Embed Size (px)
Citation preview

CEA-R-4463 - KERTESZ Claude
DOSAGE DU STRONTIUM ET St SES IMPURETES DAHS LES SOLUTIONS D'UN ATELIER PILOTE DE PURIFICATION
Sommaire.- Les solutions provenant de l'atelier pilote de purification du strontium PICPUS contiennent les éléments de la famille des alcali-no terreux et de l'argent. Cette étude définit les contrôles analytiques du procédé : elle comprend trois parties : 1] sur les solutions contenant l,« alcalino terreux et l'argent 1 des teneur» voisines, on effectue une séparation chromatograpnique sur résine cationique : l'Élut ion sMtctive des constituants est réalisée par le nalonate d'ammonium, avec gradient de concentration. On évite la précipitation du malonste d'argent en amenant l'argent 1 l'état de complexe anionique soluble. Le» fractions récupérées sont dosées par une méthode conduct!-métrique, applicable aux solutions **rés diluées ; 2} le dosage radio-chlai*ue du strontium» 90 est effectué -jar scintillation liquide. La Méthode de dosage* en présence d'yttriuM 90, comporte le tracé de deux courbes : variation du facteur de contribution de 90Y et rendement, en fonction d* l'affaiblissement lumineux. Elle est comparée a celle utilisant la séparatien chimique du "«Y ; S) pour les solutions riches en strontium, contenant les alcalino terreux st l'argent a l'état d'impuretés, on étudie un dosage par spectrographs d'émission, selon la
CEA-R-44 63 - KERTEaZ Claude
DETERMINATION OF STRONTIUM AND ITS IMPURITIES IN SOLUTIONS FROM A PILOl PURIFICATION PLANT
Su—ary.- Solutions of the pilot plant for the purification of stron-HuI"FlCPUS contain alkaline earth elements and silver. This study defines the analytical control of the chemical process. It is divided into three parts : 1) the utilization of a cationic resin chromatographic separation for solutions containing similar concentrations of alkaline earths and silver : the selective elution of the constituents is mad* using ammonium malonats with a concentration gradient. The précipitât-on of silver malonate is avoided by forming a soluble anionic complex with the silver. Determinations are performed on the recovered fractions using a coitduc tome trie method applicable to very dilute solutions i 3) the radiochemical determination of >flSr is effected using a liquid scintillator. The method used to perform determinations in the presence of *°Y involves the slotting of two curves t variation of the " D¥ contribution factor and yield, as m function of quenching. This method is compared to that using the chemical separation of 9 0 Y ; 3) in the case of strontium rich solutions containing alkaline earths and silver in the impurity state, investigations were carried out using spectrograph^ emission and the Brockpot method. A statistical method

«éthode de Breckpot. Les droites d'étalonnage du aagse'siua, calciua, baryuw sont validées par une séthode stat ist ique.
1974 - 190 p.
Cossissariat à l'Energie Atoaique - France
was used to verify the calibration corves fer sainesius; calcius, and barium.
1S74 - 100 p.
Cossisssriat a" l'Energie Atoaiçue - France

f CEAR-MW
, COMMISSARIAT A L'ENERGIE ATOMIQUE 2 • . . , , ' .
B,I mmm
Ziïtœœm'
mmmmm
DOSAGE DU STRONTIUM ET DE SES IMPURETES DANS LES SOLUTIONS
D'UN ATELIER PILOTE DE PURIFICATION
par
Claude KERTESZ
DEPARTEMENT DES RADIOELEMENTS
Centre d'Etudes Nucléaire» de Saclay
Rapport CEA-R-4463
1974 SERVICE DE DOCUMENTATION
OE.N - SAOAY B.P. n' 2, 91190 • ©F-tur-YVETTE • Frtnc*

- Rapport CEA-R-4463
Centre d'Etude» Nucléiirej de Saclay Département des Radioéléments
Service de Production des Radioéléments Section Production de Sources
DOSAGE DU STRONTIUM ET DE SES IMPURETES DANS LES SOLUTIONS D'UN ATELIER PILOTE DE PURIFICATION
par
Claude KERTESZ
Mémoire, présenté au Conservatoire National dM Arts et Métiers Centre associé du I aasji'sdoc-Romsino»
pour l'obtention du titre dlnsjénieur CNAM, spécialité : Chimie Industrielle
Mai 1974 - I

Je remercie Monsieur le Professeur ETIENNE pour la bienveillance qu'il m'a témoignée en m'autorisant A préparer cette étude au Commissariat à l'Energie Atomique.
Je suis heureux de rendre hommage à Messieurs les Professeurs du CNAM qui me font l'honneur de constituer mon jury de thèse, en particulier à Monsieur le Professeur FIETRA-SANTA, pour l'intérêt qu'il a bien voulu porter A ce travail, a Monsieur FONZES, à Monsieur BAGNOL pour leur enseignement, et A Monsieur BESSŒRE, Chef de Travaux de Chimie Analytique pour s«s indications dans la rédaction de ce mémoire.
J'exprime ma gratitude A Monsieur COUROUBLE, Chef de la Section Production des Sources - Département des Radioéléments - Centre d'Etudes Nucléaires de Saclay, qui m'a donné ce sujet d'étude, ainsi qu'A Monsieur PROSPERT, Chef de l'Atelier PUote ELAN H B, * la Hague, pour ses encouragement» et son aide puissante lors des calculs statistiques.
Je remercie Monsieur GERARD, Ingénieur A ELAN II B, pour les conseils qu'il a bien voulu me donner en Analyse Spsctrograpbique.
Je prie Monsieur LEFUVRE de trouver ici l'expression de mes remerciements, pour son accueil et les moyens mis A ma disposition dans les Laboratoires du Service des Techniques Avancées et des Applications des Radioéléments - Département des Radioéléments.
Monsieur GUILLON a plus particulièrement dirigé la premiere partie de cette étude, je l'en remercie bien vivement.
Je remercie également me* collègues FApQUET et PINTENA, pour leur amicale collaboration.
Enfin, je remercie tous mes came raie s de l'Atelier Pilote ELAN II B, pour la sympathie qu'ils m'ont toujours témoigne* et en particulier Messieurs CRE5PEL et GRELOT pour le tracé 'ies figures.

I
I
I
i;

PLAN.
INTRODUCTION
1.1. SEPARATION CRROMATOGRAPB1QUE DES ELEMENTS DE LA FAMILLE DES ALCALOTO-TERREUX EN PRESENCE D'ARGENT.
I . l . 1. Généralité.
1 .1 .2 . Principe d* la méthode 1.1.3. Etude expérimentale
1.1.3.1. Appareilla»» 1.1.3.2. Reactifi
1.1.4. Résultats - Discussion
1.2. DOSAGE DES CONSTITUANTS 1.2.1. Titrage conductimétrlque
1.2.1.1, Etude de la stabilité du complexe MY2" 1.2.1.2. Precipitation des hjdroxyues
1.2.2. Etude expérimentale dex dosages par conductimétrie de Mg, Ca, Sr, Ba 1.2.2.1. Réactifs 1.3.2.2. Conditloas opératoires 1.2.2.3. Résultats 1.2.2.4. Caractéristiques de la méthode
a) limite de sensibilité b) précision
1.2.2. s. Cas des eolations contenant Ca 2 + + Mg+
1.2.3. Doeage de l'argent 1.2.3.1. Principe 1.2.3.3. Réactifs 1.2.3.3. Mode opératoire 1.2.3.4. Résultat.

- 4 -
II. DOSAGE RADIOCHIMIQUE DU STRONTIUM 9 0
II. 1. Généralités sur la scintillation en milieu liquide
1I . I . 2 . Comptage
II. 1.3. Affaiblissement lumineux II. 1.4. Principe de la mesure (méthode de l'étalonnage externe)
n. 1.4.1. Dosage d'un isotope pur n . l . 4 .2 . Dosage d'un isotope dans un mélange i
deux composants n,2. Comparaison expérimentale des deux méthodes
II. 2.1. Dosage du Sr en présence de Y H. 2.2. Dosage du M S r après séparation de '"y n.2.3: Conclusion
IU. ANALYSE SFECTROGRAPH1QUE - METHODE GENERALE D'EXPLOITATION DES PLAQUES PHOTOGRAPHIQUE S
in. 1. Méthode de Breckpot IU. 2. Dosages envisagés
IK.2.1. Installation •ptctrographique IU.3. Dosage des alcallno-terreux en solution nitrique
III. 3.1, Influence de Ag sur le dosage des alcalino-te.*reux UI.3.2. Détermination des droites d'étalonnage et mesure
m. 4. Etude des droites d'étalonnage III.4.1. Tests de validité III. 4.2. Annexe n' 1
Annexe n* 2
IV. CONCLUSION GENERALE
V. BIBLIOGRAPHIE.

DOSAGE DU STRONTIUM ET DE SES IMPURETES DANS LES SOLUTIONS D'UN ATELIER PILOTE DE PURIFICATION
La fission d« l'uranium dans les réacteurs nucléaires puis la séparation du plutonium dana les usiner de retraitement des combustibles' irradiés, amènent l'accumulation de sous-produits de forte activité que l'on stock* sous terre, d*ns des installations complexes, en attendant leur utilisation.
Ces produits de fission constituent une source abondante d'approvisionnement en radioéléments ; leur disponibilité a entraîné dans le monde de nombreuses recherches,
lies Etats-Unis, du fait de l'importance de leur programme spatial, ont développé une production de générateurs d'électricité A radioisotopes et A conversion thsrmoélectrique (programme flNAP) : ces appareils contiennent, sous différents blindages, une source radioactive: l'absorption du rayonnement par le matériau de la source et de sa gaine, constitue une source naturelle de chaleur : cette énergie calorifique peut être convertie, en partie, en électricité à l'aide de matériaux tbarmoélsctrioues [ l ] .
Ne présentant aucune pièce mobile et tirant leur énergie d'un radioisotope, ces système- > ont une grande fiabilité et une longue durée de vie : ils sont appelés * voir croître leurs utilisations : équipement </n électricité des régions désertiques, des sites difficiles d'sccès.
Depuis 1955, plusieurs générateurs spatiaux ont été lancé» aux Etats-Unis et ont donné toute sal'sfaction. En Europe et en URSS, les générateura thermoélectriques è radioisotopes (GTJt) ont été développés è des fins civiles terrestres ou marines ; c'est le cas des programmes Hippie en Angleterre, Beta en Russie, Marguerite et Gilets en France - Glsete et Marguerite 0 sont des générateurs de mible puissance, de l'ordre de 1/10 de uatt électrique, qui alimentent des balises marines : Marguerite 20, générateur 200 fois plus puissant est utilisé dans l'exploitation des champs pétroliers sous-meiias.
aa
Le combustible isotopique de ces générateurs est le strontium , fourni par le aur-ché américain, sous la forme d'un cnuiasst chimiquement stable, le métatxtanate de strontium de formule Tl O, Sr ; par frittage, sens charge, on le transforme ea un* céramique, présentant les caractéristiques qui ea font un cssabustible de choix : insolubilité dans l'eaa de sur, point de Jusion élevé, ea même temps que période de décroissance aasss longue.
C'est pour dénsleaair un pregrasaane de tabrlcatioa de svrees pour générateurs isotopiques qu'ont été étudiées w Commissariat * l'Energie Atomique, la séparation du strontium dans les conoeatràts de produits de fission et aa purification.

L'opéntion est donc prévue en deux temped,
1) Traitement des concentrât» de produit! de tiaaion par extraction par «olvant :
La aoaftionde produit» de fission a la coBooattioa autrante (analyse type du 2/8/66) :
Ma 18,3 g l" 1
Al 22,7 " ,
Fa 15,4 "
M( 4 , 8 "
V 3 , 5 "
Ni 1,5 "
Cr 0,« "
Ca 0 ,6 g l" 1
Ce 0 , 5 "
La 1 "
Ca 0 , 5 "
Sr 0 , 3 5 "
Ba 0 , 1 5 "
HNO- 2 . 5 N
• l i e eat neutraiiaée t pil 4 par la aoude n présence de citrate de sodium qui complexe T*. Ni, U et d'acide diéthyttne trismus pentacétique (DTPA) complaxant dea terre» rare». Ill» eat «lor» aoamlM k un cycle d'extraction par aolvaat t contre courant dana une batterie de mélangeurs daeantaar» ; le eofcrent ctlliaé eat l'acide Di. ethyl haxylpboâphorique ou D, BHPA, de formule (C.H..OI, (OH)P • 0, dilué dana le dodeceae an presence de phosphate de tributyle ou TBP (0,3 M de D IBPA et 0,1» M de TBP daaa un litre de aohrant) [1].
Le rafnnat eut contient différent» produit» de flieioa et lea terre» rare» »era stocké ou envoya 1 l'atelier de vitrification : l'extrait eat souml» à une réextraction par l'acide nitrique IN.
Le atroatiam accompagné dea elcelino-torreux et du aodium paaae dana la phase ni-trique : c'est cette «dation qui aéra destinée a l'atelier pilota de purification.
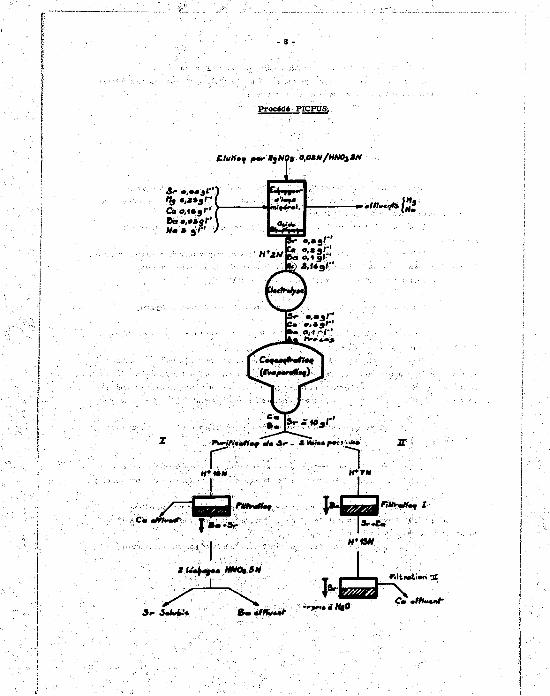
2) Purification da Mrootiua» - Procédé mi» en omvre dana le pilote chimique de purification du
strontium P1CPU8 : (3], [4].
La eolation d'extraction devrait avoir la coaapoaitioa suivante :
Sr 0.0» gl"1
Mg • . »»" Ca 0 ,15" Ba 0 ,06" HN0 3 >2N Ma 5 "
Cette solution est fixée sur une colonne chargée d'an éeanassur d'Ion mènerai, l'a
cide polyaitfiinonieae : Ce produit, obtenu par action de l'acte», chlerhyeriqne sur le pyrsaatlmouia-
te de petaaaiujn,' senne an gai qui, apraè maturation et séchage, montra une capacité d'échange
d'ion» importante, via t via i e strontium [ s ] , '

Avant utilisation/ l'échangeur eubit un prétraitement 1 l'argent. La colonne eat tnaulte «niée par une solution de Ag NO, 0,02 N dans l'acide nitrique
2 N Ï on obtient un éluat dont le compoeition eat la auivante :
Af 0,02 N ou 2,18 » f J
Sr 0.» " Ca 0,2 " Ba 0,1 "
L'expérience a montré que Mg et Na étaient trèa peu fixée par l'écbmngeur. Cet éluat eat envoyé dana une sellale d'élactrolyae à diaphragme munie d'électrodea
en toile de platine pour eéparation de l'argaat. Le dépôt cathodique remia en eolation dans l'acide nitrique servira de solution de
régénération de l'écbangeur, L'éluat débarraeaé d'argent aet conceatré dana un évaporateur et le etrontiam aéparé
dea nitrates de baryum et de calcium par différence de solubilité dans l'acide nitrique.
Fhteieurs schémas de séparatleu sont ici poasiblee [4],
1) Concentration de l'élut avec deetrviUon de l'acide nitrique au formol.
On obtient «ne aoestloa » 1 00* Ci l"1 en milieu nitrique 1 N : en principe, 11 ne doit paa sa former de précipité dana l'évaporassar.
On acidifie cette solution par l'aeide aatria.ua fumaat 0 1 , t N) : l'acidité obtenue de 13 H permet de eéparer le* nitrates de tanna et d* stroataun qui précipitent, alors que la nitrate de calcium est eofrole. Le précipité de nitrate da atrontiam at de baryum eat soumis à deux léchages coaaécutlfs dana H MO, S M qui remettent en aohition le strontium et t a 10 % du baryum . cf. schéma.
L'acidité nitrique peat être détruite par an nouveau paaaaga t l'évaporateur et réaction avec la formol. ,
3) Au cours de la concentration de l'éluat strontium ou laisse monter l'acidité vers
Par evapeeaMe» de la aeeatton obtenue, aa précipite le atreatium an milieu 13 N (Téveperaeeer utilise aet aa streeioy), le calcium restant daaa le filtrat; le eréclaété de nitrate de atreatium eet raerie par l'aaa.
Quel que sett lé précédé choisi, la solution finale a la compoeition suivante :
Procédé PICPUS.
£ /»#• • mm-*fNOt OfOtH/HNOitN
Ca o,i»3l-
M 4 MM H»T*
Cm mm—mT f i 1 • « • •
/ U 4 q u MN0.5N
I
| 5 r « C .
H'fW
5«- j«U.'« Bm^fh-S i«|0
mti»t;*n -s:
c. .m»**-

- s -
Sr 1 000 Ci 1" 1 eoit 10 gl" 1
Ba 0.8 - 1 g l" 1
Ca 0 , 1 - 0 , 2 g l" 1
Mg !
Ag ! t r i c e s
H + 1 » 5 N
EUe est traitée pour élaboration du titanate de strontium t dana la solution neutralisa
sée par l'ammoniaque, on disperse du bioxyde de titane en quantité calculée : on obtient une suspension sur laquelle on fait réagir le carbonate d'ammonium, le carbonate de strontium précipite selon la réaction :
On filtre le gateau de Ti O + CO Se,
Après séchage, broyage et calcination A 1 100 'C, on obtient le métatitanate de stron
tium, Ti O. Sr qui est à nouveau broyé, tamisé (granulométrie 0,87 m/m) puis transformé en cé
ramique dans une presse à chaud. 90 Notre étude aborde le contrôle analytique du procédé de purification du Sr mis en
oeuvres dans PICPUS, à partir de 11 fixation-élution de la colooned'aclde antimonlque : el le a
pour bot le dosage des constituants, aux différentes étapes de ce procédé dans des milieux diffé
rents par la teneur relative des composants.
C'est ainsi que pour l e s solutions d'élution de la colonne où l e s teneurs de chaque
élément «ont assez voisines, nous avons réalisé sur une résine cationlque, une séparation chroma-
tographtque des a Icalino-terreux après complexation de l'argent provenant de l'éluant. Le dosage
de chaque fraction eut réal isé au moyen d'une i i trimeirie par le se l disodique de l'acide ethylene
diamine tétracétique ou EDTA, le point de fin de réaction étant détecté par conductimétrie.
Nous avons ensuite étudié un dosage radiocbUnique du strontium 90 : l'équilibre entre
le Sr et son descendant Y étant détruit par l e s différentes opérations de purification, nous
avons mesuré l'activité du ^ S r par scintillation liquide en tenant compte de l'activité du °Y en
formation : cette méthode servira de contrôle aux mesures effectuées sur l e s solutions à haute
activité, dans un puits de comptage en chafne ulindée.
Enfin, pour l e s solutions obtenues après concentration, constituées par une matrice
de nitrate de strontium d*ns laquelle l e s autres éléments sont présents a l'état d'impuretés, nous
avons employé l'analyse spectrographique. Les courbes d'étalonnage obtenues par une méthode
statistique permettent de fournir une teneur et la précision du résultat.
Le travail peut donc se décomposer e s l e s chapitres suivants :
I. Séparation chromatographique des éléments de la famille des alcalino-terreux en présence
d'argent. Dosage des constituants
II. Dosage radîoehimique du Sr par scintillation liquide
III. Dosage des alcalino-terreux A l'état d'impuretés dans le strontium par spectrographie d'émis
sion.

- 1 0 -
1.1. SEPARATION CHROMATOGRAPHIQUE DESELEMENTS DE LA FAMILLE DESALCALENO-
TERREUX, EN PRESENCE D'ARGENT.
1.1.1. Généralités.
Le principe de la méthode est fondé sur la fixation des alcalino-terreux sur une ré
sine échangeuse de cations, suivie d'une élution par le malonate d'ammonium à des concentrations
différentes, d'après une méthode publiée par Strelow et Van Zyl [6] : le mélange, contenant 6 e ,
Mg, Ca, Sr, Ba est ÎÎXJ sur une résine Dowex AG SOWïf l : l'élution est réalisée comme suit :
Be par une solution de malonate d'ammonium 0,2 N et acide malonique 0,1 N
Mg " " 0 ,5 N
Ca " " 0 . 7 N
Sr " " 1 , 1 N
Ba par l'acide nitrique.
M. Varteressian reprenant cette étude [7 j y a introduit une originalité : i l réal ise
l'élution de Mg, Cas, Sr, Ba avec le malonate d'ammonium, en procédant non plus par paliers
de concentration, mais suivant un gradient continu, linéaire et croissant : l e s éléments alcalino-
terreux en solution aqueuse sont fixés sur résine : une pré élution avec une solution de malonate
d'ammonium 0,2 N et acide malonique 0,1 N est nécessaire pour éviter la précipitation du malona
te de baryum : les conditions optima d'élution sont ensuite déterminées expérimentalement en fai
sant varier le paramètre pente du gradient, c 'est-à-dire, l e volume d'élution, l e s bornes du
gradient de concentration ayant été fixées de 0,5 * 1, 5 N d'après l'étude [6],
Les meilleurs résulta*s sont obtenus avec les volumes suivants :
300 ml de malonate d'ammonium 0,5 N
300 ml de malonate d'ammonium l , 5 N
C - C soit une pente a - a * = r±- C - 1,5 N
600 Cj - 0,5 N
V, » V„ = 3(
On obtient en effet une séparation suffisamment rapide et efficace, avec des pics sans che
vauchement.
Partant de cette étude sur la séparation des alcalino-terreux en solution aqueuse,
nous avons adapté la méthode utilisée à leur séparation dans un milieu réel contenai-, outre ces
éléments, de l'argent en solution nitrique 2 N.
Ce problème a été résolu de la manière suivante :

- 11 -
a) Etude du comportemen v de l'ion Ag en solution aqueuse en présence de nitrates alcalino-terreux,
après fixation sur résine cationique et élution par le malonate d'ammonium ;
b) Complexation des iona Ag et mise sous forme anionique avant fixation sur la résine échangeuse
de cations. Elution des éléments alcalino-terreux ;
c) Application eu milieu acide HNO 2 N.
1 .1 .2 . Principe de la méthode.
h±*lLtÀx. La méthode employée pour la séparation des alcalino-terreux en solution
dans l'eau, par fixation sur résine cationique et élution par le malonate d'ammonium suivant un
gradient linéaire et croissant de concentration, n'est pas applicable directement aux solutions
contenant des alcalino-terreux et de l'argent, du fait de la faible solubilité du malonate d'argent.
Pour éviter, lors de relat ion, la précipitation du malonate d'argent, on opère de la
façon suivante :
Les ions Ag sont complexés avant fixation dans la prise d'essai par le sulfocyanure
d'ammonium. On obtient un composé de forme anionique qui traverse l'échangeur de cations sans
se fixer : l'argent se retrouve en totalité, après passage sur la colonne de la solution de fixation
dans laquelle on a préalablement complexé Ag : dans ce processus , l e s alcalino-terreux sont fixés
normalement et sont ensuite élues par le malonate d'ammonium avec gradient de concentration,
1 . 1 . 2 . 2 . Çojnjrteutlon^e^ÏJEKSt*
Plusieurs méthodes étaient envisageables pour éliminer le précipité de malonate
d'argent : précipitation de l'argent avant fixation à l'état d'halogénure, fixation de Ag sur une
première colonne contenant un échangeur d'ion sélectif, formation d'un complexe anionique de Ag
non fixé par la résine AG 50 W x 8,
Cette dernière méthode qui a été util isée par certains auteurs pour la séparation
de métaux t e l s que Ag, Ni, Cu, sur écb- ngeurs d'anions , nous a paru la plus intéressante [8] ,
I9J, [10J.
On sait que la réaction du sulfocyanure d'ammonium sur le nitrate d'argent donne
un précipité blanc de sulfocyanure d'argent.
Ag N 0 3 + NH 4 SCN « * Ag SCN + N H 4 H 0 3
ou
A g + + SCN" fe—ï» I Ag SCN pK * 12
Or, ce précipité ert soluble dans un excès de sulfocyanure d'ammonium suivant l e s
réactions :

