Embed Size (px)
Citation preview

Electrolytes
• Energies de formation - Potentiel rédox standard
• Energies de solvatation-Cycle de Born Haber
• Modèle de solvatation de Born
• Interactions ion-ion - Modèle de Debye-Hückel
���1

Enthalpie de formation
Enthalpie de la réaction de formation à partir des éléments dans leur forme la plus stable à une température donnée (298 K) et à pression standard
Ces valeurs peuvent être déterminées à partir de cycles thermodynamiques
Entropie standard molaire absolue définie par rapport au cristal à 0K (sauf pour les ions en solution, échelle du proton aqueux). En effet, le troisième principe stipule que l'entropie d'un corps pur est nulle à 0 K
���2
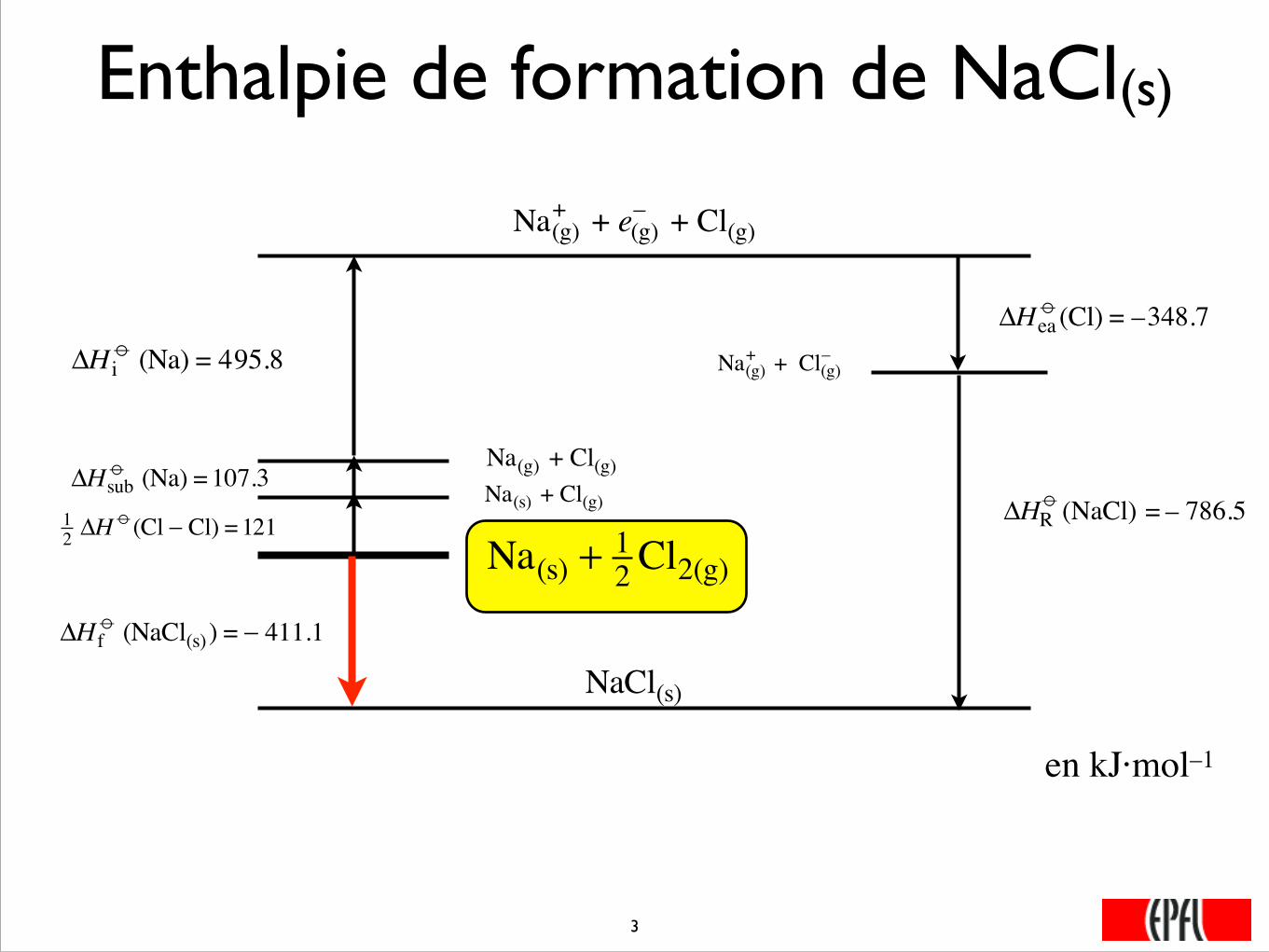
Enthalpie de formation de NaCl(s)
Na(s) + 12 Cl2(g)
ΔH io (Na) = 495.8
Na(g)+ + e(g)
– + Cl(g)
12 ΔH
o (Cl − Cl) =121Na(s) + Cl(g) ΔHsub
o (Na) =107.3Na(g) + Cl(g)
Na(g)+ + Cl(g)
– ΔHea
o (Cl) = –348.7
ΔH fo (NaCl(s) ) = − 411.1
NaCl(s)
�
ΔHRo (NaCl) = – 786.5
���3
en kJ·mol–1

Convention
So (H(aq)
+ ) = 0 J·mol–1·K–1Entropie standard molaire absolue
Il n’est pas possible de mesurer les valeurs thermodynamiques pour un ion simple, mais seulement pour un sel.
12 H2(g) → H(aq)
+ ΔH fo (H(aq)
+ ) = 0 J·mol–1
ΔGfo (H(aq)
+ ) = 0 J·mol–1
Choix arbitraire d’un zéro pour les valeurs ioniques
���4

Enthalpies de dissolution des sels
F Cl Br I OH CO NO SO
Li 4.9 -37 -48.8 -63.3 -23.6 -18.2 -2.7 -29.8
Na 1.9 3.9 -0.6 -7.5 -44.5 -26.7 20.4 -2.4
K -17.8 17.2 19.9 20.3 -57.1 -30.9 34.9 23.8
NH -1.2 14.8 16 13.7 25.7 6.6
Mg -17.7 -160 -185.6 -213.2 2.3 -25.3 -90.9 -91.2
Ca 11.5 -81.3 -103.1 -119.7 -16.7 -13.1 -19.2 -18
Réactions endothermiques���5
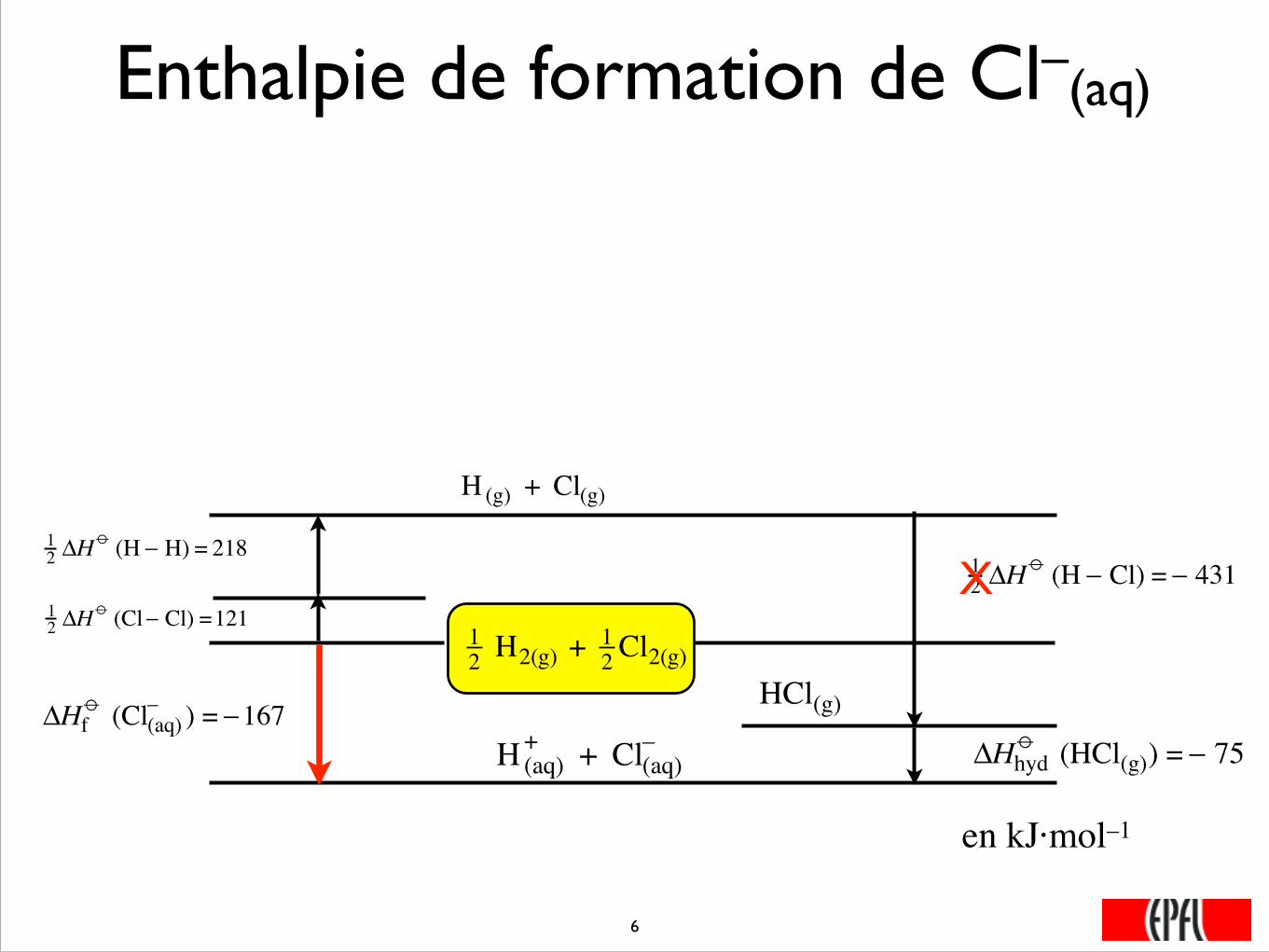
Enthalpie de formation de Cl–(aq)
12 H2(g) + 1
2 Cl2(g)
�
12 ΔH
o (Cl− Cl) =121
�
12 ΔH
o (H − H) = 218
�
H (g) + Cl(g)
�
ΔHhydo (HCl(g)) = − 75
�
H (aq)+ + Cl(aq)
–
�
ΔHfo (Cl(aq)
– ) = −167
�
12 ΔH
o (H − Cl) = − 431
�
HCl(g)
X
���6
en kJ·mol–1
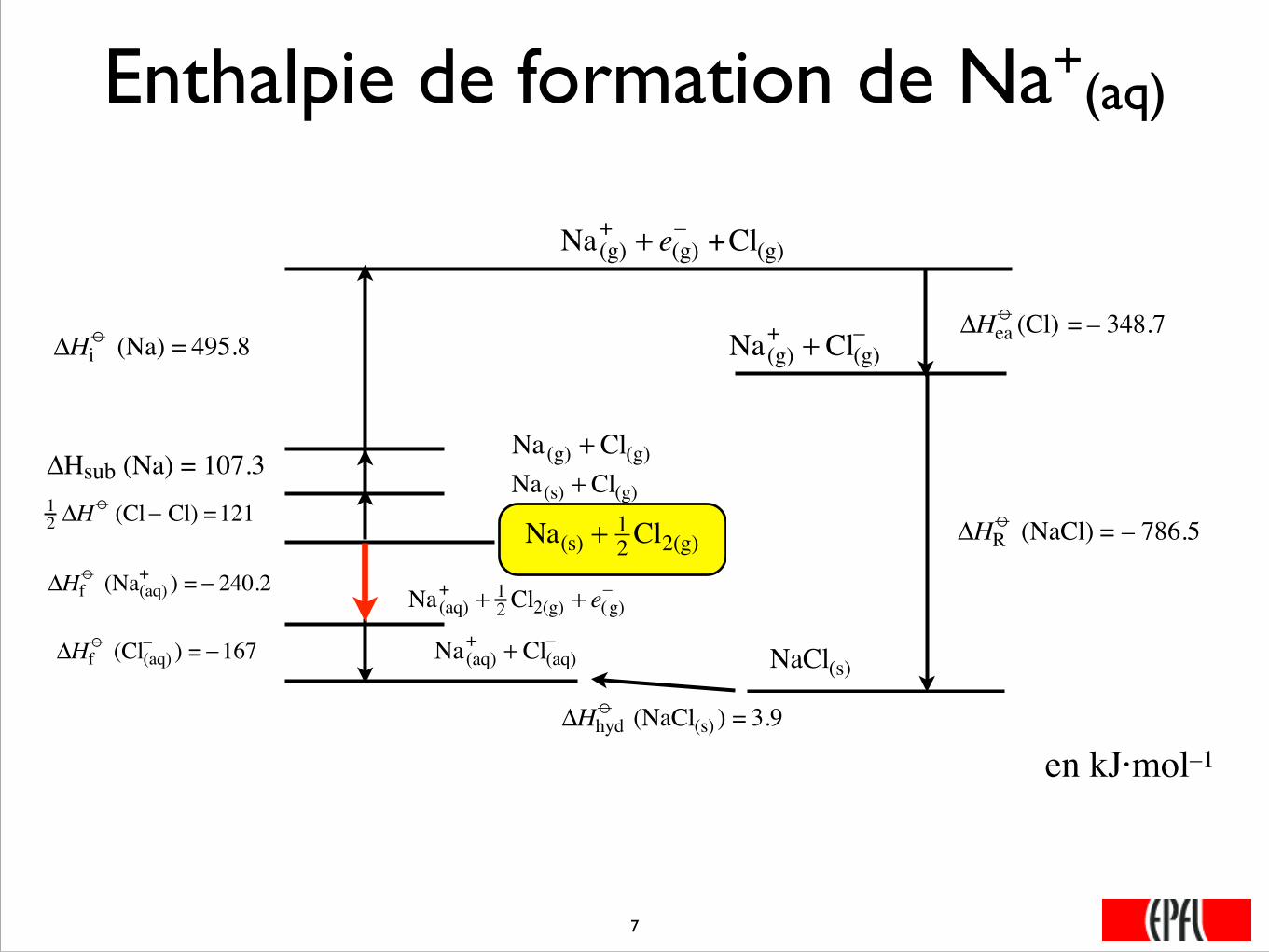
Enthalpie de formation de Na+(aq)
Na(s) + 12 Cl2(g)
�
12 ΔH
o (Cl− Cl) =121
�
Na(s) + Cl(g)ΔHsub (Na) = 107.3
�
Na(g) + Cl(g)
�
ΔHio (Na) = 495.8
�
Na(g)+ + e(g)
– +Cl(g)
�
ΔHeao (Cl) = – 348.7
�
Na(g)+ + Cl(g)
–
�
ΔHRo (NaCl) = – 786.5
�
NaCl(s)
�
Na(aq)+ + Cl(aq)
–
�
ΔHhydo (NaCl(s) ) = 3.9
�
ΔHfo (Cl(aq)
– ) = −167
�
Na(aq)+ + 1
2 Cl2(g) + e(g)–
�
ΔHfo (Na(aq)
+ ) = − 240.2
���7
en kJ·mol–1

