enzymatiques : cimétidine, ciclosporine, jus de pamplemousse

Élimination (1)• L’élimination (ou excrétion) des médicaments et
de leur métabolite est assurée par diverses voies dont la plus importante est la voie urinaire, les autres voies étant la voie biliaire et la voie pulmonaire
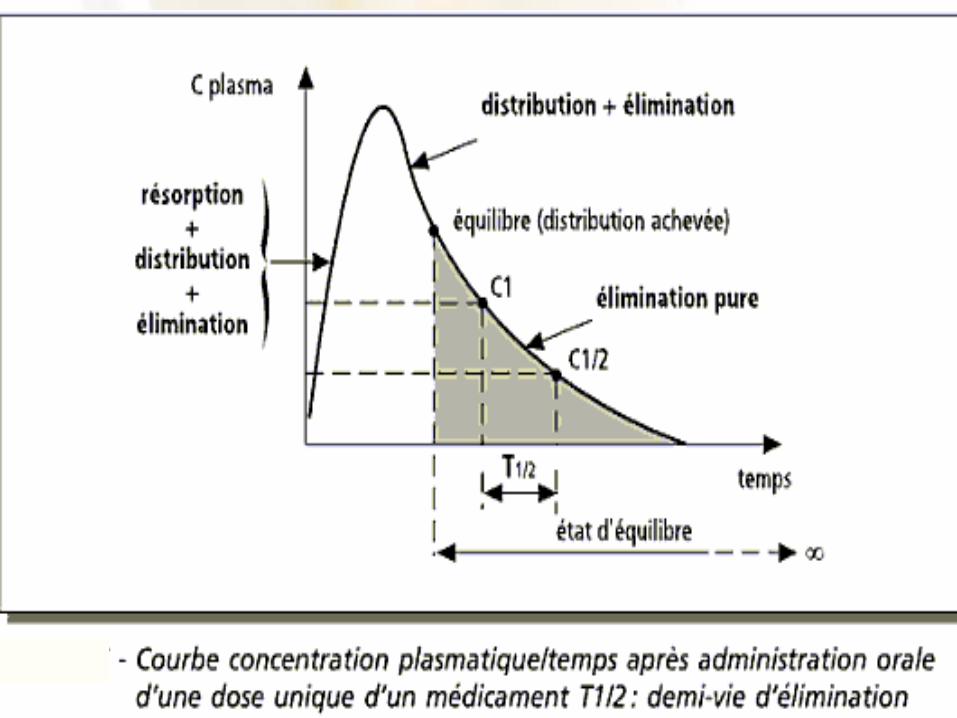
• Notion de demi-vie d’élimination– La demi-vie d’élimination d’un médicament est le
temps nécessaire à l’élimination de la moitié de la concentration d’un médicament dans le sang
• Si la concentration plasmatique de la phénytoïne est de 20 µg/mL après administration d’une dose donnée, et si le temps nécessaire pour réduire cette concentration à 10 µg/mL est de 24 heures, à condition de n’administrer aucune dose additionnelle entre-temps, la demi-vie sera dans ce cas de 24h
– La valeur de la demi-vie est fonction des processus de métabolisme et d’élimination, elle est variable d’un patient à l’autre en fonction de l’état physiopathologique individuel

