Embed Size (px)
Citation preview

ÉTUDE DE LA DIFFÉRENCIATION DES HL60 EN CELLULES DE TYPE OSTÉOCLASTES ET RÔLE DES
FACTEURS RHUMATOÏDES SUR LA RÉSORPTION OSSEUSE DES OSTÉOCLASTES
Mémoire
Murielle Patricia Nanfah Woda
Maîtrise en microbiologie-immunologie
Maître ès sciences (M.Sc.)
Québec, Canada
© Murielle Patricia Nanfah Woda, 2013


iii
RÉSUMÉ
La polyarthrite rhumatoïde est une maladie auto-immune qui touche
environ 1% de la population mondiale. Elle entraîne une inflammation
synoviale et une destruction de l’architecture articulaire. L’articulation
rhumatoïde est un milieu très inflammatoire qui est infiltré par des
cellules telles que les neutrophiles, les macrophages, les monocytes, les
lymphocytes B et T ; par des protéines telles que des cytokines, des
chimiokines, des molécules d’adhésion, par des complexes immuns, et
par des auto-anticorps comme les facteurs rhumatoïdes qui sont un
marqueur spécifique de la maladie et qui permettent d’établir un
pronostic sur son évolution. De plus, dans l’articulation rhumatoïde, il y
aura formation d’un pannus rhumatoïde avec érosion progressive du
cartilage et de l’os. Les ostéoclastes sont les cellules principalement
impliquées dans cette érosion osseuse. Afin d’effectuer des études in
vitro, les ostéoclastes sont généralement obtenus à partir de monocytes
isolés de sang humain, ce qui rend leur étude complexe car les variations
sont très grandes d’un donneur de sang à l’autre. Nous avons utilisé la
lignée cellulaire d’origine myéloïde HL60 pour obtenir des ostéoclastes et
avons caractérisé ces cellules HL60 « osteoclast-like » dans tous les
aspects de leur fonction et de leur activation. Nous avons aussi montré
que les facteurs rhumatoïdes pouvaient agir directement sur les
ostéoclastes pendant leur différenciation et pendant la résorption
osseuse. Cette étude nous a permis d’amorcer la compréhension du rôle
possible des facteurs rhumatoïdes sur la résorption osseuse dans la
polyarthrite rhumatoïde.


v
AVANT-PROPOS
Tout d’abord, je voudrais remercier Dr Patrice Poubelle qui a
accepté de me prendre dans son équipe alors que je venais de très loin,
de mon pays le Cameroun. Vous avez fait preuve d’une très grande
patience à mon égard et m’avez toujours aidé dans toutes les étapes de
ma formation. Vous êtes un excellent pédagogue et vous m’avez mis le
pied à l’étrier en ce qui concerne le monde de la recherche; avec vous j’ai
appris à aimer cela. C’est un honneur d’avoir pu travailler avec vous,
d’avoir pu apprendre toutes ces belles choses.
Je voudrais remercier aussi Dr Isabelle Allaeys, qui m’a quasiment
tout appris dans le laboratoire : tu as été avec moi à toutes les étapes de
mon apprentissage et c’est avec toi que j’ai travaillé sur mon projet de
recherche. Tu m’as appris à travailler avec méthode et organisation et je
garderai toujours cela avec moi.
Je remercie aussi Arpita Chakravarti et Daniel Rusu. Vous avez été
mes ainés du laboratoire et m’avez appris tellement de choses. J’ai
toujours pu compter sur vous à tout moment au cours de l’évolution de
mon projet. Je vous remercie pour les bons moments passés au
laboratoire et même en dehors.
J’ai aussi eu l’occasion de cotoyer dans l’équipe Martha et Simon.
Merci pour tous les moments passés ensemble dans le laboratoire.
Je voudrais aussi remercier tous mes collègues et amis des autres
équipes de l’unité de recherche. Merci pour les discussions que nous
avons eu ensemble, merci pour les moments de rire au cours de ces
années. Merci à Emmanuelle Rollet-Labelle, Louis Marois, Jean-Michel
Levesque, Sophie Proulx, Fehtia Ben Yebdri, Marie-thérèse Bawolak,
Geremy Kumbadinga, et à tous ceux dont je n’ai pas pu citer le nom.

vi
Merci à l’équipe enseignante qui m’a encadré et guidé pendant toutes les
années de ma formation : Caroline Gilbert, Sylvain Bourgoin, Jean
Sévigny, Fawzi Aoudjit, et tous ceux ceux que je n’ai pas pu citer.
Je voudrais aussi remercier toutes les personnes ressources du
CRRI, qui sont d’un soutien remarquable dans toutes les différentes
tâches du laboratoire.
Je voudrais dédicacer ce document à ma fille Annaëlle Kémila et à
mon époux Fabrice. Vous êtes le centre de ma vie, et c’est avec votre
soutient et votre amour que j’ai pu mener à bien mes travaux et terminer
ce document.
À mes parents, mes frères et soeurs vous m’avez toujours soutenu
dans tout ce que j’ai entrepris et j’ai toujours pu compter sur votre aide
inconditionnelle. Je vous remercie d’être tout simplement là.
À mes amis, je ne peux tous vous citer ici, car vous êtes nombreux,
je sais que vous pensez à moi et moi je pense très fort à chacun de vous.
Sachez que j’ai apprécié chaque moment passé ensemble, chaque
discussion, chaque marque de soutient et d’encouragement. Merci pour
votre présence.
Seigneur Dieu, à Toi soit la gloire, l’honneur et la puissance. Tu m’as
donné la force de mener à bien ces travaux et de terminer ce document,
je te rends grâce pour tes bienfaits.

vii
TABLE DES MATIÈRES
RÉSUMÉ ....................................................................................................................................... iii
AVANT-PROPOS .......................................................................................................................... v
LISTE DES FIGURES ................................................................................................................ x
LISTE DES TABLEAUX ........................................................................................................... xii
LISTE DES ABRÉVIATIONS ................................................................................................. xiii
I. INTRODUCTION ................................................................................................................. 1
1.1. LE TISSU OSSEUX .................................................................................................... 1
1.1.1. Définition .............................................................................................................. 1
1.1.2. Le remodelage osseux....................................................................................... 1
1.2. LES OSTÉOCLASTES ............................................................................................... 5
1.2.1. Origine ................................................................................................................... 5
1.2.2. Fonction ................................................................................................................ 7
1.2.3. Mécanismes moléculaires de l’activation des ostéoclastes .................. 8
1.3. LA POLYARTHRITE RHUMATOÏDE ................................................................... 11
1.3.1. Qu’est ce que la polyarthrite rhumatoïde? .............................................. 11
1.3.2. La physiologie de la polyarthrite rhumatoïde ......................................... 16
1.4. LES CELLULES HL60 ............................................................................................ 38
1.5. BUTS DE L’ÉTUDE ................................................................................................. 40
II. DÉTERMINATION DES CONDITIONS OPTIMALES DE DIFFÉRENCIATION
DES HL60 ET CARACTÉRISATIONS DES CELLULES HL60 DIFFÉRENCIÉES. 42
2.1. INTRODUCTION ....................................................................................................... 42
2.2. MATÉRIELS ET METHODES ............................................................................... 43
2.2.1. Réactifs ................................................................................................................ 43
2.2.2. Différenciation des cellules ........................................................................... 44
2.2.3. Isolement des monocytes et différenciation en ostéoclastes ............. 45
2.2.4. Immunobuvardages ........................................................................................ 46
2.2.5. Analyses par RT-PCR ..................................................................................... 48
2.2.6. Immunohistochimie ........................................................................................ 49

viii
2.2.7. Coloration TRAP ............................................................................................... 51
2.3. RÉSULTATS ............................................................................................................... 51
2.3.1. Caractéristiques morphologiques des cellules ....................................... 52
2.3.2. Présence des anneaux d’actine ................................................................... 54
2.3.3. Activité enzymatique TRAP ........................................................................... 55
2.3.4. Expression de RANK ....................................................................................... 57
2.3.5. Expression de NFATc ...................................................................................... 59
2.3.6. Expression des récepteurs de calcitonine ............................................... 61
2.3.7. Expression de MMP9 ...................................................................................... 62
2.3.8. Expression de cathepsine K ......................................................................... 63
2.3.9. Expression de DAP-12 ................................................................................... 64
2.3.10. Expression de TREM2 ................................................................................ 66
2.3.11. Expression d’OSCAR .................................................................................. 68
2.3.12. Expression de SIRP1 ................................................................................ 69
2.3.13. Expression de PIR-A ................................................................................... 70
2.3.14. Expression de CD64 ................................................................................... 71
2.4. DISCUSSION ............................................................................................................. 72
III. ACTION DES FACTEURS RHUMATOÏDES SUR LES OSTÉOCLASTES ET
LA RÉSORPTION OSSEUSE ................................................................................................ 75
3.1. INTRODUCTION ....................................................................................................... 75
3.2. MATÉRIELS ET MÉTHODES ............................................................................... 76
3.2.1. Réactifs ................................................................................................................ 76
3.2.2. Isolement des monocytes : ............................................................................ 77
3.2.3. Résorption osseuse ......................................................................................... 77
3.2.4. Coloration TRAP ............................................................................................... 78
3.3. RÉSULTATS ............................................................................................................... 79
3.3.1. Action des facteurs rhumatoïdes sur les ostéoclastes ......................... 79
3.3.2. Action des facteurs rhumatoïdes sur la résorption par les
ostéoclastes........................................................................................................................ 81
3.3.3. Activation des cellules HL60 différenciées par les facteurs
rhumatoïdes ...................................................................................................................... 83
3.4. DISCUSSION ............................................................................................................. 85

ix
IV. CONCLUSION ET PERSPECTIVES ........................................................................ 89
V. RÉFÉRENCES BIBLIOGRAPHIQUES .................................................................... 91

x
LISTE DES FIGURES
Figure 1 : Homéostasie de la structure osseuse [4] ....................................................... 4
Figure 2 : Représentation schématique de la différenciation et la fonction des
ostéoclastes en coopération avec les ostéoblastes [5]. ................................................... 6
Figure 3 : Intégration des voies RANK et ITAM par les protéines kinases Btk et
Tec [15] ........................................................................................................................................ 10
Figure 4 : Effet de la polyarthrite rhumatoïde sur les mains : radiographie des
déformations d’une main rhumatoïde [25]. .................................................................... 12
Figure 5 : Représentation globale de la pathogenèse de la PAR [39] ...................... 19
Figure 6: Citrullination des protéines [35] ...................................................................... 20
Figure 7: Modèle explicatif de l’étiologie de la PAR positive aux ACPA [38] ......... 21
Figure 8: Comparaison de l’articulation normale et l’articulation rhumatoïde
[25] ................................................................................................................................................ 23
Figure 9: Voies de signalisation des cytokines impliquées dans la polyarthrite
rhumatoïde [40]: ...................................................................................................................... 29
Figure 10 : Quelques orientations actuelles sur la pathogenèse de la PAR [65]. 30
Figure 11: Caractéristiques morphologiques des cellules HL60 différenciées
(microscopie optique 400X) .................................................................................................. 53
Figure 12: Caractéristiques morphologiques des cellules HL60 différenciées
(microscopie à fluorescence 400X) : .................................................................................. 53
Figure 13: Présence des anneaux d’actine (microscopie à fluorescence 600X)... 55
Figure 14: Expression de l’activité enzymatique TRAP (microscopie optique
200X) ........................................................................................................................................... 56
Figure 15: Expression de RANK dans les cellules HL60 différenciées ................... 58
Figure 16: Expression de NFATc ........................................................................................ 60
Figure 17: Expression des récepteurs de calcitonine .................................................. 61
Figure 18: Expression de MMP9 ......................................................................................... 62
Figure 19: Expression de cathepsine K ............................................................................ 63

xi
Figure 20: Expression de DAP-12 ...................................................................................... 65
Figure 21: Expression de TREM2 ....................................................................................... 67
Figure 22: Expression d’OSCAR ......................................................................................... 68
Figure 23: Expression de SIRP1 ....................................................................................... 69
Figure 24: Expression de PIR-A .......................................................................................... 70
Figure 25: Expression du récepteur CD64...................................................................... 71
Figure 26: Effets du facteur rhumatoïde IgM sur la différenciation des
ostéoclastes (microscopie optique 200X) ......................................................................... 80
Figure 27: Effet des facteurs rhumatoïdes sur la résorption osseuse des
ostéoclastes ............................................................................................................................... 82
Figure 28: Phosphorylation des tyrosines dans les HL60 différenciées. ............... 84
Figure 29: Protéines adaptatrices associées à leurs récepteurs
transmembranaires pour initier la transduction du signal [136]. .......................... 88

xii
LISTE DES TABLEAUX Tableau 1: Différents gènes possiblement impliqués dans la PAR et leur rôle
dans la pathogenèse de la maladie [32]. .......................................................................... 16
Tableau 2: Critères de classification de l’arthrite rhumatoïde, classification de
1987 par le collège américain de rhumatologie [77]. ................................................... 34
Tableau 3: Tableau de classification des critères diagnostics de l’arthrite
rhumatoïde établis en 2010 par le collège américain de rhumatologie et la ligue
européenne contre le rhumatisme [79]. ........................................................................... 35

xiii
LISTE DES ABRÉVIATIONS
7AAD: 7-amino actinomycine D
ADN : Acide Désoxyribonucléique
ACPA : Anti-citrullinated protein antibodies
AP-1 : Activator protein 1
CIA : Collagen type II-induced arthritis
CFU/GM : Colony Forming Unit/Granulocyte-Macrophage
1, 25(OH) 2D3 : Dihydroxyvitamine D3
DMARDs : Disease Modifying Anti-Rheumatic Drugs
DAPI: 4’6-diamidino-2 phenylindole
DAP-12 : DNAX activating protein 12
ECL : Enhanced Chemiluminescence
FBS : Fœtal Bovine Serum
FR : Facteurs rhumatoïdes
FCR : Récepteur du fragment Fc des immunoglobulines G
GM-CSF : Granulocyte/Macropage-Colony Stimulating Factor
GAPDH : Glyceraldehyde 3-phosphate déhydrogénase
HEPES : Acide N-2-hydroxy-ethylpiperazine-N'-2-ethanesulfonique
ITAM : Immunoreceptor tyrosine-based activation motif
IFN : Interféron

xiv
IL : Interleukine
MTX : Méthotrexate
MHC : Major Histocompatibility Complex
MMPs : Métalloprotéinases
M-CSF : Macrophage Colony Stimulating Factor
NFATc1 : Nuclear factor of activated T cell 1
F : Nuclear Factor B
OPG : Ostéoprotégérine
OA : Ostéoarthrite
OSCAR : Osteoclasts associated receptor
PAR : Polyarthrite rhumatoïde
PMA : Phorbol 12-myristate 13-acetate
PADs : Peptidylarginine déiminases
PTPN22 : Protein Tyrosine Phosphatase Nonreceptor 22
PFA : Paraformaldehyde
PIR-A : Paired Immunolike Receptor A
PTH : Parathormone
PVDF : Polyfluorure de vinylidène
RANK : Receptor Activator of Nuclear Factor Kappa B
RANKL : Receptor Activator of Nuclear Factor Kappa B Ligand
ROS : Reactive Oxygen Species

xv
RT-PCR : Reverse Transcriptase-Polymerase Chain Reaction
SIRP : Signal regulatory protein
TGF : Transforming growth factor
TNF : Tumor Necrosis Factor
TRAF6 : TNF receptor associated factor 6
TREM2 : Triggering receptor expressed on myeloid cells 2
TRAP : Tartrate Resistant Acid Phosphatase
TCR : T Cell Receptor
Th : T helper
TReg : T Régulatrice


1
I. INTRODUCTION
1.1. LE TISSU OSSEUX
1.1.1. Définition
L’os est un tissu conjonctif qui assure plusieurs fonctions
essentielles en dehors de la croissance :
il est responsable du maintien de l’homéostasie du calcium sérique
c’est le support mécanique des tissus mous et le site d’attachement
des muscles pour la locomotion
c’est le site majeur de l’hématopoïèse chez l’adulte
Le tissu osseux est composé de cellules et de matrice extracellulaire.
Les quatre types de cellules présents sont: les ostéoclastes responsables
de la résorption osseuse, les ostéoblastes responsables de la formation
osseuse, les cellules bordantes (ostéoblastes au repos) et les ostéocytes
(ostéoblastes différenciés). La matrice extracellulaire est composée de
phosphate de calcium qui forme les cristaux d’hydroxyapatite et de
constituants organiques tels que le collagène.
L’os possède des propriétés mécaniques étonnantes telles que rigidité,
résistance et légèreté qui sont dues à une réorganisation permanente de
la matière osseuse en réponse à des stimuli mécaniques et chimiques [1].
1.1.2. Le remodelage osseux
Le tissu osseux est un tissu vivant en constant renouvellement;
cela constitue le remodelage osseux. Ce remodelage permanent est un

2
équilibre entre d’une part la résorption et d’autre part de formation de
tissu osseux (figure 1). Ostéoclastes et ostéoblastes collaborent
étroitement dans ce processus, et constituent ce qu’on appelle l’unité de
remodelage osseux qui a une durée de vie moyenne de 200 jours. Le
remodelage osseux est constitué de trois principales phases d’inégales
longueurs, la phase de résorption qui dure environ 2 semaines, la phase
d’inversion d’environ 4 à 5 semaines, et enfin la formation qui se
poursuit pendant environ 4 mois jusqu’à ce que la nouvelle structure
osseuse soit complètement formée [2]. La résorption commence par la
migration des précurseurs ostéoclastiques à la surface de la matrice
osseuse, où ils deviendront des ostéoclastes capables de dissoudre les
cristaux d’hydroxyapatite. Ensuite survient la phase d’inversion au cours
de laquelle des cellules mononuclées de type macrophages vont
remplacer les ostéoclastes à la surface de la matrice osseuse. Ces cellules
préparent la surface pour que les ostéoblastes commencent leur travail
de formation et fournissent aussi les signaux pour la différenciation et la
migration des ostéoblastes [2]. Après cela, les ostéoblastes sont en place
et commencent la formation du nouveau tissu osseux. Lorsque cette
étape est terminée, la surface osseuse est recouverte par les cellules
bordantes qui sont des ostéoblastes au repos. À la fin de chaque cycle,
les ostéoclastes sécrètent un substrat qui sert à l’attachement des
ostéoblastes. Une période de repos commence ainsi jusqu'au début du
prochain cycle de remodelage. Durant la résorption, les ostéoclastes
libèrent certains facteurs qui permettront de réguler leur activité soit en
inhibant la fonction des ostéoclastes soit en stimulant l’activité des
ostéoblastes [3].
Le remodelage osseux est régulé à plusieurs niveaux : une
régulation systémique par des facteurs tels que l’hormone parathyroïde,
les glucocorticoïdes; une régulation locale qui passe par la régulation de
la voie de signalisation RANK/RANKL/OPG ( Receptor Activator of Nuclear

3
Factor Kappa B , récepteur activateur du facteur nucléaire Kappa B, son
ligand et Ostéoprotégérine, leurre de RANKL) et fait intervenir aussi des
facteurs de croissance tels que le TGF ( Transforming growth factor ,
facteur de transformation de croissance ), des cytokines telles que le
TNF (Tumor Necrosis Factor , facteur nécrosant des tumeurs), l’IL-10
(Interleukine 10) et des prostaglandines. Des anomalies dans le
remodelage osseux peuvent être observées dans certaines pathologies
telles que l’ostéoporose et la polyarthrite rhumatoïde.

4
Figure 1 : Homéostasie de la structure osseuse [4]
A l’intérieur du tissu osseux, ostéoblastes et ostéoclastes jouent leurs rôles respectifs de
formation et destruction. Les ostéoclastes commencent par dégrader l’os, ensuite les
puits de résorption sont remplis par la nouvelle matrice osseuse synthétisée par les
ostéoblastes. Cette matrice sera par la suite minéralisée.

5
1.2. LES OSTÉOCLASTES
Les ostéoclastes sont de grosses cellules hématopoïétiques, multi-
nucléées d’environ 50 à 100 m de diamètre. Ces cellules ont la capacité
de détruire une matrice osseuse et ont une durée de vie moyenne de 15
jours; elles mourront ensuite par apoptose. Les ostéoblastes, quand à
eux, sont responsables de la formation du tissu osseux. La
différenciation et la fonction des ostéoclastes sont étroitement liées aux
ostéoblastes [5], un contact cellule-cellule est nécessaire et nécessite
l’implication de cytokines telles que M-CSF (Macrophage-Colony
Stimulating Factor, facteur stimulant les macrophages) et RANKL
(Receptor Activator of Nuclear Factor Kappa B Ligand, ligand du récepteur
activateur du facteur nucléaire Kappa B) [6].
1.2.1. Origine
Les précurseurs des ostéoclastes sont les cellules souches
hématopoïétiques de la lignée des monocytes/macrophages (CFU/GM)
(Colony Forming Unit/Granulocyte-Macrophage, unité de formation des
colonies de granulocytes et macrophages). Des expériences utilisant le
système de coculture ont clairement établi le concept selon lequel les
ostéoblastes sont impliqués dans le processus de différentiation des
ostéoclastes [5, 7]. Les ostéoclastes sont obtenus après la fusion de
progéniteurs mononucléaires. Ces précurseurs ostéoclastiques expriment
à leur surface RANK, un membre des récepteurs de la famille du TNF qui
reconnaitra son ligand RANKL lorsqu’ils entrent en contact avec les
ostéoblastes, car les ostoblastes sont les cellules qui expriment RANKL
dans le tissu osseux. Une fois le contact effectué, ces précurseurs vont se
transformer en préostéoclastes sous l’action du M-CSF et du RANKL,
puis survient la fusion des préostéoclastes, leur activation et maturation
en ostéoclastes, comme le montre la figure 2. L’expression de RANKL par
les ostéoblastes est régulée par des facteurs tels que la 1,
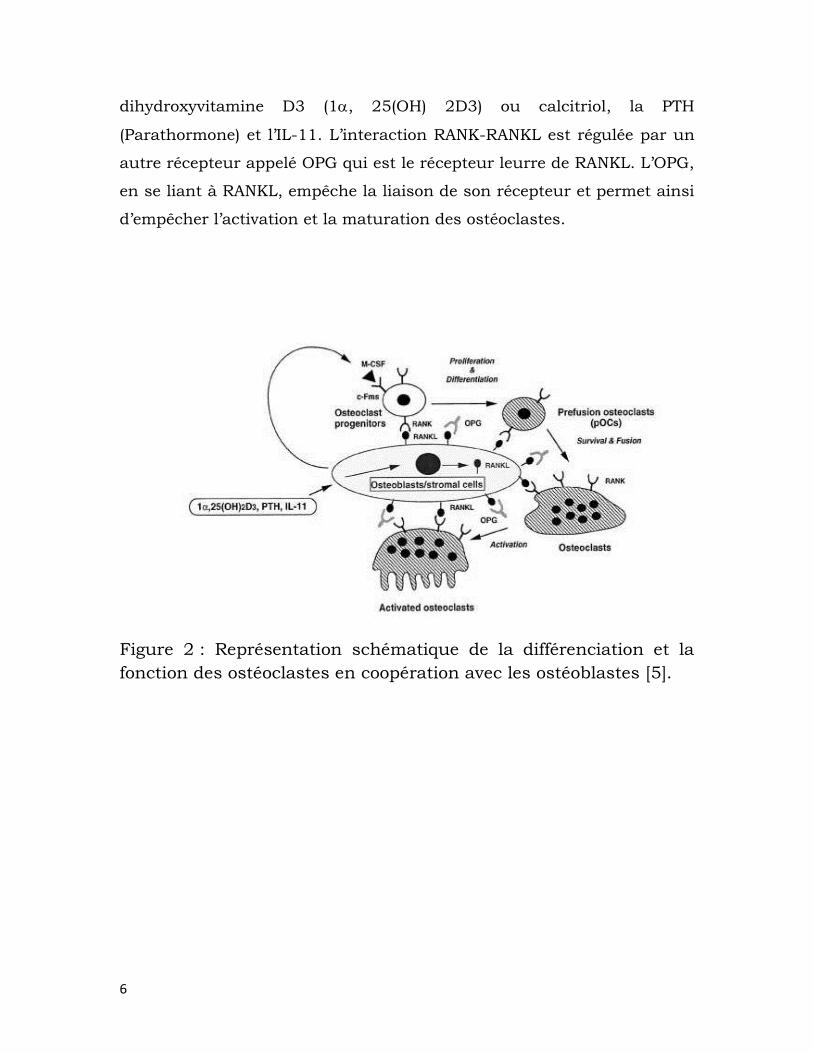
6
dihydroxyvitamine D3 (1, 25(OH) 2D3) ou calcitriol, la PTH
(Parathormone) et l’IL-11. L’interaction RANK-RANKL est régulée par un
autre récepteur appelé OPG qui est le récepteur leurre de RANKL. L’OPG,
en se liant à RANKL, empêche la liaison de son récepteur et permet ainsi
d’empêcher l’activation et la maturation des ostéoclastes.
Figure 2 : Représentation schématique de la différenciation et la
fonction des ostéoclastes en coopération avec les ostéoblastes [5].