Mesure électrochimique
12 H2(g) + Na(aq)
+ H(aq)+ + Na(s)
ΔGf
o (Na(aq)+ ) = F ENa+ /Na
o⎡⎣ ⎤⎦SHE
Les énergies Gibbs de formation des ions peuvent être obtenues de façon très précise par mesure des potentiels rédox standard.
Les deux échelles du proton sont compatibles
Entropie de formation
ΔSfo = −
∂ΔGfo
∂T⎛
⎝⎜⎞
⎠⎟ p= p o
= −dΔGf
o
dT= − nF
d E o⎡⎣ ⎤⎦SHEdT
���8

Exemple : Potentiel standard rédox de Na+/Na
ENa+ /Na
o⎡⎣ ⎤⎦SHE= − 2.714 V
ΔGfo (Na(aq)
+ ) = − 261.86 kJ ⋅mol-1 ΔH fo (Na(aq)
+ ) = − 239.7 kJ ⋅mol-1
Energie de Gibbs, entropie et enthalpie de formation
ΔSfo (Na(aq)
+ ) = 96485 ⋅0.000772 = 74.48 J ⋅mol–1 ⋅K–1
So (Na(aq)
+ ) = 60.25 J ⋅mol–1 ⋅K–1Entropie:
So (Na(s) ) = 51.45 J ⋅mol–1 ⋅K–1
So (H2(g)) = 130.684 J ⋅mol–1 ⋅K–1
12 H2(g) + Na(aq)
+ H(aq)+ + Na(s)
ΔSfo (Na(aq)
+ ) = − 51.45 − 60.25 − 12 ⋅130.684⎡⎣ ⎤⎦
= 74.14 J ⋅mol–1 ⋅K–1
d ENa+ /Nao⎡⎣ ⎤⎦SHE
dT= − 0.000772 V ⋅K–1
���9

Potentiel rédox standard
“Réaction virtuelle”
oxS + n2 H2 nH+ + redS
Eox/red
o⎡⎣ ⎤⎦SHE = ΔGfo (ox(aq)) − ΔGf
o (red(aq))⎡⎣ ⎤⎦ / nF
Exemple :Fe(aq)3+ + 1
2 H2 H+ + Fe(aq)2+
ΔGfo (Fe(aq)
3+ ) = − 4.6 kJ ⋅mol–1 ΔGf
o (Fe(aq)2+ ) = − 78.9 kJ ⋅mol–1
EFeIII /FeIIo⎡⎣ ⎤⎦SHE
= − ΔG o / F = ΔGfo (Fe(aq)
2+ ) − ΔGfo (Fe(aq)
3+ )⎡⎣ ⎤⎦ / F
= − −78900 + 4600[ ] / 96485 = 0.77V
Les potentiels rédox standard peuvent être calculés à partir des énergies Gibbs de formation
���10

Electrolytes
• Energies de formation - Potentiel rédox standard
• Energies de solvatation-Cycle de Born Haber
• Modèle de solvatation de Born
• Interactions ion-ion - Modèle de Debye-Hückel
���11

Enthalpie de solvatation
H(g)+ → H(aq)
+ ΔHhydo (H+ ) = 0 kJ ⋅mol−1
Convention : Echelle relative
Valeur à prendre avec des pincettes
Echelle absolue
ΔHhydo (H+ ) = −1130 kJ ⋅mol−1
���12

Potentiel redox absolu
1/2 H2 gaz
H+ aqueux +
électron au repos dans le videF EO/R[ ]abs
ΔGhyd(H+)
H+ en phase gazeuse +
électron au repos dans le videEnergie
d’ionisation
1/2 H2 gaz
Demi-énergie de liaison
EH+ / 1
2H2
o⎡
⎣⎢⎢
⎤
⎦⎥⎥abs
= ΔGhydo (H+)+ EI,H + 1
2 Eliaison H2(g)( )⎡⎣⎢
⎤⎦⎥ / F
EH+ / 12H2
o⎡
⎣⎢⎢
⎤
⎦⎥⎥abs
≅ 4.44± 0.05 V
Potentiel redox absolu
1/2 H2gaz
H+ aqueux+
électron au repos dans le videF EO/R[ ]abs
!Ghyd(H+)
H+ en phase gazeuse+
électron au repos dans le videEnergie
d’ionisation
1/2 H2gaz
Demi-énergiede liaison
EH+ / 12H2
o!
"#
$
%&abs
= 'Ghydo (H+) + EI,H + 1
2 Eliaison H2(g)( )!"
$% / F
EH+ / 12H2o!
"#$%&abs
' 4.44 ± 0.05 V
���13
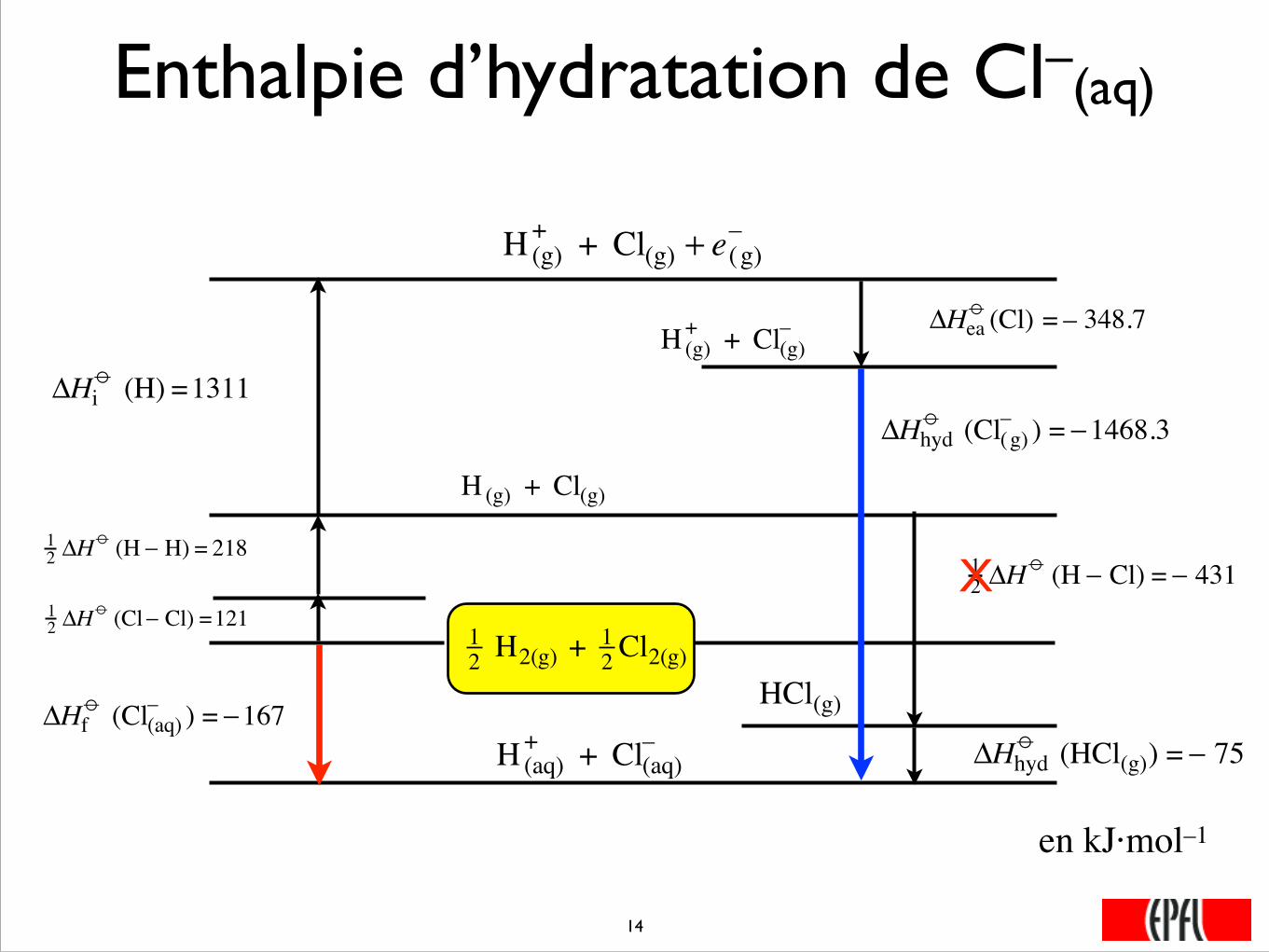
Enthalpie d’hydratation de Cl–(aq)
12 H2(g) + 1
2 Cl2(g)
�
12 ΔH
o (Cl− Cl) =121
�
12 ΔH
o (H − H) = 218
�
H (g) + Cl(g)
�
12 ΔH
o (H − Cl) = − 431
�
HCl(g)
�
ΔHhydo (HCl(g)) = − 75
�
H (aq)+ + Cl(aq)
–
�
ΔHfo (Cl(aq)
– ) = −167
�
ΔHio (H) =1311
�
H (g)+ + Cl(g) + e(g)
–
�
ΔHeao (Cl) = – 348.7
�
H (g)+ + Cl(g)
–
�
ΔHhydo (Cl(g)
– ) = −1468.3
X
���14
en kJ·mol–1
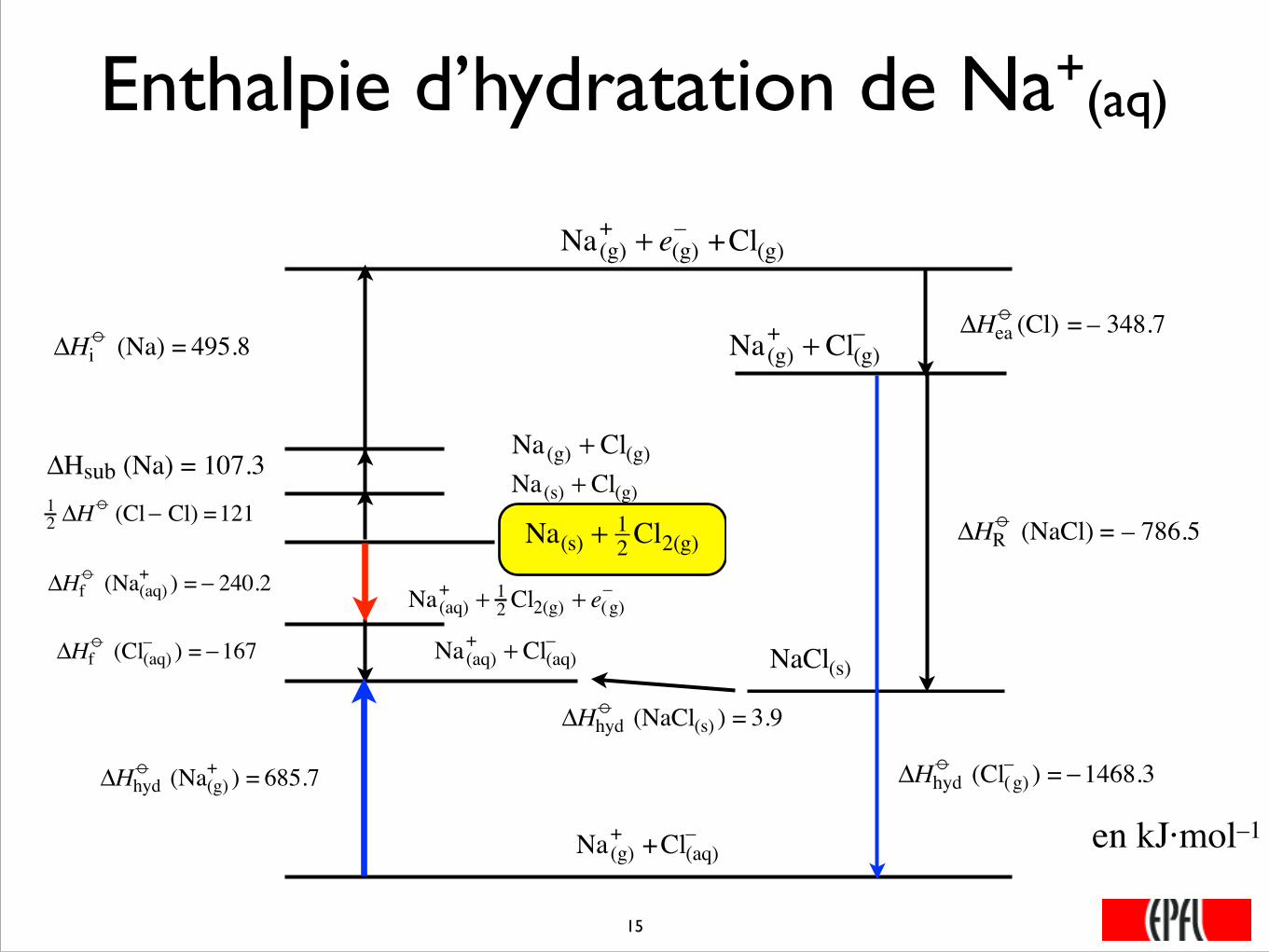
Enthalpie d’hydratation de Na+(aq)
Na(s) + 12 Cl2(g)
�
12 ΔH
o (Cl− Cl) =121
�
Na(s) + Cl(g)ΔHsub (Na) = 107.3
�
Na(g) + Cl(g)
�
ΔHio (Na) = 495.8
�
Na(g)+ + e(g)
– +Cl(g)
�
ΔHeao (Cl) = – 348.7
�
Na(g)+ + Cl(g)
–
�
ΔHRo (NaCl) = – 786.5
�
NaCl(s)
�
Na(aq)+ + Cl(aq)
–
�
ΔHhydo (NaCl(s) ) = 3.9
�
ΔHfo (Cl(aq)
– ) = −167
�
Na(aq)+ + 1
2 Cl2(g) + e(g)–
�
ΔHfo (Na(aq)
+ ) = − 240.2
�
ΔHhydo (Cl(g)
– ) = −1468.3
�
Na(g)+ +Cl(aq)
–
�
ΔHhydo (Na(g)
+ ) = 685.7
���15
en kJ·mol–1
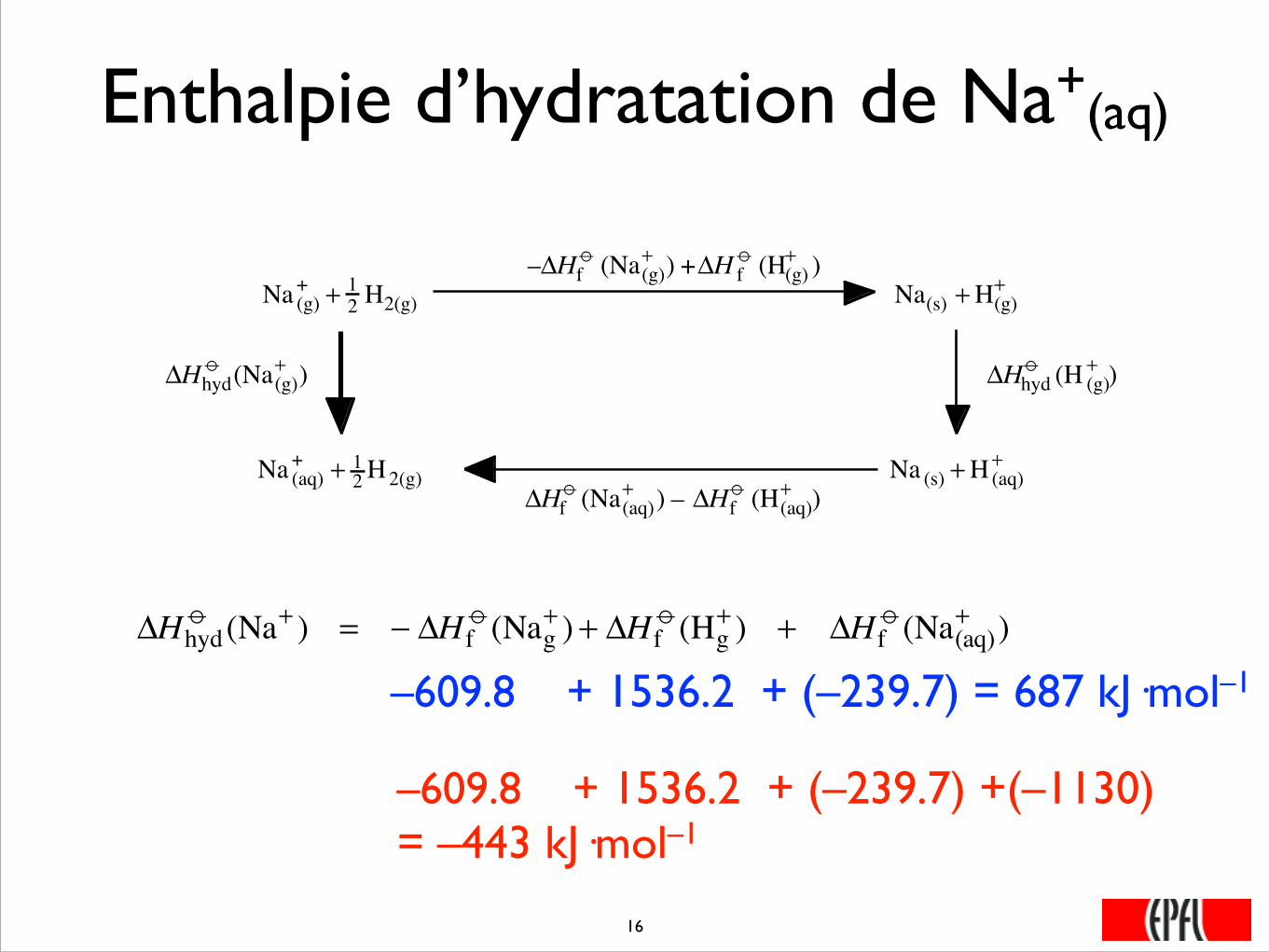
Enthalpie d’hydratation de Na+(aq)
�
ΔHhydo (Na(g)
+ )
�
–ΔHfo (Na(g)
+ ) +ΔH fo (H(g)
+ )
�
Na(g)+ + 1
2 H2(g)
�
Na(aq)+ + 1
2 H2(g)
�
Na(s) +H(g)+
�
Na(s) +H(aq)+
�
ΔHhydo (H (g)
+ )
�
ΔHfo (Na(aq)
+ ) – ΔHfo (H(aq)
+ )
ΔHhydo (Na+ ) = − ΔH f
o (Nag+ ) + ΔH f
o (Hg+ ) + ΔH f
o (Na(aq)+ )
–609.8 + 1536.2 + (–239.7) = 687 kJ·mol–1
–609.8 + 1536.2 + (–239.7) +(–1130) = –443 kJ·mol–1
���16

