Embed Size (px)
Citation preview

Étude du browning dans le cadre des
cancers associés à la cachexie
Delphine ALLOUCHE
Ophélie LESCAT
Guylène RIGNOL
Encadrées par Laurence NIETO
Master 2 Expression Génique et Protéines Recombinantes

Remerciements
Nous souhaitons dans un premier temps remercier Laurence Nieto. Un grand merci pour vos
conseils avisés, votre bienveillance et le temps que vous avez su nous consacrer malgré votre
emploi du temps si chargé.
Nous remercions Remy Poupot pour ses conseils lors des différentes présentations orales.
Nous remercions Mme Stéphanie BALOR, passionnée par son travail, qui a su répondre à nos
questions pour la préparation des échantillons pour la microscopie électronique. Merci pour
son accueil chaleureux, c’était un plaisir.
Nous remercions l’équipe 13 du Centre Méditerranéen de Médecine Moléculaire (C3M) de
Nice, L. Yvan-Charvet et V. Sarrazy pour leurs conseils concernant les souris KrasG12D.
Enfin nous remercions Laurent Paquereau, sans qui nous ne serions pas dans ce master.
Nous n’aurions pas passé quatre mois inoubliables et fait de ci belles rencontres.
Merci à tous les professeurs pour la qualité d’enseignement fourni et pour cette si belle
famille qu’est l’EGPR.
Pour finir, merci aux étudiants EGPR. Merci pour ces danses, ces fous-rires, ces tarots, cette
prise de poids (incontrôlée), cette entraide, cette bonne ambiance. Tout simplement …
Merci d’être VOUS !
Bonne lecture à tous

Résumé
La cachexie est un syndrome multifactoriel représentant près de 20% du taux de
mortalité chez les patients cancéreux. L’une des principales conséquences de la cachexie est la
perte incontrôlée des masses adipeuses et musculaires. Cet amaigrissement entraîne un
affaiblissement profond de l’organisme et une mauvaise réponse aux thérapies anti-
cancéreuses. Le tissu adipeux est principalement affecté lors des cancers associés à la cachexie
(CAC). En effet, sous l’effet de différents stimuli un phénomène de browning du tissu adipeux
blanc en beige est observé. Ce browning permet d’augmenter la production de chaleur. Ce
phénomène est possible grâce à une protéine mitochondriale : UCP1. L’équipe de Serkan Kir, a
mis en évidence la présence d’une hormone activant la transcription d’UCP1 chez des souris
présentant un modèle de tumeur pulmonaire, il s’agit de la parathyroid hormone related protein
(PTHrP). Afin de confirmer certains des résultats de l’équipe de Serkan Kir, la première étape de
notre projet sera d’observer les effets de PTHrP ; tant sur la fonctionnalité de la protéine UCP1
que sur le browning. De plus, nous souhaitons déterminer si PTHrP est libéré par d’autres types
de CAC. La seconde étape du projet, permettra de caractériser le modèle de souris déficientes
pour UCP1, largement décrit dans le cadre de l’obésité et non dans les cancers cachectiques.

Sommaire
Introduction bibliographique ............................................................................. 1
I/ La cachexie ................................................................................................................ 1
II/ Le tissu adipeux ........................................................................................................ 3
III/ Le browning ............................................................................................................. 4
Présentation d’une publication .......................................................................... 9
Projet de recherche .......................................................................................... 14
I/ Confirmer les effets de PTHrP sur la protéine UCP1 et le browning .............. 14
A/ Sur le modèle C57BL/6 ............................................................................................. 14
1) Les animaux ............................................................................................................................ 14 2) PTHrP et la cachexie ............................................................................................................... 15 3) PTHrP et le browning .............................................................................................................. 15
B/Sur le modèle Kras G12D ............................................................................................. 17
1) Les animaux .............................................................................................................................. 17 2) PTHrP et la cachexie ................................................................................................................. 17 3) PTHrP et le browning................................................................................................................ 18 4) Carctérisation de la tumeur ..................................................................................................... 18
C/ In Vitro sur d’autres modèles de cancers .................................................................. 18
II/ Effets de l’inhibition d’UCP1 sur le browning et la cachexie ......................... 20
1) Les animaux ............................................................................................................................ 20 2) Suivi de la cachexie ................................................................................................................. 21 3) Analyse du browning .............................................................................................................. 22 4) Suivi de la tumeur ................................................................................................................... 22
Conclusion et perspectives ............................................................................... 22
Bibliographie

Glossaire
ACOX1: Peroxisomal acyl-coenzyme A oxidase 1
ACSL1: Long-chain-fatty-acid--CoA ligase 1
AP2: Adipocyte protein 2
ATF-2: Activating Transcription Factor 2
ATGL: Adipose triglyceride lipase
ATP: Adenosine TriPhosphate
BAT: Brown Adipose Tissue
BTC: Betacelluline
CAC: Cancer Associated Cachexia
cAMP: cyclic Adenosine Mono Phosphate
CPT: Carnitine PalmitoylTransferase
CRE: cAMP Response Element
CREB: cAMP Response Element Binding Protein
DIO2: Type II iodothyronine deiodinase
EGF: Epidermal Growth Factor
EREG: Epiregulin
eWAT: epididymal White Adipose Tissue
FADH: Flavin Adenine Dinucleotide
FBXO32: F-box only protein 32
GLUT1-4: Glucose transporter 1-4
GPCR: G protein–coupled receptors
HB-EGF: Heparin-Binding EGF like growth factor
HOXC9: Homeobox protein Hox-C9
HSL: Hormone-sensitive lipase
iBAT: interscapular Brown Adipose Tissue
IF1: ATP synthase Inhibitor Factor 1
IgG : Immunoglobuline G
IL-6: Interleukine-6
iWAT: inguinal White Adipose Tissue
JAK: Janus kinase

LLC: Lung Lewis Carcinoma
MAPK: Mitogen-activated protein kinase
MSTN: Myostatin
MURF1: Muscle-specific RING finger protein 1
MYF5: Myogenic factor 5
NADH: Nicotinamide Adenine Dinucleotide
NE: Noradrenaline
NRF: Nuclear respiratory factor
NTB: Non Tumor Bearing
OCR: Oxygen Consumption Rate
PGC-1α: Peroxisome proliferator activated receptor gamma coactivator 1 alpha
PKA, C: Protein Kinase A, C
PPAR-γ: Persoxisome proliferator activated receptor gamma
PRDM16: Positive Regulatory Domain 16
PTH: Parathyroid hormone
PTH1R: Parathyroid hormone receptor 1
PTHrP: Parathyroid hormone related proteinSHOX2: Short stature homeobox protein 2
STAT: Signal Transducers and Activators of Transcription
TA: Tissu Adipeux
TG: Triglycérides
TGF β-1: Transforming growth factor TGF-beta1
TNFR: Tumor necrosis factor receptor
TNFα: Tumor Necrosis Factor α
TRE: Thyroid Response Element
TRIM63: E3 ubiquitin-protein ligase TRIM63
UCP1, 2, 3: Uncoupling Protein 1, 2, 3
WAT: White Adipose Tissue
β3-AR : Récepteur β3- adrénergique
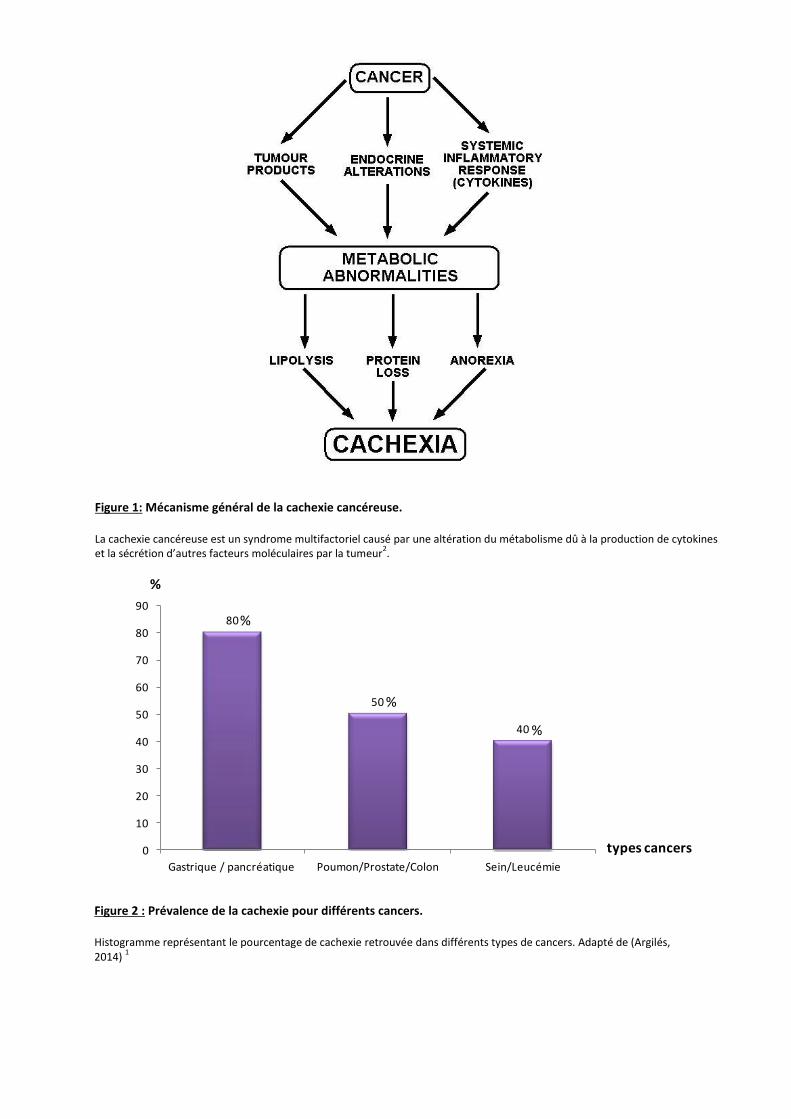
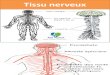
Figure 2 : Prévalence de la cachexie pour différents cancers. Histogramme représentant le pourcentage de cachexie retrouvée dans différents types de cancers. Adapté de (Argilés, 2014)
1
Figure 1: Mécanisme général de la cachexie cancéreuse. La cachexie cancéreuse est un syndrome multifactoriel causé par une altération du métabolisme dû à la production de cytokines et la sécrétion d’autres facteurs moléculaires par la tumeur
2.
80
50
40
0
10
20
30
40
50
60
70
80
90
Gastrique / pancréatique Poumon/Prostate/Colon Sein/Leucémie
%
types cancers
%
%
%

Introduction bibliographique
I/ La cachexie
Définition
La cachexie vient du grec « kakos » et « hexis » qui signifie mauvaise condition. Ce
syndrome multifactoriel est caractérisé par une perte incontrôlée des masses adipeuses et
musculaires ainsi qu’une augmentation de l’inflammation (Interleukine-6, Tumor Necrosis
Factor-α …) menant à un affaiblissement profond de l’organisme (Figure 1). Il ne faut pas
confondre la cachexie avec l’anorexie car la perte de poids ne peut ici pas être compensée par
un apport nutritionnel. On parle d’état cachectique lorsque la perte de poids est supérieure à 5%
en moins d’un an. Une autre caractéristique de la cachexie est une dérégulation du
métabolisme, il est alors observé chez les patients cachectiques une balance énergétique
négative 1,2.
La cachexie est observée dans de nombreux cancers en stades avancés mais également dans des
maladies infectieuses tels que le SIDA et la tuberculose, des maladies chroniques rénales et
cardiaques3.
La cachexie cancéreuse ou CAC (Cancer-Associated Cachexia)
Selon l’Organisation Mondiale de la Santé (OMS), 12,7 millions de cancers sont
nouvellement diagnostiqués par an. Il s’avère que, dans près de 50 % des cancers avancés, les
patients présentent une cachexie et près de 20 % des patients décèderont directement de cette
cachexie. Ceci est en partie dû à la dégradation générale de leur état, entraînant une altération
des réponses aux thérapies 4,5.
Tous les cancers ne sont pas cachectiques. Cependant, la cachexie est très fréquemment
retrouvée dans les cancers gastro-intestinaux (gastrique, pancréatique, prostatique et colique)
avec une prévalence allant de 50 % à 80 %. Pour le cancer du poumon, qui est la première cause
de mortalité par cancer dans le monde, la fréquence de la cachexie chez ces patient est près de
40% (Figure 2) 1.
1
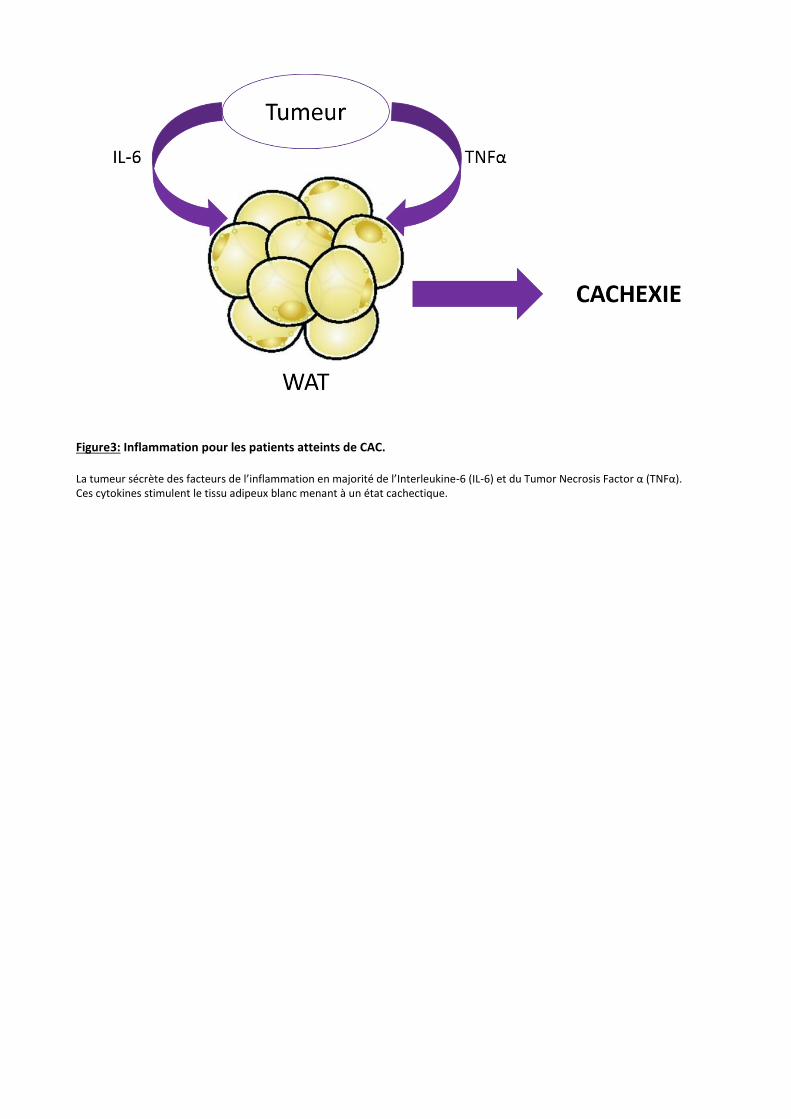
Figure3: Inflammation pour les patients atteints de CAC. La tumeur sécrète des facteurs de l’inflammation en majorité de l’Interleukine-6 (IL-6) et du Tumor Necrosis Factor α (TNFα). Ces cytokines stimulent le tissu adipeux blanc menant à un état cachectique.

