Embed Size (px)
Citation preview

0
République Algérienne Démocratique et Populaire Ministère de l’Enseignement Supérieur et de la Recherche Scientifique
Faculté des Sciences Exactes et Appliquées Département de Chimie
THESE Présentée pour obtenir le grade de
Docteur en Sciences de l’université d’Oran 1 Ahmed Benbella
Discipline : Chimie Spécialité: Chimie Physique
Option: Chimie Informatique
Présentée par
Nedjoua DRICI
Etude théorique des molécules biologiques par application des méthodes hybrides QM/MM et
Dynamique moléculaire
Soutenue le 01 juin 2017 devant le jury
Président: Mr. A. Bouyacoub Professeur Université d’Oran 1 AB
Encadreur: Mr. A. Krallafa Professeur Université d'Oran 1 AB
Examinateur: Mr. F. Hamza Reguig MCA Université d'Oran 1 AB
Examinateur: Mr. A. Rahmouni Professeur Université de Saida
Examinateur: Mr. Y. Bouhedda Professeur Université de Mascara
Examinateur: Mme. M. Sekkal Professeur Université de Sidi bel Abbes
Invité: Mr. B. Bounaceur Professeur Université d'Oran 1 AB
2016/2017

1
Remerciements
La partie dynamique moléculaire a été réalisée au sein de l’équipe Chimie Informatique,
Chimie Quantique et Simulation Numérique du Laboratoire de Chimie Physique
Macromoléculaire (LCPM) à l’université Oran1 Ahmed Benbella Algérie.
Un grand merci à mon encadreur, le Professeur Abdelghani Mohamed KRALLAFA pour sa
disponibilité, son enthousiasme pour la recherche ainsi que pour l’excellente formation en
chimie théorique que j’ai eue la chance d’avoir sous sa direction durant ma thèse et durant
mon parcours dans la recherche scientifique. Je le remercie d’avoir accepté mon encadrement
et de m’avoir laissé autant de liberté dans la conduite de mon projet de thèse.
Je remercie vivement Professeur Boumedienne BOUNACEUR directeur du Laboratoire de
Chimie Physique Macromoléculaire (LCPM) d’avoir accepté d’être parmi les membres du
jury de ma thèse, je le remercie aussi pour ses précieux conseils et son orientation
administrative, pour son assistance de près ou à distance et surtout pour son sens de
responsabilité envers les thésards.
Je remercier le Professeur Abdelatif BOUYAKOUB d’avoir accepté de présider le jury de ma
thèse. Je le remercie pour le temps qu’il a consacré à la lecture de ce manuscrit et suis très
honoré d’avoir accepté de faire partie du jury.
Mes remerciements s’adressent également aux Professeur Majda SEKKAL, Professeur Ali
RAHMOUNI, Professeur Youcef BOUHEDDA et au Docteur Farouk HAMZA-REGUIG, qui
m’ont fait l’honneur d’examiner et émettre un rapport sur ma thèse, malgré leurs nombreuses
occupations. Je suis très honorée de les compter parmi les membres de ce jury.
Je ne manquerai pas de remercier l’ensemble des membres de laboratoire LCPM-Oran pour
leurs gentillesses et leur amitié. Je ne saurais oublier d’exprimer toute ma reconnaissance
envers ma famille pour leur soutien constant et les encouragements qu’ils m’ont apporté.
Je remercie vivement l’université de Mostaganem Abdelhamid Ibn Badis de m’avoir attribué
la bourse PROFAS pour finaliser la partie QM/MM de ma thèse.
Je remercie également et vivement le consulat d’Algérie à Metz pour leur disponibilité et leur
sens de responsabilité envers les étudiants Algériens en France– un merci tout particulier à
Mme KADRI.
Les calculs QM/MM basé sur l’approche LSCF/MM ont été réalisés au sein du laboratoire de
Chimie et Biochimie Théoriques, SRSMC Structure et Réactivité des Systèmes Moléculaire
Complexes UMR 7565 de Nancy.

2

3
Résumé
L’étude des édifices moléculaires faiblements liés est d’un intérêt fondamental dans de
nombreux domaine de la chimie et de la physique, en particulier tout ce qui relève de la
biologie, de la chimie atmosphérique et de l’astrophysique. Plusieurs études expérimentales
ont été orientées vers l’étude des complexes faiblement liés afin d’apporter des réponses sur la
structure des diffèrent conformer à l’état fondamental en phase gaz et sur le rôle des liaisons
hydrogène intra et intermoléculaire.
La disponibilité des données expérimentales précises a ravivé les études théoriques dont
l’intérêt est de prédire les structures et d’analyser la nature des forces qui tiennent les
complexes faiblement liées. L’étude des interactions non-covalente continue d’être un
domaine d’activité important, ceci est prouvé par le nombre considérable de publications
récentes traitant ce domaine. Les méthodes hybrides QM-MM, développées depuis une
trentaine d’années, décrivent le centre réactionnel ou actif par des méthodes de la mécanique
quantique (QM) et son environnement par des méthodes de mécanique moléculaire (MM).
Ces méthodes se sont révélées des méthodes de choix pour l’étude de systèmes chimiques et
biochimiques contenant plus d’une centaine d’atomes. Les méthodes hybride existantes,
diffèrent dans leurs procédés de séparer les deux sous systèmes et permet de rendre compte
des interactions inter et intramoléculaires, notamment, les interactions électrostatiques à
longue portée entre les quelques atomes qui réagissent et leur environnement. Dans notre
étude nous avons choisis la méthode développée au laboratoire de Nancy :la méthode du
champ auto-cohérant locale LSCF (Local Self Consistent Field).
Nous réalisons dans cette thèse l’étude des interactions intermoléculaire d’un point de vue
orbitale localisé ou gelé. Dans un premier temps nous avons utilisé la méthode hybride basée
sur le formalisme LSCF/MM, sur un fragment peptidique contenant un ion métallique (doigt
de zinc) afin de voir de près, le rôle de ce cation dans la stabilisation du fragment protéinique
pris à l’état natif et muté. Ce rôle est évalué par le calcul de l’énergie d’interaction
supermoléculaire entre le cation métallique et ces quatre ligands coordinateurs. Nous avons
complété cette partie par une étude par dynamique moléculaire classique pour évaluer
l’importance des interactions électrostatiques dans les biomolécules activé par un ion
métallique en milieux solvatée, et l’effet de la taille du rayon de van der Waals d’un ion
métallique sur les propriétés structurale de son site de coordination ainsi que sur la
stabilisation de la structure secondaire du fragment peptidique. Cette étude a fait l’objet d’une
publication.

4
Dans un deuxième temps, nous avons appliqué la méthode de projection asymptotique qui
représente l’une des variantes du formalisme LSCF ab initio sur un ensemble de complexes
moléculaires présentés sous forme de dimères à base d’acide aminé, liés par des liaisons
hydrogène. Nous avons choisis ce genre de système moléculaire afin de mimer et d’analyser
les interactions électrostatiques due aux liaisons hydrogène engagé dans les biomolécules.
Nous nous somme focalisé sur l’étude de l’effet de polarisation mutuelle des densités
électronique au sein de chaque dimère, sans qu’il y’est le phénomène de transfert de charge de
l’un des monomère vers l’autre. Ceci revient à l’étude de l’interaction électrostatique entre
une charge ponctuelle représenté par une densité de charge gelé avec une autre densité de
charge laissée à l’état relaxé.
Pour cela, nous avons effectué pour chaque dimère, une série de calculs dans lesquelles nous
avons gelé d’une manière alternative la densité électronique de l’un des monomères, pour
qu’il soit source d’un champ externe ressenti par l’autre monomère laissé à l’état relaxé. Afin
de valider nos résultats trouvés par la méthode de projection asymptotique, nous avons pris
comme référence les travaux de K. Senthilkumar et al (J. R. Soc. Interface (2008) 5, S207–
S216) qui ont réalisé des calculs QM/MM standard et des calculs purement quantique sur le
même ensemble des dimères.
La dernière partie a été consacrée à la modélisation par dynamique moléculaire d’un ion
métallique dans son environnement protéinique. Nous avons étudié l’influence de la variation
des paramètres de van der Walls de l’ion métallique sur la dynamique des ligands du site actif.
Cette étude nous a permis de vérifier la qualité des paramètres de van der Waals de l’ion de
Zinc originalement implémentés dans le champ de force GROMOS96. La réalisation sur une
grande échelle de temps, d’une simulation de dynamique moléculaire avec le champ de force
GROMOS modifié (implication des effets de polarisation) pour explorer le chemin de
dépliement et de repliement des métalloprotéines est un objectif à atteindre.
Dans cette thèse nous avons pu mettre en évidence l’importance de la prise en compte des
effets de polarisation l’ors du traitement théorique des interactions non-liées dans de petits
systèmes biomimétiques et des molécules biologiques.
Mot clef : QM/MM, LSCF, DFT, interaction intermoléculaire, orbitale gelé, polarisation,
Champ de force GROMOS, Dynamique moléculaire, rayon de van der Waals, ion de zinc,
molécules biologiques.

5
Table des matières
Remerciements page 1
Résumé page 3
Table des matières page 5
Introduction page 8
Chapitre 1
Chimie théorique et modélisation moléculaire page 11
1.1 La mécanique quantique, Equations et principes fondateurs Page 12
1.1.1 Equation de Schrödinger page 12
1.1.2 Fonction d’onde multiélectronique et l’approximation orbitalaire page 16
1.2 Méthodes de détermination de la structure électronique page 17
1.2.1 La méthode Hartree-Fock page 17
1.2.2 Les équations de Roothaan page 19
1.3 Les méthodes post Hartree-Fock page 24
1.3.1 Interaction de configuration page 24
1.3.2 La théorie perturbationnelle : Møller-Plesset (MP) page 25
1.3.3 Les méthodes Coupled-Cluster (CC) page 26
1.4 La théorie de la fonctionnelle de la densité (DFT) page 27
1.4.1 Les équations de Kohn-Sham (KS) page 27
1.4.2 Les fonctionnelles d’échange-corrélation page 29
1.4.3 Types de fonctionnelles d’échange-corrélation page 30
1.4.3 a) L’approximation de la densité locale (LDA) page 30
1.4.3 b) L’approximation du gradient généralisé (GGA) page 31
1.4.3 c) La connexion adiabatique et les fonctionnels hybrides page 32
1.5 Principe de la Mécanique Moléculaire page 34
1.5.1 Formalisme de l´énergie potentielle page 36
1.5.2 Les interactions de valence page 37
1.5.3 Interaction des atomes non-liés page 39
1.5.3 1 Les différentes expressions des interactions de van der Waals page 39
1.5.3.2 Le potentiel électrostatique page 40
1.6 La dynamique moléculaire page 41
1.6.1 Dynamique moléculaire : Techniques numériques page 41
1.6.1.1 Intégration du temps page 41
1.6.1.2 Durée du pas de temps page 42
1.6.1.3 Conditions initiales page 43
1.6.1.4 Conditions périodiques aux limites page 43
1.6.5 Traitement des interactions électrostatiques en dynamique moléculaire page 45
Chapitre 2
Les Méthodes Hybrides QM-MM page 47
2 1 Les méthodes hybrides QM-MM page 48
2.1.1 Introduction page 48
2.2 Partitionnement QM-MM page 48
2.3 L’expression de l’énergie potentielle QM-MM page 50
2.3.1 Le schéma de couplage QM-MM soustractif page 50
2.3.2 Le schéma de couplage QM-MM Additif page 52
2.4 Effet électrostatique de l’environnement page 53
2.4.1 Le modèle Mechanical Embedding (ME) page 53
2.4.2 Le modèle Electrostatic Embedding (EE) page 54
2.4.3 Le modèle Polarized Embedding (PE) page 55
2.5 Interactions électrostatiques longue portée QM-MM page 55
2.6 Les interactions de van der Waals en QM/MM page 56
2.7 Les Interactions QM-MM liées page 57
2.8 Les coupures en QM/MM page 57

6
2.8.1 Les approches Link-Atom page 59
2.8.2 Les approches de type Orbitales localisées page 61
Chapitre 3
Méthodologie et développements
de la méthode du Champ Auto Cohérent Local (LSCF) page 64
3.1 La Méthode du Champ Auto-Coherent Local
: Local Self-Consistent Field (LSCF) : page 65
3.1.1 Principe page 65
3.2 Le Formalisme LSCF ab initio page 65
3.2.1 Orthogonalisation des orbitales localisé page 67
3.2.2 Orthogonalisation des orbitales atomiques page 68
3.2.3 Modification des équations de Roothaan page 70
3.4 La méthode LSCF/MM page 72
3.4.1 Les orbitales de liaison strictement localisées :
Strictly Localized Bond Orbital (SLBO) page 75
3.4.1.1 Les Critères externe : page 75
3.4.1.2 Les critères internes page 76
Chapitre 4
La méthode du Champ Auto cohérent locale LSCF :
Application à l’étude des interactions intermoléculaires page 79
4.1 L’interaction dans les édifices moléculaires page 80
4.2 Classification et caractères des interactions intermoléculaires page 80
4.3 Investigation expérimentale et théorique des interactions intermoléculaire. Page 81
4.3.1 Caractérisation expérimentale page 81
4.3.2 Caractérisation théorique page 82
4.3.2.1 La méthode de la supermolécule (supermolecular method) page 82
4.3.2.2 Les méthodes basées sur la théorie de perturbation page 83
4.4 Méthodes d’analyse des interactions inter moléculaire page 83
4.4.1 L’énergie électrostatique : 𝑬𝒆𝒍𝒆𝒄 page 84
4.4.2 L’énergie d’induction : 𝑬𝒊𝒏𝒅 page 84
4.4.3 L’énergie de dispersion : 𝑬𝒅𝒊𝒔𝒑 page 84
4.4.4 L’énergie de répulsion 𝑬𝒆𝒙−𝒓𝒆𝒑 page 85
4.4.5 L’énergie de transfert de charge 𝑬𝒄𝒕 page 85
4.6 Lecture dans les différentes méthodes de l’analyse de la décomposition de l’énergie
d’interaction EDA (energy decomposition analysis) page 85
4.6.1 Les méthodes variationnelles page 86
4.6.2 Méthodes perturbatives page 86
4.6.3 Autres méthodes page 87
Partie A : Rôle stabilisateur des cations métalliques dans systèmes biologique page 89
A.1 Introduction page 90
A.1.1 Le Zinc : Un élément essentiel page 91
A.1.2 Corrélation structure propriété les sites à zinc page 91
A.2 Energie d’interaction dans un site à zinc page 92
A.2.1 Présentation du système moléculaire page 92
A.2.1.1 Structure et investigation expérimentale et théorique des sites à zinc page 92
A.2.2 Description d’un ion métallique dans un champ de force page 95
A.2.2.1) le modèle liée page 95
A.2.2.2) le modèle non liée page 95
A.2.2.3) le modèle des charges cationique fictives page 95
A.3 Etude de de l’énergie d’interaction et de l’effet de mutation sur le site à zinc structurale:
Application de l’approche LSCF/MM page 96

7
A.3.1 Méthodologie page 97
A.3.2 Conclusion page 99
Partie B : Orbitales moléculaires localisées et modélisation des interactions intermoléculaires
dans les systèmes biomimétiques faiblement liées page 101
B.1 La liaison hydrogène : liaison de vie page 102
B.1.1 Propriété des liaisons hydrogène page 102
B.2 Etude théorique des complexes à liaisons hydrogènes trouvées dans des systèmes
moléculaires d’intérêt biologique par l’application de la méthode LSCF- projection
asymptotique page 103
B.2.1 La projection asymptotique page 104
B.2.2 Principe et formalisme de la projection asymptotique page 104
B.2.3 Quelques applications de l’approche de la projection asymptotique page 106
B.3 Etude de la polarisation dans les complexes à hydrogène :
application de la méthode LSCF-projection asymptotique page 107
B.3.1 La théorie de l’incorporation de densité gelée :
Frozen density embedding theory page 107
B.3.2 Système moléculaire étudié et détails calculatoires page 109
B.3.3 Résultats et discussion page 113
B.3.4 Conclusion page 121
Partie C : Investigation par dynamique moléculaire page 122
C.1 Introduction page 123
C.1.1 Paramètres de van der Waals du zinc page 123
C.1.2 Protocole de simulation page 124
C.2 Résultat et discutions page 126
C.2.1 Effet du rayon de van der Waals de zinc sur :
a) les paramètres structuraux du site de coordination page 126
b) la structure secondaire du fragment peptidique page 141
C.3 Lecture dans les champs de forces polarisables page 147
C.4. Conclusion page 149
4.7 Conclusion générale et perspectives page 150
Références bibliographiques page 153

8
Introduction
L’étude des molécules biologiques est d’un intérêt primordial pour notre compréhension de la
vie, et depuis plusieurs décennies, de très nombreuses recherches ont été menées dans ce sens,
particulièrement dans les domaines de la biochimie, de la biologie moléculaire et de la
pharmacologie. Les réactions chimiques se déroulant dans des milieux étendus comme des
macromolécules, des solides ou sur des surfaces sont plus difficiles à traiter dans un
formalisme QM/MM. En effet, la séparation entre les deux sous-systèmes nécessite de couper
une ou plusieurs liaisons chimiques. Le fragment quantique résultant de ces coupures est alors
un radical dont les propriétés sont différentes de celles du fragment dans son environnement.
Ce problème de liaisons frontières pendantes a donné lieu à de nombreux développements
méthodologiques qui ont tenté, et tentent toujours, de décrire au mieux cette frontière entre les
deux descriptions quantique et classique.
Le Champ auto-cohérent local (LSCF) est un nouveau formalisme QM/MM qui a été
développé au laboratoire de chimie théorique de Nancy. Il permet à l’aide d’une description
par orbitales de liaisons strictement localisé (SLBO) de présenter rigoureusement les liaisons
partagées entre un fragment quantique qui subit des modifications électronique remarquable
au cours d’une réaction, et son environnement décrit par mécanique moléculaire.
Le but principale de notre travail va donc être, à partir de la possibilité de calculer l’énergie
d’un fragment quantique dans l’approche du champ auto-cohérent local d’étudier les
interactions non covalente dans les fragments protéinique contenant un ion métallique ainsi
que d’avoir un aperçu sur les interactions électrostatique entre les diffèrent acides aminé dans
les biomolécules en appliquant la méthode de la projection asymptotique sur ensemble de
dimères lié par des liaisons hydrogène
Pour cela nous avons fixé comme objectifs
-Evaluer l’énergie d’interaction supermoléculaire d’une métalloprotéine, par l’application de
l’approche du champ auto-cohérent local LSCF/MM sur un doigt de zinc.
-Avoir un aperçu sur l’influence de la position de mutation sur le rôle stabilisateur de l’ion
métallique au sein des structures mutées, dans lesquelles un seul résidu cystéinates est
remplacé par un fragment Alanine.
-Valider la méthode de projection asymptotique qui est implantée dans le formalisme LSCF et
qui pourrait permettre de modéliser les interactions électrostatiques de polarisation en absence
totale du phénomène de transfert de charge. Ceci, par le traitement de deux entités
moléculaires qui interagissent d’une manière non covalentes, dont l’une d’elle, reste à l’état
relaxé tandis que l’autre est tenu à l’état gelé. Les résultats sont comparé aux valeurs trouvée

9
dans une étude antérieur, qui utilise une méthode purement quantique et une méthode mixte,
basé sur le champ de force hybride classique/quantique.
La première partie concerne les fondements théoriques des méthodes de modélisation
moléculaire et les outils méthodologiques utilisés lors de la détermination de l’état
fondamental d’un système moléculaire. Cette section est inspirée de plusieurs ouvrages de
chimie quantique de référence. Nous donnerons ici un aperçu des techniques théoriques
indépendantes du temps. Nous insistons plus particulièrement sur l’approche Hartree-Fock et
la résolution des équations de Roothaan et principales méthodes de calcul de l’énergie
électronique d’une molécule. Nous décrivons par la suite les principes généraux de la théorie
de la fonctionnelle de la densité. Enfin, nous attarderons plus amplement sur les méthodes
permettant l’étude des systèmes dont la taille rend prohibitive l’utilisation de calculs
complètement quantiques. Pour cela, outre un rappel sur les méthodes de mécanique
moléculaire, nous parlerons des méthodes hybrides, alliant mécaniques classique et quantique
(QM/MM) avec une description plus particulière de l’approche LSCF/MM et LSCF-ab initio
sur laquelle nos travaux ont été portées.
Nous consacrons la deuxième partie à la représentation de différentes forces intermoléculaires
régnante dans les édifices macromoléculaire, à la représentation des différents acides aminés
et aussi le système macromoléculaire qu’on a choisis (les métalloprotéines) et le cas
particulier du site structurale choisi.
La dernière partie est dédiée aux résultats et discussion, à la conclusion et aux perspectives et
retombés de notre travail.

10

11
Chapitre 1
Chimie théorique et modélisation moléculaire

12
1.1. La mécanique quantique, Equations et principes fondateurs
1.1.1. Equation de Schrödinger
La détermination de l’état fondamental d’un système est un objectif important pour la chimie
théorique. En effet, beaucoup de propriétés physiques observables peuvent être déduites de la
fonction d’onde de l’état fondamental. De ce fait, la qualité du modèle utilisé lors de calculs
de chimie quantique devient primordiale. Cependant, certaines approximations doivent être
faites afin de pouvoir obtenir des résultats convenables avec les ressources informatiques
actuelles. En chimie quantique, il existe trois postulats de base. Le premier est lié au principe
d’incertitude de Heisenberg qui stipule qu’il est impossible de déterminer simultanément la
position et la vitesse d’une particule. La notion de trajectoire, chère à la dynamique
moléculaire, n’a donc plus de signification et est remplacée par la notion de fonction
d’onde 𝛹(𝑟, 𝑡). La fonction d’onde (aussi appelée vecteur d'état) n’a pas de signification
physique ; par contre son carre représente la probabilité de densité de particules. Selon le
second postulat, la fonction d’onde est définie au temps (𝑡) par l’équation de Schrödinger
(Schrödinger 1926) dépendante du temps :
𝑖ħ𝜕𝛹(𝑟, 𝑡)
𝜕𝑡= ��𝛹(𝑟, 𝑡) (1.1)
Dans cette équation H est l’operateur Hamiltonien polyélectronique. Cette équation est
employée pour le calcul de propriétés moléculaires et est primordiale pour résoudre des
problèmes dépendants du temps. Qu’il s’agisse de la résolution de problèmes sur des systèmes
statiques, comme le calcul de spectres d’absorption ou sur des systèmes de dynamique ab
initio tels la dynamique d’Ehrenfest (Tully 1990), de Born-Oppenheímer (Uggerud and
Helgaker 1992) ou de Car-Parrinello (Car and Parrinello 1985; Laio et al. 2002). Les
méthodes de chimie quantique visent en premier lieu à la détermination de la fonction d’onde
du système polyélectronique que l’on veut étudier. Cela nécessite de résoudre l’équation de
Schrödinger correspondante, dans le cadre des états stationnaires ou indépendante du temps
décrite par l’équation :

13
��(𝑟)𝛹(𝑅, 𝑟) = 𝐸𝛹(𝑅, 𝑟) (1.2)
Où Ψ et E sont respectivement les vecteurs et les valeurs propres de l’opérateur Hamiltonien
indépendant du temps �� du système et (R, r) représentent respectivement l’ensemble des
vecteurs positions nucléaires et électroniques. �� Exprime toutes les interactions entre
particules composant le système et il prend la forme générale suivante :
�� = ��𝑒 + ��𝑁 + ��𝑒𝑒 + ��𝑁𝑁 + 𝑉𝑁𝑒 (1.3)
Où ��𝑒 et ��𝑁 sont les opérateurs associés respectivement à l’énergie cinétique des électrons et
des noyaux. ��𝑁𝑁 ,��𝑒𝑒 et ��𝑁𝑒 représentent les potentiels coulombiens : les deux premiers sont
des termes répulsifs tandis que le dernier renvoie à l’attraction noyaux-électrons. En unités
atomiques et pour un système à (𝑁) électrons et (𝑀) noyaux, l’Hamiltonien s’écrit :
�� = −∑∇𝑖2
2
𝑁
𝑖
−∑∇𝐴2
2𝑀𝐴
𝑀
𝐴
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
+∑∑𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐴<𝐵
𝑀
𝐴
−∑∑𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
𝑁
𝑖
(1.4)
Les indices (𝐴) et (𝐵) sont associés aux noyaux de masse (𝑀𝐴), et (𝑀𝐵) et de charge (𝑍𝐴) et
(𝑍𝐵), les distances (𝑟𝑖𝑗) entre les électrons (𝑖 , 𝑗), la distance (𝑟𝑖𝐴) entre l’électron (𝑖) et le
noyau (𝐴), et (𝑅𝐴𝐵) entre les noyaux (𝐴) et (𝐵). La présence du terme de répulsion
électronique dans l’expression du Hamiltonien rend impossible la résolution de l’équation de
Schrödinger, sauf dans le cas d’un système monoélectronique. Cela implique donc de faire
une approximation pour dépasser cet obstacle à l’étude de systèmes électroniques complexes.
La masse d’un électron étant près de 1836 fois inférieure à celle d’un proton, les mouvements
respectifs des noyaux et des électrons peuvent être découplés. On fait alors l’approximation
de Born-Oppenheimer. On peut donc se ramener à l’étude du comportement des électrons
dans le potentiel crée par les noyaux ayant une géométrie donnée. Ce dernier sera décrit par
une fonction d’onde électronique, état propre de l’Hamiltonien électronique (��𝑒) dans lequel
n’intervient plus le terme de l’équation relatif à l’énergie cinétique des noyaux. Il est à
remarquer que le terme de répulsion nucléaire, indépendant des coordonnées des électrons,
n’agit que comme correction à l’énergie électronique du système.

14
��𝑒 = −∑∇𝑖2
2
𝑁
𝑖
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
−∑∑𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
𝑁
𝑖
(1.5)
La solution de l'équation de Schrödinger correspondante, 𝛹𝑒
��𝑒𝛹𝑒 = 𝐸𝑒𝛹𝑒 (1.6)
Est la fonction d'onde électronique du système considéré décrivant le mouvement des
électrons
𝛹𝑒 = 𝛹𝑒({𝑟𝑖; 𝑅𝐴}) (1.7)
𝛹𝑒 dépend explicitement des coordonnées des électrons mais de façon paramétrique des
coordonnées nucléaires, tout comme l'énergie électronique :
𝐸𝑒 = 𝛹𝑒({𝑅𝐴}) (1.8)
L'énergie totale 𝐸 est alors donnée par la relation :
𝐸 = 𝐸𝑒 +∑∑𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐴<𝐵
𝑀
𝐴
(1.9)
Dans le cadre de l'approximation de Born-Oppenheimer, on définit l'Hamiltonien nucléaire ��
permettant de décrire le mouvement des noyaux dans le champ moyen des électrons.
�� = −∑𝛻𝐴2
2𝑀𝐴
𝑀
𝐴
+ ⟨−∑𝛻𝑖2
2
𝑁
𝑖
−∑∑𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
𝑁
𝑖
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
⟩ +∑∑𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐴<𝐵
𝑀
𝐴
(1.10)

15
= −∑𝛻𝐴2
2𝑀𝐴
𝑀
𝐴
+ 𝐸𝑒({𝑅𝐴}) +∑∑𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐴<𝐵
𝑀
𝐴
(1.11)
= −∑𝛻𝐴2
2𝑀𝐴
𝑀
𝐴
+ 𝐸 (1.12)
Les fonctions d'onde 𝛹𝑛𝑢𝑐𝑙 vérifiant l'équation de Schrödinger suivante
��𝑛𝑢𝑐𝑙𝛹𝑛𝑢𝑐𝑙 = 𝐸𝛹𝑛𝑢𝑐𝑙 (1.13)
Décrivent les mouvements de translation, rotation et vibration des noyaux.
𝛹𝑛𝑢𝑐𝑙 = 𝛹𝑛𝑢𝑐𝑙({𝑅𝐴}) (1.14)
Dans l’expression (1.13), (𝐸) donnée dans la relation (1.2) représente l’énergie totale du
système, dans le cadre de l'approximation de Born-Oppenheimer, incluant les composantes
électronique, vibrationnelle, rotationnelle et translationnelle.
𝛹({𝑟𝑖; 𝑅𝐴}) = 𝛹𝑒({𝑟𝑖; 𝑅𝐴})𝛹𝑛𝑢𝑐𝑙({𝑅𝐴}) (1.15)
Une seconde approximation importante est de se placer dans un cadre non relativiste, on fait
alors l’approximation que la vitesse de déplacement des électrons est très faible par rapport à
la vitesse de la lumière dans le vide. Cette approximation se justifie bien pour les atomes des
deux premières lignes de la classification périodique des éléments de Mendeleïev. Les effets
relativistes deviennent cependant plus importants au fur et à mesure que la charge électrique
du noyau augmente et que les électrons de cœur doivent compenser l’attraction en augmentant
leur vitesse.

16
1.1.2. Fonction d’onde multiélectronique et l’approximation orbitaire.
La mécanique quantique n’admettant pas de solution analytique exacte pour l’équation de
Schrödinger pour un nombre de particules supérieur à deux (appelé problème à (𝑛) corps),
Pour simplifier ce problème, deux autres approximations sont utilisées couramment. La
première, appelée approximation orbitale, permet de décrire le comportement individuel de
chaque électron dans le champ créé par le reste des particules du système moléculaire.
Notamment, il ressent le champ moyen des autres électrons. La deuxième consiste à négliger
le couplage existant entre le mouvement de l'électron (moment cinétique orbitalaire) et le
moment cinétique de spin. L'approximation orbitale consiste à représenter la fonction d'onde
multiélectronique du système comme le produit de fonctions d'onde monoélectronique
{𝜙𝑖}1≤𝑖≤𝑛 (1.16)
𝛹𝑒(𝑟1, 𝑟2, 𝑟3, … , 𝑟𝑛) ≡ 𝛹𝑒(1, 2, 3, … , 𝑛) = 𝜙1(1)𝜙2(2)𝜙3(3) ∙∙∙ 𝜙𝑛(𝑛) (1.17)
L'ensemble {ϕi}1≤i≤n correspond à un jeu de spinorbitales. Une spinorbitale peut s'écrire
comme le produit d'une fonction de spin 𝜎(𝑖) par une fonction spatiale 𝜑𝑖(𝑖) qui ne dépend
que de la position de l'électron (orbitale atomique (OA) pour un atome, orbitale moléculaire
(OM) dans le cas d'un système polyatomique).
𝜙𝑖 (𝑖) = 𝜙𝑖(𝜎(𝑖), 𝑟(𝑖)) = 𝜎(𝑖)𝜑𝑖(𝑖) (1.18)
Avec
𝜎(𝑖){𝛼(𝑖) 𝑝𝑜𝑢𝑟 𝑢𝑛 𝑠𝑝𝑖𝑛 =
1
2
𝛽(𝑖) 𝑝𝑜𝑢𝑟 𝑢𝑛 𝑠𝑝𝑖𝑛 = −1
2
Les électrons sont de spin demi-entier et on les qualifie de fermions. Le principe d'exclusion
de Pauli qui en découle stipule que lors de l'échange de deux particules fermioniques la
fonction d'onde du système change de signe.

17
En combinant l'approximation orbitale et le principe d'exclusion de Pauli (Pauli 1925), la
fonction d'onde électronique 𝛹𝑒 est exprimée sous la forme d'un déterminant de Slater (Slater
1929), antisymétrique pour un jeu de spinorbitales orthonormées
𝛹𝑒 =1
√𝑁
|
|
|
𝜙1(1) 𝜙2(1) 𝜙3(1) ∙ ∙ ∙ 𝜙𝑁(1)
𝜙1(2) 𝜙2(2) 𝜙3(2) ∙ ∙ ∙ 𝜙𝑁(2)
𝜙1(3) 𝜙2(3) 𝜙3(3) ∙ ∙ ∙ 𝜙𝑁(3) ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙ ∙
𝜙1(𝑁) 𝜙2(𝑁) 𝜙3(𝑁) ∙ ∙ ∙ 𝜙𝑁(𝑁)
|
|
|
= |𝜙1𝜙2𝜙3 ∙∙∙ 𝜙𝑁⟩ (1.19)
Les chiffres entre parenthèses correspondent aux coordonnées des électrons et 𝜙𝑖 est un spin
orbitale et le facteur 1
√N permet de normer le déterminant Ψe
1.2. Méthodes de détermination de la structure électronique
1.2.1. La méthode Hartree-Fock
L’Hamiltonien électronique dans le cadre de la méthode Hartree-Fock (HF) (Hartree 1928),
s'écrit :
��𝑒 = −∑∇𝑖2
2
𝑁
𝑖
−∑∑𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
𝑁
𝑖
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
(1.20)
On note ��𝑐(𝑖) l’Hamiltonien de cœur, qui correspond à l’opérateur monoélectronique
décrivant l’énergie cinétique d’un électron (i) dans le champ des noyaux M (terme
d'attraction des noyaux et de l’électron i), tandis que le reste, correspondant à la répulsion
électronique qui s'exprime sous la forme d'un opérateur biélectroniques.

18
��𝑐(𝑖) = −𝛻𝑖2
2 −∑
𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
(1.21)
L’Hamiltonien électronique devient alors en fonction de ��𝑐
��𝑒 =∑��𝑐(𝑖)
𝑁
𝑖
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
(1.22)
On se place dans le cas où les spinorbitale sont mutuellement orthogonales,
⟨𝜙𝑖|𝜙𝑗⟩ = 𝛿𝑖𝑗 (1.23)
Où 𝛿𝑖𝑗 est le symbole de Kronecker tel que 𝛿𝑖𝑗 = {0 𝑠𝑖 𝑖 ≠ 𝑗1 𝑠𝑖 𝑖 = 𝑗
L'énergie du système en utilisant les propriétés du déterminant de Slater est donnée par
𝐸𝑒 = ⟨𝛹𝑒|��𝑒|𝛹𝑒⟩
𝐸𝑒 =∑⟨𝜙𝑖(1)|��𝑐(1)|𝜙𝑖(1)⟩
𝑁
𝑖
+
∑∑[⟨𝜙𝑖(1)𝜙𝑗(2)|1
𝑟12|𝜙𝑖(1)𝜙𝑗(2)⟩ − ⟨𝜙𝑖(1)𝜙𝑗(2)|
1
𝑟12|𝜙𝑗(1)𝜙𝑖(2)⟩]
𝑁
𝑖<𝑗
𝑁
𝑖
(1.24)
��𝑐𝑖𝑖 = ⟨𝜙𝑖(1)|��𝑐(1)|𝜙𝑖(1)⟩ Représente l’intégrale monoélectronique de cœur.
L’application du principe variationnel impose que l’ensemble des spinorbitales ϕi est optimal
quand l’énergie Ee est minimale. C'est à partir de cette considération que Hartree et Fock ont
mis au point leurs célèbres équations qui permettent la détermination des spinorbitales (Fock
1930):

19
��𝜙𝑖 = 휀𝑖𝜙𝑖 (1.25)
��(1) = ��𝑐(1) +∑[2𝐽𝑖(1) − ��𝑖(1)]
𝑁 2⁄
𝑖
(1.26)
𝐽𝑖(1)𝜙𝑗(1) = 𝜙𝑗(1)∫(2)𝜙𝑖∗(2)
1
𝑟12𝜙𝑖(2)𝑑𝜏2 (1.27)
��𝑖(1)𝜙𝑗(1) = 𝜙𝑖(1)∫(2)𝜙𝑖∗(2)
1
𝑟12𝜙𝑗(2)𝑑𝜏2 (1.28)
𝐽𝑖 et ��𝑖 sont respectivement les opérateurs de Coulomb et d’échange. La méthode de Hartree-
Fock présente l’avantage de ne faire intervenir que des opérateurs monoélectronique.
Cependant, les opérateurs 𝐽𝑖(1) et ��𝑖(1) nécessitent la connaissance préalable des
spinorbitales que cette méthode est sensée fournir. Les solutions de ces équations sont
déterminées via un processus itératif du type méthode du champ cohérent SCF (Self
Consistent Field) dont on peut se placer dans le cadre du modèle HF avec contrainte de spin
RHF (Restricted Hartree-Fock), ou dans le cadre du même modèle, mais sans contrainte de
spin UHF(Unrestricted Hartree-Fock ).
1.2.2. Les équations de Roothaan
L’énergie Hartree-Fock est déterminée en résolvant les équations de Roothaan (Roothaan
1951). Les orbitales moléculaires (MO) sont obtenues par l’application de l’opérateur de Fock
à chaque orbitales et utilisé dans système linéaire défini dans l’équation Fϕi = εiϕi .
Nous nous placerons dans le cas d'un système à couches fermées possédant un nombre pair
d’électron et nous utiliserons le formalisme HF restreint ou RHF (Restricted Hartree-Fock)
pour lequel deux électrons de spin différents possèdent une partie d'espace identique.
L’approximation LCAO (Linear Combination of Atomic Orbitals) permet aux spinorbitales
moléculaires déterminées par la méthode de Hartree-Fock d’être développées sur une base de
(K) orbitales atomiques (AO) définie comme {χ}1≤≤K sous forme de combinaison linéaire.

20
𝜙𝑖 =∑𝐶𝑖𝜒
𝐾
(1.29)
Sur cette base d’OA un élément 𝜇𝜈 de la matrice de Fock s’exprime :
𝐹𝜇𝜈 = ∫𝜒∗ (1)��(1)𝜒𝜈(1) 𝑑𝜏1
= ∫𝜒∗ (1)��𝑐(1)𝜒𝜈(1) 𝑑𝜏1 +∑∫𝜒
∗
𝑁 2⁄
𝑖
(1)[2𝐽𝑖(1) − ��𝑖(1)]𝜒𝜈(1) 𝑑𝜏1
= ��𝜇𝜈𝑐 +∑2(𝜇𝜈|𝑖𝑖)
𝑁 2⁄
𝑖
− (𝜇𝑖|𝑖𝜈) (1.30)
Les intégrales biélectroniques sont définies par :
(𝜇𝜈|𝜆𝜎) = ∬𝜒∗ (1)𝜒𝜈(1)
1
𝑟12𝜒𝜆∗(2)𝜒𝜎(2)𝑑𝜏1𝑑𝜏2 (1.31)
(𝜇𝜎|𝜆𝜈) = ∬𝜒∗ (1)𝜒𝜎(1)
1
𝑟12𝜒𝜆∗(2)𝜒𝜈(2)𝑑𝜏1𝑑𝜏2 (1.32)
En introduisant l’approximation de la combinaison linéaire on obtient les équations suivantes
𝐹𝜇𝜈 = ��𝜇𝜈𝑐 +∑∑𝐶𝜆𝑖𝐶𝜎𝑖
𝜆𝜎
𝑁 2⁄
𝑖
[2(𝜇𝜈|𝜆𝜎) − (𝜇𝜎|𝜆𝜈)] (1.33)
= ��𝜇𝜈𝑐 +∑𝑃𝜆𝜎
𝑇
𝜆𝜎
[(𝜇𝜈|𝜆𝜎) −1
2 (𝜇𝜎|𝜆𝜈)]
= ��𝜇𝜈𝑐 + 𝐺𝜇𝜈
En formalisme restreint, l’élément ν de la matrice densité totale du système PT est définit
comme :

21
𝑃𝜇𝜈𝑇 = 2∑𝐶𝜇𝑖𝐶𝜈𝑖
𝑁
𝑖
(1.34)
Dans sa forme matricielle, cette expression s’écrit
𝐅 = 𝐇𝐜 + 𝐆 (1.35)
Avec F la matrice de Fock, (H) la matrice Hamiltonien de cœur et (G), la matrice obtenue par
contraction de la matrice densité avec les intégrales biélectroniques. On peut écrire l’énergie
du système sure la base des orbital atomique (AO) comme suit :
𝐸 =∑𝑃𝜇𝜈𝑇
𝑁
𝜇𝜈
𝐻𝜇𝜈 +1
2∑ 𝑃𝜇𝜈
𝑇 𝑃𝜆𝜎𝑇
𝜇𝜈𝜆𝜎
𝐺𝜇𝜈𝜆𝜎 (1.36)
Avec
𝐺𝜇𝜈𝜆𝜎 = (𝜇𝜈|𝜆𝜎) −1
2(𝜇𝜎|𝜆𝜈)
La condition de stationnarité de l'énergie par rapport aux coefficients {Ci}1≤≤N1≤i≤N
𝛿𝐸
𝛿𝐶𝑖= 0
L’introduction de multiplicateurs de Lagrange ϵi conduit à la dérivation des équations de
Roothaan. Ces dernières s’écrivent :
∑𝐹𝜇𝜈𝐶𝜈𝑖
𝑁
𝜈
=∑𝑆𝜇𝜈𝐶𝜈𝑖
𝑁
𝜈
𝜖𝑖 (1.37)
La notation sous forme matricielle est
𝐅 ∙ 𝐂 = 𝐒 ∙ 𝐂 ∙ 𝐄 (1.38)

22
S : représente la matrice de recouvrement telle dont les éléments
Sμν = ⟨χ|χν⟩ = ∫χ∗ χν dτ1
C : représente la matrice qui regroupe les coefficients des OA dans les OM tel que
(C)μi = Cμi
La matrice de recouvrement apparait dans l’équation car les AO sont des fonctions non
orthogonales entre elles. Ceci implique qu’il existe un recouvrement non nul entre les
fonctions de base. Il est donc nécessaire d’orthogonaliser ces fonctions de base afin que la
résolution des équations de Roothaan devienne un problème aux valeurs propres. Cette
orthogonalisation est réalisée grâce à une matrice X permettant une transformation unitaire.
Cette matrice est exprimée de telle que :
𝐗† ∙ 𝐒 ∙ 𝐗 = 𝕀 (1.39)
Où 𝕀 est la matrice identité. La matrice X correspond à une matrice d'orthogonalisation de la
base des orbitales atomique. Dans le formalisme SCF, la matrice d’orthogonalisation est
souvent celle de Lӧwdin avec (X = S−1 2⁄ ). La multiplication à gauche de l'expression
(F ∙ C = S ∙ C ∙ E) par (X†) ; matrice adjointe de la matrice(X), permet de se ramener à un
problème standard aux valeurs propres hermétiques :
𝐅′ ∙ 𝐂′ = 𝐂′ ∙ 𝐄 (1.40)
Avec
𝐅′ = 𝐗† ∙ 𝐅 ∙ 𝐗
𝐂 = 𝐗 ∙ 𝐂′

23
La résolution de l’équation (F′ ∙ C′ = C′ ∙ E) s’effectue par la diagonalisation de la matrice F′
dépendante des coefficients recherchés (C) . La méthode itérative appelé résolution auto-
cohérente ou SCF (Self-Consistent Field) permettant de déterminer les coefficients Cμiest
alors employée.
L'algorithme est résumé comme suit :
1-Fournir un jeu de coordonnées nucléaires ainsi que la nature des atomes et la charge totale.
2-Calcul des intégrales : Sμν Hμc et (μν|λσ)
3-Diagonaliser la matrice de recouvrement pour construire la matrice X
4-Estimation initiale de la matrice densité P grâce à une fonction d’onde d’essai initiale.
5-Construction de la matrice de Fock : F = Hc + G
6- Expression de F dans la base orthogonale (F′ = X† ∙ F ∙ X) avec (X = S−1 2⁄ )
7-Diagonalisation de F′ pour obtenir C′ et les énergies des OM.
8- Exprimer les coefficients C′ dans la base non orthogonale : (C = X ∙ C′)
9- Calcul de la nouvelle matrice densité P et calcul de l’énergie E =1
2Tr[P(Hc + F)]
10- Test de convergence sur E et / ou si non satisfait, retour au point 5.
Habituellement, on considère que la convergence du processus est atteinte lorsque la variation
de l’´énergie calculée ainsi que la différence entre les matrices densité de deux itérations
consécutives sont négligeables La fonction d’onde ainsi déterminée répond au principe
variationnel et on dit que le système a atteint sa cohérence interne.
Les méthodes de type HF ne permettent pas d'obtenir l'énergie exacte du système, même dans
le cas d'une base d'AO complète (Complete Basis Set CBS) ceci est due au traitement du
terme bioélectronique de répulsion électron-électron : (∑1
rij
ni<j ) qui n'est pris en compte que
par le biais d'une moyenne globale. La méthode HF fait partie des méthodes de champ moyen,
c'est-à-dire qu'un électron évolue dans le champ moyen crée par les autres électrons.