7
1.2.2. Fonction
Les ostéoclastes sont des cellules ostéorésorbantes très mobiles,
capables de se déplacer à travers les travées osseuses d’un site de
résorption à un autre. La résorption est un processus très important
dans la biologie osseuse. Elle est indispensable à la croissance osseuse, à
la poussée dentaire, à la guérison des fractures, et au maintien du taux
de calcium dans le sang [8]. Une fois que l’ostéoclaste a atteint la surface
osseuse, deux évènements surviennent : l’activation suivie de l’adhésion.
Lorsqu’il est activé, l’ostéoclaste se polarise fortement; l’un des pôles
entre en contact avec la surface osseuse et y adhère fortement. L’un des
mécanismes d’adhésion identifiés fait intervenir l’intrégrine V3 [9]. Elle
est très exprimée dans les ostéoclastes, que ce soit à la membrane
cytoplasmique ou à l’intérieur de certaines vacuoles. L’intégrine V3 joue
un rôle dans la forte adhésion, la migration des ostéoclastes et
l’endocytose des produits de la résorption. L’intégrine V3 joue aussi un
autre rôle non moins important dans la résorption osseuse ; en effet, elle
transmet un signal qui en fin de compte va être à l’origine de la
réorganisation du cytosquelette à l’un des pôles de la cellule,
particulièrement celui en contact avec la matrice osseuse [10]. Cette
réorganisation est à l’origine de la formation de la bordure en brosse qui
va être entourée d’une zone de scellage riche en micro-filaments d’actine.
À ce niveau, l’ostéoclaste met en place des structures d’attachement
appelées podosomes [11, 12]. L’actine se réorganise pour prendre la
forme spécifique d’un anneau et une fois l’adhésion faite, il se forme une
zone de scellage appelée lacune de Howship où aura lieu la résorption
proprement dite. À travers la bordure en brosse, l’ostéoclaste secrète des
ions H+ qui, créant un environnement acide vont permettre de dissoudre
la matrice osseuse [13]. La sécrétion des protons H+ est un processus
actif qui requiert de l’ATP. De plus, les ostéoclastes produisent des
protéases - la cathepsine K étant la plus importante - qui participent

8
elles aussi à la dégradation de la matrice osseuse [8, 14]. Outre la
cathepsine K, les métalloprotéinases (MMP) -plus précisément la MMP9-
participent elles aussi à la dégradation de la matrice osseuse. Une fois la
résorption terminée, il y a élimination des produits de dégradation de la
matrice osseuse et enfin soit apoptose des ostéoclastes soit leur retour à
l’état de repos.
1.2.3. Mécanismes moléculaires de l’activation des
ostéoclastes
L’activation des ostéoclastes passe par deux voies de signalisation
distinctes et complémentaires (figure 3). La voie principale passe par la
triade RANK/RANKL/OPG et la seconde voie est la voie de costimulation
qui fait intervenir des récepteurs à domaine ITAM (Immunoreceptor
tyrosine-based activation motif). La liaison de RANKL à son récepteur
RANK permet le recrutement de TRAF6 (TNF receptor associated factor 6,
facteur 6 associé au récepteur du TNF) qui active NFB, facteur de
transcription clé de la différenciation et de l’activation ostéoclastiques,
les MAP kinases JNK et Erk, de même que les protéines kinases Btk et
Tec. Cette liaison permet aussi le recrutement de cFos, étape préliminaire
à l’activation du complexe AP-1 [15, 16]. Les facteurs de transcription
NFB (Nuclear Factor B, facteur nucléaire B) et AP-1 (Activator protein
1, protéine activatrice 1) vont permettre, en partie, l’activation du facteur
de transcription NFATc1 (Nuclear factor of activated Tcell 1, facteur
nucléaire 1 de l’activation des cellules T) qui est le régulateur le plus
important de la différenciation des ostéoclastes [17]. La voie de
costimulation passe par les récepteurs OSCAR (Osteoclasts associated
receptor, récepteur associé aux ostéoclastes), PIR-A (Paired Immunolike
Receptor A, récepteur A couplé de type immunologique), TREM2
(Triggering receptor expressed on myeloid cells 2, récepteur déclencheur
exprimé sur les cellules myéloïdes 2) et SIRP1 (Signal regulatory protein

9
, protéine régulatrice du signal 1). Ces récepteurs sont associés à des
molécules adaptatrices : DAP-12 (DNAX activating protein 12, protéine
activatrice d’ADNX) et FCR (récepteur du fragment Fc des
immunoglobulines G). Les récepteurs et leurs molécules adaptatrices
vont envoyer un signal grâce à la phosphorylation des motifs ITAM, ce
qui recrute la tyrosine kinase Syk [18, 19]. Les ligands pour les
récepteurs de la voie de costimulation ne sont pas encore connus à ce
jour. En plus de ces deux voies de signalisation, NFATc1 est
constamment activé par la calcineurine, une phosphatase calcium-
dépendante. NFATc1 régule l’expression de nombreux gènes spécifiques
des ostéoclastes tels que ceux qui codent pour la cathepsine K, l’isoforme
5b de la phosphatase acide résistante au tartrate (TRAP) et les récepteurs
de calcitonine.

10
Figure 3 : Intégration des voies RANK et ITAM par les protéines
kinases Btk et Tec [15]

11
1.3. LA POLYARTHRITE RHUMATOÏDE
1.3.1. Qu’est ce que la polyarthrite rhumatoïde?
1.3.1.1. Définition
Les arthrites constituent toutes les formes d’inflammation aiguës
ou chroniques qui affectent les articulations sans cause reconnue. La
classification des maladies rhumatismales peut être difficile à cause de la
diversité des manifestations cliniques, on en distingue une centaine.
Parmi celles-ci, l’arthrose et la polyarthrite rhumatoïde (PAR) sont les
plus fréquentes. L’arthrose est une maladie dégénérative qui affecte le
cartilage articulaire et l’os sous-chondral. On estime qu’elle touche à peu
près 40% des personnes âgées de plus de 75 ans, ce qui en fait la
maladie articulaire à la plus forte prévalence [20, 21].
La PAR est une maladie auto-immune qui entraîne une
inflammation synoviale et une destruction de l’architecture articulaire. Le
système immunitaire attaque les tissus synoviaux qui revêtent les
articulations et parfois même d’autres organes tels que les yeux, le
poumon ou le cœur. La PAR commence généralement lentement à partir
de quelques articulations, puis elle va se propager progressivement à
d’autres articulations. Elle est la seule arthrite qui attaque les
articulations de façon symétrique. La PAR est l’une des maladies
articulaires les plus sévères, mais aussi la maladie auto-immune
systémique la plus courante. Elle affecte approximativement 1% de la
population adulte mondiale [22].
1.3.1.2. Les manifestations cliniques de la polyarthrite
rhumatoïde
La PAR est la forme d’arthropathie la plus commune ; elle a un
impact important sur la société en terme de coût et de perte de
productivité dûs aux incapacités qu’elle provoque. Les symptômes

12
prédominants sont la douleur, l’inflammation et la raideur matinale des
petites articulations (mains, poignets, pieds) [23]. La symétrie des
articulations touchées est une caractéristique de la PAR. Une
radiographie des articulations effectuée dans les premières années de la
maladie montrera une érosion évidente des os chez la plupart des
patients (plus de 70% des patients dans les deux premières années [23,
24]). Cette érosion entrainera progressivement après de nombreuses
années la déformation des articulations et les deux sont irréversibles [24]
(figure 4).
La PAR ne présente pas seulement des manifestations articulaires,
mais aussi des atteintes systémiques telles que les maladies des voies
respiratoires et les maladies cardiovasculaires.
Les manifestations et l’évolution cliniques de la maladie sont
extrêmement variables, mais dans tous les cas un traitement doit être
administré pour contrôler la maladie et éviter les handicaps qu’elle peut
entraîner [23].
Figure 4 : Effet de la polyarthrite rhumatoïde sur les mains :
radiographie des déformations d’une main rhumatoïde [25].

13
1.3.1.3. Facteurs de risque
Les causes d’apparition de la PAR ne sont pas parfaitement
déterminées. En plus des facteurs psychologiques, environnementaux et
hormonaux, des facteurs génétiques peuvent être incriminés.
Les facteurs génétiques ont une influence importante sur la
détermination de la susceptibilité à développer une PAR. Ainsi, des
études ont démontré que 60% des prédispositions d’une population à la
PAR peut être expliquée par des facteurs génétiques [23]. Peu d’études
ont été réalisées dans ce domaine, mais un risque familial a tout de
même été identifié [23, 26, 27]. Les molécules HLA de classe II sont les
facteurs génétiques les plus importants [28], soit environ 30% du facteur
génétique de la maladie [28, 29]. Les allèles HLA-DRB1 sont ceux
impliqués en particulier. Ils constituent la théorie de l’épitope partagé qui
stipule que ces allèles sont étroitement liés à la susceptibilité et à la
sévérité de la maladie. Ainsi, les individus homozygotes pour l’épitope
partagé auront un risque plus élevé de développement de l’arthrite avec
une destruction osseuse plus sévère que les individus hétérozygotes [26,
29, 30]. Les allèles HLA seraient, en fait, liés plus précisément à la
présence d’anticorps dirigés contre la fraction Fc des IgG ou contre les
peptides citrullinés, ce qui confère une plus grande sévérité aux arthrites
séropositives pour ces anticorps par rapport aux arthrites séronégatives
[27]. Les allèles HLA-DRB1 agissent aussi en déclenchant le TCR, ceci en
lui présentant l’épitope partagé.
Les gènes associés au MHC (Major histocompatibility complex,
complexe majeur d’histocompatibilité) ne sont pas les seuls impliqués
dans la PAR, d’autres gènes le sont aussi. Le gène à la plus forte
association à la maladie retrouvée est le gène PTPN22 (Protein Tyrosine
Phosphatase Nonreceptor 22) qui code pour une tyrosine phosphatase qui
exerce un rétrocontrôle négatif sur la signalisation du récepteur pour les

14
cellules T [27]. Le mécanisme de cette régulation n’implique pas les
cellules T périphériques directement mais il s’agit plutôt d’un défaut de
signalisation dans la sélection des cellules T au niveau du thymus ; il en
résulte une production de cellules T auto-réactives.
Outre les facteurs génétiques, des facteurs environnementaux sont
impliqués dans les prédispositions à la PAR. Parmi ceux-ci nous pouvons
citer le tabagisme, la consommation de caféine, l’obésité, de même que le
facteur hormonal [31].
Les gènes impliqués dans le développement de la maladie sont
connus, mais les mécanismes moléculaires eux restent encore assez peu
connus [29]. Le tableau 1 résume ces différents gènes et leurs rôles dans
la pathogenèse. Tous les facteurs cités plus haut concourent au
déclenchement de la maladie, à son évolution, de même qu’à la sévérité
des symptômes.
15
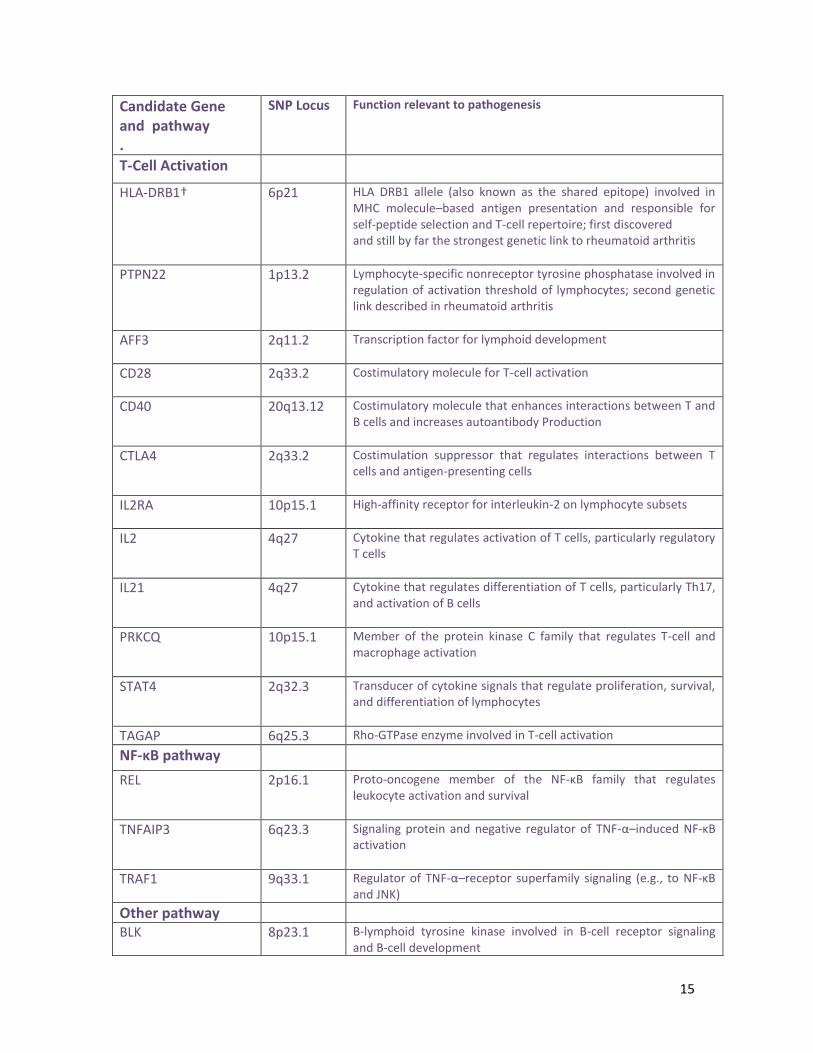
Candidate Gene and pathway .
SNP Locus Function relevant to pathogenesis
T-Cell Activation
HLA-DRB1† 6p21 HLA DRB1 allele (also known as the shared epitope) involved in MHC molecule–based antigen presentation and responsible for self-peptide selection and T-cell repertoire; first discovered and still by far the strongest genetic link to rheumatoid arthritis
PTPN22 1p13.2 Lymphocyte-specific nonreceptor tyrosine phosphatase involved in regulation of activation threshold of lymphocytes; second genetic link described in rheumatoid arthritis
AFF3 2q11.2 Transcription factor for lymphoid development
CD28 2q33.2 Costimulatory molecule for T-cell activation
CD40 20q13.12 Costimulatory molecule that enhances interactions between T and B cells and increases autoantibody Production
CTLA4 2q33.2 Costimulation suppressor that regulates interactions between T cells and antigen-presenting cells
IL2RA 10p15.1 High-affinity receptor for interleukin-2 on lymphocyte subsets
IL2 4q27 Cytokine that regulates activation of T cells, particularly regulatory T cells
IL21 4q27 Cytokine that regulates differentiation of T cells, particularly Th17, and activation of B cells
PRKCQ 10p15.1 Member of the protein kinase C family that regulates T-cell and macrophage activation
STAT4 2q32.3 Transducer of cytokine signals that regulate proliferation, survival, and differentiation of lymphocytes
TAGAP 6q25.3 Rho-GTPase enzyme involved in T-cell activation
NF-κB pathway
REL 2p16.1 Proto-oncogene member of the NF-κB family that regulates leukocyte activation and survival
TNFAIP3 6q23.3 Signaling protein and negative regulator of TNF-α–induced NF-κB activation
TRAF1 9q33.1 Regulator of TNF-α–receptor superfamily signaling (e.g., to NF-κB and JNK)
Other pathway
BLK 8p23.1 B-lymphoid tyrosine kinase involved in B-cell receptor signaling and B-cell development

16
CCL21 9q13.3 Chemokine implicated in germinal-center formation
FCGR2A 1q23.2 Low-affinity IgG Fc receptor that regulates macrophage and neutrophil activation and immune-complex clearance
PAD14 1p36.2 Enzyme that converts arginine to citrulline, creating autoantigens in rheumatoid arthritis
PRDM1 6q21 Protein that acts as a repressor of β-interferon gene expression
TNFRSF14 1p36.32 TNF-α–receptor superfamily member with proinflammatory activity
GTPase denotes guanosine triphosphatase, JNK Jun N-terminal kinase, MHC major
histocompatibility complex, NF-κB nuclear factor κB, Th17 type 17 helper T cells, and
TNF-α tumor necrosis factor α.
† Different HLA-DRB1 alleles, not only the shared epitope, are associated with
rheumatoid arthritis and with distinct immune responsesto citrullinated antigens. In
addition, HLA-DP and HLA-DQ loci (outside the HLA-DRB1 region) have been associated
with rheumatoid arthritis.
SNP : Single Nucleotid Protein
Tableau 1: Différents gènes possiblement impliqués dans la PAR et
leur rôle dans la pathogenèse de la maladie [32].
1.3.2. La physiologie de la polyarthrite rhumatoïde
1.3.2.1. La pathogenèse
La PAR est une maladie dont les causes restent encore inconnues à
ce jour, une combinaison de plusieurs facteurs permet le déclenchement
et le développement de la maladie au fil des mois. Elle peut être
caractérisée par 3 phases distinctes mais interdépendantes et activées
simultanément [33] (figure 5). La phase initiale est asymptomatique et
est caractérisée par le déclenchement de l’auto-immunité. Ensuite
suivront deux phases symptomatiques caractérisées par l’atteinte

17
articulaire, respectivement la phase intermédiaire et la phase de
destruction terminale. Ces deux phases se distinguent par leurs sévérités
et amplitudes [34].
Les facteurs génétiques jouent un rôle très important dans la
pathogenèse de la maladie surtout dans la phase initiale. Le facteur
génétique majeur est l’épitope partagé exprimant les allèles HLA-DRB1
qui agit dans le déclenchement du TCR [27], il en résulte la transmission
d’un signal inhabituel aux cellules T. Ce signal conduit à la production
de cellules T auto-réactives qui vont être activées et différenciées en Th1
et Th17 [35]. Le TCR (T Cell Receptor, récepteur pour les cellules T) ici
reconnait certains peptides spécifiques du soi. Cette reconnaissance
conduira à la production par les lymphocytes B d’auto-anticorps dont les
plus fréquemment retrouvés chez les patients sont les anticorps contre
les protéines cycliques citrullinées (ACPA : Anti-citrullinated protein
antibodies, anticorps anti protéines citullinées) et les anticorps contre les
IgG encore appelés facteurs rhumatoïdes (FR). Les anticorps de moindre
importance qui sont les anticorps contre la kératine ou contre la filagrine
sont également retrouvés dans la maladie [36]. Les auto-anticorps ne
sont pas toujours retrouvés chez les personnes atteintes de PAR mais
leur présence est de mauvais pronostic pour le malade et sont de nos
jours utilisés à des fins de diagnostic ; c’est le cas particulièrement des
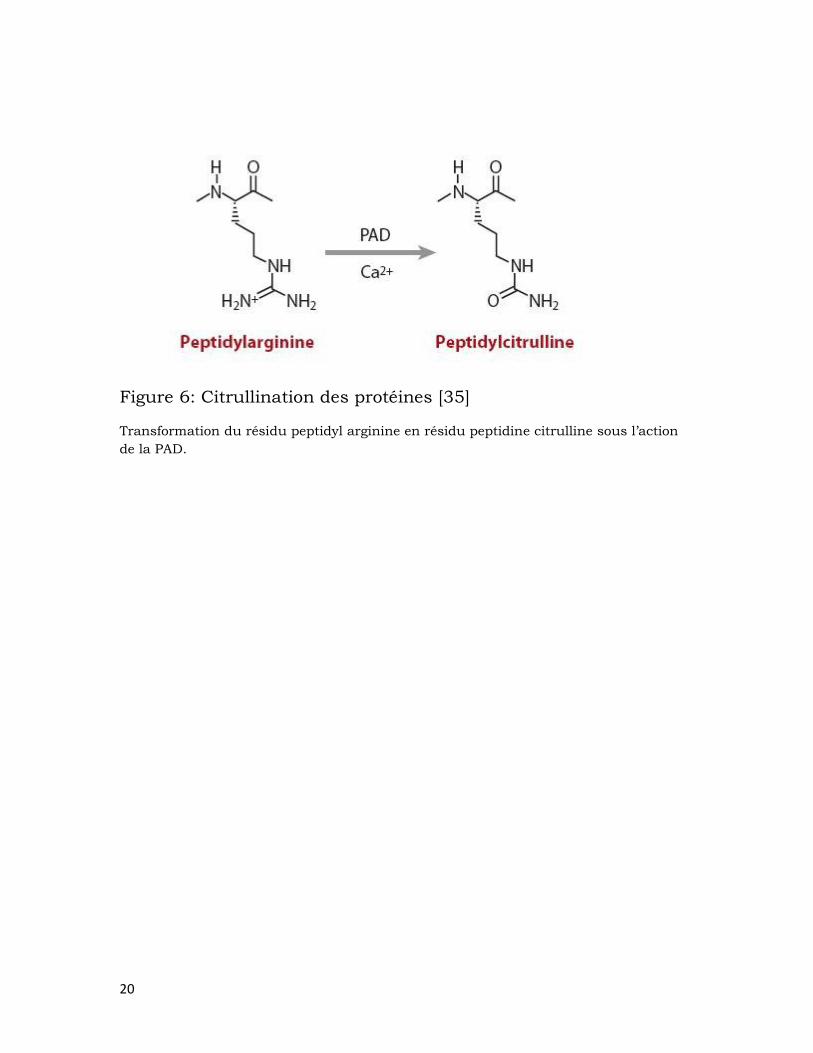
FRs. La citrullination des protéines est une modification post-
translationnelle dans laquelle les résidus peptidyl-arginine sont convertis
en peptidyl-citrulline sous l’action des peptidylarginine déiminases (PAD)
[35, 37] (figure 6). Les protéines citrullinées sont retrouvées au niveau de
plusieurs sites inflammatoires. À la suite d’un trauma, ou d’une
inflammation, il y a infiltration de cellules inflammatoires contenant des
enzymes PAD. La mort de ces cellules entraîne la libération de ces
enzymes qui, lorsqu’elles ne sont pas correctement éliminées, vont
devenir des signaux de danger surtout dans un environnement

18
immunologique tel que l’articulation rhumatoïde. C’est la perte de la
tolérance contre ces protéines qui serait en partie responsable du
déclenchement de la maladie. Des études ont démontré que le tabac
prédispose certaines personnes à la production de ACPA [37]. La figure 7
explique la pathogenèse de la PAR liée aux ACPA.
À la suite du déclenchement de l’auto-immunité, une réponse
inflammatoire pathologique est déclenchée au niveau articulaire. La
cause de cette spécificité articulaire reste encore inconnue, cependant
certaines études l’attribueraient à des infections, des traumatismes ou
même des interactions neuro-immunologiques [38, 39]. L’inflammation
se traduit par une infiltration de cellules inflammatoires, la production
de cytokines pro inflammatoires, et la formation de complexes immuns.
La troisième étape est une étape d’auto-amplification de
l’inflammation, ce qui rend la maladie chronique. Il y a amplification de
la production de cytokines pro-inflammatoires telles que le TNF-, l’IL-1
et l’IL-6, ce qui augmente le recrutement de cellules inflammatoires [38].
Il est à noter que les premiers symptômes peuvent apparaître des
années après le début de l’auto-immunité.

19
Figure 5 : Représentation globale de la pathogenèse de la PAR [39]
La phase de déclenchement de l’auto-immunité caractérisée par l’absence de
symptômes est suivie par une phase de transition caractérisée par l’atteinte articulaire,
le déclenchement de l’inflammation et l’apparition des premiers symptômes. Ensuite
viens la troisième phase caractérisée par l’amplification de l’inflammation et le début de
la destruction articulaire. Des troubles systémiques tels que les maladies
cardiovasculaires peuvent survenir à cette étape.
20
Figure 6: Citrullination des protéines [35]
Transformation du résidu peptidyl arginine en résidu peptidine citrulline sous l’action
de la PAD.