A l’heure actuelle, on ne sait ni pourquoi ni comment les cancers en stades avancés mènent à ce
syndrome. De plus, étant donné que ce syndrome est multifactoriel et que le métabolisme lié au
cancer est complexe, il n’existe pas de traitements disponibles pour lutter contre la cachexie.
L’inflammation
L’inflammation est l’un des facteurs menant à un état cachectique. Deux protéines sont
majoritairement responsables de cet état: l’interleukine-6 (IL-6) et la Tumor Necrosis Factor α
(TNFα) (Figure 3).
Le TNFα était initialement appelé cachectine. Ce facteur est libéré par la tumeur ou par
les macrophages présents dans le microenvironnement tumoral. Un des mécanismes potentiels
de l’implication du TNFα sur la cachexie est qu’il inhiberait la différenciation des myoblastes en
muscles et induirait l’apoptose des pré-adipocytes et des adipocytes.6,7 L’équipe de Tisdale a
montré que, lorsque l’on ajoute des doses croissantes en TNFα, on observe une perte de poids
et un décès précoce des animaux.8
Cependant, le TNFα n’est pas le seul facteur aggravant l’état cachectique. En effet, on retrouve
dans le sérum de ces patients un taux élevé d’interleukine-6.
L’interleukine-6 est une cytokine libérée par la tumeur et induit la voie de signalisation
Jak/STAT. Elle est responsable d’une réponse hépatique aiguë dans un CAC 7. La présence d’un
taux élevé d’IL-6 est souvent corrélée avec une tumeur de grande taille. Laine et al, expliquent
dans leur étude que lorsqu’une ablation de la tumeur chez des souris est réalisée on observe une
diminution de la concentration en interleukine-6 7.
De plus, l’équipe de Petruzzelli, a démontré que le poids des tissus adipeux blancs était
augmenté chez des souris KO pour le récepteur à l’IL-6 3.Pour finir, l’équipe d’Uomo s’est
intéressée à la cachexie liée au cancer pancréatique. Ils ont observé que ces facteurs
inflammatoires étaient surexprimés, ce qui entraînait une augmentation du taux de leptine dans
le sang, favorisant donc la lipolyse et une balance énergétique modifiée 9.
2
Figure 4 : Morphologie des différents tissus adipeux. A : Tissu adipeux brun (BAT) : Grand nombres d’adipocytes de petites tailles comportant de nombreuses vacuoles
lipidiques. B : Tissu adipeux blanc (WAT) : Adipocytes de tailles supérieures aux adipocytes du BAT. Vacuole lipidique unique
est imposante. C : Tissu adipeux beige/ « brite ».Adipocytes de tailles moyennes avec des vacuoles lipidiques uniques ou
multiples 10
Microscope optique, coloration hématoxyline-éosine

II/ Le tissu adipeux
Les différents types de tissus adipeux
Le tissu adipeux brun (BAT)
Le tissu adipeux brun est majoritairement présent chez les petits rongeurs et tous les
mammifères hibernants. Chez l’Homme, il est principalement retrouvé chez les nourrissons et
disparaît en partie avec l’âge. La fonction première de ce tissu est de produire de la chaleur par
la lipolyse de ses adipocytes grâce à un mécanisme appelé thermogénèse 11.
Ce tissu est essentiellement composé d’adipocytes de petites tailles (environ 30µm) Ces
adipocytes contiennent un noyau central, de nombreuses vacuoles lipidiques de tailles variables
mais relativement petites (comparées à celles des adipocytes blancs). Pour finir, ces adipocytes
contiennent un grand nombre de mitochondries qui contiennent du fer, donnant la couleur
brune à ce tissu (Figure 4 A) 12.
Le tissu adipeux blanc (WAT)
Le tissu adipeux blanc est majoritairement présent chez l’Homme adulte puisqu’il
représente 20 à 25% du poids corporel chez la femme et 15 à 20% du poids chez l’homme. Son
rôle consiste essentiellement à stocker les graisses sous la forme de triglycérides (TG). Il existe
deux types de localisation du WAT chez l’Homme comme chez la souris ; le tissu adipeux blanc
sous-cutané et le tissu adipeux blanc viscéral 9:
Le WAT est caractérisé par des gros adipocytes (100-150µm), contenant une large vacuole
lipidique uniloculaire qui occupe la quasi-totalité de la cellule et plaque le noyau contre la
membrane plasmique (Figure 4 B). La densité en mitochondries dans ce tissu est relativement
basse 9, 12.
Le tissu adipeux Beige / « Brite »
Récemment, il a été trouvé dans le WAT des « brown-like adipocytes ». Ces adipocytes
ont été nommés beiges ou « brite ». En conditions de cultures normales ces adipocytes sont des
adipocytes du WAT. Cependant, lorsque des stimuli de la thermogénèse sont apportés, ces
adipocytes ont tendance à se rapprocher de la morphologie des adipocytes du BAT 13. Ces
« brite » adipocytes sont caractérisés par des adipocytes de taille moyenne10, une densité en
mitochondries plus élevée que dans le WAT et des vacuoles lipidiques soit multiloculaires soit
uniloculaires (Figure 4 C). Leur localisation est essentiellement observée dans le WAT sous-
cutané. Lorsque le stimulus thermogénique n’est plus présent, les « brown-like adipocytes »
redeviennent des adipocytes blancs 15.
3

Figure 5 : Précurseurs des différents tissus adipeux.
Une cellule mésenchymateuse donne deux types de précurseurs, les MYF5
+ ou les MYF5
-(myogenic factor 5). Les
précurseurs MYF5+ ou lignée myogénique se différencient sous l’action de PRDM16 (PR domain zinc finger protein 16) en
pré-adypocytes qui finiront leurs différenciations en donnant des adipocytes bruns. S’il n’y a pas d’induction au PRDM16, des myocytes sont observés. Les précurseurs MYF5
- deviennent des pré-adipocytes blancs qui par l’action de PPAR-γ
(peroxisome proliferator-activated receptor-γ), se différencient en adipocytes blancs. (Adapté de Park et al,2014) 14
.

Les précurseurs
Lors du développement embryonnaire, on observe une différenciation en trois feuillets :
l’endoderme, le mésoderme et l’ectoderme. Les précurseurs du tissu adipeux (TA) proviennent
du mésoderme. A partir d’une cellule mésenchymateuse, différentes étapes de différenciation,
mèneront à la formation des tissus adipeux bruns et blancs.
Précurseurs du BAT : Ils proviennent du mésoderme paraxial soit la lignée myogénique. Ils
expriment l’activateur transcriptionnel MYF5 (myogenic factor 5). La différenciation en
adipocytes bruns est contrôlée entre autre par le facteur de transcription PRDM16 (PR domain
zinc finger protein 16), le co-activateur transcriptionnel PGC-1α (peroxisome proliferator
activated receptor gamma coactivator 1 alpha) et un régulateur de transcription, appelé PPAR-γ
(peroxisome proliferator-activated receptor-γ) (Figure 5) 11, 14.
Une étude a montré que, si des précurseurs d’adipocytes bruns sont déplétés en PRDM16, ils se
différencient en muscle squelettique. La différentiation inverse est possible en exprimant
PRDM16 dans des myoblastes pour qu’ils deviennent des adipocytes bruns 14.
Précurseurs du WAT : Ils proviennent du mésoderme latéral et n’expriment pas MYF5. Les pré-
adipocytes blancs se différencient en adipocytes blancs par l’action de PPAR-γ (Figure 5) 11, 14.
III/ Le browning
Le métabolisme mitochondrial
La mitochondrie est le lieu principal de synthèse de l’énergie sous forme d’ATP. Cette
synthèse se fait par différentes voies : le cycle de Krebs, la phosphorylation oxydative et la
respiration mitochondriale. Une autre voie alimente le cycle de Krebs et la respiration
mitochondriale, il s’agit de la β-oxydation ou hélice de Lynen. La glycolyse apporte du pyruvate
au cycle de Krebs, permettant de fournir les cofacteurs NADH2 et le FADH2 nécessaires pour la
phosphorylation oxydative (Figure 6) 16.
La β-oxydation est la voie principale de dégradation des acides gras. Ils sont transformés en acyl-
CoA pour passer la membrane mitochondriale. Chaque tour d’hélice permet d’alimenter le cycle
de Krebs en acétyl-CoA et en cofacteurs NADH2 et le FADH2 (Figure 6) 17.
4
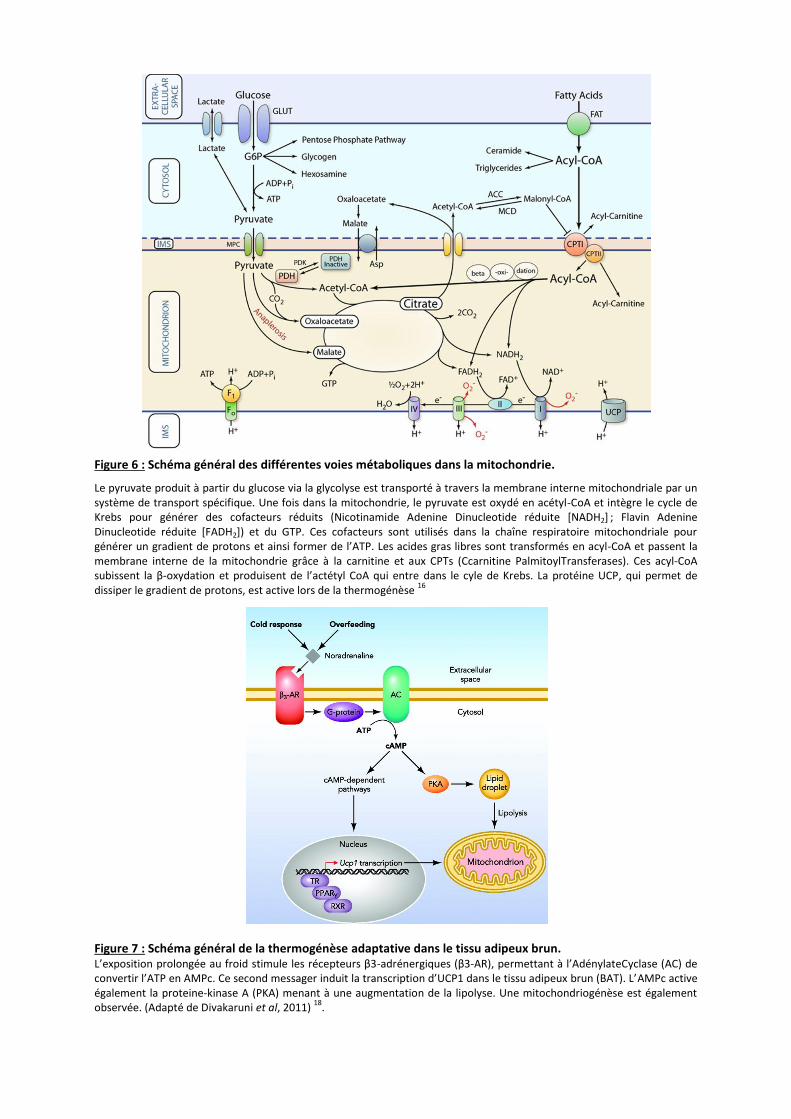
Figure 6 : Schéma général des différentes voies métaboliques dans la mitochondrie.
Le pyruvate produit à partir du glucose via la glycolyse est transporté à travers la membrane interne mitochondriale par un système de transport spécifique. Une fois dans la mitochondrie, le pyruvate est oxydé en acétyl-CoA et intègre le cycle de Krebs pour générer des cofacteurs réduits (Nicotinamide Adenine Dinucleotide réduite [NADH2] ; Flavin Adenine Dinucleotide réduite [FADH2]) et du GTP. Ces cofacteurs sont utilisés dans la chaîne respiratoire mitochondriale pour générer un gradient de protons et ainsi former de l’ATP. Les acides gras libres sont transformés en acyl-CoA et passent la membrane interne de la mitochondrie grâce à la carnitine et aux CPTs (Ccarnitine PalmitoylTransferases). Ces acyl-CoA subissent la β-oxydation et produisent de l’actétyl CoA qui entre dans le cyle de Krebs. La protéine UCP, qui permet de dissiper le gradient de protons, est active lors de la thermogénèse
16
Figure 7 : Schéma général de la thermogénèse adaptative dans le tissu adipeux brun. L’exposition prolongée au froid stimule les récepteurs β3-adrénergiques (β3-AR), permettant à l’AdénylateCyclase (AC) de convertir l’ATP en AMPc. Ce second messager induit la transcription d’UCP1 dans le tissu adipeux brun (BAT). L’AMPc active également la proteine-kinase A (PKA) menant à une augmentation de la lipolyse. Une mitochondriogénèse est également observée. (Adapté de Divakaruni et al, 2011)
18.