24
1.3 Les méthodes post Hartree-Fock
La corrélation des mouvements électroniques n’est pas prise en compte par un déterminant de
Slater construit dans l’approximation orbitalaire. En théorie, on peut prendre en compte cette
corrélation si la fonction d’onde multiélectronique tient compte des variables de position des
électrons et des variables de distance entre électrons. En pratique, cette approche ne peut être
appliquée qu’à des systèmes très petits.
L'énergie de corrélation (𝐸𝑐𝑜𝑟𝑟) est définit comme la différence entre l'énergie exacte non-
relativiste du système (휀0) et l'énergie HF (𝐸0) dans la limite d'une base supposée complète
d'AOs. Cette énergie est toujours négative puisque le principe variationnel assure que
l'énergie (𝐸0) est toujours supérieure à(휀0). Afin de se rapprocher de l’énergie exacte, les
méthodes post- Hartree-Fock ont pour but, de calculer une partie de la corrélation
électronique qu’une approche Hartree-Fock ne donne pas. Cette corrélation est ajoutée dans
les méthodes post-Hartree-Fock soit sous la forme d'une perturbation soit d'une interaction de
configuration.
𝐸𝑐𝑜𝑟𝑟 = 휀0 − 𝐸0 (1.41)
1.3.1 Interaction de configuration
La fonction d'onde multiélectronique du système est construite comme une combinaison
linéaire de plusieurs déterminants orthogonaux, obtenus à partir du déterminant de Slater
solution des équations de Hartree-Fock 𝛹0
𝛹 = 𝐶0𝛹0 +∑∑𝐶𝑖
𝑎𝛹𝑖𝑎
𝑣𝑖𝑟
𝑎
𝑜𝑐𝑐
𝑖
+∑∑𝐶𝑖𝑗𝑎𝑏𝛹𝑖𝑗
𝑎𝑏
𝑣𝑖𝑟
𝑎<𝑏
𝑜𝑐𝑐
𝑖<𝑗
+ ∑ ∑ 𝐶𝑖𝑗𝑘𝑎𝑏𝑐𝛹𝑖𝑗𝑘
𝑎𝑏𝑐
𝑣𝑖𝑟
𝑎<𝑏<𝑐
𝑜𝑐𝑐
𝑖<𝑗<𝑘
+ ∙∙∙ (1.42)
Les déterminants sont obtenus en remplaçant dans 𝛹0 une ou plusieurs spinorbitales occupées
𝜙𝑖 , 𝜙𝑗 , … par une ou plusieurs spinorbitales virtuelles 𝜙𝑎, 𝜙𝑏 , … .

25
Les déterminants 𝛹𝑖𝑎, 𝛹𝑖𝑗
𝑎𝑏 , 𝛹𝑖𝑗𝑘𝑎𝑏𝑐 correspondent respectivement aux mono, di et tri-
excitations. Par exemple 𝛹𝑖𝑎 représente l’excitation d’un électron de la spinorbital « i » vers la
spinorbitale « a »
Les coefficients de l’équation (1.42) sont alors déterminés en diagonalisant l’Hamiltonien
électronique exprimé sur la base des configurations. Cette méthode permet d’approcher de
façon variationnel les fonctions d’ondes exactes pour les différents états, excités ou
fondamental.
Plus la base d'orbitales atomiques est étendue, plus le nombre d'excitations possible est élevé.
La réalisation de toutes ces excitations conduit à un calcul Full CI qui, si la base est complète,
est alors exact. Dans la pratique, ces calculs ne sont réalisables que pour de petits systèmes en
base très limitée. Il est souvent préférable de tronquer l'espace des configurations en se
limitant aux excitations doubles (CID), simples et doubles (CISD).
1.3.2 La théorie perturbationnelle : Møller-Plesset (MP)
Cette méthode est basée sur la théorie des perturbations de Rayleigh-Schrӧdinger qui consiste
à développer en série de Taylor l’énergie du système. La corrélation électronique est
considérée comme représentant une perturbation de l'énergie Hartree-Fock l'Hamiltonien
électronique ��du système est alors définit comme étant la somme d'un Hamiltonien de
référence ��0 (étant ici l’Hamiltonien de Fock) et d'un opérateur de perturbation ��
�� = ��0 + �� (1.43)
Dans le cas de la théorie des perturbations de Møller-Plesset à l'ordre n (MPn), l'Hamiltonien
de référence est donné par la relation, où ��(𝑖) est l'opérateur de Hartree-Fock.
��0 =∑��(𝑖)
𝑛
𝑖
(1.44)
�� = ∑��(𝑖)
𝑛
𝑖
+ �� (1.45)

26
On trouve l'énergie Hartree-Fock comme égale à l'énergie non-perturbée plus la correction au
premier ordre. Dans la pratique, les méthodes Meller-Plesset à l'ordre 2 (MP2) ou à l'ordre 4
(MP4) sont les plus couramment employées. Dans La méthode MP2 qui est la plus
couramment utilisée, le développement perturbatif est fait jusqu’à l’ordre 2 et il ne fait
intervenir que les diexcitations. La plus grande partie de l’énergie de corrélation est déjà
considérer être contenu dans ce terme correctif. . L’énergie totale s’écrit alors
𝐸𝑡𝑜𝑡 = 𝐸0 + 𝐸(2) (1.46)
Avec 𝐸0 = 𝐸𝐻𝐹 et 𝐸(2) la correction de l’énergie au deuxième ordre qui est donné par la
relation suivante :
𝐸(2) = ⟨𝛹(0)|��|𝛹(1)⟩ (1.47)
𝐸(2) =∑∑|⟨𝛹0| ∑
1𝑟𝑘𝑙
𝑛𝑘<𝑙 |𝛹𝑖𝑗
𝑎𝑏⟩|2
휀𝑖 + 휀𝑗 − 휀𝑎 − 휀𝑏
𝑣𝑖𝑟
𝑎<𝑏
𝑜𝑐𝑐
𝑖<𝑗
=1
4∑∑
|(𝑖𝑗|𝑎𝑏) − (𝑖𝑎|𝑗𝑏)|2
휀𝑖 + 휀𝑗 − 휀𝑎 − 휀𝑏
𝑣𝑖𝑟
𝑎<𝑏
𝑜𝑐𝑐
𝑖<𝑗
(1.48)
La solution de l’équation MP au premier ordre, donne l’énergie de Hartree-Fock. La première
correction de perturbation au-delà du traitement Hartree-Fock qui ne s’annule pas est le terme
d’’énergie de second ordre (par le théorème de Brillouin).
1.3.3 Les méthodes Coupled-Cluster (CC)
Un moyen d'obtenir une méthode size-consistent est proposé par l'approche Coupled-Cluster
(CC) qui consiste à inclure toutes les corrections d'un type donné à l'ordre infini. Ceci
s'effectue grâce à la fonction d'onde CC :
𝛹𝐶𝐶 = 𝑒��𝛹0 (1.49)
Avec
𝑒�� = 𝕀 + �� +1
2��2 +
1
6��3 + 𝒪(��4)

27
�� est l'opérateur cluster défini par :
�� = �� 1 + �� 2 + �� 3 + ∙∙∙ +�� 𝑛
L'opérateur �� 𝑖 a pour effet de générer tous les déterminants de Slater correspondant à l’ième
excitation. Cependant, cette méthode a pour défaut de ne pas être variationelle.
1.4 La théorie de la fonctionnelle de la densité (DFT)
L’idée directrice de la théorie de la fonctionnelle de la densité (DFT) c’est qu’elle permet de
calculer l’énergie électronique des systèmes chimiques. Cette approche, introduite par
Hohenberg et Kohn (Hohenberg and Kohn 1964), stipule que toute propriété de l’état
fondamental d’un système est complètement déterminée par la densité électronique 𝜌(𝑟).
L’énergie electronique 𝐸[𝜌] apparait alors comme une fonctionelle de la densité electronique
car à chaque fonction 𝜌(𝑟) est associer une seul énergie.d’une façon analogue au principe
variationnel la densité est stationnaire pour l’état fondamental. L’introduction d’une approche
de type orbitalaire telle que
(𝑟) =∑𝜙𝑖∗
𝑛
𝑖
(𝑟)𝜙𝑖(𝑟) (1.50)
Conduit aux équations de Kohn-Sham dont la résolution peut se faire de façon analogue à
celle de Hartree-Fock
1.4.1 Les équations de Kohn-Sham (KS)
Le théorème de Hohenberg-Kohn ne donne pas la forme de la fonctionnelle, mais confirme
l’existence de celle-ci. Dans l’approche Kohn-Sham (Kohn and Sham 1965), les électrons
sont décrits comme des particules indépendantes ayant la même densité que le système réel.
Ceci permet de décomposer l’énergie totale du système 𝐸[(𝑟)] de la manière suivante :
𝐸[(𝑟)] = 𝑉[(𝑟)] + 𝑇𝑠[(𝑟)] + 𝐸𝑋𝐶[(𝑟)] (1.51)

28
Où 𝑉[(𝑟)] représente la composante électrostatique classique décomposée en contribution
noyau-électron 𝑉𝑛𝑒[(𝑟)] (énergie d’attraction entre les noyaux atomiques et les électrons) et
électron-électron 𝑉𝑒𝑒[(𝑟)] (interaction coulombiènne entre la distribution totale des charges
en 𝑟 et 𝑟′)
𝑉[(𝑟)] = 𝑉𝑛𝑒[(𝑟)] + 𝑉𝑒𝑒[(𝑟)] = −∑∫𝑍𝐴
|𝑟 − 𝑅𝐴|
𝑀
𝐴
(𝑟)𝑑𝑟 +1
2∬
(𝑟)(𝑟′)
|𝑟 − 𝑟′|𝑑𝑟𝑑𝑟′ (1.52)
Le terme 𝑇𝑠[(𝑟)] correspond à l’énergie cinétique du système sans interaction 𝑉𝑛𝑒[(𝑟)] = 0
𝑇𝑠[(𝑟)] = −1
2∑∫𝜙𝑖
∗
𝑛
𝑖
(𝑟)∇2𝜙𝑖(𝑟)𝑑𝑟 (1.53)
Le terme 𝐸𝑋𝐶[(𝑟)] regroupe la contribution de l’échange (X) et de la corrélation (C).
Cette quantité est centrale en théorie de la fonctionnelle de densité.
Les équations de Kohn-Sham sont déduite en utilisant la condition de stationnarité de 𝐸[(𝑟)]
𝛿𝐸[(𝑟)]
𝛿(𝑟)= 0 (1.54)
Les orbitales KS sont obtenues par la résolution des équations de Kohn-Sham (KS) en
appliquant un principe variationnel à l'énergie. 𝐸[(𝑟)] .les équations (KS) se présentent sous
la forme suivante
[−1
2∇2 −∑
𝑍𝐴|𝑟 − 𝑅𝐴|
+ ∫(𝑟′)
|𝑟 − 𝑟′|𝑑𝑟′ +
𝛿𝐸𝑋𝐶[(𝑟)]
𝛿(𝑟)
𝑀
𝐴
]𝜙𝑖(𝑟) = 휀𝑖𝜙𝑖(𝑟) (1.55)
Où les 휀𝑖 sont les énergies des orbitales KS. Les équations de (KS) peuvent être réécrites sous
forme d’un problème aux valeurs propres
��𝐾𝑆𝜙𝑖(𝑟) = 휀𝑖𝜙𝑖(𝑟) (1.56)

29
Ces équations se résolvent de manière auto-cohérente, tout comme les équations de HF en
développant les orbitales monoélectronique𝑠 {𝜙𝑖(𝑟)}
1≤𝑖≤𝑛 sur une base d’orbitales atomiques
{χ}1≤≤K Les équations de (KS) sont résolues par la méthode du champ auto-cohérent (SCF).
On donne une densité d’essai, comme la somme des densités atomiques par exemple, afin
d’obtenir des orbitales KS de départ. Puis ces orbitales sont utilisées pour calculer une
meilleure densité, jusqu’à ce que l’énergie totale ait convergé
𝜙𝑖(𝑟) =∑𝐶𝜇𝑖χ(𝑟)
𝐾
𝜇
(1.57)
Conduisant à
𝐅𝐊𝐒 ∙ 𝐂 = 𝐒 ∙ 𝐂 ∙ 𝐄 (1.58)
1.4.2 Les fonctionnelles d’échange-corrélation
Le potentiel d’échange-corrélation 𝑉𝑋𝐶(𝑟) est définit par la dérivé de la fonctionnelle
d’échange-corrélation 𝐸𝑋𝐶[(𝑟)]
𝑉𝑋𝐶(𝑟) =𝜕𝐸𝑋𝐶[𝜌(𝑟)]
𝜕𝜌(𝑟) (1.59)
Avec
𝐸𝑋𝐶[𝜌(𝑟)] = ∫𝜌(𝑟) 𝜖𝑋𝐶[𝜌(𝑟)]𝑑𝑟 (1.60)
Dans l’approche originelle de Kohn et Sham, l’énergie d’échange-corrélation par électron ou
par particule 𝜖𝑋𝐶 est fonction de la densité dans un gaz uniforme d’électron. Elle est définie
comme la somme des contributions d’échange et de corrélation : 𝜖𝑋𝐶 = 𝜖𝑋 + 𝜖𝐶 Où le terme
d’échange 𝜖𝑋 est associé aux interactions entre électrons de même spin, tandis que la
corrélation 𝜖𝐶 s’applique à tous les électrons

30
1.4.3 Types de fonctionnelles d’échange-corrélation
La connaissance du potentiel d’échange-corrélation intervenant dans les équations de (KS),
permet de déterminer de façon exacte la densité électronique, et donc les valeurs exactes de
l’ensemble des observable de l’état fondamentale. Cependant, la nature et la formule
mathématique précise de ce potentiel est inconnue, ce qui donne naissance à plusieurs types
d’approximation permettant d’approcher au mieux cette quantité. C’est donc le choix de la
fonctionnelle d’échange et de corrélation qui donnera la qualité du calcul DFT. Les méthodes
de construction de ces fonctionnelles reposent principalement sur le respect de certaines
conditions dont les plus importantes peuvent être trouvées dans un article de Perdew (Perdew
et al. 2004). Cependant, une deuxième approche consiste à utiliser un ajustement sur des
valeurs expérimentales de propriétés moléculaires il est possible de classer les fonctionnelles
en différents types dont la qualité théorique est croissante. Ce classement est une sorte
d’échelle de Jacob de la DFT. On distingue trois grandes familles de fonctionnelle d’échange
et de corrélation (Sousa et al. 2007) où les différentes fonctionnelles sont donc classées en
tant que fonctionnelle d’échange ou de corrélation, l’association de ces deux types donne
naissance à une fonctionnelle d’échange-corrélation.
1.4.3 a) L’approximation de la densité locale (LDA)
La méthode de la LDA (Local Density Approximation) (Kohn and Sham 1965) fait partie de
la première génération de fonctionnelles. Cette méthode est basée sur l’utilisation du modèle
du gaz uniforme d’électrons (jellium) (Brack 1993) -uniform electron gas (UEG) or
homogeneous electron gas (HEG)- pour décrire la densité électronique, qui varie de façon
lente avec la position. Elle postule qu’en chaque point (r)d’une distribution électronique
inhomogène où la densité est ρ(r), les valeurs de EX et de ϵX[ρ(r)] se comporte identiquement
au gaz uniforme d’électron. Dès lors, la densité électronique au voisinage de (r) est remplacée
par une densité électronique constante qui a la même valeur qu’en (r) . Cependant, cette
densité électronique est différente en tout point de l’éspace. L’énergie d’échange est donnée
par la formule de Dirac. Par particule, elle vaut :
𝜖𝑋𝐿𝐷𝐴[𝜌(𝑟)] = −
3
4(3
𝜋)1 3⁄
𝜌1 3⁄ (𝑟) = −𝐶𝑥𝜌1 3⁄ (𝑟) (1.61)

31
Où 𝐶𝑥 est un coefficient dépendant de la fonctionnelle. La plus ancienne est la fonctionnelle
d’énergie cinétique du gaz uniforme d’électron déterminée par Thomas-Fermi (Thomas
1927). Suivant ce principe, Dirac détermina la fonctionnelle d’échange du gaz uniforme
d’électron (Dirac 1930), ce qui donna naissance à la méthode 𝑋𝛼 de Slater en 1951 (Slater
1951). Cette fonctionnelle peut être considérée comme une fonctionnelle LDA où l’énergie de
corrélation est négligée, et l’énergie d’échange, donné par la fonctionnelle de Dirac, est
pondérée par un facteur 𝛼
𝛼
{
=
2
3 𝑝𝑜𝑢𝑟(𝐿𝐷𝐴)
= 1 𝑝𝑜𝑢𝑟 (𝑆𝑙𝑎𝑡𝑒𝑟)
=3
4 𝑝𝑜𝑢𝑟 (𝐺𝑎𝑠𝑝𝑎𝑟)
La LDA se justifie pour deux cas limites : lorsque la densité électronique varie lentement ou
lorsque la densité est très élevée. Elle trouve beaucoup d’adeptes dans l’étude des grands
systèmes, comme les métaux. Le modèle fonctionne également de façon correcte pour le
calcul de certain nombre de propriétés structurale et énergétique. Ceci est dû à une
compensation d’erreur entre l’énergie de corrélation et l’énergie d’échange. L’approximation
LDA est souvent remplacée par sa variante prenant en compte le spin des électrons LSDA
(Local Spin Density Approximation) dans laquelle l’énergie d’échange s’exprime :
𝐸𝑋𝐿𝑆𝐷𝐴 = −2
13𝐶𝑥∫(𝜌𝛼
43 + 𝜌
𝛽
43) 𝑑𝑟 (1.62)
Basé sur les travaux de Ceperley et Alder (Ceperley and Alder 1980) qui utilisèrent la
méthode Monte-Carlo quantique pour déterminer la fonctionnelle de corrélation du (jellium),
Vosko, Wilk et NNusair (VWN) trouvèrent une fonction ajustant au mieux leurs données, afin
d’obtenir une fonctionnelle de corrélation (Vosko et al. 1980).
1.4.3 b) L’approximation du gradient généralisé (GGA)
Dans système moléculaire, la densité électronique n’est pas spatialement uniforme, ce qui
limite évidemment l’application des méthodes LDA/LSDA. Rendre les fonctionnelles
d’échange et de corrélation dépendantes du gradient de la densité locale, représente une façon
de corriger le caractère non-uniforme de la densité électronique. On obtient alors des

32
fonctionnelles de seconde génération GGA (Generalized Gradient Approximation). La plupart
de ces fonctionnelles GGA sont construites en ajoutant un terme correctif à la fonctionnelle
LDA.
𝐸𝑋𝐶𝐺𝐺𝐴[𝜌(𝑟)] = 𝐸𝑋𝐶
𝐿𝑆𝐷𝐴[𝜌(𝑟)] + ∆𝐸𝑋𝐶 [|∇𝜌(𝑟)|
𝜌(𝑟)4 3⁄] (1.63)
De nombreux exemples de fonctionnelle de type GGA existent, mais les plus courantes sont
les fonctionnelles d’échange de Becke (B88) (Becke 1988), Perdew-Burke-Ernzerhof (PBE)
(Perdew et al. 1996; Perdew et al. 1997), et de Perdew et Wang (PW86) (Perdew and Yue
1986) ainsi que les fonctionnelle de corrélation de Lee,Yang et Parr (LYP) (Lee et al. 1988) et
(PW91) (Perdew et al. 1992). Une fonctionnelle d’échange est souvent associée avec une
fonctionnelle de corrélation et la nomenclature veut que l’on accole les deux noms. Par
exemple, l’utilisation de la fonctionnelle d’échange de Becke avec la fonctionnelle de
corrélation de Lee, Yang et Parr se nomme (BLYP) (Becke 1988). Dans la pratique, les
fonctionnelles GGA donnent de meilleurs résultats que les fonctionnelles LDA mais ne
conduisent pas à une description précise de toutes les propriétés des molécules. C’est
notamment le cas des spectres UV/visible pour lesquels les énergies des transitions
électronique restent sous-estimées. Il existe également des fonctionnelles meta-GGA (Tao et
al. 2003), où la fonctionnelle d’échange-corrélation dépend, en plus de la densité et de son
gradient, de la dérivée seconde de la densité électronique. Des exemples de ce type de meta-
GGA sont celles élaborées par van Voohris et Scuseria : la fonctionnelle VSXC (Voorhis and
Scuseria 1998), celle de Handy (Boese and Handy 2002) et de TPSS (Tao et al. 2003). Cette
dernière fonctionnelle donne de très bons résultats pour décrire la liaison hydrogène mais ne
décrit pas correctement les angles de torsion des molécules présentant des systèmes 𝜋
conjugués (Sancho-Garcıa and Cornil 2004).
1.4.3 c) La connexion adiabatique et les fonctionnels hybrides
Depuis les années 90, une nouvelle approche est apparue, fournissant des énergies, des
structures et des propriétés moléculaires en meilleur accord avec l’expérience que les LDA et
les GGA. Cette approche combine les traitements Hartree-Fock et ceux de la DFT sur les
effets d’échange et de corrélation aux travers des fonctionnelles hybrides. La connexion
adiabatique est un changement qui convertit un système de référence « sans interaction » en

33
un système avec interaction. On peut montrer que l’énergie d’échange et de corrélation peut
être déterminée comme :
𝐸𝑋𝐶 = ∫ ⟨𝜓(𝜆)|𝑉𝑋𝐶(𝜆)|𝜓(𝜆)⟩1
0
𝑑𝜆 (1.64)
Où 𝜆 décrit l’ampleur de l’interaction électronique, la force de couplage électronique qui varie
entre deux cas limites. Lorsque 𝜆 = 0 , l’équation correspond à la valeur de l’énergie
d’échange 𝐻𝐹 du système sans aucune interaction entre les électrons mais, mais calculée avec
les orbitales de Kohn-Sham (nommée souvent l’échange « exact »). Il n’y a pas, par
conséquent, d’énergie de corrélation. Pour 𝜆 = 1, nous avons un système réel en interaction
complète. La totalité de l’échange-corrélation est décrite par une fonctionnelle DFT.
L’intégration revient à introduire une part de l’échange exacte dans la fonctionnelle de
l’énergie afin de remédier au défaut de la correction GGA du modèle du gaz uniforme
d’électrons. L’intégration entre les deux systèmes limites se passe à densité constante et à
configuration électronique fixe, ce qui est à l’origine du terme « connexion adiabatique ».
L’équation (1.64) représente le fondement des fonctionnelles dites hybrides de type ACM1
utilisent un seul paramètre pour corriger la GGA
𝐸𝑋𝐶𝐴𝐶𝑀1 = (1 − 𝜆𝑖)𝐸𝑋
𝐷𝐹𝑇 − 𝜆𝑖𝐸𝑋𝐻𝐹 + 𝐸𝐶
𝐷𝐹𝑇 (1.65)
Le paramètre 𝜆𝑖 est souvent semi-empirique : il est ajusté par les auteurs des différents
fonctionnels hybrides pour que ces dernières s’accordent au mieux avec l’expérience. On peut
citer comme exemple B1PW91, B1LYP, PBE0 (Ernzerhof and Scuseria 1999), le pourcentage
d’échange exact (HF) dans la méthode PBE0 est de 25% (Adamo and Barone 1999) et ce
paramètre de mélange entre les parties DFT et HF est fixé uniquement par des considérations
théorique : on parle l’ACM0
𝐸𝑋𝐶𝑃𝐵𝐸0 =
3
4𝐸𝑋𝑃𝐵𝐸 +
1
4𝐸𝑋𝐻𝐹 + 𝐸𝐶
𝑃𝐵𝐸 (1.66)
Pour les hybrides ACM3, les corrections GGA sur l’échange et sur la corrélation de
fonctionnelles LDA apparaissent clairement. Ainsi pour déterminer la valeur de l’énergie
d’échange-corrélation, on ajoute une correction GGA (𝜆2 et 𝜆3) aux énergies d’échange et de

34
corrélation LDA et un certain pourcentage d’énergie d’échange exacte (𝜆1), les paramètres
empiriques, 𝜆𝑖, étant choisis pour optimiser la méthode :
𝐸𝑋𝐶 = 𝐸𝑋𝐶𝐿𝐷𝐴 + 𝜆1(𝐸𝑋
𝐻𝐹 − 𝐸𝑋𝐿𝐷𝐴) + 𝜆2∆𝐸𝑋
𝐺𝐺𝐴 + 𝜆3∆𝐸𝐶𝐺𝐺𝐴 (1.67)
en 1993, Becke (Becke 1993) a été le premier à inclure trois paramètres et à définir une
fonctionnelle du type ACM3. Il a utilisé sa fonctionnelle GGA de 1988 pour la partie
d’échange et la fonctionnelle de Perdew-Wang (Perdew and Wang 1992) pour la partie de
corrélation. Les valeurs de 𝜆1,𝜆2 et 𝜆3 valent respectivement 0.20, 0.72 et 0.81. Ces
paramètres sont tirés de résultats expérimentaux (chaleur d’atomisation, électroaffinité,
potentiel d’ionisation,…) et optimisés par la méthode des moindres carrés. La fonctionnelle la
plus utilisé actuellement est une autre ACM3 nommée B3LYP où la fonctionnelle de Perdew-
Wang, introduite dans l’ACM3 de Becke en 1993, est remplacée par la fonctionnelle de Lee-
Yang-Parr pour la partie de corrélation. Généralement, le paramètre essentiel reste celui qui
indique le pourcentage d’échange exact HF dans la fonctionnelle, c’est-à-dire𝜆1 .
L’expression de son énergie est la suivante
𝐸𝑋𝐶𝐵3𝐿𝑌𝑃 = (1 − 𝜆1 )𝐸𝑋
𝐿𝑆𝐷𝐴 + 𝜆1 𝐸𝑋𝐻𝐹 + 𝜆2∆𝐸𝑋
𝐵88 + (1 − 𝜆3)𝐸𝐶𝐿𝑆𝐷𝐴 + 𝜆3𝐸𝐶
𝐿𝑌𝑃 (1.68)
Un des problèmes fondamentaux de la DFT, est le traitement des interactions dispersives.
Malgré les améliorations apportées par les méthodes hybrides, ces interactions sont parfois
mal décrites. De nombreux papiers ont été publiés récemment pour tester des nouvelles
fonctionnelles, nous invitons le lecteur à les consulter (Jacquemin et al. 2008; Wiggins et al.
2009; Caricato et al. 2010).
1.5 Principe de la Mécanique Moléculaire
Les différentes méthodes de chimie quantique présentées précédemment, permettent une
description précise du système, et plus particulièrement de la fonction d’onde électronique. Le
cout en temps de calcul et en ressources informatique peut devenir très important même pour
des systèmes comportant un nombre d’atome très faible. Ce cout est une conséquence de la

35
résolution approchée de l’équation de Schrödinger afin de calculer l’énergie électronique pour
une configuration nucléaire donné.
Les méthodes de mécanique moléculaire est une étape importante vers le traitement complet
de système moléculaire de quelques milliers voire de millions d’atomes. Elle consiste à
modéliser la réalité moléculaire, c’est-à-dire à mettre en évidence grâce à des résultats
expérimentaux et à des calculs quantique, les grandes propriétés des atomes et des molécules
et à en déduire des modèles mathématique simples.
Ces méthodes sont basées sur l’approximation de Born-Oppenheimer et l’énergie électronique
est considérée comme une fonction paramétrique des coordonnées nucléaires, le mouvement
des électrons peut être dissocié de celui des noyaux et les atomes sont représentés par des
masses ponctuelles chargées. De plus, la distance intermoléculaire d’un système est très
supérieure à la longueur d’onde de De Broglie ce qui permet de négliger les effets quantiques
et de ne pas passer par la résolution de l’équation de Schrödinger de l’Hamiltonien
électronique.
Les paramètres de ces fonctions, sont déterminés à partir de données expérimentales ou par
des calculs ab initio de haut niveau et ajustés sur un jeu de molécules de référence. Les
molécules sont donc décrites comme des ensembles d’atomes définis par leurs paramètres.
Ces derniers interagissent entre eux par des potentiels prédéfinis suivant les lois de la
mécanique newtonienne. L’ensemble des interactions est appelé champ de forces. Il définit
une surface de potentiel modèle sur laquelle se déplacent les atomes. Les champs de forces
utilisent une fonction paramétrée, correspondant à la fonction énergie potentielle du système,
dépendant explicitement des coordonnées nucléaires. La mécanique moléculaire ne permet
pas l’étude de la formation ou de la rupture de liaisons covalentes entre les atomes, ni même
d’obtenir des informations sur des transferts d’électrons entre des atomes ou des états
électroniques excites des atomes.
L'application de cet ensemble de lois à d'autres molécules qui ressemblent plus ou moins aux
molécules de référence repose sur le principe souvent vérifiée de transférabilité des propriétés
chimiques. Par exemple la liaison carbone-hydrogène, a une longueur similaire dans toutes les
molécules organique. On peut définir un nombre restreint de paramètres sur des molécules ou
des systèmes modèles qu’on utilisera ensuite pour construire le champ de force en question. Il
est évidant que de nombreux modèles de champs de forces ont été définis en fonction des
types de systèmes étudiés. Il existe deux grandes catégories des champs de force; classique et

36
polarisable. La première catégorie est actuellement la plus employée et la plus courante de par
leur nombre mais aussi par leurs facilités à être utilisée. Les champs de force polarisable
essaient de prendre en compte la réponse électronique de l’environnement à l’aide de modèle
complexes. Les champs de force classique sont suffisants tant que la polarisation n’a pas de
contribution importante. Par contre, lorsque les effets d’induction ne peuvent plus être
négligés au long de la simulation, il est nécessaire de les prendre en compte de manière
correcte. L’importance des champs de force polarisable a été démontrée par plusieurs études
sur les métalloprotéines, la formation du cristal, et le repliement de protéines.
1.5.1 Formalisme de l´énergie potentielle
La fonction d’énergie potentielle caractérisant les champs de forces, a pour rôle de reproduire
le plus fidèlement possible toutes les itérations intermoléculaires et intramoléculaires
présentes dans le système. Il existe dans la littérature de nombreux champs de force
empiriques permettant de modéliser des systèmes de nature différente. Un certain nombre
d’entre eux ont été développés spécifiquement pour les systèmes biologiques : CHARMM
(Brooks et al. 1983; MacKerell et al. 1995) AMBER (Cornell et al. 1995), GROMOS
(Hermans et al. 1984; Ott and Meyer 1996), OPLS-AA (Kaminski et al. 2004).
L’écriture et la paramétrisation de ces champs de force diffèrent, mais ils possèdent tous des
termes de valence et des termes d’interaction non liés auxquelles chaque atome est soumis.
Ces fonctions s’expriment généralement comme la somme de potentiels de liaisons, d’angles
de valence, de dièdre, d’interaction électrostatique et de Van Der Waals (propriété d’additivité
des potentiels d’interaction).
La fonction d’énergie potentielle peut être décomposée ainsi :
𝑉𝑀𝑀(𝑟𝑁) =∑𝑉𝑏𝑏
+∑𝑉𝑎𝑎
+∑𝑉𝑑𝑑
+∑∑(𝑉𝑖𝑗𝑐 + 𝑉𝑖𝑗
𝑉𝐷𝑊)
𝑗>𝑖𝑖
(1.69)

37
1.5.2 Les interactions de valence
𝑉𝑀𝑀(𝑟𝑁) désigne l’énergie potentielle totale dépendant de la position (𝑟) des (𝑁) atomes
présents dans le système. Les termes de liaison 𝑉𝑏 et d’angle 𝑉𝑎 sont habituellement
modélisés comme des oscillateurs harmoniques ne permettant pas la formation ou la
destruction de liaisons. Dans les champs de force usuels, les potentiels de liaisons peuvent
prendre différents formes : par exemple le potentiel de Morse qui se rapproche le plus du
comportement de la liaison chimique en particulier, sa valeur pour de longues distance permet
la dissociation moléculaire, cependant le cout en temps de calculs de l’évaluation des
exponentielles est important. Le potentiel anharmonique et utilisé pour obtenir une meilleure
précision au-delà de la distance d’équilibre
𝑉𝑏 =1
2𝑘0(𝑟 − 𝑟0)
2 (1.70)
𝑉𝑏 = 𝐷𝑒{1 − 𝑒𝑥𝑝[−𝐴(𝑟 − 𝑟0)]}2 (1.71)
𝑉𝑏 =1
2𝑘2(𝑟 − 𝑟2)
2 +1
3𝑘3(𝑟 − 𝑟3)
3 +1
4𝑘4(𝑟 − 𝑟4)
4 (1.72)
Le potentiel décrivant les angles de valence est généralement représenté par le potentiel
𝑉𝑎 =1
2𝑘𝜃(𝜃 − 𝜃0)
2 (1.73)
La déviation par rapport à la position d’équilibre de liaison et d’angle est exprimée
respectivement par (𝑟 − 𝑟0), (𝜃 − 𝜃0). Les potentiels harmoniques de liaison et de l’angle est
caractérisé respectivement par les constante de force 𝑘𝑟 en 𝐾𝑐𝑎𝑙 ∙ 𝑀𝑜𝑙−1 ∙ Å−2et 𝑘𝜃 en
𝐾𝑐𝑎𝑙 ∙ 𝑀𝑜𝑙−1 Le potentiel de torsion correspondant aux dièdres 𝑉𝑑 est développé en série de
Fourier ce qui permet de reproduire le caractère périodique de ce potentiel. Les valeurs de 𝑉𝑛
permettent de reproduire le profil de torsion et ils sont exprimé en∙ 𝑀𝑜𝑙−1 , 𝑛 est le terme de
multiplicité (nombre de minima entre 0 et 3600, 𝜙 est l’angle de torsion et 𝛾 est le facteur de
phase (en degré).

38
𝑉𝑑 =∑𝑉𝑛2
𝑛
[1 + 𝑐𝑜𝑠(𝑛𝜙 − 𝛾)] (1.74)
Les dièdres impropres maintiennent une certaine disposition spatiale d’un groupe de quatre
atomes qui ne se suivent pas séquentiellement. C’est le cas par exemple de la liaison
peptidique et des cycles aromatiques dont la planéité est protégée par un angle impropre. Les
dièdres impropres empêchent également les inversions de configuration des centres
énantiomériques tels que les 𝐶 des acides aminés (à l’exception de la glycine) qui sont tous
en configuration absolue L. Dans la plupart des champs de forces, le potentiel des angles
dièdres impropres 𝑉𝑖𝑚𝑝𝑟𝑜𝑝𝑟𝑒𝑠 de constante de raideur 𝑘𝑖 et aussi considéré où (𝑑) représente
la distance entre le plan défini par les trois premières atomes et le dernier.
𝑉𝑖𝑚𝑝𝑟𝑜𝑝𝑟𝑒𝑠 = ∑1
2𝑖𝑚𝑝𝑟𝑜𝑝𝑟𝑒𝑠
𝑘𝑖𝑑2 (1.75)
L’énergie associée à la déformation des angles dièdres impropres 𝜑 (en degré) peut aussi être
décrite par un potentiel harmonique où 𝜑0est l’angle dièdre de référence. Grâce à une
constante de force 𝑘𝜑 en 𝐾𝑐𝑎𝑙 ∙ 𝑀𝑜𝑙−1 ∙ Å−2 élevée, ce terme permet de fixer la configuration
et empêcher la pyramidalisation d’un atome hybridé𝑠𝑝2.
𝑉𝑖𝑚𝑝𝑟𝑜𝑝𝑟𝑒𝑠 = ∑ 𝑘𝜑(𝜑 − 𝜑0)2
𝑖𝑚𝑝𝑟𝑜𝑝𝑟𝑒𝑠
(1.76)
Dans le cas particulier du champ de force CHARMM , l’énergie potentielle d’un système
moléculaire en interaction contient plusieurs composantes qui modélisent les forces intra et
intermoléculaires tel que Les termes d’interaction 1-3 d’Urey-Bradley qui ajoutent une
contrainte de distance entre deux atomes espacés de deux liaisons covalentes. L’expression
de ce terme dans le champ de forces CHARMM prend la forme suivante
𝑉𝑈𝐵 =∑𝑘(− 0)2
𝑈𝐵
(1.77)

39
1.5.3 Interaction des atomes non-liés
1.5.3 1 Les différentes expressions des interactions de van der Waals
Le terme de van der-Waals qui s’exprime le plus souvent par un potentiel de type Lennard-
Jones (dit potentiel 6-12) est introduit pour tenir compte des interactions à courte distance. Il
se compose d’un terme qui représente les forces d’échange-répulsion, en 𝑟𝑖𝑗−12 qui répond au
principe d’exclusion de Pauli et qui interdit aux électrons d’occuper la même région d’espace
et d’un terme attractif en 𝑟𝑖𝑗−6 qui rend compte de la dispersion ou force de London
(interaction dipôle-induit dipôle-induit).
Les interactions de van der Waals n’agissent qu’à très courte distance. Le potentiel de
Lennard-Jones peut être formulé de façon plus explicite en fonction de l’énergie d’interaction
𝜖𝑖𝑗 au minimum du puits de potentiel et de la distance 𝑟𝑚𝑖𝑛 correspondant à cette énergie.
𝑉𝑖𝑗𝑉𝐷𝑊 = 4𝜖𝑖𝑗 [(
𝜎𝑖𝑗
𝑟𝑖𝑗)
12
− (𝜎𝑖𝑗
𝑟𝑖𝑗)
6
] (1.78)
Le potentiel 12-6 de Lennard-Jones caractérisé par une partie attractive variant en 𝑟𝑖𝑗−6 et une
partie répulsive en 𝑟𝑖𝑗−12 contient deux paramètres : 𝜎𝑖𝑗 et 𝜖𝑖𝑗 représentant respectivement la
distance pour laquelle l’énergie du potentiel est nulle et la profondeur du puits, et caractérisant
les interactions entre les atomes (𝑖) et (𝑗). Ces paramètres sont calculés par la loi de de
Lorentz-Berthelot.
𝜖𝑖𝑗 = (𝜖𝑖𝑖𝜖𝑗𝑗)1 2⁄ (1.79)
𝜎𝑖𝑗 =1
2(𝜎𝑖𝑖 + 𝜎𝑗𝑗) (1.80)
Les potentiels proposés par Buckingham et collaborateurs (Buckingham et al. 1941) sont des
potentiel intermédiaires entre les potentiels de morse et Lennard-Jones, ils s’écrivent sous la
forme d’une exponentielle pour le terme répulsif et d’une fonction en 𝑟−6 pour le terme
attractif

40
𝑉𝑖𝑗 = 𝐴𝑒𝑥𝑝 (−𝑟𝑖𝑗
𝜌) −
𝐶
𝑟𝑖𝑗6 (1.81)
Le potentiel de liaison hydrogène (12-10)
𝑉𝑖𝑗 = (𝐴
𝑟𝑖𝑗12) − (
𝐵
𝑟𝑖𝑗10) (1.82)
Le potentiel (n-m)
𝑉𝑖𝑗 =𝐸0
𝑛 −𝑚[𝑚(
𝑟0𝑟𝑖𝑗)
𝑛
− 𝑛(𝑟0𝑟𝑖𝑗)
𝑚
] (1.83)
1.5.3.2 Le potentiel électrostatique
Les charges ponctuelles interagissent entre elles par un potentiel de coulomb usuel
𝑉𝑖𝑗𝐶 =
𝑞𝑖𝑞𝑗
4𝜋𝜖0𝜖𝑟𝑟𝑖𝑗 (1.84)
Où 𝜖0 est la primitivité du vide, 𝜖𝑟 la constante diélectrique du milieu considéré, et 𝑞𝑖 , 𝑞𝑗 les
charges ponctuelles sur les atomes 𝑖 et 𝑗 séparés par le distance 𝑟𝑖𝑗 .
Les méthodes de mécanique moléculaire étant par définition paramétrique, et il est évidant
que de nombreux modèles de champs de Force ont été définis en fonction des systèmes
étudiés. Dans cette étude nous avons utilisés les champs de force CHARMM27 et OPLSAA
dans les calculs LSCF/MM et le champ de force GROMOS96 pour des calculs de dynamique
moléculaire.

41
1.6 La dynamique moléculaire
Résoudre une structure consiste à déterminer les coordonnées tridimensionnelles des atomes
de la molécule d’intérêt. L’image statique des structures suggérée par la mécanique
moléculaire doit être abandonnée au profit d’une représentation dynamique des molécules où
les atomes sont en mouvement. A l’inverse de La mécanique moléculaire qui permet d’obtenir
des informations à partir d’une géométrie unique de la molécule, la dynamique moléculaire
génère une succession de conformations décrivant l’évolution du système au cours du temps.
Les fluctuations structurales, issues de la superposition de mouvements, de vitesse et
d’amplitude très différentes et qui résultent des diverses forces auxquelles les atomes sont
soumis au cours du temps, contribuent à l’activité biologique des protéines et influent sur leur
capacité à lier un substrat, une protéine partenaire ou un cofacteur. Les mouvements internes
créent une variation du volume du site de liaison du ligand et de la disposition des
groupements impliqués dans sa liaison. Ces mouvements modulent ainsi les interactions
(électrostatiques, hydrophobes et liaisons hydrogène) au sein du site de liaison de manière à
rendre la protéine plus flexible. L’exemple le plus connus est celui de l’aptitude de la
myoglobine à lier O2 et CO (Plattner and Meuwly 2008). Dans cette protéine, la réorientation
de trois chaines latérales par un mouvement de rotation locale permet au substrat de
s’échapper de la protéine (Kurplus and McCammon 1983). La mécanique et la dynamique
moléculaires partagent la même fonction d’énergie potentielle pour décrire l’ensemble des
interactions interatomiques du système étudié. Différentes techniques expérimentales
(spectroscopies de fluorescence, de RX, neutrons, RMN…etc) permettent de sonder la
dynamique des protéines sur des échelles de temps allant du femto second à la microseconde.
Pour la gamme entre la femtoseconde et la nanoseconde, une comparaison est alors possible
avec la dynamique moléculaire.
1.6.1 Dynamique moléculaire : Techniques numériques.
1.6.1.1 Intégration du temps.
Les lois définit par Newton qui permettent de dériver les trajectoires de particules classiques
soumises à des forces extérieures, sont les même utilisées lors de simulations de dynamique
moléculaire.