Energies de Gibbs & Potentiels électrochimiques
ΔGhyd
o (Cz+ )⎡⎣ ⎤⎦abs = αCz+o( )aq − µCz+
o( )g
C(g)z+ → C(aq)
z+Hydratation d’un cation
C(g)z+ + z H(aq)
+ C(aq)z+ + z H(g)
+
ΔGhyd
o (Cz+ ) = αCz+o( )aq + z µH+
o( )g − µCz+o( )g − z αH+
o( )aq
Echelle relative
ΔGhyd
o (Az– ) = αAz–o( )aq + z αH+
o( )aq − µAz–o( )g − z µH+
o( )g���17

Autres hypothèses
Hypothèse TA-TB !Hypothèse : Ferrocene/Ferricinium
Pas terrible, mais mieux que rien...
���18

Electrolytes
• Energies de formation - Potentiel rédox standard
• Energies de solvatation-Cycle de Born Haber
• Modèle de solvatation de Born
• Interactions ion-ion - Modèle de Debye-Hückel
• Paires d’ions - Modèles de Bjerrum et de Fuoss
���19
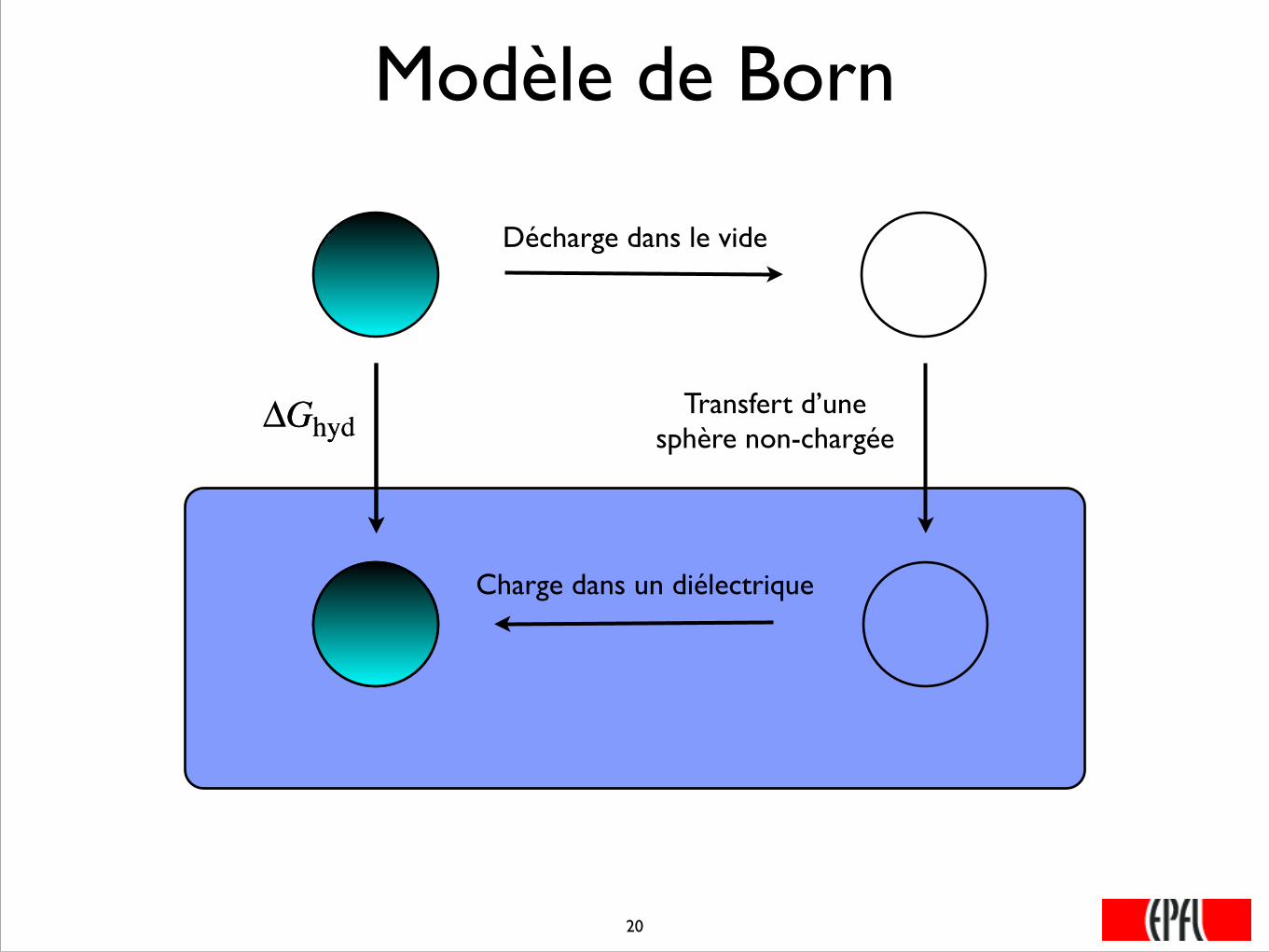
Modèle de Born
ΔGhyd
Décharge dans le vide
Charge dans un diélectrique
Transfert d’une sphère non-chargée
ΔGhyd
���20