La chaine respiratoire mitochondriale est composée de cinq complexes protéiques. Le complexe
I oxyde le NADH2 qui libère un électron. Ce dernier est capté par le Co-enzyme Q. Au niveau du
complexe II, on a également oxydation d’un autre cofacteur, le FADH2, permettant aussi la
libération d’un électron. Tout cela génère un gradient de protons dans l’espace inter-
membranaire. Le complexe V également appelé ATP synthase, capte ces protons pour les
transférer dans la matrice mitochondriale, permettant ainsi la production d’ATP (Figure 6) 18.
Lors de la thermogénèse dans le BAT, la protéine UCP1 (UnCoupling Protein1) est activée
au niveau de la membrane interne de la mitochondrie. Cette protéine permet le découplage
mitochondrial, en détournant le gradient de protons. En effet, les protons ne vont plus être
utilisés pour fabriquer de l’ATP mais de la chaleur 19, 20.
Lors d’une exposition prolongée au froid on observe un autre type de thermogénèse dite la
thermogénèse adaptative, processus également appelé browning.
La thermogénèse adaptative est activée après action d’un stimulus (exposition prolongée
au froid, signalisation β-adrénergique…). Le stimulus envoie un signal au système nerveux
sympathique induisant la libération de catécholamines dont la noradrénaline. La noradrénaline
se lie sur son récepteur β3-adrénergique. Cette fixation active l’adénylate cyclase qui transforme
l’ATP en AMP cyclique (second messager). La production d’AMPc dans un premier temps,
favorise la transcription de la thermogénine ou UCP1 dans le noyau des adipocytes. Dans un
second temps, l’AMPc induit la voie des protéines kinases A (PKA) qui phosphoryle la protéine
HSL (hormone sensitive lipase) menant à une augmentation de la lipolyse. Ceci entraine une
augmentation de la concentration en acides gras libres. La PKA induit un autre phénomène qui
est la mitochondriogénèse, soit la division d’une mitochondrie et de son patrimoine génétique
pour en donner deux. Pour pallier à l’exposition au froid, l’ensemble de ces changements mène à
une production de chaleur en découplant la chaine respiratoire mitochondriale diminuant ainsi
la synthèse d’ATP. Ce phénomène est appelé browning et se passe dans le tissu adipeux brun ou
encore dans le tissu adipeux « brite » issu du WAT (Figure 7) 18, 22.
5

Figure 8 : Régulation transcriptionnelle d’UCP1 dans le tissu adipeux brun. Lors d’un stimulus par des récepteurs β-adrénergiques, la voie des PKA est activée, induisant la transcription d’UCP1. La transcription d’UCP1 se fait grâce à différents co-activateurs transcriptionnels, qui interagissent à proximité de son promoteur. Parmi ces séquences, deux sites CRE sont retrouvés et un site PPRE. Pgc1α= Ppar coactivator 1 α; PPRE= Ppar Response Element, FA= Fatty Acid; PPAR= Peroxisome Proliferator-Activated Receptor; PRDM16= PR zinc finger protein containing 16; CRE= cAMP Response Element; CREB= cAMP Response Element Binding Protein; ATF-2= Activating Transcription Factor 2 ; MAPK= Mitogen-activated protein kinase.( Adapté de Bonet et al, 2012)
21.

La thermogénèse adaptative
Les facteurs du browning
UCP1: uncoupling protein 1
UCP1 est une protéine de 33kDa localisée dans la membrane interne de la mitochondrie
des adipocytes bruns. Elle permet la production de chaleur à partir d’un gradient de protons.
Cette protéine « court-circuite » la respiration mitochondriale et diminue donc la production
d’ATP dans la cellule. UCP1 est activée lors de la thermogénèse adaptative ou browning. La
production d’UCP1 dans les « brite » est équivalente à celle des adipocytes du BAT et ceci après
une exposition prolongée au froid 23. La biosynthèse d’UCP1 est finement contrôlée au niveau
transcriptionnel, par de nombreux co-régulateurs transcriptionnels ou facteurs de transcription
tels que PGC-1α, PRDM16 et PPAR-γ (Figure 8) 21.
PGC-1α : Avant l’identification de PRDM16 dans les adipocytes bruns, PGC-1α était considéré
comme le marqueur du BAT. C’est un co-activateur de PPAR-γ. Son expression étant stimulée
après exposition prolongée au froid, il constitue le marqueur de la thermogénèse dans le BAT ou
le « brite ». Ce co-régulateur transcriptionnel forme un complexe avec PPAR-γ et induit, grâce à
la voie de la PKA (protéine kinase A), la mitochondriogénèse via l’expression de NRF (nuclear
respiratory factor). Ce complexe induit également la transcription d’UCP1 (Figure 8) 22,24.
PRDM16 : Protéine de 140kDa permettant la différenciation en BAT en stimulant les gènes
spécifiques du BAT et en inhibant ceux du WAT 25. Lors de l’induction en adipocytes bruns,
PRDM16 s’associe à d’autres co-activateurs transcriptionnels tels que PGC-1α et PPAR-γ 26.
PPAR-γ : Facteur de transcription indispensable pour la différenciation des tissus adipeux, on le
retrouve donc dans les trois types de TA 24,27.
UCP2 et UCP3 : uncoupling protein 2-3
Ces protéines possèdent respectivement 59 et 57% d’homologie avec UCP1. UCP2 est
exprimée dans plusieurs tissus comme le système nerveux central, les reins, le pancréas ou le
tissu adipeux blanc 28,. UCP3 est exprimé principalement dans les muscles squelettiques et dans
le BAT 29. Malgré une forte homologie avec UCP1, l’activité découplante de ces protéines reste
encore à déterminer.
6

A
B
1-36 1-3688 139
Figure 9 : Structure général de PTHrP. A : Après maturation, PTHrP ((ParaThyroid Hormone Related Protein) peut produire différents peptides. La partie 1-36 est homologue à la partie 1-36 de l’hormone PTH (ParaThyroid Hormone), la partie 38-94 donnera la midregion et la partie 107-139 correspondra à l’ostéostatine. (Adapté d’Abdelsayed et al, 2004)
46
B : Homologie en 1-36 des séquences N-terminales de PTH et PTHrP. Cette homologie leur confère une fixation sur le même récepteur (PTH1R : ParaThyroid Hormone Receptor 1)

HOXC9 : homeoprotein C9
Un autre marqueur du browning est la protéine HOXC9. Cette protéine a récemment
était observée dans le WAT. L’équipe de Walden a montré, que lorsqu’une culture d’adipocytes
blancs issus du WAT sous cutané, était exposée au froid de façon prolongée, les adipocytes
« brite » présentaient une augmentation de l’expression d’HoxC9. Ce marqueur, n’est cependant
pas retrouvé dans le BAT 30. Ainsi, HoxC9 jouerait un rôle clé dans la différenciation des
adipocytes « brite » à partir d’adipocytes blancs 31,32.
SHOX2: short stature homeobox protein 2
Chez le rongeur, ce régulateur transcriptionnel est plus exprimé dans le WAT sous-cutané
que dans d’autres TA. Cependant son expression n’est pas ubiquitaire, il est retrouvé
majoritairement dans le flanc et le dos plutôt que dans la région abdominale 33. Le taux d’ARNm
de SHOX2 est élevé dans le « brite » après exposition au froid, de ce fait, SHOX2 constitue un
bon marqueur du TA beige. 30,32.
PTH/PTHrP: parathyroid hormone /Parathyroid hormone-related protein
PTH et PTHrP sont deux hormones appartenant à la grande famille des PTH. L’une
comme l’autre jouent un rôle dans le développement des os ainsi que dans l’homéostasie du
phosphate et du calcium. Cependant, ces deux hormones ne sont pas codées par les mêmes
gènes (Figure 9) 31. PTH est une hormone endocrine produite principalement par les glandes
parathyroïdiennes, sécrétée dans la circulation sanguine. Elle est codée par le gène PTH, c’est un
polypeptide de 88 acides aminés obtenu après plusieurs hydrolyses successives. 34
PTHrP est un polypeptide codé par le gène PTHLH. Il est principalement sécrété par
certaines tumeurs causant ainsi une hypercalcémie. Ce facteur paracrine contrôlant la formation
osseuse endochondrale permet également le transport d’ions calcium Il est cependant
synthétisé par certains tissus sains mais son rôle dans ces différents tissus est à ce jour peu
compris 33. La pré-hormone [1-139] est obtenue après clivages endoprotéolityques. Cette
hydrolyse entraine la production de 3 peptidiques différents appelés, PTHrP [1-36] ; Midregion
[38-94] ; Ostéostatine [107-139] (Figure 9 A) 33.
Les parties N-terminale [1-36] de PTH et PTHrP sont hautement homologues car elles
partagent 8 des 13 premiers acides aminés (AA) et leurs structures secondaires est similaire sur
les 21 AA suivants (Figure 9 B) 33 Ce fragment [1-36] permet l’interaction avec le récepteur
PTH1R. Le récepteur PTH/PTHrP est un récepteur couplé aux protéines fortement exprimé au
niveau des os et des reins 35. Ce récepteur est associé à au moins deux systèmes de transduction
du signal, les voies des PKA et PKC (Protéines Kinases A et C).32.
7
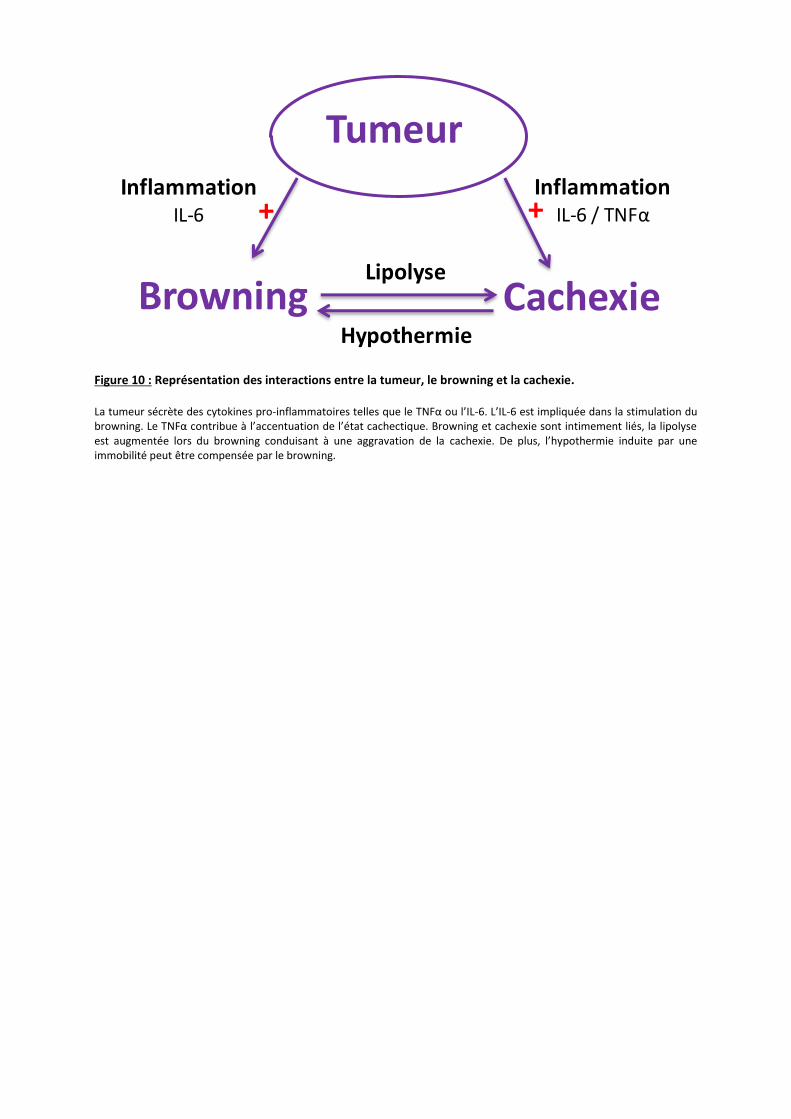
Figure 10 : Représentation des interactions entre la tumeur, le browning et la cachexie. La tumeur sécrète des cytokines pro-inflammatoires telles que le TNFα ou l’IL-6. L’IL-6 est impliquée dans la stimulation du browning. Le TNFα contribue à l’accentuation de l’état cachectique. Browning et cachexie sont intimement liés, la lipolyse est augmentée lors du browning conduisant à une aggravation de la cachexie. De plus, l’hypothermie induite par une immobilité peut être compensée par le browning.
Tumeur
Browning Cachexie
+ +Inflammation
IL-6 / TNFα
InflammationIL-6
Lipolyse
Hypothermie