42
𝑚𝑖
𝑑2𝑟𝑖(𝑡)
𝑑𝑡2= 𝑓𝑖(𝑡) (1.85)
𝑓𝑖(𝑡) = −𝜕
𝜕𝑟𝑖(𝑡)𝑉(𝑟𝑁) (1.86)
(𝑚) est la masse, (𝑟) est la position, (𝑓) est la résultante des forces et (𝑉) la fonction énergie
potentielle définissant les interactions entre les particules. Dans la pratique les équations sont
intégrées pour définir la trajectoire de chaque particule. La définition des conditions dans
lesquelles cette intégration va se dérouler est nécessaire, il convient donc de définir
l’ensemble statistique de la simulation. On peut citer :
–l’ensemble microcanonique (NVE) avec le nombre de particules, le volume et l’énergie
maintenus constants
–l’ensemble canonique (NVT) avec le nombre de particules, le volume et la température
maintenus constants
–l’ensemble Isotherme-Isobare (NPT) avec le nombre de particules, la pression et la
température maintenus constants.
–l’ensemble Grand-canonique (TV𝜇) avec la température, le volume et le potentiel chimique
maintenus constants.
1.6.1.2 Durée du pas de temps
Les équations du mouvement sont résolues numériquement et propagées grâce à des
algorithmes relativement rapides à mettre en œuvre. Néanmoins, ils imposent une restriction
sévère sur la valeur du pas d’intégration. Un pas de temps (∆𝑡) trop grand conduit à des
erreurs significatives dans l’évaluation de l’énergie et peut faire diverger la simulation. Les
fréquences de vibrations les plus rapides doivent être tenues en compte, de tel sorte que (∆𝑡)
soit 10 à 20 fois plus petit que la période de l’oscillation la plus rapide. En pratique les
liaisons 𝐶 − 𝐻 ont une période de vibration très rapide de l’ordre de (10 fs). La valeur
maximale du pas (∆𝑡) doit être le dixième de cette période, soit (1 fs). L’algorithme Shake

43
(Ryckaert et al. 1977) permet de geler certains mouvements à haute fréquence et ainsi
d’augmenter le pas d’intégration jusqu’à (2 fs).
1.6.1.3 Conditions initiales
Pour initier la simulation d’un système, l’algorithme d’intégration nécessite les coordonnées
(𝑥, 𝑦, 𝑧) et les vitesses (𝑣𝑥, 𝑣𝑦, 𝑣𝑧) de tous les atomes au temps 𝑡 = 𝑡0. Les positions
atomiques initiales 𝑟𝑖(𝑡 = 𝑡0) sont généralement issues de l’expérience (à partir d’une
structure RMN ou de cristallographie de rayons X). La seule information concernant les
vitesses initiales est la température 𝑇 de la simulation. Si on ne dispose pas de vitesses
initiales, on attribue aléatoirement les vitesses 𝑣𝑖(𝑡 = 𝑡0) à tous les atomes du système selon
une distribution de Maxwell-Boltzman centrée centrée sur la température T qui permet le
calcul des densités de probabilité 𝑓(𝑣𝑖) :
𝑓(𝑣𝑖) = √𝑚𝑖
2𝜋𝑘𝐵𝑇𝑒𝑥𝑝 (−
𝑚𝑖𝑣𝑖2
2𝑘𝐵𝑇) (1.87)
et pour chaque composante des 𝑣𝑖 :
𝑓(𝑣𝑖𝑥) = √𝑚𝑖
2𝜋𝑘𝐵𝑇𝑒𝑥𝑝 (−
𝑚𝑖𝑣𝑖𝑥2
2𝑘𝐵𝑇) (1.88)
La distribution initiale des vitesses n’est pas équilibrée. Une période d’équilibration du
système est nécessaire afin de stabiliser la simulation et d’éviter des zones ≪chaudes≫ ou
≪froides≫ dans le système.
1.6.1.4 Conditions périodiques aux limites
Lorsque l’on simule un système en solvatation explicite dans une boite, l’environnement des
atomes périphériques est déformé du fait des effets de bord comparé aux atomes situés au
centre de la boite. Pour s’en affranchir, On recourt alors aux conditions périodiques aux

44
limites PBC (periodic boundary conditions) qui consistent à répliquer un ensemble fini et
relativement restreint de particules dans une boite centrale dans les trois directions de
l’espace, de telle sorte que les forces agissant sur les particules soient équivalentes à celles
d’un système beaucoup plus grand et donc plus réaliste. La forme de la boite devient donc
importante puisque il est indispensable de pouvoir paver tout l’espace.la forme
parallélépipédique est la plus choisis, mais il est possible de trouver d’autre formes telles que
des octaèdres tronqués, et des prismes hexagonaux…etc.
Figure 1: Représentation bidimensionnelle d’une boite de simulation, avec de conditions périodiques aux
limites. Lorsqu’une molécule quitte la boite centrale (a), les images dans les cellules fantômes voisine se
déplacent de manière analogue.
Lors de la dynamique, il est possible que certaines particules quittent la boite initiale. Les
PBC permettent de conserver le nombre de particules constant en faisant entrer dans la boite
une réplique de chaque particule. Le calcul des interactions non liées devient alors
problématique puisqu’il existe un nombre infini de répliques. La convention couramment
utilisée pour limiter le nombre de calculs est l’approximation de l’image minimale dans
laquelle, chaque particule ressent la présence d’une seule réplique de chaque autre particule.
Les contraintes thermodynamiques définissent l’état macroscopique et permettent d’imposer
des valeurs des observables thermodynamiques. Le calcul de pression peut être problématique
à basse température donc la première partie de l’équilibration qui consiste à chauffer le
système est effectuée dans l’ensemble canonique (NVT). Le volume reste alors constant et la
température est réglée par un thermostat. La deuxième partie de l’équilibration ainsi que les
simulations longues sont effectuées dans l’ensemble isotherme-isobare (NPT). La valeur
moyenne de la pression ainsi que la température sont imposées par un barostat et un
thermostat, respectivement, pour reproduire les conditions de référence, e.g. des conditions
expérimentales.

45
Dans cette étude nous avons appliqué des conditions aux limites spatiales et
thermodynamiques. La forme, la taille ainsi que la périodicité du système sont imposé par les
contraintes spatiales. Une forme rectangulaire est imposée au système et les molécules d’eaux
sont ajoutées pour former la boite rectangulaire avec une dimension adaptée à la distance
limite des interactions à longue distance. Afin d’éviter les effets de taille finie et permettre
l’utilisation de la méthode PME (Particle Mesh Ewald), une périodicité infinie du système est
également imposée dans les simulations NPT.
1.6.5 Traitement des interactions électrostatiques en dynamique moléculaire
Pour la plupart des systèmes non covalents, l’interaction électrostatique est l’interaction qui
influence le plus la détermination de leur géométrie (Dykstra 1993). Une des sources
d’erreurs possible dans les champs de forces utilisés en dynamique moléculaire classique est
un traitement incorrect des interactions électrostatiques. Cela peut être dû à un mauvais choix
de distance de cut-off (Robertson et al. 2008), et/ou au modèle employé pour calculer les
interactions électrostatiques. Ainsi la plupart des champs de forces (Pearlman et al. 1995)
couramment utilisés dans des simulations de dynamique moléculaire classique et qui
permettent de modéliser un système à proximité d’une conformation locale, comme c’est le
cas pour les simulations de protéines autour de la conformation obtenue par cristallographie et
évaluent l’interaction électrostatique de manière classique à partir de multipoles. Cependant, il
a été montré que pour certains systèmes (Hobza et al. 1997; Sagui and Darden 1999) la cause
des prévisions incorrectes des simulations de dynamique moléculaire classique est l’utilisation
d’un modèle multipolaire insatisfaisant.

46

47
Chapitre 2
Les Méthodes Hybrides QM-MM

48
2 1 Les méthodes hybrides QM-MM
2.1.1 Introduction
La contribution fondatrice par Warshel et Levitt de 1976 (Warshel and Levitt 1976) marque le
début de l'ère QM/MM. Ils ont présenté le concept QM/MM, une méthode avec beaucoup de
caractéristiques que l'on considère maintenant l'élément essentiel dans le domaine de chimie
théorique.
L'approche hybride QM/MM a trouvé l'acceptation répandue seulement beaucoup plus tard,
dans les années 1990, commençant par le rapport de Field, Bash, et Karplus en 1990 (Field et
al. 1990) qui a décrit en détail le couplage des méthodes semi-empiriques au champ de force
CHARMM en évaluant soigneusement l’exactitude et l’efficacité du traitement QM/MM
contre les résultat fournis par les méthodes ab initio et les données expérimentales.
Au cours des années passée de nombreux travaux ont documentés le développement de
l'approche QM/MM ainsi que son aspect méthodologique et son application aux problèmes
biomoléculaire (Ruiz-Lopez and Rivail 1998; Lyne and Walsh 2001; Rivail 2004). L'approche
QM/MM est à ce jour établie comme un outil de valeur pour examiner non seulement des
réactions chimiques dans des phases condensées (Gao et al. 2006; Senn and Thiel 2007)
comme l’étude de la réactivité chimique dans des grands systèmes, comme les enzymes mais
aussi pour l’investigation des systèmes inorganique et organométallique (Maseras 1999;
Magistrato et al. 2002; Chatwin et al. 2006), des systemes à d’état solide (Gao 1996; Sierka
and Sauer 2005) ainsi que des processus chimique dans des solvants explicites (Rode et al.
2005).
2.2 Partitionnement QM-MM.
L'idée de base des méthodes hybride QM-MM commence par la réalisation que certaines
régions du système jouent souvent des rôles très différents dans le processus étudié. Nous
prenons comme exemple les réactions enzymatiques où les liaisons sont rompues ou formés
seulement au niveau du site actif et l’effet de l’environnement de la protéine est généralement
pris comme stérique ou électrostatique.
Avec des méthodes hybrides, chaque région est traitée avec une approche théorique
différente, Il est souvent évidant que des méthodes purement quantique, couteuses en

49
ressources informatiques et en temps sont exigées à la partie du système où l'action a lieu
tandis que pour la partie restante du système, jouant un rôle mineur et/ou secondaire sur le
phénomène étudié, peut alors être modélisée par une méthode de moindre coût, comme la
mécanique moléculaire qui autorise la prise en compte d'un nombre d'atomes important.
La représentation schématique des sous-systèmes dans l’approche QM-MM (Figure 1),
montre qu’un système macromoléculaire (𝕊) est fractionné en deux région afin d’unir les
avantages des méthodes calculatoires qui leurs sont associer.
La région intérieure (inner region 𝕀) qui présente la partie d’intérêt est traitée à un niveau
mécanique quantique QM. Tandis que la seconde partie décrivant son environnement est
appelée région extérieur ou (outer region 𝕆) et modélisé à l’aide des champs de force de
mécanique moléculaire MM.
On a utilisé le tiret entre les deux sous-systèmes (QM-MM) pour présenter une situation
générale. On notera le couplage QM/MM, si la jonction entre les deux sous-systèmes
s’effectue par l’intermédiaire d’une liaison covalente, la partition est noté QM:MM lorsque
elle correspond à un système solvant-soluté. Ce dernier est relativement simples car les deux
sous systèmes sont bien définis et interagissent exclusivement par des interactions non-liées
(électrostatique et van der Waals).
Figure 1 : Partitionnement du système (𝕊) en un sous-système intérieur (inner 𝕀) et un sous-système extérieur
(outer 𝕆 ). L’anneau gris autour du sous-système intérieur représente la région frontière.

50
2.3 L’expression de l’énergie potentielle QM-MM
Les systèmes QM-MM contiennent trois classes d'interactions :
Les interactions entre atomes dans la région QM et celles entres les atomes dans la région
MM sont relativement simple à décrire. Mais en raison de la forte interaction entre les atomes
des deux sous-systèmes l’énergie potentielle totale ne peut pas être décrite comme une simple
somme des énergies potentielles de chaque sous-système. Des précautions spéciales doivent
donc être prises en compte à la frontière, et des termes de couplage entre les atomes QM et
MM doivent être considéré. Pour cela plusieurs approches décrivant les méthodes QM-MM
ont été proposées et casées dans deux grandes catégories regroupant les schémas de couplages
soustractif et additif.
2.3.1 Le schéma de couplage QM-MM soustractif
Dans le schéma QM-MM soustractive, trois calculs indépendants sont réalisés :(i) Un calcul
MM sur le système entier (𝕊), (ii) un calcul QM sur le sous-système intérieur (𝕝), (iii) un
calcul MM sur le sous-système intérieur (𝕀). L'énergie totale du système est obtenue en
additionnant les deux premiers termes et soustrayant le dernier terme pour éviter le compte
double. De ce point de vue, l'expression d'énergie pour un schéma soustractif, peut être
considéré comme une correction ou une extrapolation. L’utilisation du concept des atomes
connectés ou (link atoms 𝕃) pour saturé la couche de valence des atomes quantique du sous-
système intérieur appelée également couche intérieur conduit à une expression d’énergie total
qui prend la forme suivante.
𝐸QM/MM(𝕊) = 𝐸MM(𝕊) + 𝐸QM(𝕀 + 𝕃) − 𝐸MM(𝕀 + 𝕃) (2.1)
Dans cette équation les indices indiquent le type de calcul effectué, ce qui est porté entre
parenthèses, représente la région d’intérêt du système
Cette méthode peut être considérée comme une méthode MM dans laquelle une région
d’intérêt a été s´éparée et traitée avec un niveau de théorie plus élevé. Dans l’équation (2.1),
aucun terme de couplage QM-MM n’apparait, ce qui simplifie fortement le modèle, évitant

51
toute modification des procédures habituelles en QM et en MM. L’étape de soustraction
corrige implicitement l’artefact dû à la présence du Link atomes.
En revanche le schéma soustractif demande un ensemble complet de paramètre MM pour le
sous-système intérieur qu’on peut appeler aussi centre réactionnel. De plus le couplage entre
les deux sous-systèmes est traité entièrement au niveau MM on s'aperçoit que les interactions
entre le centre réactionnel et son environnement ne sont prises en compte qu'au bas niveau de
théorie. Particulièrement, si ce dernier est un champ de forces classique où les contributions
électrostatique sont traitées comme de simples charges ponctuelles rigides, la fonction d'onde
du centre réactionnelle n'est pas polarisée par l'environnement seul son effet stérique et
électrostatiques évaluées au bas niveau de théorie sont prises en compte. En revanche, si les
effets électroniques sont plus importants que les effets stériques, dans ce cas il faut utiliser
soit une méthode quantique à bas niveau de calcul (QM’), soit utiliser une approche QM-MM
incluant la polarisation de la partie quantique.
Le schéma soustractif donne naissance aux approches ONIOM (our n-layered integrated
molecular orbital and molecular mechanics) développées par Morokuma et collaborateurs
(Svensson et al. 1996),qui reposent à la fois sur une partition géométrique du système et sur
une hiérarchie des niveaux théoriques utilisés, que ce soit ab initio, semi-empirique ou bien
encore mécanique moléculaires.
Le principe du calcul ONIOM peut être regroupé en deux catégories :
La méthodes IMOMO (Integrated Molecular Orbitals Molecular Orbitals) (Humbel et al.
1996) consistant à coupler deux méthodes quantiques :(QM/QM’ ou QM:QM’) typiquement,
un haut niveau ab initio et un bas niveau semi-empirique. Dans la seconde, IMOMM :
(Integrated Molecular Orbitals Molecular Mechanics) (Maseras and Morokuma 1995), le bas
niveau est traité par la mécanique moléculaire (QM/MM ou QM:MM).
Ces méthodes ont été appliquées à différentes études: réactions organiques (Svensson et al.
1996), réactions organométalliques (Matsubara et al. 1996), modélisation des surfaces
(Shoemaker and Gordon 1999). Woo, Cavallo et Ziegler ont également utilisé IMOMM pour
des simulations dynamiques moleculaire (Woo et al. 1998).

52
2.3.2 Le schéma de couplage QM-MM Additif
L’expression de l’énergie pour le schéma additif est donnée par
𝐸QM/MM(𝕊) = 𝐸MM(𝕆) + 𝐸QM(𝕀 + 𝕃) + 𝐸QM−MM(𝕀, 𝕆) (2.2)
Où le centre réactionnel (𝕀+𝕃) traité au niveau QM est incorporé dans un large sous-système
extérieur (𝕆) traité en MM. Contrairement au schéma soustractif, il y a l’apparition explicite
du terme de couplage 𝐸QM−MM(𝕀, 𝕆) qui représente un terme clef pour le traitement des
interactions entre les deux sous-systèmes (𝕀-𝕆) interagissant fortement entre eux.
Actuellement la majorité des méthodologies de calculs QM-MM adopte le schéma additif et
prend en termes d’Hamiltonien la forme suivante
�� = ��𝑀𝑀 + ��𝑄𝑀 + ��𝑄𝑀−𝑀𝑀 (2.3)
Où ��𝑀𝑀 et ��𝑄𝑀 sont respectivement les Hamiltonien de la partie MM et QM.
L’Hamiltonien ��𝑄𝑀 peut-être séparé en deux parties correspondant à la partie électronique et
nucléaire.
��𝑄𝑀 = ��𝑒𝑙𝑒𝑐𝑄𝑀 + ��𝑛𝑢𝑐𝑙
𝑄𝑀 (2.4)
Avec
��𝑒𝑙𝑒𝑐𝑄𝑀 = −∑
𝛻𝑖2
2
𝑁
𝑖
−∑∑𝑍𝐴𝑟𝑖𝐴
𝑀
𝐴
𝑁
𝑖
+∑∑1
𝑟𝑖𝑗
𝑁
𝑖<𝑗
𝑁
𝑖
(2.5)
��𝑛𝑢𝑐𝑙𝑄𝑀 =∑∑
𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐴<𝐵
𝑀
𝐴
(2.6)

53
Les interactions entres les centres QM et MM peuvent être décomposé en concordance avec
celles considérés dans un champ de force et qu’on peut écrire de manière générale sous la
forme :
𝐸𝑄𝑀−𝑀𝑀(𝕀, 𝕆) = 𝐸𝑄𝑀−𝑀𝑀𝑒𝑙𝑒𝑐 + 𝐸𝑄𝑀−𝑀𝑀
𝑉𝐷𝑊 + 𝐸𝑄𝑀−𝑀𝑀𝑏𝑜𝑛𝑑 (2.7)
Les deux premier termes sont respectivement les potentiels d’interactions de type non liée:
électron QM-charges MM et de type van der Waals entre l’ensemble des atomes QM et MM,
pendant que le troisième terme représente le potentiel d’interactions liée: noyaux QM-charges
MM.
Il faut noter que pour les atomes de la partie QM, la connaissance des paramètres {휀𝐴, 𝜎𝐴}𝐴𝜖𝑄𝑀
lors du calcul du potentiel d’interaction de van der Waals est nécessaire. Habituellement, les
paramètres du champ de force MM considéré, sont utilisées pour les atomes de la partie QM.
Cependant ceci n’est pas toujours possible et une reparamétrisation des atomes est
quelquefois indispensable (Ferré et al. 2004).
2.4 Effet électrostatique de l’environnement
La contribution électrostatique de l’environnement peut être considérée de trois manières
différentes, elle influencera donc différemment la fonction d’onde de la partie d’intérêt QM.
Ces trois modèles sont expliquées ci-dessous.
2.4.1 Le modèle Mechanical Embedding (ME)
Dans ce premier modèle, les interactions électrostatiques QM-MM sont calculées au bas
niveau de théorie, cela veut dire qu’ils sont traités comme des interactions électrostatiques
MM-MM. La disposions d’un jeu de charges classique pour les atomes quantique est alors
indispensable. Cet obstacle est résolu par l’utilisation des charges natives du champ de force
pour les atomes de la partie QM. Ceci n’est pas toujours possible car un jeu de paramètres
MM complet n’est pas toujours disponible. La limitation la plus sérieuse et la plus reconnue
de la méthode ME est que les charges de l’environnement MM ne polarisent pas la fonction
d’onde de la partie quantique c’est-à-dire que la contribution électrostatique de

54
l’environnement ne modifie absolument pas le Fockien de la partie QM. Il faut donc être
vigilant que les propriétés étudiées ne soient pas influencée par son environnement. Ceci est
particulièrement important lorsque le phénomène chimique étudié engendre un transfert de
charge c’est à dire : calculs d’affinité électronique ou de spectre d’absorption. De plus, cette
polarisation de la fonction d’onde peut, dans certain cas, modifier l’ordre énergétique d’un
ensemble d’états, comme par exemple dans le cas d’un calcul TD-DFT. La technique ME a
l’avantage d’être très simple à utiliser et à implanter. Sa méthodologie pour calculer les
interactions électrostatiques entre les sous-systèmes est utilisée dans la version originale de la
méthode IMOMM (Integrated Molecular Orbital/Molecular mechanics).
2.4.2 Le modèle Electrostatic Embedding (EE)
Le schéma EE (Electrostatic Embedding) ne requiert pas la connaissance des charges
ponctuelles de la partie QM car l’interaction électrostatique est calculée à haut niveau de la
théorie par la formule :
𝐸𝑄𝑀−𝑀𝑀𝑒𝑙𝑒𝑐 = ⟨𝛹|−∑∑
𝑞𝑀|𝑅𝑀 − 𝑟𝑖|
𝐿
𝑀𝜖𝕆
𝑁
𝑖𝜖𝕀
|𝛹⟩ (2.8)
Dans l’equation ci-dessus, les indices (i) et (M) sont respectivement pour les N électrons de la
partie QM et les (L) charges de la partie MM.
Lorsque cette interaction est ajoutée à l’Hamiltonien de cœur de la partie quantique, et donc
pris en compte lors du processus de convergence SCF, elle permet une polarisation directe de
la fonction d’onde quantique par les charges de son environnement traitée par la mécanique
moléculaire. Cependant, ce modèle surpolarise parfois la fonction d’onde lorsque les charges
ponctuelles de la partie MM sont très proches de la partie QM. C’est le cas lorsqu’une liaison
covalente lie les deux sous-systèmes.
Il est reproché au schéma EE de demander un effort au niveau du temps de calcul plus
important que dans le cas de ME. Cependant, ce surcout est souvent négligeable par rapport
au calcul QM. Ce schéma est maintenant le plus utilisé dans le cas de calculs QM /MM et
QM :MM, comme dans la nouvelle version d’IMOMM (Vreven et al. 2006) implantée dans

55
Gaussian03 dès la version C.02, et même, dans le cas de la méthode IMOMO (Hratchian et al.
2008) basé sur une partition QM:QM’.
2.4.3 Le modèle Polarized Embedding (PE)
Ce modèle permet de mettre un pas en avant et d’augmenter le niveau de sophistication par
l’introduction d’un jeu de charges MM flexibles pour décrire l’environnement qui est polarisé
à son tour par les densités de charge QM. Ce modèle peut être partitionné en deux catégories
où le modèle de charge flexible dans la région MM est polarisé par le champ électrique créé
par les densités de charge QM mais pas inversement (model C) ou par une Formulations
entièrement cohérentes qui peut inclure le modèle flexible MM dans l’Hamiltonien QM
permettant donc à une polarisation mutuelle et complète entre les deux régions (model D).
Plusieurs approches ont été développées pour spécifier la polarisation de atomes MM. parmi
les méthodes les plus populaire on peut citer : (Drude oscillators) connu aussi sous le nom de
(charge-on-aspring model) (Lamoureux and Roux 2003) , (Polarized point dipoles) (Warshel
et al. 2006) et (the fluctuating charge model) (Rappe and Goddard III 1991). Le modèle (PE)
offre un couplage plus réel à la frontière QM-MM. et remplace les champs de force
polarisable pour les biomolécules qui sont en cour de développement (Wang et al. 2000;
Vorobyov et al. 2005; Xie and Gao 2007).
2.5 Interactions électrostatiques longue portée QM-MM
Une description précise des forces électrostatiques due à l’environnement et agissant sur le
sous-système QM est essentielle pour une description plus réelle des structures et fonctions
des biomolécules. La prise en compte explicite de toutes les interactions électrostatiques ainsi
que leur zone de troncatures (cut-off) reste discutable à cause de la nature longue portée des
interactions Coulombienne (Nam et al. 2005; Schaefer et al. 2005; Riccardi et al. 2005). De
nombreuses études ont montrées que les zones de troncatures peuvent induire à de important
artefact (Riccardi et al. 2005). Le traitement du sujet d’interactions électrostatiques long porté
dans le contexte des méthodes QM-MM n’a trouvé l’attention que récemment, après avoir été
bien établi dans le domaine des simulations de dynamique moléculaire classique. On peut
souligner la méthode d’Ewald présenté par York el collaborateurs (Nam et al. 2005), la
méthode (Charge scalling) by Karplus et collaborateurs (Dinner et al. 2003), la méthode VEP

56
(Variational electrostatic projection) (Gregersen and York 2005; Gregersen and York 2006)
et la méthode GSBP (Generalized solvent boundary potential) (Schaefer et al. 2005).
2.6 Les interactions de van der Waals en QM/MM
Les interactions de répulsion à courte distance dû au nuage électronique ainsi que de la
dispersion, situé dans la région frontière QM-MM ne doivent pas être négligées, même si leur
contribution est faible. Pour en tenir compte, le terme de van der Waals est simplement ajouté
à l’énergie et n’intervent pas dans le Hamiltionien de la fonction d’onde. Ces interactions sont
typiquement décrites par le potentiel de Lennard Jones employé. Des fonctions exponentielles
peuvent être utilisées comme alternative pour représenter les forces répulsive. Il est donc
nécessaire d’assigner ou d’attribuer un type atomique exact ou similaire aux atomes QM.
Chaque atome QM est impliqué dans les interactions de van der Waals avec tous les atomes
de la région MM dont les plus proches contribueront d’une manière significative.
Dans le cas où les paramètres de van der Waals ne sont pas optimisés, seul les atomes QM qui
sont proches des atomes MM situé dans la zone frontière sont affectés. Pour surmonter ce
problème il faut juste déplacer la zone frontière un peu plus loin du centre QM. Des
considérations semblables s'appliquent à l'ambiguïté de choisir un ensemble de paramètres
fixe, par exemple, pendant une réaction. Ceci soulève alors la question si l’ensemble de
paramètre devrait être commuté d'une description de réactif à une description du produit
quelque part le long du chemin de réaction et si cette démarche présenterait des problèmes
supplémentaires plutôt que l’augmentation de la qualité du modèle. L'effet pourra simplement
être vérifié en comparant les résultats obtenus avec des paramètres différents.
Murphy et collaborateur (Murphy et al. 2000) dans leurs approche QM/MM, ont optimisé les
paramètres de van der Waals de la partie QM pour un ensemble de petit modèles de pair
d’acide aminé liée par des liaisons hydrogène. Les rayons de van der Waals obtenu sont de 5-
10% plus large que ceux utilisé dans le champ de force OPLS-AA ceci vas conduire à une
répulsion qui vas compenser la forte interaction électrostatique QM-MM causé par les charge
ponctuelle MM qui surpolarise la densité QM.
Un ensemble de paramètres optimisés pour B3LYP/AMBER a été présenté par diffèrents
groupes (Mata et al. 2008). Schaefer et collaborateurs ont montrés que les quantités
thermodynamiques de la phase condensée (l’énergie libre par exemple) calculé à partir des

57
simulations QM/MM sont plutôt insensibles envers les paramètres de van der Waals, en
revanche, il y a une certaine influence sur la structure autour de la région QM.
2.7 Les Interactions QM-MM liées
Le potentiel d’interactions liées, il est donné par la relation
𝐸𝑄𝑀−𝑀𝑀𝑏𝑜𝑛𝑑 =∑ ∑
𝑍𝐴𝑞𝐵𝑅𝐵 − 𝑅𝐴
𝐵𝜖𝑀𝑀
𝑀
𝐴
(2.9)
Où 𝑍𝐴 et 𝑞𝐵 sont respectivement les charges nucléaire et les charge ponctuelle des atomes QM
et MM situés au distance 𝑅𝐴 et 𝑅𝐵.
Pour ce qui est du potentiel d'interaction électrostatique entre les atomes QM et MM, Cette
expression dépend fortement de la méthode QM-MM utilisées pour traiter les problèmes de la
coupure d'une liaison covalente.
L’évaluation de la fonction d’onde de la partie QM doit être prise avec précaution. La coupure
simple à travers la liaison covalente QM/MM va créer un ou plusieurs électrons non apparié
au niveau du sous-système QM. En réalité, ces électrons sont apparié avec des électrons
appartenant aux atomes MM via des orbitale liante. Un certain nombre d'approches ont été
proposé comme un remède pour l'artefact de telles valences ouvertes.
2.8 Les coupures en QM/MM
On peut regrouper toutes les méthodes dans lesquelles l’interaction entre la partie quantique et
classique se fait directement, où les charges classiques sont ressenties par la partie quantique
et sont intégrées dans l’Hamiltonien de cœur. La principale différence entre ces méthodes et la
façon de traiter la coupure de certaines liaisons lors de la partition QM/MM. Par conséquence,
la coupure de la liaison covalente introduit quatres problèmes
a) la coupure d’une ou plusieurs liaisons pour créer le sous-système quantique en ferait une
espèce radicalaire dont les propriétés ne sont pas celles de ce sous-système au sein de la
molécule.

58
b) les contributions liantes des atomes MM situé à la région frontière impliquant les atomes
des deux sous-systèmes doivent être supprimées afin d’éviter un double comptage
c) une troncature de la région QM est créé lorsque la liaison pendante de l’atome 𝑄1 est saturé
d) comme dit précédemment une surpolarisation de la densité de charge QM par les charge
ponctuelle MM trop proches peut se produire.
Les solutions proposées pour résoudre ces problèmes de ce qui est appelé liaison pendante
sont assez diverses. D’une manière grossière deux groupes sont proposés :
i) La liaison est simplement complétée en ajoutant un atome monovalent.
ii) Les modifications portent sur la liaison elle-même.
Avant de détailler les différentes approches qui ont été conçue pour traiter les coupures des
liaisons covalentes en QM/MM, il faut d’abord savoir répondre à la question : où coupé ?
Pour cela des normes générales doivent être prises en compte et sont résumé ci-après
1-Minimiser le nombre de ramification par un choix approprié de la zone frontière.
2-La frontière de QM-MM devrait être pareillement éloignée de la région chimiquement
active que la taille de la partie QM.
3-Les atomes QM participant dans un processus d’association et de dissociation des liaisons
ne doivent pas être impliqués dans les termes liés (Vreven et al. 2006).
4-La liaison coupée ne doit pas être ni polaire ni être impliqué dans un système conjugué. Les
simples liaisons de type 𝐶 − 𝐶 présente le meilleur choix, en revanche la coupure selon une
liaison de type Amide est indésirable suite à son caractère partiellement double.
5-La coupure selon un groupe de charge MM qui est généralement introduit dans les champs
de force biomoléculaire doit être évité, car il va créer une charge nette artificielle en vicinité
directe avec la densité de charge QM.

59
Figure 2 : Etiquetage des atomes QM et MM d’un macrosystème QM/MM
2.8.1 Les approches Link-Atom
Le concept des atomes de connexion (Link atoms) qui sont dans la majorité des cas des
atomes d’hydrogène (HLA) (Field et al. 1990) a été introduit afin de compléter ou saturer la
couche de valence des atomes quantiques connectés à des atomes décrits par mécanique
moléculaire. Cependant, ces atomes apportent trois degrés de liberté supplémentaire au
système qui ne devraient pas être présents.
Des contraintes ont été utilisées afin d’éliminer ces degrés de liberté. L’atome (HLA) est
positionnée a une certaine distance le long de la liaison définit par le vecteur 𝑄1 → 𝐶1.
Conformément aux développements de Morokuma et collaborateurs (Svensson et al. 1996), la
distance 𝑅(𝑄1−𝐻𝐿𝐴) est modifiée suivant la relation :
𝑅(𝑄1−𝐻𝐿𝐴) = 𝑐𝐻𝐿𝐴𝑅(𝑄1−𝐶1) (2.10)
Le facteur 𝑐𝐻𝐿𝐴 peut être défini de plusieurs manières, mais il est souvent déterminé par le
rapport des rayons covalents 𝑅0 des atomes de liaison 𝑄1 − 𝐶1 . Ces rayons peuvent être pris à
partir du champ de force. La valeur du 𝑐𝐻𝐿𝐴 est proche de 0,71 dans la plupart des cas.
Alternativement, le Link atome peut être positionné à une distance constante par rapport à 𝑄1.
Avec 𝑅𝑄1 et 𝑅𝐶1 sont respectivement les coordonné cartésienne de 𝑄1 et 𝐶1 .

60
𝑐𝐻𝐿𝐴 =𝑅0(𝑄1 − 𝐻𝐿𝐴)
𝑅0(𝑄1 − 𝐶1) (2.11)
𝑐𝐻𝐿𝐴 =𝑅0(𝑄1 − 𝐻𝐿𝐴)
|𝑅𝑄1 − 𝑅𝐶1| (2.12)
En ce qui concerne le traitement des charges proches de la frontière dans le cas des méthodes
(Link atome), une surpolarisation de la fonction d’onde électronique est provoqué lorsque
l’atome 𝐶1 et du 𝐻𝐿𝐴 sont à courte distance ~0,5Å .pour pallier à cet inconvénient, la charge
de l’atome 𝐶1 est souvent mise à zéro, ainsi que dans certains cas, les charges des atomes 𝐶2
et 𝐶3. D’autre groupes conseillent de les redistribuer (Lin and Truhlar 2005; Zhang et al.
2007) ou bien encore de les délocaliser partiellement à l’aide de fonctions gaussiennes (Das et
al. 2002; Amara and Field 2003).
Antes et Thiel proposent la méthode « Adjusted Connection atoms » (Antes and Thiel 1999)
(Antes and Thiel 1999) qui est un atome quantique auquel on ajoute des paramètres classique
supplémentaires afin de connecter les deux sous-systèmes. Dans l’approche Pseudobond
(Zhang et al. 1999) une liaison carbone-carbone est mimé par une liaison (pseudo-carbone)-
carbone, l’atome de « pseudo-carbone » étant pourvu de sept électrons de valence et d’un
potentiel de cœur effectif paramétré spécialement pour décrire la liaison coupée, pendant que
l’approche quantum-capping potential (DiLabio et al. 2002), ppossèdent un atome
reparamétrisé où un potentiel de cœur effectif est employé pour reproduire les propriétés de la
liaison originelle.
Dialbo, Hurley et Christiansen proposent quant à eux de remplacer l’atome de liaison par un
pseudo-potentiel qu’ils appellent QCP (Quantum Capping Potential) (DiLabio et al. 2002),
qui ne demande que peu de modification du programme et permet d’améliorer la
représentation de la liaison coupée au niveau quantique.
Dans l’approche «Add Remove » (Swart 2003) dans laquelle la position du Link Atome
dépend de celle de l’atome classique lié à la frontière coupé, permettant ainsi de supprimer les
degrés de liberté supplémentaires introduits. De plus l’introduction de cet atome est corrigée
en retirant l’interaction que celui-ci aurait avec le sous-système quantique s’il était classique.

61
L’approche « Double link atom » proposée par Das et al (Das et al. 2002) Consiste a ajouté un
Link atom au sous-système classique, pour supprimer la polarité non physique induite par la
coupure.
2.8.2 Les approches de type Orbitales localisées
les orbitales hybrides localisées (OL) présentent une solution pour saturer la liaison pendante
et décrire au mieux la frontière QM/MM. généralement les méthodes utilisant les orbitales
localisées sont théoriquement plus réalistes que les approches Link Atom car elles décrivent de
manière quantique la distribution des charges autour de la frontière QM/MM. la délocalisation
des charges quantiques dans les orbital localisé aide à empêcher ou limiter la surpolarisation
qui est parfois présente dans les méthodes Link Atom.
La première méthode qui s’appuie sur ce principe est celle qui a été proposé par Warshel et
Levitt (Warshel and Levitt 1976): Elle consiste à utiliser des orbitales hybrides de liaison pour
décrire la liaison coupée.
Rivail et collaborateurs ont repris ces idées et ils ont formulés la méthode du champ
autochérent local ou LSCF (Local Self Consistent Field) au niveau semi-empirique (Théry et
al. 1994; Monard et al. 1996), puis au niveau ab initio (Xavier Assfeld et al. 1998; Ferré et al.
2002) que nous verrons en détail dans la chapitre suivant.
Une extension de la méthode LSCF, appelée (Generalized Hybrid Orbital) GHO (Gao et al.
1998; Pu et al. 2005) développée par Gao et dans laquelle un ensemble d’orbitales hybrides
sp3 est assigné à chaque atome frontière. L’ensemble de ces quatre orbitales forme un jeu de
quatre fonctions orthonormées. L’orbitale orientée vers la partie quantique est appelée orbitale
active et participe au processus d’optimisation de la fonction d’onde. Les trois orbitale
hybrides pointant vers les atomes MM directement connectés à l’atome frontière sont appelés
orbitales auxiliaire et sont gelées durant le cycle SCF. Ces dernières ne peuvent pas être
mélangées avec les autres orbitales.
En combinant les approches LA et GHO, Lin et collaborateur ont développé la méthode RCD
(Redistributed Charge and Dipole) (Das et al. 2002) qui consiste à utiliser un HLA et à
redistribuer la charge de l'atome M1 sur les liaisons M1-M2 (représentant les orbitales de
GHO) en conservant la charge et le dipôle de cette liaison.

62
Friesner et collaborateurs ont développé une approche nommée (frozen orbital QM /MM
interface methodology) (Philipp and Friesner 1999) qui est une variante de la méthode LSCF
et qui diffère sur des aspects techniques, en particulier, elle nécessite une paramétrisation
importante de la zone de coupure.
La méthode EFP (Effective Fragment Potential) (Kairys and Jensen 2000) défini, quant à elle,
une région tampon constituée d’orbitales localisées gelées entre la parties QM et MM.
Figure 3: Les différentes coupures en QM/MM: Link-Atome, LSCF, GHO, RCD et EFP, les atomes de la partie
quantique sont notés Q, et ceux de la partie classique M, Dans la méthode EFP, les atomes de la partie tampon
son désignées par B.

63

64
Chapitre 3
Méthodologie et développements de la méthode du
Champ Auto Cohérent Local (LSCF)

65
3.1 La Méthode du Champ Auto-Coherent Local : Local Self-Consistent Field (LSCF) :
3.1.1 Principe
Le principe fondamental de la méthode LSCF (Local Self-Consistent Field) est l’optimisation
de la fonction d’onde multiélectronique sous contrainte. Cette méthode a été développée
initialement par V.Théry dans laquelle la partie quantique était traitée à l’aide de méthode
semi-empirique (Ferenczy et al. 1992; Théry et al. 1994; Gorb et al. 1996; Monard et al.
1996). Elle fut ensuite étendue aux méthodes ab initio (Assfeld and Rivail 1996; Ferré et al.
2002; Ferré and Assfeld 2002; Ferré and Assfeld 2003; Assfeld et al. 2004; Moreau et al.
2004; Fornili et al. 2006b; Fornili et al. 2006a; Loos et al. 2007a; Loos and Assfeld 2007b;
Loos and Assfeld 2007a; Loos et al. 2007b; Loos et al. 2007c)
La méthode LSCF a été développée à Nancy pour permettre le traitement des liaisons
pendantes créées lors d’une coupure QM/MM. Cependant, l’intérêt de cette approche n’est
pas limité à ce type d’application. En effet elle peut être utilisée plus généralement pour faire
un calcul quantique de type SCF en imposant des contraintes sur certaines spinorbitales
représentant une densité électronique invariante ressentie par le reste du système. Il faut noter
qu’il n y’a pas de restriction sur la nature des spinorbitales. En effet, celles-ci peuvent être
occupées ou vacantes, et utiliser dans le formalisme restreint (RHF) ou non restreint (Pople
and Nesbet 1954) (UHF). Les modifications de la procédure itérative standard SCF pour
prendre en compte ces contraintes seront présentées dans la section suivante.
3.2 Le Formalisme LSCF ab initio
La réalisation d’un calcul LSCF, impose la définition au préalable des coefficients des
fonctions de base dans les spinorbitale devant être gelées ainsi que leurs nombre
d’occupation. L’ensemble des spinorbitales gelées est orthonormé afin d’éviter le problème de
la non-orthogonalité dans le calcul de l’énergie. Afin d’évité ce dernier, il a été imposée que
les orbitales moléculaires devant être optimisées, soient normées, mutuellement orthogonales
et orthogonales aux spinorbitales gelées. Le moyen retenu pour satisfaire cette contrainte
consiste à développer les orbitales variationnelles sur la base de fonctions présentant déjà
cette qualité. Ainsi, chaque fonction de la base initiale se voit diminuer de sa projection sur le
sous-espace défini par les spinorbitales gelées et est renormée. La base de fonctions obtenue
est orthogonale aux spinorbitales gelées mais possède au moins autant de dépendances
linéaires qu’il y a de spinorbitales gelées. Ces dépendances linéaires sont éliminées grâce à

66
l’opération d’orthogonalisation canonique. La base résultante contient des fonctions normées
mutuellement orthogonales aux spinorbitales gelées, sur lesquelles les orbitales
variationnelles pourront être construites. Il faut noter que cette nouvelle base est égale à celle
de la base initiale diminuée du nombre de spin-orbitale gelées. L’ensemble de ces opérations
peut se combiner en une seule, exprimée sous la forme d’une matrice rectangulaire. Cette
matrice a le même rôle que la matrice de Lӧwdin dans la résolution des équations de Roothan.
Les deux seules différences entre les méthodes SCF et LSCF résident dans la matrice
d’orthogonalisation et dans la définition de la matrice densité.
On considère un système à couche contenant 2𝑛 électrons et pour simplifier l’étude on se
place dans un formalisme restreint. Les orbitales moléculaire variationnelles 𝑂𝑀 du système
moléculaire sont développées sur une base contenant 𝑁 orbitale atomique 𝑂𝐴 :
|⟩, |⟩, |⟩… ≡ {|⟩}1≤≤𝑁
|𝑝⟩ =∑𝐶𝑝
𝑁
|⟩ (3.1)
Prenant un sous-système quantique relié à la partie classique par 𝐿 liaisons simples. Dans
cette approche, les liaisons chimiques entre les atomes frontières quantiques 𝑄𝑀: 𝑋𝑖 et les
atomes frontières de la partie classique 𝑀𝑀:𝑌𝑖 seront représenté par 𝐿 orbitales localisé ou
gelées (𝑂𝐿): |𝑙𝑖⟩
Il faut rappeler que les orbitales atomique |⟩, |⟩, |⟩,∙∙∙∙∙∙∙ sont définies sur tous les atomes de
la partie 𝑄𝑀 et sur l’atome frontière 𝑌𝑖 . Ce dernier est qualifié d’hybride ou d’atome quanto-
classique car il possède des orbitales de fonction atomique ainsi que tous les paramètres
propres au type d’atome qui lui a été attribué dans la partie MM.

67
3.2.1 Orthogonalisation des orbitales localisé
Les orbital localisées sont décrites comme une combinaison linaire des orbitales atomiques :
|𝑙𝑖⟩ =∑𝑎𝑖
𝑁
|⟩ (3.2)
Afin de pouvoir contraindre l’orthogonalité des orbitales moléculaire du sous-système
quantique avec les orbitales localisées, il est crucial que les orbitales moléculaire 𝑂𝑀 soient
orthogonales aux orbitales localisées 𝑂𝐿
Un moyen de faire cela est de construire les 𝑂𝑀 dans une base de fonctions atomiques à la
fois orthogonales entre elles et orthogonale aux orbitales localisées 𝑂𝐿.
La première étape de ce processus est l’orthogonalisation des 𝑂𝐿 entre elles afin d’obtenir un
jeu d’orbitales localisées orthogonale (𝑂𝐿𝑂) qui s’écrivent sous la forme :
|𝑙𝑖′⟩ = ∑𝑎𝑖
′
𝑁
|⟩ (3.3)
Il existe plusieurs méthodes permettant d’orthogonaliser les fonctions des orbitales localisé à
l’aide de la matrice d’orthogonalisation 𝐗.
La méthode d’orthogonalisation la plus commune est celle de Lӧwdin (Löwdin 1955) qui
consiste a utilisé comme matrice d’orthogonalisation 𝐗 l’inverse de la racine carré de la
matrice de recouvrement des orbitales localisées (𝑂𝐿) .
On note 𝐃 la matrice de recouvrement entre les (𝑂𝐿), 𝐷𝑃𝑄 = ⟨𝑙𝑃|𝑙𝑄⟩.
La matrice de Lӧwdin est donc égale à 𝑋 = 𝐷−1 2⁄ .
Cette procédure est employée dans le processus SCF afin de travailler dans une base
d’orbitale atomique (𝑂𝐴) mutuellement orthogonales.
La procédure d’orthogonalisation de Gram-Schmidt (GH) consiste à prendre les vecteurs un à
un, et à retrancher successivement les projections des vecteurs précédents pour former un

68
vecteur orthogonale aux précédents. Ce procédé est très utile car il permet de laisser inchangé
le premier vecteur considéré. Il est préférable d’utiliser cette procédure dans le cas d’un
nombre relativement faible de vecteurs à orthogonaliser. En effet, lorsque l’ensemble devient
trop important, des instabilités numériques peuvent intervenir.
3.2.2 Orthogonalisation des orbitales atomique
Les 𝑁 orbitales atomique du jeu de base de fonctions atomiques peuvent être ensuite
orthogonalisées à ces 𝑂𝐿𝑂 en les projetant dans le sous-espace complémentaire des 𝑂𝐿𝑂
grâce à la fonction :
|⟩ = [1 −∑𝑆𝑖2
𝐿
𝑖
]
−1 2⁄
[|⟩ −∑|𝑙𝑖′⟩⟨𝑙𝑖
′|𝜇⟩
𝐿
𝑖
] (3.4)
Où 𝑆 représente l’intégrale de recouvrement entre les orbitales localisées 𝐿𝑂 et les fonctions
de base :
𝑆𝜇𝑖 =∑𝑎𝑖′
𝑁
𝑆𝜇 (3.5)
Les fonctions orthogonales |⟩ sont déduites des fonctions |⟩ à l’aide d’une matrice 𝐌 de
dimension 𝑁 ×𝑁 dont un élément (, ) s’écrit :
𝑀 = [1 −∑𝑆𝜇𝑖2
𝐿
𝑖
]
−1 2⁄
[𝛿𝜇 −∑∑𝑎𝑖′ 𝑎𝑖
′ 𝑆𝜇
𝑁
𝐿
] (3.6)
Cet ensemble de fonctions n’est pas linéairement indépendant car elles contiennent 𝐿
dépendances linéaires dues aux 𝑂𝐿 qui sont déjà des combinaisons linaires des 𝑂𝐴.
Si la base de fonctions atomique est linéairement indépendante, la matrice de l’intégrale de
recouvrement �� entre les 𝑂𝐴 orthogonales aux 𝑂𝐿 |⟩ a 𝐿 valeurs propres nulles.