Travail de charge
dqdqdq
Sphère non chargée
w = V (q) dq0
ze
∫Travail de charge d’une sphère
V (q) =q
4πε0rPotentiel généré par une sphère chargée
wc =z2e2
8πε0rion
���21
Ion
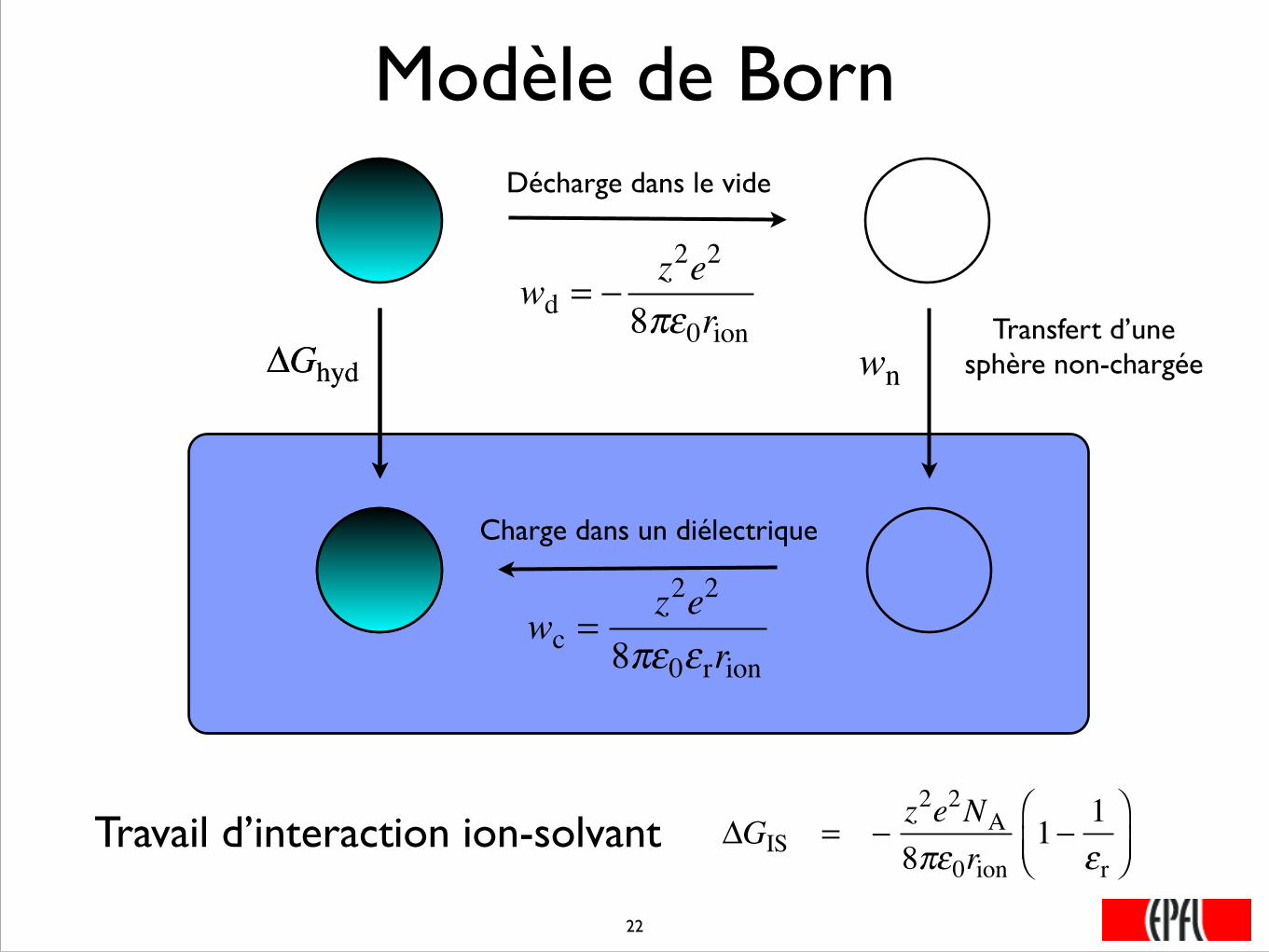
Modèle de Born
ΔGhyd
Décharge dans le vide
Charge dans un diélectrique
Transfert d’une sphère non-chargéeΔGhyd
wd = −z2e2
8πε0rionwn
Travail d’interaction ion-solvant
wc =z2e2
8πε0εrrion
ΔGIS = −z2e2NA8πε0rion
1− 1εr
⎛⎝⎜
⎞⎠⎟
���22
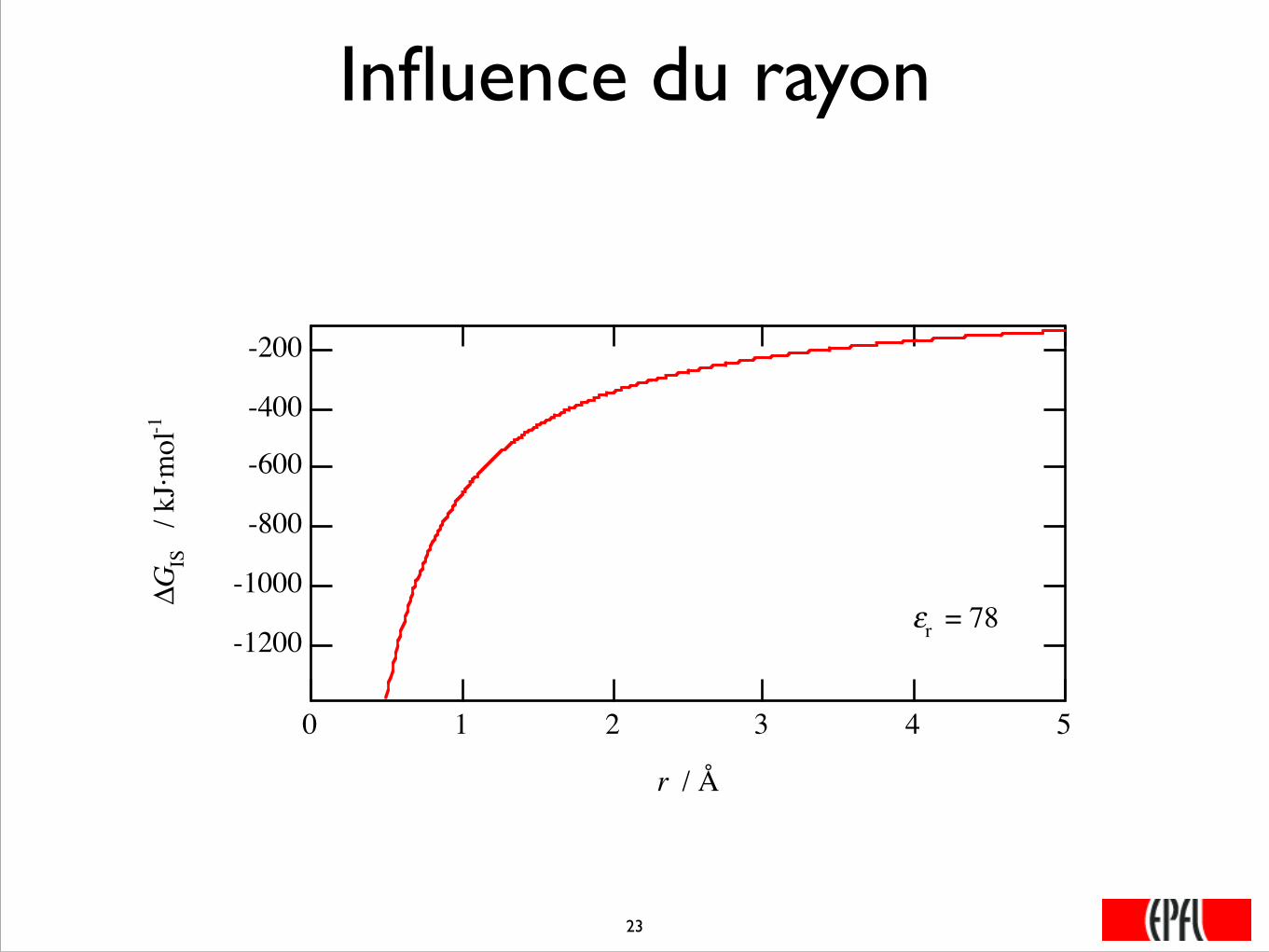
Influence du rayon
-1200
-1000
-800
-600
-400
-200
ΔGIS
/ k
J·mol
-1
543210
r / Å
εr = 78
���23
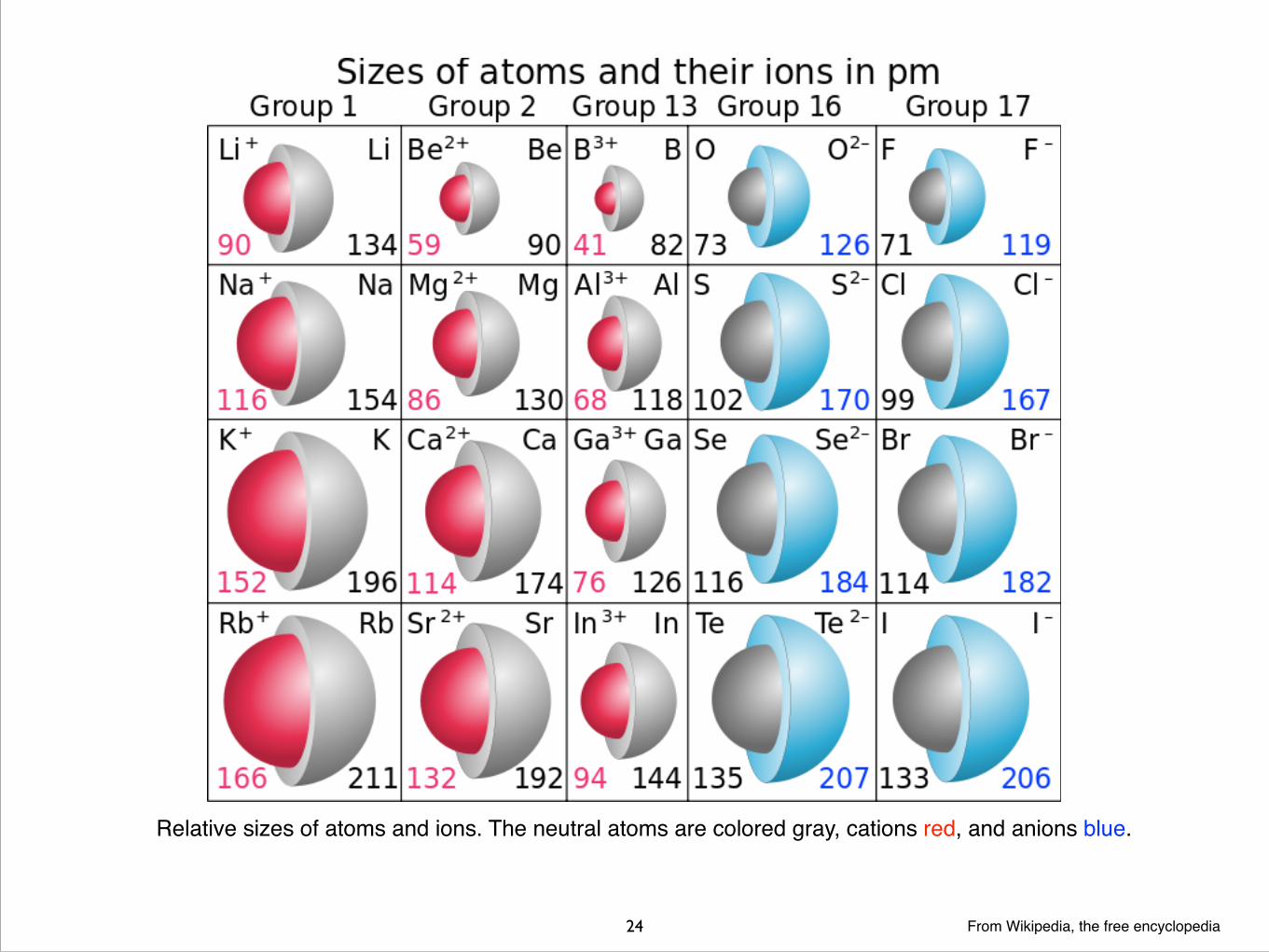
���24 From Wikipedia, the free encyclopedia
Relative sizes of atoms and ions. The neutral atoms are colored gray, cations red, and anions blue.
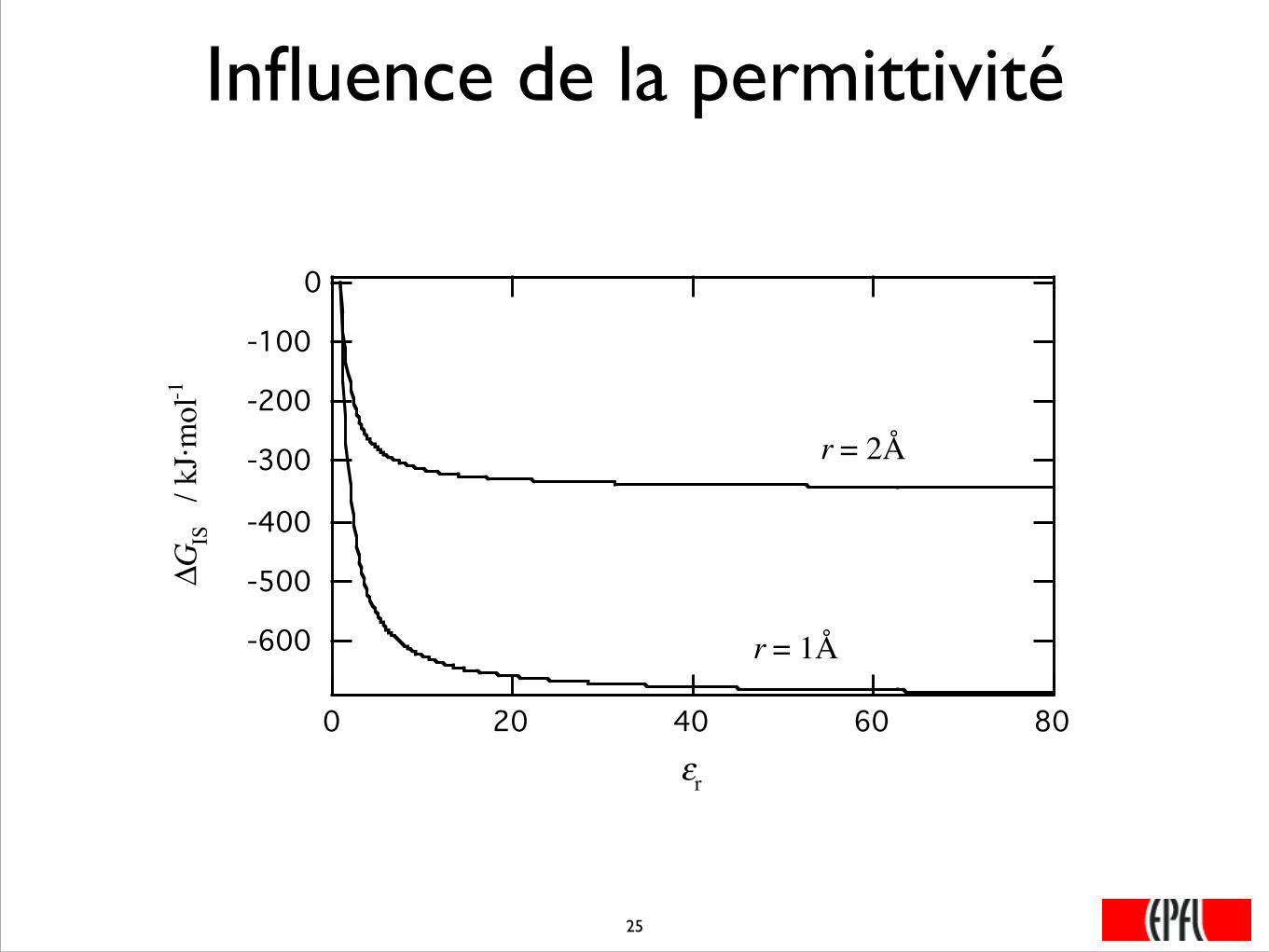
Influence de la permittivité
-600
-500
-400
-300
-200
-100
0
Δ GIS
/ k
J·mol
-1
806040200εr
r = 1Å
r = 2Å
���25
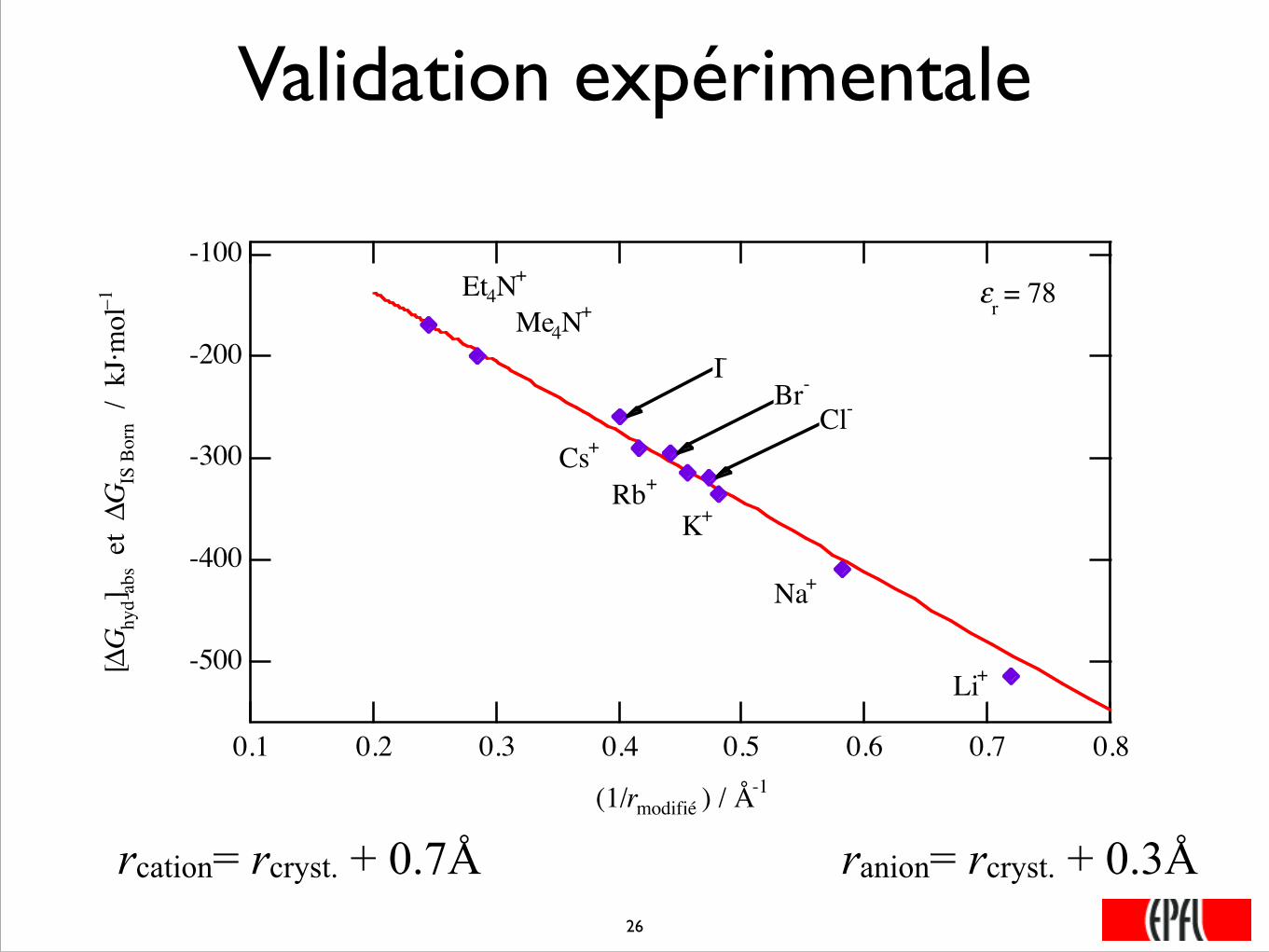
Validation expérimentale
-500
-400
-300
-200
-100
[ΔG
hyd]
abs
et ΔG
IS B
orn
/ k
J·m
ol–1
0.80.70.60.50.40.30.20.1
(1/rmodifié ) / Å-1
εr = 78
K+
I-
Rb+
Na+
Et4N+
Cs+
Li+
Br-
Cl-
Me4N+
���26
rcation= rcryst. + 0.7Å ranion= rcryst. + 0.3Å