En conclusion, la cachexie et le cancer sont intimement liés et l’étendue de la littérature
sur ce sujet le démontre. Le browning a longtemps été étudié dans le contexte de l’obésité.
Pourtant, depuis quelques années, la communauté scientifique s’intéresse à ce processus dans le
cadre des cancers et plus récemment de la cachexie.
La tumeur sécrète des médiateurs de l’inflammation (IL-6 ) qui stimulent le browning 3.
L’Il-6 ainsi que d’autres cytokines inflammatoires comme le TNFα sont également libérés par la
tumeur et accentue l’état cachectique 3,7.
Lorsque le browning est induit par la tumeur, une augmentation de la lipolyse est
observée 18,22. Ce catabolisme lipidique entraine un état cachectique 36. De plus, l’équipe de
Petruzzelli démontre que l’affaiblissement induit par la cachexie, entraine une immobilité des
souris. La production de chaleur étant alors réduite, une hypothermie est observée chez ces
souris cachectiques. Le browning permettrait ainsi de compenser cet état hypothermique 3.
Pour finir, dans cette étude des marquages d’UCP1 ont été effectués sur des
prélèvements d’adipocytes blancs de patients atteint de différents cancers. De manière
intéressante, UCP1 est constitutivement retrouvée sur des échantillons de patients atteints de
cancers CAC. Cependant, le seul cancer non cachectique ici étudié (colon sans cachexie,
probablement en stade précoce) ne présente quasiment pas de protéine UCP1. Ainsi, le
browning et les CAC paraissent étroitement liés 3.
L’équipe de Kir et al 37 a mis en évidence une nouvelle hormone libérée par la tumeur , le
PTHrP. Cette hormone jouerait un rôle dans l’induction du browning. Nous allons à présent vous
présenter les résultats de cette étude.
8
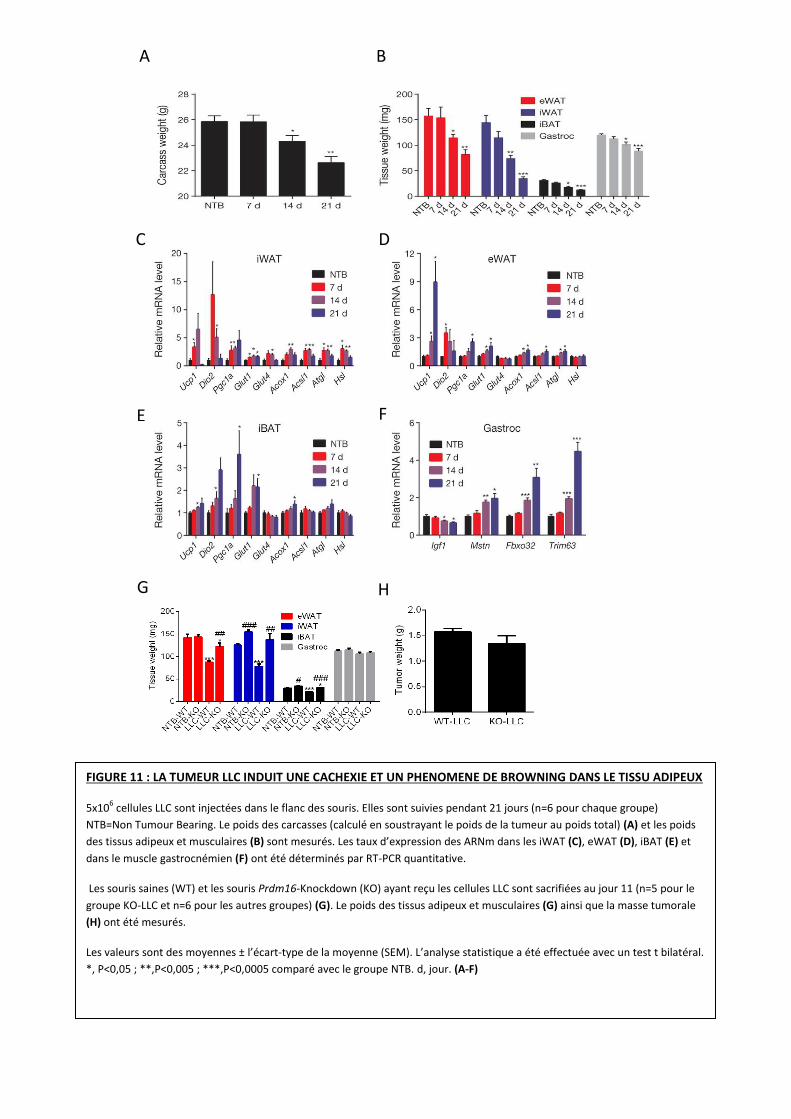
FIGURE 11 : LA TUMEUR LLC INDUIT UNE CACHEXIE ET UN PHENOMENE DE BROWNING DANS LE TISSU ADIPEUX
5x106 cellules LLC sont injectées dans le flanc des souris. Elles sont suivies pendant 21 jours (n=6 pour chaque groupe)
NTB=Non Tumour Bearing. Le poids des carcasses (calculé en soustrayant le poids de la tumeur au poids total) (A) et les poids
des tissus adipeux et musculaires (B) sont mesurés. Les taux d’expression des ARNm dans les iWAT (C), eWAT (D), iBAT (E) et
dans le muscle gastrocnémien (F) ont été déterminés par RT-PCR quantitative.
Les souris saines (WT) et les souris Prdm16-Knockdown (KO) ayant reçu les cellules LLC sont sacrifiées au jour 11 (n=5 pour le
groupe KO-LLC et n=6 pour les autres groupes) (G). Le poids des tissus adipeux et musculaires (G) ainsi que la masse tumorale
(H) ont été mesurés.
Les valeurs sont des moyennes ± l’écart-type de la moyenne (SEM). L’analyse statistique a été effectuée avec un test t bilatéral.
*, P<0,05 ; **,P<0,005 ; ***,P<0,0005 comparé avec le groupe NTB. d, jour. (A-F)
*, **, *** pour comparer le groupe NTB au groupe LLC. #, ##, ###, pour comparer le groupe WT au groupe KO. *, P<0,05 ; **,
P<0,005 ; ***,P<0,0005 ; #,P<0,05 ; ##,P<0,005 ; ###,P<0,0005. (G, H)
A
C D
F E
G H
B

Présentation d’une publication Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia37
Comme expliqué en introduction, la cachexie est un syndrome particulièrement
dévastateur caractérisé par une augmentation de l’inflammation, une perte des masses
adipeuses et musculaires ainsi qu’un dérèglement de la balance énergétique. Elle constitue donc
un facteur négatif pour les thérapies anti cancéreuses envisagées chez les patients. Les
mécanismes moléculaires liés à la cachexie sont complexes et peu compris. Une élévation des
cytokines inflammatoires est observée lors de ce syndrome. Pourtant, les thérapies anti-
cytokines ne sont pas efficaces 37.
Une activation du BAT a été décrite chez des souris et certains patients atteints de CAC. Il
n’est à ce jour par déterminé comment la tumeur induit la thermogénèse ni comment ceci
entraine une perte des masses adipeuses et musculaires. Pour comprendre de quelle manière la
tumeur stimule le browning l’équipe de Serkan Kir a utilisé un modèle murin de cancer du
poumon: Lewis Lung Carcinoma (LLC) provoquant la cachexie des souris cancéreuses.
Les auteurs ont dans un premier temps étudié les effets de la tumeur sur l’induction du
browning et de la cachexie. Pour ce faire, des cellules LLC sont injectées en sous-cutanée à des
souris syngéniques C57BL/6, on obtient alors l’induction d’une tumeur présente sous la peau
avec des caractéristiques d’une tumeur pulmonaire et le développement d’une cachexie.
Toutes les souris cancéreuses perdent du poids au cours des trois semaines avant
sacrifice (Figure 11 A), incluant une diminution des tissus adipeux et musculaires (Figure 11 B).
Différents tissus adipeux (TA) sont étudiés, le iWAT est le tissu adipeux inguinal (type sous
cutané), le eWAT est le tissu adipeux épididymaire (type viscéral). Pour finir, l’iBAT est le TA brun
inter-scapulaire, localisation majoritaire de ce tissu chez les rongeurs. La cachexie des souris
cancéreuses est aussi caractérisée par une augmentation de l’expression des gènes impliqués
dans l’atrophie musculaire comme MSTN, FBXO32 et TRIM63 (Figure 11 F).
MSTN (myostatine) est anciennement connue sous le nom de Growth différentiation
factor 8 (GDF8). C’est un facteur de croissance qui limite la croissance des tissus musculaires. Elle
fait partie de la famille des TGF β-1.
FBXO32 est également connu sous le nom d’atrogin1. Cette protéine est grandement
exprimée lors de l’atrophie musculaire. C’est un composant d’un complexe d’ubiquitination
favorisant l’adressage aux protéasomes pour la dégradation de certaines protéines.
9


TRIM63 est également nommé MURF1. Lors d’une privation en acides aminés cette
protéine adresse des protéines musculaires aux protéasomes pour alimenter les autres organes
en acides aminés. De plus, elle inhibe la synthèse de nuovo de protéines musculaires.
En réponse à la progression tumorale, tous ces types de TA présentent une
augmentation de l’expression des gènes impliqués dans la thermogénèse comme UCP1, DIO2 et
PGC1a (Figure 11 C-E). PGC1α : (Peroxisome proliferator-activated receptor gamma coactivator
1- α) est un co-activateur de PPAR-γ (Peroxisome Proliferator-Activated Receptor γ). Son
expression étant stimulée après une longue exposition au froid, il constitue le marqueur de la
thermogénèse dans le TA. De plus, il participe à la régulation transcriptionnelle d’UCP1 22,24
(Figure 8). DIO2: (Type II iodothyronine deiodinase) est responsable de la transformation de
l’hormone thyroïdienne T4 (3,5,3',5'-tetraiodothyronine) en T3 (3,5,3'-triiodothyronine). La T3 en
se fixant sur la région promotrice « Thyroid Response Element » d’UCP1 permet une activation
de la transcription d’UCP1 (Figure 8) 20.
Parallèlement, Serkan Kir et al, étudient d’autres gènes impliqués dans le métabolisme
énergétique et ces derniers voient également leur expression augmenter (Figure 11 C-E). ACSL1
joue un rôle dans la lipogenèse permettant d’augmenter le pool de lipides disponibles. HSL et
ATGL sont des lipases de la lipolyse permettant ainsi de libérer les acides gras à partir des
triglycérides pour alimenter la β-oxydation. Pour finir, ACOX1 au cours de la β-oxydation
convertit les acides gras en Acétyl Co-A pour alimenter le cycle de Krebs. GLUT 1-4 sont les
récepteurs du glucose permettant une augmentation intracellulaire du glucose pour une
conversion en pyruvate menant à une intégration dans le cycle de Krebs.
Pour déterminer si le browning aggrave l’état cachectique, les chercheurs ont utilisé des
souris déficientes pour le facteur PRDM16 38 Elles sont caractérisées par une diminution accrue
de leur potentiel thermogénique et sont résistantes au browning. Comme précédemment 5x106
cellules LLC sont injectées dans le flanc droit des souris. La perte de TA est significativement
inhibée chez les souris cancéreuses et déficientes pour PRDM16 tandis que le volume tumoral
n’est pas statistiquement différent. Par ailleurs, les masses musculaires ne sont pas affectées
(Figure 11 G, H). Nous émettons une réserve quant à l’analyse sur les masses musculaires, en
effet, les auteurs ont sacrifié ces souris à 11 jours. Ce temps d’induction du cancer et de la
cachexie n’est peut être pas le plus meilleur pour déterminer un lien entre browning et cachexie.
L’ensemble de ces résultats démontre que la tumeur induit une cachexie et un
phénomène de browning dans les tissus adipeux.
10
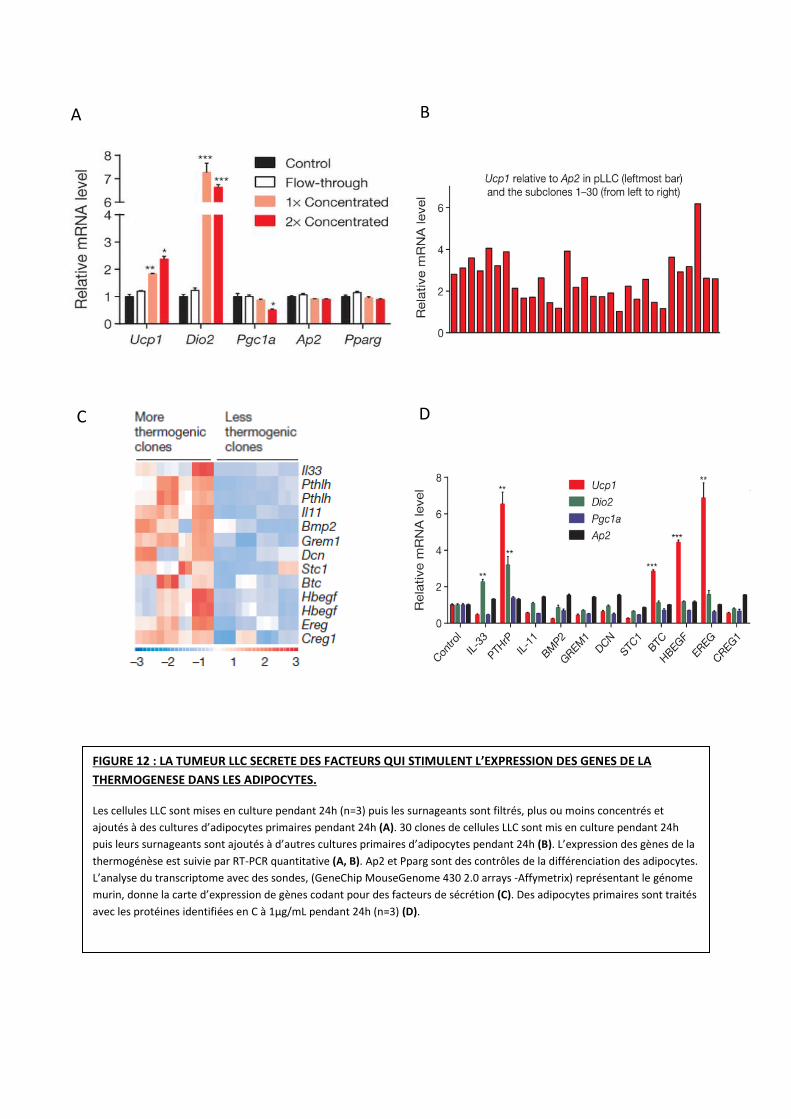
A B
D C
FIGURE 12 : LA TUMEUR LLC SECRETE DES FACTEURS QUI STIMULENT L’EXPRESSION DES GENES DE LA
THERMOGENESE DANS LES ADIPOCYTES.
Les cellules LLC sont mises en culture pendant 24h (n=3) puis les surnageants sont filtrés, plus ou moins concentrés et
ajoutés à des cultures d’adipocytes primaires pendant 24h (A). 30 clones de cellules LLC sont mis en culture pendant 24h
puis leurs surnageants sont ajoutés à d’autres cultures primaires d’adipocytes pendant 24h (B). L’expression des gènes de la
thermogénèse est suivie par RT-PCR quantitative (A, B). Ap2 et Pparg sont des contrôles de la différenciation des adipocytes.
L’analyse du transcriptome avec des sondes, (GeneChip MouseGenome 430 2.0 arrays -Affymetrix) représentant le génome
murin, donne la carte d’expression de gènes codant pour des facteurs de sécrétion (C). Des adipocytes primaires sont traités
avec les protéines identifiées en C à 1µg/mL pendant 24h (n=3) (D).
Les valeurs sont des moyennes ± l’écart-type de la moyenne (SEM). L’analyse statistique a été effectuée avec un test t
bilatéral. *, P<0,05 ; **,P<0,005 ; ***,P<0,0005 comparé avec le groupe contrôle.