69
Après avoir retiré ces valeurs propres et leurs vecteurs propres correspondant, il est possible
d’effectuer une orthogonalisation canonique au moyen d’une matrice 𝐗 de dimension
(𝑁 × (𝑁 − 𝐿)):
𝑋𝑝𝑞 = 𝑈𝑝𝑞��𝑞−12 (3.7)
Où 𝑈𝑝𝑞 est le 𝑝𝑖è𝑚𝑒 composant du 𝑞𝑖è𝑚𝑒 vecteur propre de la matrice de recouvrement �� entre
les fonctions de bases atomiques |𝜇⟩ et leurs valeurs propre correspondante ��𝑞 .
Les (𝑁 − 𝐿) fonctions |′⟩, |′⟩, |′⟩,∙∙∙∙ ≡ {|′⟩}1≤′≤𝑁−𝐿 obtenues à partir de |⟩, |⟩, |⟩,∙∙∙∙∙∙
par cette transformation sont linéairement indépendantes, mutuellement orthogonales et
orthogonales aux (𝐿𝑂) .
La matrice de dimension (𝑁 × (𝑁 − 𝐿)) qui transforme directement l’ensemble |⟩, |⟩, |⟩,∙∙∙
∙∙∙∙ en un ensemble |′⟩, |′⟩, |′⟩,∙∙∙∙ est noté par :
𝐁 = 𝐌 ∙ 𝐗 (3.8)
Cette matrice peut directement remplacer la matrice 𝐒−1
2 dans le processus SCF et permet de
réaliser l’optimisation de la fonction d’onde dans le sous-espace des (𝑁 − 𝐿) fonctions. Les
éléments de la matrice de Fock sont définit habituellement de la manière suivante :
𝐹𝜇 = 𝐻𝜇𝑐 +∑∑𝑃𝜎
𝑇
𝜎
[(𝜇|𝜎) − (𝜇𝜎|)] (3.9)
Le premier terme représente l’Hamiltonien de cœur du système, et le second terme les
intégrales biélectroniques(𝜇|𝜎). 𝑃𝑇 est la matrice densité du système que l’on peut
subdiviser en deux composantes dans le formalisme LSCF : une contribution est due aux
orbitales localisées (𝑃𝐿) et une seconde due aux orbitales moléculaires variationnelles (𝑃𝑄)

70
𝑃𝜇𝑇 = 𝑃𝜇
𝑄+ 𝑃𝜇
𝐿 (3.10)
La matrice de Fock 𝐅 de dimension (𝑁 × 𝑁) est ensuite transformée dans la base
{|′⟩}1≤≤𝑁−𝐿 à l’aide de la matrice 𝐁 pour obtenir le nouveau Fockien 𝐅′ de dimension
((𝑁 − 𝐿) × (𝑁 − 𝐿)) :
𝐅′ = 𝐁† ∙ 𝐅 ∙ 𝐁 (3.11)
Par la suite, il faut diagonaliser ce nouveau Fockien 𝐅′ pour obtenir la matrice de (𝑁 − 𝐿)
vecteurs propres 𝐶′ associés aux (𝑁 − 𝐿) valeurs propres(𝜖). On repasse ensuite dans la base
initiale grâce à la transformation inverse :
𝐂 = 𝐁 ∙ 𝐂′ (3.12)
Pour calculer la nouvelle matrice densité correspondant au orbitales moléculaire MO (𝑃𝑄) .
En ajoutant la contribution des orbitales localisées LO (𝑃𝐿), on obtient alors la matrice de
densité totale du système.
L’expression de l’énergie est donnée par :
𝐸 =∑𝑃𝑇
𝐻𝐶 +
1
2∑ 𝑃
𝑇
𝜎
𝑃𝜎𝑇 [(|𝜎) −
1
2(𝜎|)] (3.13)
3.2.3 Modification des équations de Roothaan
La condition de stationnarité de l’énergie par rapport aux coefficients de MO variationnelles
{𝐶𝜇𝑝} 1≤𝜇≤𝑁1≤𝑝≤𝑁−𝐿
𝛿𝐸
𝛿𝐶𝜇𝑝= 0 (3.14)

71
Et la contrainte d’orthogonalité entre tous les orbitales moléculaires (𝑀𝑂) et localisées (𝐿𝑂)
du système est :
∑𝑐𝜇𝑝𝜇
𝑆𝜇𝑐𝑞 = 𝛿𝑝𝑞 (3.15)
∑𝑙𝜇𝑃𝜇
𝑆𝜇𝑙𝑄 = 𝛿𝑃𝑄 (3.16)
∑𝑐𝜇𝑝𝜇
𝑆𝜇𝑙𝑄 = 0 (3.17)
Elle permet la modification des équations de Roothann
𝐅 ∙ 𝐂 = 𝐒 ∙ 𝐂 ∙ 𝐄 + 𝐒 ∙ 𝐋 ∙ 𝚲 (3.18)
Où 𝐋 représente la matrice des orbitales localisées (𝐿𝑂) et {𝚲𝑝𝑄}1≤𝑝≤(𝑁−𝐿)1≤𝑄≤𝐿
sont les
multiplicateurs de Lagrange du bloc des 𝑀𝑂 et 𝐿𝑂
𝛬𝑝𝑄 ≡ 휀𝑝𝑄 =∑𝑐𝜇𝑝𝐹𝜇𝜇
𝑙𝑄 (3.19)
En utilisant les relations 𝐂 = 𝐁 ∙ 𝐂′ et 𝐗† ∙ 𝐒 ∙ 𝐗 = 𝕝 et en multipliant à gauche par 𝐁† on
obtient
𝐅′ ∙ 𝐂′ = 𝐂′ ∙ 𝐄 (3.20)
Avec 𝐅′ = 𝐁† ∙ 𝐅 ∙ 𝐁 et 𝐶† ∙ 𝑆 ∙ 𝐿 = 𝕆 (orthogonalité entre les 𝑂𝑀 et les 𝑂𝐿). On aboutit a un
problème hermitien qui peut être résolu en diagonalisant la matrice 𝐹′.
L’algorithme LSCF implémenté dans le logicielle Gaussian03 est récapitulé comme suit :
1- Ortogonaliser mutuellement les 𝐹𝑂𝑠
2- Construire la matrice 𝐌, qui orthogonalise la base des 𝑂𝐴 aux 𝐿𝑂.

72
3- Construire la matrice. Transformer les fonctions obtenues en 2. En une base de
fonctions orthogonales et linéairement indépendantes.
4- Calculer 𝐁 = 𝐌 ∙ 𝐗
5- Définir la matrice de Fock 𝐅 dans la base originale.
6- Calculer 𝐅′ = 𝐁† ∙ 𝐅 ∙ 𝐁
7- Diagonaliser 𝐅′ : 𝛜 = 𝐂′† ∙ 𝐅 ∙ 𝐂′ où 𝜖 est la matrice diagonale des valeurs propres.
8- Transformer les vecteurs propres dans la base originale : 𝐂 = 𝐁 ∙ 𝐂′
9- Calculer 𝑃𝑄
10- Test de sortie. Si non satisfait, retour à 5.
3.4 La méthode LSCF/MM
Dans le cadre des méthodes QM/MM, l’approche LSCF est couplée avec un champ de force
classique, La notation qui est adopter pour désigner les atomes de la partie quantique (Q),
classique (C) ainsi que les atomes frontière (X) pour la partie quantique et (Y) pour la partie
classique est sont illustrer sur la figure suivante :
Figure 1: Schéma d’un système possédant une coupure QM/MM dans le formalisme LSCF.

73
Comme nous l’avons mentionné précédemment l’atome frontière (Y) a pour particularité
d’avoir à la fois des caractéristiques classiques et quantiques. Il possède les fonctions de base
de l’atome qu’il représente et apporte généralement un électron au système. Autrement dit il
sert comme un « point d’accroche » à la SLBO (Strictly Localized Bond Orbital) du coté
classique. Les interactions QM/MM dans le cadre de la méthode LSCF entre les différents
atomes des deux sous-systèmes sont résumées dans le tableau 1, 𝑍𝐴 et 𝑞𝑎 correspondent à la
charge nucléaire et à la charge ponctuelle de l’atome A. les éléments de la matrice densité
sont 𝑃𝜇, avec et sont les indices des orbitales atomique.
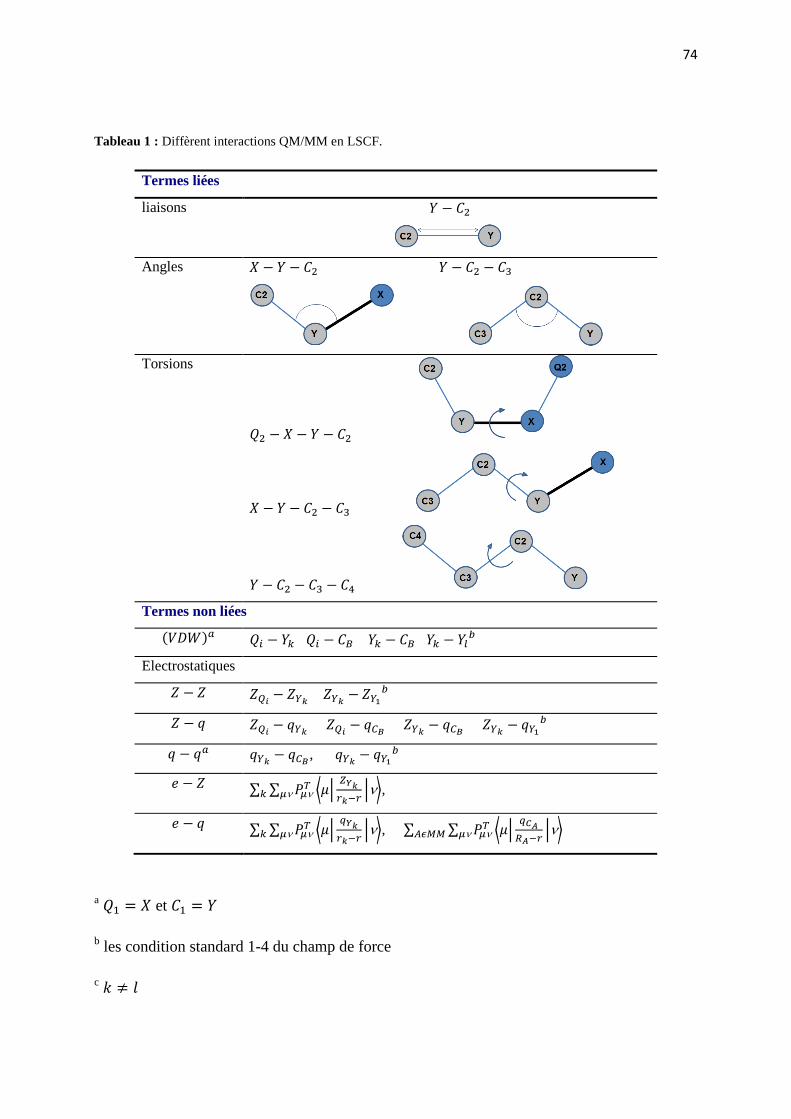
74
Tableau 1 : Diffèrent interactions QM/MM en LSCF.
Termes liées
liaisons 𝑌 − 𝐶2
Angles 𝑋 − 𝑌 − 𝐶2 𝑌 − 𝐶2 − 𝐶3
Torsions
𝑄2 − 𝑋 − 𝑌 − 𝐶2
𝑋 − 𝑌 − 𝐶2 − 𝐶3
𝑌 − 𝐶2 − 𝐶3 − 𝐶4
Termes non liées
(𝑉𝐷𝑊)𝑎 𝑄𝑖 − 𝑌𝑘 𝑄𝑖 − 𝐶𝐵 𝑌𝑘 − 𝐶𝐵 𝑌𝑘 − 𝑌𝑙𝑏
Electrostatiques
𝑍 − 𝑍 𝑍𝑄𝑖 − 𝑍𝑌𝑘 𝑍𝑌𝑘 − 𝑍𝑌1𝑏
𝑍 − 𝑞 𝑍𝑄𝑖 − 𝑞𝑌𝑘 𝑍𝑄𝑖 − 𝑞𝐶𝐵 𝑍𝑌𝑘 − 𝑞𝐶𝐵 𝑍𝑌𝑘 − 𝑞𝑌1𝑏
𝑞 − 𝑞𝑎 𝑞𝑌𝑘 − 𝑞𝐶𝐵 , 𝑞𝑌𝑘 − 𝑞𝑌1𝑏
𝑒 − 𝑍 ∑ ∑ 𝑃𝜇𝑇
𝜇𝑘 ⟨𝜇|𝑍𝑌𝑘𝑟𝑘−𝑟
|⟩,
𝑒 − 𝑞 ∑ ∑ 𝑃𝜇𝑇
𝜇𝑘 ⟨𝜇|𝑞𝑌𝑘𝑟𝑘−𝑟
|⟩, ∑ ∑ 𝑃𝜇𝑇
𝜇𝐴𝜖𝑀𝑀 ⟨𝜇|𝑞𝐶𝐴𝑅𝐴−𝑟
|⟩
a 𝑄1 = 𝑋 et 𝐶1 = 𝑌
b les condition standard 1-4 du champ de force
c 𝑘 ≠ 𝑙

75
Les calculs LSCF/MM, nécessite des paramètres supplémentaire pour assurer une description
précise de la liaison frontière, dont le potentiel s’écrit sous la forme suivante
𝐸𝑋−𝑌 = (𝐴 + 𝐵𝑟 + 𝐶𝑟2)𝑒𝐷𝑟 +𝐸
𝑟 (3.21)
Où 𝑟 représente la distance entre les atomes 𝑋. Les Cinq paramètres {𝐴, 𝐵, 𝐶, 𝐷, 𝐸} ont été
déterminé par des calculs purement quantiques. Ces paramètres dépendent du type atomique
(principalement C, N, O) ainsi que de l’hybridation (𝑠𝑝2 𝑜𝑢 𝑠𝑝3) des deux atomes définissant
la frontière QM/MM, cependant ils sont indépendant de la base.
3.4.1 Les orbitales de liaison strictement localisées : Strictly Localized Bond Orbital
(SLBO)
La frontière entre les deux sous-systèmes est décrite par une ou plusieurs orbitales de liaisons
strictement localisée que nous désignerons SLBO (Strictly Localized Bond Orbital). Ces
orbitales sont gelées grâce au formalisme LSCF, ils sont déterminés en se basant sur
l’hypothèse de la transférabilités des propriétés des liaisons, qui permet d’utiliser l’orbitale
prédéfinit sur le système complet.
La SLBO est déterminée sur un système modèle contenant la liaison chimique d’intérêt. Ils
sont données comme des paramètres du calcules QM/MM. il n’est, cependant, pas nécessaire
de réaliser un calcul sur le système complet mais juste sur un système modèle possédant la
liaison d’intérêt (nous choisirons toujours la molécule modèle la plus simple possible), par
exemple la molécule du formamide pour une liaison peptidique, la molécule d’éthane pour
traiter la liaison 𝐶𝑠𝑝3 − 𝐶𝑠𝑝3
Il existe plusieurs critère, dites externes et internes pour localiser des orbitales. De manière
générale, le choix du critère de localisation ne va pas influencer la qualité des résultats.
3.4.1.1 Les Critères externe:
Dans ce critère, les atomes pour lesquels la localisation va être effectuée sont donnés par
l’utilisateur. On peut citer les critères de:

76
a) Weinstein-Pauncz (WP)
Il consiste à maximiser le recouvrement des populations de Mulliken entre deux atomes A et
B (Weinstein and Pauncz 1968; Weinstein et al. 1971), elle est donnée par la formule.
⟨⟩WP =∑∑𝐶𝜇𝑖𝜖𝐵𝜇𝜖𝐴
𝑆𝜇𝐶𝑖 (3.22)
b) Magnasco-Perico (MP)
Il maximise le recouvrement de la population entre les atomes A et B et de la même manière
que le critère de Weinstein-Pauncz (WP) mais il prend en considération la population
électronique des deux atomes (Magnasco and Perico 1967; Magnasco and Perico 1968) il est
donnée par la relation:
⟨⟩MP = ∑ ∑ 𝐶𝜇𝑖𝜖{𝐴,𝐵}𝜇𝜖{𝐴,𝐵}
𝑆𝜇𝐶𝑖 (3.23)
3.4.1.2 Les critères internes
a) Le critère Boys-Foster
Il minimise le carré de la distance entre deux électrons, physiquement, il correspond à
déterminer un ensemble d’orbitales moins étendues dans l’espace, autrement dit il consiste à
maximiser la distance entre les barycentres des orbitales (Boys 1960; Foster and Boys 1960).
Il est donné par la fonction :
⟨⟩BF =∑⟨𝜙𝑖′𝜙𝑖
′|(𝑟2 − 𝑟1)2|𝜙𝑖
′𝜙𝑖′⟩
𝑛
𝑖
(3.24)
b) Le critère de Pipek-Mezy
Ce critère de localisation maximise la somme des charges atomiques de Mulliken (Pipek and
Mezey 1989; Pipek 1989). La charge atomique de la ième orbitale moléculaire centrée sur
l’atome (𝐴) est donné par :

77
𝑞𝑖(𝐴) =∑∑𝐶𝜇𝑖𝐶𝑖𝑆𝜇𝜇𝜖𝐴
(3.25)
La fonction maximisée est donc :
⟨⟩PM =∑[𝑞𝑖(𝐴)]2
𝑀
𝐴
(3.26)

78

79
Chapitre 4
La méthode du Champ Auto cohérent locale LSCF :
Application à l’étude des interactions
intermoléculaires

80
4.1 L’interaction dans les édifices moléculaires
Les interactions intermoléculaires, plus faibles par leur nature que des liaisons covalentes ou
intramoléculaires qui permettent ainsi de distinguer entre les molécules et les assemblages de
molécules sont déclarées souvent sous le nom « interactions non covalentes », « interactions
faibles ».Ces interactions ne modifient pas la nature des espèces moléculaire et n’influence
que le mode de d’assemblage. Parmi les interactions non-covalentes les plus connues on peut
citer les interactions de de van der waals et les interactions de type liaisons hydrogène, il y’a
aussi les interactions électrostatiques et les interactions hydrophobe.
Les interactions intermoléculaires sont responsables de la cohésion de la matière dans la
plupart des phases macroscopiques et elles conduisent à l’assemblage des systèmes
microscopiques. Ces associations sont dues aux propriétés chimiques, électriques et
géométriques des molécules et à l’inverse des liaisons covalentes, les interactions faibles
permettent une grande flexibilité aux macromolécules et des structures cellulaires. Les
interactions intermoléculaires jouent un rôle important dans des domaines aussi variés que la
physique, la chimie et la biologie. En effet, la structure des cristaux moléculaires (Cl2, CO2
etc.) et ioniques, des liquides, la compréhension des phénomènes de physisorption, et des
formes d’édifices biologiques comme les structures secondaires dans les protéines, et la
structure en hélice de l’ADN sont le résultat d’interactions intermoléculaires.
4.2 Classification et caractères des interactions intermoléculaires
Les contributions principales dans les forces responsables des phénomènes physiques
d’attractions et répulsion entre les molécules ont été identifiées, et selon la nature des
complexes étudiés, la classification des interactions intermoléculaire (London 1930;
Eisenschitz and London 1930; London 1937; Amos et al. 1985; McWeeny 1989; Stone 1996;
Ioannou et al. 1997) diffèrent en termes de l’intensité de leurs composantes. On peut classifier
ces interactions en différentes composantes : la répulsion, l’interaction électrostatique, la
polarisation et la dispersion. Selon les systèmes, les interactions dominantes seront plutôt de
type électrostatique (complexe à liaison hydrogène par exemple) ou de dispersion (complexe
de van der Waals).

81
4.3 Investigation expérimentale et théorique des interactions intermoléculaire
4.3.1 Caractérisation expérimentale
L’étude des édifices moléculaire faiblement lié est d’un intérêt fondamental dans de
nombreux domaine de la chimie et de la physique, en particulier tout ce qui révèle de la
chimie atmosphérique, de l’astrophysique et de la biologie. Plusieurs études expérimentals ont
été orientées vers l’étude des complexes faiblement liés (Beyer et al. 1984; Weber 1987) ceci,
dans le but d’apporter des reponses sur la structure des diffèrents conformeres à l’état
fondamental en phase gaz et sur le rôle des liaisons hydrogène intra et intermoléculaire.
Comme la formation d’un complexe donne lieu à des vibrations intermoléculaires, il est
également possible de caractériser expérimentalement la surface d’énergie potentielle d’un
complexe par spectroscopie infrarouge lointain (Blake et al. 1991) qui s’étend de 20cm-1
à
150 cm-1
c’est-à-dire au-delà des fréquences de vibration des liaisons chimiques usuelles.
La mesure des géométries d’équilibre et des géométries associées aux minima de la surface
d’énergie potentielle intermoléculaire peut passer par la spectroscopie micro-onde (Townes
and Schawlow 1975) qui est caractérisé par une résolution spectrale puissante et qui permet
d’obtenir les trois constante rotationnelle A, B, C d’un complexe, et à partir desquelles il est
possible de déterminer les trois moment d’inertie et la géométrie moyenne (Kisiel et al. 1992).
L’inconvénient de cette méthode réside dans la difficulté de son application dans le cas des
grands complexes, par contre les trois constantes rotationnelles peuvent sérvir de contraintes
pour améliorer la recherche de la géométrie d’équilibre (Leopold et al. 1994).
Cette méthode est habituellement appliquée, pour étudier une large variété des propriétés
moléculaires y compris les interactions non covalentes dans les clusters moléculaires et les
biomolécules en phase gazeuse (Cole and Legon 2004; Sánchez et al. 2005).
Les fréquences de vibration intermoléculaire caractérisant la géométrie d’un complexe,
peuvent aussi être détecté par la spectroscopie RAMAN à pompage d’états vibrationnels (Kim
et al. 1999) et aussi par la spectroscopie de fluorescence (Ishikawa et al. 1999).On peut citer
quelques monographies qui ont été principalement orienté vers l’étude des liaisons
hydrogène : (The Weak Hydrogen Bond) (Desiraju and Steiner 1999) par Desiraju et Steiner,
(Hydrogen Bonding) (Scheiner 1997) par Scheiner, et (An Introduction to Hydrogen Bonding)
(Jeffrey 1997) Par Jeffrey.

82
4.3.2 Caractérisation théorique
La disponibilité des données expérimentale précise a ravivé les études théoriques dont
l’intérêt est de prédire les structures et analyser la nature des forces qui tiennent les complexes
faiblement liées. L’étude des interactions non-covalentes continu d’être un domaine d’activité
important, ceci est prouvé par le nombre considérable de publications traitant ce champs
d’étude (Hobza and Zahradník 1992; Chalasinski and Szczesniak 1994; Jeziorski et al. 1994;
Jeziorski and Szalewicz 1998; Engkvist et al. 2000; Chalasinski and
Szcz&ecedil;śniak 2000) reflétant ainsi, leur importance dans des systèmes physiques
variés et dans des processus incluant les phases condensées (Allen and Tildesley 1987;
Cramer and Truhlar 1999; Karle and Huang 2003), l’ADN et les protéines (Saenger 1984;
Hunter et al. 1991), les interaction enzymes/substrat et médicament/récepteur (Politzer et al.
1985; Naray-Szabo and Ferenczy 1995) et adsorption physique (Sauer et al. 1994) etc.
Le traitement quantitatif de l’énergie d’interaction intermoléculaire 𝐸𝑖𝑛𝑡 à un niveau
d'exactitude très poussé pour des systèmes moléculaires complexes et la description de
l’équilibre infime entre ces différentes contribution (Pullman 1978; Moszynski et al. 1994;
Schütz et al. 1998; Langlet et al. 2000), présente encore un challenge pour les théoriciens.
Ceci est dû à deux raison :
La première, c’est que l’énergie d’interaction qui est défini comme la différence entre
l’énergie totale du complexe et la somme des énergies totale des espèces isolé qui le constitue,
est vraiment une fraction minuscule de l’énergies totale. Typiquement, cette fraction peut
varier de 10-7
pour les interactions faibles dans les complexes de type van der Waals et de 10-4
dans les complexes à liaison hydrogène fortement liée.
La deuxième raison, c’est que il n’y’a pas une méthode exacte pour calculer directement cette
petite fraction.
Il y a deux stratégies théoriques pour calculer l’énergie d’interaction intermoléculaire :
4.3.2.1 La méthode de la supermolécule (supermolecular method)
Qui consiste à calculer séparément l’énergie totale de chaque entité (le complexe et les
molécules individuelle) et ensuite soustraire ces énergies selon la définition de l’énergie
d’interaction 𝐸𝑖𝑛𝑡 entre des systèmes rigides, cela veut dire que les géométries des monomères
reste la même dans le complexe et à l’état individuelle. Par exemple, l’energie d’interaction

83
∆𝐸 d’un dimère 𝐴⋯𝐵 est définit comme la différence de l’énergie électronique entre le
dimère (𝐸𝐴⋯𝐵) et les monomères isolés(𝐸𝐴, 𝐸𝐵). Les energies des monomères sont calculées
dans le jeu de bases du dimère (dimer-centered basis set) en fournissant les géométries du
dimère optimisées (Boys and Bernardi 1970; Xantheas 1996).
4.3.2.2 Les méthodes basées sur la théorie de perturbation
Dans ce cas, l’énergie d’interaction peut aussi être considérée comme le résultat d’une petite
perturbation physique des monomères isolés. Ceci a permis l’application des méthodes de
calcul perturbationnel devenues des méthodes de choix, intensivement appliquées dans cette
ligne de recherche. L’approche MP2 (second-order Møller–Plesset perturbation theory) a été
appliqué avec succès, elle a fourni des énergies d’interaction précis pour les complexes à
liaison hydrogène (Xantheas and Jr 1993; Kim and Jordan 1994) et les systèmes de type
donneur accepteur entre les paires de bases de l’ADN (Šponer and Hobza 1997; Jurečka et al.
2006) mais surestime l’énergie d’interaction dans le cas de complexes dispersifs, par exemple
pour des interactions de 𝜋-staking (Hobza et al. 1996).
La méthode d’agrégats couplés de mono et diexcitations avec des excitations triples par
perturbation CCSD(T) est devenue une méthode standard pour calculer des énergies de
corrélation. La référence pour le calcul d’énergies d’interaction intermoléculaire est ainsi
CCSD(T) en base complète, i.e. CCSD(T)/CBS (CBS : Complete Basis Set extrapolation). La
théorie MBPT (The Many-Body Perturbation Theory) a été utilisée pour évaluer les termes de
corrélations (Kozmutza et al. 1996). En plus de l’évaluation de l’énergie totale d’interaction
intermoléculaire il est souvent souhaitable de comprendre son origine physique.
4.4 Méthodes d’analyse des interactions inter moléculaire
Dès 1957, Coulson propose de diviser l’énergie totale en différentes contribution pour
rationaliser les phénomènes qui entrent en jeu dans les liaisons. L’objectif des méthodes de
partition ou de décomposition de l’énergie d’interaction, est l’analyse et la compréhension de
l’origine des interactions dans les complexes non-covalent et aussi pour faciliter la conception
des potentiels intermoléculaires utilisés dans les simulations Monte Carlo et le développement
des champ de force de dynamique moléculaire.

84
L’énergie d’interaction est fréquemment traité comme étant composé de plusieurs termes
principaux: électrostatique 𝐸𝑒𝑠, polarisation (ou induction) 𝐸𝑝𝑜𝑙, dispersion 𝐸𝑑𝑖𝑠, échange-
répulsion 𝐸𝑒𝑥−𝑟𝑒𝑝 et transfert de charge (Langlet et al. 2003). Ces termes sont utilisés afin de
décrier les mouvements des densités de charge électroniques qui se produit entre les
molécules en interaction et la nature des forces qui les réunissent ensemble.
𝐸𝑖𝑛𝑡 = 𝐸𝑒𝑙𝑒𝑐 + 𝐸𝑖𝑛𝑑 + 𝐸𝑑𝑖𝑠𝑝 + 𝐸𝑒𝑥−𝑟𝑒𝑝 + 𝐸𝑐𝑡 (4.1)
4.4.1 L’énergie électrostatique : 𝑬𝒆𝒍𝒆𝒄
Représente l’ensemble des interactions coulombiennes des deux densités de charges isolées.
Cette contribution inclue les interactions entre toutes les charges permanentes et les
multipoles, comme les interactions dipole-dipole, dipole-quadripole. L’interaction
électrostatique est additive et peut être répulsive ou attractive selon l’orientation relative des
molécules. Elle constitue la plus grande partie de l’interaction intermoléculaire, avec une
décroissance comme l’inverse de la distance intermoléculaire, dans le cas où le système
interagissant contient des molécules polaires (Margenau 1939) et/ou chargées ou encore des
liaisons hydrogène.
4.4.2 L’énergie d’induction : 𝑬𝒊𝒏𝒅
Représente la stabilisation d’un système par la polarisation de ses constituants par le champ
électrique qu’ils subissent, de la part des densités de charges les avoisinant. La contribution
d’induction est une interaction non-additive, et toujours attractive.
4.4.3 L’énergie de dispersion : 𝑬𝒅𝒊𝒔𝒑
C’est une interaction qui est liée à la corrélation électronique de deux densités de charge en
interaction. Le moment dipolaire instantané d’un édifice peut polariser les édifices voisins
conduisant à une interaction entre dipôle instantané et dipôle induit. La contribution de
dispersion ne peut donc pas être saisie par la méthode Hartree-Fock. Elle est attractive et
existe dans tous les complexes. Elle est la seul interaction restante pour les complexes sans

85
moments permanents sur les constituants, à distance au-delà de la répulsion de Pauli. C’est le
cas dans les complexes de gaz rares et les systèmes où il existe des interactions de π-stacking.
4.4.4 L’énergie de répulsion 𝑬𝒆𝒙−𝒓𝒆𝒑
La répulsion vient du principe de Pauli stipulant que deux électron ne peuvent occuper le
même spin au sein d’une même orbitale. C’est une interaction toujours répulsive qui domine à
courte distance avec décroissance exponentielle en distance intermoléculaire.
4.4.5 L’énergie de transfert de charge 𝑬𝒄𝒕
La composante de transfert de charge peut devenir importante dans la région minimum du
potentielle où les fonctions d’onde électronique commence a ce recouvrir. Autrement dit,
l’énergie de transfert de charge d’un fragment moléculaire vers un autre fragment, est due au
transfert de charge de l’orbitale moléculaire occupé de l’un des fragments, vers les orbitales
moléculaire virtuelle de l’autre. Le transfert de charge a été utilisé pour expliquer, en se
basant sur des données spectroscopiques, la tendance d’association et le comportement des
complexes à liaison hydrogène (Pimentel and McClellan 1971).
Beaucoup de discussion ont été enregistré à propos de l’ampleur de chaque composante,
particulièrement au niveau des complexe à liaison hydrogène (Kollman 1977). Buckingham et
al (Buckingham and Fowler 1983; Buckingham and Fowler 1985) et Liu et al (Liu and
Dykstra 1986) ont argumenté que la géométrie des complexes faiblement lié, peut être prédit
seulement, sur la base des interactions électrostatique et de polarisation, à condition d’utiliser
des potentiel précis et qui sont associée conjointement avec des contrainte sur la distance
(rayon des sphères dure).
4.6 Lecture dans les différentes méthodes de l’analyse de la décomposition de l’énergie
d’interaction EDA (energy decomposition analysis).
L’analyse des interactions dans des systèmes complexes n’est pas une démarche facile. Les
méthodes couramment utilisées comme l’étude structurale et l’étude des charges partielles
atomiques sont très souvent prises en défaut pour expliquer une réactivité ou une stabilité
particulière. L’étude des orbitales moléculaires a pendant très longtemps été l’unique méthode
d’analyse et s’est montrée particulièrement pertinente dans de nombreux cas.

86
Par exemple, l’étude des orbitales frontières, a permis une meilleure compréhension de la
réactivité de certains complexes. Cette méthode reste encore très utilisée mais donne, parfois,
un aperçu difficile à analyser en termes physiques (énergie électrostatique, polarisation,
transfert d’une charge, répulsion…) ou chimique (liaison, paire, libre, atome…).
Les méthodes de décomposition qui seront présentées par la suite, permettent de modéliser la
sélectivité ou la réactivité enregistré entre deux ou plusieurs fragments en décomposant leurs
énergie d’interaction en composantes bien définies possédant chacune leur propre sens
physique.
De telles approches représentent aussi un outil précieux pour la paramétrisation de champs de
forces et notamment de champs de forces polarisables. En effet elles permettent d’évaluer
chaque composante de l’énergie d’interaction et donc de pouvoir la reproduire de manière
classique individuellement et indépendamment des autres.
4.6.1 Les méthodes variationnelles
Différentes méthodes théoriques ont été proposées pour la décomposition de l’énergie
d’interaction et parmi les méthodes basées sur le principe variationnelles on peut citer ; la
méthode KM : (Kitaura-Morokuma) (Kitaura and Morokuma 1976), la méthode
CSOV(Constrained Space Orbital Variation) (Bagus et al. 1984; Bagus and Illas 1992;
Piquemal et al. 2005) et la method RVS (Reduced Variational Space) (Stevens and Fink
1987). Bien que différentes dans l’approche théorique, ces méthodes mènent à des énergies
ayant un sens physique similaire. La méthode CSOV est limitée à deux fragments de par son
implémentation, par contre, la méthode RVS permet de traiter autant de fragments que l’on
souhaite. La possibilité de décomposer l’énergie d’interaction pour les complexes contenant
au moins 3 fragments permet l’accès aux effets à N-corps.
4.6.2 Méthodes perturbatives
Pour plus de trois quarts de siècle, la théorie des perturbations a constitué la base de la
compréhension de la nature, des forces interatomiques, et intermoléculaire faible .La théorie
des perturbations Rayleigh-Schrӧdinger à partir du produit des monomères seuls, permet donc
de calculer des termes d’interaction avec un sens physique. Cependant, elle ne permet pas de

87
restaurer l’antisymétrie de la fonction d’onde. De même, l’énergie d’interaction tendrait vers
l’infini à courte distance, car une contribution d’échange-répulsion manque. L’adaptation à
l’antisymétrie donne ce qui est nommé dans la littérature : la théorie de perturbation à
symétrie adaptée SAPT (Symmetry Adapted Perturbation Theory) (Rybak et al. 1987;
Jankowski et al. 1990; Moszynski et al. 1994). Qui définit les corrections à l’énergie de
perturbation par l’application d’un opérateur d’antisymétrisation qui agit sur la fonction
d’onde du dimère. Cette méthode a montré son efficacité dans l’étude de systèmes faiblement
liés tels que les dimères et les trimère d’hélium et dans l’analyse des liaisons de van der
Waals, et elle a été développé en la combinant avec La théorie de la fonctionnelle de densité
pour donner l’approche (SAPT-DFT) qui permet de déterminer tous les termes de corrélation
intramoléculaire (Williams and Chabalowski 2001; Jansen and Hesselmann 2001; Heßelmann
and Jansen 2002; Misquitta et al. 2005).
4.6.3 Autres méthodes
Le schéma de décomposition NEDA (Natural Energy Decomposition Analysis) (Glendening
and Streitwieser 1994) est basé sur la méthode des orbitales naturelles NBO (natural bond
orbital) de Weinhold and Foster (Foster and Weinhold 1980). Cette méthode découpe
l’énergie d’interaction en composante électrostatique, de transfert de charge, de polarisation,
d’échange et de déformation et elle présente une grande stabilité numérique avec
l’augmentation de la taille de la base de calcule
Dans le schéma ETS (Extended Transition State) l’énergie d’interaction totale est partitionnée
en interaction électrostatique, de Pauli, et interaction orbitalaire (Ziegler and Rauk 1977;
Bickelhaupt and Baerends 2000; te Velde et al. 2001; Mitoraj et al. 2009). Les schémas de
décompositions décrit précédemment ont été accordé à des approches de type HF et DFT.il
faut noter que l’analyse de l’énergie d’interaction pour les méthodes variationnelles est
complété par des calculs sur les supermolecule à un niveau MP2 ou CCSD(T) afin de tiré la
composante de dispersion liée à la corrélation électronique.
Le schéma définit par Vaart et Merz présente l’énergie d’interaction, comme une somme de
trois termes : électrostatique, polarisation et transfert de charge. Contrairement au schémas
basés sur des approches purement quantique la méthode de Vaart et Merz à la caractéristique
d’être appliqué aux méthodes semi empirique MNDO, AM1, PM3 (Dewar and Thiel 1977;
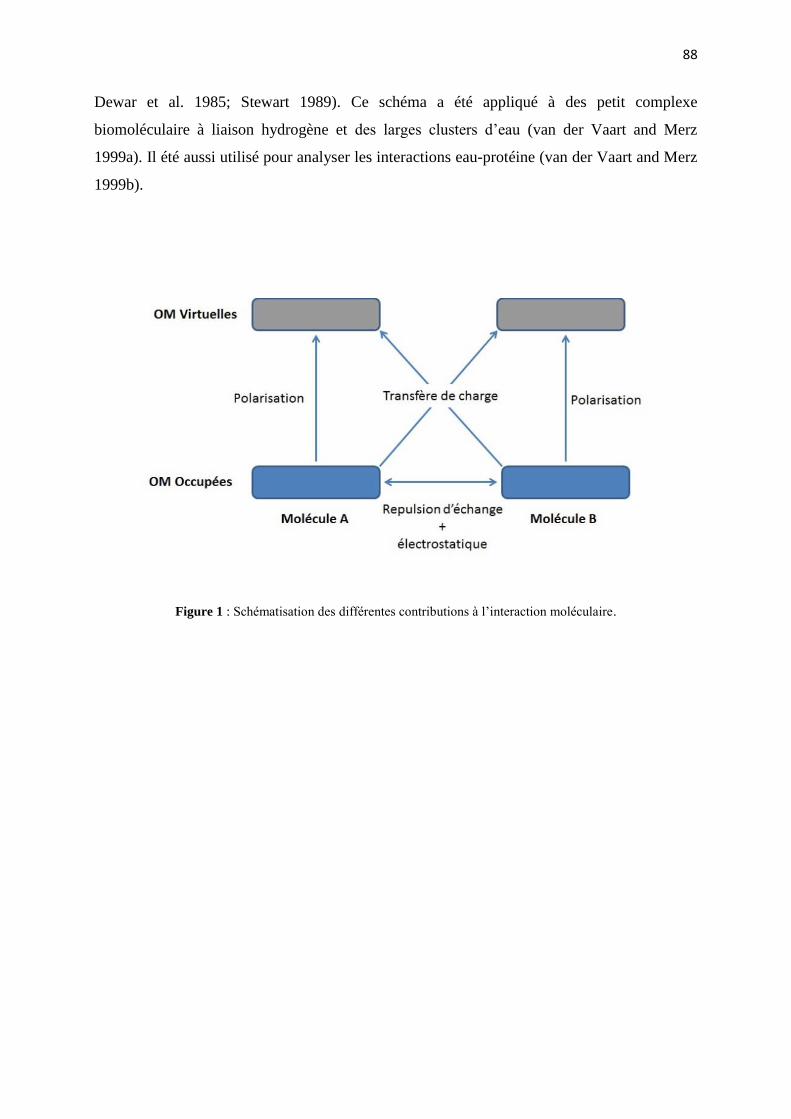
88
Dewar et al. 1985; Stewart 1989). Ce schéma a été appliqué à des petit complexe
biomoléculaire à liaison hydrogène et des larges clusters d’eau (van der Vaart and Merz
1999a). Il été aussi utilisé pour analyser les interactions eau-protéine (van der Vaart and Merz
1999b).
Figure 1 : Schématisation des différentes contributions à l’interaction moléculaire.

89
Partie A : Rôle stabilisateur des cations métalliques dans
systèmes biologique

90
A.1. Introduction :
Le corps humain est majoritairement composé d’oxygène, de carbone, d’hydrogène et d’azote
ces quatre éléments constituent 96% de notre masse totale. Les ions alcalins et halogénures, le
soufre et le potassium constituent eux même l’énorme majorité des 4 % restant. Les cations de
métaux de transition ou de métaux lourds sont, quant à eux, présents sous forme de traces et
ne dépassent jamais 0.1 %. Les ions métalliques jouent un rôle fondamental, et accomplissent
des fonctions spécifiques associées à des processus biologique chez l’ensemble des êtres
vivants.
Plus de 25000 métalloprotéines ont été recensées dans la base de données PDB (Protein Data
Bank) et elles ont toutes des propriétés catalytiques ou structurales pour lesquelles, un métal
est nécessaire (Peters et al. 2010). Par exemple le magnésium dans la chlorophylle des
organismes photosynthétiques, le cobalt dans la cobalamine (vitamine B12) essentielle au
métabolisme des acides nucléiques, le fer dans l’hémoglobine des mammifères, le cuivre dans
l’hémocyanine des mollusques et des arthropodes etc.
Les métalloprotéines, dont font partie beaucoup d’enzymes, constituent un groupe d’une
grande importance pour les organismes vivants. En effet, ces protéines sont impliquées dans
de nombreuses réactions métaboliques dans les bactéries, les plantes et chez les animaux. On
peut citer par exemple, les complexes protéiques de la chaine respiratoire de la mitochondrie
où l’on trouve de nombreux centres Fe-S ; du fer associe a des hemes, ainsi que du cuivre
localise dans la cytochome c-oxydase de même, la dégradation de l’éthanol par le corps
humain est effectuée par l’alcool deshydrogènase, Cette action est rendue possible par la
catalyse opérée par Zn(II) et la tyrosinase qui catalyse l’oxydation des phénols et
spécialement celle de la tyrosine grâce aux systèmes à cuivre de son site actif.
Les cations de métaux lourds, comme ceux du cadmium, du mercure ou du plomb, sont des
entités extrêmement toxiques. Ils se substituent aux cations natifs dans de nombreuses
enzymes et inhibent leur processus catalytique. Ainsi, le mercure, par exemple, peut causer de
sévères disfonctionnements neurologiques. En effet, le mode d’action biologique des métaux
lourds reste encore méconnu, plus particulièrement leur interaction avec leur sphère de
coordination.

91
Tableau 1: Nombre de structures protéinique correspondantes à quelques cations métallique (extrait de PDB,
juillet 2010).
Métal Nombre de structures protéiniques
Zn 5854
Mn 1412
Fe 1403
Cu 763
Cd 521
Ni 463
Co 337
Pt 44
Au 30
Ag 10
A.1.1 Le Zinc : Un élément essentiel
Le zinc est l’un des ions métalliques les plus importants dans la biologie, C’est le deuxième
élément de transition le plus abondant dans les cellules, après le fer (Christianson 1991a). Il a
des implications biologiques importantes, jouant un rôle indispensable dans une large gamme
de processus cellulaires, comme la réplication d'ADN et la transcription, (Wu and Wu 1987),
dans le métabolisme cellulaire, (Murakami and Hirano 2008) dans l’apoptose ou la mort
cellulaire (Fw 1995), et aussi comme agent catalyseur. Le zinc est inclus dans la pathogénie
de plusieurs virus y compris VIH, l’hépatite, herpès simplex, Rubéole, grippe (Lipscomb and
Sträter 1996; Wilcox 1996; Parkin 2004; Chaturvedi and Shrivastava 2005).
A.1.2 Corrélation structure propriété dans les sites à zinc
De nombreuses études antérieurs ont examinés et classifiés la géométrie de coordination, et
les préférences en termes d’acides aminé dans le site du zinc (Christianson 1991b). Patel et
collaborateurs ont estimé que la majorité (environ 82%) des ions de Zinc dans les protéines
adoptent une coordination tétraédrique et le reste sont coordinnés d’une manière soit
pentahedrique (14%) ou exahedrique (4%) (Patel et al. 2007).