Entropie d’interaction Ion-Solvant
ΔSIS = −
∂ΔGIS∂T
⎛⎝⎜⎜⎜
⎞⎠⎟⎟⎟p,µi
ΔSIS =
NAz2e2
8πε0r1εr2
∂εr∂T⎛⎝⎜⎜⎜⎞⎠⎟⎟⎟
Entropie
ΔH IS = −NAz
2e2
8πε0r1 − 1
εr−Tεr2∂εr∂T
⎡
⎣⎢
⎤
⎦⎥
Enthalpie
∂εr∂T
= − 0.3595Solution aqueuse
���27
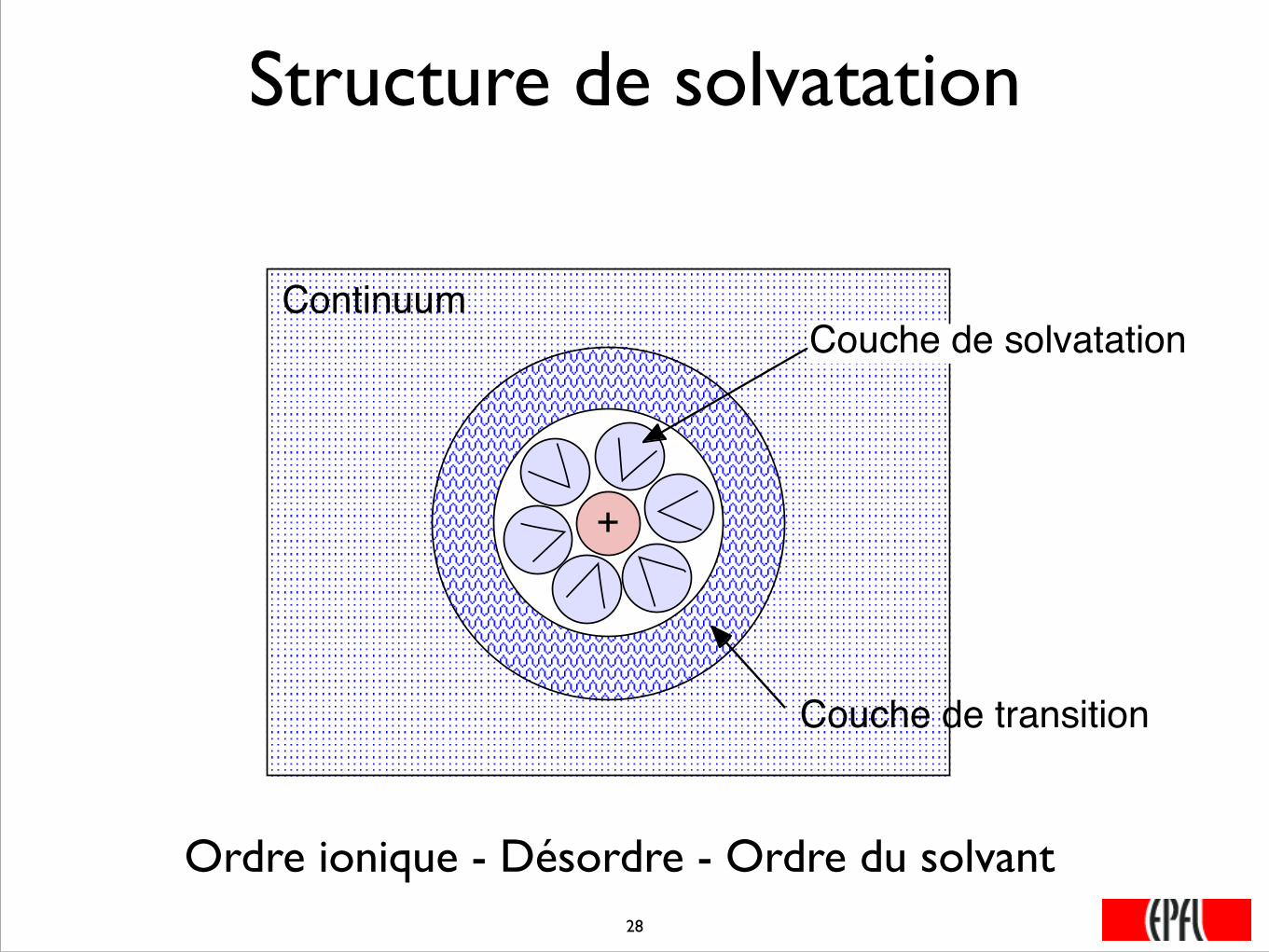
Structure de solvatation
+
Couche de solvatation
Couche de transition
Continuum
Ordre ionique - Désordre - Ordre du solvant���28
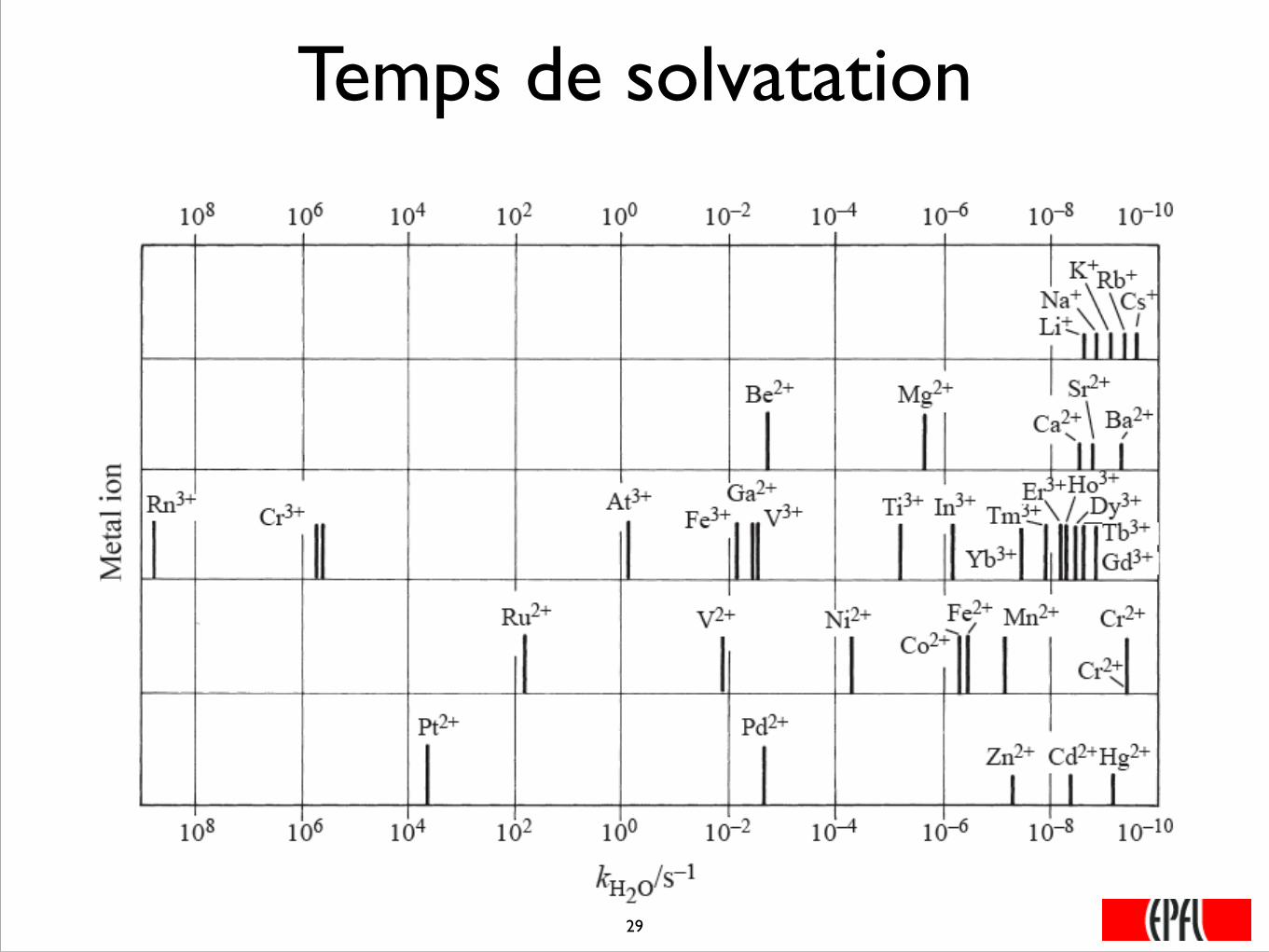
Temps de solvatation
���29

Electrolytes
• Energies de formation - Potentiel rédox standard
• Energies de solvatation-Cycle de Born Haber
• Modèle de solvatation de Born
• Interactions ion-ion - Modèle de Debye-Hückel
���30

Travail de dilution
wosm = nRT ln c2
c1
⎛
⎝⎜⎜⎜⎜
⎞
⎠⎟⎟⎟⎟
Contribution osmotique
wélec = nRT ln γ 2
γ 1
⎛
⎝⎜⎜⎜⎜
⎞
⎠⎟⎟⎟⎟
Contribution électrique
!µ1 !µ2
wdilut = nRT ln a2
a1
⎛
⎝⎜⎜⎜⎜
⎞
⎠⎟⎟⎟⎟
Energie de Gibbs de la dilution
���31

Théorie de Debye-Hückel
• Seules les forces électrostatiques sont prises en compte
• L’électrolyte est totalement dissocié
• Solvant = milieu diélectrique homogène continu
• Ions = Sphères rigides non polarisables
• Energie d’interaction électrostatique faible par rapport à l’énergie thermique
���32

Statistique de BoltzmannLa variation de population entre deux niveaux d’énergie dépend du travail pour passer d’un niveau à l’autre
n0
n1Travail
n1 = n0 exp −
ΔEkT
⎛⎝⎜⎜⎜
⎞⎠⎟⎟⎟
ΔE n1 / n0kT2kT3kT
0.370.130.05
���33

r
φ(r)
Le potentiel électrique créé par cet ion en l’absence d’autres ions est
φ(r) =zce
4πε0εrr
Densité ioniqueSoit un ion pris comme origine de coordonnées sphériques
Ni (r) = Ni∞ e −zie φ(r)−φ(∞)[ ] / kT
La densité ionique dans une coquille d’épaisseur dr est
���34

Densité volumique de charges
Ni (r) = Ni∞ e −zieφ(r) / kT
Densité ionique - Potentiel nul à l’infini
Densité volumique de charge
ρ(r) = zie Ni (r) = zie Ni∞e
i∑
i∑ −zieφ(r) / kT
zieφ(r) << kTSi on peut linéariser l’exponentielle
ρ(r) ≅ zieNi∞ 1 − zieφ(r)
kT⎡⎣⎢
⎤⎦⎥i
∑
���35

Approximation
Co-volume d’un ion (sel 1:1)
a =3
8000π NA c3 = 6 ⋅10−10 ⋅ c−1/3
exp(−zieφ(r) / kT ) 1 − zieφ(r)kTa / nm
3 0.787 0.76
Pour une concentration de 0.01 M, on peut linéariser l’exponentielle
���36

Relation charge-potentiel
ρ(r) ≅ zieNi∞ 1 − zieφ(r)
kT⎡⎣⎢
⎤⎦⎥i
∑Charge volumique
zieNi∞ = 0
i∑Electroneutralité
ρ(r) = −zi2e2Ni
∞φ(r)kT
i∑ = −
e2
kTNi
∞
i∑ zi
2⎡
⎣⎢⎢
⎤
⎦⎥⎥φ(r)Charge volumique
κ 2 =e2
ε0εr kTNi
∞zi2
i∑Constante
Relation linéaire charge-potentiel
ρ(r) = − ε0εrκ2 φ(r)
���37

Potentiel autour d’un ion
80
60
40
20
0
φ(r
) /
mV
50403020100
r / Å
c = 0.01 Ma = 2Å
V (r)φ (r)
φ(r) =zce
4πε0εr
eκa
1+κa⎡
⎣⎢⎢
⎤
⎦⎥⎥e−κ r
r≈
zce4πε0εr
e−κ r
rV (r) =
zce4πε0εr
1r
���38

Atmosphère ionique
ρ(r) =−zceκ
2
4πeκa
1+κae−κ r
rCharge volumique
Charge dans une coquille d’épaisseur dr
qsph (r) = 4πr2 ρ(r) dr
qsph (r) = − zceκ2 eκa
1+κare−κ rdr
���39
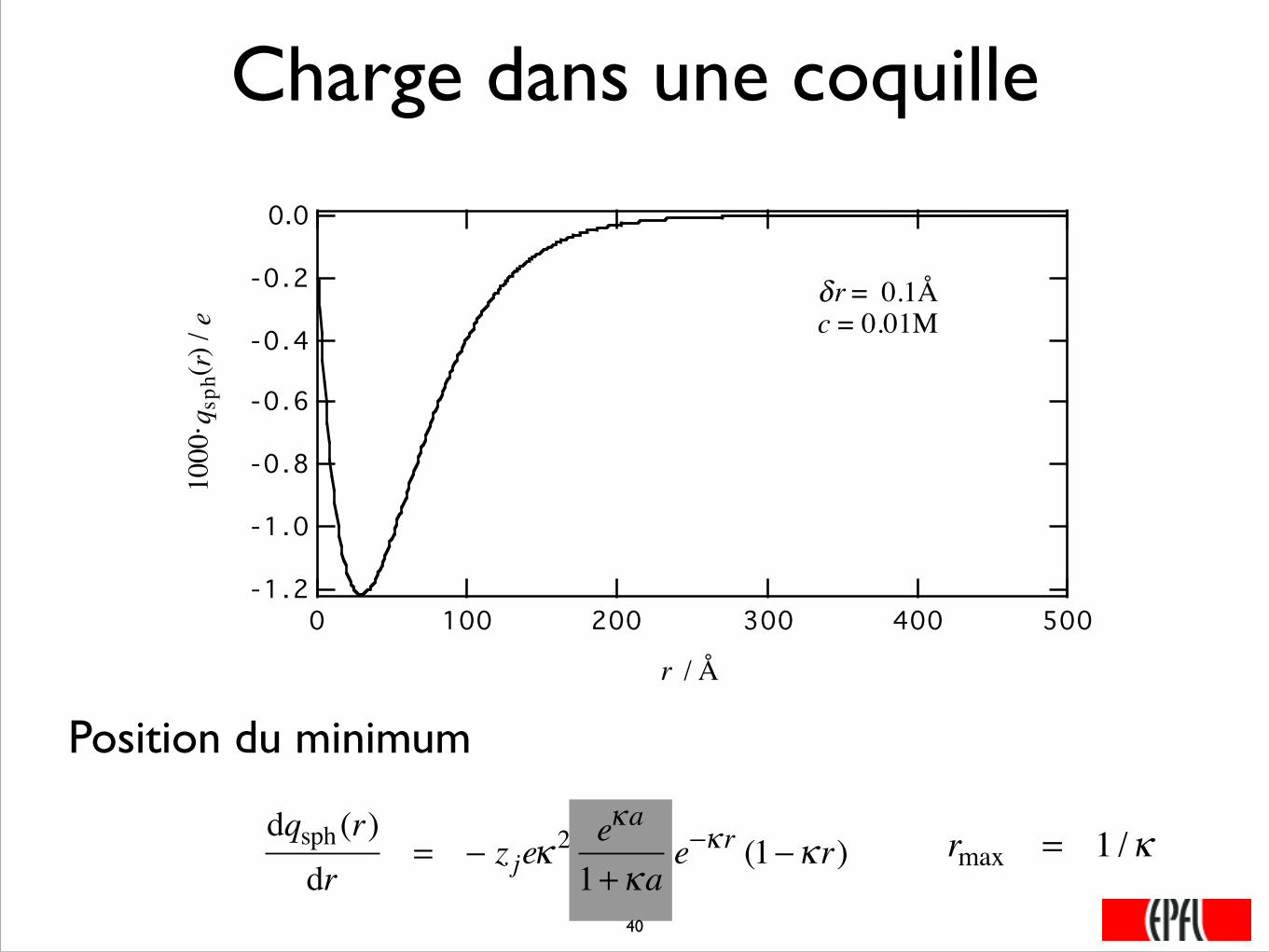
Charge dans une coquille
-1 .2
-1.0
-0.8
-0.6
-0.4
-0.2
0.010
00· q
sph(r)
/ e
5004003002001000
r / Å
δr = 0.1Åc = 0.01M
Position du minimum
rmax = 1 /κdqsph (r)dr
= − z jeκ2 eκa
1+κae−κ r (1−κ r)
���40

Les ions occupent tout l’espace
Les forces attractives isotropes résultent en une occupation totale de la solution, comme si les forces étaient répulsives.
���41