Pour comprendre comment la tumeur induit le browning du TA, l’équipe souhaite
identifier les facteurs responsables d’une augmentation de l’expression des gènes de la
thermogénèse.
Pour ce faire, le surnageant de culture LLC est filtré sur membrane de 3-KDa et
différentes fractions sont testées sur des cultures primaires d’adipocytes (prélevés à partir du TA
blanc inguinal ou du BAT inter scapulaire). Seules les fractions retenues sur filtre de 3kDa
stimulent l’expression des gènes de la thermogénèse tels qu’UCP1 et DIO2 (Figure 12 A). Ces
résultats suggèrent que les facteurs sécrétés par la tumeur LLC sont des macromolécules. De
plus, ces résultats démontrent que l’induction du browning ne nécessite pas un dialogue entre la
tumeur et les adipocytes (Figure 12 A).
D’autre part, pour identifier les facteurs qui stimulent la thermogénèse l’équipe a généré
une série de clones afin de déterminer le profil d’expression des gènes. A partir de cellules LLC
en culture, 30 clones sont obtenus et les surnageants sont récupérés pour produire 30 milieux
conditionnés indépendant puis testés sur cultures primaires d’adipocytes. De manière
intéressante les chercheurs observent une hétérogénéité induite par le microenvironnement
tumoral (UCP1 différemment exprimé en fonction des clones observés) (Figure 12 B). Les
auteurs ont choisi huit clones présentant des activités thermogéniques plus ou moins
importantes (quatre clones avec UCP1 très exprimé, quatre clones avec UCP1 moins exprimé). Le
profil d’expression de ces derniers est analysé avec des sondes (GeneChip MouseGenome 430)
représentant le génome murin. Les ARNm fortement exprimés dans les clones les plus
thermogéniques sont identifiés (Figure 12 C). Ensuite, les protéines correspondantes sont
testées sur cultures primaires d’adipocytes. Certaines induisent l’expression d’UCP1 (Figure 12
D). Ces protéines ainsi identifiées sont des membres de la famille des EGF (Epidermal Growth
Factor) comme la Betacelluline (BTC), l’Heparin-Binding EGF like growth factor (HB-EGF) et
l’Épireguline (EREG) ainsi que des protéines relatives aux hormones parathyroïdiennes
(parathyroid-hormone-related peptide: PTHrP).
Comme expliqué en introduction, PTHrP a plusieurs isoformes dont PTHrP (1-34). La PTH
(parathyroid-hormone) et la PTHrP sont homologues aux extrémités N-term (PTH 1-34, PTHrP 1-
34) leur conférant une fixation sur le même récepteur. 31-33
Ces résultats indiquent que les cellules tumorales LLC sécrètent des facteurs qui
stimulent l’expression des gènes de la thermogénèse dans les adipocytes, potentiellement les
membres de la famille des EGF et PTHrP.
C
11
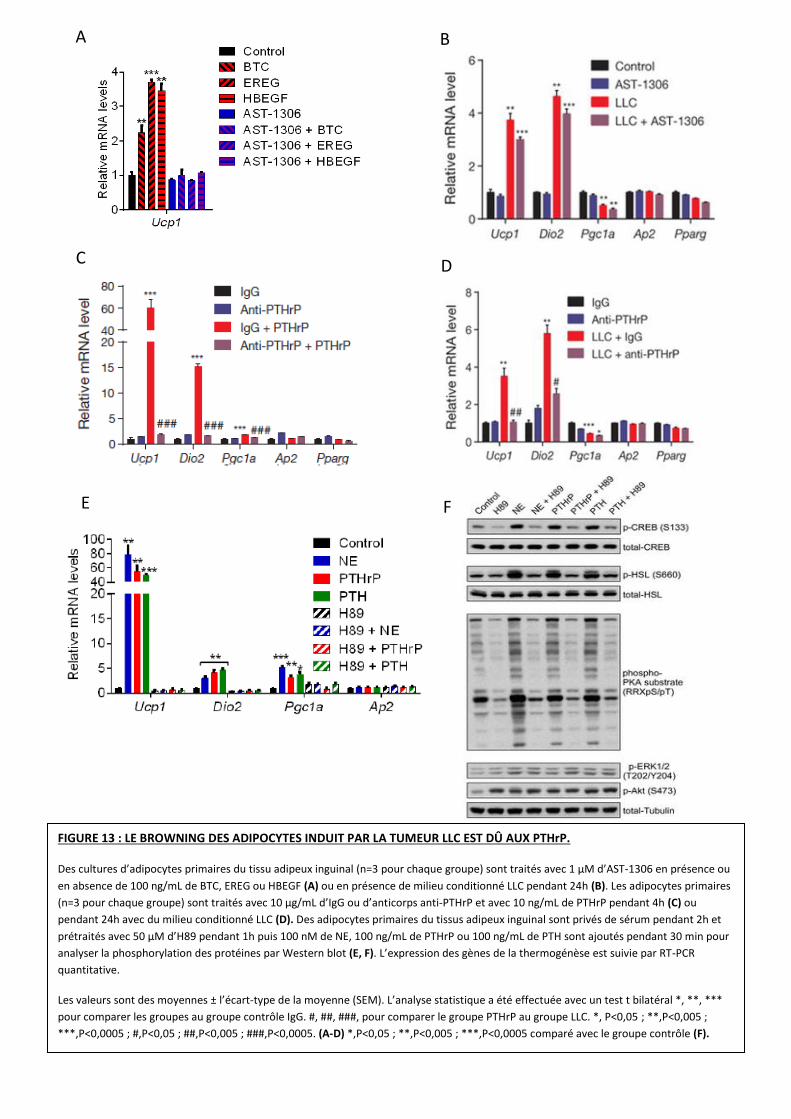
FIGURE 13 : LE BROWNING DES ADIPOCYTES INDUIT PAR LA TUMEUR LLC EST DÛ AUX PTHrP.
Des cultures d’adipocytes primaires du tissu adipeux inguinal (n=3 pour chaque groupe) sont traités avec 1 µM d’AST-1306 en présence ou
en absence de 100 ng/mL de BTC, EREG ou HBEGF (A) ou en présence de milieu conditionné LLC pendant 24h (B). Les adipocytes primaires
(n=3 pour chaque groupe) sont traités avec 10 µg/mL d’IgG ou d’anticorps anti-PTHrP et avec 10 ng/mL de PTHrP pendant 4h (C) ou
pendant 24h avec du milieu conditionné LLC (D). Des adipocytes primaires du tissus adipeux inguinal sont privés de sérum pendant 2h et
prétraités avec 50 µM d’H89 pendant 1h puis 100 nM de NE, 100 ng/mL de PTHrP ou 100 ng/mL de PTH sont ajoutés pendant 30 min pour
analyser la phosphorylation des protéines par Western blot (E, F). L’expression des gènes de la thermogénèse est suivie par RT-PCR
quantitative.
Les valeurs sont des moyennes ± l’écart-type de la moyenne (SEM). L’analyse statistique a été effectuée avec un test t bilatéral *, **, ***
pour comparer les groupes au groupe contrôle IgG. #, ##, ###, pour comparer le groupe PTHrP au groupe LLC. *, P<0,05 ; **,P<0,005 ;
***,P<0,0005 ; #,P<0,05 ; ##,P<0,005 ; ###,P<0,0005. (A-D) *,P<0,05 ; **,P<0,005 ; ***,P<0,0005 comparé avec le groupe contrôle (F).
A B
C D
E F

Aux vues de ces résultats, les chercheurs souhaitent aller plus loin en déterminant quels
facteurs sont impliqués dans cette induction de la thermogénèse.
Dans un premier temps, pour déterminer la contribution de BTC, EREG et HBEGF sur le
browning, l’équipe utilise un inhibiteur irréversible (AST-1306) des récepteurs aux EGF sur des
adipocytes primaires en culture. Cette inhibition empêche l’expression des ARNm d’UCP1
exprimés par des adipocytes non traités au milieu LLC après induction par la famille des EGFs
(Figure 13 A). Cependant, l’expression d’UCP1 n’est pas inhibée lorsqu’ AST-1306 est ajoutée aux
adipocytes cultivés en présence de milieu conditionné LLC (Figure 13 B).
Ces résultats suggèrent que les membres de la famille EGF n’ont pas de rôle crucial dans le
browning induit par la tumeur LLC.
Dans un second temps, un anticorps inhibiteur de PTHrP est utilisé pour déterminer son
implication dans l’induction des gènes de la thermogénèse en présence de la tumeur. Cet
anticorps inhibe entièrement les effets de PTHrP sur l’induction d’UCP1 et DIO2 en absence de
milieu LLC (Figure 13 C). De manière intéressante, quand les adipocytes sont traités avec le
milieu conditionné LLC et l’anticorps anti-PTHrP, l’expression d’UCP1 et DIO2 est également
inhibée (Figure 13 D).
Ces résultats indiquent que PTHrP montre un rôle dans l’induction des gènes de la
thermogénèse et donc un possible effet sur la thermogénèse.
Le récepteur aux PTHrP est couplé à une protéine G et active la voie des Protéines
Kinases A (PKA). La noradrénaline (NE) se fixe sur les récepteurs β-adrénergiques, également
connus pour activer cette voie et pour induire l’expression des gènes de la thermogénèse.
Ainsi, l’équipe a décidé d’observer cette voie de signalisation. Le traitement des cultures
primaires d’adipocytes avec NE et PTHrP stimule la phosphorylation des substrats de la PKA
CREB et HSL (Figure 13 E). Un inhibiteur de cette PKA, le H89, bloque la voie de signalisation de
PTHrP, induisant une perte de l’expression des gènes de la thermogénèse (Figure 13E, F).
Ces résultats démontrent que le browning des adipocytes induit par la tumeur est dépendant
des PTHrP. Ces derniers utilisent la voie des PKA pour induire l’expression d’UCP1 et DIO2 et
partagent des mécanismes moléculaires communs avec la voie β-adrénergique.
12
E D C
B A
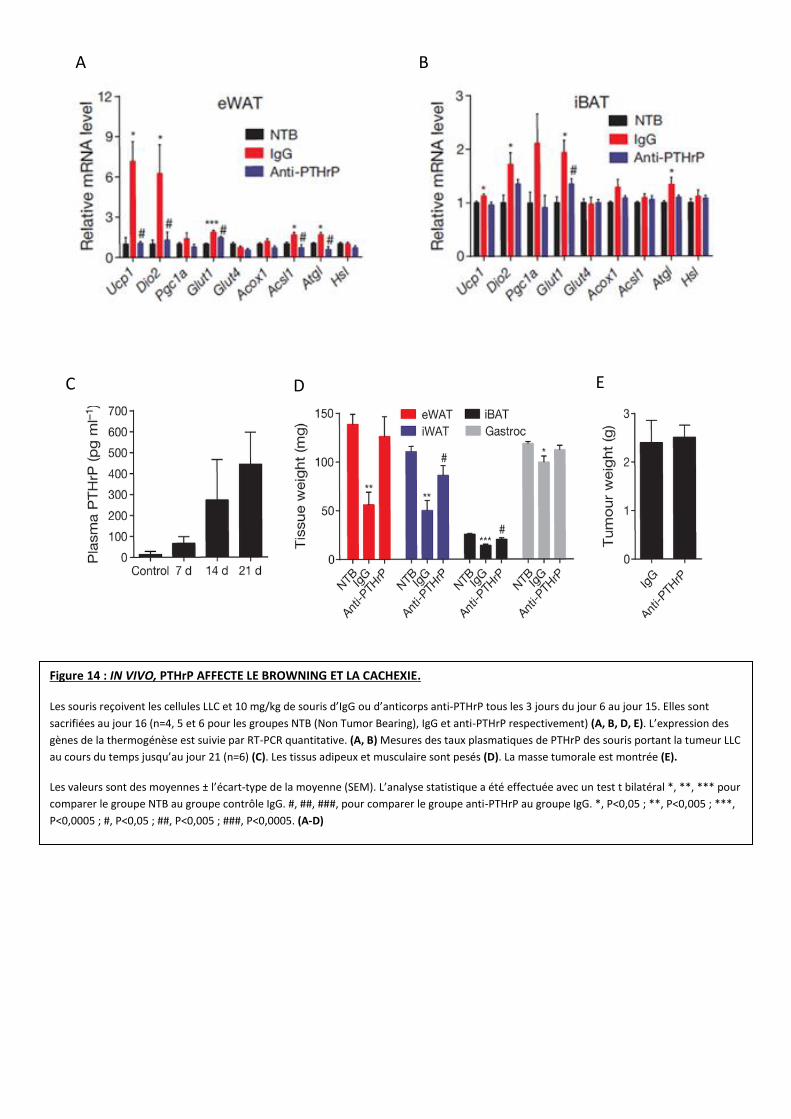
Figure 14 : IN VIVO, PTHrP AFFECTE LE BROWNING ET LA CACHEXIE.
Les souris reçoivent les cellules LLC et 10 mg/kg de souris d’IgG ou d’anticorps anti-PTHrP tous les 3 jours du jour 6 au jour 15. Elles sont
sacrifiées au jour 16 (n=4, 5 et 6 pour les groupes NTB (Non Tumor Bearing), IgG et anti-PTHrP respectivement) (A, B, D, E). L’expression des
gènes de la thermogénèse est suivie par RT-PCR quantitative. (A, B) Mesures des taux plasmatiques de PTHrP des souris portant la tumeur LLC
au cours du temps jusqu’au jour 21 (n=6) (C). Les tissus adipeux et musculaire sont pesés (D). La masse tumorale est montrée (E).
Les valeurs sont des moyennes ± l’écart-type de la moyenne (SEM). L’analyse statistique a été effectuée avec un test t bilatéral *, **, *** pour
comparer le groupe NTB au groupe contrôle IgG. #, ##, ###, pour comparer le groupe anti-PTHrP au groupe IgG. *, P<0,05 ; **, P<0,005 ; ***,
P<0,0005 ; #, P<0,05 ; ##, P<0,005 ; ###, P<0,0005. (A-D)

