Embed Size (px)
Citation preview

Prix international Henri GastautPrix international Henri Gastaut
EEG versus clinique :une rivalité apparenteMichelle Bureau505, avenue du Prado, 13008 Marseille<[email protected]>
Résumé. Après une première manifestation clinique, l’électroencéphalogramme (EEG) peut-il rivaliser avec la
clinique et apporter des informations aidant au diagnostic et à l’établissement précoce d’un pronostic ? A partir de
la classification de 1989 et de la proposition du schéma diagnostique d’Engel (2001), nous avons essayé d’évaluer les
compétences respectives de la clinique et de l’EEG. Notre expérience porte surtout sur l’évaluation précoce des
nourrissons, enfants et adolescents, moins sur les pathologies épileptiques de l’adulte. Dans les épilepsies et les
syndromes dits « généralisés », la clinique et l’EEG semblent être aussi performants. En revanche, dans les épilepsies
focales, les constatations EEG faites dès le début de l’évolution semblent apporter une aide précieuse au diagnostic
et surtout au pronostic. Dans certaines étiologies particulières, par exemple les malformations corticales focales,
l’EEG peut suggérer le diagnostic, dont la confirmation nécessite bien sûr l’IRM. Dans certaines anomalies chromo-
somiques (le chromosome 20 en anneau en est l’exemple de choix, mais d’autres cadres sont également intéres-
sants), l’EEG très particulier peut faire à lui seul le diagnostic.
Mots clés : épilepsie, EEG, évaluation clinique, diagnostic, pronostic
Abstract. EEG versus clinical information : an apparent rivalry
After a first clinical manifestation, can the EEG rivalize with the clinical evaluation in bringing the informations
needed for precise diagnosis and early prognosis ? Starting with the 1989 international classification, and the
diagnostic scheme proposed by Engel in 2001, we tried to assess the respective relevance of clinical and EEG data.
Our experience is mostly about the early assessment of infants, children and adolescents, and less about epileptic
disorders in adults. In « generalized » epilepsies and syndromes, EEG and clinical assessment appear to have equal
value. However, in focal epilepsies, early EEG findings may have a higher value in diagnosis and especially in
prognosis. There are also specific etiologies, e. g. in focal cortical dysplasias, where the EEG may suggest the
diagnosis even before MRI. In some chromosomal disorders (ring chromosome 20 being the example of choice, but
other entities are also concerned), specific EEG aspects may lead to diagnosis on their own.
Key words: epilepsy, EEG, clinical assessment, diagnosis, prognosis
Après une première manifestation clinique,l’électroencéphalogramme (EEG) peut-il riva-liser avec la clinique et apporter des infor-mations aidant au diagnostic et au pronosticprécoce ? Cette controverse est toujours d’ac-tualité, comme en témoigne l’article récent deFountain et Freeman (2006) intitulé « l’EEG estun outil clinique essentiel : le pour et le
contre » (EEG is an essential clinical tool : pro andcon). Le premier auteur suggère qu’à cause de labonne valeur prédictive des pointes intercriti-ques pour la récurrence des crises, l’EEG est unoutil utile, et le deuxième auteur souligne quel’EEG n’est pas essentiel étant donné que denombreux cliniciens ne donnent pas de trai-tement après une première crise même en
Tirés à part :M. Bureau
9 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
doi:
10.1
684/
epi.2
008.
0139
Epilepsies 2008 ; 20 (1) : 9-26

présence d’un EEG anormal. Le travail de King et al. (1998)témoigne aussi du fait que l’EEG, pratiqué très rapidement,24 heures après une première crise, contribue de manière signi-ficative à la possibilité de classer correctement la pathologie,mais ce travail a évalué parallèlement la clinique, l’EEG et larésonance magnétique (IRM).
A partir de la classification internationale de 1989 (Commis-sion, 1989) et de la proposition du schéma diagnostique deEngel (2001), nous évaluerons les compétences respectives de laclinique et de l’EEG : 1) en présence de manifestations cliniqueset/ou EEG généralisées puis, 2) en présence de signes cliniqueset/ou EEG focaux. Dans la dernière partie, nous évoqueronsl’intérêt de l’EEG dans certaines étiologies. Certaines rubriquesseront illustrées par des cas cliniques mais nous avons volontai-rement privilégié les données EEG.
Les épilepsies « généralisées » idiopathiquesou non idiopathiques et l’EEG
Les spasmes
Des spasmes survenant chez des nourrissons normaux sur leplan psychomoteur posent le problème diagnostique de leurorigine épileptique ou non épileptique. S’agit-il de spasmesinfantiles débutant un syndrome de West apparemment idiopa-thique et nécessitant l’instauration rapide d’un traitement (Du-lac et Tuxhorn, 2005) ou d’un « myoclonus bénin », termeinapproprié décrivant en fait un syndrome de spasmes bénins(Lombroso et Fejerman, 1977 ; Dravet et al., 1986 ; Pachatz etal., 1999) ? Dans les deux cas, ces spasmes peuvent survenir ensalve à la veille mais également au réveil. Un même symptômepeut donc avoir deux significations opposées quant au pronos-tic : une évolution défavorable en l’absence d’un traitement oula certitude de la guérison. Peu d’arguments cliniques permet-tent en effet de faire un diagnostic, car l’infléchissement desacquisitions et du comportement n’est pas toujours apparentd’emblée, et, dès le début des manifestations, l’enregistrementEEG de ces spasmes est l’élément primordial (figure 1).
Les accès myocloniques brefs et les pointe-ondesgénéralisées
Ils se traduisent à l’EEG par une décharge de pointe-ondesgénéralisées (PO). Le diagnostic se pose surtout entre l’épilepsiemyoclonique bénigne (EMB) (Dravet et Bureau, 2005a,b) etl’épilepsie myoclonique sévère, dorénavant syndrome de Dra-vet (Dravet et al., 2005a,b). Les PO sont très similaires, du moinsau début de la maladie (figure 2).
Lors des premiers épisodes myocloniques, la clinique peutne pas être très performante, le principal critère semblant êtrel’existence et la nature de crises fébriles chez ces nourrissons. Eneffet, les crises convulsives fébriles ou afébriles sont toujoursprésentes dans le syndrome de Dravet, souvent répétées etlongues alors que dans l’EMB, elles sont rares (26,1 % des cas(Dravet et Bureau, 2005b), simples et peu fréquentes (1 à 2 parnourrisson).
Par ailleurs, si des accès peuvent être déclenchés par desstimulations dans quelques cas d’EMB (bruit, contact ou SLI)(Guerrini et al., 1994), dans le syndrome de Dravet, seule la SLIpeut être efficace à un stade précoce dans un pourcentagevariable de cas (Dravet et al., 2005a,b).
Dans l’EMB, les PO sont pratiquement toujours accompa-gnées de secousses myocloniques alors que dans le syndrome deDravet, on peut observer des PO généralisées sans accompagne-ment clinique, et des crises et des anomalies EEG focales oumultifocales apparaissent rapidement. L’EEG est donc très utile,mais ne peut être correctement évalué qu’à la lumière desdonnées cliniques.
Les absences et leur traduction EEG
Les absences de l’épilepsie-absences se traduisent sur l’EEGpar une décharge de PO rythmiques à 3Hz (Hirsch et Panayio-topoulos, 2005). Cette décharge est la même que celle enregis-trée dans les absences myocloniques (AM) et ce n’est que l’enre-gistrement polygraphique qui permettra, en objectivant lacontraction tonique progressive associée aux secousses myoclo-niques des AM, de porter le diagnostic de cette forme particu-lière d’absences, de pronostic parfois réservé (figure 3) (Bureau etTassinari, 2005a,b). Ici, l’EEG peut être trompeur s’il n’est pasassocié à une observation clinique soigneuse ou au moins à unenregistrement vidéo.
Les absences atypiques et les décharges diffuses différentesdes PO des EGI
Les absences atypiques (figure 4) peuvent survenir chez desenfants présentant ou non des signes d’encéphalopathie. Si ellessont associées à des chutes brutales, une diminution des perfor-mances intellectuelles et des troubles du comportement, lediagnostic peut errer principalement entre deux syndromes : lesyndrome des pointe-ondes continues du sommeil (POCS) (ouESES : état de mal électrique pendant le sommeil lent) et lesyndrome de Lennox-Gastaut (Tassinari et al., 2005 ; Beauma-noir et Blume, 2005). L’enregistrement de sommeil sera primor-dial à réaliser. Il montrera dans le syndrome des POCS dèsl’endormissement (figure 5) des pointe-ondes plus ou moinslentes, généralement à 1,5-2,5 Hz, bilatérales diffuses qui persis-teront pendant toutes les phases de sommeil lent. Dans lesyndrome de Lennox-Gastaut, le sommeil mettra en évidenceles bouffées de rythmes rapides caractéristiques et, parfois, descrises toniques (figure 5) pouvant être suffisamment brèves etpeu intenses et de ce fait, passer inaperçues. Ici, l’EEG a un rôleprimordial, spécifique et irremplaçable, mais on voit poindrel’importance des enregistrements sous sommeil : il ne s’agitdonc plus d’EEG dits « de routine ».
Les crises généralisées tonico-cloniques
Une première crise généralisée tonico-clonique (CGTC) sur-venant chez un jeune adolescent doit faire évoquer avant tout lediagnostic d’épilepsie myoclonique juvénile (EMJ, Thomas etal., 2005) mais il faut toujours garder en mémoire la possibilité
M. Bureau
10Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