Distance réciproque de Debye
< ratm > = rqsph (r)zce
dra
∞
∫ = −κ 2eκa
1+κar2e−κ rdr =
1+ (1+κa)2
κ (1+κa)a
∞
∫
Rayon moyen de l’atmosphère ionique
< ratm > = 1 /κ
1/κ
���42
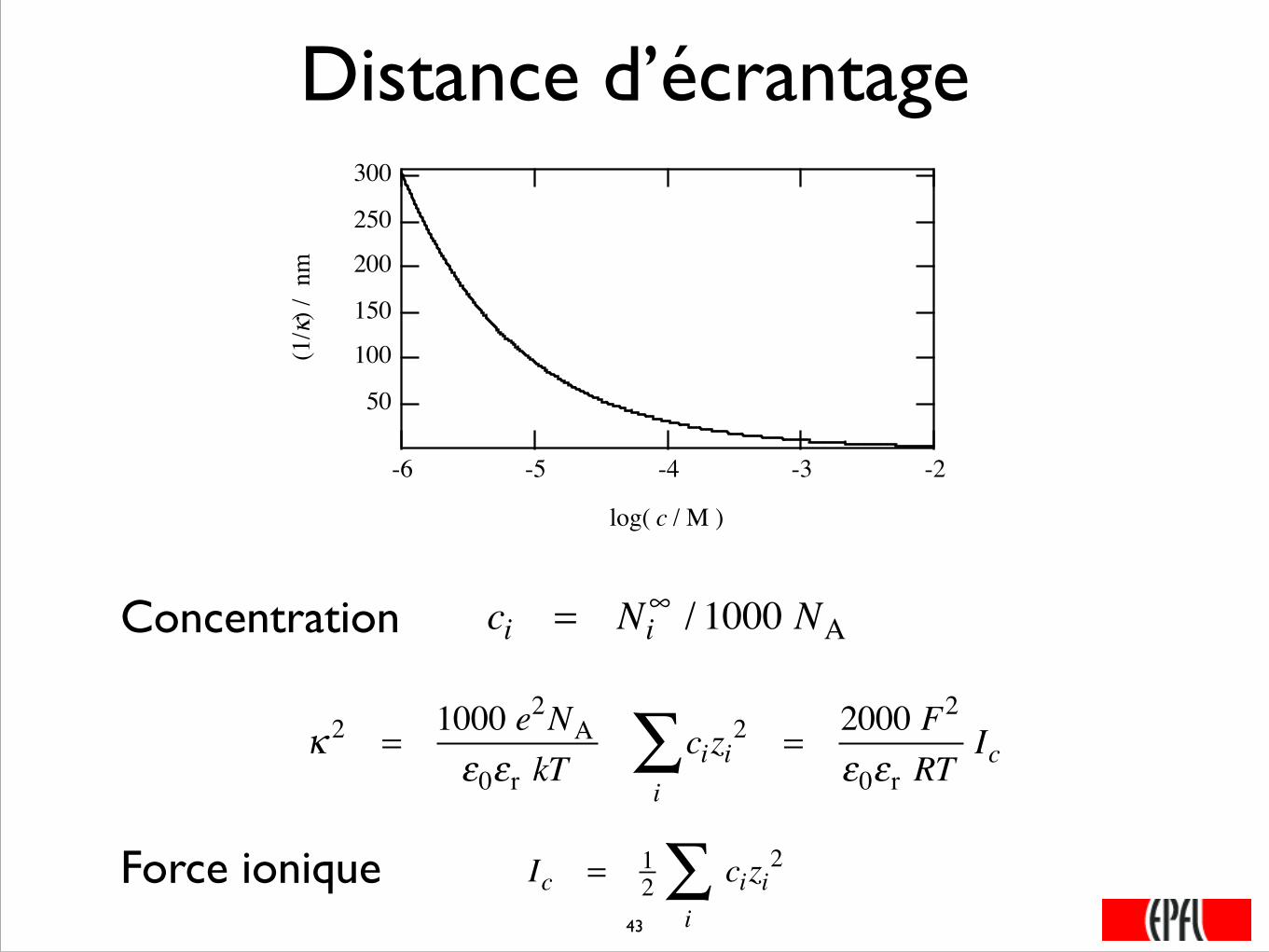
Distance d’écrantage300
250
200
150
100
50
(1/κ
) / n
m
-6 -5 -4 -3 -2
log( c / M )
κ 2 =1000 e2NAε0εr kT
cizi2
i∑ =
2000 F2
ε0εr RTIc
ci = Ni∞ / 1000 NAConcentration
Force ionique Ic = 12 cizi
2
i∑
���43
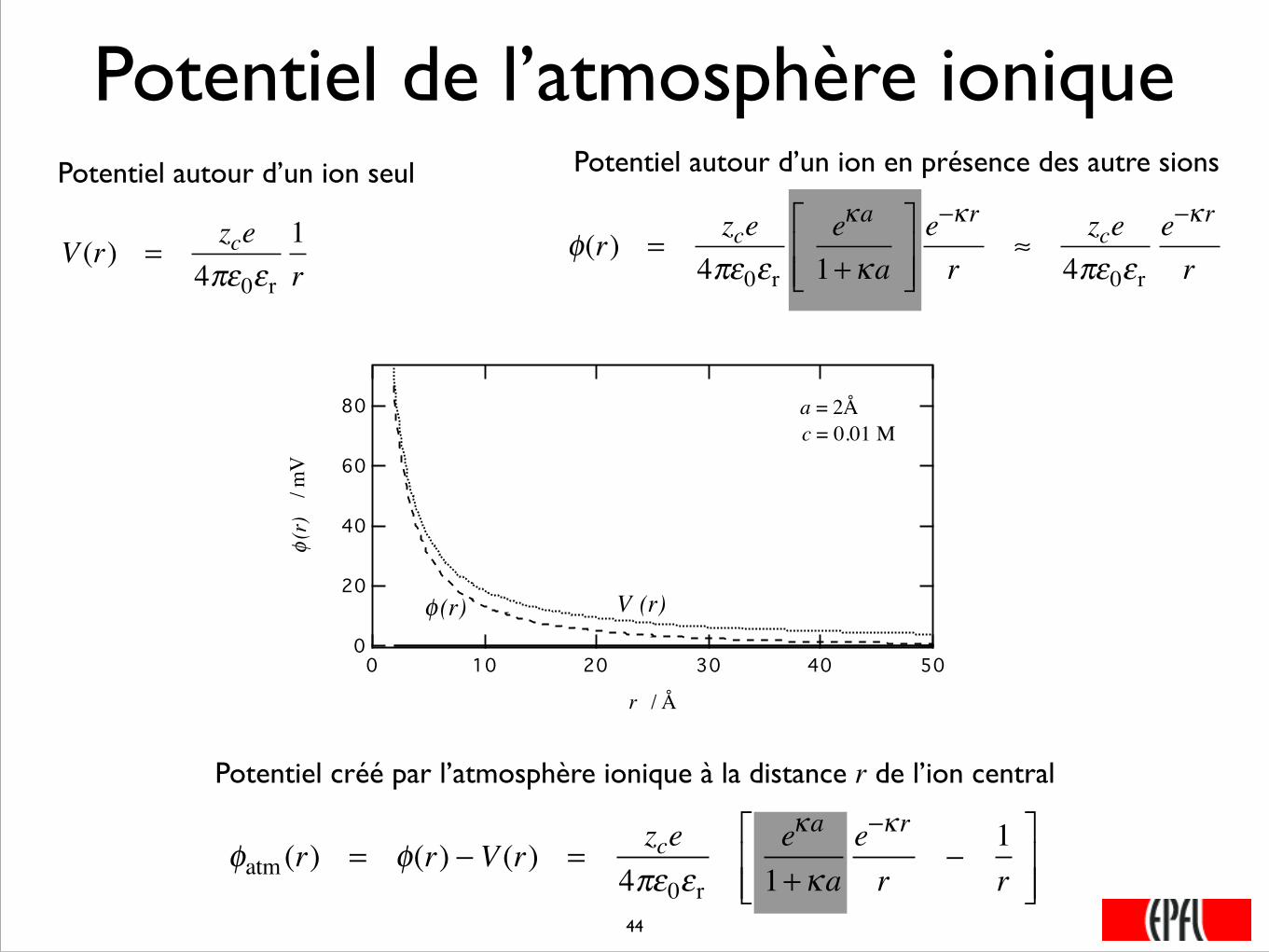
Potentiel de l’atmosphère ionique
80
60
40
20
0
φ(r
) /
mV
50403020100
r / Å
c = 0.01 Ma = 2Å
V (r)φ (r)
V (r) =zce
4πε0εr
1r
Potentiel autour d’un ion seul
φ(r) =zce
4πε0εr
eκa
1+κa⎡
⎣⎢⎢
⎤
⎦⎥⎥e−κ r
r≈
zce4πε0εr
e−κ r
r
Potentiel autour d’un ion en présence des autre sions
φatm (r) = φ(r) − V (r) =zce
4πε0εr
eκa
1+κae−κ r
r−
1r
⎡
⎣⎢⎢
⎤
⎦⎥⎥
Potentiel créé par l’atmosphère ionique à la distance r de l’ion central
���44

Coefficient d’activité ioniquelnγ i
c = −(zie)
2
8πε0εr kTκ
1+κaTravail d’interaction ion-ion
κ 2 =e2
ε0εr kTNi
∞zi2
i∑
B =κIc
= F 2000ε0εrRT
= 3.29 ⋅109 mol1/2 ⋅ l–1/2 ⋅m–1 pour l'eau à 25 °C
A =e2B
8πε0εrkT ln(10)
= 0.509 mol–1/2 ⋅ l1/2 à 25 °C
Constantes
avec
logγ ic = − zi
2 A Ic1+ aB Ic
���45

Exemple MgCl2
µMgCl2 = !µMg2+ + 2 !µCl−Potentiel chimique :
γMgCl2 = γMg2+ γ Cl−2 = γ ±
3Coefficient d’activité :
ν±3 = 1 ⋅22 = 4Coefficient stoechiométrique moyen :
cCz
+ν+
cAz–
ν _ = ν+csel( )ν+
ν−csel( )ν−
= ν±csel( )ν
µMgCl2 = ⇥µMg2+
o + 2 ⇥µCl−o⎡
⎣⎤⎦+ RT ln 4γMgCl2cMgCl2
3( )
µsel = ν+µ
Cz+
o +ν _µAz–
o⎡
⎣⎢
⎤
⎦⎥+νRT ln γ±ν±csel( )
���46