Afin de confirmer leurs résultats, l’équipe a étudié l’effet de l’inhibition de PTHrP in vivo
chez la souris. Cette inhibition bloque l’expression des gènes de la thermogénèse dans les eWAT
et iBAT (Figure 14 A, B).
De plus, près de 500 pg/mL de PTHrP plasmatique sont mesurés chez les souris LLC après 3
semaines d’induction de la tumeur (Figure 14 C). L’anticorps (Ac) anti-PTHrP (ou un contrôle IgG)
est ensuite injecté à ces dernières. De manière intéressante, les souris LLC ayant reçu l’Ac anti-
PTHrP ne perdent pas de poids, tandis que les souris LLC contrôles (IgG) démontrent une
cachexie (Figure 14 D). De surcroit, le traitement anti-PTHrP inhibe la perte des masses
adipeuses et musculaires sans changer la masse tumorale (Figure 14 E).
En partant du principe que l’inhibition des effets de PTHrP par l’anticorps n’est probablement
pas totale (présence de faux négatifs), ces résultats démontrent un rôle clé de PTHrP sur la
cachexie induite par la tumeur LLC.
L’ensemble de ces résultats démontre que PTHrP est un facteur clé de l’induction de la
cachexie associée au cancer. Qu’il est libéré par la tumeur LLC et qu’il stimule l’expression des
gènes de la thermogénèse du tissu adipeux. L’inhibition de PTHrP bloque le browning et la perte
de masses adipeuses. Ainsi, l’ensemble de ces données suggère un modèle selon lequel PTHrP
agirait seul pour induire le browning des tissus adipeux.
Afin de confirmer certains des résultats obtenus par l’équipe de Serkan Kir, nous
proposons d’utiliser le même modèle de souris LLC. Nous souhaitons aller plus loin en observant
UCP1 au niveau protéique et en approfondissant la caractérisation du browning. De plus, pour
renforcer l’étude sur PTHrP, il est indispensable d’utiliser un modèle de souris portant une réelle
tumeur pulmonaire. Nous souhaitons également déterminer si PTHrP est libéré par d’autres
types de tumeurs.
En seconde partie de projet, nous avons pour objectif d’étudier l’inhibition d’UCP1 avec
un modèle de souris KO UCP1. Ces souris devraient être déficientes pour le browning, ce qui
nous permettra d’appréhender le lien entre cancer, browning et cachexie.
13
0 8 11 14 17 20 21
8 Semaines Jours
LLC /PBS
SacrificeAnti- PTHrP
ou IgG
A
B
C57BL/6+ PBS
C57BL/6+ LLC
Anti-PTHrP
IgG
* Calorimétrieindirecte
* *
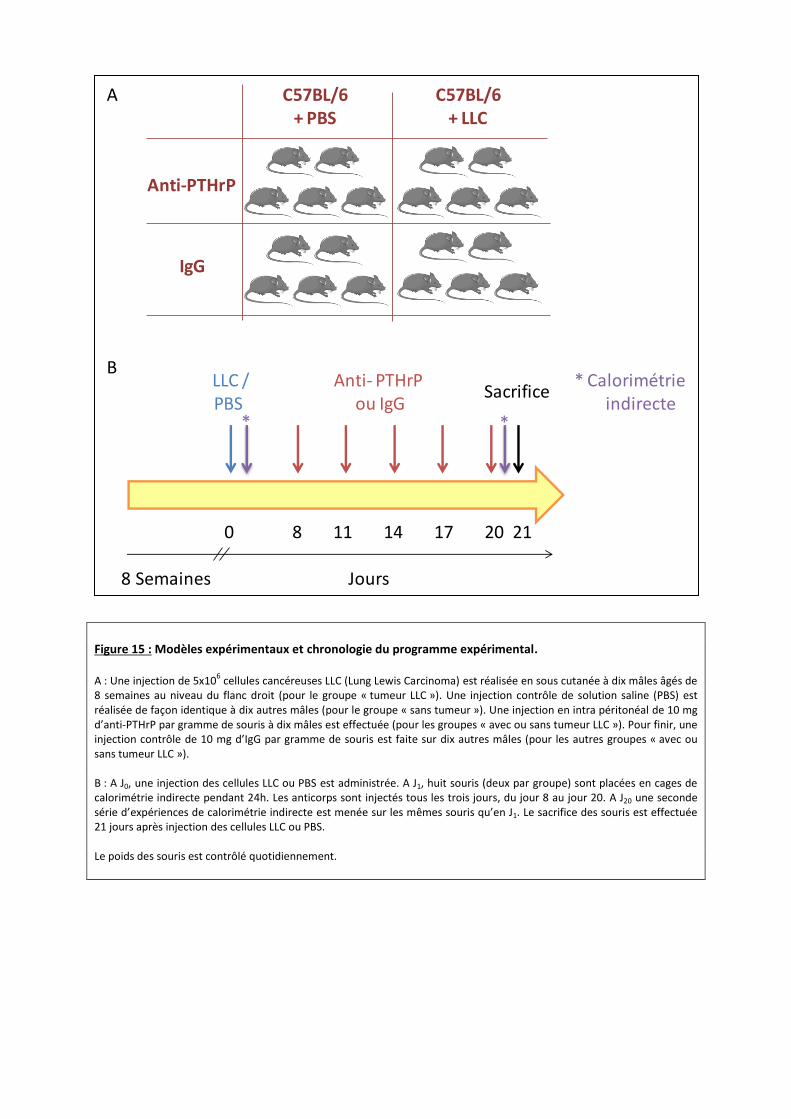
Figure 15 : Modèles expérimentaux et chronologie du programme expérimental. A : Une injection de 5x10
6 cellules cancéreuses LLC (Lung Lewis Carcinoma) est réalisée en sous cutanée à dix mâles âgés de
8 semaines au niveau du flanc droit (pour le groupe « tumeur LLC »). Une injection contrôle de solution saline (PBS) est réalisée de façon identique à dix autres mâles (pour le groupe « sans tumeur »). Une injection en intra péritonéal de 10 mg d’anti-PTHrP par gramme de souris à dix mâles est effectuée (pour les groupes « avec ou sans tumeur LLC »). Pour finir, une injection contrôle de 10 mg d’IgG par gramme de souris est faite sur dix autres mâles (pour les autres groupes « avec ou sans tumeur LLC »). B : A J0, une injection des cellules LLC ou PBS est administrée. A J1, huit souris (deux par groupe) sont placées en cages de calorimétrie indirecte pendant 24h. Les anticorps sont injectés tous les trois jours, du jour 8 au jour 20. A J20 une seconde série d’expériences de calorimétrie indirecte est menée sur les mêmes souris qu’en J1. Le sacrifice des souris est effectuée 21 jours après injection des cellules LLC ou PBS. Le poids des souris est contrôlé quotidiennement.

Projet de recherche
I/ Confirmer les effets de PTHrP sur la protéine UCP1 et le browning
A/ Sur le modèle de souris C57BL/6
Comme décrit précédemment, l’équipe de Serkan Kir observe une augmentation de
l’ARNm UCP1 après stimulation aux PTHrP. Cette augmentation est observée dans les WAT
épididymaires et inguinaux ainsi que dans le BAT. Cependant ces résultats n’ont pas étés
confirmés au niveau protéique. De plus, la caractérisation du browning doit être approfondie. En
effet, seule une étude morphologique des adipocytes a été réalisée et aucune analyse
fonctionnelle des mitochondries n’a été effectuée 37.
Ainsi, dans un premier temps, nous avons pour objectif de confirmer les effets de PTHrP
sur la protéine UCP1 et la régulation du browning.
1) Les animaux
Vingt souris mâles C57BL/6 commandées chez Jackson Laboratory seront étudiées.
Toutes les études sur les souris seront réalisées en accord avec les directives et provisions de la
Commission pour les Expérimentations Animales du Ministère Scientifique Français et les
recommandations du comité éthique local. Nous travaillerons sur des mâles maintenus dans un
cycle circadien à température ambiante (≈22°C) et leur prise alimentaire sera contrôlée.
Une injection de 5x106 cellules cancéreuses LLC (Lung Lewis Carcinoma) sera réalisée en
sous cutanée à dix mâles âgés de 8 semaines au niveau du flanc droit. Ces souris constitueront
notre modèle d’étude de la cachexie cancéreuse. Pour les souris contrôles, une injection de
solution saline (PBS) sera administrée (Figure 15 A, B).
Les cellules LLC auront préalablement été mises en culture dans du milieu Dulbecco’s Modified
Eagle Medium (DMEM) 10% SVF, en présence de pénicilline et streptomycine 37 .
Afin d’étudier les effets de PTHrP, des anticorps neutralisants seront injectés en intra
péritonéal à dix mâles (avec ou sans tumeur LLC). Les administrations seront réalisées tous les
trois jours, du jour 8 au jour 20 (après injection des cellules LLC) à raison de 10 mg d’anti-PTHrP
par gramme de souris. Un groupe de dix mâles (avec ou sans tumeur LLC) recevra 10 mg d’IgG
par gramme de souris. Le sacrifice des souris sera réalisé par dislocation cervicale 21 jours après
l’injection des cellules LLC (Figure 15 A, B).
14

Figure 16 : Programme expérimental des analyses histologiques.
Trois séries de coupes histologiques sont effectuées pour les différents groupes de souris (Figure 16 A). Trois types de tissu adipeux (TA) sont étudiés. Deux types de tissus adipeux blancs (WAT, White Adipose Tissue), sous-cutané (inguinal) et viscéral (rétro péritonéal). Un type de tissu adipeux brun (BAT, Brown Adipose Tissue), l’inter scapulaire. Chaque type de tissu adipeux est prélevé du côté droit (péri tumoral) et gauche pour comparaison. Les séries de coupes ne sont pas traitées de la même façon. Les protocoles diffèrent pour la coloration H&E et les marquages par immunohistochimie d’UCP1 et d’IF1.
WATSous cutané
Inguinal
BATInter scapulaire
WATViscéral
Rétro péritonéal
Coloration H&E
Marquage UCP1
Marquage IF1
Coloration H&E
Marquage UCP1
Marquage IF1
Coloration H&E
Marquage UCP1
Marquage IF1

2) PTHrP et cachexie
L’évolution de la cachexie sera suivie au cours du temps par des pesées journalières. Au
sacrifice les tissus adipeux blancs viscéraux (rétro péritonéaux) et sous cutanés (inguinaux) ainsi
que le tissu adipeux brun inter scapulaire seront pesés. Chaque tissu adipeux blanc sera prélevé
à partir du côté droit (péri tumoral) et du côté gauche des souris. Ces deux prélèvements nous
permettrons de déterminer si la cachexie est plus accentuée en péri tumoral. La cachexie étant
également caractérisée par une perte des masses musculaires, nous procéderons également à la
pesée des muscles gastrocnémien (mollets droit et gauche).
3) PTHrP et browning
Après sacrifice, les tissus adipeux récupérés précédemment pour les différentes pesées,
seront utilisés pour effectuer des analyses histologiques. Les tissus seront fixés dans un tampon
PBS à 3,7% formaline, inclus en paraffine, déshydratés et des sections de 6 µm seront effectuées
avec un microtome. Ces expériences seront effectuées comme expliqués dans la publication de
Petruzzelli,et al3.
Une première partie des différentes de coupes effectuées seront colorées à
l’hématoxyline-éosine. Cette étape nous permettra de caractériser les adipocytes (tailles,
morphologies et nombres). La taille (diamètre) et les quantités relatives des adipocytes de
chaque type de TA, seront déterminées manuellement grâce au logiciel Image J, à raison de 50
adipocytes par coupe. Cette première étude nous permettra de comparer les différents types de
TA de souris cachectiques cancéreuses ayant reçu (ou non) un anticorps neutralisant de PTHrP.
Nous nous attendons à observer, chez des souris cachectiques traitées sans anticorps anti-
PTHrP, des différences de tailles, nombres et morphologies des adipocytes du TA inguinal.
Certains adipocytes devraient tendre vers une morphologie du BAT soit des adipocytes plus
nombreux, plus petits avec présence de multiples vacuoles lipidiques. Les TA des souris
cachectiques traitées avec les anticorps anti-PTHrP devraient tendre vers une morphologie du
WAT viscéral (lieu non préférentiel du browning) (Figure 16).
Une seconde et troisième partie des coupes seront dédiées à une analyse
immunohistochimique des protéines UCP1 et IF1 (anticorps commandés chez Abcam). IF1 (ATP
synthase Inhibitor Factor 1) est connu pour bloquer l’ATP synthase mitochondriale dans sa
configuration inactive pour éviter que cette dernière hydrolyse l’ATP lorsque le potentiel
membranaire est trop faible. IF1 constitue donc un second marqueur du découplage
mitochondrial 40.
15


Les sections devront être déparaffinées, une étape de démasquage de l’antigène (UCP1
ou IF1) devra être effectuée pour ensuite ajouter les anticorps anti-UCP1 et anti-IF1. Finalement,
les coupes seront contre colorées à l’hématoxyline 3 (Figure 16)..
Lors de la mise en place du projet nous souhaitions observer le découplage mitochondrial
par analyses en microscopie électronique à transmission. Après une rencontre avec Mme
Stéphanie BALOR, responsable de la plateforme METI (microscopie électronique intégrative),
nous avons pris conscience de l’ampleur du travail tant sur la préparation des échantillons que
sur l’analyse des données. En effet, chaque échantillon doit être soumis à différents traitements
répartis sur une semaine.
De plus, les tissus adipeux ne peuvent pas être conservés et doivent être traités au
moment du sacrifice pour les analyses histologiques. En effet, la congélation peut entrainer une
destruction de la vacuole lipidique. Cette information est loin d’être négligeable pour une mise
en place d’un aussi grand nombre d’analyse histologiques.
Pour toutes ces raisons, une seule analyse des mitochondries sera effectuée ici pour
l’ensemble du projet.
Cette étape devrait nous permettre d’observer une différence de nombre des
mitochondries selon le type de TA observé en fonction que les souris aient été traitées avec ou
sans anti-PTHrP. De plus, la microscopie électronique nous informera sur la densité en électrons
des mitochondries après ajout d’un contrastant (osmium). Une mitochondrie active voit son
nombre de crêtes mitochondriales augmenter, et apparait plus denses aux électrons. En
comparant, les mitochondries de TA inguinaux de souris cancéreuses ayant reçu des injections
d’anticorps anti-PTHrP, par rapport à des TA inguinaux de souris contrôles (sans anticorps anti-
PTHrP) ; nous pourrons confirmer de façon fonctionnelle l’effet inhibiteur de PTHrP sur le
browning. Pour cette étude, nous procéderons de la même manière que l’équipe d’Ozaki
Kiyokazu dans le cadre de leur étude de la fonction et du phénotype du BAT 39.
Pour finir, le métabolisme général des souris sera suivi par des expériences de
calorimétrie indirecte (Figure 15 B). Cette étude nous permettra de déterminer la dépense
énergétique ainsi que la production de chaleur des souris avec ou sans inhibition des effets de
PTHrP (en présence ou en absence de tumeur). La dépense énergétique ainsi que la production
de chaleur seront calculés grâce aux mesures des échanges gazeux (consommation d’O2 et
production de CO2). Les expériences seront réalisées comme indiqué dans l’étude de Julie
Marcotorchinoa 41.
16