92
Les ligands communs pour l’ion zinc sont l’histidine, l’aspartate, le glutamate et la cystéine
présentés dans de variables combinaisons. Dans les protéines, le Zn2+
peut jouer un rôle
catalytique, par une participation directe dans les catalyses chimiques :il acte comme un
électrophile dans les réactions d’hydrolase (McCall et al. 2000). Des travaux antérieurs ont
montrés que dans les sites catalytiques, le Zn2+
est lié référentiellement aux molécules d'eau et
à l’histidine. L’eau est peu encombrante par rapport aux autres ligands et son tau de transfert
de charge vers l’ion métallique est minime, ce qui permet au Zn2+
de jouer le rôle d’un acide
de Lewis dans les catalyses, il peut aussi jouer un rôle structural important (Lee and Lim
2008), en maintenant et en stabilisant la structure de nombreux domaines protéinique, le cas
des protéines à doigt de zinc (zinc-finger proteine) (Berg and Shi 1996).
Dans les sites structuraux le zinc est préférentiellement attaché à la cystéine déprotoné Cys(-)
avec une absence totale de molécules d’eau (Auld 2001), le nombre de ligands Cys(-) attaché
au Zn+2
est supérieur ou égale à deux. Le transfert de charge sera beaucoup plus important, et
l’ion métallique ne pourra pas agir comme un acide de Lewis. En outre la répulsion stérique
entre les groupements Cys(-) préviens la coordination du cation métallique à un autre ligand.
La règle moléculaire qui gouverne cette préférence demeure encore peu claire.
A.2 Energie d’interaction dans un site à zinc
A.2.1 Présentation du système moléculaire
A.2.1.1 Structure et investigation expérimentale et théorique des sites à zinc
Les doigts de zinc sont de petits motifs structuraux trouvés dans les protéines et capable
d’ordonner en complexe un ou plusieurs ions zinc pour stabiliser leurs structure secondaire.
Ils peuvent être classés en différentes familles structurales et fonctionnent typiquement
comme des groupes d’interaction, liant l’ADN, l’ARN, lors des étapes de transcription.
Dans le cas de la protéine de la nucléocapside par exemple (Figure 1), Les doigts de zinc,
appelés encore cite de fixation ou support à zinc sont en fait des structures de la forme Cys-
X2-Cys-X4-His-X4-Cys où X désigne des acides aminés variable (Morellet et al. 1992; Darlix
et al. 1995).
Ces motifs (CCHC) sont capable de chélater le Zn2+
de manière stœchiométrique et avec une
très puissante affinité (South et al. 1990; Bess et al. 1992; Bombarda et al. 2001; Bombarda et

93
al. 2007). Le cation métallique Zn2+
est coordonné d’une manière tétraédrique par les
groupements thiols des cystéines est amino de l’histidine (Berg 1986; Green and Berg 1989).
Selon un mécanisme présentant plusieurs états intermédiaire (Bombarda et al. 2007) la liaison
du zinc est responsable de la structuration du doigt, permettant la transition d’une structure
aléatoire vers une forme repliée, fortement contrainte, à l’origine de ces fonctions biologiques
(Summers et al. 1992). La complexation de ces motifs avec le zinc est indispensable pour la
formation de protéines active. Le repliement du doigt de zinc est stabilisé seulement par la
fixation de l’ion métallique. Cette propriété lui permet d’être étudier indépendamment de la
protéine qui le contient (Berg and Shi 1996; Cox and McLendon 2000).
Figure 1 : Strucure du HIV-1 Nucleocapsid NCp7(12-53) complexé avec un fragment d’ADN.

94
Des caractéristiques importantes ont été révélées (Frankel et al. 1987; Nomura and Sugiura
2002; Blasie and Berg 2002; Bombarda et al. 2005; Zhang et al. 2009) par le biais de quelques
travaux expérimentaux et théoriques centrés sur l’identification des facteurs responsables du
repliement et la de stabilisation des doigts de zinc. La prédiction par des méthodes théorique
des structures protéinique à zinc par l’incorporation explicite et la modélisation du cofacteur
métallique (Sodhi et al. 2004; Schymkowitz et al. 2005; Kornhaber et al. 2006; Calhoun et al.
2008; Shu et al. 2008; Seebeck et al. 2008) ont permis la connaissance des positions des sites
potentielle coordonnant le Zinc dans des nouvelles séquences peptidiques synthétisé in vitro
et aussi l’amélioration de l’échantillonnage près de la conformation native (Wang et al. 2010).
Dudev and Lim ont prédit l’ordre d’attachement des quatre résidus cordonnant le zinc par des
méthodes quantique (Dudev and Lim 2007).
L’étude théorique des ions de Zinc incorporés dans les sites actifs des protéines a été un défi
de longue date. Les méthodes de mécanique quantique (Kothekar et al. 1978; Raha and Merz
2004) et les méthodes hybride mécanique quantique/mécanique moléculaire (Fatmi et al.
2006a; Fatmi et al. 2006b; Fatmi et al. 2007; Fatmi et al. 2010) ont été appliquées pour
modélisé ce genre de système, mais le cout calculatoire pour des niveaux ab initio plus poussé
ou pour des âges de simulation plus long, rend difficile l’exploration de leur espace
configurationel. D’autre part, le traitement par la mécanique classique de l’ion de zinc est
difficile à manipuler, ceci est dû au fort champ électrostatique local, ainsi que l’effet
d’induction important (Wu et al. 2010b). La difficulté vient aussi du fait que les méthodes
traditionnelles de mécanique moléculaire (Merz and Kollman 1989) sont basées sur le modèle
des charges fixe.
Cependant, la modélisation des phénomènes plus complexe comme les réactions sur sites
catalytique ainsi que le repliement couplé à des ions métalliques demande l’utilisation des
champs de forces plus développés (Halgren and Damm 2001), qui traitent les effets
électrostatique d’une manière plus explicite, afin de caractériser les effets de protonation et de
déprotonation et des effets de transfert de charge entre le cofacteur métallique et ces ligands
potentiels. Ces effets ont été abordés il y a plus de dix ans par Gresh et collaborateurs et les
champs des forces polarisables ont été développés pour inclure ces effets (Gresh 1995; Gresh
and Garmer 1996; Rogalewicz et al. 2000; Gresh et al. 2005).

95
A.2.2 Description d’un ion métallique dans un champ de force
Plusieurs approches ont été mentionnées dans la littérature, pour l’incorporation d’un atome
métallique dans un champ de force de mécanique moléculaire (Hoops et al. 1991; Bredenberg
and Nilsson 2001) (Figure 2):
A.2.2.1) Le modèle lié: Il définit les liaisons, les angles, et les angles dièdre entre le centre
métallique et ces ligands. Hancock (Hancock 1990) avait utilisé cette approches pour étudier
des systèmes contenant du Cuivre et du Nikel .Il faut noter aussi que la majorité des champs
de force dédié aux composés organiques sont développés initialement autour d’un seul angle
d’équilibre, mais dans les sites métalliques, les ligand peuvent s’arranger dans différentes
géométries et dans ce cas il est préférable de définir plusieurs valeurs pour l’angle d’équilibre.
Par exemple dans le cas d’une coordination octaédrique, l’angle à l’équilibre peut prendre les
valeurs 90o, 180
o, 270
o, mais cela conduit à un problème nommé (unique-labelling problem).
Cela devient encore plus compliqué en prenant en compte, que le même ion métallique peut
exposer plus qu'une conformation (Tétragonale contre trigonal ou quatre - contre six-
Coordination).
A.2.2.2) Le modèle non lié: Il ne définit aucun lien dans la sphère de coordination, il utilise
les interactions électrostatiques et de van der Waals pour modélisé les interactions de l’ion
métallique avec son environnement chélateur. Par exemple Donini et Kollman ont rapportés
des études basées sur cette approche pour décrire l’attache d’inhibiteurs à des matrices
métalloproteinases (Donini and Kollman 2000) et basant sur ce modèle, Sakharov et Lim ont
considérés les effets de transfert de charge entre le zinc et ces ligands ainsi que la polarisation
locale autour de l’ion du Zinc (Sakharov and Lim 2005). Dans ce modèle la charge placé sur
l’ion métallique est décrite par un nombre entier (Stote and Karplus 1995b) et le nombre de
coordination et la géométrie des doigts de Zinc de type Cys-Xn-Cys-Xn-His-Xn-His (CCHH)
et Cys-Xn-Cys-Xn-Cys-Xn-Cys (CCCC) peut être bien décrite.
A.2.2.3) Le modèle des charges cationique fictives: Il dérive du modèle non lié, place
quatre atomes cationiques fictifs autour du centre métallique d’une manière tétraédrique pour
imiter les électrons de valences du métal (Åqvist and Warshel 1992). Dans ce modèle
l’orientation des coordonnées du zinc est imposée tout le temps pendant la simulation. Les
modèles liés et semi liés rigidifient la coordination de zinc à son environnement ce qui
entraine des artéfacts sur l’échantillonnage conformationnel et sur la dynamique.

96
La variabilité de la coordination de lion de zinc peut être liée à la fonction des
métalloenzymes aux différentes étapes de la réaction (Christianson 1991a), mais l’incapacité
de donner un modèle qui peut décrire l’ion de zinc d’une manière très exacte reste une entrave
pour comprendre les fonctions multiples de cette ion.
Figure 2: Modèles d’incorporation d’un métal dans un champ de force. (M) désigne le métal, R1, R2, R3, R4
représentent les diffèrent résidus coordinateur.
A.3 Etude de l’énergie d’interaction et de l’effet de mutation sur le site à zinc
structurale: Application de l’approche LSCF/MM.
La propriété de fixation de l’ion de zinc dans les sites de coordination structurale nous a
motivés à évaluer son rôle dans la stabilisation d’une structure tridimensionnelle d’un
fragment métallo protéinique. Pour cela, nous nous somme orienté dans cette partie vers
l’évaluation de l’énergie d’interaction qui s’établie entre le cation métallique est ces diffèrent
résidus chélateurs au niveau d’un doigt de zinc examiné à l’état natif et muté en adoptant
l’approche de la super molecule et en appliquant la méthode LSCF/MM.
Dans un premier temps, nous discutons la qualité des paramètres structuraux de la sphère de
coordination de l’ion de zinc dans la structure native. Les paramètres obtenus à partir de deux
niveaux de calculs par le champ auto-cohérent local LSCF/MM ont été comparés avec ceux
qui sont issus de la structure obtenu par RMN.

97
A.3.1 Méthodologie
La structure (1DSQ.pdb) (Klein et al. 2000) correspondant au doigt de zinc, issu de de la
banque de donné des structures protéinique (RCSB) a servi comme point de départ à cette
étude. Le support de coordination du cation métallique 𝑍𝑛2+ (Zn2+
CCHC), représente le sous-
système QM. Ce dernier inclut principalement les groupements 𝐶𝛽𝐻2 − 𝑆 des trois cystéines
déprotoné et 𝐶𝛽𝐻2 − imidazole de l’histidine (Fig. 2). Le reste du fragment peptidique
représente le sous-système classique ou la partie MM, traitée en utilisant les paramètres du
champ de force CHARMM27.
Quatre orbitale de liaison strictement localisé (SLBO) ont été placées dans les frontières
QM/MM repéré entre deux atomes de carbones formant une liaison simple 𝐶𝛼 − 𝐶𝛽 (Fornili
et al. 2006a) pour traiter les liaisons pendantes. Ceci est motivé par le fait que la polarité de
cette liaison est faible, et trés proche de celle d’une liaison 𝐶 − 𝐻 utilisé par les schéma Link
atomes.
En considérant l’hypothèse de transférabilité des propriétés chimiques, l’orbitale localisée est
déterminée sur une molécule d’éthane, qui est la plus petite molécule modèle contenant ce
type de liaison formé par deux carbones hybridés sp3. Le critère de localisation ainsi que le
schéma d’orthogonalisation que nous avons utilisé sont respectivement de Boys
Foster+Pipek-Mezey et de Gram-Schmidt. Tous les calculs LSCF/MM ont été réalisés avec la
version modifiée de la suite Gaussian03 (Frisch et al. 1988) associé au programme de
mécanique moléculaire Tinker V.4.2 (Ponder and others 2004).
Nous avons utilisé la théorie de la fonctionnelle de densité DFT pour traiter la partie QM. La
fonctionnelle B3LYP qui associe la fonctionnelle d’échange de Becke à 3 paramètres à la
fonctionnelle de corrélation de Lee, Yang et Parr (Lee et al. 1988; Becke 1993) à été choisie
en concordance avec la littérature concernant des composés similaires (Ryde 1999; Minenkov
et al. 2012).
Dans les systèmes comportants des atomes lourds (3ème période et au-delà), les électrons
internes sont remplacés par un pseudo potentiel, décrivant leur interaction avec les électrons
de valence qui sont alors les seuls traités explicitement. Pour cela, nous avons choisi la base à
pseudo potentiel (LANL2DZ) (Wadt and Hay 1985; Roy et al. 2008) car elle représente bien
les systèmes moléculaires contenant un ion métallique. Nous avons également mené un calcul
supplémentaires à un niveau 6-311G, afin de vérifié l’effet de la taille de la base sur les

98
principales grandeurs géométriques du complexe de coordination ainsi que sur la qualité des
énergies d’interaction. Le but serait de comprendre le rôle de l’ion de zinc dans la stabilisation
du fragment peptidique. Les mêmes types de calculs en utilisant le même niveau de théorie
ont été appliqué à ce qu’on appelle les poches (fragment peptidique sans ion métallique) ainsi
que pour l’ion de zinc isolé. Tous les calculs LSCF/MM sont menés en phase gaz.
Figure 2 : Partition LSCF/MM adapté au site à zinc structurale étudieé.
Cette etude a fait l’objet d’une publication dont le titre est :
Titre: Effect of mutation on the stabilization energy of HIV-1 zinc finger : a hybrid local
sel-consistent field/molecular mechanics investigation.
Publiée dans le journal: JBIC Journal of Biological Inorganic Chemistry.
Authors : Nedjoua Drici, Mohamed Abdelghani Krallafa
DOI :10.1007/s00775-016-1411-6
Print ISSN 0949-8257
Online ISSN 1432-1327
Publisher Name : Springer Berlin Heidelberg
First Online : 15 Novembre 2016,
Web: http://link.springer.com/article/10.1007/s00775-016-1411-6

99
A.3.2 Conclusion
Dans cette partie nous avons mené des calculs hybrides LSCF/MM sur un complexe de
coordination d’un site à zinc structurale de type (CCHC). Pour un premier temps nous avons
effectué des calculs d’optimisation du site de coordination en utilisant différentes bases de
calcul afin de vérifier l’effet de la taille de la base sur la qualité des grandeurs géométrique du
site à zinc. Nous avons comparé les paramètres géométriques du cluster déterminé par RMN
avec ceux issus des calculs hybride LSCF/MM dans lesquels les atomes frontières (MM)
n’interagissait avec le fragment quantique que par l’intermédiaire de potentiel de van der
Waals et par l’introduction de charge ponctuelles dans l’Hamiltonien de cœur de ce dernier.
On peut voir clairement que les valeurs moyennent de distance de type (Zn2+
-S) et (Zn2+
-Nε)
calculées avec les bases (LANL2DZ), et (6-311G) sont un peu plus grandes (de quelque
dixième d’Angström), mais sont en bonne concordance avec la valeur trouvée dans la
structure RMN. Les paramètres des angles qui engagent les résidus cystéinates sont fortement
impliqués dans les processus de réarrangement du site de coordination conduisant ainsi à la
distorsion de la coordination tétraédrique du cluster métallique. Ce réarrangement est imposé
afin de diminuer les fortes interactions électrostatiques de répulsions entre les différents
résidus chélateurs. La déviation de quelque paramètres du site de coordination par rapport à la
structure RMN, est due essentiellement à l’effet de l’environnement protéinique qui n’est pas
négligé est qui est modélisé par un champ de force MM (CHARMM27) ceci est facilement
compréhensible, car le champ de force MM n’est pas paramétré pour reproduire les
géométries en phase gazeuse, mais plutôt en phase condensée.
Il est probable que la tension imposé par le squelette peptidique qui est gardé figé, sur le site
de coordination, et les interactions électrostatiques entre la densité électronique de ce dernier
avec les autres résidus du squelette peptidique qui l’enclose et qui sont représentés par des
charges ponctuelle MM, induisent à la modification de quelque paramètre géométriques du
site actif décrit au niveau QM (Bakowies and Thiel 1996) .Cette interaction permet une
polarisation du nuage électronique de la région QM par l’environnement MM.
Il est possible aussi que ces charge ponctuelle exclusivement paramétrées pour certain champ
de force l’ors d’un calcules de mécanique moléculaire, conduise à une description peut
précises de l’effet de polarisation dans la région QM. L’excès de polarisation près de la
frontière QM/MM a été suggéré (Laio et al. 2002a; Das et al. 2002; Guallar et al. 2003;
Biswas and Gogonea 2005; Dułak and Wesołowski 2006) comme une source possible d'erreur
dans les calcules

100
Il faut noter aussi que les interactions courte portée entre les électrons QM et les atomes MM,
ne sont pas décrites correctement car le terme de répulsion de Pauli n’est pas inclus dans le
potentielle d’interaction électrostatiques entre la région QM et MM (Senthilkumar et al. 2008)
Il y’avais un débat dans la littérature (Singh and Kollman 1986; Field et al. 1990;
Waszkowycz et al. 1991; Laio et al. 2002b; Das et al. 2002) sur la possibilité d’une interaction
électrostatique qui a un aspect non physique entre les atomes QM et MM situés près de la
région de partition et qui est traduite par une fuite de charge à partir de la région QM vers les
atomes MM. Le niveau de l’approche utilisé pour réaliser ces calculs peut influencer les
résultats théoriques. Les bases de calculs qui sont choisies ne font pas intervenir des fonctions
diffuses ou de polarisation qui fournissent plus de flexibilité, et qui permettent de placer une
densité électronique loin du noyau devenant ainsi sujet à une surpolarisation.
Finalement cette déviation peut aussi être due à la faible résolution de la structure RMN du
départ.Le nombre et la disposition spatiale des ligands déterminent une certaine stabilisation
du complexe de coordination incorporé dans un fragment protéinique activé par un ion
métallique. Dans les sites structuraux, l’ion de Zinc est complexé avec une très grande affinité
aux thiols, notamment aux Cystéinates. Dans les structures mutées, la perturbation la plus
forte sur l’affinité du site métallique pour l’ion de zinc est observée lorsque le résidu
Cystéinates de la partie amine est muté par un résidu non coordinateur, ceci confirme son rôle
important dans la structuration des doigts de zinc. Par contre la Cystéinates positionné dans la
partie carboxyle est la plus réactive ce qui lui permet d’être impliqué préférentiellement dans
le processus de dissociation suite à sa faible contribution à la stabilisation du site métallique.
Le site de coordination semble ainsi posséder une certaine flexibilité conformationnel qui lui
permet de réarranger de manière optimale ses résidus coordinateurs pour qu’il puisse se lier à
l’ion métallique.

101
Partie B : Orbitales moléculaires localisées et modélisation
des interactions intermoléculaires dans les systèmes
biomimétiques faiblement liées

102
B.1 La liaison hydrogène : liaison de vie.
La liaison hydrogène (Scheiner and Kar 2005) est un cas particulier de liaison non covalente,
marquant ainsi une grande importance dans le domaine de chimie et de la biochimie. Elle est
particulièrement connue pour être responsable de la structure de l’édifice tridimensionnelle de
la glace et de l’environnement d’une molécule d’eau liquide. Notons aussi que la dynamique
des liquides est influencée par sa duré d’existence. Elle joue un rôle dans les diffèrent
processus biologique, au niveau moléculaire, vu son implication dans les propriétés de l’eau
qui est le solvant biologique universel. La structure et les propriétés de beaucoup de
molécules et macromolécules biologique sont déterminées par le biais de ces liaisons qui
retienent les diffèrent segments d’une protéine, en lui donnant ainsi sa forme et ses fonctions.
C’est d’ailleurs lorsque James D.Watson et Francis H.Crick découvrirent que c’est la liaison
hydrogène qui couple les bases (adénine-thymine et cytosine-guanine), qu’ils se mirent sur la
piste de la configuration en double hélice de l’ADN.
B.1.1 Propriété des liaisons hydrogène
Une liaison hydrogène résulte de l’interaction électrostatique entre un atome d’hydrogène (H),
lié par covalence à un atome électronégatif (oxygène, azote, fluor, soufre,…) : (Donneur) et
un deuxième atome électronégatif possédant une paire d’électrons non partagée : (Accepteur).
Il existe plusieurs critères pour définir une liaison hydrogène. Le plus souvent, on utilise une
distance maximale (rmax) entre l’atome (A) accepteur de liaison hydrogene et l’atome(H),
qui peut etre combiné a un angle maximal (θmax) de déviation de la liaison par rapport au cas
idéal ou les atomes A, H et D (donneur de liaison hydrogène) sont alignées (Fig. 5.15).
Plusieurs valeurs de (rmax) associes ou non a un critère angulaire ont été testées (De Loof et
al. 1992). Les critères permettant la meilleure identification des liaisons hydrogènes par
rapport aux données expérimentales, sont : rmax < 2,4 Å et θmax > 135o (Aqvist et al.
1985). Les liaisons hydrogène sont plus fortes que les liaisons de van der Waals, et les acides
aminés polaire peuvent ainsi former des liaisons hydrogène entre eux ou avec les molécules
d’eau. La liaison hydrogène joue un rôle important dans les solvants, et possède une énergie
intermédiaire entre celle des liaisons dipôle-dipôle de Keesom, et celle des liaisons
covalentes.

103
Figure3 : Illustration des paramètres géométriques définissant les liaisons hydrogène.
B.2 Etude théorique des complexes à liaisons hydrogènes trouvées dans des systèmes
moléculaires d’intérêt biologique par l’application de la méthode LSCF- projection
asymptotique
Comme nous l’avons mentionné précédemment, l’énergie d’interaction intermoléculaire est
une observable qu’on peut interpréter par différentes décompositions qui ont un sens
physique. Buckingham (Buckingham 1967) a proposé par exemple une décomposition de
l’énergie d’interaction intermoléculaire en quatre grandes contributions : électrostatique,
induction, échange et répulsion-dispersion. L’interaction électrostatiques est l’ensemble des
interactions coulombiennes des deux densités de charges -ensemble des noyaux et électrons-
isolées. L’interaction électrostatique est additive et peut être répulsive ou attractive selon
l’orientation relative des molécules. Elle constitue la plus grande partie de l’interaction
intermoléculaire, avec une décroissance comme l’inverse de la distance intermoléculaire, dans
le cas où le système interagissant contient des molécules polaires et/ou chargées ou encore des
liaisons hydrogène.
Dans cette partie de la thèse nous utilisons la méthode de la projection asymptotique (AP) qui
représente l’une des variantes de la méthode LSCF ab initio pour décrire les interactions
électrostatiques présent dans un ensemble de complexes faiblement liées à l’aide des orbitales
localisées. Cette méthode n’est pas appliquée pour le traitement des liaisons pendantes dues
aux coupures QM/MM mais pour le traitement des orbitales moléculaires maintenues gelées
d’un résidu en interaction non covalente de type hydrogène avec un autre qui est en état de
relaxation.
Ce ci revient à stabiliser le système donneur accepteur formant le dimère, par la polarisation
de la densité de charge relaxé de l’un des monomère sous l’effet du champ électrique crée par

104
la densité de charge gelé de l’autre monomères dans l’absence totale de tout effet d’échange
d’électrons ou de transfert de charge.
L'objectif premier de cette partie sera de valider la méthode de la projection asymptotique en
prenant comme référence l’ensemble des résultats des travaux mené à l’aide de la méthode
QM:MM sans contrainte par Senthilkumar et collaborateur (Senthilkumar et al. 2008) sur un
ensembles de molécules organique formant des dimère à liaison hydrogène où la réponse
électronique est décrite lorsque la partie QM du calcul QM :MM subit une perturbation qui est
due aux charges ponctuelles de la partie MM.
B.2.1 La projection asymptotique
L’approche de la projection asymptotique (AP, Asymptotic Projection) (Glushkov and Tsaune
1985; Glushkov 1998; Glushkov 2002b; Glushkov 2002a; Glushkov et al. 2008b; Glushkov et
al. 2008a; Glushkov and Assfeld 2012) qui présente l’une des variantes de la méthode LSCF
ab initio a été développée par V.Glushkov pour orthogonaliser les orbitales gelées ou localisé
aux orbitales variationnelles. La section suivante sera consacrée à la validation de cette
approche sur un ensemble de dimères liés par des liaisons hydrogène. Par la suite les
diffèrents phénomènes de transfert de charge et de polarisation ainsi que la comparaison avec
d’autres calculs: QM/MM et QM.
B.2.2. Principe et formalisme de la projection asymptotique
La contrainte d’orthogonalité imposé au orbitales localisées ou gelées dans le formalisme
LSCF est réalisé en trois étapes
i. L’ensemble des orbitales gelées sont orthogonalisées entre elles par la méthode de
Lӧwdin:
|𝑙𝑖′⟩ = ∑𝑎𝑖
′
𝑁
|⟩ (4.2)
ii. chaque fonction de base atomique |𝜇⟩ est projetée dans le sous-espace
complémentaire des OL (OLO) par le biais de la fonction |𝜇′⟩.

105
iii. Les (𝐿) dépendances linéaires engendrées par les (𝐿) orbitales localisées (𝑂𝐿) et
présentes dans l’étape (ii) doivent être enlevées.
Les deux dernières étapes sont rassemblées en une matrice
B = M ∙ X (4.3)
L’approche (AP) est une méthode qui traite aussi des contraintes telles que les orbitales gelées
de manière totalement orthogonales au orbitales variationnelles. Son implémentation dans la
version de Gaussian03 modifiée pour le LSCF, vas nous permettre d’effectuer des
applications sur des systèmes formant des dimères à liaisons hydrogène.
Le projecteur 𝑃𝑢 peut être défini comme suit avec |⟩ et ⟨𝜇| des orbitales atomiques.
𝑃𝑢 =∑|𝑙𝑖⟩⟨𝑙𝑖|
𝑁
𝑖
(4.4)
=∑𝐶𝜇𝑖𝐶𝑖|⟩⟨𝜇| (4.5)
𝑖
Et les éléments de la matrice de Fock modifié 𝐹𝑝𝑞 𝑚𝑜𝑑 deviennent sous forme matricielle
𝐹𝑝𝑞 𝑚𝑜𝑑 = 𝐹𝑝𝑞 + ⟨𝜙𝑝|𝑃𝑢|𝜙𝑞⟩ (4.6)
= 𝐹𝑝𝑞 + (∑𝐶𝜇𝑖𝐶𝑖⟨𝜙𝑝|⟩⟨|𝜙𝑞⟩
𝑖
) (4.7)
Finalement le terme qui faisait intervenir le projecteur est calculé comme le recouvrement
orbitalaire entre les Orbitales atomiques 𝑂𝐴.
𝐹𝑝𝑞 𝑚𝑜𝑑 = 𝐹𝑝𝑞 + ∑𝐶𝜇𝑖𝐶𝑖
𝑆𝜙𝑝𝑆𝜙𝑞 (4.8)
Ce recouvrement est aussi calculé en LSCF pour l’étape 2. Le point commun entre l’approche
LSCF et AP est, en fait, l’utilisation d’un même projecteur mais utilisé de manière différente.

106
En LSCF un projecteur définit par les orbitales localisées modifie le jeu d’orbitales atomiques
pour les rendre orthogonale aux orbitales localisées (OL). Dans la Projection Asymptotique
(AP), ce même projecteur est simplement ajouté à la matrice de Fock.
B.2.3 Quelques applications de l’approche de la projection asymptotique
La Projection Asymptotique (AP) (Glushkov and Tsaune 1985; Glushkov 1998; Glushkov
2002b; Glushkov 2002a) a prouvé d’être un outil utile pour résoudre une large classe de
problèmes de la chimie quantique. Cette méthode peut être montée sous forme d’une équation
aux valeurs propres avec une contrainte d’orthogonalité imposée aux vecteurs propres.
Contrairement aux méthodes conventionnelles pour construire des fonctions d’onde des états
de trou (hole state), l’arrachement des électrons est modélisé en imposant les exigences
d’orthogonalité sur l'orbital d'un état ionisé.
Cette approche a fait l’objet de plusieurs études orientées explicitement à la prospection des
phénomènes d’excitation électronique dans les systèmes moléculaires. Par exemple dans le
travail de thèse menée par Adel Laurent, la méthode de la projection asymptotique a été testée
et validé sur un ensemble de molécules organique, cette méthode a permi aussi d’évaluer les
ionisations de cœur des acides aminés et des glycines de la Sérum-Albumine Humaine
(D.LAURENT 2010).
On peut aussi citer le travail mené par Glushkov et collaborateur (Glushkov and
Mogilevskaya 2013) pour étudier les états doublement excités et ionisés d’un ensemble de
petites molécules (𝐶𝑂,𝑁𝑂, 𝐹𝐻,𝐻2𝑂). La performance de la méthode a été démontré par les
calculs des énergies d’ionisation correspondant à l’arrachement des électrons de la sous
couche intérieur-K.
La méthode COEP (constrained optimized effective potential) (Glushkov and Assfeld 2010;
Glushkov 2013) utilise la théorie indépendante du temps pour traiter des états excités pures
par une simple implémentation de la méthode de la projection asymptotique. Contrairement
aux méthodes dépendantes du temps basés sur des approches de fonctionnelle de densité, la
méthodologie COEP a été appliquée d’une manière facile aux atomes et aux molécules pour
traiter les états excités, doublet, triplet et multiplet.

107
B.3 Etude de la polarisation dans les complexes à hydrogène : application de la méthode
LSCF-projection asymptotique
B.3.1 La théorie de l’incorporation de densité gelée : Frozen density embedding theory
L’étude quantique des systèmes moléculaire de taille réelle, est devenue un objectif pour la
chimie quantique. A cet effet, les algorithmes multi-niveaux et multi-échelles tel que les
approches QM/MM abordent généralement le problème en représentant le système comme un
ensemble de sous-systèmes dont l’interaction est approximative. Dans ce sens, le formalisme
de l’incorporation de la densité gelée (Frozen density embedding) développé par Wesolowski
et warshel (Wesolowski and Warshel 1993) est devenue une voie de recherche de choix. Cette
approche a été appliquée à une vaste gamme de problèmes chimiques, par exemple les effets
de solvants sur différents type de spectroscopie (Humbert-Droz et al. 2013), les propriétés
magnétiques (Neugebauer et al. 2005), les état excités (García-Lastra et al. 2006) et les états
de transfert de charges (Pavanello et al. 2013). Le procédé de frozen density embedding se
projette dans le cadre de la théorie de la fonctionnelle de densité des sous sous-systèmes en
interaction. Dans cette approche la densité électronique du système total est divisée en
contributions de sous-systèmes et peut être déterminée en résolvant des équations couplées
présentant un potentiel d’incorporation efficace (effective potential embedding). De cette
façon, la polarisation donnée par l’interaction des sous-systèmes est incluse. Cette subdivision
de densité totale d’électron en contributions de sous-systèmes, a conduit à l’utilisation de
l’approche de l’incorporation de densité gelée comme technique de calcul de la charge
efficace et de localisation de spin (Pavanello and Neugebauer 2011).
Dans le formalisme de l’incorporation de la densité gelée (Wesolowski and Warshel 1993) la
densité électronique du sous-système (ρI) incorporé dans un environnement donné et qui est
caractérisé par sa densité électronique (ρII) et par un ensemble de charges nucléaire (ZAII) situé
aux positions (RAII), est dérivé des équation Kohn Sham à un électron. Le potentiel effectif
dans cette équation, dérive de l’exigence que la densité totale du système moléculaire (dans
notre cas le dimère) est obtenu par un processus d’optimisation dans lequel, la densité
électronique (ρII) de l’environnement est maintenue gelée.

108
𝜌𝑡𝑜𝑡(𝑟) = 𝜌𝐼(𝑟)+ 𝜌𝐼𝐼(𝑟) (4.9)
[−∇2
2+ 𝑉𝑒𝑓𝑓
𝐾𝑆[𝜌𝐼](𝑟)+𝑉𝑒𝑓𝑓
𝑒𝑚𝑏[𝜌𝐼, 𝜌𝐼𝐼](𝑟)]𝜙𝑖
(𝐼)(𝑟) = 휀𝑖𝜙𝑖𝐼(𝑟), 𝑖 = 1,… ,𝑁𝐼 (4.10)
Dans ces équations, 𝑉𝑒𝑓𝑓𝐾𝑆 [𝜌𝐼](𝑟) represente le potentiel effectif de Kohn-sham du sous-
système isolé (I), et 𝑉𝑒𝑓𝑓𝑒𝑚𝑏[𝜌𝐼 , 𝜌𝐼𝐼](𝑟) est le potential d’integration qui prand la forme suivante:
𝑉𝑒𝑓𝑓𝑒𝑚𝑏
[𝜌𝐼, 𝜌𝐼𝐼](𝑟) =∑−𝑍𝐴𝐼𝐼
|𝑟 − 𝑅𝐴𝐼𝐼|+∫
𝜌𝐼𝐼(𝑟′)
|𝑟′ − 𝑟|𝑑𝑟′
𝐴𝐼𝐼
+𝛿𝑇𝑠[𝜌]
𝛿𝜌(𝑟)|𝜌=𝜌𝑡𝑜𝑡
−𝛿𝑇𝑠[𝜌]
𝛿𝜌(𝑟)|𝜌=𝜌𝐼
+𝛿𝐸𝑋𝐶[𝜌]
𝛿𝜌(𝑟)|𝜌=𝜌𝑡𝑜𝑡
−𝛿𝐸𝑋𝐶[𝜌]
𝛿𝜌(𝑟)|𝜌=𝜌𝐼
4.11)
Où, les fonctionnelles d’échange-corrélation (𝐸𝑋𝐶[𝜌]) et d’énergie cinétique (𝑇
𝑠[𝜌]) sont
définies dans la formulation Kohn Sham de la DFT.
Dans cette partie, nous proposons l’approche LSCF basée sur la méthode de la projection
asymptotique, pour évaluer la densité électronique de la supermolecule (dimère)
Nous commençons par choisir la densité électronique d’une molécule comme (ρI) et permettre
à la densité d’électrons gelée (ρII) de se détendre en une série d’itérations successives (voir le
schéma illustré sur la figure 4. Le processus itératif est terminé avec le même critère de
convergence (10-6
Hartree) de la procédure SCF. On fait deux séries de calculs itératifs pour
chaque dimère. Dans la première série, (ρI) dans la première itération représente la densité
électronique de l’accepteur, et dans la deuxième série (ρI) dans la première itération
représente la densité électronique du donneur.

109
Figure 4: chémat de l’évaluation itérative de la densité électronique de la supermolecule en utilisant les
évaluations des équations Kohn-sham avec contrainte sur la densité électronique.
B.3.2 Système moléculaire étudié et détails calculatoires
L’ensemble des complexes à liaison hydrogène étudiés, dans lesquelle l’un des monomère
agit comme donneur de liaison hydrogène et l’autre comme accepteur sont représentés sur la
Figure 5. On peut voir qu’un monomère peut jouer le rôle d’un donneur dans un complexe,
comme il peut jouer le rôle d’accepteur dans d’autre complexe, c’est le cas par exemple de
l’imidazole, où il acte comme donneur dans (imidazole–water, imidazole–acetate) et comme
accepteur dans (water–imidazole, phenol–imidazole). On peut voir aussi que l’ensemble des
dimères sont formés par une seule liaison hydrogène sauf pour les complexes (imidazole–
acetate, water–acetate, guanidinium–acetate), on trouve deux. Des complexes moléculaire à
l’état chargé ont également été traité tel que (imidazole–acetate, water–acetate)

110
Figure 5 : Representation des different complexes a hydrogène étudiées.
Water-N-methylacetamide Formamide-water
Imidazole-water
Water-dimer
Acetate-imidazole
Water-acetate
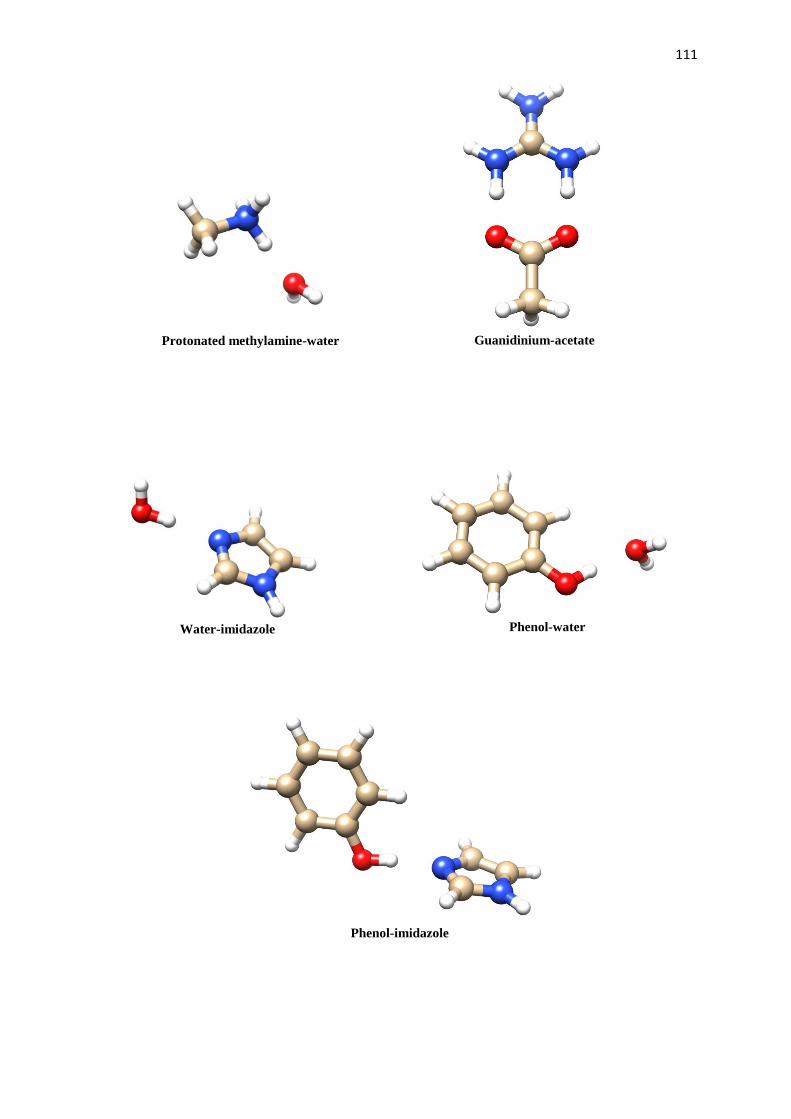
111
Water-imidazole Phenol-water
Guanidinium-acetate
Protonated methylamine-water
Phenol-imidazole

112
L’ensemble des calculs quantiques QM réalisés dans cette partie a été effectué à l'aide du
logiciel Gaussian03. L’implémentation de l’approche projection asymptotique (AP) dans la
version de Gaussian03 déjà modifiée pour le LSCF ainsi que, l’ensemble des modifications
antérieures apportées à ce code ont été réalisés principalement par X.Assfeld, V.Rooy,
N.Ferré, Y.Moreau, et P.F.Loos. Les calculs QM/MM ont été effectués par le couplage entre
le programm Gaussian03 et le programme de modélisation moléculaire Tinker 4.2. Ce
couplage a été réalisé par N.Ferré. Nous avons choisi de traiter les dimères biomimétiques
grâce à la théorie de la fonctionnelle de la densité (DFT).
L’ensemble des dimères a été initialement optimisé à un niveau purement quantique (QM) par
la fonctionnelle B3LYP qui a été choisie en accord avec la littérature (Rablen et al. 1998;
Rappé and Bernstein 2000) et combiné avec la base Split Valence triple zeta 6-311+G(d,p).
Nous avons déterminé les densités de charge pour chaque monomère à l’aide d’un calcul
effectué sur leur géométrie qui sont issus des complexes optimisés. Nous avons également
effectué pour chaque dimère deux calculs séparé de type QM:MM standards. Dans le premier
calcul, le donneur de la liaison hydrogène désigné par (D) a été traité à un niveau QM tandis
que l’accepteur qui est désigné par (A) a été traité par la méthode MM décrite par le champ de
force CHARMM27, dans le deuxième calcul les rôles sont inversés (traitement QM pour
l’accepteur et MM pour le donneur).
Dans le cadre de la méthode LSCF-projection asymptotique, la modélisation des interactions
électrostatiques pour chaque dimère est réalisée à l’aide d’un protocole calculatoire, dans le-
quel on gèle les orbitales moléculaires de l’un des monomère afin de créer une densité de
charge fixe qui vas permettre de polariser la densité de charge du monomère voisin gardé à
l’état relaxé. Cette procédure rentre dans le cadre de la théorie appelée frozen density
embedding. Nous reviendrons plus explicitement sur ce terme dans la suite de cette partie.
Contrairement au calcul QM:MM standard, cette procédure va nous permettre de modéliser
les interactions électrostatiques dans chaque dimère en l’absence totale des phénomènes de
transfert de charge.
On réalise deux séries de calculs formé de plusieurs étapes pour chaque complexe. Dans la
première série on commence par geler les orbitales moléculaires du donneur et on laisse les
orbitales moléculaires de l’accepteur à l’état relaxé. On collecte à chaque fin de calcul les
coefficients des orbitales moléculaires, obtenu sous l’effet de polarisation, pour les réutiliser
comme un nouveau (guess) dans l’étape suivante-les réinjecter dans le fichier input et les

113
déclarer gelée. Cette modification vas permettre d’inverser les rôles dans cette nouvelle étape
(donneur relaxé et accepteur gelé…et ainsi de suite) jusqu’à convergence complète de
l’énergie totale du dimère. On refait le même protocole calculatoire pour la deuxième série de
calculs, dont la première étape consiste à gelé les orbitales moléculaires de l’accepteur et
laisser celle du donneur à l’état relaxé, après on inverse jusqu’à convergence total. Il est bien
évidemment important d’utiliser le même niveau de calcul pour le dimère et les monomères
pour les calculs QM, QM/MM, et LSCF basé sur la technique de projection asymptotique. Les
résultats de cette partie ont fait l’objet d’un article qui est sous examen.
B.3.3 Résultats et discussion
Le terme de polarisation est dû à la déformation mutuelle du nuage électronique de chaque
monomère dans les dimères à hydrogène. Dans les calculs QM/MM les deux sous systèmes
QM et MM interagissent via électrostatique embeding et dans ce cas, la densité de charge QM
est totalement polarisée par la charge ponctuelle MM. dans l’approche LSCF la densité de
charge qui est gardée à l’état relaxé interagit avec le champ moyen créé par la densité de
charge gelée selon le schéma frozen density embeding.
Les charges de Mulliken, issus des calculs, QM pure, QM:MM et les deux série de calculs
LSCF désigné respectivement par LSCF-A et LSCF-D ont été effectué sur chaque dimère et
ils sont illustré sur la Figure 6. La charge de Mulliken des diffèrents dimères ( Fig. 6(a), Fig. 6
(b), Fig. 6 (c), Fig. 6 (d), Fig. 6 (e), Fig. 6 (f), Fig. 6 (g), Fig. 6 (h), Fig. 6 (i), Fig. 6 (j), Fig. 6
(k))issu des calculs QM sont utilisés comme référence et ils sont représentés par une ligne
noir solide. Les charge issues des calculs QM:MM, LSCF-A et LSCF-D sont représentées
respectivement par (triangle vide orienté vers le haut), (triangle gris orienté vers le haut), et
(triangle noire orienté vers le bas)
Nous pouvons voir à partir des différents graphique que les charges q(LSCF-A) et q(LSCF-D)
sont en bonne concordance avec les charges q(QM) et q(QM:MM) pour les dimères neutre
(Fig. 6 (a): N-methylacetamide–Water, Fig. 6 (b): Water–Formamide, Fig. 6 (f): water dimer,
Fig. 6 (g): Water–Imidazole). Ceci montre que la déformation de la densité électronique entre
le donneur et l’accepteur dans les complexes mentionnés précédemment, est bien décrite par
la méthode LSCF.
La déformation de la densité électronique sous l’effet de polarisation est beaucoup plus
importante dans le system où l’un des partenaires est chargé (Fig. 6 (c)Water–Protonated
methylamine, Fig. 6 (d) Acetate–Water and Fig. 6 (e): Acetate–Imidazole).