- 12 -
(1)
<2>
et
(3)
(4)
1)
Le produit des réactions 2, 3, 4 qui est chargé négativement ns se fixera pas sur
notre réaine échangeuse de cations. Cette propriété nous a fait retenir le sylfocyanure d'ammo
nium comme agent d« complexation de l'Ion Ag aussi bien en solution ao.ueuse qu'en solution ni
trique 2 N.
Les formes des complexes cités sont définies dans la littérature [ l l j , [12] qui, par
contre, ne donne aucun composé avec Ita éléments alcalino-terreux,dans nos conditions d'utili
sation.
1 ,1 .2 .3 . R*PJ>el>.
a) Choix de IWjnat.
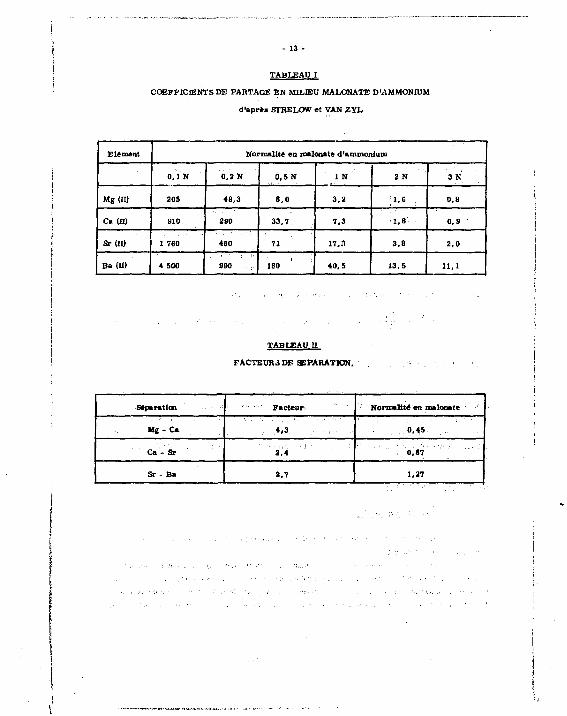
L'éluat doit être choisi de façon que l e s coefficients de partage des composés îlués
répondent aux règles suivantes [13] :
1" Rapport des coefficients de partage de deux composés voisins auss i grand que possible ;
2* Coefficients de partage pas trop granda pour que l'elution n'entraîne pas une trop grande dilu
tion des constituants.
Oh retrouve dans l'étude comparative de Strelow et Weinert [14] d'une part, l e s coef
ficients de partage pour l e s alcalino-terreux entré la résine AG 50 W x 8 et divers agents com-
plexants, d'autre part, l e s facteurs de séparation pour l e s paires d'éléments de la famille des
alcalino-terreux Mg-Cs, Ca-Sr, Sr-Ba.
L e s valeurs obtenues pour le malooate d'ammonium sont très satisfaisantes pour l e s
concentrations choisies (cf. tableaux I et II).
En outre, on peut se débarrasser dss ions malooate dans las fractions recueil l ies ;
ceux-ci se détruisent par simple chauffage, sans apparition de matière charbonneuse suivant la
réaction :
A g + +SCN" < i i AgSCN P K 1 = 4 , 7
Ag SCN + SCN" * = = * Ag (SCNÏg pK 2 = 3 , 5
Ag (SCN)~ + SCN" fe=t Ag'(SCN)*" pKg = 1,2
Ag (SCN)?" + SCN' * = * Ag (SCN)?" pK. - 0 ,2
J 4 4
1) Les pK sont ceux des équilibres prie de droite * fauche.
- 13 -
TABLEAU I
COEFFICIENTS DE PARTAGE EN MILIEU MALONATE D'AMMONIUM
d 'aprèa STHELOW «t VAN ZYL
Elément Normali té en malonate d 'ammonium
0 . 1 N 0,2 N 0 , 5 N I N 2 N 3 N
Mg(n) 20S 49 ,3 8 ,0 3 ,2 1,C 0,8
c»(n) 910 290 33,7 7 ,3 ' 1 . 8 ' 0 ,9
Sr (II) 1 760 460 71 17, a 3 . 8 2 ,0
Ba( I I ) 4 500 990 160 40 ,9 13 ,5 11,1
TABLEAU g
FACTEURS DF SEPARATION.
Séparation Normalité en malonate '
Mg - Ca 4 ,3 0,45
Ca - Sr 2 ,4 0,87
Sr - Ba 2,7 1,27

. COONH. *
/ / / - — » CH, COOH + 2 NH, ' + CO / \ c
. Remarque ; On définit ;
2+ - Le coefficient de partage, pour un cation M . entre la résine et la solution complétante com
me ie rapport des concentrations en M dans l e s deus phases [13]
| M ° + | avec M cation échangé
JlA + | R nombre de meq M a + f i x é s par g. de résine
\vr I s nombre de meq Vr par m l de solution.'
complexante
- Le facteur de séparation entre deux éléments A et B est le rapport
A K A ° B * T/_ «n fixant KL, " 10 pour l'élément le plus faiblement absorbé
^ [6].
b) Elution avec gradient de concentration.
L'utilisation d'un gradient de concentration linéaire et croissant de l'éhiat fait dé
croître régulièrement lea coefficients de partage dea composés élues (cf. tableaux I et H), L'élu-
tioh est plus rapide que par paliers de concentration. Nous avons obtenu un gradient linéaire de
concentration en utilisant deux vases communicants identiques, contenant deux volumes égaux de
solutions à des concentrations différantes : si l e débit de l'appareil est D ml , le débit entre l e s
deux vases doit être — ml .
1 ,1 .3 . Etude expérimentale.
1 .1 .3 .1 . Ap£ir«mage.
L'appareillage dérivé de celui utilisé lors de la précédente étape de l'étude (7) est
représenté sur la figure 1,
Il se compose de deux vases communicants identiques (1 1) contenant les solutions
de concentration extrême : pour obtenir un gradient de concentration croissant, l'éluant circule
du vase de concentration C . au vase de concentration C (C > C J . A la sortie du système,
l'élaat est envoyé au moyen d'une pompe doeeuse Vario Perpex, dont l e défait est soigneusement

15
réglé, aur la colonne remplie d'un échangeur de cations (résine Biorad AG "»0 W x 8 Dowex, 200-400 mesh, sous forme ammonium). L'éntat passe sur le cristal puits d'un ensemble de comptage muni d'un enregistreur, puis est recueilli dans un collecteur de fractions.
Remarques : .
1/ La colonne en verre pyre::, de 200 m/m de long et 18 m/m de diamètre interne, est équipée d'une tête d'alimentation a plusieurs voit! permettant nans aucun démontage :
a) l'introduction de la prise d'essai par une capacité de 25 ml ; b) l'admission de l'éluat sous différents débits ;
c) la mise a l'air de l'ensemble en cas de nécessité. Ce dispositif d'alimentation est raccordé à la colonne par un rodage Rotule* sans
graissage (joint d'éUnchéité en tétmfluoroéthylène), cf. figure 2 : il doit pouvoir «tre adapté sans difficulté au travail à distance - cf. flg. b.
2/ En outre, la colonne^ été munie d'une canalisation à niveau contant, empêchant la dégradation de la résine par manque de liquide ou des défauts.a l'élution par création de passages préférentiels. H est facile de rendre le système plus coinpact et moins fragile pour le travail au télémanipulateur - cf. fig, (a).
3/ Cnfin, nous ,-.vons intercalé entre l'alimentation électrique et les différents appareils - pompe, collecteur de fraction - un* horloge réglée en fonction du volume d'éluant et du débit prévu. L'élution de la colonne par passage de 600 ml d'éluat, pour un débit de 0,7 ml mn" ou 42 ml h , se pratique en un peu plus d'une nuit ( 22 ,»» 14 heures).
••-'• 1.1.3.2. JtteçUfs.
Les produits utilisés sont tous de qualité analytique HP :
- acide malonfque RP de Carlo Erba
- ammoniaque RP de Pro Lsbo - le malonate d'ammonium est obtenu directement par réaction de l'acide malonique
sur l'ammoniaque : les solutions utilisées sont 0, S N et 1,5 N ; - sulfocyanure d'emmoniam RP Pro Lsbo. solution 5 M et 1,5 M.
1.1.3.3. Ç ondjUw^j^ératojres^
La colonne est remplie de 20 g de résine Biorad à 50 % d'humidité ; sa capacité est de 5 meq g de réskie sèche, ce qui donne ans capacité totale trèa largement supérieure aux quantités d'éléments utilisées pendant nos essais.
Après lavage par 2 à 3 volume» de colonne d'eau para, la colonne est prête pour la première opération.
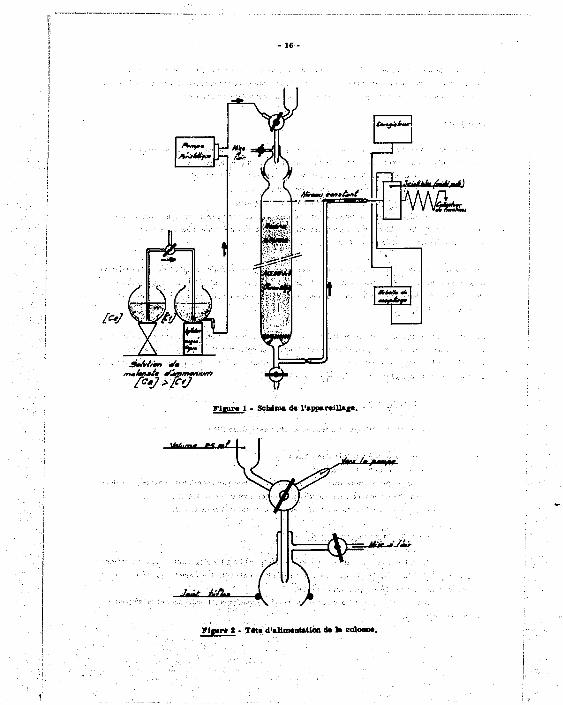
rt«ur« 1 - Schéma de l'appareillafc.
u%/.,~.. M . /
ifai' fmflmm|«r
Jiià^Jmt
Tiger* 2 - TM« d'allmtnUtlon dt 1* colour».
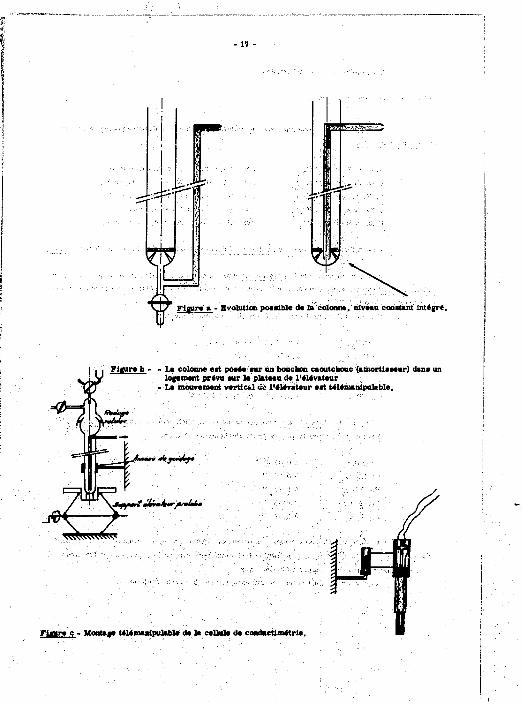
w
TJTFijjr
r w
Figure a - Evolution possible de la colonne, niveau constant intégré.
| , « Figure b - - La colonne est posée sur un bouchon caoutchouc (amortisseur) dans i logement prévu sur le plateau de l'élévateur
- Le mouvement vertical uo l'élévateur est télémanipulable.
Tïjamr/ ffir-t'"—^-
Fiaure c - Montage télémasipulable de la cellule de cooductimétrie.

- 18 -
Cinq opérations «ont effectuées :
a) Définition 6ea prises dressai :
- On fixant sur la colonne, sons un volume do 12 ml de solution aqueuse, au cours
des opérations I et V
0,3 mM de Sr (NO,) , soit 26,1 mg en Sr et 0,6,meq en Sr
0,25 mH de IC| INOJ, soit 6,1 mg en Mg et 0 ,5 meq en Mg
soit 10 mg en Ca et 0,5 meq en Ca
soit 34 mg en Ba et 0,5 meq en Ba
soit 21 ,6 m g e n A g e t 0,2 meq en Ag.
- La même prise d'essai en solution nitrique 2 N est fixée au cours de l'opération IV,
-.La prise d'essai de l'opération m représente un échantillon en vraie grandeur, en
solution dans l'eau (teneurs données par l'analyse type d'échantillons d'élution, au cours d'essais
en inactif sur le pilote PÏCPUS).
Son volume est de 10 ml d'une solution aqueuse contenant :
Sr ( N 0 3 ) 2 à 0,87 g l " 1 en Sr
Ca ( N 0 3 ) 2 à 0 ,20 " e n C a
B a ( N 0 3 ) 2 à 0,13 " e n B a
Mg ( N 0 3 ) , néant
Ag IK>3 1 2 , 1 6 " e n A g o u 0 . 0 2 N .
- De même, l'opération Y représente un échantillon réel en solution nitrique 2 H.
Sous un volume de 10 ml , la prise d'échantillon contient :
S r C N 0 3 ) 2 à O . S Ï g l " 1
C a ( N 0 3 ) 2 « 0 , 2 0 "
Ba ( N 0 3 ) 2 à 0,13 " ,
M g ( N 0 3 ) 2 à 0,18 "
Ag W0 3 a 2,16 "
Cette prise d'essai contient, en outre, des quantités négligeables de trois radioélé
ments Ag, Ba, , 5 S r , qui, par dilution isotopique permettront le repérage des fractiona
corresponns"ntes, ; ipar une mesure radioroétrique.
Ces quantités sont notées en microcuries sur la courbe d'élution V.

- 1 8 -
b '?5P*î?i? Ëïî.*le,?î!îî.ï •'
Dana lea opération» 14 IV, lea elementa Mg, Ca, Sr «ont déterminés dana lea frac-tiona reapectivea par epectroméirie d'àbaorptioh atomique (appareil Parkin, Elmer, type 303). Ba eat Identifié par précipitation du eultete, Ag par précipitation de l'iodure.
Dana l'opération V, lea élémenta Ag, Sr, Ba aont auivla en continu, en aortie de la colonne, grace k i:2ddition dana la priée d'eaaai de traceura radioactifa : lea troia émetteura T 1 1 0 A i , * S3r, I 3 3 B » ont été cbataié pour leur facilité de detection.
Remarque ': Lëa ieotopea Sr et Ba livrée aoua forme de chlorure n'ont pu être introduite dana la priée d'eaaai, mala fixée directement aur je colonne avec lavage 4 l'eau jueqù'A élimination dea ion» Cl" , afin d'éviter la précipitation de Ag Cl dana là aolution de fixation.
c) Fixation - Klution :
1' - Ser la colonne lavée à l'éeu pare, on introduit lea llémente alcalino-terreux et
l'argent. Dè« la préétation t l'acide malonique 0,1 N, on a apparition d'une précipité- cf. courbe 1.
2 - La priée d'eaaai, aoua un volume de 12 ml, contenant iee élémenta alcaUno-ter-reux «t l'argent, ait vereéé dana un bêcher de 25 ïnl': on lui ajoute, goutte * goutte, du eulfocya-nutre d'ammonium s M ; il teat 3 ml de réactif pour diaaoudre le précipité de Ag SClf, apparu avec 'la premiere goutté de NH4 SCW, ce qui amène le volume de la aohrtioa A fixer i 15 ml. La aolution incolore que l'on obtient, homogénéleée par une légère egftatioa A la main eat veraee danà la' capacité d'Introduction de la tête de colonne. Apréa rinçage au aulfocyaaure 0,1 M puia è l'eau pure, on élue directement au malonat* d'ammonium avec gradient de concentration : le précipité n'apparaît plue. L'argent eat identifié dana la fraction qui correepond au volume de finition. Cf. courbée d'éltttion II et m.
Remarque : Le paaeagé du complexe anlonlque Ag (SCN)" a évité la prééletion A l'acide malonique
O , I N . ' ; ' '••; '•' '" ' ' ' - • ' '' -
3 - La méthode de fixation e "applique A une priée d'eaaai en eolation nitrique 2 N. Aprèa complexetlon, l'argent traveree la coleune : on rince la colonne comme en (2),
puia oh effectue l'élutlon par le maloaate d'ammonium - cf. courbée délation IV et V.
1.1.4. «éaullete • Piaenerto». - ^ •
- glutloa - Lee leureee d'éeution aent oetinaea en mHant en anedeae lee volumea d'éhitioh eteadrdomee lea caaoentratiaaa en eifféreota éMmente.
L'élutlon n* I noua montré ••
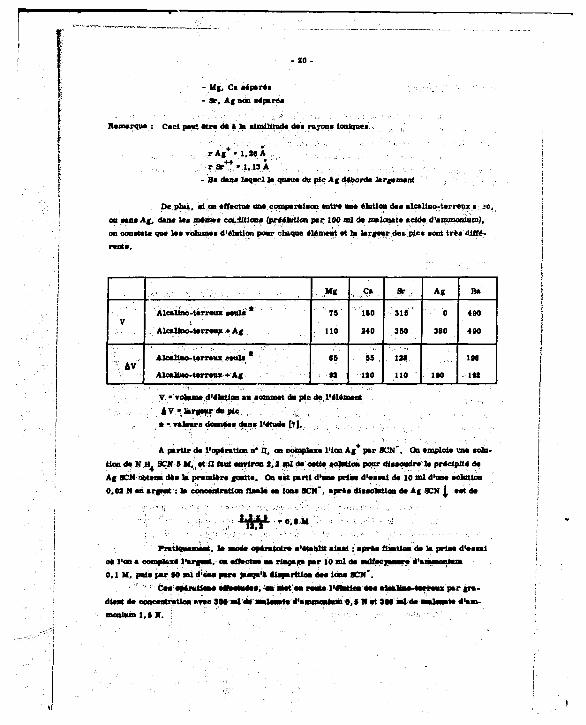
- 910 -
- Mj, Ci séparés - 3r, Ag non séparés
ltemeroue : Ceci peut être d* a te similitude d u rayons laniqu*»
r * * * - 1,13 A - Ba dans lsqerl te qu i» du pic Ag déborde largement
De plut, «i on effectue un* comparaison entre m élution dee alcalino-terreux • >c, w i u i A i , duu las mêmes conditions (préékttion par 100 ml de melouete acide d'ammonium), on constat* que tes volumes d'élntion pour chaque élément «t la. largeur decuple* sont trèa différent*.
, "« •v 9 * 3r . A« B»
V Alcalino-terreux seuls
Alcalino-terreux + Ag
' ' 7 5 '
110
110
340
315 '
, 350
0
380
490
4 M
'"•: av Alcal ino-terreuxseul*
Aloaliuo-terreux + Ag
65
*2
55 .
120
111
110 1*0
1M
in
y • volume d'élutian *» aommst du pic de, Vêlement «Vlergsurdupic . .. -fc : I
« - valeurs donnée* d u e l'étude [7].
A partir de l'opération n* H, on complexe l'ion Ag + par SUN". On emploie une sou-tkn de X H. SÇX 5 M. et i l faut environ J. 2 mi de cette solution pour dissoudre le précipité de Ag SCH obtenu dés la première goutte. On est parti d'une priée d'essai de 10 ml d'une solution 0,02 N en argent : la concentration final* en ion* SCH", après dissolution de Ag SCN | eet de
. ••;••: * ^ y » O , I M
Pratiquement, 1* mode opératoire s'établit ainsi : après fixation de la priée d'essai ou l'on a oomnlevé l'argent, on effectua un rinçage par 10 ml de selfsrjaners d'amaaontum 0,1 M, pais par M mid ;cea pure jaeou'* tieenrition des ions SCX'.
Ces eaératiens «•* aulas, eu sert en i auli l'saatlaa des aleaUne terreux par gra-dieat de concentration arec 3 W sal deaneleent» d'amnwniui o,f X et 3*0 aalde attisante d'ammonium 1,1 X. '