21
Figure 7: Modèle explicatif de l’étiologie de la PAR positive aux
ACPA [38]
1ère étape : les facteurs génétiques et environnementaux, tels le tabagisme, déclenchent
la citrullination des protéines dans les poumons et seront reconnus comme antigènes
par les cellules T. Il s’en suit la production des ACPA.
2ème étape : déclenchement de l’inflammation localisée au niveau articulaire,
recrutement des ACPA de la circulation et formation de complexes immuns
3ème étape : auto-amplification de l’inflammation avec augmentation de la production de
cytokines, d’auto-anticorps, de complexes immuns et de cellules inflammatoires.

22
1.3.2.2. L’articulation rhumatoïde
Les articulations préférentiellement atteintes par la PAR sont les
articulations synoviales, et plus fréquemment les mains, les pieds et les
genoux. L’articulation normale est composée de deux surfaces osseuses
adjacentes communes, recouvertes de cartilage et qui sont séparées par
une cavité appelée cavité articulaire. Une membrane synoviale scelle la
cavité articulaire et sécrète le liquide synovial qui est un liquide
visqueux, lubrifiant et riche en éléments nutritifs ; ce qui permet à
l’articulation d’être mobile.
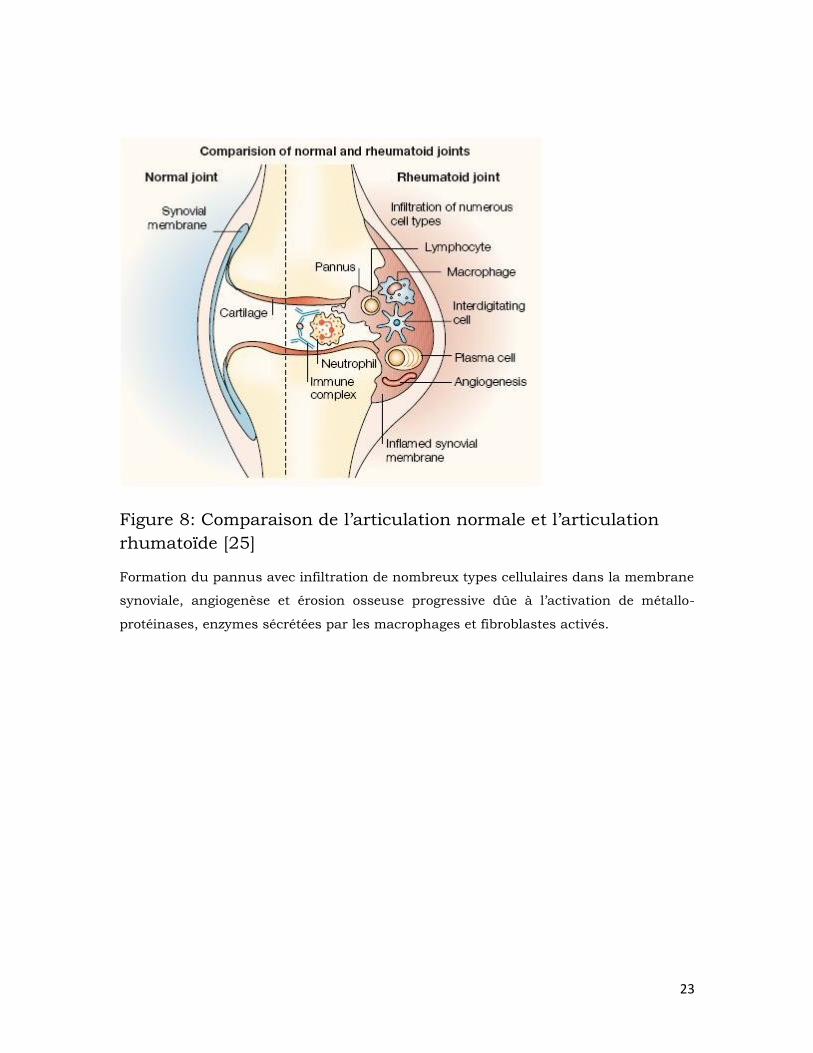
L’articulation rhumatoïde est un milieu très inflammatoire. Elle
subit des changements micro-environnementaux profonds avec une
réorganisation de la structure synoviale et activation des fibroblastes et
macrophages synoviaux [32] (figure 8). Contrairement au liquide synovial
de l’articulation normale qui est un milieu acellulaire, celui de
l’articulation rhumatoïde est infiltré de neutrophiles, macrophages,
lymphocytes T et cellules dendritiques. Progressivement la membrane
synoviale sera elle aussi envahie par plusieurs types cellulaires dont les
plus importantes sont les macrophages et les cellules T. Les leucocytes
activés ainsi que leurs protéines (cytokines, chimiokines, molécules
d’adhésion, …) vont engendrer un milieu hypoxique qui stimule
l’angiogenèse, ce qui explique l’augmentation de la vascularisation dans
les articulations des patients [40]. La membrane synoviale ainsi modifiée
forme des excroissances qui vont envahir la cavité articulaire, le cartilage
et même l’os souschondral; elle constitue désormais le pannus. Les
cellules du pannus vont progressivement causer l’érosion du cartilage et
de l’os [41].
23
Figure 8: Comparaison de l’articulation normale et l’articulation
rhumatoïde [25]
Formation du pannus avec infiltration de nombreux types cellulaires dans la membrane
synoviale, angiogenèse et érosion osseuse progressive dûe à l’activation de métallo-
protéinases, enzymes sécrétées par les macrophages et fibroblastes activés.

24
1.3.2.3. Les cellules impliquées
Plusieurs types cellulaires sont retrouvés dans l’articulation
rhumatoïde. Cependant, chacun de ces types cellulaires à lui seul ne
permet pas d’expliquer la pathogenèse de la maladie, ce sont les
différentes interactions entre ces cellules qui permettent de comprendre
la maladie [42]. Le tissu synovial rhumatoïde est composé des cellules
résidentes et des cellules qui se sont infiltrées à la suite de
l’inflammation.
L’espace articulaire normal est bordé par des synoviocytes dont il
existe deux types: le type A « macrophage-like » et le type B « fibroblast-
like » qui sont les plus nombreux [43]. Dans la PAR, la membrane
synoviale devient hyperplasique et localement invasive, entraînant la
destruction du cartilage et de l’os [44]. Les synoviocytes de type
fibroblastes vont être activés et deviendront aussi capables plus tard de
présenter des antigènes aux cellules T. Il a été démontré que les
synoviocytes de type B une fois activés sont caractérisés par un
changement de comportement et montrent certaines similarités avec les
tumeurs [45]. À la suite de cela, elles dégradent la matrice cartilagineuse
en s’accrochant fermement à sa surface et en y pénétrant profondément
jusqu’à atteindre l’os avec l’aide de certaines enzymes de dégradation
telles que les MMP et les cathepsines [46]. Cette pénétration se fait grâce
au changement de leur forme pour devenir des cellules de type
macrophage-like [47]. Les MMP préférentiellement exprimées dans le
synovium rhumatoïde sont les MMP1, MMP9, MMP13, MMP14, MMP15.
Les cathepsines B, K et L seront, elles aussi, produites et responsables
de la dégradation du collagène et des protéoglycanes. Les synoviocytes
contribuent aussi à l’inflammation en produisant des cytokines pro-
inflammatoires, des chimiokines et facteurs de croissance [46, 48]. La
matrice cartilagineuse ainsi dégradée en pannus ne sera constituée dans
un premier temps que des cellules ci-dessus citées, mais

25
progressivement elle sera envahie par les autres types cellulaires après
l’apparition des petits vaisseaux sanguins [49].
Les lymphocytes T sont l’un des types cellulaires les plus
abondamment retrouvés dans l’articulation rhumatoïde. Comme nous
l’avons vu plus haut, les cellules T sont impliquées dans la pathogenèse
de la PAR en vertu des facteurs génétiques. Plusieurs sous-types de
lymphocytes T sont présents dans le synovium influencés par les
cytokines produites. Les lymphocytes T helper (Th) sont les plus
importants et favorisent l’inflammation. Plusieurs études ont qualifiées la
PAR de « maladie à Th1 » [50]. La différenciation des cellules T naïves en
cellules Th1 est accompagnée de la production de cytokines pro-
inflammatoires telles que l’IFN, et le TNF. Le TNF semble être la
cytokine à la tête de la cascade de cytokines pro-inflammatoires. Elle est
d’autant plus importante que son blocage entraîne la diminution de
l’expression de bon nombre de cytokines telles que l’IL-8, l’IL-6, l’IL-1 et
même une diminution de l’évolution de la maladie [40]. Le TNF régule la
production des cytokines pro-inflammatoires mais peut être lui aussi
régulé par l’IL-1, le GM-CSF (Granulocyte/Macropage-Colony Stimulating
Factor, facteur stimulant les granulocytes/macrophages), l’IFN[41]. On
observe une prédominance de la production des cytokines Th1 au
détriment des cytokines Th2 telles que l’IL-4 et l’IL-13 qui elles sont anti-
inflammatoires [51]. Deux autres sous-types de cellules T sont aussi
présents dans l’articulation rhumatoïde ; il s’agit des cellules T
régulatrices (Treg) et des cellules Th17. Il existe un débalancement en
faveur des Th17. En effet elles produisent de l’IL-17, cytokine pro-
inflammatoire que l’on retrouve dans les maladies inflammatoires
chroniques [35]. L’IL-17 va agir sur bon nombre de cellules retrouvées
dans l’articulation rhumatoïde telles que les monocytes, macrophages,
fibroblastes, ostéoclastes et chondrocytes. Les cellules Treg retrouvées
dans le sang périphérique de patients atteints de PAR présentent

26
quelques anomalies ; en effet elles sont incapables d’inhiber la
production du TNF et de l’IFN [35, 52]. Les cellules T de la PAR
présentent une modification qui diminue leur activation, en effet, la
tyrosine phosphorylation de la chaîne du CD3 de ces cellules n’a pas
lieu [33]. Cette diminution de l’activation des cellules T peut aussi être
dûe à la présence de l’IL-1Ra et de TGF qui sont des inhibiteurs de
lymphocytes T.
Plusieurs études ont démontré que la PAR est une maladie qui
dépend énormément des lymphocytes B. Une déplétion de cellules B sur
des patients en utilisant un anticorps monoclonal anti-CD20 entraîne au
bout de plusieurs semaines de traitement, une baisse du taux de
facteurs rhumatoïdes jusqu’à un taux normal suivi d’une rémission de la
maladie (diminution de la raideur et du gonflement des articulations) [53,
54]. Cette expérience montre le rôle des lymphocytes B dans
l’amplification de la maladie par la production d’auto-anticorps, ce qui
dénote de leur importance dans la physiopathologie de la maladie. Elles
jouent aussi le rôle de cellules présentatrices d’antigène aux cellules T et
sécrètent des cytokines pro-inflammatoires telles que l’IL-10 et le TNF
[53].
Monocytes et macrophages sont, eux aussi, impliqués dans la
pathologie. En effet, ils sécrètent des cytokines pro-inflammatoires telles
que le TNF, l’IL-1 et l’IL-6 [37]. Le TNF et l’IL-1 sont impliqués dans la
dégradation du cartilage car elles induisent les MMP destructrices. Le
TNF stimule aussi l’expression de molécules d’adhésion par les
fibroblastes. En plus des MMP, les monocytes/macrophages sécrètent
des agents chimioattractants, ce qui leur confère un potentiel
destructeur qui contribue à la destruction articulaire dans la PAR aigüe
et chronique [55]. L’IL-1 ainsi produite régule l’inflammation en activant
les cellules B et T et en induisant l’expression de molécules d’adhésion

27
par les cellules endothéliales [56]. Le TNF et l’IL-1 semblent fonctionner
en synergie pour leurs fonctions effectrices [23]. L’IL-6 n’est pas
seulement produite par les monocytes/ macrophages, mais aussi par les
fibroblastes et lymphocytes T. L’IL-6 est responsable de l’inhibition de la
formation osseuse et de la stimulation de la résorption à travers son effet
stimulateur sur les ostéoclastes. En effet, les souris «knockout » pour le
gène de l’IL-6 ne développent pas d’érosion osseuse [41]. Cette cytokine
promeut la prolifération des cellules B, de même que la production
d’anticorps et l’activation des cellules T [40].
Les neutrophiles aussi sont retrouvés dans l’articulation synoviale
en grande quantité, il s’agit de la cellule de l’immunité innée qui est
toujours principalement impliquée dans les situations inflammatoires.
Les neutrophiles dans la PAR sont beaucoup plus que des cellules
phagocytaires, ils produisent plusieurs médiateurs qui peuvent les auto-
activer et activer d’autres cellules, ils peuvent être des cellules
présentatrices d’antigènes, de même que devenir des cellules « osteoclast-
like » [57]. Les neutrophiles sécrètent des cytokines pro-inflammatoires
telles que le TNF, des enzymes telles que les collagénases et des anions
réactifs tels que l’O2-, responsables de la destruction du cartilage [57,
58]. Les neutrophiles produisent aussi l’enzyme peptidyl-arginine
déimiase (PAD) qui est responsable de la citrullination de l’arginine d’où
la production de peptides citrullinés [59]. Les neutrophiles participent à
l’ostéo-immunologie car ceux retrouvés dans le liquide synovial
expriment à leur surface du RANK, un RANK activé par RANKL, et les
protéines de la voie de signalisation de RANK [60]. Nous savons déjà
l’importance de la voie RANK/RANKL dans l’activation de la résorption.
Les neutrophiles, de ce fait, montrent donc leur importance dans la
physiopathologie de la maladie par les interactions cellules-cellules qui
peuvent être initiées pour amplifier la résorption osseuse par les
ostéoclastes [61]. Les modèles animaux de PAR tels que les souris K/BxN

28
et l’arthrite induite au collagène (CIA : collagen type II induced arthritis)
ont été utilisés pour montrer l’importance des neutrophiles dans la
physiopathologie de la maladie. En effet, dans ces modèles animaux, on
constate une inhibition du développement de la maladie et une
amélioration de la santé des souris traitées [62, 63] lorsque les
neutrophiles sont inhibés par un anticorps anti Gr-1 (très exprimé sur
les neutrophiles).
Les cytokines et les cellules impliquées dans la PAR forment un
réseau complexe qu’il est difficile de détailler, mais elles constituent une
cible thérapeutique intéressante pour la prise en charge de la maladie
[64]. Nous les avons résumées brièvement dans les figures 9 et 10. Le
profil des cytokines retrouvées dans l’articulation est modifié avec le
temps; l’arthrite précoce a un profil de cytokines différent de celui de
l’arthrite chronique [32]. La présence de l’IL-12 par exemple serait
détectée en grande quantité dès le début de la maladie; tandis que plus
la maladie évolue, plus faible sera le taux de l’IL-12 [41].

29
Figure 9: Voies de signalisation des cytokines impliquées dans la
polyarthrite rhumatoïde [40]:
Les principales cellules impliquées dans la destruction articulaire à travers l’IL-1 et le
TNFα sont représentées, de même que les différentes cellules cibles de la plupart des
cytokines impliquées.
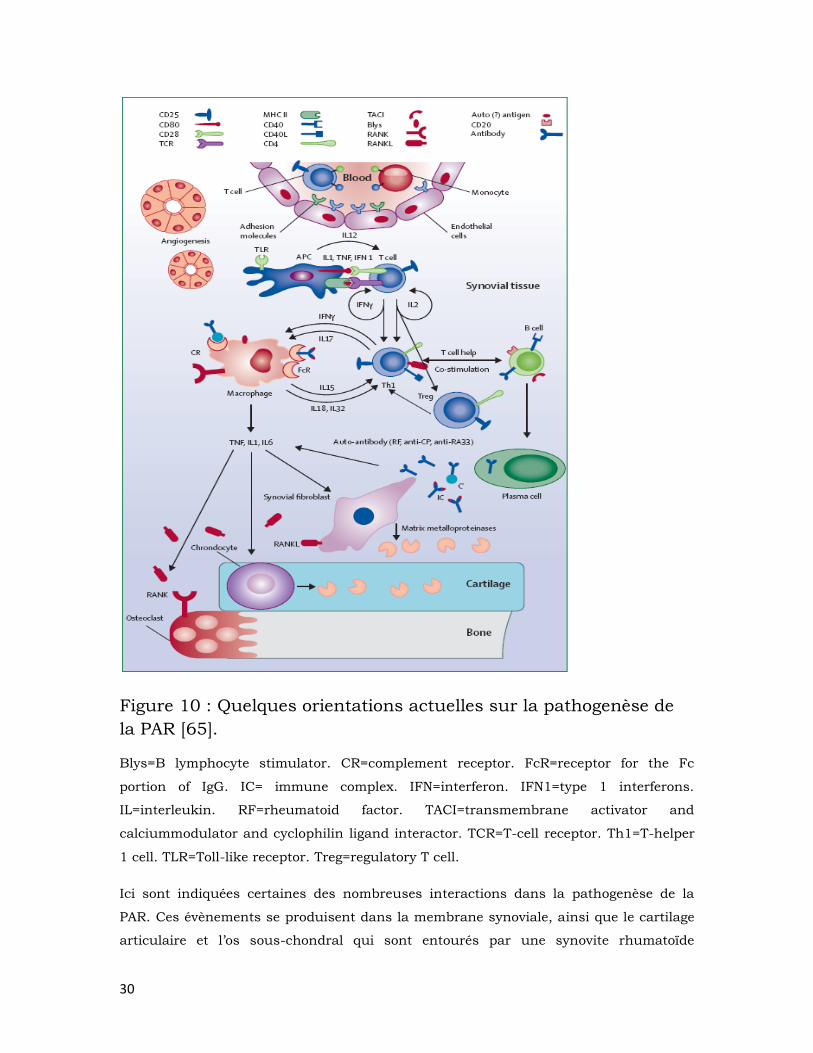
30
Figure 10 : Quelques orientations actuelles sur la pathogenèse de
la PAR [65].
Blys=B lymphocyte stimulator. CR=complement receptor. FcR=receptor for the Fc
portion of IgG. IC= immune complex. IFN=interferon. IFN1=type 1 interferons.
IL=interleukin. RF=rheumatoid factor. TACI=transmembrane activator and
calciummodulator and cyclophilin ligand interactor. TCR=T-cell receptor. Th1=T-helper
1 cell. TLR=Toll-like receptor. Treg=regulatory T cell.
Ici sont indiquées certaines des nombreuses interactions dans la pathogenèse de la
PAR. Ces évènements se produisent dans la membrane synoviale, ainsi que le cartilage
articulaire et l’os sous-chondral qui sont entourés par une synovite rhumatoïde

31
agressive. Le liquide synovial est rempli de plusieurs types cellulaires, plusieurs
cytokines et les interactions entre tous ces éléments entretiennent la pathologie.
1.3.2.4. Les auto-anticorps
Une des spécificités de la PAR, comparativement aux autres
arthrites, est la notion d’auto-immunité. En effet, on retrouve dans le
liquide synovial des malades des auto-anticorps produits par des cellules
B. Depuis la découverte du facteur rhumatoïde (FR) qui est un anticorps
dirigé contre la région Fc des IgG, plusieurs autres auto-anticorps ont été
découverts. Ces auto-anticorps peuvent être dirigés contre les
composantes du cartilage, les protéines de stress, les enzymes, les
protéines nucléaires et même les protéines citrullinées. Tous contribuent
à la pathophysiologie en formant des complexes immuns dans
l’articulation rhumatoïde. Les FR et les ACPA sont les deux auto-
anticorps utilisés comme critères diagnostiques de la maladie, bien que
les FR soient beaucoup moins spécifiques que les ACPA. En effet, on peut
retrouver les FR dans d’autres maladies auto-immunes et même dans
des conditions non auto-immunes et parfois même chez des personnes
saines. Malgré cela, le FR reste le marqueur sérologique le plus utilisé
dans la PAR [22].
1.3.2.4.1. Les anticorps anti-protéines citrullinées
Comme leur nom l’indique, les ACPA sont des anticorps dirigés contre
des épitopes qui contiennent des acides aminés citrullinés. La
citrullination est une modification post-translationnelle des résidus
arginyl en résidus citrulline sous l’action des enzymes PAD (figure 7). Les
peptides citrullinés retrouvés dans l’articulation rhumatoïde sont les
chaînes et de la fibrine, la vimentine et l’-énolase. Les ACPA sont
présents dans l’articulation rhumatoïde très tôt dans le développement
de la maladie, ils constituent donc un critère important pour le

32
diagnostic. À ce sujet, 50 à 70 % des patients diagnostiqués comme
ayant un début de PAR sont positifs pour les ACPA [38]. Les ACPA ont
une spécificité diagnostique de près de 95% et une sensibilité
approximative de 75%. La présence d’ACPA est un critère essentiel pour
établir le pronostic de la maladie. Des études ont montré une corrélation
positive entre les arthrites présentant des ACPA et la dégradation
osseuse [22, 66]. Les patients ACPA positifs présentent une plus grande
sévérité dans le développement de la maladie [67].
Les ACPA constituent un outil de prise en charge important car la
présence de ces anticorps peut être détectée plusieurs années avant
l’apparition des premiers symptômes et permettre ainsi de commencer
un traitement pour ralentir l’évolution de la maladie.
1.3.2.4.2. Les facteurs rhumatoïdes
Les facteurs rhumatoïdes ont été décrits pour la première fois par
Emil Waaler en 1940 [22] et ils ont été redécouverts plus tard par Rose
en 1948 lorsqu’il a découvert leur capacité à agglutiner les globules
rouges de mouton enrobés de sérum de lapin [68]. Depuis ces
découvertes, les FR constituent l’un des outils majeurs pour le diagnostic
de la PAR. Plusieurs techniques permettent en clinique de détecter la
présence de FR dans le liquide synovial et le sérum. Parmi elles, nous
pouvons citer : le test classique de Waaler-Rose et l’ELISA (Enzyme
Linked Immunosorbent Assay). Bien que les FRs soient les auto-anticorps
les plus recherchés pour le diagnostic, leur rôle précis dans la
pathogenèse de la PAR reste encore partiellement compris. Le FR n’est
retrouvé que chez 60 à 80 % des personnes atteintes, avec une spécificité
de 85% [69].
Le rôle physiologique des FR au cours d’une réponse immunitaire
normale est d’augmenter l’avidité et la taille des complexes immuns. De
plus, les cellules B liées aux FR vont agir comme des cellules

33
présentatrices d’antigène et peuvent présenter efficacement les anticorps
aux cellules T. Par conséquent, il est supposé que les FR du sérum et du
liquide synovial des patients atteints de PAR, et en particulier ceux
attachés aux cellules B, vont exercer ces fonctions et ainsi contribuer à
la physiopathologie de la maladie [70]. Un autre rôle important des FR
est la fixation du complément. Les complexes immuns présents dans
l’articulation rhumatoïde et la fixation du complément par les IgG des
complexes immuns sont accentués par la fixation des FR IgM. Cela est
d’autant plus important pour les complexes immuns composés par des
auto-anticorps [22].
Il existe trois sous types de FR: ceux du type IgM, type IgA et le
type IgG. Le FR de type IgM est le type principal, mais les deux autres
sont également importants et donnent des informations majeures pour le
diagnostic. Un seul patient peut posséder plus d’un sous type de FR;
dans ce cas, les sous types le plus souvent retrouvés sont les types IgM
et IgA [71-73]. Plusieurs études suggèrent que le FR de type IgA est un
marqueur plus spécifique de la PAR. Il a été également démontré que la
détection des FR permet d’établir un pronostic dans l’évolution de la
maladie; ils seraient présents très tôt dès le déclenchement de la maladie
et longtemps avant l’apparition des premiers symptômes. Les patients
avec un titre élevé et persistant de FR ont un risque plus accru de
développement et d’aggravation de la maladie; en plus, ceux qui ont les
FR de type IgM et IgA ont un risque encore plus élevé que ceux qui ont
juste un seul type de FR [74]. En effet, le titre élevé de FR est associé à
l’érosion osseuse et aux manifestations extra articulaires. Malgré
l’importance apparente des FR dans la physio-pathologie de la maladie,
on retrouve des PAR séropositives et des PAR séronégatives, ce qui
suggèrent deux mécanismes distincts d’évolution de la maladie [75, 76].
Le rôle des FR dans la formation de complexes immuns au cours
de l’évolution de la maladie a été démontré, mais cependant aucune