d’autres étiologies, en particulier les épilepsies myocloniquesprogressives (EMP) comme la maladie d’Unverricht-Lundborget la maladie de Lafora qui peuvent prendre pendant quelquetemps l’aspect d’une EMJ (Genton et al., 2005). En effet, dansl’EMJ et dans les EMP, en plus des CGTC, les patients présententdes secousses myocloniques surtout le matin au réveil. L’EEGintercritique met en évidence des décharges de PO généraliséessimilaires spontanées ou déclenchées dans certains cas par laSLI, accompagnées alors de myoclonies. Ces anomalies EEGsont donc aspécifiques (figures 6 et 7) au tout début de l’évolu-tion, car les anomalies de l’activité de fond n’apparaissent engénéral qu’au bout d’un certain temps d’évolution. En l’absence
d’antécédents familiaux, seule l’évolution clinique et EEG per-mettra le diagnostic (figure 8).
Conclusion
Dans les épilepsies « généralisées », la clinique et l’EEG ontune utilité égale dans l’évaluation d’un pronostic précoce. Ilfaut cependant souligner que l’EEG pour être performant néces-site souvent, sinon systématiquement, des enregistrements po-lygraphiques et des enregistrements de sommeil et qu’il nesaurait seul rivaliser avec la clinique.
Figure 1. A gauche : spasme bénin.Enfant de 4 mois 1/2, sans antécédent personnel ou familial présentant depuis l’âge de 3 mois, à la veille et au sommeil, des spasmes, isolés ouen série, caractérisés par un mouvement d’extension des membres supérieurs et de flexion au niveau des membres inférieurs, suivi d’une ouverturedes yeux. Ces spasmes peuvent réveiller l’enfant qui se met parfois à pleurer.Enregistrement d’un spasme survenant à l’endormissement (drowsiness). Sur l’EEG, noter seulement une réaction d’éveil et sur la polygraphie,une brève contraction tonique au niveau des muscles deltoïdes.A droite : spasme épileptique.Enfant de 6 mois présentant depuis une quinzaine de jours des spasmes avec flexion de la tête et écartement des épaules, survenant soitisolément soit en brève série. Ils surviennent surtout à la veille et parfois au réveil. Le développement psychomoteur se poursuit apparemmentnormalement.Enregistrement d’un spasme survenant au cours d’une salve au réveil de la sieste.L’activité de fond EEG montre des ondes thêta delta diffuses. Au moment du spasme : complexe lent surchargé d’une activité rapide bien visiblesur le vertex. Sur la polygraphie, brève contraction tonique au niveau des muscles deltoïdes droit et gauche.Un tracé hypsarhytmique est apparu ultérieurement.
11 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

Figure 2. A gauche : accès myoclonique chez un enfant de 14 mois, sans antécédent personnel ou familial, ayant un développementpsychomoteur normal. Début à 8 mois par des secousses myocloniques généralisées. Disparition des myoclonies sous VPA, poursuite d’undéveloppement normal. Traitement arrêté à 4 ans. Il s’agit d’une EMB. PO généralisées associées à des myoclonies visibles sur les deltoïdes.A droite : accès myoclonique chez une enfant de 2 ans, ayant présenté une crise fébrile unilatérale gauche. Par la suite, nouvelles crises fébrilesou afébriles, généralisées ou unilatérales gauches. A partir de 13 mois, secousses myocloniques et absences atypiques pluriquotidiennes,autostimulation par fixation de patterns. Ralentissement du développement psychomoteur. Inefficacité des antiépileptiques. Il s’agit d’unsyndrome de Dravet.Sur l’EEG, une première PO sans myoclonie suivie de deux autres dont la pointe est accompagnée par une myoclonie.Noter la similitude de la décharge de PO dans les deux cas.
M. Bureau
12Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

Figure 3. A gauche : PO rythmiques à 3Hz, typiques de l’absence de l’épilepsie-absences. L’enfant est en train de réaliser une hyperpnée lesbras tendus. L’enregistrement polygraphique du deltoïde droit montre une diminution du tonus due à la perte de la consigne.A droite : même décharge de PO. Noter sur les muscles deltoïdes les myoclonies rythmiques progressivement associées à une contraction toniqueresponsable d’une élévation des membres supérieurs.
Figure 4. A gauche : enfant de 8 ans née d’une grossesse gémellaire présentant une hémiparésie droite congénitale. Apparition à 6 ans 1/2d’absences atypiques et de troubles neuropsychologiques et de l’orientation spatiale. Sur l’EEG : à la veille (awake) : PO isolées diffusesprédominant sur les régions frontales et à gauche, suivies par une décharge de PO irrégulières au cours de laquelle l’enfant ralentit son activité.A droite : enfant de six ans présentant des absences atypiques, des secousses pouvant entraîner des chutes et une régression comportementale.Sur l’EEG : à la veille (awake) : à gauche : une PO isolée diffuse et à droite bouffée de PO diffuses prédominant sur les régions frontales.
13 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