ï l -
Les opérations dé fixation et rinçage sont effectuées au moyen de la pompe Vario Perpex suivant le débit de 42 ml n ' 1 fixé pour l'éluUon.
Dans les fractions récupérées apr*s fixation et lavage, la totalité de Ag est présente dans les 30 premiers ml collectés, ft. ~caractérisation est obtenue par précipitation h l'iodure de potassium dans les opérations n, HI, IV.
Dans l'opération V, 3e marquage de la prise d'essai par Ag permet de fixer le facteur de décontamination en argent, par comptage des fractions sur un spectromètre Y 400 canaux.
On volt d'une part, grâce A l'enregistrement en continu de l'activité,que, Ag traverse la colonne au bout de 30 ml ce qui correspond au volume de la prise d'essai, augmenté du volume interstitiel de la colonne ; d'autre part, après la récupération d'un volume compris entre 42 ml et 88 ml, soit
12 ml de fixation + 16 ml de volume interstitiel * 28 ml 10 ml N H. SCN 0,1 M 10 50 ml H- O de lavage ; 50
l'activité résiduelle de 1 1 0 A g est environ 10~3 %de l'activité introduite.
Remarque sur 3a dissolution du précipité Ag SCN :
Mous avons constaté expérimentalement que pour dissoudre notre précipité de Ag SCN la,; ccncentratto* en SCN~ devait atteindre 1 M dans une prise d'essai de 10 ml de Ag NO. 0,02 N (voir page 20) et nous avons cherché A interpréter t' rorlquement ce phénomène.
Apres dissolution du L , tout l'argent est en solution sous une des formes
Ag SCN, Ag (SCN)*, Ag <SCN)Jr , A g (SCN) J" (cf. page 11)
tout le précipité Ag 9CN est remplacé paries ^uUibres
t * , 8 C N i . t * ^ N - l ;
' " 1 v ' ' • ! ' . - •
[Ag (SCN),]- - ^1*11^**1*
3 K, . Kj . K,
L> ccncantntian C. i» l'argent soliiMU** **t donné» p»r
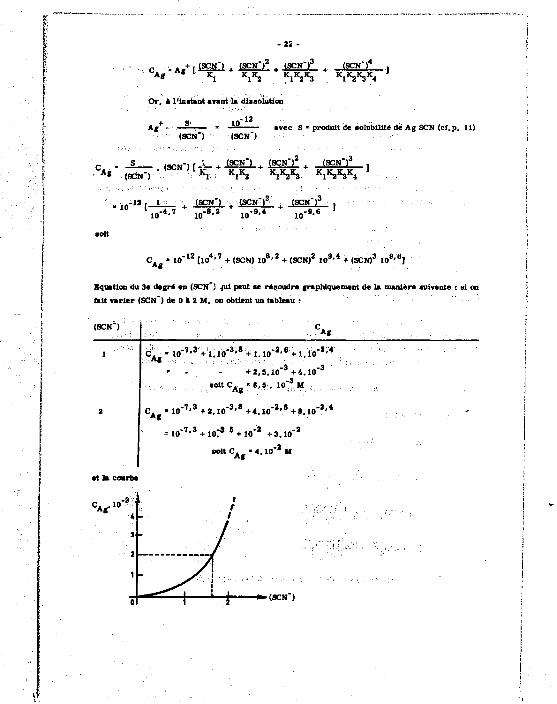
c - . / - . ( S C N " ) , (SCN")2 (sciO 3 (sen') 4 , Ag K l K i « , W S K 1 K 2 K 3 K 4
Or, A l'instant avant la dissolution
-12 A t
(8CN )
10 (SCN")
avec S « produit de solubilité de Ag SCN (ci. p . 11)
l£ + (SCN")3
c . _ . _ ^ _ , ( 8 C M - ) [->- . . IgSf l . ^ X + («H r ] A * (acN")
, , 0 - - ' r » + (SCN") + (SCN")" + (SCN-)" . " l , „ - * - 7 + , „ - 8 , 2 + , „ - 9 . 4 + , „ - » . « S
»-.?
10
C . - 1 0 " 1 Z t l O * ' 7 + (SCN) 1 0 8 ' 2 + (SCN) 2 1 0 9 ' 4 + < S C N ) 3 1 0 8 ' 8 ]
Equation du 3e degré en (SCN -) .nii peut se recoudre graphiquement de la manière suivante : si on
lait varier (SCN") de 0 à 2 M, on ObUent un Ubieau :
(SCN") . ' . : C A .
1 C . „ - 1 0 " , y - + i . l 0 " 3 ' * + 1 . 1 0 " 2 - 6 + : i . Ï 0 " , ; ' 4 ' "
+ Ï . 5 . 1 0 ' 3 + 4 . 1 0 " 3
so t tC . , " 8 ,5 . 10" 3 M Ag -
2 C A f - 1 0 " 1 ' 3 + 2 . 1 0 " 3 ' ' + 4 . 1 0 " 2 ' 5 + S . 1 0 " 2 ' *
= 1 0 " 7 ' 3 + 107* 6 + 10* 2 + 3 . 1 0 " 2
e o t t C A • 4 . 1 0 " 2 M
*»-(acN")
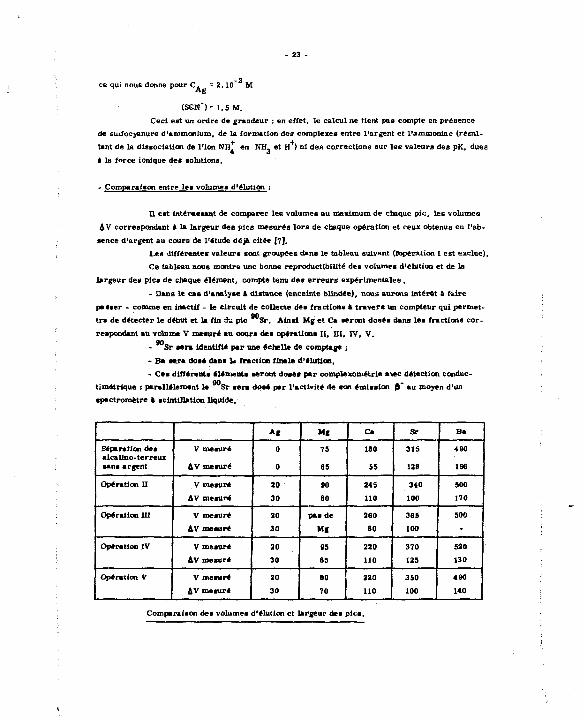
- 23 -
ce qui nous donne pour C. ~ 2.10" M
tSSN")= 1,5 M.
Ceci est un ordre de grandeur : en effet, le calcul ne tient pas compte en présence
de suifocyanure d'ammonium, de la formation des complexes entre l'argent et l'ammoniac (résul
tant de la dissociation de l'ion NH, en NH- et H ) ni des corrections sur les valeurs des pK, dues
a la force ionique de* solutions,
- Comparaison entre leu volumes d'élut ion :
H e s t intéressant de comparer l e s volumes au maximum de chaque pic, l e s volumes
&V correspondant à la largeur des pics mesurés lors de chaque opération et ceux obtenus en l'ab
sence d'argent au cours de l'étude déjà citée [7],
L e s différentes valeurs sont groupées dans le tableau suivant (Ibpératîon I es t exclue).
Ce tableau nous montre une bonne reproductibilité des volumes d'élittion et de la
largeur des pics de chaque élément, compte tenu des erreurs expérimentales,
- Dans le cas d'analyse A distance (enceinte blindée), nous aurons intérêt à faire
passer - comme en inactif - l e circuit de collecte des fractions à travers un compteur qui permet-SO
tra de détecter le début et la fin du pic Sr. Ainsi Mg et Ca seront dosés dans l e s fractions correspondant au volume V mesuré au cours des opérations IX, m , IV, V.
90
Sr aers. Identifié par une échelle de comptage ;
- Ba se,ra dosé dans la. fraction finale d'élution,
- Ces différents éléments seront dosés par complexométrie t w c détection conduc-
timétrique : parallèlement 1* Sr sera dosé par l'activité de son émission (S" au moyen d'un
spectromètre a scintillation liquide.
A t M* Ca Sr Ba
Séparation des alcalino-terreux •ana argent
V mesuré
A V mesuré
0
0
75
65
180
55
315
128
490
196
Operation II V mesuré
AV mesuré
20
30
90
80
245
110
340
100
500
170
Opération III V mesuré
AV mesuré
20
30
pas de 2S0
80
365
100
500
Opération IV V mesuré
AV mesuré
20
30
95
85
220
110
370
125
520
130
Opération V V mesuré
4 V mesuré
20
30
80
70
220
110 j
350
100
4M
140
Comparaison des volumes d'élution et largeur des pics .

OPERATION 1
Séparation, par le malonate d'ammonium avec gradient l inéaire de concentration de 0, 5 N à 1, 5 N, de pente 1/600, dee a lca î ino- ter reux en présence d 'argent .
• P r i s e d ' essa i contenant
Ba ( NO,). A g N 0 3
3'2
0,3 mM )
0 ,25 n M 0,25 mM ! dans H„0
) 2
0,25 mM
0,2 mM )
* préélution pa r acide malonique 0 ,1 N - élution par malonate d 'ammonium o,5 N a 1,5 N, 2 x 300 ml - débit 42 nil . h " 1 .
•3"«'d WOSNva SMaidVd S31
opf/Htr/mi j . -ifatikfmitjM* JE mmkintm • ' I I I I I I U *M*e jmtthial
T. /**» Jim*t mttimf * - y 4 * i ^ » » - » ~
- 1 - - | - " T fer

OPERATION II - milieu H O
Complexation de Ag pa r NH SCN - 5 M
la PE contient Sr (NO,) , 0,3 mM
Mg ( N 0 3 ) 2 0,25 mu Ca ( N 0 3 ) 2 0, 25 mM
Ba ( N 0 3 ) 2 0,25 mM
A g N O , 0,2 mM
1 - Fixation - lavage (NH 4 SCN 0,1 M + HgO)
2 - Elut ion pa r le rnalonate d'emmonium avec gradient de concentration 0, 5 à 1,5 N , pente
1/600 - débit du rnalonate 42 ml, h ,
•autJj N0SNV3 5M3MVd S31

- 26 -
- La PE contient Sr ( N 0 3 ) 2 * 0,87 gl" 1 en Sr Ba (NO a ) 2 à 0.13 g l" 1 en Ba Ca ( N 0 3 ) 2 à 0,2 g l" 1 en Ca Mg (NOj) 2 - néant
Ag NOj - 0,2 mM soit 2,16 gl" 1 en Ag
2 - Elution par malonate d'ammonium 0, 5 N à 1, 5 N -
•wtjj NOSNW synjrt sai
« w n H j r . <-A*4tf
- 27 -
Complexation de Ag par NH d SCN 5 M, LA prise d'essai contient
en solution nitrique 2 N
SCN 5 M . Sr ( N 0 3 ) 2 0,3 mM Mg ( N 0 3 ) 2 0,25 mM C . ( N 0 3 ) 2 0,25 mM B . ( N 0 3 ) 2 0,25 mM A g N O , 0,2 mM
1 - Filiation - lavage NH, SCN 0,1 M + H„0 1
2 - Elution des alcalino-terreux par malonate d'ammonium de 0,5 à 1, 5 N - T^JT
- Débit 42 ml. h" 1
.',.- Bit r | , - ^
l J t r-ft-A-Jn J§jp Jt --T-W:'
! î r "!• , i •• \ •
l 1
•W.
NH 4 S
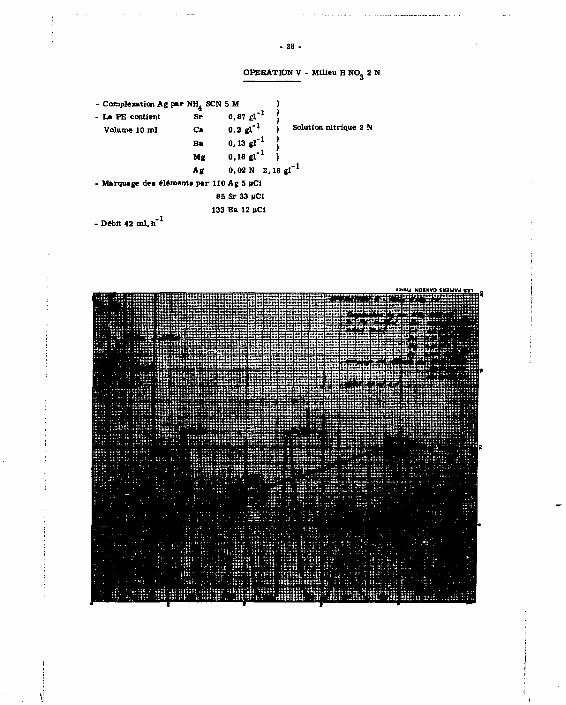
OPERATION V - Milieu H NO, 2 N
Coinplexation Ag par NH 4 S CN 5 M La PE contient Sr 0,87 El"1
Volume 10 ml Ca 0,2 g l" 1 i Solution nitrique 2 N
Ba 0,13 gl" 1
Mg 0,18 g l" 1
Ag 0,02 N 2 16 g l" 1
Marquage de« élémeol a par 110 Ag 5 MCI
89 Sr 33 |lCi
133 Ba 12 |lCi
• Débit 42 ml. h
•JHUJj HOtNvo tmtdvd « n

- 29 -
1.2. DOSAGE DES CONSTITUANTS PAK CONDDCTIMETRIE.
On a choisi la détection conductimétrique pour sa facilité de mise en œuvre : la
simplicité de l'appareillage utillaé permet d'envisager aon application pour le travail en milieu
radioactif.
L'appareillage utilisé est constitué de :
un conductimetre Métrohm E 385 B permettant de mesurer des conductivités de 0 i
3 romho (ou mS) en 6 gammes de mesure : l'appareil est muni d'un système de compensation de
0 à 7 m S ; ;
un potentiograpbe enregistreur E 436 B, qui permet par connexion avec le conducti
metre, d'enregistrer la courbe de titrage : cet aprirei l est muni de son stand de distribution auto
matique de réactif (ici l e s e l diaodique de l'acide ethylene diamine tétraacétique ou EDTA, de
formule simplifiée Na_H 2Y) ;
une cellule de conductimétrie Philips GM 4221 (constituée de 2 électrodes en platine 2
platiné de 1 cm , espacées de 1 cm.
Remarque :
Ce mode de dosage a été préféré.au titrage potentiométrique des alcalino-terreux par
le même réactif : la méthode potentiométrique permet l'obtention d'une courbe logarithmique qui
donne une bonne précision de la détermination du point équivalent : en utilisant une électrode
mercure - (mercure -EDTA) au contact d'une solution contenant des ions EDTA et une petite quan
tité du complexe Hg -EDTA, ajoutée préalablement, on pourra mesurer un potentiel d'équilibre,
indicateur de la concentration en EDTA, et tuivre le titrage des ions alcalino-terreux [ l6 j .
Cependant, la mise en otuvre de l'appareillage est délicate à adapter à l'anayse *
distance : ceci nous a encouragé à utiliser la conductimétrie pour effectuer nos dosages d'alcali-
no-terreux.
1 .2 .1 . Titrage conductimétrique.
Dans la détection du point de fin de réaction par conductimétrie. on se sert du fait
que l e s ions en solution conduisent l e courant électrique grâce i leur charge et A leur mobilité
entre deux électrodes immergée».
La courbe de titration de nitrates alcalino-terreux en solution, par complexométrie
indique la variation de conductibilité de la solution considérée': el le a la forme d'un V avec un mi
nimum distinct qui dépend de le différence de mobilité entre lea ions échangé" (cf. figure 1 , 2 , 3)
on mieux de la conductivité équivalente de ce» ions, qui est une fonction de la mobilité.
Dans la complexométrie des alcalino-terreux, l e s ions échangée août M * Mg, Ca,
Sr, B a e t H + . La réaction s'écrit :

- 3 0 -
(1) M + H, Y « t MY + 2 H 2+ 2 -M + H, Y « 1 Ml
avec X H + .» 350
X M g 2 + 63
x c . a + 60
X S r 2 + 59
X B . S + 64
• conductibilité ionique à 25 *C
La forme des courbea obtenues - elle varie avec la sensibilité de l'appareillage utilisé - pent s'interpréter de la façon suivante :
Lors du dosage d'un ion alcalino-terreux par FEDTA jusqu'au point équivalent, il y a formation d'un complexe dont la conductivité, inversement proportionnelle à la taille de l'ion est plus petite que celle de lHon à titrer, la conductivité diminue : après le point équivalent, l'ad-
1.2.1.1. Etude_deja stabilité du.çomplexe_Myf ".
2+ Au cours du titrage d'un alcalino-terreux M par le sel disodique de l'EOTA
H, Y Na où Y4" est l'anion ethylene diamine tétraeétate de formule :
- O „ C - H , C ^ J ra„-co ,"
la formaiioo du complexe métal - BDTA entrafne la libération d'ions H , qui tend à acidifier la solution
M 2 + + H2 Y2" *=
Or, la forme du complexant qui donne le plus facilement des complexes avec let ' lcalino-terreux est Y " : on peut conneftre sa proportion, à chaque pH, en solution : en effet, 1T.DTA existe en solution, sous les formes Ionisées suivante* :
C y - (Y4") + (HY3") + (H2Y*"j + (H3 Y") + (H^ Y)
connaissant les valeurs des pK de l'EOTA [15] soit 2,0 ; 2,7 ; 6,2 ; 10.3, on en déduit l'égalité :
1 io-3 io-"- 5 io- 1 8 - 2 lo-"- 2 1 2 -
sftcfaant que le constante de dissociation du complexe MY s'écrit
- 31 -
.(M2 4) (Y4') ^ <MY2")
On définira la constante 'apparente "de dissociation de ce complexe suivant les conditions du milieu
(M2*) (C v) ir* - ±
MY*"
d'où K- • * . t 1 + flQ> + <"*> * <H ' • •&> - 1 doù K D K B I H - , 0 - 3 +
1 0 -16 .5 +
1 0 - 1 9 , S
+ , „ - * l , 2 '
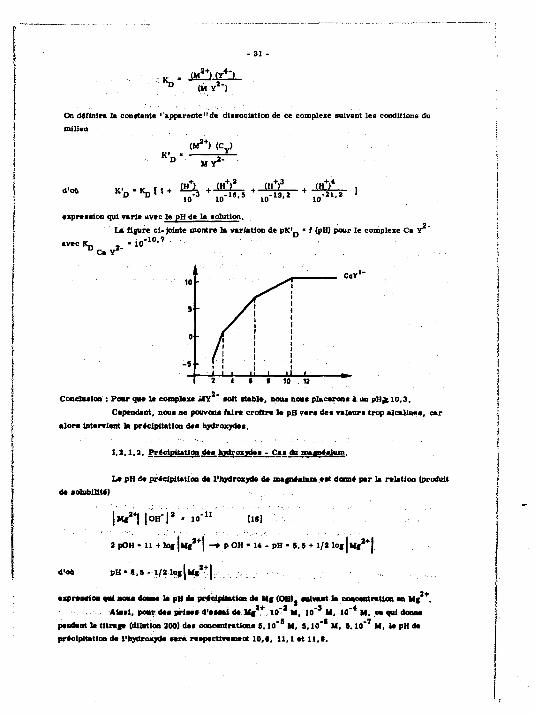
«pression qui varie avec le pH de la solution.
La figure cl-Jointe montre la variation de pK' n « f (pH) pour le complexe Ca Y -10 7 .
avec IL -10 l u ' '
^ CsY
10 ^
s ' i
0 •
• i i i
-5 t •
il i ,, 2 4 * • 10 12
Conclusion : Pour que la compte» *CY «oit stable, nous nous placerons à un pH^10,3. Cependant, noua ne pouvons faire croîtra la pB vara des valeurs trop alcalines, car
alors Intervient 1* précipitation des hydroxvdes,
1.2.1.2. Précipitation des ajdroxydos - Caa du maansaium.
Le pH de précipitation de l'hydroxrde de magnésium est donné par la relation (produit de solubilité)
|Mg 2 + | | o H " | 2 - 10 -11 lis]
2pOH« 11+log |Mg 2 + | - * pOH" 14 - pH-S,5 + l/2 1og|Mg 2 + |
d'où p H - « , 5 - l / 2 1og |s lg 2 t |
expression aaa nous donne la pH as precipttetlon de Mg (OH)j suivant la concentration an Mg"
Ainsi, pour des prises d'aaaai de Mf J + 10"'M, to" 3 M, Mf4 M, ee qui donne pendant le titrage (dilution 200) des concentration. 5.10" 5 M, S. 10'* M, 6.10~7 M, le pH de précipitation de l'hydroxrde s*™ respectivement 10,0, 11,1 et 11.*.

2+
Ces valeurs du pH ne doivent pas être dépassées pour titrer Mg : ceci est obtenu
en faisant varier la concentration en ammoniac, dans 3a solution aqueuse que l'on titre :
- pour une prise d'essai de Mg 10" M # on travaille en solution ammoniacale 0,1 M (10 ml de
N H. OH 2 ïî dans 200 ml H.O déionisée), -3
- pour une prise d'essai d'une solution 10 M, la solution ammoniacale est 0 ,05 M (10 ml NH OH N / 200 ml H,0 ) ,
-4 - pour une prise d'essai d'une solution 10 M, la solution ammoniacale est 0,005 M (10 ml
NH 4 OH 0,1 N/200 ml H 2OÎ.
2+ 2+ - Solution contenant Mg et Ca :
Pour doser Ca , en présence de Mg . , i l suffit de précipiter Mg (OH), en se pla
çant 4 un pH suffisamment élevé [16], ceci est obtenu en utilisant une base comme la diétbylamine
dont le pK_ * 10,9 est plus élevé que celui de l'ammoniaque.
La diéthylamine (C_ H . ) . NH de masse moléculi
utilisée 4 raison de 5 ml ou 3 ,55 g soit 0,05 M dans 200 ml de H_0 déionisée ; le pH obtenu avant
titrage 4 1<EDTA est de 12,15.
Conclusion :
1 - On dosera chaque élément de la famille des alcalino-erreux dans l e s fractions
séparées par échange d'ions A pH légèrement alcalins p H ^ 10. On effectuera l e s dosages conduc-
tiroétriques dans des solutions d'ammoniaque dont la concentration varie de 0,005 M 4 0,1 M en
fonction de la teneur en ions métalliques [17).
La.méthode conductimétrique permet le.dosage des alcallno-terreux dans des solu
tions très diluées (10* 4 M).
N. B . Pour fixer le pH, i l est impossible d'utiliser l e s solutions tampon habituelles dont la force
ionique élevée augmente la conductivité et empêche la mesure. •. •
2 - Dans le cas des fractions contenant 4 la fois Ca et Mg
- on peut doser C a 2 + + M g 2 + 4 pH 10 dans N H 4 OH,
- on peut doser Ca 4 pH 12,2 dans une «olution aqueuse de diétbylamine [18] 4 3 , 5 % en volume :
Mg (OH)2 précipite on dose seulement C a 8 + .
Les conditions opératoires seront définies su cours de l'étude systématique 4 laquelle
nous nous sommes l ivrés .
1 .2 .2 . Etude expérimentale des dosages par conductimétrie de Ma. Ca, 3r. Ba.
I ^ s dosages par «mô^cUinétrie conviennes p i »
diluées, lès solutions concentrées apportant une trop forte valeur de la condnetivité incompatible :

- d'une part, arec la gamme de mesures de kppareil Métrobm E 3(5 B ; - d'autre part, avec les variationa de conductlvité enregistrées pendant le dosage.
Compte tenu deateneura prévuee dene l'éluat à PICPU3 laa doeagee ont été effectuée -2 -3 -4
aur dea solution» aqueuses de nitrates de Mg, Ca, Sr, Ba de concentration 10 M, 10 M, 10 M. De plus, comme le facteur de dilution joue aur la forme de la courbe de titrage con-
ductimétriques, les solutions d'EDTA employées sont plu* concentrées que les aolution* étudiées.
1.2.2.1. Rteçtifs,
- Diéthylamine HP Prolabo Solutions titrées : Sel disodique de l'EDTA (acide ethylene diamine tétracétiqre)
1.2.2.2. .CondjMoM_e^ratoires,
-2 -2 - La prise d'essai contient 1 à 5 ml soit 10 a 5.10 mil en Mg ou Ca ou Sr ou Ba
(la solution examinée contient un serl élément de la famille des alcalrao-terreux). - Bile eat versé* dans 200 ml d'eeu déionisé* (réki ativité prix a l'abri de l'air
B Mn.cm - . - On ajoute ensuit* 10 ml de N H. OH 2 N.
-1 - On titre par l'EDTA 10 M en suivant le point de fin de réaction par conductimé-
trie. N.B. La forme de la courbe obtenue peut être améliorée en jouant aur la aenaibilité du conducti-metre, la vitesse de déroulement du papier de l'enregistreur graphique, etc.
- La prise d'essai contient 10~' à S. 10"3 mu en Mg ou Ca ou Sr, ou Ba. • Elle est versée dans 200 ml d'eau déioniaée ; • On ajoute 10 ml de N H, OH N ;
- On titre par •OTA 10 M ou 5.10"' M comme précédemment.
3) Solution lo" 4 M.
- La prise d'esssi contient de 10"4 k 5.10"4 mM en Mg, ou Ca, Sr ou Ba ; - Eli* est versée dans 200 ml d'eau déionisée ; - On ajoute X0 ml de « H. OH M/10 ; - On titre par EDTA 10 M.