34
expérience n’explique sans ambiguïté le rôle de ces facteurs dans les
évènements déclencheurs.
1.3.2.5. Les marqueurs de l’arthrite rhumatoïde
La PAR est une maladie complexe et d’origine encore inconnue à ce
jour. En effet, elle présente plusieurs aspects qui ne peuvent pas être
considérés séparément. Des chercheurs, à la suite de plusieurs études,
ont établi certains critères qui permettent de diagnostiquer la maladie et
par conséquent de la traiter. En 1987, une classification a été publiée et
les malades qui possédaient 4 de ces critères étaient considérés comme
atteints de PAR (tableau 2). Cette classification s’est basée sur les
observations cliniques de nombreux patients et, sur l’évolution des
découvertes dans le domaine.
Tableau 2: Critères de classification de l’arthrite rhumatoïde,
classification de 1987 par le collège américain de rhumatologie [77].
Cette classification s’est cependant révélée insatisfaisante pour
diagnostiquer les arthrites précoces, car ces critères sont basés sur des
manifestations qui surviennent lorsque la maladie est déjà bien installée.
Ainsi, depuis 2007 la ligue européenne contre le rhumatisme et le collège
américain de rhumatologie ont mené des études pour déterminer de
nouveaux critères de classification qui mettent l’accent sur la

35
participation des petites articulations et la séropositivité aux FR et aux
ACPA [78] (tableau 3). Cette nouvelle classification, établie en 2010,
permet de mieux diagnostiquer la maladie, plus spécifiquement et plus
rapidement afin de pouvoir ralentir sa progression.
Tableau 3: Tableau de classification des critères diagnostics de
l’arthrite rhumatoïde établis en 2010 par le collège américain de
rhumatologie et la ligue européenne contre le rhumatisme [79].
1.3.2.6. Les traitements de l’arthrite rhumatoïde
La PAR reste de nos jours une maladie complexe avec une
destruction des articulations et des complications secondaires à
l’inflammation systémique. Elle est impossible à guérir mais malgré cela,
les récentes découvertes dans la pathophysiologie ont permis de
nombreux changements dans la gestion de cette maladie. Les
médicaments de la PAR portent le nom de DMARD (Disease Modifying
Anti-Rheumatic Drugs, drogues anti-rhumatiques modifiant la maladie )
et ils ont pour rôle de réduire l’inflammation et de ralentir la progression

36
de la destruction articulaire, mais ils le font à des degrés variables [80,
81]. Les scientifiques s’accordent à dire que, plus tôt le traitement est
initié, mieux cela vaut pour les malades. Les DMARD vont être divisés en
deux groupes: les DMARD conventionnels et les DMARD biologiques [80].
Les premières thérapies incluaient aussi des corticoïdes et des anti-
inflammatoires non stéroïdiens [23]. Parmi les DMARD conventionnels, le
plus utilisé est le méthotrexate (MTX) [82]. L’utilisation du MTX présente
des effets toxiques [83], mais son administration se fait en association
avec l’acide folique qui va agir pour contrôler les effets secondaires
consécutifs à son administration tels que l’alopécie, la stomatite,
l’intolérance gastro-intestinale et la toxicité hématopoïétique [23, 84-86].
Nous pouvons citer le Leflunomide et la Sulfasalazine parmi les autres
DMARD conventionnels.
Les DMARD biologiques sont une nouvelle catégorie de
médicaments qui ciblent les anomalies spécifiques du système
immunitaire, plus précisément les molécules qui ont montré leur rôle
dans la physio-pathologie de la maladie [80]. Dans cette nouvelle
catégorie de traitements, ceux qui ciblent le TNF sont les plus employés.
Les principales drogues anti-TNF sont les suivantes : Infliximab,
Etanercept, Adalimumab, Certolizumab, Golimumab [87]. Malgré
l’efficacité de ces drogues anti-TNF, environ 30% des patients ne
répondent pas à ce traitement [80].
1.3.2.7. La résorption osseuse dans l’arthrite rhumatoïde
L’érosion osseuse représente l’un des points importants dans la
gravité de la PAR. Dans l’articulation rhumatoïde, la résorption se fait à
deux sites principaux: le cartilage et l’os sous-chondral. L’érosion du
cartilage est due aux cellules du pannus rhumatoïde [88-90]. La
résorption osseuse est le fait de deux types cellulaires dans l’arthrite : les
synoviocytes de type macrophages et les ostéoclastes. Ces derniers

37
représentent le principal type cellulaire impliqué dans ce processus [91].
Il est à noter que la résorption osseuse dans la PAR est accompagnée de
la diminution de la formation de la nouvelle structure osseuse [92], ce
qui créé un énorme débalancement dans le remodelage osseux. Les
causes de la perte osseuse dans la maladie ne sont pas parfaitement
connues à ce jour mais les changements hormonaux, la vitamine D, le
métabolisme minéral et le taux de cytokines circulants telles que l’IL-1 ,
l’IL-6 et le TNF vont être impliqués dans la pathogenèse de cette perte
osseuse [4, 93]. Ces différents facteurs vont agir à plusieurs niveaux sur
la résorption: sur les précurseurs ostéoclastiques, les ostéoclastes et
même sur les ostéoblastes [94]. La capacité de résorption, de même que
le nombre d’ostéoclastes seront accrus.
L’environnement inflammatoire de l’articulation rhumatoïde va être
à l’origine de l’augmentation du potentiel de résorption des ostéoclastes
avec un accent mis sur l’IL-1 et le TNF qui sont deux cytokines
importantes de l’inflammation. Un intérêt particulier est porté à
l’expression de RANKL chez les patients car nous savons l’importance de
la voie RANK/RANKL/OPG dans l’activation des ostéoclastes. Certaines
études ont montré que l’ARNm de RANKL, de même que la protéine, sont
exprimés dans le tissu synovial des patients atteints de PAR [95, 96], ce
qui n’est pas le cas chez les patients non atteints [92]. Dans l’articulation
rhumatoïde, RANKL n’est pas seulement exprimé par les ostéoblastes; les
cellules T activées, les synoviocytes de type fibroblastes et les
macrophages l’expriment aussi [97, 98]. Cette surexpression de RANKL
dans le milieu va entraîner une activation accrue des ostéoclastes, ainsi
qu’une augmentation du nombre d’ostéoclastes différenciés [99]. Le TNF
et le RANKL agissent de concert pour augmenter la différenciation des
ostéoclastes, tandis que l’IL-1 active les ostéoclastes déjà formés et
retarde l’apoptose normale de ces cellules. Le TNF et l’IL-1 peuvent
aussi agir sur les ostéoblastes en portant atteinte au processus de

38
formation osseuse en induisant l’apoptose des ostéoblastes [95].
L’environnement inflammatoire peut aussi agir sur les précurseurs
d’ostéoclastes en régulant en amont l’expression de RANKL sur les
cellules stromales de la moelle osseuse et en aval l’expression d’OPG par
les précurseurs d’ostéoclastes [93, 100].
Certaines études ont, de plus, démontré que les monocytes et les
macrophages présents dans l’articulation rhumatoïde pourraient être
capables de se différencier en ostéoclastes [101, 102]. En effet, Danks L
et ses collaborateurs ont montré qu’in vitro, les monocytes et
macrophages de patients atteints d’arthrite rhumatoïde sont capables de
se différencier en ostéoclastes en présence de M-CSF et de la calcitriol
[102]. Les macrophages synoviaux produisent eux aussi la calcitriol, de
même que le M-CSF et les autres facteurs indispensables à la
différenciation des ostéoclastes, ce qui leur permettrait de se différencier
en ostéoclastes, le milieu étant aussi riche en RANKL provenant des
ostéoblastes, des synoviocytes, des macrophages et des cellules T. Ceci
pourrait expliquer l’augmentation de l’activité de résorption osseuse
observée.
L’ostéoclaste est une cellule clé dans la pathologie de la PAR. Une
meilleure compréhension de leur fonctionnement dans le contexte de
cette maladie pourrait permettre le développement de nouvelles thérapies
plus spécifiques et plus efficaces.
1.4. LES CELLULES HL60
La lignée de cellule HL60 a été dérivée à partir de leucocytes du
sang périphérique d’une femme de 36 ans de race blanche avec une
leucémie aiguë promyélocytaire [103]. Elle est l’une des lignées humaines
de cellules leucémiques capable de prolifération apparemment illimitée

39
en culture en suspension [104]. En culture, ce sont des cellules rondes
ou ovoïdes qui peuvent occasionnellement présenter des pseudopodes et
être de taille variable (de 9 à 25 µm de diamètre) [103]. Elle est la
première lignée cellulaire humaine avec des caractéristiques myéloïdes à
avoir été développée [105]. Des études histochimiques utilisant des
marqueurs pour les cellules myéloïdes ont bien montré le caractère
myéloïde de ces cellules [106]. Les cellules myéloïdes HL60 semblent être
uniques quant à leur capacité de proliférer sans une source exogène
d’inducteur, elles ont juste besoin d’un milieu nutritif [106]. Les cellules
HL60 se multiplient rapidement en culture, elles ont un temps de
doublement de 24h quand les conditions de culture sont optimales.
Cette lignée cellulaire a toujours suscité un grand intérêt grâce à
sa capacité de se différencier en plusieurs types cellulaires différents. Les
conditions de culture influencent grandement la différenciation des
HL60. À la suite d’un arrêt de la division cellulaire, différentes voies
d’activation peuvent aboutir à différents types cellulaires. Une étude a
démontré que la présence de vitamine D (1,25(OH)2D3) dans le milieu de
culture entraîne la différenciation des HL60 en cellules de type
monocytes/macrophages [107, 108]; ce qui induit l’expression d’un
RANK fonctionnel dans les cellules HL60. L’expression de RANK dans ces
cellules peut être régulée de manière positive par RANKL pour augmenter
ses propres effets dans la différenciation [109]. Plusieurs études ont
démontré que les esters de phorbol sont aussi capables de différencier les
HL60 en cellules de type macrophage [110-112], notamment en leur
permettant d’acquérir une capacité d’adhérence. Les HL60 peuvent aussi
se différencier en cellules de type neutrophile si elles sont mises en
présence de différents stimuli tels que le dibutyryl-AMP ou le diméthyl
sulfoxide [112-115].
Les lignées cellulaires leucémiques ont été beaucoup utilisées au
départ pour l’étude des maladies leucémiques car elles offrent l’avantage

40
de permettre d’étudier l’induction de la différenciation des cellules
myéloïdes [116, 117]. Mais au cours de ces études, il a été constaté que
plusieurs types de composés tant physiologiques que non physiologiques
pouvaient affecter l’induction de la différenciation de ces cellules [117].
Elle est depuis utilisée pour pouvoir étudier les cellules telles que les
monocytes/macrophages qui, in vivo, découlent de cette voie de
différenciation myéloïde. L’intérêt majeur de l’utilisation de cette lignée
cellulaire pour la recherche est leur multi-potentialité et la
reproductibilité des expériences l’impliquant; c’est pourquoi elle est
largement utilisée comme modèle de différenciation lorsqu’on veut
étudier certains types cellulaires [103]. Elle est la lignée cellulaire
myéloïde la plus utilisée pour des expériences de différenciation [105].
1.5. BUTS DE L’ÉTUDE
Il a été démontré que la lignée cellulaire HL60 peut être
programmée pour se différencier en différents types cellulaires. Les
différentes voies de différenciation dépendent des stimuli ajoutés. Ainsi,
en présence de calcitriol, ces cellules sont capables de se différencier en
cellules de type macrophage [111, 118].
Nous savons qu’in vivo les ostéoclastes se différencient à partir des
précurseurs mononucléés d’origine hématopoïétique de la lignée
monocyte/macrophage. Nos différentes expériences antérieures sur les
ostéoclastes nous ont montré la difficulté d’obtenir des ostéoclastes en
laboratoire. En général, ces ostéoclastes sont obtenus en culture à partir
de monocytes isolés du sang circulant. Mais ce procédé nous a montré
ses limites car les ostéoclastes obtenus par ce procédé montrent une
variabilité dans les expériences, d’un donneur de sang à l’autre. De ce
constat et aidé par une ancienne publication de Yoneda et al. [119], nous
avons émis notre première hypothèse qui stipule que les HL60 peuvent

41
se différencier en cellules de type ostéoclaste, formant ainsi notre modèle
pour la résorption osseuse. Afin de vérifier cette hypothèse, nous nous
sommes fixés un objectif principal: il s’agit de déterminer quelles sont les
conditions optimales qui nous permettraient de différencier des HL60
pour obtenir des cellules de type ostéoclaste et de caractériser les cellules
« osteoclast-like » ainsi obtenues. De manière plus précise, de montrer la
présence des différents éléments caractéristiques qui font des cellules
HL60 différenciées des ostéoclastes proprement dit. Cet objectif
constituera la deuxième partie de ce document.
Le second point d’attention de notre travail était le rôle des FR
dans la PAR. Le rôle de ces FR a été clairement démontré dans le
diagnostic, la prédiction et la pathogenèse de la PAR par la formation de
complexes immuns avec les IgM [73, 74]. Aucune étude n’a cependant
encore montré une action des FR sur les ostéoclastes. De ceci naît notre
seconde hypothèse qui stipule que les FR augmentent la résorption
osseuse en agissant directement sur l’ostéoclaste via son récepteur Fc.
Le récepteur Fcest un des récepteurs de la voie de costimulation des
ostéoclastes ; il n’a cependant aucun ligand connu. Récepteurs et co-
récepteurs envoient un signal qui active plusieurs fonctions de
l’ostéoclaste. Pour vérifier cette hypothèse, nous avons établi un
deuxième objectif qui est de montrer l’effet des FR sur la résorption
osseuse des ostéoclastes. Cet objectif constituera la troisième partie de ce
document.

42
II. DÉTERMINATION DES CONDITIONS
OPTIMALES DE DIFFÉRENCIATION DES
HL60 ET CARACTÉRISATIONS DES
CELLULES HL60 DIFFÉRENCIÉES.
2.1. INTRODUCTION
La lignée de cellules HL60 est l’une des seules lignées humaines de
cellules leucémiques capable de prolifération illimitée [104]. En culture,
ce sont des cellules en suspension rondes ou ovoïdes qui peuvent être de
taille variable [103] et elles ont un temps de doublement de 24 heures.
Elle est la première lignée cellulaire humaine avec des caractéristiques
myéloïdes à avoir été développée [105].
Plusieurs études ont déjà démontré la capacité des HL60 à se
différencier en plusieurs types cellulaires différents [111, 118, 120]. Les
HL60 mis en présence de phorbol 12-myristate 13-acetate (PMA) passent
de l’état de cellules en suspension à celui de cellules adhérentes. Ces
cellules, incubées pendant 6h en présence de PMA, se différencient de
manière irréversible en cellules de type macrophage [111]. In vivo,
l’action de la calcitriol par l’intermédiaire des ostéoblastes, permet aux
précurseurs myéloïdes de se différencier en ostéoclastes. In vitro la
présence de calcitriol est aussi indispensable à la différenciation des
HL60 en cellules multinucléees de type macrophage [118]. Les cellules
HL60 comme les ostéoclastes expriment à leur surface les récepteurs c-
fms du M-CSF ; de ceci, il en découle que les cellules HL60 se
différencient en macrophages sous l’action du M-CSF et du PMA [121].
Plusieurs études ont suggéré que la lignée cellulaire HL60 pouvait être
utilisée pour étudier les ostéoclastes [109, 111, 121]. Ces différentes
études montrent que, tout comme lors du processus de différenciation
des ostéoclastes, les HL60 ont besoin de la présence de calcitriol, de M-

43
CSF et de RANKL pour se différencier en cellules de type ostéoclastes. De
plus, cette différenciation passe par l’activation de la voie de signalisation
NFB. Dans notre étude, nous avons trouvé les conditions optimales qui
nous ont permis d’obtenir des cellules de type ostéoclastes et nous avons
caractérisé ces cellules « osteoclast-like ».
2.2. MATÉRIELS ET METHODES
2.2.1. Réactifs
Les cellules HL60 utilisées provenaient de l’ATCC (American Type
Culture Collection) (Manassas, VA, USA). Le milieu de culture utilisé,
RPMI 1640 supplémenté avec 10% de sérum de veau fœtal (FBS), a été
obtenu de chez Wisent (St-Bruno, QC, Canada). Les monocytes et les
ostéoclastes étaient maintenus en culture dans un milieu MEM
supplémenté avec 10% de FBS obtenu de chez Wisent (St-Bruno, QC,
Canada). Le M-CSF et le RANKL recombinants ont été tous les deux
achetés chez Peprotech (Rocky Hill, NJ, USA). La calcitriol et le Phorbol
12-myristate 13-acetate (PMA) ont été obtenus de chez Sigma Chemical
Co. (St Louis, MO, USA).
Les marqueurs 4’6-diamidino-2 phenylindole (DAPI), Alexa Fluor
488 Phalloïdine et 7-amino actinomycine D (7AAD) ont été obtenus chez
Molecular Probes (Invitrogen Canada Inc. Burlington, ONT, Canada).
L’anticorps polyclonal de lapin dirigé contre la protéine RANK humaine a
été acheté chez Santa Cruz Biotechnology (Santa Cruz, CA, USA).
L’anticorps secondaire reconnaissant les immunoglobulines de lapin a
été obtenu chez Jackson Immunoresearch (West Grove, PA, USA).
L’anticorps secondaire anti-lapin couplé au fluorochrome alexa 568 a été
acheté chez Molecular Probes (Invitrogen Canada Inc. Burlington, ON,
Canada). Les anticorps monoclonaux anti-DAP12 humaine et anti-
TREM2 humaine ont été tous les deux achetés chez R&D Systems

44
(Minneapolis, MN, USA). L’anticorps polyclonal anti-Cathepsine K
humaine a été obtenu chez Biovision (Mountain View, CA, USA).
L’anticorps anti-NFATc humaine provenait de chez Imgenex (San Diego,
CA, USA). L’anticorps anti-récepteur de calcitonine humaine a été acheté
chez Serotec (Raleigh, NC, USA). L’anticorps anti-CD64 humain a été
obtenu chez EMD Biosciences (San Diego, CA, USA). Le kit pour la
détection des cellules TRAP (Tartrate Resistant Acid Phosphatase)
positives a été obtenu chez Sigma Aldrich (St Louis, MO, USA). Les
lamelles de dentine ont été préparées dans notre laboratoire à partir de
dents de morse.
Le Ficoll était de chez Wisent (St Bruno, QC, Canada). Le dextran
a été acheté chez Sigma Aldrich (Oakville, ON, Canada). Le Tween 20 a
été obtenu de chez Sigma Aldrich (Oakville, ON, Canada).
Les standards de poids moléculaires pour les protéines provenaient
de chez Fermentas (Burlington, ON, Canada). Les membranes de
polyfluorure de vinylidène (PVDF) ont été achetées chez VWR (Ville Mont
Royal, QC, Canada). Les réactifs de détection Enhanced
Chemiluminescence (ECL) étaient de chez Perkin Elmer (Montréal, QC,
Canada). Le persulfate d’amonium et l’orthovanadate ont été achetés
chez Sigma Aldrich (Oakville, ON, Canada).
Le kit Superscript™ II RT (Reverse Transcriptase) pour les PCR
(Polymerase Chain Reaction, Réaction de polymerisation en chaine) a été
acheté chez Invitrogen (Burlington, ON, Canada).
2.2.2. Différenciation des cellules
Les cellules HL60 ont été maintenues en culture dans du RPMI
supplémenté à 10% de FBS. Les cellules ont toujours été différenciées en
dessous du quinzième passage, ceci pour éviter de biaiser nos résultats
avec des cellules qui auraient changé de phénotype au cours des

45
passages, de plus le nombre de cellules mortes dans le milieu de culture
augmente au-delà du quinzième passage.
Les cellules ont été incubées à 37°C en présence de RANKL
(40ng/ml), calcitriol (10-8 M), M-CSF (25ng/ml) et PMA (10-8M) pendant
trois jours. Au bout de ces trois jours, les cellules ont été récoltées pour
effectuer les différentes analyses.
2.2.3. Isolement des monocytes et différenciation
en ostéoclastes
Les monocytes ont été isolés à partir de sang humain de donneurs
volontaires sains et recueillis dans des poches contenant de l’isocitrate
comme anticoagulant. Les monocytes ont été isolés selon la méthode
suivante : après avoir déposé du Ficoll dans des tubes de 50 ml
(température ambiante), du sang a été ajouté délicatement dans chacun
de ces tubes (le sang doit rester sur le dessus du Ficoll, ne pas agiter les
tubes). Une centrifugation à 1800 RPM (rotations par minutes) (sans
frein) a été effectuée pendant 30 minutes à la température de la pièce,
puis l’anneau de cellules à l’interface des 2 phases a été prélevé très
lentement, sans prélever de sérum ni de globules rouges. Nous avons
ensuite mélangé deux anneaux dans un tube de 50 ml et complété avec
du HBSS (Hank’s Balanced Salt Solution) 1X. Après avoir centrifugé les
tubes à 1400 RPM pendant 10 minutes à la température ambiante, les
surnageants ont été aspirés, les culots resuspendus dans deux tubes de
50 ml avec du HBSS 1X et le volume complété à 50 ml. Une
centrifugation a ensuite été effectuée à 1400 RPM pendant 10 minutes à
la température ambiante et le culot resuspendu dans 10 ml d’un
mélange de ½ Ficoll et ½ HBSS 1X déposé dans le fond du tube. Après
une centrifugation à 1000 RPM sans frein pendant 7 minutes, le culot
est resuspendu pour lavages. Ce dernier culot contient les monocytes et
lymphocytes.

46
Le procédé de différenciation des monocytes en ostéoclastes est le
suivant : le dernier culot obtenu après l’isolation des monocytes a été
resuspendu dans un petit volume de milieu MEM + 10 % FBS + 1 %
penicilline-streptomycine-fungizone. Ceci pour effectuer un comptage des
cellules. Une fois les cellules comptées, elles ont été resuspendues dans
un plus grand volume de αMEM pour les avoir à la concentration de 5
millions de cellules par milli-litres. Déposées ensuite dans une plaque de
plastique de 24 puits, à raison de 1 milli-litre par puit, les cellules ont été
laissé adhérer pendant une heure et demie dans un incubateur à 37°C.
Après l’adhérence, le milieu qui contient les lymphocytes a été enlevé car
les monocytes ont adhéré à la plaque de culture. Après un lavage avec du
PBS (Phosphate-Buffered Saline), 600 l du milieu de différenciation a
été mis dans chaque puits. Le milieu de différenciation est le suivant :
+ RANKL (40 ng/ml) + M-CSF (25ng/ml) + calcitirol (10-8M). Le
milieu de différenciation a été changé 2 fois par semaine en enlevant tout
le milieu pour le remplacer par 600 µl d’un nouveau milieu de
différenciation. Les cellules sont différenciées en ostéoclastes au bout de
10 jours. Pour être utilisées, les cellules ont été décollées des plaques de
culture à l’aide du milieu accutase : après avoir aspiré le milieu de
différenciation sur les cellules, les avoir lavées 2 fois avec du PBS, 1
milli-litre du milieu accutase a été mis dans chaque puit et laissé en
action pendant 15 à 30 minutes à 37°C. Au terme de ce temps, les
cellules ont été récupérées avec une pipette en faisant des allers et
retours avec le liquide pour bien les aider à se décoller. Les cellules ainsi
récoltées ont été ensuite lavées dans du PBS, puis comptées. Elles sont
prêtes à être utilisées
2.2.4. Immunobuvardages
Après la différenciation, les HL60 ont été lavées dans du PBS, puis
lysées dans du tampon Laemmli 1x et chauffées à 95°C pendant 10
minutes (62.5 mm Tris/HCl [pH6.8], 4% SDS [m/vol], 5% 2-

47
mercaptoethanol [vol/vol], 8.5% glycerol [vol/vol], 2.5 mM orthovanadate,
10 mM para-nitrophenylphosphate, 10 g/ml leupeptine, 10 g/ml
aprotinine, et 0.025% de bleu de bromophénol). Les ostéoclastes ont
aussi été préparés de la même manière. Les protéines ont ensuite été
séparées par électrophorèse sur des gels SDS-PAGE à 12% d’acrylamide
pour la cathepsine K, TREM2 et DAP12; 7,5% pour le CD64; 10% pour le
NFATc et les récepteurs de calcitonine. Une fois séparées selon leur taille,
les protéines ont été transférées sur membranes de PVDF à 4°C. Les sites
de liaisons non spécifiques des anticorps ont été bloqués avec une
solution de lait écrémé en poudre 5% dans du TBS-Tween (Tris-Bufferd
Saline) (Tris HCl 50 mM, NaCl 150 mM, pH 7,6; 0,1% Tween 20) pendant
20 minutes. La détection de la Cathepsine K a nécessité un blocage avec
de la gélatine de porc à 2% dans du TBS-Tween pendant 60 minutes à
37°C. Après le blocage, les différents anticorps primaires ont été ajoutés:
cathepsine K (1g/ml) pendant 1 heure à 37°C, TREM2 (1g/ml), DAP-12
(2 g/ml), NFATc (2 g/ml), récepteur de calcitonine (2g/ml) toute la
nuit à 4°C. Après trois lavages de 5 minutes dans du TBS-Tween, les
différents anticorps secondaires couplés à la HRP dilués dans du TBS-
Tween (1:20 000) ont été ajoutés et laissés pendant 30 minutes: anti IgG
de souris pour TREM2, DAP12, NFATc, Récepteurs de calcitonine et anti
IgG de chèvre pour cathepsine K. Enfin, les membranes ont été lavées
trois fois pendant 10 minutes dans du TBS-Tween. La révélation a été
effectuée à l’aide du système de détection ECL et la visualisation à l’aide
des films de révélation. Un temps d’exposition variable a été nécessaire
pour chacune des différentes protéines recherchées. Nous avons mesuré
la quantité de chaque protéine contenue dans les cellules et l’avons
exprimé en unité de densitométrie. Cette unité arbitraire correspond au
rapport de la densité optique de la bande d’électrophorèse de la protéine
concernée sur la densité optique de la bande d’électrophorèse de la
protéine référence: l’actine.