Coefficient d’activité moyen
Pour un sel
µsel = ν+ !µ+ + ν− !µ− = ν+µ+ + ν−µ− =
µselidéal + RT ln γ +
ν+γ −ν−( ) = µsel
idéal + RT lnγ sel
Coefficient d’activité moyen γ ± = γ +ν+ γ −
ν−( )1/ν = γsel1/ν
ν+z+2 + ν−z−
2
ν=
−ν−z−( )z+ + −ν+z+( )z−ν
= − z−z+ν− + ν+
ν= − z−z+
logγ ±c = z+z−
A Ic1+ a±B Ic
Conclusion
logγ ±c = −
ν+z+2
ν+ν−z−
2
ν⎛
⎝⎜⎞
⎠⎟A Ic
1+ a±B Ic
���47
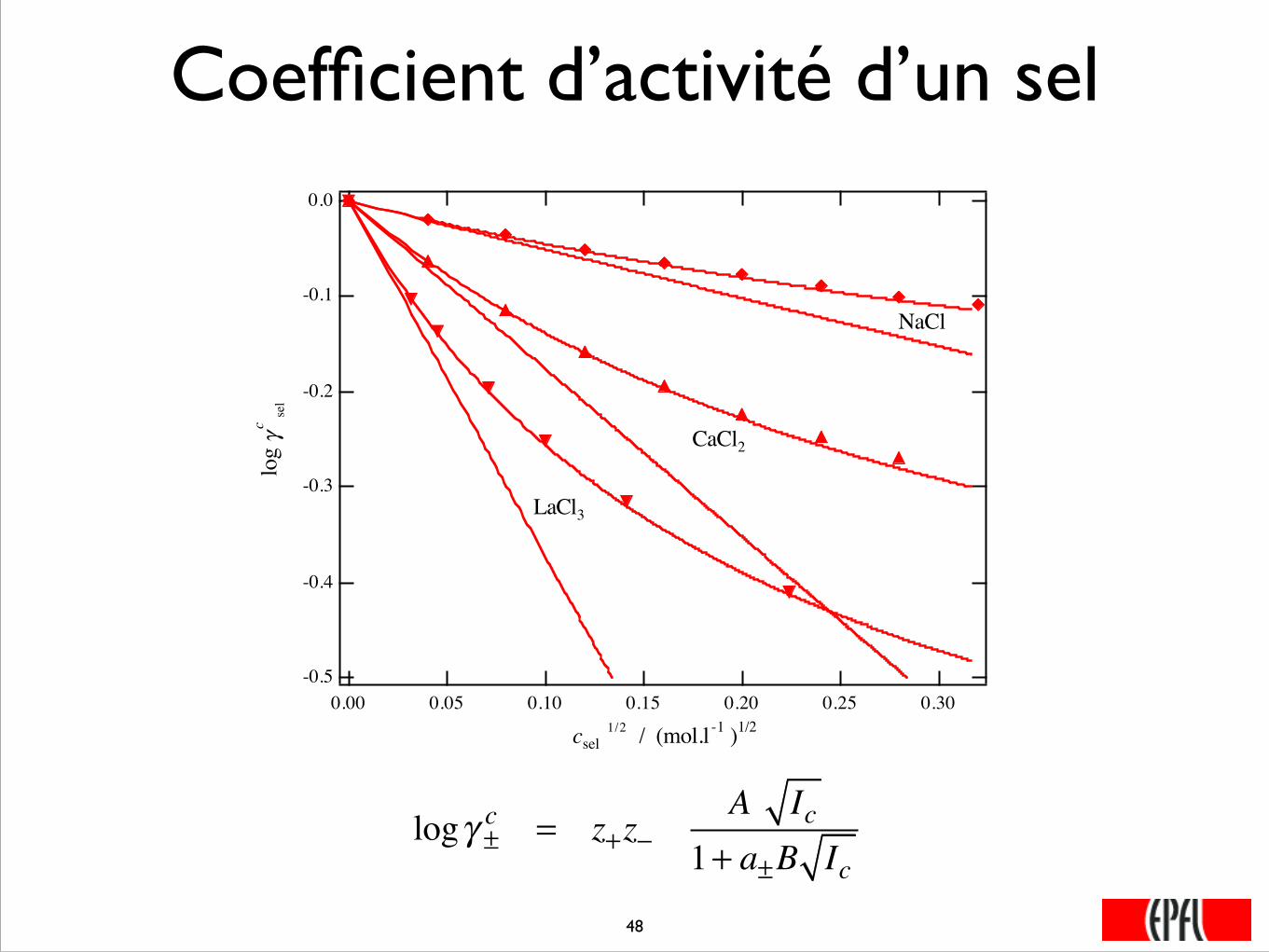
Coefficient d’activité d’un sel
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
log γc s
el
0.300.250.200.150.100.050.00
csel 1/2 / (mol.l -1 )1/2
NaCl
CaCl2
LaCl3
logγ ±c = z+z−
A Ic1+ a±B Ic
���48
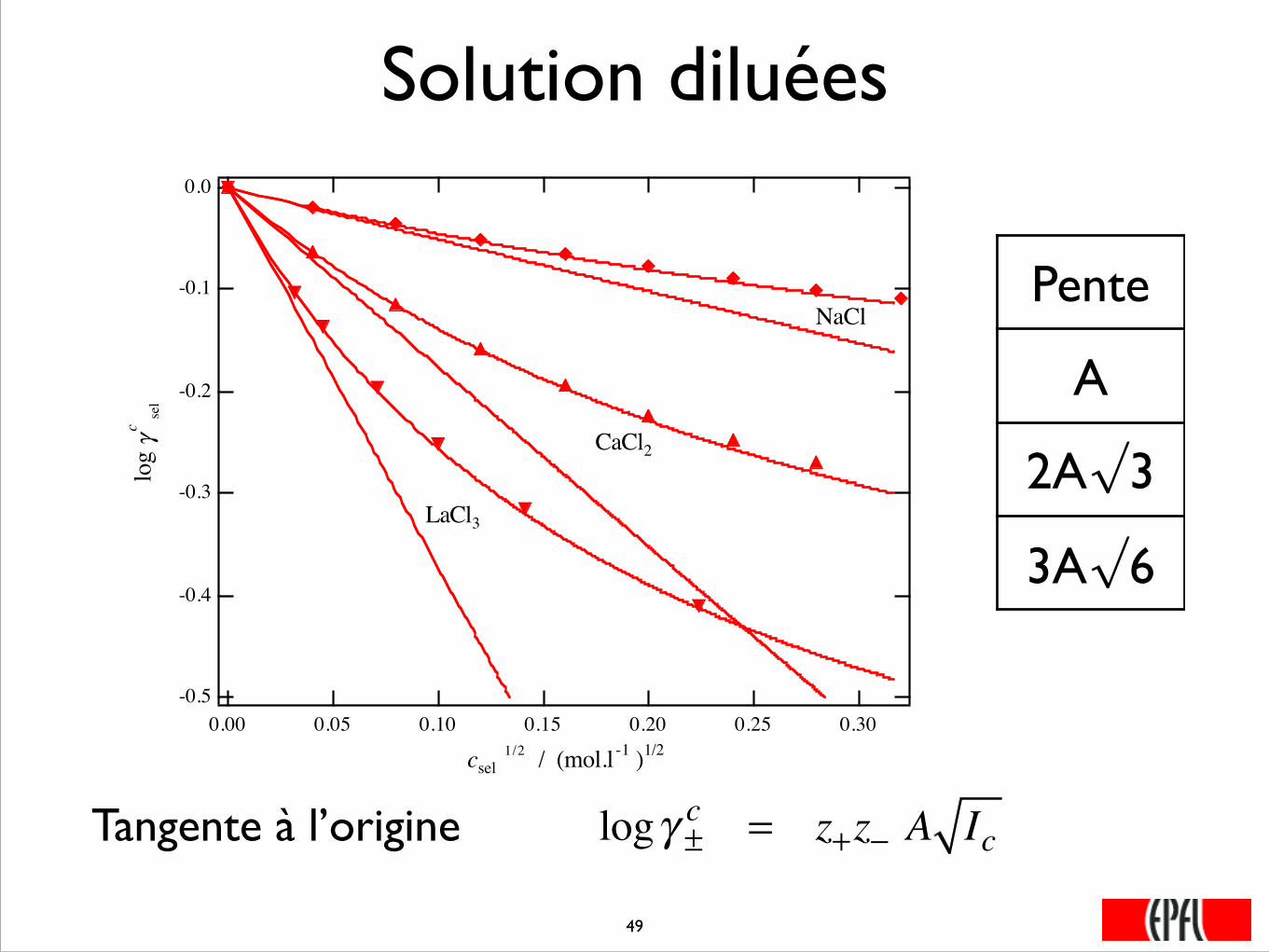
Solution diluées
logγ ±c = z+z− A IcTangente à l’origine
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
log γc s
el
0.300.250.200.150.100.050.00
csel 1/2 / (mol.l -1 )1/2
NaCl
CaCl2
LaCl3
Pente
A
2A√3
3A√6
���49
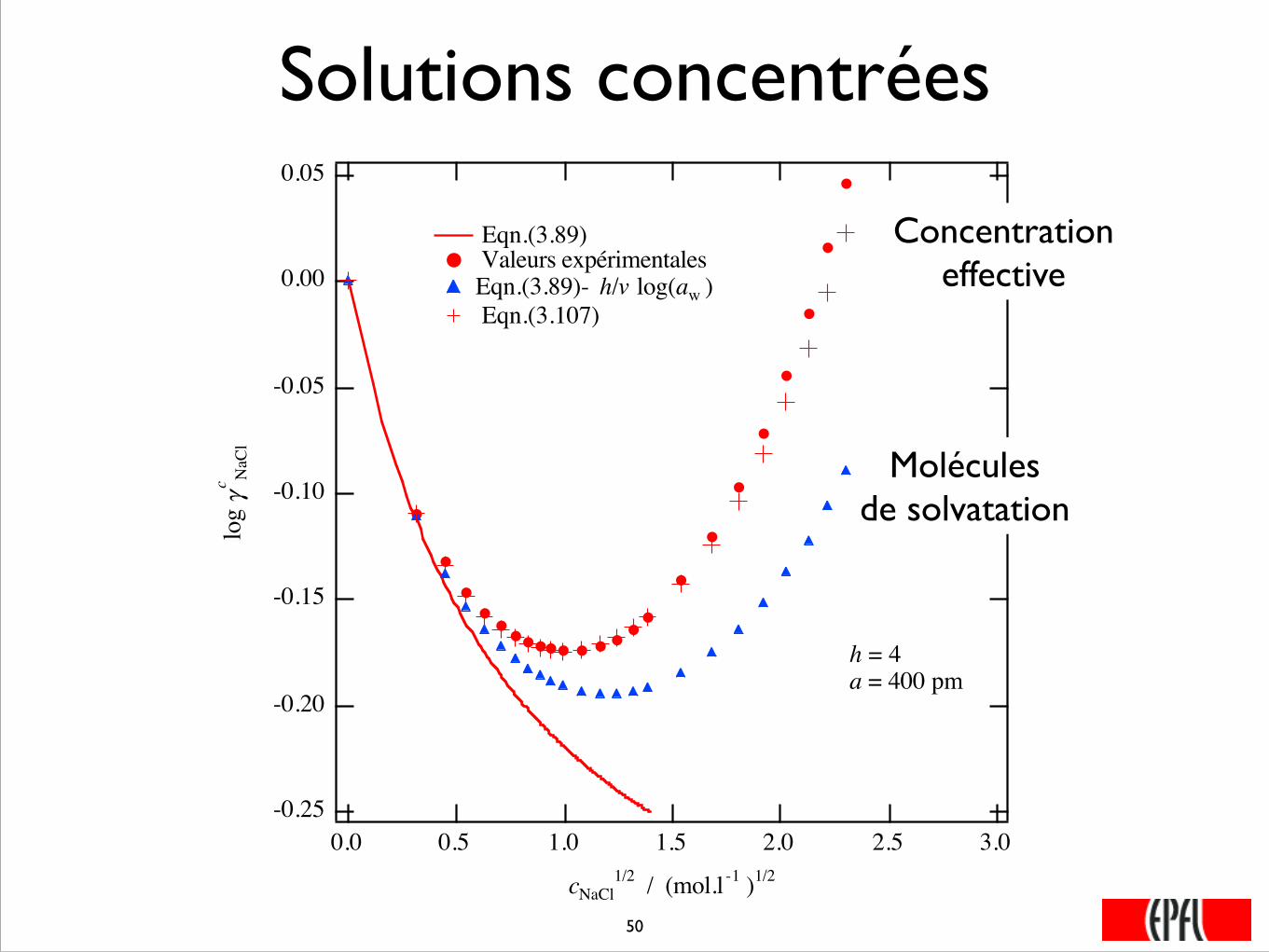
Solutions concentrées
-0.25
-0.20
-0.15
-0.10
-0.05
0.00
0.05
log γc N
aCl
3.02.52.01.51.00.50.0
cNaCl1/2 / (mol.l -1 )1/2
Eqn.(3.89) Valeurs expérimentalesEqn.(3.89)- h/v log(aw ) Eqn.(3.107)
h = 4a = 400 pm
Molécules de solvatation
Concentration effective
���50
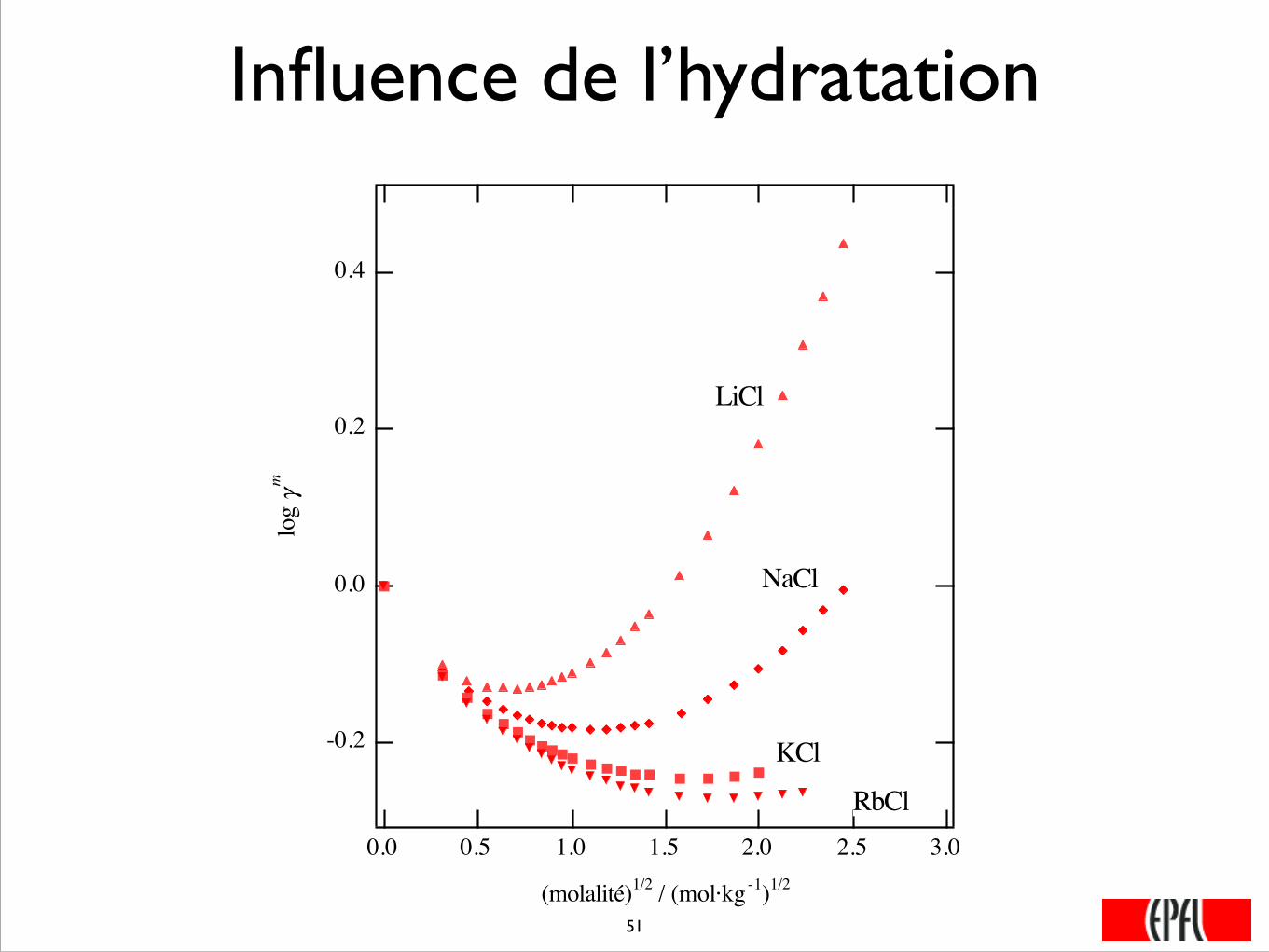
Influence de l’hydratation
0.4
0.2
0.0
-0.2
log γm
3.02.52.01.51.00.50.0
(molalité)1/2 / (mol·kg-1)1/2
LiCl
NaCl
KCl
RbCl
���51
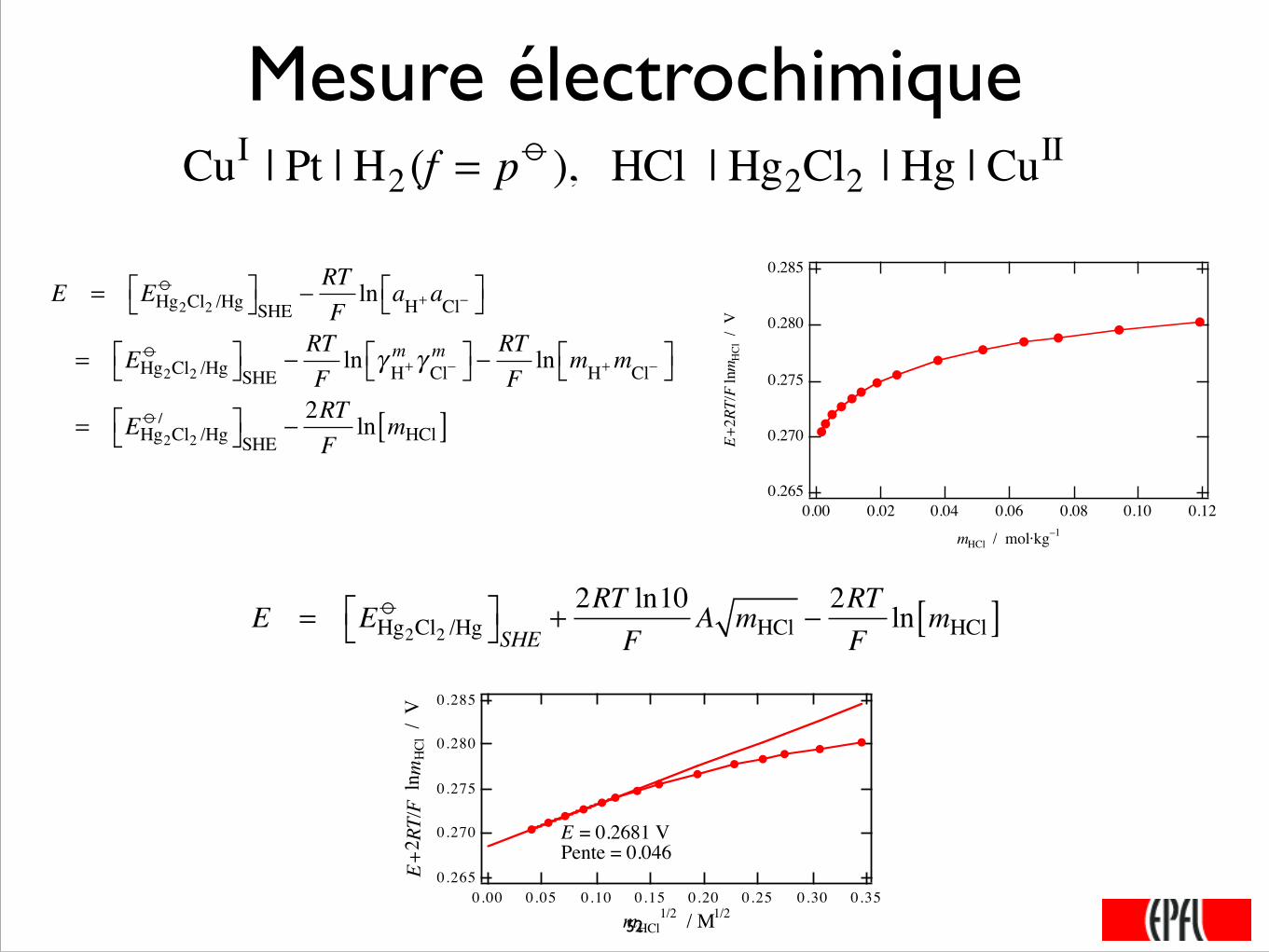
Mesure électrochimique CuI | Pt | H2(f = p o ), HCl | Hg2Cl2 | Hg | CuII
0.285
0.280
0.275
0.270
0.265
E+2RT/F
lnm H
Cl /
V
0.120.100.080.060.040.020.00
mHCl / mol·kg–1
E = EHg2Cl2 /Hgo⎡⎣ ⎤⎦SHE −
RTFln aH+aCl−⎡⎣ ⎤⎦
= EHg2Cl2 /Hgo⎡⎣ ⎤⎦SHE −
RTFln γ H+
m γ Cl−m⎡⎣ ⎤⎦ −
RTFln mH+mCl−⎡⎣ ⎤⎦
= EHg2Cl2 /Hgo /⎡⎣ ⎤⎦SHE −
2RTF
ln mHCl[ ]
E = EHg2Cl2 /Hg
o⎡⎣ ⎤⎦SHE +2RT ln10
FA mHCl −
2RTF
ln mHCl[ ]
0.285
0.280
0.275
0.270
0.265E+2 RT/F
lnm
HCl
/ V
0 .350.300.250.200.150.100.050.00
mHCl1/2 / M1/2
E = 0.2681 VPente = 0.046
���52
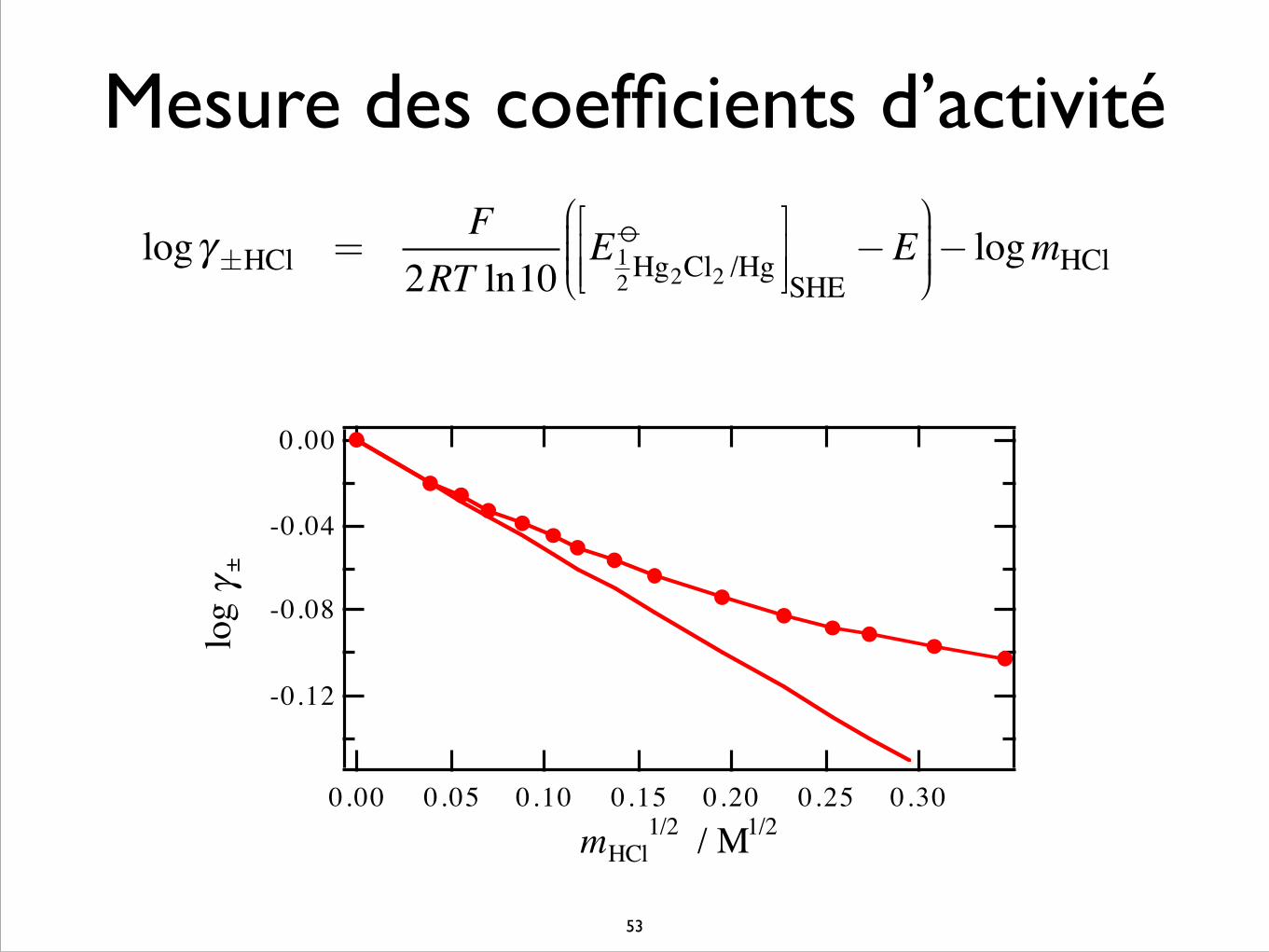
Mesure des coefficients d’activité
logγ±HCl =
F2RT ln10
E12Hg2Cl2 /Hgo⎡
⎣⎢⎢
⎤⎦⎥⎥SHE−E
⎛
⎝⎜⎜⎜
⎞
⎠⎟⎟⎟⎟− logmHCl
-0.12
-0.08
-0.04
0.00
log γ
±
0 .300.250.200.150.100.050.00
mHCl1/2
/ M1/2
���53
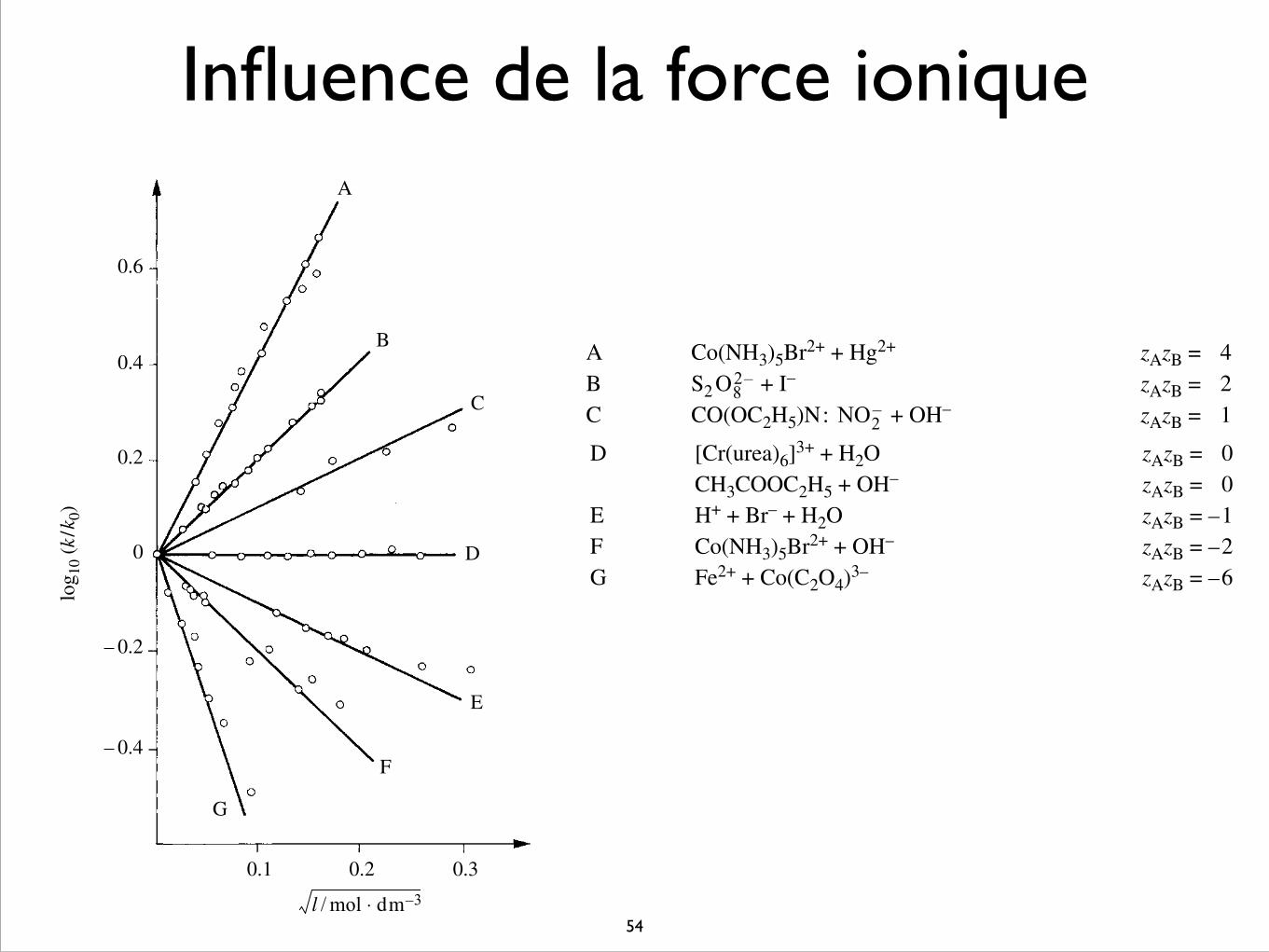
Influence de la force ionique
���54
124 Electrochimie physique et analytique
Ce graphe où le potentiel rédox standard est pris égal à 0,2681 V confirme que l’équation(3.90) n’est pas totalement respectée. Ainsi, on peut affiner l’évaluation du potentielstandard en opérant par itérations successives. La valeur finalement obtenue est0,2679 V. ■
3.4.4 Cinétique de réaction entre ions
Il a été observé expérimentalement que les vitesses de réaction de second ordreentre espèces chargées varient avec la force ionique (fig. 3.24). Quand les espècesioniques réagissantes ont une charge de même signe, la vitesse augmente avec la forceionique; par contre, lorsqu’elles ont une charge de signe opposé, l’inverse se produit.
Légende:A Co(NH3)5Br2+ + Hg2+ zAzB = 4B S2 + I– zAzB = 2C CO(OC2H5)N: + OH– zAzB = 1
O82–
NO2–
0.6
0.4
0.2
0
– 0.2
– 0.4
0.20.1 0.3
G
F
E
D
C
B
A
log 1
0 (k
/k0)
l / mol dm 3–⋅
Fig. 3.24 Variation de la constante de vitesse de réactions entre ions en fonction de la force ionique(Laidler, Chemical Kinetics, Harper & Row, New York, 1987).
124 Electrochimie physique et analytique