-34 -
1.2.3.3. R<aaltaf,
Lea reau&ata obteaua eont frupéa dana lee tableaux auivanta : il» noua ont permia de tracer lee droite» d'étalonnagea pour Mg, Ca, 9rt Sa. Lee meeurea dea volumea de réactif varient linéairement avec lea prieee d'eaaai, avec una diaperaion que l'on rencontre dana lea autrea méthode» volumétriquee.
Solution 10" 2 M.
Pria* d'aaaai Vcmmt m mlEDTA 1 0 ' 1 M Pria* d'aaaai
Eaaal N* 1 Eaaal N* 2
M * 2 +
1 ml 2 3 5
0,100 0,200 0,310 0,305
0,110 0,210 0,300 0.49
C . 2 +
1 ml 2 3 5
0,120 0,20 0,30 0,49
0,100 0,210
0,485
Solution 10' 3 M
Priât d'aaaai Volumt «n ml BDTA s . 1 0 ' 3 M Priât d'aaaai
Eaaal N> 1 . Eaaaa N* 2
M e * +
1 ml 2 3 S
0,21 0.37 O.M
. M*
0.20 0,39 0,60 0 ,99
c . 2 + 1 ml 2 3 5
0 ,20 0,42 0 ,99 1,00
0,11 0.41 0,02 1.01
S r ' * :
1 m l 2 3 S
: 0.21 • 0.42 O.M 1 . 0 2 • .• . :
0,22 0.40 0,59 1.09
B a 2 +
1 ml 2 3 5
o.io 0.3f 1,00 1,03
0.22 0.40 O.M 1,00 '
- 35 -
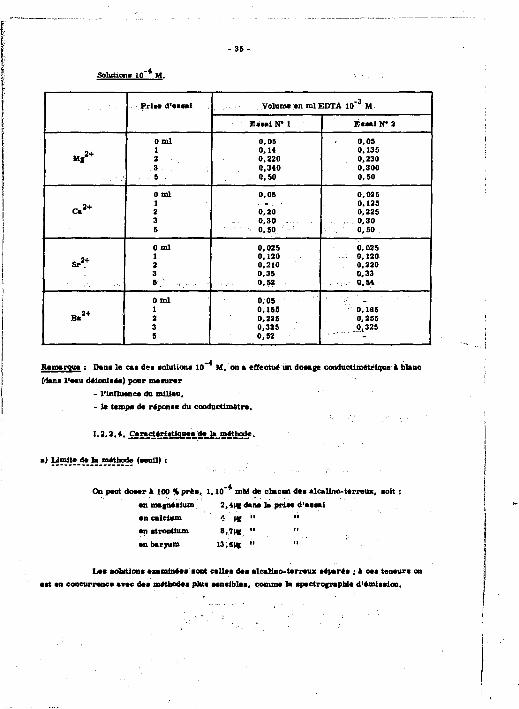
Solution» 10 M.
. Pria* d'essai Volume en mlEDTA 10" 3 M . Pria* d'essai
Essai N* 1 Essai N' 2
0 ml 0,05 0.05
M « 2 +
1 0,14 0,135 M « 2 + 2 0,320 0,230
3 0,340 0,300 5 0,50 0,50
0 ml 0,05 0,025
C . ï +
1 0,125 C . ï + 2 0,20 0.225
3 0,30 . . 0,30 5 0,50 0,50
0 ml 0,025 0,025
S r 2 +
1 0,130 0,120 S r 2 + 3 0.210 0,220
3 0,35 0,33 • : 5 , ' ' 0,52 0 ,54
„>• 0 m l 0,05 _
„>• 1 0,155 1 0,165 „>• 2 0,225 0,255 3 0,325 0,325
1 S 0,52 "" - J
Remarque : Dene le cee dee solutions 10" M, on e effectue un dosage conductlmétrlque à blanc (dene l'eau déionisée) pour mesurer
- l'Influence du milieu, - Je tempe de réponse du conductlmètre.
1.2.2.4. ^rs£éj£sUguee^lajnMude.
•' y!"*î-^"ïJ5*!b? ,ï (seuil) :
On peut doser à 100 % près, 1.10* mtl de chacun des alcalino-terreux, soit : en magnésium 2,4m c J* n" "a prise d'essai
en calcium 4 ME " " en strontium 8,7MS " "
en baryum 13,Sic " "
Les solutions examinées sont celles des alcalino-terreux séparée ; à ces.teneurs on est en concurrence avec des méthodes pats sensibles, comme la spectrofraphie d'émission.

b) Précision de la méthode :
Lorsqu'on effectue un dosage volumétrique, on détermine la quantité A d'un corps
à doser en mesurant le volume d'une solution t itrée.
Ona
A • v . t . M, v * volume de solution titrée t - titre de la solution titrée M * masse atomique du composé à doser
L'erreur commise aur A (teneur cherchée) s'écrit t
A- W t 2 M Z Av 2 +v 2 M 2 At 2 +v 2 t 2 At 2
^ « J_ . . T ,. A „2 JZ „2 *è-'-*£-+*£r+*4- ["• »]
On peut supposer que l 'erreur sur la masse atomique j * - n'est pas super
le terme -*—r- est donc négligeable. On aura alora : M 2
V AA• • - ' « - T
A V v 2 t 2
ce qui lignifie qiw la préciaion de 1» menire ««t (onction :
• de l'erreur mur le volume de réactif débité
- de l'erreur sur le titre de ce réactif.
• Ca« de» «olutiotn 10" M.
- A l :
Si on veut que l'erreur relative commise sur la lecture dea volumes débités n'excède
pas 5 %, 11 faut, s i on admet une erreur absolue dé 0,025 ml
a v * 0,025 et v * 0,5 ml
„* -Aï. . S ^ . JL V , 0 ,6 100 . . . . . . .
On a alors :

-37 -
A t -0 .0001 A t • ^
de même pour avoir -ftv . _5_
A V i o 4 10 4 1 0 °
Av = 0,025
^t io" 4 . 2
' 5 . 1 0 - 3 100
v 100
- *~ v ? 5 ^
aoit t « 10"3 At - 10*4 M AL , JiL
avec comme précédemment, une erreur relative aur le volume débité
v - 0 , 5 i v - 0 . 0 2 5 - £ * - J&-
A V 25 1Ç0 . 125
1 0 4 1 0 4 1 0 4
A - . 1 / _£». ^ i w . i«H / M U % a,. 10%.
1.2.2. 5. C»« de» «ollition» contenmnt C» * Mg.
a) Principe du doMfe :
C. 3+ + M«'+
On doae la aomme dea deux constituant a (ou de toua lea élément* de la famille des
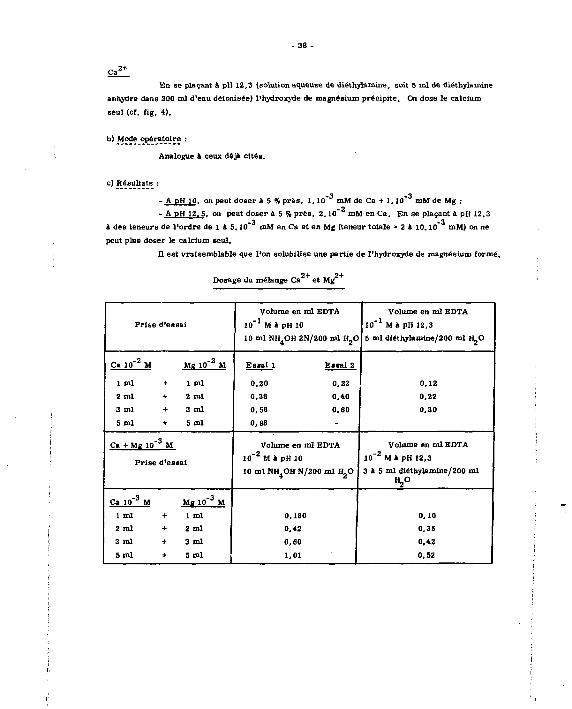
- 38 -
En se plaçant à pH 12,3 (solution aqueuse de diéthylamine, soit 5 ml de diéthylamine
anhydre dans 200 ml d'eau déionisée) l'hydroxyde de magnésium précipite. On dose le calcium
seul (cf, fig, 4) .
b) Mode opératoire :
Analogue à ceux déjà c i tés .
c) Résultats :
-3 -3 - A pH 10. on peut d o s e r a 5 % près , 1.10 mM de Ca + 1 . 1 0 mM de Mg ;
-2 - A pH 12. 5. on peut doser à 5 % près , 2 .10 mM en Ca. En se plaçant à pH 12,3
-3 -3
à des teneurs de l'ordre de 1 à 5.10 mM en Ca et en Mg (teneur totale - 2 à 10.10 mM) on ne
peut pluB doser le calcium seul,
H est vraisemblable que l'on solubilise une partie de l'hydroxyde de magnésium formé. 2+ 2+
Dosage du mélange Ca et Mg
Volume en ml EDTA Volume en ml EDTA
Prise d'essai 10" 1 M à pH 10 10" 1 M i p H 12,3
10 ml NH OH 2N/200 ml H O 5 ml diéthylmmlne/200 ml H„0
Ca 10" 2 M Me 10" z M Esaai 1 Eatai 2
1 ml + 1 ml 0,20 0,22 0,12
2 n d + 2 ml 0,38 0,40 0,22
3 ml + 3 ml 0,58 0,60 0,30
5 m l + 5 m l 0,98
C» + Me 10" 3 M
Prise d'eBSai
Volume en ml EDTA
10~ 2 M » pH 10
10 ml NH.OH N/200 ml H O
Volume en ml EDTA
10" 2 M a p H 12,3
3 à 5 ml diéthylamine/200 ml
«2°
Ca 10" 3 M Me 10" 3 M
1 ml + 1 ml 0,180 0,10
2 ml + 2 ml 0,42 0,35
3 ml + 3 ml 0,60 0,42
5 ml + 5 m l 1,01 0,52

1.2.3. Dosage de l'argent.
Les dosages ont été effectués sur des solutions contenant :
A g N O a 2,16 g l" 1 ou 0,02 N
Sr 0.87 »
Ba 0,13 " dans H NO, ? N
Ca 0.2 "
A g N 0 3 5,40 g!" 1 OU 0 , 0 5 »
&• 0,87 "
Ba 0,13 " dans H NO 2 N
Ca 0,2 "
A g N 0 3 0,216 g l ' 1 OU0.002K
Sr 0,87 "
Ba 0,13 " dana H NOj 2 N
Ca 0,2 "
1.2.3.1. Principe,
Elimination dea ione H par mlae A aec. Remiae en aolution dana l'eau.
Précipitation de l'iodure d'argent (I Ag + | .11 + |= 10" 1 6) aulvant la réaction :
Ag + + N0 3" + K* + 1 + • Agll + N0 3" + K +
Le point de tin de reaction eat aulvi par conductlmétrie, en effet, la concentration dea ion» NO," reate constante : avant le point équivalent, dea lone Ag (A • 62) «ont remplacéa par dea ion» K (A » 74) la conductlvlte varie peu. Aprèa le point equivalent, la conductibilité augmente par addition de K + (A - 74) et I" (A - 77) cf. Hg. 5.
1.2.3.2. B*acUfa.
Solution» titrées d'iodore de potaasium 10*1 N et 5. l u ' 3 N.
1.2.3.3. Mode opératoire,
La priée d'eaaai, en aolution nitrique 2 N contient de 50 ug A 25 mg d'argent en présence d'éléments elcallno-terreux : elle est versée en bêcher pyrex 250 ml et mise A sec.
Aprèa reprise dana 200 ml d'eau délonlaee, on titre par l'iodure de potassium 10" N selon teneur. La fin de précipitation est suivie par conductimétrie.
- 40 -
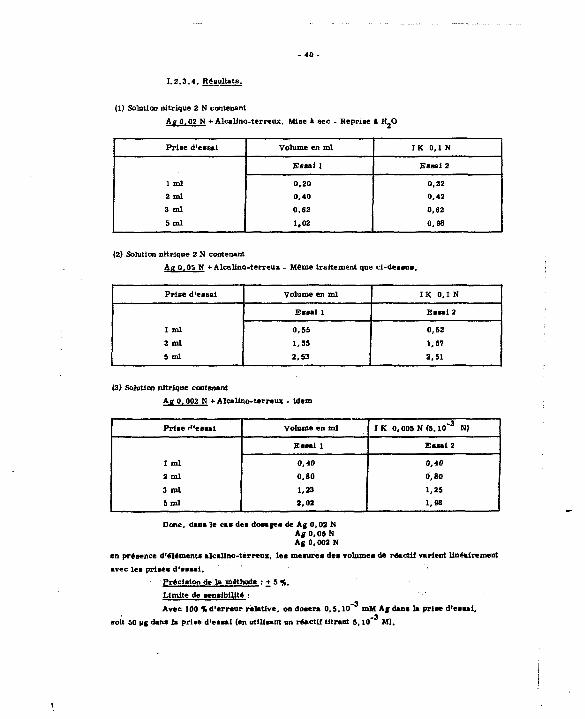
1 .2 .3 .4 . Résultats.
(lï Solution nitrique 2 N contenant
Ag 0.02 N + Alcalino-terreux, Mise a sec - Reprise » H.O
Prise d'essai Volume en ml I K 0 , 1 N
1 ml
E s s a i 1 Essa i 2
1 ml 0,20 0,22
2 ml 0,40 0,42
3 ml 0,62 0,62
5 m l 1,02 0,98
(2) Solution nitrique 2 N contenant
Ag 0,05 N + Alcalino-terreux - Même traitement que cl-dea«us.
Frise d'essai Volume en ml I K 0,1 N
1 ml
Essa i 1 Essa i 2
1 ml 0,55 0,52
2 m l 1,55 1,57
5 m l 2,53 2,51
(3) Solution nitrique contenant
Ag Q, 002 N + Alcalino-terreux - idem
Prise i*'es*ai Volume en ml I K 0.005 N (5 .10" 3 N)
1 ml
Essai 1 Essa i 2
1 ml 0 ,40 0,40
2 m l 0,80 0,80
3 m l 1,23 1,25
5 m l 2 ,02 1,98
Donc, osas le cas des dosages de Ag 0,02 N Ag 0,05 If Ag 0,002 N
en présence d'éléments alcalino-terreux, l e s mesures des volumes de réactif varient linéairement
avec l e s prises d'essai .
Précision de la méthode : + 5 f,_
Limite de sensibilité : _3
Avec 100 % d'erreur relative, on dosera 0 ,5 ,10 mM A f dans la prise d'essai, -3
soit 50 ug dans la prise d'essai (en utilisant un réactif titrant 5,10 M),

Figure 1
Courbe de titrage de Mg (NO.L î o "
«-3 , par EDTA 10 M {burette 5 ml)
- Sensibil ité condactimètre 1 us
- Compensation 6
- Déroulen ent du papier 2
i H i L-^fefi^r^ lllIWIlllllllllllilIfîîtlflfiftîtlIfîffî iiiiiiiiiiiniiiiuiiiiiiiiniiiiiiiiiiiiiiiiitiiiiiiiiiiiiiiiiiiiiniiiiiiiiiiiiiiiiiiiiiiiiiiiiHiiiiiiiiiiiiiiiiiiiiiiiiiiiiiii iiiiiiiiiiiiniiilliiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiir*'<fiiiiiiiiiii!iiiiHiiiiiiiiiiiliiiiiniiiiiiiiu{iiiiiifiiiiiij
ifiiiiiiilliiiiillliiiiliiilHlliiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiii<iiiuiiiiiiiiMiiiiiiiiiii:'<iiiiiiiiiiiiiiiiiiiiilllllllllll! •iMniiillllUllllllllllllIIIIItlllllMllUIUIIMIllllllIIIIlllllllMIIIIIIIIIIIIIIIIIIIIIIIIIK^oilllllIllllllllllIIIHIIini »iiiiiiiiiiiiiiiiiiiiiiiiiiNiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiuiiiiiiiiiiiiiiiMiiiiiiiiiiiiiiiiiiiiiit;:Miiiiiii!!!Miiiiiiiiii
HiiiiiiiiiiHiHfiiMtiiHiiiiimiiitiiiiiiiifiiiiiiiifiiiitiwifiiiiiiiiiiiiifiiittiiiiHtiitiiiitiiiniHiimiiiimHii Ï IIIIHIIIIlilllllllllllllllllllllIIIIIIIIIIIIIIItlIIIIIMIIIMlIllllllllllllllllllllllllllllIIIIIIIIIIIlIIIIllIllllllllllllll I ,î*l
si i m m imtiiiniHtiiniMti iiimifmmimnmiimiiiiimmtimt.~niiiiiiimiimiiiiiiiiitiimiimiiiiiiiii i min iiiiiiiiiii i iiiiiini iiiiiiiiiiiiiiiriiiiiiiiiiiii!niiriiiiiiiiiiiiii.->iiiiiiiiiiiiiiiiiiiiiiriiiiiiiiiiiiiiii <i i min iiijiiiiiiiii iiiiiiiii iiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiîiiiiiiiiii.:<MiiiiiiiiiiiiMiiiiiiiiiiiiiiiiiiiiii i imniiiiiiimiiii iiiiiiin iiiiHiiiHiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiHiiiiiiii(,;Miiiiiiiiiiiniiiiiiiiiiiiiiuiiii I III1II tlftllttlllll Itillllll llllllMllllllHlllllllllllllllllIIIIlIllllllllllllltlIIIIIl»'MltlllllltHIIIIIIIIIÏI|Hllii i min HtntiiiiiM iiniiiit iiiiHiiiHMiuttninmiiHiiituniniiiHimtniiiiiHiiik:'iiiiiiiiiiiiiiiiiiti|H|iii i nun Miiiiniiiii iiiiiiiii iiiiiiiitiiiiiiiiMiiiiiiiniiiHiiiiiiiiuiiiuiiiiiiiiiiiiiiiiiit.'iiiiiiiiiiiiiiH)h|hii •K I I I I I iiiiiiiiiiiii ihiiiiii iiiiiMiiiiiiiiiiiiiiiiniiiiiiiMiiiiiiiiiiiiliiMiiiiiiiiiiMiiiiiii,MiiiiiiuM|i|Q|ii H iiiiiiiiiiiiiiiiiiiiMiiiiiMiiiiiiiiiitiiii'iiiiiiniiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiMiiiiiiiiiiiiiii.'Miuiiuniti ifiiiiiiiitiiiitiiiiuiiiiiniiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiHiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiuiiiiiiiit iiinr m •
•iiuiuiiiiiiiiiiiiiiiiiiiifiiiiiiiiifiiiiiiiiiiiiiiiiiuiiiHiiiiiiiintiiiifiiiiHiiiHiimiuiiiifUiiuui'iiiuiiiuiui • limMHHIMIIIlIllllllimtlllUllltltlllllllllillllMllltllllWIIIttHltttllltllHtUIMIimillfHiiflHmiftllUillItt
sp

Figure 2 Courbe de titrage de Ba (NO ) 10"3 M P«r EPTA 5.10~3 M ( burette 10 ml ).
1 i L ' . » i ' ' ' i i i '~ ' ! L ' ' _
• . T T ' I r i — . ' ^ j T j s - r - f r * * - -I ' ' i L • ' i .>r ' J «fi. '_ ' . . ' I l . ,-. Jt{--\ l - L - l Y l - l -• ' j j .L i • •-,. _j.-»!H ' M I ' ' ' i _
,i i i - i-i- "i J V r 7 f ; ,ft'i fl-i •. i.i > i i-i il nh-.yi'Çtr*!. -l.'ri'ïi^.vii-'h
1 J l il ' J J=T" l*?1 îîijiâ-13'
- Sensibilité conductimètre 1 us
. Compensation 7
. Déroulement du papier 3
1 - Prie* d'étui 2 ml 2 - Prise d'essai 5 ml
Figure 3 - 3 , Courbe de titrage de Sr (HOjlj » M w r EDTA 8.10 M ( buretta 10 ml )
- Sensibilité conductimètre 3 us
- Compensation 7 - Déroulement du papier
3
1 - Pris* d'essai 2 ml 2 - Pris* d'essai 3 ml 3 - Pris* d'essai 5 ml

- 4 3 -
Figure 4
Dosage par EDTA 10*' M du mélange Mg (NO ) 10" Z M (burette 5 ml)
Ca ( N 0 3 ) 2 10" 2 M
a) Prise dressai 5 ml. M g 2 + + 5 ml C a 2 + dans 200 ml H.O + 5 ml dtéthylamlne
3 ml, M g 2 + + 3 ml C a 2 + <• " •• " >•
a 1 ) Prise d'essai i ml. M g 2 + + 6 ml. C a 2 + dans 200 ml H O + 10 ml diéthylamine
M a')
b') 3 ml. M g 2 + + 3 ml C a 2 *
Sensibilité 300 *.a
Compensation g
Déroulement du papier 7
- Stn»Ubilité 300 iiê
- Compeasttloo 7
- DéroulenMDt du papier 7
\

Figure 5
D o n e e Ag NO- 8.10" W en pretence
per IK 5.10 N (burette 10 ml)
- Sensibilité 30 lie
- Compeneetion 5
- Déroulement du pépier 4
Sr 0.B7 gl Ba 0,136 gl" Ca 0,2 g l" 1
r » i » *
1 - Priée d ' u n i 3 ml 2 - Priée d't ini s ml.

- 45 -
90 A partir de la teneur en Sr. Q eat donc essentiel de disposer d'une méthode d'analyse de l'Isotope radioactif.
90 Le Sr est un émetteur 0 , d'énergie maximum E * 0,54 Mev de période
T * 28,5 ans {31J. Dana la plupart de «es applications, i l est utilisé * l'équilibre avec son dea-fln
cendant radioactif l e Y, émetteur & , d'énergie maximum E * 2,27 Mev et de période
T « 84,2 heures.
On admet que l'équilibre 9 0 S r - M Y est atteint au bout de 10 période», soit 640 heu-90 00
res (au bout de 640 heures, on a autant de Y que de Sr). Cependant, dane l'ensemble des opérations de purification, l'équilibre est détruit : dans la fraction strontium récupérée, on dosera
90 90 le Sr en tenant compte de la contribution du Y en formation : on pourra également effectuer l e
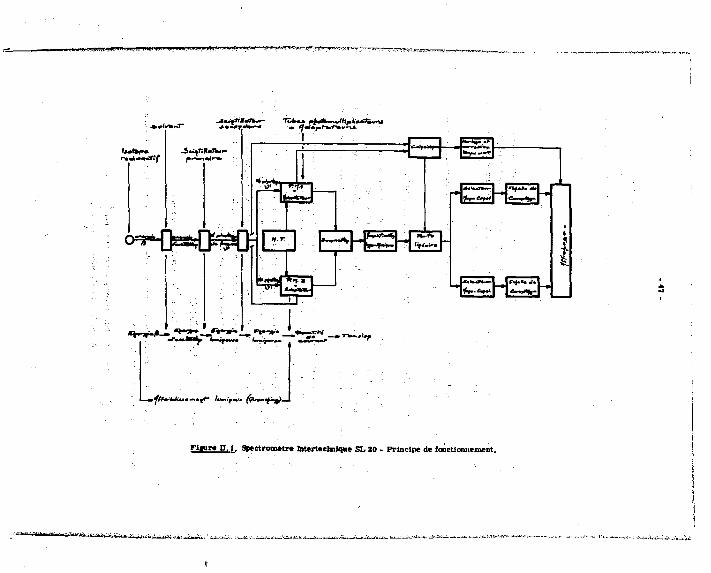
90 dosage après une séparation préalable du yttrium. Noua étudierons ces deux méthodes, en utilisant pour nos mesures d'activité un spectromètre Intertechnique SL 20 * scintillation liquide.
II. 1. Généralités sur 1» scintillation en milieu liquide.
La méthode de comptage 0 * par scintillation liquide est d'un développement a s sez
récent, puisque la plupart des travaux publiés le sont lors de la dernière décennie {22} [23],
Son avantage essentiel a i t la détection «t le dosage des émetteurs de faible énergie,
grace s l'élimination des phénomènes d'absorption. En outre, el le présente plusieurs caracté
ristiques intéressantes :
- grand rendement de détection, ce qui permet de travailler avec dee temps de comptage réduits ;
- possibilité de sélectionner deux Isotopes suivant leur énergie. De plus , la préparation de l 'é
chantillon'eat t rès simple puisque l'on travaille sur des solutions,
rotions essentiel les :
I n scintillation liquide, le matériau radioactif est incorporé a une subsUnce scintil
lante (ou sclntniateur) grées 4 un solvant approprié. Le tout constitue la "«citation scintillante".
- Le solvant, outre son role habituel, doit favoriser le transfert d'énergie entre le
scintilkrteur e t le produit radioactif.
On utilise de préférence, : des' hydrocarbures arométiquea a liaisons it délocs l isées ,
facilement excitables : toluène pour l e s échantillons organiques, mélange dioxane + napbtalèné
pour l e s échantillons aqueux.
- Le scihtilleteùr est une substance fluorescente sous l'effet d'une radiation t ' :
l'énergie des photons émis doit être compatible avec le spectre de réponse de la pbotocatbode.
Pour arriver a ce résultat, en incorpore éventuellement au scintlllateur primaire, un scintillateur

secondaire, ainsi dénommé d'après l'ordre dans lequel s'effectuent les transferts d'énergie. Notre scintUlateur primaire est le PFO ou 2,5 diphényl oxazole * 6 pour 1 000 dans dioxane + napfatalène.
V Notre «clntUlateur aecondalra «at le dimethyl POPOP ou 1 - 4 dt(a,4 methyl
S phenyl oxacolyl benzène) à 0,3/1 000.
C H , ^ CH,
• V y. Remarque : 1* PPO utilisé seul donnerait une longeur d'onde trop courte pour la plage de sensibilité des pbotomultiplicateurs [18]. L'addition de dimethyl POPOP déplace l'émission vers de plus grandes longueurs d'oode. Dans la solution scintillante ainsi constituée, l'émetteur Û" provoque une excitation des molécules du scintillsteur et la production de photons de fluorescence,
11.1,2. Comptage.
, Avec le spectromètre à scintillation liquide Intertschnique SL 30, l'échantillon à mesurer est placé entre deux tubes pbotomultiplicateurs montés en coïncidence (système qui élimine les émissions parasites et, en particulier, réduit la valeur du bruit de fond de l'appareil). Les impulsions électriques engendrées par l'impnct des photons de fluorescence sur les photo-cathodes, sont transmises à un amplificateur, logarithmique, à travers un circuit de sommation qui restitue une Impulsion dont l'amplitude est la somme des deux premières (sensibilité accrue). La sortie de l'amplificateur est dirigée vers deux sélecteurs d'amplitude correspondant A 2 voies de mesure A et B, munies chacune d'un seuil inférieur et d'un seuil supérieur : le réglage manuel de ces seuils permet une sélection des énergies et, donc, la mesure d'activité sur un mélange de 2 Isotopes (ça» du W S r + ^Y).
n.1,3, A^^]^sejaentJ^mineux,
Dans les mesures avec les sdntlllateurs liquides, intervient un phénomène appelé affaiblissement lumineux ou Quenching.
Le Quenching résulte d'une interruption du transfert d'énergie au sein de la solution scintillante et occasionne un glissement du spectre vers les basses énergies. U est d'origine physique ou chimique.
Le Quenching physique est dt à la non transparence de la solution scintillante et des parois du flacon de comptage.
"••« •' .ak.,M'lï*» "!",»l.,'U. M»!l ' M ' ** i l l * i,K*.PM«ïJM..'« J'M
o=H]^0^]?
* * - * » * - -
—^WtPm-'h/£â*m'—{f /vmipw* ffl'*"^'".»}—
Fiinre n , l . Spectrometry biterteclnique SL 20 - Principe de fonctionnement.
V