Figure 5. Mêmes enfants que sur la figure 4. Sommeil montrant à gauche dès l’endormissement (drowsiness) des PO continues et à droite unebrève crise tonique accompagnée d’une contraction tonique sur le deltoïde droit et une modification de la respiration. Ces crises n’avaient pasété notées par les parents.
Figure 6. Garçon de 14 ans ayant présenté une crise généralisée tonico-clonique une quinzaine de jours auparavant. A gauche : tracé de veille(awake) montrant une décharge de PO généralisées.A droite : une décharge de polypointes ondes déclenchées par la SLI, accompagnées de myoclonies visibles sur le deltoïde droit. Sur la cliniqueet les constatations EEG, le diagnostic d’EMJ a été porté. Ce diagnostic a été confirmé par un enregistrement de sieste qui a permis d’enregistrerdes secousses myocloniques au réveil (voir figure 8, à gauche). A l’interrogatoire, le patient révèle que, depuis quelque temps, il lui arrivait lematin de laisser tomber les objets qu’il tenait.
M. Bureau
14Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

Les épilepsies et les anomalies EEG focales
Les épilepsies focales idiopathiques
Épilepsie du nourrisson à pointes et PO centraleset vertex pendant le sommeil
A partir d’une étude rétrospective portant sur 50 cas (Bureauet Maton, 1998), nous avons essayé d’évaluer la valeur de l’EEGdans le pronostic précoce des épilepsies focales non idiopathi-ques. Les critères d’inclusion dans cette étude étaient : un débutavant 10 ans, un premier EEG dans la première année suivant lapremière crise, un suivi EEG régulier au cours des deux premiè-res années (au minimum 5 EEG de veille et 2 de sommeil), uneévolution électroclinique supérieure à 5 ans (moyenne : 8 ans6 mois). Nous avons comparé l’EEG avec la clinique. Dans 88 %des cas, il y avait une bonne concordance entre l’EEG et laclinique et pour 12 %, les cas classés cliniquement défavorables
ont eu une bonne évolution. En effet, les crises cliniques lais-saient prévoir une évolution défavorable (Dulac et al., 1989) :leur début se situait à un âge moyen de 9 mois (extrêmes entre 1et 20 mois), elles comportaient un arrêt de l’activité, une fixitédu regard ou une révulsion oculaire, une perte du contact, dessignes végétatifs (pâleur, cyanose) avec ou sans mouvements dedéglutition ou de mâchonnement, une hyper ou une hypoto-nie. La généralisation était exceptionnelle. Bien entendu, cessymptômes n’étaient pas tous présents au cours de la mêmecrise ou chez le même enfant. Dans 30 % des cas, il existaitplusieurs types de crises. Or malgré ces signes apparemmentpéjoratifs, l’évolution a été favorable même en l’absence detraitement avec une guérison complète vers l’âge de 3-4 ansdans tous les cas. Des cas ayant un début précoce et le mêmetype de crise (soit partielle complexe soit avec une généralisa-tion secondaire) ainsi qu’une évolution favorable ont été rap-
Figure 7. Fille de 13 ans 4 mois : apparition à 13 ans de crises convulsives généralisées.A gauche : activités thêta, l’enfant a les yeux ouverts. PO en brèves bouffées généralisées.A droite : la SLI provoque une décharge de PO diffuses mais à prédominance postérieure. A la fin de la décharge, l’enfant ferme les yeux et l’onnote une activité alpha. Un enregistrement de longue durée permettra ultérieurement de mettre en évidence (figure 8), outre les déchargesdiffuses, des anomalies postérieures. A un interrogatoire plus approfondi, la jeune fille signale qu’avant même l’apparition des crises convulsives,elle avait de temps en temps des manifestations visuelles à type de phospènes. Le diagnostic de maladie de Lafora est alors évoqué, diagnosticqui sera confirmé par la biopsie de peau.
15 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