48
2.2.5. Analyses par RT-PCR
Après la différenciation, les cellules ont été lavées dans du PBS,
l’ARN total a été isolé suite à la lyse des cellules dans du Trizol (environ 2
millions de cellules dans 1 milli-litre de trizol). La réaction de reverse
transcriptase (RT) a été effectuée suivant le protocole du fabricant du kit
Superscript™ II RT avec 2µg d’ARN. Les ADN complémentaires (ADNc)
obtenus ont été amplifiés par une réaction de polymérase en chaîne
(PCR) en utilisant des paires d’amorces spécifiques pour chacun des
gènes ciblés. Chaque PCR a été effectuée avec 2 µl d’ADNc obtenu après
la reverse transcription, 10 µM d’amorces spécifiques soit 1µl; 100 µM de
dNTPs soit 0,5 µl ; 2,5µl de tampon PCR 10X (200 mM Tris-HCl pH 8,4,
500 mM KCl); 0,5 U d’enzyme Taq polymerase de chez Feldan; de l’eau
distillée pour obtenir un volume final de 25 µl. Le nombre de cycles
effectués a été de 30. La température de dénaturation a été de 90°C, celle
de l’hybridation des amorces a été de 60°C et celle de l’élongation, de
72°C. Le gène de la GAPDH (Glyceraldehyde 3-phosphate déhydrogénase)
humaine a été utilisé comme gène référence. Les différents produits de la
PCR ont été séparés par électrophorèse sur un gel d’agarose à 1% dans
du tampon TAE (Tris Acetyl EDTA), en présence du bromure d’éthidium.
Nous avons quantifié les différents ADNc qui correspondent à
l’expression des gènes et les avons exprimés en unité de densitométrie.
Cette unité arbitraire correspond au rapport de la densité optique de
chaque bande d’électrophorèse du gène concerné sur la densité optique
de la bande d’électrophorèse du gène référence GAPDH. Les séquences
des amorces des différents gènes recherchés ont été les suivantes :
TREM2 S (sens): 5’-GTC-TTG-CCC-CTA-TGA-CTC-CA-3’ AS (anti sens): 5’-CTG-GTA-GAG-ACC-CGC-ATC-AT-3’
S: 5’-CCA-AGC-ATC-CTA-TCC-CTC-AA-3’ AS: 5’-ATG-CAT-TTT-GCA-GGC-AGA-AC-3’

49
DAP-12 S: 5’-ACT-GAG-ACC-GAG-TCG-CCT-TA-3’
AS: 5’-TCT-CTC-AGG-GAT-GGC-ACT-CT-3’
PIR-A S: 5’-GCT-CAG-TAG-CAC-AAC-CAC-CA-3’ AS: 5’-TTA-GTC-CGC-TGC-TGA-CCT-TT-3’
OSCAR S: 5’-CCC-AGC-TTC-ATA-CCA-CCC-TA-3’ AS: 5’-GAA-GAG-AAG-GGG-AGC-GAT-CT-3’
Cathepsine K S: 5’-TTC-TGC-TGC-TAC-CTG-TGG-TG-3’ AS: 5’-GCC-TCA-AGG-TTA-TGG-ATG-GA-3’
MMP9 S: 5’-TTG-ACA-GCG-ACA-AGA-AGT-CG-3’ AS: 5’-GCC-ATT-CAC-GTC-GTC-CTT-AT-3’
NFATc S: 5’-AGA-AAG-CGA-AGC-CAG-TAC-CA-3’ AS: 5’-CGG-TCT-CAC-TAA-CGG-GAC-AT-3’
RANK S: 5’-TTG-CAG-CTC-AAC-AAG-GAG-AC-3’ AS: 5’-AGC-TGG-CAG-AGA-AGA-ACT-GC-3’
GAPDH S: 5’-CGA-GAT-CCC-TCC-AAA-ATC-AA-3’ AS: 5’-TTC-ACA-CCC-ATG-ACG-AAC-AT-3’
2.2.6. Immunohistochimie
Marquage de l’actine
Les cellules (2 millions de cellules/ml) ont été différenciées pendant
3 jours dans des plaques de 6 puits sur des lamelles de verre. Après la
différenciation, nous avons effectué deux lavages de 5 minutes dans du
PBS puis les cellules ont été fixées dans du paraformaldéhyde (PFA) 4%
pendant 15 minutes. Après la perméabilisation des cellules avec une
solution de PBS contenant 1% de BSA (Bovine Serum Albumin) et 0,1%
de triton X100 pendant 20 à 30 minutes, nous avons procédé au

50
marquage des cellules avec l’Alexa Phalloidine 488 (5 U/ml dans du PBS-
BSA-Triton X100) pendant 30 minutes à la température ambiante et à
l’abri de la lumière. Ensuite, nous avons procédé à la contre coloration
pour marquer les noyaux avec un marqueur d’ADN moléculaire ; soit
avec le DAPI (30 nM dans du tampon de blocage) pendant 5 minutes, soit
avec le 7AAD (40 g/ml dans de l’eau) pendant 45 minutes. Les
marquages ont été faits à l’abri de la lumière. La visualisation des
cellules a ensuite été effectuée au microscope optique à fluorescence en
utilisant les filtres appropriés aux différents fluorochromes après avoir
déposé sur les cellules de l’huile de montage ; aux grossissements 40X et
60X. Pour la visualisation des anneaux d’actine, nous avons utilisé un
microscope confocal. Les longueurs d’onde (excitation/émission) étaient
de 358/461 nm pour le DAPI, 546/647 nm pour 7AAD et 495 /518 nm
pour l’Alexa Phalloïdine 488.
Marquage de RANK:
Après différenciation, lavages et fixation comme précédemment, les
cellules ont été perméabilisées avec de la saponine 0,3% dans du PBS
pendant 20 à 30 minutes. Ensuite les sites de liaison non spécifiques des
anticorps ont été bloqués avec 10% de sérum humain dans du PBS
pendant 1 heure à temperature ambiante, dans une chambre humide
pour éviter que les lamelles ne sèchent. Après l’heure de blocage, la
solution a été aspirée et l’anticorps primaire RANK humain (7,5 g/ml)
ou l’isotype IgG1 ont été déposés et laissés toute la nuit à 4°C, toujours
dans une chambre humide. Le lendemain, l’anticorps primaire et
l’isotype ont été aspirés et les cellules ont subit 3 lavages de 5 minutes
dans du PBS. Puis l’anticorps secondaire chèvre anti lapin Alexa 568 a
été déposé sur les cellules pendant 1 heure à température ambiante, à
l’abri de la lumière. Un marquage de l’ADN nucléaire a aussi été effectué
avec du DAPI pendant 5 minutes, toujours à l’abri de la lumière. Les
cellules ont ensuite été visualisées en utilisant un microscope optique à

51
fluorescence en utilisant les filtres appropriés aux différents
fluorochromes.
2.2.7. Coloration TRAP
Les cellules ont été différenciées pendant 3 jours sur des lamelles
de dentine placées dans des plaques de 96 puits (300 000 cellules dans
200 l de milieu de différenciation). Les lamelles de dentine ont été au
préalable imbibées du milieu RPMI pendant au moins 1 heure. Le milieu
de différenciation contenait du RANKL (40 ng/ml), de la calcitriol (10-8
M), du M-CSF (25ng/ml) et du PMA (10-8M). Les cellules ont été colorées
selon les instructions du fabricant du kit TRAP. Le principe de cette
coloration est le suivant: les cellules après avoir été lavées, ont été fixées
avec une solution d’acétone et citrate ; ensuite elles ont été incubées avec
40 mM d’une solution de tartrate. Cette coloration permet de révéler
l’activité enzymatique (phosphatase) à pH acide en présence de tartrate.
Les ostéoclastes sont des cellules dont la phosphatase acide est
résistante au tartrate et dans ce cas, l’activité enzymatique de la TRAP se
traduit par la formation d’un précipité rouge brique à la suite de la
coloration.
2.3. RÉSULTATS
Nous avons vérifié si les HL60 une fois différenciées remplissaient
tous les critères des ostéoclastes. Pour cela, nous avons effectué
plusieurs expériences pour vérifier la morphologie et la fonction de ces
cellules. Comme caractéristiques morphologiques nous avons vérifié la
taille et le nombre de noyaux et comme caractéristiques fonctionelles,
nous avons recherché la présence des récepteurs RANK, récepteurs de
calcitonine, OSCAR, PIR-A, TREM2, SIRP1 et la molécule adaptatrice
DAP12, clés de la différenciation et de l’activation des ostéoclastes in

52
vivo ; des molécules MMP9 et cathepsine K, du facteur de transcription
NFATc . Nous avons aussi recherché la présence des anneaux d’actine et
de l’activité enzymatique TRAP qui sont deux éléments caractéristiques
de l’activation des ostéoclastes.
2.3.1. Caractéristiques morphologiques des cellules
Lorsqu’on observe les cellules HL60 en culture, déjà au bout de 2
jours on observe l’allongement des cellules et leur adhésion à la plaque
de culture, ce qui constitue de grands changements dans la cellule
d’origine. Au bout des trois jours de différenciation, on a observé au
microscope optique que les cellules ont complètement changé de forme ;
elles sont devenues adhérentes et plus grosses (figure 11). On a pu aussi
observer la présence de plusieurs noyaux. Il est à noter que ce ne sont
pas toutes les cellules incubées qui se différencient, environ 60% se
transforment.
Un marquage de ces cellules à la phalloïdine pour le cytosquelette
d’actine et au DAPI pour les noyaux, nous a permis de mieux les
observer et de confirmer ces changements (figure 12).
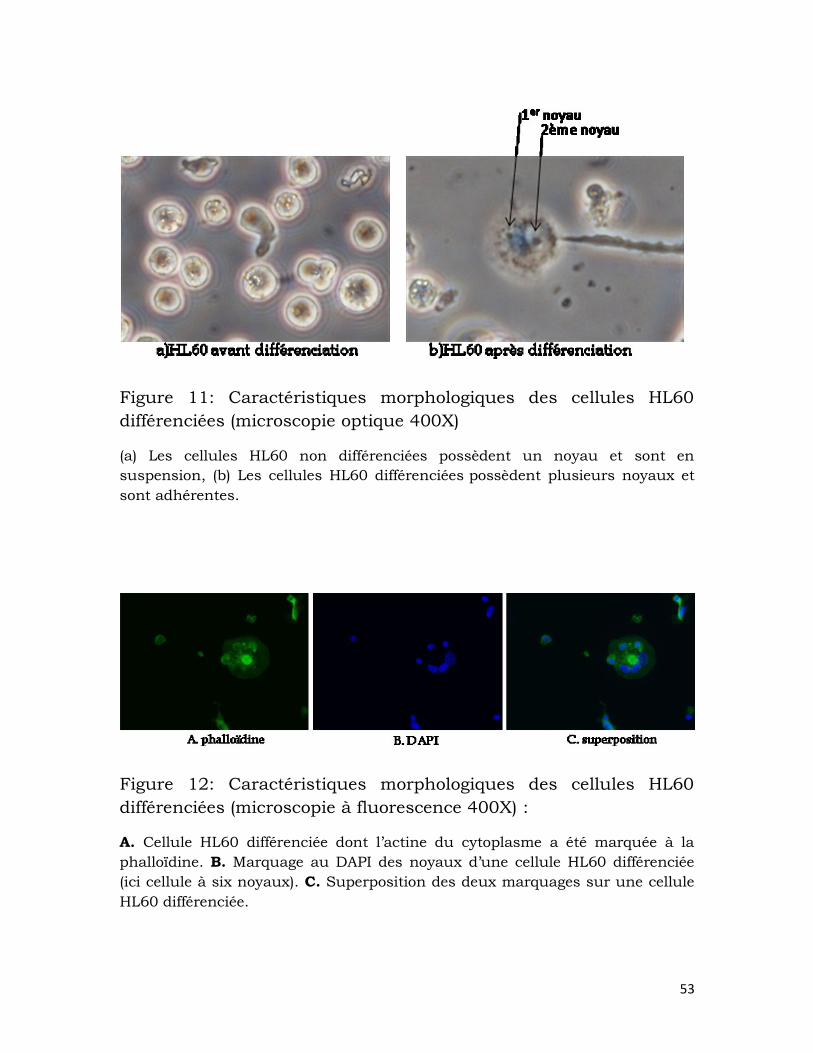
53
Figure 11: Caractéristiques morphologiques des cellules HL60
différenciées (microscopie optique 400X)
(a) Les cellules HL60 non différenciées possèdent un noyau et sont en
suspension, (b) Les cellules HL60 différenciées possèdent plusieurs noyaux et
sont adhérentes.
Figure 12: Caractéristiques morphologiques des cellules HL60
différenciées (microscopie à fluorescence 400X) :
A. Cellule HL60 différenciée dont l’actine du cytoplasme a été marquée à la
phalloïdine. B. Marquage au DAPI des noyaux d’une cellule HL60 différenciée
(ici cellule à six noyaux). C. Superposition des deux marquages sur une cellule
HL60 différenciée.

54
2.3.2. Présence des anneaux d’actine
Les anneaux d’actine sont des éléments importants de la
résorption car ils sont nécessaires à l’adhésion ferme des cellules à la
surface de résorption et permettent ainsi de former une zone de scellage
dans laquelle aura lieu la résorption proprement dite. Afin de déterminer
si les HL60, une fois différenciées étaient capables de former des
anneaux d’actine, nous avons fait une expérience d’immuno-histochimie
qui nous a permis de marquer l’actine et les noyaux des cellules. Puis
grâce à une microscopie confocale, nous avons pu observer la présence
de ces anneaux. Les anneaux d’actine sur les ostéoclastes se forment
lorsque les cellules adhèrent à la matrice osseuse. Nous avons observé la
présence de ces anneaux sur des cellules HL60 seulement après leur
différenciation et adhésion sur les lamelles de verre. Les cellules non
différenciées n’étant pas adhérentes, nous n’avons donc pas effectué
l’expérience sur ces cellules. La microscopie confocale nous a permis
d’observer les cellules en coupes transversales. Les anneaux étaient
visibles sur la coupe la plus basale, c'est-à-dire la plus proche de la
lamelle de verre (figure 13). La présence de ces anneaux d’actine nous
suggère que, comme les ostéoclastes, les HL60 différenciées pourraient
être capables d’adhérer fermement à une matrice osseuse et de former
une zone de scellage dans laquelle la résorption aura lieu.

55
Figure 13: Présence des anneaux d’actine (microscopie à
fluorescence 600X)
Cellules HL60 différenciées sur lamelles de verre dont l’actine forme un anneau
à la base de la cellule.
2.3.3. Activité enzymatique TRAP
Nous avons cherché à déterminer si les HL60 une fois différenciées
pouvaient présenter l’activité enzymatique TRAP comme les ostéoclastes.
Pour cela, les cellules ont été différenciées sur des lamelles de dentine
pendant trois jours. Nous avons utilisé comme contrôle négatif des
cellules HL60 non différenciées que nous avons fixées sur les lamelles de
dentine par cytospin à une vitesse de 500 RPM et comme contrôle positif
des monocytes différenciées en ostéoclastes sur une plaque en plastique.
Nous avons effectué cette expérience avec des HL60 sur dentine
car nous pensons que les cellules HL60 doivent être en présence d’une
matrice osseuse pour exprimer la TRAP. Comme présenté dans la figure
14, nous avons pu observer que les HL60 différenciées sur lamelles de
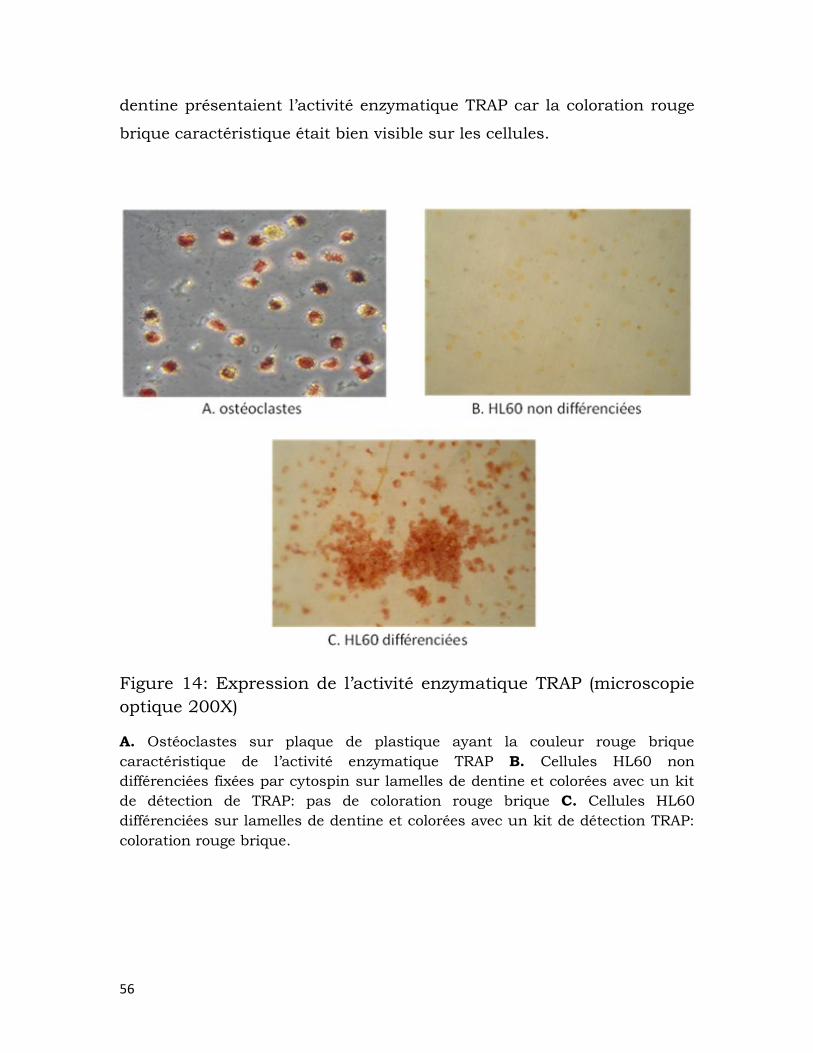
56
dentine présentaient l’activité enzymatique TRAP car la coloration rouge
brique caractéristique était bien visible sur les cellules.
Figure 14: Expression de l’activité enzymatique TRAP (microscopie
optique 200X)
A. Ostéoclastes sur plaque de plastique ayant la couleur rouge brique
caractéristique de l’activité enzymatique TRAP B. Cellules HL60 non
différenciées fixées par cytospin sur lamelles de dentine et colorées avec un kit
de détection de TRAP: pas de coloration rouge brique C. Cellules HL60
différenciées sur lamelles de dentine et colorées avec un kit de détection TRAP:
coloration rouge brique.

57
2.3.4. Expression de RANK
L’interaction RANK-RANKL est essentielle à la différenciation et à
l’activation des ostéoclastes. Selon la littérature, nous savons que le
récepteur RANK n’est pas constitutivement présent dans les cellules
HL60 [109]. Des études ont démontré qu’il était induit par la présence de
calcitriol; cette induction pourrait même être dépendante du temps et de
la dose [109]. Ainsi, nous avons vérifié si la différenciation de nos cellules
HL60 permettait bien de détecter la présence de l’ARNm de RANK par
PCR ou de la protéine par immuno-histochimie. Comme observé dans la
figure 15, nous avons pu confirmer l’expression de RANK par les cellules
HL60 différenciées. Le niveau d’expression du gène RANK est environ 6
fois plus élevé dans les cellules différenciées que dans les cellules non
différenciées. Les résultats entre les deux populations de HL60 non
différenciées et différenciées sont statistiquement significatifs (p< 0,05)
avec un test statistique t non apparié (n=5).
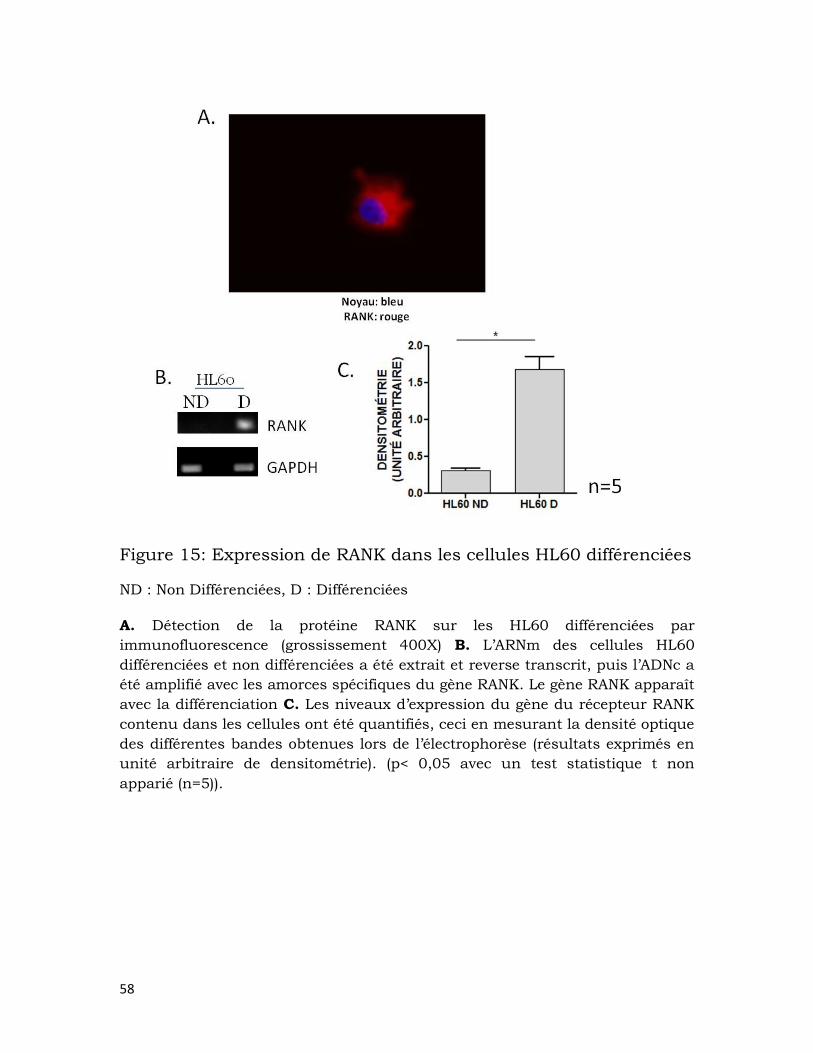
58
Figure 15: Expression de RANK dans les cellules HL60 différenciées
ND : Non Différenciées, D : Différenciées
A. Détection de la protéine RANK sur les HL60 différenciées par
immunofluorescence (grossissement 400X) B. L’ARNm des cellules HL60
différenciées et non différenciées a été extrait et reverse transcrit, puis l’ADNc a
été amplifié avec les amorces spécifiques du gène RANK. Le gène RANK apparaît
avec la différenciation C. Les niveaux d’expression du gène du récepteur RANK
contenu dans les cellules ont été quantifiés, ceci en mesurant la densité optique
des différentes bandes obtenues lors de l’électrophorèse (résultats exprimés en
unité arbitraire de densitométrie). (p< 0,05 avec un test statistique t non
apparié (n=5)).