Le Quenching chimique a deux origines [24] : la désactivation non radiative de l'excitateur et le tranefert de l'excitation à dea aubatancee autre» que le ncintillateur (l'oxygène, en particulier) [24} [25], Dana le cas préeent,. nos eaaaia noua ont montré que le Quenching augmentait avec l'acidité d«a aolutlona à doaer.
L'exlatence de cee divers proceaaua d'affaiblissement lumineux conduit a obtenir pour une même activité, dea rendements de comptage varieblee. "
1) Pour doaer un isotope pur, on devra determiner la courbe du rendement en fonction de l'affai-bllssement lumineux ; 2) Pour doaer un isotope dans un mélange (à 2 composante), on devra déterminer le facteur de
contribution que l'élément le plus énergétique amena dana le comptage du moins énergétique (cas 90 00
du mélange Sr - Y) sachant que ce facteur dépend de l'affaibliaeement lumineux. Cette contribution une foia déduite, on déterminera la courbe du rendement.
Remarque : L'élément le plue énergétique est dosé dans une zone, appartenant aux hautes énergiea, libre d'interférence relative de l'élément le moins énergétique.
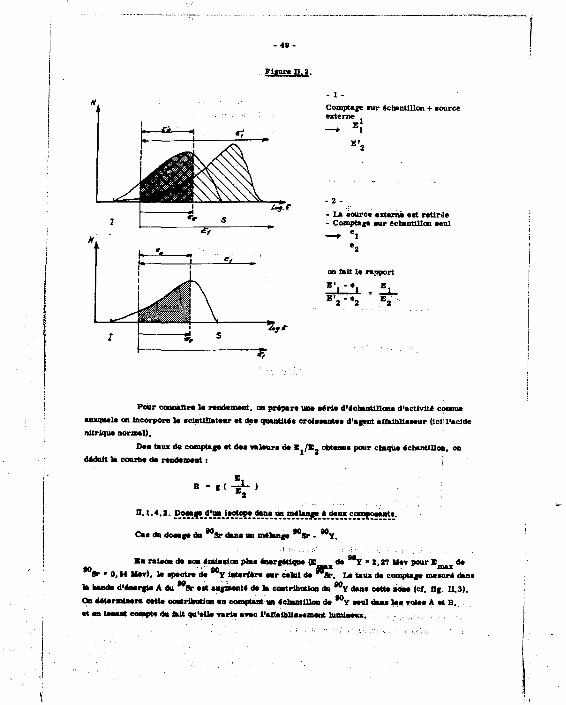
n.1.4. Principe de la masure (méthode de l'étalonnage externe).
Le comptage de l'échantillon eat effectué dana la voie de meaure qui couvre la bande
d'énergie de l'émetteur 0" (bande déterminée par réglage manuel dea seuils Inférieur et eupé-20 90
rieur). Pour un mélange de deux isotopes ( Sr et Y par exemple) on devra compter dana las deux voiea de mesure A et B correspondantea. On obtient ainsi le (ou las) taux de comptage N de l'échantillon.
Pour déterminer l'affaibliaeement lumineux qui correspond à ce taux de comptage et accéder ainsi au rendement de comptage, on utilise une source étalon externe ; dans le SL 20, celle-ci est une pastille de Ce que l'on place près de h solution scintillante : cette source T crée dea électrons Compton qui e'ajoutent a l'émissioD eT à meaurer ; du fait de l'affaiblissement,
137 le spectre du Ce est décalé : en repéra ce décalage par le rapport dea taux de comptages dus
137 au seul e s dana deux voies contigees, incluses dans le.spectre de cet isotope (cf. fig. H.2). Chaque affaiblissement sera ainsi caractérisé par un rapport JGJ/*«I • , et I - étant déterminés par la différence des comptages avec et sans étalon externe .{M],
II. 1.4.1. Ooeajsa d'anisotoptpur.
Ces de *°Sr après séparation du *°Y. Courbe de rendement.
Le comptage sera effectué dans une seule dee voles : on obtient un taux de comptage N qui est lié au rendement par l'égalité : ''
N 1 lt " X où N • taux de comptage en coups me
A • activité an désintégrations ma"',
F i g u r e ^ .
t*.r I r f .
r—i U ^
7**
Comptage nir échantillon + source externe
— E î
- 2 -- La âourc* externa eat retirée - Comptage «ur échantillon aeul —* 'l
on fait le rapport E • - ' - •
E' - e t 1
Pour connaîtra la rendement, on prepare une eéria d'échantillon* d'activité connue auxquale on incorpore le «clntUlateur et dee quantité» croieeantee d'agent aifeibliaaeur (ici: l'acide nitrique normal).
De* taux de comptage et dea valeur* de E. /E- obtenu! pour chaque échantillon, on déduit la courba de rendement :
f (
n. 1.4.3. j>°*R d j^jL l ^9*. d > A*. u ?.^ < J!*iy* * d > a x comaoaant»
Ca* du doaag* du M S r daaa un mélange *°Sr - M Y .
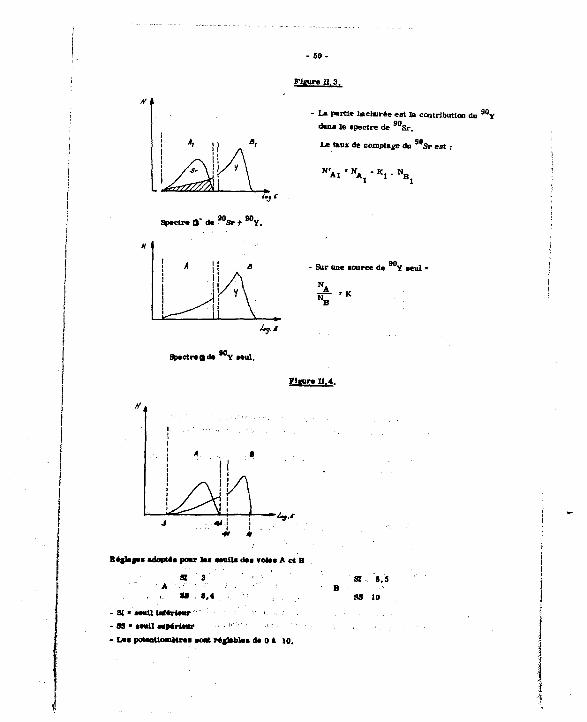
En ralaoa de aoa émiealon pan énergétique (E de ar - O.M Mer), 1* apactr* de M Y Interfere car celui de *Sr. Le taux de comptage meauré dana
la banda d'énergie A du *** eat augmenté de la contribution du W Y dan* cette K M (ct. «g. II. 3). On aétarauaera oetta contribution en comptant un échantillon de *°Y aeul dana laa voie* A at B. et en tenant compte du mit qu'elle varia avec l'attaibliaaament lumineux.
50-
FiEure n,3.
«I La partie hachurée est la contribution du 9 0 Y dana le apectre de 9 0 Sr .
Le taux de comptage du 9 0 S r est :
Spectra fl" da '"sr + 9 0 Y .
/«,./
SpactraQda ™ï aaul.
- Sur une aource de ^V aeul *
N.
Flwre 11.4.
' * » • *
Régla»»» adopMa poar laa senila daa volaa A et B
a 3 A
« «,4
- 31 - eeatl UeMriemr - SS - aavil supérieur
- Laa potentiomètraa aoajt réglable a de 0 a 10.
SI 8,5

- 51 -
90 90 90
La source de Y est obtenue à partir d'une solution nitrique de Sr et y , par
précipitation à l'ammoniaque et redissolution de l'hydroxyde par l'acide nitrique. Le filtrat con
tient M S r .
Une aliquote de chaque solution, incorporée au scintillateur, permet de fixer les
seuils inférieur et supérieur des bandes d'énergie A et B (fig. II.4). 90 On prépare ensuite une série de Y, d'affaiblissement croissant. Le comptage de
chaque échantillon dans l e s voies A et B permet d'évaluer le facteur de contribution K = -g- .
Le comptage de la source externe vue à travers la solution scintillante dans l e s deux voies pré
réglées E . et E_ donne pour chaque valeur de K une valeur correspondante de E . / E _ , n est
a lors possible de tracer une courbe expérimentale
E i K ' I ( - E T - '
E 2
B - Courbe de rendement :
an an A partir d'une solution étalon de Sr + Y, on prépare une série progressivement
90 affaiblie. La mesure dans les voies A et B donne les taux de comptage - N . pour le Sr augmen-
té de la contribution de " Y ; . „ - N B pour le 8 U Y .
E . On détermine ensuite l'affaiblissement correspondant •— .
S C*M valeurs permettent de calculer l e s taux de comptage dus au W S r seul :
[2]
E (K ttt t i ré de la courbe, pour chaque valeur de - j p ) puis l e rendement B , d'où une courbe
de rendement 2
B l E 2
C - Echantillon inconnu :
Les deux courbes K * f ( -=*- ) E 2
sont exploitées de la façon suivante :
- l e comptage de la solution 1 doser donne :

- 52 -
90 - une valeur du taux de comptage dane la zone A ( Sr) N .
90
- une valeur du taux de comptage dans la zone B ( Y) N -
- une-jfaleur du rapportE. /E_ donnant l'affaiblissement,
- la courbe K = f (-5— ) donne la valeur du facteur K pour l'affaiblissement de l'échantillon. 2 90
On obtient le taux de comptage N . du Sr seul (cf. égalité 2) B i
• De même, la courbe R = g (-=r~ ) permet d'accéder au rendement de comptage, d'où l'activité -1 2
en désintégration mn A = & A R
On passe aisément A l'activité en curies puisque
II. 2. Comparaison expérimentale des deux méthodes.
90 90 II. 2 . 1 . Doraj£ j iu_ — Sj^ej i j^se jwej i e ¥•
Mode opératoire :
A) La substance scintillante est constituée à partir de produits de qualité "scintillation
liquide". Elle a la composition suivante [27] :
PPO (Packard Instrument Company) 6 g
Dimethyl POPOP (Merck - Darmstadt) 300 mg
Naphtalène (Merck - Darmstadt) 100 g
Dioxane (Merck - Darmstadt) Q sp 1 1
Elle est conservée A l'abri de la lumière.
Pour l e s mesures , e l le es t mélangée A raison de 15 ml par prise d'essai de 0,2 ml
dans des flacons "Packard" type scintillation liquide en verre exempt de potassium (interférence
d u 4 0 K ) .
B ) 5*earat i o n -* i J^SX *î -4.U 1?X* Qfl Qn on
Sur une solution étalon de Sr + V Y, dont l'activité en Sr est de l'ordre de 1 A -1 90 10 mci ml en Sr, on précipite l'bydroxyde d'yttrium par l'ammoniaque 6 N, en présence d'yt-
trium entraîneur (solution A 10 gl~ ) : le strontium 90 reste dans le filtrat.
Le précipité d'hydroxyde d'yttrium, après lavage A l'eau jusqu'A pH 7 est remis en
solution par l'acide nitrique N.
C) QiyV^?P3êVLS!L4^J^SïH.:
~~ " * gn Une série d'échantillons est préparée A partir de 0,2 ml de solution de Y séparé
et de volume de 0,2 A 2 ml de H NO- 0,1 N auxquels on mélange le scintillateur :

- 53 -
Les différentes valeurs expérimentales sont groupées dans le tableau I et permettent E j
de tracer la courbe K * f (-=— J (cf. courbe établie à partir d'une prise d'essai d'activité égale
A 4 nci en 9 0 S r + 9 0 Y ) (prise %'essai de 0,2 ml) .
D) P>Jer«ywiiOT.4w.rffodppitat :
Une sér ie affaiblie es t constituée par 8 pr ises d'essai de 0 ,2 ml d'une solution éta-Qn fin - 1
Ion de B U S r + avY a 200 nci ml x et des volumes croissants de H NOu 0,1 N. 3 E i
Les résultats donnent le tableau n et la courbe R * g ( - ~ ~ J. E 2
E ) ÇpDtrÇte. d jJR. ffiéthjD(te_ : ejse.mpte ife. £ p**ge_: 90 90 -1
A partir d'une solution étalon Sr + Y d'activité égale à 100 nci ml , on prépare
4 échantillons de 20 nci .
Les comptages de chaque échantillon donnent l e s résultats suivants (bruit de fond de
l'appareil déduit) :
N A 1 73 455
N A 2 73 393
N A 3 73 675
N A 4 73 200
N B 1 5 624
N B 2 0 196
H B 3 .> 516
N B 4 5 509
dans l e s voies E . et E .
V E 2 4,91
V E 2 4,88
V E 2 4,90
E x / E 2 4,88
5 536
±- ' 4,9 B 2
Cette valeur reportée - dana la courbe K * f (•=—J Indique un facteur de contribution
D'où le taux de comptage du Sr aeul
'73 4 3 0 - 6,8 . 5 536 - 3S 785
E l • dans la courbe R * g (-=r~) un rendement de 0 ,83 .
et l'activité de l'échantillon

- 54 -
ou si 1 nci « 37.60 = 2 2 2 0 d é s . m n
A * 19,4 nci .
Précision de la mesure.
Le résultat 19,4 nci pour une activité de 20 nci est obtenu avec une erreur
20 20 7 P '
Cette erreur est due :
- aux erreurs statistiques sur l e s comptages,
- aux erreurs de lecture sur les abaques.
Analysons l'erreur statistique sur le comptage N* :
On aait que ai le nombre N de coups comptés est grand et s i le temps de comptage
est court par rapport à la période du radioélément, l'écart type sur le comptage est
9 * \J N
avec'pour une probabilité de 95 % un intervalle de confiance de la mesure
N + 2ff
A N ' A - A N A + A ( K . N B )
Ona -.
Û N A * 2 V N 1 A + N 2 A * 2 V 7 3 5 1 0 + 8 0 " 5 4 2
K . N B K N B
JaJL K
•ur notre valeur de K intrapolée d'une courbe peut être estimée par le calcul, sur une valeur de K
expérimentale proche (cf. tableau I)
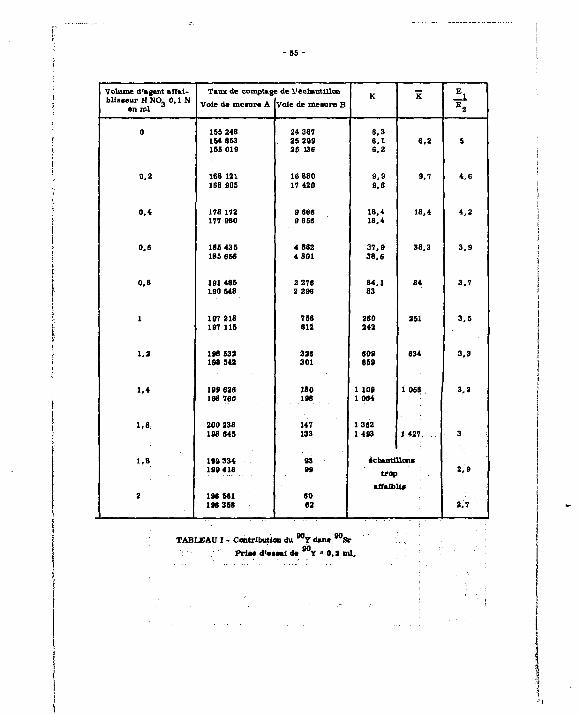
Volume d'agent affai-b l iaaeurHNO, 0,1 N
en m l
Taux de comptage de l'échantillon Vole de mesure A Voie de meeure B
0,8
1,2
1,6
1,8
155 248 154 853 155 018
168 121 168 905
178 172 177 980
185 435 185 656
191 485 190 548
197 218 197 115
198 532 198 542
199 626 198 760
200 238 198 645
199 334 199 418
196 561 198 358
24 387 25 299 25 136
16 880 17 420
9 666 9 656
4 882 4 801
2 276 2 296
758 812
326 301
180 198
147 133
93 99
60 62
6.3 6 ,1 6 ,2
9,6
18,4 18,4
3 7 , 9 38 ,6
84 ,1 83
280 242
609 85»
1 109 1 004
1362 1 493
9,7
251
1 427
échantillons trop
affaibli»
3,9
3.5
3,3
2,9
90 Pria* d'aaaai da Y * 0,2 ml.
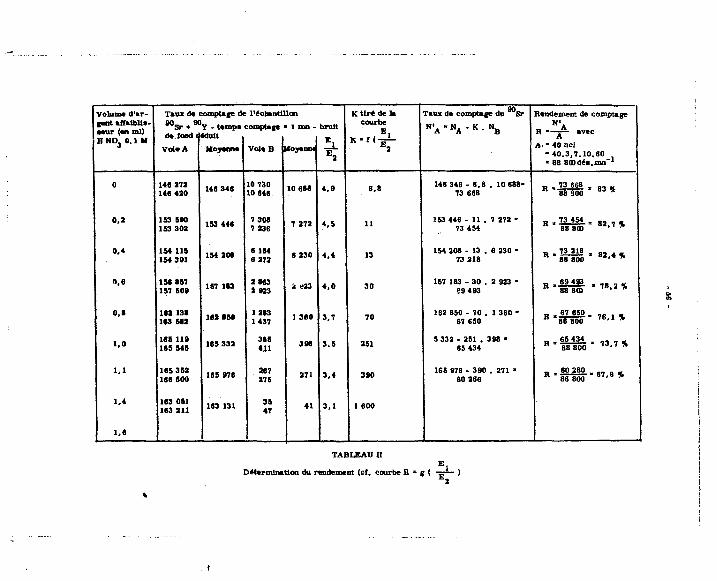
Volume d'argent affeiblie-eeur (en ml)
Taux de comptage de l'échantillon Sr + Y - tempe comptage " l s » - bru»
de. fond déduit _
h. Voie A Moyenne Voie B Moyen»
K tiré de la courbe
E l
90 Taux de comptage du Sr ' N A - K . N. B
Rendement de comptage »'•
avec A . - 40 nci - 4 0 . 3 , 7 . 1 0 . 6 0 * 88 8<Ddéa.mn~
146 272 146 430
153 690 153 302
154 IIS 154 301
156 157 157 50»
1*2 132 1(3 562
1(5 11» 115 545
165 352 166 600
163 051 163 211
157 163
162 250
165 332
165 276
10 730 10 646
7 306 7 236
6 164 6 272
2S63 2 223
1213 1437
365 «U
267 275
35 4T
10 666
6 230
2 823
1 3 6 0
386
271
4 , 9
4.0
3 , 5
6.8
251
390
146 346 - 6.8 . 10 f 73 668
153 446 - 11 . 7 272 • 73 454
154 208 - 13 . 6 230 • 73 218
157 183 - 30 . 2 923 • 69 493
162 850 - 70 . 1 360 • 67 650
5 332 - 251 . 398 • 65 434
165 976 - 390 . 271 ' 60 286
8 3 *
K , 73 454 . g
" 88 800 • " • 7 "
R . 73 21» . . . * -R «TBÔÔ M ' 4 *
, 69 493
88 800
TABULAI! H
Détermination du rendement {cf. courbe R ' S<
r
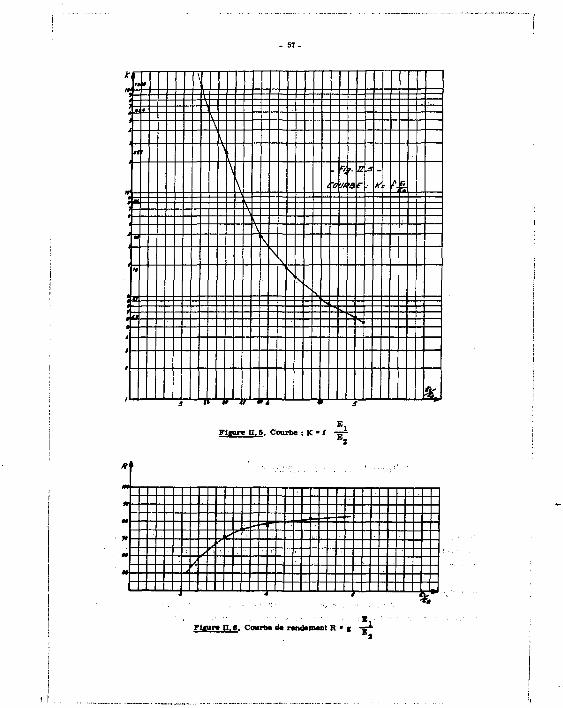
Ira»
Tr i '""" " M '.v ~^-v \ 3
\-. Y . L.
< .* . — — , ç _ _ _
i - J , L. 3
! !sj \ k
g m 2 : * — i
;a_ i EEEEEEzEi=EEEEEEE— s
' ' ' ' ' ' J ' i n r1-; r - •» i - 1 J %. Figure n. S. Courbe : K ' 1 -=p
* • •
J J 4 H i ii '
Wgir» n.g. Courbe de rendement R - g
1

- 58-
pour K * 6,2 155 019 . 155 099 - 80 25 136 25 144 - 8
V 185 179 . \T! AIL . «f V 185 » » + V 2» 152 . 2 394 2 145 „ _2_ K ' 155 019 25 136 ' 155 019 25 136 — 100
de même ^B . . JT . - .544 + 8 . . 74 . 2.7
N B 5 544 - 8 " 5 536 100
ï i ô + f s o - - ^
A(K . N B ) - - i j j . 6,2 . 5 536 - 1 613
AN' AN" - 542 + 1 613 * 2 155 et —
Cette erreur de 6 % eat tout & fait acceptable pour léa doeagea uauela : on remarque cependant dana le tableau I que l'erreur aur la valeur de K augmente avec l'affaiblieeecaent donné
Et par le rapport E .
La détermination de K a donc une part prépondérante dana la.préciaion de la meaure.
- Expreaeion du réaultat :
19,4 + 1,2 ncl.
H.ï .2. i>^aaj(e_du_^Sr-anr»£ **XfIS!iSSJS^Xx
A " ?5'iîïft*dî.îllL."*Jï'.,?iiîîts,J-
L'yttrium M eat précipité en préeence d'entraîneur aoua forme d'hydroxyde : le etrontium 90 eat purifié dana le eurnefeant par extraction i la taénoyl trifluoro acétone (ou TTA) dana l'hcxone a pH 9,!. n eat enauitc remia en aolution nitrique par réaxtraction dana H NO,3 N : dea aliquotea aont incorporéea a la aolution eclntillante au dioxane (cf. II, 2.1. ) pour comptage.
Remarque : • • La TTA donne avec 1* etrontium un chelate extractible dana l'hexone. Le rendement
d'extraction eat de H, 5 % k pH », 5. n décroît vera lea pH Inférieur» [29] [30],

B - Réactifs.
1 - Entraîneur .Y (NO,). 1 10 gl"1 en yttrium en milieu H N0 3 N ;
5 - Acetate d'ammonium N ; 6 - TTA 0,05 M dana l'isobutylméthylcétone (Hexooe).
C - Mode opératoire,
90 La précipitation de Y se fait de la manière suivante : On place dans un tube à centrifuger de 15 ml
- la prise d'essai, - 1 ml d'entraîneur yttrium sous forme nitrate, à 10 gl" en yttrium. On complète A 5 ml par l'eau déionisée et ajoute 5 ml d'ammoniaque 6 N, Le préci
pité est séparé par centrlfagatlon : on reprend ensuite, dans une ampoule à décanter 5 ml du surnageant que l'on amène » pH 9,5 par 0,2 ml H NO, 10 N + 1 ml d'acétate d'ammonium 1 N, On y ajoute S ml de la solution de TTA 0,05 M. Aprèa agitation (5 nu) et décantation, on centrifuge la pfaase organique : on en prélève 3 soi auxquels on incorpore 6 ml de H NO, 3 N [28],
Après nouvelle agitation et décantation, on récupère une phase nitrique qui contient SO Sr et que l'on incorpore à la solution scintillante. Les opérations décrites sont menées sur une solution de teneur coswe (tracé de la courbe de rendementlpuis sur l'échantillon inconnu.
* Mesure de l'actlvitt PET- scintillation llouide :
_ La solution scintillante est celle utilisée en n .2 .1 . On trace la courbe de rendement i flo fin -1 no
R « g <-==- ) à partir d'une solution étalon Sr + Y i 1 uci ml en Sr sur laquelle on a effec-2 on
tué la séparation du Sr. La figure II. 7 nous montre la courbe d'étalonnage obtenue à partir d'une série affai
blie constituée de la façon suivante : - 3 prélèvements non affaiblis d« 0,1 ml en phase H NO 3 N + 0,3 ml H.O (de façon A ttre en
milieu nitrique normal) sont incorporés à 15 ml de solution scintillante. L'activité de chaque prélèvement compte tenu de la dilution due * la méthode de séparation est 10 naaocuries.
- Les échantillon hunea de H NO, N de 0 * 1,5 ml par 1/10 de ml soit 0.1, 0 ,2 . . . soit la échantillons par série. Pour chique échantillon on obtient :
- un taux de comptage : on accède ainsi au rendement — N mu B - B—— r