Élimination (2)• La demi-vie d’élimination est un paramètre
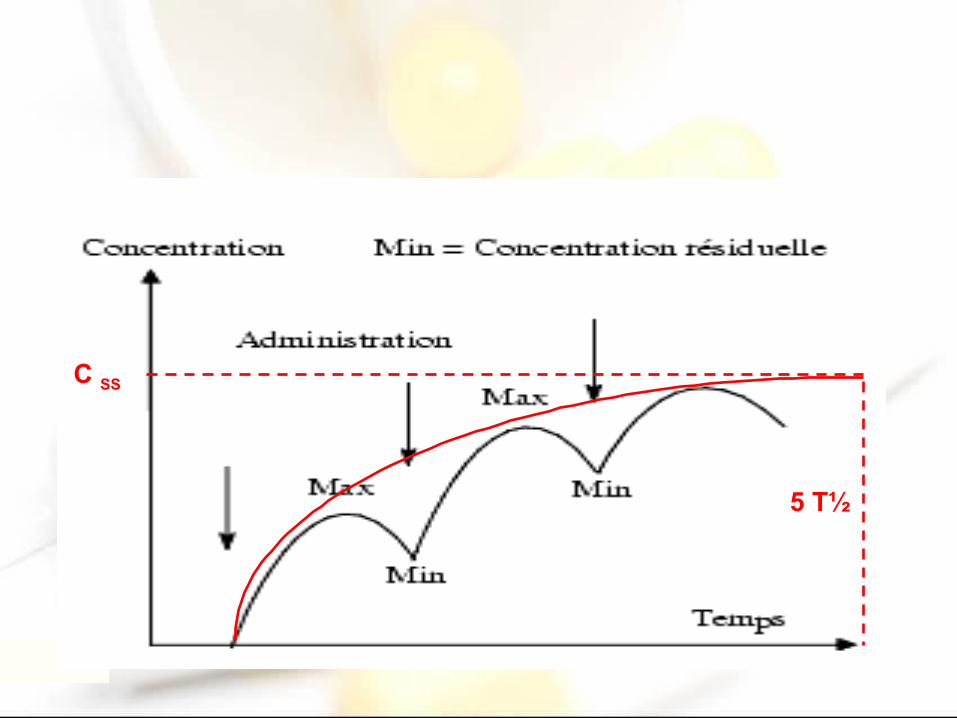
pharmacocinétique important à connaître. Il conditionne en particulier le nombre de prises par 24 h. Plus la demi-vie d’un médicament est courte, plus le nombre de prises sera important et la fréquence d’administration rapprochée
• Lors de la mise en route d’un traitement oral à long terme, le médicament s’accumule dans l’organisme jusqu’au moment où la vitesse d’élimination égale la vitesse de résorption+distribution. L’état d ’équilibre est atteint au bout d’un temps égal à 5 fois la demi-vie
• Les dosages plasmatiques nécessaires au suivi thérapeutique doivent être pratiquée, pour être interprétables, lorsque l’état d’équilibre est atteint. Après tout changement de posologie, il faut attendre le nouvel état d’équilibre pour prélever– Exemple : pour la phénytoïne dont la demi-vie est de 24h, il faut
attendre 5 à 6 jours pour obtenir l’état d’équilibre
5 T½
C SS

Élimination (3)• Élimination rénale (1)
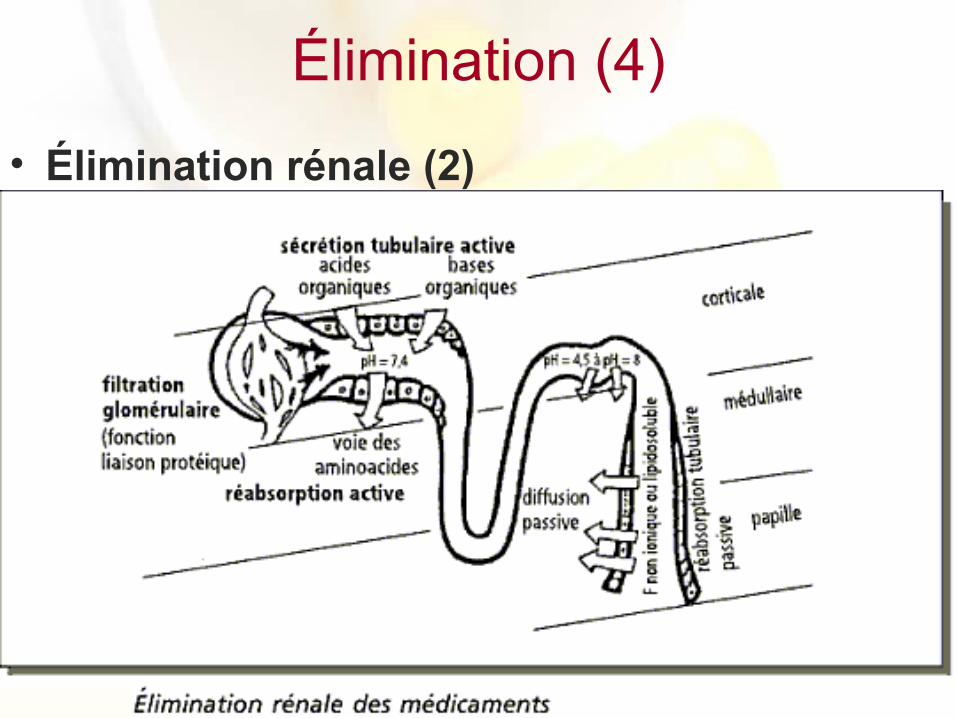
– L’élimination des médicaments par le rein est la résultat de phénomènes complexes au niveau du néphron, unité fonctionnelle du rein
• Filtration glomérulaire : le glomérule se comporte comme un filtre laissant passer tous les composés d’un poids moléculaire < 60 000 d, ce qui est le cas de tous les médicaments, à l’exception des médicaments liés aux protéines plasmatiques
• Sécrétion tubulaire active : processus de transport actif situé au niveau du tube contourné proximal, consommant de l’énergie et permettant le passage de médicaments, sous forme ionisée (acides et bases organiques faibles), de la corticale vers le tube contourné
• Résorption tubulaire : les médicaments présents dans le tubule peuvent être réabsorbés dans la circulation générale, par diffusion passive pour la fraction non ionique ou liposoluble au niveau du tube distal, ou par transport actif au niveau du tube proximal.
Élimination (4)
• Élimination rénale (2)