59
2.3.5. Expression de NFATc
Nous avons recherché par PCR et par immunobuvardage la
présence du facteur de transcription NFATc dans les cellules HL60
différenciées. NFATc régule l’expression de nombreux gènes spécifiques
des ostéoclastes. Nous avons utilisé comme contrôle positif des
ostéoclastes différenciés à partir de monocytes. La figure 16 montre
l’expression de ce facteur de transcription autant dans les cellules
différenciées que non différenciées. Les cellules différenciées et non
différenciées ont environ les mêmes niveaux d’expression du gène NFATc.
Les cellules différenciées expriment environ deux fois moins de protéine
que les non différenciées, mais elle reste abondante comme dans les
ostéoclastes. Les résultats entre les deux populations de HL60 non
différenciées et différenciées sont statistiquement significatifs (p< 0,05)
avec un test statistique t non apparié (n=5 pour le gène et n=3 pour la
protéine).

60
Figure 16: Expression de NFATc
ND: Non Différenciées ; D: Différenciées ; OCs : ostéoclastes
WB= immunobuvardage
A. Expression du gène du facteur de transcription NFATc dans les cellules
HL60 différenciées et non différenciées B. Détection de l’expression de la
protéine NFATc dans les cellules différenciées et non différenciées C. En
mesurant la densité optique des différentes bandes obtenues à la suite de
l’électrophorèse, nous avons pu quantifier les niveaux d’expression du gène
NFATc dans les cellules, les résultats sont exprimés en unité de densitométrie
D. La quantité de protéine a aussi été obtenue en mesurant la densité optique
des différentes bandes d’électrophorèse. Résultats exprimés en unité de
densitométrie.

61
2.3.6. Expression des récepteurs de calcitonine
Nous avons recherché par immunobuvardage la présence des
récepteurs de calcitonine dans les cellules HL60 différenciées, car ils sont
un récepteur spécifique de l’activation des ostéoclastes. Nous avons
utilisé comme contrôle positif des ostéoclastes différenciés à partir de
monocytes. La figure 17 montre l’expression de ces récepteurs autant
dans les cellules différenciées que non différenciées. La quantité de
protéine augmente à la suite de la différenciation; mais cette
augmentation n’est pas significative (test statistique t non apparié n=3,
p>0,05).
Figure 17: Expression des récepteurs de calcitonine
ND: Non Différenciées ; D: Différenciées ; OCs : ostéoclastes
A. Détection par immunobuvardage de l’expression de la protéine des
récepteurs de calcitonine dans les cellules HL60 différenciées comme dans les
ostéoclastes B. Mesure de la densité optique (densitométrie) des bandes
obtenues suite à l’électrophorèse (n=3)
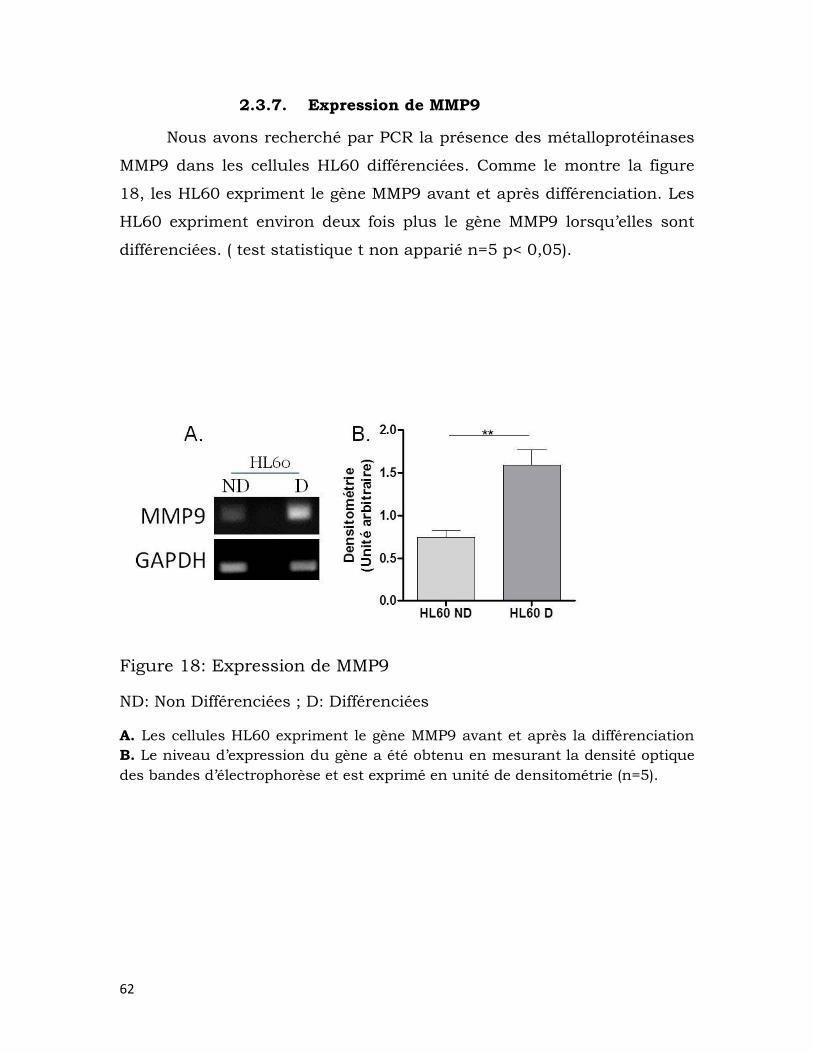
62
2.3.7. Expression de MMP9
Nous avons recherché par PCR la présence des métalloprotéinases
MMP9 dans les cellules HL60 différenciées. Comme le montre la figure
18, les HL60 expriment le gène MMP9 avant et après différenciation. Les
HL60 expriment environ deux fois plus le gène MMP9 lorsqu’elles sont
différenciées. ( test statistique t non apparié n=5 p< 0,05).
Figure 18: Expression de MMP9
ND: Non Différenciées ; D: Différenciées
A. Les cellules HL60 expriment le gène MMP9 avant et après la différenciation
B. Le niveau d’expression du gène a été obtenu en mesurant la densité optique
des bandes d’électrophorèse et est exprimé en unité de densitométrie (n=5).

63
2.3.8. Expression de cathepsine K
La présence de la cathepsine K dans les cellules HL60 différenciées
a été recherchée en effectuant des immunobuvardages. La cathepsine K a
été retrouvée autant dans les cellules HL60 non différenciées que dans
les différenciées (figure 19). Les HL60 expriment légèrement plus de
protéine cathepsine K quand elles se différencient que lorsqu’elles ne
sont pas différenciées, mais la différence n’est pas significative entre les
deux groupes (test statistique t non apparié n=3, p>0,05).
Figure 19: Expression de cathepsine K
ND: Non Différenciées ; D: Différenciées ; OCs : ostéoclastes
A. Les HL60 produisent la cathepsine K que les cellules soient différenciées ou
non, comme dans les ostéoclastes B. La quantité de protéines a été obtenue en
mesurant la densité optique des bandes d’électrophorèse et est exprimée en
unité de densitométrie (n=3).

64
2.3.9. Expression de DAP-12
L’expression du corécepteur DAP-12 a été investiguée dans les
cellules HL60 différenciées et non différenciées. Nous avons par PCR et
par immunobuvardage trouvé que les HL60 expriment bel et bien ce
facteur de costimulation (figure 20). Les cellules différenciées ont un
niveau d’expression du gène DAP12 environ 0,5 fois plus élevé que les
cellules non différenciées; mais cette différence n’est pas significative.
Pour ce qui est de la protéine, les cellules différenciées en expriment
environ 1,5 fois moins que les non différenciées mais elle reste quand
même abondante. Les différences entre les deux populations de HL60
non différenciées et différenciées n’étaient pas statistiquement
significatives (test statistique t non apparié (n=5 pour l’ARNm et n=4
pour la protéine), p>0,05).

65
Figure 20: Expression de DAP-12
ND: Non Différenciées ; D: Différenciées ; OCs : ostéoclastes
WB= immunobuvardage
A.Détection par PCR de l’expression du gène du corécepteur DAP-12 dans les
cellules HL60 différenciées et non différenciées B. Détection par
immunobuvardage de la protéine DAP-12 dans les cellules différenciées et non
différenciées C. En mesurant la densité optique des différentes bandes obtenues
suite à l’électrophorèse, nous avons pu quantifier l’expression du gène DAP-12
dans les cellules, les résultats sont exprimés en unité de densitométrie (n=5) D.
La quantité de protéine a aussi été obtenue en mesurant la densité optique des
différentes bandes d’électrophorèse. Résultats exprimés en unité de
densitométrie (n=3).

66
2.3.10. Expression de TREM2
Le co-récepteur TREM2 a été recherché dans les cellules HL60
différenciées et non différenciées. En effectuant des expériences
d’immunobuvardage et de PCR, nous avons trouvé que les HL60
expriment ce corécepteur (figure 21) tant sur le plan de l’ARNm que de la
protéine. Les cellules différenciées ont un niveau d’expression du gène
TREM2 environ 2 fois plus important que celui des cellules non
différenciées; et cette différence est statistiquement significative (p<
0,05). Les cellules différenciées et non différenciées expriment
approximativement les même quantités de protéines. Nous avons effectué
des tests statistiques t non apparié (n=5 pour l’ARNm et n=3 pour la
protéine).

67
Figure 21: Expression de TREM2
ND: Non Différenciées ; D: Différenciées ; OCs : ostéoclastes
WB=immunobuvardage
A. Détection par PCR de l’expression du gène du corécepteur TREM2 dans les
cellules HL60 différenciées B. Détection de la protéine TREM2 par western blot
dans les cellules différenciées et non différenciées comme dans les ostéoclastes
C. En mesurant la densité optique des différentes bandes obtenues suite à
l’électrophorèse, nous avons pu mesurer les niveaux d’expression du gène
TREM2 dans les cellules, les résultats sont exprimés en unité de densitométrie
(n=5) D. La quantité de protéine a aussi été obtenue en mesurant la densité
optique des différentes bandes d’électrophorèse. Résultats exprimés en unité de
densitométrie (n=3).
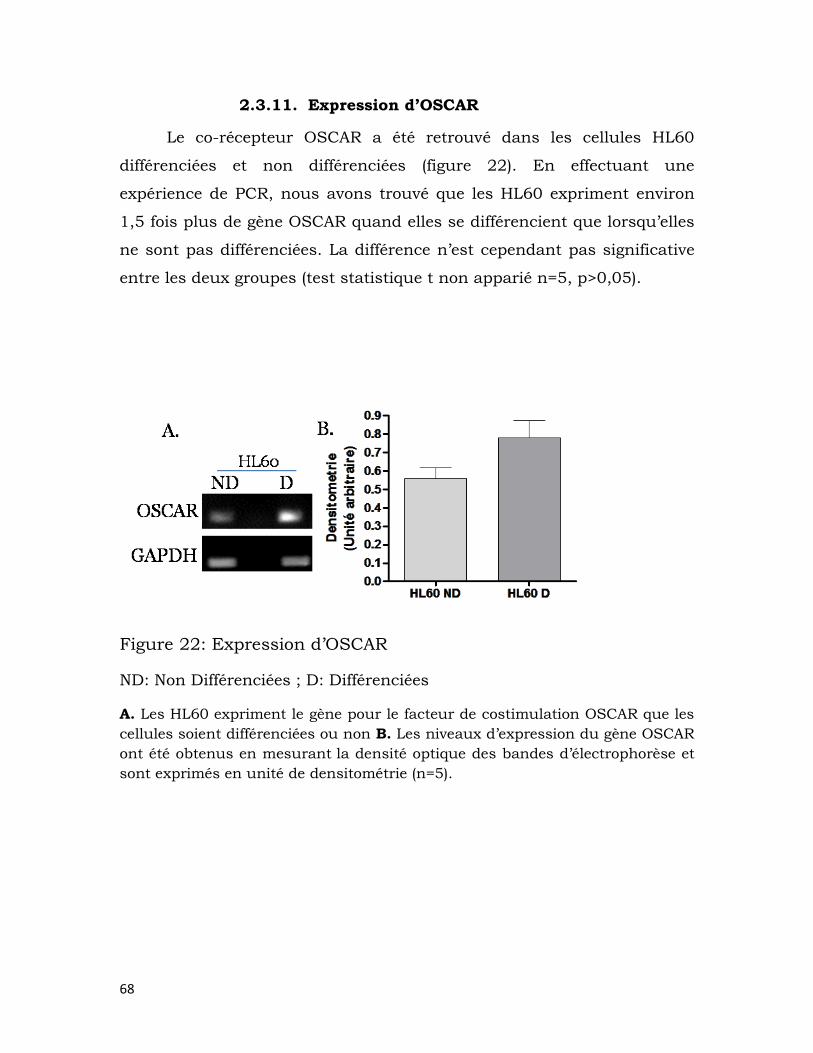
68
2.3.11. Expression d’OSCAR
Le co-récepteur OSCAR a été retrouvé dans les cellules HL60
différenciées et non différenciées (figure 22). En effectuant une
expérience de PCR, nous avons trouvé que les HL60 expriment environ
1,5 fois plus de gène OSCAR quand elles se différencient que lorsqu’elles
ne sont pas différenciées. La différence n’est cependant pas significative
entre les deux groupes (test statistique t non apparié n=5, p>0,05).
Figure 22: Expression d’OSCAR
ND: Non Différenciées ; D: Différenciées
A. Les HL60 expriment le gène pour le facteur de costimulation OSCAR que les
cellules soient différenciées ou non B. Les niveaux d’expression du gène OSCAR
ont été obtenus en mesurant la densité optique des bandes d’électrophorèse et
sont exprimés en unité de densitométrie (n=5).

69
2.3.12. Expression de SIRP1
L’expression du co-récepteur SIRP1 a été démontrée dans les
cellules HL60 (figure 23). L’ARNm a été retrouvée par PCR que ce soit
dans les cellules non différenciées ou dans les cellules différenciées. Les
HL60 expriment environ 2 fois moins de gène SIRP1 quand elles se
différencient que lorsqu’elles ne sont pas différenciées. Cette différence
est statistiquement significative mais la quantité d’ARNm dans les
cellules différenciées reste importante (test statistique t non apparié
n=5 ; p< 0,05).
Figure 23: Expression de SIRP1
ND: Non Différenciées ; D: Différenciées
A. Les HL60 expriment le gène pour le facteur de costimulation SIRP1 que les
cellules soient différenciées ou non B. Les niveaux d’expression du gène ont été
obtenus en mesurant la densité optique des bandes d’électrophorèse et sont
exprimés en unité de densitométrie (n=5).

70
2.3.13. Expression de PIR-A
Le co-récepteur PIR-A a été recherché dans les cellules HL60. Sa
détection a pu se faire en effectuant une expérience de PCR. L’ARNm de
ce récepteur a ainsi été trouvé tant dans les cellules HL60 différenciées
que dans les cellules non différenciées (figure 24). Les HL60 expriment
environ 1,5 fois plus de gène PIRA quand elles se différencient que
lorsqu’elles ne sont pas différenciées. Cette différence n’est cependant
pas statistiquement significative (test statistique t non apparié n=5,
p>0,05).
Figure 24: Expression de PIR-A
ND: Non Différenciées ; D: Différenciées
A. La PCR nous montre que les HL60 expriment le gène du facteur de
costimulation PIR-A que les cellules soient différenciées ou non B. Les niveaux
d’expression du gène PIR-A ont été obtenus en mesurant la densité optique des
bandes d’électrophorèse et sont exprimés en unité de densitométrie (n=5).
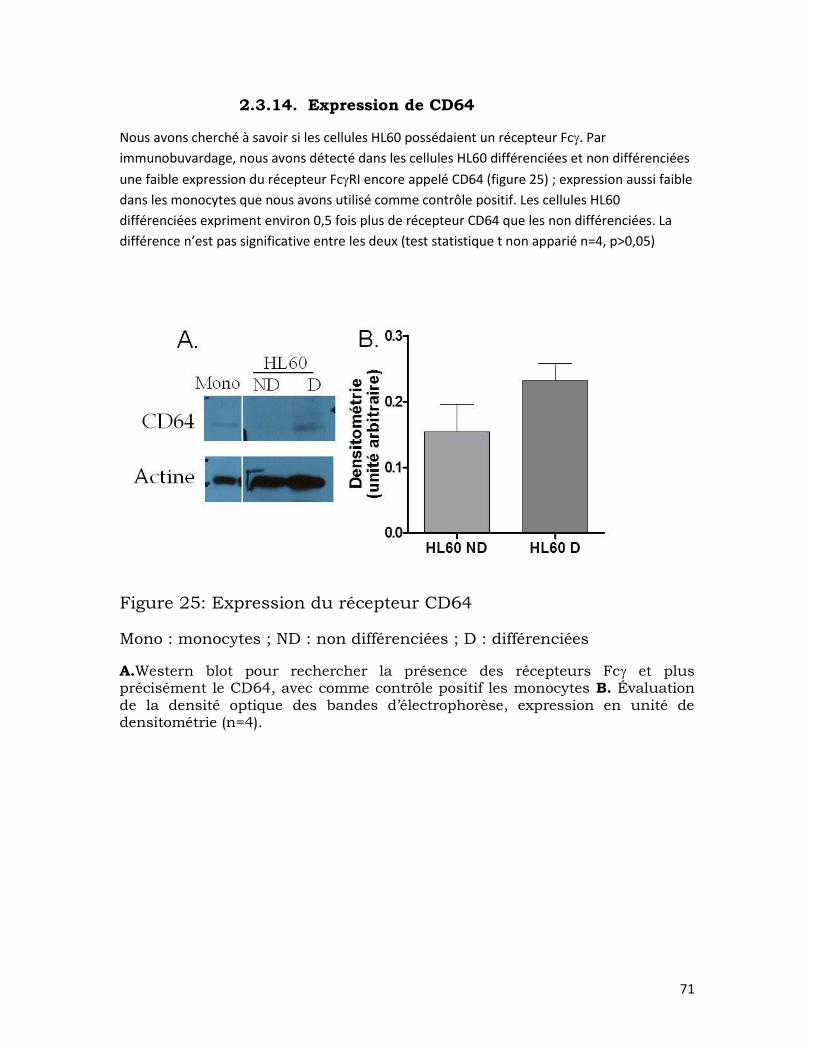
71
2.3.14. Expression de CD64
Nous avons cherché à savoir si les cellules HL60 possédaient un récepteur Fc. Par
immunobuvardage, nous avons détecté dans les cellules HL60 différenciées et non différenciées
une faible expression du récepteur FcRI encore appelé CD64 (figure 25) ; expression aussi faible
dans les monocytes que nous avons utilisé comme contrôle positif. Les cellules HL60
différenciées expriment environ 0,5 fois plus de récepteur CD64 que les non différenciées. La
différence n’est pas significative entre les deux (test statistique t non apparié n=4, p>0,05)
Figure 25: Expression du récepteur CD64
Mono : monocytes ; ND : non différenciées ; D : différenciées
A.Western blot pour rechercher la présence des récepteurs Fc et plus précisément le CD64, avec comme contrôle positif les monocytes B. Évaluation de la densité optique des bandes d’électrophorèse, expression en unité de densitométrie (n=4).

72
2.4. DISCUSSION
Notre étude nous a permis de trouver les conditions optimales de
différenciation des cellules HL60 en cellules de type « osteoclast-like » et
de pouvoir ainsi définir un modèle de différenciation pour l’obtention
d’ostéoclastes. Nos résultats suggèrent que le traitement des cellules
HL60 avec des facteurs tels que la calcitriol, le M-CSF, le RANKL et le
PMA est essentiel à cette différenciation. Ces cellules différenciées ainsi
obtenues deviennent, tant sur le plan morphologique que fonctionnel,
très proches des ostéoclastes. Sur le plan morphologique, on constate
l’apparition de plusieurs noyaux, l’augmentation de la taille des cellules,
et de la capacité d’adhérence. Sur le plan fonctionnel, elles expriment,
tant en matière de protéines que d’ARNm, les molécules MMP9 et
cathepsine K, le facteur de transcription NFATc, les récepteurs RANK,
récepteurs de calcitonine, OSCAR, PIR-A, TREM2, SIRP1 et la molécule
adaptatrice DAP12, clés de la différenciation et de l’activation des
ostéoclastes in vivo. Elles expriment aussi le récepteur Fc qui est un des
récepteurs de co-stimulation des ostéoclastes. L’implication réelle de ce
récepteur n’a pas encore été démontrée mais on pourrait supposer que,
comme chez le neutrophile, il participe à l’immunité en se liant à des
immunoglobulines qui ont déjà des antigènes attachés à elles. Les
cellules HL60 différenciées possèdent aussi une activité enzymatique
TRAP et ont la possibilité de réorganiser leur cytosquelette d’actine de
manière à former des anneaux.
À ce jour, les modèles de différenciation d’ostéoclastes utilisés dans
la littérature sont les modèles à partir de cellules de souris [122, 123],
tels que la lignée cellulaire RAW utilisée dans notre laboratoire et les
modèles humains qui eux sont obtenus à la suite de la différenciation in
vitro de monocytes isolés de sang humain, ce qui peut quelques fois
aboutir à une irrégularité des résultats d’un donneur de sang à l’autre.
Les cellules HL60 constituent de ce fait le modèle humain par excellence

73
pour l’étude des ostéoclastes. Plusieurs études ont permis de montrer la
capacité de différenciation des cellules HL60 en ostéoclastes [108, 118,
119]. Cependant le plus souvent ces études étaient menées pour étudier
un aspect particulier de l’ostéoclaste. C’est le cas par exemple de Kido et
ses collaborateurs qui ont étudié l’induction de l’expression de RANK sur
des cellules HL60 différenciées en ostéoclastes [109]. De même, Hillstrom
Shapiro et ses collaborateurs ont étudié l’expression de l’anhydrase
carbonique II sur les HL60 différenciées en ostéoclastes [124]. Nous
n’avons trouvé aucune étude qui caractérise la cellule HL60 « osteoclast-
like » dans tous les aspects de son activation et de sa fonction ; notre
étude a ainsi permis de combler ce vide. Malgré le fait que nous n’avons
pas pu quantifier le nombre de cellules HL60 qui se différencient, nous
avons pu remarquer pendant nos expériences que les HL60 arrêtent leur
prolifération en présence des facteurs de différenciation, car nous
n’avons pas observé pas le doublement habituel de ces cellules en 24
heures de culture. McCarthy et ses collaborateurs l’ont d’ailleurs
démontré dans leur étude [107]. En effet, ils ont trouvé qu’en présence de
vitamine D3 le nombre de cellules HL60 diminuait avec le temps et avec
l’augmentation de la concentration (à des concentrations élevées la
prolifération s’arrête en 48 heures). Le mécanisme de différenciation
pourrait impliquer la fusion comme c’est le cas des précurseurs
myéloïdes in vivo car on observe de nombreux amas cellulaires après la
différenciation, de même qu’une augmentation de la taille des cellules.
Plusieurs récepteurs sont impliqués dans la fusion des précurseurs
d’ostéoclastes ; on peut citer parmi eux: le DC-STAMP (« Dentritic Cell-
Specific Transmembrane Protein », protéine transmembranaire spécifique
des cellules dendritiques), la cadhérine E et le MFR (« Macrophage Fusion
Receptor », récepteur de fusion des macrophages) [125, 126]. Il serait
juste de penser que les HL60 pourraient posséder certains de ces
récepteurs indispensables à la fusion des précurseurs d’ostéoclastes.