- un rapport E. /E- . On peut donc tracer la courbe de rendement (fig. H,7).
90 • Le tableau m donne un exemple de dosage effectué sur une solution inconnue de Sr séparé de
- Contrôle de la méthode :
90 80 "1 90 Sur une solution de Sr + "Y à 4,1 (ici ml en Sr, on prélève 2 prises d'eBeaia
distinctes sur lesquelles on applique la méthode de séparation : on mesure l'activité 0~ de 2 séries de 5 échantillon* en phase nitrique dissous dans la solution scintillante ; l * s valeurs du rapport E./E» obtenues avec les taux de comptage sont reportées mur la courbe de rendement. On en déduit l'activité A - -g- en dés. mn"1
On eiprimar* alternent l'activité en ne* <nanocuries} ; Le tableau m nous donne les
résultats obtenus
avec R • 0,97 N j • 23 796
5 ^ - 2 3 964
A - Jii î i 1 0 .97
• 2 4 MO
2 0,97 • 24 599
La prise d'essai que l'on compte m subi la dilution suivante :
Zî ± î y • . • c • e ' . 2
V, - pria* d'essai initiait - 0,5 ml a « volume d. dilution pour precipitation - 10 ml
b • reprit* du surnageant • 5 ml c * volume de la phase d ^«.traction TTA/hexone » 5 ml d • reprise phase organ qu». > 3 ml e - volume phase de reextraction nitrique 3 N > s ml V, * reprise pour comptage.
0.5 5 3 ' • ' . , . 0,05 ', 0.25 10 * 5 * 5 - ' 20 100
A i " 257Îo"ooo ' " » S 1 4 0 0 ° « « • • m n " 1 °» « • « ""* "i1

-61 -
3 a s / 10* 4 42 - 4 11 90
L'erreur sur la meaure ' ' * 8 % «'expliqua par la crotaaancc de Y : la aéparation du strontium prend environ 1 heure, temps auquel on doit ajouter la preparation des flacone de comptage et le tempa de leur refroidissement (le comptage a été effectué dans un appareil muni d'une régulation de température a + 15 *C) on peut estimer le temps écoulé de la précipitation jusqu'au comptage, * 3 heures,
SO &0
La courbe joint* [31] montre la formation de Y par rapport au Sr initial : au
bout de 3 1 4 heures, nous avons déjà 4 » S % de W Y formé (et. fig. II.8).
- soit 4 ,11 X 0,04 « 0,1«44 «cl
que l'on doit déduire de l'activité Sr : 4,42 - 0,16 " 4,26 uci. On a alora une erreur de :
4,11 4,11 " .
n.2.3. Conclusion.
S0 90 La méthode de dosage du Sr sans séparation du Y est rapide dès lors que sont
On Ml
établies la courbe de variation du facteur de contribution de Y dans " s r , B,
K - f ( - a 1 - ) • »
puis la courbe de rendement
T.
R-!(-r"> 2
Le résultat dépend d* deux facteurs que l'on détermine sur deux abaques, pour un affaiblissement I J / E J donné : la masure du (acteur K eat très importante pour la précision de la mesure et les courbes K et R doivent être tracées avec beaucoup de soin.
Pour des échantillons peu affaiblie, on obtient une précision satisfaisante : la méthode semble donc convenir pour les dosages de routine où la rapidité de réponse est important* ; on la pratiquera couramment sur des échantillons tels que les effluents du procédé.
Pour lea échantillons tela que solutions d'éhstion, concentrate d* strontium, servant A l'établiasement du biles de production, on aura intérêt * pratiquer - après dilution - la method* avec aéparation chimique de l'vttrium : la mesure du strontium M set alora possible t partir de la seul* courbe du rendement de comptage en fonction du Qa—cbinf.
La séparation doit cependant (ire mené* assez rapidement de façon t éviter l'interférence de l'vttrium en formation sur la mesure.
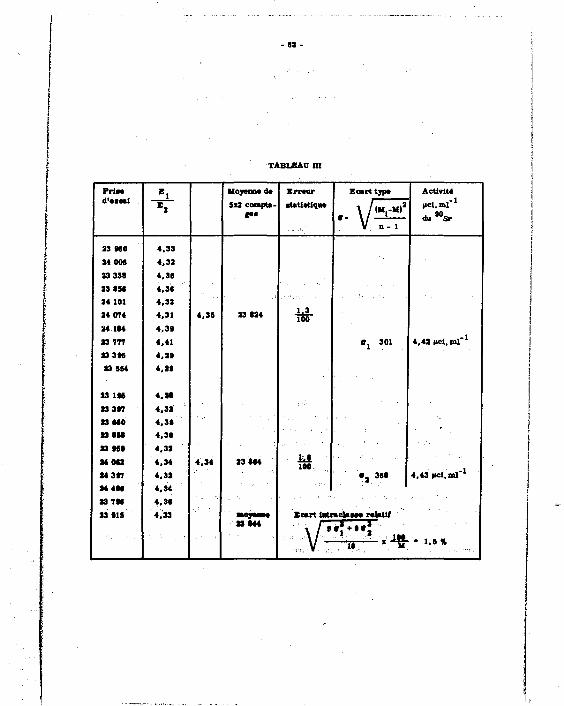
TABLEAU m j !
PriM d'atatl
B l
« 2
Moyenoada Sx2 coctpt»-
6»«
Erraur j Ecart typa Activité
ixci.ml"
Ai**Sr
PriM d'atatl
B l
« 2
Moyenoada Sx2 coctpt»-
6»« •Uttatiqa* \/<"r»a
Activité
ixci.ml"
Ai**Sr
PriM d'atatl
B l
« 2
Moyenoada Sx2 coctpt»-
6»« •Uttatiqa*
V - i
Activité
ixci.ml"
Ai**Sr
23 MO 4,33
24 006 4 ,32
23 33S 4 ,36
23 696 4,36 24 101 4 ,33 24 074 4 ,31 4 ,38 23 624 1.3
100 24 164 4 ,36
23 777 4 ,41 Oj 301 4,4S MCI. ml" 1
23 3 M 4 ,26 23 SS4 4 ,26
13 1M 4 .26
23 317 4 . 3 2 23 660 4 , 3 6 23 616 4,36
23 666 4 ,33
34 062
24 367
4 ,34
4 .32
4 .34 23 664 ' U. • . 366 4 .43 pet. ml" 1
24 4M 4 ,34 • •••••- 1 ' ' "
23 766 4 ,36 23 615 4 .33 • ajaaai
23 644 Ecart iatradaaaa rakt i f 23 615 4 .33 • ajaaai
23 644 V " ' » ' ' ^ » 1 , 5%
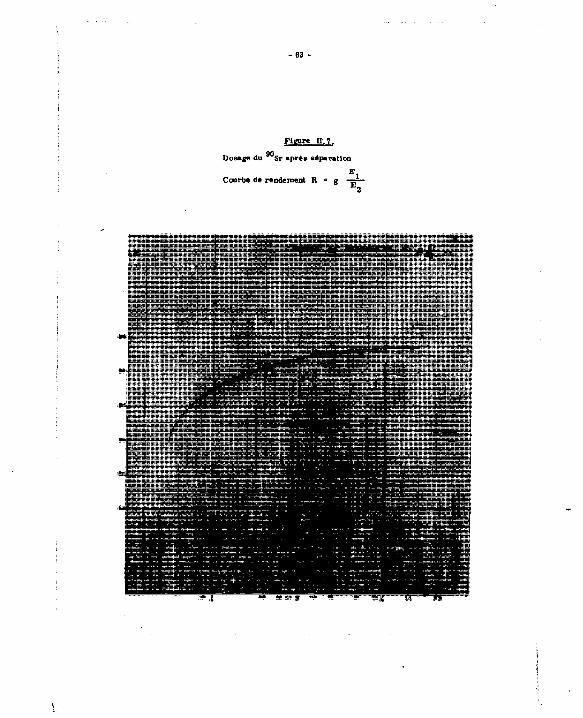
Figure II. 7 T
,90, Dosage du Sr «près séparation
Courbe de rendement R « g
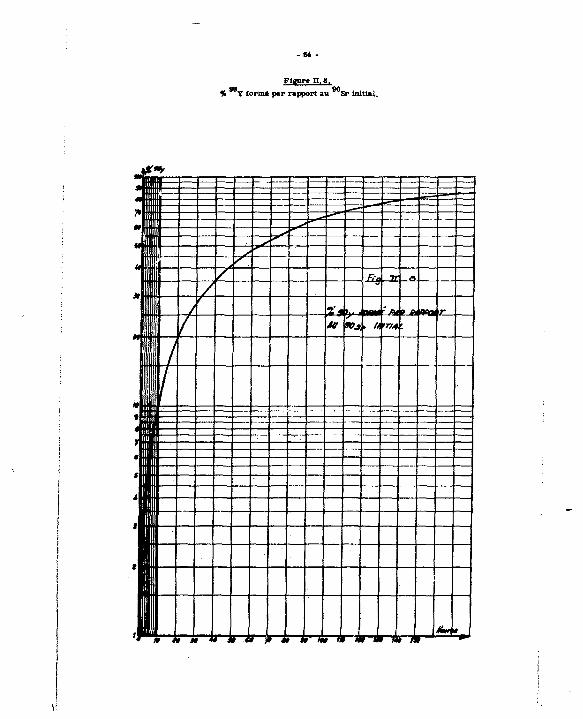
- M -
Figure n. g. * " y formé p»r rapport au " s r Initiai.
«£ «y — —
= r - -
Inj - -
Mu
a 1 !, 1 R-<5 7T a
/ Î 1 '.' • . A W *
» A « » i r
i l / «tt W j . A W * :
1/ ill
* D t M
/ •
ê M -: — —
n 1
'™ n » « F-i n n >"" r-S - i " * n r l n ni n r- * . !*.
f

III. l . ANALYiŒ) SPECTROGRAFHÏQUE - METHODE GENERALE D'EXPLOITATION DES
PLAQUES PHOTOGRAPHIQT^S.
Les méthode* de spectrograph^ d'émission impliquent la comparaison des échantil
lons A doser A des échantillons de référence.
L'analyse se fait par la méthode du standard interne introduite par GERLACH [32],
où C e i t lu différence de densité optique entre deux raies homologues qui est une fonction de con
centration.
Ainsi , dans le système binaire constitué par l'étalon interne A et le corps à doser B,
la relation entre l'intensité des raies et In concentration est de la forme :
I_ L. et L sont l e s intensités de deux raies homologues de A et
_ I . B et de B, respectivement
K et m sont des constantes
soit log J& - Tog Ig - log I A - m log C + K [2]
Comme, d'autre part, le noircissement photographique est relié A l'éclalrement at
teignant la plaque par l'expression
Do Y » facteur de contraste - pente de la droite caractéristique T * 1 0 « E d'émul.ion
Do * densité optique aa noircissement
E * K • éclairexnent avec
I ' intensité lumineuse de la raie
t • temps d'exposition
- Do * T i ogE = Y log It est l'équation de la courbe "caractéristique" de plaque -
soit D o , la densité optique obtenue avec la raie de l'étalon interne A
D o . * t log 1. t
D o B - T log Ig t
i t • d e , i l vient :
Do„
'A
égalité qui nous donne la relation entre '.a différence des densités optiques et la concentration.

La comparaison échantillons-étalon peut se faire à partir de spectres enregistrés
sur la même plaque.
Elle peut se faire également après calcul d'un facteur analytique et report eur une
courbe d'étalonnage (ou "courbe fixe" des spectrographistes).
Cette courbe établie une fois pour toutes, est obtenue au moyen d'un grand nombre
de "poses" enregistrées indépendamment de F échantillons et traitées statistiquement : el le per
met d'estimer une teneur et le domaine de confiance de cette estimation.
Pour nos dosages d'éléments alcallno-terreux à l'état d'impuretés dane des solutions
de strontium, nous avons utilisé la méthode dès ci.urbce fixes ou de BRECKPOT [33] dans laquelle
on détermine ;
- d'une part, la caractéristique de plaque pour les longueurs d'onde considérées ;
- d'autre part, la "différence d'efficience A s " entre la caractéristique de la raie de l'étalon in
terne et celle de la raie de chaque corps à doser.
Pour un élément donné, Ca, par exemple, on tracera la courbe d'étalonnage : diffé
rence d'efficience
A S c en fonction de la concentration en Ca.
étalon interne
III. ï . 1. Méthode de BRECKPOT - Obtention de la caractéristique de plaque.
L'obtentioi de la caractéristique ou courbe d<? réponse de 1'emulsion photographique
est fondée sur la densltométrie des spectres dégradés que l'on obtient en interposant, un secteur
tournant à échelons, entre la source de lumière et la fente d'entrée du spectrograph,
IM caractéristique sera tracée en portant en ordonnée la densité optique et en absc is
se le logarithme de r éclaire ment ou des termes qui y sont directement re l iés .
Sachant que le secteur tournant, dont l e s angles au centre des échelons successifs
croissent suivant une progression géométrique, permet d'obtenir une variation définie des temps
d'éclairé ment, on portera en abscisse des unités arbitraires qui sont, dans notre cas , le rang
de chaque échelon - cf. annexe n* 2.
La méthode demande l'éclairage uniforme de la fente d'entrée du spectrograph©
(cf. schéma) : el le exige que la vi tesse de rotation du secteur soit suffisante pour s'affranchir de
l'effet etroboscopique.
Remarque :
La caractéristique obtenue préserve, en général, une partie rectiligne entre l e s deux
régions de sous exposition et sur exposition.
L'emploi de la transformée de KAISER [34] à la place de la densité optique augmente
la partie rectiligne de la caractéristique.
Lorsque l'on photcmètre une raie, on éclaire l e s différents points de 1'emulsion par
un flux lumineux.
On a :
\
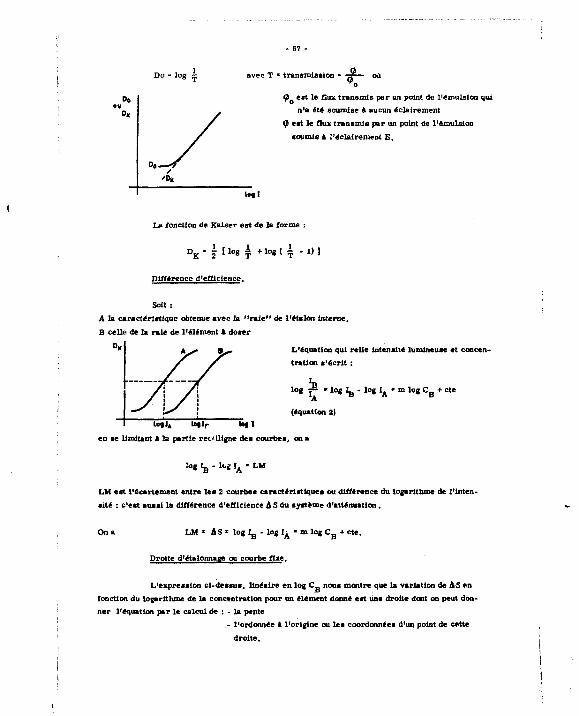
67 -
Oo u
0»
Do = log ç
0».
/
avec T * transmission * -j=£— où "o
<p est le flux transmis par un point de l'émulsion qui n'a été soumise & aucun éclajrement
Ç est le Stuc transmis par un point de l'émulsion soumis à l'éclairement E,
l.fl
La fonction de Kaiser est de la forme :
D R * | [ log i + 10g ( ç - 1) ]
Différence d'efficience.
Soit :
A la caractéristique obtenue avec la "raie" de l'étalon interne
B celle de la raie de l'élément a doser
D, L'équation qui relie Intensité lumineuse et concentration s'écrit :
log | - « log 1g - log I A « m log C + cte
(équation 2!
leaJA bal r Isa I en se limitant A la partie rec*tligne des courbes, on a
log Ig - log ! A - LM
LM est l'écartement entre les 2 courbes caractéristiques ou différence du logarithme de l'intensité : c'est aussi la différence d'efficience & S du système d'atténuation.
On a LM - AS » log L. - log I A - m log C + cte.
Droite d'étalonnage ou courbe fUe.
L'expression ci-dessus, linéaire en log C_ nous montre que la variation de AS en fonction du logarithme de la concentration pour un élément donné eat une droite dont on peut donner l'équation par le calcul de ; - la pente
- l'ordonnée à l'origine ou les coordonnées d'un point de cette droite.

- 68
Si on utilise du papier semi-logarithmique, on construit graphiquement l e s droites :
A S = m log C + cte
On porte : en ordonnée, la différence d'efficience
en abscisse , la gamme des teneurs étudiées.
Pour un élément donné, le calcium, par exemple, on tracera ainsi la droite d'étalon
nage : différence d'efficience en fonction de la concentration en calcium [36] , Cette courbe du Ca
sera établie pour diverses teneurs en Sr 0 - 5 - 10 gl~ [36].
Le faisceau de courbes obtenues sera utilisé compte tenu de la teneur en strontium,
tirée de son activité (le strontium est mesuré par comptage 0").
AinBi, pour doser Mg, Ca, Ba dans des solutions de strontium 0 - 5 - 10 gl~ (cas
du dosage d'impuretés dans une solution dont le teneur en constituant principal influe sur la
sortie des raies) on sera amené 1 constituer une gamme d'étalons Mg, Ca, Ba : dans nos e s s a i s ,
cette gamme est de 5 concentrations par élément dans chacune des solutions de strontium 0 - 5 -
10 g l" 1 .
Ce qui fait 15 étalons, chaque étalon faisant l'objet d'une plaque a 7 poses soit
105 spectres .
Dans chacun de ces spectres , on utilise la raie de l'étalon Interne + la raie de chaque
élément, soit 4 raies et donc 420 raies , avec en moyenne 3 échelons par raie, soit 1 260 photome
tries qui serviront A la détermination des faisceaux de droites d'étalonnage.
Ce grand nombre de données a été exploité sur une calculatrice Hewlett Packard
9 100 B, grâce à un programme de calcul [37] qui permet d'obtenir :
1) l'équation des caractéristiques pour chaque raie par la méthode des moindres carrés ;
2) l e s différences d'efficience pour chaque pose, leur moyenne , leur variance : l e s AS sont u -
terminées pour l e s points correspondant A une transmission de 20 pour 100.
3) l'équation de la droite d'étalonnage (moindres carrés) et l e s éléments permettant de contrôler
la validité de la régression.
Remarque :
Le programme est prévu pour effectuer la transformation de la Transmission en den-
t ité Kaiser.
III,2. Dosages envisagés,,
Le dosage de Mg, Ca, Ba par spectrographie d'émission est étudié pour des solutions
de Sr <NO.)„ d'acidité nitrique 1 N, d'activité de l'ordre de 1 000 Ci l - 1 , soit une teneur en stron-J * _ i
tium voisine de 10 gl . Ceci correspond aux teneurs attendues dans l e s solutions industrielles
obtenues après concentration de l'éluat et précipitation sélective des impuretés,
La teneur an Sr total se détermine d'après l e s comptages $ ' . On admet qu'un Ci de M S r représente 7 mg de W S r et 11 mg de Sr total [3«],

- 69 -
m. 2.1. Installation spectrographique utilisée.
/de focale Spectrographs HSV de 2 m./équipé de 2 réseaux. Noua avons utilisé pour nos essaie
-1 " °
le réseau de 1 200 traita mm .blazé pour 3 000 A. Dispersion de 4,2 A /mm. Réseau orienté pour un domaine de 2 400 A 4 500 A .
L'appareil est équipé en photographie, et peut recevoir 2 plaques 9 x 24 cm ; nous avons travaillé avec des emulsions orthochromatiques Eastman Kodak Spectral Analysis n* 1 qui conviennent au domaine spectral employé.
, L'éclairage de la fente du spectrographe est du type a image intermédiaire [36].Dans /montage,
ce montage, l'image dea électrodes est formée, non sur la fente du apectrographe, maia A une distance auffisante pour éclairer la fente de façon homogène sur toute sa hauteur (cette distance dans notre cas est 162 mm, voir schéma n* 1).
Cet éclairage peut être effectué A partir de 2 sources distinctes : - î porte électrodes classique pour échantillona inactif s ; - I porte électrodes télécommandé Intégré dans une enceinte en plomb pour les échantillons radio
actifs. L'utilisation d'un système de miroirs plans et aphérlquea permet la transmission de
la lumière. Le schéma n' 1 montre le chemin optique depuis lea 2 porte électrodes jusqu'à la
fente di spectrographe. - le dispositif atténuateur employé est un secteur tournant de marque Jsrrel-Ash de raison r • 1,58 (log r • 0,2) ; le» résultats obtenus avec cet appareil permettent une bonne définition de la courbe caractéristique d'émulsion. - le générateur électrique : c'est un ensemble "France et Gams" des Ets. Durr, qui permet l'arc continu, l'arc Jnterraptif et l'étincelle haute fréquence. Nos dosages des éléments alcallna-terreux ont été réalisés avec cette dernière forme d'excitation. Traitement des plaque» photographique» ;
Le» emulsion» utilisée» sont des Kodak du type spectral Analysis n* 1. Elles sont traitées de la façon auivante : - développement par le bain Kodak D 19 - fixage tannant par le sain Kodak F 5.
L'étude dea spectres enregistré* et leur pbotométrle est réalisée grâce A un projecteur Zeiss S P 2 et un photomètre rapide Zeiss H,
III. 3. Dosage de» alcallno-tarreux an aolutlon nitrique.
Nous avoas introduit comme étalon Interne, le béryllium, premier élément de la famille des alcalino-terreux, absent dea solutions A examiner.
Pour simplifier-le mode opératoire et éviter les erreurs dues A un changement de milieu, les dosage» «ont fait» directement es milieu nitrique, sur électrodes de graphite.
Lea éléments alcalino-terreux se comportent comme des réfractairea t ils demandent une forte énergie d'excitation fournie par l'étincelle HF [39],
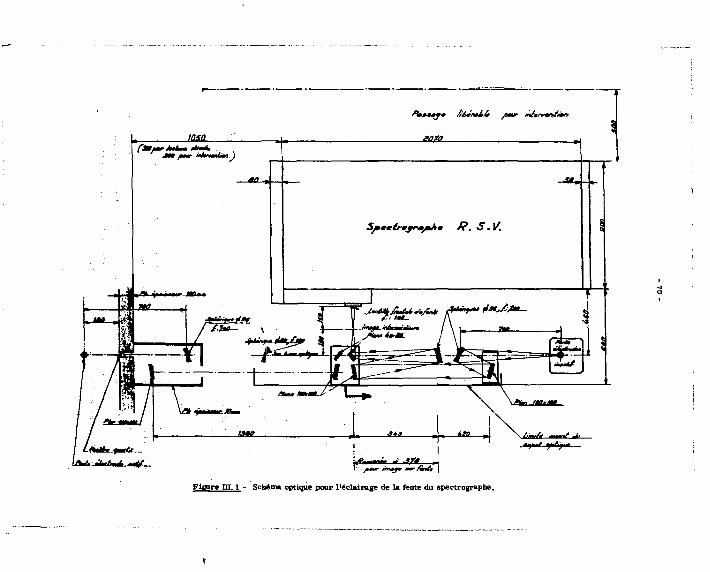
«ft—™ * AH .1 f*r *t»*J* Mir fintt |
Figure in. 1 - Schéma optique pour l'éclairage de la fente du âpectrograpbe.
\
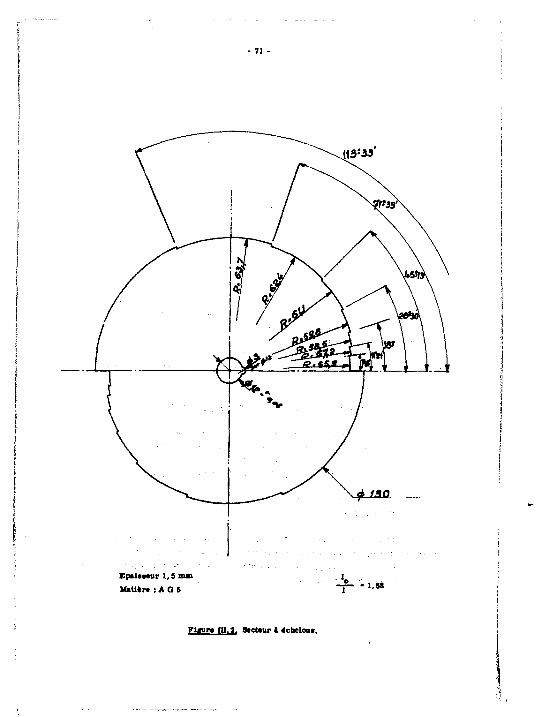
EpaiaMur 1,5 mm
MmUèr* : A G S
Figura m. a. Sactaur 4 achetai*.