portés par Watanabe et al., (1987, 1990, 1993) et Okumura et al.,(2006).
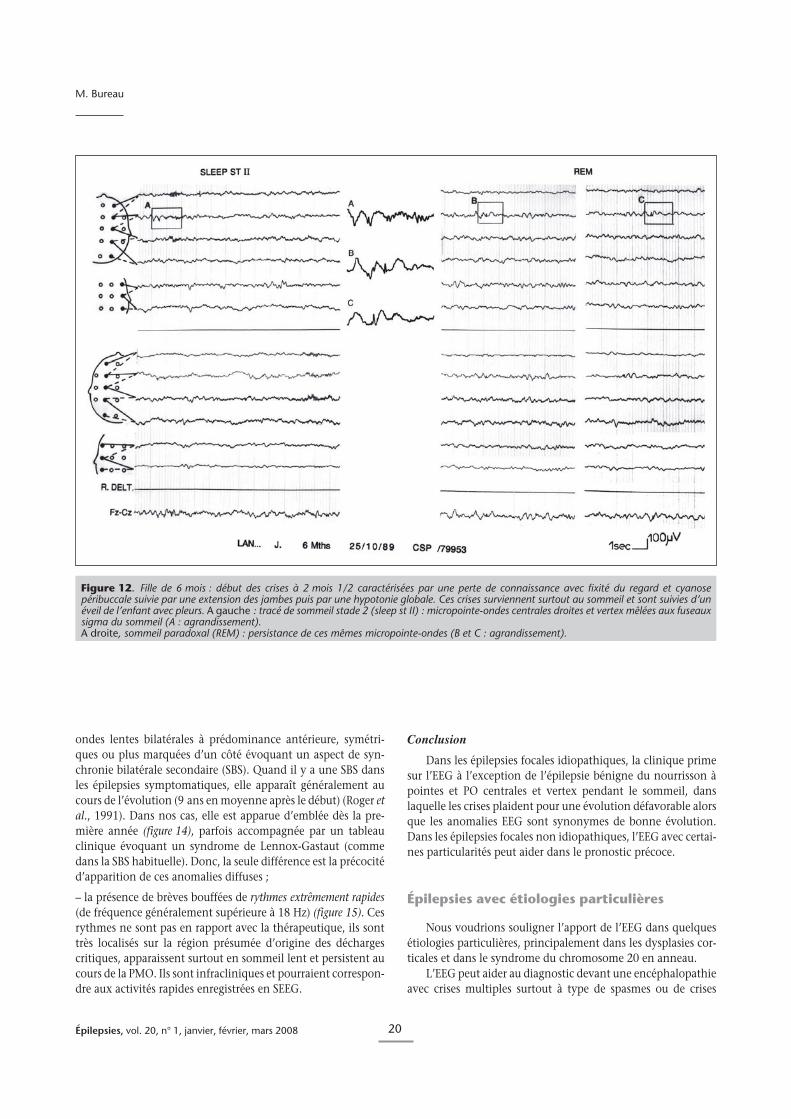
Dans nos cas, l’EEG montrait des anomalies très particuliè-res au cours du sommeil (Bureau et Maton, 1998 ; Bureau et al.,1998 ; Bureau et al., 2002). L’EEG de veille était normal danstous les cas et au cours du sommeil sont apparues des pointes debas voltage suivies ou non par une onde lente également peuample, localisées sur la région fronto-centrale et le vertex surl’un ou les deux hémisphères de façon simultanée ou indépen-dante. Ces anomalies étaient présentes pendant toutes les pha-ses de sommeil y compris pendant la phase de sommeil para-doxal (PMO). Elles sont souvent difficiles à mettre en évidencecar mêlées aux fuseaux et aux K complexes (figure 12). Cesmêmes anomalies ont été également observées par Capovilla etal., (2000) chez des nourrissons présentant la même sémiologieclinique. Nos cas et ceux de Capovilla se rapprochent de trèsprès des cas de Watanabe et al. (1987 ; 1993) mais ces auteursn’ont pas mentionné d’enregistrement de sommeil.
Ici, l’EEG seul a donc permis de définir une forme d’épilepsieignorée du nourrisson, et a permis de mettre en évidence lapossibilité d’épilepsies focales bénignes à début précoce, trèscontestée auparavant.
L’épilepsie à pointes centro-temporales (EPCT)La clinique de l’EPCT est souvent évocatrice (Dalla Bernar-
dina et al., 2005). Les crises typiques sont généralement noctur-nes, caractérisées par des secousses cloniques intéressant l’hémi-face, occupant la langue, les lèvres et la joue, parfois associées àune déviation tonique de la bouche intéressant les lèvres et lalangue, ainsi que les muscles pharyngés et laryngés, entraînantun arrêt de la parole et une sialorrhée. Ces crises surviennententre 3 et 14 ans avec un pic de fréquence entre 5 et 8 ans, chezdes enfants normaux, ayant une incidence élevée d’antécédentsfamiliaux d’épilepsie bénigne (Genetic Collaborative ItalianGroup, 1993). Les crises disparaissent avant l’adolescence.
L’EEG dans ce contexte clinique est également évocateur(figure 9). Il montre lors de la veille des pointes plus ou moinslentes focales (centro-temporales), diphasiques, négatives, gé-néralement amples et qui sont activées durant toutes les phasesde sommeil. Cependant, ces mêmes anomalies peuvent s’obser-ver dans d’autres entités cliniques (tumeurs cérébrales, syn-drome de l’X fragile, dysplasie corticale focale (Van der Meij etal., 1993 ; Panayiotopoulos, 2005) et également chez des en-fants ne présentant pas d’épilepsie mais quelques troubles du
Figure 8. A gauche, même patient que sur la figure 6. Au réveil (awakening) de la sieste, deux bouffées de PO plus ou moins régulières,associées à des secousses myocloniques généralisées cliniquement et visibles sur cet enregistrement sur le muscle deltoïde droit.A droite, même patiente que figure 7. Pointes rapides suivies d’une onde lente sur les deux régions temporo-pariéto-occipitales.
M. Bureau
16Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

comportement, des difficultés d’apprentissage, des manifesta-tions de type vagal (figure 9, voire même en l’absence de toutesymptomatologie. De telles anomalies sont détectées chez une pro-portion d’enfants témoins de la classe d’âge concernée qui varie peud’un auteur à l’autre (2,1 % pour Eeg-Olofsson et al., 1971 ; 2,4 %pour Cavazzuti et al., 1980 ; 3 % pour Lerman et Kivity-Ephraim, 1981 ; 3,5 % pour Okubo et al., 1994). Ici, l’EEG « faitle diagnostic », mais il faut savoir se méfier de certains pièges.
L’épilepsie occipitale bénigne à débutprécoce ou syndrome de Panayiotopoulos (SP)
Les crises débutent entre 1 et 14 ans (pic entre 4 et 5 ans),sont rares et même souvent uniques. Elles sont très caractéristi-
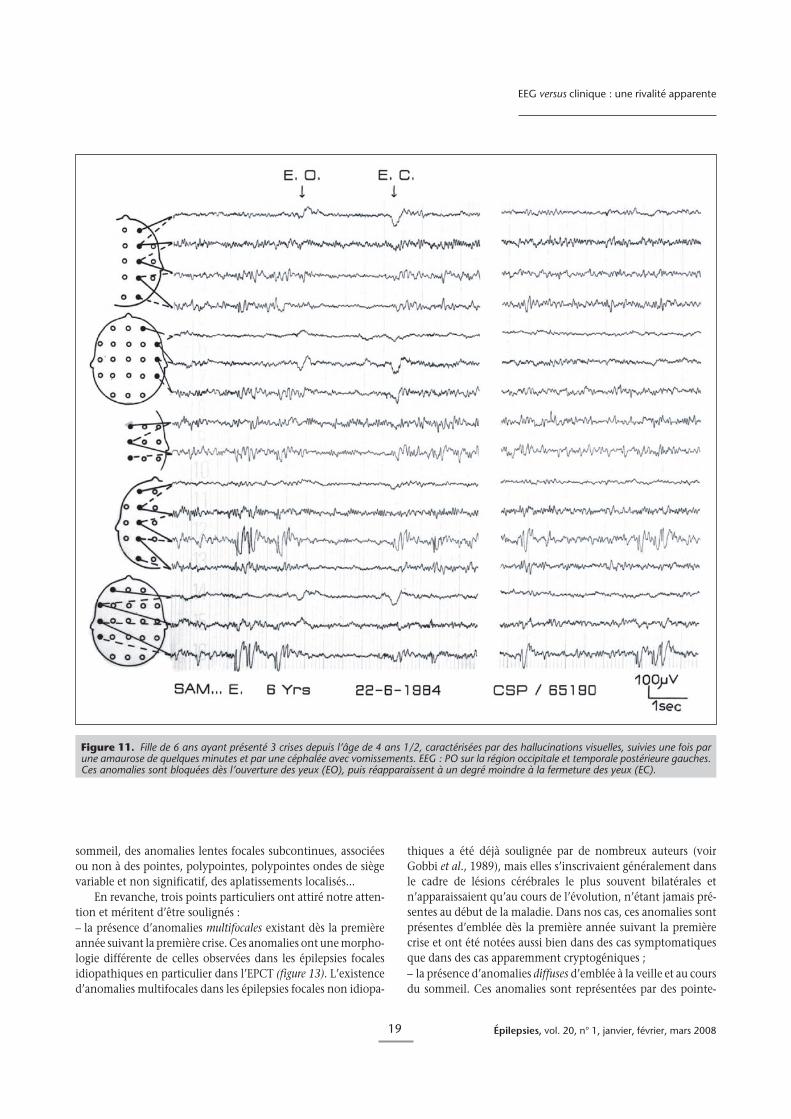
ques, le plus souvent nocturnes et comportent au début dessignes végétatifs importants avec vomissements, un change-ment de comportement, suivis parfois d’une déviation des yeuxet d’un trouble de la conscience. Leur durée est dans la moitiédes cas longue, allant de 30 minutes à plusieurs heures. Si lescrises sont caractéristiques, l’EEG intercritique est très variable(Panayiotopoulos, 1999, 2005 ; Covanis et al., 2005). Il montredans 68 % des cas des paroxysmes occipitaux à type de pointeslentes de haut voltage associées dans 64 % des cas à des pointesmultifocales extra-occipitales. Dans 32 % des cas, on n’observejamais d’anomalies occipitales mais soit des pointes multifoca-les (sans anomalies occipitales), soit des bouffées d’ondes lentesdiffuses (figure 10) associées à des pointes de bas voltage, soitenfin un EEG normal. Dans ce syndrome, sans doute relative-ment fréquent, l’EEG est donc potentiellement peu informatif.
Figure 9. A gauche : garçon de 7 ans ayant présenté 3 crises nocturnes avec secousses cloniques de l’hémiface droite, hypersalivation et bruitsde gorge. Une fois, les clonies se sont étendues au bras droit. Tracé EEG de veille (awake) montrant des pointes centro-temporales gauches et unepointe isolée indépendante à droite.A droite : garçon de 10 ans ayant eu 2 crises fébriles simples à 15 mois et présentant des troubles discrets du comportement et des problèmesscolaires. EEG de veille (w) montrant des pointes centro-temporales droites.
17 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