114
De grande différence sont enregistré entre les charge q(LSCF) et q(QM) pour les systèmes
dans lesquels le cycle imidazole agit une fois comme un donneur (Fig. 6 (g): Water–
Imidazole) et une fois comme accepteur (Fig. 6 (h): Imidazole–Water). On peut aussi voir de
larges différences entre les charges q(LSCF) et q(QM) dans les systèmes π-conjuguées (Fig. 6
(i): Water–Phenol and Fig. 6 (j): Imidazole–Phenol)).
Dans le cas de la paire d’ion guanidinium-acetate, on peut voir que les charge q(LSCF-A)
diffère peut par rapport au charges q(QM) comparé aux charges q(LSCF-D). La grande
déviation des q(LSCF) par rapport à la référence q(QM) observé dans les complexes chargé et
π-conjuguées est due essentiellement à l’excès des effets de polarisations qui sont due
essentiellement aux interactions électrostatique courte portée. Cette déviation peut également
être attribuée à la nature de la liaison hydrogène établie dans les complexes contenant des
espèces chargées (Izgorodina and MacFarlane 2011) . Alkorta et collaborateurs (Alkorta et al.
2016) ont démontrés que la nature des liaisons hydrogène dans les complexes chargés est
directement liée aux composantes de répulsion qui permet de retrouver la dépendance des
profils énergétiques avec la distance dans les système chargé. Nos résultats indiquent que
l’approche LSCF basée sur le formalisme projection asymptotique est raisonnablement
précise surtout pour les complexes neutres, cependant, d’autres développements sont
nécessaires, particulièrement, dans le cas d’un large recouvrement entre les densités
d’électrons des fragments en interaction.

115
Figure 6 : Representation graphique des charge de Mulliken issus des calculs QM, QM:MM, LSCF-A et LSCF-
D et représenté respectivement par ligne solide, triangle vide orienté vers le haut, triangle gris orienté vers le
haut, et triangle noire orienté vers le bas

116
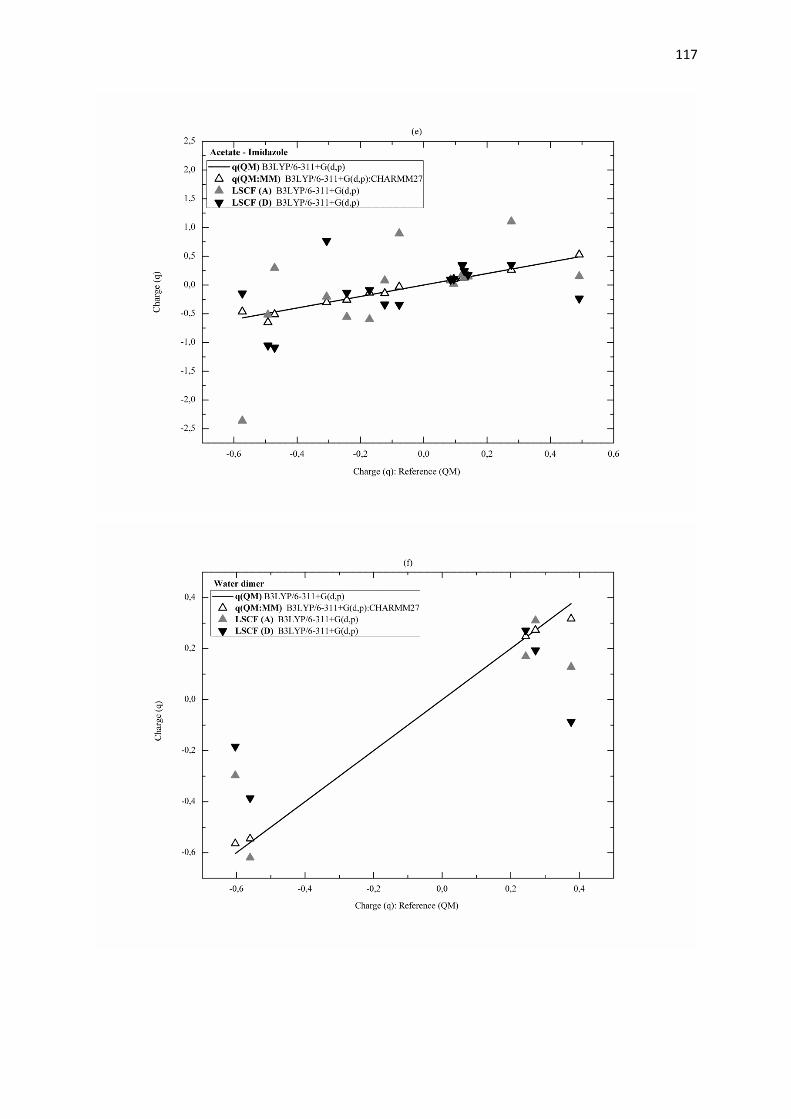
117

118
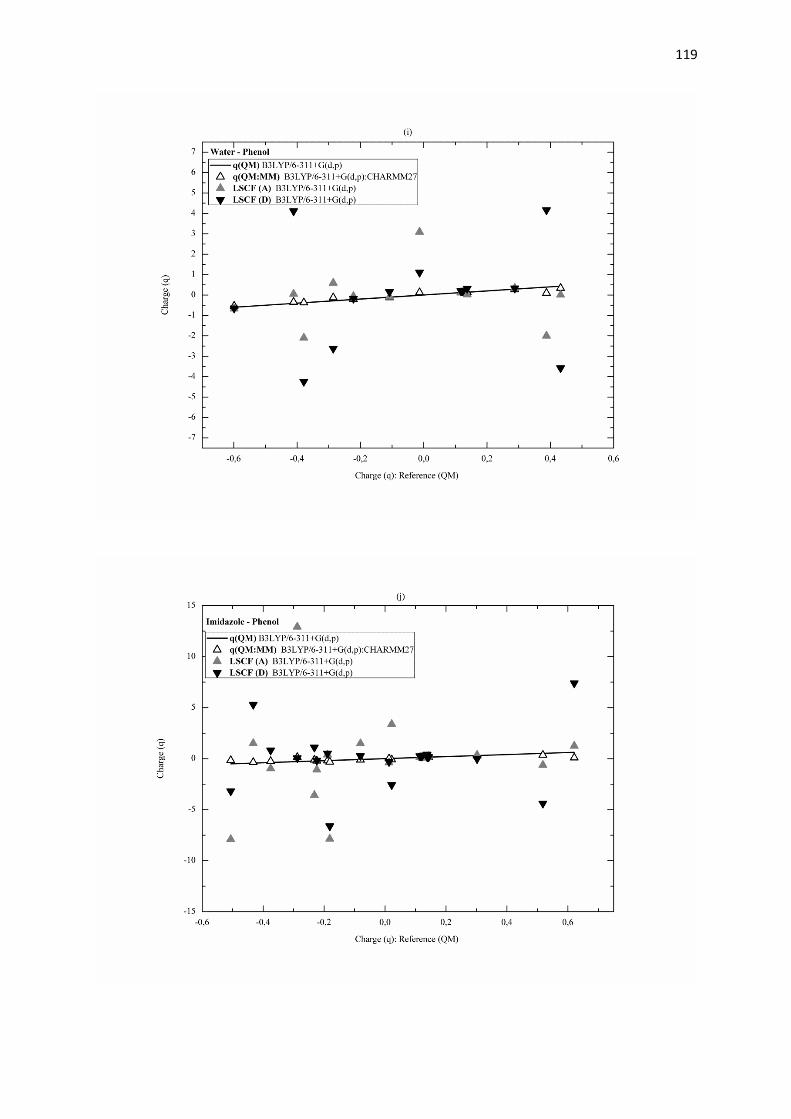
119
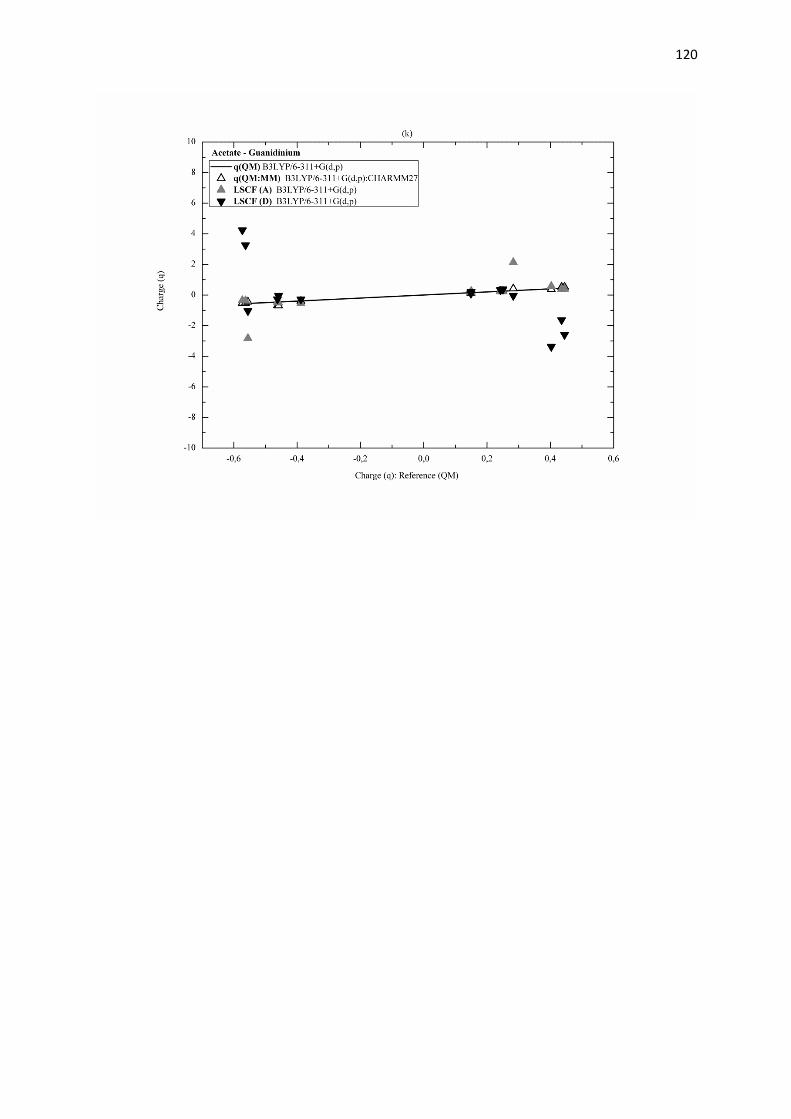
120

121
B.3.4 Conclusion
Dans cette partie, l’effet de polarisation d’un ensemble de complexes biomimétique à liaison
hydrogène de natures neutre et chargés a été étudié par la méthode LSCF. Cette approche a
été utilisée pour mesurer la délocalisation de la densité électronique intermoléculaire d’un
fragment incorporé dans le champ créé par la densité électronique gelée d’un autre fragment.
Nous avons trouvé que la méthode LSCF basée sur la théorie de l’incorporation de densité
gelée est capable de nous fournir une description raisonnable de la réponse de polarisation
dans les complexes neutre, ce qui n’est pas le cas pour les complexes chargés et π-conjugués
Une modélisation fiable et réaliste des effets électrostatique à courte portée, dans les systèmes
chargé à liaison hydrogène par la méthode LSCF, suggère que la distance entre les sous –
systèmes gelés et relaxés soit manipulé avec précaution.

122
Partie C: Investigation par dynamique moléculaire

123
C. 1 Introduction
Un nombre d'études expérimentales et théoriques ont démontré l'importance des interactions
longues portées protéine-ion dans lequel des contributions significatives interviennent pour
des distances aussi grandes que (15 Å) (Russell and Fersht 1987; Bashford et al. 1988). Nous
avons effectué une étude par dynamique moléculaire qui fait intervenir un champ de force
classique, afin d’avoir un aperçu de l’effet du rayon de van der Waals de l’ion de zinc sur
l’évolution des paramètres géométrique du complexe de coordination situé dans le fragment
peptidique. Pour cela nous avons adopté le modèle non liée, présentée par Stote et Karplus qui
a été largement utilisé suite à sa simplicité, pour examiner la structure et la dynamique des
protéines à Zinc autour de leur état natife. Le modèle non lié ne prédéfinit pas des paramètres
structuraux (liaison, angles…etc) et place une charge formelle sur l’ion métallique (Koca et
al. 2003).
C.1.1 Paramètres de van der Waals du zinc
Plusieurs valeurs de paramètres de van der Waals ont été proposées dans la littérature, pour
traiter l’ion de zinc à l’état solvatée ou trouvé dans des environnements biologiques. Il faut
noter que les paramètres de van der Waals peuvent être combiné différemment et injecté dans
différents champs de force selon la nature du système étudié. Par exemple les paramètres de
de van der Waals permettant de reproduire l’énergie libre de solvatation trouvé
expérimentalement pour l’ion de zinc ainsi que pour 22 autre dication, sont de l’ordre de
(σ=1.57 Å, ε= 0.183 Kcal/mol) (Sakharov and Lim 2005; Babu and Lim 2006). Ces
paramètres ont été adopté par Li est collaborateurs (Li et al. 2008) et combiné avec le champ
de force modifier AMBER ff03 pour le traitement du phénomène de repliement du motif de
zinc Cys2His2. Dans l’étude par dynamique moléculaire des effets de polarisations du zinc
dans la région 1-16 de l’amyloïde peptide mené par Huang et collaborateur , deux ensemble
de paramètre ont été utilisé pour l’ion de zinc : celle du champ de force CHARMM22 (σ=1.09
Å, ε=0.25 Kcal/mol) et celles qui ont été trouvés à partir d’un ajustement des paramètres de
CHARMM22 : (σ=0.88 Å, ε= 0.183 Kcal/mol) et qui permettent de reproduire l’énergie libre
d’hydratation de l’ion de zinc. Hopps et collaborateur ont proposé des valeurs (σ=1.1 Å,
ε=0.0125 Kcal/mol) (Hoops et al. 1991) qui ont été introduite dans le champ de force
AMBER pour modélisé l’ion de zinc incorporé dans l’enzyme HCAII (human carbonic
anhydrase II). Les valeurs (σ=1.4 Å, ε=0.01 Kcal/mol) ont également été combinées avec
AMBER lors de l’étude par mécanique moléculaires de l’enzyme (Alcohol dehydrogenase)

124
(Ryde 1995a; Ryde 1995b; Ryde 1996). Les paramètres (σ=1.21 Å, ε=0.23 Kcal/mol) (Wu et
al. 2011) ont été injecter dans le champ de force SLEF (short-long effective Functions force
field) qui présente une stratégie pratique de surmonter le défi célèbre de modéliser le zinc
divalent dans les métalloprotéines.
Les paramètres du champ de force GROMOS96-ff53A6 ont été choisi pour les systèmes
étudiés. Cependant deux ensembles de paramètres de van der Waals ont été discuté. Comme
indiqué dans la Table 2, nous avons utilisé pour l’ion de zinc, les paramètres adopté dans les
travaux de Stote et Kraplus (Stote and Karplus 1995a) ainsi que celles qui sont issus du
champ de force CHARMM22 (MacKerell et al. 1998). Ces dernière, ont la caractéristique de
reproduire le nombre de coordination et les distance Zn-O trouvées expérimentalement dans
la première sphère de coordination du zinc solvatée dans l’eau, ainsi que la reproduction de
son énergie libre d’hydratation trouvé expérimentalement.
Table 2: Paramètres de Van der Waals de l’ion de zinc testés
VDW1 VDW2
ε (Kcal/mol) 0.25 0.25
σ (Å) 1.95 1.09
C.1.2 Protocole de simulation
La structure du fragment protéinique du départ est issue de la base PDB (proteine data bank)
sous le code 1DSQ.pdb. Les résidus Cystéine et l’azote NE du résidu histidine cordinnant
l’ion de Zinc, ont été dans leur état déprotoné. Pour l’ensemble des paramètres VDW1 et
VDW2, le fragment peptidique a été solvatée dans une boite cubique en présence de
molécules d’eau du type SPC. La taille de la boite d’eau a été définie de telle sorte que la
distance entre la proteine et murs soit de (9 Å) (Figure. 7). Nous avons considéré que les
systèmes étudiés se trouvaient à un pH neutre. Dès lors, les résidus aspartate et glutamate ont
été déprotoné tandis que les acides aminés ; lysine et argénine sont protoné et le but d’avoir la
neutralité électrique du système, des contre ion (Na+, Cl
-) ont été ajoutés et distribués d’une
manière aléatoire autour du fragment peptidique. Chaque système a été relaxé pendant 10 ps
après avoir été minimisé par la méthode (steepest descent). Deux simulations indépendantes
par dynamique moléculaire de 100 ps ont été réalisées avec le logiciel GROMACS (Scott et

125
al. 1999) combiné avec le champ de force GROMOS96 (van Gunsteren et al. 2002) à la
température 300K dans un ensemble NPT. Les interactions électrostatiques sont
préférablement traitées par la méthode PME (particle-mesh Ewald) (Darden et al. 1993;
Essmann et al. 1995) lors de la simulation des systèmes contenant un ion métallique, ceci afin
de compenser la limitation des champs de force classiques a traité explicitement ces effets
électroniques et aussi pour mieux géré les fortes interactions électrostatiques de longues
portées.
Figure 7: Boite de simulation de du fragment peptidique (1DSQ.pdb) dans l’eau.

126
C.2 Résultat et discutions
C.2.1 Effet du rayon de van der Waals de zinc sur :
a) les paramètres structuraux du site de coordination
Les paramètres de distance formés entre l’ion métallique et les atomes chélateurs, ainsi que la
distribution des angles dans le complexe de coordination, présentes les éléments importants
des dynamiques moléculaires effectuées sur la structure native. L’évolution du comportement
dynamique du site de coordination va nous permettre de vérifier la qualité des paramètres de
van der Waals de l’ion de zinc, injecté dans le champ de force GROMOS96, et leur aptitude à
maintenir la structure protéinique comparable à la structure du départ. La Figure 8 représente
l’évolution dans le temps des distances (Zn2+
-S) et (Zn2+
-NE). Les fluctuations pendant la
simulation de chaque distance, ont été représentées dans différents graphique pour les deux
ensembles de paramètres de van der Waals : VDW1 et VDW2.

127
Figure 8 : Evolution dans le temps des distances de type Zn-S, et Zn-Nε du site de coordination dans les
structures native modélisé pour les deux série de paramètres de van der waals de l’ion de Zinc VDW1 et VDW2.
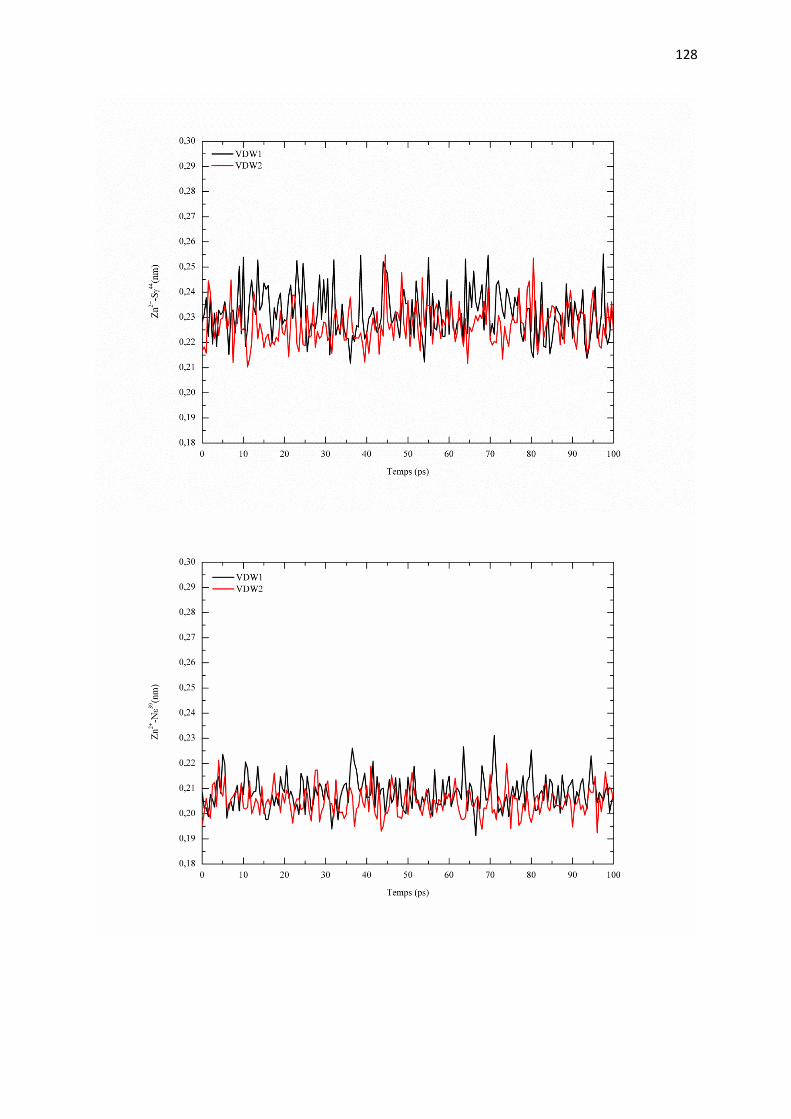
128

129
Les distances entre l’ion de zinc et les différents atomes coordinateurs, ainsi que les angles
formé autour de cet ion sont listé dans la Table 3, est comparé à la structure de référencé
RMN (PDB-ID : 1DSQ.pdb), nous avons également comparé ces paramètres structuraux du
site de coordination, à d’autres données qui sont issues de le la littérature (Alberts et al. 1998)
et trouvé dans des structure protéiniques et de petits complexes à zinc, répertoriés
respectivement dans les bases de données PDB (protein data bank) et la CSD (Cambridge
Crystal Database) (Table 4).

130
Table 3 : Paramètres géométriques du site de coordination de la structure native calculé par dynamique
moléculaire pour les deux valeurs du rayon de de Van der Waals de l’ion de zinc et comparé avec la structure de
référence RMN. Les distance sont en (Å) et les angles sont en degré (º).
Structure Native : (1DSQ.pdb) Dynamique Moléculaire
Paramètres
structurales
RMN VDW1 VDW2
(Zn2+
-S31
) 2,339 2.321 (-0,018) 2.254 (-0,085)
(Zn2+
-S34
) 2.326 (-0,013) 2.277 (-0,062)
(Zn2+
-S44
) 2.315 (-0,024) 2.269 (-0,07)
(Zn2+
-NE39
) 2.082 2.0823 (0,0003) 2.0481 (-0,0339)
(NE39
-Zn2+
-S31
) 97.46 92,02 (-5,44) 92,91 (-4,55)
(NE39
-Zn2+
-S34
) 91,38 (-6,08) 171,12 (73,66)
(NE39
-Zn2+
-S44
) 90,59 (-6,87) 95,47 (-1,99)
(S31
-Zn2+
-S34
) 113.86 89,28 (-24,58) 87,75 (-26,11)
(S31
-Zn2+
-S44
) 91,05 (-22,81) 90,82 (-23,04)
(S34
-Zn2+
-S44
) 172,31 (58,45) 90,69 (-23,17)
RMSDDistance Zn-S, 0.018 0.073
RMSDDistance Zn-NE 0,0003 0,0339
RMSDAngle NE-Zn-S 6.15 42.62
RMSDAngle S-Zn-S 38.90 24.14
Table 4 : Valeurs moyennes des paramètres géométriques du site coordination de la structure native comparé
avec la structure de référence 1DSQ.pdb pour les deux valeurs du rayon de van der Waals de l’ion de zinc. Les
distance sont en (Å) et les angles sont en degré (º).
Paramètres
structurales
Dynamique Moléculaire RMN
(1DSQ.pd
b)
Structures
protéinique
PDB
Petits
complexes à
zinc CSD VDW1 VDW2
(Zn2+
-S) 2.320
(-0,019)
(-0,03)
(0,02)
2.266
(-0,073)
(-0,084)
(-0,034)
2,339 2.35 2.30
(Zn2+
-NE) 2.0823
(0,0003)
(-0,0077)
(0,0623)
2.0481
(-0,0339)
(-0,0419)
(0,0281)
2.082 2.09 2.02
(NE2-Zn2+
-
S)
91,33
(-6,13)
(-16)
119.83
(22,37)
(12,83)
97.46 107 -
(S-Zn2+
-S) 117.54
(3,68)
(7,54)
89.75
(-24,11)
(-20,25)
113.86 110 -

131
On peut voir clairement (Table 4) que les distances de type (Zn2+
-S) et (Zn2+
-NE) sont
beaucoup plus proches des valeurs expérimentales pour le jeu de paramètres VDW1 :
(σ=1.95Å) et fluctuent respectivement autour d’une valeur moyenne de (2.3Å et 2.08Å), avec
une déviation respective de l’ordre de (0.019Å et 0.0003Å).
Dans le but de maintenir l’ion de zinc dans son environnement protéinique, un rétrécissement
important des distance formé entre l’ion zinc et ces atomes chélateur est enregistré pour le jeu
de paramètre VDW2 (σ=1.09Å). Les distances dans ce cas, sont autour de (2.2Å) pour les
(Zn2+
-S) et de (2.04 Å) pour les (Zn2+
-NE) (Table 4). Cette contraction est probablement due,
à la réduction de l’impact des effets d’induction de l’ion de zinc sur la totalité du fragment
peptidique, et leurs limitations à la première sphère de coordination. De plus, la diminution de
l’ensemble des paramètres de distance (Zn2+
-S) et (Zn2+
-NE) vas induire à de fortes
interactions de répulsion, surtout entre les atomes de soufre des résidus cystéinates.
Nous avons préféré de représenter les angles de type (S-Zn2+
-S) et (NE-Zn2+
-S ) sous forme
de distribution (Figure 9(a), Figure 9(b)). Ce qui va nous permettre de voir clairement leur
variation en fonction de la taille du rayon de van der waals de l’ion métallique.
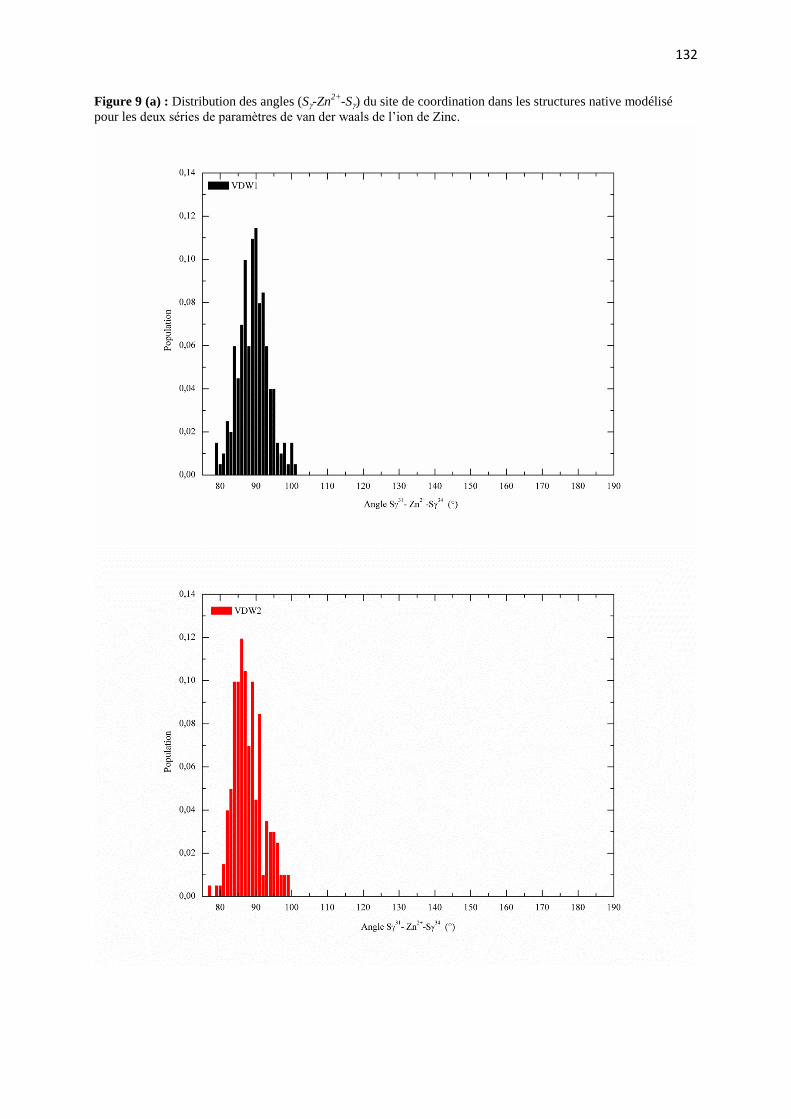
132
Figure 9 (a) : Distribution des angles (S-Zn2+
-S) du site de coordination dans les structures native modélisé
pour les deux séries de paramètres de van der waals de l’ion de Zinc.
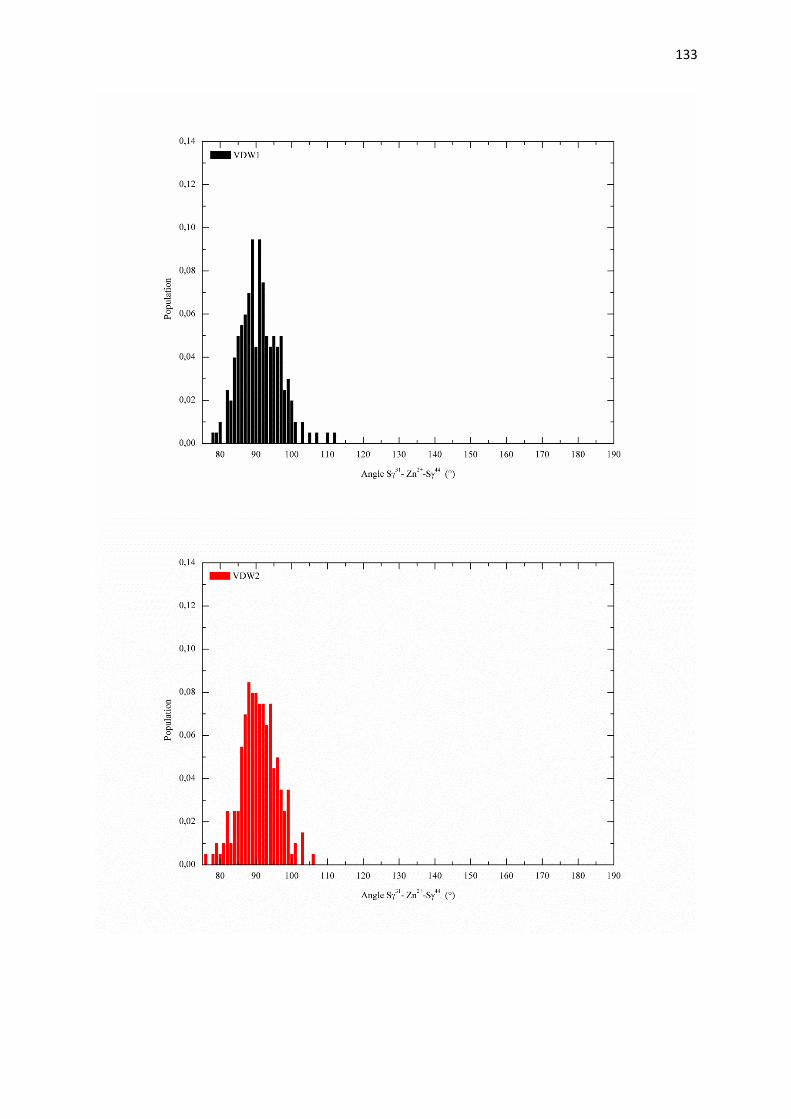
133

134

135
Figure 9(b): Distribution des angles (NE2-Zn2+
-S) du site de coordination dans les structures native modélisé
pour les deux séries de paramètres de van der waals de l’ion de Zinc.

136

137

138
Pour les jeux de paramètres VDW1, où le rayon de zinc est de l’ordre de (σ=1.95Å), on
remarque que l’ensemble des angles (S-Zn2+
-S) sont sujet d’une forte distorsion, suite à un
réarrangement interne du complexe de coordination. Ce dernier passe à la géométrie en mode
octaédrique (Figure 10) suite à la capture et l’insertion de deux molécules d’eau dans la
première sphère de coordination de l’ion de zinc. Selon la Table 3 on peut voir que l’angle
(S34
-Zn2+
-S44
) formé par les résidus Cystéinates en position (34) et (44) est le plus concerné
par cette distorsion car il est les plus exposé au solvant (Khandogin et al. 2003). Les angles de
type (NE39
-Zn2+
-S) fluctuent autour d’une valeur moyenne de (91.33º) ce qui montre que le
cycle imidazole du résidu histidine pointe verticalement sur le plan moyen formé par les trois
cystéinates (Figure 10(a). Le réarrangement conformationnel du site de coordination dans ce
cas, favorisera l’adoption d’une géométrie octaédrique (Figure 10b).
Quant aux jeux de paramètres VDW2 (σ=1.09Å), un élargissement important de l’angle
(NE39
-Zn2+
-S34
) (Table 3) permettra l’introduction d’une seul molécule d’eau à la première
sphère de coordination de zinc (Figure10c). Il y’a une réarrangent des angles (S-Zn2+
-S)
autour d’une valeur moyenne de (89.75º) qui permettra dans un deuxième temps de réduire les
fortes interactions de répulsion entre les résidus coordinateur ce réarrangement
conformationnel du site de coordination favorisera l’adoption d’une géométrie bipyramidale à
base triangulaire (Figure 10d)
La présence des molécules d’eau dans la première sphère de coordination du zinc ainsi que la
distorsion de la géométrie du complexe de coordination, a été déjà observée dans des études
antérieur, réalisé par dynamique moléculaire sur des motifs à zinc (Hok Hei 2012; Zhang et
al. 2012). La non prise en considération des effets quantique de polarisation et de transfert de
charges dans les champs de forces classique représente la cause directe de la présence des
molécules d’eau dans le site de coordination (Li et al. 2008). De plus, le réarrangement
conformationnel du site de coordination, n’est qu’une conséquence directe du modèle non lié
(Donini and Kollman 2000) proposé par Stote et Karplus et adopté par un champ de force
purement classique, dans lequel l’interaction de l’ion de zinc avec son environnement (Stote
and Karplus 1995) est représenté exclusivement par les termes électrostatiques et de van der
Waals.
Ce modèle permet de surmonter la restriction conformationnel, imposé par le modèle liée
(Vedani et al. 1986; Vedani and Huhta 1990; Hoops et al. 1991; Ryde 1995a), mais il se
trouve face à des difficultés due aux fortes interactions électrostatiques créé par la charge

139
formelle de l’ion de zinc (Zn2+
) qui est gardé égale à +2 durant la simulation (Makinen et al.
1989). Il a été reporté que le traitement de l’ion de zinc par un champ de force classique qui
adapte le modèle non lié, conduit préférentiellement à une géométrie de coordination en mode
octaédrique, et dans certain cas l’ion de zinc s’échappe de la sphère de coordination (Zhang et
al. 2012). Ce modèle est incapable de décrire proprement le nombre de coordination et
l’énergie en même temps (Koca et al. 2003).
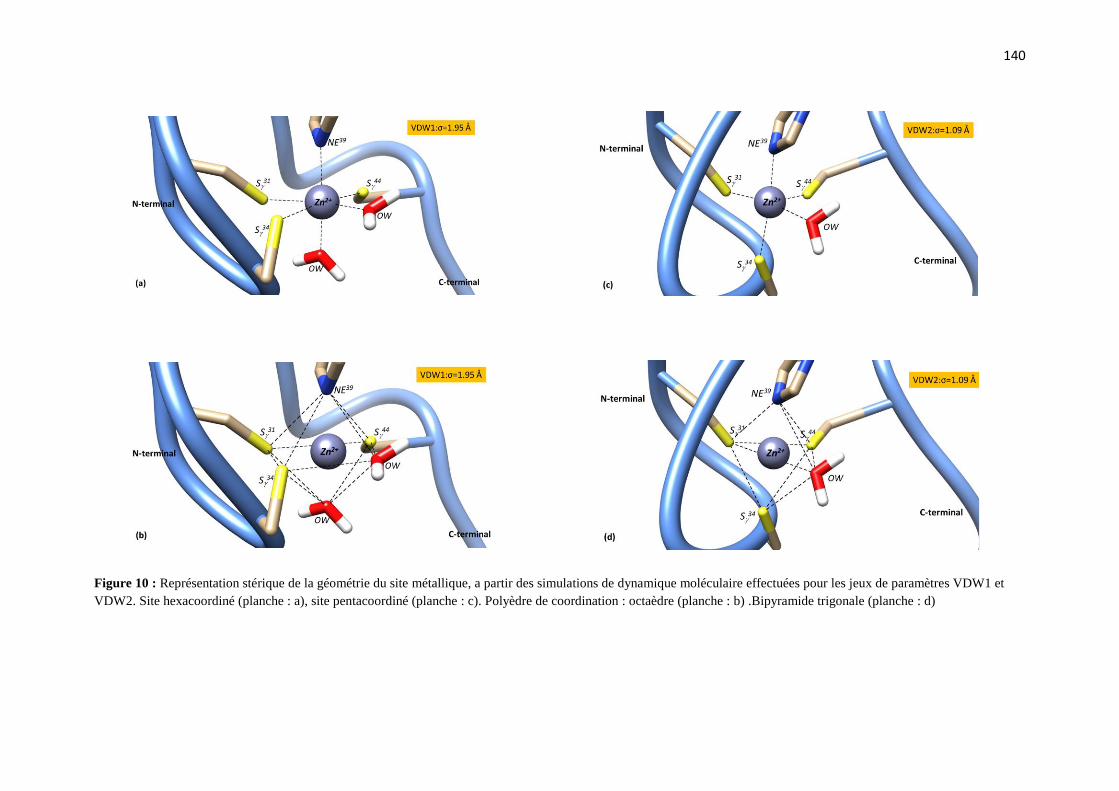
140
Figure 10 : Représentation stérique de la géométrie du site métallique, a partir des simulations de dynamique moléculaire effectuées pour les jeux de paramètres VDW1 et
VDW2. Site hexacoordiné (planche : a), site pentacoordiné (planche : c). Polyèdre de coordination : octaèdre (planche : b) .Bipyramide trigonale (planche : d)

141
b) la structure secondaire du fragment peptidique
Nous avons également évalué en fonction de l’âge de simulation le deplacement quadratique
moyen RMSD (Root-Mean-Square Deviations) du squelette polypeptidique, qui représente
l’une des propriétés fondamentales permettant de mesurer la stabilité de la structure étudiée
pendant la simulation, ainsi que sa proximité de la structure de référence, déterminée par
RMN.
Les trajectoires RMSD des deux simulations effectuées, pour les deux ensembles WDV1 et
VDW2 sont portés en fonction du temps sur la figure 11. Ces deux profiles vont nous
permettre de connaitre, l’influence de la taille du rayon métallique sur le comportement
structurale et la stabilité du fragment peptidique. Les deplacement quadratiques moyens sont
respectivement de l’ordre de (1.68 Å) et (2.07 Å) pour les jeux de paramètres VDW1 et
VDW2.
On peut voir clairement que le jeu de paramètres VDW1 ne perturbe pas d’une manière
importante la structure secondaire du polypeptide où les feuillets β antiparallèle appelé encore
(β-hairpin) de la partie amine du polypeptide (N-terminal) et l’hélice α de la partie carboxyle
(C-terminal) reste globalement intacte avec un RMSD de (1.68 Å), ceci démontre
l’importance des effets d’induction de l’ion de zinc sur la stabilisation de la structure
secondaire du fragment peptidique. On peut aussi remarquer la présence de deux transitions
conformationnelles qui se produisent respectivement à (20 ps) et à (50 ps) traduisant des
changements conformationnels dus aux réorientations des résidus coordinateurs et la
déstabilisation du cœur hydrophobe.
Par contre, de significatifs changements conformationnel du squelette polypeptidique sont
enregistrés et mesurés par une augmentation du RMSD à (2.07 Å), lors de l’utilisation de
l’ensemble de paramètres VDW2. Ceci est certainement dû au fait que les fortes interactions
courtes porté qui sont établie entre l’ion zinc et les résidus coordinateurs réduisent
significativement le degré de de liberté ou la flexibilité du complexe de coordination, afin de
de maintenir le cœur hydrophobique le plus intacte possible. La rigidité du complexe de
coordination est confirmée par la présence de deux transitions conformationnelle très écarté
enregistré respectivement à (35 ps) et à (85 ps). Dans ce cas le squelette polypeptidique sera
plus flexible et imposera des changements important capable de déstabilisé sa structure
secondaire.

142
Figure 11 : Trajectoire de l’écart quadratique moyen du squelette peptidique pour l’ensemble des paramètres de
van der Waals de l’ion de zinc : WDV1 e WDV2.

143
Les variations des RMSF (Root Mean Square Fluctuation, fluctuation quadratique moyenne)
au cours de la simulation du fragment peptidique correspendant au doigt de zinc de la NCp7
(1DSQ.pdb) sont reportées sur la Figure 12. Les RMSF sont calculés à partir des positions des
atomes du fragment peptedique (Figure 12(a)) et des atomes Cα des aminoacides (Figure
12(b)). Les RMSF des atomes des residus formant feuillet-β et et l’helice-α sont
particulièrement tres elevé (RMSF compris entre 0.20-0.40 nm). Ceci est du essentielement a
la grande flecxibilité de ces deux structures secondaires et les extrimités N- et C-terminales du
fragment peptidique sout l’effet du champ electrique créé par l’ion de zinc. L’influence de la
taille du rayon de l’ion de zinc sur la dynamique des residus coordinateurs peut etre distingué
clairement a partir de la Figure 12 (b). Les residus coordinateur Cys31 et Cys34 sont
beaucoup influencé par la grande felexibilité du backbone sous l’effet du rayon VDW2
comparé aux résidus cordinateur His39 et Cys44. Les residues Cys31 et Cys34 sont beaucoup
plus inclus dans le réarengement interne de la sphère de coordination dans le but de minimiser
les fortes interactions de répulsion entre les sulfures, ce qui n’est pas le cas pour le residus
Histidine dont la dynamique est régit par sa libre rotation autour de sa liaison Cα-Cβ lui
donne beaucoup plus d’independance de la dynamique du backbone. On voit aussi une
remarquable superposition des deux tracés RMSF dans la zone Cys44 localisés sur l’hilce-α
qui est destabilisée. Le rôle hydrophobe très important du groupement phenyl et son
interaction avec les residus non polaire situés dans la la boucle (loop region) reduit
considerablement les fluctuations des atomes Cα et maintent les regions des feuillet-β le plus
intact possible. Nous avons également fait une superposition des deux dernières
configurations issues des deux simulations (Figure 13) pour voir de près les changements
enregistrés dans les structures secondaires.

144
Figure 12 : Fluctuation quadratique moyenne a) des atomes du fragment peptidique et b) des atomes du
backbone, calculée pour les trajectoires de dynamique moleculaire pour les paramètres de lion de zinc VDW1 et
VDW2.