Ce graphe où le potentiel rédox standard est pris égal à 0,2681 V confirme que l’équation(3.90) n’est pas totalement respectée. Ainsi, on peut affiner l’évaluation du potentielstandard en opérant par itérations successives. La valeur finalement obtenue est0,2679 V. ■
3.4.4 Cinétique de réaction entre ions
Il a été observé expérimentalement que les vitesses de réaction de second ordreentre espèces chargées varient avec la force ionique (fig. 3.24). Quand les espècesioniques réagissantes ont une charge de même signe, la vitesse augmente avec la forceionique; par contre, lorsqu’elles ont une charge de signe opposé, l’inverse se produit.
Légende:A Co(NH3)5Br2+ + Hg2+ zAzB = 4B S2 + I– zAzB = 2C CO(OC2H5)N: + OH– zAzB = 1
O82–
NO2–
0.6
0.4
0.2
0
– 0.2
– 0.4
0.20.1 0.3
G
F
E
D
C
B
A
log 1
0 (k
/k0)
l / mol dm 3–⋅
Fig. 3.24 Variation de la constante de vitesse de réactions entre ions en fonction de la force ionique(Laidler, Chemical Kinetics, Harper & Row, New York, 1987).
Solutions électrolytiques 125
D [Cr(urea)6]3+ + H2O zAzB = 0CH3COOC2H5 + OH– zAzB = 0
E H+ + Br– + H2O zAzB = –1F Co(NH3)5Br2+ + OH– zAzB = –2G Fe2+ + Co(C2O4)3– zAzB = –6
Brönsted a suggéré que la constante de vitesse d’une réaction entre deux espè-ces A et B en solution s’écrive
(3.112)
où ko est la constante de vitesse pour une solution idéale, et où est le coefficientd’activité du complexe activé ionisé.
En appliquant les équations de Debye-Hückel pour exprimer les coefficientsd’activités des ions (3.90), on obtient
(3.113)
Cette équation a été testée sur plusieurs systèmes et décrit en général assez bien lesphénomènes observés.
Les résultats de ces mesures cinétiques illustrent bien l’effet d’écrantage. Eneffet, il est clair que pour que deux ions de même signe puissent réagir, il est néces-saire d’écranter ces deux charges autant que possible pour favoriser la réaction.Ainsi, une augmentation de la force ionique abaisse la distance d’écrantage et parconséquent augmente la cinétique de réactions entre deux ions de même signe.Inversement, deux ions de signe opposé interagiront à plus longue distance dans dessolutions diluées, et, dans ce cas, un accroissement de la force ionique défavorise laréaction.
3.4.5 Commentaires
La théorie de Debye-Hückel est une théorie basée sur l’électrostatique qui per-met, malgré l’hypothèse considérant le solvant comme un milieu homogène ayantune constante diélectrique égale à celle du solvant en l’absence des ions, de prédireles coefficients d’activités ioniques moyens. Ainsi, malgré la complexité du sys-tème, ce modèle de mécanique statistique donne une équation à un paramètre(éq. (3.89)) qui prédit assez bien le comportement de solutions électrolytiquesjusqu’à des concentrations de l’ordre de 0,1 M.
Un aspect conceptuel très important de cette théorie est la notion de distanceréciproque κ qui montre que les interactions électrostatiques sont écrantées par laprésence de l’électrolyte.
k ko A B= γ γγ #
γ #
log log ( ) log/ /10 10
2 2 2 1 210
1 22k k A z z z z I k Az z IoA B A B
oA B= − + − +[ ] = +







![so asa' tee accru,cci [Q 11] auq [Q OUS a. soot' Õ(Ho) ms ...auuoabPSLS woqqa auq bP040!OkJlSa410kJ coqsa. ous 0k 4ps bLomswa WOLSOASL .1psas a4nq!sa cn.s naskr-ll 40 nal!qqs a4sllak,](https://img.pdfslide.fr/doc/110x75/5e9c719aca614d4bd52f4a99/so-asa-tee-accrucci-q-11-auq-q-ous-a-soot-ho-ms-auuoabpsls-woqqa.jpg)





