On utilise les raies d'étincelle
Be 3 131,1 A
Mg 2 852,1 A
Ba 3 891,8 A
Ca « 228.T A
- Concentration» utllieéea pour le tracé dea droitea d'étalonnage : 5 gammée contenant chacune
Mg 3 - 8 - 12 - 25 - 50mgl" 1
Ca 15 - 30 - 60 - 125 • 250 Ba 30 - 80 - 125 - 250 - 500 - 1 000
1* en milieu H N 0 3 N à 0,5 mg l" 1 en 3e et 0 g de Sr 2° n n 5
3- " " 10
Remarque :
Toute» cea «olution» «ont en milieu nitrique normal, a l'exception de la aolution de béryllium. Le béryllium métallique •'attaquant trèa difficilement par lea acidea oxygénée, eat dlaaoua par l'acide chlorhydrique [40] ; l'adoption du milieu nitrique eat néceaaatre ai on veut do-aer Ag dana la aolution a examiner : or, en préeence d'iona Cl" apportée par l'étalon Be, on a un précipité. Il noua a paru intértaeant de vérifier l'influence de Ag eonble ou non, aur lea doaagea dea alcalino-terreux.
111,3.1. influence de Ag aur le doaage dea alcalino-terreux.
Noua «von» comparé - aana doaer l'argent - lea différence» d'efficience AS, leur moyenne et leur variance, obtenue» en préeence de quantité croiaaante d'Ag,
Pour cela noua avone préparé une aolution S :
1) S-Ca 50 ï , Ba2ST, Mg 5 Ï , Be 0 ,5 ïen milieu HNOjN, puie 2) S + Ag 10 t 3)S+Agl00Y 4 > S + A g l 0 0 0 ï
L'examen dea pUquea A 1 poeea enregietréea dana le domaine 2 300 - 4 800 R aur lea aolution» S et S + Ag 1 000 noua donne lea réaultata conalgnée dana le tableau ni. 1.
Cet peut en déduire que la préeence d'Ag à cea teneura ne modifie paa lea valeur» de
A S, compte tenu de l'erreur atatietique.
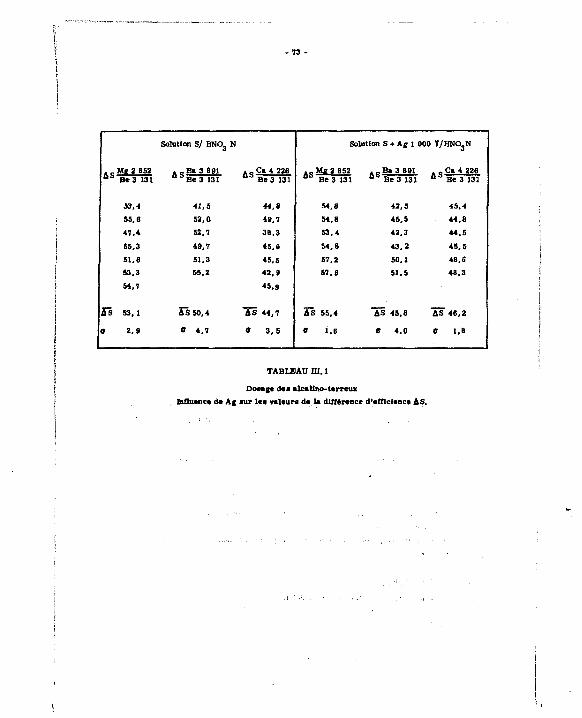
Solution S/ HN03 N Solution S + Ag l »00 T/HN03N
M g 2 852 a* Be 3 131
> . E > ] 891 Û S B e 3 131
. _ Ca 4 226 . „ Mg 3 852 à S Be 3 131
. „B»3 891 Û S Be 3 131
. „ Ca 4 226 6 Ù B e 3 131
M g 2 852 a* Be 3 131
> . E > ] 891 Û S B e 3 131 0 " Be 3 131
. „ Mg 3 852 à S Be 3 131
. „B»3 891 Û S Be 3 131
. „ Ca 4 226 6 Ù B e 3 131
S3,4 41, S 44,8 54.8 42,5 45.4
55.6 52,0 4», 7 54.8 45,5 44,8
47,4 52,7 38,3 53,4 42,3 44.5
65,3 49,7 45,6 54,8 43,2 45,5 SI. 6 51,3 45,5 57,2 50,1 48,6 53,3 55,2 42,8 57.8 51.5 48,3
54,7 45.9
2S S3, 1 Ï S 50,4 "AS 44,7 AS 55,4 "ÏS 45,8 "ES 46,2
0 2,9 0 4,7 « 3,5 0 1,6 O 4.0 0 1,8
TABLEAU UI. 1
Dotage des alcalino-terreux Influence de Ag *ur le» valeurs de la différence d'efficience As.

- 74 -
Remarque : La présence du précipité Ag Cl dana lea solutions 3 et 4 eat sans influence sur le
dosage de Mg, Ca, Ba comme le montrent les valeurs de Ks pour les trois alcalino-terreux : ceci nous a permis d'étudier leur dosage sur dea solutions ne contenant pas d'argent.
ni,3,2, ^^term^^ion^s^^ro^te^^^JaJonna^e^m^Bure.
Principe :
Dépôt de la aolution nitrique dea étalons additionnée de Be aur électrodes graphite. (On opère de la même façon pour les échantillons A doser).
Emiesion'd'une étincelle BF entre ces électrodes - Enregiatrement dea apectrea aur plaque photographique, denaitomélri*, détermination dea teneura au moyen d'un faiaceau de droites d'étalonnage.
Matériel : - Electrodea de graphita Kingadc-rf ^ 8,15 mm imprégnées par une solution de
graisse Apiéxon N 3/1000 dana le toluene . - Plaques photographiques Eastman Kodak SA 1 - Pipettes Eppendorf de 50 microlitres - Séchoir 4 électrodes en graphite : la plaque en graphite est pourvue de logements
pour él«ctrod«a : le chauffage eat obtenu par un* réaiatance incorporée av«c régulation de température.
Mod* opératoire ;
a) Dépôt sur électrodes d* graphite (sécha «lectrod* régulé A 100 *C) » raison d* 50 microlitres aur chaque électrode. Un* foi* séché** les électrode* «ont placées sur 1* port* électrode» deux par deux, avec un* entrode d* 2 mm. Fair* 7 poaea par étalon.
b) Excitation - Générateur d'étincelles haut* fréquence, intensité 10 A. Capacité 9 000 pF,
Self «0 wH. Duré* «0 a. Réglages spectrographiqses.
- réseau 1 200 traits mm" . - angl» 102*7, ce qui correspond A un* région spactrala d* 2 600 A 4 600 A. - largeur d* fente 40 microns. - hauteur d* fente 10 mm permettant l'usage d'un dispositif atténuateur A 7 échelons d* 1,5 mm/échelon.
c) Traitomentojti Jd*qu*a :
- développement : durée 4 madens le bein Kodak D 19
température : 20 *C. - rinçage A l'eau.

7 5 -
- fixage dana le bain Kodak F 5 : durée 10 mn - lavage à l'eau courante : 10 mn - séchage : séchoir à plaques à courant d'air à 35"C.
d) Exploitation dea spectres :
Après pbotométrie des spectres correspondant à chaque solution étalon et rédaction de la feuille de densitométrie, on détermine la différence d'efficience A S entre la raie du Be et chacune des raies Mg, Ba, Ca.
Pour chaque élément alcalino-terreux, on trace enauite les dr n*~s <r '-talonnage dana chaque solution de strontium examinée, suivant la méthode de Breckpot décrite précédemment (cf. fig. III. 3, 4, 5).
L'exploitation de ces droites peut être effectuée graphiquement ou au moyen d'un programme de calcul, '
1) méthode graphique :
De l'activité d'un échantillon inconnu, on connaît la teneur en strontium. Par interpolation entre les teneurs en Sr dea étalons (0 - 5 - 10 gl" ). on tracera une
droite correspondant * la solution examinée. Oh reportera la moyenne AS dea AS obtenues.
On en déduira les teneurs en Mg, Ca, Ba, En reportant l'écart type des différents A S par rapport * la moyenne T s , on obtien
dra un intervalle de confiance de la mesure,
2) méthode par le calcul :
Cette méthode est tirée du volume "Etalonnage" des méthodes statistiques en Analyse, publiées par la CETAMA {Commission d'Etablissement des méthodes d'Analyse du CEA)[41].
La concentration peut être déduite de l'équation
y • AS • mx + p avec x* f (log C)
Connaissant AS, on tire x, puis C • concentration cherchée. Un calcul de variance sur la valeur moyenne de y • 7 3 , permet de déterminer
l'intervalle de confiance de la mesure
c + Ac
Pour obtenir une meilleure estimation du résultat, on procède * :
1) une validation de la droite de régression obtenue au moyen de trois tests de variance ;

2) un calcul de variance utlliaant toutes lea déterminations de A S (et non phis de la . oyenne AS) pour arriver au résultat.
Cette deuxième méthode fait l'objet de l'étude ci-apré» :
TU. 4. Etude dea droitaa d'étalonnage.
Las droitaa d'étalonnag* obtenu**, dont nous donnons le* équation*, sont le* droite* d* réfreaaion établi** par la method* de* moindre* carré*, à partir d*i meaurea expérimentale* (cf. tableaux m. 2, 3).
C** équation* *oat d* la forme :
y* As • p + mx où x - 125 log C 135 e*t une constante introduite pour dilater l'échelle dea concentration*.
m.4.1 . 2e*UjJe^UditejdeJa_rfgrej*ion.
Pour vérifier que le* valeurs expérimental** «ont compatible* av*c l'existence d'une droit* de réfresslon, il faut étudier la liaison entre x et y et effectuer une analyse de variance [éz].
Les fluctuation* d* y : (y, - y ) sont décomposées en deux parties : - variations due* t la régression y 1, - y - variations residual]** autour d* la droit* de
réfresaion ^ - y'â
1\
Js T TV» 7 — . ^ . \
1 1 1
- 1 - *
7 *l x
Ona :
variance total*
i»
n - 1 s? -2 n - 1
y, • vatoers *xpérim*ntal*a de y • AS
y • valeur moyeu* n " nombre d* détermination
loi à D - 1 degré* de liberté.
- variance do* a la régression
s? - z ( Ï -J-S 1 -»» lu , - : )* i , J j x, • vateiir* d* x pour chaque concentration
x • mbyesm de* valeurs
loi à Vj - l degré de liberté
Figura III. 3
Droites d'étalonnage du magnésium
5 iHié*- ,•»: !Jii--i*,-iiii#jia»SÉ;fiiÊns;!!Sll!)l)!!l • _:ini:liP' ;\ •N*-^ ÏBilIJiltaiSIrdii-fc-^SiîiiiBli!
. •;• !" * •,: r-Stm iiiSiiilBiSElSSiSS! « ^ SiyaiSi'njiS: < i|..--.;;• i, ^Bi;^iii^!ipi|li|l^:i!nt*,aljl..*ii5.|ji*!;ig
" « ' i i l S l i i î i : ":;»'• !'': !;• •"!•. ,> •••„ „.,• • • • , . .
lW-ri

- 78
Figure m, 4,
Droites d'étalonnage ou baryum
Figure HI. 5-
Droi tes d'étalonnage du calcium.
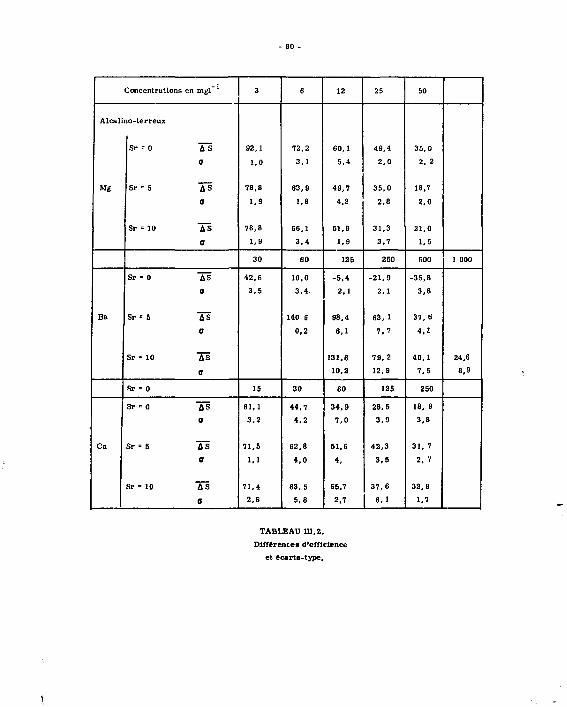
- 80 -
Concentrations en mgl" 3 6 12 25 50
Alcali
Mg
no-terreux
Sr = 0 T s 0
Sr = 5 Ts 0
Sr - 10 "ÏS
a
92,1
1,0
78,8
1,8
78,B
1,9
72.2
3 .1
63 ,9
1,8
66,1
3 , 4
60,1
5,4
49,7
4,2
51,8
1,9
49,4
2 .0
35 ,0
2.8
31 ,3
3 .7
35 ,0
2, 2
18,7
2 ,0
21,0
1,5
30 60 125 250 500 1 000
Ba
Sr = 0 Ts 0
Sr * 5 T i c
Sr " 10 TS a
42,6
3 ,5
16,0
3 ,4 .
140 6
0,2
- 5 , 4
2,1
98,4
6,1
131,6
10,2
-21 ,9
2 ,1
63, 1
7 , 7
79, 2
12.9
-35 ,8
3 ,8
37, 6
4 ,1
40 ,1
7,5
24,6
8,9
Sr = 0 15 30 60 125 250
Ca
Sr = 0 Ts a
Sr = 5 T s a
Sr = 10 T s a
61,1
3 ,2
71 ,5
1,1
71 ,4
2 ,6
44 ,7
4 , 2
62,8
4 , 0
6 3 , 5
5 ,8
34 ,9
7 ,0
51,6
4,
55,7
2,7
28,5
3 , 9
42 ,3
3 , 5
37 ,6
6 ,1
18, 9
3 ,8
31, 7
2, ï
32 ,9
1,7
TABLEAU ID. 2. Différences d'efficience
et écarte-type.
\
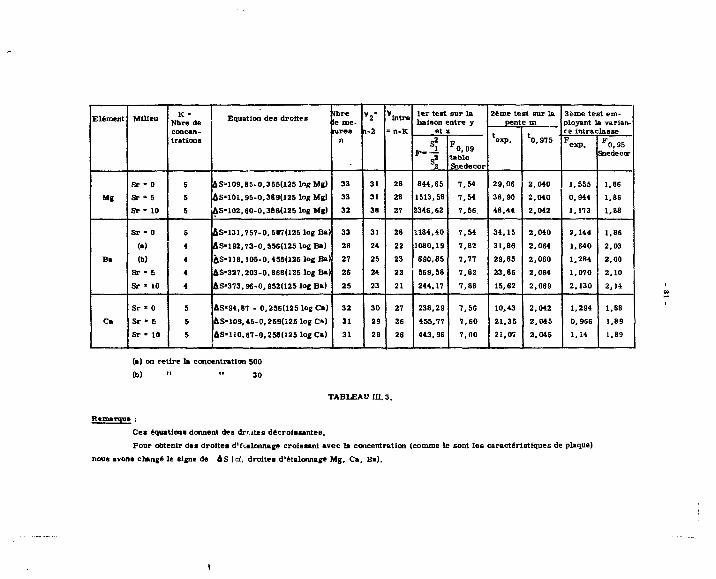
Elément Milieu K -Nbre de concentrations
Ibre v * Equation dee droites . _ 2
rares 1-2 n
V intra
- n-K
1er test sur la liaison entre y
2ème test sur la pente m
3ème test employant la varianElément Milieu K -
Nbre de concentrations
Ibre v * Equation dee droites . _ 2
rares 1-2 n
V intra
- n-K et x t exp. 'o ,975 ce intr&classe
Elément Milieu K -Nbre de concentrations
Ibre v * Equation dee droites . _ 2
rares 1-2 n
V intra
- n-K
F 0.99
table Snedecor
t exp. 'o ,975 P exp. F 0,35 Snedecor
Mg
Sr - 0
* » 5
Sr - 10
5
5
S
ttS=109,85-0,355(125 log Mg)
&S"101,35-0.389(125 log Mg)
ÛS-102,60-0,386(125 log Mg)
33
33
32
31
31
30
28
28
27
844,65
1513,58
2346,62
7.54
7 ,54
7 ,56
29,06
38,90
48,44
2.040
2,040
2,042
1,555
0.944
1,173
1,86
1,86
1,88
Ba
Sr - 0
(») (b)
Sr = 5
Sr » 10
5
4
4
4
4
&S-131,757-0,507(125 log B»)
SS-192,73-0,556(125 log Ba)
ûS-116.105-0.455(125 log Ba)
&S-327.203-0,868(125 log Ba)
6.S-373,96-0,952(125 log Ba)
33
26
27
26
25
31
24
25
24
23
28
22
23
22
21
1184.40
1080,19
690.85
569.58
244.17
7 ,54
7 ,82
7,77
7 ,82
7,88
34,15
31,86
29.85
23.86
15,62
2,040
2,064
2,060
2,064
2,069
2,144
1,640
1,284
1,070
2 .130
1.86
2,03
2,00
2,10
2,14
Ca
Sr « 0
Sr • 5
Sr - 10
5
5
5
AS-94,87 - 0,256(125 log C»)
D.S-109,45-0,259(125 log Ca)
nSMIO,67-0.258(125 log Ca)
32
31
31
30
29
29
27
26
26
238.29
455,77
443, 96
7,56
7 ,60
7 ,00
10,43
21,35
21,07
2,042
2 ,045
2,045
1,294
0.966
1,14
1,88
1,89
1.89
(a) on retire la concentration 500
(b) " •' 30
TABLEAU IU. 3.
Remarque :
Ces équations donnent des drr ttes décroiasantes.
Four obtenir des droites d'étalonnage croissant avec la concentration (comme le sont les caractéristiques de plaque)
noue avons changé le signe de AS (si\ droites d'étalonnage Mg, Ca, Ba).
I

- variance résiduelle
I <»i-y',)2 2 ( y , - y ) 2 - m 2 I f e - î ) 2
i _ . __! L J
loi a V = n - 2 degrés de liberté. 2
Nous avons appliqué les trois tests de validité de la régression suivant les indications
fournies dans le volume - Etalonnage des méthodes statistiques en Chimie Analytique de la CE TA
MA [41].
1) Test utilisant l'analyse de variance : on détermine un rapport
s?
Pour vérifier qu'il existe bien une raltlon r.rtre x et y , ce rapport F , doit être supé
rieur à une limite F . donnée par la u b l e de Snedecor [41], Cette table donne l e s valeurs de
F . . pour la valeur de Po choisie * 0,99 , en fonction des nombres de degré de liberté 1 1* T
Dans le tableau m . 3 , nous avons groupé l e s différentes valeurs de F , trouvées pour
nos droites de régression : dans tous ces cas :
Il y a bien une relation entre y • âS et z - probabilité 89 %,
2) Test étudiant la pente m de la droite :
Une estimation de la variance de m est donnée par :
= n- avec si * variance résiduelle de y.
1 ' L'intervalle de confiance, autour de m est défini par la loi de Student
avec t „ f valeur du coefficient t dam la table de
2 Student pour un aeuil de confiance ft
(« • 5 % pour une probabilité P * 95 %. )
Le teat aéra ilgnificatif et la pente trouvée «era acceptée ai :

- 83 -
dent les coefficients m trouvés dans les équations des droites d'étalonnage avec une probabilité de 95 %,
3) Test employant la variance intra classe :
Pour vérifier si les droites calculées sont bien les droites de régression, on détermine le rapport :
2 2 S. où S_ est la variance résiduelle des y, =ÛS, autour de la
expér -2 droite de régression intra est la variance intra classe
Si n est le nombre total de déterminations
y i l ' y i 2 ' * " yiK v *- '* u r - d c v d e moyenne y, pour chaque x.
<y„ - y , ) 2 + (y4- - y , ) 2 + . . . + <yÏK. - y v >2
intra
Le rapport ^ « ^ , ^ , ^ 1 d o i t * t r * inférieur [41] à la valeur F p donnée par les tables de Snedecor pour V. • VY,t «t V ' n - 2 degrés de liberté, et pour une probabilité choisie P * 0.95. Si F < F—, la droite de régression est bien représentative de la variation étudiée.
exp. P • r
Si F > Fp, on ne peut pas dire que la courbe d'étalonnage est représentée par la. droite de régression.
Les valeurs de F trouvées sont consignées dans le tableau III. 3. : elles permettent de tirer les conclusions suivantes :
Cas du magnésium :
Mf en milieu Sr - 0 d o œ i j M d e c o n c t n t r l l t i o o ^ v à l t
U, " S r - B . 1 - 1 a - . - W - Î B - S O m , ! - 1
M( " Sr - 10 g f *
Lee trois droite» de régre»ion •ont lea droite» d'étilonnife (probabilité 95 %).

- 84 -
Cas du calcium ;
Ca en milieu Sr - 0 domaine de concentration étudié c * " *-«H" is-ao-eo-m^BOmgi- 1
Ca " Sr - 10 gl
Les trois droites de régression tracées sent également validées.
Cas du baryum :
Pour le domaine de concentration : 30 - 60 - 125 - 250 - 500 mgl"
expérimental P
L'équation de la droite correspondante n'est pas validée ; noue avons donc calculé
l'équation de cette droite en supprimant alternativement l e s teneurs en début et fin de gamme, soit:
- l e s ÙS obtenues avec la concentration 500 mgl"
" » " 30 mgl" 1
Nous retiendrons comme équation de la droite d'étalonnage, celle pour laquelle
expér imenta l " * m m J œ u m s F - 1 .M4, c'est-à-dire :
A S * 1 1 6 , 1 0 - 0,455 (125 log | B a | ) ((b) dans le tableau III.3.)
1
Pour l e s concentration
représentée par l'équation trouvée.
Pour l e s concentrations 3 a • 60 - 12b - 250 - 500 mgl" , la droite d'étalonnage est
La droite de régression est également validée pour les domaines de concentrations
125 - 250 - 500 - 1 000 mgl" 1 .
On remarque la baisse de sensibilité dans le dosage du baryum, en fonction de la
teneur en strontium - inconvénient mineur puisqu'on attend Ba/Sr 8 - 10 %.
Le calcul confirme bien l'expérience : i l es t difficile de photomètrer les raies très
du fc»
opéré.
faibles du baryum à des concentrations Inférieures à 125 mgl" , dans l e s conditions où nous avons
4) Utili lation des droites d'étalonnage : Précision de la mesure.
Cbaque droite d'étalonnage est définie par l'équation : y * A S * mx + p.
Un calcul de variance effectué sur AS permet de définir la variance sur at * S , puis
une loi de Student
avec P - 0,95
t • variab
qui donne l'intervalle de confiance sur x, donc sur la concentration C.
% - S x * *P t • variable de Student

- « 5 -
On trouvera ainsi la précision de la mesure pour cbaque teneur en Mg, Ca, Ba dans chaque solution de strontium examinée.
Elle est de 10 à 15 % pour le magnésium, le calcium 12 A 20 % pour le baryum
- Cf. Annexe 1 - Calcul de C + &C dans le cas d'un AS considéré sur la droite du magnésium, en milieu strontium 5 gl .