145
Figure 13 : Superposition et comparaison de la structure secondaire des deux dernières configurations, pour les
simulations VDW1 (beige claire) VDW2 (bleu).

146
le traitement des protéines à zinc par dynamique moléculaire classique néglige la
redistribution des électron sur l’ion métallique et ces résidus coordinateurs à chaque pas de la
simulation, car d’un point de vue physique, le champ électrique fort créé par l’ion de zinc,
peut non seulement inciter des dipôles atomiques, mais conduit aussi à la polarisation
dramatique des résidus latéraux polarisable tel que le cycle imidazole dans l’histidine, et le
cycle benzène dans le tryptophane ou la phénylalanine (Garmer and Gresh 1994; Sakharov
and Lim 2005; Li et al. 2008). De plus, les électrons de valence des atomes coordinateurs, tel
que l’azote et le soufre déprotoné, trouvés respectivement dans les residues histidine et
cystéine, sont fortement délocalisé sous l’effet du champ créé par l’ion de zinc et ils ont une
certaine probabilité de présence dans les orbitales de ce dernier. En conséquence, les charges
sont transférées d’atomes coordinateurs au zinc (Hoops et al. 1991) .Comme le témoigne le
travail de Sakharov and Lim (Sakharov and Lim 2005), le champ électrostatique local fort et
l’effet d’induction (Wu et al. 2010a; Jiajing Zhang 2012) et de transfert de charges, présente
un grand défi pour la modélisation du Zn2+
(Sakharov and Lim 2005; Li et al. 2008; Sakharov
and Lim 2009) incorporé dans les biomolécules par un champ de force classique qui utilise
des charges fixe (Merz and Kollman 1989; Wang et al. 2010). La simulation par dynamique
moléculaire classique, nous a permis de voir de manière qualitative, l’influence de la taille du
rayon métallique, sur les effets électrostatiques ainsi que sur le comportement structurale du
site à zinc dans un milieu solvatée. Les paramètres de van der Waals de l’ion de zinc adopté
originailement pour le champ de force CHARMM22 et celle qui ont été proposé par Stote et
Karplus sont capables de donner des distance de coordination raisonnable et comparable à
celles trouvé dans la structure RMN.
Le choix des paramètres de van der Waals appropriés pour l’ion de zinc est important pour
une coordination correcte dans les sites à zinc. La stabilisation du site de coordination et de la
structure secondaire dans le fragment à zinc ainsi que la conservation du caractère
hydrophobe de son cœur, est lié directement à la contribution de cet ion métallique à la zone
d’écrantage de ces voisins (résidus coordinateur)
Pour un grand rayon le caractère hydrophile de l’ion de zinc l’emporte sur le caractère
hydrophobe des résidus Cystéinates ce qui déstabilisera son site de coordination, pendant que
son environnement protéinique reste généralement intact. En revanche, un petit rayon va
tronquer les interactions électrostatiques longues portées de l’ion métallique et réduira sa zone
d’action, ce qui maintiendra beaucoup plus le cœur hydrophobe et déstabilisera son
environnement protéinique.

147
Dans cette partie nous nous sommes interessé à la dynamique des ligands coordinateurs sous
les effets electrostatiques de l’ion de zinc. Cet age reduit de la simulation est imposé aussi par
la taille du système etudié et le modèle non lié qu’on a adopté pour incorporer un ion
métalique dans un champ de force classique. L’utilisation d’un champ de force polarisable vas
etre notre prochain objectif pour étudier les propriétés structurales (depliment et repliement)
de la NCp7 sur un echelle de temps qui depasse les 2ns.
C.3 Lecture dans les champs de forces polarisables
Les effets stabilisateurs d’induction (transfert de charge et polarisation) ne peuvent pas être
capturés par une simple fonction d’énergie potentielle contenant le terme conventionnel de
Coulomb qui est basé sur des charges fixe, et le terme de van der Waals présenté par le
potentiel de Lennard Jhones. Beaucoup d’études ont suggérées qu’il est important de
représenter l'impact global de l’ion de zinc sur son environnement par l’inclusion des effets
de polarisation et de transfert de charge dans le modèle de l’énergie potentielle (Peraro et al.
2007; Sakharov and Lim 2009; Lu et al. 2010) ceci afin d’investir correctement les effets
d’interactions électrostatiques de l’ion métallique dans son environnement protéinique.
Plusieurs modèles de potentiels qui tiennent compte de la dépendance entre la structure, la
polarisation et transfert de charge ont été proposées. Par exemple, le modèle SIBFA (sum of
interactions between fragments ab initio computed) (Tiraboschi et al. 2000; Antony et al.
2005) a été l’un des premiers potentiels développés pour modéliser l’interaction du zinc avec
les molécules biologique. Ce modèle est caractérisé par l’incorporation rigoureuse le la
polarisation et du transfert de charge dans l’expression de l’énergie potentiel. La
paramétrisation du champ de force SIBFA est relativement complexe mais en parallèle, il
donne des résultats comparables à des calculs ab initio de niveau très poussé.
Sakharov and Lim ont menée des travaux basés sur le model non lié présenté par Stote et
Karplus et ils ont considérés deux effet d’induction, celui de la polarisation qui a été présenté
par le model des dipôles ponctuels et celui de transfert de charge qui a été considéré par une
redistribution des charges du zinc ainsi que ces atomes coordinateurs à chaque pas de la
simulation (Kaminski and Jorgensen 1999; Sakharov and Lim 2005). Ils ont inclus tous les
composantes qui dérivent de la décomposition de Kitora-Morocuma dans le but d’une
description complète de l’énergie d’interaction non covalente.(Kitaura and Morokuma 1976).
Par exemple les composantes de dispersion et de polarisation ont été ajouté au potentiel de

148
Lennard-Jones 6-12 tandis que le potentiel de coulomb regroupe le terme d’interaction
électrostatique des charges ponctuelles ainsi que l’effet de transfert de charges. L’inclusion
empirique des effets quantiques à la fonction d’énergie potentielle, combiné aux paramètres
de van der Waals appropriés, leur a permis de capturer avec succès la géométrie de
coordination, observé expérimentalement dans les sites à zinc structurales (Sakharov and Lim
2005).
Sakharov et Lim ont aussi introduit la dépendance entre la distance et la charge partielle du
zinc afin de considérer les effets de polarisations et de transfert de charge (CTPOL) (Sakharov
and Lim 2009). Les simulations de dynamique moléculaires qui utilisent ce modèle, reproduit
la structure tétraédrique observé expérimentalement dans les doigts de zinc de type Cys2His2,
et Cys4 et aussi dans les protéines à site à zinc multiples.
Ren et collaborateur ont tenu compte de l’effet de polarisation avec le model AMOEBA
(Atomic Multipole Optimized Energetics for Biomolecular Simulation) (Jiajing Zhang 2012).
Leur simulations de dynamique moléculaire appliquées sur les sites à zinc de type Cys2His2
ainsi que sur des enzymes à zinc, ont conduit à des résultats raisonnables.
Zhu et collaborateurs ont présenté l’approche APPC (adaptive polarized protein-specific
charge) (Li et al. 2011) qui inclut le transfert de charge et la polarisation pour étudier la
dynamique des métalloprotéines et qui consiste à l’exécution périodique d’un calcule
purement quantique sur les sites à zinc afin de mettre à jour leurs distribution de charge. Sur
la base de cette méthode, la structure est la dynamique des métalloprotéines possédant deux
cite à zincs, ont été correctement décrit, cependant le calcule basé sur l’approche quantique
est numériquement couteux pour un âge de simulation long.
Le modèle SCF (Semi fluctuating Charge), (Ji et al. 2012) a été développé pour prendre en
compte les effets de polarisation dans les biomolécules. Les paramètres dans ce modèle ont
été issus des propriétés électroniques et déterminées à partir des calculs quantiques effectués
sur grands nombre de structure modèle dans la phase gaz et en solution. Enfin, Le champ de
force QPCT (calibrated polarizable-charge transfer force field) (Zhu et al. 2013) a été
proposé pour décrire la dynamique de l’interaction entre l’ion de zinc et ces ligands dans la
protéine

149
C.4 Conclusion
Les simulations de métaux de transition sont souvent problématiques à cause de la nécessité
de modéliser leur diverse structure de coordination. En raison de la forte polarisabilité des
métaux de transition, leur charges et leur propriétés de coordination varient facilement
(Scheiner and Kar 2005). Le nombre de ligands coordinateurs peut varier aussi bien que la
structure de coordination d’un nombre donné de ligands. Par exemple les coordinations de
géométrie tétraédrique ou carré plat, peuvent être adopté par des métaux tétra coordinnés,
tandis que les métaux à Cinq coordinants, adoptent une géométrie en pyramide à base carré ou
une géométrie bipyramidale à base triangulaire.
La raison de ces difficultés vient du fait que, les paramètres associés aux fonctions d’énergies
potentielle décrivant les termes liés (liaisons, angles, dièdres… etc) et les termes non liés
(électrostatiques, van der Waals… etc) pour les systèmes moléculaires impliquant des ions de
métaux de transition, ne sont pas incorporés d’une manière standard dans les principales
bibliothèques des champs de force. Par exemple, et dans le cadre du modèle lié, la fonction
d’énergie potentielle décrivant les angles de valence doit avoir plus d'un angle idéal pour bien
décrire un ion métallique dans son site de coordination. Un développement de Taylor autour
d'une valeur unique ne convient pas. Généralement les champs de force les plus précis sont
développés pour des complexes de coordination ou des métalloprotéines bien spécifiques.
Le champ de force classique GROMOS96 originailement orienté vers la simulation des
protéines et des aminoacides peut être appliqué sur des protéines activé par un ion métallique,
mais pour des âges de simulation courte, car la charge de l’ion métallique gardée fixe dans les
champs de force classique, peut affecter significativement la structure globale de la protéine
pour un âge de simulation important. L’inclusion des termes énergétiques traitant,
explicitement les phénomènes de transfert de charge et de polarisation entre le cation
métallique et les acides aminés dans les champs de force de mécanique moléculaire est
nécessaire pour décrire avec précision les conformations du site actif obtenues
expérimentalement. Le développement d’un champ de force beaucoup plus précis et
transférable pour l’ensemble des protéines à zinc est reste l’objet d’intensif effort.

150
4.7 Conclusion générale et perspectives
Le but de cette thèse est d’appliquer la méthode du Champ Auto-Cohérent Local ou Local
Self-Consistent Field (LSCF) au système biologique et biomimétique. Le principe
fondamental de la méthode LSCF, développée initialement pour des calculs semi-empirique
puis ab initio, est l’optimisation de la fonction d’onde multiélectronique sous contrainte. En
effet, dans cette dernière, il est possible de geler une partie des spinorbitales durant le
processus SCF. Ces spinorbitales sont fixées par l’utilisateur et représentent une densité
électronique invariante ressentie par le reste du système.
Dans un premier temps nous avons utilisé l’approche LSCF/MM qui représente une des
méthodes hybride QM/MM. Cette approche a l’avantage de remplacer les liaisons frontières
par des orbitales de liaison strictement localisées ou Strictly Localised Bond orbital (SLBO)
afin d’avoir une description précise de la frontière QM/MM. La SLBO est localisée entre
deux atomes, l’un appartenant à la partie quantique et l’autre à la partie classique. L’atome de
la partie classique possède un caractère hybride. En effet, ce dernier possède certains
paramètres MM du champ de force utilisé, mais aussi un jeu de fonctions de base, identique à
celui qu’il aurait s’il était dans la partie QM.
La SLBO est déterminée sur un système modèle contenant la liaison chimique d’intérêt, et
appliquée sur le système réel en accord avec le principe de transférabilité, après rotation et
renormation. La SLBO peut être obtenue grâce aux nombreux critères de localisation
disponible telle que Pipek-Mezey, Boys-Foster, Edmiston-Ruedenberg, Weinstein-Pauncz,
Magnasco-Perico ou ELMO (Extremely Localized Molecular Orbitals).
Dans un premier temps, nous avons appliqué la méthode hybride LSCF/MM sur un système
moléculaire de grande taille contenant un ion métallique afin d’avoir une idée sur le rôle
stabilisateur de l’ion zinc sur la structure native et mutée. Cela a fait l’objet d’une publication
intitulé (Effect of mutation on the stabilization energy of HIV-1 zinc finger : a hybrid local
self-consistent field/molecular mechanics investigation). Un axe prometteur, est l’utilisation
des champs de forces polarisables, comme AMOEBA intégré dans le logiciel Tinker, au sein
des méthodes mixtes QM/MM. ceci a pour but de prendre en compte à la fois la polarisation
de la fonction d’onde ab initio causé par l’environnement classique, mais aussi l’influence
que peut engendrer la partie quantique sur les multipoles atomiques de la partie classique qui
peut avoir un rôle important dans la compréhension des certains phénomène ayant lieu dans
les systèmes biologiques
Dans un deuxième temps, nous avons appliqué l’approche de la projection asymptotique qui
représente l’une des variantes de la méthode LSCF ab initio sur un ensemble de complexes à

151
liaison hydrogène, dans lesquels l’un des monomères agit comme donneur de liaison
hydrogène et l’autre comme accepteur. Cette approche possède une philosophie QM/MM qui
consiste à appliquer des calculs tournant selon un processus appelée (freez-and thaw cycles).
Les résultats ont montré que cette méthode est prometteuse dans le cas des complexes à
hydrogène neutre. Elle a permis de décrire les effets de polarisation dans un cadre purement
quantique dans lequel la modélisation des interactions électrostatiques pour chaque dimère est
réalisée à l’aide d’un protocole calculatoire, dans lequel on gèle les orbitales moléculaires de
l’un des monomères afin de créer une densité de charge fixe qui vas permettre de polariser la
densité de charge du monomère voisin gardé à l’état relaxé. Nous avons aussi constaté que
cette méthode doit être utilisé avec précaution et le paramètre distance entre les deux
partenaires doit être pris en considération lors du traitement des complexes à hydrogène
chargé. Cette partie fait l’objet d’une publication qui est encore sous examen
La dernière partie concerne la modélisation d’un ion métallique dans son environnement
protéinique en utilisant le modèle non lié. Pour cela nous avons utilisé le champ de force
classique GROMOS96 implémenté dans le programme Gromacs pour modéliser l’influence
de la variation des paramètres de van der Walls de l’ion métallique sur la dynamique des
ligands dans la sphère de coordination. Cette étude nous a donc menés vers la validation des
paramètres de van der Walls de l’ion de zinc originailement implémenter dans le champ de
force GROMOS96. La réalisation d’un long âge de dynamique moléculaire basé sur un
champ de force GROMOS96 modifié d’une manière à tenir en compte des effets de
polarisation causé l’environnement protéinique présente un objectif à atteindre.
Cette thèse a montré l’importance de la prise en compte des effets de polarisation l’ors du
traitement theorique des interactions non-lié dans des petits systèmes biomimetique et des
molecules biologique.

152

153
Références bibliographiques

154
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable
parameters: The PBE0 model. J Chem Phys 110:6158–6170. doi: 10.1063/1.478522
Alberts IL, Nadassy K, Wodak SJ (1998) Analysis of zinc binding sites in protein crystal
structures. Protein Sci A Publ Protein Soc 7:1700–1716. doi: 10.1002/pro.5560070805
Alkorta I, Mata I, Molins E, Espinosa E (2016) Charged versus Neutral Hydrogen-Bonded
Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem - A Eur J
22:9226–9234. doi: 10.1002/chem.201600788
Allen FH, Bellard S, Brice MD, et al (1979) The Cambridge Crystallographic Data Centre:
computer-based search, retrieval, analysis and display of information. Acta Crystallogr
Sect B 35:2331–2339. doi: 10.1107/S0567740879009249
Allen MP, Tildesley DJ (1987) Computer Simulation of Liquids, Oxford Uni. Oxford
Amara P, Field MJ (2003) Evaluation of an ab initio quantum mechanical/molecular
mechanical hybrid-potential link-atom method. Theor Chem Acc 109:43–52. doi:
10.1007/s00214-002-0413-3
Amos RD, Handy NC, Knowles PJ, et al (1985) AB-initio prediction of properties of carbon
dioxide, ammonia, and carbon dioxide...ammonia. J Phys Chem 89:2186–2192. doi:
10.1021/j100257a010
Antes I, Thiel W (1999) Adjusted Connection Atoms for Combined Quantum Mechanical and
Molecular Mechanical Methods. J Phys Chem A 103:9290–9295. doi:
10.1021/jp991771w
Antony J, Piquemal J-P, Gresh N (2005) Complexes of thiomandelate and captopril
mercaptocarboxylate inhibitors to metallo-β-lactamase by polarizable molecular
mechanics. Validation on model binding sites by quantum chemistry. J Comput Chem
26:1131–1147. doi: 10.1002/jcc.20245
Aqvist J, van Gunsteren WF, Leijonmarck M, Tapia O (1985) A molecular dynamics study of
the C-terminal fragment of the L7/L12 ribosomal protein. Secondary structure motion in
a 150 picosecond trajectory. J Mol Biol 183:461–477.
Åqvist J, Warshel A (1992) Computer simulation of the initial proton transfer step in human
carbonic anhydrase I. J Mol Biol 224:7–14. doi: 10.1016/0022-2836(92)90572-2
Assfeld X, Ferré N, Rivail J-L (2004) Electrostatic interactions in peptides. Polarisation
effects due to an α-helix. Theor Chem Acc 111:328–334. doi: 10.1007/s00214-003-
0513-8
Assfeld X, Rivail J-L (1996) Quantum chemical computations on parts of large molecules: the
ab initio local self consistent field method. Chem Phys Lett 263:100–106. doi:
10.1016/S0009-2614(96)01165-7
Auld DS (2001) Zinc coordination sphere in biochemical zinc sites. In: Maret W (ed).
Springer Netherlands, pp 85–127

155
Babu CS, Lim C (2006) Empirical Force Fields for Biologically Active Divalent Metal
Cations in Water†. J Phys Chem A 110:691–699. doi: 10.1021/jp054177x
Bagus PS, Hermann K, Jr CWB (1984) A new analysis of charge transfer and polarization for
ligand–metal bonding: Model studies of Al4CO and Al4NH3. J Chem Phys 80:4378–
4386. doi: 10.1063/1.447215
Bagus PS, Illas F (1992) Decomposition of the chemisorption bond by constrained variations:
Order of the variations and construction of the variational spaces. J Chem Phys 96:8962–
8970. doi: 10.1063/1.462875
Bakowies D, Thiel W (1996) Hybrid Models for Combined Quantum Mechanical and
Molecular Mechanical Approaches. J Phys Chem 100:10580–10594. doi:
10.1021/jp9536514
Bashford D, Karplus M, Canters GW (1988) Electrostatic effects of charge perturbations
introduced by metal oxidation in proteins. A theoretical analysis. J Mol Biol 203:507–
510.
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic
behavior. Phys Rev A 38:3098–3100. doi: 10.1103/PhysRevA.38.3098
Becke AD (1993) Density‐functional thermochemistry. III. The role of exact exchange. J
Chem Phys 98:5648–5652. doi: 10.1063/1.464913
Becke AD (1993) Density‐functional thermochemistry. III. The role of exact exchange. J
Chem Phys 98:5648–5652. doi: 10.1063/1.464913
Berg JM (1986) Potential metal-binding domains in nucleic acid binding proteins. Science
(80- ) 232:485–487. doi: 10.1126/science.2421409
Berg JM, Shi Y (1996) The Galvanization of Biology: A Growing Appreciation for the Roles
of Zinc. Science (80- ) 271:1081–1085. doi: 10.1126/science.271.5252.1081
Berg JM, Shi Y (1996) The Galvanization of Biology: A Growing Appreciation for the Roles
of Zinc. Science (80- ) 271:1081–1085. doi: 10.1126/science.271.5252.1081
Bernstein FC, Koetzle TF, Williams GJ, et al (1977) The Protein Data Bank: a computer-
based archival file for macromolecular structures. J Mol Biol 112:535–542.
Bess JW, Powell PJ, Issaq HJ, et al (1992) Tightly bound zinc in human immunodeficiency
virus type 1, human T-cell leukemia virus type I, and other retroviruses. J Virol 66:840–
847.
Beyer A, Karpfen A, Schuster P (1984) Energy surfaces of hydrogen-bonded complexes in
the vapor phase. In: Schuster P (ed). Springer Berlin Heidelberg, pp 1–40
Bickelhaupt FM, Baerends EJ (2000) Kohn-Sham Density Functional Theory: Predicting and
Understanding Chemistry. In: Lipkowitz KB, Boyd DB (eds). John Wiley & Sons, Inc.,
pp 1–86

156
Biswas PK, Gogonea V (2005) A regularized and renormalized electrostatic coupling
Hamiltonian for hybrid quantum-mechanical–molecular-mechanical calculations. J
Chem Phys 123:164114. doi: 10.1063/1.2064907
Blake GA, Laughlin KB, Cohen RC, et al (1991) Tunable far infrared laser spectrometers.
Rev Sci Instrum 62:1693–1700. doi: 10.1063/1.1142409
Blasie CA, Berg JM (2002) Structure-Based Thermodynamic Analysis of a Coupled Metal
Binding−Protein Folding Reaction Involving a Zinc Finger Peptide†. Biochemistry
41:15068–15073. doi: 10.1021/bi026621h
Boese AD, Handy NC (2002) New exchange-correlation density functionals: The role of the
kinetic-energy density. J Chem Phys 116:9559–9569. doi: 10.1063/1.1476309
Bombarda E, Grell E, Roques BP, Mély Y (2007) Molecular mechanism of the Zn2+-induced
folding of the distal CCHC finger motif of the HIV-1 nucleocapsid protein. Biophys J
93:208–217. doi: 10.1529/biophysj.106.101378
Bombarda E, Grell E, Roques BP, Mély Y (2007) Molecular mechanism of the Zn2+-induced
folding of the distal CCHC finger motif of the HIV-1 nucleocapsid protein. Biophys J
93:208–217. doi: 10.1529/biophysj.106.101378
Bombarda E, Grell E, Roques BP, Mély Y (2007) Molecular mechanism of the Zn2+-induced
folding of the distal CCHC finger motif of the HIV-1 nucleocapsid protein. Biophys J
93:208–217. doi: 10.1529/biophysj.106.101378
Bombarda E, Morellet N, Cherradi H, et al (2001) Determination of the pKa of the four Zn2+-
coordinating residues of the distal finger motif of the HIV-1 nucleocapsid protein:
Consequences on the binding of Zn2+. J Mol Biol 310:659–672. doi:
10.1006/jmbi.2001.4770
Bombarda E, Morellet N, Cherradi H, et al (2001) Determination of the pK(a) of the four
Zn2+-coordinating residues of the distal finger motif of the HIV-1 nucleocapsid protein:
consequences on the binding of Zn2+. J Mol Biol 310:659–672. doi:
10.1006/jmbi.2001.4770
Bombarda E, Roques BP, Mély Y, Grell E (2005) Mechanism of zinc coordination by point-
mutated structures of the distal CCHC binding motif of the HIV-1 NCp7 protein.
Biochemistry 44:7315–7325. doi: 10.1021/bi047349+
Bombarda E, Roques BP, Mély Y, Grell E (2005) Mechanism of zinc coordination by point-
mutated structures of the distal CCHC binding motif of the HIV-1 NCp7 protein.
Biochemistry 44:7315–7325. doi: 10.1021/bi047349+
Boys SF (1960) Construction of Some Molecular Orbitals to Be Approximately Invariant for
Changes from One Molecule to Another. Rev Mod Phys 32:296–299. doi:
10.1103/RevModPhys.32.296

157
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences
of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566.
doi: 10.1080/00268977000101561
Brack M (1993) The physics of simple metal clusters: self-consistent jellium model and
semiclassical approaches. Rev Mod Phys 65:677–732. doi: 10.1103/RevModPhys.65.677
Bredenberg J, Nilsson L (2001) Modeling zinc sulfhydryl bonds in zinc fingers. Int J
Quantum Chem 83:230–244. doi: 10.1002/qua.1214
Brooks BR, Bruccoleri RE, Olafson BD, et al (1983) CHARMM: A program for
macromolecular energy, minimization, and dynamics calculations. J Comput Chem
4:187–217. doi: 10.1002/jcc.540040211
Buckingham AD (1967) Permanent and Induced Molecular Moments and Long-Range
Intermolecular Forces. In: Hirschfelder JO (ed). John Wiley & Sons, Inc., pp 107–142
Buckingham AD, Fowler PW (1983) Do electrostatic interactions predict structures of van der
Waals molecules? J Chem Phys 79:6426–6428. doi: 10.1063/1.445721
Buckingham AD, Fowler PW (1985) A model for the geometries of Van der Waals
complexes. Can J Chem 63:2018–2025. doi: 10.1139/v85-334
Buckingham RA, Hamilton J, Massey HSW (1941) The Low-Temperature Properties of
Gaseous Helium. II. Proc R Soc London Ser A Math Phys Sci 179:103–122. doi:
10.1098/rspa.1941.0082
Calhoun JR, Liu W, Spiegel K, et al (2008) Solution NMR structure of a designed
metalloprotein and complementary molecular dynamics refinement. Struct (London,
Engl 1993) 16:210–215. doi: 10.1016/j.str.2007.11.011
Car R, Parrinello M (1985) Unified Approach for Molecular Dynamics and Density-
Functional Theory. Phys Rev Lett 55:2471–2474. doi: 10.1103/PhysRevLett.55.2471
Caricato M, Trucks GW, Frisch MJ, Wiberg KB (2010) Electronic Transition Energies: A
Study of the Performance of a Large Range of Single Reference Density Functional and
Wave Function Methods on Valence and Rydberg States Compared to Experiment. J
Chem Theory Comput 6:370–383. doi: 10.1021/ct9005129
Ceperley DM, Alder BJ (1980) Ground State of the Electron Gas by a Stochastic Method.
Phys Rev Lett 45:566–569. doi: 10.1103/PhysRevLett.45.566
Chalasinski G, Szcz&ecedil;śniak MM (2000) State of the Art and Challenges of the
ab Initio Theory of Intermolecular Interactions. Chem Rev 100:4227–4252.
Chalasinski G, Szczesniak MM (1994) Origins of Structure and Energetics of van der Waals
Clusters from ab Initio Calculations. Chem Rev 94:1723–1765. doi:
10.1021/cr00031a001

158
Chaturvedi UC, Shrivastava R (2005) Interaction of viral proteins with metal ions: role in
maintaining the structure and functions of viruses. FEMS Immunol Med Microbiol
43:105–114. doi: 10.1016/j.femsim.2004.11.004
Chatwin SL, Davidson MG, Doherty C, et al (2006) H−X Bond Activation via Hydrogen
Transfer to Hydride in Ruthenium N-Heterocyclic Carbene Complexes: Density
Functional and Synthetic Studies. Organometallics 25:99–110. doi: 10.1021/om0507427
Christianson DW (1991) Structural biology of zinc. Adv Protein Chem 42:281–355.
Christianson DW (1991) Structural biology of zinc. Adv Protein Chem 42:281–355.
Christianson DW (1991) Structural biology of zinc. Adv Protein Chem 42:281–355.
Cole GC, Legon AC (2004) The nature of the complex formed between pyridine and
hydrogen bromide in the gas phase: an experimental approach using rotational
spectroscopy. J Chem Phys 121:10467–10473. doi: 10.1063/1.1809577
Cornell WD, Cieplak P, Bayly CI, et al (1995) A Second Generation Force Field for the
Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc
117:5179–5197. doi: 10.1021/ja00124a002
Cox EH, McLendon GL (2000) Zinc-dependent protein folding. Curr Opin Chem Biol 4:162–
165. doi: 10.1016/S1367-5931(99)00070-8
Cramer CJ, Truhlar DG (1999) Implicit Solvation Models: Equilibria, Structure, Spectra, and
Dynamics. Chem Rev 99:2161–2200.
D.LAURENT A (2010) Etude de phénomènes électroniques de macromolécules à l’aide de
méthodes hybrides QM-MM.
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N⋅log(N) method for Ewald
sums in large systems. J Chem Phys 98:10089–10092. doi: 10.1063/1.464397
Darlix J-L, Lapadat-Tapolsky M, de Rocquigny H, Roques BP (1995) First Glimpses at
Structure-function Relationships of the Nucleocapsid Protein of Retroviruses. J Mol Biol
254:523–537. doi: 10.1006/jmbi.1995.0635
Das D, Eurenius KP, Billings EM, et al (2002) Optimization of quantum mechanical
molecular mechanical partitioning schemes: Gaussian delocalization of molecular
mechanical charges and the double link atom method. J Chem Phys 117:10534–10547.
doi: 10.1063/1.1520134
Das D, Eurenius KP, Billings EM, et al (2002) Optimization of quantum mechanical
molecular mechanical partitioning schemes: Gaussian delocalization of molecular
mechanical charges and the double link atom method. J Chem Phys 117:10534–10547.
doi: 10.1063/1.1520134
De Loof H, Nilsson L, Rigler R (1992) Molecular dynamics simulation of galanin in aqueous
and nonaqueous solution. J Am Chem Soc 114:4028–4035. doi: 10.1021/ja00037a002

159
Desiraju GR, Steiner T (1999) The Weak hydrogen bond: in structural chemistry and biology.
Oxford University Press, Oxford
Dewar MJS, Thiel W (1977) Ground states of molecules. 38. The MNDO method.
Approximations and parameters. J Am Chem Soc 99:4899–4907. doi:
10.1021/ja00457a004
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) Development and use of quantum
mechanical molecular models. 76. AM1: a new general purpose quantum mechanical
molecular model. J Am Chem Soc 107:3902–3909. doi: 10.1021/ja00299a024
DiLabio GA, Hurley MM, Christiansen PA (2002) Simple one-electron quantum capping
potentials for use in hybrid QM/MM studies of biological molecules. J Chem Phys
116:9578–9584. doi: 10.1063/1.1477182
Dinner AR, Lopez X, Karplus M (2003) A charge-scaling method to treat solvent in QM/MM
simulations. Theor Chem Acc 109:118–124. doi: 10.1007/s00214-002-0417-z
Dirac P a. M (1930) Note on Exchange Phenomena in the Thomas Atom. Math Proc
Cambridge Philos Soc 26:376–385. doi: 10.1017/S0305004100016108
Donini OA, Kollman PA (2000) Calculation and prediction of binding free energies for the
matrix metalloproteinases. J Med Chem 43:4180–4188.
Dudev T, Lim C (2007) All-electron calculations of the nucleation structures in metal-induced
zinc-finger folding: role of the Peptide backbone. J Am Chem Soc 129:12497–12504.
doi: 10.1021/ja073322c
Dułak M, Wesołowski TA (2006) On the electron leak problem in orbital-free embedding
calculations. J Chem Phys 124:164101. doi: 10.1063/1.2189228
Dykstra CE (1993) Electrostatic interaction potentials in molecular force fields. Chem Rev
93:2339–2353. doi: 10.1021/cr00023a001
Eisenschitz R, London F (1930) Über das Verhältnis der van der Waalsschen Kräfte zu den
homöopolaren Bindungskräften. Zeitschrift für Phys 60:491–527. doi:
10.1007/BF01341258
Engkvist O, Åstrand P-O, Karlström G, et al (2000) Accurate intermolecular potentials
obtained from molecular wave functions: Bridging the gap between quantum chemistry
and molecular simulations. Chem Rev 100:4087–4108.
Ernzerhof M, Scuseria GE (1999) Assessment of the Perdew–Burke–Ernzerhof exchange-
correlation functional. J Chem Phys 110:5029–5036. doi: 10.1063/1.478401
Essmann U, Perera L, Berkowitz ML, et al (1995) A smooth particle mesh {Ewald} method. J
Chem Phys 103:8577–8593. doi: 10.1063/1.470117
Fatmi MQ, Hofer TS, Randolf BR, Rode BM (2006) Structure and dynamics of the
[Zn(NH3)(H2O)5]2+ complex in aqueous solution obtained by an ab initio QM/MM

160
molecular dynamics study. Phys Chem Chem Phys 8:1675–1681. doi:
10.1039/B518223A
Fatmi MQ, Hofer TS, Randolf BR, Rode BM (2006) Temperature Effects on the Structural
and Dynamical Properties of the Zn(II)−Water Complex in Aqueous Solution: A
QM/MM Molecular Dynamics Study. J Phys Chem B 110:616–621. doi:
10.1021/jp0546655
Fatmi MQ, Hofer TS, Randolf BR, Rode BM (2007) Stability of different zinc(II)-diamine
complexes in aqueous solution with respect to structure and dynamics: a QM/MM MD
study. J Phys Chem B 111:151–158. doi: 10.1021/jp0654213
Fatmi MQ, Hofer TS, Rode BM (2010) The stability of [Zn(NH3)4]2+ in water: A quantum
mechanical/molecular mechanical molecular dynamics study. Phys Chem Chem Phys
12:9713–9718. doi: 10.1039/C002021D
Ferenczy GG, Rivail J-L, Surján PR, Náray-Szabó G (1992) NDDO fragment self-consistent
field approximation for large electronic systems. J Comput Chem 13:830–837. doi:
10.1002/jcc.540130706
Ferré N, Assfeld X (2002) Application of the local self-consistent-field method to core-
ionized and core-excited molecules, polymers, and proteins: True orthogonality between
ground and excited states. J Chem Phys 117:4119–4125. doi: 10.1063/1.1496462
Ferré N, Assfeld X (2003) A new three-layer hybrid method (LSCF/MM/Madelung) devoted
to the study of chemical reactivity in zeolites. Preliminary results. J Mol Struct
THEOCHEM 632:83–90. doi: 10.1016/S0166-1280(03)00290-2
Ferré N, Assfeld X, Rivail J-L (2002) Specific force field parameters determination for the
hybrid ab initio QM/MM LSCF method. J Comput Chem 23:610–624. doi:
10.1002/jcc.10058
Ferré N, Assfeld X, Rivail J-L (2002) Specific force field parameters determination for the
hybrid ab initio QM/MM LSCF method. J Comput Chem 23:610–624. doi:
10.1002/jcc.10058
Ferré N, Cembran A, Garavelli M, Olivucci M (2004) Complete-active-space self-consistent-
field/Amber parameterization of the Lys296–retinal–Glu113 rhodopsin chromophore-
counterion system. Theor Chem Acc 112:335–341. doi: 10.1007/s00214-004-0593-0
Field MJ, Bash PA, Karplus M (1990) A combined quantum mechanical and molecular
mechanical potential for molecular dynamics simulations. J Comput Chem 11:700–733.
doi: 10.1002/jcc.540110605
Field MJ, Bash PA, Karplus M (1990) A combined quantum mechanical and molecular
mechanical potential for molecular dynamics simulations. J Comput Chem 11:700–733.
doi: 10.1002/jcc.540110605
Fock V (1930) Näherungsmethode zur Lösung des quantenmechanischen
Mehrkörperproblems. Zeitschrift für Phys 61:126–148. doi: 10.1007/BF01340294

161
Fornili A, Loos P-F, Sironi M, Assfeld X (2006) Frozen core orbitals as an alternative to
specific frontier bond potential in hybrid Quantum Mechanics/Molecular Mechanics
methods. Chem Phys Lett 427:236–240. doi: 10.1016/j.cplett.2006.06.095
Fornili A, Loos P-F, Sironi M, Assfeld X (2006) Frozen core orbitals as an alternative to
specific frontier bond potential in hybrid Quantum Mechanics/Molecular Mechanics
methods. Chem Phys Lett 427:236–240. doi: 10.1016/j.cplett.2006.06.095
Fornili A, Moreau Y, Sironi M, Assfeld X (2006) On the suitability of strictly localized
orbitals for hybrid QM/MM calculations. J Comput Chem 27:515–523. doi:
10.1002/jcc.20366
Foster JM, Boys SF (1960) Canonical Configurational Interaction Procedure. Rev Mod Phys
32:300–302. doi: 10.1103/RevModPhys.32.300
Foster JP, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218. doi:
10.1021/ja00544a007
Frankel AD, Berg JM, Pabo CO (1987) Metal-dependent folding of a single zinc finger from
transcription factor IIIA. Proc Natl Acad Sci U S A 84:4841–4845.
Frisch M, Trucks G, Schlegel H, et al (1988) GAUSSIAN 03, Revision B1, Gaussian, Inc.,
Pittsburgh, PA, 2003;(b) AD Becke. Phys Rev A 38:3098.
Fw S (1995) The influence of zinc on apoptosis. Ann Clin Lab Sci 25:134–142.
Gao J (1996) Reviews in Computational Chemistry, VCH. K. B. Lipkowitz, D. B. Boyd, New
York
Gao J, Amara P, Alhambra C, Field MJ (1998) A Generalized Hybrid Orbital (GHO) Method
for the Treatment of Boundary Atoms in Combined QM/MM Calculations. J Phys Chem
A 102:4714–4721. doi: 10.1021/jp9809890
Gao J, Ma S, Major DT, et al (2006) Mechanisms and free energies of enzymatic reactions.
Chem Rev 106:3188–3209. doi: 10.1021/cr050293k
García-Lastra JM, Wesolowski T, Barriuso MT, et al (2006) Optical and vibrational
properties of MnF 6 4− complexes in cubic fluoroperovskites: insight through
embedding calculations using Kohn–Sham equations with constrained electron density. J
Phys Condens Matter 18:1519.
Glendening ED, Streitwieser A (1994) Natural energy decomposition analysis: An energy
partitioning procedure for molecular interactions with application to weak hydrogen
bonding, strong ionic, and moderate donor–acceptor interactions. J Chem Phys
100:2900–2909. doi: 10.1063/1.466432
Glushkov VN (1998) Open-shell Møller–Plesset perturbation theory based on the asymptotic
method of obtaining SCF orbitals. Chem Phys Lett 287:189–194. doi: 10.1016/S0009-
2614(98)00142-0

162
Glushkov VN (2002) An eigenvalue problem with limitations in a finite movable basis. Opt
Spectrosc 93:11–18. doi: 10.1134/1.1496717
Glushkov VN (2002) Spin-Unrestricted Formalism for a Partially Restricted Hartree–Fock
Approach. J Math Chem 31:91–103. doi: 10.1023/A:1015486430744
Glushkov VN (2013) Excited and core-ionized state calculations with a local potential
expressed in terms of the external potential. Int J Quantum Chem 113:637–642. doi:
10.1002/qua.24019
Glushkov VN, Assfeld X (2010) Doubly, triply, and multiply excited states from a
constrained optimized effective potential method. J Chem Phys 132:204106. doi:
10.1063/1.3443777
Glushkov VN, Assfeld X (2012) On orthogonality constrained multiple core-hole states and
optimized effective potential method. J Comput Chem 33:2058–2066. doi:
10.1002/jcc.23041
Glushkov VN, Gidopoulos NI, Wilson S (2008) Alternative Technique for the Constrained
Variational Problem Based on an Asymptotic Projection Method: I. Basics. In: Wilson S,
Grout PJ, Maruani J, et al. (eds). Springer Netherlands, pp 429–450
Glushkov VN, Gidopoulos NI, Wilson S (2008) Alternative Technique for the Constrained
Variational Problem Based on an Asymptotic Projection Method: II. Applications to
Open-Shell Self-Consistent Field Theory. In: Wilson S, Grout PJ, Maruani J, et al. (eds)
Frontiers in Quantum Systems in Chemistry and Physics. Springer Netherlands,
Dordrecht, pp 451–489
Glushkov VN, Mogilevskaya N V. (2013) The Hartree-Fock method with orthogonality
restrictions for doubly excited and ionized states. Opt Spectrosc 114:161–166. doi:
10.1134/S0030400X13020100
Glushkov VN, Tsaune AY (1985) Unconditional minimization in eigenvalue problems with
additional conditions. 25:298–301.
Gorb LG, Rivail J-L, Thery V, Rinaldi D (1996) Modification of the local self-consistent field
method for modeling surface reactivity of covalent solids. Int J Quantum Chem 60:1525–
1536. doi: 10.1002/(SICI)1097-461X(1996)60:7<1525::AID-QUA34>3.0.CO;2-#
Green LM, Berg JM (1989) A retroviral Cys-Xaa2-Cys-Xaa4-His-Xaa4-Cys peptide binds
metal ions: spectroscopic studies and a proposed three-dimensional structure. Proc Natl
Acad Sci 86:4047–4051.
Gregersen BA, York DM (2005) Variational electrostatic projection (VEP) methods for
efficient modeling of the macromolecular electrostatic and solvation environment in
activated dynamics simulations. J Phys Chem B 109:536–556. doi: 10.1021/jp0469968
Gregersen BA, York DM (2006) A charge-scaling implementation of the variational
electrostatic projection method. J Comput Chem 27:103–115. doi: 10.1002/jcc.20318

163
Gresh N (1995) Energetics of Zn2+ binding to a series of biologically relevant ligands: A
molecular mechanics investigation grounded on ab initio self-consistent field
supermolecular computations. J Comput Chem 16:856–882. doi: 10.1002/jcc.540160705
Gresh N, Garmer DR (1996) Comparative binding energetics of Mg2+, Ca2+, Zn2+, and
Cd2+ to biologically relevant ligands: Combined ab initio SCF supermolecule and
molecular mechanics investigation. J Comput Chem 17:1481–1495. doi:
10.1002/(SICI)1096-987X(199609)17:12<1481::AID-JCC7>3.0.CO;2-G
Gresh N, Piquemal J-P, Krauss M (2005) Representation of Zn(II) complexes in polarizable
molecular mechanics. Further refinements of the electrostatic and short-range
contributions. Comparisons with parallel ab initio computations. J Comput Chem
26:1113–1130. doi: 10.1002/jcc.20244
Guallar V, Baik M-H, Lippard SJ, Friesner RA (2003) Peripheral heme substituents control
the hydrogen-atom abstraction chemistry in cytochromes P450. Proc Natl Acad Sci
100:6998–7002. doi: 10.1073/pnas.0732000100
Halgren TA (1992) The representation of van der Waals (vdW) interactions in molecular
mechanics force fields: potential form, combination rules, and vdW parameters. J Am
Chem Soc 114:7827–7843. doi: 10.1021/ja00046a032
Halgren TA, Damm W (2001) Polarizable force fields. Curr Opin Struct Biol 11:236–242.
doi: 10.1016/S0959-440X(00)00196-2
Hancock RD (1990) Molecular mechanics calculations and metal ion recognition. Acc Chem
Res 23:253–257. doi: 10.1021/ar00176a003
Hartree DR (1928) The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part
II. Some Results and Discussion. Math Proc Cambridge Philos Soc 24:111–132. doi:
10.1017/S0305004100011920
Hathout Y, Fabris D, Han MS, et al (1996) Characterization of intermediates in the oxidation
of zinc fingers in human immunodeficiency virus type 1 nucleocapsid protein {P}7.
Drug Metab Dispos 24:1395–1400.
Hermans J, Berendsen HJC, Van Gunsteren WF, Postma JPM (1984) A consistent empirical
potential for water–protein interactions. Biopolymers 23:1513–1518. doi:
10.1002/bip.360230807
Heßelmann A, Jansen G (2002) First-order intermolecular interaction energies from Kohn–
Sham orbitals. Chem Phys Lett 357:464–470. doi: 10.1016/S0009-2614(02)00538-9
Hobza P, Kabeláč M, Šponer J, et al (1997) Performance of empirical potentials (AMBER,
CFF95, CVFF, CHARMM, OPLS, POLTEV), semiempirical quantum chemical
methods (AM1, MNDO/M, PM3), and ab initio Hartree–Fock method for interaction of
DNA bases: Comparison with nonempir. J Comput Chem 18:1136–1150. doi:
10.1002/(SICI)1096-987X(19970715)18:9<1136::AID-JCC3>3.0.CO;2-S