Élimination (5)• Élimination biliaire
– Après passage hépatique, la fraction du médicament non métabolisé peut retourner dans la circulation générale ou être excrétée par la bile. Il peut exister dans ce dernier cas un cycle entéro-hépatique : le médicament excrété dans la bile arrive dans l’intestin et peut être à nouveau être absorbé, repasser dans le sang et revenir au foie
– La voie d’élimination biliaire est généralement une voie accessoire (certains médicaments ont cependant une élimination biliaire prédominante), mais qui peutdevenir prépondérante si la voie principale d’élimination (rénale) est défaillante

Élimination (6)• Facteurs pouvant modifier l’élimination
– Certains facteurs physiopathologique ou exogènes peuvent modifier l’élimination des médicaments
• Facteurs physiopathologiques– L’insuffisance rénale entraîne une diminution de l’élimination des
médicaments et en favorise l’accumulation nécessitant une adaptation posologique en fonction du degré de l’insuffisance rénale qui peut être mesurée par certains examen biologiques (créatininémie, clairance de la créatinine)
– Toute modification de fixation protéique des médicaments, en augmentant la fraction libre, accélère l’élimination
• Facteurs exogènes– Toute modification du pH urinaire est susceptible de modifier la
réabsorption tubulaire passive, intervenant sur la fraction liposoluble non ionisée du médicament
» L’alcalinisation des urines par du bicarbonate de sodium est un traitement habituel des intoxications par barbituriques (acides faible).

Notions essentielles (1)• La pharmacocinétique d’un médicament
dans l’organisme comporte quatre étapes : résorption, distribution, biotransformation (métabolisme), élimination
• La résorption correspond au passage d’un médicament dans la circulation générale à partir de son lieu d’administration. Plusieurs mécanismes entrent en jeu dans la résorption en permettant le passage des membranes cellulaires; la diffusion passive est le mode de transfert le plus courant

Notions essentielles (2)• La biodisponibilité d’un médicament est la fraction
(exprimée en %) de la dose administrée qui atteint la circulation générale. Par définition, la biodisponibilité est de 100% après administration par voie intraveineuse; Pour les autres voies, la biodisponibilité est fonction de la quantité résorbée et de la quantité éliminée par les « effet de « premier passage » notamment hépatique.
• La distribution est l’étape correspondant à la diffusion du médicament dans l’organisme au niveau plasmatique et tissulaire. Dans le plasma, le médicament se trouve sous deux formes :– Forme liée aux protéines plasmatiques : cette fraction
circulante est inactive pharmacologiquement et non diffusibles vers les tissus. Elle constitue une « réserve » de principe actif qui est progressivement libéré (fixation réversible)
– Forme libre, active et diffusible

Notions essentielles (3)• La biotransformation correspond à la transformation
enzymatique, essentiellement hépatique, du médicament en métabolite (le plus souvent inactif) plus hydrosoluble, donc plus facile à éliminer au niveau rénal ou biliaire
• Certains médicaments dits « inducteurs enzymatiques » peuvent accélérer le métabolisme hépatique d’autres médicaments associés
• La demi-vie d’élimination (T1/2) correspond au temps nécessaire à la diminution de moitié de la concentration sanguine d’un médicament. Il s’agit d’un paramètre théorique important pour définir un nombre de prises par jour ou préciser le temps nécessaire pour parvenir à l’état d’équilibre en début de traitement
• L’élimination rénale d’un médicament ou de son métabolite est la résultante de mécanismes complexes au niveau du néphron : filtration glomérulaire, sécrétion et réabsorption tubulaires. En cas d’insuffisance rénale, la posologie de certains médicaments ou leur fréquence d’administration doivent être adaptées en fonction de la valeur de la clairance à la créatinine