L’épilepsie occipitale idiopathique à début tardif de typeGastaut
Le début est plus tardif que dans le SP entre 3 et 15 ans (picà 8 ans). Les crises sont généralement fréquentes, diurnes, du-rant de quelques secondes à quelques minutes, caractérisées pardes hallucinations visuelles simples, une cécité, une possibledéviation de la tête et des yeux pouvant progresser en hémicon-vulsions. Une céphalée postcritique diffuse ou unilatérale s’ob-serve dans la moitié des cas.
L’EEG typique intercritique montre des PO de haut voltage(200-300 lV), répétées rythmiquement à 2-3 Hz, en bouffées surles régions occipitales et temporales postérieures sur l’un ou lesdeux hémisphères de façon indépendante ou simultanée. CesPO sont bloquées par l’ouverture des yeux et réapparaissent dèsleur fermeture (figure 11) (Gastaut, 1982 ; Gastaut, 1984 ; Beau-manoir, 1983). Mais ce type d’anomalies a été décrit dansd’autres pathologies, en particulier dans des épilepsies sympto-matiques. Aicardi et Newton (1987) ont rapporté 21 cas corres-
pondant aux critères EEG parmi lesquels 7 avaient une épilepsiesévère avec plusieurs types de crises, dont 4 avaient une épilep-sie lésionnelle certaine. Gobbi et al. (1988) ont retrouvé cesmêmes anomalies dans des cas d’épilepsie avec calcificationsoccipitales bilatérales. Par ailleurs dans la série de HerranzTanarro et al. (1984), portant sur 31 enfants ayant le patterntypique, 10 n’ont jamais présenté de crises.
Ici, l’EEG montre des anomalies très évocatrices du diagnos-tic d’épilepsie occipitale, mais peut induire en erreur quant à lanature idiopathique ou autre de l’épilepsie.
Les épilepsies focales non idiopathiques
Au cours de la même étude effectuée en 1998, nous avonsessayé de dégager dans les épilepsies focales non idiopathiquesdes éléments EEG qui permettraient d’établir un pronostic pré-coce d’évolution défavorable.
Nous avons retrouvé les critères classiques à savoir uneactivité de fond anormale asymétrique à la veille et au cours du
Figure 10. Fille de 10 ans ayant présenté à 2 reprises (à 5 et 7 ans) un épisode prolongé nocturne avec perte du contact, convulsions peut-êtrelatéralisées lors du premier épisode, état nauséeux avec vomissements. Ces épisodes ont duré entre 20 à 30 minutes à la suite desquels l’enfants’est rendormie. Développement psychomoteur et comportement normaux.A gauche : ondes lentes temporales gauches et bouffée d’ondes lentes assez diffuses associées à quelques pointes maximales sur la régioncentrale et temporale moyenne gauche diffusant discrètement à droite. A droite : une bouffée d’ondes lentes mêlées à quelques pointes surl’hémisphère gauche et se voyant à un degré moindre à droite.
M. Bureau
18Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
sommeil, des anomalies lentes focales subcontinues, associéesou non à des pointes, polypointes, polypointes ondes de siègevariable et non significatif, des aplatissements localisés...
En revanche, trois points particuliers ont attiré notre atten-tion et méritent d’être soulignés :– la présence d’anomalies multifocales existant dès la premièreannée suivant la première crise. Ces anomalies ont une morpho-logie différente de celles observées dans les épilepsies focalesidiopathiques en particulier dans l’EPCT (figure 13). L’existenced’anomalies multifocales dans les épilepsies focales non idiopa-
thiques a été déjà soulignée par de nombreux auteurs (voirGobbi et al., 1989), mais elles s’inscrivaient généralement dansle cadre de lésions cérébrales le plus souvent bilatérales etn’apparaissaient qu’au cours de l’évolution, n’étant jamais pré-sentes au début de la maladie. Dans nos cas, ces anomalies sontprésentes d’emblée dès la première année suivant la premièrecrise et ont été notées aussi bien dans des cas symptomatiquesque dans des cas apparemment cryptogéniques ;– la présence d’anomalies diffuses d’emblée à la veille et au coursdu sommeil. Ces anomalies sont représentées par des pointe-
Figure 11. Fille de 6 ans ayant présenté 3 crises depuis l’âge de 4 ans 1/2, caractérisées par des hallucinations visuelles, suivies une fois parune amaurose de quelques minutes et par une céphalée avec vomissements. EEG : PO sur la région occipitale et temporale postérieure gauches.Ces anomalies sont bloquées dès l’ouverture des yeux (EO), puis réapparaissent à un degré moindre à la fermeture des yeux (EC).
19 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente
ondes lentes bilatérales à prédominance antérieure, symétri-ques ou plus marquées d’un côté évoquant un aspect de syn-chronie bilatérale secondaire (SBS). Quand il y a une SBS dansles épilepsies symptomatiques, elle apparaît généralement aucours de l’évolution (9 ans en moyenne après le début) (Roger etal., 1991). Dans nos cas, elle est apparue d’emblée dès la pre-mière année (figure 14), parfois accompagnée par un tableauclinique évoquant un syndrome de Lennox-Gastaut (commedans la SBS habituelle). Donc, la seule différence est la précocitéd’apparition de ces anomalies diffuses ;
– la présence de brèves bouffées de rythmes extrêmement rapides(de fréquence généralement supérieure à 18 Hz) (figure 15). Cesrythmes ne sont pas en rapport avec la thérapeutique, ils sonttrès localisés sur la région présumée d’origine des déchargescritiques, apparaissent surtout en sommeil lent et persistent aucours de la PMO. Ils sont infracliniques et pourraient correspon-dre aux activités rapides enregistrées en SEEG.
Conclusion
Dans les épilepsies focales idiopathiques, la clinique primesur l’EEG à l’exception de l’épilepsie bénigne du nourrisson àpointes et PO centrales et vertex pendant le sommeil, danslaquelle les crises plaident pour une évolution défavorable alorsque les anomalies EEG sont synonymes de bonne évolution.Dans les épilepsies focales non idiopathiques, l’EEG avec certai-nes particularités peut aider dans le pronostic précoce.
Épilepsies avec étiologies particulières
Nous voudrions souligner l’apport de l’EEG dans quelquesétiologies particulières, principalement dans les dysplasies cor-ticales et dans le syndrome du chromosome 20 en anneau.
L’EEG peut aider au diagnostic devant une encéphalopathieavec crises multiples surtout à type de spasmes ou de crises
Figure 12. Fille de 6 mois : début des crises à 2 mois 1/2 caractérisées par une perte de connaissance avec fixité du regard et cyanosepéribuccale suivie par une extension des jambes puis par une hypotonie globale. Ces crises surviennent surtout au sommeil et sont suivies d’unéveil de l’enfant avec pleurs. A gauche : tracé de sommeil stade 2 (sleep st II) : micropointe-ondes centrales droites et vertex mêlées aux fuseauxsigma du sommeil (A : agrandissement).A droite, sommeil paradoxal (REM) : persistance de ces mêmes micropointe-ondes (B et C : agrandissement).
M. Bureau
20Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