A
B
0 1 2 3 4 5 6 7 8
Semaines
Inhalation Adénovirus Sacrifice
Prélèvements sanguins
Agés de 8 semaines
Anti- PTHrP ou IgG
Anti-PTHrP
IgG
Culture Adipocytes
KrasG12D
Adéno videKrasG12D
Adéno CRE
Figure 17 : Modèles expérimentaux et chronologie du programme expérimental. A : Toutes les souris portent la mutation Kras
G12D comportant une cassette stop bordée de deux sites LoxP reconnus par
l’enzyme CRE recombinase. Une solution d’adénovirus à 2,5x107 PFU exprimant l’enzyme CRE recombinase est administrée
par inhalation à dix mâles KrasG12D
âgés de 8 semaines (groupe cancer du poumon). Les souris sont anesthésiées au
Vetflurane 5% et inspirent la solution par inhalation réflexe au réveil. Une solution contrôle d’adénovirus sans CRE
recombinase est administrée par inhalation à dix autres mâles KrasG12D
âgés de 8 semaines (groupe sans cancer).
10 mg d’anti-PTHrP par gramme de souris sont injectés en intra péritonéal à dix mâles (groupe avec ou sans cancer du
poumon). Pour le contrôle, 10 mg d’IgG par gramme de souris sont injectés à dix autres mâles (avec ou sans cancer du
poumon).
B : A S0, les inhalations d’adénovirus sont effectuées. Aux semaines 0, 2, 4, 6 et 8 différents échantillons sanguins à partir de
la veine saphène sont prélevés. Les anticorps sont injectés tous les trois jours, de la semaine 6 à la semaine 8. Le sacrifice
des souris est effectué huit semaines après l’inhalation de l’adénovirus. Au sacrifice, le tissu adipeux blancs inguinal de deux
souris contrôle est prélevé pour cultures primaires d’adipocytes.
Le poids des souris est contrôlé chaque semaine.

B/ Sur le modèle de souris KrasG12D
L’objectif de cette partie, est de vérifier que PTHrP agit sur UCP1 et donc influe le
browning à partir d’un modèle présentant une tumeur localisée, cette fois ci aux poumons.
1) Les animaux
Pour des raisons budgétaires, deux couples de souris KrasG12D transgéniques seront
commandés chez Jackson Laboratory 42. L’élevage sera effectué en interne afin d’obtenir vingt-
deux mâles. Ces souris portent la mutation KrasG12D comportant une cassette stop bordée de
deux sites LoxP reconnus par l’enzyme CRE recombinase. L’inhalation d’un adénovirus exprimant
l’enzyme CRE recombinase nous permet d’induire une tumeur localisée au poumon (Genecust,
Luxembourg).
Dix mâles âgés de huit semaines seront anesthésiés au Vetflurane 5% et inspireront une
solution d’adénovirus à 2,5x107 PFU par inhalation réflexe à leur réveil.
Ces mâles « KrasG12D/Adéno-CRE » constitueront notre modèle de souris cancéreuses
cachectiques. Afin d’être dans un état de cancer avancé et ainsi d’observer une cachexie plus
marquée, les souris seront sacrifiées huit semaines après inhalation.
Comme en partie I/A, dix souris (5 KrasG12D/Adéno-CRE + 5 KrasG12D/Adéno-vide)
recevront des injections d’anticorps neutralisant PTHrP six semaines après inhalation de
l’adénovirus. Les injections seront réalisées tous les trois jours de la semaine 6 à la semaine 8 à
raison de 10 mg d’anti-PTHrP par gramme de souris. Le groupe contrôle recevra 10 mg d’IgG par
gramme de souris (Figure 17).
Deux souris contrôles seront dédiées à des cultures primaires d’adipocytes, que nous
décrirons plus tard (Figure 17).
2) PTHrP et cachexie
La cachexie sera étudiée comme précédemment (suivi du poids corporel chaque semaine
et pesées des tissus adipeux et musculaires au sacrifice). Nous avons également souhaité suivre
l’inflammation, qui est une des caractéristiques indispensables pour confirmer une cachexie.
Pour ce faire, des prélèvements sanguins réalisés à partir de la veine saphène seront effectués
toutes les deux semaines (Figure 17). Le sang sera centrifugé et le plasma conservé à -80°C pour
effectuer des dosages de TNFα et IL-6 par test ELISA.
17


3) PTHrP et browning
La caractérisation du browning sera effectuée comme précédemment (Figure 16). Des
coupes histologiques seront faites pour étudier la morphologie des TA et marquer UCP1 et IF1.
Pour aller plus loin dans la caractérisation du browning, nous souhaitons étudier deux
marqueurs moléculaires des adipocytes beiges: Hoxc9 et Shox2. Ainsi, après sacrifice, environ
100 mg de TA blancs (viscéraux et sous cutanés) et brun seront prélevés et conservés à -80°C
pour réaliser l’extraction des ARN en temps voulu. Les RT-qPCR seront effectuées comme décrit
par l’équipe de Waldén30.
Le plasma récolté pour le suivi de l’inflammation sera également utilisé pour doser
PTHrP. Nous pourrons ainsi suivre son évolution en fonction de l’avancé du cancer par test ELISA.
D’après l’étude menée par Serkan Kir et Al, sur le modèle de souris LLC, la concentration
plasmatique en PTHrP atteint environ 500 pg/mL 21 jours après injection des cellules LLC aux
souris 37. Comme nous ne travaillons pas sur le même modèle, il nous est difficile de comparer
les temps de développement des cancers. Il faudra donc vérifier que la concentration
plasmatique en PTHrP soit supérieure ou égale à 500 pg/mL avant de procéder à son inhibition
en semaine six. Si les concentrations plasmatiques correspondent, les anticorps seront injectés
pendant 14 jours à partir de la semaine 6 (Figure 17). Si besoin est, nous attendrons.
4) Caractérisation des tumeurs
Nos analyses sur ce modèle seront complétées par une observation des tumeurs
développées par les souris ayant reçu les traitements précédemment décrits. Au sacrifice, les
poumons seront pesés, les nodules comptés puis les poumons seront inclus en paraffine pour
des analyses immunohistochimiques avec un marquage au KI67.
C/ In vitro, sur d’autres types de cancers
Pour des raisons éthiques et pratiques, nous n’effectuerons pas cette étape in vivo mais
travaillerons sur des cultures primaires d’adipocytes à partir de deux souris contrôles sacrifiées
en partie I/B. La membrane plasmique de l’adipocyte étant très fragile et vulnérable à la pression
mécanique, l’adipocyte mature éclate au bout de 24 à 48 heures en condition de culture
classique. Par conséquent, la plupart des études sur les adipocytes sont réalisées sur des pré-
adipocytes différenciés in vitro 43. Nous procéderons de la même manière que l’équipe de Serkan
Kir, le WAT sous-cutané (inguinal) et le BAT seront récupérés pour réaliser des cultures primaires
d’adipocytes 37
18
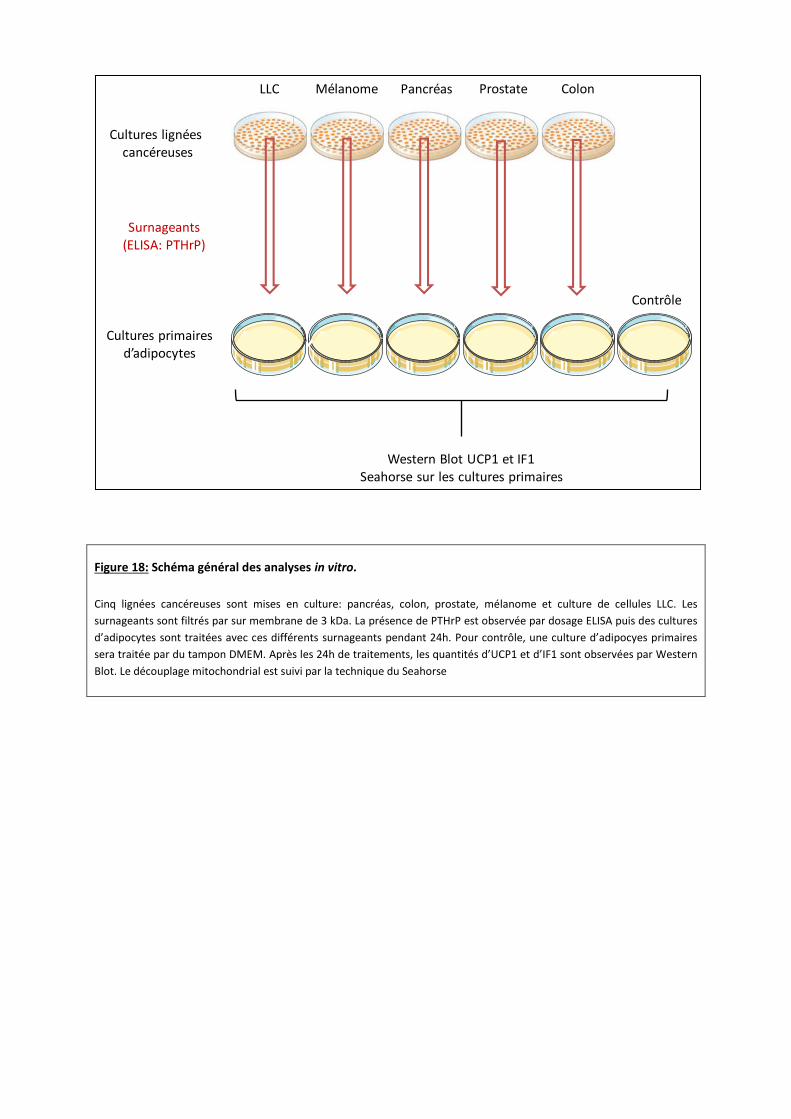
Figure 18: Schéma général des analyses in vitro.
Cinq lignées cancéreuses sont mises en culture: pancréas, colon, prostate, mélanome et culture de cellules LLC. Les
surnageants sont filtrés par sur membrane de 3 kDa. La présence de PTHrP est observée par dosage ELISA puis des cultures
d’adipocytes sont traitées avec ces différents surnageants pendant 24h. Pour contrôle, une culture d’adipocyes primaires
sera traitée par du tampon DMEM. Après les 24h de traitements, les quantités d’UCP1 et d’IF1 sont observées par Western
Blot. Le découplage mitochondrial est suivi par la technique du Seahorse
Cultures lignées cancéreuses
Mélanome Pancréas Prostate Colon
Surnageants(ELISA: PTHrP)
Cultures primairesd’adipocytes
Contrôle
Western Blot UCP1 et IF1Seahorse sur les cultures primaires
LLC

Parallèlement, différentes lignées tumorales seront mises en culture. Comme expliqué en
introduction, les cancers du pancréas, de la prostate, du colon ainsi que les mélanomes évoluent
vers une cachexie 1,36. Pour contrôle positif, nous étudierons également la lignée tumorale LLC 37.
Nous utiliserons la lignée « LTPA » pour l’étude du cancer pancréatique. La lignée « PNEC30 »
pour l’étude du cancer de la prostate. La lignée « B16 » pour le mélanome et enfin, la lignée
« CT26, CL25 » pour l’étude du cancer du colon. Les lignées tumorales seront commandées chez
LGC Standards-ATCC. Les cellules seront maintenues dans du milieu DMEM à 10% SVF, en
présence de pénicilline et streptomycine 37. Au bout de 3 jours, ce milieu sera filtré sur
membrane de 3 kDa comme expliqué dans l’étude de Serkan Kir 37. Les milieux conditionnés
obtenus contiendront donc les molécules solubles et les vésicules sécrétées par les cellules
tumorales, dont PTHrP. Les cultures d’adipocytes seront traitées avec ces différents milieux
conditionnés pendant 24h (Figure 18).
Dans un premier temps nous observerons la présence de PTHrP par dosage ELISA à partir
des surnageants des différentes lignées cellulaires cancéreuses avant dépôt sur les cultures
d’adipocytes primaires. 7
Après les 24h de traitement des adipocytes par les surnageants de cultures cancéreuses,
les quantités relatives d’UCP1 et d’IF1 seront observées par western blot de façon identique que
Serkan Kir et al 30.
Pour finir, nous caractériserons le découplage mitochondrial des adipocytes blancs et
bruns présents dans les cultures primaires par la technique du Seahorse. Cette expérience nous
permettra de mesurer la consommation d’oxygène (OCR) en temps réel. Pour n’observer que les
effets de la β-oxydation, il est nécessaire de bloquer la glycolyse par ajout d’iodoacétate. Nous
procéderons de la même manière que l’étude en cours de publication de L. Nieto
(communication personnelle).
En conclusion, cette première partie de notre projet nous permettra de confirmer que
PTHrP active la transcription d’UCP1 et que cet ARNm est bien traduit en protéine. Nous saurons
également si PTHrP est libéré par une tumeur localisée aux poumons tout en conservant ses
propriétés (modèle KrasG12D). Enfin, nous pourrons déterminer grâce aux différentes lignées
tumorales en culture si PTHrP est cancer spécifique.
Pour finir, la caractérisation du browning après inhibition des effets de PTHrP nous
permettra de confirmer les conséquences d’une réduction du browning sur l’état cachectique
des souris cancéreuses présentant une tumeur localisée au poumon (modèle KrasG12D).
19