74
La différenciation des cellules HL60 pourrait impliquer l’activation
de la voie RANK/RANKL. Dans le même ordre d’idée, l’expression du
facteur de transcription NFATc sous-entend une activation en amont de
la voie de signalisation de RANK: recrutement de TRAF6 suivi de
l’activation de NFB et des MAP kinases JNK et Erk (figure 3). D’autant
plus qu’une étude a démontré que les souris déficientes en NFATc1
étaient incapables d’initier l’ostéoclastogenèse [127]. L’activation de cette
voie de signalisation est responsable de l’expression de toutes les
molécules spécifiques des ostéoclastes et de la résorption osseuse, ceci
s’est vérifié dans les HL60 « osteoclast-like » avec l’expression de MMP9,
de la cathepsine K, des récepteurs de calcitonine, de l’activité
enzymatique TRAP et la formation des anneaux d’actine. Yoneda et ses
collaborateurs ont démontré que les récepteurs de calcitonine
apparaissent avec la différenciation des HL60 en cellules « osteoclast-
like » et que cette différenciation était inhibée par la calcitonine de
saumon, de même que la résorption osseuse [119]. L’expression des
molécules de costimulation (DAP12, TREM2, SIRP1, OSCAR, FcR, PIR-
A) par les HL60 « osteoclast-like » vient corroborer le constat selon lequel
ces cellules remplissent vraiment tous les critères d’un ostéoclaste.
Les récepteurs Fcγ sont les récepteurs pour la fraction Fc des IgG.
Il en existe trois : FcRI ou CD64, FcRII ou CD32 et FcRIII ou CD16
[128]. Nous n’avons trouvé dans notre étude que le récepteur CD64; dans
les cellules non différenciées en faible quantité et en plus grande quantité
après la différenciation. Certaines études ont montré que les cellules
HL60 expriment déjà un récepteur pour les IgG [129, 130], tout comme
les monocytes/macrophages, cependant ces études ne précisaient pas
exactement lequel des trois récepteurs Fc.

75
III. ACTION DES FACTEURS RHUMATOÏDES SUR
LES OSTÉOCLASTES ET LA RÉSORPTION
OSSEUSE
3.1. INTRODUCTION
Les FR sont un des bio marqueurs indispensables pour le
diagnostic de la PAR. Ils sont retrouvés dans le liquide synovial, la
membrane synoviale, le sang, de même que dans les autres fluides et
tissus des patients atteints de PAR et ils participent à la pathogenèse de
la maladie. Il en existe trois types: IgM, IgG et IgA. Les FR interviennent
dans la formation des complexes immuns, la présentation antigénique et
l’activation du complément. Les complexes immuns, et particulièrement
ceux formés avec les FR, sont en partie à l’origine de l’activation du
processus inflammatoire [131]. L’érosion osseuse peut toucher à la fois
les arthrites séropositives pour les FR, de même que les arthrites
séronégatives. Cependant, une corrélation a été retrouvée entre le taux
d’érosion osseuse et le taux de FR détecté; le titre élevé de FR chez un
patient est signe d’un développement de la maladie vers des érosions
osseuses et même des manifestations extra-articulaires. Il n’a pas été
clairement établi lequel est le type causant le plus d’érosions osseuses,
mais les auteurs d’une étude pensent que le FR de type IgA pourrait
corréler avec de fortes érosions osseuses [132].
Les cellules du milieu synovial peuvent exprimer des récepteurs
pour la fraction Fc des IgG appelé FcR ; cela pourrait permettre
d’expliquer le rôle des FR de type IgG dans la maladie [131].
L’importance des FR dans la pathologie est établie: présentation
antigénique avec formation d’auto-anticorps et activation du
complément. Cependant aucune étude n’explique la corrélation
retrouvée entre le taux élevé de FR et l’érosion osseuse. Les ostéoclastes

76
possèdent à leur surface plusieurs co-récepteurs dont le récepteur Fc
[15] qui pourrait lier les FR sous la forme de complexe. D’où notre
hypothèse qui stipule que les FR augmentent la résorption osseuse en
agissant directement sur l’ostéoclaste via ce récepteur. Nous nous
sommes donc fixé comme objectif de montrer l’effet des FR sur la
résorption osseuse des ostéoclastes. Dans notre étude, nous avons
incubé des FR directement sur des ostéoclastes pour observer leurs effets
sur les cellules et sur la résorption osseuse.
3.2. MATÉRIELS ET MÉTHODES
3.2.1. Réactifs
Le milieu de culture utilisé, MEM et le sérum de veau fœtal (FBS)
ont été obtenu de chez Wisent (St-Bruno, QC, Canada). Le M-CSF et le
RANKL recombinants ont été tous les deux achetés chez Peprotech
(Rocky Hill, NJ, USA). La 1 dihydroxyvitamine D3 et le phorbol 12-
myristate 13-acétate (PMA) ont été obtenus de chez Sigma Chemical Co.
(St Louis, MO, USA). Les facteurs rhumatoïdes de type IgG, IgM ont été
obtenus d’une collaboration avec le Dr Marianna Newkirk (Université
McGill, Montréal). Les concentrations des différents facteurs rhumatoïdes
sont les suivantes: IgM FR: 50 µg/ml; IgA FR: 50 µg/ml ; celle de l’IgG:
10 µg/ml. L’anticorps de souris anti tyrosine-phosphorylation humaine a
été obtenu chez Upstate Biotechnology Inc. (Billerica, MA, USA) et était
utilisé à la concentration de 2 g/ml. L’anticorps IgM humaine provenait
de Sigma Chemical Co. (St Louis, MO, USA) et était utilisé à la
concentration de 50 g/ml.
Les lamelles d’OSTEOLOGIC utilisées comme matrice pour la
résorption des ostéoclastes ont été achetées chez BD Biosciences
(Mississauga, ON, Canada). Les lamelles d’OSTEOLOGIC consistent en

77
des micromolécules de phosphate de calcium disposées en fine lamelles
sur des lames de verre.
3.2.2. Isolement des monocytes :
Les monocytes ont été isolés à partir de sang humain de donneurs
volontaires sains recueilli dans des poches contenant de l’isocitrate
comme anticoagulant. Les monocytes ont été isolés selon la même
méthode que décrite dans le paragraphe 2.2.3.
La différenciation des monocytes en ostéoclastes a aussi suivi le
même protocole que dans le paragraphe sus-cité. Nous avons aussi
différencié des ostéoclastes en présence des FR. Nous avons ajouté soit le
FR IgM (50 g/ml), soit l’anticorps IgM (50 g/ml) qui nous servait de
contrôle. Les FR étaient ajoutés au milieu de différenciation et le
processus de différenciation se faisait en 10 jours avec un changement
de milieu 2 fois par semaine.
3.2.3. Résorption osseuse
Une fois décollés des plaques de culture et lavés, les ostéoclastes ont
été comptés et resuspendus dans le milieu MEM pour les mettre à la
concentration de 100 000 ostéoclastes /200 l. Un volume de 200 l a
été ajouté dans chacun des puits d’OSTEOLOGIC. Après avoir déposé
les 200l dans chacun des puits, les cellules ont été incubées pendant
24h pour permettre aux ostéoclastes d’adhérer à la matrice.
Après 24 heures, le milieu de culture a été enlevé et remplacé par un
milieu de différenciation (MEM/FBS + RANKL (40 ng/ml) + M-CSF (25
ng/ml) + vitamine D3 (10-8M)) auquel ont été ajoutés ou non les FR ou les
isotypes contrôle en fonction des conditions à tester. Les cellules ont
été laissées pour la résorption pendant 12 jours avec des changements
de milieu tous les trois ou quatre jours. Les conditions testées sur les
cellules ont été les suivantes :

78
Milieu de culture (MEM +FBS)
Milieu de différenciation (MEM/FBS + RANKL (40 ng/ml) + M-
CSF (25ng/ml) + vitamine D3 (10-8M))
Milieu de différenciation + IgM FR
Milieu de différenciation + complexe IgG – IgM FR (IgG et IgM
incubés ensemble pendant 30 minutes à 37°C et au bout de ce
temps incubés avec les ostéoclastes)
Milieu de différenciation + IgG (incubé avec les ostéoclastes
pendant 30 minutes à 37°C) + IgM FR rajoutés au bout des 30
minutes.
Les FR sont des immunoglobulines qui pourraient se lier aux
récepteurs Fc retrouvés sur les ostéoclastes. Nous ne savons pas si les
ostéoclastes possèdent les récepteurs pour les IgM FR alors que nous
savons avec certitude qu’ils possèdent les récepteurs pour les IgG (FcR).
Nous avons de ce constat choisi de faire des essais avec ou sans
complexes immuns (IgG – IgM FR) car nous émettons l’hypothèse que les
IgG peuvent servir de ponts entre les IgM FR et le récepteur Fc.
Au bout des 12 jours de résorption, les milieux de culture ont été
enlevés des plaques d’OSTEOLOGIC et ces dernières lavées avec une
solution d’hypochlorite de sodium diluée à 10% dans de l’eau : 2 lavages
pendant 10 minutes chacun en agitant de temps en temps. Chaque puits
a ensuite été bien rincé avec de l’eau distillée (4 ou 5 lavages). Les
plaques ont été laissées séchées à l’air libre et ensuite observées au
microscope optique pour voir la résorption.
3.2.4. Coloration TRAP
Les cellules, après avoir été différenciées, ont été lavées puis fixées
avec une solution de mélange acétone et citrate. Elles ont ensuite été

79
incubées avec 40 mM d’une solution de tartrate et enfin colorées avec
une solution d’hématoxyline pour colorer les noyaux.
3.3. RÉSULTATS
3.3.1. Action des facteurs rhumatoïdes sur les
ostéoclastes
Puisque nous savons que les FR jouent un rôle important dans la
résorption osseuse au cours de la PAR, nous avons cherché à savoir si
ces FR influencaient directement les ostéoclastes au cours de leur
processus de différenciation. Nous avons incubé des monocytes en cours
de différenciation en ostéoclastes soit avec le FR IgM (IgM FR), soit avec
l’anticorps IgM comme contrôle négatif. Une fois les ostéoclastes
différenciés, nous les avons ensuite colorés avec une coloration TRAP et
une contre coloration à l’haematoxyline pour bien observer les noyaux. A
la fin du processus de différenciation, nous avons remarqué une grande
différence entre les cellules incubées avec l’IgM FR et celles différenciées
normalement. Bien que la coloration TRAP soit positive sur les cellules
différenciées avec et sans FR, les FR permettaient d’obtenir des cellules
beaucoup plus grosses et avec un nombre plus élevé de noyaux (figure
26). De plus, l’incubation des cellules avec l’anticorps IgM qui n’est pas
un FR avait plutôt un effet négatif (inhibition de la différenciation ou peut
être toxicité), car aucune cellule n’a réussi à terminer le processus de
différenciation et elles se sont toutes décollées de la plaque de culture.

80
Figure 26: Effets du facteur rhumatoïde IgM sur la différenciation
des ostéoclastes (microscopie optique 200X)
A. Les monocytes ont été incubés avec le milieu de différenciation (MEM +
RANKL (40 ng/ml) + MCSF (25ng/ml) + vitamine D3 (10-8M) et différenciés en
ostéoclastes pendant 12 jours. Les ostéoclastes obtenus étaient positifs à la
coloration TRAP. Les flèches indiquent les noyaux B. Monocytes incubés avec le
milieu de différenciation et les facteurs rhumatoïdes IgM et différenciés en
ostéoclastes pendant 12 jours. Les flèches indiquent les noyaux C. Monocytes
incubés avec le milieu de différenciation et l’anticorps IgM qui n’est pas un
facteur rhumatoïde.

81
3.3.2. Action des facteurs rhumatoïdes sur la
résorption par les ostéoclastes
Nous avons cherché à savoir si les FR, en plus d’augmenter la taille
des ostéoclastes, augmentaient aussi leur activité de résorption. Nous
avons pour cela différencié des ostéoclastes et les avons incubé sur une
lamelle d’OSTEOLOGIC pour la résorption et évalué les surfaces
moyennes de résorption. La figure 27 montre l’histogramme des surfaces
moyenne de résorption osseuse avec et sans FR. Nous avons testé
plusieurs concentrations avec le FR IgM: 10 g/ml; 30 g/ml et 50
g/ml; et trouvé un effet sur la surface de résorption qui est dépendant
de la dose de FR IgM-FR. La surface de résorption augmente avec la dose
de FR. L’incubation des cellules avec IgM-FR en complexes immuns avec
IgG, a aussi un impact sur la résorption. En effet les surfaces de
résorption étaient plus importantes en présence de FR avec complexes
immuns comparativement à la résorption sans FR. Nous avons testé la
résorption avec deux types de complexes immuns; premier type :
anticorps IgG ajouté 30 minutes avant IgM-FR et deuxième type :
complexe IgG-IgMFR constitué avant et rajouté ensuite sur les cellules.
La différence entre les deux types de complexes immuns était visible: la
surface de résorption était plus importante avec le premier type qu’avec
le second type de complexes immuns. Le FR IgA-FR quant à lui avait
plutôt un effet inhibiteur sur la résorption.

82
0
2500
5000
7500
10000
CTL- M
M + IgM RF
10ug/ml 30ug/ml 50ug/mlM
+
IgM
M
+
IgA RF
M +
complexe
IgG-IgMRF
M+
IgG avant
+ IgMRF
su
rface m
oyen
ne
de r
éso
rpti
on
en
pix
els
(un
ité a
rbit
rair
e)
Figure 27: Effet des facteurs rhumatoïdes sur la résorption osseuse
des ostéoclastes
CTL- : contrôle négatif, milieu de culture MEM
M : milieu de différenciation : MEM + RANKL (40 ng/ml) + MCSF (25ng/ml) + vitamine
D3 (10-8M)
Ostéoclastes incubés sur lamelles d’OSTEOLOGIC pour la résorption avec et
sans facteurs rhumatoïdes. Résultats correspondants à une seule expérience.
(IgM FR=IgM RF)
Nous avons effectué plusieurs autres expériences pour confirmer
nos premiers résultats et effectuer des calculs statistiques. La tendance
se confirme quant à l’impact des FR sur la résorption osseuse.
Cependant, les ostéoclastes utilisés sont des cellules différenciées à
partir de monocytes isolés de sang humain, ce qui donnait des variations
très importantes entre les résultats d’un donneur à l’autre. En effet, les
ordres de grandeur des surfaces de résorption sont très différents d’un
donneur à l’autre. Pour avoir des résultats significatifs, il faudrait un
nombre très élevé de donneurs de sang. Nous avons, de ce fait, pensé
tester l’effet des FR sur les HL60 différenciées en ostéoclastes.

83
3.3.3. Activation des cellules HL60 différenciées par
les facteurs rhumatoïdes
Nous avons démontré que les cellules HL60 étaient capables de se
différencier en des cellules qui remplissaient plusieurs critères des
ostéoclastes. Les ostéoclastes possèdent à leur surface des récepteurs Fc
qui sont des récepteurs dont l’activation aboutit à la phosphorylation des
protéines sur résidus tyrosine. Les FR pourraient donc avoir un effet sur
les cellules HL60 différenciées en se liant possiblement à ces récepteurs.
La phosphorylation des tyrosines est un des signes de l’activation des
cellules. La stimulation des cellules HL60 différenciées par des FR IgM-
FR pourrait éventuellement stimuler la production de protéines, soit en
passant par la voie de costimulation seule, soit de concert avec la voie
principale d’activation des ostéoclastes RANK-RANKL. En stimulant les
cellules à des temps différents (5 minutes, 10 minutes et 30 minutes), on
observe des profils différents de phosphorylation (figure 28 A). Le profil
des protéines phosphorylées sur résidus tyrosine montre plusieurs
bandes qui ne sont pas modifiées en présence de RANKL; par contre en
présence du mélange RANKL avec le FR IgM-FR ou avec le FR IgM-FR
seul, les bandes s’intensifient au cours du temps, ce qui montre une
activation des cellules surtout aux temps 5 minutes et 10 minutes par
rapport au véhicule. La stimulation avec l’anticorps IgM contrôle a plutôt
pour effet de diminuer l’activation des cellules avec le temps.
L’histogramme de la densitométrie globale des bandes d’électrophorèse
de l’immunobuvardage ciblant la phosphorylation des tyrosines montre
une différence entre la stimulation RANKL + IgM FR et IgM-FR avec la
stimulation RANKL (figure 28 B).

84
Figure 28: Phosphorylation des tyrosines dans les HL60
différenciées.
A. Les HL60 différenciées ont été incubés pendant 5, 10 ou 30 minutes soit avec le véhicule seul (Ve), soit avec RANKL, RANKL+IgM RF, IgM RF ou IgM. L’immunobuvardage a été fait avec un anticorps primaire de souris anti-tyrosine phosphorylée humaine suivi de l’anticorps secondaire couplé à la HRP chèvre anti-souris. B. Mesure de la densité optique des bandes d’électrophorèse. Résultats obtenus pour deux expériences indépendantes.
(IgM RF=IgM FR).

85
3.4. DISCUSSION
Dans cette étude nous avons montré que les FR agissaient sur la
différenciation des ostéoclastes en l’accentuant, de même qu’ils
accentuent aussi la résorption osseuse. Nous avons aussi montré que les
cellules HL60 différenciées en ostéoclastes étaient activées par les FR
IgM. Nous avons testé les FR IgM à plusieurs concentrations, les FR IgA,
de même que les complexes immuns IgG-IgM FR sur l’activité de
résorption par les ostéoclastes. Nous n’avons pas pu réaliser assez
d’expériences pour faire une analyse statistique de nos résultats, mais
nous avons observé que les FR augmentent la résorption; ce qui pourrait
expliquer l’augmentation de l’érosion osseuse observée chez les patients
atteints de PAR. Cette étude constitue un premier pas vers la
compréhension de l’action des FR sur les ostéoclastes. Peu d’études
montrent l’action des FR directement sur les ostéoclastes, alors que nous
savons leur importance dans l’évolution de l’érosion osseuse chez les
patients atteints de PAR.
L’importance du récepteur Fc dans la physiologie osseuse et la
pathologie rhumatoïde a été soulignée par certains auteurs. En effet, il a
été démontré que les souris déficientes pour les adaptateurs DAP12 et
FcR de la voie de costimulation étaient incapables d’initier
l’ostéoclastogenèse et donc la résorption osseuse [133, 134]. Ochi et ses
collaborateurs ont démontré que dans un contexte inflammatoire, la
résorption osseuse est dépendante de l’augmentation de la formation
d’ostéoclastes à travers le récepteur Fc associé à un récepteur tel que
PIR-A [135]. Les adaptateurs DAP12 et FcR comme le montre la figure
29, à travers leur motif ITAM, transmettent un signal d’activation des
ostéoclastes. Leur fonction dépend non seulement des récepteurs
auxquels ils sont associés, de leurs ligands (encore inconnus), mais aussi
du contexte dans lequel ils sont activés [136]. Nous pensons que les FR
pourraient constituer des ligands activateurs pour le récepteur Fcγ.

86
Nous avons constaté que l’IgM FR entraîne une augmentation de la
résorption osseuse qu’il soit incubé seul ou sous forme de complexes
immuns. Par contre, l’IgA FR a plutôt un effet inhibiteur sur la résorption
alors que nous savons que la présence du FR IgA corrèle avec des
érosions osseuses importantes [132]. L’IgM FR pourrait être sous sa
forme de complexe immun pour se lier au récepteur Fcγ. Ce récepteur ne
peut lier que la fraction Fc d’une IgG, alors ce pont serait peut être
nécessaire pour la co-stimulation des ostéoclastes, d’autant plus que
l’importance des complexes immuns, surtout ceux associés aux FR a
déjà été démontrée dans la pathologie [137, 138]. Certains auteurs ont
montré que ces complexes immuns jouent un rôle dans la stimulation de
monocytes/macrophages pour la production du TNF via la stimulation
du récepteur Fc [131, 139]. Les cellules HL60 « osteoclast-like » et les
ostéoclastes possèdent peut être le récepteur Fcµ, récepteur pour la
fraction Fc d’un IgM sur lequel l’IgM FR pourrait se lier directement afin
d’agir sur la cellule, au vue de l’action de ce FR sur la résorption. Le
problème majeur que pose l’utilisation de FR humains est leur pureté
(mélange d’IgG et d’IgM, communication du Dr M. Newkirk). En effet, le
sérum sanguin et le liquide synovial sont des milieux très riches en toute
sorte de facteurs qui peuvent influencer la résorption osseuse, alors
l’extraction des FR purs reste une opération complexe.
Nous avons constaté la formation de trous de résorption plus gros
en présence de FR, ce phénomène peut être le fait d’une suractivation
des ostéoclastes ou alors de la présence d’un plus grand nombre
d’ostéoclastes actifs. On peut donc penser que les cellules deviennent
plus « agressives ». Les FR stimuleraient peut être la fusion des
ostéoclastes en stimulant l’expression de récepteurs de fusion tels que le
DC-STAMP, la cadherine E, etc... Dans tous les cas, ce constat suscite de
nombreuses questions qui restent à élucider.

87
L’activation de la phosphorylation des protéines sur résidus
tyrosine des cellules HL60 différenciées par les FR IgM FR, montre une
fois de plus l’intérêt de l’utilisation de ces cellules pour étudier les
ostéoclastes. L’activité des ostéoclastes est en grande partie régulée par
la phosphorylation des protéines tyrosines et parmi elles nous avons les
protéines tyrosines kinases et les protéines tyrosines phosphatases[140].
Les protéines tyrosines kinases impliqués dans l’ostéoclastogenèse et
l’activation des ostéoclastes sont celles de la famille des tyrosines kinases
non-réceptrices et parmi elles les tyrosines kinases Btk et Tec sont les
plus exprimées[141]. Parmi les protéines tyrosines phosphatases, les
plus connues dans les ostéoclastes sont : PTPRO, PTP, SHP-1, et PTP-
PEST[142].
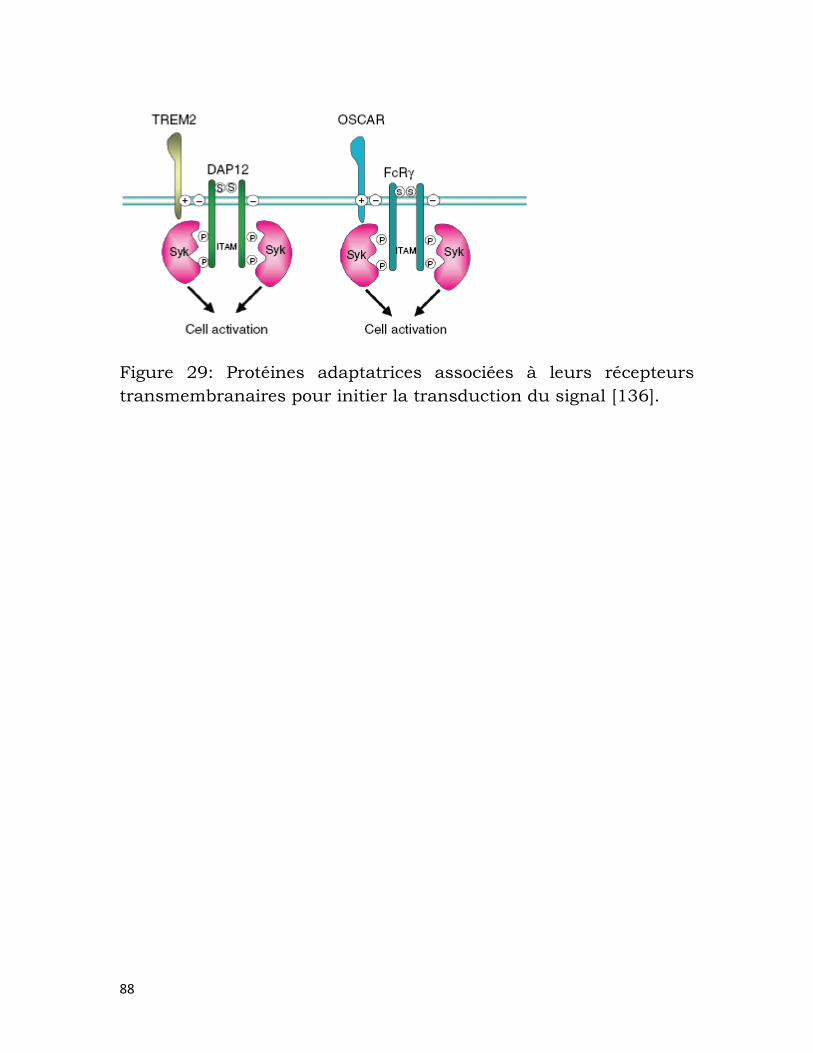
88
Figure 29: Protéines adaptatrices associées à leurs récepteurs
transmembranaires pour initier la transduction du signal [136].