toniques. Effectué précocement, il pourra montrer des rythmesde grande amplitude évoquant une agyrie-pachygyrie (Dalla Ber-nardina et al., 1996). Si ce pattern est bien évident dans le jeuneâge, son amplitude décroît au cours de l’évolution (figure 16) etle diagnostic sera alors plus aléatoire.
De même, en présence de crises focales ne posant pas deproblème quant à leur séméiologie clinique, l’EEG est volon-tiers, dans notre expérience, contributif dans le diagnostic étio-logique notamment dans les dysplasies corticales focales avec laprésence d’activités rapides localisées (figure 17).
Enfin, dans le syndrome du chromosome 20 en anneau, l’EEGintercritique est aspécifique montrant quelques ondes lentesthêta uni- ou bilatérales. L’EEG devient caractéristique, et mêmespécifique (Inoue et al., 1997 ; Canevini et al., 1998 ; Roubertieet al., 2000), lors des états d’obnubilation prolongés en mon-trant des rythmes lents relativement amples, autour de 2-3 Hz,auxquels se superposent des pointes ou des PO prédominant surles régions antérieures (figure 18) mais, cependant, cet aspectEEG particulier ne s’observe qu’à partir de l’âge de 4 à 5 ans(Ville et al., 2006).
Conclusion
Un diagnostic et un pronostic précoces reposent à la fois surla clinique et sur l’EEG, les deux sont indissociables. En fait larivalité n’est qu’apparente, les deux sont complémentaires. Ilfaut, bien évidemment, commencer par la clinique qui estprimordiale, faire l’EEG en fonction des questions posées par laclinique et ne l’interpréter qu’en fonction des données appor-tées par cette dernière. Le poids respectif de la clinique et del’EEG peut varier en fonction du syndrome, et plus générale-ment de la situation particulière du patient. Il est bien évidentque l’évaluation clinique doit être complète et rigoureuse, etque l’examen EEG doit s’adapter aux questions soulevées, etinclure, au-delà d’un EEG dit « de routine », des éléments poly-graphiques, et un recueil des tracés sous sommeil. Si les moyensmodernes, en particulier la neuroimagerie, ont permis et per-mettent toujours d’affiner le diagnostic, ils n’ont pas remis encause le couple parfois en désaccord mais inséparable que for-ment la clinique et l’EEG. M
Figure 13. Fille de 5 ans : début à 4 ans par des crises convulsives généralisées, puis rapidement apparaissent des crises partielles complexeset des crises focales simples motrices du membre supérieur droit. Imagerie normale. Persistance de crises fréquentes (follow-up : 14 ans).EEG : anomalies multifocales : à gauche : sommeil stade 2 (sleep stade II) : polypointes rapides postérieures gauches, bouffée de PO dégradéesautour des régions centrales et du vertex, pointes rapides pariétales droites. Au milieu, sommeil lent (slow sleep) : pointes et PO avec l’élémentpointe rapide sur la région temporale droite et indépendamment sur la région temporale postérieure gauche. A droite : sommeil en PMO (REM) :pointes et PO pariétales droites et temporo-occipitales gauches.
21 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

Figure 15. Garçon de 9 ans ayant présenté ses premières crises à l’âge de 6 ans 1/2 de type partiel complexe : s’arrête de jouer, a le regardfixe, se frotte les mains, est crispé. Persistance des crises après 7 ans d’évolution. A gauche : sommeil stade 2 (sleep stade II) : bouffées derythmes rapides en frontal gauche (agrandissement B). Noter la différence de fréquence entre cette activité rapide et les spindles (agrandisse-ment A). Au milieu (à droite), tracé de sommeil lent (slow sleep), les rythmes rapides sont moins évidents et redeviennent plus évidents en PMO(REM) (à la fin de l’extrait).
Figure 14. Garçon de 5 ans 6 mois ayant présenté sa première crise à l’âge de 5 ans, apparemment de type clonique, et par la suite des crisescaractérisées par une fixité du regard, une hypotonie avec cyanose. Développement psychomoteur normal avant l’apparition des crises. Imagerienormale. A gauche : PO et ondes lentes temporales droites suivies par une décharge de PO irrégulières plus diffuse. A droite : décharge de POdiffuse précédée par des ondes lentes et des pointes fronto-temporales gauches.
M. Bureau
22Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

Figure 16. Evolution de l’EEG chez une fille ayant eu d’emblée un retard des acquisitions et des crises à type de spasmes apparues à l’âge de 18 mois.De gauche à droite : activité rapide de grande amplitude (alpha like) à 3 ans et 8 mois et à 7 ans, diminuant progressivement en amplitude eten fréquence (13 ans et 6 mois, 20 ans) au cours de l’évolution. Sur le premier EEG réalisé au centre Saint-Paul, le diagnostic de pachygyrie a étéposé et a été confirmé par l’IRM.
Figure 17. Enfant de 8 ans et 6 mois suivi pour épilepsie frontale.L’EEG met en évidence une activité lente sur les régions frontales mais nettement plus ample à droite où elle est surchargée de rythmes rapidesplus évidents qu’à gauche (agrandissements : A) frontal droit et B) frontal gauche).
23 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