UH.2. Annexe 1
Toua lea calculs relatif» aux droltea de regression ont été effectuée sur une calculatrice HP 9100 B : 1* déroulement du programme de calcul peut être rulvi aur l'exemple de la droite d'étalonnage du Mg (solution de Sr a 5 fl" 1 {H N 0 3 H).
Lea réaultata sont réauméa dana le tableau III. 4. On en déduit :
. - - i f f i p i . 49.64 (I)
. ï - l ^ S J O . ,34,63 VU
V n ^ x y . - X x l y y _ _ - 2.(x t - xi ( y j - y) ^ * S- - 2. x t y t - x 2.y 4
• 113 511,85 - 134,63 . 1 638,01 - - 37 014,09 (m)
„T_a T_2
• G«3 401,60 - 134,63 , 44 420,80 • 95 365,35 (IV)
v - 3 * y i y i y 3 - y y"y,
' 95 «80,94 - 49,64 . 1 638,01 • 14 675, 58
Pente de ]a droite dea moindres carrée
m . ML - -37 013.43 IV «5 365,35
3' -l3WJio£Jejle_drolte
y • 49,64 • 0,38» (X - U4.63) • 101,95- 0,389 x.
3' - lvî*.iLvi&&ÈJsJi.âs!Ssj
- a) Variance due a la régreaaion : S* • m £(x , - x)
.{Ih^L^L. .Jt . <»<W .14 35.04 2 ( x - î ) 8 W «»Ï6$,3S 14 385,0*
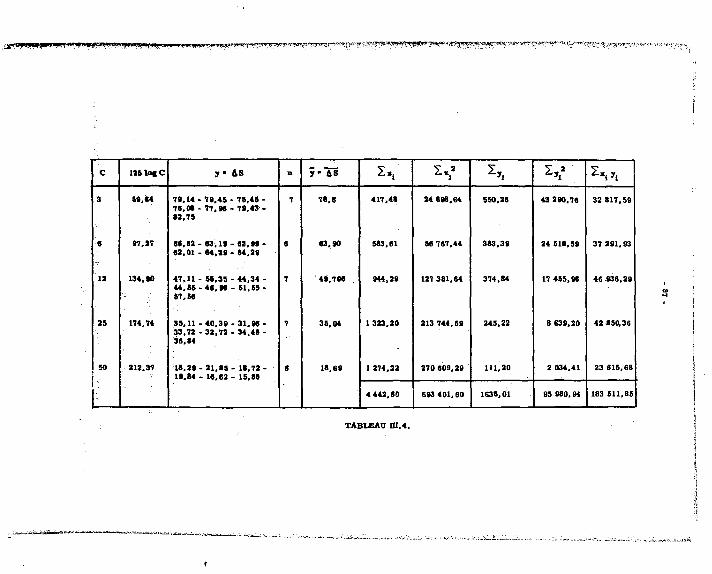
rgt&mXMfWJWW- 'milff9ms^V^Vh^>^amf9^^WM''m,w'W «gwa-y^r***;*^^^ J v q ^ r e ^ r v ^ r ^ ^ r ^ - T - ^ "'"•X-^-^i^^r.
c 125 log C y As n y - AS 2*/ V ^ V ^*ih
3 5», «4 78,14 - 78,45 - 75,45 -76,08-77. »8 - 72,43--(2,75
7 78.6 417,41 24 898,64 550.26 43 290.76 32 «17,59
« 47.27 ««,•2 - «3 ,1* - 62.88-62,01 - «4 ,22- «4,2»
S «3,90 5(3,61 56 787,44 3(3,39 24 510,59 37 291,93
12 134,50 47,11 - 55 .35-44 ,34-44.55-4C,M - 51,55-57,5«
7 '49,706 944,28 127 381,64 374,(4 17 455,98 46 936.29
25 174,74 35,11 -40,38 -31,96 -33,72 - 32,72 - 34,48 -36,(4
7 35,04 1 323.20 213 744,58 245,22 8 639,20 42 (50,36
50 212,3? 1 5 . 2 » - 2 1 , ( 5 - K . 7 2 -1S.84 - 15.62 - 15,15
6 16.69 1 274,22 270 609,29 111,20 2 034,41 23 615,68 212,3? 1 5 . 2 » - 2 1 , ( 5 - K . 7 2 -1S.84 - 15.62 - 15,15
6 16.69
4 442,60 693 401,60 1638,01 95 980.94 183 511,85
TABLEAU m.4.

Variance réiiduclle :
"2 n-2 „« , ^-(yj-y) - m <»!-»> _ 14 675.58- 19 3B5.04 . . . . 2 n-2 31 "' "
M F j ' 1 4 3f*'fM ' * 5 1 3 - 5*. On a bien Fj > P 0 9 g - 7,54 (valeur donnée par la table
de Snedecor, pour V • 1 et V > 31 degréa de liberté.
Variance eur m (pente de la droite)
m .2 ï ) 2
9.49 m .2 ï ) 2 95 265,25
S m * 8, M . i o - 3 d'où
t » m " 0.389 t » m "
9,98 . 10 .3" ' M , S
0 n > t > t . > 2,04 (tablede Studentlpou.'i> - O.SSct V >31 defrét de liberté. Le teat eat eignittcatif.
2 y* Variance iiitricla.ee : (n - K) . &?„„. - 2 yf4 - 2 j-J—
• c ) ' i j
«. sL„ • « *o.»* - [Wfif+p»3.™3
(347,94^. <»45,H)3
+ (!»,?»>' ) 7 , 7 8 '
Rapport F e x p (Snedecor) : F » -fg^§j- " 0.943
S o i t F . „ < F „ . . - 1,86 (table abedecor pour V._._ - 28 et v . "31). La droite calculée eat eap. o, so intra 2 bien la arolte de regreaalon.
- Calcul de la concentration et de l'Intervalle de confiance :
Sur la droite d'étalonnage du magn4alum, en mill u Sr ' 5 gl'1
y - AS • - 0,389 X + 101.95
Noua cherchon» la concentration correspondante a 5s" • 35,04 CàS obtenu pour la concentration 25 mgi"1 en Mf). Cf. Ubleau HI. 2.

" 5 S - y - 3 5 , 0 4 X» 172,20
La variance «ur y est estimée par
** 2 S? • S? ( rr~ + z * ( * " " ^ , ) m t Su - varfrnc* résiduelle
y 2 o 2 <*,- *) s
n « nombre de mesurée correspondant a la détermination de T s .
B y " ' " l 7 33 95 255,25 1 , 1 M
S « 1,3351 et S x - — S m e S x - variance sur x m * pente de la droite
\ • - » • » • •
D'après la loi de Student, l'intervalle de confiance sur x est :
x + S x . t j _ &_ pour IIM probabilité de »5 % (a ' 5 %), la valeur tg m
3 donné* par la tabled* Student est t * 2,04 (V-31 de.
grés de liberté)
S x . t 0 « 6 - 3 . 4 3 . 2,04-7,00 o
Soit x + û x " 172,20 + 7.00
comme x - 136 log C
tx • 125 | lof (C + Ac) - log C [ - 125 lof (1 + -&r- )
lof (1 + ~ - ) • J j -0 ,055- lOf 1.131
•^S- » 0,13»
Dans 95 f des cas, le résultat obtenu comme moyenne de 7 mesures sera écarté de
: la valeur vraie de moins de 14 £ On a , lof C • i j | * î - l ,S i - lof ,*s,I C - 23,t
4C • 25,* . 0,15» » . 1 , 1 . . . . . . . Pour un échantillon k 25 mfl~ *n Mj (Sr • 5 gl" ) le'résultat trouvé sur là droite de
régression est : 2 4 + 3 m».

- 90 -
Annexe 2
Détermination dea caractéri«tique», de» difference» d'cffieience.de leur moyenne et l'écart type correspondant.
A partir de» plaque» à 7 pose», on détermine pour chaque élément et pour létalon interne le» droites de régression qui représentent le» caractéristique» d'emulsion pour chaque rue.
L'écart entre le» caractéristique» d'emulsion pour la raie de l'étalon interne et celle
du corps examiné est A S : on en exprima la moyenne AS et l'écart type
Noua alloua traiter, en example, cea différente calcula pour une xolutiou de Ca
15 m» l" 1 dana le atrontium a S g l" 1 - l'étalon intern* eat Be 0, S mg l* 1 .
Bëinarquea :
On rappelle que 1» «acteur tournant a 7 échelon» et que tea tranamiaaione meeuréea au photomètre ne sont prieea an compte, qu'entre lea valeura 50/1 000 ou 0,050 et 050/ 1 000 ou 0,*6.
On a effectué le changement de variable* auivaat :
x • N x 30 N : numéro d'identification de l'échelon
y • D. x 100 Ce changement de variable permet d'avoir pour pente dea caractéristique*,
de* valeura voiaina* d* 1 à 2 (cf. remarque 1).
Calcul de la première droite de nSgreaaion pour Be (étalon interne) par la méthode
dea moindrea carrée.
N* échelon>N Traaemiaelon T X • 20 M y - *00. j [ log i - log ( i . "3 4 0,0M «0 123,03
5 0,100 100 f7,7i
« 0,200 120 «5,05
7 0,47» 140 vt.u
DWaawreea - y et x représentée* de* quantité* aana rajjoort avec celles désignées par cette notation deaa l'autre partie du texte.

- 91
Calcul de la droite : «on équation est de la forme
{ x - z ) tv -? (x - x?
Nombre d* pointa de la droit* n » 4
2jr > 304,64 y «76,159
- • x - 4 4 0 x » U 0
£-x* • 50 400 et n x 2 - 48 400
2(x . x) - ^x* - n x 2 - 2 000 -
2 y * -29459,82 et II y 2 «23 200,4
^•(y - y) 2 -^-y 2 - o y* - 6 259.33
X x y - 3 0 003,48 et II x y - 33 509.76
2 ( x - x ) (y- y) - - 3 506,28
On en tire la pente de la droite ; m (*-*>(? - ,Y> » . t t 7 M
2(x-x)
l'ordonnée 4 l'origine p * y • m x « 298,004
Oo pent étalement calculer le coefficient de correlation £ de la droit* de régreealoi trouvée ; l'accord Idéal entre la drolta expérimental» et la droite de régreaalon eet réaliaé pour r - 1 ; dana le caa prêtent
. / v . « = T J 3 558,57 y 2(x - x)'I(y . y)2 •
991
On *p4r* de façon ideation* pour le calcium 15 mal"
N T x y
1 0,045 20 133,88
2 0,165 40 74.34 3 0,455 60 21.02 4 0,750 80 - 17,51

Nombre de points de la droite n z 4
Iy-. 211,42
u. 200
ZJ * 12 000
V - 2 4 747,75
2 „ * 5 499,41
- S 071.81 2 000
p - 179,65
1 - 5 071.81 r = 5 093,66
y 52,86
! » 50
n ï 2 - 10 000 d'où Z(x - î ) S * 2 000
n y 2 - 1 1 175,16 £(y - y) 2 * 12 972.68
n x y - 10 571.22 A(x - x)(y - y) =-5 071,81
Ce calcul ect mené «vec les 7 poses de cbique plaque photographique. On en tire un faisceau de droites caractéristiques pour Be et pour Ca.
Calcul de la différence d'efficience AS.
On calcule les absciases sur les 2 caractéristiques au point correspondant è une transmission de 20 %. La différence d'absc'sse est AS.
Si T - 20 %
y l 0 0 . i | a o g ( ^ - ) + l o g ( - ^ , - l ) |
y - 65
Ainsi, pourla première droite de l'étalon Be, on aura y * m a + p
J 0 0 * = * -'«tUSr*-•"••»•» x_ * abscisse du pt correspon-* dant à une transmission 200
sur la droite du Be de même pour le Ca
200 , «5-179.66 -Ca - 2.536 * 45,211
On fait de même pour les 7 droites
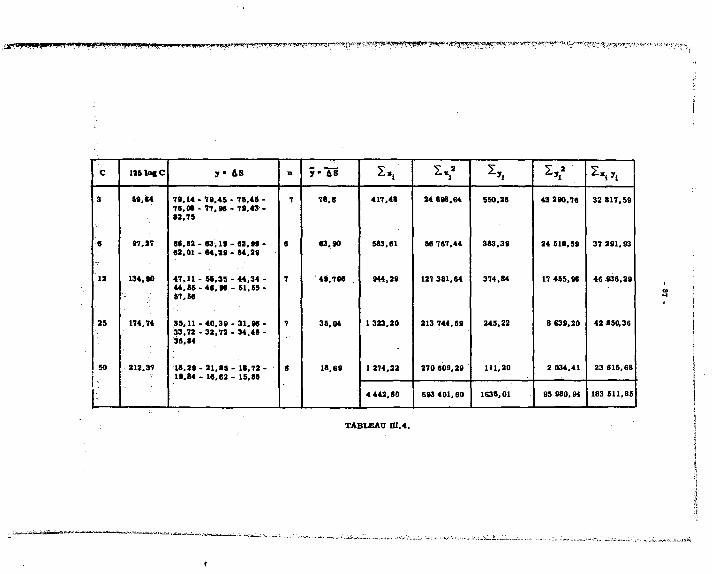
rgt&mXMfWJWW- 'milff9ms^V^Vh^>^amf9^^WM''m,w'W «gwa-y^r***;*^^^ J v q ^ r e ^ r v ^ r ^ ^ r ^ - T - ^ "'"•X-^-^i^^r.
c 125 log C y As n y - AS 2*/ V ^ V ^*ih
3 5», «4 78,14 - 78,45 - 75,45 -76,08-77. »8 - 72,43--(2,75
7 78.6 417,41 24 898,64 550.26 43 290.76 32 «17,59
« 47.27 ««,•2 - «3 ,1* - 62.88-62,01 - «4 ,22- «4,2»
S «3,90 5(3,61 56 787,44 3(3,39 24 510,59 37 291,93
12 134,50 47,11 - 55 .35-44 ,34-44.55-4C,M - 51,55-57,5«
7 '49,706 944,28 127 381,64 374,(4 17 455,98 46 936.29
25 174,74 35,11 -40,38 -31,96 -33,72 - 32,72 - 34,48 -36,(4
7 35,04 1 323.20 213 744,58 245,22 8 639,20 42 (50,36
50 212,3? 1 5 . 2 » - 2 1 , ( 5 - K . 7 2 -1S.84 - 15.62 - 15,15
6 16.69 1 274,22 270 609,29 111,20 2 034,41 23 615,68 212,3? 1 5 . 2 » - 2 1 , ( 5 - K . 7 2 -1S.84 - 15.62 - 15,15
6 16.69
4 442,60 693 401,60 1638,01 95 980.94 183 511,85
TABLEAU m.4.
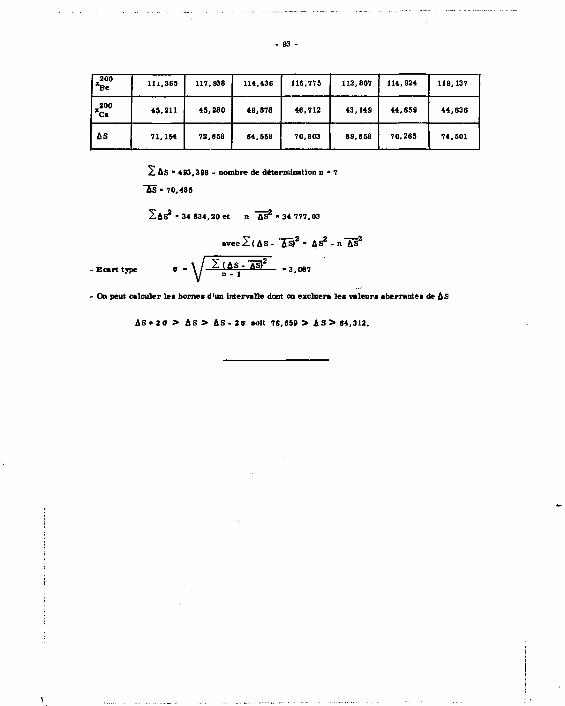
200 ^ e 111.365 117,938 114,436 116.775 112,807 114, 924 119.137
v 8 0 0 Ca 45.211 45,280 49,878 46,712 43,149 44,659 44.636
AS 71.154 72,658 64,558 70.603 69.658 70.265 74,501
Z AS * 493,396 - nombre de détermination n * 1
"SE - 70,485
Z A S 2 " 34 834,20 et n "AS* " 34 777,03
avec Z ( A S - A S ) 2 - A ^ - n " ^
- Ecart type '\p^ ai "3,087
• On peut calculer les bornes d'un intervalle dont on exclue» les valeurs aberrantea de As
AS+ 2 0- > AS > AS - 2ff soit 76,659 > AS > 64,312.
\

- 94 -
V. CONCLUSION GENERALE.
L'étude que nous avons effectuée nous a permis de définir l e s méthodes d'analyse et
l'appareillage nécessaire au contrôle du procédé de purification du strontium envisagé dans l ' ins
tallation FICPUS.
La première partie nous a permis , compte tenu des résultats précédant le début de
nos travaux, de déterminer les conditions d'une séparation des éléments de la famille des alcalino-
terreux en présence d'argent dans l e s solutions nitriques du procédé.
La formation d'un complexe argent-sulfocyanure chargé négativement, empêche la
fixation de ce composé sur la résine Biorad AG 50 W x 8 Dowex, sous forme NH. : U séparation
chromatograpbique des éléments de la famille des alcalino-terreux est ensuite, réalisée par élu-
tion au malonate d'ammonium avec gradient de concentration : l e contrôle de la composition des
fractions montre un excellent rendement de la séparation.
Four le dosnge des différentes fractions, nous avons utilisé une méthode volumétri-
que, facile A appliquer en milieu radioactif et permettant la mesure des faiblee teneurs : nous avons
choisi la conductimétrie et défini l e s conditions opératoires pour l e s alcalino-terreux et l'argent,
La deuxième partie est consacrée A la mesure de l'activité du strontium 90 par scin
tillation liquide. Cette technique caractérisée par un grand rendement de détection est d'un grand
intérêt pratique : on doit avant tout s'affranchir des phénomènes de "Quenching" dans la solution
scintillante, ce qui est réalisé avec la méthode de l'étalonnage externe.
Noua avons successivement effectué le dosage du Sr en présence de Y et le dosa
ge du Sr après séparation chimique.
La première méthode réclame la constitution de deux abaques exprimant l'une, la 90 90
variation de contribution du Y dans Sr en fonction du Quenching, l'autre, la variation du ren
dement de comptage. El le est simple puisque ne demandant aucun traitement chimique de l'échan
tillon : cependant, l'utilisation des deux abaques amène des risques d'erreur.
La méthode avec séparation donne des résultats trvts reproductibles : nous l'utilise
rons pour le dosage des échantillons a forte teneur (éluat, concentrât , . . ). La séparation doit être
menée rapidement pour éviter l'interférence de 1'yttrium, en formation sur la mesure.
Enfin, nous avons dosé le magnésium, calcium, baryum à l'état d'impuretés dans l e s
solutions de nitrate de strontium, de fin du procédé, par spectrographie d'émission.
Ces dosages sont effectués A partir des solutions nitriques sans changement de mi
lieu, sur électrodes de graphite. L'impression de l'émulsion photographique est effectuée en pré
sence d'un dispositif atténuateur à échelons et l'exploitation des spectres obtenus réalisée selon
la méthode de BRECKPOT : la différence d'efficience As entre la caractéristique de la raie de
l'étalon interne et celle de la raie de l'élément A doser est une fonction de la concentration. L'in
troduction de ce facteur analytique indépendant permet de s'affranchir des facteurs plaque et
développement. La validité des droites d'étalonnage a été contrôlée dans des milieux contenant du
strontium A 0 - 5 - 10 gl" par une analyse de variance.
La précision obtenue pour Mg, Ca et Ba peut s'améliorer soit en faisant un plus grand
nombre de poses , soit en effectuant un changement de milieu des solutions de départ ; l e s résul
tats que nous avons obtenus sont, en tous cas , tout A fait conformes aux demandes de l'exploitant.

BIBLIOGRAPHIE.
[1] FREUND J.
Les générateurs d*électricité à radioisotopes et à conversion thermoélectrique -
Division atomique de la SNECMA.
[2] GALAUD G.
Extraction du strontium par D EHPA - Note technique DR/SFF N* 609 - Départe
ment des Radioéléments - CEN SA CLAY
[3] GALAUD G.
Récupération du strontium sur acide antimonique - Note technique DR/SPF
tr «58-59 - Département des Radioéléments CEN SACLAY
[4] GALAUD G.
Séparation rtrontlum-alcallno-terreux dans PICPUS - Note technique DR/SPF
N* 89S-59 - Département des Radioéléments CEN SACLAY
[S] D'HONT et BAESTLE L.
Medel. KoninckViaamAca. Belg. 196S, 27-7
[«J STRELOW et VAN ZYL
Anal. Chlœ. AcU n> 40, 1968, 145-163
[7] VARTERESSIAN
Séparation chromatographique des alcalino-terreux. Note technique DR/STAAR
N' 103 - Département des Radioéléments CEN SACLAY
[«J TREMILLON
Séparation par l e s rés ines échanges d'ions p. 83-94, Gauthier Villars

- 96 -
[9] TURNER J . B . , PHILIP P . at DAY R.
Anal. Chim. Act» 26, 1962, M-98
[10] MARCUS Y.
Acta cHffl. Scand. 11, 1SS7. 4
[11] PASCAL
Traité de Chimie Minérale T III - p . «37, Maaaon Ed.
[12] CHARLCT G.
Aialyae Qualitative et Réaction* en Sofeitien, Maaaon Ed.
[13] TREMILLGN
Séparation par lea réeine* échmnfeuae* d'iona, GautbierB Villar*
[14] STRELOW et WEINERT
Talanta, 1870, 1 » 12
[IS] CHARLOT G.
Anal. Qualitative et Réactione en aolution, Maaaon Ed.
[16] CHARLCT G.
Méthode* de la Chimie Analytique - Maaaon Ed.
[17] VYDRA at KARLIK
Chim. Uaty , l t M . 1741-53
[18] [IS] CHARLCT G.
Méthode» de la Chimie analytique - Anal. Quant. Maaaon Ed .
[20] TREMILLON
Cour» de Chimie Analytique - Méthode* de meaur* quantitative - OISTN -
CEA SACLAY
[21] PANNETIER
Tabla daa radiolaotopaa
[22]' BBtKS J . B .
Theory and Pratice of Scintillation Countinf - Pergamon Près* London 67
[23] introduction to ecintillation countinf. Intertechnique, rat. 13-M-062 Pfeiair France

[24] ROBF.RFROID M. I* scintillation en milieu liquide - Université Catholique de Lou vain.
[25] KERR, HAVES, OTT
Inter Journal Appl. Radioisotopes 1, 284, 1957
[26] Manuel d'utilisation du apectrometre SL 20 P - Documentation Intertecbnique
[27] RAPKINE. Solvants and scintillation - Documentation Intertecbnique
[28] BAUDE L., CUNQ M. J., MBERT J. C. Rapport SPU n* F 9/66 CEA MARCOULE
[29] JOHNSON W.C.
Anelyt. Chim. 38, 954-55, 1966
[30] KIBA, OHASHI, MOEDA
Bull. Soc. Chim. Japon 33, 6, 818-21, I960
[31] AUCHAPT J. Doaafe du strontium dans lea effluenta, S.E, Pu MARCOULE
[32] GERLACH W. Die chemieche Emission Spectralanalyse
[33] BRBCKPOT L'analyse apectrochimique par la méthode du secteur A échelons • SpectroChim. Act», 1939, 1, 137-163
[34] CETAMA Calibrate dea émuleiona - méthode n* 74, Dunod Ed.
[35] Projet CETAMA GT S Dosage spectrographiq» des impuretés dana le plutonium - en préparation
[36] GERARD R. Dosage des alcalins dans le césium - Note technique DR/SPF 71-08 - Elan IIB -CEA LA BAGUE

[37] PROSPERT J. Programmai HP/n" 7-8-» - Calcul daa différence» d'efficience As, "ÏS, variance -Droit** d'étalonnage - Elan S B - CIA LA HAGUE
[38] IXPEVRE J, Not* technique DR/SPF n* 8*1 - Département dea Radioélément* - CEN SACLAY
[3»] Technique* de l'Ingénieur Meeurea et iVnalya* - p. 1140- 1,8
[40] PASCAL Trait* de Chimie Minérale - Propriété» chimique* du glucinium - MaieonEd.
[41] Méthode* Statlatiquea en Chimie Analytique - CETAMA - vol, IV et I - 2, Dunod Ed.
Inline» i*i» K 17 turn 1973





