89
IV. CONCLUSION ET PERSPECTIVES
Cette étude nous a permis de développer un modèle valide
d’ostéoclastes in vitro qui permettra d’étudier différentes conditions de
stimulation ou d’inhibition des fonctions ostéoclastiques. En effet, les
cellules HL60 « osteoclast-like » répondent à tous les critères d’un
ostéoclaste ; tant sur le plan de la morphologie que de l’activation. Nous
avons obtenu des cellules multi-nucléées, adhérentes et plus grosses; ces
cellules expriment les récepteurs RANK, les récepteurs de la calcitonine,
OSCAR, PIR-A, TREM2, SIRP1, DAP12 et FcR, le facteur de
transcription NFATc, la TRAP et sont capables de former des anneaux
d’actine. Il serait intéressant de calculer le pourcentage de cellules
différenciées sur le nombre total de cellules mises en culture, ainsi cela
pourrait aider à optimiser les expériences utilisant ce modèle car
l’apoptose ou la concentration cellulaires influencent peut être les
conditions de culture. La fusion semble être le procédé par lequel les
HL60 se différencient en ostéoclastes, il serait intéressant de vérifier les
récepteurs de fusion retrouvés dans les HL60. L’utilisation des FR sur les
cellules HL60 différenciées permettrait de valider le modèle et confirmer
nos premiers résultats obtenus sur la résorption osseuse par des
ostéoclastes en présence de FR. En effet, nous avons aussi montré
l’action des FR sur la différenciation des ostéoclastes et sur leur fonction
de résorption ; d’autres expériences doivent être cependant menées pour
valider cette fonction de résorption. Beaucoup reste encore à faire pour
comprendre l’impact des FR sur les ostéoclastes, mais les premiers
résultats de cette étude nous encouragent à poursuivre les expériences
dans le but d’obtenir des résultats statistiquement significatifs. Dans
cette étude il n’a été question que des facteurs rhumatoïdes IgM et IgA,
mais il serait important d’aller voir l’effet de chacune des trois classes de
FR sur la résorption osseuse. L’importance apparente du récepteur Fc
dans l’arthrite rhumatoïde, nous interpelle sur la question des sous-

90
types de ce récepteur qui peuvent être présents. Certes nous n’avons
trouvé que le CD64 (FcRI) dans les HL60, mais il serait intéressant
d’utiliser plusieurs techniques différentes pour cette recherche, car les
autres sous-types semblent être impliqués dans la maladie. Il serait
aussi interessant d’aller rechercher la présence des récepteurs pour les
IgM (récepteurs Fc), au vue de l’importance du FR IgM sur la résorption
dans l’arthrite rhumatoïde.

91
V. RÉFÉRENCES BIBLIOGRAPHIQUES
1. Naili, S., et al., Bone remodeling. J Mech Behav Biomed Mater. 4(6): p. 827-8. 2. Hadjidakis, D.J. and Androulakis, II, Bone remodeling. Ann N Y Acad Sci, 2006. 1092: p.
385-96. 3. Crockett, J.C., et al., Bone remodelling at a glance. J Cell Sci. 124(Pt 7): p. 991-8. 4. Redlich, K. and J.S. Smolen, Inflammatory bone loss: pathogenesis and therapeutic
intervention. Nat Rev Drug Discov. 11(3): p. 234-50. 5. Suda, T., et al., Modulation of osteoclast differentiation and function by the new
members of the tumor necrosis factor receptor and ligand families. Endocr Rev, 1999. 20(3): p. 345-57.
6. Takahashi, N., et al., Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front Biosci. 16: p. 21-30.
7. Kobayashi, Y., N. Udagawa, and N. Takahashi, Action of RANKL and OPG for osteoclastogenesis. Crit Rev Eukaryot Gene Expr, 2009. 19(1): p. 61-72.
8. Vaananen, H.K., et al., The cell biology of osteoclast function. J Cell Sci, 2000. 113 ( Pt 3): p. 377-81.
9. Gay, C.V., V.R. Gilman, and T. Sugiyama, Perspectives on osteoblast and osteoclast function. Poult Sci, 2000. 79(7): p. 1005-8.
10. Teitelbaum, S.L., Bone resorption by osteoclasts. Science, 2000. 289(5484): p. 1504-8. 11. Lakkakorpi, P.T., et al., Spatial organization of microfilaments and vitronectin receptor,
alpha v beta 3, in osteoclasts. A study using confocal laser scanning microscopy. J Cell Sci, 1993. 104 ( Pt 3): p. 663-70.
12. Akisaka, T., H. Yoshida, and R. Suzuki, The ruffled border and attachment regions of the apposing membrane of resorbing osteoclasts as visualized from the cytoplasmic face of the membrane. J Electron Microsc (Tokyo), 2006. 55(2): p. 53-61.
13. Neutzsky-Wulff, A.V., et al., Alterations in osteoclast function and phenotype induced by different inhibitors of bone resorption--implications for osteoclast quality. BMC Musculoskelet Disord. 11: p. 109.
14. Saftig, P., et al., Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A, 1998. 95(23): p. 13453-8.
15. Gallois, A., et al., [Osteoimmunology: an integrated vision of immune and bone systems]. Med Sci (Paris), 2009. 25(3): p. 259-65.
16. Katagiri, T. and N. Takahashi, Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis, 2002. 8(3): p. 147-59.
17. Fretz, J.A., et al., Receptor activator of nuclear factor-kappaB ligand-induced nuclear factor of activated T cells (C1) autoregulates its own expression in osteoclasts and mediates the up-regulation of tartrate-resistant acid phosphatase. Mol Endocrinol, 2008. 22(3): p. 737-50.
18. Takayanagi, H., Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol, 2009. 5(12): p. 667-76.
19. Leibbrandt, A. and J.M. Penninger, RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci, 2008. 1143: p. 123-50.
20. Valdes, A.M. and T.D. Spector, Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 7(1): p. 23-32.

92
21. Sangha, O., Epidemiology of rheumatic diseases. Rheumatology (Oxford), 2000. 39 Suppl 2: p. 3-12.
22. Steiner, G., Auto-antibodies and autoreactive T-cells in rheumatoid arthritis: pathogenetic players and diagnostic tools. Clin Rev Allergy Immunol, 2007. 32(1): p. 23-36.
23. Lee, D.M. and M.E. Weinblatt, Rheumatoid arthritis. Lancet, 2001. 358(9285): p. 903-11. 24. O'Dell, J.R., Therapeutic strategies for rheumatoid arthritis. N Engl J Med, 2004. 350(25):
p. 2591-602. 25. Feldmann, M., Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev
Immunol, 2002. 2(5): p. 364-71. 26. Silman, A.J. and J.E. Pearson, Epidemiology and genetics of rheumatoid arthritis. Arthritis
Res, 2002. 4 Suppl 3: p. S265-72. 27. Goronzy, J.J. and C.M. Weyand, Developments in the scientific understanding of
rheumatoid arthritis. Arthritis Res Ther, 2009. 11(5): p. 249. 28. van der Helm-van Mil, A.H., J.Z. Wesoly, and T.W. Huizinga, Understanding the genetic
contribution to rheumatoid arthritis. Curr Opin Rheumatol, 2005. 17(3): p. 299-304. 29. Newton, J.L., et al., A review of the MHC genetics of rheumatoid arthritis. Genes Immun,
2004. 5(3): p. 151-7. 30. Dieude, P. and F. Cornelis, Genetic basis of rheumatoid arthritis. Joint Bone Spine, 2005.
72(6): p. 520-6. 31. Klareskog, L., L. Padyukov, and L. Alfredsson, Smoking as a trigger for inflammatory
rheumatic diseases. Curr Opin Rheumatol, 2007. 19(1): p. 49-54. 32. McInnes, I.B. and G. Schett, The pathogenesis of rheumatoid arthritis. N Engl J Med.
365(23): p. 2205-19. 33. Firestein, G.S. and N.J. Zvaifler, How important are T cells in chronic rheumatoid
synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum, 2002. 46(2): p. 298-308.
34. Harris, E.D., Jr., Rheumatoid arthritis. Pathophysiology and implications for therapy. N Engl J Med, 1990. 322(18): p. 1277-89.
35. Imboden, J.B., The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol, 2009. 4: p. 417-34.
36. Nielen, M.M., et al., Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum, 2004. 50(2): p. 380-6.
37. Cooles, F.A. and J.D. Isaacs, Pathophysiology of rheumatoid arthritis. Curr Opin Rheumatol. 23(3): p. 233-40.
38. Klareskog, L., et al., Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol, 2008. 26: p. 651-75.
39. McInnes, I.B. and G. Schett, Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol, 2007. 7(6): p. 429-42.
40. Choy, E.H. and G.S. Panayi, Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med, 2001. 344(12): p. 907-16.
41. Feldmann, M., F.M. Brennan, and R.N. Maini, Role of cytokines in rheumatoid arthritis. Annu Rev Immunol, 1996. 14: p. 397-440.
42. Fox, D.A., et al., Cell-cell interactions in rheumatoid arthritis synovium. Rheum Dis Clin North Am. 36(2): p. 311-23.
43. Walsh, D.A., et al., Focally regulated endothelial proliferation and cell death in human synovium. Am J Pathol, 1998. 152(3): p. 691-702.

93
44. Karouzakis, E., et al., Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett, 2006. 106(1): p. 8-13.
45. Pap, T., et al., Are fibroblasts involved in joint destruction? Ann Rheum Dis, 2005. 64 Suppl 4: p. iv52-4.
46. Neumann, E., et al., Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med. 16(10): p. 458-68.
47. Shiozawa, S., et al., Pathogenesis of joint destruction in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz). 59(2): p. 89-95.
48. Firestein, G.S., Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum, 1996. 39(11): p. 1781-90.
49. Muller-Ladner, U., et al., Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol, 2005. 1(2): p. 102-10.
50. Boissier, M.C., et al., Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine, 2008. 75(4): p. 373-5.
51. Scrivo, R., et al., The immunology of rheumatoid arthritis. Ann N Y Acad Sci, 2007. 1108: p. 312-22.
52. Ehrenstein, M.R., et al., Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med, 2004. 200(3): p. 277-85.
53. Edwards, J.C. and G. Cambridge, Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology (Oxford), 2001. 40(2): p. 205-11.
54. Tran, C.N., S.K. Lundy, and D.A. Fox, Synovial biology and T cells in rheumatoid arthritis. Pathophysiology, 2005. 12(3): p. 183-9.
55. Kinne, R.W., B. Stuhlmuller, and G.R. Burmester, Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther, 2007. 9(6): p. 224.
56. Dayer, J.M., The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford), 2003. 42 Suppl 2: p. ii3-10.
57. Cascao, R., et al., Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun Rev. 9(8): p. 531-5.
58. Abramson, S., et al., The neutrophil in rheumatoid arthritis: its role and the inhibition of its activation by nonsteroidal antiinflammatory drugs. Semin Arthritis Rheum, 1983. 13(1 Suppl 1): p. 148-53.
59. Vossenaar, E.R., et al., Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum, 2003. 48(9): p. 2489-500.
60. Poubelle, P.E., et al., Differential expression of RANK, RANK-L, and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Res Ther, 2007. 9(2): p. R25.
61. Chakravarti, A., et al., Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood, 2009. 114(8): p. 1633-44.
62. Wipke, B.T. and P.M. Allen, Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol, 2001. 167(3): p. 1601-8.
63. Tanaka, D., et al., Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology, 2006. 119(2): p. 195-202.
64. Brennan, F. and J. Beech, Update on cytokines in rheumatoid arthritis. Curr Opin Rheumatol, 2007. 19(3): p. 296-301.
65. Smolen, J.S., et al., New therapies for treatment of rheumatoid arthritis. Lancet, 2007. 370(9602): p. 1861-74.

94
66. Lee, W. and M.H. Weisman, The predictive power of anti-cyclic citrullinated peptide antibodies: window into understanding gene/ environment/immunity interactions. J Rheumatol, 2006. 33(7): p. 1216-8.
67. Agrawal, S., R. Misra, and A. Aggarwal, Autoantibodies in rheumatoid arthritis: association with severity of disease in established RA. Clin Rheumatol, 2007. 26(2): p. 201-4.
68. Firestein, G.S., Evolving concepts of rheumatoid arthritis. Nature, 2003. 423(6937): p. 356-61.
69. Nishimura, K., et al., Meta-analysis: diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann Intern Med, 2007. 146(11): p. 797-808.
70. Newkirk, M.M., Rheumatoid factors: host resistance or autoimmunity? Clin Immunol, 2002. 104(1): p. 1-13.
71. Halldorsdottir, H.D., et al., A prospective study on the incidence of rheumatoid arthritis among people with persistent increase of rheumatoid factor. Ann Rheum Dis, 2000. 59(2): p. 149-51.
72. Mannik, M. and F.A. Nardella, IgG rheumatoid factors and self-association of these antibodies. Clin Rheum Dis, 1985. 11(3): p. 551-72.
73. Mannik, M., Rheumatoid factors in the pathogenesis of rheumatoid arthritis. J Rheumatol Suppl, 1992. 32: p. 46-9.
74. Newkirk, M.M., Rheumatoid factors: what do they tell us? J Rheumatol, 2002. 29(10): p. 2034-40.
75. Westwood, O.M., P.N. Nelson, and F.C. Hay, Rheumatoid factors: what's new? Rheumatology (Oxford), 2006. 45(4): p. 379-85.
76. Aho, K., et al., Rheumatoid factors and rheumatoid arthritis. Scand J Rheumatol Suppl, 1988. 74: p. 41-4.
77. Arnett, F.C., et al., The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum, 1988. 31(3): p. 315-24.
78. Kaneko, Y., et al., Sensitivity and specificity of 2010 rheumatoid arthritis classification criteria. Rheumatology (Oxford). 50(7): p. 1268-74.
79. Aletaha, D., et al., 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62(9): p. 2569-81.
80. Kahlenberg, J.M. and D.A. Fox, Advances in the medical treatment of rheumatoid arthritis. Hand Clin. 27(1): p. 11-20.
81. Emery, P. and T. Dorner, Optimising treatment in rheumatoid arthritis: a review of potential biological markers of response. Ann Rheum Dis. 70(12): p. 2063-70.
82. Cutolo, M., et al., Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis, 2001. 60(8): p. 729-35.
83. Alarcon, G.S., I.C. Tracy, and W.D. Blackburn, Jr., Methotrexate in rheumatoid arthritis. Toxic effects as the major factor in limiting long-term treatment. Arthritis Rheum, 1989. 32(6): p. 671-6.
84. Agarwal, S.K., Core management principles in rheumatoid arthritis to help guide managed care professionals. J Manag Care Pharm. 17(9 Suppl B): p. S03-8.
85. O'Dell, J.R., et al., Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications. N Engl J Med, 1996. 334(20): p. 1287-91.

95
86. Lipsky, P.E., et al., Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med, 2000. 343(22): p. 1594-602.
87. Agarwal, S.K., Biologic agents in rheumatoid arthritis: an update for managed care professionals. J Manag Care Pharm. 17(9 Suppl B): p. S14-8.
88. Bromley, M. and D.E. Woolley, Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum, 1984. 27(9): p. 968-75.
89. Bromley, M. and D.E. Woolley, Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheum, 1984. 27(8): p. 857-63.
90. Sun, H.B., Mechanical loading, cartilage degradation, and arthritis. Ann N Y Acad Sci. 1211: p. 37-50.
91. Gravallese, E.M., et al., Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol, 1998. 152(4): p. 943-51.
92. Goldring, S.R., Bone and joint destruction in rheumatoid arthritis: what is really happening? J Rheumatol Suppl, 2002. 65: p. 44-8.
93. Hirayama, T., et al., Osteoclast formation and activity in the pathogenesis of osteoporosis in rheumatoid arthritis. Rheumatology (Oxford), 2002. 41(11): p. 1232-9.
94. Gravallese, E.M., Bone destruction in arthritis. Ann Rheum Dis, 2002. 61 Suppl 2: p. ii84-6.
95. Goldring, S.R., Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology (Oxford), 2003. 42 Suppl 2: p. ii11-6.
96. Gravallese, E.M., et al., Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum, 2000. 43(2): p. 250-8.
97. Bezerra, M.C., et al., RANK, RANKL and osteoprotegerin in arthritic bone loss. Braz J Med Biol Res, 2005. 38(2): p. 161-70.
98. Sato, K. and H. Takayanagi, Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr Opin Rheumatol, 2006. 18(4): p. 419-26.
99. Haynes, D.R., et al., Osteoprotegerin and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint. Rheumatology (Oxford), 2001. 40(6): p. 623-30.
100. Maruotti, N., et al., Osteoclastogenesis and arthritis. Clin Exp Med. 11(3): p. 137-45. 101. Fujikawa, Y., et al., Human osteoclast formation and bone resorption by monocytes and
synovial macrophages in rheumatoid arthritis. Ann Rheum Dis, 1996. 55(11): p. 816-22. 102. Danks, L., et al., Synovial macrophage-osteoclast differentiation in inflammatory
arthritis. Ann Rheum Dis, 2002. 61(10): p. 916-21. 103. Fleck, R.A., S. Romero-Steiner, and M.H. Nahm, Use of HL-60 cell line to measure opsonic
capacity of pneumococcal antibodies. Clin Diagn Lab Immunol, 2005. 12(1): p. 19-27. 104. Schumpp, B. and E.J. Schlaeger, Optimization of culture conditions for high cell density
proliferation of HL-60 human promyelocytic leukemia cells. J Cell Sci, 1990. 97 ( Pt 4): p. 639-47.
105. Breitman, T.R., Growth and differentiation of human myeloid leukemia cell line HL60. Methods Enzymol, 1990. 190: p. 118-30.
106. Collins, S.J., R.C. Gallo, and R.E. Gallagher, Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature, 1977. 270(5635): p. 347-9.
107. McCarthy, D.M., et al., 1,25-dihydroxyvitamin D3 inhibits proliferation of human promyelocytic leukaemia (HL60) cells and induces monocyte-macrophage differentiation in HL60 and normal human bone marrow cells. Leuk Res, 1983. 7(1): p. 51-5.

96
108. Andersson, G. and E.K. Johansson, Adhesion of human myelomonocytic (HL-60) cells induced by 1,25-dihydroxyvitamin D3 and phorbol myristate acetate is dependent on osteopontin synthesis and the alpha v beta 3 integrin. Connect Tissue Res, 1996. 35(1-4): p. 163-71.
109. Kido, S., et al., Expression of RANK is dependent upon differentiation into the macrophage/osteoclast lineage: induction by 1alpha,25-dihydroxyvitamin D3 and TPA in a human myelomonocytic cell line, HL60. Bone, 2003. 32(6): p. 621-9.
110. Ryves, W.J., et al., HL-60 cell differentiation induced by phorbol- and 12-deoxyphorbol-esters. Carcinogenesis, 1994. 15(11): p. 2501-6.
111. Fontana, J.A., D.A. Colbert, and A.B. Deisseroth, Identification of a population of bipotent stem cells in the HL60 human promyelocytic leukemia cell line. Proc Natl Acad Sci U S A, 1981. 78(6): p. 3863-6.
112. Olins, A.L., et al., Retinoic acid differentiation of HL-60 cells promotes cytoskeletal polarization. Exp Cell Res, 2000. 254(1): p. 130-42.
113. Jian, P., et al., Retinoic acid induces HL-60 cell differentiation via the upregulation of miR-663. J Hematol Oncol. 4: p. 20.
114. Yen, A., et al., Control of HL-60 cell differentiation lineage specificity, a late event occurring after precommitment. Cancer Res, 1987. 47(1): p. 129-34.
115. Janick-Buckner, D., A.B. Barua, and J.A. Olson, Induction of HL-60 cell differentiation by water-soluble and nitrogen-containing conjugates of retinoic acid and retinol. Faseb J, 1991. 5(3): p. 320-5.
116. Koeffler, H.P., Induction of differentiation of human acute myelogenous leukemia cells: therapeutic implications. Blood, 1983. 62(4): p. 709-21.
117. Hozumi, M., Established leukemia cell lines: their role in the understanding and control of leukemia proliferation. Crit Rev Oncol Hematol, 1985. 3(3): p. 235-77.
118. Roodman, G.D., et al., 1,25-Dihydroxyvitamin D3 causes formation of multinucleated cells with several osteoclast characteristics in cultures of primate marrow. Proc Natl Acad Sci U S A, 1985. 82(23): p. 8213-7.
119. Yoneda, T., et al., Differentiation of HL-60 cells into cells with the osteoclast phenotype. Endocrinology, 1991. 129(2): p. 683-9.
120. Fibach, E., et al., Modulation of the maturation of human leukemic promyelocytes (HL-60) to granulocytes or macrophages. Leuk Res, 1982. 6(6): p. 781-90.
121. Wu, J., et al., The role of the c-fms oncogene in the regulation of HL-60 cell differentiation. Oncogene, 1990. 5(6): p. 873-7.
122. Amano, S., et al., A novel osteoclast precursor cell line, 4B12, recapitulates the features of primary osteoclast differentiation and function: enhanced transfection efficiency before and after differentiation. J Cell Physiol, 2009. 221(1): p. 40-53.
123. Chambers, T.J., et al., Generation of osteoclast-inductive and osteoclastogenic cell lines from the H-2KbtsA58 transgenic mouse. Proc Natl Acad Sci U S A, 1993. 90(12): p. 5578-82.
124. Hillstrom Shapiro, L., et al., Carbonic anhydrase II is induced in HL-60 cells by 1,25-dihydroxyvitamin D3: a model for osteoclast gene regulation. FEBS Lett, 1989. 249(2): p. 307-10.
125. Miyamoto, T., Regulators of osteoclast differentiation and cell-cell fusion. Keio J Med. 60(4): p. 101-5.
126. Yagi, M., et al., DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med, 2005. 202(3): p. 345-51.

97
127. Takayanagi, H., The role of NFAT in osteoclast formation. Ann N Y Acad Sci, 2007. 1116: p. 227-37.
128. Nimmerjahn, F. and J.V. Ravetch, Fcgamma receptors as regulators of immune responses. Nat Rev Immunol, 2008. 8(1): p. 34-47.
129. Raychaudhuri, G., D. McCool, and R.H. Painter, Human IgG1 and its Fc fragment bind with different affinities to the Fc receptors on the human U937, HL-60 and ML-1 cell lines. Mol Immunol, 1985. 22(9): p. 1009-19.
130. McCool, D., B.K. Birshtein, and R.H. Painter, Structural requirements of immunoglobulin G for binding to the Fc gamma receptors of the human tumor cell lines U937, HL-60, ML-1, and K562. J Immunol, 1985. 135(3): p. 1975-80.
131. Edwards, J.C. and G. Cambridge, Rheumatoid arthritis: the predictable effect of small immune complexes in which antibody is also antigen. Br J Rheumatol, 1998. 37(2): p. 126-30.
132. Arnason, J.A., et al., Relation between bone erosions and rheumatoid factor isotypes. Ann Rheum Dis, 1987. 46(5): p. 380-4.
133. Mocsai, A., et al., The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A, 2004. 101(16): p. 6158-63.
134. Koga, T., et al., Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature, 2004. 428(6984): p. 758-63.
135. Ochi, S., et al., Pathological role of osteoclast costimulation in arthritis-induced bone loss. Proc Natl Acad Sci U S A, 2007. 104(27): p. 11394-9.
136. Humphrey, M.B., L.L. Lanier, and M.C. Nakamura, Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev, 2005. 208: p. 50-65.
137. Duquesnoy, B., et al., [Circulating immune complexes and anti-IgG antibodies in rheumatoid disease. Value of C3d assay]. Rev Med Interne, 1981. 2(1): p. 11-6.
138. Reeback, J.S., et al., Circulating immune complexes and rheumatoid arthritis: a comparison of different assay methods and their early predictive value for disease activity and outcome. Ann Rheum Dis, 1985. 44(2): p. 79-82.
139. Mathsson, L., et al., Immune complexes from rheumatoid arthritis synovial fluid induce FcgammaRIIa dependent and rheumatoid factor correlated production of tumour necrosis factor-alpha by peripheral blood mononuclear cells. Arthritis Res Ther, 2006. 8(3): p. R64.
140. Sheng, M.H. and K.H. Lau, Role of protein-tyrosine phosphatases in regulation of osteoclastic activity. Cell Mol Life Sci, 2009. 66(11-12): p. 1946-61.
141. Shinohara, M., et al., Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell, 2008. 132(5): p. 794-806.
142. Granot-Attas, S., H. Knobler, and A. Elson, Protein tyrosine phosphatases in osteoclasts. Crit Rev Eukaryot Gene Expr, 2007. 17(1): p. 49-71.





