Remerciements : Je remercie la Ligue Française Contrel’Epilepsie et les Laboratoires Sanofi-Aventis de m’avoir décernéle Prix International Henri Gastaut. Je remercie également PierreGenton pour son aide précieuse dans la rédaction de cet article,ainsi qu’Arielle Crespel et Philippe Gélisse pour leur soutieniconographique.
Références
Aicardi J, Newton R. Clinical findings in children with occipital spikewave complexes suppressed by eye opening. In : Andermann F, Lugaresi E,eds. Migraine and epilepsy. Boston-London : Butterworths, 1987 : 111-24.
Beaumanoir A. Infantile epilepsy with occipital focus and goodprognosis. Eur Neurol 1983 ; 22 : 43-52.
Beaumanoir A, Blume W. Le syndrome de Lennox-Gastaut. In :Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Lessyndromes épileptiques de l’enfant et de l’adolescent, 4e ed. Montrouge : JohnLibbey Eurotext, 2005 : 125-48.
Bureau M, Maton B. Valeur de l’EEG dans le pronostic précoce desépilesies partielles non idiopathiques de l’enfant. In : Bureau M, Kahane P,Munari C, eds. Epilepsies partielles graves pharmaco-résistantes de l’enfant :stratégies diagnostiques et traitements chirurgicaux. Montrouge : John LibbeyEurotext, 1998 : 67-78.
Bureau M, Tassinari CA. Myoclonic absences : The seizure and thesyndrome. In : Delgado-Escueta AV, Guerrini R, Medina MT, Genton P,Bureau M, Dravet C, eds. Advances in Neurology : Myoclonic epilepsies.Philadelphia : Lippincott Williams &Wilkins, 2005 : 185-96 ; (95).
Bureau M, Tassinari CA. Le syndrome des absences myocloniques. In :Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Lessyndromes épileptiques de l’enfant et de l’adolescent, 4ème ed. Montrouge : JohnLibbey Eurotext, 2005 : 337-44.
Figure 18. Enfant de 10 ans : présentant des crises partielles complexes avec frayeur et parfois hallucinations depuis l’âge de 6 ans, associéesà partir de l’âge de 8 ans à des états d’obnubilation quotidiens pouvant durer plusieurs heures et à une une baisse des performances scolaires.Enregistrement EEG au cours d’un état d’absence : en A), 30 secondes après le début de l’état : activité lente de grande amplitude revêtant unaspect de pointes lentes sur les régions temporales ; en B), 20 secondes plus tard, le tracé est identique à celui de A ; en C), 20 secondes après :fin de la décharge avec un ralentissement des anomalies sous forme d’ondes lentes angulaires qui disparaissent brutalement, laissant place à uneactivité peu ample ; noter la présence d’une pointe unique postérieure droite.
M. Bureau
24Epilepsies, vol. 20, n° 1, janvier, février, mars 2008