Figure 19 : Modèles expérimentaux et chronologie du programme expérimental.
A : Toutes les souris sont invalidées pour le gène UCP1. L’njections de 5x106 cellules cancéreuses LLC (Lung Lewis
Carcinoma) est réalisée en sous cutanée pour dix mâles âgés de 8 semaines au niveau du flanc droit (groupe « KO
UCP1/tumeur LLC »). L’injection contrôle de solution saline (PBS) est administrée de façon identique à dix autres mâles
(groupe « KO UCP1 sans tumeur »).
B : A S0, une injection des cellules LLC ou PBS est effectuée. Le lendemain, huit souris (deux par groupe) sont placées en
cages de calorimétrie indirecte pendant 24h. Toutes les semaines, différents échantillons sanguins sont prélevés à partir de
la veine saphène. Deux temps de sacrifices seront effectués en semaines 3 et 6. D’autres expériences de calorimétrie
indirecte sur les mêmes souris qu’en J1 seront mises en place ces deux temps de sacrifices.
Le poids des souris est contrôlé trois fois par semaines.
A
B
KO UCP1+ PBS
KO UCP1+ LLC
Sacrifice 21 jours
Sacrifice 42 jours
8 Semaines Semaines
LLC /PBS
Sacrifice
0 1 2 3 4 5 6
Prélèvements sanguins
Sacrifice
* Calorimétrieindirecte
* * *

II/ Effets de l’inhibition d’UCP1 sur le browning et la cachexie
Pour cette seconde partie de notre projet, nous souhaitons déterminer si UCP1 joue un
rôle majeur dans le browning et plus particulièrement, si une inhibition du browning améliore
l’état cachectique des souris.
Pour ce faire, nous étudierons des souris KO UCP1. D’après la littérature, ces dernières
sont largement étudiées dans le cadre de l’obésité44 mais aucun de ces modèles n’a été étudié
pour les cancers associés à la cachexie. Si nos résultats démontrent qu’un KO UCP1 entraine
l’inhibition complète du browning, ces souris pourront être utilisées comme modèle de choix
pour étudier les effets de l’altération du browning dans le cadre des CAC.
1) Les animaux
Pour des raisons budgétaires, deux couples de souris KO UCP1 seront commandés chez
Jackson Laboratory. L’élevage sera effectué en interne afin d’obtenir vingt mâles. Puisque nous
souhaitons observer l’effet de l’inhibition d’UCP1 sur la cachexie cancéreuse, l’induction d’un
cancer aux souris KO UCP1 est nécessaire. Les croiser avec une lignée KrasG12D représenterait un
élevage beaucoup trop important dans le cadre de notre projet. Nous avons donc décidé
d’injecter 5x106 cellules LLC dans le flanc droit de dix souris mâles âgées de 8 semaines en sous
cutané, comme décrit en partie I/A/1.
Dix autres mâles recevront des injections de PBS et constitueront notre groupe « KO UCP1/sans
tumeur LLC ». Pour finir, un premier temps de sacrifice sera effectué trois semaines après
injection des cellules LLC (5 souris KO UCP1/sans tumeur LLC et 5 souris KO UCP1/ tumeur LLC) ;
un second temps de sacrifice six semaines après injection (5 souris KO UCP1/sans tumeur LLC et
5 souris KO UCP1/ tumeur LLC) (Figure 19).
En effet, lors de l’étude dirigée par Serkan Kir les souris C57BL/6 ayant reçu les cellules LLC
étaient sacrifiées 16 ou 21 jours après injection. Lors des pesées, les muscles gastrocnémiens ne
présentaient pas de différences significatives de poids avant et après induction de la tumeur 37.
Nous pensons que le temps de sacrifice choisi était un peu tôt pour observer une cachexie
prononcée. C’est pour cela que nous mettons en place deux temps de sacrifice pour cette
seconde partie du projet.
20


2) Suivi de la cachexie
L’évolution de la cachexie sera suivie au cours du temps à raison de trois pesées par
semaine. Au sacrifice, nous procéderons à la pesée des tissus adipeux blancs viscéraux et sous
cutanés (côtés gauche et droit, péri tumoral) et du tissu adipeux brun inter-scapulaire. Nous
pèserons également les muscles gastrocnémiens (mollets gauche et droit).
L’état inflammatoire sera caractérisé à partir de prélèvements sanguins effectués toutes
les semaines. Le sang sera centrifugé et le plasma conservé à -80°C pour procéder aux dosages
de TNFα et IL-6 par test ELISA en temps voulu.
3) Analyse du browning
Dans un premier temps nous devons nous assurer que les souris KO UCP1 sont bien
déficientes pour le browning. En effet, Jackson Laboratory (le seul laboratoire à commercialiser
ces lignées) décrit cette lignée comme étant sensible au froid, démontrant un métabolisme
altéré et étant résistante à l’obésité induite par l’alimentation.
Les TA blancs viscéraux (rétro péritonéaux) et sous cutanés (inguinaux) ainsi que le TA
brun seront récupérés après sacrifice pour procéder à des analyses histologiques.
Comme en partie I/A/3, trois séries de coupes histologiques seront effectuées. Nous
déterminerons la morphologie des TA et nous étudierons la présente d’UCP1 et d’IF1 (Figure 16).
Les résultats obtenus seront comparés avec les résultats obtenus en partie I/A sur les souris
cancéreuses LLC n’étant pas déficientes pour UCP1.
D’autre part, nous avons pour objectif de déterminer si UCP1 est la seule protéine
responsable du browning. En effet, comme expliqué dans notre introduction, il existe deux
autres protéines découplantes, UCP2 et UCP3. Ainsi, si nous observons un maintien du browning
même après le KO d’UCP1, nous effectuerons des Western Blot d’UCP2 et d’UCP3.
Pour finir, le métabolisme général des souris sera suivi par des expériences de
calorimétrie indirecte. Cette étude nous permettra de déterminer la dépense énergétique ainsi
que la production de chaleur des souris KO UCP1 (plus ou moins cancéreuses). La dépense
énergétique et la production de chaleur seront calculées grâce aux mesures des échanges gazeux
(consommation d’O2 et production de CO2). Les résultats obtenus seront comparés avec les
résultats de la partie I/A. Les expériences seront réalisées comme indiqué dans l’étude de Julie
Marcotorchinoa et. Al 41.
21


4) Suivi de la tumeur
La tumeur sera suivie au cours du temps par palpation et sera pesée après sacrifice des
souris. L’observation de la tumeur nous permettra de déterminer si une inhibition du browning
entraine une diminution de la masse tumorale. Nous pourrons ainsi, à partir du modèle KO
UCP1, confirmer ou infirmer les résultats de l’équipe de Serkan Kir où la progression tumorale
était inchangée 37.
Conclusion et perspectives
A la fin de notre projet, nous devrions pouvoir mettre en évidence l’effet de PTHrP sur la
protéine UCP1. La caractérisation du browning devrait nous permettre d’affirmer que PTHrP
stimule bien le browning en activant UCP1. Il nous paraissait également indispensable de
confirmer l’action de PTHrP dans le cadre d’une tumeur localisée aux poumons, tant sur le
browning que sur l’état cachectique des souris. Enfin, le projet permettra de déterminer si PTHrP
est une hormone cancer spécifique.
De plus, la caractérisation de souris KO UCP1 devrait permettre de déterminer si cette
protéine est la seule responsable du browning. Et de ce fait, si ces souris KO UCP1 sont bien
déficientes pour le browning.
Longtemps observé dans le cadre de l’obésité, le browning parait être une voie d’étude
prometteuse pour les cancers associés à la cachexie. De nombreuses études cliniques portent
sur PTHrP dans le cadre de l’hypercalcémie et de l’ostéoporose (clinicaltrials.gov). Cependant,
l’étude de son rôle dans l’induction du browning et par conséquent l’aggravation de la cachexie
est très récente 45.
Suite à l’inhibition (ou du moins la diminution) du browning, l’état général des souris
devrait être amélioré. Il parait indispensable de tester différentes thérapies anti-cancéreuses sur
les souris KO UCP1 afin de voir si celles-ci répondent mieux aux traitements. Ces résultats
permettraient alors de confirmer la pertinence des recherches sur le browning et la cachexie.
Enfin, l’enjeu public de cette étude est d’inhiber le browning de patients atteint de
cancers associés à la cachexie pour améliorer leurs réponses aux thérapies.
22

Bibliographie
1. Argilés, J. M., Busquets, S., Stemmler, B. & López-Soriano, F. J. Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer (2014). doi:10.1038/nrc3829
2. Argilés, J. M. et al. Targets in clinical oncology: the metabolic environment of the patient. Front. Biosci. 12, 3024–51 (2007).
3. Petruzzelli, M. et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 20, 433–47 (2014).
4. Ambrus, J. L., Ambrus, C. M., Mink, I. B. & Pickren, J. W. Causes of death in cancer patients. J. Med. 6, 61–64 (1975).
5. Von Haehling, S. & Anker, S. D. Cachexia as a major underestimated and unmet medical need: facts and numbers. J. Cachexia. Sarcopenia Muscle 1, 1–5 (2010).
6. Argilés, J. M., Busquets, S. & López-Soriano, F. J. Anti-inflammatory therapies in cancer cachexia. Eur. J. Pharmacol. 668 Suppl, S81–6 (2011).
7. Laine, A., Iyengar, P. & Pandita, T. K. The role of inflammatory pathways in cancer-associated cachexia and radiation resistance. Mol. Cancer Res. 11, 967–72 (2013).
8. Tisdale, M. J. Biology of Cachexia. JNCI J. Natl. Cancer Inst. 89, 1763–1773 (1997).
9. Uomo, G., Gallucci, F. & Rabitti, P. G. Anorexia-cachexia syndrome in pancreatic cancer: recent development in research and management. JOP 7, 157–62 (2006).
10. Keipert, S. & Jastroch, M. Brite/beige fat and UCP1 - is it thermogenesis? Biochim. Biophys. Acta 1837, 1075–82 (2014).
11. Harms, M. & Seale, P. Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19, 1252–63 (2013).
12. Bartelt, A. & Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 10, 24–36 (2014).
13. Carobbio, S., Rosen, B. & Vidal-Puig, A. Adipogenesis: new insights into brown adipose tissue differentiation. J. Mol. Endocrinol. 51, T75–85 (2013).
14. Park, A., Kim, W. K. & Bae, K.-H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 6, 33–42 (2014).
15. Beranger, G. E. et al. In vitro brown and “brite”/“beige” adipogenesis: human cellular models and molecular aspects. Biochim. Biophys. Acta 1831, 905–14 (2013).
16. Doenst, T., Nguyen, T. D. & Abel, E. D. Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 113, 709–24 (2013).

17. Tsoli, M. & Robertson, G. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 24, 174–83 (2013).
18. Divakaruni, A. S. & Brand, M. D. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda). 26, 192–205 (2011).
19. Lee, Y.-H., Jung, Y.-S. & Choi, D. Recent advance in brown adipose physiology and its therapeutic potential. Exp. Mol. Med. 46, e78 (2014).
20. Giralt, M. & Villarroya, F. White, brown, beige/brite: different adipose cells for different functions? Endocrinology 154, 2992–3000 (2013).
21. Bonet, M. L., Oliver, P. & Palou, A. Pharmacological and nutritional agents promoting browning of white adipose tissue. Biochim. Biophys. Acta 1831, 969–85 (2013).
22. Yao, X., Shan, S., Zhang, Y. & Ying, H. Recent progress in the study of brown adipose tissue. Cell Biosci. 1, 35 (2011).
23. Hoang, T., Smith, M. D. & Jelokhani-Niaraki, M. Expression, folding, and proton transport activity of human uncoupling protein-1 (UCP1) in lipid membranes: evidence for associated functional forms. J. Biol. Chem. 288, 36244–58 (2013).
24. Lo, K. A. & Sun, L. Turning WAT into BAT: a review on regulators controlling the browning of white adipocytes. Biosci. Rep. 33, (2013).
25. Frühbeck, G., Sesma, P. & Burrell, M. A. PRDM16: the interconvertible adipo-myocyte switch. Trends Cell Biol. 19, 141–6 (2009).
26. Seale, P. et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–7 (2008).
27. Janani, C. & Ranjitha Kumari, B. D. PPAR gamma gene - A review. Diabetes Metab. Syndr. (2014). doi:10.1016/j.dsx.2014.09.015
28. Yamada, M. et al. Genomic organization and promoter function of the mouse uncoupling protein 2 (UCP2) gene. FEBS Lett. 432, 65–9 (1998).
29. Brand, M. D. & Esteves, T. C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2, 85–93 (2005).
30. Waldén, T. B., Hansen, I. R., Timmons, J. A., Cannon, B. & Nedergaard, J. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am. J. Physiol. Endocrinol. Metab. 302, E19–31 (2012).
31. Petrovic, N. et al. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocyt. J. Biol. Chem. 285, 7153–64 (2010).

32. Servera, M., López, N., Serra, F. & Palou, A. Expression of “brown-in-white” adipocyte biomarkers shows gender differences and the influence of early dietary exposure. Genes Nutr. 9, 372 (2014).
33. Yamamoto, Y. et al. Adipose depots possess unique developmental gene signatures. Obesity (Silver Spring). 18, 872–8 (2010).
34. Pioszak, A. A., Parker, N. R., Gardella, T. J. & Xu, H. E. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J. Biol. Chem. 284, 28382–91 (2009).
35. Mannstadt, M., Jüppner, H. & Gardella, T. J. Receptors for PTH and PTHrP: their biological importance and functional properties. Am. J. Physiol. 277, F665–75 (1999).
36. Das, S. K. et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–8 (2011).
37. Kir, S. et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513, 100–4 (2014).
38. Cohen, P. et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–16 (2014).
39. Ozaki, K., Sano, T., Tsuji, N., Matsuura, T. & Narama, I. Carnitine is necessary to maintain the phenotype and function of brown adipose tissue. Lab. Invest. 91, 704–10 (2011).
40. Sánchez-Aragó, M. et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis 2, e46 (2013).
41. Marcotorchino, J. et al. Vitamin D protects against diet-induced obesity by enhancing fatty acid oxidation. J. Nutr. Biochem. 25, 1077–83 (2014).
42. Jackson, E. L. et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–8 (2001).
43. Mercier, M. P2142 AdipoPhYt, leader en culture 3D d’adipocytes matures humains. Diabetes Metab. 39, A101 (2013).
44. Inokuma, K. et al. Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. Am. J. Physiol. Endocrinol. Metab. 290, E1014–21 (2006).
45. Holmes, D. Metabolism: WAT browning--key feature of cancer-associated cachexia. Nat. Rev. Endocrinol. 10, 578 (2014).
46. Abdelsayed, R. A., Vartanian, R. K., Smith, K. K. & Ibrahim, N. A. Parathyroid hormone-related protein (PTHrP) expression in ameloblastoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 97, 208–19 (2004).