164
Hobza P, Selzle HL, Schlag EW (1996) Potential Energy Surface for the Benzene Dimer.
Results of ab Initio CCSD(T) Calculations Show Two Nearly Isoenergetic Structures: T-
Shaped and Parallel-Displaced. J Phys Chem 100:18790–18794. doi: 10.1021/jp961239y
Hobza P, Zahradník R (1992) An essay on the theory and calculations of intermolecular
interactions. Int J Quantum Chem 42:581–590. doi: 10.1002/qua.560420407
Hohenberg P, Kohn W (1964) Inhomogeneous {Electron} {Gas}. Phys Rev 136:B864--B871.
doi: 10.1103/PhysRev.136.B864
Hok Hei T (2012) Towards accurate calculations of Zn2+ binding free energies in zinc finger
proteins.
Hoops SC, Anderson KW, Merz KM (1991) Force field design for metalloproteins. J Am
Chem Soc 113:8262–8270. doi: 10.1021/ja00022a010
Hratchian HP, Parandekar P V., Raghavachari K, et al (2008) QM:QM electronic embedding
using Mulliken atomic charges: Energies and analytic gradients in an ONIOM
framework. J Chem Phys 128:34107. doi: 10.1063/1.2814164
Humbel S, Sieber S, Morokuma K (1996) The IMOMO method: Integration of different
levels of molecular orbital approximations for geometry optimization of large systems:
Test for n‐butane conformation and SN2 reaction: RCl+Cl−. J Chem Phys 105:1959–
1967. doi: 10.1063/1.472065
Humbert-Droz M, Zhou X, Shedge S V, Wesolowski TA (2013) How to choose the frozen
density in Frozen-Density Embedding Theory-based numerical simulations of local
excitations? Theor Chem Acc 133:1–20. doi: 10.1007/s00214-013-1405-1
Hunter CA, Singh J, Thornton JM (1991) Pi-pi interactions: the geometry and energetics of
phenylalanine-phenylalanine interactions in proteins. J Mol Biol 218:837–846.
Ioannou AG, Colwell SM, Amos RD (1997) The calculation of frequency-dependent
polarizabilities using current density functional theory. Chem Phys Lett 278:278–284.
doi: 10.1016/S0009-2614(97)00978-0
Ishikawa S, Ebata T, Mikami N (1999) Structures and the vibrational relaxations of size-
selected benzonitrile–(H2O)n=1–3 and –(CH3OH)n=1–3 clusters studied by
fluorescence detected Raman and infrared spectroscopies. J Chem Phys 110:9504–9515.
doi: 10.1063/1.478915
Izgorodina EI, MacFarlane DR (2011) Nature of Hydrogen Bonding in Charged Hydrogen-
Bonded Complexes and Imidazolium-Based Ionic Liquids. J Phys Chem B 115:14659–
14667. doi: 10.1021/jp208150b
Jacquemin D, Perpète EA, Scuseria GE, et al (2008) TD-DFT Performance for the Visible
Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J
Chem Theory Comput 4:123–135. doi: 10.1021/ct700187z

165
Jankowski P, Jeziorski B, Rybak S, Szalewicz K (1990) Symmetry‐adapted perturbation
theory calculation of the intra‐atomic correlation contribution to the short‐range
repulsion of helium atoms. J Chem Phys 92:7441–7447. doi: 10.1063/1.458230
Jansen G, Hesselmann A (2001) Comment on “Using Kohn−Sham Orbitals in Symmetry-
Adapted Perturbation Theory To Investigate Intermolecular Interactions.” J Phys Chem
A 105:11156–11157. doi: 10.1021/jp0112774
Jeffrey GA (1997) An Introduction to Hydrogen Bonding, Oxford Uni. : New Yor
Jeziorski B, Moszynski R, Szalewicz K (1994) Perturbation Theory Approach to
Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem Rev
94:1887–1930. doi: 10.1021/cr00031a008
Jeziorski B, Szalewicz K (1998) Encyclopedia of Computational Chemistry. New York
Ji CG, Xiao X, Zhang JZH (2012) Studying the Effect of Site-Specific Hydrophobicity and
Polarization on Hydrogen Bond Energy of Protein Using a Polarizable Method. J Chem
Theory Comput 8:2157–2164. doi: 10.1021/ct300252d
Jurečka P, Šponer J, Černý J, Hobza P (2006) Benchmark database of accurate (MP2 and
CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA
base pairs, and amino acid pairs. Phys Chem Chem Phys 8:1985–1993. doi:
10.1039/B600027D
Kairys V, Jensen JH (2000) QM/MM Boundaries Across Covalent Bonds: A Frozen
Localized Molecular Orbital-Based Approach for the Effective Fragment Potential
Method. J Phys Chem A 104:6656–6665. doi: 10.1021/jp000887l
Kaminski GA, Jorgensen WL (1999) Host–guest chemistry of rotaxanes and catenanes:
application of a polarizable all-atom force field to cyclobis(paraquat-p-phenylene)
complexes with disubstituted benzenes and biphenyls†. J Chem Soc Perkin Trans 2
2365–2375. doi: 10.1039/A905160K
Kaminski GA, Stern HA, Berne BJ, Friesner RA (2004) Development of an Accurate and
Robust Polarizable Molecular Mechanics Force Field from ab Initio Quantum Chemistry.
J Phys Chem A 108:621–627. doi: 10.1021/jp0301103
Karle J, Huang L (2003) The glue that holds crystals together: a review. J Mol Struct 647:9–
16. doi: 10.1016/S0022-2860(02)00516-1
Khandogin J, Musier-Forsyth K, York DM (2003) Insights into the Regioselectivity and
RNA-binding Affinity of HIV-1 Nucleocapsid Protein from Linear-scaling Quantum
Methods. J Mol Biol 330:993–1004. doi: 10.1016/S0022-2836(03)00658-2
Kim K, Jordan KD (1994) Comparison of Density Functional and MP2 Calculations on the
Water Monomer and Dimer. J Phys Chem 98:10089–10094. doi: 10.1021/j100091a024

166
Kim W, Schaeffer MW, Lee S, et al (1999) Intermolecular vibrations of naphthalene trimer by
ionization-detected stimulated Raman spectroscopy. J Chem Phys 110:11264–11276.
doi: 10.1063/1.479067
Kisiel Z, Fowler PW, Legon AC (1992) Investigation of the rotational spectrum of the
hydrogen-bonded dimer CF2CH2...HCl. J Chem Soc Faraday Trans 88:3385. doi:
10.1039/ft9928803385
Kitaura K, Morokuma K (1976) A new energy decomposition scheme for molecular
interactions within the Hartree-Fock approximation. Int J Quantum Chem 10:325–340.
doi: 10.1002/qua.560100211
Kitaura K, Morokuma K (1976) A new energy decomposition scheme for molecular
interactions within the Hartree-Fock approximation. Int J Quantum Chem 10:325–340.
doi: 10.1002/qua.560100211
Klein DJ, Johnson PE, Zollars ES, et al (2000) The NMR structure of the nucleocapsid
protein from the mouse mammary tumor virus reveals unusual folding of the C-terminal
zinc knuckle. Biochemistry 39:1604–1612.
Koca J, Zhan C-G, Rittenhouse RC, Ornstein RL (2003) Coordination number of zinc ions in
the phosphotriesterase active site by molecular dynamics and quantum mechanics. J
Comput Chem 24:368–378. doi: 10.1002/jcc.10217
Kohn W, Sham LJ (1965) Self-Consistent Equations Including Exchange and Correlation
Effects. Phys Rev 140:A1133–A1138. doi: 10.1103/PhysRev.140.A1133
Kollman PA (1977) Hydrogen Bonding and Donor—Acceptor Interactions. In: III HFS (ed).
Springer US, pp 109–152
Kornhaber GJ, Snyder D, Moseley HNB, Montelione GT (2006) Identification of zinc-ligated
cysteine residues based on 13Calpha and 13Cbeta chemical shift data. J Biomol NMR
34:259–269. doi: 10.1007/s10858-006-0027-5
Kothekar V, Pullman A, Demoulin D (1978) Ab initio molecular-orbital study of the binding
of ZnII with SH2 and SH−. Int J Quantum Chem 14:779–791. doi:
10.1002/qua.560140610
Kozmutza C, Kapuy E, Evleth EM, et al (1996) Application of the localized representation for
studying interaction energies. Int J Quantum Chem 57:775–780. doi:
10.1002/(SICI)1097-461X(1996)57:4<775::AID-QUA25>3.0.CO;2-Y
Kurplus M, McCammon JA (1983) Dynamics of Proteins: Elements and Function. Annu Rev
Biochem 52:263–300. doi: 10.1146/annurev.bi.52.070183.001403
Laio A, VandeVondele J, Rothlisberger U (2002) A Hamiltonian electrostatic coupling
scheme for hybrid Car–Parrinello molecular dynamics simulations. J Chem Phys
116:6941–6947. doi: 10.1063/1.1462041

167
Lamoureux G, Roux B (2003) Modeling induced polarization with classical Drude oscillators:
Theory and molecular dynamics simulation algorithm. J Chem Phys 119:3025–3039.
doi: 10.1063/1.1589749
Langlet J, Bergès J, Caillet J, Kozelka J (2000) Hydration of platinum(II) complexes: a
molecular mechanics study using atom-based force-field parameters. Theor Chem Acc
104:247–251. doi: 10.1007/s002140000147
Langlet J, Caillet J, Bergès J, Reinhardt P (2003) Comparison of two ways to decompose
intermolecular interactions for hydrogen-bonded dimer systems. J Chem Phys 118:6157–
6166. doi: 10.1063/1.1558473
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy
formula into a functional of the electron density. Phys Rev B 37:785–789. doi:
10.1103/PhysRevB.37.785
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy
formula into a functional of the electron density. Phys Rev B 37:785–789. doi:
10.1103/PhysRevB.37.785
Lee Y-M, Lim C (2008) Physical basis of structural and catalytic Zn-binding sites in proteins.
J Mol Biol 379:545–553. doi: 10.1016/j.jmb.2008.04.004
Leopold KR, Fraser GT, Novick SE, Klemperer W (1994) Current Themes in Microwave and
Infrared Spectroscopy of Weakly Bound Complexes. Chem Rev 94:1807–1827. doi:
10.1021/cr00031a004
Li W, Zhang J, Wang J, Wang W (2008) Metal-Coupled Folding of Cys2His2 Zinc-Finger. J
Am Chem Soc 130:892–900. doi: 10.1021/ja075302g
Li YL, Mei Y, Zhang DW, et al (2011) Structure and Dynamics of a Dizinc Metalloprotein:
Effect of Charge Transfer and Polarization. J Phys Chem B 115:10154–10162. doi:
10.1021/jp203505v
Lin H, Truhlar DG (2005) Redistributed Charge and Dipole Schemes for Combined Quantum
Mechanical and Molecular Mechanical Calculations. J Phys Chem A 109:3991–4004.
doi: 10.1021/jp0446332
Lipscomb WN, Sträter N (1996) Recent Advances in Zinc Enzymology. Chem Rev 96:2375–
2434. doi: 10.1021/cr950042j
Liu S, Dykstra CE (1986) Electrical influence on monomer orientation in hydrogen bonded
and other weakly bonded complexes. Chem Phys 107:343–349. doi: 10.1016/0301-
0104(86)85012-1
London F (1930) Zur Theorie und Systematik der Molekularkräfte. Z Phys 63:245–279.
London F (1937) The General Theory of Molecular Forces. Trans Faraday Soc 33:8–26.

168
Loos P-F, Assfeld X (2007) Core-ionized and core-excited states of macromolecules. Int J
Quantum Chem 107:2243–2252. doi: 10.1002/qua.21410
Loos P-F, Assfeld X (2007) Self-Consistent Strictly Localized Orbitals. J Chem Theory
Comput 3:1047–1053. doi: 10.1021/ct6003214
Loos P-F, Assfeld X, others (2007) On The Frontier Bond Location In The QM/MM
Description Of Peptides And Proteins. pp 308–315
Loos P-F, Assfeld X, Rivail J-L (2007) Intramolecular interactions and cis peptidic bonds.
Theor Chem Acc 118:165–171. doi: 10.1007/s00214-007-0258-x
Loos P-F, Fornili A, Sironi M, Assfeld X (2007) Removing extra frontier parameters in
QM/MM methods : a tentative with the Local Self-Consistent Field approach. Comput
Lett 3:473–486. doi: 10.1163/157404007782913309
Löwdin P-O (1955) Quantum Theory of Many-Particle Systems. I. Physical Interpretations by
Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the
Method of Configurational Interaction. Phys Rev 97:1474–1489. doi:
10.1103/PhysRev.97.1474
Lu Y, Mei Y, Zhang JZH, Zhang D (2010) Communications: Electron polarization critically
stabilizes the Mg2+ complex in the catalytic core domain of HIV-1 integrase. J Chem
Phys 132:131101. doi: 10.1063/1.3360769
Lyne PD, Walsh OA (2001) Computational Biochemistry and Biophysics, Dekker. O. M.
Becker, M. Watanabe, New York
MacKerell AD, Bashford D, Bellott M, et al (1998) All-atom empirical potential for
molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616.
doi: 10.1021/jp973084f
MacKerell AD, Wiorkiewicz-Kuczera J, Karplus M (1995) An all-atom empirical energy
function for the simulation of nucleic acids. J Am Chem Soc 117:11946–11975. doi:
10.1021/ja00153a017
Magistrato A, Togni A, Rӧthlisberger U, Woo TK (2002) Computational Modeling of
Homogeneous Catalysis, Springer. F. Maseras, A. Lledos, Dordrecht
Magnasco V, Perico A (1967) Uniform Localization of Atomic and Molecular Orbitals. I. J
Chem Phys 47:971–981. doi: 10.1063/1.1712065
Magnasco V, Perico A (1968) Uniform Localization of Atomic and Molecular Orbitals. II. J
Chem Phys 48:800–808. doi: 10.1063/1.1668714
Makinen MW, Troyer JM, van der Werff H, et al (1989) Dynamical structure of
carboxypeptidase A. J Mol Biol 207:201–216.
Maseras F (1999) Organometallic Bonding and Reactivity: Fundamental Studies, Springer. J.
M. Brown, P. Hofmann, Berlin

169
Maseras F, Morokuma K (1995) IMOMM: A new integrated ab initio + molecular mechanics
geometry optimization scheme of equilibrium structures and transition states. J Comput
Chem 16:1170–1179. doi: 10.1002/jcc.540160911
Mata RA, Werner H-J, Thiel S, Thiel W (2008) Toward accurate barriers for enzymatic
reactions: QM/MM case study on p-hydroxybenzoate hydroxylase. J Chem Phys
128:25104. doi: 10.1063/1.2823055
Matsubara T, Maseras F, Koga N, Morokuma K (1996) Application of the New “Integrated
MO + MM” (IMOMM) Method to the Organometallic Reaction Pt(PR3)2 + H2 (R = H,
Me, t-Bu, and Ph). J Phys Chem 100:2573–2580. doi: 10.1021/jp951762x
McCall KA, Huang C, Fierke CA (2000) Function and Mechanism of Zinc Metalloenzymes. J
Nutr 130:1437S–1446S.
McWeeny R (1989) Methods of molecular quantum mechanics, 2nd ed. Academic Press,
London ; San Diego
Merz KM, Kollman PA (1989) Free energy perturbation simulations of the inhibition of
thermolysin: prediction of the free energy of binding of a new inhibitor. J Am Chem Soc
111:5649–5658. doi: 10.1021/ja00197a022
Merz KM, Kollman PA (1989) Free energy perturbation simulations of the inhibition of
thermolysin: prediction of the free energy of binding of a new inhibitor. J Am Chem Soc
111:5649–5658. doi: 10.1021/ja00197a022
Minenkov Y, Singstad Å, Occhipinti G, Jensen VR (2012) The accuracy of DFT-optimized
geometries of functional transition metal compounds: a validation study of catalysts for
olefin metathesis and other reactions in the homogeneous phase. Dalt Trans 41:5526–
5541. doi: 10.1039/C2DT12232D
Misquitta AJ, Podeszwa R, Jeziorski B, Szalewicz K (2005) Intermolecular potentials based
on symmetry-adapted perturbation theory with dispersion energies from time-dependent
density-functional calculations. J Chem Phys 123:214103. doi: 10.1063/1.2135288
Mitoraj MP, Michalak A, Ziegler T (2009) A Combined Charge and Energy Decomposition
Scheme for Bond Analysis. J Chem Theory Comput 5:962–975. doi: 10.1021/ct800503d
Monard G, Loos M, Théry V, et al (1996) Hybrid classical quantum force field for modeling
very large molecules. Int J Quantum Chem 58:153–159. doi: 10.1002/(SICI)1097-
461X(1996)58:2<153::AID-QUA4>3.0.CO;2-X
Monard G, Loos M, Théry V, et al (1996) Hybrid classical quantum force field for modeling
very large molecules. Int J Quantum Chem 58:153–159. doi: 10.1002/(SICI)1097-
461X(1996)58:2<153::AID-QUA4>3.0.CO;2-X
Moreau Y, Loos P-F, Assfeld X (2004) Solvent effects on the asymmetric Diels–Alder
reaction between cyclopentadiene and (−)-menthyl acrylate revisited with the three-layer
hybrid local self-consistent field/molecular mechanics/self-consistent reaction field
method. Theor Chem Acc 112:228–239. doi: 10.1007/s00214-004-0581-4

170
Morellet N, Jullian N, De Rocquigny H, et al (1992) Determination of the structure of the
nucleocapsid protein NCp7 from the human immunodeficiency virus type 1 by 1H NMR.
EMBO J 11:3059–3065.
Moszynski R, Jeziorski B, Rybak S, et al (1994) Many‐body theory of exchange effects in
intermolecular interactions. Density matrix approach and applications to He–F−, He–HF,
H2–HF, and Ar–H2 dimers. J Chem Phys 100:5080–5092. doi: 10.1063/1.467225
Moszynski R, Jeziorski B, Rybak S, et al (1994) Many‐body theory of exchange effects in
intermolecular interactions. Density matrix approach and applications to He–F−, He–HF,
H2–HF, and Ar–H2 dimers. J Chem Phys 100:5080–5092. doi: 10.1063/1.467225
Murakami M, Hirano T (2008) Intracellular zinc homeostasis and zinc signaling. Cancer Sci
99:1515–1522. doi: 10.1111/j.1349-7006.2008.00854.x
Murphy RB, Philipp DM, Friesner RA (2000) A mixed quantum mechanics/molecular
mechanics (QM/MM) method for large-scale modeling of chemistry in protein
environments. J Comput Chem 21:1442–1457. doi: 10.1002/1096-
987X(200012)21:16<1442::AID-JCC3>3.0.CO;2-O
Nam K, Gao J, York DM (2005) An Efficient Linear-Scaling Ewald Method for Long-Range
Electrostatic Interactions in Combined QM/MM Calculations. J Chem Theory Comput
1:2–13. doi: 10.1021/ct049941i
Naray-Szabo G, Ferenczy GG (1995) Molecular Electrostatics. Chem Rev 95:829–847. doi:
10.1021/cr00036a002
Neugebauer J, Louwerse MJ, Belanzoni P, et al (2005) Modeling solvent effects on electron-
spin-resonance hyperfine couplings by frozen-density embedding. J Chem Phys. doi:
http://dx.doi.org/10.1063/1.2033749
Nomura A, Sugiura Y (2002) Contribution of Individual Zinc Ligands to Metal Binding and
Peptide Folding of Zinc Finger Peptides. Inorg Chem 41:3693–3698. doi:
10.1021/ic025557p
Ott K-H, Meyer B (1996) Parametrization of GROMOS force field for oligosaccharides and
assessment of efficiency of molecular dynamics simulations. J Comput Chem 17:1068–
1084. doi: 10.1002/(SICI)1096-987X(199606)17:8<1068::AID-JCC14>3.0.CO;2-A
Parkin G (2004) Synthetic Analogues Relevant to the Structure and Function of Zinc
Enzymes. Chem Rev 104:699–768. doi: 10.1021/cr0206263
Patel K, Kumar A, Durani S (2007) Analysis of the structural consensus of the zinc
coordination centers of metalloprotein structures. Biochim Biophys Acta - Proteins
Proteomics 1774:1247–1253. doi: 10.1016/j.bbapap.2007.07.010
Pauli W (1925) Über den Zusammenhang des Abschlusses der Elektronengruppen im Atom
mit der Komplexstruktur der Spektren. Zeitschrift für Phys 31:765–783. doi:
10.1007/BF02980631

171
Pavanello M, Van Voorhis T, Visscher L, Neugebauer J (2013) An accurate and linear-scaling
method for calculating charge-transfer excitation energies and diabatic couplings. J
Chem Phys. doi: http://dx.doi.org/10.1063/1.4789418
Pearlman DA, Case DA, Caldwell JW, et al (1995) AMBER, a package of computer
programs for applying molecular mechanics, normal mode analysis, molecular dynamics
and free energy calculations to simulate the structural and energetic properties of
molecules. Comput Phys Commun 91:1–41. doi: 10.1016/0010-4655(95)00041-D
Peraro MD, Spiegel K, Lamoureux G, et al (2007) Modeling the charge distribution at metal
sites in proteins for molecular dynamics simulations. J Struct Biol 157:444–453. doi:
10.1016/j.jsb.2006.10.019
Perdew JP, Burke K, Ernzerhof M (1996) Generalized Gradient Approximation Made Simple.
Phys Rev Lett 77:3865–3868. doi: 10.1103/PhysRevLett.77.3865
Perdew JP, Burke K, Ernzerhof M (1997) Generalized Gradient Approximation Made Simple.
Phys Rev Lett 78:1396–1396. doi: 10.1103/PhysRevLett.78.1396
Perdew JP, Chevary JA, Vosko SH, et al (1992) Atoms, molecules, solids, and surfaces:
Applications of the generalized gradient approximation for exchange and correlation.
Phys Rev B 46:6671–6687. doi: 10.1103/PhysRevB.46.6671
Perdew JP, Tao J, Staroverov VN, Scuseria GE (2004) Meta-generalized gradient
approximation: Explanation of a realistic nonempirical density functional. J Chem Phys
120:6898–6911. doi: 10.1063/1.1665298
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas
correlation energy. Phys Rev B 45:13244–13249. doi: 10.1103/PhysRevB.45.13244
Perdew JP, Yue W (1986) Accurate and simple density functional for the electronic exchange
energy: Generalized gradient approximation. Phys Rev B 33:8800–8802. doi:
10.1103/PhysRevB.33.8800
Peters MB, Yang Y, Wang B, et al (2010) Structural Survey of Zinc-Containing Proteins and
Development of the Zinc AMBER Force Field (ZAFF). J Chem Theory Comput 6:2935–
2947. doi: 10.1021/ct1002626
Peters MB, Yang Y, Wang B, et al (2010) Structural Survey of Zinc-Containing Proteins and
Development of the Zinc AMBER Force Field (ZAFF). J Chem Theory Comput 6:2935–
2947. doi: 10.1021/ct1002626
Philipp DM, Friesner RA (1999) Mixed ab initio QM/MM modeling using frozen orbitals and
tests with alanine dipeptide and tetrapeptide. J Comput Chem 20:1468–1494. doi:
10.1002/(SICI)1096-987X(19991115)20:14<1468::AID-JCC2>3.0.CO;2-0
Pimentel GC, McClellan AL (1971) Hydrogen Bonding. Annu Rev Phys Chem 22:347–385.
doi: 10.1146/annurev.pc.22.100171.002023

172
Pipek J (1989) Localization measure and maximum delocalization in molecular systems. Int J
Quantum Chem 36:487–501. doi: 10.1002/qua.560360405
Pipek J, Mezey PG (1989) A fast intrinsic localization procedure applicable for abinitio and
semiempirical linear combination of atomic orbital wave functions. J Chem Phys
90:4916–4926. doi: 10.1063/1.456588
Piquemal J-P, Marquez A, Parisel O, Giessner–Prettre C (2005) A CSOV study of the
difference between HF and DFT intermolecular interaction energy values: The
importance of the charge transfer contribution. J Comput Chem 26:1052–1062. doi:
10.1002/jcc.20242
Plattner N, Meuwly M (2008) The Role of Higher CO-Multipole Moments in Understanding
the Dynamics of Photodissociated Carbonmonoxide in Myoglobin. Biophys J 94:2505–
2515. doi: 10.1529/biophysj.107.120519
Politzer P, Laurence PR, Jayasuriya K (1985) Molecular electrostatic potentials: an effective
tool for the elucidation of biochemical phenomena. Environ Health Perspect 61:191–202.
Ponder JW, others (2004) TINKER: Software tools for molecular design.
Pople JA, Nesbet RK (1954) Self‐Consistent Orbitals for Radicals. J Chem Phys 22:571–572.
doi: 10.1063/1.1740120
Pu J, Gao J, Truhlar DG (2005) Generalized Hybrid-Orbital Method for Combining Density
Functional Theory with Molecular Mechanicals. ChemPhysChem 6:1853–1865. doi:
10.1002/cphc.200400602
Pullman B (1978) Intermolecular Interactions: From Diatomics to Biopolymers, John Wiley.
New York
Rablen PR, Lockman JW, Jorgensen WL (1998) Ab Initio Study of Hydrogen-Bonded
Complexes of Small Organic Molecules with Water. J Phys Chem A 102:3782–3797.
doi: 10.1021/jp980708o
Raha K, Merz KMJ (2004) A quantum mechanics-based scoring function: study of zinc ion-
mediated ligand binding. J Am Chem Soc 126:1020–1021. doi: 10.1021/ja038496i
Rappé AK, Bernstein ER (2000) Ab Initio Calculation of Nonbonded Interactions: Are We
There Yet? J Phys Chem A 104:6117–6128. doi: 10.1021/jp0008997
Rappe AK, Goddard III WA (1991) Charge equilibration for molecular dynamics simulations.
J Phys Chem 95:3358–3363. doi: 10.1021/j100161a070
Riccardi D, Schaefer P, Cui Q (2005) pKa Calculations in Solution and Proteins with
QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM
Protocols. J Phys Chem B 109:17715–17733. doi: 10.1021/jp0517192
Rivail J-L (2004) Computational Medicinal Chemistry for Drug Discovery, Dekker. P.
Bultinck, H. DeWinter, L. Wilfried, J. P. Tollenaere, New York

173
Robertson A, Luttmann E, Pande VS (2008) Effects of long-range electrostatic forces on
simulated protein folding kinetics. J Comput Chem 29:694–700. doi: 10.1002/jcc.20828
Rode BM, Schwenk CF, Hofer TS, Randolf BR (2005) Coordination and ligand exchange
dynamics of solvated metal ions. Coord Chem Rev 249:2993–3006. doi:
10.1016/j.ccr.2005.03.032
Rogalewicz F, Ohanessian G, Gresh N (2000) Interaction of neutral and zwitterionic glycine
with Zn2+ in gas phase: ab initio and SIBFA molecular mechanics calculations. J
Comput Chem 21:963–973. doi: 10.1002/1096-987X(200008)21:11<963::AID-
JCC6>3.0.CO;2-3
Roothaan CCJ (1951) New Developments in Molecular Orbital Theory. Rev Mod Phys
23:69–89. doi: 10.1103/RevModPhys.23.69
Roy LE, Hay PJ, Martin RL (2008) Revised Basis Sets for the LANL Effective Core
Potentials. J Chem Theory Comput 4:1029–1031. doi: 10.1021/ct8000409
Ruiz-Lopez M., Rivail J. (1998) Encyclopedia of Computational Chemistry, P.v.R. Sch.
Chichester
Russell AJ, Fersht AR (1987) Rational modification of enzyme catalysis by engineering
surface charge. Nature 328:496–500. doi: 10.1038/328496a0
Rybak S, Szalewicz K, Jeziorski B, Jaszunski M (1987) Intraatomic correlation effects for the
He–He dispersion and exchange–dispersion energies using explicitly correlated Gaussian
geminals. J Chem Phys 86:5652–5659. doi: 10.1063/1.452542
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian
equations of motion of a system with constraints: molecular dynamics of n-alkanes. J
Comput Phys 23:327–341. doi: 10.1016/0021-9991(77)90098-5
Ryde U (1995) Molecular dynamics simulations of alcohol dehydrogenase with a four- or
five-coordinate catalytic zinc ion. Proteins 21:40–56. doi: 10.1002/prot.340210106
Ryde U (1995) On the role of Glu-68 in alcohol dehydrogenase. Protein Sci A Publ Protein
Soc 4:1124–1132. doi: 10.1002/pro.5560040611
Ryde U (1996) The coordination of the catalytic zinc in alcohol dehydrogenase studied by
combined quantum-chemical and molecular mechanics calculations. J Comput Aided
Mol Des 10:153–164.
Ryde U (1999) Carboxylate binding modes in zinc proteins: A theoretical study. Biophys J
77:2777–2787.
Saenger W (1984) Principles of nucleic acid structure, Springer-V. New York
Sagui C, Darden TA (1999) Molecular dynamics simulations of biomolecules: long-range
electrostatic effects. Annu Rev Biophys Biomol Struct 28:155–179. doi:
10.1146/annurev.biophys.28.1.155

174
Sakharov D V., Lim C (2005) Zn Protein Simulations Including Charge Transfer and Local
Polarization Effects. J Am Chem Soc 127:4921–4929. doi: 10.1021/ja0429115
Sakharov D V., Lim C (2009) Force fields including charge transfer and local polarization
effects: Application to proteins containing multi/heavy metal ions. J Comput Chem
30:191–202. doi: 10.1002/jcc.21048
Sánchez R, Blanco S, Lesarri A, et al (2005) The rotational spectrum of the 3,3-
dimethyloxetane⋯hydrogen chloride complex. Chem Phys Lett 401:259–265. doi:
10.1016/j.cplett.2004.11.095
Sancho-Garcıa JC, Cornil J (2004) Assessment of recently developed exchange-correlation
functionals for the description of torsion potentials in π-conjugated molecules. J Chem
Phys 121:3096–3101. doi: 10.1063/1.1774976
Sauer J, Ugliengo P, Garrone E, Saunders VR (1994) Theoretical Study of van der Waals
Complexes at Surface Sites in Comparison with the Experiment. Chem Rev 94:2095–
2160. doi: 10.1021/cr00031a014
Schaefer P, Riccardi D, Cui Q (2005) Reliable treatment of electrostatics in combined
QM/MM simulation of macromolecules. J Chem Phys 123:14905. doi:
10.1063/1.1940047
Scheiner S (1997) Hydrogen Bonding. A Theoretical Perspective, Oxford Uni.
Scheiner S, Kar T (2005) Effect of Solvent upon CH···O Hydrogen Bonds with Implications
for Protein Folding. J Phys Chem B 109:3681–3689. doi: 10.1021/jp0446736
Scheiner S, Kar T (2005) Effect of Solvent upon CH···O Hydrogen Bonds with Implications
for Protein Folding. J Phys Chem B 109:3681–3689. doi: 10.1021/jp0446736
Schrödinger E (1926) An Undulatory Theory of the Mechanics of Atoms and Molecules. Phys
Rev 28:1049–1070. doi: 10.1103/PhysRev.28.1049
Schütz M, Rauhut G, Werner H-J (1998) Local Treatment of Electron Correlation in
Molecular Clusters: Structures and Stabilities of (H2O)n, n = 2−4. J Phys Chem A
102:5997–6003. doi: 10.1021/jp981168y
Schymkowitz JWH, Rousseau F, Martins IC, et al (2005) Prediction of water and metal
binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci U S
A 102:10147–10152. doi: 10.1073/pnas.0501980102
Scott WRP, Hünenberger PH, Tironi IG, et al (1999) The GROMOS Biomolecular
Simulation Program Package. J Phys Chem A 103:3596–3607. doi: 10.1021/jp984217f
Seebeck B, Reulecke I, Kämper A, Rarey M (2008) Modeling of metal interaction geometries
for protein-ligand docking. Proteins 71:1237–1254. doi: 10.1002/prot.21818
Senn HM, Thiel W (2007) Atomistic Approaches in Modern Biology, Springer. M. Reiher,
Berlin

175
Senthilkumar K, Mujika JI, Ranaghan KE, et al (2008) Analysis of polarization in QM/MM
modelling of biologically relevant hydrogen bonds. J R Soc Interface 5:S207–S216. doi:
10.1098/rsif.2008.0243.focus
Shoemaker JR, Gordon MS (1999) SIMOMM: An Integrated Molecular Orbital/Molecular
Mechanics Optimization Scheme for Surfaces. J Phys Chem A 103:3245–3251. doi:
10.1021/jp982600e
Shu N, Zhou T, Hovmöller S (2008) Prediction of zinc-binding sites in proteins from
sequence. Bioinformatics 24:775–782. doi: 10.1093/bioinformatics/btm618
Sierka M, Sauer J (2005) Handbook of Materials Modeling, Springer. S. Yip, Dordrecht
Singh UC, Kollman PA (1986) A combined ab initio quantum mechanical and molecular
mechanical method for carrying out simulations on complex molecular systems:
Applications to the CH3Cl + Cl− exchange reaction and gas phase protonation of
polyethers. J Comput Chem 7:718–730. doi: 10.1002/jcc.540070604
Slater JC (1929) The Theory of Complex Spectra. Phys Rev 34:1293–1322. doi:
10.1103/PhysRev.34.1293
Slater JC (1951) A Simplification of the Hartree-Fock Method. Phys Rev 81:385–390. doi:
10.1103/PhysRev.81.385
Sodhi JS, Bryson K, McGuffin LJ, et al (2004) Predicting Metal-binding Site Residues in
Low-resolution Structural Models. J Mol Biol 342:307–320. doi:
10.1016/j.jmb.2004.07.019
Sousa SF, Fernandes PA, Ramos MJ (2007) General Performance of Density Functionals. J
Phys Chem A 111:10439–10452. doi: 10.1021/jp0734474
South TL, Blake PR, Sowder RC, et al (1990) The nucleocapsid protein isolated from HIV-1
particles binds zinc and forms retroviral-type zinc fingers. Biochemistry 29:7786–7789.
doi: 10.1021/bi00486a002
Šponer J, Hobza P (1997) MP2 and CCSD(T) study on hydrogen bonding, aromatic stacking
and nonaromatic stacking. Chem Phys Lett 267:263–270. doi: 10.1016/S0009-
2614(97)00118-8
Stevens WJ, Fink WH (1987) Frozen fragment reduced variational space analysis of hydrogen
bonding interactions. Application to the water dimer. Chem Phys Lett 139:15–22. doi:
10.1016/0009-2614(87)80143-4
Stewart JJP (1989) Optimization of parameters for semiempirical methods I. Method. J
Comput Chem 10:209–220. doi: 10.1002/jcc.540100208
Stone AJ (1996) The theory of intermolecular forces. Clarendon Press, Oxford, Oxford, New
York

176
Stote RH, Karplus M (1995) Zinc binding in proteins and solution: a simple but accurate
nonbonded representation. Proteins 23:12–31. doi: 10.1002/prot.340230104
Summers MF, Henderson LE, Chance MR, et al (1992) Nucleocapsid zinc fingers detected in
retroviruses: EXAFS studies of intact viruses and the solution-state structure of the
nucleocapsid protein from HIV-1. Protein Sci a Publ Protein Soc 1:563–574. doi:
10.1002/pro.5560010502
Svensson M, Humbel S, Froese RDJ, et al (1996) ONIOM: A Multilayered Integrated MO +
MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test
for Diels−Alder Reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. J Phys Chem
100:19357–19363. doi: 10.1021/jp962071j
Swart M (2003) AddRemove: A new link model for use in QM/MM studies. Int J Quantum
Chem 91:177–183. doi: 10.1002/qua.10463
Tao J, Perdew JP, Staroverov VN, Scuseria GE (2003) Climbing the density functional
ladder: nonempirical meta-generalized gradient approximation designed for molecules
and solids. Phys Rev Lett 91:146401.
Te Velde G, Bickelhaupt FM, Baerends EJ, et al (2001) Chemistry with ADF. J Comput
Chem 22:931–967. doi: 10.1002/jcc.1056
Théry V, Rinaldi D, Rivail J-L, et al (1994) Quantum mechanical computations on very large
molecular systems: The local self-consistent field method. J Comput Chem 15:269–282.
doi: 10.1002/jcc.540150303
Théry V, Rinaldi D, Rivail J-L, et al (1994) Quantum mechanical computations on very large
molecular systems: The local self-consistent field method. J Comput Chem 15:269–282.
doi: 10.1002/jcc.540150303
Thomas LH (1927) The calculation of atomic fields. Math Proc Cambridge Philos Soc
23:542–548. doi: 10.1017/S0305004100011683
Tiraboschi G, Gresh N, Giessner-Prettre C, et al (2000) Parallel ab initio and molecular
mechanics investigation of polycoordinated Zn(II) complexes with model hard and soft
ligands: Variations of binding energy and of its components with number and charges of
ligands. J Comput Chem 21:1011–1039. doi: 10.1002/1096-
987X(200009)21:12<1011::AID-JCC1>3.0.CO;2-B
Townes CH, Schawlow AL (1975) Microwave Spectroscopy. International, Dover Publ. John
Wiley And Sons LTD
Tully JC (1990) Molecular dynamics with electronic transitions. J Chem Phys 93:1061–1071.
doi: 10.1063/1.459170
Uggerud E, Helgaker T (1992) Dynamics of the reaction CH2OH+ .fwdarw. CHO+ + H2.
Translational energy release from ab initio trajectory calculations. J Am Chem Soc
114:4265–4268. doi: 10.1021/ja00037a033

177
van der Vaart A, Merz KM (1999) Divide and Conquer Interaction Energy Decomposition. J
Phys Chem A 103:3321–3329. doi: 10.1021/jp9844967
van der Vaart A, Merz KM (1999) The Role of Polarization and Charge Transfer in the
Solvation of Biomolecules. J Am Chem Soc 121:9182–9190. doi: 10.1021/ja9912325
van Gunsteren WF, Daura X, Mark AE (2002) GROMOS Force Field. John Wiley & Sons,
Ltd,
Vedani A, Dobler M, Dunitz JD (1986) An empirical potential function for metal centers:
Application to molecular mechanics calculations on metalloproteins. J Comput Chem
7:701–710. doi: 10.1002/jcc.540070602
Vedani A, Huhta DW (1990) A new force field for modeling metalloproteins. J Am Chem
Soc 112:4759–4767. doi: 10.1021/ja00168a021
Voorhis T Van, Scuseria GE (1998) A novel form for the exchange-correlation energy
functional. J Chem Phys 109:400–410. doi: 10.1063/1.476577
Vorobyov I V., Anisimov VM, MacKerell AD (2005) Polarizable Empirical Force Field for
Alkanes Based on the Classical Drude Oscillator Model. J Phys Chem B 109:18988–
18999. doi: 10.1021/jp053182y
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation
energies for local spin density calculations: a critical analysis. Can J Phys 58:1200–1211.
doi: 10.1139/p80-159
Vreven T, Byun KS, Komáromi I, et al (2006) Combining Quantum Mechanics Methods with
Molecular Mechanics Methods in ONIOM. J Chem Theory Comput 2:815–826. doi:
10.1021/ct050289g
Wadt WR, Hay PJ (1985) Ab initio effective core potentials for molecular calculations.
Potentials for main group elements Na to Bi. J Chem Phys 82:284–298. doi:
10.1063/1.448800
Wang C, Vernon R, Lange O, et al (2010) Prediction of structures of zinc-binding proteins
through explicit modeling of metal coordination geometry. Protein Sci 19:494–506. doi:
10.1002/pro.327
Wang J, Cieplak P, Kollman PA (2000) How well does a restrained electrostatic potential
(RESP) model perform in calculating conformational energies of organic and biological
molecules? J Comput Chem 21:1049–1074. doi: 10.1002/1096-
987X(200009)21:12<1049::AID-JCC3>3.0.CO;2-F
Warshel A, Levitt M (1976) Theoretical studies of enzymic reactions: dielectric, electrostatic
and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol
103:227–249.
Warshel A, Sharma PK, Kato M, et al (2006) Electrostatic basis for enzyme catalysis. Chem
Rev 106:3210–3235. doi: 10.1021/cr0503106

178
Waszkowycz B, Hillier IH, Gensmantel N, Payling DW (1991) Combined quantum
mechanical–molecular mechanical study of catalysis by the enzyme phospholipase A2:
an investigation of the potential energy surface for amide hydrolysis. J Chem Soc Perkin
Trans 2 2025–2032. doi: 10.1039/P29910002025
Weber A (1987) Structure and Dynamics of Weakly Bound Molecular Complexes, Alfons
Web. Dordrecht
Weinstein H, Pauncz R (1968) Molecular orbital set determined by a localization procedure.
Symp Faraday Soc 2:23–31. doi: 10.1039/SF9680200023
Weinstein H, Pauncz R, Cohen M (1971) Localized Molecular Orbitals. In: D.R. Bates and
Immanuel Esterman (ed). Academic Press, pp 97–140
Wesolowski TA, Warshel A (1993) Frozen density functional approach for ab initio
calculations of solvated molecules. J Phys Chem 97:8050–8053. doi:
10.1021/j100132a040
Wiggins P, Williams JAG, Tozer DJ (2009) Excited state surfaces in density functional
theory: A new twist on an old problem. J Chem Phys 131:91101. doi:
10.1063/1.3222641
Wilcox DE (1996) Binuclear Metallohydrolases. Chem Rev 96:2435–2458. doi:
10.1021/cr950043b
Williams HL, Chabalowski CF (2001) Using Kohn−Sham Orbitals in Symmetry-Adapted
Perturbation Theory to Investigate Intermolecular Interactions. J Phys Chem A 105:646–
659. doi: 10.1021/jp003883p
Woo TK, Cavallo L, Ziegler T (1998) Implementation of the IMOMM methodology for
performing combined QM/MM molecular dynamics simulations and frequency
calculations. Theor Chem Acc 100:307–313. doi: 10.1007/s002140050391
Wu FYH, Wu CW (1987) Zinc in DNA Replication and Transcription. Annu Rev Nutr
7:251–272. doi: 10.1146/annurev.nu.07.070187.001343
Wu JC, Piquemal J-P, Chaudret R, et al (2010) Polarizable molecular dynamics simulation of
{Zn}({II}) in water using the {AMOEBA} force field. J Chem Theory Comput 6:2059–
2070. doi: 10.1021/ct100091j
Wu R, Lu Z, Cao Z, Zhang Y (2011) A Transferable Nonbonded Pairwise Force Field to
Model Zinc Interactions in Metalloproteins. J Chem Theory Comput 7:433–443. doi:
10.1021/ct100525r
Xantheas SS (1996) On the importance of the fragment relaxation energy terms in the
estimation of the basis set superposition error correction to the intermolecular interaction
energy. J Chem Phys 104:8821–8824. doi: 10.1063/1.471605

179
Xantheas SS, Jr THD (1993) Abinitio studies of cyclic water clusters (H2O)n, n=1–6. I.
Optimal structures and vibrational spectra. J Chem Phys 99:8774–8792. doi:
10.1063/1.465599
Xavier Assfeld, Nicolas Ferré, Jean-Louis Rivail (1998) The Local Self-Consistent Field
Principles and Applications to Combined Quantum Mechanical-Molecular Mechanical
Computations on Biomacromolecular Systems. American Chemical Society, pp 234–249
Xie W, Gao J (2007) Design of a Next Generation Force Field: The X-POL Potential. J Chem
Theory Comput 3:1890–1900. doi: 10.1021/ct700167b
Zhang J, Li W, Wang J, et al (2009) Protein folding simulations: From coarse-grained model
to all-atom model. IUBMB Life 61:627–643. doi: 10.1002/iub.223
Zhang J, Yang W, Piquemal J-P, Ren P (2012) Modeling Structural Coordination and Ligand
Binding in Zinc Proteins with a Polarizable Potential. J Chem Theory Comput 8:1314–
1324. doi: 10.1021/ct200812y
Zhang Y, Lee T-S, Yang W (1999) A pseudobond approach to combining quantum
mechanical and molecular mechanical methods. J Chem Phys 110:46–54. doi:
10.1063/1.478083
Zhang Y, Lin H, Truhlar DG (2007) Self-Consistent Polarization of the Boundary in the
Redistributed Charge and Dipole Scheme for Combined Quantum-Mechanical and
Molecular-Mechanical Calculations. J Chem Theory Comput 3:1378–1398. doi:
10.1021/ct7000107
Zhu T, Xiao X, Ji C, Zhang JZH (2013) A New Quantum Calibrated Force Field for Zinc–
Protein Complex. J Chem Theory Comput 9:1788–1798. doi: 10.1021/ct301091z
Ziegler T, Rauk A (1977) On the calculation of bonding energies by the Hartree Fock Slater
method. Theor Chim Acta 46:1–10. doi: 10.1007/BF00551648





