Bureau M, Cokar O, Maton B, Genton P, Dravet C. Sleep-related lowvoltage rolandic and vertex spikes : an EEG marker of benignity in infancy-onset focal epilepsies. Epileptic Disord 2002 ; 4 : 15-22.
Bureau M, Kaleli O, Maton B, Dravet C. EEG correlates of benign focalepilepsy in early childhood. Epilepsia 1998 ; 39(suppl 2) : 91-2.
Canevini MP, Sgro V, Zuffardi O, et al. Chromosome 20 ring : achromosomal disorder associated with a particular electroclinical pattern.Epilepsia 1998 ; 39 : 942-51.
Capovilla G, Beccaria F. Benign partial epilepsy in infancy and earlychildhood with vertex spikes and waves during sleep : a new epileptic form.Brain Dev 2000 ; 22 : 93-9.
Cavazzuti GB, Cappella L, Nalin A. Longitudinal study of epileptiformEEG patterns in normal children. Epilepsia 1980 ; 21 : 43-55.
Commission on Classification and Terminology of the InternationalLeague Against Epilepsy. Proposal for revised classification of epilepsies andepileptic syndromes. Epilepsia 1989 ; 30 : 289-99.
Covanis A, Ferrie CD, Koutroumanidis M, Oguni H,Panayiotopoulos CP. Le syndrome de Panayiotopoulos et l’épilepsieoccipitale idiopathique de l’enfant de type Gastaut. In : Roger J, Bureau M,Dravet C, Genton P, Tassinari CA, Wolf P, eds. Les syndromes épileptiques del’enfant et de l’adolescent 4e ed. Montrouge : John Libbey Eurotext, 2005 :227-54.
Dalla Bernardina B, Pérez-Jiménez A, Fontana E, et al.Electroencephalographic findings associated with cortical dysplasias. In :Guerrini R, Andermann F, Canapicchi R, Roger J, Zifkin BG, Pfanner P, eds.Dysplasias of Cerebral Cortex and Epilepsy. Philadelphia-New York :Lippincott-Raven, 1996 : 235-45.
Dalla Bernardina B, Sgrò V, Fejerman N. Epilepsie à pointes centro-temporales et syndromes apparentés. In : Roger J, Bureau M, Dravet C,Genton P, Tassinari CA, Wolf P, eds. Les syndromes épileptiques de l’enfant etde l’adolescent 4e ed. Montrouge : John Libbey Eurotext, 2005 : 203-25.
Dravet C, Giraud N, Bureau M, Roger J, Gobbi G, Dalla Bernardina B.Benign myoclonus of early infancy or benign non-epileptic spasms.Neuropediatrics 1986 ; 17 : 33-8.
Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. In :Delgado-Escueta V, Guerrini R, Medina MT, Genton P, Bureau M, Dravet C,eds. Advances in Neurology, Myoclonic Epilepsies. Philadelphia : LippincottWilliams & Wilkins, 2005 : 127-37 ; (95).
Dravet C, Bureau M. Epilepsie myoclonique bénigne du nourrisson.In : Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Lessyndromes épileptiques de l’enfant et de l’adolescent, 4e ed. Montrouge : JohnLibbey Eurotext, 2005 : 77-88.
Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Epilepsiemyoclonique sévère du nourrisson (syndrome de Dravet). In : Roger J,Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Les syndromesépileptiques de l’enfant et de l’adolescent 4e ed. Montrouge : John LibbeyEurotext, 2005 : 89-113.
Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severemyoclonic epilepsy in infancy (Dravet syndrome). In : Delgado-Escueta V,Guerrini R, Medina MT, Genton P, Bureau M, Dravet C, eds. Advances inNeurology, vol 95, Myoclonic Epilepsies. Philadelphia : Lippincott Williams &Wilkins, 2005 : 71-102.
Dulac O, Tuxhorn I. Spasmes infantiles et syndrome de West. In :Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Lessyndromes épileptiques de l’enfant et de l’adolescent, 4e ed. Montrouge : JohnLibbey Eurotext, 2005 : 53-72.
Dulac O, Cusmai R, de Oliveira K. Is there a partial benign epilepsy ininfancy? Epilepsia 1989 ; 30 : 798-801.
Eeg-Olofsson O, Petersen I, Selden U. The develpment of theelectroencephalogram in normal children from the age of 1 through 15years. Neuropadatrie 1971 ; 2 : 375-404.
Engel Jr. J. A proposed diagnostic scheme for people with epilepticseizures and with epilepsy : report of the ILAE Task Force on Classificationand Terminology. Epilepsia 2001 ; 42 : 1-8.
Fountain NB, Freeman J-M. EEG is an essential clinical too : pro andcon. Epilepsia 2006 ; 47(suppl 1) : 23-5.
Gastaut H. L’épilepsie bénigne de l’enfant à pointes-ondes occipitales.Rev EEG Neurophysiol Clin 1982 ; 12 : 179-201.
Gastaut H. Epilepsie bénigne de l’enfance of childhood avecparoxysmes occipitaux. In : Roger J, Dravet C, Bureau M, Dreifuss FE,Wolf P, eds. Les syndromes épileptiques de l’enfant et de l’adolescent. London :John Libbey, 1984 : 163-74.
Genetic CGILAE. Concordance of clinical forms of epilepsy in familieswith several affectedmembers. Epilepsia 1993 ; 34 : 819-26.
Genton P, Malafosse A, Moilard B, et al. Les épilepsies myocloniquesprogressives. In : Roger J, Bureau M, Dravet C, Genton P, Tassinari CA,Wolf P, eds. Les syndromes épileptiques de l’enfant et de l’adolescent, 4e ed.Montrouge : John Libbey Eurotext, 2005 : 441-66.
Gobbi G, Sorrent G, Santucci M, et al. Epilepsy with bilateral occipitalcalcifications : a benign onset with progressive severity. Neurology 1988 ;38 : 913-20.
Gobbi G, Tassinari C, Roger J, Bureau M, Dravet C, Salas-Puig X.Particularités électroencéphalographiques des épilepsies partiellessymptomatiques de l’enfant. Neurophysiol Clin 1989 ; 19 : 209-18.
Guerrini R, Dravet C, Gobbi G, Ricci S, Dulac O. Idiopathicgeneralized epilepsies with myoclonus in infancy and childhood. In :Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R,eds. Idiopathic generalized epilepsies : clinical, experimental, and genetic aspects.London, Paris : John Libbey Eurotext Ltd, 1994 : 267-80.
Herranz Tanarro FJ, Saenz Lope E, Cristobal Sassot S. La pointe ondeoccipitale avec et sans éplepsie bénigne chez l’enfant. Rev ElectroencephalogrNeurophysiol Clin 1984 ; 14 : 1-17.
Hirsch E, Panayiotopoulos CP. Epilepsie-absences de l’enfance etsyndromes apparentés. In : Roger J, Bureau M, Dravet C, Genton P,Tassinari CA, Wolf P, eds. Les syndromes épileptiques de l’enfant et del’adolescent, 4e ed. Montrouge : John Libbey Eurotext, 2005 : 315-36.
Inoue Y, Fujiwara T, Matsuda K, et al. Ring chromosome 20 and nonconvulsive status epilepticus. A new epileptic syndrome. Brain 1997 ; 120 :939-53.
King MA, Newton MR, Jackson GD, et al. Epileptology of the first-seizure presentation : a clinical, electroencephalographic, and magneticresonance imaging study of 300 consecutive patients. Lancet 1998 ; 352 :1007-11.
Lerman P, Kivity-Ephraim S. Focal epileptic EEG discharges in childrennot suffering from clinical epilepsy : etiology, clinical significance, andmanagement. Epilepsia 1981 ; 22 : 551-8.
Lombroso CT, Fejerman N. Benign myoclonus of early infancy. AnnNeurol 1977 ; 1 : 138-43.
Okubo Y, Matsuura M, Asai T, et al. Epileptiform EEG discharges inhealthy children : prevalence, emotional and behavioural correlates andgenetic influences. Epilepsia 1994 ; 35 : 832-41.
Okumura A, Watanabe K, Negoro T, et al. Long-term follow-up ofpatients with benign partial epilepsy in infancy. Epilesia 2006 ; 47 : 181-5.
Pachatz C, Fusco L, Vigevano F. Benign myoclonus of early infancy.Epileptic Disord 1999 ; 1 : 57-61.
Panayiotopoulos CP. Benign childhood partial seizures and relatedepileptic syndromes. London : John Libbey, 1999.
Panayiotopoulos CT. The epilespsies : seizures, syndromes andmanagement. Oxford : Bladon Medical Publishing, 2005.
Roger J, Bureau M, Gobbi G, Tassinari CA, Dravet C. Les épilepsiespartielles sévères de l’enfant. Epilepsies 1991 ; 2(2) : 191-8.
25 Epilepsies, vol. 20, n° 1, janvier, février, mars 2008
EEG versus clinique : une rivalité apparente

Roubertie A, Petit J, Genton P. Chromosome 20 en anneau : unsyndrome épileptique identifiable. Rev Neurol 2000 ; 156 : 149-53.
Tassinari CA, Rubboli G, Volpi L, Billard C, Bureau M. Etat de malélectrique épileptique pendant le sommeil lent (ESES ou POCS) incluantl’aphasie épileptique acquise (syndrome de Landau-Kleffner). In : Roger J,Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Les syndromesépileptiques de l’enfant et de l’adolescent, 4ème ed. Montrouge : John LibbeyEurotext, 2005 : 295-314.
Thomas P, Genton P, Gélisse P, Wolf P. Epilepsie myocloniquejuvénile. In : Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P,eds. Les syndromes épileptiques de l’enfant et de l’adolescent, 4e ed. Montrouge :John Libbey Eurotext, 2005 : 367-89.
Van der Meij W, Wieneke GH, Van Huffelen AC, Schenk-Rootlieb AJF,Willemse J. Identical morphology of the rolandic spike-and-wave complexin different clinical entities. Epilepsia 1993 ; 34 : 540-50.
Ville D, Kaminska A, Bahi-Buisson N, et al. Early pattren of epilespy inthe ring chromosome 20 syndrome. Epilepsia 2006 ; 47 : 543-9.
Watanabe K, Yamamoto N, Negoro T, et al. Benign complex partialepilepsies in infancy. Pediatr Neurol 1987 ; 3 : 208-11.
Watanabe K, Yamamoto N, Negoro T, Takahashi I, Aso K, Maehara M.Benign infantile epilepsy with complex partial seizures. J Clin Neurophysiol1990 ; 7 : 409-16.
Watanabe K, Negoro T, Aso K. Benign partial epilepsy with secondarilygeneralized seizures in infancy. Epilepsia 1993 ; 34 : 635-8.
M. Bureau
26Epilepsies, vol. 20, n° 1, janvier, février, mars 2008